User login
2019 at a glance: Hem-onc U.S. drug approvals
The rapid development and identification of novel drugs has translated into innovative therapies in hematology and oncology. The aim of this piece is to present newly approved drugs and expanded indications to serve as a reference guide for practicing clinicians.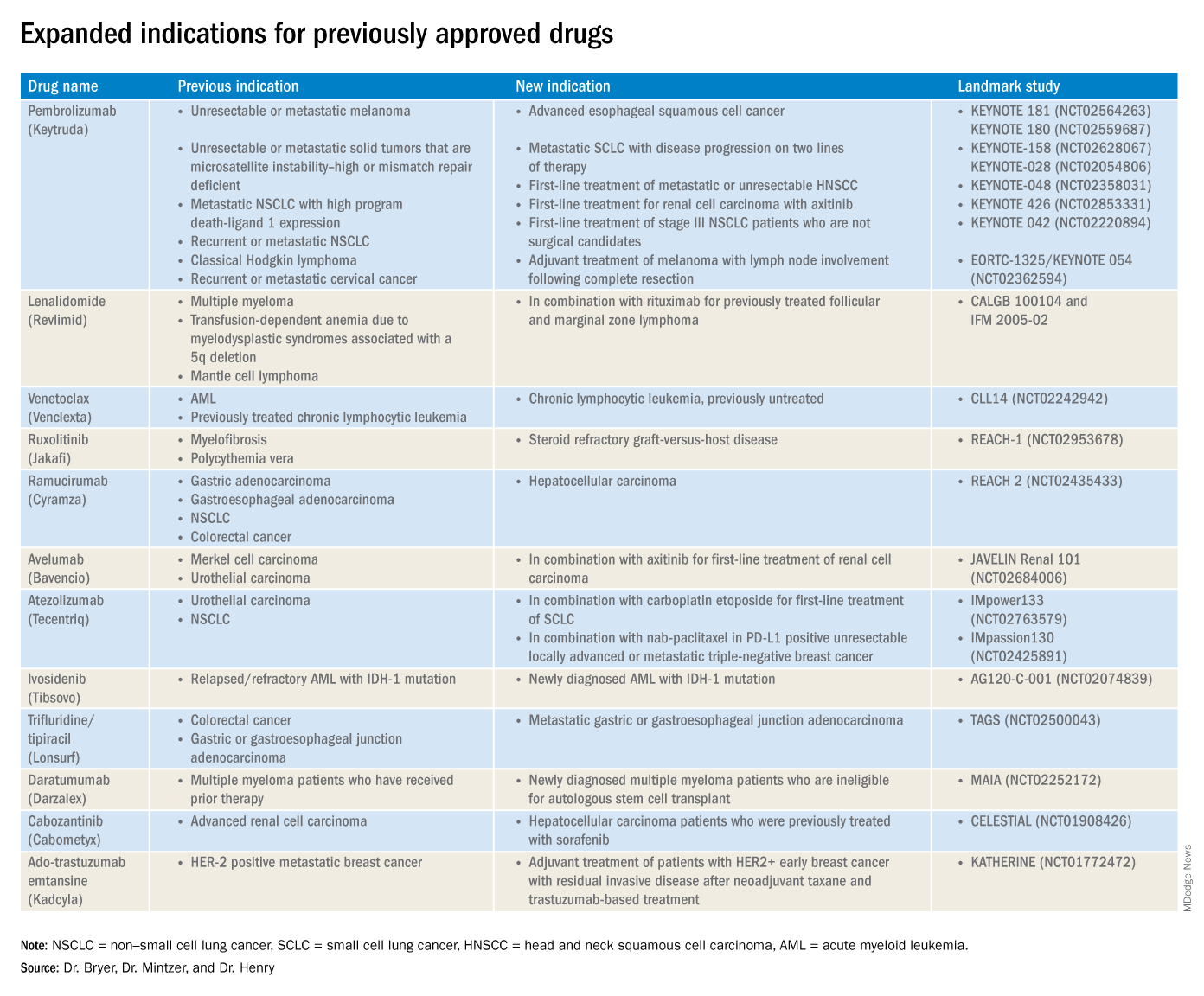
This article reviews therapies that were newly approved so far in 2019, as well as those previously approved whose indications were expanded this past year. The list highlights the most clinically important approvals, as well as adverse events that are unique or especially severe.
New approvals
Fedratinib (Inrebic)
Class: JAK2 and FLT3 selective kinase inhibitor.
