User login
Pediatric cholestatic liver disease: Successful transition of care
Thanks to advances in medical science and our understanding of inherited and acquired liver disease, many more children with acquired or congenital liver disease survive into adulthood than they did 2 decades ago. Improvements in immunosuppression and surgery have increased the chances of pediatric liver transplant recipients reaching adulthood, with a survival rate of 75% at 15 to 20 years.1

With the growing number of adult patients with pediatric-onset liver disease, internists and adult hepatologists need to be aware of these liver diseases and develop expertise to manage this challenging group of patients. Moreover, young adults with pediatric-onset chronic liver disease pose distinct challenges such as pregnancy, adherence to medical regimens, and psychosocial changes in life.
These patients need a “transition of care” rather than a “transfer of care.” Transition of care is a multifaceted process that takes the medical, educational, and psychosocial needs of the patient into consideration before switching their care to adult care physicians, whereas transfer of care is simply an administrative process of change to adult care without previous knowledge of the patients.2

BILIARY ATRESIA
Biliary atresia is a progressive inflammatory fibrosclerosing cholangiopathy of unknown cause. Its prevalence varies with geographic location, ranging from 1 in 6,000 to 1 in 19,000, with the highest prevalence in Taiwan.3
Biliary atresia usually presents within the first few weeks of life, with progressive cholestasis leading to failure to thrive and to fat-soluble vitamin deficiency. Approximately 20% of patients have congenital splenic, gastrointestinal, genitourinary, cardiac, and venous malformations.4,5 Untreated, biliary atresia progresses to end-stage liver disease and death within 2 years.
The first-line treatment for biliary atresia is to establish biliary outflow with the Kasai procedure (hepatic portoenterostomy), in which a jejunal limb is anastomosed in a Roux-en-Y with the liver. The outcomes of the Kasai procedure depend on the timing of surgery, so timely diagnosis of biliary atresia is crucial. When the Kasai procedure is performed within 60 days of birth, biliary flow is achieved in up to 70% of patients; but if performed after 90 days, biliary flow is achieved in fewer than 25%.6
Long-term outcomes of biliary atresia in patients with their native liver have been reported in a few studies.
In a French study,7 743 patients with biliary atresia underwent the Kasai procedure at a median age of 60 days. Survival rates were 57.1% at 2 years, 37.9% at 5 years, 32.4% at 10 years, and 28.5% at 15 years. In other studies,4–9 the 20-year transplant-free survival rate ranged from 23% to 46%. Therefore, at least one-third of children with biliary atresia survive to adulthood with their native liver.
Implications of biliary atresia in adulthood
Although the Kasai procedure improves biliary outflow, up to 70% of patients develop complications of biliary atresia such as progressive fibrosis, cirrhosis, portal hypertension, cholangitis, and hepatocellular carcinoma, even after a successful Kasai procedure.10
Portal hypertension with evidence of splenomegaly, thrombocytopenia, or ascites is found in two-thirds of long-term survivors of biliary atresia with a native liver, with variceal hemorrhage occurring in 30%.11 Therefore, patients with biliary atresia who have evidence of portal hypertension should be screened for varices with upper endoscopy on an annual basis. Management of variceal hemorrhage in these patients includes the use of octreotide, antibiotics, variceal ligation, and sclerotherapy; primary prophylaxis can be achieved with beta-blockers and endoscopic variceal ligation.12
Cholangitis is frequent, occurring in 40% to 60% of biliary atresia patients after the Kasai procedure, and about one-fourth of these patients have multiple episodes.13 The number of episodes of cholangitis negatively affects transplant-free survival.14 Patients with cholangitis should be adequately treated with oral or intravenous antibiotics depending on the severity of presentation. The role of prophylaxis with antibiotics remains unclear.15
Pulmonary complications such as hepatopulmonary syndrome and portopulmonary hypertension can also occur in biliary atresia patients with a native liver. It is important for physicians to be aware of these complications and to screen for them, for example, with agitated saline echocardiography for hepatopulmonary syndrome and with echocardiography for portopulmonary hypertension. Timely screening is crucial, as the outcome of liver transplant depends on the severity at the time of transplant in these conditions, especially portopulmonary hypertension.
Hepatocellular carcinoma has been rarely reported in children with biliary atresia,16 so well-defined guidelines for screening in young adults with biliary atresia are lacking. Most centers recommend screening with ultrasonography of the abdomen and alpha-fetoprotein measurement every 6 months or annually starting soon after the Kasai procedure, since hepatocellular carcinoma has been reported in children as young as age 2.16
Transplant. Adult hepatologists are faced with the challenging task of deciding when it is time for transplant, balancing the long-term complications of biliary atresia with the risk of long-term immunosuppression after transplant. In addition, young adults with these complications may have preserved synthetic function, resulting in low Model for End-Stage Liver Disease (MELD) scores, which may complicate the process of listing for transplant.
Neurocognitive deficits are reported in children with biliary atresia,17 but young adults with biliary atresia generally have reasonable cognitive function and prospects for education and employment.
Pregnancy with successful outcomes has been reported.8
ALAGILLE SYNDROME
Alagille syndrome is an autosomal-dominant multisystemic disease caused by mutations in the JAG1 gene (accounting for > 95% of cases) and the NOTCH2 gene, with highly variable expression.18
Extrahepatic manifestations include butterfly vertebral defects, facial dysmorphism (eg, deep-set and low-set eyes, with characteristic “triangular” facies), posterior embryotoxon (a congenital defect of the eye characterized by an opaque ring around the margin of the cornea), peripheral pulmonary stenosis, renal abnormalities, and vascular malformations.
Hepatic manifestations vary from asymptomatic laboratory abnormalities to progressive cholestasis starting in early infancy with intractable pruritus, xanthomas, failure to thrive, and end-stage liver disease requiring liver transplant in childhood in 15% to 20% of patients.19
Implications of Alagille syndrome in adulthood
Transplant. Interestingly, the phenotype of hepatic disease is already established in childhood, with minimal or no progression in adulthood. Most children with minimal liver disease experience spontaneous resolution, whereas those with significant cholestasis might ultimately develop progressive liver fibrosis or cirrhosis requiring liver transplant in childhood. Only a small subset of children with minimal cholestasis progress to end-stage liver disease in late childhood or early adulthood.20 Therefore, liver transplant for progressive liver disease from significant cholestasis almost always occurs in childhood, usually between ages 1 and 4.21
In a retrospective study comparing posttransplant outcomes in children with Alagille syndrome and biliary atresia, 1-year patient survival was excellent overall in children with Alagille syndrome, although slightly lower than in children with biliary atresia, most likely owing to extrahepatic morbidities of Alagille syndrome and especially the use of immunosuppression in those with renal disease.21 Similarly, 1- and 5-year patient and graft survival outcomes of liver transplant in adults with Alagille syndrome were also excellent compared with those who received a liver transplant in childhood for Alagille syndrome or in adulthood for biliary atresia.22
Hepatocellular carcinoma has occurred in these patients in the absence of cirrhosis, which makes implementation of prognostic and surveillance strategies almost impossible to design for them. Annual ultrasonography with alpha-fetoprotein testing might be applicable in Alagille syndrome patients. However, deciding which patients should undergo this testing and when it should start will be challenging, given the paucity of data.
Cardiovascular disease. Cardiac phenotype is also mostly established in childhood, with the pulmonary vasculature being most commonly involved.19 In contrast, renal and other vascular abnormalities can manifest in adulthood. Renal manifestations vary and include structural anomalies such as hyperechoic kidneys or renal cysts, which can manifest in childhood, and some abnormalities such as hypertension and renal artery stenosis that can manifest in adulthood.23,24
Vasculopathy is reported to involve the intracranial, renal, and intra-abdominal blood vessels.25 Neurovascular accidents such as stroke and intracranial hemorrhage can occur at any age, with significant rates of morbidity and death.26 Therefore, some experts recommend magnetic resonance angiography every 5 years and before any major intervention to prevent these devastating complications.20
Pregnancy. Successful pregnancies have been reported. Preexisting cardiac and hepatic disease can complicate pregnancy depending on the severity of the disease. Because of the autosomal-dominant pattern of inheritance, infants have a 50% risk of the disease, so genetic counseling should be seriously considered before conception.27 Prenatal diagnosis is possible, but the lack of genotype-phenotype correlation precludes its use in clinical practice.
PROGRESSIVE FAMILIAL INTRAHEPATIC CHOLESTASIS
Progressive familial intrahepatic cholestasis (PFIC) is a heterogeneous group of autosomal-recessive conditions associated with disruption of bile formation causing cholestatic liver disease in infants and young children. Three types have been described, depending on the genetic mutation in the hepatobiliary transport pathway:
- PFIC 1 (Byler disease) is caused by impaired bile salt secretion due to mutations in the ATP8B1 gene encoding for the familial intrahepatic cholestasis 1 (FIC 1) protein
- PFIC 2 is caused by impaired bile salt secretion due to mutations in the ABCB11 gene encoding for the bile salt export pump (BSEP) protein
- PFIC 3 is caused by impaired biliary phospholipid secretion due to a defect in ABCB4 encoding for multidrug resistance 3 (MDR3) protein.28
PFIC 1 and 2 manifest with low gamma-glutamyl transferase (GGT) cholestasis, whereas PFIC 3 presents with high GGT cholestasis.
PFIC 1 and PFIC 2 usually cause cholestasis in early infancy, but PFIC 3 can cause cholestasis in late infancy, childhood, and even adulthood.
Because ATP8B1 is expressed in other tissues, PFIC 1 is characterized by extrahepatic manifestations such as sensorineural hearing loss, growth failure, severe diarrhea, and pancreatic insufficiency.
Implications of PFIC in adulthood
PFIC 1 and 2 (low-GGT cholestasis) are usually progressive and often lead to end-stage liver disease and cirrhosis before adulthood. Therefore, almost all patients with PFIC 1 and 2 undergo liver transplant or at least a biliary diversion procedure before reaching adulthood. Intractable pruritus is one of the most challenging clinical manifestations in patients with PFIC.
First-line management is pharmacologic and includes ursodeoxycholic acid, antihistamines (eg, hydroxyzine), bile acid sequestrants (eg, cholestyramine, colestipol), naltrexone, and rifampin, but these have limited efficacy.10
Most patients, especially those with PFIC 1 and 2, undergo a biliary diversion procedure such as partial external biliary diversion (cholecystojejunocutaneostomy), ileal exclusion, or partial internal biliary diversion (cholecystojejunocolic anastomosis) to decrease enterohepatic circulation of bile salts. The efficacy of these procedures is very limited in patients with established cirrhosis. Excessive losses of bile can occur through the biliary stoma, leading to dehydration in patients with external biliary diversion. In patients who are not candidates for biliary diversion, endoscopic nasobiliary drainage of pancreatobiliary secretions could be achieved by placing a catheter in the common bile duct; this has been reported to be effective in relieving cholestasis in a few cases.29
Liver transplant is needed in patients with progressive liver disease and intractable pruritus despite medical management and biliary diversion. Unlike in biliary atresia, liver transplant is not curative in PFIC 1, due to extrahepatic manifestations: patients with PFIC 1 can still have intractable diarrhea and pancreatitis after liver transplant. More importantly, allograft steatohepatitis with further progression to cirrhosis can occur after liver transplant in patients with PFIC 1. Interestingly, biliary diversion has been reported to improve graft steatosis and diarrhea after liver transplant.30
Although graft survival after transplant is good in patients with PFIC 2, recurrence of low-GGT cholestasis has been reported and is believed to be due to the formation of anti-bile salt export pump (anti-BSEP) antibodies by the host immune system in response to exposure to new proteins from the transplant graft.31
Cancer. The risk of malignancy, especially hepatocellular carcinoma, is also increased in PFIC 2, affecting nearly 15% of patients. Therefore, standard hepatocellular carcinoma surveillance with ultrasonography or alpha-fetoprotein testing or both is recommended in patients with PFIC 2. Cholangiocarcinoma and pancreatic adenocarcinoma have also been reported in patients with PFIC 2.20
Incomplete penetrance of mutations in ATP8B1 and ABCB11 can cause recurrent episodes of cholestasis and pruritus with asymptomatic periods between episodes, referred to as benign recurrent intrahepatic cholestasis. Prognosis is usually good, with no progression to cirrhosis.32
Pregnancy. In contrast to FIC 1 and BSEP deficiency, MDR3 defects lead to a wide phenotypic spectrum depending on the type of mutation. Heterozygous mutation is associated with increased risk of development of cholestasis during pregnancy, which typically presents with generalized pruritus in the third trimester and is associated with adverse fetal outcomes. Intrahepatic cholestasis of pregnancy is usually treated with ursodeoxycholic acid, with reported improvement in pruritus, liver function, and pregnancy outcomes.33
In adults, drug-induced liver injury and idiopathic cirrhosis have also been described with MDR3 defects. Intrahepatic lithiasis and cholesterol gallstones can also occur with MDR3 defects as a result of impaired secretion of biliary phospholipid.32 Despite intrahepatic cholestasis of pregnancy, successful outcomes have been reported in women with PFIC.20
OTHER CHILDHOOD-ONSET INHERITED CHOLESTATIC DISEASES
Cystic fibrosis-associated liver disease
Nearly 40% of patients with cystic fibrosis develop liver disease.34 Cystic fibrosis-associated liver disease encompasses a broad clinical spectrum including asymptomatic elevation of aminotransferases, neonatal cholestasis, hepatic steatosis, focal biliary cirrhosis, and multilobar cirrhosis. Cirrhosis and portal hypertension can occur in 5% to 10% of patients and is the third-leading cause of death in patients with cystic fibrosis.35
Risk factors for cystic fibrosis-associated liver disease include male sex, meconium ileus, and severe CFTR gene mutation (class I–III) with pancreatic insufficiency. Cystic fibrosis-related cirrhosis is more frequent in children and adolescents, whereas noncirrhotic portal hypertension and intrahepatic cholangiopathies are more common in adults.36
Limited available studies support treatment with ursodeoxycholic acid in patients with cholestasis to delay the progression of liver disease, but the impact of this drug on long-term outcome is unknown.29
Most patients remain in compensated cirrhosis for many years before progressing to decompensated cirrhosis requiring liver transplant. Other indications for liver transplant include recurrent intractable variceal bleeding, hepatopulmonary syndrome, and portopulmonary hypertension. Combined liver and lung transplant may be considered in patients with advanced liver and lung disease. Outcomes after isolated liver or liver-lung transplant in cystic fibrosis patients have been comparable to those in patients with other liver diseases.37
Defects in bile acid synthesis
Inherited defects of enzymes required for the synthesis of primary bile acids from cholesterol can cause cholestasis from impaired bile flow and production of hepatotoxic aberrant bile acids. The clinical presentation varies depending on the enzymatic defect and can range from liver disease of varying severity to neurologic manifestations. Idiopathic late-onset cholestasis and cirrhosis of unknown etiology have been reported in adults with bile acid synthesis defects.38,39 Therefore, this diagnosis should be considered in cases of cryptogenic cirrhosis and other cholestatic features.
Treatment with primary bile acids (cholic acid) has been effective in most patients with defective bile acid synthesis.
Primary sclerosing cholangitis
Primary sclerosing cholangitis is characterized by progressive obliteration of intrahepatic and extrahepatic bile ducts and is most commonly seen in patients with inflammatory bowel disease. Sclerosing cholangitis can also be secondary to other diseases in children such as immunodeficiency syndromes, Langerhans cell histiocytosis, cystic fibrosis, or sickle cell anemia.40 Neonatal sclerosing cholangitis is a rare autosomal-recessive disease characterized by a severe form of cholangiopathy in neonates and young infants requiring transplant. It can be associated with Kabuki syndrome and neonatal ichthyosis-sclerosing cholangitis syndrome.
Treatment options are limited. Ursodeoxycholic acid and oral vancomycin have variable efficacy. Liver transplant is needed in patients with decompensated cirrhosis. Patients with primary sclerosing cholangitis, especially adults, are at higher risk of developing cholangiocarcinoma, and therefore screening with ultrasonography or magnetic resonance imaging every 6 to 12 months is recommended.
The risk of preterm and cesarean deliveries may be elevated in women with primary sclerosing cholangitis, though data are limited.33
PEDIATRIC LIVER TRANSPLANT RECIPIENTS WHO SURVIVE INTO ADULTHOOD
Adolescent rebellion poses risks
Outcomes of liver transplant in children and adolescents have improved tremendously in the past 2 decades with advances in surgical techniques, pre- and postoperative management, organ preservation, and immunosuppression. Now, most pediatric liver transplant recipients survive into adulthood, creating a unique challenge for internists and adult care hepatologists.41
In rebellious adolescents and young adults, risk-taking behavior, nonadherence to immunosuppressive medications, alcohol intake, and substance abuse increase the risk of graft rejection and loss. Current immunosuppressive drugs such as calcineurin inhibitors (tacrolimus, cyclosporine), mycophenolate mofetil, sirolimus, and corticosteroids have drastically decreased rejection rates in compliant patients.41 Educating patients on the importance of taking their medications and avoiding alcohol and drug abuse is especially important for adolescents and young adults, as rates of nonadherence are high in these age groups.
Although pregnancy is usually successful after liver transplant, it should be considered high-risk due to reported complications such as graft rejection, diabetes, preeclampsia, sepsis, prematurity, and low birth weight. Conception should be avoided for at least 1 year after transplant.42 Appropriate counseling with regard to pregnancy and contraception is important.
There is no consensus on breastfeeding, but it is considered safe in women on low-dose calcineurin inhibitors.43
Life is better with a new liver, but patients have special needs
Liver transplant is life-saving and improves quality of life. However, long-term pediatric liver transplant recipients face challenges such as strict adherence to medications and follow-up visits, avoiding exposure to infections, and fear of graft rejection.
Chronic liver disease in children leads to failure to thrive, growth failure, and even delayed puberty, which resolve in most patients after liver transplant before adulthood in the absence of other comorbidities.44 However, these patients are reported to have lower psychosocial functioning and more psychiatric disorders such as anxiety or posttraumatic disorder.41,44
Therefore, a psychologist or other mental health professional should be part of the management team from the time of pretransplant assessment to identify mental health problems and the need for adjustments before liver transplant. Ongoing psychosocial assessment after liver transplant is equally important to identify risks such as drug or alcohol abuse, depression, posttraumatic stress disorder, and medication nonadherence, all of which can negatively affect posttransplant outcome.45
In addition, assessment of family functioning and structure is important for good long-term outcomes posttransplant; therefore, a social worker should also be a part of the transplant team. Psyschosocial assessment tools can identify high-risk candidates who would benefit from earlier intervention to avoid any negative impact posttransplant.
Neurocognitive development can be delayed in children with chronic liver disease, and the delay may persist even after liver transplant, with reported impairments in intellectual ability, language, verbal, and visuospatial functioning skills.41 In spite of this, a recent study found that more than half the study patients were employed at a median follow-up of 24 years from liver transplant and a median age of 27.46
Remarkably, pediatric liver transplant recipients have reported quality of life comparable to that in the general population,47 and even better than in patients with other chronic illnesses.48
Long-term medical comorbidities in pediatric liver transplant recipients
Favorable outcomes such as long-term survival and good quality of life in pediatric liver transplant recipients are lessened by late complications such as portal vein thrombosis or biliary strictures needing interventions, chronic graft rejection, adverse effects of immunosuppression, and recurrence of the disease.
Split-liver transplant—splitting a deceased-donor allograft to provide grafts for 2 recipients—has revolutionized liver transplant by increasing the donor pool and thereby decreasing waitlist mortality rates, especially in pediatric candidates. Despite this advantage, split-liver transplant is technically challenging and associated with increased perioperative complications compared with whole-liver transplant, especially in adult recipients. Recently, experienced centers have reported favorable outcomes with split-liver transplant comparable to those with whole-liver transplant; therefore, split-liver transplant should be considered after careful evaluation of donor organ and recipient clinical status.49
Old age in the recipient can also adversely affect liver transplant outcomes.50
Interestingly, even in patients whose clinical course is unremarkable and biochemical values are normal, graft hepatitis or fibrosis of unknown cause with progression to cirrhosis has been described in the decade after transplant.41
Chronic rejection with eventual graft loss may be related to nonadherence in adolescents and can be reduced with use of an additional immunosuppressant such as sirolimus or mycophenolate. Chronic kidney disease can occur in about one-third of liver transplant recipients secondary to renal disease associated with primary disease (like Alagille syndrome), hepatorenal syndrome, and most importantly, use of calcineurin inhibitors.45
Components of the metabolic syndrome such as type 2 diabetes, obesity, nonalcoholic fatty liver disease, hypertension, and dyslipidemia are also seen in long-term pediatric liver transplant survivors. Internists are advised to screen for these comorbidities so that interventions can be applied early to improve long-term health outcomes and graft survival.
Of importance, multiple studies have shown a 2-fold increase in the rates of de novo malignancy in liver transplant recipients, including solid-organ and lymphoproliferative cancers, probably due to long-term immunosuppression. Posttransplant lymphoproliferative disorder occurs at lower rates than with other solid-organ transplants; its incidence is greatest in pediatric patients and in the first 12 to 18 months after transplant.51
TRANSITION TO ADULT CARE
While the number of patients with childhood-onset liver disease and pediatric liver transplant recipients who survive into adulthood is increasing, there are no established guidelines or formal models for transitioning these patients into adult care. Consequently, studies on transitional process have examined various issues such as patient and parent frustration, poor medical knowledge among patients during transition, lack of parental facilitation, and inadequate knowledge on disease process among adult-care hepatologists.52–54

A prolonged period of transition up to age 25 is preferred in complicated cases. Distinctive consideration for transition should include those with neurocognitive developmental delay from underlying disease or hepatic encephalopathy before transplant. These patients need additional support and time to achieve independence in health management before transition.57 Validated questionnaires are available to assess readiness to transition into adult care,58 implying that the decision to transition should not be based solely on age.
- Kelly DA, Bucuvalas JC, Alonso EM, et al; American Association for the Study of Liver Diseases; American Society of Transplantation. Long-term medical management of the pediatric patient after liver transplantation: 2013 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Liver Transpl 2013; 19(8):798–825. doi:10.1002/lt.23697
- Rosen DS, Blum RW, Britto M, Sawyer SM, Siegel DM; Society for Adolescent Medicine. Transition to adult health care for adolescents and young adults with chronic conditions: position paper of the Society for Adolescent Medicine. J Adolesc Health 2003; 33(4):309–311. pmid:14519573
- Fawaz R, Baumann U, Ekong U, et al. Guideline for the evaluation of cholestatic jaundice in infants: joint recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr 2017; 64(1):154–168. doi:10.1097/MPG.0000000000001334
- Vajro P, Ferrante L, Lenta S, Mandato C, Persico M. Management of adults with paediatric-onset chronic liver disease: strategic issues for transition care. Dig Liver Dis 2014; 46(4):295–301. doi:10.1016/j.dld.2013.10.018
- Davenport M, Tizzard SA, Underhill J, Mieli-Vergani G, Portmann B, Hadzic N. The biliary atresia splenic malformation syndrome: a 28-year single-center retrospective study. J Pediatr 2006; 149(3):393–400. doi:10.1016/j.jpeds.2006.05.030
- Balistreri WF, Bezerra JA. Whatever happened to “neonatal hepatitis?” Clin Liver Dis 2006; 10(1):27–53. doi:10.1016/j.cld.2005.10.008
- Serinet MO, Wildhaber BE, Broué P, et al. Impact of age at Kasai operation on its results in late childhood and adolescence: a rational basis for biliary atresia screening. Pediatrics 2009; 123(5):1280–1286. doi:10.1542/peds.2008-1949
- de Vries W, Homan-Van der Veen J, Hulscher JB, Hoekstra-Weebers JE, Houwen RH, Verkade HJ; Netherlands Study Group of Biliary Atresia Registry. Twenty-year transplant-free survival rate among patients with biliary atresia. Clin Gastroenterol Hepatol 2011; 9(12):1086–1091. doi:10.1016/j.cgh.2011.07.024
- Lykavieris P, Chardot C, Sokhn M, Gauthier F, Valayer J, Bernard O. Outcome in adulthood of biliary atresia: a study of 63 patients who survived for over 20 years with their native liver. Hepatology 2005; 41(2):366–371. doi:10.1002/hep.20547
- Joshi D, Gupta N, Samyn M, Deheragoda M, Dobbels F, Heneghan MA. The management of childhood liver diseases in adulthood. J Hepatol 2017; 66(3):631–644. doi:10.1016/j.jhep.2016.11.013
- Shneider BL, Abel B, Haber B, et al; Childhood Liver Disease Research and Education Network. Portal hypertension in children and young adults with biliary atresia. J Pediatr Gastroenterol Nutr 2012; 55(5):567–573. doi:10.1097/MPG.0b013e31826eb0cf
- Garcia-Tsao G, Abraldes JG, Berzigotti A, Bosch J. Portal hypertensive bleeding in cirrhosis: risk stratification, diagnosis, and management: 2016 practice guidance by the American Association for the Study of Liver Diseases. Hepatology 2017; 65(1):310–335. doi:10.1002/hep.28906
- Shneider BL, Brown MB, Haber B, et al; Biliary Atresia Research Consortium. A multicenter study of the outcome of biliary atresia in the United States, 1997 to 2000. J Pediatr 2006; 148(4):467–474. doi:10.1016/j.jpeds.2005.12.054
- Hung PY, Chen CC, Chen WJ, et al. Long-term prognosis of patients with biliary atresia: a 25 year summary. J Pediatr Gastroenterol Nutr 2006; 42(2):190–195. doi:10.1097/01.mpg.0000189339.92891.64
- Verkade HJ, Bezerra JA, Davenport M, et al. Biliary atresia and other cholestatic childhood diseases: advances and future challenges. J Hepatol 2016; 65(3):631–642. doi:10.1016/j.jhep.2016.04.032
- Hadžic N, Quaglia A, Portmann B, et al. Hepatocellular carcinoma in biliary atresia: King’s College Hospital experience. J Pediatr 2011; 159(4):617–622.e1. doi:10.1016/j.jpeds.2011.03.004
- Sokol RJ, Shepherd RW, Superina R, Bezerra JA, Robuck P, Hoofnagle JH. Screening and outcomes in biliary atresia: summary of a National Institutes of Health workshop. Hepatology 2007; 46(2):566–581. doi:10.1002/hep.21790
- Li L, Krantz ID, Deng Y, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet 1997; 16(3):243–251. doi:10.1038/ng0797-243
- Saleh M, Kamath BM, Chitayat D. Alagille syndrome: clinical perspectives. Appl Clin Genet 2016; 9:75–82. doi:10.2147/TACG.S86420
- Bass LM, Kamath BM. Inherited disorders of cholestasis in adulthood. Clinical Liver Disease 2013; 2(5):200–203. doi:10.1002/cld.245
- Kamath BM, Yin W, Miller H, Anand R, Rand EB, Alonso E, Bucuvalas J; Studies of Pediatric Liver Transplantation. Outcomes of liver transplantation for patients with Alagille syndrome: the studies of pediatric liver transplantation experience. Liver Transpl 2012; 18(8):940–948. doi:10.1002/lt.23437
- Arnon R, Annunziato R, Schiano T, et al. Orthotopic liver transplantation for adults with Alagille syndrome. Clin Transplant 2012; 26(2):E94–E100. doi:10.1111/j.1399-0012.2011.01574.x
- Salem JE, Bruguiere E, Iserin L, Guiochon-Mantel A, Plouin PF. Hypertension and aortorenal disease in Alagille syndrome. J Hypertens 2012; 30(7):1300–1306. doi:10.1097/HJH.0b013e3283531e1f
- Kamath BM, Podkameni G, Hutchinson AL, et al. Renal anomalies in Alagille syndrome: a disease-defining feature. Am J Med Genet A 2012; 158A(1):85–89. doi:10.1002/ajmg.a.34369
- Kamath BM, Bason L, Piccoli DA, Krantz ID, Spinner NB. Consequences of JAG1 mutations. J Med Genet 2003; 40(12):891–895. pmid:14684686
- Emerick KM, Krantz ID, Kamath BM, et al. Intracranial vascular abnormalities in patients with Alagille syndrome. J Pediatr Gastroenterol Nutr 2005; 41(1):99–107. pmid:15990638
- Ferrarese A, Senzolo M, Burra P. Successful pregnancy in Alagille syndrome. Dig Liver Dis 2015; 47(1):86–87. doi:10.1016/j.dld.2014.08.047
- Davit-Spraul A, Fabre M, Branchereau S, et al. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology 2010; 51(5):1645–1655. doi:10.1002/hep.23539
- Zellos A, Lykopoulou L, Polydorou A, et al. Nasobiliary drainage in an episode of intrahepatic cholestasis in a child with mild ABCB11 disease. J Pediatr Gastroenterol Nutr 2012; 55(1):88–90. doi:10.1097/MPG.0b013e31822f2bda
- Alrabadi LS, Morotti RA, Valentino PL, Rodriguez-Davalos MI, Ekong UD, Emre SH. Biliary drainage as treatment for allograft steatosis following liver transplantation for PFIC-1 disease: a single-center experience. Pediatr Transplant 2018; 22(4):e13184. doi:10.1111/petr.13184
- Kubitz R, Dröge C, Kluge S, et al. Autoimmune BSEP disease: disease recurrence after liver transplantation for progressive familial intrahepatic cholestasis. Clin Rev Allergy Immunol 2015; 48(2–3):273–284. doi:10.1007/s12016-014-8457-4
- Jacquemin E. Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol 2012; 36(suppl 1):S26–S35. doi:10.1016/S2210-7401(12)70018-9
- Pataia V, Dixon PH, Williamson C. Pregnancy and bile acid disorders. Am J Physiol Gastrointest Liver Physiol 2017; 313(1):G1–G6. doi:10.1152/ajpgi.00028.2017
- Lamireau T, Monnereau S, Martin S, Marcotte JE, Winnock M, Alvarez F. Epidemiology of liver disease in cystic fibrosis: a longitudinal study. J Hepatol 2004; 41(6):920–925. doi:10.1016/j.jhep.2004.08.006
- Bolia R, Ooi CY, Lewindon P, et al. Practical approach to the gastrointestinal manifestations of cystic fibrosis. J Paediatr Child Health 2018; 54(6):609–619. doi:10.1111/jpc.13921
- Debray D, Kelly D, Houwen R, Strandvik B, Colombo C. Best practice guidance for the diagnosis and management of cystic fibrosis-associated liver disease. J Cyst Fibros 2011; 10(suppl 2):S29–S36. doi:10.1016/S1569-1993(11)60006-4
- Fridell JA, Bond GJ, Mazariegos G V, et al. Liver transplantation in children with cystic fibrosis: a long-term longitudinal review of a single center’s experience. J Pediatr Surg 2003; 38(8):1152–1156. pmid:12891484
- Fischler B, Bodin K, Stjernman H, et al. Cholestatic liver disease in adults may be due to an inherited defect in bile acid biosynthesis. J Intern Med 2007; 262(2):254–262. doi:10.1111/j.1365-2796.2007.01814.x
- Molho-Pessach V, Rios JJ, Xing C, Setchell KD, Cohen JC, Hobbs HH. Homozygosity mapping identifies a bile acid biosynthetic defect in an adult with cirrhosis of unknown etiology. Hepatology 2012; 55(4):1139–1145. doi:10.1002/hep.24781
- Mieli-Vergani G, Vergani D. Sclerosing cholangitis in children and adolescents. Clin Liver Dis 2016; 20(1):99–111. doi:10.1016/j.cld.2015.08.008
- Kelly D, Wray J. The adolescent liver transplant patient. Clin Liver Dis 2014; 18(3):613–632. doi:10.1016/j.cld.2014.05.006
- Westbrook RH, Yeoman AD, Agarwal K, et al. Outcomes of pregnancy following liver transplantation: the King’s College Hospital experience. Liver Transpl. 2015; 21(9):1153–1159. doi:10.1002/lt.24182
- Hammoud GM, Almashhrawi AA, Ahmed KT, Rahman R, Ibdah JA. Liver diseases in pregnancy: liver transplantation in pregnancy. World J Gastroenterol 2013; 19(43):7647–7651. doi:10.3748/wjg.v19.i43.7647
- Codoner-Franch P, Bernard O, Alvarez F. Long-term follow-up of growth in height after successful liver transplantation. J Pediatr 1994; 124(3):368–373. pmid:8120704
- Shemesh E. Assessment and management of psychosocial challenges in pediatric liver transplantation. Liver Transpl 2008; 14(9):1229–1236. doi:10.1002/lt.21582
- Martinelli J, Habes D, Majed L, et al. Long-term outcome of liver transplantation in childhood: a study of 20-year survivors. Am J Transplant 2018; 18(7):1680–1689. doi:10.1111/ajt.14626
- Roblin E, Audhuy F, Boillot O, Rivet C, Lachaux A. Long-term quality of life after pediatric liver transplantation. Arch Pediatr 2012; 19(10):1039–1052. French. doi:10.1016/j.arcped.2012.06.020
- Duffy JP, Kao K, Ko CY, et al. Long-term patient outcome and quality of life after liver transplantation: analysis of 20-year survivors. Ann Surg 2010; 252(4):652–661. doi:10.1097/SLA.0b013e3181f5f23a
- Hackl C, Schmidt KM, Süsal C, Döhler B, Zidek M, Schlitt HJ. Split liver transplantation: Current developments. World J Gastroenterol 2018; 24(47):5312–5321. doi:10.3748/wjg.v24.i47.5312
- Durand F, Levitsky J, Cauchy F, Gilgenkrantz H, Soubrane O, Francoz C. Age and liver transplantation. J Hepatol 2019; 70(4):745–758. doi:10.1016/j.jhep.2018.12.009
- Chandok N, Watt KD. Burden of de novo malignancy in the liver transplant recipient. Liver Transpl 2012; 18(11):1277–1289. doi:10.1002/lt.23531
- Ferrarese A, Germani G, Lazzaro S, et al. Short-term outcomes of paediatric liver transplant recipients after transition to Adult Healthcare Service. Liver Int 2018; 38(7):1316–1321. doi:10.1111/liv.13655
- Wright J, Elwell L, McDonagh JE, Kelly DA, Wray J. “Are these adult doctors gonna know me?” Experiences of transition for young people with a liver transplant. Pediatr Transplant 2016; 20(7):912–920. doi:10.1111/petr.12777
- Heldman MR, Sohn MW, Gordon EJ, et al. National survey of adult transplant hepatologists on the pediatric-to-adult care transition after liver transplantation. Liver Transpl 2015; 21(2):213–223. doi:10.1002/lt.24044
- Vajro P, Fischler B, Burra P, et al. The health care transition of youth with liver disease into the adult health system. J Pediatr Gastroenterol Nutr 2018; 66(6):976–990. doi:10.1097/MPG.0000000000001965
- Fredericks EM, Lopez MJ. Transition of the adolescent transplant patient to adult care. Clin Liver Dis (Hoboken) 2013; 2(5):223–226. doi:10.1002/cld.243
- Kaufman M. Transition of cognitively delayed adolescent organ transplant recipients to adult care. Pediatr Transplant 2006; 10(4):413–417. doi:10.1111/j.1399-3046.2006.00491.x
- Sawicki GS, Lukens-Bull K, Yin X, et al. Measuring the transition readiness of youth with special healthcare needs: validation of the TRAQ—Transition Readiness Assessment Questionnaire. J Pediatr Psychol 2011; 36(2):160–171. doi:10.1093/jpepsy/jsp128
Thanks to advances in medical science and our understanding of inherited and acquired liver disease, many more children with acquired or congenital liver disease survive into adulthood than they did 2 decades ago. Improvements in immunosuppression and surgery have increased the chances of pediatric liver transplant recipients reaching adulthood, with a survival rate of 75% at 15 to 20 years.1
With the growing number of adult patients with pediatric-onset liver disease, internists and adult hepatologists need to be aware of these liver diseases and develop expertise to manage this challenging group of patients. Moreover, young adults with pediatric-onset chronic liver disease pose distinct challenges such as pregnancy, adherence to medical regimens, and psychosocial changes in life.
These patients need a “transition of care” rather than a “transfer of care.” Transition of care is a multifaceted process that takes the medical, educational, and psychosocial needs of the patient into consideration before switching their care to adult care physicians, whereas transfer of care is simply an administrative process of change to adult care without previous knowledge of the patients.2
BILIARY ATRESIA
Biliary atresia is a progressive inflammatory fibrosclerosing cholangiopathy of unknown cause. Its prevalence varies with geographic location, ranging from 1 in 6,000 to 1 in 19,000, with the highest prevalence in Taiwan.3
Biliary atresia usually presents within the first few weeks of life, with progressive cholestasis leading to failure to thrive and to fat-soluble vitamin deficiency. Approximately 20% of patients have congenital splenic, gastrointestinal, genitourinary, cardiac, and venous malformations.4,5 Untreated, biliary atresia progresses to end-stage liver disease and death within 2 years.
The first-line treatment for biliary atresia is to establish biliary outflow with the Kasai procedure (hepatic portoenterostomy), in which a jejunal limb is anastomosed in a Roux-en-Y with the liver. The outcomes of the Kasai procedure depend on the timing of surgery, so timely diagnosis of biliary atresia is crucial. When the Kasai procedure is performed within 60 days of birth, biliary flow is achieved in up to 70% of patients; but if performed after 90 days, biliary flow is achieved in fewer than 25%.6
Long-term outcomes of biliary atresia in patients with their native liver have been reported in a few studies.
In a French study,7 743 patients with biliary atresia underwent the Kasai procedure at a median age of 60 days. Survival rates were 57.1% at 2 years, 37.9% at 5 years, 32.4% at 10 years, and 28.5% at 15 years. In other studies,4–9 the 20-year transplant-free survival rate ranged from 23% to 46%. Therefore, at least one-third of children with biliary atresia survive to adulthood with their native liver.
Implications of biliary atresia in adulthood
Although the Kasai procedure improves biliary outflow, up to 70% of patients develop complications of biliary atresia such as progressive fibrosis, cirrhosis, portal hypertension, cholangitis, and hepatocellular carcinoma, even after a successful Kasai procedure.10
Portal hypertension with evidence of splenomegaly, thrombocytopenia, or ascites is found in two-thirds of long-term survivors of biliary atresia with a native liver, with variceal hemorrhage occurring in 30%.11 Therefore, patients with biliary atresia who have evidence of portal hypertension should be screened for varices with upper endoscopy on an annual basis. Management of variceal hemorrhage in these patients includes the use of octreotide, antibiotics, variceal ligation, and sclerotherapy; primary prophylaxis can be achieved with beta-blockers and endoscopic variceal ligation.12
Cholangitis is frequent, occurring in 40% to 60% of biliary atresia patients after the Kasai procedure, and about one-fourth of these patients have multiple episodes.13 The number of episodes of cholangitis negatively affects transplant-free survival.14 Patients with cholangitis should be adequately treated with oral or intravenous antibiotics depending on the severity of presentation. The role of prophylaxis with antibiotics remains unclear.15
Pulmonary complications such as hepatopulmonary syndrome and portopulmonary hypertension can also occur in biliary atresia patients with a native liver. It is important for physicians to be aware of these complications and to screen for them, for example, with agitated saline echocardiography for hepatopulmonary syndrome and with echocardiography for portopulmonary hypertension. Timely screening is crucial, as the outcome of liver transplant depends on the severity at the time of transplant in these conditions, especially portopulmonary hypertension.
Hepatocellular carcinoma has been rarely reported in children with biliary atresia,16 so well-defined guidelines for screening in young adults with biliary atresia are lacking. Most centers recommend screening with ultrasonography of the abdomen and alpha-fetoprotein measurement every 6 months or annually starting soon after the Kasai procedure, since hepatocellular carcinoma has been reported in children as young as age 2.16
Transplant. Adult hepatologists are faced with the challenging task of deciding when it is time for transplant, balancing the long-term complications of biliary atresia with the risk of long-term immunosuppression after transplant. In addition, young adults with these complications may have preserved synthetic function, resulting in low Model for End-Stage Liver Disease (MELD) scores, which may complicate the process of listing for transplant.
Neurocognitive deficits are reported in children with biliary atresia,17 but young adults with biliary atresia generally have reasonable cognitive function and prospects for education and employment.
Pregnancy with successful outcomes has been reported.8
ALAGILLE SYNDROME
Alagille syndrome is an autosomal-dominant multisystemic disease caused by mutations in the JAG1 gene (accounting for > 95% of cases) and the NOTCH2 gene, with highly variable expression.18
Extrahepatic manifestations include butterfly vertebral defects, facial dysmorphism (eg, deep-set and low-set eyes, with characteristic “triangular” facies), posterior embryotoxon (a congenital defect of the eye characterized by an opaque ring around the margin of the cornea), peripheral pulmonary stenosis, renal abnormalities, and vascular malformations.
Hepatic manifestations vary from asymptomatic laboratory abnormalities to progressive cholestasis starting in early infancy with intractable pruritus, xanthomas, failure to thrive, and end-stage liver disease requiring liver transplant in childhood in 15% to 20% of patients.19
Implications of Alagille syndrome in adulthood
Transplant. Interestingly, the phenotype of hepatic disease is already established in childhood, with minimal or no progression in adulthood. Most children with minimal liver disease experience spontaneous resolution, whereas those with significant cholestasis might ultimately develop progressive liver fibrosis or cirrhosis requiring liver transplant in childhood. Only a small subset of children with minimal cholestasis progress to end-stage liver disease in late childhood or early adulthood.20 Therefore, liver transplant for progressive liver disease from significant cholestasis almost always occurs in childhood, usually between ages 1 and 4.21
In a retrospective study comparing posttransplant outcomes in children with Alagille syndrome and biliary atresia, 1-year patient survival was excellent overall in children with Alagille syndrome, although slightly lower than in children with biliary atresia, most likely owing to extrahepatic morbidities of Alagille syndrome and especially the use of immunosuppression in those with renal disease.21 Similarly, 1- and 5-year patient and graft survival outcomes of liver transplant in adults with Alagille syndrome were also excellent compared with those who received a liver transplant in childhood for Alagille syndrome or in adulthood for biliary atresia.22
Hepatocellular carcinoma has occurred in these patients in the absence of cirrhosis, which makes implementation of prognostic and surveillance strategies almost impossible to design for them. Annual ultrasonography with alpha-fetoprotein testing might be applicable in Alagille syndrome patients. However, deciding which patients should undergo this testing and when it should start will be challenging, given the paucity of data.
Cardiovascular disease. Cardiac phenotype is also mostly established in childhood, with the pulmonary vasculature being most commonly involved.19 In contrast, renal and other vascular abnormalities can manifest in adulthood. Renal manifestations vary and include structural anomalies such as hyperechoic kidneys or renal cysts, which can manifest in childhood, and some abnormalities such as hypertension and renal artery stenosis that can manifest in adulthood.23,24
Vasculopathy is reported to involve the intracranial, renal, and intra-abdominal blood vessels.25 Neurovascular accidents such as stroke and intracranial hemorrhage can occur at any age, with significant rates of morbidity and death.26 Therefore, some experts recommend magnetic resonance angiography every 5 years and before any major intervention to prevent these devastating complications.20
Pregnancy. Successful pregnancies have been reported. Preexisting cardiac and hepatic disease can complicate pregnancy depending on the severity of the disease. Because of the autosomal-dominant pattern of inheritance, infants have a 50% risk of the disease, so genetic counseling should be seriously considered before conception.27 Prenatal diagnosis is possible, but the lack of genotype-phenotype correlation precludes its use in clinical practice.
PROGRESSIVE FAMILIAL INTRAHEPATIC CHOLESTASIS
Progressive familial intrahepatic cholestasis (PFIC) is a heterogeneous group of autosomal-recessive conditions associated with disruption of bile formation causing cholestatic liver disease in infants and young children. Three types have been described, depending on the genetic mutation in the hepatobiliary transport pathway:
- PFIC 1 (Byler disease) is caused by impaired bile salt secretion due to mutations in the ATP8B1 gene encoding for the familial intrahepatic cholestasis 1 (FIC 1) protein
- PFIC 2 is caused by impaired bile salt secretion due to mutations in the ABCB11 gene encoding for the bile salt export pump (BSEP) protein
- PFIC 3 is caused by impaired biliary phospholipid secretion due to a defect in ABCB4 encoding for multidrug resistance 3 (MDR3) protein.28
PFIC 1 and 2 manifest with low gamma-glutamyl transferase (GGT) cholestasis, whereas PFIC 3 presents with high GGT cholestasis.
PFIC 1 and PFIC 2 usually cause cholestasis in early infancy, but PFIC 3 can cause cholestasis in late infancy, childhood, and even adulthood.
Because ATP8B1 is expressed in other tissues, PFIC 1 is characterized by extrahepatic manifestations such as sensorineural hearing loss, growth failure, severe diarrhea, and pancreatic insufficiency.
Implications of PFIC in adulthood
PFIC 1 and 2 (low-GGT cholestasis) are usually progressive and often lead to end-stage liver disease and cirrhosis before adulthood. Therefore, almost all patients with PFIC 1 and 2 undergo liver transplant or at least a biliary diversion procedure before reaching adulthood. Intractable pruritus is one of the most challenging clinical manifestations in patients with PFIC.
First-line management is pharmacologic and includes ursodeoxycholic acid, antihistamines (eg, hydroxyzine), bile acid sequestrants (eg, cholestyramine, colestipol), naltrexone, and rifampin, but these have limited efficacy.10
Most patients, especially those with PFIC 1 and 2, undergo a biliary diversion procedure such as partial external biliary diversion (cholecystojejunocutaneostomy), ileal exclusion, or partial internal biliary diversion (cholecystojejunocolic anastomosis) to decrease enterohepatic circulation of bile salts. The efficacy of these procedures is very limited in patients with established cirrhosis. Excessive losses of bile can occur through the biliary stoma, leading to dehydration in patients with external biliary diversion. In patients who are not candidates for biliary diversion, endoscopic nasobiliary drainage of pancreatobiliary secretions could be achieved by placing a catheter in the common bile duct; this has been reported to be effective in relieving cholestasis in a few cases.29
Liver transplant is needed in patients with progressive liver disease and intractable pruritus despite medical management and biliary diversion. Unlike in biliary atresia, liver transplant is not curative in PFIC 1, due to extrahepatic manifestations: patients with PFIC 1 can still have intractable diarrhea and pancreatitis after liver transplant. More importantly, allograft steatohepatitis with further progression to cirrhosis can occur after liver transplant in patients with PFIC 1. Interestingly, biliary diversion has been reported to improve graft steatosis and diarrhea after liver transplant.30
Although graft survival after transplant is good in patients with PFIC 2, recurrence of low-GGT cholestasis has been reported and is believed to be due to the formation of anti-bile salt export pump (anti-BSEP) antibodies by the host immune system in response to exposure to new proteins from the transplant graft.31
Cancer. The risk of malignancy, especially hepatocellular carcinoma, is also increased in PFIC 2, affecting nearly 15% of patients. Therefore, standard hepatocellular carcinoma surveillance with ultrasonography or alpha-fetoprotein testing or both is recommended in patients with PFIC 2. Cholangiocarcinoma and pancreatic adenocarcinoma have also been reported in patients with PFIC 2.20
Incomplete penetrance of mutations in ATP8B1 and ABCB11 can cause recurrent episodes of cholestasis and pruritus with asymptomatic periods between episodes, referred to as benign recurrent intrahepatic cholestasis. Prognosis is usually good, with no progression to cirrhosis.32
Pregnancy. In contrast to FIC 1 and BSEP deficiency, MDR3 defects lead to a wide phenotypic spectrum depending on the type of mutation. Heterozygous mutation is associated with increased risk of development of cholestasis during pregnancy, which typically presents with generalized pruritus in the third trimester and is associated with adverse fetal outcomes. Intrahepatic cholestasis of pregnancy is usually treated with ursodeoxycholic acid, with reported improvement in pruritus, liver function, and pregnancy outcomes.33
In adults, drug-induced liver injury and idiopathic cirrhosis have also been described with MDR3 defects. Intrahepatic lithiasis and cholesterol gallstones can also occur with MDR3 defects as a result of impaired secretion of biliary phospholipid.32 Despite intrahepatic cholestasis of pregnancy, successful outcomes have been reported in women with PFIC.20
OTHER CHILDHOOD-ONSET INHERITED CHOLESTATIC DISEASES
Cystic fibrosis-associated liver disease
Nearly 40% of patients with cystic fibrosis develop liver disease.34 Cystic fibrosis-associated liver disease encompasses a broad clinical spectrum including asymptomatic elevation of aminotransferases, neonatal cholestasis, hepatic steatosis, focal biliary cirrhosis, and multilobar cirrhosis. Cirrhosis and portal hypertension can occur in 5% to 10% of patients and is the third-leading cause of death in patients with cystic fibrosis.35
Risk factors for cystic fibrosis-associated liver disease include male sex, meconium ileus, and severe CFTR gene mutation (class I–III) with pancreatic insufficiency. Cystic fibrosis-related cirrhosis is more frequent in children and adolescents, whereas noncirrhotic portal hypertension and intrahepatic cholangiopathies are more common in adults.36
Limited available studies support treatment with ursodeoxycholic acid in patients with cholestasis to delay the progression of liver disease, but the impact of this drug on long-term outcome is unknown.29
Most patients remain in compensated cirrhosis for many years before progressing to decompensated cirrhosis requiring liver transplant. Other indications for liver transplant include recurrent intractable variceal bleeding, hepatopulmonary syndrome, and portopulmonary hypertension. Combined liver and lung transplant may be considered in patients with advanced liver and lung disease. Outcomes after isolated liver or liver-lung transplant in cystic fibrosis patients have been comparable to those in patients with other liver diseases.37
Defects in bile acid synthesis
Inherited defects of enzymes required for the synthesis of primary bile acids from cholesterol can cause cholestasis from impaired bile flow and production of hepatotoxic aberrant bile acids. The clinical presentation varies depending on the enzymatic defect and can range from liver disease of varying severity to neurologic manifestations. Idiopathic late-onset cholestasis and cirrhosis of unknown etiology have been reported in adults with bile acid synthesis defects.38,39 Therefore, this diagnosis should be considered in cases of cryptogenic cirrhosis and other cholestatic features.
Treatment with primary bile acids (cholic acid) has been effective in most patients with defective bile acid synthesis.
Primary sclerosing cholangitis
Primary sclerosing cholangitis is characterized by progressive obliteration of intrahepatic and extrahepatic bile ducts and is most commonly seen in patients with inflammatory bowel disease. Sclerosing cholangitis can also be secondary to other diseases in children such as immunodeficiency syndromes, Langerhans cell histiocytosis, cystic fibrosis, or sickle cell anemia.40 Neonatal sclerosing cholangitis is a rare autosomal-recessive disease characterized by a severe form of cholangiopathy in neonates and young infants requiring transplant. It can be associated with Kabuki syndrome and neonatal ichthyosis-sclerosing cholangitis syndrome.
Treatment options are limited. Ursodeoxycholic acid and oral vancomycin have variable efficacy. Liver transplant is needed in patients with decompensated cirrhosis. Patients with primary sclerosing cholangitis, especially adults, are at higher risk of developing cholangiocarcinoma, and therefore screening with ultrasonography or magnetic resonance imaging every 6 to 12 months is recommended.
The risk of preterm and cesarean deliveries may be elevated in women with primary sclerosing cholangitis, though data are limited.33
PEDIATRIC LIVER TRANSPLANT RECIPIENTS WHO SURVIVE INTO ADULTHOOD
Adolescent rebellion poses risks
Outcomes of liver transplant in children and adolescents have improved tremendously in the past 2 decades with advances in surgical techniques, pre- and postoperative management, organ preservation, and immunosuppression. Now, most pediatric liver transplant recipients survive into adulthood, creating a unique challenge for internists and adult care hepatologists.41
In rebellious adolescents and young adults, risk-taking behavior, nonadherence to immunosuppressive medications, alcohol intake, and substance abuse increase the risk of graft rejection and loss. Current immunosuppressive drugs such as calcineurin inhibitors (tacrolimus, cyclosporine), mycophenolate mofetil, sirolimus, and corticosteroids have drastically decreased rejection rates in compliant patients.41 Educating patients on the importance of taking their medications and avoiding alcohol and drug abuse is especially important for adolescents and young adults, as rates of nonadherence are high in these age groups.
Although pregnancy is usually successful after liver transplant, it should be considered high-risk due to reported complications such as graft rejection, diabetes, preeclampsia, sepsis, prematurity, and low birth weight. Conception should be avoided for at least 1 year after transplant.42 Appropriate counseling with regard to pregnancy and contraception is important.
There is no consensus on breastfeeding, but it is considered safe in women on low-dose calcineurin inhibitors.43
Life is better with a new liver, but patients have special needs
Liver transplant is life-saving and improves quality of life. However, long-term pediatric liver transplant recipients face challenges such as strict adherence to medications and follow-up visits, avoiding exposure to infections, and fear of graft rejection.
Chronic liver disease in children leads to failure to thrive, growth failure, and even delayed puberty, which resolve in most patients after liver transplant before adulthood in the absence of other comorbidities.44 However, these patients are reported to have lower psychosocial functioning and more psychiatric disorders such as anxiety or posttraumatic disorder.41,44
Therefore, a psychologist or other mental health professional should be part of the management team from the time of pretransplant assessment to identify mental health problems and the need for adjustments before liver transplant. Ongoing psychosocial assessment after liver transplant is equally important to identify risks such as drug or alcohol abuse, depression, posttraumatic stress disorder, and medication nonadherence, all of which can negatively affect posttransplant outcome.45
In addition, assessment of family functioning and structure is important for good long-term outcomes posttransplant; therefore, a social worker should also be a part of the transplant team. Psyschosocial assessment tools can identify high-risk candidates who would benefit from earlier intervention to avoid any negative impact posttransplant.
Neurocognitive development can be delayed in children with chronic liver disease, and the delay may persist even after liver transplant, with reported impairments in intellectual ability, language, verbal, and visuospatial functioning skills.41 In spite of this, a recent study found that more than half the study patients were employed at a median follow-up of 24 years from liver transplant and a median age of 27.46
Remarkably, pediatric liver transplant recipients have reported quality of life comparable to that in the general population,47 and even better than in patients with other chronic illnesses.48
Long-term medical comorbidities in pediatric liver transplant recipients
Favorable outcomes such as long-term survival and good quality of life in pediatric liver transplant recipients are lessened by late complications such as portal vein thrombosis or biliary strictures needing interventions, chronic graft rejection, adverse effects of immunosuppression, and recurrence of the disease.
Split-liver transplant—splitting a deceased-donor allograft to provide grafts for 2 recipients—has revolutionized liver transplant by increasing the donor pool and thereby decreasing waitlist mortality rates, especially in pediatric candidates. Despite this advantage, split-liver transplant is technically challenging and associated with increased perioperative complications compared with whole-liver transplant, especially in adult recipients. Recently, experienced centers have reported favorable outcomes with split-liver transplant comparable to those with whole-liver transplant; therefore, split-liver transplant should be considered after careful evaluation of donor organ and recipient clinical status.49
Old age in the recipient can also adversely affect liver transplant outcomes.50
Interestingly, even in patients whose clinical course is unremarkable and biochemical values are normal, graft hepatitis or fibrosis of unknown cause with progression to cirrhosis has been described in the decade after transplant.41
Chronic rejection with eventual graft loss may be related to nonadherence in adolescents and can be reduced with use of an additional immunosuppressant such as sirolimus or mycophenolate. Chronic kidney disease can occur in about one-third of liver transplant recipients secondary to renal disease associated with primary disease (like Alagille syndrome), hepatorenal syndrome, and most importantly, use of calcineurin inhibitors.45
Components of the metabolic syndrome such as type 2 diabetes, obesity, nonalcoholic fatty liver disease, hypertension, and dyslipidemia are also seen in long-term pediatric liver transplant survivors. Internists are advised to screen for these comorbidities so that interventions can be applied early to improve long-term health outcomes and graft survival.
