User login
A Dermatology Hospitalist Team’s Response to the Inpatient Consult Flowchart
To the Editor:
We read with interest the Cutis article by Dobkin et al1 (Cutis. 2022;109:218-220) regarding guidelines for inpatient and emergency department dermatology consultations. We agree with the authors that dermatology training is lacking in other medical specialties, which makes it challenging for teams to assess the appropriateness of a dermatology consultation in the inpatient setting. Inpatient dermatology consultation can be utilized in a hospital system to aid in rapid and accurate diagnosis, avoid inappropriate therapies, and decrease length of stay2 and readmission rates3 while providing education to the primary teams. This is precisely why in many instances the availability of inpatient dermatology consultation is so important because nondermatologists often are unable to determine whether a rash is life-threatening, benign, or something in between. From the perspective of dermatology hospitalists, there is room for improvement in the flowchart Dobkin et al1 presented to guide inpatient dermatology consultation.
To have a productive relationship with our internal medicine, surgery, pediatrics, psychiatry, and other hospital-based colleagues, we must keep an open mind when a consultation is received. We feel that the flowchart proposed by Dobkin et al1 presents too narrow a viewpoint on the utility of inpatient dermatology. It rests on assertions that other teams will be able to determine the appropriate dermatologic diagnosis without involving a dermatologist, which often is not the case.
We disagree with several recommendations in the flowchart, the first being the assertion that patients who are “hemodynamically unstable due to [a] nondermatologic problem (eg, intubated on pressors, febrile, and hypotensive)” are not appropriate for inpatient dermatology consultation.1 Although dermatologists do not commonly encounter patients with critical illness in the outpatient clinic, dermatology consultation can be extremely helpful and even lifesaving in the inpatient setting. It is unrealistic to expect the primary teams to know whether cutaneous manifestations potentially could be related to the patient’s overall clinical picture. On the contrary, we would encourage the primary team in charge of a hemodynamically unstable patient to consult dermatology at the first sign of an unexplained rash. Take for example an acutely ill patient who develops retiform purpura. There are well-established dermatology guidelines for the workup of retiform purpura,4 including prompt biopsy and assessment of broad, potentially life-threatening differential diagnoses from calciphylaxis to angioinvasive fungal infection. In this scenario, the dermatology consultant may render the correct diagnosis and recommend immediate treatment that could be lifesaving.
Secondly, we do not agree with the recommendation that a patient in hospice care is not appropriate for inpatient dermatology consultation. Patients receiving hospice or palliative care have high rates of potentially symptomatic cutaneous diseases,5 including intertrigo and dermatitis—comprising stasis, seborrheic, and contact dermatitis.6 Although aggressive intervention for asymptomatic benign or malignant skin conditions may not be in line with their goals of care, an inpatient dermatology consultation can reduce symptoms and improve quality of life. This population also is one that is unlikely to be able to attend an outpatient dermatology clinic appointment and therefore are good candidates for inpatient consultation.
Lastly, we want to highlight the difference between a stable chronic dermatologic disease and an acute flare that may occur while a patient is hospitalized, regardless of whether it is the reason for admission. For example, a patient with psoriasis affecting limited body surface area who is hospitalized for a myocardial infarction is not appropriate for a dermatology consultation. However, if that same patient develops erythroderma while they are hospitalized for cardiac monitoring, it would certainly be appropriate for dermatology to be consulted. Additionally, there are times when a chronic skin disease is the reason for hospitalization; dermatology, although technically a consulting service, would be the primary decision-maker for the patient’s care in this situation. In these scenarios, it is important for the patient to be able to establish care for long-term outpatient management of their condition; however, it is prudent to involve dermatology while the patient is acutely hospitalized to guide their treatment plan until they are able to see a dermatologist after discharge.
In conclusion, we believe that hospital dermatology is a valuable tool that can be utilized in many different scenarios. Although there are certainly situations more appropriate for outpatient dermatology referral, we would caution against overly simplified algorithms that could discourage valuable inpatient dermatology consultations. It often is worth a conversation with your dermatology consultant (when available at an institution) to determine the best course of action for each patient. Additionally, we recognize the need for more formalized guidelines on when to pursue inpatient dermatology consultation. We are members of the Society of Dermatology Hospitalists and encourage readers to reference their website, which provides additional resources on inpatient dermatology (https://societydermatologyhospitalists.com/inpatient-dermatology-literature/).
Authors’ Response
We appreciate the letter in response to our commentary on the appropriateness of inpatient dermatology consultations. It is the continued refining and re-evaluation of concepts such as these that allow our field to grow and improve knowledge and patient care.
We sought to provide a nonpatronizing yet simple consultation flowchart that would help guide triage of patients in need or not in need of dermatologic evaluation by the inpatient teams. Understandably, the impressions of our flowchart have been variable based on different readers’ medical backgrounds and experiences. It is certainly possible that our flowchart lacked certain exceptions and oversimplified certain concepts, and we welcome further refining of this flowchart to better guide inpatient dermatology consultations.
We do, however, disagree that the primary team would not know whether a patient is intubated in the intensive care unit for a dermatology reason. If the patient is in such a status, it would be pertinent for the primary team to conduct a timely workup that could include consultations until a diagnosis is made. We were not implying that every dermatology consultation in the intensive care unit is unwarranted, especially if it can lead to a primary dermatologic diagnosis. We do believe that a thorough history could elicit an allergy or other chronic skin condition that could save resources and spending within a hospital. Likewise, psoriasis comes in many different presentations, and although we do not believe a consultation for chronic psoriatic plaques is appropriate in the hospital, it is absolutely appropriate for a patient who is erythrodermic from any cause.
Our flowchart was intended to be the first step to providing education on when consultations are appropriate, and further refinement will be necessary.
Hershel Dobkin, MD; Timothy Blackwell, BS; Robin Ashinoff, MD
Drs. Dobkin and Ashinoff are from Hackensack University Medical Center, New Jersey. Mr. Blackwell is from the Rowan University School of Osteopathic Medicine, Stratford, New Jersey.
The authors report no conflict of interest.
Correspondence: Hershel Dobkin, MD, Hackensack University Medical Center, 30 Prospect Ave, Hackensack, NJ 07601 (hersheldobkinpublic@gmail.com).
- Dobkin H, Blackwell T, Ashinoff R. When are inpatient and emergency dermatologic consultations appropriate? Cutis. 2022;109:218-220. doi:10.12788/cutis.0492
- Ko LN, Garza-Mayers AC, St John J, et al. Effect of dermatology consultation on outcomes for patients with presumed cellulitis: a randomized clinical trial. JAMA Dermatol. 2018;154:529-536. doi:10.1001/jamadermatol.2017.6196
- Hu L, Haynes H, Ferrazza D, et al. Impact of specialist consultations on inpatient admissions for dermatology-specific and related DRGs. J Gen Intern Med. 2013;28:1477-1482. doi:10.1007/s11606-013-2440-2
- Georgesen C, Fox LP, Harp J. Retiform purpura: a diagnostic approach. J Am Acad Dermatol. 2020;82:783-796. doi:10.1016/j.jaad.2019.07.112
- Pisano C, Paladichuk H, Keeling B. Dermatology in palliative medicine [published online October 14, 2021]. BMJ Support Palliat Care. doi:10.1136/bmjspcare-2021-003342
- Barnabé C, Daeninck P. “Beauty is only skin deep”: prevalence of dermatologic disease on a palliative care unit. J Pain Symptom Manage. 2005;29:419-422. doi:10.1016/j.jpainsymman.2004.08.009
To the Editor:
We read with interest the Cutis article by Dobkin et al1 (Cutis. 2022;109:218-220) regarding guidelines for inpatient and emergency department dermatology consultations. We agree with the authors that dermatology training is lacking in other medical specialties, which makes it challenging for teams to assess the appropriateness of a dermatology consultation in the inpatient setting. Inpatient dermatology consultation can be utilized in a hospital system to aid in rapid and accurate diagnosis, avoid inappropriate therapies, and decrease length of stay2 and readmission rates3 while providing education to the primary teams. This is precisely why in many instances the availability of inpatient dermatology consultation is so important because nondermatologists often are unable to determine whether a rash is life-threatening, benign, or something in between. From the perspective of dermatology hospitalists, there is room for improvement in the flowchart Dobkin et al1 presented to guide inpatient dermatology consultation.
To have a productive relationship with our internal medicine, surgery, pediatrics, psychiatry, and other hospital-based colleagues, we must keep an open mind when a consultation is received. We feel that the flowchart proposed by Dobkin et al1 presents too narrow a viewpoint on the utility of inpatient dermatology. It rests on assertions that other teams will be able to determine the appropriate dermatologic diagnosis without involving a dermatologist, which often is not the case.
We disagree with several recommendations in the flowchart, the first being the assertion that patients who are “hemodynamically unstable due to [a] nondermatologic problem (eg, intubated on pressors, febrile, and hypotensive)” are not appropriate for inpatient dermatology consultation.1 Although dermatologists do not commonly encounter patients with critical illness in the outpatient clinic, dermatology consultation can be extremely helpful and even lifesaving in the inpatient setting. It is unrealistic to expect the primary teams to know whether cutaneous manifestations potentially could be related to the patient’s overall clinical picture. On the contrary, we would encourage the primary team in charge of a hemodynamically unstable patient to consult dermatology at the first sign of an unexplained rash. Take for example an acutely ill patient who develops retiform purpura. There are well-established dermatology guidelines for the workup of retiform purpura,4 including prompt biopsy and assessment of broad, potentially life-threatening differential diagnoses from calciphylaxis to angioinvasive fungal infection. In this scenario, the dermatology consultant may render the correct diagnosis and recommend immediate treatment that could be lifesaving.
Secondly, we do not agree with the recommendation that a patient in hospice care is not appropriate for inpatient dermatology consultation. Patients receiving hospice or palliative care have high rates of potentially symptomatic cutaneous diseases,5 including intertrigo and dermatitis—comprising stasis, seborrheic, and contact dermatitis.6 Although aggressive intervention for asymptomatic benign or malignant skin conditions may not be in line with their goals of care, an inpatient dermatology consultation can reduce symptoms and improve quality of life. This population also is one that is unlikely to be able to attend an outpatient dermatology clinic appointment and therefore are good candidates for inpatient consultation.
Lastly, we want to highlight the difference between a stable chronic dermatologic disease and an acute flare that may occur while a patient is hospitalized, regardless of whether it is the reason for admission. For example, a patient with psoriasis affecting limited body surface area who is hospitalized for a myocardial infarction is not appropriate for a dermatology consultation. However, if that same patient develops erythroderma while they are hospitalized for cardiac monitoring, it would certainly be appropriate for dermatology to be consulted. Additionally, there are times when a chronic skin disease is the reason for hospitalization; dermatology, although technically a consulting service, would be the primary decision-maker for the patient’s care in this situation. In these scenarios, it is important for the patient to be able to establish care for long-term outpatient management of their condition; however, it is prudent to involve dermatology while the patient is acutely hospitalized to guide their treatment plan until they are able to see a dermatologist after discharge.
In conclusion, we believe that hospital dermatology is a valuable tool that can be utilized in many different scenarios. Although there are certainly situations more appropriate for outpatient dermatology referral, we would caution against overly simplified algorithms that could discourage valuable inpatient dermatology consultations. It often is worth a conversation with your dermatology consultant (when available at an institution) to determine the best course of action for each patient. Additionally, we recognize the need for more formalized guidelines on when to pursue inpatient dermatology consultation. We are members of the Society of Dermatology Hospitalists and encourage readers to reference their website, which provides additional resources on inpatient dermatology (https://societydermatologyhospitalists.com/inpatient-dermatology-literature/).
Authors’ Response
We appreciate the letter in response to our commentary on the appropriateness of inpatient dermatology consultations. It is the continued refining and re-evaluation of concepts such as these that allow our field to grow and improve knowledge and patient care.
We sought to provide a nonpatronizing yet simple consultation flowchart that would help guide triage of patients in need or not in need of dermatologic evaluation by the inpatient teams. Understandably, the impressions of our flowchart have been variable based on different readers’ medical backgrounds and experiences. It is certainly possible that our flowchart lacked certain exceptions and oversimplified certain concepts, and we welcome further refining of this flowchart to better guide inpatient dermatology consultations.
We do, however, disagree that the primary team would not know whether a patient is intubated in the intensive care unit for a dermatology reason. If the patient is in such a status, it would be pertinent for the primary team to conduct a timely workup that could include consultations until a diagnosis is made. We were not implying that every dermatology consultation in the intensive care unit is unwarranted, especially if it can lead to a primary dermatologic diagnosis. We do believe that a thorough history could elicit an allergy or other chronic skin condition that could save resources and spending within a hospital. Likewise, psoriasis comes in many different presentations, and although we do not believe a consultation for chronic psoriatic plaques is appropriate in the hospital, it is absolutely appropriate for a patient who is erythrodermic from any cause.
Our flowchart was intended to be the first step to providing education on when consultations are appropriate, and further refinement will be necessary.
Hershel Dobkin, MD; Timothy Blackwell, BS; Robin Ashinoff, MD
Drs. Dobkin and Ashinoff are from Hackensack University Medical Center, New Jersey. Mr. Blackwell is from the Rowan University School of Osteopathic Medicine, Stratford, New Jersey.
The authors report no conflict of interest.
Correspondence: Hershel Dobkin, MD, Hackensack University Medical Center, 30 Prospect Ave, Hackensack, NJ 07601 (hersheldobkinpublic@gmail.com).
To the Editor:
We read with interest the Cutis article by Dobkin et al1 (Cutis. 2022;109:218-220) regarding guidelines for inpatient and emergency department dermatology consultations. We agree with the authors that dermatology training is lacking in other medical specialties, which makes it challenging for teams to assess the appropriateness of a dermatology consultation in the inpatient setting. Inpatient dermatology consultation can be utilized in a hospital system to aid in rapid and accurate diagnosis, avoid inappropriate therapies, and decrease length of stay2 and readmission rates3 while providing education to the primary teams. This is precisely why in many instances the availability of inpatient dermatology consultation is so important because nondermatologists often are unable to determine whether a rash is life-threatening, benign, or something in between. From the perspective of dermatology hospitalists, there is room for improvement in the flowchart Dobkin et al1 presented to guide inpatient dermatology consultation.
To have a productive relationship with our internal medicine, surgery, pediatrics, psychiatry, and other hospital-based colleagues, we must keep an open mind when a consultation is received. We feel that the flowchart proposed by Dobkin et al1 presents too narrow a viewpoint on the utility of inpatient dermatology. It rests on assertions that other teams will be able to determine the appropriate dermatologic diagnosis without involving a dermatologist, which often is not the case.
We disagree with several recommendations in the flowchart, the first being the assertion that patients who are “hemodynamically unstable due to [a] nondermatologic problem (eg, intubated on pressors, febrile, and hypotensive)” are not appropriate for inpatient dermatology consultation.1 Although dermatologists do not commonly encounter patients with critical illness in the outpatient clinic, dermatology consultation can be extremely helpful and even lifesaving in the inpatient setting. It is unrealistic to expect the primary teams to know whether cutaneous manifestations potentially could be related to the patient’s overall clinical picture. On the contrary, we would encourage the primary team in charge of a hemodynamically unstable patient to consult dermatology at the first sign of an unexplained rash. Take for example an acutely ill patient who develops retiform purpura. There are well-established dermatology guidelines for the workup of retiform purpura,4 including prompt biopsy and assessment of broad, potentially life-threatening differential diagnoses from calciphylaxis to angioinvasive fungal infection. In this scenario, the dermatology consultant may render the correct diagnosis and recommend immediate treatment that could be lifesaving.
Secondly, we do not agree with the recommendation that a patient in hospice care is not appropriate for inpatient dermatology consultation. Patients receiving hospice or palliative care have high rates of potentially symptomatic cutaneous diseases,5 including intertrigo and dermatitis—comprising stasis, seborrheic, and contact dermatitis.6 Although aggressive intervention for asymptomatic benign or malignant skin conditions may not be in line with their goals of care, an inpatient dermatology consultation can reduce symptoms and improve quality of life. This population also is one that is unlikely to be able to attend an outpatient dermatology clinic appointment and therefore are good candidates for inpatient consultation.
Lastly, we want to highlight the difference between a stable chronic dermatologic disease and an acute flare that may occur while a patient is hospitalized, regardless of whether it is the reason for admission. For example, a patient with psoriasis affecting limited body surface area who is hospitalized for a myocardial infarction is not appropriate for a dermatology consultation. However, if that same patient develops erythroderma while they are hospitalized for cardiac monitoring, it would certainly be appropriate for dermatology to be consulted. Additionally, there are times when a chronic skin disease is the reason for hospitalization; dermatology, although technically a consulting service, would be the primary decision-maker for the patient’s care in this situation. In these scenarios, it is important for the patient to be able to establish care for long-term outpatient management of their condition; however, it is prudent to involve dermatology while the patient is acutely hospitalized to guide their treatment plan until they are able to see a dermatologist after discharge.
In conclusion, we believe that hospital dermatology is a valuable tool that can be utilized in many different scenarios. Although there are certainly situations more appropriate for outpatient dermatology referral, we would caution against overly simplified algorithms that could discourage valuable inpatient dermatology consultations. It often is worth a conversation with your dermatology consultant (when available at an institution) to determine the best course of action for each patient. Additionally, we recognize the need for more formalized guidelines on when to pursue inpatient dermatology consultation. We are members of the Society of Dermatology Hospitalists and encourage readers to reference their website, which provides additional resources on inpatient dermatology (https://societydermatologyhospitalists.com/inpatient-dermatology-literature/).
Authors’ Response
We appreciate the letter in response to our commentary on the appropriateness of inpatient dermatology consultations. It is the continued refining and re-evaluation of concepts such as these that allow our field to grow and improve knowledge and patient care.
We sought to provide a nonpatronizing yet simple consultation flowchart that would help guide triage of patients in need or not in need of dermatologic evaluation by the inpatient teams. Understandably, the impressions of our flowchart have been variable based on different readers’ medical backgrounds and experiences. It is certainly possible that our flowchart lacked certain exceptions and oversimplified certain concepts, and we welcome further refining of this flowchart to better guide inpatient dermatology consultations.
We do, however, disagree that the primary team would not know whether a patient is intubated in the intensive care unit for a dermatology reason. If the patient is in such a status, it would be pertinent for the primary team to conduct a timely workup that could include consultations until a diagnosis is made. We were not implying that every dermatology consultation in the intensive care unit is unwarranted, especially if it can lead to a primary dermatologic diagnosis. We do believe that a thorough history could elicit an allergy or other chronic skin condition that could save resources and spending within a hospital. Likewise, psoriasis comes in many different presentations, and although we do not believe a consultation for chronic psoriatic plaques is appropriate in the hospital, it is absolutely appropriate for a patient who is erythrodermic from any cause.
Our flowchart was intended to be the first step to providing education on when consultations are appropriate, and further refinement will be necessary.
Hershel Dobkin, MD; Timothy Blackwell, BS; Robin Ashinoff, MD
Drs. Dobkin and Ashinoff are from Hackensack University Medical Center, New Jersey. Mr. Blackwell is from the Rowan University School of Osteopathic Medicine, Stratford, New Jersey.
The authors report no conflict of interest.
Correspondence: Hershel Dobkin, MD, Hackensack University Medical Center, 30 Prospect Ave, Hackensack, NJ 07601 (hersheldobkinpublic@gmail.com).
- Dobkin H, Blackwell T, Ashinoff R. When are inpatient and emergency dermatologic consultations appropriate? Cutis. 2022;109:218-220. doi:10.12788/cutis.0492
- Ko LN, Garza-Mayers AC, St John J, et al. Effect of dermatology consultation on outcomes for patients with presumed cellulitis: a randomized clinical trial. JAMA Dermatol. 2018;154:529-536. doi:10.1001/jamadermatol.2017.6196
- Hu L, Haynes H, Ferrazza D, et al. Impact of specialist consultations on inpatient admissions for dermatology-specific and related DRGs. J Gen Intern Med. 2013;28:1477-1482. doi:10.1007/s11606-013-2440-2
- Georgesen C, Fox LP, Harp J. Retiform purpura: a diagnostic approach. J Am Acad Dermatol. 2020;82:783-796. doi:10.1016/j.jaad.2019.07.112
- Pisano C, Paladichuk H, Keeling B. Dermatology in palliative medicine [published online October 14, 2021]. BMJ Support Palliat Care. doi:10.1136/bmjspcare-2021-003342
- Barnabé C, Daeninck P. “Beauty is only skin deep”: prevalence of dermatologic disease on a palliative care unit. J Pain Symptom Manage. 2005;29:419-422. doi:10.1016/j.jpainsymman.2004.08.009
- Dobkin H, Blackwell T, Ashinoff R. When are inpatient and emergency dermatologic consultations appropriate? Cutis. 2022;109:218-220. doi:10.12788/cutis.0492
- Ko LN, Garza-Mayers AC, St John J, et al. Effect of dermatology consultation on outcomes for patients with presumed cellulitis: a randomized clinical trial. JAMA Dermatol. 2018;154:529-536. doi:10.1001/jamadermatol.2017.6196
- Hu L, Haynes H, Ferrazza D, et al. Impact of specialist consultations on inpatient admissions for dermatology-specific and related DRGs. J Gen Intern Med. 2013;28:1477-1482. doi:10.1007/s11606-013-2440-2
- Georgesen C, Fox LP, Harp J. Retiform purpura: a diagnostic approach. J Am Acad Dermatol. 2020;82:783-796. doi:10.1016/j.jaad.2019.07.112
- Pisano C, Paladichuk H, Keeling B. Dermatology in palliative medicine [published online October 14, 2021]. BMJ Support Palliat Care. doi:10.1136/bmjspcare-2021-003342
- Barnabé C, Daeninck P. “Beauty is only skin deep”: prevalence of dermatologic disease on a palliative care unit. J Pain Symptom Manage. 2005;29:419-422. doi:10.1016/j.jpainsymman.2004.08.009
Differences in Underrepresented in Medicine Applicant Backgrounds and Outcomes in the 2020-2021 Dermatology Residency Match
Dermatology is one of the least diverse medical specialties with only 3% of dermatologists being Black and 4% Latinx.1 Leading dermatology organizations have called for specialty-wide efforts to improve diversity, with a particular focus on the resident selection process.2,3 Medical students who are underrepresented in medicine (UIM)(ie, those who identify as Black, Latinx, Native American, or Pacific Islander) face many potential barriers in applying to dermatology programs, including financial limitations, lack of support and mentorship, and less exposure to the specialty.1,2,4 The COVID-19 pandemic introduced additional challenges in the residency application process with limitations on clinical, research, and volunteer experiences; decreased opportunities for in-person mentorship and away rotations; and a shift to virtual recruitment. Although there has been increased emphasis on recruiting diverse candidates to dermatology, the COVID-19 pandemic may have exacerbated existing barriers for UIM applicants.
We surveyed dermatology residency program directors (PDs) and applicants to evaluate how UIM students approach and fare in the dermatology residency application process as well as the effects of COVID-19 on the most recent application cycle. Herein, we report the results of our surveys with a focus on racial differences in the application process.
Methods
We administered 2 anonymous online surveys—one to 115 PDs through the Association of Professors of Dermatology (APD) email listserve and another to applicants who participated in the 2020-2021 dermatology residency application cycle through the Dermatology Interest Group Association (DIGA) listserve. The surveys were distributed from March 29 through May 23, 2021. There was no way to determine the number of dermatology applicants on the DIGA listserve. The surveys were reviewed and approved by the University of Southern California (Los Angeles, California) institutional review board (approval #UP-21-00118).
Participants were not required to answer every survey question; response rates varied by question. Survey responses with less than 10% completion were excluded from analysis. Data were collected, analyzed, and stored using Qualtrics, a secure online survey platform. The test of equal or given proportions in R studio was used to determine statistically significant differences between variables (P<.05 indicated statistical significance).
Results
The PD survey received 79 complete responses (83.5% complete responses, 73.8% response rate) and the applicant survey received 232 complete responses (83.6% complete responses).
Applicant Characteristics—Applicant characteristics are provided in the eTable; 13.2% and 8.4% of participants were Black and Latinx (including those who identify as Hispanic/Latino), respectively. Only 0.8% of respondents identified as American Indian or Alaskan Native and were excluded from the analysis due to the limited sample size. Those who identified as White, Asian, multiple races, or other and those who preferred not to answer were considered non-UIM participants.
Differences in family background were observed in our cohort, with UIM candidates more likely to have experienced disadvantages, defined as being the first in their family to attend college/graduate school, growing up in a rural area, being a first-generation immigrant, or qualifying as low income. Underrepresented in medicine applicants also were less likely to have a dermatology program at their medical school (both Black and Latinx) and to have been elected to honor societies such as Alpha Omega Alpha and the Gold Humanism Honor Society (Black only).
Underrepresented in medicine applicants were more likely to complete a research gap year (eTable). Most applicants who took research years did so to improve their chances of matching, regardless of their race/ethnicity. For those who did not complete a research year, Black applicants (46.7%) were more likely to base that decision on financial limitations compared to non-UIMs (18.6%, P<.0001). Interestingly, in the PD survey, only 4.5% of respondents considered completion of a research year extremely or very important when compiling rank lists.

Application Process and Match Outcomes—The Table highlights differences in how UIM applicants approached the application process. Black but not Latinx applicants were less likely to be first-time applicants to dermatology compared to non-UIM applicants. Black applicants (8.3%) were significantly less likely to apply to more than 100 programs compared to non-UIM applicants (29.5%, P=.0002). Underrepresented in medicine applicants received greater numbers of interviews despite applying to fewer programs overall.

There also were differences in how UIM candidates approached their rank lists, with Black and Latinx applicants prioritizing diversity of patient populations and program faculty as well as program missions and values (Figure).

In our cohort, UIM candidates were more likely than non-UIM to match, and Black applicants were most likely to match at one of their top 3 choices (Table). In the PD survey, 77.6% of PDs considered contribution to diversity an important factor when compiling their rank lists.
Comment
Applicant Background—Dermatology is a competitive specialty with a challenging application process2 that has been further complicated by the COVID-19 pandemic. Our study elucidated how the 2020-2021 application cycle affected UIM dermatology applicants. Prior studies have found that UIM medical students were more likely to come from lower socioeconomic backgrounds; financial constraints pose a major barrier for UIM and low-income students interested in dermatology.4-6 We found this to be true in our cohort, as Black and Latinx applicants were significantly more likely to come from disadvantaged backgrounds (P<.000008 and P=.006, respectively). Additionally, we found that Black applicants were more likely than any other group to indicate financial concerns as their primary reason for not taking a research gap year.
Although most applicants who completed a research year did so to increase their chances of matching, a higher percentage of UIMs took research years compared to non-UIM applicants. This finding could indicate greater anxiety about matching among UIM applicants vs their non-UIM counterparts. Black students have faced discrimination in clinical grading,7 have perceived racial discrimination in residency interviews,8,9 and have shown to be less likely to be elected to medical school honor societies.10 We found that UIM applicants were more likely to pursue a research year compared to other applicants,11 possibly because they felt additional pressure to enhance their applications or because UIM candidates were less likely to have a home dermatology program. Expansion of mentorship programs, visiting student electives, and grants for UIMs may alleviate the need for these candidates to complete a research year and reduce disparities in the application process.
Factors Influencing Rank Lists for Applicants—In our cohort, UIMs were significantly more likely to rank diversity of patients (P<.0001 for Black applicants and P=.04 for Latinx applicants) and faculty (P<.001 for Black applicants and P<.001 for Latinx applicants) as important factors in choosing a residency program. Historically, dermatology has been disproportionately White in its physician workforce and patient population.1,12 Students with lower incomes or who identify as minorities cite the lack of diversity in dermatology as a considerable barrier to pursuing a career in the specialty.4,5 Service learning, pipeline programs aimed at early exposure to dermatology, and increased access to care for diverse patient populations are important measures to improve diversity in the dermatology workforce.13-15 Residency programs should consider how to incorporate these aspects into didactic and clinical curricula to better recruit diverse candidates to the field.
Equity in the Application Process—We found that Black applicants were more likely than non-UIM applicants to be reapplicants to dermatology; however, Black applicants in our study also were more likely to receive more interview invites, match into dermatology, and match into one of their top 3 programs. These findings are interesting, particularly given concerns about equity in the application process. It is possible that Black applicants who overcome barriers to applying to dermatology ultimately are more successful applicants. Recently, there has been an increased focus in the field on diversifying dermatology, which was further intensified last year.2,3 Indicative of this shift, our PD survey showed that most programs reported that applicants’ contributions to diversity were important factors in the application process. Additionally, an emphasis by PDs on a holistic review of applications coupled with direct advocacy for increased representation may have contributed to the increased match rates for UIM applicants reported in our survey.
Latinx Applicants—Our study showed differences in how Latinx candidates fared in the application process; although Latinx applicants were more likely than their non-Latinx counterparts to match into dermatology, they were less likely than non-Latinx applicants to match into one of their top 3 programs. Given that Latinx encompasses ethnicity, not race, there may be a difference in how intentional focus on and advocacy for increasing diversity in dermatology affected different UIM applicant groups. Both race and ethnicity are social constructs rather than scientific categorizations; thus, it is difficult in survey studies such as ours to capture the intersectionality present across and between groups. Lastly, it is possible that the respondents to our applicant survey are not representative of the full cohort of UIM applicants.
Study Limitations—A major limitation of our study was that we did not have a method of reaching all dermatology applicants. Although our study shows promising results suggestive of increased diversity in the last application cycle, release of the National Resident Matching Program results from 2020-2021 with racially stratified data will be imperative to assess equity in the match process for all specialties and to confirm the generalizability of our results.
- Pandya AG, Alexis AF, Berger TG, et al. Increasing racial and ethnic diversity in dermatology: a call to action. J Am Acad Dermatol. 2016;74:584-587. doi:10.1016/j.jaad.2015.10.044
- Chen A, Shinkai K. Rethinking how we select dermatology applicants—turning the tide. JAMA Dermatol. 2017;153:259-260. doi:10.1001/jamadermatol.2016.4683
- American Academy of Dermatology Association. Diversity In Dermatology: Diversity Committee Approved Plan 2021-2023. Published January 26, 2021. Accessed July 26, 2022. https://assets.ctfassets.net/1ny4yoiyrqia/xQgnCE6ji5skUlcZQHS2b/65f0a9072811e11afcc33d043e02cd4d/DEI_Plan.pdf
- Vasquez R, Jeong H, Florez-Pollack S, et al. What are the barriers faced by under-represented minorities applying to dermatology? a qualitative cross-sectional study of applicants applying to a large dermatologyresidency program. J Am Acad Dermatol. 2020;83:1770-1773. doi:10.1016/j.jaad.2020.03.067
- Jones VA, Clark KA, Patel PM, et al. Considerations for dermatology residency applicants underrepresented in medicine amid the COVID-19 pandemic. J Am Acad Dermatol. 2020;83:E247.doi:10.1016/j.jaad.2020.05.141
- Soliman YS, Rzepecki AK, Guzman AK, et al. Understanding perceived barriers of minority medical students pursuing a career in dermatology. JAMA Dermatol. 2019;155:252-254. doi:10.1001/jamadermatol.2018.4813
- Grbic D, Jones DJ, Case ST. The role of socioeconomic status in medical school admissions: validation of a socioeconomic indicator for use in medical school admissions. Acad Med. 2015;90:953-960. doi:10.1097/ACM.0000000000000653
- Low D, Pollack SW, Liao ZC, et al. Racial/ethnic disparities in clinical grading in medical school. Teach Learn Med. 2019;31:487-496. doi:10.1080/10401334.2019.1597724
- Ellis J, Otugo O, Landry A, et al. Interviewed while Black [published online November 11, 2020]. N Engl J Med. 2020;383:2401-2404. doi:10.1056/NEJMp2023999
- Anthony Douglas II, Hendrix J. Black medical student considerations in the era of virtual interviews. Ann Surg. 2021;274:232-233. doi:10.1097/SLA.0000000000004946
- Boatright D, Ross D, O’Connor P, et al. Racial disparities in medical student membership in the Alpha Omega Alpha honor society. JAMA Intern Med. 2017;177:659. doi:10.1001/jamainternmed.2016.9623
- Runge M, Renati S, Helfrich Y. 16146 dermatology residency applicants: how many pursue a dedicated research year or dual-degree, and do their stats differ [published online December 1, 2020]? J Am Acad Dermatol. doi:10.1016/j.jaad.2020.06.304
- Stern RS. Dermatologists and office-based care of dermatologic disease in the 21st century. J Investig Dermatol Symp Proc. 2004;9:126-130. doi:10.1046/j.1087-0024.2003.09108.x
- Oyesanya T, Grossberg AL, Okoye GA. Increasing minority representation in the dermatology department: the Johns Hopkins experience. JAMA Dermatol. 2018;154:1133-1134. doi:10.1001/jamadermatol.2018.2018
- Humphrey VS, James AJ. The importance of service learning in dermatology residency: an actionable approach to improve resident education and skin health equity. Cutis. 2021;107:120-122. doi:10.12788/cutis.0199
Dermatology is one of the least diverse medical specialties with only 3% of dermatologists being Black and 4% Latinx.1 Leading dermatology organizations have called for specialty-wide efforts to improve diversity, with a particular focus on the resident selection process.2,3 Medical students who are underrepresented in medicine (UIM)(ie, those who identify as Black, Latinx, Native American, or Pacific Islander) face many potential barriers in applying to dermatology programs, including financial limitations, lack of support and mentorship, and less exposure to the specialty.1,2,4 The COVID-19 pandemic introduced additional challenges in the residency application process with limitations on clinical, research, and volunteer experiences; decreased opportunities for in-person mentorship and away rotations; and a shift to virtual recruitment. Although there has been increased emphasis on recruiting diverse candidates to dermatology, the COVID-19 pandemic may have exacerbated existing barriers for UIM applicants.
