User login
In reply: Acute liver failure
In Reply: We thank Dr. Homler for bringing hepatitis D as a potential cause of acute liver failure to our attention.
Hepatitis D virus, first described in the 1970s, requires the hepatitis B surface antigen (HBsAg) capsid to enter the hepatocyte and, thus, can only cause liver injury when the patient is also infected simultaneously with hepatitis B virus.1 Hepatitis D virus can cause either coinfection (presence of immunoglobulin M anti-HB core antibody with or without HBsAg) or superinfection (presence of HBsAg without immunoglobulin M anti-HB core antibody) with hepatitis B virus. In India, coinfection has been reported to be the cause of acute liver failure in about 4% of all patients, and superinfection in 4.5%.2
While simultaneous treatment for hepatitis D and B viruses with pegylated interferon and any of the agents used for treatment of hepatitis B has been successful in treating chronic hepatitis, it has not been proven useful in patients with acute liver failure, and liver transplant remains the only treatment option.3
- Rizzetto M. The adventure of delta. Liver Int 2016; 36(suppl 1):135–140.
- Irshad M, Acharya SK. Hepatitis D virus (HDV) infection in severe forms of liver diseases in North India. Eur J Gastroenterol Hepatol 1996; 8:995–998.
- Noureddin M, Gish R. Hepatitis delta: epidemiology, diagnosis and management 36 years after discovery. Curr Gastroenterol Rep 2014; 16:365.
In Reply: We thank Dr. Homler for bringing hepatitis D as a potential cause of acute liver failure to our attention.
Hepatitis D virus, first described in the 1970s, requires the hepatitis B surface antigen (HBsAg) capsid to enter the hepatocyte and, thus, can only cause liver injury when the patient is also infected simultaneously with hepatitis B virus.1 Hepatitis D virus can cause either coinfection (presence of immunoglobulin M anti-HB core antibody with or without HBsAg) or superinfection (presence of HBsAg without immunoglobulin M anti-HB core antibody) with hepatitis B virus. In India, coinfection has been reported to be the cause of acute liver failure in about 4% of all patients, and superinfection in 4.5%.2
While simultaneous treatment for hepatitis D and B viruses with pegylated interferon and any of the agents used for treatment of hepatitis B has been successful in treating chronic hepatitis, it has not been proven useful in patients with acute liver failure, and liver transplant remains the only treatment option.3
In Reply: We thank Dr. Homler for bringing hepatitis D as a potential cause of acute liver failure to our attention.
Hepatitis D virus, first described in the 1970s, requires the hepatitis B surface antigen (HBsAg) capsid to enter the hepatocyte and, thus, can only cause liver injury when the patient is also infected simultaneously with hepatitis B virus.1 Hepatitis D virus can cause either coinfection (presence of immunoglobulin M anti-HB core antibody with or without HBsAg) or superinfection (presence of HBsAg without immunoglobulin M anti-HB core antibody) with hepatitis B virus. In India, coinfection has been reported to be the cause of acute liver failure in about 4% of all patients, and superinfection in 4.5%.2
While simultaneous treatment for hepatitis D and B viruses with pegylated interferon and any of the agents used for treatment of hepatitis B has been successful in treating chronic hepatitis, it has not been proven useful in patients with acute liver failure, and liver transplant remains the only treatment option.3
- Rizzetto M. The adventure of delta. Liver Int 2016; 36(suppl 1):135–140.
- Irshad M, Acharya SK. Hepatitis D virus (HDV) infection in severe forms of liver diseases in North India. Eur J Gastroenterol Hepatol 1996; 8:995–998.
- Noureddin M, Gish R. Hepatitis delta: epidemiology, diagnosis and management 36 years after discovery. Curr Gastroenterol Rep 2014; 16:365.
- Rizzetto M. The adventure of delta. Liver Int 2016; 36(suppl 1):135–140.
- Irshad M, Acharya SK. Hepatitis D virus (HDV) infection in severe forms of liver diseases in North India. Eur J Gastroenterol Hepatol 1996; 8:995–998.
- Noureddin M, Gish R. Hepatitis delta: epidemiology, diagnosis and management 36 years after discovery. Curr Gastroenterol Rep 2014; 16:365.
A guide to managing acute liver failure
When the liver fails, it usually fails gradually. The sudden (acute) onset of liver failure, while less common, demands prompt management, with transfer to an intensive care unit, specific treatment depending on the cause, and consideration of liver transplant, without which the mortality rate is high.
This article reviews the definition, epidemiology, etiology, and management of acute liver failure.
DEFINITIONS
Acute liver failure is defined as a syndrome of acute hepatitis with evidence of abnormal coagulation (eg, an international normalized ratio > 1.5) complicated by the development of mental alteration (encephalopathy) within 26 weeks of the onset of illness in a patient without a history of liver disease.1 In general, patients have no evidence of underlying chronic liver disease, but there are exceptions; patients with Wilson disease, vertically acquired hepatitis B virus infection, or autoimmune hepatitis can present with acute liver failure superimposed on chronic liver disease or even cirrhosis.
The term acute liver failure has replaced older terms such as fulminant hepatic failure, hyperacute liver failure, and subacute liver failure, which were used for prognostic purposes. Patients with hyperacute liver failure (defined as development of encephalopathy within 7 days of onset of illness) generally have a good prognosis with medical management, whereas those with subacute liver failure (defined as development of encephalopathy within 5 to 26 weeks of onset of illness) have a poor prognosis without liver transplant.2,3
NEARLY 2,000 CASES A YEAR
There are nearly 2,000 cases of acute liver failure each year in the United States, and it accounts for 6% of all deaths due to liver disease.4 It is more common in women than in men, and more common in white people than in other races. The peak incidence is at a fairly young age, ie, 35 to 45 years.
CAUSES
The most common cause of acute liver failure in the United States and other Western countries is acetaminophen toxicity, followed by viral hepatitis. In contrast, viral hepatitis is the most common cause in developing countries.5
Acetaminophen toxicity
Patients with acetaminophen-induced liver failure tend to be younger than other patients with acute liver failure.1 Nearly half of them present after intentionally taking a single large dose, while the rest present with unintentional toxicity while taking acetaminophen for pain relief on a long-term basis and ingesting more than the recommended dose.6
After ingestion, 52% to 57% of acetaminophen is converted to glucuronide conjugates, and 30% to 44% is converted to sulfate conjugates. These compounds are nontoxic, water-soluble, and rapidly excreted in the urine.
However, about 5% to 10% of ingested acetaminophen is shunted to the cytochrome P450 system. P450 2E1 is the main isoenzyme involved in acetaminophen metabolism, but 1A2, 3A4, and 2A6 also contribute.7,8 P450 2E1 is the same isoenzyme responsible for ethanol metabolism and is inducible. Thus, regular alcohol consumption can increase P450 2E1 activity, setting the stage under certain circumstances for increased acetaminophen metabolism through this pathway.

Metabolism of acetaminophen through the cytochrome P450 pathway results in production of N-acetyl-p-benzoquinone imine (NAPQI), the compound that damages the liver. NAPQI is rendered nontoxic by binding to glutathione, forming NAPQI-glutathione adducts. Glutathione capacity is limited, however. With too much acetaminophen, glutathione becomes depleted and NAPQI accumulates, binds with proteins to form adducts, and leads to necrosis of hepatocytes (Figure 1).9,10
Acetylcysteine, used in treating acetaminophen toxicity, is a substrate for glutathione synthesis and ultimately increases the amount of glutathione available to bind NAPQI and prevent damage to hepatocytes.11
Acetaminophen is a dose-related toxin. Most ingestions leading to acute liver failure exceed 10 g/day (> 150 mg/kg/day). Moderate chronic ingestion, eg, 4 g/day, usually leads to transient mild elevation of liver enzymes in healthy individuals12 but can in rare cases cause acute liver failure.13

Whitcomb and Block14 retrospectively identified 49 patients who presented with acetaminophen-induced hepatotoxicity in 1987 through 1993; 21 (43%) had been taking acetaminophen for therapeutic purposes. All 49 patients took more than the recommended limit of 4 g/day, many of them while fasting and some while using alcohol. Acute liver failure was seen with ingestion of more than 12 g/day—or more than 10 g/day in alcohol users. The authors attributed the increased risk to activation of cytochrome P450 2E1 by alcohol and depletion of glutathione stores by starvation or alcohol abuse.
Advice to patients taking acetaminophen is given in Table 1.
Other drugs and supplements

A number of other drugs and herbal supplements can also cause acute liver failure (Table 2), the most common being antimicrobial and antiepileptic drugs.15 Of the antimicrobials, antitubercular drugs (especially isoniazid) are believed to be the most common causes, followed by trimethoprim-sulfamethoxazole. Phenytoin is the antiepileptic drug most often implicated in acute liver failure.
Statins can also cause acute liver failure, especially when combined with other hepatotoxic agents.16
The herbal supplements and weight-loss agents Hydroxycut and Herbalife have both been reported to cause acute liver failure, with patients presenting with either the hepatocellular or the cholestatic pattern of liver injury.17 The exact chemical in these supplements that causes liver injury has not yet been determined.
The National Institutes of Health maintains a database of cases of liver failure due to medications and supplements at livertox.nih.gov. The database includes the pattern of hepatic injury, mechanism of injury, management, and outcomes.
Viral hepatitis
Hepatitis B virus is the most common viral cause of acute liver failure and is responsible for about 8% of cases.18
Patients with chronic hepatitis B virus infection—as evidenced by positive hepatitis B surface antigen—can develop acute liver failure if the infection is reactivated by the use of immunosuppressive drugs for solid-organ or bone-marrow transplant or medications such as anti-tumor necrosis agents, rituximab, or chemotherapy. These patients should be treated prophylactically with a nucleoside analogue, which should be continued for 6 months after immunosuppressive therapy is completed.
Hepatitis A virus is responsible for about 4% of cases.18
Hepatitis C virus rarely causes acute liver failure, especially in the absence of hepatitis A and hepatitis B.3,19
Hepatitis E virus, which is endemic in areas of Asia and Africa, can cause liver disease in pregnant women and in young adults who have concomitant liver disease from another cause. It tends to cause acute liver failure more frequently in pregnant women than in the rest of the population and carries a mortality rate of more than 20% in this subgroup.
