User login
Recognizing and treating cutaneous signs of liver disease
Dysfunction in the body’s second largest organ, the liver, often yields changes in the body’s largest organ, the skin. If we can recognize these manifestations early, we are better able to promptly diagnose and treat the underlying liver disease, as well as the skin lesions.
The liver has many jobs: synthesizing proteins such as clotting factors, complements, and albumin; neutralizing toxins; and metabolizing lipids and carbohydrates. Insults to the liver can compromise any of these functions, affecting visceral organs, joints, gastrointestinal tissues, and the skin. Dermatologic signs of specific liver diseases include alopecia and vitiligo associated with autoimmune hepatitis, and xanthelasma in chronic cholestatic liver disease.
This article reviews the important cutaneous manifestations of specific liver diseases. We focus first on skin conditions that may represent liver disease, and then we discuss several major liver diseases and their typical cutaneous manifestations.
JAUNDICE AND HYPERBILIRUBINEMIA
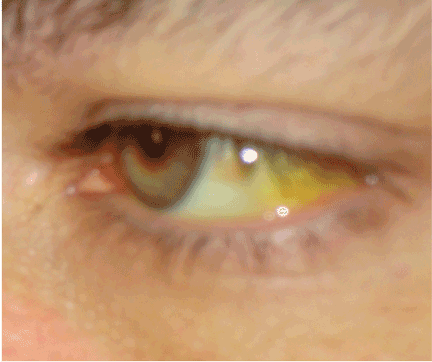
Establishing whether the excess bilirubin is conjugated or unconjugated gives a clue as to whether the cause is prehepatic, intrahepatic, or posthepatic.3–8 One of the liver’s main functions is to conjugate bilirubin into a secretable form. Prehepatic causes of jaundice include hemolysis and ineffective erythropoiesis, both of which lead to higher levels of circulating unconjugated bilirubin.4 Intrahepatic causes of jaundice can lead to both unconjugated and conjugated hyperbilirubinemia.4,8 Posthepatic causes such as bile duct obstruction primarily result in conjugated hyperbilirubinemia.4
PRURITUS AND PRURIGO NODULARIS
Pruritus can be multifactorial or the result of a specific dermatologic or systemic condition.9 A thorough history and physical examination are warranted to rule out hepatic or systemic causes of itching.10
The liver neutralizes toxins and filters bile salts. If its function is impaired, these materials can accumulate in the body, and deposition in the skin causes irritation and itching.11,12 In cholestatic liver disorders such as primary sclerosing cholangitis and obstructive gallstone disease, pruritus tends to be generalized, but worse on the hands and feet.13
Although the severity of pruritus is not directly associated with the level of bile salts and toxic substances, lowering bile salt levels can mitigate symptoms.11
Treatment. Pruritus due to liver disease is particularly resistant to therapy.
In a strategy described by Mela et al for managing pruritus in chronic liver disease,14 the initial treatment is the anion exchange resin cholestyramine (Questran) at a starting dose of 4 g/day, gradually increased to 24 g/day in two doses at mealtimes.
If the pruritus does not respond adequately to cholestyramine or the patient cannot tolerate the drug, then the antituberculosis drug rifampin (Rifadin) can be tried. Rifampin promotes metabolism of endogenous pruritogens and has been effective against cholestatic pruritus when started at 150 mg/day and increased up to 600 mg/day, depending on the clinical response.14
Third-line drug therapies include opioid antagonists such as naltrexone (ReVia) and nalmefene (Revex).14,15
Plasmapheresis can be considered if drug therapy fails.16 Experimental therapies include albumin dialysis using the molecular adsorbent recirculating system (a form of artificial liver support), antioxidant treatment, and bright-light therapy.15 Liver transplantation, when appropriate, also resolves cholestatic pruritus.14
Prurigo nodularis
Prurigo nodularis, distinguished by firm, crusty nodules, is associated with viral infections (eg, hepatitis C, human immunodeficiency virus), bacterial infections, and kidney dysfunction.17,18 The lesions are intensely pruritic and often lead to persistent scratching, excoriation, and, ultimately, diffuse scarring.19
Treatment. Although the exact cause of prurigo nodularis is not known and no cure exists, corticosteroid or antihistamine ointments control the symptoms in most patients with hepatitis.19 Low doses of thalidomide (Thalomid), a tumor necrosis factor antagonist, have also been used safely and effectively.18,19
SUPERFICIAL VASCULAR SIGNS
Spider angiomas
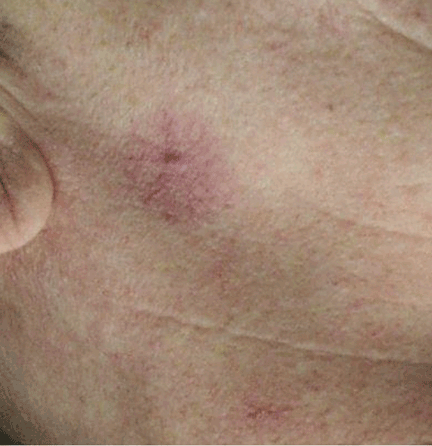
Spider angiomas can occur anywhere on the body, but they occur most often on the face and the trunk.21,23 A key feature is that they disappear when pressure is applied and reappear when pressure is removed.23,24 Biopsy is rarely necessary for diagnosis.
These lesions occur with elevated estrogen levels, such as in cirrhosis, during estrogen therapy, or during pregnancy.25–28 Although spider angiomas are common in pregnant women and in children, adults with spider angiomas deserve a workup for liver dysfunction.29
Given their innocuous nature and asymptomatic course, spider angiomas themselves require no medical treatment.
Bier spots
Bier spots are small, irregularly shaped, hypopigmented patches on the arms and legs. They are likely due to venous stasis associated with functional damage to the small vessels of the skin.30
Since Bier spots are a sign of liver disease, they must be distinguished from true pigmentation disorders. A key distinguishing feature is that Bier spots disappear when pressure is applied. Also, raising the affected limb from a dependent position causes the hypopigmented macules of Bier spots to disappear, which is not the case in true pigmentation disorders.10,30
Paper-money skin
Paper-money skin (or “dollar-paper” markings) describes the condition in which the upper trunk is covered with many randomly scattered, needle-thin superficial capillaries. It often occurs in association with spider angiomas. The name comes from the resemblance the thread-like capillaries have to the finely chopped silk threads in American dollar bills.10,31 The condition is commonly seen in patients with alcoholic cirrhosis and may improve with hemodialysis.31
PALMAR ERYTHEMA
Palmar erythema is a florid, crimson coloration of the palms of the hands and the fingertips. It can occur anywhere on the palm and fingers but is most common on the hypothenar eminence. It can occur in a number of liver conditions but most often with cirrhosis.32 Hepatic compromise, as seen in alcoholic liver disease, disrupts the body’s androgen balance, causing local vasodilation and erythema.32,33 Although the exact mechanism remains unknown, research suggests that prostacyclins and nitric oxide play a role, as both are increased in liver disease.32,33
XANTHELASMA
Xanthelasma—a localized cholesterol deposit beneath the skin and especially beneath the eyelids—is a common manifestation of hypercholesterolemia. Xanthelasma often presents as a painless, yellowish, soft plaque with well-defined borders,34 which may enlarge over the course of weeks.
Several liver diseases can lead to various forms of secondary dyslipoproteinemia.35 The most common dyslipoproteinemias in liver disease are hypertriglyceridemia and low levels of high-density lipoprotein cholesterol, and both of these often accompany fatty liver disease.36 Hypercholesterolemia is a common feature of primary biliary cirrhosis and other forms of cholestatic liver disease.37 Studies suggest that the total plasma cholesterol level is elevated in as many as 50% of patients with compromised liver function.38
Treatment. The underlying hyperlipidemia is treated with cholesterol-lowering drugs. Laser treatment and surgical excision have proven efficacious in treating the lesions.39
OTHER CUTANEOUS FINDINGS IN LIVER DISEASE
Bleeding and bruising. Liver disease can cause hypersplenism and thrombocytopenia, in addition to a decrease in clotting factors. These may present with a myriad of cutaneous symptoms, including purpura, bleeding gums, and easy bruising and bleeding, even from minor trauma.40–42
Hyperpigmentation of the skin may accompany hemochromatosis, alcoholic liver disease, and cirrhosis.43–45
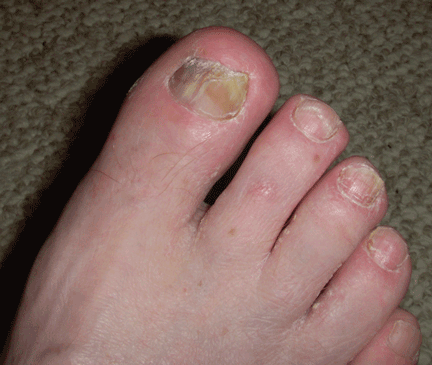
“Terry’s nails,” in which the proximal two-thirds of the nail plate turns powdery white with a ground-glass opacity, may develop in patients with advanced cirrhosis.48
ALCOHOLIC CIRRHOSIS AND THE SKIN
The cutaneous changes associated with alcoholic cirrhosis are more widely recognized than those due to other forms of liver dysfunction. In the United States, approximately 3 million people have alcoholic cirrhosis, the second-leading reason for liver transplantation.49,50
As the body’s main site of alcohol metabolism, the liver is the organ most affected by excessive alcohol intake, which can lead to end-stage liver disease secondary to alcoholic cirrhosis.41,51 The characteristic feature of cirrhosis is advanced fibrous scarring of parenchymal tissue and the formation of regenerative nodules with increased resistance to blood flow throughout the organ.41,52 The insufficient blood flow damages vital structures in the liver and compromises liver function. For example, liver cirrhosis leads to defective hepatic synthesis of clotting factors and results in bleeding disorders.
Cutaneous lesions often accompany alcoholic cirrhosis and have been detected in up to 43% of people with chronic alcoholism.53 Skin changes in alcoholic cirrhosis can be of great diagnostic value. The combined prevalence of spider angiomas, palmar erythema, and Dupuytren contracture in alcoholic cirrhosis was found to be 72%. Paper-money skin and Dupuytren contracture are more distinct lesions for alcoholic cirrhosis.31 Recognizing these skin changes contributes to the diagnosis and staging of liver cirrhosis.51,52
Dupuytren contracture
Dupuytren contracture is characterized by progressive fibrosis and thickening of tendons in the palmar fascia, the connective tissue that lies beneath the skin of the palms.54 Over time, as fibrotic involvement expands across the fascia, rampant stiffness of the joints ensues, sometimes to a point where the fingers cannot fully flex or extend.54
Although the exact cause of Dupuytren contracture is unknown, it appears to be associated with excess alcohol consumption and can be found in patients with alcoholic cirrhosis.54,55 These patients often present with painless stiffness of the fingers, curling of fingers, and loss of motion in involved fingers.54 Surgery in the form of limited fasciectomy has been curative in such patients.54
Disseminated superficial porokeratosis
Porokeratosis is a keratinization disorder of clonal origin that presents as a linear configuration of white scaly papules that coalesce into plaques throughout the body.56 Although it most commonly afflicts fair-skinned people, patients with alcoholic cirrhosis have a much greater susceptibility than the general population.57,58
A recent study58 documented that the lesions completely resolved when liver function improved, thus underlining the relationship between the two conditions. Since immunosuppression has been linked to eruption of the lesion, the fact that both humoral and cell-mediated immune responses are impaired in alcoholic liver disease provides another dimension to the association between porokeratosis and alcoholic cirrhosis.58
These lesions can transform into squamous cell carcinoma.59 The risk of widespread metastases in squamous cell carcinoma highlights the importance of dermatologic consultation in such patients.59
HEPATITIS C AND THE SKIN
Extrahepatic manifestations have been documented in up to 74% of people with hepatitis C virus infection.60 In addition to parasthesias, arthralgias, and myalgias, hepatitis C has a significant association with porphyria cutanea tarda, lichen planus, vitiligo, sialadenitis, urticarial vasculitis, corneal ulcers, xerosis, pruritus, and prurigo nodularis.60–64 Although the primary causative agents of sialadenitis are bacteria, viruses such as hepatitis C have been implicated as a cause of chronic sialadenitis with associated xerostomia.65
Patients with hepatitis C being treated with interferons also present with cutaneous manifestations such as hyperkeratosis and vasculitis.63
Porphyria cutanea tarda
Porphyria cutanea tarda is the most common of the porphyrias, disorders distinguished by deficiencies or defects in one or more of the enzymes responsible for hepatic production of heme.66 If these enzymes are impaired, heme precursors such as porphyrins accumulate.66
Porphyria cutanea tarda results from a deficiency of the hepatic enzyme uroporphyrin decarboxylase. In the absence of this enzyme, shortwave visible light activates uroporphyrin deposited in the skin, resulting in a photochemical reaction that generates reactive oxygen species that lead to the characteristic skin blistering.
Although porphyria cutanea tarda is associated with liver disease in general, recent studies confirm that patients with hepatitis C are at particularly high risk.67 Those with the disorder often present with skin photosensitivity. 68 Many develop blisters on sun-exposed skin, including the dorsal aspects of the hands and forearms and on the neck and face. Chronic porphyria cutanea tarda can lead to scarring, alopecia, and skin ulceration.69 As the blisters heal, keratin-filled milial cysts may develop in the areas of ulceration.
The condition is also commonly associated with melasma-like hyperpigmentation and hypertrichosis in sun-exposed areas of the head and neck. People of Northern European ancestry may be more at risk than the general population because of a presumed genetic susceptibility.70
Treatment. Because many patients with porphyria cutanea tarda have iron overload, they need to restrict foods rich in iron and to avoid alcohol.71,72 Severe cases may necessitate iron removal via phlebotomy or antimalarial therapy. Patients with porphyria cutanea tarda induced by hepatitis C should have their bodily iron stores depleted before starting antiviral therapy.60
Lichen planus
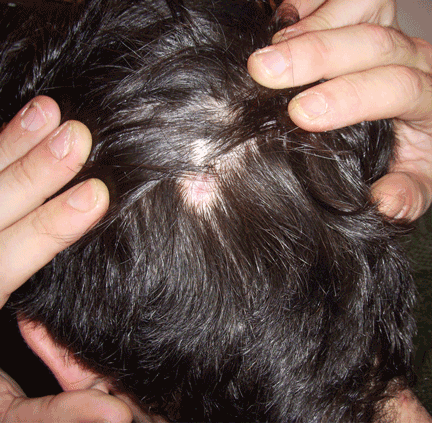
Lichen planopilaris is a subset of lichen planus that causes scaling and atrophy of the scalp and permanent hair loss (Figure 4).73
Interferon-induced vitiligo
Vitiligo is an autoimmune disease in which melanocytes in the skin are destroyed, with resulting depigmentation in affected areas.75 Although it has no specific association with liver disease, it has been linked to treatments for hepatitis C such as interferons.76 Interferon-induced vitiligo often completely resolves when interferon is stopped.77
Typical findings include aggregations of irregularly shaped white patches in a focal or segmental pattern.78 The diagnosis is based on the medical history, physical examination, and sometimes skin biopsy.
HEMOCHROMATOSIS
Hemochromatosis or “bronze diabetes” is a devastating multisystem disease with a relentless course. It is among the most common genetic disorders of metabolism, and results in deposition of iron in tissues and organs throughout the body, including the liver, usually in patients ages 30 to 40.
As iron stores increase in tissues and organs, multiorgan failure and associated complications may ensue. In addition, surplus iron stores can also result in widespread bronze discoloration of skin exposed to the sun. Hemochromatosis also results in loss of body hair, ichthyosiform alterations, and koilonychia.79
Treatments that lower serum iron levels can reverse the cutaneous manifestations of the disorder and minimize the risk of organ failure.
Since the condition is inherited in an autosomal-recessive pattern, family members of patients should consider being screened.80
Hyperpigmentation in hemochromatosis
Hyperpigmentation is an early sign of hemochromatosis, affecting up to 90% of patients. Usually, sun-exposed areas of the body are the most prone and take on a grayish or brownish-bronze hue.81 Cutaneous iron deposits injure vital skin structures, initiating a process that culminates in enhanced melanin production by melanocytes.82 Exposure to ultraviolet light may have synergistic effects with iron, hastening the process of hyperpigmentation. As a result of this synergistic effect, many patients with hemochromatosis notice tanning with minimal sun exposure.
Although organ function can improve immediately with phlebotomy to reduce iron stores, skin hyperpigmentation does not immediately resolve.81,82
Ichthyosiform alterations in hemochromatosis
Ichthyosiform changes, in which the skin takes on the appearance of fish scales,83 can be seen in patients with hemochromatosis.80 Affected areas typically become extremely dry. Treatment includes topical hydrating creams and ointments. Avoiding sunlight is paramount, as sunlight exposure may exacerbate the condition.83
- Morioka D, Togo S, Kumamoto T, et al. Six consecutive cases of successful adult ABO-incompatible living donor liver transplantation: a proposal for grading the severity of antibody-mediated rejection. Transplantation 2008; 85:171–178.
- Bertini G, Rubaltelli FF. Non-invasive bilirubinometry in neonatal jaundice. Semin Neonatol 2002; 7:129–133.
- Clementi M, Di Gianantonio E, Fabris L, et al. Inheritance of hyperbilirubinemia: evidence for a major autosomal recessive gene. Dig Liver Dis 2007; 39:351–355.
- Roche SP, Kobos R. Jaundice in the adult patient. Am Fam Physician 2004; 69:299–304.
- Odemis B, Parlak E, Basar O, Yüksel O, Sahin B. Biliary tract obstruction secondary to malignant lymphoma: experience at a referral center. Dig Dis Sci 2007; 52:2323–2332.
- Caddy GR, Tham TC. Gallstone disease: symptoms, diagnosis and endoscopic management of common bile duct stones. Best Pract Res Clin Gastroenterol 2006; 20:1085–1101.
- Heathcote EJ. Diagnosis and management of cholestatic liver disease. Clin Gastroenterol Hepatol 2007; 5:776–782.
- Faust TW, Reddy KR. Postoperative jaundice. Clin Liver Dis 2004; 8:151–166.
- Maticic M, Poljak M, Lunder T, Rener-Sitar K, Stojanovic L. Lichen planus and other cutaneous manifestations in chronic hepatitis C: pre- and post-interferon-based treatment prevalence vary in a cohort of patients from low hepatitis C virus endemic area. J Eur Acad Dermatol Venereol 2008; 22:779–788.
- Polat M, Oztas P, Ilhan MN, Yalçin B, Alli N. Generalized pruritus: a prospective study concerning etiology. Am J Clin Dermatol 2008; 9:39–44.
- Gaspari R, Avolio AW, Zileri Dal Verme L, et al. Molecular adsorbent recirculating system in liver transplantation: safety and efficacy. Transplant Proc 2006; 38:3544–3551.
- Dasgupta R, Saha I, Pal S, et al. Immunosuppression, hepatotoxicity and depression of antioxidant status by arecoline in albino mice. Toxicology 2006; 227:94–104.
- Mayo MJ, Handem I, Saldana S, Jacobe H, Getachew Y, Rush AJ. Sertraline as a first-line treatment for cholestatic pruritus. Hepatology 2007; 45:666–674.
- Mela M, Mancuso A, Burroughs AK. Review article: pruritus in cholestatic and other liver diseases. Aliment Pharmacol Ther 2003; 17:857–870.
- Montero JL, Pozo JC, Barrera P, et al. Treatment of refractory cholestatic pruritus with molecular adsorbent recirculating system (MARS). Transplant Proc 2006; 38:2511–2513.
- Neff GW, O'Brien CB, Reddy KR, et al. Preliminary observation with dronabinol in patients with intractable pruritus secondary to cholestatic liver disease. Am J Gastroenterol 2002; 97:2117–2119.
- Neri S, Raciti C, D’Angelo G, Ierna D, Bruno CM. Hyde’s prurigo nodularis and chronic HCV hepatitis. J Hepatol 1998; 28:161–164.
- Brown MA, George CR, Dunstan CR, Kalowski S, Corrigan AB. Prurigo nodularis and aluminium overload in maintenance haemodialysis. Lancet 1992; 340:48.
- Stander S, Luger T, Metze D. Treatment of prurigo nodularis with topical capsaicin. J Am Acad Dermatol 2001; 44:471–478.
- Requena L, Sangueza OP. Cutaneous vascular anomalies. Part I. Hamartomas, malformations, and dilation of preexisting vessels. J Am Acad Dermatol 1997; 37:523–549.
- Khasnis A, Gokula RM. Spider nevus. J Postgrad Med 2002; 48:307–309.
- Kaul V, Friedenberg FK, Braitman LE, et al. Development and validation of a model to diagnose cirrhosis in patients with hepatitis C. Am J Gastroenterol 2002; 97:2623–2628.
- Banyai AL. There is more than surface appearance to skin spiders. Chest 1971; 60:48.
- Errickson CV, Matus NR. Skin disorders of pregnancy. Am Fam Physician 1994; 49:605–610.
- Li CP, Lee FY, Hwang SJ, et al. Spider angiomas in patients with liver cirrhosis: role of alcoholism and impaired liver function. Scand J Gastroenterol 1999; 34:520–523.
- Li CP, Lee FY, Hwang SJ, et al. Role of substance P in the pathogenesis of spider angiomas in patients with nonalcoholic liver cirrhosis. Am J Gastroenterol 1999; 94:502–507.
- Sadick NS, Niedt GW. A study of estrogen and progesterone receptors in spider telangiectasias of the lower extremities. J Dermatol Surg Oncol 1990; 16:620–623.
- Henry F, Quatresooz P, Valverde-Lopez JC, Pierard GE. Blood vessel changes during pregnancy: a review. Am J Clin Dermatol 2006; 7:65–69.
- Finn SM, Rowland M, Lawlor F, et al. The significance of cutaneous spider naevi in children. Arch Dis Child 2006; 91:604–605.
- Peyrot I, Boulinguez S, Sparsa A, Le Meur Y, Bonnetblanc JM, Bedane C. Bier’s white spots associated with scleroderma renal crisis. Clin Exp Dermatol 2007; 32:165–167.
- Satoh T, Yokozeki H, Nishioka K. Vascular spiders and paper money skin improved by hemodialysis. Dermatology 2002; 205:73–74.
- Serrao R, Zirwas M, English JC. Palmar erythema. Am J Clin Dermatol 2007; 8:347–356.
- Matsumoto M, Ohki K, Nagai I, Oshibuchi T. Lung traction causes an increase in plasma prostacyclin concentration and decrease in mean arterial blood pressure. Anesth Analg 1992; 75:773–776.
- Otto AI, Horvath I, Feldmann J. Multiple firm, painless erythematous papules with a yellowish hue. Arch Dermatol 2005; 141:1595–1600.
- Gandelman G, Aronow WS, Weiss MB. Resolving hyperlipidemia after liver transplantation in a patient with primary sclerosing cholangitis. Am J Ther 2006; 13:171–174.
- Assy N, Kaita K, Mymin D, Levy C, Rosser B, Minuk G. Fatty infiltration of liver in hyperlipidemic patients. Dig Dis Sci 2000; 45:1929–1934.
- Allocca M, Crosignani A, Gritti A, et al. Hypercholesterolaemia is not associated with early atherosclerotic lesions in primary biliary cirrhosis. Gut 2006; 55:1795–1800.
- Dickson E, Fleming C, Ludwig J. Primary biliary cirrhosis. In:Popper H, Schaffner F, editors. Progress in Liver Diseases. New York: Grune and Stratton; 1978:487.
- Elner VM, Mintz R, Demirci H, Hassan AS. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Trans Am Ophthalmol Soc 2005; 103:69–73.
- Craxi A, Camma C, Giunta M. Clinical aspects of bleeding complications in cirrhotic patients. Blood Coagul Fibrinolysis 2000; 11( suppl 1):S75–579.
- Kajihara M, Okazaki Y, Kato S, et al. Evaluation of platelet kinetics in patients with liver cirrhosis: similarity to idiopathic thrombocytopenic purpura. J Gastroenterol Hepatol 2007; 22:112–118.
- Levine N. Patient reports six-month history of minimally pruritic purple dots on legs. Non-blanching macules developed over six months. Geriatrics 2006; 61:22.
- Barton JC, Rao SV, Pereira NM, et al. Juvenile hemochromatosis in the southeastern United States: a report of seven cases in two kinships. Blood Cells Mol Dis 2002; 29:104–115.
- Smith AG, Shuster S, Bomford A, Williams R. Plasma immunoreactive beta-melanocyte-stimulating hormone in chronic liver disease and fulminant hepatic failure. J Invest Dermatol 1978; 70:326–327.
- Barton JC, McDonnell SM, Adams PC, et al. Management of hemochromatosis. Hemochromatosis Management Working Group. Ann Intern Med 1998; 129:932–939.
- Bahnsen M, Gluud C, Johnsen SG, et al. Pituitary-testicular function in patients with alcoholic cirrhosis of the liver. Eur J Clin Invest 1981; 11:473–479.
- Kumar N, Aggarwal SR, Anand BS. Comparison of truncal hair distribution in alcoholic liver disease and alcohol-related chronic pancreatitis. J Gastroenterol Hepatol 2001; 16:855–856.
- Holzberg M, Walker HK. Terry's nails: revised definition and new correlations. Lancet 1984; 1:896–899.
- Mandayam S, Jamal MM, Morgan TR. Epidemiology of alcoholic liver disease. Semin Liver Dis 2004; 24:217–232.
- Belle SH, Beringer KC, Detre KM. Liver transplantation in the United States: results from the National Pitt-UNOS Liver Transplant Registry. United Network for Organ Sharing Clin Transpl 1994:19–35.
- Dunn W, Xu R, Schwimmer JB. Modest wine drinking and decreased prevalence of suspected nonalcoholic fatty liver disease. Hepatology 2008; 47:1947–1954.
- Afford SC, Fisher NC, Neil DA, et al. Distinct patterns of chemokine expression are associated with leukocyte recruitment in alcoholic hepatitis and alcoholic cirrhosis. J Pathol 1998; 186:82–89.
- Evstaf’ev VV, Levin MM. Dermatologic pathology in chronic alcoholics. Vestn Dermatol Venerol 1989; 8:72–74.
- Jerosch-Herold C, Shepstone L, Chojnowski AJ, Larson D. Splinting after contracture release for Dupuytren's contracture (SCoRD): protocol of a pragmatic, multi-centre, randomized controlled trial. BMC Musculoskelet Disord 2008; 9:62.
- Houghton S, Holdstock G, Cockerell R, Wright R. Dupuytren’s contracture, chronic liver disease and IgA immune complexes. Liver 1983; 3:220–224.
- Ibbotson SH. Disseminated superficial porokeratosis: what is the association with ultraviolet radiation? Clin Exp Dermatol 1996; 21:48–50.
- Kono T, Kobayashi H, Ishii M, Nishiguchi S, Taniguchi S. Synchronous development of disseminated superficial porokeratosis and hepatitis C virus-related hepatocellular carcinoma. J Am Acad Dermatol 2000; 43:966–968.
- Park BS, Moon SE, Kim JA. Disseminated superficial porokeratosis in a patient with chronic liver disease. J Dermatol 1997; 24:485–487.
- Murata Y, Kumano K, Takai T. Type 2 segmental manifestation of disseminated superficial porokeratosis showing a systematized pattern of involvement and pronounced cancer proneness. Eur J Dermatol 2001; 11:191–194.
- Galossi A, Guarisco R, Bellis L, Puoti C. Extrahepatic manifestations of chronic HCV infection. J Gastrointestin Liver Dis 2007; 16:65–73.
- El-Serag HB, Hampel H, Yeh C, Rabeneck L. Extrahepatic manifestations of hepatitis C among United States male veterans. Hepatology 2002; 36:1439–1445.
- Stefanova-Petrova DV, Tzvetanska AH, Naumova EJ, et al. Chronic hepatitis C virus infection: prevalence of extrahepatic manifestations and association with cryoglobulinemia in Bulgarian patients. World J Gastroenterol 2007; 13:6518–6528.
- Vassilopoulos D, Calabrese LH. Extrahepatic immunological complications of hepatitis C virus infection. AIDS 2005; 19( suppl 3):S123–S127.