We surveyed dermatology residency program directors (PDs) and applicants to evaluate how UIM students approach and fare in the dermatology residency application process as well as the effects of COVID-19 on the most recent application cycle. Herein, we report the results of our surveys with a focus on racial differences in the application process.
Methods
We administered 2 anonymous online surveys—one to 115 PDs through the Association of Professors of Dermatology (APD) email listserve and another to applicants who participated in the 2020-2021 dermatology residency application cycle through the Dermatology Interest Group Association (DIGA) listserve. The surveys were distributed from March 29 through May 23, 2021. There was no way to determine the number of dermatology applicants on the DIGA listserve. The surveys were reviewed and approved by the University of Southern California (Los Angeles, California) institutional review board (approval #UP-21-00118).
Participants were not required to answer every survey question; response rates varied by question. Survey responses with less than 10% completion were excluded from analysis. Data were collected, analyzed, and stored using Qualtrics, a secure online survey platform. The test of equal or given proportions in R studio was used to determine statistically significant differences between variables (P<.05 indicated statistical significance).
Results
The PD survey received 79 complete responses (83.5% complete responses, 73.8% response rate) and the applicant survey received 232 complete responses (83.6% complete responses).
Applicant Characteristics—Applicant characteristics are provided in the eTable; 13.2% and 8.4% of participants were Black and Latinx (including those who identify as Hispanic/Latino), respectively. Only 0.8% of respondents identified as American Indian or Alaskan Native and were excluded from the analysis due to the limited sample size. Those who identified as White, Asian, multiple races, or other and those who preferred not to answer were considered non-UIM participants.
Differences in family background were observed in our cohort, with UIM candidates more likely to have experienced disadvantages, defined as being the first in their family to attend college/graduate school, growing up in a rural area, being a first-generation immigrant, or qualifying as low income. Underrepresented in medicine applicants also were less likely to have a dermatology program at their medical school (both Black and Latinx) and to have been elected to honor societies such as Alpha Omega Alpha and the Gold Humanism Honor Society (Black only).
Underrepresented in medicine applicants were more likely to complete a research gap year (eTable). Most applicants who took research years did so to improve their chances of matching, regardless of their race/ethnicity. For those who did not complete a research year, Black applicants (46.7%) were more likely to base that decision on financial limitations compared to non-UIMs (18.6%, P<.0001). Interestingly, in the PD survey, only 4.5% of respondents considered completion of a research year extremely or very important when compiling rank lists.
Application Process and Match Outcomes—The Table highlights differences in how UIM applicants approached the application process. Black but not Latinx applicants were less likely to be first-time applicants to dermatology compared to non-UIM applicants. Black applicants (8.3%) were significantly less likely to apply to more than 100 programs compared to non-UIM applicants (29.5%, P=.0002). Underrepresented in medicine applicants received greater numbers of interviews despite applying to fewer programs overall.
There also were differences in how UIM candidates approached their rank lists, with Black and Latinx applicants prioritizing diversity of patient populations and program faculty as well as program missions and values (Figure).
In our cohort, UIM candidates were more likely than non-UIM to match, and Black applicants were most likely to match at one of their top 3 choices (Table). In the PD survey, 77.6% of PDs considered contribution to diversity an important factor when compiling their rank lists.
Comment
Applicant Background—Dermatology is a competitive specialty with a challenging application process2 that has been further complicated by the COVID-19 pandemic. Our study elucidated how the 2020-2021 application cycle affected UIM dermatology applicants. Prior studies have found that UIM medical students were more likely to come from lower socioeconomic backgrounds; financial constraints pose a major barrier for UIM and low-income students interested in dermatology.4-6 We found this to be true in our cohort, as Black and Latinx applicants were significantly more likely to come from disadvantaged backgrounds (P<.000008 and P=.006, respectively). Additionally, we found that Black applicants were more likely than any other group to indicate financial concerns as their primary reason for not taking a research gap year.
Although most applicants who completed a research year did so to increase their chances of matching, a higher percentage of UIMs took research years compared to non-UIM applicants. This finding could indicate greater anxiety about matching among UIM applicants vs their non-UIM counterparts. Black students have faced discrimination in clinical grading,7 have perceived racial discrimination in residency interviews,8,9 and have shown to be less likely to be elected to medical school honor societies.10 We found that UIM applicants were more likely to pursue a research year compared to other applicants,11 possibly because they felt additional pressure to enhance their applications or because UIM candidates were less likely to have a home dermatology program. Expansion of mentorship programs, visiting student electives, and grants for UIMs may alleviate the need for these candidates to complete a research year and reduce disparities in the application process.
Factors Influencing Rank Lists for Applicants—In our cohort, UIMs were significantly more likely to rank diversity of patients (P<.0001 for Black applicants and P=.04 for Latinx applicants) and faculty (P<.001 for Black applicants and P<.001 for Latinx applicants) as important factors in choosing a residency program. Historically, dermatology has been disproportionately White in its physician workforce and patient population.1,12 Students with lower incomes or who identify as minorities cite the lack of diversity in dermatology as a considerable barrier to pursuing a career in the specialty.4,5 Service learning, pipeline programs aimed at early exposure to dermatology, and increased access to care for diverse patient populations are important measures to improve diversity in the dermatology workforce.13-15 Residency programs should consider how to incorporate these aspects into didactic and clinical curricula to better recruit diverse candidates to the field.
Equity in the Application Process—We found that Black applicants were more likely than non-UIM applicants to be reapplicants to dermatology; however, Black applicants in our study also were more likely to receive more interview invites, match into dermatology, and match into one of their top 3 programs. These findings are interesting, particularly given concerns about equity in the application process. It is possible that Black applicants who overcome barriers to applying to dermatology ultimately are more successful applicants. Recently, there has been an increased focus in the field on diversifying dermatology, which was further intensified last year.2,3 Indicative of this shift, our PD survey showed that most programs reported that applicants’ contributions to diversity were important factors in the application process. Additionally, an emphasis by PDs on a holistic review of applications coupled with direct advocacy for increased representation may have contributed to the increased match rates for UIM applicants reported in our survey.
Latinx Applicants—Our study showed differences in how Latinx candidates fared in the application process; although Latinx applicants were more likely than their non-Latinx counterparts to match into dermatology, they were less likely than non-Latinx applicants to match into one of their top 3 programs. Given that Latinx encompasses ethnicity, not race, there may be a difference in how intentional focus on and advocacy for increasing diversity in dermatology affected different UIM applicant groups. Both race and ethnicity are social constructs rather than scientific categorizations; thus, it is difficult in survey studies such as ours to capture the intersectionality present across and between groups. Lastly, it is possible that the respondents to our applicant survey are not representative of the full cohort of UIM applicants.
Study Limitations—A major limitation of our study was that we did not have a method of reaching all dermatology applicants. Although our study shows promising results suggestive of increased diversity in the last application cycle, release of the National Resident Matching Program results from 2020-2021 with racially stratified data will be imperative to assess equity in the match process for all specialties and to confirm the generalizability of our results.
Dermatology is one of the least diverse medical specialties with only 3% of dermatologists being Black and 4% Latinx.1 Leading dermatology organizations have called for specialty-wide efforts to improve diversity, with a particular focus on the resident selection process.2,3 Medical students who are underrepresented in medicine (UIM)(ie, those who identify as Black, Latinx, Native American, or Pacific Islander) face many potential barriers in applying to dermatology programs, including financial limitations, lack of support and mentorship, and less exposure to the specialty.1,2,4 The COVID-19 pandemic introduced additional challenges in the residency application process with limitations on clinical, research, and volunteer experiences; decreased opportunities for in-person mentorship and away rotations; and a shift to virtual recruitment. Although there has been increased emphasis on recruiting diverse candidates to dermatology, the COVID-19 pandemic may have exacerbated existing barriers for UIM applicants.
We surveyed dermatology residency program directors (PDs) and applicants to evaluate how UIM students approach and fare in the dermatology residency application process as well as the effects of COVID-19 on the most recent application cycle. Herein, we report the results of our surveys with a focus on racial differences in the application process.
Methods
We administered 2 anonymous online surveys—one to 115 PDs through the Association of Professors of Dermatology (APD) email listserve and another to applicants who participated in the 2020-2021 dermatology residency application cycle through the Dermatology Interest Group Association (DIGA) listserve. The surveys were distributed from March 29 through May 23, 2021. There was no way to determine the number of dermatology applicants on the DIGA listserve. The surveys were reviewed and approved by the University of Southern California (Los Angeles, California) institutional review board (approval #UP-21-00118).
Participants were not required to answer every survey question; response rates varied by question. Survey responses with less than 10% completion were excluded from analysis. Data were collected, analyzed, and stored using Qualtrics, a secure online survey platform. The test of equal or given proportions in R studio was used to determine statistically significant differences between variables (P<.05 indicated statistical significance).
Results
The PD survey received 79 complete responses (83.5% complete responses, 73.8% response rate) and the applicant survey received 232 complete responses (83.6% complete responses).
Applicant Characteristics—Applicant characteristics are provided in the eTable; 13.2% and 8.4% of participants were Black and Latinx (including those who identify as Hispanic/Latino), respectively. Only 0.8% of respondents identified as American Indian or Alaskan Native and were excluded from the analysis due to the limited sample size. Those who identified as White, Asian, multiple races, or other and those who preferred not to answer were considered non-UIM participants.
Differences in family background were observed in our cohort, with UIM candidates more likely to have experienced disadvantages, defined as being the first in their family to attend college/graduate school, growing up in a rural area, being a first-generation immigrant, or qualifying as low income. Underrepresented in medicine applicants also were less likely to have a dermatology program at their medical school (both Black and Latinx) and to have been elected to honor societies such as Alpha Omega Alpha and the Gold Humanism Honor Society (Black only).
Underrepresented in medicine applicants were more likely to complete a research gap year (eTable). Most applicants who took research years did so to improve their chances of matching, regardless of their race/ethnicity. For those who did not complete a research year, Black applicants (46.7%) were more likely to base that decision on financial limitations compared to non-UIMs (18.6%, P<.0001). Interestingly, in the PD survey, only 4.5% of respondents considered completion of a research year extremely or very important when compiling rank lists.
Application Process and Match Outcomes—The Table highlights differences in how UIM applicants approached the application process. Black but not Latinx applicants were less likely to be first-time applicants to dermatology compared to non-UIM applicants. Black applicants (8.3%) were significantly less likely to apply to more than 100 programs compared to non-UIM applicants (29.5%, P=.0002). Underrepresented in medicine applicants received greater numbers of interviews despite applying to fewer programs overall.
There also were differences in how UIM candidates approached their rank lists, with Black and Latinx applicants prioritizing diversity of patient populations and program faculty as well as program missions and values (Figure).
In our cohort, UIM candidates were more likely than non-UIM to match, and Black applicants were most likely to match at one of their top 3 choices (Table). In the PD survey, 77.6% of PDs considered contribution to diversity an important factor when compiling their rank lists.
Comment
Applicant Background—Dermatology is a competitive specialty with a challenging application process2 that has been further complicated by the COVID-19 pandemic. Our study elucidated how the 2020-2021 application cycle affected UIM dermatology applicants. Prior studies have found that UIM medical students were more likely to come from lower socioeconomic backgrounds; financial constraints pose a major barrier for UIM and low-income students interested in dermatology.4-6 We found this to be true in our cohort, as Black and Latinx applicants were significantly more likely to come from disadvantaged backgrounds (P<.000008 and P=.006, respectively). Additionally, we found that Black applicants were more likely than any other group to indicate financial concerns as their primary reason for not taking a research gap year.
Although most applicants who completed a research year did so to increase their chances of matching, a higher percentage of UIMs took research years compared to non-UIM applicants. This finding could indicate greater anxiety about matching among UIM applicants vs their non-UIM counterparts. Black students have faced discrimination in clinical grading,7 have perceived racial discrimination in residency interviews,8,9 and have shown to be less likely to be elected to medical school honor societies.10 We found that UIM applicants were more likely to pursue a research year compared to other applicants,11 possibly because they felt additional pressure to enhance their applications or because UIM candidates were less likely to have a home dermatology program. Expansion of mentorship programs, visiting student electives, and grants for UIMs may alleviate the need for these candidates to complete a research year and reduce disparities in the application process.
Factors Influencing Rank Lists for Applicants—In our cohort, UIMs were significantly more likely to rank diversity of patients (P<.0001 for Black applicants and P=.04 for Latinx applicants) and faculty (P<.001 for Black applicants and P<.001 for Latinx applicants) as important factors in choosing a residency program. Historically, dermatology has been disproportionately White in its physician workforce and patient population.1,12 Students with lower incomes or who identify as minorities cite the lack of diversity in dermatology as a considerable barrier to pursuing a career in the specialty.4,5 Service learning, pipeline programs aimed at early exposure to dermatology, and increased access to care for diverse patient populations are important measures to improve diversity in the dermatology workforce.13-15 Residency programs should consider how to incorporate these aspects into didactic and clinical curricula to better recruit diverse candidates to the field.
Equity in the Application Process—We found that Black applicants were more likely than non-UIM applicants to be reapplicants to dermatology; however, Black applicants in our study also were more likely to receive more interview invites, match into dermatology, and match into one of their top 3 programs. These findings are interesting, particularly given concerns about equity in the application process. It is possible that Black applicants who overcome barriers to applying to dermatology ultimately are more successful applicants. Recently, there has been an increased focus in the field on diversifying dermatology, which was further intensified last year.2,3 Indicative of this shift, our PD survey showed that most programs reported that applicants’ contributions to diversity were important factors in the application process. Additionally, an emphasis by PDs on a holistic review of applications coupled with direct advocacy for increased representation may have contributed to the increased match rates for UIM applicants reported in our survey.
Latinx Applicants—Our study showed differences in how Latinx candidates fared in the application process; although Latinx applicants were more likely than their non-Latinx counterparts to match into dermatology, they were less likely than non-Latinx applicants to match into one of their top 3 programs. Given that Latinx encompasses ethnicity, not race, there may be a difference in how intentional focus on and advocacy for increasing diversity in dermatology affected different UIM applicant groups. Both race and ethnicity are social constructs rather than scientific categorizations; thus, it is difficult in survey studies such as ours to capture the intersectionality present across and between groups. Lastly, it is possible that the respondents to our applicant survey are not representative of the full cohort of UIM applicants.
Study Limitations—A major limitation of our study was that we did not have a method of reaching all dermatology applicants. Although our study shows promising results suggestive of increased diversity in the last application cycle, release of the National Resident Matching Program results from 2020-2021 with racially stratified data will be imperative to assess equity in the match process for all specialties and to confirm the generalizability of our results.
- Pandya AG, Alexis AF, Berger TG, et al. Increasing racial and ethnic diversity in dermatology: a call to action. J Am Acad Dermatol. 2016;74:584-587. doi:10.1016/j.jaad.2015.10.044
- Chen A, Shinkai K. Rethinking how we select dermatology applicants—turning the tide. JAMA Dermatol. 2017;153:259-260. doi:10.1001/jamadermatol.2016.4683
- American Academy of Dermatology Association. Diversity In Dermatology: Diversity Committee Approved Plan 2021-2023. Published January 26, 2021. Accessed July 26, 2022. https://assets.ctfassets.net/1ny4yoiyrqia/xQgnCE6ji5skUlcZQHS2b/65f0a9072811e11afcc33d043e02cd4d/DEI_Plan.pdf
- Vasquez R, Jeong H, Florez-Pollack S, et al. What are the barriers faced by under-represented minorities applying to dermatology? a qualitative cross-sectional study of applicants applying to a large dermatologyresidency program. J Am Acad Dermatol. 2020;83:1770-1773. doi:10.1016/j.jaad.2020.03.067
- Jones VA, Clark KA, Patel PM, et al. Considerations for dermatology residency applicants underrepresented in medicine amid the COVID-19 pandemic. J Am Acad Dermatol. 2020;83:E247.doi:10.1016/j.jaad.2020.05.141
- Soliman YS, Rzepecki AK, Guzman AK, et al. Understanding perceived barriers of minority medical students pursuing a career in dermatology. JAMA Dermatol. 2019;155:252-254. doi:10.1001/jamadermatol.2018.4813
- Grbic D, Jones DJ, Case ST. The role of socioeconomic status in medical school admissions: validation of a socioeconomic indicator for use in medical school admissions. Acad Med. 2015;90:953-960. doi:10.1097/ACM.0000000000000653
- Low D, Pollack SW, Liao ZC, et al. Racial/ethnic disparities in clinical grading in medical school. Teach Learn Med. 2019;31:487-496. doi:10.1080/10401334.2019.1597724
- Ellis J, Otugo O, Landry A, et al. Interviewed while Black [published online November 11, 2020]. N Engl J Med. 2020;383:2401-2404. doi:10.1056/NEJMp2023999
- Anthony Douglas II, Hendrix J. Black medical student considerations in the era of virtual interviews. Ann Surg. 2021;274:232-233. doi:10.1097/SLA.0000000000004946
- Boatright D, Ross D, O’Connor P, et al. Racial disparities in medical student membership in the Alpha Omega Alpha honor society. JAMA Intern Med. 2017;177:659. doi:10.1001/jamainternmed.2016.9623
- Runge M, Renati S, Helfrich Y. 16146 dermatology residency applicants: how many pursue a dedicated research year or dual-degree, and do their stats differ [published online December 1, 2020]? J Am Acad Dermatol. doi:10.1016/j.jaad.2020.06.304
- Stern RS. Dermatologists and office-based care of dermatologic disease in the 21st century. J Investig Dermatol Symp Proc. 2004;9:126-130. doi:10.1046/j.1087-0024.2003.09108.x
- Oyesanya T, Grossberg AL, Okoye GA. Increasing minority representation in the dermatology department: the Johns Hopkins experience. JAMA Dermatol. 2018;154:1133-1134. doi:10.1001/jamadermatol.2018.2018
- Humphrey VS, James AJ. The importance of service learning in dermatology residency: an actionable approach to improve resident education and skin health equity. Cutis. 2021;107:120-122. doi:10.12788/cutis.0199
- Pandya AG, Alexis AF, Berger TG, et al. Increasing racial and ethnic diversity in dermatology: a call to action. J Am Acad Dermatol. 2016;74:584-587. doi:10.1016/j.jaad.2015.10.044
- Chen A, Shinkai K. Rethinking how we select dermatology applicants—turning the tide. JAMA Dermatol. 2017;153:259-260. doi:10.1001/jamadermatol.2016.4683
- American Academy of Dermatology Association. Diversity In Dermatology: Diversity Committee Approved Plan 2021-2023. Published January 26, 2021. Accessed July 26, 2022. https://assets.ctfassets.net/1ny4yoiyrqia/xQgnCE6ji5skUlcZQHS2b/65f0a9072811e11afcc33d043e02cd4d/DEI_Plan.pdf
- Vasquez R, Jeong H, Florez-Pollack S, et al. What are the barriers faced by under-represented minorities applying to dermatology? a qualitative cross-sectional study of applicants applying to a large dermatologyresidency program. J Am Acad Dermatol. 2020;83:1770-1773. doi:10.1016/j.jaad.2020.03.067
- Jones VA, Clark KA, Patel PM, et al. Considerations for dermatology residency applicants underrepresented in medicine amid the COVID-19 pandemic. J Am Acad Dermatol. 2020;83:E247.doi:10.1016/j.jaad.2020.05.141
- Soliman YS, Rzepecki AK, Guzman AK, et al. Understanding perceived barriers of minority medical students pursuing a career in dermatology. JAMA Dermatol. 2019;155:252-254. doi:10.1001/jamadermatol.2018.4813
- Grbic D, Jones DJ, Case ST. The role of socioeconomic status in medical school admissions: validation of a socioeconomic indicator for use in medical school admissions. Acad Med. 2015;90:953-960. doi:10.1097/ACM.0000000000000653
- Low D, Pollack SW, Liao ZC, et al. Racial/ethnic disparities in clinical grading in medical school. Teach Learn Med. 2019;31:487-496. doi:10.1080/10401334.2019.1597724
- Ellis J, Otugo O, Landry A, et al. Interviewed while Black [published online November 11, 2020]. N Engl J Med. 2020;383:2401-2404. doi:10.1056/NEJMp2023999
- Anthony Douglas II, Hendrix J. Black medical student considerations in the era of virtual interviews. Ann Surg. 2021;274:232-233. doi:10.1097/SLA.0000000000004946
- Boatright D, Ross D, O’Connor P, et al. Racial disparities in medical student membership in the Alpha Omega Alpha honor society. JAMA Intern Med. 2017;177:659. doi:10.1001/jamainternmed.2016.9623
- Runge M, Renati S, Helfrich Y. 16146 dermatology residency applicants: how many pursue a dedicated research year or dual-degree, and do their stats differ [published online December 1, 2020]? J Am Acad Dermatol. doi:10.1016/j.jaad.2020.06.304
- Stern RS. Dermatologists and office-based care of dermatologic disease in the 21st century. J Investig Dermatol Symp Proc. 2004;9:126-130. doi:10.1046/j.1087-0024.2003.09108.x
- Oyesanya T, Grossberg AL, Okoye GA. Increasing minority representation in the dermatology department: the Johns Hopkins experience. JAMA Dermatol. 2018;154:1133-1134. doi:10.1001/jamadermatol.2018.2018
- Humphrey VS, James AJ. The importance of service learning in dermatology residency: an actionable approach to improve resident education and skin health equity. Cutis. 2021;107:120-122. doi:10.12788/cutis.0199
Practice Points
- Underrepresented in medicine (UIM) dermatology residency applicants (Black and Latinx) are more likely to come from disadvantaged backgrounds and to have financial concerns about the residency application process.
- When choosing a dermatology residency program, diversity of patients and faculty are more important to UIM dermatology residency applicants than to their non-UIM counterparts.
- Increased awareness of and focus on a holistic review process by dermatology residency programs may contribute to higher rates of matching among Black applicants in our study.
Residency Roundup: Introducing a New Partnership Between Cutis and the APD-RPDS
We are excited to announce a new partnership between Cutis and the Association of Professors of Dermatology Residency Program Directors Section (APD-RPDS). The new APD-RPDS column Residency Roundup will contain quarterly communications and submissions that we hope will facilitate greater dissemination of information that is useful to the dermatology teaching community.
The APD is a group of academic dermatologists whose membership comprises chairs, chiefs, residency and fellowship program directors, and teaching faculty. Each fall, the group convenes in Chicago, Illinois, for a 2-day meeting centered around departmental and program leadership with a focus on education. The APD-RPDS was formed in 2020 and is led by a steering committee of 9 members, including our current Chair, Ilana S. Rosman, MD (Washington University School of Medicine, St. Louis, Missouri), and Vice Chair, Jo-Ann M. Latkowski, MD (New York University, New York). Committee members are elected from and by the APD membership and must serve in program leadership at their home programs. The APD-RPDS helps plan and coordinate breakout sessions and lectures at the annual APD meeting, which typically relate to program director duties, changing policies within the American Board of Dermatology or Accreditation Council for Graduate Medical Education, ideas for future growth, and changes in our specialty and in resident education. Members of the APD-RPDS have access to the APD listserv, a valuable resource for discussing issues affecting residency training. We also have work groups led by our members, which include diversity, equity, and inclusion; resource development; communications; and the annual survey. To join the APD, the RPDS, and/or any of our workgroups, please reach out to us or visit the APD website (https://www.dermatologyprofessors.org).
We look forward to welcoming and expediently reviewing members’ submissions to the new Residency Roundup column falling into 2 principal categories within the scope of dermatologic recruitment, didactic education, and clinical training. The first category will feature novel tools, programs, and platforms to improve dermatology training through collaboration. This could entail a description of a new platform designed for sharing resources among programs and specialties to enhance learning for trainees and faculty alike. For example, if a database is created that contains prerecorded lectures pertaining to alopecia, a potential article submission might introduce the database and provide information on what topics are covered and how to access these lectures for readers worldwide. Likewise, if a new technology emerges that allows for easier collaboration among programs, a possible submission would introduce the technology and discuss its potential benefits to trainees, faculty, and practicing dermatologists.
Secondly and more commonly, we anticipate the Residency Roundup column will feature articles that delve into the critical issues and challenges currently impacting recruitment, training, and administration in dermatology residency programs. Specific topics may include but are not limited to recruitment of underrepresented in medicine applicants to dermatology, technological advances to improve teaching methods within training programs, surveys delving into the dermatology match process, and educational gaps or future directions in the specialty. The column occasionally may be used to disseminate information from our section of the APD, including consensus statements or editorials related to changes implemented in the dermatology residency application process. A prospective editorial on this subject could explore varying viewpoints of implemented and proposed changes as well as the reasons behind the changes.
Our group is collaborative, and our aim is to improve education, equity, management of program director responsibilities, and the dermatology application process for programs and applicants alike. With your input, experience, and varied perspectives, we look forward to moving the field of dermatology to a better future by working together.
We are excited to announce a new partnership between Cutis and the Association of Professors of Dermatology Residency Program Directors Section (APD-RPDS). The new APD-RPDS column Residency Roundup will contain quarterly communications and submissions that we hope will facilitate greater dissemination of information that is useful to the dermatology teaching community.
The APD is a group of academic dermatologists whose membership comprises chairs, chiefs, residency and fellowship program directors, and teaching faculty. Each fall, the group convenes in Chicago, Illinois, for a 2-day meeting centered around departmental and program leadership with a focus on education. The APD-RPDS was formed in 2020 and is led by a steering committee of 9 members, including our current Chair, Ilana S. Rosman, MD (Washington University School of Medicine, St. Louis, Missouri), and Vice Chair, Jo-Ann M. Latkowski, MD (New York University, New York). Committee members are elected from and by the APD membership and must serve in program leadership at their home programs. The APD-RPDS helps plan and coordinate breakout sessions and lectures at the annual APD meeting, which typically relate to program director duties, changing policies within the American Board of Dermatology or Accreditation Council for Graduate Medical Education, ideas for future growth, and changes in our specialty and in resident education. Members of the APD-RPDS have access to the APD listserv, a valuable resource for discussing issues affecting residency training. We also have work groups led by our members, which include diversity, equity, and inclusion; resource development; communications; and the annual survey. To join the APD, the RPDS, and/or any of our workgroups, please reach out to us or visit the APD website (https://www.dermatologyprofessors.org).
We look forward to welcoming and expediently reviewing members’ submissions to the new Residency Roundup column falling into 2 principal categories within the scope of dermatologic recruitment, didactic education, and clinical training. The first category will feature novel tools, programs, and platforms to improve dermatology training through collaboration. This could entail a description of a new platform designed for sharing resources among programs and specialties to enhance learning for trainees and faculty alike. For example, if a database is created that contains prerecorded lectures pertaining to alopecia, a potential article submission might introduce the database and provide information on what topics are covered and how to access these lectures for readers worldwide. Likewise, if a new technology emerges that allows for easier collaboration among programs, a possible submission would introduce the technology and discuss its potential benefits to trainees, faculty, and practicing dermatologists.
Secondly and more commonly, we anticipate the Residency Roundup column will feature articles that delve into the critical issues and challenges currently impacting recruitment, training, and administration in dermatology residency programs. Specific topics may include but are not limited to recruitment of underrepresented in medicine applicants to dermatology, technological advances to improve teaching methods within training programs, surveys delving into the dermatology match process, and educational gaps or future directions in the specialty. The column occasionally may be used to disseminate information from our section of the APD, including consensus statements or editorials related to changes implemented in the dermatology residency application process. A prospective editorial on this subject could explore varying viewpoints of implemented and proposed changes as well as the reasons behind the changes.
Our group is collaborative, and our aim is to improve education, equity, management of program director responsibilities, and the dermatology application process for programs and applicants alike. With your input, experience, and varied perspectives, we look forward to moving the field of dermatology to a better future by working together.
We are excited to announce a new partnership between Cutis and the Association of Professors of Dermatology Residency Program Directors Section (APD-RPDS). The new APD-RPDS column Residency Roundup will contain quarterly communications and submissions that we hope will facilitate greater dissemination of information that is useful to the dermatology teaching community.
The APD is a group of academic dermatologists whose membership comprises chairs, chiefs, residency and fellowship program directors, and teaching faculty. Each fall, the group convenes in Chicago, Illinois, for a 2-day meeting centered around departmental and program leadership with a focus on education. The APD-RPDS was formed in 2020 and is led by a steering committee of 9 members, including our current Chair, Ilana S. Rosman, MD (Washington University School of Medicine, St. Louis, Missouri), and Vice Chair, Jo-Ann M. Latkowski, MD (New York University, New York). Committee members are elected from and by the APD membership and must serve in program leadership at their home programs. The APD-RPDS helps plan and coordinate breakout sessions and lectures at the annual APD meeting, which typically relate to program director duties, changing policies within the American Board of Dermatology or Accreditation Council for Graduate Medical Education, ideas for future growth, and changes in our specialty and in resident education. Members of the APD-RPDS have access to the APD listserv, a valuable resource for discussing issues affecting residency training. We also have work groups led by our members, which include diversity, equity, and inclusion; resource development; communications; and the annual survey. To join the APD, the RPDS, and/or any of our workgroups, please reach out to us or visit the APD website (https://www.dermatologyprofessors.org).
We look forward to welcoming and expediently reviewing members’ submissions to the new Residency Roundup column falling into 2 principal categories within the scope of dermatologic recruitment, didactic education, and clinical training. The first category will feature novel tools, programs, and platforms to improve dermatology training through collaboration. This could entail a description of a new platform designed for sharing resources among programs and specialties to enhance learning for trainees and faculty alike. For example, if a database is created that contains prerecorded lectures pertaining to alopecia, a potential article submission might introduce the database and provide information on what topics are covered and how to access these lectures for readers worldwide. Likewise, if a new technology emerges that allows for easier collaboration among programs, a possible submission would introduce the technology and discuss its potential benefits to trainees, faculty, and practicing dermatologists.
Secondly and more commonly, we anticipate the Residency Roundup column will feature articles that delve into the critical issues and challenges currently impacting recruitment, training, and administration in dermatology residency programs. Specific topics may include but are not limited to recruitment of underrepresented in medicine applicants to dermatology, technological advances to improve teaching methods within training programs, surveys delving into the dermatology match process, and educational gaps or future directions in the specialty. The column occasionally may be used to disseminate information from our section of the APD, including consensus statements or editorials related to changes implemented in the dermatology residency application process. A prospective editorial on this subject could explore varying viewpoints of implemented and proposed changes as well as the reasons behind the changes.
Our group is collaborative, and our aim is to improve education, equity, management of program director responsibilities, and the dermatology application process for programs and applicants alike. With your input, experience, and varied perspectives, we look forward to moving the field of dermatology to a better future by working together.
Scleral Plaques in Nephrogenic Systemic Fibrosis
To the Editor:
A 44-year-old man with a history of systemic lupus erythematosus (SLE) complicated by lupus nephritis, end-stage renal disease, and antiphospholipid syndrome was evaluated for progressive skin tightening over the last 3 years, predominantly on the hands but also involving the feet, legs, and arms. Physical examination revealed multiple flesh-colored to hypopigmented, bound-down, indurated, fissured plaques over the distal upper and lower extremities, most prominent over the hands (Figure 1). Yellow plaques appeared on the lateral sclera of both eyes (Figure 2). A diagnosis of nephrogenic systemic fibrosis (NSF) was supported by typical findings on punch biopsy, including a proliferation of dermal fibroblasts with thickened collagen bundles and mucin deposition.

