User login
Efficacy of Etanercept in the Treatment of Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis
Regarded as dermatologic emergencies, Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) represent a spectrum of blistering skin diseases that have a high mortality rate. Because of a misguided immune response to medications or infections, CD8+ T lymphocytes release proinflammatory cytokines, giving rise to the extensive epidermal destruction seen in SJS and TEN. The exact pathogenesis of SJS and TEN is still poorly defined, but studies have proposed that T cells mediate keratinocyte (KC) apoptosis through perforin and granzyme release and activation of the Fas/Fas ligand (FasL). Functioning as a transmembrane death receptor in the tumor necrosis factor (TNF) superfamily, Fas (CD95) activates Fas-associated death domain protein, caspases, and nucleases, resulting in organized cell destruction. Likewise, perforin and granzymes also have been shown to play a similar role in apoptosis via activation of caspases.1
Evidence for the role of TNF-α in SJS and TEN has been supported by findings of elevated levels of TNF-α within the blister fluid, serum, and KC cell surface. Additionally, TNF-α has been shown to upregulate inducible nitric oxide synthase in KCs, causing an accumulation of nitric oxide and subsequent FasL-mediated cell death.1-3 Notably, studies have demonstrated a relative lack of lymphocytes in the tissue of TEN patients despite the extensive destruction that is observed, thus emphasizing the importance of amplification and cell signaling via inflammatory mediators such as TNF-α.1 In this proposed model, T cells release IFN-γ, causing KCs to release TNF-α that subsequently promotes the upregulation of the aforementioned FasL.1 Tumor necrosis factor α also may promote increased MHC class I complex deposition on KC surfaces that may play a role in perforin and granzyme-mediated apoptosis of KCs.1
There is still debate on the standard of care for the treatment of SJS and TEN, attributed to the absence of randomized controlled trials and the rarity of the disease as well as the numerous conflicting studies evaluating potential treatments.1,4 Despite conflicting data to support their use, supportive care and intravenous immunoglobulin (IVIG) continue to be common treatments for SJS and TEN in hospitals worldwide. Elucidation of the role of TNF-α has prompted the use of infliximab and etanercept. In a case series of Italian patients with TEN (average SCORTEN, 3.6) treated with the TNF-α antagonist etanercept, no mortality was observed, which was well below the calculated expected mortality of 46.9%.2 Our retrospective study compared the use of a TNF antagonist to other therapies in the treatment of SJS/TEN. Our data suggest that etanercept is a lifesaving and disease-modifying therapy.
Methods
Twenty-two patients with SJS/TEN were included in this analysis. This included all patients who carried a clinical diagnosis of SJS/TEN with a confirmatory biopsy at our 2 university centers—University of California, Los Angeles, and Keck-LA County-Norris Hospital at the University of Southern California, Los Angeles—from 2013 to 2016. The diagnosis was rendered when a clinical diagnosis of SJS/TEN was given by a dermatologist and a confirmatory biopsy was performed. Every patient given a diagnosis of SJS/TEN at either university system from 2015 onward received an injection of etanercept given the positive results reported by Paradisi et al.2
The 9 patients who presented from 2013 to 2014 to our 2 hospital systems and were given a diagnosis of SJS/TEN received either IVIG or supportive care alone and had an average body surface area (BSA) affected of 23%. The 13 patients who presented from 2015 to 2016 were treated with etanercept in the form of a 50-mg subcutaneous injection given once to the right upper arm. Of this group, 4 patients received dual therapy with both IVIG and etanercept. In the etanercept-treated group (etanercept alone and etanercept plus IVIG), the average BSA affected was 30%. At the time of preliminary diagnosis, all patient medications were evaluated for a possible temporal relationship to the onset of rash and were discontinued if felt to be causative. The causative agent and treatment course for each patient is summarized in Table 1.
Patients were monitored daily in the hospital for improvement, and time to re-epithelialization was measured. Re-epithelialization was defined as progressive healing with residual lesions (erosions, ulcers, or bullae) covering no more than 5% BSA and was contingent on the patient having no new lesions within 24 hours.5 SCORe of Toxic Epidermal Necrosis (SCORTEN), a validated severity-of-illness score,6 was calculated by giving 1 point for each of the following criteria at the time of diagnosis: age ≥40 years, concurrent malignancy, heart rate ≥120 beats/min, serum blood urea nitrogen >27 mg/dL, serum bicarbonate <20 mEq/L, serum glucose >250 mg/dL, and detached or compromised BSA >10%. The total SCORTEN was correlated with the following risk of mortality as supported by prior validation studies: SCORTEN of 0 to 1, 3.2%; SCORTEN of 2, 12.1%; SCORTEN of 3, 35.3%; SCORTEN of 4, 58.3%; SCORTEN of ≥5, >90%.
Results
A total of 13 patients received etanercept. The mean SCORTEN was 2.2. The observed mortality was 0%, which was markedly lower than the predicted mortality of 24.3% (as determined by linear interpolation). Of this cohort, 9 patients received etanercept alone (mean SCORTEN of 2.1, predicted mortality of 22.9%), whereas 4 patients received a combination of etanercept and IVIG (mean SCORTEN of 2.3, predicted mortality of 27.2%).
The 4 patients who received both etanercept and IVIG received dual therapy for varying reasons. In patient 2 (Table 1), the perceived severity of this case ultimately led to the decision to start IVIG in addition to etanercept, resulting in rapid recovery and discharge after only 1 week of hospitalization. Intravenous immunoglobulin also was given in patient 3 (SCORTEN of 4) and patient 6 (SCORTEN of 2) for progression of disease despite administration of etanercept, with subsequent cessation of progression after the addition of the second agent (IVIG). Patient 12 might have done well on etanercept monotherapy but was administered IVIG as a precautionary measure because of hospital treatment algorithms.
Nine patients did not receive etanercept. Of this group, 5 received IVIG and 4 were managed with supportive care alone. The average SCORTEN for this group was 2.4, only slightly higher than the group that received etanercept (Table 2). The mortality rate in this group was 33%, which was higher than the predicted mortality of 28.1%.
Re-epithelialization data were available for 8 patients who received etanercept. The average time to re-epithelialization for these patients was 8.9 days and ranged from 3 to 19 days. Of these patients, 2 received both IVIG and etanercept, with an average time to re-epithelialization of 13 days. For the 6 patients who received etanercept alone, the average time to re-epithelialization was 7.5 days. Re-epithelialization data were not available for any of the patients who received only IVIG or supportive care but to our recollection ranged from 14 to 21 days.
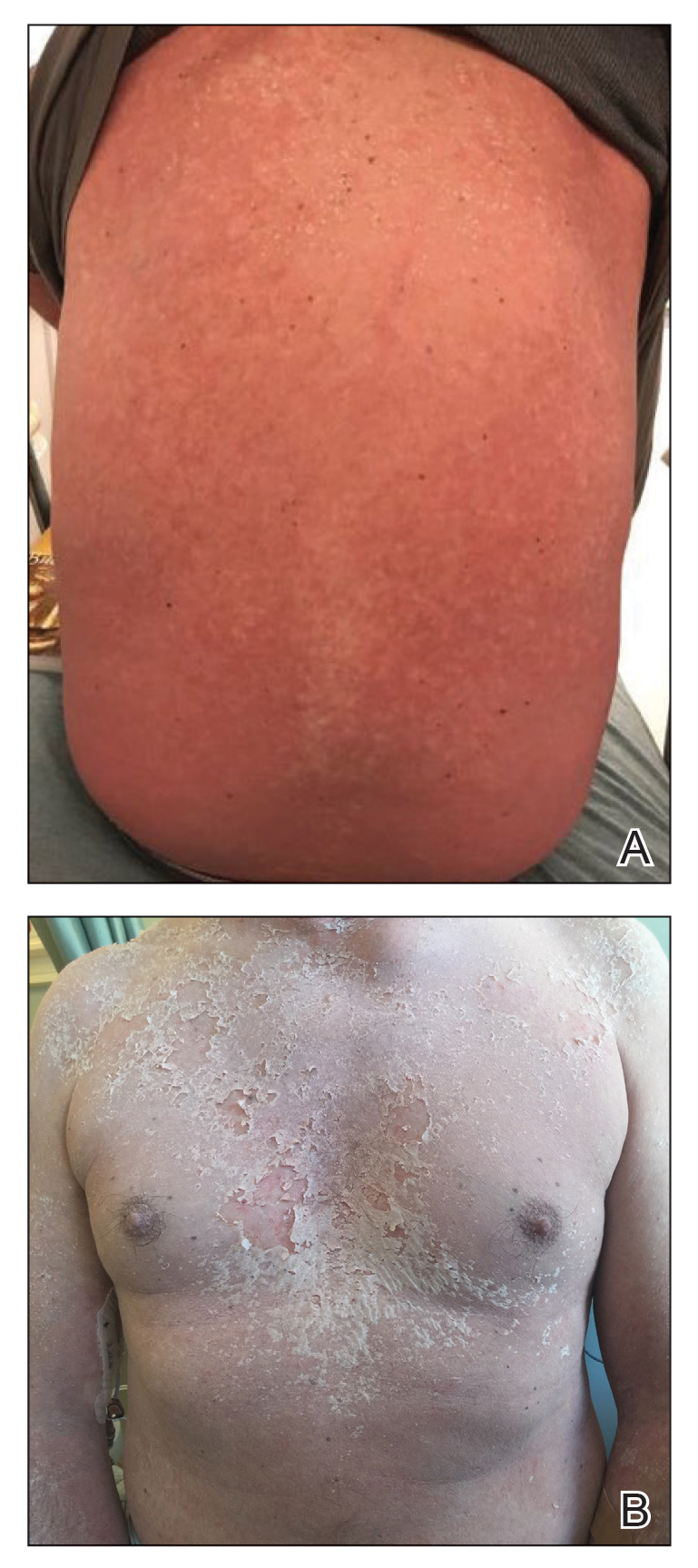
The clinical course of the 13 patients after the administration of a single dose of etanercept was remarkable, as there was complete absence of mortality and an increase in speed of recovery in most patients receiving this intervention (time to re-epithelialization, 3–19 days). We also observed another interesting trend from our patients treated with etanercept, which was the suggestion that treatment with etanercept may be less effective if IVIG and/or steroids are given prior to etanercept; likewise, treatment is more effective when etanercept is given quickly. For patients 1, 4, 5, 7, 9, and 11 (as shown in Table 1), no prior IVIG therapy or other immunosuppressive therapy had been given before etanercept was administered. In these 6 patients, the average time to re-epithelialization after etanercept administration was 7.5 days; average time to re-epithelialization, unfortunately, is not available for the patients who were not treated with etanercept. In addition, as shown in the Figure, it was noted in some patients that the depth of denudation was markedly more superficial than what would typically be clinically observed with TEN after administration of other immunomodulatory therapies such as IVIG or prednisone or with supportive care alone. In these 2 patients with superficial desquamation—patients 7 and 9—etanercept notably was given within 6 hours of onset of skin pain.