- Hsing AW, Zhang M, Rashid A, et al. Hepatitis B and C virus infection and the risk of biliary tract cancer: a population-based study in China. Int J Cancer 2008; 122:1849–1853.
- Madrid C, Courtois B, Duran D. Chronic sialadenitis revealing hepatitis C: a case report. Med Oral 2004; 9:328–332.
- Lançoni G, Ravinal RC, Costa RS, Roselino AM. Mast cells and transforming growth factor-beta expression: a possible relationship in the development of porphyria cutanea tarda skin lesions. Int J Dermatol 2008; 47:575–581.
- Toll A, Celis R, Ozalla MD, Bruguera M, Herrero C, Ercilla MG. The prevalence of HFE C282Y gene mutation is increased in Spanish patients with porphyria cutanea tarda without hepatitis C virus infection. J Eur Acad Dermatol Venereol 2006; 20:1201–1206.
- Badminton MN, Elder GH. Management of acute and cutaneous porphyrias. Int J Clin Pract 2002; 56:272–278.
- Jackson JM, Callen JP. Scarring alopecia and sclerodermatous changes of the scalp in a patient with hepatitis C infection. J Am Acad Dermatol 1998; 39:824–826.
- Mortimore M, Merryweather-Clarke AT, Robson KJ, Powell LW. The haemochromatosis gene: a global perspective and implications for the Asia-Pacific region. J Gastroenterol Hepatol 1999; 14:838–843.
- Shehan JM, Huerter CJ. Porphyria cutanea tarda associated with an acute gastrointestinal bleed: the roles of supplemental iron and blood transfusion. Cutis 2001; 68:147–150.
- Lambrecht RW, Thapar M, Bonkovsky HL. Genetic aspects of porphyria cutanea tarda. Semin Liver Dis 2007; 27:99–108.
- d’Ovidio R, Sgarra C, Conserva A, Angelotti UF, Erriquez R, Foti C. Alterated integrin expression in lichen planopilaris. Head Face Med 2007; 3:11.
- Chuang TY, Stitle L, Brashear R, Lewis C. Hepatitis C virus and lichen planus: a case-control study of 340 patients. J Am Acad Dermatol 1999; 41:787–789.
- Kemp EH, Gavalas NG, Gawkrodger DJ, Weetman AP. Autoantibody responses to melanocytes in the depigmenting skin disease vitiligo. Autoimmun Rev 2007; 6:138–142.
- Tomasiewicz K, Modrzewska R, Semczuk G. Vitiligo associated with pegylated interferon and ribavirin treatment of patients with chronic hepatitis C: a case report. Adv Ther 2006; 23:139–142.
- Simsek H, Savas C, Akkiz H, Telatar H. Interferon-induced vitiligo in a patient with chronic viral hepatitis C infection. Dermatology 1996; 193:65–66.
- Mulekar SV, Al Issa A, Asaad M, Ghwish B, Al Eisa A. Mixed vitiligo. J Cutan Med Surg. 2006; 10:104–107.
- Waalen J, Felitti V, Gelbart T, Ho NJ, Beutler E. Prevalence of hemochromatosis-related symptoms among individuals with mutations in the HFE gene. Mayo Clin Proc 2002; 77:522–530.
- Walker AP, Tucker DC, Hall MA, et al. Results communication and patient education after screening for possible hemochromatosis and iron overload: experience from the HEIRS study of a large ethnically and linguistically diverse group. Genet Med 2007; 9:778–791.
- Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, cafe au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician 2003; 68:1955–1960.
- Tsuji T. Experimental hemosiderosis: relationship between skin pigmentation and hemosiderin. Acta Derm Venereol 1980; 60:109–114.
- Oji V, Traupe H. Ichthyoses: differential diagnosis and molecular genetics. Eur J Dermatol 2006; 16:349–359.
Dysfunction in the body’s second largest organ, the liver, often yields changes in the body’s largest organ, the skin. If we can recognize these manifestations early, we are better able to promptly diagnose and treat the underlying liver disease, as well as the skin lesions.
The liver has many jobs: synthesizing proteins such as clotting factors, complements, and albumin; neutralizing toxins; and metabolizing lipids and carbohydrates. Insults to the liver can compromise any of these functions, affecting visceral organs, joints, gastrointestinal tissues, and the skin. Dermatologic signs of specific liver diseases include alopecia and vitiligo associated with autoimmune hepatitis, and xanthelasma in chronic cholestatic liver disease.
This article reviews the important cutaneous manifestations of specific liver diseases. We focus first on skin conditions that may represent liver disease, and then we discuss several major liver diseases and their typical cutaneous manifestations.
JAUNDICE AND HYPERBILIRUBINEMIA
Establishing whether the excess bilirubin is conjugated or unconjugated gives a clue as to whether the cause is prehepatic, intrahepatic, or posthepatic.3–8 One of the liver’s main functions is to conjugate bilirubin into a secretable form. Prehepatic causes of jaundice include hemolysis and ineffective erythropoiesis, both of which lead to higher levels of circulating unconjugated bilirubin.4 Intrahepatic causes of jaundice can lead to both unconjugated and conjugated hyperbilirubinemia.4,8 Posthepatic causes such as bile duct obstruction primarily result in conjugated hyperbilirubinemia.4
PRURITUS AND PRURIGO NODULARIS
Pruritus can be multifactorial or the result of a specific dermatologic or systemic condition.9 A thorough history and physical examination are warranted to rule out hepatic or systemic causes of itching.10
The liver neutralizes toxins and filters bile salts. If its function is impaired, these materials can accumulate in the body, and deposition in the skin causes irritation and itching.11,12 In cholestatic liver disorders such as primary sclerosing cholangitis and obstructive gallstone disease, pruritus tends to be generalized, but worse on the hands and feet.13
Although the severity of pruritus is not directly associated with the level of bile salts and toxic substances, lowering bile salt levels can mitigate symptoms.11
Treatment. Pruritus due to liver disease is particularly resistant to therapy.
In a strategy described by Mela et al for managing pruritus in chronic liver disease,14 the initial treatment is the anion exchange resin cholestyramine (Questran) at a starting dose of 4 g/day, gradually increased to 24 g/day in two doses at mealtimes.
If the pruritus does not respond adequately to cholestyramine or the patient cannot tolerate the drug, then the antituberculosis drug rifampin (Rifadin) can be tried. Rifampin promotes metabolism of endogenous pruritogens and has been effective against cholestatic pruritus when started at 150 mg/day and increased up to 600 mg/day, depending on the clinical response.14
Third-line drug therapies include opioid antagonists such as naltrexone (ReVia) and nalmefene (Revex).14,15
Plasmapheresis can be considered if drug therapy fails.16 Experimental therapies include albumin dialysis using the molecular adsorbent recirculating system (a form of artificial liver support), antioxidant treatment, and bright-light therapy.15 Liver transplantation, when appropriate, also resolves cholestatic pruritus.14
Prurigo nodularis
Prurigo nodularis, distinguished by firm, crusty nodules, is associated with viral infections (eg, hepatitis C, human immunodeficiency virus), bacterial infections, and kidney dysfunction.17,18 The lesions are intensely pruritic and often lead to persistent scratching, excoriation, and, ultimately, diffuse scarring.19
Treatment. Although the exact cause of prurigo nodularis is not known and no cure exists, corticosteroid or antihistamine ointments control the symptoms in most patients with hepatitis.19 Low doses of thalidomide (Thalomid), a tumor necrosis factor antagonist, have also been used safely and effectively.18,19
SUPERFICIAL VASCULAR SIGNS
Spider angiomas
Spider angiomas can occur anywhere on the body, but they occur most often on the face and the trunk.21,23 A key feature is that they disappear when pressure is applied and reappear when pressure is removed.23,24 Biopsy is rarely necessary for diagnosis.
These lesions occur with elevated estrogen levels, such as in cirrhosis, during estrogen therapy, or during pregnancy.25–28 Although spider angiomas are common in pregnant women and in children, adults with spider angiomas deserve a workup for liver dysfunction.29
Given their innocuous nature and asymptomatic course, spider angiomas themselves require no medical treatment.
Bier spots
Bier spots are small, irregularly shaped, hypopigmented patches on the arms and legs. They are likely due to venous stasis associated with functional damage to the small vessels of the skin.30
Since Bier spots are a sign of liver disease, they must be distinguished from true pigmentation disorders. A key distinguishing feature is that Bier spots disappear when pressure is applied. Also, raising the affected limb from a dependent position causes the hypopigmented macules of Bier spots to disappear, which is not the case in true pigmentation disorders.10,30
Paper-money skin
Paper-money skin (or “dollar-paper” markings) describes the condition in which the upper trunk is covered with many randomly scattered, needle-thin superficial capillaries. It often occurs in association with spider angiomas. The name comes from the resemblance the thread-like capillaries have to the finely chopped silk threads in American dollar bills.10,31 The condition is commonly seen in patients with alcoholic cirrhosis and may improve with hemodialysis.31
PALMAR ERYTHEMA
Palmar erythema is a florid, crimson coloration of the palms of the hands and the fingertips. It can occur anywhere on the palm and fingers but is most common on the hypothenar eminence. It can occur in a number of liver conditions but most often with cirrhosis.32 Hepatic compromise, as seen in alcoholic liver disease, disrupts the body’s androgen balance, causing local vasodilation and erythema.32,33 Although the exact mechanism remains unknown, research suggests that prostacyclins and nitric oxide play a role, as both are increased in liver disease.32,33
XANTHELASMA
Xanthelasma—a localized cholesterol deposit beneath the skin and especially beneath the eyelids—is a common manifestation of hypercholesterolemia. Xanthelasma often presents as a painless, yellowish, soft plaque with well-defined borders,34 which may enlarge over the course of weeks.
Several liver diseases can lead to various forms of secondary dyslipoproteinemia.35 The most common dyslipoproteinemias in liver disease are hypertriglyceridemia and low levels of high-density lipoprotein cholesterol, and both of these often accompany fatty liver disease.36 Hypercholesterolemia is a common feature of primary biliary cirrhosis and other forms of cholestatic liver disease.37 Studies suggest that the total plasma cholesterol level is elevated in as many as 50% of patients with compromised liver function.38
Treatment. The underlying hyperlipidemia is treated with cholesterol-lowering drugs. Laser treatment and surgical excision have proven efficacious in treating the lesions.39
OTHER CUTANEOUS FINDINGS IN LIVER DISEASE
Bleeding and bruising. Liver disease can cause hypersplenism and thrombocytopenia, in addition to a decrease in clotting factors. These may present with a myriad of cutaneous symptoms, including purpura, bleeding gums, and easy bruising and bleeding, even from minor trauma.40–42
Hyperpigmentation of the skin may accompany hemochromatosis, alcoholic liver disease, and cirrhosis.43–45
“Terry’s nails,” in which the proximal two-thirds of the nail plate turns powdery white with a ground-glass opacity, may develop in patients with advanced cirrhosis.48
ALCOHOLIC CIRRHOSIS AND THE SKIN
The cutaneous changes associated with alcoholic cirrhosis are more widely recognized than those due to other forms of liver dysfunction. In the United States, approximately 3 million people have alcoholic cirrhosis, the second-leading reason for liver transplantation.49,50
As the body’s main site of alcohol metabolism, the liver is the organ most affected by excessive alcohol intake, which can lead to end-stage liver disease secondary to alcoholic cirrhosis.41,51 The characteristic feature of cirrhosis is advanced fibrous scarring of parenchymal tissue and the formation of regenerative nodules with increased resistance to blood flow throughout the organ.41,52 The insufficient blood flow damages vital structures in the liver and compromises liver function. For example, liver cirrhosis leads to defective hepatic synthesis of clotting factors and results in bleeding disorders.
Cutaneous lesions often accompany alcoholic cirrhosis and have been detected in up to 43% of people with chronic alcoholism.53 Skin changes in alcoholic cirrhosis can be of great diagnostic value. The combined prevalence of spider angiomas, palmar erythema, and Dupuytren contracture in alcoholic cirrhosis was found to be 72%. Paper-money skin and Dupuytren contracture are more distinct lesions for alcoholic cirrhosis.31 Recognizing these skin changes contributes to the diagnosis and staging of liver cirrhosis.51,52
Dupuytren contracture
Dupuytren contracture is characterized by progressive fibrosis and thickening of tendons in the palmar fascia, the connective tissue that lies beneath the skin of the palms.54 Over time, as fibrotic involvement expands across the fascia, rampant stiffness of the joints ensues, sometimes to a point where the fingers cannot fully flex or extend.54
Although the exact cause of Dupuytren contracture is unknown, it appears to be associated with excess alcohol consumption and can be found in patients with alcoholic cirrhosis.54,55 These patients often present with painless stiffness of the fingers, curling of fingers, and loss of motion in involved fingers.54 Surgery in the form of limited fasciectomy has been curative in such patients.54
Disseminated superficial porokeratosis
Porokeratosis is a keratinization disorder of clonal origin that presents as a linear configuration of white scaly papules that coalesce into plaques throughout the body.56 Although it most commonly afflicts fair-skinned people, patients with alcoholic cirrhosis have a much greater susceptibility than the general population.57,58
A recent study58 documented that the lesions completely resolved when liver function improved, thus underlining the relationship between the two conditions. Since immunosuppression has been linked to eruption of the lesion, the fact that both humoral and cell-mediated immune responses are impaired in alcoholic liver disease provides another dimension to the association between porokeratosis and alcoholic cirrhosis.58
These lesions can transform into squamous cell carcinoma.59 The risk of widespread metastases in squamous cell carcinoma highlights the importance of dermatologic consultation in such patients.59
HEPATITIS C AND THE SKIN
Extrahepatic manifestations have been documented in up to 74% of people with hepatitis C virus infection.60 In addition to parasthesias, arthralgias, and myalgias, hepatitis C has a significant association with porphyria cutanea tarda, lichen planus, vitiligo, sialadenitis, urticarial vasculitis, corneal ulcers, xerosis, pruritus, and prurigo nodularis.60–64 Although the primary causative agents of sialadenitis are bacteria, viruses such as hepatitis C have been implicated as a cause of chronic sialadenitis with associated xerostomia.65
Patients with hepatitis C being treated with interferons also present with cutaneous manifestations such as hyperkeratosis and vasculitis.63
Porphyria cutanea tarda
Porphyria cutanea tarda is the most common of the porphyrias, disorders distinguished by deficiencies or defects in one or more of the enzymes responsible for hepatic production of heme.66 If these enzymes are impaired, heme precursors such as porphyrins accumulate.66
Porphyria cutanea tarda results from a deficiency of the hepatic enzyme uroporphyrin decarboxylase. In the absence of this enzyme, shortwave visible light activates uroporphyrin deposited in the skin, resulting in a photochemical reaction that generates reactive oxygen species that lead to the characteristic skin blistering.
Although porphyria cutanea tarda is associated with liver disease in general, recent studies confirm that patients with hepatitis C are at particularly high risk.67 Those with the disorder often present with skin photosensitivity. 68 Many develop blisters on sun-exposed skin, including the dorsal aspects of the hands and forearms and on the neck and face. Chronic porphyria cutanea tarda can lead to scarring, alopecia, and skin ulceration.69 As the blisters heal, keratin-filled milial cysts may develop in the areas of ulceration.
The condition is also commonly associated with melasma-like hyperpigmentation and hypertrichosis in sun-exposed areas of the head and neck. People of Northern European ancestry may be more at risk than the general population because of a presumed genetic susceptibility.70
Treatment. Because many patients with porphyria cutanea tarda have iron overload, they need to restrict foods rich in iron and to avoid alcohol.71,72 Severe cases may necessitate iron removal via phlebotomy or antimalarial therapy. Patients with porphyria cutanea tarda induced by hepatitis C should have their bodily iron stores depleted before starting antiviral therapy.60
Lichen planus
Lichen planopilaris is a subset of lichen planus that causes scaling and atrophy of the scalp and permanent hair loss (Figure 4).73
Interferon-induced vitiligo
Vitiligo is an autoimmune disease in which melanocytes in the skin are destroyed, with resulting depigmentation in affected areas.75 Although it has no specific association with liver disease, it has been linked to treatments for hepatitis C such as interferons.76 Interferon-induced vitiligo often completely resolves when interferon is stopped.77
Typical findings include aggregations of irregularly shaped white patches in a focal or segmental pattern.78 The diagnosis is based on the medical history, physical examination, and sometimes skin biopsy.
HEMOCHROMATOSIS
Hemochromatosis or “bronze diabetes” is a devastating multisystem disease with a relentless course. It is among the most common genetic disorders of metabolism, and results in deposition of iron in tissues and organs throughout the body, including the liver, usually in patients ages 30 to 40.
As iron stores increase in tissues and organs, multiorgan failure and associated complications may ensue. In addition, surplus iron stores can also result in widespread bronze discoloration of skin exposed to the sun. Hemochromatosis also results in loss of body hair, ichthyosiform alterations, and koilonychia.79
Treatments that lower serum iron levels can reverse the cutaneous manifestations of the disorder and minimize the risk of organ failure.
Since the condition is inherited in an autosomal-recessive pattern, family members of patients should consider being screened.80
Hyperpigmentation in hemochromatosis
Hyperpigmentation is an early sign of hemochromatosis, affecting up to 90% of patients. Usually, sun-exposed areas of the body are the most prone and take on a grayish or brownish-bronze hue.81 Cutaneous iron deposits injure vital skin structures, initiating a process that culminates in enhanced melanin production by melanocytes.82 Exposure to ultraviolet light may have synergistic effects with iron, hastening the process of hyperpigmentation. As a result of this synergistic effect, many patients with hemochromatosis notice tanning with minimal sun exposure.
Although organ function can improve immediately with phlebotomy to reduce iron stores, skin hyperpigmentation does not immediately resolve.81,82
Ichthyosiform alterations in hemochromatosis
Ichthyosiform changes, in which the skin takes on the appearance of fish scales,83 can be seen in patients with hemochromatosis.80 Affected areas typically become extremely dry. Treatment includes topical hydrating creams and ointments. Avoiding sunlight is paramount, as sunlight exposure may exacerbate the condition.83
Dysfunction in the body’s second largest organ, the liver, often yields changes in the body’s largest organ, the skin. If we can recognize these manifestations early, we are better able to promptly diagnose and treat the underlying liver disease, as well as the skin lesions.
The liver has many jobs: synthesizing proteins such as clotting factors, complements, and albumin; neutralizing toxins; and metabolizing lipids and carbohydrates. Insults to the liver can compromise any of these functions, affecting visceral organs, joints, gastrointestinal tissues, and the skin. Dermatologic signs of specific liver diseases include alopecia and vitiligo associated with autoimmune hepatitis, and xanthelasma in chronic cholestatic liver disease.
This article reviews the important cutaneous manifestations of specific liver diseases. We focus first on skin conditions that may represent liver disease, and then we discuss several major liver diseases and their typical cutaneous manifestations.
JAUNDICE AND HYPERBILIRUBINEMIA
Establishing whether the excess bilirubin is conjugated or unconjugated gives a clue as to whether the cause is prehepatic, intrahepatic, or posthepatic.3–8 One of the liver’s main functions is to conjugate bilirubin into a secretable form. Prehepatic causes of jaundice include hemolysis and ineffective erythropoiesis, both of which lead to higher levels of circulating unconjugated bilirubin.4 Intrahepatic causes of jaundice can lead to both unconjugated and conjugated hyperbilirubinemia.4,8 Posthepatic causes such as bile duct obstruction primarily result in conjugated hyperbilirubinemia.4
PRURITUS AND PRURIGO NODULARIS
Pruritus can be multifactorial or the result of a specific dermatologic or systemic condition.9 A thorough history and physical examination are warranted to rule out hepatic or systemic causes of itching.10
The liver neutralizes toxins and filters bile salts. If its function is impaired, these materials can accumulate in the body, and deposition in the skin causes irritation and itching.11,12 In cholestatic liver disorders such as primary sclerosing cholangitis and obstructive gallstone disease, pruritus tends to be generalized, but worse on the hands and feet.13
Although the severity of pruritus is not directly associated with the level of bile salts and toxic substances, lowering bile salt levels can mitigate symptoms.11
Treatment. Pruritus due to liver disease is particularly resistant to therapy.
In a strategy described by Mela et al for managing pruritus in chronic liver disease,14 the initial treatment is the anion exchange resin cholestyramine (Questran) at a starting dose of 4 g/day, gradually increased to 24 g/day in two doses at mealtimes.
If the pruritus does not respond adequately to cholestyramine or the patient cannot tolerate the drug, then the antituberculosis drug rifampin (Rifadin) can be tried. Rifampin promotes metabolism of endogenous pruritogens and has been effective against cholestatic pruritus when started at 150 mg/day and increased up to 600 mg/day, depending on the clinical response.14
Third-line drug therapies include opioid antagonists such as naltrexone (ReVia) and nalmefene (Revex).14,15
Plasmapheresis can be considered if drug therapy fails.16 Experimental therapies include albumin dialysis using the molecular adsorbent recirculating system (a form of artificial liver support), antioxidant treatment, and bright-light therapy.15 Liver transplantation, when appropriate, also resolves cholestatic pruritus.14
Prurigo nodularis
Prurigo nodularis, distinguished by firm, crusty nodules, is associated with viral infections (eg, hepatitis C, human immunodeficiency virus), bacterial infections, and kidney dysfunction.17,18 The lesions are intensely pruritic and often lead to persistent scratching, excoriation, and, ultimately, diffuse scarring.19
Treatment. Although the exact cause of prurigo nodularis is not known and no cure exists, corticosteroid or antihistamine ointments control the symptoms in most patients with hepatitis.19 Low doses of thalidomide (Thalomid), a tumor necrosis factor antagonist, have also been used safely and effectively.18,19
SUPERFICIAL VASCULAR SIGNS
Spider angiomas
Spider angiomas can occur anywhere on the body, but they occur most often on the face and the trunk.21,23 A key feature is that they disappear when pressure is applied and reappear when pressure is removed.23,24 Biopsy is rarely necessary for diagnosis.
These lesions occur with elevated estrogen levels, such as in cirrhosis, during estrogen therapy, or during pregnancy.25–28 Although spider angiomas are common in pregnant women and in children, adults with spider angiomas deserve a workup for liver dysfunction.29
Given their innocuous nature and asymptomatic course, spider angiomas themselves require no medical treatment.
Bier spots
Bier spots are small, irregularly shaped, hypopigmented patches on the arms and legs. They are likely due to venous stasis associated with functional damage to the small vessels of the skin.30
Since Bier spots are a sign of liver disease, they must be distinguished from true pigmentation disorders. A key distinguishing feature is that Bier spots disappear when pressure is applied. Also, raising the affected limb from a dependent position causes the hypopigmented macules of Bier spots to disappear, which is not the case in true pigmentation disorders.10,30
Paper-money skin
Paper-money skin (or “dollar-paper” markings) describes the condition in which the upper trunk is covered with many randomly scattered, needle-thin superficial capillaries. It often occurs in association with spider angiomas. The name comes from the resemblance the thread-like capillaries have to the finely chopped silk threads in American dollar bills.10,31 The condition is commonly seen in patients with alcoholic cirrhosis and may improve with hemodialysis.31
PALMAR ERYTHEMA
Palmar erythema is a florid, crimson coloration of the palms of the hands and the fingertips. It can occur anywhere on the palm and fingers but is most common on the hypothenar eminence. It can occur in a number of liver conditions but most often with cirrhosis.32 Hepatic compromise, as seen in alcoholic liver disease, disrupts the body’s androgen balance, causing local vasodilation and erythema.32,33 Although the exact mechanism remains unknown, research suggests that prostacyclins and nitric oxide play a role, as both are increased in liver disease.32,33
XANTHELASMA
Xanthelasma—a localized cholesterol deposit beneath the skin and especially beneath the eyelids—is a common manifestation of hypercholesterolemia. Xanthelasma often presents as a painless, yellowish, soft plaque with well-defined borders,34 which may enlarge over the course of weeks.
Several liver diseases can lead to various forms of secondary dyslipoproteinemia.35 The most common dyslipoproteinemias in liver disease are hypertriglyceridemia and low levels of high-density lipoprotein cholesterol, and both of these often accompany fatty liver disease.36 Hypercholesterolemia is a common feature of primary biliary cirrhosis and other forms of cholestatic liver disease.37 Studies suggest that the total plasma cholesterol level is elevated in as many as 50% of patients with compromised liver function.38
Treatment. The underlying hyperlipidemia is treated with cholesterol-lowering drugs. Laser treatment and surgical excision have proven efficacious in treating the lesions.39
OTHER CUTANEOUS FINDINGS IN LIVER DISEASE
Bleeding and bruising. Liver disease can cause hypersplenism and thrombocytopenia, in addition to a decrease in clotting factors. These may present with a myriad of cutaneous symptoms, including purpura, bleeding gums, and easy bruising and bleeding, even from minor trauma.40–42
Hyperpigmentation of the skin may accompany hemochromatosis, alcoholic liver disease, and cirrhosis.43–45
“Terry’s nails,” in which the proximal two-thirds of the nail plate turns powdery white with a ground-glass opacity, may develop in patients with advanced cirrhosis.48
ALCOHOLIC CIRRHOSIS AND THE SKIN
The cutaneous changes associated with alcoholic cirrhosis are more widely recognized than those due to other forms of liver dysfunction. In the United States, approximately 3 million people have alcoholic cirrhosis, the second-leading reason for liver transplantation.49,50
As the body’s main site of alcohol metabolism, the liver is the organ most affected by excessive alcohol intake, which can lead to end-stage liver disease secondary to alcoholic cirrhosis.41,51 The characteristic feature of cirrhosis is advanced fibrous scarring of parenchymal tissue and the formation of regenerative nodules with increased resistance to blood flow throughout the organ.41,52 The insufficient blood flow damages vital structures in the liver and compromises liver function. For example, liver cirrhosis leads to defective hepatic synthesis of clotting factors and results in bleeding disorders.
Cutaneous lesions often accompany alcoholic cirrhosis and have been detected in up to 43% of people with chronic alcoholism.53 Skin changes in alcoholic cirrhosis can be of great diagnostic value. The combined prevalence of spider angiomas, palmar erythema, and Dupuytren contracture in alcoholic cirrhosis was found to be 72%. Paper-money skin and Dupuytren contracture are more distinct lesions for alcoholic cirrhosis.31 Recognizing these skin changes contributes to the diagnosis and staging of liver cirrhosis.51,52
Dupuytren contracture
Dupuytren contracture is characterized by progressive fibrosis and thickening of tendons in the palmar fascia, the connective tissue that lies beneath the skin of the palms.54 Over time, as fibrotic involvement expands across the fascia, rampant stiffness of the joints ensues, sometimes to a point where the fingers cannot fully flex or extend.54
Although the exact cause of Dupuytren contracture is unknown, it appears to be associated with excess alcohol consumption and can be found in patients with alcoholic cirrhosis.54,55 These patients often present with painless stiffness of the fingers, curling of fingers, and loss of motion in involved fingers.54 Surgery in the form of limited fasciectomy has been curative in such patients.54
Disseminated superficial porokeratosis
Porokeratosis is a keratinization disorder of clonal origin that presents as a linear configuration of white scaly papules that coalesce into plaques throughout the body.56 Although it most commonly afflicts fair-skinned people, patients with alcoholic cirrhosis have a much greater susceptibility than the general population.57,58
A recent study58 documented that the lesions completely resolved when liver function improved, thus underlining the relationship between the two conditions. Since immunosuppression has been linked to eruption of the lesion, the fact that both humoral and cell-mediated immune responses are impaired in alcoholic liver disease provides another dimension to the association between porokeratosis and alcoholic cirrhosis.58
These lesions can transform into squamous cell carcinoma.59 The risk of widespread metastases in squamous cell carcinoma highlights the importance of dermatologic consultation in such patients.59
HEPATITIS C AND THE SKIN
Extrahepatic manifestations have been documented in up to 74% of people with hepatitis C virus infection.60 In addition to parasthesias, arthralgias, and myalgias, hepatitis C has a significant association with porphyria cutanea tarda, lichen planus, vitiligo, sialadenitis, urticarial vasculitis, corneal ulcers, xerosis, pruritus, and prurigo nodularis.60–64 Although the primary causative agents of sialadenitis are bacteria, viruses such as hepatitis C have been implicated as a cause of chronic sialadenitis with associated xerostomia.65
Patients with hepatitis C being treated with interferons also present with cutaneous manifestations such as hyperkeratosis and vasculitis.63
Porphyria cutanea tarda
Porphyria cutanea tarda is the most common of the porphyrias, disorders distinguished by deficiencies or defects in one or more of the enzymes responsible for hepatic production of heme.66 If these enzymes are impaired, heme precursors such as porphyrins accumulate.66
Porphyria cutanea tarda results from a deficiency of the hepatic enzyme uroporphyrin decarboxylase. In the absence of this enzyme, shortwave visible light activates uroporphyrin deposited in the skin, resulting in a photochemical reaction that generates reactive oxygen species that lead to the characteristic skin blistering.