Disease: Intermediate or high-risk primary or secondary (postpolycythemia vera or postessential thrombocythemia) myelofibrosis.
Dose: 400 mg orally once daily, with or without food.
Adverse events (AEs): Black box warning: Fatal encephalopathy, including Wernicke’s (thiamine level monitoring suggested).
Trials: In JAKARTA (NCT01437787), 37% of patients achieved a 35% or greater reduction in spleen volume and 40% received a 50% or greater reduction in myelofibrosis-related symptoms. In Jakarta-2, there was a 55% spleen response in patients resistant or intolerant to ruxolitinib.
Entrectinib (Rozlytrek)
Class: Tropomyosin receptor tyrosine kinase inhibitor.
Disease: Solid tumors that have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion and for ROS-1 positive non–small cell lung cancer (NSCLC).
Dose: 600 mg orally once daily.
AEs: Heart failure, QT prolongation, skeletal fractures, hepatotoxicity, central nervous system effects, and hyperuricemia.
Trial: ALKA, STARTRK-1 (NCT02097810) and STARTRK-2 (NCT02568267): Overall response rate of 57% for NTRK positive patients; response rate of 77% in ROS-1 positive NSCLC.
Pexidartinib (Turalio)
Class: Small molecule tyrosine kinase inhibitor targeting CSF1R.
Disease: Symptomatic tenosynovial giant cell tumor.
Dose: 400 mg orally twice daily without food.
AEs: Black box warning on hepatotoxicity.
Trial: ENLIVEN (NCT02371369): Overall response rate of 38% at 25 weeks, with a 15% complete response rate and a 23% partial response rate.
Darolutamide (Nubeqa)
Class: Androgen receptor inhibitor.
Disease: Nonmetastatic castration-resistant prostate cancer.
Dose: 600 mg orally twice daily with food with concomitant androgen deprivation therapy.
AEs: Fatigue, extremity pain, and rash.
Trial: ARAMIS (NCT02200614): Median metastasis free survival was 40.4 months for patients with darolutamide, compared with 18.4 months for controls.
Selinexor (Xpovio)
Class: Reversible inhibitor of nuclear export of tumor suppressor proteins, growth regulators, and mRNAs of oncogenic proteins.
Disease: Relapsed or refractory multiple myeloma. Indicated for patients who have received at least four prior therapies, including at least two immunomodulatory agents and an anti-CD38 monoclonal antibody.
Dose: 80 mg orally in combination with oral dexamethasone on days 1 and 3 of each week.
AEs: Thrombocytopenia, fatigue, pancytopenia, and hyponatremia.
Trial: STORM (NCT02336815): Overall response rate 25.3% with a median time to first response of 4 weeks and 3.8-month median duration of response.
Polatuzumab vedotin-piiq (Polivy)
Class: CD79b-directed antibody-drug conjugate.
Disease: Relapsed or refractory diffuse large B-cell lymphoma. Indicated for patients who have had at least two prior therapies.
Dose: 1.8 mg/kg intravenous infusion every 21 days for six cycles in combination with bendamustine and a rituximab product.
AEs: Pancytopenia, peripheral neuropathy.
Trial: GO29365 (NCT02257567): Complete response rate was 40% for polatuzumab vedotin-piiq plus bendamustine/rituximab, compared with 18% with bendamustine/rituximab alone.*
Caplacizumab-yhdp (Cablivi)
Class: Monoclonal antibody fragment directed against von Willebrand factor.
Disease: Thrombotic thrombocytopenic purpura.
Dose: 11 mg IV initially, then daily subcutaneously; in combination with plasma exchange and immunosuppressive therapy.
AEs: Epistaxis, headache, and gingival bleeding.
Trial: Hercules trial (NCT02553317): More rapid normalization of platelets, lower incidence of composite TTP-related death, and lower rate of recurrence when added to plasma exchange and steroids.
Alpelisib (Piqray)
Class: Phosphatidylinositol-3-kinase (PI3K) inhibitor.
Disease: Hormone receptor positive HER2-negative PIK3CA-mutated, advanced or metastatic breast cancer.
Dose: 300 mg orally once daily with food with concomitant fulvestrant.
AEs: Hyperglycemia, pancytopenia.
Trial: SOLAR-1 (NCT02437318): 11-month progression-free survival among patients treated with alpelisib and fulvestrant, compared with 5.7 months in fulvestrant alone control arm; overall response rate of 36% versus 16%, respectively.