Comment
There is no definitive gold standard treatment of SJS, SJS/TEN overlap, or TEN. However, generally agreed upon management includes immediate discontinuation of the offending medication and supportive therapy with aggressive electrolyte replacement and wound care. Management in a burn unit or intensive care unit is recommended in severe cases. Contention over the efficacy of various medications in the treatment of SJS and TEN continues and largely is due to the rarity of SJS and TEN; studies are small and almost all lack randomization. Therapies that have been used include high-dose steroids, IVIG, plasmapheresis, cyclophosphamide, cyclosporine A, and TNF inhibitors (eg, etanercept, infliximab).1
Evidence for the use of anti–TNF-α antibodies has been limited thus far, with most of the literature focusing on infliximab and etanercept. Adalimumab, a fully humanized clonal antibody, has no reported cases in the dermatologic literature for use in patients with SJS/TEN. Two case reports of adalimumab paradoxically causing SJS have been documented. In both cases, adalimumab was stopped and patients responded to intravenous corticosteroids and infliximab.7,8 Similarly, thalidomide has not proven to be a promising anti–TNF-α agent for the treatment of SJS/TEN. In the only attempted randomized controlled trial for SJS and TEN, thalidomide appeared to increase mortality, eventuating in this trial being terminated prior to the planned end date.9Infliximab and etanercept have several case reports and a few case series highlighting potentially efficacious application of TNF-α inhibitors for the treatment of SJS/TEN.10-13 In 2002, Fischer et al10 reported the first case of TEN treated successfully with a single dose of infliximab 5 mg/kg. Kreft et al14 reported on etoricoxib-induced TEN that was treated with infliximab 5 mg/kg, which led to re-epithelialization within 5 weeks (notably a 5-week re-epithelialization time is not necessarily an improvement).
In 2005, Hunger et al3 demonstrated TNF-α’s release by KCs in the epidermis and by inflammatory cells in the dermis of a TEN patient. Twenty-four hours after the administration of infliximab 5 mg/kg in these patients, TNF-α was found to be below normal and epidermal detachment ceased.3 Wojtkietwicz et al13 demonstrated benefit following an infusion of infliximab 5 mg/kg in a patient whose disease continued to progress despite treatment with dexamethasone and 1.8 g/kg of IVIG.
Then 2 subsequent case series added further support for the efficacy of infliximab in the treatment of TEN. Patmanidis et al15 and Gaitanis et al16 reported similar results in 4 patients, each treated with infliximab 5 mg/kg immediately followed by initiation of high-dose IVIG (2 g/kg over 5 days). Zárate-Correa et al17 reported a 0% mortality rate and near-complete re-epithelialization after 5 to 14 days in 4 patients treated with a single 300-mg dose of infliximab.
However, the success of infliximab in the treatment of TEN has been countered by the pilot study by Paquet et al,18 which compared the efficacy of 150 mg/kg of N-acetylcysteine alone vs adding infliximab 5 mg/kg to treat 10 TEN patients. The study demonstrated no benefit at 48 hours in the group given infliximab, the time frame in which prior case reports touting infliximab’s benefit claimed the benefit was observed. Similarly, there was no effect on mortality for either treatment modality as assessed by illness auxiliary score.18
Evidence in support of the use of etanercept in the treatment of SJS/TEN is mounting, and some centers have begun to use it as the first-choice therapy for SJS/TEN. The first case was reported by Famularo et al,19 in which a patient with TEN was given 2 doses of etanercept 25 mg after failure to improve with prednisolone 1 mg/kg. The patient showed near-complete and rapid re-epithelization in 6 days before death due to disseminated intravascular coagulation 10 days after admission.19 Gubinelli et al20 and Sadighha21 independently reported cases of TEN and TEN/acute generalized exanthematous pustulosis overlap treated with a total of 50 mg of etanercept, demonstrating rapid cessation of lesion progression. Didona et al22 found similar benefit using etanercept 50 mg to treat TEN secondary to rituximab after failure to improve with prednisone and cyclophosphamide. Treatment of TEN with etanercept in an HIV-positive patient also has been reported. Lee et al23 described a patient who was administered 50-mg and 25-mg injections on days 3 and 5 of hospitalization, respectively, with re-epithelialization occurring by day 8. Finally, Owczarczyk-Saczonek et al24 reported a case of SJS in a patient with a 4-year history of etanercept and sulfasalazine treatment of rheumatoid arthritis; sulfasalazine was stopped, but this patient was continued on etanercept until resolution of skin and mucosal symptoms. However, it is important to consider the possibility of publication bias among these cases selected for their positive outcomes.
Perhaps the most compelling literature regarding the use of etanercept for TEN was described in a case series by Paradisi et al.2 This study included 10 patients with TEN, all of whom demonstrated complete re-epithelialization shortly after receiving etanercept 50 mg. Average SCORTEN was 3.6 with a range of 2 to 6. Eight patients in this study had severe comorbidities and all 10 patients survived, with a time to re-epithelialization ranging from 7 to 20 days.2 Additionally, a randomized controlled trial showed that 38 etanercept-treated patients had improved mortality (P=.266) and re-epithelialization time (P=.01) compared to patients treated with intravenous methylprednisolone.25Limitations to our study are similar to other reports of SJS/TEN and included the small number of cases and lack of randomization. Additionally, we do not have data available for all patients for time between onset of disease and treatment initiation. Because of these challenges, data presented in this case series is observational only. Additionally, the patients treated with etanercept alone had a slightly lower SCORTEN compared to the group that received IVIG or supportive care alone (2.1 and 2.4 respectively). However, the etanercept-only group actually had higher involvement of epidermal detachment (33%) compared to the non-etanercept group (23%).
Conclusion
Although treatment with etanercept lacks the support of a randomized controlled trial, similar to all other treatments currently used for SJS and TEN, preliminary reports highlight a benefit in disease progression and improvement in time to re-epithelialization. In particular, if etanercept 50 mg subcutaneously is given as monotherapy or is given early in the disease course (prior to other therapies being attempted and ideally within 6 hours of presentation), our data suggest an even greater trend toward improved mortality and decreased time to re-epithelialization. Additionally, our findings may suggest that in some patients, etanercept monotherapy is not an adequate intervention but the addition of IVIG may be helpful; however, the senior author (S.W.) notes anecdotally that in his experience with the patients treated at the University of California Los Angeles, the order of administration of combination therapies—etanercept followed by IVIG—was important in addition to the choice of therapy. These findings are promising enough to warrant a multicenter randomized controlled trial comparing the efficacy of etanercept to other more commonly used treatments for this spectrum of disease, including IVIG and/or cyclosporine. Based on the data presented in this case series, including the 13 patients who received etanercept and had a 0% mortality rate, etanercept may be viewed as a targeted therapeutic intervention for patients with SJS and TEN.
- Pereira FA, Mudgil AV, Rosmarin DM. Toxic epidermal necrolysis. J Am Acad Dermatol. 2007;56:181-200.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Hunger RE, Hunziker T, Buettiker U, et al. Rapid resolution of toxic epidermal necrolysis with anti-TNF-α treatment. J Allergy Clin Immunol. 2005;116:923-924.
- Worswick S, Cotliar J. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of treatment options. Dermatol Ther. 2011;24:207-218.
- Wallace AB. The exposure treatment of burns. Lancet Lond Engl. 1951;1:501-504.
- Bastuji-Garin S, Fouchard N, Bertocchi M, et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149-153.
- Mounach A, Rezqi A, Nouijai A, et al. Stevens-Johnson syndrome complicating adalimumab therapy in rheumatoid arthritis disease. Rheumatol Int. 2013;33:1351-1353.
- Salama M, Lawrance I-C. Stevens-Johnson syndrome complicating adalimumab therapy in Crohn’s disease. World J Gastroenterol. 2009;15:4449-4452.
- Wolkenstein P, Latarjet J, Roujeau JC, et al. Randomised comparison of thalidomide versus placebo in toxic epidermal necrolysis. Lancet Lond Engl. 1998;352:1586-1589.
- Fischer M, Fiedler E, Marsch WC, et al Antitumour necrosis factor-α antibodies (infliximab) in the treatment of a patient with toxic epidermal necrolysis. Br J Dermatol. 2002;146:707-709.
- Meiss F, Helmbold P, Meykadeh N, et al. Overlap of acute generalized exanthematous pustulosis and toxic epidermal necrolysis: response to antitumour necrosis factor-alpha antibody infliximab: report of three cases. J Eur Acad Dermatol Venereol. 2007;21:717-719.
- Al-Shouli S, Abouchala N, Bogusz MJ, et al. Toxic epidermal necrolysis associated with high intake of sildenafil and its response to infliximab. Acta Derm Venereol. 2005;85:534-535.
- Wojtkiewicz A, Wysocki M, Fortuna J, et al. Beneficial and rapid effect of infliximab on the course of toxic epidermal necrolysis. Acta Derm Venereol. 2008;88:420-421.
- Kreft B, Wohlrab J, Bramsiepe I, et al. Etoricoxib-induced toxic epidermal necrolysis: successful treatment with infliximab. J Dermatol. 2010;37:904-906.
- Patmanidis K, Sidiras A, Dolianitis K, et al. Combination of infliximab and high-dose intravenous immunoglobulin for toxic epidermal necrolysis: successful treatment of an elderly patient. Case Rep Dermatol Med. 2012;2012:915314.
- Gaitanis G, Spyridonos P, Patmanidis K, et al. Treatment of toxic epidermal necrolysis with the combination of infliximab and high-dose intravenous immunoglobulin. Dermatol Basel Switz. 2012;224:134-139.