Nephrogenic systemic fibrosis, also known as nephrogenic fibrosing dermopathy, is characterized by fibrotic plaques and nodules that tend to be bilateral.1 The chronic course of this disease often is accompanied by flexion contractures. Yellow scleral plaques caused by calcium phosphate deposition are present in up to 75% of cases and are more specific to a diagnosis of NSF in patients younger than 45 years.1,2 A strong association exists between NSF and gadolinium contrast agents in patients with acute renal failure; our patient later confirmed multiple gadolinium exposures years prior. Deposits of gadolinium have even been found in NSF skin lesions.2

- Stone JH. A Clinician’s Pearls & Myths in Rheumatology. Springer London; 2009.
- Barker-Griffith A, Goldberg J, Abraham JL. Ocular pathologic features and gadolinium deposition in nephrogenic systemic fibrosis. Arch Ophthalmol. 2011;129:661-663.
To the Editor:
A 44-year-old man with a history of systemic lupus erythematosus (SLE) complicated by lupus nephritis, end-stage renal disease, and antiphospholipid syndrome was evaluated for progressive skin tightening over the last 3 years, predominantly on the hands but also involving the feet, legs, and arms. Physical examination revealed multiple flesh-colored to hypopigmented, bound-down, indurated, fissured plaques over the distal upper and lower extremities, most prominent over the hands (Figure 1). Yellow plaques appeared on the lateral sclera of both eyes (Figure 2). A diagnosis of nephrogenic systemic fibrosis (NSF) was supported by typical findings on punch biopsy, including a proliferation of dermal fibroblasts with thickened collagen bundles and mucin deposition.
Nephrogenic systemic fibrosis, also known as nephrogenic fibrosing dermopathy, is characterized by fibrotic plaques and nodules that tend to be bilateral.1 The chronic course of this disease often is accompanied by flexion contractures. Yellow scleral plaques caused by calcium phosphate deposition are present in up to 75% of cases and are more specific to a diagnosis of NSF in patients younger than 45 years.1,2 A strong association exists between NSF and gadolinium contrast agents in patients with acute renal failure; our patient later confirmed multiple gadolinium exposures years prior. Deposits of gadolinium have even been found in NSF skin lesions.2
To the Editor:
A 44-year-old man with a history of systemic lupus erythematosus (SLE) complicated by lupus nephritis, end-stage renal disease, and antiphospholipid syndrome was evaluated for progressive skin tightening over the last 3 years, predominantly on the hands but also involving the feet, legs, and arms. Physical examination revealed multiple flesh-colored to hypopigmented, bound-down, indurated, fissured plaques over the distal upper and lower extremities, most prominent over the hands (Figure 1). Yellow plaques appeared on the lateral sclera of both eyes (Figure 2). A diagnosis of nephrogenic systemic fibrosis (NSF) was supported by typical findings on punch biopsy, including a proliferation of dermal fibroblasts with thickened collagen bundles and mucin deposition.
Nephrogenic systemic fibrosis, also known as nephrogenic fibrosing dermopathy, is characterized by fibrotic plaques and nodules that tend to be bilateral.1 The chronic course of this disease often is accompanied by flexion contractures. Yellow scleral plaques caused by calcium phosphate deposition are present in up to 75% of cases and are more specific to a diagnosis of NSF in patients younger than 45 years.1,2 A strong association exists between NSF and gadolinium contrast agents in patients with acute renal failure; our patient later confirmed multiple gadolinium exposures years prior. Deposits of gadolinium have even been found in NSF skin lesions.2
- Stone JH. A Clinician’s Pearls & Myths in Rheumatology. Springer London; 2009.
- Barker-Griffith A, Goldberg J, Abraham JL. Ocular pathologic features and gadolinium deposition in nephrogenic systemic fibrosis. Arch Ophthalmol. 2011;129:661-663.
- Stone JH. A Clinician’s Pearls & Myths in Rheumatology. Springer London; 2009.
- Barker-Griffith A, Goldberg J, Abraham JL. Ocular pathologic features and gadolinium deposition in nephrogenic systemic fibrosis. Arch Ophthalmol. 2011;129:661-663.
Practice Points
- It is important to examine the eyes in a patient with sclerotic skin changes on physical examination.
- The presence of yellow scleral plaques strongly is associated with a diagnosis of nephrogenic systemic fibrosis.

A Fatal Case of Hemophagocytic Lymphohistiocytosis Secondary to Anti-MDA5–Positive Dermatomyositis
To the Editor:
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by bilateral, symmetrical, proximal muscle weakness and classic cutaneous manifestations.1 Patients with antibodies directed against melanoma differentiation–associated gene 5, MDA5, have a distinct presentation due to vasculopathy with more severe cutaneous ulcerations, palmar papules, alopecia, and an elevated risk of rapidly progressive interstitial lung disease.2 A ferritin level greater than 1600 ng/mL portends an increased risk for pulmonary disease and therefore can be of prognostic value.3 Further, patients with anti-MDA5 DM are at a lower risk of malignancy and are more likely to test negative for antinuclear antibodies in comparison to other patients with DM.2,4
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a potentially lethal condition whereby uncontrolled activation of histiocytes in the reticuloendothelial system causes hemophagocytosis and a hyperinflammatory state. Patients present with fever, splenomegaly, cytopenia, and hyperferritinemia.5 Autoimmune‐associated hemophagocytic syndrome (AAHS) describes HLH that develops in association with autoimmune conditions, most commonly systemic lupus erythematosus and adult-onset Still disease. Cases reported in association with DM exist but are few in number, and there is no standard-of-care treatment.6 We report a case of a woman with anti-MDA5 DM complicated by HLH and DM-associated liver injury.
A 50-year-old woman presented as a direct admit from the rheumatology clinic for diffuse muscle weakness of 8 months’ duration, 40-pound unintentional weight loss, pruritic rash, bilateral joint pains, dry eyes, dry mouth, and altered mental status. Four months prior, she presented to an outside hospital and was given a diagnosis of probable Sjögren syndrome and autoimmune hepatitis vs drug-induced liver injury. At that time, a workup was notable for antibodies against Sjögren syndrome–related antigen A, anti–smooth muscle antibodies, and transaminitis. Ultrasonography of the right upper quadrant revealed hepatic steatosis. The patient was started on oral prednisone and pilocarpine but had been off all medications for 1 month when she presented to our hospital.
On hospital admission, physical examination revealed a violaceous heliotrope rash; a v-sign on the chest; shawl sign; palmar papules with pits at the fingertips; and periungual erythema and ulcerations along the metacarpophalangeal joints, elbows, lateral feet, and upper eyelids (Figure 1). Laboratory workup showed the following results: white blood cell count, 4100/μL (reference range, 4000–11,000/μL); hemoglobin, 11.6 g/dL (reference range, 12–16 g/dL); platelet count, 100,000/μL (reference range, 150,000–450,000/μL); lactate dehydrogenase, 510 U/L (reference range, 80–225 U/L); alkaline phosphatase (ALP), 766 U/L (reference range, 30–120 U/L); alanine aminotransferase (ALT), 88 U/L (reference range, 10–40 U/L); aspartate aminotransferase (AST), 544 U/L (reference range, 10–40 U/L); total bilirubin, 4.2 mg/dL (reference range, 0.3–1.0 mg/dL); direct bilirubin, 3.7 mg/dL (reference range, 0.1–0.3 mg/dL); aldolase, 20.2 U/L (reference range, 1–7.5 U/L), creatine kinase, 180 U/L (reference range, 30–135 U/L); γ-glutamyltransferase (GGT), 2743 U/L (reference range, 8–40 U/L); high sensitivity C-reactive protein, 122.9 mg/L (low-risk reference range, <1.0 mg/L); triglycerides, 534 mg/dL (reference range, <150 mg/dL); ferritin, 3784 ng/mL (reference range, 24–307 ng/mL); antinuclear antibody, negative titer; antimitochondrial antibody, negative titer; soluble IL-2 receptor (CD25), 7000 U/mL (reference range, 189–846 U/mL); anti-Sjögren syndrome–related antigen A antibody, positive.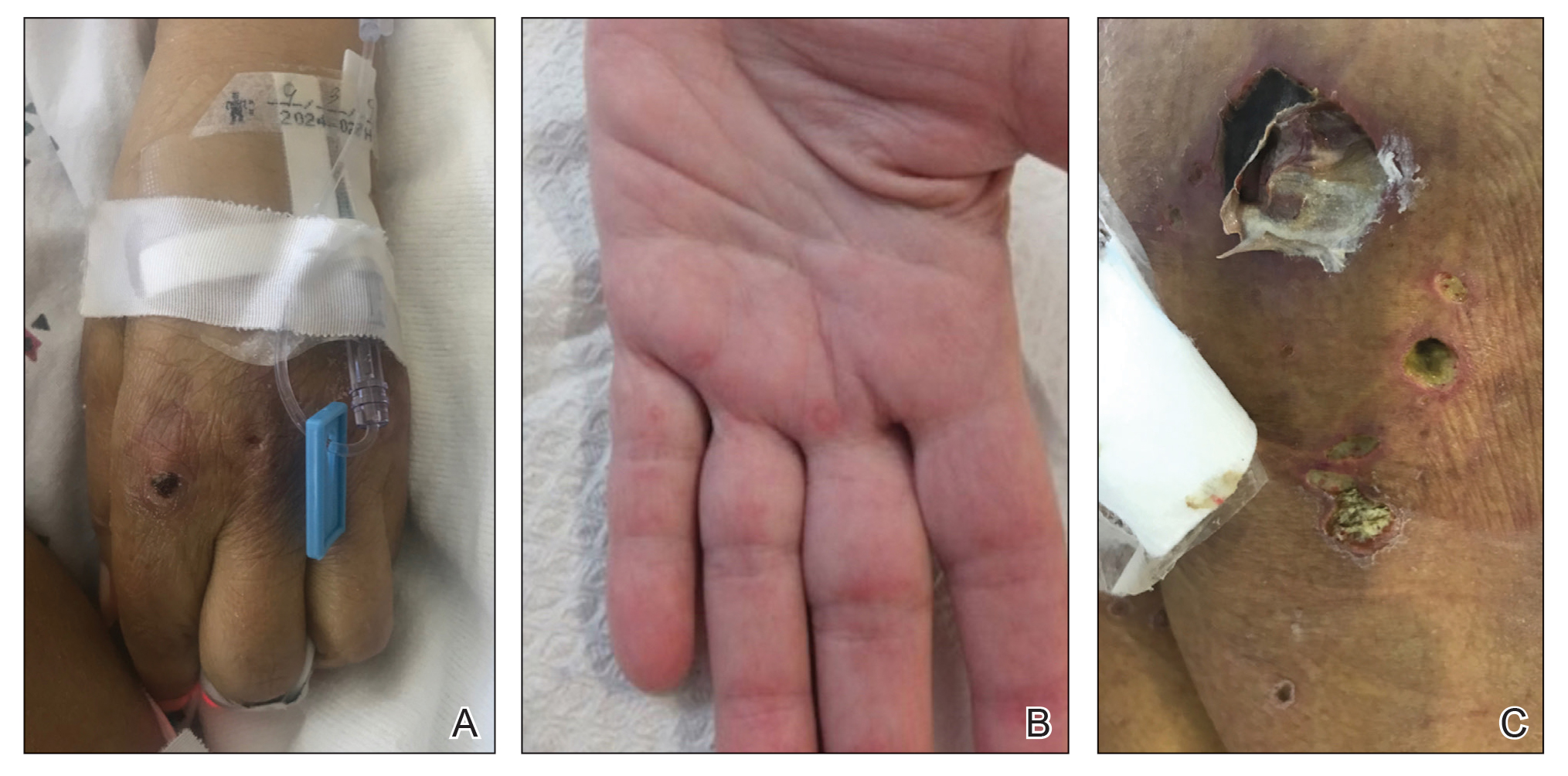
Magnetic resonance imaging of the shoulders showed diffuse soft-tissue edema. Computed tomography (CT) of the chest demonstrated parabronchial thickening and parenchymal bands suggestive of DM. An age-appropriate malignancy workup was negative, and results from a liver biopsy showed diffuse steatosis with no histologic evidence of autoimmune hepatitis. Punch biopsy results from a plaque on the left knee revealed vacuolar interface dermatitis with increased dermal mucin on colloidal iron staining, indicative of connective tissue disease (Figure 2). The patient was treated with intravenous (IV) methylprednisolone 250 mg twice daily for 2 days followed by oral prednisone 50 mg daily with IV immunoglobulin (IVIG) 0.4 mg/kg daily for 5 days. The patient’s symptoms improved, and she was discharged on oral prednisone 50 mg and mycophenolate mofetil 1000 mg twice daily with a plan for outpatient IVIG.
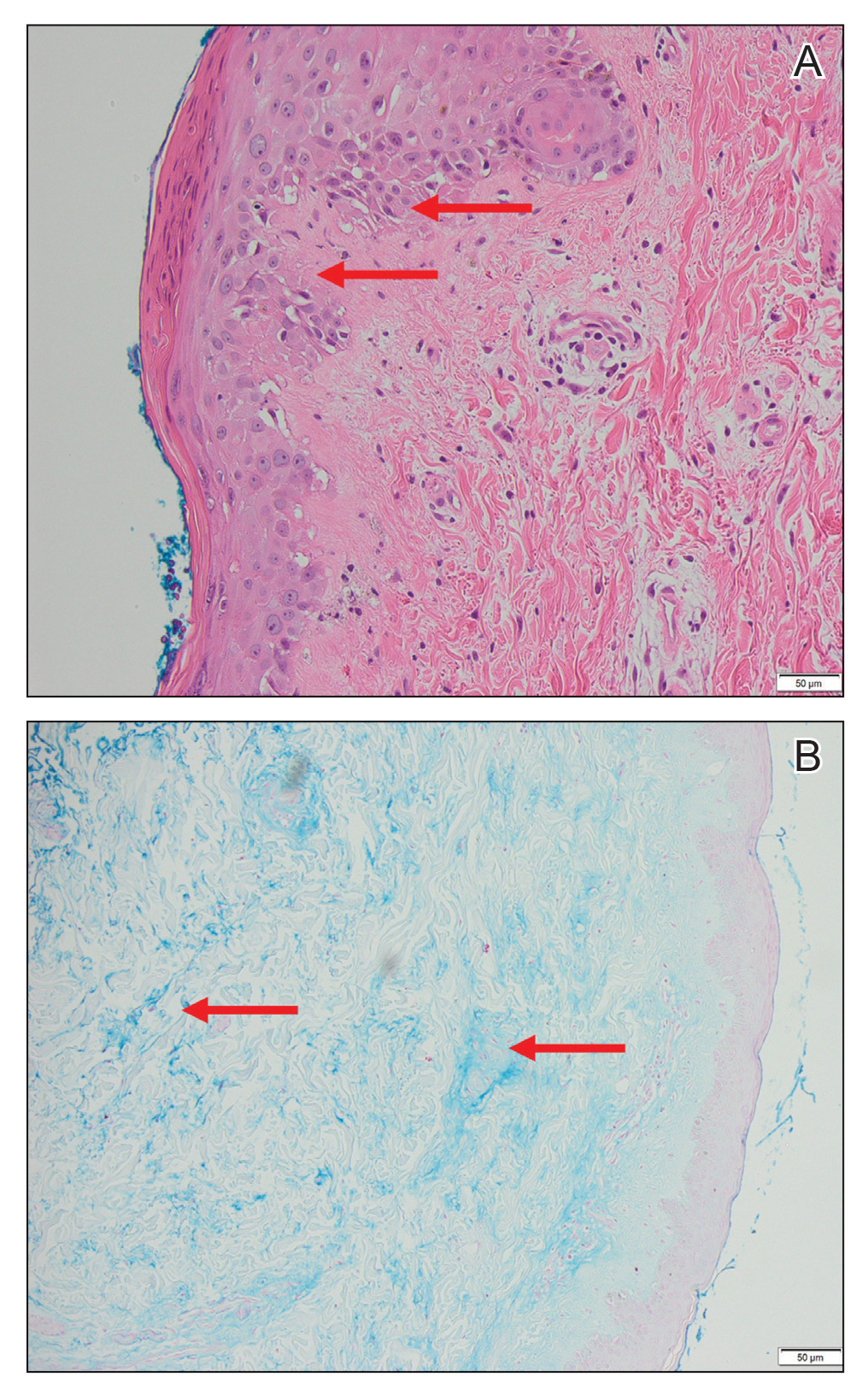
Two days after discharge, the patient was re-admitted for worsening muscle weakness; recalcitrant rash; new-onset hypophonia, dysphagia, and odynophagia; and intermittent fevers. Myositis panel results were positive for MDA5. Additionally, workup for HLH, which was initiated during the first hospital admission, revealed that she met 6 of 8 diagnostic criteria: intermittent fevers (maximum temperature, 38.2 °C), splenomegaly (12.6 cm on CT scan of abdomen), cytopenia in 2 cell lines (anemia, thrombocytopenia), hypertriglyceridemia, hyperferritinemia, and elevated IL-2 receptor (CD25). Based on these findings, the patient was diagnosed with anti-MDA5 DM associated with HLH.
The patient was started on IV methylprednisolone 1000 mg daily and received 1 rituximab infusion. Two days later, she experienced worsening fever with tachycardia, and a chest radiograph showed bibasilar infiltrates concerning for aspiration pneumonia, with sputum cultures growing Staphylococcus aureus. Due to the infection, the dosage of methylprednisolone was decreased to 16 mg 3 times daily and rituximab was stopped. The hematology department was consulted for the patient’s HLH, and due to her profound weakness and sepsis, the decision was made to hold initiation of etoposide, which, in addition to glucocorticoids, is considered first-line therapy for HLH. She subsequently experienced worsening hypoxia requiring intubation and received a second course of IVIG. Two days later, CT of the chest revealed progressive ground-glass opacities in the lower lobes of the lungs. The patient was then started on plasmapheresis every other day, hydroxychloroquine 200 mg daily, and IV methylprednisolone 1000 mg daily. Over the subsequent 6 days, she developed worsening renal failure, liver dysfunction, profound thrombocytopenia (13/μL), and acidemia. After extensive discussion with her family, the patient was transitioned to comfort care, and she died 33 days after the initial admission to our hospital.
Our case is a collection of several rare presentations: anti-MDA5 DM, with HLH and AAHS as complications of anti-MDA5 DM, and DM-associated liver injury. Anti-MDA5 DM is frequently refractory to conventional therapy, including high-dose glucocorticoids, cyclophosphamide, oral tacrolimus, and cyclosporine, and there currently is no single treatment algorithm.2 Lake and colleagues7 highlighted the importance of personalizing treatment of anti-MDA5 DM, as it can be one of the most aggressive rheumatologic diseases. We initially chose to treat our patient with high-dose methylprednisolone, IVIG, and rituximab. Kampylafka et al8 performed a retrospective analysis of the use of IVIG for DM as compared to standard therapy and demonstrated improved muscle and cutaneous involvement from a collection of 50 patients. Case reports have specifically revealed efficacy for the use of IVIG in patients with anti-MDA5 DM.9,10 Additionally, rituximab—an anti–B lymphocyte therapy—has been shown to be an effective supplemental therapy for cases of aggressive anti-MDA5 DM with associated interstitial lung disease, especially when conventional therapy has failed.11,12 Our patient’s sepsis secondary to S aureus pneumonia limited her to only receiving 1 dose of rituximab.
One promising treatment approach for anti-MDA5 DM recently published by Tsuji et al13 involves the use of combination therapy. In this prospective multicenter trial, patients were initially treated with a combination of high-dose glucocorticoids, oral tacrolimus, and IV cyclophosphamide. Plasmapheresis was then started for patients without symptomatic improvement. This method was compared to the more traditional step-up approach of high-dose steroids followed by another immunosuppressant. At 1-year follow-up, the combination therapy group demonstrated an 85% survival rate compared to 33% of historical controls.13
We suspect that our patient developed HLH and AAHS secondary to her underlying anti-MDA5 DM. Kumakura and Murakawa6 reported that among 116 cases of AAHS, 6.9% of cases were associated with DM, most commonly anti-Jo-1 DM. Hemophagocytic lymphohistiocytosis associated with anti-MDA5 DM has been described in only a few cases.14-16 The diagnosis of HLH is critical, as the treatments for HLH and DM differ. Both diseases manifest with hyperferritinemia—greater than 500 ng/mL in the case of HLH and 3784 ng/mL in our patient. Therefore, HLH can be easily overlooked. It is possible the rates of HLH associated with anti-MDA5 DM are higher than reported given their similar presentations.
Analogous to our case, Fujita et al15 reported a case of HLH associated with anti-MDA5 DM successfully treated with IV cyclophosphamide pulse therapy and plasmapheresis. The rationale for using plasmapheresis in anti-MDA5 DM is based on its success in patients with other antibody-mediated conditions such as Goodpasture syndrome and granulomatosis with polyangiitis.7 It is thought to expedite response to traditional treatment, and in the case described by Fujita et al,15 the patient received plasmapheresis 6 times total over the course of 9 days. The patient’s clinical symptoms, as well as platelet levels, liver enzymes, and ferritin value, improved.15 Our patient received 3 days of plasmapheresis with no improvement when the decision was made to discontinue plasmapheresis given her worsening clinical state.
Additionally, our patient had elevated hepatic enzymes (ALT, AST, ALP, GGT), and results of a liver biopsy demonstrated diffuse steatosis. We speculate her transaminitis was a complication of anti-MDA5 DM. Hepatocellular damage accompanying DM has been investigated in multiple studies and is most often defined as an elevated ALT.17-20 Improvement in ALT levels has been seen with DM treatment. However, investigators note that creatine kinase (CK) values often do not correlate with the resolution of the transaminitis, suggesting that CK denotes muscle damage whereas ALT represents separate liver damage.18-21
Nagashima et al22 highlighted that among 50 patients with DM without malignancy, only 20% presented with a transaminitis or elevated bilirubin. However, among those with liver injury, all were positive for antibodies against MDA5.22 The patients with anti-MDA5 DM liver dysfunction had higher ALT, ALP, and GGT levels compared to those without liver dysfunction. Similarly, in a retrospective review of 14 patients with anti-MDA5 DM, Gono and colleagues3 found elevated GGT levels and lower CK levels in comparison to patients with anti-aminoacyl-transfer RNA synthetase DM. Although liver enzymes can be elevated in patients with DM secondary to muscle damage, the authors argue that the specificity of GGT to the liver suggests intrinsic liver damage.3
The mechanism behind liver disease in anti-MDA5 DM is unclear, but it is hypothesized to be similar to nonalcoholic steatohepatitis.22 Other studies have revealed drug-induced hepatitis, hepatic congestion, nonspecific reactive hepatitis, metastatic liver tumor, primary biliary cholangitis, and autoimmune hepatitis as the etiology behind liver disease in their patients with DM.17-19 Liver biopsy results from patients with anti-MDA5 DM most commonly reveal hepatic steatosis, as seen in our patient, as well as hepatocyte ballooning and increased pigmented macrophages.22
We presented a case of anti-MDA5 DM complicated by HLH. Our patient had a fatal outcome despite aggressive treatment with high-dose methylprednisolone, IVIG, rituximab, and plasmapheresis. It is accepted that anti-MDA5 DM affects the lungs and skin, and our patient’s presentation also suggests liver involvement. In our case, onset of symptoms to fatality was approximately 1 year. It is essential to consider the diagnosis of HLH in all cases of anti-MDA5 DM given clinical disease overlap. Our patient could have benefited from earlier disease recognition and thus earlier aggressive therapy.
1. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-347.
2. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785.
3. Gono T, Kawaguchi Y, Satoh T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49:1713-1719.
4. Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34.
5. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. 2017;49:20-26.
6. Kumakura S, Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheum. 2014;66:2297-2307.
7. Lake M, George G, Summer R. Time to personalize the treatment of anti-MDA-5 associated lung disease. Ann Rheum Dis. 2019;78:E52.
8. Kampylafka EI, Kosmidis ML, Panagiotakos DB, et al. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol. 2012;30:397-401.
9. Koguchi-Yoshioka H, Okiyama N, Iwamoto K, et al. Intravenous immunoglobulin contributes to the control of antimelanoma differentiation-associated protein 5 antibody-associated dermatomyositis with palmar violaceous macules/papules. Br J Dermatol. 2017;177:1442-1446.
10. Hamada-Ode K, Taniguchi Y, Kimata T, et al. High-dose intravenous immunoglobulin therapy for rapidly progressive interstitial pneumonitis accompanied by anti-melanoma differentiation-associated gene 5 antibody-positive amyopathic dermatomyositis. Eur J Rheumatol. 2015;2:83-85.
11. So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989.
12. Koichi Y, Aya Y, Megumi U, et al. A case of anti-MDA5-positive rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis ameliorated by rituximab, in addition to standard immunosuppressive treatment. Mod Rheumatol. 2017;27:536-540.
13. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. 2020;72:488-498.
14. Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune-associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246.
15. Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478.
16. Gono T, Miyake K, Kawaguchi Y, et al. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology (Oxford). 2012;51:1336-1338.
17. Wada T, Abe G, Kudou, T, et al. Liver damage in patients with polymyositis and dermatomyositis. Kitasato Med Journal. 2016;46:40-46.
18. Takahashi A, Abe K, Yokokawa J, et al. Clinical features of liver dysfunction in collagen diseases. Hepatol Res. 2010;40:1092-1097.
19. Matsumoto T, Kobayashi S, Shimizu H, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000;20:366-373.
20. Shi Q, Niu J, Huang X, et al. Do muscle enzyme changes forecast liver injury in polymyositis/dermatomyositis patients treated with methylprednisolone and methotrexate? Ann Clin Lab Sci. 2016;46:266-269.
21. Noda S, Asano Y, Tamaki Z, et al. A case of dermatomyositis with “liver disease associated with rheumatoid diseases” positive for anti-liver-kidney microsome-1 antibody. Clin Rheumatol. 2010;29:941-943.
22. Nagashima T, Kamata Y, Iwamoto M, et al. Liver dysfunction in anti-melanoma differentiation-associated gene 5 antibody-positive patients with dermatomyositis. Rheumatol Int. 2019;39:901-909.
To the Editor:
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by bilateral, symmetrical, proximal muscle weakness and classic cutaneous manifestations.1 Patients with antibodies directed against melanoma differentiation–associated gene 5, MDA5, have a distinct presentation due to vasculopathy with more severe cutaneous ulcerations, palmar papules, alopecia, and an elevated risk of rapidly progressive interstitial lung disease.2 A ferritin level greater than 1600 ng/mL portends an increased risk for pulmonary disease and therefore can be of prognostic value.3 Further, patients with anti-MDA5 DM are at a lower risk of malignancy and are more likely to test negative for antinuclear antibodies in comparison to other patients with DM.2,4
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a potentially lethal condition whereby uncontrolled activation of histiocytes in the reticuloendothelial system causes hemophagocytosis and a hyperinflammatory state. Patients present with fever, splenomegaly, cytopenia, and hyperferritinemia.5 Autoimmune‐associated hemophagocytic syndrome (AAHS) describes HLH that develops in association with autoimmune conditions, most commonly systemic lupus erythematosus and adult-onset Still disease. Cases reported in association with DM exist but are few in number, and there is no standard-of-care treatment.6 We report a case of a woman with anti-MDA5 DM complicated by HLH and DM-associated liver injury.
A 50-year-old woman presented as a direct admit from the rheumatology clinic for diffuse muscle weakness of 8 months’ duration, 40-pound unintentional weight loss, pruritic rash, bilateral joint pains, dry eyes, dry mouth, and altered mental status. Four months prior, she presented to an outside hospital and was given a diagnosis of probable Sjögren syndrome and autoimmune hepatitis vs drug-induced liver injury. At that time, a workup was notable for antibodies against Sjögren syndrome–related antigen A, anti–smooth muscle antibodies, and transaminitis. Ultrasonography of the right upper quadrant revealed hepatic steatosis. The patient was started on oral prednisone and pilocarpine but had been off all medications for 1 month when she presented to our hospital.
On hospital admission, physical examination revealed a violaceous heliotrope rash; a v-sign on the chest; shawl sign; palmar papules with pits at the fingertips; and periungual erythema and ulcerations along the metacarpophalangeal joints, elbows, lateral feet, and upper eyelids (Figure 1). Laboratory workup showed the following results: white blood cell count, 4100/μL (reference range, 4000–11,000/μL); hemoglobin, 11.6 g/dL (reference range, 12–16 g/dL); platelet count, 100,000/μL (reference range, 150,000–450,000/μL); lactate dehydrogenase, 510 U/L (reference range, 80–225 U/L); alkaline phosphatase (ALP), 766 U/L (reference range, 30–120 U/L); alanine aminotransferase (ALT), 88 U/L (reference range, 10–40 U/L); aspartate aminotransferase (AST), 544 U/L (reference range, 10–40 U/L); total bilirubin, 4.2 mg/dL (reference range, 0.3–1.0 mg/dL); direct bilirubin, 3.7 mg/dL (reference range, 0.1–0.3 mg/dL); aldolase, 20.2 U/L (reference range, 1–7.5 U/L), creatine kinase, 180 U/L (reference range, 30–135 U/L); γ-glutamyltransferase (GGT), 2743 U/L (reference range, 8–40 U/L); high sensitivity C-reactive protein, 122.9 mg/L (low-risk reference range, <1.0 mg/L); triglycerides, 534 mg/dL (reference range, <150 mg/dL); ferritin, 3784 ng/mL (reference range, 24–307 ng/mL); antinuclear antibody, negative titer; antimitochondrial antibody, negative titer; soluble IL-2 receptor (CD25), 7000 U/mL (reference range, 189–846 U/mL); anti-Sjögren syndrome–related antigen A antibody, positive.
Magnetic resonance imaging of the shoulders showed diffuse soft-tissue edema. Computed tomography (CT) of the chest demonstrated parabronchial thickening and parenchymal bands suggestive of DM. An age-appropriate malignancy workup was negative, and results from a liver biopsy showed diffuse steatosis with no histologic evidence of autoimmune hepatitis. Punch biopsy results from a plaque on the left knee revealed vacuolar interface dermatitis with increased dermal mucin on colloidal iron staining, indicative of connective tissue disease (Figure 2). The patient was treated with intravenous (IV) methylprednisolone 250 mg twice daily for 2 days followed by oral prednisone 50 mg daily with IV immunoglobulin (IVIG) 0.4 mg/kg daily for 5 days. The patient’s symptoms improved, and she was discharged on oral prednisone 50 mg and mycophenolate mofetil 1000 mg twice daily with a plan for outpatient IVIG.
Two days after discharge, the patient was re-admitted for worsening muscle weakness; recalcitrant rash; new-onset hypophonia, dysphagia, and odynophagia; and intermittent fevers. Myositis panel results were positive for MDA5. Additionally, workup for HLH, which was initiated during the first hospital admission, revealed that she met 6 of 8 diagnostic criteria: intermittent fevers (maximum temperature, 38.2 °C), splenomegaly (12.6 cm on CT scan of abdomen), cytopenia in 2 cell lines (anemia, thrombocytopenia), hypertriglyceridemia, hyperferritinemia, and elevated IL-2 receptor (CD25). Based on these findings, the patient was diagnosed with anti-MDA5 DM associated with HLH.
The patient was started on IV methylprednisolone 1000 mg daily and received 1 rituximab infusion. Two days later, she experienced worsening fever with tachycardia, and a chest radiograph showed bibasilar infiltrates concerning for aspiration pneumonia, with sputum cultures growing Staphylococcus aureus. Due to the infection, the dosage of methylprednisolone was decreased to 16 mg 3 times daily and rituximab was stopped. The hematology department was consulted for the patient’s HLH, and due to her profound weakness and sepsis, the decision was made to hold initiation of etoposide, which, in addition to glucocorticoids, is considered first-line therapy for HLH. She subsequently experienced worsening hypoxia requiring intubation and received a second course of IVIG. Two days later, CT of the chest revealed progressive ground-glass opacities in the lower lobes of the lungs. The patient was then started on plasmapheresis every other day, hydroxychloroquine 200 mg daily, and IV methylprednisolone 1000 mg daily. Over the subsequent 6 days, she developed worsening renal failure, liver dysfunction, profound thrombocytopenia (13/μL), and acidemia. After extensive discussion with her family, the patient was transitioned to comfort care, and she died 33 days after the initial admission to our hospital.
