User login
In reply: Acute liver failure
In Reply: We thank Dr. Homler for bringing hepatitis D as a potential cause of acute liver failure to our attention.
Hepatitis D virus, first described in the 1970s, requires the hepatitis B surface antigen (HBsAg) capsid to enter the hepatocyte and, thus, can only cause liver injury when the patient is also infected simultaneously with hepatitis B virus.1 Hepatitis D virus can cause either coinfection (presence of immunoglobulin M anti-HB core antibody with or without HBsAg) or superinfection (presence of HBsAg without immunoglobulin M anti-HB core antibody) with hepatitis B virus. In India, coinfection has been reported to be the cause of acute liver failure in about 4% of all patients, and superinfection in 4.5%.2
While simultaneous treatment for hepatitis D and B viruses with pegylated interferon and any of the agents used for treatment of hepatitis B has been successful in treating chronic hepatitis, it has not been proven useful in patients with acute liver failure, and liver transplant remains the only treatment option.3
- Rizzetto M. The adventure of delta. Liver Int 2016; 36(suppl 1):135–140.
- Irshad M, Acharya SK. Hepatitis D virus (HDV) infection in severe forms of liver diseases in North India. Eur J Gastroenterol Hepatol 1996; 8:995–998.
- Noureddin M, Gish R. Hepatitis delta: epidemiology, diagnosis and management 36 years after discovery. Curr Gastroenterol Rep 2014; 16:365.
In Reply: We thank Dr. Homler for bringing hepatitis D as a potential cause of acute liver failure to our attention.
Hepatitis D virus, first described in the 1970s, requires the hepatitis B surface antigen (HBsAg) capsid to enter the hepatocyte and, thus, can only cause liver injury when the patient is also infected simultaneously with hepatitis B virus.1 Hepatitis D virus can cause either coinfection (presence of immunoglobulin M anti-HB core antibody with or without HBsAg) or superinfection (presence of HBsAg without immunoglobulin M anti-HB core antibody) with hepatitis B virus. In India, coinfection has been reported to be the cause of acute liver failure in about 4% of all patients, and superinfection in 4.5%.2
While simultaneous treatment for hepatitis D and B viruses with pegylated interferon and any of the agents used for treatment of hepatitis B has been successful in treating chronic hepatitis, it has not been proven useful in patients with acute liver failure, and liver transplant remains the only treatment option.3
In Reply: We thank Dr. Homler for bringing hepatitis D as a potential cause of acute liver failure to our attention.
Hepatitis D virus, first described in the 1970s, requires the hepatitis B surface antigen (HBsAg) capsid to enter the hepatocyte and, thus, can only cause liver injury when the patient is also infected simultaneously with hepatitis B virus.1 Hepatitis D virus can cause either coinfection (presence of immunoglobulin M anti-HB core antibody with or without HBsAg) or superinfection (presence of HBsAg without immunoglobulin M anti-HB core antibody) with hepatitis B virus. In India, coinfection has been reported to be the cause of acute liver failure in about 4% of all patients, and superinfection in 4.5%.2
While simultaneous treatment for hepatitis D and B viruses with pegylated interferon and any of the agents used for treatment of hepatitis B has been successful in treating chronic hepatitis, it has not been proven useful in patients with acute liver failure, and liver transplant remains the only treatment option.3
- Rizzetto M. The adventure of delta. Liver Int 2016; 36(suppl 1):135–140.
- Irshad M, Acharya SK. Hepatitis D virus (HDV) infection in severe forms of liver diseases in North India. Eur J Gastroenterol Hepatol 1996; 8:995–998.
- Noureddin M, Gish R. Hepatitis delta: epidemiology, diagnosis and management 36 years after discovery. Curr Gastroenterol Rep 2014; 16:365.
- Rizzetto M. The adventure of delta. Liver Int 2016; 36(suppl 1):135–140.
- Irshad M, Acharya SK. Hepatitis D virus (HDV) infection in severe forms of liver diseases in North India. Eur J Gastroenterol Hepatol 1996; 8:995–998.
- Noureddin M, Gish R. Hepatitis delta: epidemiology, diagnosis and management 36 years after discovery. Curr Gastroenterol Rep 2014; 16:365.
A guide to managing acute liver failure
When the liver fails, it usually fails gradually. The sudden (acute) onset of liver failure, while less common, demands prompt management, with transfer to an intensive care unit, specific treatment depending on the cause, and consideration of liver transplant, without which the mortality rate is high.
This article reviews the definition, epidemiology, etiology, and management of acute liver failure.
DEFINITIONS
Acute liver failure is defined as a syndrome of acute hepatitis with evidence of abnormal coagulation (eg, an international normalized ratio > 1.5) complicated by the development of mental alteration (encephalopathy) within 26 weeks of the onset of illness in a patient without a history of liver disease.1 In general, patients have no evidence of underlying chronic liver disease, but there are exceptions; patients with Wilson disease, vertically acquired hepatitis B virus infection, or autoimmune hepatitis can present with acute liver failure superimposed on chronic liver disease or even cirrhosis.
The term acute liver failure has replaced older terms such as fulminant hepatic failure, hyperacute liver failure, and subacute liver failure, which were used for prognostic purposes. Patients with hyperacute liver failure (defined as development of encephalopathy within 7 days of onset of illness) generally have a good prognosis with medical management, whereas those with subacute liver failure (defined as development of encephalopathy within 5 to 26 weeks of onset of illness) have a poor prognosis without liver transplant.2,3
NEARLY 2,000 CASES A YEAR
There are nearly 2,000 cases of acute liver failure each year in the United States, and it accounts for 6% of all deaths due to liver disease.4 It is more common in women than in men, and more common in white people than in other races. The peak incidence is at a fairly young age, ie, 35 to 45 years.
CAUSES
The most common cause of acute liver failure in the United States and other Western countries is acetaminophen toxicity, followed by viral hepatitis. In contrast, viral hepatitis is the most common cause in developing countries.5
Acetaminophen toxicity
Patients with acetaminophen-induced liver failure tend to be younger than other patients with acute liver failure.1 Nearly half of them present after intentionally taking a single large dose, while the rest present with unintentional toxicity while taking acetaminophen for pain relief on a long-term basis and ingesting more than the recommended dose.6
After ingestion, 52% to 57% of acetaminophen is converted to glucuronide conjugates, and 30% to 44% is converted to sulfate conjugates. These compounds are nontoxic, water-soluble, and rapidly excreted in the urine.
However, about 5% to 10% of ingested acetaminophen is shunted to the cytochrome P450 system. P450 2E1 is the main isoenzyme involved in acetaminophen metabolism, but 1A2, 3A4, and 2A6 also contribute.7,8 P450 2E1 is the same isoenzyme responsible for ethanol metabolism and is inducible. Thus, regular alcohol consumption can increase P450 2E1 activity, setting the stage under certain circumstances for increased acetaminophen metabolism through this pathway.

Metabolism of acetaminophen through the cytochrome P450 pathway results in production of N-acetyl-p-benzoquinone imine (NAPQI), the compound that damages the liver. NAPQI is rendered nontoxic by binding to glutathione, forming NAPQI-glutathione adducts. Glutathione capacity is limited, however. With too much acetaminophen, glutathione becomes depleted and NAPQI accumulates, binds with proteins to form adducts, and leads to necrosis of hepatocytes (Figure 1).9,10
Acetylcysteine, used in treating acetaminophen toxicity, is a substrate for glutathione synthesis and ultimately increases the amount of glutathione available to bind NAPQI and prevent damage to hepatocytes.11
Acetaminophen is a dose-related toxin. Most ingestions leading to acute liver failure exceed 10 g/day (> 150 mg/kg/day). Moderate chronic ingestion, eg, 4 g/day, usually leads to transient mild elevation of liver enzymes in healthy individuals12 but can in rare cases cause acute liver failure.13

Whitcomb and Block14 retrospectively identified 49 patients who presented with acetaminophen-induced hepatotoxicity in 1987 through 1993; 21 (43%) had been taking acetaminophen for therapeutic purposes. All 49 patients took more than the recommended limit of 4 g/day, many of them while fasting and some while using alcohol. Acute liver failure was seen with ingestion of more than 12 g/day—or more than 10 g/day in alcohol users. The authors attributed the increased risk to activation of cytochrome P450 2E1 by alcohol and depletion of glutathione stores by starvation or alcohol abuse.
Advice to patients taking acetaminophen is given in Table 1.
Other drugs and supplements

A number of other drugs and herbal supplements can also cause acute liver failure (Table 2), the most common being antimicrobial and antiepileptic drugs.15 Of the antimicrobials, antitubercular drugs (especially isoniazid) are believed to be the most common causes, followed by trimethoprim-sulfamethoxazole. Phenytoin is the antiepileptic drug most often implicated in acute liver failure.
Statins can also cause acute liver failure, especially when combined with other hepatotoxic agents.16
The herbal supplements and weight-loss agents Hydroxycut and Herbalife have both been reported to cause acute liver failure, with patients presenting with either the hepatocellular or the cholestatic pattern of liver injury.17 The exact chemical in these supplements that causes liver injury has not yet been determined.
The National Institutes of Health maintains a database of cases of liver failure due to medications and supplements at livertox.nih.gov. The database includes the pattern of hepatic injury, mechanism of injury, management, and outcomes.
Viral hepatitis
Hepatitis B virus is the most common viral cause of acute liver failure and is responsible for about 8% of cases.18
Patients with chronic hepatitis B virus infection—as evidenced by positive hepatitis B surface antigen—can develop acute liver failure if the infection is reactivated by the use of immunosuppressive drugs for solid-organ or bone-marrow transplant or medications such as anti-tumor necrosis agents, rituximab, or chemotherapy. These patients should be treated prophylactically with a nucleoside analogue, which should be continued for 6 months after immunosuppressive therapy is completed.
Hepatitis A virus is responsible for about 4% of cases.18
Hepatitis C virus rarely causes acute liver failure, especially in the absence of hepatitis A and hepatitis B.3,19
Hepatitis E virus, which is endemic in areas of Asia and Africa, can cause liver disease in pregnant women and in young adults who have concomitant liver disease from another cause. It tends to cause acute liver failure more frequently in pregnant women than in the rest of the population and carries a mortality rate of more than 20% in this subgroup.
TT (transfusion-transmitted) virus was reported in the 1990s to cause acute liver failure in about 27% of patients in whom no other cause could be found.20
Other rare viral causes of acute liver failure include Epstein-Barr virus, cytomegalovirus, and herpes simplex virus types 1, 2, and 6.
Other causes
Other causes of acute liver failure include ischemic hepatitis, autoimmune hepatitis, Wilson disease, Budd-Chiari syndrome, and HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome.
MANY PATIENTS NEED LIVER TRANSPLANT

Many patients with acute liver failure ultimately require orthotopic liver transplant,21 especially if they present with severe encephalopathy. Other aspects of treatment vary according to the cause of liver failure (Table 3).
SPECIFIC MANAGEMENT
Management of acetaminophen toxicity
If the time of ingestion is known, checking the acetaminophen level can help determine the cause of acute liver failure and also predict the risk of hepatotoxicity, based on the work of Rumack and Matthew.22 Calculators are available, eg, http://reference.medscape.com/calculator/acetaminophen-toxicity.
If a patient presents with acute liver failure several days after ingesting acetaminophen, the level can be in the nontoxic range, however. In this scenario, measuring acetaminophen-protein adducts can help establish acetaminophen toxicity as the cause, as the adducts last longer in the serum and provide 100% sensitivity and specificity.23 While most laboratories can rapidly measure acetaminophen levels, only a few can measure acetaminophen-protein adducts, and thus this test is not used clinically.
Acetylcysteine is the main drug used for acetaminophen toxicity. Ideally, it should be given within 8 hours of acetaminophen ingestion, but giving it later is also useful.1
Acetylcysteine is available in oral and intravenous forms, the latter for patients who have encephalopathy or cannot tolerate oral intake due to repeated episodes of vomiting.24,25 The oral form is much less costly and is thus preferred over intravenous acetylcysteine in patients who can tolerate oral intake. Intravenous acetylcysteine should be given in a loading dose of 150 mg/kg in 5% dextrose over 15 minutes, followed by a maintenance dose of 50 mg/kg over 4 hours and then 100 mg/kg given over 16 hours.1 No dose adjustment is needed in patients who have renal toxicity (acetaminophen can also be toxic to the kidneys).
Most patients with acetaminophen-induced liver failure survive with medical management alone and do not need a liver transplant.3,26 Cirrhosis does not occur in these patients.
Management of viral acute liver failure
When patients present with acute liver failure, it is necessary to look for a viral cause by serologic testing, including hepatitis A virus IgM antibody, hepatitis B surface antigen, and hepatitis B core IgM antibody.
Hepatitis B can become reactivated in immunocompromised patients, and therefore the hepatitis B virus DNA level should be checked. Detection of hepatitis B virus DNA in a patient previously known to have undetectable hepatitis B virus DNA confirms hepatitis B reactivation.
Patients with hepatitis B-induced acute liver failure should be treated with entecavir or tenofovir. Although this treatment may not change the course of acute liver failure or accelerate the recovery, it can prevent reinfection in the transplanted liver if liver transplant becomes indicated.27–29
Herpes simplex virus should be suspected in patients presenting with anicteric hepatitis with fever. Polymerase chain reaction testing for herpes simplex virus should be done,30 and if positive, patients should be given intravenous acyclovir.31 Despite treatment, herpes simplex virus disease is associated with a very poor prognosis without liver transplant.
Autoimmune hepatitis
The autoantibodies usually seen in autoimmune hepatitis are antinuclear antibody, antismooth muscle antibody, and anti-liver-kidney microsomal antibody, and patients need to be tested for them.
The diagnosis of autoimmune hepatitis can be challenging, as these autoimmune markers can be negative in 5% of patients. Liver biopsy becomes essential to establish the diagnosis in that setting.32
Guidelines advise starting prednisone 40 to 60 mg/day and placing the patient on the liver transplant list.1
Wilson disease
Although it is an uncommon cause of liver failure, Wilson disease needs special attention because it has a poor prognosis. The mortality rate in acute liver failure from Wilson disease reaches 100% without liver transplant.
Wilson disease is caused by a genetic defect that allows copper to accumulate in the liver and other organs. However, diagnosing Wilson disease as the cause of acute liver failure can be challenging because elevated serum and urine copper levels are not specific to Wilson disease and can be seen in patients with acute liver failure from any cause. In addition, the ceruloplasmin level is usually normal or high because it is an acute-phase reactant. Accumulation of copper in the liver parenchyma is usually patchy; therefore, qualitative copper staining on random liver biopsy samples provides low diagnostic yield. Quantitative copper on liver biopsy is the gold standard test to establish the diagnosis, but the test is time-consuming. Kayser-Fleischer rings around the iris are considered pathognomic for Wilson disease when seen with acute liver failure, but they are seen in only about 50% of patients.33
A unique feature of acute Wilson disease is that most patients have very high bilirubin levels and low alkaline phosphatase levels. An alkaline phosphatase-to-bilirubin ratio less than 2 in patients with acute liver failure is highly suggestive of Wilson disease.34
Another clue to the diagnosis is that patients with Wilson disease tend to develop Coombs-negative hemolytic anemia, which leads to a disproportionate elevation in aminotransferase levels, with aspartate aminotransferase being higher than alanine aminotransferase.
Once Wilson disease is suspected, the patient should be listed for liver transplant because death is almost certain without it. For patients awaiting liver transplant, the American Association for the Study of Liver Diseases guidelines recommend certain measures to lower the serum copper level such as albumin dialysis, continuous hemofiltration, plasmapheresis, and plasma exchange,1 but the evidence supporting their use is limited.
NONSPECIFIC MANAGEMENT
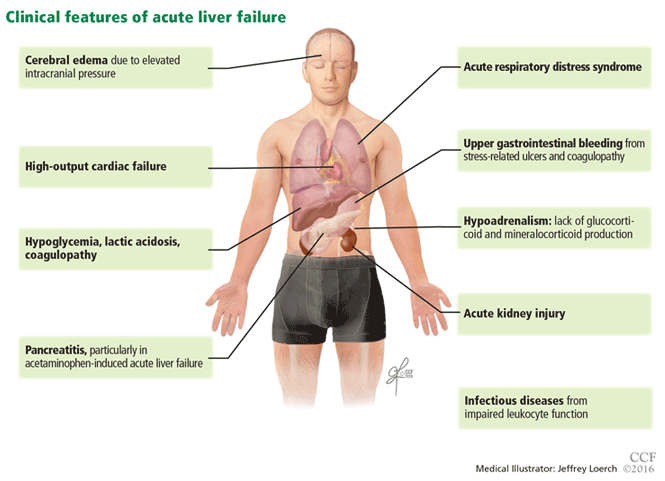
Acute liver failure can affect a number of organs and systems in addition to the liver (Figure 2).
General considerations
Because their condition can rapidly deteriorate, patients with acute liver failure are best managed in intensive care.
Patients who present to a center that does not have the facilities for liver transplant should be transferred to a transplant center as soon as possible, preferably by air. If the patient may not be able to protect the airway, endotracheal intubation should be performed before transfer.
The major causes of death in patients with acute liver failure are cerebral edema and infection. Gastrointestinal bleeding was a major cause of death in the past, but with prophylactic use of histamine H2 receptor blockers and proton pump inhibitors, the incidence of gastrointestinal bleeding has been significantly reduced.
Although initially used only in patients with acetaminophen-induced liver failure, acetylcysteine has also shown benefit in patients with acute liver failure from other causes. In patients with grade 1 or 2 encephalopathy on a scale of 0 (minimal) to 4 (comatose), the transplant-free survival rate is higher when acetylcysteine is given compared with placebo, but this benefit does not extend to patients with a higher grade of encephalopathy.35
Cerebral edema and intracranial hypertension
Cerebral edema is the leading cause of death in patients with acute liver failure, and it develops in nearly 40% of patients.36
The mechanism by which cerebral edema develops is not well understood. Some have proposed that ammonia is converted to glutamine, which causes cerebral edema either directly by its osmotic effect37,38 or indirectly by decreasing other osmolytes, thereby promoting water retention.39
Cerebral edema leads to intracranial hypertension, which can ultimately cause cerebral herniation and death. Because of the high mortality rate associated with cerebral edema, invasive devices were extensively used in the past to monitor intracranial pressure. However, in light of known complications of these devices, including bleeding,40 and lack of evidence of long-term benefit in terms of mortality rates, their use has come under debate.
Treatments. Many treatments are available for cerebral edema and intracranial hypertension. The first step is to elevate the head of the bed about 30 degrees. In addition, hyponatremia should be corrected, as it can worsen cerebral edema.41 If patients are intubated, maintaining a hypercapneic state is advisable to decrease the intracranial pressure.
Of the two pharmacologic options, mannitol is more often used.42 It is given as a bolus dose of 0.5 to 1 g/kg intravenously if the serum osmolality is less than 320 mOsm/L.1 Given the risk of fluid overload with mannitol, caution must be exercised in patients with renal dysfunction. The other pharmacologic option is 3% hypertonic saline.
Therapeutic hypothermia is a newer treatment for cerebral edema. Lowering the body temperature to 32 to 33°C (89.6 to 91.4°F) using cooling blankets decreases intracranial pressure and cerebral blood flow and improves the cerebral perfusion pressure.43 With this treatment, patients should be closely monitored for side effects of infection, coagulopathy, and cardiac arrythmias.1
l-ornithine l-aspartate was successfully used to prevent brain edema in rats, but in humans, no benefit was seen compared with placebo.44,45 The underlying basis for this experimental treatment is that supplemental ornithine and aspartate should increase glutamate synthesis, which should increase the activity of enzyme glutamine synthetase in skeletal muscles. With the increase in enzyme activity, conversion of ammonia to glutamine should increase, thereby decreasing ammonia circulation and thus decreasing cerebral edema.
Patients with cerebral edema have a high incidence of seizures, but prophylactic antiseizure medications such as phenytoin have not been proven to be beneficial.46
Infection
Nearly 80% of patients with acute liver failure develop an infectious complication, which can be attributed to a state of immunodeficiency.47
The respiratory and urinary tracts are the most common sources of infection.48 In patients with bacteremia, Enterococcus species and coagulase-negative Staphylococcus species49 are the commonly isolated organisms. Also, in patients with acute liver failure, fungal infections account for 30% of all infections.50
Infected patients often develop worsening of their encephalopathy51 without fever or elevated white blood cell count.49,52 Thus, in any patient in whom encephalopathy is worsening, an evaluation must be done to rule out infection. In these patients, systemic inflammatory response syndrome is an independent risk factor for death.53
Despite the high mortality rate with infection, whether using antibiotics prophylactically in acute liver failure is beneficial is controversial.54,55
Gastrointestinal bleeding
The current prevalence of upper gastrointestinal bleeding in acute liver failure patients is about 1.5%.56 Coagulopathy and endotracheal intubation are the main risk factors for upper gastrointestinal bleeding in these patients.57 The most common source of bleeding is stress ulcers in the stomach. The ulcers develop from a combination of factors, including decreased blood flow to the mucosa causing ischemia and hypoperfusion-reperfusion injury.
Pharmacologic inhibition of gastric acid secretion has been shown to reduce upper gastrointestinal bleeding in acute liver failure. A histamine H2 receptor blocker or proton pump inhibitor should be given to prevent gastrointestinal bleeding in patients with acute liver failure.1,58
EXPERIMENTAL TREATMENTS
Artificial liver support systems
Membranes and dialysate solutions have been developed to remove toxic substances that are normally metabolized by the liver. Two of these—the molecular adsorbent recycling system (MARS) and the extracorporeal liver assist device (ELAD)—were developed in the late 1990s. MARS consisted of a highly permeable hollow fiber membrane mixed with albumin, and ELAD consisted of porcine hepatocytes attached to microcarriers in the extracapillary space of the hollow fiber membrane. Both systems allowed for transfer of water-soluble and protein-bound toxins in the blood across the membrane and into the dialysate.59 The clinical benefit offered by these devices is controversial,60–62 thus limiting their use to experimental purposes only.
Hepatocyte transplant
Use of hepatocyte transplant as a bridge to liver transplant was tested in 1970s, first in rats and later in humans.63 By reducing the blood ammonia level and improving cerebral perfusion pressure and cardiac function, replacement of 1% to 2% of the total liver cell mass by transplanted hepatocytes acts as a bridge to orthotopic liver transplant.64,65
PROGNOSIS
Different criteria have been used to identify patients with poor prognosis who may eventually need to undergo liver transplant.
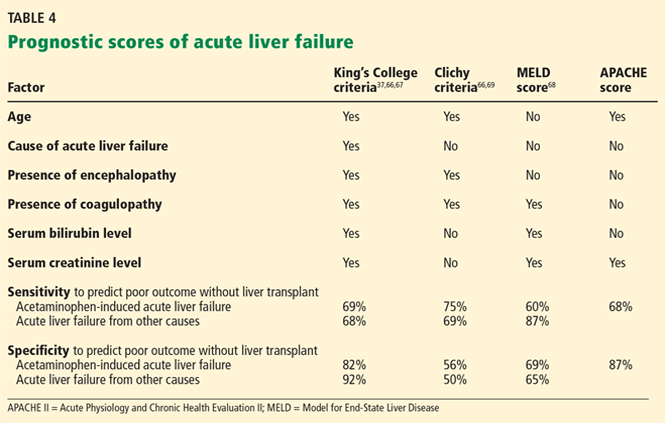
The King’s College criteria system is the most commonly used for prognosis (Table 4).37,66–69 Its main drawback is that it is applicable only in patients with encephalopathy, and when patients reach this stage, their condition often deteriorates rapidly, and they die while awaiting liver transplant.37,66,67
The Model for End-Stage Liver Disease (MELD) score is an alternative to the King’s College criteria. A high MELD score on admission signifies advanced disease, and patients with a high MELD score tend to have a worse prognosis than those with a low score.68
The Acute Physiology and Chronic Health Evaluation (APACHE) II score can also be used, as it is more sensitive than the King’s College criteria.6
The Clichy criteria66,69 can also be used.
Liver biopsy. In addition to helping establish the cause of acute liver failure, liver biopsy can also be used as a prognostic tool. Hepatocellular necrosis greater than 70% on the biopsy predicts death with a specificity of 90% and a sensitivity of 56%.70
Hypophosphatemia has been reported to indicate recovering liver function in patients with acute liver failure.71 As the liver regenerates, its energy requirement increases. To supply the energy, adenosine triphosphate production increases, and phosphorus shifts from the extracellular to the intracellular compartment to meet the need for extra phosphorus during this process. A serum phosphorus level of 2.9 mg/dL or higher appears to indicate a poor prognosis in patients with acute liver failure, as it signifies that adequate hepatocyte regeneration is not occurring.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 342:273–275.
- Ostapowicz G, Fontana RJ, Schiodt FV, et al; US Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 2008; 47:1401–1415.
- Acharya SK, Panda SK, Saxena A, Gupta SD. Acute hepatic failure in India: a perspective from the East. J Gastroenterol Hepatol 2000; 15:473–479.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Patten CJ, Thomas PE, Guy RL, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 1993; 6:511–518.
- Chen W, Koenigs LL, Thompson SJ, et al. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol 1998; 11:295-301.
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187:211–217.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 1983; 71:980–991.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- Schiødt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med 1997; 337:1112–1117.
- Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994; 272:1845–1850.
- Chalasani N, Fontana RJ, Bonkovsky HL, et al; Drug Induced Liver Injury Network (DILIN). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008; 135:1924–1934 e1–4
- Reuben A, Koch DG, Lee WM; Acute Liver Failure Study Group. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology 2010; 52:2065–2076.
- Stevens T, Qadri A, Zein NN. Two patients with acute liver injury associated with use of the herbal weight-loss supplement hydroxycut. Ann Intern Med 2005; 142:477–478.
- Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol 2015; 62(suppl 1):S112–S120.
- Schiodt FV, Davern TJ, Shakil AO, McGuire B, Samuel G, Lee WM. Viral hepatitis-related acute liver failure. Am J Gastroenterol 2003; 98:448–453.
- Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology 1998; 28:839–842.
- Bismuth H, Samuel D, Gugenheim J, et al. Emergency liver transplantation for fulminant hepatitis. Ann Intern Med 1987; 107:337–341.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Davern TJ 2nd, James LP, Hinson JA, et al; Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006; 130:687–694.
- Perry HE, Shannon MW. Efficacy of oral versus intravenous N-acetylcysteine in acetaminophen overdose: results of an open-label, clinical trial. J Pediatr 1998; 132:149–152.
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557–1562.
- Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology 1995; 109:1907–1916.
- Tsang SW, Chan HL, Leung NW, et al. Lamivudine treatment for fulminant hepatic failure due to acute exacerbation of chronic hepatitis B infection. Aliment Pharmacol Ther 2001; 15:1737–1744.
- Yu JW, Sun LJ, Yan BZ, Kang P, Zhao YH. Lamivudine treatment is associated with improved survival in fulminant hepatitis B. Liver Int 2011; 31:499–506.
- Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 2011; 53:774–780.
- Pinna AD, Rakela J, Demetris AJ, Fung JJ. Five cases of fulminant hepatitis due to herpes simplex virus in adults. Dig Dis Sci 2002; 47:750–754.
- Farr RW, Short S, Weissman D. Fulminant hepatitis during herpes simplex virus infection in apparently immunocompetent adults: report of two cases and review of the literature. Clin Infect Dis 1997; 24:1191–1194.
- Czaja AJ, Freese DK; American Association for the Study of Liver Disease. Diagnosis and treatment of autoimmune hepatitis. Hepatology 2002; 36:479–497.
- Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003; 37:1475–1492.
- Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991; 100:1129–1134.
- Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009; 137:856–864.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology 1992; 15:449–453.
- Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 1994; 107:1475–1480.
- Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet 1993; 341:157–158.
- Cordoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol 1998; 29:589–594.
- Canalese J, Gimson AE, Davis C, Mellon PJ, Davis M, Williams R. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut 1982; 23:625–629.
- Jalan R, SW OD, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 1999; 354:1164–1168.
- Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 1999; 30:636–640.
- Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology 2009; 136:2159–2168.
- Bhatia V, Batra Y, Acharya SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol 2004; 41:89–96.
- Canalese J, Gove CD, Gimson AE, Wilkinson SP, Wardle EN, Williams R. Reticuloendothelial system and hepatocytic function in fulminant hepatic failure. Gut 1982; 23:265–269.
- Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology 1990; 11:49–53.
- Rolando N, Wade JJ, Stangou A, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg 1996; 2:8–13.
- Rolando N, Harvey F, Brahm J, et al. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol 1991; 12:1–9.
- Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology 2003; 125:755–764.
- Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis 1996; 16:389–402.
- Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32:734–739.
- Rolando N, Gimson A, Wade J, Philpott- Howard J, Casewell M, Williams R. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology 1993; 17:196–201.
- Karvellas CJ, Cavazos J, Battenhouse H, et al; US Acute Liver Failure Study Group. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol 2014; 12:1942–1949.
- Acharya SK, Dasarathy S, Kumer TL, et al. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology 1996; 23:1148–1155.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- MacDougall BR, Williams R. H2-receptor antagonist in the prevention of acute gastrointestinal hemorrhage in fulminant hepatic failure: a controlled trial. Gastroenterology 1978; 74:464–465.
- Stange J, Mitzner SR, Risler T, et al. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs 1999; 23:319–330.
- Vaid A, Chewich H, Balk EM, Jaber BL. Molecular adsorbent recirculating system as artificial support therapy for liver failure: a meta-analysis. ASAIO J 2012; 58:51–59.
- Khuroo MS, Khuroo MS, Farahat KL. Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: a meta-analysis. Liver Transpl 2004; 10:1099–1106.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Sommer BG, Sutherland DE, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation for treatment of D-galactosamine-induced acute liver failure in rats. Transplant Proc 1979; 11:578–584.
- Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology 1988; 8:1006–1009.
- Strom SC, Fisher RA, Thompson MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 1997; 63:559–569.
- Pauwels A, Mostefa-Kara N, Florent C, Levy VG. Emergency liver transplantation for acute liver failure. Evaluation of London and Clichy criteria. J Hepatol 1993; 17:124–127.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King's criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Larsen FS. MELD score as a predictor of liver failure and death in patients with acetaminophen-induced liver injury. Hepatology 2007; 45:789–796.
- Bernuau J, Goudeau A, Poynard T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology 1986; 6:648–651.
- Donaldson BW, Gopinath R, Wanless IR, et al. The role of transjugular liver biopsy in fulminant liver failure: relation to other prognostic indicators. Hepatology 1993; 18:1370–1376.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
When the liver fails, it usually fails gradually. The sudden (acute) onset of liver failure, while less common, demands prompt management, with transfer to an intensive care unit, specific treatment depending on the cause, and consideration of liver transplant, without which the mortality rate is high.
This article reviews the definition, epidemiology, etiology, and management of acute liver failure.
DEFINITIONS
Acute liver failure is defined as a syndrome of acute hepatitis with evidence of abnormal coagulation (eg, an international normalized ratio > 1.5) complicated by the development of mental alteration (encephalopathy) within 26 weeks of the onset of illness in a patient without a history of liver disease.1 In general, patients have no evidence of underlying chronic liver disease, but there are exceptions; patients with Wilson disease, vertically acquired hepatitis B virus infection, or autoimmune hepatitis can present with acute liver failure superimposed on chronic liver disease or even cirrhosis.
The term acute liver failure has replaced older terms such as fulminant hepatic failure, hyperacute liver failure, and subacute liver failure, which were used for prognostic purposes. Patients with hyperacute liver failure (defined as development of encephalopathy within 7 days of onset of illness) generally have a good prognosis with medical management, whereas those with subacute liver failure (defined as development of encephalopathy within 5 to 26 weeks of onset of illness) have a poor prognosis without liver transplant.2,3
NEARLY 2,000 CASES A YEAR
There are nearly 2,000 cases of acute liver failure each year in the United States, and it accounts for 6% of all deaths due to liver disease.4 It is more common in women than in men, and more common in white people than in other races. The peak incidence is at a fairly young age, ie, 35 to 45 years.
CAUSES
The most common cause of acute liver failure in the United States and other Western countries is acetaminophen toxicity, followed by viral hepatitis. In contrast, viral hepatitis is the most common cause in developing countries.5
Acetaminophen toxicity
Patients with acetaminophen-induced liver failure tend to be younger than other patients with acute liver failure.1 Nearly half of them present after intentionally taking a single large dose, while the rest present with unintentional toxicity while taking acetaminophen for pain relief on a long-term basis and ingesting more than the recommended dose.6
After ingestion, 52% to 57% of acetaminophen is converted to glucuronide conjugates, and 30% to 44% is converted to sulfate conjugates. These compounds are nontoxic, water-soluble, and rapidly excreted in the urine.
However, about 5% to 10% of ingested acetaminophen is shunted to the cytochrome P450 system. P450 2E1 is the main isoenzyme involved in acetaminophen metabolism, but 1A2, 3A4, and 2A6 also contribute.7,8 P450 2E1 is the same isoenzyme responsible for ethanol metabolism and is inducible. Thus, regular alcohol consumption can increase P450 2E1 activity, setting the stage under certain circumstances for increased acetaminophen metabolism through this pathway.
Metabolism of acetaminophen through the cytochrome P450 pathway results in production of N-acetyl-p-benzoquinone imine (NAPQI), the compound that damages the liver. NAPQI is rendered nontoxic by binding to glutathione, forming NAPQI-glutathione adducts. Glutathione capacity is limited, however. With too much acetaminophen, glutathione becomes depleted and NAPQI accumulates, binds with proteins to form adducts, and leads to necrosis of hepatocytes (Figure 1).9,10
Acetylcysteine, used in treating acetaminophen toxicity, is a substrate for glutathione synthesis and ultimately increases the amount of glutathione available to bind NAPQI and prevent damage to hepatocytes.11
Acetaminophen is a dose-related toxin. Most ingestions leading to acute liver failure exceed 10 g/day (> 150 mg/kg/day). Moderate chronic ingestion, eg, 4 g/day, usually leads to transient mild elevation of liver enzymes in healthy individuals12 but can in rare cases cause acute liver failure.13
Whitcomb and Block14 retrospectively identified 49 patients who presented with acetaminophen-induced hepatotoxicity in 1987 through 1993; 21 (43%) had been taking acetaminophen for therapeutic purposes. All 49 patients took more than the recommended limit of 4 g/day, many of them while fasting and some while using alcohol. Acute liver failure was seen with ingestion of more than 12 g/day—or more than 10 g/day in alcohol users. The authors attributed the increased risk to activation of cytochrome P450 2E1 by alcohol and depletion of glutathione stores by starvation or alcohol abuse.
Advice to patients taking acetaminophen is given in Table 1.
Other drugs and supplements
A number of other drugs and herbal supplements can also cause acute liver failure (Table 2), the most common being antimicrobial and antiepileptic drugs.15 Of the antimicrobials, antitubercular drugs (especially isoniazid) are believed to be the most common causes, followed by trimethoprim-sulfamethoxazole. Phenytoin is the antiepileptic drug most often implicated in acute liver failure.
Statins can also cause acute liver failure, especially when combined with other hepatotoxic agents.16
The herbal supplements and weight-loss agents Hydroxycut and Herbalife have both been reported to cause acute liver failure, with patients presenting with either the hepatocellular or the cholestatic pattern of liver injury.17 The exact chemical in these supplements that causes liver injury has not yet been determined.
The National Institutes of Health maintains a database of cases of liver failure due to medications and supplements at livertox.nih.gov. The database includes the pattern of hepatic injury, mechanism of injury, management, and outcomes.
Viral hepatitis
Hepatitis B virus is the most common viral cause of acute liver failure and is responsible for about 8% of cases.18
Patients with chronic hepatitis B virus infection—as evidenced by positive hepatitis B surface antigen—can develop acute liver failure if the infection is reactivated by the use of immunosuppressive drugs for solid-organ or bone-marrow transplant or medications such as anti-tumor necrosis agents, rituximab, or chemotherapy. These patients should be treated prophylactically with a nucleoside analogue, which should be continued for 6 months after immunosuppressive therapy is completed.
Hepatitis A virus is responsible for about 4% of cases.18
Hepatitis C virus rarely causes acute liver failure, especially in the absence of hepatitis A and hepatitis B.3,19
Hepatitis E virus, which is endemic in areas of Asia and Africa, can cause liver disease in pregnant women and in young adults who have concomitant liver disease from another cause. It tends to cause acute liver failure more frequently in pregnant women than in the rest of the population and carries a mortality rate of more than 20% in this subgroup.
TT (transfusion-transmitted) virus was reported in the 1990s to cause acute liver failure in about 27% of patients in whom no other cause could be found.20
Other rare viral causes of acute liver failure include Epstein-Barr virus, cytomegalovirus, and herpes simplex virus types 1, 2, and 6.
Other causes
Other causes of acute liver failure include ischemic hepatitis, autoimmune hepatitis, Wilson disease, Budd-Chiari syndrome, and HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome.
MANY PATIENTS NEED LIVER TRANSPLANT
Many patients with acute liver failure ultimately require orthotopic liver transplant,21 especially if they present with severe encephalopathy. Other aspects of treatment vary according to the cause of liver failure (Table 3).
SPECIFIC MANAGEMENT
Management of acetaminophen toxicity
If the time of ingestion is known, checking the acetaminophen level can help determine the cause of acute liver failure and also predict the risk of hepatotoxicity, based on the work of Rumack and Matthew.22 Calculators are available, eg, http://reference.medscape.com/calculator/acetaminophen-toxicity.
If a patient presents with acute liver failure several days after ingesting acetaminophen, the level can be in the nontoxic range, however. In this scenario, measuring acetaminophen-protein adducts can help establish acetaminophen toxicity as the cause, as the adducts last longer in the serum and provide 100% sensitivity and specificity.23 While most laboratories can rapidly measure acetaminophen levels, only a few can measure acetaminophen-protein adducts, and thus this test is not used clinically.
Acetylcysteine is the main drug used for acetaminophen toxicity. Ideally, it should be given within 8 hours of acetaminophen ingestion, but giving it later is also useful.1
Acetylcysteine is available in oral and intravenous forms, the latter for patients who have encephalopathy or cannot tolerate oral intake due to repeated episodes of vomiting.24,25 The oral form is much less costly and is thus preferred over intravenous acetylcysteine in patients who can tolerate oral intake. Intravenous acetylcysteine should be given in a loading dose of 150 mg/kg in 5% dextrose over 15 minutes, followed by a maintenance dose of 50 mg/kg over 4 hours and then 100 mg/kg given over 16 hours.1 No dose adjustment is needed in patients who have renal toxicity (acetaminophen can also be toxic to the kidneys).
Most patients with acetaminophen-induced liver failure survive with medical management alone and do not need a liver transplant.3,26 Cirrhosis does not occur in these patients.
Management of viral acute liver failure
When patients present with acute liver failure, it is necessary to look for a viral cause by serologic testing, including hepatitis A virus IgM antibody, hepatitis B surface antigen, and hepatitis B core IgM antibody.
Hepatitis B can become reactivated in immunocompromised patients, and therefore the hepatitis B virus DNA level should be checked. Detection of hepatitis B virus DNA in a patient previously known to have undetectable hepatitis B virus DNA confirms hepatitis B reactivation.
Patients with hepatitis B-induced acute liver failure should be treated with entecavir or tenofovir. Although this treatment may not change the course of acute liver failure or accelerate the recovery, it can prevent reinfection in the transplanted liver if liver transplant becomes indicated.27–29
Herpes simplex virus should be suspected in patients presenting with anicteric hepatitis with fever. Polymerase chain reaction testing for herpes simplex virus should be done,30 and if positive, patients should be given intravenous acyclovir.31 Despite treatment, herpes simplex virus disease is associated with a very poor prognosis without liver transplant.
Autoimmune hepatitis
The autoantibodies usually seen in autoimmune hepatitis are antinuclear antibody, antismooth muscle antibody, and anti-liver-kidney microsomal antibody, and patients need to be tested for them.
The diagnosis of autoimmune hepatitis can be challenging, as these autoimmune markers can be negative in 5% of patients. Liver biopsy becomes essential to establish the diagnosis in that setting.32
Guidelines advise starting prednisone 40 to 60 mg/day and placing the patient on the liver transplant list.1
Wilson disease
Although it is an uncommon cause of liver failure, Wilson disease needs special attention because it has a poor prognosis. The mortality rate in acute liver failure from Wilson disease reaches 100% without liver transplant.
Wilson disease is caused by a genetic defect that allows copper to accumulate in the liver and other organs. However, diagnosing Wilson disease as the cause of acute liver failure can be challenging because elevated serum and urine copper levels are not specific to Wilson disease and can be seen in patients with acute liver failure from any cause. In addition, the ceruloplasmin level is usually normal or high because it is an acute-phase reactant. Accumulation of copper in the liver parenchyma is usually patchy; therefore, qualitative copper staining on random liver biopsy samples provides low diagnostic yield. Quantitative copper on liver biopsy is the gold standard test to establish the diagnosis, but the test is time-consuming. Kayser-Fleischer rings around the iris are considered pathognomic for Wilson disease when seen with acute liver failure, but they are seen in only about 50% of patients.33
A unique feature of acute Wilson disease is that most patients have very high bilirubin levels and low alkaline phosphatase levels. An alkaline phosphatase-to-bilirubin ratio less than 2 in patients with acute liver failure is highly suggestive of Wilson disease.34
Another clue to the diagnosis is that patients with Wilson disease tend to develop Coombs-negative hemolytic anemia, which leads to a disproportionate elevation in aminotransferase levels, with aspartate aminotransferase being higher than alanine aminotransferase.
Once Wilson disease is suspected, the patient should be listed for liver transplant because death is almost certain without it. For patients awaiting liver transplant, the American Association for the Study of Liver Diseases guidelines recommend certain measures to lower the serum copper level such as albumin dialysis, continuous hemofiltration, plasmapheresis, and plasma exchange,1 but the evidence supporting their use is limited.
NONSPECIFIC MANAGEMENT
Acute liver failure can affect a number of organs and systems in addition to the liver (Figure 2).
General considerations
Because their condition can rapidly deteriorate, patients with acute liver failure are best managed in intensive care.
Patients who present to a center that does not have the facilities for liver transplant should be transferred to a transplant center as soon as possible, preferably by air. If the patient may not be able to protect the airway, endotracheal intubation should be performed before transfer.
The major causes of death in patients with acute liver failure are cerebral edema and infection. Gastrointestinal bleeding was a major cause of death in the past, but with prophylactic use of histamine H2 receptor blockers and proton pump inhibitors, the incidence of gastrointestinal bleeding has been significantly reduced.
Although initially used only in patients with acetaminophen-induced liver failure, acetylcysteine has also shown benefit in patients with acute liver failure from other causes. In patients with grade 1 or 2 encephalopathy on a scale of 0 (minimal) to 4 (comatose), the transplant-free survival rate is higher when acetylcysteine is given compared with placebo, but this benefit does not extend to patients with a higher grade of encephalopathy.35
Cerebral edema and intracranial hypertension
Cerebral edema is the leading cause of death in patients with acute liver failure, and it develops in nearly 40% of patients.36
The mechanism by which cerebral edema develops is not well understood. Some have proposed that ammonia is converted to glutamine, which causes cerebral edema either directly by its osmotic effect37,38 or indirectly by decreasing other osmolytes, thereby promoting water retention.39
Cerebral edema leads to intracranial hypertension, which can ultimately cause cerebral herniation and death. Because of the high mortality rate associated with cerebral edema, invasive devices were extensively used in the past to monitor intracranial pressure. However, in light of known complications of these devices, including bleeding,40 and lack of evidence of long-term benefit in terms of mortality rates, their use has come under debate.
Treatments. Many treatments are available for cerebral edema and intracranial hypertension. The first step is to elevate the head of the bed about 30 degrees. In addition, hyponatremia should be corrected, as it can worsen cerebral edema.41 If patients are intubated, maintaining a hypercapneic state is advisable to decrease the intracranial pressure.
Of the two pharmacologic options, mannitol is more often used.42 It is given as a bolus dose of 0.5 to 1 g/kg intravenously if the serum osmolality is less than 320 mOsm/L.1 Given the risk of fluid overload with mannitol, caution must be exercised in patients with renal dysfunction. The other pharmacologic option is 3% hypertonic saline.
Therapeutic hypothermia is a newer treatment for cerebral edema. Lowering the body temperature to 32 to 33°C (89.6 to 91.4°F) using cooling blankets decreases intracranial pressure and cerebral blood flow and improves the cerebral perfusion pressure.43 With this treatment, patients should be closely monitored for side effects of infection, coagulopathy, and cardiac arrythmias.1
l-ornithine l-aspartate was successfully used to prevent brain edema in rats, but in humans, no benefit was seen compared with placebo.44,45 The underlying basis for this experimental treatment is that supplemental ornithine and aspartate should increase glutamate synthesis, which should increase the activity of enzyme glutamine synthetase in skeletal muscles. With the increase in enzyme activity, conversion of ammonia to glutamine should increase, thereby decreasing ammonia circulation and thus decreasing cerebral edema.