Of importance, multiple studies have shown a 2-fold increase in the rates of de novo malignancy in liver transplant recipients, including solid-organ and lymphoproliferative cancers, probably due to long-term immunosuppression. Posttransplant lymphoproliferative disorder occurs at lower rates than with other solid-organ transplants; its incidence is greatest in pediatric patients and in the first 12 to 18 months after transplant.51
TRANSITION TO ADULT CARE
While the number of patients with childhood-onset liver disease and pediatric liver transplant recipients who survive into adulthood is increasing, there are no established guidelines or formal models for transitioning these patients into adult care. Consequently, studies on transitional process have examined various issues such as patient and parent frustration, poor medical knowledge among patients during transition, lack of parental facilitation, and inadequate knowledge on disease process among adult-care hepatologists.52–54
A prolonged period of transition up to age 25 is preferred in complicated cases. Distinctive consideration for transition should include those with neurocognitive developmental delay from underlying disease or hepatic encephalopathy before transplant. These patients need additional support and time to achieve independence in health management before transition.57 Validated questionnaires are available to assess readiness to transition into adult care,58 implying that the decision to transition should not be based solely on age.
Thanks to advances in medical science and our understanding of inherited and acquired liver disease, many more children with acquired or congenital liver disease survive into adulthood than they did 2 decades ago. Improvements in immunosuppression and surgery have increased the chances of pediatric liver transplant recipients reaching adulthood, with a survival rate of 75% at 15 to 20 years.1
With the growing number of adult patients with pediatric-onset liver disease, internists and adult hepatologists need to be aware of these liver diseases and develop expertise to manage this challenging group of patients. Moreover, young adults with pediatric-onset chronic liver disease pose distinct challenges such as pregnancy, adherence to medical regimens, and psychosocial changes in life.
These patients need a “transition of care” rather than a “transfer of care.” Transition of care is a multifaceted process that takes the medical, educational, and psychosocial needs of the patient into consideration before switching their care to adult care physicians, whereas transfer of care is simply an administrative process of change to adult care without previous knowledge of the patients.2
BILIARY ATRESIA
Biliary atresia is a progressive inflammatory fibrosclerosing cholangiopathy of unknown cause. Its prevalence varies with geographic location, ranging from 1 in 6,000 to 1 in 19,000, with the highest prevalence in Taiwan.3
Biliary atresia usually presents within the first few weeks of life, with progressive cholestasis leading to failure to thrive and to fat-soluble vitamin deficiency. Approximately 20% of patients have congenital splenic, gastrointestinal, genitourinary, cardiac, and venous malformations.4,5 Untreated, biliary atresia progresses to end-stage liver disease and death within 2 years.
The first-line treatment for biliary atresia is to establish biliary outflow with the Kasai procedure (hepatic portoenterostomy), in which a jejunal limb is anastomosed in a Roux-en-Y with the liver. The outcomes of the Kasai procedure depend on the timing of surgery, so timely diagnosis of biliary atresia is crucial. When the Kasai procedure is performed within 60 days of birth, biliary flow is achieved in up to 70% of patients; but if performed after 90 days, biliary flow is achieved in fewer than 25%.6
Long-term outcomes of biliary atresia in patients with their native liver have been reported in a few studies.
In a French study,7 743 patients with biliary atresia underwent the Kasai procedure at a median age of 60 days. Survival rates were 57.1% at 2 years, 37.9% at 5 years, 32.4% at 10 years, and 28.5% at 15 years. In other studies,4–9 the 20-year transplant-free survival rate ranged from 23% to 46%. Therefore, at least one-third of children with biliary atresia survive to adulthood with their native liver.
Implications of biliary atresia in adulthood
Although the Kasai procedure improves biliary outflow, up to 70% of patients develop complications of biliary atresia such as progressive fibrosis, cirrhosis, portal hypertension, cholangitis, and hepatocellular carcinoma, even after a successful Kasai procedure.10
Portal hypertension with evidence of splenomegaly, thrombocytopenia, or ascites is found in two-thirds of long-term survivors of biliary atresia with a native liver, with variceal hemorrhage occurring in 30%.11 Therefore, patients with biliary atresia who have evidence of portal hypertension should be screened for varices with upper endoscopy on an annual basis. Management of variceal hemorrhage in these patients includes the use of octreotide, antibiotics, variceal ligation, and sclerotherapy; primary prophylaxis can be achieved with beta-blockers and endoscopic variceal ligation.12
Cholangitis is frequent, occurring in 40% to 60% of biliary atresia patients after the Kasai procedure, and about one-fourth of these patients have multiple episodes.13 The number of episodes of cholangitis negatively affects transplant-free survival.14 Patients with cholangitis should be adequately treated with oral or intravenous antibiotics depending on the severity of presentation. The role of prophylaxis with antibiotics remains unclear.15
Pulmonary complications such as hepatopulmonary syndrome and portopulmonary hypertension can also occur in biliary atresia patients with a native liver. It is important for physicians to be aware of these complications and to screen for them, for example, with agitated saline echocardiography for hepatopulmonary syndrome and with echocardiography for portopulmonary hypertension. Timely screening is crucial, as the outcome of liver transplant depends on the severity at the time of transplant in these conditions, especially portopulmonary hypertension.
Hepatocellular carcinoma has been rarely reported in children with biliary atresia,16 so well-defined guidelines for screening in young adults with biliary atresia are lacking. Most centers recommend screening with ultrasonography of the abdomen and alpha-fetoprotein measurement every 6 months or annually starting soon after the Kasai procedure, since hepatocellular carcinoma has been reported in children as young as age 2.16
Transplant. Adult hepatologists are faced with the challenging task of deciding when it is time for transplant, balancing the long-term complications of biliary atresia with the risk of long-term immunosuppression after transplant. In addition, young adults with these complications may have preserved synthetic function, resulting in low Model for End-Stage Liver Disease (MELD) scores, which may complicate the process of listing for transplant.
Neurocognitive deficits are reported in children with biliary atresia,17 but young adults with biliary atresia generally have reasonable cognitive function and prospects for education and employment.
Pregnancy with successful outcomes has been reported.8
ALAGILLE SYNDROME
Alagille syndrome is an autosomal-dominant multisystemic disease caused by mutations in the JAG1 gene (accounting for > 95% of cases) and the NOTCH2 gene, with highly variable expression.18
Extrahepatic manifestations include butterfly vertebral defects, facial dysmorphism (eg, deep-set and low-set eyes, with characteristic “triangular” facies), posterior embryotoxon (a congenital defect of the eye characterized by an opaque ring around the margin of the cornea), peripheral pulmonary stenosis, renal abnormalities, and vascular malformations.
Hepatic manifestations vary from asymptomatic laboratory abnormalities to progressive cholestasis starting in early infancy with intractable pruritus, xanthomas, failure to thrive, and end-stage liver disease requiring liver transplant in childhood in 15% to 20% of patients.19
Implications of Alagille syndrome in adulthood
Transplant. Interestingly, the phenotype of hepatic disease is already established in childhood, with minimal or no progression in adulthood. Most children with minimal liver disease experience spontaneous resolution, whereas those with significant cholestasis might ultimately develop progressive liver fibrosis or cirrhosis requiring liver transplant in childhood. Only a small subset of children with minimal cholestasis progress to end-stage liver disease in late childhood or early adulthood.20 Therefore, liver transplant for progressive liver disease from significant cholestasis almost always occurs in childhood, usually between ages 1 and 4.21
In a retrospective study comparing posttransplant outcomes in children with Alagille syndrome and biliary atresia, 1-year patient survival was excellent overall in children with Alagille syndrome, although slightly lower than in children with biliary atresia, most likely owing to extrahepatic morbidities of Alagille syndrome and especially the use of immunosuppression in those with renal disease.21 Similarly, 1- and 5-year patient and graft survival outcomes of liver transplant in adults with Alagille syndrome were also excellent compared with those who received a liver transplant in childhood for Alagille syndrome or in adulthood for biliary atresia.22
Hepatocellular carcinoma has occurred in these patients in the absence of cirrhosis, which makes implementation of prognostic and surveillance strategies almost impossible to design for them. Annual ultrasonography with alpha-fetoprotein testing might be applicable in Alagille syndrome patients. However, deciding which patients should undergo this testing and when it should start will be challenging, given the paucity of data.
Cardiovascular disease. Cardiac phenotype is also mostly established in childhood, with the pulmonary vasculature being most commonly involved.19 In contrast, renal and other vascular abnormalities can manifest in adulthood. Renal manifestations vary and include structural anomalies such as hyperechoic kidneys or renal cysts, which can manifest in childhood, and some abnormalities such as hypertension and renal artery stenosis that can manifest in adulthood.23,24
Vasculopathy is reported to involve the intracranial, renal, and intra-abdominal blood vessels.25 Neurovascular accidents such as stroke and intracranial hemorrhage can occur at any age, with significant rates of morbidity and death.26 Therefore, some experts recommend magnetic resonance angiography every 5 years and before any major intervention to prevent these devastating complications.20
Pregnancy. Successful pregnancies have been reported. Preexisting cardiac and hepatic disease can complicate pregnancy depending on the severity of the disease. Because of the autosomal-dominant pattern of inheritance, infants have a 50% risk of the disease, so genetic counseling should be seriously considered before conception.27 Prenatal diagnosis is possible, but the lack of genotype-phenotype correlation precludes its use in clinical practice.
PROGRESSIVE FAMILIAL INTRAHEPATIC CHOLESTASIS
Progressive familial intrahepatic cholestasis (PFIC) is a heterogeneous group of autosomal-recessive conditions associated with disruption of bile formation causing cholestatic liver disease in infants and young children. Three types have been described, depending on the genetic mutation in the hepatobiliary transport pathway:
- PFIC 1 (Byler disease) is caused by impaired bile salt secretion due to mutations in the ATP8B1 gene encoding for the familial intrahepatic cholestasis 1 (FIC 1) protein
- PFIC 2 is caused by impaired bile salt secretion due to mutations in the ABCB11 gene encoding for the bile salt export pump (BSEP) protein
- PFIC 3 is caused by impaired biliary phospholipid secretion due to a defect in ABCB4 encoding for multidrug resistance 3 (MDR3) protein.28
PFIC 1 and 2 manifest with low gamma-glutamyl transferase (GGT) cholestasis, whereas PFIC 3 presents with high GGT cholestasis.
PFIC 1 and PFIC 2 usually cause cholestasis in early infancy, but PFIC 3 can cause cholestasis in late infancy, childhood, and even adulthood.
Because ATP8B1 is expressed in other tissues, PFIC 1 is characterized by extrahepatic manifestations such as sensorineural hearing loss, growth failure, severe diarrhea, and pancreatic insufficiency.
Implications of PFIC in adulthood
PFIC 1 and 2 (low-GGT cholestasis) are usually progressive and often lead to end-stage liver disease and cirrhosis before adulthood. Therefore, almost all patients with PFIC 1 and 2 undergo liver transplant or at least a biliary diversion procedure before reaching adulthood. Intractable pruritus is one of the most challenging clinical manifestations in patients with PFIC.
First-line management is pharmacologic and includes ursodeoxycholic acid, antihistamines (eg, hydroxyzine), bile acid sequestrants (eg, cholestyramine, colestipol), naltrexone, and rifampin, but these have limited efficacy.10
Most patients, especially those with PFIC 1 and 2, undergo a biliary diversion procedure such as partial external biliary diversion (cholecystojejunocutaneostomy), ileal exclusion, or partial internal biliary diversion (cholecystojejunocolic anastomosis) to decrease enterohepatic circulation of bile salts. The efficacy of these procedures is very limited in patients with established cirrhosis. Excessive losses of bile can occur through the biliary stoma, leading to dehydration in patients with external biliary diversion. In patients who are not candidates for biliary diversion, endoscopic nasobiliary drainage of pancreatobiliary secretions could be achieved by placing a catheter in the common bile duct; this has been reported to be effective in relieving cholestasis in a few cases.29
Liver transplant is needed in patients with progressive liver disease and intractable pruritus despite medical management and biliary diversion. Unlike in biliary atresia, liver transplant is not curative in PFIC 1, due to extrahepatic manifestations: patients with PFIC 1 can still have intractable diarrhea and pancreatitis after liver transplant. More importantly, allograft steatohepatitis with further progression to cirrhosis can occur after liver transplant in patients with PFIC 1. Interestingly, biliary diversion has been reported to improve graft steatosis and diarrhea after liver transplant.30
Although graft survival after transplant is good in patients with PFIC 2, recurrence of low-GGT cholestasis has been reported and is believed to be due to the formation of anti-bile salt export pump (anti-BSEP) antibodies by the host immune system in response to exposure to new proteins from the transplant graft.31
Cancer. The risk of malignancy, especially hepatocellular carcinoma, is also increased in PFIC 2, affecting nearly 15% of patients. Therefore, standard hepatocellular carcinoma surveillance with ultrasonography or alpha-fetoprotein testing or both is recommended in patients with PFIC 2. Cholangiocarcinoma and pancreatic adenocarcinoma have also been reported in patients with PFIC 2.20
Incomplete penetrance of mutations in ATP8B1 and ABCB11 can cause recurrent episodes of cholestasis and pruritus with asymptomatic periods between episodes, referred to as benign recurrent intrahepatic cholestasis. Prognosis is usually good, with no progression to cirrhosis.32
Pregnancy. In contrast to FIC 1 and BSEP deficiency, MDR3 defects lead to a wide phenotypic spectrum depending on the type of mutation. Heterozygous mutation is associated with increased risk of development of cholestasis during pregnancy, which typically presents with generalized pruritus in the third trimester and is associated with adverse fetal outcomes. Intrahepatic cholestasis of pregnancy is usually treated with ursodeoxycholic acid, with reported improvement in pruritus, liver function, and pregnancy outcomes.33
In adults, drug-induced liver injury and idiopathic cirrhosis have also been described with MDR3 defects. Intrahepatic lithiasis and cholesterol gallstones can also occur with MDR3 defects as a result of impaired secretion of biliary phospholipid.32 Despite intrahepatic cholestasis of pregnancy, successful outcomes have been reported in women with PFIC.20
OTHER CHILDHOOD-ONSET INHERITED CHOLESTATIC DISEASES
Cystic fibrosis-associated liver disease
Nearly 40% of patients with cystic fibrosis develop liver disease.34 Cystic fibrosis-associated liver disease encompasses a broad clinical spectrum including asymptomatic elevation of aminotransferases, neonatal cholestasis, hepatic steatosis, focal biliary cirrhosis, and multilobar cirrhosis. Cirrhosis and portal hypertension can occur in 5% to 10% of patients and is the third-leading cause of death in patients with cystic fibrosis.35
Risk factors for cystic fibrosis-associated liver disease include male sex, meconium ileus, and severe CFTR gene mutation (class I–III) with pancreatic insufficiency. Cystic fibrosis-related cirrhosis is more frequent in children and adolescents, whereas noncirrhotic portal hypertension and intrahepatic cholangiopathies are more common in adults.36
Limited available studies support treatment with ursodeoxycholic acid in patients with cholestasis to delay the progression of liver disease, but the impact of this drug on long-term outcome is unknown.29
Most patients remain in compensated cirrhosis for many years before progressing to decompensated cirrhosis requiring liver transplant. Other indications for liver transplant include recurrent intractable variceal bleeding, hepatopulmonary syndrome, and portopulmonary hypertension. Combined liver and lung transplant may be considered in patients with advanced liver and lung disease. Outcomes after isolated liver or liver-lung transplant in cystic fibrosis patients have been comparable to those in patients with other liver diseases.37
Defects in bile acid synthesis
Inherited defects of enzymes required for the synthesis of primary bile acids from cholesterol can cause cholestasis from impaired bile flow and production of hepatotoxic aberrant bile acids. The clinical presentation varies depending on the enzymatic defect and can range from liver disease of varying severity to neurologic manifestations. Idiopathic late-onset cholestasis and cirrhosis of unknown etiology have been reported in adults with bile acid synthesis defects.38,39 Therefore, this diagnosis should be considered in cases of cryptogenic cirrhosis and other cholestatic features.
Treatment with primary bile acids (cholic acid) has been effective in most patients with defective bile acid synthesis.
Primary sclerosing cholangitis
Primary sclerosing cholangitis is characterized by progressive obliteration of intrahepatic and extrahepatic bile ducts and is most commonly seen in patients with inflammatory bowel disease. Sclerosing cholangitis can also be secondary to other diseases in children such as immunodeficiency syndromes, Langerhans cell histiocytosis, cystic fibrosis, or sickle cell anemia.40 Neonatal sclerosing cholangitis is a rare autosomal-recessive disease characterized by a severe form of cholangiopathy in neonates and young infants requiring transplant. It can be associated with Kabuki syndrome and neonatal ichthyosis-sclerosing cholangitis syndrome.
Treatment options are limited. Ursodeoxycholic acid and oral vancomycin have variable efficacy. Liver transplant is needed in patients with decompensated cirrhosis. Patients with primary sclerosing cholangitis, especially adults, are at higher risk of developing cholangiocarcinoma, and therefore screening with ultrasonography or magnetic resonance imaging every 6 to 12 months is recommended.
The risk of preterm and cesarean deliveries may be elevated in women with primary sclerosing cholangitis, though data are limited.33
PEDIATRIC LIVER TRANSPLANT RECIPIENTS WHO SURVIVE INTO ADULTHOOD
Adolescent rebellion poses risks
Outcomes of liver transplant in children and adolescents have improved tremendously in the past 2 decades with advances in surgical techniques, pre- and postoperative management, organ preservation, and immunosuppression. Now, most pediatric liver transplant recipients survive into adulthood, creating a unique challenge for internists and adult care hepatologists.41
In rebellious adolescents and young adults, risk-taking behavior, nonadherence to immunosuppressive medications, alcohol intake, and substance abuse increase the risk of graft rejection and loss. Current immunosuppressive drugs such as calcineurin inhibitors (tacrolimus, cyclosporine), mycophenolate mofetil, sirolimus, and corticosteroids have drastically decreased rejection rates in compliant patients.41 Educating patients on the importance of taking their medications and avoiding alcohol and drug abuse is especially important for adolescents and young adults, as rates of nonadherence are high in these age groups.
Although pregnancy is usually successful after liver transplant, it should be considered high-risk due to reported complications such as graft rejection, diabetes, preeclampsia, sepsis, prematurity, and low birth weight. Conception should be avoided for at least 1 year after transplant.42 Appropriate counseling with regard to pregnancy and contraception is important.
There is no consensus on breastfeeding, but it is considered safe in women on low-dose calcineurin inhibitors.43
Life is better with a new liver, but patients have special needs
Liver transplant is life-saving and improves quality of life. However, long-term pediatric liver transplant recipients face challenges such as strict adherence to medications and follow-up visits, avoiding exposure to infections, and fear of graft rejection.
Chronic liver disease in children leads to failure to thrive, growth failure, and even delayed puberty, which resolve in most patients after liver transplant before adulthood in the absence of other comorbidities.44 However, these patients are reported to have lower psychosocial functioning and more psychiatric disorders such as anxiety or posttraumatic disorder.41,44
Therefore, a psychologist or other mental health professional should be part of the management team from the time of pretransplant assessment to identify mental health problems and the need for adjustments before liver transplant. Ongoing psychosocial assessment after liver transplant is equally important to identify risks such as drug or alcohol abuse, depression, posttraumatic stress disorder, and medication nonadherence, all of which can negatively affect posttransplant outcome.45
In addition, assessment of family functioning and structure is important for good long-term outcomes posttransplant; therefore, a social worker should also be a part of the transplant team. Psyschosocial assessment tools can identify high-risk candidates who would benefit from earlier intervention to avoid any negative impact posttransplant.
Neurocognitive development can be delayed in children with chronic liver disease, and the delay may persist even after liver transplant, with reported impairments in intellectual ability, language, verbal, and visuospatial functioning skills.41 In spite of this, a recent study found that more than half the study patients were employed at a median follow-up of 24 years from liver transplant and a median age of 27.46
Remarkably, pediatric liver transplant recipients have reported quality of life comparable to that in the general population,47 and even better than in patients with other chronic illnesses.48
Long-term medical comorbidities in pediatric liver transplant recipients
Favorable outcomes such as long-term survival and good quality of life in pediatric liver transplant recipients are lessened by late complications such as portal vein thrombosis or biliary strictures needing interventions, chronic graft rejection, adverse effects of immunosuppression, and recurrence of the disease.
Split-liver transplant—splitting a deceased-donor allograft to provide grafts for 2 recipients—has revolutionized liver transplant by increasing the donor pool and thereby decreasing waitlist mortality rates, especially in pediatric candidates. Despite this advantage, split-liver transplant is technically challenging and associated with increased perioperative complications compared with whole-liver transplant, especially in adult recipients. Recently, experienced centers have reported favorable outcomes with split-liver transplant comparable to those with whole-liver transplant; therefore, split-liver transplant should be considered after careful evaluation of donor organ and recipient clinical status.49
Old age in the recipient can also adversely affect liver transplant outcomes.50
Interestingly, even in patients whose clinical course is unremarkable and biochemical values are normal, graft hepatitis or fibrosis of unknown cause with progression to cirrhosis has been described in the decade after transplant.41
Chronic rejection with eventual graft loss may be related to nonadherence in adolescents and can be reduced with use of an additional immunosuppressant such as sirolimus or mycophenolate. Chronic kidney disease can occur in about one-third of liver transplant recipients secondary to renal disease associated with primary disease (like Alagille syndrome), hepatorenal syndrome, and most importantly, use of calcineurin inhibitors.45
Components of the metabolic syndrome such as type 2 diabetes, obesity, nonalcoholic fatty liver disease, hypertension, and dyslipidemia are also seen in long-term pediatric liver transplant survivors. Internists are advised to screen for these comorbidities so that interventions can be applied early to improve long-term health outcomes and graft survival.
Of importance, multiple studies have shown a 2-fold increase in the rates of de novo malignancy in liver transplant recipients, including solid-organ and lymphoproliferative cancers, probably due to long-term immunosuppression. Posttransplant lymphoproliferative disorder occurs at lower rates than with other solid-organ transplants; its incidence is greatest in pediatric patients and in the first 12 to 18 months after transplant.51
TRANSITION TO ADULT CARE
While the number of patients with childhood-onset liver disease and pediatric liver transplant recipients who survive into adulthood is increasing, there are no established guidelines or formal models for transitioning these patients into adult care. Consequently, studies on transitional process have examined various issues such as patient and parent frustration, poor medical knowledge among patients during transition, lack of parental facilitation, and inadequate knowledge on disease process among adult-care hepatologists.52–54
A prolonged period of transition up to age 25 is preferred in complicated cases. Distinctive consideration for transition should include those with neurocognitive developmental delay from underlying disease or hepatic encephalopathy before transplant. These patients need additional support and time to achieve independence in health management before transition.57 Validated questionnaires are available to assess readiness to transition into adult care,58 implying that the decision to transition should not be based solely on age.
- Kelly DA, Bucuvalas JC, Alonso EM, et al; American Association for the Study of Liver Diseases; American Society of Transplantation. Long-term medical management of the pediatric patient after liver transplantation: 2013 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Liver Transpl 2013; 19(8):798–825. doi:10.1002/lt.23697
- Rosen DS, Blum RW, Britto M, Sawyer SM, Siegel DM; Society for Adolescent Medicine. Transition to adult health care for adolescents and young adults with chronic conditions: position paper of the Society for Adolescent Medicine. J Adolesc Health 2003; 33(4):309–311. pmid:14519573
- Fawaz R, Baumann U, Ekong U, et al. Guideline for the evaluation of cholestatic jaundice in infants: joint recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr 2017; 64(1):154–168. doi:10.1097/MPG.0000000000001334
- Vajro P, Ferrante L, Lenta S, Mandato C, Persico M. Management of adults with paediatric-onset chronic liver disease: strategic issues for transition care. Dig Liver Dis 2014; 46(4):295–301. doi:10.1016/j.dld.2013.10.018
- Davenport M, Tizzard SA, Underhill J, Mieli-Vergani G, Portmann B, Hadzic N. The biliary atresia splenic malformation syndrome: a 28-year single-center retrospective study. J Pediatr 2006; 149(3):393–400. doi:10.1016/j.jpeds.2006.05.030
- Balistreri WF, Bezerra JA. Whatever happened to “neonatal hepatitis?” Clin Liver Dis 2006; 10(1):27–53. doi:10.1016/j.cld.2005.10.008
- Serinet MO, Wildhaber BE, Broué P, et al. Impact of age at Kasai operation on its results in late childhood and adolescence: a rational basis for biliary atresia screening. Pediatrics 2009; 123(5):1280–1286. doi:10.1542/peds.2008-1949
- de Vries W, Homan-Van der Veen J, Hulscher JB, Hoekstra-Weebers JE, Houwen RH, Verkade HJ; Netherlands Study Group of Biliary Atresia Registry. Twenty-year transplant-free survival rate among patients with biliary atresia. Clin Gastroenterol Hepatol 2011; 9(12):1086–1091. doi:10.1016/j.cgh.2011.07.024
- Lykavieris P, Chardot C, Sokhn M, Gauthier F, Valayer J, Bernard O. Outcome in adulthood of biliary atresia: a study of 63 patients who survived for over 20 years with their native liver. Hepatology 2005; 41(2):366–371. doi:10.1002/hep.20547
- Joshi D, Gupta N, Samyn M, Deheragoda M, Dobbels F, Heneghan MA. The management of childhood liver diseases in adulthood. J Hepatol 2017; 66(3):631–644. doi:10.1016/j.jhep.2016.11.013
- Shneider BL, Abel B, Haber B, et al; Childhood Liver Disease Research and Education Network. Portal hypertension in children and young adults with biliary atresia. J Pediatr Gastroenterol Nutr 2012; 55(5):567–573. doi:10.1097/MPG.0b013e31826eb0cf
- Garcia-Tsao G, Abraldes JG, Berzigotti A, Bosch J. Portal hypertensive bleeding in cirrhosis: risk stratification, diagnosis, and management: 2016 practice guidance by the American Association for the Study of Liver Diseases. Hepatology 2017; 65(1):310–335. doi:10.1002/hep.28906
- Shneider BL, Brown MB, Haber B, et al; Biliary Atresia Research Consortium. A multicenter study of the outcome of biliary atresia in the United States, 1997 to 2000. J Pediatr 2006; 148(4):467–474. doi:10.1016/j.jpeds.2005.12.054
- Hung PY, Chen CC, Chen WJ, et al. Long-term prognosis of patients with biliary atresia: a 25 year summary. J Pediatr Gastroenterol Nutr 2006; 42(2):190–195. doi:10.1097/01.mpg.0000189339.92891.64
- Verkade HJ, Bezerra JA, Davenport M, et al. Biliary atresia and other cholestatic childhood diseases: advances and future challenges. J Hepatol 2016; 65(3):631–642. doi:10.1016/j.jhep.2016.04.032
- Hadžic N, Quaglia A, Portmann B, et al. Hepatocellular carcinoma in biliary atresia: King’s College Hospital experience. J Pediatr 2011; 159(4):617–622.e1. doi:10.1016/j.jpeds.2011.03.004
- Sokol RJ, Shepherd RW, Superina R, Bezerra JA, Robuck P, Hoofnagle JH. Screening and outcomes in biliary atresia: summary of a National Institutes of Health workshop. Hepatology 2007; 46(2):566–581. doi:10.1002/hep.21790
- Li L, Krantz ID, Deng Y, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet 1997; 16(3):243–251. doi:10.1038/ng0797-243
- Saleh M, Kamath BM, Chitayat D. Alagille syndrome: clinical perspectives. Appl Clin Genet 2016; 9:75–82. doi:10.2147/TACG.S86420
- Bass LM, Kamath BM. Inherited disorders of cholestasis in adulthood. Clinical Liver Disease 2013; 2(5):200–203. doi:10.1002/cld.245
- Kamath BM, Yin W, Miller H, Anand R, Rand EB, Alonso E, Bucuvalas J; Studies of Pediatric Liver Transplantation. Outcomes of liver transplantation for patients with Alagille syndrome: the studies of pediatric liver transplantation experience. Liver Transpl 2012; 18(8):940–948. doi:10.1002/lt.23437
- Arnon R, Annunziato R, Schiano T, et al. Orthotopic liver transplantation for adults with Alagille syndrome. Clin Transplant 2012; 26(2):E94–E100. doi:10.1111/j.1399-0012.2011.01574.x
- Salem JE, Bruguiere E, Iserin L, Guiochon-Mantel A, Plouin PF. Hypertension and aortorenal disease in Alagille syndrome. J Hypertens 2012; 30(7):1300–1306. doi:10.1097/HJH.0b013e3283531e1f
- Kamath BM, Podkameni G, Hutchinson AL, et al. Renal anomalies in Alagille syndrome: a disease-defining feature. Am J Med Genet A 2012; 158A(1):85–89. doi:10.1002/ajmg.a.34369
- Kamath BM, Bason L, Piccoli DA, Krantz ID, Spinner NB. Consequences of JAG1 mutations. J Med Genet 2003; 40(12):891–895. pmid:14684686
- Emerick KM, Krantz ID, Kamath BM, et al. Intracranial vascular abnormalities in patients with Alagille syndrome. J Pediatr Gastroenterol Nutr 2005; 41(1):99–107. pmid:15990638
- Ferrarese A, Senzolo M, Burra P. Successful pregnancy in Alagille syndrome. Dig Liver Dis 2015; 47(1):86–87. doi:10.1016/j.dld.2014.08.047
- Davit-Spraul A, Fabre M, Branchereau S, et al. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology 2010; 51(5):1645–1655. doi:10.1002/hep.23539
- Zellos A, Lykopoulou L, Polydorou A, et al. Nasobiliary drainage in an episode of intrahepatic cholestasis in a child with mild ABCB11 disease. J Pediatr Gastroenterol Nutr 2012; 55(1):88–90. doi:10.1097/MPG.0b013e31822f2bda
- Alrabadi LS, Morotti RA, Valentino PL, Rodriguez-Davalos MI, Ekong UD, Emre SH. Biliary drainage as treatment for allograft steatosis following liver transplantation for PFIC-1 disease: a single-center experience. Pediatr Transplant 2018; 22(4):e13184. doi:10.1111/petr.13184
- Kubitz R, Dröge C, Kluge S, et al. Autoimmune BSEP disease: disease recurrence after liver transplantation for progressive familial intrahepatic cholestasis. Clin Rev Allergy Immunol 2015; 48(2–3):273–284. doi:10.1007/s12016-014-8457-4
- Jacquemin E. Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol 2012; 36(suppl 1):S26–S35. doi:10.1016/S2210-7401(12)70018-9
- Pataia V, Dixon PH, Williamson C. Pregnancy and bile acid disorders. Am J Physiol Gastrointest Liver Physiol 2017; 313(1):G1–G6. doi:10.1152/ajpgi.00028.2017
- Lamireau T, Monnereau S, Martin S, Marcotte JE, Winnock M, Alvarez F. Epidemiology of liver disease in cystic fibrosis: a longitudinal study. J Hepatol 2004; 41(6):920–925. doi:10.1016/j.jhep.2004.08.006
- Bolia R, Ooi CY, Lewindon P, et al. Practical approach to the gastrointestinal manifestations of cystic fibrosis. J Paediatr Child Health 2018; 54(6):609–619. doi:10.1111/jpc.13921
- Debray D, Kelly D, Houwen R, Strandvik B, Colombo C. Best practice guidance for the diagnosis and management of cystic fibrosis-associated liver disease. J Cyst Fibros 2011; 10(suppl 2):S29–S36. doi:10.1016/S1569-1993(11)60006-4
- Fridell JA, Bond GJ, Mazariegos G V, et al. Liver transplantation in children with cystic fibrosis: a long-term longitudinal review of a single center’s experience. J Pediatr Surg 2003; 38(8):1152–1156. pmid:12891484
- Fischler B, Bodin K, Stjernman H, et al. Cholestatic liver disease in adults may be due to an inherited defect in bile acid biosynthesis. J Intern Med 2007; 262(2):254–262. doi:10.1111/j.1365-2796.2007.01814.x
- Molho-Pessach V, Rios JJ, Xing C, Setchell KD, Cohen JC, Hobbs HH. Homozygosity mapping identifies a bile acid biosynthetic defect in an adult with cirrhosis of unknown etiology. Hepatology 2012; 55(4):1139–1145. doi:10.1002/hep.24781
- Mieli-Vergani G, Vergani D. Sclerosing cholangitis in children and adolescents. Clin Liver Dis 2016; 20(1):99–111. doi:10.1016/j.cld.2015.08.008
- Kelly D, Wray J. The adolescent liver transplant patient. Clin Liver Dis 2014; 18(3):613–632. doi:10.1016/j.cld.2014.05.006
- Westbrook RH, Yeoman AD, Agarwal K, et al. Outcomes of pregnancy following liver transplantation: the King’s College Hospital experience. Liver Transpl. 2015; 21(9):1153–1159. doi:10.1002/lt.24182
- Hammoud GM, Almashhrawi AA, Ahmed KT, Rahman R, Ibdah JA. Liver diseases in pregnancy: liver transplantation in pregnancy. World J Gastroenterol 2013; 19(43):7647–7651. doi:10.3748/wjg.v19.i43.7647
- Codoner-Franch P, Bernard O, Alvarez F. Long-term follow-up of growth in height after successful liver transplantation. J Pediatr 1994; 124(3):368–373. pmid:8120704
- Shemesh E. Assessment and management of psychosocial challenges in pediatric liver transplantation. Liver Transpl 2008; 14(9):1229–1236. doi:10.1002/lt.21582
- Martinelli J, Habes D, Majed L, et al. Long-term outcome of liver transplantation in childhood: a study of 20-year survivors. Am J Transplant 2018; 18(7):1680–1689. doi:10.1111/ajt.14626
- Roblin E, Audhuy F, Boillot O, Rivet C, Lachaux A. Long-term quality of life after pediatric liver transplantation. Arch Pediatr 2012; 19(10):1039–1052. French. doi:10.1016/j.arcped.2012.06.020
- Duffy JP, Kao K, Ko CY, et al. Long-term patient outcome and quality of life after liver transplantation: analysis of 20-year survivors. Ann Surg 2010; 252(4):652–661. doi:10.1097/SLA.0b013e3181f5f23a
- Hackl C, Schmidt KM, Süsal C, Döhler B, Zidek M, Schlitt HJ. Split liver transplantation: Current developments. World J Gastroenterol 2018; 24(47):5312–5321. doi:10.3748/wjg.v24.i47.5312
- Durand F, Levitsky J, Cauchy F, Gilgenkrantz H, Soubrane O, Francoz C. Age and liver transplantation. J Hepatol 2019; 70(4):745–758. doi:10.1016/j.jhep.2018.12.009
- Chandok N, Watt KD. Burden of de novo malignancy in the liver transplant recipient. Liver Transpl 2012; 18(11):1277–1289. doi:10.1002/lt.23531
- Ferrarese A, Germani G, Lazzaro S, et al. Short-term outcomes of paediatric liver transplant recipients after transition to Adult Healthcare Service. Liver Int 2018; 38(7):1316–1321. doi:10.1111/liv.13655
- Wright J, Elwell L, McDonagh JE, Kelly DA, Wray J. “Are these adult doctors gonna know me?” Experiences of transition for young people with a liver transplant. Pediatr Transplant 2016; 20(7):912–920. doi:10.1111/petr.12777
- Heldman MR, Sohn MW, Gordon EJ, et al. National survey of adult transplant hepatologists on the pediatric-to-adult care transition after liver transplantation. Liver Transpl 2015; 21(2):213–223. doi:10.1002/lt.24044
- Vajro P, Fischler B, Burra P, et al. The health care transition of youth with liver disease into the adult health system. J Pediatr Gastroenterol Nutr 2018; 66(6):976–990. doi:10.1097/MPG.0000000000001965
- Fredericks EM, Lopez MJ. Transition of the adolescent transplant patient to adult care. Clin Liver Dis (Hoboken) 2013; 2(5):223–226. doi:10.1002/cld.243
- Kaufman M. Transition of cognitively delayed adolescent organ transplant recipients to adult care. Pediatr Transplant 2006; 10(4):413–417. doi:10.1111/j.1399-3046.2006.00491.x
- Sawicki GS, Lukens-Bull K, Yin X, et al. Measuring the transition readiness of youth with special healthcare needs: validation of the TRAQ—Transition Readiness Assessment Questionnaire. J Pediatr Psychol 2011; 36(2):160–171. doi:10.1093/jpepsy/jsp128
- Kelly DA, Bucuvalas JC, Alonso EM, et al; American Association for the Study of Liver Diseases; American Society of Transplantation. Long-term medical management of the pediatric patient after liver transplantation: 2013 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Liver Transpl 2013; 19(8):798–825. doi:10.1002/lt.23697
- Rosen DS, Blum RW, Britto M, Sawyer SM, Siegel DM; Society for Adolescent Medicine. Transition to adult health care for adolescents and young adults with chronic conditions: position paper of the Society for Adolescent Medicine. J Adolesc Health 2003; 33(4):309–311. pmid:14519573
- Fawaz R, Baumann U, Ekong U, et al. Guideline for the evaluation of cholestatic jaundice in infants: joint recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr 2017; 64(1):154–168. doi:10.1097/MPG.0000000000001334
- Vajro P, Ferrante L, Lenta S, Mandato C, Persico M. Management of adults with paediatric-onset chronic liver disease: strategic issues for transition care. Dig Liver Dis 2014; 46(4):295–301. doi:10.1016/j.dld.2013.10.018
- Davenport M, Tizzard SA, Underhill J, Mieli-Vergani G, Portmann B, Hadzic N. The biliary atresia splenic malformation syndrome: a 28-year single-center retrospective study. J Pediatr 2006; 149(3):393–400. doi:10.1016/j.jpeds.2006.05.030
- Balistreri WF, Bezerra JA. Whatever happened to “neonatal hepatitis?” Clin Liver Dis 2006; 10(1):27–53. doi:10.1016/j.cld.2005.10.008
- Serinet MO, Wildhaber BE, Broué P, et al. Impact of age at Kasai operation on its results in late childhood and adolescence: a rational basis for biliary atresia screening. Pediatrics 2009; 123(5):1280–1286. doi:10.1542/peds.2008-1949
- de Vries W, Homan-Van der Veen J, Hulscher JB, Hoekstra-Weebers JE, Houwen RH, Verkade HJ; Netherlands Study Group of Biliary Atresia Registry. Twenty-year transplant-free survival rate among patients with biliary atresia. Clin Gastroenterol Hepatol 2011; 9(12):1086–1091. doi:10.1016/j.cgh.2011.07.024
- Lykavieris P, Chardot C, Sokhn M, Gauthier F, Valayer J, Bernard O. Outcome in adulthood of biliary atresia: a study of 63 patients who survived for over 20 years with their native liver. Hepatology 2005; 41(2):366–371. doi:10.1002/hep.20547
- Joshi D, Gupta N, Samyn M, Deheragoda M, Dobbels F, Heneghan MA. The management of childhood liver diseases in adulthood. J Hepatol 2017; 66(3):631–644. doi:10.1016/j.jhep.2016.11.013
- Shneider BL, Abel B, Haber B, et al; Childhood Liver Disease Research and Education Network. Portal hypertension in children and young adults with biliary atresia. J Pediatr Gastroenterol Nutr 2012; 55(5):567–573. doi:10.1097/MPG.0b013e31826eb0cf
- Garcia-Tsao G, Abraldes JG, Berzigotti A, Bosch J. Portal hypertensive bleeding in cirrhosis: risk stratification, diagnosis, and management: 2016 practice guidance by the American Association for the Study of Liver Diseases. Hepatology 2017; 65(1):310–335. doi:10.1002/hep.28906
- Shneider BL, Brown MB, Haber B, et al; Biliary Atresia Research Consortium. A multicenter study of the outcome of biliary atresia in the United States, 1997 to 2000. J Pediatr 2006; 148(4):467–474. doi:10.1016/j.jpeds.2005.12.054
- Hung PY, Chen CC, Chen WJ, et al. Long-term prognosis of patients with biliary atresia: a 25 year summary. J Pediatr Gastroenterol Nutr 2006; 42(2):190–195. doi:10.1097/01.mpg.0000189339.92891.64
- Verkade HJ, Bezerra JA, Davenport M, et al. Biliary atresia and other cholestatic childhood diseases: advances and future challenges. J Hepatol 2016; 65(3):631–642. doi:10.1016/j.jhep.2016.04.032
- Hadžic N, Quaglia A, Portmann B, et al. Hepatocellular carcinoma in biliary atresia: King’s College Hospital experience. J Pediatr 2011; 159(4):617–622.e1. doi:10.1016/j.jpeds.2011.03.004
- Sokol RJ, Shepherd RW, Superina R, Bezerra JA, Robuck P, Hoofnagle JH. Screening and outcomes in biliary atresia: summary of a National Institutes of Health workshop. Hepatology 2007; 46(2):566–581. doi:10.1002/hep.21790
- Li L, Krantz ID, Deng Y, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet 1997; 16(3):243–251. doi:10.1038/ng0797-243
- Saleh M, Kamath BM, Chitayat D. Alagille syndrome: clinical perspectives. Appl Clin Genet 2016; 9:75–82. doi:10.2147/TACG.S86420
- Bass LM, Kamath BM. Inherited disorders of cholestasis in adulthood. Clinical Liver Disease 2013; 2(5):200–203. doi:10.1002/cld.245
- Kamath BM, Yin W, Miller H, Anand R, Rand EB, Alonso E, Bucuvalas J; Studies of Pediatric Liver Transplantation. Outcomes of liver transplantation for patients with Alagille syndrome: the studies of pediatric liver transplantation experience. Liver Transpl 2012; 18(8):940–948. doi:10.1002/lt.23437
- Arnon R, Annunziato R, Schiano T, et al. Orthotopic liver transplantation for adults with Alagille syndrome. Clin Transplant 2012; 26(2):E94–E100. doi:10.1111/j.1399-0012.2011.01574.x
- Salem JE, Bruguiere E, Iserin L, Guiochon-Mantel A, Plouin PF. Hypertension and aortorenal disease in Alagille syndrome. J Hypertens 2012; 30(7):1300–1306. doi:10.1097/HJH.0b013e3283531e1f
- Kamath BM, Podkameni G, Hutchinson AL, et al. Renal anomalies in Alagille syndrome: a disease-defining feature. Am J Med Genet A 2012; 158A(1):85–89. doi:10.1002/ajmg.a.34369
- Kamath BM, Bason L, Piccoli DA, Krantz ID, Spinner NB. Consequences of JAG1 mutations. J Med Genet 2003; 40(12):891–895. pmid:14684686
- Emerick KM, Krantz ID, Kamath BM, et al. Intracranial vascular abnormalities in patients with Alagille syndrome. J Pediatr Gastroenterol Nutr 2005; 41(1):99–107. pmid:15990638
- Ferrarese A, Senzolo M, Burra P. Successful pregnancy in Alagille syndrome. Dig Liver Dis 2015; 47(1):86–87. doi:10.1016/j.dld.2014.08.047
- Davit-Spraul A, Fabre M, Branchereau S, et al. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology 2010; 51(5):1645–1655. doi:10.1002/hep.23539
- Zellos A, Lykopoulou L, Polydorou A, et al. Nasobiliary drainage in an episode of intrahepatic cholestasis in a child with mild ABCB11 disease. J Pediatr Gastroenterol Nutr 2012; 55(1):88–90. doi:10.1097/MPG.0b013e31822f2bda
- Alrabadi LS, Morotti RA, Valentino PL, Rodriguez-Davalos MI, Ekong UD, Emre SH. Biliary drainage as treatment for allograft steatosis following liver transplantation for PFIC-1 disease: a single-center experience. Pediatr Transplant 2018; 22(4):e13184. doi:10.1111/petr.13184
- Kubitz R, Dröge C, Kluge S, et al. Autoimmune BSEP disease: disease recurrence after liver transplantation for progressive familial intrahepatic cholestasis. Clin Rev Allergy Immunol 2015; 48(2–3):273–284. doi:10.1007/s12016-014-8457-4
- Jacquemin E. Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol 2012; 36(suppl 1):S26–S35. doi:10.1016/S2210-7401(12)70018-9
- Pataia V, Dixon PH, Williamson C. Pregnancy and bile acid disorders. Am J Physiol Gastrointest Liver Physiol 2017; 313(1):G1–G6. doi:10.1152/ajpgi.00028.2017
- Lamireau T, Monnereau S, Martin S, Marcotte JE, Winnock M, Alvarez F. Epidemiology of liver disease in cystic fibrosis: a longitudinal study. J Hepatol 2004; 41(6):920–925. doi:10.1016/j.jhep.2004.08.006
- Bolia R, Ooi CY, Lewindon P, et al. Practical approach to the gastrointestinal manifestations of cystic fibrosis. J Paediatr Child Health 2018; 54(6):609–619. doi:10.1111/jpc.13921
- Debray D, Kelly D, Houwen R, Strandvik B, Colombo C. Best practice guidance for the diagnosis and management of cystic fibrosis-associated liver disease. J Cyst Fibros 2011; 10(suppl 2):S29–S36. doi:10.1016/S1569-1993(11)60006-4
- Fridell JA, Bond GJ, Mazariegos G V, et al. Liver transplantation in children with cystic fibrosis: a long-term longitudinal review of a single center’s experience. J Pediatr Surg 2003; 38(8):1152–1156. pmid:12891484
- Fischler B, Bodin K, Stjernman H, et al. Cholestatic liver disease in adults may be due to an inherited defect in bile acid biosynthesis. J Intern Med 2007; 262(2):254–262. doi:10.1111/j.1365-2796.2007.01814.x
- Molho-Pessach V, Rios JJ, Xing C, Setchell KD, Cohen JC, Hobbs HH. Homozygosity mapping identifies a bile acid biosynthetic defect in an adult with cirrhosis of unknown etiology. Hepatology 2012; 55(4):1139–1145. doi:10.1002/hep.24781
- Mieli-Vergani G, Vergani D. Sclerosing cholangitis in children and adolescents. Clin Liver Dis 2016; 20(1):99–111. doi:10.1016/j.cld.2015.08.008
- Kelly D, Wray J. The adolescent liver transplant patient. Clin Liver Dis 2014; 18(3):613–632. doi:10.1016/j.cld.2014.05.006
- Westbrook RH, Yeoman AD, Agarwal K, et al. Outcomes of pregnancy following liver transplantation: the King’s College Hospital experience. Liver Transpl. 2015; 21(9):1153–1159. doi:10.1002/lt.24182
- Hammoud GM, Almashhrawi AA, Ahmed KT, Rahman R, Ibdah JA. Liver diseases in pregnancy: liver transplantation in pregnancy. World J Gastroenterol 2013; 19(43):7647–7651. doi:10.3748/wjg.v19.i43.7647
- Codoner-Franch P, Bernard O, Alvarez F. Long-term follow-up of growth in height after successful liver transplantation. J Pediatr 1994; 124(3):368–373. pmid:8120704
- Shemesh E. Assessment and management of psychosocial challenges in pediatric liver transplantation. Liver Transpl 2008; 14(9):1229–1236. doi:10.1002/lt.21582
- Martinelli J, Habes D, Majed L, et al. Long-term outcome of liver transplantation in childhood: a study of 20-year survivors. Am J Transplant 2018; 18(7):1680–1689. doi:10.1111/ajt.14626
- Roblin E, Audhuy F, Boillot O, Rivet C, Lachaux A. Long-term quality of life after pediatric liver transplantation. Arch Pediatr 2012; 19(10):1039–1052. French. doi:10.1016/j.arcped.2012.06.020
- Duffy JP, Kao K, Ko CY, et al. Long-term patient outcome and quality of life after liver transplantation: analysis of 20-year survivors. Ann Surg 2010; 252(4):652–661. doi:10.1097/SLA.0b013e3181f5f23a
- Hackl C, Schmidt KM, Süsal C, Döhler B, Zidek M, Schlitt HJ. Split liver transplantation: Current developments. World J Gastroenterol 2018; 24(47):5312–5321. doi:10.3748/wjg.v24.i47.5312
- Durand F, Levitsky J, Cauchy F, Gilgenkrantz H, Soubrane O, Francoz C. Age and liver transplantation. J Hepatol 2019; 70(4):745–758. doi:10.1016/j.jhep.2018.12.009
- Chandok N, Watt KD. Burden of de novo malignancy in the liver transplant recipient. Liver Transpl 2012; 18(11):1277–1289. doi:10.1002/lt.23531
- Ferrarese A, Germani G, Lazzaro S, et al. Short-term outcomes of paediatric liver transplant recipients after transition to Adult Healthcare Service. Liver Int 2018; 38(7):1316–1321. doi:10.1111/liv.13655
- Wright J, Elwell L, McDonagh JE, Kelly DA, Wray J. “Are these adult doctors gonna know me?” Experiences of transition for young people with a liver transplant. Pediatr Transplant 2016; 20(7):912–920. doi:10.1111/petr.12777
- Heldman MR, Sohn MW, Gordon EJ, et al. National survey of adult transplant hepatologists on the pediatric-to-adult care transition after liver transplantation. Liver Transpl 2015; 21(2):213–223. doi:10.1002/lt.24044
- Vajro P, Fischler B, Burra P, et al. The health care transition of youth with liver disease into the adult health system. J Pediatr Gastroenterol Nutr 2018; 66(6):976–990. doi:10.1097/MPG.0000000000001965
- Fredericks EM, Lopez MJ. Transition of the adolescent transplant patient to adult care. Clin Liver Dis (Hoboken) 2013; 2(5):223–226. doi:10.1002/cld.243
- Kaufman M. Transition of cognitively delayed adolescent organ transplant recipients to adult care. Pediatr Transplant 2006; 10(4):413–417. doi:10.1111/j.1399-3046.2006.00491.x
- Sawicki GS, Lukens-Bull K, Yin X, et al. Measuring the transition readiness of youth with special healthcare needs: validation of the TRAQ—Transition Readiness Assessment Questionnaire. J Pediatr Psychol 2011; 36(2):160–171. doi:10.1093/jpepsy/jsp128
KEY POINTS
- The causes of cholestasis in children are different from those in adults, with genetic inherited causes more common in childhood.
- Cholestasis in children can be caused by biliary tract obstruction such as in biliary atresia or defects in forming and excreting bile acids and other components of bile.
- With the growing number of people with childhood-onset liver disease surviving into adulthood, it is important for internists to be aware of unique problems and challenges in continuing management of this population.
- In addition to medical comorbidities, these patients may also have impaired psychosocial functioning and quality of life.
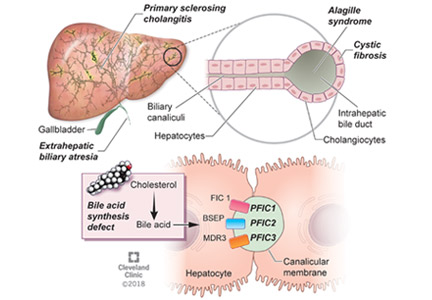
In reply: Acute liver failure
In Reply: We thank Dr. Homler for bringing hepatitis D as a potential cause of acute liver failure to our attention.
Hepatitis D virus, first described in the 1970s, requires the hepatitis B surface antigen (HBsAg) capsid to enter the hepatocyte and, thus, can only cause liver injury when the patient is also infected simultaneously with hepatitis B virus.1 Hepatitis D virus can cause either coinfection (presence of immunoglobulin M anti-HB core antibody with or without HBsAg) or superinfection (presence of HBsAg without immunoglobulin M anti-HB core antibody) with hepatitis B virus. In India, coinfection has been reported to be the cause of acute liver failure in about 4% of all patients, and superinfection in 4.5%.2
While simultaneous treatment for hepatitis D and B viruses with pegylated interferon and any of the agents used for treatment of hepatitis B has been successful in treating chronic hepatitis, it has not been proven useful in patients with acute liver failure, and liver transplant remains the only treatment option.3
- Rizzetto M. The adventure of delta. Liver Int 2016; 36(suppl 1):135–140.
- Irshad M, Acharya SK. Hepatitis D virus (HDV) infection in severe forms of liver diseases in North India. Eur J Gastroenterol Hepatol 1996; 8:995–998.
- Noureddin M, Gish R. Hepatitis delta: epidemiology, diagnosis and management 36 years after discovery. Curr Gastroenterol Rep 2014; 16:365.
In Reply: We thank Dr. Homler for bringing hepatitis D as a potential cause of acute liver failure to our attention.
Hepatitis D virus, first described in the 1970s, requires the hepatitis B surface antigen (HBsAg) capsid to enter the hepatocyte and, thus, can only cause liver injury when the patient is also infected simultaneously with hepatitis B virus.1 Hepatitis D virus can cause either coinfection (presence of immunoglobulin M anti-HB core antibody with or without HBsAg) or superinfection (presence of HBsAg without immunoglobulin M anti-HB core antibody) with hepatitis B virus. In India, coinfection has been reported to be the cause of acute liver failure in about 4% of all patients, and superinfection in 4.5%.2
While simultaneous treatment for hepatitis D and B viruses with pegylated interferon and any of the agents used for treatment of hepatitis B has been successful in treating chronic hepatitis, it has not been proven useful in patients with acute liver failure, and liver transplant remains the only treatment option.3
In Reply: We thank Dr. Homler for bringing hepatitis D as a potential cause of acute liver failure to our attention.
Hepatitis D virus, first described in the 1970s, requires the hepatitis B surface antigen (HBsAg) capsid to enter the hepatocyte and, thus, can only cause liver injury when the patient is also infected simultaneously with hepatitis B virus.1 Hepatitis D virus can cause either coinfection (presence of immunoglobulin M anti-HB core antibody with or without HBsAg) or superinfection (presence of HBsAg without immunoglobulin M anti-HB core antibody) with hepatitis B virus. In India, coinfection has been reported to be the cause of acute liver failure in about 4% of all patients, and superinfection in 4.5%.2
While simultaneous treatment for hepatitis D and B viruses with pegylated interferon and any of the agents used for treatment of hepatitis B has been successful in treating chronic hepatitis, it has not been proven useful in patients with acute liver failure, and liver transplant remains the only treatment option.3
- Rizzetto M. The adventure of delta. Liver Int 2016; 36(suppl 1):135–140.
- Irshad M, Acharya SK. Hepatitis D virus (HDV) infection in severe forms of liver diseases in North India. Eur J Gastroenterol Hepatol 1996; 8:995–998.
- Noureddin M, Gish R. Hepatitis delta: epidemiology, diagnosis and management 36 years after discovery. Curr Gastroenterol Rep 2014; 16:365.
- Rizzetto M. The adventure of delta. Liver Int 2016; 36(suppl 1):135–140.
- Irshad M, Acharya SK. Hepatitis D virus (HDV) infection in severe forms of liver diseases in North India. Eur J Gastroenterol Hepatol 1996; 8:995–998.
- Noureddin M, Gish R. Hepatitis delta: epidemiology, diagnosis and management 36 years after discovery. Curr Gastroenterol Rep 2014; 16:365.
A guide to managing acute liver failure
When the liver fails, it usually fails gradually. The sudden (acute) onset of liver failure, while less common, demands prompt management, with transfer to an intensive care unit, specific treatment depending on the cause, and consideration of liver transplant, without which the mortality rate is high.
This article reviews the definition, epidemiology, etiology, and management of acute liver failure.
DEFINITIONS
Acute liver failure is defined as a syndrome of acute hepatitis with evidence of abnormal coagulation (eg, an international normalized ratio > 1.5) complicated by the development of mental alteration (encephalopathy) within 26 weeks of the onset of illness in a patient without a history of liver disease.1 In general, patients have no evidence of underlying chronic liver disease, but there are exceptions; patients with Wilson disease, vertically acquired hepatitis B virus infection, or autoimmune hepatitis can present with acute liver failure superimposed on chronic liver disease or even cirrhosis.
The term acute liver failure has replaced older terms such as fulminant hepatic failure, hyperacute liver failure, and subacute liver failure, which were used for prognostic purposes. Patients with hyperacute liver failure (defined as development of encephalopathy within 7 days of onset of illness) generally have a good prognosis with medical management, whereas those with subacute liver failure (defined as development of encephalopathy within 5 to 26 weeks of onset of illness) have a poor prognosis without liver transplant.2,3
NEARLY 2,000 CASES A YEAR
There are nearly 2,000 cases of acute liver failure each year in the United States, and it accounts for 6% of all deaths due to liver disease.4 It is more common in women than in men, and more common in white people than in other races. The peak incidence is at a fairly young age, ie, 35 to 45 years.