Erdafitinib (Balversa)
Class: Fibroblast growth factor receptor kinase inhibitor.
Disease: Locally advanced or metastatic urothelial carcinoma with FGFR3 or FGFR2 mutations.
Dose: 8 mg orally once daily, with or without food.
AEs: Ocular disorders including retinopathy or retinal detachment.
Trial: BLC2001 (NCT02365597): Objective response rate of 32.2%, with a complete response in 2.3% of patients and partial response in 29.9% of patients.
Biosimilar approvals
Trastuzumab and hyaluronidase-oysk (Herceptin Hylecta)
Biosimilar to: Trastuzumab.
Indication: HER2-overexpressing breast cancer.
Dr. Bryer is a resident in the department of internal medicine at the University of Pennsylvania, Philadelphia. Dr. Mintzer is chief of hematology-oncology at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania. Dr. Henry is a hematologist-oncologist at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania.
*Correction, 11/7/2019: An earlier version of this article misstated the drug combination in the GO29365 trial.
The rapid development and identification of novel drugs has translated into innovative therapies in hematology and oncology. The aim of this piece is to present newly approved drugs and expanded indications to serve as a reference guide for practicing clinicians.
This article reviews therapies that were newly approved so far in 2019, as well as those previously approved whose indications were expanded this past year. The list highlights the most clinically important approvals, as well as adverse events that are unique or especially severe.
New approvals
Fedratinib (Inrebic)
Class: JAK2 and FLT3 selective kinase inhibitor.
Disease: Intermediate or high-risk primary or secondary (postpolycythemia vera or postessential thrombocythemia) myelofibrosis.
Dose: 400 mg orally once daily, with or without food.
Adverse events (AEs): Black box warning: Fatal encephalopathy, including Wernicke’s (thiamine level monitoring suggested).
Trials: In JAKARTA (NCT01437787), 37% of patients achieved a 35% or greater reduction in spleen volume and 40% received a 50% or greater reduction in myelofibrosis-related symptoms. In Jakarta-2, there was a 55% spleen response in patients resistant or intolerant to ruxolitinib.
Entrectinib (Rozlytrek)
Class: Tropomyosin receptor tyrosine kinase inhibitor.
Disease: Solid tumors that have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion and for ROS-1 positive non–small cell lung cancer (NSCLC).
Dose: 600 mg orally once daily.
AEs: Heart failure, QT prolongation, skeletal fractures, hepatotoxicity, central nervous system effects, and hyperuricemia.
Trial: ALKA, STARTRK-1 (NCT02097810) and STARTRK-2 (NCT02568267): Overall response rate of 57% for NTRK positive patients; response rate of 77% in ROS-1 positive NSCLC.
Pexidartinib (Turalio)
Class: Small molecule tyrosine kinase inhibitor targeting CSF1R.
Disease: Symptomatic tenosynovial giant cell tumor.
Dose: 400 mg orally twice daily without food.
AEs: Black box warning on hepatotoxicity.
Trial: ENLIVEN (NCT02371369): Overall response rate of 38% at 25 weeks, with a 15% complete response rate and a 23% partial response rate.
Darolutamide (Nubeqa)
Class: Androgen receptor inhibitor.
Disease: Nonmetastatic castration-resistant prostate cancer.
Dose: 600 mg orally twice daily with food with concomitant androgen deprivation therapy.
AEs: Fatigue, extremity pain, and rash.
Trial: ARAMIS (NCT02200614): Median metastasis free survival was 40.4 months for patients with darolutamide, compared with 18.4 months for controls.
Selinexor (Xpovio)
Class: Reversible inhibitor of nuclear export of tumor suppressor proteins, growth regulators, and mRNAs of oncogenic proteins.
Disease: Relapsed or refractory multiple myeloma. Indicated for patients who have received at least four prior therapies, including at least two immunomodulatory agents and an anti-CD38 monoclonal antibody.
Dose: 80 mg orally in combination with oral dexamethasone on days 1 and 3 of each week.
AEs: Thrombocytopenia, fatigue, pancytopenia, and hyponatremia.
Trial: STORM (NCT02336815): Overall response rate 25.3% with a median time to first response of 4 weeks and 3.8-month median duration of response.
Polatuzumab vedotin-piiq (Polivy)
Class: CD79b-directed antibody-drug conjugate.
Disease: Relapsed or refractory diffuse large B-cell lymphoma. Indicated for patients who have had at least two prior therapies.
