User login
Eruptive Syringoma Manifesting as a Widespread Rash in 3 Patients
To the Editor:
Syringoma is a relatively common benign adnexal neoplasm originating in the ducts of eccrine sweat glands. It can be
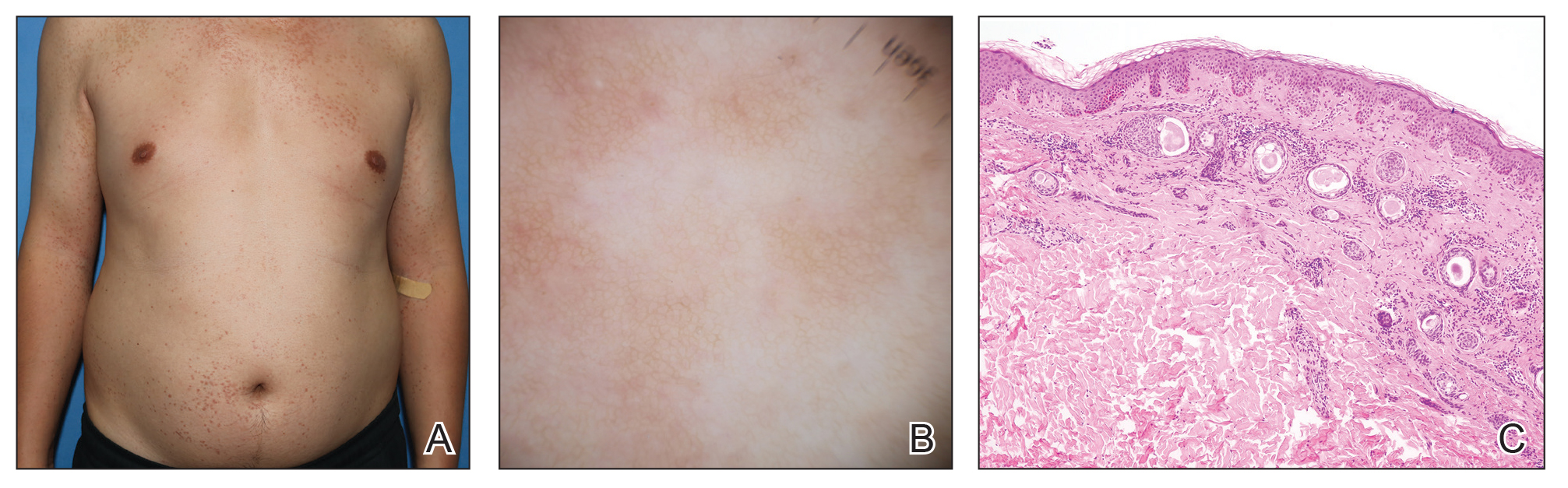
A 28-year-old man presented with multiple asymptomatic papules on the trunk and upper arms of 20 years’ duration (patient 1). He had been diagnosed with Darier disease 3 years prior to the current presentation and was treated with oral and topical retinoic acid without a response. After 3 months of oral treatment, the retinoic acid was stopped due to elevated liver enzymes. Physical examination at the current presentation revealed multiple smooth, firm, nonfused, 1- to 4-mm
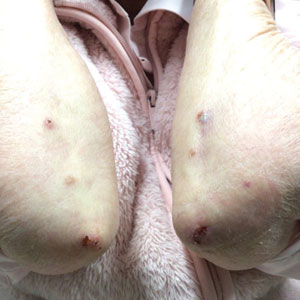
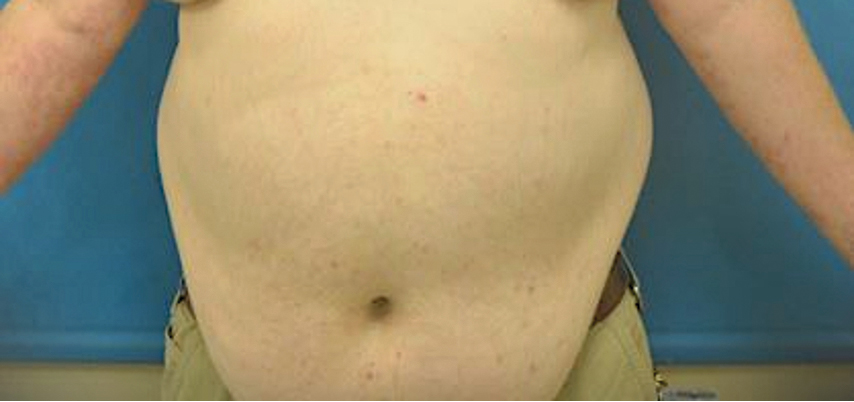
A 27-year-old woman presented with widespread asymptomatic papules

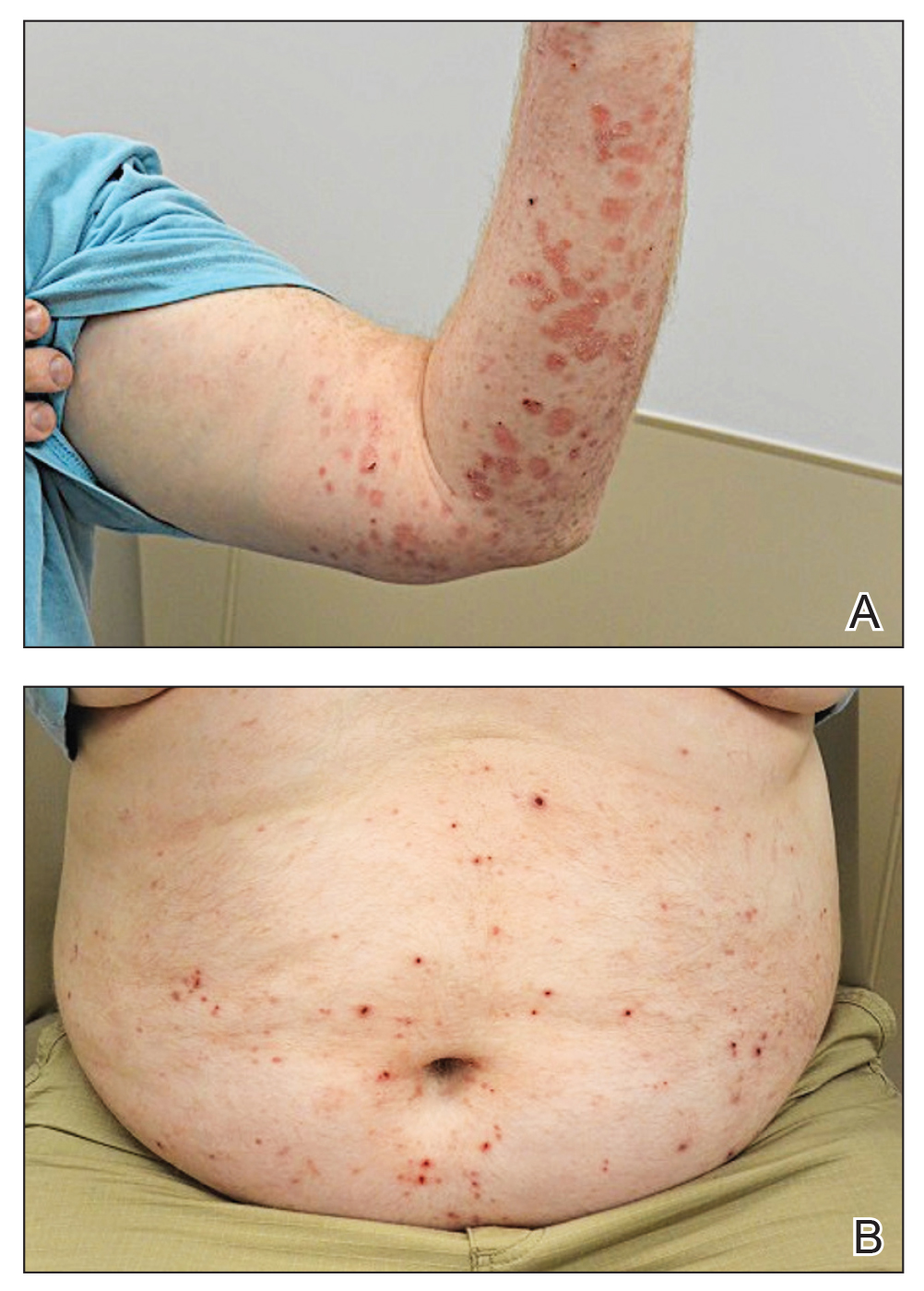
A 43-year-old man who was otherwise healthy presented with brownish flat-topped papules on the chest and abdomen of 19 years’ duration (Figure 3A)(patient 3). The lesions had remained stable and did not progress. He denied any treatment. 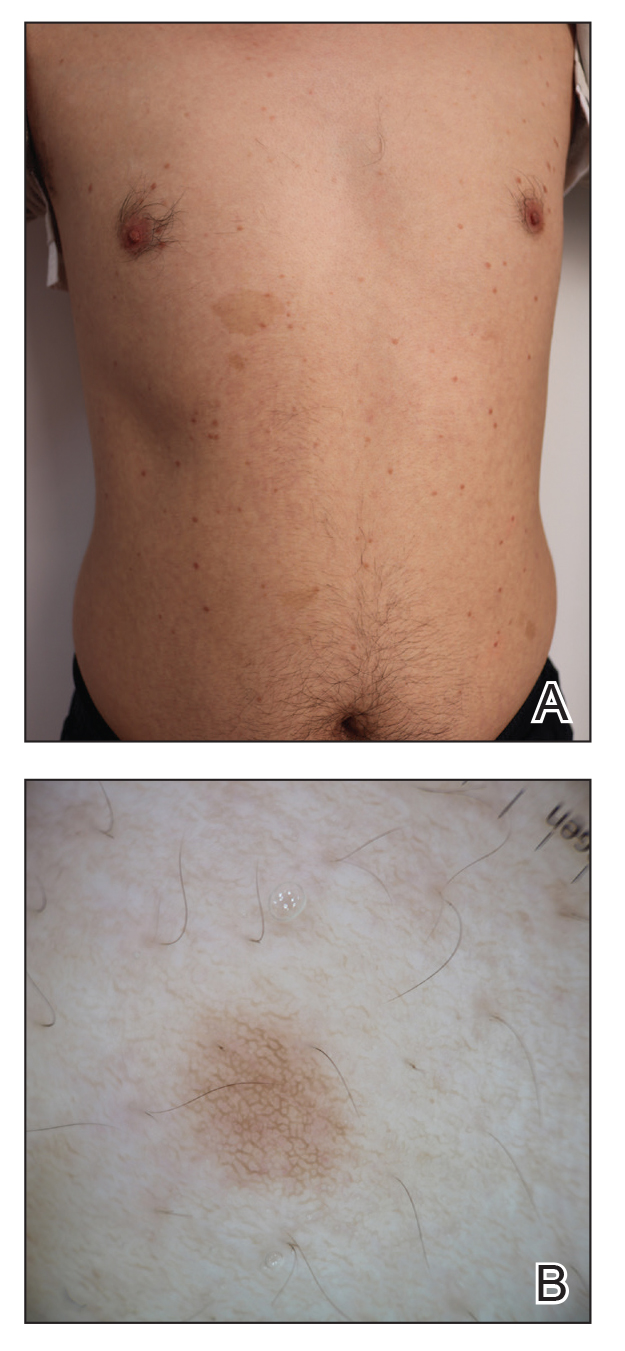
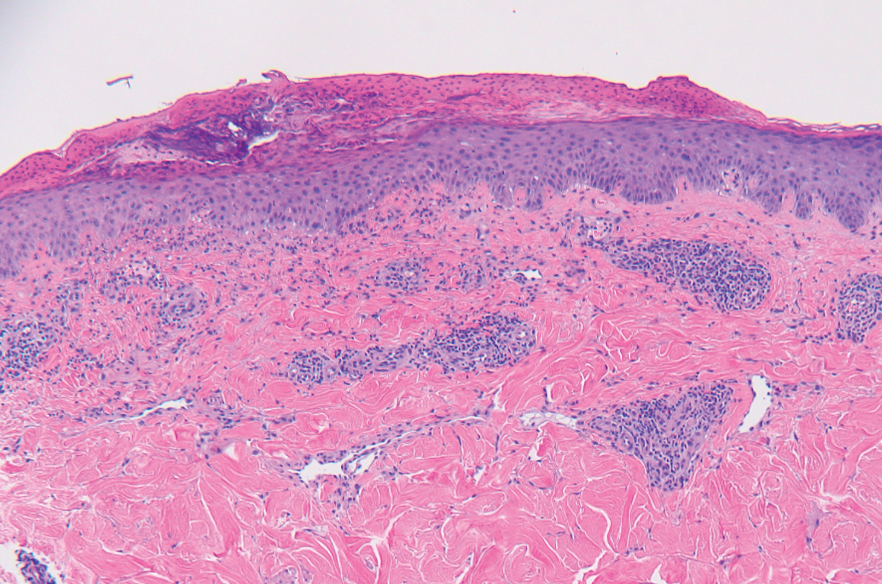
All 3 patients demonstrated classic histopathologic features of syringoma, and none had a family history of similar skin lesions. The clinical and dermoscopic findings along with the histopathology in all 3 patients were consistent with ES.
The pathogenesis of ES is
- Williams K, Shinkai K. Evaluation and management of the patient with multiple syringomas: a systematic review of the literature. J Am Acad Dermatol. 2016;74:1234-1240.e1239. doi:10.1016/j.jaad.2015.12.006
- Jacquet L, Darier J.
Hidradénomes éruptifs, I.épithéliomes adenoids des glandes sudoripares ou adénomes sudoripares. Ann Dermatol Venerol. 1887;8:317-323. - Huang A, Taylor G, Liebman TN. Generalized eruptive syringomas. Dermatol Online J. 2017;23:13030/qt0hb8q22g..
- Maeda T, Natsuga K, Nishie W, et al. Extensive eruptive syringoma after liver transplantation. Acta Derm Venereol. 2018;98:119-120. doi:10.2340/00015555-2814
- Lerner TH, Barr RJ, Dolezal JF, et al. Syringomatous hyperplasia and eccrine squamous syringometaplasia associated with benoxaprofen therapy. Arch Dermatol. 1987;123:1202-1204. doi:10.1001/archderm.1987.01660330113022
- Ozturk F, Ermertcan AT, Bilac C, et al.
A case report of postpubertal eruptive syringoma triggered with antiepileptic drugs. J Drugs Dermatol. 2010;9:707-710. - Guitart J, Rosenbaum MM, Requena L. ‘Eruptive syringoma’: a misnomer for a reactive eccrine gland ductal proliferation? J Cutan Pathol. 2003;30:202-205. doi:10.1034/j.1600-0560.2003.00023.x
- Dupre A, Carrere S, Bonafe JL, et al. Eruptive generalized syringomas, milium and atrophoderma vermiculata. Nicolau and Balus’ syndrome (author’s transl). Dermatologica. 1981;162:281-286.
- Schepis C, Torre V, Siragusa M, et al. Eruptive syringomas with calcium deposits in a young woman with Down’s syndrome. Dermatology. 2001;203:345-347. doi:10.1159/000051788
- Samia AM, Donthi D, Nenow J, et al. A case study and review of literature of eruptive syringoma in a six-year-old. Cureus. 2021;13:E14634. doi:10.7759/cureus.14634
- Soler-Carrillo J, Estrach T, Mascaró JM. Eruptive syringoma: 27 new cases and review of the literature. J Eur Acad Dermatol Venereol. 2001;15:242-246. doi:10.1046/j.1468-3083.2001.00235.x
- Aleissa M, Aljarbou O, AlJasser MI. Dermoscopy of eruptive syringoma. Skin Appendage Disord. 2021;7:401-403. doi:10.1159/000515443
- Botsali A, Caliskan E, Coskun A, et al. Eruptive syringoma: two cases with dermoscopic features. Skin Appendage Disord. 2020;6:319-322. doi:10.1159/000508656
- Dutra Rezende H, Madia ACT, Elias BM, et al. Comment on: eruptive syringoma—two cases with dermoscopic features. Skin Appendage Disord. 2022;8:81-82. doi:10.1159/000518158
- Silva-Hirschberg C, Cabrera R, Rollán MP, et al. Darier disease: the use of dermoscopy in monitoring acitretin treatment. An Bras Dermatol. 2022;97:644-647. doi:10.1016/j.abd.2021.05.021
- Singal A, Kaur I, Jakhar D. Fox-Fordyce disease: dermoscopic perspective. Skin Appendage Disord. 2020;6:247-249. doi:10.1159/000508201
- Brau Javier CN, Morales A, Sanchez JL. Histopathology attributes of Fox-Fordyce disease. Int J Dermatol. 2012;51:1313-1318. doi:10.1159/000508201
- Horie K, Shinkuma S, Fujita Y, et al. Efficacy of N-(3,4-dimethoxycinnamoyl)-anthranilic acid (tranilast) against eruptive syringoma: report of two cases and review of published work. J Dermatol. 2012;39:1044-1046. doi:10.1111/j.1346-8138.2012.01612.x
- Sanchez TS, Dauden E, Casas AP, et al. Eruptive pruritic syringomas: treatment with topical atropine. J Am Acad Dermatol. 2001;44:148-149. doi:10.1067/mjd.2001.109854
To the Editor:
Syringoma is a relatively common benign adnexal neoplasm originating in the ducts of eccrine sweat glands. It can be
A 28-year-old man presented with multiple asymptomatic papules on the trunk and upper arms of 20 years’ duration (patient 1). He had been diagnosed with Darier disease 3 years prior to the current presentation and was treated with oral and topical retinoic acid without a response. After 3 months of oral treatment, the retinoic acid was stopped due to elevated liver enzymes. Physical examination at the current presentation revealed multiple smooth, firm, nonfused, 1- to 4-mm
A 27-year-old woman presented with widespread asymptomatic papules
A 43-year-old man who was otherwise healthy presented with brownish flat-topped papules on the chest and abdomen of 19 years’ duration (Figure 3A)(patient 3). The lesions had remained stable and did not progress. He denied any treatment.
All 3 patients demonstrated classic histopathologic features of syringoma, and none had a family history of similar skin lesions. The clinical and dermoscopic findings along with the histopathology in all 3 patients were consistent with ES.
The pathogenesis of ES is
To the Editor:
Syringoma is a relatively common benign adnexal neoplasm originating in the ducts of eccrine sweat glands. It can be
A 28-year-old man presented with multiple asymptomatic papules on the trunk and upper arms of 20 years’ duration (patient 1). He had been diagnosed with Darier disease 3 years prior to the current presentation and was treated with oral and topical retinoic acid without a response. After 3 months of oral treatment, the retinoic acid was stopped due to elevated liver enzymes. Physical examination at the current presentation revealed multiple smooth, firm, nonfused, 1- to 4-mm
A 27-year-old woman presented with widespread asymptomatic papules
A 43-year-old man who was otherwise healthy presented with brownish flat-topped papules on the chest and abdomen of 19 years’ duration (Figure 3A)(patient 3). The lesions had remained stable and did not progress. He denied any treatment.
All 3 patients demonstrated classic histopathologic features of syringoma, and none had a family history of similar skin lesions. The clinical and dermoscopic findings along with the histopathology in all 3 patients were consistent with ES.
The pathogenesis of ES is
- Williams K, Shinkai K. Evaluation and management of the patient with multiple syringomas: a systematic review of the literature. J Am Acad Dermatol. 2016;74:1234-1240.e1239. doi:10.1016/j.jaad.2015.12.006
- Jacquet L, Darier J.
Hidradénomes éruptifs, I.épithéliomes adenoids des glandes sudoripares ou adénomes sudoripares. Ann Dermatol Venerol. 1887;8:317-323. - Huang A, Taylor G, Liebman TN. Generalized eruptive syringomas. Dermatol Online J. 2017;23:13030/qt0hb8q22g..
- Maeda T, Natsuga K, Nishie W, et al. Extensive eruptive syringoma after liver transplantation. Acta Derm Venereol. 2018;98:119-120. doi:10.2340/00015555-2814
- Lerner TH, Barr RJ, Dolezal JF, et al. Syringomatous hyperplasia and eccrine squamous syringometaplasia associated with benoxaprofen therapy. Arch Dermatol. 1987;123:1202-1204. doi:10.1001/archderm.1987.01660330113022
- Ozturk F, Ermertcan AT, Bilac C, et al.
A case report of postpubertal eruptive syringoma triggered with antiepileptic drugs. J Drugs Dermatol. 2010;9:707-710. - Guitart J, Rosenbaum MM, Requena L. ‘Eruptive syringoma’: a misnomer for a reactive eccrine gland ductal proliferation? J Cutan Pathol. 2003;30:202-205. doi:10.1034/j.1600-0560.2003.00023.x
- Dupre A, Carrere S, Bonafe JL, et al. Eruptive generalized syringomas, milium and atrophoderma vermiculata. Nicolau and Balus’ syndrome (author’s transl). Dermatologica. 1981;162:281-286.
- Schepis C, Torre V, Siragusa M, et al. Eruptive syringomas with calcium deposits in a young woman with Down’s syndrome. Dermatology. 2001;203:345-347. doi:10.1159/000051788
- Samia AM, Donthi D, Nenow J, et al. A case study and review of literature of eruptive syringoma in a six-year-old. Cureus. 2021;13:E14634. doi:10.7759/cureus.14634
- Soler-Carrillo J, Estrach T, Mascaró JM. Eruptive syringoma: 27 new cases and review of the literature. J Eur Acad Dermatol Venereol. 2001;15:242-246. doi:10.1046/j.1468-3083.2001.00235.x
- Aleissa M, Aljarbou O, AlJasser MI. Dermoscopy of eruptive syringoma. Skin Appendage Disord. 2021;7:401-403. doi:10.1159/000515443
- Botsali A, Caliskan E, Coskun A, et al. Eruptive syringoma: two cases with dermoscopic features. Skin Appendage Disord. 2020;6:319-322. doi:10.1159/000508656
- Dutra Rezende H, Madia ACT, Elias BM, et al. Comment on: eruptive syringoma—two cases with dermoscopic features. Skin Appendage Disord. 2022;8:81-82. doi:10.1159/000518158
- Silva-Hirschberg C, Cabrera R, Rollán MP, et al. Darier disease: the use of dermoscopy in monitoring acitretin treatment. An Bras Dermatol. 2022;97:644-647. doi:10.1016/j.abd.2021.05.021
- Singal A, Kaur I, Jakhar D. Fox-Fordyce disease: dermoscopic perspective. Skin Appendage Disord. 2020;6:247-249. doi:10.1159/000508201
- Brau Javier CN, Morales A, Sanchez JL. Histopathology attributes of Fox-Fordyce disease. Int J Dermatol. 2012;51:1313-1318. doi:10.1159/000508201
- Horie K, Shinkuma S, Fujita Y, et al. Efficacy of N-(3,4-dimethoxycinnamoyl)-anthranilic acid (tranilast) against eruptive syringoma: report of two cases and review of published work. J Dermatol. 2012;39:1044-1046. doi:10.1111/j.1346-8138.2012.01612.x
- Sanchez TS, Dauden E, Casas AP, et al. Eruptive pruritic syringomas: treatment with topical atropine. J Am Acad Dermatol. 2001;44:148-149. doi:10.1067/mjd.2001.109854
- Williams K, Shinkai K. Evaluation and management of the patient with multiple syringomas: a systematic review of the literature. J Am Acad Dermatol. 2016;74:1234-1240.e1239. doi:10.1016/j.jaad.2015.12.006
- Jacquet L, Darier J.
Hidradénomes éruptifs, I.épithéliomes adenoids des glandes sudoripares ou adénomes sudoripares. Ann Dermatol Venerol. 1887;8:317-323. - Huang A, Taylor G, Liebman TN. Generalized eruptive syringomas. Dermatol Online J. 2017;23:13030/qt0hb8q22g..
- Maeda T, Natsuga K, Nishie W, et al. Extensive eruptive syringoma after liver transplantation. Acta Derm Venereol. 2018;98:119-120. doi:10.2340/00015555-2814
- Lerner TH, Barr RJ, Dolezal JF, et al. Syringomatous hyperplasia and eccrine squamous syringometaplasia associated with benoxaprofen therapy. Arch Dermatol. 1987;123:1202-1204. doi:10.1001/archderm.1987.01660330113022
- Ozturk F, Ermertcan AT, Bilac C, et al.
A case report of postpubertal eruptive syringoma triggered with antiepileptic drugs. J Drugs Dermatol. 2010;9:707-710. - Guitart J, Rosenbaum MM, Requena L. ‘Eruptive syringoma’: a misnomer for a reactive eccrine gland ductal proliferation? J Cutan Pathol. 2003;30:202-205. doi:10.1034/j.1600-0560.2003.00023.x
- Dupre A, Carrere S, Bonafe JL, et al. Eruptive generalized syringomas, milium and atrophoderma vermiculata. Nicolau and Balus’ syndrome (author’s transl). Dermatologica. 1981;162:281-286.
- Schepis C, Torre V, Siragusa M, et al. Eruptive syringomas with calcium deposits in a young woman with Down’s syndrome. Dermatology. 2001;203:345-347. doi:10.1159/000051788
- Samia AM, Donthi D, Nenow J, et al. A case study and review of literature of eruptive syringoma in a six-year-old. Cureus. 2021;13:E14634. doi:10.7759/cureus.14634
- Soler-Carrillo J, Estrach T, Mascaró JM. Eruptive syringoma: 27 new cases and review of the literature. J Eur Acad Dermatol Venereol. 2001;15:242-246. doi:10.1046/j.1468-3083.2001.00235.x
- Aleissa M, Aljarbou O, AlJasser MI. Dermoscopy of eruptive syringoma. Skin Appendage Disord. 2021;7:401-403. doi:10.1159/000515443
- Botsali A, Caliskan E, Coskun A, et al. Eruptive syringoma: two cases with dermoscopic features. Skin Appendage Disord. 2020;6:319-322. doi:10.1159/000508656
- Dutra Rezende H, Madia ACT, Elias BM, et al. Comment on: eruptive syringoma—two cases with dermoscopic features. Skin Appendage Disord. 2022;8:81-82. doi:10.1159/000518158
- Silva-Hirschberg C, Cabrera R, Rollán MP, et al. Darier disease: the use of dermoscopy in monitoring acitretin treatment. An Bras Dermatol. 2022;97:644-647. doi:10.1016/j.abd.2021.05.021
- Singal A, Kaur I, Jakhar D. Fox-Fordyce disease: dermoscopic perspective. Skin Appendage Disord. 2020;6:247-249. doi:10.1159/000508201
- Brau Javier CN, Morales A, Sanchez JL. Histopathology attributes of Fox-Fordyce disease. Int J Dermatol. 2012;51:1313-1318. doi:10.1159/000508201
- Horie K, Shinkuma S, Fujita Y, et al. Efficacy of N-(3,4-dimethoxycinnamoyl)-anthranilic acid (tranilast) against eruptive syringoma: report of two cases and review of published work. J Dermatol. 2012;39:1044-1046. doi:10.1111/j.1346-8138.2012.01612.x
- Sanchez TS, Dauden E, Casas AP, et al. Eruptive pruritic syringomas: treatment with topical atropine. J Am Acad Dermatol. 2001;44:148-149. doi:10.1067/mjd.2001.109854
Practice Points
- Eruptive syringoma (ES) is a benign cutaneous adnexal neoplasm that typically does not require treatment.
- Dermoscopy and biopsy are helpful for the diagnosis of ES, which often is missed or misdiagnosed clinically.
Saxophone Penis: A Forgotten Manifestation of Hidradenitis Suppurativa
To the Editor:
Hidradenitis suppurativa (HS) is a multifactorial chronic inflammatory skin disease affecting 1% to 4% of Europeans. It is characterized by recurrent inflamed nodules, abscesses, and sinus tracts in intertriginous regions.1 The genital area is affected in 11% of cases2 and usually is connected to severe forms of HS in both men and women.3 The prevalence of HS-associated genital lymphedema remains unknown.
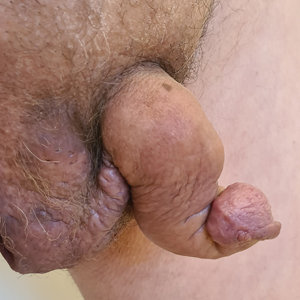
Saxophone penis is a specific penile malformation characterized by a saxophone shape due to inflammation of the major penile lymphatic vessels that cause fibrosis of the surrounding connective tissue. Poor blood flow further causes contracture and distortion of the penile axis.4 Saxophone penis also has been associated with primary lymphedema, lymphogranuloma venereum, filariasis,5 and administration of paraffin injections.6 We describe 3 men with HS who presented with saxophone penis.
A 33-year-old man with Hurley stage III HS presented with a medical history of groin lesions and progressive penoscrotal edema of 13 years’ duration. He had a body mass index (BMI) of 37, no family history of HS or comorbidities, and a 15-year history of smoking 20 cigarettes per day. After repeated surgical drainage of the HS lesions as well as antibiotic treatment with clindamycin 600 mg/d and rifampicin 600 mg/d, the patient was kept on a maintenance therapy with adalimumab 40 mg/wk. Due to lack of response, treatment was discontinued at week 16. Clindamycin and rifampicin 300 mg were immediately reintroduced with no benefit on the genital lesions. The patient underwent genital reconstruction, including penile degloving, scrotoplasty, infrapubic fat pad removal, and perineoplasty (Figure 1). The patient currently is not undergoing any therapies.
A 55-year-old man presented with Hurley stage II HS of 33 years’ duration. He had a BMI of 52; a history of hypertension, hyperuricemia, severe hip and knee osteoarthritis, and orchiopexy in childhood; a smoking history of 40 cigarettes per day; and an alcohol consumption history of 200 mL per day since 18 years of age. He had radical excision of axillary lesions 8 years prior. One year later, he was treated with concomitant clindamycin and rifampicin 300 mg twice daily for 3 months with no desirable effects. Adalimumab 40 mg/wk was initiated. After 12 weeks of treatment, he experienced 80% improvement in all areas except the genital region. He continued adalimumab for 3 years with good clinical response in all HS-affected sites except the genital region.
A 66-year-old man presented with Hurley stage III HS of 37 years’ duration. He had a smoking history of 10 cigarettes per day for 30 years, a BMI of 24.6, and a medical history of long-standing hypertension and hypothyroidism. A 3-month course of clindamycin and rifampicin 600 mg/d was ineffective; adalimumab 40 mg/wk was initiated. All affected areas improved, except for the saxophone penis. He continues his fifth year of therapy with adalimumab (Figure 2).

Hidradenitis suppurativa is associated with chronic pain, purulent malodor, and scarring with structural deformity. Repetitive inflammation causes fibrosis, scar formation, and soft-tissue destruction of lymphatic vessels, leading to lymphedema; primary lymphedema of the genitals in men has been reported to result in a saxophone penis.4
The only approved biologic treatments for moderate to severe HS are the tumor necrosis factor α inhibitor adalimumab and anti-IL-17 secukinumab.1 All 3 of our patients with HS were treated with adalimumab with reasonable success; however, the penile condition remained refractory, which we speculate may be due to adalimumab’s ability to control only active inflammatory lesions but not scars or fibrotic tissue.7 Higher adalimumab dosages were unlikely to be beneficial for their penile condition; some improvements have been reported following fluoroquinolone therapy. To our knowledge, there is no effective medical treatment for saxophone penis. However, surgery showed good results in one of our patients. Among our 3 adalimumab-treated patients, only 1 patient had corrective surgery that resulted in improvement in the penile deformity, further confirming adalimumab’s limited role in genital lymphedema.7 Extensive resection of the lymphedematous tissue, scrotoplasty, and Charles procedure are treatment options.8
Genital lymphedema has been associated with lymphangiectasia, lymphangioma circumscriptum, infections, and neoplasms such as lymphangiosarcoma and squamous cell carcinoma.9 Our patients reported discomfort, hygiene issues, and swelling. One patient reported micturition, and 2 patients reported sexual dysfunction.
Saxophone penis remains a disabling sequela of HS. Early diagnosis and treatment of HS may help prevent development of this condition.
- Lee EY, Alhusayen R, Lansang P, et al. What is hidradenitis suppurativa? Can Fam Physician. 2017;63:114-120.
- Fertitta L, Hotz C, Wolkenstein P, et al. Efficacy and satisfaction of surgical treatment for hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2020;34:839-845.
- Micieli R, Alavi A. Lymphedema in patients with hidradenitis suppurativa: a systematic review of published literature. Int J Dermatol. 2018;57:1471-1480.
- Maatouk I, Moutran R. Saxophone penis. JAMA Dermatol. 2013;149:802.
- Koley S, Mandal RK. Saxophone penis after unilateral inguinal bubo of lymphogranuloma venereum. Indian J Sex Transm Dis AIDS. 2013;34:149-151.
- D’Antuono A, Lambertini M, Gaspari V, et al. Visual dermatology: self-induced chronic saxophone penis due to paraffin injections. J Cutan Med Surg. 2019;23:330.
- Musumeci ML, Scilletta A, Sorci F, et al. Genital lymphedema associated with hidradenitis suppurativa unresponsive to adalimumab treatment. JAAD Case Rep. 2019;5:326-328.
- Jain V, Singh S, Garge S, et al. Saxophone penis due to primary lymphoedema. J Indian Assoc Pediatr Surg. 2009;14:230-231.
- Moosbrugger EA, Mutasim DF. Hidradenitis suppurativa complicated by severe lymphedema and lymphangiectasias. J Am Acad Dermatol. 2011;64:1223-1224.
To the Editor:
Hidradenitis suppurativa (HS) is a multifactorial chronic inflammatory skin disease affecting 1% to 4% of Europeans. It is characterized by recurrent inflamed nodules, abscesses, and sinus tracts in intertriginous regions.1 The genital area is affected in 11% of cases2 and usually is connected to severe forms of HS in both men and women.3 The prevalence of HS-associated genital lymphedema remains unknown.
Saxophone penis is a specific penile malformation characterized by a saxophone shape due to inflammation of the major penile lymphatic vessels that cause fibrosis of the surrounding connective tissue. Poor blood flow further causes contracture and distortion of the penile axis.4 Saxophone penis also has been associated with primary lymphedema, lymphogranuloma venereum, filariasis,5 and administration of paraffin injections.6 We describe 3 men with HS who presented with saxophone penis.
A 33-year-old man with Hurley stage III HS presented with a medical history of groin lesions and progressive penoscrotal edema of 13 years’ duration. He had a body mass index (BMI) of 37, no family history of HS or comorbidities, and a 15-year history of smoking 20 cigarettes per day. After repeated surgical drainage of the HS lesions as well as antibiotic treatment with clindamycin 600 mg/d and rifampicin 600 mg/d, the patient was kept on a maintenance therapy with adalimumab 40 mg/wk. Due to lack of response, treatment was discontinued at week 16. Clindamycin and rifampicin 300 mg were immediately reintroduced with no benefit on the genital lesions. The patient underwent genital reconstruction, including penile degloving, scrotoplasty, infrapubic fat pad removal, and perineoplasty (Figure 1). The patient currently is not undergoing any therapies.
A 55-year-old man presented with Hurley stage II HS of 33 years’ duration. He had a BMI of 52; a history of hypertension, hyperuricemia, severe hip and knee osteoarthritis, and orchiopexy in childhood; a smoking history of 40 cigarettes per day; and an alcohol consumption history of 200 mL per day since 18 years of age. He had radical excision of axillary lesions 8 years prior. One year later, he was treated with concomitant clindamycin and rifampicin 300 mg twice daily for 3 months with no desirable effects. Adalimumab 40 mg/wk was initiated. After 12 weeks of treatment, he experienced 80% improvement in all areas except the genital region. He continued adalimumab for 3 years with good clinical response in all HS-affected sites except the genital region.
A 66-year-old man presented with Hurley stage III HS of 37 years’ duration. He had a smoking history of 10 cigarettes per day for 30 years, a BMI of 24.6, and a medical history of long-standing hypertension and hypothyroidism. A 3-month course of clindamycin and rifampicin 600 mg/d was ineffective; adalimumab 40 mg/wk was initiated. All affected areas improved, except for the saxophone penis. He continues his fifth year of therapy with adalimumab (Figure 2).
Hidradenitis suppurativa is associated with chronic pain, purulent malodor, and scarring with structural deformity. Repetitive inflammation causes fibrosis, scar formation, and soft-tissue destruction of lymphatic vessels, leading to lymphedema; primary lymphedema of the genitals in men has been reported to result in a saxophone penis.4
The only approved biologic treatments for moderate to severe HS are the tumor necrosis factor α inhibitor adalimumab and anti-IL-17 secukinumab.1 All 3 of our patients with HS were treated with adalimumab with reasonable success; however, the penile condition remained refractory, which we speculate may be due to adalimumab’s ability to control only active inflammatory lesions but not scars or fibrotic tissue.7 Higher adalimumab dosages were unlikely to be beneficial for their penile condition; some improvements have been reported following fluoroquinolone therapy. To our knowledge, there is no effective medical treatment for saxophone penis. However, surgery showed good results in one of our patients. Among our 3 adalimumab-treated patients, only 1 patient had corrective surgery that resulted in improvement in the penile deformity, further confirming adalimumab’s limited role in genital lymphedema.7 Extensive resection of the lymphedematous tissue, scrotoplasty, and Charles procedure are treatment options.8
Genital lymphedema has been associated with lymphangiectasia, lymphangioma circumscriptum, infections, and neoplasms such as lymphangiosarcoma and squamous cell carcinoma.9 Our patients reported discomfort, hygiene issues, and swelling. One patient reported micturition, and 2 patients reported sexual dysfunction.
Saxophone penis remains a disabling sequela of HS. Early diagnosis and treatment of HS may help prevent development of this condition.
To the Editor:
Hidradenitis suppurativa (HS) is a multifactorial chronic inflammatory skin disease affecting 1% to 4% of Europeans. It is characterized by recurrent inflamed nodules, abscesses, and sinus tracts in intertriginous regions.1 The genital area is affected in 11% of cases2 and usually is connected to severe forms of HS in both men and women.3 The prevalence of HS-associated genital lymphedema remains unknown.
Saxophone penis is a specific penile malformation characterized by a saxophone shape due to inflammation of the major penile lymphatic vessels that cause fibrosis of the surrounding connective tissue. Poor blood flow further causes contracture and distortion of the penile axis.4 Saxophone penis also has been associated with primary lymphedema, lymphogranuloma venereum, filariasis,5 and administration of paraffin injections.6 We describe 3 men with HS who presented with saxophone penis.
A 33-year-old man with Hurley stage III HS presented with a medical history of groin lesions and progressive penoscrotal edema of 13 years’ duration. He had a body mass index (BMI) of 37, no family history of HS or comorbidities, and a 15-year history of smoking 20 cigarettes per day. After repeated surgical drainage of the HS lesions as well as antibiotic treatment with clindamycin 600 mg/d and rifampicin 600 mg/d, the patient was kept on a maintenance therapy with adalimumab 40 mg/wk. Due to lack of response, treatment was discontinued at week 16. Clindamycin and rifampicin 300 mg were immediately reintroduced with no benefit on the genital lesions. The patient underwent genital reconstruction, including penile degloving, scrotoplasty, infrapubic fat pad removal, and perineoplasty (Figure 1). The patient currently is not undergoing any therapies.