TT (transfusion-transmitted) virus was reported in the 1990s to cause acute liver failure in about 27% of patients in whom no other cause could be found.20
Other rare viral causes of acute liver failure include Epstein-Barr virus, cytomegalovirus, and herpes simplex virus types 1, 2, and 6.
Other causes
Other causes of acute liver failure include ischemic hepatitis, autoimmune hepatitis, Wilson disease, Budd-Chiari syndrome, and HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome.
MANY PATIENTS NEED LIVER TRANSPLANT

Many patients with acute liver failure ultimately require orthotopic liver transplant,21 especially if they present with severe encephalopathy. Other aspects of treatment vary according to the cause of liver failure (Table 3).
SPECIFIC MANAGEMENT
Management of acetaminophen toxicity
If the time of ingestion is known, checking the acetaminophen level can help determine the cause of acute liver failure and also predict the risk of hepatotoxicity, based on the work of Rumack and Matthew.22 Calculators are available, eg, http://reference.medscape.com/calculator/acetaminophen-toxicity.
If a patient presents with acute liver failure several days after ingesting acetaminophen, the level can be in the nontoxic range, however. In this scenario, measuring acetaminophen-protein adducts can help establish acetaminophen toxicity as the cause, as the adducts last longer in the serum and provide 100% sensitivity and specificity.23 While most laboratories can rapidly measure acetaminophen levels, only a few can measure acetaminophen-protein adducts, and thus this test is not used clinically.
Acetylcysteine is the main drug used for acetaminophen toxicity. Ideally, it should be given within 8 hours of acetaminophen ingestion, but giving it later is also useful.1
Acetylcysteine is available in oral and intravenous forms, the latter for patients who have encephalopathy or cannot tolerate oral intake due to repeated episodes of vomiting.24,25 The oral form is much less costly and is thus preferred over intravenous acetylcysteine in patients who can tolerate oral intake. Intravenous acetylcysteine should be given in a loading dose of 150 mg/kg in 5% dextrose over 15 minutes, followed by a maintenance dose of 50 mg/kg over 4 hours and then 100 mg/kg given over 16 hours.1 No dose adjustment is needed in patients who have renal toxicity (acetaminophen can also be toxic to the kidneys).
Most patients with acetaminophen-induced liver failure survive with medical management alone and do not need a liver transplant.3,26 Cirrhosis does not occur in these patients.
Management of viral acute liver failure
When patients present with acute liver failure, it is necessary to look for a viral cause by serologic testing, including hepatitis A virus IgM antibody, hepatitis B surface antigen, and hepatitis B core IgM antibody.
Hepatitis B can become reactivated in immunocompromised patients, and therefore the hepatitis B virus DNA level should be checked. Detection of hepatitis B virus DNA in a patient previously known to have undetectable hepatitis B virus DNA confirms hepatitis B reactivation.
Patients with hepatitis B-induced acute liver failure should be treated with entecavir or tenofovir. Although this treatment may not change the course of acute liver failure or accelerate the recovery, it can prevent reinfection in the transplanted liver if liver transplant becomes indicated.27–29
Herpes simplex virus should be suspected in patients presenting with anicteric hepatitis with fever. Polymerase chain reaction testing for herpes simplex virus should be done,30 and if positive, patients should be given intravenous acyclovir.31 Despite treatment, herpes simplex virus disease is associated with a very poor prognosis without liver transplant.
Autoimmune hepatitis
The autoantibodies usually seen in autoimmune hepatitis are antinuclear antibody, antismooth muscle antibody, and anti-liver-kidney microsomal antibody, and patients need to be tested for them.
The diagnosis of autoimmune hepatitis can be challenging, as these autoimmune markers can be negative in 5% of patients. Liver biopsy becomes essential to establish the diagnosis in that setting.32
Guidelines advise starting prednisone 40 to 60 mg/day and placing the patient on the liver transplant list.1
Wilson disease
Although it is an uncommon cause of liver failure, Wilson disease needs special attention because it has a poor prognosis. The mortality rate in acute liver failure from Wilson disease reaches 100% without liver transplant.
Wilson disease is caused by a genetic defect that allows copper to accumulate in the liver and other organs. However, diagnosing Wilson disease as the cause of acute liver failure can be challenging because elevated serum and urine copper levels are not specific to Wilson disease and can be seen in patients with acute liver failure from any cause. In addition, the ceruloplasmin level is usually normal or high because it is an acute-phase reactant. Accumulation of copper in the liver parenchyma is usually patchy; therefore, qualitative copper staining on random liver biopsy samples provides low diagnostic yield. Quantitative copper on liver biopsy is the gold standard test to establish the diagnosis, but the test is time-consuming. Kayser-Fleischer rings around the iris are considered pathognomic for Wilson disease when seen with acute liver failure, but they are seen in only about 50% of patients.33
A unique feature of acute Wilson disease is that most patients have very high bilirubin levels and low alkaline phosphatase levels. An alkaline phosphatase-to-bilirubin ratio less than 2 in patients with acute liver failure is highly suggestive of Wilson disease.34
Another clue to the diagnosis is that patients with Wilson disease tend to develop Coombs-negative hemolytic anemia, which leads to a disproportionate elevation in aminotransferase levels, with aspartate aminotransferase being higher than alanine aminotransferase.
Once Wilson disease is suspected, the patient should be listed for liver transplant because death is almost certain without it. For patients awaiting liver transplant, the American Association for the Study of Liver Diseases guidelines recommend certain measures to lower the serum copper level such as albumin dialysis, continuous hemofiltration, plasmapheresis, and plasma exchange,1 but the evidence supporting their use is limited.
NONSPECIFIC MANAGEMENT
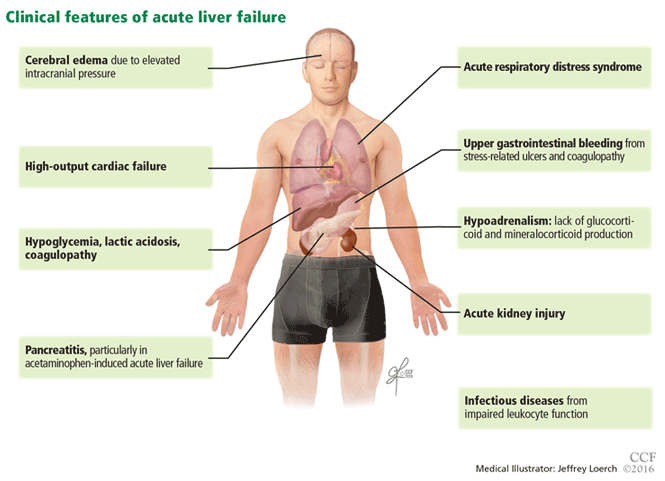
Acute liver failure can affect a number of organs and systems in addition to the liver (Figure 2).
General considerations
Because their condition can rapidly deteriorate, patients with acute liver failure are best managed in intensive care.
Patients who present to a center that does not have the facilities for liver transplant should be transferred to a transplant center as soon as possible, preferably by air. If the patient may not be able to protect the airway, endotracheal intubation should be performed before transfer.
The major causes of death in patients with acute liver failure are cerebral edema and infection. Gastrointestinal bleeding was a major cause of death in the past, but with prophylactic use of histamine H2 receptor blockers and proton pump inhibitors, the incidence of gastrointestinal bleeding has been significantly reduced.
Although initially used only in patients with acetaminophen-induced liver failure, acetylcysteine has also shown benefit in patients with acute liver failure from other causes. In patients with grade 1 or 2 encephalopathy on a scale of 0 (minimal) to 4 (comatose), the transplant-free survival rate is higher when acetylcysteine is given compared with placebo, but this benefit does not extend to patients with a higher grade of encephalopathy.35
Cerebral edema and intracranial hypertension
Cerebral edema is the leading cause of death in patients with acute liver failure, and it develops in nearly 40% of patients.36
The mechanism by which cerebral edema develops is not well understood. Some have proposed that ammonia is converted to glutamine, which causes cerebral edema either directly by its osmotic effect37,38 or indirectly by decreasing other osmolytes, thereby promoting water retention.39
Cerebral edema leads to intracranial hypertension, which can ultimately cause cerebral herniation and death. Because of the high mortality rate associated with cerebral edema, invasive devices were extensively used in the past to monitor intracranial pressure. However, in light of known complications of these devices, including bleeding,40 and lack of evidence of long-term benefit in terms of mortality rates, their use has come under debate.
Treatments. Many treatments are available for cerebral edema and intracranial hypertension. The first step is to elevate the head of the bed about 30 degrees. In addition, hyponatremia should be corrected, as it can worsen cerebral edema.41 If patients are intubated, maintaining a hypercapneic state is advisable to decrease the intracranial pressure.
Of the two pharmacologic options, mannitol is more often used.42 It is given as a bolus dose of 0.5 to 1 g/kg intravenously if the serum osmolality is less than 320 mOsm/L.1 Given the risk of fluid overload with mannitol, caution must be exercised in patients with renal dysfunction. The other pharmacologic option is 3% hypertonic saline.
Therapeutic hypothermia is a newer treatment for cerebral edema. Lowering the body temperature to 32 to 33°C (89.6 to 91.4°F) using cooling blankets decreases intracranial pressure and cerebral blood flow and improves the cerebral perfusion pressure.43 With this treatment, patients should be closely monitored for side effects of infection, coagulopathy, and cardiac arrythmias.1
l-ornithine l-aspartate was successfully used to prevent brain edema in rats, but in humans, no benefit was seen compared with placebo.44,45 The underlying basis for this experimental treatment is that supplemental ornithine and aspartate should increase glutamate synthesis, which should increase the activity of enzyme glutamine synthetase in skeletal muscles. With the increase in enzyme activity, conversion of ammonia to glutamine should increase, thereby decreasing ammonia circulation and thus decreasing cerebral edema.