- Zárate-Correa LC, Carrillo-Gómez DC, Ramírez-Escobar AF, et al. Toxic epidermal necrolysis successfully treated with infliximab. J Investig Allergol Clin Immunol. 2013;23:61-63.
- Paquet P, Jennes S, Rousseau AF, et al. Effect of N-acetylcysteine combined with infliximab on toxic epidermal necrolysis. a proof-of-concept study. Burns J Int Soc Burn Inj. 2014;40:1707-1712.
- Famularo G, Dona BD, Canzona F, et al. Etanercept for toxic epidermal necrolysis. Ann Pharmacother. 2007;41:1083-1084.
- Gubinelli E, Canzona F, Tonanzi T, et al. Toxic epidermal necrolysis successfully treated with etanercept. J Dermatol. 2009;36:150-153.
- Sadighha A. Etanercept in the treatment of a patient with acute generalized exanthematous pustulosis/toxic epidermal necrolysis: definition of a new model based on translational research. Int J Dermatol. 2009;48:913-914.
- Didona D, Paolino G, Garcovich S, et al. Successful use of etanercept in a case of toxic epidermal necrolysis induced by rituximab. J Eur Acad Dermatol Venereol. 2016;30:E83-E84.
- Lee Y-Y, Ko J-H, Wei C-H, et al. Use of etanercept to treat toxic epidermal necrolysis in a human immunodeficiency virus-positive patient. Dermatol Sin. 2013;31:78-81.
- Owczarczyk-Saczonek A, Zdanowska N, Znajewska-Pander A, et al. Stevens-Johnson syndrome in a patient with rheumatoid arthritis during long-term etanercept therapy. J Dermatol Case Rep. 2016;10:14-16.
- Wang CW, Yang LY, Chen CB, et al. Randomized, controlled trial of TNF-α antagonist in CTL mediated severe cutaneous adverse reactions. J Clin Invest. 2018;128:985-996.
Regarded as dermatologic emergencies, Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) represent a spectrum of blistering skin diseases that have a high mortality rate. Because of a misguided immune response to medications or infections, CD8+ T lymphocytes release proinflammatory cytokines, giving rise to the extensive epidermal destruction seen in SJS and TEN. The exact pathogenesis of SJS and TEN is still poorly defined, but studies have proposed that T cells mediate keratinocyte (KC) apoptosis through perforin and granzyme release and activation of the Fas/Fas ligand (FasL). Functioning as a transmembrane death receptor in the tumor necrosis factor (TNF) superfamily, Fas (CD95) activates Fas-associated death domain protein, caspases, and nucleases, resulting in organized cell destruction. Likewise, perforin and granzymes also have been shown to play a similar role in apoptosis via activation of caspases.1
Evidence for the role of TNF-α in SJS and TEN has been supported by findings of elevated levels of TNF-α within the blister fluid, serum, and KC cell surface. Additionally, TNF-α has been shown to upregulate inducible nitric oxide synthase in KCs, causing an accumulation of nitric oxide and subsequent FasL-mediated cell death.1-3 Notably, studies have demonstrated a relative lack of lymphocytes in the tissue of TEN patients despite the extensive destruction that is observed, thus emphasizing the importance of amplification and cell signaling via inflammatory mediators such as TNF-α.1 In this proposed model, T cells release IFN-γ, causing KCs to release TNF-α that subsequently promotes the upregulation of the aforementioned FasL.1 Tumor necrosis factor α also may promote increased MHC class I complex deposition on KC surfaces that may play a role in perforin and granzyme-mediated apoptosis of KCs.1
There is still debate on the standard of care for the treatment of SJS and TEN, attributed to the absence of randomized controlled trials and the rarity of the disease as well as the numerous conflicting studies evaluating potential treatments.1,4 Despite conflicting data to support their use, supportive care and intravenous immunoglobulin (IVIG) continue to be common treatments for SJS and TEN in hospitals worldwide. Elucidation of the role of TNF-α has prompted the use of infliximab and etanercept. In a case series of Italian patients with TEN (average SCORTEN, 3.6) treated with the TNF-α antagonist etanercept, no mortality was observed, which was well below the calculated expected mortality of 46.9%.2 Our retrospective study compared the use of a TNF antagonist to other therapies in the treatment of SJS/TEN. Our data suggest that etanercept is a lifesaving and disease-modifying therapy.
Methods
Twenty-two patients with SJS/TEN were included in this analysis. This included all patients who carried a clinical diagnosis of SJS/TEN with a confirmatory biopsy at our 2 university centers—University of California, Los Angeles, and Keck-LA County-Norris Hospital at the University of Southern California, Los Angeles—from 2013 to 2016. The diagnosis was rendered when a clinical diagnosis of SJS/TEN was given by a dermatologist and a confirmatory biopsy was performed. Every patient given a diagnosis of SJS/TEN at either university system from 2015 onward received an injection of etanercept given the positive results reported by Paradisi et al.2
The 9 patients who presented from 2013 to 2014 to our 2 hospital systems and were given a diagnosis of SJS/TEN received either IVIG or supportive care alone and had an average body surface area (BSA) affected of 23%. The 13 patients who presented from 2015 to 2016 were treated with etanercept in the form of a 50-mg subcutaneous injection given once to the right upper arm. Of this group, 4 patients received dual therapy with both IVIG and etanercept. In the etanercept-treated group (etanercept alone and etanercept plus IVIG), the average BSA affected was 30%. At the time of preliminary diagnosis, all patient medications were evaluated for a possible temporal relationship to the onset of rash and were discontinued if felt to be causative. The causative agent and treatment course for each patient is summarized in Table 1.
Patients were monitored daily in the hospital for improvement, and time to re-epithelialization was measured. Re-epithelialization was defined as progressive healing with residual lesions (erosions, ulcers, or bullae) covering no more than 5% BSA and was contingent on the patient having no new lesions within 24 hours.5 SCORe of Toxic Epidermal Necrosis (SCORTEN), a validated severity-of-illness score,6 was calculated by giving 1 point for each of the following criteria at the time of diagnosis: age ≥40 years, concurrent malignancy, heart rate ≥120 beats/min, serum blood urea nitrogen >27 mg/dL, serum bicarbonate <20 mEq/L, serum glucose >250 mg/dL, and detached or compromised BSA >10%. The total SCORTEN was correlated with the following risk of mortality as supported by prior validation studies: SCORTEN of 0 to 1, 3.2%; SCORTEN of 2, 12.1%; SCORTEN of 3, 35.3%; SCORTEN of 4, 58.3%; SCORTEN of ≥5, >90%.
Results
A total of 13 patients received etanercept. The mean SCORTEN was 2.2. The observed mortality was 0%, which was markedly lower than the predicted mortality of 24.3% (as determined by linear interpolation). Of this cohort, 9 patients received etanercept alone (mean SCORTEN of 2.1, predicted mortality of 22.9%), whereas 4 patients received a combination of etanercept and IVIG (mean SCORTEN of 2.3, predicted mortality of 27.2%).
The 4 patients who received both etanercept and IVIG received dual therapy for varying reasons. In patient 2 (Table 1), the perceived severity of this case ultimately led to the decision to start IVIG in addition to etanercept, resulting in rapid recovery and discharge after only 1 week of hospitalization. Intravenous immunoglobulin also was given in patient 3 (SCORTEN of 4) and patient 6 (SCORTEN of 2) for progression of disease despite administration of etanercept, with subsequent cessation of progression after the addition of the second agent (IVIG). Patient 12 might have done well on etanercept monotherapy but was administered IVIG as a precautionary measure because of hospital treatment algorithms.
Nine patients did not receive etanercept. Of this group, 5 received IVIG and 4 were managed with supportive care alone. The average SCORTEN for this group was 2.4, only slightly higher than the group that received etanercept (Table 2). The mortality rate in this group was 33%, which was higher than the predicted mortality of 28.1%.
Re-epithelialization data were available for 8 patients who received etanercept. The average time to re-epithelialization for these patients was 8.9 days and ranged from 3 to 19 days. Of these patients, 2 received both IVIG and etanercept, with an average time to re-epithelialization of 13 days. For the 6 patients who received etanercept alone, the average time to re-epithelialization was 7.5 days. Re-epithelialization data were not available for any of the patients who received only IVIG or supportive care but to our recollection ranged from 14 to 21 days.
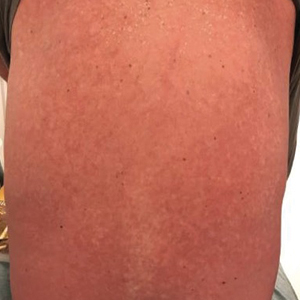
The clinical course of the 13 patients after the administration of a single dose of etanercept was remarkable, as there was complete absence of mortality and an increase in speed of recovery in most patients receiving this intervention (time to re-epithelialization, 3–19 days). We also observed another interesting trend from our patients treated with etanercept, which was the suggestion that treatment with etanercept may be less effective if IVIG and/or steroids are given prior to etanercept; likewise, treatment is more effective when etanercept is given quickly. For patients 1, 4, 5, 7, 9, and 11 (as shown in Table 1), no prior IVIG therapy or other immunosuppressive therapy had been given before etanercept was administered. In these 6 patients, the average time to re-epithelialization after etanercept administration was 7.5 days; average time to re-epithelialization, unfortunately, is not available for the patients who were not treated with etanercept. In addition, as shown in the Figure, it was noted in some patients that the depth of denudation was markedly more superficial than what would typically be clinically observed with TEN after administration of other immunomodulatory therapies such as IVIG or prednisone or with supportive care alone. In these 2 patients with superficial desquamation—patients 7 and 9—etanercept notably was given within 6 hours of onset of skin pain.