A 55-year-old man presented with Hurley stage II HS of 33 years’ duration. He had a BMI of 52; a history of hypertension, hyperuricemia, severe hip and knee osteoarthritis, and orchiopexy in childhood; a smoking history of 40 cigarettes per day; and an alcohol consumption history of 200 mL per day since 18 years of age. He had radical excision of axillary lesions 8 years prior. One year later, he was treated with concomitant clindamycin and rifampicin 300 mg twice daily for 3 months with no desirable effects. Adalimumab 40 mg/wk was initiated. After 12 weeks of treatment, he experienced 80% improvement in all areas except the genital region. He continued adalimumab for 3 years with good clinical response in all HS-affected sites except the genital region.
A 66-year-old man presented with Hurley stage III HS of 37 years’ duration. He had a smoking history of 10 cigarettes per day for 30 years, a BMI of 24.6, and a medical history of long-standing hypertension and hypothyroidism. A 3-month course of clindamycin and rifampicin 600 mg/d was ineffective; adalimumab 40 mg/wk was initiated. All affected areas improved, except for the saxophone penis. He continues his fifth year of therapy with adalimumab (Figure 2).
Hidradenitis suppurativa is associated with chronic pain, purulent malodor, and scarring with structural deformity. Repetitive inflammation causes fibrosis, scar formation, and soft-tissue destruction of lymphatic vessels, leading to lymphedema; primary lymphedema of the genitals in men has been reported to result in a saxophone penis.4
The only approved biologic treatments for moderate to severe HS are the tumor necrosis factor α inhibitor adalimumab and anti-IL-17 secukinumab.1 All 3 of our patients with HS were treated with adalimumab with reasonable success; however, the penile condition remained refractory, which we speculate may be due to adalimumab’s ability to control only active inflammatory lesions but not scars or fibrotic tissue.7 Higher adalimumab dosages were unlikely to be beneficial for their penile condition; some improvements have been reported following fluoroquinolone therapy. To our knowledge, there is no effective medical treatment for saxophone penis. However, surgery showed good results in one of our patients. Among our 3 adalimumab-treated patients, only 1 patient had corrective surgery that resulted in improvement in the penile deformity, further confirming adalimumab’s limited role in genital lymphedema.7 Extensive resection of the lymphedematous tissue, scrotoplasty, and Charles procedure are treatment options.8
Genital lymphedema has been associated with lymphangiectasia, lymphangioma circumscriptum, infections, and neoplasms such as lymphangiosarcoma and squamous cell carcinoma.9 Our patients reported discomfort, hygiene issues, and swelling. One patient reported micturition, and 2 patients reported sexual dysfunction.
Saxophone penis remains a disabling sequela of HS. Early diagnosis and treatment of HS may help prevent development of this condition.
- Lee EY, Alhusayen R, Lansang P, et al. What is hidradenitis suppurativa? Can Fam Physician. 2017;63:114-120.
- Fertitta L, Hotz C, Wolkenstein P, et al. Efficacy and satisfaction of surgical treatment for hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2020;34:839-845.
- Micieli R, Alavi A. Lymphedema in patients with hidradenitis suppurativa: a systematic review of published literature. Int J Dermatol. 2018;57:1471-1480.
- Maatouk I, Moutran R. Saxophone penis. JAMA Dermatol. 2013;149:802.
- Koley S, Mandal RK. Saxophone penis after unilateral inguinal bubo of lymphogranuloma venereum. Indian J Sex Transm Dis AIDS. 2013;34:149-151.
- D’Antuono A, Lambertini M, Gaspari V, et al. Visual dermatology: self-induced chronic saxophone penis due to paraffin injections. J Cutan Med Surg. 2019;23:330.
- Musumeci ML, Scilletta A, Sorci F, et al. Genital lymphedema associated with hidradenitis suppurativa unresponsive to adalimumab treatment. JAAD Case Rep. 2019;5:326-328.
- Jain V, Singh S, Garge S, et al. Saxophone penis due to primary lymphoedema. J Indian Assoc Pediatr Surg. 2009;14:230-231.
- Moosbrugger EA, Mutasim DF. Hidradenitis suppurativa complicated by severe lymphedema and lymphangiectasias. J Am Acad Dermatol. 2011;64:1223-1224.
- Lee EY, Alhusayen R, Lansang P, et al. What is hidradenitis suppurativa? Can Fam Physician. 2017;63:114-120.
- Fertitta L, Hotz C, Wolkenstein P, et al. Efficacy and satisfaction of surgical treatment for hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2020;34:839-845.
- Micieli R, Alavi A. Lymphedema in patients with hidradenitis suppurativa: a systematic review of published literature. Int J Dermatol. 2018;57:1471-1480.
- Maatouk I, Moutran R. Saxophone penis. JAMA Dermatol. 2013;149:802.
- Koley S, Mandal RK. Saxophone penis after unilateral inguinal bubo of lymphogranuloma venereum. Indian J Sex Transm Dis AIDS. 2013;34:149-151.
- D’Antuono A, Lambertini M, Gaspari V, et al. Visual dermatology: self-induced chronic saxophone penis due to paraffin injections. J Cutan Med Surg. 2019;23:330.
- Musumeci ML, Scilletta A, Sorci F, et al. Genital lymphedema associated with hidradenitis suppurativa unresponsive to adalimumab treatment. JAAD Case Rep. 2019;5:326-328.
- Jain V, Singh S, Garge S, et al. Saxophone penis due to primary lymphoedema. J Indian Assoc Pediatr Surg. 2009;14:230-231.
- Moosbrugger EA, Mutasim DF. Hidradenitis suppurativa complicated by severe lymphedema and lymphangiectasias. J Am Acad Dermatol. 2011;64:1223-1224.
Practice Points
- Hidradenitis suppurativa (HS) is a multifactorial chronic inflammatory skin disease.
- Saxophone penis is a specific penile malformation characterized by a saxophone shape due to inflammation.
- Repetitive inflammation within the context of HS may cause structural deformity of the penis, resulting in a saxophone penis.
- Early diagnosis and treatment of HS may help prevent development of this condition.
Anti-Smith and Anti–Double-Stranded DNA Antibodies in a Patient With Henoch-Schönlein Purpura Following COVID-19 Vaccination
To the Editor:
Henoch-Schönlein purpura (HSP)(also known as IgA vasculitis) is a small vessel vasculitis characterized by deposition of IgA in small vessels, resulting in the development of purpura on the legs. Based on the European Alliance of Associations for Rheumatology criteria,1 the patient also must have at least 1 of the following: arthritis, arthralgia, abdominal pain, leukocytoclastic vasculitis with IgA deposition, or kidney involvement. The disease can be triggered by infection—with more than 75% of patients reporting an antecedent upper respiratory tract infection2—as well as medications, circulating immune complexes, certain foods, vaccines, and rarely cancer.3,4 The disease more commonly occurs in children but also can affect adults.
Several cases of HSP have been reported following COVID-19 vaccination.5 We report a case of HSP developing days after the messenger RNA Pfizer-BioNTech COVID-19 vaccine booster that was associated with anti-Smith and anti–double-stranded DNA (dsDNA) antibodies as well as antineutrophil cytoplasmic antibodies (ANCAs).
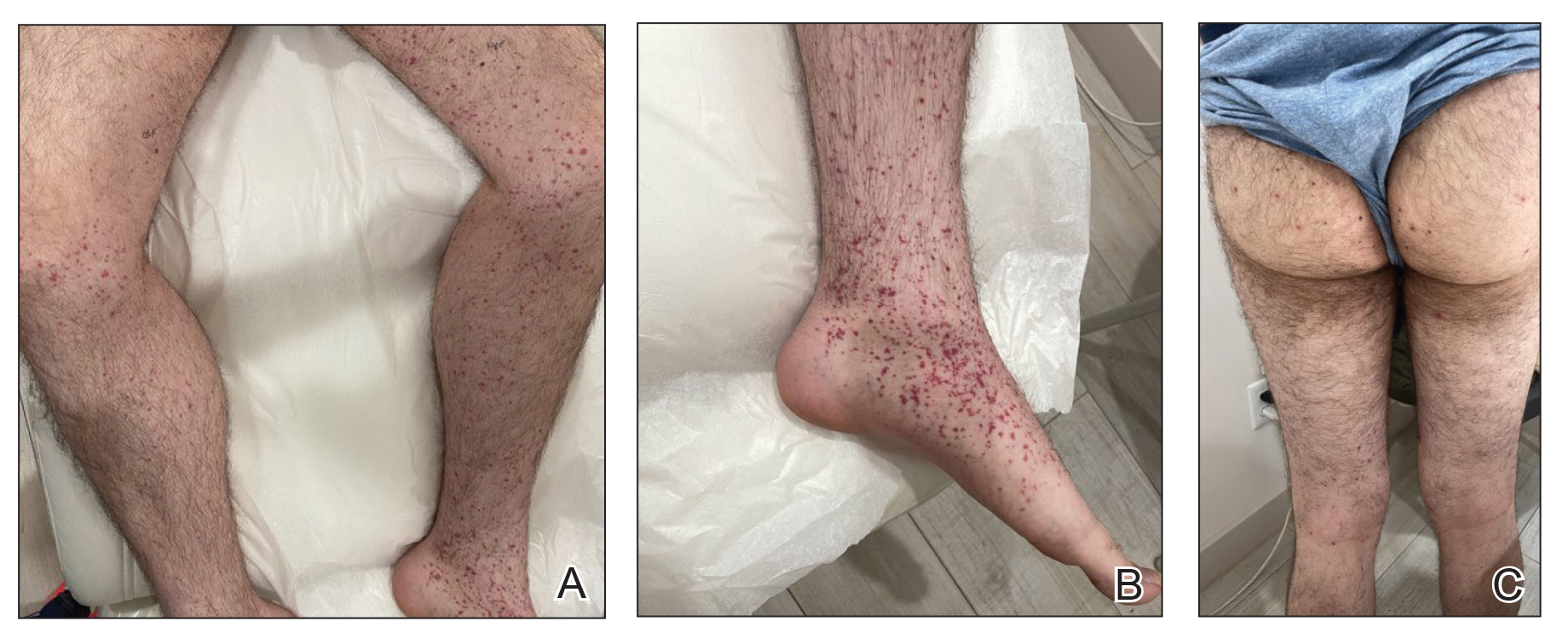
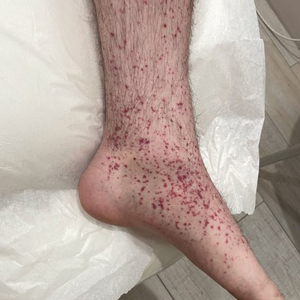
A 24-year-old man presented to dermatology with a rash of 3 weeks’ duration that first appeared 1 week after receiving his second booster of the messenger RNA Pfizer-BioNTech COVID-19 vaccine. Physical examination revealed petechiae with nonblanching erythematous macules and papules covering the legs below the knees (Figure 1) as well as the back of the right arm. A few days later, he developed arthralgia in the knees, hands, and feet. The patient denied any recent infections as well as respiratory and urinary tract symptoms. Approximately 10 days after the rash appeared, he developed epigastric abdominal pain that gradually worsened and sought care from his primary care physician, who ordered computed tomography and referred him for endoscopy. Computed tomography with and without contrast was suspicious for colitis. Colonoscopy and endoscopy were unremarkable. Laboratory tests were notable for elevated white blood cell count (17.08×103/µL [reference range, 3.66–10.60×103/µL]), serum IgA (437 mg/dL [reference range, 70–400 mg/dL]), C-reactive protein (1.5 mg/dL [reference range, <0.5 mg/dL]), anti-Smith antibody (28.1 CU [reference range, <20 CU), positive antinuclear antibody with titer (1:160 [reference range, <1:80]), anti-dsDNA (40.4 IU/mL [reference range, <27 IU/mL]), and cytoplasmic ANCA (c-ANCA) titer (1:320 [reference range, <1:20]). Blood urea nitrogen, creatinine, and estimated glomerular filtration rate were all within reference range. Urinalysis with microscopic examination was notable for 2 to 5 red blood cells per high-power field (reference range, 0) and proteinuria of 1+ (reference range, negative for protein).
The patient’s rash progressively worsened over the next few weeks, spreading proximally on the legs to the buttocks and the back of both elbows. A repeat complete blood cell count showed resolution of the leukocytosis. Two biopsies were taken from a lesion on the left proximal thigh: 1 for hematoxylin and eosin stain for histopathologic examination and 1 for direct immunofluorescence examination.
The patient was preliminarily diagnosed with HSP, and dermatology prescribed oral tofacitinib 5 mg twice daily for 5 days, which was supposed to be increased to 10 mg twice daily on the sixth day of treatment; however, the patient discontinued the medication after 4 days based on his primary care physician’s recommendation due to clotting concerns. The rash and arthralgia temporarily improved for 1 week, then relapsed.
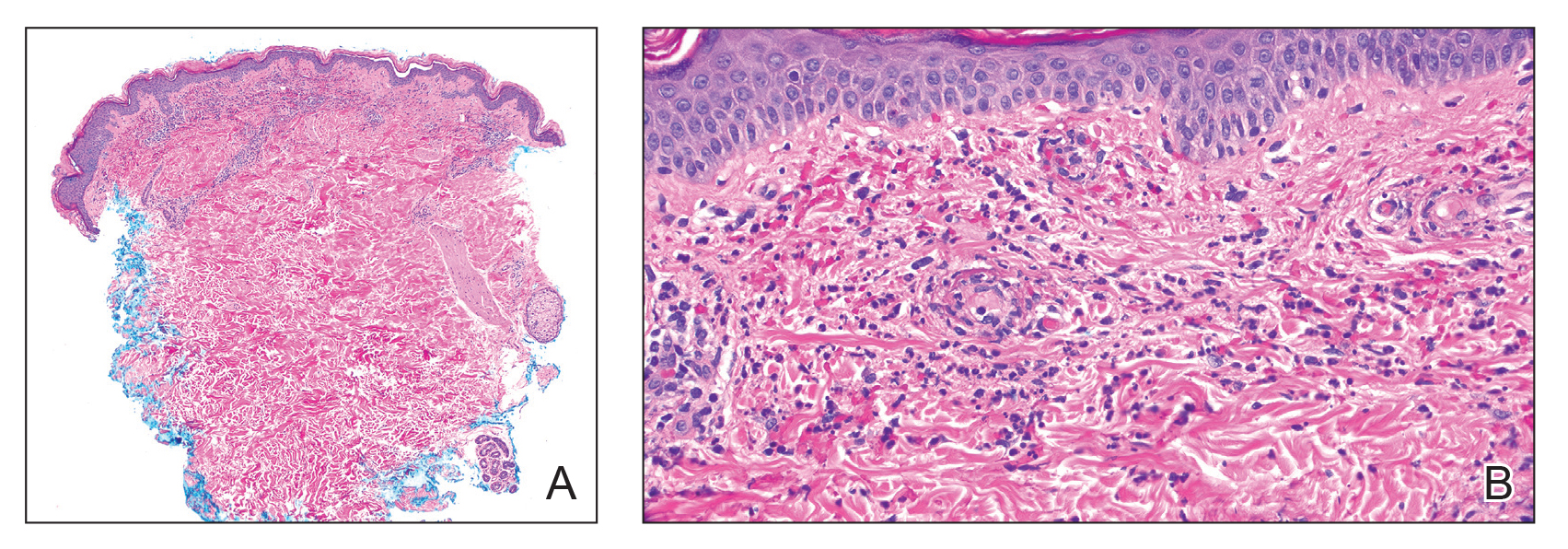
Histopathology revealed neutrophils surrounding and infiltrating small dermal blood vessel walls as well as associated neutrophilic debris and erythrocytes, consistent with leukocytoclastic vasculitis (Figure 2). Direct immunofluorescence was negative for IgA antibodies. His primary care physician, in consultation with his dermatologist, then started the patient on oral prednisone 70 mg once daily for 7 days with a plan to taper. Three days after prednisone was started, the arthralgia and abdominal pain resolved, and the rash became lighter in color. After 1 week, the rash resolved completely.
Due to the unusual antibodies, the patient was referred to a rheumatologist, who repeated the blood tests approximately 1 week after the patient started prednisone. The tests were negative for anti-Smith, anti-dsDNA, and c-ANCA but showed an elevated atypical perinuclear ANCA (p-ANCA) titer of 1:80 (reference range [negative], <1:20). A repeat urinalysis was unremarkable. The patient slowly tapered the prednisone over the course of 3 months and was subsequently lost to follow-up. The rash and other symptoms had not recurred as of the patient’s last physician contact. The most recent laboratory results showed a white blood cell count of 14.0×103/µL (reference range, 3.4–10.8×103/µL), likely due to the prednisone; blood urea nitrogen, creatinine, and estimated glomerular filtration rate were within reference range. The urinalysis was notable for occult blood and was negative for protein. C-reactive protein was 1 mg/dL (reference range, 0–10 mg/dL); p-ANCA, c-ANCA, and atypical p-ANCA, as well as antinuclear antibody, were negative. As of his last follow-up, the patient felt well.
The major differential diagnoses for our patient included HSP, ANCA vasculitis, and systemic lupus erythematosus. Although ANCA vasculitis has been reported after SARS-CoV-2 infection,6 the lack of pulmonary symptoms made this diagnosis unlikely.7 Although our patient initially had elevated anti-Smith and anti-dsDNA antibodies as well as mild renal involvement, he fulfilled at most only 2 of the 11 criteria necessary for diagnosing lupus: malar rash, discoid rash (includes alopecia), photosensitivity, ocular ulcers, nonerosive arthritis, serositis, renal disorder (protein >500 mg/24 h, red blood cells, casts), neurologic disorder (seizures, psychosis), hematologic disorders (hemolytic anemia, leukopenia), ANA, and immunologic disorder (anti-Smith). Four of the 11 criteria are necessary for the diagnosis of lupus.8
Torraca et al7 reported a case of HSP with positive c-ANCA (1:640) in a patient lacking pulmonary symptoms who was diagnosed with HSP. Cytoplasmic ANCA is not a typical finding in HSP. However, the additional findings of anti-Smith, anti-dsDNA, and mildly elevated atypical p-ANCA antibodies in our patient were unexpected and could be explained by the proposed pathogenesis of HSP—an overzealous immune response resulting in aberrant antibody complex deposition with ensuing complement activation.5,9 Production of these additional antibodies could be part of the overzealous response to COVID-19 vaccination.
Of all the COVID-19 vaccines, messenger RNA–based vaccines have been associated with the majority of cutaneous reactions, including local injection-site reactions (most common), delayed local reactions, urticaria, angioedema, morbilliform eruption, herpes zoster eruption, bullous eruptions, dermal filler reactions, chilblains, and pityriasis rosea. Less common reactions have included acute generalized exanthematous pustulosis, Stevens-Johnson syndrome, erythema multiforme, Sweet Syndrome, lichen planus, papulovesicular eruptions, pityriasis rosea–like eruptions, generalized annular lesions, facial pustular neutrophilic eruptions, and flares of underlying autoimmune skin conditions.10 Multiple cases of HSP have been reported following COVID-19 vaccination from all the major vaccine companies.5
In our patient, laboratory tests were repeated by a rheumatologist and were negative for anti-Smith and anti-dsDNA antibodies as well as c-ANCA, most likely because he started taking prednisone approximately 1 week prior, which may have resulted in decreased antibodies. Also, the patient’s symptoms resolved after 1 week of steroid therapy. Therefore, the diagnosis is most consistent with HSP associated with COVID-19 vaccination. The clinical presentation, microscopic hematuria and proteinuria, and histopathology were consistent with the European Alliance of Associations for Rheumatology criteria for HSP.1
Although direct immunofluorescence typically is positive for IgA deposition on biopsies, it can be negative for IgA, especially in lesions that are biopsied more than 7 days after their appearance, as shown in our case; a negative IgA on immunofluorescence does not rule out HSP.4 Elevated serum IgA is seen in more than 50% of cases of HSP.11 Although the disease typically is self-limited, glucocorticoids are used if the disease course is prolonged or if there is evidence of kidney involvement.9 The unique combination of anti-Smith and anti-dsDNA antibodies as well as ANCAs associated with HSP with negative IgA on direct immunofluorescence has been reported with lupus.12 Clinicians should be aware of COVID-19 vaccine–associated HSP that is negative for IgA deposition and positive for anti-Smith and anti-dsDNA antibodies as well as ANCAs.
Acknowledgment—We thank our patient for granting permission to publish this information.
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936-941. doi:10.1136/ard.2005.046300
- Rai A, Nast C, Adler S. Henoch–Schönlein purpura nephritis. J Am Soc Nephrol. 1999;10:2637-2644.
- Casini F, Magenes VC, De Sanctis M, et al. Henoch-Schönlein purpura following COVID-19 vaccine in a child: a case report. Ital J Pediatr. 2022;48:158. doi:10.1186/s13052-022-01351-1
- Poudel P, Adams SH, Mirchia K, et al. IgA negative immunofluorescence in diagnoses of adult-onset Henoch-Schönlein purpura. Proc (Bayl Univ Med Cent). 2020;33:436-437. doi:10.1080/08998280.2020.1770526
- Maronese CA, Zelin E, Avallone G, et al. Cutaneous vasculitis and vasculopathy in the era of COVID-19 pandemic. Front Med (Lausanne). 2022;9:996288. doi:10.3389/fmed.2022.996288
- Bryant MC, Spencer LT, Yalcindag A. A case of ANCA-associated vasculitis in a 16-year-old female following SARS-COV-2 infection and a systematic review of the literature. Pediatr Rheumatol Online J. 2022;20:65. doi:10.1186/s12969-022-00727-1
- Torraca PFS, Castro BC, Hans Filho G. Henoch-Schönlein purpura with c-ANCA antibody in adult. An Bras Dermatol. 2016;91:667-669. doi:10.1590/abd1806-4841.20164181
- Agabegi SS, Agabegi ED. Step-Up to Medicine. 4th ed. Wolters Kluwer; 2015.
- Ball-Burack MR, Kosowsky JM. A Case of leukocytoclastic vasculitis following SARS-CoV-2 vaccination. J Emerg Med. 2022;63:E62-E65. doi:10.1016/j.jemermed.2021.10.005
- Tan SW, Tam YC, Pang SM. Cutaneous reactions to COVID-19 vaccines: a review. JAAD Int. 2022;7:178-186. doi:10.1016/j.jdin.2022.01.011
- Calviño MC, Llorca J, García-Porrúa C, et al. Henoch-Schönlein purpura in children from northwestern Spain: a 20-year epidemiologic and clinical study. Medicine (Baltimore). 2001;80:279-290.
- Hu P, Huang BY, Zhang DD, et al. Henoch-Schönlein purpura in a pediatric patient with lupus. Arch Med Sci. 2017;13:689-690. doi:10.5114/aoms.2017.67288
To the Editor:
Henoch-Schönlein purpura (HSP)(also known as IgA vasculitis) is a small vessel vasculitis characterized by deposition of IgA in small vessels, resulting in the development of purpura on the legs. Based on the European Alliance of Associations for Rheumatology criteria,1 the patient also must have at least 1 of the following: arthritis, arthralgia, abdominal pain, leukocytoclastic vasculitis with IgA deposition, or kidney involvement. The disease can be triggered by infection—with more than 75% of patients reporting an antecedent upper respiratory tract infection2—as well as medications, circulating immune complexes, certain foods, vaccines, and rarely cancer.3,4 The disease more commonly occurs in children but also can affect adults.
Several cases of HSP have been reported following COVID-19 vaccination.5 We report a case of HSP developing days after the messenger RNA Pfizer-BioNTech COVID-19 vaccine booster that was associated with anti-Smith and anti–double-stranded DNA (dsDNA) antibodies as well as antineutrophil cytoplasmic antibodies (ANCAs).
A 24-year-old man presented to dermatology with a rash of 3 weeks’ duration that first appeared 1 week after receiving his second booster of the messenger RNA Pfizer-BioNTech COVID-19 vaccine. Physical examination revealed petechiae with nonblanching erythematous macules and papules covering the legs below the knees (Figure 1) as well as the back of the right arm. A few days later, he developed arthralgia in the knees, hands, and feet. The patient denied any recent infections as well as respiratory and urinary tract symptoms. Approximately 10 days after the rash appeared, he developed epigastric abdominal pain that gradually worsened and sought care from his primary care physician, who ordered computed tomography and referred him for endoscopy. Computed tomography with and without contrast was suspicious for colitis. Colonoscopy and endoscopy were unremarkable. Laboratory tests were notable for elevated white blood cell count (17.08×103/µL [reference range, 3.66–10.60×103/µL]), serum IgA (437 mg/dL [reference range, 70–400 mg/dL]), C-reactive protein (1.5 mg/dL [reference range, <0.5 mg/dL]), anti-Smith antibody (28.1 CU [reference range, <20 CU), positive antinuclear antibody with titer (1:160 [reference range, <1:80]), anti-dsDNA (40.4 IU/mL [reference range, <27 IU/mL]), and cytoplasmic ANCA (c-ANCA) titer (1:320 [reference range, <1:20]). Blood urea nitrogen, creatinine, and estimated glomerular filtration rate were all within reference range. Urinalysis with microscopic examination was notable for 2 to 5 red blood cells per high-power field (reference range, 0) and proteinuria of 1+ (reference range, negative for protein).
The patient’s rash progressively worsened over the next few weeks, spreading proximally on the legs to the buttocks and the back of both elbows. A repeat complete blood cell count showed resolution of the leukocytosis. Two biopsies were taken from a lesion on the left proximal thigh: 1 for hematoxylin and eosin stain for histopathologic examination and 1 for direct immunofluorescence examination.
The patient was preliminarily diagnosed with HSP, and dermatology prescribed oral tofacitinib 5 mg twice daily for 5 days, which was supposed to be increased to 10 mg twice daily on the sixth day of treatment; however, the patient discontinued the medication after 4 days based on his primary care physician’s recommendation due to clotting concerns. The rash and arthralgia temporarily improved for 1 week, then relapsed.
Histopathology revealed neutrophils surrounding and infiltrating small dermal blood vessel walls as well as associated neutrophilic debris and erythrocytes, consistent with leukocytoclastic vasculitis (Figure 2). Direct immunofluorescence was negative for IgA antibodies. His primary care physician, in consultation with his dermatologist, then started the patient on oral prednisone 70 mg once daily for 7 days with a plan to taper. Three days after prednisone was started, the arthralgia and abdominal pain resolved, and the rash became lighter in color. After 1 week, the rash resolved completely.
Due to the unusual antibodies, the patient was referred to a rheumatologist, who repeated the blood tests approximately 1 week after the patient started prednisone. The tests were negative for anti-Smith, anti-dsDNA, and c-ANCA but showed an elevated atypical perinuclear ANCA (p-ANCA) titer of 1:80 (reference range [negative], <1:20). A repeat urinalysis was unremarkable. The patient slowly tapered the prednisone over the course of 3 months and was subsequently lost to follow-up. The rash and other symptoms had not recurred as of the patient’s last physician contact. The most recent laboratory results showed a white blood cell count of 14.0×103/µL (reference range, 3.4–10.8×103/µL), likely due to the prednisone; blood urea nitrogen, creatinine, and estimated glomerular filtration rate were within reference range. The urinalysis was notable for occult blood and was negative for protein. C-reactive protein was 1 mg/dL (reference range, 0–10 mg/dL); p-ANCA, c-ANCA, and atypical p-ANCA, as well as antinuclear antibody, were negative. As of his last follow-up, the patient felt well.
The major differential diagnoses for our patient included HSP, ANCA vasculitis, and systemic lupus erythematosus. Although ANCA vasculitis has been reported after SARS-CoV-2 infection,6 the lack of pulmonary symptoms made this diagnosis unlikely.7 Although our patient initially had elevated anti-Smith and anti-dsDNA antibodies as well as mild renal involvement, he fulfilled at most only 2 of the 11 criteria necessary for diagnosing lupus: malar rash, discoid rash (includes alopecia), photosensitivity, ocular ulcers, nonerosive arthritis, serositis, renal disorder (protein >500 mg/24 h, red blood cells, casts), neurologic disorder (seizures, psychosis), hematologic disorders (hemolytic anemia, leukopenia), ANA, and immunologic disorder (anti-Smith). Four of the 11 criteria are necessary for the diagnosis of lupus.8
Torraca et al7 reported a case of HSP with positive c-ANCA (1:640) in a patient lacking pulmonary symptoms who was diagnosed with HSP. Cytoplasmic ANCA is not a typical finding in HSP. However, the additional findings of anti-Smith, anti-dsDNA, and mildly elevated atypical p-ANCA antibodies in our patient were unexpected and could be explained by the proposed pathogenesis of HSP—an overzealous immune response resulting in aberrant antibody complex deposition with ensuing complement activation.5,9 Production of these additional antibodies could be part of the overzealous response to COVID-19 vaccination.
Of all the COVID-19 vaccines, messenger RNA–based vaccines have been associated with the majority of cutaneous reactions, including local injection-site reactions (most common), delayed local reactions, urticaria, angioedema, morbilliform eruption, herpes zoster eruption, bullous eruptions, dermal filler reactions, chilblains, and pityriasis rosea. Less common reactions have included acute generalized exanthematous pustulosis, Stevens-Johnson syndrome, erythema multiforme, Sweet Syndrome, lichen planus, papulovesicular eruptions, pityriasis rosea–like eruptions, generalized annular lesions, facial pustular neutrophilic eruptions, and flares of underlying autoimmune skin conditions.10 Multiple cases of HSP have been reported following COVID-19 vaccination from all the major vaccine companies.5
In our patient, laboratory tests were repeated by a rheumatologist and were negative for anti-Smith and anti-dsDNA antibodies as well as c-ANCA, most likely because he started taking prednisone approximately 1 week prior, which may have resulted in decreased antibodies. Also, the patient’s symptoms resolved after 1 week of steroid therapy. Therefore, the diagnosis is most consistent with HSP associated with COVID-19 vaccination. The clinical presentation, microscopic hematuria and proteinuria, and histopathology were consistent with the European Alliance of Associations for Rheumatology criteria for HSP.1
Although direct immunofluorescence typically is positive for IgA deposition on biopsies, it can be negative for IgA, especially in lesions that are biopsied more than 7 days after their appearance, as shown in our case; a negative IgA on immunofluorescence does not rule out HSP.4 Elevated serum IgA is seen in more than 50% of cases of HSP.11 Although the disease typically is self-limited, glucocorticoids are used if the disease course is prolonged or if there is evidence of kidney involvement.9 The unique combination of anti-Smith and anti-dsDNA antibodies as well as ANCAs associated with HSP with negative IgA on direct immunofluorescence has been reported with lupus.12 Clinicians should be aware of COVID-19 vaccine–associated HSP that is negative for IgA deposition and positive for anti-Smith and anti-dsDNA antibodies as well as ANCAs.
Acknowledgment—We thank our patient for granting permission to publish this information.
To the Editor:
Henoch-Schönlein purpura (HSP)(also known as IgA vasculitis) is a small vessel vasculitis characterized by deposition of IgA in small vessels, resulting in the development of purpura on the legs. Based on the European Alliance of Associations for Rheumatology criteria,1 the patient also must have at least 1 of the following: arthritis, arthralgia, abdominal pain, leukocytoclastic vasculitis with IgA deposition, or kidney involvement. The disease can be triggered by infection—with more than 75% of patients reporting an antecedent upper respiratory tract infection2—as well as medications, circulating immune complexes, certain foods, vaccines, and rarely cancer.3,4 The disease more commonly occurs in children but also can affect adults.
Several cases of HSP have been reported following COVID-19 vaccination.5 We report a case of HSP developing days after the messenger RNA Pfizer-BioNTech COVID-19 vaccine booster that was associated with anti-Smith and anti–double-stranded DNA (dsDNA) antibodies as well as antineutrophil cytoplasmic antibodies (ANCAs).
A 24-year-old man presented to dermatology with a rash of 3 weeks’ duration that first appeared 1 week after receiving his second booster of the messenger RNA Pfizer-BioNTech COVID-19 vaccine. Physical examination revealed petechiae with nonblanching erythematous macules and papules covering the legs below the knees (Figure 1) as well as the back of the right arm. A few days later, he developed arthralgia in the knees, hands, and feet. The patient denied any recent infections as well as respiratory and urinary tract symptoms. Approximately 10 days after the rash appeared, he developed epigastric abdominal pain that gradually worsened and sought care from his primary care physician, who ordered computed tomography and referred him for endoscopy. Computed tomography with and without contrast was suspicious for colitis. Colonoscopy and endoscopy were unremarkable. Laboratory tests were notable for elevated white blood cell count (17.08×103/µL [reference range, 3.66–10.60×103/µL]), serum IgA (437 mg/dL [reference range, 70–400 mg/dL]), C-reactive protein (1.5 mg/dL [reference range, <0.5 mg/dL]), anti-Smith antibody (28.1 CU [reference range, <20 CU), positive antinuclear antibody with titer (1:160 [reference range, <1:80]), anti-dsDNA (40.4 IU/mL [reference range, <27 IU/mL]), and cytoplasmic ANCA (c-ANCA) titer (1:320 [reference range, <1:20]). Blood urea nitrogen, creatinine, and estimated glomerular filtration rate were all within reference range. Urinalysis with microscopic examination was notable for 2 to 5 red blood cells per high-power field (reference range, 0) and proteinuria of 1+ (reference range, negative for protein).
The patient’s rash progressively worsened over the next few weeks, spreading proximally on the legs to the buttocks and the back of both elbows. A repeat complete blood cell count showed resolution of the leukocytosis. Two biopsies were taken from a lesion on the left proximal thigh: 1 for hematoxylin and eosin stain for histopathologic examination and 1 for direct immunofluorescence examination.
The patient was preliminarily diagnosed with HSP, and dermatology prescribed oral tofacitinib 5 mg twice daily for 5 days, which was supposed to be increased to 10 mg twice daily on the sixth day of treatment; however, the patient discontinued the medication after 4 days based on his primary care physician’s recommendation due to clotting concerns. The rash and arthralgia temporarily improved for 1 week, then relapsed.
Histopathology revealed neutrophils surrounding and infiltrating small dermal blood vessel walls as well as associated neutrophilic debris and erythrocytes, consistent with leukocytoclastic vasculitis (Figure 2). Direct immunofluorescence was negative for IgA antibodies. His primary care physician, in consultation with his dermatologist, then started the patient on oral prednisone 70 mg once daily for 7 days with a plan to taper. Three days after prednisone was started, the arthralgia and abdominal pain resolved, and the rash became lighter in color. After 1 week, the rash resolved completely.
Due to the unusual antibodies, the patient was referred to a rheumatologist, who repeated the blood tests approximately 1 week after the patient started prednisone. The tests were negative for anti-Smith, anti-dsDNA, and c-ANCA but showed an elevated atypical perinuclear ANCA (p-ANCA) titer of 1:80 (reference range [negative], <1:20). A repeat urinalysis was unremarkable. The patient slowly tapered the prednisone over the course of 3 months and was subsequently lost to follow-up. The rash and other symptoms had not recurred as of the patient’s last physician contact. The most recent laboratory results showed a white blood cell count of 14.0×103/µL (reference range, 3.4–10.8×103/µL), likely due to the prednisone; blood urea nitrogen, creatinine, and estimated glomerular filtration rate were within reference range. The urinalysis was notable for occult blood and was negative for protein. C-reactive protein was 1 mg/dL (reference range, 0–10 mg/dL); p-ANCA, c-ANCA, and atypical p-ANCA, as well as antinuclear antibody, were negative. As of his last follow-up, the patient felt well.
The major differential diagnoses for our patient included HSP, ANCA vasculitis, and systemic lupus erythematosus. Although ANCA vasculitis has been reported after SARS-CoV-2 infection,6 the lack of pulmonary symptoms made this diagnosis unlikely.7 Although our patient initially had elevated anti-Smith and anti-dsDNA antibodies as well as mild renal involvement, he fulfilled at most only 2 of the 11 criteria necessary for diagnosing lupus: malar rash, discoid rash (includes alopecia), photosensitivity, ocular ulcers, nonerosive arthritis, serositis, renal disorder (protein >500 mg/24 h, red blood cells, casts), neurologic disorder (seizures, psychosis), hematologic disorders (hemolytic anemia, leukopenia), ANA, and immunologic disorder (anti-Smith). Four of the 11 criteria are necessary for the diagnosis of lupus.8
Torraca et al7 reported a case of HSP with positive c-ANCA (1:640) in a patient lacking pulmonary symptoms who was diagnosed with HSP. Cytoplasmic ANCA is not a typical finding in HSP. However, the additional findings of anti-Smith, anti-dsDNA, and mildly elevated atypical p-ANCA antibodies in our patient were unexpected and could be explained by the proposed pathogenesis of HSP—an overzealous immune response resulting in aberrant antibody complex deposition with ensuing complement activation.5,9 Production of these additional antibodies could be part of the overzealous response to COVID-19 vaccination.
Of all the COVID-19 vaccines, messenger RNA–based vaccines have been associated with the majority of cutaneous reactions, including local injection-site reactions (most common), delayed local reactions, urticaria, angioedema, morbilliform eruption, herpes zoster eruption, bullous eruptions, dermal filler reactions, chilblains, and pityriasis rosea. Less common reactions have included acute generalized exanthematous pustulosis, Stevens-Johnson syndrome, erythema multiforme, Sweet Syndrome, lichen planus, papulovesicular eruptions, pityriasis rosea–like eruptions, generalized annular lesions, facial pustular neutrophilic eruptions, and flares of underlying autoimmune skin conditions.10 Multiple cases of HSP have been reported following COVID-19 vaccination from all the major vaccine companies.5
In our patient, laboratory tests were repeated by a rheumatologist and were negative for anti-Smith and anti-dsDNA antibodies as well as c-ANCA, most likely because he started taking prednisone approximately 1 week prior, which may have resulted in decreased antibodies. Also, the patient’s symptoms resolved after 1 week of steroid therapy. Therefore, the diagnosis is most consistent with HSP associated with COVID-19 vaccination. The clinical presentation, microscopic hematuria and proteinuria, and histopathology were consistent with the European Alliance of Associations for Rheumatology criteria for HSP.1
Although direct immunofluorescence typically is positive for IgA deposition on biopsies, it can be negative for IgA, especially in lesions that are biopsied more than 7 days after their appearance, as shown in our case; a negative IgA on immunofluorescence does not rule out HSP.4 Elevated serum IgA is seen in more than 50% of cases of HSP.11 Although the disease typically is self-limited, glucocorticoids are used if the disease course is prolonged or if there is evidence of kidney involvement.9 The unique combination of anti-Smith and anti-dsDNA antibodies as well as ANCAs associated with HSP with negative IgA on direct immunofluorescence has been reported with lupus.12 Clinicians should be aware of COVID-19 vaccine–associated HSP that is negative for IgA deposition and positive for anti-Smith and anti-dsDNA antibodies as well as ANCAs.
Acknowledgment—We thank our patient for granting permission to publish this information.
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936-941. doi:10.1136/ard.2005.046300
- Rai A, Nast C, Adler S. Henoch–Schönlein purpura nephritis. J Am Soc Nephrol. 1999;10:2637-2644.
