User login
Permanent Alopecia in Breast Cancer Patients: Role of Taxanes and Endocrine Therapies
Anagen effluvium during chemotherapy is common, typically beginning within 1 month of treatment onset and resolving by 6 months after the final course.1 Permanent chemotherapy-induced alopecia (PCIA), in which hair loss persists beyond 6 months after chemotherapy without recovery to original density, was first reported in patients following high-dose chemotherapy regimens for allogeneic bone marrow transplantation.2 There are now increasing reports of PCIA in patients with breast cancer; at least 400 such cases have been documented.3-16 In addition to chemotherapy, patients often receive adjuvant endocrine therapy with selective estrogen receptor modulators, aromatase inhibitors, or gonadotropin-releasing hormone agonists.5-16 Endocrine therapies also can lead to alopecia, but their role in PCIA has not been well defined.15,16 We describe 3 patients with breast cancer who experienced PCIA following chemotherapy with taxanes with or without endocrine therapies. We also review the literature on non–bone marrow transplantation PCIA to better characterize this entity and explore the role of endocrine therapies in PCIA.
Case Reports
Patient 1
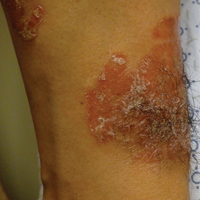
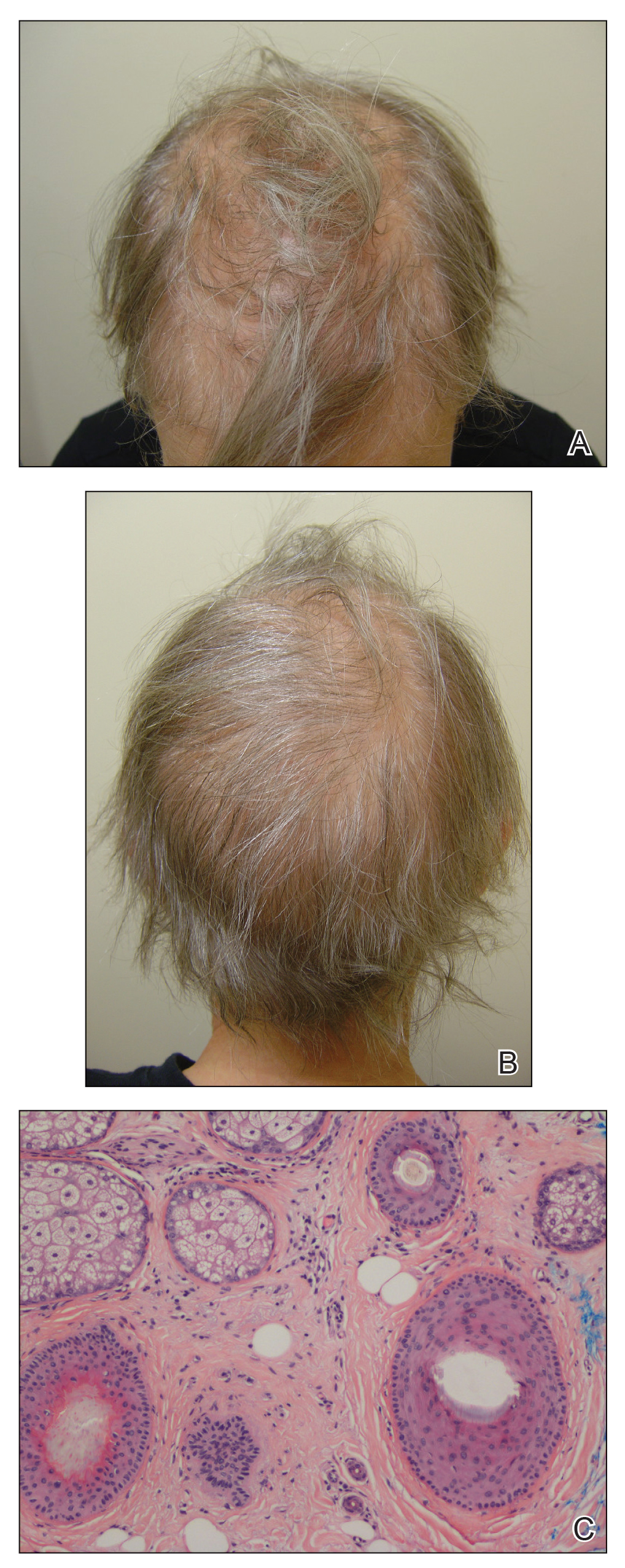
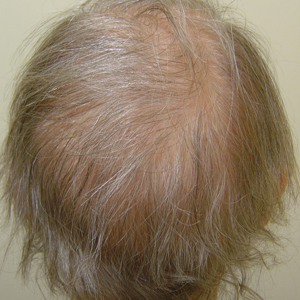
A 62-year-old woman with a history of stage II invasive ductal carcinoma presented with persistent hair loss 5 years after completing chemotherapy. She underwent 6 cycles of docetaxel and carboplatin along with radiation therapy as well as 1 year of trastuzumab and did not receive endocrine therapy. At the current presentation, she reported patchy hair regrowth that gradually filled in but failed to return to full density. Physical examination revealed the hair was diffusely thin, especially bitemporally (Figures 1A and 1B), and she did not experience any loss of body hair. She had no family history of hair loss. Her medical history was notable for hypertension, chronic obstructive bronchitis, osteopenia, and depression. Her thyroid stimulating hormone (TSH) level was within reference range. Medications included lisinopril, metoprolol, escitalopram, and trazodone. A biopsy from the occipital scalp showed nonscarring alopecia with variation of hair follicle size, a decreased number of hair follicles, and a decreased anagen to telogen ratio (Figure 1C). She was treated with clobetasol solution and minoxidil solution 5% for 1 year with mild improvement. She experienced no further hair loss but did not regain original hair density.
Patient 2
A 35-year-old woman with a history of stage II invasive ductal carcinoma presented with persistent hair loss 10 months after chemotherapy. She underwent 4 cycles of doxorubicin and cyclophosphamide followed by 4 cycles of paclitaxel and was started on trastuzumab. Tamoxifen was initiated 1 month after completing chemotherapy. She received radiation therapy the following month and continued trastuzumab for 1 year. At the current presentation, the patient noted that hair regrowth had started 1 month after the last course of chemotherapy but had progressed slowly. She denied body hair loss. Physical examination revealed diffuse thinning, especially over the crown, with scattered broken hairs throughout the scalp and several miniaturized hairs over the crown. She was evaluated as grade 3 on the Sinclair clinical grading scale used to evaluate female pattern hair loss (FPHL).17 Her family history was remarkable for FPHL in her maternal grandmother. She had no notable medical history, her TSH was normal, and she was taking tamoxifen and trastuzumab. Biopsy was not performed. The patient was started on minoxidil solution 2% and had mild improvement with no further broken-off hairs after 10 months. At that point, she was evaluated as grade 2 to 3 on the Sinclair scale.17
Patient 3
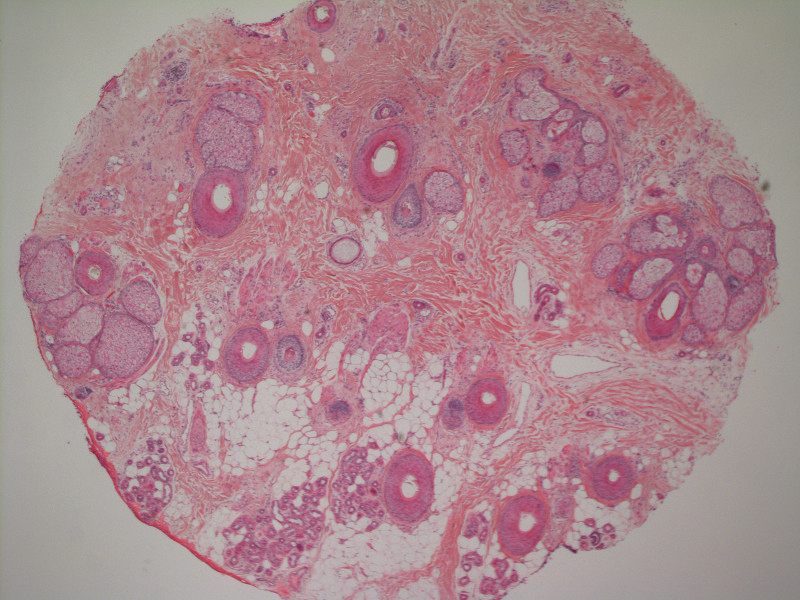
A 51-year-old woman with a history of papillary carcinoma and extensive ductal carcinoma in situ presented with persistent hair loss for 3.5 years following chemotherapy for recurrent breast cancer. After her initial diagnosis in the left breast, she received cyclophosphamide, methotrexate, and 5-fluorouracil but did not receive endocrine therapy. Her hair thinned during chemotherapy but returned to normal density within 1 year. She had a recurrence of the cancer in the right breast 14 years later and received 6 cycles of chemotherapy with cyclophosphamide and docetaxel followed by radiation therapy. After this course, her hair loss incompletely recovered. One year after chemotherapy, she underwent bilateral salpingo-oophorectomy and started anastrozole. Three months later, she noticed increased shedding and progressive thinning of the hair. Physical examination revealed diffuse thinning that was most pronounced over the crown. She also experienced lateral thinning of the eyebrows, decreased eyelashes, and dystrophic fingernails. Fluocinonide solution was discontinued by the patient due to scalp burning. She had a brother with bitemporal recession. Her medical history was notable for Hashimoto thyroiditis, vitamin D deficiency, and peripheral neuropathy. Her TSH occasionally was elevated, and she was intermittently on levothyroxine; however, her free T4 was maintained within reference range on all records. Her medications at the time of evaluation were anastrozole and gabapentin. Biopsies taken from the right and left temporal scalp revealed decreased follicle density with a majority of follicles in anagen, scattered miniaturized follicles, and a mild perivascular and perifollicular lymphoid infiltrate. Mild dermal fibrosis was present without evidence of frank scarring (Figure 2). She declined treatment, and there was no change in her condition over 3 years of follow-up.
Comment
Classification of Chemotherapy-Induced Hair Loss
Chemotherapy-induced alopecia is typically an anagen effluvium that is reversed within 6 months following the final course of chemotherapy. When incomplete regrowth persists, the patient is considered to have PCIA.1 The pathophysiology of PCIA is unclear.
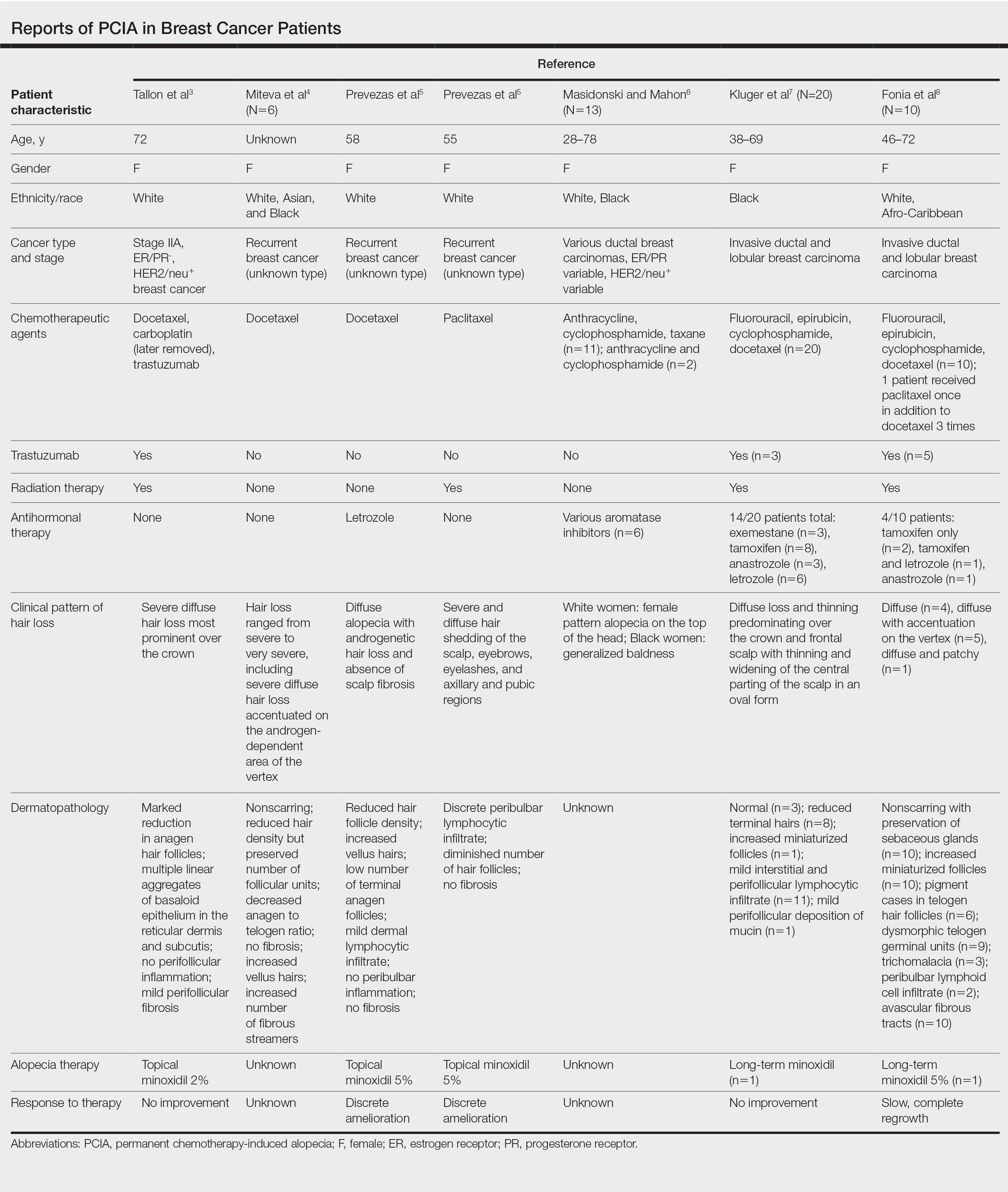
Traditional grading for chemotherapy-induced alopecia does not account for the patterns of loss seen in PCIA, of which the most common appears to be a female pattern with accentuated hair loss in androgen-dependent regions of the scalp.18 Other patterns include a diffuse type with body hair loss, patchy alopecia, and complete alopecia with or without body hair loss (Table).3-8 Whether these patterns all can be attributed to chemotherapy remains to be explored.
Breast Cancer Therapies Causing PCIA
The main agents thought to be responsible for PCIA in breast cancer patients are taxanes. The role of endocrine therapies has not been well explored. Trastuzumab lacks several of the common side effects of chemotherapy due to its specificity for the HER2/neu receptor and has not been found to increase the rate of hair loss when combined with standard chemotherapy.19,20 Although radiation therapy has the potential to damage hair follicles, and a dose-dependent relationship has been described for temporary and permanent alopecia at irradiated sites, permanent alopecia predominantly has been reported with cranial radiation used in the treatment of intracranial malignancies.21 The role of radiation therapy of the breasts in PCIA is unclear, as its inclusion in therapy has not been consistently reported in the literature.
Docetaxel is known to cause chemotherapy-induced alopecia, with an 83.4% incidence in phase 2 trials; however, it also appears to be related to PCIA.20 A PubMed search of articles indexed for MEDLINE was performed using the terms permanent chemotherapy induced alopecia, chemotherapy, docetaxel, endocrine therapies, hair loss, alopecia, and breast cancer. More than 400 cases of PCIA related to chemotherapy in breast cancer patients have been reported in the literature from a combination of case reports/series, retrospective surveys, and at least one prospective study. Data from some of the more detailed reports (n=52) are summarized in the Table. In the single-center, 3-year prospective study of women given adjuvant taxane-based or non–taxane-based chemotherapy, those who received taxane therapy were more likely to develop PCIA (odds ratio, 8.01).9
All 3 of our patients received taxanes. Interestingly, patient 3 underwent 2 rounds of chemotherapy 14 years apart and experienced full regrowth of the hair after the first course of taxane-free chemotherapy but experienced persistent hair loss following docetaxel treatment. Adjuvant endocrine therapies also may contribute to PCIA. A review of the side effects of endocrine therapies revealed an incidence of alopecia that was higher than expected; tamoxifen was the greatest offender. Additionally, using endocrine treatments in combination was found to have a synergistic effect on alopecia.18 Adjuvant endocrine therapy was used in patients 2 and 3. Although endocrine therapies appear to have a milder effect on hair loss compared to chemotherapy, these medications are continued for a longer duration, potentially contributing to the severity of hair loss and prolonging the time to regrowth.
Furthermore, endocrine therapies used in breast cancer treatment decrease estrogen levels or antagonize estrogen receptors, creating an environment of relative hyperandrogenism that may contribute to FPHL in genetically susceptible women.18 Although taxanes may cause irreversible hair loss in these patients, the action of endocrine therapies on the remaining hair follicles may affect the typical female pattern seen clinically. Patients 2 and 3 who presented with FPHL received adjuvant endocrine therapies and had positive family history, while patient 1 did not. Of note, patient 3 experienced worsening hair loss following the addition of anastrozole, which suggests a contribution of endocrine therapy to her PCIA. Our limited cases do not allow for evaluation of a worsened outcome with the combination of taxanes and endocrine therapies; however, we suggest further evaluation for a synergistic effect that may be contributing to PCIA.
Conclusion
Permanent alopecia in breast cancer patients appears to be a true potential adverse effect of taxanes and endocrine therapies, and it is important to characterize it appropriately so that its mechanism can be understood and appropriate treatment and counseling can take place. Although it may not influence clinical decision-making, patients should be informed that hair loss with chemotherapy can be permanent. Treatment with scalp cooling can reduce the risk for severe chemotherapy-induced alopecia, but it is unclear if it reduces risk for PCIA.12,15 Topical or oral minoxidil may be helpful in the treatment of PCIA once it has developed.7,8,15,22 Better characterization of these cases may elucidate risk factors for developing permanent alopecia, allowing for more appropriate risk stratification, counseling, and treatment.
- Dorr VJ. A practitioner’s guide to cancer-related alopecia. Semin Oncol. 1998;25:562-570.
- Machado M, Moreb JS, Khan SA. Six cases of permanent alopecia after various conditioning regimens commonly used in hematopoietic stem cell transplantation. Bone Marrow Transplant. 2007;40:979-982.
- Tallon B, Blanchard E, Goldberg LJ. Permanent chemotherapy-induced alopecia: case report and review of the literature. J Am Acad Dermatol. 2010;63:333-336.
- Miteva M, Misciali C, Fanti PA, et al. Permanent alopecia after systemic chemotherapy: a clinicopathological study of 10 cases. Am J Dermatopathol. 2011;33:345-350.
- Prevezas C, Matard B, Pinquier L, et al. Irreversible and severe alopecia following docetaxel or paclitaxel cytotoxic therapy for breast cancer. Br J Dermatol. 2009;160:883-885.
- Masidonski P, Mahon SM. Permanent alopecia in women being treated for breast cancer. Clin J Oncol Nurs. 2009;13:13-14.
- Kluger N, Jacot W, Frouin E, et al. Permanent scalp alopecia related to breast cancer chemotherapy by sequential fluorouracil/epirubicin/cyclophosphamide (FEC) and docetaxel: a prospective study of 20 patients. Ann Oncol. 2012;23:2879-2884.
- Fonia A, Cota C, Setterfield JF, et al. Permanent alopecia in patients with breast cancer after taxane chemotherapy and adjuvant hormonal therapy: clinicopathologic findings in a cohort of 10 patients. J Am Acad Dermatol. 2017;76:948-957.
- Kang D, Kim IR, Choi EK, et al. Permanent chemotherapy-induced alopecia in patients with breast cancer: a 3-year prospective cohort study [published online August 17, 2018]. Oncologist. 2019;24:414-420.