Comment
There is no definitive gold standard treatment of SJS, SJS/TEN overlap, or TEN. However, generally agreed upon management includes immediate discontinuation of the offending medication and supportive therapy with aggressive electrolyte replacement and wound care. Management in a burn unit or intensive care unit is recommended in severe cases. Contention over the efficacy of various medications in the treatment of SJS and TEN continues and largely is due to the rarity of SJS and TEN; studies are small and almost all lack randomization. Therapies that have been used include high-dose steroids, IVIG, plasmapheresis, cyclophosphamide, cyclosporine A, and TNF inhibitors (eg, etanercept, infliximab).1
Evidence for the use of anti–TNF-α antibodies has been limited thus far, with most of the literature focusing on infliximab and etanercept. Adalimumab, a fully humanized clonal antibody, has no reported cases in the dermatologic literature for use in patients with SJS/TEN. Two case reports of adalimumab paradoxically causing SJS have been documented. In both cases, adalimumab was stopped and patients responded to intravenous corticosteroids and infliximab.7,8 Similarly, thalidomide has not proven to be a promising anti–TNF-α agent for the treatment of SJS/TEN. In the only attempted randomized controlled trial for SJS and TEN, thalidomide appeared to increase mortality, eventuating in this trial being terminated prior to the planned end date.9Infliximab and etanercept have several case reports and a few case series highlighting potentially efficacious application of TNF-α inhibitors for the treatment of SJS/TEN.10-13 In 2002, Fischer et al10 reported the first case of TEN treated successfully with a single dose of infliximab 5 mg/kg. Kreft et al14 reported on etoricoxib-induced TEN that was treated with infliximab 5 mg/kg, which led to re-epithelialization within 5 weeks (notably a 5-week re-epithelialization time is not necessarily an improvement).
In 2005, Hunger et al3 demonstrated TNF-α’s release by KCs in the epidermis and by inflammatory cells in the dermis of a TEN patient. Twenty-four hours after the administration of infliximab 5 mg/kg in these patients, TNF-α was found to be below normal and epidermal detachment ceased.3 Wojtkietwicz et al13 demonstrated benefit following an infusion of infliximab 5 mg/kg in a patient whose disease continued to progress despite treatment with dexamethasone and 1.8 g/kg of IVIG.
Then 2 subsequent case series added further support for the efficacy of infliximab in the treatment of TEN. Patmanidis et al15 and Gaitanis et al16 reported similar results in 4 patients, each treated with infliximab 5 mg/kg immediately followed by initiation of high-dose IVIG (2 g/kg over 5 days). Zárate-Correa et al17 reported a 0% mortality rate and near-complete re-epithelialization after 5 to 14 days in 4 patients treated with a single 300-mg dose of infliximab.
However, the success of infliximab in the treatment of TEN has been countered by the pilot study by Paquet et al,18 which compared the efficacy of 150 mg/kg of N-acetylcysteine alone vs adding infliximab 5 mg/kg to treat 10 TEN patients. The study demonstrated no benefit at 48 hours in the group given infliximab, the time frame in which prior case reports touting infliximab’s benefit claimed the benefit was observed. Similarly, there was no effect on mortality for either treatment modality as assessed by illness auxiliary score.18
Evidence in support of the use of etanercept in the treatment of SJS/TEN is mounting, and some centers have begun to use it as the first-choice therapy for SJS/TEN. The first case was reported by Famularo et al,19 in which a patient with TEN was given 2 doses of etanercept 25 mg after failure to improve with prednisolone 1 mg/kg. The patient showed near-complete and rapid re-epithelization in 6 days before death due to disseminated intravascular coagulation 10 days after admission.19 Gubinelli et al20 and Sadighha21 independently reported cases of TEN and TEN/acute generalized exanthematous pustulosis overlap treated with a total of 50 mg of etanercept, demonstrating rapid cessation of lesion progression. Didona et al22 found similar benefit using etanercept 50 mg to treat TEN secondary to rituximab after failure to improve with prednisone and cyclophosphamide. Treatment of TEN with etanercept in an HIV-positive patient also has been reported. Lee et al23 described a patient who was administered 50-mg and 25-mg injections on days 3 and 5 of hospitalization, respectively, with re-epithelialization occurring by day 8. Finally, Owczarczyk-Saczonek et al24 reported a case of SJS in a patient with a 4-year history of etanercept and sulfasalazine treatment of rheumatoid arthritis; sulfasalazine was stopped, but this patient was continued on etanercept until resolution of skin and mucosal symptoms. However, it is important to consider the possibility of publication bias among these cases selected for their positive outcomes.
Perhaps the most compelling literature regarding the use of etanercept for TEN was described in a case series by Paradisi et al.2 This study included 10 patients with TEN, all of whom demonstrated complete re-epithelialization shortly after receiving etanercept 50 mg. Average SCORTEN was 3.6 with a range of 2 to 6. Eight patients in this study had severe comorbidities and all 10 patients survived, with a time to re-epithelialization ranging from 7 to 20 days.2 Additionally, a randomized controlled trial showed that 38 etanercept-treated patients had improved mortality (P=.266) and re-epithelialization time (P=.01) compared to patients treated with intravenous methylprednisolone.25Limitations to our study are similar to other reports of SJS/TEN and included the small number of cases and lack of randomization. Additionally, we do not have data available for all patients for time between onset of disease and treatment initiation. Because of these challenges, data presented in this case series is observational only. Additionally, the patients treated with etanercept alone had a slightly lower SCORTEN compared to the group that received IVIG or supportive care alone (2.1 and 2.4 respectively). However, the etanercept-only group actually had higher involvement of epidermal detachment (33%) compared to the non-etanercept group (23%).
Conclusion
Although treatment with etanercept lacks the support of a randomized controlled trial, similar to all other treatments currently used for SJS and TEN, preliminary reports highlight a benefit in disease progression and improvement in time to re-epithelialization. In particular, if etanercept 50 mg subcutaneously is given as monotherapy or is given early in the disease course (prior to other therapies being attempted and ideally within 6 hours of presentation), our data suggest an even greater trend toward improved mortality and decreased time to re-epithelialization. Additionally, our findings may suggest that in some patients, etanercept monotherapy is not an adequate intervention but the addition of IVIG may be helpful; however, the senior author (S.W.) notes anecdotally that in his experience with the patients treated at the University of California Los Angeles, the order of administration of combination therapies—etanercept followed by IVIG—was important in addition to the choice of therapy. These findings are promising enough to warrant a multicenter randomized controlled trial comparing the efficacy of etanercept to other more commonly used treatments for this spectrum of disease, including IVIG and/or cyclosporine. Based on the data presented in this case series, including the 13 patients who received etanercept and had a 0% mortality rate, etanercept may be viewed as a targeted therapeutic intervention for patients with SJS and TEN.
Regarded as dermatologic emergencies, Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) represent a spectrum of blistering skin diseases that have a high mortality rate. Because of a misguided immune response to medications or infections, CD8+ T lymphocytes release proinflammatory cytokines, giving rise to the extensive epidermal destruction seen in SJS and TEN. The exact pathogenesis of SJS and TEN is still poorly defined, but studies have proposed that T cells mediate keratinocyte (KC) apoptosis through perforin and granzyme release and activation of the Fas/Fas ligand (FasL). Functioning as a transmembrane death receptor in the tumor necrosis factor (TNF) superfamily, Fas (CD95) activates Fas-associated death domain protein, caspases, and nucleases, resulting in organized cell destruction. Likewise, perforin and granzymes also have been shown to play a similar role in apoptosis via activation of caspases.1
Evidence for the role of TNF-α in SJS and TEN has been supported by findings of elevated levels of TNF-α within the blister fluid, serum, and KC cell surface. Additionally, TNF-α has been shown to upregulate inducible nitric oxide synthase in KCs, causing an accumulation of nitric oxide and subsequent FasL-mediated cell death.1-3 Notably, studies have demonstrated a relative lack of lymphocytes in the tissue of TEN patients despite the extensive destruction that is observed, thus emphasizing the importance of amplification and cell signaling via inflammatory mediators such as TNF-α.1 In this proposed model, T cells release IFN-γ, causing KCs to release TNF-α that subsequently promotes the upregulation of the aforementioned FasL.1 Tumor necrosis factor α also may promote increased MHC class I complex deposition on KC surfaces that may play a role in perforin and granzyme-mediated apoptosis of KCs.1
There is still debate on the standard of care for the treatment of SJS and TEN, attributed to the absence of randomized controlled trials and the rarity of the disease as well as the numerous conflicting studies evaluating potential treatments.1,4 Despite conflicting data to support their use, supportive care and intravenous immunoglobulin (IVIG) continue to be common treatments for SJS and TEN in hospitals worldwide. Elucidation of the role of TNF-α has prompted the use of infliximab and etanercept. In a case series of Italian patients with TEN (average SCORTEN, 3.6) treated with the TNF-α antagonist etanercept, no mortality was observed, which was well below the calculated expected mortality of 46.9%.2 Our retrospective study compared the use of a TNF antagonist to other therapies in the treatment of SJS/TEN. Our data suggest that etanercept is a lifesaving and disease-modifying therapy.
Methods
Twenty-two patients with SJS/TEN were included in this analysis. This included all patients who carried a clinical diagnosis of SJS/TEN with a confirmatory biopsy at our 2 university centers—University of California, Los Angeles, and Keck-LA County-Norris Hospital at the University of Southern California, Los Angeles—from 2013 to 2016. The diagnosis was rendered when a clinical diagnosis of SJS/TEN was given by a dermatologist and a confirmatory biopsy was performed. Every patient given a diagnosis of SJS/TEN at either university system from 2015 onward received an injection of etanercept given the positive results reported by Paradisi et al.2
The 9 patients who presented from 2013 to 2014 to our 2 hospital systems and were given a diagnosis of SJS/TEN received either IVIG or supportive care alone and had an average body surface area (BSA) affected of 23%. The 13 patients who presented from 2015 to 2016 were treated with etanercept in the form of a 50-mg subcutaneous injection given once to the right upper arm. Of this group, 4 patients received dual therapy with both IVIG and etanercept. In the etanercept-treated group (etanercept alone and etanercept plus IVIG), the average BSA affected was 30%. At the time of preliminary diagnosis, all patient medications were evaluated for a possible temporal relationship to the onset of rash and were discontinued if felt to be causative. The causative agent and treatment course for each patient is summarized in Table 1.
Patients were monitored daily in the hospital for improvement, and time to re-epithelialization was measured. Re-epithelialization was defined as progressive healing with residual lesions (erosions, ulcers, or bullae) covering no more than 5% BSA and was contingent on the patient having no new lesions within 24 hours.5 SCORe of Toxic Epidermal Necrosis (SCORTEN), a validated severity-of-illness score,6 was calculated by giving 1 point for each of the following criteria at the time of diagnosis: age ≥40 years, concurrent malignancy, heart rate ≥120 beats/min, serum blood urea nitrogen >27 mg/dL, serum bicarbonate <20 mEq/L, serum glucose >250 mg/dL, and detached or compromised BSA >10%. The total SCORTEN was correlated with the following risk of mortality as supported by prior validation studies: SCORTEN of 0 to 1, 3.2%; SCORTEN of 2, 12.1%; SCORTEN of 3, 35.3%; SCORTEN of 4, 58.3%; SCORTEN of ≥5, >90%.