Dose: 1.8 mg/kg intravenous infusion every 21 days for six cycles in combination with bendamustine and a rituximab product.
AEs: Pancytopenia, peripheral neuropathy.
Trial: GO29365 (NCT02257567): Complete response rate was 40% for polatuzumab vedotin-piiq plus bendamustine/rituximab, compared with 18% with bendamustine/rituximab alone.*
Caplacizumab-yhdp (Cablivi)
Class: Monoclonal antibody fragment directed against von Willebrand factor.
Disease: Thrombotic thrombocytopenic purpura.
Dose: 11 mg IV initially, then daily subcutaneously; in combination with plasma exchange and immunosuppressive therapy.
AEs: Epistaxis, headache, and gingival bleeding.
Trial: Hercules trial (NCT02553317): More rapid normalization of platelets, lower incidence of composite TTP-related death, and lower rate of recurrence when added to plasma exchange and steroids.
Alpelisib (Piqray)
Class: Phosphatidylinositol-3-kinase (PI3K) inhibitor.
Disease: Hormone receptor positive HER2-negative PIK3CA-mutated, advanced or metastatic breast cancer.
Dose: 300 mg orally once daily with food with concomitant fulvestrant.
AEs: Hyperglycemia, pancytopenia.
Trial: SOLAR-1 (NCT02437318): 11-month progression-free survival among patients treated with alpelisib and fulvestrant, compared with 5.7 months in fulvestrant alone control arm; overall response rate of 36% versus 16%, respectively.
Erdafitinib (Balversa)
Class: Fibroblast growth factor receptor kinase inhibitor.
Disease: Locally advanced or metastatic urothelial carcinoma with FGFR3 or FGFR2 mutations.
Dose: 8 mg orally once daily, with or without food.
AEs: Ocular disorders including retinopathy or retinal detachment.
Trial: BLC2001 (NCT02365597): Objective response rate of 32.2%, with a complete response in 2.3% of patients and partial response in 29.9% of patients.
Biosimilar approvals
Trastuzumab and hyaluronidase-oysk (Herceptin Hylecta)
Biosimilar to: Trastuzumab.
Indication: HER2-overexpressing breast cancer.
Dr. Bryer is a resident in the department of internal medicine at the University of Pennsylvania, Philadelphia. Dr. Mintzer is chief of hematology-oncology at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania. Dr. Henry is a hematologist-oncologist at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania.
*Correction, 11/7/2019: An earlier version of this article misstated the drug combination in the GO29365 trial.
The rapid development and identification of novel drugs has translated into innovative therapies in hematology and oncology. The aim of this piece is to present newly approved drugs and expanded indications to serve as a reference guide for practicing clinicians.
This article reviews therapies that were newly approved so far in 2019, as well as those previously approved whose indications were expanded this past year. The list highlights the most clinically important approvals, as well as adverse events that are unique or especially severe.
New approvals
Fedratinib (Inrebic)
Class: JAK2 and FLT3 selective kinase inhibitor.
Disease: Intermediate or high-risk primary or secondary (postpolycythemia vera or postessential thrombocythemia) myelofibrosis.
Dose: 400 mg orally once daily, with or without food.
Adverse events (AEs): Black box warning: Fatal encephalopathy, including Wernicke’s (thiamine level monitoring suggested).
Trials: In JAKARTA (NCT01437787), 37% of patients achieved a 35% or greater reduction in spleen volume and 40% received a 50% or greater reduction in myelofibrosis-related symptoms. In Jakarta-2, there was a 55% spleen response in patients resistant or intolerant to ruxolitinib.
Entrectinib (Rozlytrek)
Class: Tropomyosin receptor tyrosine kinase inhibitor.
Disease: Solid tumors that have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion and for ROS-1 positive non–small cell lung cancer (NSCLC).
Dose: 600 mg orally once daily.
AEs: Heart failure, QT prolongation, skeletal fractures, hepatotoxicity, central nervous system effects, and hyperuricemia.
Trial: ALKA, STARTRK-1 (NCT02097810) and STARTRK-2 (NCT02568267): Overall response rate of 57% for NTRK positive patients; response rate of 77% in ROS-1 positive NSCLC.
Pexidartinib (Turalio)
Class: Small molecule tyrosine kinase inhibitor targeting CSF1R.
Disease: Symptomatic tenosynovial giant cell tumor.
Dose: 400 mg orally twice daily without food.
AEs: Black box warning on hepatotoxicity.