Our case is a collection of several rare presentations: anti-MDA5 DM, with HLH and AAHS as complications of anti-MDA5 DM, and DM-associated liver injury. Anti-MDA5 DM is frequently refractory to conventional therapy, including high-dose glucocorticoids, cyclophosphamide, oral tacrolimus, and cyclosporine, and there currently is no single treatment algorithm.2 Lake and colleagues7 highlighted the importance of personalizing treatment of anti-MDA5 DM, as it can be one of the most aggressive rheumatologic diseases. We initially chose to treat our patient with high-dose methylprednisolone, IVIG, and rituximab. Kampylafka et al8 performed a retrospective analysis of the use of IVIG for DM as compared to standard therapy and demonstrated improved muscle and cutaneous involvement from a collection of 50 patients. Case reports have specifically revealed efficacy for the use of IVIG in patients with anti-MDA5 DM.9,10 Additionally, rituximab—an anti–B lymphocyte therapy—has been shown to be an effective supplemental therapy for cases of aggressive anti-MDA5 DM with associated interstitial lung disease, especially when conventional therapy has failed.11,12 Our patient’s sepsis secondary to S aureus pneumonia limited her to only receiving 1 dose of rituximab.
One promising treatment approach for anti-MDA5 DM recently published by Tsuji et al13 involves the use of combination therapy. In this prospective multicenter trial, patients were initially treated with a combination of high-dose glucocorticoids, oral tacrolimus, and IV cyclophosphamide. Plasmapheresis was then started for patients without symptomatic improvement. This method was compared to the more traditional step-up approach of high-dose steroids followed by another immunosuppressant. At 1-year follow-up, the combination therapy group demonstrated an 85% survival rate compared to 33% of historical controls.13
We suspect that our patient developed HLH and AAHS secondary to her underlying anti-MDA5 DM. Kumakura and Murakawa6 reported that among 116 cases of AAHS, 6.9% of cases were associated with DM, most commonly anti-Jo-1 DM. Hemophagocytic lymphohistiocytosis associated with anti-MDA5 DM has been described in only a few cases.14-16 The diagnosis of HLH is critical, as the treatments for HLH and DM differ. Both diseases manifest with hyperferritinemia—greater than 500 ng/mL in the case of HLH and 3784 ng/mL in our patient. Therefore, HLH can be easily overlooked. It is possible the rates of HLH associated with anti-MDA5 DM are higher than reported given their similar presentations.
Analogous to our case, Fujita et al15 reported a case of HLH associated with anti-MDA5 DM successfully treated with IV cyclophosphamide pulse therapy and plasmapheresis. The rationale for using plasmapheresis in anti-MDA5 DM is based on its success in patients with other antibody-mediated conditions such as Goodpasture syndrome and granulomatosis with polyangiitis.7 It is thought to expedite response to traditional treatment, and in the case described by Fujita et al,15 the patient received plasmapheresis 6 times total over the course of 9 days. The patient’s clinical symptoms, as well as platelet levels, liver enzymes, and ferritin value, improved.15 Our patient received 3 days of plasmapheresis with no improvement when the decision was made to discontinue plasmapheresis given her worsening clinical state.
Additionally, our patient had elevated hepatic enzymes (ALT, AST, ALP, GGT), and results of a liver biopsy demonstrated diffuse steatosis. We speculate her transaminitis was a complication of anti-MDA5 DM. Hepatocellular damage accompanying DM has been investigated in multiple studies and is most often defined as an elevated ALT.17-20 Improvement in ALT levels has been seen with DM treatment. However, investigators note that creatine kinase (CK) values often do not correlate with the resolution of the transaminitis, suggesting that CK denotes muscle damage whereas ALT represents separate liver damage.18-21
Nagashima et al22 highlighted that among 50 patients with DM without malignancy, only 20% presented with a transaminitis or elevated bilirubin. However, among those with liver injury, all were positive for antibodies against MDA5.22 The patients with anti-MDA5 DM liver dysfunction had higher ALT, ALP, and GGT levels compared to those without liver dysfunction. Similarly, in a retrospective review of 14 patients with anti-MDA5 DM, Gono and colleagues3 found elevated GGT levels and lower CK levels in comparison to patients with anti-aminoacyl-transfer RNA synthetase DM. Although liver enzymes can be elevated in patients with DM secondary to muscle damage, the authors argue that the specificity of GGT to the liver suggests intrinsic liver damage.3
The mechanism behind liver disease in anti-MDA5 DM is unclear, but it is hypothesized to be similar to nonalcoholic steatohepatitis.22 Other studies have revealed drug-induced hepatitis, hepatic congestion, nonspecific reactive hepatitis, metastatic liver tumor, primary biliary cholangitis, and autoimmune hepatitis as the etiology behind liver disease in their patients with DM.17-19 Liver biopsy results from patients with anti-MDA5 DM most commonly reveal hepatic steatosis, as seen in our patient, as well as hepatocyte ballooning and increased pigmented macrophages.22
We presented a case of anti-MDA5 DM complicated by HLH. Our patient had a fatal outcome despite aggressive treatment with high-dose methylprednisolone, IVIG, rituximab, and plasmapheresis. It is accepted that anti-MDA5 DM affects the lungs and skin, and our patient’s presentation also suggests liver involvement. In our case, onset of symptoms to fatality was approximately 1 year. It is essential to consider the diagnosis of HLH in all cases of anti-MDA5 DM given clinical disease overlap. Our patient could have benefited from earlier disease recognition and thus earlier aggressive therapy.
To the Editor:
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by bilateral, symmetrical, proximal muscle weakness and classic cutaneous manifestations.1 Patients with antibodies directed against melanoma differentiation–associated gene 5, MDA5, have a distinct presentation due to vasculopathy with more severe cutaneous ulcerations, palmar papules, alopecia, and an elevated risk of rapidly progressive interstitial lung disease.2 A ferritin level greater than 1600 ng/mL portends an increased risk for pulmonary disease and therefore can be of prognostic value.3 Further, patients with anti-MDA5 DM are at a lower risk of malignancy and are more likely to test negative for antinuclear antibodies in comparison to other patients with DM.2,4
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a potentially lethal condition whereby uncontrolled activation of histiocytes in the reticuloendothelial system causes hemophagocytosis and a hyperinflammatory state. Patients present with fever, splenomegaly, cytopenia, and hyperferritinemia.5 Autoimmune‐associated hemophagocytic syndrome (AAHS) describes HLH that develops in association with autoimmune conditions, most commonly systemic lupus erythematosus and adult-onset Still disease. Cases reported in association with DM exist but are few in number, and there is no standard-of-care treatment.6 We report a case of a woman with anti-MDA5 DM complicated by HLH and DM-associated liver injury.
A 50-year-old woman presented as a direct admit from the rheumatology clinic for diffuse muscle weakness of 8 months’ duration, 40-pound unintentional weight loss, pruritic rash, bilateral joint pains, dry eyes, dry mouth, and altered mental status. Four months prior, she presented to an outside hospital and was given a diagnosis of probable Sjögren syndrome and autoimmune hepatitis vs drug-induced liver injury. At that time, a workup was notable for antibodies against Sjögren syndrome–related antigen A, anti–smooth muscle antibodies, and transaminitis. Ultrasonography of the right upper quadrant revealed hepatic steatosis. The patient was started on oral prednisone and pilocarpine but had been off all medications for 1 month when she presented to our hospital.
On hospital admission, physical examination revealed a violaceous heliotrope rash; a v-sign on the chest; shawl sign; palmar papules with pits at the fingertips; and periungual erythema and ulcerations along the metacarpophalangeal joints, elbows, lateral feet, and upper eyelids (Figure 1). Laboratory workup showed the following results: white blood cell count, 4100/μL (reference range, 4000–11,000/μL); hemoglobin, 11.6 g/dL (reference range, 12–16 g/dL); platelet count, 100,000/μL (reference range, 150,000–450,000/μL); lactate dehydrogenase, 510 U/L (reference range, 80–225 U/L); alkaline phosphatase (ALP), 766 U/L (reference range, 30–120 U/L); alanine aminotransferase (ALT), 88 U/L (reference range, 10–40 U/L); aspartate aminotransferase (AST), 544 U/L (reference range, 10–40 U/L); total bilirubin, 4.2 mg/dL (reference range, 0.3–1.0 mg/dL); direct bilirubin, 3.7 mg/dL (reference range, 0.1–0.3 mg/dL); aldolase, 20.2 U/L (reference range, 1–7.5 U/L), creatine kinase, 180 U/L (reference range, 30–135 U/L); γ-glutamyltransferase (GGT), 2743 U/L (reference range, 8–40 U/L); high sensitivity C-reactive protein, 122.9 mg/L (low-risk reference range, <1.0 mg/L); triglycerides, 534 mg/dL (reference range, <150 mg/dL); ferritin, 3784 ng/mL (reference range, 24–307 ng/mL); antinuclear antibody, negative titer; antimitochondrial antibody, negative titer; soluble IL-2 receptor (CD25), 7000 U/mL (reference range, 189–846 U/mL); anti-Sjögren syndrome–related antigen A antibody, positive.
Magnetic resonance imaging of the shoulders showed diffuse soft-tissue edema. Computed tomography (CT) of the chest demonstrated parabronchial thickening and parenchymal bands suggestive of DM. An age-appropriate malignancy workup was negative, and results from a liver biopsy showed diffuse steatosis with no histologic evidence of autoimmune hepatitis. Punch biopsy results from a plaque on the left knee revealed vacuolar interface dermatitis with increased dermal mucin on colloidal iron staining, indicative of connective tissue disease (Figure 2). The patient was treated with intravenous (IV) methylprednisolone 250 mg twice daily for 2 days followed by oral prednisone 50 mg daily with IV immunoglobulin (IVIG) 0.4 mg/kg daily for 5 days. The patient’s symptoms improved, and she was discharged on oral prednisone 50 mg and mycophenolate mofetil 1000 mg twice daily with a plan for outpatient IVIG.
Two days after discharge, the patient was re-admitted for worsening muscle weakness; recalcitrant rash; new-onset hypophonia, dysphagia, and odynophagia; and intermittent fevers. Myositis panel results were positive for MDA5. Additionally, workup for HLH, which was initiated during the first hospital admission, revealed that she met 6 of 8 diagnostic criteria: intermittent fevers (maximum temperature, 38.2 °C), splenomegaly (12.6 cm on CT scan of abdomen), cytopenia in 2 cell lines (anemia, thrombocytopenia), hypertriglyceridemia, hyperferritinemia, and elevated IL-2 receptor (CD25). Based on these findings, the patient was diagnosed with anti-MDA5 DM associated with HLH.
The patient was started on IV methylprednisolone 1000 mg daily and received 1 rituximab infusion. Two days later, she experienced worsening fever with tachycardia, and a chest radiograph showed bibasilar infiltrates concerning for aspiration pneumonia, with sputum cultures growing Staphylococcus aureus. Due to the infection, the dosage of methylprednisolone was decreased to 16 mg 3 times daily and rituximab was stopped. The hematology department was consulted for the patient’s HLH, and due to her profound weakness and sepsis, the decision was made to hold initiation of etoposide, which, in addition to glucocorticoids, is considered first-line therapy for HLH. She subsequently experienced worsening hypoxia requiring intubation and received a second course of IVIG. Two days later, CT of the chest revealed progressive ground-glass opacities in the lower lobes of the lungs. The patient was then started on plasmapheresis every other day, hydroxychloroquine 200 mg daily, and IV methylprednisolone 1000 mg daily. Over the subsequent 6 days, she developed worsening renal failure, liver dysfunction, profound thrombocytopenia (13/μL), and acidemia. After extensive discussion with her family, the patient was transitioned to comfort care, and she died 33 days after the initial admission to our hospital.
Our case is a collection of several rare presentations: anti-MDA5 DM, with HLH and AAHS as complications of anti-MDA5 DM, and DM-associated liver injury. Anti-MDA5 DM is frequently refractory to conventional therapy, including high-dose glucocorticoids, cyclophosphamide, oral tacrolimus, and cyclosporine, and there currently is no single treatment algorithm.2 Lake and colleagues7 highlighted the importance of personalizing treatment of anti-MDA5 DM, as it can be one of the most aggressive rheumatologic diseases. We initially chose to treat our patient with high-dose methylprednisolone, IVIG, and rituximab. Kampylafka et al8 performed a retrospective analysis of the use of IVIG for DM as compared to standard therapy and demonstrated improved muscle and cutaneous involvement from a collection of 50 patients. Case reports have specifically revealed efficacy for the use of IVIG in patients with anti-MDA5 DM.9,10 Additionally, rituximab—an anti–B lymphocyte therapy—has been shown to be an effective supplemental therapy for cases of aggressive anti-MDA5 DM with associated interstitial lung disease, especially when conventional therapy has failed.11,12 Our patient’s sepsis secondary to S aureus pneumonia limited her to only receiving 1 dose of rituximab.
One promising treatment approach for anti-MDA5 DM recently published by Tsuji et al13 involves the use of combination therapy. In this prospective multicenter trial, patients were initially treated with a combination of high-dose glucocorticoids, oral tacrolimus, and IV cyclophosphamide. Plasmapheresis was then started for patients without symptomatic improvement. This method was compared to the more traditional step-up approach of high-dose steroids followed by another immunosuppressant. At 1-year follow-up, the combination therapy group demonstrated an 85% survival rate compared to 33% of historical controls.13
We suspect that our patient developed HLH and AAHS secondary to her underlying anti-MDA5 DM. Kumakura and Murakawa6 reported that among 116 cases of AAHS, 6.9% of cases were associated with DM, most commonly anti-Jo-1 DM. Hemophagocytic lymphohistiocytosis associated with anti-MDA5 DM has been described in only a few cases.14-16 The diagnosis of HLH is critical, as the treatments for HLH and DM differ. Both diseases manifest with hyperferritinemia—greater than 500 ng/mL in the case of HLH and 3784 ng/mL in our patient. Therefore, HLH can be easily overlooked. It is possible the rates of HLH associated with anti-MDA5 DM are higher than reported given their similar presentations.
Analogous to our case, Fujita et al15 reported a case of HLH associated with anti-MDA5 DM successfully treated with IV cyclophosphamide pulse therapy and plasmapheresis. The rationale for using plasmapheresis in anti-MDA5 DM is based on its success in patients with other antibody-mediated conditions such as Goodpasture syndrome and granulomatosis with polyangiitis.7 It is thought to expedite response to traditional treatment, and in the case described by Fujita et al,15 the patient received plasmapheresis 6 times total over the course of 9 days. The patient’s clinical symptoms, as well as platelet levels, liver enzymes, and ferritin value, improved.15 Our patient received 3 days of plasmapheresis with no improvement when the decision was made to discontinue plasmapheresis given her worsening clinical state.
Additionally, our patient had elevated hepatic enzymes (ALT, AST, ALP, GGT), and results of a liver biopsy demonstrated diffuse steatosis. We speculate her transaminitis was a complication of anti-MDA5 DM. Hepatocellular damage accompanying DM has been investigated in multiple studies and is most often defined as an elevated ALT.17-20 Improvement in ALT levels has been seen with DM treatment. However, investigators note that creatine kinase (CK) values often do not correlate with the resolution of the transaminitis, suggesting that CK denotes muscle damage whereas ALT represents separate liver damage.18-21
Nagashima et al22 highlighted that among 50 patients with DM without malignancy, only 20% presented with a transaminitis or elevated bilirubin. However, among those with liver injury, all were positive for antibodies against MDA5.22 The patients with anti-MDA5 DM liver dysfunction had higher ALT, ALP, and GGT levels compared to those without liver dysfunction. Similarly, in a retrospective review of 14 patients with anti-MDA5 DM, Gono and colleagues3 found elevated GGT levels and lower CK levels in comparison to patients with anti-aminoacyl-transfer RNA synthetase DM. Although liver enzymes can be elevated in patients with DM secondary to muscle damage, the authors argue that the specificity of GGT to the liver suggests intrinsic liver damage.3
The mechanism behind liver disease in anti-MDA5 DM is unclear, but it is hypothesized to be similar to nonalcoholic steatohepatitis.22 Other studies have revealed drug-induced hepatitis, hepatic congestion, nonspecific reactive hepatitis, metastatic liver tumor, primary biliary cholangitis, and autoimmune hepatitis as the etiology behind liver disease in their patients with DM.17-19 Liver biopsy results from patients with anti-MDA5 DM most commonly reveal hepatic steatosis, as seen in our patient, as well as hepatocyte ballooning and increased pigmented macrophages.22
We presented a case of anti-MDA5 DM complicated by HLH. Our patient had a fatal outcome despite aggressive treatment with high-dose methylprednisolone, IVIG, rituximab, and plasmapheresis. It is accepted that anti-MDA5 DM affects the lungs and skin, and our patient’s presentation also suggests liver involvement. In our case, onset of symptoms to fatality was approximately 1 year. It is essential to consider the diagnosis of HLH in all cases of anti-MDA5 DM given clinical disease overlap. Our patient could have benefited from earlier disease recognition and thus earlier aggressive therapy.
1. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-347.
2. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785.
3. Gono T, Kawaguchi Y, Satoh T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49:1713-1719.
4. Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34.
5. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. 2017;49:20-26.
6. Kumakura S, Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheum. 2014;66:2297-2307.
7. Lake M, George G, Summer R. Time to personalize the treatment of anti-MDA-5 associated lung disease. Ann Rheum Dis. 2019;78:E52.
8. Kampylafka EI, Kosmidis ML, Panagiotakos DB, et al. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol. 2012;30:397-401.
9. Koguchi-Yoshioka H, Okiyama N, Iwamoto K, et al. Intravenous immunoglobulin contributes to the control of antimelanoma differentiation-associated protein 5 antibody-associated dermatomyositis with palmar violaceous macules/papules. Br J Dermatol. 2017;177:1442-1446.
10. Hamada-Ode K, Taniguchi Y, Kimata T, et al. High-dose intravenous immunoglobulin therapy for rapidly progressive interstitial pneumonitis accompanied by anti-melanoma differentiation-associated gene 5 antibody-positive amyopathic dermatomyositis. Eur J Rheumatol. 2015;2:83-85.
11. So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989.
12. Koichi Y, Aya Y, Megumi U, et al. A case of anti-MDA5-positive rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis ameliorated by rituximab, in addition to standard immunosuppressive treatment. Mod Rheumatol. 2017;27:536-540.
13. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. 2020;72:488-498.
14. Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune-associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246.
15. Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478.
16. Gono T, Miyake K, Kawaguchi Y, et al. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology (Oxford). 2012;51:1336-1338.
17. Wada T, Abe G, Kudou, T, et al. Liver damage in patients with polymyositis and dermatomyositis. Kitasato Med Journal. 2016;46:40-46.
18. Takahashi A, Abe K, Yokokawa J, et al. Clinical features of liver dysfunction in collagen diseases. Hepatol Res. 2010;40:1092-1097.
19. Matsumoto T, Kobayashi S, Shimizu H, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000;20:366-373.
20. Shi Q, Niu J, Huang X, et al. Do muscle enzyme changes forecast liver injury in polymyositis/dermatomyositis patients treated with methylprednisolone and methotrexate? Ann Clin Lab Sci. 2016;46:266-269.
21. Noda S, Asano Y, Tamaki Z, et al. A case of dermatomyositis with “liver disease associated with rheumatoid diseases” positive for anti-liver-kidney microsome-1 antibody. Clin Rheumatol. 2010;29:941-943.
22. Nagashima T, Kamata Y, Iwamoto M, et al. Liver dysfunction in anti-melanoma differentiation-associated gene 5 antibody-positive patients with dermatomyositis. Rheumatol Int. 2019;39:901-909.
1. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-347.
2. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785.
3. Gono T, Kawaguchi Y, Satoh T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49:1713-1719.
4. Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34.
5. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. 2017;49:20-26.
6. Kumakura S, Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheum. 2014;66:2297-2307.
7. Lake M, George G, Summer R. Time to personalize the treatment of anti-MDA-5 associated lung disease. Ann Rheum Dis. 2019;78:E52.
8. Kampylafka EI, Kosmidis ML, Panagiotakos DB, et al. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol. 2012;30:397-401.
9. Koguchi-Yoshioka H, Okiyama N, Iwamoto K, et al. Intravenous immunoglobulin contributes to the control of antimelanoma differentiation-associated protein 5 antibody-associated dermatomyositis with palmar violaceous macules/papules. Br J Dermatol. 2017;177:1442-1446.
10. Hamada-Ode K, Taniguchi Y, Kimata T, et al. High-dose intravenous immunoglobulin therapy for rapidly progressive interstitial pneumonitis accompanied by anti-melanoma differentiation-associated gene 5 antibody-positive amyopathic dermatomyositis. Eur J Rheumatol. 2015;2:83-85.
11. So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989.
12. Koichi Y, Aya Y, Megumi U, et al. A case of anti-MDA5-positive rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis ameliorated by rituximab, in addition to standard immunosuppressive treatment. Mod Rheumatol. 2017;27:536-540.
13. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. 2020;72:488-498.
14. Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune-associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246.
15. Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478.
16. Gono T, Miyake K, Kawaguchi Y, et al. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology (Oxford). 2012;51:1336-1338.
17. Wada T, Abe G, Kudou, T, et al. Liver damage in patients with polymyositis and dermatomyositis. Kitasato Med Journal. 2016;46:40-46.
18. Takahashi A, Abe K, Yokokawa J, et al. Clinical features of liver dysfunction in collagen diseases. Hepatol Res. 2010;40:1092-1097.
19. Matsumoto T, Kobayashi S, Shimizu H, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000;20:366-373.
20. Shi Q, Niu J, Huang X, et al. Do muscle enzyme changes forecast liver injury in polymyositis/dermatomyositis patients treated with methylprednisolone and methotrexate? Ann Clin Lab Sci. 2016;46:266-269.
21. Noda S, Asano Y, Tamaki Z, et al. A case of dermatomyositis with “liver disease associated with rheumatoid diseases” positive for anti-liver-kidney microsome-1 antibody. Clin Rheumatol. 2010;29:941-943.
22. Nagashima T, Kamata Y, Iwamoto M, et al. Liver dysfunction in anti-melanoma differentiation-associated gene 5 antibody-positive patients with dermatomyositis. Rheumatol Int. 2019;39:901-909.
PRACTICE POINTS
- Anti-MDA5 (melanoma differentiation–associated gene 5 antibody)–positive dermatomyositis associated with hemophagocytic lymphohistiocytosis is a rare and aggressive condition associated with a poor prognosis, and there is no standard treatment.
- Dermatomyositis-associated liver injury is not well defined.
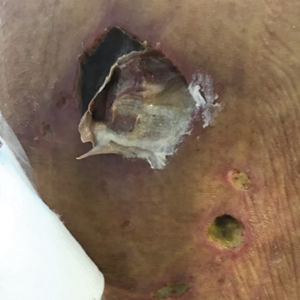
Efficacy of Etanercept in the Treatment of Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis
Regarded as dermatologic emergencies, Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) represent a spectrum of blistering skin diseases that have a high mortality rate. Because of a misguided immune response to medications or infections, CD8+ T lymphocytes release proinflammatory cytokines, giving rise to the extensive epidermal destruction seen in SJS and TEN. The exact pathogenesis of SJS and TEN is still poorly defined, but studies have proposed that T cells mediate keratinocyte (KC) apoptosis through perforin and granzyme release and activation of the Fas/Fas ligand (FasL). Functioning as a transmembrane death receptor in the tumor necrosis factor (TNF) superfamily, Fas (CD95) activates Fas-associated death domain protein, caspases, and nucleases, resulting in organized cell destruction. Likewise, perforin and granzymes also have been shown to play a similar role in apoptosis via activation of caspases.1
Evidence for the role of TNF-α in SJS and TEN has been supported by findings of elevated levels of TNF-α within the blister fluid, serum, and KC cell surface. Additionally, TNF-α has been shown to upregulate inducible nitric oxide synthase in KCs, causing an accumulation of nitric oxide and subsequent FasL-mediated cell death.1-3 Notably, studies have demonstrated a relative lack of lymphocytes in the tissue of TEN patients despite the extensive destruction that is observed, thus emphasizing the importance of amplification and cell signaling via inflammatory mediators such as TNF-α.1 In this proposed model, T cells release IFN-γ, causing KCs to release TNF-α that subsequently promotes the upregulation of the aforementioned FasL.1 Tumor necrosis factor α also may promote increased MHC class I complex deposition on KC surfaces that may play a role in perforin and granzyme-mediated apoptosis of KCs.1
There is still debate on the standard of care for the treatment of SJS and TEN, attributed to the absence of randomized controlled trials and the rarity of the disease as well as the numerous conflicting studies evaluating potential treatments.1,4 Despite conflicting data to support their use, supportive care and intravenous immunoglobulin (IVIG) continue to be common treatments for SJS and TEN in hospitals worldwide. Elucidation of the role of TNF-α has prompted the use of infliximab and etanercept. In a case series of Italian patients with TEN (average SCORTEN, 3.6) treated with the TNF-α antagonist etanercept, no mortality was observed, which was well below the calculated expected mortality of 46.9%.2 Our retrospective study compared the use of a TNF antagonist to other therapies in the treatment of SJS/TEN. Our data suggest that etanercept is a lifesaving and disease-modifying therapy.
Methods
Twenty-two patients with SJS/TEN were included in this analysis. This included all patients who carried a clinical diagnosis of SJS/TEN with a confirmatory biopsy at our 2 university centers—University of California, Los Angeles, and Keck-LA County-Norris Hospital at the University of Southern California, Los Angeles—from 2013 to 2016. The diagnosis was rendered when a clinical diagnosis of SJS/TEN was given by a dermatologist and a confirmatory biopsy was performed. Every patient given a diagnosis of SJS/TEN at either university system from 2015 onward received an injection of etanercept given the positive results reported by Paradisi et al.2
The 9 patients who presented from 2013 to 2014 to our 2 hospital systems and were given a diagnosis of SJS/TEN received either IVIG or supportive care alone and had an average body surface area (BSA) affected of 23%. The 13 patients who presented from 2015 to 2016 were treated with etanercept in the form of a 50-mg subcutaneous injection given once to the right upper arm. Of this group, 4 patients received dual therapy with both IVIG and etanercept. In the etanercept-treated group (etanercept alone and etanercept plus IVIG), the average BSA affected was 30%. At the time of preliminary diagnosis, all patient medications were evaluated for a possible temporal relationship to the onset of rash and were discontinued if felt to be causative. The causative agent and treatment course for each patient is summarized in Table 1.
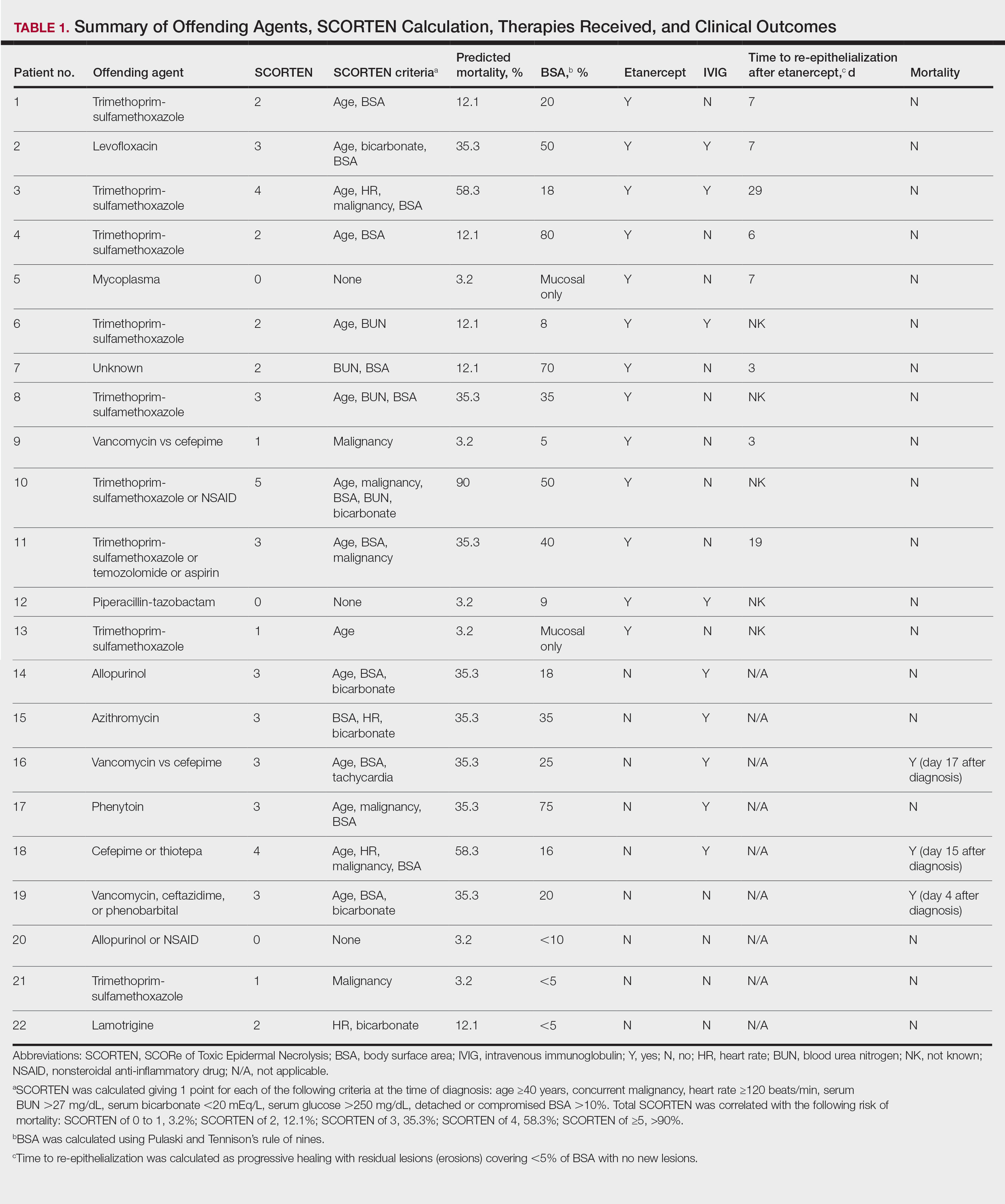
Patients were monitored daily in the hospital for improvement, and time to re-epithelialization was measured. Re-epithelialization was defined as progressive healing with residual lesions (erosions, ulcers, or bullae) covering no more than 5% BSA and was contingent on the patient having no new lesions within 24 hours.5 SCORe of Toxic Epidermal Necrosis (SCORTEN), a validated severity-of-illness score,6 was calculated by giving 1 point for each of the following criteria at the time of diagnosis: age ≥40 years, concurrent malignancy, heart rate ≥120 beats/min, serum blood urea nitrogen >27 mg/dL, serum bicarbonate <20 mEq/L, serum glucose >250 mg/dL, and detached or compromised BSA >10%. The total SCORTEN was correlated with the following risk of mortality as supported by prior validation studies: SCORTEN of 0 to 1, 3.2%; SCORTEN of 2, 12.1%; SCORTEN of 3, 35.3%; SCORTEN of 4, 58.3%; SCORTEN of ≥5, >90%.
Results
A total of 13 patients received etanercept. The mean SCORTEN was 2.2. The observed mortality was 0%, which was markedly lower than the predicted mortality of 24.3% (as determined by linear interpolation). Of this cohort, 9 patients received etanercept alone (mean SCORTEN of 2.1, predicted mortality of 22.9%), whereas 4 patients received a combination of etanercept and IVIG (mean SCORTEN of 2.3, predicted mortality of 27.2%).
The 4 patients who received both etanercept and IVIG received dual therapy for varying reasons. In patient 2 (Table 1), the perceived severity of this case ultimately led to the decision to start IVIG in addition to etanercept, resulting in rapid recovery and discharge after only 1 week of hospitalization. Intravenous immunoglobulin also was given in patient 3 (SCORTEN of 4) and patient 6 (SCORTEN of 2) for progression of disease despite administration of etanercept, with subsequent cessation of progression after the addition of the second agent (IVIG). Patient 12 might have done well on etanercept monotherapy but was administered IVIG as a precautionary measure because of hospital treatment algorithms.
Nine patients did not receive etanercept. Of this group, 5 received IVIG and 4 were managed with supportive care alone. The average SCORTEN for this group was 2.4, only slightly higher than the group that received etanercept (Table 2). The mortality rate in this group was 33%, which was higher than the predicted mortality of 28.1%.