Patients with cerebral edema have a high incidence of seizures, but prophylactic antiseizure medications such as phenytoin have not been proven to be beneficial.46
Infection
Nearly 80% of patients with acute liver failure develop an infectious complication, which can be attributed to a state of immunodeficiency.47
The respiratory and urinary tracts are the most common sources of infection.48 In patients with bacteremia, Enterococcus species and coagulase-negative Staphylococcus species49 are the commonly isolated organisms. Also, in patients with acute liver failure, fungal infections account for 30% of all infections.50
Infected patients often develop worsening of their encephalopathy51 without fever or elevated white blood cell count.49,52 Thus, in any patient in whom encephalopathy is worsening, an evaluation must be done to rule out infection. In these patients, systemic inflammatory response syndrome is an independent risk factor for death.53
Despite the high mortality rate with infection, whether using antibiotics prophylactically in acute liver failure is beneficial is controversial.54,55
Gastrointestinal bleeding
The current prevalence of upper gastrointestinal bleeding in acute liver failure patients is about 1.5%.56 Coagulopathy and endotracheal intubation are the main risk factors for upper gastrointestinal bleeding in these patients.57 The most common source of bleeding is stress ulcers in the stomach. The ulcers develop from a combination of factors, including decreased blood flow to the mucosa causing ischemia and hypoperfusion-reperfusion injury.
Pharmacologic inhibition of gastric acid secretion has been shown to reduce upper gastrointestinal bleeding in acute liver failure. A histamine H2 receptor blocker or proton pump inhibitor should be given to prevent gastrointestinal bleeding in patients with acute liver failure.1,58
EXPERIMENTAL TREATMENTS
Artificial liver support systems
Membranes and dialysate solutions have been developed to remove toxic substances that are normally metabolized by the liver. Two of these—the molecular adsorbent recycling system (MARS) and the extracorporeal liver assist device (ELAD)—were developed in the late 1990s. MARS consisted of a highly permeable hollow fiber membrane mixed with albumin, and ELAD consisted of porcine hepatocytes attached to microcarriers in the extracapillary space of the hollow fiber membrane. Both systems allowed for transfer of water-soluble and protein-bound toxins in the blood across the membrane and into the dialysate.59 The clinical benefit offered by these devices is controversial,60–62 thus limiting their use to experimental purposes only.
Hepatocyte transplant
Use of hepatocyte transplant as a bridge to liver transplant was tested in 1970s, first in rats and later in humans.63 By reducing the blood ammonia level and improving cerebral perfusion pressure and cardiac function, replacement of 1% to 2% of the total liver cell mass by transplanted hepatocytes acts as a bridge to orthotopic liver transplant.64,65
PROGNOSIS
Different criteria have been used to identify patients with poor prognosis who may eventually need to undergo liver transplant.
The King’s College criteria system is the most commonly used for prognosis (Table 4).37,66–69 Its main drawback is that it is applicable only in patients with encephalopathy, and when patients reach this stage, their condition often deteriorates rapidly, and they die while awaiting liver transplant.37,66,67
The Model for End-Stage Liver Disease (MELD) score is an alternative to the King’s College criteria. A high MELD score on admission signifies advanced disease, and patients with a high MELD score tend to have a worse prognosis than those with a low score.68
The Acute Physiology and Chronic Health Evaluation (APACHE) II score can also be used, as it is more sensitive than the King’s College criteria.6
The Clichy criteria66,69 can also be used.
Liver biopsy. In addition to helping establish the cause of acute liver failure, liver biopsy can also be used as a prognostic tool. Hepatocellular necrosis greater than 70% on the biopsy predicts death with a specificity of 90% and a sensitivity of 56%.70
Hypophosphatemia has been reported to indicate recovering liver function in patients with acute liver failure.71 As the liver regenerates, its energy requirement increases. To supply the energy, adenosine triphosphate production increases, and phosphorus shifts from the extracellular to the intracellular compartment to meet the need for extra phosphorus during this process. A serum phosphorus level of 2.9 mg/dL or higher appears to indicate a poor prognosis in patients with acute liver failure, as it signifies that adequate hepatocyte regeneration is not occurring.
When the liver fails, it usually fails gradually. The sudden (acute) onset of liver failure, while less common, demands prompt management, with transfer to an intensive care unit, specific treatment depending on the cause, and consideration of liver transplant, without which the mortality rate is high.
This article reviews the definition, epidemiology, etiology, and management of acute liver failure.
DEFINITIONS
Acute liver failure is defined as a syndrome of acute hepatitis with evidence of abnormal coagulation (eg, an international normalized ratio > 1.5) complicated by the development of mental alteration (encephalopathy) within 26 weeks of the onset of illness in a patient without a history of liver disease.1 In general, patients have no evidence of underlying chronic liver disease, but there are exceptions; patients with Wilson disease, vertically acquired hepatitis B virus infection, or autoimmune hepatitis can present with acute liver failure superimposed on chronic liver disease or even cirrhosis.
The term acute liver failure has replaced older terms such as fulminant hepatic failure, hyperacute liver failure, and subacute liver failure, which were used for prognostic purposes. Patients with hyperacute liver failure (defined as development of encephalopathy within 7 days of onset of illness) generally have a good prognosis with medical management, whereas those with subacute liver failure (defined as development of encephalopathy within 5 to 26 weeks of onset of illness) have a poor prognosis without liver transplant.2,3
NEARLY 2,000 CASES A YEAR
There are nearly 2,000 cases of acute liver failure each year in the United States, and it accounts for 6% of all deaths due to liver disease.4 It is more common in women than in men, and more common in white people than in other races. The peak incidence is at a fairly young age, ie, 35 to 45 years.
CAUSES
The most common cause of acute liver failure in the United States and other Western countries is acetaminophen toxicity, followed by viral hepatitis. In contrast, viral hepatitis is the most common cause in developing countries.5
Acetaminophen toxicity
Patients with acetaminophen-induced liver failure tend to be younger than other patients with acute liver failure.1 Nearly half of them present after intentionally taking a single large dose, while the rest present with unintentional toxicity while taking acetaminophen for pain relief on a long-term basis and ingesting more than the recommended dose.6
After ingestion, 52% to 57% of acetaminophen is converted to glucuronide conjugates, and 30% to 44% is converted to sulfate conjugates. These compounds are nontoxic, water-soluble, and rapidly excreted in the urine.
However, about 5% to 10% of ingested acetaminophen is shunted to the cytochrome P450 system. P450 2E1 is the main isoenzyme involved in acetaminophen metabolism, but 1A2, 3A4, and 2A6 also contribute.7,8 P450 2E1 is the same isoenzyme responsible for ethanol metabolism and is inducible. Thus, regular alcohol consumption can increase P450 2E1 activity, setting the stage under certain circumstances for increased acetaminophen metabolism through this pathway.
Metabolism of acetaminophen through the cytochrome P450 pathway results in production of N-acetyl-p-benzoquinone imine (NAPQI), the compound that damages the liver. NAPQI is rendered nontoxic by binding to glutathione, forming NAPQI-glutathione adducts. Glutathione capacity is limited, however. With too much acetaminophen, glutathione becomes depleted and NAPQI accumulates, binds with proteins to form adducts, and leads to necrosis of hepatocytes (Figure 1).9,10
Acetylcysteine, used in treating acetaminophen toxicity, is a substrate for glutathione synthesis and ultimately increases the amount of glutathione available to bind NAPQI and prevent damage to hepatocytes.11
Acetaminophen is a dose-related toxin. Most ingestions leading to acute liver failure exceed 10 g/day (> 150 mg/kg/day). Moderate chronic ingestion, eg, 4 g/day, usually leads to transient mild elevation of liver enzymes in healthy individuals12 but can in rare cases cause acute liver failure.13
Whitcomb and Block14 retrospectively identified 49 patients who presented with acetaminophen-induced hepatotoxicity in 1987 through 1993; 21 (43%) had been taking acetaminophen for therapeutic purposes. All 49 patients took more than the recommended limit of 4 g/day, many of them while fasting and some while using alcohol. Acute liver failure was seen with ingestion of more than 12 g/day—or more than 10 g/day in alcohol users. The authors attributed the increased risk to activation of cytochrome P450 2E1 by alcohol and depletion of glutathione stores by starvation or alcohol abuse.
Advice to patients taking acetaminophen is given in Table 1.
Other drugs and supplements
A number of other drugs and herbal supplements can also cause acute liver failure (Table 2), the most common being antimicrobial and antiepileptic drugs.15 Of the antimicrobials, antitubercular drugs (especially isoniazid) are believed to be the most common causes, followed by trimethoprim-sulfamethoxazole. Phenytoin is the antiepileptic drug most often implicated in acute liver failure.
Statins can also cause acute liver failure, especially when combined with other hepatotoxic agents.16
The herbal supplements and weight-loss agents Hydroxycut and Herbalife have both been reported to cause acute liver failure, with patients presenting with either the hepatocellular or the cholestatic pattern of liver injury.17 The exact chemical in these supplements that causes liver injury has not yet been determined.
The National Institutes of Health maintains a database of cases of liver failure due to medications and supplements at livertox.nih.gov. The database includes the pattern of hepatic injury, mechanism of injury, management, and outcomes.
Viral hepatitis
Hepatitis B virus is the most common viral cause of acute liver failure and is responsible for about 8% of cases.18
Patients with chronic hepatitis B virus infection—as evidenced by positive hepatitis B surface antigen—can develop acute liver failure if the infection is reactivated by the use of immunosuppressive drugs for solid-organ or bone-marrow transplant or medications such as anti-tumor necrosis agents, rituximab, or chemotherapy. These patients should be treated prophylactically with a nucleoside analogue, which should be continued for 6 months after immunosuppressive therapy is completed.
Hepatitis A virus is responsible for about 4% of cases.18
Hepatitis C virus rarely causes acute liver failure, especially in the absence of hepatitis A and hepatitis B.3,19
Hepatitis E virus, which is endemic in areas of Asia and Africa, can cause liver disease in pregnant women and in young adults who have concomitant liver disease from another cause. It tends to cause acute liver failure more frequently in pregnant women than in the rest of the population and carries a mortality rate of more than 20% in this subgroup.
TT (transfusion-transmitted) virus was reported in the 1990s to cause acute liver failure in about 27% of patients in whom no other cause could be found.20
Other rare viral causes of acute liver failure include Epstein-Barr virus, cytomegalovirus, and herpes simplex virus types 1, 2, and 6.
Other causes
Other causes of acute liver failure include ischemic hepatitis, autoimmune hepatitis, Wilson disease, Budd-Chiari syndrome, and HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome.
MANY PATIENTS NEED LIVER TRANSPLANT
Many patients with acute liver failure ultimately require orthotopic liver transplant,21 especially if they present with severe encephalopathy. Other aspects of treatment vary according to the cause of liver failure (Table 3).
SPECIFIC MANAGEMENT
Management of acetaminophen toxicity
If the time of ingestion is known, checking the acetaminophen level can help determine the cause of acute liver failure and also predict the risk of hepatotoxicity, based on the work of Rumack and Matthew.22 Calculators are available, eg, http://reference.medscape.com/calculator/acetaminophen-toxicity.
If a patient presents with acute liver failure several days after ingesting acetaminophen, the level can be in the nontoxic range, however. In this scenario, measuring acetaminophen-protein adducts can help establish acetaminophen toxicity as the cause, as the adducts last longer in the serum and provide 100% sensitivity and specificity.23 While most laboratories can rapidly measure acetaminophen levels, only a few can measure acetaminophen-protein adducts, and thus this test is not used clinically.
Acetylcysteine is the main drug used for acetaminophen toxicity. Ideally, it should be given within 8 hours of acetaminophen ingestion, but giving it later is also useful.1
Acetylcysteine is available in oral and intravenous forms, the latter for patients who have encephalopathy or cannot tolerate oral intake due to repeated episodes of vomiting.24,25 The oral form is much less costly and is thus preferred over intravenous acetylcysteine in patients who can tolerate oral intake. Intravenous acetylcysteine should be given in a loading dose of 150 mg/kg in 5% dextrose over 15 minutes, followed by a maintenance dose of 50 mg/kg over 4 hours and then 100 mg/kg given over 16 hours.1 No dose adjustment is needed in patients who have renal toxicity (acetaminophen can also be toxic to the kidneys).
Most patients with acetaminophen-induced liver failure survive with medical management alone and do not need a liver transplant.3,26 Cirrhosis does not occur in these patients.
Management of viral acute liver failure
When patients present with acute liver failure, it is necessary to look for a viral cause by serologic testing, including hepatitis A virus IgM antibody, hepatitis B surface antigen, and hepatitis B core IgM antibody.
Hepatitis B can become reactivated in immunocompromised patients, and therefore the hepatitis B virus DNA level should be checked. Detection of hepatitis B virus DNA in a patient previously known to have undetectable hepatitis B virus DNA confirms hepatitis B reactivation.
Patients with hepatitis B-induced acute liver failure should be treated with entecavir or tenofovir. Although this treatment may not change the course of acute liver failure or accelerate the recovery, it can prevent reinfection in the transplanted liver if liver transplant becomes indicated.27–29
Herpes simplex virus should be suspected in patients presenting with anicteric hepatitis with fever. Polymerase chain reaction testing for herpes simplex virus should be done,30 and if positive, patients should be given intravenous acyclovir.31 Despite treatment, herpes simplex virus disease is associated with a very poor prognosis without liver transplant.
Autoimmune hepatitis
The autoantibodies usually seen in autoimmune hepatitis are antinuclear antibody, antismooth muscle antibody, and anti-liver-kidney microsomal antibody, and patients need to be tested for them.
The diagnosis of autoimmune hepatitis can be challenging, as these autoimmune markers can be negative in 5% of patients. Liver biopsy becomes essential to establish the diagnosis in that setting.32
Guidelines advise starting prednisone 40 to 60 mg/day and placing the patient on the liver transplant list.1
Wilson disease
Although it is an uncommon cause of liver failure, Wilson disease needs special attention because it has a poor prognosis. The mortality rate in acute liver failure from Wilson disease reaches 100% without liver transplant.
Wilson disease is caused by a genetic defect that allows copper to accumulate in the liver and other organs. However, diagnosing Wilson disease as the cause of acute liver failure can be challenging because elevated serum and urine copper levels are not specific to Wilson disease and can be seen in patients with acute liver failure from any cause. In addition, the ceruloplasmin level is usually normal or high because it is an acute-phase reactant. Accumulation of copper in the liver parenchyma is usually patchy; therefore, qualitative copper staining on random liver biopsy samples provides low diagnostic yield. Quantitative copper on liver biopsy is the gold standard test to establish the diagnosis, but the test is time-consuming. Kayser-Fleischer rings around the iris are considered pathognomic for Wilson disease when seen with acute liver failure, but they are seen in only about 50% of patients.33
A unique feature of acute Wilson disease is that most patients have very high bilirubin levels and low alkaline phosphatase levels. An alkaline phosphatase-to-bilirubin ratio less than 2 in patients with acute liver failure is highly suggestive of Wilson disease.34
Another clue to the diagnosis is that patients with Wilson disease tend to develop Coombs-negative hemolytic anemia, which leads to a disproportionate elevation in aminotransferase levels, with aspartate aminotransferase being higher than alanine aminotransferase.
Once Wilson disease is suspected, the patient should be listed for liver transplant because death is almost certain without it. For patients awaiting liver transplant, the American Association for the Study of Liver Diseases guidelines recommend certain measures to lower the serum copper level such as albumin dialysis, continuous hemofiltration, plasmapheresis, and plasma exchange,1 but the evidence supporting their use is limited.
NONSPECIFIC MANAGEMENT
Acute liver failure can affect a number of organs and systems in addition to the liver (Figure 2).
General considerations
Because their condition can rapidly deteriorate, patients with acute liver failure are best managed in intensive care.
Patients who present to a center that does not have the facilities for liver transplant should be transferred to a transplant center as soon as possible, preferably by air. If the patient may not be able to protect the airway, endotracheal intubation should be performed before transfer.
The major causes of death in patients with acute liver failure are cerebral edema and infection. Gastrointestinal bleeding was a major cause of death in the past, but with prophylactic use of histamine H2 receptor blockers and proton pump inhibitors, the incidence of gastrointestinal bleeding has been significantly reduced.
Although initially used only in patients with acetaminophen-induced liver failure, acetylcysteine has also shown benefit in patients with acute liver failure from other causes. In patients with grade 1 or 2 encephalopathy on a scale of 0 (minimal) to 4 (comatose), the transplant-free survival rate is higher when acetylcysteine is given compared with placebo, but this benefit does not extend to patients with a higher grade of encephalopathy.35
Cerebral edema and intracranial hypertension
Cerebral edema is the leading cause of death in patients with acute liver failure, and it develops in nearly 40% of patients.36
The mechanism by which cerebral edema develops is not well understood. Some have proposed that ammonia is converted to glutamine, which causes cerebral edema either directly by its osmotic effect37,38 or indirectly by decreasing other osmolytes, thereby promoting water retention.39
Cerebral edema leads to intracranial hypertension, which can ultimately cause cerebral herniation and death. Because of the high mortality rate associated with cerebral edema, invasive devices were extensively used in the past to monitor intracranial pressure. However, in light of known complications of these devices, including bleeding,40 and lack of evidence of long-term benefit in terms of mortality rates, their use has come under debate.
Treatments. Many treatments are available for cerebral edema and intracranial hypertension. The first step is to elevate the head of the bed about 30 degrees. In addition, hyponatremia should be corrected, as it can worsen cerebral edema.41 If patients are intubated, maintaining a hypercapneic state is advisable to decrease the intracranial pressure.
Of the two pharmacologic options, mannitol is more often used.42 It is given as a bolus dose of 0.5 to 1 g/kg intravenously if the serum osmolality is less than 320 mOsm/L.1 Given the risk of fluid overload with mannitol, caution must be exercised in patients with renal dysfunction. The other pharmacologic option is 3% hypertonic saline.
Therapeutic hypothermia is a newer treatment for cerebral edema. Lowering the body temperature to 32 to 33°C (89.6 to 91.4°F) using cooling blankets decreases intracranial pressure and cerebral blood flow and improves the cerebral perfusion pressure.43 With this treatment, patients should be closely monitored for side effects of infection, coagulopathy, and cardiac arrythmias.1
l-ornithine l-aspartate was successfully used to prevent brain edema in rats, but in humans, no benefit was seen compared with placebo.44,45 The underlying basis for this experimental treatment is that supplemental ornithine and aspartate should increase glutamate synthesis, which should increase the activity of enzyme glutamine synthetase in skeletal muscles. With the increase in enzyme activity, conversion of ammonia to glutamine should increase, thereby decreasing ammonia circulation and thus decreasing cerebral edema.
Patients with cerebral edema have a high incidence of seizures, but prophylactic antiseizure medications such as phenytoin have not been proven to be beneficial.46
Infection
Nearly 80% of patients with acute liver failure develop an infectious complication, which can be attributed to a state of immunodeficiency.47
The respiratory and urinary tracts are the most common sources of infection.48 In patients with bacteremia, Enterococcus species and coagulase-negative Staphylococcus species49 are the commonly isolated organisms. Also, in patients with acute liver failure, fungal infections account for 30% of all infections.50
Infected patients often develop worsening of their encephalopathy51 without fever or elevated white blood cell count.49,52 Thus, in any patient in whom encephalopathy is worsening, an evaluation must be done to rule out infection. In these patients, systemic inflammatory response syndrome is an independent risk factor for death.53
Despite the high mortality rate with infection, whether using antibiotics prophylactically in acute liver failure is beneficial is controversial.54,55
Gastrointestinal bleeding
The current prevalence of upper gastrointestinal bleeding in acute liver failure patients is about 1.5%.56 Coagulopathy and endotracheal intubation are the main risk factors for upper gastrointestinal bleeding in these patients.57 The most common source of bleeding is stress ulcers in the stomach. The ulcers develop from a combination of factors, including decreased blood flow to the mucosa causing ischemia and hypoperfusion-reperfusion injury.
Pharmacologic inhibition of gastric acid secretion has been shown to reduce upper gastrointestinal bleeding in acute liver failure. A histamine H2 receptor blocker or proton pump inhibitor should be given to prevent gastrointestinal bleeding in patients with acute liver failure.1,58
EXPERIMENTAL TREATMENTS
Artificial liver support systems
Membranes and dialysate solutions have been developed to remove toxic substances that are normally metabolized by the liver. Two of these—the molecular adsorbent recycling system (MARS) and the extracorporeal liver assist device (ELAD)—were developed in the late 1990s. MARS consisted of a highly permeable hollow fiber membrane mixed with albumin, and ELAD consisted of porcine hepatocytes attached to microcarriers in the extracapillary space of the hollow fiber membrane. Both systems allowed for transfer of water-soluble and protein-bound toxins in the blood across the membrane and into the dialysate.59 The clinical benefit offered by these devices is controversial,60–62 thus limiting their use to experimental purposes only.
Hepatocyte transplant
Use of hepatocyte transplant as a bridge to liver transplant was tested in 1970s, first in rats and later in humans.63 By reducing the blood ammonia level and improving cerebral perfusion pressure and cardiac function, replacement of 1% to 2% of the total liver cell mass by transplanted hepatocytes acts as a bridge to orthotopic liver transplant.64,65
PROGNOSIS
Different criteria have been used to identify patients with poor prognosis who may eventually need to undergo liver transplant.
The King’s College criteria system is the most commonly used for prognosis (Table 4).37,66–69 Its main drawback is that it is applicable only in patients with encephalopathy, and when patients reach this stage, their condition often deteriorates rapidly, and they die while awaiting liver transplant.37,66,67
The Model for End-Stage Liver Disease (MELD) score is an alternative to the King’s College criteria. A high MELD score on admission signifies advanced disease, and patients with a high MELD score tend to have a worse prognosis than those with a low score.68
The Acute Physiology and Chronic Health Evaluation (APACHE) II score can also be used, as it is more sensitive than the King’s College criteria.6
The Clichy criteria66,69 can also be used.
Liver biopsy. In addition to helping establish the cause of acute liver failure, liver biopsy can also be used as a prognostic tool. Hepatocellular necrosis greater than 70% on the biopsy predicts death with a specificity of 90% and a sensitivity of 56%.70
Hypophosphatemia has been reported to indicate recovering liver function in patients with acute liver failure.71 As the liver regenerates, its energy requirement increases. To supply the energy, adenosine triphosphate production increases, and phosphorus shifts from the extracellular to the intracellular compartment to meet the need for extra phosphorus during this process. A serum phosphorus level of 2.9 mg/dL or higher appears to indicate a poor prognosis in patients with acute liver failure, as it signifies that adequate hepatocyte regeneration is not occurring.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 342:273–275.
- Ostapowicz G, Fontana RJ, Schiodt FV, et al; US Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 2008; 47:1401–1415.
- Acharya SK, Panda SK, Saxena A, Gupta SD. Acute hepatic failure in India: a perspective from the East. J Gastroenterol Hepatol 2000; 15:473–479.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Patten CJ, Thomas PE, Guy RL, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 1993; 6:511–518.
- Chen W, Koenigs LL, Thompson SJ, et al. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol 1998; 11:295-301.
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187:211–217.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 1983; 71:980–991.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- Schiødt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med 1997; 337:1112–1117.
- Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994; 272:1845–1850.
- Chalasani N, Fontana RJ, Bonkovsky HL, et al; Drug Induced Liver Injury Network (DILIN). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008; 135:1924–1934 e1–4
- Reuben A, Koch DG, Lee WM; Acute Liver Failure Study Group. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology 2010; 52:2065–2076.
- Stevens T, Qadri A, Zein NN. Two patients with acute liver injury associated with use of the herbal weight-loss supplement hydroxycut. Ann Intern Med 2005; 142:477–478.
- Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol 2015; 62(suppl 1):S112–S120.
- Schiodt FV, Davern TJ, Shakil AO, McGuire B, Samuel G, Lee WM. Viral hepatitis-related acute liver failure. Am J Gastroenterol 2003; 98:448–453.
- Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology 1998; 28:839–842.
- Bismuth H, Samuel D, Gugenheim J, et al. Emergency liver transplantation for fulminant hepatitis. Ann Intern Med 1987; 107:337–341.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Davern TJ 2nd, James LP, Hinson JA, et al; Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006; 130:687–694.
- Perry HE, Shannon MW. Efficacy of oral versus intravenous N-acetylcysteine in acetaminophen overdose: results of an open-label, clinical trial. J Pediatr 1998; 132:149–152.
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557–1562.
- Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology 1995; 109:1907–1916.
- Tsang SW, Chan HL, Leung NW, et al. Lamivudine treatment for fulminant hepatic failure due to acute exacerbation of chronic hepatitis B infection. Aliment Pharmacol Ther 2001; 15:1737–1744.
- Yu JW, Sun LJ, Yan BZ, Kang P, Zhao YH. Lamivudine treatment is associated with improved survival in fulminant hepatitis B. Liver Int 2011; 31:499–506.
- Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 2011; 53:774–780.
- Pinna AD, Rakela J, Demetris AJ, Fung JJ. Five cases of fulminant hepatitis due to herpes simplex virus in adults. Dig Dis Sci 2002; 47:750–754.
- Farr RW, Short S, Weissman D. Fulminant hepatitis during herpes simplex virus infection in apparently immunocompetent adults: report of two cases and review of the literature. Clin Infect Dis 1997; 24:1191–1194.
- Czaja AJ, Freese DK; American Association for the Study of Liver Disease. Diagnosis and treatment of autoimmune hepatitis. Hepatology 2002; 36:479–497.
- Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003; 37:1475–1492.
- Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991; 100:1129–1134.
- Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009; 137:856–864.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology 1992; 15:449–453.
- Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 1994; 107:1475–1480.
- Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet 1993; 341:157–158.
- Cordoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol 1998; 29:589–594.
- Canalese J, Gimson AE, Davis C, Mellon PJ, Davis M, Williams R. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut 1982; 23:625–629.
- Jalan R, SW OD, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 1999; 354:1164–1168.
- Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 1999; 30:636–640.
- Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology 2009; 136:2159–2168.
- Bhatia V, Batra Y, Acharya SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol 2004; 41:89–96.
- Canalese J, Gove CD, Gimson AE, Wilkinson SP, Wardle EN, Williams R. Reticuloendothelial system and hepatocytic function in fulminant hepatic failure. Gut 1982; 23:265–269.
- Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology 1990; 11:49–53.
- Rolando N, Wade JJ, Stangou A, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg 1996; 2:8–13.
- Rolando N, Harvey F, Brahm J, et al. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol 1991; 12:1–9.
- Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology 2003; 125:755–764.
- Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis 1996; 16:389–402.
- Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32:734–739.
- Rolando N, Gimson A, Wade J, Philpott- Howard J, Casewell M, Williams R. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology 1993; 17:196–201.
- Karvellas CJ, Cavazos J, Battenhouse H, et al; US Acute Liver Failure Study Group. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol 2014; 12:1942–1949.
- Acharya SK, Dasarathy S, Kumer TL, et al. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology 1996; 23:1148–1155.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- MacDougall BR, Williams R. H2-receptor antagonist in the prevention of acute gastrointestinal hemorrhage in fulminant hepatic failure: a controlled trial. Gastroenterology 1978; 74:464–465.
- Stange J, Mitzner SR, Risler T, et al. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs 1999; 23:319–330.
- Vaid A, Chewich H, Balk EM, Jaber BL. Molecular adsorbent recirculating system as artificial support therapy for liver failure: a meta-analysis. ASAIO J 2012; 58:51–59.
- Khuroo MS, Khuroo MS, Farahat KL. Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: a meta-analysis. Liver Transpl 2004; 10:1099–1106.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Sommer BG, Sutherland DE, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation for treatment of D-galactosamine-induced acute liver failure in rats. Transplant Proc 1979; 11:578–584.
- Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology 1988; 8:1006–1009.
- Strom SC, Fisher RA, Thompson MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 1997; 63:559–569.
- Pauwels A, Mostefa-Kara N, Florent C, Levy VG. Emergency liver transplantation for acute liver failure. Evaluation of London and Clichy criteria. J Hepatol 1993; 17:124–127.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King's criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Larsen FS. MELD score as a predictor of liver failure and death in patients with acetaminophen-induced liver injury. Hepatology 2007; 45:789–796.
- Bernuau J, Goudeau A, Poynard T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology 1986; 6:648–651.
- Donaldson BW, Gopinath R, Wanless IR, et al. The role of transjugular liver biopsy in fulminant liver failure: relation to other prognostic indicators. Hepatology 1993; 18:1370–1376.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 342:273–275.
- Ostapowicz G, Fontana RJ, Schiodt FV, et al; US Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 2008; 47:1401–1415.
- Acharya SK, Panda SK, Saxena A, Gupta SD. Acute hepatic failure in India: a perspective from the East. J Gastroenterol Hepatol 2000; 15:473–479.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Patten CJ, Thomas PE, Guy RL, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 1993; 6:511–518.
- Chen W, Koenigs LL, Thompson SJ, et al. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol 1998; 11:295-301.
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187:211–217.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 1983; 71:980–991.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- Schiødt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med 1997; 337:1112–1117.
- Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994; 272:1845–1850.
- Chalasani N, Fontana RJ, Bonkovsky HL, et al; Drug Induced Liver Injury Network (DILIN). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008; 135:1924–1934 e1–4
- Reuben A, Koch DG, Lee WM; Acute Liver Failure Study Group. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology 2010; 52:2065–2076.
- Stevens T, Qadri A, Zein NN. Two patients with acute liver injury associated with use of the herbal weight-loss supplement hydroxycut. Ann Intern Med 2005; 142:477–478.
- Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol 2015; 62(suppl 1):S112–S120.
- Schiodt FV, Davern TJ, Shakil AO, McGuire B, Samuel G, Lee WM. Viral hepatitis-related acute liver failure. Am J Gastroenterol 2003; 98:448–453.
- Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology 1998; 28:839–842.
- Bismuth H, Samuel D, Gugenheim J, et al. Emergency liver transplantation for fulminant hepatitis. Ann Intern Med 1987; 107:337–341.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Davern TJ 2nd, James LP, Hinson JA, et al; Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006; 130:687–694.
- Perry HE, Shannon MW. Efficacy of oral versus intravenous N-acetylcysteine in acetaminophen overdose: results of an open-label, clinical trial. J Pediatr 1998; 132:149–152.
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557–1562.
- Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology 1995; 109:1907–1916.
- Tsang SW, Chan HL, Leung NW, et al. Lamivudine treatment for fulminant hepatic failure due to acute exacerbation of chronic hepatitis B infection. Aliment Pharmacol Ther 2001; 15:1737–1744.
- Yu JW, Sun LJ, Yan BZ, Kang P, Zhao YH. Lamivudine treatment is associated with improved survival in fulminant hepatitis B. Liver Int 2011; 31:499–506.
- Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 2011; 53:774–780.
- Pinna AD, Rakela J, Demetris AJ, Fung JJ. Five cases of fulminant hepatitis due to herpes simplex virus in adults. Dig Dis Sci 2002; 47:750–754.
- Farr RW, Short S, Weissman D. Fulminant hepatitis during herpes simplex virus infection in apparently immunocompetent adults: report of two cases and review of the literature. Clin Infect Dis 1997; 24:1191–1194.
- Czaja AJ, Freese DK; American Association for the Study of Liver Disease. Diagnosis and treatment of autoimmune hepatitis. Hepatology 2002; 36:479–497.
- Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003; 37:1475–1492.
- Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991; 100:1129–1134.
- Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009; 137:856–864.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology 1992; 15:449–453.
- Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 1994; 107:1475–1480.
- Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet 1993; 341:157–158.
- Cordoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol 1998; 29:589–594.
- Canalese J, Gimson AE, Davis C, Mellon PJ, Davis M, Williams R. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut 1982; 23:625–629.
- Jalan R, SW OD, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 1999; 354:1164–1168.
- Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 1999; 30:636–640.
- Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology 2009; 136:2159–2168.
- Bhatia V, Batra Y, Acharya SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol 2004; 41:89–96.
- Canalese J, Gove CD, Gimson AE, Wilkinson SP, Wardle EN, Williams R. Reticuloendothelial system and hepatocytic function in fulminant hepatic failure. Gut 1982; 23:265–269.
- Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology 1990; 11:49–53.
- Rolando N, Wade JJ, Stangou A, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg 1996; 2:8–13.
- Rolando N, Harvey F, Brahm J, et al. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol 1991; 12:1–9.
- Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology 2003; 125:755–764.
- Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis 1996; 16:389–402.
- Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32:734–739.
- Rolando N, Gimson A, Wade J, Philpott- Howard J, Casewell M, Williams R. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology 1993; 17:196–201.
- Karvellas CJ, Cavazos J, Battenhouse H, et al; US Acute Liver Failure Study Group. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol 2014; 12:1942–1949.
- Acharya SK, Dasarathy S, Kumer TL, et al. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology 1996; 23:1148–1155.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- MacDougall BR, Williams R. H2-receptor antagonist in the prevention of acute gastrointestinal hemorrhage in fulminant hepatic failure: a controlled trial. Gastroenterology 1978; 74:464–465.
- Stange J, Mitzner SR, Risler T, et al. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs 1999; 23:319–330.
- Vaid A, Chewich H, Balk EM, Jaber BL. Molecular adsorbent recirculating system as artificial support therapy for liver failure: a meta-analysis. ASAIO J 2012; 58:51–59.
- Khuroo MS, Khuroo MS, Farahat KL. Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: a meta-analysis. Liver Transpl 2004; 10:1099–1106.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Sommer BG, Sutherland DE, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation for treatment of D-galactosamine-induced acute liver failure in rats. Transplant Proc 1979; 11:578–584.
- Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology 1988; 8:1006–1009.
- Strom SC, Fisher RA, Thompson MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 1997; 63:559–569.
- Pauwels A, Mostefa-Kara N, Florent C, Levy VG. Emergency liver transplantation for acute liver failure. Evaluation of London and Clichy criteria. J Hepatol 1993; 17:124–127.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King's criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Larsen FS. MELD score as a predictor of liver failure and death in patients with acetaminophen-induced liver injury. Hepatology 2007; 45:789–796.
- Bernuau J, Goudeau A, Poynard T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology 1986; 6:648–651.
- Donaldson BW, Gopinath R, Wanless IR, et al. The role of transjugular liver biopsy in fulminant liver failure: relation to other prognostic indicators. Hepatology 1993; 18:1370–1376.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
KEY POINTS
- In the United States, the most common cause of acute liver failure is acetaminophen toxicity, followed by viral hepatitis.
- Testing for the cause of acute liver failure needs to start as soon as possible so that specific treatment can be initiated and the patient can be placed on the transplant list if needed.
- Acetylcysteine and either a proton pump inhibitor or a histamine H2 receptor blocker should be given to all patients with acute liver failure. Liver transplant is the cornerstone of therapy in patients not responding to other treatments.
- There are a number of prognostic scores for acute liver failure, but each has limitations.
Hepatitis C virus: Prevention, screening, and interpretation of assays
Screening for hepatitis C virus (HCV) infection in high-risk populations can identify, early on, people at risk of progressive liver disease who may benefit from antiviral therapy and counseling. The US Centers for Disease Control and Prevention (CDC) recommends that all people be assessed for HCV risk factors and that those with risk factors be screened for HCV antibodies (anti-HCV),1 and members of the national societies of gastroenterology and hepatology have endorsed this recommendation.2
Unfortunately, rates at which primary care patients are assessed for risk factors and the rates at which patients at higher risk are screened remain below the goals set by the CDC.3–6 All health care practitioners need to understand how to establish or exclude a diagnosis of HCV infection and to interpret the tests correctly.
WHY SCREEN FOR HCV?
HCV infection is a major public health problem and a leading cause of chronic liver disease. In the United States, an estimated 3.2 million persons (1.3% of the population) have been infected.7 However, in the inner-city primary care setting the rate of HCV infection is as high as 8%, and in Veterans Administration populations it is 17%.8,9 The worldwide prevalence of HCV infection is 2.0%, corresponding to 140 million persons.
Screening of blood products has led to a decline in the incidence of acute hepatitis C since the late 1980s, although rates have reached a plateau in recent years (Figure 1).10
Approximately 20% of patients infected with HCV develop a serious sequela, such as severe fibrosis, cirrhosis, end-stage liver disease, or hepatocellular carcinoma. Currently, HCV infection causes an estimated 8,000 to 10,000 deaths annually in the United States, and that number is predicted to triple in the next 10 to 20 years. Furthermore, HCV-related disease is the leading indication for liver transplantation in the United States, and it is estimated to cost $600 million to $1 billion annually in medical expenses and loss of work.8
Screening can reduce adverse outcomes
HCV screening has several potential benefits. By detecting HCV infection early, screening facilitates virologic suppression, as treatment earlier in the course of the disease is more effective than later.11,12 Further, early diagnosis together with patient education and subsequent lifestyle modifications may reduce the risk of transmission of HCV infection to other people.13,14
Antiviral therapy with pegylated interferons and ribavirin can cure hepatitis C in up to 90% of cases, depending on the viral genotype15–17 (see discussion of HCV genotypes below). In addition, treatment slows the progression of fibrosis.18 The incidence of hepatocellular carcinoma is lower in patients who achieve a sustained virologic response to antiviral therapy.19 Finally, antiviral therapy prolongs survival.20
New drug therapies are being developed and may, we hope, be even more effective than current drugs. Inhibitors of HCV-specific enzymes such as NS3/4 protease, combined with pegylated interferons and ribavirin, are in phase III clinical trials. These drugs are expected to be available for clinical practice within the next 2 years.21–23 Additionally, nitazoxanide (Alinia), an inducer of eIF2a and PKR phosphorylation, has been shown to increase the treatment response to HCV genotype 4. Studies24 are currently under way in patients infected with HCV genotype 1.
Screening is cost-effective
The National Hepatitis Surveillance Program25 calculated the cost of screening for HCV to be $1,246 per case detected. However, a more vigorous analysis of the same data using several different models to incorporate risk factors based on history revealed costs between $357 and $1,047 per case detected. This compares favorably with the cost of screening for other diseases that physicians routinely screen for.
Antiviral combination therapy for chronic hepatitis C has been shown to be effective in terms of quality-adjusted life-years gained and cost-effectiveness in several studies.26–28
HOW TO SCREEN
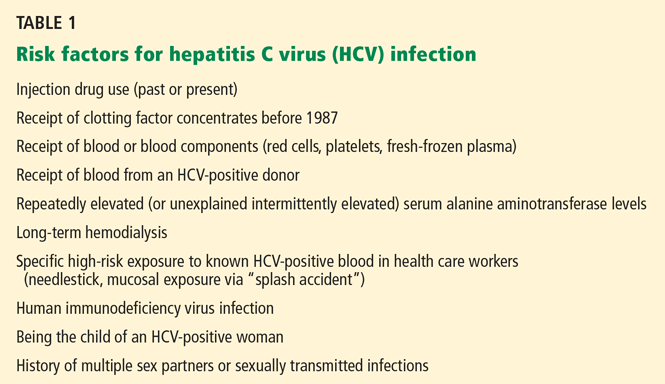
To test everyone in the general population would be neither cost-effective nor practical, which is why the CDC recommends that serologic screening for HCV infection be done only in people who have well-established risk factors for it.1,5
Therefore, screening should begin by obtaining a relevant medical history as part of a routine health evaluation. But how should this be done?
McGinn et al29 asked 1,000 patients attending an inner-city clinic to fill out a 27-item questionnaire assessing five “domains” of risk factors for HCV: work, medical, exposure, personal care, and social history. Afterward, they tested all 1,000 patients. They found that the risk factors that best predicted positive results on testing were in three domains: medical (eg, blood transfusions, dialysis, other medical procedures, and elevated liver enzymes), exposure (past contact with another person’s blood), and social history (eg, illicit drug use, incarceration, and sexual activity).
The National Hepatitis Surveillance Program25 explored the cost and yield of several screening strategies for hepatitis C, ie, testing only in patients who had a greater than 7% likelihood of infection based on an empirically derived mathematical model; testing only if significant risk factors were revealed in a simple questionnaire; or testing only if the alanine aminotransferase (ALT) level was elevated. The predictive mathematical model was the most effective and efficient means of deciding who should be tested.
Unfortunately, such a model is too cumbersome to be clinically applicable, and clinical prediction tools for HCV screening have been underused.
GROUPS AT HIGH RISK OF HCV
Groups at risk of HCV infection can be classified as being at high, intermediate, or low risk. The American Association for the Study of Liver Diseases2 rates the level of evidence for screening in all of the following risk groups as class I (ie, there is evidence or general agreement that it is beneficial, useful, and effective) and level B (ie, the data are derived from non-randomized studies).
Intravenous drug abusers
Intravenous drug abuse is the strongest independent risk factor for HCV infection.30–33 It has been the main route of HCV infection over the past decades and currently accounts for 60% of HCV transmission in the United States.7,10,34–37
Hemophilia patients treated with clotting factor concentrates produced before 1987
HCV seroprevalence is very high in patients with hemophilia who received infusions of plasma-derived clotting factor concentrates before 1987.38 In these patients, the HCV genotypes are predominantly 1 and 3, and to a lesser extent genotype 2.39,40 These genotypes likely reflect the prior exposures of the plasma donors.41 (See discussion of HCV genotypes below.) Individuals receiving clotting factor concentrates prepared from plasma pools were at high risk of HCV infection until effective procedures to inactivate viruses were introduced in 1985 (factor VIII) and 1987 (factor IX).42
People infected with HIV
About 25% of people infected with human immunodeficiency virus (HIV) in the Western world also have chronic HCV infection.43 Progression of liver disease is accelerated in HIV-HCV coinfection, and the risk of cirrhosis is twice as high.44
However, about 6% of HIV-positive patients fail to develop HCV antibodies when infected. Thus, HCV RNA should be assessed in HIV patients with unexplained liver disease who are negative for anti-HCV.45
The distribution of HCV genotypes in HIV-infected patients reflects the route of transmission. Genotype 1b accounts for 66% of posttransfusion HCV infections, while genotypes 1a and 3a are more common in intravenous drug users.
GROUPS AT INTERMEDIATE RISK OF HCV
Recipients of blood transfusions before 1992
Before 1992, blood transfusions carried a risk of HCV infection of up to 7% with each unit transfused. Prospective studies of transfusion recipients in the United States found that rates of posttransfusion hepatitis in the 1960s exceeded 20%,36 since most patients received multiple units of blood.
In the mid-1970s, before HCV had been identified, available diagnostic tests indicated that 90% of cases of posttransfusion hepatitis were not caused by hepatitis A or hepatitis B viruses. By this time, the move to all-volunteer blood donors instead of paid donors had reduced the risk of posttransfusion hepatitis to 10%.22,37,46
Although non-A, non-B hepatitis was first recognized because of its association with blood transfusion, population-based sentinel surveillance showed that it accounted for 15% to 20% of cases of community-acquired viral hepatitis in the United States.35 The advent of molecular cloning in 1988 indicated that non-A, non-B hepatitis was primarily caused by HCV.47–52
Screening of blood has reduced the rate of posttransfusion hepatitis C by a factor of about 10,000, to a current rate of 1 per million transfusions.53 The few cases that still occur are due to newly infected people donating blood before they have developed antibodies to the virus, which can take up to 8 weeks.54
Recipients of solid-organ transplants before 1992
Before organ donors were screened for HCV, recipients of solid-organ transplants from infected donors had a high risk of acquiring HCV infection. Transmission rates in different cohorts ranged from 30% to 80%.55 In an attempt to improve the safety of organ transplantation, many transplant centers now screen donors for anti-HCV and test for HCV RNA for verification.
A related problem is pre-existing HCV infection in transplant recipients. Izopet et al56 reported that, in renal transplant recipients with preexisting HCV infection, the HCV RNA titer rose about 10 times (1 log) higher after transplantation, owing to the immunosuppressive drugs that transplant recipients must take. Although this higher viral load does not affect the progression of fibrosis in all patients, the effect of immunosuppressive therapy on liver disease results in a worse outcome for some, and it reduces survival beginning in the second decade after kidney transplantation.56
Additionally, treatment of HCV infection in transplant recipients may pose a challenge, as those receiving immunosuppressive therapy with tacrolimus (Prograf) or cyclosporine (Sandimmune) may develop some degree of renal insufficiency, complicating the use of ribavirin (Rebetol) and subjecting patients to a higher risk of severe anemia. Furthermore, interferon therapy increases the risk of renal allograft rejection and, accordingly, is not often used in renal transplant recipients.
Patients with unexplained elevated aminotransferase levels
HCV infection affects an estimated 1.8% of the general population, but the rate is much higher in people with ALT levels over 40 U/L. Most patients with chronic hepatitis C have no symptoms or only mild symptoms and minimally elevated levels of ALT and aspartate aminotransferase (AST)—ie, two to five times higher than the upper limit of normal.
The first step in the workup of aminotransferase elevations is to confirm the abnormality by repeating the blood test. If an elevation is confirmed, further investigation is warranted. A directed history and physical examination is important and may disclose risk factors, raising clinical suspicion of a particular disease.
Some caveats: The proportion of patients with HCV viremia who have abnormally high aminotransferase levels ranges between only 54% and 66%.57–59 In patients with risk factors for HCV infection and abnormal liver enzyme levels, HCV infection is probable but not certain. Also, liver enzyme tests do not reveal the extent of hepatic injury or reflect the true status of hepatic function.60
Infants born to infected mothers
Children born to HCV-positive women should be tested for anti-HCV no sooner than age 12 months, when passively transferred maternal anti-HCV declines below detectable levels. If earlier diagnosis of HCV infection is desired, a real-time polymerase chain reaction (PCR) test for HCV RNA can be done at or after the infant's first “well-child” visit at age 1 to 2 months.
If positive for either anti-HCV or HCV RNA, children should be evaluated for liver disease, and those with persistently elevated ALT levels should be referred to a specialist for medical management.2,5
GROUPS AT LOW RISK OF HCV
People who have had sexual relations with multiple or infected partners
Sexual activity is associated with a low but measurable risk of transmission of HCV. Large population-based studies, including the National Hepatitis Surveillance Program,25 found an independent association between HCV infection and having sexual relations with multiple partners or with a partner who is infected with HCV.