Trial: ENLIVEN (NCT02371369): Overall response rate of 38% at 25 weeks, with a 15% complete response rate and a 23% partial response rate.
Darolutamide (Nubeqa)
Class: Androgen receptor inhibitor.
Disease: Nonmetastatic castration-resistant prostate cancer.
Dose: 600 mg orally twice daily with food with concomitant androgen deprivation therapy.
AEs: Fatigue, extremity pain, and rash.
Trial: ARAMIS (NCT02200614): Median metastasis free survival was 40.4 months for patients with darolutamide, compared with 18.4 months for controls.
Selinexor (Xpovio)
Class: Reversible inhibitor of nuclear export of tumor suppressor proteins, growth regulators, and mRNAs of oncogenic proteins.
Disease: Relapsed or refractory multiple myeloma. Indicated for patients who have received at least four prior therapies, including at least two immunomodulatory agents and an anti-CD38 monoclonal antibody.
Dose: 80 mg orally in combination with oral dexamethasone on days 1 and 3 of each week.
AEs: Thrombocytopenia, fatigue, pancytopenia, and hyponatremia.
Trial: STORM (NCT02336815): Overall response rate 25.3% with a median time to first response of 4 weeks and 3.8-month median duration of response.
Polatuzumab vedotin-piiq (Polivy)
Class: CD79b-directed antibody-drug conjugate.
Disease: Relapsed or refractory diffuse large B-cell lymphoma. Indicated for patients who have had at least two prior therapies.
Dose: 1.8 mg/kg intravenous infusion every 21 days for six cycles in combination with bendamustine and a rituximab product.
AEs: Pancytopenia, peripheral neuropathy.
Trial: GO29365 (NCT02257567): Complete response rate was 40% for polatuzumab vedotin-piiq plus bendamustine/rituximab, compared with 18% with bendamustine/rituximab alone.*
Caplacizumab-yhdp (Cablivi)
Class: Monoclonal antibody fragment directed against von Willebrand factor.
Disease: Thrombotic thrombocytopenic purpura.
Dose: 11 mg IV initially, then daily subcutaneously; in combination with plasma exchange and immunosuppressive therapy.
AEs: Epistaxis, headache, and gingival bleeding.
Trial: Hercules trial (NCT02553317): More rapid normalization of platelets, lower incidence of composite TTP-related death, and lower rate of recurrence when added to plasma exchange and steroids.
Alpelisib (Piqray)
Class: Phosphatidylinositol-3-kinase (PI3K) inhibitor.
Disease: Hormone receptor positive HER2-negative PIK3CA-mutated, advanced or metastatic breast cancer.
Dose: 300 mg orally once daily with food with concomitant fulvestrant.
AEs: Hyperglycemia, pancytopenia.
Trial: SOLAR-1 (NCT02437318): 11-month progression-free survival among patients treated with alpelisib and fulvestrant, compared with 5.7 months in fulvestrant alone control arm; overall response rate of 36% versus 16%, respectively.
Erdafitinib (Balversa)
Class: Fibroblast growth factor receptor kinase inhibitor.
Disease: Locally advanced or metastatic urothelial carcinoma with FGFR3 or FGFR2 mutations.
Dose: 8 mg orally once daily, with or without food.
AEs: Ocular disorders including retinopathy or retinal detachment.
Trial: BLC2001 (NCT02365597): Objective response rate of 32.2%, with a complete response in 2.3% of patients and partial response in 29.9% of patients.
Biosimilar approvals
Trastuzumab and hyaluronidase-oysk (Herceptin Hylecta)
Biosimilar to: Trastuzumab.
Indication: HER2-overexpressing breast cancer.
Dr. Bryer is a resident in the department of internal medicine at the University of Pennsylvania, Philadelphia. Dr. Mintzer is chief of hematology-oncology at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania. Dr. Henry is a hematologist-oncologist at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania.
*Correction, 11/7/2019: An earlier version of this article misstated the drug combination in the GO29365 trial.
Opioid use disorder is a chronic condition that needs ongoing therapy
Think of opioid use disorder as a chronic condition that requires continuous treatment.
This advice comes from Mitra Ahadpour, MD, who is the deputy director of the Office of Translational Sciences at the Food and Drug Administration’s Center for Drug Evaluation and Research and an addiction medicine specialist.
In an interview, Dr. Ahadpour discusses prescribing requirements and the administration of drugs for medication-assisted treatment, which is associated with improved social functioning and reduced risks for death from overdoses and by contracting HIV.