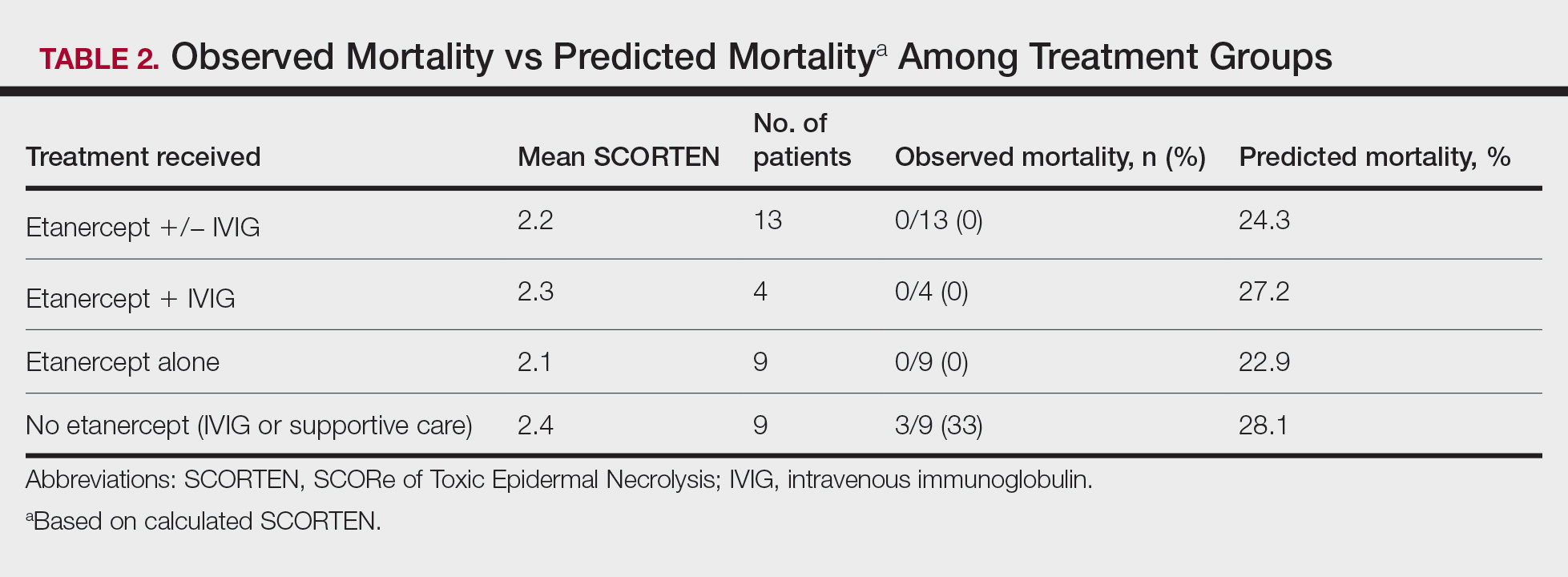
Re-epithelialization data were available for 8 patients who received etanercept. The average time to re-epithelialization for these patients was 8.9 days and ranged from 3 to 19 days. Of these patients, 2 received both IVIG and etanercept, with an average time to re-epithelialization of 13 days. For the 6 patients who received etanercept alone, the average time to re-epithelialization was 7.5 days. Re-epithelialization data were not available for any of the patients who received only IVIG or supportive care but to our recollection ranged from 14 to 21 days.
The clinical course of the 13 patients after the administration of a single dose of etanercept was remarkable, as there was complete absence of mortality and an increase in speed of recovery in most patients receiving this intervention (time to re-epithelialization, 3–19 days). We also observed another interesting trend from our patients treated with etanercept, which was the suggestion that treatment with etanercept may be less effective if IVIG and/or steroids are given prior to etanercept; likewise, treatment is more effective when etanercept is given quickly. For patients 1, 4, 5, 7, 9, and 11 (as shown in Table 1), no prior IVIG therapy or other immunosuppressive therapy had been given before etanercept was administered. In these 6 patients, the average time to re-epithelialization after etanercept administration was 7.5 days; average time to re-epithelialization, unfortunately, is not available for the patients who were not treated with etanercept. In addition, as shown in the Figure, it was noted in some patients that the depth of denudation was markedly more superficial than what would typically be clinically observed with TEN after administration of other immunomodulatory therapies such as IVIG or prednisone or with supportive care alone. In these 2 patients with superficial desquamation—patients 7 and 9—etanercept notably was given within 6 hours of onset of skin pain.
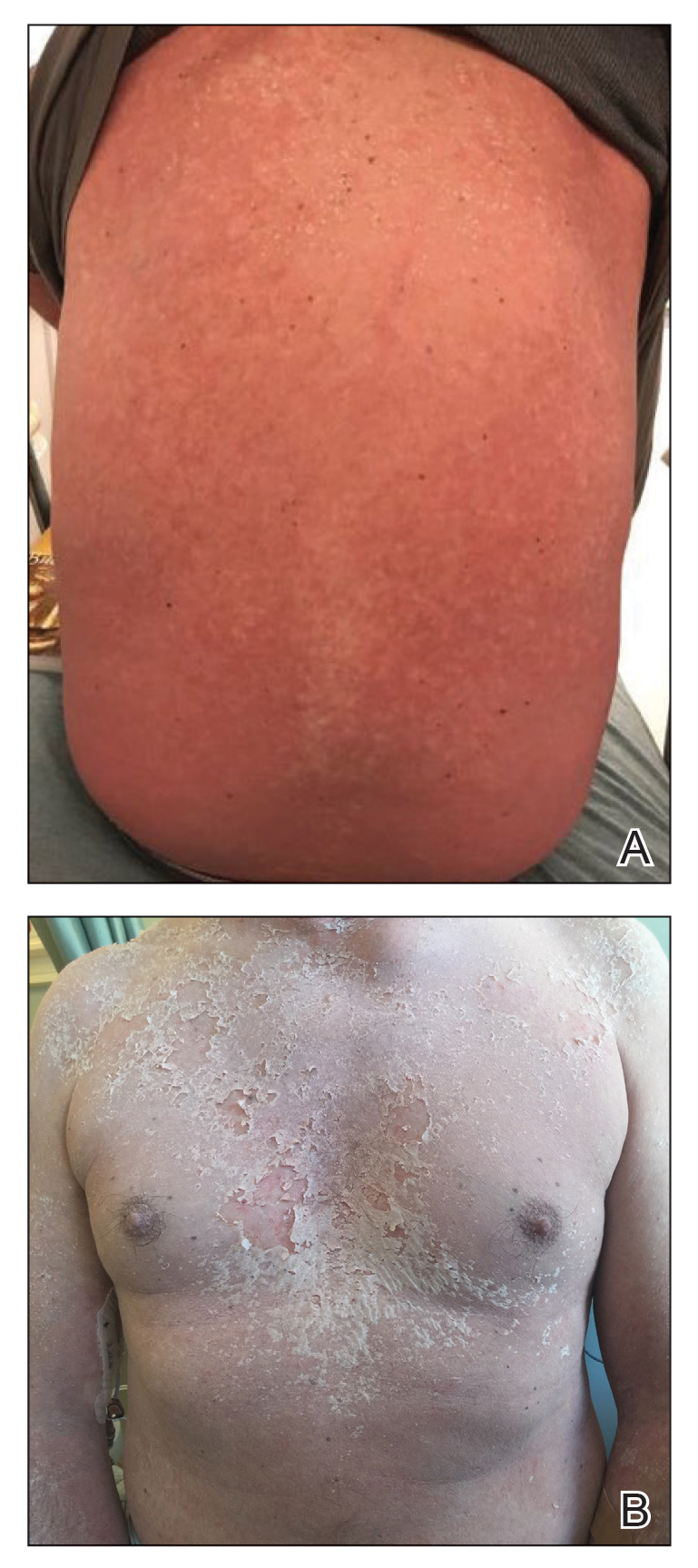
Comment
There is no definitive gold standard treatment of SJS, SJS/TEN overlap, or TEN. However, generally agreed upon management includes immediate discontinuation of the offending medication and supportive therapy with aggressive electrolyte replacement and wound care. Management in a burn unit or intensive care unit is recommended in severe cases. Contention over the efficacy of various medications in the treatment of SJS and TEN continues and largely is due to the rarity of SJS and TEN; studies are small and almost all lack randomization. Therapies that have been used include high-dose steroids, IVIG, plasmapheresis, cyclophosphamide, cyclosporine A, and TNF inhibitors (eg, etanercept, infliximab).1
Evidence for the use of anti–TNF-α antibodies has been limited thus far, with most of the literature focusing on infliximab and etanercept. Adalimumab, a fully humanized clonal antibody, has no reported cases in the dermatologic literature for use in patients with SJS/TEN. Two case reports of adalimumab paradoxically causing SJS have been documented. In both cases, adalimumab was stopped and patients responded to intravenous corticosteroids and infliximab.7,8 Similarly, thalidomide has not proven to be a promising anti–TNF-α agent for the treatment of SJS/TEN. In the only attempted randomized controlled trial for SJS and TEN, thalidomide appeared to increase mortality, eventuating in this trial being terminated prior to the planned end date.9Infliximab and etanercept have several case reports and a few case series highlighting potentially efficacious application of TNF-α inhibitors for the treatment of SJS/TEN.10-13 In 2002, Fischer et al10 reported the first case of TEN treated successfully with a single dose of infliximab 5 mg/kg. Kreft et al14 reported on etoricoxib-induced TEN that was treated with infliximab 5 mg/kg, which led to re-epithelialization within 5 weeks (notably a 5-week re-epithelialization time is not necessarily an improvement).
In 2005, Hunger et al3 demonstrated TNF-α’s release by KCs in the epidermis and by inflammatory cells in the dermis of a TEN patient. Twenty-four hours after the administration of infliximab 5 mg/kg in these patients, TNF-α was found to be below normal and epidermal detachment ceased.3 Wojtkietwicz et al13 demonstrated benefit following an infusion of infliximab 5 mg/kg in a patient whose disease continued to progress despite treatment with dexamethasone and 1.8 g/kg of IVIG.
Then 2 subsequent case series added further support for the efficacy of infliximab in the treatment of TEN. Patmanidis et al15 and Gaitanis et al16 reported similar results in 4 patients, each treated with infliximab 5 mg/kg immediately followed by initiation of high-dose IVIG (2 g/kg over 5 days). Zárate-Correa et al17 reported a 0% mortality rate and near-complete re-epithelialization after 5 to 14 days in 4 patients treated with a single 300-mg dose of infliximab.
However, the success of infliximab in the treatment of TEN has been countered by the pilot study by Paquet et al,18 which compared the efficacy of 150 mg/kg of N-acetylcysteine alone vs adding infliximab 5 mg/kg to treat 10 TEN patients. The study demonstrated no benefit at 48 hours in the group given infliximab, the time frame in which prior case reports touting infliximab’s benefit claimed the benefit was observed. Similarly, there was no effect on mortality for either treatment modality as assessed by illness auxiliary score.18
Evidence in support of the use of etanercept in the treatment of SJS/TEN is mounting, and some centers have begun to use it as the first-choice therapy for SJS/TEN. The first case was reported by Famularo et al,19 in which a patient with TEN was given 2 doses of etanercept 25 mg after failure to improve with prednisolone 1 mg/kg. The patient showed near-complete and rapid re-epithelization in 6 days before death due to disseminated intravascular coagulation 10 days after admission.19 Gubinelli et al20 and Sadighha21 independently reported cases of TEN and TEN/acute generalized exanthematous pustulosis overlap treated with a total of 50 mg of etanercept, demonstrating rapid cessation of lesion progression. Didona et al22 found similar benefit using etanercept 50 mg to treat TEN secondary to rituximab after failure to improve with prednisone and cyclophosphamide. Treatment of TEN with etanercept in an HIV-positive patient also has been reported. Lee et al23 described a patient who was administered 50-mg and 25-mg injections on days 3 and 5 of hospitalization, respectively, with re-epithelialization occurring by day 8. Finally, Owczarczyk-Saczonek et al24 reported a case of SJS in a patient with a 4-year history of etanercept and sulfasalazine treatment of rheumatoid arthritis; sulfasalazine was stopped, but this patient was continued on etanercept until resolution of skin and mucosal symptoms. However, it is important to consider the possibility of publication bias among these cases selected for their positive outcomes.
Perhaps the most compelling literature regarding the use of etanercept for TEN was described in a case series by Paradisi et al.2 This study included 10 patients with TEN, all of whom demonstrated complete re-epithelialization shortly after receiving etanercept 50 mg. Average SCORTEN was 3.6 with a range of 2 to 6. Eight patients in this study had severe comorbidities and all 10 patients survived, with a time to re-epithelialization ranging from 7 to 20 days.2 Additionally, a randomized controlled trial showed that 38 etanercept-treated patients had improved mortality (P=.266) and re-epithelialization time (P=.01) compared to patients treated with intravenous methylprednisolone.25Limitations to our study are similar to other reports of SJS/TEN and included the small number of cases and lack of randomization. Additionally, we do not have data available for all patients for time between onset of disease and treatment initiation. Because of these challenges, data presented in this case series is observational only. Additionally, the patients treated with etanercept alone had a slightly lower SCORTEN compared to the group that received IVIG or supportive care alone (2.1 and 2.4 respectively). However, the etanercept-only group actually had higher involvement of epidermal detachment (33%) compared to the non-etanercept group (23%).
Conclusion
Although treatment with etanercept lacks the support of a randomized controlled trial, similar to all other treatments currently used for SJS and TEN, preliminary reports highlight a benefit in disease progression and improvement in time to re-epithelialization. In particular, if etanercept 50 mg subcutaneously is given as monotherapy or is given early in the disease course (prior to other therapies being attempted and ideally within 6 hours of presentation), our data suggest an even greater trend toward improved mortality and decreased time to re-epithelialization. Additionally, our findings may suggest that in some patients, etanercept monotherapy is not an adequate intervention but the addition of IVIG may be helpful; however, the senior author (S.W.) notes anecdotally that in his experience with the patients treated at the University of California Los Angeles, the order of administration of combination therapies—etanercept followed by IVIG—was important in addition to the choice of therapy. These findings are promising enough to warrant a multicenter randomized controlled trial comparing the efficacy of etanercept to other more commonly used treatments for this spectrum of disease, including IVIG and/or cyclosporine. Based on the data presented in this case series, including the 13 patients who received etanercept and had a 0% mortality rate, etanercept may be viewed as a targeted therapeutic intervention for patients with SJS and TEN.
- Pereira FA, Mudgil AV, Rosmarin DM. Toxic epidermal necrolysis. J Am Acad Dermatol. 2007;56:181-200.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Hunger RE, Hunziker T, Buettiker U, et al. Rapid resolution of toxic epidermal necrolysis with anti-TNF-α treatment. J Allergy Clin Immunol. 2005;116:923-924.
- Worswick S, Cotliar J. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of treatment options. Dermatol Ther. 2011;24:207-218.
- Wallace AB. The exposure treatment of burns. Lancet Lond Engl. 1951;1:501-504.
- Bastuji-Garin S, Fouchard N, Bertocchi M, et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149-153.
- Mounach A, Rezqi A, Nouijai A, et al. Stevens-Johnson syndrome complicating adalimumab therapy in rheumatoid arthritis disease. Rheumatol Int. 2013;33:1351-1353.
- Salama M, Lawrance I-C. Stevens-Johnson syndrome complicating adalimumab therapy in Crohn’s disease. World J Gastroenterol. 2009;15:4449-4452.
- Wolkenstein P, Latarjet J, Roujeau JC, et al. Randomised comparison of thalidomide versus placebo in toxic epidermal necrolysis. Lancet Lond Engl. 1998;352:1586-1589.
- Fischer M, Fiedler E, Marsch WC, et al Antitumour necrosis factor-α antibodies (infliximab) in the treatment of a patient with toxic epidermal necrolysis. Br J Dermatol. 2002;146:707-709.
- Meiss F, Helmbold P, Meykadeh N, et al. Overlap of acute generalized exanthematous pustulosis and toxic epidermal necrolysis: response to antitumour necrosis factor-alpha antibody infliximab: report of three cases. J Eur Acad Dermatol Venereol. 2007;21:717-719.
- Al-Shouli S, Abouchala N, Bogusz MJ, et al. Toxic epidermal necrolysis associated with high intake of sildenafil and its response to infliximab. Acta Derm Venereol. 2005;85:534-535.
- Wojtkiewicz A, Wysocki M, Fortuna J, et al. Beneficial and rapid effect of infliximab on the course of toxic epidermal necrolysis. Acta Derm Venereol. 2008;88:420-421.
- Kreft B, Wohlrab J, Bramsiepe I, et al. Etoricoxib-induced toxic epidermal necrolysis: successful treatment with infliximab. J Dermatol. 2010;37:904-906.
- Patmanidis K, Sidiras A, Dolianitis K, et al. Combination of infliximab and high-dose intravenous immunoglobulin for toxic epidermal necrolysis: successful treatment of an elderly patient. Case Rep Dermatol Med. 2012;2012:915314.
- Gaitanis G, Spyridonos P, Patmanidis K, et al. Treatment of toxic epidermal necrolysis with the combination of infliximab and high-dose intravenous immunoglobulin. Dermatol Basel Switz. 2012;224:134-139.
- Zárate-Correa LC, Carrillo-Gómez DC, Ramírez-Escobar AF, et al. Toxic epidermal necrolysis successfully treated with infliximab. J Investig Allergol Clin Immunol. 2013;23:61-63.
- Paquet P, Jennes S, Rousseau AF, et al. Effect of N-acetylcysteine combined with infliximab on toxic epidermal necrolysis. a proof-of-concept study. Burns J Int Soc Burn Inj. 2014;40:1707-1712.
- Famularo G, Dona BD, Canzona F, et al. Etanercept for toxic epidermal necrolysis. Ann Pharmacother. 2007;41:1083-1084.
- Gubinelli E, Canzona F, Tonanzi T, et al. Toxic epidermal necrolysis successfully treated with etanercept. J Dermatol. 2009;36:150-153.
- Sadighha A. Etanercept in the treatment of a patient with acute generalized exanthematous pustulosis/toxic epidermal necrolysis: definition of a new model based on translational research. Int J Dermatol. 2009;48:913-914.
- Didona D, Paolino G, Garcovich S, et al. Successful use of etanercept in a case of toxic epidermal necrolysis induced by rituximab. J Eur Acad Dermatol Venereol. 2016;30:E83-E84.
- Lee Y-Y, Ko J-H, Wei C-H, et al. Use of etanercept to treat toxic epidermal necrolysis in a human immunodeficiency virus-positive patient. Dermatol Sin. 2013;31:78-81.
- Owczarczyk-Saczonek A, Zdanowska N, Znajewska-Pander A, et al. Stevens-Johnson syndrome in a patient with rheumatoid arthritis during long-term etanercept therapy. J Dermatol Case Rep. 2016;10:14-16.
- Wang CW, Yang LY, Chen CB, et al. Randomized, controlled trial of TNF-α antagonist in CTL mediated severe cutaneous adverse reactions. J Clin Invest. 2018;128:985-996.
Regarded as dermatologic emergencies, Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) represent a spectrum of blistering skin diseases that have a high mortality rate. Because of a misguided immune response to medications or infections, CD8+ T lymphocytes release proinflammatory cytokines, giving rise to the extensive epidermal destruction seen in SJS and TEN. The exact pathogenesis of SJS and TEN is still poorly defined, but studies have proposed that T cells mediate keratinocyte (KC) apoptosis through perforin and granzyme release and activation of the Fas/Fas ligand (FasL). Functioning as a transmembrane death receptor in the tumor necrosis factor (TNF) superfamily, Fas (CD95) activates Fas-associated death domain protein, caspases, and nucleases, resulting in organized cell destruction. Likewise, perforin and granzymes also have been shown to play a similar role in apoptosis via activation of caspases.1
Evidence for the role of TNF-α in SJS and TEN has been supported by findings of elevated levels of TNF-α within the blister fluid, serum, and KC cell surface. Additionally, TNF-α has been shown to upregulate inducible nitric oxide synthase in KCs, causing an accumulation of nitric oxide and subsequent FasL-mediated cell death.1-3 Notably, studies have demonstrated a relative lack of lymphocytes in the tissue of TEN patients despite the extensive destruction that is observed, thus emphasizing the importance of amplification and cell signaling via inflammatory mediators such as TNF-α.1 In this proposed model, T cells release IFN-γ, causing KCs to release TNF-α that subsequently promotes the upregulation of the aforementioned FasL.1 Tumor necrosis factor α also may promote increased MHC class I complex deposition on KC surfaces that may play a role in perforin and granzyme-mediated apoptosis of KCs.1
There is still debate on the standard of care for the treatment of SJS and TEN, attributed to the absence of randomized controlled trials and the rarity of the disease as well as the numerous conflicting studies evaluating potential treatments.1,4 Despite conflicting data to support their use, supportive care and intravenous immunoglobulin (IVIG) continue to be common treatments for SJS and TEN in hospitals worldwide. Elucidation of the role of TNF-α has prompted the use of infliximab and etanercept. In a case series of Italian patients with TEN (average SCORTEN, 3.6) treated with the TNF-α antagonist etanercept, no mortality was observed, which was well below the calculated expected mortality of 46.9%.2 Our retrospective study compared the use of a TNF antagonist to other therapies in the treatment of SJS/TEN. Our data suggest that etanercept is a lifesaving and disease-modifying therapy.
Methods
Twenty-two patients with SJS/TEN were included in this analysis. This included all patients who carried a clinical diagnosis of SJS/TEN with a confirmatory biopsy at our 2 university centers—University of California, Los Angeles, and Keck-LA County-Norris Hospital at the University of Southern California, Los Angeles—from 2013 to 2016. The diagnosis was rendered when a clinical diagnosis of SJS/TEN was given by a dermatologist and a confirmatory biopsy was performed. Every patient given a diagnosis of SJS/TEN at either university system from 2015 onward received an injection of etanercept given the positive results reported by Paradisi et al.2
The 9 patients who presented from 2013 to 2014 to our 2 hospital systems and were given a diagnosis of SJS/TEN received either IVIG or supportive care alone and had an average body surface area (BSA) affected of 23%. The 13 patients who presented from 2015 to 2016 were treated with etanercept in the form of a 50-mg subcutaneous injection given once to the right upper arm. Of this group, 4 patients received dual therapy with both IVIG and etanercept. In the etanercept-treated group (etanercept alone and etanercept plus IVIG), the average BSA affected was 30%. At the time of preliminary diagnosis, all patient medications were evaluated for a possible temporal relationship to the onset of rash and were discontinued if felt to be causative. The causative agent and treatment course for each patient is summarized in Table 1.
Patients were monitored daily in the hospital for improvement, and time to re-epithelialization was measured. Re-epithelialization was defined as progressive healing with residual lesions (erosions, ulcers, or bullae) covering no more than 5% BSA and was contingent on the patient having no new lesions within 24 hours.5 SCORe of Toxic Epidermal Necrosis (SCORTEN), a validated severity-of-illness score,6 was calculated by giving 1 point for each of the following criteria at the time of diagnosis: age ≥40 years, concurrent malignancy, heart rate ≥120 beats/min, serum blood urea nitrogen >27 mg/dL, serum bicarbonate <20 mEq/L, serum glucose >250 mg/dL, and detached or compromised BSA >10%. The total SCORTEN was correlated with the following risk of mortality as supported by prior validation studies: SCORTEN of 0 to 1, 3.2%; SCORTEN of 2, 12.1%; SCORTEN of 3, 35.3%; SCORTEN of 4, 58.3%; SCORTEN of ≥5, >90%.
Results
A total of 13 patients received etanercept. The mean SCORTEN was 2.2. The observed mortality was 0%, which was markedly lower than the predicted mortality of 24.3% (as determined by linear interpolation). Of this cohort, 9 patients received etanercept alone (mean SCORTEN of 2.1, predicted mortality of 22.9%), whereas 4 patients received a combination of etanercept and IVIG (mean SCORTEN of 2.3, predicted mortality of 27.2%).
The 4 patients who received both etanercept and IVIG received dual therapy for varying reasons. In patient 2 (Table 1), the perceived severity of this case ultimately led to the decision to start IVIG in addition to etanercept, resulting in rapid recovery and discharge after only 1 week of hospitalization. Intravenous immunoglobulin also was given in patient 3 (SCORTEN of 4) and patient 6 (SCORTEN of 2) for progression of disease despite administration of etanercept, with subsequent cessation of progression after the addition of the second agent (IVIG). Patient 12 might have done well on etanercept monotherapy but was administered IVIG as a precautionary measure because of hospital treatment algorithms.
Nine patients did not receive etanercept. Of this group, 5 received IVIG and 4 were managed with supportive care alone. The average SCORTEN for this group was 2.4, only slightly higher than the group that received etanercept (Table 2). The mortality rate in this group was 33%, which was higher than the predicted mortality of 28.1%.
Re-epithelialization data were available for 8 patients who received etanercept. The average time to re-epithelialization for these patients was 8.9 days and ranged from 3 to 19 days. Of these patients, 2 received both IVIG and etanercept, with an average time to re-epithelialization of 13 days. For the 6 patients who received etanercept alone, the average time to re-epithelialization was 7.5 days. Re-epithelialization data were not available for any of the patients who received only IVIG or supportive care but to our recollection ranged from 14 to 21 days.
The clinical course of the 13 patients after the administration of a single dose of etanercept was remarkable, as there was complete absence of mortality and an increase in speed of recovery in most patients receiving this intervention (time to re-epithelialization, 3–19 days). We also observed another interesting trend from our patients treated with etanercept, which was the suggestion that treatment with etanercept may be less effective if IVIG and/or steroids are given prior to etanercept; likewise, treatment is more effective when etanercept is given quickly. For patients 1, 4, 5, 7, 9, and 11 (as shown in Table 1), no prior IVIG therapy or other immunosuppressive therapy had been given before etanercept was administered. In these 6 patients, the average time to re-epithelialization after etanercept administration was 7.5 days; average time to re-epithelialization, unfortunately, is not available for the patients who were not treated with etanercept. In addition, as shown in the Figure, it was noted in some patients that the depth of denudation was markedly more superficial than what would typically be clinically observed with TEN after administration of other immunomodulatory therapies such as IVIG or prednisone or with supportive care alone. In these 2 patients with superficial desquamation—patients 7 and 9—etanercept notably was given within 6 hours of onset of skin pain.
Comment
There is no definitive gold standard treatment of SJS, SJS/TEN overlap, or TEN. However, generally agreed upon management includes immediate discontinuation of the offending medication and supportive therapy with aggressive electrolyte replacement and wound care. Management in a burn unit or intensive care unit is recommended in severe cases. Contention over the efficacy of various medications in the treatment of SJS and TEN continues and largely is due to the rarity of SJS and TEN; studies are small and almost all lack randomization. Therapies that have been used include high-dose steroids, IVIG, plasmapheresis, cyclophosphamide, cyclosporine A, and TNF inhibitors (eg, etanercept, infliximab).1
Evidence for the use of anti–TNF-α antibodies has been limited thus far, with most of the literature focusing on infliximab and etanercept. Adalimumab, a fully humanized clonal antibody, has no reported cases in the dermatologic literature for use in patients with SJS/TEN. Two case reports of adalimumab paradoxically causing SJS have been documented. In both cases, adalimumab was stopped and patients responded to intravenous corticosteroids and infliximab.7,8 Similarly, thalidomide has not proven to be a promising anti–TNF-α agent for the treatment of SJS/TEN. In the only attempted randomized controlled trial for SJS and TEN, thalidomide appeared to increase mortality, eventuating in this trial being terminated prior to the planned end date.9Infliximab and etanercept have several case reports and a few case series highlighting potentially efficacious application of TNF-α inhibitors for the treatment of SJS/TEN.10-13 In 2002, Fischer et al10 reported the first case of TEN treated successfully with a single dose of infliximab 5 mg/kg. Kreft et al14 reported on etoricoxib-induced TEN that was treated with infliximab 5 mg/kg, which led to re-epithelialization within 5 weeks (notably a 5-week re-epithelialization time is not necessarily an improvement).
In 2005, Hunger et al3 demonstrated TNF-α’s release by KCs in the epidermis and by inflammatory cells in the dermis of a TEN patient. Twenty-four hours after the administration of infliximab 5 mg/kg in these patients, TNF-α was found to be below normal and epidermal detachment ceased.3 Wojtkietwicz et al13 demonstrated benefit following an infusion of infliximab 5 mg/kg in a patient whose disease continued to progress despite treatment with dexamethasone and 1.8 g/kg of IVIG.
Then 2 subsequent case series added further support for the efficacy of infliximab in the treatment of TEN. Patmanidis et al15 and Gaitanis et al16 reported similar results in 4 patients, each treated with infliximab 5 mg/kg immediately followed by initiation of high-dose IVIG (2 g/kg over 5 days). Zárate-Correa et al17 reported a 0% mortality rate and near-complete re-epithelialization after 5 to 14 days in 4 patients treated with a single 300-mg dose of infliximab.
However, the success of infliximab in the treatment of TEN has been countered by the pilot study by Paquet et al,18 which compared the efficacy of 150 mg/kg of N-acetylcysteine alone vs adding infliximab 5 mg/kg to treat 10 TEN patients. The study demonstrated no benefit at 48 hours in the group given infliximab, the time frame in which prior case reports touting infliximab’s benefit claimed the benefit was observed. Similarly, there was no effect on mortality for either treatment modality as assessed by illness auxiliary score.18
Evidence in support of the use of etanercept in the treatment of SJS/TEN is mounting, and some centers have begun to use it as the first-choice therapy for SJS/TEN. The first case was reported by Famularo et al,19 in which a patient with TEN was given 2 doses of etanercept 25 mg after failure to improve with prednisolone 1 mg/kg. The patient showed near-complete and rapid re-epithelization in 6 days before death due to disseminated intravascular coagulation 10 days after admission.19 Gubinelli et al20 and Sadighha21 independently reported cases of TEN and TEN/acute generalized exanthematous pustulosis overlap treated with a total of 50 mg of etanercept, demonstrating rapid cessation of lesion progression. Didona et al22 found similar benefit using etanercept 50 mg to treat TEN secondary to rituximab after failure to improve with prednisone and cyclophosphamide. Treatment of TEN with etanercept in an HIV-positive patient also has been reported. Lee et al23 described a patient who was administered 50-mg and 25-mg injections on days 3 and 5 of hospitalization, respectively, with re-epithelialization occurring by day 8. Finally, Owczarczyk-Saczonek et al24 reported a case of SJS in a patient with a 4-year history of etanercept and sulfasalazine treatment of rheumatoid arthritis; sulfasalazine was stopped, but this patient was continued on etanercept until resolution of skin and mucosal symptoms. However, it is important to consider the possibility of publication bias among these cases selected for their positive outcomes.
Perhaps the most compelling literature regarding the use of etanercept for TEN was described in a case series by Paradisi et al.2 This study included 10 patients with TEN, all of whom demonstrated complete re-epithelialization shortly after receiving etanercept 50 mg. Average SCORTEN was 3.6 with a range of 2 to 6. Eight patients in this study had severe comorbidities and all 10 patients survived, with a time to re-epithelialization ranging from 7 to 20 days.2 Additionally, a randomized controlled trial showed that 38 etanercept-treated patients had improved mortality (P=.266) and re-epithelialization time (P=.01) compared to patients treated with intravenous methylprednisolone.25Limitations to our study are similar to other reports of SJS/TEN and included the small number of cases and lack of randomization. Additionally, we do not have data available for all patients for time between onset of disease and treatment initiation. Because of these challenges, data presented in this case series is observational only. Additionally, the patients treated with etanercept alone had a slightly lower SCORTEN compared to the group that received IVIG or supportive care alone (2.1 and 2.4 respectively). However, the etanercept-only group actually had higher involvement of epidermal detachment (33%) compared to the non-etanercept group (23%).
Conclusion
Although treatment with etanercept lacks the support of a randomized controlled trial, similar to all other treatments currently used for SJS and TEN, preliminary reports highlight a benefit in disease progression and improvement in time to re-epithelialization. In particular, if etanercept 50 mg subcutaneously is given as monotherapy or is given early in the disease course (prior to other therapies being attempted and ideally within 6 hours of presentation), our data suggest an even greater trend toward improved mortality and decreased time to re-epithelialization. Additionally, our findings may suggest that in some patients, etanercept monotherapy is not an adequate intervention but the addition of IVIG may be helpful; however, the senior author (S.W.) notes anecdotally that in his experience with the patients treated at the University of California Los Angeles, the order of administration of combination therapies—etanercept followed by IVIG—was important in addition to the choice of therapy. These findings are promising enough to warrant a multicenter randomized controlled trial comparing the efficacy of etanercept to other more commonly used treatments for this spectrum of disease, including IVIG and/or cyclosporine. Based on the data presented in this case series, including the 13 patients who received etanercept and had a 0% mortality rate, etanercept may be viewed as a targeted therapeutic intervention for patients with SJS and TEN.