The CDC reported that 15% to 20% of patients with acute hepatitis C had a history of sexual exposure but no other risk factors. Two-thirds of them had an anti-HCV-positive sexual partner, and one-third reported having had more than two partners in the 6 months before illness.5
More data are needed to determine the risk of and the factors related to transmission of HCV between long-term steady partners as well as in persons with high-risk sexual practices, including whether other sexually transmitted diseases promote transmission of HCV by influencing viral load or modifying mucosal barriers.
Health care workers exposed to HCV, eg, by needlestick
The prevalence of HCV infection in health care workers is no greater than that in the general population, averaging 1% to 2%, and is actually 10 times lower than that of hepatitis B virus infection.47,48,61,62
However, within the disciplines, some groups have a higher prevalence of HCV infection, suggesting that some occupations carry a higher risk. In two US studies, the prevalence of HCV infection was higher in oral surgeons (2.0% and 9.3%) than in other dentists (0.7% and 0.97%).63,64
In a single study that evaluated risk factors for infection, a history of needlestick injury was the only occupational risk factor that was independently associated with HCV infection.65 The average incidence of anti-HCV seroconversion after a needlestick or after an injury with a sharp object contaminated by an HCV-positive source is 1.8% (range 0%–7%).66–69
Although no studies of incidence have documented transmission via mucous membrane or nonintact skin exposures, transmission of HCV from blood splashes to the conjunctiva have been described.70,71

It is worth noting that exposure to blood from unclean needles used in tattooing or body piercing also confers a risk of HCV infection.
SEROLOGIC SCREENING TESTS FOR HCV
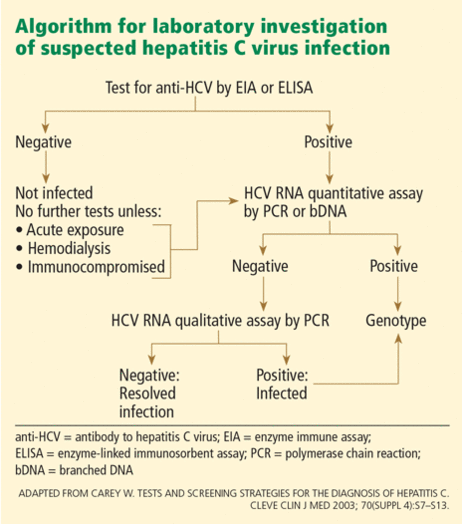
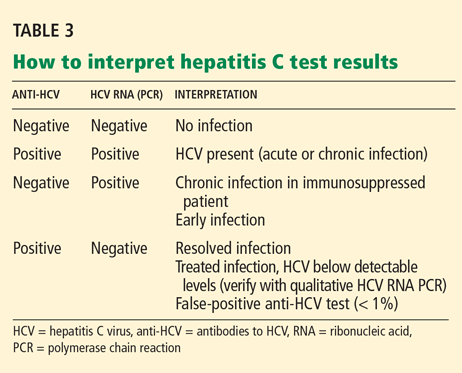
- Serologic assays that detect specific antibody to HCV (anti-HCV)
- Molecular assays that detect viral RNA.
Initial serologic screening tests for anti-HCV
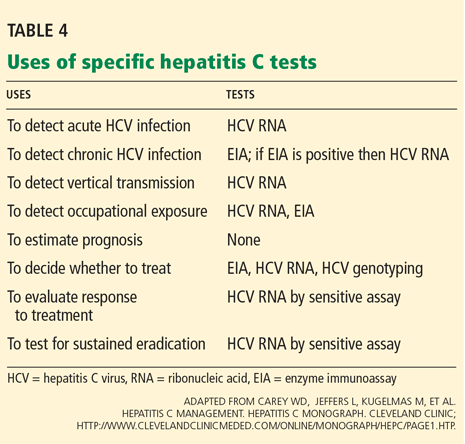
Two EIAs are approved for clinical use:
- Abbott HCV EIA 2.0 (Abbott Laboratories, Abbott Park, IL)
- Ortho HCV Version 3.0 enzyme-linked immunosorbent assay (ELISA) (Ortho-Clinical Diagnostics, Rochester, NY).
One enhanced chemiluminescence immunoassay is also approved:
- Vitros Anti-HCV assay (Ortho-Clinical Diagnostics). In practical terms, this test is equivalent to the two EIAs, and the discussion below about EIAs applies to this test as well.
These third-generation tests are highly sensitive (> 99%) and specific (99%) in immunocompetent patients, and eliminate the need for a confirmatory immunoblot assay in patients with clinical liver disease, particularly those with risk factors for HCV infection.
False-positive results are rare now, but they were common with earlier generations of these assays. Most false-positive results occur in patients with autoimmune liver disease or hypergammaglobulinemia who have normal liver enzyme levels and no risk factors for HCV infection. In fact, all positive anti-HCV results should be followed up with an HCV RNA test.
False-negative results are also uncommon, usually occurring only in immunosuppressed patients (eg, organ transplant recipients and HIV-positive patients) and in patients on long-term hemodialysis. Therefore, patients with a history of hemodialysis should be considered for an HCV RNA assay rather than an EIA. Measurement of ALT will not be useful because ALT levels are lower in patients with end-stage renal disease. In most other clinical situations, the HCV EIA is an outstanding screening test for HCV infection because of its high sensitivity and relatively low cost (< $50).
Although the specificity of these tests is good, the predictive value of a positive result varies substantially by the pretest probability of HCV infection. For example, in a group of injection-drug users who are very likely to have ongoing or remote infection, all positive HCV EIA results are likely truly positive.74 On the other hand, in healthy blood donors, up to half of all positive third-generation EIA tests are falsely positive.75
Important points
- A positive anti-HCV antibody test does not distinguish acute from chronic disease or active from past infection, nor is it a sign of immunity or protection.
- A positive anti-HCV EIA requires HCV RNA measurement to discriminate between current infection on the one hand, and either resolved HCV infection or a false-positive result on the other.
- A positive EIA anti-HCV test is a marker that hepatitis C may be present, and it must be followed by confirmatory HCV RNA testing.
- Physicians should be mindful of the potential tribulations associated with false-positive tests. A false-positive test may result in harm to patients that is difficult to measure, such as anxiety, labeling in the medical record, and detrimental effects on close relationships.
CONFIRMATORY TESTING WITH ASSAYS FOR HCV RNA
As stated above, a positive result on an anti-HCV EIA needs to be confirmed with an assay for HCV RNA, of which there are two types, ie, qualitative and quantitative.
Each involves trade-offs. Qualitative assays are more sensitive and detect more cases, but they provide no information about the amount of virus (viral load). Quantitative assays are less sensitive, so a negative result does not completely exclude hepatitis C, although they can still can detect 95% of cases. They do, however, measure the viral load.
Therefore, the type of test to use depends on the patient’s risk profile, the goals of testing, and the setting in which future care will be provided. The primary objective when a patient has a positive EIA test is to determine whether he or she has ongoing infection, a goal most expeditiously achieved using a qualitative assay. However, since a quantitative assay can detect the vast majority of cases of active HCV infection, many clinicians select this as the test of first choice when the probability of HCV is high (eg, in a patient with risk factors and abnormal liver tests). If the pretest probability is low, a qualitative assay is the better choice.
Many commercial assays are available for detecting (qualitative assays) or measuring (quantitative assays) HCV RNA.
Qualitative HCV RNA assays
The approved qualitative assays are:
- Amplicor HCV Test, version 2.0 (Roche Molecular Diagnostics, Pleasanton, CA)
- Cobas Amplicor HCV Test, version 2.0 (Roche Molecular Diagnostics)
- Ampliscreen (Roche Molecular Diagnostics)
- Versant HCV RNA Qualitative Assay (Siemens Healthcare Diagnostics, Deerfield, IL)
- Procleix HIV-1/HCV Assay (Chiron, Emeryville, CA).
Quantitative HCV RNA assays
The approved quantitative assays are:
- Amplicor HCV Monitor (Roche Molecular Diagnostics)
- Cobas Amplicor HCV Monitor, version 2.0 (Roche Molecular Diagnostics)
- Versant HCV RNA 3.0 Assay (bDNA) (Siemens Healthcare Diagnostics)
- Cobas Taqman HCV Test (Roche Molecular Diagnostics).
Quantitative tests use target amplification with PCR, transcription-mediated amplification (TMA), or a signal amplification technique such as a branched DNA (bDNA) assay. The sensitivity varies for different types of amplification. TMA assays appear to be the most sensitive for detecting HCV RNA.
The latest innovation is real-time PCR, which shortens the typical time for PCR processing from 1.5 hours to 35 minutes. It may also detect relapsed HCV infection earlier than regular PCR. With the recent availability of real-time PCR assays, which have sensitivities of 10 to 50 IU/mL, many experts feel there is no longer a need for qualitative assays.74 In fact, many laboratories no longer offer qualitative testing. The Cleveland Clinic laboratory has recently stopped offering this test.
Because RNA testing is widely available, the recombinant immunoblot assay (RIBA) has become obsolete in diagnosing HCV infection, except in special circumstances. Currently, the primary purpose of RIBA testing is to distinguish between resolved HCV infection (EIA-positive, HCV RNA-negative, RIBA-positive) and a false-positive EIA (EIA-positive, HCV RNA-negative, RIBA-negative).
In summary, patients suspected of having acute or chronic HCV infection should first be tested for anti-HCV. Subsequently, HCV RNA testing should be performed in:
- Patients with a positive anti-HCV test
- Patients for whom antiviral treatment is being considered (using a sensitive quantitative assay)
- Patients with unexplained liver disease whose anti-HCV test is negative and who are immunocompromised or suspected of having acute HCV infection.
Significance of the HCV viral load
The significance of the HCV viral load is widely misunderstood. The amount of virus in the blood does not correlate with symptoms, histologic liver injury, or the stage or aggressiveness of disease. Its sole importance is in relation to therapy.
The HCV viral load, measured before treatment, helps predict the likelihood of a treatment response: the lower the pretreatment viral load, the more likely that the patient will respond to current HCV therapies.
Additionally, the pretreatment viral load serves as a baseline for comparison with subsequent measurements during treatment. Patients with HCV genotype 1 who do not achieve more than a 2-log (99%) reduction in viral load by the 12th week of treatment (an early virologic response) have a low response rate, and treatment should generally be stopped, given its cost and side effects.76 However, measuring the viral load to detect an early virologic response is less helpful in patients with HCV genotype 2 or 3 infection, since these patients require only 24 weeks of therapy and most of them clear the virus by week 12 and respond to therapy.
Additionally, patients with genotype 2 or 3 and those with a viral load of less than 600,000 IU/mL have been found to achieve higher rates of sustained virologic response.15 A sustained virologic response is defined as the absence of HCV RNA 24 weeks after stopping treatment and is now considered to be the best predictor of long-term treatment response. A sustained virologic response is generally regarded as a “virologic cure.”
HCV GENOTYPE AFFECTS SUCCESS AND DURATION OF TREATMENT
HCV has at least six major genotypes.1,3–6 Several genotypes are subclassified as “a” or “b” (ie, genotype 1a or 1b); however, these distinctions are of little clinical use.
In the laboratory, HCV genotypes are identified by restriction fragment length polymorphism, by direct sequence analysis, or by reverse hybridization. Once the HCV genotype has been identified, there is no need to repeat the test.
Different genotypes are more common in some areas of the world than in others. Genotype 1 is the one most common in the United States (accounting for 70% to 75% of cases), followed by genotypes 2 and 3 (25%–30%). Genotype 4 is most common in Egypt and the Arabian peninsula.
HCV genotyping is important because it can help predict the likelihood of a response to treatment and in planning the dose and duration of therapy.77 For example, treatment with pegylated interferon plus ribavirin is predicted to work approximately 50% of the time for people with genotype 1, but 80% to 90% of the time for people with genotypes 2 or 3.15–17,78 Additionally, patients with genotype 1 need 12 months of therapy to achieve maximum benefit, whereas those with genotypes 2 and 3 require treatment for only 6 months to achieve maximum benefit.
- Alter MJ, Seeff LB, Bacon BR, Thomas DL, Rigsby MO, Di Bisceglie AM. Testing for hepatitis C virus infection should be routine for persons at increased risk for infection. Ann Intern Med 2004; 141:715–717.
- Ghany MG, Strader DB, Thomas DL, Seeff LB; American Association for the Study of Liver Diseases. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology 2009; 49:1335–1374.
- Shehab TM, Orrego M, Chunduri R, Lok AS. Identification and management of hepatitis C patients in primary care clinics. Am J Gastroenterol 2003; 98:639–644.
- Shehab TM, Sonnad SS, Lok AS. Management of hepatitis C patients by primary care physicians in the USA: results of a national survey. J Viral Hepat 2001; 8:377–383.
- US Centers for Disease Control and Prevention. Recommendations for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. MMWR Recomm Rep 1998; 47:1–39.
- US Centers for Disease Control and Prevention. National prevention strategy: a comprehensive strategy for the prevention and control of hepatitis C virus infection and its consequences; summer 2001. http://www.cdc.gov/hepatitis/HCV/Strategy/NatHep-CPrevStrategy.htm. Accessed August 8, 2010.
- Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med 2006; 144:705–714.
- Kim WR. The burden of hepatitis C in the United States. Hepatology 2002; 36(suppl 1):S30–S34.
- Lau DT, Kleiner DE, Ghany MG, Park Y, Schmid P, Hoofnagle JH. 10-Year follow-up after interferon-alpha therapy for chronic hepatitis C. Hepatology 1998; 28:1121–1127.
- Daniels D, Grytdal S, Wasley A; US Centers for Disease Control and Prevention. Surveillance for acute viral hepatitis—United States, 2007. MMWR Surveill Summ 2009; 58:1–27.
- Thomson BJ, Kwong G, Ratib S, et al; Trent HCV Study Group. Response rates to combination therapy for chronic HCV infection in a clinical setting and derivation of probability tables for individual patient management. J Viral Hepat 2008; 15:271–278.
- Hayashi N, Takehara T. Antiviral therapy for chronic hepatitis C: past, present, and future. J Gastroenterol 2006; 41:17–27.
- Gordon FD. Cost-effectiveness of screening patients for hepatitis C. Am J Med 1999; 107:36S–40S.
- Hill L, Henry B, Schweikert S; Prevention Practice Committee, American College of Preventive Medicine. Screening for chronic hepatitis C: American College of Preventive Medicine practice policy statement. Am J Prev Med 2005; 28:327–330.
- Hadziyannis SJ, Sette H, Morgan TR, et al; PEGASYS International Study Group. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann Intern Med 2004; 140:346–355.
- Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 2001; 358:958–965.
- Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 2002; 347:975–982.
- Cammà C, Di Bona D, Schepis F, et al. Effect of peginterferon alfa-2a on liver histology in chronic hepatitis C: a meta-analysis of individual patient data. Hepatology 2004; 39:333–342.
- Yoshida H, Tateishi R, Arakawa Y, et al. Benefit of interferon therapy in hepatocellular carcinoma prevention for individual patients with chronic hepatitis C. Gut 2004; 53:425–430.
- Yoshida H, Arakawa Y, Sata M, et al. Interferon therapy prolonged life expectancy among chronic hepatitis C patients. Gastroenterology 2002; 123:483–491.
- Hézode C, Forestier N, Dusheiko G, et al; PROVE2 Study Team. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N Engl J Med 2009; 360:1839–1850.
- McHutchison JG, Everson GT, Gordon SC, et al; PROVE1 Study Team. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009; 360:1827–1838.
- Berman K, Kwo PY. Boceprevir, an NS3 protease inhibitor of HCV. Clin Liver Dis 2009; 13:429–439.
- Rossignol JF, Elfert A, Keeffe EB. Treatment of chronic hepatitis C using a 4-week lead-in with nitazoxanide before peginterferon plus nitazoxanide. J Clin Gastroenterol 2009 Dec 31; epub ahead of print.
- Lapane KL, Jakiche AF, Sugano D, Weng CS, Carey WD. Hepatitis C infection risk analysis: who should be screened? Comparison of multiple screening strategies based on the National Hepatitis Surveillance Program. Am J Gastroenterol 1998; 93:591–596.
- Wong JB, Davis GL, McHutchison JG, Manns MP, Albrecht JK; International Hepatitis Interventional Therapy Group. Economic and clinical effects of evaluating rapid viral response to peginterferon alfa-2b plus ribavirin for the initial treatment of chronic hepatitis C. Am J Gastroenterol 2003; 98:2354–2362.
- Salomon JA, Weinstein MC, Hammitt JK, Goldie SJ. Cost-effectiveness of treatment for chronic hepatitis C infection in an evolving patient population. JAMA 2003; 290:228–237.
- Sullivan SD, Jensen DM, Bernstein DE, et al. Cost-effectiveness of combination peginterferon alpha-2a and ribavirin compared with interferon alpha-2b and ribavirin in patients with chronic hepatitis C. Am J Gastroenterol 2004; 99:1490–1496.
- McGinn T, O’Connor-Moore N, Alfandre D, Gardenier D, Wisnivesky J. Validation of a hepatitis C screening tool in primary care. Arch Intern Med 2008; 168:2009–2013.
- Kaur S, Rybicki L, Bacon BR, Gollan JL, Rustgi VK, Carey WD. Performance characteristics and results of a large-scale screening program for viral hepatitis and risk factors associated with exposure to viral hepatitis B and C: results of the National Hepatitis Screening Survey. National Hepatitis Surveillance Group. Hepatology 1996; 24:979–986.
- Cheung RC. Epidemiology of hepatitis C virus infection in American veterans. Am J Gastroenterol 2000; 95:740–747.
- Austin GE, Jensen B, Leete J, et al. Prevalence of hepatitis C virus seropositivity among hospitalized US veterans. Am J Med Sci 2000; 319:353–359.
- Yawn BP, Wollan P, Gazzuola L, Kim WR. Diagnosis and 10-year follow-up of a community-based hepatitis C cohort. J Fam Pract 2002; 51:135–140.
- Garfein RS, Doherty MC, Monterroso ER, Thomas DL, Nelson KE, Vlahov D. Prevalence and incidence of hepatitis C virus infection among young adult injection drug users. J Acquir Immune Defic Syndr Hum Retrovirol 1998; 18(suppl 1):S11–S19.
- Alter MJ. The epidemiology of acute and chronic hepatitis C. Clin Liver Dis 1997; 1:559–568,
- Alter MJ, Hadler SC, Judson FN, et al. Risk factors for acute non-A, non-B hepatitis in the United States and association with hepatitis C virus infection. JAMA 1990; 264:2231–2235.
- Wasley A, Miller JT, Finelli L; Centers for Disease Control and Prevention (CDC). Surveillance for acute viral hepatitis—United States, 2005. MMWR Surveill Summ 2007; 56:1–24.
- Goedert JJ, Chen BE, Preiss L, Aledort LM, Rosenberg PS. Reconstruction of the hepatitis C virus epidemic in the US hemophilia population, 1940–1990. Am J Epidemiol 2007; 165:1443–1453.
- Eyster ME, Sherman KE, Goedert JJ, Katsoulidou A, Hatzakis A. Prevalence and changes in hepatitis C virus genotypes among multitransfused persons with hemophilia. The Multicenter Hemophilia Cohort Study. J Infect Dis 1999; 179:1062–1069.
- Yee TT, Griffioen A, Sabin CA, Dusheiko G, Lee CA. The natural history of HCV in a cohort of haemophilic patients infected between 1961 and 1985. Gut 2000; 47:845–851.
- Lee C, Dusheiko G. The natural history and antiviral treatment of hepatitis C in haemophilia. Haemophilia 2002; 8:322–329.
- Makris M, Garson JA, Ring CJ, Tuke PW, Tedder RS, Preston FE. Hepatitis C viral RNA in clotting factor concentrates and the development of hepatitis in recipients. Blood 1993; 81:1898–1902.
- Sherman KE, Rouster SD, Chung RT, Rajicic N. Hepatitis C virus prevalence among patients infected with human immunodeficiency virus: a cross-sectional analysis of the US adult AIDS Clinical Trials Group. Clin Infect Dis 2002; 34:831–837.
- Sulkowski MS. The HIV-coinfected patient: managing viral hepatitis. J Acquir Immune Defic Syndr 2007; 45(suppl 2):S36–S37.
- Bonacini M, Lin HJ, Hollinger FB. Effect of coexisting HIV-1 infection on the diagnosis and evaluation of hepatitis C virus. J Acquir Immune Defic Syndr 2001; 26:340–344.
- Garfein RS, Vlahov D, Galai N, Doherty MC, Nelson KE. Viral infections in short-term injection drug users: the prevalence of the hepatitis C, hepatitis B, human immunodeficiency, and human T-lymphotropic viruses. Am J Public Health 1996; 86:655–661.
- Bell J, Batey RG, Farrell GC, Crewe EB, Cunningham AL, Byth K. Hepatitis C virus in intravenous drug users. Med J Aust 1990; 153:274–276.
- Villano SA, Vlahov D, Nelson KE, Lyles CM, Cohn S, Thomas DL. Incidence and risk factors for hepatitis C among injection drug users in Baltimore, Maryland. J Clin Microbiol 1997; 35:3274–3277.
- Patrick DM, Tyndall MW, Cornelisse PG, et al. Incidence of hepatitis C virus infection among injection drug users during an outbreak of HIV infection. CMAJ 2001; 165:889–895.
- Seeff LB, Wright EC, Zimmerman HJ, McCollum RW. VA cooperative study of post-transfusion hepatitis, 1969-1974: incidence and characteristics of hepatitis and responsible risk factors. Am J Med Sci 1975; 270:355–362.
- Feinstone SM, Kapikian AZ, Purcell RH, Alter HJ, Holland PV. Transfusion-associated hepatitis not due to viral hepatitis type A or B. N Engl J Med 1975; 292:767–770.
- Alter HJ, Holland PV, Purcell RH, et al. Posttransfusion hepatitis after exclusion of commercial and hepatitis-B antigen-positive donors. Ann Intern Med. 1972; 77:691–699.
- Blajchman MA, Vamvakas EC. The continuing risk of transfusion-transmitted infections. N Engl J Med 2006; 355:1303–1305.
- Lauer GM, Walker BD. Hepatitis C virus infection. N Engl J Med 2001; 345:41–52.
- Roth D, Zucker K, Cirocco R, et al. The impact of hepatitis C virus infection on renal allograft recipients. Kidney Int 1994; 45:238–244.
- Izopet J, Rostaing L, Sandres K, et al. Longitudinal analysis of hepatitis C virus replication and liver fibrosis progression in renal transplant recipients. J Infect Dis 2000; 181:852–858.
- Dubois F, Desenclos JC, Mariotte N, Goudeau A. Hepatitis C in a French population-based survey, 1994: seroprevalence, frequency of viremia, genotype distribution, and risk factors. The Collaborative Study Group. Hepatology 1997; 25:1490–1496.
- Bellentani S, Pozzato G, Saccoccio G, et al. Clinical course and risk factors of hepatitis C virus related liver disease in the general population: report from the Dionysos study. Gut 1999; 44:874–880.
- Alberti A, Noventa F, Benvegnù L, Boccato S, Gatta A. Prevalence of liver disease in a population of asymptomatic persons with hepatitis C virus infection. Ann Intern Med 2002; 137:961–964.
- Shiffman ML, Diago M, Tran A, et al. Chronic hepatitis C in patients with persistently normal alanine transaminase levels. Clin Gastroenterol Hepatol 2006; 4:645–652.
- Stary A, Kopp W, Hofmann H, Heller-Vitouch C, Kunz C. Seroepidemiologic study of hepatitis C virus in sexually transmitted disease risk groups. Sex Transm Dis 1992; 19:252–258.
- Weinstock HS, Bolan G, Reingold AL, Polish LB. Hepatitis C virus infection among patients attending a clinic for sexually transmitted diseases. JAMA 1993; 269:392–394.
- Thomas DL, Gruninger SE, Siew C, Joy ED, Quinn TC. Occupational risk of hepatitis C infections among general dentists and oral surgeons in North America. Am J Med 1996; 100:41–45.
- Klein RS, Freeman K, Taylor PE, Stevens CE. Occupational risk for hepatitis C virus infection among New York City dentists. Lancet 1991; 338:1539–1542.
- Polish LB, Tong MJ, Co RL, Coleman PJ, Alter MJ. Risk factors for hepatitis C virus infection among health care personnel in a community hospital. Am J Infect Control 1993; 21:196–200.
- Alter MJ. Occupational exposure to hepatitis C virus: a dilemma. Infect Control Hosp Epidemiol 1994; 15:742–744.
- Lanphear BP, Linnemann CC, Cannon CG, DeRonde MM, Pendy L, Kerley LM. Hepatitis C virus infection in healthcare workers: risk of exposure and infection. Infect Control Hosp Epidemiol 1994; 15:745–750.
- Puro V, Petrosillo N, Ippolito G. Risk of hepatitis C seroconversion after occupational exposures in health care workers. Italian Study Group on Occupational Risk of HIV and Other Bloodborne Infections. Am J Infect Control 1995; 23:273–277.
- Mitsui T, Iwano K, Masuko K, et al. Hepatitis C virus infection in medical personnel after needlestick accident. Hepatology 1992; 16:1109–1114.
- Sartori M, La Terra G, Aglietta M, Manzin A, Navino C, Verzetti G. Transmission of hepatitis C via blood splash into conjunctiva. Scand J Infect Dis 1993; 25:270–271.
- Ippolito G, Puro V, Petrosillo N, De Carli G, Micheloni G, Magliano E. Simultaneous infection with HIV and hepatitis C virus following occupational conjunctival blood exposure. JAMA 1998; 280:28.
- Carey W. Tests and screening strategies for the diagnosis of hepatitis C. Cleve Clin J Med 2003; 70(suppl 4):S7–S13.
- Carey WD, Jeffers L, Kugelmas M, et al; Hepatitis C management. Hepatitis C Monograph. Cleveland Clinic; www.clevelandclinicmeded.com/online/monograph/hepc/page1.htm. Accessed 7/30/2010.
- Scott JD, Gretch DR. Molecular diagnostics of hepatitis C virus infection: a systematic review. JAMA 2007; 297:724–732.
- Bowden DS, Berzsenyi MD. Chronic hepatitis C virus infection: genotyping and its clinical role. Future Microbiol 2006; 1:103–112.
- Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 2002; 347:975–982.
- Poynard T, McHutchison J, Davis GL, et al. Impact of interferon alfa-2b and ribavirin on progression of liver fibrosis in patients with chronic hepatitis C. Hepatology 2000; 32:1131–1137.
- Zeuzem S. Interferon-based therapy for chronic hepatitis C: current and future perspectives. Nat Clin Pract Gastroenterol Hepatol 2008; 5:610–622.
Screening for hepatitis C virus (HCV) infection in high-risk populations can identify, early on, people at risk of progressive liver disease who may benefit from antiviral therapy and counseling. The US Centers for Disease Control and Prevention (CDC) recommends that all people be assessed for HCV risk factors and that those with risk factors be screened for HCV antibodies (anti-HCV),1 and members of the national societies of gastroenterology and hepatology have endorsed this recommendation.2
Unfortunately, rates at which primary care patients are assessed for risk factors and the rates at which patients at higher risk are screened remain below the goals set by the CDC.3–6 All health care practitioners need to understand how to establish or exclude a diagnosis of HCV infection and to interpret the tests correctly.
WHY SCREEN FOR HCV?
HCV infection is a major public health problem and a leading cause of chronic liver disease. In the United States, an estimated 3.2 million persons (1.3% of the population) have been infected.7 However, in the inner-city primary care setting the rate of HCV infection is as high as 8%, and in Veterans Administration populations it is 17%.8,9 The worldwide prevalence of HCV infection is 2.0%, corresponding to 140 million persons.
Screening of blood products has led to a decline in the incidence of acute hepatitis C since the late 1980s, although rates have reached a plateau in recent years (Figure 1).10
Approximately 20% of patients infected with HCV develop a serious sequela, such as severe fibrosis, cirrhosis, end-stage liver disease, or hepatocellular carcinoma. Currently, HCV infection causes an estimated 8,000 to 10,000 deaths annually in the United States, and that number is predicted to triple in the next 10 to 20 years. Furthermore, HCV-related disease is the leading indication for liver transplantation in the United States, and it is estimated to cost $600 million to $1 billion annually in medical expenses and loss of work.8
Screening can reduce adverse outcomes
HCV screening has several potential benefits. By detecting HCV infection early, screening facilitates virologic suppression, as treatment earlier in the course of the disease is more effective than later.11,12 Further, early diagnosis together with patient education and subsequent lifestyle modifications may reduce the risk of transmission of HCV infection to other people.13,14
Antiviral therapy with pegylated interferons and ribavirin can cure hepatitis C in up to 90% of cases, depending on the viral genotype15–17 (see discussion of HCV genotypes below). In addition, treatment slows the progression of fibrosis.18 The incidence of hepatocellular carcinoma is lower in patients who achieve a sustained virologic response to antiviral therapy.19 Finally, antiviral therapy prolongs survival.20
New drug therapies are being developed and may, we hope, be even more effective than current drugs. Inhibitors of HCV-specific enzymes such as NS3/4 protease, combined with pegylated interferons and ribavirin, are in phase III clinical trials. These drugs are expected to be available for clinical practice within the next 2 years.21–23 Additionally, nitazoxanide (Alinia), an inducer of eIF2a and PKR phosphorylation, has been shown to increase the treatment response to HCV genotype 4. Studies24 are currently under way in patients infected with HCV genotype 1.
Screening is cost-effective
The National Hepatitis Surveillance Program25 calculated the cost of screening for HCV to be $1,246 per case detected. However, a more vigorous analysis of the same data using several different models to incorporate risk factors based on history revealed costs between $357 and $1,047 per case detected. This compares favorably with the cost of screening for other diseases that physicians routinely screen for.
Antiviral combination therapy for chronic hepatitis C has been shown to be effective in terms of quality-adjusted life-years gained and cost-effectiveness in several studies.26–28
HOW TO SCREEN
To test everyone in the general population would be neither cost-effective nor practical, which is why the CDC recommends that serologic screening for HCV infection be done only in people who have well-established risk factors for it.1,5
Therefore, screening should begin by obtaining a relevant medical history as part of a routine health evaluation. But how should this be done?
McGinn et al29 asked 1,000 patients attending an inner-city clinic to fill out a 27-item questionnaire assessing five “domains” of risk factors for HCV: work, medical, exposure, personal care, and social history. Afterward, they tested all 1,000 patients. They found that the risk factors that best predicted positive results on testing were in three domains: medical (eg, blood transfusions, dialysis, other medical procedures, and elevated liver enzymes), exposure (past contact with another person’s blood), and social history (eg, illicit drug use, incarceration, and sexual activity).
The National Hepatitis Surveillance Program25 explored the cost and yield of several screening strategies for hepatitis C, ie, testing only in patients who had a greater than 7% likelihood of infection based on an empirically derived mathematical model; testing only if significant risk factors were revealed in a simple questionnaire; or testing only if the alanine aminotransferase (ALT) level was elevated. The predictive mathematical model was the most effective and efficient means of deciding who should be tested.
Unfortunately, such a model is too cumbersome to be clinically applicable, and clinical prediction tools for HCV screening have been underused.
GROUPS AT HIGH RISK OF HCV
Groups at risk of HCV infection can be classified as being at high, intermediate, or low risk. The American Association for the Study of Liver Diseases2 rates the level of evidence for screening in all of the following risk groups as class I (ie, there is evidence or general agreement that it is beneficial, useful, and effective) and level B (ie, the data are derived from non-randomized studies).
Intravenous drug abusers
Intravenous drug abuse is the strongest independent risk factor for HCV infection.30–33 It has been the main route of HCV infection over the past decades and currently accounts for 60% of HCV transmission in the United States.7,10,34–37
Hemophilia patients treated with clotting factor concentrates produced before 1987
HCV seroprevalence is very high in patients with hemophilia who received infusions of plasma-derived clotting factor concentrates before 1987.38 In these patients, the HCV genotypes are predominantly 1 and 3, and to a lesser extent genotype 2.39,40 These genotypes likely reflect the prior exposures of the plasma donors.41 (See discussion of HCV genotypes below.) Individuals receiving clotting factor concentrates prepared from plasma pools were at high risk of HCV infection until effective procedures to inactivate viruses were introduced in 1985 (factor VIII) and 1987 (factor IX).42
People infected with HIV
About 25% of people infected with human immunodeficiency virus (HIV) in the Western world also have chronic HCV infection.43 Progression of liver disease is accelerated in HIV-HCV coinfection, and the risk of cirrhosis is twice as high.44
However, about 6% of HIV-positive patients fail to develop HCV antibodies when infected. Thus, HCV RNA should be assessed in HIV patients with unexplained liver disease who are negative for anti-HCV.45
The distribution of HCV genotypes in HIV-infected patients reflects the route of transmission. Genotype 1b accounts for 66% of posttransfusion HCV infections, while genotypes 1a and 3a are more common in intravenous drug users.
GROUPS AT INTERMEDIATE RISK OF HCV
Recipients of blood transfusions before 1992
Before 1992, blood transfusions carried a risk of HCV infection of up to 7% with each unit transfused. Prospective studies of transfusion recipients in the United States found that rates of posttransfusion hepatitis in the 1960s exceeded 20%,36 since most patients received multiple units of blood.
In the mid-1970s, before HCV had been identified, available diagnostic tests indicated that 90% of cases of posttransfusion hepatitis were not caused by hepatitis A or hepatitis B viruses. By this time, the move to all-volunteer blood donors instead of paid donors had reduced the risk of posttransfusion hepatitis to 10%.22,37,46
Although non-A, non-B hepatitis was first recognized because of its association with blood transfusion, population-based sentinel surveillance showed that it accounted for 15% to 20% of cases of community-acquired viral hepatitis in the United States.35 The advent of molecular cloning in 1988 indicated that non-A, non-B hepatitis was primarily caused by HCV.47–52
Screening of blood has reduced the rate of posttransfusion hepatitis C by a factor of about 10,000, to a current rate of 1 per million transfusions.53 The few cases that still occur are due to newly infected people donating blood before they have developed antibodies to the virus, which can take up to 8 weeks.54
Recipients of solid-organ transplants before 1992
Before organ donors were screened for HCV, recipients of solid-organ transplants from infected donors had a high risk of acquiring HCV infection. Transmission rates in different cohorts ranged from 30% to 80%.55 In an attempt to improve the safety of organ transplantation, many transplant centers now screen donors for anti-HCV and test for HCV RNA for verification.
A related problem is pre-existing HCV infection in transplant recipients. Izopet et al56 reported that, in renal transplant recipients with preexisting HCV infection, the HCV RNA titer rose about 10 times (1 log) higher after transplantation, owing to the immunosuppressive drugs that transplant recipients must take. Although this higher viral load does not affect the progression of fibrosis in all patients, the effect of immunosuppressive therapy on liver disease results in a worse outcome for some, and it reduces survival beginning in the second decade after kidney transplantation.56
Additionally, treatment of HCV infection in transplant recipients may pose a challenge, as those receiving immunosuppressive therapy with tacrolimus (Prograf) or cyclosporine (Sandimmune) may develop some degree of renal insufficiency, complicating the use of ribavirin (Rebetol) and subjecting patients to a higher risk of severe anemia. Furthermore, interferon therapy increases the risk of renal allograft rejection and, accordingly, is not often used in renal transplant recipients.
Patients with unexplained elevated aminotransferase levels
HCV infection affects an estimated 1.8% of the general population, but the rate is much higher in people with ALT levels over 40 U/L. Most patients with chronic hepatitis C have no symptoms or only mild symptoms and minimally elevated levels of ALT and aspartate aminotransferase (AST)—ie, two to five times higher than the upper limit of normal.
The first step in the workup of aminotransferase elevations is to confirm the abnormality by repeating the blood test. If an elevation is confirmed, further investigation is warranted. A directed history and physical examination is important and may disclose risk factors, raising clinical suspicion of a particular disease.
Some caveats: The proportion of patients with HCV viremia who have abnormally high aminotransferase levels ranges between only 54% and 66%.57–59 In patients with risk factors for HCV infection and abnormal liver enzyme levels, HCV infection is probable but not certain. Also, liver enzyme tests do not reveal the extent of hepatic injury or reflect the true status of hepatic function.60
Infants born to infected mothers
Children born to HCV-positive women should be tested for anti-HCV no sooner than age 12 months, when passively transferred maternal anti-HCV declines below detectable levels. If earlier diagnosis of HCV infection is desired, a real-time polymerase chain reaction (PCR) test for HCV RNA can be done at or after the infant's first “well-child” visit at age 1 to 2 months.
If positive for either anti-HCV or HCV RNA, children should be evaluated for liver disease, and those with persistently elevated ALT levels should be referred to a specialist for medical management.2,5
GROUPS AT LOW RISK OF HCV
People who have had sexual relations with multiple or infected partners
Sexual activity is associated with a low but measurable risk of transmission of HCV. Large population-based studies, including the National Hepatitis Surveillance Program,25 found an independent association between HCV infection and having sexual relations with multiple partners or with a partner who is infected with HCV.
The CDC reported that 15% to 20% of patients with acute hepatitis C had a history of sexual exposure but no other risk factors. Two-thirds of them had an anti-HCV-positive sexual partner, and one-third reported having had more than two partners in the 6 months before illness.5
More data are needed to determine the risk of and the factors related to transmission of HCV between long-term steady partners as well as in persons with high-risk sexual practices, including whether other sexually transmitted diseases promote transmission of HCV by influencing viral load or modifying mucosal barriers.
Health care workers exposed to HCV, eg, by needlestick
The prevalence of HCV infection in health care workers is no greater than that in the general population, averaging 1% to 2%, and is actually 10 times lower than that of hepatitis B virus infection.47,48,61,62
However, within the disciplines, some groups have a higher prevalence of HCV infection, suggesting that some occupations carry a higher risk. In two US studies, the prevalence of HCV infection was higher in oral surgeons (2.0% and 9.3%) than in other dentists (0.7% and 0.97%).63,64
In a single study that evaluated risk factors for infection, a history of needlestick injury was the only occupational risk factor that was independently associated with HCV infection.65 The average incidence of anti-HCV seroconversion after a needlestick or after an injury with a sharp object contaminated by an HCV-positive source is 1.8% (range 0%–7%).66–69
Although no studies of incidence have documented transmission via mucous membrane or nonintact skin exposures, transmission of HCV from blood splashes to the conjunctiva have been described.70,71
It is worth noting that exposure to blood from unclean needles used in tattooing or body piercing also confers a risk of HCV infection.
SEROLOGIC SCREENING TESTS FOR HCV
- Serologic assays that detect specific antibody to HCV (anti-HCV)
- Molecular assays that detect viral RNA.
Initial serologic screening tests for anti-HCV
Two EIAs are approved for clinical use:
- Abbott HCV EIA 2.0 (Abbott Laboratories, Abbott Park, IL)
- Ortho HCV Version 3.0 enzyme-linked immunosorbent assay (ELISA) (Ortho-Clinical Diagnostics, Rochester, NY).
One enhanced chemiluminescence immunoassay is also approved:
- Vitros Anti-HCV assay (Ortho-Clinical Diagnostics). In practical terms, this test is equivalent to the two EIAs, and the discussion below about EIAs applies to this test as well.
These third-generation tests are highly sensitive (> 99%) and specific (99%) in immunocompetent patients, and eliminate the need for a confirmatory immunoblot assay in patients with clinical liver disease, particularly those with risk factors for HCV infection.
False-positive results are rare now, but they were common with earlier generations of these assays. Most false-positive results occur in patients with autoimmune liver disease or hypergammaglobulinemia who have normal liver enzyme levels and no risk factors for HCV infection. In fact, all positive anti-HCV results should be followed up with an HCV RNA test.
False-negative results are also uncommon, usually occurring only in immunosuppressed patients (eg, organ transplant recipients and HIV-positive patients) and in patients on long-term hemodialysis. Therefore, patients with a history of hemodialysis should be considered for an HCV RNA assay rather than an EIA. Measurement of ALT will not be useful because ALT levels are lower in patients with end-stage renal disease. In most other clinical situations, the HCV EIA is an outstanding screening test for HCV infection because of its high sensitivity and relatively low cost (< $50).
Although the specificity of these tests is good, the predictive value of a positive result varies substantially by the pretest probability of HCV infection. For example, in a group of injection-drug users who are very likely to have ongoing or remote infection, all positive HCV EIA results are likely truly positive.74 On the other hand, in healthy blood donors, up to half of all positive third-generation EIA tests are falsely positive.75
Important points
- A positive anti-HCV antibody test does not distinguish acute from chronic disease or active from past infection, nor is it a sign of immunity or protection.
- A positive anti-HCV EIA requires HCV RNA measurement to discriminate between current infection on the one hand, and either resolved HCV infection or a false-positive result on the other.
- A positive EIA anti-HCV test is a marker that hepatitis C may be present, and it must be followed by confirmatory HCV RNA testing.
- Physicians should be mindful of the potential tribulations associated with false-positive tests. A false-positive test may result in harm to patients that is difficult to measure, such as anxiety, labeling in the medical record, and detrimental effects on close relationships.
CONFIRMATORY TESTING WITH ASSAYS FOR HCV RNA
As stated above, a positive result on an anti-HCV EIA needs to be confirmed with an assay for HCV RNA, of which there are two types, ie, qualitative and quantitative.
Each involves trade-offs. Qualitative assays are more sensitive and detect more cases, but they provide no information about the amount of virus (viral load). Quantitative assays are less sensitive, so a negative result does not completely exclude hepatitis C, although they can still can detect 95% of cases. They do, however, measure the viral load.
Therefore, the type of test to use depends on the patient’s risk profile, the goals of testing, and the setting in which future care will be provided. The primary objective when a patient has a positive EIA test is to determine whether he or she has ongoing infection, a goal most expeditiously achieved using a qualitative assay. However, since a quantitative assay can detect the vast majority of cases of active HCV infection, many clinicians select this as the test of first choice when the probability of HCV is high (eg, in a patient with risk factors and abnormal liver tests). If the pretest probability is low, a qualitative assay is the better choice.
Many commercial assays are available for detecting (qualitative assays) or measuring (quantitative assays) HCV RNA.
Qualitative HCV RNA assays
The approved qualitative assays are:
- Amplicor HCV Test, version 2.0 (Roche Molecular Diagnostics, Pleasanton, CA)
- Cobas Amplicor HCV Test, version 2.0 (Roche Molecular Diagnostics)
- Ampliscreen (Roche Molecular Diagnostics)
- Versant HCV RNA Qualitative Assay (Siemens Healthcare Diagnostics, Deerfield, IL)
- Procleix HIV-1/HCV Assay (Chiron, Emeryville, CA).
Quantitative HCV RNA assays
The approved quantitative assays are:
- Amplicor HCV Monitor (Roche Molecular Diagnostics)
- Cobas Amplicor HCV Monitor, version 2.0 (Roche Molecular Diagnostics)
- Versant HCV RNA 3.0 Assay (bDNA) (Siemens Healthcare Diagnostics)
- Cobas Taqman HCV Test (Roche Molecular Diagnostics).
Quantitative tests use target amplification with PCR, transcription-mediated amplification (TMA), or a signal amplification technique such as a branched DNA (bDNA) assay. The sensitivity varies for different types of amplification. TMA assays appear to be the most sensitive for detecting HCV RNA.
The latest innovation is real-time PCR, which shortens the typical time for PCR processing from 1.5 hours to 35 minutes. It may also detect relapsed HCV infection earlier than regular PCR. With the recent availability of real-time PCR assays, which have sensitivities of 10 to 50 IU/mL, many experts feel there is no longer a need for qualitative assays.74 In fact, many laboratories no longer offer qualitative testing. The Cleveland Clinic laboratory has recently stopped offering this test.
Because RNA testing is widely available, the recombinant immunoblot assay (RIBA) has become obsolete in diagnosing HCV infection, except in special circumstances. Currently, the primary purpose of RIBA testing is to distinguish between resolved HCV infection (EIA-positive, HCV RNA-negative, RIBA-positive) and a false-positive EIA (EIA-positive, HCV RNA-negative, RIBA-negative).
In summary, patients suspected of having acute or chronic HCV infection should first be tested for anti-HCV. Subsequently, HCV RNA testing should be performed in:
- Patients with a positive anti-HCV test
- Patients for whom antiviral treatment is being considered (using a sensitive quantitative assay)
- Patients with unexplained liver disease whose anti-HCV test is negative and who are immunocompromised or suspected of having acute HCV infection.
Significance of the HCV viral load
The significance of the HCV viral load is widely misunderstood. The amount of virus in the blood does not correlate with symptoms, histologic liver injury, or the stage or aggressiveness of disease. Its sole importance is in relation to therapy.
The HCV viral load, measured before treatment, helps predict the likelihood of a treatment response: the lower the pretreatment viral load, the more likely that the patient will respond to current HCV therapies.
Additionally, the pretreatment viral load serves as a baseline for comparison with subsequent measurements during treatment. Patients with HCV genotype 1 who do not achieve more than a 2-log (99%) reduction in viral load by the 12th week of treatment (an early virologic response) have a low response rate, and treatment should generally be stopped, given its cost and side effects.76 However, measuring the viral load to detect an early virologic response is less helpful in patients with HCV genotype 2 or 3 infection, since these patients require only 24 weeks of therapy and most of them clear the virus by week 12 and respond to therapy.
Additionally, patients with genotype 2 or 3 and those with a viral load of less than 600,000 IU/mL have been found to achieve higher rates of sustained virologic response.15 A sustained virologic response is defined as the absence of HCV RNA 24 weeks after stopping treatment and is now considered to be the best predictor of long-term treatment response. A sustained virologic response is generally regarded as a “virologic cure.”