- Casini F, Magenes VC, De Sanctis M, et al. Henoch-Schönlein purpura following COVID-19 vaccine in a child: a case report. Ital J Pediatr. 2022;48:158. doi:10.1186/s13052-022-01351-1
- Poudel P, Adams SH, Mirchia K, et al. IgA negative immunofluorescence in diagnoses of adult-onset Henoch-Schönlein purpura. Proc (Bayl Univ Med Cent). 2020;33:436-437. doi:10.1080/08998280.2020.1770526
- Maronese CA, Zelin E, Avallone G, et al. Cutaneous vasculitis and vasculopathy in the era of COVID-19 pandemic. Front Med (Lausanne). 2022;9:996288. doi:10.3389/fmed.2022.996288
- Bryant MC, Spencer LT, Yalcindag A. A case of ANCA-associated vasculitis in a 16-year-old female following SARS-COV-2 infection and a systematic review of the literature. Pediatr Rheumatol Online J. 2022;20:65. doi:10.1186/s12969-022-00727-1
- Torraca PFS, Castro BC, Hans Filho G. Henoch-Schönlein purpura with c-ANCA antibody in adult. An Bras Dermatol. 2016;91:667-669. doi:10.1590/abd1806-4841.20164181
- Agabegi SS, Agabegi ED. Step-Up to Medicine. 4th ed. Wolters Kluwer; 2015.
- Ball-Burack MR, Kosowsky JM. A Case of leukocytoclastic vasculitis following SARS-CoV-2 vaccination. J Emerg Med. 2022;63:E62-E65. doi:10.1016/j.jemermed.2021.10.005
- Tan SW, Tam YC, Pang SM. Cutaneous reactions to COVID-19 vaccines: a review. JAAD Int. 2022;7:178-186. doi:10.1016/j.jdin.2022.01.011
- Calviño MC, Llorca J, García-Porrúa C, et al. Henoch-Schönlein purpura in children from northwestern Spain: a 20-year epidemiologic and clinical study. Medicine (Baltimore). 2001;80:279-290.
- Hu P, Huang BY, Zhang DD, et al. Henoch-Schönlein purpura in a pediatric patient with lupus. Arch Med Sci. 2017;13:689-690. doi:10.5114/aoms.2017.67288
- Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936-941. doi:10.1136/ard.2005.046300
- Rai A, Nast C, Adler S. Henoch–Schönlein purpura nephritis. J Am Soc Nephrol. 1999;10:2637-2644.
- Casini F, Magenes VC, De Sanctis M, et al. Henoch-Schönlein purpura following COVID-19 vaccine in a child: a case report. Ital J Pediatr. 2022;48:158. doi:10.1186/s13052-022-01351-1
- Poudel P, Adams SH, Mirchia K, et al. IgA negative immunofluorescence in diagnoses of adult-onset Henoch-Schönlein purpura. Proc (Bayl Univ Med Cent). 2020;33:436-437. doi:10.1080/08998280.2020.1770526
- Maronese CA, Zelin E, Avallone G, et al. Cutaneous vasculitis and vasculopathy in the era of COVID-19 pandemic. Front Med (Lausanne). 2022;9:996288. doi:10.3389/fmed.2022.996288
- Bryant MC, Spencer LT, Yalcindag A. A case of ANCA-associated vasculitis in a 16-year-old female following SARS-COV-2 infection and a systematic review of the literature. Pediatr Rheumatol Online J. 2022;20:65. doi:10.1186/s12969-022-00727-1
- Torraca PFS, Castro BC, Hans Filho G. Henoch-Schönlein purpura with c-ANCA antibody in adult. An Bras Dermatol. 2016;91:667-669. doi:10.1590/abd1806-4841.20164181
- Agabegi SS, Agabegi ED. Step-Up to Medicine. 4th ed. Wolters Kluwer; 2015.
- Ball-Burack MR, Kosowsky JM. A Case of leukocytoclastic vasculitis following SARS-CoV-2 vaccination. J Emerg Med. 2022;63:E62-E65. doi:10.1016/j.jemermed.2021.10.005
- Tan SW, Tam YC, Pang SM. Cutaneous reactions to COVID-19 vaccines: a review. JAAD Int. 2022;7:178-186. doi:10.1016/j.jdin.2022.01.011
- Calviño MC, Llorca J, García-Porrúa C, et al. Henoch-Schönlein purpura in children from northwestern Spain: a 20-year epidemiologic and clinical study. Medicine (Baltimore). 2001;80:279-290.
- Hu P, Huang BY, Zhang DD, et al. Henoch-Schönlein purpura in a pediatric patient with lupus. Arch Med Sci. 2017;13:689-690. doi:10.5114/aoms.2017.67288
Practice Points
- Dermatologists should be vigilant for Henoch-Schönlein purpura (HSP) despite negative direct immunofluorescence of IgA deposition and unusual antibodies.
- Messenger RNA–based COVID-19 vaccines are associated with various cutaneous reactions, including HSP.
- Anti-Smith and anti–double-stranded DNA antibodies typically are not associated with HSP but may be seen in patients with coexisting systemic lupus erythematosus.
Cyclosporine for Recalcitrant Bullous Pemphigoid Induced by Nivolumab Therapy for Malignant Melanoma
To the Editor:
Immune checkpoint inhibitors have revolutionized the treatment of advanced-stage melanoma, with remarkably improved progression-free survival.1 Anti–programmed death receptor 1 (anti–PD-1) therapies, such as nivolumab and pembrolizumab, are a class of checkpoint inhibitors that have been approved by the US Food and Drug Administration for unresectable metastatic melanoma. Anti–PD-1 agents block the interaction of programmed death-ligand 1 (PD-L1) found on tumor cells with the PD-1 receptor on T cells, facilitating a positive immune response.2
Although these therapies have demonstrated notable antitumor efficacy, they also give rise to numerous immune-related adverse events (irAEs). As many as 70% of patients treated with PD-1/PD-L1 inhibitors experience some type of organ system irAE, of which 30% to 40% are cutaneous.3-6 Dermatologic adverse events are the most common irAEs, specifically spongiotic dermatitis, lichenoid dermatitis, pruritus, and vitiligo.7 Bullous pemphigoid (BP), an autoimmune bullous skin disorder caused by autoantibodies to basement membrane zone antigens, is a rare but potentially serious cutaneous irAE.8 Systemic corticosteroids commonly are used to treat immune checkpoint inhibitor–induced BP; other options include tetracyclines for maintenance therapy and rituximab for corticosteroid-refractory BP associated with anti-PD-1.9 We present a case of recalcitrant BP secondary to nivolumab therapy in a patient with metastatic melanoma who had near-complete resolution of BP following 2 months of cyclosporine.
A 41-year-old man presented with a generalized papular skin eruption of 1 month’s duration. He had a history of stage IIIC malignant melanoma of the lower right leg with positive sentinel lymph node biopsy. The largest lymph node deposit was 0.03 mm without extracapsular extension. Whole-body positron emission tomography–computed tomography showed no evidence of distant disease. The patient was treated with wide local excision with clear surgical margins plus 12 cycles of nivolumab, which was discontinued due to colitis. Four months after the final cycle of nivolumab, the patient developed widespread erythematous papules with hemorrhagic yellow crusting and no mucosal involvement. He was referred to dermatology by his primary oncologist for further evaluation.
A punch biopsy from the abdomen showed parakeratosis with leukocytoclasis and a superficial dermal infiltrate of neutrophils and eosinophils (Figure 1). Direct immunofluorescence revealed linear basement membrane deposits of IgG and C3, consistent with subepidermal blistering disease. Indirect immunofluorescence demonstrated trace IgG and IgG4 antibodies localized to the epidermal roof of salt-split skin and was negative for IgA antibodies. An enzyme-linked immunoassay was positive for BP antigen 2 (BP180) antibodies (98.4 U/mL [positive, ≥9 U/mL]) and negative for BP antigen 1 (BP230) antibodies (4.3 U/mL [positive, ≥9 U/mL]). Overall, these findings were consistent with a diagnosis of BP.
The patient was treated with prednisone 60 mg daily with initial response; however, there was disease recurrence with tapering. Doxycycline 100 mg twice daily and nicotinamide 500 mg twice daily were added as steroid-sparing agents, as prednisone was discontinued due to mood changes. Three months after the prednisone taper, the patient continued to develop new blisters. He completed treatment with doxycycline and nicotinamide. Rituximab 375 mg weekly was then initiated for 4 weeks.
At 2-week follow-up after completing the rituximab course, the patient reported worsening symptoms and presented with new bullae on the abdomen and upper extremities (Figure 2). Because of the recent history of mood changes while taking prednisone, a trial of cyclosporine 100 mg twice daily (1.37 mg/kg/d) was initiated, with notable improvement within 2 weeks of treatment. After 2 months of cyclosporine, approximately 90% of the rash had resolved with a few tense bullae remaining on the left frontal scalp but no new flares (Figure 3). One month after treatment ended, the patient remained clear of lesions without relapse.
Programmed death receptor 1 inhibitors have shown dramatic efficacy for a growing number of solid and hematologic malignancies, especially malignant melanoma. However, their use is accompanied by nonspecific activation of the immune system, resulting in a variety of adverse events, many manifesting on the skin. Several cases of BP in patients treated with PD-1/PD-L1 inhibitors have been reported.9 Cutaneous irAEs usually manifest within 3 weeks of initiation of PD-1 inhibitor therapy; however, the onset of BP typically occurs later at approximately 21 weeks.4,9 Our patient developed cutaneous manifestations 4 months after cessation of nivolumab.
Bullous pemphigoid classically manifests with pruritus and tense bullae. Notably, our patient’s clinical presentation included a widespread eruption of papules without bullae, which was similar to a review by Tsiogka et al,9 which reported that one-third of patients first present with a nonspecific cutaneous eruption. Bullous pemphigoid induced by anti–PD-1 may manifest differently than traditional BP, illuminating the importance of a thorough diagnostic workup.
Although the pathogenesis of immune checkpoint inhibitor–induced BP has not been fully elucidated, it is hypothesized to be caused by increased T cell cytotoxic activity leading to tumor lysis and release of numerous autoantigens. These autoantigens cause priming of abnormal T cells that can lead to further tissue damage in peripheral tissue and to generation of aberrant B cells and subsequent autoantibodies such as BP180 in germinal centers.4,10,11
Cyclosporine is a calcineurin inhibitor that reduces synthesis of IL-2, resulting in reduced cell activation.12 Therefore, cyclosporine may alleviate BP in patients who are being treated, or were previously treated, with an immune checkpoint inhibitor by suppressing T cell–mediated immune reaction and may be a rapid alternative for patients who cannot tolerate systemic steroids.
Treatment options for mild to moderate cases of BP include topical corticosteroids and antihistamines, while severe cases may require high-dose systemic corticosteroids. In recalcitrant cases, rituximab infusion with or without intravenous immunoglobulin often is utilized.8,13 The use of cyclosporine for various bullous disorders, including pemphigus vulgaris and epidermolysis bullosa acquisita, has been described.14 In recent years there has been a shift away from the use of cyclosporine for these conditions following the introduction of rituximab, a monoclonal antibody directed against the CD20 antigen on B lymphocytes. We utilized cyclosporine in our patient after he experienced worsening symptoms 1 month after completing treatment with rituximab.
Improvement from rituximab therapy may be delayed because it can take months to deplete CD20+ B lymphocytes from circulation, which may necessitate additional immunosuppressants or re-treatment with rituximab.15,16 In these instances, cyclosporine may be beneficial as a low-cost alternative in patients who are unable to tolerate systemic steroids, with a relatively good safety profile. The dosage of cyclosporine prescribed to the patient was chosen based on Joint American Academy of Dermatology–National Psoriasis Foundation management guidelines for psoriasis with systemic nonbiologic therapies, which recommends an initial dosage of 1 to 3 mg/kg/d in 2 divided doses.17
As immunotherapy for treating various cancers gains popularity, the frequency of dermatologic irAEs will increase. Therefore, dermatologists must be aware of the array of cutaneous manifestations, such as BP, and potential treatment options. When first-line and second-line therapies are contraindicated or do not provide notable improvement, cyclosporine may be an effective alternative for immune checkpoint inhibitor–induced BP.
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34. doi:10.1056/NEJMoa1504030
- Alsaab HO, Sau S, Alzhrani R, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol. 2017;8:561. doi:10.3389/fphar.2017.00561
- Puzanov I, Diab A, Abdallah K, et al; . Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunother Cancer. 2017;5:95. doi:10.1186/s40425-017-0300-z
- Geisler AN, Phillips GS, Barrios DM, et al. Immune checkpoint inhibitor-related dermatologic adverse events. J Am Acad Dermatol. 2020;83:1255-1268. doi:10.1016/j.jaad.2020.03.132
- Villadolid J, Amin A. Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities. Transl Lung Cancer Res. 2015;4:560-575. doi:10.3978/j.issn.2218-6751.2015.06.06
- Kumar V, Chaudhary N, Garg M, et al. Current diagnosis and management of immune related adverse events (irAEs) induced by immune checkpoint inhibitor therapy. Front Pharmacol. 2017;8:49. doi:10.3389/fphar.2017.00049
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25. doi:10.1016/j.ejca.2016.02.010
- Schauer F, Rafei-Shamsabadi D, Mai S, et al. Hemidesmosomal reactivity and treatment recommendations in immune checkpoint inhibitor-induced bullous pemphigoid—a retrospective, monocentric study. Front Immunol. 2022;13:953546. doi:10.3389/fimmu.2022.953546
- Tsiogka A, Bauer JW, Patsatsi A. Bullous pemphigoid associated with anti-programmed cell death protein 1 and anti-programmed cell death ligand 1 therapy: a review of the literature. Acta Derm Venereol. 2021;101:adv00377. doi:10.2340/00015555-3740
- Lopez AT, Khanna T, Antonov N, et al. A review of bullous pemphigoid associated with PD-1 and PD-L1 inhibitors. Int J Dermatol. 2018;57:664-669. doi:10.1111/ijd.13984
- Yang H, Yao Z, Zhou X, et al. Immune-related adverse events of checkpoint inhibitors: insights into immunological dysregulation. Clin Immunol. 2020;213:108377. doi:10.1016/j.clim.2020.108377
- Russell G, Graveley R, Seid J, et al. Mechanisms of action of cyclosporine and effects on connective tissues. Semin Arthritis Rheum. 1992;21(6 suppl 3):16-22. doi:10.1016/0049-0172(92)90009-3
- Ahmed AR, Shetty S, Kaveri S, et al. Treatment of recalcitrant bullous pemphigoid (BP) with a novel protocol: a retrospective study with a 6-year follow-up. J Am Acad Dermatol. 2016;74:700-708.e3. doi:10.1016/j.jaad.2015.11.030
- Amor KT, Ryan C, Menter A. The use of cyclosporine in dermatology: part I. J Am Acad Dermatol. 2010;63:925-946. doi:10.1016/j.jaad.2010.02.063
- Schmidt E, Hunzelmann N, Zillikens D, et al. Rituximab in refractory autoimmune bullous diseases. Clin Exp Dermatol. 2006;31:503-508. doi:10.1111/j.1365-2230.2006.02151.x
- Kasperkiewicz M, Shimanovich I, Ludwig RJ, et al. Rituximab for treatment-refractory pemphigus and pemphigoid: a case series of 17 patients. J Am Acad Dermatol. 2011;65:552-558.
- Menter A, Gelfand JM, Connor C, et al. Joint American Academy of Dermatology–National Psoriasis Foundation guidelines of care for the management of psoriasis with systemic nonbiologic therapies. J Am Acad Dermatol. 2020;82:1445-1486. doi:10.1016/j.jaad.2020.02.044
To the Editor:
Immune checkpoint inhibitors have revolutionized the treatment of advanced-stage melanoma, with remarkably improved progression-free survival.1 Anti–programmed death receptor 1 (anti–PD-1) therapies, such as nivolumab and pembrolizumab, are a class of checkpoint inhibitors that have been approved by the US Food and Drug Administration for unresectable metastatic melanoma. Anti–PD-1 agents block the interaction of programmed death-ligand 1 (PD-L1) found on tumor cells with the PD-1 receptor on T cells, facilitating a positive immune response.2
Although these therapies have demonstrated notable antitumor efficacy, they also give rise to numerous immune-related adverse events (irAEs). As many as 70% of patients treated with PD-1/PD-L1 inhibitors experience some type of organ system irAE, of which 30% to 40% are cutaneous.3-6 Dermatologic adverse events are the most common irAEs, specifically spongiotic dermatitis, lichenoid dermatitis, pruritus, and vitiligo.7 Bullous pemphigoid (BP), an autoimmune bullous skin disorder caused by autoantibodies to basement membrane zone antigens, is a rare but potentially serious cutaneous irAE.8 Systemic corticosteroids commonly are used to treat immune checkpoint inhibitor–induced BP; other options include tetracyclines for maintenance therapy and rituximab for corticosteroid-refractory BP associated with anti-PD-1.9 We present a case of recalcitrant BP secondary to nivolumab therapy in a patient with metastatic melanoma who had near-complete resolution of BP following 2 months of cyclosporine.
A 41-year-old man presented with a generalized papular skin eruption of 1 month’s duration. He had a history of stage IIIC malignant melanoma of the lower right leg with positive sentinel lymph node biopsy. The largest lymph node deposit was 0.03 mm without extracapsular extension. Whole-body positron emission tomography–computed tomography showed no evidence of distant disease. The patient was treated with wide local excision with clear surgical margins plus 12 cycles of nivolumab, which was discontinued due to colitis. Four months after the final cycle of nivolumab, the patient developed widespread erythematous papules with hemorrhagic yellow crusting and no mucosal involvement. He was referred to dermatology by his primary oncologist for further evaluation.
A punch biopsy from the abdomen showed parakeratosis with leukocytoclasis and a superficial dermal infiltrate of neutrophils and eosinophils (Figure 1). Direct immunofluorescence revealed linear basement membrane deposits of IgG and C3, consistent with subepidermal blistering disease. Indirect immunofluorescence demonstrated trace IgG and IgG4 antibodies localized to the epidermal roof of salt-split skin and was negative for IgA antibodies. An enzyme-linked immunoassay was positive for BP antigen 2 (BP180) antibodies (98.4 U/mL [positive, ≥9 U/mL]) and negative for BP antigen 1 (BP230) antibodies (4.3 U/mL [positive, ≥9 U/mL]). Overall, these findings were consistent with a diagnosis of BP.
The patient was treated with prednisone 60 mg daily with initial response; however, there was disease recurrence with tapering. Doxycycline 100 mg twice daily and nicotinamide 500 mg twice daily were added as steroid-sparing agents, as prednisone was discontinued due to mood changes. Three months after the prednisone taper, the patient continued to develop new blisters. He completed treatment with doxycycline and nicotinamide. Rituximab 375 mg weekly was then initiated for 4 weeks.
At 2-week follow-up after completing the rituximab course, the patient reported worsening symptoms and presented with new bullae on the abdomen and upper extremities (Figure 2). Because of the recent history of mood changes while taking prednisone, a trial of cyclosporine 100 mg twice daily (1.37 mg/kg/d) was initiated, with notable improvement within 2 weeks of treatment. After 2 months of cyclosporine, approximately 90% of the rash had resolved with a few tense bullae remaining on the left frontal scalp but no new flares (Figure 3). One month after treatment ended, the patient remained clear of lesions without relapse.
Programmed death receptor 1 inhibitors have shown dramatic efficacy for a growing number of solid and hematologic malignancies, especially malignant melanoma. However, their use is accompanied by nonspecific activation of the immune system, resulting in a variety of adverse events, many manifesting on the skin. Several cases of BP in patients treated with PD-1/PD-L1 inhibitors have been reported.9 Cutaneous irAEs usually manifest within 3 weeks of initiation of PD-1 inhibitor therapy; however, the onset of BP typically occurs later at approximately 21 weeks.4,9 Our patient developed cutaneous manifestations 4 months after cessation of nivolumab.
Bullous pemphigoid classically manifests with pruritus and tense bullae. Notably, our patient’s clinical presentation included a widespread eruption of papules without bullae, which was similar to a review by Tsiogka et al,9 which reported that one-third of patients first present with a nonspecific cutaneous eruption. Bullous pemphigoid induced by anti–PD-1 may manifest differently than traditional BP, illuminating the importance of a thorough diagnostic workup.
Although the pathogenesis of immune checkpoint inhibitor–induced BP has not been fully elucidated, it is hypothesized to be caused by increased T cell cytotoxic activity leading to tumor lysis and release of numerous autoantigens. These autoantigens cause priming of abnormal T cells that can lead to further tissue damage in peripheral tissue and to generation of aberrant B cells and subsequent autoantibodies such as BP180 in germinal centers.4,10,11
Cyclosporine is a calcineurin inhibitor that reduces synthesis of IL-2, resulting in reduced cell activation.12 Therefore, cyclosporine may alleviate BP in patients who are being treated, or were previously treated, with an immune checkpoint inhibitor by suppressing T cell–mediated immune reaction and may be a rapid alternative for patients who cannot tolerate systemic steroids.
Treatment options for mild to moderate cases of BP include topical corticosteroids and antihistamines, while severe cases may require high-dose systemic corticosteroids. In recalcitrant cases, rituximab infusion with or without intravenous immunoglobulin often is utilized.8,13 The use of cyclosporine for various bullous disorders, including pemphigus vulgaris and epidermolysis bullosa acquisita, has been described.14 In recent years there has been a shift away from the use of cyclosporine for these conditions following the introduction of rituximab, a monoclonal antibody directed against the CD20 antigen on B lymphocytes. We utilized cyclosporine in our patient after he experienced worsening symptoms 1 month after completing treatment with rituximab.
Improvement from rituximab therapy may be delayed because it can take months to deplete CD20+ B lymphocytes from circulation, which may necessitate additional immunosuppressants or re-treatment with rituximab.15,16 In these instances, cyclosporine may be beneficial as a low-cost alternative in patients who are unable to tolerate systemic steroids, with a relatively good safety profile. The dosage of cyclosporine prescribed to the patient was chosen based on Joint American Academy of Dermatology–National Psoriasis Foundation management guidelines for psoriasis with systemic nonbiologic therapies, which recommends an initial dosage of 1 to 3 mg/kg/d in 2 divided doses.17
As immunotherapy for treating various cancers gains popularity, the frequency of dermatologic irAEs will increase. Therefore, dermatologists must be aware of the array of cutaneous manifestations, such as BP, and potential treatment options. When first-line and second-line therapies are contraindicated or do not provide notable improvement, cyclosporine may be an effective alternative for immune checkpoint inhibitor–induced BP.
To the Editor:
Immune checkpoint inhibitors have revolutionized the treatment of advanced-stage melanoma, with remarkably improved progression-free survival.1 Anti–programmed death receptor 1 (anti–PD-1) therapies, such as nivolumab and pembrolizumab, are a class of checkpoint inhibitors that have been approved by the US Food and Drug Administration for unresectable metastatic melanoma. Anti–PD-1 agents block the interaction of programmed death-ligand 1 (PD-L1) found on tumor cells with the PD-1 receptor on T cells, facilitating a positive immune response.2
Although these therapies have demonstrated notable antitumor efficacy, they also give rise to numerous immune-related adverse events (irAEs). As many as 70% of patients treated with PD-1/PD-L1 inhibitors experience some type of organ system irAE, of which 30% to 40% are cutaneous.3-6 Dermatologic adverse events are the most common irAEs, specifically spongiotic dermatitis, lichenoid dermatitis, pruritus, and vitiligo.7 Bullous pemphigoid (BP), an autoimmune bullous skin disorder caused by autoantibodies to basement membrane zone antigens, is a rare but potentially serious cutaneous irAE.8 Systemic corticosteroids commonly are used to treat immune checkpoint inhibitor–induced BP; other options include tetracyclines for maintenance therapy and rituximab for corticosteroid-refractory BP associated with anti-PD-1.9 We present a case of recalcitrant BP secondary to nivolumab therapy in a patient with metastatic melanoma who had near-complete resolution of BP following 2 months of cyclosporine.
A 41-year-old man presented with a generalized papular skin eruption of 1 month’s duration. He had a history of stage IIIC malignant melanoma of the lower right leg with positive sentinel lymph node biopsy. The largest lymph node deposit was 0.03 mm without extracapsular extension. Whole-body positron emission tomography–computed tomography showed no evidence of distant disease. The patient was treated with wide local excision with clear surgical margins plus 12 cycles of nivolumab, which was discontinued due to colitis. Four months after the final cycle of nivolumab, the patient developed widespread erythematous papules with hemorrhagic yellow crusting and no mucosal involvement. He was referred to dermatology by his primary oncologist for further evaluation.
A punch biopsy from the abdomen showed parakeratosis with leukocytoclasis and a superficial dermal infiltrate of neutrophils and eosinophils (Figure 1). Direct immunofluorescence revealed linear basement membrane deposits of IgG and C3, consistent with subepidermal blistering disease. Indirect immunofluorescence demonstrated trace IgG and IgG4 antibodies localized to the epidermal roof of salt-split skin and was negative for IgA antibodies. An enzyme-linked immunoassay was positive for BP antigen 2 (BP180) antibodies (98.4 U/mL [positive, ≥9 U/mL]) and negative for BP antigen 1 (BP230) antibodies (4.3 U/mL [positive, ≥9 U/mL]). Overall, these findings were consistent with a diagnosis of BP.
The patient was treated with prednisone 60 mg daily with initial response; however, there was disease recurrence with tapering. Doxycycline 100 mg twice daily and nicotinamide 500 mg twice daily were added as steroid-sparing agents, as prednisone was discontinued due to mood changes. Three months after the prednisone taper, the patient continued to develop new blisters. He completed treatment with doxycycline and nicotinamide. Rituximab 375 mg weekly was then initiated for 4 weeks.
At 2-week follow-up after completing the rituximab course, the patient reported worsening symptoms and presented with new bullae on the abdomen and upper extremities (Figure 2). Because of the recent history of mood changes while taking prednisone, a trial of cyclosporine 100 mg twice daily (1.37 mg/kg/d) was initiated, with notable improvement within 2 weeks of treatment. After 2 months of cyclosporine, approximately 90% of the rash had resolved with a few tense bullae remaining on the left frontal scalp but no new flares (Figure 3). One month after treatment ended, the patient remained clear of lesions without relapse.
Programmed death receptor 1 inhibitors have shown dramatic efficacy for a growing number of solid and hematologic malignancies, especially malignant melanoma. However, their use is accompanied by nonspecific activation of the immune system, resulting in a variety of adverse events, many manifesting on the skin. Several cases of BP in patients treated with PD-1/PD-L1 inhibitors have been reported.9 Cutaneous irAEs usually manifest within 3 weeks of initiation of PD-1 inhibitor therapy; however, the onset of BP typically occurs later at approximately 21 weeks.4,9 Our patient developed cutaneous manifestations 4 months after cessation of nivolumab.
Bullous pemphigoid classically manifests with pruritus and tense bullae. Notably, our patient’s clinical presentation included a widespread eruption of papules without bullae, which was similar to a review by Tsiogka et al,9 which reported that one-third of patients first present with a nonspecific cutaneous eruption. Bullous pemphigoid induced by anti–PD-1 may manifest differently than traditional BP, illuminating the importance of a thorough diagnostic workup.
Although the pathogenesis of immune checkpoint inhibitor–induced BP has not been fully elucidated, it is hypothesized to be caused by increased T cell cytotoxic activity leading to tumor lysis and release of numerous autoantigens. These autoantigens cause priming of abnormal T cells that can lead to further tissue damage in peripheral tissue and to generation of aberrant B cells and subsequent autoantibodies such as BP180 in germinal centers.4,10,11
Cyclosporine is a calcineurin inhibitor that reduces synthesis of IL-2, resulting in reduced cell activation.12 Therefore, cyclosporine may alleviate BP in patients who are being treated, or were previously treated, with an immune checkpoint inhibitor by suppressing T cell–mediated immune reaction and may be a rapid alternative for patients who cannot tolerate systemic steroids.
Treatment options for mild to moderate cases of BP include topical corticosteroids and antihistamines, while severe cases may require high-dose systemic corticosteroids. In recalcitrant cases, rituximab infusion with or without intravenous immunoglobulin often is utilized.8,13 The use of cyclosporine for various bullous disorders, including pemphigus vulgaris and epidermolysis bullosa acquisita, has been described.14 In recent years there has been a shift away from the use of cyclosporine for these conditions following the introduction of rituximab, a monoclonal antibody directed against the CD20 antigen on B lymphocytes. We utilized cyclosporine in our patient after he experienced worsening symptoms 1 month after completing treatment with rituximab.
Improvement from rituximab therapy may be delayed because it can take months to deplete CD20+ B lymphocytes from circulation, which may necessitate additional immunosuppressants or re-treatment with rituximab.15,16 In these instances, cyclosporine may be beneficial as a low-cost alternative in patients who are unable to tolerate systemic steroids, with a relatively good safety profile. The dosage of cyclosporine prescribed to the patient was chosen based on Joint American Academy of Dermatology–National Psoriasis Foundation management guidelines for psoriasis with systemic nonbiologic therapies, which recommends an initial dosage of 1 to 3 mg/kg/d in 2 divided doses.17
As immunotherapy for treating various cancers gains popularity, the frequency of dermatologic irAEs will increase. Therefore, dermatologists must be aware of the array of cutaneous manifestations, such as BP, and potential treatment options. When first-line and second-line therapies are contraindicated or do not provide notable improvement, cyclosporine may be an effective alternative for immune checkpoint inhibitor–induced BP.
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34. doi:10.1056/NEJMoa1504030
- Alsaab HO, Sau S, Alzhrani R, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol. 2017;8:561. doi:10.3389/fphar.2017.00561
- Puzanov I, Diab A, Abdallah K, et al; . Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunother Cancer. 2017;5:95. doi:10.1186/s40425-017-0300-z
- Geisler AN, Phillips GS, Barrios DM, et al. Immune checkpoint inhibitor-related dermatologic adverse events. J Am Acad Dermatol. 2020;83:1255-1268. doi:10.1016/j.jaad.2020.03.132
- Villadolid J, Amin A. Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities. Transl Lung Cancer Res. 2015;4:560-575. doi:10.3978/j.issn.2218-6751.2015.06.06
- Kumar V, Chaudhary N, Garg M, et al. Current diagnosis and management of immune related adverse events (irAEs) induced by immune checkpoint inhibitor therapy. Front Pharmacol. 2017;8:49. doi:10.3389/fphar.2017.00049
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25. doi:10.1016/j.ejca.2016.02.010
- Schauer F, Rafei-Shamsabadi D, Mai S, et al. Hemidesmosomal reactivity and treatment recommendations in immune checkpoint inhibitor-induced bullous pemphigoid—a retrospective, monocentric study. Front Immunol. 2022;13:953546. doi:10.3389/fimmu.2022.953546
- Tsiogka A, Bauer JW, Patsatsi A. Bullous pemphigoid associated with anti-programmed cell death protein 1 and anti-programmed cell death ligand 1 therapy: a review of the literature. Acta Derm Venereol. 2021;101:adv00377. doi:10.2340/00015555-3740
- Lopez AT, Khanna T, Antonov N, et al. A review of bullous pemphigoid associated with PD-1 and PD-L1 inhibitors. Int J Dermatol. 2018;57:664-669. doi:10.1111/ijd.13984
- Yang H, Yao Z, Zhou X, et al. Immune-related adverse events of checkpoint inhibitors: insights into immunological dysregulation. Clin Immunol. 2020;213:108377. doi:10.1016/j.clim.2020.108377
- Russell G, Graveley R, Seid J, et al. Mechanisms of action of cyclosporine and effects on connective tissues. Semin Arthritis Rheum. 1992;21(6 suppl 3):16-22. doi:10.1016/0049-0172(92)90009-3
- Ahmed AR, Shetty S, Kaveri S, et al. Treatment of recalcitrant bullous pemphigoid (BP) with a novel protocol: a retrospective study with a 6-year follow-up. J Am Acad Dermatol. 2016;74:700-708.e3. doi:10.1016/j.jaad.2015.11.030
- Amor KT, Ryan C, Menter A. The use of cyclosporine in dermatology: part I. J Am Acad Dermatol. 2010;63:925-946. doi:10.1016/j.jaad.2010.02.063
- Schmidt E, Hunzelmann N, Zillikens D, et al. Rituximab in refractory autoimmune bullous diseases. Clin Exp Dermatol. 2006;31:503-508. doi:10.1111/j.1365-2230.2006.02151.x
- Kasperkiewicz M, Shimanovich I, Ludwig RJ, et al. Rituximab for treatment-refractory pemphigus and pemphigoid: a case series of 17 patients. J Am Acad Dermatol. 2011;65:552-558.
- Menter A, Gelfand JM, Connor C, et al. Joint American Academy of Dermatology–National Psoriasis Foundation guidelines of care for the management of psoriasis with systemic nonbiologic therapies. J Am Acad Dermatol. 2020;82:1445-1486. doi:10.1016/j.jaad.2020.02.044
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23-34. doi:10.1056/NEJMoa1504030
- Alsaab HO, Sau S, Alzhrani R, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol. 2017;8:561. doi:10.3389/fphar.2017.00561
- Puzanov I, Diab A, Abdallah K, et al; . Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunother Cancer. 2017;5:95. doi:10.1186/s40425-017-0300-z
- Geisler AN, Phillips GS, Barrios DM, et al. Immune checkpoint inhibitor-related dermatologic adverse events. J Am Acad Dermatol. 2020;83:1255-1268. doi:10.1016/j.jaad.2020.03.132
- Villadolid J, Amin A. Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities. Transl Lung Cancer Res. 2015;4:560-575. doi:10.3978/j.issn.2218-6751.2015.06.06
- Kumar V, Chaudhary N, Garg M, et al. Current diagnosis and management of immune related adverse events (irAEs) induced by immune checkpoint inhibitor therapy. Front Pharmacol. 2017;8:49. doi:10.3389/fphar.2017.00049
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25. doi:10.1016/j.ejca.2016.02.010
- Schauer F, Rafei-Shamsabadi D, Mai S, et al. Hemidesmosomal reactivity and treatment recommendations in immune checkpoint inhibitor-induced bullous pemphigoid—a retrospective, monocentric study. Front Immunol. 2022;13:953546. doi:10.3389/fimmu.2022.953546
- Tsiogka A, Bauer JW, Patsatsi A. Bullous pemphigoid associated with anti-programmed cell death protein 1 and anti-programmed cell death ligand 1 therapy: a review of the literature. Acta Derm Venereol. 2021;101:adv00377. doi:10.2340/00015555-3740
- Lopez AT, Khanna T, Antonov N, et al. A review of bullous pemphigoid associated with PD-1 and PD-L1 inhibitors. Int J Dermatol. 2018;57:664-669. doi:10.1111/ijd.13984
- Yang H, Yao Z, Zhou X, et al. Immune-related adverse events of checkpoint inhibitors: insights into immunological dysregulation. Clin Immunol. 2020;213:108377. doi:10.1016/j.clim.2020.108377
- Russell G, Graveley R, Seid J, et al. Mechanisms of action of cyclosporine and effects on connective tissues. Semin Arthritis Rheum. 1992;21(6 suppl 3):16-22. doi:10.1016/0049-0172(92)90009-3
- Ahmed AR, Shetty S, Kaveri S, et al. Treatment of recalcitrant bullous pemphigoid (BP) with a novel protocol: a retrospective study with a 6-year follow-up. J Am Acad Dermatol. 2016;74:700-708.e3. doi:10.1016/j.jaad.2015.11.030
- Amor KT, Ryan C, Menter A. The use of cyclosporine in dermatology: part I. J Am Acad Dermatol. 2010;63:925-946. doi:10.1016/j.jaad.2010.02.063
- Schmidt E, Hunzelmann N, Zillikens D, et al. Rituximab in refractory autoimmune bullous diseases. Clin Exp Dermatol. 2006;31:503-508. doi:10.1111/j.1365-2230.2006.02151.x
- Kasperkiewicz M, Shimanovich I, Ludwig RJ, et al. Rituximab for treatment-refractory pemphigus and pemphigoid: a case series of 17 patients. J Am Acad Dermatol. 2011;65:552-558.
- Menter A, Gelfand JM, Connor C, et al. Joint American Academy of Dermatology–National Psoriasis Foundation guidelines of care for the management of psoriasis with systemic nonbiologic therapies. J Am Acad Dermatol. 2020;82:1445-1486. doi:10.1016/j.jaad.2020.02.044
Practice Points
- Bullous pemphigoid is a rare dermatologic immune-related adverse event that can occur secondary to anti–programmed death receptor 1 therapy.
- For cases of immune checkpoint inhibitor–induced bullous pemphigoid that are recalcitrant to corticosteroids and rituximab, cyclosporine might be an effective alternative.
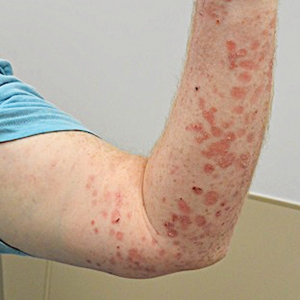
Mycobacterium interjectum Infection in an Immunocompetent Host Following Contact With Aquarium Fish
To the Editor:
A 48-year-old man presented with nodular lesions in a sporotrichoid pattern on the right hand and forearm of 3 months’ duration (Figure). There were no lymphadeno-pathies, and he had no notable medical history. He denied fever and other systemic symptoms. The patient recently had manipulated a warm water fish aquarium. Although he did not recall a clear injury, inadvertent mild trauma was a possibility. He denied other contact or trauma in relation to animals or vegetables.
Histopathology from a punch biopsy of the forearm revealed a granulomatous infiltrate with necrosis at the deep dermis level at the interface with the subcutaneous cellular tissue that was composed of mainly epithelioid cells with a few multinucleated giant cells. No acid-fast bacilli or fungi were observed with special stains.
A polymerase chain reaction assay for atypical mycobacteria was positive for Mycobacterium interjectum. The culture of the skin biopsy was negative for fungi and mycobacteria after long incubation (6 weeks) on 2 occasions, and an antibiogram was not available. Complementary tests including hemogram, HIV serology, and chest and upper extremity radiographs did not reveal any abnormalities.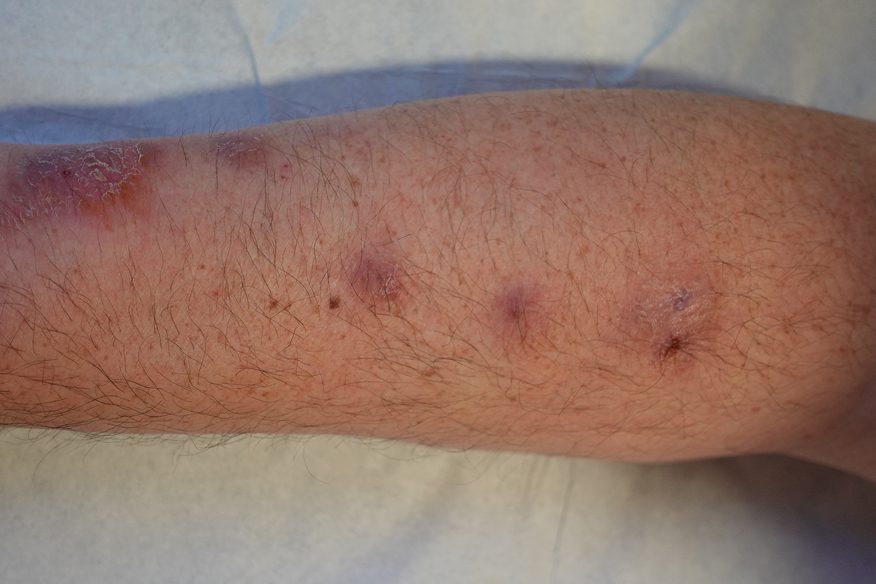
The patient was treated with rifampicin 600 mg/d, clarithromycin 500 mg every 12 hours, and co-trimoxazole 160/800 mg every 12 hours for 9 months with some resolution but persistence of some residual scarring lesions. There was no recurrence at 6-month follow-up.