These treatments – which include various formulations of The interview includes her recommendations for recognizing at-risk patients and avoiding potential drug-drug interactions associated with using these drugs.
Click here to read the article at the FDA website.
Think of opioid use disorder as a chronic condition that requires continuous treatment.
This advice comes from Mitra Ahadpour, MD, who is the deputy director of the Office of Translational Sciences at the Food and Drug Administration’s Center for Drug Evaluation and Research and an addiction medicine specialist.
In an interview, Dr. Ahadpour discusses prescribing requirements and the administration of drugs for medication-assisted treatment, which is associated with improved social functioning and reduced risks for death from overdoses and by contracting HIV.
These treatments – which include various formulations of The interview includes her recommendations for recognizing at-risk patients and avoiding potential drug-drug interactions associated with using these drugs.
Click here to read the article at the FDA website.
Think of opioid use disorder as a chronic condition that requires continuous treatment.
This advice comes from Mitra Ahadpour, MD, who is the deputy director of the Office of Translational Sciences at the Food and Drug Administration’s Center for Drug Evaluation and Research and an addiction medicine specialist.
In an interview, Dr. Ahadpour discusses prescribing requirements and the administration of drugs for medication-assisted treatment, which is associated with improved social functioning and reduced risks for death from overdoses and by contracting HIV.
These treatments – which include various formulations of The interview includes her recommendations for recognizing at-risk patients and avoiding potential drug-drug interactions associated with using these drugs.
Click here to read the article at the FDA website.
FDA announces plan for biosimilar innovation and competition
Some of the actions include tools to enhance public information about the FDA’s evaluation of biosimilars, including more information about approved biological products in the Purple Book; exploring the potential for entering into new data sharing agreements with foreign regulators to facilitate the increased use of non–U.S.-licensed comparator products in certain studies to support a biosimilar application; releasing a series of videos that explain key concepts about biosimilar and interchangeable products; and requesting information from the public on additional policy steps the FDA should consider for enhancing the biosimilar program.
The FDA’s Biosimilar Action Plan is available here.
Some of the actions include tools to enhance public information about the FDA’s evaluation of biosimilars, including more information about approved biological products in the Purple Book; exploring the potential for entering into new data sharing agreements with foreign regulators to facilitate the increased use of non–U.S.-licensed comparator products in certain studies to support a biosimilar application; releasing a series of videos that explain key concepts about biosimilar and interchangeable products; and requesting information from the public on additional policy steps the FDA should consider for enhancing the biosimilar program.
The FDA’s Biosimilar Action Plan is available here.
Some of the actions include tools to enhance public information about the FDA’s evaluation of biosimilars, including more information about approved biological products in the Purple Book; exploring the potential for entering into new data sharing agreements with foreign regulators to facilitate the increased use of non–U.S.-licensed comparator products in certain studies to support a biosimilar application; releasing a series of videos that explain key concepts about biosimilar and interchangeable products; and requesting information from the public on additional policy steps the FDA should consider for enhancing the biosimilar program.
The FDA’s Biosimilar Action Plan is available here.
CDC now offering CME course on HPV vaccination
The Centers for Disease Control and Prevention is now offering a CME course to educate clinicians about the importance of human papillomavirus (HPV) vaccination in protecting adolescents from certain types of cancer and to provide them with the skills and resources to make effective HPV vaccine recommendations.
The course is a Web-on-demand video that will teach clinicians how to be successful in making HPV vaccination recommendations, how to communicate HPV vaccination information to parents and patients, and how to properly answer parents’ questions. Currently, the CDC recommends the HPV vaccine for adolescents at 11- to 12-years-of-age. The CDC hopes this will reduce missed opportunities to protect patients against HPV.
Speakers in the video include Alix Casler, MD, of the Orlando Family Physician Association; Linda Fu, MD, MS, of Children’s National Health System in Washington; Todd Wolynn, MD, president and CEO of Kids Plus Pediatrics, Pittsburgh; and Wendy Sue Swanson, MD, MBE, a pediatrician who is chief of digital innovation at Seattle Children’s Hospital.
The course was initiated in Jan. 16, 2018, and will continue until Jan. 16, 2020. Anyone who provides immunization to patients can participate.
The course is called “Routinely Recommending Cancer Prevention: HPV Vaccination at 11 and 12 as a Standard of Care.” Read more about the course on and download it from the CDC’s website.