CAUSES
The most common cause of acute liver failure in the United States and other Western countries is acetaminophen toxicity, followed by viral hepatitis. In contrast, viral hepatitis is the most common cause in developing countries.5
Acetaminophen toxicity
Patients with acetaminophen-induced liver failure tend to be younger than other patients with acute liver failure.1 Nearly half of them present after intentionally taking a single large dose, while the rest present with unintentional toxicity while taking acetaminophen for pain relief on a long-term basis and ingesting more than the recommended dose.6
After ingestion, 52% to 57% of acetaminophen is converted to glucuronide conjugates, and 30% to 44% is converted to sulfate conjugates. These compounds are nontoxic, water-soluble, and rapidly excreted in the urine.
However, about 5% to 10% of ingested acetaminophen is shunted to the cytochrome P450 system. P450 2E1 is the main isoenzyme involved in acetaminophen metabolism, but 1A2, 3A4, and 2A6 also contribute.7,8 P450 2E1 is the same isoenzyme responsible for ethanol metabolism and is inducible. Thus, regular alcohol consumption can increase P450 2E1 activity, setting the stage under certain circumstances for increased acetaminophen metabolism through this pathway.

Metabolism of acetaminophen through the cytochrome P450 pathway results in production of N-acetyl-p-benzoquinone imine (NAPQI), the compound that damages the liver. NAPQI is rendered nontoxic by binding to glutathione, forming NAPQI-glutathione adducts. Glutathione capacity is limited, however. With too much acetaminophen, glutathione becomes depleted and NAPQI accumulates, binds with proteins to form adducts, and leads to necrosis of hepatocytes (Figure 1).9,10
Acetylcysteine, used in treating acetaminophen toxicity, is a substrate for glutathione synthesis and ultimately increases the amount of glutathione available to bind NAPQI and prevent damage to hepatocytes.11
Acetaminophen is a dose-related toxin. Most ingestions leading to acute liver failure exceed 10 g/day (> 150 mg/kg/day). Moderate chronic ingestion, eg, 4 g/day, usually leads to transient mild elevation of liver enzymes in healthy individuals12 but can in rare cases cause acute liver failure.13

Whitcomb and Block14 retrospectively identified 49 patients who presented with acetaminophen-induced hepatotoxicity in 1987 through 1993; 21 (43%) had been taking acetaminophen for therapeutic purposes. All 49 patients took more than the recommended limit of 4 g/day, many of them while fasting and some while using alcohol. Acute liver failure was seen with ingestion of more than 12 g/day—or more than 10 g/day in alcohol users. The authors attributed the increased risk to activation of cytochrome P450 2E1 by alcohol and depletion of glutathione stores by starvation or alcohol abuse.
Advice to patients taking acetaminophen is given in Table 1.
Other drugs and supplements

A number of other drugs and herbal supplements can also cause acute liver failure (Table 2), the most common being antimicrobial and antiepileptic drugs.15 Of the antimicrobials, antitubercular drugs (especially isoniazid) are believed to be the most common causes, followed by trimethoprim-sulfamethoxazole. Phenytoin is the antiepileptic drug most often implicated in acute liver failure.
Statins can also cause acute liver failure, especially when combined with other hepatotoxic agents.16
The herbal supplements and weight-loss agents Hydroxycut and Herbalife have both been reported to cause acute liver failure, with patients presenting with either the hepatocellular or the cholestatic pattern of liver injury.17 The exact chemical in these supplements that causes liver injury has not yet been determined.
The National Institutes of Health maintains a database of cases of liver failure due to medications and supplements at livertox.nih.gov. The database includes the pattern of hepatic injury, mechanism of injury, management, and outcomes.
Viral hepatitis
Hepatitis B virus is the most common viral cause of acute liver failure and is responsible for about 8% of cases.18
Patients with chronic hepatitis B virus infection—as evidenced by positive hepatitis B surface antigen—can develop acute liver failure if the infection is reactivated by the use of immunosuppressive drugs for solid-organ or bone-marrow transplant or medications such as anti-tumor necrosis agents, rituximab, or chemotherapy. These patients should be treated prophylactically with a nucleoside analogue, which should be continued for 6 months after immunosuppressive therapy is completed.
Hepatitis A virus is responsible for about 4% of cases.18
Hepatitis C virus rarely causes acute liver failure, especially in the absence of hepatitis A and hepatitis B.3,19
Hepatitis E virus, which is endemic in areas of Asia and Africa, can cause liver disease in pregnant women and in young adults who have concomitant liver disease from another cause. It tends to cause acute liver failure more frequently in pregnant women than in the rest of the population and carries a mortality rate of more than 20% in this subgroup.
TT (transfusion-transmitted) virus was reported in the 1990s to cause acute liver failure in about 27% of patients in whom no other cause could be found.20
Other rare viral causes of acute liver failure include Epstein-Barr virus, cytomegalovirus, and herpes simplex virus types 1, 2, and 6.
Other causes
Other causes of acute liver failure include ischemic hepatitis, autoimmune hepatitis, Wilson disease, Budd-Chiari syndrome, and HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome.
MANY PATIENTS NEED LIVER TRANSPLANT

Many patients with acute liver failure ultimately require orthotopic liver transplant,21 especially if they present with severe encephalopathy. Other aspects of treatment vary according to the cause of liver failure (Table 3).
SPECIFIC MANAGEMENT
Management of acetaminophen toxicity
If the time of ingestion is known, checking the acetaminophen level can help determine the cause of acute liver failure and also predict the risk of hepatotoxicity, based on the work of Rumack and Matthew.22 Calculators are available, eg, http://reference.medscape.com/calculator/acetaminophen-toxicity.
If a patient presents with acute liver failure several days after ingesting acetaminophen, the level can be in the nontoxic range, however. In this scenario, measuring acetaminophen-protein adducts can help establish acetaminophen toxicity as the cause, as the adducts last longer in the serum and provide 100% sensitivity and specificity.23 While most laboratories can rapidly measure acetaminophen levels, only a few can measure acetaminophen-protein adducts, and thus this test is not used clinically.
Acetylcysteine is the main drug used for acetaminophen toxicity. Ideally, it should be given within 8 hours of acetaminophen ingestion, but giving it later is also useful.1
Acetylcysteine is available in oral and intravenous forms, the latter for patients who have encephalopathy or cannot tolerate oral intake due to repeated episodes of vomiting.24,25 The oral form is much less costly and is thus preferred over intravenous acetylcysteine in patients who can tolerate oral intake. Intravenous acetylcysteine should be given in a loading dose of 150 mg/kg in 5% dextrose over 15 minutes, followed by a maintenance dose of 50 mg/kg over 4 hours and then 100 mg/kg given over 16 hours.1 No dose adjustment is needed in patients who have renal toxicity (acetaminophen can also be toxic to the kidneys).
Most patients with acetaminophen-induced liver failure survive with medical management alone and do not need a liver transplant.3,26 Cirrhosis does not occur in these patients.
Management of viral acute liver failure
When patients present with acute liver failure, it is necessary to look for a viral cause by serologic testing, including hepatitis A virus IgM antibody, hepatitis B surface antigen, and hepatitis B core IgM antibody.
Hepatitis B can become reactivated in immunocompromised patients, and therefore the hepatitis B virus DNA level should be checked. Detection of hepatitis B virus DNA in a patient previously known to have undetectable hepatitis B virus DNA confirms hepatitis B reactivation.
Patients with hepatitis B-induced acute liver failure should be treated with entecavir or tenofovir. Although this treatment may not change the course of acute liver failure or accelerate the recovery, it can prevent reinfection in the transplanted liver if liver transplant becomes indicated.27–29
Herpes simplex virus should be suspected in patients presenting with anicteric hepatitis with fever. Polymerase chain reaction testing for herpes simplex virus should be done,30 and if positive, patients should be given intravenous acyclovir.31 Despite treatment, herpes simplex virus disease is associated with a very poor prognosis without liver transplant.
Autoimmune hepatitis
The autoantibodies usually seen in autoimmune hepatitis are antinuclear antibody, antismooth muscle antibody, and anti-liver-kidney microsomal antibody, and patients need to be tested for them.
The diagnosis of autoimmune hepatitis can be challenging, as these autoimmune markers can be negative in 5% of patients. Liver biopsy becomes essential to establish the diagnosis in that setting.32
Guidelines advise starting prednisone 40 to 60 mg/day and placing the patient on the liver transplant list.1
Wilson disease
Although it is an uncommon cause of liver failure, Wilson disease needs special attention because it has a poor prognosis. The mortality rate in acute liver failure from Wilson disease reaches 100% without liver transplant.
Wilson disease is caused by a genetic defect that allows copper to accumulate in the liver and other organs. However, diagnosing Wilson disease as the cause of acute liver failure can be challenging because elevated serum and urine copper levels are not specific to Wilson disease and can be seen in patients with acute liver failure from any cause. In addition, the ceruloplasmin level is usually normal or high because it is an acute-phase reactant. Accumulation of copper in the liver parenchyma is usually patchy; therefore, qualitative copper staining on random liver biopsy samples provides low diagnostic yield. Quantitative copper on liver biopsy is the gold standard test to establish the diagnosis, but the test is time-consuming. Kayser-Fleischer rings around the iris are considered pathognomic for Wilson disease when seen with acute liver failure, but they are seen in only about 50% of patients.33
A unique feature of acute Wilson disease is that most patients have very high bilirubin levels and low alkaline phosphatase levels. An alkaline phosphatase-to-bilirubin ratio less than 2 in patients with acute liver failure is highly suggestive of Wilson disease.34
Another clue to the diagnosis is that patients with Wilson disease tend to develop Coombs-negative hemolytic anemia, which leads to a disproportionate elevation in aminotransferase levels, with aspartate aminotransferase being higher than alanine aminotransferase.
Once Wilson disease is suspected, the patient should be listed for liver transplant because death is almost certain without it. For patients awaiting liver transplant, the American Association for the Study of Liver Diseases guidelines recommend certain measures to lower the serum copper level such as albumin dialysis, continuous hemofiltration, plasmapheresis, and plasma exchange,1 but the evidence supporting their use is limited.
NONSPECIFIC MANAGEMENT
Acute liver failure can affect a number of organs and systems in addition to the liver (Figure 2).
General considerations
Because their condition can rapidly deteriorate, patients with acute liver failure are best managed in intensive care.
Patients who present to a center that does not have the facilities for liver transplant should be transferred to a transplant center as soon as possible, preferably by air. If the patient may not be able to protect the airway, endotracheal intubation should be performed before transfer.
The major causes of death in patients with acute liver failure are cerebral edema and infection. Gastrointestinal bleeding was a major cause of death in the past, but with prophylactic use of histamine H2 receptor blockers and proton pump inhibitors, the incidence of gastrointestinal bleeding has been significantly reduced.
Although initially used only in patients with acetaminophen-induced liver failure, acetylcysteine has also shown benefit in patients with acute liver failure from other causes. In patients with grade 1 or 2 encephalopathy on a scale of 0 (minimal) to 4 (comatose), the transplant-free survival rate is higher when acetylcysteine is given compared with placebo, but this benefit does not extend to patients with a higher grade of encephalopathy.35
Cerebral edema and intracranial hypertension
Cerebral edema is the leading cause of death in patients with acute liver failure, and it develops in nearly 40% of patients.36
The mechanism by which cerebral edema develops is not well understood. Some have proposed that ammonia is converted to glutamine, which causes cerebral edema either directly by its osmotic effect37,38 or indirectly by decreasing other osmolytes, thereby promoting water retention.39
Cerebral edema leads to intracranial hypertension, which can ultimately cause cerebral herniation and death. Because of the high mortality rate associated with cerebral edema, invasive devices were extensively used in the past to monitor intracranial pressure. However, in light of known complications of these devices, including bleeding,40 and lack of evidence of long-term benefit in terms of mortality rates, their use has come under debate.
Treatments. Many treatments are available for cerebral edema and intracranial hypertension. The first step is to elevate the head of the bed about 30 degrees. In addition, hyponatremia should be corrected, as it can worsen cerebral edema.41 If patients are intubated, maintaining a hypercapneic state is advisable to decrease the intracranial pressure.
Of the two pharmacologic options, mannitol is more often used.42 It is given as a bolus dose of 0.5 to 1 g/kg intravenously if the serum osmolality is less than 320 mOsm/L.1 Given the risk of fluid overload with mannitol, caution must be exercised in patients with renal dysfunction. The other pharmacologic option is 3% hypertonic saline.
Therapeutic hypothermia is a newer treatment for cerebral edema. Lowering the body temperature to 32 to 33°C (89.6 to 91.4°F) using cooling blankets decreases intracranial pressure and cerebral blood flow and improves the cerebral perfusion pressure.43 With this treatment, patients should be closely monitored for side effects of infection, coagulopathy, and cardiac arrythmias.1
l-ornithine l-aspartate was successfully used to prevent brain edema in rats, but in humans, no benefit was seen compared with placebo.44,45 The underlying basis for this experimental treatment is that supplemental ornithine and aspartate should increase glutamate synthesis, which should increase the activity of enzyme glutamine synthetase in skeletal muscles. With the increase in enzyme activity, conversion of ammonia to glutamine should increase, thereby decreasing ammonia circulation and thus decreasing cerebral edema.
Patients with cerebral edema have a high incidence of seizures, but prophylactic antiseizure medications such as phenytoin have not been proven to be beneficial.46
Infection
Nearly 80% of patients with acute liver failure develop an infectious complication, which can be attributed to a state of immunodeficiency.47
The respiratory and urinary tracts are the most common sources of infection.48 In patients with bacteremia, Enterococcus species and coagulase-negative Staphylococcus species49 are the commonly isolated organisms. Also, in patients with acute liver failure, fungal infections account for 30% of all infections.50
Infected patients often develop worsening of their encephalopathy51 without fever or elevated white blood cell count.49,52 Thus, in any patient in whom encephalopathy is worsening, an evaluation must be done to rule out infection. In these patients, systemic inflammatory response syndrome is an independent risk factor for death.53
Despite the high mortality rate with infection, whether using antibiotics prophylactically in acute liver failure is beneficial is controversial.54,55
Gastrointestinal bleeding
The current prevalence of upper gastrointestinal bleeding in acute liver failure patients is about 1.5%.56 Coagulopathy and endotracheal intubation are the main risk factors for upper gastrointestinal bleeding in these patients.57 The most common source of bleeding is stress ulcers in the stomach. The ulcers develop from a combination of factors, including decreased blood flow to the mucosa causing ischemia and hypoperfusion-reperfusion injury.
Pharmacologic inhibition of gastric acid secretion has been shown to reduce upper gastrointestinal bleeding in acute liver failure. A histamine H2 receptor blocker or proton pump inhibitor should be given to prevent gastrointestinal bleeding in patients with acute liver failure.1,58
EXPERIMENTAL TREATMENTS
Artificial liver support systems
Membranes and dialysate solutions have been developed to remove toxic substances that are normally metabolized by the liver. Two of these—the molecular adsorbent recycling system (MARS) and the extracorporeal liver assist device (ELAD)—were developed in the late 1990s. MARS consisted of a highly permeable hollow fiber membrane mixed with albumin, and ELAD consisted of porcine hepatocytes attached to microcarriers in the extracapillary space of the hollow fiber membrane. Both systems allowed for transfer of water-soluble and protein-bound toxins in the blood across the membrane and into the dialysate.59 The clinical benefit offered by these devices is controversial,60–62 thus limiting their use to experimental purposes only.
Hepatocyte transplant
Use of hepatocyte transplant as a bridge to liver transplant was tested in 1970s, first in rats and later in humans.63 By reducing the blood ammonia level and improving cerebral perfusion pressure and cardiac function, replacement of 1% to 2% of the total liver cell mass by transplanted hepatocytes acts as a bridge to orthotopic liver transplant.64,65
PROGNOSIS
Different criteria have been used to identify patients with poor prognosis who may eventually need to undergo liver transplant.
The King’s College criteria system is the most commonly used for prognosis (Table 4).37,66–69 Its main drawback is that it is applicable only in patients with encephalopathy, and when patients reach this stage, their condition often deteriorates rapidly, and they die while awaiting liver transplant.37,66,67
The Model for End-Stage Liver Disease (MELD) score is an alternative to the King’s College criteria. A high MELD score on admission signifies advanced disease, and patients with a high MELD score tend to have a worse prognosis than those with a low score.68
The Acute Physiology and Chronic Health Evaluation (APACHE) II score can also be used, as it is more sensitive than the King’s College criteria.6
The Clichy criteria66,69 can also be used.
Liver biopsy. In addition to helping establish the cause of acute liver failure, liver biopsy can also be used as a prognostic tool. Hepatocellular necrosis greater than 70% on the biopsy predicts death with a specificity of 90% and a sensitivity of 56%.70
Hypophosphatemia has been reported to indicate recovering liver function in patients with acute liver failure.71 As the liver regenerates, its energy requirement increases. To supply the energy, adenosine triphosphate production increases, and phosphorus shifts from the extracellular to the intracellular compartment to meet the need for extra phosphorus during this process. A serum phosphorus level of 2.9 mg/dL or higher appears to indicate a poor prognosis in patients with acute liver failure, as it signifies that adequate hepatocyte regeneration is not occurring.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 342:273–275.
- Ostapowicz G, Fontana RJ, Schiodt FV, et al; US Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 2008; 47:1401–1415.
- Acharya SK, Panda SK, Saxena A, Gupta SD. Acute hepatic failure in India: a perspective from the East. J Gastroenterol Hepatol 2000; 15:473–479.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Patten CJ, Thomas PE, Guy RL, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 1993; 6:511–518.
- Chen W, Koenigs LL, Thompson SJ, et al. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol 1998; 11:295-301.
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187:211–217.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 1983; 71:980–991.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- Schiødt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med 1997; 337:1112–1117.
- Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994; 272:1845–1850.
- Chalasani N, Fontana RJ, Bonkovsky HL, et al; Drug Induced Liver Injury Network (DILIN). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008; 135:1924–1934 e1–4
- Reuben A, Koch DG, Lee WM; Acute Liver Failure Study Group. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology 2010; 52:2065–2076.
- Stevens T, Qadri A, Zein NN. Two patients with acute liver injury associated with use of the herbal weight-loss supplement hydroxycut. Ann Intern Med 2005; 142:477–478.
- Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol 2015; 62(suppl 1):S112–S120.
- Schiodt FV, Davern TJ, Shakil AO, McGuire B, Samuel G, Lee WM. Viral hepatitis-related acute liver failure. Am J Gastroenterol 2003; 98:448–453.
- Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology 1998; 28:839–842.
- Bismuth H, Samuel D, Gugenheim J, et al. Emergency liver transplantation for fulminant hepatitis. Ann Intern Med 1987; 107:337–341.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Davern TJ 2nd, James LP, Hinson JA, et al; Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006; 130:687–694.
- Perry HE, Shannon MW. Efficacy of oral versus intravenous N-acetylcysteine in acetaminophen overdose: results of an open-label, clinical trial. J Pediatr 1998; 132:149–152.
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557–1562.
- Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology 1995; 109:1907–1916.
- Tsang SW, Chan HL, Leung NW, et al. Lamivudine treatment for fulminant hepatic failure due to acute exacerbation of chronic hepatitis B infection. Aliment Pharmacol Ther 2001; 15:1737–1744.
- Yu JW, Sun LJ, Yan BZ, Kang P, Zhao YH. Lamivudine treatment is associated with improved survival in fulminant hepatitis B. Liver Int 2011; 31:499–506.
- Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 2011; 53:774–780.
- Pinna AD, Rakela J, Demetris AJ, Fung JJ. Five cases of fulminant hepatitis due to herpes simplex virus in adults. Dig Dis Sci 2002; 47:750–754.
- Farr RW, Short S, Weissman D. Fulminant hepatitis during herpes simplex virus infection in apparently immunocompetent adults: report of two cases and review of the literature. Clin Infect Dis 1997; 24:1191–1194.
- Czaja AJ, Freese DK; American Association for the Study of Liver Disease. Diagnosis and treatment of autoimmune hepatitis. Hepatology 2002; 36:479–497.
- Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003; 37:1475–1492.
- Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991; 100:1129–1134.
- Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009; 137:856–864.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology 1992; 15:449–453.
- Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 1994; 107:1475–1480.
- Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet 1993; 341:157–158.
- Cordoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol 1998; 29:589–594.
- Canalese J, Gimson AE, Davis C, Mellon PJ, Davis M, Williams R. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut 1982; 23:625–629.
- Jalan R, SW OD, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 1999; 354:1164–1168.
- Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 1999; 30:636–640.
- Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology 2009; 136:2159–2168.
- Bhatia V, Batra Y, Acharya SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol 2004; 41:89–96.
- Canalese J, Gove CD, Gimson AE, Wilkinson SP, Wardle EN, Williams R. Reticuloendothelial system and hepatocytic function in fulminant hepatic failure. Gut 1982; 23:265–269.
- Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology 1990; 11:49–53.
- Rolando N, Wade JJ, Stangou A, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg 1996; 2:8–13.
- Rolando N, Harvey F, Brahm J, et al. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol 1991; 12:1–9.
- Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology 2003; 125:755–764.
- Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis 1996; 16:389–402.
- Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32:734–739.
- Rolando N, Gimson A, Wade J, Philpott- Howard J, Casewell M, Williams R. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology 1993; 17:196–201.
- Karvellas CJ, Cavazos J, Battenhouse H, et al; US Acute Liver Failure Study Group. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol 2014; 12:1942–1949.
- Acharya SK, Dasarathy S, Kumer TL, et al. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology 1996; 23:1148–1155.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- MacDougall BR, Williams R. H2-receptor antagonist in the prevention of acute gastrointestinal hemorrhage in fulminant hepatic failure: a controlled trial. Gastroenterology 1978; 74:464–465.
- Stange J, Mitzner SR, Risler T, et al. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs 1999; 23:319–330.
- Vaid A, Chewich H, Balk EM, Jaber BL. Molecular adsorbent recirculating system as artificial support therapy for liver failure: a meta-analysis. ASAIO J 2012; 58:51–59.
- Khuroo MS, Khuroo MS, Farahat KL. Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: a meta-analysis. Liver Transpl 2004; 10:1099–1106.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Sommer BG, Sutherland DE, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation for treatment of D-galactosamine-induced acute liver failure in rats. Transplant Proc 1979; 11:578–584.
- Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology 1988; 8:1006–1009.
- Strom SC, Fisher RA, Thompson MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 1997; 63:559–569.
- Pauwels A, Mostefa-Kara N, Florent C, Levy VG. Emergency liver transplantation for acute liver failure. Evaluation of London and Clichy criteria. J Hepatol 1993; 17:124–127.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King's criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Larsen FS. MELD score as a predictor of liver failure and death in patients with acetaminophen-induced liver injury. Hepatology 2007; 45:789–796.
- Bernuau J, Goudeau A, Poynard T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology 1986; 6:648–651.
- Donaldson BW, Gopinath R, Wanless IR, et al. The role of transjugular liver biopsy in fulminant liver failure: relation to other prognostic indicators. Hepatology 1993; 18:1370–1376.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
When the liver fails, it usually fails gradually. The sudden (acute) onset of liver failure, while less common, demands prompt management, with transfer to an intensive care unit, specific treatment depending on the cause, and consideration of liver transplant, without which the mortality rate is high.
This article reviews the definition, epidemiology, etiology, and management of acute liver failure.
DEFINITIONS
Acute liver failure is defined as a syndrome of acute hepatitis with evidence of abnormal coagulation (eg, an international normalized ratio > 1.5) complicated by the development of mental alteration (encephalopathy) within 26 weeks of the onset of illness in a patient without a history of liver disease.1 In general, patients have no evidence of underlying chronic liver disease, but there are exceptions; patients with Wilson disease, vertically acquired hepatitis B virus infection, or autoimmune hepatitis can present with acute liver failure superimposed on chronic liver disease or even cirrhosis.
The term acute liver failure has replaced older terms such as fulminant hepatic failure, hyperacute liver failure, and subacute liver failure, which were used for prognostic purposes. Patients with hyperacute liver failure (defined as development of encephalopathy within 7 days of onset of illness) generally have a good prognosis with medical management, whereas those with subacute liver failure (defined as development of encephalopathy within 5 to 26 weeks of onset of illness) have a poor prognosis without liver transplant.2,3
NEARLY 2,000 CASES A YEAR
There are nearly 2,000 cases of acute liver failure each year in the United States, and it accounts for 6% of all deaths due to liver disease.4 It is more common in women than in men, and more common in white people than in other races. The peak incidence is at a fairly young age, ie, 35 to 45 years.
CAUSES
The most common cause of acute liver failure in the United States and other Western countries is acetaminophen toxicity, followed by viral hepatitis. In contrast, viral hepatitis is the most common cause in developing countries.5
Acetaminophen toxicity
Patients with acetaminophen-induced liver failure tend to be younger than other patients with acute liver failure.1 Nearly half of them present after intentionally taking a single large dose, while the rest present with unintentional toxicity while taking acetaminophen for pain relief on a long-term basis and ingesting more than the recommended dose.6
After ingestion, 52% to 57% of acetaminophen is converted to glucuronide conjugates, and 30% to 44% is converted to sulfate conjugates. These compounds are nontoxic, water-soluble, and rapidly excreted in the urine.
However, about 5% to 10% of ingested acetaminophen is shunted to the cytochrome P450 system. P450 2E1 is the main isoenzyme involved in acetaminophen metabolism, but 1A2, 3A4, and 2A6 also contribute.7,8 P450 2E1 is the same isoenzyme responsible for ethanol metabolism and is inducible. Thus, regular alcohol consumption can increase P450 2E1 activity, setting the stage under certain circumstances for increased acetaminophen metabolism through this pathway.
Metabolism of acetaminophen through the cytochrome P450 pathway results in production of N-acetyl-p-benzoquinone imine (NAPQI), the compound that damages the liver. NAPQI is rendered nontoxic by binding to glutathione, forming NAPQI-glutathione adducts. Glutathione capacity is limited, however. With too much acetaminophen, glutathione becomes depleted and NAPQI accumulates, binds with proteins to form adducts, and leads to necrosis of hepatocytes (Figure 1).9,10
Acetylcysteine, used in treating acetaminophen toxicity, is a substrate for glutathione synthesis and ultimately increases the amount of glutathione available to bind NAPQI and prevent damage to hepatocytes.11
Acetaminophen is a dose-related toxin. Most ingestions leading to acute liver failure exceed 10 g/day (> 150 mg/kg/day). Moderate chronic ingestion, eg, 4 g/day, usually leads to transient mild elevation of liver enzymes in healthy individuals12 but can in rare cases cause acute liver failure.13
Whitcomb and Block14 retrospectively identified 49 patients who presented with acetaminophen-induced hepatotoxicity in 1987 through 1993; 21 (43%) had been taking acetaminophen for therapeutic purposes. All 49 patients took more than the recommended limit of 4 g/day, many of them while fasting and some while using alcohol. Acute liver failure was seen with ingestion of more than 12 g/day—or more than 10 g/day in alcohol users. The authors attributed the increased risk to activation of cytochrome P450 2E1 by alcohol and depletion of glutathione stores by starvation or alcohol abuse.
Advice to patients taking acetaminophen is given in Table 1.
Other drugs and supplements
A number of other drugs and herbal supplements can also cause acute liver failure (Table 2), the most common being antimicrobial and antiepileptic drugs.15 Of the antimicrobials, antitubercular drugs (especially isoniazid) are believed to be the most common causes, followed by trimethoprim-sulfamethoxazole. Phenytoin is the antiepileptic drug most often implicated in acute liver failure.
Statins can also cause acute liver failure, especially when combined with other hepatotoxic agents.16
The herbal supplements and weight-loss agents Hydroxycut and Herbalife have both been reported to cause acute liver failure, with patients presenting with either the hepatocellular or the cholestatic pattern of liver injury.17 The exact chemical in these supplements that causes liver injury has not yet been determined.
The National Institutes of Health maintains a database of cases of liver failure due to medications and supplements at livertox.nih.gov. The database includes the pattern of hepatic injury, mechanism of injury, management, and outcomes.
Viral hepatitis
Hepatitis B virus is the most common viral cause of acute liver failure and is responsible for about 8% of cases.18
Patients with chronic hepatitis B virus infection—as evidenced by positive hepatitis B surface antigen—can develop acute liver failure if the infection is reactivated by the use of immunosuppressive drugs for solid-organ or bone-marrow transplant or medications such as anti-tumor necrosis agents, rituximab, or chemotherapy. These patients should be treated prophylactically with a nucleoside analogue, which should be continued for 6 months after immunosuppressive therapy is completed.
Hepatitis A virus is responsible for about 4% of cases.18
Hepatitis C virus rarely causes acute liver failure, especially in the absence of hepatitis A and hepatitis B.3,19
Hepatitis E virus, which is endemic in areas of Asia and Africa, can cause liver disease in pregnant women and in young adults who have concomitant liver disease from another cause. It tends to cause acute liver failure more frequently in pregnant women than in the rest of the population and carries a mortality rate of more than 20% in this subgroup.
TT (transfusion-transmitted) virus was reported in the 1990s to cause acute liver failure in about 27% of patients in whom no other cause could be found.20
Other rare viral causes of acute liver failure include Epstein-Barr virus, cytomegalovirus, and herpes simplex virus types 1, 2, and 6.
Other causes
Other causes of acute liver failure include ischemic hepatitis, autoimmune hepatitis, Wilson disease, Budd-Chiari syndrome, and HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome.
MANY PATIENTS NEED LIVER TRANSPLANT
Many patients with acute liver failure ultimately require orthotopic liver transplant,21 especially if they present with severe encephalopathy. Other aspects of treatment vary according to the cause of liver failure (Table 3).
SPECIFIC MANAGEMENT
Management of acetaminophen toxicity
If the time of ingestion is known, checking the acetaminophen level can help determine the cause of acute liver failure and also predict the risk of hepatotoxicity, based on the work of Rumack and Matthew.22 Calculators are available, eg, http://reference.medscape.com/calculator/acetaminophen-toxicity.
If a patient presents with acute liver failure several days after ingesting acetaminophen, the level can be in the nontoxic range, however. In this scenario, measuring acetaminophen-protein adducts can help establish acetaminophen toxicity as the cause, as the adducts last longer in the serum and provide 100% sensitivity and specificity.23 While most laboratories can rapidly measure acetaminophen levels, only a few can measure acetaminophen-protein adducts, and thus this test is not used clinically.
Acetylcysteine is the main drug used for acetaminophen toxicity. Ideally, it should be given within 8 hours of acetaminophen ingestion, but giving it later is also useful.1
Acetylcysteine is available in oral and intravenous forms, the latter for patients who have encephalopathy or cannot tolerate oral intake due to repeated episodes of vomiting.24,25 The oral form is much less costly and is thus preferred over intravenous acetylcysteine in patients who can tolerate oral intake. Intravenous acetylcysteine should be given in a loading dose of 150 mg/kg in 5% dextrose over 15 minutes, followed by a maintenance dose of 50 mg/kg over 4 hours and then 100 mg/kg given over 16 hours.1 No dose adjustment is needed in patients who have renal toxicity (acetaminophen can also be toxic to the kidneys).
Most patients with acetaminophen-induced liver failure survive with medical management alone and do not need a liver transplant.3,26 Cirrhosis does not occur in these patients.
Management of viral acute liver failure
When patients present with acute liver failure, it is necessary to look for a viral cause by serologic testing, including hepatitis A virus IgM antibody, hepatitis B surface antigen, and hepatitis B core IgM antibody.
Hepatitis B can become reactivated in immunocompromised patients, and therefore the hepatitis B virus DNA level should be checked. Detection of hepatitis B virus DNA in a patient previously known to have undetectable hepatitis B virus DNA confirms hepatitis B reactivation.
Patients with hepatitis B-induced acute liver failure should be treated with entecavir or tenofovir. Although this treatment may not change the course of acute liver failure or accelerate the recovery, it can prevent reinfection in the transplanted liver if liver transplant becomes indicated.27–29
Herpes simplex virus should be suspected in patients presenting with anicteric hepatitis with fever. Polymerase chain reaction testing for herpes simplex virus should be done,30 and if positive, patients should be given intravenous acyclovir.31 Despite treatment, herpes simplex virus disease is associated with a very poor prognosis without liver transplant.
Autoimmune hepatitis
The autoantibodies usually seen in autoimmune hepatitis are antinuclear antibody, antismooth muscle antibody, and anti-liver-kidney microsomal antibody, and patients need to be tested for them.
The diagnosis of autoimmune hepatitis can be challenging, as these autoimmune markers can be negative in 5% of patients. Liver biopsy becomes essential to establish the diagnosis in that setting.32
Guidelines advise starting prednisone 40 to 60 mg/day and placing the patient on the liver transplant list.1
Wilson disease
Although it is an uncommon cause of liver failure, Wilson disease needs special attention because it has a poor prognosis. The mortality rate in acute liver failure from Wilson disease reaches 100% without liver transplant.
Wilson disease is caused by a genetic defect that allows copper to accumulate in the liver and other organs. However, diagnosing Wilson disease as the cause of acute liver failure can be challenging because elevated serum and urine copper levels are not specific to Wilson disease and can be seen in patients with acute liver failure from any cause. In addition, the ceruloplasmin level is usually normal or high because it is an acute-phase reactant. Accumulation of copper in the liver parenchyma is usually patchy; therefore, qualitative copper staining on random liver biopsy samples provides low diagnostic yield. Quantitative copper on liver biopsy is the gold standard test to establish the diagnosis, but the test is time-consuming. Kayser-Fleischer rings around the iris are considered pathognomic for Wilson disease when seen with acute liver failure, but they are seen in only about 50% of patients.33
A unique feature of acute Wilson disease is that most patients have very high bilirubin levels and low alkaline phosphatase levels. An alkaline phosphatase-to-bilirubin ratio less than 2 in patients with acute liver failure is highly suggestive of Wilson disease.34
Another clue to the diagnosis is that patients with Wilson disease tend to develop Coombs-negative hemolytic anemia, which leads to a disproportionate elevation in aminotransferase levels, with aspartate aminotransferase being higher than alanine aminotransferase.
Once Wilson disease is suspected, the patient should be listed for liver transplant because death is almost certain without it. For patients awaiting liver transplant, the American Association for the Study of Liver Diseases guidelines recommend certain measures to lower the serum copper level such as albumin dialysis, continuous hemofiltration, plasmapheresis, and plasma exchange,1 but the evidence supporting their use is limited.
NONSPECIFIC MANAGEMENT
Acute liver failure can affect a number of organs and systems in addition to the liver (Figure 2).
General considerations
Because their condition can rapidly deteriorate, patients with acute liver failure are best managed in intensive care.
Patients who present to a center that does not have the facilities for liver transplant should be transferred to a transplant center as soon as possible, preferably by air. If the patient may not be able to protect the airway, endotracheal intubation should be performed before transfer.
The major causes of death in patients with acute liver failure are cerebral edema and infection. Gastrointestinal bleeding was a major cause of death in the past, but with prophylactic use of histamine H2 receptor blockers and proton pump inhibitors, the incidence of gastrointestinal bleeding has been significantly reduced.
Although initially used only in patients with acetaminophen-induced liver failure, acetylcysteine has also shown benefit in patients with acute liver failure from other causes. In patients with grade 1 or 2 encephalopathy on a scale of 0 (minimal) to 4 (comatose), the transplant-free survival rate is higher when acetylcysteine is given compared with placebo, but this benefit does not extend to patients with a higher grade of encephalopathy.35
Cerebral edema and intracranial hypertension
Cerebral edema is the leading cause of death in patients with acute liver failure, and it develops in nearly 40% of patients.36
The mechanism by which cerebral edema develops is not well understood. Some have proposed that ammonia is converted to glutamine, which causes cerebral edema either directly by its osmotic effect37,38 or indirectly by decreasing other osmolytes, thereby promoting water retention.39
Cerebral edema leads to intracranial hypertension, which can ultimately cause cerebral herniation and death. Because of the high mortality rate associated with cerebral edema, invasive devices were extensively used in the past to monitor intracranial pressure. However, in light of known complications of these devices, including bleeding,40 and lack of evidence of long-term benefit in terms of mortality rates, their use has come under debate.
Treatments. Many treatments are available for cerebral edema and intracranial hypertension. The first step is to elevate the head of the bed about 30 degrees. In addition, hyponatremia should be corrected, as it can worsen cerebral edema.41 If patients are intubated, maintaining a hypercapneic state is advisable to decrease the intracranial pressure.
Of the two pharmacologic options, mannitol is more often used.42 It is given as a bolus dose of 0.5 to 1 g/kg intravenously if the serum osmolality is less than 320 mOsm/L.1 Given the risk of fluid overload with mannitol, caution must be exercised in patients with renal dysfunction. The other pharmacologic option is 3% hypertonic saline.
Therapeutic hypothermia is a newer treatment for cerebral edema. Lowering the body temperature to 32 to 33°C (89.6 to 91.4°F) using cooling blankets decreases intracranial pressure and cerebral blood flow and improves the cerebral perfusion pressure.43 With this treatment, patients should be closely monitored for side effects of infection, coagulopathy, and cardiac arrythmias.1
l-ornithine l-aspartate was successfully used to prevent brain edema in rats, but in humans, no benefit was seen compared with placebo.44,45 The underlying basis for this experimental treatment is that supplemental ornithine and aspartate should increase glutamate synthesis, which should increase the activity of enzyme glutamine synthetase in skeletal muscles. With the increase in enzyme activity, conversion of ammonia to glutamine should increase, thereby decreasing ammonia circulation and thus decreasing cerebral edema.
Patients with cerebral edema have a high incidence of seizures, but prophylactic antiseizure medications such as phenytoin have not been proven to be beneficial.46
Infection
Nearly 80% of patients with acute liver failure develop an infectious complication, which can be attributed to a state of immunodeficiency.47
The respiratory and urinary tracts are the most common sources of infection.48 In patients with bacteremia, Enterococcus species and coagulase-negative Staphylococcus species49 are the commonly isolated organisms. Also, in patients with acute liver failure, fungal infections account for 30% of all infections.50
Infected patients often develop worsening of their encephalopathy51 without fever or elevated white blood cell count.49,52 Thus, in any patient in whom encephalopathy is worsening, an evaluation must be done to rule out infection. In these patients, systemic inflammatory response syndrome is an independent risk factor for death.53
Despite the high mortality rate with infection, whether using antibiotics prophylactically in acute liver failure is beneficial is controversial.54,55
Gastrointestinal bleeding
The current prevalence of upper gastrointestinal bleeding in acute liver failure patients is about 1.5%.56 Coagulopathy and endotracheal intubation are the main risk factors for upper gastrointestinal bleeding in these patients.57 The most common source of bleeding is stress ulcers in the stomach. The ulcers develop from a combination of factors, including decreased blood flow to the mucosa causing ischemia and hypoperfusion-reperfusion injury.
Pharmacologic inhibition of gastric acid secretion has been shown to reduce upper gastrointestinal bleeding in acute liver failure. A histamine H2 receptor blocker or proton pump inhibitor should be given to prevent gastrointestinal bleeding in patients with acute liver failure.1,58
EXPERIMENTAL TREATMENTS
Artificial liver support systems
Membranes and dialysate solutions have been developed to remove toxic substances that are normally metabolized by the liver. Two of these—the molecular adsorbent recycling system (MARS) and the extracorporeal liver assist device (ELAD)—were developed in the late 1990s. MARS consisted of a highly permeable hollow fiber membrane mixed with albumin, and ELAD consisted of porcine hepatocytes attached to microcarriers in the extracapillary space of the hollow fiber membrane. Both systems allowed for transfer of water-soluble and protein-bound toxins in the blood across the membrane and into the dialysate.59 The clinical benefit offered by these devices is controversial,60–62 thus limiting their use to experimental purposes only.
Hepatocyte transplant
Use of hepatocyte transplant as a bridge to liver transplant was tested in 1970s, first in rats and later in humans.63 By reducing the blood ammonia level and improving cerebral perfusion pressure and cardiac function, replacement of 1% to 2% of the total liver cell mass by transplanted hepatocytes acts as a bridge to orthotopic liver transplant.64,65
PROGNOSIS
Different criteria have been used to identify patients with poor prognosis who may eventually need to undergo liver transplant.
The King’s College criteria system is the most commonly used for prognosis (Table 4).37,66–69 Its main drawback is that it is applicable only in patients with encephalopathy, and when patients reach this stage, their condition often deteriorates rapidly, and they die while awaiting liver transplant.37,66,67
The Model for End-Stage Liver Disease (MELD) score is an alternative to the King’s College criteria. A high MELD score on admission signifies advanced disease, and patients with a high MELD score tend to have a worse prognosis than those with a low score.68
The Acute Physiology and Chronic Health Evaluation (APACHE) II score can also be used, as it is more sensitive than the King’s College criteria.6
The Clichy criteria66,69 can also be used.
Liver biopsy. In addition to helping establish the cause of acute liver failure, liver biopsy can also be used as a prognostic tool. Hepatocellular necrosis greater than 70% on the biopsy predicts death with a specificity of 90% and a sensitivity of 56%.70
Hypophosphatemia has been reported to indicate recovering liver function in patients with acute liver failure.71 As the liver regenerates, its energy requirement increases. To supply the energy, adenosine triphosphate production increases, and phosphorus shifts from the extracellular to the intracellular compartment to meet the need for extra phosphorus during this process. A serum phosphorus level of 2.9 mg/dL or higher appears to indicate a poor prognosis in patients with acute liver failure, as it signifies that adequate hepatocyte regeneration is not occurring.
When the liver fails, it usually fails gradually. The sudden (acute) onset of liver failure, while less common, demands prompt management, with transfer to an intensive care unit, specific treatment depending on the cause, and consideration of liver transplant, without which the mortality rate is high.
This article reviews the definition, epidemiology, etiology, and management of acute liver failure.
DEFINITIONS
Acute liver failure is defined as a syndrome of acute hepatitis with evidence of abnormal coagulation (eg, an international normalized ratio > 1.5) complicated by the development of mental alteration (encephalopathy) within 26 weeks of the onset of illness in a patient without a history of liver disease.1 In general, patients have no evidence of underlying chronic liver disease, but there are exceptions; patients with Wilson disease, vertically acquired hepatitis B virus infection, or autoimmune hepatitis can present with acute liver failure superimposed on chronic liver disease or even cirrhosis.
The term acute liver failure has replaced older terms such as fulminant hepatic failure, hyperacute liver failure, and subacute liver failure, which were used for prognostic purposes. Patients with hyperacute liver failure (defined as development of encephalopathy within 7 days of onset of illness) generally have a good prognosis with medical management, whereas those with subacute liver failure (defined as development of encephalopathy within 5 to 26 weeks of onset of illness) have a poor prognosis without liver transplant.2,3
NEARLY 2,000 CASES A YEAR
There are nearly 2,000 cases of acute liver failure each year in the United States, and it accounts for 6% of all deaths due to liver disease.4 It is more common in women than in men, and more common in white people than in other races. The peak incidence is at a fairly young age, ie, 35 to 45 years.
CAUSES
The most common cause of acute liver failure in the United States and other Western countries is acetaminophen toxicity, followed by viral hepatitis. In contrast, viral hepatitis is the most common cause in developing countries.5
Acetaminophen toxicity
Patients with acetaminophen-induced liver failure tend to be younger than other patients with acute liver failure.1 Nearly half of them present after intentionally taking a single large dose, while the rest present with unintentional toxicity while taking acetaminophen for pain relief on a long-term basis and ingesting more than the recommended dose.6
After ingestion, 52% to 57% of acetaminophen is converted to glucuronide conjugates, and 30% to 44% is converted to sulfate conjugates. These compounds are nontoxic, water-soluble, and rapidly excreted in the urine.
However, about 5% to 10% of ingested acetaminophen is shunted to the cytochrome P450 system. P450 2E1 is the main isoenzyme involved in acetaminophen metabolism, but 1A2, 3A4, and 2A6 also contribute.7,8 P450 2E1 is the same isoenzyme responsible for ethanol metabolism and is inducible. Thus, regular alcohol consumption can increase P450 2E1 activity, setting the stage under certain circumstances for increased acetaminophen metabolism through this pathway.
Metabolism of acetaminophen through the cytochrome P450 pathway results in production of N-acetyl-p-benzoquinone imine (NAPQI), the compound that damages the liver. NAPQI is rendered nontoxic by binding to glutathione, forming NAPQI-glutathione adducts. Glutathione capacity is limited, however. With too much acetaminophen, glutathione becomes depleted and NAPQI accumulates, binds with proteins to form adducts, and leads to necrosis of hepatocytes (Figure 1).9,10
Acetylcysteine, used in treating acetaminophen toxicity, is a substrate for glutathione synthesis and ultimately increases the amount of glutathione available to bind NAPQI and prevent damage to hepatocytes.11
Acetaminophen is a dose-related toxin. Most ingestions leading to acute liver failure exceed 10 g/day (> 150 mg/kg/day). Moderate chronic ingestion, eg, 4 g/day, usually leads to transient mild elevation of liver enzymes in healthy individuals12 but can in rare cases cause acute liver failure.13
Whitcomb and Block14 retrospectively identified 49 patients who presented with acetaminophen-induced hepatotoxicity in 1987 through 1993; 21 (43%) had been taking acetaminophen for therapeutic purposes. All 49 patients took more than the recommended limit of 4 g/day, many of them while fasting and some while using alcohol. Acute liver failure was seen with ingestion of more than 12 g/day—or more than 10 g/day in alcohol users. The authors attributed the increased risk to activation of cytochrome P450 2E1 by alcohol and depletion of glutathione stores by starvation or alcohol abuse.
Advice to patients taking acetaminophen is given in Table 1.
Other drugs and supplements
A number of other drugs and herbal supplements can also cause acute liver failure (Table 2), the most common being antimicrobial and antiepileptic drugs.15 Of the antimicrobials, antitubercular drugs (especially isoniazid) are believed to be the most common causes, followed by trimethoprim-sulfamethoxazole. Phenytoin is the antiepileptic drug most often implicated in acute liver failure.
Statins can also cause acute liver failure, especially when combined with other hepatotoxic agents.16
The herbal supplements and weight-loss agents Hydroxycut and Herbalife have both been reported to cause acute liver failure, with patients presenting with either the hepatocellular or the cholestatic pattern of liver injury.17 The exact chemical in these supplements that causes liver injury has not yet been determined.
The National Institutes of Health maintains a database of cases of liver failure due to medications and supplements at livertox.nih.gov. The database includes the pattern of hepatic injury, mechanism of injury, management, and outcomes.
Viral hepatitis
Hepatitis B virus is the most common viral cause of acute liver failure and is responsible for about 8% of cases.18
Patients with chronic hepatitis B virus infection—as evidenced by positive hepatitis B surface antigen—can develop acute liver failure if the infection is reactivated by the use of immunosuppressive drugs for solid-organ or bone-marrow transplant or medications such as anti-tumor necrosis agents, rituximab, or chemotherapy. These patients should be treated prophylactically with a nucleoside analogue, which should be continued for 6 months after immunosuppressive therapy is completed.
Hepatitis A virus is responsible for about 4% of cases.18
Hepatitis C virus rarely causes acute liver failure, especially in the absence of hepatitis A and hepatitis B.3,19
Hepatitis E virus, which is endemic in areas of Asia and Africa, can cause liver disease in pregnant women and in young adults who have concomitant liver disease from another cause. It tends to cause acute liver failure more frequently in pregnant women than in the rest of the population and carries a mortality rate of more than 20% in this subgroup.
TT (transfusion-transmitted) virus was reported in the 1990s to cause acute liver failure in about 27% of patients in whom no other cause could be found.20
Other rare viral causes of acute liver failure include Epstein-Barr virus, cytomegalovirus, and herpes simplex virus types 1, 2, and 6.
Other causes
Other causes of acute liver failure include ischemic hepatitis, autoimmune hepatitis, Wilson disease, Budd-Chiari syndrome, and HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome.
MANY PATIENTS NEED LIVER TRANSPLANT
Many patients with acute liver failure ultimately require orthotopic liver transplant,21 especially if they present with severe encephalopathy. Other aspects of treatment vary according to the cause of liver failure (Table 3).
SPECIFIC MANAGEMENT
Management of acetaminophen toxicity
If the time of ingestion is known, checking the acetaminophen level can help determine the cause of acute liver failure and also predict the risk of hepatotoxicity, based on the work of Rumack and Matthew.22 Calculators are available, eg, http://reference.medscape.com/calculator/acetaminophen-toxicity.
If a patient presents with acute liver failure several days after ingesting acetaminophen, the level can be in the nontoxic range, however. In this scenario, measuring acetaminophen-protein adducts can help establish acetaminophen toxicity as the cause, as the adducts last longer in the serum and provide 100% sensitivity and specificity.23 While most laboratories can rapidly measure acetaminophen levels, only a few can measure acetaminophen-protein adducts, and thus this test is not used clinically.
Acetylcysteine is the main drug used for acetaminophen toxicity. Ideally, it should be given within 8 hours of acetaminophen ingestion, but giving it later is also useful.1
Acetylcysteine is available in oral and intravenous forms, the latter for patients who have encephalopathy or cannot tolerate oral intake due to repeated episodes of vomiting.24,25 The oral form is much less costly and is thus preferred over intravenous acetylcysteine in patients who can tolerate oral intake. Intravenous acetylcysteine should be given in a loading dose of 150 mg/kg in 5% dextrose over 15 minutes, followed by a maintenance dose of 50 mg/kg over 4 hours and then 100 mg/kg given over 16 hours.1 No dose adjustment is needed in patients who have renal toxicity (acetaminophen can also be toxic to the kidneys).
Most patients with acetaminophen-induced liver failure survive with medical management alone and do not need a liver transplant.3,26 Cirrhosis does not occur in these patients.
Management of viral acute liver failure
When patients present with acute liver failure, it is necessary to look for a viral cause by serologic testing, including hepatitis A virus IgM antibody, hepatitis B surface antigen, and hepatitis B core IgM antibody.
Hepatitis B can become reactivated in immunocompromised patients, and therefore the hepatitis B virus DNA level should be checked. Detection of hepatitis B virus DNA in a patient previously known to have undetectable hepatitis B virus DNA confirms hepatitis B reactivation.
Patients with hepatitis B-induced acute liver failure should be treated with entecavir or tenofovir. Although this treatment may not change the course of acute liver failure or accelerate the recovery, it can prevent reinfection in the transplanted liver if liver transplant becomes indicated.27–29
Herpes simplex virus should be suspected in patients presenting with anicteric hepatitis with fever. Polymerase chain reaction testing for herpes simplex virus should be done,30 and if positive, patients should be given intravenous acyclovir.31 Despite treatment, herpes simplex virus disease is associated with a very poor prognosis without liver transplant.
Autoimmune hepatitis
The autoantibodies usually seen in autoimmune hepatitis are antinuclear antibody, antismooth muscle antibody, and anti-liver-kidney microsomal antibody, and patients need to be tested for them.
The diagnosis of autoimmune hepatitis can be challenging, as these autoimmune markers can be negative in 5% of patients. Liver biopsy becomes essential to establish the diagnosis in that setting.32
Guidelines advise starting prednisone 40 to 60 mg/day and placing the patient on the liver transplant list.1
Wilson disease
Although it is an uncommon cause of liver failure, Wilson disease needs special attention because it has a poor prognosis. The mortality rate in acute liver failure from Wilson disease reaches 100% without liver transplant.
Wilson disease is caused by a genetic defect that allows copper to accumulate in the liver and other organs. However, diagnosing Wilson disease as the cause of acute liver failure can be challenging because elevated serum and urine copper levels are not specific to Wilson disease and can be seen in patients with acute liver failure from any cause. In addition, the ceruloplasmin level is usually normal or high because it is an acute-phase reactant. Accumulation of copper in the liver parenchyma is usually patchy; therefore, qualitative copper staining on random liver biopsy samples provides low diagnostic yield. Quantitative copper on liver biopsy is the gold standard test to establish the diagnosis, but the test is time-consuming. Kayser-Fleischer rings around the iris are considered pathognomic for Wilson disease when seen with acute liver failure, but they are seen in only about 50% of patients.33
A unique feature of acute Wilson disease is that most patients have very high bilirubin levels and low alkaline phosphatase levels. An alkaline phosphatase-to-bilirubin ratio less than 2 in patients with acute liver failure is highly suggestive of Wilson disease.34
Another clue to the diagnosis is that patients with Wilson disease tend to develop Coombs-negative hemolytic anemia, which leads to a disproportionate elevation in aminotransferase levels, with aspartate aminotransferase being higher than alanine aminotransferase.
Once Wilson disease is suspected, the patient should be listed for liver transplant because death is almost certain without it. For patients awaiting liver transplant, the American Association for the Study of Liver Diseases guidelines recommend certain measures to lower the serum copper level such as albumin dialysis, continuous hemofiltration, plasmapheresis, and plasma exchange,1 but the evidence supporting their use is limited.
NONSPECIFIC MANAGEMENT
Acute liver failure can affect a number of organs and systems in addition to the liver (Figure 2).
General considerations
Because their condition can rapidly deteriorate, patients with acute liver failure are best managed in intensive care.
Patients who present to a center that does not have the facilities for liver transplant should be transferred to a transplant center as soon as possible, preferably by air. If the patient may not be able to protect the airway, endotracheal intubation should be performed before transfer.
The major causes of death in patients with acute liver failure are cerebral edema and infection. Gastrointestinal bleeding was a major cause of death in the past, but with prophylactic use of histamine H2 receptor blockers and proton pump inhibitors, the incidence of gastrointestinal bleeding has been significantly reduced.
Although initially used only in patients with acetaminophen-induced liver failure, acetylcysteine has also shown benefit in patients with acute liver failure from other causes. In patients with grade 1 or 2 encephalopathy on a scale of 0 (minimal) to 4 (comatose), the transplant-free survival rate is higher when acetylcysteine is given compared with placebo, but this benefit does not extend to patients with a higher grade of encephalopathy.35
Cerebral edema and intracranial hypertension
Cerebral edema is the leading cause of death in patients with acute liver failure, and it develops in nearly 40% of patients.36
The mechanism by which cerebral edema develops is not well understood. Some have proposed that ammonia is converted to glutamine, which causes cerebral edema either directly by its osmotic effect37,38 or indirectly by decreasing other osmolytes, thereby promoting water retention.39
Cerebral edema leads to intracranial hypertension, which can ultimately cause cerebral herniation and death. Because of the high mortality rate associated with cerebral edema, invasive devices were extensively used in the past to monitor intracranial pressure. However, in light of known complications of these devices, including bleeding,40 and lack of evidence of long-term benefit in terms of mortality rates, their use has come under debate.
Treatments. Many treatments are available for cerebral edema and intracranial hypertension. The first step is to elevate the head of the bed about 30 degrees. In addition, hyponatremia should be corrected, as it can worsen cerebral edema.41 If patients are intubated, maintaining a hypercapneic state is advisable to decrease the intracranial pressure.
Of the two pharmacologic options, mannitol is more often used.42 It is given as a bolus dose of 0.5 to 1 g/kg intravenously if the serum osmolality is less than 320 mOsm/L.1 Given the risk of fluid overload with mannitol, caution must be exercised in patients with renal dysfunction. The other pharmacologic option is 3% hypertonic saline.
Therapeutic hypothermia is a newer treatment for cerebral edema. Lowering the body temperature to 32 to 33°C (89.6 to 91.4°F) using cooling blankets decreases intracranial pressure and cerebral blood flow and improves the cerebral perfusion pressure.43 With this treatment, patients should be closely monitored for side effects of infection, coagulopathy, and cardiac arrythmias.1
l-ornithine l-aspartate was successfully used to prevent brain edema in rats, but in humans, no benefit was seen compared with placebo.44,45 The underlying basis for this experimental treatment is that supplemental ornithine and aspartate should increase glutamate synthesis, which should increase the activity of enzyme glutamine synthetase in skeletal muscles. With the increase in enzyme activity, conversion of ammonia to glutamine should increase, thereby decreasing ammonia circulation and thus decreasing cerebral edema.