Mycobacterium interjectum is a rare, slow-growing, scotochromogenic mycobacteria. Case reports usually refer to lymphadenitis in healthy children and pulmonary infections in immunocompromised or immunocompetent adults.1,2 A case of M interjectum with cutaneous involvement was reported by Fukuoka et al,3 with ulcerated nodules and abscesses on the leg identified in an immunocompromised patient. Our patient did not present with any cause of immunosuppression or clear injury predisposing him to infection. This microorganism has been detected in water, soil,3 and aquarium fish,4 the latter being the most likely source of infection in our patient. Given its slow growth rate and the need for a specific polymerase chain reaction assay, which is not widely available, M interjectum infection may be underdiagnosed.
No standard antibiotic regimen has been established, but M interjectum has proven to be a multidrug-resistant bacterium with frequent therapy failures. Treatment options have ranged from standard tuberculostatic therapy to combination therapy with medications such as amikacin, levofloxacin, rifampicin, and co-trimoxazole.1 Because an antibiogram was not available for our patient, empiric treatment with rifampicin, clarithromycin, and co-trimoxazole was prescribed for 9 months, with satisfactory response and tolerance. These drugs were selected because of their susceptibility profile in the literature.1,5
- Sotello D, Hata DJ, Reza M, et al. Disseminated Mycobacterium interjectum infection with bacteremia, hepatic and pulmonary involvement associated with a long-term catheter infection. Case Rep Infect Dis. 2017;2017:1-5.
- Dholakia YN. Mycobacterium interjectum isolated from an immunocompetent host with lung infection. Int J Mycobacteriol. 2017;6:401-403.
- Fukuoka M, Matsumura Y, Kore-eda S, et al. Cutaneous infection due to Mycobacterium interjectum in an immunosuppressed patient with microscopic polyangiitis. Br J Dermatol. 2008;159:1382-1384.
- Zanoni RG, Florio D, Fioravanti ML, et al. Occurrence of Mycobacterium spp. in ornamental fish in Italy. J Fish Dis. 2008;31:433-441.
- Emler S, Rochat T, Rohner P, et al. Chronic destructive lung disease associated with a novel mycobacterium. Am J Respir Crit Care Med. 1994;150:261-265.
To the Editor:
A 48-year-old man presented with nodular lesions in a sporotrichoid pattern on the right hand and forearm of 3 months’ duration (Figure). There were no lymphadeno-pathies, and he had no notable medical history. He denied fever and other systemic symptoms. The patient recently had manipulated a warm water fish aquarium. Although he did not recall a clear injury, inadvertent mild trauma was a possibility. He denied other contact or trauma in relation to animals or vegetables.
Histopathology from a punch biopsy of the forearm revealed a granulomatous infiltrate with necrosis at the deep dermis level at the interface with the subcutaneous cellular tissue that was composed of mainly epithelioid cells with a few multinucleated giant cells. No acid-fast bacilli or fungi were observed with special stains.
A polymerase chain reaction assay for atypical mycobacteria was positive for Mycobacterium interjectum. The culture of the skin biopsy was negative for fungi and mycobacteria after long incubation (6 weeks) on 2 occasions, and an antibiogram was not available. Complementary tests including hemogram, HIV serology, and chest and upper extremity radiographs did not reveal any abnormalities.
The patient was treated with rifampicin 600 mg/d, clarithromycin 500 mg every 12 hours, and co-trimoxazole 160/800 mg every 12 hours for 9 months with some resolution but persistence of some residual scarring lesions. There was no recurrence at 6-month follow-up.
Mycobacterium interjectum is a rare, slow-growing, scotochromogenic mycobacteria. Case reports usually refer to lymphadenitis in healthy children and pulmonary infections in immunocompromised or immunocompetent adults.1,2 A case of M interjectum with cutaneous involvement was reported by Fukuoka et al,3 with ulcerated nodules and abscesses on the leg identified in an immunocompromised patient. Our patient did not present with any cause of immunosuppression or clear injury predisposing him to infection. This microorganism has been detected in water, soil,3 and aquarium fish,4 the latter being the most likely source of infection in our patient. Given its slow growth rate and the need for a specific polymerase chain reaction assay, which is not widely available, M interjectum infection may be underdiagnosed.
No standard antibiotic regimen has been established, but M interjectum has proven to be a multidrug-resistant bacterium with frequent therapy failures. Treatment options have ranged from standard tuberculostatic therapy to combination therapy with medications such as amikacin, levofloxacin, rifampicin, and co-trimoxazole.1 Because an antibiogram was not available for our patient, empiric treatment with rifampicin, clarithromycin, and co-trimoxazole was prescribed for 9 months, with satisfactory response and tolerance. These drugs were selected because of their susceptibility profile in the literature.1,5
To the Editor:
A 48-year-old man presented with nodular lesions in a sporotrichoid pattern on the right hand and forearm of 3 months’ duration (Figure). There were no lymphadeno-pathies, and he had no notable medical history. He denied fever and other systemic symptoms. The patient recently had manipulated a warm water fish aquarium. Although he did not recall a clear injury, inadvertent mild trauma was a possibility. He denied other contact or trauma in relation to animals or vegetables.
Histopathology from a punch biopsy of the forearm revealed a granulomatous infiltrate with necrosis at the deep dermis level at the interface with the subcutaneous cellular tissue that was composed of mainly epithelioid cells with a few multinucleated giant cells. No acid-fast bacilli or fungi were observed with special stains.
A polymerase chain reaction assay for atypical mycobacteria was positive for Mycobacterium interjectum. The culture of the skin biopsy was negative for fungi and mycobacteria after long incubation (6 weeks) on 2 occasions, and an antibiogram was not available. Complementary tests including hemogram, HIV serology, and chest and upper extremity radiographs did not reveal any abnormalities.
The patient was treated with rifampicin 600 mg/d, clarithromycin 500 mg every 12 hours, and co-trimoxazole 160/800 mg every 12 hours for 9 months with some resolution but persistence of some residual scarring lesions. There was no recurrence at 6-month follow-up.
Mycobacterium interjectum is a rare, slow-growing, scotochromogenic mycobacteria. Case reports usually refer to lymphadenitis in healthy children and pulmonary infections in immunocompromised or immunocompetent adults.1,2 A case of M interjectum with cutaneous involvement was reported by Fukuoka et al,3 with ulcerated nodules and abscesses on the leg identified in an immunocompromised patient. Our patient did not present with any cause of immunosuppression or clear injury predisposing him to infection. This microorganism has been detected in water, soil,3 and aquarium fish,4 the latter being the most likely source of infection in our patient. Given its slow growth rate and the need for a specific polymerase chain reaction assay, which is not widely available, M interjectum infection may be underdiagnosed.
No standard antibiotic regimen has been established, but M interjectum has proven to be a multidrug-resistant bacterium with frequent therapy failures. Treatment options have ranged from standard tuberculostatic therapy to combination therapy with medications such as amikacin, levofloxacin, rifampicin, and co-trimoxazole.1 Because an antibiogram was not available for our patient, empiric treatment with rifampicin, clarithromycin, and co-trimoxazole was prescribed for 9 months, with satisfactory response and tolerance. These drugs were selected because of their susceptibility profile in the literature.1,5
- Sotello D, Hata DJ, Reza M, et al. Disseminated Mycobacterium interjectum infection with bacteremia, hepatic and pulmonary involvement associated with a long-term catheter infection. Case Rep Infect Dis. 2017;2017:1-5.
- Dholakia YN. Mycobacterium interjectum isolated from an immunocompetent host with lung infection. Int J Mycobacteriol. 2017;6:401-403.
- Fukuoka M, Matsumura Y, Kore-eda S, et al. Cutaneous infection due to Mycobacterium interjectum in an immunosuppressed patient with microscopic polyangiitis. Br J Dermatol. 2008;159:1382-1384.
- Zanoni RG, Florio D, Fioravanti ML, et al. Occurrence of Mycobacterium spp. in ornamental fish in Italy. J Fish Dis. 2008;31:433-441.
- Emler S, Rochat T, Rohner P, et al. Chronic destructive lung disease associated with a novel mycobacterium. Am J Respir Crit Care Med. 1994;150:261-265.
- Sotello D, Hata DJ, Reza M, et al. Disseminated Mycobacterium interjectum infection with bacteremia, hepatic and pulmonary involvement associated with a long-term catheter infection. Case Rep Infect Dis. 2017;2017:1-5.
- Dholakia YN. Mycobacterium interjectum isolated from an immunocompetent host with lung infection. Int J Mycobacteriol. 2017;6:401-403.
- Fukuoka M, Matsumura Y, Kore-eda S, et al. Cutaneous infection due to Mycobacterium interjectum in an immunosuppressed patient with microscopic polyangiitis. Br J Dermatol. 2008;159:1382-1384.
- Zanoni RG, Florio D, Fioravanti ML, et al. Occurrence of Mycobacterium spp. in ornamental fish in Italy. J Fish Dis. 2008;31:433-441.
- Emler S, Rochat T, Rohner P, et al. Chronic destructive lung disease associated with a novel mycobacterium. Am J Respir Crit Care Med. 1994;150:261-265.
Practice Points
- Mycobacterium interjectum can cause cutaneous nodules in a sporotrichoid or lymphocutaneous pattern and may affect immunocompromised and immunocompetent patients.
- This mycobacteria has been detected in water, soil, and aquarium fish. The latter could be a source of infection and should be taken into account in the anamnesis.
- There is no established therapeutic regimen for M interjectum infection. Combination therapy with rifampicin, clarithromycin, and co-trimoxazole could be an option, though it must always be adapted to an antibiogram if results are available.
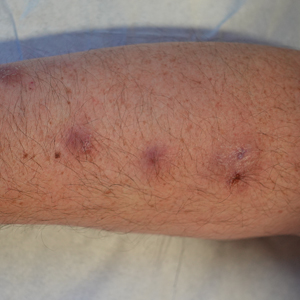
Histiocytoid Pyoderma Gangrenosum: A Challenging Case With Features of Sweet Syndrome
To the Editor:
Neutrophilic dermatoses—a group of inflammatory cutaneous conditions—include acute febrile neutrophilic dermatosis (Sweet syndrome), pyoderma gangrenosum, and neutrophilic dermatosis of the dorsal hands. Histopathology shows a dense dermal infiltrate of mature neutrophils. In 2005, the histiocytoid subtype of Sweet syndrome was introduced with histopathologic findings of a dermal infiltrate composed of immature myeloid cells that resemble histiocytes in appearance but stain strongly with neutrophil markers on immunohistochemistry.1 We present a case of histiocytoid pyoderma gangrenosum with histopathology that showed a dense dermal histiocytoid infiltrate with strong positivity for neutrophil markers on immunohistochemistry.
An 85-year-old man was seen by dermatology in the inpatient setting for a new-onset painful abdominal wound. He had a medical history of myelodysplastic syndrome (MDS), high-grade invasive papillary urothelial carcinoma of the bladder, and a recent diagnosis of low-grade invasive ascending colon adenocarcinoma. Ten days prior he underwent a right colectomy without intraoperative complications that was followed by septic shock. Workup with urinalysis and urine culture showed minimal pyuria with Pseudomonas aeruginosa. Additional studies, including blood cultures, abdominal wound cultures, computed tomography of the abdomen and pelvis, renal ultrasound, and chest radiographs, were unremarkable and showed no signs of surgical site infection, intra-abdominal or pelvic abscess formation, or pulmonary embolism. Broad-spectrum antibiotics—vancomycin and piperacillin-tazobactam—were started. Persistent fever (Tmax of 102.3 °F [39.1 °C]) and leukocytosis (45.3×109/L [4.2–10×109/L]) despite antibiotic therapy, increasing pressor requirements, and progressive painful erythema and purulence at the abdominal surgical site led to debridement of the wound by the general surgery team on day 9 following the initial surgery due to suspected necrotizing infection. Within 24 hours, dermatology was consulted for continued rapid expansion of the wound. Physical examination of the abdomen revealed a large, well-demarcated, pink-red, indurated, ulcerated plaque with clear to purulent exudate and superficial erosions with violaceous undermined borders extending centrifugally from the abdominal surgical incision line (Figure 1A). Two punch biopsies sent for histopathologic evaluation and tissue culture showed dermal edema with a dense histiocytic infiltrate with nodular foci and admixed mature neutrophils to a lesser degree (Figure 2). Special staining was negative for bacteria, fungi, and mycobacteria. Immunohistochemistry revealed positive staining of the dermal inflammatory infiltrate with CD68, myeloperoxidase, and lysozyme, as well as negative staining with CD34 (Figure 3). These findings were suggestive of a histiocytoid neutrophilic dermatosis such as Sweet syndrome or pyoderma gangrenosum. Due to the morphology of the solitary lesion and the abrupt exacerbation shortly after surgical intervention, the patient was diagnosed with histiocytoid pyoderma gangrenosum. At the same time, the patient’s septic shock was treated with intravenous hydrocortisone (100 mg 3 times daily) for 2 days and also achieved a prompt response in the cutaneous symptoms (Figure 1B).
Sweet syndrome and pyoderma gangrenosum are considered distinct neutrophilic dermatoses that rarely coexist but share several clinical and histopathologic features, which can become a diagnostic challenge.2 Both conditions can manifest clinically as abrupt-onset, tender, erythematous papules; vesiculopustular lesions; or bullae with ulcerative changes. They also exhibit pathergy; present with systemic symptoms such as pyrexia, malaise, and joint pain; are associated with underlying systemic conditions such as infections and/or malignancy; demonstrate a dense neutrophilic infiltrate in the dermis on histopathology; and respond promptly to systemic corticosteroids.2-6 Bullous Sweet syndrome, which can present as vesicles, pustules, or bullae that progress to superficial ulcerations, may represent a variant of neutrophilic dermatosis characterized by features seen in both Sweet syndrome and pyoderma gangrenosum, suggesting that these 2 conditions may be on a spectrum.5Clinical features such as erythema with a blue, gray, or purple hue; undermined and ragged borders; and healing of skin lesions with atrophic or cribriform scarring may favor pyoderma gangrenosum, whereas a dull red or plum color and resolution of lesions without scarring may support the diagnosis of Sweet syndrome.7 Although both conditions can exhibit pathergy secondary to minor skin trauma such as venipuncture and biopsies,2,3,5,8 Sweet syndrome rarely has been described to develop after surgery in a patient without a known history of the condition.9 In contrast, postsurgical pyoderma gangrenosum has been well described as secondary to the pathergy phenomenon.5
Our patient was favored to have pyoderma gangrenosum given the solitary lesion, its abrupt development after surgery, and the morphology of the lesion that exhibited a large violaceous to red ulcerative and exudative plaque with undermined borders with atrophic scarring. In patients with skin disease that cannot be distinguished with certainty as either Sweet syndrome or pyoderma gangrenosum, it is essential to recognize that, as neutrophilic dermatoses, both conditions can be managed with either the first-line treatment option of high-dose systemic steroids or one of the shared alternative first-line or second-line steroid-sparing treatments, such as dapsone and cyclosporine.2
Although the exact pathogenesis of pyoderma gangrenosum remains to be fully understood, paraneoplastic pyoderma gangrenosum is a frequently described phenomenon.10,11 Our patient’s history of multiple malignancies, both solid and hematologic, supports the likelihood of malignancy-induced pyoderma gangrenosum; however, given his history of MDS, several other conditions were ruled out prior to making the diagnosis of pyoderma gangrenosum.
Classically, neutrophilic dermatoses such as pyoderma gangrenosum have a dense dermal neutrophilic infiltrate. Concurrent myeloproliferative disorders can alter the maturation of leukocytes, subsequently leading to an atypical appearance of the inflammatory cells on histopathology. Further, in the setting of myeloproliferative disorders, conditions such as leukemia cutis, in which there can be a cutaneous infiltrate of immature or mature myeloid or lymphocytic cells, must be considered. To ensure our patient’s abdominal skin changes were not a cutaneous manifestation of hematologic malignancy, immunohistochemical staining with CD20 and CD3 was performed and showed only the rare presence of B and T lymphocytes, respectively. Staining with CD34 for lymphocytic and myeloid progenitor cells was negative in the dermal infiltrate and further reduced the likelihood of leukemia cutis. Alternatively, patients can have aleukemic cutaneous myeloid sarcoma or leukemia cutis without an underlying hematologic condition or with latent peripheral blood or bone marrow myeloproliferative disorder, but our patient’s history of MDS eliminated this possibility.12 After exclusion of cutaneous infiltration by malignant leukocytes, our patient was diagnosed with histiocytoid neutrophilic dermatosis.
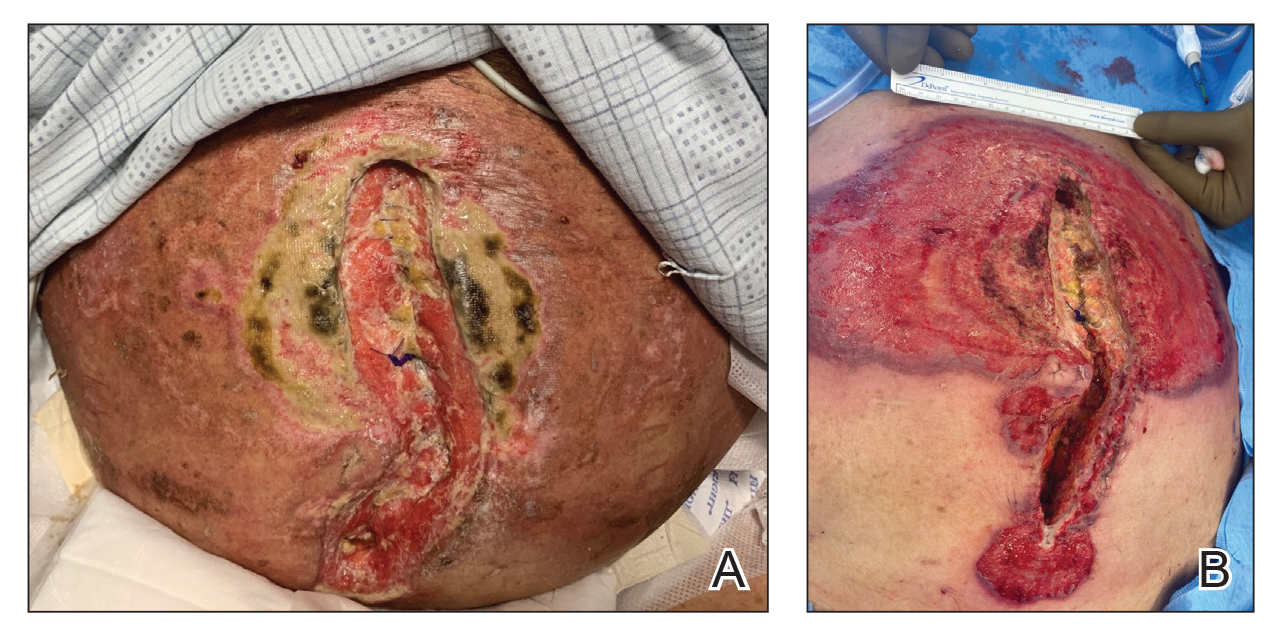
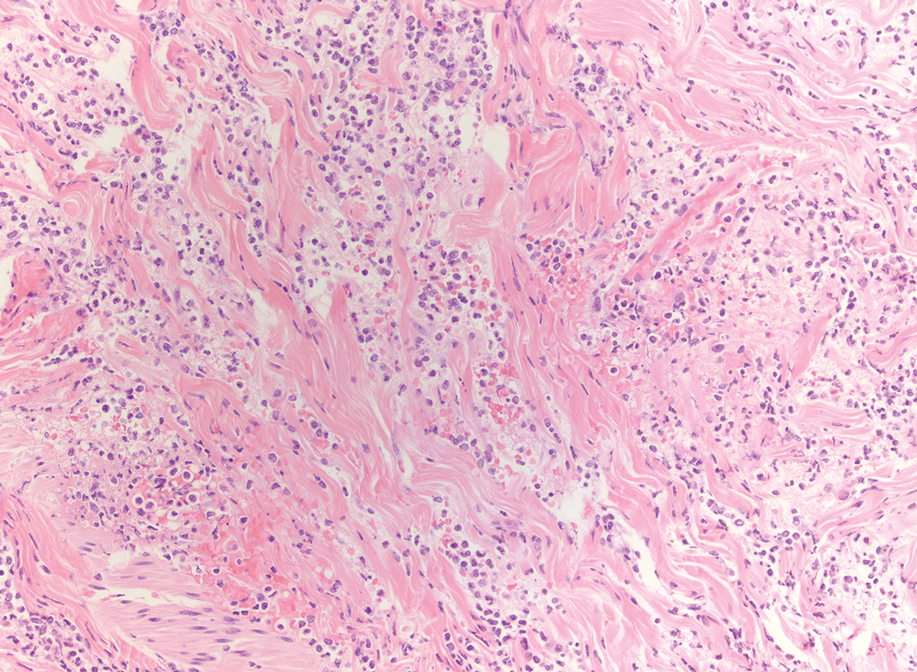
Multiple reports have described histiocytoid Sweet syndrome, in which there is a dense dermal histiocytoid infiltrate on histopathology that demonstrates myeloid lineage with immunologic staining.1,13 The typical pattern of histiocytoid Sweet syndrome includes a predominantly unaffected epidermis with papillary dermal edema, an absence of vasculitis, and a dense dermal infiltrate primarily composed of immature histiocytelike mononuclear cells with a basophilic elongated, twisted, or kidney-shaped nucleus and pale eosinophilic cytoplasm.1,13 In an analogous manner, Morin et al12 described a patient with congenital hypogammaglobulinemia who presented with lesions that clinically resembled pyoderma gangrenosum but revealed a dense dermal infiltrate mostly made of large immature histiocytoid mononuclear cells on histopathology, consistent with the histopathologic features observed in histiocytoid Sweet syndrome. The patient ultimately was diagnosed with histiocytoid pyoderma gangrenosum. Similarly, we believe that our patient also developed histiocytoid pyoderma gangrenosum. As with histiocytoid Sweet syndrome, this diagnosis is based on histopathologic and immunohistochemical findings of a dense dermal infiltrate composed of histiocyte-resembling immature neutrophils.
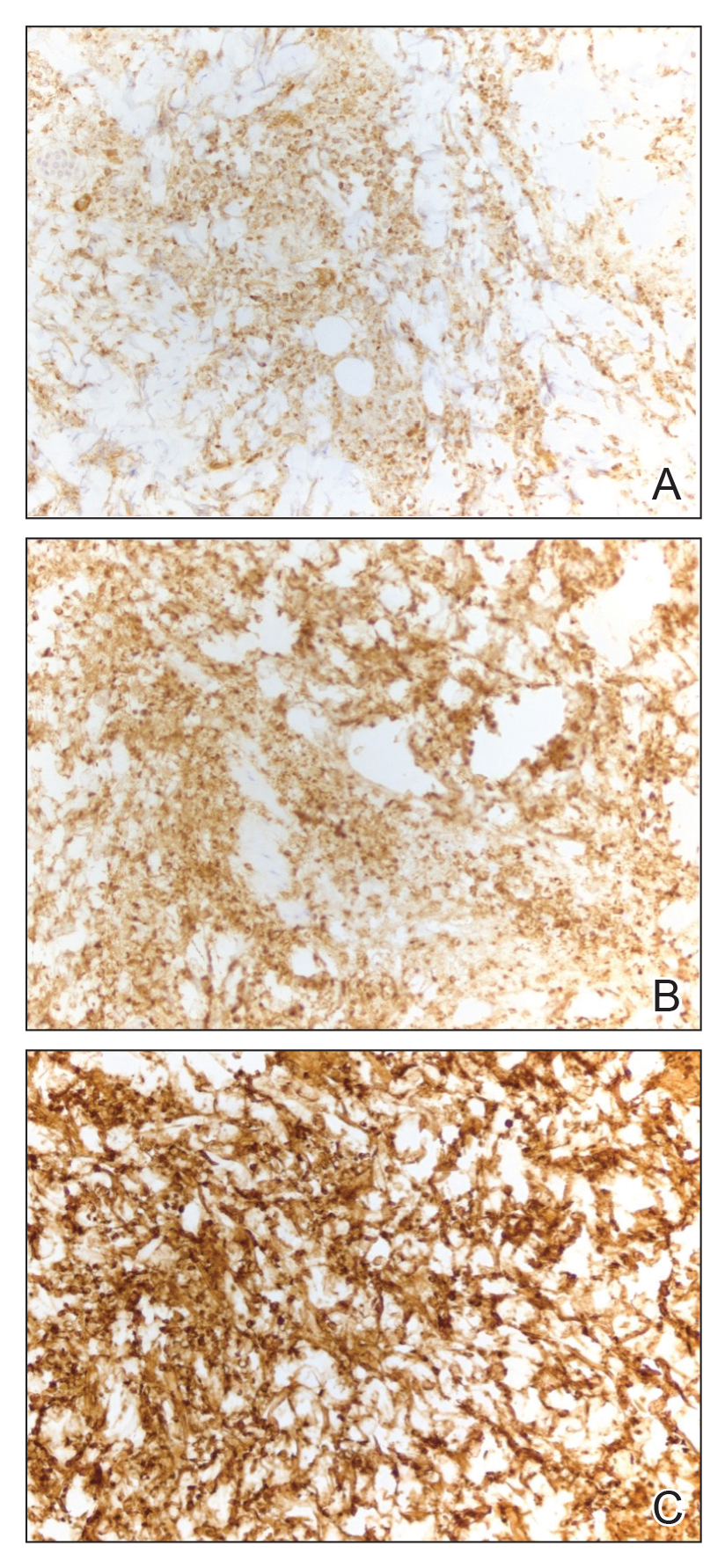
Typically, pyoderma gangrenosum responds promptly to treatment with systemic corticosteroids.4 Steroid-sparing agents such as cyclosporine, azathioprine, dapsone, and tumor necrosis factor α inhibitors also may be used.4,10 In the setting of MDS, clearance of pyoderma gangrenosum has been reported upon treatment of the underlying malignancy,14 high-dose systemic corticosteroids,11,15 cyclosporine with systemic steroids,16 thalidomide,17 combination therapy with thalidomide and interferon alfa-2a,18 and ustekinumab with vacuum-assisted closure therapy.19 Our patient’s histiocytoid pyoderma gangrenosum in the setting of solid and hematologic malignancy cleared rapidly with high-dose systemic hydrocortisone.
In the setting of malignancy, as in our patient, neutrophilic dermatoses may develop from an aberrant immune system or tumor-induced cytokine dysregulation that leads to increased neutrophil production or dysfunction.4,10,11 Although our patient’s MDS may have contributed to the atypical appearance of the dermal inflammatory infiltrate, it is unclear whether the hematologic disorder increased his risk for the histiocytoid variant of neutrophilic dermatoses. Alegría-Landa et al13 reported that histiocytoid Sweet syndrome is associated with hematologic malignancy at a similar frequency as classic Sweet syndrome. It is unknown if histiocytoid pyoderma gangrenosum would have a strong association with hematologic malignancy. Future reports may elucidate a better understanding of the histiocytoid subtype of pyoderma gangrenosum and its clinical implications.
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842.
- Cohen PR. Neutrophilic dermatoses: a review of current treatment options. Am J Clin Dermatol. 2009;10:301-312.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Braswell SF, Kostopoulos TC, Ortega-Loayza AG. Pathophysiology of pyoderma gangrenosum (PG): an updated review. J Am Acad Dermatol. 2015;73:691-698.
- Wallach D, Vignon-Pennamen MD. Pyoderma gangrenosum and Sweet syndrome: the prototypic neutrophilic dermatoses. Br J Dermatol. 2018;178:595-602.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and Sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Lear JT, Atherton MT, Byrne JP. Neutrophilic dermatoses: pyoderma gangrenosum and Sweet’s syndrome. Postgrad Med. 1997;73:65-68.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006.
- Minocha R, Sebaratnam DF, Choi JY. Sweet’s syndrome following surgery: cutaneous trauma as a possible aetiological co-factor in neutrophilic dermatoses. Australas J Dermatol. 2015;56:E74-E76.
- Shah M, Sachdeva M, Gefri A, et al. Paraneoplastic pyoderma gangrenosum in solid organ malignancy: a literature review. Int J Dermatol. 2020;59:154-158.
- Montagnon CM, Fracica EA, Patel AA, et al. Pyoderma gangrenosum in hematologic malignancies: a systematic review. J Am Acad Dermatol. 2020;82:1346-1359.
- Morin CB, Côté B, Belisle A. An interesting case of pyoderma gangrenosum with immature histiocytoid neutrophils. J Cutan Pathol. 2018;45:63-66.
- Alegría-Landa V, Rodríguez-Pinilla SM, Santos-Briz A, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153:651-659.
- Saleh MFM, Saunthararajah Y. Severe pyoderma gangrenosum caused by myelodysplastic syndrome successfully treated with decitabine administered by a noncytotoxic regimen. Clin Case Rep. 2017;5:2025-2027.
- Yamauchi R, Ishida K, Iwashima Y, et al. Successful treatment of pyoderma gangrenosum that developed in a patient with myelodysplastic syndrome. J Infect Chemother. 2003;9:268-271.
- Ha JW, Hahm JE, Kim KS, et al. A case of pyoderma gangrenosum with myelodysplastic syndrome. Ann Dermatol. 2018;30:392-393.
- Malkan UY, Gunes G, Eliacik E, et al. Treatment of pyoderma gangrenosum with thalidomide in a myelodysplastic syndrome case. Int J Med Case Rep. 2016;9:61-64.
- Koca E, Duman AE, Cetiner D, et al. Successful treatment of myelodysplastic syndrome-induced pyoderma gangrenosum. Neth J Med. 2006;64:422-424.
- Nieto D, Sendagorta E, Rueda JM, et al. Successful treatment with ustekinumab and vacuum-assisted closure therapy in recalcitrant myelodysplastic syndrome-associated pyoderma gangrenosum: case report and literature review. Clin Exp Dermatol. 2019;44:116-119.
To the Editor:
Neutrophilic dermatoses—a group of inflammatory cutaneous conditions—include acute febrile neutrophilic dermatosis (Sweet syndrome), pyoderma gangrenosum, and neutrophilic dermatosis of the dorsal hands. Histopathology shows a dense dermal infiltrate of mature neutrophils. In 2005, the histiocytoid subtype of Sweet syndrome was introduced with histopathologic findings of a dermal infiltrate composed of immature myeloid cells that resemble histiocytes in appearance but stain strongly with neutrophil markers on immunohistochemistry.1 We present a case of histiocytoid pyoderma gangrenosum with histopathology that showed a dense dermal histiocytoid infiltrate with strong positivity for neutrophil markers on immunohistochemistry.
An 85-year-old man was seen by dermatology in the inpatient setting for a new-onset painful abdominal wound. He had a medical history of myelodysplastic syndrome (MDS), high-grade invasive papillary urothelial carcinoma of the bladder, and a recent diagnosis of low-grade invasive ascending colon adenocarcinoma. Ten days prior he underwent a right colectomy without intraoperative complications that was followed by septic shock. Workup with urinalysis and urine culture showed minimal pyuria with Pseudomonas aeruginosa. Additional studies, including blood cultures, abdominal wound cultures, computed tomography of the abdomen and pelvis, renal ultrasound, and chest radiographs, were unremarkable and showed no signs of surgical site infection, intra-abdominal or pelvic abscess formation, or pulmonary embolism. Broad-spectrum antibiotics—vancomycin and piperacillin-tazobactam—were started. Persistent fever (Tmax of 102.3 °F [39.1 °C]) and leukocytosis (45.3×109/L [4.2–10×109/L]) despite antibiotic therapy, increasing pressor requirements, and progressive painful erythema and purulence at the abdominal surgical site led to debridement of the wound by the general surgery team on day 9 following the initial surgery due to suspected necrotizing infection. Within 24 hours, dermatology was consulted for continued rapid expansion of the wound. Physical examination of the abdomen revealed a large, well-demarcated, pink-red, indurated, ulcerated plaque with clear to purulent exudate and superficial erosions with violaceous undermined borders extending centrifugally from the abdominal surgical incision line (Figure 1A). Two punch biopsies sent for histopathologic evaluation and tissue culture showed dermal edema with a dense histiocytic infiltrate with nodular foci and admixed mature neutrophils to a lesser degree (Figure 2). Special staining was negative for bacteria, fungi, and mycobacteria. Immunohistochemistry revealed positive staining of the dermal inflammatory infiltrate with CD68, myeloperoxidase, and lysozyme, as well as negative staining with CD34 (Figure 3). These findings were suggestive of a histiocytoid neutrophilic dermatosis such as Sweet syndrome or pyoderma gangrenosum. Due to the morphology of the solitary lesion and the abrupt exacerbation shortly after surgical intervention, the patient was diagnosed with histiocytoid pyoderma gangrenosum. At the same time, the patient’s septic shock was treated with intravenous hydrocortisone (100 mg 3 times daily) for 2 days and also achieved a prompt response in the cutaneous symptoms (Figure 1B).
Sweet syndrome and pyoderma gangrenosum are considered distinct neutrophilic dermatoses that rarely coexist but share several clinical and histopathologic features, which can become a diagnostic challenge.2 Both conditions can manifest clinically as abrupt-onset, tender, erythematous papules; vesiculopustular lesions; or bullae with ulcerative changes. They also exhibit pathergy; present with systemic symptoms such as pyrexia, malaise, and joint pain; are associated with underlying systemic conditions such as infections and/or malignancy; demonstrate a dense neutrophilic infiltrate in the dermis on histopathology; and respond promptly to systemic corticosteroids.2-6 Bullous Sweet syndrome, which can present as vesicles, pustules, or bullae that progress to superficial ulcerations, may represent a variant of neutrophilic dermatosis characterized by features seen in both Sweet syndrome and pyoderma gangrenosum, suggesting that these 2 conditions may be on a spectrum.5Clinical features such as erythema with a blue, gray, or purple hue; undermined and ragged borders; and healing of skin lesions with atrophic or cribriform scarring may favor pyoderma gangrenosum, whereas a dull red or plum color and resolution of lesions without scarring may support the diagnosis of Sweet syndrome.7 Although both conditions can exhibit pathergy secondary to minor skin trauma such as venipuncture and biopsies,2,3,5,8 Sweet syndrome rarely has been described to develop after surgery in a patient without a known history of the condition.9 In contrast, postsurgical pyoderma gangrenosum has been well described as secondary to the pathergy phenomenon.5
Our patient was favored to have pyoderma gangrenosum given the solitary lesion, its abrupt development after surgery, and the morphology of the lesion that exhibited a large violaceous to red ulcerative and exudative plaque with undermined borders with atrophic scarring. In patients with skin disease that cannot be distinguished with certainty as either Sweet syndrome or pyoderma gangrenosum, it is essential to recognize that, as neutrophilic dermatoses, both conditions can be managed with either the first-line treatment option of high-dose systemic steroids or one of the shared alternative first-line or second-line steroid-sparing treatments, such as dapsone and cyclosporine.2
Although the exact pathogenesis of pyoderma gangrenosum remains to be fully understood, paraneoplastic pyoderma gangrenosum is a frequently described phenomenon.10,11 Our patient’s history of multiple malignancies, both solid and hematologic, supports the likelihood of malignancy-induced pyoderma gangrenosum; however, given his history of MDS, several other conditions were ruled out prior to making the diagnosis of pyoderma gangrenosum.
Classically, neutrophilic dermatoses such as pyoderma gangrenosum have a dense dermal neutrophilic infiltrate. Concurrent myeloproliferative disorders can alter the maturation of leukocytes, subsequently leading to an atypical appearance of the inflammatory cells on histopathology. Further, in the setting of myeloproliferative disorders, conditions such as leukemia cutis, in which there can be a cutaneous infiltrate of immature or mature myeloid or lymphocytic cells, must be considered. To ensure our patient’s abdominal skin changes were not a cutaneous manifestation of hematologic malignancy, immunohistochemical staining with CD20 and CD3 was performed and showed only the rare presence of B and T lymphocytes, respectively. Staining with CD34 for lymphocytic and myeloid progenitor cells was negative in the dermal infiltrate and further reduced the likelihood of leukemia cutis. Alternatively, patients can have aleukemic cutaneous myeloid sarcoma or leukemia cutis without an underlying hematologic condition or with latent peripheral blood or bone marrow myeloproliferative disorder, but our patient’s history of MDS eliminated this possibility.12 After exclusion of cutaneous infiltration by malignant leukocytes, our patient was diagnosed with histiocytoid neutrophilic dermatosis.
Multiple reports have described histiocytoid Sweet syndrome, in which there is a dense dermal histiocytoid infiltrate on histopathology that demonstrates myeloid lineage with immunologic staining.1,13 The typical pattern of histiocytoid Sweet syndrome includes a predominantly unaffected epidermis with papillary dermal edema, an absence of vasculitis, and a dense dermal infiltrate primarily composed of immature histiocytelike mononuclear cells with a basophilic elongated, twisted, or kidney-shaped nucleus and pale eosinophilic cytoplasm.1,13 In an analogous manner, Morin et al12 described a patient with congenital hypogammaglobulinemia who presented with lesions that clinically resembled pyoderma gangrenosum but revealed a dense dermal infiltrate mostly made of large immature histiocytoid mononuclear cells on histopathology, consistent with the histopathologic features observed in histiocytoid Sweet syndrome. The patient ultimately was diagnosed with histiocytoid pyoderma gangrenosum. Similarly, we believe that our patient also developed histiocytoid pyoderma gangrenosum. As with histiocytoid Sweet syndrome, this diagnosis is based on histopathologic and immunohistochemical findings of a dense dermal infiltrate composed of histiocyte-resembling immature neutrophils.
Typically, pyoderma gangrenosum responds promptly to treatment with systemic corticosteroids.4 Steroid-sparing agents such as cyclosporine, azathioprine, dapsone, and tumor necrosis factor α inhibitors also may be used.4,10 In the setting of MDS, clearance of pyoderma gangrenosum has been reported upon treatment of the underlying malignancy,14 high-dose systemic corticosteroids,11,15 cyclosporine with systemic steroids,16 thalidomide,17 combination therapy with thalidomide and interferon alfa-2a,18 and ustekinumab with vacuum-assisted closure therapy.19 Our patient’s histiocytoid pyoderma gangrenosum in the setting of solid and hematologic malignancy cleared rapidly with high-dose systemic hydrocortisone.