Although porphyria cutanea tarda is associated with liver disease in general, recent studies confirm that patients with hepatitis C are at particularly high risk.67 Those with the disorder often present with skin photosensitivity. 68 Many develop blisters on sun-exposed skin, including the dorsal aspects of the hands and forearms and on the neck and face. Chronic porphyria cutanea tarda can lead to scarring, alopecia, and skin ulceration.69 As the blisters heal, keratin-filled milial cysts may develop in the areas of ulceration.
The condition is also commonly associated with melasma-like hyperpigmentation and hypertrichosis in sun-exposed areas of the head and neck. People of Northern European ancestry may be more at risk than the general population because of a presumed genetic susceptibility.70
Treatment. Because many patients with porphyria cutanea tarda have iron overload, they need to restrict foods rich in iron and to avoid alcohol.71,72 Severe cases may necessitate iron removal via phlebotomy or antimalarial therapy. Patients with porphyria cutanea tarda induced by hepatitis C should have their bodily iron stores depleted before starting antiviral therapy.60
Lichen planus
Lichen planopilaris is a subset of lichen planus that causes scaling and atrophy of the scalp and permanent hair loss (Figure 4).73
Interferon-induced vitiligo
Vitiligo is an autoimmune disease in which melanocytes in the skin are destroyed, with resulting depigmentation in affected areas.75 Although it has no specific association with liver disease, it has been linked to treatments for hepatitis C such as interferons.76 Interferon-induced vitiligo often completely resolves when interferon is stopped.77
Typical findings include aggregations of irregularly shaped white patches in a focal or segmental pattern.78 The diagnosis is based on the medical history, physical examination, and sometimes skin biopsy.
HEMOCHROMATOSIS
Hemochromatosis or “bronze diabetes” is a devastating multisystem disease with a relentless course. It is among the most common genetic disorders of metabolism, and results in deposition of iron in tissues and organs throughout the body, including the liver, usually in patients ages 30 to 40.
As iron stores increase in tissues and organs, multiorgan failure and associated complications may ensue. In addition, surplus iron stores can also result in widespread bronze discoloration of skin exposed to the sun. Hemochromatosis also results in loss of body hair, ichthyosiform alterations, and koilonychia.79
Treatments that lower serum iron levels can reverse the cutaneous manifestations of the disorder and minimize the risk of organ failure.
Since the condition is inherited in an autosomal-recessive pattern, family members of patients should consider being screened.80
Hyperpigmentation in hemochromatosis
Hyperpigmentation is an early sign of hemochromatosis, affecting up to 90% of patients. Usually, sun-exposed areas of the body are the most prone and take on a grayish or brownish-bronze hue.81 Cutaneous iron deposits injure vital skin structures, initiating a process that culminates in enhanced melanin production by melanocytes.82 Exposure to ultraviolet light may have synergistic effects with iron, hastening the process of hyperpigmentation. As a result of this synergistic effect, many patients with hemochromatosis notice tanning with minimal sun exposure.
Although organ function can improve immediately with phlebotomy to reduce iron stores, skin hyperpigmentation does not immediately resolve.81,82
Ichthyosiform alterations in hemochromatosis
Ichthyosiform changes, in which the skin takes on the appearance of fish scales,83 can be seen in patients with hemochromatosis.80 Affected areas typically become extremely dry. Treatment includes topical hydrating creams and ointments. Avoiding sunlight is paramount, as sunlight exposure may exacerbate the condition.83
- Morioka D, Togo S, Kumamoto T, et al. Six consecutive cases of successful adult ABO-incompatible living donor liver transplantation: a proposal for grading the severity of antibody-mediated rejection. Transplantation 2008; 85:171–178.
- Bertini G, Rubaltelli FF. Non-invasive bilirubinometry in neonatal jaundice. Semin Neonatol 2002; 7:129–133.
- Clementi M, Di Gianantonio E, Fabris L, et al. Inheritance of hyperbilirubinemia: evidence for a major autosomal recessive gene. Dig Liver Dis 2007; 39:351–355.
- Roche SP, Kobos R. Jaundice in the adult patient. Am Fam Physician 2004; 69:299–304.
- Odemis B, Parlak E, Basar O, Yüksel O, Sahin B. Biliary tract obstruction secondary to malignant lymphoma: experience at a referral center. Dig Dis Sci 2007; 52:2323–2332.
- Caddy GR, Tham TC. Gallstone disease: symptoms, diagnosis and endoscopic management of common bile duct stones. Best Pract Res Clin Gastroenterol 2006; 20:1085–1101.
- Heathcote EJ. Diagnosis and management of cholestatic liver disease. Clin Gastroenterol Hepatol 2007; 5:776–782.
- Faust TW, Reddy KR. Postoperative jaundice. Clin Liver Dis 2004; 8:151–166.
- Maticic M, Poljak M, Lunder T, Rener-Sitar K, Stojanovic L. Lichen planus and other cutaneous manifestations in chronic hepatitis C: pre- and post-interferon-based treatment prevalence vary in a cohort of patients from low hepatitis C virus endemic area. J Eur Acad Dermatol Venereol 2008; 22:779–788.
- Polat M, Oztas P, Ilhan MN, Yalçin B, Alli N. Generalized pruritus: a prospective study concerning etiology. Am J Clin Dermatol 2008; 9:39–44.
- Gaspari R, Avolio AW, Zileri Dal Verme L, et al. Molecular adsorbent recirculating system in liver transplantation: safety and efficacy. Transplant Proc 2006; 38:3544–3551.
- Dasgupta R, Saha I, Pal S, et al. Immunosuppression, hepatotoxicity and depression of antioxidant status by arecoline in albino mice. Toxicology 2006; 227:94–104.
- Mayo MJ, Handem I, Saldana S, Jacobe H, Getachew Y, Rush AJ. Sertraline as a first-line treatment for cholestatic pruritus. Hepatology 2007; 45:666–674.
- Mela M, Mancuso A, Burroughs AK. Review article: pruritus in cholestatic and other liver diseases. Aliment Pharmacol Ther 2003; 17:857–870.
- Montero JL, Pozo JC, Barrera P, et al. Treatment of refractory cholestatic pruritus with molecular adsorbent recirculating system (MARS). Transplant Proc 2006; 38:2511–2513.
- Neff GW, O'Brien CB, Reddy KR, et al. Preliminary observation with dronabinol in patients with intractable pruritus secondary to cholestatic liver disease. Am J Gastroenterol 2002; 97:2117–2119.
- Neri S, Raciti C, D’Angelo G, Ierna D, Bruno CM. Hyde’s prurigo nodularis and chronic HCV hepatitis. J Hepatol 1998; 28:161–164.
- Brown MA, George CR, Dunstan CR, Kalowski S, Corrigan AB. Prurigo nodularis and aluminium overload in maintenance haemodialysis. Lancet 1992; 340:48.
- Stander S, Luger T, Metze D. Treatment of prurigo nodularis with topical capsaicin. J Am Acad Dermatol 2001; 44:471–478.
- Requena L, Sangueza OP. Cutaneous vascular anomalies. Part I. Hamartomas, malformations, and dilation of preexisting vessels. J Am Acad Dermatol 1997; 37:523–549.
- Khasnis A, Gokula RM. Spider nevus. J Postgrad Med 2002; 48:307–309.
- Kaul V, Friedenberg FK, Braitman LE, et al. Development and validation of a model to diagnose cirrhosis in patients with hepatitis C. Am J Gastroenterol 2002; 97:2623–2628.
- Banyai AL. There is more than surface appearance to skin spiders. Chest 1971; 60:48.
- Errickson CV, Matus NR. Skin disorders of pregnancy. Am Fam Physician 1994; 49:605–610.
- Li CP, Lee FY, Hwang SJ, et al. Spider angiomas in patients with liver cirrhosis: role of alcoholism and impaired liver function. Scand J Gastroenterol 1999; 34:520–523.
- Li CP, Lee FY, Hwang SJ, et al. Role of substance P in the pathogenesis of spider angiomas in patients with nonalcoholic liver cirrhosis. Am J Gastroenterol 1999; 94:502–507.
- Sadick NS, Niedt GW. A study of estrogen and progesterone receptors in spider telangiectasias of the lower extremities. J Dermatol Surg Oncol 1990; 16:620–623.
- Henry F, Quatresooz P, Valverde-Lopez JC, Pierard GE. Blood vessel changes during pregnancy: a review. Am J Clin Dermatol 2006; 7:65–69.
- Finn SM, Rowland M, Lawlor F, et al. The significance of cutaneous spider naevi in children. Arch Dis Child 2006; 91:604–605.
- Peyrot I, Boulinguez S, Sparsa A, Le Meur Y, Bonnetblanc JM, Bedane C. Bier’s white spots associated with scleroderma renal crisis. Clin Exp Dermatol 2007; 32:165–167.
- Satoh T, Yokozeki H, Nishioka K. Vascular spiders and paper money skin improved by hemodialysis. Dermatology 2002; 205:73–74.
- Serrao R, Zirwas M, English JC. Palmar erythema. Am J Clin Dermatol 2007; 8:347–356.
- Matsumoto M, Ohki K, Nagai I, Oshibuchi T. Lung traction causes an increase in plasma prostacyclin concentration and decrease in mean arterial blood pressure. Anesth Analg 1992; 75:773–776.
- Otto AI, Horvath I, Feldmann J. Multiple firm, painless erythematous papules with a yellowish hue. Arch Dermatol 2005; 141:1595–1600.
- Gandelman G, Aronow WS, Weiss MB. Resolving hyperlipidemia after liver transplantation in a patient with primary sclerosing cholangitis. Am J Ther 2006; 13:171–174.
- Assy N, Kaita K, Mymin D, Levy C, Rosser B, Minuk G. Fatty infiltration of liver in hyperlipidemic patients. Dig Dis Sci 2000; 45:1929–1934.
- Allocca M, Crosignani A, Gritti A, et al. Hypercholesterolaemia is not associated with early atherosclerotic lesions in primary biliary cirrhosis. Gut 2006; 55:1795–1800.
- Dickson E, Fleming C, Ludwig J. Primary biliary cirrhosis. In:Popper H, Schaffner F, editors. Progress in Liver Diseases. New York: Grune and Stratton; 1978:487.
- Elner VM, Mintz R, Demirci H, Hassan AS. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Trans Am Ophthalmol Soc 2005; 103:69–73.
- Craxi A, Camma C, Giunta M. Clinical aspects of bleeding complications in cirrhotic patients. Blood Coagul Fibrinolysis 2000; 11( suppl 1):S75–579.
- Kajihara M, Okazaki Y, Kato S, et al. Evaluation of platelet kinetics in patients with liver cirrhosis: similarity to idiopathic thrombocytopenic purpura. J Gastroenterol Hepatol 2007; 22:112–118.
- Levine N. Patient reports six-month history of minimally pruritic purple dots on legs. Non-blanching macules developed over six months. Geriatrics 2006; 61:22.
- Barton JC, Rao SV, Pereira NM, et al. Juvenile hemochromatosis in the southeastern United States: a report of seven cases in two kinships. Blood Cells Mol Dis 2002; 29:104–115.
- Smith AG, Shuster S, Bomford A, Williams R. Plasma immunoreactive beta-melanocyte-stimulating hormone in chronic liver disease and fulminant hepatic failure. J Invest Dermatol 1978; 70:326–327.
- Barton JC, McDonnell SM, Adams PC, et al. Management of hemochromatosis. Hemochromatosis Management Working Group. Ann Intern Med 1998; 129:932–939.
- Bahnsen M, Gluud C, Johnsen SG, et al. Pituitary-testicular function in patients with alcoholic cirrhosis of the liver. Eur J Clin Invest 1981; 11:473–479.
- Kumar N, Aggarwal SR, Anand BS. Comparison of truncal hair distribution in alcoholic liver disease and alcohol-related chronic pancreatitis. J Gastroenterol Hepatol 2001; 16:855–856.
- Holzberg M, Walker HK. Terry's nails: revised definition and new correlations. Lancet 1984; 1:896–899.
- Mandayam S, Jamal MM, Morgan TR. Epidemiology of alcoholic liver disease. Semin Liver Dis 2004; 24:217–232.
- Belle SH, Beringer KC, Detre KM. Liver transplantation in the United States: results from the National Pitt-UNOS Liver Transplant Registry. United Network for Organ Sharing Clin Transpl 1994:19–35.
- Dunn W, Xu R, Schwimmer JB. Modest wine drinking and decreased prevalence of suspected nonalcoholic fatty liver disease. Hepatology 2008; 47:1947–1954.
- Afford SC, Fisher NC, Neil DA, et al. Distinct patterns of chemokine expression are associated with leukocyte recruitment in alcoholic hepatitis and alcoholic cirrhosis. J Pathol 1998; 186:82–89.
- Evstaf’ev VV, Levin MM. Dermatologic pathology in chronic alcoholics. Vestn Dermatol Venerol 1989; 8:72–74.
- Jerosch-Herold C, Shepstone L, Chojnowski AJ, Larson D. Splinting after contracture release for Dupuytren's contracture (SCoRD): protocol of a pragmatic, multi-centre, randomized controlled trial. BMC Musculoskelet Disord 2008; 9:62.
- Houghton S, Holdstock G, Cockerell R, Wright R. Dupuytren’s contracture, chronic liver disease and IgA immune complexes. Liver 1983; 3:220–224.
- Ibbotson SH. Disseminated superficial porokeratosis: what is the association with ultraviolet radiation? Clin Exp Dermatol 1996; 21:48–50.
- Kono T, Kobayashi H, Ishii M, Nishiguchi S, Taniguchi S. Synchronous development of disseminated superficial porokeratosis and hepatitis C virus-related hepatocellular carcinoma. J Am Acad Dermatol 2000; 43:966–968.
- Park BS, Moon SE, Kim JA. Disseminated superficial porokeratosis in a patient with chronic liver disease. J Dermatol 1997; 24:485–487.
- Murata Y, Kumano K, Takai T. Type 2 segmental manifestation of disseminated superficial porokeratosis showing a systematized pattern of involvement and pronounced cancer proneness. Eur J Dermatol 2001; 11:191–194.
- Galossi A, Guarisco R, Bellis L, Puoti C. Extrahepatic manifestations of chronic HCV infection. J Gastrointestin Liver Dis 2007; 16:65–73.
- El-Serag HB, Hampel H, Yeh C, Rabeneck L. Extrahepatic manifestations of hepatitis C among United States male veterans. Hepatology 2002; 36:1439–1445.
- Stefanova-Petrova DV, Tzvetanska AH, Naumova EJ, et al. Chronic hepatitis C virus infection: prevalence of extrahepatic manifestations and association with cryoglobulinemia in Bulgarian patients. World J Gastroenterol 2007; 13:6518–6528.
- Vassilopoulos D, Calabrese LH. Extrahepatic immunological complications of hepatitis C virus infection. AIDS 2005; 19( suppl 3):S123–S127.
- Hsing AW, Zhang M, Rashid A, et al. Hepatitis B and C virus infection and the risk of biliary tract cancer: a population-based study in China. Int J Cancer 2008; 122:1849–1853.
- Madrid C, Courtois B, Duran D. Chronic sialadenitis revealing hepatitis C: a case report. Med Oral 2004; 9:328–332.
- Lançoni G, Ravinal RC, Costa RS, Roselino AM. Mast cells and transforming growth factor-beta expression: a possible relationship in the development of porphyria cutanea tarda skin lesions. Int J Dermatol 2008; 47:575–581.
- Toll A, Celis R, Ozalla MD, Bruguera M, Herrero C, Ercilla MG. The prevalence of HFE C282Y gene mutation is increased in Spanish patients with porphyria cutanea tarda without hepatitis C virus infection. J Eur Acad Dermatol Venereol 2006; 20:1201–1206.
- Badminton MN, Elder GH. Management of acute and cutaneous porphyrias. Int J Clin Pract 2002; 56:272–278.
- Jackson JM, Callen JP. Scarring alopecia and sclerodermatous changes of the scalp in a patient with hepatitis C infection. J Am Acad Dermatol 1998; 39:824–826.
- Mortimore M, Merryweather-Clarke AT, Robson KJ, Powell LW. The haemochromatosis gene: a global perspective and implications for the Asia-Pacific region. J Gastroenterol Hepatol 1999; 14:838–843.
- Shehan JM, Huerter CJ. Porphyria cutanea tarda associated with an acute gastrointestinal bleed: the roles of supplemental iron and blood transfusion. Cutis 2001; 68:147–150.
- Lambrecht RW, Thapar M, Bonkovsky HL. Genetic aspects of porphyria cutanea tarda. Semin Liver Dis 2007; 27:99–108.
- d’Ovidio R, Sgarra C, Conserva A, Angelotti UF, Erriquez R, Foti C. Alterated integrin expression in lichen planopilaris. Head Face Med 2007; 3:11.
- Chuang TY, Stitle L, Brashear R, Lewis C. Hepatitis C virus and lichen planus: a case-control study of 340 patients. J Am Acad Dermatol 1999; 41:787–789.
- Kemp EH, Gavalas NG, Gawkrodger DJ, Weetman AP. Autoantibody responses to melanocytes in the depigmenting skin disease vitiligo. Autoimmun Rev 2007; 6:138–142.
- Tomasiewicz K, Modrzewska R, Semczuk G. Vitiligo associated with pegylated interferon and ribavirin treatment of patients with chronic hepatitis C: a case report. Adv Ther 2006; 23:139–142.
- Simsek H, Savas C, Akkiz H, Telatar H. Interferon-induced vitiligo in a patient with chronic viral hepatitis C infection. Dermatology 1996; 193:65–66.
- Mulekar SV, Al Issa A, Asaad M, Ghwish B, Al Eisa A. Mixed vitiligo. J Cutan Med Surg. 2006; 10:104–107.
- Waalen J, Felitti V, Gelbart T, Ho NJ, Beutler E. Prevalence of hemochromatosis-related symptoms among individuals with mutations in the HFE gene. Mayo Clin Proc 2002; 77:522–530.
- Walker AP, Tucker DC, Hall MA, et al. Results communication and patient education after screening for possible hemochromatosis and iron overload: experience from the HEIRS study of a large ethnically and linguistically diverse group. Genet Med 2007; 9:778–791.
- Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, cafe au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician 2003; 68:1955–1960.
- Tsuji T. Experimental hemosiderosis: relationship between skin pigmentation and hemosiderin. Acta Derm Venereol 1980; 60:109–114.
- Oji V, Traupe H. Ichthyoses: differential diagnosis and molecular genetics. Eur J Dermatol 2006; 16:349–359.
- Morioka D, Togo S, Kumamoto T, et al. Six consecutive cases of successful adult ABO-incompatible living donor liver transplantation: a proposal for grading the severity of antibody-mediated rejection. Transplantation 2008; 85:171–178.
- Bertini G, Rubaltelli FF. Non-invasive bilirubinometry in neonatal jaundice. Semin Neonatol 2002; 7:129–133.
- Clementi M, Di Gianantonio E, Fabris L, et al. Inheritance of hyperbilirubinemia: evidence for a major autosomal recessive gene. Dig Liver Dis 2007; 39:351–355.
- Roche SP, Kobos R. Jaundice in the adult patient. Am Fam Physician 2004; 69:299–304.
- Odemis B, Parlak E, Basar O, Yüksel O, Sahin B. Biliary tract obstruction secondary to malignant lymphoma: experience at a referral center. Dig Dis Sci 2007; 52:2323–2332.
- Caddy GR, Tham TC. Gallstone disease: symptoms, diagnosis and endoscopic management of common bile duct stones. Best Pract Res Clin Gastroenterol 2006; 20:1085–1101.
- Heathcote EJ. Diagnosis and management of cholestatic liver disease. Clin Gastroenterol Hepatol 2007; 5:776–782.
- Faust TW, Reddy KR. Postoperative jaundice. Clin Liver Dis 2004; 8:151–166.
- Maticic M, Poljak M, Lunder T, Rener-Sitar K, Stojanovic L. Lichen planus and other cutaneous manifestations in chronic hepatitis C: pre- and post-interferon-based treatment prevalence vary in a cohort of patients from low hepatitis C virus endemic area. J Eur Acad Dermatol Venereol 2008; 22:779–788.
- Polat M, Oztas P, Ilhan MN, Yalçin B, Alli N. Generalized pruritus: a prospective study concerning etiology. Am J Clin Dermatol 2008; 9:39–44.
- Gaspari R, Avolio AW, Zileri Dal Verme L, et al. Molecular adsorbent recirculating system in liver transplantation: safety and efficacy. Transplant Proc 2006; 38:3544–3551.
- Dasgupta R, Saha I, Pal S, et al. Immunosuppression, hepatotoxicity and depression of antioxidant status by arecoline in albino mice. Toxicology 2006; 227:94–104.
- Mayo MJ, Handem I, Saldana S, Jacobe H, Getachew Y, Rush AJ. Sertraline as a first-line treatment for cholestatic pruritus. Hepatology 2007; 45:666–674.
- Mela M, Mancuso A, Burroughs AK. Review article: pruritus in cholestatic and other liver diseases. Aliment Pharmacol Ther 2003; 17:857–870.
- Montero JL, Pozo JC, Barrera P, et al. Treatment of refractory cholestatic pruritus with molecular adsorbent recirculating system (MARS). Transplant Proc 2006; 38:2511–2513.
- Neff GW, O'Brien CB, Reddy KR, et al. Preliminary observation with dronabinol in patients with intractable pruritus secondary to cholestatic liver disease. Am J Gastroenterol 2002; 97:2117–2119.
- Neri S, Raciti C, D’Angelo G, Ierna D, Bruno CM. Hyde’s prurigo nodularis and chronic HCV hepatitis. J Hepatol 1998; 28:161–164.
- Brown MA, George CR, Dunstan CR, Kalowski S, Corrigan AB. Prurigo nodularis and aluminium overload in maintenance haemodialysis. Lancet 1992; 340:48.
- Stander S, Luger T, Metze D. Treatment of prurigo nodularis with topical capsaicin. J Am Acad Dermatol 2001; 44:471–478.
- Requena L, Sangueza OP. Cutaneous vascular anomalies. Part I. Hamartomas, malformations, and dilation of preexisting vessels. J Am Acad Dermatol 1997; 37:523–549.
- Khasnis A, Gokula RM. Spider nevus. J Postgrad Med 2002; 48:307–309.
- Kaul V, Friedenberg FK, Braitman LE, et al. Development and validation of a model to diagnose cirrhosis in patients with hepatitis C. Am J Gastroenterol 2002; 97:2623–2628.
- Banyai AL. There is more than surface appearance to skin spiders. Chest 1971; 60:48.
- Errickson CV, Matus NR. Skin disorders of pregnancy. Am Fam Physician 1994; 49:605–610.
- Li CP, Lee FY, Hwang SJ, et al. Spider angiomas in patients with liver cirrhosis: role of alcoholism and impaired liver function. Scand J Gastroenterol 1999; 34:520–523.
- Li CP, Lee FY, Hwang SJ, et al. Role of substance P in the pathogenesis of spider angiomas in patients with nonalcoholic liver cirrhosis. Am J Gastroenterol 1999; 94:502–507.
- Sadick NS, Niedt GW. A study of estrogen and progesterone receptors in spider telangiectasias of the lower extremities. J Dermatol Surg Oncol 1990; 16:620–623.
- Henry F, Quatresooz P, Valverde-Lopez JC, Pierard GE. Blood vessel changes during pregnancy: a review. Am J Clin Dermatol 2006; 7:65–69.
- Finn SM, Rowland M, Lawlor F, et al. The significance of cutaneous spider naevi in children. Arch Dis Child 2006; 91:604–605.
- Peyrot I, Boulinguez S, Sparsa A, Le Meur Y, Bonnetblanc JM, Bedane C. Bier’s white spots associated with scleroderma renal crisis. Clin Exp Dermatol 2007; 32:165–167.
- Satoh T, Yokozeki H, Nishioka K. Vascular spiders and paper money skin improved by hemodialysis. Dermatology 2002; 205:73–74.
- Serrao R, Zirwas M, English JC. Palmar erythema. Am J Clin Dermatol 2007; 8:347–356.
- Matsumoto M, Ohki K, Nagai I, Oshibuchi T. Lung traction causes an increase in plasma prostacyclin concentration and decrease in mean arterial blood pressure. Anesth Analg 1992; 75:773–776.
- Otto AI, Horvath I, Feldmann J. Multiple firm, painless erythematous papules with a yellowish hue. Arch Dermatol 2005; 141:1595–1600.
- Gandelman G, Aronow WS, Weiss MB. Resolving hyperlipidemia after liver transplantation in a patient with primary sclerosing cholangitis. Am J Ther 2006; 13:171–174.
- Assy N, Kaita K, Mymin D, Levy C, Rosser B, Minuk G. Fatty infiltration of liver in hyperlipidemic patients. Dig Dis Sci 2000; 45:1929–1934.
- Allocca M, Crosignani A, Gritti A, et al. Hypercholesterolaemia is not associated with early atherosclerotic lesions in primary biliary cirrhosis. Gut 2006; 55:1795–1800.
- Dickson E, Fleming C, Ludwig J. Primary biliary cirrhosis. In:Popper H, Schaffner F, editors. Progress in Liver Diseases. New York: Grune and Stratton; 1978:487.
- Elner VM, Mintz R, Demirci H, Hassan AS. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Trans Am Ophthalmol Soc 2005; 103:69–73.
- Craxi A, Camma C, Giunta M. Clinical aspects of bleeding complications in cirrhotic patients. Blood Coagul Fibrinolysis 2000; 11( suppl 1):S75–579.
- Kajihara M, Okazaki Y, Kato S, et al. Evaluation of platelet kinetics in patients with liver cirrhosis: similarity to idiopathic thrombocytopenic purpura. J Gastroenterol Hepatol 2007; 22:112–118.
- Levine N. Patient reports six-month history of minimally pruritic purple dots on legs. Non-blanching macules developed over six months. Geriatrics 2006; 61:22.
- Barton JC, Rao SV, Pereira NM, et al. Juvenile hemochromatosis in the southeastern United States: a report of seven cases in two kinships. Blood Cells Mol Dis 2002; 29:104–115.
- Smith AG, Shuster S, Bomford A, Williams R. Plasma immunoreactive beta-melanocyte-stimulating hormone in chronic liver disease and fulminant hepatic failure. J Invest Dermatol 1978; 70:326–327.
- Barton JC, McDonnell SM, Adams PC, et al. Management of hemochromatosis. Hemochromatosis Management Working Group. Ann Intern Med 1998; 129:932–939.
- Bahnsen M, Gluud C, Johnsen SG, et al. Pituitary-testicular function in patients with alcoholic cirrhosis of the liver. Eur J Clin Invest 1981; 11:473–479.
- Kumar N, Aggarwal SR, Anand BS. Comparison of truncal hair distribution in alcoholic liver disease and alcohol-related chronic pancreatitis. J Gastroenterol Hepatol 2001; 16:855–856.
- Holzberg M, Walker HK. Terry's nails: revised definition and new correlations. Lancet 1984; 1:896–899.
- Mandayam S, Jamal MM, Morgan TR. Epidemiology of alcoholic liver disease. Semin Liver Dis 2004; 24:217–232.
- Belle SH, Beringer KC, Detre KM. Liver transplantation in the United States: results from the National Pitt-UNOS Liver Transplant Registry. United Network for Organ Sharing Clin Transpl 1994:19–35.
- Dunn W, Xu R, Schwimmer JB. Modest wine drinking and decreased prevalence of suspected nonalcoholic fatty liver disease. Hepatology 2008; 47:1947–1954.