Patients with cerebral edema have a high incidence of seizures, but prophylactic antiseizure medications such as phenytoin have not been proven to be beneficial.46
Infection
Nearly 80% of patients with acute liver failure develop an infectious complication, which can be attributed to a state of immunodeficiency.47
The respiratory and urinary tracts are the most common sources of infection.48 In patients with bacteremia, Enterococcus species and coagulase-negative Staphylococcus species49 are the commonly isolated organisms. Also, in patients with acute liver failure, fungal infections account for 30% of all infections.50
Infected patients often develop worsening of their encephalopathy51 without fever or elevated white blood cell count.49,52 Thus, in any patient in whom encephalopathy is worsening, an evaluation must be done to rule out infection. In these patients, systemic inflammatory response syndrome is an independent risk factor for death.53
Despite the high mortality rate with infection, whether using antibiotics prophylactically in acute liver failure is beneficial is controversial.54,55
Gastrointestinal bleeding
The current prevalence of upper gastrointestinal bleeding in acute liver failure patients is about 1.5%.56 Coagulopathy and endotracheal intubation are the main risk factors for upper gastrointestinal bleeding in these patients.57 The most common source of bleeding is stress ulcers in the stomach. The ulcers develop from a combination of factors, including decreased blood flow to the mucosa causing ischemia and hypoperfusion-reperfusion injury.
Pharmacologic inhibition of gastric acid secretion has been shown to reduce upper gastrointestinal bleeding in acute liver failure. A histamine H2 receptor blocker or proton pump inhibitor should be given to prevent gastrointestinal bleeding in patients with acute liver failure.1,58
EXPERIMENTAL TREATMENTS
Artificial liver support systems
Membranes and dialysate solutions have been developed to remove toxic substances that are normally metabolized by the liver. Two of these—the molecular adsorbent recycling system (MARS) and the extracorporeal liver assist device (ELAD)—were developed in the late 1990s. MARS consisted of a highly permeable hollow fiber membrane mixed with albumin, and ELAD consisted of porcine hepatocytes attached to microcarriers in the extracapillary space of the hollow fiber membrane. Both systems allowed for transfer of water-soluble and protein-bound toxins in the blood across the membrane and into the dialysate.59 The clinical benefit offered by these devices is controversial,60–62 thus limiting their use to experimental purposes only.
Hepatocyte transplant
Use of hepatocyte transplant as a bridge to liver transplant was tested in 1970s, first in rats and later in humans.63 By reducing the blood ammonia level and improving cerebral perfusion pressure and cardiac function, replacement of 1% to 2% of the total liver cell mass by transplanted hepatocytes acts as a bridge to orthotopic liver transplant.64,65
PROGNOSIS
Different criteria have been used to identify patients with poor prognosis who may eventually need to undergo liver transplant.
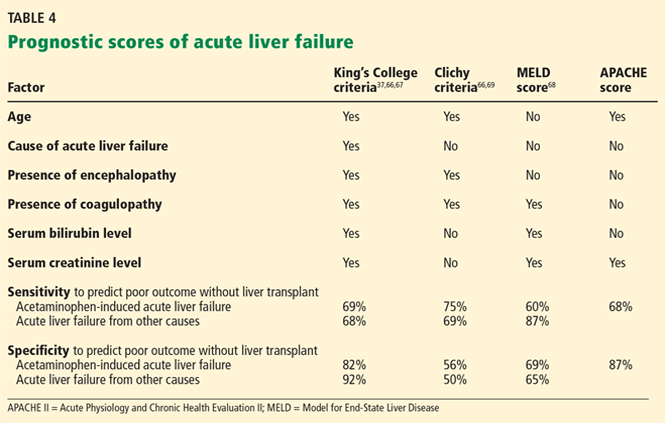
The King’s College criteria system is the most commonly used for prognosis (Table 4).37,66–69 Its main drawback is that it is applicable only in patients with encephalopathy, and when patients reach this stage, their condition often deteriorates rapidly, and they die while awaiting liver transplant.37,66,67
The Model for End-Stage Liver Disease (MELD) score is an alternative to the King’s College criteria. A high MELD score on admission signifies advanced disease, and patients with a high MELD score tend to have a worse prognosis than those with a low score.68
The Acute Physiology and Chronic Health Evaluation (APACHE) II score can also be used, as it is more sensitive than the King’s College criteria.6
The Clichy criteria66,69 can also be used.
Liver biopsy. In addition to helping establish the cause of acute liver failure, liver biopsy can also be used as a prognostic tool. Hepatocellular necrosis greater than 70% on the biopsy predicts death with a specificity of 90% and a sensitivity of 56%.70
Hypophosphatemia has been reported to indicate recovering liver function in patients with acute liver failure.71 As the liver regenerates, its energy requirement increases. To supply the energy, adenosine triphosphate production increases, and phosphorus shifts from the extracellular to the intracellular compartment to meet the need for extra phosphorus during this process. A serum phosphorus level of 2.9 mg/dL or higher appears to indicate a poor prognosis in patients with acute liver failure, as it signifies that adequate hepatocyte regeneration is not occurring.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 342:273–275.
- Ostapowicz G, Fontana RJ, Schiodt FV, et al; US Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 2008; 47:1401–1415.
- Acharya SK, Panda SK, Saxena A, Gupta SD. Acute hepatic failure in India: a perspective from the East. J Gastroenterol Hepatol 2000; 15:473–479.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Patten CJ, Thomas PE, Guy RL, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 1993; 6:511–518.
- Chen W, Koenigs LL, Thompson SJ, et al. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol 1998; 11:295-301.
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187:211–217.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 1983; 71:980–991.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- Schiødt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med 1997; 337:1112–1117.
- Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994; 272:1845–1850.
- Chalasani N, Fontana RJ, Bonkovsky HL, et al; Drug Induced Liver Injury Network (DILIN). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008; 135:1924–1934 e1–4
- Reuben A, Koch DG, Lee WM; Acute Liver Failure Study Group. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology 2010; 52:2065–2076.
- Stevens T, Qadri A, Zein NN. Two patients with acute liver injury associated with use of the herbal weight-loss supplement hydroxycut. Ann Intern Med 2005; 142:477–478.
- Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol 2015; 62(suppl 1):S112–S120.
- Schiodt FV, Davern TJ, Shakil AO, McGuire B, Samuel G, Lee WM. Viral hepatitis-related acute liver failure. Am J Gastroenterol 2003; 98:448–453.
- Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology 1998; 28:839–842.
- Bismuth H, Samuel D, Gugenheim J, et al. Emergency liver transplantation for fulminant hepatitis. Ann Intern Med 1987; 107:337–341.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Davern TJ 2nd, James LP, Hinson JA, et al; Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006; 130:687–694.
- Perry HE, Shannon MW. Efficacy of oral versus intravenous N-acetylcysteine in acetaminophen overdose: results of an open-label, clinical trial. J Pediatr 1998; 132:149–152.
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557–1562.
- Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology 1995; 109:1907–1916.
- Tsang SW, Chan HL, Leung NW, et al. Lamivudine treatment for fulminant hepatic failure due to acute exacerbation of chronic hepatitis B infection. Aliment Pharmacol Ther 2001; 15:1737–1744.
- Yu JW, Sun LJ, Yan BZ, Kang P, Zhao YH. Lamivudine treatment is associated with improved survival in fulminant hepatitis B. Liver Int 2011; 31:499–506.
- Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 2011; 53:774–780.
- Pinna AD, Rakela J, Demetris AJ, Fung JJ. Five cases of fulminant hepatitis due to herpes simplex virus in adults. Dig Dis Sci 2002; 47:750–754.
- Farr RW, Short S, Weissman D. Fulminant hepatitis during herpes simplex virus infection in apparently immunocompetent adults: report of two cases and review of the literature. Clin Infect Dis 1997; 24:1191–1194.
- Czaja AJ, Freese DK; American Association for the Study of Liver Disease. Diagnosis and treatment of autoimmune hepatitis. Hepatology 2002; 36:479–497.
- Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003; 37:1475–1492.
- Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991; 100:1129–1134.
- Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009; 137:856–864.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology 1992; 15:449–453.
- Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 1994; 107:1475–1480.
- Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet 1993; 341:157–158.
- Cordoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol 1998; 29:589–594.
- Canalese J, Gimson AE, Davis C, Mellon PJ, Davis M, Williams R. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut 1982; 23:625–629.
- Jalan R, SW OD, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 1999; 354:1164–1168.
- Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 1999; 30:636–640.
- Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology 2009; 136:2159–2168.
- Bhatia V, Batra Y, Acharya SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol 2004; 41:89–96.
- Canalese J, Gove CD, Gimson AE, Wilkinson SP, Wardle EN, Williams R. Reticuloendothelial system and hepatocytic function in fulminant hepatic failure. Gut 1982; 23:265–269.
- Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology 1990; 11:49–53.
- Rolando N, Wade JJ, Stangou A, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg 1996; 2:8–13.
- Rolando N, Harvey F, Brahm J, et al. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol 1991; 12:1–9.
- Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology 2003; 125:755–764.
- Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis 1996; 16:389–402.
- Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32:734–739.
- Rolando N, Gimson A, Wade J, Philpott- Howard J, Casewell M, Williams R. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology 1993; 17:196–201.
- Karvellas CJ, Cavazos J, Battenhouse H, et al; US Acute Liver Failure Study Group. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol 2014; 12:1942–1949.
- Acharya SK, Dasarathy S, Kumer TL, et al. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology 1996; 23:1148–1155.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- MacDougall BR, Williams R. H2-receptor antagonist in the prevention of acute gastrointestinal hemorrhage in fulminant hepatic failure: a controlled trial. Gastroenterology 1978; 74:464–465.
- Stange J, Mitzner SR, Risler T, et al. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs 1999; 23:319–330.
- Vaid A, Chewich H, Balk EM, Jaber BL. Molecular adsorbent recirculating system as artificial support therapy for liver failure: a meta-analysis. ASAIO J 2012; 58:51–59.
- Khuroo MS, Khuroo MS, Farahat KL. Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: a meta-analysis. Liver Transpl 2004; 10:1099–1106.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Sommer BG, Sutherland DE, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation for treatment of D-galactosamine-induced acute liver failure in rats. Transplant Proc 1979; 11:578–584.
- Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology 1988; 8:1006–1009.
- Strom SC, Fisher RA, Thompson MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 1997; 63:559–569.