Regarded as dermatologic emergencies, Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) represent a spectrum of blistering skin diseases that have a high mortality rate. Because of a misguided immune response to medications or infections, CD8+ T lymphocytes release proinflammatory cytokines, giving rise to the extensive epidermal destruction seen in SJS and TEN. The exact pathogenesis of SJS and TEN is still poorly defined, but studies have proposed that T cells mediate keratinocyte (KC) apoptosis through perforin and granzyme release and activation of the Fas/Fas ligand (FasL). Functioning as a transmembrane death receptor in the tumor necrosis factor (TNF) superfamily, Fas (CD95) activates Fas-associated death domain protein, caspases, and nucleases, resulting in organized cell destruction. Likewise, perforin and granzymes also have been shown to play a similar role in apoptosis via activation of caspases.1
Evidence for the role of TNF-α in SJS and TEN has been supported by findings of elevated levels of TNF-α within the blister fluid, serum, and KC cell surface. Additionally, TNF-α has been shown to upregulate inducible nitric oxide synthase in KCs, causing an accumulation of nitric oxide and subsequent FasL-mediated cell death.1-3 Notably, studies have demonstrated a relative lack of lymphocytes in the tissue of TEN patients despite the extensive destruction that is observed, thus emphasizing the importance of amplification and cell signaling via inflammatory mediators such as TNF-α.1 In this proposed model, T cells release IFN-γ, causing KCs to release TNF-α that subsequently promotes the upregulation of the aforementioned FasL.1 Tumor necrosis factor α also may promote increased MHC class I complex deposition on KC surfaces that may play a role in perforin and granzyme-mediated apoptosis of KCs.1
There is still debate on the standard of care for the treatment of SJS and TEN, attributed to the absence of randomized controlled trials and the rarity of the disease as well as the numerous conflicting studies evaluating potential treatments.1,4 Despite conflicting data to support their use, supportive care and intravenous immunoglobulin (IVIG) continue to be common treatments for SJS and TEN in hospitals worldwide. Elucidation of the role of TNF-α has prompted the use of infliximab and etanercept. In a case series of Italian patients with TEN (average SCORTEN, 3.6) treated with the TNF-α antagonist etanercept, no mortality was observed, which was well below the calculated expected mortality of 46.9%.2 Our retrospective study compared the use of a TNF antagonist to other therapies in the treatment of SJS/TEN. Our data suggest that etanercept is a lifesaving and disease-modifying therapy.
Methods
Twenty-two patients with SJS/TEN were included in this analysis. This included all patients who carried a clinical diagnosis of SJS/TEN with a confirmatory biopsy at our 2 university centers—University of California, Los Angeles, and Keck-LA County-Norris Hospital at the University of Southern California, Los Angeles—from 2013 to 2016. The diagnosis was rendered when a clinical diagnosis of SJS/TEN was given by a dermatologist and a confirmatory biopsy was performed. Every patient given a diagnosis of SJS/TEN at either university system from 2015 onward received an injection of etanercept given the positive results reported by Paradisi et al.2
The 9 patients who presented from 2013 to 2014 to our 2 hospital systems and were given a diagnosis of SJS/TEN received either IVIG or supportive care alone and had an average body surface area (BSA) affected of 23%. The 13 patients who presented from 2015 to 2016 were treated with etanercept in the form of a 50-mg subcutaneous injection given once to the right upper arm. Of this group, 4 patients received dual therapy with both IVIG and etanercept. In the etanercept-treated group (etanercept alone and etanercept plus IVIG), the average BSA affected was 30%. At the time of preliminary diagnosis, all patient medications were evaluated for a possible temporal relationship to the onset of rash and were discontinued if felt to be causative. The causative agent and treatment course for each patient is summarized in Table 1.
Patients were monitored daily in the hospital for improvement, and time to re-epithelialization was measured. Re-epithelialization was defined as progressive healing with residual lesions (erosions, ulcers, or bullae) covering no more than 5% BSA and was contingent on the patient having no new lesions within 24 hours.5 SCORe of Toxic Epidermal Necrosis (SCORTEN), a validated severity-of-illness score,6 was calculated by giving 1 point for each of the following criteria at the time of diagnosis: age ≥40 years, concurrent malignancy, heart rate ≥120 beats/min, serum blood urea nitrogen >27 mg/dL, serum bicarbonate <20 mEq/L, serum glucose >250 mg/dL, and detached or compromised BSA >10%. The total SCORTEN was correlated with the following risk of mortality as supported by prior validation studies: SCORTEN of 0 to 1, 3.2%; SCORTEN of 2, 12.1%; SCORTEN of 3, 35.3%; SCORTEN of 4, 58.3%; SCORTEN of ≥5, >90%.
Results
A total of 13 patients received etanercept. The mean SCORTEN was 2.2. The observed mortality was 0%, which was markedly lower than the predicted mortality of 24.3% (as determined by linear interpolation). Of this cohort, 9 patients received etanercept alone (mean SCORTEN of 2.1, predicted mortality of 22.9%), whereas 4 patients received a combination of etanercept and IVIG (mean SCORTEN of 2.3, predicted mortality of 27.2%).
The 4 patients who received both etanercept and IVIG received dual therapy for varying reasons. In patient 2 (Table 1), the perceived severity of this case ultimately led to the decision to start IVIG in addition to etanercept, resulting in rapid recovery and discharge after only 1 week of hospitalization. Intravenous immunoglobulin also was given in patient 3 (SCORTEN of 4) and patient 6 (SCORTEN of 2) for progression of disease despite administration of etanercept, with subsequent cessation of progression after the addition of the second agent (IVIG). Patient 12 might have done well on etanercept monotherapy but was administered IVIG as a precautionary measure because of hospital treatment algorithms.
Nine patients did not receive etanercept. Of this group, 5 received IVIG and 4 were managed with supportive care alone. The average SCORTEN for this group was 2.4, only slightly higher than the group that received etanercept (Table 2). The mortality rate in this group was 33%, which was higher than the predicted mortality of 28.1%.
Re-epithelialization data were available for 8 patients who received etanercept. The average time to re-epithelialization for these patients was 8.9 days and ranged from 3 to 19 days. Of these patients, 2 received both IVIG and etanercept, with an average time to re-epithelialization of 13 days. For the 6 patients who received etanercept alone, the average time to re-epithelialization was 7.5 days. Re-epithelialization data were not available for any of the patients who received only IVIG or supportive care but to our recollection ranged from 14 to 21 days.
The clinical course of the 13 patients after the administration of a single dose of etanercept was remarkable, as there was complete absence of mortality and an increase in speed of recovery in most patients receiving this intervention (time to re-epithelialization, 3–19 days). We also observed another interesting trend from our patients treated with etanercept, which was the suggestion that treatment with etanercept may be less effective if IVIG and/or steroids are given prior to etanercept; likewise, treatment is more effective when etanercept is given quickly. For patients 1, 4, 5, 7, 9, and 11 (as shown in Table 1), no prior IVIG therapy or other immunosuppressive therapy had been given before etanercept was administered. In these 6 patients, the average time to re-epithelialization after etanercept administration was 7.5 days; average time to re-epithelialization, unfortunately, is not available for the patients who were not treated with etanercept. In addition, as shown in the Figure, it was noted in some patients that the depth of denudation was markedly more superficial than what would typically be clinically observed with TEN after administration of other immunomodulatory therapies such as IVIG or prednisone or with supportive care alone. In these 2 patients with superficial desquamation—patients 7 and 9—etanercept notably was given within 6 hours of onset of skin pain.
Comment
There is no definitive gold standard treatment of SJS, SJS/TEN overlap, or TEN. However, generally agreed upon management includes immediate discontinuation of the offending medication and supportive therapy with aggressive electrolyte replacement and wound care. Management in a burn unit or intensive care unit is recommended in severe cases. Contention over the efficacy of various medications in the treatment of SJS and TEN continues and largely is due to the rarity of SJS and TEN; studies are small and almost all lack randomization. Therapies that have been used include high-dose steroids, IVIG, plasmapheresis, cyclophosphamide, cyclosporine A, and TNF inhibitors (eg, etanercept, infliximab).1
Evidence for the use of anti–TNF-α antibodies has been limited thus far, with most of the literature focusing on infliximab and etanercept. Adalimumab, a fully humanized clonal antibody, has no reported cases in the dermatologic literature for use in patients with SJS/TEN. Two case reports of adalimumab paradoxically causing SJS have been documented. In both cases, adalimumab was stopped and patients responded to intravenous corticosteroids and infliximab.7,8 Similarly, thalidomide has not proven to be a promising anti–TNF-α agent for the treatment of SJS/TEN. In the only attempted randomized controlled trial for SJS and TEN, thalidomide appeared to increase mortality, eventuating in this trial being terminated prior to the planned end date.9Infliximab and etanercept have several case reports and a few case series highlighting potentially efficacious application of TNF-α inhibitors for the treatment of SJS/TEN.10-13 In 2002, Fischer et al10 reported the first case of TEN treated successfully with a single dose of infliximab 5 mg/kg. Kreft et al14 reported on etoricoxib-induced TEN that was treated with infliximab 5 mg/kg, which led to re-epithelialization within 5 weeks (notably a 5-week re-epithelialization time is not necessarily an improvement).
In 2005, Hunger et al3 demonstrated TNF-α’s release by KCs in the epidermis and by inflammatory cells in the dermis of a TEN patient. Twenty-four hours after the administration of infliximab 5 mg/kg in these patients, TNF-α was found to be below normal and epidermal detachment ceased.3 Wojtkietwicz et al13 demonstrated benefit following an infusion of infliximab 5 mg/kg in a patient whose disease continued to progress despite treatment with dexamethasone and 1.8 g/kg of IVIG.
Then 2 subsequent case series added further support for the efficacy of infliximab in the treatment of TEN. Patmanidis et al15 and Gaitanis et al16 reported similar results in 4 patients, each treated with infliximab 5 mg/kg immediately followed by initiation of high-dose IVIG (2 g/kg over 5 days). Zárate-Correa et al17 reported a 0% mortality rate and near-complete re-epithelialization after 5 to 14 days in 4 patients treated with a single 300-mg dose of infliximab.
However, the success of infliximab in the treatment of TEN has been countered by the pilot study by Paquet et al,18 which compared the efficacy of 150 mg/kg of N-acetylcysteine alone vs adding infliximab 5 mg/kg to treat 10 TEN patients. The study demonstrated no benefit at 48 hours in the group given infliximab, the time frame in which prior case reports touting infliximab’s benefit claimed the benefit was observed. Similarly, there was no effect on mortality for either treatment modality as assessed by illness auxiliary score.18
Evidence in support of the use of etanercept in the treatment of SJS/TEN is mounting, and some centers have begun to use it as the first-choice therapy for SJS/TEN. The first case was reported by Famularo et al,19 in which a patient with TEN was given 2 doses of etanercept 25 mg after failure to improve with prednisolone 1 mg/kg. The patient showed near-complete and rapid re-epithelization in 6 days before death due to disseminated intravascular coagulation 10 days after admission.19 Gubinelli et al20 and Sadighha21 independently reported cases of TEN and TEN/acute generalized exanthematous pustulosis overlap treated with a total of 50 mg of etanercept, demonstrating rapid cessation of lesion progression. Didona et al22 found similar benefit using etanercept 50 mg to treat TEN secondary to rituximab after failure to improve with prednisone and cyclophosphamide. Treatment of TEN with etanercept in an HIV-positive patient also has been reported. Lee et al23 described a patient who was administered 50-mg and 25-mg injections on days 3 and 5 of hospitalization, respectively, with re-epithelialization occurring by day 8. Finally, Owczarczyk-Saczonek et al24 reported a case of SJS in a patient with a 4-year history of etanercept and sulfasalazine treatment of rheumatoid arthritis; sulfasalazine was stopped, but this patient was continued on etanercept until resolution of skin and mucosal symptoms. However, it is important to consider the possibility of publication bias among these cases selected for their positive outcomes.
Perhaps the most compelling literature regarding the use of etanercept for TEN was described in a case series by Paradisi et al.2 This study included 10 patients with TEN, all of whom demonstrated complete re-epithelialization shortly after receiving etanercept 50 mg. Average SCORTEN was 3.6 with a range of 2 to 6. Eight patients in this study had severe comorbidities and all 10 patients survived, with a time to re-epithelialization ranging from 7 to 20 days.2 Additionally, a randomized controlled trial showed that 38 etanercept-treated patients had improved mortality (P=.266) and re-epithelialization time (P=.01) compared to patients treated with intravenous methylprednisolone.25Limitations to our study are similar to other reports of SJS/TEN and included the small number of cases and lack of randomization. Additionally, we do not have data available for all patients for time between onset of disease and treatment initiation. Because of these challenges, data presented in this case series is observational only. Additionally, the patients treated with etanercept alone had a slightly lower SCORTEN compared to the group that received IVIG or supportive care alone (2.1 and 2.4 respectively). However, the etanercept-only group actually had higher involvement of epidermal detachment (33%) compared to the non-etanercept group (23%).
Conclusion
Although treatment with etanercept lacks the support of a randomized controlled trial, similar to all other treatments currently used for SJS and TEN, preliminary reports highlight a benefit in disease progression and improvement in time to re-epithelialization. In particular, if etanercept 50 mg subcutaneously is given as monotherapy or is given early in the disease course (prior to other therapies being attempted and ideally within 6 hours of presentation), our data suggest an even greater trend toward improved mortality and decreased time to re-epithelialization. Additionally, our findings may suggest that in some patients, etanercept monotherapy is not an adequate intervention but the addition of IVIG may be helpful; however, the senior author (S.W.) notes anecdotally that in his experience with the patients treated at the University of California Los Angeles, the order of administration of combination therapies—etanercept followed by IVIG—was important in addition to the choice of therapy. These findings are promising enough to warrant a multicenter randomized controlled trial comparing the efficacy of etanercept to other more commonly used treatments for this spectrum of disease, including IVIG and/or cyclosporine. Based on the data presented in this case series, including the 13 patients who received etanercept and had a 0% mortality rate, etanercept may be viewed as a targeted therapeutic intervention for patients with SJS and TEN.
- Pereira FA, Mudgil AV, Rosmarin DM. Toxic epidermal necrolysis. J Am Acad Dermatol. 2007;56:181-200.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Hunger RE, Hunziker T, Buettiker U, et al. Rapid resolution of toxic epidermal necrolysis with anti-TNF-α treatment. J Allergy Clin Immunol. 2005;116:923-924.
- Worswick S, Cotliar J. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of treatment options. Dermatol Ther. 2011;24:207-218.
- Wallace AB. The exposure treatment of burns. Lancet Lond Engl. 1951;1:501-504.
- Bastuji-Garin S, Fouchard N, Bertocchi M, et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149-153.
- Mounach A, Rezqi A, Nouijai A, et al. Stevens-Johnson syndrome complicating adalimumab therapy in rheumatoid arthritis disease. Rheumatol Int. 2013;33:1351-1353.
- Salama M, Lawrance I-C. Stevens-Johnson syndrome complicating adalimumab therapy in Crohn’s disease. World J Gastroenterol. 2009;15:4449-4452.
- Wolkenstein P, Latarjet J, Roujeau JC, et al. Randomised comparison of thalidomide versus placebo in toxic epidermal necrolysis. Lancet Lond Engl. 1998;352:1586-1589.
- Fischer M, Fiedler E, Marsch WC, et al Antitumour necrosis factor-α antibodies (infliximab) in the treatment of a patient with toxic epidermal necrolysis. Br J Dermatol. 2002;146:707-709.
- Meiss F, Helmbold P, Meykadeh N, et al. Overlap of acute generalized exanthematous pustulosis and toxic epidermal necrolysis: response to antitumour necrosis factor-alpha antibody infliximab: report of three cases. J Eur Acad Dermatol Venereol. 2007;21:717-719.
- Al-Shouli S, Abouchala N, Bogusz MJ, et al. Toxic epidermal necrolysis associated with high intake of sildenafil and its response to infliximab. Acta Derm Venereol. 2005;85:534-535.
- Wojtkiewicz A, Wysocki M, Fortuna J, et al. Beneficial and rapid effect of infliximab on the course of toxic epidermal necrolysis. Acta Derm Venereol. 2008;88:420-421.
- Kreft B, Wohlrab J, Bramsiepe I, et al. Etoricoxib-induced toxic epidermal necrolysis: successful treatment with infliximab. J Dermatol. 2010;37:904-906.
- Patmanidis K, Sidiras A, Dolianitis K, et al. Combination of infliximab and high-dose intravenous immunoglobulin for toxic epidermal necrolysis: successful treatment of an elderly patient. Case Rep Dermatol Med. 2012;2012:915314.
- Gaitanis G, Spyridonos P, Patmanidis K, et al. Treatment of toxic epidermal necrolysis with the combination of infliximab and high-dose intravenous immunoglobulin. Dermatol Basel Switz. 2012;224:134-139.
- Zárate-Correa LC, Carrillo-Gómez DC, Ramírez-Escobar AF, et al. Toxic epidermal necrolysis successfully treated with infliximab. J Investig Allergol Clin Immunol. 2013;23:61-63.
- Paquet P, Jennes S, Rousseau AF, et al. Effect of N-acetylcysteine combined with infliximab on toxic epidermal necrolysis. a proof-of-concept study. Burns J Int Soc Burn Inj. 2014;40:1707-1712.
- Famularo G, Dona BD, Canzona F, et al. Etanercept for toxic epidermal necrolysis. Ann Pharmacother. 2007;41:1083-1084.
- Gubinelli E, Canzona F, Tonanzi T, et al. Toxic epidermal necrolysis successfully treated with etanercept. J Dermatol. 2009;36:150-153.
- Sadighha A. Etanercept in the treatment of a patient with acute generalized exanthematous pustulosis/toxic epidermal necrolysis: definition of a new model based on translational research. Int J Dermatol. 2009;48:913-914.
- Didona D, Paolino G, Garcovich S, et al. Successful use of etanercept in a case of toxic epidermal necrolysis induced by rituximab. J Eur Acad Dermatol Venereol. 2016;30:E83-E84.
- Lee Y-Y, Ko J-H, Wei C-H, et al. Use of etanercept to treat toxic epidermal necrolysis in a human immunodeficiency virus-positive patient. Dermatol Sin. 2013;31:78-81.
- Owczarczyk-Saczonek A, Zdanowska N, Znajewska-Pander A, et al. Stevens-Johnson syndrome in a patient with rheumatoid arthritis during long-term etanercept therapy. J Dermatol Case Rep. 2016;10:14-16.
- Wang CW, Yang LY, Chen CB, et al. Randomized, controlled trial of TNF-α antagonist in CTL mediated severe cutaneous adverse reactions. J Clin Invest. 2018;128:985-996.
- Pereira FA, Mudgil AV, Rosmarin DM. Toxic epidermal necrolysis. J Am Acad Dermatol. 2007;56:181-200.
- Paradisi A, Abeni D, Bergamo F, et al. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-283.
- Hunger RE, Hunziker T, Buettiker U, et al. Rapid resolution of toxic epidermal necrolysis with anti-TNF-α treatment. J Allergy Clin Immunol. 2005;116:923-924.
- Worswick S, Cotliar J. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of treatment options. Dermatol Ther. 2011;24:207-218.
- Wallace AB. The exposure treatment of burns. Lancet Lond Engl. 1951;1:501-504.
- Bastuji-Garin S, Fouchard N, Bertocchi M, et al. SCORTEN: a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149-153.
- Mounach A, Rezqi A, Nouijai A, et al. Stevens-Johnson syndrome complicating adalimumab therapy in rheumatoid arthritis disease. Rheumatol Int. 2013;33:1351-1353.
- Salama M, Lawrance I-C. Stevens-Johnson syndrome complicating adalimumab therapy in Crohn’s disease. World J Gastroenterol. 2009;15:4449-4452.
- Wolkenstein P, Latarjet J, Roujeau JC, et al. Randomised comparison of thalidomide versus placebo in toxic epidermal necrolysis. Lancet Lond Engl. 1998;352:1586-1589.
- Fischer M, Fiedler E, Marsch WC, et al Antitumour necrosis factor-α antibodies (infliximab) in the treatment of a patient with toxic epidermal necrolysis. Br J Dermatol. 2002;146:707-709.
- Meiss F, Helmbold P, Meykadeh N, et al. Overlap of acute generalized exanthematous pustulosis and toxic epidermal necrolysis: response to antitumour necrosis factor-alpha antibody infliximab: report of three cases. J Eur Acad Dermatol Venereol. 2007;21:717-719.
- Al-Shouli S, Abouchala N, Bogusz MJ, et al. Toxic epidermal necrolysis associated with high intake of sildenafil and its response to infliximab. Acta Derm Venereol. 2005;85:534-535.
- Wojtkiewicz A, Wysocki M, Fortuna J, et al. Beneficial and rapid effect of infliximab on the course of toxic epidermal necrolysis. Acta Derm Venereol. 2008;88:420-421.
- Kreft B, Wohlrab J, Bramsiepe I, et al. Etoricoxib-induced toxic epidermal necrolysis: successful treatment with infliximab. J Dermatol. 2010;37:904-906.
- Patmanidis K, Sidiras A, Dolianitis K, et al. Combination of infliximab and high-dose intravenous immunoglobulin for toxic epidermal necrolysis: successful treatment of an elderly patient. Case Rep Dermatol Med. 2012;2012:915314.
- Gaitanis G, Spyridonos P, Patmanidis K, et al. Treatment of toxic epidermal necrolysis with the combination of infliximab and high-dose intravenous immunoglobulin. Dermatol Basel Switz. 2012;224:134-139.
- Zárate-Correa LC, Carrillo-Gómez DC, Ramírez-Escobar AF, et al. Toxic epidermal necrolysis successfully treated with infliximab. J Investig Allergol Clin Immunol. 2013;23:61-63.
- Paquet P, Jennes S, Rousseau AF, et al. Effect of N-acetylcysteine combined with infliximab on toxic epidermal necrolysis. a proof-of-concept study. Burns J Int Soc Burn Inj. 2014;40:1707-1712.
- Famularo G, Dona BD, Canzona F, et al. Etanercept for toxic epidermal necrolysis. Ann Pharmacother. 2007;41:1083-1084.
- Gubinelli E, Canzona F, Tonanzi T, et al. Toxic epidermal necrolysis successfully treated with etanercept. J Dermatol. 2009;36:150-153.
- Sadighha A. Etanercept in the treatment of a patient with acute generalized exanthematous pustulosis/toxic epidermal necrolysis: definition of a new model based on translational research. Int J Dermatol. 2009;48:913-914.
- Didona D, Paolino G, Garcovich S, et al. Successful use of etanercept in a case of toxic epidermal necrolysis induced by rituximab. J Eur Acad Dermatol Venereol. 2016;30:E83-E84.
- Lee Y-Y, Ko J-H, Wei C-H, et al. Use of etanercept to treat toxic epidermal necrolysis in a human immunodeficiency virus-positive patient. Dermatol Sin. 2013;31:78-81.
- Owczarczyk-Saczonek A, Zdanowska N, Znajewska-Pander A, et al. Stevens-Johnson syndrome in a patient with rheumatoid arthritis during long-term etanercept therapy. J Dermatol Case Rep. 2016;10:14-16.
- Wang CW, Yang LY, Chen CB, et al. Randomized, controlled trial of TNF-α antagonist in CTL mediated severe cutaneous adverse reactions. J Clin Invest. 2018;128:985-996.
Practice Points
- Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are life-threatening dermatologic emergencies without a universally accepted treatment.
- Results of this study support the use of single-dose subcutaneous etanercept 50 mg as a potentially lifesaving therapy for patients with SJS/TEN.
Stevens-Johnson Syndrome/Toxic Epidermal Necrolysis Overlap Syndrome in a Patient With Relapsing Polychondritis
To the Editor:
Relapsing polychondritis (RP) is a chronic, progressive, and episodic systemic inflammatory disease that primarily affects the cartilaginous structures of the ears and nose. Involvement of other proteoglycan-rich structures such as the joints, eyes, inner ears, blood vessels, heart, and kidneys also may be seen. Dermatologic manifestations occur in 35% to 50% of patients and may be the presenting sign in up to 12% of cases.1 The most commonly reported dermatologic findings include oral aphthosis, erythema nodosum, and purpura with vasculitic changes. Less commonly reported associations include Sweet syndrome, pyoderma gangrenosum, panniculitis, erythema elevatum diutinum, erythema annulare centrifugum, and erythema multiforme.1
A 43-year-old woman who was otherwise healthy developed new-onset tenderness and swelling of the left pinna while on vacation. She was treated with trimethoprim-sulfamethoxazole, clindamycin, and levofloxacin for presumed auricular cellulitis. The patient developed a fever; sore throat; and a progressive, pruritic, blistering rash on the face, torso, bilateral extremities, palms, and soles 1 day after completing the antibiotic course. After 5 days of unremitting symptoms despite oral, intramuscular, and topical steroids, the patient presented to the emergency department. Physical examination revealed diffuse, tender, erythematous to violaceous macules with varying degrees of coalescence on the chest, back, and extremities. Scattered flaccid bullae and erosions of the oral and genital mucosa also were seen. Laboratory analysis was notable only for a urinary tract infection with Klebsiella pneumoniae. A punch biopsy demonstrated full-thickness necrosis of the epidermis with subepidermal bullae and a mild to moderate lymphocytic infiltrate with rare eosinophils, consistent with a diagnosis of Stevens-Johnson syndrome (SJS). Because of the body surface area involved (20%) and the recent history of trimethoprim-sulfamethoxazole use, a diagnosis of SJS/toxic epidermal necrolysis (TEN) overlap syndrome was made. The patient was successfully treated with subcutaneous etanercept (50 mg), supportive care, and cephalexin for the urinary tract infection.
Approximately 5 weeks after discharge from the hospital, the patient was evaluated for new-onset pain and swelling of the right ear (Figure) in conjunction with recent tenderness and depression of the superior septal structure of the nose. A punch biopsy of the ear revealed mild perichondral inflammation without vasculitic changes and a superficial, deep perivascular, and periadnexal lymphoplasmacytic inflammatory infiltrate with scattered eosinophils. A diagnosis of RP was made, as the patient met Damiani and Levine’s2 criteria with bilateral auricular inflammation, ocular inflammation, and nasal chondritis.

Although the exact pathogenesis of RP remains unclear, there is strong evidence to suggest an underlying autoimmune etiology.3 Autoantibodies against type II collagen, in addition to other minor collagen and cartilage proteins, such as cartilage oligomeric matrix proteins and matrilin-1, are seen in a subset of patients. Titers have been reported to correlate with disease activity.3,4 Direct immunofluorescence also has demonstrated plentiful CD4+ T cells, as well as IgM, IgA, IgG, and C3 deposits in the inflamed cartilage of patients with RP.3 Additionally, approximately 30% of patients with RP will have another autoimmune disease, and more than 50% of patients with RP carry the HLA-DR4 antigen.3 Alternatively, SJS and TEN are not reported in association with autoimmune diseases and are believed to be CD8+ T-cell driven. Some HLA-B subtypes have been found in strong association with SJS and TEN, suggesting the role of a potential genetic susceptibility.5
We report a unique case of SJS/TEN overlap syndrome occurring in a patient with RP.1 Although the association may be coincidental, it is well known that patients with lupus erythematosus are predisposed to the development of SJS and TEN. Therefore, a shared underlying genetic predisposition or immune system hyperactivity secondary to active RP is a possible explanation for our patient’s unique presentation.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Damiani JM, Levine HL. Relapsing polychondritis—report of ten cases. Laryngoscope. 1979;89:929-46.
- Puéchal X, Terrier B, Mouthon L, et al. Relapsing polychondritis. Joint Bone Spine. 2014;81:118-24.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis. Autoimmun Rev. 2014;13:90-95.
- Harr T, French L. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;16;5:39.
To the Editor:
Relapsing polychondritis (RP) is a chronic, progressive, and episodic systemic inflammatory disease that primarily affects the cartilaginous structures of the ears and nose. Involvement of other proteoglycan-rich structures such as the joints, eyes, inner ears, blood vessels, heart, and kidneys also may be seen. Dermatologic manifestations occur in 35% to 50% of patients and may be the presenting sign in up to 12% of cases.1 The most commonly reported dermatologic findings include oral aphthosis, erythema nodosum, and purpura with vasculitic changes. Less commonly reported associations include Sweet syndrome, pyoderma gangrenosum, panniculitis, erythema elevatum diutinum, erythema annulare centrifugum, and erythema multiforme.1
A 43-year-old woman who was otherwise healthy developed new-onset tenderness and swelling of the left pinna while on vacation. She was treated with trimethoprim-sulfamethoxazole, clindamycin, and levofloxacin for presumed auricular cellulitis. The patient developed a fever; sore throat; and a progressive, pruritic, blistering rash on the face, torso, bilateral extremities, palms, and soles 1 day after completing the antibiotic course. After 5 days of unremitting symptoms despite oral, intramuscular, and topical steroids, the patient presented to the emergency department. Physical examination revealed diffuse, tender, erythematous to violaceous macules with varying degrees of coalescence on the chest, back, and extremities. Scattered flaccid bullae and erosions of the oral and genital mucosa also were seen. Laboratory analysis was notable only for a urinary tract infection with Klebsiella pneumoniae. A punch biopsy demonstrated full-thickness necrosis of the epidermis with subepidermal bullae and a mild to moderate lymphocytic infiltrate with rare eosinophils, consistent with a diagnosis of Stevens-Johnson syndrome (SJS). Because of the body surface area involved (20%) and the recent history of trimethoprim-sulfamethoxazole use, a diagnosis of SJS/toxic epidermal necrolysis (TEN) overlap syndrome was made. The patient was successfully treated with subcutaneous etanercept (50 mg), supportive care, and cephalexin for the urinary tract infection.
Approximately 5 weeks after discharge from the hospital, the patient was evaluated for new-onset pain and swelling of the right ear (Figure) in conjunction with recent tenderness and depression of the superior septal structure of the nose. A punch biopsy of the ear revealed mild perichondral inflammation without vasculitic changes and a superficial, deep perivascular, and periadnexal lymphoplasmacytic inflammatory infiltrate with scattered eosinophils. A diagnosis of RP was made, as the patient met Damiani and Levine’s2 criteria with bilateral auricular inflammation, ocular inflammation, and nasal chondritis.
Although the exact pathogenesis of RP remains unclear, there is strong evidence to suggest an underlying autoimmune etiology.3 Autoantibodies against type II collagen, in addition to other minor collagen and cartilage proteins, such as cartilage oligomeric matrix proteins and matrilin-1, are seen in a subset of patients. Titers have been reported to correlate with disease activity.3,4 Direct immunofluorescence also has demonstrated plentiful CD4+ T cells, as well as IgM, IgA, IgG, and C3 deposits in the inflamed cartilage of patients with RP.3 Additionally, approximately 30% of patients with RP will have another autoimmune disease, and more than 50% of patients with RP carry the HLA-DR4 antigen.3 Alternatively, SJS and TEN are not reported in association with autoimmune diseases and are believed to be CD8+ T-cell driven. Some HLA-B subtypes have been found in strong association with SJS and TEN, suggesting the role of a potential genetic susceptibility.5
We report a unique case of SJS/TEN overlap syndrome occurring in a patient with RP.1 Although the association may be coincidental, it is well known that patients with lupus erythematosus are predisposed to the development of SJS and TEN. Therefore, a shared underlying genetic predisposition or immune system hyperactivity secondary to active RP is a possible explanation for our patient’s unique presentation.