HCV GENOTYPE AFFECTS SUCCESS AND DURATION OF TREATMENT
HCV has at least six major genotypes.1,3–6 Several genotypes are subclassified as “a” or “b” (ie, genotype 1a or 1b); however, these distinctions are of little clinical use.
In the laboratory, HCV genotypes are identified by restriction fragment length polymorphism, by direct sequence analysis, or by reverse hybridization. Once the HCV genotype has been identified, there is no need to repeat the test.
Different genotypes are more common in some areas of the world than in others. Genotype 1 is the one most common in the United States (accounting for 70% to 75% of cases), followed by genotypes 2 and 3 (25%–30%). Genotype 4 is most common in Egypt and the Arabian peninsula.
HCV genotyping is important because it can help predict the likelihood of a response to treatment and in planning the dose and duration of therapy.77 For example, treatment with pegylated interferon plus ribavirin is predicted to work approximately 50% of the time for people with genotype 1, but 80% to 90% of the time for people with genotypes 2 or 3.15–17,78 Additionally, patients with genotype 1 need 12 months of therapy to achieve maximum benefit, whereas those with genotypes 2 and 3 require treatment for only 6 months to achieve maximum benefit.
Screening for hepatitis C virus (HCV) infection in high-risk populations can identify, early on, people at risk of progressive liver disease who may benefit from antiviral therapy and counseling. The US Centers for Disease Control and Prevention (CDC) recommends that all people be assessed for HCV risk factors and that those with risk factors be screened for HCV antibodies (anti-HCV),1 and members of the national societies of gastroenterology and hepatology have endorsed this recommendation.2
Unfortunately, rates at which primary care patients are assessed for risk factors and the rates at which patients at higher risk are screened remain below the goals set by the CDC.3–6 All health care practitioners need to understand how to establish or exclude a diagnosis of HCV infection and to interpret the tests correctly.
WHY SCREEN FOR HCV?
HCV infection is a major public health problem and a leading cause of chronic liver disease. In the United States, an estimated 3.2 million persons (1.3% of the population) have been infected.7 However, in the inner-city primary care setting the rate of HCV infection is as high as 8%, and in Veterans Administration populations it is 17%.8,9 The worldwide prevalence of HCV infection is 2.0%, corresponding to 140 million persons.
Screening of blood products has led to a decline in the incidence of acute hepatitis C since the late 1980s, although rates have reached a plateau in recent years (Figure 1).10
Approximately 20% of patients infected with HCV develop a serious sequela, such as severe fibrosis, cirrhosis, end-stage liver disease, or hepatocellular carcinoma. Currently, HCV infection causes an estimated 8,000 to 10,000 deaths annually in the United States, and that number is predicted to triple in the next 10 to 20 years. Furthermore, HCV-related disease is the leading indication for liver transplantation in the United States, and it is estimated to cost $600 million to $1 billion annually in medical expenses and loss of work.8
Screening can reduce adverse outcomes
HCV screening has several potential benefits. By detecting HCV infection early, screening facilitates virologic suppression, as treatment earlier in the course of the disease is more effective than later.11,12 Further, early diagnosis together with patient education and subsequent lifestyle modifications may reduce the risk of transmission of HCV infection to other people.13,14
Antiviral therapy with pegylated interferons and ribavirin can cure hepatitis C in up to 90% of cases, depending on the viral genotype15–17 (see discussion of HCV genotypes below). In addition, treatment slows the progression of fibrosis.18 The incidence of hepatocellular carcinoma is lower in patients who achieve a sustained virologic response to antiviral therapy.19 Finally, antiviral therapy prolongs survival.20
New drug therapies are being developed and may, we hope, be even more effective than current drugs. Inhibitors of HCV-specific enzymes such as NS3/4 protease, combined with pegylated interferons and ribavirin, are in phase III clinical trials. These drugs are expected to be available for clinical practice within the next 2 years.21–23 Additionally, nitazoxanide (Alinia), an inducer of eIF2a and PKR phosphorylation, has been shown to increase the treatment response to HCV genotype 4. Studies24 are currently under way in patients infected with HCV genotype 1.
Screening is cost-effective
The National Hepatitis Surveillance Program25 calculated the cost of screening for HCV to be $1,246 per case detected. However, a more vigorous analysis of the same data using several different models to incorporate risk factors based on history revealed costs between $357 and $1,047 per case detected. This compares favorably with the cost of screening for other diseases that physicians routinely screen for.
Antiviral combination therapy for chronic hepatitis C has been shown to be effective in terms of quality-adjusted life-years gained and cost-effectiveness in several studies.26–28
HOW TO SCREEN
To test everyone in the general population would be neither cost-effective nor practical, which is why the CDC recommends that serologic screening for HCV infection be done only in people who have well-established risk factors for it.1,5
Therefore, screening should begin by obtaining a relevant medical history as part of a routine health evaluation. But how should this be done?
McGinn et al29 asked 1,000 patients attending an inner-city clinic to fill out a 27-item questionnaire assessing five “domains” of risk factors for HCV: work, medical, exposure, personal care, and social history. Afterward, they tested all 1,000 patients. They found that the risk factors that best predicted positive results on testing were in three domains: medical (eg, blood transfusions, dialysis, other medical procedures, and elevated liver enzymes), exposure (past contact with another person’s blood), and social history (eg, illicit drug use, incarceration, and sexual activity).
The National Hepatitis Surveillance Program25 explored the cost and yield of several screening strategies for hepatitis C, ie, testing only in patients who had a greater than 7% likelihood of infection based on an empirically derived mathematical model; testing only if significant risk factors were revealed in a simple questionnaire; or testing only if the alanine aminotransferase (ALT) level was elevated. The predictive mathematical model was the most effective and efficient means of deciding who should be tested.
Unfortunately, such a model is too cumbersome to be clinically applicable, and clinical prediction tools for HCV screening have been underused.
GROUPS AT HIGH RISK OF HCV
Groups at risk of HCV infection can be classified as being at high, intermediate, or low risk. The American Association for the Study of Liver Diseases2 rates the level of evidence for screening in all of the following risk groups as class I (ie, there is evidence or general agreement that it is beneficial, useful, and effective) and level B (ie, the data are derived from non-randomized studies).
Intravenous drug abusers
Intravenous drug abuse is the strongest independent risk factor for HCV infection.30–33 It has been the main route of HCV infection over the past decades and currently accounts for 60% of HCV transmission in the United States.7,10,34–37
Hemophilia patients treated with clotting factor concentrates produced before 1987
HCV seroprevalence is very high in patients with hemophilia who received infusions of plasma-derived clotting factor concentrates before 1987.38 In these patients, the HCV genotypes are predominantly 1 and 3, and to a lesser extent genotype 2.39,40 These genotypes likely reflect the prior exposures of the plasma donors.41 (See discussion of HCV genotypes below.) Individuals receiving clotting factor concentrates prepared from plasma pools were at high risk of HCV infection until effective procedures to inactivate viruses were introduced in 1985 (factor VIII) and 1987 (factor IX).42
People infected with HIV
About 25% of people infected with human immunodeficiency virus (HIV) in the Western world also have chronic HCV infection.43 Progression of liver disease is accelerated in HIV-HCV coinfection, and the risk of cirrhosis is twice as high.44
However, about 6% of HIV-positive patients fail to develop HCV antibodies when infected. Thus, HCV RNA should be assessed in HIV patients with unexplained liver disease who are negative for anti-HCV.45
The distribution of HCV genotypes in HIV-infected patients reflects the route of transmission. Genotype 1b accounts for 66% of posttransfusion HCV infections, while genotypes 1a and 3a are more common in intravenous drug users.
GROUPS AT INTERMEDIATE RISK OF HCV
Recipients of blood transfusions before 1992
Before 1992, blood transfusions carried a risk of HCV infection of up to 7% with each unit transfused. Prospective studies of transfusion recipients in the United States found that rates of posttransfusion hepatitis in the 1960s exceeded 20%,36 since most patients received multiple units of blood.
In the mid-1970s, before HCV had been identified, available diagnostic tests indicated that 90% of cases of posttransfusion hepatitis were not caused by hepatitis A or hepatitis B viruses. By this time, the move to all-volunteer blood donors instead of paid donors had reduced the risk of posttransfusion hepatitis to 10%.22,37,46
Although non-A, non-B hepatitis was first recognized because of its association with blood transfusion, population-based sentinel surveillance showed that it accounted for 15% to 20% of cases of community-acquired viral hepatitis in the United States.35 The advent of molecular cloning in 1988 indicated that non-A, non-B hepatitis was primarily caused by HCV.47–52
Screening of blood has reduced the rate of posttransfusion hepatitis C by a factor of about 10,000, to a current rate of 1 per million transfusions.53 The few cases that still occur are due to newly infected people donating blood before they have developed antibodies to the virus, which can take up to 8 weeks.54
Recipients of solid-organ transplants before 1992
Before organ donors were screened for HCV, recipients of solid-organ transplants from infected donors had a high risk of acquiring HCV infection. Transmission rates in different cohorts ranged from 30% to 80%.55 In an attempt to improve the safety of organ transplantation, many transplant centers now screen donors for anti-HCV and test for HCV RNA for verification.
A related problem is pre-existing HCV infection in transplant recipients. Izopet et al56 reported that, in renal transplant recipients with preexisting HCV infection, the HCV RNA titer rose about 10 times (1 log) higher after transplantation, owing to the immunosuppressive drugs that transplant recipients must take. Although this higher viral load does not affect the progression of fibrosis in all patients, the effect of immunosuppressive therapy on liver disease results in a worse outcome for some, and it reduces survival beginning in the second decade after kidney transplantation.56
Additionally, treatment of HCV infection in transplant recipients may pose a challenge, as those receiving immunosuppressive therapy with tacrolimus (Prograf) or cyclosporine (Sandimmune) may develop some degree of renal insufficiency, complicating the use of ribavirin (Rebetol) and subjecting patients to a higher risk of severe anemia. Furthermore, interferon therapy increases the risk of renal allograft rejection and, accordingly, is not often used in renal transplant recipients.
Patients with unexplained elevated aminotransferase levels
HCV infection affects an estimated 1.8% of the general population, but the rate is much higher in people with ALT levels over 40 U/L. Most patients with chronic hepatitis C have no symptoms or only mild symptoms and minimally elevated levels of ALT and aspartate aminotransferase (AST)—ie, two to five times higher than the upper limit of normal.
The first step in the workup of aminotransferase elevations is to confirm the abnormality by repeating the blood test. If an elevation is confirmed, further investigation is warranted. A directed history and physical examination is important and may disclose risk factors, raising clinical suspicion of a particular disease.
Some caveats: The proportion of patients with HCV viremia who have abnormally high aminotransferase levels ranges between only 54% and 66%.57–59 In patients with risk factors for HCV infection and abnormal liver enzyme levels, HCV infection is probable but not certain. Also, liver enzyme tests do not reveal the extent of hepatic injury or reflect the true status of hepatic function.60
Infants born to infected mothers
Children born to HCV-positive women should be tested for anti-HCV no sooner than age 12 months, when passively transferred maternal anti-HCV declines below detectable levels. If earlier diagnosis of HCV infection is desired, a real-time polymerase chain reaction (PCR) test for HCV RNA can be done at or after the infant's first “well-child” visit at age 1 to 2 months.
If positive for either anti-HCV or HCV RNA, children should be evaluated for liver disease, and those with persistently elevated ALT levels should be referred to a specialist for medical management.2,5
GROUPS AT LOW RISK OF HCV
People who have had sexual relations with multiple or infected partners
Sexual activity is associated with a low but measurable risk of transmission of HCV. Large population-based studies, including the National Hepatitis Surveillance Program,25 found an independent association between HCV infection and having sexual relations with multiple partners or with a partner who is infected with HCV.
The CDC reported that 15% to 20% of patients with acute hepatitis C had a history of sexual exposure but no other risk factors. Two-thirds of them had an anti-HCV-positive sexual partner, and one-third reported having had more than two partners in the 6 months before illness.5
More data are needed to determine the risk of and the factors related to transmission of HCV between long-term steady partners as well as in persons with high-risk sexual practices, including whether other sexually transmitted diseases promote transmission of HCV by influencing viral load or modifying mucosal barriers.
Health care workers exposed to HCV, eg, by needlestick
The prevalence of HCV infection in health care workers is no greater than that in the general population, averaging 1% to 2%, and is actually 10 times lower than that of hepatitis B virus infection.47,48,61,62
However, within the disciplines, some groups have a higher prevalence of HCV infection, suggesting that some occupations carry a higher risk. In two US studies, the prevalence of HCV infection was higher in oral surgeons (2.0% and 9.3%) than in other dentists (0.7% and 0.97%).63,64
In a single study that evaluated risk factors for infection, a history of needlestick injury was the only occupational risk factor that was independently associated with HCV infection.65 The average incidence of anti-HCV seroconversion after a needlestick or after an injury with a sharp object contaminated by an HCV-positive source is 1.8% (range 0%–7%).66–69
Although no studies of incidence have documented transmission via mucous membrane or nonintact skin exposures, transmission of HCV from blood splashes to the conjunctiva have been described.70,71
It is worth noting that exposure to blood from unclean needles used in tattooing or body piercing also confers a risk of HCV infection.
SEROLOGIC SCREENING TESTS FOR HCV
- Serologic assays that detect specific antibody to HCV (anti-HCV)
- Molecular assays that detect viral RNA.
Initial serologic screening tests for anti-HCV
Two EIAs are approved for clinical use:
- Abbott HCV EIA 2.0 (Abbott Laboratories, Abbott Park, IL)
- Ortho HCV Version 3.0 enzyme-linked immunosorbent assay (ELISA) (Ortho-Clinical Diagnostics, Rochester, NY).
One enhanced chemiluminescence immunoassay is also approved:
- Vitros Anti-HCV assay (Ortho-Clinical Diagnostics). In practical terms, this test is equivalent to the two EIAs, and the discussion below about EIAs applies to this test as well.
These third-generation tests are highly sensitive (> 99%) and specific (99%) in immunocompetent patients, and eliminate the need for a confirmatory immunoblot assay in patients with clinical liver disease, particularly those with risk factors for HCV infection.
False-positive results are rare now, but they were common with earlier generations of these assays. Most false-positive results occur in patients with autoimmune liver disease or hypergammaglobulinemia who have normal liver enzyme levels and no risk factors for HCV infection. In fact, all positive anti-HCV results should be followed up with an HCV RNA test.
False-negative results are also uncommon, usually occurring only in immunosuppressed patients (eg, organ transplant recipients and HIV-positive patients) and in patients on long-term hemodialysis. Therefore, patients with a history of hemodialysis should be considered for an HCV RNA assay rather than an EIA. Measurement of ALT will not be useful because ALT levels are lower in patients with end-stage renal disease. In most other clinical situations, the HCV EIA is an outstanding screening test for HCV infection because of its high sensitivity and relatively low cost (< $50).
Although the specificity of these tests is good, the predictive value of a positive result varies substantially by the pretest probability of HCV infection. For example, in a group of injection-drug users who are very likely to have ongoing or remote infection, all positive HCV EIA results are likely truly positive.74 On the other hand, in healthy blood donors, up to half of all positive third-generation EIA tests are falsely positive.75
Important points
- A positive anti-HCV antibody test does not distinguish acute from chronic disease or active from past infection, nor is it a sign of immunity or protection.
- A positive anti-HCV EIA requires HCV RNA measurement to discriminate between current infection on the one hand, and either resolved HCV infection or a false-positive result on the other.
- A positive EIA anti-HCV test is a marker that hepatitis C may be present, and it must be followed by confirmatory HCV RNA testing.
- Physicians should be mindful of the potential tribulations associated with false-positive tests. A false-positive test may result in harm to patients that is difficult to measure, such as anxiety, labeling in the medical record, and detrimental effects on close relationships.
CONFIRMATORY TESTING WITH ASSAYS FOR HCV RNA
As stated above, a positive result on an anti-HCV EIA needs to be confirmed with an assay for HCV RNA, of which there are two types, ie, qualitative and quantitative.
Each involves trade-offs. Qualitative assays are more sensitive and detect more cases, but they provide no information about the amount of virus (viral load). Quantitative assays are less sensitive, so a negative result does not completely exclude hepatitis C, although they can still can detect 95% of cases. They do, however, measure the viral load.
Therefore, the type of test to use depends on the patient’s risk profile, the goals of testing, and the setting in which future care will be provided. The primary objective when a patient has a positive EIA test is to determine whether he or she has ongoing infection, a goal most expeditiously achieved using a qualitative assay. However, since a quantitative assay can detect the vast majority of cases of active HCV infection, many clinicians select this as the test of first choice when the probability of HCV is high (eg, in a patient with risk factors and abnormal liver tests). If the pretest probability is low, a qualitative assay is the better choice.
Many commercial assays are available for detecting (qualitative assays) or measuring (quantitative assays) HCV RNA.
Qualitative HCV RNA assays
The approved qualitative assays are:
- Amplicor HCV Test, version 2.0 (Roche Molecular Diagnostics, Pleasanton, CA)
- Cobas Amplicor HCV Test, version 2.0 (Roche Molecular Diagnostics)
- Ampliscreen (Roche Molecular Diagnostics)
- Versant HCV RNA Qualitative Assay (Siemens Healthcare Diagnostics, Deerfield, IL)
- Procleix HIV-1/HCV Assay (Chiron, Emeryville, CA).
Quantitative HCV RNA assays
The approved quantitative assays are:
- Amplicor HCV Monitor (Roche Molecular Diagnostics)
- Cobas Amplicor HCV Monitor, version 2.0 (Roche Molecular Diagnostics)
- Versant HCV RNA 3.0 Assay (bDNA) (Siemens Healthcare Diagnostics)
- Cobas Taqman HCV Test (Roche Molecular Diagnostics).
Quantitative tests use target amplification with PCR, transcription-mediated amplification (TMA), or a signal amplification technique such as a branched DNA (bDNA) assay. The sensitivity varies for different types of amplification. TMA assays appear to be the most sensitive for detecting HCV RNA.
The latest innovation is real-time PCR, which shortens the typical time for PCR processing from 1.5 hours to 35 minutes. It may also detect relapsed HCV infection earlier than regular PCR. With the recent availability of real-time PCR assays, which have sensitivities of 10 to 50 IU/mL, many experts feel there is no longer a need for qualitative assays.74 In fact, many laboratories no longer offer qualitative testing. The Cleveland Clinic laboratory has recently stopped offering this test.
Because RNA testing is widely available, the recombinant immunoblot assay (RIBA) has become obsolete in diagnosing HCV infection, except in special circumstances. Currently, the primary purpose of RIBA testing is to distinguish between resolved HCV infection (EIA-positive, HCV RNA-negative, RIBA-positive) and a false-positive EIA (EIA-positive, HCV RNA-negative, RIBA-negative).
In summary, patients suspected of having acute or chronic HCV infection should first be tested for anti-HCV. Subsequently, HCV RNA testing should be performed in:
- Patients with a positive anti-HCV test
- Patients for whom antiviral treatment is being considered (using a sensitive quantitative assay)
- Patients with unexplained liver disease whose anti-HCV test is negative and who are immunocompromised or suspected of having acute HCV infection.
Significance of the HCV viral load
The significance of the HCV viral load is widely misunderstood. The amount of virus in the blood does not correlate with symptoms, histologic liver injury, or the stage or aggressiveness of disease. Its sole importance is in relation to therapy.
The HCV viral load, measured before treatment, helps predict the likelihood of a treatment response: the lower the pretreatment viral load, the more likely that the patient will respond to current HCV therapies.
Additionally, the pretreatment viral load serves as a baseline for comparison with subsequent measurements during treatment. Patients with HCV genotype 1 who do not achieve more than a 2-log (99%) reduction in viral load by the 12th week of treatment (an early virologic response) have a low response rate, and treatment should generally be stopped, given its cost and side effects.76 However, measuring the viral load to detect an early virologic response is less helpful in patients with HCV genotype 2 or 3 infection, since these patients require only 24 weeks of therapy and most of them clear the virus by week 12 and respond to therapy.
Additionally, patients with genotype 2 or 3 and those with a viral load of less than 600,000 IU/mL have been found to achieve higher rates of sustained virologic response.15 A sustained virologic response is defined as the absence of HCV RNA 24 weeks after stopping treatment and is now considered to be the best predictor of long-term treatment response. A sustained virologic response is generally regarded as a “virologic cure.”
HCV GENOTYPE AFFECTS SUCCESS AND DURATION OF TREATMENT
HCV has at least six major genotypes.1,3–6 Several genotypes are subclassified as “a” or “b” (ie, genotype 1a or 1b); however, these distinctions are of little clinical use.
In the laboratory, HCV genotypes are identified by restriction fragment length polymorphism, by direct sequence analysis, or by reverse hybridization. Once the HCV genotype has been identified, there is no need to repeat the test.
Different genotypes are more common in some areas of the world than in others. Genotype 1 is the one most common in the United States (accounting for 70% to 75% of cases), followed by genotypes 2 and 3 (25%–30%). Genotype 4 is most common in Egypt and the Arabian peninsula.
HCV genotyping is important because it can help predict the likelihood of a response to treatment and in planning the dose and duration of therapy.77 For example, treatment with pegylated interferon plus ribavirin is predicted to work approximately 50% of the time for people with genotype 1, but 80% to 90% of the time for people with genotypes 2 or 3.15–17,78 Additionally, patients with genotype 1 need 12 months of therapy to achieve maximum benefit, whereas those with genotypes 2 and 3 require treatment for only 6 months to achieve maximum benefit.
- Alter MJ, Seeff LB, Bacon BR, Thomas DL, Rigsby MO, Di Bisceglie AM. Testing for hepatitis C virus infection should be routine for persons at increased risk for infection. Ann Intern Med 2004; 141:715–717.
- Ghany MG, Strader DB, Thomas DL, Seeff LB; American Association for the Study of Liver Diseases. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology 2009; 49:1335–1374.
- Shehab TM, Orrego M, Chunduri R, Lok AS. Identification and management of hepatitis C patients in primary care clinics. Am J Gastroenterol 2003; 98:639–644.
- Shehab TM, Sonnad SS, Lok AS. Management of hepatitis C patients by primary care physicians in the USA: results of a national survey. J Viral Hepat 2001; 8:377–383.
- US Centers for Disease Control and Prevention. Recommendations for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. MMWR Recomm Rep 1998; 47:1–39.
- US Centers for Disease Control and Prevention. National prevention strategy: a comprehensive strategy for the prevention and control of hepatitis C virus infection and its consequences; summer 2001. http://www.cdc.gov/hepatitis/HCV/Strategy/NatHep-CPrevStrategy.htm. Accessed August 8, 2010.
- Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med 2006; 144:705–714.
- Kim WR. The burden of hepatitis C in the United States. Hepatology 2002; 36(suppl 1):S30–S34.
- Lau DT, Kleiner DE, Ghany MG, Park Y, Schmid P, Hoofnagle JH. 10-Year follow-up after interferon-alpha therapy for chronic hepatitis C. Hepatology 1998; 28:1121–1127.
- Daniels D, Grytdal S, Wasley A; US Centers for Disease Control and Prevention. Surveillance for acute viral hepatitis—United States, 2007. MMWR Surveill Summ 2009; 58:1–27.
- Thomson BJ, Kwong G, Ratib S, et al; Trent HCV Study Group. Response rates to combination therapy for chronic HCV infection in a clinical setting and derivation of probability tables for individual patient management. J Viral Hepat 2008; 15:271–278.
- Hayashi N, Takehara T. Antiviral therapy for chronic hepatitis C: past, present, and future. J Gastroenterol 2006; 41:17–27.
- Gordon FD. Cost-effectiveness of screening patients for hepatitis C. Am J Med 1999; 107:36S–40S.
- Hill L, Henry B, Schweikert S; Prevention Practice Committee, American College of Preventive Medicine. Screening for chronic hepatitis C: American College of Preventive Medicine practice policy statement. Am J Prev Med 2005; 28:327–330.
- Hadziyannis SJ, Sette H, Morgan TR, et al; PEGASYS International Study Group. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann Intern Med 2004; 140:346–355.
- Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 2001; 358:958–965.
- Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 2002; 347:975–982.
- Cammà C, Di Bona D, Schepis F, et al. Effect of peginterferon alfa-2a on liver histology in chronic hepatitis C: a meta-analysis of individual patient data. Hepatology 2004; 39:333–342.
- Yoshida H, Tateishi R, Arakawa Y, et al. Benefit of interferon therapy in hepatocellular carcinoma prevention for individual patients with chronic hepatitis C. Gut 2004; 53:425–430.
- Yoshida H, Arakawa Y, Sata M, et al. Interferon therapy prolonged life expectancy among chronic hepatitis C patients. Gastroenterology 2002; 123:483–491.
- Hézode C, Forestier N, Dusheiko G, et al; PROVE2 Study Team. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N Engl J Med 2009; 360:1839–1850.
- McHutchison JG, Everson GT, Gordon SC, et al; PROVE1 Study Team. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009; 360:1827–1838.
- Berman K, Kwo PY. Boceprevir, an NS3 protease inhibitor of HCV. Clin Liver Dis 2009; 13:429–439.
- Rossignol JF, Elfert A, Keeffe EB. Treatment of chronic hepatitis C using a 4-week lead-in with nitazoxanide before peginterferon plus nitazoxanide. J Clin Gastroenterol 2009 Dec 31; epub ahead of print.
- Lapane KL, Jakiche AF, Sugano D, Weng CS, Carey WD. Hepatitis C infection risk analysis: who should be screened? Comparison of multiple screening strategies based on the National Hepatitis Surveillance Program. Am J Gastroenterol 1998; 93:591–596.
- Wong JB, Davis GL, McHutchison JG, Manns MP, Albrecht JK; International Hepatitis Interventional Therapy Group. Economic and clinical effects of evaluating rapid viral response to peginterferon alfa-2b plus ribavirin for the initial treatment of chronic hepatitis C. Am J Gastroenterol 2003; 98:2354–2362.
- Salomon JA, Weinstein MC, Hammitt JK, Goldie SJ. Cost-effectiveness of treatment for chronic hepatitis C infection in an evolving patient population. JAMA 2003; 290:228–237.
- Sullivan SD, Jensen DM, Bernstein DE, et al. Cost-effectiveness of combination peginterferon alpha-2a and ribavirin compared with interferon alpha-2b and ribavirin in patients with chronic hepatitis C. Am J Gastroenterol 2004; 99:1490–1496.
- McGinn T, O’Connor-Moore N, Alfandre D, Gardenier D, Wisnivesky J. Validation of a hepatitis C screening tool in primary care. Arch Intern Med 2008; 168:2009–2013.
- Kaur S, Rybicki L, Bacon BR, Gollan JL, Rustgi VK, Carey WD. Performance characteristics and results of a large-scale screening program for viral hepatitis and risk factors associated with exposure to viral hepatitis B and C: results of the National Hepatitis Screening Survey. National Hepatitis Surveillance Group. Hepatology 1996; 24:979–986.
- Cheung RC. Epidemiology of hepatitis C virus infection in American veterans. Am J Gastroenterol 2000; 95:740–747.
- Austin GE, Jensen B, Leete J, et al. Prevalence of hepatitis C virus seropositivity among hospitalized US veterans. Am J Med Sci 2000; 319:353–359.
- Yawn BP, Wollan P, Gazzuola L, Kim WR. Diagnosis and 10-year follow-up of a community-based hepatitis C cohort. J Fam Pract 2002; 51:135–140.
- Garfein RS, Doherty MC, Monterroso ER, Thomas DL, Nelson KE, Vlahov D. Prevalence and incidence of hepatitis C virus infection among young adult injection drug users. J Acquir Immune Defic Syndr Hum Retrovirol 1998; 18(suppl 1):S11–S19.
- Alter MJ. The epidemiology of acute and chronic hepatitis C. Clin Liver Dis 1997; 1:559–568,
- Alter MJ, Hadler SC, Judson FN, et al. Risk factors for acute non-A, non-B hepatitis in the United States and association with hepatitis C virus infection. JAMA 1990; 264:2231–2235.
- Wasley A, Miller JT, Finelli L; Centers for Disease Control and Prevention (CDC). Surveillance for acute viral hepatitis—United States, 2005. MMWR Surveill Summ 2007; 56:1–24.
- Goedert JJ, Chen BE, Preiss L, Aledort LM, Rosenberg PS. Reconstruction of the hepatitis C virus epidemic in the US hemophilia population, 1940–1990. Am J Epidemiol 2007; 165:1443–1453.
- Eyster ME, Sherman KE, Goedert JJ, Katsoulidou A, Hatzakis A. Prevalence and changes in hepatitis C virus genotypes among multitransfused persons with hemophilia. The Multicenter Hemophilia Cohort Study. J Infect Dis 1999; 179:1062–1069.
- Yee TT, Griffioen A, Sabin CA, Dusheiko G, Lee CA. The natural history of HCV in a cohort of haemophilic patients infected between 1961 and 1985. Gut 2000; 47:845–851.
- Lee C, Dusheiko G. The natural history and antiviral treatment of hepatitis C in haemophilia. Haemophilia 2002; 8:322–329.
- Makris M, Garson JA, Ring CJ, Tuke PW, Tedder RS, Preston FE. Hepatitis C viral RNA in clotting factor concentrates and the development of hepatitis in recipients. Blood 1993; 81:1898–1902.
- Sherman KE, Rouster SD, Chung RT, Rajicic N. Hepatitis C virus prevalence among patients infected with human immunodeficiency virus: a cross-sectional analysis of the US adult AIDS Clinical Trials Group. Clin Infect Dis 2002; 34:831–837.
- Sulkowski MS. The HIV-coinfected patient: managing viral hepatitis. J Acquir Immune Defic Syndr 2007; 45(suppl 2):S36–S37.
- Bonacini M, Lin HJ, Hollinger FB. Effect of coexisting HIV-1 infection on the diagnosis and evaluation of hepatitis C virus. J Acquir Immune Defic Syndr 2001; 26:340–344.
- Garfein RS, Vlahov D, Galai N, Doherty MC, Nelson KE. Viral infections in short-term injection drug users: the prevalence of the hepatitis C, hepatitis B, human immunodeficiency, and human T-lymphotropic viruses. Am J Public Health 1996; 86:655–661.
- Bell J, Batey RG, Farrell GC, Crewe EB, Cunningham AL, Byth K. Hepatitis C virus in intravenous drug users. Med J Aust 1990; 153:274–276.
- Villano SA, Vlahov D, Nelson KE, Lyles CM, Cohn S, Thomas DL. Incidence and risk factors for hepatitis C among injection drug users in Baltimore, Maryland. J Clin Microbiol 1997; 35:3274–3277.
- Patrick DM, Tyndall MW, Cornelisse PG, et al. Incidence of hepatitis C virus infection among injection drug users during an outbreak of HIV infection. CMAJ 2001; 165:889–895.
- Seeff LB, Wright EC, Zimmerman HJ, McCollum RW. VA cooperative study of post-transfusion hepatitis, 1969-1974: incidence and characteristics of hepatitis and responsible risk factors. Am J Med Sci 1975; 270:355–362.
- Feinstone SM, Kapikian AZ, Purcell RH, Alter HJ, Holland PV. Transfusion-associated hepatitis not due to viral hepatitis type A or B. N Engl J Med 1975; 292:767–770.
- Alter HJ, Holland PV, Purcell RH, et al. Posttransfusion hepatitis after exclusion of commercial and hepatitis-B antigen-positive donors. Ann Intern Med. 1972; 77:691–699.
- Blajchman MA, Vamvakas EC. The continuing risk of transfusion-transmitted infections. N Engl J Med 2006; 355:1303–1305.
- Lauer GM, Walker BD. Hepatitis C virus infection. N Engl J Med 2001; 345:41–52.
- Roth D, Zucker K, Cirocco R, et al. The impact of hepatitis C virus infection on renal allograft recipients. Kidney Int 1994; 45:238–244.
- Izopet J, Rostaing L, Sandres K, et al. Longitudinal analysis of hepatitis C virus replication and liver fibrosis progression in renal transplant recipients. J Infect Dis 2000; 181:852–858.
- Dubois F, Desenclos JC, Mariotte N, Goudeau A. Hepatitis C in a French population-based survey, 1994: seroprevalence, frequency of viremia, genotype distribution, and risk factors. The Collaborative Study Group. Hepatology 1997; 25:1490–1496.
- Bellentani S, Pozzato G, Saccoccio G, et al. Clinical course and risk factors of hepatitis C virus related liver disease in the general population: report from the Dionysos study. Gut 1999; 44:874–880.
- Alberti A, Noventa F, Benvegnù L, Boccato S, Gatta A. Prevalence of liver disease in a population of asymptomatic persons with hepatitis C virus infection. Ann Intern Med 2002; 137:961–964.
- Shiffman ML, Diago M, Tran A, et al. Chronic hepatitis C in patients with persistently normal alanine transaminase levels. Clin Gastroenterol Hepatol 2006; 4:645–652.
- Stary A, Kopp W, Hofmann H, Heller-Vitouch C, Kunz C. Seroepidemiologic study of hepatitis C virus in sexually transmitted disease risk groups. Sex Transm Dis 1992; 19:252–258.
- Weinstock HS, Bolan G, Reingold AL, Polish LB. Hepatitis C virus infection among patients attending a clinic for sexually transmitted diseases. JAMA 1993; 269:392–394.
- Thomas DL, Gruninger SE, Siew C, Joy ED, Quinn TC. Occupational risk of hepatitis C infections among general dentists and oral surgeons in North America. Am J Med 1996; 100:41–45.
- Klein RS, Freeman K, Taylor PE, Stevens CE. Occupational risk for hepatitis C virus infection among New York City dentists. Lancet 1991; 338:1539–1542.
- Polish LB, Tong MJ, Co RL, Coleman PJ, Alter MJ. Risk factors for hepatitis C virus infection among health care personnel in a community hospital. Am J Infect Control 1993; 21:196–200.
- Alter MJ. Occupational exposure to hepatitis C virus: a dilemma. Infect Control Hosp Epidemiol 1994; 15:742–744.
- Lanphear BP, Linnemann CC, Cannon CG, DeRonde MM, Pendy L, Kerley LM. Hepatitis C virus infection in healthcare workers: risk of exposure and infection. Infect Control Hosp Epidemiol 1994; 15:745–750.
- Puro V, Petrosillo N, Ippolito G. Risk of hepatitis C seroconversion after occupational exposures in health care workers. Italian Study Group on Occupational Risk of HIV and Other Bloodborne Infections. Am J Infect Control 1995; 23:273–277.
- Mitsui T, Iwano K, Masuko K, et al. Hepatitis C virus infection in medical personnel after needlestick accident. Hepatology 1992; 16:1109–1114.
- Sartori M, La Terra G, Aglietta M, Manzin A, Navino C, Verzetti G. Transmission of hepatitis C via blood splash into conjunctiva. Scand J Infect Dis 1993; 25:270–271.
- Ippolito G, Puro V, Petrosillo N, De Carli G, Micheloni G, Magliano E. Simultaneous infection with HIV and hepatitis C virus following occupational conjunctival blood exposure. JAMA 1998; 280:28.
- Carey W. Tests and screening strategies for the diagnosis of hepatitis C. Cleve Clin J Med 2003; 70(suppl 4):S7–S13.
- Carey WD, Jeffers L, Kugelmas M, et al; Hepatitis C management. Hepatitis C Monograph. Cleveland Clinic; www.clevelandclinicmeded.com/online/monograph/hepc/page1.htm. Accessed 7/30/2010.
- Scott JD, Gretch DR. Molecular diagnostics of hepatitis C virus infection: a systematic review. JAMA 2007; 297:724–732.
- Bowden DS, Berzsenyi MD. Chronic hepatitis C virus infection: genotyping and its clinical role. Future Microbiol 2006; 1:103–112.
- Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 2002; 347:975–982.
- Poynard T, McHutchison J, Davis GL, et al. Impact of interferon alfa-2b and ribavirin on progression of liver fibrosis in patients with chronic hepatitis C. Hepatology 2000; 32:1131–1137.
- Zeuzem S. Interferon-based therapy for chronic hepatitis C: current and future perspectives. Nat Clin Pract Gastroenterol Hepatol 2008; 5:610–622.
- Alter MJ, Seeff LB, Bacon BR, Thomas DL, Rigsby MO, Di Bisceglie AM. Testing for hepatitis C virus infection should be routine for persons at increased risk for infection. Ann Intern Med 2004; 141:715–717.
- Ghany MG, Strader DB, Thomas DL, Seeff LB; American Association for the Study of Liver Diseases. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology 2009; 49:1335–1374.
- Shehab TM, Orrego M, Chunduri R, Lok AS. Identification and management of hepatitis C patients in primary care clinics. Am J Gastroenterol 2003; 98:639–644.
- Shehab TM, Sonnad SS, Lok AS. Management of hepatitis C patients by primary care physicians in the USA: results of a national survey. J Viral Hepat 2001; 8:377–383.
- US Centers for Disease Control and Prevention. Recommendations for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. MMWR Recomm Rep 1998; 47:1–39.
- US Centers for Disease Control and Prevention. National prevention strategy: a comprehensive strategy for the prevention and control of hepatitis C virus infection and its consequences; summer 2001. http://www.cdc.gov/hepatitis/HCV/Strategy/NatHep-CPrevStrategy.htm. Accessed August 8, 2010.
- Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med 2006; 144:705–714.
- Kim WR. The burden of hepatitis C in the United States. Hepatology 2002; 36(suppl 1):S30–S34.
- Lau DT, Kleiner DE, Ghany MG, Park Y, Schmid P, Hoofnagle JH. 10-Year follow-up after interferon-alpha therapy for chronic hepatitis C. Hepatology 1998; 28:1121–1127.
- Daniels D, Grytdal S, Wasley A; US Centers for Disease Control and Prevention. Surveillance for acute viral hepatitis—United States, 2007. MMWR Surveill Summ 2009; 58:1–27.
- Thomson BJ, Kwong G, Ratib S, et al; Trent HCV Study Group. Response rates to combination therapy for chronic HCV infection in a clinical setting and derivation of probability tables for individual patient management. J Viral Hepat 2008; 15:271–278.
- Hayashi N, Takehara T. Antiviral therapy for chronic hepatitis C: past, present, and future. J Gastroenterol 2006; 41:17–27.
- Gordon FD. Cost-effectiveness of screening patients for hepatitis C. Am J Med 1999; 107:36S–40S.
- Hill L, Henry B, Schweikert S; Prevention Practice Committee, American College of Preventive Medicine. Screening for chronic hepatitis C: American College of Preventive Medicine practice policy statement. Am J Prev Med 2005; 28:327–330.
- Hadziyannis SJ, Sette H, Morgan TR, et al; PEGASYS International Study Group. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann Intern Med 2004; 140:346–355.
- Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 2001; 358:958–965.
- Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 2002; 347:975–982.
- Cammà C, Di Bona D, Schepis F, et al. Effect of peginterferon alfa-2a on liver histology in chronic hepatitis C: a meta-analysis of individual patient data. Hepatology 2004; 39:333–342.
- Yoshida H, Tateishi R, Arakawa Y, et al. Benefit of interferon therapy in hepatocellular carcinoma prevention for individual patients with chronic hepatitis C. Gut 2004; 53:425–430.
- Yoshida H, Arakawa Y, Sata M, et al. Interferon therapy prolonged life expectancy among chronic hepatitis C patients. Gastroenterology 2002; 123:483–491.
- Hézode C, Forestier N, Dusheiko G, et al; PROVE2 Study Team. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N Engl J Med 2009; 360:1839–1850.
- McHutchison JG, Everson GT, Gordon SC, et al; PROVE1 Study Team. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009; 360:1827–1838.
- Berman K, Kwo PY. Boceprevir, an NS3 protease inhibitor of HCV. Clin Liver Dis 2009; 13:429–439.
- Rossignol JF, Elfert A, Keeffe EB. Treatment of chronic hepatitis C using a 4-week lead-in with nitazoxanide before peginterferon plus nitazoxanide. J Clin Gastroenterol 2009 Dec 31; epub ahead of print.
- Lapane KL, Jakiche AF, Sugano D, Weng CS, Carey WD. Hepatitis C infection risk analysis: who should be screened? Comparison of multiple screening strategies based on the National Hepatitis Surveillance Program. Am J Gastroenterol 1998; 93:591–596.
- Wong JB, Davis GL, McHutchison JG, Manns MP, Albrecht JK; International Hepatitis Interventional Therapy Group. Economic and clinical effects of evaluating rapid viral response to peginterferon alfa-2b plus ribavirin for the initial treatment of chronic hepatitis C. Am J Gastroenterol 2003; 98:2354–2362.
- Salomon JA, Weinstein MC, Hammitt JK, Goldie SJ. Cost-effectiveness of treatment for chronic hepatitis C infection in an evolving patient population. JAMA 2003; 290:228–237.
- Sullivan SD, Jensen DM, Bernstein DE, et al. Cost-effectiveness of combination peginterferon alpha-2a and ribavirin compared with interferon alpha-2b and ribavirin in patients with chronic hepatitis C. Am J Gastroenterol 2004; 99:1490–1496.
- McGinn T, O’Connor-Moore N, Alfandre D, Gardenier D, Wisnivesky J. Validation of a hepatitis C screening tool in primary care. Arch Intern Med 2008; 168:2009–2013.
- Kaur S, Rybicki L, Bacon BR, Gollan JL, Rustgi VK, Carey WD. Performance characteristics and results of a large-scale screening program for viral hepatitis and risk factors associated with exposure to viral hepatitis B and C: results of the National Hepatitis Screening Survey. National Hepatitis Surveillance Group. Hepatology 1996; 24:979–986.
- Cheung RC. Epidemiology of hepatitis C virus infection in American veterans. Am J Gastroenterol 2000; 95:740–747.
- Austin GE, Jensen B, Leete J, et al. Prevalence of hepatitis C virus seropositivity among hospitalized US veterans. Am J Med Sci 2000; 319:353–359.
- Yawn BP, Wollan P, Gazzuola L, Kim WR. Diagnosis and 10-year follow-up of a community-based hepatitis C cohort. J Fam Pract 2002; 51:135–140.
- Garfein RS, Doherty MC, Monterroso ER, Thomas DL, Nelson KE, Vlahov D. Prevalence and incidence of hepatitis C virus infection among young adult injection drug users. J Acquir Immune Defic Syndr Hum Retrovirol 1998; 18(suppl 1):S11–S19.
- Alter MJ. The epidemiology of acute and chronic hepatitis C. Clin Liver Dis 1997; 1:559–568,
- Alter MJ, Hadler SC, Judson FN, et al. Risk factors for acute non-A, non-B hepatitis in the United States and association with hepatitis C virus infection. JAMA 1990; 264:2231–2235.
- Wasley A, Miller JT, Finelli L; Centers for Disease Control and Prevention (CDC). Surveillance for acute viral hepatitis—United States, 2005. MMWR Surveill Summ 2007; 56:1–24.
- Goedert JJ, Chen BE, Preiss L, Aledort LM, Rosenberg PS. Reconstruction of the hepatitis C virus epidemic in the US hemophilia population, 1940–1990. Am J Epidemiol 2007; 165:1443–1453.
- Eyster ME, Sherman KE, Goedert JJ, Katsoulidou A, Hatzakis A. Prevalence and changes in hepatitis C virus genotypes among multitransfused persons with hemophilia. The Multicenter Hemophilia Cohort Study. J Infect Dis 1999; 179:1062–1069.
- Yee TT, Griffioen A, Sabin CA, Dusheiko G, Lee CA. The natural history of HCV in a cohort of haemophilic patients infected between 1961 and 1985. Gut 2000; 47:845–851.
- Lee C, Dusheiko G. The natural history and antiviral treatment of hepatitis C in haemophilia. Haemophilia 2002; 8:322–329.
- Makris M, Garson JA, Ring CJ, Tuke PW, Tedder RS, Preston FE. Hepatitis C viral RNA in clotting factor concentrates and the development of hepatitis in recipients. Blood 1993; 81:1898–1902.
- Sherman KE, Rouster SD, Chung RT, Rajicic N. Hepatitis C virus prevalence among patients infected with human immunodeficiency virus: a cross-sectional analysis of the US adult AIDS Clinical Trials Group. Clin Infect Dis 2002; 34:831–837.
- Sulkowski MS. The HIV-coinfected patient: managing viral hepatitis. J Acquir Immune Defic Syndr 2007; 45(suppl 2):S36–S37.
- Bonacini M, Lin HJ, Hollinger FB. Effect of coexisting HIV-1 infection on the diagnosis and evaluation of hepatitis C virus. J Acquir Immune Defic Syndr 2001; 26:340–344.
- Garfein RS, Vlahov D, Galai N, Doherty MC, Nelson KE. Viral infections in short-term injection drug users: the prevalence of the hepatitis C, hepatitis B, human immunodeficiency, and human T-lymphotropic viruses. Am J Public Health 1996; 86:655–661.
- Bell J, Batey RG, Farrell GC, Crewe EB, Cunningham AL, Byth K. Hepatitis C virus in intravenous drug users. Med J Aust 1990; 153:274–276.
- Villano SA, Vlahov D, Nelson KE, Lyles CM, Cohn S, Thomas DL. Incidence and risk factors for hepatitis C among injection drug users in Baltimore, Maryland. J Clin Microbiol 1997; 35:3274–3277.
- Patrick DM, Tyndall MW, Cornelisse PG, et al. Incidence of hepatitis C virus infection among injection drug users during an outbreak of HIV infection. CMAJ 2001; 165:889–895.
- Seeff LB, Wright EC, Zimmerman HJ, McCollum RW. VA cooperative study of post-transfusion hepatitis, 1969-1974: incidence and characteristics of hepatitis and responsible risk factors. Am J Med Sci 1975; 270:355–362.