- Pauwels A, Mostefa-Kara N, Florent C, Levy VG. Emergency liver transplantation for acute liver failure. Evaluation of London and Clichy criteria. J Hepatol 1993; 17:124–127.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King's criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Larsen FS. MELD score as a predictor of liver failure and death in patients with acetaminophen-induced liver injury. Hepatology 2007; 45:789–796.
- Bernuau J, Goudeau A, Poynard T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology 1986; 6:648–651.
- Donaldson BW, Gopinath R, Wanless IR, et al. The role of transjugular liver biopsy in fulminant liver failure: relation to other prognostic indicators. Hepatology 1993; 18:1370–1376.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
When the liver fails, it usually fails gradually. The sudden (acute) onset of liver failure, while less common, demands prompt management, with transfer to an intensive care unit, specific treatment depending on the cause, and consideration of liver transplant, without which the mortality rate is high.
This article reviews the definition, epidemiology, etiology, and management of acute liver failure.
DEFINITIONS
Acute liver failure is defined as a syndrome of acute hepatitis with evidence of abnormal coagulation (eg, an international normalized ratio > 1.5) complicated by the development of mental alteration (encephalopathy) within 26 weeks of the onset of illness in a patient without a history of liver disease.1 In general, patients have no evidence of underlying chronic liver disease, but there are exceptions; patients with Wilson disease, vertically acquired hepatitis B virus infection, or autoimmune hepatitis can present with acute liver failure superimposed on chronic liver disease or even cirrhosis.
The term acute liver failure has replaced older terms such as fulminant hepatic failure, hyperacute liver failure, and subacute liver failure, which were used for prognostic purposes. Patients with hyperacute liver failure (defined as development of encephalopathy within 7 days of onset of illness) generally have a good prognosis with medical management, whereas those with subacute liver failure (defined as development of encephalopathy within 5 to 26 weeks of onset of illness) have a poor prognosis without liver transplant.2,3
NEARLY 2,000 CASES A YEAR
There are nearly 2,000 cases of acute liver failure each year in the United States, and it accounts for 6% of all deaths due to liver disease.4 It is more common in women than in men, and more common in white people than in other races. The peak incidence is at a fairly young age, ie, 35 to 45 years.
CAUSES
The most common cause of acute liver failure in the United States and other Western countries is acetaminophen toxicity, followed by viral hepatitis. In contrast, viral hepatitis is the most common cause in developing countries.5
Acetaminophen toxicity
Patients with acetaminophen-induced liver failure tend to be younger than other patients with acute liver failure.1 Nearly half of them present after intentionally taking a single large dose, while the rest present with unintentional toxicity while taking acetaminophen for pain relief on a long-term basis and ingesting more than the recommended dose.6
After ingestion, 52% to 57% of acetaminophen is converted to glucuronide conjugates, and 30% to 44% is converted to sulfate conjugates. These compounds are nontoxic, water-soluble, and rapidly excreted in the urine.
However, about 5% to 10% of ingested acetaminophen is shunted to the cytochrome P450 system. P450 2E1 is the main isoenzyme involved in acetaminophen metabolism, but 1A2, 3A4, and 2A6 also contribute.7,8 P450 2E1 is the same isoenzyme responsible for ethanol metabolism and is inducible. Thus, regular alcohol consumption can increase P450 2E1 activity, setting the stage under certain circumstances for increased acetaminophen metabolism through this pathway.
Metabolism of acetaminophen through the cytochrome P450 pathway results in production of N-acetyl-p-benzoquinone imine (NAPQI), the compound that damages the liver. NAPQI is rendered nontoxic by binding to glutathione, forming NAPQI-glutathione adducts. Glutathione capacity is limited, however. With too much acetaminophen, glutathione becomes depleted and NAPQI accumulates, binds with proteins to form adducts, and leads to necrosis of hepatocytes (Figure 1).9,10
Acetylcysteine, used in treating acetaminophen toxicity, is a substrate for glutathione synthesis and ultimately increases the amount of glutathione available to bind NAPQI and prevent damage to hepatocytes.11
Acetaminophen is a dose-related toxin. Most ingestions leading to acute liver failure exceed 10 g/day (> 150 mg/kg/day). Moderate chronic ingestion, eg, 4 g/day, usually leads to transient mild elevation of liver enzymes in healthy individuals12 but can in rare cases cause acute liver failure.13
Whitcomb and Block14 retrospectively identified 49 patients who presented with acetaminophen-induced hepatotoxicity in 1987 through 1993; 21 (43%) had been taking acetaminophen for therapeutic purposes. All 49 patients took more than the recommended limit of 4 g/day, many of them while fasting and some while using alcohol. Acute liver failure was seen with ingestion of more than 12 g/day—or more than 10 g/day in alcohol users. The authors attributed the increased risk to activation of cytochrome P450 2E1 by alcohol and depletion of glutathione stores by starvation or alcohol abuse.
Advice to patients taking acetaminophen is given in Table 1.
Other drugs and supplements
A number of other drugs and herbal supplements can also cause acute liver failure (Table 2), the most common being antimicrobial and antiepileptic drugs.15 Of the antimicrobials, antitubercular drugs (especially isoniazid) are believed to be the most common causes, followed by trimethoprim-sulfamethoxazole. Phenytoin is the antiepileptic drug most often implicated in acute liver failure.
Statins can also cause acute liver failure, especially when combined with other hepatotoxic agents.16
The herbal supplements and weight-loss agents Hydroxycut and Herbalife have both been reported to cause acute liver failure, with patients presenting with either the hepatocellular or the cholestatic pattern of liver injury.17 The exact chemical in these supplements that causes liver injury has not yet been determined.
The National Institutes of Health maintains a database of cases of liver failure due to medications and supplements at livertox.nih.gov. The database includes the pattern of hepatic injury, mechanism of injury, management, and outcomes.
Viral hepatitis
Hepatitis B virus is the most common viral cause of acute liver failure and is responsible for about 8% of cases.18
Patients with chronic hepatitis B virus infection—as evidenced by positive hepatitis B surface antigen—can develop acute liver failure if the infection is reactivated by the use of immunosuppressive drugs for solid-organ or bone-marrow transplant or medications such as anti-tumor necrosis agents, rituximab, or chemotherapy. These patients should be treated prophylactically with a nucleoside analogue, which should be continued for 6 months after immunosuppressive therapy is completed.
Hepatitis A virus is responsible for about 4% of cases.18
Hepatitis C virus rarely causes acute liver failure, especially in the absence of hepatitis A and hepatitis B.3,19
Hepatitis E virus, which is endemic in areas of Asia and Africa, can cause liver disease in pregnant women and in young adults who have concomitant liver disease from another cause. It tends to cause acute liver failure more frequently in pregnant women than in the rest of the population and carries a mortality rate of more than 20% in this subgroup.
TT (transfusion-transmitted) virus was reported in the 1990s to cause acute liver failure in about 27% of patients in whom no other cause could be found.20
Other rare viral causes of acute liver failure include Epstein-Barr virus, cytomegalovirus, and herpes simplex virus types 1, 2, and 6.
Other causes
Other causes of acute liver failure include ischemic hepatitis, autoimmune hepatitis, Wilson disease, Budd-Chiari syndrome, and HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome.
MANY PATIENTS NEED LIVER TRANSPLANT
Many patients with acute liver failure ultimately require orthotopic liver transplant,21 especially if they present with severe encephalopathy. Other aspects of treatment vary according to the cause of liver failure (Table 3).
SPECIFIC MANAGEMENT
Management of acetaminophen toxicity
If the time of ingestion is known, checking the acetaminophen level can help determine the cause of acute liver failure and also predict the risk of hepatotoxicity, based on the work of Rumack and Matthew.22 Calculators are available, eg, http://reference.medscape.com/calculator/acetaminophen-toxicity.
If a patient presents with acute liver failure several days after ingesting acetaminophen, the level can be in the nontoxic range, however. In this scenario, measuring acetaminophen-protein adducts can help establish acetaminophen toxicity as the cause, as the adducts last longer in the serum and provide 100% sensitivity and specificity.23 While most laboratories can rapidly measure acetaminophen levels, only a few can measure acetaminophen-protein adducts, and thus this test is not used clinically.
Acetylcysteine is the main drug used for acetaminophen toxicity. Ideally, it should be given within 8 hours of acetaminophen ingestion, but giving it later is also useful.1
Acetylcysteine is available in oral and intravenous forms, the latter for patients who have encephalopathy or cannot tolerate oral intake due to repeated episodes of vomiting.24,25 The oral form is much less costly and is thus preferred over intravenous acetylcysteine in patients who can tolerate oral intake. Intravenous acetylcysteine should be given in a loading dose of 150 mg/kg in 5% dextrose over 15 minutes, followed by a maintenance dose of 50 mg/kg over 4 hours and then 100 mg/kg given over 16 hours.1 No dose adjustment is needed in patients who have renal toxicity (acetaminophen can also be toxic to the kidneys).
Most patients with acetaminophen-induced liver failure survive with medical management alone and do not need a liver transplant.3,26 Cirrhosis does not occur in these patients.
Management of viral acute liver failure
When patients present with acute liver failure, it is necessary to look for a viral cause by serologic testing, including hepatitis A virus IgM antibody, hepatitis B surface antigen, and hepatitis B core IgM antibody.
Hepatitis B can become reactivated in immunocompromised patients, and therefore the hepatitis B virus DNA level should be checked. Detection of hepatitis B virus DNA in a patient previously known to have undetectable hepatitis B virus DNA confirms hepatitis B reactivation.
Patients with hepatitis B-induced acute liver failure should be treated with entecavir or tenofovir. Although this treatment may not change the course of acute liver failure or accelerate the recovery, it can prevent reinfection in the transplanted liver if liver transplant becomes indicated.27–29
Herpes simplex virus should be suspected in patients presenting with anicteric hepatitis with fever. Polymerase chain reaction testing for herpes simplex virus should be done,30 and if positive, patients should be given intravenous acyclovir.31 Despite treatment, herpes simplex virus disease is associated with a very poor prognosis without liver transplant.
Autoimmune hepatitis
The autoantibodies usually seen in autoimmune hepatitis are antinuclear antibody, antismooth muscle antibody, and anti-liver-kidney microsomal antibody, and patients need to be tested for them.