To the Editor:
Relapsing polychondritis (RP) is a chronic, progressive, and episodic systemic inflammatory disease that primarily affects the cartilaginous structures of the ears and nose. Involvement of other proteoglycan-rich structures such as the joints, eyes, inner ears, blood vessels, heart, and kidneys also may be seen. Dermatologic manifestations occur in 35% to 50% of patients and may be the presenting sign in up to 12% of cases.1 The most commonly reported dermatologic findings include oral aphthosis, erythema nodosum, and purpura with vasculitic changes. Less commonly reported associations include Sweet syndrome, pyoderma gangrenosum, panniculitis, erythema elevatum diutinum, erythema annulare centrifugum, and erythema multiforme.1
A 43-year-old woman who was otherwise healthy developed new-onset tenderness and swelling of the left pinna while on vacation. She was treated with trimethoprim-sulfamethoxazole, clindamycin, and levofloxacin for presumed auricular cellulitis. The patient developed a fever; sore throat; and a progressive, pruritic, blistering rash on the face, torso, bilateral extremities, palms, and soles 1 day after completing the antibiotic course. After 5 days of unremitting symptoms despite oral, intramuscular, and topical steroids, the patient presented to the emergency department. Physical examination revealed diffuse, tender, erythematous to violaceous macules with varying degrees of coalescence on the chest, back, and extremities. Scattered flaccid bullae and erosions of the oral and genital mucosa also were seen. Laboratory analysis was notable only for a urinary tract infection with Klebsiella pneumoniae. A punch biopsy demonstrated full-thickness necrosis of the epidermis with subepidermal bullae and a mild to moderate lymphocytic infiltrate with rare eosinophils, consistent with a diagnosis of Stevens-Johnson syndrome (SJS). Because of the body surface area involved (20%) and the recent history of trimethoprim-sulfamethoxazole use, a diagnosis of SJS/toxic epidermal necrolysis (TEN) overlap syndrome was made. The patient was successfully treated with subcutaneous etanercept (50 mg), supportive care, and cephalexin for the urinary tract infection.
Approximately 5 weeks after discharge from the hospital, the patient was evaluated for new-onset pain and swelling of the right ear (Figure) in conjunction with recent tenderness and depression of the superior septal structure of the nose. A punch biopsy of the ear revealed mild perichondral inflammation without vasculitic changes and a superficial, deep perivascular, and periadnexal lymphoplasmacytic inflammatory infiltrate with scattered eosinophils. A diagnosis of RP was made, as the patient met Damiani and Levine’s2 criteria with bilateral auricular inflammation, ocular inflammation, and nasal chondritis.
Although the exact pathogenesis of RP remains unclear, there is strong evidence to suggest an underlying autoimmune etiology.3 Autoantibodies against type II collagen, in addition to other minor collagen and cartilage proteins, such as cartilage oligomeric matrix proteins and matrilin-1, are seen in a subset of patients. Titers have been reported to correlate with disease activity.3,4 Direct immunofluorescence also has demonstrated plentiful CD4+ T cells, as well as IgM, IgA, IgG, and C3 deposits in the inflamed cartilage of patients with RP.3 Additionally, approximately 30% of patients with RP will have another autoimmune disease, and more than 50% of patients with RP carry the HLA-DR4 antigen.3 Alternatively, SJS and TEN are not reported in association with autoimmune diseases and are believed to be CD8+ T-cell driven. Some HLA-B subtypes have been found in strong association with SJS and TEN, suggesting the role of a potential genetic susceptibility.5
We report a unique case of SJS/TEN overlap syndrome occurring in a patient with RP.1 Although the association may be coincidental, it is well known that patients with lupus erythematosus are predisposed to the development of SJS and TEN. Therefore, a shared underlying genetic predisposition or immune system hyperactivity secondary to active RP is a possible explanation for our patient’s unique presentation.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Damiani JM, Levine HL. Relapsing polychondritis—report of ten cases. Laryngoscope. 1979;89:929-46.
- Puéchal X, Terrier B, Mouthon L, et al. Relapsing polychondritis. Joint Bone Spine. 2014;81:118-24.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis. Autoimmun Rev. 2014;13:90-95.
- Harr T, French L. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;16;5:39.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Damiani JM, Levine HL. Relapsing polychondritis—report of ten cases. Laryngoscope. 1979;89:929-46.
- Puéchal X, Terrier B, Mouthon L, et al. Relapsing polychondritis. Joint Bone Spine. 2014;81:118-24.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis. Autoimmun Rev. 2014;13:90-95.
- Harr T, French L. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;16;5:39.
Practice Points
- The clinical presentation of relapsing polychondritis (RP) may demonstrate cutaneous manifestations other than the typical inflammation of cartilage-rich structures.
- Approximately 30% of patients with RP will have another autoimmune disease.
Residency Training During the #MeToo Movement
The #MeToo movement that took hold in the wake of the Harvey Weinstein allegations in 2017 likely will be considered one of the major cultural touchpoints of the 2010s. Although activism within the entertainment industry initially drew attention to this movement, it is understood that virtually no workplace is immune to sexual misconduct. Many medical professionals acknowledge #MeToo as a catchy hashtag summarizing a problem that has long been recognized in the field of medicine but often has been inadequately addressed.1 As dermatology residency program directors (PDs) at the University of Southern California (USC) Keck School of Medicine (Los Angeles, California), we have seen the considerable impact that recent high-profile allegations of sexual assault have had at our institution, leading us to take part in institutional and departmental initiatives and reflections that we believe have strengthened the culture within our residency program and positioned us to be proactive in addressing this critical issue.
Before we discuss the efforts to combat sexual misconduct and gender inequality at USC and within our dermatology department, it is worth reflecting on where we stand as a specialty with regard to gender representation. A recent JAMA Dermatology article reported that in 1970 only 10.8% of dermatology academic faculty were women but by 2018 that number had skyrocketed to 51.2%; however, in contrast to this overall increase, only 19.4% of dermatology department chairs in 2018 were women.2 Although we have made large strides as a field, this discrepancy indicates that we still have a long way to go to achieve gender equality.
Although dermatology as a specialty is working toward gender equality, we believe it is crucial to consider this issue in the context of the entire field of medicine, particularly because academic physicians and trainees often interface with a myriad of specialties. It is well known that women in medicine are more likely to be victims of sexual harassment or assault in the workplace and that subsequent issues with imposter syndrome and/or depression are more prevalent in female physicians.3,4 Gender inequality and sexism, among other factors, can make it difficult for women to obtain and maintain leadership positions and can negatively impact the culture of an academic institution in numerous downstream ways.
We also know that academic environments in medicine have a higher prevalence of gender equality issues than in private practice or in settings where medicine is practiced without trainees due to the hierarchical nature of training and the necessary differences in experience between trainees and faculty.3 Furthermore, because trainees form and solidify their professional identities during graduate medical education (GME) training, it is a prime time to emphasize the importance of gender equality and establish zero tolerance policies for workplace abuse and transgressions.5
The data and our personal experiences delineate a clear need for continued vigilance regarding gender equality issues both in dermatology as a specialty and in medicine in general. As PDs, we feel fortunate to have worked in conjunction with our GME committee and our dermatology department to solidify and create policies that work to promote a culture of gender equality. Herein, we will outline some of these efforts with the hope that other academic institutions may consider implementing these programs to protect members of their community from harassment, sexual violence, and gender discrimination.
Create a SAFE Committee
At the institutional level, our GME committee has created the SAFE (Safety, Fairness & Equity) committee under the leadership of Lawrence Opas, MD. The SAFE committee is headed by a female faculty physician and includes members of the medical community who have the influence to affect change and a commitment to protect vulnerable populations. Members include the Chief Medical Officer, the Designated Institutional Officer, the Director of Resident Wellness, and the Dean of the Keck School of Medicine at USC. The SAFE committee serves as a 24/7 reporting resource whereby trainees can report any issues relating to harassment in the workplace via a telephone hotline or online platform. Issues brought to this committee are immediately dealt with and reviewed at monthly GME meetings to keep institutional PDs up-to-date on issues pertaining to sexual harassment and assault within our workplace. The SAFE committee also has departmental resident liaisons who bring information to residents and help guide them to appropriate resources.
Emphasize Resident Wellness
Along with the development of robust reporting resources, our institution has continued to build upon a culture that places a strong emphasis on resident wellness. One of the most meaningful efforts over the last 5 years has included recruitment of a clinical psychologist, Tobi Fishel, PhD, to serve as our institution’s Director of Wellness. She is available to meet confidentially with our residents and helps to serve as a link between trainees and the GME committee.
Our dermatology department takes a tremendous amount of pride in its culture. We are fortunate to have David Peng, MD, MPH, Chair, and Stefani Takahashi, MD, Vice Chair of Education, working daily to create an environment that values teamwork, selflessness, and wellness. We have been continuously grateful for their leadership and guidance in addressing the allegations of sexual assault and harassment that arose at USC over the past several years. Our department has a zero tolerance policy for sexual harassment or harassment of any kind, and we have taken important steps to ensure and promote a safe environment for our trainees, many of which are focused on communication. We try to avoid assumptions and encourage both residents and faculty to explicitly state their experiences and opinions in general but also in relation to instances of potential misconduct.
Encourage Communication
When allegations of sexual misconduct in the workplace were made at our institution, we prioritized immediate in-person communication with our residents to reinforce our zero tolerance policy and to remind them that we are available should any similar issues arise in our department. It was of equal value to remind our trainees of potential resources, such as the SAFE committee, to whom they could bring their concerns if they were not comfortable communicating directly with us. Although we hoped that our trainees understood that we would not be tolerant of any form of harassment based on our past actions and communications, we felt that it was helpful to explicitly delineate this by laying out other avenues of support on a regular basis with them. By ensuring there is a space for a dialogue with others, if needed, our institution and department have provided an extra layer of security for our trainees. Multiple channels of support are crucial to ensure trainee safety.
Dr. Peng also created a workplace safety committee that includes several female faculty members. The committee regularly shares and highlights institutional and departmental resources as they pertain to gender equality and safety within the workplace and also has considerable faculty overlap with our departmental diversity committee. Together, these committees work toward the common goal of fostering an environment in which all members of our department feel comfortable voicing concerns, and we are best able to recruit and retain a diverse faculty.
As PDs, we work to reinforce departmental and institutional messages in our daily communication with residents. We have found that ensuring frequent and varied interactions—quarterly meetings, biannual evaluations, faculty-led didactics 2 half-days per week, and weekly clinical interactions—with our trainees can help to create a culture where they feel comfortable bringing up issues, be they routine clinical operations questions or issues relating to their professional identity. We hope it also has created the space for them to approach us with any issues pertaining to harassment should they ever arise, and we are grateful to know that even if this comfort does not exist, our institution and department have other resources for them.
Final Thoughts
Although some of the measures discussed here were reactionary, many predated the recent institutional concerns and allegations at USC. We hope and believe that the culture we foster within our department has helped our trainees feel safe and cared for during a time of institutional turbulence. We also believe that taking similar proactive measures may benefit the overall culture and foster the development of diverse physicians and leadership at other institutions. In conjunction with reworking legislation and implementing institutional safeguards, the long-term goals of taking these proactive measures are to promote gender equality and workplace safety and to cultivate and retain effective female leadership in medical institutions and training programs.
We feel incredibly fortunate to be part of a specialty in which gender equality has long been considered and sought after. We also are proud to be members of the Association of Professors of Dermatology, which has addressed issues such as diversity and gender equality in a transparent and head-on manner and continues to do so. As a specialty, we hope we can support our trainees in their professional growth and help to cultivate sensitive physicians who will care for an increasingly diverse population and better support each other in their own career development.
- Ladika S. Sexual harassment: health care, it is #youtoo. Manag Care. 2018;27:14-17.
- Xierali IM, Nivet MA, Pandya AG. US dermatology department faculty diversity trends by sex and underrepresented-in-medicine status, 1970 to 2018 [published online January 8, 2020]. JAMA Dermatol. doi:10.1001/jamadermatol.2019.4297.
- Minkina N. Can #MeToo abolish sexual harassment and discrimination in medicine? Lancet. 2019;394:383-384.
- Dzau VJ, Johnson PA. Ending sexual harassment in academic medicine. N Engl J Med. 2018;379:1589-1591.
- Nothnagle M, Reis S, Goldman RE, et al. Fostering professional formation in residency: development and evaluation of the “forum” seminar series. Teach Learn Med. 2014;26:230-238.
The #MeToo movement that took hold in the wake of the Harvey Weinstein allegations in 2017 likely will be considered one of the major cultural touchpoints of the 2010s. Although activism within the entertainment industry initially drew attention to this movement, it is understood that virtually no workplace is immune to sexual misconduct. Many medical professionals acknowledge #MeToo as a catchy hashtag summarizing a problem that has long been recognized in the field of medicine but often has been inadequately addressed.1 As dermatology residency program directors (PDs) at the University of Southern California (USC) Keck School of Medicine (Los Angeles, California), we have seen the considerable impact that recent high-profile allegations of sexual assault have had at our institution, leading us to take part in institutional and departmental initiatives and reflections that we believe have strengthened the culture within our residency program and positioned us to be proactive in addressing this critical issue.
Before we discuss the efforts to combat sexual misconduct and gender inequality at USC and within our dermatology department, it is worth reflecting on where we stand as a specialty with regard to gender representation. A recent JAMA Dermatology article reported that in 1970 only 10.8% of dermatology academic faculty were women but by 2018 that number had skyrocketed to 51.2%; however, in contrast to this overall increase, only 19.4% of dermatology department chairs in 2018 were women.2 Although we have made large strides as a field, this discrepancy indicates that we still have a long way to go to achieve gender equality.
Although dermatology as a specialty is working toward gender equality, we believe it is crucial to consider this issue in the context of the entire field of medicine, particularly because academic physicians and trainees often interface with a myriad of specialties. It is well known that women in medicine are more likely to be victims of sexual harassment or assault in the workplace and that subsequent issues with imposter syndrome and/or depression are more prevalent in female physicians.3,4 Gender inequality and sexism, among other factors, can make it difficult for women to obtain and maintain leadership positions and can negatively impact the culture of an academic institution in numerous downstream ways.
We also know that academic environments in medicine have a higher prevalence of gender equality issues than in private practice or in settings where medicine is practiced without trainees due to the hierarchical nature of training and the necessary differences in experience between trainees and faculty.3 Furthermore, because trainees form and solidify their professional identities during graduate medical education (GME) training, it is a prime time to emphasize the importance of gender equality and establish zero tolerance policies for workplace abuse and transgressions.5
The data and our personal experiences delineate a clear need for continued vigilance regarding gender equality issues both in dermatology as a specialty and in medicine in general. As PDs, we feel fortunate to have worked in conjunction with our GME committee and our dermatology department to solidify and create policies that work to promote a culture of gender equality. Herein, we will outline some of these efforts with the hope that other academic institutions may consider implementing these programs to protect members of their community from harassment, sexual violence, and gender discrimination.
Create a SAFE Committee
At the institutional level, our GME committee has created the SAFE (Safety, Fairness & Equity) committee under the leadership of Lawrence Opas, MD. The SAFE committee is headed by a female faculty physician and includes members of the medical community who have the influence to affect change and a commitment to protect vulnerable populations. Members include the Chief Medical Officer, the Designated Institutional Officer, the Director of Resident Wellness, and the Dean of the Keck School of Medicine at USC. The SAFE committee serves as a 24/7 reporting resource whereby trainees can report any issues relating to harassment in the workplace via a telephone hotline or online platform. Issues brought to this committee are immediately dealt with and reviewed at monthly GME meetings to keep institutional PDs up-to-date on issues pertaining to sexual harassment and assault within our workplace. The SAFE committee also has departmental resident liaisons who bring information to residents and help guide them to appropriate resources.
Emphasize Resident Wellness
Along with the development of robust reporting resources, our institution has continued to build upon a culture that places a strong emphasis on resident wellness. One of the most meaningful efforts over the last 5 years has included recruitment of a clinical psychologist, Tobi Fishel, PhD, to serve as our institution’s Director of Wellness. She is available to meet confidentially with our residents and helps to serve as a link between trainees and the GME committee.
Our dermatology department takes a tremendous amount of pride in its culture. We are fortunate to have David Peng, MD, MPH, Chair, and Stefani Takahashi, MD, Vice Chair of Education, working daily to create an environment that values teamwork, selflessness, and wellness. We have been continuously grateful for their leadership and guidance in addressing the allegations of sexual assault and harassment that arose at USC over the past several years. Our department has a zero tolerance policy for sexual harassment or harassment of any kind, and we have taken important steps to ensure and promote a safe environment for our trainees, many of which are focused on communication. We try to avoid assumptions and encourage both residents and faculty to explicitly state their experiences and opinions in general but also in relation to instances of potential misconduct.
Encourage Communication
When allegations of sexual misconduct in the workplace were made at our institution, we prioritized immediate in-person communication with our residents to reinforce our zero tolerance policy and to remind them that we are available should any similar issues arise in our department. It was of equal value to remind our trainees of potential resources, such as the SAFE committee, to whom they could bring their concerns if they were not comfortable communicating directly with us. Although we hoped that our trainees understood that we would not be tolerant of any form of harassment based on our past actions and communications, we felt that it was helpful to explicitly delineate this by laying out other avenues of support on a regular basis with them. By ensuring there is a space for a dialogue with others, if needed, our institution and department have provided an extra layer of security for our trainees. Multiple channels of support are crucial to ensure trainee safety.
Dr. Peng also created a workplace safety committee that includes several female faculty members. The committee regularly shares and highlights institutional and departmental resources as they pertain to gender equality and safety within the workplace and also has considerable faculty overlap with our departmental diversity committee. Together, these committees work toward the common goal of fostering an environment in which all members of our department feel comfortable voicing concerns, and we are best able to recruit and retain a diverse faculty.
As PDs, we work to reinforce departmental and institutional messages in our daily communication with residents. We have found that ensuring frequent and varied interactions—quarterly meetings, biannual evaluations, faculty-led didactics 2 half-days per week, and weekly clinical interactions—with our trainees can help to create a culture where they feel comfortable bringing up issues, be they routine clinical operations questions or issues relating to their professional identity. We hope it also has created the space for them to approach us with any issues pertaining to harassment should they ever arise, and we are grateful to know that even if this comfort does not exist, our institution and department have other resources for them.
Final Thoughts
Although some of the measures discussed here were reactionary, many predated the recent institutional concerns and allegations at USC. We hope and believe that the culture we foster within our department has helped our trainees feel safe and cared for during a time of institutional turbulence. We also believe that taking similar proactive measures may benefit the overall culture and foster the development of diverse physicians and leadership at other institutions. In conjunction with reworking legislation and implementing institutional safeguards, the long-term goals of taking these proactive measures are to promote gender equality and workplace safety and to cultivate and retain effective female leadership in medical institutions and training programs.
We feel incredibly fortunate to be part of a specialty in which gender equality has long been considered and sought after. We also are proud to be members of the Association of Professors of Dermatology, which has addressed issues such as diversity and gender equality in a transparent and head-on manner and continues to do so. As a specialty, we hope we can support our trainees in their professional growth and help to cultivate sensitive physicians who will care for an increasingly diverse population and better support each other in their own career development.
The #MeToo movement that took hold in the wake of the Harvey Weinstein allegations in 2017 likely will be considered one of the major cultural touchpoints of the 2010s. Although activism within the entertainment industry initially drew attention to this movement, it is understood that virtually no workplace is immune to sexual misconduct. Many medical professionals acknowledge #MeToo as a catchy hashtag summarizing a problem that has long been recognized in the field of medicine but often has been inadequately addressed.1 As dermatology residency program directors (PDs) at the University of Southern California (USC) Keck School of Medicine (Los Angeles, California), we have seen the considerable impact that recent high-profile allegations of sexual assault have had at our institution, leading us to take part in institutional and departmental initiatives and reflections that we believe have strengthened the culture within our residency program and positioned us to be proactive in addressing this critical issue.
Before we discuss the efforts to combat sexual misconduct and gender inequality at USC and within our dermatology department, it is worth reflecting on where we stand as a specialty with regard to gender representation. A recent JAMA Dermatology article reported that in 1970 only 10.8% of dermatology academic faculty were women but by 2018 that number had skyrocketed to 51.2%; however, in contrast to this overall increase, only 19.4% of dermatology department chairs in 2018 were women.2 Although we have made large strides as a field, this discrepancy indicates that we still have a long way to go to achieve gender equality.
Although dermatology as a specialty is working toward gender equality, we believe it is crucial to consider this issue in the context of the entire field of medicine, particularly because academic physicians and trainees often interface with a myriad of specialties. It is well known that women in medicine are more likely to be victims of sexual harassment or assault in the workplace and that subsequent issues with imposter syndrome and/or depression are more prevalent in female physicians.3,4 Gender inequality and sexism, among other factors, can make it difficult for women to obtain and maintain leadership positions and can negatively impact the culture of an academic institution in numerous downstream ways.
We also know that academic environments in medicine have a higher prevalence of gender equality issues than in private practice or in settings where medicine is practiced without trainees due to the hierarchical nature of training and the necessary differences in experience between trainees and faculty.3 Furthermore, because trainees form and solidify their professional identities during graduate medical education (GME) training, it is a prime time to emphasize the importance of gender equality and establish zero tolerance policies for workplace abuse and transgressions.5
The data and our personal experiences delineate a clear need for continued vigilance regarding gender equality issues both in dermatology as a specialty and in medicine in general. As PDs, we feel fortunate to have worked in conjunction with our GME committee and our dermatology department to solidify and create policies that work to promote a culture of gender equality. Herein, we will outline some of these efforts with the hope that other academic institutions may consider implementing these programs to protect members of their community from harassment, sexual violence, and gender discrimination.
Create a SAFE Committee
At the institutional level, our GME committee has created the SAFE (Safety, Fairness & Equity) committee under the leadership of Lawrence Opas, MD. The SAFE committee is headed by a female faculty physician and includes members of the medical community who have the influence to affect change and a commitment to protect vulnerable populations. Members include the Chief Medical Officer, the Designated Institutional Officer, the Director of Resident Wellness, and the Dean of the Keck School of Medicine at USC. The SAFE committee serves as a 24/7 reporting resource whereby trainees can report any issues relating to harassment in the workplace via a telephone hotline or online platform. Issues brought to this committee are immediately dealt with and reviewed at monthly GME meetings to keep institutional PDs up-to-date on issues pertaining to sexual harassment and assault within our workplace. The SAFE committee also has departmental resident liaisons who bring information to residents and help guide them to appropriate resources.
Emphasize Resident Wellness
Along with the development of robust reporting resources, our institution has continued to build upon a culture that places a strong emphasis on resident wellness. One of the most meaningful efforts over the last 5 years has included recruitment of a clinical psychologist, Tobi Fishel, PhD, to serve as our institution’s Director of Wellness. She is available to meet confidentially with our residents and helps to serve as a link between trainees and the GME committee.
Our dermatology department takes a tremendous amount of pride in its culture. We are fortunate to have David Peng, MD, MPH, Chair, and Stefani Takahashi, MD, Vice Chair of Education, working daily to create an environment that values teamwork, selflessness, and wellness. We have been continuously grateful for their leadership and guidance in addressing the allegations of sexual assault and harassment that arose at USC over the past several years. Our department has a zero tolerance policy for sexual harassment or harassment of any kind, and we have taken important steps to ensure and promote a safe environment for our trainees, many of which are focused on communication. We try to avoid assumptions and encourage both residents and faculty to explicitly state their experiences and opinions in general but also in relation to instances of potential misconduct.
Encourage Communication
When allegations of sexual misconduct in the workplace were made at our institution, we prioritized immediate in-person communication with our residents to reinforce our zero tolerance policy and to remind them that we are available should any similar issues arise in our department. It was of equal value to remind our trainees of potential resources, such as the SAFE committee, to whom they could bring their concerns if they were not comfortable communicating directly with us. Although we hoped that our trainees understood that we would not be tolerant of any form of harassment based on our past actions and communications, we felt that it was helpful to explicitly delineate this by laying out other avenues of support on a regular basis with them. By ensuring there is a space for a dialogue with others, if needed, our institution and department have provided an extra layer of security for our trainees. Multiple channels of support are crucial to ensure trainee safety.
Dr. Peng also created a workplace safety committee that includes several female faculty members. The committee regularly shares and highlights institutional and departmental resources as they pertain to gender equality and safety within the workplace and also has considerable faculty overlap with our departmental diversity committee. Together, these committees work toward the common goal of fostering an environment in which all members of our department feel comfortable voicing concerns, and we are best able to recruit and retain a diverse faculty.
As PDs, we work to reinforce departmental and institutional messages in our daily communication with residents. We have found that ensuring frequent and varied interactions—quarterly meetings, biannual evaluations, faculty-led didactics 2 half-days per week, and weekly clinical interactions—with our trainees can help to create a culture where they feel comfortable bringing up issues, be they routine clinical operations questions or issues relating to their professional identity. We hope it also has created the space for them to approach us with any issues pertaining to harassment should they ever arise, and we are grateful to know that even if this comfort does not exist, our institution and department have other resources for them.
Final Thoughts
Although some of the measures discussed here were reactionary, many predated the recent institutional concerns and allegations at USC. We hope and believe that the culture we foster within our department has helped our trainees feel safe and cared for during a time of institutional turbulence. We also believe that taking similar proactive measures may benefit the overall culture and foster the development of diverse physicians and leadership at other institutions. In conjunction with reworking legislation and implementing institutional safeguards, the long-term goals of taking these proactive measures are to promote gender equality and workplace safety and to cultivate and retain effective female leadership in medical institutions and training programs.
We feel incredibly fortunate to be part of a specialty in which gender equality has long been considered and sought after. We also are proud to be members of the Association of Professors of Dermatology, which has addressed issues such as diversity and gender equality in a transparent and head-on manner and continues to do so. As a specialty, we hope we can support our trainees in their professional growth and help to cultivate sensitive physicians who will care for an increasingly diverse population and better support each other in their own career development.
- Ladika S. Sexual harassment: health care, it is #youtoo. Manag Care. 2018;27:14-17.
- Xierali IM, Nivet MA, Pandya AG. US dermatology department faculty diversity trends by sex and underrepresented-in-medicine status, 1970 to 2018 [published online January 8, 2020]. JAMA Dermatol. doi:10.1001/jamadermatol.2019.4297.
- Minkina N. Can #MeToo abolish sexual harassment and discrimination in medicine? Lancet. 2019;394:383-384.
- Dzau VJ, Johnson PA. Ending sexual harassment in academic medicine. N Engl J Med. 2018;379:1589-1591.
- Nothnagle M, Reis S, Goldman RE, et al. Fostering professional formation in residency: development and evaluation of the “forum” seminar series. Teach Learn Med. 2014;26:230-238.
- Ladika S. Sexual harassment: health care, it is #youtoo. Manag Care. 2018;27:14-17.
- Xierali IM, Nivet MA, Pandya AG. US dermatology department faculty diversity trends by sex and underrepresented-in-medicine status, 1970 to 2018 [published online January 8, 2020]. JAMA Dermatol. doi:10.1001/jamadermatol.2019.4297.
- Minkina N. Can #MeToo abolish sexual harassment and discrimination in medicine? Lancet. 2019;394:383-384.
- Dzau VJ, Johnson PA. Ending sexual harassment in academic medicine. N Engl J Med. 2018;379:1589-1591.
- Nothnagle M, Reis S, Goldman RE, et al. Fostering professional formation in residency: development and evaluation of the “forum” seminar series. Teach Learn Med. 2014;26:230-238.

Paraneoplastic Dermatomyositis Presenting With Interesting Cutaneous Findings
To the Editor:
We report an interesting clinical case of dermatomyositis (DM) that presented with an associated malignancy (small cell lung cancer). This patient also had an unusual clinical finding of predominantly unilateral, confluent, erythematous papules on the knee, a cutaneous sign that is seldom described in the DM literature. This case serves to reinforce the classic findings and associations of DM, in addition to the uncommon manifestation of predominantly unilateral papules on the knee.
A 68-year-old woman presented with several cutaneous manifestations including the classic findings of photo distributed erythema on the arms and face, a heliotrope rash, Gottron papules, and confluent pink papules on the left knee (Figure 1). The patient also had one of the more rare manifestations of DM, flagellate erythema on the back (Figure 2). She had a history of breast cancer and was found to have metastatic small cell lung cancer at the time of the DM diagnosis.
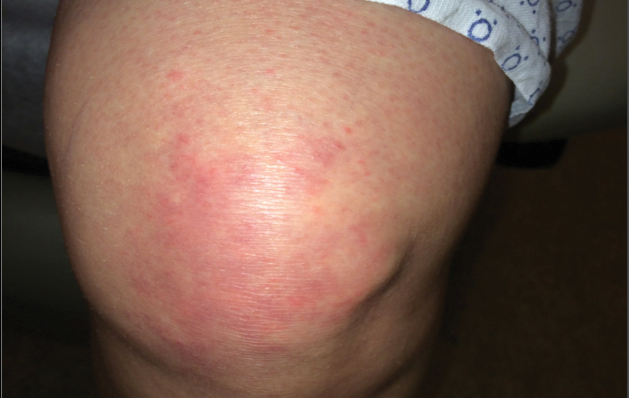
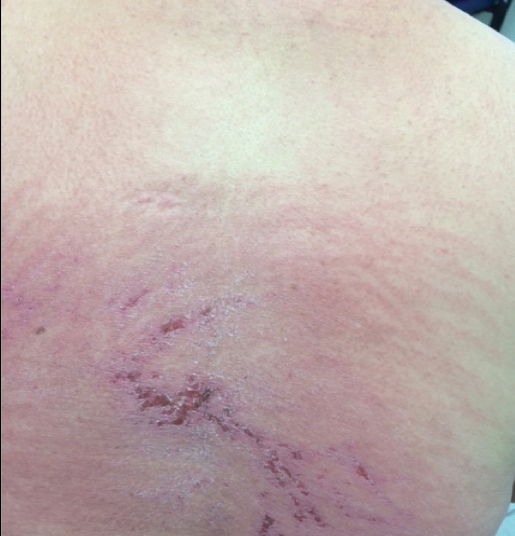
A punch biopsy from an area of flagellate erythema on the back revealed an interface dermatitis with a superficial, perivascular, lymphocyte-predominant inflammatory infiltrate (Figure 3). Alcian blue and colloidal iron stains revealed a marked increase in papillary dermal mucin. With the characteristic changes on skin biopsy and the classic skin findings present in our patient, we felt confident diagnosing her with DM. At the time of diagnosis, the patient also was found to have metastatic small cell lung cancer, suggesting a true paraneoplastic relationship.
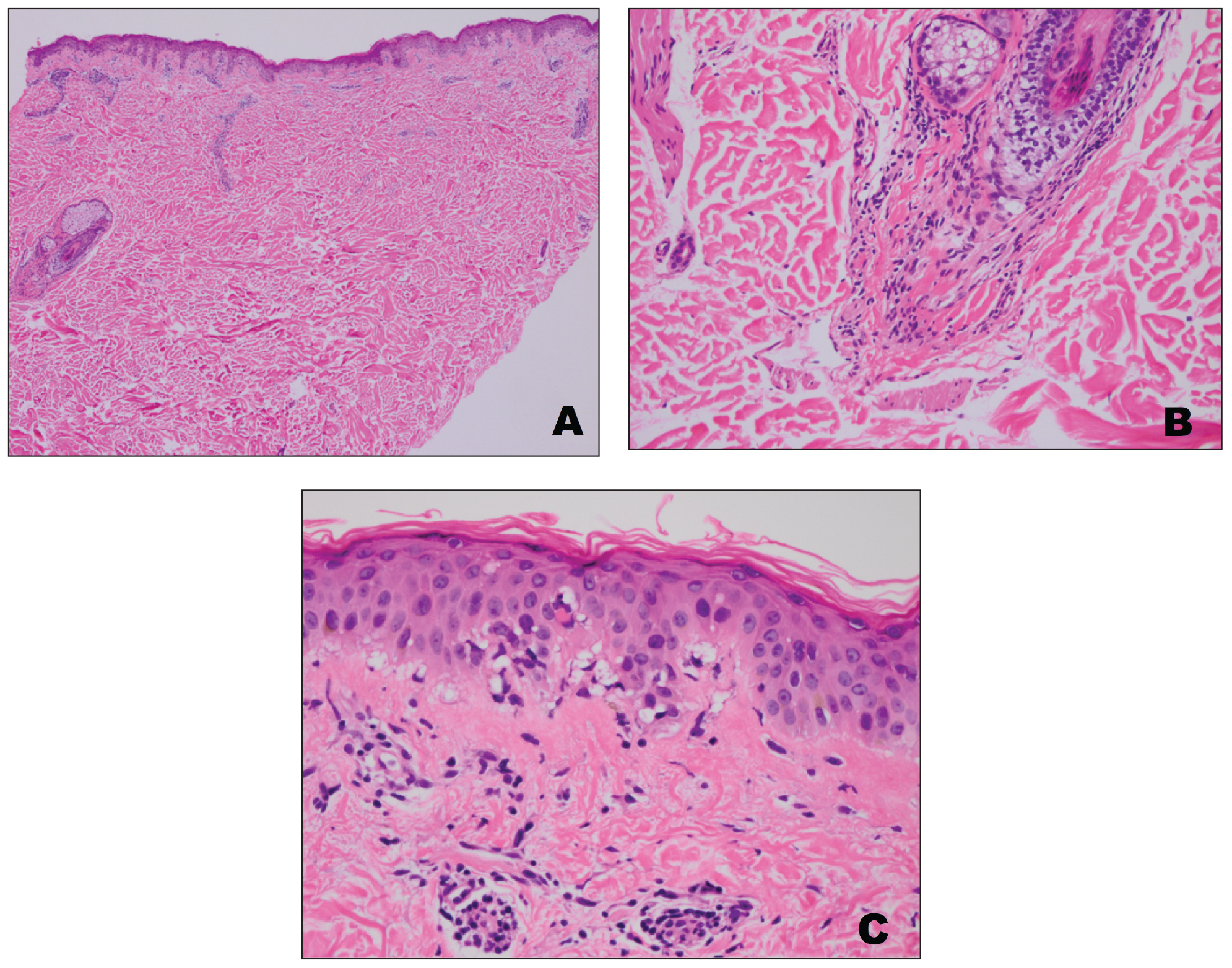

The association of DM and amyopathic DM with internal malignancy is well known. Bohan and Peter1 noted an overall figure ranging from 15% to 34% with an increased frequency in patients with skin and muscle involvement.1 Hill et al5 examined this link in a population-based study that identified corresponding malignancies. Specifically, they noted cancers to arise most frequently in the airway (eg, lung, trachea, bronchus), ovaries, breasts, colorectal region, and stomach.5 There also has been work performed to identify if certain dermatologic findings may be associated with a higher risk of malignancy.6,7 A meta-analysis by Wang et al6 showed that Gottron sign did not have an association with cancer, but findings of cutaneous necrosis did have an association. It is unknown if the specific cutaneous findings in our patient, including the predominantly unilateral papules on the knee, may have been a clue to the underlying malignancy.