- Feinstone SM, Kapikian AZ, Purcell RH, Alter HJ, Holland PV. Transfusion-associated hepatitis not due to viral hepatitis type A or B. N Engl J Med 1975; 292:767–770.
- Alter HJ, Holland PV, Purcell RH, et al. Posttransfusion hepatitis after exclusion of commercial and hepatitis-B antigen-positive donors. Ann Intern Med. 1972; 77:691–699.
- Blajchman MA, Vamvakas EC. The continuing risk of transfusion-transmitted infections. N Engl J Med 2006; 355:1303–1305.
- Lauer GM, Walker BD. Hepatitis C virus infection. N Engl J Med 2001; 345:41–52.
- Roth D, Zucker K, Cirocco R, et al. The impact of hepatitis C virus infection on renal allograft recipients. Kidney Int 1994; 45:238–244.
- Izopet J, Rostaing L, Sandres K, et al. Longitudinal analysis of hepatitis C virus replication and liver fibrosis progression in renal transplant recipients. J Infect Dis 2000; 181:852–858.
- Dubois F, Desenclos JC, Mariotte N, Goudeau A. Hepatitis C in a French population-based survey, 1994: seroprevalence, frequency of viremia, genotype distribution, and risk factors. The Collaborative Study Group. Hepatology 1997; 25:1490–1496.
- Bellentani S, Pozzato G, Saccoccio G, et al. Clinical course and risk factors of hepatitis C virus related liver disease in the general population: report from the Dionysos study. Gut 1999; 44:874–880.
- Alberti A, Noventa F, Benvegnù L, Boccato S, Gatta A. Prevalence of liver disease in a population of asymptomatic persons with hepatitis C virus infection. Ann Intern Med 2002; 137:961–964.
- Shiffman ML, Diago M, Tran A, et al. Chronic hepatitis C in patients with persistently normal alanine transaminase levels. Clin Gastroenterol Hepatol 2006; 4:645–652.
- Stary A, Kopp W, Hofmann H, Heller-Vitouch C, Kunz C. Seroepidemiologic study of hepatitis C virus in sexually transmitted disease risk groups. Sex Transm Dis 1992; 19:252–258.
- Weinstock HS, Bolan G, Reingold AL, Polish LB. Hepatitis C virus infection among patients attending a clinic for sexually transmitted diseases. JAMA 1993; 269:392–394.
- Thomas DL, Gruninger SE, Siew C, Joy ED, Quinn TC. Occupational risk of hepatitis C infections among general dentists and oral surgeons in North America. Am J Med 1996; 100:41–45.
- Klein RS, Freeman K, Taylor PE, Stevens CE. Occupational risk for hepatitis C virus infection among New York City dentists. Lancet 1991; 338:1539–1542.
- Polish LB, Tong MJ, Co RL, Coleman PJ, Alter MJ. Risk factors for hepatitis C virus infection among health care personnel in a community hospital. Am J Infect Control 1993; 21:196–200.
- Alter MJ. Occupational exposure to hepatitis C virus: a dilemma. Infect Control Hosp Epidemiol 1994; 15:742–744.
- Lanphear BP, Linnemann CC, Cannon CG, DeRonde MM, Pendy L, Kerley LM. Hepatitis C virus infection in healthcare workers: risk of exposure and infection. Infect Control Hosp Epidemiol 1994; 15:745–750.
- Puro V, Petrosillo N, Ippolito G. Risk of hepatitis C seroconversion after occupational exposures in health care workers. Italian Study Group on Occupational Risk of HIV and Other Bloodborne Infections. Am J Infect Control 1995; 23:273–277.
- Mitsui T, Iwano K, Masuko K, et al. Hepatitis C virus infection in medical personnel after needlestick accident. Hepatology 1992; 16:1109–1114.
- Sartori M, La Terra G, Aglietta M, Manzin A, Navino C, Verzetti G. Transmission of hepatitis C via blood splash into conjunctiva. Scand J Infect Dis 1993; 25:270–271.
- Ippolito G, Puro V, Petrosillo N, De Carli G, Micheloni G, Magliano E. Simultaneous infection with HIV and hepatitis C virus following occupational conjunctival blood exposure. JAMA 1998; 280:28.
- Carey W. Tests and screening strategies for the diagnosis of hepatitis C. Cleve Clin J Med 2003; 70(suppl 4):S7–S13.
- Carey WD, Jeffers L, Kugelmas M, et al; Hepatitis C management. Hepatitis C Monograph. Cleveland Clinic; www.clevelandclinicmeded.com/online/monograph/hepc/page1.htm. Accessed 7/30/2010.
- Scott JD, Gretch DR. Molecular diagnostics of hepatitis C virus infection: a systematic review. JAMA 2007; 297:724–732.
- Bowden DS, Berzsenyi MD. Chronic hepatitis C virus infection: genotyping and its clinical role. Future Microbiol 2006; 1:103–112.
- Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 2002; 347:975–982.
- Poynard T, McHutchison J, Davis GL, et al. Impact of interferon alfa-2b and ribavirin on progression of liver fibrosis in patients with chronic hepatitis C. Hepatology 2000; 32:1131–1137.
- Zeuzem S. Interferon-based therapy for chronic hepatitis C: current and future perspectives. Nat Clin Pract Gastroenterol Hepatol 2008; 5:610–622.
KEY POINTS
- Patients who should be screened include intravenous drug abusers, people infected with human immunodeficiency virus, patients with unexplained elevated alanine aminotransferase levels, infants born to infected mothers, and people with infected sexual partners.
- Patients at risk of HCV infection should be tested for anti-HCV antibody using an enzyme immunoassay (EIA).
- Positive results on anti-HCV EIA testing should be confirmed with an assay for HCV RNA.
- HCV genotyping can help predict the response to therapy. Genotypes 2 or 3 are more likely to respond to therapy than genotype 1.
Noninvasive tests for liver disease, fibrosis, and cirrhosis: Is liver biopsy obsolete?
Primary care physicians and specialists alike often encounter patients with chronic liver disease. Fortunately, these days we need to resort to liver biopsy less often than in the past.
The purpose of this review is to provide a critical assessment of the growing number of noninvasive tests available for diagnosing liver disease and assessing hepatic fibrosis, and to discuss the implications of these advances related to the indications for needle liver biopsy.
WHEN IS LIVER BIOPSY USEFUL?
In diagnosis
Needle liver biopsy for diagnosis remains important in cases of:
Diagnostic uncertainty (eg, in patients with atypical features)
Coexisting disorders (eg, human immunodeficiency virus [HIV] and hepatitis C virus infection, or alcoholic liver disease and hepatitis C)
An overlapping syndrome (eg, primary biliary cirrhosis with autoimmune hepatitis).
Fatty liver. Needle liver biopsy can distinguish between benign steatosis and progressive steatohepatitis in a patient with a fatty liver found on imaging, subject to the limitations of sampling error.
Because fatty liver disease is common and proven treatments are few, no consensus has emerged about which patients with suspected fatty liver disease should undergo needle biopsy. Many specialists eschew needle biopsy and treat the underlying risk factors of metabolic syndrome, reserving biopsy for patients with findings that raise the concern of cirrhosis.
Hereditary disorders, eg, hemochromatosis, alpha-1 antitrypsin deficiency, and Wilson disease.
In management
Periodic needle biopsy is also valuable in the management of a few diseases.
In autoimmune hepatitis, monitoring the plasma cell score on liver biopsy may help predict relapse when a physician is considering reducing or discontinuing immunosuppressive therapy.1
After liver transplantation, a liver biopsy is highly valuable to assess for rejection and the presence and intensity of disease recurrence.
PROBLEMS WITH LIVER BIOPSY
Liver biopsy is invasive and can cause significant complications. Nearly 30% of patients report having substantial pain after liver biopsy, and some experience serious complications such as pneumothorax, bleeding, or puncture of the biliary tree. In rare cases, patients die of bleeding.2
Furthermore, hepatic pathology, particularly fibrosis, is not always uniformly distributed. Surgical wedge biopsy provides adequate tissue volume to overcome this problem. Needle biopsy, on the other hand, provides a much smaller volume of tissue (1/50,000 of the total mass of the liver).3
As examples of the resulting sampling errors that can occur, consider the two most common chronic liver diseases: hepatitis C and fatty liver disease.
Regev et al4 performed laparoscopically guided biopsy of the right and left hepatic lobes in a series of 124 patients with chronic hepatitis C. Biopsy samples from the right and left lobes differed in the intensity of inflammation in 24.2% of cases, and in the intensity of fibrosis in 33.1%. Differences of more than one grade of inflammation or stage of fibrosis were uncommon. However, in 14.5%, cirrhosis was diagnosed in one lobe but not the other.
In a study in patients with nonalcoholic fatty liver disease, Ratziu et al5 found that none of the features characteristic of nonalcoholic steatohepatitis were highly concordant in paired liver biopsies. Clearly, needle liver biopsy is far from an ideal test.
Increasingly, liver diseases can be diagnosed precisely with laboratory tests, imaging studies, or both. Thus, needle liver biopsy is playing a lesser role in diagnosis.
ADVANCES IN NONINVASIVE DIAGNOSIS OF LIVER DISEASE
Over the past 30 years, substantial strides have been made in our ability to make certain diagnoses through noninvasive means.
Blood tests can be used to diagnose viral hepatitis A, B, and C and many cases of hemochromatosis and primary biliary cirrhosis. For a detailed discussion of how blood tests are used in diagnosing liver diseases, see www.clevelandclinicmeded.com/medicalpubs/diseasemanagement/hepatology/guide-to-common-liver-tests/.
Imaging studies. Primary sclerosing cholangitis can be diagnosed with an imaging study, ie, magnetic resonance cholangiopancreatography (MRCP) or endoscopic retrograde cholangiopancreatography (ERCP). The value of needle biopsy in these patients is limited to assessing the degree of fibrosis to help with management of the disease and, less often, to discovering other liver pathologies.6
Most benign space-occupying liver lesions, both cystic and solid, can be fully characterized by imaging, especially in patients who have no underlying chronic liver disease, and no biopsy is needed. Whether biopsy should be performed to investigate liver lesions depends on the clinical scenario; the topic is beyond the scope of this paper but has been reviewed in detail by Rockey et al.2
CAN NONINVASIVE TESTS DETECT HEPATIC FIBROSIS?
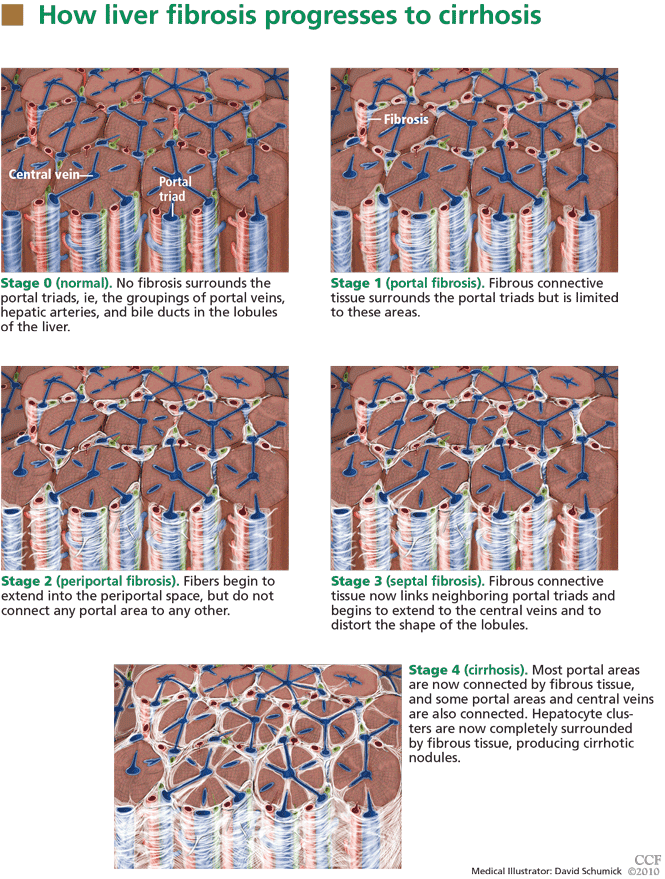
Cirrhosis (stage 4 fibrosis) results in nodular transformation of the liver and impedance of portal blood flow, setting the stage for portal hypertension and its sequelae. Knowing whether cirrhosis is present is important in subsequent management.
In advanced cases, cirrhosis is associated with typical clinical manifestations and laboratory and radiographic findings. In such cases, needle biopsy will add little. However, in most cases, particularly early in the course, clinical, laboratory, and radiologic correlates of cirrhosis are absent. In one study of patients with hepatitis C, 27% had cirrhosis, but in only a small number would cirrhosis have been apparent from clinical signs and laboratory and imaging studies.6
Since a major contemporary role for liver biopsy is in assessing the degree of fibrosis, it is reasonable to ask if newer noninvasive means are available to estimate hepatic fibrosis. The remainder of this review focuses on assessing our increasing ability to stage the degree of fibrosis (including the presence or absence of cirrhosis) by noninvasive means.
Clinical features point to cirrhosis, but not earlier fibrosis
Clinical manifestations help point to the diagnosis of cirrhosis but not to earlier stages of fibrosis.
For example, if a patient is known to have liver disease, the findings of ascites, splenomegaly, or asterixis mean that cirrhosis is highly probable. Similarly, hypersplenism (splenomegaly with a decrease in circulating blood cells but a normal to hyperactive bone marrow) in a patient with liver test abnormalities almost always represents portal hypertension due to cirrhosis, although other, nonhepatic causes are possible, such as congestive heart failure and constrictive pericarditis.
These features generally emerge late in the course of cirrhosis. The absence of such stigmata certainly does not preclude the presence of cirrhosis. Thus, these clinical signs have a high positive predictive value but a low negative predictive value, making them insufficient by themselves to diagnose or stage liver disease.
Laboratory tests are of limited value in assessing the degree of fibrosis
Standard liver tests are of limited value in assessing the degree of fibrosis.
Usual laboratory tests. At one end of the spectrum, anemia, thrombocytopenia, and leukopenia in the presence of liver disease correlate with cirrhosis. At the other end, a serum ferritin concentration of less than 1,000 mg/mL in a patient with hemochromatosis and no confounding features such as hepatitis C, HIV infection, or heavy alcohol use strongly predicts that the patient does not have significant hepatic fibrosis.8
Bilirubin elevation is a late finding in cirrhosis, but in cholestatic diseases bilirubin may be elevated before cirrhosis occurs.
Albumin is made exclusively in the liver, and its concentration falls as liver function worsens with progressive cirrhosis.
The prothrombin time increases as the liver loses its ability to synthesize clotting factors in cirrhosis. Coagulopathy correlates with the degree of liver disease.
Hyponatremia due to impaired ability to excrete free water is seen in patients with cirrhosis and ascites.
In summary, the usual laboratory tests related to liver disease are imprecise and, when abnormal, often indicate not just the presence of cirrhosis, but impending or actual decompensation.
Newer serologic markers, alone or in combination, have been proposed as aids in determining the degree of fibrosis or cirrhosis in the liver. Direct markers of fibrosis measure the turnover or metabolism of extracellular matrix. Indirect markers of fibrosis reflect alterations in hepatic function (see below).
Parkes et al9 reviewed 10 different panels of serum markers of hepatic fibrosis in chronic hepatitis C. Only 35% of patients had fibrosis adequately ruled in or ruled out by these panels, and the stage of fibrosis could not be adequately determined.
These serologic markers have not been validated in other chronic liver diseases or in liver disease due to multiple causes. Thus, although they show promise for use by the general internist, they need to be validated in patients with disease and in normal reference populations before they are ready for “prime time.”
Direct serologic markers of fibrosis
Direct serologic markers of fibrosis include those associated with matrix deposition—eg, procollagen type III amino-terminal peptide (P3NP), type I and IV collagens, laminin, hyaluronic acid, and chondrex.
P3NP is the most widely studied marker of hepatic fibrosis. It is elevated in both acute and chronic liver diseases; serum levels reflect the histologic stage of hepatic fibrosis in various chronic liver diseases, including alcoholic, viral, and primary biliary cirrhosis.10–12 Successful treatment of autoimmune hepatitis has been shown to lead to reductions of P3NP levels.13
Other direct markers of fibrosis are those associated with matrix degradation, ie, matrix metalloproteinases 2 and 3 (MMP-2, MMP-3) and tissue inhibitors of metalloproteinases 1 and 2 (TIMP-1, TIMP-2). Levels of MMP-2 proenzymes and active enzymes are increased in liver disease, but studies are inconsistent in correlating serum levels of MMP-2 to the degree of hepatic fibrosis.14,15 These tests are not commercially available, and the components are not readily available in most clinical laboratories.
Indirect serologic markers of fibrosis
Some indirect markers are readily available:
The AST:ALT ratio. The normal ratio of aspartate aminotransferase (AST) to alanine aminotransferase (ALT) is approximately 0.8. A ratio greater than 1.0 provides evidence of cirrhosis. However, findings have been inconsistent.
The AST:platelet ratio index (APRI), a commonly used index, is calculated by the following formula:
![]()
In studies of hepatitis C and hepatitis C-HIV, the APRI has shown a sensitivity of 37% to 80% and a specificity of 45% to 98%, depending on the cutoff value and whether a diagnosis of severe fibrosis or cirrhosis was being tested.16–19 These sensitivities and specificities are disappointing and do not provide information equal to that provided by needle liver biopsy in most patients with chronic liver disease.
The combination of prothrombin, gamma glutamyl, and apolipoprotein AI levels (PGA index) has been validated in patients with many types of chronic liver disease, and its accuracy for detecting cirrhosis is highest (66%–72%) in patients with alcoholic liver disease.20,21
FibroIndex uses the platelet count, AST level, and gamma globulin level to detect significant fibrosis in chronic hepatitis C, but its accuracy has yet to be validated.22
The FIB-4 index is based on four independent predictors of fibrosis, ie, age, the platelet count, AST level, and ALT level. It has shown good accuracy for detecting advanced fibrosis in two studies in patients with hepatitis C.23,24
Fibrometer (based on the platelet count; the prothrombin index; the levels of AST, alfa-2 macroglobulin, hyaluronate, and blood urea nitrogen; and age) predicted fibrosis well in chronic viral hepatitis.25,26
Fibrotest and Fibrosure are proprietary commercial tests available in many laboratories. They employ a mathematical formula to predict fibrosis (characterized as mild, significant, or indeterminate) using the levels of alpha-2 macroglobulin, alpha-2 globulin, gamma globulin, apolipoprotein A1, gamma glutamyl transferase, and total bilirubin. For detecting significant fibrosis, these tests are reported to have a sensitivity of about 75% and a specificity of 85%.27–29
ActiTest incorporates the ALT level into the Fibrotest to reflect liver fibrosis and necro-inflammatory activity.
A meta-analysis showed that Fibrotest and ActiTest could be reliable alternatives to liver biopsy in patients with chronic hepatitis C.30 The area under the receiver operator characteristic curve for the diagnosis of significant fibrosis ranged from 0.73 to 0.87; for the diagnosis of significant histologic activity it ranged from 0.75 to 0.86. Fibrotest had a negative predictive value for excluding significant fibrosis of 91% with a cutoff of 0.31. ActiTest’s negative predictive value for excluding significant necrosis was 85% with a cutoff of 0.36. None of these serum tests have become part of standard of practice for diagnosing fibrosis or cirrhosis.
The Sequential Algorithm for Fibrosis Evaluation (SAFE) combines the APRI and Fibrotest-Fibrosure tests in a sequential fashion to test for fibrosis and cirrhosis. In a large multicenter study31 validating this algorithm to detect significant fibrosis (stage F2 or greater by the F0–F4 METAVIR scoring system32), its accuracy was 90.1%, the area under the receiver operating characteristic curve was 0.89 (95% CI 0.87–0.90), and it reduced the number of liver biopsies needed by 46.5%. When the algorithm was used to detect cirrhosis, its accuracy was 92.5%, the area under the curve was 0.92 (95% CI 0.89–0.94), and it reduced the number of liver biopsies needed by 81.5%.
Another algorithm was developed to simultaneously detect significant fibrosis and cirrhosis. It had a 97.4% accuracy, but 64% of patients still required a liver biopsy.31
SAFE algorithms have the potential to reduce the number of needle biopsies needed to assess the degree of hepatic fibrosis.
CONVENTIONAL IMAGING STUDIES ARE NOT SENSITIVE FOR FIBROSIS
Standard imaging studies often show findings of cirrhosis but are not particularly sensitive, with a low negative predictive value.
Ultrasonography can show a small, nodular liver in advanced cirrhosis, but surface nodularity or increased echogenicity can be seen in hepatic steatosis as well as in cirrhosis. In one study,33 ultrasonography identified diffuse parenchymal disease but could not reliably distinguish fat from fibrosis or diagnose cirrhosis.
Often, in cirrhosis, the right lobe of the liver is atrophied and the caudate or left lobes are hypertrophied. Efforts to use the ratio of the widths of the lobes to diagnose cirrhosis have shown varying performance characterstics.34,35
One study of the splenic artery pulsatility index has shown this to be an accurate predictor of cirrhosis.36
Computed tomography provides information similar to that of ultrasonography, and it can identify complications of cirrhosis, including portal hypertension and ascites. On the other hand, it costs more and it exposes the patient to radiation and contrast media.
ELASTOGRAPHY, A PROMISING TEST
Hepatic elastography, a method for estimating liver stiffness, is an exciting recent development in the noninvasive measurement of hepatic fibrosis. Currently, elastography can be accomplished by ultrasound or magnetic resonance.
Ultrasound elastography
The FibroScan device (EchoSens, Paris, France) uses a mild-amplitude, low-frequency (50-Hz) vibration transmitted through the liver.37 It induces an elastic shear wave that is detected by pulse-echo ultrasonography as the wave propagates through the organ.
The velocity of the wave correlates with tissue stiffness: the wave travels faster through denser, fibrotic tissue.38,39
Ultrasound elastography (also called transient elastography) can sample a much larger area than liver biopsy can, providing a better understanding of the entire hepatic parenchyma. 40 Moreover, it can be repeated often without risk. This device is in widespread use in many parts of the world, but it is not yet approved in the United States.
A meta-analysis of 50 studies assessed the overall performance of ultrasound elastography for diagnosing liver fibrosis.41 The areas under the receiver operating characteristic curve were as follows:
- For significant fibrosis: 0.84 (95% CI 0.82–0.86)
- For severe fibrosis: 0.89 (95% CI 0.88–0.91)
- For cirrhosis: 0.94 (95% CI 0.93–0.95).
The type of underlying liver disease influenced the diagnosis of significant fibrosis, which was diagnosed most consistently in patients with hepatitis C. The authors concluded that ultrasound elastography had excellent diagnostic accuracy for diagnosing cirrhosis irrespective of the underlying liver disease, while the diagnosis of significant fibrosis had higher variation, which was dependent on the underlying liver disease.
A meta-analysis of nine studies42 showed ultrasound elastography to have a sensitivity of 87% (95% CI 84%–90%) and a specificity of 91% (95% CI 89%–92%) for the diagnosis of cirrhosis. In seven of the nine studies, it diagnosed stage II to IV fibrosis with 70% sensitivity (95% CI 67%–73%) and 84% specificity (95% CI 80%–88%).
Limitations. Ultrasound elastography is less effective in obese patients, as the adipose tissue attenuates the elastic wave, and it has not been reliable in patients with acute viral hepatitis.43 Male sex, body mass index greater than 30, and metabolic syndrome seem to increase liver stiffness, thus limiting the use of this test.44
Until more data are available, the ultimate value of ultrasound elastography in reducing the number of liver biopsies needed remains unknown. However, this test shows potential as a reliable and noninvasive way to assess the degree of fibrosis in patients with liver disease.
Magnetic resonance elastography
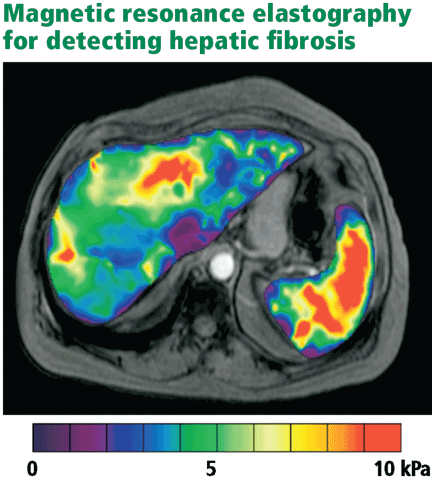
Studies have shown a magnetic resonance scoring system that distinguishes Child-Pugh grade A cirrhosis from other grades to be 93% sensitive and 82% specific.45
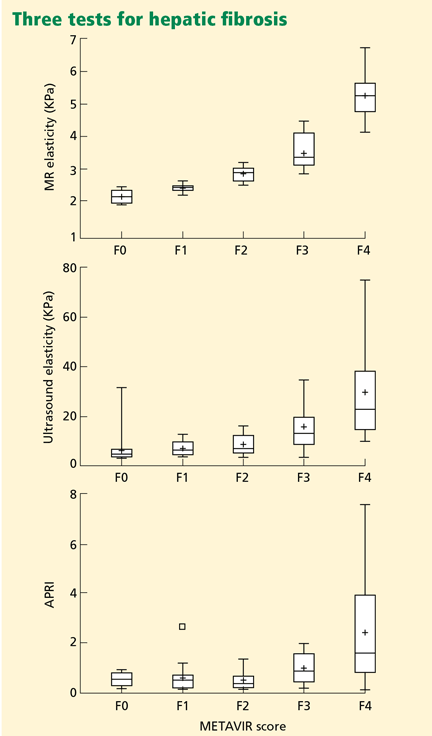
Cost may limit the use of magnetic resonance elastography, and some patients may be unable to tolerate the procedure because of claustrophobia. It seems clear, though, that this test currently has the most promise in reducing the need for liver biopsy for grading the severity of hepatic fibrosis.
WHERE ARE WE NOW?
The importance of liver biopsy in arriving at a diagnosis of diffuse parenchymal liver disease is being diminished by accurate blood testing strategies for chronic viral hepatitis, autoimmune hepatitis, and primary biliary cirrhosis. Further, imaging tests are superior to liver biopsy in the diagnosis of primary sclerosing cholangitis.
However, many cases remain in which diagnostic confusion exists even after suitable laboratory testing and imaging studies. Diagnosing infiltrative disease (eg, amyloidosis, sarcoidosis), separating benign fatty liver disease from steatohepatitis, and evaluating liver parenchyma after liver transplantation are best accomplished by liver biopsy.
While needle biopsy is still the mainstay in diagnosing hepatic fibrosis, its days of dominance seem limited as technology improves. When physical examination or standard laboratory tests reveal clear-cut signs of portal hypertension, liver biopsy will seldom add useful information. Similarly, when imaging studies provide compelling evidence of cirrhosis and portal hypertension, needle biopsy is not warranted.
The SAFE algorithms warrant further evaluation in all chronic liver diseases, as they may help decrease the number of liver biopsies required. And we believe elastography will play an ever-increasing role in the assessment of hepatic fibrosis and will significantly reduce the need for biopsy in patients with liver disease.
- Verma S, Gunuwan B, Mendler M, Govindrajan S, Redeker A. Factors predicting relapse and poor outcome in type I autoimmune hepatitis: role of cirrhosis development, patterns of transaminases during remission and plasma cell activity in the liver biopsy. Am J Gastroenterol 2004; 99:1510–1516.
- Rockey DC, Caldwell SH, Goodman ZD, Nelson RC, Smith AD; American Association for the Study of Liver Diseases. Liver biopsy. Hepatology 2009; 49:1017–1044.
- Bravo AA, Sheth SG, Chopra S. Liver biopsy. N Engl J Med 2001; 344:495–500.
- Regev A, Berho M, Jeffers LJ, et al. Sampling error and intraobserver variation in liver biopsy in patients with chronic HCV infection. Am J Gastroenterol 2002; 97:2614–2618.
- Ratziu V, Charlotte F, Heurtier A, et al; LIDO Study Group Sampling variability of liver biopsy in nonalcoholic fatty liver disease. Gastroenterology 2005; 128:1898–1906.
- Saadeh S, Cammell G, Carey WD, Younossi Z, Barnes D, Easley K. The role of liver biopsy in chronic hepatitis C. Hepatology 2001; 33:196–200.
- Batts KP, Ludwig J. Chronic hepatitis. An update on terminology and reporting. Am J Surg Pathol 1995; 19:1409–1417.
- Morrison ED, Brandhagen DJ, Phatak PD, et al. Serum ferritin level predicts advanced hepatic fibrosis among U.S. patients with phenotypic hemochromatosis. Ann Intern Med 2003; 138:627–633.
- Parkes J, Guha IN, Roderick P, Rosenberg W. Performance of serum marker panels for liver fibrosis in chronic hepatitis C. J Hepatol 2006; 44:462–474.
- Montalto G, Soresi M, Aragona F, et al. Procollagen III and laminin in chronic viral hepatopathies. Presse Med 1996; 25:59–62.
- Teare JP, Sherman D, Greenfield SM, et al. Comparison of serum procollagen III peptide concentrations and PGA index for assessment of hepatic fibrosis. Lancet 1993; 342:895–898.
- Trinchet JC, Hartmann DJ, Pateron D, et al. Serum type I collagen and N-terminal peptide of type III procollagen in chronic hepatitis. Relationship to liver histology and conventional liver tests. J Hepatol 1991; 12:139–144.
- McCullough AJ, Stassen WN, Wiesner RH, Czaja AJ. Serial determinations of the amino-terminal peptide of type III procollagen in severe chronic active hepatitis. J Lab Clin Med 1987; 109:55–61.
- Takahara T, Furui K, Funaki J, et al. Increased expression of matrix metalloproteinase-II in experimental liver fibrosis in rats. Hepatology 1995; 21:787–795.
- Takahara T, Furui K, Yata Y, et al. Dual expression of matrix metalloproteinase-2 and membrane-type 1-matrix metalloproteinase in fibrotic human livers. Hepatology 1997; 26:1521–1529.
- Wai CT, Greenson JK, Fontana RJ, et al. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology 2003; 38:518–526.
- Kelleher TB, Mehta SH, Bhaskar R, et al. Prediction of hepatic fibrosis in HIV/HCV co-infected patients using serum fibrosis markers: the SHASTA index. J Hepatol 2005; 43:78–84.
- Islam S, Antonsson L, Westin J, Lagging M. Cirrhosis in hepatitis C virus-infected patients can be excluded using an index of standard biochemical serum markers. Scand J Gastroenterol 2005; 40:867–872.
- Lackner C, Struber G, Liegl B, et al. Comparison and validation of simple noninvasive tests for prediction of fibrosis in chronic hepatitis C. Hepatology 2005; 41:1376–1382.
- Poynard T, Aubert A, Bedossa P, et al. A simple biological index for detection of alcoholic liver disease in drinkers. Gastroenterology 1991; 100:1397–1402.
- Oberti F, Valsesia E, Pilette C, et al. Noninvasive diagnosis of hepatic fibrosis or cirrhosis. Gastroenterology 1997; 113:1609–1616.
- Koda M, Matunaga Y, Kawakami M, Kishimoto Y, Suou T, Murawaki Y. FibroIndex, a practical index for predicting significant fibrosis in patients with chronic hepatitis C. Hepatology 2007; 45:297–306.
- Vallet-Pichard A, Mallet V, Nalpas B, et al. FIB-4: an inexpensive and accurate marker of fibrosis in HCV infection. Comparison with liver biopsy and fibrotest. Hepatology 2007; 46:32–36.
- Sterling RK, Lissen E, Clumeck N, et al; APRI COT Clinical Investigators. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology 2006; 43:1317–1325.
- Calès P, Oberti F, Michalak S, et al. A novel panel of blood markers to assess the degree of liver fibrosis. Hepatology 2005; 42:1373–1381.
- Leroy V, Hilleret MN, Sturm N, et al. Prospective comparison of six non-invasive scores for the diagnosis of liver fibrosis in chronic hepatitis C. J Hepatol 2007; 46:775–782.
- Myers RP, De Torres M, Imbert-Bismut F, Ratziu V, Charlotte F, Poynard T; MULTIVIRC Group. Biochemical markers of fibrosis in patients with chronic hepatitis C: a comparison with prothrombin time, platelet count, and age-platelet index. Dig Dis Sci 2003; 48:146–153.
- Rossi E, Adams L, Prins A, et al. Validation of the FibroTest biochemical markers score in assessing liver fibrosis in hepatitis C patients. Clin Chem 2003; 49:450–454.
- Halfon P, Bourliere M, Deydier R, et al. Independent prospective multicenter validation of biochemical markers (fibrotest-actitest) for the prediction of liver fibrosis and activity in patients with chronic hepatitis C: the fibropaca study. Am J Gastroenterol 2006; 101:547–555.
- Poynard T, Imbert-Bismut F, Munteanu M, et al. Overview of the diagnostic value of biochemical markers of liver fibrosis (FibroTest, HCV FibroSure) and necrosis (ActiTest) in patients with chronic hepatitis C. Comp Hepatol 2004; 3:8.
- Sebastiani G, Halfon P, Castera L, et al. SAFE biopsy: a validated method for large-scale staging of liver fibrosis in chronic hepatitis C. Hepatology 2009; 49:1821–1827.
- The French METAVIR Cooperative Study Group. Intraobserver and interobserver variations in liver biopsy interpretations in patients with chronic hepatitis C. Hepatology 1994; 20:15–20.
- Sanford NL, Walsh P, Matis C, Baddeley H, Powell LW. Is ultrasonography useful in the assessment of diffuse parenchymal liver disease? Gastroenterology 1985; 89:186–191.
- Harbin WP, Robert NJ, Ferrucci JT. Diagnosis of cirrhosis based on regional changes in hepatic morphology: a radiological and pathological analysis. Radiology 1980; 135:273–283.
- Giorgio A, Amoroso P, Lettieri G, et al. Cirrhosis: value of caudate to right lobe ratio in diagnosis with US. Radiology 1986; 161:443–445.
- Liu CH, Hsu SJ, Lin JW, et al. Noninvasive diagnosis of hepatic fibrosis in patients with chronic hepatitis C by splenic Doppler impedance index. Clin Gastroenterol Hepatol 2007; 5:1199–1206.
- Talawalkar JA. Elastography for detecting hepatic fibrosis: options and considerations. Gastroenterology 2008; 135:299–302.
- Sandrin L, Fourquet B, Hasquenoph JM, et al. Transient elastography: a new noninvasive method for assessment of hepatic fibrosis. Ultrasound Med Biol 2003; 29:1705–1713.
- Kettaneh A, Marcellin P, Douvin C, et al. Features associated with success rate and performance of FibroScan measurements for the diagnosis of cirrhosis in HCV patients: a prospective study of 935 patients. J Hepatol 2007; 46:628–634.
- Ziol M, Handra-Luca A, Kettaneh A, et al. Noninvasive assessment of liver fibrosis by measurement of stiffness in patients with chronic hepatitis C. Hepatology 2005; 41:48–54.
- Friedrich-Rust M, Ong MF, Martens S, et al. Performance of transient elastography for the staging of liver fibrosis: a meta-analysis. Gastroenterology 2008; 134:960–974.
- Talwalkar JA, Kurtz DM, Schoenleber SJ, West CP, Montori VM. Ultrasound-based transient elastography for the detection of hepatic fibrosis: systematic review and meta-analysis. Clin Gastroenterol Hepatol 2007; 5:1214–1220.
- Arena U, Vizzutti F, Corti G, et al. Acute viral hepatitis increases liver stiffness values measured by transient elastography. Hepatology 2008; 47:380–384.
- Roulot D, Czernichow S, Le Clésiau H, Costes JL, Vergnaud AC, Beaugrand M. Liver stiffness values in apparently healthy subjects: influence of gender and metabolic syndrome. J Hepatol 2008; 48:606–613.
- Ito K, Mitchell DG, Hann HW, et al. Viral-induced cirrhosis: grading of severity using MR imaging. AJR Am J Roentgenol 1999; 173:591–596.
- Huwart L, Sempoux C, Vicaut E, et al. Magnetic resonance elastography for the noninvasive staging of liver fibrosis. Gastroenterology 2008; 135:32–40.
Primary care physicians and specialists alike often encounter patients with chronic liver disease. Fortunately, these days we need to resort to liver biopsy less often than in the past.
The purpose of this review is to provide a critical assessment of the growing number of noninvasive tests available for diagnosing liver disease and assessing hepatic fibrosis, and to discuss the implications of these advances related to the indications for needle liver biopsy.
WHEN IS LIVER BIOPSY USEFUL?
In diagnosis
Needle liver biopsy for diagnosis remains important in cases of:
Diagnostic uncertainty (eg, in patients with atypical features)
Coexisting disorders (eg, human immunodeficiency virus [HIV] and hepatitis C virus infection, or alcoholic liver disease and hepatitis C)
An overlapping syndrome (eg, primary biliary cirrhosis with autoimmune hepatitis).
Fatty liver. Needle liver biopsy can distinguish between benign steatosis and progressive steatohepatitis in a patient with a fatty liver found on imaging, subject to the limitations of sampling error.
Because fatty liver disease is common and proven treatments are few, no consensus has emerged about which patients with suspected fatty liver disease should undergo needle biopsy. Many specialists eschew needle biopsy and treat the underlying risk factors of metabolic syndrome, reserving biopsy for patients with findings that raise the concern of cirrhosis.
Hereditary disorders, eg, hemochromatosis, alpha-1 antitrypsin deficiency, and Wilson disease.
In management
Periodic needle biopsy is also valuable in the management of a few diseases.
In autoimmune hepatitis, monitoring the plasma cell score on liver biopsy may help predict relapse when a physician is considering reducing or discontinuing immunosuppressive therapy.1
After liver transplantation, a liver biopsy is highly valuable to assess for rejection and the presence and intensity of disease recurrence.
PROBLEMS WITH LIVER BIOPSY
Liver biopsy is invasive and can cause significant complications. Nearly 30% of patients report having substantial pain after liver biopsy, and some experience serious complications such as pneumothorax, bleeding, or puncture of the biliary tree. In rare cases, patients die of bleeding.2
Furthermore, hepatic pathology, particularly fibrosis, is not always uniformly distributed. Surgical wedge biopsy provides adequate tissue volume to overcome this problem. Needle biopsy, on the other hand, provides a much smaller volume of tissue (1/50,000 of the total mass of the liver).3
As examples of the resulting sampling errors that can occur, consider the two most common chronic liver diseases: hepatitis C and fatty liver disease.
Regev et al4 performed laparoscopically guided biopsy of the right and left hepatic lobes in a series of 124 patients with chronic hepatitis C. Biopsy samples from the right and left lobes differed in the intensity of inflammation in 24.2% of cases, and in the intensity of fibrosis in 33.1%. Differences of more than one grade of inflammation or stage of fibrosis were uncommon. However, in 14.5%, cirrhosis was diagnosed in one lobe but not the other.
In a study in patients with nonalcoholic fatty liver disease, Ratziu et al5 found that none of the features characteristic of nonalcoholic steatohepatitis were highly concordant in paired liver biopsies. Clearly, needle liver biopsy is far from an ideal test.
Increasingly, liver diseases can be diagnosed precisely with laboratory tests, imaging studies, or both. Thus, needle liver biopsy is playing a lesser role in diagnosis.
ADVANCES IN NONINVASIVE DIAGNOSIS OF LIVER DISEASE
Over the past 30 years, substantial strides have been made in our ability to make certain diagnoses through noninvasive means.
Blood tests can be used to diagnose viral hepatitis A, B, and C and many cases of hemochromatosis and primary biliary cirrhosis. For a detailed discussion of how blood tests are used in diagnosing liver diseases, see www.clevelandclinicmeded.com/medicalpubs/diseasemanagement/hepatology/guide-to-common-liver-tests/.
Imaging studies. Primary sclerosing cholangitis can be diagnosed with an imaging study, ie, magnetic resonance cholangiopancreatography (MRCP) or endoscopic retrograde cholangiopancreatography (ERCP). The value of needle biopsy in these patients is limited to assessing the degree of fibrosis to help with management of the disease and, less often, to discovering other liver pathologies.6
Most benign space-occupying liver lesions, both cystic and solid, can be fully characterized by imaging, especially in patients who have no underlying chronic liver disease, and no biopsy is needed. Whether biopsy should be performed to investigate liver lesions depends on the clinical scenario; the topic is beyond the scope of this paper but has been reviewed in detail by Rockey et al.2
CAN NONINVASIVE TESTS DETECT HEPATIC FIBROSIS?
Cirrhosis (stage 4 fibrosis) results in nodular transformation of the liver and impedance of portal blood flow, setting the stage for portal hypertension and its sequelae. Knowing whether cirrhosis is present is important in subsequent management.
In advanced cases, cirrhosis is associated with typical clinical manifestations and laboratory and radiographic findings. In such cases, needle biopsy will add little. However, in most cases, particularly early in the course, clinical, laboratory, and radiologic correlates of cirrhosis are absent. In one study of patients with hepatitis C, 27% had cirrhosis, but in only a small number would cirrhosis have been apparent from clinical signs and laboratory and imaging studies.6
Since a major contemporary role for liver biopsy is in assessing the degree of fibrosis, it is reasonable to ask if newer noninvasive means are available to estimate hepatic fibrosis. The remainder of this review focuses on assessing our increasing ability to stage the degree of fibrosis (including the presence or absence of cirrhosis) by noninvasive means.
Clinical features point to cirrhosis, but not earlier fibrosis
Clinical manifestations help point to the diagnosis of cirrhosis but not to earlier stages of fibrosis.
For example, if a patient is known to have liver disease, the findings of ascites, splenomegaly, or asterixis mean that cirrhosis is highly probable. Similarly, hypersplenism (splenomegaly with a decrease in circulating blood cells but a normal to hyperactive bone marrow) in a patient with liver test abnormalities almost always represents portal hypertension due to cirrhosis, although other, nonhepatic causes are possible, such as congestive heart failure and constrictive pericarditis.
These features generally emerge late in the course of cirrhosis. The absence of such stigmata certainly does not preclude the presence of cirrhosis. Thus, these clinical signs have a high positive predictive value but a low negative predictive value, making them insufficient by themselves to diagnose or stage liver disease.
Laboratory tests are of limited value in assessing the degree of fibrosis
Standard liver tests are of limited value in assessing the degree of fibrosis.
Usual laboratory tests. At one end of the spectrum, anemia, thrombocytopenia, and leukopenia in the presence of liver disease correlate with cirrhosis. At the other end, a serum ferritin concentration of less than 1,000 mg/mL in a patient with hemochromatosis and no confounding features such as hepatitis C, HIV infection, or heavy alcohol use strongly predicts that the patient does not have significant hepatic fibrosis.8
Bilirubin elevation is a late finding in cirrhosis, but in cholestatic diseases bilirubin may be elevated before cirrhosis occurs.
Albumin is made exclusively in the liver, and its concentration falls as liver function worsens with progressive cirrhosis.
The prothrombin time increases as the liver loses its ability to synthesize clotting factors in cirrhosis. Coagulopathy correlates with the degree of liver disease.
Hyponatremia due to impaired ability to excrete free water is seen in patients with cirrhosis and ascites.
In summary, the usual laboratory tests related to liver disease are imprecise and, when abnormal, often indicate not just the presence of cirrhosis, but impending or actual decompensation.
Newer serologic markers, alone or in combination, have been proposed as aids in determining the degree of fibrosis or cirrhosis in the liver. Direct markers of fibrosis measure the turnover or metabolism of extracellular matrix. Indirect markers of fibrosis reflect alterations in hepatic function (see below).
Parkes et al9 reviewed 10 different panels of serum markers of hepatic fibrosis in chronic hepatitis C. Only 35% of patients had fibrosis adequately ruled in or ruled out by these panels, and the stage of fibrosis could not be adequately determined.
These serologic markers have not been validated in other chronic liver diseases or in liver disease due to multiple causes. Thus, although they show promise for use by the general internist, they need to be validated in patients with disease and in normal reference populations before they are ready for “prime time.”
Direct serologic markers of fibrosis
Direct serologic markers of fibrosis include those associated with matrix deposition—eg, procollagen type III amino-terminal peptide (P3NP), type I and IV collagens, laminin, hyaluronic acid, and chondrex.
P3NP is the most widely studied marker of hepatic fibrosis. It is elevated in both acute and chronic liver diseases; serum levels reflect the histologic stage of hepatic fibrosis in various chronic liver diseases, including alcoholic, viral, and primary biliary cirrhosis.10–12 Successful treatment of autoimmune hepatitis has been shown to lead to reductions of P3NP levels.13
Other direct markers of fibrosis are those associated with matrix degradation, ie, matrix metalloproteinases 2 and 3 (MMP-2, MMP-3) and tissue inhibitors of metalloproteinases 1 and 2 (TIMP-1, TIMP-2). Levels of MMP-2 proenzymes and active enzymes are increased in liver disease, but studies are inconsistent in correlating serum levels of MMP-2 to the degree of hepatic fibrosis.14,15 These tests are not commercially available, and the components are not readily available in most clinical laboratories.
Indirect serologic markers of fibrosis
Some indirect markers are readily available:
The AST:ALT ratio. The normal ratio of aspartate aminotransferase (AST) to alanine aminotransferase (ALT) is approximately 0.8. A ratio greater than 1.0 provides evidence of cirrhosis. However, findings have been inconsistent.