- Afford SC, Fisher NC, Neil DA, et al. Distinct patterns of chemokine expression are associated with leukocyte recruitment in alcoholic hepatitis and alcoholic cirrhosis. J Pathol 1998; 186:82–89.
- Evstaf’ev VV, Levin MM. Dermatologic pathology in chronic alcoholics. Vestn Dermatol Venerol 1989; 8:72–74.
- Jerosch-Herold C, Shepstone L, Chojnowski AJ, Larson D. Splinting after contracture release for Dupuytren's contracture (SCoRD): protocol of a pragmatic, multi-centre, randomized controlled trial. BMC Musculoskelet Disord 2008; 9:62.
- Houghton S, Holdstock G, Cockerell R, Wright R. Dupuytren’s contracture, chronic liver disease and IgA immune complexes. Liver 1983; 3:220–224.
- Ibbotson SH. Disseminated superficial porokeratosis: what is the association with ultraviolet radiation? Clin Exp Dermatol 1996; 21:48–50.
- Kono T, Kobayashi H, Ishii M, Nishiguchi S, Taniguchi S. Synchronous development of disseminated superficial porokeratosis and hepatitis C virus-related hepatocellular carcinoma. J Am Acad Dermatol 2000; 43:966–968.
- Park BS, Moon SE, Kim JA. Disseminated superficial porokeratosis in a patient with chronic liver disease. J Dermatol 1997; 24:485–487.
- Murata Y, Kumano K, Takai T. Type 2 segmental manifestation of disseminated superficial porokeratosis showing a systematized pattern of involvement and pronounced cancer proneness. Eur J Dermatol 2001; 11:191–194.
- Galossi A, Guarisco R, Bellis L, Puoti C. Extrahepatic manifestations of chronic HCV infection. J Gastrointestin Liver Dis 2007; 16:65–73.
- El-Serag HB, Hampel H, Yeh C, Rabeneck L. Extrahepatic manifestations of hepatitis C among United States male veterans. Hepatology 2002; 36:1439–1445.
- Stefanova-Petrova DV, Tzvetanska AH, Naumova EJ, et al. Chronic hepatitis C virus infection: prevalence of extrahepatic manifestations and association with cryoglobulinemia in Bulgarian patients. World J Gastroenterol 2007; 13:6518–6528.
- Vassilopoulos D, Calabrese LH. Extrahepatic immunological complications of hepatitis C virus infection. AIDS 2005; 19( suppl 3):S123–S127.
- Hsing AW, Zhang M, Rashid A, et al. Hepatitis B and C virus infection and the risk of biliary tract cancer: a population-based study in China. Int J Cancer 2008; 122:1849–1853.
- Madrid C, Courtois B, Duran D. Chronic sialadenitis revealing hepatitis C: a case report. Med Oral 2004; 9:328–332.
- Lançoni G, Ravinal RC, Costa RS, Roselino AM. Mast cells and transforming growth factor-beta expression: a possible relationship in the development of porphyria cutanea tarda skin lesions. Int J Dermatol 2008; 47:575–581.
- Toll A, Celis R, Ozalla MD, Bruguera M, Herrero C, Ercilla MG. The prevalence of HFE C282Y gene mutation is increased in Spanish patients with porphyria cutanea tarda without hepatitis C virus infection. J Eur Acad Dermatol Venereol 2006; 20:1201–1206.
- Badminton MN, Elder GH. Management of acute and cutaneous porphyrias. Int J Clin Pract 2002; 56:272–278.
- Jackson JM, Callen JP. Scarring alopecia and sclerodermatous changes of the scalp in a patient with hepatitis C infection. J Am Acad Dermatol 1998; 39:824–826.
- Mortimore M, Merryweather-Clarke AT, Robson KJ, Powell LW. The haemochromatosis gene: a global perspective and implications for the Asia-Pacific region. J Gastroenterol Hepatol 1999; 14:838–843.
- Shehan JM, Huerter CJ. Porphyria cutanea tarda associated with an acute gastrointestinal bleed: the roles of supplemental iron and blood transfusion. Cutis 2001; 68:147–150.
- Lambrecht RW, Thapar M, Bonkovsky HL. Genetic aspects of porphyria cutanea tarda. Semin Liver Dis 2007; 27:99–108.
- d’Ovidio R, Sgarra C, Conserva A, Angelotti UF, Erriquez R, Foti C. Alterated integrin expression in lichen planopilaris. Head Face Med 2007; 3:11.
- Chuang TY, Stitle L, Brashear R, Lewis C. Hepatitis C virus and lichen planus: a case-control study of 340 patients. J Am Acad Dermatol 1999; 41:787–789.
- Kemp EH, Gavalas NG, Gawkrodger DJ, Weetman AP. Autoantibody responses to melanocytes in the depigmenting skin disease vitiligo. Autoimmun Rev 2007; 6:138–142.
- Tomasiewicz K, Modrzewska R, Semczuk G. Vitiligo associated with pegylated interferon and ribavirin treatment of patients with chronic hepatitis C: a case report. Adv Ther 2006; 23:139–142.
- Simsek H, Savas C, Akkiz H, Telatar H. Interferon-induced vitiligo in a patient with chronic viral hepatitis C infection. Dermatology 1996; 193:65–66.
- Mulekar SV, Al Issa A, Asaad M, Ghwish B, Al Eisa A. Mixed vitiligo. J Cutan Med Surg. 2006; 10:104–107.
- Waalen J, Felitti V, Gelbart T, Ho NJ, Beutler E. Prevalence of hemochromatosis-related symptoms among individuals with mutations in the HFE gene. Mayo Clin Proc 2002; 77:522–530.
- Walker AP, Tucker DC, Hall MA, et al. Results communication and patient education after screening for possible hemochromatosis and iron overload: experience from the HEIRS study of a large ethnically and linguistically diverse group. Genet Med 2007; 9:778–791.
- Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, cafe au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician 2003; 68:1955–1960.
- Tsuji T. Experimental hemosiderosis: relationship between skin pigmentation and hemosiderin. Acta Derm Venereol 1980; 60:109–114.
- Oji V, Traupe H. Ichthyoses: differential diagnosis and molecular genetics. Eur J Dermatol 2006; 16:349–359.
KEY POINTS
- Pruritus due to liver disease is particularly resistant to therapy. Cholestyramine (Questran) 4 g/day, gradually increased to 24 g/day, is one option. If the pruritus does not respond or the patient cannot tolerate cholestyramine, rifampin (Rifadin) can be tried.
- Spider angiomas, Bier spots, and “paper-money” skin are all superficial vascular problems that may be related to liver disease.
- Cutaneous lesions often accompany alcoholic cirrhosis and have been detected in up to 43% of people with chronic alcoholism. The combination of spider angiomas, palmar erythema, and Dupuytren contracture is common in alcoholic cirrhosis.
- Although porphyria cutanea tarda is associated with liver disease in general, recent studies show that patients with hepatitis C are at particularly high risk.
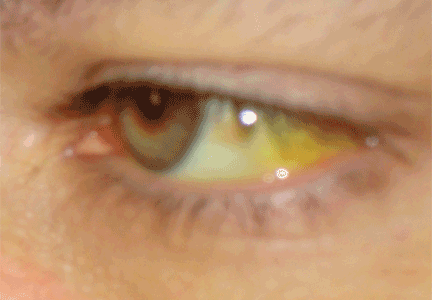
Hepatitis B: A strategy for evaluation and management
Hepatitis b virus (HBV) infection is sometimes challenging to manage because the disease has several stages and many clinical scenarios. HBV-infected patients are a very heterogeneous group, and we cannot apply a single management approach to all.
An understanding of the natural history of HBV infection and its diagnosis, which we reviewed in last month’s issue of this Journal1, is critical to understanding how to manage HBV infection.
In this article, we will review the principles of HBV management in adults, including those on immunosuppressant therapy and pregnant women, and guidelines for HBV vaccination.
WORKUP FOR HBV INFECTION
Once the diagnosis of HBV infection is made,1 a full management strategy should be formulated, as outlined below.
History
When and how did the patient acquire HBV? This information is important to know when making treatment decisions. For example, most acute, adult-onset cases (eg, acquired recently via sexual contact or parenteral drug abuse) resolve spontaneously within a few months, whereas most chronic cases (defined as being positive for HBV surface antigen for more than 6 months) were acquired at birth or in early childhood. Therefore, we should try to determine if the patient’s mother, siblings, household contacts, and sexual partners are positive for HBV surface antigen or have risk factors for HBV infection1; those without infection or immunity to HBV should be vaccinated.
People at risk of HBV infection include:
- Parenteral drug users
- People with multiple sexual partners
- Household contacts and sexual partners of people positive for HBV surface antigen
- Infants born to HBV-infected mothers
- Patients and staff in custodial institutions for the developmentally disabled
- Recipients of certain plasma-derived products (including patients with congenital coagulation defects)
- Hemodialysis patients
- Health and public-safety workers who have contact with blood
- People born in areas where HBV is endemic, and their children.1
Does the patient have risk factors for other infections? Especially look for risk factors for human immunodeficiency virus (HIV) infection (eg, intravenous drug users and men having sex with men) and hepatitis D virus (intravenous drug users and patients from countries where hepatitis D virus infection is common, particularly Eastern Europe, Mediterranean countries, and the Amazon basin).
Does the patient have other modifiable risk factors for progressive liver disease, particularly alcohol abuse and obesity?
Does the patient have symptoms or signs of cirrhosis or hepatocellular carcinoma? Symptoms and signs that involve multiple systems could be extrahepatic manifestations of HBV infection, such as polyarteritis nodosa, which causes abdominal pain, arthralgia, hypertension, and asymmetric polyneuropathy.
Baseline laboratory evaluation
At baseline we should obtain a complete blood count, blood urea nitrogen level, serum creatinine level, liver profile, prothrombin time, urinalysis, and HBV serologic markers. In addition, HBV DNA can be detected in the serum at levels as low as 60 IU/mL, and it should be measured in the initial evaluation to establish a baseline before starting antiviral therapy in patients with chronic HBV infection and subsequently to monitor the response.
All patients with chronic HBV infection should also be tested for serologic markers of hepatitis A and hepatitis C; patients at risk of HIV and hepatitis D should also be tested for these diseases.
Not all patients need liver biopsy
Liver biopsy is the most accurate tool for staging the degree of HBV-related hepatic fibrosis in patients who have no obvious clinical manifestations of cirrhosis.
Not all patients with HBV infection need a biopsy, however. In patients with acute HBV infection, liver biopsy has no benefit except if concomitant pathology (eg, iron overload, nonalcoholic steatohepatitis, or alcoholic steatohepatitis) is suspected. In patients with chronic hepatitis B, liver biopsy is helpful when the viral load alone does not provide sufficient guidance for treatment, eg, when the viral load is less than 2 × 104 IU/mL in a patient positive for hepatitis e antigen or less than 2 × 103 IU/mL in a patient negative for hepatitis e antigen. (The presence of e antigen is a marker of HBV replication and infectivity.1) Biopsy should also be considered in those who have been infected a long time (eg, more than 10 years), because they may have occult cirrhosis, and if they do they may need to undergo antiviral treatment, endoscopy to look for varices, and surveillance for liver cancer.
In some situations it is easy to decide whether antiviral therapy is indicated without resorting to liver biopsy.
We would treat:
- A patient positive for HBV e antigen for more than 6 months, whose HBV DNA level is higher than 2 × 104 IU/mL and whose alanine aminotransferase (ALT) level is high
- A patient with HBV for more than 6 months who is negative for e antigen and who has an HBV DNA level higher than 2 × 103 IU/mL and elevated ALT
- A patient with compensated HBV cirrhosis and an HBV DNA level higher than 2 × 103 IU/mL
- A patient with HBV cirrhosis with decompensation and any detectable HBV DNA.
We would not treat:
- An HBV carrier with a normal ALT level and an HBV DNA level that is lower than 2 × 104 IU/mL or undetectable.
If a patient does not fit into one of these categories but has HBV DNA, a liver biopsy showing significant necroinflammation or fibrosis would be an indication for treatment.
ANTIVIRAL THERAPY
Below, we summarize the main principles of anti-HBV therapy, emphasizing whether to treat and with which agent. Treatment of HBV infection in patients who are also infected with HIV or hepatitis C virus and in those with resistant or refractory hepatitis B is not within the scope of this article.
Acute infection rarely needs treatment
Acute, adult-acquired HBV infection is self-limited in most cases,1 and antiviral therapy is not routinely indicated.
In the rare cases of acute liver failure related to acute HBV infection, use of a nucleoside or nucleotide analogue reverse transcriptase inhibitor (nucleoside/nucleotide analogues) has been recommended, although no properly designed studies have been done.2,3 This recommendation is based on anecdotal experience, the relative safety of the antiviral agents, the serious nature of acute liver failure, and the possible need for emergency liver transplantation that requires prophylaxis against recurrence.
The nucleoside/nucleotide analogues that have been recommended in acute liver failure are lamivudine (Epivir), telbivudine (Tyzeka), and entecavir (Baraclude)—but not adefovir (Hepsera), which has a slow action and potential nephrotoxicity. Interferon drugs are contraindicated because they frequently cause side effects and can worsen hepatitis.4
Patients with acute liver failure should be referred promptly to a liver transplant center, and other management measures should also be started in a timely fashion.
In chronic HBV infection, treatment decisions are individualized
In chronic hepatitis B (ie, lasting > 6 months), treatment decisions should be based on the patient’s clinical situation and test results. The route and duration of infection (if known), history of previous hepatitis flares, ALT levels, current and previous HBV serologic test results and DNA levels, findings on liver biopsy (if previously done), and clinically suspected cirrhosis are all important to consider when deciding whether antiviral therapy is needed.

For many patients with chronic HBV infection, observation without antiviral therapy is warranted, eg:
- Young patients (< 30 years old) who acquired HBV at birth and who have persistently normal ALT levels with no evidence of advanced liver disease, regardless of their HBV DNA level (immune tolerance phase)
- Chronic inactive carriers who have no e antigen, persistently normal ALT levels, and very low or undetectable levels of HBV DNA without evidence of significant liver injury.
These patients can be managed by internists by close monitoring for hepatitis flares with serial ALT measurements along with other general management measures.
Antiviral agents for chronic HBV infection
An ideal agent for treating hepatitis B does not exist. Trade-offs are the essence of agent selection.
Interferons, the first drugs shown to be effective against HBV, can in some respects be considered the best available initial choice, especially in patients positive for hepatitis e antigen. Interferons have numerous side effects but, unlike all the other options, they have a well-defined duration of treatment (4–6 months in patients positive for e antigen). The principal goal of this therapy is disappearance of e antigen.
Interferon-based therapy is not recommended in patients with cirrhosis, however, because of the risk of hepatic decompensation associated with interferon-related flares of hepatitis.4
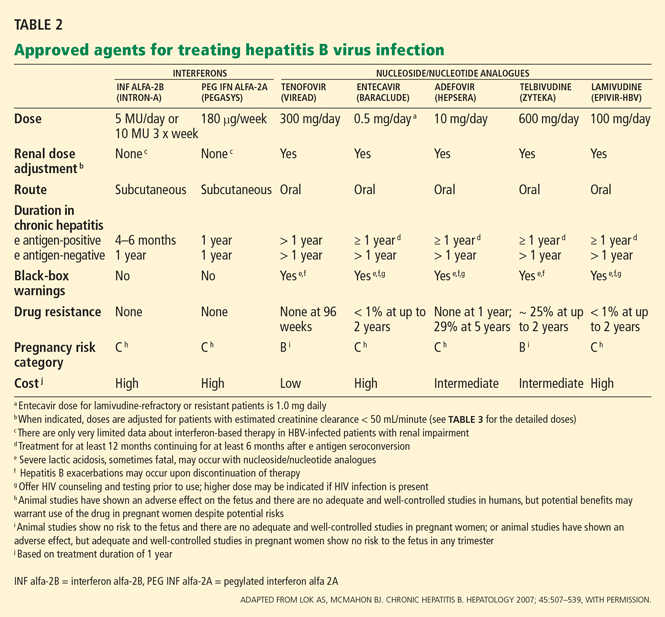
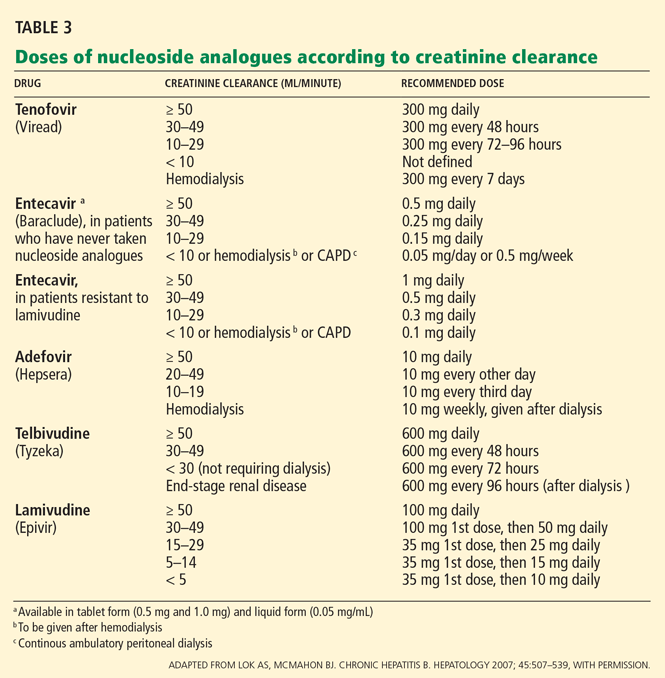
Although single-agent antiviral therapy may someday be replaced by a multidrug regimen, the data so far are not sufficiently robust to recommend multidrug regimens except possibly in cases of established drug resistance.
Adjunctive management
Vaccinations. All patients with chronic hepatitis B should be vaccinated against hepatitis A if serologic testing indicates they have no immunity to it. Influenza and pneumococcal vaccines are recommended for all patients with chronic liver disease.6
Alcohol rehabilitation. Patients who abuse alcohol should be counseled, and many need consultation with a psychosocial care provider for alcohol rehabilitation.
Smoking cessation. Cigarette smoking is linked to a higher risk of hepatocellular carcinoma in patients with chronic liver disease, including chronic HBV infection.7 Therefore, smokers should be counseled to quit.
Surveillance for hepatocellular carcinoma. Hepatocellular carcinoma can occur in patients with chronic hepatitis B, in most cases on top of cirrhosis, although important exceptions exist. The American Association for the Study of Liver Diseases recommends surveillance for hepatocellular carcinoma in all HBV carriers with cirrhosis and in the following groups regardless of whether they have cirrhosis8:
- Asian men age 40 and older
- Asian women age 50 and older
- African patients age 20 and older
- Patients with a family history of hepatocellular carcinoma
- Possibly, those with high HBV DNA levels and ongoing inflammatory activity.
In the United States, liver ultrasonography and alpha fetoprotein measurement every 6 to 12 months is a reasonable strategy.
If there is evidence of cirrhosis, esophagogastroduodenoscopy is recommended to screen for esophageal and gastric varices.
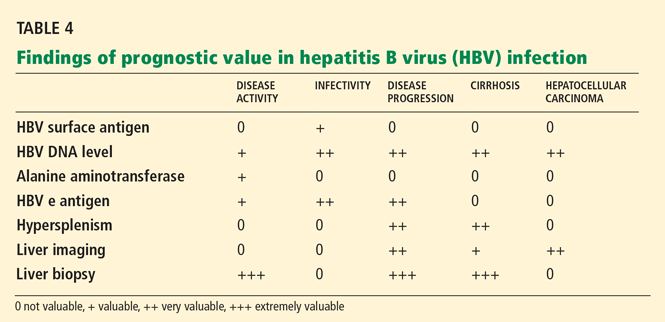
SCREEN BEFORE CHEMOTHERAPY OR IMMUNOSUPPRESSIVE THERAPY
When patients who are positive for HBV surface antigen undergo immunosuppressive therapy or cancer chemotherapy, from 20% to 50% develop reactivated HBV infection with high HBV viral loads. Even patients who have resolved hepatitis B (ie, negative for HBV surface antigen and positive for surface antibody) may experience hepatitis B reactivation, with serious consequences. Hepatic decompensation and death have been reported during and after chemotherapy, especially in patients with cirrhosis.9 Therefore, patients at risk of HBV infection should be screened for it before starting these therapies.4 Furthermore, perhaps all patients about to undergo anticancer therapies that include anti-B-cell or anti-T-cell therapies or hematopoietic stem cell transplantation should be screened.9
Recent data indicate that many oncologists have not been screening for HBV.10 Hence, more effort is needed to make this important testing routine in this setting.
The initial tests in these patients should be liver chemistry tests, HBV surface antigen, HBV surface antibody, and HBV core antibody. In those who test positive for surface antigen, one should test for e antigen, e antibody, and HBV DNA.
Patients with indications for anti-HBV therapy (Table 1) should receive antiviral therapy. Otherwise, those positive for surface antigen should start taking anti-HBV medication at the start of chemotherapy or immunosuppressive therapy and should continue taking it until 6 months after the chemotherapy or immunosuppressive therapy is finished.4 Some experts also recommend starting anti-HBV therapy 7 days before the chemotherapy or immunosuppressive therapy and continuing it for 1 year afterward.10 Those with HBV DNA levels higher than 2 × 103 IU/mL should continue HBV therapy until they reach the same treatment end points as for immunocompetent patients as outlined above.4
Because we have little information on patients who are negative for surface antigen and who have antibodies against surface antigen and core antigen, we cannot make an unequivocal recommendation for anti-HBV therapy in this group.11 Rather, these patients should be monitored during immunosuppressive treatment, preferably with liver chemistry tests and HBV DNA titers, and antiviral drugs should be given as a deferred therapy upon evidence of HBV reactivation.9 Few cases of fatal hepatic failure in patients with this serologic pattern receiving rituximab (Rituxan) have been reported.12–14
With their small risk of drug resistance and rapid onset of action, entecavir or tenofovir may be the preferred anti-HBV therapy in patients undergoing immunosuppression or chemotherapy, especially in those requiring prolonged immunosuppressive therapy (longer than 12 months). In those requiring shorter courses, lamivudine or telbivudine is a possible alternative.4
OUTCOMES OF LIVER TRANSPLANTATION HAVE IMPROVED IN HBV PATIENTS
The early results of liver transplantation for HBV were discouraging because many patients developed rapidly progressive recurrent disease (fibrosing cholestatic hepatitis) and died within 12 to 18 months after the operation.15 However, patients with HBV are now treated perioperatively with lamivudine or adefovir combined with prolonged administration of hepatitis B immune globulin, and their survival now exceeds that of patients who receive transplants for many other conditions.16
Like patients with cirrhosis due to other causes, those with HBV-related cirrhosis who have any of the following should be referred for liver transplantation evaluation16:

- A Child-Turcotte-Pugh score of 7 or higher (Table 5).
- A major complication of cirrhosis such as ascites, variceal bleeding, hepatocellular carcinoma, or hepatic encephalopathy.
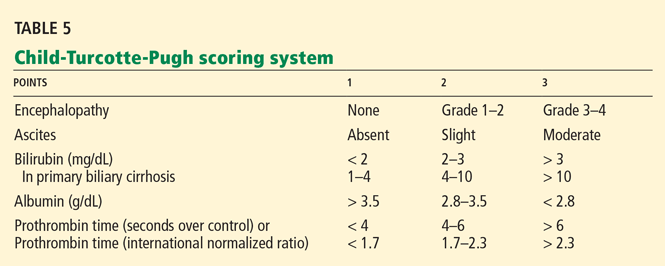
PREVENTING VERTICAL TRANSMISSION
The major problem in young women with chronic HBV infection is the risk of vertical (mother-to-infant) transmission at delivery. The risk varies, depending on the viral load and e antigen status of the mother at the time of delivery; if she is positive for e antigen, the risk of HBV infection in the newborn is 70% to 90% by the age of 6 months if the newborn does not receive postexposure immunoprophylaxis; if the mother is positive for surface antigen but negative for e antigen, the risk of chronic infection is less than 10%, even without postexposure immunoprophylaxis.17
All women should be tested for HBV surface antigen early in pregnancy each time they become pregnant. If a patient tests negative early in pregnancy but continues behaviors that put her at risk of HBV infection (eg, having multiple sexual partners, having had a sex partner positive for surface antigen, using injection drugs, or contracting any sexually transmitted disease), she should be retested at the time of admission to the hospital for delivery.17 This also includes women who were not screened prenatally and those with clinical hepatitis.
Vaccine and immune globulin for the infant
If the mother is positive for HBV surface antigen, the infant should receive single-antigen HBV vaccine and hepatitis B immune globulin within 12 hours of birth, given at different injection sites.17 The second dose of vaccine should be given at age 1 to 2 months and the third at age 6 months (but not before age 24 weeks). The response to vaccination should be ascertained by testing for surface antigen and surface antibody after completion of the vaccine series, at age 9 to 18 months.
Maternal HBV infection does not contraindicate breastfeeding, as studies suggest that breastfeeding by a mother positive for surface antigen does not increase the infant’s risk of acquiring HBV infection.18
Which HBV therapy for a pregnant woman?
Some evidence supports antiviral therapy with nucleoside/nucleotide analogues in pregnant women who have viral loads of 106 IU/mL or higher. Lamivudine is safe in pregnancy and, together with immunization of the infant, reduces HBV transmission. Interferon-based therapy is contraindicated in pregnant women (and in women who may want to become pregnant) because of interferon’s antiproliferative effects. Nucleoside/nucleotide analogues classified as category B (eg, lamivudine, telbivudine, and tenofovir) could be used when the benefit of treating the pregnant mother outweighs the risk to the mother or fetus,2 although the possible effects of tenofovir on bone density argue against its use during pregnancy or breastfeeding.19
VACCINATION HAS REDUCED THE INCIDENCE OF ACUTE HEPATITIS B
HBV vaccination, a major achievement in HBV management, has played a big role in reducing the incidence of acute HBV infection, especially in children and adolescents.20
The currently available vaccines in the United States contain HBV surface antigen derived through recombinant DNA technology from yeast.21 Two single-antigen vaccines are available in the United States, under the brand names Recombivax HB and Engerix B. Of the three licensed combination vaccines, one (Twinrix) is used in adults, and two (Comvax and Pediarix) are used in infants and young children. Twinrix contains recombinant HBV surface antigens and inactivated hepatitis A virus and it is recommended for people age 18 years and older and at risk of both HBV and hepatitis A infections.20
Vaccinate all infants
All infants should be vaccinated against HBV as part of the recommended childhood immunization schedule. The vaccine is given on a three-dose schedule at birth and again at 1 month and 6 months of age.16 All children and adolescents under age 19 who have not previously received HBV vaccine should be vaccinated at any age with an appropriate dose and schedule.16
Vaccinate adults at risk—or who ask for it

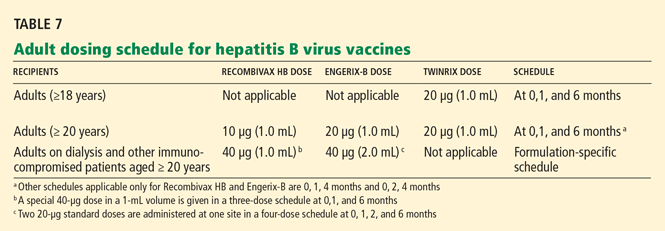
Vaccination is less effective in older people
The three-dose vaccine series given intramuscularly initially, then again at 1 month and 6 months, produces a protective antibody response in approximately 30% to 55% of healthy adults under age 40 after the first dose, 75% after the second dose, and more than 90% after the third dose.21,22 After age 40, however, the proportion of persons who have a protective antibody response after three doses declines to less than 90%, and by age 60, protective levels of antibody develop in only 75%.23
Other factors that lower the response to vaccination are smoking, obesity, genetic factors, and immune suppression.20
Postvaccination serologic testing for immunity is not necessary after routine vaccination of adults, but it is recommended for patients whose subsequent clinical management depends on knowledge of their immune status, such as health care workers who have contact with patients or blood and are at ongoing risk of injuries with sharp instruments or needlesticks; chronic hemodialysis patients and people infected with HIV or otherwise immunocompromised; and sex partners or needle-sharing partners of people positive for HBV surface antigen.20 A protective concentration of HBV surface antibody measured 1 to 2 months after completion of the vaccine series is defined as 10 mIU/mL. Further periodic testing to document persistence of protective levels of surface antibody is not indicated.