- Chan J, Adderley H, Alameddine M, et al. Permanent hair loss associated with taxane chemotherapy use in breast cancer: a retrospective survey at two tertiary UK cancer centres [published online December 22, 2020]. Eur J Cancer Care (Engl). doi:10.1111/ecc.13395
- Bourgeois H, Denis F, Kerbrat P, et al. Long term persistent alopecia and suboptimal hair regrowth after adjuvant chemotherapy for breast cancer: alert for an emerging side effect: ALOPERS Observatory. Cancer Res. 2009;69(24 suppl). doi:10.1158/0008-5472.SABCS-09-3174
- Bertrand M, Mailliez A, Vercambre S, et al. Permanent chemotherapy induced alopecia in early breast cancer patients after (neo)adjuvant chemotherapy: long term follow up. Cancer Res. 2013;73(24 suppl). doi:10.1158/0008-5472.SABCS13-P3-09-15
- Kim S, Park HS, Kim JY, et al. Irreversible chemotherapy-induced alopecia in breast cancer patient. Cancer Res. 2016;76(4 suppl). doi:10.1158/1538-7445.SABCS15-P1-15-04
- Thorp NJ, Swift F, Arundell D, et al. Long term hair loss in patients with early breast cancer receiving docetaxel chemotherapy. Cancer Res. 2015;75(9 suppl). doi:10.1158/1538-7445.SABCS14-P5-17-04
- Freites-Martinez A, Shapiro J, van den Hurk C, et al. Hair disorders in cancer survivors. J Am Acad Dermatol. 2019;80:1199-1213.
- Freites-Martinez A, Chan D, Sibaud V, et al. Assessment of quality of life and treatment outcomes of patients with persistent postchemotherapy alopecia. JAMA Dermatol. 2019;155:724-728.
- Sinclair R, Jolley D, Mallari R, et al. The reliability of horizontally sectioned scalp biopsies in the diagnosis of chronic diffuse telogen hair loss in women. J Am Acad Dermatol. 2004;51:189-199.
- Saggar V, Wu S, Dickler MN, et al. Alopecia with endocrine therapies in patients with cancer. Oncologist. 2013;18:1126-1134.
- Yeager CE, Olsen EA. Treatment of chemotherapy-induced alopecia. Dermatol Ther. 2011;24:432-442.
- Baselga J. Clinical trials of single-agent trastuzumab (Herceptin). Semin Oncol. 2000;27(5 suppl 9):20-26.
- Lawenda BD, Gagne HM, Gierga DP, et al. Permanent alopecia after cranial irradiation: dose-response relationship. Int J Radiat Oncol Biol Phys. 2004;60:879-887.
- Yang X, Thai KE. Treatment of permanent chemotherapy-induced alopecia with low dose oral minoxidil [published online May 13, 2015]. Australas J Dermatol. 2016;57:E130-E132.
Anagen effluvium during chemotherapy is common, typically beginning within 1 month of treatment onset and resolving by 6 months after the final course.1 Permanent chemotherapy-induced alopecia (PCIA), in which hair loss persists beyond 6 months after chemotherapy without recovery to original density, was first reported in patients following high-dose chemotherapy regimens for allogeneic bone marrow transplantation.2 There are now increasing reports of PCIA in patients with breast cancer; at least 400 such cases have been documented.3-16 In addition to chemotherapy, patients often receive adjuvant endocrine therapy with selective estrogen receptor modulators, aromatase inhibitors, or gonadotropin-releasing hormone agonists.5-16 Endocrine therapies also can lead to alopecia, but their role in PCIA has not been well defined.15,16 We describe 3 patients with breast cancer who experienced PCIA following chemotherapy with taxanes with or without endocrine therapies. We also review the literature on non–bone marrow transplantation PCIA to better characterize this entity and explore the role of endocrine therapies in PCIA.
Case Reports
Patient 1
A 62-year-old woman with a history of stage II invasive ductal carcinoma presented with persistent hair loss 5 years after completing chemotherapy. She underwent 6 cycles of docetaxel and carboplatin along with radiation therapy as well as 1 year of trastuzumab and did not receive endocrine therapy. At the current presentation, she reported patchy hair regrowth that gradually filled in but failed to return to full density. Physical examination revealed the hair was diffusely thin, especially bitemporally (Figures 1A and 1B), and she did not experience any loss of body hair. She had no family history of hair loss. Her medical history was notable for hypertension, chronic obstructive bronchitis, osteopenia, and depression. Her thyroid stimulating hormone (TSH) level was within reference range. Medications included lisinopril, metoprolol, escitalopram, and trazodone. A biopsy from the occipital scalp showed nonscarring alopecia with variation of hair follicle size, a decreased number of hair follicles, and a decreased anagen to telogen ratio (Figure 1C). She was treated with clobetasol solution and minoxidil solution 5% for 1 year with mild improvement. She experienced no further hair loss but did not regain original hair density.
Patient 2
A 35-year-old woman with a history of stage II invasive ductal carcinoma presented with persistent hair loss 10 months after chemotherapy. She underwent 4 cycles of doxorubicin and cyclophosphamide followed by 4 cycles of paclitaxel and was started on trastuzumab. Tamoxifen was initiated 1 month after completing chemotherapy. She received radiation therapy the following month and continued trastuzumab for 1 year. At the current presentation, the patient noted that hair regrowth had started 1 month after the last course of chemotherapy but had progressed slowly. She denied body hair loss. Physical examination revealed diffuse thinning, especially over the crown, with scattered broken hairs throughout the scalp and several miniaturized hairs over the crown. She was evaluated as grade 3 on the Sinclair clinical grading scale used to evaluate female pattern hair loss (FPHL).17 Her family history was remarkable for FPHL in her maternal grandmother. She had no notable medical history, her TSH was normal, and she was taking tamoxifen and trastuzumab. Biopsy was not performed. The patient was started on minoxidil solution 2% and had mild improvement with no further broken-off hairs after 10 months. At that point, she was evaluated as grade 2 to 3 on the Sinclair scale.17
Patient 3
A 51-year-old woman with a history of papillary carcinoma and extensive ductal carcinoma in situ presented with persistent hair loss for 3.5 years following chemotherapy for recurrent breast cancer. After her initial diagnosis in the left breast, she received cyclophosphamide, methotrexate, and 5-fluorouracil but did not receive endocrine therapy. Her hair thinned during chemotherapy but returned to normal density within 1 year. She had a recurrence of the cancer in the right breast 14 years later and received 6 cycles of chemotherapy with cyclophosphamide and docetaxel followed by radiation therapy. After this course, her hair loss incompletely recovered. One year after chemotherapy, she underwent bilateral salpingo-oophorectomy and started anastrozole. Three months later, she noticed increased shedding and progressive thinning of the hair. Physical examination revealed diffuse thinning that was most pronounced over the crown. She also experienced lateral thinning of the eyebrows, decreased eyelashes, and dystrophic fingernails. Fluocinonide solution was discontinued by the patient due to scalp burning. She had a brother with bitemporal recession. Her medical history was notable for Hashimoto thyroiditis, vitamin D deficiency, and peripheral neuropathy. Her TSH occasionally was elevated, and she was intermittently on levothyroxine; however, her free T4 was maintained within reference range on all records. Her medications at the time of evaluation were anastrozole and gabapentin. Biopsies taken from the right and left temporal scalp revealed decreased follicle density with a majority of follicles in anagen, scattered miniaturized follicles, and a mild perivascular and perifollicular lymphoid infiltrate. Mild dermal fibrosis was present without evidence of frank scarring (Figure 2). She declined treatment, and there was no change in her condition over 3 years of follow-up.
Comment
Classification of Chemotherapy-Induced Hair Loss
Chemotherapy-induced alopecia is typically an anagen effluvium that is reversed within 6 months following the final course of chemotherapy. When incomplete regrowth persists, the patient is considered to have PCIA.1 The pathophysiology of PCIA is unclear.
Traditional grading for chemotherapy-induced alopecia does not account for the patterns of loss seen in PCIA, of which the most common appears to be a female pattern with accentuated hair loss in androgen-dependent regions of the scalp.18 Other patterns include a diffuse type with body hair loss, patchy alopecia, and complete alopecia with or without body hair loss (Table).3-8 Whether these patterns all can be attributed to chemotherapy remains to be explored.
Breast Cancer Therapies Causing PCIA
The main agents thought to be responsible for PCIA in breast cancer patients are taxanes. The role of endocrine therapies has not been well explored. Trastuzumab lacks several of the common side effects of chemotherapy due to its specificity for the HER2/neu receptor and has not been found to increase the rate of hair loss when combined with standard chemotherapy.19,20 Although radiation therapy has the potential to damage hair follicles, and a dose-dependent relationship has been described for temporary and permanent alopecia at irradiated sites, permanent alopecia predominantly has been reported with cranial radiation used in the treatment of intracranial malignancies.21 The role of radiation therapy of the breasts in PCIA is unclear, as its inclusion in therapy has not been consistently reported in the literature.
Docetaxel is known to cause chemotherapy-induced alopecia, with an 83.4% incidence in phase 2 trials; however, it also appears to be related to PCIA.20 A PubMed search of articles indexed for MEDLINE was performed using the terms permanent chemotherapy induced alopecia, chemotherapy, docetaxel, endocrine therapies, hair loss, alopecia, and breast cancer. More than 400 cases of PCIA related to chemotherapy in breast cancer patients have been reported in the literature from a combination of case reports/series, retrospective surveys, and at least one prospective study. Data from some of the more detailed reports (n=52) are summarized in the Table. In the single-center, 3-year prospective study of women given adjuvant taxane-based or non–taxane-based chemotherapy, those who received taxane therapy were more likely to develop PCIA (odds ratio, 8.01).9
All 3 of our patients received taxanes. Interestingly, patient 3 underwent 2 rounds of chemotherapy 14 years apart and experienced full regrowth of the hair after the first course of taxane-free chemotherapy but experienced persistent hair loss following docetaxel treatment. Adjuvant endocrine therapies also may contribute to PCIA. A review of the side effects of endocrine therapies revealed an incidence of alopecia that was higher than expected; tamoxifen was the greatest offender. Additionally, using endocrine treatments in combination was found to have a synergistic effect on alopecia.18 Adjuvant endocrine therapy was used in patients 2 and 3. Although endocrine therapies appear to have a milder effect on hair loss compared to chemotherapy, these medications are continued for a longer duration, potentially contributing to the severity of hair loss and prolonging the time to regrowth.
Furthermore, endocrine therapies used in breast cancer treatment decrease estrogen levels or antagonize estrogen receptors, creating an environment of relative hyperandrogenism that may contribute to FPHL in genetically susceptible women.18 Although taxanes may cause irreversible hair loss in these patients, the action of endocrine therapies on the remaining hair follicles may affect the typical female pattern seen clinically. Patients 2 and 3 who presented with FPHL received adjuvant endocrine therapies and had positive family history, while patient 1 did not. Of note, patient 3 experienced worsening hair loss following the addition of anastrozole, which suggests a contribution of endocrine therapy to her PCIA. Our limited cases do not allow for evaluation of a worsened outcome with the combination of taxanes and endocrine therapies; however, we suggest further evaluation for a synergistic effect that may be contributing to PCIA.
Conclusion
Permanent alopecia in breast cancer patients appears to be a true potential adverse effect of taxanes and endocrine therapies, and it is important to characterize it appropriately so that its mechanism can be understood and appropriate treatment and counseling can take place. Although it may not influence clinical decision-making, patients should be informed that hair loss with chemotherapy can be permanent. Treatment with scalp cooling can reduce the risk for severe chemotherapy-induced alopecia, but it is unclear if it reduces risk for PCIA.12,15 Topical or oral minoxidil may be helpful in the treatment of PCIA once it has developed.7,8,15,22 Better characterization of these cases may elucidate risk factors for developing permanent alopecia, allowing for more appropriate risk stratification, counseling, and treatment.
Anagen effluvium during chemotherapy is common, typically beginning within 1 month of treatment onset and resolving by 6 months after the final course.1 Permanent chemotherapy-induced alopecia (PCIA), in which hair loss persists beyond 6 months after chemotherapy without recovery to original density, was first reported in patients following high-dose chemotherapy regimens for allogeneic bone marrow transplantation.2 There are now increasing reports of PCIA in patients with breast cancer; at least 400 such cases have been documented.3-16 In addition to chemotherapy, patients often receive adjuvant endocrine therapy with selective estrogen receptor modulators, aromatase inhibitors, or gonadotropin-releasing hormone agonists.5-16 Endocrine therapies also can lead to alopecia, but their role in PCIA has not been well defined.15,16 We describe 3 patients with breast cancer who experienced PCIA following chemotherapy with taxanes with or without endocrine therapies. We also review the literature on non–bone marrow transplantation PCIA to better characterize this entity and explore the role of endocrine therapies in PCIA.
Case Reports
Patient 1
A 62-year-old woman with a history of stage II invasive ductal carcinoma presented with persistent hair loss 5 years after completing chemotherapy. She underwent 6 cycles of docetaxel and carboplatin along with radiation therapy as well as 1 year of trastuzumab and did not receive endocrine therapy. At the current presentation, she reported patchy hair regrowth that gradually filled in but failed to return to full density. Physical examination revealed the hair was diffusely thin, especially bitemporally (Figures 1A and 1B), and she did not experience any loss of body hair. She had no family history of hair loss. Her medical history was notable for hypertension, chronic obstructive bronchitis, osteopenia, and depression. Her thyroid stimulating hormone (TSH) level was within reference range. Medications included lisinopril, metoprolol, escitalopram, and trazodone. A biopsy from the occipital scalp showed nonscarring alopecia with variation of hair follicle size, a decreased number of hair follicles, and a decreased anagen to telogen ratio (Figure 1C). She was treated with clobetasol solution and minoxidil solution 5% for 1 year with mild improvement. She experienced no further hair loss but did not regain original hair density.
Patient 2
A 35-year-old woman with a history of stage II invasive ductal carcinoma presented with persistent hair loss 10 months after chemotherapy. She underwent 4 cycles of doxorubicin and cyclophosphamide followed by 4 cycles of paclitaxel and was started on trastuzumab. Tamoxifen was initiated 1 month after completing chemotherapy. She received radiation therapy the following month and continued trastuzumab for 1 year. At the current presentation, the patient noted that hair regrowth had started 1 month after the last course of chemotherapy but had progressed slowly. She denied body hair loss. Physical examination revealed diffuse thinning, especially over the crown, with scattered broken hairs throughout the scalp and several miniaturized hairs over the crown. She was evaluated as grade 3 on the Sinclair clinical grading scale used to evaluate female pattern hair loss (FPHL).17 Her family history was remarkable for FPHL in her maternal grandmother. She had no notable medical history, her TSH was normal, and she was taking tamoxifen and trastuzumab. Biopsy was not performed. The patient was started on minoxidil solution 2% and had mild improvement with no further broken-off hairs after 10 months. At that point, she was evaluated as grade 2 to 3 on the Sinclair scale.17
Patient 3
A 51-year-old woman with a history of papillary carcinoma and extensive ductal carcinoma in situ presented with persistent hair loss for 3.5 years following chemotherapy for recurrent breast cancer. After her initial diagnosis in the left breast, she received cyclophosphamide, methotrexate, and 5-fluorouracil but did not receive endocrine therapy. Her hair thinned during chemotherapy but returned to normal density within 1 year. She had a recurrence of the cancer in the right breast 14 years later and received 6 cycles of chemotherapy with cyclophosphamide and docetaxel followed by radiation therapy. After this course, her hair loss incompletely recovered. One year after chemotherapy, she underwent bilateral salpingo-oophorectomy and started anastrozole. Three months later, she noticed increased shedding and progressive thinning of the hair. Physical examination revealed diffuse thinning that was most pronounced over the crown. She also experienced lateral thinning of the eyebrows, decreased eyelashes, and dystrophic fingernails. Fluocinonide solution was discontinued by the patient due to scalp burning. She had a brother with bitemporal recession. Her medical history was notable for Hashimoto thyroiditis, vitamin D deficiency, and peripheral neuropathy. Her TSH occasionally was elevated, and she was intermittently on levothyroxine; however, her free T4 was maintained within reference range on all records. Her medications at the time of evaluation were anastrozole and gabapentin. Biopsies taken from the right and left temporal scalp revealed decreased follicle density with a majority of follicles in anagen, scattered miniaturized follicles, and a mild perivascular and perifollicular lymphoid infiltrate. Mild dermal fibrosis was present without evidence of frank scarring (Figure 2). She declined treatment, and there was no change in her condition over 3 years of follow-up.
Comment
Classification of Chemotherapy-Induced Hair Loss
Chemotherapy-induced alopecia is typically an anagen effluvium that is reversed within 6 months following the final course of chemotherapy. When incomplete regrowth persists, the patient is considered to have PCIA.1 The pathophysiology of PCIA is unclear.
Traditional grading for chemotherapy-induced alopecia does not account for the patterns of loss seen in PCIA, of which the most common appears to be a female pattern with accentuated hair loss in androgen-dependent regions of the scalp.18 Other patterns include a diffuse type with body hair loss, patchy alopecia, and complete alopecia with or without body hair loss (Table).3-8 Whether these patterns all can be attributed to chemotherapy remains to be explored.
Breast Cancer Therapies Causing PCIA
The main agents thought to be responsible for PCIA in breast cancer patients are taxanes. The role of endocrine therapies has not been well explored. Trastuzumab lacks several of the common side effects of chemotherapy due to its specificity for the HER2/neu receptor and has not been found to increase the rate of hair loss when combined with standard chemotherapy.19,20 Although radiation therapy has the potential to damage hair follicles, and a dose-dependent relationship has been described for temporary and permanent alopecia at irradiated sites, permanent alopecia predominantly has been reported with cranial radiation used in the treatment of intracranial malignancies.21 The role of radiation therapy of the breasts in PCIA is unclear, as its inclusion in therapy has not been consistently reported in the literature.
Docetaxel is known to cause chemotherapy-induced alopecia, with an 83.4% incidence in phase 2 trials; however, it also appears to be related to PCIA.20 A PubMed search of articles indexed for MEDLINE was performed using the terms permanent chemotherapy induced alopecia, chemotherapy, docetaxel, endocrine therapies, hair loss, alopecia, and breast cancer. More than 400 cases of PCIA related to chemotherapy in breast cancer patients have been reported in the literature from a combination of case reports/series, retrospective surveys, and at least one prospective study. Data from some of the more detailed reports (n=52) are summarized in the Table. In the single-center, 3-year prospective study of women given adjuvant taxane-based or non–taxane-based chemotherapy, those who received taxane therapy were more likely to develop PCIA (odds ratio, 8.01).9
All 3 of our patients received taxanes. Interestingly, patient 3 underwent 2 rounds of chemotherapy 14 years apart and experienced full regrowth of the hair after the first course of taxane-free chemotherapy but experienced persistent hair loss following docetaxel treatment. Adjuvant endocrine therapies also may contribute to PCIA. A review of the side effects of endocrine therapies revealed an incidence of alopecia that was higher than expected; tamoxifen was the greatest offender. Additionally, using endocrine treatments in combination was found to have a synergistic effect on alopecia.18 Adjuvant endocrine therapy was used in patients 2 and 3. Although endocrine therapies appear to have a milder effect on hair loss compared to chemotherapy, these medications are continued for a longer duration, potentially contributing to the severity of hair loss and prolonging the time to regrowth.
Furthermore, endocrine therapies used in breast cancer treatment decrease estrogen levels or antagonize estrogen receptors, creating an environment of relative hyperandrogenism that may contribute to FPHL in genetically susceptible women.18 Although taxanes may cause irreversible hair loss in these patients, the action of endocrine therapies on the remaining hair follicles may affect the typical female pattern seen clinically. Patients 2 and 3 who presented with FPHL received adjuvant endocrine therapies and had positive family history, while patient 1 did not. Of note, patient 3 experienced worsening hair loss following the addition of anastrozole, which suggests a contribution of endocrine therapy to her PCIA. Our limited cases do not allow for evaluation of a worsened outcome with the combination of taxanes and endocrine therapies; however, we suggest further evaluation for a synergistic effect that may be contributing to PCIA.
Conclusion
Permanent alopecia in breast cancer patients appears to be a true potential adverse effect of taxanes and endocrine therapies, and it is important to characterize it appropriately so that its mechanism can be understood and appropriate treatment and counseling can take place. Although it may not influence clinical decision-making, patients should be informed that hair loss with chemotherapy can be permanent. Treatment with scalp cooling can reduce the risk for severe chemotherapy-induced alopecia, but it is unclear if it reduces risk for PCIA.12,15 Topical or oral minoxidil may be helpful in the treatment of PCIA once it has developed.7,8,15,22 Better characterization of these cases may elucidate risk factors for developing permanent alopecia, allowing for more appropriate risk stratification, counseling, and treatment.
- Dorr VJ. A practitioner’s guide to cancer-related alopecia. Semin Oncol. 1998;25:562-570.
- Machado M, Moreb JS, Khan SA. Six cases of permanent alopecia after various conditioning regimens commonly used in hematopoietic stem cell transplantation. Bone Marrow Transplant. 2007;40:979-982.
- Tallon B, Blanchard E, Goldberg LJ. Permanent chemotherapy-induced alopecia: case report and review of the literature. J Am Acad Dermatol. 2010;63:333-336.