The AST:platelet ratio index (APRI), a commonly used index, is calculated by the following formula:
![]()
In studies of hepatitis C and hepatitis C-HIV, the APRI has shown a sensitivity of 37% to 80% and a specificity of 45% to 98%, depending on the cutoff value and whether a diagnosis of severe fibrosis or cirrhosis was being tested.16–19 These sensitivities and specificities are disappointing and do not provide information equal to that provided by needle liver biopsy in most patients with chronic liver disease.
The combination of prothrombin, gamma glutamyl, and apolipoprotein AI levels (PGA index) has been validated in patients with many types of chronic liver disease, and its accuracy for detecting cirrhosis is highest (66%–72%) in patients with alcoholic liver disease.20,21
FibroIndex uses the platelet count, AST level, and gamma globulin level to detect significant fibrosis in chronic hepatitis C, but its accuracy has yet to be validated.22
The FIB-4 index is based on four independent predictors of fibrosis, ie, age, the platelet count, AST level, and ALT level. It has shown good accuracy for detecting advanced fibrosis in two studies in patients with hepatitis C.23,24
Fibrometer (based on the platelet count; the prothrombin index; the levels of AST, alfa-2 macroglobulin, hyaluronate, and blood urea nitrogen; and age) predicted fibrosis well in chronic viral hepatitis.25,26
Fibrotest and Fibrosure are proprietary commercial tests available in many laboratories. They employ a mathematical formula to predict fibrosis (characterized as mild, significant, or indeterminate) using the levels of alpha-2 macroglobulin, alpha-2 globulin, gamma globulin, apolipoprotein A1, gamma glutamyl transferase, and total bilirubin. For detecting significant fibrosis, these tests are reported to have a sensitivity of about 75% and a specificity of 85%.27–29
ActiTest incorporates the ALT level into the Fibrotest to reflect liver fibrosis and necro-inflammatory activity.
A meta-analysis showed that Fibrotest and ActiTest could be reliable alternatives to liver biopsy in patients with chronic hepatitis C.30 The area under the receiver operator characteristic curve for the diagnosis of significant fibrosis ranged from 0.73 to 0.87; for the diagnosis of significant histologic activity it ranged from 0.75 to 0.86. Fibrotest had a negative predictive value for excluding significant fibrosis of 91% with a cutoff of 0.31. ActiTest’s negative predictive value for excluding significant necrosis was 85% with a cutoff of 0.36. None of these serum tests have become part of standard of practice for diagnosing fibrosis or cirrhosis.
The Sequential Algorithm for Fibrosis Evaluation (SAFE) combines the APRI and Fibrotest-Fibrosure tests in a sequential fashion to test for fibrosis and cirrhosis. In a large multicenter study31 validating this algorithm to detect significant fibrosis (stage F2 or greater by the F0–F4 METAVIR scoring system32), its accuracy was 90.1%, the area under the receiver operating characteristic curve was 0.89 (95% CI 0.87–0.90), and it reduced the number of liver biopsies needed by 46.5%. When the algorithm was used to detect cirrhosis, its accuracy was 92.5%, the area under the curve was 0.92 (95% CI 0.89–0.94), and it reduced the number of liver biopsies needed by 81.5%.
Another algorithm was developed to simultaneously detect significant fibrosis and cirrhosis. It had a 97.4% accuracy, but 64% of patients still required a liver biopsy.31
SAFE algorithms have the potential to reduce the number of needle biopsies needed to assess the degree of hepatic fibrosis.
CONVENTIONAL IMAGING STUDIES ARE NOT SENSITIVE FOR FIBROSIS
Standard imaging studies often show findings of cirrhosis but are not particularly sensitive, with a low negative predictive value.
Ultrasonography can show a small, nodular liver in advanced cirrhosis, but surface nodularity or increased echogenicity can be seen in hepatic steatosis as well as in cirrhosis. In one study,33 ultrasonography identified diffuse parenchymal disease but could not reliably distinguish fat from fibrosis or diagnose cirrhosis.
Often, in cirrhosis, the right lobe of the liver is atrophied and the caudate or left lobes are hypertrophied. Efforts to use the ratio of the widths of the lobes to diagnose cirrhosis have shown varying performance characterstics.34,35
One study of the splenic artery pulsatility index has shown this to be an accurate predictor of cirrhosis.36
Computed tomography provides information similar to that of ultrasonography, and it can identify complications of cirrhosis, including portal hypertension and ascites. On the other hand, it costs more and it exposes the patient to radiation and contrast media.
ELASTOGRAPHY, A PROMISING TEST
Hepatic elastography, a method for estimating liver stiffness, is an exciting recent development in the noninvasive measurement of hepatic fibrosis. Currently, elastography can be accomplished by ultrasound or magnetic resonance.
Ultrasound elastography
The FibroScan device (EchoSens, Paris, France) uses a mild-amplitude, low-frequency (50-Hz) vibration transmitted through the liver.37 It induces an elastic shear wave that is detected by pulse-echo ultrasonography as the wave propagates through the organ.
The velocity of the wave correlates with tissue stiffness: the wave travels faster through denser, fibrotic tissue.38,39
Ultrasound elastography (also called transient elastography) can sample a much larger area than liver biopsy can, providing a better understanding of the entire hepatic parenchyma. 40 Moreover, it can be repeated often without risk. This device is in widespread use in many parts of the world, but it is not yet approved in the United States.
A meta-analysis of 50 studies assessed the overall performance of ultrasound elastography for diagnosing liver fibrosis.41 The areas under the receiver operating characteristic curve were as follows:
- For significant fibrosis: 0.84 (95% CI 0.82–0.86)
- For severe fibrosis: 0.89 (95% CI 0.88–0.91)
- For cirrhosis: 0.94 (95% CI 0.93–0.95).
The type of underlying liver disease influenced the diagnosis of significant fibrosis, which was diagnosed most consistently in patients with hepatitis C. The authors concluded that ultrasound elastography had excellent diagnostic accuracy for diagnosing cirrhosis irrespective of the underlying liver disease, while the diagnosis of significant fibrosis had higher variation, which was dependent on the underlying liver disease.
A meta-analysis of nine studies42 showed ultrasound elastography to have a sensitivity of 87% (95% CI 84%–90%) and a specificity of 91% (95% CI 89%–92%) for the diagnosis of cirrhosis. In seven of the nine studies, it diagnosed stage II to IV fibrosis with 70% sensitivity (95% CI 67%–73%) and 84% specificity (95% CI 80%–88%).
Limitations. Ultrasound elastography is less effective in obese patients, as the adipose tissue attenuates the elastic wave, and it has not been reliable in patients with acute viral hepatitis.43 Male sex, body mass index greater than 30, and metabolic syndrome seem to increase liver stiffness, thus limiting the use of this test.44
Until more data are available, the ultimate value of ultrasound elastography in reducing the number of liver biopsies needed remains unknown. However, this test shows potential as a reliable and noninvasive way to assess the degree of fibrosis in patients with liver disease.
Magnetic resonance elastography
Studies have shown a magnetic resonance scoring system that distinguishes Child-Pugh grade A cirrhosis from other grades to be 93% sensitive and 82% specific.45
Cost may limit the use of magnetic resonance elastography, and some patients may be unable to tolerate the procedure because of claustrophobia. It seems clear, though, that this test currently has the most promise in reducing the need for liver biopsy for grading the severity of hepatic fibrosis.
WHERE ARE WE NOW?
The importance of liver biopsy in arriving at a diagnosis of diffuse parenchymal liver disease is being diminished by accurate blood testing strategies for chronic viral hepatitis, autoimmune hepatitis, and primary biliary cirrhosis. Further, imaging tests are superior to liver biopsy in the diagnosis of primary sclerosing cholangitis.
However, many cases remain in which diagnostic confusion exists even after suitable laboratory testing and imaging studies. Diagnosing infiltrative disease (eg, amyloidosis, sarcoidosis), separating benign fatty liver disease from steatohepatitis, and evaluating liver parenchyma after liver transplantation are best accomplished by liver biopsy.
While needle biopsy is still the mainstay in diagnosing hepatic fibrosis, its days of dominance seem limited as technology improves. When physical examination or standard laboratory tests reveal clear-cut signs of portal hypertension, liver biopsy will seldom add useful information. Similarly, when imaging studies provide compelling evidence of cirrhosis and portal hypertension, needle biopsy is not warranted.
The SAFE algorithms warrant further evaluation in all chronic liver diseases, as they may help decrease the number of liver biopsies required. And we believe elastography will play an ever-increasing role in the assessment of hepatic fibrosis and will significantly reduce the need for biopsy in patients with liver disease.
Primary care physicians and specialists alike often encounter patients with chronic liver disease. Fortunately, these days we need to resort to liver biopsy less often than in the past.
The purpose of this review is to provide a critical assessment of the growing number of noninvasive tests available for diagnosing liver disease and assessing hepatic fibrosis, and to discuss the implications of these advances related to the indications for needle liver biopsy.
WHEN IS LIVER BIOPSY USEFUL?
In diagnosis
Needle liver biopsy for diagnosis remains important in cases of:
Diagnostic uncertainty (eg, in patients with atypical features)
Coexisting disorders (eg, human immunodeficiency virus [HIV] and hepatitis C virus infection, or alcoholic liver disease and hepatitis C)
An overlapping syndrome (eg, primary biliary cirrhosis with autoimmune hepatitis).
Fatty liver. Needle liver biopsy can distinguish between benign steatosis and progressive steatohepatitis in a patient with a fatty liver found on imaging, subject to the limitations of sampling error.
Because fatty liver disease is common and proven treatments are few, no consensus has emerged about which patients with suspected fatty liver disease should undergo needle biopsy. Many specialists eschew needle biopsy and treat the underlying risk factors of metabolic syndrome, reserving biopsy for patients with findings that raise the concern of cirrhosis.
Hereditary disorders, eg, hemochromatosis, alpha-1 antitrypsin deficiency, and Wilson disease.
In management
Periodic needle biopsy is also valuable in the management of a few diseases.
In autoimmune hepatitis, monitoring the plasma cell score on liver biopsy may help predict relapse when a physician is considering reducing or discontinuing immunosuppressive therapy.1
After liver transplantation, a liver biopsy is highly valuable to assess for rejection and the presence and intensity of disease recurrence.
PROBLEMS WITH LIVER BIOPSY
Liver biopsy is invasive and can cause significant complications. Nearly 30% of patients report having substantial pain after liver biopsy, and some experience serious complications such as pneumothorax, bleeding, or puncture of the biliary tree. In rare cases, patients die of bleeding.2
Furthermore, hepatic pathology, particularly fibrosis, is not always uniformly distributed. Surgical wedge biopsy provides adequate tissue volume to overcome this problem. Needle biopsy, on the other hand, provides a much smaller volume of tissue (1/50,000 of the total mass of the liver).3
As examples of the resulting sampling errors that can occur, consider the two most common chronic liver diseases: hepatitis C and fatty liver disease.
Regev et al4 performed laparoscopically guided biopsy of the right and left hepatic lobes in a series of 124 patients with chronic hepatitis C. Biopsy samples from the right and left lobes differed in the intensity of inflammation in 24.2% of cases, and in the intensity of fibrosis in 33.1%. Differences of more than one grade of inflammation or stage of fibrosis were uncommon. However, in 14.5%, cirrhosis was diagnosed in one lobe but not the other.
In a study in patients with nonalcoholic fatty liver disease, Ratziu et al5 found that none of the features characteristic of nonalcoholic steatohepatitis were highly concordant in paired liver biopsies. Clearly, needle liver biopsy is far from an ideal test.
Increasingly, liver diseases can be diagnosed precisely with laboratory tests, imaging studies, or both. Thus, needle liver biopsy is playing a lesser role in diagnosis.
ADVANCES IN NONINVASIVE DIAGNOSIS OF LIVER DISEASE
Over the past 30 years, substantial strides have been made in our ability to make certain diagnoses through noninvasive means.
Blood tests can be used to diagnose viral hepatitis A, B, and C and many cases of hemochromatosis and primary biliary cirrhosis. For a detailed discussion of how blood tests are used in diagnosing liver diseases, see www.clevelandclinicmeded.com/medicalpubs/diseasemanagement/hepatology/guide-to-common-liver-tests/.
Imaging studies. Primary sclerosing cholangitis can be diagnosed with an imaging study, ie, magnetic resonance cholangiopancreatography (MRCP) or endoscopic retrograde cholangiopancreatography (ERCP). The value of needle biopsy in these patients is limited to assessing the degree of fibrosis to help with management of the disease and, less often, to discovering other liver pathologies.6
Most benign space-occupying liver lesions, both cystic and solid, can be fully characterized by imaging, especially in patients who have no underlying chronic liver disease, and no biopsy is needed. Whether biopsy should be performed to investigate liver lesions depends on the clinical scenario; the topic is beyond the scope of this paper but has been reviewed in detail by Rockey et al.2
CAN NONINVASIVE TESTS DETECT HEPATIC FIBROSIS?
Cirrhosis (stage 4 fibrosis) results in nodular transformation of the liver and impedance of portal blood flow, setting the stage for portal hypertension and its sequelae. Knowing whether cirrhosis is present is important in subsequent management.
In advanced cases, cirrhosis is associated with typical clinical manifestations and laboratory and radiographic findings. In such cases, needle biopsy will add little. However, in most cases, particularly early in the course, clinical, laboratory, and radiologic correlates of cirrhosis are absent. In one study of patients with hepatitis C, 27% had cirrhosis, but in only a small number would cirrhosis have been apparent from clinical signs and laboratory and imaging studies.6
Since a major contemporary role for liver biopsy is in assessing the degree of fibrosis, it is reasonable to ask if newer noninvasive means are available to estimate hepatic fibrosis. The remainder of this review focuses on assessing our increasing ability to stage the degree of fibrosis (including the presence or absence of cirrhosis) by noninvasive means.
Clinical features point to cirrhosis, but not earlier fibrosis
Clinical manifestations help point to the diagnosis of cirrhosis but not to earlier stages of fibrosis.
For example, if a patient is known to have liver disease, the findings of ascites, splenomegaly, or asterixis mean that cirrhosis is highly probable. Similarly, hypersplenism (splenomegaly with a decrease in circulating blood cells but a normal to hyperactive bone marrow) in a patient with liver test abnormalities almost always represents portal hypertension due to cirrhosis, although other, nonhepatic causes are possible, such as congestive heart failure and constrictive pericarditis.
These features generally emerge late in the course of cirrhosis. The absence of such stigmata certainly does not preclude the presence of cirrhosis. Thus, these clinical signs have a high positive predictive value but a low negative predictive value, making them insufficient by themselves to diagnose or stage liver disease.
Laboratory tests are of limited value in assessing the degree of fibrosis
Standard liver tests are of limited value in assessing the degree of fibrosis.
Usual laboratory tests. At one end of the spectrum, anemia, thrombocytopenia, and leukopenia in the presence of liver disease correlate with cirrhosis. At the other end, a serum ferritin concentration of less than 1,000 mg/mL in a patient with hemochromatosis and no confounding features such as hepatitis C, HIV infection, or heavy alcohol use strongly predicts that the patient does not have significant hepatic fibrosis.8
Bilirubin elevation is a late finding in cirrhosis, but in cholestatic diseases bilirubin may be elevated before cirrhosis occurs.
Albumin is made exclusively in the liver, and its concentration falls as liver function worsens with progressive cirrhosis.
The prothrombin time increases as the liver loses its ability to synthesize clotting factors in cirrhosis. Coagulopathy correlates with the degree of liver disease.
Hyponatremia due to impaired ability to excrete free water is seen in patients with cirrhosis and ascites.
In summary, the usual laboratory tests related to liver disease are imprecise and, when abnormal, often indicate not just the presence of cirrhosis, but impending or actual decompensation.
Newer serologic markers, alone or in combination, have been proposed as aids in determining the degree of fibrosis or cirrhosis in the liver. Direct markers of fibrosis measure the turnover or metabolism of extracellular matrix. Indirect markers of fibrosis reflect alterations in hepatic function (see below).
Parkes et al9 reviewed 10 different panels of serum markers of hepatic fibrosis in chronic hepatitis C. Only 35% of patients had fibrosis adequately ruled in or ruled out by these panels, and the stage of fibrosis could not be adequately determined.
These serologic markers have not been validated in other chronic liver diseases or in liver disease due to multiple causes. Thus, although they show promise for use by the general internist, they need to be validated in patients with disease and in normal reference populations before they are ready for “prime time.”
Direct serologic markers of fibrosis
Direct serologic markers of fibrosis include those associated with matrix deposition—eg, procollagen type III amino-terminal peptide (P3NP), type I and IV collagens, laminin, hyaluronic acid, and chondrex.
P3NP is the most widely studied marker of hepatic fibrosis. It is elevated in both acute and chronic liver diseases; serum levels reflect the histologic stage of hepatic fibrosis in various chronic liver diseases, including alcoholic, viral, and primary biliary cirrhosis.10–12 Successful treatment of autoimmune hepatitis has been shown to lead to reductions of P3NP levels.13
Other direct markers of fibrosis are those associated with matrix degradation, ie, matrix metalloproteinases 2 and 3 (MMP-2, MMP-3) and tissue inhibitors of metalloproteinases 1 and 2 (TIMP-1, TIMP-2). Levels of MMP-2 proenzymes and active enzymes are increased in liver disease, but studies are inconsistent in correlating serum levels of MMP-2 to the degree of hepatic fibrosis.14,15 These tests are not commercially available, and the components are not readily available in most clinical laboratories.
Indirect serologic markers of fibrosis
Some indirect markers are readily available:
The AST:ALT ratio. The normal ratio of aspartate aminotransferase (AST) to alanine aminotransferase (ALT) is approximately 0.8. A ratio greater than 1.0 provides evidence of cirrhosis. However, findings have been inconsistent.
The AST:platelet ratio index (APRI), a commonly used index, is calculated by the following formula:
![]()
In studies of hepatitis C and hepatitis C-HIV, the APRI has shown a sensitivity of 37% to 80% and a specificity of 45% to 98%, depending on the cutoff value and whether a diagnosis of severe fibrosis or cirrhosis was being tested.16–19 These sensitivities and specificities are disappointing and do not provide information equal to that provided by needle liver biopsy in most patients with chronic liver disease.
The combination of prothrombin, gamma glutamyl, and apolipoprotein AI levels (PGA index) has been validated in patients with many types of chronic liver disease, and its accuracy for detecting cirrhosis is highest (66%–72%) in patients with alcoholic liver disease.20,21
FibroIndex uses the platelet count, AST level, and gamma globulin level to detect significant fibrosis in chronic hepatitis C, but its accuracy has yet to be validated.22
The FIB-4 index is based on four independent predictors of fibrosis, ie, age, the platelet count, AST level, and ALT level. It has shown good accuracy for detecting advanced fibrosis in two studies in patients with hepatitis C.23,24
Fibrometer (based on the platelet count; the prothrombin index; the levels of AST, alfa-2 macroglobulin, hyaluronate, and blood urea nitrogen; and age) predicted fibrosis well in chronic viral hepatitis.25,26
Fibrotest and Fibrosure are proprietary commercial tests available in many laboratories. They employ a mathematical formula to predict fibrosis (characterized as mild, significant, or indeterminate) using the levels of alpha-2 macroglobulin, alpha-2 globulin, gamma globulin, apolipoprotein A1, gamma glutamyl transferase, and total bilirubin. For detecting significant fibrosis, these tests are reported to have a sensitivity of about 75% and a specificity of 85%.27–29
ActiTest incorporates the ALT level into the Fibrotest to reflect liver fibrosis and necro-inflammatory activity.
A meta-analysis showed that Fibrotest and ActiTest could be reliable alternatives to liver biopsy in patients with chronic hepatitis C.30 The area under the receiver operator characteristic curve for the diagnosis of significant fibrosis ranged from 0.73 to 0.87; for the diagnosis of significant histologic activity it ranged from 0.75 to 0.86. Fibrotest had a negative predictive value for excluding significant fibrosis of 91% with a cutoff of 0.31. ActiTest’s negative predictive value for excluding significant necrosis was 85% with a cutoff of 0.36. None of these serum tests have become part of standard of practice for diagnosing fibrosis or cirrhosis.
The Sequential Algorithm for Fibrosis Evaluation (SAFE) combines the APRI and Fibrotest-Fibrosure tests in a sequential fashion to test for fibrosis and cirrhosis. In a large multicenter study31 validating this algorithm to detect significant fibrosis (stage F2 or greater by the F0–F4 METAVIR scoring system32), its accuracy was 90.1%, the area under the receiver operating characteristic curve was 0.89 (95% CI 0.87–0.90), and it reduced the number of liver biopsies needed by 46.5%. When the algorithm was used to detect cirrhosis, its accuracy was 92.5%, the area under the curve was 0.92 (95% CI 0.89–0.94), and it reduced the number of liver biopsies needed by 81.5%.
Another algorithm was developed to simultaneously detect significant fibrosis and cirrhosis. It had a 97.4% accuracy, but 64% of patients still required a liver biopsy.31
SAFE algorithms have the potential to reduce the number of needle biopsies needed to assess the degree of hepatic fibrosis.
CONVENTIONAL IMAGING STUDIES ARE NOT SENSITIVE FOR FIBROSIS
Standard imaging studies often show findings of cirrhosis but are not particularly sensitive, with a low negative predictive value.
Ultrasonography can show a small, nodular liver in advanced cirrhosis, but surface nodularity or increased echogenicity can be seen in hepatic steatosis as well as in cirrhosis. In one study,33 ultrasonography identified diffuse parenchymal disease but could not reliably distinguish fat from fibrosis or diagnose cirrhosis.
Often, in cirrhosis, the right lobe of the liver is atrophied and the caudate or left lobes are hypertrophied. Efforts to use the ratio of the widths of the lobes to diagnose cirrhosis have shown varying performance characterstics.34,35
One study of the splenic artery pulsatility index has shown this to be an accurate predictor of cirrhosis.36
Computed tomography provides information similar to that of ultrasonography, and it can identify complications of cirrhosis, including portal hypertension and ascites. On the other hand, it costs more and it exposes the patient to radiation and contrast media.
ELASTOGRAPHY, A PROMISING TEST
Hepatic elastography, a method for estimating liver stiffness, is an exciting recent development in the noninvasive measurement of hepatic fibrosis. Currently, elastography can be accomplished by ultrasound or magnetic resonance.
Ultrasound elastography
The FibroScan device (EchoSens, Paris, France) uses a mild-amplitude, low-frequency (50-Hz) vibration transmitted through the liver.37 It induces an elastic shear wave that is detected by pulse-echo ultrasonography as the wave propagates through the organ.
The velocity of the wave correlates with tissue stiffness: the wave travels faster through denser, fibrotic tissue.38,39
Ultrasound elastography (also called transient elastography) can sample a much larger area than liver biopsy can, providing a better understanding of the entire hepatic parenchyma. 40 Moreover, it can be repeated often without risk. This device is in widespread use in many parts of the world, but it is not yet approved in the United States.
A meta-analysis of 50 studies assessed the overall performance of ultrasound elastography for diagnosing liver fibrosis.41 The areas under the receiver operating characteristic curve were as follows:
- For significant fibrosis: 0.84 (95% CI 0.82–0.86)
- For severe fibrosis: 0.89 (95% CI 0.88–0.91)
- For cirrhosis: 0.94 (95% CI 0.93–0.95).
The type of underlying liver disease influenced the diagnosis of significant fibrosis, which was diagnosed most consistently in patients with hepatitis C. The authors concluded that ultrasound elastography had excellent diagnostic accuracy for diagnosing cirrhosis irrespective of the underlying liver disease, while the diagnosis of significant fibrosis had higher variation, which was dependent on the underlying liver disease.
A meta-analysis of nine studies42 showed ultrasound elastography to have a sensitivity of 87% (95% CI 84%–90%) and a specificity of 91% (95% CI 89%–92%) for the diagnosis of cirrhosis. In seven of the nine studies, it diagnosed stage II to IV fibrosis with 70% sensitivity (95% CI 67%–73%) and 84% specificity (95% CI 80%–88%).
Limitations. Ultrasound elastography is less effective in obese patients, as the adipose tissue attenuates the elastic wave, and it has not been reliable in patients with acute viral hepatitis.43 Male sex, body mass index greater than 30, and metabolic syndrome seem to increase liver stiffness, thus limiting the use of this test.44
Until more data are available, the ultimate value of ultrasound elastography in reducing the number of liver biopsies needed remains unknown. However, this test shows potential as a reliable and noninvasive way to assess the degree of fibrosis in patients with liver disease.
Magnetic resonance elastography
Studies have shown a magnetic resonance scoring system that distinguishes Child-Pugh grade A cirrhosis from other grades to be 93% sensitive and 82% specific.45
Cost may limit the use of magnetic resonance elastography, and some patients may be unable to tolerate the procedure because of claustrophobia. It seems clear, though, that this test currently has the most promise in reducing the need for liver biopsy for grading the severity of hepatic fibrosis.
WHERE ARE WE NOW?
The importance of liver biopsy in arriving at a diagnosis of diffuse parenchymal liver disease is being diminished by accurate blood testing strategies for chronic viral hepatitis, autoimmune hepatitis, and primary biliary cirrhosis. Further, imaging tests are superior to liver biopsy in the diagnosis of primary sclerosing cholangitis.
However, many cases remain in which diagnostic confusion exists even after suitable laboratory testing and imaging studies. Diagnosing infiltrative disease (eg, amyloidosis, sarcoidosis), separating benign fatty liver disease from steatohepatitis, and evaluating liver parenchyma after liver transplantation are best accomplished by liver biopsy.
While needle biopsy is still the mainstay in diagnosing hepatic fibrosis, its days of dominance seem limited as technology improves. When physical examination or standard laboratory tests reveal clear-cut signs of portal hypertension, liver biopsy will seldom add useful information. Similarly, when imaging studies provide compelling evidence of cirrhosis and portal hypertension, needle biopsy is not warranted.
The SAFE algorithms warrant further evaluation in all chronic liver diseases, as they may help decrease the number of liver biopsies required. And we believe elastography will play an ever-increasing role in the assessment of hepatic fibrosis and will significantly reduce the need for biopsy in patients with liver disease.
- Verma S, Gunuwan B, Mendler M, Govindrajan S, Redeker A. Factors predicting relapse and poor outcome in type I autoimmune hepatitis: role of cirrhosis development, patterns of transaminases during remission and plasma cell activity in the liver biopsy. Am J Gastroenterol 2004; 99:1510–1516.
- Rockey DC, Caldwell SH, Goodman ZD, Nelson RC, Smith AD; American Association for the Study of Liver Diseases. Liver biopsy. Hepatology 2009; 49:1017–1044.
- Bravo AA, Sheth SG, Chopra S. Liver biopsy. N Engl J Med 2001; 344:495–500.
- Regev A, Berho M, Jeffers LJ, et al. Sampling error and intraobserver variation in liver biopsy in patients with chronic HCV infection. Am J Gastroenterol 2002; 97:2614–2618.
- Ratziu V, Charlotte F, Heurtier A, et al; LIDO Study Group Sampling variability of liver biopsy in nonalcoholic fatty liver disease. Gastroenterology 2005; 128:1898–1906.
- Saadeh S, Cammell G, Carey WD, Younossi Z, Barnes D, Easley K. The role of liver biopsy in chronic hepatitis C. Hepatology 2001; 33:196–200.
- Batts KP, Ludwig J. Chronic hepatitis. An update on terminology and reporting. Am J Surg Pathol 1995; 19:1409–1417.
- Morrison ED, Brandhagen DJ, Phatak PD, et al. Serum ferritin level predicts advanced hepatic fibrosis among U.S. patients with phenotypic hemochromatosis. Ann Intern Med 2003; 138:627–633.
- Parkes J, Guha IN, Roderick P, Rosenberg W. Performance of serum marker panels for liver fibrosis in chronic hepatitis C. J Hepatol 2006; 44:462–474.
- Montalto G, Soresi M, Aragona F, et al. Procollagen III and laminin in chronic viral hepatopathies. Presse Med 1996; 25:59–62.
- Teare JP, Sherman D, Greenfield SM, et al. Comparison of serum procollagen III peptide concentrations and PGA index for assessment of hepatic fibrosis. Lancet 1993; 342:895–898.
- Trinchet JC, Hartmann DJ, Pateron D, et al. Serum type I collagen and N-terminal peptide of type III procollagen in chronic hepatitis. Relationship to liver histology and conventional liver tests. J Hepatol 1991; 12:139–144.
- McCullough AJ, Stassen WN, Wiesner RH, Czaja AJ. Serial determinations of the amino-terminal peptide of type III procollagen in severe chronic active hepatitis. J Lab Clin Med 1987; 109:55–61.
- Takahara T, Furui K, Funaki J, et al. Increased expression of matrix metalloproteinase-II in experimental liver fibrosis in rats. Hepatology 1995; 21:787–795.
- Takahara T, Furui K, Yata Y, et al. Dual expression of matrix metalloproteinase-2 and membrane-type 1-matrix metalloproteinase in fibrotic human livers. Hepatology 1997; 26:1521–1529.
- Wai CT, Greenson JK, Fontana RJ, et al. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology 2003; 38:518–526.
- Kelleher TB, Mehta SH, Bhaskar R, et al. Prediction of hepatic fibrosis in HIV/HCV co-infected patients using serum fibrosis markers: the SHASTA index. J Hepatol 2005; 43:78–84.
- Islam S, Antonsson L, Westin J, Lagging M. Cirrhosis in hepatitis C virus-infected patients can be excluded using an index of standard biochemical serum markers. Scand J Gastroenterol 2005; 40:867–872.
- Lackner C, Struber G, Liegl B, et al. Comparison and validation of simple noninvasive tests for prediction of fibrosis in chronic hepatitis C. Hepatology 2005; 41:1376–1382.
- Poynard T, Aubert A, Bedossa P, et al. A simple biological index for detection of alcoholic liver disease in drinkers. Gastroenterology 1991; 100:1397–1402.
- Oberti F, Valsesia E, Pilette C, et al. Noninvasive diagnosis of hepatic fibrosis or cirrhosis. Gastroenterology 1997; 113:1609–1616.
- Koda M, Matunaga Y, Kawakami M, Kishimoto Y, Suou T, Murawaki Y. FibroIndex, a practical index for predicting significant fibrosis in patients with chronic hepatitis C. Hepatology 2007; 45:297–306.
- Vallet-Pichard A, Mallet V, Nalpas B, et al. FIB-4: an inexpensive and accurate marker of fibrosis in HCV infection. Comparison with liver biopsy and fibrotest. Hepatology 2007; 46:32–36.
- Sterling RK, Lissen E, Clumeck N, et al; APRI COT Clinical Investigators. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology 2006; 43:1317–1325.
- Calès P, Oberti F, Michalak S, et al. A novel panel of blood markers to assess the degree of liver fibrosis. Hepatology 2005; 42:1373–1381.
- Leroy V, Hilleret MN, Sturm N, et al. Prospective comparison of six non-invasive scores for the diagnosis of liver fibrosis in chronic hepatitis C. J Hepatol 2007; 46:775–782.
- Myers RP, De Torres M, Imbert-Bismut F, Ratziu V, Charlotte F, Poynard T; MULTIVIRC Group. Biochemical markers of fibrosis in patients with chronic hepatitis C: a comparison with prothrombin time, platelet count, and age-platelet index. Dig Dis Sci 2003; 48:146–153.
- Rossi E, Adams L, Prins A, et al. Validation of the FibroTest biochemical markers score in assessing liver fibrosis in hepatitis C patients. Clin Chem 2003; 49:450–454.
- Halfon P, Bourliere M, Deydier R, et al. Independent prospective multicenter validation of biochemical markers (fibrotest-actitest) for the prediction of liver fibrosis and activity in patients with chronic hepatitis C: the fibropaca study. Am J Gastroenterol 2006; 101:547–555.
- Poynard T, Imbert-Bismut F, Munteanu M, et al. Overview of the diagnostic value of biochemical markers of liver fibrosis (FibroTest, HCV FibroSure) and necrosis (ActiTest) in patients with chronic hepatitis C. Comp Hepatol 2004; 3:8.
- Sebastiani G, Halfon P, Castera L, et al. SAFE biopsy: a validated method for large-scale staging of liver fibrosis in chronic hepatitis C. Hepatology 2009; 49:1821–1827.
- The French METAVIR Cooperative Study Group. Intraobserver and interobserver variations in liver biopsy interpretations in patients with chronic hepatitis C. Hepatology 1994; 20:15–20.
- Sanford NL, Walsh P, Matis C, Baddeley H, Powell LW. Is ultrasonography useful in the assessment of diffuse parenchymal liver disease? Gastroenterology 1985; 89:186–191.
- Harbin WP, Robert NJ, Ferrucci JT. Diagnosis of cirrhosis based on regional changes in hepatic morphology: a radiological and pathological analysis. Radiology 1980; 135:273–283.
- Giorgio A, Amoroso P, Lettieri G, et al. Cirrhosis: value of caudate to right lobe ratio in diagnosis with US. Radiology 1986; 161:443–445.
- Liu CH, Hsu SJ, Lin JW, et al. Noninvasive diagnosis of hepatic fibrosis in patients with chronic hepatitis C by splenic Doppler impedance index. Clin Gastroenterol Hepatol 2007; 5:1199–1206.
- Talawalkar JA. Elastography for detecting hepatic fibrosis: options and considerations. Gastroenterology 2008; 135:299–302.
- Sandrin L, Fourquet B, Hasquenoph JM, et al. Transient elastography: a new noninvasive method for assessment of hepatic fibrosis. Ultrasound Med Biol 2003; 29:1705–1713.
- Kettaneh A, Marcellin P, Douvin C, et al. Features associated with success rate and performance of FibroScan measurements for the diagnosis of cirrhosis in HCV patients: a prospective study of 935 patients. J Hepatol 2007; 46:628–634.
- Ziol M, Handra-Luca A, Kettaneh A, et al. Noninvasive assessment of liver fibrosis by measurement of stiffness in patients with chronic hepatitis C. Hepatology 2005; 41:48–54.
- Friedrich-Rust M, Ong MF, Martens S, et al. Performance of transient elastography for the staging of liver fibrosis: a meta-analysis. Gastroenterology 2008; 134:960–974.
- Talwalkar JA, Kurtz DM, Schoenleber SJ, West CP, Montori VM. Ultrasound-based transient elastography for the detection of hepatic fibrosis: systematic review and meta-analysis. Clin Gastroenterol Hepatol 2007; 5:1214–1220.
- Arena U, Vizzutti F, Corti G, et al. Acute viral hepatitis increases liver stiffness values measured by transient elastography. Hepatology 2008; 47:380–384.
- Roulot D, Czernichow S, Le Clésiau H, Costes JL, Vergnaud AC, Beaugrand M. Liver stiffness values in apparently healthy subjects: influence of gender and metabolic syndrome. J Hepatol 2008; 48:606–613.
- Ito K, Mitchell DG, Hann HW, et al. Viral-induced cirrhosis: grading of severity using MR imaging. AJR Am J Roentgenol 1999; 173:591–596.
- Huwart L, Sempoux C, Vicaut E, et al. Magnetic resonance elastography for the noninvasive staging of liver fibrosis. Gastroenterology 2008; 135:32–40.
- Verma S, Gunuwan B, Mendler M, Govindrajan S, Redeker A. Factors predicting relapse and poor outcome in type I autoimmune hepatitis: role of cirrhosis development, patterns of transaminases during remission and plasma cell activity in the liver biopsy. Am J Gastroenterol 2004; 99:1510–1516.
- Rockey DC, Caldwell SH, Goodman ZD, Nelson RC, Smith AD; American Association for the Study of Liver Diseases. Liver biopsy. Hepatology 2009; 49:1017–1044.
- Bravo AA, Sheth SG, Chopra S. Liver biopsy. N Engl J Med 2001; 344:495–500.
- Regev A, Berho M, Jeffers LJ, et al. Sampling error and intraobserver variation in liver biopsy in patients with chronic HCV infection. Am J Gastroenterol 2002; 97:2614–2618.
- Ratziu V, Charlotte F, Heurtier A, et al; LIDO Study Group Sampling variability of liver biopsy in nonalcoholic fatty liver disease. Gastroenterology 2005; 128:1898–1906.
- Saadeh S, Cammell G, Carey WD, Younossi Z, Barnes D, Easley K. The role of liver biopsy in chronic hepatitis C. Hepatology 2001; 33:196–200.
- Batts KP, Ludwig J. Chronic hepatitis. An update on terminology and reporting. Am J Surg Pathol 1995; 19:1409–1417.
- Morrison ED, Brandhagen DJ, Phatak PD, et al. Serum ferritin level predicts advanced hepatic fibrosis among U.S. patients with phenotypic hemochromatosis. Ann Intern Med 2003; 138:627–633.
- Parkes J, Guha IN, Roderick P, Rosenberg W. Performance of serum marker panels for liver fibrosis in chronic hepatitis C. J Hepatol 2006; 44:462–474.
- Montalto G, Soresi M, Aragona F, et al. Procollagen III and laminin in chronic viral hepatopathies. Presse Med 1996; 25:59–62.
- Teare JP, Sherman D, Greenfield SM, et al. Comparison of serum procollagen III peptide concentrations and PGA index for assessment of hepatic fibrosis. Lancet 1993; 342:895–898.
- Trinchet JC, Hartmann DJ, Pateron D, et al. Serum type I collagen and N-terminal peptide of type III procollagen in chronic hepatitis. Relationship to liver histology and conventional liver tests. J Hepatol 1991; 12:139–144.
- McCullough AJ, Stassen WN, Wiesner RH, Czaja AJ. Serial determinations of the amino-terminal peptide of type III procollagen in severe chronic active hepatitis. J Lab Clin Med 1987; 109:55–61.
- Takahara T, Furui K, Funaki J, et al. Increased expression of matrix metalloproteinase-II in experimental liver fibrosis in rats. Hepatology 1995; 21:787–795.
- Takahara T, Furui K, Yata Y, et al. Dual expression of matrix metalloproteinase-2 and membrane-type 1-matrix metalloproteinase in fibrotic human livers. Hepatology 1997; 26:1521–1529.
- Wai CT, Greenson JK, Fontana RJ, et al. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology 2003; 38:518–526.
- Kelleher TB, Mehta SH, Bhaskar R, et al. Prediction of hepatic fibrosis in HIV/HCV co-infected patients using serum fibrosis markers: the SHASTA index. J Hepatol 2005; 43:78–84.
- Islam S, Antonsson L, Westin J, Lagging M. Cirrhosis in hepatitis C virus-infected patients can be excluded using an index of standard biochemical serum markers. Scand J Gastroenterol 2005; 40:867–872.
- Lackner C, Struber G, Liegl B, et al. Comparison and validation of simple noninvasive tests for prediction of fibrosis in chronic hepatitis C. Hepatology 2005; 41:1376–1382.
- Poynard T, Aubert A, Bedossa P, et al. A simple biological index for detection of alcoholic liver disease in drinkers. Gastroenterology 1991; 100:1397–1402.
- Oberti F, Valsesia E, Pilette C, et al. Noninvasive diagnosis of hepatic fibrosis or cirrhosis. Gastroenterology 1997; 113:1609–1616.
- Koda M, Matunaga Y, Kawakami M, Kishimoto Y, Suou T, Murawaki Y. FibroIndex, a practical index for predicting significant fibrosis in patients with chronic hepatitis C. Hepatology 2007; 45:297–306.
- Vallet-Pichard A, Mallet V, Nalpas B, et al. FIB-4: an inexpensive and accurate marker of fibrosis in HCV infection. Comparison with liver biopsy and fibrotest. Hepatology 2007; 46:32–36.
- Sterling RK, Lissen E, Clumeck N, et al; APRI COT Clinical Investigators. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology 2006; 43:1317–1325.
- Calès P, Oberti F, Michalak S, et al. A novel panel of blood markers to assess the degree of liver fibrosis. Hepatology 2005; 42:1373–1381.
- Leroy V, Hilleret MN, Sturm N, et al. Prospective comparison of six non-invasive scores for the diagnosis of liver fibrosis in chronic hepatitis C. J Hepatol 2007; 46:775–782.
- Myers RP, De Torres M, Imbert-Bismut F, Ratziu V, Charlotte F, Poynard T; MULTIVIRC Group. Biochemical markers of fibrosis in patients with chronic hepatitis C: a comparison with prothrombin time, platelet count, and age-platelet index. Dig Dis Sci 2003; 48:146–153.
- Rossi E, Adams L, Prins A, et al. Validation of the FibroTest biochemical markers score in assessing liver fibrosis in hepatitis C patients. Clin Chem 2003; 49:450–454.
- Halfon P, Bourliere M, Deydier R, et al. Independent prospective multicenter validation of biochemical markers (fibrotest-actitest) for the prediction of liver fibrosis and activity in patients with chronic hepatitis C: the fibropaca study. Am J Gastroenterol 2006; 101:547–555.
- Poynard T, Imbert-Bismut F, Munteanu M, et al. Overview of the diagnostic value of biochemical markers of liver fibrosis (FibroTest, HCV FibroSure) and necrosis (ActiTest) in patients with chronic hepatitis C. Comp Hepatol 2004; 3:8.
- Sebastiani G, Halfon P, Castera L, et al. SAFE biopsy: a validated method for large-scale staging of liver fibrosis in chronic hepatitis C. Hepatology 2009; 49:1821–1827.
- The French METAVIR Cooperative Study Group. Intraobserver and interobserver variations in liver biopsy interpretations in patients with chronic hepatitis C. Hepatology 1994; 20:15–20.
- Sanford NL, Walsh P, Matis C, Baddeley H, Powell LW. Is ultrasonography useful in the assessment of diffuse parenchymal liver disease? Gastroenterology 1985; 89:186–191.
- Harbin WP, Robert NJ, Ferrucci JT. Diagnosis of cirrhosis based on regional changes in hepatic morphology: a radiological and pathological analysis. Radiology 1980; 135:273–283.
- Giorgio A, Amoroso P, Lettieri G, et al. Cirrhosis: value of caudate to right lobe ratio in diagnosis with US. Radiology 1986; 161:443–445.
- Liu CH, Hsu SJ, Lin JW, et al. Noninvasive diagnosis of hepatic fibrosis in patients with chronic hepatitis C by splenic Doppler impedance index. Clin Gastroenterol Hepatol 2007; 5:1199–1206.
- Talawalkar JA. Elastography for detecting hepatic fibrosis: options and considerations. Gastroenterology 2008; 135:299–302.
- Sandrin L, Fourquet B, Hasquenoph JM, et al. Transient elastography: a new noninvasive method for assessment of hepatic fibrosis. Ultrasound Med Biol 2003; 29:1705–1713.
- Kettaneh A, Marcellin P, Douvin C, et al. Features associated with success rate and performance of FibroScan measurements for the diagnosis of cirrhosis in HCV patients: a prospective study of 935 patients. J Hepatol 2007; 46:628–634.
- Ziol M, Handra-Luca A, Kettaneh A, et al. Noninvasive assessment of liver fibrosis by measurement of stiffness in patients with chronic hepatitis C. Hepatology 2005; 41:48–54.
- Friedrich-Rust M, Ong MF, Martens S, et al. Performance of transient elastography for the staging of liver fibrosis: a meta-analysis. Gastroenterology 2008; 134:960–974.
- Talwalkar JA, Kurtz DM, Schoenleber SJ, West CP, Montori VM. Ultrasound-based transient elastography for the detection of hepatic fibrosis: systematic review and meta-analysis. Clin Gastroenterol Hepatol 2007; 5:1214–1220.
- Arena U, Vizzutti F, Corti G, et al. Acute viral hepatitis increases liver stiffness values measured by transient elastography. Hepatology 2008; 47:380–384.
- Roulot D, Czernichow S, Le Clésiau H, Costes JL, Vergnaud AC, Beaugrand M. Liver stiffness values in apparently healthy subjects: influence of gender and metabolic syndrome. J Hepatol 2008; 48:606–613.
- Ito K, Mitchell DG, Hann HW, et al. Viral-induced cirrhosis: grading of severity using MR imaging. AJR Am J Roentgenol 1999; 173:591–596.
- Huwart L, Sempoux C, Vicaut E, et al. Magnetic resonance elastography for the noninvasive staging of liver fibrosis. Gastroenterology 2008; 135:32–40.
KEY POINTS
- Liver biopsy remains an important tool in the evaluation and management of liver disease.
- The role of liver biopsy for diagnosis of chronic liver disease has diminished, owing to accurate blood tests and imaging studies.
- Noninvasive tests for assessing the degree of hepatic fibrosis are showing more promise and may further reduce the need for liver biopsy. Elastography, in particular, shows promise in measuring hepatic fibrosis.
- Liver biopsy is still needed if laboratory testing and imaging studies are inconclusive.
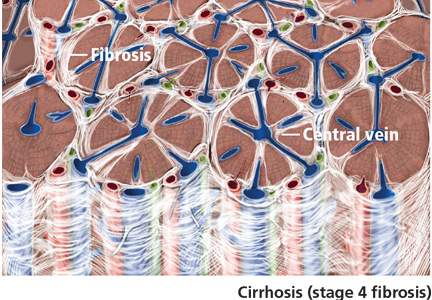
The prevalence and natural history of hepatitis B in the 21st century
Hepatitis B virus (HBV) infection is highly prevalent worldwide and is a major cause of morbidity and death. Two billion people globally have been infected with HBV, 350 to 400 million are chronic carriers, and tens of millions of new cases occur annually. Of those infected, 15% to 40% develop HBV complications, namely cirrhosis or hepatocellular carcinoma (HCC).1–3
The high prevalence of HBV infection represents an enormous failure of public health, considering that HBV immunization has been available for an entire generation, and where it has been employed it has been highly effective at reducing the incidence of HBV infection. Immunization, however, has been underused.