If the first series does not ‘take’
Patients who do not respond to the primary vaccine series should complete a second three-dose series, with doses at 0, 1, and 6 months. Serologic testing is done 1 to 2 months after finishing the second series.
Patients who do not have protective levels of HBV surface antibody after revaccination by the appropriate schedule in the deltoid muscle (< 5% of those receiving six doses of hepatitis B vaccine) either are primary nonresponders or are infected with HBV.20 Therefore, they should be tested for HBV surface antigen. If this test is negative, then they should be considered susceptible to HBV infection and should be counseled accordingly.
Contraindications and precautions
HBV vaccination is contraindicated in people with a history of hypersensitivity to baker’s yeast or to a previous dose of HBV vaccine.20 Patients with moderate or severe acute illness at the time the shot is scheduled should wait until they recover before getting HBV vaccine. Pregnancy is not a contraindication.20
- Elgouhari HM, Abu-Rajab Tamimi T, Carey WD. Hepatitis B virus infection: understanding its epidemiology, course, and diagnosis. Cleve Clin J Med 2008; 75:881–889.
- Stravitz RT, Kramer AH, Davern T, et al. Intensive care of patients with acute liver failure: recommendations of the U.S. Acute Liver Failure Study Group. Crit Care Med 2007; 35:2498–2508.
- Hoofnagle JH, Doo E, Liang TJ, Fleischer R, Lok AS. Management of hepatitis B: summary of a clinical research workshop. Hepatology 2007; 45:1056–1075.
- Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Keeffe EB, Dieterich DT, Han SH, et al. A treatment algorithm for the management of chronic hepatitis B virus infection in the United States. Clin Gastroenterol Hepatol 2004; 2:87–106.
- Jacobs RJ, Meyerhoff AS, Saab S. Immunization needs of chronic liver disease patients seen in primary care versus specialist settings. Dig Dis Sci 2005; 50:1525–1531.
- Wang LY, You SL, Lu SN, et al. Risk of hepatocellular carcinoma and habits of alcohol drinking, betel quid chewing and cigarette smoking: a cohort of 2416 HBsAg–seropositive and 9421 HBsAg–seronegative male residents in Taiwan. Cancer Causes Control 2003; 14:241–250.
- Bruix J, Sherman M; Practice Guidelines Committee, American Association for the Study of Liver Diseases. Management of hepatocellular carcinoma. Hepatology 2005; 42:1208–1236.
- Yeo W, Johnson PJ. Diagnosis, prevention and management of hepatitis B virus reactivation during anticancer therapy. Hepatology 2006; 43:209–220.
- Tran T, Oh M, Poordad F, Martin P. Screening for hepatitis B in chemotherapy patients: survey of current oncology practices [abstract]. Hepatology 2007; 46:978A.
- Kohrt HE, Ouyang DL, Keeffe EB. Antiviral prophylaxis for chemotherapy–induced reactivation of chronic hepatitis B virus infection. Clin Liver Dis 2007; 11:965–991.
- Westhoff TH, Jochimsen F, Schmittel A, et al. Fatal hepatitis B virus reactivation by an escape mutant following rituximab therapy. Blood 2003; 102:1930.
- Sarrecchia C, Cappelli A, Aiello P. HBV reactivation with fatal fulminating hepatitis during rituximab treatment in a subject negative for HBsAg and positive for HBsAb and HBcAb. J Infect Chemother 2005; 11:189–191.
- Law JK, Ho JK, Hoskins PJ, Erb SR, Steinbrecher UP, Yoshida FM. Fatal reactivation of hepatitis B post-chemotherapy for lymphoma in a hepatitis B surface antigen-negative, hepatitis B core antibody-positive patient: potential implications for future prophylaxis recommendations. Leuk Lymphoma 2005; 46:1085–1089.
- Todo S, Demetris AJ, Van Thiel D, Teperman L, Fung JJ, Starzl TE. Orthotopic liver transplantation for patients with hepatitis B virus-related liver disease. Hepatology 1991; 13:619–626.
- Murray KF, Carithers RLAASLD. AASLD practice guidelines: Evaluation of the patient for liver transplantation. Hepatology 2005; 41:1407–1432.
- Mast EE, Margolis HS, Fiore AE, et al. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP) part 1: immunization of infants, children, and adolescents. MMWR Recomm Rep 2005; 54:1–31.
- Beasley RP, Stevens CE, Shiao IS, Meng HC. Evidence against breast–feeding as a mechanism for vertical transmission of hepatitis B. Lancet 1975; 2:740–741.
- Parsonage MJ, Wilkins EG, Snowden N, Issa BG, Savage MW. The development of hypophosphataemic osteomalacia with myopathy in two patients with HIV infection receiving tenofovir therapy. HIV Med 2005; 6:341–346.
- Mast EE, Weinbaum CM, Fiore AE, et al. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP) Part II: immunization of adults. MMWR Recomm Rep 2006; 55:1–33.
- Andre FE. Summary of safety and efficacy data on a yeast–derived hepatitis B vaccine. Am J Med 1989; 87:14S–20S.
- Zajac BA, West DJ, McAleer WJ, Scolnick EM. Overview of clinical studies with hepatitis B vaccine made by recombinant DNA. J Infect 1986; 13( suppl A):39–45.
- Averhoff F, Mahoney F, Coleman P, Schatz G, Hurwitz E, Margolis H. Immunogenicity of hepatitis B vaccines: implications for persons at occupational risk for hepatitis B virus infection. Am J Prev Med 1998; 15:1–8.
Hepatitis b virus (HBV) infection is sometimes challenging to manage because the disease has several stages and many clinical scenarios. HBV-infected patients are a very heterogeneous group, and we cannot apply a single management approach to all.
An understanding of the natural history of HBV infection and its diagnosis, which we reviewed in last month’s issue of this Journal1, is critical to understanding how to manage HBV infection.
In this article, we will review the principles of HBV management in adults, including those on immunosuppressant therapy and pregnant women, and guidelines for HBV vaccination.
WORKUP FOR HBV INFECTION
Once the diagnosis of HBV infection is made,1 a full management strategy should be formulated, as outlined below.
History
When and how did the patient acquire HBV? This information is important to know when making treatment decisions. For example, most acute, adult-onset cases (eg, acquired recently via sexual contact or parenteral drug abuse) resolve spontaneously within a few months, whereas most chronic cases (defined as being positive for HBV surface antigen for more than 6 months) were acquired at birth or in early childhood. Therefore, we should try to determine if the patient’s mother, siblings, household contacts, and sexual partners are positive for HBV surface antigen or have risk factors for HBV infection1; those without infection or immunity to HBV should be vaccinated.
People at risk of HBV infection include:
- Parenteral drug users
- People with multiple sexual partners
- Household contacts and sexual partners of people positive for HBV surface antigen
- Infants born to HBV-infected mothers
- Patients and staff in custodial institutions for the developmentally disabled
- Recipients of certain plasma-derived products (including patients with congenital coagulation defects)
- Hemodialysis patients
- Health and public-safety workers who have contact with blood
- People born in areas where HBV is endemic, and their children.1
Does the patient have risk factors for other infections? Especially look for risk factors for human immunodeficiency virus (HIV) infection (eg, intravenous drug users and men having sex with men) and hepatitis D virus (intravenous drug users and patients from countries where hepatitis D virus infection is common, particularly Eastern Europe, Mediterranean countries, and the Amazon basin).
Does the patient have other modifiable risk factors for progressive liver disease, particularly alcohol abuse and obesity?
Does the patient have symptoms or signs of cirrhosis or hepatocellular carcinoma? Symptoms and signs that involve multiple systems could be extrahepatic manifestations of HBV infection, such as polyarteritis nodosa, which causes abdominal pain, arthralgia, hypertension, and asymmetric polyneuropathy.
Baseline laboratory evaluation
At baseline we should obtain a complete blood count, blood urea nitrogen level, serum creatinine level, liver profile, prothrombin time, urinalysis, and HBV serologic markers. In addition, HBV DNA can be detected in the serum at levels as low as 60 IU/mL, and it should be measured in the initial evaluation to establish a baseline before starting antiviral therapy in patients with chronic HBV infection and subsequently to monitor the response.
All patients with chronic HBV infection should also be tested for serologic markers of hepatitis A and hepatitis C; patients at risk of HIV and hepatitis D should also be tested for these diseases.
Not all patients need liver biopsy
Liver biopsy is the most accurate tool for staging the degree of HBV-related hepatic fibrosis in patients who have no obvious clinical manifestations of cirrhosis.
Not all patients with HBV infection need a biopsy, however. In patients with acute HBV infection, liver biopsy has no benefit except if concomitant pathology (eg, iron overload, nonalcoholic steatohepatitis, or alcoholic steatohepatitis) is suspected. In patients with chronic hepatitis B, liver biopsy is helpful when the viral load alone does not provide sufficient guidance for treatment, eg, when the viral load is less than 2 × 104 IU/mL in a patient positive for hepatitis e antigen or less than 2 × 103 IU/mL in a patient negative for hepatitis e antigen. (The presence of e antigen is a marker of HBV replication and infectivity.1) Biopsy should also be considered in those who have been infected a long time (eg, more than 10 years), because they may have occult cirrhosis, and if they do they may need to undergo antiviral treatment, endoscopy to look for varices, and surveillance for liver cancer.
In some situations it is easy to decide whether antiviral therapy is indicated without resorting to liver biopsy.
We would treat:
- A patient positive for HBV e antigen for more than 6 months, whose HBV DNA level is higher than 2 × 104 IU/mL and whose alanine aminotransferase (ALT) level is high
- A patient with HBV for more than 6 months who is negative for e antigen and who has an HBV DNA level higher than 2 × 103 IU/mL and elevated ALT
- A patient with compensated HBV cirrhosis and an HBV DNA level higher than 2 × 103 IU/mL
- A patient with HBV cirrhosis with decompensation and any detectable HBV DNA.
We would not treat:
- An HBV carrier with a normal ALT level and an HBV DNA level that is lower than 2 × 104 IU/mL or undetectable.
If a patient does not fit into one of these categories but has HBV DNA, a liver biopsy showing significant necroinflammation or fibrosis would be an indication for treatment.
ANTIVIRAL THERAPY
Below, we summarize the main principles of anti-HBV therapy, emphasizing whether to treat and with which agent. Treatment of HBV infection in patients who are also infected with HIV or hepatitis C virus and in those with resistant or refractory hepatitis B is not within the scope of this article.
Acute infection rarely needs treatment
Acute, adult-acquired HBV infection is self-limited in most cases,1 and antiviral therapy is not routinely indicated.
In the rare cases of acute liver failure related to acute HBV infection, use of a nucleoside or nucleotide analogue reverse transcriptase inhibitor (nucleoside/nucleotide analogues) has been recommended, although no properly designed studies have been done.2,3 This recommendation is based on anecdotal experience, the relative safety of the antiviral agents, the serious nature of acute liver failure, and the possible need for emergency liver transplantation that requires prophylaxis against recurrence.
The nucleoside/nucleotide analogues that have been recommended in acute liver failure are lamivudine (Epivir), telbivudine (Tyzeka), and entecavir (Baraclude)—but not adefovir (Hepsera), which has a slow action and potential nephrotoxicity. Interferon drugs are contraindicated because they frequently cause side effects and can worsen hepatitis.4
Patients with acute liver failure should be referred promptly to a liver transplant center, and other management measures should also be started in a timely fashion.
In chronic HBV infection, treatment decisions are individualized
In chronic hepatitis B (ie, lasting > 6 months), treatment decisions should be based on the patient’s clinical situation and test results. The route and duration of infection (if known), history of previous hepatitis flares, ALT levels, current and previous HBV serologic test results and DNA levels, findings on liver biopsy (if previously done), and clinically suspected cirrhosis are all important to consider when deciding whether antiviral therapy is needed.
For many patients with chronic HBV infection, observation without antiviral therapy is warranted, eg:
- Young patients (< 30 years old) who acquired HBV at birth and who have persistently normal ALT levels with no evidence of advanced liver disease, regardless of their HBV DNA level (immune tolerance phase)
- Chronic inactive carriers who have no e antigen, persistently normal ALT levels, and very low or undetectable levels of HBV DNA without evidence of significant liver injury.
These patients can be managed by internists by close monitoring for hepatitis flares with serial ALT measurements along with other general management measures.
Antiviral agents for chronic HBV infection
An ideal agent for treating hepatitis B does not exist. Trade-offs are the essence of agent selection.
Interferons, the first drugs shown to be effective against HBV, can in some respects be considered the best available initial choice, especially in patients positive for hepatitis e antigen. Interferons have numerous side effects but, unlike all the other options, they have a well-defined duration of treatment (4–6 months in patients positive for e antigen). The principal goal of this therapy is disappearance of e antigen.
Interferon-based therapy is not recommended in patients with cirrhosis, however, because of the risk of hepatic decompensation associated with interferon-related flares of hepatitis.4
Although single-agent antiviral therapy may someday be replaced by a multidrug regimen, the data so far are not sufficiently robust to recommend multidrug regimens except possibly in cases of established drug resistance.
Adjunctive management
Vaccinations. All patients with chronic hepatitis B should be vaccinated against hepatitis A if serologic testing indicates they have no immunity to it. Influenza and pneumococcal vaccines are recommended for all patients with chronic liver disease.6
Alcohol rehabilitation. Patients who abuse alcohol should be counseled, and many need consultation with a psychosocial care provider for alcohol rehabilitation.
Smoking cessation. Cigarette smoking is linked to a higher risk of hepatocellular carcinoma in patients with chronic liver disease, including chronic HBV infection.7 Therefore, smokers should be counseled to quit.
Surveillance for hepatocellular carcinoma. Hepatocellular carcinoma can occur in patients with chronic hepatitis B, in most cases on top of cirrhosis, although important exceptions exist. The American Association for the Study of Liver Diseases recommends surveillance for hepatocellular carcinoma in all HBV carriers with cirrhosis and in the following groups regardless of whether they have cirrhosis8:
- Asian men age 40 and older
- Asian women age 50 and older
- African patients age 20 and older
- Patients with a family history of hepatocellular carcinoma
- Possibly, those with high HBV DNA levels and ongoing inflammatory activity.
In the United States, liver ultrasonography and alpha fetoprotein measurement every 6 to 12 months is a reasonable strategy.
If there is evidence of cirrhosis, esophagogastroduodenoscopy is recommended to screen for esophageal and gastric varices.
SCREEN BEFORE CHEMOTHERAPY OR IMMUNOSUPPRESSIVE THERAPY
When patients who are positive for HBV surface antigen undergo immunosuppressive therapy or cancer chemotherapy, from 20% to 50% develop reactivated HBV infection with high HBV viral loads. Even patients who have resolved hepatitis B (ie, negative for HBV surface antigen and positive for surface antibody) may experience hepatitis B reactivation, with serious consequences. Hepatic decompensation and death have been reported during and after chemotherapy, especially in patients with cirrhosis.9 Therefore, patients at risk of HBV infection should be screened for it before starting these therapies.4 Furthermore, perhaps all patients about to undergo anticancer therapies that include anti-B-cell or anti-T-cell therapies or hematopoietic stem cell transplantation should be screened.9
Recent data indicate that many oncologists have not been screening for HBV.10 Hence, more effort is needed to make this important testing routine in this setting.
The initial tests in these patients should be liver chemistry tests, HBV surface antigen, HBV surface antibody, and HBV core antibody. In those who test positive for surface antigen, one should test for e antigen, e antibody, and HBV DNA.
Patients with indications for anti-HBV therapy (Table 1) should receive antiviral therapy. Otherwise, those positive for surface antigen should start taking anti-HBV medication at the start of chemotherapy or immunosuppressive therapy and should continue taking it until 6 months after the chemotherapy or immunosuppressive therapy is finished.4 Some experts also recommend starting anti-HBV therapy 7 days before the chemotherapy or immunosuppressive therapy and continuing it for 1 year afterward.10 Those with HBV DNA levels higher than 2 × 103 IU/mL should continue HBV therapy until they reach the same treatment end points as for immunocompetent patients as outlined above.4
Because we have little information on patients who are negative for surface antigen and who have antibodies against surface antigen and core antigen, we cannot make an unequivocal recommendation for anti-HBV therapy in this group.11 Rather, these patients should be monitored during immunosuppressive treatment, preferably with liver chemistry tests and HBV DNA titers, and antiviral drugs should be given as a deferred therapy upon evidence of HBV reactivation.9 Few cases of fatal hepatic failure in patients with this serologic pattern receiving rituximab (Rituxan) have been reported.12–14
With their small risk of drug resistance and rapid onset of action, entecavir or tenofovir may be the preferred anti-HBV therapy in patients undergoing immunosuppression or chemotherapy, especially in those requiring prolonged immunosuppressive therapy (longer than 12 months). In those requiring shorter courses, lamivudine or telbivudine is a possible alternative.4
OUTCOMES OF LIVER TRANSPLANTATION HAVE IMPROVED IN HBV PATIENTS
The early results of liver transplantation for HBV were discouraging because many patients developed rapidly progressive recurrent disease (fibrosing cholestatic hepatitis) and died within 12 to 18 months after the operation.15 However, patients with HBV are now treated perioperatively with lamivudine or adefovir combined with prolonged administration of hepatitis B immune globulin, and their survival now exceeds that of patients who receive transplants for many other conditions.16
Like patients with cirrhosis due to other causes, those with HBV-related cirrhosis who have any of the following should be referred for liver transplantation evaluation16:
- A Child-Turcotte-Pugh score of 7 or higher (Table 5).
- A major complication of cirrhosis such as ascites, variceal bleeding, hepatocellular carcinoma, or hepatic encephalopathy.
PREVENTING VERTICAL TRANSMISSION
The major problem in young women with chronic HBV infection is the risk of vertical (mother-to-infant) transmission at delivery. The risk varies, depending on the viral load and e antigen status of the mother at the time of delivery; if she is positive for e antigen, the risk of HBV infection in the newborn is 70% to 90% by the age of 6 months if the newborn does not receive postexposure immunoprophylaxis; if the mother is positive for surface antigen but negative for e antigen, the risk of chronic infection is less than 10%, even without postexposure immunoprophylaxis.17
All women should be tested for HBV surface antigen early in pregnancy each time they become pregnant. If a patient tests negative early in pregnancy but continues behaviors that put her at risk of HBV infection (eg, having multiple sexual partners, having had a sex partner positive for surface antigen, using injection drugs, or contracting any sexually transmitted disease), she should be retested at the time of admission to the hospital for delivery.17 This also includes women who were not screened prenatally and those with clinical hepatitis.
Vaccine and immune globulin for the infant
If the mother is positive for HBV surface antigen, the infant should receive single-antigen HBV vaccine and hepatitis B immune globulin within 12 hours of birth, given at different injection sites.17 The second dose of vaccine should be given at age 1 to 2 months and the third at age 6 months (but not before age 24 weeks). The response to vaccination should be ascertained by testing for surface antigen and surface antibody after completion of the vaccine series, at age 9 to 18 months.
Maternal HBV infection does not contraindicate breastfeeding, as studies suggest that breastfeeding by a mother positive for surface antigen does not increase the infant’s risk of acquiring HBV infection.18
Which HBV therapy for a pregnant woman?
Some evidence supports antiviral therapy with nucleoside/nucleotide analogues in pregnant women who have viral loads of 106 IU/mL or higher. Lamivudine is safe in pregnancy and, together with immunization of the infant, reduces HBV transmission. Interferon-based therapy is contraindicated in pregnant women (and in women who may want to become pregnant) because of interferon’s antiproliferative effects. Nucleoside/nucleotide analogues classified as category B (eg, lamivudine, telbivudine, and tenofovir) could be used when the benefit of treating the pregnant mother outweighs the risk to the mother or fetus,2 although the possible effects of tenofovir on bone density argue against its use during pregnancy or breastfeeding.19
VACCINATION HAS REDUCED THE INCIDENCE OF ACUTE HEPATITIS B
HBV vaccination, a major achievement in HBV management, has played a big role in reducing the incidence of acute HBV infection, especially in children and adolescents.20
The currently available vaccines in the United States contain HBV surface antigen derived through recombinant DNA technology from yeast.21 Two single-antigen vaccines are available in the United States, under the brand names Recombivax HB and Engerix B. Of the three licensed combination vaccines, one (Twinrix) is used in adults, and two (Comvax and Pediarix) are used in infants and young children. Twinrix contains recombinant HBV surface antigens and inactivated hepatitis A virus and it is recommended for people age 18 years and older and at risk of both HBV and hepatitis A infections.20
Vaccinate all infants
All infants should be vaccinated against HBV as part of the recommended childhood immunization schedule. The vaccine is given on a three-dose schedule at birth and again at 1 month and 6 months of age.16 All children and adolescents under age 19 who have not previously received HBV vaccine should be vaccinated at any age with an appropriate dose and schedule.16
Vaccinate adults at risk—or who ask for it
Vaccination is less effective in older people
The three-dose vaccine series given intramuscularly initially, then again at 1 month and 6 months, produces a protective antibody response in approximately 30% to 55% of healthy adults under age 40 after the first dose, 75% after the second dose, and more than 90% after the third dose.21,22 After age 40, however, the proportion of persons who have a protective antibody response after three doses declines to less than 90%, and by age 60, protective levels of antibody develop in only 75%.23
Other factors that lower the response to vaccination are smoking, obesity, genetic factors, and immune suppression.20
Postvaccination serologic testing for immunity is not necessary after routine vaccination of adults, but it is recommended for patients whose subsequent clinical management depends on knowledge of their immune status, such as health care workers who have contact with patients or blood and are at ongoing risk of injuries with sharp instruments or needlesticks; chronic hemodialysis patients and people infected with HIV or otherwise immunocompromised; and sex partners or needle-sharing partners of people positive for HBV surface antigen.20 A protective concentration of HBV surface antibody measured 1 to 2 months after completion of the vaccine series is defined as 10 mIU/mL. Further periodic testing to document persistence of protective levels of surface antibody is not indicated.
If the first series does not ‘take’
Patients who do not respond to the primary vaccine series should complete a second three-dose series, with doses at 0, 1, and 6 months. Serologic testing is done 1 to 2 months after finishing the second series.
Patients who do not have protective levels of HBV surface antibody after revaccination by the appropriate schedule in the deltoid muscle (< 5% of those receiving six doses of hepatitis B vaccine) either are primary nonresponders or are infected with HBV.20 Therefore, they should be tested for HBV surface antigen. If this test is negative, then they should be considered susceptible to HBV infection and should be counseled accordingly.
Contraindications and precautions
HBV vaccination is contraindicated in people with a history of hypersensitivity to baker’s yeast or to a previous dose of HBV vaccine.20 Patients with moderate or severe acute illness at the time the shot is scheduled should wait until they recover before getting HBV vaccine. Pregnancy is not a contraindication.20
Hepatitis b virus (HBV) infection is sometimes challenging to manage because the disease has several stages and many clinical scenarios. HBV-infected patients are a very heterogeneous group, and we cannot apply a single management approach to all.
An understanding of the natural history of HBV infection and its diagnosis, which we reviewed in last month’s issue of this Journal1, is critical to understanding how to manage HBV infection.
In this article, we will review the principles of HBV management in adults, including those on immunosuppressant therapy and pregnant women, and guidelines for HBV vaccination.
WORKUP FOR HBV INFECTION
Once the diagnosis of HBV infection is made,1 a full management strategy should be formulated, as outlined below.
History
When and how did the patient acquire HBV? This information is important to know when making treatment decisions. For example, most acute, adult-onset cases (eg, acquired recently via sexual contact or parenteral drug abuse) resolve spontaneously within a few months, whereas most chronic cases (defined as being positive for HBV surface antigen for more than 6 months) were acquired at birth or in early childhood. Therefore, we should try to determine if the patient’s mother, siblings, household contacts, and sexual partners are positive for HBV surface antigen or have risk factors for HBV infection1; those without infection or immunity to HBV should be vaccinated.
People at risk of HBV infection include:
- Parenteral drug users
- People with multiple sexual partners
- Household contacts and sexual partners of people positive for HBV surface antigen
- Infants born to HBV-infected mothers
- Patients and staff in custodial institutions for the developmentally disabled
- Recipients of certain plasma-derived products (including patients with congenital coagulation defects)
- Hemodialysis patients
- Health and public-safety workers who have contact with blood
- People born in areas where HBV is endemic, and their children.1
Does the patient have risk factors for other infections? Especially look for risk factors for human immunodeficiency virus (HIV) infection (eg, intravenous drug users and men having sex with men) and hepatitis D virus (intravenous drug users and patients from countries where hepatitis D virus infection is common, particularly Eastern Europe, Mediterranean countries, and the Amazon basin).
Does the patient have other modifiable risk factors for progressive liver disease, particularly alcohol abuse and obesity?
Does the patient have symptoms or signs of cirrhosis or hepatocellular carcinoma? Symptoms and signs that involve multiple systems could be extrahepatic manifestations of HBV infection, such as polyarteritis nodosa, which causes abdominal pain, arthralgia, hypertension, and asymmetric polyneuropathy.
Baseline laboratory evaluation
At baseline we should obtain a complete blood count, blood urea nitrogen level, serum creatinine level, liver profile, prothrombin time, urinalysis, and HBV serologic markers. In addition, HBV DNA can be detected in the serum at levels as low as 60 IU/mL, and it should be measured in the initial evaluation to establish a baseline before starting antiviral therapy in patients with chronic HBV infection and subsequently to monitor the response.
All patients with chronic HBV infection should also be tested for serologic markers of hepatitis A and hepatitis C; patients at risk of HIV and hepatitis D should also be tested for these diseases.
Not all patients need liver biopsy
Liver biopsy is the most accurate tool for staging the degree of HBV-related hepatic fibrosis in patients who have no obvious clinical manifestations of cirrhosis.
Not all patients with HBV infection need a biopsy, however. In patients with acute HBV infection, liver biopsy has no benefit except if concomitant pathology (eg, iron overload, nonalcoholic steatohepatitis, or alcoholic steatohepatitis) is suspected. In patients with chronic hepatitis B, liver biopsy is helpful when the viral load alone does not provide sufficient guidance for treatment, eg, when the viral load is less than 2 × 104 IU/mL in a patient positive for hepatitis e antigen or less than 2 × 103 IU/mL in a patient negative for hepatitis e antigen. (The presence of e antigen is a marker of HBV replication and infectivity.1) Biopsy should also be considered in those who have been infected a long time (eg, more than 10 years), because they may have occult cirrhosis, and if they do they may need to undergo antiviral treatment, endoscopy to look for varices, and surveillance for liver cancer.
In some situations it is easy to decide whether antiviral therapy is indicated without resorting to liver biopsy.
We would treat:
- A patient positive for HBV e antigen for more than 6 months, whose HBV DNA level is higher than 2 × 104 IU/mL and whose alanine aminotransferase (ALT) level is high
- A patient with HBV for more than 6 months who is negative for e antigen and who has an HBV DNA level higher than 2 × 103 IU/mL and elevated ALT
- A patient with compensated HBV cirrhosis and an HBV DNA level higher than 2 × 103 IU/mL
- A patient with HBV cirrhosis with decompensation and any detectable HBV DNA.
We would not treat:
- An HBV carrier with a normal ALT level and an HBV DNA level that is lower than 2 × 104 IU/mL or undetectable.
If a patient does not fit into one of these categories but has HBV DNA, a liver biopsy showing significant necroinflammation or fibrosis would be an indication for treatment.
ANTIVIRAL THERAPY
Below, we summarize the main principles of anti-HBV therapy, emphasizing whether to treat and with which agent. Treatment of HBV infection in patients who are also infected with HIV or hepatitis C virus and in those with resistant or refractory hepatitis B is not within the scope of this article.
Acute infection rarely needs treatment
Acute, adult-acquired HBV infection is self-limited in most cases,1 and antiviral therapy is not routinely indicated.
In the rare cases of acute liver failure related to acute HBV infection, use of a nucleoside or nucleotide analogue reverse transcriptase inhibitor (nucleoside/nucleotide analogues) has been recommended, although no properly designed studies have been done.2,3 This recommendation is based on anecdotal experience, the relative safety of the antiviral agents, the serious nature of acute liver failure, and the possible need for emergency liver transplantation that requires prophylaxis against recurrence.