The diagnosis of autoimmune hepatitis can be challenging, as these autoimmune markers can be negative in 5% of patients. Liver biopsy becomes essential to establish the diagnosis in that setting.32
Guidelines advise starting prednisone 40 to 60 mg/day and placing the patient on the liver transplant list.1
Wilson disease
Although it is an uncommon cause of liver failure, Wilson disease needs special attention because it has a poor prognosis. The mortality rate in acute liver failure from Wilson disease reaches 100% without liver transplant.
Wilson disease is caused by a genetic defect that allows copper to accumulate in the liver and other organs. However, diagnosing Wilson disease as the cause of acute liver failure can be challenging because elevated serum and urine copper levels are not specific to Wilson disease and can be seen in patients with acute liver failure from any cause. In addition, the ceruloplasmin level is usually normal or high because it is an acute-phase reactant. Accumulation of copper in the liver parenchyma is usually patchy; therefore, qualitative copper staining on random liver biopsy samples provides low diagnostic yield. Quantitative copper on liver biopsy is the gold standard test to establish the diagnosis, but the test is time-consuming. Kayser-Fleischer rings around the iris are considered pathognomic for Wilson disease when seen with acute liver failure, but they are seen in only about 50% of patients.33
A unique feature of acute Wilson disease is that most patients have very high bilirubin levels and low alkaline phosphatase levels. An alkaline phosphatase-to-bilirubin ratio less than 2 in patients with acute liver failure is highly suggestive of Wilson disease.34
Another clue to the diagnosis is that patients with Wilson disease tend to develop Coombs-negative hemolytic anemia, which leads to a disproportionate elevation in aminotransferase levels, with aspartate aminotransferase being higher than alanine aminotransferase.
Once Wilson disease is suspected, the patient should be listed for liver transplant because death is almost certain without it. For patients awaiting liver transplant, the American Association for the Study of Liver Diseases guidelines recommend certain measures to lower the serum copper level such as albumin dialysis, continuous hemofiltration, plasmapheresis, and plasma exchange,1 but the evidence supporting their use is limited.
NONSPECIFIC MANAGEMENT
Acute liver failure can affect a number of organs and systems in addition to the liver (Figure 2).
General considerations
Because their condition can rapidly deteriorate, patients with acute liver failure are best managed in intensive care.
Patients who present to a center that does not have the facilities for liver transplant should be transferred to a transplant center as soon as possible, preferably by air. If the patient may not be able to protect the airway, endotracheal intubation should be performed before transfer.
The major causes of death in patients with acute liver failure are cerebral edema and infection. Gastrointestinal bleeding was a major cause of death in the past, but with prophylactic use of histamine H2 receptor blockers and proton pump inhibitors, the incidence of gastrointestinal bleeding has been significantly reduced.
Although initially used only in patients with acetaminophen-induced liver failure, acetylcysteine has also shown benefit in patients with acute liver failure from other causes. In patients with grade 1 or 2 encephalopathy on a scale of 0 (minimal) to 4 (comatose), the transplant-free survival rate is higher when acetylcysteine is given compared with placebo, but this benefit does not extend to patients with a higher grade of encephalopathy.35
Cerebral edema and intracranial hypertension
Cerebral edema is the leading cause of death in patients with acute liver failure, and it develops in nearly 40% of patients.36
The mechanism by which cerebral edema develops is not well understood. Some have proposed that ammonia is converted to glutamine, which causes cerebral edema either directly by its osmotic effect37,38 or indirectly by decreasing other osmolytes, thereby promoting water retention.39
Cerebral edema leads to intracranial hypertension, which can ultimately cause cerebral herniation and death. Because of the high mortality rate associated with cerebral edema, invasive devices were extensively used in the past to monitor intracranial pressure. However, in light of known complications of these devices, including bleeding,40 and lack of evidence of long-term benefit in terms of mortality rates, their use has come under debate.
Treatments. Many treatments are available for cerebral edema and intracranial hypertension. The first step is to elevate the head of the bed about 30 degrees. In addition, hyponatremia should be corrected, as it can worsen cerebral edema.41 If patients are intubated, maintaining a hypercapneic state is advisable to decrease the intracranial pressure.
Of the two pharmacologic options, mannitol is more often used.42 It is given as a bolus dose of 0.5 to 1 g/kg intravenously if the serum osmolality is less than 320 mOsm/L.1 Given the risk of fluid overload with mannitol, caution must be exercised in patients with renal dysfunction. The other pharmacologic option is 3% hypertonic saline.
Therapeutic hypothermia is a newer treatment for cerebral edema. Lowering the body temperature to 32 to 33°C (89.6 to 91.4°F) using cooling blankets decreases intracranial pressure and cerebral blood flow and improves the cerebral perfusion pressure.43 With this treatment, patients should be closely monitored for side effects of infection, coagulopathy, and cardiac arrythmias.1
l-ornithine l-aspartate was successfully used to prevent brain edema in rats, but in humans, no benefit was seen compared with placebo.44,45 The underlying basis for this experimental treatment is that supplemental ornithine and aspartate should increase glutamate synthesis, which should increase the activity of enzyme glutamine synthetase in skeletal muscles. With the increase in enzyme activity, conversion of ammonia to glutamine should increase, thereby decreasing ammonia circulation and thus decreasing cerebral edema.
Patients with cerebral edema have a high incidence of seizures, but prophylactic antiseizure medications such as phenytoin have not been proven to be beneficial.46
Infection
Nearly 80% of patients with acute liver failure develop an infectious complication, which can be attributed to a state of immunodeficiency.47
The respiratory and urinary tracts are the most common sources of infection.48 In patients with bacteremia, Enterococcus species and coagulase-negative Staphylococcus species49 are the commonly isolated organisms. Also, in patients with acute liver failure, fungal infections account for 30% of all infections.50
Infected patients often develop worsening of their encephalopathy51 without fever or elevated white blood cell count.49,52 Thus, in any patient in whom encephalopathy is worsening, an evaluation must be done to rule out infection. In these patients, systemic inflammatory response syndrome is an independent risk factor for death.53
Despite the high mortality rate with infection, whether using antibiotics prophylactically in acute liver failure is beneficial is controversial.54,55
Gastrointestinal bleeding
The current prevalence of upper gastrointestinal bleeding in acute liver failure patients is about 1.5%.56 Coagulopathy and endotracheal intubation are the main risk factors for upper gastrointestinal bleeding in these patients.57 The most common source of bleeding is stress ulcers in the stomach. The ulcers develop from a combination of factors, including decreased blood flow to the mucosa causing ischemia and hypoperfusion-reperfusion injury.
Pharmacologic inhibition of gastric acid secretion has been shown to reduce upper gastrointestinal bleeding in acute liver failure. A histamine H2 receptor blocker or proton pump inhibitor should be given to prevent gastrointestinal bleeding in patients with acute liver failure.1,58
EXPERIMENTAL TREATMENTS
Artificial liver support systems
Membranes and dialysate solutions have been developed to remove toxic substances that are normally metabolized by the liver. Two of these—the molecular adsorbent recycling system (MARS) and the extracorporeal liver assist device (ELAD)—were developed in the late 1990s. MARS consisted of a highly permeable hollow fiber membrane mixed with albumin, and ELAD consisted of porcine hepatocytes attached to microcarriers in the extracapillary space of the hollow fiber membrane. Both systems allowed for transfer of water-soluble and protein-bound toxins in the blood across the membrane and into the dialysate.59 The clinical benefit offered by these devices is controversial,60–62 thus limiting their use to experimental purposes only.
Hepatocyte transplant
Use of hepatocyte transplant as a bridge to liver transplant was tested in 1970s, first in rats and later in humans.63 By reducing the blood ammonia level and improving cerebral perfusion pressure and cardiac function, replacement of 1% to 2% of the total liver cell mass by transplanted hepatocytes acts as a bridge to orthotopic liver transplant.64,65
PROGNOSIS
Different criteria have been used to identify patients with poor prognosis who may eventually need to undergo liver transplant.
The King’s College criteria system is the most commonly used for prognosis (Table 4).37,66–69 Its main drawback is that it is applicable only in patients with encephalopathy, and when patients reach this stage, their condition often deteriorates rapidly, and they die while awaiting liver transplant.37,66,67
The Model for End-Stage Liver Disease (MELD) score is an alternative to the King’s College criteria. A high MELD score on admission signifies advanced disease, and patients with a high MELD score tend to have a worse prognosis than those with a low score.68
The Acute Physiology and Chronic Health Evaluation (APACHE) II score can also be used, as it is more sensitive than the King’s College criteria.6
The Clichy criteria66,69 can also be used.
Liver biopsy. In addition to helping establish the cause of acute liver failure, liver biopsy can also be used as a prognostic tool. Hepatocellular necrosis greater than 70% on the biopsy predicts death with a specificity of 90% and a sensitivity of 56%.70
Hypophosphatemia has been reported to indicate recovering liver function in patients with acute liver failure.71 As the liver regenerates, its energy requirement increases. To supply the energy, adenosine triphosphate production increases, and phosphorus shifts from the extracellular to the intracellular compartment to meet the need for extra phosphorus during this process. A serum phosphorus level of 2.9 mg/dL or higher appears to indicate a poor prognosis in patients with acute liver failure, as it signifies that adequate hepatocyte regeneration is not occurring.
When the liver fails, it usually fails gradually. The sudden (acute) onset of liver failure, while less common, demands prompt management, with transfer to an intensive care unit, specific treatment depending on the cause, and consideration of liver transplant, without which the mortality rate is high.
This article reviews the definition, epidemiology, etiology, and management of acute liver failure.
DEFINITIONS
Acute liver failure is defined as a syndrome of acute hepatitis with evidence of abnormal coagulation (eg, an international normalized ratio > 1.5) complicated by the development of mental alteration (encephalopathy) within 26 weeks of the onset of illness in a patient without a history of liver disease.1 In general, patients have no evidence of underlying chronic liver disease, but there are exceptions; patients with Wilson disease, vertically acquired hepatitis B virus infection, or autoimmune hepatitis can present with acute liver failure superimposed on chronic liver disease or even cirrhosis.