Patients with cerebral edema have a high incidence of seizures, but prophylactic antiseizure medications such as phenytoin have not been proven to be beneficial.46
Infection
Nearly 80% of patients with acute liver failure develop an infectious complication, which can be attributed to a state of immunodeficiency.47
The respiratory and urinary tracts are the most common sources of infection.48 In patients with bacteremia, Enterococcus species and coagulase-negative Staphylococcus species49 are the commonly isolated organisms. Also, in patients with acute liver failure, fungal infections account for 30% of all infections.50
Infected patients often develop worsening of their encephalopathy51 without fever or elevated white blood cell count.49,52 Thus, in any patient in whom encephalopathy is worsening, an evaluation must be done to rule out infection. In these patients, systemic inflammatory response syndrome is an independent risk factor for death.53
Despite the high mortality rate with infection, whether using antibiotics prophylactically in acute liver failure is beneficial is controversial.54,55
Gastrointestinal bleeding
The current prevalence of upper gastrointestinal bleeding in acute liver failure patients is about 1.5%.56 Coagulopathy and endotracheal intubation are the main risk factors for upper gastrointestinal bleeding in these patients.57 The most common source of bleeding is stress ulcers in the stomach. The ulcers develop from a combination of factors, including decreased blood flow to the mucosa causing ischemia and hypoperfusion-reperfusion injury.
Pharmacologic inhibition of gastric acid secretion has been shown to reduce upper gastrointestinal bleeding in acute liver failure. A histamine H2 receptor blocker or proton pump inhibitor should be given to prevent gastrointestinal bleeding in patients with acute liver failure.1,58
EXPERIMENTAL TREATMENTS
Artificial liver support systems
Membranes and dialysate solutions have been developed to remove toxic substances that are normally metabolized by the liver. Two of these—the molecular adsorbent recycling system (MARS) and the extracorporeal liver assist device (ELAD)—were developed in the late 1990s. MARS consisted of a highly permeable hollow fiber membrane mixed with albumin, and ELAD consisted of porcine hepatocytes attached to microcarriers in the extracapillary space of the hollow fiber membrane. Both systems allowed for transfer of water-soluble and protein-bound toxins in the blood across the membrane and into the dialysate.59 The clinical benefit offered by these devices is controversial,60–62 thus limiting their use to experimental purposes only.
Hepatocyte transplant
Use of hepatocyte transplant as a bridge to liver transplant was tested in 1970s, first in rats and later in humans.63 By reducing the blood ammonia level and improving cerebral perfusion pressure and cardiac function, replacement of 1% to 2% of the total liver cell mass by transplanted hepatocytes acts as a bridge to orthotopic liver transplant.64,65
PROGNOSIS
Different criteria have been used to identify patients with poor prognosis who may eventually need to undergo liver transplant.
The King’s College criteria system is the most commonly used for prognosis (Table 4).37,66–69 Its main drawback is that it is applicable only in patients with encephalopathy, and when patients reach this stage, their condition often deteriorates rapidly, and they die while awaiting liver transplant.37,66,67
The Model for End-Stage Liver Disease (MELD) score is an alternative to the King’s College criteria. A high MELD score on admission signifies advanced disease, and patients with a high MELD score tend to have a worse prognosis than those with a low score.68
The Acute Physiology and Chronic Health Evaluation (APACHE) II score can also be used, as it is more sensitive than the King’s College criteria.6
The Clichy criteria66,69 can also be used.
Liver biopsy. In addition to helping establish the cause of acute liver failure, liver biopsy can also be used as a prognostic tool. Hepatocellular necrosis greater than 70% on the biopsy predicts death with a specificity of 90% and a sensitivity of 56%.70
Hypophosphatemia has been reported to indicate recovering liver function in patients with acute liver failure.71 As the liver regenerates, its energy requirement increases. To supply the energy, adenosine triphosphate production increases, and phosphorus shifts from the extracellular to the intracellular compartment to meet the need for extra phosphorus during this process. A serum phosphorus level of 2.9 mg/dL or higher appears to indicate a poor prognosis in patients with acute liver failure, as it signifies that adequate hepatocyte regeneration is not occurring.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 342:273–275.
- Ostapowicz G, Fontana RJ, Schiodt FV, et al; US Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 2008; 47:1401–1415.
- Acharya SK, Panda SK, Saxena A, Gupta SD. Acute hepatic failure in India: a perspective from the East. J Gastroenterol Hepatol 2000; 15:473–479.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Patten CJ, Thomas PE, Guy RL, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 1993; 6:511–518.
- Chen W, Koenigs LL, Thompson SJ, et al. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol 1998; 11:295-301.
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187:211–217.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 1983; 71:980–991.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- Schiødt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med 1997; 337:1112–1117.
- Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994; 272:1845–1850.
- Chalasani N, Fontana RJ, Bonkovsky HL, et al; Drug Induced Liver Injury Network (DILIN). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008; 135:1924–1934 e1–4
- Reuben A, Koch DG, Lee WM; Acute Liver Failure Study Group. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology 2010; 52:2065–2076.
- Stevens T, Qadri A, Zein NN. Two patients with acute liver injury associated with use of the herbal weight-loss supplement hydroxycut. Ann Intern Med 2005; 142:477–478.
- Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol 2015; 62(suppl 1):S112–S120.
- Schiodt FV, Davern TJ, Shakil AO, McGuire B, Samuel G, Lee WM. Viral hepatitis-related acute liver failure. Am J Gastroenterol 2003; 98:448–453.
- Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology 1998; 28:839–842.
- Bismuth H, Samuel D, Gugenheim J, et al. Emergency liver transplantation for fulminant hepatitis. Ann Intern Med 1987; 107:337–341.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Davern TJ 2nd, James LP, Hinson JA, et al; Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006; 130:687–694.
- Perry HE, Shannon MW. Efficacy of oral versus intravenous N-acetylcysteine in acetaminophen overdose: results of an open-label, clinical trial. J Pediatr 1998; 132:149–152.
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557–1562.
- Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology 1995; 109:1907–1916.
- Tsang SW, Chan HL, Leung NW, et al. Lamivudine treatment for fulminant hepatic failure due to acute exacerbation of chronic hepatitis B infection. Aliment Pharmacol Ther 2001; 15:1737–1744.
- Yu JW, Sun LJ, Yan BZ, Kang P, Zhao YH. Lamivudine treatment is associated with improved survival in fulminant hepatitis B. Liver Int 2011; 31:499–506.
- Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 2011; 53:774–780.
- Pinna AD, Rakela J, Demetris AJ, Fung JJ. Five cases of fulminant hepatitis due to herpes simplex virus in adults. Dig Dis Sci 2002; 47:750–754.
- Farr RW, Short S, Weissman D. Fulminant hepatitis during herpes simplex virus infection in apparently immunocompetent adults: report of two cases and review of the literature. Clin Infect Dis 1997; 24:1191–1194.
- Czaja AJ, Freese DK; American Association for the Study of Liver Disease. Diagnosis and treatment of autoimmune hepatitis. Hepatology 2002; 36:479–497.
- Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003; 37:1475–1492.
- Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991; 100:1129–1134.
- Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009; 137:856–864.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology 1992; 15:449–453.
- Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 1994; 107:1475–1480.
- Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet 1993; 341:157–158.
- Cordoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol 1998; 29:589–594.
- Canalese J, Gimson AE, Davis C, Mellon PJ, Davis M, Williams R. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut 1982; 23:625–629.
- Jalan R, SW OD, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 1999; 354:1164–1168.
- Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 1999; 30:636–640.
- Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology 2009; 136:2159–2168.
- Bhatia V, Batra Y, Acharya SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol 2004; 41:89–96.
- Canalese J, Gove CD, Gimson AE, Wilkinson SP, Wardle EN, Williams R. Reticuloendothelial system and hepatocytic function in fulminant hepatic failure. Gut 1982; 23:265–269.
- Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology 1990; 11:49–53.
- Rolando N, Wade JJ, Stangou A, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg 1996; 2:8–13.
- Rolando N, Harvey F, Brahm J, et al. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol 1991; 12:1–9.
- Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology 2003; 125:755–764.
- Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis 1996; 16:389–402.
- Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32:734–739.
- Rolando N, Gimson A, Wade J, Philpott- Howard J, Casewell M, Williams R. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology 1993; 17:196–201.
- Karvellas CJ, Cavazos J, Battenhouse H, et al; US Acute Liver Failure Study Group. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol 2014; 12:1942–1949.
- Acharya SK, Dasarathy S, Kumer TL, et al. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology 1996; 23:1148–1155.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- MacDougall BR, Williams R. H2-receptor antagonist in the prevention of acute gastrointestinal hemorrhage in fulminant hepatic failure: a controlled trial. Gastroenterology 1978; 74:464–465.
- Stange J, Mitzner SR, Risler T, et al. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs 1999; 23:319–330.
- Vaid A, Chewich H, Balk EM, Jaber BL. Molecular adsorbent recirculating system as artificial support therapy for liver failure: a meta-analysis. ASAIO J 2012; 58:51–59.
- Khuroo MS, Khuroo MS, Farahat KL. Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: a meta-analysis. Liver Transpl 2004; 10:1099–1106.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Sommer BG, Sutherland DE, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation for treatment of D-galactosamine-induced acute liver failure in rats. Transplant Proc 1979; 11:578–584.
- Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology 1988; 8:1006–1009.
- Strom SC, Fisher RA, Thompson MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 1997; 63:559–569.
- Pauwels A, Mostefa-Kara N, Florent C, Levy VG. Emergency liver transplantation for acute liver failure. Evaluation of London and Clichy criteria. J Hepatol 1993; 17:124–127.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King's criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Larsen FS. MELD score as a predictor of liver failure and death in patients with acetaminophen-induced liver injury. Hepatology 2007; 45:789–796.
- Bernuau J, Goudeau A, Poynard T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology 1986; 6:648–651.
- Donaldson BW, Gopinath R, Wanless IR, et al. The role of transjugular liver biopsy in fulminant liver failure: relation to other prognostic indicators. Hepatology 1993; 18:1370–1376.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 342:273–275.
- Ostapowicz G, Fontana RJ, Schiodt FV, et al; US Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 2008; 47:1401–1415.
- Acharya SK, Panda SK, Saxena A, Gupta SD. Acute hepatic failure in India: a perspective from the East. J Gastroenterol Hepatol 2000; 15:473–479.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Patten CJ, Thomas PE, Guy RL, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 1993; 6:511–518.
- Chen W, Koenigs LL, Thompson SJ, et al. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol 1998; 11:295-301.
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187:211–217.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 1983; 71:980–991.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- Schiødt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med 1997; 337:1112–1117.
- Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994; 272:1845–1850.
- Chalasani N, Fontana RJ, Bonkovsky HL, et al; Drug Induced Liver Injury Network (DILIN). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008; 135:1924–1934 e1–4
- Reuben A, Koch DG, Lee WM; Acute Liver Failure Study Group. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology 2010; 52:2065–2076.
- Stevens T, Qadri A, Zein NN. Two patients with acute liver injury associated with use of the herbal weight-loss supplement hydroxycut. Ann Intern Med 2005; 142:477–478.
- Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol 2015; 62(suppl 1):S112–S120.
- Schiodt FV, Davern TJ, Shakil AO, McGuire B, Samuel G, Lee WM. Viral hepatitis-related acute liver failure. Am J Gastroenterol 2003; 98:448–453.
- Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology 1998; 28:839–842.
- Bismuth H, Samuel D, Gugenheim J, et al. Emergency liver transplantation for fulminant hepatitis. Ann Intern Med 1987; 107:337–341.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Davern TJ 2nd, James LP, Hinson JA, et al; Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006; 130:687–694.
- Perry HE, Shannon MW. Efficacy of oral versus intravenous N-acetylcysteine in acetaminophen overdose: results of an open-label, clinical trial. J Pediatr 1998; 132:149–152.
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557–1562.
- Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology 1995; 109:1907–1916.
- Tsang SW, Chan HL, Leung NW, et al. Lamivudine treatment for fulminant hepatic failure due to acute exacerbation of chronic hepatitis B infection. Aliment Pharmacol Ther 2001; 15:1737–1744.
- Yu JW, Sun LJ, Yan BZ, Kang P, Zhao YH. Lamivudine treatment is associated with improved survival in fulminant hepatitis B. Liver Int 2011; 31:499–506.
- Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 2011; 53:774–780.
- Pinna AD, Rakela J, Demetris AJ, Fung JJ. Five cases of fulminant hepatitis due to herpes simplex virus in adults. Dig Dis Sci 2002; 47:750–754.
- Farr RW, Short S, Weissman D. Fulminant hepatitis during herpes simplex virus infection in apparently immunocompetent adults: report of two cases and review of the literature. Clin Infect Dis 1997; 24:1191–1194.
- Czaja AJ, Freese DK; American Association for the Study of Liver Disease. Diagnosis and treatment of autoimmune hepatitis. Hepatology 2002; 36:479–497.
- Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003; 37:1475–1492.
- Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991; 100:1129–1134.
- Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009; 137:856–864.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology 1992; 15:449–453.
- Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 1994; 107:1475–1480.
- Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet 1993; 341:157–158.
- Cordoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol 1998; 29:589–594.
- Canalese J, Gimson AE, Davis C, Mellon PJ, Davis M, Williams R. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut 1982; 23:625–629.
- Jalan R, SW OD, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 1999; 354:1164–1168.
- Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 1999; 30:636–640.
- Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology 2009; 136:2159–2168.
- Bhatia V, Batra Y, Acharya SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol 2004; 41:89–96.
- Canalese J, Gove CD, Gimson AE, Wilkinson SP, Wardle EN, Williams R. Reticuloendothelial system and hepatocytic function in fulminant hepatic failure. Gut 1982; 23:265–269.
- Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology 1990; 11:49–53.
- Rolando N, Wade JJ, Stangou A, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg 1996; 2:8–13.
- Rolando N, Harvey F, Brahm J, et al. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol 1991; 12:1–9.
- Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology 2003; 125:755–764.
- Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis 1996; 16:389–402.
- Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32:734–739.
- Rolando N, Gimson A, Wade J, Philpott- Howard J, Casewell M, Williams R. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology 1993; 17:196–201.
- Karvellas CJ, Cavazos J, Battenhouse H, et al; US Acute Liver Failure Study Group. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol 2014; 12:1942–1949.
- Acharya SK, Dasarathy S, Kumer TL, et al. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology 1996; 23:1148–1155.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- MacDougall BR, Williams R. H2-receptor antagonist in the prevention of acute gastrointestinal hemorrhage in fulminant hepatic failure: a controlled trial. Gastroenterology 1978; 74:464–465.
- Stange J, Mitzner SR, Risler T, et al. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs 1999; 23:319–330.
- Vaid A, Chewich H, Balk EM, Jaber BL. Molecular adsorbent recirculating system as artificial support therapy for liver failure: a meta-analysis. ASAIO J 2012; 58:51–59.
- Khuroo MS, Khuroo MS, Farahat KL. Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: a meta-analysis. Liver Transpl 2004; 10:1099–1106.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Sommer BG, Sutherland DE, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation for treatment of D-galactosamine-induced acute liver failure in rats. Transplant Proc 1979; 11:578–584.
- Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology 1988; 8:1006–1009.
- Strom SC, Fisher RA, Thompson MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 1997; 63:559–569.
- Pauwels A, Mostefa-Kara N, Florent C, Levy VG. Emergency liver transplantation for acute liver failure. Evaluation of London and Clichy criteria. J Hepatol 1993; 17:124–127.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King's criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Larsen FS. MELD score as a predictor of liver failure and death in patients with acetaminophen-induced liver injury. Hepatology 2007; 45:789–796.
- Bernuau J, Goudeau A, Poynard T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology 1986; 6:648–651.
- Donaldson BW, Gopinath R, Wanless IR, et al. The role of transjugular liver biopsy in fulminant liver failure: relation to other prognostic indicators. Hepatology 1993; 18:1370–1376.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
KEY POINTS
- In the United States, the most common cause of acute liver failure is acetaminophen toxicity, followed by viral hepatitis.
- Testing for the cause of acute liver failure needs to start as soon as possible so that specific treatment can be initiated and the patient can be placed on the transplant list if needed.
- Acetylcysteine and either a proton pump inhibitor or a histamine H2 receptor blocker should be given to all patients with acute liver failure. Liver transplant is the cornerstone of therapy in patients not responding to other treatments.
- There are a number of prognostic scores for acute liver failure, but each has limitations.
Long-term management of liver transplant recipients: A review for the internist
Since 1963, when Starzl et al performed the first successful liver transplantation,1 outcomes of this life-saving procedure have continued to improve. Long-term survival rates have increased markedly: the current 5-year rate is 73.8% and the 10-year rate is 60%.2
This success means that internists will be caring for a greater number of liver transplant recipients and managing their long-term problems, such as hypertension, diabetes mellitus, dyslipidemia, obesity, metabolic syndrome, cardiovascular disease, renal insufficiency, osteoporosis, cancer, and gout.
This review will discuss these complications, focusing on the role the primary care physician assumes beyond the first year after transplantation.
ROLE OF THE PRIMARY CARE PHYSICIAN
Hepatologists, primary care physicians, and surgeons share the care of transplant recipients. The first several weeks after transplantation require close follow-up by the hepatologist and transplantation team, with particular attention paid to the patient’s overall health and well-being, medication compliance, and biochemical and immunosuppression monitoring.
After the first year, the primary care physician assumes a greater role, becoming the main provider of the patient’s care.3,4 Good communication between the transplant center and the primary care physician should lead to a smooth transition.4 Although the hepatologist continues to manage immunosuppressive drugs, allograft rejections, and biliary complications, the primary care physician manages most of the long-term complications and thus needs to be aware of the common ones and feel comfortable managing them. Aims during visits are to screen for and detect common complications and manage them appropriately, in addition to performing annual physical examinations and routine health care. A reasonable interval for liver transplant recipients to visit their primary care physician is every 6 months.
IMMUNOSUPPRESSANT MEDICATIONS
Multiple agents are used for immunosuppression after liver transplantation:
- Calcineurin inhibitors (cyclosporine and tacrolimus)
- Antimetabolites (mycophenolate mofetil, azathioprine, and mycophenolate sodium)
- Mammalian target of rapamycin (mTOR) inhibitors (sirolimus and everolimus)
- Corticosteroids.
![]()
Table 1 lists their common side effects.
Most centers use a combination of two to four immunosuppressants as induction therapy in the immediate posttransplant period, then taper the doses and eliminate all but a calcineurin inhibitor and an antimetabolite. For example, some start with a combination of tacrolimus, mycophenolate mofetil, and a corticosteroid. The choice in the immediate posttransplant period is frequently made by the transplant center in cooperation with the hepatologist. By the time primary care physicians see these patients, they usually are on a calcineurin inhibitor alone or a calcineurin inhibitor plus mycophenolate mofetil.
Calcineurin inhibitors
Cyclosporine is metabolized by the cytochrome CYP3A4 pathway. With an average half-life of 15 hours, it is given orally, usually every 12 hours.
The dosage is adjusted according to the trough level. Higher levels are needed in the initial posttransplant period to prevent graft rejection, whereas lower levels are preferred later to decrease the occurrence and severity of adverse effects. Typical long-term trough levels are 50 to 100 ng/mL. Levels should be checked more often if an acute illness develops or the patient starts taking a potentially interfering drug.
Of importance: the dosage should be based on trough levels and not on random levels. Levels are often falsely high if blood samples are not drawn at the trough level. Repeating the measurement and making sure the sample is drawn at the trough level, ie, 12 hours after the last dose, is advised in this condition.
Cyclosporine causes widespread vasoconstriction resulting in decreased renal blood flow and systemic hypertension, often within a few days of starting it. Other important adverse effects include renal insufficiency, dyslipidemia, neurotoxicity (headache, tremor, seizure), and diabetes.
Tacrolimus is superior to cyclosporine in terms of survival, graft loss, acute rejection, and steroid-resistant rejection in the first year.5 Currently, it is the agent used most often for maintenance immunosuppression after liver transplantation.
Like cyclosporine, tacrolimus is metabolized in the liver by CYP3A4. Satisfactory trough levels after 1 year are 4 to 6 ng/mL.
The adverse effects of tacrolimus are similar to those of cyclosporine, but diabetes mellitus is more common with tacrolimus. Bone marrow suppression may occur more often with tacrolimus as well.
Antimetabolites
Antimetabolites are generally not potent enough to be used alone.
Mycophenolate mofetil causes adverse effects that include bone marrow suppression and gastrointestinal symptoms such as gastritis, diarrhea, and abdominal pain.
Azathioprine, infrequently used in transplantation in the United States, is nevertheless sometimes substituted for mycophenolate mofetil in pregnant women, as it seems safer for use in pregnancy.
Serum levels of azathioprine and mycophenolate mofetil are not routinely monitored.
mTOR inhibitors
Sirolimus and everolimus are mTOR inhibitors, inhibiting proliferation of lymphocytes.6,7
Unlike calcineurin inhibitors, mTOR inhibitors are not associated with nephrotoxicity, neurotoxicity, renal dysfunction, hypertension, or diabetes. Sirolimus is considered an alternative to calcineurin inhibitors or, in some instances, used as add-on-therapy to lower the dose of the calcineurin inhibitor.
However, sirolimus carries a potential risk of hepatic artery thrombosis, a life-threatening complication.8 This has led the US Food and Drug Administration (FDA) to require sirolimus to carry a black-box warning, and most transplant centers avoid using it in the first 30 days after transplantation.
Dyslipidemia is perhaps the most common adverse effect of sirolimus. Others include dose-related cytopenia and wound dehiscence.9
Everolimus has yet to be established for use in liver transplantation, although safety trials have been published.10,11 The FDA currently recommends against using it in the first 30 days after liver transplantation.
Both sirolimus and everolimus are metabolized by CYP3A4, which is the same metabolic pathway used by cyclosporine and tacrolimus. Hence, drugs that inhibit CYP3A4 may significantly impair clearance of both sirolimus and everolimus.
Corticosteroids
Corticosteroids have been the cornerstone of immunosuppression and remain the first line of treatment for acute allograft rejection. High intravenous doses of corticosteroids are usually started in the peritransplant period and are then switched to oral doses, which are tapered and continued with a fixed dose such as 20 mg of prednisone daily for 3 to 6 months after transplantation. However, some transplant centers keep patients on prednisone 5 mg/day indefinitely.
Adverse effects of corticosteroids include diabetes, salt and fluid retention, hypertension, hyperlipidemia, cosmetic changes (acne, cervical fat pad or “buffalo hump”), delayed wound healing, susceptibility to infection, cataracts, osteopenia, and potential adrenal suppression.12 There is concern that the use of these drugs may increase hepatitis C virus replication in patients who received a liver transplant for hepatitis C cirrhosis. Randomized trials have yielded conflicting results.13–15
Drug interactions
Certain drugs can affect the metabolism of calcineurin inhibitors and mTOR inhibitors by inducing CYP3A4, which results in decreasing the levels of the immunosuppressive drugs, or by inhibiting CYP3A4, which has the opposite effect.
Medications that can decrease the levels of calcineurin inhibitors and mTOR inhibitors:
- Anticonvulsants (carbamazepine, phenobarbital, phenytoin)
- Antibiotics (rifampin, isoniazid)
- St John’s wort.
Medications that can increase the levels of calcineurin inhibitors and mTOR inhibitors:
- Antifungals (fluconazole, ketoconazole, itraconazole, voriconazole, aspofungin)
- Antibiotics (azithromycin, erythromycin, clarithromycin)
- Nondihydropyridine calcium channel blockers (diltiazem, verapamil).16
Selected antibiotics are generally well tolerated, such as penicillins, cephalosporins, quinolones, sulfonamides, and topical antifungal agents.
LONG-TERM COMPLICATIONS
![]()
Figure 1 summarizes the common long-term complications of liver transplantation.
Hypertension
The prevalence of hypertension after liver transplantation is 40% to 85%, which is markedly higher than in patients with chronic liver disease before liver transplantation.17,18
One of the factors contributing to this increase is the use of immunosuppressive medications. Of these drugs, cyclosporine seems to be the one that most often causes an increase in both the incidence and the severity of hypertension, as it produces widespread vasoconstriction.19 Corticosteroids cause hypertension through their mineralocorticoid effects.
The diagnostic cutoffs for hypertension (ie, 140/90 mm Hg) and the treatment goals in posttransplant patients are similar to those in the general population. However, at our institution we target a blood pressure of less than 130/80 mm Hg in transplant patients because they have a high prevalence of other cardiovascular risk factors such as diabetes, obesity, and renal insufficiency.20
Dihydropyridine calcium channel blockers such as amlodipine and nifedipine are considered the best first-line agents because they dilate renal afferent arterioles, an effect that may counteract the vasoconstriction mediated by calcineurin inhibitors. Nondihydropyridine calcium channel blockers such as diltiazem and verapamil tend to have more marked negative inotropic effects and are not recommended in liver transplant recipients because they increase the levels of calcineurin inhibitors.21
Diuretics (eg, furosemide) might be the second-line agents, especially in patients with peripheral edema.16 One should be vigilant for hyperuricemia if thiazide agents are used.
Angiotensin-converting-enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) are typically avoided in the early posttransplant period, but they can be started later and have additional benefits in patients with diabetes and congestive heart failure. Starting ACE inhibitors is acceptable in these patients unless there is a contraindication such as allergy to ACE inhibitors, hypotension, history of bilateral renal artery stenosis, significant hyperkalemia, or acute kidney injury. Monitor the serum potassium level closely for hyperkalemia in patients concurrently using calcineurin inhibitors.
Alpha-blockers and beta-blockers can be used as add-on therapy in patients with uncontrolled hypertension with the exception of carvedilol, because it increases the levels of calcineurin inhibitors.22
Blood pressure monitoring by the primary care physician is recommended every 6 months after the early posttransplant period, or more frequently when changes in treatment are being considered.
If hypertension continues to be inadequately controlled despite treatment, changing the immunosuppressive drugs or decreasing the doses can be considered, but the transplant hepatologist must be involved in this decision.23,24
Diabetes mellitus
The prevalence of diabetes mellitus is higher in liver transplant recipients than in the general population, reaching 30% to 40%.17,25 In addition to preexisting diabetes, 15% of liver transplant recipients develop new-onset diabetes.26,27
Risk factors for developing diabetes after liver transplantation include African American or Hispanic ethnicity, obesity, family history, pretransplant diabetes, hepatitis C virus infection, use of corticosteroids, and use of calcineurin inhibitors (tacrolimus more than cyclosporine) and sirolimus.26
In addition to increasing the risk of cardiovascular disease and other diseases, diabetes decreases both patient and graft survival after liver transplantation.28
The management of diabetes and the treatment target after transplantation should follow the American Diabetes Association guidelines for the treatment of type 2 diabetes mellitus.29 Lifestyle modifications, diet, and exercise are as important for transplant patients as for the nontransplant population. Insulin therapy is usually needed in the early posttransplant period to control blood glucose levels well, especially with the high doses of corticosteroids used during the first few weeks. No trials to date have compared oral agents in posttransplant patients. Therefore, the choice of oral hypoglycemic agents should be individualized on the basis of the patient’s characteristics and comorbidities.
We recommend that primary care providers screen all liver transplant recipients for diabetes regardless of their pretransplant status. This can be done by obtaining regular fasting blood glucose levels or a hemoglobin A1c level every 6 months. Additionally, liver transplant recipients diagnosed with diabetes require annual eye examinations to look for cataracts and diabetes-related changes.30
Dyslipidemia
On November 12, 2013, the American College of Cardiology and the American Heart Association (ACC/AHA) released new clinical practice guidelines for treating blood cholesterol levels.31,32 According to these new guidelines, there are four groups of patients for whom treatment with statins is clearly indicated:
- Patients with cardiovascular disease
- Patients with low-density lipoprotein cholesterol (LDL-C) levels ≥ 190 mg/dL
- Patients 40 to 75 years old with type 2 diabetes
- Patients 40 to 75 years old with an estimated 10-year risk of cardiovascular disease of 7.5% or greater.
Liver transplant recipients should be evaluated on an individual basis to see if they fit in any of the four groups and if statin treatment therefore needs to be initiated.
A few things need to be kept in mind. First, the incidence of dyslipidemia after liver transplantation is estimated to be 45% to 69%. Risk factors include obesity, diabetes mellitus, cholestatic liver disease, and immunosuppressant medications.33 Sirolimus has a significant and well-documented association with dyslipidemia. Cyclosporine and corticosteroids are also strongly associated with dyslipidemia. Tacrolimus has a minor effect, and mycophenolate mofetil and azathioprine have no significant effects on serum lipid levels.16
Second, of the seven currently marketed statins, pravastatin and fluvastatin are preferred in liver transplant recipients because they are not metabolized by the same cytochrome CYP3A4 pathway that metabolizes calcineurin inhibitors and sirolimus.34 The doses of 40 to 80 mg daily of pravastatin or 40 mg twice daily of fluvastatin lower low-density lipoprotein cholesterol (LDL-C) levels by approximately 30% to 35%. However, these two agents are considered “moderate-intensity” statins according to the new ACC/AHA guidelines. The only two “high-intensity” statins are atorvastatin (40–80 mg) and rosuvastatin (20–40 mg), but they are both metabolized by CYP3A4. Therefore it is prudent to avoid them with the concurrent use of a calcineurin inhibitor or tacrolimus.
Gemfibrozil does not lower LDL-C and should not be used concomitantly with statins due to unacceptable risk of rhabdomyolysis and myopathy. Fenofibrates are usually avoided due to potential nephrotoxicity in patients receiving cyclosporine. Bile acid sequestrants (cholestyramine, colestipol, colesevelam) can decrease plasma mycophenolate mofetil levels by 35%.16,35 Thus, these agents should be avoided if mycophenolate mofetil is used.
It is reasonable to screen all liver transplant recipients with a fasting lipid profile at 3, 6, and 12 months after transplantation and annually thereafter. Creatine kinase should be measured if the patient complains of severe muscle pain or weakness but not on a routine basis.
Obesity
Approximately one-third of patients who are of normal weight at the time of transplantation will become obese afterward.18,25 Corticosteroid use is an important risk factor for posttransplant obesity, and tapering these drugs helps reduce weight.36 Patients treated with cyclosporine are more likely to gain weight than those who receive tacrolimus.37
Of importance: nonalcoholic fatty liver disease, currently the most common cause of chronic liver disease in adults, is rapidly increasing as an indication for liver transplantation. In fact, the proportion of liver transplantation procedures for nonalcoholic steatohepatitis-related cirrhosis increased from 1.2% in 2001 to 9.7% in 2009, and nonalcoholic steatohepatitis is expected to become the leading indication for liver transplantation in the next 20 years. And because nonalcoholic fatty liver disease is directly linked to obesity, the prevalence of obesity as a complication of liver transplantation will most likely increase in the near future.
Overweight liver transplant recipients may have great difficulty losing weight. Treatment starts with patient education on caloric restriction and exercise. If traditional measures fail to result in adequate weight loss, additional options include switching from cyclosporine to tacrolimus.23
Bariatric surgery may become an option for posttransplant patients. In a recent case series from the University of Minnesota, Al-Nowaylati et al38 reported their experience with seven patients who underwent orthotopic liver transplantation and then open Roux-en-Y gastric bypass. After bariatric surgery, the patients’ mean body mass index declined significantly, and glycemic control and high-density lipoprotein cholesterol (HDL-C) levels improved. However, one patient died of multiple organ failure, to which the bariatric surgery might have contributed.38
Heimbach et al39 conducted a study in patients referred for liver transplantation for whom a rigorous noninvasive weight-loss program before transplantation had failed. The researchers performed combined liver transplantation and sleeve gastrectomy in seven carefully selected patients who had failed to achieve weight loss to a body mass index less than 35 kg/m2 before transplantation. All seven patients lost weight, decreasing their mean body mass index from 49 kg/m2 before the procedure to 29 kg/m2 at last follow-up, and none of them developed posttransplant diabetes or steatosis.
At this time, there is not enough evidence to recommend concurrent orthotopic liver transplantation plus bariatric surgery, or combined orthotopic liver transplantation and sleeve gastrectomy. More study is needed to further evaluate these advanced approaches.
Posttransplant metabolic syndrome
Metabolic syndrome is common after liver transplantation and is strongly associated with increased morbidity in this patient population.40,41 The general definition of metabolic syndrome includes a combination of at least three of the following: hypertension, insulin resistance, hypertriglyceridemia, low HDL-C, and obesity.
The prevalence of metabolic syndrome is higher in patients after liver transplantation than in nontransplant patients. In a review of 252 liver transplant recipients, 52% were diagnosed with posttransplant metabolic syndrome, but only 5% had had it pretransplant.42
Careful screening for posttransplant metabolic syndrome and early recognition of risk factors are important. Nevertheless, the treatment of this condition depends on treating its components according to recommended guidelines.41
Cardiovascular disease
The incidence of cardiovascular morbidity and death is increased after liver transplantation.24 In addition, after liver transplantation, cardiovascular disease is a major cause of death unrelated to liver disease. It accounts for 12% to 16% of deaths and is the third most common cause of late mortality after liver transplantation.43 Of note, a recent study by our group demonstrated that patients undergoing liver transplantation for nonalcoholic steatohepatitis had a significantly higher risk of a cardiovascular event during the 3 years after transplantation than patients undergoing liver transplantation for cholestatic liver disease.44
Risk factors for cardiovascular disease after liver transplantation include older age at transplantation, male sex, posttransplant diabetes, posttransplant hypertension, and the use of mycophenolate mofetil.44 Modifying the risk factors is essential in decreasing the risk of cardiovascular events.
It is reasonable to perform dobutamine stress testing every 3 to 5 years in patients with multiple risk factors for cardiovascular disease, or more frequently in those with preexisting coronary artery disease.45,46
Malignancy
The risk of several malignancies increases after liver transplantation. Liver transplant recipients have an incidence of cancer 2.1 to 4.3 times greater than age- and sex-matched controls.24,47–49
Skin cancers are the most common and account for almost 40% of malignancies in organ transplant recipients.50 Whereas basal cell carcinoma is more common in the general population, squamous cell carcinoma is equally common in liver transplant recipients.
Multiple clinical studies have linked calcineurin inhibitors and azathioprine to the development of skin cancer. Annual skin examinations in addition to avoiding other risk factors such as smoking and sun exposure are generally recommended. Changing the immunosuppressants to sirolimus in high-risk patients may lower their chance of developing skin cancer.51,52
Patients with ulcerative colitis who undergo liver transplantation because of sclerosing cholangitis are at higher risk of colon cancer and require annual colonoscopy with surveillance biopsies. Patients who undergo transplantation for alcoholic liver disease seem to have a higher risk of pulmonary and oropharyngeal cancers.53,54
It is important that transplant patients adhere to recommended cancer screening guidelines, in view of their increased risk. Studies have shown improved overall survival in liver transplant recipients who underwent intensive cancer surveillance.55
Renal insufficiency
Renal insufficiency is a well-recognized complication of liver transplantation and is associated with an increased long-term death rate.56,57
The incidence of renal insufficiency increases dramatically over time. Ojo et al,57 in a study of almost 37,000 liver transplant recipients, found that the incidence of chronic kidney disease (defined as an estimated glomerular filtration rate < 30 mL/min/1.73 m2) was 13.9% at 3 years, 18% at 5 years, and approximately 26% at 10 years.
Risk factors include the use of calcineurin inhibitors (both cyclosporine and tacrolimus), older age, female sex, lower pretransplant glomerular filtration rate, postoperative acute renal failure, diabetes, hypertension, hepatitis C virus infection, and transplantation before 1998.58,59 Replacing a calcineurin inhibitor with mycophenolate mofetil or sirolimus may be considered with communication with the transplant center, as mycophenolate mofetil or sirolimus are associated with a lower risk of renal injury.60–64
Starting 1 year after liver transplantation, primary care providers should screen for renal dysfunction by obtaining kidney function tests every 6 months, including urinalysis and microalbuminuria assessment. Equations for estimating the glomerular filtration rate used in practice, such as the Modification of Diet in Renal Disease Study equation, rely mainly on serum creatinine, which may lead to overestimating renal function in some circumstances. Therefore, other equations can be used to confirm the estimated glomerular filtration rate measured by creatinine clearance, and to more accurately evaluate kidney function. Calculators are available at www.kidney.org/professionals/kdoqi/gfr_calculator.
All liver transplant recipients should avoid nonsteroidal anti-inflammatory drugs and nephrotoxic medications, and should have their hypertension and diabetes adequately controlled.
Bone diseases
Osteopenia is another major complication of liver transplantation. One-third of liver transplant recipients have a bone mineral density below the fracture threshold.65
Multiple factors contribute to increased bone loss after transplantation, including use of corticosteroids, use of calcineurin inhibitors (cyclosporine, tacrolimus), poor nutrition, vitamin D deficiency, immobility, sarcopenia (reduced muscle mass), hypogonadism, smoking, and alcohol abuse.66 Even at low doses of less than 7.5 mg per day, corticosteroids inhibit osteoblast activity and increase bone resorption.
Studies have reported rapid bone loss at around 6 months after transplantation.67–69 However, long-term follow-up of bone mineral density up to 15 years after transplantation revealed an improvement mainly in the 2nd postoperative year, with no deterioration afterward.65
High-risk patients need to be identified early with appropriate screening and evaluation. Evaluation includes dual-energy x-ray absorptiometry and serum levels of calcium, phosphorous, parathyroid hormone, testosterone (men), estradiol (women), and alpha-25-hydroxyvitamin D. These tests are typically done before transplantation and then every other year afterward.
We recommend a daily dose of calcitriol and a calcium supplement to all our liver transplant patients.70 If osteoporosis (a T-score 2.5 or more standard deviations below the mean) or a fragility fracture occurs, then the patient may benefit from an oral bisphosphonate. Calcitonin has also been shown to improve bone mineral density in patients with osteoporosis after liver transplantation.71
Hyperuricemia and gout
Although hyperuricemia is common in liver transplant recipients (reported in approximately 47%), the development of clinical gout is less common (6%).72 Asymptomatic hyperuricemia requires observation only and is not usually treated in liver transplant recipients.
Acute attacks of gout are typically managed with colchicine 0.6 mg every 2 hours, up to five doses. Prednisone can be considered if symptoms persist despite treatment with colchicine. Allopurinol in an initial dose of 100 mg daily is used as maintenance therapy to reduce production of uric acid.73 However, because of the potential for drug interactions, the combination of azathioprine and allopurinol should be avoided.
Psychiatric complications and quality of life
Depression is common in liver transplant recipients, significantly more so in patients who received a transplant because of hepatitis C.74 The type of immunosuppressant is not associated with the incidence of depression. When indicated, the internist may start the patient on a selective serotonin reuptake inhibitor such as citalopram 20 mg daily, as these medications are usually effective and well tolerated in liver transplant patients.73
Liver transplantation has a major positive effect on quality of life. Most patients with end-stage liver disease have poor quality of life before transplantation, but this seems to improve notably afterward. A meta-analysis showed significant improvement in posttransplant physical health, sexual functioning, daily activities, and social functioning compared with before transplantation.75
Alcohol abuse and smoking
Patients who underwent liver transplantation because of alcoholic liver disease should be advised to abstain from alcohol.19 Patients who underwent the procedure for a different indication are advised to avoid excessive alcohol intake, as it is proven to lower the survival rate.20 Alcohol recidivism and smoking (including marijuana) are major problems, and internists are best positioned to address these issues and treat them.
Vaccinations
All liver transplant recipients should be vaccinated against influenza, pneumococcal infection, and tetanus. Hepatitis A and B vaccines are typically given before transplantation. In general, live vaccines such as measles-mumps-rubella and varicella are not recommended after any solid organ transplant.76
A study in Germany showed that immunization rates were too low in solid-organ transplant recipients, and almost 90% of patients were not adequately informed about immunizations.77 Hence, there may be room for improvement, and primary care providers should take the lead toward better outcomes in this regard.
Recurrence of the primary liver disease after transplantation
Different primary liver diseases recur with different frequencies.
Hepatitis C has the highest rate of recurrence of the liver diseases.78,79 Reinfection with hepatitis C virus after liver transplantation is almost universal and can follow different patterns. One of the most aggressive patterns is fibrosing cholestatic hepatitis, which frequently leads to graft failure and death, and hence necessitates urgent detection and treatment.
Hepatocellular carcinoma also has a high recurrence rate.80 Surveillance with liver ultrasonography or computed tomography is required every 6 months for the first 5 years after liver transplantation.
Other liver diseases. Nonalcoholic steatohepatitis, primary biliary cirrhosis, primary sclerosing cholangitis, autoimmune hepatitis, and hepatitis B infection also tend to recur after liver transplantation.46,81 On the other hand, alpha-1 antitrypsin deficiency, Wilson disease, hemochromatosis, and metabolic disorders are “cured” after liver transplantation.
It is important to detect any increase in liver enzymes above baseline. An elevation of 1.5 times the upper limit of normal or more should trigger further investigation.
Allograft dysfunction
A number of complications can develop in the liver allograft and result in abnormal liver function tests and, if not treated, graft failure. The most common causes of late graft dysfunction include recurrence of primary liver disease, biliary complications, and chronic rejection.46
Vascular complications include hepatic artery thrombosis and stenosis and are usually evaluated by liver ultrasonography and Doppler scan of the hepatic artery and venous structures.24
Biliary strictures give a cholestatic picture, with elevated bilirubin and greater elevation of alkaline phosphatase than of alanine aminotransferase and aspartate aminotransferase. Strictures are usually treated by endoscopic dilation and stenting, but they may eventually require surgery.
Late acute cellular rejections occur in 10% to 20% of cases and are a risk factor for chronic rejection. Liver biopsy is needed to make the diagnosis, and pulsed doses of corticosteroids remain the backbone of treatment therapy.
Chronic rejection is not common, occurring in 3% to 4% of liver transplant recipients.46 Treatment is based on increasing immunosuppression and ensuring compliance with prescribed medications. However, chronic rejection may not respond well, and repeat transplantation may be the last resort for some patients.
WHEN TO REFER TO THE HEPATOLOGIST
Some situations require referral to the hepatologist or the transplant center. In general, the following are best managed by a hepatologist: adjustment of immunosuppressive drugs and dosages, allograft dysfunction, vascular and biliary complications, progressing renal dysfunction, and recurrence of primary liver disease. Early communication with a hepatologist and the transplant center is recommended in these cases.
- Starzl TE, Marchioro TL, Vonkaulla KN, Hermann G, Brittain RS, Waddell WR. Homotransplantation of the liver in humans. Surg Gynecol Obstet 1963; 117:659–676.
- Matas AJ, Smith JM, Skeans MA, et al. OPTN/SRTR 2011 annual data report: kidney. Am J Transplant 2013; 13(suppl 1):11–46.
- McCashland TM. Posttransplantation care: role of the primary care physician versus transplant center. Liver Transpl 2001; 7(suppl 1):S2–S12.
- Heller JC, Prochazka AV, Everson GT, Forman LM. Long-term management after liver transplantation: primary care physician versus hepatologist. Liver Transpl 2009; 15:1330–1335.
- McAlister VC, Haddad E, Renouf E, Malthaner RA, Kjaer MS, Gluud LL. Cyclosporin versus tacrolimus as primary immunosuppressant after liver transplantation: a meta-analysis. Am J Transplant 2006; 6:1578–1585.
- Neuhaus P, Klupp J, Langrehr JM. mTOR inhibitors: an overview. Liver Transpl 2001; 7:473–484.
- Sehgal SN. Rapamune (RAPA, rapamycin, sirolimus): mechanism of action immunosuppressive effect results from blockade of signal transduction and inhibition of cell cycle progression. Clin Biochem 1998; 31:335–340.
- Asrani SK, Wiesner RH, Trotter JF, et al. De novo sirolimus and reduced-dose tacrolimus versus standard-dose tacrolimus after liver transplantation: the 2000-2003 phase II prospective randomized trial. Am J Transplant 2014; 14:356–366.
- Montalbano M, Neff GW, Yamashiki N, et al. A retrospective review of liver transplant patients treated with sirolimus from a single center: an analysis of sirolimus-related complications. Transplantation 2004; 78:264–268.
- Everson GT. Everolimus and mTOR inhibitors in liver transplantation: opening the “box.” Liver Transpl 2006; 12:1571–1573.
- Levy G, Schmidli H, Punch J, et al. Safety, tolerability, and efficacy of everolimus in de novo liver transplant recipients: 12- and 36-month results. Liver Transpl 2006; 12:1640–1648.
- Toniutto P, Fabris C, Fumolo E, et al. Prevalence and risk factors for delayed adrenal insufficiency after liver transplantation. Liver Transpl 2008; 14:1014–1019.
- Klintmalm GB, Davis GL, Teperman L, et al. A randomized, multicenter study comparing steroid-free immunosuppression and standard immunosuppression for liver transplant recipients with chronic hepatitis C. Liver Transpl 2011; 17:1394–1403.
- Llado L, Fabregat J, Castellote J, et al. Impact of immunosuppression without steroids on rejection and hepatitis C virus evolution after liver transplantation: results of a prospective randomized study. Liver Transpl 2008; 14:1752–1760.
- Lake JR. Immunosuppression and outcomes of patients transplanted for hepatitis C. J Hepatol 2006; 44:627–629.
- Sohn AJ, Jeon H, Ahn J. Primary care of the liver transplant recipient. Prim Care 2011; 38:499–514.
- Laish I, Braun M, Mor E, Sulkes J, Harif Y, Ben Ari Z. Metabolic syndrome in liver transplant recipients: prevalence, risk factors, and association with cardiovascular events. Liver Transpl 2011; 17:15–22.
- Stegall MD, Everson G, Schroter G, Bilir B, Karrer F, Kam I. Metabolic complications after liver transplantation. Diabetes, hypercholesterolemia, hypertension, and obesity. Transplantation 1995; 60:1057–1060.
- Textor SC, Canzanello VJ, Taler SJ, et al. Cyclosporine-induced hypertension after transplantation. Mayo Clin Proc 1994; 69:1182–1193.
- Prevention, detection, evaluation, and treatment of hypertension. The Sixth Report of the Joint National Committee. National Institutes of Health-National Heart, Lung, and Blood Institute. National High Blood Pressure Education Programme. Indian Heart J 1999; 51:381–396.
- Frishman WH. Calcium channel blockers: differences between subclasses. Am J Cardiovasc Drugs 2007; 7(suppl 1):17–23.
- Galioto A, Semplicini A, Zanus G, et al. Nifedipine versus carvedilol in the treatment of de novo arterial hypertension after liver transplantation: results of a controlled clinical trial. Liver Transpl 2008; 14:1020–1028.
- Neal DA, Gimson AE, Gibbs P, Alexander GJ. Beneficial effects of converting liver transplant recipients from cyclosporine to tacrolimus on blood pressure, serum lipids, and weight. Liver Transpl 2001; 7:533–539.
- Singh S, Watt KD. Long-term medical management of the liver transplant recipient: what the primary care physician needs to know. Mayo Clin Proc 2012; 87:779–790.
- Bianchi G, Marchesini G, Marzocchi R, Pinna AD, Zoli M. Metabolic syndrome in liver transplantation: relation to etiology and immunosuppression. Liver Transpl 2008; 14:1648–1654.
- Lane JT, Dagogo-Jack S. Approach to the patient with new-onset diabetes after transplant (NODAT). J Clin Endocrinol Metab 2011; 96:3289–3297.
- Wilkinson A, Davidson J, Dotta F, et al. Guidelines for the treatment and management of new-onset diabetes after transplantation. Clin Transplant 2005; 19:291–298.
- Moon JI, Barbeito R, Faradji RN, Gaynor JJ, Tzakis AG. Negative impact of new-onset diabetes mellitus on patient and graft survival after liver transplantation: long-term follow up. Transplantation 2006; 82:1625–1628.
- American Diabetes Association. Standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S11–S63.
- Marchetti P. New-onset diabetes after liver transplantation: from pathogenesis to management. Liver Transpl 2005; 11:612–620.
- Stone NJ, Robinson J, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129(suppl 2):S1–S45.
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Gisbert C, Prieto M, Berenguer M, et al. Hyperlipidemia in liver transplant recipients: prevalence and risk factors. Liver Transpl Surg 1997; 3:416–422.
- Asberg A. Interactions between cyclosporin and lipid-lowering drugs: implications for organ transplant recipients. Drugs 2003; 63:367–378.
- Bullingham RE, Nicholls AJ, Kamm BR. Clinical pharmacokinetics of mycophenolate mofetil. Clin Pharmacokinet 1998; 34:429–455.
- Everhart JE, Lombardero M, Lake JR, Wiesner RH, Zetterman RK, Hoofnagle JH. Weight change and obesity after liver transplantation: incidence and risk factors. Liver Transpl Surg 1998; 4:285–296.
- Canzanello VJ, Schwartz L, Taler SJ, et al. Evolution of cardiovascular risk after liver transplantation: a comparison of cyclosporine A and tacrolimus (FK506). Liver Transpl Surg 1997; 3:1–9.
- Al-Nowaylati AR, Al-Haddad BJ, Dorman RB, et al. Gastric bypass after liver transplantation. Liver Transpl 2013; 19:1324–1329.
- Heimbach JK, Watt KD, Poterucha JJ, et al. Combined liver transplantation and gastric sleeve resection for patients with medically complicated obesity and end-stage liver disease. Am J Transplant 2013; 13:363–368.
- Watt KD, Charlton MR. Metabolic syndrome and liver transplantation: a review and guide to management. J Hepatol 2010; 53:199–206.
- Pagadala M, Dasarathy S, Eghtesad B, McCullough AJ. Posttransplant metabolic syndrome: an epidemic waiting to happen. Liver Transpl 2009; 15:1662–1670.
- Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009; 120:1640–1645.
- Watt KD, Pedersen RA, Kremers WK, Heimbach JK, Charlton MR. Evolution of causes and risk factors for mortality post-liver transplant: results of the NIDDK long-term follow-up study. Am J Transplant 2010; 10:1420–1427.
- Albeldawi M, Aggarwal A, Madhwal S, et al. Cumulative risk of cardiovascular events after orthotopic liver transplantation. Liver Transpl 2012; 18:370–375.
- Gibbons RJ, Balady GJ, Bricker JT, et al. ACC/AHA 2002 guideline update for exercise testing: summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Update the 1997 Exercise Testing Guidelines). Circulation 2002; 106:1883–1892.
- Aberg F, Isoniemi H, Höckerstedt K. Long-term results of liver transplantation. Scand J Surg 2011; 100:14–21.
- Aberg F, Pukkala E, Höckerstedt K, Sankila R, Isoniemi H. Risk of malignant neoplasms after liver transplantation: a population-based study. Liver Transpl 2008; 14:1428–1436.
- Mells G, Neuberger J. Long-term care of the liver allograft recipient. Semin Liver Dis 2009; 29:102–120.
- Herrero JI. De novo malignancies following liver transplantation: impact and recommendations. Liver Transpl 2009; 15(suppl 2):S90–S94.
- Euvrard S, Kanitakis J, Claudy A. Skin cancers after organ transplantation. N Engl J Med 2003; 348:1681–1691.
- Euvrard S, Morelon E, Rostaing L, et al; TUMORAPA Study Group. Sirolimus and secondary skin-cancer prevention in kidney transplantation. N Engl J Med 2012; 367:329–339.
- Salgo R, Gossmann J, Schofer H, et al. Switch to a sirolimus-based immunosuppression in long-term renal transplant recipients: reduced rate of (pre-)malignancies and nonmelanoma skin cancer in a prospective, randomized, assessor-blinded, controlled clinical trial. Am J Transplant 2010; 10:1385–1393.
- Narumi S, Roberts JP, Emond JC, Lake J, Ascher NL. Liver transplantation for sclerosing cholangitis. Hepatology 1995; 22:451–457.
- Knechtle SJ, D’Alessandro AM, Harms BA, Pirsch JD, Belzer FO, Kalayoglu M. Relationships between sclerosing cholangitis, inflammatory bowel disease, and cancer in patients undergoing liver transplantation. Surgery 1995; 118:615–620.
- Finkenstedt A, Graziadei IW, Oberaigner W, et al. Extensive surveillance promotes early diagnosis and improved survival of de novo malignancies in liver transplant recipients. Am J Transplant 2009; 9:2355–2361.
- Fisher NC, Nightingale PG, Gunson BK, Lipkin GW, Neuberger JM. Chronic renal failure following liver transplantation: a retrospective analysis. Transplantation 1998; 66:59–66.
- Ojo AO, Held PJ, Port FK, et al. Chronic renal failure after transplantation of a nonrenal organ. N Engl J Med 2003; 349:931–940.
- Klintmalm GB, Gonwa TA. Nephrotoxicity associated with cyclosporine and FK506. Liver Transpl Surg 1995; 1:11–19.
- A comparison of tacrolimus (FK506) and cyclosporine for immunosuppression in liver transplantation. The US Multicenter FK506 Liver Study Group. N Engl J Med 1994; 331:1110–1115.
- Neff GW, Montalbano M, Slapak-Green G, et al. Sirolimus therapy in orthotopic liver transplant recipients with calcineurin inhibitor related chronic renal insufficiency. Transplant Proc 2003; 35:3029–3031.
- Cotterell AH, Fisher RA, King AL, et al. Calcineurin inhibitor-induced chronic nephrotoxicity in liver transplant patients is reversible using rapamycin as the primary immunosuppressive agent. Clin Transplant 2002; 16(suppl 7):49–51.
- Manzia TM, De Liguori Carino N, Orlando G, et al. Use of mycophenolate mofetil in liver transplantation: a literature review. Transplant Proc 2005; 37:2616–2617.
- Schlitt HJ, Barkmann A, Boker KH, et al. Replacement of calcineurin inhibitors with mycophenolate mofetil in liver-transplant patients with renal dysfunction: a randomised controlled study. Lancet 2001; 357:587–591.
- Hodge EE, Reich DJ, Clavien PA, Kim-Schluger L. Use of mycophenolate mofetil in liver transplant recipients experiencing renal dysfunction on cyclosporine or tacrolimus-randomized, prospective, multicenter study results. Transplant Proc 2002; 34:1546–1547.
- Hamburg SM, Piers DA, van den Berg AP, Slooff MJ, Haagsma EB. Bone mineral density in the long term after liver transplantation. Osteoporos Int 2000; 11:600–606.
- Maalouf NM, Shane E. Osteoporosis after solid organ transplantation. J Clin Endocrinol Metab 2005; 90:2456–2465.
- Crosbie OM, Freaney R, McKenna MJ, Curry MP, Hegarty JE. Predicting bone loss following orthotopic liver transplantation. Gut 1999; 44:430–434.
- Giannini S, Nobile M, Ciuffreda M, et al. Long-term persistence of low bone density in orthotopic liver transplantation. Osteoporos Int 2000; 11:417–424.
- Monegal A, Navasa M, Guanabens N, et al. Bone disease after liver transplantation: a long-term prospective study of bone mass changes, hormonal status and histomorphometric characteristics. Osteoporos Int 2001; 12:484–492.
- Neuhaus R, Lohmann R, Platz KP, et al. Treatment of osteoporosis after liver transplantation. Transplant Proc 1995; 27:1226–1227.
- Valero MA, Loinaz C, Larrodera L, Leon M, Moreno E, Hawkins F. Calcitonin and bisphosphonates treatment in bone loss after liver transplantation. Calcif Tissue Int 1995; 57:15–19.
- Neal DA, Tom BD, Gimson AE, Gibbs P, Alexander GJ. Hyperuricemia, gout, and renal function after liver transplantation. Transplantation 2001; 72:1689–1691.
- Schiff ER, Sorrell MF, Maddrey WC, editors. Schiff’s Diseases of the Liver. 10th ed. Philadephia, PA: Lippincott Williams & Wilkins; 2007.
- Tombazzi CR, Waters B, Shokouh-Amiri MH, Vera SR, Riely CA. Neuropsychiatric complications after liver transplantation: role of immunosuppression and hepatitis C. Dig Dis Sci 2006; 51:1079–1081.