The nucleoside/nucleotide analogues that have been recommended in acute liver failure are lamivudine (Epivir), telbivudine (Tyzeka), and entecavir (Baraclude)—but not adefovir (Hepsera), which has a slow action and potential nephrotoxicity. Interferon drugs are contraindicated because they frequently cause side effects and can worsen hepatitis.4
Patients with acute liver failure should be referred promptly to a liver transplant center, and other management measures should also be started in a timely fashion.
In chronic HBV infection, treatment decisions are individualized
In chronic hepatitis B (ie, lasting > 6 months), treatment decisions should be based on the patient’s clinical situation and test results. The route and duration of infection (if known), history of previous hepatitis flares, ALT levels, current and previous HBV serologic test results and DNA levels, findings on liver biopsy (if previously done), and clinically suspected cirrhosis are all important to consider when deciding whether antiviral therapy is needed.
For many patients with chronic HBV infection, observation without antiviral therapy is warranted, eg:
- Young patients (< 30 years old) who acquired HBV at birth and who have persistently normal ALT levels with no evidence of advanced liver disease, regardless of their HBV DNA level (immune tolerance phase)
- Chronic inactive carriers who have no e antigen, persistently normal ALT levels, and very low or undetectable levels of HBV DNA without evidence of significant liver injury.
These patients can be managed by internists by close monitoring for hepatitis flares with serial ALT measurements along with other general management measures.
Antiviral agents for chronic HBV infection
An ideal agent for treating hepatitis B does not exist. Trade-offs are the essence of agent selection.
Interferons, the first drugs shown to be effective against HBV, can in some respects be considered the best available initial choice, especially in patients positive for hepatitis e antigen. Interferons have numerous side effects but, unlike all the other options, they have a well-defined duration of treatment (4–6 months in patients positive for e antigen). The principal goal of this therapy is disappearance of e antigen.
Interferon-based therapy is not recommended in patients with cirrhosis, however, because of the risk of hepatic decompensation associated with interferon-related flares of hepatitis.4
Although single-agent antiviral therapy may someday be replaced by a multidrug regimen, the data so far are not sufficiently robust to recommend multidrug regimens except possibly in cases of established drug resistance.
Adjunctive management
Vaccinations. All patients with chronic hepatitis B should be vaccinated against hepatitis A if serologic testing indicates they have no immunity to it. Influenza and pneumococcal vaccines are recommended for all patients with chronic liver disease.6
Alcohol rehabilitation. Patients who abuse alcohol should be counseled, and many need consultation with a psychosocial care provider for alcohol rehabilitation.
Smoking cessation. Cigarette smoking is linked to a higher risk of hepatocellular carcinoma in patients with chronic liver disease, including chronic HBV infection.7 Therefore, smokers should be counseled to quit.
Surveillance for hepatocellular carcinoma. Hepatocellular carcinoma can occur in patients with chronic hepatitis B, in most cases on top of cirrhosis, although important exceptions exist. The American Association for the Study of Liver Diseases recommends surveillance for hepatocellular carcinoma in all HBV carriers with cirrhosis and in the following groups regardless of whether they have cirrhosis8:
- Asian men age 40 and older
- Asian women age 50 and older
- African patients age 20 and older
- Patients with a family history of hepatocellular carcinoma
- Possibly, those with high HBV DNA levels and ongoing inflammatory activity.
In the United States, liver ultrasonography and alpha fetoprotein measurement every 6 to 12 months is a reasonable strategy.
If there is evidence of cirrhosis, esophagogastroduodenoscopy is recommended to screen for esophageal and gastric varices.
SCREEN BEFORE CHEMOTHERAPY OR IMMUNOSUPPRESSIVE THERAPY
When patients who are positive for HBV surface antigen undergo immunosuppressive therapy or cancer chemotherapy, from 20% to 50% develop reactivated HBV infection with high HBV viral loads. Even patients who have resolved hepatitis B (ie, negative for HBV surface antigen and positive for surface antibody) may experience hepatitis B reactivation, with serious consequences. Hepatic decompensation and death have been reported during and after chemotherapy, especially in patients with cirrhosis.9 Therefore, patients at risk of HBV infection should be screened for it before starting these therapies.4 Furthermore, perhaps all patients about to undergo anticancer therapies that include anti-B-cell or anti-T-cell therapies or hematopoietic stem cell transplantation should be screened.9
Recent data indicate that many oncologists have not been screening for HBV.10 Hence, more effort is needed to make this important testing routine in this setting.
The initial tests in these patients should be liver chemistry tests, HBV surface antigen, HBV surface antibody, and HBV core antibody. In those who test positive for surface antigen, one should test for e antigen, e antibody, and HBV DNA.
Patients with indications for anti-HBV therapy (Table 1) should receive antiviral therapy. Otherwise, those positive for surface antigen should start taking anti-HBV medication at the start of chemotherapy or immunosuppressive therapy and should continue taking it until 6 months after the chemotherapy or immunosuppressive therapy is finished.4 Some experts also recommend starting anti-HBV therapy 7 days before the chemotherapy or immunosuppressive therapy and continuing it for 1 year afterward.10 Those with HBV DNA levels higher than 2 × 103 IU/mL should continue HBV therapy until they reach the same treatment end points as for immunocompetent patients as outlined above.4
Because we have little information on patients who are negative for surface antigen and who have antibodies against surface antigen and core antigen, we cannot make an unequivocal recommendation for anti-HBV therapy in this group.11 Rather, these patients should be monitored during immunosuppressive treatment, preferably with liver chemistry tests and HBV DNA titers, and antiviral drugs should be given as a deferred therapy upon evidence of HBV reactivation.9 Few cases of fatal hepatic failure in patients with this serologic pattern receiving rituximab (Rituxan) have been reported.12–14
With their small risk of drug resistance and rapid onset of action, entecavir or tenofovir may be the preferred anti-HBV therapy in patients undergoing immunosuppression or chemotherapy, especially in those requiring prolonged immunosuppressive therapy (longer than 12 months). In those requiring shorter courses, lamivudine or telbivudine is a possible alternative.4
OUTCOMES OF LIVER TRANSPLANTATION HAVE IMPROVED IN HBV PATIENTS
The early results of liver transplantation for HBV were discouraging because many patients developed rapidly progressive recurrent disease (fibrosing cholestatic hepatitis) and died within 12 to 18 months after the operation.15 However, patients with HBV are now treated perioperatively with lamivudine or adefovir combined with prolonged administration of hepatitis B immune globulin, and their survival now exceeds that of patients who receive transplants for many other conditions.16
Like patients with cirrhosis due to other causes, those with HBV-related cirrhosis who have any of the following should be referred for liver transplantation evaluation16:
- A Child-Turcotte-Pugh score of 7 or higher (Table 5).
- A major complication of cirrhosis such as ascites, variceal bleeding, hepatocellular carcinoma, or hepatic encephalopathy.
PREVENTING VERTICAL TRANSMISSION
The major problem in young women with chronic HBV infection is the risk of vertical (mother-to-infant) transmission at delivery. The risk varies, depending on the viral load and e antigen status of the mother at the time of delivery; if she is positive for e antigen, the risk of HBV infection in the newborn is 70% to 90% by the age of 6 months if the newborn does not receive postexposure immunoprophylaxis; if the mother is positive for surface antigen but negative for e antigen, the risk of chronic infection is less than 10%, even without postexposure immunoprophylaxis.17
All women should be tested for HBV surface antigen early in pregnancy each time they become pregnant. If a patient tests negative early in pregnancy but continues behaviors that put her at risk of HBV infection (eg, having multiple sexual partners, having had a sex partner positive for surface antigen, using injection drugs, or contracting any sexually transmitted disease), she should be retested at the time of admission to the hospital for delivery.17 This also includes women who were not screened prenatally and those with clinical hepatitis.
Vaccine and immune globulin for the infant
If the mother is positive for HBV surface antigen, the infant should receive single-antigen HBV vaccine and hepatitis B immune globulin within 12 hours of birth, given at different injection sites.17 The second dose of vaccine should be given at age 1 to 2 months and the third at age 6 months (but not before age 24 weeks). The response to vaccination should be ascertained by testing for surface antigen and surface antibody after completion of the vaccine series, at age 9 to 18 months.
Maternal HBV infection does not contraindicate breastfeeding, as studies suggest that breastfeeding by a mother positive for surface antigen does not increase the infant’s risk of acquiring HBV infection.18
Which HBV therapy for a pregnant woman?
Some evidence supports antiviral therapy with nucleoside/nucleotide analogues in pregnant women who have viral loads of 106 IU/mL or higher. Lamivudine is safe in pregnancy and, together with immunization of the infant, reduces HBV transmission. Interferon-based therapy is contraindicated in pregnant women (and in women who may want to become pregnant) because of interferon’s antiproliferative effects. Nucleoside/nucleotide analogues classified as category B (eg, lamivudine, telbivudine, and tenofovir) could be used when the benefit of treating the pregnant mother outweighs the risk to the mother or fetus,2 although the possible effects of tenofovir on bone density argue against its use during pregnancy or breastfeeding.19
VACCINATION HAS REDUCED THE INCIDENCE OF ACUTE HEPATITIS B
HBV vaccination, a major achievement in HBV management, has played a big role in reducing the incidence of acute HBV infection, especially in children and adolescents.20
The currently available vaccines in the United States contain HBV surface antigen derived through recombinant DNA technology from yeast.21 Two single-antigen vaccines are available in the United States, under the brand names Recombivax HB and Engerix B. Of the three licensed combination vaccines, one (Twinrix) is used in adults, and two (Comvax and Pediarix) are used in infants and young children. Twinrix contains recombinant HBV surface antigens and inactivated hepatitis A virus and it is recommended for people age 18 years and older and at risk of both HBV and hepatitis A infections.20
Vaccinate all infants
All infants should be vaccinated against HBV as part of the recommended childhood immunization schedule. The vaccine is given on a three-dose schedule at birth and again at 1 month and 6 months of age.16 All children and adolescents under age 19 who have not previously received HBV vaccine should be vaccinated at any age with an appropriate dose and schedule.16
Vaccinate adults at risk—or who ask for it
Vaccination is less effective in older people
The three-dose vaccine series given intramuscularly initially, then again at 1 month and 6 months, produces a protective antibody response in approximately 30% to 55% of healthy adults under age 40 after the first dose, 75% after the second dose, and more than 90% after the third dose.21,22 After age 40, however, the proportion of persons who have a protective antibody response after three doses declines to less than 90%, and by age 60, protective levels of antibody develop in only 75%.23
Other factors that lower the response to vaccination are smoking, obesity, genetic factors, and immune suppression.20
Postvaccination serologic testing for immunity is not necessary after routine vaccination of adults, but it is recommended for patients whose subsequent clinical management depends on knowledge of their immune status, such as health care workers who have contact with patients or blood and are at ongoing risk of injuries with sharp instruments or needlesticks; chronic hemodialysis patients and people infected with HIV or otherwise immunocompromised; and sex partners or needle-sharing partners of people positive for HBV surface antigen.20 A protective concentration of HBV surface antibody measured 1 to 2 months after completion of the vaccine series is defined as 10 mIU/mL. Further periodic testing to document persistence of protective levels of surface antibody is not indicated.
If the first series does not ‘take’
Patients who do not respond to the primary vaccine series should complete a second three-dose series, with doses at 0, 1, and 6 months. Serologic testing is done 1 to 2 months after finishing the second series.
Patients who do not have protective levels of HBV surface antibody after revaccination by the appropriate schedule in the deltoid muscle (< 5% of those receiving six doses of hepatitis B vaccine) either are primary nonresponders or are infected with HBV.20 Therefore, they should be tested for HBV surface antigen. If this test is negative, then they should be considered susceptible to HBV infection and should be counseled accordingly.
Contraindications and precautions
HBV vaccination is contraindicated in people with a history of hypersensitivity to baker’s yeast or to a previous dose of HBV vaccine.20 Patients with moderate or severe acute illness at the time the shot is scheduled should wait until they recover before getting HBV vaccine. Pregnancy is not a contraindication.20
- Elgouhari HM, Abu-Rajab Tamimi T, Carey WD. Hepatitis B virus infection: understanding its epidemiology, course, and diagnosis. Cleve Clin J Med 2008; 75:881–889.
- Stravitz RT, Kramer AH, Davern T, et al. Intensive care of patients with acute liver failure: recommendations of the U.S. Acute Liver Failure Study Group. Crit Care Med 2007; 35:2498–2508.
- Hoofnagle JH, Doo E, Liang TJ, Fleischer R, Lok AS. Management of hepatitis B: summary of a clinical research workshop. Hepatology 2007; 45:1056–1075.
- Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Keeffe EB, Dieterich DT, Han SH, et al. A treatment algorithm for the management of chronic hepatitis B virus infection in the United States. Clin Gastroenterol Hepatol 2004; 2:87–106.
- Jacobs RJ, Meyerhoff AS, Saab S. Immunization needs of chronic liver disease patients seen in primary care versus specialist settings. Dig Dis Sci 2005; 50:1525–1531.
- Wang LY, You SL, Lu SN, et al. Risk of hepatocellular carcinoma and habits of alcohol drinking, betel quid chewing and cigarette smoking: a cohort of 2416 HBsAg–seropositive and 9421 HBsAg–seronegative male residents in Taiwan. Cancer Causes Control 2003; 14:241–250.
- Bruix J, Sherman M; Practice Guidelines Committee, American Association for the Study of Liver Diseases. Management of hepatocellular carcinoma. Hepatology 2005; 42:1208–1236.
- Yeo W, Johnson PJ. Diagnosis, prevention and management of hepatitis B virus reactivation during anticancer therapy. Hepatology 2006; 43:209–220.
- Tran T, Oh M, Poordad F, Martin P. Screening for hepatitis B in chemotherapy patients: survey of current oncology practices [abstract]. Hepatology 2007; 46:978A.
- Kohrt HE, Ouyang DL, Keeffe EB. Antiviral prophylaxis for chemotherapy–induced reactivation of chronic hepatitis B virus infection. Clin Liver Dis 2007; 11:965–991.
- Westhoff TH, Jochimsen F, Schmittel A, et al. Fatal hepatitis B virus reactivation by an escape mutant following rituximab therapy. Blood 2003; 102:1930.
- Sarrecchia C, Cappelli A, Aiello P. HBV reactivation with fatal fulminating hepatitis during rituximab treatment in a subject negative for HBsAg and positive for HBsAb and HBcAb. J Infect Chemother 2005; 11:189–191.
- Law JK, Ho JK, Hoskins PJ, Erb SR, Steinbrecher UP, Yoshida FM. Fatal reactivation of hepatitis B post-chemotherapy for lymphoma in a hepatitis B surface antigen-negative, hepatitis B core antibody-positive patient: potential implications for future prophylaxis recommendations. Leuk Lymphoma 2005; 46:1085–1089.
- Todo S, Demetris AJ, Van Thiel D, Teperman L, Fung JJ, Starzl TE. Orthotopic liver transplantation for patients with hepatitis B virus-related liver disease. Hepatology 1991; 13:619–626.
- Murray KF, Carithers RLAASLD. AASLD practice guidelines: Evaluation of the patient for liver transplantation. Hepatology 2005; 41:1407–1432.
- Mast EE, Margolis HS, Fiore AE, et al. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP) part 1: immunization of infants, children, and adolescents. MMWR Recomm Rep 2005; 54:1–31.
- Beasley RP, Stevens CE, Shiao IS, Meng HC. Evidence against breast–feeding as a mechanism for vertical transmission of hepatitis B. Lancet 1975; 2:740–741.
- Parsonage MJ, Wilkins EG, Snowden N, Issa BG, Savage MW. The development of hypophosphataemic osteomalacia with myopathy in two patients with HIV infection receiving tenofovir therapy. HIV Med 2005; 6:341–346.
- Mast EE, Weinbaum CM, Fiore AE, et al. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP) Part II: immunization of adults. MMWR Recomm Rep 2006; 55:1–33.
- Andre FE. Summary of safety and efficacy data on a yeast–derived hepatitis B vaccine. Am J Med 1989; 87:14S–20S.
- Zajac BA, West DJ, McAleer WJ, Scolnick EM. Overview of clinical studies with hepatitis B vaccine made by recombinant DNA. J Infect 1986; 13( suppl A):39–45.
- Averhoff F, Mahoney F, Coleman P, Schatz G, Hurwitz E, Margolis H. Immunogenicity of hepatitis B vaccines: implications for persons at occupational risk for hepatitis B virus infection. Am J Prev Med 1998; 15:1–8.
- Elgouhari HM, Abu-Rajab Tamimi T, Carey WD. Hepatitis B virus infection: understanding its epidemiology, course, and diagnosis. Cleve Clin J Med 2008; 75:881–889.
- Stravitz RT, Kramer AH, Davern T, et al. Intensive care of patients with acute liver failure: recommendations of the U.S. Acute Liver Failure Study Group. Crit Care Med 2007; 35:2498–2508.
- Hoofnagle JH, Doo E, Liang TJ, Fleischer R, Lok AS. Management of hepatitis B: summary of a clinical research workshop. Hepatology 2007; 45:1056–1075.
- Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Keeffe EB, Dieterich DT, Han SH, et al. A treatment algorithm for the management of chronic hepatitis B virus infection in the United States. Clin Gastroenterol Hepatol 2004; 2:87–106.
- Jacobs RJ, Meyerhoff AS, Saab S. Immunization needs of chronic liver disease patients seen in primary care versus specialist settings. Dig Dis Sci 2005; 50:1525–1531.
- Wang LY, You SL, Lu SN, et al. Risk of hepatocellular carcinoma and habits of alcohol drinking, betel quid chewing and cigarette smoking: a cohort of 2416 HBsAg–seropositive and 9421 HBsAg–seronegative male residents in Taiwan. Cancer Causes Control 2003; 14:241–250.
- Bruix J, Sherman M; Practice Guidelines Committee, American Association for the Study of Liver Diseases. Management of hepatocellular carcinoma. Hepatology 2005; 42:1208–1236.
- Yeo W, Johnson PJ. Diagnosis, prevention and management of hepatitis B virus reactivation during anticancer therapy. Hepatology 2006; 43:209–220.
- Tran T, Oh M, Poordad F, Martin P. Screening for hepatitis B in chemotherapy patients: survey of current oncology practices [abstract]. Hepatology 2007; 46:978A.
- Kohrt HE, Ouyang DL, Keeffe EB. Antiviral prophylaxis for chemotherapy–induced reactivation of chronic hepatitis B virus infection. Clin Liver Dis 2007; 11:965–991.
- Westhoff TH, Jochimsen F, Schmittel A, et al. Fatal hepatitis B virus reactivation by an escape mutant following rituximab therapy. Blood 2003; 102:1930.
- Sarrecchia C, Cappelli A, Aiello P. HBV reactivation with fatal fulminating hepatitis during rituximab treatment in a subject negative for HBsAg and positive for HBsAb and HBcAb. J Infect Chemother 2005; 11:189–191.
- Law JK, Ho JK, Hoskins PJ, Erb SR, Steinbrecher UP, Yoshida FM. Fatal reactivation of hepatitis B post-chemotherapy for lymphoma in a hepatitis B surface antigen-negative, hepatitis B core antibody-positive patient: potential implications for future prophylaxis recommendations. Leuk Lymphoma 2005; 46:1085–1089.
- Todo S, Demetris AJ, Van Thiel D, Teperman L, Fung JJ, Starzl TE. Orthotopic liver transplantation for patients with hepatitis B virus-related liver disease. Hepatology 1991; 13:619–626.
- Murray KF, Carithers RLAASLD. AASLD practice guidelines: Evaluation of the patient for liver transplantation. Hepatology 2005; 41:1407–1432.
- Mast EE, Margolis HS, Fiore AE, et al. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP) part 1: immunization of infants, children, and adolescents. MMWR Recomm Rep 2005; 54:1–31.
- Beasley RP, Stevens CE, Shiao IS, Meng HC. Evidence against breast–feeding as a mechanism for vertical transmission of hepatitis B. Lancet 1975; 2:740–741.
- Parsonage MJ, Wilkins EG, Snowden N, Issa BG, Savage MW. The development of hypophosphataemic osteomalacia with myopathy in two patients with HIV infection receiving tenofovir therapy. HIV Med 2005; 6:341–346.
- Mast EE, Weinbaum CM, Fiore AE, et al. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP) Part II: immunization of adults. MMWR Recomm Rep 2006; 55:1–33.
- Andre FE. Summary of safety and efficacy data on a yeast–derived hepatitis B vaccine. Am J Med 1989; 87:14S–20S.
- Zajac BA, West DJ, McAleer WJ, Scolnick EM. Overview of clinical studies with hepatitis B vaccine made by recombinant DNA. J Infect 1986; 13( suppl A):39–45.
- Averhoff F, Mahoney F, Coleman P, Schatz G, Hurwitz E, Margolis H. Immunogenicity of hepatitis B vaccines: implications for persons at occupational risk for hepatitis B virus infection. Am J Prev Med 1998; 15:1–8.
KEY POINTS
- Patients with HBV infection should be screened for hepatocellular carcinoma, especially if they have cirrhosis.
- Nucleoside and nucleotide analogue reverse transcriptase inhibitors are easy to use and therefore are usually the first-line therapy. Problems with these agents are that the optimal treatment duration is not known, and that drug resistance can emerge.
- Patients with advanced liver disease or hepatocellular carcinoma should be referred promptly for possible liver transplantation.
- Candidates for immunosuppressant therapy or cytotoxic chemotherapy should be screened for HBV, as this therapy can cause a potentially fatal flare of HBV.
- People at risk should be vaccinated; many have not been.
Hepatitis B virus infection: Understanding its epidemiology, course, and diagnosis
Our knowledge about hepatitis B and related diseases has dramatically increased since the discovery of the causative virus, HBV, in 1963. Despite effective vaccination, hepatitis B still constitutes a major public health problem.
In two parts, this comprehensive review will highlight a practical clinical approach to HBV infection. In this first part, we discuss the epidemiology, natural history, and diagnosis of HBV infection. In the second part, to be published in the next issue of this journal, we will review the general principles of its management, its management in patients on immunosuppressant therapy and in pregnant women, and HBV vaccination.
COMMON IN ASIA, LESS SO IN AMERICA
More than 2 billion people—one-third of the world’s population—alive today have been infected with HBV at some time in their life, and of these, about 350 million remain infected.1 Every year, 1 million people die of HBV-related cirrhosis or hepatocellular carcinoma, which means that HBV takes a life every 30 seconds.2
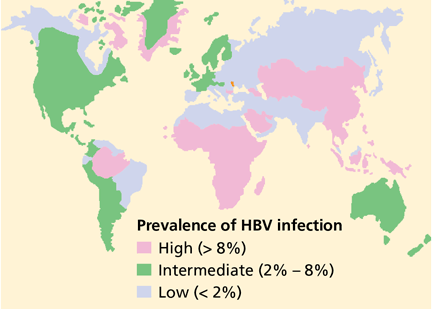
The incidence of acute hepatitis B in the United States has declined from 8.5 per 100,000 population in 1990 to 2.1 per 100,000 population in 2004, with the greatest declines (94%) in children and adolescents, coincident with an increase in hepatitis B vaccination in these age groups.5 Despite these advances, HBV still causes a considerable number of cases of cirrhosis, cancer, and death—about 5,000 deaths each year in the United States.
HBV HAS FOUR GENES, EIGHT GENOTYPES
HBV is a DNA virus of the Hepadnaviridae family. Its genome is double-stranded with four genes, each one encoding a specific structural protein or proteins6,7:
- S gene, for the viral envelope (surface antigen)
- C gene, for both the nucleocapsid (core) antigen and the pre-core (e) antigen
- X gene, for two regulatory proteins required for HBV replication
- P gene, for DNA polymerase.

Eight genotypes of HBV (labeled A though H) have been identified.13,14 All eight have been found in the United States, but genotype A accounts for 35% of cases, genotype B for 22%, and genotype C for 31%.15
The clinical significance of HBV genotypes is not as clear as that of hepatitis C virus genotypes. Although recent data have suggested that different HBV genotypes may be associated with different rates of progression of liver disease and different rates of response to interferon therapy,13 these data were not enough to recommend routine testing for HBV genotypes in clinical practice.16
In HBV infection, the virus itself does not injure liver cells. Rather, the damage of hepatitis is immune-mediated and begins to appear as the host’s immune system attempts to clear the virus.
MARKERS OF HBV INFECTION
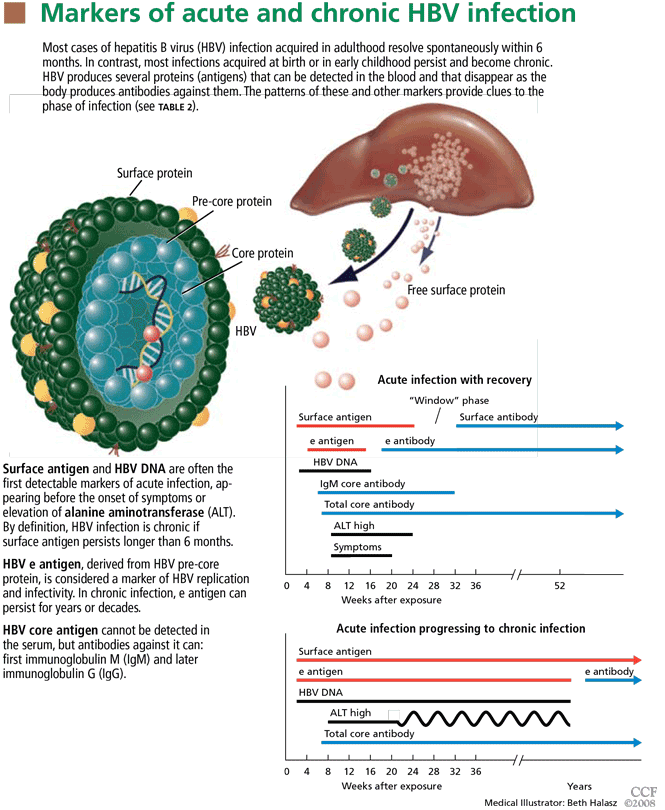
HBV surface antigen and HBV DNA are often the first detectable markers of acute infection, appearing before the onset of symptoms or before elevation of alanine aminotransferase (ALT) occurs. By definition, an HBV infection is chronic if surface antigen persists longer than 6 months.
HBV e antigen, derived from pre-core protein, is considered a marker of HBV replication and infectivity. In chronic infection, e antigen can persist for years or decades.
HBV core antigen cannot be detected in the serum, but antibodies against it can, first immunoglobulin M (IgM) and later immunoglobulin G (IgG).
TRANSMISSION: VERTICAL OR HORIZONTAL
Because HBV replicates profusely and produces high titers in the blood (108 to 1,010 virions/ mL), any parenteral or mucosal exposure to infected blood poses a high risk of HBV acquisition. The risk of HBV transmission from a single needlestick is 1% to 6% if the blood is positive for HBV surface antigen but negative for HBV e antigen, and 22% to 40% if positive for both antigens.17–19 Saliva, nasopharyngeal fluid, breast milk, semen, urine, and cervical secretions can also harbor HBV.20
Worldwide, perinatal (vertical) transmission is the predominant mode of HBV transmission, whereas intravenous drug abuse and unprotected sexual intercourse are the main routes of infection in areas of low prevalence, such as the United States. In sub-Saharan Africa, Alaska, and Mediterranean countries, transmission of HBV usually occurs horizontally during childhood, presumably via contact with nonintact skin.21–24 Saliva has also been thought to be the route of HBV transmission in sporadic cases through human bites.25
People at risk of HBV infection include:
- Parenteral drug users
- People with multiple sexual partners
- Household contacts and sexual partners of people who are positive for HBV surface antigen
- Infants born to HBV-infected mothers
- Patients and staff in custodial institutions for the developmentally disabled
- Recipients of certain plasma-derived products (including patients with congenital coagulation defects)
- Hemodialysis patients
- Health and public-safety workers who have contact with blood
- People born in areas where HBV is endemic, and their children.