Results
A total of 13 patients received etanercept. The mean SCORTEN was 2.2. The observed mortality was 0%, which was markedly lower than the predicted mortality of 24.3% (as determined by linear interpolation). Of this cohort, 9 patients received etanercept alone (mean SCORTEN of 2.1, predicted mortality of 22.9%), whereas 4 patients received a combination of etanercept and IVIG (mean SCORTEN of 2.3, predicted mortality of 27.2%).
The 4 patients who received both etanercept and IVIG received dual therapy for varying reasons. In patient 2 (Table 1), the perceived severity of this case ultimately led to the decision to start IVIG in addition to etanercept, resulting in rapid recovery and discharge after only 1 week of hospitalization. Intravenous immunoglobulin also was given in patient 3 (SCORTEN of 4) and patient 6 (SCORTEN of 2) for progression of disease despite administration of etanercept, with subsequent cessation of progression after the addition of the second agent (IVIG). Patient 12 might have done well on etanercept monotherapy but was administered IVIG as a precautionary measure because of hospital treatment algorithms.
Nine patients did not receive etanercept. Of this group, 5 received IVIG and 4 were managed with supportive care alone. The average SCORTEN for this group was 2.4, only slightly higher than the group that received etanercept (Table 2). The mortality rate in this group was 33%, which was higher than the predicted mortality of 28.1%.
Re-epithelialization data were available for 8 patients who received etanercept. The average time to re-epithelialization for these patients was 8.9 days and ranged from 3 to 19 days. Of these patients, 2 received both IVIG and etanercept, with an average time to re-epithelialization of 13 days. For the 6 patients who received etanercept alone, the average time to re-epithelialization was 7.5 days. Re-epithelialization data were not available for any of the patients who received only IVIG or supportive care but to our recollection ranged from 14 to 21 days.
The clinical course of the 13 patients after the administration of a single dose of etanercept was remarkable, as there was complete absence of mortality and an increase in speed of recovery in most patients receiving this intervention (time to re-epithelialization, 3–19 days). We also observed another interesting trend from our patients treated with etanercept, which was the suggestion that treatment with etanercept may be less effective if IVIG and/or steroids are given prior to etanercept; likewise, treatment is more effective when etanercept is given quickly. For patients 1, 4, 5, 7, 9, and 11 (as shown in Table 1), no prior IVIG therapy or other immunosuppressive therapy had been given before etanercept was administered. In these 6 patients, the average time to re-epithelialization after etanercept administration was 7.5 days; average time to re-epithelialization, unfortunately, is not available for the patients who were not treated with etanercept. In addition, as shown in the Figure, it was noted in some patients that the depth of denudation was markedly more superficial than what would typically be clinically observed with TEN after administration of other immunomodulatory therapies such as IVIG or prednisone or with supportive care alone. In these 2 patients with superficial desquamation—patients 7 and 9—etanercept notably was given within 6 hours of onset of skin pain.
Comment
There is no definitive gold standard treatment of SJS, SJS/TEN overlap, or TEN. However, generally agreed upon management includes immediate discontinuation of the offending medication and supportive therapy with aggressive electrolyte replacement and wound care. Management in a burn unit or intensive care unit is recommended in severe cases. Contention over the efficacy of various medications in the treatment of SJS and TEN continues and largely is due to the rarity of SJS and TEN; studies are small and almost all lack randomization. Therapies that have been used include high-dose steroids, IVIG, plasmapheresis, cyclophosphamide, cyclosporine A, and TNF inhibitors (eg, etanercept, infliximab).1
Evidence for the use of anti–TNF-α antibodies has been limited thus far, with most of the literature focusing on infliximab and etanercept. Adalimumab, a fully humanized clonal antibody, has no reported cases in the dermatologic literature for use in patients with SJS/TEN. Two case reports of adalimumab paradoxically causing SJS have been documented. In both cases, adalimumab was stopped and patients responded to intravenous corticosteroids and infliximab.7,8 Similarly, thalidomide has not proven to be a promising anti–TNF-α agent for the treatment of SJS/TEN. In the only attempted randomized controlled trial for SJS and TEN, thalidomide appeared to increase mortality, eventuating in this trial being terminated prior to the planned end date.9Infliximab and etanercept have several case reports and a few case series highlighting potentially efficacious application of TNF-α inhibitors for the treatment of SJS/TEN.10-13 In 2002, Fischer et al10 reported the first case of TEN treated successfully with a single dose of infliximab 5 mg/kg. Kreft et al14 reported on etoricoxib-induced TEN that was treated with infliximab 5 mg/kg, which led to re-epithelialization within 5 weeks (notably a 5-week re-epithelialization time is not necessarily an improvement).
In 2005, Hunger et al3 demonstrated TNF-α’s release by KCs in the epidermis and by inflammatory cells in the dermis of a TEN patient. Twenty-four hours after the administration of infliximab 5 mg/kg in these patients, TNF-α was found to be below normal and epidermal detachment ceased.3 Wojtkietwicz et al13 demonstrated benefit following an infusion of infliximab 5 mg/kg in a patient whose disease continued to progress despite treatment with dexamethasone and 1.8 g/kg of IVIG.
Then 2 subsequent case series added further support for the efficacy of infliximab in the treatment of TEN. Patmanidis et al15 and Gaitanis et al16 reported similar results in 4 patients, each treated with infliximab 5 mg/kg immediately followed by initiation of high-dose IVIG (2 g/kg over 5 days). Zárate-Correa et al17 reported a 0% mortality rate and near-complete re-epithelialization after 5 to 14 days in 4 patients treated with a single 300-mg dose of infliximab.
However, the success of infliximab in the treatment of TEN has been countered by the pilot study by Paquet et al,18 which compared the efficacy of 150 mg/kg of N-acetylcysteine alone vs adding infliximab 5 mg/kg to treat 10 TEN patients. The study demonstrated no benefit at 48 hours in the group given infliximab, the time frame in which prior case reports touting infliximab’s benefit claimed the benefit was observed. Similarly, there was no effect on mortality for either treatment modality as assessed by illness auxiliary score.18
Evidence in support of the use of etanercept in the treatment of SJS/TEN is mounting, and some centers have begun to use it as the first-choice therapy for SJS/TEN. The first case was reported by Famularo et al,19 in which a patient with TEN was given 2 doses of etanercept 25 mg after failure to improve with prednisolone 1 mg/kg. The patient showed near-complete and rapid re-epithelization in 6 days before death due to disseminated intravascular coagulation 10 days after admission.19 Gubinelli et al20 and Sadighha21 independently reported cases of TEN and TEN/acute generalized exanthematous pustulosis overlap treated with a total of 50 mg of etanercept, demonstrating rapid cessation of lesion progression. Didona et al22 found similar benefit using etanercept 50 mg to treat TEN secondary to rituximab after failure to improve with prednisone and cyclophosphamide. Treatment of TEN with etanercept in an HIV-positive patient also has been reported. Lee et al23 described a patient who was administered 50-mg and 25-mg injections on days 3 and 5 of hospitalization, respectively, with re-epithelialization occurring by day 8. Finally, Owczarczyk-Saczonek et al24 reported a case of SJS in a patient with a 4-year history of etanercept and sulfasalazine treatment of rheumatoid arthritis; sulfasalazine was stopped, but this patient was continued on etanercept until resolution of skin and mucosal symptoms. However, it is important to consider the possibility of publication bias among these cases selected for their positive outcomes.
Perhaps the most compelling literature regarding the use of etanercept for TEN was described in a case series by Paradisi et al.2 This study included 10 patients with TEN, all of whom demonstrated complete re-epithelialization shortly after receiving etanercept 50 mg. Average SCORTEN was 3.6 with a range of 2 to 6. Eight patients in this study had severe comorbidities and all 10 patients survived, with a time to re-epithelialization ranging from 7 to 20 days.2 Additionally, a randomized controlled trial showed that 38 etanercept-treated patients had improved mortality (P=.266) and re-epithelialization time (P=.01) compared to patients treated with intravenous methylprednisolone.25Limitations to our study are similar to other reports of SJS/TEN and included the small number of cases and lack of randomization. Additionally, we do not have data available for all patients for time between onset of disease and treatment initiation. Because of these challenges, data presented in this case series is observational only. Additionally, the patients treated with etanercept alone had a slightly lower SCORTEN compared to the group that received IVIG or supportive care alone (2.1 and 2.4 respectively). However, the etanercept-only group actually had higher involvement of epidermal detachment (33%) compared to the non-etanercept group (23%).
Conclusion
Although treatment with etanercept lacks the support of a randomized controlled trial, similar to all other treatments currently used for SJS and TEN, preliminary reports highlight a benefit in disease progression and improvement in time to re-epithelialization. In particular, if etanercept 50 mg subcutaneously is given as monotherapy or is given early in the disease course (prior to other therapies being attempted and ideally within 6 hours of presentation), our data suggest an even greater trend toward improved mortality and decreased time to re-epithelialization. Additionally, our findings may suggest that in some patients, etanercept monotherapy is not an adequate intervention but the addition of IVIG may be helpful; however, the senior author (S.W.) notes anecdotally that in his experience with the patients treated at the University of California Los Angeles, the order of administration of combination therapies—etanercept followed by IVIG—was important in addition to the choice of therapy. These findings are promising enough to warrant a multicenter randomized controlled trial comparing the efficacy of etanercept to other more commonly used treatments for this spectrum of disease, including IVIG and/or cyclosporine. Based on the data presented in this case series, including the 13 patients who received etanercept and had a 0% mortality rate, etanercept may be viewed as a targeted therapeutic intervention for patients with SJS and TEN.
- Pereira FA, Mudgil AV, Rosmarin DM. Toxic epidermal necrolysis. J Am Acad Dermatol. 2007;56:181-200.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Hunger RE, Hunziker T, Buettiker U, et al. Rapid resolution of toxic epidermal necrolysis with anti-TNF-α treatment. J Allergy Clin Immunol. 2005;116:923-924.
- Worswick S, Cotliar J. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of treatment options. Dermatol Ther. 2011;24:207-218.