- Miteva M, Misciali C, Fanti PA, et al. Permanent alopecia after systemic chemotherapy: a clinicopathological study of 10 cases. Am J Dermatopathol. 2011;33:345-350.
- Prevezas C, Matard B, Pinquier L, et al. Irreversible and severe alopecia following docetaxel or paclitaxel cytotoxic therapy for breast cancer. Br J Dermatol. 2009;160:883-885.
- Masidonski P, Mahon SM. Permanent alopecia in women being treated for breast cancer. Clin J Oncol Nurs. 2009;13:13-14.
- Kluger N, Jacot W, Frouin E, et al. Permanent scalp alopecia related to breast cancer chemotherapy by sequential fluorouracil/epirubicin/cyclophosphamide (FEC) and docetaxel: a prospective study of 20 patients. Ann Oncol. 2012;23:2879-2884.
- Fonia A, Cota C, Setterfield JF, et al. Permanent alopecia in patients with breast cancer after taxane chemotherapy and adjuvant hormonal therapy: clinicopathologic findings in a cohort of 10 patients. J Am Acad Dermatol. 2017;76:948-957.
- Kang D, Kim IR, Choi EK, et al. Permanent chemotherapy-induced alopecia in patients with breast cancer: a 3-year prospective cohort study [published online August 17, 2018]. Oncologist. 2019;24:414-420.
- Chan J, Adderley H, Alameddine M, et al. Permanent hair loss associated with taxane chemotherapy use in breast cancer: a retrospective survey at two tertiary UK cancer centres [published online December 22, 2020]. Eur J Cancer Care (Engl). doi:10.1111/ecc.13395
- Bourgeois H, Denis F, Kerbrat P, et al. Long term persistent alopecia and suboptimal hair regrowth after adjuvant chemotherapy for breast cancer: alert for an emerging side effect: ALOPERS Observatory. Cancer Res. 2009;69(24 suppl). doi:10.1158/0008-5472.SABCS-09-3174
- Bertrand M, Mailliez A, Vercambre S, et al. Permanent chemotherapy induced alopecia in early breast cancer patients after (neo)adjuvant chemotherapy: long term follow up. Cancer Res. 2013;73(24 suppl). doi:10.1158/0008-5472.SABCS13-P3-09-15
- Kim S, Park HS, Kim JY, et al. Irreversible chemotherapy-induced alopecia in breast cancer patient. Cancer Res. 2016;76(4 suppl). doi:10.1158/1538-7445.SABCS15-P1-15-04
- Thorp NJ, Swift F, Arundell D, et al. Long term hair loss in patients with early breast cancer receiving docetaxel chemotherapy. Cancer Res. 2015;75(9 suppl). doi:10.1158/1538-7445.SABCS14-P5-17-04
- Freites-Martinez A, Shapiro J, van den Hurk C, et al. Hair disorders in cancer survivors. J Am Acad Dermatol. 2019;80:1199-1213.
- Freites-Martinez A, Chan D, Sibaud V, et al. Assessment of quality of life and treatment outcomes of patients with persistent postchemotherapy alopecia. JAMA Dermatol. 2019;155:724-728.
- Sinclair R, Jolley D, Mallari R, et al. The reliability of horizontally sectioned scalp biopsies in the diagnosis of chronic diffuse telogen hair loss in women. J Am Acad Dermatol. 2004;51:189-199.
- Saggar V, Wu S, Dickler MN, et al. Alopecia with endocrine therapies in patients with cancer. Oncologist. 2013;18:1126-1134.
- Yeager CE, Olsen EA. Treatment of chemotherapy-induced alopecia. Dermatol Ther. 2011;24:432-442.
- Baselga J. Clinical trials of single-agent trastuzumab (Herceptin). Semin Oncol. 2000;27(5 suppl 9):20-26.
- Lawenda BD, Gagne HM, Gierga DP, et al. Permanent alopecia after cranial irradiation: dose-response relationship. Int J Radiat Oncol Biol Phys. 2004;60:879-887.
- Yang X, Thai KE. Treatment of permanent chemotherapy-induced alopecia with low dose oral minoxidil [published online May 13, 2015]. Australas J Dermatol. 2016;57:E130-E132.
- Dorr VJ. A practitioner’s guide to cancer-related alopecia. Semin Oncol. 1998;25:562-570.
- Machado M, Moreb JS, Khan SA. Six cases of permanent alopecia after various conditioning regimens commonly used in hematopoietic stem cell transplantation. Bone Marrow Transplant. 2007;40:979-982.
- Tallon B, Blanchard E, Goldberg LJ. Permanent chemotherapy-induced alopecia: case report and review of the literature. J Am Acad Dermatol. 2010;63:333-336.
- Miteva M, Misciali C, Fanti PA, et al. Permanent alopecia after systemic chemotherapy: a clinicopathological study of 10 cases. Am J Dermatopathol. 2011;33:345-350.
- Prevezas C, Matard B, Pinquier L, et al. Irreversible and severe alopecia following docetaxel or paclitaxel cytotoxic therapy for breast cancer. Br J Dermatol. 2009;160:883-885.
- Masidonski P, Mahon SM. Permanent alopecia in women being treated for breast cancer. Clin J Oncol Nurs. 2009;13:13-14.
- Kluger N, Jacot W, Frouin E, et al. Permanent scalp alopecia related to breast cancer chemotherapy by sequential fluorouracil/epirubicin/cyclophosphamide (FEC) and docetaxel: a prospective study of 20 patients. Ann Oncol. 2012;23:2879-2884.
- Fonia A, Cota C, Setterfield JF, et al. Permanent alopecia in patients with breast cancer after taxane chemotherapy and adjuvant hormonal therapy: clinicopathologic findings in a cohort of 10 patients. J Am Acad Dermatol. 2017;76:948-957.
- Kang D, Kim IR, Choi EK, et al. Permanent chemotherapy-induced alopecia in patients with breast cancer: a 3-year prospective cohort study [published online August 17, 2018]. Oncologist. 2019;24:414-420.
- Chan J, Adderley H, Alameddine M, et al. Permanent hair loss associated with taxane chemotherapy use in breast cancer: a retrospective survey at two tertiary UK cancer centres [published online December 22, 2020]. Eur J Cancer Care (Engl). doi:10.1111/ecc.13395
- Bourgeois H, Denis F, Kerbrat P, et al. Long term persistent alopecia and suboptimal hair regrowth after adjuvant chemotherapy for breast cancer: alert for an emerging side effect: ALOPERS Observatory. Cancer Res. 2009;69(24 suppl). doi:10.1158/0008-5472.SABCS-09-3174
- Bertrand M, Mailliez A, Vercambre S, et al. Permanent chemotherapy induced alopecia in early breast cancer patients after (neo)adjuvant chemotherapy: long term follow up. Cancer Res. 2013;73(24 suppl). doi:10.1158/0008-5472.SABCS13-P3-09-15
- Kim S, Park HS, Kim JY, et al. Irreversible chemotherapy-induced alopecia in breast cancer patient. Cancer Res. 2016;76(4 suppl). doi:10.1158/1538-7445.SABCS15-P1-15-04
- Thorp NJ, Swift F, Arundell D, et al. Long term hair loss in patients with early breast cancer receiving docetaxel chemotherapy. Cancer Res. 2015;75(9 suppl). doi:10.1158/1538-7445.SABCS14-P5-17-04
- Freites-Martinez A, Shapiro J, van den Hurk C, et al. Hair disorders in cancer survivors. J Am Acad Dermatol. 2019;80:1199-1213.
- Freites-Martinez A, Chan D, Sibaud V, et al. Assessment of quality of life and treatment outcomes of patients with persistent postchemotherapy alopecia. JAMA Dermatol. 2019;155:724-728.
- Sinclair R, Jolley D, Mallari R, et al. The reliability of horizontally sectioned scalp biopsies in the diagnosis of chronic diffuse telogen hair loss in women. J Am Acad Dermatol. 2004;51:189-199.
- Saggar V, Wu S, Dickler MN, et al. Alopecia with endocrine therapies in patients with cancer. Oncologist. 2013;18:1126-1134.
- Yeager CE, Olsen EA. Treatment of chemotherapy-induced alopecia. Dermatol Ther. 2011;24:432-442.
- Baselga J. Clinical trials of single-agent trastuzumab (Herceptin). Semin Oncol. 2000;27(5 suppl 9):20-26.
- Lawenda BD, Gagne HM, Gierga DP, et al. Permanent alopecia after cranial irradiation: dose-response relationship. Int J Radiat Oncol Biol Phys. 2004;60:879-887.
- Yang X, Thai KE. Treatment of permanent chemotherapy-induced alopecia with low dose oral minoxidil [published online May 13, 2015]. Australas J Dermatol. 2016;57:E130-E132.
Practice Points
- Permanent chemotherapy-induced alopecia (PCIA) is defined as hair loss that persists beyond 6 months after treatment with chemotherapy. It may be complicated by the addition of endocrine therapies.
- Patients and clinicians should be aware that PCIA can occur and appears to be a higher risk with taxane therapy.
Bothersome Blisters: Localized Epidermolysis Bullosa Simplex
To the Editor:
Epidermolysis bullosa (EB) was first described in 1886, with the first classification scheme proposed in 1962 utilizing transmission electron microscopy (TEM) findings to delineate categories: epidermolytic (EB simplex [EBS]), lucidolytic (junctional EB), and dermolytic (dystrophic EB).1 Localized EBS (EBS-loc) is an autosomal-dominant disorder caused by negative mutations in keratin-5 and keratin-14, proteins expressed in the intermediate filaments of basal keratinocytes, which result in fragility of the skin in response to minor trauma.2 The incidence of EBS-loc is approximately 10 to 30 cases per million live births, with the age of presentation typically between the first and third decades of life.3,4 Because EBS-loc is the most common and often mildest form of EB, not all patients present for medical evaluation and true prevalence may be underestimated.4 We report a case of EBS-loc.
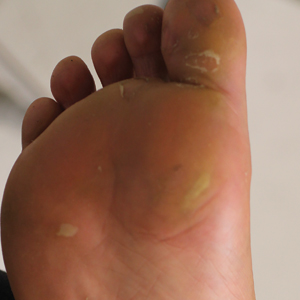
A 26-year-old woman with no notable medical history presented to the dermatology clinic for evaluation of skin blisters that had been intermittently present since infancy. The blisters primarily occurred on the feet, but she did occasionally develop blisters on the hands, knees, and elbows and at sites of friction or trauma (eg, bra line, medial thighs) following exercise. The blisters were worsened by heat and tight-fitting shoes. Because of the painful nature of the blisters, she would lance them with a needle. On the medial thighs, she utilized nonstick and gauze bandage roll dressings to minimize friction. A review of systems was positive for hyperhidrosis. Her family history revealed multiple family members with blisters involving the feet and areas of friction or trauma for 4 generations with no known diagnosis.
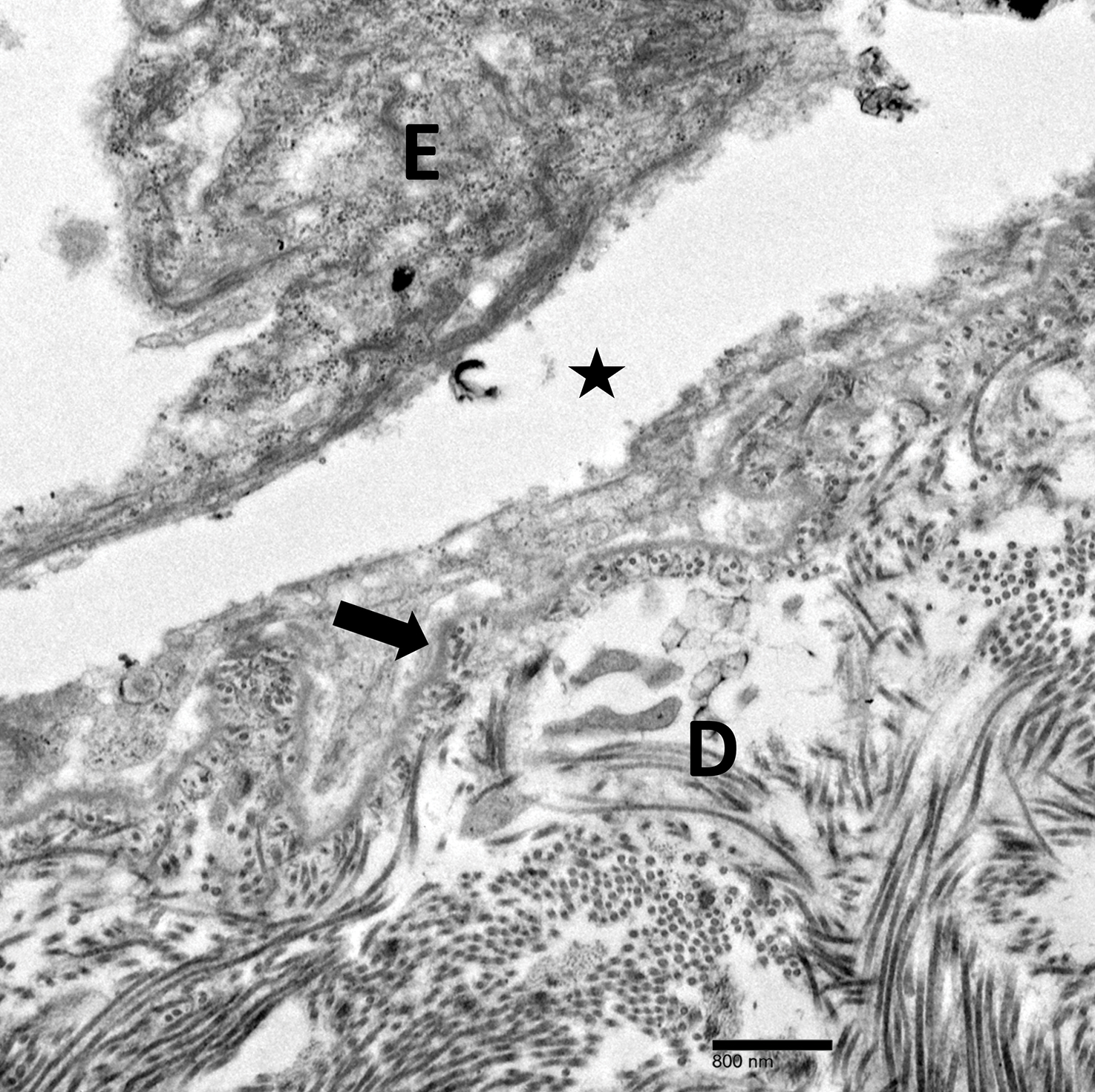
Physical examination revealed multiple tense bullae and calluses scattered over the bilateral plantar and distal dorsal feet with a few healing, superficially eroded, erythematous papules and plaques on the bilateral medial thighs (Figure 1). A biopsy from an induced blister on the right dorsal second toe was performed and sent in glutaraldehyde to the Epidermolysis Bullosa Clinic at Stanford University (Redwood City, California) for electron microscopy, which revealed lysis within the basal keratinocytes through the tonofilaments with continuous and intact lamina densa and lamina lucida (Figure 2). In this clinical context with the relevant family history, the findings were consistent with the diagnosis of EBS-loc (formerly Weber-Cockayne syndrome).2

Skin manifestations of EBS-loc typically consist of friction-induced blisters, erosions, and calluses primarily on the palms and soles, often associated with hyperhidrosis and worsening of symptoms in summer months and hot temperatures.3 Milia, atrophic scarring, and dystrophic nails are uncommon.1 Extracutaneous involvement is rare with the exception of oral cavity erosions, which typically are asymptomatic and usually are only seen during infancy.1
Light microscopy does not have a notable role in diagnosis of classic forms of inherited EB unless another autoimmune blistering disorder is suspected.2,5 Both TEM and immunofluorescence mapping are used to diagnose EB.1 DNA mutational analysis is not considered a first-line diagnostic test for EB given it is a costly labor-intensive technique with limited access at present, but it may be considered in settings of prenatal diagnosis or in vitro fertilization.1 Biopsy of a freshly induced blister should be performed, as early reepithelialization of an existing blister makes it difficult to establish the level of cleavage.5 Applying firm pressure using a pencil eraser and rotating it on intact skin induces a subclinical blister. Two punch biopsies (4 mm) at the edge of the blister with one-third lesional and two-thirds perilesional skin should be obtained, with one biopsy sent for immunofluorescence mapping in Michel fixative and the other for TEM in glutaraldehyde.3,5 Transmission electron microscopy of an induced blister in EBS-loc shows cleavage within the most inferior portion of the basilar keratinocyte.2 Immunofluorescence mapping with anti–epidermal basement membrane monoclonal antibodies can distinguish between EB subtypes and assess expression of specific skin-associated proteins on both a qualitative or semiquantitative basis, providing insight on which structural protein is mutated.1,5
No specific treatments are available for EBS-loc. Mainstays of treatment include prevention of mechanical trauma and secondary infection. Hyperhidrosis of thepalms and soles may be treated with topical aluminum chloride hexahydrate or injections of botulinum toxin type A.2,6 Patients have normal life expectancy, though some cases may have complications with substantial morbidity.1 Awareness of this disease, its clinical course, and therapeutic options will allow physicians to more appropriately counsel patients on the disease process.
Localized EBS may be more common than previously thought, as not all patients seek medical care. Given its impact on patient quality of life, it is important for clinicians to recognize EBS-loc. Although no specific treatments are available, wound care counseling and explanation of the genetics of the disease should be provided to patients.
- Fine JD, Eady RA, Bauer EA, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58:931-950.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Limited; 2012.
- Eichenfield LF, Frieden IJ, Mathes EF, et al, eds. Neonatal and Infant Dermatology. 3rd ed. New York, NY: Elsevier Health Sciences; 2015.
- Spitz JL. Genodermatoses: A Clinical Guide to Genetic Skin Disorders. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005.
- Epidermolysis bullosa. Stanford Medicine website. http://med.stanford.edu/dermatopathology/dermpath-services/epiderm.html. Accessed April 3, 2019.
- Abitbol RJ, Zhou LH. Treatment of epidermolysis bullosa simplex, Weber-Cockayne type, with botulinum toxin type A. Arch Dermatol. 2009;145:13-15.
To the Editor:
Epidermolysis bullosa (EB) was first described in 1886, with the first classification scheme proposed in 1962 utilizing transmission electron microscopy (TEM) findings to delineate categories: epidermolytic (EB simplex [EBS]), lucidolytic (junctional EB), and dermolytic (dystrophic EB).1 Localized EBS (EBS-loc) is an autosomal-dominant disorder caused by negative mutations in keratin-5 and keratin-14, proteins expressed in the intermediate filaments of basal keratinocytes, which result in fragility of the skin in response to minor trauma.2 The incidence of EBS-loc is approximately 10 to 30 cases per million live births, with the age of presentation typically between the first and third decades of life.3,4 Because EBS-loc is the most common and often mildest form of EB, not all patients present for medical evaluation and true prevalence may be underestimated.4 We report a case of EBS-loc.
A 26-year-old woman with no notable medical history presented to the dermatology clinic for evaluation of skin blisters that had been intermittently present since infancy. The blisters primarily occurred on the feet, but she did occasionally develop blisters on the hands, knees, and elbows and at sites of friction or trauma (eg, bra line, medial thighs) following exercise. The blisters were worsened by heat and tight-fitting shoes. Because of the painful nature of the blisters, she would lance them with a needle. On the medial thighs, she utilized nonstick and gauze bandage roll dressings to minimize friction. A review of systems was positive for hyperhidrosis. Her family history revealed multiple family members with blisters involving the feet and areas of friction or trauma for 4 generations with no known diagnosis.