The Centers for Disease Control and Prevention is now offering a CME course to educate clinicians about the importance of human papillomavirus (HPV) vaccination in protecting adolescents from certain types of cancer and to provide them with the skills and resources to make effective HPV vaccine recommendations.
The course is a Web-on-demand video that will teach clinicians how to be successful in making HPV vaccination recommendations, how to communicate HPV vaccination information to parents and patients, and how to properly answer parents’ questions. Currently, the CDC recommends the HPV vaccine for adolescents at 11- to 12-years-of-age. The CDC hopes this will reduce missed opportunities to protect patients against HPV.
Speakers in the video include Alix Casler, MD, of the Orlando Family Physician Association; Linda Fu, MD, MS, of Children’s National Health System in Washington; Todd Wolynn, MD, president and CEO of Kids Plus Pediatrics, Pittsburgh; and Wendy Sue Swanson, MD, MBE, a pediatrician who is chief of digital innovation at Seattle Children’s Hospital.
The course was initiated in Jan. 16, 2018, and will continue until Jan. 16, 2020. Anyone who provides immunization to patients can participate.
The course is called “Routinely Recommending Cancer Prevention: HPV Vaccination at 11 and 12 as a Standard of Care.” Read more about the course on and download it from the CDC’s website.
The Centers for Disease Control and Prevention is now offering a CME course to educate clinicians about the importance of human papillomavirus (HPV) vaccination in protecting adolescents from certain types of cancer and to provide them with the skills and resources to make effective HPV vaccine recommendations.
The course is a Web-on-demand video that will teach clinicians how to be successful in making HPV vaccination recommendations, how to communicate HPV vaccination information to parents and patients, and how to properly answer parents’ questions. Currently, the CDC recommends the HPV vaccine for adolescents at 11- to 12-years-of-age. The CDC hopes this will reduce missed opportunities to protect patients against HPV.
Speakers in the video include Alix Casler, MD, of the Orlando Family Physician Association; Linda Fu, MD, MS, of Children’s National Health System in Washington; Todd Wolynn, MD, president and CEO of Kids Plus Pediatrics, Pittsburgh; and Wendy Sue Swanson, MD, MBE, a pediatrician who is chief of digital innovation at Seattle Children’s Hospital.
The course was initiated in Jan. 16, 2018, and will continue until Jan. 16, 2020. Anyone who provides immunization to patients can participate.
The course is called “Routinely Recommending Cancer Prevention: HPV Vaccination at 11 and 12 as a Standard of Care.” Read more about the course on and download it from the CDC’s website.
CDC releases website for ME/CFS information
The Centers for Disease Control and Prevention has released a website for health care providers about myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS).
The website is designed to help clinicians by offering information about how they can better assess and help their patients manage the illness. The website includes information about the presentation and clinical course of ME/CFS, diagnosis, clinical care, understanding historical case definitions and criteria, and free continuing education.
The website’s resource section includes the National Academy of Medicine report published in 2015, as well as primers and clinical practice guidelines.
Currently, it is estimated that 836,000 to 2.5 million Americans have ME/CFS. Roughly 90% of people who have ME/CFS have not been diagnosed.
The Centers for Disease Control and Prevention has released a website for health care providers about myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS).
The website is designed to help clinicians by offering information about how they can better assess and help their patients manage the illness. The website includes information about the presentation and clinical course of ME/CFS, diagnosis, clinical care, understanding historical case definitions and criteria, and free continuing education.
The website’s resource section includes the National Academy of Medicine report published in 2015, as well as primers and clinical practice guidelines.
Currently, it is estimated that 836,000 to 2.5 million Americans have ME/CFS. Roughly 90% of people who have ME/CFS have not been diagnosed.
The Centers for Disease Control and Prevention has released a website for health care providers about myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS).
The website is designed to help clinicians by offering information about how they can better assess and help their patients manage the illness. The website includes information about the presentation and clinical course of ME/CFS, diagnosis, clinical care, understanding historical case definitions and criteria, and free continuing education.
The website’s resource section includes the National Academy of Medicine report published in 2015, as well as primers and clinical practice guidelines.
Currently, it is estimated that 836,000 to 2.5 million Americans have ME/CFS. Roughly 90% of people who have ME/CFS have not been diagnosed.
Adult soft tissue sarcoma: Professional resources from the National Cancer Institute
Adult Soft Tissue Sarcoma Treatment (PDQ®)–Health Professional Version
General Information About Adult Soft Tissue Sarcoma
Incidence and Mortality
Estimated new cases and deaths from soft tissue sarcoma in the United States in 2018:[1]
- New cases: 13,040.