This supplement to the Cleveland Clinic Journal of Medicine, derived from a live symposium, aims to enhance awareness of the natural history of HBV infection and clarify its management recommendations with illustrative case histories. The supplement starts with a brief review of HBV terminology, natural history, and epidemiology.
CHRONIC HBV INFECTION TERMINOLOGY
Familiarity with the terms commonly used to describe chronic HBV infection will help clinicians in the management of the disease4:
- Chronic HBV infection is defined as presence of hepatitis B surface antigen (HBsAg) for more than 6 months. Those with infection may also express another antigen, HB e antigen (HBeAg), a marker of heightened infectivity. At the same time, those who are HBeAg positive are better responders to antiviral therapy compared with those who are HBeAg negative.
- An inactive HBsAg carrier is an individual who is HBsAg positive with a very low level of circulating virus, liver enzyme levels within normal limits, and a low likelihood of having chronic progressive disease.
- Resolved HBV infection is defined as previous HBV infection with no remaining evidence of active disease. Such individuals test negative for HBsAg and positive for antibody to HBsAg (anti-HBs) and to HB core antigen (anti-HBc). They also have no detectable viral load, or HBV DNA, in their blood. In most instances, they are protected from reinfection.
- Reactivation is the reappearance of HBV infection in someone who is known to be an inactive HBsAg carrier or whose previous HBV infection had resolved (see “Case: Recurrence despite anti-HBs and HBsAg negativity”).
- HBeAg seroconversion is the transition from HBeAg-positive to HBeAg-negative status and development of antibody to HBeAg (anti-HBe), usually accompanied by less active liver disease and lower viral loads.
- HBeAg clearance is disappearance of HBeAg without the development of anti-HBe; reactivation or reversion to HBeAg-positive status can occur.

GEOGRAPHIC DISTRIBUTION OF CHRONIC HBV INFECTION
The global prevalence of HBV varies widely. Regions are divided into areas of low, intermediate, and high prevalence, defined as follows4:
- High prevalence implies that at least 8% of the population is currently infected, with a lifetime likelihood of active or resolved infection greater than 60%. About 45% of the world’s population lives in regions of high prevalence. Among this group, early childhood infections are common, with the virus usually transmitted from mother to infant during the perinatal period.
- Intermediate prevalence is defined as 2% to 7%, with a lifetime risk of infection of 20% to 60%. These regions represent about 43% of the global population. In intermediate-prevalence areas, infections occur in all age groups.
- Low prevalence is defined as less than 2% and represents only 12% of the global population. In these regions, the lifetime risk of infection is less than 20%.
North America is a low-prevalence area except for the northern rim, where Inuit and Yupik Eskimos have a high prevalence, and communities that have a substantial immigrant population from high-prevalence areas, such as sub-Saharan Africa and many parts of Asia.
Chronic HBV infection in the United States
Approximately 1.25 million individuals in the United States are HBsAg carriers.2,4 In Asian Americans and Alaskan natives, the prevalence of HBsAg positivity, or chronic disease, is 5% to 15%.5,6 Similarly, US health statistics sources estimate that among those who are chronically infected, approximately half are Asian American.7 As the Asian American population continues to increase (1.5 million to 7 million from 1970 to 19905,8; 11.9 million in the 2000 US Census8), the total prevalence of chronic HBV infection will increase as well.
NATURAL HISTORY OF CHRONIC HBV INFECTION
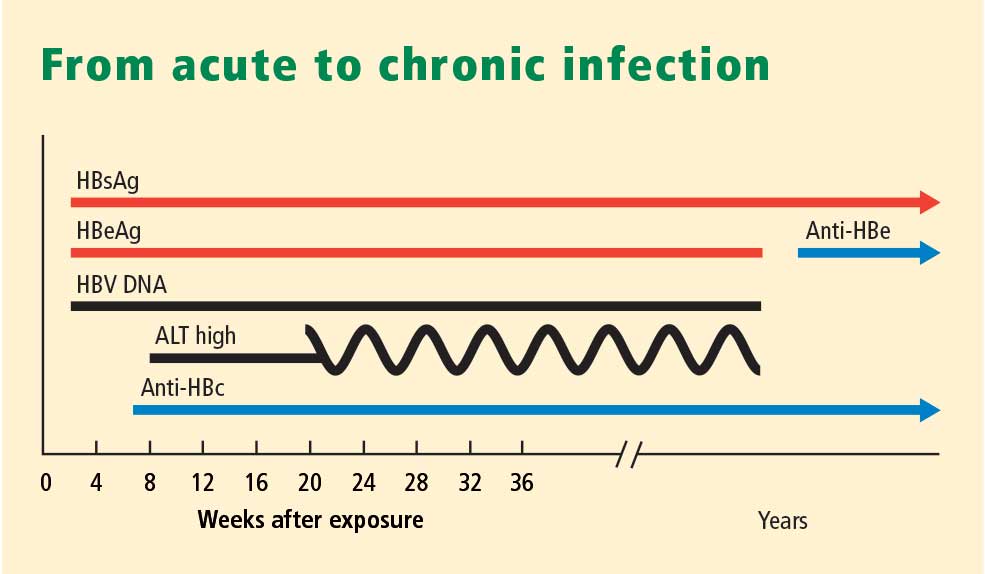
Chronic HBV usually causes microinflammatory changes that evoke a fibrotic response in the liver, and many infected individuals will eventually develop cirrhosis and are at risk for the development of HCC. Inactive HBsAg carriers often bypass the development of cirrhosis but remain at risk for HCC if their viral load is very high. This is particularly true when infection is acquired in infancy.
The age at acquisition of HBV has a large impact on the likelihood of the disease becoming chronic. The chance of chronic infection is 90% or greater among neonates who become infected with HBV through perinatal transmission. Exposure during adolescence or young adulthood is associated with a 95% or greater likelihood that the disease will be self-limiting.
The typical North American patient with HBV acquires the infection as an adolescent or young adult and is not at risk of HCC unless cirrhosis develops. In most patients who acquire the disease in adolescence or adulthood, the infection resolves after weeks or a few months and they are not at risk of either cirrhosis or HCC. However, an individual such as the one described in the accompanying case, who becomes immunocompromised, is at risk of reactivation of HBV infection (see “Case revisited”).

HBV MODES OF TRANSMISSION
In low-prevalence areas, such as most of North America, most cases of HBV infection are acquired during adolescence to midadulthood, a period during which behaviors that increase the risk of HBV infection (ie, intravenous drug abuse or unprotected sexual activity) are most likely.9,10 Sex workers and homosexuals are at particular risk of sexual transmission of HBV. Intravenous drug abusers and health workers are at risk of parenteral transmission.
In high-prevalence areas, HBV is mostly transmitted during the perinatal period from mother to infant, conferring a high likelihood of chronicity.9,10 Mothers who are HBsAg positive, particularly those who are also HBeAg positive, are much more likely than others to transmit HBV to their offspring.
FACTORS THAT INFLUENCE THE COURSE OF HBV INFECTION
Viral load has emerged as the most significant factor implicated in the development of cirrhosis or HCC. Iloeje et al11 found that viral load predicted progression to cirrhosis among a cohort of nearly 4,000 Taiwanese. Other factors that can influence the course of HBV infection include age at onset, male sex, and comorbidities (ie, alcohol use, human immunodeficiency virus infection, hepatitis C virus infection). Core promoter and precore mutants may affect the likelihood of developing HCC. A genetic signature that predisposes liver cells to proliferate, termed field effects, may also lead to the development of HCC. The influence of smoking and diabetes on the development of HCC in HBV-infected individuals is not well documented.
Reduction or elimination of measurable virus is the current holy grail of treatment; available antiviral therapies are potent tools that lower viral load with the hope of reducing the likelihood of either cirrhosis or HCC.
HBV genotypes may be implicated in the progression of liver disease or the risk of development of HCC. HBV genotypes differ by region and may correlate with ethnicity and disease progression. In a study of 694 US patients with chronic HBV, Chu et al12 found that genotypes A and C were associated with a higher prevalence of HBsAg positivity than other genotypes. Genotypes B and C were the most common among Asian American patients, while genotype A was the most common among Caucasian and African American patients. The authors suggested that HBV genotypes may explain the heterogeneity in the manifestation of the disease.
- Lavanchy D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J Viral Hepat 2004; 11:97–107.
- Hepatitis B Foundation. Statistics. Hepatitis B Foundation Web site. http://www.hepb.org/hepb/statistics.htm. Published 2003–2008. Accessed January 9, 2009.
- Hepatitis Foundation International. The ABC’s of hepatitis. Hepatitis Foundation International Web site. http://www.hepfi.org/living/liv_abc.html. Published 2003. Accessed January 9, 2009.
- Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Tong MJ, Hwang S-J. Hepatitis B virus infection in Asian Americans. Gastroenterol Clin North Am 1994; 23:523–536.
- McMahon BJ, Schoenberg S, Bulkow L, et al. Seroprevalence of hepatitis B viral markers in 52,000 Alaska natives. Am J Epidemiol 1993; 138:544–549.
- US Department of Health and Human Services. Hepatitis and Asian Americans. The Office of Minority Health Web site. http://www.omhrc.gov/templates/content.aspx?lvl=3&lvlid=541&ID=6495. Updated May 5, 2008. Accessed January 12, 2009.
- Barnes JS, Bennett CE. The Asian population: 2000. Census 2000 brief. United States Census 2000 Web site. http://www.census.gov/prod/2002pubs/c2kbr01-16.pdf. Published February 2002. Accessed January 12, 2009.
- Lok AS, McMahon BJ; Practice Guidelines Committee, American Association for the Study of Liver Diseases. Chronic hepatitis B. Hepatology 2001; 34:1225–1241.
- Lok AS, Heathcote EJ, Hoofnagle JH. Management of hepatitis B: 2000—summary of a workshop. Gastroenterology 2001; 120:1828–1853.
- Iloeje UH, Yang H-I, Su J, Jen C-L, You S-L, Chen C-J, and The Risk Evaluation of Viral Load Elevation and Associated Liver Disease/Cancer-in HBV (the REVEAL-HBV) Study Group. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology 2006; 130:678–686.
- Chu CJ, Keeffe EB, Han SH, et al. Hepatitis B virus genotypes in the United States: results of a nationwide study. Gastroenterology 2003; 125:444–451.
Hepatitis B virus (HBV) infection is highly prevalent worldwide and is a major cause of morbidity and death. Two billion people globally have been infected with HBV, 350 to 400 million are chronic carriers, and tens of millions of new cases occur annually. Of those infected, 15% to 40% develop HBV complications, namely cirrhosis or hepatocellular carcinoma (HCC).1–3
The high prevalence of HBV infection represents an enormous failure of public health, considering that HBV immunization has been available for an entire generation, and where it has been employed it has been highly effective at reducing the incidence of HBV infection. Immunization, however, has been underused.
This supplement to the Cleveland Clinic Journal of Medicine, derived from a live symposium, aims to enhance awareness of the natural history of HBV infection and clarify its management recommendations with illustrative case histories. The supplement starts with a brief review of HBV terminology, natural history, and epidemiology.
CHRONIC HBV INFECTION TERMINOLOGY
Familiarity with the terms commonly used to describe chronic HBV infection will help clinicians in the management of the disease4:
- Chronic HBV infection is defined as presence of hepatitis B surface antigen (HBsAg) for more than 6 months. Those with infection may also express another antigen, HB e antigen (HBeAg), a marker of heightened infectivity. At the same time, those who are HBeAg positive are better responders to antiviral therapy compared with those who are HBeAg negative.
- An inactive HBsAg carrier is an individual who is HBsAg positive with a very low level of circulating virus, liver enzyme levels within normal limits, and a low likelihood of having chronic progressive disease.
- Resolved HBV infection is defined as previous HBV infection with no remaining evidence of active disease. Such individuals test negative for HBsAg and positive for antibody to HBsAg (anti-HBs) and to HB core antigen (anti-HBc). They also have no detectable viral load, or HBV DNA, in their blood. In most instances, they are protected from reinfection.
- Reactivation is the reappearance of HBV infection in someone who is known to be an inactive HBsAg carrier or whose previous HBV infection had resolved (see “Case: Recurrence despite anti-HBs and HBsAg negativity”).
- HBeAg seroconversion is the transition from HBeAg-positive to HBeAg-negative status and development of antibody to HBeAg (anti-HBe), usually accompanied by less active liver disease and lower viral loads.
- HBeAg clearance is disappearance of HBeAg without the development of anti-HBe; reactivation or reversion to HBeAg-positive status can occur.
GEOGRAPHIC DISTRIBUTION OF CHRONIC HBV INFECTION
The global prevalence of HBV varies widely. Regions are divided into areas of low, intermediate, and high prevalence, defined as follows4:
- High prevalence implies that at least 8% of the population is currently infected, with a lifetime likelihood of active or resolved infection greater than 60%. About 45% of the world’s population lives in regions of high prevalence. Among this group, early childhood infections are common, with the virus usually transmitted from mother to infant during the perinatal period.
- Intermediate prevalence is defined as 2% to 7%, with a lifetime risk of infection of 20% to 60%. These regions represent about 43% of the global population. In intermediate-prevalence areas, infections occur in all age groups.
- Low prevalence is defined as less than 2% and represents only 12% of the global population. In these regions, the lifetime risk of infection is less than 20%.
North America is a low-prevalence area except for the northern rim, where Inuit and Yupik Eskimos have a high prevalence, and communities that have a substantial immigrant population from high-prevalence areas, such as sub-Saharan Africa and many parts of Asia.
Chronic HBV infection in the United States
Approximately 1.25 million individuals in the United States are HBsAg carriers.2,4 In Asian Americans and Alaskan natives, the prevalence of HBsAg positivity, or chronic disease, is 5% to 15%.5,6 Similarly, US health statistics sources estimate that among those who are chronically infected, approximately half are Asian American.7 As the Asian American population continues to increase (1.5 million to 7 million from 1970 to 19905,8; 11.9 million in the 2000 US Census8), the total prevalence of chronic HBV infection will increase as well.
NATURAL HISTORY OF CHRONIC HBV INFECTION
Chronic HBV usually causes microinflammatory changes that evoke a fibrotic response in the liver, and many infected individuals will eventually develop cirrhosis and are at risk for the development of HCC. Inactive HBsAg carriers often bypass the development of cirrhosis but remain at risk for HCC if their viral load is very high. This is particularly true when infection is acquired in infancy.
The age at acquisition of HBV has a large impact on the likelihood of the disease becoming chronic. The chance of chronic infection is 90% or greater among neonates who become infected with HBV through perinatal transmission. Exposure during adolescence or young adulthood is associated with a 95% or greater likelihood that the disease will be self-limiting.
The typical North American patient with HBV acquires the infection as an adolescent or young adult and is not at risk of HCC unless cirrhosis develops. In most patients who acquire the disease in adolescence or adulthood, the infection resolves after weeks or a few months and they are not at risk of either cirrhosis or HCC. However, an individual such as the one described in the accompanying case, who becomes immunocompromised, is at risk of reactivation of HBV infection (see “Case revisited”).
HBV MODES OF TRANSMISSION
In low-prevalence areas, such as most of North America, most cases of HBV infection are acquired during adolescence to midadulthood, a period during which behaviors that increase the risk of HBV infection (ie, intravenous drug abuse or unprotected sexual activity) are most likely.9,10 Sex workers and homosexuals are at particular risk of sexual transmission of HBV. Intravenous drug abusers and health workers are at risk of parenteral transmission.
In high-prevalence areas, HBV is mostly transmitted during the perinatal period from mother to infant, conferring a high likelihood of chronicity.9,10 Mothers who are HBsAg positive, particularly those who are also HBeAg positive, are much more likely than others to transmit HBV to their offspring.
FACTORS THAT INFLUENCE THE COURSE OF HBV INFECTION
Viral load has emerged as the most significant factor implicated in the development of cirrhosis or HCC. Iloeje et al11 found that viral load predicted progression to cirrhosis among a cohort of nearly 4,000 Taiwanese. Other factors that can influence the course of HBV infection include age at onset, male sex, and comorbidities (ie, alcohol use, human immunodeficiency virus infection, hepatitis C virus infection). Core promoter and precore mutants may affect the likelihood of developing HCC. A genetic signature that predisposes liver cells to proliferate, termed field effects, may also lead to the development of HCC. The influence of smoking and diabetes on the development of HCC in HBV-infected individuals is not well documented.
Reduction or elimination of measurable virus is the current holy grail of treatment; available antiviral therapies are potent tools that lower viral load with the hope of reducing the likelihood of either cirrhosis or HCC.
HBV genotypes may be implicated in the progression of liver disease or the risk of development of HCC. HBV genotypes differ by region and may correlate with ethnicity and disease progression. In a study of 694 US patients with chronic HBV, Chu et al12 found that genotypes A and C were associated with a higher prevalence of HBsAg positivity than other genotypes. Genotypes B and C were the most common among Asian American patients, while genotype A was the most common among Caucasian and African American patients. The authors suggested that HBV genotypes may explain the heterogeneity in the manifestation of the disease.
Hepatitis B virus (HBV) infection is highly prevalent worldwide and is a major cause of morbidity and death. Two billion people globally have been infected with HBV, 350 to 400 million are chronic carriers, and tens of millions of new cases occur annually. Of those infected, 15% to 40% develop HBV complications, namely cirrhosis or hepatocellular carcinoma (HCC).1–3
The high prevalence of HBV infection represents an enormous failure of public health, considering that HBV immunization has been available for an entire generation, and where it has been employed it has been highly effective at reducing the incidence of HBV infection. Immunization, however, has been underused.
This supplement to the Cleveland Clinic Journal of Medicine, derived from a live symposium, aims to enhance awareness of the natural history of HBV infection and clarify its management recommendations with illustrative case histories. The supplement starts with a brief review of HBV terminology, natural history, and epidemiology.
CHRONIC HBV INFECTION TERMINOLOGY
Familiarity with the terms commonly used to describe chronic HBV infection will help clinicians in the management of the disease4:
- Chronic HBV infection is defined as presence of hepatitis B surface antigen (HBsAg) for more than 6 months. Those with infection may also express another antigen, HB e antigen (HBeAg), a marker of heightened infectivity. At the same time, those who are HBeAg positive are better responders to antiviral therapy compared with those who are HBeAg negative.
- An inactive HBsAg carrier is an individual who is HBsAg positive with a very low level of circulating virus, liver enzyme levels within normal limits, and a low likelihood of having chronic progressive disease.
- Resolved HBV infection is defined as previous HBV infection with no remaining evidence of active disease. Such individuals test negative for HBsAg and positive for antibody to HBsAg (anti-HBs) and to HB core antigen (anti-HBc). They also have no detectable viral load, or HBV DNA, in their blood. In most instances, they are protected from reinfection.
- Reactivation is the reappearance of HBV infection in someone who is known to be an inactive HBsAg carrier or whose previous HBV infection had resolved (see “Case: Recurrence despite anti-HBs and HBsAg negativity”).
- HBeAg seroconversion is the transition from HBeAg-positive to HBeAg-negative status and development of antibody to HBeAg (anti-HBe), usually accompanied by less active liver disease and lower viral loads.
- HBeAg clearance is disappearance of HBeAg without the development of anti-HBe; reactivation or reversion to HBeAg-positive status can occur.
GEOGRAPHIC DISTRIBUTION OF CHRONIC HBV INFECTION
The global prevalence of HBV varies widely. Regions are divided into areas of low, intermediate, and high prevalence, defined as follows4:
- High prevalence implies that at least 8% of the population is currently infected, with a lifetime likelihood of active or resolved infection greater than 60%. About 45% of the world’s population lives in regions of high prevalence. Among this group, early childhood infections are common, with the virus usually transmitted from mother to infant during the perinatal period.
- Intermediate prevalence is defined as 2% to 7%, with a lifetime risk of infection of 20% to 60%. These regions represent about 43% of the global population. In intermediate-prevalence areas, infections occur in all age groups.
- Low prevalence is defined as less than 2% and represents only 12% of the global population. In these regions, the lifetime risk of infection is less than 20%.
North America is a low-prevalence area except for the northern rim, where Inuit and Yupik Eskimos have a high prevalence, and communities that have a substantial immigrant population from high-prevalence areas, such as sub-Saharan Africa and many parts of Asia.
Chronic HBV infection in the United States
Approximately 1.25 million individuals in the United States are HBsAg carriers.2,4 In Asian Americans and Alaskan natives, the prevalence of HBsAg positivity, or chronic disease, is 5% to 15%.5,6 Similarly, US health statistics sources estimate that among those who are chronically infected, approximately half are Asian American.7 As the Asian American population continues to increase (1.5 million to 7 million from 1970 to 19905,8; 11.9 million in the 2000 US Census8), the total prevalence of chronic HBV infection will increase as well.
NATURAL HISTORY OF CHRONIC HBV INFECTION
Chronic HBV usually causes microinflammatory changes that evoke a fibrotic response in the liver, and many infected individuals will eventually develop cirrhosis and are at risk for the development of HCC. Inactive HBsAg carriers often bypass the development of cirrhosis but remain at risk for HCC if their viral load is very high. This is particularly true when infection is acquired in infancy.
The age at acquisition of HBV has a large impact on the likelihood of the disease becoming chronic. The chance of chronic infection is 90% or greater among neonates who become infected with HBV through perinatal transmission. Exposure during adolescence or young adulthood is associated with a 95% or greater likelihood that the disease will be self-limiting.
The typical North American patient with HBV acquires the infection as an adolescent or young adult and is not at risk of HCC unless cirrhosis develops. In most patients who acquire the disease in adolescence or adulthood, the infection resolves after weeks or a few months and they are not at risk of either cirrhosis or HCC. However, an individual such as the one described in the accompanying case, who becomes immunocompromised, is at risk of reactivation of HBV infection (see “Case revisited”).
HBV MODES OF TRANSMISSION
In low-prevalence areas, such as most of North America, most cases of HBV infection are acquired during adolescence to midadulthood, a period during which behaviors that increase the risk of HBV infection (ie, intravenous drug abuse or unprotected sexual activity) are most likely.9,10 Sex workers and homosexuals are at particular risk of sexual transmission of HBV. Intravenous drug abusers and health workers are at risk of parenteral transmission.
In high-prevalence areas, HBV is mostly transmitted during the perinatal period from mother to infant, conferring a high likelihood of chronicity.9,10 Mothers who are HBsAg positive, particularly those who are also HBeAg positive, are much more likely than others to transmit HBV to their offspring.
FACTORS THAT INFLUENCE THE COURSE OF HBV INFECTION
Viral load has emerged as the most significant factor implicated in the development of cirrhosis or HCC. Iloeje et al11 found that viral load predicted progression to cirrhosis among a cohort of nearly 4,000 Taiwanese. Other factors that can influence the course of HBV infection include age at onset, male sex, and comorbidities (ie, alcohol use, human immunodeficiency virus infection, hepatitis C virus infection). Core promoter and precore mutants may affect the likelihood of developing HCC. A genetic signature that predisposes liver cells to proliferate, termed field effects, may also lead to the development of HCC. The influence of smoking and diabetes on the development of HCC in HBV-infected individuals is not well documented.
Reduction or elimination of measurable virus is the current holy grail of treatment; available antiviral therapies are potent tools that lower viral load with the hope of reducing the likelihood of either cirrhosis or HCC.
HBV genotypes may be implicated in the progression of liver disease or the risk of development of HCC. HBV genotypes differ by region and may correlate with ethnicity and disease progression. In a study of 694 US patients with chronic HBV, Chu et al12 found that genotypes A and C were associated with a higher prevalence of HBsAg positivity than other genotypes. Genotypes B and C were the most common among Asian American patients, while genotype A was the most common among Caucasian and African American patients. The authors suggested that HBV genotypes may explain the heterogeneity in the manifestation of the disease.
- Lavanchy D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J Viral Hepat 2004; 11:97–107.
- Hepatitis B Foundation. Statistics. Hepatitis B Foundation Web site. http://www.hepb.org/hepb/statistics.htm. Published 2003–2008. Accessed January 9, 2009.
- Hepatitis Foundation International. The ABC’s of hepatitis. Hepatitis Foundation International Web site. http://www.hepfi.org/living/liv_abc.html. Published 2003. Accessed January 9, 2009.
- Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Tong MJ, Hwang S-J. Hepatitis B virus infection in Asian Americans. Gastroenterol Clin North Am 1994; 23:523–536.
- McMahon BJ, Schoenberg S, Bulkow L, et al. Seroprevalence of hepatitis B viral markers in 52,000 Alaska natives. Am J Epidemiol 1993; 138:544–549.
- US Department of Health and Human Services. Hepatitis and Asian Americans. The Office of Minority Health Web site. http://www.omhrc.gov/templates/content.aspx?lvl=3&lvlid=541&ID=6495. Updated May 5, 2008. Accessed January 12, 2009.
- Barnes JS, Bennett CE. The Asian population: 2000. Census 2000 brief. United States Census 2000 Web site. http://www.census.gov/prod/2002pubs/c2kbr01-16.pdf. Published February 2002. Accessed January 12, 2009.
- Lok AS, McMahon BJ; Practice Guidelines Committee, American Association for the Study of Liver Diseases. Chronic hepatitis B. Hepatology 2001; 34:1225–1241.
- Lok AS, Heathcote EJ, Hoofnagle JH. Management of hepatitis B: 2000—summary of a workshop. Gastroenterology 2001; 120:1828–1853.
- Iloeje UH, Yang H-I, Su J, Jen C-L, You S-L, Chen C-J, and The Risk Evaluation of Viral Load Elevation and Associated Liver Disease/Cancer-in HBV (the REVEAL-HBV) Study Group. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology 2006; 130:678–686.
- Chu CJ, Keeffe EB, Han SH, et al. Hepatitis B virus genotypes in the United States: results of a nationwide study. Gastroenterology 2003; 125:444–451.
- Lavanchy D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J Viral Hepat 2004; 11:97–107.
- Hepatitis B Foundation. Statistics. Hepatitis B Foundation Web site. http://www.hepb.org/hepb/statistics.htm. Published 2003–2008. Accessed January 9, 2009.
- Hepatitis Foundation International. The ABC’s of hepatitis. Hepatitis Foundation International Web site. http://www.hepfi.org/living/liv_abc.html. Published 2003. Accessed January 9, 2009.
- Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Tong MJ, Hwang S-J. Hepatitis B virus infection in Asian Americans. Gastroenterol Clin North Am 1994; 23:523–536.
- McMahon BJ, Schoenberg S, Bulkow L, et al. Seroprevalence of hepatitis B viral markers in 52,000 Alaska natives. Am J Epidemiol 1993; 138:544–549.
- US Department of Health and Human Services. Hepatitis and Asian Americans. The Office of Minority Health Web site. http://www.omhrc.gov/templates/content.aspx?lvl=3&lvlid=541&ID=6495. Updated May 5, 2008. Accessed January 12, 2009.
- Barnes JS, Bennett CE. The Asian population: 2000. Census 2000 brief. United States Census 2000 Web site. http://www.census.gov/prod/2002pubs/c2kbr01-16.pdf. Published February 2002. Accessed January 12, 2009.
- Lok AS, McMahon BJ; Practice Guidelines Committee, American Association for the Study of Liver Diseases. Chronic hepatitis B. Hepatology 2001; 34:1225–1241.
- Lok AS, Heathcote EJ, Hoofnagle JH. Management of hepatitis B: 2000—summary of a workshop. Gastroenterology 2001; 120:1828–1853.
- Iloeje UH, Yang H-I, Su J, Jen C-L, You S-L, Chen C-J, and The Risk Evaluation of Viral Load Elevation and Associated Liver Disease/Cancer-in HBV (the REVEAL-HBV) Study Group. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology 2006; 130:678–686.
- Chu CJ, Keeffe EB, Han SH, et al. Hepatitis B virus genotypes in the United States: results of a nationwide study. Gastroenterology 2003; 125:444–451.
KEY POINTS
- The prevalence of chronic HBV infection in the United States is expected to increase as Asian immigrants constitute a larger proportion of the US population.
- The chance of chronic infection is 90% or greater with perinatal transmission; conversely, the risk of chronic disease is less than 10% with adult-acquired infection.
- In addition to viral load, predictors of disease progression include age at onset, male sex, and comorbidities.
Hepatitis B virus infection: Understanding its epidemiology, course, and diagnosis
Our knowledge about hepatitis B and related diseases has dramatically increased since the discovery of the causative virus, HBV, in 1963. Despite effective vaccination, hepatitis B still constitutes a major public health problem.
In two parts, this comprehensive review will highlight a practical clinical approach to HBV infection. In this first part, we discuss the epidemiology, natural history, and diagnosis of HBV infection. In the second part, to be published in the next issue of this journal, we will review the general principles of its management, its management in patients on immunosuppressant therapy and in pregnant women, and HBV vaccination.
COMMON IN ASIA, LESS SO IN AMERICA
More than 2 billion people—one-third of the world’s population—alive today have been infected with HBV at some time in their life, and of these, about 350 million remain infected.1 Every year, 1 million people die of HBV-related cirrhosis or hepatocellular carcinoma, which means that HBV takes a life every 30 seconds.2
The incidence of acute hepatitis B in the United States has declined from 8.5 per 100,000 population in 1990 to 2.1 per 100,000 population in 2004, with the greatest declines (94%) in children and adolescents, coincident with an increase in hepatitis B vaccination in these age groups.5 Despite these advances, HBV still causes a considerable number of cases of cirrhosis, cancer, and death—about 5,000 deaths each year in the United States.
HBV HAS FOUR GENES, EIGHT GENOTYPES
HBV is a DNA virus of the Hepadnaviridae family. Its genome is double-stranded with four genes, each one encoding a specific structural protein or proteins6,7:
- S gene, for the viral envelope (surface antigen)
- C gene, for both the nucleocapsid (core) antigen and the pre-core (e) antigen
- X gene, for two regulatory proteins required for HBV replication
- P gene, for DNA polymerase.

Eight genotypes of HBV (labeled A though H) have been identified.13,14 All eight have been found in the United States, but genotype A accounts for 35% of cases, genotype B for 22%, and genotype C for 31%.15
The clinical significance of HBV genotypes is not as clear as that of hepatitis C virus genotypes. Although recent data have suggested that different HBV genotypes may be associated with different rates of progression of liver disease and different rates of response to interferon therapy,13 these data were not enough to recommend routine testing for HBV genotypes in clinical practice.16
In HBV infection, the virus itself does not injure liver cells. Rather, the damage of hepatitis is immune-mediated and begins to appear as the host’s immune system attempts to clear the virus.
MARKERS OF HBV INFECTION
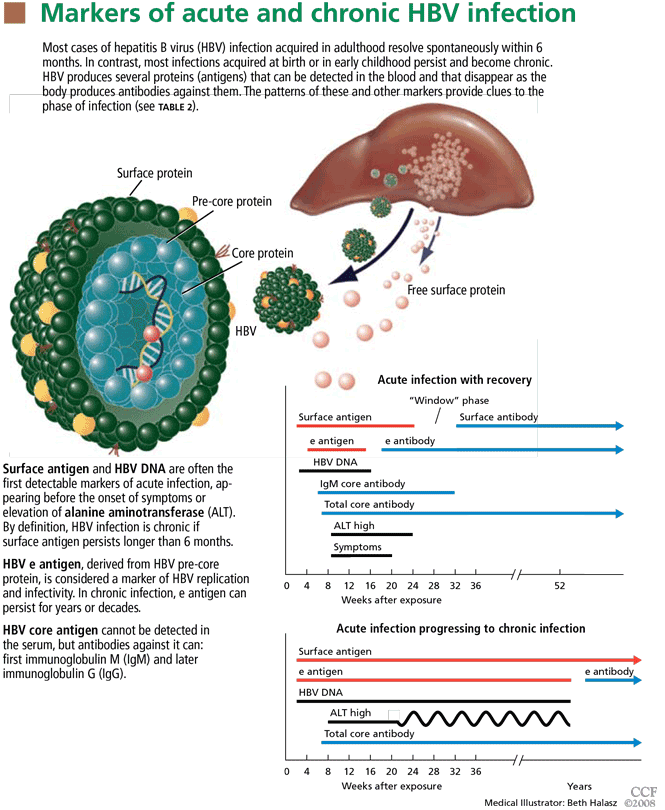
HBV surface antigen and HBV DNA are often the first detectable markers of acute infection, appearing before the onset of symptoms or before elevation of alanine aminotransferase (ALT) occurs. By definition, an HBV infection is chronic if surface antigen persists longer than 6 months.
HBV e antigen, derived from pre-core protein, is considered a marker of HBV replication and infectivity. In chronic infection, e antigen can persist for years or decades.
HBV core antigen cannot be detected in the serum, but antibodies against it can, first immunoglobulin M (IgM) and later immunoglobulin G (IgG).
TRANSMISSION: VERTICAL OR HORIZONTAL
Because HBV replicates profusely and produces high titers in the blood (108 to 1,010 virions/ mL), any parenteral or mucosal exposure to infected blood poses a high risk of HBV acquisition. The risk of HBV transmission from a single needlestick is 1% to 6% if the blood is positive for HBV surface antigen but negative for HBV e antigen, and 22% to 40% if positive for both antigens.17–19 Saliva, nasopharyngeal fluid, breast milk, semen, urine, and cervical secretions can also harbor HBV.20
Worldwide, perinatal (vertical) transmission is the predominant mode of HBV transmission, whereas intravenous drug abuse and unprotected sexual intercourse are the main routes of infection in areas of low prevalence, such as the United States. In sub-Saharan Africa, Alaska, and Mediterranean countries, transmission of HBV usually occurs horizontally during childhood, presumably via contact with nonintact skin.21–24 Saliva has also been thought to be the route of HBV transmission in sporadic cases through human bites.25
People at risk of HBV infection include:
- Parenteral drug users
- People with multiple sexual partners
- Household contacts and sexual partners of people who are positive for HBV surface antigen
- Infants born to HBV-infected mothers
- Patients and staff in custodial institutions for the developmentally disabled
- Recipients of certain plasma-derived products (including patients with congenital coagulation defects)
- Hemodialysis patients
- Health and public-safety workers who have contact with blood
- People born in areas where HBV is endemic, and their children.
These people—as well as all pregnant women, patients infected with hepatitis C virus or human immunodeficiency virus, and patients with chronically elevated ALT or aspartate aminotransferase (AST) levels—should be screened for HBV infection with serologic markers.
CLINICAL MANIFESTATIONS VARY
HBV infection, acute or chronic, has variable manifestations. During the acute stage, HBV infection can manifest as anicteric (subclinical) hepatitis, icteric hepatitis, or, rarely, acute fulminant hepatitis. Chronic HBV infection can be asymptomatic (the HBV surface antigen carrier state), or it can be manifested by symptoms and signs of cirrhosis or hepatocellular carcinoma or both. Extrahepatic manifestations, including serum sickness, polyarteritis nodosa, essential mixed cryoglobulinemia, membranous glomerulonephritis, and aplastic anemia, have been reported in patients with HBV infection.26
Acute hepatitis B
The incubation period of HBV ranges from 2 weeks to 4 months. Initially, patients complain of fatigue, malaise, anorexia, right upper quad-rant discomfort, or flu-like symptoms (coryza, photophobia, headache, and myalgia); then jaundice becomes apparent, usually within 10 days of the onset of symptoms. Low-grade fever, jaundice, and mildly tender hepatomegaly are the most common signs. Generalized lymphadenopathy is not a feature of acute HBV infection. If the patient also has hepatitis D virus infection or underlying liver disease (eg, alcoholic liver disease), then acute HBV infection may be more severe.
In the acute phase, ALT and AST levels rise, sometimes to values above 1,000 IU/L. In icteric hepatitis, bilirubin levels also rise, usually after the ALT level does. Although the peak ALT level reflects the hepatocellular injury, it has no prognostic value. With recovery, ALT levels normalize in 1 to 4 months.
Acute fulminant hepatitis B occurs in 0.1% to 0.5% of patients, and causes about 10% of cases of acute liver failure in the United States.27 Patients typically present with rapidly progressive acute hepatitis characterized by signs of liver failure, such as coagulopathy, encephalopathy, and cerebral edema.
In the so-called window phase, laboratory testing may not reveal HBV surface antigen because of early clearance but shows IgM antibody against the HBV core antigen. HBV DNA may be low or undetected.
Chronic hepatitis B
Chronic hepatitis B is usually diagnosed as a result of a workup for abnormal liver function tests or as a result of screening patients at risk for HBV infection. Many patients with chronic hepatitis B have no symptoms or have nonspecific symptoms such as fatigue or right upper quadrant discomfort.
Acute exacerbations due to HBV e antigen seroreversion (ie, in which e antigen reappears) occasionally occur in patients with chronic hepatitis B. Most of these exacerbations are asymptomatic, but occasionally an acute hepatitis-like clinical picture with detectable IgM antibody against the core antigen occurs, leading to misdiagnosis of acute HBV infection in patients not previously known to have chronic HBV infection.28
In late cases, signs of cirrhosis such as jaundice, ascites, splenomegaly, pedal edema, encephalopathy, or variceal bleeding can be present.
Hepatocellular carcinoma should be suspected in cirrhotic patients with new-onset right upper quadrant pain, rapidly developing ascites, a palpable liver mass, or hepatic encephalopathy. Other nonspecific features of hepatocellular carcinoma include watery diarrhea, hypoglycemia, and certain cutaneous manifestations such as acanthosis nigricans and the Leser-Trelat sign (multiple pruritic seborrheic keratoses of sudden onset).
In chronic hepatitis B, liver enzyme levels can be normal, even in patients with wellcompensated cirrhosis. ALT levels may range from normal to five times higher than normal. Thrombocytopenia, hypoalbuminemia, direct hyperbilirubinemia, and prolonged prothrombin time suggest cirrhosis.
Findings of chronic hepatitis B on liver biopsy range from minimal inflammation to cirrhosis. The most characteristic histologic feature of chronic HBV infection is the “ground-glass hepatocyte,” which is due to intracellular accumulation of HBV surface antigen. 29
FEW ADULTS (BUT MANY CHILDREN) REMAIN CHRONICALLY INFECTED
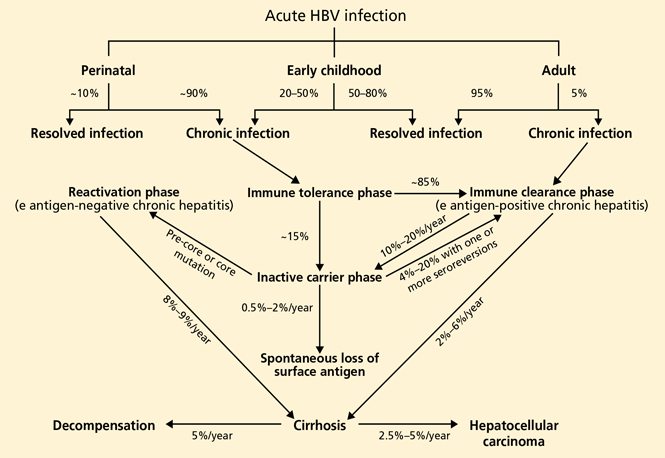
HBV surface antigen can be detected in the blood approximately 2 to 4 weeks after inoculation. Simultaneously, HBV DNA, usually in very high levels, is also detectable in the blood. However, in the rare cases of acute fulminant hepatitis, HBV DNA levels can be low or undetectable at the time of presentation because the immune system mounts a robust response with extensive damage to HBVinfected hepatocytes.
The rate of spontaneous recovery from acute HBV infection varies, depending on the patient’s age at the time of HBV acquisition and the patient’s immune status. Fewer than 5% of immunocompetent adults infected with HBV remain chronically infected, defined as being positive for HBV surface antigen for more than 6 months. On the other hand, 80% to 90% of infected infants and about 20% to 50% of children 1 to 5 years old at the time of acute infection remain chronically infected.21
Four phases of chronic HBV infection

The immune tolerance phase, the initial phase of chronic HBV infection, is seen almost exclusively in those who acquired HBV infection vertically or during early childhood. Although patients have high HBV DNA levels, they do not have significant liver disease. This discrepancy is thought to be related to the immune tolerance to HBV; however, the exact mechanism of that tolerance is unclear.31
Only 15% of those with immune tolerance have spontaneous HBV e antigen seroconversion (ie, loss of e antigen and appearance of anti-e antibody) within 20 years after infection. 32
The immune clearance phase (HBV e antigen-positive chronic hepatitis) appears about 20 to 30 years after the onset of the immune tolerance phase in patients who acquire HBV early in life. It is also often seen in patients with infections acquired late in childhood or in adulthood.
This phase marks the start of an immunemediated process aimed at clearing the viral infection, but it also leads to concomitant hepatocellular injury. Spontaneous clearance of the e antigen increases in this phase to an annual rate of 10% to 20%.32,33 The strongest predictors of spontaneous e antigen seroconversion are old age, an elevated ALT level, and an acute exacerbation.26
Although ALT levels are elevated and there is evidence on liver biopsy of chronic active hepatitis, this phase is usually asymptomatic. Rarely, however, it presents with an acute flare of hepatitis, sometimes accompanied by IgM antibodies against the HBV core antigen (in low titer), leading to an incorrect diagnosis of acute HBV infection.
Depending on the duration of the chronic hepatitis and the frequency and severity of flares, about 12% to 20% of patients in the immune-clearance phase develop serious liver disease within 5 years.31
The inactive carrier phase following HBV e antigen seroconversion is characterized by undetectable or low HBV DNA levels (< 1,000 copies/mL), normal ALT levels, and minimal or no necroinflammation on liver biopsy. 30 Such patients should be followed with serial testing, as 4% to 20% of them spontaneously revert to being positive for e antigen at least once.16 On the other hand, only 0.5% to 2% of surface antigen carriers in western countries clear themselves of surface antigen yearly, but up to half of those who clear the surface antigen have low-level HBV viremia. 34
The reactivation phase (HBV e antigennegative chronic hepatitis) is seen in some HBV-infected patients, especially those from Asia and southern Europe, in whom the virus has a spontaneous pre-core or core mutation that makes infected cells unable to secrete the e antigen. Although these patients have no e antigen in their blood, they do have intermittent or persistent elevation of ALT, elevated HBV DNA, and histopathologic findings of chronic hepatitis. Compared with those in the immune clearance phase, patients in the reactivation phase tend to be older and to have lower HBV DNA levels but advanced hepatic damage.
Immunity to HBV infection is characterized by loss of HBV surface antigen, DNA, e antigen, and anti-core antigen IgM with development of anti-surface antigen antibody and anti-core antigen IgG (total anti-core antigen antibody). The presence of anti-surface antigen antibody and anti-core antigen IgG together differentiates natural immunity through resolved infection from that which is acquired through vaccination, which is denoted by isolated anti-surface antigen antibody.

Cirrhosis, liver failure, cancer
Cirrhosis, hepatic decompensation, and hepatocellular carcinoma are the major long-term complications of HBV infection. In untreated patients, the annual rate of progression to cirrhosis has been estimated to be 2% to 6% in patients with HBV e antigen-positive chronic hepatitis and 8% to 9% in those with e antigen- negative chronic hepatitis.30
The likely explanation for these surprising cirrhosis rates is that e antigen-negative chronic hepatitis usually represents a late stage of the disease, and patients in this phase are usually older and have more advanced liver disease.
Subsequently, the annual rate of progression from compensated cirrhosis to hepatic decompensation has been estimated to be about 5%.35
Across all the stages described above, a high serum HBV DNA level has been shown to be a strong predictor of progression to cirrhosis in patients with chronic HBV infection. In a population-based prospective cohort study of 3,582 untreated HBV-infected patients in Taiwan, Iloeje et al36 found that, compared with patients with serum HBV DNA levels lower than 104 copies/mL, those with levels of 104 or higher had an adjusted relative risk of cirrhosis of 2.5. The relative risk rose to 5.9 with HBV DNA levels of 105 or higher, and 9.8 with levels of 106 copies/mL or higher. More studies in different patient populations are needed for confirmation.

The most important risk factor for hepatocellular carcinoma in HBV-infected patients is cirrhosis, but this cancer can also develop in noncirrhotic livers.37 The annual rate of hepatocelluar carcinoma has been estimated to be higher (2.5%–3%) in patients with cirrhosis than in noncirrhotic carriers (0.5%–1%).30,35–38 Risk factors for cirrhosis and hepatocellular carcinoma are summarized in Table 4.16,30
- World Health Organization. Hepatitis B. www.who.int/csr/disease/hepatitis/whocdscsrlyo20022/en. Accessed 11/10/2008.
- Center for Disease Control and Prevention. HBV a silent killer. www.cdc.gov/ncidod/diseases/hepatitis/b/hbv_silent_killer. Accessed 2/19/2007.
- McQuillan GM, Coleman PJ, Kruszon-Moran D, Moyer LA, Lambert SB, Margolis HS. Prevalence of hepatitis B virus infection in the United States: the National Health and Nutrition Examination Surveys, 1976 through 1994. Am J Public Health 1999; 89:14–18.
- Armstrong GL, Mast EE, Wojczynski M, Margolis HS. Childhood hepatitis B virus infections in the United States before hepatitis B immunization. Pediatrics 2001; 108:1123–1128.
- Mast EE, Weinbaum CM, Fiore AE, et al. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP). Part II: immunization of adults. MMWR Recomm Rep 2006 Dec 8; 55(RR-16):1–33.
- Gish RG, Gadano AC. Chronic hepatitis B: current epidemiology in the Americas and implications for management. J Viral Hepatol 2006; 13:787–798.
- Wei Y, Tiollais PK. Molecular biology of hepatitis B virus. Clin Liver Dis 1999; 3:189–219.
- Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev 2000; 64:51–68.