- Wallace AB. The exposure treatment of burns. Lancet Lond Engl. 1951;1:501-504.
- Bastuji-Garin S, Fouchard N, Bertocchi M, et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149-153.
- Mounach A, Rezqi A, Nouijai A, et al. Stevens-Johnson syndrome complicating adalimumab therapy in rheumatoid arthritis disease. Rheumatol Int. 2013;33:1351-1353.
- Salama M, Lawrance I-C. Stevens-Johnson syndrome complicating adalimumab therapy in Crohn’s disease. World J Gastroenterol. 2009;15:4449-4452.
- Wolkenstein P, Latarjet J, Roujeau JC, et al. Randomised comparison of thalidomide versus placebo in toxic epidermal necrolysis. Lancet Lond Engl. 1998;352:1586-1589.
- Fischer M, Fiedler E, Marsch WC, et al Antitumour necrosis factor-α antibodies (infliximab) in the treatment of a patient with toxic epidermal necrolysis. Br J Dermatol. 2002;146:707-709.
- Meiss F, Helmbold P, Meykadeh N, et al. Overlap of acute generalized exanthematous pustulosis and toxic epidermal necrolysis: response to antitumour necrosis factor-alpha antibody infliximab: report of three cases. J Eur Acad Dermatol Venereol. 2007;21:717-719.
- Al-Shouli S, Abouchala N, Bogusz MJ, et al. Toxic epidermal necrolysis associated with high intake of sildenafil and its response to infliximab. Acta Derm Venereol. 2005;85:534-535.
- Wojtkiewicz A, Wysocki M, Fortuna J, et al. Beneficial and rapid effect of infliximab on the course of toxic epidermal necrolysis. Acta Derm Venereol. 2008;88:420-421.
- Kreft B, Wohlrab J, Bramsiepe I, et al. Etoricoxib-induced toxic epidermal necrolysis: successful treatment with infliximab. J Dermatol. 2010;37:904-906.
- Patmanidis K, Sidiras A, Dolianitis K, et al. Combination of infliximab and high-dose intravenous immunoglobulin for toxic epidermal necrolysis: successful treatment of an elderly patient. Case Rep Dermatol Med. 2012;2012:915314.
- Gaitanis G, Spyridonos P, Patmanidis K, et al. Treatment of toxic epidermal necrolysis with the combination of infliximab and high-dose intravenous immunoglobulin. Dermatol Basel Switz. 2012;224:134-139.
- Zárate-Correa LC, Carrillo-Gómez DC, Ramírez-Escobar AF, et al. Toxic epidermal necrolysis successfully treated with infliximab. J Investig Allergol Clin Immunol. 2013;23:61-63.
- Paquet P, Jennes S, Rousseau AF, et al. Effect of N-acetylcysteine combined with infliximab on toxic epidermal necrolysis. a proof-of-concept study. Burns J Int Soc Burn Inj. 2014;40:1707-1712.
- Famularo G, Dona BD, Canzona F, et al. Etanercept for toxic epidermal necrolysis. Ann Pharmacother. 2007;41:1083-1084.
- Gubinelli E, Canzona F, Tonanzi T, et al. Toxic epidermal necrolysis successfully treated with etanercept. J Dermatol. 2009;36:150-153.
- Sadighha A. Etanercept in the treatment of a patient with acute generalized exanthematous pustulosis/toxic epidermal necrolysis: definition of a new model based on translational research. Int J Dermatol. 2009;48:913-914.
- Didona D, Paolino G, Garcovich S, et al. Successful use of etanercept in a case of toxic epidermal necrolysis induced by rituximab. J Eur Acad Dermatol Venereol. 2016;30:E83-E84.
- Lee Y-Y, Ko J-H, Wei C-H, et al. Use of etanercept to treat toxic epidermal necrolysis in a human immunodeficiency virus-positive patient. Dermatol Sin. 2013;31:78-81.
- Owczarczyk-Saczonek A, Zdanowska N, Znajewska-Pander A, et al. Stevens-Johnson syndrome in a patient with rheumatoid arthritis during long-term etanercept therapy. J Dermatol Case Rep. 2016;10:14-16.
- Wang CW, Yang LY, Chen CB, et al. Randomized, controlled trial of TNF-α antagonist in CTL mediated severe cutaneous adverse reactions. J Clin Invest. 2018;128:985-996.
- Pereira FA, Mudgil AV, Rosmarin DM. Toxic epidermal necrolysis. J Am Acad Dermatol. 2007;56:181-200.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Hunger RE, Hunziker T, Buettiker U, et al. Rapid resolution of toxic epidermal necrolysis with anti-TNF-α treatment. J Allergy Clin Immunol. 2005;116:923-924.
- Worswick S, Cotliar J. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of treatment options. Dermatol Ther. 2011;24:207-218.
- Wallace AB. The exposure treatment of burns. Lancet Lond Engl. 1951;1:501-504.
- Bastuji-Garin S, Fouchard N, Bertocchi M, et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149-153.
- Mounach A, Rezqi A, Nouijai A, et al. Stevens-Johnson syndrome complicating adalimumab therapy in rheumatoid arthritis disease. Rheumatol Int. 2013;33:1351-1353.
- Salama M, Lawrance I-C. Stevens-Johnson syndrome complicating adalimumab therapy in Crohn’s disease. World J Gastroenterol. 2009;15:4449-4452.
- Wolkenstein P, Latarjet J, Roujeau JC, et al. Randomised comparison of thalidomide versus placebo in toxic epidermal necrolysis. Lancet Lond Engl. 1998;352:1586-1589.
- Fischer M, Fiedler E, Marsch WC, et al Antitumour necrosis factor-α antibodies (infliximab) in the treatment of a patient with toxic epidermal necrolysis. Br J Dermatol. 2002;146:707-709.
- Meiss F, Helmbold P, Meykadeh N, et al. Overlap of acute generalized exanthematous pustulosis and toxic epidermal necrolysis: response to antitumour necrosis factor-alpha antibody infliximab: report of three cases. J Eur Acad Dermatol Venereol. 2007;21:717-719.
- Al-Shouli S, Abouchala N, Bogusz MJ, et al. Toxic epidermal necrolysis associated with high intake of sildenafil and its response to infliximab. Acta Derm Venereol. 2005;85:534-535.
- Wojtkiewicz A, Wysocki M, Fortuna J, et al. Beneficial and rapid effect of infliximab on the course of toxic epidermal necrolysis. Acta Derm Venereol. 2008;88:420-421.
- Kreft B, Wohlrab J, Bramsiepe I, et al. Etoricoxib-induced toxic epidermal necrolysis: successful treatment with infliximab. J Dermatol. 2010;37:904-906.
- Patmanidis K, Sidiras A, Dolianitis K, et al. Combination of infliximab and high-dose intravenous immunoglobulin for toxic epidermal necrolysis: successful treatment of an elderly patient. Case Rep Dermatol Med. 2012;2012:915314.
- Gaitanis G, Spyridonos P, Patmanidis K, et al. Treatment of toxic epidermal necrolysis with the combination of infliximab and high-dose intravenous immunoglobulin. Dermatol Basel Switz. 2012;224:134-139.
- Zárate-Correa LC, Carrillo-Gómez DC, Ramírez-Escobar AF, et al. Toxic epidermal necrolysis successfully treated with infliximab. J Investig Allergol Clin Immunol. 2013;23:61-63.
- Paquet P, Jennes S, Rousseau AF, et al. Effect of N-acetylcysteine combined with infliximab on toxic epidermal necrolysis. a proof-of-concept study. Burns J Int Soc Burn Inj. 2014;40:1707-1712.
- Famularo G, Dona BD, Canzona F, et al. Etanercept for toxic epidermal necrolysis. Ann Pharmacother. 2007;41:1083-1084.
- Gubinelli E, Canzona F, Tonanzi T, et al. Toxic epidermal necrolysis successfully treated with etanercept. J Dermatol. 2009;36:150-153.
- Sadighha A. Etanercept in the treatment of a patient with acute generalized exanthematous pustulosis/toxic epidermal necrolysis: definition of a new model based on translational research. Int J Dermatol. 2009;48:913-914.
- Didona D, Paolino G, Garcovich S, et al. Successful use of etanercept in a case of toxic epidermal necrolysis induced by rituximab. J Eur Acad Dermatol Venereol. 2016;30:E83-E84.
- Lee Y-Y, Ko J-H, Wei C-H, et al. Use of etanercept to treat toxic epidermal necrolysis in a human immunodeficiency virus-positive patient. Dermatol Sin. 2013;31:78-81.
- Owczarczyk-Saczonek A, Zdanowska N, Znajewska-Pander A, et al. Stevens-Johnson syndrome in a patient with rheumatoid arthritis during long-term etanercept therapy. J Dermatol Case Rep. 2016;10:14-16.
- Wang CW, Yang LY, Chen CB, et al. Randomized, controlled trial of TNF-α antagonist in CTL mediated severe cutaneous adverse reactions. J Clin Invest. 2018;128:985-996.
Practice Points
- Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are life-threatening dermatologic emergencies without a universally accepted treatment.
- Results of this study support the use of single-dose subcutaneous etanercept 50 mg as a potentially lifesaving therapy for patients with SJS/TEN.
Chilblain Lupus Erythematosus Presenting With Bilateral Hemorrhagic Bullae of Distal Halluces
To the Editor:
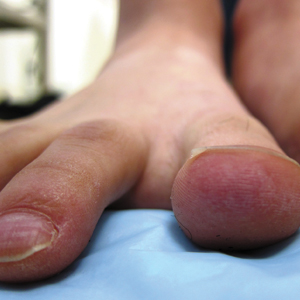
A 20-year-old man with no notable medical history presented to our dermatology clinic for evaluation of mildly painful, hemorrhagic bullae on the bilateral halluces of 1 month’s duration. On initial presentation the patient reported the lesions developed after wearing a new pair of tight-fitting shoes, suggesting a diagnosis of trauma-induced bullae. The patient was instructed to wear loose-fitting shoes and to follow up in 6 weeks to assess for improvement. At follow-up the bullae had resolved with residual violaceous patches on the bilateral distal halluces. He additionally developed a faint retiform erythematous patch on the left distal toe (Figure 1). The patient also had reticulate erythematous patches on the dorsal aspects of the hands extending to the forearms and legs resembling livedo reticularis. The patient was unsure if the skin lesions were triggered or worsened by cold exposure and reported that he smoked half a pack of cigarettes daily. At this time, the differential diagnosis still included trauma; however, there was concern for either embolic, thrombotic, or connective-tissue disease. A 4-mm punch biopsy of the left distal hallux demonstrated basal vacuolar interface dermatitis with superficial and deep perivascular inflammation and deep periadnexal mucin deposition (Figure 2) consistent with lupus dermatitis.