In the setting of malignancy, as in our patient, neutrophilic dermatoses may develop from an aberrant immune system or tumor-induced cytokine dysregulation that leads to increased neutrophil production or dysfunction.4,10,11 Although our patient’s MDS may have contributed to the atypical appearance of the dermal inflammatory infiltrate, it is unclear whether the hematologic disorder increased his risk for the histiocytoid variant of neutrophilic dermatoses. Alegría-Landa et al13 reported that histiocytoid Sweet syndrome is associated with hematologic malignancy at a similar frequency as classic Sweet syndrome. It is unknown if histiocytoid pyoderma gangrenosum would have a strong association with hematologic malignancy. Future reports may elucidate a better understanding of the histiocytoid subtype of pyoderma gangrenosum and its clinical implications.
To the Editor:
Neutrophilic dermatoses—a group of inflammatory cutaneous conditions—include acute febrile neutrophilic dermatosis (Sweet syndrome), pyoderma gangrenosum, and neutrophilic dermatosis of the dorsal hands. Histopathology shows a dense dermal infiltrate of mature neutrophils. In 2005, the histiocytoid subtype of Sweet syndrome was introduced with histopathologic findings of a dermal infiltrate composed of immature myeloid cells that resemble histiocytes in appearance but stain strongly with neutrophil markers on immunohistochemistry.1 We present a case of histiocytoid pyoderma gangrenosum with histopathology that showed a dense dermal histiocytoid infiltrate with strong positivity for neutrophil markers on immunohistochemistry.
An 85-year-old man was seen by dermatology in the inpatient setting for a new-onset painful abdominal wound. He had a medical history of myelodysplastic syndrome (MDS), high-grade invasive papillary urothelial carcinoma of the bladder, and a recent diagnosis of low-grade invasive ascending colon adenocarcinoma. Ten days prior he underwent a right colectomy without intraoperative complications that was followed by septic shock. Workup with urinalysis and urine culture showed minimal pyuria with Pseudomonas aeruginosa. Additional studies, including blood cultures, abdominal wound cultures, computed tomography of the abdomen and pelvis, renal ultrasound, and chest radiographs, were unremarkable and showed no signs of surgical site infection, intra-abdominal or pelvic abscess formation, or pulmonary embolism. Broad-spectrum antibiotics—vancomycin and piperacillin-tazobactam—were started. Persistent fever (Tmax of 102.3 °F [39.1 °C]) and leukocytosis (45.3×109/L [4.2–10×109/L]) despite antibiotic therapy, increasing pressor requirements, and progressive painful erythema and purulence at the abdominal surgical site led to debridement of the wound by the general surgery team on day 9 following the initial surgery due to suspected necrotizing infection. Within 24 hours, dermatology was consulted for continued rapid expansion of the wound. Physical examination of the abdomen revealed a large, well-demarcated, pink-red, indurated, ulcerated plaque with clear to purulent exudate and superficial erosions with violaceous undermined borders extending centrifugally from the abdominal surgical incision line (Figure 1A). Two punch biopsies sent for histopathologic evaluation and tissue culture showed dermal edema with a dense histiocytic infiltrate with nodular foci and admixed mature neutrophils to a lesser degree (Figure 2). Special staining was negative for bacteria, fungi, and mycobacteria. Immunohistochemistry revealed positive staining of the dermal inflammatory infiltrate with CD68, myeloperoxidase, and lysozyme, as well as negative staining with CD34 (Figure 3). These findings were suggestive of a histiocytoid neutrophilic dermatosis such as Sweet syndrome or pyoderma gangrenosum. Due to the morphology of the solitary lesion and the abrupt exacerbation shortly after surgical intervention, the patient was diagnosed with histiocytoid pyoderma gangrenosum. At the same time, the patient’s septic shock was treated with intravenous hydrocortisone (100 mg 3 times daily) for 2 days and also achieved a prompt response in the cutaneous symptoms (Figure 1B).
Sweet syndrome and pyoderma gangrenosum are considered distinct neutrophilic dermatoses that rarely coexist but share several clinical and histopathologic features, which can become a diagnostic challenge.2 Both conditions can manifest clinically as abrupt-onset, tender, erythematous papules; vesiculopustular lesions; or bullae with ulcerative changes. They also exhibit pathergy; present with systemic symptoms such as pyrexia, malaise, and joint pain; are associated with underlying systemic conditions such as infections and/or malignancy; demonstrate a dense neutrophilic infiltrate in the dermis on histopathology; and respond promptly to systemic corticosteroids.2-6 Bullous Sweet syndrome, which can present as vesicles, pustules, or bullae that progress to superficial ulcerations, may represent a variant of neutrophilic dermatosis characterized by features seen in both Sweet syndrome and pyoderma gangrenosum, suggesting that these 2 conditions may be on a spectrum.5Clinical features such as erythema with a blue, gray, or purple hue; undermined and ragged borders; and healing of skin lesions with atrophic or cribriform scarring may favor pyoderma gangrenosum, whereas a dull red or plum color and resolution of lesions without scarring may support the diagnosis of Sweet syndrome.7 Although both conditions can exhibit pathergy secondary to minor skin trauma such as venipuncture and biopsies,2,3,5,8 Sweet syndrome rarely has been described to develop after surgery in a patient without a known history of the condition.9 In contrast, postsurgical pyoderma gangrenosum has been well described as secondary to the pathergy phenomenon.5
Our patient was favored to have pyoderma gangrenosum given the solitary lesion, its abrupt development after surgery, and the morphology of the lesion that exhibited a large violaceous to red ulcerative and exudative plaque with undermined borders with atrophic scarring. In patients with skin disease that cannot be distinguished with certainty as either Sweet syndrome or pyoderma gangrenosum, it is essential to recognize that, as neutrophilic dermatoses, both conditions can be managed with either the first-line treatment option of high-dose systemic steroids or one of the shared alternative first-line or second-line steroid-sparing treatments, such as dapsone and cyclosporine.2
Although the exact pathogenesis of pyoderma gangrenosum remains to be fully understood, paraneoplastic pyoderma gangrenosum is a frequently described phenomenon.10,11 Our patient’s history of multiple malignancies, both solid and hematologic, supports the likelihood of malignancy-induced pyoderma gangrenosum; however, given his history of MDS, several other conditions were ruled out prior to making the diagnosis of pyoderma gangrenosum.
Classically, neutrophilic dermatoses such as pyoderma gangrenosum have a dense dermal neutrophilic infiltrate. Concurrent myeloproliferative disorders can alter the maturation of leukocytes, subsequently leading to an atypical appearance of the inflammatory cells on histopathology. Further, in the setting of myeloproliferative disorders, conditions such as leukemia cutis, in which there can be a cutaneous infiltrate of immature or mature myeloid or lymphocytic cells, must be considered. To ensure our patient’s abdominal skin changes were not a cutaneous manifestation of hematologic malignancy, immunohistochemical staining with CD20 and CD3 was performed and showed only the rare presence of B and T lymphocytes, respectively. Staining with CD34 for lymphocytic and myeloid progenitor cells was negative in the dermal infiltrate and further reduced the likelihood of leukemia cutis. Alternatively, patients can have aleukemic cutaneous myeloid sarcoma or leukemia cutis without an underlying hematologic condition or with latent peripheral blood or bone marrow myeloproliferative disorder, but our patient’s history of MDS eliminated this possibility.12 After exclusion of cutaneous infiltration by malignant leukocytes, our patient was diagnosed with histiocytoid neutrophilic dermatosis.
Multiple reports have described histiocytoid Sweet syndrome, in which there is a dense dermal histiocytoid infiltrate on histopathology that demonstrates myeloid lineage with immunologic staining.1,13 The typical pattern of histiocytoid Sweet syndrome includes a predominantly unaffected epidermis with papillary dermal edema, an absence of vasculitis, and a dense dermal infiltrate primarily composed of immature histiocytelike mononuclear cells with a basophilic elongated, twisted, or kidney-shaped nucleus and pale eosinophilic cytoplasm.1,13 In an analogous manner, Morin et al12 described a patient with congenital hypogammaglobulinemia who presented with lesions that clinically resembled pyoderma gangrenosum but revealed a dense dermal infiltrate mostly made of large immature histiocytoid mononuclear cells on histopathology, consistent with the histopathologic features observed in histiocytoid Sweet syndrome. The patient ultimately was diagnosed with histiocytoid pyoderma gangrenosum. Similarly, we believe that our patient also developed histiocytoid pyoderma gangrenosum. As with histiocytoid Sweet syndrome, this diagnosis is based on histopathologic and immunohistochemical findings of a dense dermal infiltrate composed of histiocyte-resembling immature neutrophils.
Typically, pyoderma gangrenosum responds promptly to treatment with systemic corticosteroids.4 Steroid-sparing agents such as cyclosporine, azathioprine, dapsone, and tumor necrosis factor α inhibitors also may be used.4,10 In the setting of MDS, clearance of pyoderma gangrenosum has been reported upon treatment of the underlying malignancy,14 high-dose systemic corticosteroids,11,15 cyclosporine with systemic steroids,16 thalidomide,17 combination therapy with thalidomide and interferon alfa-2a,18 and ustekinumab with vacuum-assisted closure therapy.19 Our patient’s histiocytoid pyoderma gangrenosum in the setting of solid and hematologic malignancy cleared rapidly with high-dose systemic hydrocortisone.
In the setting of malignancy, as in our patient, neutrophilic dermatoses may develop from an aberrant immune system or tumor-induced cytokine dysregulation that leads to increased neutrophil production or dysfunction.4,10,11 Although our patient’s MDS may have contributed to the atypical appearance of the dermal inflammatory infiltrate, it is unclear whether the hematologic disorder increased his risk for the histiocytoid variant of neutrophilic dermatoses. Alegría-Landa et al13 reported that histiocytoid Sweet syndrome is associated with hematologic malignancy at a similar frequency as classic Sweet syndrome. It is unknown if histiocytoid pyoderma gangrenosum would have a strong association with hematologic malignancy. Future reports may elucidate a better understanding of the histiocytoid subtype of pyoderma gangrenosum and its clinical implications.
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842.
- Cohen PR. Neutrophilic dermatoses: a review of current treatment options. Am J Clin Dermatol. 2009;10:301-312.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Braswell SF, Kostopoulos TC, Ortega-Loayza AG. Pathophysiology of pyoderma gangrenosum (PG): an updated review. J Am Acad Dermatol. 2015;73:691-698.
- Wallach D, Vignon-Pennamen MD. Pyoderma gangrenosum and Sweet syndrome: the prototypic neutrophilic dermatoses. Br J Dermatol. 2018;178:595-602.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and Sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Lear JT, Atherton MT, Byrne JP. Neutrophilic dermatoses: pyoderma gangrenosum and Sweet’s syndrome. Postgrad Med. 1997;73:65-68.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006.
- Minocha R, Sebaratnam DF, Choi JY. Sweet’s syndrome following surgery: cutaneous trauma as a possible aetiological co-factor in neutrophilic dermatoses. Australas J Dermatol. 2015;56:E74-E76.
- Shah M, Sachdeva M, Gefri A, et al. Paraneoplastic pyoderma gangrenosum in solid organ malignancy: a literature review. Int J Dermatol. 2020;59:154-158.
- Montagnon CM, Fracica EA, Patel AA, et al. Pyoderma gangrenosum in hematologic malignancies: a systematic review. J Am Acad Dermatol. 2020;82:1346-1359.
- Morin CB, Côté B, Belisle A. An interesting case of pyoderma gangrenosum with immature histiocytoid neutrophils. J Cutan Pathol. 2018;45:63-66.
- Alegría-Landa V, Rodríguez-Pinilla SM, Santos-Briz A, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153:651-659.
- Saleh MFM, Saunthararajah Y. Severe pyoderma gangrenosum caused by myelodysplastic syndrome successfully treated with decitabine administered by a noncytotoxic regimen. Clin Case Rep. 2017;5:2025-2027.
- Yamauchi R, Ishida K, Iwashima Y, et al. Successful treatment of pyoderma gangrenosum that developed in a patient with myelodysplastic syndrome. J Infect Chemother. 2003;9:268-271.
- Ha JW, Hahm JE, Kim KS, et al. A case of pyoderma gangrenosum with myelodysplastic syndrome. Ann Dermatol. 2018;30:392-393.
- Malkan UY, Gunes G, Eliacik E, et al. Treatment of pyoderma gangrenosum with thalidomide in a myelodysplastic syndrome case. Int J Med Case Rep. 2016;9:61-64.
- Koca E, Duman AE, Cetiner D, et al. Successful treatment of myelodysplastic syndrome-induced pyoderma gangrenosum. Neth J Med. 2006;64:422-424.
- Nieto D, Sendagorta E, Rueda JM, et al. Successful treatment with ustekinumab and vacuum-assisted closure therapy in recalcitrant myelodysplastic syndrome-associated pyoderma gangrenosum: case report and literature review. Clin Exp Dermatol. 2019;44:116-119.
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842.
- Cohen PR. Neutrophilic dermatoses: a review of current treatment options. Am J Clin Dermatol. 2009;10:301-312.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Braswell SF, Kostopoulos TC, Ortega-Loayza AG. Pathophysiology of pyoderma gangrenosum (PG): an updated review. J Am Acad Dermatol. 2015;73:691-698.
- Wallach D, Vignon-Pennamen MD. Pyoderma gangrenosum and Sweet syndrome: the prototypic neutrophilic dermatoses. Br J Dermatol. 2018;178:595-602.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and Sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Lear JT, Atherton MT, Byrne JP. Neutrophilic dermatoses: pyoderma gangrenosum and Sweet’s syndrome. Postgrad Med. 1997;73:65-68.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006.
- Minocha R, Sebaratnam DF, Choi JY. Sweet’s syndrome following surgery: cutaneous trauma as a possible aetiological co-factor in neutrophilic dermatoses. Australas J Dermatol. 2015;56:E74-E76.
- Shah M, Sachdeva M, Gefri A, et al. Paraneoplastic pyoderma gangrenosum in solid organ malignancy: a literature review. Int J Dermatol. 2020;59:154-158.
- Montagnon CM, Fracica EA, Patel AA, et al. Pyoderma gangrenosum in hematologic malignancies: a systematic review. J Am Acad Dermatol. 2020;82:1346-1359.
- Morin CB, Côté B, Belisle A. An interesting case of pyoderma gangrenosum with immature histiocytoid neutrophils. J Cutan Pathol. 2018;45:63-66.
- Alegría-Landa V, Rodríguez-Pinilla SM, Santos-Briz A, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153:651-659.
- Saleh MFM, Saunthararajah Y. Severe pyoderma gangrenosum caused by myelodysplastic syndrome successfully treated with decitabine administered by a noncytotoxic regimen. Clin Case Rep. 2017;5:2025-2027.
- Yamauchi R, Ishida K, Iwashima Y, et al. Successful treatment of pyoderma gangrenosum that developed in a patient with myelodysplastic syndrome. J Infect Chemother. 2003;9:268-271.
- Ha JW, Hahm JE, Kim KS, et al. A case of pyoderma gangrenosum with myelodysplastic syndrome. Ann Dermatol. 2018;30:392-393.
- Malkan UY, Gunes G, Eliacik E, et al. Treatment of pyoderma gangrenosum with thalidomide in a myelodysplastic syndrome case. Int J Med Case Rep. 2016;9:61-64.
- Koca E, Duman AE, Cetiner D, et al. Successful treatment of myelodysplastic syndrome-induced pyoderma gangrenosum. Neth J Med. 2006;64:422-424.
- Nieto D, Sendagorta E, Rueda JM, et al. Successful treatment with ustekinumab and vacuum-assisted closure therapy in recalcitrant myelodysplastic syndrome-associated pyoderma gangrenosum: case report and literature review. Clin Exp Dermatol. 2019;44:116-119.
Practice Points:
- Dermatologists and dermatopathologists should be aware of the histiocytoid variant of pyoderma gangrenosum, which can clinical and histologic features that overlap with histiocytoid Sweet syndrome.
- When considering a diagnosis of histiocytoid neutrophilic dermatoses, leukemia cutis or aleukemic cutaneous myeloid sarcoma should be ruled out.
- Similar to histiocytoid Sweet syndrome and neutrophilic dermatoses in the setting of hematologic or solid organ malignancy, histiocytoid pyoderma gangrenosum may respond well to high-dose systemic corticosteroids.
Extensive Multidrug-Resistant Dermatophytosis From Trichophyton indotineae
To the Editor:
Historically, commonly available antifungal medications have been effective for treating dermatophytosis (tinea). However, recent tinea outbreaks caused by Trichophyton indotineae—a dermatophyte often resistant to terbinafine and sometimes to other antifungals—have been reported in South Asia, Europe, the Middle East, Southeast Asia, and Australia.1-5
Three confirmed cases of T indotineae dermatophytosis in the United States were reported in 2023 in New York3,6; a fourth confirmed case was reported in 2024 in Pennsylvania.7 Post hoc laboratory testing of fungal isolates in New York in 2022 and 2023 identified an additional 11 cases.8 We present a case of extensive multidrug-resistant tinea caused by T indotineae in a man in California.
An otherwise healthy 65-year-old man who had traveled to Europe in the past 3 months presented to his primary care physician with a widespread pruritic rash (Figure 1). He was treated with 2 weeks of oral terbinafine 250 mg/d and topical medicines, including clotrimazole cream 1%, fluocinonide ointment 0.05%, and clobetasol ointment 0.05% without improvement. Subsequently, 2 weeks of oral griseofulvin microsize 500 mg/d also proved ineffective. An antibody test was negative for HIV. His hemoglobin A1c was 6.2% (reference range, ≤5.6%). The patient was referred to dermatology.
Erythematous plaques—many scaly throughout and some annular with central clearing—were present on the arms, legs, and torso as well as in the groin. Honey crust was present on some plaques on the leg. A potassium hydroxide preparation showed abundant fungal hyphae. Material for fungal and bacterial cultures was collected. The patient was treated again with oral terbinafine 250 mg/d, an oral prednisone taper starting at 60 mg/d for a presumed id reaction, and various oral antihistamines for pruritus; all were ineffective. A bacterial culture showed only mixed skin flora. Oral fluconazole 200 mg/d was prescribed. A skin biopsy specimen showed compact orthokeratosis and parakeratosis of the stratum corneum with few neutrophils and focal pustule formation (Figure 2). Superficial perivascular inflammation, including lymphocytes, histiocytes, and few neutrophils, was present. A periodic acid–Schiff stain showed fungal hyphae in the stratum corneum and a hair follicle (Figure 3). After approximately 2 weeks, mold was identified in the fungal culture. Approximately 2 weeks thereafter, the organism was reported as Trichophyton species.
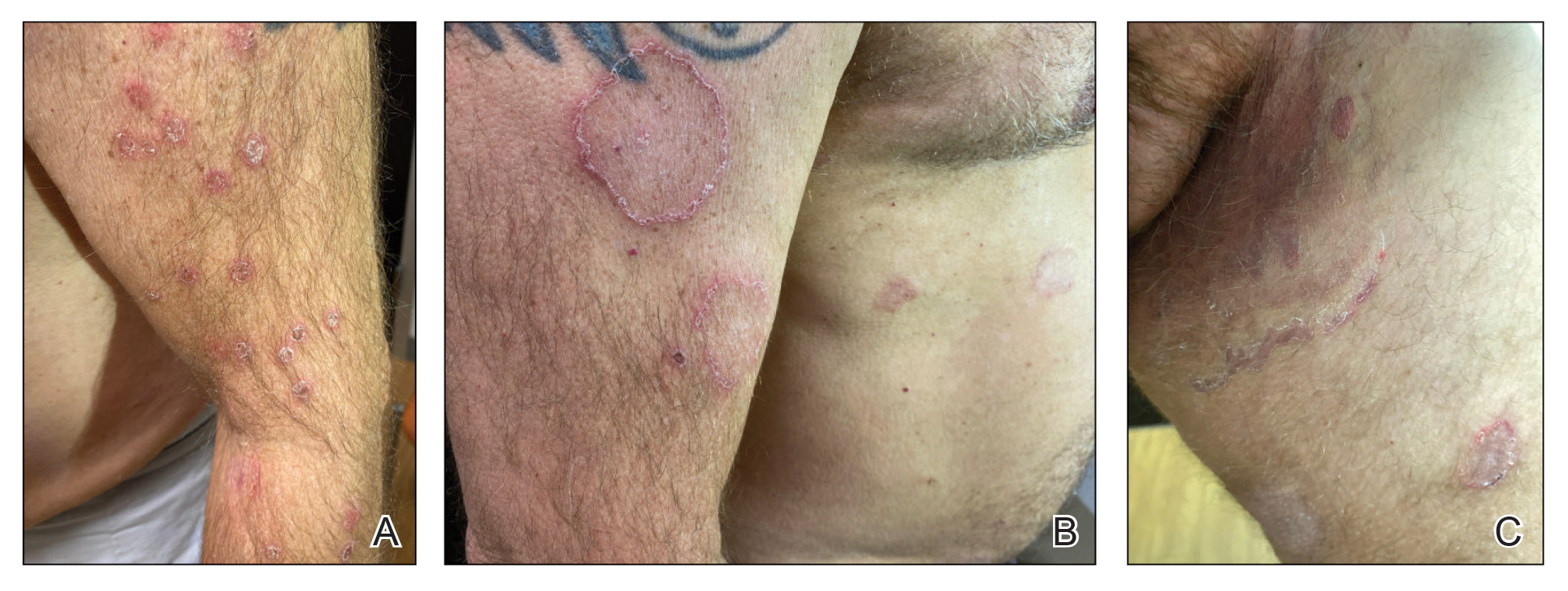
The rash did not improve; resistance to terbinafine, griseofulvin, and fluconazole was suspected clinically. The fungal isolate was sent to a reference laboratory (University of Texas Health Science Center, San Antonio). Meanwhile, oral itraconazole 200 mg twice daily and ketoconazole cream 2% were prescribed; the rash began to improve. A serum itraconazole trough level obtained 4 days after treatment initiation was 0.5 μg/mL (reference range, ≥0.6 μg/mL). The evening itraconazole dose was increased to 300 mg; a subsequent trough level was 0.8 μg/mL.
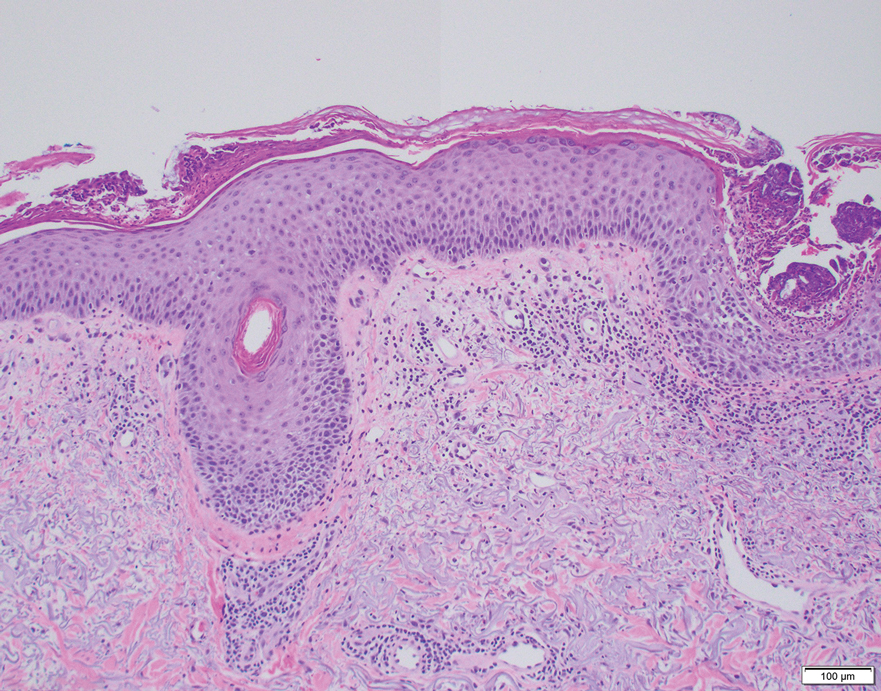
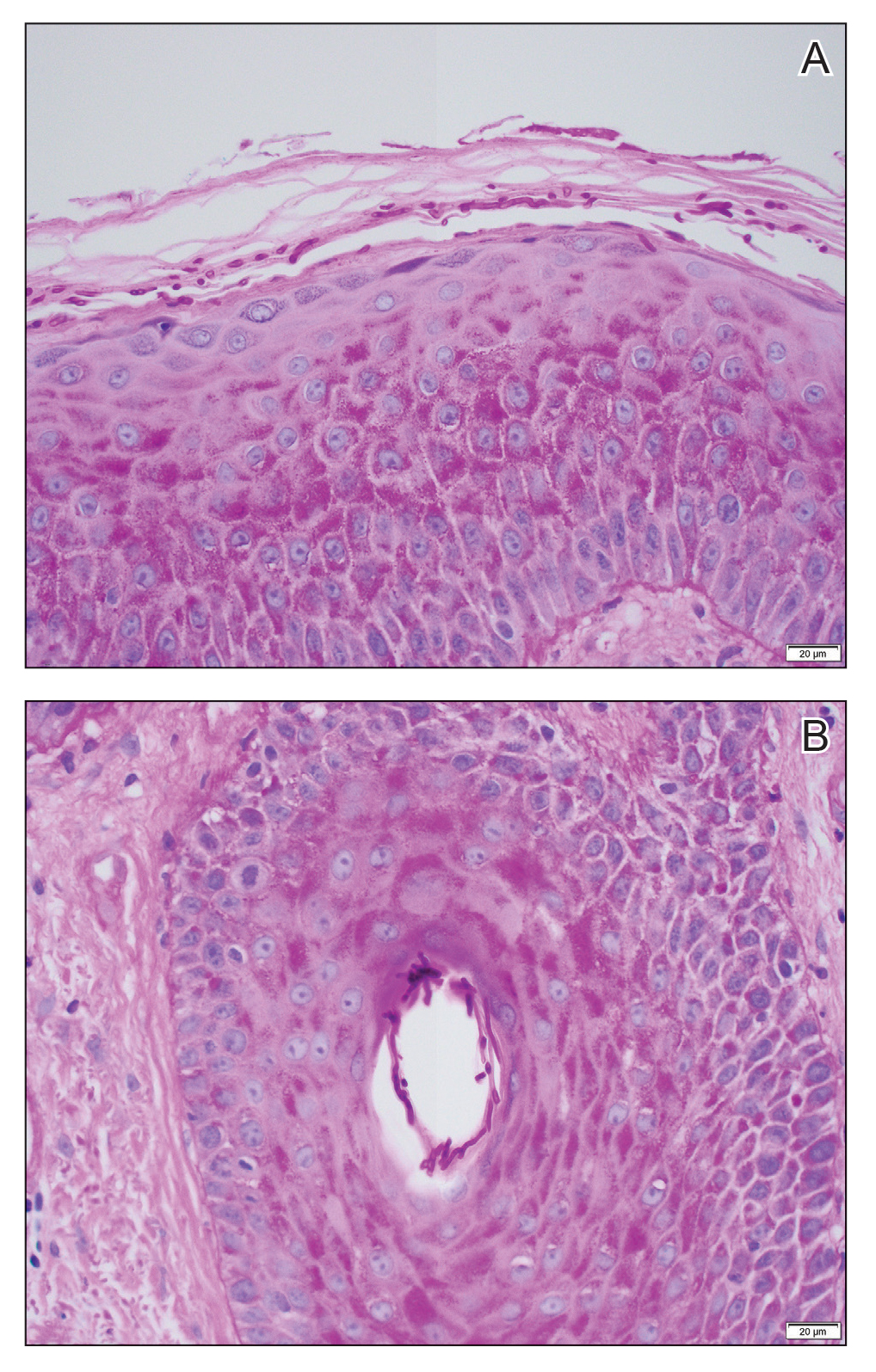
Approximately 1 month after the fungal isolate was sent to the reference laboratory, T indotineae was confirmed based on polymerase chain reaction (PCR) testing of internal transcribed spacer region sequences. Minimum inhibitory concentrations (MICs) obtained through antifungal susceptibility testing (AFST) were reported for fluconazole (8 μg/mL), griseofulvin (2 μg/mL), itraconazole (≤0.03 μg/mL), posaconazole (≤0.03 μg/mL), terbinafine (≥2 μg/mL), and voriconazole (0.125 μg/mL).
Approximately 7 weeks after itraconazole and ketoconazole were started, the rash had completely resolved. Nearly 8 months later (at the time this article was written), the rash had not recurred.
We report a unique case of T indotineae in a patient residing in California. Post hoc laboratory testing of dermatophyte isolates sent to the University of Texas reference laboratory identified terbinafine-resistant T indotineae specimens from the United States and Canada dating to 2017; clinical characteristics of patients from whom those isolates were obtained were unavailable.9
Trichophyton indotineae dermatophytosis typically is more extensive, inflamed, and pruritic, as well as likely more contagious, than tinea caused by other dermatophytes.5 Previously called Trichophyton mentagrophytes genotype VIII when first isolated in 2017, the pathogen was renamed T indotineae in 2020 after important genetic differences were discovered between it and other T mentagrophytes species.5 The emergence of T indotineae has been attributed to concomitant use of topical steroids and antifungals,5,10 inappropriate prescribing of antifungals,5 and nonadherence to antifungal treatment.5
Likely risk factors for T indotineae infection include suboptimal hygiene, overcrowded conditions, hot and humid environments, and tight-fitting synthetic clothing.4 Transmission from family members appears common,5 especially when fomites are shared.4 A case reported in Pennsylvania likely was acquired through sexual contact.7 Travel to South Asia has been associated with acquisition of T indotineae infection,3,5-7 though our patient and some others had not traveled there.3,8 It is not clear whether immunosuppression and diabetes mellitus are associated with T indotineae infection.4,5,8Trichophyton indotineae also can affect animals,11 though zoonotic transmission has not been reported.4
Not all T indotineae isolates are resistant to one or more antifungals; furthermore, antifungal resistance in other dermatophyte species has been reported.5 Terbinafine resistance in T indotineae is conferred by mutations in the gene encoding squalene epoxidase, which helps synthesize ergosterol—a component of the cell membrane in fungi.2,4,5,12 Although clinical cut-points for MIC obtained by AFST are not well established, T indotineae MICs for terbinafine of 0.5 μg/mL or more correlate with resistance.9 Resistance to azoles has been linked to overexpression of transporter genes, which increase azole efflux from cells, as well as to mutations in the gene encoding lanosterol 14α demethylase.4,12,13
Potassium hydroxide preparations and fungal cultures cannot differentiate T indotineae from other dermatophytes that typically cause tinea.5,14 Histopathologic findings in our case were no different than those of non–T indotineae dermatophytes. Only molecular testing using PCR assays to sequence internal transcribed spacer genes can confirm T indotineae infection. However, PCR assays and AFST are not available in many US laboratories.5 Matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry has shown promise in distinguishing T indotineae from other dermatophytes, though its clinical use is limited and it cannot assess terbinafine sensitivity.15,16 Clinicians in the United States who want to test specimens from cases suspicious for T indotineae infection should contact their local or state health department or the Centers for Disease Control and Prevention for assistance.3,5
Systemic treatment typically is necessary for T indotineae infection.5 Combinations of oral and topical azoles have been used, as well as topical ciclopirox, amorolfine (not available in the United States), and luliconazole.1,5,17-21
Itraconazole has emerged as the treatment of choice for T indotineae tinea, typically at 200 mg/d and often for courses of more than 3 months.5 Testing for serum itraconazole trough levels, as done for our patient, typically is not recommended. Clinicians should counsel patients to take itraconazole with high-fat foods and an acidic beverage to increase bioavailability.5 Potential adverse effects of itraconazole include heart failure and numerous drug-drug interactions.5,22 Patients with T indotineae dermatophytosis should avoid sharing personal belongings and having skin-to-skin contact of affected areas with others.4
Dermatologists who suspect T indotineae infection should work with public health agencies that can assist with testing and undertake infection surveillance, prevention, and control.5,23 Challenges to diagnosing and managing T indotineae infection include lack of awareness among dermatology providers, the need for specialized laboratory testing to confirm infection, lack of established clinical cut-points for MICs from AFST, the need for longer duration of treatment vs what is needed for typical tinea, and potential challenges with insurance coverage for testing and treatment. Empiric treatment with itraconazole should be considered when terbinafine-resistant dermatophytosis is suspected or when terbinafine-resistant T indotineae infection is confirmed.
Acknowledgments—Jeremy Gold, MD; Dallas J. Smith, PharmD; and Shawn Lockhart, PhD, all of the Centers for Disease Control and Prevention, Mycotic Diseases Branch (Atlanta, Georgia), provided helpful comments to the authors in preparing the manuscript of this article.
- Uhrlaß S, Verma SB, Gräser Y, al. Trichophyton indotineae—an emerging pathogen causing recalcitrant dermatophytoses in India and worldwide—a multidimensional perspective. J Fungi (Basel). 2022;8:757. doi:10.3390/jof8070757
- Jabet A, Brun S, Normand A-C, et al. Extensive dermatophytosis caused by terbinafine-resistant Trichophyton indotineae, France. Emerg Infect Dis. 2022;28:229-233. doi:10.3201/eid2801.210883
- Caplan AS, Chaturvedi S, Zhu Y, et al. Notes from the field. First reported U.S. cases of tinea caused by Trichophyton indotineae—New York City, December 2021-March 2023. MMWR Morb Mortal Wkly Rep. 2023;72:536-537. doi:10.15585/mmwr.mm7219a4
- Jabet A, Normand A-C, Brun S, et al. Trichophyton indotineae, from epidemiology to therapeutic. J Mycol Med. 2023;33:101383. doi:10.1016/j.mycmed.2023.101383
- Hill RC, Caplan AS, Elewski B, et al. Expert panel review of skin and hair dermatophytoses in an era of antifungal resistance. Am J Clin Dermatol. 2024;25:359-389. doi:10.1007/s40257-024-00848-1
- Caplan AS, Zakhem GA, Pomeranz MK. Trichophyton mentagrophytes internal transcribed spacer genotype VIII. JAMA Dermatol. 2023;159:1130. doi:10.1001/jamadermatol.2023.2645
- Spivack S, Gold JAW, Lockhart SR, et al. Potential sexual transmission of antifungal-resistant Trichophyton indotineae. Emerg Infect Dis. 2024;30:807-809. doi:10.3201/eid3004.240115
- Caplan AS, Todd GC, Zhu Y, et al. Clinical course, antifungal susceptibility, and genomic sequencing of Trichophyton indotineae. JAMA Dermatol. Published online May 15, 2024. doi:10.1001/jamadermatol.2024.1126
- Cañete-Gibas CF, Mele J, Patterson HP, et al. Terbinafine-resistant dermatophytes and the presence of Trichophyton indotineae in North America. J Clin Microbiol. 2023;61:e0056223. doi:10.1128/jcm.00562-23
- Gupta AK, Venkataraman M, Hall DC, et al. The emergence of Trichophyton indotineae: implications for clinical practice. Int J Dermatol. 2023;62:857-861.
- Oladzad V, Nasrollahi Omran A, Haghani I, et al. Multi-drug resistance Trichophyton indotineae in a stray dog. Res Vet Sci. 2024;166:105105. doi:10.1016/j.rvsc.2023.105105
- Martinez-Rossi NM, Bitencourt TA, Peres NTA, et al. Dermatophyte resistance to antifungal drugs: mechanisms and prospectus. Front Microbiol. 2018;9:1108. doi:10.3389/fmicb.2018.01108
- Sacheli R, Hayette MP. Antifungal resistance in dermatophytes: genetic considerations, clinical presentations and alternative therapies. J Fungi (Basel). 2021;711:983. doi:10.3390/jof7110983
- Gupta AK, Cooper EA. Dermatophytosis (tinea) and other superficial fungal infections. In: Hospenthal DR, Rinaldi MG, eds. Diagnosis and Treatment of Human Mycoses. Humana Press; 2008:355-381.
- Normand A-C, Moreno-Sabater A, Jabet A, et al. MALDI-TOF mass spectrometry online identification of Trichophyton indotineae using the MSI-2 application. J Fungi (Basel). 2022;8:1103. doi:10.3390/jof8101103
- De Paepe R, Normand A-C, Uhrlaß S, et al. Resistance profile, terbinafine resistance screening and MALDI-TOF MS identification of the emerging pathogen Trichophyton indotineae. Mycopathologia. 2024;189:29. doi:10.1007/s11046-024-00835-4
- Rajagopalan M, Inamadar A, Mittal A, et al. Expert consensus on the management of dermatophytosis in India (ECTODERM India). BMC Dermatol. 2018;18:6. doi:10.1186/s12895-018-0073-1
- Verma SB, Panda S, Nenoff P, et al. The unprecedented epidemic-like scenario of dermatophytosis in India: III. Antifungal resistance and treatment options. Indian J Dermatol Venereol Leprol. 2021;87:468-482. doi:10.25259/IJDVL_303_20
- Shaw D, Singh S, Dogra S, et al. MIC and upper limit of wild-type distribution for 13 antifungal agents against a Trichophyton mentagrophytes–Trichophyton interdigitale complex of Indian origin. Antimicrob Agents Chemother. 2020;64:E01964-19. doi:10.1128/AAC.01964-19
- Burmester A, Hipler U-C, Uhrlaß S, et al. Indian Trichophyton mentagrophytes squalene epoxidase erg1 double mutants show high proportion of combined fluconazole and terbinafine resistance. Mycoses. 2020;63:1175-1180. doi:10.1111/myc.13150
- Khurana A, Agarwal A, Agrawal D, et al. Effect of different itraconazole dosing regimens on cure rates, treatment duration, safety, and relapse rates in adult patients with tinea corporis/cruris: a randomized clinical trial. JAMA Dermatol. 2022;158:1269-1278. doi:10.1001/jamadermatol.2022.3745
- Itraconazole capsule. DailyMed [Internet]. Updated June 3, 2024. Accessed June 19, 2024. https://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=2ab38a8a-3708-4b97-9f7f-8e554a15348d
- Bui TS, Katz KA. Resistant Trichophyton indotineae dermatophytosis—an emerging pandemic, now in the US. JAMA Dermatol. Published online May 15, 2024. doi:10.1001/jamadermatol.2024.1125
To the Editor:
Historically, commonly available antifungal medications have been effective for treating dermatophytosis (tinea). However, recent tinea outbreaks caused by Trichophyton indotineae—a dermatophyte often resistant to terbinafine and sometimes to other antifungals—have been reported in South Asia, Europe, the Middle East, Southeast Asia, and Australia.1-5
Three confirmed cases of T indotineae dermatophytosis in the United States were reported in 2023 in New York3,6; a fourth confirmed case was reported in 2024 in Pennsylvania.7 Post hoc laboratory testing of fungal isolates in New York in 2022 and 2023 identified an additional 11 cases.8 We present a case of extensive multidrug-resistant tinea caused by T indotineae in a man in California.
An otherwise healthy 65-year-old man who had traveled to Europe in the past 3 months presented to his primary care physician with a widespread pruritic rash (Figure 1). He was treated with 2 weeks of oral terbinafine 250 mg/d and topical medicines, including clotrimazole cream 1%, fluocinonide ointment 0.05%, and clobetasol ointment 0.05% without improvement. Subsequently, 2 weeks of oral griseofulvin microsize 500 mg/d also proved ineffective. An antibody test was negative for HIV. His hemoglobin A1c was 6.2% (reference range, ≤5.6%). The patient was referred to dermatology.