- Bravata DM, Olkin I, Barnato AE, Keeffe EB, Owens DK. Health-related quality of life after liver transplantation: a meta-analysis. Liver Transpl Surg 1999; 5:318–331.
- Danziger-Isakov L, Kumar D; AST Infectious Diseases Community of Practice. Vaccination in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):311–317.
- Chesi C, Gunther M, Huzly D, et al. Immunization of liver and renal transplant recipients: a seroepidemiological and sociodemographic survey. Transpl Infect Dis 2009; 11:507–512.
- Berenguer M, Lopez-Labrador FX, Wright TL. Hepatitis C and liver transplantation. J Hepatol 2001; 35:666–678.
- Forman LM, Lewis JD, Berlin JA, Feldman HI, Lucey MR. The association between hepatitis C infection and survival after orthotopic liver transplantation. Gastroenterology 2002; 122:889–896.
- Benten D, Staufer K, Sterneck M. Orthotopic liver transplantation and what to do during follow-up: recommendations for the practitioner. Nat Clin Pract Gastroenterol Hepatol 2009; 6:23–36.
- Kotlyar DS, Campbell MS, Reddy KR. Recurrence of diseases following orthotopic liver transplantation. Am J Gastroenterol 2006; 101:1370–1378.
Since 1963, when Starzl et al performed the first successful liver transplantation,1 outcomes of this life-saving procedure have continued to improve. Long-term survival rates have increased markedly: the current 5-year rate is 73.8% and the 10-year rate is 60%.2
This success means that internists will be caring for a greater number of liver transplant recipients and managing their long-term problems, such as hypertension, diabetes mellitus, dyslipidemia, obesity, metabolic syndrome, cardiovascular disease, renal insufficiency, osteoporosis, cancer, and gout.
This review will discuss these complications, focusing on the role the primary care physician assumes beyond the first year after transplantation.
ROLE OF THE PRIMARY CARE PHYSICIAN
Hepatologists, primary care physicians, and surgeons share the care of transplant recipients. The first several weeks after transplantation require close follow-up by the hepatologist and transplantation team, with particular attention paid to the patient’s overall health and well-being, medication compliance, and biochemical and immunosuppression monitoring.
After the first year, the primary care physician assumes a greater role, becoming the main provider of the patient’s care.3,4 Good communication between the transplant center and the primary care physician should lead to a smooth transition.4 Although the hepatologist continues to manage immunosuppressive drugs, allograft rejections, and biliary complications, the primary care physician manages most of the long-term complications and thus needs to be aware of the common ones and feel comfortable managing them. Aims during visits are to screen for and detect common complications and manage them appropriately, in addition to performing annual physical examinations and routine health care. A reasonable interval for liver transplant recipients to visit their primary care physician is every 6 months.
IMMUNOSUPPRESSANT MEDICATIONS
Multiple agents are used for immunosuppression after liver transplantation:
- Calcineurin inhibitors (cyclosporine and tacrolimus)
- Antimetabolites (mycophenolate mofetil, azathioprine, and mycophenolate sodium)
- Mammalian target of rapamycin (mTOR) inhibitors (sirolimus and everolimus)
- Corticosteroids.
![]()
Table 1 lists their common side effects.
Most centers use a combination of two to four immunosuppressants as induction therapy in the immediate posttransplant period, then taper the doses and eliminate all but a calcineurin inhibitor and an antimetabolite. For example, some start with a combination of tacrolimus, mycophenolate mofetil, and a corticosteroid. The choice in the immediate posttransplant period is frequently made by the transplant center in cooperation with the hepatologist. By the time primary care physicians see these patients, they usually are on a calcineurin inhibitor alone or a calcineurin inhibitor plus mycophenolate mofetil.
Calcineurin inhibitors
Cyclosporine is metabolized by the cytochrome CYP3A4 pathway. With an average half-life of 15 hours, it is given orally, usually every 12 hours.
The dosage is adjusted according to the trough level. Higher levels are needed in the initial posttransplant period to prevent graft rejection, whereas lower levels are preferred later to decrease the occurrence and severity of adverse effects. Typical long-term trough levels are 50 to 100 ng/mL. Levels should be checked more often if an acute illness develops or the patient starts taking a potentially interfering drug.
Of importance: the dosage should be based on trough levels and not on random levels. Levels are often falsely high if blood samples are not drawn at the trough level. Repeating the measurement and making sure the sample is drawn at the trough level, ie, 12 hours after the last dose, is advised in this condition.
Cyclosporine causes widespread vasoconstriction resulting in decreased renal blood flow and systemic hypertension, often within a few days of starting it. Other important adverse effects include renal insufficiency, dyslipidemia, neurotoxicity (headache, tremor, seizure), and diabetes.
Tacrolimus is superior to cyclosporine in terms of survival, graft loss, acute rejection, and steroid-resistant rejection in the first year.5 Currently, it is the agent used most often for maintenance immunosuppression after liver transplantation.
Like cyclosporine, tacrolimus is metabolized in the liver by CYP3A4. Satisfactory trough levels after 1 year are 4 to 6 ng/mL.
The adverse effects of tacrolimus are similar to those of cyclosporine, but diabetes mellitus is more common with tacrolimus. Bone marrow suppression may occur more often with tacrolimus as well.
Antimetabolites
Antimetabolites are generally not potent enough to be used alone.
Mycophenolate mofetil causes adverse effects that include bone marrow suppression and gastrointestinal symptoms such as gastritis, diarrhea, and abdominal pain.
Azathioprine, infrequently used in transplantation in the United States, is nevertheless sometimes substituted for mycophenolate mofetil in pregnant women, as it seems safer for use in pregnancy.
Serum levels of azathioprine and mycophenolate mofetil are not routinely monitored.
mTOR inhibitors
Sirolimus and everolimus are mTOR inhibitors, inhibiting proliferation of lymphocytes.6,7
Unlike calcineurin inhibitors, mTOR inhibitors are not associated with nephrotoxicity, neurotoxicity, renal dysfunction, hypertension, or diabetes. Sirolimus is considered an alternative to calcineurin inhibitors or, in some instances, used as add-on-therapy to lower the dose of the calcineurin inhibitor.
However, sirolimus carries a potential risk of hepatic artery thrombosis, a life-threatening complication.8 This has led the US Food and Drug Administration (FDA) to require sirolimus to carry a black-box warning, and most transplant centers avoid using it in the first 30 days after transplantation.
Dyslipidemia is perhaps the most common adverse effect of sirolimus. Others include dose-related cytopenia and wound dehiscence.9
Everolimus has yet to be established for use in liver transplantation, although safety trials have been published.10,11 The FDA currently recommends against using it in the first 30 days after liver transplantation.
Both sirolimus and everolimus are metabolized by CYP3A4, which is the same metabolic pathway used by cyclosporine and tacrolimus. Hence, drugs that inhibit CYP3A4 may significantly impair clearance of both sirolimus and everolimus.
Corticosteroids
Corticosteroids have been the cornerstone of immunosuppression and remain the first line of treatment for acute allograft rejection. High intravenous doses of corticosteroids are usually started in the peritransplant period and are then switched to oral doses, which are tapered and continued with a fixed dose such as 20 mg of prednisone daily for 3 to 6 months after transplantation. However, some transplant centers keep patients on prednisone 5 mg/day indefinitely.
Adverse effects of corticosteroids include diabetes, salt and fluid retention, hypertension, hyperlipidemia, cosmetic changes (acne, cervical fat pad or “buffalo hump”), delayed wound healing, susceptibility to infection, cataracts, osteopenia, and potential adrenal suppression.12 There is concern that the use of these drugs may increase hepatitis C virus replication in patients who received a liver transplant for hepatitis C cirrhosis. Randomized trials have yielded conflicting results.13–15
Drug interactions
Certain drugs can affect the metabolism of calcineurin inhibitors and mTOR inhibitors by inducing CYP3A4, which results in decreasing the levels of the immunosuppressive drugs, or by inhibiting CYP3A4, which has the opposite effect.
Medications that can decrease the levels of calcineurin inhibitors and mTOR inhibitors:
- Anticonvulsants (carbamazepine, phenobarbital, phenytoin)
- Antibiotics (rifampin, isoniazid)
- St John’s wort.
Medications that can increase the levels of calcineurin inhibitors and mTOR inhibitors:
- Antifungals (fluconazole, ketoconazole, itraconazole, voriconazole, aspofungin)
- Antibiotics (azithromycin, erythromycin, clarithromycin)
- Nondihydropyridine calcium channel blockers (diltiazem, verapamil).16
Selected antibiotics are generally well tolerated, such as penicillins, cephalosporins, quinolones, sulfonamides, and topical antifungal agents.
LONG-TERM COMPLICATIONS
![]()
Figure 1 summarizes the common long-term complications of liver transplantation.
Hypertension
The prevalence of hypertension after liver transplantation is 40% to 85%, which is markedly higher than in patients with chronic liver disease before liver transplantation.17,18
One of the factors contributing to this increase is the use of immunosuppressive medications. Of these drugs, cyclosporine seems to be the one that most often causes an increase in both the incidence and the severity of hypertension, as it produces widespread vasoconstriction.19 Corticosteroids cause hypertension through their mineralocorticoid effects.
The diagnostic cutoffs for hypertension (ie, 140/90 mm Hg) and the treatment goals in posttransplant patients are similar to those in the general population. However, at our institution we target a blood pressure of less than 130/80 mm Hg in transplant patients because they have a high prevalence of other cardiovascular risk factors such as diabetes, obesity, and renal insufficiency.20
Dihydropyridine calcium channel blockers such as amlodipine and nifedipine are considered the best first-line agents because they dilate renal afferent arterioles, an effect that may counteract the vasoconstriction mediated by calcineurin inhibitors. Nondihydropyridine calcium channel blockers such as diltiazem and verapamil tend to have more marked negative inotropic effects and are not recommended in liver transplant recipients because they increase the levels of calcineurin inhibitors.21
Diuretics (eg, furosemide) might be the second-line agents, especially in patients with peripheral edema.16 One should be vigilant for hyperuricemia if thiazide agents are used.
Angiotensin-converting-enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) are typically avoided in the early posttransplant period, but they can be started later and have additional benefits in patients with diabetes and congestive heart failure. Starting ACE inhibitors is acceptable in these patients unless there is a contraindication such as allergy to ACE inhibitors, hypotension, history of bilateral renal artery stenosis, significant hyperkalemia, or acute kidney injury. Monitor the serum potassium level closely for hyperkalemia in patients concurrently using calcineurin inhibitors.
Alpha-blockers and beta-blockers can be used as add-on therapy in patients with uncontrolled hypertension with the exception of carvedilol, because it increases the levels of calcineurin inhibitors.22
Blood pressure monitoring by the primary care physician is recommended every 6 months after the early posttransplant period, or more frequently when changes in treatment are being considered.
If hypertension continues to be inadequately controlled despite treatment, changing the immunosuppressive drugs or decreasing the doses can be considered, but the transplant hepatologist must be involved in this decision.23,24
Diabetes mellitus
The prevalence of diabetes mellitus is higher in liver transplant recipients than in the general population, reaching 30% to 40%.17,25 In addition to preexisting diabetes, 15% of liver transplant recipients develop new-onset diabetes.26,27
Risk factors for developing diabetes after liver transplantation include African American or Hispanic ethnicity, obesity, family history, pretransplant diabetes, hepatitis C virus infection, use of corticosteroids, and use of calcineurin inhibitors (tacrolimus more than cyclosporine) and sirolimus.26
In addition to increasing the risk of cardiovascular disease and other diseases, diabetes decreases both patient and graft survival after liver transplantation.28
The management of diabetes and the treatment target after transplantation should follow the American Diabetes Association guidelines for the treatment of type 2 diabetes mellitus.29 Lifestyle modifications, diet, and exercise are as important for transplant patients as for the nontransplant population. Insulin therapy is usually needed in the early posttransplant period to control blood glucose levels well, especially with the high doses of corticosteroids used during the first few weeks. No trials to date have compared oral agents in posttransplant patients. Therefore, the choice of oral hypoglycemic agents should be individualized on the basis of the patient’s characteristics and comorbidities.
We recommend that primary care providers screen all liver transplant recipients for diabetes regardless of their pretransplant status. This can be done by obtaining regular fasting blood glucose levels or a hemoglobin A1c level every 6 months. Additionally, liver transplant recipients diagnosed with diabetes require annual eye examinations to look for cataracts and diabetes-related changes.30
Dyslipidemia
On November 12, 2013, the American College of Cardiology and the American Heart Association (ACC/AHA) released new clinical practice guidelines for treating blood cholesterol levels.31,32 According to these new guidelines, there are four groups of patients for whom treatment with statins is clearly indicated:
- Patients with cardiovascular disease
- Patients with low-density lipoprotein cholesterol (LDL-C) levels ≥ 190 mg/dL
- Patients 40 to 75 years old with type 2 diabetes
- Patients 40 to 75 years old with an estimated 10-year risk of cardiovascular disease of 7.5% or greater.
Liver transplant recipients should be evaluated on an individual basis to see if they fit in any of the four groups and if statin treatment therefore needs to be initiated.
A few things need to be kept in mind. First, the incidence of dyslipidemia after liver transplantation is estimated to be 45% to 69%. Risk factors include obesity, diabetes mellitus, cholestatic liver disease, and immunosuppressant medications.33 Sirolimus has a significant and well-documented association with dyslipidemia. Cyclosporine and corticosteroids are also strongly associated with dyslipidemia. Tacrolimus has a minor effect, and mycophenolate mofetil and azathioprine have no significant effects on serum lipid levels.16
Second, of the seven currently marketed statins, pravastatin and fluvastatin are preferred in liver transplant recipients because they are not metabolized by the same cytochrome CYP3A4 pathway that metabolizes calcineurin inhibitors and sirolimus.34 The doses of 40 to 80 mg daily of pravastatin or 40 mg twice daily of fluvastatin lower low-density lipoprotein cholesterol (LDL-C) levels by approximately 30% to 35%. However, these two agents are considered “moderate-intensity” statins according to the new ACC/AHA guidelines. The only two “high-intensity” statins are atorvastatin (40–80 mg) and rosuvastatin (20–40 mg), but they are both metabolized by CYP3A4. Therefore it is prudent to avoid them with the concurrent use of a calcineurin inhibitor or tacrolimus.
Gemfibrozil does not lower LDL-C and should not be used concomitantly with statins due to unacceptable risk of rhabdomyolysis and myopathy. Fenofibrates are usually avoided due to potential nephrotoxicity in patients receiving cyclosporine. Bile acid sequestrants (cholestyramine, colestipol, colesevelam) can decrease plasma mycophenolate mofetil levels by 35%.16,35 Thus, these agents should be avoided if mycophenolate mofetil is used.
It is reasonable to screen all liver transplant recipients with a fasting lipid profile at 3, 6, and 12 months after transplantation and annually thereafter. Creatine kinase should be measured if the patient complains of severe muscle pain or weakness but not on a routine basis.
Obesity
Approximately one-third of patients who are of normal weight at the time of transplantation will become obese afterward.18,25 Corticosteroid use is an important risk factor for posttransplant obesity, and tapering these drugs helps reduce weight.36 Patients treated with cyclosporine are more likely to gain weight than those who receive tacrolimus.37
Of importance: nonalcoholic fatty liver disease, currently the most common cause of chronic liver disease in adults, is rapidly increasing as an indication for liver transplantation. In fact, the proportion of liver transplantation procedures for nonalcoholic steatohepatitis-related cirrhosis increased from 1.2% in 2001 to 9.7% in 2009, and nonalcoholic steatohepatitis is expected to become the leading indication for liver transplantation in the next 20 years. And because nonalcoholic fatty liver disease is directly linked to obesity, the prevalence of obesity as a complication of liver transplantation will most likely increase in the near future.
Overweight liver transplant recipients may have great difficulty losing weight. Treatment starts with patient education on caloric restriction and exercise. If traditional measures fail to result in adequate weight loss, additional options include switching from cyclosporine to tacrolimus.23
Bariatric surgery may become an option for posttransplant patients. In a recent case series from the University of Minnesota, Al-Nowaylati et al38 reported their experience with seven patients who underwent orthotopic liver transplantation and then open Roux-en-Y gastric bypass. After bariatric surgery, the patients’ mean body mass index declined significantly, and glycemic control and high-density lipoprotein cholesterol (HDL-C) levels improved. However, one patient died of multiple organ failure, to which the bariatric surgery might have contributed.38
Heimbach et al39 conducted a study in patients referred for liver transplantation for whom a rigorous noninvasive weight-loss program before transplantation had failed. The researchers performed combined liver transplantation and sleeve gastrectomy in seven carefully selected patients who had failed to achieve weight loss to a body mass index less than 35 kg/m2 before transplantation. All seven patients lost weight, decreasing their mean body mass index from 49 kg/m2 before the procedure to 29 kg/m2 at last follow-up, and none of them developed posttransplant diabetes or steatosis.
At this time, there is not enough evidence to recommend concurrent orthotopic liver transplantation plus bariatric surgery, or combined orthotopic liver transplantation and sleeve gastrectomy. More study is needed to further evaluate these advanced approaches.
Posttransplant metabolic syndrome
Metabolic syndrome is common after liver transplantation and is strongly associated with increased morbidity in this patient population.40,41 The general definition of metabolic syndrome includes a combination of at least three of the following: hypertension, insulin resistance, hypertriglyceridemia, low HDL-C, and obesity.
The prevalence of metabolic syndrome is higher in patients after liver transplantation than in nontransplant patients. In a review of 252 liver transplant recipients, 52% were diagnosed with posttransplant metabolic syndrome, but only 5% had had it pretransplant.42
Careful screening for posttransplant metabolic syndrome and early recognition of risk factors are important. Nevertheless, the treatment of this condition depends on treating its components according to recommended guidelines.41
Cardiovascular disease
The incidence of cardiovascular morbidity and death is increased after liver transplantation.24 In addition, after liver transplantation, cardiovascular disease is a major cause of death unrelated to liver disease. It accounts for 12% to 16% of deaths and is the third most common cause of late mortality after liver transplantation.43 Of note, a recent study by our group demonstrated that patients undergoing liver transplantation for nonalcoholic steatohepatitis had a significantly higher risk of a cardiovascular event during the 3 years after transplantation than patients undergoing liver transplantation for cholestatic liver disease.44
Risk factors for cardiovascular disease after liver transplantation include older age at transplantation, male sex, posttransplant diabetes, posttransplant hypertension, and the use of mycophenolate mofetil.44 Modifying the risk factors is essential in decreasing the risk of cardiovascular events.
It is reasonable to perform dobutamine stress testing every 3 to 5 years in patients with multiple risk factors for cardiovascular disease, or more frequently in those with preexisting coronary artery disease.45,46
Malignancy
The risk of several malignancies increases after liver transplantation. Liver transplant recipients have an incidence of cancer 2.1 to 4.3 times greater than age- and sex-matched controls.24,47–49
Skin cancers are the most common and account for almost 40% of malignancies in organ transplant recipients.50 Whereas basal cell carcinoma is more common in the general population, squamous cell carcinoma is equally common in liver transplant recipients.
Multiple clinical studies have linked calcineurin inhibitors and azathioprine to the development of skin cancer. Annual skin examinations in addition to avoiding other risk factors such as smoking and sun exposure are generally recommended. Changing the immunosuppressants to sirolimus in high-risk patients may lower their chance of developing skin cancer.51,52
Patients with ulcerative colitis who undergo liver transplantation because of sclerosing cholangitis are at higher risk of colon cancer and require annual colonoscopy with surveillance biopsies. Patients who undergo transplantation for alcoholic liver disease seem to have a higher risk of pulmonary and oropharyngeal cancers.53,54
It is important that transplant patients adhere to recommended cancer screening guidelines, in view of their increased risk. Studies have shown improved overall survival in liver transplant recipients who underwent intensive cancer surveillance.55
Renal insufficiency
Renal insufficiency is a well-recognized complication of liver transplantation and is associated with an increased long-term death rate.56,57
The incidence of renal insufficiency increases dramatically over time. Ojo et al,57 in a study of almost 37,000 liver transplant recipients, found that the incidence of chronic kidney disease (defined as an estimated glomerular filtration rate < 30 mL/min/1.73 m2) was 13.9% at 3 years, 18% at 5 years, and approximately 26% at 10 years.
Risk factors include the use of calcineurin inhibitors (both cyclosporine and tacrolimus), older age, female sex, lower pretransplant glomerular filtration rate, postoperative acute renal failure, diabetes, hypertension, hepatitis C virus infection, and transplantation before 1998.58,59 Replacing a calcineurin inhibitor with mycophenolate mofetil or sirolimus may be considered with communication with the transplant center, as mycophenolate mofetil or sirolimus are associated with a lower risk of renal injury.60–64
Starting 1 year after liver transplantation, primary care providers should screen for renal dysfunction by obtaining kidney function tests every 6 months, including urinalysis and microalbuminuria assessment. Equations for estimating the glomerular filtration rate used in practice, such as the Modification of Diet in Renal Disease Study equation, rely mainly on serum creatinine, which may lead to overestimating renal function in some circumstances. Therefore, other equations can be used to confirm the estimated glomerular filtration rate measured by creatinine clearance, and to more accurately evaluate kidney function. Calculators are available at www.kidney.org/professionals/kdoqi/gfr_calculator.
All liver transplant recipients should avoid nonsteroidal anti-inflammatory drugs and nephrotoxic medications, and should have their hypertension and diabetes adequately controlled.
Bone diseases
Osteopenia is another major complication of liver transplantation. One-third of liver transplant recipients have a bone mineral density below the fracture threshold.65
Multiple factors contribute to increased bone loss after transplantation, including use of corticosteroids, use of calcineurin inhibitors (cyclosporine, tacrolimus), poor nutrition, vitamin D deficiency, immobility, sarcopenia (reduced muscle mass), hypogonadism, smoking, and alcohol abuse.66 Even at low doses of less than 7.5 mg per day, corticosteroids inhibit osteoblast activity and increase bone resorption.
Studies have reported rapid bone loss at around 6 months after transplantation.67–69 However, long-term follow-up of bone mineral density up to 15 years after transplantation revealed an improvement mainly in the 2nd postoperative year, with no deterioration afterward.65
High-risk patients need to be identified early with appropriate screening and evaluation. Evaluation includes dual-energy x-ray absorptiometry and serum levels of calcium, phosphorous, parathyroid hormone, testosterone (men), estradiol (women), and alpha-25-hydroxyvitamin D. These tests are typically done before transplantation and then every other year afterward.
We recommend a daily dose of calcitriol and a calcium supplement to all our liver transplant patients.70 If osteoporosis (a T-score 2.5 or more standard deviations below the mean) or a fragility fracture occurs, then the patient may benefit from an oral bisphosphonate. Calcitonin has also been shown to improve bone mineral density in patients with osteoporosis after liver transplantation.71
Hyperuricemia and gout
Although hyperuricemia is common in liver transplant recipients (reported in approximately 47%), the development of clinical gout is less common (6%).72 Asymptomatic hyperuricemia requires observation only and is not usually treated in liver transplant recipients.
Acute attacks of gout are typically managed with colchicine 0.6 mg every 2 hours, up to five doses. Prednisone can be considered if symptoms persist despite treatment with colchicine. Allopurinol in an initial dose of 100 mg daily is used as maintenance therapy to reduce production of uric acid.73 However, because of the potential for drug interactions, the combination of azathioprine and allopurinol should be avoided.
Psychiatric complications and quality of life
Depression is common in liver transplant recipients, significantly more so in patients who received a transplant because of hepatitis C.74 The type of immunosuppressant is not associated with the incidence of depression. When indicated, the internist may start the patient on a selective serotonin reuptake inhibitor such as citalopram 20 mg daily, as these medications are usually effective and well tolerated in liver transplant patients.73
Liver transplantation has a major positive effect on quality of life. Most patients with end-stage liver disease have poor quality of life before transplantation, but this seems to improve notably afterward. A meta-analysis showed significant improvement in posttransplant physical health, sexual functioning, daily activities, and social functioning compared with before transplantation.75
Alcohol abuse and smoking
Patients who underwent liver transplantation because of alcoholic liver disease should be advised to abstain from alcohol.19 Patients who underwent the procedure for a different indication are advised to avoid excessive alcohol intake, as it is proven to lower the survival rate.20 Alcohol recidivism and smoking (including marijuana) are major problems, and internists are best positioned to address these issues and treat them.
Vaccinations
All liver transplant recipients should be vaccinated against influenza, pneumococcal infection, and tetanus. Hepatitis A and B vaccines are typically given before transplantation. In general, live vaccines such as measles-mumps-rubella and varicella are not recommended after any solid organ transplant.76
A study in Germany showed that immunization rates were too low in solid-organ transplant recipients, and almost 90% of patients were not adequately informed about immunizations.77 Hence, there may be room for improvement, and primary care providers should take the lead toward better outcomes in this regard.
Recurrence of the primary liver disease after transplantation
Different primary liver diseases recur with different frequencies.
Hepatitis C has the highest rate of recurrence of the liver diseases.78,79 Reinfection with hepatitis C virus after liver transplantation is almost universal and can follow different patterns. One of the most aggressive patterns is fibrosing cholestatic hepatitis, which frequently leads to graft failure and death, and hence necessitates urgent detection and treatment.
Hepatocellular carcinoma also has a high recurrence rate.80 Surveillance with liver ultrasonography or computed tomography is required every 6 months for the first 5 years after liver transplantation.
Other liver diseases. Nonalcoholic steatohepatitis, primary biliary cirrhosis, primary sclerosing cholangitis, autoimmune hepatitis, and hepatitis B infection also tend to recur after liver transplantation.46,81 On the other hand, alpha-1 antitrypsin deficiency, Wilson disease, hemochromatosis, and metabolic disorders are “cured” after liver transplantation.
It is important to detect any increase in liver enzymes above baseline. An elevation of 1.5 times the upper limit of normal or more should trigger further investigation.
Allograft dysfunction
A number of complications can develop in the liver allograft and result in abnormal liver function tests and, if not treated, graft failure. The most common causes of late graft dysfunction include recurrence of primary liver disease, biliary complications, and chronic rejection.46
Vascular complications include hepatic artery thrombosis and stenosis and are usually evaluated by liver ultrasonography and Doppler scan of the hepatic artery and venous structures.24
Biliary strictures give a cholestatic picture, with elevated bilirubin and greater elevation of alkaline phosphatase than of alanine aminotransferase and aspartate aminotransferase. Strictures are usually treated by endoscopic dilation and stenting, but they may eventually require surgery.
Late acute cellular rejections occur in 10% to 20% of cases and are a risk factor for chronic rejection. Liver biopsy is needed to make the diagnosis, and pulsed doses of corticosteroids remain the backbone of treatment therapy.
Chronic rejection is not common, occurring in 3% to 4% of liver transplant recipients.46 Treatment is based on increasing immunosuppression and ensuring compliance with prescribed medications. However, chronic rejection may not respond well, and repeat transplantation may be the last resort for some patients.
WHEN TO REFER TO THE HEPATOLOGIST
Some situations require referral to the hepatologist or the transplant center. In general, the following are best managed by a hepatologist: adjustment of immunosuppressive drugs and dosages, allograft dysfunction, vascular and biliary complications, progressing renal dysfunction, and recurrence of primary liver disease. Early communication with a hepatologist and the transplant center is recommended in these cases.
Since 1963, when Starzl et al performed the first successful liver transplantation,1 outcomes of this life-saving procedure have continued to improve. Long-term survival rates have increased markedly: the current 5-year rate is 73.8% and the 10-year rate is 60%.2
This success means that internists will be caring for a greater number of liver transplant recipients and managing their long-term problems, such as hypertension, diabetes mellitus, dyslipidemia, obesity, metabolic syndrome, cardiovascular disease, renal insufficiency, osteoporosis, cancer, and gout.
This review will discuss these complications, focusing on the role the primary care physician assumes beyond the first year after transplantation.
ROLE OF THE PRIMARY CARE PHYSICIAN
Hepatologists, primary care physicians, and surgeons share the care of transplant recipients. The first several weeks after transplantation require close follow-up by the hepatologist and transplantation team, with particular attention paid to the patient’s overall health and well-being, medication compliance, and biochemical and immunosuppression monitoring.
After the first year, the primary care physician assumes a greater role, becoming the main provider of the patient’s care.3,4 Good communication between the transplant center and the primary care physician should lead to a smooth transition.4 Although the hepatologist continues to manage immunosuppressive drugs, allograft rejections, and biliary complications, the primary care physician manages most of the long-term complications and thus needs to be aware of the common ones and feel comfortable managing them. Aims during visits are to screen for and detect common complications and manage them appropriately, in addition to performing annual physical examinations and routine health care. A reasonable interval for liver transplant recipients to visit their primary care physician is every 6 months.
IMMUNOSUPPRESSANT MEDICATIONS
Multiple agents are used for immunosuppression after liver transplantation:
- Calcineurin inhibitors (cyclosporine and tacrolimus)
- Antimetabolites (mycophenolate mofetil, azathioprine, and mycophenolate sodium)
- Mammalian target of rapamycin (mTOR) inhibitors (sirolimus and everolimus)
- Corticosteroids.
![]()
Table 1 lists their common side effects.
Most centers use a combination of two to four immunosuppressants as induction therapy in the immediate posttransplant period, then taper the doses and eliminate all but a calcineurin inhibitor and an antimetabolite. For example, some start with a combination of tacrolimus, mycophenolate mofetil, and a corticosteroid. The choice in the immediate posttransplant period is frequently made by the transplant center in cooperation with the hepatologist. By the time primary care physicians see these patients, they usually are on a calcineurin inhibitor alone or a calcineurin inhibitor plus mycophenolate mofetil.
Calcineurin inhibitors
Cyclosporine is metabolized by the cytochrome CYP3A4 pathway. With an average half-life of 15 hours, it is given orally, usually every 12 hours.
The dosage is adjusted according to the trough level. Higher levels are needed in the initial posttransplant period to prevent graft rejection, whereas lower levels are preferred later to decrease the occurrence and severity of adverse effects. Typical long-term trough levels are 50 to 100 ng/mL. Levels should be checked more often if an acute illness develops or the patient starts taking a potentially interfering drug.
Of importance: the dosage should be based on trough levels and not on random levels. Levels are often falsely high if blood samples are not drawn at the trough level. Repeating the measurement and making sure the sample is drawn at the trough level, ie, 12 hours after the last dose, is advised in this condition.
Cyclosporine causes widespread vasoconstriction resulting in decreased renal blood flow and systemic hypertension, often within a few days of starting it. Other important adverse effects include renal insufficiency, dyslipidemia, neurotoxicity (headache, tremor, seizure), and diabetes.
Tacrolimus is superior to cyclosporine in terms of survival, graft loss, acute rejection, and steroid-resistant rejection in the first year.5 Currently, it is the agent used most often for maintenance immunosuppression after liver transplantation.
Like cyclosporine, tacrolimus is metabolized in the liver by CYP3A4. Satisfactory trough levels after 1 year are 4 to 6 ng/mL.
The adverse effects of tacrolimus are similar to those of cyclosporine, but diabetes mellitus is more common with tacrolimus. Bone marrow suppression may occur more often with tacrolimus as well.
Antimetabolites
Antimetabolites are generally not potent enough to be used alone.
Mycophenolate mofetil causes adverse effects that include bone marrow suppression and gastrointestinal symptoms such as gastritis, diarrhea, and abdominal pain.
Azathioprine, infrequently used in transplantation in the United States, is nevertheless sometimes substituted for mycophenolate mofetil in pregnant women, as it seems safer for use in pregnancy.
Serum levels of azathioprine and mycophenolate mofetil are not routinely monitored.
mTOR inhibitors
Sirolimus and everolimus are mTOR inhibitors, inhibiting proliferation of lymphocytes.6,7
Unlike calcineurin inhibitors, mTOR inhibitors are not associated with nephrotoxicity, neurotoxicity, renal dysfunction, hypertension, or diabetes. Sirolimus is considered an alternative to calcineurin inhibitors or, in some instances, used as add-on-therapy to lower the dose of the calcineurin inhibitor.
However, sirolimus carries a potential risk of hepatic artery thrombosis, a life-threatening complication.8 This has led the US Food and Drug Administration (FDA) to require sirolimus to carry a black-box warning, and most transplant centers avoid using it in the first 30 days after transplantation.
Dyslipidemia is perhaps the most common adverse effect of sirolimus. Others include dose-related cytopenia and wound dehiscence.9
Everolimus has yet to be established for use in liver transplantation, although safety trials have been published.10,11 The FDA currently recommends against using it in the first 30 days after liver transplantation.
Both sirolimus and everolimus are metabolized by CYP3A4, which is the same metabolic pathway used by cyclosporine and tacrolimus. Hence, drugs that inhibit CYP3A4 may significantly impair clearance of both sirolimus and everolimus.
Corticosteroids
Corticosteroids have been the cornerstone of immunosuppression and remain the first line of treatment for acute allograft rejection. High intravenous doses of corticosteroids are usually started in the peritransplant period and are then switched to oral doses, which are tapered and continued with a fixed dose such as 20 mg of prednisone daily for 3 to 6 months after transplantation. However, some transplant centers keep patients on prednisone 5 mg/day indefinitely.
Adverse effects of corticosteroids include diabetes, salt and fluid retention, hypertension, hyperlipidemia, cosmetic changes (acne, cervical fat pad or “buffalo hump”), delayed wound healing, susceptibility to infection, cataracts, osteopenia, and potential adrenal suppression.12 There is concern that the use of these drugs may increase hepatitis C virus replication in patients who received a liver transplant for hepatitis C cirrhosis. Randomized trials have yielded conflicting results.13–15
Drug interactions
Certain drugs can affect the metabolism of calcineurin inhibitors and mTOR inhibitors by inducing CYP3A4, which results in decreasing the levels of the immunosuppressive drugs, or by inhibiting CYP3A4, which has the opposite effect.
Medications that can decrease the levels of calcineurin inhibitors and mTOR inhibitors:
- Anticonvulsants (carbamazepine, phenobarbital, phenytoin)
- Antibiotics (rifampin, isoniazid)
- St John’s wort.
Medications that can increase the levels of calcineurin inhibitors and mTOR inhibitors:
- Antifungals (fluconazole, ketoconazole, itraconazole, voriconazole, aspofungin)
- Antibiotics (azithromycin, erythromycin, clarithromycin)
- Nondihydropyridine calcium channel blockers (diltiazem, verapamil).16
Selected antibiotics are generally well tolerated, such as penicillins, cephalosporins, quinolones, sulfonamides, and topical antifungal agents.
LONG-TERM COMPLICATIONS
![]()
Figure 1 summarizes the common long-term complications of liver transplantation.
Hypertension
The prevalence of hypertension after liver transplantation is 40% to 85%, which is markedly higher than in patients with chronic liver disease before liver transplantation.17,18
One of the factors contributing to this increase is the use of immunosuppressive medications. Of these drugs, cyclosporine seems to be the one that most often causes an increase in both the incidence and the severity of hypertension, as it produces widespread vasoconstriction.19 Corticosteroids cause hypertension through their mineralocorticoid effects.
The diagnostic cutoffs for hypertension (ie, 140/90 mm Hg) and the treatment goals in posttransplant patients are similar to those in the general population. However, at our institution we target a blood pressure of less than 130/80 mm Hg in transplant patients because they have a high prevalence of other cardiovascular risk factors such as diabetes, obesity, and renal insufficiency.20
Dihydropyridine calcium channel blockers such as amlodipine and nifedipine are considered the best first-line agents because they dilate renal afferent arterioles, an effect that may counteract the vasoconstriction mediated by calcineurin inhibitors. Nondihydropyridine calcium channel blockers such as diltiazem and verapamil tend to have more marked negative inotropic effects and are not recommended in liver transplant recipients because they increase the levels of calcineurin inhibitors.21
Diuretics (eg, furosemide) might be the second-line agents, especially in patients with peripheral edema.16 One should be vigilant for hyperuricemia if thiazide agents are used.
Angiotensin-converting-enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) are typically avoided in the early posttransplant period, but they can be started later and have additional benefits in patients with diabetes and congestive heart failure. Starting ACE inhibitors is acceptable in these patients unless there is a contraindication such as allergy to ACE inhibitors, hypotension, history of bilateral renal artery stenosis, significant hyperkalemia, or acute kidney injury. Monitor the serum potassium level closely for hyperkalemia in patients concurrently using calcineurin inhibitors.
Alpha-blockers and beta-blockers can be used as add-on therapy in patients with uncontrolled hypertension with the exception of carvedilol, because it increases the levels of calcineurin inhibitors.22
Blood pressure monitoring by the primary care physician is recommended every 6 months after the early posttransplant period, or more frequently when changes in treatment are being considered.
If hypertension continues to be inadequately controlled despite treatment, changing the immunosuppressive drugs or decreasing the doses can be considered, but the transplant hepatologist must be involved in this decision.23,24
Diabetes mellitus
The prevalence of diabetes mellitus is higher in liver transplant recipients than in the general population, reaching 30% to 40%.17,25 In addition to preexisting diabetes, 15% of liver transplant recipients develop new-onset diabetes.26,27
Risk factors for developing diabetes after liver transplantation include African American or Hispanic ethnicity, obesity, family history, pretransplant diabetes, hepatitis C virus infection, use of corticosteroids, and use of calcineurin inhibitors (tacrolimus more than cyclosporine) and sirolimus.26
In addition to increasing the risk of cardiovascular disease and other diseases, diabetes decreases both patient and graft survival after liver transplantation.28
The management of diabetes and the treatment target after transplantation should follow the American Diabetes Association guidelines for the treatment of type 2 diabetes mellitus.29 Lifestyle modifications, diet, and exercise are as important for transplant patients as for the nontransplant population. Insulin therapy is usually needed in the early posttransplant period to control blood glucose levels well, especially with the high doses of corticosteroids used during the first few weeks. No trials to date have compared oral agents in posttransplant patients. Therefore, the choice of oral hypoglycemic agents should be individualized on the basis of the patient’s characteristics and comorbidities.
We recommend that primary care providers screen all liver transplant recipients for diabetes regardless of their pretransplant status. This can be done by obtaining regular fasting blood glucose levels or a hemoglobin A1c level every 6 months. Additionally, liver transplant recipients diagnosed with diabetes require annual eye examinations to look for cataracts and diabetes-related changes.30
Dyslipidemia
On November 12, 2013, the American College of Cardiology and the American Heart Association (ACC/AHA) released new clinical practice guidelines for treating blood cholesterol levels.31,32 According to these new guidelines, there are four groups of patients for whom treatment with statins is clearly indicated:
- Patients with cardiovascular disease
- Patients with low-density lipoprotein cholesterol (LDL-C) levels ≥ 190 mg/dL
- Patients 40 to 75 years old with type 2 diabetes
- Patients 40 to 75 years old with an estimated 10-year risk of cardiovascular disease of 7.5% or greater.
Liver transplant recipients should be evaluated on an individual basis to see if they fit in any of the four groups and if statin treatment therefore needs to be initiated.
A few things need to be kept in mind. First, the incidence of dyslipidemia after liver transplantation is estimated to be 45% to 69%. Risk factors include obesity, diabetes mellitus, cholestatic liver disease, and immunosuppressant medications.33 Sirolimus has a significant and well-documented association with dyslipidemia. Cyclosporine and corticosteroids are also strongly associated with dyslipidemia. Tacrolimus has a minor effect, and mycophenolate mofetil and azathioprine have no significant effects on serum lipid levels.16
Second, of the seven currently marketed statins, pravastatin and fluvastatin are preferred in liver transplant recipients because they are not metabolized by the same cytochrome CYP3A4 pathway that metabolizes calcineurin inhibitors and sirolimus.34 The doses of 40 to 80 mg daily of pravastatin or 40 mg twice daily of fluvastatin lower low-density lipoprotein cholesterol (LDL-C) levels by approximately 30% to 35%. However, these two agents are considered “moderate-intensity” statins according to the new ACC/AHA guidelines. The only two “high-intensity” statins are atorvastatin (40–80 mg) and rosuvastatin (20–40 mg), but they are both metabolized by CYP3A4. Therefore it is prudent to avoid them with the concurrent use of a calcineurin inhibitor or tacrolimus.
Gemfibrozil does not lower LDL-C and should not be used concomitantly with statins due to unacceptable risk of rhabdomyolysis and myopathy. Fenofibrates are usually avoided due to potential nephrotoxicity in patients receiving cyclosporine. Bile acid sequestrants (cholestyramine, colestipol, colesevelam) can decrease plasma mycophenolate mofetil levels by 35%.16,35 Thus, these agents should be avoided if mycophenolate mofetil is used.
It is reasonable to screen all liver transplant recipients with a fasting lipid profile at 3, 6, and 12 months after transplantation and annually thereafter. Creatine kinase should be measured if the patient complains of severe muscle pain or weakness but not on a routine basis.
Obesity
Approximately one-third of patients who are of normal weight at the time of transplantation will become obese afterward.18,25 Corticosteroid use is an important risk factor for posttransplant obesity, and tapering these drugs helps reduce weight.36 Patients treated with cyclosporine are more likely to gain weight than those who receive tacrolimus.37
Of importance: nonalcoholic fatty liver disease, currently the most common cause of chronic liver disease in adults, is rapidly increasing as an indication for liver transplantation. In fact, the proportion of liver transplantation procedures for nonalcoholic steatohepatitis-related cirrhosis increased from 1.2% in 2001 to 9.7% in 2009, and nonalcoholic steatohepatitis is expected to become the leading indication for liver transplantation in the next 20 years. And because nonalcoholic fatty liver disease is directly linked to obesity, the prevalence of obesity as a complication of liver transplantation will most likely increase in the near future.
Overweight liver transplant recipients may have great difficulty losing weight. Treatment starts with patient education on caloric restriction and exercise. If traditional measures fail to result in adequate weight loss, additional options include switching from cyclosporine to tacrolimus.23
Bariatric surgery may become an option for posttransplant patients. In a recent case series from the University of Minnesota, Al-Nowaylati et al38 reported their experience with seven patients who underwent orthotopic liver transplantation and then open Roux-en-Y gastric bypass. After bariatric surgery, the patients’ mean body mass index declined significantly, and glycemic control and high-density lipoprotein cholesterol (HDL-C) levels improved. However, one patient died of multiple organ failure, to which the bariatric surgery might have contributed.38
Heimbach et al39 conducted a study in patients referred for liver transplantation for whom a rigorous noninvasive weight-loss program before transplantation had failed. The researchers performed combined liver transplantation and sleeve gastrectomy in seven carefully selected patients who had failed to achieve weight loss to a body mass index less than 35 kg/m2 before transplantation. All seven patients lost weight, decreasing their mean body mass index from 49 kg/m2 before the procedure to 29 kg/m2 at last follow-up, and none of them developed posttransplant diabetes or steatosis.
At this time, there is not enough evidence to recommend concurrent orthotopic liver transplantation plus bariatric surgery, or combined orthotopic liver transplantation and sleeve gastrectomy. More study is needed to further evaluate these advanced approaches.
Posttransplant metabolic syndrome
Metabolic syndrome is common after liver transplantation and is strongly associated with increased morbidity in this patient population.40,41 The general definition of metabolic syndrome includes a combination of at least three of the following: hypertension, insulin resistance, hypertriglyceridemia, low HDL-C, and obesity.
The prevalence of metabolic syndrome is higher in patients after liver transplantation than in nontransplant patients. In a review of 252 liver transplant recipients, 52% were diagnosed with posttransplant metabolic syndrome, but only 5% had had it pretransplant.42
Careful screening for posttransplant metabolic syndrome and early recognition of risk factors are important. Nevertheless, the treatment of this condition depends on treating its components according to recommended guidelines.41
Cardiovascular disease
The incidence of cardiovascular morbidity and death is increased after liver transplantation.24 In addition, after liver transplantation, cardiovascular disease is a major cause of death unrelated to liver disease. It accounts for 12% to 16% of deaths and is the third most common cause of late mortality after liver transplantation.43 Of note, a recent study by our group demonstrated that patients undergoing liver transplantation for nonalcoholic steatohepatitis had a significantly higher risk of a cardiovascular event during the 3 years after transplantation than patients undergoing liver transplantation for cholestatic liver disease.44
Risk factors for cardiovascular disease after liver transplantation include older age at transplantation, male sex, posttransplant diabetes, posttransplant hypertension, and the use of mycophenolate mofetil.44 Modifying the risk factors is essential in decreasing the risk of cardiovascular events.
It is reasonable to perform dobutamine stress testing every 3 to 5 years in patients with multiple risk factors for cardiovascular disease, or more frequently in those with preexisting coronary artery disease.45,46
Malignancy
The risk of several malignancies increases after liver transplantation. Liver transplant recipients have an incidence of cancer 2.1 to 4.3 times greater than age- and sex-matched controls.24,47–49
Skin cancers are the most common and account for almost 40% of malignancies in organ transplant recipients.50 Whereas basal cell carcinoma is more common in the general population, squamous cell carcinoma is equally common in liver transplant recipients.
Multiple clinical studies have linked calcineurin inhibitors and azathioprine to the development of skin cancer. Annual skin examinations in addition to avoiding other risk factors such as smoking and sun exposure are generally recommended. Changing the immunosuppressants to sirolimus in high-risk patients may lower their chance of developing skin cancer.51,52
Patients with ulcerative colitis who undergo liver transplantation because of sclerosing cholangitis are at higher risk of colon cancer and require annual colonoscopy with surveillance biopsies. Patients who undergo transplantation for alcoholic liver disease seem to have a higher risk of pulmonary and oropharyngeal cancers.53,54
It is important that transplant patients adhere to recommended cancer screening guidelines, in view of their increased risk. Studies have shown improved overall survival in liver transplant recipients who underwent intensive cancer surveillance.55
Renal insufficiency
Renal insufficiency is a well-recognized complication of liver transplantation and is associated with an increased long-term death rate.56,57
The incidence of renal insufficiency increases dramatically over time. Ojo et al,57 in a study of almost 37,000 liver transplant recipients, found that the incidence of chronic kidney disease (defined as an estimated glomerular filtration rate < 30 mL/min/1.73 m2) was 13.9% at 3 years, 18% at 5 years, and approximately 26% at 10 years.
Risk factors include the use of calcineurin inhibitors (both cyclosporine and tacrolimus), older age, female sex, lower pretransplant glomerular filtration rate, postoperative acute renal failure, diabetes, hypertension, hepatitis C virus infection, and transplantation before 1998.58,59 Replacing a calcineurin inhibitor with mycophenolate mofetil or sirolimus may be considered with communication with the transplant center, as mycophenolate mofetil or sirolimus are associated with a lower risk of renal injury.60–64
Starting 1 year after liver transplantation, primary care providers should screen for renal dysfunction by obtaining kidney function tests every 6 months, including urinalysis and microalbuminuria assessment. Equations for estimating the glomerular filtration rate used in practice, such as the Modification of Diet in Renal Disease Study equation, rely mainly on serum creatinine, which may lead to overestimating renal function in some circumstances. Therefore, other equations can be used to confirm the estimated glomerular filtration rate measured by creatinine clearance, and to more accurately evaluate kidney function. Calculators are available at www.kidney.org/professionals/kdoqi/gfr_calculator.
All liver transplant recipients should avoid nonsteroidal anti-inflammatory drugs and nephrotoxic medications, and should have their hypertension and diabetes adequately controlled.
Bone diseases
Osteopenia is another major complication of liver transplantation. One-third of liver transplant recipients have a bone mineral density below the fracture threshold.65
Multiple factors contribute to increased bone loss after transplantation, including use of corticosteroids, use of calcineurin inhibitors (cyclosporine, tacrolimus), poor nutrition, vitamin D deficiency, immobility, sarcopenia (reduced muscle mass), hypogonadism, smoking, and alcohol abuse.66 Even at low doses of less than 7.5 mg per day, corticosteroids inhibit osteoblast activity and increase bone resorption.
Studies have reported rapid bone loss at around 6 months after transplantation.67–69 However, long-term follow-up of bone mineral density up to 15 years after transplantation revealed an improvement mainly in the 2nd postoperative year, with no deterioration afterward.65
High-risk patients need to be identified early with appropriate screening and evaluation. Evaluation includes dual-energy x-ray absorptiometry and serum levels of calcium, phosphorous, parathyroid hormone, testosterone (men), estradiol (women), and alpha-25-hydroxyvitamin D. These tests are typically done before transplantation and then every other year afterward.
We recommend a daily dose of calcitriol and a calcium supplement to all our liver transplant patients.70 If osteoporosis (a T-score 2.5 or more standard deviations below the mean) or a fragility fracture occurs, then the patient may benefit from an oral bisphosphonate. Calcitonin has also been shown to improve bone mineral density in patients with osteoporosis after liver transplantation.71
Hyperuricemia and gout
Although hyperuricemia is common in liver transplant recipients (reported in approximately 47%), the development of clinical gout is less common (6%).72 Asymptomatic hyperuricemia requires observation only and is not usually treated in liver transplant recipients.
Acute attacks of gout are typically managed with colchicine 0.6 mg every 2 hours, up to five doses. Prednisone can be considered if symptoms persist despite treatment with colchicine. Allopurinol in an initial dose of 100 mg daily is used as maintenance therapy to reduce production of uric acid.73 However, because of the potential for drug interactions, the combination of azathioprine and allopurinol should be avoided.
Psychiatric complications and quality of life
Depression is common in liver transplant recipients, significantly more so in patients who received a transplant because of hepatitis C.74 The type of immunosuppressant is not associated with the incidence of depression. When indicated, the internist may start the patient on a selective serotonin reuptake inhibitor such as citalopram 20 mg daily, as these medications are usually effective and well tolerated in liver transplant patients.73
Liver transplantation has a major positive effect on quality of life. Most patients with end-stage liver disease have poor quality of life before transplantation, but this seems to improve notably afterward. A meta-analysis showed significant improvement in posttransplant physical health, sexual functioning, daily activities, and social functioning compared with before transplantation.75
Alcohol abuse and smoking
Patients who underwent liver transplantation because of alcoholic liver disease should be advised to abstain from alcohol.19 Patients who underwent the procedure for a different indication are advised to avoid excessive alcohol intake, as it is proven to lower the survival rate.20 Alcohol recidivism and smoking (including marijuana) are major problems, and internists are best positioned to address these issues and treat them.
Vaccinations
All liver transplant recipients should be vaccinated against influenza, pneumococcal infection, and tetanus. Hepatitis A and B vaccines are typically given before transplantation. In general, live vaccines such as measles-mumps-rubella and varicella are not recommended after any solid organ transplant.76
A study in Germany showed that immunization rates were too low in solid-organ transplant recipients, and almost 90% of patients were not adequately informed about immunizations.77 Hence, there may be room for improvement, and primary care providers should take the lead toward better outcomes in this regard.
Recurrence of the primary liver disease after transplantation
Different primary liver diseases recur with different frequencies.
Hepatitis C has the highest rate of recurrence of the liver diseases.78,79 Reinfection with hepatitis C virus after liver transplantation is almost universal and can follow different patterns. One of the most aggressive patterns is fibrosing cholestatic hepatitis, which frequently leads to graft failure and death, and hence necessitates urgent detection and treatment.
Hepatocellular carcinoma also has a high recurrence rate.80 Surveillance with liver ultrasonography or computed tomography is required every 6 months for the first 5 years after liver transplantation.
Other liver diseases. Nonalcoholic steatohepatitis, primary biliary cirrhosis, primary sclerosing cholangitis, autoimmune hepatitis, and hepatitis B infection also tend to recur after liver transplantation.46,81 On the other hand, alpha-1 antitrypsin deficiency, Wilson disease, hemochromatosis, and metabolic disorders are “cured” after liver transplantation.
It is important to detect any increase in liver enzymes above baseline. An elevation of 1.5 times the upper limit of normal or more should trigger further investigation.
Allograft dysfunction
A number of complications can develop in the liver allograft and result in abnormal liver function tests and, if not treated, graft failure. The most common causes of late graft dysfunction include recurrence of primary liver disease, biliary complications, and chronic rejection.46
Vascular complications include hepatic artery thrombosis and stenosis and are usually evaluated by liver ultrasonography and Doppler scan of the hepatic artery and venous structures.24
Biliary strictures give a cholestatic picture, with elevated bilirubin and greater elevation of alkaline phosphatase than of alanine aminotransferase and aspartate aminotransferase. Strictures are usually treated by endoscopic dilation and stenting, but they may eventually require surgery.
Late acute cellular rejections occur in 10% to 20% of cases and are a risk factor for chronic rejection. Liver biopsy is needed to make the diagnosis, and pulsed doses of corticosteroids remain the backbone of treatment therapy.
Chronic rejection is not common, occurring in 3% to 4% of liver transplant recipients.46 Treatment is based on increasing immunosuppression and ensuring compliance with prescribed medications. However, chronic rejection may not respond well, and repeat transplantation may be the last resort for some patients.
WHEN TO REFER TO THE HEPATOLOGIST
Some situations require referral to the hepatologist or the transplant center. In general, the following are best managed by a hepatologist: adjustment of immunosuppressive drugs and dosages, allograft dysfunction, vascular and biliary complications, progressing renal dysfunction, and recurrence of primary liver disease. Early communication with a hepatologist and the transplant center is recommended in these cases.
- Starzl TE, Marchioro TL, Vonkaulla KN, Hermann G, Brittain RS, Waddell WR. Homotransplantation of the liver in humans. Surg Gynecol Obstet 1963; 117:659–676.
- Matas AJ, Smith JM, Skeans MA, et al. OPTN/SRTR 2011 annual data report: kidney. Am J Transplant 2013; 13(suppl 1):11–46.
- McCashland TM. Posttransplantation care: role of the primary care physician versus transplant center. Liver Transpl 2001; 7(suppl 1):S2–S12.
- Heller JC, Prochazka AV, Everson GT, Forman LM. Long-term management after liver transplantation: primary care physician versus hepatologist. Liver Transpl 2009; 15:1330–1335.
- McAlister VC, Haddad E, Renouf E, Malthaner RA, Kjaer MS, Gluud LL. Cyclosporin versus tacrolimus as primary immunosuppressant after liver transplantation: a meta-analysis. Am J Transplant 2006; 6:1578–1585.
- Neuhaus P, Klupp J, Langrehr JM. mTOR inhibitors: an overview. Liver Transpl 2001; 7:473–484.
- Sehgal SN. Rapamune (RAPA, rapamycin, sirolimus): mechanism of action immunosuppressive effect results from blockade of signal transduction and inhibition of cell cycle progression. Clin Biochem 1998; 31:335–340.
- Asrani SK, Wiesner RH, Trotter JF, et al. De novo sirolimus and reduced-dose tacrolimus versus standard-dose tacrolimus after liver transplantation: the 2000-2003 phase II prospective randomized trial. Am J Transplant 2014; 14:356–366.
- Montalbano M, Neff GW, Yamashiki N, et al. A retrospective review of liver transplant patients treated with sirolimus from a single center: an analysis of sirolimus-related complications. Transplantation 2004; 78:264–268.
- Everson GT. Everolimus and mTOR inhibitors in liver transplantation: opening the “box.” Liver Transpl 2006; 12:1571–1573.
- Levy G, Schmidli H, Punch J, et al. Safety, tolerability, and efficacy of everolimus in de novo liver transplant recipients: 12- and 36-month results. Liver Transpl 2006; 12:1640–1648.
- Toniutto P, Fabris C, Fumolo E, et al. Prevalence and risk factors for delayed adrenal insufficiency after liver transplantation. Liver Transpl 2008; 14:1014–1019.
- Klintmalm GB, Davis GL, Teperman L, et al. A randomized, multicenter study comparing steroid-free immunosuppression and standard immunosuppression for liver transplant recipients with chronic hepatitis C. Liver Transpl 2011; 17:1394–1403.
- Llado L, Fabregat J, Castellote J, et al. Impact of immunosuppression without steroids on rejection and hepatitis C virus evolution after liver transplantation: results of a prospective randomized study. Liver Transpl 2008; 14:1752–1760.
- Lake JR. Immunosuppression and outcomes of patients transplanted for hepatitis C. J Hepatol 2006; 44:627–629.