These people—as well as all pregnant women, patients infected with hepatitis C virus or human immunodeficiency virus, and patients with chronically elevated ALT or aspartate aminotransferase (AST) levels—should be screened for HBV infection with serologic markers.
CLINICAL MANIFESTATIONS VARY
HBV infection, acute or chronic, has variable manifestations. During the acute stage, HBV infection can manifest as anicteric (subclinical) hepatitis, icteric hepatitis, or, rarely, acute fulminant hepatitis. Chronic HBV infection can be asymptomatic (the HBV surface antigen carrier state), or it can be manifested by symptoms and signs of cirrhosis or hepatocellular carcinoma or both. Extrahepatic manifestations, including serum sickness, polyarteritis nodosa, essential mixed cryoglobulinemia, membranous glomerulonephritis, and aplastic anemia, have been reported in patients with HBV infection.26
Acute hepatitis B
The incubation period of HBV ranges from 2 weeks to 4 months. Initially, patients complain of fatigue, malaise, anorexia, right upper quad-rant discomfort, or flu-like symptoms (coryza, photophobia, headache, and myalgia); then jaundice becomes apparent, usually within 10 days of the onset of symptoms. Low-grade fever, jaundice, and mildly tender hepatomegaly are the most common signs. Generalized lymphadenopathy is not a feature of acute HBV infection. If the patient also has hepatitis D virus infection or underlying liver disease (eg, alcoholic liver disease), then acute HBV infection may be more severe.
In the acute phase, ALT and AST levels rise, sometimes to values above 1,000 IU/L. In icteric hepatitis, bilirubin levels also rise, usually after the ALT level does. Although the peak ALT level reflects the hepatocellular injury, it has no prognostic value. With recovery, ALT levels normalize in 1 to 4 months.
Acute fulminant hepatitis B occurs in 0.1% to 0.5% of patients, and causes about 10% of cases of acute liver failure in the United States.27 Patients typically present with rapidly progressive acute hepatitis characterized by signs of liver failure, such as coagulopathy, encephalopathy, and cerebral edema.
In the so-called window phase, laboratory testing may not reveal HBV surface antigen because of early clearance but shows IgM antibody against the HBV core antigen. HBV DNA may be low or undetected.
Chronic hepatitis B
Chronic hepatitis B is usually diagnosed as a result of a workup for abnormal liver function tests or as a result of screening patients at risk for HBV infection. Many patients with chronic hepatitis B have no symptoms or have nonspecific symptoms such as fatigue or right upper quadrant discomfort.
Acute exacerbations due to HBV e antigen seroreversion (ie, in which e antigen reappears) occasionally occur in patients with chronic hepatitis B. Most of these exacerbations are asymptomatic, but occasionally an acute hepatitis-like clinical picture with detectable IgM antibody against the core antigen occurs, leading to misdiagnosis of acute HBV infection in patients not previously known to have chronic HBV infection.28
In late cases, signs of cirrhosis such as jaundice, ascites, splenomegaly, pedal edema, encephalopathy, or variceal bleeding can be present.
Hepatocellular carcinoma should be suspected in cirrhotic patients with new-onset right upper quadrant pain, rapidly developing ascites, a palpable liver mass, or hepatic encephalopathy. Other nonspecific features of hepatocellular carcinoma include watery diarrhea, hypoglycemia, and certain cutaneous manifestations such as acanthosis nigricans and the Leser-Trelat sign (multiple pruritic seborrheic keratoses of sudden onset).
In chronic hepatitis B, liver enzyme levels can be normal, even in patients with wellcompensated cirrhosis. ALT levels may range from normal to five times higher than normal. Thrombocytopenia, hypoalbuminemia, direct hyperbilirubinemia, and prolonged prothrombin time suggest cirrhosis.
Findings of chronic hepatitis B on liver biopsy range from minimal inflammation to cirrhosis. The most characteristic histologic feature of chronic HBV infection is the “ground-glass hepatocyte,” which is due to intracellular accumulation of HBV surface antigen. 29
FEW ADULTS (BUT MANY CHILDREN) REMAIN CHRONICALLY INFECTED
HBV surface antigen can be detected in the blood approximately 2 to 4 weeks after inoculation. Simultaneously, HBV DNA, usually in very high levels, is also detectable in the blood. However, in the rare cases of acute fulminant hepatitis, HBV DNA levels can be low or undetectable at the time of presentation because the immune system mounts a robust response with extensive damage to HBVinfected hepatocytes.
The rate of spontaneous recovery from acute HBV infection varies, depending on the patient’s age at the time of HBV acquisition and the patient’s immune status. Fewer than 5% of immunocompetent adults infected with HBV remain chronically infected, defined as being positive for HBV surface antigen for more than 6 months. On the other hand, 80% to 90% of infected infants and about 20% to 50% of children 1 to 5 years old at the time of acute infection remain chronically infected.21
Four phases of chronic HBV infection

The immune tolerance phase, the initial phase of chronic HBV infection, is seen almost exclusively in those who acquired HBV infection vertically or during early childhood. Although patients have high HBV DNA levels, they do not have significant liver disease. This discrepancy is thought to be related to the immune tolerance to HBV; however, the exact mechanism of that tolerance is unclear.31
Only 15% of those with immune tolerance have spontaneous HBV e antigen seroconversion (ie, loss of e antigen and appearance of anti-e antibody) within 20 years after infection. 32
The immune clearance phase (HBV e antigen-positive chronic hepatitis) appears about 20 to 30 years after the onset of the immune tolerance phase in patients who acquire HBV early in life. It is also often seen in patients with infections acquired late in childhood or in adulthood.
This phase marks the start of an immunemediated process aimed at clearing the viral infection, but it also leads to concomitant hepatocellular injury. Spontaneous clearance of the e antigen increases in this phase to an annual rate of 10% to 20%.32,33 The strongest predictors of spontaneous e antigen seroconversion are old age, an elevated ALT level, and an acute exacerbation.26
Although ALT levels are elevated and there is evidence on liver biopsy of chronic active hepatitis, this phase is usually asymptomatic. Rarely, however, it presents with an acute flare of hepatitis, sometimes accompanied by IgM antibodies against the HBV core antigen (in low titer), leading to an incorrect diagnosis of acute HBV infection.
Depending on the duration of the chronic hepatitis and the frequency and severity of flares, about 12% to 20% of patients in the immune-clearance phase develop serious liver disease within 5 years.31
The inactive carrier phase following HBV e antigen seroconversion is characterized by undetectable or low HBV DNA levels (< 1,000 copies/mL), normal ALT levels, and minimal or no necroinflammation on liver biopsy. 30 Such patients should be followed with serial testing, as 4% to 20% of them spontaneously revert to being positive for e antigen at least once.16 On the other hand, only 0.5% to 2% of surface antigen carriers in western countries clear themselves of surface antigen yearly, but up to half of those who clear the surface antigen have low-level HBV viremia. 34
The reactivation phase (HBV e antigennegative chronic hepatitis) is seen in some HBV-infected patients, especially those from Asia and southern Europe, in whom the virus has a spontaneous pre-core or core mutation that makes infected cells unable to secrete the e antigen. Although these patients have no e antigen in their blood, they do have intermittent or persistent elevation of ALT, elevated HBV DNA, and histopathologic findings of chronic hepatitis. Compared with those in the immune clearance phase, patients in the reactivation phase tend to be older and to have lower HBV DNA levels but advanced hepatic damage.
Immunity to HBV infection is characterized by loss of HBV surface antigen, DNA, e antigen, and anti-core antigen IgM with development of anti-surface antigen antibody and anti-core antigen IgG (total anti-core antigen antibody). The presence of anti-surface antigen antibody and anti-core antigen IgG together differentiates natural immunity through resolved infection from that which is acquired through vaccination, which is denoted by isolated anti-surface antigen antibody.

Cirrhosis, liver failure, cancer
Cirrhosis, hepatic decompensation, and hepatocellular carcinoma are the major long-term complications of HBV infection. In untreated patients, the annual rate of progression to cirrhosis has been estimated to be 2% to 6% in patients with HBV e antigen-positive chronic hepatitis and 8% to 9% in those with e antigen- negative chronic hepatitis.30
The likely explanation for these surprising cirrhosis rates is that e antigen-negative chronic hepatitis usually represents a late stage of the disease, and patients in this phase are usually older and have more advanced liver disease.
Subsequently, the annual rate of progression from compensated cirrhosis to hepatic decompensation has been estimated to be about 5%.35
Across all the stages described above, a high serum HBV DNA level has been shown to be a strong predictor of progression to cirrhosis in patients with chronic HBV infection. In a population-based prospective cohort study of 3,582 untreated HBV-infected patients in Taiwan, Iloeje et al36 found that, compared with patients with serum HBV DNA levels lower than 104 copies/mL, those with levels of 104 or higher had an adjusted relative risk of cirrhosis of 2.5. The relative risk rose to 5.9 with HBV DNA levels of 105 or higher, and 9.8 with levels of 106 copies/mL or higher. More studies in different patient populations are needed for confirmation.

The most important risk factor for hepatocellular carcinoma in HBV-infected patients is cirrhosis, but this cancer can also develop in noncirrhotic livers.37 The annual rate of hepatocelluar carcinoma has been estimated to be higher (2.5%–3%) in patients with cirrhosis than in noncirrhotic carriers (0.5%–1%).30,35–38 Risk factors for cirrhosis and hepatocellular carcinoma are summarized in Table 4.16,30
- World Health Organization. Hepatitis B. www.who.int/csr/disease/hepatitis/whocdscsrlyo20022/en. Accessed 11/10/2008.
- Center for Disease Control and Prevention. HBV a silent killer. www.cdc.gov/ncidod/diseases/hepatitis/b/hbv_silent_killer. Accessed 2/19/2007.
- McQuillan GM, Coleman PJ, Kruszon-Moran D, Moyer LA, Lambert SB, Margolis HS. Prevalence of hepatitis B virus infection in the United States: the National Health and Nutrition Examination Surveys, 1976 through 1994. Am J Public Health 1999; 89:14–18.
- Armstrong GL, Mast EE, Wojczynski M, Margolis HS. Childhood hepatitis B virus infections in the United States before hepatitis B immunization. Pediatrics 2001; 108:1123–1128.
- Mast EE, Weinbaum CM, Fiore AE, et al. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP). Part II: immunization of adults. MMWR Recomm Rep 2006 Dec 8; 55(RR-16):1–33.
- Gish RG, Gadano AC. Chronic hepatitis B: current epidemiology in the Americas and implications for management. J Viral Hepatol 2006; 13:787–798.
- Wei Y, Tiollais PK. Molecular biology of hepatitis B virus. Clin Liver Dis 1999; 3:189–219.
- Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev 2000; 64:51–68.
- Hunt CM, McGill JM, Allen MI, Condreay LD. Clinical relevance of hepatitis B viral mutations. Hepatology 2000; 3:1037–1044.
- Allen MI, Deslauriers M, Andrews CW, et al. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Lamivudine Clinical Investigation Group. Hepatology 1998; 27:1670–1677.
- Hadziyannis SJ, Vassilopoulos D. Hepatitis B e antigen- negative chronic hepatitis B. Hepatology 2001; 34:617–624.
- Wai CT, Fontana RJ. Clinical significance of hepatitis B virus genotypes, variants, and mutants. Clin Liver Dis 2004; 8:321–352.
- Fung SK, Lok AS. Hepatitis B virus genotypes: do they play a role in the outcome of HBV infection? Hepatology 2004; 40:790–792.
- Norder H, Courouce AM, Coursaget P, et al. Genetic diversity of hepatitis B virus strains derived worldwide: genotypes, subgenotypes, and HBsAg subtypes. Intervirology 2004; 47:289–309.
- Chu CJ, Keeffe EB, Han SH, et al. Hepatitis B virus genotypes in the United States: results of a nationwide study. Gastroenterology 2003; 125:444–451.
- Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Mast EE, Alter MJ. Prevention of hepatitis B virus infection among health-care workers. In:Ellis RE, editor. Hepatitis B Vaccines in Clinical Practice. New York: Marcel Dekker, 1993:295–307.
- Werner BG, Grady GF. Accidental hepatitis-B-surface-antigen-positive inoculations: use of e antigen to estimate infectivity. Ann Intern Med 1982; 97:367–369.
- Gerberding JL. Management of occupational exposures to blood-borne viruses. N Engl J Med 1995; 332:444–451.
- Kidd-Ljunggren K, Holmberg A, Blackberg J, Lindqvist B. High levels of hepatitis B virus DNA in body fluids from chronic carriers. J Hosp Infect 2006; 64:352–357.
- McMahon BJ, Alward WL, Hall DB, et al. Acute hepatitis B virus infection: relation of age to the clinical expression of disease and subsequent development of the carrier state. J Infect Dis 1985; 151:599–603.
- Dusheiko GM, Brink BA, Conradie JD, Marimuthu T, Sher R. Regional prevalence of hepatitis B, delta, and human immunodeficiency virus infection in southern Africa: a large population survey. Am J Epidemiol 1989; 129:138–45.
- Bortolotti F, Guido M, Bartolacci S, et al. Chronic hepatitis B in children after e antigen seroclearance: final report of a 29-year longitudinal study. Hepatology 2006; 43:556–562.
- Moreno MR, Otero M, Millan A, et al. Clinical and histological outcome after hepatitis B e antigen to antibody seroconversion in children with chronic hepatitis B. Hepatology 1999:572–575.
- Hui AY, Hung LC, Tse PC, Leung WK, Chan PK, Chan HL. Transmission of hepatitis B by human bite--confirmation by detection of virus in saliva and full genome sequencing. J Clin Virol 2005; 33:254–256.
- Cacoub P, Saadoun D, Bourlière M, et al. Hepatitis B virus genotypes and extrahepatic manifestations. J Hepatol 2005; 43:764–770.
- Schiodt FV, Atillasoy E, Shakil AO, et al. Etiology and outcome for 295 patients with acute liver failure in the United States. Liver Transplant Surg 1999; 5:29–34.
- Chu CM, Liaw YF, Pao CC, Huang MJ. The etiology of acute hepatitis superimposed upon previously unrecognized asymptomatic HBsAg carriers. Hepatology 1989; 9:452–456.
- Gerber MA, Hadziyannis S, Vissoulis C, et al. Electron microscopy and immunoelectronmicroscopy of cytoplasmic hepatitis B antigen in hepatocytes. Am J Pathol 1974; 75:489–502.
- Yim HJ, Lok AS. Natural history of chronic hepatitis B virus infection: what we knew in 1981 and what we know in 2005. Hepatology 2006; 43:S173–S181.
- Pungpapong S, Kim WR, Poterucha JJ. Natural history of hepatitis B virus infection: an update for clinicians. Mayo Clin Proc 2007; 82:967–975.
- Lok AS, Lai CL, Wu PC, Leung EK, Lam TS. Spontaneous hepatitis B e antigen to antibody seroconversion and reversion in Chinese patients with chronic hepatitis B virus infection. Gastroenterology 1987; 92:1839–1843.
- McMahon BJ, Holck P, Bulkow L, Snowball M. Serologic and clinical outcomes in 1536 Alaska Natives chronically infected with hepatitis B virus. Ann Intern Med 2001; 135:759–768.
- McMahon BJ. Epidemiology and natural history of hepatitis B. Semin Liver Dis 2005; 25( suppl 1):3–8.
- Benvegnu L, Gios M, Boccato S, Alberti A. Natural history of compensated viral cirrhosis: a prospective study on the incidence and hierarchy of major complications. Gut 2004; 53:744–749.
- Iloeje UH, Yang HI, Su J, Jen CL, You SL, Chen CJ. Risk evaluation of viral load elevation and associated liver disease/cancer in HBV. The REVEAL-HBV Study Group. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology 2006; 130:678–686.
- Bosch FX, Ribes J, Cleries R, Diaz M. Epidemiology of hepatocellular carcinoma. Clin Liver Dis 2005; 9:191–211.
- Fattovich G. Natural history of hepatitis B. J Hepatol 2003; 39:S50–S58.
Our knowledge about hepatitis B and related diseases has dramatically increased since the discovery of the causative virus, HBV, in 1963. Despite effective vaccination, hepatitis B still constitutes a major public health problem.
In two parts, this comprehensive review will highlight a practical clinical approach to HBV infection. In this first part, we discuss the epidemiology, natural history, and diagnosis of HBV infection. In the second part, to be published in the next issue of this journal, we will review the general principles of its management, its management in patients on immunosuppressant therapy and in pregnant women, and HBV vaccination.
COMMON IN ASIA, LESS SO IN AMERICA
More than 2 billion people—one-third of the world’s population—alive today have been infected with HBV at some time in their life, and of these, about 350 million remain infected.1 Every year, 1 million people die of HBV-related cirrhosis or hepatocellular carcinoma, which means that HBV takes a life every 30 seconds.2
The incidence of acute hepatitis B in the United States has declined from 8.5 per 100,000 population in 1990 to 2.1 per 100,000 population in 2004, with the greatest declines (94%) in children and adolescents, coincident with an increase in hepatitis B vaccination in these age groups.5 Despite these advances, HBV still causes a considerable number of cases of cirrhosis, cancer, and death—about 5,000 deaths each year in the United States.
HBV HAS FOUR GENES, EIGHT GENOTYPES
HBV is a DNA virus of the Hepadnaviridae family. Its genome is double-stranded with four genes, each one encoding a specific structural protein or proteins6,7:
- S gene, for the viral envelope (surface antigen)
- C gene, for both the nucleocapsid (core) antigen and the pre-core (e) antigen
- X gene, for two regulatory proteins required for HBV replication
- P gene, for DNA polymerase.
Eight genotypes of HBV (labeled A though H) have been identified.13,14 All eight have been found in the United States, but genotype A accounts for 35% of cases, genotype B for 22%, and genotype C for 31%.15
The clinical significance of HBV genotypes is not as clear as that of hepatitis C virus genotypes. Although recent data have suggested that different HBV genotypes may be associated with different rates of progression of liver disease and different rates of response to interferon therapy,13 these data were not enough to recommend routine testing for HBV genotypes in clinical practice.16
In HBV infection, the virus itself does not injure liver cells. Rather, the damage of hepatitis is immune-mediated and begins to appear as the host’s immune system attempts to clear the virus.
MARKERS OF HBV INFECTION
HBV surface antigen and HBV DNA are often the first detectable markers of acute infection, appearing before the onset of symptoms or before elevation of alanine aminotransferase (ALT) occurs. By definition, an HBV infection is chronic if surface antigen persists longer than 6 months.
HBV e antigen, derived from pre-core protein, is considered a marker of HBV replication and infectivity. In chronic infection, e antigen can persist for years or decades.
HBV core antigen cannot be detected in the serum, but antibodies against it can, first immunoglobulin M (IgM) and later immunoglobulin G (IgG).
TRANSMISSION: VERTICAL OR HORIZONTAL
Because HBV replicates profusely and produces high titers in the blood (108 to 1,010 virions/ mL), any parenteral or mucosal exposure to infected blood poses a high risk of HBV acquisition. The risk of HBV transmission from a single needlestick is 1% to 6% if the blood is positive for HBV surface antigen but negative for HBV e antigen, and 22% to 40% if positive for both antigens.17–19 Saliva, nasopharyngeal fluid, breast milk, semen, urine, and cervical secretions can also harbor HBV.20
Worldwide, perinatal (vertical) transmission is the predominant mode of HBV transmission, whereas intravenous drug abuse and unprotected sexual intercourse are the main routes of infection in areas of low prevalence, such as the United States. In sub-Saharan Africa, Alaska, and Mediterranean countries, transmission of HBV usually occurs horizontally during childhood, presumably via contact with nonintact skin.21–24 Saliva has also been thought to be the route of HBV transmission in sporadic cases through human bites.25
People at risk of HBV infection include:
- Parenteral drug users
- People with multiple sexual partners
- Household contacts and sexual partners of people who are positive for HBV surface antigen
- Infants born to HBV-infected mothers
- Patients and staff in custodial institutions for the developmentally disabled
- Recipients of certain plasma-derived products (including patients with congenital coagulation defects)
- Hemodialysis patients
- Health and public-safety workers who have contact with blood
- People born in areas where HBV is endemic, and their children.
These people—as well as all pregnant women, patients infected with hepatitis C virus or human immunodeficiency virus, and patients with chronically elevated ALT or aspartate aminotransferase (AST) levels—should be screened for HBV infection with serologic markers.
CLINICAL MANIFESTATIONS VARY
HBV infection, acute or chronic, has variable manifestations. During the acute stage, HBV infection can manifest as anicteric (subclinical) hepatitis, icteric hepatitis, or, rarely, acute fulminant hepatitis. Chronic HBV infection can be asymptomatic (the HBV surface antigen carrier state), or it can be manifested by symptoms and signs of cirrhosis or hepatocellular carcinoma or both. Extrahepatic manifestations, including serum sickness, polyarteritis nodosa, essential mixed cryoglobulinemia, membranous glomerulonephritis, and aplastic anemia, have been reported in patients with HBV infection.26
Acute hepatitis B
The incubation period of HBV ranges from 2 weeks to 4 months. Initially, patients complain of fatigue, malaise, anorexia, right upper quad-rant discomfort, or flu-like symptoms (coryza, photophobia, headache, and myalgia); then jaundice becomes apparent, usually within 10 days of the onset of symptoms. Low-grade fever, jaundice, and mildly tender hepatomegaly are the most common signs. Generalized lymphadenopathy is not a feature of acute HBV infection. If the patient also has hepatitis D virus infection or underlying liver disease (eg, alcoholic liver disease), then acute HBV infection may be more severe.
In the acute phase, ALT and AST levels rise, sometimes to values above 1,000 IU/L. In icteric hepatitis, bilirubin levels also rise, usually after the ALT level does. Although the peak ALT level reflects the hepatocellular injury, it has no prognostic value. With recovery, ALT levels normalize in 1 to 4 months.
Acute fulminant hepatitis B occurs in 0.1% to 0.5% of patients, and causes about 10% of cases of acute liver failure in the United States.27 Patients typically present with rapidly progressive acute hepatitis characterized by signs of liver failure, such as coagulopathy, encephalopathy, and cerebral edema.
In the so-called window phase, laboratory testing may not reveal HBV surface antigen because of early clearance but shows IgM antibody against the HBV core antigen. HBV DNA may be low or undetected.
Chronic hepatitis B
Chronic hepatitis B is usually diagnosed as a result of a workup for abnormal liver function tests or as a result of screening patients at risk for HBV infection. Many patients with chronic hepatitis B have no symptoms or have nonspecific symptoms such as fatigue or right upper quadrant discomfort.
Acute exacerbations due to HBV e antigen seroreversion (ie, in which e antigen reappears) occasionally occur in patients with chronic hepatitis B. Most of these exacerbations are asymptomatic, but occasionally an acute hepatitis-like clinical picture with detectable IgM antibody against the core antigen occurs, leading to misdiagnosis of acute HBV infection in patients not previously known to have chronic HBV infection.28
In late cases, signs of cirrhosis such as jaundice, ascites, splenomegaly, pedal edema, encephalopathy, or variceal bleeding can be present.
Hepatocellular carcinoma should be suspected in cirrhotic patients with new-onset right upper quadrant pain, rapidly developing ascites, a palpable liver mass, or hepatic encephalopathy. Other nonspecific features of hepatocellular carcinoma include watery diarrhea, hypoglycemia, and certain cutaneous manifestations such as acanthosis nigricans and the Leser-Trelat sign (multiple pruritic seborrheic keratoses of sudden onset).
In chronic hepatitis B, liver enzyme levels can be normal, even in patients with wellcompensated cirrhosis. ALT levels may range from normal to five times higher than normal. Thrombocytopenia, hypoalbuminemia, direct hyperbilirubinemia, and prolonged prothrombin time suggest cirrhosis.
Findings of chronic hepatitis B on liver biopsy range from minimal inflammation to cirrhosis. The most characteristic histologic feature of chronic HBV infection is the “ground-glass hepatocyte,” which is due to intracellular accumulation of HBV surface antigen. 29
FEW ADULTS (BUT MANY CHILDREN) REMAIN CHRONICALLY INFECTED
HBV surface antigen can be detected in the blood approximately 2 to 4 weeks after inoculation. Simultaneously, HBV DNA, usually in very high levels, is also detectable in the blood. However, in the rare cases of acute fulminant hepatitis, HBV DNA levels can be low or undetectable at the time of presentation because the immune system mounts a robust response with extensive damage to HBVinfected hepatocytes.
The rate of spontaneous recovery from acute HBV infection varies, depending on the patient’s age at the time of HBV acquisition and the patient’s immune status. Fewer than 5% of immunocompetent adults infected with HBV remain chronically infected, defined as being positive for HBV surface antigen for more than 6 months. On the other hand, 80% to 90% of infected infants and about 20% to 50% of children 1 to 5 years old at the time of acute infection remain chronically infected.21
Four phases of chronic HBV infection
The immune tolerance phase, the initial phase of chronic HBV infection, is seen almost exclusively in those who acquired HBV infection vertically or during early childhood. Although patients have high HBV DNA levels, they do not have significant liver disease. This discrepancy is thought to be related to the immune tolerance to HBV; however, the exact mechanism of that tolerance is unclear.31
Only 15% of those with immune tolerance have spontaneous HBV e antigen seroconversion (ie, loss of e antigen and appearance of anti-e antibody) within 20 years after infection. 32
The immune clearance phase (HBV e antigen-positive chronic hepatitis) appears about 20 to 30 years after the onset of the immune tolerance phase in patients who acquire HBV early in life. It is also often seen in patients with infections acquired late in childhood or in adulthood.
This phase marks the start of an immunemediated process aimed at clearing the viral infection, but it also leads to concomitant hepatocellular injury. Spontaneous clearance of the e antigen increases in this phase to an annual rate of 10% to 20%.32,33 The strongest predictors of spontaneous e antigen seroconversion are old age, an elevated ALT level, and an acute exacerbation.26
Although ALT levels are elevated and there is evidence on liver biopsy of chronic active hepatitis, this phase is usually asymptomatic. Rarely, however, it presents with an acute flare of hepatitis, sometimes accompanied by IgM antibodies against the HBV core antigen (in low titer), leading to an incorrect diagnosis of acute HBV infection.
Depending on the duration of the chronic hepatitis and the frequency and severity of flares, about 12% to 20% of patients in the immune-clearance phase develop serious liver disease within 5 years.31
The inactive carrier phase following HBV e antigen seroconversion is characterized by undetectable or low HBV DNA levels (< 1,000 copies/mL), normal ALT levels, and minimal or no necroinflammation on liver biopsy. 30 Such patients should be followed with serial testing, as 4% to 20% of them spontaneously revert to being positive for e antigen at least once.16 On the other hand, only 0.5% to 2% of surface antigen carriers in western countries clear themselves of surface antigen yearly, but up to half of those who clear the surface antigen have low-level HBV viremia. 34
The reactivation phase (HBV e antigennegative chronic hepatitis) is seen in some HBV-infected patients, especially those from Asia and southern Europe, in whom the virus has a spontaneous pre-core or core mutation that makes infected cells unable to secrete the e antigen. Although these patients have no e antigen in their blood, they do have intermittent or persistent elevation of ALT, elevated HBV DNA, and histopathologic findings of chronic hepatitis. Compared with those in the immune clearance phase, patients in the reactivation phase tend to be older and to have lower HBV DNA levels but advanced hepatic damage.
Immunity to HBV infection is characterized by loss of HBV surface antigen, DNA, e antigen, and anti-core antigen IgM with development of anti-surface antigen antibody and anti-core antigen IgG (total anti-core antigen antibody). The presence of anti-surface antigen antibody and anti-core antigen IgG together differentiates natural immunity through resolved infection from that which is acquired through vaccination, which is denoted by isolated anti-surface antigen antibody.
Cirrhosis, liver failure, cancer
Cirrhosis, hepatic decompensation, and hepatocellular carcinoma are the major long-term complications of HBV infection. In untreated patients, the annual rate of progression to cirrhosis has been estimated to be 2% to 6% in patients with HBV e antigen-positive chronic hepatitis and 8% to 9% in those with e antigen- negative chronic hepatitis.30
The likely explanation for these surprising cirrhosis rates is that e antigen-negative chronic hepatitis usually represents a late stage of the disease, and patients in this phase are usually older and have more advanced liver disease.