(H&E, original magnification ×400) with deep periadnexal mucin deposition (B)(colloidal iron, original magnification ×40).")
Serologic workup revealed increased antinuclear antibody titers of 1:320 (reference range, <1:40) and anti-Ro/Sjögren syndrome antigen antibodies of 86 (reference range, <20). There was no elevation in anti–double-stranded DNA, anti-Smith, antiribonucleoprotein, or anticardiolipin antibodies. Complement levels also were within reference range. Furthermore, the patient denied a history of Raynaud phenomenon, photosensitivity, oral ulcers, joint pain, shortness of breath, pleuritic chest pain, arthritis, blood clots, or any other systemic symptoms. Additional evaluation by the rheumatology department did not support criteria for systemic lupus erythematosus (SLE). In the context of the clinical presentation, histologic findings, and serologic markers, a diagnosis of chilblain lupus erythematosus (CHLE) was made. He was counseled on sun protection and smoking cessation and declined systemic therapy citing concern for side effects. Follow-up with the dermatology and rheumatology departments was advised.
Cutaneous lupus erythematosus (CLE) comprises various forms of lupus, including acute cutaneous lupus, subacute cutaneous lupus, and chronic cutaneous lupus. Chilblain lupus erythematosus is a rare subset of chronic CLE that first was described in 18881 and is characterized by tender violaceous papules and plaques that typically present in an acral distribution (ie, fingers, toes, nose, cheeks, ears). The skin lesions often are triggered or exacerbated by cold temperatures and dampness. As the lesions evolve, they can ulcerate, fissure, become hyperkeratotic, or result in atrophic plaques with scarring.2,3 A subset of patients also may have concurrent Raynaud phenomenon.1 Up to 20% of patients will eventually develop SLE, especially those patients with concurrent discoid lupus erythematosus, warranting close long-term follow-up.3 Serologic studies can reveal antinuclear antibodies, anti-Ro/Sjögren syndrome antigen antibodies, rheumatic factor, and anti–double-stranded DNA antibodies.1,4 Hypergammaglobulinemia also is a common finding in patients with CHLE, affecting more than two-thirds of patients.1 Typical features of CHLE seen on histopathology include interface dermatitis, perivascular lymphocytic infiltrate, apoptotic keratinocytes, lichenoid tissue reaction, and increased dermal mucin.1,4
Chilblain lupus erythematosus most commonly presents sporadically; however, there is a familial form that has been previously described.5 Sporadic CHLE usually occurs in middle-aged females, in contrast to familial CHLE, which presents in early childhood.1 The pathogenesis of the sporadic form is poorly understood, but it is thought to be stimulated by vasoconstriction or microvascular injury provoked by cold exposure. Furthermore, hypergammaglobulinemia and the presence of autoantibodies may contribute to the pathogenesis by increasing blood viscosity.1 The
Several drugs including thiazides, terbinafine, calcium channel blockers, angiotensin-converting enzyme inhibitors, and chemotherapeutic agents have been reported to trigger CHLE.4 Tumor necrosis factor α inhibitors have been shown to precipitate CHLE.6 Of note, drug-induced CHLE usually is limited to the skin and has not been shown to progress to SLE.6 Lebeau et al4 described a patient with breast cancer and preexisting CHLE that flared while the patient received docetaxel therapy, suggesting that certain drugs may not only induce but also may aggravate CHLE.
Many of the therapies that are effective in SLE such as antimalarial agents (ie, chloroquine, hydroxychloroquine) often are less efficacious in treating the lesions of CHLE.1 However, these patients often can be managed successfully by physical protection from the cold environment.1 Calcium channel blockers such as nifedipine also have been implicated, as they counteract vasoconstriction, which is thought to contribute to the pathogenesis of CHLE.1 Topical and systemic steroids also have been used to treat CHLE. Dapsone and pentoxifylline are other treatment modalities that have been effective in select cases of CHLE.5 Boehm and Bieber7 reported near resolution of CHLE with mycophenolate mofetil in an elderly woman with skin lesions that had been refractory to systemic steroids, antimalarial agents, azathioprine, dapsone, and pentoxifylline, suggesting that mycophenolate mofetil may be a therapeutic option for recalcitrant cases of CHLE. Local immunosuppressive agents such as tacrolimus also can be considered in treatment-refractory disease.
Chilblain lupus erythematosus is a rare chronic form of CLE that typically occurs sporadically but also has a familial form that has been described in several families. It most commonly is observed in middle-aged women, but we describe a case in a young man. Although CHLE typically does not respond well to traditional lupus therapies used in the management of SLE, good effects have been observed with cold avoidance, calcium channel blockers, and topical or oral steroids. For treatment-refractory cases, mycophenolate mofetil and other immunosuppressive agents have been shown to be effective.
- Hedrich CM, Fiebig B, Hauck FH, et al. Chilblain lupus erythematosus—a review of literature. Clin Rheumatol. 2008;27:949-954.
- Kuhn A, Lehmann P, Ruzicka T, eds. Cutaneous Lupus Erythematosus. Berlin, Germany: Springer; 2005.
- Obermoser G, Sontheimer RD, Zelger B. Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus. 2010;19:1050-1070.
- Lebeau S, També S, Sallam MA, et al. Docetaxel-induced relapse of subacute cutaneous lupus erythematosus and chilblain lupus. J Dtsch Dermatol Ges. 2013;11:871-874.
- Günther C, Hillebrand M, Brunk J, et al. Systemic involvement in TREX1-associated familial chilblain lupus. J Am Acad Dermatol. 2013;69:179-181.
- Sifuentes Giraldo WA, Ahijón Lana M, García Villanueva MJ, et al. Chilblain lupus induced by TNF-α antagonists: a case report and literature review. Clin Rheumatol. 2012;31:563-568.
- Boehm I, Bieber T. Chilblain lupus erythematosus Hutchinson: successful treatment with mycophenolate mofetil. Arch Dermatol. 2001;137:235-236.
To the Editor:
A 20-year-old man with no notable medical history presented to our dermatology clinic for evaluation of mildly painful, hemorrhagic bullae on the bilateral halluces of 1 month’s duration. On initial presentation the patient reported the lesions developed after wearing a new pair of tight-fitting shoes, suggesting a diagnosis of trauma-induced bullae. The patient was instructed to wear loose-fitting shoes and to follow up in 6 weeks to assess for improvement. At follow-up the bullae had resolved with residual violaceous patches on the bilateral distal halluces. He additionally developed a faint retiform erythematous patch on the left distal toe (Figure 1). The patient also had reticulate erythematous patches on the dorsal aspects of the hands extending to the forearms and legs resembling livedo reticularis. The patient was unsure if the skin lesions were triggered or worsened by cold exposure and reported that he smoked half a pack of cigarettes daily. At this time, the differential diagnosis still included trauma; however, there was concern for either embolic, thrombotic, or connective-tissue disease. A 4-mm punch biopsy of the left distal hallux demonstrated basal vacuolar interface dermatitis with superficial and deep perivascular inflammation and deep periadnexal mucin deposition (Figure 2) consistent with lupus dermatitis.
Serologic workup revealed increased antinuclear antibody titers of 1:320 (reference range, <1:40) and anti-Ro/Sjögren syndrome antigen antibodies of 86 (reference range, <20). There was no elevation in anti–double-stranded DNA, anti-Smith, antiribonucleoprotein, or anticardiolipin antibodies. Complement levels also were within reference range. Furthermore, the patient denied a history of Raynaud phenomenon, photosensitivity, oral ulcers, joint pain, shortness of breath, pleuritic chest pain, arthritis, blood clots, or any other systemic symptoms. Additional evaluation by the rheumatology department did not support criteria for systemic lupus erythematosus (SLE). In the context of the clinical presentation, histologic findings, and serologic markers, a diagnosis of chilblain lupus erythematosus (CHLE) was made. He was counseled on sun protection and smoking cessation and declined systemic therapy citing concern for side effects. Follow-up with the dermatology and rheumatology departments was advised.
Cutaneous lupus erythematosus (CLE) comprises various forms of lupus, including acute cutaneous lupus, subacute cutaneous lupus, and chronic cutaneous lupus. Chilblain lupus erythematosus is a rare subset of chronic CLE that first was described in 18881 and is characterized by tender violaceous papules and plaques that typically present in an acral distribution (ie, fingers, toes, nose, cheeks, ears). The skin lesions often are triggered or exacerbated by cold temperatures and dampness. As the lesions evolve, they can ulcerate, fissure, become hyperkeratotic, or result in atrophic plaques with scarring.2,3 A subset of patients also may have concurrent Raynaud phenomenon.1 Up to 20% of patients will eventually develop SLE, especially those patients with concurrent discoid lupus erythematosus, warranting close long-term follow-up.3 Serologic studies can reveal antinuclear antibodies, anti-Ro/Sjögren syndrome antigen antibodies, rheumatic factor, and anti–double-stranded DNA antibodies.1,4 Hypergammaglobulinemia also is a common finding in patients with CHLE, affecting more than two-thirds of patients.1 Typical features of CHLE seen on histopathology include interface dermatitis, perivascular lymphocytic infiltrate, apoptotic keratinocytes, lichenoid tissue reaction, and increased dermal mucin.1,4
Chilblain lupus erythematosus most commonly presents sporadically; however, there is a familial form that has been previously described.5 Sporadic CHLE usually occurs in middle-aged females, in contrast to familial CHLE, which presents in early childhood.1 The pathogenesis of the sporadic form is poorly understood, but it is thought to be stimulated by vasoconstriction or microvascular injury provoked by cold exposure. Furthermore, hypergammaglobulinemia and the presence of autoantibodies may contribute to the pathogenesis by increasing blood viscosity.1 The
Several drugs including thiazides, terbinafine, calcium channel blockers, angiotensin-converting enzyme inhibitors, and chemotherapeutic agents have been reported to trigger CHLE.4 Tumor necrosis factor α inhibitors have been shown to precipitate CHLE.6 Of note, drug-induced CHLE usually is limited to the skin and has not been shown to progress to SLE.6 Lebeau et al4 described a patient with breast cancer and preexisting CHLE that flared while the patient received docetaxel therapy, suggesting that certain drugs may not only induce but also may aggravate CHLE.