- Sohn AJ, Jeon H, Ahn J. Primary care of the liver transplant recipient. Prim Care 2011; 38:499–514.
- Laish I, Braun M, Mor E, Sulkes J, Harif Y, Ben Ari Z. Metabolic syndrome in liver transplant recipients: prevalence, risk factors, and association with cardiovascular events. Liver Transpl 2011; 17:15–22.
- Stegall MD, Everson G, Schroter G, Bilir B, Karrer F, Kam I. Metabolic complications after liver transplantation. Diabetes, hypercholesterolemia, hypertension, and obesity. Transplantation 1995; 60:1057–1060.
- Textor SC, Canzanello VJ, Taler SJ, et al. Cyclosporine-induced hypertension after transplantation. Mayo Clin Proc 1994; 69:1182–1193.
- Prevention, detection, evaluation, and treatment of hypertension. The Sixth Report of the Joint National Committee. National Institutes of Health-National Heart, Lung, and Blood Institute. National High Blood Pressure Education Programme. Indian Heart J 1999; 51:381–396.
- Frishman WH. Calcium channel blockers: differences between subclasses. Am J Cardiovasc Drugs 2007; 7(suppl 1):17–23.
- Galioto A, Semplicini A, Zanus G, et al. Nifedipine versus carvedilol in the treatment of de novo arterial hypertension after liver transplantation: results of a controlled clinical trial. Liver Transpl 2008; 14:1020–1028.
- Neal DA, Gimson AE, Gibbs P, Alexander GJ. Beneficial effects of converting liver transplant recipients from cyclosporine to tacrolimus on blood pressure, serum lipids, and weight. Liver Transpl 2001; 7:533–539.
- Singh S, Watt KD. Long-term medical management of the liver transplant recipient: what the primary care physician needs to know. Mayo Clin Proc 2012; 87:779–790.
- Bianchi G, Marchesini G, Marzocchi R, Pinna AD, Zoli M. Metabolic syndrome in liver transplantation: relation to etiology and immunosuppression. Liver Transpl 2008; 14:1648–1654.
- Lane JT, Dagogo-Jack S. Approach to the patient with new-onset diabetes after transplant (NODAT). J Clin Endocrinol Metab 2011; 96:3289–3297.
- Wilkinson A, Davidson J, Dotta F, et al. Guidelines for the treatment and management of new-onset diabetes after transplantation. Clin Transplant 2005; 19:291–298.
- Moon JI, Barbeito R, Faradji RN, Gaynor JJ, Tzakis AG. Negative impact of new-onset diabetes mellitus on patient and graft survival after liver transplantation: long-term follow up. Transplantation 2006; 82:1625–1628.
- American Diabetes Association. Standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S11–S63.
- Marchetti P. New-onset diabetes after liver transplantation: from pathogenesis to management. Liver Transpl 2005; 11:612–620.
- Stone NJ, Robinson J, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129(suppl 2):S1–S45.
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Gisbert C, Prieto M, Berenguer M, et al. Hyperlipidemia in liver transplant recipients: prevalence and risk factors. Liver Transpl Surg 1997; 3:416–422.
- Asberg A. Interactions between cyclosporin and lipid-lowering drugs: implications for organ transplant recipients. Drugs 2003; 63:367–378.
- Bullingham RE, Nicholls AJ, Kamm BR. Clinical pharmacokinetics of mycophenolate mofetil. Clin Pharmacokinet 1998; 34:429–455.
- Everhart JE, Lombardero M, Lake JR, Wiesner RH, Zetterman RK, Hoofnagle JH. Weight change and obesity after liver transplantation: incidence and risk factors. Liver Transpl Surg 1998; 4:285–296.
- Canzanello VJ, Schwartz L, Taler SJ, et al. Evolution of cardiovascular risk after liver transplantation: a comparison of cyclosporine A and tacrolimus (FK506). Liver Transpl Surg 1997; 3:1–9.
- Al-Nowaylati AR, Al-Haddad BJ, Dorman RB, et al. Gastric bypass after liver transplantation. Liver Transpl 2013; 19:1324–1329.
- Heimbach JK, Watt KD, Poterucha JJ, et al. Combined liver transplantation and gastric sleeve resection for patients with medically complicated obesity and end-stage liver disease. Am J Transplant 2013; 13:363–368.
- Watt KD, Charlton MR. Metabolic syndrome and liver transplantation: a review and guide to management. J Hepatol 2010; 53:199–206.
- Pagadala M, Dasarathy S, Eghtesad B, McCullough AJ. Posttransplant metabolic syndrome: an epidemic waiting to happen. Liver Transpl 2009; 15:1662–1670.
- Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009; 120:1640–1645.
- Watt KD, Pedersen RA, Kremers WK, Heimbach JK, Charlton MR. Evolution of causes and risk factors for mortality post-liver transplant: results of the NIDDK long-term follow-up study. Am J Transplant 2010; 10:1420–1427.
- Albeldawi M, Aggarwal A, Madhwal S, et al. Cumulative risk of cardiovascular events after orthotopic liver transplantation. Liver Transpl 2012; 18:370–375.
- Gibbons RJ, Balady GJ, Bricker JT, et al. ACC/AHA 2002 guideline update for exercise testing: summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Update the 1997 Exercise Testing Guidelines). Circulation 2002; 106:1883–1892.
- Aberg F, Isoniemi H, Höckerstedt K. Long-term results of liver transplantation. Scand J Surg 2011; 100:14–21.
- Aberg F, Pukkala E, Höckerstedt K, Sankila R, Isoniemi H. Risk of malignant neoplasms after liver transplantation: a population-based study. Liver Transpl 2008; 14:1428–1436.
- Mells G, Neuberger J. Long-term care of the liver allograft recipient. Semin Liver Dis 2009; 29:102–120.
- Herrero JI. De novo malignancies following liver transplantation: impact and recommendations. Liver Transpl 2009; 15(suppl 2):S90–S94.
- Euvrard S, Kanitakis J, Claudy A. Skin cancers after organ transplantation. N Engl J Med 2003; 348:1681–1691.
- Euvrard S, Morelon E, Rostaing L, et al; TUMORAPA Study Group. Sirolimus and secondary skin-cancer prevention in kidney transplantation. N Engl J Med 2012; 367:329–339.
- Salgo R, Gossmann J, Schofer H, et al. Switch to a sirolimus-based immunosuppression in long-term renal transplant recipients: reduced rate of (pre-)malignancies and nonmelanoma skin cancer in a prospective, randomized, assessor-blinded, controlled clinical trial. Am J Transplant 2010; 10:1385–1393.
- Narumi S, Roberts JP, Emond JC, Lake J, Ascher NL. Liver transplantation for sclerosing cholangitis. Hepatology 1995; 22:451–457.
- Knechtle SJ, D’Alessandro AM, Harms BA, Pirsch JD, Belzer FO, Kalayoglu M. Relationships between sclerosing cholangitis, inflammatory bowel disease, and cancer in patients undergoing liver transplantation. Surgery 1995; 118:615–620.
- Finkenstedt A, Graziadei IW, Oberaigner W, et al. Extensive surveillance promotes early diagnosis and improved survival of de novo malignancies in liver transplant recipients. Am J Transplant 2009; 9:2355–2361.
- Fisher NC, Nightingale PG, Gunson BK, Lipkin GW, Neuberger JM. Chronic renal failure following liver transplantation: a retrospective analysis. Transplantation 1998; 66:59–66.
- Ojo AO, Held PJ, Port FK, et al. Chronic renal failure after transplantation of a nonrenal organ. N Engl J Med 2003; 349:931–940.
- Klintmalm GB, Gonwa TA. Nephrotoxicity associated with cyclosporine and FK506. Liver Transpl Surg 1995; 1:11–19.
- A comparison of tacrolimus (FK506) and cyclosporine for immunosuppression in liver transplantation. The US Multicenter FK506 Liver Study Group. N Engl J Med 1994; 331:1110–1115.
- Neff GW, Montalbano M, Slapak-Green G, et al. Sirolimus therapy in orthotopic liver transplant recipients with calcineurin inhibitor related chronic renal insufficiency. Transplant Proc 2003; 35:3029–3031.
- Cotterell AH, Fisher RA, King AL, et al. Calcineurin inhibitor-induced chronic nephrotoxicity in liver transplant patients is reversible using rapamycin as the primary immunosuppressive agent. Clin Transplant 2002; 16(suppl 7):49–51.
- Manzia TM, De Liguori Carino N, Orlando G, et al. Use of mycophenolate mofetil in liver transplantation: a literature review. Transplant Proc 2005; 37:2616–2617.
- Schlitt HJ, Barkmann A, Boker KH, et al. Replacement of calcineurin inhibitors with mycophenolate mofetil in liver-transplant patients with renal dysfunction: a randomised controlled study. Lancet 2001; 357:587–591.
- Hodge EE, Reich DJ, Clavien PA, Kim-Schluger L. Use of mycophenolate mofetil in liver transplant recipients experiencing renal dysfunction on cyclosporine or tacrolimus-randomized, prospective, multicenter study results. Transplant Proc 2002; 34:1546–1547.
- Hamburg SM, Piers DA, van den Berg AP, Slooff MJ, Haagsma EB. Bone mineral density in the long term after liver transplantation. Osteoporos Int 2000; 11:600–606.
- Maalouf NM, Shane E. Osteoporosis after solid organ transplantation. J Clin Endocrinol Metab 2005; 90:2456–2465.
- Crosbie OM, Freaney R, McKenna MJ, Curry MP, Hegarty JE. Predicting bone loss following orthotopic liver transplantation. Gut 1999; 44:430–434.
- Giannini S, Nobile M, Ciuffreda M, et al. Long-term persistence of low bone density in orthotopic liver transplantation. Osteoporos Int 2000; 11:417–424.
- Monegal A, Navasa M, Guanabens N, et al. Bone disease after liver transplantation: a long-term prospective study of bone mass changes, hormonal status and histomorphometric characteristics. Osteoporos Int 2001; 12:484–492.
- Neuhaus R, Lohmann R, Platz KP, et al. Treatment of osteoporosis after liver transplantation. Transplant Proc 1995; 27:1226–1227.
- Valero MA, Loinaz C, Larrodera L, Leon M, Moreno E, Hawkins F. Calcitonin and bisphosphonates treatment in bone loss after liver transplantation. Calcif Tissue Int 1995; 57:15–19.
- Neal DA, Tom BD, Gimson AE, Gibbs P, Alexander GJ. Hyperuricemia, gout, and renal function after liver transplantation. Transplantation 2001; 72:1689–1691.
- Schiff ER, Sorrell MF, Maddrey WC, editors. Schiff’s Diseases of the Liver. 10th ed. Philadephia, PA: Lippincott Williams & Wilkins; 2007.
- Tombazzi CR, Waters B, Shokouh-Amiri MH, Vera SR, Riely CA. Neuropsychiatric complications after liver transplantation: role of immunosuppression and hepatitis C. Dig Dis Sci 2006; 51:1079–1081.
- Bravata DM, Olkin I, Barnato AE, Keeffe EB, Owens DK. Health-related quality of life after liver transplantation: a meta-analysis. Liver Transpl Surg 1999; 5:318–331.
- Danziger-Isakov L, Kumar D; AST Infectious Diseases Community of Practice. Vaccination in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):311–317.
- Chesi C, Gunther M, Huzly D, et al. Immunization of liver and renal transplant recipients: a seroepidemiological and sociodemographic survey. Transpl Infect Dis 2009; 11:507–512.
- Berenguer M, Lopez-Labrador FX, Wright TL. Hepatitis C and liver transplantation. J Hepatol 2001; 35:666–678.
- Forman LM, Lewis JD, Berlin JA, Feldman HI, Lucey MR. The association between hepatitis C infection and survival after orthotopic liver transplantation. Gastroenterology 2002; 122:889–896.
- Benten D, Staufer K, Sterneck M. Orthotopic liver transplantation and what to do during follow-up: recommendations for the practitioner. Nat Clin Pract Gastroenterol Hepatol 2009; 6:23–36.
- Kotlyar DS, Campbell MS, Reddy KR. Recurrence of diseases following orthotopic liver transplantation. Am J Gastroenterol 2006; 101:1370–1378.
- Starzl TE, Marchioro TL, Vonkaulla KN, Hermann G, Brittain RS, Waddell WR. Homotransplantation of the liver in humans. Surg Gynecol Obstet 1963; 117:659–676.
- Matas AJ, Smith JM, Skeans MA, et al. OPTN/SRTR 2011 annual data report: kidney. Am J Transplant 2013; 13(suppl 1):11–46.
- McCashland TM. Posttransplantation care: role of the primary care physician versus transplant center. Liver Transpl 2001; 7(suppl 1):S2–S12.
- Heller JC, Prochazka AV, Everson GT, Forman LM. Long-term management after liver transplantation: primary care physician versus hepatologist. Liver Transpl 2009; 15:1330–1335.
- McAlister VC, Haddad E, Renouf E, Malthaner RA, Kjaer MS, Gluud LL. Cyclosporin versus tacrolimus as primary immunosuppressant after liver transplantation: a meta-analysis. Am J Transplant 2006; 6:1578–1585.
- Neuhaus P, Klupp J, Langrehr JM. mTOR inhibitors: an overview. Liver Transpl 2001; 7:473–484.
- Sehgal SN. Rapamune (RAPA, rapamycin, sirolimus): mechanism of action immunosuppressive effect results from blockade of signal transduction and inhibition of cell cycle progression. Clin Biochem 1998; 31:335–340.
- Asrani SK, Wiesner RH, Trotter JF, et al. De novo sirolimus and reduced-dose tacrolimus versus standard-dose tacrolimus after liver transplantation: the 2000-2003 phase II prospective randomized trial. Am J Transplant 2014; 14:356–366.
- Montalbano M, Neff GW, Yamashiki N, et al. A retrospective review of liver transplant patients treated with sirolimus from a single center: an analysis of sirolimus-related complications. Transplantation 2004; 78:264–268.
- Everson GT. Everolimus and mTOR inhibitors in liver transplantation: opening the “box.” Liver Transpl 2006; 12:1571–1573.
- Levy G, Schmidli H, Punch J, et al. Safety, tolerability, and efficacy of everolimus in de novo liver transplant recipients: 12- and 36-month results. Liver Transpl 2006; 12:1640–1648.
- Toniutto P, Fabris C, Fumolo E, et al. Prevalence and risk factors for delayed adrenal insufficiency after liver transplantation. Liver Transpl 2008; 14:1014–1019.
- Klintmalm GB, Davis GL, Teperman L, et al. A randomized, multicenter study comparing steroid-free immunosuppression and standard immunosuppression for liver transplant recipients with chronic hepatitis C. Liver Transpl 2011; 17:1394–1403.
- Llado L, Fabregat J, Castellote J, et al. Impact of immunosuppression without steroids on rejection and hepatitis C virus evolution after liver transplantation: results of a prospective randomized study. Liver Transpl 2008; 14:1752–1760.
- Lake JR. Immunosuppression and outcomes of patients transplanted for hepatitis C. J Hepatol 2006; 44:627–629.
- Sohn AJ, Jeon H, Ahn J. Primary care of the liver transplant recipient. Prim Care 2011; 38:499–514.
- Laish I, Braun M, Mor E, Sulkes J, Harif Y, Ben Ari Z. Metabolic syndrome in liver transplant recipients: prevalence, risk factors, and association with cardiovascular events. Liver Transpl 2011; 17:15–22.
- Stegall MD, Everson G, Schroter G, Bilir B, Karrer F, Kam I. Metabolic complications after liver transplantation. Diabetes, hypercholesterolemia, hypertension, and obesity. Transplantation 1995; 60:1057–1060.
- Textor SC, Canzanello VJ, Taler SJ, et al. Cyclosporine-induced hypertension after transplantation. Mayo Clin Proc 1994; 69:1182–1193.
- Prevention, detection, evaluation, and treatment of hypertension. The Sixth Report of the Joint National Committee. National Institutes of Health-National Heart, Lung, and Blood Institute. National High Blood Pressure Education Programme. Indian Heart J 1999; 51:381–396.
- Frishman WH. Calcium channel blockers: differences between subclasses. Am J Cardiovasc Drugs 2007; 7(suppl 1):17–23.
- Galioto A, Semplicini A, Zanus G, et al. Nifedipine versus carvedilol in the treatment of de novo arterial hypertension after liver transplantation: results of a controlled clinical trial. Liver Transpl 2008; 14:1020–1028.
- Neal DA, Gimson AE, Gibbs P, Alexander GJ. Beneficial effects of converting liver transplant recipients from cyclosporine to tacrolimus on blood pressure, serum lipids, and weight. Liver Transpl 2001; 7:533–539.
- Singh S, Watt KD. Long-term medical management of the liver transplant recipient: what the primary care physician needs to know. Mayo Clin Proc 2012; 87:779–790.
- Bianchi G, Marchesini G, Marzocchi R, Pinna AD, Zoli M. Metabolic syndrome in liver transplantation: relation to etiology and immunosuppression. Liver Transpl 2008; 14:1648–1654.
- Lane JT, Dagogo-Jack S. Approach to the patient with new-onset diabetes after transplant (NODAT). J Clin Endocrinol Metab 2011; 96:3289–3297.
- Wilkinson A, Davidson J, Dotta F, et al. Guidelines for the treatment and management of new-onset diabetes after transplantation. Clin Transplant 2005; 19:291–298.
- Moon JI, Barbeito R, Faradji RN, Gaynor JJ, Tzakis AG. Negative impact of new-onset diabetes mellitus on patient and graft survival after liver transplantation: long-term follow up. Transplantation 2006; 82:1625–1628.
- American Diabetes Association. Standards of medical care in diabetes—2012. Diabetes Care 2012; 35(suppl 1):S11–S63.
- Marchetti P. New-onset diabetes after liver transplantation: from pathogenesis to management. Liver Transpl 2005; 11:612–620.
- Stone NJ, Robinson J, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129(suppl 2):S1–S45.
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Gisbert C, Prieto M, Berenguer M, et al. Hyperlipidemia in liver transplant recipients: prevalence and risk factors. Liver Transpl Surg 1997; 3:416–422.
- Asberg A. Interactions between cyclosporin and lipid-lowering drugs: implications for organ transplant recipients. Drugs 2003; 63:367–378.
- Bullingham RE, Nicholls AJ, Kamm BR. Clinical pharmacokinetics of mycophenolate mofetil. Clin Pharmacokinet 1998; 34:429–455.
- Everhart JE, Lombardero M, Lake JR, Wiesner RH, Zetterman RK, Hoofnagle JH. Weight change and obesity after liver transplantation: incidence and risk factors. Liver Transpl Surg 1998; 4:285–296.
- Canzanello VJ, Schwartz L, Taler SJ, et al. Evolution of cardiovascular risk after liver transplantation: a comparison of cyclosporine A and tacrolimus (FK506). Liver Transpl Surg 1997; 3:1–9.
- Al-Nowaylati AR, Al-Haddad BJ, Dorman RB, et al. Gastric bypass after liver transplantation. Liver Transpl 2013; 19:1324–1329.
- Heimbach JK, Watt KD, Poterucha JJ, et al. Combined liver transplantation and gastric sleeve resection for patients with medically complicated obesity and end-stage liver disease. Am J Transplant 2013; 13:363–368.
- Watt KD, Charlton MR. Metabolic syndrome and liver transplantation: a review and guide to management. J Hepatol 2010; 53:199–206.
- Pagadala M, Dasarathy S, Eghtesad B, McCullough AJ. Posttransplant metabolic syndrome: an epidemic waiting to happen. Liver Transpl 2009; 15:1662–1670.
- Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009; 120:1640–1645.
- Watt KD, Pedersen RA, Kremers WK, Heimbach JK, Charlton MR. Evolution of causes and risk factors for mortality post-liver transplant: results of the NIDDK long-term follow-up study. Am J Transplant 2010; 10:1420–1427.
- Albeldawi M, Aggarwal A, Madhwal S, et al. Cumulative risk of cardiovascular events after orthotopic liver transplantation. Liver Transpl 2012; 18:370–375.
- Gibbons RJ, Balady GJ, Bricker JT, et al. ACC/AHA 2002 guideline update for exercise testing: summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Update the 1997 Exercise Testing Guidelines). Circulation 2002; 106:1883–1892.
- Aberg F, Isoniemi H, Höckerstedt K. Long-term results of liver transplantation. Scand J Surg 2011; 100:14–21.
- Aberg F, Pukkala E, Höckerstedt K, Sankila R, Isoniemi H. Risk of malignant neoplasms after liver transplantation: a population-based study. Liver Transpl 2008; 14:1428–1436.
- Mells G, Neuberger J. Long-term care of the liver allograft recipient. Semin Liver Dis 2009; 29:102–120.
- Herrero JI. De novo malignancies following liver transplantation: impact and recommendations. Liver Transpl 2009; 15(suppl 2):S90–S94.
- Euvrard S, Kanitakis J, Claudy A. Skin cancers after organ transplantation. N Engl J Med 2003; 348:1681–1691.
- Euvrard S, Morelon E, Rostaing L, et al; TUMORAPA Study Group. Sirolimus and secondary skin-cancer prevention in kidney transplantation. N Engl J Med 2012; 367:329–339.
- Salgo R, Gossmann J, Schofer H, et al. Switch to a sirolimus-based immunosuppression in long-term renal transplant recipients: reduced rate of (pre-)malignancies and nonmelanoma skin cancer in a prospective, randomized, assessor-blinded, controlled clinical trial. Am J Transplant 2010; 10:1385–1393.
- Narumi S, Roberts JP, Emond JC, Lake J, Ascher NL. Liver transplantation for sclerosing cholangitis. Hepatology 1995; 22:451–457.
- Knechtle SJ, D’Alessandro AM, Harms BA, Pirsch JD, Belzer FO, Kalayoglu M. Relationships between sclerosing cholangitis, inflammatory bowel disease, and cancer in patients undergoing liver transplantation. Surgery 1995; 118:615–620.
- Finkenstedt A, Graziadei IW, Oberaigner W, et al. Extensive surveillance promotes early diagnosis and improved survival of de novo malignancies in liver transplant recipients. Am J Transplant 2009; 9:2355–2361.
- Fisher NC, Nightingale PG, Gunson BK, Lipkin GW, Neuberger JM. Chronic renal failure following liver transplantation: a retrospective analysis. Transplantation 1998; 66:59–66.
- Ojo AO, Held PJ, Port FK, et al. Chronic renal failure after transplantation of a nonrenal organ. N Engl J Med 2003; 349:931–940.
- Klintmalm GB, Gonwa TA. Nephrotoxicity associated with cyclosporine and FK506. Liver Transpl Surg 1995; 1:11–19.
- A comparison of tacrolimus (FK506) and cyclosporine for immunosuppression in liver transplantation. The US Multicenter FK506 Liver Study Group. N Engl J Med 1994; 331:1110–1115.
- Neff GW, Montalbano M, Slapak-Green G, et al. Sirolimus therapy in orthotopic liver transplant recipients with calcineurin inhibitor related chronic renal insufficiency. Transplant Proc 2003; 35:3029–3031.
- Cotterell AH, Fisher RA, King AL, et al. Calcineurin inhibitor-induced chronic nephrotoxicity in liver transplant patients is reversible using rapamycin as the primary immunosuppressive agent. Clin Transplant 2002; 16(suppl 7):49–51.
- Manzia TM, De Liguori Carino N, Orlando G, et al. Use of mycophenolate mofetil in liver transplantation: a literature review. Transplant Proc 2005; 37:2616–2617.
- Schlitt HJ, Barkmann A, Boker KH, et al. Replacement of calcineurin inhibitors with mycophenolate mofetil in liver-transplant patients with renal dysfunction: a randomised controlled study. Lancet 2001; 357:587–591.
- Hodge EE, Reich DJ, Clavien PA, Kim-Schluger L. Use of mycophenolate mofetil in liver transplant recipients experiencing renal dysfunction on cyclosporine or tacrolimus-randomized, prospective, multicenter study results. Transplant Proc 2002; 34:1546–1547.
- Hamburg SM, Piers DA, van den Berg AP, Slooff MJ, Haagsma EB. Bone mineral density in the long term after liver transplantation. Osteoporos Int 2000; 11:600–606.
- Maalouf NM, Shane E. Osteoporosis after solid organ transplantation. J Clin Endocrinol Metab 2005; 90:2456–2465.
- Crosbie OM, Freaney R, McKenna MJ, Curry MP, Hegarty JE. Predicting bone loss following orthotopic liver transplantation. Gut 1999; 44:430–434.
- Giannini S, Nobile M, Ciuffreda M, et al. Long-term persistence of low bone density in orthotopic liver transplantation. Osteoporos Int 2000; 11:417–424.
- Monegal A, Navasa M, Guanabens N, et al. Bone disease after liver transplantation: a long-term prospective study of bone mass changes, hormonal status and histomorphometric characteristics. Osteoporos Int 2001; 12:484–492.
- Neuhaus R, Lohmann R, Platz KP, et al. Treatment of osteoporosis after liver transplantation. Transplant Proc 1995; 27:1226–1227.
- Valero MA, Loinaz C, Larrodera L, Leon M, Moreno E, Hawkins F. Calcitonin and bisphosphonates treatment in bone loss after liver transplantation. Calcif Tissue Int 1995; 57:15–19.
- Neal DA, Tom BD, Gimson AE, Gibbs P, Alexander GJ. Hyperuricemia, gout, and renal function after liver transplantation. Transplantation 2001; 72:1689–1691.
- Schiff ER, Sorrell MF, Maddrey WC, editors. Schiff’s Diseases of the Liver. 10th ed. Philadephia, PA: Lippincott Williams & Wilkins; 2007.
- Tombazzi CR, Waters B, Shokouh-Amiri MH, Vera SR, Riely CA. Neuropsychiatric complications after liver transplantation: role of immunosuppression and hepatitis C. Dig Dis Sci 2006; 51:1079–1081.
- Bravata DM, Olkin I, Barnato AE, Keeffe EB, Owens DK. Health-related quality of life after liver transplantation: a meta-analysis. Liver Transpl Surg 1999; 5:318–331.
- Danziger-Isakov L, Kumar D; AST Infectious Diseases Community of Practice. Vaccination in solid organ transplantation. Am J Transplant 2013; 13(suppl 4):311–317.
- Chesi C, Gunther M, Huzly D, et al. Immunization of liver and renal transplant recipients: a seroepidemiological and sociodemographic survey. Transpl Infect Dis 2009; 11:507–512.
- Berenguer M, Lopez-Labrador FX, Wright TL. Hepatitis C and liver transplantation. J Hepatol 2001; 35:666–678.
- Forman LM, Lewis JD, Berlin JA, Feldman HI, Lucey MR. The association between hepatitis C infection and survival after orthotopic liver transplantation. Gastroenterology 2002; 122:889–896.
- Benten D, Staufer K, Sterneck M. Orthotopic liver transplantation and what to do during follow-up: recommendations for the practitioner. Nat Clin Pract Gastroenterol Hepatol 2009; 6:23–36.
- Kotlyar DS, Campbell MS, Reddy KR. Recurrence of diseases following orthotopic liver transplantation. Am J Gastroenterol 2006; 101:1370–1378.
KEY POINTS
- Tacrolimus and cyclosporine are the most commonly used immunosuppressive agents in liver transplant recipients. Adverse effects include hypertension, hypercholesterolemia, diabetes (more common with tacrolimus), renal insufficiency, and osteoporosis.
- Hypertension affects 40% to 85% of liver transplant patients. Dihydropyridine calcium channel blockers (eg, amlodipine, nifedipine) are the first-line agents.
- Cardiovascular disease is the third most common cause of death after liver transplantation. Modifying risk factors is essential.
- Skin cancers account for 40% of all cancers after liver transplantation. Intensive screening is required and has been proven to lower the risk of death.
Genetics and hepatitis C: It’s good to be ‘CC’
What a difference a single nucleotide can make! The human genome contains more than 3 billion base pairs. Yet having a different nucleotide in only one pair can make a big difference in how we respond to a disease or its treatment.
Specifically, in hepatitis C virus infection, people born with the nucleotide cytosine (C) at location rs12979860 in both alleles of the gene that codes for interleukin 28B (the IL28B CC genotype) can count themselves luckier than those born with thymine (T) in this location in one of their alleles (the CT genotype) or both of their alleles (the TT genotype). Those with the CC genotype are more likely to clear the virus spontaneously, and even if the infection persists, it is less likely to progress to liver cancer and more likely to respond to treatment with interferon.
Here, we review the IL28B polymorphism and its implications in treating hepatitis C.
GENETIC POLYMORPHISM AND HUMAN DISEASE
Of the 3 billion base pairs of nucleotides, fewer than 1% differ between individuals, but this 1% is responsible for the diversity of human beings. Differences in genetic sequences among individuals are called genetic polymorphisms. A single-nucleotide polymorphism is a DNA sequence variation that occurs in a single nucleotide in the genome. For example, two sequenced DNA fragments from different individuals, AAGCCTA and AAGCTTA, contain a difference in a single nucleotide.
Genetic variations such as these underlie some of the differences in our susceptibility to disease, the severity of illness we develop, and our response to treatments. Therefore, identifying genetic polymorphisms may shed light on biologic pathways involved in diseases and may uncover new targets for therapy.1
Genome-wide association studies have looked at hundreds of thousands of single-nucleotide polymorphisms to try to identify most of the common genetic differences among people and relate them to common chronic diseases such as coronary artery disease,2 type 2 diabetes,3 stroke,4 breast cancer,5 rheumatoid arthritis,6 Alzheimer disease,7 and, more recently, hepatitis C virus infection.8
HEPATITIS C VIRUS: A MAJOR CAUSE OF LIVER DISEASE
Hepatitis C virus infection is a major cause of chronic liver disease and hepatocellular carcinoma and has become the most common indication for liver transplantation in the United States.9
This virus has six distinct genotypes throughout the world, with multiple subtypes in each genotype. (A genotype is a classification of a virus based on its RNA.9) In this review, we will focus on genotype 1; hence, “hepatitis C virus” will refer to hepatitis C virus genotype 1.
Our knowledge of the biology, pathogenesis, and treatment of hepatitis C has been advancing. Originally, fewer than 50% of patients responded to therapy with the combination of pegylated interferon and ribavirin,10,11 but since 2011 the response rate has increased to approximately 70% with the approval of the protease inhibitors telaprevir and boceprevir, used in combination with pegylated interferon and ribavirin.12–15
Unfortunately, interferon-based treatment is often complicated by side effects such as fatigue, influenza-like symptoms, hematologic abnormalities, and neuropsychiatric symptoms. An accurate way to predict response would help patients make informed decisions about antiviral treatment, taking into account the risk and possible benefit for individual patients.
GENETIC POLYMORPHISM AND HEPATITIS C VIRUS INFECTION
Genome-wide association studies have identified single-nucleotide polymorphisms in the IL28B gene that are associated with differences in response to hepatitis C treatment.8
Studying 565,759 polymorphisms in 1,137 patients, researchers at Duke University identified a single-nucleotide polymorphism at location rs12979860 in IL28B (Figure 1) that was strongly associated with response to combination therapy with pegylated interferon and ribavirin.8 The chance of cure with this standard treatment is twice as high in patients who are homozygous for cytosine in this location (the CC genotype) than in those who are heterozygous (CT) or homozygous for thymine in this location (the TT genotype) (Table 1).

Adding one of the new protease inhibitors, telaprevir or boceprevir, to the standard hepatitis C treatment substantially improves the cure rates in all three IL28B genotypes, but especially in people with CT or TT, in whom the response rate almost triples with the addition of one of these drugs. Those with the CC genotype (who are more likely to be cured with pegylated interferon and ribavirin alone) also achieve an increase (although minimal) in cure rates when a protease inhibitor is included in the regimen (TABLE 1).13–15 Thus, it remains unclear if adding a protease inhibitor to pegylated interferon plus ribavirin in patients with the IL28B CC genotype translates into added effectiveness worth the additional cost of the protease inhibitor in previously untreated patients.
Additionally, the effect of the IL28B genotype on telaprevir-based triple therapy has been disputed in more recent studies. In a subgroup analysis of the results of a trial that evaluated telaprevir in the treatment of hepatitis C, researchers found that sustained virologic response rates were significantly higher in the telaprevir group, and this was similar across the different IL28B polymorphisms.16
The favorable IL28B CC genotype is associated with higher rates of rapid virologic response to antiviral therapy.13–15 Of note, almost all patients who achieve a rapid virologic response do well, with a high rate of sustained virologic response even after a shorter duration of therapy (24 vs 48 weeks). Therefore, in addition to predicting response to interferon before starting treatment, the IL28B CC genotype may also identify patients who need only a shorter duration of therapy.
Interestingly, the C allele is much more frequent in white than in African American populations, an important observation that explains the racial difference in response to hepatitis C therapy.8
Two other research groups, from Asia and Australia, performed independent genome-wide association studies that identified different single-nucleotide polymorphisms (eg, rs8099917) in the same IL28B gene as predictors of response to treatment in patients with hepatitis C virus infection.17,18 These findings may be explained by linkage disequilibrium, which means that these single-nucleotide polymorphisms are found more frequently together in the same patient due to their proximity to each other. In this review, we will focus on the rs12979860 polymorphism; hence “IL28B genotype” will refer to the single-nucleotide polymorphism at rs12979860, unless otherwise specified.
The favorable CC genotype is less common in African Americans than in patients of other ethnicities.19 Moreover, although IL28B CC is associated with a better response rate to interferon-based antiviral therapy across all ethnicities, those of African American descent with the CC genotype are less likely to achieve a sustained virologic response than white or Hispanic Americans.8
BIOLOGIC ASSOCIATION: IL28B POLYMORPHISM AND HEPATITIS C
The interferon lambda family consists of three cytokines:
- Interleukin 29 (interferon lambda 1)
- Interleukin 28A (interferon lambda 2)
- Interleukin 28B (interferon lambda 3).
Production of these three molecules can be triggered by viral infection, and they induce antiviral activity through both innate and adaptive immune pathways. They signal through the IL10R-IL28R receptor complex.20–22 This receptor activates the JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway, which regulates a large number of interferon-stimulated genes, primarily through the interferon-stimulated response element (Figure 2).
A 2013 study found that interferon-stimulated gene expression levels in patients with normal livers were highest in those with the CC genotype, intermediate with CT, and lowest with TT. Interestingly, this pattern was reversed in those with hepatitis C virus infection, indicating a relationship between the IL28B genotype and gene expression before infection.23
The mechanism underlying the association between the IL28B polymorphism and response to hepatitis C treatment is not well understood. The unfavorable TT genotype seems to lead to continuous activation of a subset of interferon-stimulated genes in the presence of intracellular hepatitis C viral RNA. But this level of expression is not sufficient to eliminate the virus from the cells. Instead, it might lead to up-regulation of interferon-inhibitory molecules that suppress JAK-STAT signaling, thereby reducing sensitivity to interferon signaling. Therefore, the hepatocyte not only cannot clear the virus by itself, but also cannot induce strong interferon-stimulated gene expression when interferon is given during therapy.20–22
The recently identified ss469425590 polymorphism, which is located in close proximity to rs12979860 in the IL28B gene, is particularly interesting, as it suggests a possible molecular mechanism. The delta G frameshift variant creates a novel gene called IFNL4, which is transiently activated in response to hepatitis C virus infection.24IFNL4 stimulates STAT1 and STAT2 phosphorylation and induces the expression of interferon-stimulated genes. Increased interferon-stimulated gene expression has been shown to be associated with decreased response to pegylated interferon-ribavirin treatment. These observations suggest that the ss469425590 delta G allele is responsible for the increased activation of interferon-stimulated genes and the lower sustained virologic response rate observed in patients who receive pegylated interferon-ribavirin treatment. It is possible that the activation of interferon-stimulated genes in patients with the ss469425590 delta G/delta G genotype reduces interferon-stimulated gene responsiveness to interferon alpha, which normally activates interferon-stimulated genes and inhibits hepatitis C progression.24
IL28B POLYMORPHISM AND ACUTE HEPATITIS C VIRUS INFECTION
From 70% to 80% of acute hepatitis C virus infections persist and become chronic, while 20% to 30% spontaneously resolve. Epidemiologic, viral, and host factors have been associated with the differences in viral clearance or persistence, and studies have found that a strong host immune response against the virus favors viral clearance. Thus, variation in the genes involved in the immune response may contribute to one’s ability to clear the virus. Consistent with these observations, recent studies have shown that the polymorphism in the IL28 gene region encoding interferon lambda 3 strongly predicts spontaneous resolution of acute hepatitis C virus infection. People who have the IL28B CC genotype are three times more likely to spontaneously clear the virus than those with the CT or TT genotype (Figure 3).24
IL28B POLYMORPHISM AND THE NATURAL HISTORY OF HEPATITIS C
In people in whom hepatitis C virus infection persists, up to 20% develop progressive liver fibrosis and eventually cirrhosis over 10 to 20 years.19,25,26 The speed at which fibrosis develops in these patients is variable and unpredictable.25 The relationship between IL28B polymorphisms and hepatic fibrosis in patients with chronic hepatitis C virus infection has not been clearly established, although a study indicated that in patients with a known date of infection, the IL28B genotype is not associated with progression of hepatic fibrosis.27 Obstacles in this field of study are that it is difficult to determine accurately when the patient contracted the virus, and that serial liver biopsies are needed to investigate the progression of hepatic fibrosis.
Patients with chronic hepatitis C virus infection are also at higher risk of hepatocellular carcinoma compared with the general population.28 An analysis of explanted livers of patients with hepatitis C found that the prevalence of hepatocellular carcinoma in those with the unfavorable TT genotype was significantly higher than with the other genotypes.29 Similarly, an earlier study demonstrated that patients with hepatitis C-associated hepatocellular carcinoma carried the T allele more frequently.30 As with other aspects of IL28B associations with hepatitis C, these findings indicate that the C allele confers a certain degree of protection.
An important implication of these relationships is that they may eventually help identify patients at greater risk, who therefore need earlier intervention.
IL28B POLYMORPHISM AND LIVER TRANSPLANTATION
Hepatitis C virus infection always recurs after liver transplantation, with serious consequences that include cirrhosis and liver failure. Recurrent hepatitis C virus infection has become an important reason for repeat transplantation in the United States.
Results of treatment with pegylated interferon and ribavirin for recurrent hepatitis C after liver transplantation have been disappointing, with response rates lower than 30% and significant side effects.31 Identifying the factors that predict the response to therapy allows for better selection of treatment candidates.
Similar to the way the IL28B genotype predicts response to antiviral therapy in the nontransplant setting, the IL28B genotypes of both the recipient and the donor are strongly and independently associated with response to interferon-based treatment in patients with hepatitis C after liver transplantation. The IL28B CC genotype in either the recipient or the donor is associated with a higher rate of response to pegylated interferon and ribavirin combination therapy after liver transplantation.30,32 For example, the response rate to therapy after liver transplantation reaches 86% in CC-donor and CC-recipient livers, compared with 0% in TT-donor and TT-recipient livers.
Additionally, the IL28B genotype of the recipient may determine the severity of histologic recurrence of hepatitis C, as indicated by progressive hepatic fibrosis. A recipient IL28B TT genotype is associated with more severe histologic recurrence of hepatitis C.33
These data suggest that CC donor livers might be preferentially allocated to patients with hepatitis C virus infection.
IL28B AND OTHER FACTORS IN HEPATITIS C VIRUS INFECTION
Although it is tempting to think that the IL28B polymorphism is the sole predictor of response to antiviral therapy, it is but one of several known factors in the virus and the host.
While IL28B polymorphisms are the most important predictor of sustained virologic response with an interferon-based regimen, a rapid virologic response (undetectable viral load at 4 weeks) had superior predictive value and specificity in one study.34 In fact, for patients with chronic hepatitis C infection who achieved a rapid virologic response with pegylated interferon and ribavirin, the IL28B polymorphism had no effect on the rate of sustained virologic response. However, it did predict a sustained virologic response in the group who did not achieve rapid virologic response.
In a study of patients with acute hepatitis C infection,35 jaundice and the IL28 rs12979860 CC genotype both predicted spontaneous clearance. The best predictor of viral persistence was the combination of the CT or TT genotype plus the absence of jaundice, which had a predictive value of 98%.
IL28B AND THE FUTURE OF HEPATITIS C VIRUS THERAPY
New oral agents were recently approved for treating hepatitis C. As of November 2014, these included simeprevir, sofosbuvir, and ledipasvir.
Simeprevir is a second-generation NS3/4A protease inhibitor approved for use in combination with pegylated interferon and ribavirin. A recent phase 3 trial evaluating simeprevir in patients who had relapsed after prior therapy found sustained virologic response rates to be higher with simeprevir than with placebo, irrespective of IL28B status.36 This finding was similar to that of a trial of telaprevir.16
Sofosbuvir is a nucleotide analogue NS5B polymerase inhibitor that becomes incorporated into the growing RNA, inducing a chain termination event.37 In phase 3 trials,38,39 researchers found an initial rapid decrease in viral load for patients treated with this agent regardless of IL28B status.
In the NEUTRINO trial (Sofosbuvir With Peginterferon Alfa 2a and Ribavirin for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1, 4, 5, or 6 HCV Infection),38 which used sofusbuvir in combination with interferon and ribavirin, the rate of sustained virologic response was higher in those with the favorable CC genotype (98%) than with a non-CC genotype (87%).
In COSMOS (A Study of TMC435 in Combination With PSI-7977 [GS7977] in Chronic Hepatitis C Genotype 1-Infected Prior Null Responders to Peginterferon/Ribavirin Therapy or HCV Treatment-Naive Patients),39 which used a combination of simeprevir, sofosbuvir, and ribavirin, the rate of sustained virologic response was higher in those with the CC genotype (100%) than with the TT genotype (83%; Table 1).
These new medications have radically changed the landscape of hepatitis C therapy and have also unlocked the potential for developing completely interferon-free regimens.
Other new interferon-free regimens such as ledipasvir, daclatasvir, and asunaprevir promise high rates of sustained virologic response, which makes the utility of testing for IL28B polymorphisms to predict sustained virologic response very much diminished (Table 1).40,41 However, these new drugs are expected to be expensive, and IL28B polymorphisms may be used to identify candidates who are more likely to respond to pegylated interferon and ribavirin, particularly in resource-poor settings and in developing countries. Additionally, patients who have contraindications to these newer therapies will still likely need an interferon-based regimen, and thus the IL28B polymorphism will still be important in predicting treatment response and prognosis.
IL28B WILL STILL BE RELEVANT IN THE INTERFERON-FREE AGE
The IL28B polymorphism is a strong predictor of spontaneous clearance of hepatitis C virus and responsiveness to interferon-based therapy, and testing for it has demonstrated a great potential to improve patient care. IL28B testing has become available for clinical use and may optimize the outcome of hepatitis C treatment by helping us to select the best treatment for individual patients and minimizing the duration of therapy and the side effects associated with interferon-based antiviral medications.
As newer therapies have shifted toward interferon-free regimens that offer very high sustained virologic response rates, the usefulness of IL28B polymorphism as a clinical test to predict the response rate to antiviral therapy is minimized substantially. It may remain clinically relevant in resource-poor settings and in developing countries, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated. This does not minimize the lesson we learned from the discovery of the IL28B gene and the impact on our understanding of the pathogenesis of hepatitis C virus infection.
- Attia J, Ioannidis JP, Thakkinstian A, et al. How to use an article about genetic association: A: background concepts. JAMA 2009; 301:74–81.
- Samani NJ, Erdmann J, Hall AS, et al; WTCCC and the Cardiogenics Consortium. Genomewide association analysis of coronary artery disease. N Engl J Med 2007; 357:443–453.
- Zeggini E, Weedon MN, Lindgren CM, et al; Wellcome Trust Case Control Consortium (WTCCC). Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007; 316:1336–1341.
- Matarín M, Brown WM, Scholz S, et al. A genome-wide genotyping study in patients with ischaemic stroke: initial analysis and data release. Lancet Neurol 2007; 6:414–420.
- Easton DF, Pooley KA, Dunning AM, et al; AOCS Management Group. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007; 447:1087–1093.
- Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—a genomewide study. N Engl J Med 2007; 357:1199–1209.
- Coon KD, Myers AJ, Craig DW, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 2007; 68:613–618.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance (letter). Nature 2009; 461:399–401.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005-1019.
- Hanouneh IA, Feldstein AE, Lopez R, et al. Clinical significance of metabolic syndrome in the setting of chronic hepatitis C virus infection. Clin Gastroenterol Hepatol 2008; 6:584–589.
- Elgouhari HM, Zein CO, Hanouneh I, Feldstein AE, Zein NN. Diabetes mellitus is associated with impaired response to antiviral therapy in chronic hepatitis C infection. Dig Dis Sci 2009; 54:2699–2705.
- Alkhouri N, Zein NN. Protease inhibitors: silver bullets for chronic hepatitis C infection? Cleve Clin J Med 2012; 79:213–222.
- McHutchison JG, Everson GT, Gordon SC, et al; PROVE1 Study Team. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009; 360:1827–1838.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Jacobson IM, Catlett I, Marcellin P, et al. Telaprevir substantially improved SVR rates across all IL28B genotypes in the ADVANCE trial. J Hepatol 2011; 54(suppl 1):S542–S543.
- Pol S, Aerssens J, Zeuzem S, et al. Limited impact of IL28B genotype on response rates in telaprevir-treated patients with prior treatment failure. J Hepatol 2013; 58:883–889.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009; 41:1105–1109.
- Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009; 461:798–801.
- Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 2009; 119:1745–1754.
- Marcello T, Grakoui A, Barba-Spaeth G, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006; 131:1887–1898.
- Doyle SE, Schreckhise H, Khuu-Duong K, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006; 44:896–906.
- Raglow Z, Thoma-Perry C, Gilroy R, Wan YJ. IL28B genotype and the expression of ISGs in normal liver. Liver Int 2013; 33:991–998.
- Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet 2013; 45:164–171.
- Hanouneh IA, Zein NN, Askar M, Lopez R, John B. Interleukin-28B polymorphisms are associated with fibrosing cholestatic hepatitis in recurrent hepatitis C after liver transplantation. Clin Transplant 2012; 26:E335–E336.
- Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 1997; 349:825–832.
- Thomas DL, Astemborski J, Rai RM, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA 2000; 284:450–456.
- Bochud PY, Cai T, Overbeck K, et al; Swiss Hepatitis C Cohort Study Group. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol 2009; 51:655–666.
- Marabita F, Aghemo A, De Nicola S, et al. Genetic variation in the interleukin-28B gene is not associated with fibrosis progression in patients with chronic hepatitis C and known date of infection. Hepatology 2011; 54:1127–1134.
- Fabris C, Falleti E, Cussigh A, et al. IL-28B rs12979860 C/T allele distribution in patients with liver cirrhosis: role in the course of chronic viral hepatitis and the development of HCC. J Hepatol 2011; 54:716–722.
- Eurich D, Boas-Knoop S, Bahra M, et al. Role of IL28B polymorphism in the development of hepatitis C virus-induced hepatocellular carcinoma, graft fibrosis, and posttransplant antiviral therapy. Transplantation 2012; 93:644–649.
- Hanouneh IA, Miller C, Aucejo F, Lopez R, Quinn MK, Zein NN. Recurrent hepatitis C after liver transplantation: on-treatment prediction of response to peginterferon/ribavirin therapy. Liver Transpl 2008; 14:53–58.
- Charlton MR, Thompson A, Veldt BJ, et al. Interleukin-28B polymorphisms are associated with histological recurrence and treatment response following liver transplantation in patients with hepatitis C virus infection. Hepatology 2011; 53:317–324.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e18.
- Beinhardt S, Payer BA, Datz C, et al. A diagnostic score for the prediction of spontaneous resolution of acute hepatitis C virus infection. J Hepatol 2013; 59:972–977.
- Forns X, Lawitz E, Zeuzem S, et al. Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a phase 3 trial. Gastroenterology 2014; 146:1669–1679.e3.
- Sofia MJ, Bao D, Chang W, et al. Discovery of a ß-d-2’-deoxy-2’-ß-fluoro-2’-ß-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem 2010; 53:7202–7218.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Sulkowski MS, Jacobson IM, Ghalib R, et al. Once-daily simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in HCV genotype 1 prior null responders with metavir F0-2: COSMOS study subgroup analysis. 49th EASL, April 2014, London. Oral abstract O7. www.natap.org/2014/EASL/EASL_46.htm. Accesed January 9, 2015.
- Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 2012; 366:216–224.
- Afdhal N, Zeuzem S, Kwo P, et al; ION-1 Investigators. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 2014; 370:1889–1898.
What a difference a single nucleotide can make! The human genome contains more than 3 billion base pairs. Yet having a different nucleotide in only one pair can make a big difference in how we respond to a disease or its treatment.
Specifically, in hepatitis C virus infection, people born with the nucleotide cytosine (C) at location rs12979860 in both alleles of the gene that codes for interleukin 28B (the IL28B CC genotype) can count themselves luckier than those born with thymine (T) in this location in one of their alleles (the CT genotype) or both of their alleles (the TT genotype). Those with the CC genotype are more likely to clear the virus spontaneously, and even if the infection persists, it is less likely to progress to liver cancer and more likely to respond to treatment with interferon.
Here, we review the IL28B polymorphism and its implications in treating hepatitis C.
GENETIC POLYMORPHISM AND HUMAN DISEASE
Of the 3 billion base pairs of nucleotides, fewer than 1% differ between individuals, but this 1% is responsible for the diversity of human beings. Differences in genetic sequences among individuals are called genetic polymorphisms. A single-nucleotide polymorphism is a DNA sequence variation that occurs in a single nucleotide in the genome. For example, two sequenced DNA fragments from different individuals, AAGCCTA and AAGCTTA, contain a difference in a single nucleotide.
Genetic variations such as these underlie some of the differences in our susceptibility to disease, the severity of illness we develop, and our response to treatments. Therefore, identifying genetic polymorphisms may shed light on biologic pathways involved in diseases and may uncover new targets for therapy.1
Genome-wide association studies have looked at hundreds of thousands of single-nucleotide polymorphisms to try to identify most of the common genetic differences among people and relate them to common chronic diseases such as coronary artery disease,2 type 2 diabetes,3 stroke,4 breast cancer,5 rheumatoid arthritis,6 Alzheimer disease,7 and, more recently, hepatitis C virus infection.8
HEPATITIS C VIRUS: A MAJOR CAUSE OF LIVER DISEASE
Hepatitis C virus infection is a major cause of chronic liver disease and hepatocellular carcinoma and has become the most common indication for liver transplantation in the United States.9
This virus has six distinct genotypes throughout the world, with multiple subtypes in each genotype. (A genotype is a classification of a virus based on its RNA.9) In this review, we will focus on genotype 1; hence, “hepatitis C virus” will refer to hepatitis C virus genotype 1.
Our knowledge of the biology, pathogenesis, and treatment of hepatitis C has been advancing. Originally, fewer than 50% of patients responded to therapy with the combination of pegylated interferon and ribavirin,10,11 but since 2011 the response rate has increased to approximately 70% with the approval of the protease inhibitors telaprevir and boceprevir, used in combination with pegylated interferon and ribavirin.12–15
Unfortunately, interferon-based treatment is often complicated by side effects such as fatigue, influenza-like symptoms, hematologic abnormalities, and neuropsychiatric symptoms. An accurate way to predict response would help patients make informed decisions about antiviral treatment, taking into account the risk and possible benefit for individual patients.
GENETIC POLYMORPHISM AND HEPATITIS C VIRUS INFECTION
Genome-wide association studies have identified single-nucleotide polymorphisms in the IL28B gene that are associated with differences in response to hepatitis C treatment.8
Studying 565,759 polymorphisms in 1,137 patients, researchers at Duke University identified a single-nucleotide polymorphism at location rs12979860 in IL28B (Figure 1) that was strongly associated with response to combination therapy with pegylated interferon and ribavirin.8 The chance of cure with this standard treatment is twice as high in patients who are homozygous for cytosine in this location (the CC genotype) than in those who are heterozygous (CT) or homozygous for thymine in this location (the TT genotype) (Table 1).
Adding one of the new protease inhibitors, telaprevir or boceprevir, to the standard hepatitis C treatment substantially improves the cure rates in all three IL28B genotypes, but especially in people with CT or TT, in whom the response rate almost triples with the addition of one of these drugs. Those with the CC genotype (who are more likely to be cured with pegylated interferon and ribavirin alone) also achieve an increase (although minimal) in cure rates when a protease inhibitor is included in the regimen (TABLE 1).13–15 Thus, it remains unclear if adding a protease inhibitor to pegylated interferon plus ribavirin in patients with the IL28B CC genotype translates into added effectiveness worth the additional cost of the protease inhibitor in previously untreated patients.
Additionally, the effect of the IL28B genotype on telaprevir-based triple therapy has been disputed in more recent studies. In a subgroup analysis of the results of a trial that evaluated telaprevir in the treatment of hepatitis C, researchers found that sustained virologic response rates were significantly higher in the telaprevir group, and this was similar across the different IL28B polymorphisms.16
The favorable IL28B CC genotype is associated with higher rates of rapid virologic response to antiviral therapy.13–15 Of note, almost all patients who achieve a rapid virologic response do well, with a high rate of sustained virologic response even after a shorter duration of therapy (24 vs 48 weeks). Therefore, in addition to predicting response to interferon before starting treatment, the IL28B CC genotype may also identify patients who need only a shorter duration of therapy.
Interestingly, the C allele is much more frequent in white than in African American populations, an important observation that explains the racial difference in response to hepatitis C therapy.8
Two other research groups, from Asia and Australia, performed independent genome-wide association studies that identified different single-nucleotide polymorphisms (eg, rs8099917) in the same IL28B gene as predictors of response to treatment in patients with hepatitis C virus infection.17,18 These findings may be explained by linkage disequilibrium, which means that these single-nucleotide polymorphisms are found more frequently together in the same patient due to their proximity to each other. In this review, we will focus on the rs12979860 polymorphism; hence “IL28B genotype” will refer to the single-nucleotide polymorphism at rs12979860, unless otherwise specified.
The favorable CC genotype is less common in African Americans than in patients of other ethnicities.19 Moreover, although IL28B CC is associated with a better response rate to interferon-based antiviral therapy across all ethnicities, those of African American descent with the CC genotype are less likely to achieve a sustained virologic response than white or Hispanic Americans.8
BIOLOGIC ASSOCIATION: IL28B POLYMORPHISM AND HEPATITIS C
The interferon lambda family consists of three cytokines:
- Interleukin 29 (interferon lambda 1)
- Interleukin 28A (interferon lambda 2)
- Interleukin 28B (interferon lambda 3).
Production of these three molecules can be triggered by viral infection, and they induce antiviral activity through both innate and adaptive immune pathways. They signal through the IL10R-IL28R receptor complex.20–22 This receptor activates the JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway, which regulates a large number of interferon-stimulated genes, primarily through the interferon-stimulated response element (Figure 2).