Subsequently, the annual rate of progression from compensated cirrhosis to hepatic decompensation has been estimated to be about 5%.35
Across all the stages described above, a high serum HBV DNA level has been shown to be a strong predictor of progression to cirrhosis in patients with chronic HBV infection. In a population-based prospective cohort study of 3,582 untreated HBV-infected patients in Taiwan, Iloeje et al36 found that, compared with patients with serum HBV DNA levels lower than 104 copies/mL, those with levels of 104 or higher had an adjusted relative risk of cirrhosis of 2.5. The relative risk rose to 5.9 with HBV DNA levels of 105 or higher, and 9.8 with levels of 106 copies/mL or higher. More studies in different patient populations are needed for confirmation.
The most important risk factor for hepatocellular carcinoma in HBV-infected patients is cirrhosis, but this cancer can also develop in noncirrhotic livers.37 The annual rate of hepatocelluar carcinoma has been estimated to be higher (2.5%–3%) in patients with cirrhosis than in noncirrhotic carriers (0.5%–1%).30,35–38 Risk factors for cirrhosis and hepatocellular carcinoma are summarized in Table 4.16,30
Our knowledge about hepatitis B and related diseases has dramatically increased since the discovery of the causative virus, HBV, in 1963. Despite effective vaccination, hepatitis B still constitutes a major public health problem.
In two parts, this comprehensive review will highlight a practical clinical approach to HBV infection. In this first part, we discuss the epidemiology, natural history, and diagnosis of HBV infection. In the second part, to be published in the next issue of this journal, we will review the general principles of its management, its management in patients on immunosuppressant therapy and in pregnant women, and HBV vaccination.
COMMON IN ASIA, LESS SO IN AMERICA
More than 2 billion people—one-third of the world’s population—alive today have been infected with HBV at some time in their life, and of these, about 350 million remain infected.1 Every year, 1 million people die of HBV-related cirrhosis or hepatocellular carcinoma, which means that HBV takes a life every 30 seconds.2
The incidence of acute hepatitis B in the United States has declined from 8.5 per 100,000 population in 1990 to 2.1 per 100,000 population in 2004, with the greatest declines (94%) in children and adolescents, coincident with an increase in hepatitis B vaccination in these age groups.5 Despite these advances, HBV still causes a considerable number of cases of cirrhosis, cancer, and death—about 5,000 deaths each year in the United States.
HBV HAS FOUR GENES, EIGHT GENOTYPES
HBV is a DNA virus of the Hepadnaviridae family. Its genome is double-stranded with four genes, each one encoding a specific structural protein or proteins6,7:
- S gene, for the viral envelope (surface antigen)
- C gene, for both the nucleocapsid (core) antigen and the pre-core (e) antigen
- X gene, for two regulatory proteins required for HBV replication
- P gene, for DNA polymerase.
Eight genotypes of HBV (labeled A though H) have been identified.13,14 All eight have been found in the United States, but genotype A accounts for 35% of cases, genotype B for 22%, and genotype C for 31%.15
The clinical significance of HBV genotypes is not as clear as that of hepatitis C virus genotypes. Although recent data have suggested that different HBV genotypes may be associated with different rates of progression of liver disease and different rates of response to interferon therapy,13 these data were not enough to recommend routine testing for HBV genotypes in clinical practice.16
In HBV infection, the virus itself does not injure liver cells. Rather, the damage of hepatitis is immune-mediated and begins to appear as the host’s immune system attempts to clear the virus.
MARKERS OF HBV INFECTION
HBV surface antigen and HBV DNA are often the first detectable markers of acute infection, appearing before the onset of symptoms or before elevation of alanine aminotransferase (ALT) occurs. By definition, an HBV infection is chronic if surface antigen persists longer than 6 months.
HBV e antigen, derived from pre-core protein, is considered a marker of HBV replication and infectivity. In chronic infection, e antigen can persist for years or decades.
HBV core antigen cannot be detected in the serum, but antibodies against it can, first immunoglobulin M (IgM) and later immunoglobulin G (IgG).
TRANSMISSION: VERTICAL OR HORIZONTAL
Because HBV replicates profusely and produces high titers in the blood (108 to 1,010 virions/ mL), any parenteral or mucosal exposure to infected blood poses a high risk of HBV acquisition. The risk of HBV transmission from a single needlestick is 1% to 6% if the blood is positive for HBV surface antigen but negative for HBV e antigen, and 22% to 40% if positive for both antigens.17–19 Saliva, nasopharyngeal fluid, breast milk, semen, urine, and cervical secretions can also harbor HBV.20
Worldwide, perinatal (vertical) transmission is the predominant mode of HBV transmission, whereas intravenous drug abuse and unprotected sexual intercourse are the main routes of infection in areas of low prevalence, such as the United States. In sub-Saharan Africa, Alaska, and Mediterranean countries, transmission of HBV usually occurs horizontally during childhood, presumably via contact with nonintact skin.21–24 Saliva has also been thought to be the route of HBV transmission in sporadic cases through human bites.25
People at risk of HBV infection include:
- Parenteral drug users
- People with multiple sexual partners
- Household contacts and sexual partners of people who are positive for HBV surface antigen
- Infants born to HBV-infected mothers
- Patients and staff in custodial institutions for the developmentally disabled
- Recipients of certain plasma-derived products (including patients with congenital coagulation defects)
- Hemodialysis patients
- Health and public-safety workers who have contact with blood
- People born in areas where HBV is endemic, and their children.
These people—as well as all pregnant women, patients infected with hepatitis C virus or human immunodeficiency virus, and patients with chronically elevated ALT or aspartate aminotransferase (AST) levels—should be screened for HBV infection with serologic markers.
CLINICAL MANIFESTATIONS VARY
HBV infection, acute or chronic, has variable manifestations. During the acute stage, HBV infection can manifest as anicteric (subclinical) hepatitis, icteric hepatitis, or, rarely, acute fulminant hepatitis. Chronic HBV infection can be asymptomatic (the HBV surface antigen carrier state), or it can be manifested by symptoms and signs of cirrhosis or hepatocellular carcinoma or both. Extrahepatic manifestations, including serum sickness, polyarteritis nodosa, essential mixed cryoglobulinemia, membranous glomerulonephritis, and aplastic anemia, have been reported in patients with HBV infection.26
Acute hepatitis B
The incubation period of HBV ranges from 2 weeks to 4 months. Initially, patients complain of fatigue, malaise, anorexia, right upper quad-rant discomfort, or flu-like symptoms (coryza, photophobia, headache, and myalgia); then jaundice becomes apparent, usually within 10 days of the onset of symptoms. Low-grade fever, jaundice, and mildly tender hepatomegaly are the most common signs. Generalized lymphadenopathy is not a feature of acute HBV infection. If the patient also has hepatitis D virus infection or underlying liver disease (eg, alcoholic liver disease), then acute HBV infection may be more severe.
In the acute phase, ALT and AST levels rise, sometimes to values above 1,000 IU/L. In icteric hepatitis, bilirubin levels also rise, usually after the ALT level does. Although the peak ALT level reflects the hepatocellular injury, it has no prognostic value. With recovery, ALT levels normalize in 1 to 4 months.
Acute fulminant hepatitis B occurs in 0.1% to 0.5% of patients, and causes about 10% of cases of acute liver failure in the United States.27 Patients typically present with rapidly progressive acute hepatitis characterized by signs of liver failure, such as coagulopathy, encephalopathy, and cerebral edema.
In the so-called window phase, laboratory testing may not reveal HBV surface antigen because of early clearance but shows IgM antibody against the HBV core antigen. HBV DNA may be low or undetected.
Chronic hepatitis B
Chronic hepatitis B is usually diagnosed as a result of a workup for abnormal liver function tests or as a result of screening patients at risk for HBV infection. Many patients with chronic hepatitis B have no symptoms or have nonspecific symptoms such as fatigue or right upper quadrant discomfort.
Acute exacerbations due to HBV e antigen seroreversion (ie, in which e antigen reappears) occasionally occur in patients with chronic hepatitis B. Most of these exacerbations are asymptomatic, but occasionally an acute hepatitis-like clinical picture with detectable IgM antibody against the core antigen occurs, leading to misdiagnosis of acute HBV infection in patients not previously known to have chronic HBV infection.28
In late cases, signs of cirrhosis such as jaundice, ascites, splenomegaly, pedal edema, encephalopathy, or variceal bleeding can be present.
Hepatocellular carcinoma should be suspected in cirrhotic patients with new-onset right upper quadrant pain, rapidly developing ascites, a palpable liver mass, or hepatic encephalopathy. Other nonspecific features of hepatocellular carcinoma include watery diarrhea, hypoglycemia, and certain cutaneous manifestations such as acanthosis nigricans and the Leser-Trelat sign (multiple pruritic seborrheic keratoses of sudden onset).
In chronic hepatitis B, liver enzyme levels can be normal, even in patients with wellcompensated cirrhosis. ALT levels may range from normal to five times higher than normal. Thrombocytopenia, hypoalbuminemia, direct hyperbilirubinemia, and prolonged prothrombin time suggest cirrhosis.
Findings of chronic hepatitis B on liver biopsy range from minimal inflammation to cirrhosis. The most characteristic histologic feature of chronic HBV infection is the “ground-glass hepatocyte,” which is due to intracellular accumulation of HBV surface antigen. 29
FEW ADULTS (BUT MANY CHILDREN) REMAIN CHRONICALLY INFECTED
HBV surface antigen can be detected in the blood approximately 2 to 4 weeks after inoculation. Simultaneously, HBV DNA, usually in very high levels, is also detectable in the blood. However, in the rare cases of acute fulminant hepatitis, HBV DNA levels can be low or undetectable at the time of presentation because the immune system mounts a robust response with extensive damage to HBVinfected hepatocytes.
The rate of spontaneous recovery from acute HBV infection varies, depending on the patient’s age at the time of HBV acquisition and the patient’s immune status. Fewer than 5% of immunocompetent adults infected with HBV remain chronically infected, defined as being positive for HBV surface antigen for more than 6 months. On the other hand, 80% to 90% of infected infants and about 20% to 50% of children 1 to 5 years old at the time of acute infection remain chronically infected.21
Four phases of chronic HBV infection
The immune tolerance phase, the initial phase of chronic HBV infection, is seen almost exclusively in those who acquired HBV infection vertically or during early childhood. Although patients have high HBV DNA levels, they do not have significant liver disease. This discrepancy is thought to be related to the immune tolerance to HBV; however, the exact mechanism of that tolerance is unclear.31
Only 15% of those with immune tolerance have spontaneous HBV e antigen seroconversion (ie, loss of e antigen and appearance of anti-e antibody) within 20 years after infection. 32
The immune clearance phase (HBV e antigen-positive chronic hepatitis) appears about 20 to 30 years after the onset of the immune tolerance phase in patients who acquire HBV early in life. It is also often seen in patients with infections acquired late in childhood or in adulthood.
This phase marks the start of an immunemediated process aimed at clearing the viral infection, but it also leads to concomitant hepatocellular injury. Spontaneous clearance of the e antigen increases in this phase to an annual rate of 10% to 20%.32,33 The strongest predictors of spontaneous e antigen seroconversion are old age, an elevated ALT level, and an acute exacerbation.26
Although ALT levels are elevated and there is evidence on liver biopsy of chronic active hepatitis, this phase is usually asymptomatic. Rarely, however, it presents with an acute flare of hepatitis, sometimes accompanied by IgM antibodies against the HBV core antigen (in low titer), leading to an incorrect diagnosis of acute HBV infection.
Depending on the duration of the chronic hepatitis and the frequency and severity of flares, about 12% to 20% of patients in the immune-clearance phase develop serious liver disease within 5 years.31
The inactive carrier phase following HBV e antigen seroconversion is characterized by undetectable or low HBV DNA levels (< 1,000 copies/mL), normal ALT levels, and minimal or no necroinflammation on liver biopsy. 30 Such patients should be followed with serial testing, as 4% to 20% of them spontaneously revert to being positive for e antigen at least once.16 On the other hand, only 0.5% to 2% of surface antigen carriers in western countries clear themselves of surface antigen yearly, but up to half of those who clear the surface antigen have low-level HBV viremia. 34
The reactivation phase (HBV e antigennegative chronic hepatitis) is seen in some HBV-infected patients, especially those from Asia and southern Europe, in whom the virus has a spontaneous pre-core or core mutation that makes infected cells unable to secrete the e antigen. Although these patients have no e antigen in their blood, they do have intermittent or persistent elevation of ALT, elevated HBV DNA, and histopathologic findings of chronic hepatitis. Compared with those in the immune clearance phase, patients in the reactivation phase tend to be older and to have lower HBV DNA levels but advanced hepatic damage.
Immunity to HBV infection is characterized by loss of HBV surface antigen, DNA, e antigen, and anti-core antigen IgM with development of anti-surface antigen antibody and anti-core antigen IgG (total anti-core antigen antibody). The presence of anti-surface antigen antibody and anti-core antigen IgG together differentiates natural immunity through resolved infection from that which is acquired through vaccination, which is denoted by isolated anti-surface antigen antibody.
Cirrhosis, liver failure, cancer
Cirrhosis, hepatic decompensation, and hepatocellular carcinoma are the major long-term complications of HBV infection. In untreated patients, the annual rate of progression to cirrhosis has been estimated to be 2% to 6% in patients with HBV e antigen-positive chronic hepatitis and 8% to 9% in those with e antigen- negative chronic hepatitis.30
The likely explanation for these surprising cirrhosis rates is that e antigen-negative chronic hepatitis usually represents a late stage of the disease, and patients in this phase are usually older and have more advanced liver disease.
Subsequently, the annual rate of progression from compensated cirrhosis to hepatic decompensation has been estimated to be about 5%.35
Across all the stages described above, a high serum HBV DNA level has been shown to be a strong predictor of progression to cirrhosis in patients with chronic HBV infection. In a population-based prospective cohort study of 3,582 untreated HBV-infected patients in Taiwan, Iloeje et al36 found that, compared with patients with serum HBV DNA levels lower than 104 copies/mL, those with levels of 104 or higher had an adjusted relative risk of cirrhosis of 2.5. The relative risk rose to 5.9 with HBV DNA levels of 105 or higher, and 9.8 with levels of 106 copies/mL or higher. More studies in different patient populations are needed for confirmation.
The most important risk factor for hepatocellular carcinoma in HBV-infected patients is cirrhosis, but this cancer can also develop in noncirrhotic livers.37 The annual rate of hepatocelluar carcinoma has been estimated to be higher (2.5%–3%) in patients with cirrhosis than in noncirrhotic carriers (0.5%–1%).30,35–38 Risk factors for cirrhosis and hepatocellular carcinoma are summarized in Table 4.16,30
- World Health Organization. Hepatitis B. www.who.int/csr/disease/hepatitis/whocdscsrlyo20022/en. Accessed 11/10/2008.
- Center for Disease Control and Prevention. HBV a silent killer. www.cdc.gov/ncidod/diseases/hepatitis/b/hbv_silent_killer. Accessed 2/19/2007.
- McQuillan GM, Coleman PJ, Kruszon-Moran D, Moyer LA, Lambert SB, Margolis HS. Prevalence of hepatitis B virus infection in the United States: the National Health and Nutrition Examination Surveys, 1976 through 1994. Am J Public Health 1999; 89:14–18.
- Armstrong GL, Mast EE, Wojczynski M, Margolis HS. Childhood hepatitis B virus infections in the United States before hepatitis B immunization. Pediatrics 2001; 108:1123–1128.
- Mast EE, Weinbaum CM, Fiore AE, et al. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP). Part II: immunization of adults. MMWR Recomm Rep 2006 Dec 8; 55(RR-16):1–33.
- Gish RG, Gadano AC. Chronic hepatitis B: current epidemiology in the Americas and implications for management. J Viral Hepatol 2006; 13:787–798.
- Wei Y, Tiollais PK. Molecular biology of hepatitis B virus. Clin Liver Dis 1999; 3:189–219.
- Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev 2000; 64:51–68.
- Hunt CM, McGill JM, Allen MI, Condreay LD. Clinical relevance of hepatitis B viral mutations. Hepatology 2000; 3:1037–1044.
- Allen MI, Deslauriers M, Andrews CW, et al. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Lamivudine Clinical Investigation Group. Hepatology 1998; 27:1670–1677.
- Hadziyannis SJ, Vassilopoulos D. Hepatitis B e antigen- negative chronic hepatitis B. Hepatology 2001; 34:617–624.
- Wai CT, Fontana RJ. Clinical significance of hepatitis B virus genotypes, variants, and mutants. Clin Liver Dis 2004; 8:321–352.
- Fung SK, Lok AS. Hepatitis B virus genotypes: do they play a role in the outcome of HBV infection? Hepatology 2004; 40:790–792.
- Norder H, Courouce AM, Coursaget P, et al. Genetic diversity of hepatitis B virus strains derived worldwide: genotypes, subgenotypes, and HBsAg subtypes. Intervirology 2004; 47:289–309.
- Chu CJ, Keeffe EB, Han SH, et al. Hepatitis B virus genotypes in the United States: results of a nationwide study. Gastroenterology 2003; 125:444–451.
- Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Mast EE, Alter MJ. Prevention of hepatitis B virus infection among health-care workers. In:Ellis RE, editor. Hepatitis B Vaccines in Clinical Practice. New York: Marcel Dekker, 1993:295–307.
- Werner BG, Grady GF. Accidental hepatitis-B-surface-antigen-positive inoculations: use of e antigen to estimate infectivity. Ann Intern Med 1982; 97:367–369.
- Gerberding JL. Management of occupational exposures to blood-borne viruses. N Engl J Med 1995; 332:444–451.
- Kidd-Ljunggren K, Holmberg A, Blackberg J, Lindqvist B. High levels of hepatitis B virus DNA in body fluids from chronic carriers. J Hosp Infect 2006; 64:352–357.
- McMahon BJ, Alward WL, Hall DB, et al. Acute hepatitis B virus infection: relation of age to the clinical expression of disease and subsequent development of the carrier state. J Infect Dis 1985; 151:599–603.
- Dusheiko GM, Brink BA, Conradie JD, Marimuthu T, Sher R. Regional prevalence of hepatitis B, delta, and human immunodeficiency virus infection in southern Africa: a large population survey. Am J Epidemiol 1989; 129:138–45.
- Bortolotti F, Guido M, Bartolacci S, et al. Chronic hepatitis B in children after e antigen seroclearance: final report of a 29-year longitudinal study. Hepatology 2006; 43:556–562.
- Moreno MR, Otero M, Millan A, et al. Clinical and histological outcome after hepatitis B e antigen to antibody seroconversion in children with chronic hepatitis B. Hepatology 1999:572–575.
- Hui AY, Hung LC, Tse PC, Leung WK, Chan PK, Chan HL. Transmission of hepatitis B by human bite--confirmation by detection of virus in saliva and full genome sequencing. J Clin Virol 2005; 33:254–256.
- Cacoub P, Saadoun D, Bourlière M, et al. Hepatitis B virus genotypes and extrahepatic manifestations. J Hepatol 2005; 43:764–770.
- Schiodt FV, Atillasoy E, Shakil AO, et al. Etiology and outcome for 295 patients with acute liver failure in the United States. Liver Transplant Surg 1999; 5:29–34.
- Chu CM, Liaw YF, Pao CC, Huang MJ. The etiology of acute hepatitis superimposed upon previously unrecognized asymptomatic HBsAg carriers. Hepatology 1989; 9:452–456.
- Gerber MA, Hadziyannis S, Vissoulis C, et al. Electron microscopy and immunoelectronmicroscopy of cytoplasmic hepatitis B antigen in hepatocytes. Am J Pathol 1974; 75:489–502.
- Yim HJ, Lok AS. Natural history of chronic hepatitis B virus infection: what we knew in 1981 and what we know in 2005. Hepatology 2006; 43:S173–S181.
- Pungpapong S, Kim WR, Poterucha JJ. Natural history of hepatitis B virus infection: an update for clinicians. Mayo Clin Proc 2007; 82:967–975.
- Lok AS, Lai CL, Wu PC, Leung EK, Lam TS. Spontaneous hepatitis B e antigen to antibody seroconversion and reversion in Chinese patients with chronic hepatitis B virus infection. Gastroenterology 1987; 92:1839–1843.
- McMahon BJ, Holck P, Bulkow L, Snowball M. Serologic and clinical outcomes in 1536 Alaska Natives chronically infected with hepatitis B virus. Ann Intern Med 2001; 135:759–768.
- McMahon BJ. Epidemiology and natural history of hepatitis B. Semin Liver Dis 2005; 25( suppl 1):3–8.
- Benvegnu L, Gios M, Boccato S, Alberti A. Natural history of compensated viral cirrhosis: a prospective study on the incidence and hierarchy of major complications. Gut 2004; 53:744–749.
- Iloeje UH, Yang HI, Su J, Jen CL, You SL, Chen CJ. Risk evaluation of viral load elevation and associated liver disease/cancer in HBV. The REVEAL-HBV Study Group. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology 2006; 130:678–686.
- Bosch FX, Ribes J, Cleries R, Diaz M. Epidemiology of hepatocellular carcinoma. Clin Liver Dis 2005; 9:191–211.
- Fattovich G. Natural history of hepatitis B. J Hepatol 2003; 39:S50–S58.
- World Health Organization. Hepatitis B. www.who.int/csr/disease/hepatitis/whocdscsrlyo20022/en. Accessed 11/10/2008.
- Center for Disease Control and Prevention. HBV a silent killer. www.cdc.gov/ncidod/diseases/hepatitis/b/hbv_silent_killer. Accessed 2/19/2007.
- McQuillan GM, Coleman PJ, Kruszon-Moran D, Moyer LA, Lambert SB, Margolis HS. Prevalence of hepatitis B virus infection in the United States: the National Health and Nutrition Examination Surveys, 1976 through 1994. Am J Public Health 1999; 89:14–18.
- Armstrong GL, Mast EE, Wojczynski M, Margolis HS. Childhood hepatitis B virus infections in the United States before hepatitis B immunization. Pediatrics 2001; 108:1123–1128.
- Mast EE, Weinbaum CM, Fiore AE, et al. A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP). Part II: immunization of adults. MMWR Recomm Rep 2006 Dec 8; 55(RR-16):1–33.
- Gish RG, Gadano AC. Chronic hepatitis B: current epidemiology in the Americas and implications for management. J Viral Hepatol 2006; 13:787–798.
- Wei Y, Tiollais PK. Molecular biology of hepatitis B virus. Clin Liver Dis 1999; 3:189–219.
- Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev 2000; 64:51–68.
- Hunt CM, McGill JM, Allen MI, Condreay LD. Clinical relevance of hepatitis B viral mutations. Hepatology 2000; 3:1037–1044.
- Allen MI, Deslauriers M, Andrews CW, et al. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Lamivudine Clinical Investigation Group. Hepatology 1998; 27:1670–1677.
- Hadziyannis SJ, Vassilopoulos D. Hepatitis B e antigen- negative chronic hepatitis B. Hepatology 2001; 34:617–624.
- Wai CT, Fontana RJ. Clinical significance of hepatitis B virus genotypes, variants, and mutants. Clin Liver Dis 2004; 8:321–352.
- Fung SK, Lok AS. Hepatitis B virus genotypes: do they play a role in the outcome of HBV infection? Hepatology 2004; 40:790–792.
- Norder H, Courouce AM, Coursaget P, et al. Genetic diversity of hepatitis B virus strains derived worldwide: genotypes, subgenotypes, and HBsAg subtypes. Intervirology 2004; 47:289–309.
- Chu CJ, Keeffe EB, Han SH, et al. Hepatitis B virus genotypes in the United States: results of a nationwide study. Gastroenterology 2003; 125:444–451.
- Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45:507–539.
- Mast EE, Alter MJ. Prevention of hepatitis B virus infection among health-care workers. In:Ellis RE, editor. Hepatitis B Vaccines in Clinical Practice. New York: Marcel Dekker, 1993:295–307.
- Werner BG, Grady GF. Accidental hepatitis-B-surface-antigen-positive inoculations: use of e antigen to estimate infectivity. Ann Intern Med 1982; 97:367–369.
- Gerberding JL. Management of occupational exposures to blood-borne viruses. N Engl J Med 1995; 332:444–451.
- Kidd-Ljunggren K, Holmberg A, Blackberg J, Lindqvist B. High levels of hepatitis B virus DNA in body fluids from chronic carriers. J Hosp Infect 2006; 64:352–357.
- McMahon BJ, Alward WL, Hall DB, et al. Acute hepatitis B virus infection: relation of age to the clinical expression of disease and subsequent development of the carrier state. J Infect Dis 1985; 151:599–603.
- Dusheiko GM, Brink BA, Conradie JD, Marimuthu T, Sher R. Regional prevalence of hepatitis B, delta, and human immunodeficiency virus infection in southern Africa: a large population survey. Am J Epidemiol 1989; 129:138–45.
- Bortolotti F, Guido M, Bartolacci S, et al. Chronic hepatitis B in children after e antigen seroclearance: final report of a 29-year longitudinal study. Hepatology 2006; 43:556–562.
- Moreno MR, Otero M, Millan A, et al. Clinical and histological outcome after hepatitis B e antigen to antibody seroconversion in children with chronic hepatitis B. Hepatology 1999:572–575.
- Hui AY, Hung LC, Tse PC, Leung WK, Chan PK, Chan HL. Transmission of hepatitis B by human bite--confirmation by detection of virus in saliva and full genome sequencing. J Clin Virol 2005; 33:254–256.
- Cacoub P, Saadoun D, Bourlière M, et al. Hepatitis B virus genotypes and extrahepatic manifestations. J Hepatol 2005; 43:764–770.
- Schiodt FV, Atillasoy E, Shakil AO, et al. Etiology and outcome for 295 patients with acute liver failure in the United States. Liver Transplant Surg 1999; 5:29–34.
- Chu CM, Liaw YF, Pao CC, Huang MJ. The etiology of acute hepatitis superimposed upon previously unrecognized asymptomatic HBsAg carriers. Hepatology 1989; 9:452–456.
- Gerber MA, Hadziyannis S, Vissoulis C, et al. Electron microscopy and immunoelectronmicroscopy of cytoplasmic hepatitis B antigen in hepatocytes. Am J Pathol 1974; 75:489–502.
- Yim HJ, Lok AS. Natural history of chronic hepatitis B virus infection: what we knew in 1981 and what we know in 2005. Hepatology 2006; 43:S173–S181.
- Pungpapong S, Kim WR, Poterucha JJ. Natural history of hepatitis B virus infection: an update for clinicians. Mayo Clin Proc 2007; 82:967–975.
- Lok AS, Lai CL, Wu PC, Leung EK, Lam TS. Spontaneous hepatitis B e antigen to antibody seroconversion and reversion in Chinese patients with chronic hepatitis B virus infection. Gastroenterology 1987; 92:1839–1843.
- McMahon BJ, Holck P, Bulkow L, Snowball M. Serologic and clinical outcomes in 1536 Alaska Natives chronically infected with hepatitis B virus. Ann Intern Med 2001; 135:759–768.
- McMahon BJ. Epidemiology and natural history of hepatitis B. Semin Liver Dis 2005; 25( suppl 1):3–8.
- Benvegnu L, Gios M, Boccato S, Alberti A. Natural history of compensated viral cirrhosis: a prospective study on the incidence and hierarchy of major complications. Gut 2004; 53:744–749.
- Iloeje UH, Yang HI, Su J, Jen CL, You SL, Chen CJ. Risk evaluation of viral load elevation and associated liver disease/cancer in HBV. The REVEAL-HBV Study Group. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology 2006; 130:678–686.
- Bosch FX, Ribes J, Cleries R, Diaz M. Epidemiology of hepatocellular carcinoma. Clin Liver Dis 2005; 9:191–211.
- Fattovich G. Natural history of hepatitis B. J Hepatol 2003; 39:S50–S58.
KEY POINTS
- HBV infection is much more likely to persist and become chronic if it is acquired at birth or in early childhood rather than during adulthood.
- Chronic HBV infection is defined as persistence of HBV surface antigen in the serum for more than 6 months.
- Although many cases of chronic HBV infection resolve spontaneously, some progress to cirrhosis, hepatocellular carcinoma, and death.