Physical examination revealed multiple tense bullae and calluses scattered over the bilateral plantar and distal dorsal feet with a few healing, superficially eroded, erythematous papules and plaques on the bilateral medial thighs (Figure 1). A biopsy from an induced blister on the right dorsal second toe was performed and sent in glutaraldehyde to the Epidermolysis Bullosa Clinic at Stanford University (Redwood City, California) for electron microscopy, which revealed lysis within the basal keratinocytes through the tonofilaments with continuous and intact lamina densa and lamina lucida (Figure 2). In this clinical context with the relevant family history, the findings were consistent with the diagnosis of EBS-loc (formerly Weber-Cockayne syndrome).2
Skin manifestations of EBS-loc typically consist of friction-induced blisters, erosions, and calluses primarily on the palms and soles, often associated with hyperhidrosis and worsening of symptoms in summer months and hot temperatures.3 Milia, atrophic scarring, and dystrophic nails are uncommon.1 Extracutaneous involvement is rare with the exception of oral cavity erosions, which typically are asymptomatic and usually are only seen during infancy.1
Light microscopy does not have a notable role in diagnosis of classic forms of inherited EB unless another autoimmune blistering disorder is suspected.2,5 Both TEM and immunofluorescence mapping are used to diagnose EB.1 DNA mutational analysis is not considered a first-line diagnostic test for EB given it is a costly labor-intensive technique with limited access at present, but it may be considered in settings of prenatal diagnosis or in vitro fertilization.1 Biopsy of a freshly induced blister should be performed, as early reepithelialization of an existing blister makes it difficult to establish the level of cleavage.5 Applying firm pressure using a pencil eraser and rotating it on intact skin induces a subclinical blister. Two punch biopsies (4 mm) at the edge of the blister with one-third lesional and two-thirds perilesional skin should be obtained, with one biopsy sent for immunofluorescence mapping in Michel fixative and the other for TEM in glutaraldehyde.3,5 Transmission electron microscopy of an induced blister in EBS-loc shows cleavage within the most inferior portion of the basilar keratinocyte.2 Immunofluorescence mapping with anti–epidermal basement membrane monoclonal antibodies can distinguish between EB subtypes and assess expression of specific skin-associated proteins on both a qualitative or semiquantitative basis, providing insight on which structural protein is mutated.1,5
No specific treatments are available for EBS-loc. Mainstays of treatment include prevention of mechanical trauma and secondary infection. Hyperhidrosis of thepalms and soles may be treated with topical aluminum chloride hexahydrate or injections of botulinum toxin type A.2,6 Patients have normal life expectancy, though some cases may have complications with substantial morbidity.1 Awareness of this disease, its clinical course, and therapeutic options will allow physicians to more appropriately counsel patients on the disease process.
Localized EBS may be more common than previously thought, as not all patients seek medical care. Given its impact on patient quality of life, it is important for clinicians to recognize EBS-loc. Although no specific treatments are available, wound care counseling and explanation of the genetics of the disease should be provided to patients.
To the Editor:
Epidermolysis bullosa (EB) was first described in 1886, with the first classification scheme proposed in 1962 utilizing transmission electron microscopy (TEM) findings to delineate categories: epidermolytic (EB simplex [EBS]), lucidolytic (junctional EB), and dermolytic (dystrophic EB).1 Localized EBS (EBS-loc) is an autosomal-dominant disorder caused by negative mutations in keratin-5 and keratin-14, proteins expressed in the intermediate filaments of basal keratinocytes, which result in fragility of the skin in response to minor trauma.2 The incidence of EBS-loc is approximately 10 to 30 cases per million live births, with the age of presentation typically between the first and third decades of life.3,4 Because EBS-loc is the most common and often mildest form of EB, not all patients present for medical evaluation and true prevalence may be underestimated.4 We report a case of EBS-loc.
A 26-year-old woman with no notable medical history presented to the dermatology clinic for evaluation of skin blisters that had been intermittently present since infancy. The blisters primarily occurred on the feet, but she did occasionally develop blisters on the hands, knees, and elbows and at sites of friction or trauma (eg, bra line, medial thighs) following exercise. The blisters were worsened by heat and tight-fitting shoes. Because of the painful nature of the blisters, she would lance them with a needle. On the medial thighs, she utilized nonstick and gauze bandage roll dressings to minimize friction. A review of systems was positive for hyperhidrosis. Her family history revealed multiple family members with blisters involving the feet and areas of friction or trauma for 4 generations with no known diagnosis.
Physical examination revealed multiple tense bullae and calluses scattered over the bilateral plantar and distal dorsal feet with a few healing, superficially eroded, erythematous papules and plaques on the bilateral medial thighs (Figure 1). A biopsy from an induced blister on the right dorsal second toe was performed and sent in glutaraldehyde to the Epidermolysis Bullosa Clinic at Stanford University (Redwood City, California) for electron microscopy, which revealed lysis within the basal keratinocytes through the tonofilaments with continuous and intact lamina densa and lamina lucida (Figure 2). In this clinical context with the relevant family history, the findings were consistent with the diagnosis of EBS-loc (formerly Weber-Cockayne syndrome).2
Skin manifestations of EBS-loc typically consist of friction-induced blisters, erosions, and calluses primarily on the palms and soles, often associated with hyperhidrosis and worsening of symptoms in summer months and hot temperatures.3 Milia, atrophic scarring, and dystrophic nails are uncommon.1 Extracutaneous involvement is rare with the exception of oral cavity erosions, which typically are asymptomatic and usually are only seen during infancy.1
Light microscopy does not have a notable role in diagnosis of classic forms of inherited EB unless another autoimmune blistering disorder is suspected.2,5 Both TEM and immunofluorescence mapping are used to diagnose EB.1 DNA mutational analysis is not considered a first-line diagnostic test for EB given it is a costly labor-intensive technique with limited access at present, but it may be considered in settings of prenatal diagnosis or in vitro fertilization.1 Biopsy of a freshly induced blister should be performed, as early reepithelialization of an existing blister makes it difficult to establish the level of cleavage.5 Applying firm pressure using a pencil eraser and rotating it on intact skin induces a subclinical blister. Two punch biopsies (4 mm) at the edge of the blister with one-third lesional and two-thirds perilesional skin should be obtained, with one biopsy sent for immunofluorescence mapping in Michel fixative and the other for TEM in glutaraldehyde.3,5 Transmission electron microscopy of an induced blister in EBS-loc shows cleavage within the most inferior portion of the basilar keratinocyte.2 Immunofluorescence mapping with anti–epidermal basement membrane monoclonal antibodies can distinguish between EB subtypes and assess expression of specific skin-associated proteins on both a qualitative or semiquantitative basis, providing insight on which structural protein is mutated.1,5
No specific treatments are available for EBS-loc. Mainstays of treatment include prevention of mechanical trauma and secondary infection. Hyperhidrosis of thepalms and soles may be treated with topical aluminum chloride hexahydrate or injections of botulinum toxin type A.2,6 Patients have normal life expectancy, though some cases may have complications with substantial morbidity.1 Awareness of this disease, its clinical course, and therapeutic options will allow physicians to more appropriately counsel patients on the disease process.
Localized EBS may be more common than previously thought, as not all patients seek medical care. Given its impact on patient quality of life, it is important for clinicians to recognize EBS-loc. Although no specific treatments are available, wound care counseling and explanation of the genetics of the disease should be provided to patients.
- Fine JD, Eady RA, Bauer EA, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58:931-950.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Limited; 2012.
- Eichenfield LF, Frieden IJ, Mathes EF, et al, eds. Neonatal and Infant Dermatology. 3rd ed. New York, NY: Elsevier Health Sciences; 2015.
- Spitz JL. Genodermatoses: A Clinical Guide to Genetic Skin Disorders. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005.
- Epidermolysis bullosa. Stanford Medicine website. http://med.stanford.edu/dermatopathology/dermpath-services/epiderm.html. Accessed April 3, 2019.
- Abitbol RJ, Zhou LH. Treatment of epidermolysis bullosa simplex, Weber-Cockayne type, with botulinum toxin type A. Arch Dermatol. 2009;145:13-15.
- Fine JD, Eady RA, Bauer EA, et al. The classification of inherited epidermolysis bullosa (EB): report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58:931-950.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Limited; 2012.
- Eichenfield LF, Frieden IJ, Mathes EF, et al, eds. Neonatal and Infant Dermatology. 3rd ed. New York, NY: Elsevier Health Sciences; 2015.
- Spitz JL. Genodermatoses: A Clinical Guide to Genetic Skin Disorders. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005.
- Epidermolysis bullosa. Stanford Medicine website. http://med.stanford.edu/dermatopathology/dermpath-services/epiderm.html. Accessed April 3, 2019.
- Abitbol RJ, Zhou LH. Treatment of epidermolysis bullosa simplex, Weber-Cockayne type, with botulinum toxin type A. Arch Dermatol. 2009;145:13-15.
Practice Points
- Localized epidermolysis bullosa simplex (formerly Weber-Cockayne syndrome) presents with flaccid bullae and erosions predominantly on the hands and feet, most commonly related to mechanical friction and heat.
- It is inherited in an autosomal-dominant fashion with defects in keratin-5 and keratin-14.
- Biopsy of a freshly induced blister should be examined by transmission electron microscopy or immunofluorescence mapping.
- Treatment is focused on wound management and infection control of the blisters.
Ecthyma Gangrenosum Due to Pseudomonas fluorescens
To the Editor:
A 50-year-old female farmer with diabetes mellitus, paroxysmal atrial fibrillation, and treatment-refractory systemic lupus erythematosus presented with worsening erythema, ecchymoses, edema, and tenderness in the bilateral legs of 3 weeks’ duration. The patient was taking oral methylprednisolone 12 mg daily (8 mg in the morning, 4 mg in the evening) for systemic lupus erythematosus. She previously was treated with mycophenolate mofetil, mycophenolic acid, methotrexate, azathioprine, hydroxychloroquine, etanercept, and cyclosporine without success. Cyclophosphamide was helpful in the past, but the last dose was more than 1 year prior to the current presentation. Physical examination showed no fever and 1+ pitting edema to the mid shin. Multiple warm, tender, erythematous to gray plaques were present on the bilateral lower extremities, and a 2-cm ulcerated plaque with a violaceous border was present on the medial surface of the lower left leg (Figure 1). The surrounding erythematous tissue was markedly tender to palpation. No popliteal or inguinal lymphadenopathy was appreciated.
...")
Punch biopsies were obtained from the periphery and center of the ulcerated plaque on the left leg. Histopathologic analysis revealed an ulcerated necrotic epidermis with scant diffuse acute and chronic inflammation (Figure 2A). Leukocytoclastic vasculitis was present at the periphery of the lesion (Figure 2B). Colloidal iron stain revealed a marked increase in dermal mucin. Gram stain showed both gram-positive and gram-negative organisms (Figure 2C). Fungal and hyphal elements were seen in the superficial epidermis. Tissue cultures revealed a predominance of Pseudomonas fluorescens, along with Candida albicans, Klebsiella oxytoca, and Staphylococcus and Enterococcus species. Bacterial and fungal blood cultures were negative.
(H&E, original magnification ×100); biopsy from the periphery of the lesion showed leukocytoclastic vasculitis (B)...")
The patient was treated with ciprofloxacin, vancomycin, and voriconazole based on culture sensitivities. Although double coverage often is recommended for pseudomonal infections,1 the patient could not be started on a second antipseudomonal agent due to multiple severe antibiotic allergies. She continued home administration of methylprednisolone in the setting of active lupus; additional immunosuppression was avoided. Over the course of 1 week, the patient’s preexisting ulcerated plaque on the medial surface of the lower left leg gradually improved, and no new lesions developed. Ciprofloxacin, vancomycin, and voriconazole were continued along with insulin, aspirin, warfarin, metoprolol, furosemide, and bumetanide at discharge. The patient subsequently was readmitted to the hospital several more times over the next 4 months for multiple bacterial infections and ultimately died of overwhelming septic shock several months later.
Ecthyma gangrenosum (EG) is a rare cutaneous infection that results from either direct inoculation or hematogenous dissemination. It classically is caused by infection with Pseudomonas aeruginosa in immunocompromised or neutropenic patients. However, other bacteria and fungi, mucormycosis, and herpes simplex virus also have been reported to cause EG.1 Skin lesions often start as erythematous or purpuric macules, develop into vesicles and bullae, and eventually become necrotic ulcers with central eschars.2 Histopathologic findings reveal necrotizing hemorrhagic vasculitis; gram-negative rods often are found in the medial and adventitial walls of deeper vessels.3,4 The case mortality rate is high, ranging from 15% in nonbacteremic patients to 38% to 96% in patients with bacteremia.3
The leukocytoclastic vasculitis seen on biopsy in our patient was a reaction pattern, likely a direct result of the soft tissue infection. Biopsy showed hyphal or pseudohyphal elements in the superficial epidermis, corresponding to the positive C albicans growth on fungal culture. Candida albicans has been reported to cause lesions that mimic bacterial EG.1 However, the marked predominance of P fluorescens on biopsy and culture suggests that the Candida likely were opportunistic and managed to invade secondary to the vascular damage caused by P fluorescens.
Pseudomonas fluorescens is an aerobic gram-negative rod-shaped bacterium found in soil that rarely is implicated in human disease. This bacterium is unable to ferment lactose and grows best on MacConkey agar between 30°C and 37°C but also can grow at temperatures as low as 4°C.5 The ability of P fluorescens to rapidly proliferate at low temperatures (ie, in refrigerated blood products, saline solutions, water dispensers, ice baths, humidifier water) is thought to explain a number of reported clinical consequences, ranging from asymptomatic colonization to fatal bacteremia.6-10 This opportunistic pathogen also has been linked to Crohn disease and has been reported to cause pelvic inflammatory disease with the use of intrauterine contraception devices and nosocomial respiratory tract infections due to contaminated spirometers.11-14 In our case, the patient was part of a family of farmers and worked in an agricultural setting. She often handled the produce and worked at the family’s produce stand at the local farmer’s market. Her exposure to soil and soil pathogens may have been the source of the P fluorescens infection.
This case introduces P fluorescens as a causative agent of EG, suggests that exposure to agricultural products may predispose an immunosuppressed patient to this type of infection, and emphasizes the importance of timely diagnosis through tissue culture and histopathology so that immunosuppressive medications can be withheld and appropriate antibiotics can be initiated.
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl):S114-S117.
- Güçlüer H, Ergun T, Demirçay Z. Ecthyma gangrenosum. Int J Dermatol. 1999;38:299-302.
- Solowski NL, Yao FB, Agarwal A, et al. Ecthyma gangrenosum: a rare cutaneous manifestation of a potentially fatal disease. Ann Otol Rhinol Laryngol. 2004;113:462-464.
- Lobo I, Pinto A, Ferreira M, et al. Non-pseudomonal ecthyma gangrenosum present in diclofenac-induced agranulocytosis. Eur J Dermatol. 2008;18:350-551.
- Pappas G, Karavasilis V, Christou L, et al. Pseudomonas fluorescens infections in clinical practice. Scand J Infect Dis. 2006;38:68-70.
- Gershman MD, Kennedy DJ, Noble-Wang J, et al. Multistate outbreak of Pseudomonas fluorescens bloodstream infection after exposure to contaminated heparinized saline flush prepared by a compounding pharmacy. Clin Infect Dis. 2008;47:1372-1378.
- Hsueh P, Teng L, Pan H, et al. Outbreak of Pseudomonas fluorescens bacteremia among oncology patients. J Clin Microbiol. 1998;36:2914-2917.
- Wong V, Levi K, Baddal B, et al. Spread of Pseudomonas fluorescens due to contaminated drinking water in a bone marrow transplant unit. J Clin Microbiol. 2011;49:2093-2096.
- Benito N, Mirelis B, Galvez ML, et al. Outbreak of Pseudomonas fluorescens bloodstream infection in a coronary care unit. J Hosp Infect. 2012;82:286-289.
- Redding PJ, McWalter PW. Pseudomonas fluorescens cross-infection due to contaminated humidifier water. Br Med J. 1980;281:275.
- Landers CJ, Cohavy O, Misra R, et al. Selected loss of tolerance evidenced by Crohn’s disease-associated immune responses to auto- and microbial antigens. Gastroenterology. 2002;123:689-699.
- Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn’s disease associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567-6575.
- Foulon W, Naessens A, Lauwers S, et al. Pelvic inflammatory disease due to Pseudomonas fluorescens in patient wearing an intrauterine device. Lancet. 1981;2:358-359.
- Burgos F, Torres A, González J, et al. Bacterial colonization as a potential source of nosocomial respiratory infections in 2 types of spirometer. Eur Respir J. 1996;9:2612-2617.
To the Editor:
A 50-year-old female farmer with diabetes mellitus, paroxysmal atrial fibrillation, and treatment-refractory systemic lupus erythematosus presented with worsening erythema, ecchymoses, edema, and tenderness in the bilateral legs of 3 weeks’ duration. The patient was taking oral methylprednisolone 12 mg daily (8 mg in the morning, 4 mg in the evening) for systemic lupus erythematosus. She previously was treated with mycophenolate mofetil, mycophenolic acid, methotrexate, azathioprine, hydroxychloroquine, etanercept, and cyclosporine without success. Cyclophosphamide was helpful in the past, but the last dose was more than 1 year prior to the current presentation. Physical examination showed no fever and 1+ pitting edema to the mid shin. Multiple warm, tender, erythematous to gray plaques were present on the bilateral lower extremities, and a 2-cm ulcerated plaque with a violaceous border was present on the medial surface of the lower left leg (Figure 1). The surrounding erythematous tissue was markedly tender to palpation. No popliteal or inguinal lymphadenopathy was appreciated.
Punch biopsies were obtained from the periphery and center of the ulcerated plaque on the left leg. Histopathologic analysis revealed an ulcerated necrotic epidermis with scant diffuse acute and chronic inflammation (Figure 2A). Leukocytoclastic vasculitis was present at the periphery of the lesion (Figure 2B). Colloidal iron stain revealed a marked increase in dermal mucin. Gram stain showed both gram-positive and gram-negative organisms (Figure 2C). Fungal and hyphal elements were seen in the superficial epidermis. Tissue cultures revealed a predominance of Pseudomonas fluorescens, along with Candida albicans, Klebsiella oxytoca, and Staphylococcus and Enterococcus species. Bacterial and fungal blood cultures were negative.
The patient was treated with ciprofloxacin, vancomycin, and voriconazole based on culture sensitivities. Although double coverage often is recommended for pseudomonal infections,1 the patient could not be started on a second antipseudomonal agent due to multiple severe antibiotic allergies. She continued home administration of methylprednisolone in the setting of active lupus; additional immunosuppression was avoided. Over the course of 1 week, the patient’s preexisting ulcerated plaque on the medial surface of the lower left leg gradually improved, and no new lesions developed. Ciprofloxacin, vancomycin, and voriconazole were continued along with insulin, aspirin, warfarin, metoprolol, furosemide, and bumetanide at discharge. The patient subsequently was readmitted to the hospital several more times over the next 4 months for multiple bacterial infections and ultimately died of overwhelming septic shock several months later.
Ecthyma gangrenosum (EG) is a rare cutaneous infection that results from either direct inoculation or hematogenous dissemination. It classically is caused by infection with Pseudomonas aeruginosa in immunocompromised or neutropenic patients. However, other bacteria and fungi, mucormycosis, and herpes simplex virus also have been reported to cause EG.1 Skin lesions often start as erythematous or purpuric macules, develop into vesicles and bullae, and eventually become necrotic ulcers with central eschars.2 Histopathologic findings reveal necrotizing hemorrhagic vasculitis; gram-negative rods often are found in the medial and adventitial walls of deeper vessels.3,4 The case mortality rate is high, ranging from 15% in nonbacteremic patients to 38% to 96% in patients with bacteremia.3
The leukocytoclastic vasculitis seen on biopsy in our patient was a reaction pattern, likely a direct result of the soft tissue infection. Biopsy showed hyphal or pseudohyphal elements in the superficial epidermis, corresponding to the positive C albicans growth on fungal culture. Candida albicans has been reported to cause lesions that mimic bacterial EG.1 However, the marked predominance of P fluorescens on biopsy and culture suggests that the Candida likely were opportunistic and managed to invade secondary to the vascular damage caused by P fluorescens.
Pseudomonas fluorescens is an aerobic gram-negative rod-shaped bacterium found in soil that rarely is implicated in human disease. This bacterium is unable to ferment lactose and grows best on MacConkey agar between 30°C and 37°C but also can grow at temperatures as low as 4°C.5 The ability of P fluorescens to rapidly proliferate at low temperatures (ie, in refrigerated blood products, saline solutions, water dispensers, ice baths, humidifier water) is thought to explain a number of reported clinical consequences, ranging from asymptomatic colonization to fatal bacteremia.6-10 This opportunistic pathogen also has been linked to Crohn disease and has been reported to cause pelvic inflammatory disease with the use of intrauterine contraception devices and nosocomial respiratory tract infections due to contaminated spirometers.11-14 In our case, the patient was part of a family of farmers and worked in an agricultural setting. She often handled the produce and worked at the family’s produce stand at the local farmer’s market. Her exposure to soil and soil pathogens may have been the source of the P fluorescens infection.
This case introduces P fluorescens as a causative agent of EG, suggests that exposure to agricultural products may predispose an immunosuppressed patient to this type of infection, and emphasizes the importance of timely diagnosis through tissue culture and histopathology so that immunosuppressive medications can be withheld and appropriate antibiotics can be initiated.