Erythematous plaques—many scaly throughout and some annular with central clearing—were present on the arms, legs, and torso as well as in the groin. Honey crust was present on some plaques on the leg. A potassium hydroxide preparation showed abundant fungal hyphae. Material for fungal and bacterial cultures was collected. The patient was treated again with oral terbinafine 250 mg/d, an oral prednisone taper starting at 60 mg/d for a presumed id reaction, and various oral antihistamines for pruritus; all were ineffective. A bacterial culture showed only mixed skin flora. Oral fluconazole 200 mg/d was prescribed. A skin biopsy specimen showed compact orthokeratosis and parakeratosis of the stratum corneum with few neutrophils and focal pustule formation (Figure 2). Superficial perivascular inflammation, including lymphocytes, histiocytes, and few neutrophils, was present. A periodic acid–Schiff stain showed fungal hyphae in the stratum corneum and a hair follicle (Figure 3). After approximately 2 weeks, mold was identified in the fungal culture. Approximately 2 weeks thereafter, the organism was reported as Trichophyton species.
The rash did not improve; resistance to terbinafine, griseofulvin, and fluconazole was suspected clinically. The fungal isolate was sent to a reference laboratory (University of Texas Health Science Center, San Antonio). Meanwhile, oral itraconazole 200 mg twice daily and ketoconazole cream 2% were prescribed; the rash began to improve. A serum itraconazole trough level obtained 4 days after treatment initiation was 0.5 μg/mL (reference range, ≥0.6 μg/mL). The evening itraconazole dose was increased to 300 mg; a subsequent trough level was 0.8 μg/mL.
Approximately 1 month after the fungal isolate was sent to the reference laboratory, T indotineae was confirmed based on polymerase chain reaction (PCR) testing of internal transcribed spacer region sequences. Minimum inhibitory concentrations (MICs) obtained through antifungal susceptibility testing (AFST) were reported for fluconazole (8 μg/mL), griseofulvin (2 μg/mL), itraconazole (≤0.03 μg/mL), posaconazole (≤0.03 μg/mL), terbinafine (≥2 μg/mL), and voriconazole (0.125 μg/mL).
Approximately 7 weeks after itraconazole and ketoconazole were started, the rash had completely resolved. Nearly 8 months later (at the time this article was written), the rash had not recurred.
We report a unique case of T indotineae in a patient residing in California. Post hoc laboratory testing of dermatophyte isolates sent to the University of Texas reference laboratory identified terbinafine-resistant T indotineae specimens from the United States and Canada dating to 2017; clinical characteristics of patients from whom those isolates were obtained were unavailable.9
Trichophyton indotineae dermatophytosis typically is more extensive, inflamed, and pruritic, as well as likely more contagious, than tinea caused by other dermatophytes.5 Previously called Trichophyton mentagrophytes genotype VIII when first isolated in 2017, the pathogen was renamed T indotineae in 2020 after important genetic differences were discovered between it and other T mentagrophytes species.5 The emergence of T indotineae has been attributed to concomitant use of topical steroids and antifungals,5,10 inappropriate prescribing of antifungals,5 and nonadherence to antifungal treatment.5
Likely risk factors for T indotineae infection include suboptimal hygiene, overcrowded conditions, hot and humid environments, and tight-fitting synthetic clothing.4 Transmission from family members appears common,5 especially when fomites are shared.4 A case reported in Pennsylvania likely was acquired through sexual contact.7 Travel to South Asia has been associated with acquisition of T indotineae infection,3,5-7 though our patient and some others had not traveled there.3,8 It is not clear whether immunosuppression and diabetes mellitus are associated with T indotineae infection.4,5,8Trichophyton indotineae also can affect animals,11 though zoonotic transmission has not been reported.4
Not all T indotineae isolates are resistant to one or more antifungals; furthermore, antifungal resistance in other dermatophyte species has been reported.5 Terbinafine resistance in T indotineae is conferred by mutations in the gene encoding squalene epoxidase, which helps synthesize ergosterol—a component of the cell membrane in fungi.2,4,5,12 Although clinical cut-points for MIC obtained by AFST are not well established, T indotineae MICs for terbinafine of 0.5 μg/mL or more correlate with resistance.9 Resistance to azoles has been linked to overexpression of transporter genes, which increase azole efflux from cells, as well as to mutations in the gene encoding lanosterol 14α demethylase.4,12,13
Potassium hydroxide preparations and fungal cultures cannot differentiate T indotineae from other dermatophytes that typically cause tinea.5,14 Histopathologic findings in our case were no different than those of non–T indotineae dermatophytes. Only molecular testing using PCR assays to sequence internal transcribed spacer genes can confirm T indotineae infection. However, PCR assays and AFST are not available in many US laboratories.5 Matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry has shown promise in distinguishing T indotineae from other dermatophytes, though its clinical use is limited and it cannot assess terbinafine sensitivity.15,16 Clinicians in the United States who want to test specimens from cases suspicious for T indotineae infection should contact their local or state health department or the Centers for Disease Control and Prevention for assistance.3,5
Systemic treatment typically is necessary for T indotineae infection.5 Combinations of oral and topical azoles have been used, as well as topical ciclopirox, amorolfine (not available in the United States), and luliconazole.1,5,17-21
Itraconazole has emerged as the treatment of choice for T indotineae tinea, typically at 200 mg/d and often for courses of more than 3 months.5 Testing for serum itraconazole trough levels, as done for our patient, typically is not recommended. Clinicians should counsel patients to take itraconazole with high-fat foods and an acidic beverage to increase bioavailability.5 Potential adverse effects of itraconazole include heart failure and numerous drug-drug interactions.5,22 Patients with T indotineae dermatophytosis should avoid sharing personal belongings and having skin-to-skin contact of affected areas with others.4
Dermatologists who suspect T indotineae infection should work with public health agencies that can assist with testing and undertake infection surveillance, prevention, and control.5,23 Challenges to diagnosing and managing T indotineae infection include lack of awareness among dermatology providers, the need for specialized laboratory testing to confirm infection, lack of established clinical cut-points for MICs from AFST, the need for longer duration of treatment vs what is needed for typical tinea, and potential challenges with insurance coverage for testing and treatment. Empiric treatment with itraconazole should be considered when terbinafine-resistant dermatophytosis is suspected or when terbinafine-resistant T indotineae infection is confirmed.
Acknowledgments—Jeremy Gold, MD; Dallas J. Smith, PharmD; and Shawn Lockhart, PhD, all of the Centers for Disease Control and Prevention, Mycotic Diseases Branch (Atlanta, Georgia), provided helpful comments to the authors in preparing the manuscript of this article.
To the Editor:
Historically, commonly available antifungal medications have been effective for treating dermatophytosis (tinea). However, recent tinea outbreaks caused by Trichophyton indotineae—a dermatophyte often resistant to terbinafine and sometimes to other antifungals—have been reported in South Asia, Europe, the Middle East, Southeast Asia, and Australia.1-5
Three confirmed cases of T indotineae dermatophytosis in the United States were reported in 2023 in New York3,6; a fourth confirmed case was reported in 2024 in Pennsylvania.7 Post hoc laboratory testing of fungal isolates in New York in 2022 and 2023 identified an additional 11 cases.8 We present a case of extensive multidrug-resistant tinea caused by T indotineae in a man in California.
An otherwise healthy 65-year-old man who had traveled to Europe in the past 3 months presented to his primary care physician with a widespread pruritic rash (Figure 1). He was treated with 2 weeks of oral terbinafine 250 mg/d and topical medicines, including clotrimazole cream 1%, fluocinonide ointment 0.05%, and clobetasol ointment 0.05% without improvement. Subsequently, 2 weeks of oral griseofulvin microsize 500 mg/d also proved ineffective. An antibody test was negative for HIV. His hemoglobin A1c was 6.2% (reference range, ≤5.6%). The patient was referred to dermatology.
Erythematous plaques—many scaly throughout and some annular with central clearing—were present on the arms, legs, and torso as well as in the groin. Honey crust was present on some plaques on the leg. A potassium hydroxide preparation showed abundant fungal hyphae. Material for fungal and bacterial cultures was collected. The patient was treated again with oral terbinafine 250 mg/d, an oral prednisone taper starting at 60 mg/d for a presumed id reaction, and various oral antihistamines for pruritus; all were ineffective. A bacterial culture showed only mixed skin flora. Oral fluconazole 200 mg/d was prescribed. A skin biopsy specimen showed compact orthokeratosis and parakeratosis of the stratum corneum with few neutrophils and focal pustule formation (Figure 2). Superficial perivascular inflammation, including lymphocytes, histiocytes, and few neutrophils, was present. A periodic acid–Schiff stain showed fungal hyphae in the stratum corneum and a hair follicle (Figure 3). After approximately 2 weeks, mold was identified in the fungal culture. Approximately 2 weeks thereafter, the organism was reported as Trichophyton species.
The rash did not improve; resistance to terbinafine, griseofulvin, and fluconazole was suspected clinically. The fungal isolate was sent to a reference laboratory (University of Texas Health Science Center, San Antonio). Meanwhile, oral itraconazole 200 mg twice daily and ketoconazole cream 2% were prescribed; the rash began to improve. A serum itraconazole trough level obtained 4 days after treatment initiation was 0.5 μg/mL (reference range, ≥0.6 μg/mL). The evening itraconazole dose was increased to 300 mg; a subsequent trough level was 0.8 μg/mL.
Approximately 1 month after the fungal isolate was sent to the reference laboratory, T indotineae was confirmed based on polymerase chain reaction (PCR) testing of internal transcribed spacer region sequences. Minimum inhibitory concentrations (MICs) obtained through antifungal susceptibility testing (AFST) were reported for fluconazole (8 μg/mL), griseofulvin (2 μg/mL), itraconazole (≤0.03 μg/mL), posaconazole (≤0.03 μg/mL), terbinafine (≥2 μg/mL), and voriconazole (0.125 μg/mL).
Approximately 7 weeks after itraconazole and ketoconazole were started, the rash had completely resolved. Nearly 8 months later (at the time this article was written), the rash had not recurred.
We report a unique case of T indotineae in a patient residing in California. Post hoc laboratory testing of dermatophyte isolates sent to the University of Texas reference laboratory identified terbinafine-resistant T indotineae specimens from the United States and Canada dating to 2017; clinical characteristics of patients from whom those isolates were obtained were unavailable.9
Trichophyton indotineae dermatophytosis typically is more extensive, inflamed, and pruritic, as well as likely more contagious, than tinea caused by other dermatophytes.5 Previously called Trichophyton mentagrophytes genotype VIII when first isolated in 2017, the pathogen was renamed T indotineae in 2020 after important genetic differences were discovered between it and other T mentagrophytes species.5 The emergence of T indotineae has been attributed to concomitant use of topical steroids and antifungals,5,10 inappropriate prescribing of antifungals,5 and nonadherence to antifungal treatment.5
Likely risk factors for T indotineae infection include suboptimal hygiene, overcrowded conditions, hot and humid environments, and tight-fitting synthetic clothing.4 Transmission from family members appears common,5 especially when fomites are shared.4 A case reported in Pennsylvania likely was acquired through sexual contact.7 Travel to South Asia has been associated with acquisition of T indotineae infection,3,5-7 though our patient and some others had not traveled there.3,8 It is not clear whether immunosuppression and diabetes mellitus are associated with T indotineae infection.4,5,8Trichophyton indotineae also can affect animals,11 though zoonotic transmission has not been reported.4
Not all T indotineae isolates are resistant to one or more antifungals; furthermore, antifungal resistance in other dermatophyte species has been reported.5 Terbinafine resistance in T indotineae is conferred by mutations in the gene encoding squalene epoxidase, which helps synthesize ergosterol—a component of the cell membrane in fungi.2,4,5,12 Although clinical cut-points for MIC obtained by AFST are not well established, T indotineae MICs for terbinafine of 0.5 μg/mL or more correlate with resistance.9 Resistance to azoles has been linked to overexpression of transporter genes, which increase azole efflux from cells, as well as to mutations in the gene encoding lanosterol 14α demethylase.4,12,13
Potassium hydroxide preparations and fungal cultures cannot differentiate T indotineae from other dermatophytes that typically cause tinea.5,14 Histopathologic findings in our case were no different than those of non–T indotineae dermatophytes. Only molecular testing using PCR assays to sequence internal transcribed spacer genes can confirm T indotineae infection. However, PCR assays and AFST are not available in many US laboratories.5 Matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry has shown promise in distinguishing T indotineae from other dermatophytes, though its clinical use is limited and it cannot assess terbinafine sensitivity.15,16 Clinicians in the United States who want to test specimens from cases suspicious for T indotineae infection should contact their local or state health department or the Centers for Disease Control and Prevention for assistance.3,5
Systemic treatment typically is necessary for T indotineae infection.5 Combinations of oral and topical azoles have been used, as well as topical ciclopirox, amorolfine (not available in the United States), and luliconazole.1,5,17-21
Itraconazole has emerged as the treatment of choice for T indotineae tinea, typically at 200 mg/d and often for courses of more than 3 months.5 Testing for serum itraconazole trough levels, as done for our patient, typically is not recommended. Clinicians should counsel patients to take itraconazole with high-fat foods and an acidic beverage to increase bioavailability.5 Potential adverse effects of itraconazole include heart failure and numerous drug-drug interactions.5,22 Patients with T indotineae dermatophytosis should avoid sharing personal belongings and having skin-to-skin contact of affected areas with others.4
Dermatologists who suspect T indotineae infection should work with public health agencies that can assist with testing and undertake infection surveillance, prevention, and control.5,23 Challenges to diagnosing and managing T indotineae infection include lack of awareness among dermatology providers, the need for specialized laboratory testing to confirm infection, lack of established clinical cut-points for MICs from AFST, the need for longer duration of treatment vs what is needed for typical tinea, and potential challenges with insurance coverage for testing and treatment. Empiric treatment with itraconazole should be considered when terbinafine-resistant dermatophytosis is suspected or when terbinafine-resistant T indotineae infection is confirmed.
Acknowledgments—Jeremy Gold, MD; Dallas J. Smith, PharmD; and Shawn Lockhart, PhD, all of the Centers for Disease Control and Prevention, Mycotic Diseases Branch (Atlanta, Georgia), provided helpful comments to the authors in preparing the manuscript of this article.
- Uhrlaß S, Verma SB, Gräser Y, al. Trichophyton indotineae—an emerging pathogen causing recalcitrant dermatophytoses in India and worldwide—a multidimensional perspective. J Fungi (Basel). 2022;8:757. doi:10.3390/jof8070757
- Jabet A, Brun S, Normand A-C, et al. Extensive dermatophytosis caused by terbinafine-resistant Trichophyton indotineae, France. Emerg Infect Dis. 2022;28:229-233. doi:10.3201/eid2801.210883
- Caplan AS, Chaturvedi S, Zhu Y, et al. Notes from the field. First reported U.S. cases of tinea caused by Trichophyton indotineae—New York City, December 2021-March 2023. MMWR Morb Mortal Wkly Rep. 2023;72:536-537. doi:10.15585/mmwr.mm7219a4
- Jabet A, Normand A-C, Brun S, et al. Trichophyton indotineae, from epidemiology to therapeutic. J Mycol Med. 2023;33:101383. doi:10.1016/j.mycmed.2023.101383
- Hill RC, Caplan AS, Elewski B, et al. Expert panel review of skin and hair dermatophytoses in an era of antifungal resistance. Am J Clin Dermatol. 2024;25:359-389. doi:10.1007/s40257-024-00848-1
- Caplan AS, Zakhem GA, Pomeranz MK. Trichophyton mentagrophytes internal transcribed spacer genotype VIII. JAMA Dermatol. 2023;159:1130. doi:10.1001/jamadermatol.2023.2645
- Spivack S, Gold JAW, Lockhart SR, et al. Potential sexual transmission of antifungal-resistant Trichophyton indotineae. Emerg Infect Dis. 2024;30:807-809. doi:10.3201/eid3004.240115
- Caplan AS, Todd GC, Zhu Y, et al. Clinical course, antifungal susceptibility, and genomic sequencing of Trichophyton indotineae. JAMA Dermatol. Published online May 15, 2024. doi:10.1001/jamadermatol.2024.1126
- Cañete-Gibas CF, Mele J, Patterson HP, et al. Terbinafine-resistant dermatophytes and the presence of Trichophyton indotineae in North America. J Clin Microbiol. 2023;61:e0056223. doi:10.1128/jcm.00562-23
- Gupta AK, Venkataraman M, Hall DC, et al. The emergence of Trichophyton indotineae: implications for clinical practice. Int J Dermatol. 2023;62:857-861.
- Oladzad V, Nasrollahi Omran A, Haghani I, et al. Multi-drug resistance Trichophyton indotineae in a stray dog. Res Vet Sci. 2024;166:105105. doi:10.1016/j.rvsc.2023.105105
- Martinez-Rossi NM, Bitencourt TA, Peres NTA, et al. Dermatophyte resistance to antifungal drugs: mechanisms and prospectus. Front Microbiol. 2018;9:1108. doi:10.3389/fmicb.2018.01108
- Sacheli R, Hayette MP. Antifungal resistance in dermatophytes: genetic considerations, clinical presentations and alternative therapies. J Fungi (Basel). 2021;711:983. doi:10.3390/jof7110983
- Gupta AK, Cooper EA. Dermatophytosis (tinea) and other superficial fungal infections. In: Hospenthal DR, Rinaldi MG, eds. Diagnosis and Treatment of Human Mycoses. Humana Press; 2008:355-381.
- Normand A-C, Moreno-Sabater A, Jabet A, et al. MALDI-TOF mass spectrometry online identification of Trichophyton indotineae using the MSI-2 application. J Fungi (Basel). 2022;8:1103. doi:10.3390/jof8101103
- De Paepe R, Normand A-C, Uhrlaß S, et al. Resistance profile, terbinafine resistance screening and MALDI-TOF MS identification of the emerging pathogen Trichophyton indotineae. Mycopathologia. 2024;189:29. doi:10.1007/s11046-024-00835-4
- Rajagopalan M, Inamadar A, Mittal A, et al. Expert consensus on the management of dermatophytosis in India (ECTODERM India). BMC Dermatol. 2018;18:6. doi:10.1186/s12895-018-0073-1
- Verma SB, Panda S, Nenoff P, et al. The unprecedented epidemic-like scenario of dermatophytosis in India: III. Antifungal resistance and treatment options. Indian J Dermatol Venereol Leprol. 2021;87:468-482. doi:10.25259/IJDVL_303_20
- Shaw D, Singh S, Dogra S, et al. MIC and upper limit of wild-type distribution for 13 antifungal agents against a Trichophyton mentagrophytes–Trichophyton interdigitale complex of Indian origin. Antimicrob Agents Chemother. 2020;64:E01964-19. doi:10.1128/AAC.01964-19
- Burmester A, Hipler U-C, Uhrlaß S, et al. Indian Trichophyton mentagrophytes squalene epoxidase erg1 double mutants show high proportion of combined fluconazole and terbinafine resistance. Mycoses. 2020;63:1175-1180. doi:10.1111/myc.13150
- Khurana A, Agarwal A, Agrawal D, et al. Effect of different itraconazole dosing regimens on cure rates, treatment duration, safety, and relapse rates in adult patients with tinea corporis/cruris: a randomized clinical trial. JAMA Dermatol. 2022;158:1269-1278. doi:10.1001/jamadermatol.2022.3745
- Itraconazole capsule. DailyMed [Internet]. Updated June 3, 2024. Accessed June 19, 2024. https://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=2ab38a8a-3708-4b97-9f7f-8e554a15348d
- Bui TS, Katz KA. Resistant Trichophyton indotineae dermatophytosis—an emerging pandemic, now in the US. JAMA Dermatol. Published online May 15, 2024. doi:10.1001/jamadermatol.2024.1125
- Uhrlaß S, Verma SB, Gräser Y, al. Trichophyton indotineae—an emerging pathogen causing recalcitrant dermatophytoses in India and worldwide—a multidimensional perspective. J Fungi (Basel). 2022;8:757. doi:10.3390/jof8070757
- Jabet A, Brun S, Normand A-C, et al. Extensive dermatophytosis caused by terbinafine-resistant Trichophyton indotineae, France. Emerg Infect Dis. 2022;28:229-233. doi:10.3201/eid2801.210883
- Caplan AS, Chaturvedi S, Zhu Y, et al. Notes from the field. First reported U.S. cases of tinea caused by Trichophyton indotineae—New York City, December 2021-March 2023. MMWR Morb Mortal Wkly Rep. 2023;72:536-537. doi:10.15585/mmwr.mm7219a4
- Jabet A, Normand A-C, Brun S, et al. Trichophyton indotineae, from epidemiology to therapeutic. J Mycol Med. 2023;33:101383. doi:10.1016/j.mycmed.2023.101383
- Hill RC, Caplan AS, Elewski B, et al. Expert panel review of skin and hair dermatophytoses in an era of antifungal resistance. Am J Clin Dermatol. 2024;25:359-389. doi:10.1007/s40257-024-00848-1
- Caplan AS, Zakhem GA, Pomeranz MK. Trichophyton mentagrophytes internal transcribed spacer genotype VIII. JAMA Dermatol. 2023;159:1130. doi:10.1001/jamadermatol.2023.2645
- Spivack S, Gold JAW, Lockhart SR, et al. Potential sexual transmission of antifungal-resistant Trichophyton indotineae. Emerg Infect Dis. 2024;30:807-809. doi:10.3201/eid3004.240115
- Caplan AS, Todd GC, Zhu Y, et al. Clinical course, antifungal susceptibility, and genomic sequencing of Trichophyton indotineae. JAMA Dermatol. Published online May 15, 2024. doi:10.1001/jamadermatol.2024.1126
- Cañete-Gibas CF, Mele J, Patterson HP, et al. Terbinafine-resistant dermatophytes and the presence of Trichophyton indotineae in North America. J Clin Microbiol. 2023;61:e0056223. doi:10.1128/jcm.00562-23
- Gupta AK, Venkataraman M, Hall DC, et al. The emergence of Trichophyton indotineae: implications for clinical practice. Int J Dermatol. 2023;62:857-861.
- Oladzad V, Nasrollahi Omran A, Haghani I, et al. Multi-drug resistance Trichophyton indotineae in a stray dog. Res Vet Sci. 2024;166:105105. doi:10.1016/j.rvsc.2023.105105
- Martinez-Rossi NM, Bitencourt TA, Peres NTA, et al. Dermatophyte resistance to antifungal drugs: mechanisms and prospectus. Front Microbiol. 2018;9:1108. doi:10.3389/fmicb.2018.01108
- Sacheli R, Hayette MP. Antifungal resistance in dermatophytes: genetic considerations, clinical presentations and alternative therapies. J Fungi (Basel). 2021;711:983. doi:10.3390/jof7110983
- Gupta AK, Cooper EA. Dermatophytosis (tinea) and other superficial fungal infections. In: Hospenthal DR, Rinaldi MG, eds. Diagnosis and Treatment of Human Mycoses. Humana Press; 2008:355-381.
- Normand A-C, Moreno-Sabater A, Jabet A, et al. MALDI-TOF mass spectrometry online identification of Trichophyton indotineae using the MSI-2 application. J Fungi (Basel). 2022;8:1103. doi:10.3390/jof8101103
- De Paepe R, Normand A-C, Uhrlaß S, et al. Resistance profile, terbinafine resistance screening and MALDI-TOF MS identification of the emerging pathogen Trichophyton indotineae. Mycopathologia. 2024;189:29. doi:10.1007/s11046-024-00835-4
- Rajagopalan M, Inamadar A, Mittal A, et al. Expert consensus on the management of dermatophytosis in India (ECTODERM India). BMC Dermatol. 2018;18:6. doi:10.1186/s12895-018-0073-1
- Verma SB, Panda S, Nenoff P, et al. The unprecedented epidemic-like scenario of dermatophytosis in India: III. Antifungal resistance and treatment options. Indian J Dermatol Venereol Leprol. 2021;87:468-482. doi:10.25259/IJDVL_303_20
- Shaw D, Singh S, Dogra S, et al. MIC and upper limit of wild-type distribution for 13 antifungal agents against a Trichophyton mentagrophytes–Trichophyton interdigitale complex of Indian origin. Antimicrob Agents Chemother. 2020;64:E01964-19. doi:10.1128/AAC.01964-19
- Burmester A, Hipler U-C, Uhrlaß S, et al. Indian Trichophyton mentagrophytes squalene epoxidase erg1 double mutants show high proportion of combined fluconazole and terbinafine resistance. Mycoses. 2020;63:1175-1180. doi:10.1111/myc.13150
- Khurana A, Agarwal A, Agrawal D, et al. Effect of different itraconazole dosing regimens on cure rates, treatment duration, safety, and relapse rates in adult patients with tinea corporis/cruris: a randomized clinical trial. JAMA Dermatol. 2022;158:1269-1278. doi:10.1001/jamadermatol.2022.3745
- Itraconazole capsule. DailyMed [Internet]. Updated June 3, 2024. Accessed June 19, 2024. https://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=2ab38a8a-3708-4b97-9f7f-8e554a15348d
- Bui TS, Katz KA. Resistant Trichophyton indotineae dermatophytosis—an emerging pandemic, now in the US. JAMA Dermatol. Published online May 15, 2024. doi:10.1001/jamadermatol.2024.1125
Practice Points
- Trichophyton indotineae can cause extensive dermatophytosis that often is resistant to terbinafine and in some cases to other antifungals.
- Only molecular testing, which is not widely available, can distinguish T indotineae from other dermatophytes.
- Suspected or confirmed cases of T indotineae dermatophytosis should be reported to public health agencies to provide assistance with testing, as well as surveillance, prevention, and control of infection.
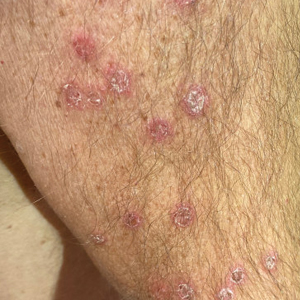
Treatment of Infantile Hemangiomas in Concomitant Tuberous Sclerosis Complex Should Prompt Evaluation for Cardiac Rhabdomyomas Prior to Initiation of Propranolol
To the Editor:
Cardiac rhabdomyomas are benign hamartomas that are common in patients with tuberous sclerosis complex (TSC).1 We describe a patient who presented with large infantile hemangiomas (IHs) and hypopigmented macules, which prompted further testing that eventually showed concomitant multiple cardiac rhabdomyomas in the context of TSC.
A 5-week-old girl—who was born at 38 weeks and 3 days’ gestation via uncomplicated vaginal delivery—was referred to our pediatric dermatology clinic for evaluation of multiple erythematous lesions on the scalp and left buttock that were first noticed 2 weeks prior to presentation. There was a family history of seizures in the patient’s mother. The patient’s older brother did not have similar symptoms.
Physical examination revealed 2 nonulcerating erythematous nodules on the middle and posterior left vertex scalp that measured 2.5×2 cm (Figure 1A) as well as 1 bright red plaque on the left buttock (Figure 1B). Five hypopigmented macules, ranging from 5 mm to 1.5 cm in diameter, also were detected on the left thorax (Figure 2A) as well as the middle and lower back (Figure 2B). These findings, along with the history of seizures in the patient’s mother, prompted further evaluation of the family history, which uncovered TSC in the patient’s mother, maternal aunt, and maternal grandmother.
The large IHs on the scalp did not pose concerns for potential functional impairment but were still considered high risk for permanent alopecia based on clinical practice guidelines for the management of IH.2 Treatment with oral propranolol was recommended; however, because of a strong suspicion of TSC due to the presence of 5 hypopigmented macules measuring more than 5 mm in diameter (≥3 hypopigmented macules of ≥5 mm is one of the major criterion for TSC), the patient was referred to cardiology prior to initiation of propranolol.
Echocardiography revealed 3 intracardiac masses measuring 4 to 5 mm in diameter in the left ventricle (LV), along the interventricular septum and the LV posterior wall. These masses were consistent with rhabdomyomas (Figure 3)—a major criterion for TSC—which had not been detected by prenatal ultrasonography. No obstruction to LV inflow or outflow was observed. Additionally, no arrhythmias were detected on electrocardiography.
The patient was cleared for propranolol, which was slowly uptitrated to 2 mg/kg/d. She completed the course without adverse effects. The treatment of IH was successful with substantial reduction in size over the following months until clearance. She also was referred to neurology for magnetic resonance imaging of the brain, which showed a 3-mm subependymal nodule in the lateral right ventricle, another major feature of TSC.
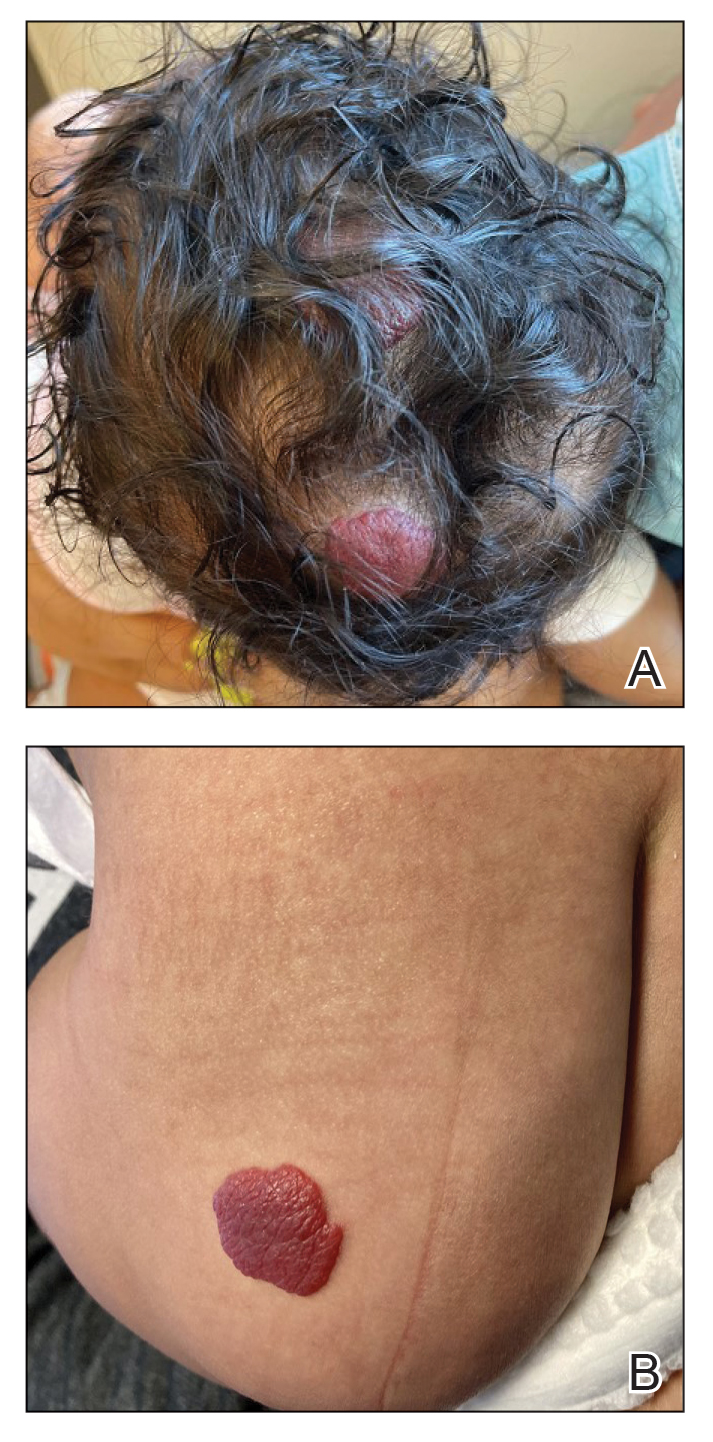
Cardiac rhabdomyomas are benign hamartomas that affect as many as 80% of patients with TSC1 and are primarily localized in the ventricles. Although cardiac rhabdomyomas usually regress over time, they can compromise ventricular function or valvular function, or both, and result in outflow obstruction, arrhythmias, and Wolff- Parkinson-White syndrome.3 Surgical resection may be needed in patients whose condition is refractory to medical management for heart failure.
The pathophysiologic mechanism behind the natural involution of cardiac rhabdomyomas has not been fully elucidated. It has been hypothesized that these masses stem from the inability of rhabdomyoma cells to divide after birth due to their embryonic myocyte derivation.4
According to the TSC diagnostic criteria from the Tuberous Sclerosis Complex International Consensus Group, at least 2 major features or 1 major and 2 minor features are required to make a definitive diagnosis of TSC. Cutaneous signs represent more than one-third of major features of TSC; almost all patients with TSC have skin findings.5
Identification of pathogenic mutations in either TSC1 (on chromosome 9q34.3, encoding for hamartin) or TSC2 (on chromosome 16p13.3, encoding for tuberin), resulting in constitutive activation of mammalian target of rapamycin and subsequent increased cell growth, is sufficient for a definitive diagnosis of TSC. However, mutations cannot be identified by conventional genetic testing in as many as one-quarter of patients with TSC; therefore, a negative result does not exclude TSC if the patient meets clinical diagnostic criteria.
Although a cardiology workup is indicated prior to initiating propranolol in the presence of possible cardiac rhabdomyomas, most of those lesions are hemodynamically stable and do not require treatment. There also is no contraindication for β-blocker therapy. In fact, propranolol has been reported as a successful treatment in rhabdomyoma-associated arrhythmias in children.6 Notably, obstructive cardiac rhabdomyomas have been successfully treated with mammalian target of rapamycin inhibitors, such as sirolimus7 and everolimus.8
Baseline cardiology screening with echocardiography prior to initiating propranolol for treatment of IH is not routinely indicated in babies with uncomplicated IH. However, in a patient with TSC, cardiology screening is necessary to rule out rhabdomyomas with associated arrhythmias or obstructed blood flow, or both, prior to initiating treatment.
We presented a case of concomitant IH and TSC in a patient with cardiac rhabdomyomas. The manifestation of large IHs in our patient prompted further testing that revealed multiple cardiac rhabdomyomas in the context of TSC. It is imperative for cardiologists, cardiac surgeons, and dermatologists to be familiar with the TSC diagnostic criteria so that they can reach a prompt diagnosis and make appropriate referrals for further evaluation of cardiac, neurologic, and ophthalmologic signs.
- Frudit P, Vitturi BK, Navarro FC, et al. Multiple cardiac rhabdomyomas in tuberous sclerosis complex: case report and review of the literature. Autops Case Rep. 2019;9:e2019125. doi:10.4322/acr.2019.125
- Krowchuk DP, Frieden IJ, Mancini AJ, et al; Subcommittee on the Management of Infantile Hemangiomas. Clinical practice guideline for the management of infantile hemangiomas. Pediatrics. 2019;143:e20183475. doi:10.1542/peds.2018-3475
- Venugopalan P, Babu JS, Al-Bulushi A. Right atrial rhabdomyoma acting as the substrate for Wolff-Parkinson-White syndrome in a 3-month-old infant. Acta Cardiol. 2005;60:543-545. doi:10.2143/AC.60.5.2004977
- DiMario FJ Jr, Diana D, Leopold H, et al. Evolution of cardiac rhabdomyoma in tuberous sclerosis complex. Clin Pediatr (Phila). 1996;35:615-619. doi:10.1177/000992289603501202
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254. doi:10.1016/j.pediatrneurol.2013.08.001
- Kathare PA, Muthuswamy KS, Sadasivan J, et al. Incessant ventricular tachycardia due to multiple cardiac rhabdomyomas in an infant with tuberous sclerosis. Indian Heart J. 2013;65:111-113. doi:10.1016/j.ihj.2012.12.003
- Breathnach C, Pears J, Franklin O, et al. Rapid regression of left ventricular outflow tract rhabdomyoma after sirolimus therapy. Pediatrics. 2014;134:e1199-e1202. doi:10.1542/peds.2013-3293
- Chang J-S, Chiou P-Y, Yao S-H, et al. Regression of neonatal cardiac rhabdomyoma in two months through low-dose everolimus therapy: a report of three cases. Pediatr Cardiol. 2017;38:1478-1484. doi:10.1007/s00246-017-1688-4
To the Editor:
Cardiac rhabdomyomas are benign hamartomas that are common in patients with tuberous sclerosis complex (TSC).1 We describe a patient who presented with large infantile hemangiomas (IHs) and hypopigmented macules, which prompted further testing that eventually showed concomitant multiple cardiac rhabdomyomas in the context of TSC.
A 5-week-old girl—who was born at 38 weeks and 3 days’ gestation via uncomplicated vaginal delivery—was referred to our pediatric dermatology clinic for evaluation of multiple erythematous lesions on the scalp and left buttock that were first noticed 2 weeks prior to presentation. There was a family history of seizures in the patient’s mother. The patient’s older brother did not have similar symptoms.
Physical examination revealed 2 nonulcerating erythematous nodules on the middle and posterior left vertex scalp that measured 2.5×2 cm (Figure 1A) as well as 1 bright red plaque on the left buttock (Figure 1B). Five hypopigmented macules, ranging from 5 mm to 1.5 cm in diameter, also were detected on the left thorax (Figure 2A) as well as the middle and lower back (Figure 2B). These findings, along with the history of seizures in the patient’s mother, prompted further evaluation of the family history, which uncovered TSC in the patient’s mother, maternal aunt, and maternal grandmother.
The large IHs on the scalp did not pose concerns for potential functional impairment but were still considered high risk for permanent alopecia based on clinical practice guidelines for the management of IH.2 Treatment with oral propranolol was recommended; however, because of a strong suspicion of TSC due to the presence of 5 hypopigmented macules measuring more than 5 mm in diameter (≥3 hypopigmented macules of ≥5 mm is one of the major criterion for TSC), the patient was referred to cardiology prior to initiation of propranolol.
Echocardiography revealed 3 intracardiac masses measuring 4 to 5 mm in diameter in the left ventricle (LV), along the interventricular septum and the LV posterior wall. These masses were consistent with rhabdomyomas (Figure 3)—a major criterion for TSC—which had not been detected by prenatal ultrasonography. No obstruction to LV inflow or outflow was observed. Additionally, no arrhythmias were detected on electrocardiography.
The patient was cleared for propranolol, which was slowly uptitrated to 2 mg/kg/d. She completed the course without adverse effects. The treatment of IH was successful with substantial reduction in size over the following months until clearance. She also was referred to neurology for magnetic resonance imaging of the brain, which showed a 3-mm subependymal nodule in the lateral right ventricle, another major feature of TSC.
Cardiac rhabdomyomas are benign hamartomas that affect as many as 80% of patients with TSC1 and are primarily localized in the ventricles. Although cardiac rhabdomyomas usually regress over time, they can compromise ventricular function or valvular function, or both, and result in outflow obstruction, arrhythmias, and Wolff- Parkinson-White syndrome.3 Surgical resection may be needed in patients whose condition is refractory to medical management for heart failure.