A 2013 study found that interferon-stimulated gene expression levels in patients with normal livers were highest in those with the CC genotype, intermediate with CT, and lowest with TT. Interestingly, this pattern was reversed in those with hepatitis C virus infection, indicating a relationship between the IL28B genotype and gene expression before infection.23
The mechanism underlying the association between the IL28B polymorphism and response to hepatitis C treatment is not well understood. The unfavorable TT genotype seems to lead to continuous activation of a subset of interferon-stimulated genes in the presence of intracellular hepatitis C viral RNA. But this level of expression is not sufficient to eliminate the virus from the cells. Instead, it might lead to up-regulation of interferon-inhibitory molecules that suppress JAK-STAT signaling, thereby reducing sensitivity to interferon signaling. Therefore, the hepatocyte not only cannot clear the virus by itself, but also cannot induce strong interferon-stimulated gene expression when interferon is given during therapy.20–22
The recently identified ss469425590 polymorphism, which is located in close proximity to rs12979860 in the IL28B gene, is particularly interesting, as it suggests a possible molecular mechanism. The delta G frameshift variant creates a novel gene called IFNL4, which is transiently activated in response to hepatitis C virus infection.24IFNL4 stimulates STAT1 and STAT2 phosphorylation and induces the expression of interferon-stimulated genes. Increased interferon-stimulated gene expression has been shown to be associated with decreased response to pegylated interferon-ribavirin treatment. These observations suggest that the ss469425590 delta G allele is responsible for the increased activation of interferon-stimulated genes and the lower sustained virologic response rate observed in patients who receive pegylated interferon-ribavirin treatment. It is possible that the activation of interferon-stimulated genes in patients with the ss469425590 delta G/delta G genotype reduces interferon-stimulated gene responsiveness to interferon alpha, which normally activates interferon-stimulated genes and inhibits hepatitis C progression.24
IL28B POLYMORPHISM AND ACUTE HEPATITIS C VIRUS INFECTION
From 70% to 80% of acute hepatitis C virus infections persist and become chronic, while 20% to 30% spontaneously resolve. Epidemiologic, viral, and host factors have been associated with the differences in viral clearance or persistence, and studies have found that a strong host immune response against the virus favors viral clearance. Thus, variation in the genes involved in the immune response may contribute to one’s ability to clear the virus. Consistent with these observations, recent studies have shown that the polymorphism in the IL28 gene region encoding interferon lambda 3 strongly predicts spontaneous resolution of acute hepatitis C virus infection. People who have the IL28B CC genotype are three times more likely to spontaneously clear the virus than those with the CT or TT genotype (Figure 3).24
IL28B POLYMORPHISM AND THE NATURAL HISTORY OF HEPATITIS C
In people in whom hepatitis C virus infection persists, up to 20% develop progressive liver fibrosis and eventually cirrhosis over 10 to 20 years.19,25,26 The speed at which fibrosis develops in these patients is variable and unpredictable.25 The relationship between IL28B polymorphisms and hepatic fibrosis in patients with chronic hepatitis C virus infection has not been clearly established, although a study indicated that in patients with a known date of infection, the IL28B genotype is not associated with progression of hepatic fibrosis.27 Obstacles in this field of study are that it is difficult to determine accurately when the patient contracted the virus, and that serial liver biopsies are needed to investigate the progression of hepatic fibrosis.
Patients with chronic hepatitis C virus infection are also at higher risk of hepatocellular carcinoma compared with the general population.28 An analysis of explanted livers of patients with hepatitis C found that the prevalence of hepatocellular carcinoma in those with the unfavorable TT genotype was significantly higher than with the other genotypes.29 Similarly, an earlier study demonstrated that patients with hepatitis C-associated hepatocellular carcinoma carried the T allele more frequently.30 As with other aspects of IL28B associations with hepatitis C, these findings indicate that the C allele confers a certain degree of protection.
An important implication of these relationships is that they may eventually help identify patients at greater risk, who therefore need earlier intervention.
IL28B POLYMORPHISM AND LIVER TRANSPLANTATION
Hepatitis C virus infection always recurs after liver transplantation, with serious consequences that include cirrhosis and liver failure. Recurrent hepatitis C virus infection has become an important reason for repeat transplantation in the United States.
Results of treatment with pegylated interferon and ribavirin for recurrent hepatitis C after liver transplantation have been disappointing, with response rates lower than 30% and significant side effects.31 Identifying the factors that predict the response to therapy allows for better selection of treatment candidates.
Similar to the way the IL28B genotype predicts response to antiviral therapy in the nontransplant setting, the IL28B genotypes of both the recipient and the donor are strongly and independently associated with response to interferon-based treatment in patients with hepatitis C after liver transplantation. The IL28B CC genotype in either the recipient or the donor is associated with a higher rate of response to pegylated interferon and ribavirin combination therapy after liver transplantation.30,32 For example, the response rate to therapy after liver transplantation reaches 86% in CC-donor and CC-recipient livers, compared with 0% in TT-donor and TT-recipient livers.
Additionally, the IL28B genotype of the recipient may determine the severity of histologic recurrence of hepatitis C, as indicated by progressive hepatic fibrosis. A recipient IL28B TT genotype is associated with more severe histologic recurrence of hepatitis C.33
These data suggest that CC donor livers might be preferentially allocated to patients with hepatitis C virus infection.
IL28B AND OTHER FACTORS IN HEPATITIS C VIRUS INFECTION
Although it is tempting to think that the IL28B polymorphism is the sole predictor of response to antiviral therapy, it is but one of several known factors in the virus and the host.
While IL28B polymorphisms are the most important predictor of sustained virologic response with an interferon-based regimen, a rapid virologic response (undetectable viral load at 4 weeks) had superior predictive value and specificity in one study.34 In fact, for patients with chronic hepatitis C infection who achieved a rapid virologic response with pegylated interferon and ribavirin, the IL28B polymorphism had no effect on the rate of sustained virologic response. However, it did predict a sustained virologic response in the group who did not achieve rapid virologic response.
In a study of patients with acute hepatitis C infection,35 jaundice and the IL28 rs12979860 CC genotype both predicted spontaneous clearance. The best predictor of viral persistence was the combination of the CT or TT genotype plus the absence of jaundice, which had a predictive value of 98%.
IL28B AND THE FUTURE OF HEPATITIS C VIRUS THERAPY
New oral agents were recently approved for treating hepatitis C. As of November 2014, these included simeprevir, sofosbuvir, and ledipasvir.
Simeprevir is a second-generation NS3/4A protease inhibitor approved for use in combination with pegylated interferon and ribavirin. A recent phase 3 trial evaluating simeprevir in patients who had relapsed after prior therapy found sustained virologic response rates to be higher with simeprevir than with placebo, irrespective of IL28B status.36 This finding was similar to that of a trial of telaprevir.16
Sofosbuvir is a nucleotide analogue NS5B polymerase inhibitor that becomes incorporated into the growing RNA, inducing a chain termination event.37 In phase 3 trials,38,39 researchers found an initial rapid decrease in viral load for patients treated with this agent regardless of IL28B status.
In the NEUTRINO trial (Sofosbuvir With Peginterferon Alfa 2a and Ribavirin for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1, 4, 5, or 6 HCV Infection),38 which used sofusbuvir in combination with interferon and ribavirin, the rate of sustained virologic response was higher in those with the favorable CC genotype (98%) than with a non-CC genotype (87%).
In COSMOS (A Study of TMC435 in Combination With PSI-7977 [GS7977] in Chronic Hepatitis C Genotype 1-Infected Prior Null Responders to Peginterferon/Ribavirin Therapy or HCV Treatment-Naive Patients),39 which used a combination of simeprevir, sofosbuvir, and ribavirin, the rate of sustained virologic response was higher in those with the CC genotype (100%) than with the TT genotype (83%; Table 1).
These new medications have radically changed the landscape of hepatitis C therapy and have also unlocked the potential for developing completely interferon-free regimens.
Other new interferon-free regimens such as ledipasvir, daclatasvir, and asunaprevir promise high rates of sustained virologic response, which makes the utility of testing for IL28B polymorphisms to predict sustained virologic response very much diminished (Table 1).40,41 However, these new drugs are expected to be expensive, and IL28B polymorphisms may be used to identify candidates who are more likely to respond to pegylated interferon and ribavirin, particularly in resource-poor settings and in developing countries. Additionally, patients who have contraindications to these newer therapies will still likely need an interferon-based regimen, and thus the IL28B polymorphism will still be important in predicting treatment response and prognosis.
IL28B WILL STILL BE RELEVANT IN THE INTERFERON-FREE AGE
The IL28B polymorphism is a strong predictor of spontaneous clearance of hepatitis C virus and responsiveness to interferon-based therapy, and testing for it has demonstrated a great potential to improve patient care. IL28B testing has become available for clinical use and may optimize the outcome of hepatitis C treatment by helping us to select the best treatment for individual patients and minimizing the duration of therapy and the side effects associated with interferon-based antiviral medications.
As newer therapies have shifted toward interferon-free regimens that offer very high sustained virologic response rates, the usefulness of IL28B polymorphism as a clinical test to predict the response rate to antiviral therapy is minimized substantially. It may remain clinically relevant in resource-poor settings and in developing countries, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated. This does not minimize the lesson we learned from the discovery of the IL28B gene and the impact on our understanding of the pathogenesis of hepatitis C virus infection.
What a difference a single nucleotide can make! The human genome contains more than 3 billion base pairs. Yet having a different nucleotide in only one pair can make a big difference in how we respond to a disease or its treatment.
Specifically, in hepatitis C virus infection, people born with the nucleotide cytosine (C) at location rs12979860 in both alleles of the gene that codes for interleukin 28B (the IL28B CC genotype) can count themselves luckier than those born with thymine (T) in this location in one of their alleles (the CT genotype) or both of their alleles (the TT genotype). Those with the CC genotype are more likely to clear the virus spontaneously, and even if the infection persists, it is less likely to progress to liver cancer and more likely to respond to treatment with interferon.
Here, we review the IL28B polymorphism and its implications in treating hepatitis C.
GENETIC POLYMORPHISM AND HUMAN DISEASE
Of the 3 billion base pairs of nucleotides, fewer than 1% differ between individuals, but this 1% is responsible for the diversity of human beings. Differences in genetic sequences among individuals are called genetic polymorphisms. A single-nucleotide polymorphism is a DNA sequence variation that occurs in a single nucleotide in the genome. For example, two sequenced DNA fragments from different individuals, AAGCCTA and AAGCTTA, contain a difference in a single nucleotide.
Genetic variations such as these underlie some of the differences in our susceptibility to disease, the severity of illness we develop, and our response to treatments. Therefore, identifying genetic polymorphisms may shed light on biologic pathways involved in diseases and may uncover new targets for therapy.1
Genome-wide association studies have looked at hundreds of thousands of single-nucleotide polymorphisms to try to identify most of the common genetic differences among people and relate them to common chronic diseases such as coronary artery disease,2 type 2 diabetes,3 stroke,4 breast cancer,5 rheumatoid arthritis,6 Alzheimer disease,7 and, more recently, hepatitis C virus infection.8
HEPATITIS C VIRUS: A MAJOR CAUSE OF LIVER DISEASE
Hepatitis C virus infection is a major cause of chronic liver disease and hepatocellular carcinoma and has become the most common indication for liver transplantation in the United States.9
This virus has six distinct genotypes throughout the world, with multiple subtypes in each genotype. (A genotype is a classification of a virus based on its RNA.9) In this review, we will focus on genotype 1; hence, “hepatitis C virus” will refer to hepatitis C virus genotype 1.
Our knowledge of the biology, pathogenesis, and treatment of hepatitis C has been advancing. Originally, fewer than 50% of patients responded to therapy with the combination of pegylated interferon and ribavirin,10,11 but since 2011 the response rate has increased to approximately 70% with the approval of the protease inhibitors telaprevir and boceprevir, used in combination with pegylated interferon and ribavirin.12–15
Unfortunately, interferon-based treatment is often complicated by side effects such as fatigue, influenza-like symptoms, hematologic abnormalities, and neuropsychiatric symptoms. An accurate way to predict response would help patients make informed decisions about antiviral treatment, taking into account the risk and possible benefit for individual patients.
GENETIC POLYMORPHISM AND HEPATITIS C VIRUS INFECTION
Genome-wide association studies have identified single-nucleotide polymorphisms in the IL28B gene that are associated with differences in response to hepatitis C treatment.8
Studying 565,759 polymorphisms in 1,137 patients, researchers at Duke University identified a single-nucleotide polymorphism at location rs12979860 in IL28B (Figure 1) that was strongly associated with response to combination therapy with pegylated interferon and ribavirin.8 The chance of cure with this standard treatment is twice as high in patients who are homozygous for cytosine in this location (the CC genotype) than in those who are heterozygous (CT) or homozygous for thymine in this location (the TT genotype) (Table 1).
Adding one of the new protease inhibitors, telaprevir or boceprevir, to the standard hepatitis C treatment substantially improves the cure rates in all three IL28B genotypes, but especially in people with CT or TT, in whom the response rate almost triples with the addition of one of these drugs. Those with the CC genotype (who are more likely to be cured with pegylated interferon and ribavirin alone) also achieve an increase (although minimal) in cure rates when a protease inhibitor is included in the regimen (TABLE 1).13–15 Thus, it remains unclear if adding a protease inhibitor to pegylated interferon plus ribavirin in patients with the IL28B CC genotype translates into added effectiveness worth the additional cost of the protease inhibitor in previously untreated patients.
Additionally, the effect of the IL28B genotype on telaprevir-based triple therapy has been disputed in more recent studies. In a subgroup analysis of the results of a trial that evaluated telaprevir in the treatment of hepatitis C, researchers found that sustained virologic response rates were significantly higher in the telaprevir group, and this was similar across the different IL28B polymorphisms.16
The favorable IL28B CC genotype is associated with higher rates of rapid virologic response to antiviral therapy.13–15 Of note, almost all patients who achieve a rapid virologic response do well, with a high rate of sustained virologic response even after a shorter duration of therapy (24 vs 48 weeks). Therefore, in addition to predicting response to interferon before starting treatment, the IL28B CC genotype may also identify patients who need only a shorter duration of therapy.
Interestingly, the C allele is much more frequent in white than in African American populations, an important observation that explains the racial difference in response to hepatitis C therapy.8
Two other research groups, from Asia and Australia, performed independent genome-wide association studies that identified different single-nucleotide polymorphisms (eg, rs8099917) in the same IL28B gene as predictors of response to treatment in patients with hepatitis C virus infection.17,18 These findings may be explained by linkage disequilibrium, which means that these single-nucleotide polymorphisms are found more frequently together in the same patient due to their proximity to each other. In this review, we will focus on the rs12979860 polymorphism; hence “IL28B genotype” will refer to the single-nucleotide polymorphism at rs12979860, unless otherwise specified.
The favorable CC genotype is less common in African Americans than in patients of other ethnicities.19 Moreover, although IL28B CC is associated with a better response rate to interferon-based antiviral therapy across all ethnicities, those of African American descent with the CC genotype are less likely to achieve a sustained virologic response than white or Hispanic Americans.8
BIOLOGIC ASSOCIATION: IL28B POLYMORPHISM AND HEPATITIS C
The interferon lambda family consists of three cytokines:
- Interleukin 29 (interferon lambda 1)
- Interleukin 28A (interferon lambda 2)
- Interleukin 28B (interferon lambda 3).
Production of these three molecules can be triggered by viral infection, and they induce antiviral activity through both innate and adaptive immune pathways. They signal through the IL10R-IL28R receptor complex.20–22 This receptor activates the JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway, which regulates a large number of interferon-stimulated genes, primarily through the interferon-stimulated response element (Figure 2).
A 2013 study found that interferon-stimulated gene expression levels in patients with normal livers were highest in those with the CC genotype, intermediate with CT, and lowest with TT. Interestingly, this pattern was reversed in those with hepatitis C virus infection, indicating a relationship between the IL28B genotype and gene expression before infection.23
The mechanism underlying the association between the IL28B polymorphism and response to hepatitis C treatment is not well understood. The unfavorable TT genotype seems to lead to continuous activation of a subset of interferon-stimulated genes in the presence of intracellular hepatitis C viral RNA. But this level of expression is not sufficient to eliminate the virus from the cells. Instead, it might lead to up-regulation of interferon-inhibitory molecules that suppress JAK-STAT signaling, thereby reducing sensitivity to interferon signaling. Therefore, the hepatocyte not only cannot clear the virus by itself, but also cannot induce strong interferon-stimulated gene expression when interferon is given during therapy.20–22
The recently identified ss469425590 polymorphism, which is located in close proximity to rs12979860 in the IL28B gene, is particularly interesting, as it suggests a possible molecular mechanism. The delta G frameshift variant creates a novel gene called IFNL4, which is transiently activated in response to hepatitis C virus infection.24IFNL4 stimulates STAT1 and STAT2 phosphorylation and induces the expression of interferon-stimulated genes. Increased interferon-stimulated gene expression has been shown to be associated with decreased response to pegylated interferon-ribavirin treatment. These observations suggest that the ss469425590 delta G allele is responsible for the increased activation of interferon-stimulated genes and the lower sustained virologic response rate observed in patients who receive pegylated interferon-ribavirin treatment. It is possible that the activation of interferon-stimulated genes in patients with the ss469425590 delta G/delta G genotype reduces interferon-stimulated gene responsiveness to interferon alpha, which normally activates interferon-stimulated genes and inhibits hepatitis C progression.24
IL28B POLYMORPHISM AND ACUTE HEPATITIS C VIRUS INFECTION
From 70% to 80% of acute hepatitis C virus infections persist and become chronic, while 20% to 30% spontaneously resolve. Epidemiologic, viral, and host factors have been associated with the differences in viral clearance or persistence, and studies have found that a strong host immune response against the virus favors viral clearance. Thus, variation in the genes involved in the immune response may contribute to one’s ability to clear the virus. Consistent with these observations, recent studies have shown that the polymorphism in the IL28 gene region encoding interferon lambda 3 strongly predicts spontaneous resolution of acute hepatitis C virus infection. People who have the IL28B CC genotype are three times more likely to spontaneously clear the virus than those with the CT or TT genotype (Figure 3).24
IL28B POLYMORPHISM AND THE NATURAL HISTORY OF HEPATITIS C
In people in whom hepatitis C virus infection persists, up to 20% develop progressive liver fibrosis and eventually cirrhosis over 10 to 20 years.19,25,26 The speed at which fibrosis develops in these patients is variable and unpredictable.25 The relationship between IL28B polymorphisms and hepatic fibrosis in patients with chronic hepatitis C virus infection has not been clearly established, although a study indicated that in patients with a known date of infection, the IL28B genotype is not associated with progression of hepatic fibrosis.27 Obstacles in this field of study are that it is difficult to determine accurately when the patient contracted the virus, and that serial liver biopsies are needed to investigate the progression of hepatic fibrosis.
Patients with chronic hepatitis C virus infection are also at higher risk of hepatocellular carcinoma compared with the general population.28 An analysis of explanted livers of patients with hepatitis C found that the prevalence of hepatocellular carcinoma in those with the unfavorable TT genotype was significantly higher than with the other genotypes.29 Similarly, an earlier study demonstrated that patients with hepatitis C-associated hepatocellular carcinoma carried the T allele more frequently.30 As with other aspects of IL28B associations with hepatitis C, these findings indicate that the C allele confers a certain degree of protection.
An important implication of these relationships is that they may eventually help identify patients at greater risk, who therefore need earlier intervention.
IL28B POLYMORPHISM AND LIVER TRANSPLANTATION
Hepatitis C virus infection always recurs after liver transplantation, with serious consequences that include cirrhosis and liver failure. Recurrent hepatitis C virus infection has become an important reason for repeat transplantation in the United States.
Results of treatment with pegylated interferon and ribavirin for recurrent hepatitis C after liver transplantation have been disappointing, with response rates lower than 30% and significant side effects.31 Identifying the factors that predict the response to therapy allows for better selection of treatment candidates.
Similar to the way the IL28B genotype predicts response to antiviral therapy in the nontransplant setting, the IL28B genotypes of both the recipient and the donor are strongly and independently associated with response to interferon-based treatment in patients with hepatitis C after liver transplantation. The IL28B CC genotype in either the recipient or the donor is associated with a higher rate of response to pegylated interferon and ribavirin combination therapy after liver transplantation.30,32 For example, the response rate to therapy after liver transplantation reaches 86% in CC-donor and CC-recipient livers, compared with 0% in TT-donor and TT-recipient livers.
Additionally, the IL28B genotype of the recipient may determine the severity of histologic recurrence of hepatitis C, as indicated by progressive hepatic fibrosis. A recipient IL28B TT genotype is associated with more severe histologic recurrence of hepatitis C.33
These data suggest that CC donor livers might be preferentially allocated to patients with hepatitis C virus infection.
IL28B AND OTHER FACTORS IN HEPATITIS C VIRUS INFECTION
Although it is tempting to think that the IL28B polymorphism is the sole predictor of response to antiviral therapy, it is but one of several known factors in the virus and the host.
While IL28B polymorphisms are the most important predictor of sustained virologic response with an interferon-based regimen, a rapid virologic response (undetectable viral load at 4 weeks) had superior predictive value and specificity in one study.34 In fact, for patients with chronic hepatitis C infection who achieved a rapid virologic response with pegylated interferon and ribavirin, the IL28B polymorphism had no effect on the rate of sustained virologic response. However, it did predict a sustained virologic response in the group who did not achieve rapid virologic response.
In a study of patients with acute hepatitis C infection,35 jaundice and the IL28 rs12979860 CC genotype both predicted spontaneous clearance. The best predictor of viral persistence was the combination of the CT or TT genotype plus the absence of jaundice, which had a predictive value of 98%.
IL28B AND THE FUTURE OF HEPATITIS C VIRUS THERAPY
New oral agents were recently approved for treating hepatitis C. As of November 2014, these included simeprevir, sofosbuvir, and ledipasvir.
Simeprevir is a second-generation NS3/4A protease inhibitor approved for use in combination with pegylated interferon and ribavirin. A recent phase 3 trial evaluating simeprevir in patients who had relapsed after prior therapy found sustained virologic response rates to be higher with simeprevir than with placebo, irrespective of IL28B status.36 This finding was similar to that of a trial of telaprevir.16
Sofosbuvir is a nucleotide analogue NS5B polymerase inhibitor that becomes incorporated into the growing RNA, inducing a chain termination event.37 In phase 3 trials,38,39 researchers found an initial rapid decrease in viral load for patients treated with this agent regardless of IL28B status.
In the NEUTRINO trial (Sofosbuvir With Peginterferon Alfa 2a and Ribavirin for 12 Weeks in Treatment-Naive Subjects With Chronic Genotype 1, 4, 5, or 6 HCV Infection),38 which used sofusbuvir in combination with interferon and ribavirin, the rate of sustained virologic response was higher in those with the favorable CC genotype (98%) than with a non-CC genotype (87%).
In COSMOS (A Study of TMC435 in Combination With PSI-7977 [GS7977] in Chronic Hepatitis C Genotype 1-Infected Prior Null Responders to Peginterferon/Ribavirin Therapy or HCV Treatment-Naive Patients),39 which used a combination of simeprevir, sofosbuvir, and ribavirin, the rate of sustained virologic response was higher in those with the CC genotype (100%) than with the TT genotype (83%; Table 1).
These new medications have radically changed the landscape of hepatitis C therapy and have also unlocked the potential for developing completely interferon-free regimens.
Other new interferon-free regimens such as ledipasvir, daclatasvir, and asunaprevir promise high rates of sustained virologic response, which makes the utility of testing for IL28B polymorphisms to predict sustained virologic response very much diminished (Table 1).40,41 However, these new drugs are expected to be expensive, and IL28B polymorphisms may be used to identify candidates who are more likely to respond to pegylated interferon and ribavirin, particularly in resource-poor settings and in developing countries. Additionally, patients who have contraindications to these newer therapies will still likely need an interferon-based regimen, and thus the IL28B polymorphism will still be important in predicting treatment response and prognosis.
IL28B WILL STILL BE RELEVANT IN THE INTERFERON-FREE AGE
The IL28B polymorphism is a strong predictor of spontaneous clearance of hepatitis C virus and responsiveness to interferon-based therapy, and testing for it has demonstrated a great potential to improve patient care. IL28B testing has become available for clinical use and may optimize the outcome of hepatitis C treatment by helping us to select the best treatment for individual patients and minimizing the duration of therapy and the side effects associated with interferon-based antiviral medications.
As newer therapies have shifted toward interferon-free regimens that offer very high sustained virologic response rates, the usefulness of IL28B polymorphism as a clinical test to predict the response rate to antiviral therapy is minimized substantially. It may remain clinically relevant in resource-poor settings and in developing countries, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated. This does not minimize the lesson we learned from the discovery of the IL28B gene and the impact on our understanding of the pathogenesis of hepatitis C virus infection.
- Attia J, Ioannidis JP, Thakkinstian A, et al. How to use an article about genetic association: A: background concepts. JAMA 2009; 301:74–81.
- Samani NJ, Erdmann J, Hall AS, et al; WTCCC and the Cardiogenics Consortium. Genomewide association analysis of coronary artery disease. N Engl J Med 2007; 357:443–453.
- Zeggini E, Weedon MN, Lindgren CM, et al; Wellcome Trust Case Control Consortium (WTCCC). Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007; 316:1336–1341.
- Matarín M, Brown WM, Scholz S, et al. A genome-wide genotyping study in patients with ischaemic stroke: initial analysis and data release. Lancet Neurol 2007; 6:414–420.
- Easton DF, Pooley KA, Dunning AM, et al; AOCS Management Group. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007; 447:1087–1093.
- Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—a genomewide study. N Engl J Med 2007; 357:1199–1209.
- Coon KD, Myers AJ, Craig DW, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 2007; 68:613–618.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance (letter). Nature 2009; 461:399–401.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005-1019.
- Hanouneh IA, Feldstein AE, Lopez R, et al. Clinical significance of metabolic syndrome in the setting of chronic hepatitis C virus infection. Clin Gastroenterol Hepatol 2008; 6:584–589.
- Elgouhari HM, Zein CO, Hanouneh I, Feldstein AE, Zein NN. Diabetes mellitus is associated with impaired response to antiviral therapy in chronic hepatitis C infection. Dig Dis Sci 2009; 54:2699–2705.
- Alkhouri N, Zein NN. Protease inhibitors: silver bullets for chronic hepatitis C infection? Cleve Clin J Med 2012; 79:213–222.
- McHutchison JG, Everson GT, Gordon SC, et al; PROVE1 Study Team. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009; 360:1827–1838.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Jacobson IM, Catlett I, Marcellin P, et al. Telaprevir substantially improved SVR rates across all IL28B genotypes in the ADVANCE trial. J Hepatol 2011; 54(suppl 1):S542–S543.
- Pol S, Aerssens J, Zeuzem S, et al. Limited impact of IL28B genotype on response rates in telaprevir-treated patients with prior treatment failure. J Hepatol 2013; 58:883–889.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009; 41:1105–1109.
- Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009; 461:798–801.
- Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 2009; 119:1745–1754.
- Marcello T, Grakoui A, Barba-Spaeth G, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006; 131:1887–1898.
- Doyle SE, Schreckhise H, Khuu-Duong K, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006; 44:896–906.
- Raglow Z, Thoma-Perry C, Gilroy R, Wan YJ. IL28B genotype and the expression of ISGs in normal liver. Liver Int 2013; 33:991–998.
- Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet 2013; 45:164–171.
- Hanouneh IA, Zein NN, Askar M, Lopez R, John B. Interleukin-28B polymorphisms are associated with fibrosing cholestatic hepatitis in recurrent hepatitis C after liver transplantation. Clin Transplant 2012; 26:E335–E336.
- Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 1997; 349:825–832.
- Thomas DL, Astemborski J, Rai RM, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA 2000; 284:450–456.
- Bochud PY, Cai T, Overbeck K, et al; Swiss Hepatitis C Cohort Study Group. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol 2009; 51:655–666.
- Marabita F, Aghemo A, De Nicola S, et al. Genetic variation in the interleukin-28B gene is not associated with fibrosis progression in patients with chronic hepatitis C and known date of infection. Hepatology 2011; 54:1127–1134.
- Fabris C, Falleti E, Cussigh A, et al. IL-28B rs12979860 C/T allele distribution in patients with liver cirrhosis: role in the course of chronic viral hepatitis and the development of HCC. J Hepatol 2011; 54:716–722.
- Eurich D, Boas-Knoop S, Bahra M, et al. Role of IL28B polymorphism in the development of hepatitis C virus-induced hepatocellular carcinoma, graft fibrosis, and posttransplant antiviral therapy. Transplantation 2012; 93:644–649.
- Hanouneh IA, Miller C, Aucejo F, Lopez R, Quinn MK, Zein NN. Recurrent hepatitis C after liver transplantation: on-treatment prediction of response to peginterferon/ribavirin therapy. Liver Transpl 2008; 14:53–58.
- Charlton MR, Thompson A, Veldt BJ, et al. Interleukin-28B polymorphisms are associated with histological recurrence and treatment response following liver transplantation in patients with hepatitis C virus infection. Hepatology 2011; 53:317–324.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e18.
- Beinhardt S, Payer BA, Datz C, et al. A diagnostic score for the prediction of spontaneous resolution of acute hepatitis C virus infection. J Hepatol 2013; 59:972–977.
- Forns X, Lawitz E, Zeuzem S, et al. Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a phase 3 trial. Gastroenterology 2014; 146:1669–1679.e3.
- Sofia MJ, Bao D, Chang W, et al. Discovery of a ß-d-2’-deoxy-2’-ß-fluoro-2’-ß-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem 2010; 53:7202–7218.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Sulkowski MS, Jacobson IM, Ghalib R, et al. Once-daily simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in HCV genotype 1 prior null responders with metavir F0-2: COSMOS study subgroup analysis. 49th EASL, April 2014, London. Oral abstract O7. www.natap.org/2014/EASL/EASL_46.htm. Accesed January 9, 2015.
- Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 2012; 366:216–224.
- Afdhal N, Zeuzem S, Kwo P, et al; ION-1 Investigators. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 2014; 370:1889–1898.
- Attia J, Ioannidis JP, Thakkinstian A, et al. How to use an article about genetic association: A: background concepts. JAMA 2009; 301:74–81.
- Samani NJ, Erdmann J, Hall AS, et al; WTCCC and the Cardiogenics Consortium. Genomewide association analysis of coronary artery disease. N Engl J Med 2007; 357:443–453.
- Zeggini E, Weedon MN, Lindgren CM, et al; Wellcome Trust Case Control Consortium (WTCCC). Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007; 316:1336–1341.
- Matarín M, Brown WM, Scholz S, et al. A genome-wide genotyping study in patients with ischaemic stroke: initial analysis and data release. Lancet Neurol 2007; 6:414–420.
- Easton DF, Pooley KA, Dunning AM, et al; AOCS Management Group. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007; 447:1087–1093.
- Plenge RM, Seielstad M, Padyukov L, et al. TRAF1-C5 as a risk locus for rheumatoid arthritis—a genomewide study. N Engl J Med 2007; 357:1199–1209.
- Coon KD, Myers AJ, Craig DW, et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 2007; 68:613–618.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance (letter). Nature 2009; 461:399–401.
- Ali A, Zein NN. Hepatitis C infection: a systemic disease with extrahepatic manifestations. Cleve Clin J Med 2005; 72:1005-1019.
- Hanouneh IA, Feldstein AE, Lopez R, et al. Clinical significance of metabolic syndrome in the setting of chronic hepatitis C virus infection. Clin Gastroenterol Hepatol 2008; 6:584–589.
- Elgouhari HM, Zein CO, Hanouneh I, Feldstein AE, Zein NN. Diabetes mellitus is associated with impaired response to antiviral therapy in chronic hepatitis C infection. Dig Dis Sci 2009; 54:2699–2705.
- Alkhouri N, Zein NN. Protease inhibitors: silver bullets for chronic hepatitis C infection? Cleve Clin J Med 2012; 79:213–222.
- McHutchison JG, Everson GT, Gordon SC, et al; PROVE1 Study Team. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med 2009; 360:1827–1838.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Jacobson IM, Catlett I, Marcellin P, et al. Telaprevir substantially improved SVR rates across all IL28B genotypes in the ADVANCE trial. J Hepatol 2011; 54(suppl 1):S542–S543.
- Pol S, Aerssens J, Zeuzem S, et al. Limited impact of IL28B genotype on response rates in telaprevir-treated patients with prior treatment failure. J Hepatol 2013; 58:883–889.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Tanaka Y, Nishida N, Sugiyama M, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet 2009; 41:1105–1109.
- Thomas DL, Thio CL, Martin MP, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009; 461:798–801.
- Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 2009; 119:1745–1754.
- Marcello T, Grakoui A, Barba-Spaeth G, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006; 131:1887–1898.
- Doyle SE, Schreckhise H, Khuu-Duong K, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006; 44:896–906.
- Raglow Z, Thoma-Perry C, Gilroy R, Wan YJ. IL28B genotype and the expression of ISGs in normal liver. Liver Int 2013; 33:991–998.
- Prokunina-Olsson L, Muchmore B, Tang W, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet 2013; 45:164–171.
- Hanouneh IA, Zein NN, Askar M, Lopez R, John B. Interleukin-28B polymorphisms are associated with fibrosing cholestatic hepatitis in recurrent hepatitis C after liver transplantation. Clin Transplant 2012; 26:E335–E336.
- Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet 1997; 349:825–832.
- Thomas DL, Astemborski J, Rai RM, et al. The natural history of hepatitis C virus infection: host, viral, and environmental factors. JAMA 2000; 284:450–456.
- Bochud PY, Cai T, Overbeck K, et al; Swiss Hepatitis C Cohort Study Group. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol 2009; 51:655–666.
- Marabita F, Aghemo A, De Nicola S, et al. Genetic variation in the interleukin-28B gene is not associated with fibrosis progression in patients with chronic hepatitis C and known date of infection. Hepatology 2011; 54:1127–1134.
- Fabris C, Falleti E, Cussigh A, et al. IL-28B rs12979860 C/T allele distribution in patients with liver cirrhosis: role in the course of chronic viral hepatitis and the development of HCC. J Hepatol 2011; 54:716–722.
- Eurich D, Boas-Knoop S, Bahra M, et al. Role of IL28B polymorphism in the development of hepatitis C virus-induced hepatocellular carcinoma, graft fibrosis, and posttransplant antiviral therapy. Transplantation 2012; 93:644–649.
- Hanouneh IA, Miller C, Aucejo F, Lopez R, Quinn MK, Zein NN. Recurrent hepatitis C after liver transplantation: on-treatment prediction of response to peginterferon/ribavirin therapy. Liver Transpl 2008; 14:53–58.
- Charlton MR, Thompson A, Veldt BJ, et al. Interleukin-28B polymorphisms are associated with histological recurrence and treatment response following liver transplantation in patients with hepatitis C virus infection. Hepatology 2011; 53:317–324.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e18.
- Beinhardt S, Payer BA, Datz C, et al. A diagnostic score for the prediction of spontaneous resolution of acute hepatitis C virus infection. J Hepatol 2013; 59:972–977.
- Forns X, Lawitz E, Zeuzem S, et al. Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a phase 3 trial. Gastroenterology 2014; 146:1669–1679.e3.
- Sofia MJ, Bao D, Chang W, et al. Discovery of a ß-d-2’-deoxy-2’-ß-fluoro-2’-ß-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J Med Chem 2010; 53:7202–7218.
- Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013; 368:1878–1887.
- Sulkowski MS, Jacobson IM, Ghalib R, et al. Once-daily simeprevir (TMC435) plus sofosbuvir (GS-7977) with or without ribavirin in HCV genotype 1 prior null responders with metavir F0-2: COSMOS study subgroup analysis. 49th EASL, April 2014, London. Oral abstract O7. www.natap.org/2014/EASL/EASL_46.htm. Accesed January 9, 2015.
- Lok AS, Gardiner DF, Lawitz E, et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N Engl J Med 2012; 366:216–224.
- Afdhal N, Zeuzem S, Kwo P, et al; ION-1 Investigators. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 2014; 370:1889–1898.
KEY POINTS
- In IL28B, the rs12979860 location can be occupied by either cytosine (C) or thymine (T). The CC genotype is more favorable than the CT or TT genotype.
- Testing for the IL28B polymorphism is currently available and allows for better outcomes through proper selection of treatment, particularly with interferon-based treatment.
- Although newer therapies have shifted toward regimens that do not use interferon, the IL28B polymorphism remains clinically significant, especially in light of the potentially prohibitive costs of the newer regimens, and for patients in whom these treatments are contraindicated.
Protease inhibitors: Silver bullets for chronic hepatitis C infection?
The treatment of hepatitis c virus (HCV) infection is on the brink of major changes with the recent approval of the first direct-acting antiviral agents, the protease inhibitors boceprevir (Victrelis) and telaprevir (Incivek).
Both drugs were approved by the US Food and Drug Administration (FDA) Advisory Panel for Chronic Hepatitis C in May 2011 and are believed to significantly improve treatment outcomes for patients with HCV genotype 1 infection.
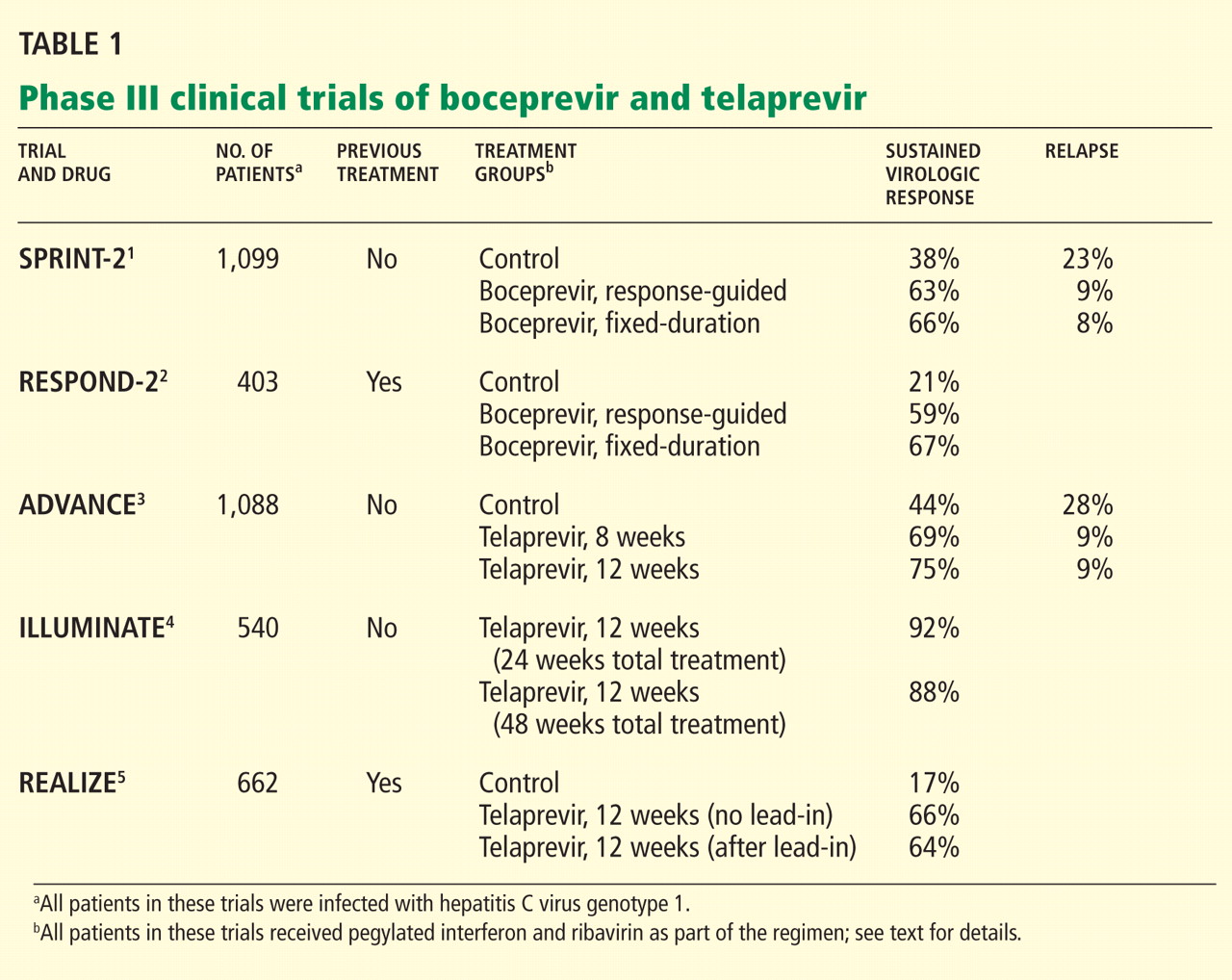
A MAJOR PUBLIC HEALTH PROBLEM
HCV infection is a major public health problem. Nearly 4 million people in the United States are infected.6,7 Most patients with acute HCV infection become chronically infected, and up to 25% eventually develop cirrhosis and its complications, making HCV infection the leading indication for liver transplantation.8–10
Chronic HCV infection has a large global impact, with 180 million people affected across all economic and social groups.11 The highest prevalence of HCV has been reported in Egypt (14%), in part due to the use of inadequately sterilized needles in mass programs to treat endemic schistosomiasis. In developed countries, hepatocellular carcinoma associated with HCV has the fastest growing cancer-related death rate.12
CURRENTLY, FEWER THAN 50% OF PATIENTS ARE CURED
The goal of HCV treatment is to eradicate the virus. However, most infected patients (especially in the United States and Europe) are infected with HCV genotype 1, which is the most difficult genotype to treat.
Successful treatment of HCV is defined as achieving a sustained virologic response—ie, the absence of detectable HCV RNA in the serum 24 weeks after completion of therapy. Once a sustained virologic response is achieved, lifetime “cure” of HCV infection is expected in more than 99% of patients.13
The current standard therapy for HCV, pegylated interferon plus ribavirin for 48 weeks, is effective in only 40% to 50% of patients with genotype 1 infection.14 Therefore, assessing predictors of response before starting treatment can help select patients who are most likely to benefit from therapy.
Viral factors associated with a sustained virologic response include HCV genotypes other than genotype 1 and a low baseline viral load.
Beneficial patient-related factors include younger age, nonblack ethnicity, low body weight (≤ 75 kg), low body mass index, absence of insulin resistance, and absence of advanced fibrosis or cirrhosis.
More recently, a single-nucleotide polymorphism near the interleukin 28B (IL28B) gene, coding for interferon lambda 3, was found to be associated with a twofold difference in the rates of sustained virologic response: patients with the favorable genotype CC were two times more likely to achieve a sustained virologic response than patients with the CT or TT genotypes.15–17
PROTEASE INHIBITORS: MECHANISM OF ACTION
NS3/4A protease inhibitors rely on the principle of end-product inhibition, in which the cleavage product of the protease (a peptide) acts to inhibit the enzyme activity; this is why they are called peptidomimetics. The active site of the NS3/4A protease is a shallow groove composed of three highly conserved amino acid residues, which may explain why protease inhibitors display high antiviral efficacy but pose a low barrier to the development of resistance.20
Protease inhibitors are prone to resistance
The development of viral resistance to protease inhibitors has been a major drawback to their use in patients with chronic HCV infection.21
HCV is a highly variable virus with many genetically distinct but closely related quasispecies circulating in the blood at any given time. Drug-resistant, mutated variants preexist within the patient’s quasispecies, but only in small quantities because of their lesser replication fitness compared with the wild-type virus.22 When direct-acting antiviral therapy is started, the quantity of the wild-type virus decreases and the mutated virus gains replication fitness. Using protease inhibitors as monotherapy selects resistant viral populations rapidly within a few days or weeks.
HCV subtypes 1a and 1b may have different resistance profiles. With genotype 1a, some resistance-associated amino acid substitutions require only one nucleotide change, but with genotype 1b, two nucleotide changes are needed, making resistance less frequent in patients with HCV genotype 1b.23
BOCEPREVIR
Boceprevir is a specific inhibitor of the HCV viral protease NS3/4A.
In phase 3 clinical trials, boceprevir 800 mg three times a day was used with pegylated interferon alfa-2b (PegIntron) 1.5 μg/kg/week and ribavirin (Rebetol) 600 to 1,400 mg daily according to body weight.
Before patients started taking boceprevir, they went through a 4-week lead-in phase, during which they received pegylated interferon and ribavirin. This schedule appeared to reduce the incidence of viral breakthrough in phase 2 trials, and it produced higher rates of sustained virologic response and lower relapse rates compared with triple therapy without a lead-in phase.
Rapid virologic response was defined as undetectable HCV RNA at week 4 of boceprevir therapy (week 8 of the whole regimen).
Boceprevir in previously untreated patients with HCV genotype 1: The SPRINT-2 trial
The Serine Protease Inhibitor Therapy 2 (SPRINT-2) trial1 included more than 1,000 previously untreated adults with HCV genotype 1 infection (938 nonblack patients and 159 black patients; two other nonblack patients did not receive any study drug and were not included in the analysis). In this double-blind trial, patients were randomized into three groups:
- The control group received the standard of care with pegylated interferon and ribavirin for 48 weeks
- The response-guided therapy group received boceprevir plus pegylated interferon and ribavirin for 24 weeks after the 4-week lead-in phase; if HCV RNA was undetectable from week 8 to week 24, treatment was considered complete, but if HCV RNA was detectable at any point from week 8 to week 24, pegylated interferon and ribavirin were continued for a total of 48 weeks.
- The fixed-duration therapy group received boceprevir, pegylated interferon, and ribavirin for 44 weeks after the lead-in period.
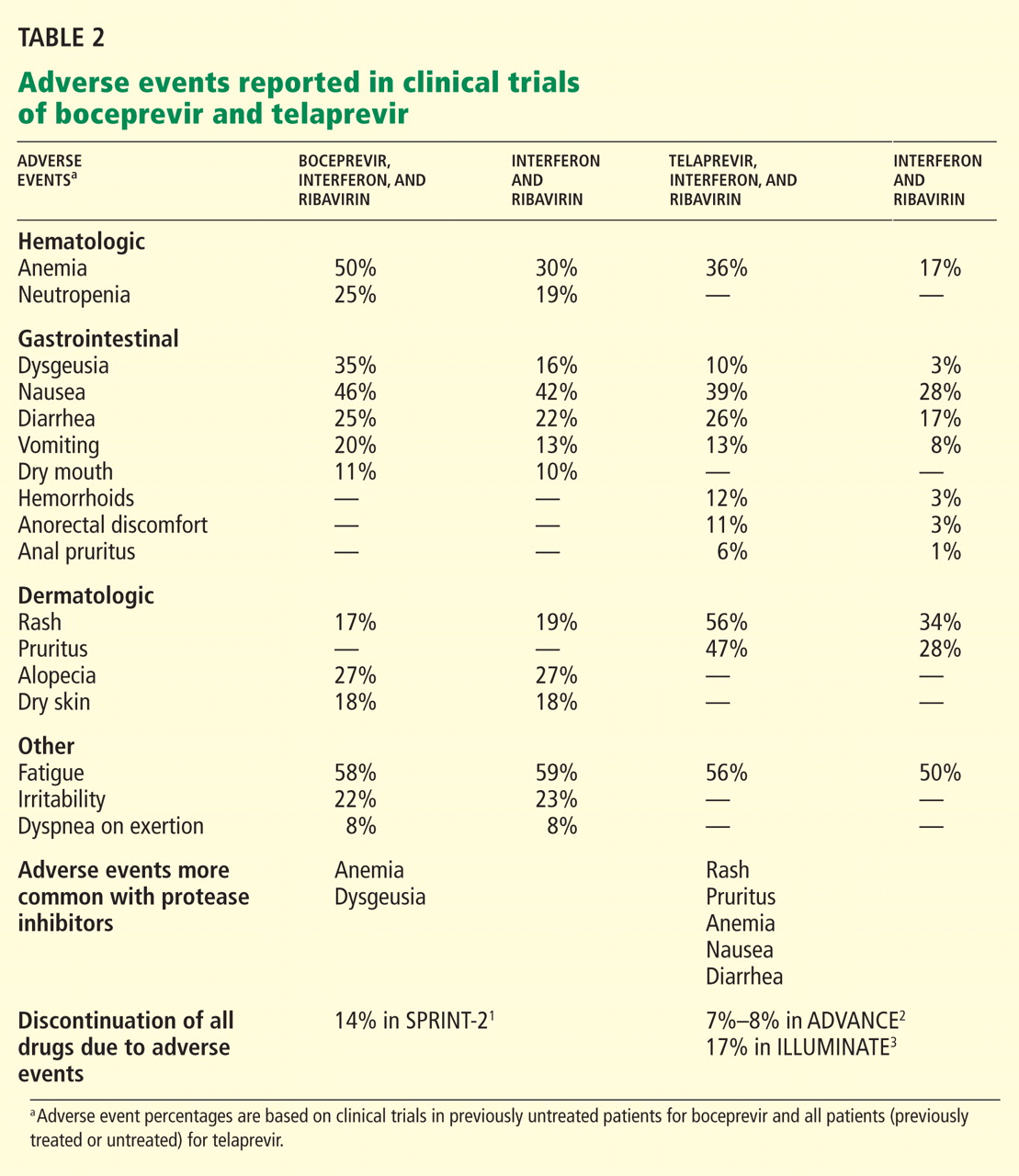
The rate of relapse was 8% and 9% in the boceprevir groups vs 23% in the control group. Patients in the boceprevir groups who had a decrease in HCV RNA of less than 1 log10 during the lead-in phase were found to have a significantly higher rate of boceprevirresistant variants than those who achieved a decrease of HCV RNA of 1 log10 or more.
Boceprevir in previously treated patients with HCV genotype 1: The RESPOND-2 trial
The Retreatment With HCV Serine Protease Inhibitor Boceprevir and PegIntron/Rebetol 2) (RESPOND-2) trial2 was designed to assess the efficacy of combined boceprevir, pegylated interferon, and ribavirin for repeat treatment of patients with HCV genotype 1. These patients had previously undergone standard treatment and had a reduction of 2 log10 or more in HCV RNA after 12 weeks of therapy but with detectable HCV RNA during the therapy period or had had a relapse (defined as undetectable HCV RNA at the end of a previous course of therapy with HCV RNA positivity thereafter). Importantly, null-responders (those who had a reduction of less than 2 log10 in HCV RNA after 12 weeks of therapy) were excluded from this trial.
After a lead-in period of interferon-ribavirin treatment for 4 weeks, 403 patients were assigned to one of three treatment groups:
- Pegylated interferon and ribavirin for 44 weeks (the control group)
- Boceprevir, pegylated interferon, and ribavirin in a response-guided regimen
- Boceprevir, pegylated interferon, and ribavirin for 44 weeks (the fixed-duration group).
Sustained virologic response was achieved in only 21% of patients in the control group. Adding boceprevir increased the rate to 59% in the response-guided therapy group and to 67% in the fixed-duration group. Previous relapsers had better rates than partial responders (69%–75% vs 40%–52%).
Importantly, patients who had a poor response to pegylated interferon and ribavirin during the lead-in phase (defined as having less than a 1-log decrease in the virus before starting boceprevir) had significantly lower rates of sustained virologic response and higher rates of resistance-associated virus variants.
Side effects of boceprevir
Overall, boceprevir is well tolerated. The most common side effects of triple therapy are those usually seen with pegylated interferon and ribavirin, such as flulike symptoms and fatigue (Table 2). However, anemia was more frequent in the boceprevir groups in both SPRINT-2 and RESPOND-2 (45%–50% compared with 20%–29% in the control groups). Erythropoietin was allowed in these studies and was used in about 40% of patients.
The other common side effect associated with boceprevir was dysgeusia (alteration of taste). Dysgeusia was reported by approximately 40% of patients; however, most dysgeusia events were mild to moderate in intensity and did not lead to treatment cessation.
In the SPRINT-2 trial,1 the study drugs had to be discontinued in 12% to 16% of patients in the boceprevir groups because of adverse events, which was similar to the rate (16%) in the control group. Erythropoietin was allowed in this trial, and it was used in 43% of patients in the boceprevir groups compared with 24% in the control group, with discontinuation owing to anemia occurring in 2% and 1% of cases, respectively.
TELAPREVIR
Telaprevir, the other protease NS3/4A inhibitor, has also shown efficacy over current standard therapy in phase 3 clinical trials. It was used in a dose of 750 mg three times a day with pegylated interferon alfa-2a (Pegasys) 180 μg per week and ribavirin (Copegus) 1,000 to 1,200 mg daily according to body weight. A lead-in phase with pegylated interferon and ribavirin was not applied with telaprevir, as it was in the boceprevir trials. Extended rapid virologic response was defined as an undetectable HCV RNA at weeks 4 and 12 of therapy.
Telaprevir in previously untreated patients with HCV genotype 1
The ADVANCE study3 was a double-blind randomized trial assessing the efficacy and safety of telaprevir in combination with pegylated interferon and ribavirin in more than 1,000 previously untreated patients. The three treatment groups received:
- Telaprevir, pegylated interferon, and ribavirin for 8 weeks, followed by pegylated interferon and ribavirin alone for 16 weeks in patients who achieved an extended rapid virologic response (total duration of 24 weeks) or 40 weeks in patients who did not (total duration of 48 weeks)
- Telaprevir, pegylated interferon, and ribavirin for 12 weeks, followed by pegylated interferon-ribavirin alone for 12 (total of 24 weeks) or 36 weeks (total of 48 weeks) according to extended rapid virologic response
- Standard care with pegylated interferon and ribavirin for 48 weeks.
The rate of sustained virologic response was 69% in the group that received telaprevir for 8 weeks and 75% in the group that received it for 12 weeks compared with 44% in the control group (P < .0001 for both) (Table 2). Patients infected with HCV genotype 1b had a higher sustained virologic response rate (79%) than those infected with HCV genotype 1a (71%).
Sustained virologic response rates were lower in black patients and patients with bridging fibrosis or cirrhosis, but were still significantly higher in the telaprevir groups than in the control group. The results of this subset analysis were limited by small numbers of patients in each category.
In total, 57% of those who received telaprevir for 8 weeks and 58% of those who received it for 12 weeks achieved an extended rapid virologic response and were able to cut the duration of their therapy in half (from 48 weeks to 24 weeks).
The relapse rates were 9% in the telaprevir groups and 28% in the control group.
The rate of virologic failure was lower in patients who received triple therapy than in those who received interferon-ribavirin alone (8% in the group that got telaprevir for 12 weeks and 13% in the group that got it for 8 weeks, vs 32% in the control group). The failure rate was also lower in patients with HCV genotype 1b infection than in those with genotype 1a.
The ILLUMINATE study4 (Illustrating the Effects of Combination Therapy With Telaprevir) investigated whether longer duration of treatment than that given in the ADVANCE trial increased the rate of sustained virologic response. Previously untreated patients received telaprevir, interferon, and ribavirin for 12 weeks, and those who achieved an extended rapid virologic response were randomized at week 20 to continue interferonribavirin treatment for 24 or 48 weeks of total treatment.
The sustained virologic response rates in patients who achieved an extended rapid virologic response were 92% in the group that received pegylated interferon and ribavirin for 12 weeks, and 88% in those who received it for 48 weeks. Thus, the results of this study support the use of response-guided therapy for telaprevir-based regimens.
Telaprevir in previously treated patients with HCV genotype 1: The REALIZE trial
In this phase 3 placebo-controlled trial,5 622 patients with prior relapse, partial response, or null response were randomly allocated into one of three groups:
- Telaprevir for 12 weeks plus pegylated interferon and ribavirin for 48 weeks
- Lead-in for 4 weeks followed by 12 weeks of triple therapy and another 32 weeks of pegylated interferon and ribavirin
- Pegylated interferon and ribavirin for 48 weeks (the control group).
The overall sustained virologic response rates were 66% and 64%, respectively, in the telaprevir groups vs 17% in the control group (P < .0001). The sustained virologic response rates in the telaprevir groups were 83% to 88% in prior relapsers, 54% to 59% in partial responders, and 29% to 33% in null-responders. Of note, patients did not benefit from the lead-in phase.
This was the only trial to investigate the response to triple therapy in null-responders, a group in which treatment has been considered hopeless. A response rate of approximately 31% was encouraging, especially if we compare it with the 5% response rate achieved with the current standard of care with pegylated interferon and ribavirin.
Telaprevir side effects
As with boceprevir-based triple therapy, the most common adverse events were related to pegylated interferon (Table 2).
Nearly 50% of patients who receive telaprevir develop a skin rash that is primarily eczematous, can be managed with topical steroids, and usually resolves when telaprevir is discontinued. Severe rashes occurred in 3% to 6% of patients in the ADVANCE trial,3 and three suspected cases of Stevens-Johnson syndrome have been reported to the FDA.
Other side effects that were more frequent with telaprevir included pruritus, nausea, diarrhea, and anemia. On average, the hemoglobin level decreased by an additional 1 g/dL in the telaprevir treatment groups compared with the groups that received only pegylated interferon-ribavirin. Erythropoietin use was not allowed in the phase 3 telaprevir studies, and anemia was managed by ribavirin dose reduction.