The term acute liver failure has replaced older terms such as fulminant hepatic failure, hyperacute liver failure, and subacute liver failure, which were used for prognostic purposes. Patients with hyperacute liver failure (defined as development of encephalopathy within 7 days of onset of illness) generally have a good prognosis with medical management, whereas those with subacute liver failure (defined as development of encephalopathy within 5 to 26 weeks of onset of illness) have a poor prognosis without liver transplant.2,3
NEARLY 2,000 CASES A YEAR
There are nearly 2,000 cases of acute liver failure each year in the United States, and it accounts for 6% of all deaths due to liver disease.4 It is more common in women than in men, and more common in white people than in other races. The peak incidence is at a fairly young age, ie, 35 to 45 years.
CAUSES
The most common cause of acute liver failure in the United States and other Western countries is acetaminophen toxicity, followed by viral hepatitis. In contrast, viral hepatitis is the most common cause in developing countries.5
Acetaminophen toxicity
Patients with acetaminophen-induced liver failure tend to be younger than other patients with acute liver failure.1 Nearly half of them present after intentionally taking a single large dose, while the rest present with unintentional toxicity while taking acetaminophen for pain relief on a long-term basis and ingesting more than the recommended dose.6
After ingestion, 52% to 57% of acetaminophen is converted to glucuronide conjugates, and 30% to 44% is converted to sulfate conjugates. These compounds are nontoxic, water-soluble, and rapidly excreted in the urine.
However, about 5% to 10% of ingested acetaminophen is shunted to the cytochrome P450 system. P450 2E1 is the main isoenzyme involved in acetaminophen metabolism, but 1A2, 3A4, and 2A6 also contribute.7,8 P450 2E1 is the same isoenzyme responsible for ethanol metabolism and is inducible. Thus, regular alcohol consumption can increase P450 2E1 activity, setting the stage under certain circumstances for increased acetaminophen metabolism through this pathway.
Metabolism of acetaminophen through the cytochrome P450 pathway results in production of N-acetyl-p-benzoquinone imine (NAPQI), the compound that damages the liver. NAPQI is rendered nontoxic by binding to glutathione, forming NAPQI-glutathione adducts. Glutathione capacity is limited, however. With too much acetaminophen, glutathione becomes depleted and NAPQI accumulates, binds with proteins to form adducts, and leads to necrosis of hepatocytes (Figure 1).9,10
Acetylcysteine, used in treating acetaminophen toxicity, is a substrate for glutathione synthesis and ultimately increases the amount of glutathione available to bind NAPQI and prevent damage to hepatocytes.11
Acetaminophen is a dose-related toxin. Most ingestions leading to acute liver failure exceed 10 g/day (> 150 mg/kg/day). Moderate chronic ingestion, eg, 4 g/day, usually leads to transient mild elevation of liver enzymes in healthy individuals12 but can in rare cases cause acute liver failure.13
Whitcomb and Block14 retrospectively identified 49 patients who presented with acetaminophen-induced hepatotoxicity in 1987 through 1993; 21 (43%) had been taking acetaminophen for therapeutic purposes. All 49 patients took more than the recommended limit of 4 g/day, many of them while fasting and some while using alcohol. Acute liver failure was seen with ingestion of more than 12 g/day—or more than 10 g/day in alcohol users. The authors attributed the increased risk to activation of cytochrome P450 2E1 by alcohol and depletion of glutathione stores by starvation or alcohol abuse.
Advice to patients taking acetaminophen is given in Table 1.
Other drugs and supplements
A number of other drugs and herbal supplements can also cause acute liver failure (Table 2), the most common being antimicrobial and antiepileptic drugs.15 Of the antimicrobials, antitubercular drugs (especially isoniazid) are believed to be the most common causes, followed by trimethoprim-sulfamethoxazole. Phenytoin is the antiepileptic drug most often implicated in acute liver failure.
Statins can also cause acute liver failure, especially when combined with other hepatotoxic agents.16
The herbal supplements and weight-loss agents Hydroxycut and Herbalife have both been reported to cause acute liver failure, with patients presenting with either the hepatocellular or the cholestatic pattern of liver injury.17 The exact chemical in these supplements that causes liver injury has not yet been determined.
The National Institutes of Health maintains a database of cases of liver failure due to medications and supplements at livertox.nih.gov. The database includes the pattern of hepatic injury, mechanism of injury, management, and outcomes.
Viral hepatitis
Hepatitis B virus is the most common viral cause of acute liver failure and is responsible for about 8% of cases.18
Patients with chronic hepatitis B virus infection—as evidenced by positive hepatitis B surface antigen—can develop acute liver failure if the infection is reactivated by the use of immunosuppressive drugs for solid-organ or bone-marrow transplant or medications such as anti-tumor necrosis agents, rituximab, or chemotherapy. These patients should be treated prophylactically with a nucleoside analogue, which should be continued for 6 months after immunosuppressive therapy is completed.
Hepatitis A virus is responsible for about 4% of cases.18
Hepatitis C virus rarely causes acute liver failure, especially in the absence of hepatitis A and hepatitis B.3,19
Hepatitis E virus, which is endemic in areas of Asia and Africa, can cause liver disease in pregnant women and in young adults who have concomitant liver disease from another cause. It tends to cause acute liver failure more frequently in pregnant women than in the rest of the population and carries a mortality rate of more than 20% in this subgroup.
TT (transfusion-transmitted) virus was reported in the 1990s to cause acute liver failure in about 27% of patients in whom no other cause could be found.20
Other rare viral causes of acute liver failure include Epstein-Barr virus, cytomegalovirus, and herpes simplex virus types 1, 2, and 6.
Other causes
Other causes of acute liver failure include ischemic hepatitis, autoimmune hepatitis, Wilson disease, Budd-Chiari syndrome, and HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome.
MANY PATIENTS NEED LIVER TRANSPLANT
Many patients with acute liver failure ultimately require orthotopic liver transplant,21 especially if they present with severe encephalopathy. Other aspects of treatment vary according to the cause of liver failure (Table 3).
SPECIFIC MANAGEMENT
Management of acetaminophen toxicity
If the time of ingestion is known, checking the acetaminophen level can help determine the cause of acute liver failure and also predict the risk of hepatotoxicity, based on the work of Rumack and Matthew.22 Calculators are available, eg, http://reference.medscape.com/calculator/acetaminophen-toxicity.
If a patient presents with acute liver failure several days after ingesting acetaminophen, the level can be in the nontoxic range, however. In this scenario, measuring acetaminophen-protein adducts can help establish acetaminophen toxicity as the cause, as the adducts last longer in the serum and provide 100% sensitivity and specificity.23 While most laboratories can rapidly measure acetaminophen levels, only a few can measure acetaminophen-protein adducts, and thus this test is not used clinically.
Acetylcysteine is the main drug used for acetaminophen toxicity. Ideally, it should be given within 8 hours of acetaminophen ingestion, but giving it later is also useful.1
Acetylcysteine is available in oral and intravenous forms, the latter for patients who have encephalopathy or cannot tolerate oral intake due to repeated episodes of vomiting.24,25 The oral form is much less costly and is thus preferred over intravenous acetylcysteine in patients who can tolerate oral intake. Intravenous acetylcysteine should be given in a loading dose of 150 mg/kg in 5% dextrose over 15 minutes, followed by a maintenance dose of 50 mg/kg over 4 hours and then 100 mg/kg given over 16 hours.1 No dose adjustment is needed in patients who have renal toxicity (acetaminophen can also be toxic to the kidneys).
Most patients with acetaminophen-induced liver failure survive with medical management alone and do not need a liver transplant.3,26 Cirrhosis does not occur in these patients.
Management of viral acute liver failure
When patients present with acute liver failure, it is necessary to look for a viral cause by serologic testing, including hepatitis A virus IgM antibody, hepatitis B surface antigen, and hepatitis B core IgM antibody.
Hepatitis B can become reactivated in immunocompromised patients, and therefore the hepatitis B virus DNA level should be checked. Detection of hepatitis B virus DNA in a patient previously known to have undetectable hepatitis B virus DNA confirms hepatitis B reactivation.
Patients with hepatitis B-induced acute liver failure should be treated with entecavir or tenofovir. Although this treatment may not change the course of acute liver failure or accelerate the recovery, it can prevent reinfection in the transplanted liver if liver transplant becomes indicated.27–29
Herpes simplex virus should be suspected in patients presenting with anicteric hepatitis with fever. Polymerase chain reaction testing for herpes simplex virus should be done,30 and if positive, patients should be given intravenous acyclovir.31 Despite treatment, herpes simplex virus disease is associated with a very poor prognosis without liver transplant.
Autoimmune hepatitis
The autoantibodies usually seen in autoimmune hepatitis are antinuclear antibody, antismooth muscle antibody, and anti-liver-kidney microsomal antibody, and patients need to be tested for them.
The diagnosis of autoimmune hepatitis can be challenging, as these autoimmune markers can be negative in 5% of patients. Liver biopsy becomes essential to establish the diagnosis in that setting.32
Guidelines advise starting prednisone 40 to 60 mg/day and placing the patient on the liver transplant list.1
Wilson disease
Although it is an uncommon cause of liver failure, Wilson disease needs special attention because it has a poor prognosis. The mortality rate in acute liver failure from Wilson disease reaches 100% without liver transplant.
Wilson disease is caused by a genetic defect that allows copper to accumulate in the liver and other organs. However, diagnosing Wilson disease as the cause of acute liver failure can be challenging because elevated serum and urine copper levels are not specific to Wilson disease and can be seen in patients with acute liver failure from any cause. In addition, the ceruloplasmin level is usually normal or high because it is an acute-phase reactant. Accumulation of copper in the liver parenchyma is usually patchy; therefore, qualitative copper staining on random liver biopsy samples provides low diagnostic yield. Quantitative copper on liver biopsy is the gold standard test to establish the diagnosis, but the test is time-consuming. Kayser-Fleischer rings around the iris are considered pathognomic for Wilson disease when seen with acute liver failure, but they are seen in only about 50% of patients.33
A unique feature of acute Wilson disease is that most patients have very high bilirubin levels and low alkaline phosphatase levels. An alkaline phosphatase-to-bilirubin ratio less than 2 in patients with acute liver failure is highly suggestive of Wilson disease.34
Another clue to the diagnosis is that patients with Wilson disease tend to develop Coombs-negative hemolytic anemia, which leads to a disproportionate elevation in aminotransferase levels, with aspartate aminotransferase being higher than alanine aminotransferase.