To the Editor:
A 50-year-old female farmer with diabetes mellitus, paroxysmal atrial fibrillation, and treatment-refractory systemic lupus erythematosus presented with worsening erythema, ecchymoses, edema, and tenderness in the bilateral legs of 3 weeks’ duration. The patient was taking oral methylprednisolone 12 mg daily (8 mg in the morning, 4 mg in the evening) for systemic lupus erythematosus. She previously was treated with mycophenolate mofetil, mycophenolic acid, methotrexate, azathioprine, hydroxychloroquine, etanercept, and cyclosporine without success. Cyclophosphamide was helpful in the past, but the last dose was more than 1 year prior to the current presentation. Physical examination showed no fever and 1+ pitting edema to the mid shin. Multiple warm, tender, erythematous to gray plaques were present on the bilateral lower extremities, and a 2-cm ulcerated plaque with a violaceous border was present on the medial surface of the lower left leg (Figure 1). The surrounding erythematous tissue was markedly tender to palpation. No popliteal or inguinal lymphadenopathy was appreciated.
Punch biopsies were obtained from the periphery and center of the ulcerated plaque on the left leg. Histopathologic analysis revealed an ulcerated necrotic epidermis with scant diffuse acute and chronic inflammation (Figure 2A). Leukocytoclastic vasculitis was present at the periphery of the lesion (Figure 2B). Colloidal iron stain revealed a marked increase in dermal mucin. Gram stain showed both gram-positive and gram-negative organisms (Figure 2C). Fungal and hyphal elements were seen in the superficial epidermis. Tissue cultures revealed a predominance of Pseudomonas fluorescens, along with Candida albicans, Klebsiella oxytoca, and Staphylococcus and Enterococcus species. Bacterial and fungal blood cultures were negative.
The patient was treated with ciprofloxacin, vancomycin, and voriconazole based on culture sensitivities. Although double coverage often is recommended for pseudomonal infections,1 the patient could not be started on a second antipseudomonal agent due to multiple severe antibiotic allergies. She continued home administration of methylprednisolone in the setting of active lupus; additional immunosuppression was avoided. Over the course of 1 week, the patient’s preexisting ulcerated plaque on the medial surface of the lower left leg gradually improved, and no new lesions developed. Ciprofloxacin, vancomycin, and voriconazole were continued along with insulin, aspirin, warfarin, metoprolol, furosemide, and bumetanide at discharge. The patient subsequently was readmitted to the hospital several more times over the next 4 months for multiple bacterial infections and ultimately died of overwhelming septic shock several months later.
Ecthyma gangrenosum (EG) is a rare cutaneous infection that results from either direct inoculation or hematogenous dissemination. It classically is caused by infection with Pseudomonas aeruginosa in immunocompromised or neutropenic patients. However, other bacteria and fungi, mucormycosis, and herpes simplex virus also have been reported to cause EG.1 Skin lesions often start as erythematous or purpuric macules, develop into vesicles and bullae, and eventually become necrotic ulcers with central eschars.2 Histopathologic findings reveal necrotizing hemorrhagic vasculitis; gram-negative rods often are found in the medial and adventitial walls of deeper vessels.3,4 The case mortality rate is high, ranging from 15% in nonbacteremic patients to 38% to 96% in patients with bacteremia.3
The leukocytoclastic vasculitis seen on biopsy in our patient was a reaction pattern, likely a direct result of the soft tissue infection. Biopsy showed hyphal or pseudohyphal elements in the superficial epidermis, corresponding to the positive C albicans growth on fungal culture. Candida albicans has been reported to cause lesions that mimic bacterial EG.1 However, the marked predominance of P fluorescens on biopsy and culture suggests that the Candida likely were opportunistic and managed to invade secondary to the vascular damage caused by P fluorescens.
Pseudomonas fluorescens is an aerobic gram-negative rod-shaped bacterium found in soil that rarely is implicated in human disease. This bacterium is unable to ferment lactose and grows best on MacConkey agar between 30°C and 37°C but also can grow at temperatures as low as 4°C.5 The ability of P fluorescens to rapidly proliferate at low temperatures (ie, in refrigerated blood products, saline solutions, water dispensers, ice baths, humidifier water) is thought to explain a number of reported clinical consequences, ranging from asymptomatic colonization to fatal bacteremia.6-10 This opportunistic pathogen also has been linked to Crohn disease and has been reported to cause pelvic inflammatory disease with the use of intrauterine contraception devices and nosocomial respiratory tract infections due to contaminated spirometers.11-14 In our case, the patient was part of a family of farmers and worked in an agricultural setting. She often handled the produce and worked at the family’s produce stand at the local farmer’s market. Her exposure to soil and soil pathogens may have been the source of the P fluorescens infection.
This case introduces P fluorescens as a causative agent of EG, suggests that exposure to agricultural products may predispose an immunosuppressed patient to this type of infection, and emphasizes the importance of timely diagnosis through tissue culture and histopathology so that immunosuppressive medications can be withheld and appropriate antibiotics can be initiated.
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl):S114-S117.
- Güçlüer H, Ergun T, Demirçay Z. Ecthyma gangrenosum. Int J Dermatol. 1999;38:299-302.
- Solowski NL, Yao FB, Agarwal A, et al. Ecthyma gangrenosum: a rare cutaneous manifestation of a potentially fatal disease. Ann Otol Rhinol Laryngol. 2004;113:462-464.
- Lobo I, Pinto A, Ferreira M, et al. Non-pseudomonal ecthyma gangrenosum present in diclofenac-induced agranulocytosis. Eur J Dermatol. 2008;18:350-551.
- Pappas G, Karavasilis V, Christou L, et al. Pseudomonas fluorescens infections in clinical practice. Scand J Infect Dis. 2006;38:68-70.
- Gershman MD, Kennedy DJ, Noble-Wang J, et al. Multistate outbreak of Pseudomonas fluorescens bloodstream infection after exposure to contaminated heparinized saline flush prepared by a compounding pharmacy. Clin Infect Dis. 2008;47:1372-1378.
- Hsueh P, Teng L, Pan H, et al. Outbreak of Pseudomonas fluorescens bacteremia among oncology patients. J Clin Microbiol. 1998;36:2914-2917.
- Wong V, Levi K, Baddal B, et al. Spread of Pseudomonas fluorescens due to contaminated drinking water in a bone marrow transplant unit. J Clin Microbiol. 2011;49:2093-2096.
- Benito N, Mirelis B, Galvez ML, et al. Outbreak of Pseudomonas fluorescens bloodstream infection in a coronary care unit. J Hosp Infect. 2012;82:286-289.
- Redding PJ, McWalter PW. Pseudomonas fluorescens cross-infection due to contaminated humidifier water. Br Med J. 1980;281:275.
- Landers CJ, Cohavy O, Misra R, et al. Selected loss of tolerance evidenced by Crohn’s disease-associated immune responses to auto- and microbial antigens. Gastroenterology. 2002;123:689-699.
- Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn’s disease associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567-6575.
- Foulon W, Naessens A, Lauwers S, et al. Pelvic inflammatory disease due to Pseudomonas fluorescens in patient wearing an intrauterine device. Lancet. 1981;2:358-359.
- Burgos F, Torres A, González J, et al. Bacterial colonization as a potential source of nosocomial respiratory infections in 2 types of spirometer. Eur Respir J. 1996;9:2612-2617.
- Reich HL, Williams Fadeyi D, Naik NS, et al. Nonpseudomonal ecthyma gangrenosum. J Am Acad Dermatol. 2004;50(5 suppl):S114-S117.
- Güçlüer H, Ergun T, Demirçay Z. Ecthyma gangrenosum. Int J Dermatol. 1999;38:299-302.
- Solowski NL, Yao FB, Agarwal A, et al. Ecthyma gangrenosum: a rare cutaneous manifestation of a potentially fatal disease. Ann Otol Rhinol Laryngol. 2004;113:462-464.
- Lobo I, Pinto A, Ferreira M, et al. Non-pseudomonal ecthyma gangrenosum present in diclofenac-induced agranulocytosis. Eur J Dermatol. 2008;18:350-551.
- Pappas G, Karavasilis V, Christou L, et al. Pseudomonas fluorescens infections in clinical practice. Scand J Infect Dis. 2006;38:68-70.
- Gershman MD, Kennedy DJ, Noble-Wang J, et al. Multistate outbreak of Pseudomonas fluorescens bloodstream infection after exposure to contaminated heparinized saline flush prepared by a compounding pharmacy. Clin Infect Dis. 2008;47:1372-1378.
- Hsueh P, Teng L, Pan H, et al. Outbreak of Pseudomonas fluorescens bacteremia among oncology patients. J Clin Microbiol. 1998;36:2914-2917.
- Wong V, Levi K, Baddal B, et al. Spread of Pseudomonas fluorescens due to contaminated drinking water in a bone marrow transplant unit. J Clin Microbiol. 2011;49:2093-2096.
- Benito N, Mirelis B, Galvez ML, et al. Outbreak of Pseudomonas fluorescens bloodstream infection in a coronary care unit. J Hosp Infect. 2012;82:286-289.
- Redding PJ, McWalter PW. Pseudomonas fluorescens cross-infection due to contaminated humidifier water. Br Med J. 1980;281:275.
- Landers CJ, Cohavy O, Misra R, et al. Selected loss of tolerance evidenced by Crohn’s disease-associated immune responses to auto- and microbial antigens. Gastroenterology. 2002;123:689-699.
- Wei B, Huang T, Dalwadi H, et al. Pseudomonas fluorescens encodes the Crohn’s disease associated I2 sequence and T-cell superantigen. Infect Immun. 2002;70:6567-6575.
- Foulon W, Naessens A, Lauwers S, et al. Pelvic inflammatory disease due to Pseudomonas fluorescens in patient wearing an intrauterine device. Lancet. 1981;2:358-359.
- Burgos F, Torres A, González J, et al. Bacterial colonization as a potential source of nosocomial respiratory infections in 2 types of spirometer. Eur Respir J. 1996;9:2612-2617.
Practice Points
- Immunocompromised patients with a high exposure to agricultural products may be at increased risk for systemic infection by Pseudomonas fluorescens.
- Pseudomonas fluorescens is an opportunistic pathogen that can cause ecthyma gangrenosum, which necessitates rapid diagnosis and treatment to prevent mortality.
Chilblain Lupus Erythematosus Presenting With Bilateral Hemorrhagic Bullae of Distal Halluces
To the Editor:
A 20-year-old man with no notable medical history presented to our dermatology clinic for evaluation of mildly painful, hemorrhagic bullae on the bilateral halluces of 1 month’s duration. On initial presentation the patient reported the lesions developed after wearing a new pair of tight-fitting shoes, suggesting a diagnosis of trauma-induced bullae. The patient was instructed to wear loose-fitting shoes and to follow up in 6 weeks to assess for improvement. At follow-up the bullae had resolved with residual violaceous patches on the bilateral distal halluces. He additionally developed a faint retiform erythematous patch on the left distal toe (Figure 1). The patient also had reticulate erythematous patches on the dorsal aspects of the hands extending to the forearms and legs resembling livedo reticularis. The patient was unsure if the skin lesions were triggered or worsened by cold exposure and reported that he smoked half a pack of cigarettes daily. At this time, the differential diagnosis still included trauma; however, there was concern for either embolic, thrombotic, or connective-tissue disease. A 4-mm punch biopsy of the left distal hallux demonstrated basal vacuolar interface dermatitis with superficial and deep perivascular inflammation and deep periadnexal mucin deposition (Figure 2) consistent with lupus dermatitis.

(H&E, original magnification ×400) with deep periadnexal mucin deposition (B)(colloidal iron, original magnification ×40).")
Serologic workup revealed increased antinuclear antibody titers of 1:320 (reference range, <1:40) and anti-Ro/Sjögren syndrome antigen antibodies of 86 (reference range, <20). There was no elevation in anti–double-stranded DNA, anti-Smith, antiribonucleoprotein, or anticardiolipin antibodies. Complement levels also were within reference range. Furthermore, the patient denied a history of Raynaud phenomenon, photosensitivity, oral ulcers, joint pain, shortness of breath, pleuritic chest pain, arthritis, blood clots, or any other systemic symptoms. Additional evaluation by the rheumatology department did not support criteria for systemic lupus erythematosus (SLE). In the context of the clinical presentation, histologic findings, and serologic markers, a diagnosis of chilblain lupus erythematosus (CHLE) was made. He was counseled on sun protection and smoking cessation and declined systemic therapy citing concern for side effects. Follow-up with the dermatology and rheumatology departments was advised.
Cutaneous lupus erythematosus (CLE) comprises various forms of lupus, including acute cutaneous lupus, subacute cutaneous lupus, and chronic cutaneous lupus. Chilblain lupus erythematosus is a rare subset of chronic CLE that first was described in 18881 and is characterized by tender violaceous papules and plaques that typically present in an acral distribution (ie, fingers, toes, nose, cheeks, ears). The skin lesions often are triggered or exacerbated by cold temperatures and dampness. As the lesions evolve, they can ulcerate, fissure, become hyperkeratotic, or result in atrophic plaques with scarring.2,3 A subset of patients also may have concurrent Raynaud phenomenon.1 Up to 20% of patients will eventually develop SLE, especially those patients with concurrent discoid lupus erythematosus, warranting close long-term follow-up.3 Serologic studies can reveal antinuclear antibodies, anti-Ro/Sjögren syndrome antigen antibodies, rheumatic factor, and anti–double-stranded DNA antibodies.1,4 Hypergammaglobulinemia also is a common finding in patients with CHLE, affecting more than two-thirds of patients.1 Typical features of CHLE seen on histopathology include interface dermatitis, perivascular lymphocytic infiltrate, apoptotic keratinocytes, lichenoid tissue reaction, and increased dermal mucin.1,4
Chilblain lupus erythematosus most commonly presents sporadically; however, there is a familial form that has been previously described.5 Sporadic CHLE usually occurs in middle-aged females, in contrast to familial CHLE, which presents in early childhood.1 The pathogenesis of the sporadic form is poorly understood, but it is thought to be stimulated by vasoconstriction or microvascular injury provoked by cold exposure. Furthermore, hypergammaglobulinemia and the presence of autoantibodies may contribute to the pathogenesis by increasing blood viscosity.1 The
Several drugs including thiazides, terbinafine, calcium channel blockers, angiotensin-converting enzyme inhibitors, and chemotherapeutic agents have been reported to trigger CHLE.4 Tumor necrosis factor α inhibitors have been shown to precipitate CHLE.6 Of note, drug-induced CHLE usually is limited to the skin and has not been shown to progress to SLE.6 Lebeau et al4 described a patient with breast cancer and preexisting CHLE that flared while the patient received docetaxel therapy, suggesting that certain drugs may not only induce but also may aggravate CHLE.
Many of the therapies that are effective in SLE such as antimalarial agents (ie, chloroquine, hydroxychloroquine) often are less efficacious in treating the lesions of CHLE.1 However, these patients often can be managed successfully by physical protection from the cold environment.1 Calcium channel blockers such as nifedipine also have been implicated, as they counteract vasoconstriction, which is thought to contribute to the pathogenesis of CHLE.1 Topical and systemic steroids also have been used to treat CHLE. Dapsone and pentoxifylline are other treatment modalities that have been effective in select cases of CHLE.5 Boehm and Bieber7 reported near resolution of CHLE with mycophenolate mofetil in an elderly woman with skin lesions that had been refractory to systemic steroids, antimalarial agents, azathioprine, dapsone, and pentoxifylline, suggesting that mycophenolate mofetil may be a therapeutic option for recalcitrant cases of CHLE. Local immunosuppressive agents such as tacrolimus also can be considered in treatment-refractory disease.
Chilblain lupus erythematosus is a rare chronic form of CLE that typically occurs sporadically but also has a familial form that has been described in several families. It most commonly is observed in middle-aged women, but we describe a case in a young man. Although CHLE typically does not respond well to traditional lupus therapies used in the management of SLE, good effects have been observed with cold avoidance, calcium channel blockers, and topical or oral steroids. For treatment-refractory cases, mycophenolate mofetil and other immunosuppressive agents have been shown to be effective.
- Hedrich CM, Fiebig B, Hauck FH, et al. Chilblain lupus erythematosus—a review of literature. Clin Rheumatol. 2008;27:949-954.
- Kuhn A, Lehmann P, Ruzicka T, eds. Cutaneous Lupus Erythematosus. Berlin, Germany: Springer; 2005.
- Obermoser G, Sontheimer RD, Zelger B. Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus. 2010;19:1050-1070.
- Lebeau S, També S, Sallam MA, et al. Docetaxel-induced relapse of subacute cutaneous lupus erythematosus and chilblain lupus. J Dtsch Dermatol Ges. 2013;11:871-874.
- Günther C, Hillebrand M, Brunk J, et al. Systemic involvement in TREX1-associated familial chilblain lupus. J Am Acad Dermatol. 2013;69:179-181.
- Sifuentes Giraldo WA, Ahijón Lana M, García Villanueva MJ, et al. Chilblain lupus induced by TNF-α antagonists: a case report and literature review. Clin Rheumatol. 2012;31:563-568.
- Boehm I, Bieber T. Chilblain lupus erythematosus Hutchinson: successful treatment with mycophenolate mofetil. Arch Dermatol. 2001;137:235-236.
To the Editor:
A 20-year-old man with no notable medical history presented to our dermatology clinic for evaluation of mildly painful, hemorrhagic bullae on the bilateral halluces of 1 month’s duration. On initial presentation the patient reported the lesions developed after wearing a new pair of tight-fitting shoes, suggesting a diagnosis of trauma-induced bullae. The patient was instructed to wear loose-fitting shoes and to follow up in 6 weeks to assess for improvement. At follow-up the bullae had resolved with residual violaceous patches on the bilateral distal halluces. He additionally developed a faint retiform erythematous patch on the left distal toe (Figure 1). The patient also had reticulate erythematous patches on the dorsal aspects of the hands extending to the forearms and legs resembling livedo reticularis. The patient was unsure if the skin lesions were triggered or worsened by cold exposure and reported that he smoked half a pack of cigarettes daily. At this time, the differential diagnosis still included trauma; however, there was concern for either embolic, thrombotic, or connective-tissue disease. A 4-mm punch biopsy of the left distal hallux demonstrated basal vacuolar interface dermatitis with superficial and deep perivascular inflammation and deep periadnexal mucin deposition (Figure 2) consistent with lupus dermatitis.
Serologic workup revealed increased antinuclear antibody titers of 1:320 (reference range, <1:40) and anti-Ro/Sjögren syndrome antigen antibodies of 86 (reference range, <20). There was no elevation in anti–double-stranded DNA, anti-Smith, antiribonucleoprotein, or anticardiolipin antibodies. Complement levels also were within reference range. Furthermore, the patient denied a history of Raynaud phenomenon, photosensitivity, oral ulcers, joint pain, shortness of breath, pleuritic chest pain, arthritis, blood clots, or any other systemic symptoms. Additional evaluation by the rheumatology department did not support criteria for systemic lupus erythematosus (SLE). In the context of the clinical presentation, histologic findings, and serologic markers, a diagnosis of chilblain lupus erythematosus (CHLE) was made. He was counseled on sun protection and smoking cessation and declined systemic therapy citing concern for side effects. Follow-up with the dermatology and rheumatology departments was advised.
Cutaneous lupus erythematosus (CLE) comprises various forms of lupus, including acute cutaneous lupus, subacute cutaneous lupus, and chronic cutaneous lupus. Chilblain lupus erythematosus is a rare subset of chronic CLE that first was described in 18881 and is characterized by tender violaceous papules and plaques that typically present in an acral distribution (ie, fingers, toes, nose, cheeks, ears). The skin lesions often are triggered or exacerbated by cold temperatures and dampness. As the lesions evolve, they can ulcerate, fissure, become hyperkeratotic, or result in atrophic plaques with scarring.2,3 A subset of patients also may have concurrent Raynaud phenomenon.1 Up to 20% of patients will eventually develop SLE, especially those patients with concurrent discoid lupus erythematosus, warranting close long-term follow-up.3 Serologic studies can reveal antinuclear antibodies, anti-Ro/Sjögren syndrome antigen antibodies, rheumatic factor, and anti–double-stranded DNA antibodies.1,4 Hypergammaglobulinemia also is a common finding in patients with CHLE, affecting more than two-thirds of patients.1 Typical features of CHLE seen on histopathology include interface dermatitis, perivascular lymphocytic infiltrate, apoptotic keratinocytes, lichenoid tissue reaction, and increased dermal mucin.1,4
Chilblain lupus erythematosus most commonly presents sporadically; however, there is a familial form that has been previously described.5 Sporadic CHLE usually occurs in middle-aged females, in contrast to familial CHLE, which presents in early childhood.1 The pathogenesis of the sporadic form is poorly understood, but it is thought to be stimulated by vasoconstriction or microvascular injury provoked by cold exposure. Furthermore, hypergammaglobulinemia and the presence of autoantibodies may contribute to the pathogenesis by increasing blood viscosity.1 The
Several drugs including thiazides, terbinafine, calcium channel blockers, angiotensin-converting enzyme inhibitors, and chemotherapeutic agents have been reported to trigger CHLE.4 Tumor necrosis factor α inhibitors have been shown to precipitate CHLE.6 Of note, drug-induced CHLE usually is limited to the skin and has not been shown to progress to SLE.6 Lebeau et al4 described a patient with breast cancer and preexisting CHLE that flared while the patient received docetaxel therapy, suggesting that certain drugs may not only induce but also may aggravate CHLE.