In the ADVANCE trial,3 study drugs were discontinued owing to adverse events in 7% to 8% of the patients in the telaprevir groups compared with 4% in the control group. In the ILLUMINATE trial,4 17% of patients had to permanently discontinue all study drugs due to adverse events.
FDA-APPROVED TREATMENT REGIMENS FOR BOCEPREVIR AND TELAPREVIR
For treatment algorithms, see the eFigures that accompany this article online.
Boceprevir in previously untreated patients
- Week 0—Start pegylated interferon and ribavirin
- Week 4—Add boceprevir
- Week 8—Measure HCV RNA
- Week 12—Measure HCV RNA; stop treatment if it is more than 100 IU/mL
- Week 24—Measure HCV RNA; stop treatment if it is detectable
- Week 28—Stop all treatment if HCV RNA was undetectable at weeks 8 and 24
- Week 36—Measure HCV RNA; stop boceprevir
- Week 48—Stop all treatment (eFigure 1).
Boceprevir in previously treated patients
- Week 0—Start pegylated interferon and ribavirin
- Week 4—Add boceprevir
- Week 8—Measure HCV RNA
- Week 12—Measure HCV RNA; stop treatment if it is more than 100 IU/mL
- Week 24—Measure HCV RNA; stop treatment if it is detectable
- Week 36—if HCV RNA was not detectable at week 8, stop all treatment now; if HCV RNA was detectable at week 8, stop boceprevir now but continue pegylated interferon and ribavirin
- Week 48—Stop all treatment (eFigure 2).
Telaprevir in previously untreated patients and prior relapsers
- Week 0—start telaprevir, pegylated interferon, and ribavirin
- Week 4—measure HCV RNA; stop all treatment if it is more than 1,000 IU/mL
- Week 12—Stop telaprevir; measure HCV RNA; stop all treatment if HCV RNA is more than 1,000 IU/mL
- Week 24—Stop pegylated interferon and ribavirin if HCV RNA was undetectable at week 12; measure HCV RNA and stop treatment if it is detectable; otherwise, continue pegylated interferon and ribavirin
- Week 48—Stop all treatment (eFigure 3).
Telaprevir in patients who previously achieved a partial or null response
- Week 0—Start telaprevir, pegylated interferon, and ribavirin
- Week 4—Measure HCV RNA; stop treatment if it is more than 1,000 IU/mL
- Week 12—Measure HCV RNA; stop all treatment if it is more than 1,000 IU/mL; if less than 1,000 IU/mL then stop telaprevir but continue pegylated interferon and ribavirin
- Week 24—Measure HCV RNA; stop treatment if HCV RNA is detectable
- Week 48—Stop all treatment (eFigure 4).
Drug interactions with boceprevir and telaprevir
Both boceprevir and telaprevir inhibit cytochrome P450 3A (CYP3A) and thus are contraindicated in combination with drugs highly dependent on CYP3A for clearance and with drugs for which elevated plasma concentrations are associated with serious adverse events, such as atorvastatin (Lipitor), simvastatin (Zocor), sildenafil (Viagra), midazolam (Versed), and St. John’s wort. Giving potent inducers of CYP3A with boceprevir or telaprevir may lead to lower exposure and loss of efficacy of both protease inhibitors.
EMERGING THERAPIES FOR HCV
Thanks to a better understanding of the biology of HCV infection, the effort to develop new therapeutic agents started to focus on targeting specific steps of the viral life cycle, including attachment, entry into cells, replication, and release.24
Currently, more than 50 clinical trials are evaluating new direct-acting antivirals to treat HCV infection.25 Monoclonal and polyclonal antibodies that target the molecular process involved in HCV attachment and entry are being developed.26 The nonstructural protein NS5B (RNA polymerase) is intimately involved in viral replication and represents a promising target.27 Several nucleosides and nonnucleoside protease inhibitors have already entered clinical trials.
The low fidelity of the HCV replication machinery leads to a very high mutation rate, thus enabling the virus to quickly develop mutations that resist agents targeting viral enzymes.28 Therefore, a novel approach is to target host cofactors that are essential for HCV replication. An intriguing study by Lanford et al29 demonstrated that antagonizing microRNA-122 (the most abundant microRNA in the liver and an essential cofactor for viral RNA replication) by the oligonucleotide SPC3649 caused marked and prolonged reduction of HCV viremia in chronically infected chimpanzees.29
Although we are still in the early stages of drug development, the future holds great promise for newer drugs to improve the sustained virologic response, shorten the duration of treatment, improve tolerability with interferon-sparing regimens, and decrease viral resistance.
FUTURE PERSPECTIVES
With the introduction of the first direct-acting antiviral medications for HCV (boceprevir and telaprevir), 2011 will be marked as the year that changed hepatitis C treatment for the better. Triple therapy with pegylated interferon, ribavirin, and either boceprevir or telaprevir has the potential for increasing the rate of sustained virologic response to around 70% in previously untreated patients and 65% in previously treated patients who are infected with HCV genotype 1. The IL28B polymorphisms appear to play a role in the rate of sustained virologic response achieved with triple therapy, with preliminary data showing a better response rate in patients who have the CC genotype.17
These drugs will add up to $50,000 to the cost of treating hepatitis C virus infection, depending on the drug used and the length of treatment. However, they may be well worth it if they prevent liver failure and the need for transplantation.
Many questions remain, such as how to use these new regimens to treat special patient populations—for example, those with a recurrence of HCV infection after liver transplantation, those co-infected with HCV and human immunodeficiency virus, and those infected with HCV genotypes other than genotype 1.
Other direct-acting antiviral agents that specifically target the replication cycle of HCV are currently in clinical development. In fact, the future has already started with the release of the Interferon-Free Regimen for the Management of HCV (INFORM-1) study results.30 This was the first trial to evaluate an interferon-free regimen for patients with chronic HCV infection using two direct-acting antiviral drugs (the protease inhibitor danoprevir and the polymerase inhibitor RG7128), with promising results.
- Poordad F, McCone J, Bacon BR, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med 2011; 364:1195–1206.
- Bacon BR, Gordon SC, Lawitz E, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med 2011; 364:1207–1217.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; for the ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Sherman KE, Flamm SL, Afdhal NH, et al; for the ILLUMINATE Study Team. Response-guided telaprevir combination treatment for hepatitis C virus infection. N Engl J Med 2011; 365:1014–1024.
- Zeuzem S, Andreone P, Pol S, et al; for the REALIZE Study Team. Telaprevir for retreatment of HCV infection. N Engl J Med 2011; 364:2417–2428.
- Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med 2006; 144:705–714.
- Mitchell AE, Colvin HM, Palmer Beasley R. Institute of Medicine recommendations for the prevention and control of hepatitis B and C. Hepatology 2010; 51:729–733.
- Kim WR. The burden of hepatitis C in the United States. Hepatology 2002; 36:S30–S34.
- Marcellin P, Asselah T, Boyer N. Fibrosis and disease progression in hepatitis C. Hepatology 2002; 36:S47–S56.
- Seeff LB. Natural history of chronic hepatitis C. Hepatology 2002; 36:S35–S46.
- Lavanchy D. The global burden of hepatitis C. Liver Int 2009; 29(suppl 1):74–81.
- National Institutes of Health Consensus Development Conference Statement: Management of hepatitis C: 2002—June 10–12, 2002. Hepatology 2002; 36:S3–S20.
- Pearlman BL, Traub N. Sustained virologic response to antiviral therapy for chronic hepatitis C virus infection: a cure and so much more. Clin Infect Dis 2011; 52:889–900.
- Hoofnagle JH, Seeff LB. Peginterferon and ribavirin for chronic hepatitis C. N Engl J Med 2006; 355:2444–2451.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009; 461:399–401.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e118.
- Nielsen SU, Bassendine MF, Burt AD, Bevitt DJ, Toms GL. Characterization of the genome and structural proteins of hepatitis C virus resolved from infected human liver. J Gen Virol 2004; 85:1497–1507.
- Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. Structural biology of hepatitis C virus. Hepatology 2004; 39:5–19.
- Nelson DR. The role of triple therapy with protease inhibitors in hepatitis C virus genotype 1 naive patients. Liver Int 2011; 31(suppl 1):53–57.
- Pawlotsky JM. Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology 2011; 53:1742–1751.
- Monto A, Schooley RT, Lai JC, et al. Lessons from HIV therapy applied to viral hepatitis therapy: summary of a workshop. Am J Gastroenterol 2010; 105:989–1004.
- McCown MF, Rajyaguru S, Kular S, Cammack N, Najera I. GT-1a or GT-1b subtype-specific resistance profiles for hepatitis C virus inhibitors telaprevir and HCV-796. Antimicrob Agents Chemother 2009; 53:2129–2132.
- Cholongitas E, Papatheodoridis GV. Review article: novel therapeutic options for chronic hepatitis C. Aliment Pharmacol Ther 2008; 27:866–884.
- Naggie S, Patel K, McHutchison J. Hepatitis C virus directly acting antivirals: current developments with NS3/4A HCV serine protease inhibitors. J Antimicrob Chemother 2010; 65:2063–2069.
- Mir HM, Birerdinc A, Younossi ZM. Monoclonal and polyclonal antibodies against the HCV envelope proteins. Clin Liver Dis 2009; 13:477–486.
- Birerdinc A, Younossi ZM. Emerging therapies for hepatitis C virus. Expert Opin Emerg Drugs 2010; 15:535–544.
- Khattab MA. Targeting host factors: a novel rationale for the management of hepatitis C virus. World J Gastroenterol 2009; 15:3472–3479.
- Lanford RE, Hildebrandt-Eriksen ES, Petri A, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010; 327:198–201.
- Gane EJ, Roberts SK, Stedman CA, et al. Oral combination therapy with a nucleoside polymerase inhibitor (RG7128) and danoprevir for chronic hepatitis C genotype 1 infection (INFORM-1): a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 2010; 376:1467–1475.
The treatment of hepatitis c virus (HCV) infection is on the brink of major changes with the recent approval of the first direct-acting antiviral agents, the protease inhibitors boceprevir (Victrelis) and telaprevir (Incivek).
Both drugs were approved by the US Food and Drug Administration (FDA) Advisory Panel for Chronic Hepatitis C in May 2011 and are believed to significantly improve treatment outcomes for patients with HCV genotype 1 infection.
A MAJOR PUBLIC HEALTH PROBLEM
HCV infection is a major public health problem. Nearly 4 million people in the United States are infected.6,7 Most patients with acute HCV infection become chronically infected, and up to 25% eventually develop cirrhosis and its complications, making HCV infection the leading indication for liver transplantation.8–10
Chronic HCV infection has a large global impact, with 180 million people affected across all economic and social groups.11 The highest prevalence of HCV has been reported in Egypt (14%), in part due to the use of inadequately sterilized needles in mass programs to treat endemic schistosomiasis. In developed countries, hepatocellular carcinoma associated with HCV has the fastest growing cancer-related death rate.12
CURRENTLY, FEWER THAN 50% OF PATIENTS ARE CURED
The goal of HCV treatment is to eradicate the virus. However, most infected patients (especially in the United States and Europe) are infected with HCV genotype 1, which is the most difficult genotype to treat.
Successful treatment of HCV is defined as achieving a sustained virologic response—ie, the absence of detectable HCV RNA in the serum 24 weeks after completion of therapy. Once a sustained virologic response is achieved, lifetime “cure” of HCV infection is expected in more than 99% of patients.13
The current standard therapy for HCV, pegylated interferon plus ribavirin for 48 weeks, is effective in only 40% to 50% of patients with genotype 1 infection.14 Therefore, assessing predictors of response before starting treatment can help select patients who are most likely to benefit from therapy.
Viral factors associated with a sustained virologic response include HCV genotypes other than genotype 1 and a low baseline viral load.
Beneficial patient-related factors include younger age, nonblack ethnicity, low body weight (≤ 75 kg), low body mass index, absence of insulin resistance, and absence of advanced fibrosis or cirrhosis.
More recently, a single-nucleotide polymorphism near the interleukin 28B (IL28B) gene, coding for interferon lambda 3, was found to be associated with a twofold difference in the rates of sustained virologic response: patients with the favorable genotype CC were two times more likely to achieve a sustained virologic response than patients with the CT or TT genotypes.15–17
PROTEASE INHIBITORS: MECHANISM OF ACTION
NS3/4A protease inhibitors rely on the principle of end-product inhibition, in which the cleavage product of the protease (a peptide) acts to inhibit the enzyme activity; this is why they are called peptidomimetics. The active site of the NS3/4A protease is a shallow groove composed of three highly conserved amino acid residues, which may explain why protease inhibitors display high antiviral efficacy but pose a low barrier to the development of resistance.20
Protease inhibitors are prone to resistance
The development of viral resistance to protease inhibitors has been a major drawback to their use in patients with chronic HCV infection.21
HCV is a highly variable virus with many genetically distinct but closely related quasispecies circulating in the blood at any given time. Drug-resistant, mutated variants preexist within the patient’s quasispecies, but only in small quantities because of their lesser replication fitness compared with the wild-type virus.22 When direct-acting antiviral therapy is started, the quantity of the wild-type virus decreases and the mutated virus gains replication fitness. Using protease inhibitors as monotherapy selects resistant viral populations rapidly within a few days or weeks.
HCV subtypes 1a and 1b may have different resistance profiles. With genotype 1a, some resistance-associated amino acid substitutions require only one nucleotide change, but with genotype 1b, two nucleotide changes are needed, making resistance less frequent in patients with HCV genotype 1b.23
BOCEPREVIR
Boceprevir is a specific inhibitor of the HCV viral protease NS3/4A.
In phase 3 clinical trials, boceprevir 800 mg three times a day was used with pegylated interferon alfa-2b (PegIntron) 1.5 μg/kg/week and ribavirin (Rebetol) 600 to 1,400 mg daily according to body weight.
Before patients started taking boceprevir, they went through a 4-week lead-in phase, during which they received pegylated interferon and ribavirin. This schedule appeared to reduce the incidence of viral breakthrough in phase 2 trials, and it produced higher rates of sustained virologic response and lower relapse rates compared with triple therapy without a lead-in phase.
Rapid virologic response was defined as undetectable HCV RNA at week 4 of boceprevir therapy (week 8 of the whole regimen).
Boceprevir in previously untreated patients with HCV genotype 1: The SPRINT-2 trial
The Serine Protease Inhibitor Therapy 2 (SPRINT-2) trial1 included more than 1,000 previously untreated adults with HCV genotype 1 infection (938 nonblack patients and 159 black patients; two other nonblack patients did not receive any study drug and were not included in the analysis). In this double-blind trial, patients were randomized into three groups:
- The control group received the standard of care with pegylated interferon and ribavirin for 48 weeks
- The response-guided therapy group received boceprevir plus pegylated interferon and ribavirin for 24 weeks after the 4-week lead-in phase; if HCV RNA was undetectable from week 8 to week 24, treatment was considered complete, but if HCV RNA was detectable at any point from week 8 to week 24, pegylated interferon and ribavirin were continued for a total of 48 weeks.
- The fixed-duration therapy group received boceprevir, pegylated interferon, and ribavirin for 44 weeks after the lead-in period.
The rate of relapse was 8% and 9% in the boceprevir groups vs 23% in the control group. Patients in the boceprevir groups who had a decrease in HCV RNA of less than 1 log10 during the lead-in phase were found to have a significantly higher rate of boceprevirresistant variants than those who achieved a decrease of HCV RNA of 1 log10 or more.
Boceprevir in previously treated patients with HCV genotype 1: The RESPOND-2 trial
The Retreatment With HCV Serine Protease Inhibitor Boceprevir and PegIntron/Rebetol 2) (RESPOND-2) trial2 was designed to assess the efficacy of combined boceprevir, pegylated interferon, and ribavirin for repeat treatment of patients with HCV genotype 1. These patients had previously undergone standard treatment and had a reduction of 2 log10 or more in HCV RNA after 12 weeks of therapy but with detectable HCV RNA during the therapy period or had had a relapse (defined as undetectable HCV RNA at the end of a previous course of therapy with HCV RNA positivity thereafter). Importantly, null-responders (those who had a reduction of less than 2 log10 in HCV RNA after 12 weeks of therapy) were excluded from this trial.
After a lead-in period of interferon-ribavirin treatment for 4 weeks, 403 patients were assigned to one of three treatment groups:
- Pegylated interferon and ribavirin for 44 weeks (the control group)
- Boceprevir, pegylated interferon, and ribavirin in a response-guided regimen
- Boceprevir, pegylated interferon, and ribavirin for 44 weeks (the fixed-duration group).
Sustained virologic response was achieved in only 21% of patients in the control group. Adding boceprevir increased the rate to 59% in the response-guided therapy group and to 67% in the fixed-duration group. Previous relapsers had better rates than partial responders (69%–75% vs 40%–52%).
Importantly, patients who had a poor response to pegylated interferon and ribavirin during the lead-in phase (defined as having less than a 1-log decrease in the virus before starting boceprevir) had significantly lower rates of sustained virologic response and higher rates of resistance-associated virus variants.
Side effects of boceprevir
Overall, boceprevir is well tolerated. The most common side effects of triple therapy are those usually seen with pegylated interferon and ribavirin, such as flulike symptoms and fatigue (Table 2). However, anemia was more frequent in the boceprevir groups in both SPRINT-2 and RESPOND-2 (45%–50% compared with 20%–29% in the control groups). Erythropoietin was allowed in these studies and was used in about 40% of patients.
The other common side effect associated with boceprevir was dysgeusia (alteration of taste). Dysgeusia was reported by approximately 40% of patients; however, most dysgeusia events were mild to moderate in intensity and did not lead to treatment cessation.
In the SPRINT-2 trial,1 the study drugs had to be discontinued in 12% to 16% of patients in the boceprevir groups because of adverse events, which was similar to the rate (16%) in the control group. Erythropoietin was allowed in this trial, and it was used in 43% of patients in the boceprevir groups compared with 24% in the control group, with discontinuation owing to anemia occurring in 2% and 1% of cases, respectively.
TELAPREVIR
Telaprevir, the other protease NS3/4A inhibitor, has also shown efficacy over current standard therapy in phase 3 clinical trials. It was used in a dose of 750 mg three times a day with pegylated interferon alfa-2a (Pegasys) 180 μg per week and ribavirin (Copegus) 1,000 to 1,200 mg daily according to body weight. A lead-in phase with pegylated interferon and ribavirin was not applied with telaprevir, as it was in the boceprevir trials. Extended rapid virologic response was defined as an undetectable HCV RNA at weeks 4 and 12 of therapy.
Telaprevir in previously untreated patients with HCV genotype 1
The ADVANCE study3 was a double-blind randomized trial assessing the efficacy and safety of telaprevir in combination with pegylated interferon and ribavirin in more than 1,000 previously untreated patients. The three treatment groups received:
- Telaprevir, pegylated interferon, and ribavirin for 8 weeks, followed by pegylated interferon and ribavirin alone for 16 weeks in patients who achieved an extended rapid virologic response (total duration of 24 weeks) or 40 weeks in patients who did not (total duration of 48 weeks)
- Telaprevir, pegylated interferon, and ribavirin for 12 weeks, followed by pegylated interferon-ribavirin alone for 12 (total of 24 weeks) or 36 weeks (total of 48 weeks) according to extended rapid virologic response
- Standard care with pegylated interferon and ribavirin for 48 weeks.
The rate of sustained virologic response was 69% in the group that received telaprevir for 8 weeks and 75% in the group that received it for 12 weeks compared with 44% in the control group (P < .0001 for both) (Table 2). Patients infected with HCV genotype 1b had a higher sustained virologic response rate (79%) than those infected with HCV genotype 1a (71%).
Sustained virologic response rates were lower in black patients and patients with bridging fibrosis or cirrhosis, but were still significantly higher in the telaprevir groups than in the control group. The results of this subset analysis were limited by small numbers of patients in each category.
In total, 57% of those who received telaprevir for 8 weeks and 58% of those who received it for 12 weeks achieved an extended rapid virologic response and were able to cut the duration of their therapy in half (from 48 weeks to 24 weeks).
The relapse rates were 9% in the telaprevir groups and 28% in the control group.
The rate of virologic failure was lower in patients who received triple therapy than in those who received interferon-ribavirin alone (8% in the group that got telaprevir for 12 weeks and 13% in the group that got it for 8 weeks, vs 32% in the control group). The failure rate was also lower in patients with HCV genotype 1b infection than in those with genotype 1a.
The ILLUMINATE study4 (Illustrating the Effects of Combination Therapy With Telaprevir) investigated whether longer duration of treatment than that given in the ADVANCE trial increased the rate of sustained virologic response. Previously untreated patients received telaprevir, interferon, and ribavirin for 12 weeks, and those who achieved an extended rapid virologic response were randomized at week 20 to continue interferonribavirin treatment for 24 or 48 weeks of total treatment.
The sustained virologic response rates in patients who achieved an extended rapid virologic response were 92% in the group that received pegylated interferon and ribavirin for 12 weeks, and 88% in those who received it for 48 weeks. Thus, the results of this study support the use of response-guided therapy for telaprevir-based regimens.
Telaprevir in previously treated patients with HCV genotype 1: The REALIZE trial
In this phase 3 placebo-controlled trial,5 622 patients with prior relapse, partial response, or null response were randomly allocated into one of three groups:
- Telaprevir for 12 weeks plus pegylated interferon and ribavirin for 48 weeks
- Lead-in for 4 weeks followed by 12 weeks of triple therapy and another 32 weeks of pegylated interferon and ribavirin
- Pegylated interferon and ribavirin for 48 weeks (the control group).
The overall sustained virologic response rates were 66% and 64%, respectively, in the telaprevir groups vs 17% in the control group (P < .0001). The sustained virologic response rates in the telaprevir groups were 83% to 88% in prior relapsers, 54% to 59% in partial responders, and 29% to 33% in null-responders. Of note, patients did not benefit from the lead-in phase.
This was the only trial to investigate the response to triple therapy in null-responders, a group in which treatment has been considered hopeless. A response rate of approximately 31% was encouraging, especially if we compare it with the 5% response rate achieved with the current standard of care with pegylated interferon and ribavirin.
Telaprevir side effects
As with boceprevir-based triple therapy, the most common adverse events were related to pegylated interferon (Table 2).
Nearly 50% of patients who receive telaprevir develop a skin rash that is primarily eczematous, can be managed with topical steroids, and usually resolves when telaprevir is discontinued. Severe rashes occurred in 3% to 6% of patients in the ADVANCE trial,3 and three suspected cases of Stevens-Johnson syndrome have been reported to the FDA.
Other side effects that were more frequent with telaprevir included pruritus, nausea, diarrhea, and anemia. On average, the hemoglobin level decreased by an additional 1 g/dL in the telaprevir treatment groups compared with the groups that received only pegylated interferon-ribavirin. Erythropoietin use was not allowed in the phase 3 telaprevir studies, and anemia was managed by ribavirin dose reduction.
In the ADVANCE trial,3 study drugs were discontinued owing to adverse events in 7% to 8% of the patients in the telaprevir groups compared with 4% in the control group. In the ILLUMINATE trial,4 17% of patients had to permanently discontinue all study drugs due to adverse events.
FDA-APPROVED TREATMENT REGIMENS FOR BOCEPREVIR AND TELAPREVIR
For treatment algorithms, see the eFigures that accompany this article online.
Boceprevir in previously untreated patients
- Week 0—Start pegylated interferon and ribavirin
- Week 4—Add boceprevir
- Week 8—Measure HCV RNA
- Week 12—Measure HCV RNA; stop treatment if it is more than 100 IU/mL
- Week 24—Measure HCV RNA; stop treatment if it is detectable
- Week 28—Stop all treatment if HCV RNA was undetectable at weeks 8 and 24
- Week 36—Measure HCV RNA; stop boceprevir
- Week 48—Stop all treatment (eFigure 1).
Boceprevir in previously treated patients
- Week 0—Start pegylated interferon and ribavirin
- Week 4—Add boceprevir
- Week 8—Measure HCV RNA
- Week 12—Measure HCV RNA; stop treatment if it is more than 100 IU/mL
- Week 24—Measure HCV RNA; stop treatment if it is detectable
- Week 36—if HCV RNA was not detectable at week 8, stop all treatment now; if HCV RNA was detectable at week 8, stop boceprevir now but continue pegylated interferon and ribavirin
- Week 48—Stop all treatment (eFigure 2).
Telaprevir in previously untreated patients and prior relapsers
- Week 0—start telaprevir, pegylated interferon, and ribavirin
- Week 4—measure HCV RNA; stop all treatment if it is more than 1,000 IU/mL
- Week 12—Stop telaprevir; measure HCV RNA; stop all treatment if HCV RNA is more than 1,000 IU/mL
- Week 24—Stop pegylated interferon and ribavirin if HCV RNA was undetectable at week 12; measure HCV RNA and stop treatment if it is detectable; otherwise, continue pegylated interferon and ribavirin
- Week 48—Stop all treatment (eFigure 3).
Telaprevir in patients who previously achieved a partial or null response
- Week 0—Start telaprevir, pegylated interferon, and ribavirin
- Week 4—Measure HCV RNA; stop treatment if it is more than 1,000 IU/mL
- Week 12—Measure HCV RNA; stop all treatment if it is more than 1,000 IU/mL; if less than 1,000 IU/mL then stop telaprevir but continue pegylated interferon and ribavirin
- Week 24—Measure HCV RNA; stop treatment if HCV RNA is detectable
- Week 48—Stop all treatment (eFigure 4).
Drug interactions with boceprevir and telaprevir
Both boceprevir and telaprevir inhibit cytochrome P450 3A (CYP3A) and thus are contraindicated in combination with drugs highly dependent on CYP3A for clearance and with drugs for which elevated plasma concentrations are associated with serious adverse events, such as atorvastatin (Lipitor), simvastatin (Zocor), sildenafil (Viagra), midazolam (Versed), and St. John’s wort. Giving potent inducers of CYP3A with boceprevir or telaprevir may lead to lower exposure and loss of efficacy of both protease inhibitors.
EMERGING THERAPIES FOR HCV
Thanks to a better understanding of the biology of HCV infection, the effort to develop new therapeutic agents started to focus on targeting specific steps of the viral life cycle, including attachment, entry into cells, replication, and release.24
Currently, more than 50 clinical trials are evaluating new direct-acting antivirals to treat HCV infection.25 Monoclonal and polyclonal antibodies that target the molecular process involved in HCV attachment and entry are being developed.26 The nonstructural protein NS5B (RNA polymerase) is intimately involved in viral replication and represents a promising target.27 Several nucleosides and nonnucleoside protease inhibitors have already entered clinical trials.
The low fidelity of the HCV replication machinery leads to a very high mutation rate, thus enabling the virus to quickly develop mutations that resist agents targeting viral enzymes.28 Therefore, a novel approach is to target host cofactors that are essential for HCV replication. An intriguing study by Lanford et al29 demonstrated that antagonizing microRNA-122 (the most abundant microRNA in the liver and an essential cofactor for viral RNA replication) by the oligonucleotide SPC3649 caused marked and prolonged reduction of HCV viremia in chronically infected chimpanzees.29
Although we are still in the early stages of drug development, the future holds great promise for newer drugs to improve the sustained virologic response, shorten the duration of treatment, improve tolerability with interferon-sparing regimens, and decrease viral resistance.
FUTURE PERSPECTIVES
With the introduction of the first direct-acting antiviral medications for HCV (boceprevir and telaprevir), 2011 will be marked as the year that changed hepatitis C treatment for the better. Triple therapy with pegylated interferon, ribavirin, and either boceprevir or telaprevir has the potential for increasing the rate of sustained virologic response to around 70% in previously untreated patients and 65% in previously treated patients who are infected with HCV genotype 1. The IL28B polymorphisms appear to play a role in the rate of sustained virologic response achieved with triple therapy, with preliminary data showing a better response rate in patients who have the CC genotype.17
These drugs will add up to $50,000 to the cost of treating hepatitis C virus infection, depending on the drug used and the length of treatment. However, they may be well worth it if they prevent liver failure and the need for transplantation.
Many questions remain, such as how to use these new regimens to treat special patient populations—for example, those with a recurrence of HCV infection after liver transplantation, those co-infected with HCV and human immunodeficiency virus, and those infected with HCV genotypes other than genotype 1.
Other direct-acting antiviral agents that specifically target the replication cycle of HCV are currently in clinical development. In fact, the future has already started with the release of the Interferon-Free Regimen for the Management of HCV (INFORM-1) study results.30 This was the first trial to evaluate an interferon-free regimen for patients with chronic HCV infection using two direct-acting antiviral drugs (the protease inhibitor danoprevir and the polymerase inhibitor RG7128), with promising results.
The treatment of hepatitis c virus (HCV) infection is on the brink of major changes with the recent approval of the first direct-acting antiviral agents, the protease inhibitors boceprevir (Victrelis) and telaprevir (Incivek).
Both drugs were approved by the US Food and Drug Administration (FDA) Advisory Panel for Chronic Hepatitis C in May 2011 and are believed to significantly improve treatment outcomes for patients with HCV genotype 1 infection.
A MAJOR PUBLIC HEALTH PROBLEM
HCV infection is a major public health problem. Nearly 4 million people in the United States are infected.6,7 Most patients with acute HCV infection become chronically infected, and up to 25% eventually develop cirrhosis and its complications, making HCV infection the leading indication for liver transplantation.8–10
Chronic HCV infection has a large global impact, with 180 million people affected across all economic and social groups.11 The highest prevalence of HCV has been reported in Egypt (14%), in part due to the use of inadequately sterilized needles in mass programs to treat endemic schistosomiasis. In developed countries, hepatocellular carcinoma associated with HCV has the fastest growing cancer-related death rate.12
CURRENTLY, FEWER THAN 50% OF PATIENTS ARE CURED
The goal of HCV treatment is to eradicate the virus. However, most infected patients (especially in the United States and Europe) are infected with HCV genotype 1, which is the most difficult genotype to treat.
Successful treatment of HCV is defined as achieving a sustained virologic response—ie, the absence of detectable HCV RNA in the serum 24 weeks after completion of therapy. Once a sustained virologic response is achieved, lifetime “cure” of HCV infection is expected in more than 99% of patients.13
The current standard therapy for HCV, pegylated interferon plus ribavirin for 48 weeks, is effective in only 40% to 50% of patients with genotype 1 infection.14 Therefore, assessing predictors of response before starting treatment can help select patients who are most likely to benefit from therapy.
Viral factors associated with a sustained virologic response include HCV genotypes other than genotype 1 and a low baseline viral load.
Beneficial patient-related factors include younger age, nonblack ethnicity, low body weight (≤ 75 kg), low body mass index, absence of insulin resistance, and absence of advanced fibrosis or cirrhosis.
More recently, a single-nucleotide polymorphism near the interleukin 28B (IL28B) gene, coding for interferon lambda 3, was found to be associated with a twofold difference in the rates of sustained virologic response: patients with the favorable genotype CC were two times more likely to achieve a sustained virologic response than patients with the CT or TT genotypes.15–17
PROTEASE INHIBITORS: MECHANISM OF ACTION
NS3/4A protease inhibitors rely on the principle of end-product inhibition, in which the cleavage product of the protease (a peptide) acts to inhibit the enzyme activity; this is why they are called peptidomimetics. The active site of the NS3/4A protease is a shallow groove composed of three highly conserved amino acid residues, which may explain why protease inhibitors display high antiviral efficacy but pose a low barrier to the development of resistance.20
Protease inhibitors are prone to resistance
The development of viral resistance to protease inhibitors has been a major drawback to their use in patients with chronic HCV infection.21
HCV is a highly variable virus with many genetically distinct but closely related quasispecies circulating in the blood at any given time. Drug-resistant, mutated variants preexist within the patient’s quasispecies, but only in small quantities because of their lesser replication fitness compared with the wild-type virus.22 When direct-acting antiviral therapy is started, the quantity of the wild-type virus decreases and the mutated virus gains replication fitness. Using protease inhibitors as monotherapy selects resistant viral populations rapidly within a few days or weeks.
HCV subtypes 1a and 1b may have different resistance profiles. With genotype 1a, some resistance-associated amino acid substitutions require only one nucleotide change, but with genotype 1b, two nucleotide changes are needed, making resistance less frequent in patients with HCV genotype 1b.23
BOCEPREVIR
Boceprevir is a specific inhibitor of the HCV viral protease NS3/4A.
In phase 3 clinical trials, boceprevir 800 mg three times a day was used with pegylated interferon alfa-2b (PegIntron) 1.5 μg/kg/week and ribavirin (Rebetol) 600 to 1,400 mg daily according to body weight.
Before patients started taking boceprevir, they went through a 4-week lead-in phase, during which they received pegylated interferon and ribavirin. This schedule appeared to reduce the incidence of viral breakthrough in phase 2 trials, and it produced higher rates of sustained virologic response and lower relapse rates compared with triple therapy without a lead-in phase.
Rapid virologic response was defined as undetectable HCV RNA at week 4 of boceprevir therapy (week 8 of the whole regimen).
Boceprevir in previously untreated patients with HCV genotype 1: The SPRINT-2 trial
The Serine Protease Inhibitor Therapy 2 (SPRINT-2) trial1 included more than 1,000 previously untreated adults with HCV genotype 1 infection (938 nonblack patients and 159 black patients; two other nonblack patients did not receive any study drug and were not included in the analysis). In this double-blind trial, patients were randomized into three groups:
- The control group received the standard of care with pegylated interferon and ribavirin for 48 weeks
- The response-guided therapy group received boceprevir plus pegylated interferon and ribavirin for 24 weeks after the 4-week lead-in phase; if HCV RNA was undetectable from week 8 to week 24, treatment was considered complete, but if HCV RNA was detectable at any point from week 8 to week 24, pegylated interferon and ribavirin were continued for a total of 48 weeks.
- The fixed-duration therapy group received boceprevir, pegylated interferon, and ribavirin for 44 weeks after the lead-in period.
The rate of relapse was 8% and 9% in the boceprevir groups vs 23% in the control group. Patients in the boceprevir groups who had a decrease in HCV RNA of less than 1 log10 during the lead-in phase were found to have a significantly higher rate of boceprevirresistant variants than those who achieved a decrease of HCV RNA of 1 log10 or more.
Boceprevir in previously treated patients with HCV genotype 1: The RESPOND-2 trial
The Retreatment With HCV Serine Protease Inhibitor Boceprevir and PegIntron/Rebetol 2) (RESPOND-2) trial2 was designed to assess the efficacy of combined boceprevir, pegylated interferon, and ribavirin for repeat treatment of patients with HCV genotype 1. These patients had previously undergone standard treatment and had a reduction of 2 log10 or more in HCV RNA after 12 weeks of therapy but with detectable HCV RNA during the therapy period or had had a relapse (defined as undetectable HCV RNA at the end of a previous course of therapy with HCV RNA positivity thereafter). Importantly, null-responders (those who had a reduction of less than 2 log10 in HCV RNA after 12 weeks of therapy) were excluded from this trial.
After a lead-in period of interferon-ribavirin treatment for 4 weeks, 403 patients were assigned to one of three treatment groups:
- Pegylated interferon and ribavirin for 44 weeks (the control group)
- Boceprevir, pegylated interferon, and ribavirin in a response-guided regimen
- Boceprevir, pegylated interferon, and ribavirin for 44 weeks (the fixed-duration group).
Sustained virologic response was achieved in only 21% of patients in the control group. Adding boceprevir increased the rate to 59% in the response-guided therapy group and to 67% in the fixed-duration group. Previous relapsers had better rates than partial responders (69%–75% vs 40%–52%).
Importantly, patients who had a poor response to pegylated interferon and ribavirin during the lead-in phase (defined as having less than a 1-log decrease in the virus before starting boceprevir) had significantly lower rates of sustained virologic response and higher rates of resistance-associated virus variants.
Side effects of boceprevir
Overall, boceprevir is well tolerated. The most common side effects of triple therapy are those usually seen with pegylated interferon and ribavirin, such as flulike symptoms and fatigue (Table 2). However, anemia was more frequent in the boceprevir groups in both SPRINT-2 and RESPOND-2 (45%–50% compared with 20%–29% in the control groups). Erythropoietin was allowed in these studies and was used in about 40% of patients.
The other common side effect associated with boceprevir was dysgeusia (alteration of taste). Dysgeusia was reported by approximately 40% of patients; however, most dysgeusia events were mild to moderate in intensity and did not lead to treatment cessation.
In the SPRINT-2 trial,1 the study drugs had to be discontinued in 12% to 16% of patients in the boceprevir groups because of adverse events, which was similar to the rate (16%) in the control group. Erythropoietin was allowed in this trial, and it was used in 43% of patients in the boceprevir groups compared with 24% in the control group, with discontinuation owing to anemia occurring in 2% and 1% of cases, respectively.
TELAPREVIR
Telaprevir, the other protease NS3/4A inhibitor, has also shown efficacy over current standard therapy in phase 3 clinical trials. It was used in a dose of 750 mg three times a day with pegylated interferon alfa-2a (Pegasys) 180 μg per week and ribavirin (Copegus) 1,000 to 1,200 mg daily according to body weight. A lead-in phase with pegylated interferon and ribavirin was not applied with telaprevir, as it was in the boceprevir trials. Extended rapid virologic response was defined as an undetectable HCV RNA at weeks 4 and 12 of therapy.
Telaprevir in previously untreated patients with HCV genotype 1
The ADVANCE study3 was a double-blind randomized trial assessing the efficacy and safety of telaprevir in combination with pegylated interferon and ribavirin in more than 1,000 previously untreated patients. The three treatment groups received:
- Telaprevir, pegylated interferon, and ribavirin for 8 weeks, followed by pegylated interferon and ribavirin alone for 16 weeks in patients who achieved an extended rapid virologic response (total duration of 24 weeks) or 40 weeks in patients who did not (total duration of 48 weeks)
- Telaprevir, pegylated interferon, and ribavirin for 12 weeks, followed by pegylated interferon-ribavirin alone for 12 (total of 24 weeks) or 36 weeks (total of 48 weeks) according to extended rapid virologic response
- Standard care with pegylated interferon and ribavirin for 48 weeks.
The rate of sustained virologic response was 69% in the group that received telaprevir for 8 weeks and 75% in the group that received it for 12 weeks compared with 44% in the control group (P < .0001 for both) (Table 2). Patients infected with HCV genotype 1b had a higher sustained virologic response rate (79%) than those infected with HCV genotype 1a (71%).
Sustained virologic response rates were lower in black patients and patients with bridging fibrosis or cirrhosis, but were still significantly higher in the telaprevir groups than in the control group. The results of this subset analysis were limited by small numbers of patients in each category.
In total, 57% of those who received telaprevir for 8 weeks and 58% of those who received it for 12 weeks achieved an extended rapid virologic response and were able to cut the duration of their therapy in half (from 48 weeks to 24 weeks).
The relapse rates were 9% in the telaprevir groups and 28% in the control group.
The rate of virologic failure was lower in patients who received triple therapy than in those who received interferon-ribavirin alone (8% in the group that got telaprevir for 12 weeks and 13% in the group that got it for 8 weeks, vs 32% in the control group). The failure rate was also lower in patients with HCV genotype 1b infection than in those with genotype 1a.
The ILLUMINATE study4 (Illustrating the Effects of Combination Therapy With Telaprevir) investigated whether longer duration of treatment than that given in the ADVANCE trial increased the rate of sustained virologic response. Previously untreated patients received telaprevir, interferon, and ribavirin for 12 weeks, and those who achieved an extended rapid virologic response were randomized at week 20 to continue interferonribavirin treatment for 24 or 48 weeks of total treatment.
The sustained virologic response rates in patients who achieved an extended rapid virologic response were 92% in the group that received pegylated interferon and ribavirin for 12 weeks, and 88% in those who received it for 48 weeks. Thus, the results of this study support the use of response-guided therapy for telaprevir-based regimens.
Telaprevir in previously treated patients with HCV genotype 1: The REALIZE trial
In this phase 3 placebo-controlled trial,5 622 patients with prior relapse, partial response, or null response were randomly allocated into one of three groups:
- Telaprevir for 12 weeks plus pegylated interferon and ribavirin for 48 weeks
- Lead-in for 4 weeks followed by 12 weeks of triple therapy and another 32 weeks of pegylated interferon and ribavirin
- Pegylated interferon and ribavirin for 48 weeks (the control group).
The overall sustained virologic response rates were 66% and 64%, respectively, in the telaprevir groups vs 17% in the control group (P < .0001). The sustained virologic response rates in the telaprevir groups were 83% to 88% in prior relapsers, 54% to 59% in partial responders, and 29% to 33% in null-responders. Of note, patients did not benefit from the lead-in phase.
This was the only trial to investigate the response to triple therapy in null-responders, a group in which treatment has been considered hopeless. A response rate of approximately 31% was encouraging, especially if we compare it with the 5% response rate achieved with the current standard of care with pegylated interferon and ribavirin.
Telaprevir side effects
As with boceprevir-based triple therapy, the most common adverse events were related to pegylated interferon (Table 2).
Nearly 50% of patients who receive telaprevir develop a skin rash that is primarily eczematous, can be managed with topical steroids, and usually resolves when telaprevir is discontinued. Severe rashes occurred in 3% to 6% of patients in the ADVANCE trial,3 and three suspected cases of Stevens-Johnson syndrome have been reported to the FDA.
Other side effects that were more frequent with telaprevir included pruritus, nausea, diarrhea, and anemia. On average, the hemoglobin level decreased by an additional 1 g/dL in the telaprevir treatment groups compared with the groups that received only pegylated interferon-ribavirin. Erythropoietin use was not allowed in the phase 3 telaprevir studies, and anemia was managed by ribavirin dose reduction.
In the ADVANCE trial,3 study drugs were discontinued owing to adverse events in 7% to 8% of the patients in the telaprevir groups compared with 4% in the control group. In the ILLUMINATE trial,4 17% of patients had to permanently discontinue all study drugs due to adverse events.
FDA-APPROVED TREATMENT REGIMENS FOR BOCEPREVIR AND TELAPREVIR
For treatment algorithms, see the eFigures that accompany this article online.
Boceprevir in previously untreated patients
- Week 0—Start pegylated interferon and ribavirin
- Week 4—Add boceprevir
- Week 8—Measure HCV RNA
- Week 12—Measure HCV RNA; stop treatment if it is more than 100 IU/mL
- Week 24—Measure HCV RNA; stop treatment if it is detectable
- Week 28—Stop all treatment if HCV RNA was undetectable at weeks 8 and 24
- Week 36—Measure HCV RNA; stop boceprevir
- Week 48—Stop all treatment (eFigure 1).
Boceprevir in previously treated patients
- Week 0—Start pegylated interferon and ribavirin
- Week 4—Add boceprevir
- Week 8—Measure HCV RNA
- Week 12—Measure HCV RNA; stop treatment if it is more than 100 IU/mL
- Week 24—Measure HCV RNA; stop treatment if it is detectable
- Week 36—if HCV RNA was not detectable at week 8, stop all treatment now; if HCV RNA was detectable at week 8, stop boceprevir now but continue pegylated interferon and ribavirin
- Week 48—Stop all treatment (eFigure 2).
Telaprevir in previously untreated patients and prior relapsers
- Week 0—start telaprevir, pegylated interferon, and ribavirin
- Week 4—measure HCV RNA; stop all treatment if it is more than 1,000 IU/mL
- Week 12—Stop telaprevir; measure HCV RNA; stop all treatment if HCV RNA is more than 1,000 IU/mL
- Week 24—Stop pegylated interferon and ribavirin if HCV RNA was undetectable at week 12; measure HCV RNA and stop treatment if it is detectable; otherwise, continue pegylated interferon and ribavirin
- Week 48—Stop all treatment (eFigure 3).
Telaprevir in patients who previously achieved a partial or null response
- Week 0—Start telaprevir, pegylated interferon, and ribavirin
- Week 4—Measure HCV RNA; stop treatment if it is more than 1,000 IU/mL
- Week 12—Measure HCV RNA; stop all treatment if it is more than 1,000 IU/mL; if less than 1,000 IU/mL then stop telaprevir but continue pegylated interferon and ribavirin
- Week 24—Measure HCV RNA; stop treatment if HCV RNA is detectable
- Week 48—Stop all treatment (eFigure 4).
Drug interactions with boceprevir and telaprevir
Both boceprevir and telaprevir inhibit cytochrome P450 3A (CYP3A) and thus are contraindicated in combination with drugs highly dependent on CYP3A for clearance and with drugs for which elevated plasma concentrations are associated with serious adverse events, such as atorvastatin (Lipitor), simvastatin (Zocor), sildenafil (Viagra), midazolam (Versed), and St. John’s wort. Giving potent inducers of CYP3A with boceprevir or telaprevir may lead to lower exposure and loss of efficacy of both protease inhibitors.
EMERGING THERAPIES FOR HCV
Thanks to a better understanding of the biology of HCV infection, the effort to develop new therapeutic agents started to focus on targeting specific steps of the viral life cycle, including attachment, entry into cells, replication, and release.24
Currently, more than 50 clinical trials are evaluating new direct-acting antivirals to treat HCV infection.25 Monoclonal and polyclonal antibodies that target the molecular process involved in HCV attachment and entry are being developed.26 The nonstructural protein NS5B (RNA polymerase) is intimately involved in viral replication and represents a promising target.27 Several nucleosides and nonnucleoside protease inhibitors have already entered clinical trials.
The low fidelity of the HCV replication machinery leads to a very high mutation rate, thus enabling the virus to quickly develop mutations that resist agents targeting viral enzymes.28 Therefore, a novel approach is to target host cofactors that are essential for HCV replication. An intriguing study by Lanford et al29 demonstrated that antagonizing microRNA-122 (the most abundant microRNA in the liver and an essential cofactor for viral RNA replication) by the oligonucleotide SPC3649 caused marked and prolonged reduction of HCV viremia in chronically infected chimpanzees.29
Although we are still in the early stages of drug development, the future holds great promise for newer drugs to improve the sustained virologic response, shorten the duration of treatment, improve tolerability with interferon-sparing regimens, and decrease viral resistance.
FUTURE PERSPECTIVES
With the introduction of the first direct-acting antiviral medications for HCV (boceprevir and telaprevir), 2011 will be marked as the year that changed hepatitis C treatment for the better. Triple therapy with pegylated interferon, ribavirin, and either boceprevir or telaprevir has the potential for increasing the rate of sustained virologic response to around 70% in previously untreated patients and 65% in previously treated patients who are infected with HCV genotype 1. The IL28B polymorphisms appear to play a role in the rate of sustained virologic response achieved with triple therapy, with preliminary data showing a better response rate in patients who have the CC genotype.17
These drugs will add up to $50,000 to the cost of treating hepatitis C virus infection, depending on the drug used and the length of treatment. However, they may be well worth it if they prevent liver failure and the need for transplantation.
Many questions remain, such as how to use these new regimens to treat special patient populations—for example, those with a recurrence of HCV infection after liver transplantation, those co-infected with HCV and human immunodeficiency virus, and those infected with HCV genotypes other than genotype 1.
Other direct-acting antiviral agents that specifically target the replication cycle of HCV are currently in clinical development. In fact, the future has already started with the release of the Interferon-Free Regimen for the Management of HCV (INFORM-1) study results.30 This was the first trial to evaluate an interferon-free regimen for patients with chronic HCV infection using two direct-acting antiviral drugs (the protease inhibitor danoprevir and the polymerase inhibitor RG7128), with promising results.
- Poordad F, McCone J, Bacon BR, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med 2011; 364:1195–1206.
- Bacon BR, Gordon SC, Lawitz E, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med 2011; 364:1207–1217.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; for the ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Sherman KE, Flamm SL, Afdhal NH, et al; for the ILLUMINATE Study Team. Response-guided telaprevir combination treatment for hepatitis C virus infection. N Engl J Med 2011; 365:1014–1024.
- Zeuzem S, Andreone P, Pol S, et al; for the REALIZE Study Team. Telaprevir for retreatment of HCV infection. N Engl J Med 2011; 364:2417–2428.
- Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med 2006; 144:705–714.
- Mitchell AE, Colvin HM, Palmer Beasley R. Institute of Medicine recommendations for the prevention and control of hepatitis B and C. Hepatology 2010; 51:729–733.
- Kim WR. The burden of hepatitis C in the United States. Hepatology 2002; 36:S30–S34.
- Marcellin P, Asselah T, Boyer N. Fibrosis and disease progression in hepatitis C. Hepatology 2002; 36:S47–S56.
- Seeff LB. Natural history of chronic hepatitis C. Hepatology 2002; 36:S35–S46.
- Lavanchy D. The global burden of hepatitis C. Liver Int 2009; 29(suppl 1):74–81.
- National Institutes of Health Consensus Development Conference Statement: Management of hepatitis C: 2002—June 10–12, 2002. Hepatology 2002; 36:S3–S20.
- Pearlman BL, Traub N. Sustained virologic response to antiviral therapy for chronic hepatitis C virus infection: a cure and so much more. Clin Infect Dis 2011; 52:889–900.
- Hoofnagle JH, Seeff LB. Peginterferon and ribavirin for chronic hepatitis C. N Engl J Med 2006; 355:2444–2451.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009; 461:399–401.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e118.
- Nielsen SU, Bassendine MF, Burt AD, Bevitt DJ, Toms GL. Characterization of the genome and structural proteins of hepatitis C virus resolved from infected human liver. J Gen Virol 2004; 85:1497–1507.
- Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. Structural biology of hepatitis C virus. Hepatology 2004; 39:5–19.
- Nelson DR. The role of triple therapy with protease inhibitors in hepatitis C virus genotype 1 naive patients. Liver Int 2011; 31(suppl 1):53–57.
- Pawlotsky JM. Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology 2011; 53:1742–1751.
- Monto A, Schooley RT, Lai JC, et al. Lessons from HIV therapy applied to viral hepatitis therapy: summary of a workshop. Am J Gastroenterol 2010; 105:989–1004.
- McCown MF, Rajyaguru S, Kular S, Cammack N, Najera I. GT-1a or GT-1b subtype-specific resistance profiles for hepatitis C virus inhibitors telaprevir and HCV-796. Antimicrob Agents Chemother 2009; 53:2129–2132.
- Cholongitas E, Papatheodoridis GV. Review article: novel therapeutic options for chronic hepatitis C. Aliment Pharmacol Ther 2008; 27:866–884.
- Naggie S, Patel K, McHutchison J. Hepatitis C virus directly acting antivirals: current developments with NS3/4A HCV serine protease inhibitors. J Antimicrob Chemother 2010; 65:2063–2069.
- Mir HM, Birerdinc A, Younossi ZM. Monoclonal and polyclonal antibodies against the HCV envelope proteins. Clin Liver Dis 2009; 13:477–486.
- Birerdinc A, Younossi ZM. Emerging therapies for hepatitis C virus. Expert Opin Emerg Drugs 2010; 15:535–544.
- Khattab MA. Targeting host factors: a novel rationale for the management of hepatitis C virus. World J Gastroenterol 2009; 15:3472–3479.
- Lanford RE, Hildebrandt-Eriksen ES, Petri A, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010; 327:198–201.
- Gane EJ, Roberts SK, Stedman CA, et al. Oral combination therapy with a nucleoside polymerase inhibitor (RG7128) and danoprevir for chronic hepatitis C genotype 1 infection (INFORM-1): a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 2010; 376:1467–1475.
- Poordad F, McCone J, Bacon BR, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med 2011; 364:1195–1206.
- Bacon BR, Gordon SC, Lawitz E, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med 2011; 364:1207–1217.
- Jacobson IM, McHutchison JG, Dusheiko G, et al; for the ADVANCE Study Team. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med 2011; 364:2405–2416.
- Sherman KE, Flamm SL, Afdhal NH, et al; for the ILLUMINATE Study Team. Response-guided telaprevir combination treatment for hepatitis C virus infection. N Engl J Med 2011; 365:1014–1024.
- Zeuzem S, Andreone P, Pol S, et al; for the REALIZE Study Team. Telaprevir for retreatment of HCV infection. N Engl J Med 2011; 364:2417–2428.
- Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med 2006; 144:705–714.
- Mitchell AE, Colvin HM, Palmer Beasley R. Institute of Medicine recommendations for the prevention and control of hepatitis B and C. Hepatology 2010; 51:729–733.
- Kim WR. The burden of hepatitis C in the United States. Hepatology 2002; 36:S30–S34.
- Marcellin P, Asselah T, Boyer N. Fibrosis and disease progression in hepatitis C. Hepatology 2002; 36:S47–S56.
- Seeff LB. Natural history of chronic hepatitis C. Hepatology 2002; 36:S35–S46.
- Lavanchy D. The global burden of hepatitis C. Liver Int 2009; 29(suppl 1):74–81.
- National Institutes of Health Consensus Development Conference Statement: Management of hepatitis C: 2002—June 10–12, 2002. Hepatology 2002; 36:S3–S20.
- Pearlman BL, Traub N. Sustained virologic response to antiviral therapy for chronic hepatitis C virus infection: a cure and so much more. Clin Infect Dis 2011; 52:889–900.
- Hoofnagle JH, Seeff LB. Peginterferon and ribavirin for chronic hepatitis C. N Engl J Med 2006; 355:2444–2451.
- Ge D, Fellay J, Thompson AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009; 461:399–401.
- Suppiah V, Moldovan M, Ahlenstiel G, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet 2009; 41:1100–1104.
- Thompson AJ, Muir AJ, Sulkowski MS, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology 2010; 139:120–129.e118.
- Nielsen SU, Bassendine MF, Burt AD, Bevitt DJ, Toms GL. Characterization of the genome and structural proteins of hepatitis C virus resolved from infected human liver. J Gen Virol 2004; 85:1497–1507.
- Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. Structural biology of hepatitis C virus. Hepatology 2004; 39:5–19.
- Nelson DR. The role of triple therapy with protease inhibitors in hepatitis C virus genotype 1 naive patients. Liver Int 2011; 31(suppl 1):53–57.
- Pawlotsky JM. Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology 2011; 53:1742–1751.
- Monto A, Schooley RT, Lai JC, et al. Lessons from HIV therapy applied to viral hepatitis therapy: summary of a workshop. Am J Gastroenterol 2010; 105:989–1004.
- McCown MF, Rajyaguru S, Kular S, Cammack N, Najera I. GT-1a or GT-1b subtype-specific resistance profiles for hepatitis C virus inhibitors telaprevir and HCV-796. Antimicrob Agents Chemother 2009; 53:2129–2132.
- Cholongitas E, Papatheodoridis GV. Review article: novel therapeutic options for chronic hepatitis C. Aliment Pharmacol Ther 2008; 27:866–884.
- Naggie S, Patel K, McHutchison J. Hepatitis C virus directly acting antivirals: current developments with NS3/4A HCV serine protease inhibitors. J Antimicrob Chemother 2010; 65:2063–2069.
- Mir HM, Birerdinc A, Younossi ZM. Monoclonal and polyclonal antibodies against the HCV envelope proteins. Clin Liver Dis 2009; 13:477–486.
- Birerdinc A, Younossi ZM. Emerging therapies for hepatitis C virus. Expert Opin Emerg Drugs 2010; 15:535–544.
- Khattab MA. Targeting host factors: a novel rationale for the management of hepatitis C virus. World J Gastroenterol 2009; 15:3472–3479.
- Lanford RE, Hildebrandt-Eriksen ES, Petri A, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010; 327:198–201.
- Gane EJ, Roberts SK, Stedman CA, et al. Oral combination therapy with a nucleoside polymerase inhibitor (RG7128) and danoprevir for chronic hepatitis C genotype 1 infection (INFORM-1): a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 2010; 376:1467–1475.
KEY POINTS
- Standard care with the combination of pegylated interferon and ribavirin produces a sustained virologic response in about 40% of patients infected with HCV genotype 1, the most prevalent genotype in North America.
- New phase 3 trials showed that the addition of an oral protease inhibitor (boceprevir or telaprevir) increased the sustained virologic response rates to 70% in patients infected with HCV genotype 1.
- Boceprevir and telaprevir must be used in combination with pegylated interferon and ribavirin; they should not be used as monotherapy because of concern about the development of drug-resistant mutations.
- The main side effects of boceprevir were anemia and dysgeusia. Adverse events associated with telaprevir included rash, pruritus, anemia, and diarrhea.