The pathophysiologic mechanism behind the natural involution of cardiac rhabdomyomas has not been fully elucidated. It has been hypothesized that these masses stem from the inability of rhabdomyoma cells to divide after birth due to their embryonic myocyte derivation.4
According to the TSC diagnostic criteria from the Tuberous Sclerosis Complex International Consensus Group, at least 2 major features or 1 major and 2 minor features are required to make a definitive diagnosis of TSC. Cutaneous signs represent more than one-third of major features of TSC; almost all patients with TSC have skin findings.5
Identification of pathogenic mutations in either TSC1 (on chromosome 9q34.3, encoding for hamartin) or TSC2 (on chromosome 16p13.3, encoding for tuberin), resulting in constitutive activation of mammalian target of rapamycin and subsequent increased cell growth, is sufficient for a definitive diagnosis of TSC. However, mutations cannot be identified by conventional genetic testing in as many as one-quarter of patients with TSC; therefore, a negative result does not exclude TSC if the patient meets clinical diagnostic criteria.
Although a cardiology workup is indicated prior to initiating propranolol in the presence of possible cardiac rhabdomyomas, most of those lesions are hemodynamically stable and do not require treatment. There also is no contraindication for β-blocker therapy. In fact, propranolol has been reported as a successful treatment in rhabdomyoma-associated arrhythmias in children.6 Notably, obstructive cardiac rhabdomyomas have been successfully treated with mammalian target of rapamycin inhibitors, such as sirolimus7 and everolimus.8
Baseline cardiology screening with echocardiography prior to initiating propranolol for treatment of IH is not routinely indicated in babies with uncomplicated IH. However, in a patient with TSC, cardiology screening is necessary to rule out rhabdomyomas with associated arrhythmias or obstructed blood flow, or both, prior to initiating treatment.
We presented a case of concomitant IH and TSC in a patient with cardiac rhabdomyomas. The manifestation of large IHs in our patient prompted further testing that revealed multiple cardiac rhabdomyomas in the context of TSC. It is imperative for cardiologists, cardiac surgeons, and dermatologists to be familiar with the TSC diagnostic criteria so that they can reach a prompt diagnosis and make appropriate referrals for further evaluation of cardiac, neurologic, and ophthalmologic signs.
To the Editor:
Cardiac rhabdomyomas are benign hamartomas that are common in patients with tuberous sclerosis complex (TSC).1 We describe a patient who presented with large infantile hemangiomas (IHs) and hypopigmented macules, which prompted further testing that eventually showed concomitant multiple cardiac rhabdomyomas in the context of TSC.
A 5-week-old girl—who was born at 38 weeks and 3 days’ gestation via uncomplicated vaginal delivery—was referred to our pediatric dermatology clinic for evaluation of multiple erythematous lesions on the scalp and left buttock that were first noticed 2 weeks prior to presentation. There was a family history of seizures in the patient’s mother. The patient’s older brother did not have similar symptoms.
Physical examination revealed 2 nonulcerating erythematous nodules on the middle and posterior left vertex scalp that measured 2.5×2 cm (Figure 1A) as well as 1 bright red plaque on the left buttock (Figure 1B). Five hypopigmented macules, ranging from 5 mm to 1.5 cm in diameter, also were detected on the left thorax (Figure 2A) as well as the middle and lower back (Figure 2B). These findings, along with the history of seizures in the patient’s mother, prompted further evaluation of the family history, which uncovered TSC in the patient’s mother, maternal aunt, and maternal grandmother.
The large IHs on the scalp did not pose concerns for potential functional impairment but were still considered high risk for permanent alopecia based on clinical practice guidelines for the management of IH.2 Treatment with oral propranolol was recommended; however, because of a strong suspicion of TSC due to the presence of 5 hypopigmented macules measuring more than 5 mm in diameter (≥3 hypopigmented macules of ≥5 mm is one of the major criterion for TSC), the patient was referred to cardiology prior to initiation of propranolol.
Echocardiography revealed 3 intracardiac masses measuring 4 to 5 mm in diameter in the left ventricle (LV), along the interventricular septum and the LV posterior wall. These masses were consistent with rhabdomyomas (Figure 3)—a major criterion for TSC—which had not been detected by prenatal ultrasonography. No obstruction to LV inflow or outflow was observed. Additionally, no arrhythmias were detected on electrocardiography.
The patient was cleared for propranolol, which was slowly uptitrated to 2 mg/kg/d. She completed the course without adverse effects. The treatment of IH was successful with substantial reduction in size over the following months until clearance. She also was referred to neurology for magnetic resonance imaging of the brain, which showed a 3-mm subependymal nodule in the lateral right ventricle, another major feature of TSC.
Cardiac rhabdomyomas are benign hamartomas that affect as many as 80% of patients with TSC1 and are primarily localized in the ventricles. Although cardiac rhabdomyomas usually regress over time, they can compromise ventricular function or valvular function, or both, and result in outflow obstruction, arrhythmias, and Wolff- Parkinson-White syndrome.3 Surgical resection may be needed in patients whose condition is refractory to medical management for heart failure.
The pathophysiologic mechanism behind the natural involution of cardiac rhabdomyomas has not been fully elucidated. It has been hypothesized that these masses stem from the inability of rhabdomyoma cells to divide after birth due to their embryonic myocyte derivation.4
According to the TSC diagnostic criteria from the Tuberous Sclerosis Complex International Consensus Group, at least 2 major features or 1 major and 2 minor features are required to make a definitive diagnosis of TSC. Cutaneous signs represent more than one-third of major features of TSC; almost all patients with TSC have skin findings.5
Identification of pathogenic mutations in either TSC1 (on chromosome 9q34.3, encoding for hamartin) or TSC2 (on chromosome 16p13.3, encoding for tuberin), resulting in constitutive activation of mammalian target of rapamycin and subsequent increased cell growth, is sufficient for a definitive diagnosis of TSC. However, mutations cannot be identified by conventional genetic testing in as many as one-quarter of patients with TSC; therefore, a negative result does not exclude TSC if the patient meets clinical diagnostic criteria.
Although a cardiology workup is indicated prior to initiating propranolol in the presence of possible cardiac rhabdomyomas, most of those lesions are hemodynamically stable and do not require treatment. There also is no contraindication for β-blocker therapy. In fact, propranolol has been reported as a successful treatment in rhabdomyoma-associated arrhythmias in children.6 Notably, obstructive cardiac rhabdomyomas have been successfully treated with mammalian target of rapamycin inhibitors, such as sirolimus7 and everolimus.8
Baseline cardiology screening with echocardiography prior to initiating propranolol for treatment of IH is not routinely indicated in babies with uncomplicated IH. However, in a patient with TSC, cardiology screening is necessary to rule out rhabdomyomas with associated arrhythmias or obstructed blood flow, or both, prior to initiating treatment.
We presented a case of concomitant IH and TSC in a patient with cardiac rhabdomyomas. The manifestation of large IHs in our patient prompted further testing that revealed multiple cardiac rhabdomyomas in the context of TSC. It is imperative for cardiologists, cardiac surgeons, and dermatologists to be familiar with the TSC diagnostic criteria so that they can reach a prompt diagnosis and make appropriate referrals for further evaluation of cardiac, neurologic, and ophthalmologic signs.
- Frudit P, Vitturi BK, Navarro FC, et al. Multiple cardiac rhabdomyomas in tuberous sclerosis complex: case report and review of the literature. Autops Case Rep. 2019;9:e2019125. doi:10.4322/acr.2019.125
- Krowchuk DP, Frieden IJ, Mancini AJ, et al; Subcommittee on the Management of Infantile Hemangiomas. Clinical practice guideline for the management of infantile hemangiomas. Pediatrics. 2019;143:e20183475. doi:10.1542/peds.2018-3475
- Venugopalan P, Babu JS, Al-Bulushi A. Right atrial rhabdomyoma acting as the substrate for Wolff-Parkinson-White syndrome in a 3-month-old infant. Acta Cardiol. 2005;60:543-545. doi:10.2143/AC.60.5.2004977
- DiMario FJ Jr, Diana D, Leopold H, et al. Evolution of cardiac rhabdomyoma in tuberous sclerosis complex. Clin Pediatr (Phila). 1996;35:615-619. doi:10.1177/000992289603501202
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254. doi:10.1016/j.pediatrneurol.2013.08.001
- Kathare PA, Muthuswamy KS, Sadasivan J, et al. Incessant ventricular tachycardia due to multiple cardiac rhabdomyomas in an infant with tuberous sclerosis. Indian Heart J. 2013;65:111-113. doi:10.1016/j.ihj.2012.12.003
- Breathnach C, Pears J, Franklin O, et al. Rapid regression of left ventricular outflow tract rhabdomyoma after sirolimus therapy. Pediatrics. 2014;134:e1199-e1202. doi:10.1542/peds.2013-3293
- Chang J-S, Chiou P-Y, Yao S-H, et al. Regression of neonatal cardiac rhabdomyoma in two months through low-dose everolimus therapy: a report of three cases. Pediatr Cardiol. 2017;38:1478-1484. doi:10.1007/s00246-017-1688-4
- Frudit P, Vitturi BK, Navarro FC, et al. Multiple cardiac rhabdomyomas in tuberous sclerosis complex: case report and review of the literature. Autops Case Rep. 2019;9:e2019125. doi:10.4322/acr.2019.125
- Krowchuk DP, Frieden IJ, Mancini AJ, et al; Subcommittee on the Management of Infantile Hemangiomas. Clinical practice guideline for the management of infantile hemangiomas. Pediatrics. 2019;143:e20183475. doi:10.1542/peds.2018-3475
- Venugopalan P, Babu JS, Al-Bulushi A. Right atrial rhabdomyoma acting as the substrate for Wolff-Parkinson-White syndrome in a 3-month-old infant. Acta Cardiol. 2005;60:543-545. doi:10.2143/AC.60.5.2004977
- DiMario FJ Jr, Diana D, Leopold H, et al. Evolution of cardiac rhabdomyoma in tuberous sclerosis complex. Clin Pediatr (Phila). 1996;35:615-619. doi:10.1177/000992289603501202
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254. doi:10.1016/j.pediatrneurol.2013.08.001
- Kathare PA, Muthuswamy KS, Sadasivan J, et al. Incessant ventricular tachycardia due to multiple cardiac rhabdomyomas in an infant with tuberous sclerosis. Indian Heart J. 2013;65:111-113. doi:10.1016/j.ihj.2012.12.003
- Breathnach C, Pears J, Franklin O, et al. Rapid regression of left ventricular outflow tract rhabdomyoma after sirolimus therapy. Pediatrics. 2014;134:e1199-e1202. doi:10.1542/peds.2013-3293
- Chang J-S, Chiou P-Y, Yao S-H, et al. Regression of neonatal cardiac rhabdomyoma in two months through low-dose everolimus therapy: a report of three cases. Pediatr Cardiol. 2017;38:1478-1484. doi:10.1007/s00246-017-1688-4
Practice Points
- Dermatologists may see patients with infantile hemangiomas (IHs) and tuberous sclerosis complex (TSC); therefore, they should be familiar with TSC diagnostic criteria to reach a prompt diagnosis and make appropriate referrals.
- Cardiologic evaluation is not routinely required prior to systemic treatment of IH, but knowledge of cardiac findings in TSC should prompt cardiologic clearance prior to β-blocker initiation.
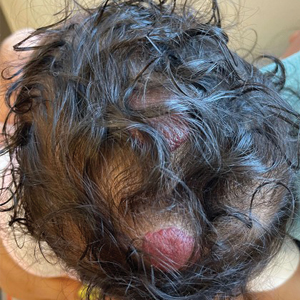
Hypopigmented Cutaneous Langerhans Cell Histiocytosis in a Hispanic Infant
To the Editor:
Langerhans cell histiocytosis (LCH) is a rare inflammatory neoplasia caused by accumulation of clonal Langerhans cells in 1 or more organs. The clinical spectrum is diverse, ranging from mild, single-organ involvement that may resolve spontaneously to severe progressive multisystem disease that can be fatal. It is most prevalent in children, affecting an estimated 4 to 5 children for every 1 million annually, with male predominance.1 The pathogenesis is driven by activating mutations in the mitogen-activated protein kinase pathway, with the BRAF V600E mutation detected in most LCH patients, resulting in proliferation of pathologic Langerhans cells and dysregulated expression of inflammatory cytokines in LCH lesions.2 A biopsy of lesional tissue is required for definitive diagnosis. Histopathology reveals a mixed inflammatory infiltrate and characteristic mononuclear cells with reniform nuclei that are positive for CD1a and CD207 proteins on immunohistochemical staining.3
Langerhans cell histiocytosis is categorized by the extent of organ involvement. It commonly affects the bones, skin, pituitary gland, liver, lungs, bone marrow, and lymph nodes.4 Single-system LCH involves a single organ with unifocal or multifocal lesions; multisystem LCH involves 2 or more organs and has a worse prognosis if risk organs (eg, liver, spleen, bone marrow) are involved.4
Skin lesions are reported in more than half of LCH cases and are the most common initial manifestation in patients younger than 2 years.4 Cutaneous findings are highly variable, which poses a diagnostic challenge. Common morphologies include erythematous papules, pustules, papulovesicles, scaly plaques, erosions, and petechiae. Lesions can be solitary or widespread and favor the trunk, head, and face.4 We describe an atypical case of hypopigmented cutaneous LCH and review the literature on this morphology in patients with skin of color.
A 7-month-old Hispanic male infant who was otherwise healthy presented with numerous hypopigmented macules and pink papules on the trunk and groin that had progressed since birth. A review of systems was unremarkable. Physical examination revealed 1- to 3-mm, discrete, hypopigmented macules intermixed with 1- to 2-mm pearly pink papules scattered on the back, chest, abdomen, and inguinal folds (Figure 1). Some lesions appeared koebnerized; however, the parents denied a history of scratching or trauma.
Histopathology of a lesion in the inguinal fold showed aggregates of mononuclear cells with reniform nuclei and abundant amphophilic cytoplasm in the papillary dermis, with focal extension into the epidermis. Scattered eosinophils and multinucleated giant cells were present in the dermal inflammatory infiltrate (Figure 2). Immunohistochemical staining was positive for CD1a (Figure 3) and S-100 protein (Figure 4). Although epidermal Langerhans cell collections also can be seen in allergic contact dermatitis,5 predominant involvement of the papillary dermis and the presence of multinucleated giant cells are characteristic of LCH.4 Given these findings, which were consistent with LCH, the dermatopathology deemed BRAF V600E immunostaining unnecessary for diagnostic purposes.
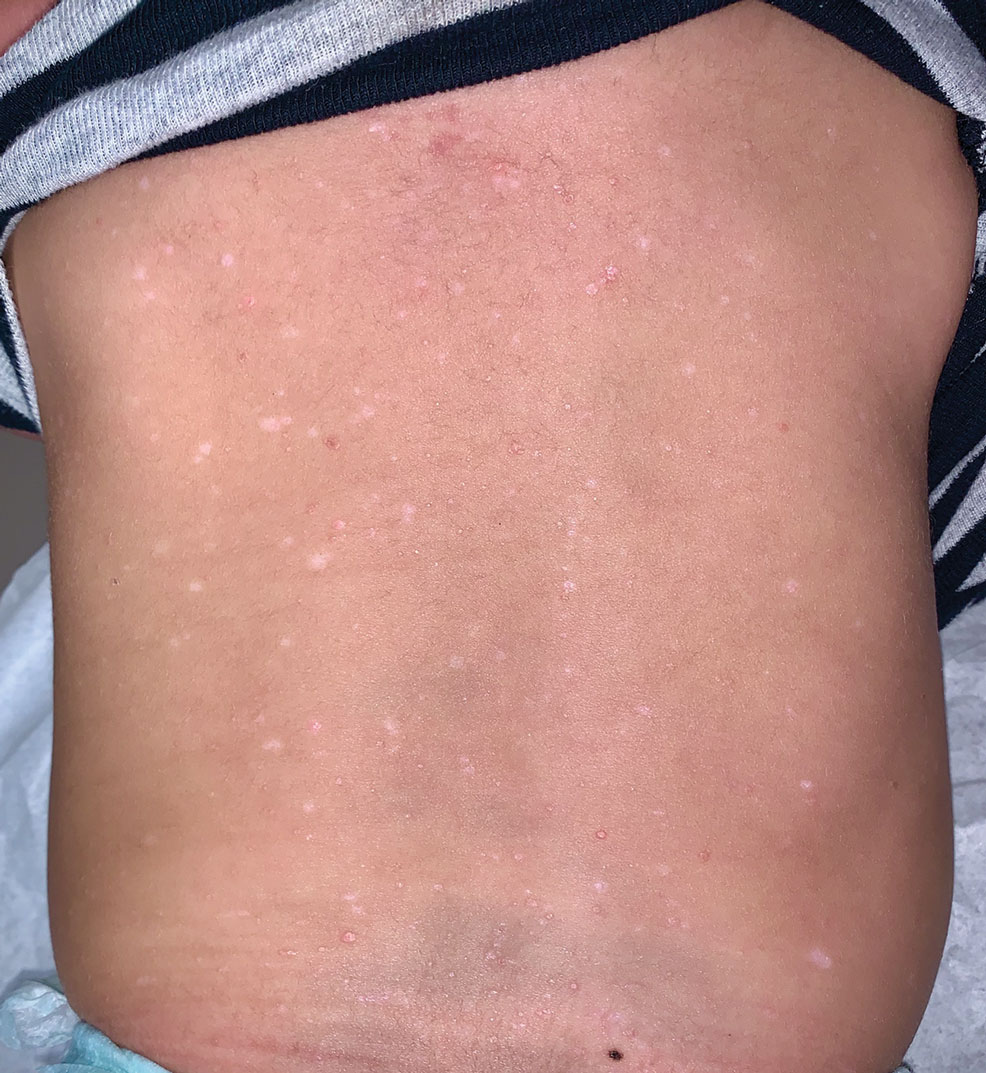
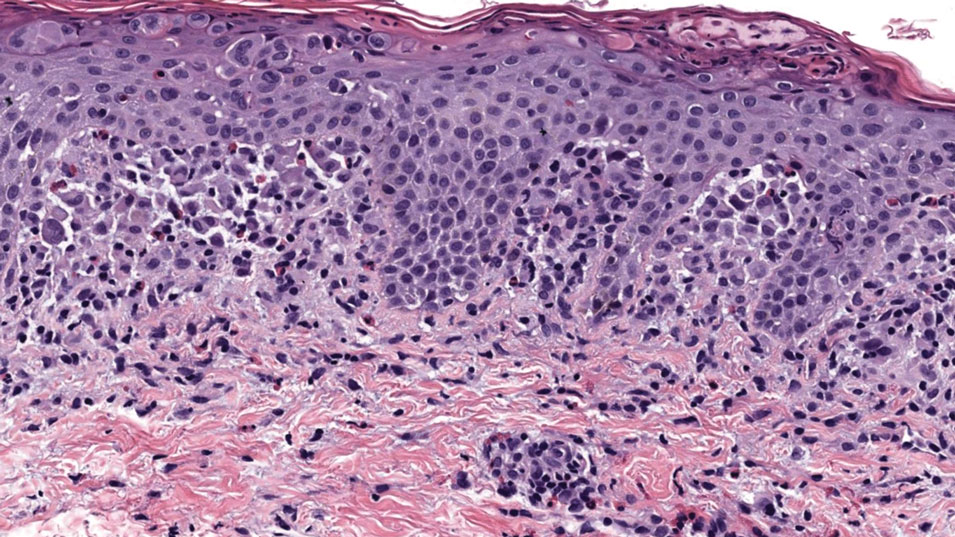
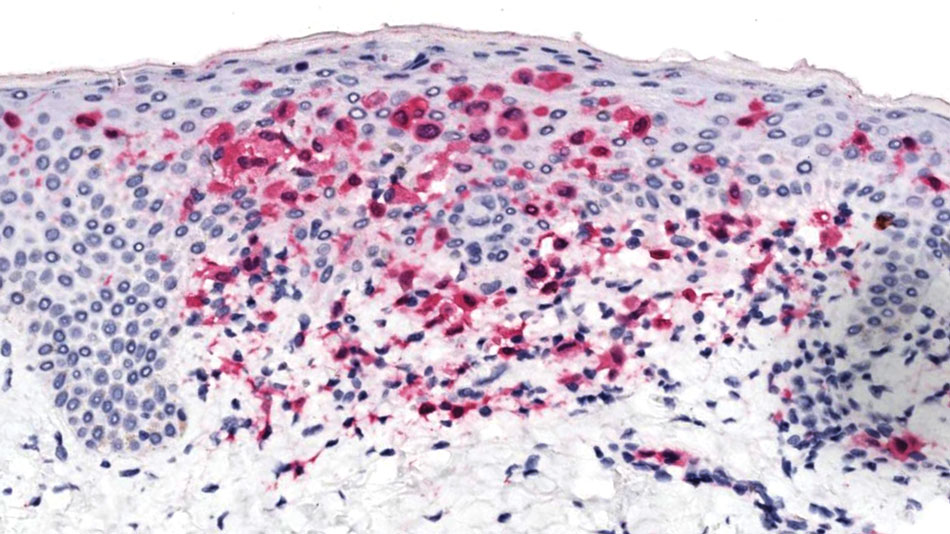
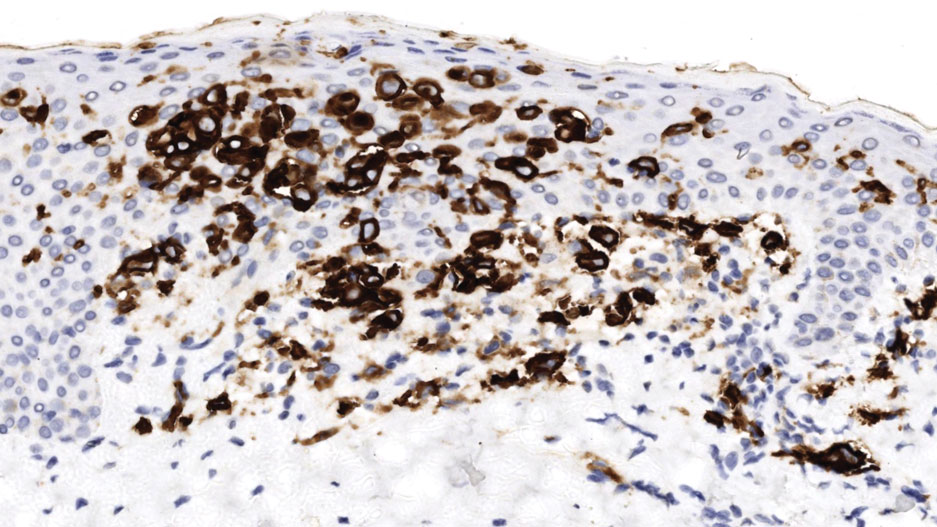
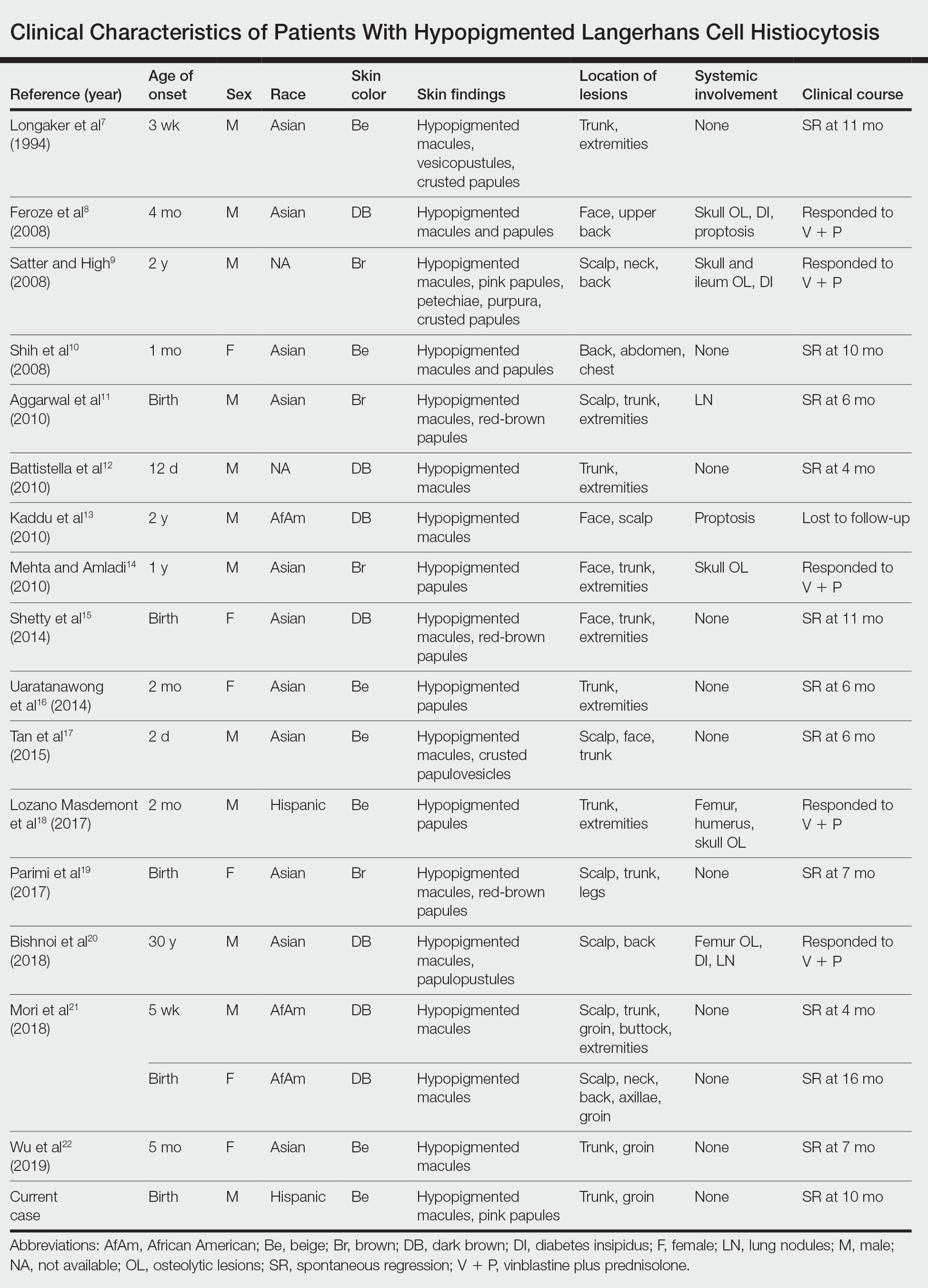
The patient was referred to the hematology and oncology department to undergo thorough evaluation for extracutaneous involvement. The workup included a complete blood cell count, liver function testing, electrolyte assessment, skeletal survey, chest radiography, and ultrasonography of the liver and spleen. All results were negative, suggesting a diagnosis of single-system cutaneous LCH.
Three months later, the patient presented to dermatology with spontaneous regression of all skin lesions. Continued follow-up—every 6 months for 5 years—was recommended to monitor for disease recurrence or progression to multisystem disease.
Cutaneous LCH is a clinically heterogeneous disease with the potential for multisystem involvement and long-term sequelae; therefore, timely diagnosis is paramount to optimize outcomes. However, delayed diagnosis is common because of the spectrum of skin findings that can mimic common pediatric dermatoses, such as seborrheic dermatitis, atopic dermatitis, and diaper dermatitis.4 In one study, the median time from onset of skin lesions to diagnostic biopsy was longer than 3 months (maximum, 5 years).6 Our patient was referred to dermatology 7 months after onset of hypopigmented macules, a rarely reported cutaneous manifestation of LCH.
A PubMed search of articles indexed for MEDLINE from 1994 to 2019 using the terms Langerhans cell histiocytotis and hypopigmented yielded 17 cases of LCH presenting as hypopigmented skin lesions (Table).7-22 All cases occurred in patients with skin of color (ie, patients of Asian, Hispanic, or African descent). Hypopigmented macules were the only cutaneous manifestation in 10 (59%) cases. Lesions most commonly were distributed on the trunk (16/17 [94%]) and extremities (8/17 [47%]). The median age of onset was 1 month; 76% (13/17) of patients developed skin lesions before 1 year of age, indicating that this morphology may be more common in newborns. In most patients, the diagnosis was single-system cutaneous LCH; they exhibited spontaneous regression by 8 months of age on average, suggesting that this variant may be associated with a better prognosis. Mori and colleagues21 hypothesized that hypopigmented lesions may represent the resolving stage of active LCH based on histopathologic findings of dermal pallor and fibrosis in a hypopigmented LCH lesion. However, systemic involvement was reported in 7 cases of hypopigmented LCH, highlighting the importance of assessing for multisystem disease regardless of cutaneous morphology.21Langerhans cell histiocytosis should be considered in the differential diagnosis when evaluating hypopigmented skin eruptions in infants with darker skin types. Prompt diagnosis of this atypical variant requires a higher index of suspicion because of its rarity and the polymorphic nature of cutaneous LCH. This morphology may go undiagnosed in the setting of mild or spontaneously resolving disease; notwithstanding, accurate diagnosis and longitudinal surveillance are necessary given the potential for progressive systemic involvement.
1. Guyot-Goubin A, Donadieu J, Barkaoui M, et al. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000–2004. Pediatr Blood Cancer. 2008;51:71-75. doi:10.1002/pbc.21498
2. Badalian-Very G, Vergilio J-A, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919-1923. doi:10.1182/blood-2010-04-279083
3. Haupt R, Minkov M, Astigarraga I, et al; Euro Histio Network. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work‐up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
4. Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: history, classification, pathobiology, clinical manifestations, and prognosis. J Am Acad Dermatol. 2018;78:1035-1044. doi:10.1016/j.jaad.2017.05.059
5. Rosa G, Fernandez AP, Vij A, et al. Langerhans cell collections, but not eosinophils, are clues to a diagnosis of allergic contact dermatitis in appropriate skin biopsies. J Cutan Pathol. 2016;43:498-504. doi:10.1111/cup.12707
6. Simko SJ, Garmezy B, Abhyankar H, et al. Differentiating skin-limited and multisystem Langerhans cell histiocytosis. J Pediatr. 2014;165:990-996. doi:10.1016/j.jpeds.2014.07.063
7. Longaker MA, Frieden IJ, LeBoit PE, et al. Congenital “self-healing” Langerhans cell histiocytosis: the need for long-term follow-up. J Am Acad Dermatol. 1994;31(5, pt 2):910-916. doi:10.1016/s0190-9622(94)70258-6
8. Feroze K, Unni M, Jayasree MG, et al. Langerhans cell histiocytosis presenting with hypopigmented macules. Indian J Dermatol Venereol Leprol. 2008;74:670-672. doi:10.4103/0378-6323.45128
9. Satter EK, High WA. Langerhans cell histiocytosis: a case report and summary of the current recommendations of the Histiocyte Society. Dermatol Online J. 2008;14:3.
10. Chang SL, Shih IH, Kuo TT, et al. Congenital self-healing reticulohistiocytosis presenting as hypopigmented macules and papules in a neonate. Dermatologica Sinica 2008;26:80-84.
11. Aggarwal V, Seth A, Jain M, et al. Congenital Langerhans cell histiocytosis with skin and lung involvement: spontaneous regression. Indian J Pediatr. 2010;77:811-812.
12. Battistella M, Fraitag S, Teillac DH, et al. Neonatal and early infantile cutaneous Langerhans cell histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol. 2010;146:149-156. doi:10.1001/archdermatol.2009.360
13. Kaddu S, Mulyowa G, Kovarik C. Hypopigmented scaly, scalp and facial lesions and disfiguring exopthalmus. Clin Exp Dermatol. 2010;3:E52-E53. doi:10.1111/j.1365-2230.2009.03336.x
14. Mehta B, Amladi S. Langerhans cell histiocytosis presenting as hypopigmented papules. Pediatr Dermatol. 2010;27:215-217. doi:10.1111/j.1525-1470.2010.01104.x
15. Shetty S, Monappa V, Pai K, et al. Congenital self-healing reticulohistiocytosis: a case report. Our Dermatol Online. 2014;5:264-266.
16. Uaratanawong R, Kootiratrakarn T, Sudtikoonaseth P, et al. Congenital self-healing reticulohistiocytosis presented with multiple hypopigmented flat-topped papules: a case report and review of literatures. J Med Assoc Thai. 2014;97:993-997.
17. Tan Q, Gan LQ, Wang H. Congenital self-healing Langerhans cell histiocytosis in a male neonate. Indian J Dermatol Venereol Leprol. 2015;81:75-77. doi:10.4103/0378-6323.148587
18. Lozano Masdemont B, Gómez‐Recuero Muñoz L, Villanueva Álvarez‐Santullano A, et al. Langerhans cell histiocytosis mimicking lichen nitidus with bone involvement. Australas J Dermatol. 2017;58:231-233. doi:10.1111/ajd.12467
19. Parimi LR, You J, Hong L, et al. Congenital self-healing reticulohistiocytosis with spontaneous regression. An Bras Dermatol. 2017;92:553-555. doi:10.1590/abd1806-4841.20175432
20. Bishnoi A, De D, Khullar G, et al. Hypopigmented and acneiform lesions: an unusual initial presentation of adult-onset multisystem Langerhans cell histiocytosis. Indian J Dermatol Venereol Leprol. 2018;84:621-626. doi:10.4103/ijdvl.IJDVL_639_17
21. Mori S, Adar T, Kazlouskaya V, et al. Cutaneous Langerhans cell histiocytosis presenting with hypopigmented lesions: report of two cases and review of literature. Pediatr Dermatol. 2018;35:502-506. doi:10.1111/pde.13509
22. Wu X, Huang J, Jiang L, et al. Congenital self‐healing reticulohistiocytosis with BRAF V600E mutation in an infant. Clin Exp Dermatol. 2019;44:647-650. doi:10.1111/ced.13880
To the Editor:
Langerhans cell histiocytosis (LCH) is a rare inflammatory neoplasia caused by accumulation of clonal Langerhans cells in 1 or more organs. The clinical spectrum is diverse, ranging from mild, single-organ involvement that may resolve spontaneously to severe progressive multisystem disease that can be fatal. It is most prevalent in children, affecting an estimated 4 to 5 children for every 1 million annually, with male predominance.1 The pathogenesis is driven by activating mutations in the mitogen-activated protein kinase pathway, with the BRAF V600E mutation detected in most LCH patients, resulting in proliferation of pathologic Langerhans cells and dysregulated expression of inflammatory cytokines in LCH lesions.2 A biopsy of lesional tissue is required for definitive diagnosis. Histopathology reveals a mixed inflammatory infiltrate and characteristic mononuclear cells with reniform nuclei that are positive for CD1a and CD207 proteins on immunohistochemical staining.3
Langerhans cell histiocytosis is categorized by the extent of organ involvement. It commonly affects the bones, skin, pituitary gland, liver, lungs, bone marrow, and lymph nodes.4 Single-system LCH involves a single organ with unifocal or multifocal lesions; multisystem LCH involves 2 or more organs and has a worse prognosis if risk organs (eg, liver, spleen, bone marrow) are involved.4
Skin lesions are reported in more than half of LCH cases and are the most common initial manifestation in patients younger than 2 years.4 Cutaneous findings are highly variable, which poses a diagnostic challenge. Common morphologies include erythematous papules, pustules, papulovesicles, scaly plaques, erosions, and petechiae. Lesions can be solitary or widespread and favor the trunk, head, and face.4 We describe an atypical case of hypopigmented cutaneous LCH and review the literature on this morphology in patients with skin of color.
A 7-month-old Hispanic male infant who was otherwise healthy presented with numerous hypopigmented macules and pink papules on the trunk and groin that had progressed since birth. A review of systems was unremarkable. Physical examination revealed 1- to 3-mm, discrete, hypopigmented macules intermixed with 1- to 2-mm pearly pink papules scattered on the back, chest, abdomen, and inguinal folds (Figure 1). Some lesions appeared koebnerized; however, the parents denied a history of scratching or trauma.
Histopathology of a lesion in the inguinal fold showed aggregates of mononuclear cells with reniform nuclei and abundant amphophilic cytoplasm in the papillary dermis, with focal extension into the epidermis. Scattered eosinophils and multinucleated giant cells were present in the dermal inflammatory infiltrate (Figure 2). Immunohistochemical staining was positive for CD1a (Figure 3) and S-100 protein (Figure 4). Although epidermal Langerhans cell collections also can be seen in allergic contact dermatitis,5 predominant involvement of the papillary dermis and the presence of multinucleated giant cells are characteristic of LCH.4 Given these findings, which were consistent with LCH, the dermatopathology deemed BRAF V600E immunostaining unnecessary for diagnostic purposes.
The patient was referred to the hematology and oncology department to undergo thorough evaluation for extracutaneous involvement. The workup included a complete blood cell count, liver function testing, electrolyte assessment, skeletal survey, chest radiography, and ultrasonography of the liver and spleen. All results were negative, suggesting a diagnosis of single-system cutaneous LCH.
Three months later, the patient presented to dermatology with spontaneous regression of all skin lesions. Continued follow-up—every 6 months for 5 years—was recommended to monitor for disease recurrence or progression to multisystem disease.
Cutaneous LCH is a clinically heterogeneous disease with the potential for multisystem involvement and long-term sequelae; therefore, timely diagnosis is paramount to optimize outcomes. However, delayed diagnosis is common because of the spectrum of skin findings that can mimic common pediatric dermatoses, such as seborrheic dermatitis, atopic dermatitis, and diaper dermatitis.4 In one study, the median time from onset of skin lesions to diagnostic biopsy was longer than 3 months (maximum, 5 years).6 Our patient was referred to dermatology 7 months after onset of hypopigmented macules, a rarely reported cutaneous manifestation of LCH.
A PubMed search of articles indexed for MEDLINE from 1994 to 2019 using the terms Langerhans cell histiocytotis and hypopigmented yielded 17 cases of LCH presenting as hypopigmented skin lesions (Table).7-22 All cases occurred in patients with skin of color (ie, patients of Asian, Hispanic, or African descent). Hypopigmented macules were the only cutaneous manifestation in 10 (59%) cases. Lesions most commonly were distributed on the trunk (16/17 [94%]) and extremities (8/17 [47%]). The median age of onset was 1 month; 76% (13/17) of patients developed skin lesions before 1 year of age, indicating that this morphology may be more common in newborns. In most patients, the diagnosis was single-system cutaneous LCH; they exhibited spontaneous regression by 8 months of age on average, suggesting that this variant may be associated with a better prognosis. Mori and colleagues21 hypothesized that hypopigmented lesions may represent the resolving stage of active LCH based on histopathologic findings of dermal pallor and fibrosis in a hypopigmented LCH lesion. However, systemic involvement was reported in 7 cases of hypopigmented LCH, highlighting the importance of assessing for multisystem disease regardless of cutaneous morphology.21Langerhans cell histiocytosis should be considered in the differential diagnosis when evaluating hypopigmented skin eruptions in infants with darker skin types. Prompt diagnosis of this atypical variant requires a higher index of suspicion because of its rarity and the polymorphic nature of cutaneous LCH. This morphology may go undiagnosed in the setting of mild or spontaneously resolving disease; notwithstanding, accurate diagnosis and longitudinal surveillance are necessary given the potential for progressive systemic involvement.