Many of the therapies that are effective in SLE such as antimalarial agents (ie, chloroquine, hydroxychloroquine) often are less efficacious in treating the lesions of CHLE.1 However, these patients often can be managed successfully by physical protection from the cold environment.1 Calcium channel blockers such as nifedipine also have been implicated, as they counteract vasoconstriction, which is thought to contribute to the pathogenesis of CHLE.1 Topical and systemic steroids also have been used to treat CHLE. Dapsone and pentoxifylline are other treatment modalities that have been effective in select cases of CHLE.5 Boehm and Bieber7 reported near resolution of CHLE with mycophenolate mofetil in an elderly woman with skin lesions that had been refractory to systemic steroids, antimalarial agents, azathioprine, dapsone, and pentoxifylline, suggesting that mycophenolate mofetil may be a therapeutic option for recalcitrant cases of CHLE. Local immunosuppressive agents such as tacrolimus also can be considered in treatment-refractory disease.
Chilblain lupus erythematosus is a rare chronic form of CLE that typically occurs sporadically but also has a familial form that has been described in several families. It most commonly is observed in middle-aged women, but we describe a case in a young man. Although CHLE typically does not respond well to traditional lupus therapies used in the management of SLE, good effects have been observed with cold avoidance, calcium channel blockers, and topical or oral steroids. For treatment-refractory cases, mycophenolate mofetil and other immunosuppressive agents have been shown to be effective.
To the Editor:
A 20-year-old man with no notable medical history presented to our dermatology clinic for evaluation of mildly painful, hemorrhagic bullae on the bilateral halluces of 1 month’s duration. On initial presentation the patient reported the lesions developed after wearing a new pair of tight-fitting shoes, suggesting a diagnosis of trauma-induced bullae. The patient was instructed to wear loose-fitting shoes and to follow up in 6 weeks to assess for improvement. At follow-up the bullae had resolved with residual violaceous patches on the bilateral distal halluces. He additionally developed a faint retiform erythematous patch on the left distal toe (Figure 1). The patient also had reticulate erythematous patches on the dorsal aspects of the hands extending to the forearms and legs resembling livedo reticularis. The patient was unsure if the skin lesions were triggered or worsened by cold exposure and reported that he smoked half a pack of cigarettes daily. At this time, the differential diagnosis still included trauma; however, there was concern for either embolic, thrombotic, or connective-tissue disease. A 4-mm punch biopsy of the left distal hallux demonstrated basal vacuolar interface dermatitis with superficial and deep perivascular inflammation and deep periadnexal mucin deposition (Figure 2) consistent with lupus dermatitis.
Serologic workup revealed increased antinuclear antibody titers of 1:320 (reference range, <1:40) and anti-Ro/Sjögren syndrome antigen antibodies of 86 (reference range, <20). There was no elevation in anti–double-stranded DNA, anti-Smith, antiribonucleoprotein, or anticardiolipin antibodies. Complement levels also were within reference range. Furthermore, the patient denied a history of Raynaud phenomenon, photosensitivity, oral ulcers, joint pain, shortness of breath, pleuritic chest pain, arthritis, blood clots, or any other systemic symptoms. Additional evaluation by the rheumatology department did not support criteria for systemic lupus erythematosus (SLE). In the context of the clinical presentation, histologic findings, and serologic markers, a diagnosis of chilblain lupus erythematosus (CHLE) was made. He was counseled on sun protection and smoking cessation and declined systemic therapy citing concern for side effects. Follow-up with the dermatology and rheumatology departments was advised.
Cutaneous lupus erythematosus (CLE) comprises various forms of lupus, including acute cutaneous lupus, subacute cutaneous lupus, and chronic cutaneous lupus. Chilblain lupus erythematosus is a rare subset of chronic CLE that first was described in 18881 and is characterized by tender violaceous papules and plaques that typically present in an acral distribution (ie, fingers, toes, nose, cheeks, ears). The skin lesions often are triggered or exacerbated by cold temperatures and dampness. As the lesions evolve, they can ulcerate, fissure, become hyperkeratotic, or result in atrophic plaques with scarring.2,3 A subset of patients also may have concurrent Raynaud phenomenon.1 Up to 20% of patients will eventually develop SLE, especially those patients with concurrent discoid lupus erythematosus, warranting close long-term follow-up.3 Serologic studies can reveal antinuclear antibodies, anti-Ro/Sjögren syndrome antigen antibodies, rheumatic factor, and anti–double-stranded DNA antibodies.1,4 Hypergammaglobulinemia also is a common finding in patients with CHLE, affecting more than two-thirds of patients.1 Typical features of CHLE seen on histopathology include interface dermatitis, perivascular lymphocytic infiltrate, apoptotic keratinocytes, lichenoid tissue reaction, and increased dermal mucin.1,4
Chilblain lupus erythematosus most commonly presents sporadically; however, there is a familial form that has been previously described.5 Sporadic CHLE usually occurs in middle-aged females, in contrast to familial CHLE, which presents in early childhood.1 The pathogenesis of the sporadic form is poorly understood, but it is thought to be stimulated by vasoconstriction or microvascular injury provoked by cold exposure. Furthermore, hypergammaglobulinemia and the presence of autoantibodies may contribute to the pathogenesis by increasing blood viscosity.1 The
Several drugs including thiazides, terbinafine, calcium channel blockers, angiotensin-converting enzyme inhibitors, and chemotherapeutic agents have been reported to trigger CHLE.4 Tumor necrosis factor α inhibitors have been shown to precipitate CHLE.6 Of note, drug-induced CHLE usually is limited to the skin and has not been shown to progress to SLE.6 Lebeau et al4 described a patient with breast cancer and preexisting CHLE that flared while the patient received docetaxel therapy, suggesting that certain drugs may not only induce but also may aggravate CHLE.
Many of the therapies that are effective in SLE such as antimalarial agents (ie, chloroquine, hydroxychloroquine) often are less efficacious in treating the lesions of CHLE.1 However, these patients often can be managed successfully by physical protection from the cold environment.1 Calcium channel blockers such as nifedipine also have been implicated, as they counteract vasoconstriction, which is thought to contribute to the pathogenesis of CHLE.1 Topical and systemic steroids also have been used to treat CHLE. Dapsone and pentoxifylline are other treatment modalities that have been effective in select cases of CHLE.5 Boehm and Bieber7 reported near resolution of CHLE with mycophenolate mofetil in an elderly woman with skin lesions that had been refractory to systemic steroids, antimalarial agents, azathioprine, dapsone, and pentoxifylline, suggesting that mycophenolate mofetil may be a therapeutic option for recalcitrant cases of CHLE. Local immunosuppressive agents such as tacrolimus also can be considered in treatment-refractory disease.
Chilblain lupus erythematosus is a rare chronic form of CLE that typically occurs sporadically but also has a familial form that has been described in several families. It most commonly is observed in middle-aged women, but we describe a case in a young man. Although CHLE typically does not respond well to traditional lupus therapies used in the management of SLE, good effects have been observed with cold avoidance, calcium channel blockers, and topical or oral steroids. For treatment-refractory cases, mycophenolate mofetil and other immunosuppressive agents have been shown to be effective.
- Hedrich CM, Fiebig B, Hauck FH, et al. Chilblain lupus erythematosus—a review of literature. Clin Rheumatol. 2008;27:949-954.
- Kuhn A, Lehmann P, Ruzicka T, eds. Cutaneous Lupus Erythematosus. Berlin, Germany: Springer; 2005.
- Obermoser G, Sontheimer RD, Zelger B. Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus. 2010;19:1050-1070.
- Lebeau S, També S, Sallam MA, et al. Docetaxel-induced relapse of subacute cutaneous lupus erythematosus and chilblain lupus. J Dtsch Dermatol Ges. 2013;11:871-874.
- Günther C, Hillebrand M, Brunk J, et al. Systemic involvement in TREX1-associated familial chilblain lupus. J Am Acad Dermatol. 2013;69:179-181.
- Sifuentes Giraldo WA, Ahijón Lana M, García Villanueva MJ, et al. Chilblain lupus induced by TNF-α antagonists: a case report and literature review. Clin Rheumatol. 2012;31:563-568.
- Boehm I, Bieber T. Chilblain lupus erythematosus Hutchinson: successful treatment with mycophenolate mofetil. Arch Dermatol. 2001;137:235-236.
- Hedrich CM, Fiebig B, Hauck FH, et al. Chilblain lupus erythematosus—a review of literature. Clin Rheumatol. 2008;27:949-954.
- Kuhn A, Lehmann P, Ruzicka T, eds. Cutaneous Lupus Erythematosus. Berlin, Germany: Springer; 2005.
- Obermoser G, Sontheimer RD, Zelger B. Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus. 2010;19:1050-1070.
- Lebeau S, També S, Sallam MA, et al. Docetaxel-induced relapse of subacute cutaneous lupus erythematosus and chilblain lupus. J Dtsch Dermatol Ges. 2013;11:871-874.
- Günther C, Hillebrand M, Brunk J, et al. Systemic involvement in TREX1-associated familial chilblain lupus. J Am Acad Dermatol. 2013;69:179-181.
- Sifuentes Giraldo WA, Ahijón Lana M, García Villanueva MJ, et al. Chilblain lupus induced by TNF-α antagonists: a case report and literature review. Clin Rheumatol. 2012;31:563-568.
- Boehm I, Bieber T. Chilblain lupus erythematosus Hutchinson: successful treatment with mycophenolate mofetil. Arch Dermatol. 2001;137:235-236.
Practice Points
- Up to 20% of patients with chilblain lupus erythematosus (CHLE) will develop systemic lupus erythematosus (SLE), necessitating close long-term follow-up.
- Medications such as antihypertensives, antifungals, chemotherapeutic agents, and tumor necrosis factor 11α inhibitors have been reported to trigger CHLE.
- Chilblain lupus erythematosus is less responsive to traditional antimalarial agents commonly used to treat SLE.
- Management of CHLE includes physical protection from cold environments, calcium channel blockers, topical and systemic steroids, and pentoxifylline, among other treatment modalities.
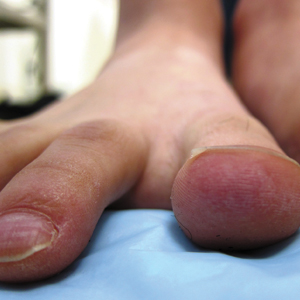
Recovery of Hair in the Psoriatic Plaques of a Patient With Coexistent Alopecia Universalis
To the Editor:
Both alopecia areata (AA) and psoriasis vulgaris are chronic relapsing autoimmune diseases, with AA causing nonscarring hair loss in approximately 0.1% to 0.2%1 of the population with a lifetime risk of 1.7%,2 and psoriasis more broadly impacting 1.5% to 2% of the population.3 The helper T cell (TH1) cytokine milieu is pathogenic in both conditions.4-6 IFN-γ knockout mice, unlike their wild-type counterparts, do not exhibit AA.7 Psoriasis is notably improved by IL-10 injections, which dampen the TH1 response.8 Distinct from AA, TH17 and TH22 cells have been implicated as key players in psoriasis pathogenesis, along with the associated IL-17 and IL-22 cytokines.9-12
Few cases of patients with concurrent AA and psoriasis have been described. Interestingly, these cases document normal hair regrowth in the areas of psoriasis.13-16 These cases may offer unique insight into the immune factors driving each disease. We describe a case of a man with both alopecia universalis (AU) and psoriasis who developed hair regrowth in some of the psoriatic plaques.
A 34-year-old man with concurrent AU and psoriasis who had not used any systemic or topical medication for either condition in the last year presented to our clinic seeking treatment. The patient had a history of alopecia totalis as a toddler that completely resolved by 4 years of age with the use of squaric acid dibutylester (SADBE). At 31 years of age, the alopecia recurred and was localized to the scalp. It was partially responsive to intralesional triamcinolone acetonide. The patient’s alopecia worsened over the 2 years following recurrence, ultimately progressing to AU. Two months after the alopecia recurrence, he developed the first psoriatic plaques. As the plaque psoriasis progressed, systemic therapy was initiated, first methotrexate and then etanercept. Shortly after developing AU, he lost his health insurance and discontinued all therapy. The patient’s psoriasis began to recur approximately 3 months after stopping etanercept. He was not using any other psoriasis medications. At that time, he noted terminal hair regrowth within some of the psoriatic plaques. No terminal hairs grew outside of the psoriatic plaques, and all regions with growth had previously been without hair for an extended period of time. The patient presented to our clinic approximately 1 year later. He had no other medical conditions and no relevant family history.
On initial physical examination, he had nonscarring hair loss involving nearly 100% of the body with psoriatic plaques on approximately 30% of the body surface area. Regions of terminal hair growth were confined to some but not all of the psoriatic plaques (Figure). Interestingly, the terminal hairs were primarily localized to the thickest central regions of the plaques. The patient’s psoriasis was treated with a combination of topical clobetasol and calcipotriene. In addition, he was started on tacrolimus ointment to the face and eyebrows for the AA. Maintenance of terminal hair within a region of topically treated psoriasis on the forearm persisted at the 2-month follow-up despite complete clearance of the corresponding psoriatic plaque. A small psoriatic plaque on the scalp cleared early with topical therapy without noticeable hair regrowth. The patient subsequently was started on contact immunotherapy with SADBE and intralesional triamcinolone acetonide for the scalp alopecia without satisfactory response. He decided to discontinue further attempts at treating the alopecia and requested to be restarted on etanercept therapy for recalcitrant psoriatic plaques. His psoriasis responded well to this therapy and he continues to be followed in our psoriasis clinic. One year after clearance of the treated psoriatic plaques, the corresponding terminal hairs persist.
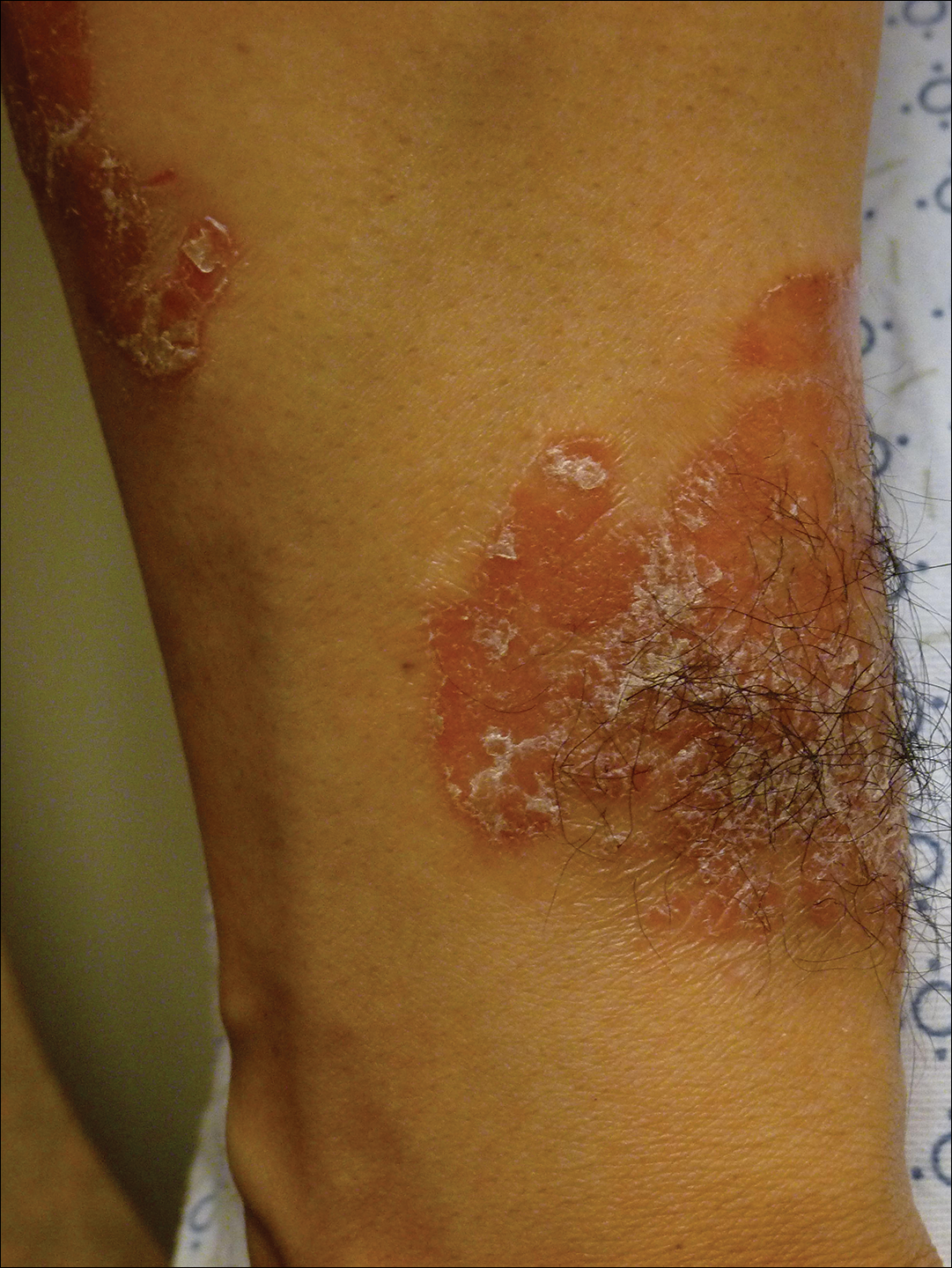
Contact immunotherapy, most commonly with diphenylcyclopropenone or SADBE, is reported to have a 50% to 60% success rate in extensive AA, with a broad range of 9% to 87%17; however, randomized controlled trials testing the efficacy of contact immunotherapy are lacking. Although the mechanism of action of these topical sensitizers is not clearly delineated, it has been postulated that by inducing a new type of inflammatory response in the region, the immunologic milieu is changed, allowing the hair to grow. Some proposed mechanisms include promoting perifollicular lymphocyte apoptosis, preventing new recruitment of autoreactive lymphocytes, and allowing for the correction of aberrant major histocompatibility complex expression on the hair matrix epithelium to regain follicle immune privilege.18-20
Iatrogenic immunotherapy may work analogously to the natural immune system deviation demonstrated in our patient. Psoriasis and AA are believed to form competing immune cells and cytokine milieus, thus explaining how an individual with AA could regain normal hair growth in areas of psoriasis.15,16 The Renbök phenomenon, or reverse Köbner phenomenon, coined by Happle et al13 can be used to describe both the iatrogenic and natural cases of dermatologic disease improvement in response to secondary insults.14
A complex cascade of immune cells and cytokines coordinate AA pathogenesis. In the acute stage of AA, an inflammatory infiltrate of CD4+ T cells, CD8+ T cells, and antigen-presenting cells target anagen phase follicles, with a higher CD4+:CD8+ ratio in clinically active disease.21-23 Subcutaneous injections of either CD4+ or CD8+ lymphocyte subsets from mice with AA into normal-haired mice induces disease. However, CD8+ T cell injections rapidly produce apparent hair loss, whereas CD4+ T cells cause hair loss after several weeks, suggesting that CD8+ T cells directly modulate AA hair loss and CD4+ T cells act as an aide.24 The growth, differentiation, and survival of CD8+ T cells are stimulated by IL-2 and IFN-γ. Alopecia areata biopsies demonstrate a prevalence of TH1 cytokines, and patients with localized AA, alopecia totalis, and AU have notably higher serum IFN-γ levels compared to controls.25 In murine models, IL-1α and IL-1β increase during the catagen phase of the hair cycle and peak during the telogen phase.26 Excessive IL-1β expression is detected in the early stages of human disease, and certain IL-1β polymorphisms are associated with severe forms of AA.26 The role of tumor necrosis factor (TNF) α in AA is not well understood. In vitro studies show it inhibits hair growth, suggesting the cytokine may play a role in AA.27 However, anti–TNF-α therapy is not effective in AA, and case reports propose these therapies rarely induce AA.28-31
The TH1 response is likewise critical to psoriatic plaque development. IFN-γ and TNF-α are overexpressed in psoriatic plaques.32 IFN-γ has an antiproliferative and differentiation-inducing effect on normal keratinocytes, but psoriatic epithelial cells in vitro respond differently to the cytokine with a notably diminished growth inhibition.33,34 One explanation for the role of IFN-γ is that it stimulates dendritic cells to produce IL-1 and IL-23.35 IL-23 activates TH17 cells36; TH1 and TH17 conditions produce IL-22 whose serum level correlates with disease severity.37-39 IL-22 induces keratinocyte proliferation and migration and inhibits keratinocyte differentiation, helping account for hallmarks of the disease.40 Patients with psoriasis have increased levels of TH1, TH17, and TH22 cells, as well as their associated cytokines, in the skin and blood compared to controls.4,11,32,39,41
Alopecia areata and psoriasis are regulated by complex and still not entirely understood immune interactions. The fact that many of the same therapies are used to treat both diseases emphasizes both their overlapping characteristics and the lack of targeted therapy. It is unclear if and how the topical or systemic therapies used in our patient to treat one disease affected the natural history of the other condition. It is important to highlight, however, that the patient had not been treated for months when he developed the psoriatic plaques with hair regrowth. Other case reports also document hair regrowth in untreated plaques,13,16 making it unlikely to be a side effect of the medication regimen. For both psoriasis and AA, the immune cell composition and cytokine levels in the skin or serum vary throughout a patient’s disease course depending on severity of disease or response to treatment.6,39,42,43 Therefore, we hypothesize that the 2 conditions interact in a similarly distinct manner based on each disease’s stage and intensity in the patient. Both our patient’s course thus far and the various presentations described by other groups support this hypothesis. Our patient had a small region of psoriasis on the scalp that cleared without any terminal hair growth. He also had larger plaques on the forearms that developed hair growth most predominantly within the thicker regions of the plaques. His unique presentation highlights the fluidity of the immune factors driving psoriasis vulgaris and AA.