- Hunt CM, McGill JM, Allen MI, Condreay LD. Clinical relevance of hepatitis B viral mutations. Hepatology 2000; 3:1037–1044.
- Allen MI, Deslauriers M, Andrews CW, et al. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Lamivudine Clinical Investigation Group. Hepatology 1998; 27:1670–1677.
- Hadziyannis SJ, Vassilopoulos D. Hepatitis B e antigen- negative chronic hepatitis B. Hepatology 2001; 34:617–624.
- Wai CT, Fontana RJ. Clinical significance of hepatitis B virus genotypes, variants, and mutants. Clin Liver Dis 2004; 8:321–352.
- Fung SK, Lok AS. Hepatitis B virus genotypes: do they play a role in the outcome of HBV infection? Hepatology 2004; 40:790–792.
- Norder H, Courouce AM, Coursaget P, et al. Genetic diversity of hepatitis B virus strains derived worldwide: genotypes, subgenotypes, and HBsAg subtypes. Intervirology 2004; 47:289–309.
- Chu CJ, Keeffe EB, Han SH, et al. Hepatitis B virus genotypes in the United States: results of a nationwide study. Gastroenterology 2003; 125:444–451.
- Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Mast EE, Alter MJ. Prevention of hepatitis B virus infection among health-care workers. In:Ellis RE, editor. Hepatitis B Vaccines in Clinical Practice. New York: Marcel Dekker, 1993:295–307.
- Werner BG, Grady GF. Accidental hepatitis-B-surface-antigen-positive inoculations: use of e antigen to estimate infectivity. Ann Intern Med 1982; 97:367–369.
- Gerberding JL. Management of occupational exposures to blood-borne viruses. N Engl J Med 1995; 332:444–451.
- Kidd-Ljunggren K, Holmberg A, Blackberg J, Lindqvist B. High levels of hepatitis B virus DNA in body fluids from chronic carriers. J Hosp Infect 2006; 64:352–357.
- McMahon BJ, Alward WL, Hall DB, et al. Acute hepatitis B virus infection: relation of age to the clinical expression of disease and subsequent development of the carrier state. J Infect Dis 1985; 151:599–603.
- Dusheiko GM, Brink BA, Conradie JD, Marimuthu T, Sher R. Regional prevalence of hepatitis B, delta, and human immunodeficiency virus infection in southern Africa: a large population survey. Am J Epidemiol 1989; 129:138–45.
- Bortolotti F, Guido M, Bartolacci S, et al. Chronic hepatitis B in children after e antigen seroclearance: final report of a 29-year longitudinal study. Hepatology 2006; 43:556–562.
- Moreno MR, Otero M, Millan A, et al. Clinical and histological outcome after hepatitis B e antigen to antibody seroconversion in children with chronic hepatitis B. Hepatology 1999:572–575.
- Hui AY, Hung LC, Tse PC, Leung WK, Chan PK, Chan HL. Transmission of hepatitis B by human bite--confirmation by detection of virus in saliva and full genome sequencing. J Clin Virol 2005; 33:254–256.
- Cacoub P, Saadoun D, Bourlière M, et al. Hepatitis B virus genotypes and extrahepatic manifestations. J Hepatol 2005; 43:764–770.
- Schiodt FV, Atillasoy E, Shakil AO, et al. Etiology and outcome for 295 patients with acute liver failure in the United States. Liver Transplant Surg 1999; 5:29–34.
- Chu CM, Liaw YF, Pao CC, Huang MJ. The etiology of acute hepatitis superimposed upon previously unrecognized asymptomatic HBsAg carriers. Hepatology 1989; 9:452–456.
- Gerber MA, Hadziyannis S, Vissoulis C, et al. Electron microscopy and immunoelectronmicroscopy of cytoplasmic hepatitis B antigen in hepatocytes. Am J Pathol 1974; 75:489–502.
- Yim HJ, Lok AS. Natural history of chronic hepatitis B virus infection: what we knew in 1981 and what we know in 2005. Hepatology 2006; 43:S173–S181.
- Pungpapong S, Kim WR, Poterucha JJ. Natural history of hepatitis B virus infection: an update for clinicians. Mayo Clin Proc 2007; 82:967–975.
- Lok AS, Lai CL, Wu PC, Leung EK, Lam TS. Spontaneous hepatitis B e antigen to antibody seroconversion and reversion in Chinese patients with chronic hepatitis B virus infection. Gastroenterology 1987; 92:1839–1843.
- McMahon BJ, Holck P, Bulkow L, Snowball M. Serologic and clinical outcomes in 1536 Alaska Natives chronically infected with hepatitis B virus. Ann Intern Med 2001; 135:759–768.
- McMahon BJ. Epidemiology and natural history of hepatitis B. Semin Liver Dis 2005; 25( suppl 1):3–8.
- Benvegnu L, Gios M, Boccato S, Alberti A. Natural history of compensated viral cirrhosis: a prospective study on the incidence and hierarchy of major complications. Gut 2004; 53:744–749.
- Iloeje UH, Yang HI, Su J, Jen CL, You SL, Chen CJ. Risk evaluation of viral load elevation and associated liver disease/cancer in HBV. The REVEAL-HBV Study Group. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology 2006; 130:678–686.
- Bosch FX, Ribes J, Cleries R, Diaz M. Epidemiology of hepatocellular carcinoma. Clin Liver Dis 2005; 9:191–211.
- Fattovich G. Natural history of hepatitis B. J Hepatol 2003; 39:S50–S58.
Our knowledge about hepatitis B and related diseases has dramatically increased since the discovery of the causative virus, HBV, in 1963. Despite effective vaccination, hepatitis B still constitutes a major public health problem.
In two parts, this comprehensive review will highlight a practical clinical approach to HBV infection. In this first part, we discuss the epidemiology, natural history, and diagnosis of HBV infection. In the second part, to be published in the next issue of this journal, we will review the general principles of its management, its management in patients on immunosuppressant therapy and in pregnant women, and HBV vaccination.
COMMON IN ASIA, LESS SO IN AMERICA
More than 2 billion people—one-third of the world’s population—alive today have been infected with HBV at some time in their life, and of these, about 350 million remain infected.1 Every year, 1 million people die of HBV-related cirrhosis or hepatocellular carcinoma, which means that HBV takes a life every 30 seconds.2
The incidence of acute hepatitis B in the United States has declined from 8.5 per 100,000 population in 1990 to 2.1 per 100,000 population in 2004, with the greatest declines (94%) in children and adolescents, coincident with an increase in hepatitis B vaccination in these age groups.5 Despite these advances, HBV still causes a considerable number of cases of cirrhosis, cancer, and death—about 5,000 deaths each year in the United States.
HBV HAS FOUR GENES, EIGHT GENOTYPES
HBV is a DNA virus of the Hepadnaviridae family. Its genome is double-stranded with four genes, each one encoding a specific structural protein or proteins6,7:
- S gene, for the viral envelope (surface antigen)
- C gene, for both the nucleocapsid (core) antigen and the pre-core (e) antigen
- X gene, for two regulatory proteins required for HBV replication
- P gene, for DNA polymerase.
Eight genotypes of HBV (labeled A though H) have been identified.13,14 All eight have been found in the United States, but genotype A accounts for 35% of cases, genotype B for 22%, and genotype C for 31%.15
The clinical significance of HBV genotypes is not as clear as that of hepatitis C virus genotypes. Although recent data have suggested that different HBV genotypes may be associated with different rates of progression of liver disease and different rates of response to interferon therapy,13 these data were not enough to recommend routine testing for HBV genotypes in clinical practice.16
In HBV infection, the virus itself does not injure liver cells. Rather, the damage of hepatitis is immune-mediated and begins to appear as the host’s immune system attempts to clear the virus.
MARKERS OF HBV INFECTION
HBV surface antigen and HBV DNA are often the first detectable markers of acute infection, appearing before the onset of symptoms or before elevation of alanine aminotransferase (ALT) occurs. By definition, an HBV infection is chronic if surface antigen persists longer than 6 months.
HBV e antigen, derived from pre-core protein, is considered a marker of HBV replication and infectivity. In chronic infection, e antigen can persist for years or decades.
HBV core antigen cannot be detected in the serum, but antibodies against it can, first immunoglobulin M (IgM) and later immunoglobulin G (IgG).
TRANSMISSION: VERTICAL OR HORIZONTAL
Because HBV replicates profusely and produces high titers in the blood (108 to 1,010 virions/ mL), any parenteral or mucosal exposure to infected blood poses a high risk of HBV acquisition. The risk of HBV transmission from a single needlestick is 1% to 6% if the blood is positive for HBV surface antigen but negative for HBV e antigen, and 22% to 40% if positive for both antigens.17–19 Saliva, nasopharyngeal fluid, breast milk, semen, urine, and cervical secretions can also harbor HBV.20
Worldwide, perinatal (vertical) transmission is the predominant mode of HBV transmission, whereas intravenous drug abuse and unprotected sexual intercourse are the main routes of infection in areas of low prevalence, such as the United States. In sub-Saharan Africa, Alaska, and Mediterranean countries, transmission of HBV usually occurs horizontally during childhood, presumably via contact with nonintact skin.21–24 Saliva has also been thought to be the route of HBV transmission in sporadic cases through human bites.25
People at risk of HBV infection include:
- Parenteral drug users
- People with multiple sexual partners
- Household contacts and sexual partners of people who are positive for HBV surface antigen
- Infants born to HBV-infected mothers
- Patients and staff in custodial institutions for the developmentally disabled
- Recipients of certain plasma-derived products (including patients with congenital coagulation defects)
- Hemodialysis patients
- Health and public-safety workers who have contact with blood
- People born in areas where HBV is endemic, and their children.
These people—as well as all pregnant women, patients infected with hepatitis C virus or human immunodeficiency virus, and patients with chronically elevated ALT or aspartate aminotransferase (AST) levels—should be screened for HBV infection with serologic markers.
CLINICAL MANIFESTATIONS VARY
HBV infection, acute or chronic, has variable manifestations. During the acute stage, HBV infection can manifest as anicteric (subclinical) hepatitis, icteric hepatitis, or, rarely, acute fulminant hepatitis. Chronic HBV infection can be asymptomatic (the HBV surface antigen carrier state), or it can be manifested by symptoms and signs of cirrhosis or hepatocellular carcinoma or both. Extrahepatic manifestations, including serum sickness, polyarteritis nodosa, essential mixed cryoglobulinemia, membranous glomerulonephritis, and aplastic anemia, have been reported in patients with HBV infection.26
Acute hepatitis B
The incubation period of HBV ranges from 2 weeks to 4 months. Initially, patients complain of fatigue, malaise, anorexia, right upper quad-rant discomfort, or flu-like symptoms (coryza, photophobia, headache, and myalgia); then jaundice becomes apparent, usually within 10 days of the onset of symptoms. Low-grade fever, jaundice, and mildly tender hepatomegaly are the most common signs. Generalized lymphadenopathy is not a feature of acute HBV infection. If the patient also has hepatitis D virus infection or underlying liver disease (eg, alcoholic liver disease), then acute HBV infection may be more severe.
In the acute phase, ALT and AST levels rise, sometimes to values above 1,000 IU/L. In icteric hepatitis, bilirubin levels also rise, usually after the ALT level does. Although the peak ALT level reflects the hepatocellular injury, it has no prognostic value. With recovery, ALT levels normalize in 1 to 4 months.
Acute fulminant hepatitis B occurs in 0.1% to 0.5% of patients, and causes about 10% of cases of acute liver failure in the United States.27 Patients typically present with rapidly progressive acute hepatitis characterized by signs of liver failure, such as coagulopathy, encephalopathy, and cerebral edema.
In the so-called window phase, laboratory testing may not reveal HBV surface antigen because of early clearance but shows IgM antibody against the HBV core antigen. HBV DNA may be low or undetected.
Chronic hepatitis B
Chronic hepatitis B is usually diagnosed as a result of a workup for abnormal liver function tests or as a result of screening patients at risk for HBV infection. Many patients with chronic hepatitis B have no symptoms or have nonspecific symptoms such as fatigue or right upper quadrant discomfort.
Acute exacerbations due to HBV e antigen seroreversion (ie, in which e antigen reappears) occasionally occur in patients with chronic hepatitis B. Most of these exacerbations are asymptomatic, but occasionally an acute hepatitis-like clinical picture with detectable IgM antibody against the core antigen occurs, leading to misdiagnosis of acute HBV infection in patients not previously known to have chronic HBV infection.28
In late cases, signs of cirrhosis such as jaundice, ascites, splenomegaly, pedal edema, encephalopathy, or variceal bleeding can be present.
Hepatocellular carcinoma should be suspected in cirrhotic patients with new-onset right upper quadrant pain, rapidly developing ascites, a palpable liver mass, or hepatic encephalopathy. Other nonspecific features of hepatocellular carcinoma include watery diarrhea, hypoglycemia, and certain cutaneous manifestations such as acanthosis nigricans and the Leser-Trelat sign (multiple pruritic seborrheic keratoses of sudden onset).
In chronic hepatitis B, liver enzyme levels can be normal, even in patients with wellcompensated cirrhosis. ALT levels may range from normal to five times higher than normal. Thrombocytopenia, hypoalbuminemia, direct hyperbilirubinemia, and prolonged prothrombin time suggest cirrhosis.
Findings of chronic hepatitis B on liver biopsy range from minimal inflammation to cirrhosis. The most characteristic histologic feature of chronic HBV infection is the “ground-glass hepatocyte,” which is due to intracellular accumulation of HBV surface antigen. 29
FEW ADULTS (BUT MANY CHILDREN) REMAIN CHRONICALLY INFECTED
HBV surface antigen can be detected in the blood approximately 2 to 4 weeks after inoculation. Simultaneously, HBV DNA, usually in very high levels, is also detectable in the blood. However, in the rare cases of acute fulminant hepatitis, HBV DNA levels can be low or undetectable at the time of presentation because the immune system mounts a robust response with extensive damage to HBVinfected hepatocytes.
The rate of spontaneous recovery from acute HBV infection varies, depending on the patient’s age at the time of HBV acquisition and the patient’s immune status. Fewer than 5% of immunocompetent adults infected with HBV remain chronically infected, defined as being positive for HBV surface antigen for more than 6 months. On the other hand, 80% to 90% of infected infants and about 20% to 50% of children 1 to 5 years old at the time of acute infection remain chronically infected.21
Four phases of chronic HBV infection
The immune tolerance phase, the initial phase of chronic HBV infection, is seen almost exclusively in those who acquired HBV infection vertically or during early childhood. Although patients have high HBV DNA levels, they do not have significant liver disease. This discrepancy is thought to be related to the immune tolerance to HBV; however, the exact mechanism of that tolerance is unclear.31
Only 15% of those with immune tolerance have spontaneous HBV e antigen seroconversion (ie, loss of e antigen and appearance of anti-e antibody) within 20 years after infection. 32
The immune clearance phase (HBV e antigen-positive chronic hepatitis) appears about 20 to 30 years after the onset of the immune tolerance phase in patients who acquire HBV early in life. It is also often seen in patients with infections acquired late in childhood or in adulthood.
This phase marks the start of an immunemediated process aimed at clearing the viral infection, but it also leads to concomitant hepatocellular injury. Spontaneous clearance of the e antigen increases in this phase to an annual rate of 10% to 20%.32,33 The strongest predictors of spontaneous e antigen seroconversion are old age, an elevated ALT level, and an acute exacerbation.26
Although ALT levels are elevated and there is evidence on liver biopsy of chronic active hepatitis, this phase is usually asymptomatic. Rarely, however, it presents with an acute flare of hepatitis, sometimes accompanied by IgM antibodies against the HBV core antigen (in low titer), leading to an incorrect diagnosis of acute HBV infection.
Depending on the duration of the chronic hepatitis and the frequency and severity of flares, about 12% to 20% of patients in the immune-clearance phase develop serious liver disease within 5 years.31
The inactive carrier phase following HBV e antigen seroconversion is characterized by undetectable or low HBV DNA levels (< 1,000 copies/mL), normal ALT levels, and minimal or no necroinflammation on liver biopsy. 30 Such patients should be followed with serial testing, as 4% to 20% of them spontaneously revert to being positive for e antigen at least once.16 On the other hand, only 0.5% to 2% of surface antigen carriers in western countries clear themselves of surface antigen yearly, but up to half of those who clear the surface antigen have low-level HBV viremia. 34
The reactivation phase (HBV e antigennegative chronic hepatitis) is seen in some HBV-infected patients, especially those from Asia and southern Europe, in whom the virus has a spontaneous pre-core or core mutation that makes infected cells unable to secrete the e antigen. Although these patients have no e antigen in their blood, they do have intermittent or persistent elevation of ALT, elevated HBV DNA, and histopathologic findings of chronic hepatitis. Compared with those in the immune clearance phase, patients in the reactivation phase tend to be older and to have lower HBV DNA levels but advanced hepatic damage.
Immunity to HBV infection is characterized by loss of HBV surface antigen, DNA, e antigen, and anti-core antigen IgM with development of anti-surface antigen antibody and anti-core antigen IgG (total anti-core antigen antibody). The presence of anti-surface antigen antibody and anti-core antigen IgG together differentiates natural immunity through resolved infection from that which is acquired through vaccination, which is denoted by isolated anti-surface antigen antibody.
Cirrhosis, liver failure, cancer
Cirrhosis, hepatic decompensation, and hepatocellular carcinoma are the major long-term complications of HBV infection. In untreated patients, the annual rate of progression to cirrhosis has been estimated to be 2% to 6% in patients with HBV e antigen-positive chronic hepatitis and 8% to 9% in those with e antigen- negative chronic hepatitis.30
The likely explanation for these surprising cirrhosis rates is that e antigen-negative chronic hepatitis usually represents a late stage of the disease, and patients in this phase are usually older and have more advanced liver disease.
Subsequently, the annual rate of progression from compensated cirrhosis to hepatic decompensation has been estimated to be about 5%.35
Across all the stages described above, a high serum HBV DNA level has been shown to be a strong predictor of progression to cirrhosis in patients with chronic HBV infection. In a population-based prospective cohort study of 3,582 untreated HBV-infected patients in Taiwan, Iloeje et al36 found that, compared with patients with serum HBV DNA levels lower than 104 copies/mL, those with levels of 104 or higher had an adjusted relative risk of cirrhosis of 2.5. The relative risk rose to 5.9 with HBV DNA levels of 105 or higher, and 9.8 with levels of 106 copies/mL or higher. More studies in different patient populations are needed for confirmation.
The most important risk factor for hepatocellular carcinoma in HBV-infected patients is cirrhosis, but this cancer can also develop in noncirrhotic livers.37 The annual rate of hepatocelluar carcinoma has been estimated to be higher (2.5%–3%) in patients with cirrhosis than in noncirrhotic carriers (0.5%–1%).30,35–38 Risk factors for cirrhosis and hepatocellular carcinoma are summarized in Table 4.16,30
Our knowledge about hepatitis B and related diseases has dramatically increased since the discovery of the causative virus, HBV, in 1963. Despite effective vaccination, hepatitis B still constitutes a major public health problem.
In two parts, this comprehensive review will highlight a practical clinical approach to HBV infection. In this first part, we discuss the epidemiology, natural history, and diagnosis of HBV infection. In the second part, to be published in the next issue of this journal, we will review the general principles of its management, its management in patients on immunosuppressant therapy and in pregnant women, and HBV vaccination.
COMMON IN ASIA, LESS SO IN AMERICA
More than 2 billion people—one-third of the world’s population—alive today have been infected with HBV at some time in their life, and of these, about 350 million remain infected.1 Every year, 1 million people die of HBV-related cirrhosis or hepatocellular carcinoma, which means that HBV takes a life every 30 seconds.2
The incidence of acute hepatitis B in the United States has declined from 8.5 per 100,000 population in 1990 to 2.1 per 100,000 population in 2004, with the greatest declines (94%) in children and adolescents, coincident with an increase in hepatitis B vaccination in these age groups.5 Despite these advances, HBV still causes a considerable number of cases of cirrhosis, cancer, and death—about 5,000 deaths each year in the United States.
HBV HAS FOUR GENES, EIGHT GENOTYPES
HBV is a DNA virus of the Hepadnaviridae family. Its genome is double-stranded with four genes, each one encoding a specific structural protein or proteins6,7:
- S gene, for the viral envelope (surface antigen)
- C gene, for both the nucleocapsid (core) antigen and the pre-core (e) antigen
- X gene, for two regulatory proteins required for HBV replication
- P gene, for DNA polymerase.
Eight genotypes of HBV (labeled A though H) have been identified.13,14 All eight have been found in the United States, but genotype A accounts for 35% of cases, genotype B for 22%, and genotype C for 31%.15
The clinical significance of HBV genotypes is not as clear as that of hepatitis C virus genotypes. Although recent data have suggested that different HBV genotypes may be associated with different rates of progression of liver disease and different rates of response to interferon therapy,13 these data were not enough to recommend routine testing for HBV genotypes in clinical practice.16
In HBV infection, the virus itself does not injure liver cells. Rather, the damage of hepatitis is immune-mediated and begins to appear as the host’s immune system attempts to clear the virus.
MARKERS OF HBV INFECTION
HBV surface antigen and HBV DNA are often the first detectable markers of acute infection, appearing before the onset of symptoms or before elevation of alanine aminotransferase (ALT) occurs. By definition, an HBV infection is chronic if surface antigen persists longer than 6 months.
HBV e antigen, derived from pre-core protein, is considered a marker of HBV replication and infectivity. In chronic infection, e antigen can persist for years or decades.
HBV core antigen cannot be detected in the serum, but antibodies against it can, first immunoglobulin M (IgM) and later immunoglobulin G (IgG).
TRANSMISSION: VERTICAL OR HORIZONTAL
Because HBV replicates profusely and produces high titers in the blood (108 to 1,010 virions/ mL), any parenteral or mucosal exposure to infected blood poses a high risk of HBV acquisition. The risk of HBV transmission from a single needlestick is 1% to 6% if the blood is positive for HBV surface antigen but negative for HBV e antigen, and 22% to 40% if positive for both antigens.17–19 Saliva, nasopharyngeal fluid, breast milk, semen, urine, and cervical secretions can also harbor HBV.20
Worldwide, perinatal (vertical) transmission is the predominant mode of HBV transmission, whereas intravenous drug abuse and unprotected sexual intercourse are the main routes of infection in areas of low prevalence, such as the United States. In sub-Saharan Africa, Alaska, and Mediterranean countries, transmission of HBV usually occurs horizontally during childhood, presumably via contact with nonintact skin.21–24 Saliva has also been thought to be the route of HBV transmission in sporadic cases through human bites.25
People at risk of HBV infection include:
- Parenteral drug users
- People with multiple sexual partners
- Household contacts and sexual partners of people who are positive for HBV surface antigen
- Infants born to HBV-infected mothers
- Patients and staff in custodial institutions for the developmentally disabled
- Recipients of certain plasma-derived products (including patients with congenital coagulation defects)
- Hemodialysis patients
- Health and public-safety workers who have contact with blood
- People born in areas where HBV is endemic, and their children.
These people—as well as all pregnant women, patients infected with hepatitis C virus or human immunodeficiency virus, and patients with chronically elevated ALT or aspartate aminotransferase (AST) levels—should be screened for HBV infection with serologic markers.
CLINICAL MANIFESTATIONS VARY
HBV infection, acute or chronic, has variable manifestations. During the acute stage, HBV infection can manifest as anicteric (subclinical) hepatitis, icteric hepatitis, or, rarely, acute fulminant hepatitis. Chronic HBV infection can be asymptomatic (the HBV surface antigen carrier state), or it can be manifested by symptoms and signs of cirrhosis or hepatocellular carcinoma or both. Extrahepatic manifestations, including serum sickness, polyarteritis nodosa, essential mixed cryoglobulinemia, membranous glomerulonephritis, and aplastic anemia, have been reported in patients with HBV infection.26
Acute hepatitis B
The incubation period of HBV ranges from 2 weeks to 4 months. Initially, patients complain of fatigue, malaise, anorexia, right upper quad-rant discomfort, or flu-like symptoms (coryza, photophobia, headache, and myalgia); then jaundice becomes apparent, usually within 10 days of the onset of symptoms. Low-grade fever, jaundice, and mildly tender hepatomegaly are the most common signs. Generalized lymphadenopathy is not a feature of acute HBV infection. If the patient also has hepatitis D virus infection or underlying liver disease (eg, alcoholic liver disease), then acute HBV infection may be more severe.
In the acute phase, ALT and AST levels rise, sometimes to values above 1,000 IU/L. In icteric hepatitis, bilirubin levels also rise, usually after the ALT level does. Although the peak ALT level reflects the hepatocellular injury, it has no prognostic value. With recovery, ALT levels normalize in 1 to 4 months.
Acute fulminant hepatitis B occurs in 0.1% to 0.5% of patients, and causes about 10% of cases of acute liver failure in the United States.27 Patients typically present with rapidly progressive acute hepatitis characterized by signs of liver failure, such as coagulopathy, encephalopathy, and cerebral edema.
In the so-called window phase, laboratory testing may not reveal HBV surface antigen because of early clearance but shows IgM antibody against the HBV core antigen. HBV DNA may be low or undetected.
Chronic hepatitis B
Chronic hepatitis B is usually diagnosed as a result of a workup for abnormal liver function tests or as a result of screening patients at risk for HBV infection. Many patients with chronic hepatitis B have no symptoms or have nonspecific symptoms such as fatigue or right upper quadrant discomfort.
Acute exacerbations due to HBV e antigen seroreversion (ie, in which e antigen reappears) occasionally occur in patients with chronic hepatitis B. Most of these exacerbations are asymptomatic, but occasionally an acute hepatitis-like clinical picture with detectable IgM antibody against the core antigen occurs, leading to misdiagnosis of acute HBV infection in patients not previously known to have chronic HBV infection.28
In late cases, signs of cirrhosis such as jaundice, ascites, splenomegaly, pedal edema, encephalopathy, or variceal bleeding can be present.
Hepatocellular carcinoma should be suspected in cirrhotic patients with new-onset right upper quadrant pain, rapidly developing ascites, a palpable liver mass, or hepatic encephalopathy. Other nonspecific features of hepatocellular carcinoma include watery diarrhea, hypoglycemia, and certain cutaneous manifestations such as acanthosis nigricans and the Leser-Trelat sign (multiple pruritic seborrheic keratoses of sudden onset).
In chronic hepatitis B, liver enzyme levels can be normal, even in patients with wellcompensated cirrhosis. ALT levels may range from normal to five times higher than normal. Thrombocytopenia, hypoalbuminemia, direct hyperbilirubinemia, and prolonged prothrombin time suggest cirrhosis.
Findings of chronic hepatitis B on liver biopsy range from minimal inflammation to cirrhosis. The most characteristic histologic feature of chronic HBV infection is the “ground-glass hepatocyte,” which is due to intracellular accumulation of HBV surface antigen. 29
FEW ADULTS (BUT MANY CHILDREN) REMAIN CHRONICALLY INFECTED
HBV surface antigen can be detected in the blood approximately 2 to 4 weeks after inoculation. Simultaneously, HBV DNA, usually in very high levels, is also detectable in the blood. However, in the rare cases of acute fulminant hepatitis, HBV DNA levels can be low or undetectable at the time of presentation because the immune system mounts a robust response with extensive damage to HBVinfected hepatocytes.
The rate of spontaneous recovery from acute HBV infection varies, depending on the patient’s age at the time of HBV acquisition and the patient’s immune status. Fewer than 5% of immunocompetent adults infected with HBV remain chronically infected, defined as being positive for HBV surface antigen for more than 6 months. On the other hand, 80% to 90% of infected infants and about 20% to 50% of children 1 to 5 years old at the time of acute infection remain chronically infected.21
Four phases of chronic HBV infection
The immune tolerance phase, the initial phase of chronic HBV infection, is seen almost exclusively in those who acquired HBV infection vertically or during early childhood. Although patients have high HBV DNA levels, they do not have significant liver disease. This discrepancy is thought to be related to the immune tolerance to HBV; however, the exact mechanism of that tolerance is unclear.31
Only 15% of those with immune tolerance have spontaneous HBV e antigen seroconversion (ie, loss of e antigen and appearance of anti-e antibody) within 20 years after infection. 32
The immune clearance phase (HBV e antigen-positive chronic hepatitis) appears about 20 to 30 years after the onset of the immune tolerance phase in patients who acquire HBV early in life. It is also often seen in patients with infections acquired late in childhood or in adulthood.
This phase marks the start of an immunemediated process aimed at clearing the viral infection, but it also leads to concomitant hepatocellular injury. Spontaneous clearance of the e antigen increases in this phase to an annual rate of 10% to 20%.32,33 The strongest predictors of spontaneous e antigen seroconversion are old age, an elevated ALT level, and an acute exacerbation.26
Although ALT levels are elevated and there is evidence on liver biopsy of chronic active hepatitis, this phase is usually asymptomatic. Rarely, however, it presents with an acute flare of hepatitis, sometimes accompanied by IgM antibodies against the HBV core antigen (in low titer), leading to an incorrect diagnosis of acute HBV infection.
Depending on the duration of the chronic hepatitis and the frequency and severity of flares, about 12% to 20% of patients in the immune-clearance phase develop serious liver disease within 5 years.31
The inactive carrier phase following HBV e antigen seroconversion is characterized by undetectable or low HBV DNA levels (< 1,000 copies/mL), normal ALT levels, and minimal or no necroinflammation on liver biopsy. 30 Such patients should be followed with serial testing, as 4% to 20% of them spontaneously revert to being positive for e antigen at least once.16 On the other hand, only 0.5% to 2% of surface antigen carriers in western countries clear themselves of surface antigen yearly, but up to half of those who clear the surface antigen have low-level HBV viremia. 34
The reactivation phase (HBV e antigennegative chronic hepatitis) is seen in some HBV-infected patients, especially those from Asia and southern Europe, in whom the virus has a spontaneous pre-core or core mutation that makes infected cells unable to secrete the e antigen. Although these patients have no e antigen in their blood, they do have intermittent or persistent elevation of ALT, elevated HBV DNA, and histopathologic findings of chronic hepatitis. Compared with those in the immune clearance phase, patients in the reactivation phase tend to be older and to have lower HBV DNA levels but advanced hepatic damage.
Immunity to HBV infection is characterized by loss of HBV surface antigen, DNA, e antigen, and anti-core antigen IgM with development of anti-surface antigen antibody and anti-core antigen IgG (total anti-core antigen antibody). The presence of anti-surface antigen antibody and anti-core antigen IgG together differentiates natural immunity through resolved infection from that which is acquired through vaccination, which is denoted by isolated anti-surface antigen antibody.
Cirrhosis, liver failure, cancer
Cirrhosis, hepatic decompensation, and hepatocellular carcinoma are the major long-term complications of HBV infection. In untreated patients, the annual rate of progression to cirrhosis has been estimated to be 2% to 6% in patients with HBV e antigen-positive chronic hepatitis and 8% to 9% in those with e antigen- negative chronic hepatitis.30
The likely explanation for these surprising cirrhosis rates is that e antigen-negative chronic hepatitis usually represents a late stage of the disease, and patients in this phase are usually older and have more advanced liver disease.
Subsequently, the annual rate of progression from compensated cirrhosis to hepatic decompensation has been estimated to be about 5%.35
Across all the stages described above, a high serum HBV DNA level has been shown to be a strong predictor of progression to cirrhosis in patients with chronic HBV infection. In a population-based prospective cohort study of 3,582 untreated HBV-infected patients in Taiwan, Iloeje et al36 found that, compared with patients with serum HBV DNA levels lower than 104 copies/mL, those with levels of 104 or higher had an adjusted relative risk of cirrhosis of 2.5. The relative risk rose to 5.9 with HBV DNA levels of 105 or higher, and 9.8 with levels of 106 copies/mL or higher. More studies in different patient populations are needed for confirmation.
The most important risk factor for hepatocellular carcinoma in HBV-infected patients is cirrhosis, but this cancer can also develop in noncirrhotic livers.37 The annual rate of hepatocelluar carcinoma has been estimated to be higher (2.5%–3%) in patients with cirrhosis than in noncirrhotic carriers (0.5%–1%).30,35–38 Risk factors for cirrhosis and hepatocellular carcinoma are summarized in Table 4.16,30
- World Health Organization. Hepatitis B. www.who.int/csr/disease/hepatitis/whocdscsrlyo20022/en. Accessed 11/10/2008.
- Center for Disease Control and Prevention. HBV a silent killer. www.cdc.gov/ncidod/diseases/hepatitis/b/hbv_silent_killer. Accessed 2/19/2007.
- McQuillan GM, Coleman PJ, Kruszon-Moran D, Moyer LA, Lambert SB, Margolis HS. Prevalence of hepatitis B virus infection in the United States: the National Health and Nutrition Examination Surveys, 1976 through 1994. Am J Public Health 1999; 89:14–18.
- Armstrong GL, Mast EE, Wojczynski M, Margolis HS. Childhood hepatitis B virus infections in the United States before hepatitis B immunization. Pediatrics 2001; 108:1123–1128.
- Mast EE, Weinbaum CM, Fiore AE, et al. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP). Part II: immunization of adults. MMWR Recomm Rep 2006 Dec 8; 55(RR-16):1–33.
- Gish RG, Gadano AC. Chronic hepatitis B: current epidemiology in the Americas and implications for management. J Viral Hepatol 2006; 13:787–798.
- Wei Y, Tiollais PK. Molecular biology of hepatitis B virus. Clin Liver Dis 1999; 3:189–219.
- Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev 2000; 64:51–68.
- Hunt CM, McGill JM, Allen MI, Condreay LD. Clinical relevance of hepatitis B viral mutations. Hepatology 2000; 3:1037–1044.
- Allen MI, Deslauriers M, Andrews CW, et al. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Lamivudine Clinical Investigation Group. Hepatology 1998; 27:1670–1677.
- Hadziyannis SJ, Vassilopoulos D. Hepatitis B e antigen- negative chronic hepatitis B. Hepatology 2001; 34:617–624.
- Wai CT, Fontana RJ. Clinical significance of hepatitis B virus genotypes, variants, and mutants. Clin Liver Dis 2004; 8:321–352.
- Fung SK, Lok AS. Hepatitis B virus genotypes: do they play a role in the outcome of HBV infection? Hepatology 2004; 40:790–792.
- Norder H, Courouce AM, Coursaget P, et al. Genetic diversity of hepatitis B virus strains derived worldwide: genotypes, subgenotypes, and HBsAg subtypes. Intervirology 2004; 47:289–309.
- Chu CJ, Keeffe EB, Han SH, et al. Hepatitis B virus genotypes in the United States: results of a nationwide study. Gastroenterology 2003; 125:444–451.
- Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Mast EE, Alter MJ. Prevention of hepatitis B virus infection among health-care workers. In:Ellis RE, editor. Hepatitis B Vaccines in Clinical Practice. New York: Marcel Dekker, 1993:295–307.
- Werner BG, Grady GF. Accidental hepatitis-B-surface-antigen-positive inoculations: use of e antigen to estimate infectivity. Ann Intern Med 1982; 97:367–369.
- Gerberding JL. Management of occupational exposures to blood-borne viruses. N Engl J Med 1995; 332:444–451.
- Kidd-Ljunggren K, Holmberg A, Blackberg J, Lindqvist B. High levels of hepatitis B virus DNA in body fluids from chronic carriers. J Hosp Infect 2006; 64:352–357.
- McMahon BJ, Alward WL, Hall DB, et al. Acute hepatitis B virus infection: relation of age to the clinical expression of disease and subsequent development of the carrier state. J Infect Dis 1985; 151:599–603.
- Dusheiko GM, Brink BA, Conradie JD, Marimuthu T, Sher R. Regional prevalence of hepatitis B, delta, and human immunodeficiency virus infection in southern Africa: a large population survey. Am J Epidemiol 1989; 129:138–45.
- Bortolotti F, Guido M, Bartolacci S, et al. Chronic hepatitis B in children after e antigen seroclearance: final report of a 29-year longitudinal study. Hepatology 2006; 43:556–562.
- Moreno MR, Otero M, Millan A, et al. Clinical and histological outcome after hepatitis B e antigen to antibody seroconversion in children with chronic hepatitis B. Hepatology 1999:572–575.
- Hui AY, Hung LC, Tse PC, Leung WK, Chan PK, Chan HL. Transmission of hepatitis B by human bite--confirmation by detection of virus in saliva and full genome sequencing. J Clin Virol 2005; 33:254–256.
- Cacoub P, Saadoun D, Bourlière M, et al. Hepatitis B virus genotypes and extrahepatic manifestations. J Hepatol 2005; 43:764–770.
- Schiodt FV, Atillasoy E, Shakil AO, et al. Etiology and outcome for 295 patients with acute liver failure in the United States. Liver Transplant Surg 1999; 5:29–34.
- Chu CM, Liaw YF, Pao CC, Huang MJ. The etiology of acute hepatitis superimposed upon previously unrecognized asymptomatic HBsAg carriers. Hepatology 1989; 9:452–456.
- Gerber MA, Hadziyannis S, Vissoulis C, et al. Electron microscopy and immunoelectronmicroscopy of cytoplasmic hepatitis B antigen in hepatocytes. Am J Pathol 1974; 75:489–502.
- Yim HJ, Lok AS. Natural history of chronic hepatitis B virus infection: what we knew in 1981 and what we know in 2005. Hepatology 2006; 43:S173–S181.
- Pungpapong S, Kim WR, Poterucha JJ. Natural history of hepatitis B virus infection: an update for clinicians. Mayo Clin Proc 2007; 82:967–975.
- Lok AS, Lai CL, Wu PC, Leung EK, Lam TS. Spontaneous hepatitis B e antigen to antibody seroconversion and reversion in Chinese patients with chronic hepatitis B virus infection. Gastroenterology 1987; 92:1839–1843.
- McMahon BJ, Holck P, Bulkow L, Snowball M. Serologic and clinical outcomes in 1536 Alaska Natives chronically infected with hepatitis B virus. Ann Intern Med 2001; 135:759–768.
- McMahon BJ. Epidemiology and natural history of hepatitis B. Semin Liver Dis 2005; 25( suppl 1):3–8.
- Benvegnu L, Gios M, Boccato S, Alberti A. Natural history of compensated viral cirrhosis: a prospective study on the incidence and hierarchy of major complications. Gut 2004; 53:744–749.
- Iloeje UH, Yang HI, Su J, Jen CL, You SL, Chen CJ. Risk evaluation of viral load elevation and associated liver disease/cancer in HBV. The REVEAL-HBV Study Group. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology 2006; 130:678–686.
- Bosch FX, Ribes J, Cleries R, Diaz M. Epidemiology of hepatocellular carcinoma. Clin Liver Dis 2005; 9:191–211.
- Fattovich G. Natural history of hepatitis B. J Hepatol 2003; 39:S50–S58.
- World Health Organization. Hepatitis B. www.who.int/csr/disease/hepatitis/whocdscsrlyo20022/en. Accessed 11/10/2008.
- Center for Disease Control and Prevention. HBV a silent killer. www.cdc.gov/ncidod/diseases/hepatitis/b/hbv_silent_killer. Accessed 2/19/2007.
- McQuillan GM, Coleman PJ, Kruszon-Moran D, Moyer LA, Lambert SB, Margolis HS. Prevalence of hepatitis B virus infection in the United States: the National Health and Nutrition Examination Surveys, 1976 through 1994. Am J Public Health 1999; 89:14–18.
- Armstrong GL, Mast EE, Wojczynski M, Margolis HS. Childhood hepatitis B virus infections in the United States before hepatitis B immunization. Pediatrics 2001; 108:1123–1128.
- Mast EE, Weinbaum CM, Fiore AE, et al. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP). Part II: immunization of adults. MMWR Recomm Rep 2006 Dec 8; 55(RR-16):1–33.
- Gish RG, Gadano AC. Chronic hepatitis B: current epidemiology in the Americas and implications for management. J Viral Hepatol 2006; 13:787–798.
- Wei Y, Tiollais PK. Molecular biology of hepatitis B virus. Clin Liver Dis 1999; 3:189–219.
- Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev 2000; 64:51–68.
- Hunt CM, McGill JM, Allen MI, Condreay LD. Clinical relevance of hepatitis B viral mutations. Hepatology 2000; 3:1037–1044.
- Allen MI, Deslauriers M, Andrews CW, et al. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Lamivudine Clinical Investigation Group. Hepatology 1998; 27:1670–1677.
- Hadziyannis SJ, Vassilopoulos D. Hepatitis B e antigen- negative chronic hepatitis B. Hepatology 2001; 34:617–624.
- Wai CT, Fontana RJ. Clinical significance of hepatitis B virus genotypes, variants, and mutants. Clin Liver Dis 2004; 8:321–352.
- Fung SK, Lok AS. Hepatitis B virus genotypes: do they play a role in the outcome of HBV infection? Hepatology 2004; 40:790–792.
- Norder H, Courouce AM, Coursaget P, et al. Genetic diversity of hepatitis B virus strains derived worldwide: genotypes, subgenotypes, and HBsAg subtypes. Intervirology 2004; 47:289–309.
- Chu CJ, Keeffe EB, Han SH, et al. Hepatitis B virus genotypes in the United States: results of a nationwide study. Gastroenterology 2003; 125:444–451.
- Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Mast EE, Alter MJ. Prevention of hepatitis B virus infection among health-care workers. In:Ellis RE, editor. Hepatitis B Vaccines in Clinical Practice. New York: Marcel Dekker, 1993:295–307.
- Werner BG, Grady GF. Accidental hepatitis-B-surface-antigen-positive inoculations: use of e antigen to estimate infectivity. Ann Intern Med 1982; 97:367–369.
- Gerberding JL. Management of occupational exposures to blood-borne viruses. N Engl J Med 1995; 332:444–451.
- Kidd-Ljunggren K, Holmberg A, Blackberg J, Lindqvist B. High levels of hepatitis B virus DNA in body fluids from chronic carriers. J Hosp Infect 2006; 64:352–357.
- McMahon BJ, Alward WL, Hall DB, et al. Acute hepatitis B virus infection: relation of age to the clinical expression of disease and subsequent development of the carrier state. J Infect Dis 1985; 151:599–603.
- Dusheiko GM, Brink BA, Conradie JD, Marimuthu T, Sher R. Regional prevalence of hepatitis B, delta, and human immunodeficiency virus infection in southern Africa: a large population survey. Am J Epidemiol 1989; 129:138–45.
- Bortolotti F, Guido M, Bartolacci S, et al. Chronic hepatitis B in children after e antigen seroclearance: final report of a 29-year longitudinal study. Hepatology 2006; 43:556–562.
- Moreno MR, Otero M, Millan A, et al. Clinical and histological outcome after hepatitis B e antigen to antibody seroconversion in children with chronic hepatitis B. Hepatology 1999:572–575.
- Hui AY, Hung LC, Tse PC, Leung WK, Chan PK, Chan HL. Transmission of hepatitis B by human bite--confirmation by detection of virus in saliva and full genome sequencing. J Clin Virol 2005; 33:254–256.
- Cacoub P, Saadoun D, Bourlière M, et al. Hepatitis B virus genotypes and extrahepatic manifestations. J Hepatol 2005; 43:764–770.
- Schiodt FV, Atillasoy E, Shakil AO, et al. Etiology and outcome for 295 patients with acute liver failure in the United States. Liver Transplant Surg 1999; 5:29–34.
- Chu CM, Liaw YF, Pao CC, Huang MJ. The etiology of acute hepatitis superimposed upon previously unrecognized asymptomatic HBsAg carriers. Hepatology 1989; 9:452–456.
- Gerber MA, Hadziyannis S, Vissoulis C, et al. Electron microscopy and immunoelectronmicroscopy of cytoplasmic hepatitis B antigen in hepatocytes. Am J Pathol 1974; 75:489–502.
- Yim HJ, Lok AS. Natural history of chronic hepatitis B virus infection: what we knew in 1981 and what we know in 2005. Hepatology 2006; 43:S173–S181.
- Pungpapong S, Kim WR, Poterucha JJ. Natural history of hepatitis B virus infection: an update for clinicians. Mayo Clin Proc 2007; 82:967–975.
- Lok AS, Lai CL, Wu PC, Leung EK, Lam TS. Spontaneous hepatitis B e antigen to antibody seroconversion and reversion in Chinese patients with chronic hepatitis B virus infection. Gastroenterology 1987; 92:1839–1843.
- McMahon BJ, Holck P, Bulkow L, Snowball M. Serologic and clinical outcomes in 1536 Alaska Natives chronically infected with hepatitis B virus. Ann Intern Med 2001; 135:759–768.
- McMahon BJ. Epidemiology and natural history of hepatitis B. Semin Liver Dis 2005; 25( suppl 1):3–8.
- Benvegnu L, Gios M, Boccato S, Alberti A. Natural history of compensated viral cirrhosis: a prospective study on the incidence and hierarchy of major complications. Gut 2004; 53:744–749.
- Iloeje UH, Yang HI, Su J, Jen CL, You SL, Chen CJ. Risk evaluation of viral load elevation and associated liver disease/cancer in HBV. The REVEAL-HBV Study Group. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology 2006; 130:678–686.
- Bosch FX, Ribes J, Cleries R, Diaz M. Epidemiology of hepatocellular carcinoma. Clin Liver Dis 2005; 9:191–211.
- Fattovich G. Natural history of hepatitis B. J Hepatol 2003; 39:S50–S58.
KEY POINTS
- HBV infection is much more likely to persist and become chronic if it is acquired at birth or in early childhood rather than during adulthood.
- Chronic HBV infection is defined as persistence of HBV surface antigen in the serum for more than 6 months.
- Although many cases of chronic HBV infection resolve spontaneously, some progress to cirrhosis, hepatocellular carcinoma, and death.