Once Wilson disease is suspected, the patient should be listed for liver transplant because death is almost certain without it. For patients awaiting liver transplant, the American Association for the Study of Liver Diseases guidelines recommend certain measures to lower the serum copper level such as albumin dialysis, continuous hemofiltration, plasmapheresis, and plasma exchange,1 but the evidence supporting their use is limited.
NONSPECIFIC MANAGEMENT
Acute liver failure can affect a number of organs and systems in addition to the liver (Figure 2).
General considerations
Because their condition can rapidly deteriorate, patients with acute liver failure are best managed in intensive care.
Patients who present to a center that does not have the facilities for liver transplant should be transferred to a transplant center as soon as possible, preferably by air. If the patient may not be able to protect the airway, endotracheal intubation should be performed before transfer.
The major causes of death in patients with acute liver failure are cerebral edema and infection. Gastrointestinal bleeding was a major cause of death in the past, but with prophylactic use of histamine H2 receptor blockers and proton pump inhibitors, the incidence of gastrointestinal bleeding has been significantly reduced.
Although initially used only in patients with acetaminophen-induced liver failure, acetylcysteine has also shown benefit in patients with acute liver failure from other causes. In patients with grade 1 or 2 encephalopathy on a scale of 0 (minimal) to 4 (comatose), the transplant-free survival rate is higher when acetylcysteine is given compared with placebo, but this benefit does not extend to patients with a higher grade of encephalopathy.35
Cerebral edema and intracranial hypertension
Cerebral edema is the leading cause of death in patients with acute liver failure, and it develops in nearly 40% of patients.36
The mechanism by which cerebral edema develops is not well understood. Some have proposed that ammonia is converted to glutamine, which causes cerebral edema either directly by its osmotic effect37,38 or indirectly by decreasing other osmolytes, thereby promoting water retention.39
Cerebral edema leads to intracranial hypertension, which can ultimately cause cerebral herniation and death. Because of the high mortality rate associated with cerebral edema, invasive devices were extensively used in the past to monitor intracranial pressure. However, in light of known complications of these devices, including bleeding,40 and lack of evidence of long-term benefit in terms of mortality rates, their use has come under debate.
Treatments. Many treatments are available for cerebral edema and intracranial hypertension. The first step is to elevate the head of the bed about 30 degrees. In addition, hyponatremia should be corrected, as it can worsen cerebral edema.41 If patients are intubated, maintaining a hypercapneic state is advisable to decrease the intracranial pressure.
Of the two pharmacologic options, mannitol is more often used.42 It is given as a bolus dose of 0.5 to 1 g/kg intravenously if the serum osmolality is less than 320 mOsm/L.1 Given the risk of fluid overload with mannitol, caution must be exercised in patients with renal dysfunction. The other pharmacologic option is 3% hypertonic saline.
Therapeutic hypothermia is a newer treatment for cerebral edema. Lowering the body temperature to 32 to 33°C (89.6 to 91.4°F) using cooling blankets decreases intracranial pressure and cerebral blood flow and improves the cerebral perfusion pressure.43 With this treatment, patients should be closely monitored for side effects of infection, coagulopathy, and cardiac arrythmias.1
l-ornithine l-aspartate was successfully used to prevent brain edema in rats, but in humans, no benefit was seen compared with placebo.44,45 The underlying basis for this experimental treatment is that supplemental ornithine and aspartate should increase glutamate synthesis, which should increase the activity of enzyme glutamine synthetase in skeletal muscles. With the increase in enzyme activity, conversion of ammonia to glutamine should increase, thereby decreasing ammonia circulation and thus decreasing cerebral edema.
Patients with cerebral edema have a high incidence of seizures, but prophylactic antiseizure medications such as phenytoin have not been proven to be beneficial.46
Infection
Nearly 80% of patients with acute liver failure develop an infectious complication, which can be attributed to a state of immunodeficiency.47
The respiratory and urinary tracts are the most common sources of infection.48 In patients with bacteremia, Enterococcus species and coagulase-negative Staphylococcus species49 are the commonly isolated organisms. Also, in patients with acute liver failure, fungal infections account for 30% of all infections.50
Infected patients often develop worsening of their encephalopathy51 without fever or elevated white blood cell count.49,52 Thus, in any patient in whom encephalopathy is worsening, an evaluation must be done to rule out infection. In these patients, systemic inflammatory response syndrome is an independent risk factor for death.53
Despite the high mortality rate with infection, whether using antibiotics prophylactically in acute liver failure is beneficial is controversial.54,55
Gastrointestinal bleeding
The current prevalence of upper gastrointestinal bleeding in acute liver failure patients is about 1.5%.56 Coagulopathy and endotracheal intubation are the main risk factors for upper gastrointestinal bleeding in these patients.57 The most common source of bleeding is stress ulcers in the stomach. The ulcers develop from a combination of factors, including decreased blood flow to the mucosa causing ischemia and hypoperfusion-reperfusion injury.
Pharmacologic inhibition of gastric acid secretion has been shown to reduce upper gastrointestinal bleeding in acute liver failure. A histamine H2 receptor blocker or proton pump inhibitor should be given to prevent gastrointestinal bleeding in patients with acute liver failure.1,58
EXPERIMENTAL TREATMENTS
Artificial liver support systems
Membranes and dialysate solutions have been developed to remove toxic substances that are normally metabolized by the liver. Two of these—the molecular adsorbent recycling system (MARS) and the extracorporeal liver assist device (ELAD)—were developed in the late 1990s. MARS consisted of a highly permeable hollow fiber membrane mixed with albumin, and ELAD consisted of porcine hepatocytes attached to microcarriers in the extracapillary space of the hollow fiber membrane. Both systems allowed for transfer of water-soluble and protein-bound toxins in the blood across the membrane and into the dialysate.59 The clinical benefit offered by these devices is controversial,60–62 thus limiting their use to experimental purposes only.
Hepatocyte transplant
Use of hepatocyte transplant as a bridge to liver transplant was tested in 1970s, first in rats and later in humans.63 By reducing the blood ammonia level and improving cerebral perfusion pressure and cardiac function, replacement of 1% to 2% of the total liver cell mass by transplanted hepatocytes acts as a bridge to orthotopic liver transplant.64,65
PROGNOSIS
Different criteria have been used to identify patients with poor prognosis who may eventually need to undergo liver transplant.
The King’s College criteria system is the most commonly used for prognosis (Table 4).37,66–69 Its main drawback is that it is applicable only in patients with encephalopathy, and when patients reach this stage, their condition often deteriorates rapidly, and they die while awaiting liver transplant.37,66,67
The Model for End-Stage Liver Disease (MELD) score is an alternative to the King’s College criteria. A high MELD score on admission signifies advanced disease, and patients with a high MELD score tend to have a worse prognosis than those with a low score.68
The Acute Physiology and Chronic Health Evaluation (APACHE) II score can also be used, as it is more sensitive than the King’s College criteria.6
The Clichy criteria66,69 can also be used.
Liver biopsy. In addition to helping establish the cause of acute liver failure, liver biopsy can also be used as a prognostic tool. Hepatocellular necrosis greater than 70% on the biopsy predicts death with a specificity of 90% and a sensitivity of 56%.70
Hypophosphatemia has been reported to indicate recovering liver function in patients with acute liver failure.71 As the liver regenerates, its energy requirement increases. To supply the energy, adenosine triphosphate production increases, and phosphorus shifts from the extracellular to the intracellular compartment to meet the need for extra phosphorus during this process. A serum phosphorus level of 2.9 mg/dL or higher appears to indicate a poor prognosis in patients with acute liver failure, as it signifies that adequate hepatocyte regeneration is not occurring.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 342:273–275.
- Ostapowicz G, Fontana RJ, Schiodt FV, et al; US Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 2008; 47:1401–1415.
- Acharya SK, Panda SK, Saxena A, Gupta SD. Acute hepatic failure in India: a perspective from the East. J Gastroenterol Hepatol 2000; 15:473–479.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Patten CJ, Thomas PE, Guy RL, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 1993; 6:511–518.
- Chen W, Koenigs LL, Thompson SJ, et al. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol 1998; 11:295-301.
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187:211–217.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 1983; 71:980–991.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- Schiødt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med 1997; 337:1112–1117.
- Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994; 272:1845–1850.
- Chalasani N, Fontana RJ, Bonkovsky HL, et al; Drug Induced Liver Injury Network (DILIN). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008; 135:1924–1934 e1–4
- Reuben A, Koch DG, Lee WM; Acute Liver Failure Study Group. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology 2010; 52:2065–2076.
- Stevens T, Qadri A, Zein NN. Two patients with acute liver injury associated with use of the herbal weight-loss supplement hydroxycut. Ann Intern Med 2005; 142:477–478.
- Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol 2015; 62(suppl 1):S112–S120.
- Schiodt FV, Davern TJ, Shakil AO, McGuire B, Samuel G, Lee WM. Viral hepatitis-related acute liver failure. Am J Gastroenterol 2003; 98:448–453.
- Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology 1998; 28:839–842.
- Bismuth H, Samuel D, Gugenheim J, et al. Emergency liver transplantation for fulminant hepatitis. Ann Intern Med 1987; 107:337–341.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Davern TJ 2nd, James LP, Hinson JA, et al; Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006; 130:687–694.
- Perry HE, Shannon MW. Efficacy of oral versus intravenous N-acetylcysteine in acetaminophen overdose: results of an open-label, clinical trial. J Pediatr 1998; 132:149–152.
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557–1562.
- Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology 1995; 109:1907–1916.
- Tsang SW, Chan HL, Leung NW, et al. Lamivudine treatment for fulminant hepatic failure due to acute exacerbation of chronic hepatitis B infection. Aliment Pharmacol Ther 2001; 15:1737–1744.
- Yu JW, Sun LJ, Yan BZ, Kang P, Zhao YH. Lamivudine treatment is associated with improved survival in fulminant hepatitis B. Liver Int 2011; 31:499–506.
- Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 2011; 53:774–780.