- Safavi K. Prevalence of alopecia areata in the First National Health and Nutrition Examination Survey. Arch Dermatol. 1992;128:702.
- Safavi KH, Muller SA, Suman VJ, et al. Incidence of alopecia areata in Olmsted County, Minnesota, 1975 through 1989. Mayo Clin Proc. 1995;70:628-633.
- Wolff K, Johnson RA. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 6th ed. New York, NY: McGraw-Hill; 2009.
- Austin LM, Ozawa M, Kikuchi T, et al. The majority of epidermal T cells in psoriasis vulgaris lesions can produce type 1 cytokines, interferon-gamma, interleukin-2, and tumor necrosis factor-alpha, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: a type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J Invest Dermatol. 1999;113:752-759.
- Ghoreishi M, Martinka M, Dutz JP. Type 1 interferon signature in the scalp lesions of alopecia areata. Br J Dermatol. 2010;163:57-62.
- Rossi A, Cantisani C, Carlesimo M, et al. Serum concentrations of IL-2, IL-6, IL-12 and TNF-α in patients with alopecia areata. Int J Immunopathol Pharmacol. 2012;25:781-788.
- Freyschmidt-Paul P, McElwee KJ, Hoffmann R, et al. Interferon-gamma-deficient mice are resistant to the development of alopecia areata. Br J Dermatol. 2006;155:515-521.
- Reich K, Garbe C, Blaschke V, et al. Response of psoriasis to interleukin-10 is associated with suppression of cutaneous type 1 inflammation, downregulation of the epidermal interleukin-8/CXCR2 pathway and normalization of keratinocyte maturation. J Invest Dermatol. 2001;116:319-329.
- Teunissen MB, Koomen CW, de Waal Malefyt R, et al. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. 1998;111:645-649.
- Zheng Y, Danilenko DM, Valdez P, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648-651.
- Boniface K, Guignouard E, Pedretti N, et al. A role for T cell-derived interleukin 22 in psoriatic skin inflammation. Clin Exp Immunol. 2007;150:407-415.
- Zaba LC, Suárez-Fariñas M, Fuentes-Duculan J, et al. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J Allergy Clin Immunol. 2009;124:1022-1030.e395.
- Happle R, van der Steen PHM, Perret CM. The Renbök phenomenon: an inverse Köebner reaction observed in alopecia areata. Eur J Dermatol. 1991;2:39-40.
- Ito T, Hashizume H, Takigawa M. Contact immunotherapy-induced Renbök phenomenon in a patient with alopecia areata and psoriasis vulgaris. Eur J Dermatol. 2010;20:126-127.
- Criado PR, Valente NY, Michalany NS, et al. An unusual association between scalp psoriasis and ophiasic alopecia areata: the Renbök phenomenon. Clin Exp Dermatol. 2007;32:320-321.
- Harris JE, Seykora JT, Lee RA. Renbök phenomenon and contact sensitization in a patient with alopecia universalis. Arch Dermatol. 2010;146:422-425.
- Alkhalifah A. Topical and intralesional therapies for alopecia areata. Dermatol Ther. 2011;24:355-363.
- Herbst V, Zöller M, Kissling S, et al. Diphenylcyclopropenone treatment of alopecia areata induces apoptosis of perifollicular lymphocytes. Eur J Dermatol. 2006;16:537-542.
- Zöller M, Freyschmidt-Paul P, Vitacolonna M, et al. Chronic delayed-type hypersensitivity reaction as a means to treat alopecia areata. Clin Exp Immunol. 2004;135:398-408.
- Bröcker EB, Echternacht-Happle K, Hamm H, et al. Abnormal expression of class I and class II major histocompatibility antigens in alopecia areata: modulation by topical immunotherapy. J Invest Dermatol. 1987;88:564-568.
- Todes-Taylor N, Turner R, Wood GS, et al. T cell subpopulations in alopecia areata. J Am Acad Dermatol. 1984;11:216-223.
- Perret C, Wiesner-Menzel L, Happle R. Immunohistochemical analysis of T-cell subsets in the peribulbar and intrabulbar infiltrates of alopecia areata. Acta Derm Venereol. 1984;64:26-30.
- Wiesner-Menzel L, Happle R. Intrabulbar and peribulbar accumulation of dendritic OKT 6-positive cells in alopecia areata. Arch Dermatol Res. 1984;276:333-334.
- McElwee KJ, Freyschmidt-Paul P, Hoffmann R, et al. Transfer of CD8+ cells induces localized hair loss whereas CD4+/CD25– cells promote systemic alopecia areata and CD4+/CD25+ cells blockade disease onset in the C3H/HeJ mouse model. J Invest Dermatol. 2005;124:947-957.
- Arca E, Muşabak U, Akar A, et al. Interferon-gamma in alopecia areata. Eur J Dermatol. 2004;14:33-36.
- Hoffmann R. The potential role of cytokines and T cells in alopecia areata. J Investig Dermatol Symp Proc. 1999;4:235-238.
- Philpott MP, Sanders DA, Bowen J, et al. Effects of interleukins, colony-stimulating factor and tumour necrosis factor on human hair follicle growth in vitro: a possible role for interleukin-1 and tumour necrosis factor-alpha in alopecia areata. Br J Dermatol. 1996;135:942-948.
- Le Bidre E, Chaby G, Martin L, et al. Alopecia areata during anti-TNF alpha therapy: nine cases. Ann Dermatol Venereol. 2011;138:285-293.
- Ferran M, Calvet J, Almirall M, et al. Alopecia areata as another immune-mediated disease developed in patients treated with tumour necrosis factor-α blocker agents: report of five cases and review of the literature. J Eur Acad Dermatol Venereol. 2011;25:479-484.
- Pan Y, Rao NA. Alopecia areata during etanercept therapy. Ocul Immunol Inflamm. 2009;17:127-129.
- Pelivani N, Hassan AS, Braathen LR, et al. Alopecia areata universalis elicited during treatment with adalimumab. Dermatology. 2008;216:320-323.
- Uyemura K, Yamamura M, Fivenson DF, et al. The cytokine network in lesional and lesion-free psoriatic skin is characterized by a T-helper type 1 cell-mediated response. J Invest Dermatol. 1993;101:701-705.
- Baker BS, Powles AV, Valdimarsson H, et al. An altered response by psoriatic keratinocytes to gamma interferon. Scan J Immunol. 1988;28:735-740.
- Jackson M, Howie SE, Weller R, et al. Psoriatic keratinocytes show reduced IRF-1 and STAT-1alpha activation in response to gamma-IFN. FASEB J. 1999;13:495-502.
- Perera GK, Di Meglio P, Nestle FO. Psoriasis. Annu Rev Pathol. 2012;7:385-422.
- McGeachy MJ, Chen Y, Tato CM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10:314-324.
- Volpe E, Servant N, Zollinger R, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9:650-657.
- Boniface K, Blumenschein WM, Brovont-Porth K, et al. Human Th17 cells comprise heterogeneous subsets including IFN-gamma-producing cells with distinct properties from the Th1 lineage. J Immunol. 2010;185:679-687.
- Kagami S, Rizzo HL, Lee JJ, et al. Circulating Th17, Th22, and Th1 cells are increased in psoriasis. J Invest Dermatol. 2010;130:1373-1383.
- Boniface K, Bernard FX, Garcia M, et al. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. 2005;174:3695-3702.
- Harper EG, Guo C, Rizzo H, et al. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Invest Dermatol. 2009;129:2175-2183.
- Bowcock AM, Krueger JG. Getting under the skin: the immunogenetics of psoriasis. Nat Rev Immunol. 2005;5:699-711.
- Hoffmann R, Wenzel E, Huth A, et al. Cytokine mRNA levels in alopecia areata before and after treatment with the contact allergen diphenylcyclopropenone. J Invest Dermatol. 1994;103:530-533.
To the Editor:
Both alopecia areata (AA) and psoriasis vulgaris are chronic relapsing autoimmune diseases, with AA causing nonscarring hair loss in approximately 0.1% to 0.2%1 of the population with a lifetime risk of 1.7%,2 and psoriasis more broadly impacting 1.5% to 2% of the population.3 The helper T cell (TH1) cytokine milieu is pathogenic in both conditions.4-6 IFN-γ knockout mice, unlike their wild-type counterparts, do not exhibit AA.7 Psoriasis is notably improved by IL-10 injections, which dampen the TH1 response.8 Distinct from AA, TH17 and TH22 cells have been implicated as key players in psoriasis pathogenesis, along with the associated IL-17 and IL-22 cytokines.9-12
Few cases of patients with concurrent AA and psoriasis have been described. Interestingly, these cases document normal hair regrowth in the areas of psoriasis.13-16 These cases may offer unique insight into the immune factors driving each disease. We describe a case of a man with both alopecia universalis (AU) and psoriasis who developed hair regrowth in some of the psoriatic plaques.
A 34-year-old man with concurrent AU and psoriasis who had not used any systemic or topical medication for either condition in the last year presented to our clinic seeking treatment. The patient had a history of alopecia totalis as a toddler that completely resolved by 4 years of age with the use of squaric acid dibutylester (SADBE). At 31 years of age, the alopecia recurred and was localized to the scalp. It was partially responsive to intralesional triamcinolone acetonide. The patient’s alopecia worsened over the 2 years following recurrence, ultimately progressing to AU. Two months after the alopecia recurrence, he developed the first psoriatic plaques. As the plaque psoriasis progressed, systemic therapy was initiated, first methotrexate and then etanercept. Shortly after developing AU, he lost his health insurance and discontinued all therapy. The patient’s psoriasis began to recur approximately 3 months after stopping etanercept. He was not using any other psoriasis medications. At that time, he noted terminal hair regrowth within some of the psoriatic plaques. No terminal hairs grew outside of the psoriatic plaques, and all regions with growth had previously been without hair for an extended period of time. The patient presented to our clinic approximately 1 year later. He had no other medical conditions and no relevant family history.
On initial physical examination, he had nonscarring hair loss involving nearly 100% of the body with psoriatic plaques on approximately 30% of the body surface area. Regions of terminal hair growth were confined to some but not all of the psoriatic plaques (Figure). Interestingly, the terminal hairs were primarily localized to the thickest central regions of the plaques. The patient’s psoriasis was treated with a combination of topical clobetasol and calcipotriene. In addition, he was started on tacrolimus ointment to the face and eyebrows for the AA. Maintenance of terminal hair within a region of topically treated psoriasis on the forearm persisted at the 2-month follow-up despite complete clearance of the corresponding psoriatic plaque. A small psoriatic plaque on the scalp cleared early with topical therapy without noticeable hair regrowth. The patient subsequently was started on contact immunotherapy with SADBE and intralesional triamcinolone acetonide for the scalp alopecia without satisfactory response. He decided to discontinue further attempts at treating the alopecia and requested to be restarted on etanercept therapy for recalcitrant psoriatic plaques. His psoriasis responded well to this therapy and he continues to be followed in our psoriasis clinic. One year after clearance of the treated psoriatic plaques, the corresponding terminal hairs persist.
Contact immunotherapy, most commonly with diphenylcyclopropenone or SADBE, is reported to have a 50% to 60% success rate in extensive AA, with a broad range of 9% to 87%17; however, randomized controlled trials testing the efficacy of contact immunotherapy are lacking. Although the mechanism of action of these topical sensitizers is not clearly delineated, it has been postulated that by inducing a new type of inflammatory response in the region, the immunologic milieu is changed, allowing the hair to grow. Some proposed mechanisms include promoting perifollicular lymphocyte apoptosis, preventing new recruitment of autoreactive lymphocytes, and allowing for the correction of aberrant major histocompatibility complex expression on the hair matrix epithelium to regain follicle immune privilege.18-20
Iatrogenic immunotherapy may work analogously to the natural immune system deviation demonstrated in our patient. Psoriasis and AA are believed to form competing immune cells and cytokine milieus, thus explaining how an individual with AA could regain normal hair growth in areas of psoriasis.15,16 The Renbök phenomenon, or reverse Köbner phenomenon, coined by Happle et al13 can be used to describe both the iatrogenic and natural cases of dermatologic disease improvement in response to secondary insults.14
A complex cascade of immune cells and cytokines coordinate AA pathogenesis. In the acute stage of AA, an inflammatory infiltrate of CD4+ T cells, CD8+ T cells, and antigen-presenting cells target anagen phase follicles, with a higher CD4+:CD8+ ratio in clinically active disease.21-23 Subcutaneous injections of either CD4+ or CD8+ lymphocyte subsets from mice with AA into normal-haired mice induces disease. However, CD8+ T cell injections rapidly produce apparent hair loss, whereas CD4+ T cells cause hair loss after several weeks, suggesting that CD8+ T cells directly modulate AA hair loss and CD4+ T cells act as an aide.24 The growth, differentiation, and survival of CD8+ T cells are stimulated by IL-2 and IFN-γ. Alopecia areata biopsies demonstrate a prevalence of TH1 cytokines, and patients with localized AA, alopecia totalis, and AU have notably higher serum IFN-γ levels compared to controls.25 In murine models, IL-1α and IL-1β increase during the catagen phase of the hair cycle and peak during the telogen phase.26 Excessive IL-1β expression is detected in the early stages of human disease, and certain IL-1β polymorphisms are associated with severe forms of AA.26 The role of tumor necrosis factor (TNF) α in AA is not well understood. In vitro studies show it inhibits hair growth, suggesting the cytokine may play a role in AA.27 However, anti–TNF-α therapy is not effective in AA, and case reports propose these therapies rarely induce AA.28-31
The TH1 response is likewise critical to psoriatic plaque development. IFN-γ and TNF-α are overexpressed in psoriatic plaques.32 IFN-γ has an antiproliferative and differentiation-inducing effect on normal keratinocytes, but psoriatic epithelial cells in vitro respond differently to the cytokine with a notably diminished growth inhibition.33,34 One explanation for the role of IFN-γ is that it stimulates dendritic cells to produce IL-1 and IL-23.35 IL-23 activates TH17 cells36; TH1 and TH17 conditions produce IL-22 whose serum level correlates with disease severity.37-39 IL-22 induces keratinocyte proliferation and migration and inhibits keratinocyte differentiation, helping account for hallmarks of the disease.40 Patients with psoriasis have increased levels of TH1, TH17, and TH22 cells, as well as their associated cytokines, in the skin and blood compared to controls.4,11,32,39,41
Alopecia areata and psoriasis are regulated by complex and still not entirely understood immune interactions. The fact that many of the same therapies are used to treat both diseases emphasizes both their overlapping characteristics and the lack of targeted therapy. It is unclear if and how the topical or systemic therapies used in our patient to treat one disease affected the natural history of the other condition. It is important to highlight, however, that the patient had not been treated for months when he developed the psoriatic plaques with hair regrowth. Other case reports also document hair regrowth in untreated plaques,13,16 making it unlikely to be a side effect of the medication regimen. For both psoriasis and AA, the immune cell composition and cytokine levels in the skin or serum vary throughout a patient’s disease course depending on severity of disease or response to treatment.6,39,42,43 Therefore, we hypothesize that the 2 conditions interact in a similarly distinct manner based on each disease’s stage and intensity in the patient. Both our patient’s course thus far and the various presentations described by other groups support this hypothesis. Our patient had a small region of psoriasis on the scalp that cleared without any terminal hair growth. He also had larger plaques on the forearms that developed hair growth most predominantly within the thicker regions of the plaques. His unique presentation highlights the fluidity of the immune factors driving psoriasis vulgaris and AA.
To the Editor:
Both alopecia areata (AA) and psoriasis vulgaris are chronic relapsing autoimmune diseases, with AA causing nonscarring hair loss in approximately 0.1% to 0.2%1 of the population with a lifetime risk of 1.7%,2 and psoriasis more broadly impacting 1.5% to 2% of the population.3 The helper T cell (TH1) cytokine milieu is pathogenic in both conditions.4-6 IFN-γ knockout mice, unlike their wild-type counterparts, do not exhibit AA.7 Psoriasis is notably improved by IL-10 injections, which dampen the TH1 response.8 Distinct from AA, TH17 and TH22 cells have been implicated as key players in psoriasis pathogenesis, along with the associated IL-17 and IL-22 cytokines.9-12
Few cases of patients with concurrent AA and psoriasis have been described. Interestingly, these cases document normal hair regrowth in the areas of psoriasis.13-16 These cases may offer unique insight into the immune factors driving each disease. We describe a case of a man with both alopecia universalis (AU) and psoriasis who developed hair regrowth in some of the psoriatic plaques.
A 34-year-old man with concurrent AU and psoriasis who had not used any systemic or topical medication for either condition in the last year presented to our clinic seeking treatment. The patient had a history of alopecia totalis as a toddler that completely resolved by 4 years of age with the use of squaric acid dibutylester (SADBE). At 31 years of age, the alopecia recurred and was localized to the scalp. It was partially responsive to intralesional triamcinolone acetonide. The patient’s alopecia worsened over the 2 years following recurrence, ultimately progressing to AU. Two months after the alopecia recurrence, he developed the first psoriatic plaques. As the plaque psoriasis progressed, systemic therapy was initiated, first methotrexate and then etanercept. Shortly after developing AU, he lost his health insurance and discontinued all therapy. The patient’s psoriasis began to recur approximately 3 months after stopping etanercept. He was not using any other psoriasis medications. At that time, he noted terminal hair regrowth within some of the psoriatic plaques. No terminal hairs grew outside of the psoriatic plaques, and all regions with growth had previously been without hair for an extended period of time. The patient presented to our clinic approximately 1 year later. He had no other medical conditions and no relevant family history.
On initial physical examination, he had nonscarring hair loss involving nearly 100% of the body with psoriatic plaques on approximately 30% of the body surface area. Regions of terminal hair growth were confined to some but not all of the psoriatic plaques (Figure). Interestingly, the terminal hairs were primarily localized to the thickest central regions of the plaques. The patient’s psoriasis was treated with a combination of topical clobetasol and calcipotriene. In addition, he was started on tacrolimus ointment to the face and eyebrows for the AA. Maintenance of terminal hair within a region of topically treated psoriasis on the forearm persisted at the 2-month follow-up despite complete clearance of the corresponding psoriatic plaque. A small psoriatic plaque on the scalp cleared early with topical therapy without noticeable hair regrowth. The patient subsequently was started on contact immunotherapy with SADBE and intralesional triamcinolone acetonide for the scalp alopecia without satisfactory response. He decided to discontinue further attempts at treating the alopecia and requested to be restarted on etanercept therapy for recalcitrant psoriatic plaques. His psoriasis responded well to this therapy and he continues to be followed in our psoriasis clinic. One year after clearance of the treated psoriatic plaques, the corresponding terminal hairs persist.