To the Editor:
Langerhans cell histiocytosis (LCH) is a rare inflammatory neoplasia caused by accumulation of clonal Langerhans cells in 1 or more organs. The clinical spectrum is diverse, ranging from mild, single-organ involvement that may resolve spontaneously to severe progressive multisystem disease that can be fatal. It is most prevalent in children, affecting an estimated 4 to 5 children for every 1 million annually, with male predominance.1 The pathogenesis is driven by activating mutations in the mitogen-activated protein kinase pathway, with the BRAF V600E mutation detected in most LCH patients, resulting in proliferation of pathologic Langerhans cells and dysregulated expression of inflammatory cytokines in LCH lesions.2 A biopsy of lesional tissue is required for definitive diagnosis. Histopathology reveals a mixed inflammatory infiltrate and characteristic mononuclear cells with reniform nuclei that are positive for CD1a and CD207 proteins on immunohistochemical staining.3
Langerhans cell histiocytosis is categorized by the extent of organ involvement. It commonly affects the bones, skin, pituitary gland, liver, lungs, bone marrow, and lymph nodes.4 Single-system LCH involves a single organ with unifocal or multifocal lesions; multisystem LCH involves 2 or more organs and has a worse prognosis if risk organs (eg, liver, spleen, bone marrow) are involved.4
Skin lesions are reported in more than half of LCH cases and are the most common initial manifestation in patients younger than 2 years.4 Cutaneous findings are highly variable, which poses a diagnostic challenge. Common morphologies include erythematous papules, pustules, papulovesicles, scaly plaques, erosions, and petechiae. Lesions can be solitary or widespread and favor the trunk, head, and face.4 We describe an atypical case of hypopigmented cutaneous LCH and review the literature on this morphology in patients with skin of color.
A 7-month-old Hispanic male infant who was otherwise healthy presented with numerous hypopigmented macules and pink papules on the trunk and groin that had progressed since birth. A review of systems was unremarkable. Physical examination revealed 1- to 3-mm, discrete, hypopigmented macules intermixed with 1- to 2-mm pearly pink papules scattered on the back, chest, abdomen, and inguinal folds (Figure 1). Some lesions appeared koebnerized; however, the parents denied a history of scratching or trauma.
Histopathology of a lesion in the inguinal fold showed aggregates of mononuclear cells with reniform nuclei and abundant amphophilic cytoplasm in the papillary dermis, with focal extension into the epidermis. Scattered eosinophils and multinucleated giant cells were present in the dermal inflammatory infiltrate (Figure 2). Immunohistochemical staining was positive for CD1a (Figure 3) and S-100 protein (Figure 4). Although epidermal Langerhans cell collections also can be seen in allergic contact dermatitis,5 predominant involvement of the papillary dermis and the presence of multinucleated giant cells are characteristic of LCH.4 Given these findings, which were consistent with LCH, the dermatopathology deemed BRAF V600E immunostaining unnecessary for diagnostic purposes.
The patient was referred to the hematology and oncology department to undergo thorough evaluation for extracutaneous involvement. The workup included a complete blood cell count, liver function testing, electrolyte assessment, skeletal survey, chest radiography, and ultrasonography of the liver and spleen. All results were negative, suggesting a diagnosis of single-system cutaneous LCH.
Three months later, the patient presented to dermatology with spontaneous regression of all skin lesions. Continued follow-up—every 6 months for 5 years—was recommended to monitor for disease recurrence or progression to multisystem disease.
Cutaneous LCH is a clinically heterogeneous disease with the potential for multisystem involvement and long-term sequelae; therefore, timely diagnosis is paramount to optimize outcomes. However, delayed diagnosis is common because of the spectrum of skin findings that can mimic common pediatric dermatoses, such as seborrheic dermatitis, atopic dermatitis, and diaper dermatitis.4 In one study, the median time from onset of skin lesions to diagnostic biopsy was longer than 3 months (maximum, 5 years).6 Our patient was referred to dermatology 7 months after onset of hypopigmented macules, a rarely reported cutaneous manifestation of LCH.
A PubMed search of articles indexed for MEDLINE from 1994 to 2019 using the terms Langerhans cell histiocytotis and hypopigmented yielded 17 cases of LCH presenting as hypopigmented skin lesions (Table).7-22 All cases occurred in patients with skin of color (ie, patients of Asian, Hispanic, or African descent). Hypopigmented macules were the only cutaneous manifestation in 10 (59%) cases. Lesions most commonly were distributed on the trunk (16/17 [94%]) and extremities (8/17 [47%]). The median age of onset was 1 month; 76% (13/17) of patients developed skin lesions before 1 year of age, indicating that this morphology may be more common in newborns. In most patients, the diagnosis was single-system cutaneous LCH; they exhibited spontaneous regression by 8 months of age on average, suggesting that this variant may be associated with a better prognosis. Mori and colleagues21 hypothesized that hypopigmented lesions may represent the resolving stage of active LCH based on histopathologic findings of dermal pallor and fibrosis in a hypopigmented LCH lesion. However, systemic involvement was reported in 7 cases of hypopigmented LCH, highlighting the importance of assessing for multisystem disease regardless of cutaneous morphology.21Langerhans cell histiocytosis should be considered in the differential diagnosis when evaluating hypopigmented skin eruptions in infants with darker skin types. Prompt diagnosis of this atypical variant requires a higher index of suspicion because of its rarity and the polymorphic nature of cutaneous LCH. This morphology may go undiagnosed in the setting of mild or spontaneously resolving disease; notwithstanding, accurate diagnosis and longitudinal surveillance are necessary given the potential for progressive systemic involvement.
1. Guyot-Goubin A, Donadieu J, Barkaoui M, et al. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000–2004. Pediatr Blood Cancer. 2008;51:71-75. doi:10.1002/pbc.21498
2. Badalian-Very G, Vergilio J-A, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919-1923. doi:10.1182/blood-2010-04-279083
3. Haupt R, Minkov M, Astigarraga I, et al; Euro Histio Network. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work‐up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
4. Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: history, classification, pathobiology, clinical manifestations, and prognosis. J Am Acad Dermatol. 2018;78:1035-1044. doi:10.1016/j.jaad.2017.05.059
5. Rosa G, Fernandez AP, Vij A, et al. Langerhans cell collections, but not eosinophils, are clues to a diagnosis of allergic contact dermatitis in appropriate skin biopsies. J Cutan Pathol. 2016;43:498-504. doi:10.1111/cup.12707
6. Simko SJ, Garmezy B, Abhyankar H, et al. Differentiating skin-limited and multisystem Langerhans cell histiocytosis. J Pediatr. 2014;165:990-996. doi:10.1016/j.jpeds.2014.07.063
7. Longaker MA, Frieden IJ, LeBoit PE, et al. Congenital “self-healing” Langerhans cell histiocytosis: the need for long-term follow-up. J Am Acad Dermatol. 1994;31(5, pt 2):910-916. doi:10.1016/s0190-9622(94)70258-6
8. Feroze K, Unni M, Jayasree MG, et al. Langerhans cell histiocytosis presenting with hypopigmented macules. Indian J Dermatol Venereol Leprol. 2008;74:670-672. doi:10.4103/0378-6323.45128
9. Satter EK, High WA. Langerhans cell histiocytosis: a case report and summary of the current recommendations of the Histiocyte Society. Dermatol Online J. 2008;14:3.
10. Chang SL, Shih IH, Kuo TT, et al. Congenital self-healing reticulohistiocytosis presenting as hypopigmented macules and papules in a neonate. Dermatologica Sinica 2008;26:80-84.
11. Aggarwal V, Seth A, Jain M, et al. Congenital Langerhans cell histiocytosis with skin and lung involvement: spontaneous regression. Indian J Pediatr. 2010;77:811-812.
12. Battistella M, Fraitag S, Teillac DH, et al. Neonatal and early infantile cutaneous Langerhans cell histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol. 2010;146:149-156. doi:10.1001/archdermatol.2009.360
13. Kaddu S, Mulyowa G, Kovarik C. Hypopigmented scaly, scalp and facial lesions and disfiguring exopthalmus. Clin Exp Dermatol. 2010;3:E52-E53. doi:10.1111/j.1365-2230.2009.03336.x
14. Mehta B, Amladi S. Langerhans cell histiocytosis presenting as hypopigmented papules. Pediatr Dermatol. 2010;27:215-217. doi:10.1111/j.1525-1470.2010.01104.x
15. Shetty S, Monappa V, Pai K, et al. Congenital self-healing reticulohistiocytosis: a case report. Our Dermatol Online. 2014;5:264-266.
16. Uaratanawong R, Kootiratrakarn T, Sudtikoonaseth P, et al. Congenital self-healing reticulohistiocytosis presented with multiple hypopigmented flat-topped papules: a case report and review of literatures. J Med Assoc Thai. 2014;97:993-997.
17. Tan Q, Gan LQ, Wang H. Congenital self-healing Langerhans cell histiocytosis in a male neonate. Indian J Dermatol Venereol Leprol. 2015;81:75-77. doi:10.4103/0378-6323.148587
18. Lozano Masdemont B, Gómez‐Recuero Muñoz L, Villanueva Álvarez‐Santullano A, et al. Langerhans cell histiocytosis mimicking lichen nitidus with bone involvement. Australas J Dermatol. 2017;58:231-233. doi:10.1111/ajd.12467
19. Parimi LR, You J, Hong L, et al. Congenital self-healing reticulohistiocytosis with spontaneous regression. An Bras Dermatol. 2017;92:553-555. doi:10.1590/abd1806-4841.20175432
20. Bishnoi A, De D, Khullar G, et al. Hypopigmented and acneiform lesions: an unusual initial presentation of adult-onset multisystem Langerhans cell histiocytosis. Indian J Dermatol Venereol Leprol. 2018;84:621-626. doi:10.4103/ijdvl.IJDVL_639_17
21. Mori S, Adar T, Kazlouskaya V, et al. Cutaneous Langerhans cell histiocytosis presenting with hypopigmented lesions: report of two cases and review of literature. Pediatr Dermatol. 2018;35:502-506. doi:10.1111/pde.13509
22. Wu X, Huang J, Jiang L, et al. Congenital self‐healing reticulohistiocytosis with BRAF V600E mutation in an infant. Clin Exp Dermatol. 2019;44:647-650. doi:10.1111/ced.13880
1. Guyot-Goubin A, Donadieu J, Barkaoui M, et al. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000–2004. Pediatr Blood Cancer. 2008;51:71-75. doi:10.1002/pbc.21498
2. Badalian-Very G, Vergilio J-A, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919-1923. doi:10.1182/blood-2010-04-279083
3. Haupt R, Minkov M, Astigarraga I, et al; Euro Histio Network. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work‐up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184. doi:10.1002/pbc.24367
4. Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: history, classification, pathobiology, clinical manifestations, and prognosis. J Am Acad Dermatol. 2018;78:1035-1044. doi:10.1016/j.jaad.2017.05.059
5. Rosa G, Fernandez AP, Vij A, et al. Langerhans cell collections, but not eosinophils, are clues to a diagnosis of allergic contact dermatitis in appropriate skin biopsies. J Cutan Pathol. 2016;43:498-504. doi:10.1111/cup.12707
6. Simko SJ, Garmezy B, Abhyankar H, et al. Differentiating skin-limited and multisystem Langerhans cell histiocytosis. J Pediatr. 2014;165:990-996. doi:10.1016/j.jpeds.2014.07.063
7. Longaker MA, Frieden IJ, LeBoit PE, et al. Congenital “self-healing” Langerhans cell histiocytosis: the need for long-term follow-up. J Am Acad Dermatol. 1994;31(5, pt 2):910-916. doi:10.1016/s0190-9622(94)70258-6
8. Feroze K, Unni M, Jayasree MG, et al. Langerhans cell histiocytosis presenting with hypopigmented macules. Indian J Dermatol Venereol Leprol. 2008;74:670-672. doi:10.4103/0378-6323.45128
9. Satter EK, High WA. Langerhans cell histiocytosis: a case report and summary of the current recommendations of the Histiocyte Society. Dermatol Online J. 2008;14:3.
10. Chang SL, Shih IH, Kuo TT, et al. Congenital self-healing reticulohistiocytosis presenting as hypopigmented macules and papules in a neonate. Dermatologica Sinica 2008;26:80-84.
11. Aggarwal V, Seth A, Jain M, et al. Congenital Langerhans cell histiocytosis with skin and lung involvement: spontaneous regression. Indian J Pediatr. 2010;77:811-812.
12. Battistella M, Fraitag S, Teillac DH, et al. Neonatal and early infantile cutaneous Langerhans cell histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol. 2010;146:149-156. doi:10.1001/archdermatol.2009.360
13. Kaddu S, Mulyowa G, Kovarik C. Hypopigmented scaly, scalp and facial lesions and disfiguring exopthalmus. Clin Exp Dermatol. 2010;3:E52-E53. doi:10.1111/j.1365-2230.2009.03336.x
14. Mehta B, Amladi S. Langerhans cell histiocytosis presenting as hypopigmented papules. Pediatr Dermatol. 2010;27:215-217. doi:10.1111/j.1525-1470.2010.01104.x
15. Shetty S, Monappa V, Pai K, et al. Congenital self-healing reticulohistiocytosis: a case report. Our Dermatol Online. 2014;5:264-266.
16. Uaratanawong R, Kootiratrakarn T, Sudtikoonaseth P, et al. Congenital self-healing reticulohistiocytosis presented with multiple hypopigmented flat-topped papules: a case report and review of literatures. J Med Assoc Thai. 2014;97:993-997.
17. Tan Q, Gan LQ, Wang H. Congenital self-healing Langerhans cell histiocytosis in a male neonate. Indian J Dermatol Venereol Leprol. 2015;81:75-77. doi:10.4103/0378-6323.148587
18. Lozano Masdemont B, Gómez‐Recuero Muñoz L, Villanueva Álvarez‐Santullano A, et al. Langerhans cell histiocytosis mimicking lichen nitidus with bone involvement. Australas J Dermatol. 2017;58:231-233. doi:10.1111/ajd.12467
19. Parimi LR, You J, Hong L, et al. Congenital self-healing reticulohistiocytosis with spontaneous regression. An Bras Dermatol. 2017;92:553-555. doi:10.1590/abd1806-4841.20175432
20. Bishnoi A, De D, Khullar G, et al. Hypopigmented and acneiform lesions: an unusual initial presentation of adult-onset multisystem Langerhans cell histiocytosis. Indian J Dermatol Venereol Leprol. 2018;84:621-626. doi:10.4103/ijdvl.IJDVL_639_17
21. Mori S, Adar T, Kazlouskaya V, et al. Cutaneous Langerhans cell histiocytosis presenting with hypopigmented lesions: report of two cases and review of literature. Pediatr Dermatol. 2018;35:502-506. doi:10.1111/pde.13509
22. Wu X, Huang J, Jiang L, et al. Congenital self‐healing reticulohistiocytosis with BRAF V600E mutation in an infant. Clin Exp Dermatol. 2019;44:647-650. doi:10.1111/ced.13880
Practice Points
- Dermatologists should be aware of the hypopigmented variant of cutaneous Langerhans cell histiocytosis (LCH), which has been reported exclusively in patients with skin of color.
- Langerhans cell histiocytosis should be included in the differential diagnosis of hypopigmented macules, which may be the only cutaneous manifestation or may coincide with typical lesions of LCH.
- Hypopigmented cutaneous LCH may be more common in newborns and associated with a better prognosis.
Reactive Granulomatous Dermatitis: Variability of the Predominant Inflammatory Cell Type
To the Editor:
The term palisaded neutrophilic and granulomatous dermatitis (PNGD) has been proposed to encompass various conditions, including Winkelmann granuloma and superficial ulcerating rheumatoid necrobiosis. More recently, PNGD has been classified along with interstitial granulomatous dermatitis and interstitial granulomatous drug reaction under a unifying rubric of reactive granulomatous dermatitis (RGD).1-4 The diagnosis of RGD can be challenging because of a range of clinical and histopathologic features as well as variable nomenclature.1-3,5
Palisaded neutrophilic and granulomatous dermatitis classically manifests with papules and small plaques on the extensor extremities, with histopathology showing characteristic necrobiosis with both neutrophils and histiocytes.1,2,6 We report 6 cases of RGD, including an index case in which a predominance of neutrophils in the infiltrate impeded the diagnosis.
An 85-year-old woman (the index patient) presented with a several-week history of asymmetric crusted papules on the right upper extremity—3 lesions on the elbow and forearm and 1 lesion on a finger. She was an avid gardener with severe rheumatoid arthritis treated with Janus kinase (JAK) inhibitor therapy. An initial biopsy of the elbow revealed a dense infiltrate of neutrophils and sparse eosinophils within the dermis. Special stains for bacterial, fungal, and acid-fast organisms were negative.
Because infection with sporotrichoid spread remained high in the differential diagnosis, the JAK inhibitor was discontinued and an antifungal agent was initiated. Given the persistence of the lesions, a subsequent biopsy of the right finger revealed scarce neutrophils and predominant histiocytes with rare foci of degenerated collagen. Sporotrichosis remained the leading diagnosis for these unilateral lesions. The patient subsequently developed additional crusted papules on the left arm (Figure 1). A biopsy of a left elbow lesion revealed palisades of histiocytes around degenerated collagen and collections of neutrophils compatible with RGD (Figures 2 and 3). Incidentally, the patient also presented with bilateral lower extremity palpable purpura, with a biopsy showing leukocytoclastic vasculitis. Antifungal therapy was discontinued and JAK inhibitor therapy resumed, with partial resolution of both the arm and right finger lesions and complete resolution of the lower extremity palpable purpura over several months.
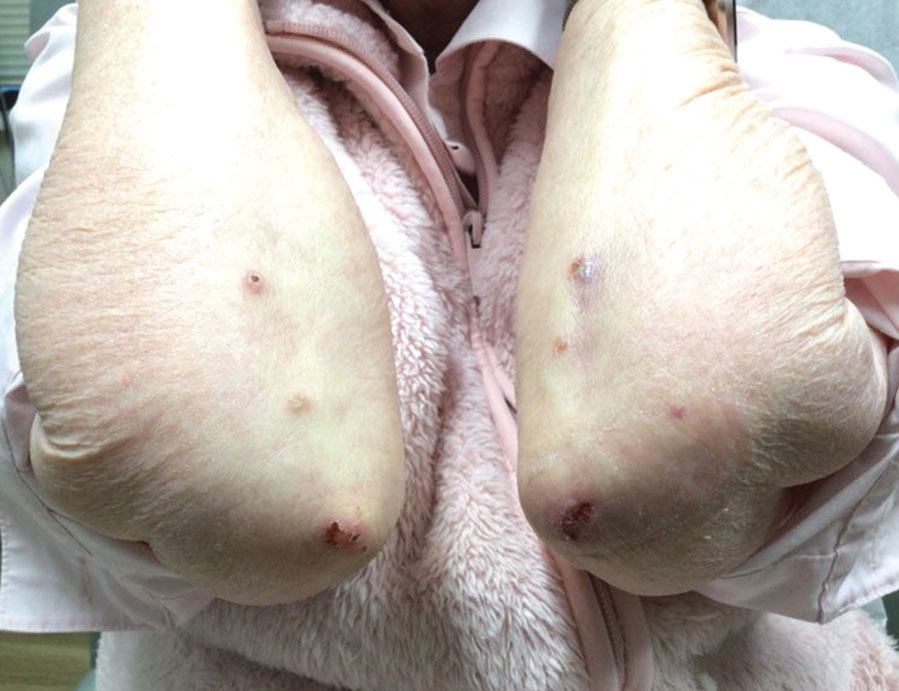
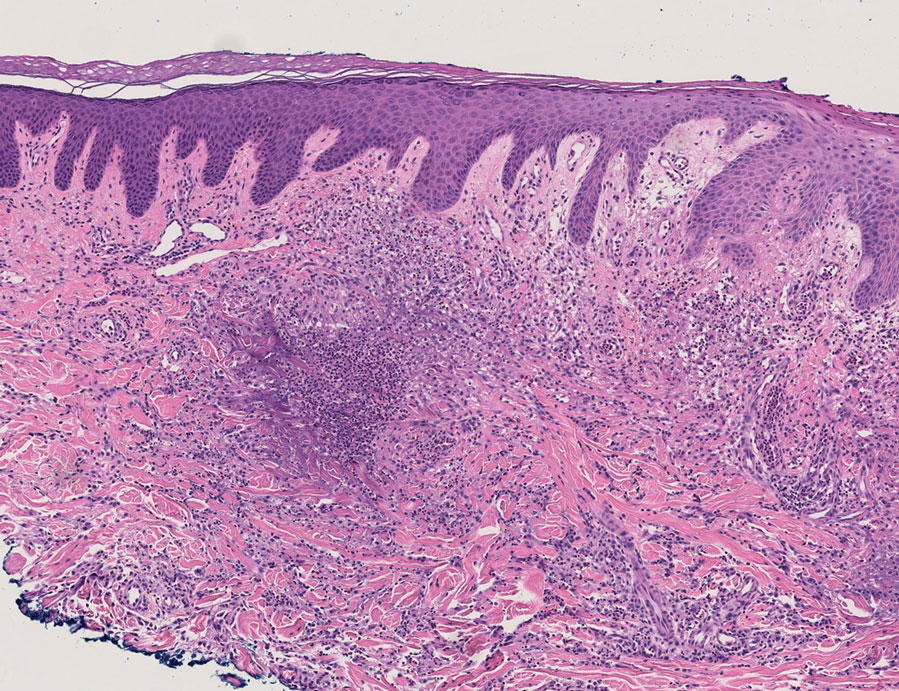
The dense neutrophilic infiltrate and asymmetric presentation seen in our index patient’s initial biopsy hindered categorization of the cutaneous findings as RGD in association with her rheumatoid arthritis rather than as an infectious process. To ascertain whether diagnosis also was difficult in other cases of RGD, we conducted a search of the Yale Dermatopathology database for the diagnosis palisaded neutrophilic and granulomatous dermatitis, a term consistently used at our institution over the past decade. This study was approved by the institutional review board of Yale University (New Haven, Connecticut), and informed consent was waived. The search covered a 10-year period; 13 patients were found. Eight patients were eliminated because further clinical information or follow-up could not be obtained, leaving 5 additional cases (Table). The 8 eliminated cases were consultations submitted to the laboratory by outside pathologists from other institutions.
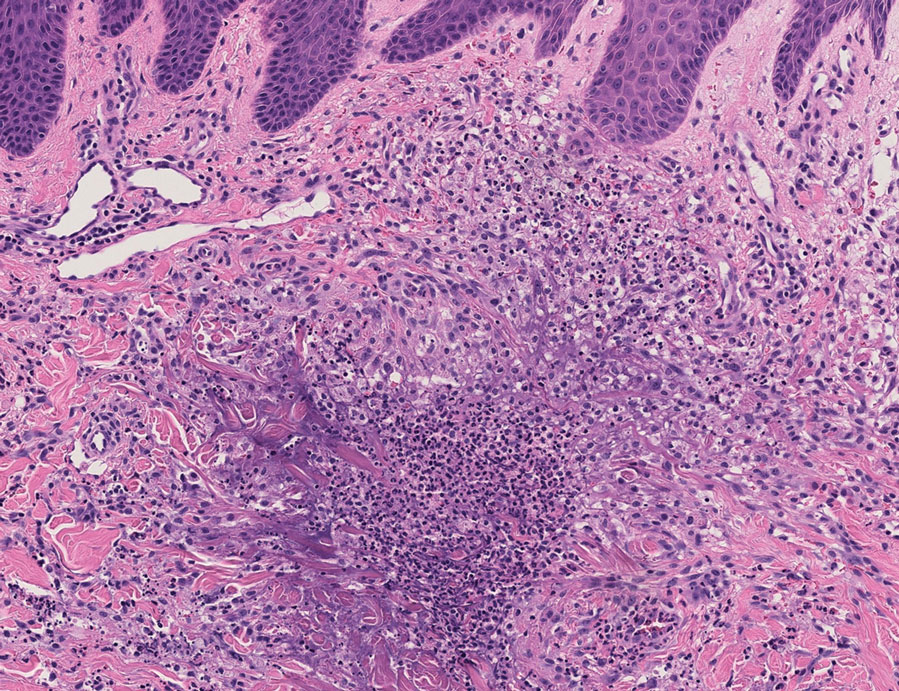
In one case (patient 5), the diagnosis of RGD was delayed for 7 years from first documentation of an RGD-compatible neutrophil-predominant infiltrate (Table). In 3 other cases, PNGD was in the clinical differential diagnosis. In patient 6 with known eosinophilic granulomatosis with polyangiitis, biopsy findings included a mixed inflammatory infiltrate with eosinophils, and the clinical and histopathologic findings were deemed compatible with RGD by group consensus at Grand Rounds.
In practice, a consistent unifying nomenclature has not been achieved for RGD and the diseases it encompasses—PNGD, interstitial granulomatous dermatitis, and interstitial granulomatous drug reaction. In this small series, a diagnosis of PNGD was given in the dermatopathology report only when biopsy specimens were characterized by histiocytes, neutrophils, and necrobiosis. Histopathology reports for neutrophil-predominant, histiocyte-predominant, and eosinophil-predominant cases did not mention PNGD or RGD, though potential association with systemic disease generally was noted.
Given the variability in the predominant inflammatory cell type in these patients, adding a qualifier to the histopathologic diagnosis—“RGD, eosinophil rich,” “RGD, histiocyte rich,” or “RGD, neutrophil rich”1—would underscore the range of inflammatory cells in this entity. Employing this terminology rather than stating a solely descriptive diagnosis such as neutrophilic infiltrate, which may bias clinicians toward an infectious process, would aid in the association of a given rash with systemic disease and may prevent unnecessary tissue sampling. Indeed, 3 patients in this small series underwent more than 2 biopsies; multiple procedures might have been avoided had there been better communication about the spectrum of inflammatory cells compatible with RGD.
The inflammatory infiltrate in biopsy specimens of RGD can be solely neutrophil or histiocyte predominant or even have prominent eosinophils depending on the stage of disease. Awareness of variability in the predominant inflammatory cell in RGD may facilitate an accurate diagnosis as well as an association with any underlying autoimmune process, thereby allowing better management and treatment.1
- Rosenbach M, English JC. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387. doi:10.1016/j.det.2015.03.005
- Wanat KA, Caplan A, Messenger E, et al. Reactive granulomatous dermatitis: a useful and encompassing term. JAAD Intl. 2022;7:126-128. doi:10.1016/j.jdin.2022.03.004
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283. doi:10.1001/archderm.1994.01690100062010
- Dykman CJ, Galens GJ, Good AE. Linear subcutaneous bands in rheumatoid arthritis: an unusual form of rheumatoid granuloma. Ann Intern Med. 1965;63:134-140. doi:10.7326/0003-4819-63-1-134
- Rodríguez-Garijo N, Bielsa I, Mascaró JM Jr, et al. Reactive granulomatous dermatitis as a histological pattern including manifestations of interstitial granulomatous dermatitis and palisaded neutrophilic and granulomtous dermatitis: a study of 52 patients. J Eur Acad Dermatol Venereol. 2021;35:988-994. doi:10.1111/jdv.17010
- Kalen JE, Shokeen D, Ramos-Caro F, et al. Palisaded neutrophilic granulomatous dermatitis: spectrum of histologic findings in a single patient. JAAD Case Rep. 2017;3:425. doi:10.1016/j.jdcr.2017.06.010
To the Editor:
The term palisaded neutrophilic and granulomatous dermatitis (PNGD) has been proposed to encompass various conditions, including Winkelmann granuloma and superficial ulcerating rheumatoid necrobiosis. More recently, PNGD has been classified along with interstitial granulomatous dermatitis and interstitial granulomatous drug reaction under a unifying rubric of reactive granulomatous dermatitis (RGD).1-4 The diagnosis of RGD can be challenging because of a range of clinical and histopathologic features as well as variable nomenclature.1-3,5
Palisaded neutrophilic and granulomatous dermatitis classically manifests with papules and small plaques on the extensor extremities, with histopathology showing characteristic necrobiosis with both neutrophils and histiocytes.1,2,6 We report 6 cases of RGD, including an index case in which a predominance of neutrophils in the infiltrate impeded the diagnosis.
An 85-year-old woman (the index patient) presented with a several-week history of asymmetric crusted papules on the right upper extremity—3 lesions on the elbow and forearm and 1 lesion on a finger. She was an avid gardener with severe rheumatoid arthritis treated with Janus kinase (JAK) inhibitor therapy. An initial biopsy of the elbow revealed a dense infiltrate of neutrophils and sparse eosinophils within the dermis. Special stains for bacterial, fungal, and acid-fast organisms were negative.
Because infection with sporotrichoid spread remained high in the differential diagnosis, the JAK inhibitor was discontinued and an antifungal agent was initiated. Given the persistence of the lesions, a subsequent biopsy of the right finger revealed scarce neutrophils and predominant histiocytes with rare foci of degenerated collagen. Sporotrichosis remained the leading diagnosis for these unilateral lesions. The patient subsequently developed additional crusted papules on the left arm (Figure 1). A biopsy of a left elbow lesion revealed palisades of histiocytes around degenerated collagen and collections of neutrophils compatible with RGD (Figures 2 and 3). Incidentally, the patient also presented with bilateral lower extremity palpable purpura, with a biopsy showing leukocytoclastic vasculitis. Antifungal therapy was discontinued and JAK inhibitor therapy resumed, with partial resolution of both the arm and right finger lesions and complete resolution of the lower extremity palpable purpura over several months.
The dense neutrophilic infiltrate and asymmetric presentation seen in our index patient’s initial biopsy hindered categorization of the cutaneous findings as RGD in association with her rheumatoid arthritis rather than as an infectious process. To ascertain whether diagnosis also was difficult in other cases of RGD, we conducted a search of the Yale Dermatopathology database for the diagnosis palisaded neutrophilic and granulomatous dermatitis, a term consistently used at our institution over the past decade. This study was approved by the institutional review board of Yale University (New Haven, Connecticut), and informed consent was waived. The search covered a 10-year period; 13 patients were found. Eight patients were eliminated because further clinical information or follow-up could not be obtained, leaving 5 additional cases (Table). The 8 eliminated cases were consultations submitted to the laboratory by outside pathologists from other institutions.
In one case (patient 5), the diagnosis of RGD was delayed for 7 years from first documentation of an RGD-compatible neutrophil-predominant infiltrate (Table). In 3 other cases, PNGD was in the clinical differential diagnosis. In patient 6 with known eosinophilic granulomatosis with polyangiitis, biopsy findings included a mixed inflammatory infiltrate with eosinophils, and the clinical and histopathologic findings were deemed compatible with RGD by group consensus at Grand Rounds.
In practice, a consistent unifying nomenclature has not been achieved for RGD and the diseases it encompasses—PNGD, interstitial granulomatous dermatitis, and interstitial granulomatous drug reaction. In this small series, a diagnosis of PNGD was given in the dermatopathology report only when biopsy specimens were characterized by histiocytes, neutrophils, and necrobiosis. Histopathology reports for neutrophil-predominant, histiocyte-predominant, and eosinophil-predominant cases did not mention PNGD or RGD, though potential association with systemic disease generally was noted.
Given the variability in the predominant inflammatory cell type in these patients, adding a qualifier to the histopathologic diagnosis—“RGD, eosinophil rich,” “RGD, histiocyte rich,” or “RGD, neutrophil rich”1—would underscore the range of inflammatory cells in this entity. Employing this terminology rather than stating a solely descriptive diagnosis such as neutrophilic infiltrate, which may bias clinicians toward an infectious process, would aid in the association of a given rash with systemic disease and may prevent unnecessary tissue sampling. Indeed, 3 patients in this small series underwent more than 2 biopsies; multiple procedures might have been avoided had there been better communication about the spectrum of inflammatory cells compatible with RGD.
The inflammatory infiltrate in biopsy specimens of RGD can be solely neutrophil or histiocyte predominant or even have prominent eosinophils depending on the stage of disease. Awareness of variability in the predominant inflammatory cell in RGD may facilitate an accurate diagnosis as well as an association with any underlying autoimmune process, thereby allowing better management and treatment.1
To the Editor:
The term palisaded neutrophilic and granulomatous dermatitis (PNGD) has been proposed to encompass various conditions, including Winkelmann granuloma and superficial ulcerating rheumatoid necrobiosis. More recently, PNGD has been classified along with interstitial granulomatous dermatitis and interstitial granulomatous drug reaction under a unifying rubric of reactive granulomatous dermatitis (RGD).1-4 The diagnosis of RGD can be challenging because of a range of clinical and histopathologic features as well as variable nomenclature.1-3,5
Palisaded neutrophilic and granulomatous dermatitis classically manifests with papules and small plaques on the extensor extremities, with histopathology showing characteristic necrobiosis with both neutrophils and histiocytes.1,2,6 We report 6 cases of RGD, including an index case in which a predominance of neutrophils in the infiltrate impeded the diagnosis.
An 85-year-old woman (the index patient) presented with a several-week history of asymmetric crusted papules on the right upper extremity—3 lesions on the elbow and forearm and 1 lesion on a finger. She was an avid gardener with severe rheumatoid arthritis treated with Janus kinase (JAK) inhibitor therapy. An initial biopsy of the elbow revealed a dense infiltrate of neutrophils and sparse eosinophils within the dermis. Special stains for bacterial, fungal, and acid-fast organisms were negative.
Because infection with sporotrichoid spread remained high in the differential diagnosis, the JAK inhibitor was discontinued and an antifungal agent was initiated. Given the persistence of the lesions, a subsequent biopsy of the right finger revealed scarce neutrophils and predominant histiocytes with rare foci of degenerated collagen. Sporotrichosis remained the leading diagnosis for these unilateral lesions. The patient subsequently developed additional crusted papules on the left arm (Figure 1). A biopsy of a left elbow lesion revealed palisades of histiocytes around degenerated collagen and collections of neutrophils compatible with RGD (Figures 2 and 3). Incidentally, the patient also presented with bilateral lower extremity palpable purpura, with a biopsy showing leukocytoclastic vasculitis. Antifungal therapy was discontinued and JAK inhibitor therapy resumed, with partial resolution of both the arm and right finger lesions and complete resolution of the lower extremity palpable purpura over several months.
The dense neutrophilic infiltrate and asymmetric presentation seen in our index patient’s initial biopsy hindered categorization of the cutaneous findings as RGD in association with her rheumatoid arthritis rather than as an infectious process. To ascertain whether diagnosis also was difficult in other cases of RGD, we conducted a search of the Yale Dermatopathology database for the diagnosis palisaded neutrophilic and granulomatous dermatitis, a term consistently used at our institution over the past decade. This study was approved by the institutional review board of Yale University (New Haven, Connecticut), and informed consent was waived. The search covered a 10-year period; 13 patients were found. Eight patients were eliminated because further clinical information or follow-up could not be obtained, leaving 5 additional cases (Table). The 8 eliminated cases were consultations submitted to the laboratory by outside pathologists from other institutions.
In one case (patient 5), the diagnosis of RGD was delayed for 7 years from first documentation of an RGD-compatible neutrophil-predominant infiltrate (Table). In 3 other cases, PNGD was in the clinical differential diagnosis. In patient 6 with known eosinophilic granulomatosis with polyangiitis, biopsy findings included a mixed inflammatory infiltrate with eosinophils, and the clinical and histopathologic findings were deemed compatible with RGD by group consensus at Grand Rounds.
In practice, a consistent unifying nomenclature has not been achieved for RGD and the diseases it encompasses—PNGD, interstitial granulomatous dermatitis, and interstitial granulomatous drug reaction. In this small series, a diagnosis of PNGD was given in the dermatopathology report only when biopsy specimens were characterized by histiocytes, neutrophils, and necrobiosis. Histopathology reports for neutrophil-predominant, histiocyte-predominant, and eosinophil-predominant cases did not mention PNGD or RGD, though potential association with systemic disease generally was noted.
Given the variability in the predominant inflammatory cell type in these patients, adding a qualifier to the histopathologic diagnosis—“RGD, eosinophil rich,” “RGD, histiocyte rich,” or “RGD, neutrophil rich”1—would underscore the range of inflammatory cells in this entity. Employing this terminology rather than stating a solely descriptive diagnosis such as neutrophilic infiltrate, which may bias clinicians toward an infectious process, would aid in the association of a given rash with systemic disease and may prevent unnecessary tissue sampling. Indeed, 3 patients in this small series underwent more than 2 biopsies; multiple procedures might have been avoided had there been better communication about the spectrum of inflammatory cells compatible with RGD.
The inflammatory infiltrate in biopsy specimens of RGD can be solely neutrophil or histiocyte predominant or even have prominent eosinophils depending on the stage of disease. Awareness of variability in the predominant inflammatory cell in RGD may facilitate an accurate diagnosis as well as an association with any underlying autoimmune process, thereby allowing better management and treatment.1
- Rosenbach M, English JC. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387. doi:10.1016/j.det.2015.03.005
- Wanat KA, Caplan A, Messenger E, et al. Reactive granulomatous dermatitis: a useful and encompassing term. JAAD Intl. 2022;7:126-128. doi:10.1016/j.jdin.2022.03.004
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283. doi:10.1001/archderm.1994.01690100062010
- Dykman CJ, Galens GJ, Good AE. Linear subcutaneous bands in rheumatoid arthritis: an unusual form of rheumatoid granuloma. Ann Intern Med. 1965;63:134-140. doi:10.7326/0003-4819-63-1-134
- Rodríguez-Garijo N, Bielsa I, Mascaró JM Jr, et al. Reactive granulomatous dermatitis as a histological pattern including manifestations of interstitial granulomatous dermatitis and palisaded neutrophilic and granulomtous dermatitis: a study of 52 patients. J Eur Acad Dermatol Venereol. 2021;35:988-994. doi:10.1111/jdv.17010
- Kalen JE, Shokeen D, Ramos-Caro F, et al. Palisaded neutrophilic granulomatous dermatitis: spectrum of histologic findings in a single patient. JAAD Case Rep. 2017;3:425. doi:10.1016/j.jdcr.2017.06.010
- Rosenbach M, English JC. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387. doi:10.1016/j.det.2015.03.005
- Wanat KA, Caplan A, Messenger E, et al. Reactive granulomatous dermatitis: a useful and encompassing term. JAAD Intl. 2022;7:126-128. doi:10.1016/j.jdin.2022.03.004
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283. doi:10.1001/archderm.1994.01690100062010
- Dykman CJ, Galens GJ, Good AE. Linear subcutaneous bands in rheumatoid arthritis: an unusual form of rheumatoid granuloma. Ann Intern Med. 1965;63:134-140. doi:10.7326/0003-4819-63-1-134
- Rodríguez-Garijo N, Bielsa I, Mascaró JM Jr, et al. Reactive granulomatous dermatitis as a histological pattern including manifestations of interstitial granulomatous dermatitis and palisaded neutrophilic and granulomtous dermatitis: a study of 52 patients. J Eur Acad Dermatol Venereol. 2021;35:988-994. doi:10.1111/jdv.17010
- Kalen JE, Shokeen D, Ramos-Caro F, et al. Palisaded neutrophilic granulomatous dermatitis: spectrum of histologic findings in a single patient. JAAD Case Rep. 2017;3:425. doi:10.1016/j.jdcr.2017.06.010
Practice Points
- The term reactive granulomatous dermatitis (RGD) provides a unifying rubric for palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, and interstitial granulomatous drug reaction.
- Reactive granulomatous dermatitis can have a variable infiltrate that includes neutrophils, histiocytes, and/or eosinophils.
- Awareness of the variability in inflammatory cell type is important for the diagnosis of RGD.