In summary, we believe that our patient presented with the classic manifestations of DM in addition to the curious cutaneous sign of predominantly unilateral, confluent, erythematous papules on the knee, a clinical finding that may aid in the diagnosis of DM and also may alert the clinician to a possible underlying malignancy.
- Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344-347.
- Santmyire-Rosenberger B, Dugan EM. Skin involvement in dermatomyositis. Curr Opin Rheumatol. 2003;15:714-722.
- Callen JP. Dermatomyositis. Lancet. 2000;355:53-57.
- Lister RK, Cooper ES, Paige DG. Papules and pustules of the elbows and knees: an uncommon clinical sign of dermatomyositis in oriental children. Pediatr Dermatol. 2000;17:37-40.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Wang J, Guo G, Chen G, et al. Meta‐analysis of the association of dermatomyositis and polymyositis with cancer. Br J Dermatol. 2013;169:838-847.
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case–control study. Br J Dermatol. 2001;144:825-831.
To the Editor:
We report an interesting clinical case of dermatomyositis (DM) that presented with an associated malignancy (small cell lung cancer). This patient also had an unusual clinical finding of predominantly unilateral, confluent, erythematous papules on the knee, a cutaneous sign that is seldom described in the DM literature. This case serves to reinforce the classic findings and associations of DM, in addition to the uncommon manifestation of predominantly unilateral papules on the knee.
A 68-year-old woman presented with several cutaneous manifestations including the classic findings of photo distributed erythema on the arms and face, a heliotrope rash, Gottron papules, and confluent pink papules on the left knee (Figure 1). The patient also had one of the more rare manifestations of DM, flagellate erythema on the back (Figure 2). She had a history of breast cancer and was found to have metastatic small cell lung cancer at the time of the DM diagnosis.
A punch biopsy from an area of flagellate erythema on the back revealed an interface dermatitis with a superficial, perivascular, lymphocyte-predominant inflammatory infiltrate (Figure 3). Alcian blue and colloidal iron stains revealed a marked increase in papillary dermal mucin. With the characteristic changes on skin biopsy and the classic skin findings present in our patient, we felt confident diagnosing her with DM. At the time of diagnosis, the patient also was found to have metastatic small cell lung cancer, suggesting a true paraneoplastic relationship.
The association of DM and amyopathic DM with internal malignancy is well known. Bohan and Peter1 noted an overall figure ranging from 15% to 34% with an increased frequency in patients with skin and muscle involvement.1 Hill et al5 examined this link in a population-based study that identified corresponding malignancies. Specifically, they noted cancers to arise most frequently in the airway (eg, lung, trachea, bronchus), ovaries, breasts, colorectal region, and stomach.5 There also has been work performed to identify if certain dermatologic findings may be associated with a higher risk of malignancy.6,7 A meta-analysis by Wang et al6 showed that Gottron sign did not have an association with cancer, but findings of cutaneous necrosis did have an association. It is unknown if the specific cutaneous findings in our patient, including the predominantly unilateral papules on the knee, may have been a clue to the underlying malignancy.
In summary, we believe that our patient presented with the classic manifestations of DM in addition to the curious cutaneous sign of predominantly unilateral, confluent, erythematous papules on the knee, a clinical finding that may aid in the diagnosis of DM and also may alert the clinician to a possible underlying malignancy.
To the Editor:
We report an interesting clinical case of dermatomyositis (DM) that presented with an associated malignancy (small cell lung cancer). This patient also had an unusual clinical finding of predominantly unilateral, confluent, erythematous papules on the knee, a cutaneous sign that is seldom described in the DM literature. This case serves to reinforce the classic findings and associations of DM, in addition to the uncommon manifestation of predominantly unilateral papules on the knee.
A 68-year-old woman presented with several cutaneous manifestations including the classic findings of photo distributed erythema on the arms and face, a heliotrope rash, Gottron papules, and confluent pink papules on the left knee (Figure 1). The patient also had one of the more rare manifestations of DM, flagellate erythema on the back (Figure 2). She had a history of breast cancer and was found to have metastatic small cell lung cancer at the time of the DM diagnosis.
A punch biopsy from an area of flagellate erythema on the back revealed an interface dermatitis with a superficial, perivascular, lymphocyte-predominant inflammatory infiltrate (Figure 3). Alcian blue and colloidal iron stains revealed a marked increase in papillary dermal mucin. With the characteristic changes on skin biopsy and the classic skin findings present in our patient, we felt confident diagnosing her with DM. At the time of diagnosis, the patient also was found to have metastatic small cell lung cancer, suggesting a true paraneoplastic relationship.
The association of DM and amyopathic DM with internal malignancy is well known. Bohan and Peter1 noted an overall figure ranging from 15% to 34% with an increased frequency in patients with skin and muscle involvement.1 Hill et al5 examined this link in a population-based study that identified corresponding malignancies. Specifically, they noted cancers to arise most frequently in the airway (eg, lung, trachea, bronchus), ovaries, breasts, colorectal region, and stomach.5 There also has been work performed to identify if certain dermatologic findings may be associated with a higher risk of malignancy.6,7 A meta-analysis by Wang et al6 showed that Gottron sign did not have an association with cancer, but findings of cutaneous necrosis did have an association. It is unknown if the specific cutaneous findings in our patient, including the predominantly unilateral papules on the knee, may have been a clue to the underlying malignancy.
In summary, we believe that our patient presented with the classic manifestations of DM in addition to the curious cutaneous sign of predominantly unilateral, confluent, erythematous papules on the knee, a clinical finding that may aid in the diagnosis of DM and also may alert the clinician to a possible underlying malignancy.
- Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344-347.
- Santmyire-Rosenberger B, Dugan EM. Skin involvement in dermatomyositis. Curr Opin Rheumatol. 2003;15:714-722.
- Callen JP. Dermatomyositis. Lancet. 2000;355:53-57.
- Lister RK, Cooper ES, Paige DG. Papules and pustules of the elbows and knees: an uncommon clinical sign of dermatomyositis in oriental children. Pediatr Dermatol. 2000;17:37-40.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Wang J, Guo G, Chen G, et al. Meta‐analysis of the association of dermatomyositis and polymyositis with cancer. Br J Dermatol. 2013;169:838-847.
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case–control study. Br J Dermatol. 2001;144:825-831.
- Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344-347.
- Santmyire-Rosenberger B, Dugan EM. Skin involvement in dermatomyositis. Curr Opin Rheumatol. 2003;15:714-722.
- Callen JP. Dermatomyositis. Lancet. 2000;355:53-57.
- Lister RK, Cooper ES, Paige DG. Papules and pustules of the elbows and knees: an uncommon clinical sign of dermatomyositis in oriental children. Pediatr Dermatol. 2000;17:37-40.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Wang J, Guo G, Chen G, et al. Meta‐analysis of the association of dermatomyositis and polymyositis with cancer. Br J Dermatol. 2013;169:838-847.
- Chen YJ, Wu CY, Shen JL. Predicting factors of malignancy in dermatomyositis and polymyositis: a case–control study. Br J Dermatol. 2001;144:825-831.
Practice Points
- Dermatomyositis has myriad cutaneous features including the shawl sign, the heliotrope sign, and Gottron papules.
- Less commonly, patients can present with the Holster sign (poikiloderma of the lateral thighs).
- Even less commonly, as in this report, patients can present with a psoriasiform papular eruption on the knees or with flagellate erythema on the back.
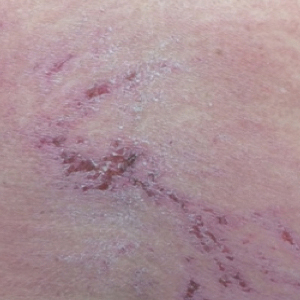
Acrodermatitis Enteropathica in a Patient With Short Bowel Syndrome
To the Editor:
Acrodermatitis enteropathica (AE) is an inherited defect in zinc absorption that leads to hypozincemia. Its clinical presentation can vary based on serum zinc level and ranges from periorificial erosive dermatitis to psoriasiform dermatitis.1 Recognition of the cutaneous manifestations of zinc deficiency can lead to early intervention with zinc supplementation and prevention of long-term morbidity and even mortality. In our case, the coexistence of a bullous acral dermatosis with the additional feature of extensor digital dermatitis with fissuring suggests a diagnosis of AE and can alert the astute clinician to the need for testing of serum zinc levels and/or treatment with zinc supplementation. Causes of acquired zinc deficiency that have been reported in the literature include eating disorders such as anorexia nervosa and bulimia nervosa, Crohn disease, food allergy, intestinal parasitic infestations, and an inborn error of metabolism known as nonketotic hyperglycemia (Table).2-4
RELATED ARTICLE: Acquired Acrodermatitis Enteropathica Secondary to Alcoholism

A 42-year-old woman with a medical history of rheumatoid arthritis and short bowel syndrome due to multiple small bowel obstructions with subsequent bowel resections who was on chronic total parenteral nutrition (TPN) presented with bullae on the hands, shins, and feet. The patient initially noticed small erythematous macules on the hands and feet months prior to presentation. Three weeks prior to presentation, bullae started to form on the hands, mostly between the web spaces; dorsal aspects of the feet; and anterior aspects of the shins. The patient denied any oral ulcers. One day prior to presentation the patient was seen at an outside hospital and was started on prednisone 5 mg daily, oral clindamycin, mupirocin ointment, and nystatin-triamcinolone cream. These medications failed to improve her condition. On review of systems, the patient denied any fever, chills, eye pain, or dysuria.
Upon initial presentation the patient appeared weak and fatigued, though vital signs were normal. Physical examination revealed multiple flaccid bullae in the web spaces of the hands and shallow erosions with hemorrhagic crusts on the bilateral wrists. She also had violaceous patches in the extensor creases of the metacarpophalangeal, proximal interphalangeal, and distal interphalangeal joints, which were strikingly symmetric (Figure 1). Prominent flaccid bullae and shallow erosions with hemorrhagic crusts also were present on the bilateral shins and dorsal aspects of the feet (Figure 2). No oral ulcers were present. A punch biopsy from the dorsal aspect of the left foot revealed psoriasiform hyperplasia of the epidermis with prominent ballooning degeneration and hyperkeratosis/parakeratosis (Figure 3); a periodic acid–Schiff stain was negative for fungal organisms.
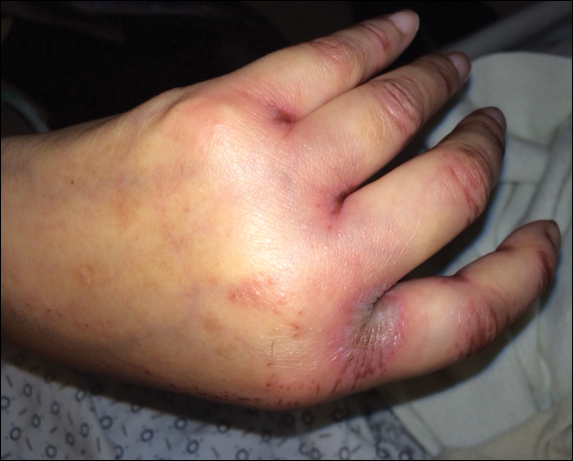
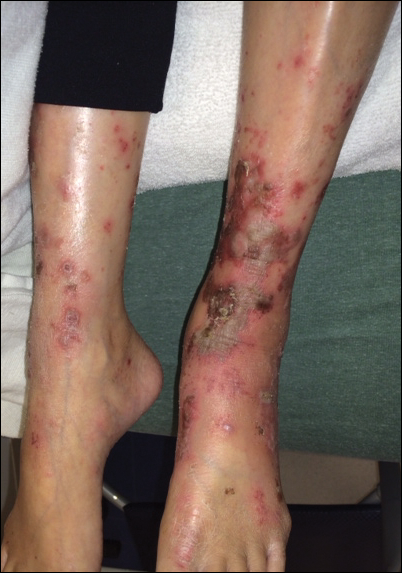
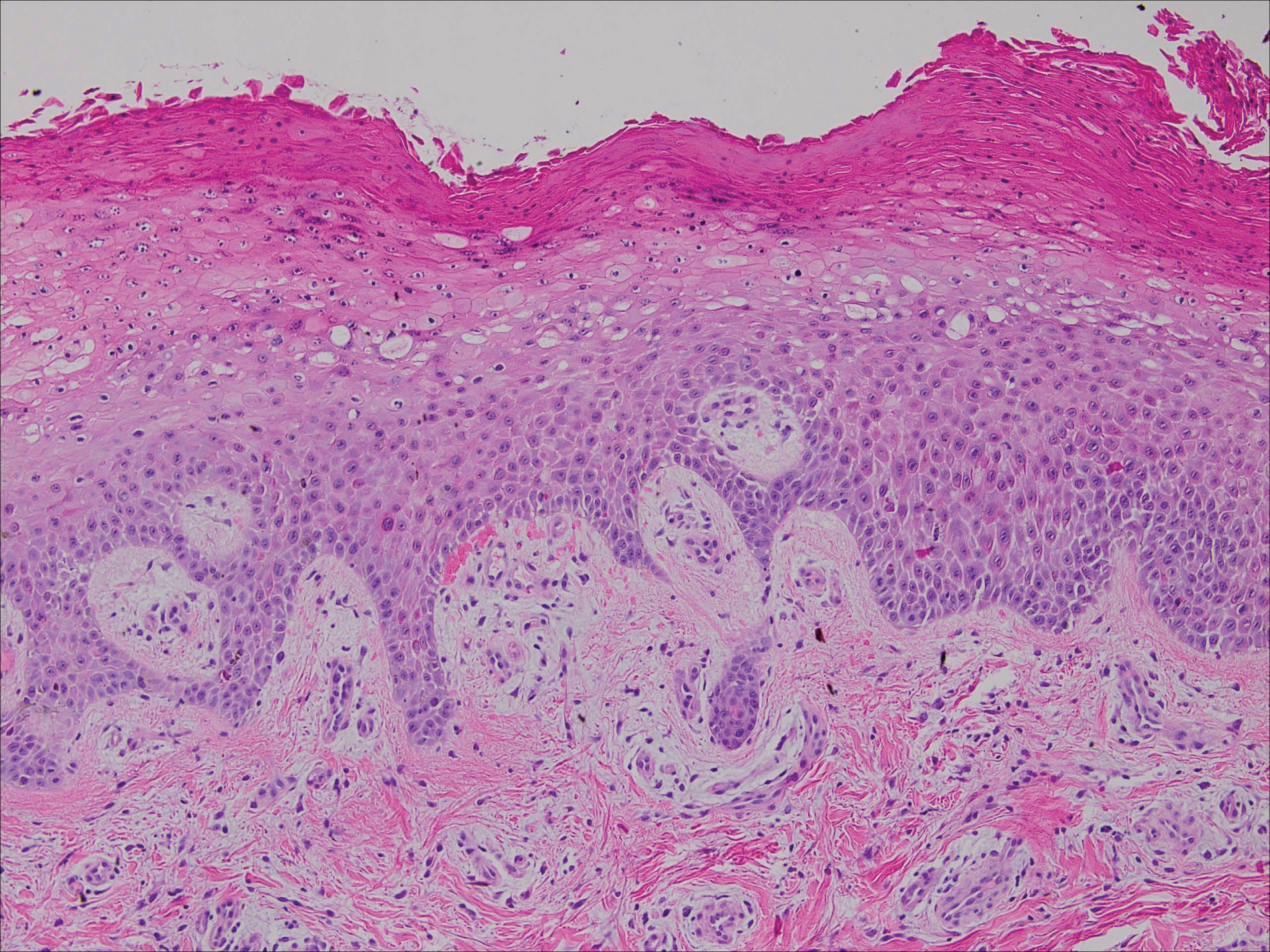
Given the biopsy results and clinical presentation, a nutritional deficiency was suspected and serum levels of zinc, vitamin B1, vitamin B2, and vitamin B3 were assessed. Vitamins B1, B2
Zinc is an essential trace element and can be found in high concentration in foods such as shellfish, green vegetables, legumes, nuts, and whole grains.6 The majority of zinc is absorbed in the jejunum; as such, many cases of acquired zinc deficiency leading to AE are dueto disorders that affect the small intestine.2 Conditions that may lead to poor gastrointestinal zinc absorption include alcoholism, eating disorders, TPN, burns, surgery, and malignancies.2,7
Diagnosis typically is made based on characteristic clinical features, biopsy results, and a measurement of the serum zinc concentration. Although a low serum zinc level supports the diagnosis, serum zinc concentration is not a reliable indicator of body zinc stores and a normal serum zinc concentration does not rule out AE. The gold standard for diagnosis is the resolution of lesions after zinc supplementation.1 Notably, because the production of alkaline phosphatase is dependent on zinc, levels of this enzyme also may be low in cases of AE,6 as in our patient.
The clinical manifestations of AE can vary greatly; patients may initially present with eczematous pink scaly plaques, which may subsequently become vesicular, bullous, pustular, or desquamative. The lesions may develop over the arms and legs as well as the anogenital and periorificial areas.5 Other notable manifestations that may present early in the course of AE include angular cheilitis followed by paronychia. In patients who are not promptly treated, long-term zinc deficiency may lead to growth delay, mental slowing, poor wound healing, anemia, and anorexia.5 Of note, deficiencies of branched-chain amino acids and essential fatty acids may appear clinically similar to AE.2
Zinc replacement is the treatment of choice for patients with AE due to dietary deficiency, and replacement therapy should begin with 0.5 to 1 mg/kg daily of elemental zinc.5 Response to acquired AE with zinc supplementation often is rapid. Lesions tend to resolve within days to weeks depending on the degree of deficiency.2
Although AE is an uncommon dermatosis in the United States, it is an important diagnosis to make because its clinical features are fairly specific and early zinc supplementation allows for full resolution of the disease without permanent sequelae. The diagnosis of AE should be strongly considered when features of an acral bullous dermatosis are combined with a fissured dermatitis of extensor joints of the hands or elbows. It is particularly important to recognize that alcoholics, burn victims, postsurgical patients, and those with malignancies and eating disorders are at an increased risk for developing this nutritional deficiency.
- Kumar P, Lal NR, Mondal AK, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Suchithra N, Sreejith P, Pappachan JM, et al. Acrodermatitis enteropathica-like skin eruption in a case of short bowel syndrome following jejuno-transverse colon anastomosis. Dermatol Online J. 2007;13:20.
- Sundaram A, Koutkia P, Apovian CM. Nutritional management of short bowel syndrome in adults. J Clin Gastroenterol. 2002;34:207-220.
- Griffin IJ, Kim SC, Hicks PD, et al. Zinc metabolism in adolescents with Crohn’s disease. Pediatr Res. 2004;56:235-239.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism [published online October 30, 2006]. J Am Acad Dermatol. 2007;56:116-124.
- Cheshire H, Stather P, Vorster J. Acquired acrodermatitis enteropathica due to zinc deficiency in a patient with pre-existing Darier’s disease. J Dermatol Case Rep. 2009;3:41-43.
- Strumia R. Dermatologic signs in patients with eating disorders. Am J Clin Dermatol. 2005;6:165-173.
To the Editor:
Acrodermatitis enteropathica (AE) is an inherited defect in zinc absorption that leads to hypozincemia. Its clinical presentation can vary based on serum zinc level and ranges from periorificial erosive dermatitis to psoriasiform dermatitis.1 Recognition of the cutaneous manifestations of zinc deficiency can lead to early intervention with zinc supplementation and prevention of long-term morbidity and even mortality. In our case, the coexistence of a bullous acral dermatosis with the additional feature of extensor digital dermatitis with fissuring suggests a diagnosis of AE and can alert the astute clinician to the need for testing of serum zinc levels and/or treatment with zinc supplementation. Causes of acquired zinc deficiency that have been reported in the literature include eating disorders such as anorexia nervosa and bulimia nervosa, Crohn disease, food allergy, intestinal parasitic infestations, and an inborn error of metabolism known as nonketotic hyperglycemia (Table).2-4
RELATED ARTICLE: Acquired Acrodermatitis Enteropathica Secondary to Alcoholism
A 42-year-old woman with a medical history of rheumatoid arthritis and short bowel syndrome due to multiple small bowel obstructions with subsequent bowel resections who was on chronic total parenteral nutrition (TPN) presented with bullae on the hands, shins, and feet. The patient initially noticed small erythematous macules on the hands and feet months prior to presentation. Three weeks prior to presentation, bullae started to form on the hands, mostly between the web spaces; dorsal aspects of the feet; and anterior aspects of the shins. The patient denied any oral ulcers. One day prior to presentation the patient was seen at an outside hospital and was started on prednisone 5 mg daily, oral clindamycin, mupirocin ointment, and nystatin-triamcinolone cream. These medications failed to improve her condition. On review of systems, the patient denied any fever, chills, eye pain, or dysuria.
Upon initial presentation the patient appeared weak and fatigued, though vital signs were normal. Physical examination revealed multiple flaccid bullae in the web spaces of the hands and shallow erosions with hemorrhagic crusts on the bilateral wrists. She also had violaceous patches in the extensor creases of the metacarpophalangeal, proximal interphalangeal, and distal interphalangeal joints, which were strikingly symmetric (Figure 1). Prominent flaccid bullae and shallow erosions with hemorrhagic crusts also were present on the bilateral shins and dorsal aspects of the feet (Figure 2). No oral ulcers were present. A punch biopsy from the dorsal aspect of the left foot revealed psoriasiform hyperplasia of the epidermis with prominent ballooning degeneration and hyperkeratosis/parakeratosis (Figure 3); a periodic acid–Schiff stain was negative for fungal organisms.
Given the biopsy results and clinical presentation, a nutritional deficiency was suspected and serum levels of zinc, vitamin B1, vitamin B2, and vitamin B3 were assessed. Vitamins B1, B2
Zinc is an essential trace element and can be found in high concentration in foods such as shellfish, green vegetables, legumes, nuts, and whole grains.6 The majority of zinc is absorbed in the jejunum; as such, many cases of acquired zinc deficiency leading to AE are dueto disorders that affect the small intestine.2 Conditions that may lead to poor gastrointestinal zinc absorption include alcoholism, eating disorders, TPN, burns, surgery, and malignancies.2,7
Diagnosis typically is made based on characteristic clinical features, biopsy results, and a measurement of the serum zinc concentration. Although a low serum zinc level supports the diagnosis, serum zinc concentration is not a reliable indicator of body zinc stores and a normal serum zinc concentration does not rule out AE. The gold standard for diagnosis is the resolution of lesions after zinc supplementation.1 Notably, because the production of alkaline phosphatase is dependent on zinc, levels of this enzyme also may be low in cases of AE,6 as in our patient.
The clinical manifestations of AE can vary greatly; patients may initially present with eczematous pink scaly plaques, which may subsequently become vesicular, bullous, pustular, or desquamative. The lesions may develop over the arms and legs as well as the anogenital and periorificial areas.5 Other notable manifestations that may present early in the course of AE include angular cheilitis followed by paronychia. In patients who are not promptly treated, long-term zinc deficiency may lead to growth delay, mental slowing, poor wound healing, anemia, and anorexia.5 Of note, deficiencies of branched-chain amino acids and essential fatty acids may appear clinically similar to AE.2
Zinc replacement is the treatment of choice for patients with AE due to dietary deficiency, and replacement therapy should begin with 0.5 to 1 mg/kg daily of elemental zinc.5 Response to acquired AE with zinc supplementation often is rapid. Lesions tend to resolve within days to weeks depending on the degree of deficiency.2
Although AE is an uncommon dermatosis in the United States, it is an important diagnosis to make because its clinical features are fairly specific and early zinc supplementation allows for full resolution of the disease without permanent sequelae. The diagnosis of AE should be strongly considered when features of an acral bullous dermatosis are combined with a fissured dermatitis of extensor joints of the hands or elbows. It is particularly important to recognize that alcoholics, burn victims, postsurgical patients, and those with malignancies and eating disorders are at an increased risk for developing this nutritional deficiency.
To the Editor:
Acrodermatitis enteropathica (AE) is an inherited defect in zinc absorption that leads to hypozincemia. Its clinical presentation can vary based on serum zinc level and ranges from periorificial erosive dermatitis to psoriasiform dermatitis.1 Recognition of the cutaneous manifestations of zinc deficiency can lead to early intervention with zinc supplementation and prevention of long-term morbidity and even mortality. In our case, the coexistence of a bullous acral dermatosis with the additional feature of extensor digital dermatitis with fissuring suggests a diagnosis of AE and can alert the astute clinician to the need for testing of serum zinc levels and/or treatment with zinc supplementation. Causes of acquired zinc deficiency that have been reported in the literature include eating disorders such as anorexia nervosa and bulimia nervosa, Crohn disease, food allergy, intestinal parasitic infestations, and an inborn error of metabolism known as nonketotic hyperglycemia (Table).2-4
RELATED ARTICLE: Acquired Acrodermatitis Enteropathica Secondary to Alcoholism
A 42-year-old woman with a medical history of rheumatoid arthritis and short bowel syndrome due to multiple small bowel obstructions with subsequent bowel resections who was on chronic total parenteral nutrition (TPN) presented with bullae on the hands, shins, and feet. The patient initially noticed small erythematous macules on the hands and feet months prior to presentation. Three weeks prior to presentation, bullae started to form on the hands, mostly between the web spaces; dorsal aspects of the feet; and anterior aspects of the shins. The patient denied any oral ulcers. One day prior to presentation the patient was seen at an outside hospital and was started on prednisone 5 mg daily, oral clindamycin, mupirocin ointment, and nystatin-triamcinolone cream. These medications failed to improve her condition. On review of systems, the patient denied any fever, chills, eye pain, or dysuria.
Upon initial presentation the patient appeared weak and fatigued, though vital signs were normal. Physical examination revealed multiple flaccid bullae in the web spaces of the hands and shallow erosions with hemorrhagic crusts on the bilateral wrists. She also had violaceous patches in the extensor creases of the metacarpophalangeal, proximal interphalangeal, and distal interphalangeal joints, which were strikingly symmetric (Figure 1). Prominent flaccid bullae and shallow erosions with hemorrhagic crusts also were present on the bilateral shins and dorsal aspects of the feet (Figure 2). No oral ulcers were present. A punch biopsy from the dorsal aspect of the left foot revealed psoriasiform hyperplasia of the epidermis with prominent ballooning degeneration and hyperkeratosis/parakeratosis (Figure 3); a periodic acid–Schiff stain was negative for fungal organisms.
Given the biopsy results and clinical presentation, a nutritional deficiency was suspected and serum levels of zinc, vitamin B1, vitamin B2, and vitamin B3 were assessed. Vitamins B1, B2
Zinc is an essential trace element and can be found in high concentration in foods such as shellfish, green vegetables, legumes, nuts, and whole grains.6 The majority of zinc is absorbed in the jejunum; as such, many cases of acquired zinc deficiency leading to AE are dueto disorders that affect the small intestine.2 Conditions that may lead to poor gastrointestinal zinc absorption include alcoholism, eating disorders, TPN, burns, surgery, and malignancies.2,7
Diagnosis typically is made based on characteristic clinical features, biopsy results, and a measurement of the serum zinc concentration. Although a low serum zinc level supports the diagnosis, serum zinc concentration is not a reliable indicator of body zinc stores and a normal serum zinc concentration does not rule out AE. The gold standard for diagnosis is the resolution of lesions after zinc supplementation.1 Notably, because the production of alkaline phosphatase is dependent on zinc, levels of this enzyme also may be low in cases of AE,6 as in our patient.
The clinical manifestations of AE can vary greatly; patients may initially present with eczematous pink scaly plaques, which may subsequently become vesicular, bullous, pustular, or desquamative. The lesions may develop over the arms and legs as well as the anogenital and periorificial areas.5 Other notable manifestations that may present early in the course of AE include angular cheilitis followed by paronychia. In patients who are not promptly treated, long-term zinc deficiency may lead to growth delay, mental slowing, poor wound healing, anemia, and anorexia.5 Of note, deficiencies of branched-chain amino acids and essential fatty acids may appear clinically similar to AE.2
Zinc replacement is the treatment of choice for patients with AE due to dietary deficiency, and replacement therapy should begin with 0.5 to 1 mg/kg daily of elemental zinc.5 Response to acquired AE with zinc supplementation often is rapid. Lesions tend to resolve within days to weeks depending on the degree of deficiency.2
Although AE is an uncommon dermatosis in the United States, it is an important diagnosis to make because its clinical features are fairly specific and early zinc supplementation allows for full resolution of the disease without permanent sequelae. The diagnosis of AE should be strongly considered when features of an acral bullous dermatosis are combined with a fissured dermatitis of extensor joints of the hands or elbows. It is particularly important to recognize that alcoholics, burn victims, postsurgical patients, and those with malignancies and eating disorders are at an increased risk for developing this nutritional deficiency.
- Kumar P, Lal NR, Mondal AK, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Suchithra N, Sreejith P, Pappachan JM, et al. Acrodermatitis enteropathica-like skin eruption in a case of short bowel syndrome following jejuno-transverse colon anastomosis. Dermatol Online J. 2007;13:20.
- Sundaram A, Koutkia P, Apovian CM. Nutritional management of short bowel syndrome in adults. J Clin Gastroenterol. 2002;34:207-220.
- Griffin IJ, Kim SC, Hicks PD, et al. Zinc metabolism in adolescents with Crohn’s disease. Pediatr Res. 2004;56:235-239.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism [published online October 30, 2006]. J Am Acad Dermatol. 2007;56:116-124.
- Cheshire H, Stather P, Vorster J. Acquired acrodermatitis enteropathica due to zinc deficiency in a patient with pre-existing Darier’s disease. J Dermatol Case Rep. 2009;3:41-43.
- Strumia R. Dermatologic signs in patients with eating disorders. Am J Clin Dermatol. 2005;6:165-173.
- Kumar P, Lal NR, Mondal AK, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Suchithra N, Sreejith P, Pappachan JM, et al. Acrodermatitis enteropathica-like skin eruption in a case of short bowel syndrome following jejuno-transverse colon anastomosis. Dermatol Online J. 2007;13:20.
- Sundaram A, Koutkia P, Apovian CM. Nutritional management of short bowel syndrome in adults. J Clin Gastroenterol. 2002;34:207-220.
- Griffin IJ, Kim SC, Hicks PD, et al. Zinc metabolism in adolescents with Crohn’s disease. Pediatr Res. 2004;56:235-239.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism [published online October 30, 2006]. J Am Acad Dermatol. 2007;56:116-124.
- Cheshire H, Stather P, Vorster J. Acquired acrodermatitis enteropathica due to zinc deficiency in a patient with pre-existing Darier’s disease. J Dermatol Case Rep. 2009;3:41-43.
- Strumia R. Dermatologic signs in patients with eating disorders. Am J Clin Dermatol. 2005;6:165-173.
Practice Points
- Acrodermatitis enteropathica can be a manifestation of zinc deficiency.
- Acrodermatitis enteropathica should be considered in patients with poor intestinal absorption of nutrients.