Contact immunotherapy, most commonly with diphenylcyclopropenone or SADBE, is reported to have a 50% to 60% success rate in extensive AA, with a broad range of 9% to 87%17; however, randomized controlled trials testing the efficacy of contact immunotherapy are lacking. Although the mechanism of action of these topical sensitizers is not clearly delineated, it has been postulated that by inducing a new type of inflammatory response in the region, the immunologic milieu is changed, allowing the hair to grow. Some proposed mechanisms include promoting perifollicular lymphocyte apoptosis, preventing new recruitment of autoreactive lymphocytes, and allowing for the correction of aberrant major histocompatibility complex expression on the hair matrix epithelium to regain follicle immune privilege.18-20
Iatrogenic immunotherapy may work analogously to the natural immune system deviation demonstrated in our patient. Psoriasis and AA are believed to form competing immune cells and cytokine milieus, thus explaining how an individual with AA could regain normal hair growth in areas of psoriasis.15,16 The Renbök phenomenon, or reverse Köbner phenomenon, coined by Happle et al13 can be used to describe both the iatrogenic and natural cases of dermatologic disease improvement in response to secondary insults.14
A complex cascade of immune cells and cytokines coordinate AA pathogenesis. In the acute stage of AA, an inflammatory infiltrate of CD4+ T cells, CD8+ T cells, and antigen-presenting cells target anagen phase follicles, with a higher CD4+:CD8+ ratio in clinically active disease.21-23 Subcutaneous injections of either CD4+ or CD8+ lymphocyte subsets from mice with AA into normal-haired mice induces disease. However, CD8+ T cell injections rapidly produce apparent hair loss, whereas CD4+ T cells cause hair loss after several weeks, suggesting that CD8+ T cells directly modulate AA hair loss and CD4+ T cells act as an aide.24 The growth, differentiation, and survival of CD8+ T cells are stimulated by IL-2 and IFN-γ. Alopecia areata biopsies demonstrate a prevalence of TH1 cytokines, and patients with localized AA, alopecia totalis, and AU have notably higher serum IFN-γ levels compared to controls.25 In murine models, IL-1α and IL-1β increase during the catagen phase of the hair cycle and peak during the telogen phase.26 Excessive IL-1β expression is detected in the early stages of human disease, and certain IL-1β polymorphisms are associated with severe forms of AA.26 The role of tumor necrosis factor (TNF) α in AA is not well understood. In vitro studies show it inhibits hair growth, suggesting the cytokine may play a role in AA.27 However, anti–TNF-α therapy is not effective in AA, and case reports propose these therapies rarely induce AA.28-31
The TH1 response is likewise critical to psoriatic plaque development. IFN-γ and TNF-α are overexpressed in psoriatic plaques.32 IFN-γ has an antiproliferative and differentiation-inducing effect on normal keratinocytes, but psoriatic epithelial cells in vitro respond differently to the cytokine with a notably diminished growth inhibition.33,34 One explanation for the role of IFN-γ is that it stimulates dendritic cells to produce IL-1 and IL-23.35 IL-23 activates TH17 cells36; TH1 and TH17 conditions produce IL-22 whose serum level correlates with disease severity.37-39 IL-22 induces keratinocyte proliferation and migration and inhibits keratinocyte differentiation, helping account for hallmarks of the disease.40 Patients with psoriasis have increased levels of TH1, TH17, and TH22 cells, as well as their associated cytokines, in the skin and blood compared to controls.4,11,32,39,41
Alopecia areata and psoriasis are regulated by complex and still not entirely understood immune interactions. The fact that many of the same therapies are used to treat both diseases emphasizes both their overlapping characteristics and the lack of targeted therapy. It is unclear if and how the topical or systemic therapies used in our patient to treat one disease affected the natural history of the other condition. It is important to highlight, however, that the patient had not been treated for months when he developed the psoriatic plaques with hair regrowth. Other case reports also document hair regrowth in untreated plaques,13,16 making it unlikely to be a side effect of the medication regimen. For both psoriasis and AA, the immune cell composition and cytokine levels in the skin or serum vary throughout a patient’s disease course depending on severity of disease or response to treatment.6,39,42,43 Therefore, we hypothesize that the 2 conditions interact in a similarly distinct manner based on each disease’s stage and intensity in the patient. Both our patient’s course thus far and the various presentations described by other groups support this hypothesis. Our patient had a small region of psoriasis on the scalp that cleared without any terminal hair growth. He also had larger plaques on the forearms that developed hair growth most predominantly within the thicker regions of the plaques. His unique presentation highlights the fluidity of the immune factors driving psoriasis vulgaris and AA.
- Safavi K. Prevalence of alopecia areata in the First National Health and Nutrition Examination Survey. Arch Dermatol. 1992;128:702.
- Safavi KH, Muller SA, Suman VJ, et al. Incidence of alopecia areata in Olmsted County, Minnesota, 1975 through 1989. Mayo Clin Proc. 1995;70:628-633.
- Wolff K, Johnson RA. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 6th ed. New York, NY: McGraw-Hill; 2009.
- Austin LM, Ozawa M, Kikuchi T, et al. The majority of epidermal T cells in psoriasis vulgaris lesions can produce type 1 cytokines, interferon-gamma, interleukin-2, and tumor necrosis factor-alpha, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: a type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J Invest Dermatol. 1999;113:752-759.
- Ghoreishi M, Martinka M, Dutz JP. Type 1 interferon signature in the scalp lesions of alopecia areata. Br J Dermatol. 2010;163:57-62.
- Rossi A, Cantisani C, Carlesimo M, et al. Serum concentrations of IL-2, IL-6, IL-12 and TNF-α in patients with alopecia areata. Int J Immunopathol Pharmacol. 2012;25:781-788.
- Freyschmidt-Paul P, McElwee KJ, Hoffmann R, et al. Interferon-gamma-deficient mice are resistant to the development of alopecia areata. Br J Dermatol. 2006;155:515-521.
- Reich K, Garbe C, Blaschke V, et al. Response of psoriasis to interleukin-10 is associated with suppression of cutaneous type 1 inflammation, downregulation of the epidermal interleukin-8/CXCR2 pathway and normalization of keratinocyte maturation. J Invest Dermatol. 2001;116:319-329.
- Teunissen MB, Koomen CW, de Waal Malefyt R, et al. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. 1998;111:645-649.
- Zheng Y, Danilenko DM, Valdez P, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648-651.
- Boniface K, Guignouard E, Pedretti N, et al. A role for T cell-derived interleukin 22 in psoriatic skin inflammation. Clin Exp Immunol. 2007;150:407-415.
- Zaba LC, Suárez-Fariñas M, Fuentes-Duculan J, et al. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J Allergy Clin Immunol. 2009;124:1022-1030.e395.
- Happle R, van der Steen PHM, Perret CM. The Renbök phenomenon: an inverse Köebner reaction observed in alopecia areata. Eur J Dermatol. 1991;2:39-40.
- Ito T, Hashizume H, Takigawa M. Contact immunotherapy-induced Renbök phenomenon in a patient with alopecia areata and psoriasis vulgaris. Eur J Dermatol. 2010;20:126-127.
- Criado PR, Valente NY, Michalany NS, et al. An unusual association between scalp psoriasis and ophiasic alopecia areata: the Renbök phenomenon. Clin Exp Dermatol. 2007;32:320-321.
- Harris JE, Seykora JT, Lee RA. Renbök phenomenon and contact sensitization in a patient with alopecia universalis. Arch Dermatol. 2010;146:422-425.
- Alkhalifah A. Topical and intralesional therapies for alopecia areata. Dermatol Ther. 2011;24:355-363.
- Herbst V, Zöller M, Kissling S, et al. Diphenylcyclopropenone treatment of alopecia areata induces apoptosis of perifollicular lymphocytes. Eur J Dermatol. 2006;16:537-542.
- Zöller M, Freyschmidt-Paul P, Vitacolonna M, et al. Chronic delayed-type hypersensitivity reaction as a means to treat alopecia areata. Clin Exp Immunol. 2004;135:398-408.
- Bröcker EB, Echternacht-Happle K, Hamm H, et al. Abnormal expression of class I and class II major histocompatibility antigens in alopecia areata: modulation by topical immunotherapy. J Invest Dermatol. 1987;88:564-568.
- Todes-Taylor N, Turner R, Wood GS, et al. T cell subpopulations in alopecia areata. J Am Acad Dermatol. 1984;11:216-223.
- Perret C, Wiesner-Menzel L, Happle R. Immunohistochemical analysis of T-cell subsets in the peribulbar and intrabulbar infiltrates of alopecia areata. Acta Derm Venereol. 1984;64:26-30.
- Wiesner-Menzel L, Happle R. Intrabulbar and peribulbar accumulation of dendritic OKT 6-positive cells in alopecia areata. Arch Dermatol Res. 1984;276:333-334.
- McElwee KJ, Freyschmidt-Paul P, Hoffmann R, et al. Transfer of CD8+ cells induces localized hair loss whereas CD4+/CD25– cells promote systemic alopecia areata and CD4+/CD25+ cells blockade disease onset in the C3H/HeJ mouse model. J Invest Dermatol. 2005;124:947-957.
- Arca E, Muşabak U, Akar A, et al. Interferon-gamma in alopecia areata. Eur J Dermatol. 2004;14:33-36.
- Hoffmann R. The potential role of cytokines and T cells in alopecia areata. J Investig Dermatol Symp Proc. 1999;4:235-238.
- Philpott MP, Sanders DA, Bowen J, et al. Effects of interleukins, colony-stimulating factor and tumour necrosis factor on human hair follicle growth in vitro: a possible role for interleukin-1 and tumour necrosis factor-alpha in alopecia areata. Br J Dermatol. 1996;135:942-948.
- Le Bidre E, Chaby G, Martin L, et al. Alopecia areata during anti-TNF alpha therapy: nine cases. Ann Dermatol Venereol. 2011;138:285-293.
- Ferran M, Calvet J, Almirall M, et al. Alopecia areata as another immune-mediated disease developed in patients treated with tumour necrosis factor-α blocker agents: report of five cases and review of the literature. J Eur Acad Dermatol Venereol. 2011;25:479-484.
- Pan Y, Rao NA. Alopecia areata during etanercept therapy. Ocul Immunol Inflamm. 2009;17:127-129.
- Pelivani N, Hassan AS, Braathen LR, et al. Alopecia areata universalis elicited during treatment with adalimumab. Dermatology. 2008;216:320-323.
- Uyemura K, Yamamura M, Fivenson DF, et al. The cytokine network in lesional and lesion-free psoriatic skin is characterized by a T-helper type 1 cell-mediated response. J Invest Dermatol. 1993;101:701-705.
- Baker BS, Powles AV, Valdimarsson H, et al. An altered response by psoriatic keratinocytes to gamma interferon. Scan J Immunol. 1988;28:735-740.
- Jackson M, Howie SE, Weller R, et al. Psoriatic keratinocytes show reduced IRF-1 and STAT-1alpha activation in response to gamma-IFN. FASEB J. 1999;13:495-502.
- Perera GK, Di Meglio P, Nestle FO. Psoriasis. Annu Rev Pathol. 2012;7:385-422.
- McGeachy MJ, Chen Y, Tato CM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10:314-324.
- Volpe E, Servant N, Zollinger R, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9:650-657.
- Boniface K, Blumenschein WM, Brovont-Porth K, et al. Human Th17 cells comprise heterogeneous subsets including IFN-gamma-producing cells with distinct properties from the Th1 lineage. J Immunol. 2010;185:679-687.
- Kagami S, Rizzo HL, Lee JJ, et al. Circulating Th17, Th22, and Th1 cells are increased in psoriasis. J Invest Dermatol. 2010;130:1373-1383.
- Boniface K, Bernard FX, Garcia M, et al. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. 2005;174:3695-3702.
- Harper EG, Guo C, Rizzo H, et al. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Invest Dermatol. 2009;129:2175-2183.
- Bowcock AM, Krueger JG. Getting under the skin: the immunogenetics of psoriasis. Nat Rev Immunol. 2005;5:699-711.
- Hoffmann R, Wenzel E, Huth A, et al. Cytokine mRNA levels in alopecia areata before and after treatment with the contact allergen diphenylcyclopropenone. J Invest Dermatol. 1994;103:530-533.
- Safavi K. Prevalence of alopecia areata in the First National Health and Nutrition Examination Survey. Arch Dermatol. 1992;128:702.
- Safavi KH, Muller SA, Suman VJ, et al. Incidence of alopecia areata in Olmsted County, Minnesota, 1975 through 1989. Mayo Clin Proc. 1995;70:628-633.
- Wolff K, Johnson RA. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 6th ed. New York, NY: McGraw-Hill; 2009.
- Austin LM, Ozawa M, Kikuchi T, et al. The majority of epidermal T cells in psoriasis vulgaris lesions can produce type 1 cytokines, interferon-gamma, interleukin-2, and tumor necrosis factor-alpha, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: a type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J Invest Dermatol. 1999;113:752-759.
- Ghoreishi M, Martinka M, Dutz JP. Type 1 interferon signature in the scalp lesions of alopecia areata. Br J Dermatol. 2010;163:57-62.
- Rossi A, Cantisani C, Carlesimo M, et al. Serum concentrations of IL-2, IL-6, IL-12 and TNF-α in patients with alopecia areata. Int J Immunopathol Pharmacol. 2012;25:781-788.
- Freyschmidt-Paul P, McElwee KJ, Hoffmann R, et al. Interferon-gamma-deficient mice are resistant to the development of alopecia areata. Br J Dermatol. 2006;155:515-521.
- Reich K, Garbe C, Blaschke V, et al. Response of psoriasis to interleukin-10 is associated with suppression of cutaneous type 1 inflammation, downregulation of the epidermal interleukin-8/CXCR2 pathway and normalization of keratinocyte maturation. J Invest Dermatol. 2001;116:319-329.
- Teunissen MB, Koomen CW, de Waal Malefyt R, et al. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. 1998;111:645-649.
- Zheng Y, Danilenko DM, Valdez P, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648-651.
- Boniface K, Guignouard E, Pedretti N, et al. A role for T cell-derived interleukin 22 in psoriatic skin inflammation. Clin Exp Immunol. 2007;150:407-415.
- Zaba LC, Suárez-Fariñas M, Fuentes-Duculan J, et al. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J Allergy Clin Immunol. 2009;124:1022-1030.e395.
- Happle R, van der Steen PHM, Perret CM. The Renbök phenomenon: an inverse Köebner reaction observed in alopecia areata. Eur J Dermatol. 1991;2:39-40.
- Ito T, Hashizume H, Takigawa M. Contact immunotherapy-induced Renbök phenomenon in a patient with alopecia areata and psoriasis vulgaris. Eur J Dermatol. 2010;20:126-127.
- Criado PR, Valente NY, Michalany NS, et al. An unusual association between scalp psoriasis and ophiasic alopecia areata: the Renbök phenomenon. Clin Exp Dermatol. 2007;32:320-321.
- Harris JE, Seykora JT, Lee RA. Renbök phenomenon and contact sensitization in a patient with alopecia universalis. Arch Dermatol. 2010;146:422-425.
- Alkhalifah A. Topical and intralesional therapies for alopecia areata. Dermatol Ther. 2011;24:355-363.
- Herbst V, Zöller M, Kissling S, et al. Diphenylcyclopropenone treatment of alopecia areata induces apoptosis of perifollicular lymphocytes. Eur J Dermatol. 2006;16:537-542.
- Zöller M, Freyschmidt-Paul P, Vitacolonna M, et al. Chronic delayed-type hypersensitivity reaction as a means to treat alopecia areata. Clin Exp Immunol. 2004;135:398-408.
- Bröcker EB, Echternacht-Happle K, Hamm H, et al. Abnormal expression of class I and class II major histocompatibility antigens in alopecia areata: modulation by topical immunotherapy. J Invest Dermatol. 1987;88:564-568.
- Todes-Taylor N, Turner R, Wood GS, et al. T cell subpopulations in alopecia areata. J Am Acad Dermatol. 1984;11:216-223.
- Perret C, Wiesner-Menzel L, Happle R. Immunohistochemical analysis of T-cell subsets in the peribulbar and intrabulbar infiltrates of alopecia areata. Acta Derm Venereol. 1984;64:26-30.
- Wiesner-Menzel L, Happle R. Intrabulbar and peribulbar accumulation of dendritic OKT 6-positive cells in alopecia areata. Arch Dermatol Res. 1984;276:333-334.
- McElwee KJ, Freyschmidt-Paul P, Hoffmann R, et al. Transfer of CD8+ cells induces localized hair loss whereas CD4+/CD25– cells promote systemic alopecia areata and CD4+/CD25+ cells blockade disease onset in the C3H/HeJ mouse model. J Invest Dermatol. 2005;124:947-957.
- Arca E, Muşabak U, Akar A, et al. Interferon-gamma in alopecia areata. Eur J Dermatol. 2004;14:33-36.
- Hoffmann R. The potential role of cytokines and T cells in alopecia areata. J Investig Dermatol Symp Proc. 1999;4:235-238.
- Philpott MP, Sanders DA, Bowen J, et al. Effects of interleukins, colony-stimulating factor and tumour necrosis factor on human hair follicle growth in vitro: a possible role for interleukin-1 and tumour necrosis factor-alpha in alopecia areata. Br J Dermatol. 1996;135:942-948.
- Le Bidre E, Chaby G, Martin L, et al. Alopecia areata during anti-TNF alpha therapy: nine cases. Ann Dermatol Venereol. 2011;138:285-293.
- Ferran M, Calvet J, Almirall M, et al. Alopecia areata as another immune-mediated disease developed in patients treated with tumour necrosis factor-α blocker agents: report of five cases and review of the literature. J Eur Acad Dermatol Venereol. 2011;25:479-484.
- Pan Y, Rao NA. Alopecia areata during etanercept therapy. Ocul Immunol Inflamm. 2009;17:127-129.
- Pelivani N, Hassan AS, Braathen LR, et al. Alopecia areata universalis elicited during treatment with adalimumab. Dermatology. 2008;216:320-323.
- Uyemura K, Yamamura M, Fivenson DF, et al. The cytokine network in lesional and lesion-free psoriatic skin is characterized by a T-helper type 1 cell-mediated response. J Invest Dermatol. 1993;101:701-705.
- Baker BS, Powles AV, Valdimarsson H, et al. An altered response by psoriatic keratinocytes to gamma interferon. Scan J Immunol. 1988;28:735-740.
- Jackson M, Howie SE, Weller R, et al. Psoriatic keratinocytes show reduced IRF-1 and STAT-1alpha activation in response to gamma-IFN. FASEB J. 1999;13:495-502.
- Perera GK, Di Meglio P, Nestle FO. Psoriasis. Annu Rev Pathol. 2012;7:385-422.
- McGeachy MJ, Chen Y, Tato CM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10:314-324.
- Volpe E, Servant N, Zollinger R, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9:650-657.
- Boniface K, Blumenschein WM, Brovont-Porth K, et al. Human Th17 cells comprise heterogeneous subsets including IFN-gamma-producing cells with distinct properties from the Th1 lineage. J Immunol. 2010;185:679-687.
- Kagami S, Rizzo HL, Lee JJ, et al. Circulating Th17, Th22, and Th1 cells are increased in psoriasis. J Invest Dermatol. 2010;130:1373-1383.
- Boniface K, Bernard FX, Garcia M, et al. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. 2005;174:3695-3702.
- Harper EG, Guo C, Rizzo H, et al. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Invest Dermatol. 2009;129:2175-2183.
- Bowcock AM, Krueger JG. Getting under the skin: the immunogenetics of psoriasis. Nat Rev Immunol. 2005;5:699-711.
- Hoffmann R, Wenzel E, Huth A, et al. Cytokine mRNA levels in alopecia areata before and after treatment with the contact allergen diphenylcyclopropenone. J Invest Dermatol. 1994;103:530-533.
Practice Points
- The Renbök phenomenon, or reverse Köbner phenomenon, describes cases where secondary insults improve dermatologic disease.
- Current evidence suggests that alopecia areata (AA) is driven by a helper T cell (TH1) response whereas psoriasis vulgaris is driven by TH1, TH17, and TH22.
- Patients with concurrent AA and psoriasis can develop normal hair regrowth confined to the psoriatic plaques. Developing methods to artificially alter the cytokine milieu in affected skin may lead to new therapeutic options for each condition.