- Pinna AD, Rakela J, Demetris AJ, Fung JJ. Five cases of fulminant hepatitis due to herpes simplex virus in adults. Dig Dis Sci 2002; 47:750–754.
- Farr RW, Short S, Weissman D. Fulminant hepatitis during herpes simplex virus infection in apparently immunocompetent adults: report of two cases and review of the literature. Clin Infect Dis 1997; 24:1191–1194.
- Czaja AJ, Freese DK; American Association for the Study of Liver Disease. Diagnosis and treatment of autoimmune hepatitis. Hepatology 2002; 36:479–497.
- Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003; 37:1475–1492.
- Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991; 100:1129–1134.
- Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009; 137:856–864.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology 1992; 15:449–453.
- Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 1994; 107:1475–1480.
- Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet 1993; 341:157–158.
- Cordoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol 1998; 29:589–594.
- Canalese J, Gimson AE, Davis C, Mellon PJ, Davis M, Williams R. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut 1982; 23:625–629.
- Jalan R, SW OD, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 1999; 354:1164–1168.
- Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 1999; 30:636–640.
- Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology 2009; 136:2159–2168.
- Bhatia V, Batra Y, Acharya SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol 2004; 41:89–96.
- Canalese J, Gove CD, Gimson AE, Wilkinson SP, Wardle EN, Williams R. Reticuloendothelial system and hepatocytic function in fulminant hepatic failure. Gut 1982; 23:265–269.
- Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology 1990; 11:49–53.
- Rolando N, Wade JJ, Stangou A, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg 1996; 2:8–13.
- Rolando N, Harvey F, Brahm J, et al. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol 1991; 12:1–9.
- Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology 2003; 125:755–764.
- Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis 1996; 16:389–402.
- Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32:734–739.
- Rolando N, Gimson A, Wade J, Philpott- Howard J, Casewell M, Williams R. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology 1993; 17:196–201.
- Karvellas CJ, Cavazos J, Battenhouse H, et al; US Acute Liver Failure Study Group. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol 2014; 12:1942–1949.
- Acharya SK, Dasarathy S, Kumer TL, et al. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology 1996; 23:1148–1155.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- MacDougall BR, Williams R. H2-receptor antagonist in the prevention of acute gastrointestinal hemorrhage in fulminant hepatic failure: a controlled trial. Gastroenterology 1978; 74:464–465.
- Stange J, Mitzner SR, Risler T, et al. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs 1999; 23:319–330.
- Vaid A, Chewich H, Balk EM, Jaber BL. Molecular adsorbent recirculating system as artificial support therapy for liver failure: a meta-analysis. ASAIO J 2012; 58:51–59.
- Khuroo MS, Khuroo MS, Farahat KL. Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: a meta-analysis. Liver Transpl 2004; 10:1099–1106.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Sommer BG, Sutherland DE, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation for treatment of D-galactosamine-induced acute liver failure in rats. Transplant Proc 1979; 11:578–584.
- Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology 1988; 8:1006–1009.
- Strom SC, Fisher RA, Thompson MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 1997; 63:559–569.
- Pauwels A, Mostefa-Kara N, Florent C, Levy VG. Emergency liver transplantation for acute liver failure. Evaluation of London and Clichy criteria. J Hepatol 1993; 17:124–127.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King's criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Larsen FS. MELD score as a predictor of liver failure and death in patients with acetaminophen-induced liver injury. Hepatology 2007; 45:789–796.
- Bernuau J, Goudeau A, Poynard T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology 1986; 6:648–651.
- Donaldson BW, Gopinath R, Wanless IR, et al. The role of transjugular liver biopsy in fulminant liver failure: relation to other prognostic indicators. Hepatology 1993; 18:1370–1376.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 342:273–275.
- Ostapowicz G, Fontana RJ, Schiodt FV, et al; US Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 2008; 47:1401–1415.
- Acharya SK, Panda SK, Saxena A, Gupta SD. Acute hepatic failure in India: a perspective from the East. J Gastroenterol Hepatol 2000; 15:473–479.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Patten CJ, Thomas PE, Guy RL, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 1993; 6:511–518.
- Chen W, Koenigs LL, Thompson SJ, et al. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol 1998; 11:295-301.
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187:211–217.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 1983; 71:980–991.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- Schiødt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med 1997; 337:1112–1117.
- Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994; 272:1845–1850.
- Chalasani N, Fontana RJ, Bonkovsky HL, et al; Drug Induced Liver Injury Network (DILIN). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008; 135:1924–1934 e1–4
- Reuben A, Koch DG, Lee WM; Acute Liver Failure Study Group. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology 2010; 52:2065–2076.
- Stevens T, Qadri A, Zein NN. Two patients with acute liver injury associated with use of the herbal weight-loss supplement hydroxycut. Ann Intern Med 2005; 142:477–478.
- Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol 2015; 62(suppl 1):S112–S120.
- Schiodt FV, Davern TJ, Shakil AO, McGuire B, Samuel G, Lee WM. Viral hepatitis-related acute liver failure. Am J Gastroenterol 2003; 98:448–453.
- Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology 1998; 28:839–842.
- Bismuth H, Samuel D, Gugenheim J, et al. Emergency liver transplantation for fulminant hepatitis. Ann Intern Med 1987; 107:337–341.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Davern TJ 2nd, James LP, Hinson JA, et al; Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006; 130:687–694.
- Perry HE, Shannon MW. Efficacy of oral versus intravenous N-acetylcysteine in acetaminophen overdose: results of an open-label, clinical trial. J Pediatr 1998; 132:149–152.
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557–1562.
- Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology 1995; 109:1907–1916.
- Tsang SW, Chan HL, Leung NW, et al. Lamivudine treatment for fulminant hepatic failure due to acute exacerbation of chronic hepatitis B infection. Aliment Pharmacol Ther 2001; 15:1737–1744.
- Yu JW, Sun LJ, Yan BZ, Kang P, Zhao YH. Lamivudine treatment is associated with improved survival in fulminant hepatitis B. Liver Int 2011; 31:499–506.
- Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 2011; 53:774–780.
- Pinna AD, Rakela J, Demetris AJ, Fung JJ. Five cases of fulminant hepatitis due to herpes simplex virus in adults. Dig Dis Sci 2002; 47:750–754.
- Farr RW, Short S, Weissman D. Fulminant hepatitis during herpes simplex virus infection in apparently immunocompetent adults: report of two cases and review of the literature. Clin Infect Dis 1997; 24:1191–1194.
- Czaja AJ, Freese DK; American Association for the Study of Liver Disease. Diagnosis and treatment of autoimmune hepatitis. Hepatology 2002; 36:479–497.
- Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003; 37:1475–1492.
- Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991; 100:1129–1134.
- Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009; 137:856–864.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology 1992; 15:449–453.
- Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 1994; 107:1475–1480.
- Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet 1993; 341:157–158.
- Cordoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol 1998; 29:589–594.
- Canalese J, Gimson AE, Davis C, Mellon PJ, Davis M, Williams R. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut 1982; 23:625–629.
- Jalan R, SW OD, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 1999; 354:1164–1168.
- Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 1999; 30:636–640.
- Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology 2009; 136:2159–2168.
- Bhatia V, Batra Y, Acharya SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol 2004; 41:89–96.
- Canalese J, Gove CD, Gimson AE, Wilkinson SP, Wardle EN, Williams R. Reticuloendothelial system and hepatocytic function in fulminant hepatic failure. Gut 1982; 23:265–269.
- Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology 1990; 11:49–53.
- Rolando N, Wade JJ, Stangou A, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg 1996; 2:8–13.
- Rolando N, Harvey F, Brahm J, et al. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol 1991; 12:1–9.
- Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology 2003; 125:755–764.
- Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis 1996; 16:389–402.
- Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32:734–739.
- Rolando N, Gimson A, Wade J, Philpott- Howard J, Casewell M, Williams R. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology 1993; 17:196–201.
- Karvellas CJ, Cavazos J, Battenhouse H, et al; US Acute Liver Failure Study Group. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol 2014; 12:1942–1949.
- Acharya SK, Dasarathy S, Kumer TL, et al. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology 1996; 23:1148–1155.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- MacDougall BR, Williams R. H2-receptor antagonist in the prevention of acute gastrointestinal hemorrhage in fulminant hepatic failure: a controlled trial. Gastroenterology 1978; 74:464–465.
- Stange J, Mitzner SR, Risler T, et al. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs 1999; 23:319–330.
- Vaid A, Chewich H, Balk EM, Jaber BL. Molecular adsorbent recirculating system as artificial support therapy for liver failure: a meta-analysis. ASAIO J 2012; 58:51–59.
- Khuroo MS, Khuroo MS, Farahat KL. Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: a meta-analysis. Liver Transpl 2004; 10:1099–1106.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Sommer BG, Sutherland DE, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation for treatment of D-galactosamine-induced acute liver failure in rats. Transplant Proc 1979; 11:578–584.
- Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology 1988; 8:1006–1009.
- Strom SC, Fisher RA, Thompson MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 1997; 63:559–569.
- Pauwels A, Mostefa-Kara N, Florent C, Levy VG. Emergency liver transplantation for acute liver failure. Evaluation of London and Clichy criteria. J Hepatol 1993; 17:124–127.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King's criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Larsen FS. MELD score as a predictor of liver failure and death in patients with acetaminophen-induced liver injury. Hepatology 2007; 45:789–796.
- Bernuau J, Goudeau A, Poynard T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology 1986; 6:648–651.
- Donaldson BW, Gopinath R, Wanless IR, et al. The role of transjugular liver biopsy in fulminant liver failure: relation to other prognostic indicators. Hepatology 1993; 18:1370–1376.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
KEY POINTS
- In the United States, the most common cause of acute liver failure is acetaminophen toxicity, followed by viral hepatitis.
- Testing for the cause of acute liver failure needs to start as soon as possible so that specific treatment can be initiated and the patient can be placed on the transplant list if needed.
- Acetylcysteine and either a proton pump inhibitor or a histamine H2 receptor blocker should be given to all patients with acute liver failure. Liver transplant is the cornerstone of therapy in patients not responding to other treatments.
- There are a number of prognostic scores for acute liver failure, but each has limitations.