Many of the therapies that are effective in SLE such as antimalarial agents (ie, chloroquine, hydroxychloroquine) often are less efficacious in treating the lesions of CHLE.1 However, these patients often can be managed successfully by physical protection from the cold environment.1 Calcium channel blockers such as nifedipine also have been implicated, as they counteract vasoconstriction, which is thought to contribute to the pathogenesis of CHLE.1 Topical and systemic steroids also have been used to treat CHLE. Dapsone and pentoxifylline are other treatment modalities that have been effective in select cases of CHLE.5 Boehm and Bieber7 reported near resolution of CHLE with mycophenolate mofetil in an elderly woman with skin lesions that had been refractory to systemic steroids, antimalarial agents, azathioprine, dapsone, and pentoxifylline, suggesting that mycophenolate mofetil may be a therapeutic option for recalcitrant cases of CHLE. Local immunosuppressive agents such as tacrolimus also can be considered in treatment-refractory disease.
Chilblain lupus erythematosus is a rare chronic form of CLE that typically occurs sporadically but also has a familial form that has been described in several families. It most commonly is observed in middle-aged women, but we describe a case in a young man. Although CHLE typically does not respond well to traditional lupus therapies used in the management of SLE, good effects have been observed with cold avoidance, calcium channel blockers, and topical or oral steroids. For treatment-refractory cases, mycophenolate mofetil and other immunosuppressive agents have been shown to be effective.
To the Editor:
A 20-year-old man with no notable medical history presented to our dermatology clinic for evaluation of mildly painful, hemorrhagic bullae on the bilateral halluces of 1 month’s duration. On initial presentation the patient reported the lesions developed after wearing a new pair of tight-fitting shoes, suggesting a diagnosis of trauma-induced bullae. The patient was instructed to wear loose-fitting shoes and to follow up in 6 weeks to assess for improvement. At follow-up the bullae had resolved with residual violaceous patches on the bilateral distal halluces. He additionally developed a faint retiform erythematous patch on the left distal toe (Figure 1). The patient also had reticulate erythematous patches on the dorsal aspects of the hands extending to the forearms and legs resembling livedo reticularis. The patient was unsure if the skin lesions were triggered or worsened by cold exposure and reported that he smoked half a pack of cigarettes daily. At this time, the differential diagnosis still included trauma; however, there was concern for either embolic, thrombotic, or connective-tissue disease. A 4-mm punch biopsy of the left distal hallux demonstrated basal vacuolar interface dermatitis with superficial and deep perivascular inflammation and deep periadnexal mucin deposition (Figure 2) consistent with lupus dermatitis.
Serologic workup revealed increased antinuclear antibody titers of 1:320 (reference range, <1:40) and anti-Ro/Sjögren syndrome antigen antibodies of 86 (reference range, <20). There was no elevation in anti–double-stranded DNA, anti-Smith, antiribonucleoprotein, or anticardiolipin antibodies. Complement levels also were within reference range. Furthermore, the patient denied a history of Raynaud phenomenon, photosensitivity, oral ulcers, joint pain, shortness of breath, pleuritic chest pain, arthritis, blood clots, or any other systemic symptoms. Additional evaluation by the rheumatology department did not support criteria for systemic lupus erythematosus (SLE). In the context of the clinical presentation, histologic findings, and serologic markers, a diagnosis of chilblain lupus erythematosus (CHLE) was made. He was counseled on sun protection and smoking cessation and declined systemic therapy citing concern for side effects. Follow-up with the dermatology and rheumatology departments was advised.
Cutaneous lupus erythematosus (CLE) comprises various forms of lupus, including acute cutaneous lupus, subacute cutaneous lupus, and chronic cutaneous lupus. Chilblain lupus erythematosus is a rare subset of chronic CLE that first was described in 18881 and is characterized by tender violaceous papules and plaques that typically present in an acral distribution (ie, fingers, toes, nose, cheeks, ears). The skin lesions often are triggered or exacerbated by cold temperatures and dampness. As the lesions evolve, they can ulcerate, fissure, become hyperkeratotic, or result in atrophic plaques with scarring.2,3 A subset of patients also may have concurrent Raynaud phenomenon.1 Up to 20% of patients will eventually develop SLE, especially those patients with concurrent discoid lupus erythematosus, warranting close long-term follow-up.3 Serologic studies can reveal antinuclear antibodies, anti-Ro/Sjögren syndrome antigen antibodies, rheumatic factor, and anti–double-stranded DNA antibodies.1,4 Hypergammaglobulinemia also is a common finding in patients with CHLE, affecting more than two-thirds of patients.1 Typical features of CHLE seen on histopathology include interface dermatitis, perivascular lymphocytic infiltrate, apoptotic keratinocytes, lichenoid tissue reaction, and increased dermal mucin.1,4
Chilblain lupus erythematosus most commonly presents sporadically; however, there is a familial form that has been previously described.5 Sporadic CHLE usually occurs in middle-aged females, in contrast to familial CHLE, which presents in early childhood.1 The pathogenesis of the sporadic form is poorly understood, but it is thought to be stimulated by vasoconstriction or microvascular injury provoked by cold exposure. Furthermore, hypergammaglobulinemia and the presence of autoantibodies may contribute to the pathogenesis by increasing blood viscosity.1 The
Several drugs including thiazides, terbinafine, calcium channel blockers, angiotensin-converting enzyme inhibitors, and chemotherapeutic agents have been reported to trigger CHLE.4 Tumor necrosis factor α inhibitors have been shown to precipitate CHLE.6 Of note, drug-induced CHLE usually is limited to the skin and has not been shown to progress to SLE.6 Lebeau et al4 described a patient with breast cancer and preexisting CHLE that flared while the patient received docetaxel therapy, suggesting that certain drugs may not only induce but also may aggravate CHLE.
Many of the therapies that are effective in SLE such as antimalarial agents (ie, chloroquine, hydroxychloroquine) often are less efficacious in treating the lesions of CHLE.1 However, these patients often can be managed successfully by physical protection from the cold environment.1 Calcium channel blockers such as nifedipine also have been implicated, as they counteract vasoconstriction, which is thought to contribute to the pathogenesis of CHLE.1 Topical and systemic steroids also have been used to treat CHLE. Dapsone and pentoxifylline are other treatment modalities that have been effective in select cases of CHLE.5 Boehm and Bieber7 reported near resolution of CHLE with mycophenolate mofetil in an elderly woman with skin lesions that had been refractory to systemic steroids, antimalarial agents, azathioprine, dapsone, and pentoxifylline, suggesting that mycophenolate mofetil may be a therapeutic option for recalcitrant cases of CHLE. Local immunosuppressive agents such as tacrolimus also can be considered in treatment-refractory disease.
Chilblain lupus erythematosus is a rare chronic form of CLE that typically occurs sporadically but also has a familial form that has been described in several families. It most commonly is observed in middle-aged women, but we describe a case in a young man. Although CHLE typically does not respond well to traditional lupus therapies used in the management of SLE, good effects have been observed with cold avoidance, calcium channel blockers, and topical or oral steroids. For treatment-refractory cases, mycophenolate mofetil and other immunosuppressive agents have been shown to be effective.
- Hedrich CM, Fiebig B, Hauck FH, et al. Chilblain lupus erythematosus—a review of literature. Clin Rheumatol. 2008;27:949-954.
- Kuhn A, Lehmann P, Ruzicka T, eds. Cutaneous Lupus Erythematosus. Berlin, Germany: Springer; 2005.
- Obermoser G, Sontheimer RD, Zelger B. Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus. 2010;19:1050-1070.
- Lebeau S, També S, Sallam MA, et al. Docetaxel-induced relapse of subacute cutaneous lupus erythematosus and chilblain lupus. J Dtsch Dermatol Ges. 2013;11:871-874.
- Günther C, Hillebrand M, Brunk J, et al. Systemic involvement in TREX1-associated familial chilblain lupus. J Am Acad Dermatol. 2013;69:179-181.
- Sifuentes Giraldo WA, Ahijón Lana M, García Villanueva MJ, et al. Chilblain lupus induced by TNF-α antagonists: a case report and literature review. Clin Rheumatol. 2012;31:563-568.
- Boehm I, Bieber T. Chilblain lupus erythematosus Hutchinson: successful treatment with mycophenolate mofetil. Arch Dermatol. 2001;137:235-236.
- Hedrich CM, Fiebig B, Hauck FH, et al. Chilblain lupus erythematosus—a review of literature. Clin Rheumatol. 2008;27:949-954.
- Kuhn A, Lehmann P, Ruzicka T, eds. Cutaneous Lupus Erythematosus. Berlin, Germany: Springer; 2005.
- Obermoser G, Sontheimer RD, Zelger B. Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus. 2010;19:1050-1070.
- Lebeau S, També S, Sallam MA, et al. Docetaxel-induced relapse of subacute cutaneous lupus erythematosus and chilblain lupus. J Dtsch Dermatol Ges. 2013;11:871-874.
- Günther C, Hillebrand M, Brunk J, et al. Systemic involvement in TREX1-associated familial chilblain lupus. J Am Acad Dermatol. 2013;69:179-181.
- Sifuentes Giraldo WA, Ahijón Lana M, García Villanueva MJ, et al. Chilblain lupus induced by TNF-α antagonists: a case report and literature review. Clin Rheumatol. 2012;31:563-568.
- Boehm I, Bieber T. Chilblain lupus erythematosus Hutchinson: successful treatment with mycophenolate mofetil. Arch Dermatol. 2001;137:235-236.
Practice Points
- Up to 20% of patients with chilblain lupus erythematosus (CHLE) will develop systemic lupus erythematosus (SLE), necessitating close long-term follow-up.
- Medications such as antihypertensives, antifungals, chemotherapeutic agents, and tumor necrosis factor 11α inhibitors have been reported to trigger CHLE.
- Chilblain lupus erythematosus is less responsive to traditional antimalarial agents commonly used to treat SLE.
- Management of CHLE includes physical protection from cold environments, calcium channel blockers, topical and systemic steroids, and pentoxifylline, among other treatment modalities.