- Deaths: 5,150.
Soft tissue sarcomas are malignant tumors that arise in any of the mesodermal tissues of the extremities (50%), trunk and retroperitoneum (40%), or head and neck (10%). The reported international incidence rates range from 1.8 to 5 per 100,000 individuals per year.[2]
Risk Factors and Genetic Factors
The risk of sporadic soft tissue sarcomas is increased by previous radiation therapy and, in the case of lymphangiosarcoma, by chronic lymphedema. The chemicals Thorotrast (thorium dioxide), vinyl chloride, and arsenic are also established carcinogens for hepatic angiosarcomas.[3-5]
Soft tissue sarcomas occur with greater frequency in patients with the following inherited syndromes:[3-5]
- Nevoid basal cell carcinoma syndrome (Gorlin syndrome: PTC gene mutation).
- Gardner syndrome (APC mutation).
- Li-Fraumeni syndrome (p53 mutation).
- Tuberous sclerosis (Bourneville disease: TSC1 or TSC2 mutation).
- von Recklinghausen disease (neurofibromatosis type 1: NF1 mutation).
- Werner syndrome (adult progeria: WRN mutation).
Diagnosis
Soft tissue sarcomas may be heterogeneous, so adequate tissue should be obtained via either core-needle or incisional biopsy for microscopic examination to determine histologic type and tumor grade. Careful planning of the initial biopsy is important to avoid compromising subsequent curative resection. Since the selection of treatment is determined by the grade of the tumor, it is essential to have a careful review of the biopsy tissue by a pathologist who is experienced in diagnosing sarcomas. Complete staging and treatment planning by a multidisciplinary team of cancer specialists is required to determine the optimal treatment for patients with this disease.
There is evidence that at least some favorable clinical outcomes may be associated with referral to a specialized sarcoma treatment center. In a population-based consecutive series of 375 soft tissue sarcoma patients in Sweden, local recurrence rates of resected tumors were higher in patients who were not referred to the specialized center: in 35 of 78 (45%) patients not referred; in 24 of 102 (24%) patients referred after initial surgery or incisional biopsy; and in 36 of 195 (18%) patients referred before any surgical procedure (P = .0001 for the difference between those never referred vs. those referred before any surgical procedure).[6][Level of evidence: 3iDii] However, there were no statistically significant differences in death from sarcoma between the groups of patients.
Prognostic Factors
The prognosis for patients with adult soft tissue sarcomas depends on several factors, including:[3-5,7,8]
- Patient’s age.
- Size, sarcoma subtype, histologic grade, mitotic activity, and stage of the tumor.
Factors associated with a poorer prognosis include the following:[9]
- Age older than 60 years.
- Tumors larger than 5 cm in greatest dimension.
- High-grade histology with high mitotic activity.
- Positive margins after resection.[10]
Although low-grade tumors are usually curable by surgery alone, higher-grade sarcomas (as determined by the mitotic index and by the presence of hemorrhage and necrosis) are associated with higher local-treatment failure rates and increased metastatic potential.
Surveillance for Relapse
A retrospective review included 174 consecutive patients with a soft tissue sarcoma of the limb who underwent follow-up by oncologists at a single center from 2003 to 2009.[11] The rate and site of recurrence and mode of detection were analyzed. Eighty-two patients (47%) experienced relapse. Isolated local recurrences occurred in 26 patients and local relapse with synchronous pulmonary metastases occurred in 5 patients. Local recurrences were detected clinically in 30 of the 31 patients; magnetic resonance imaging identified only one local recurrence. Twenty-eight patients developed isolated lung metastases; in 9 patients, the lung metastases were amenable to resections, 7 of whom were free of disease after treatment. Lung metastases were detected by chest x-ray in 19 patients, by computed tomography scanning in 3 patients, and clinically in 11 patients. Twenty-three patients developed nonpulmonary metastases. More than 80% of the relapses occurred in the first 2 years of follow-up; however, later recurrences were also observed.[11][Level of evidence: 3iiDi] This study supports imaging surveillance for detection of lung metastases, whereas local recurrences at the primary site were usually detected by clinical examination. The impact of picking up metastases from overall survival or quality-of-life data is unknown.
Related Summaries
Other PDQ summaries containing information about soft tissue sarcoma include:
References
- American Cancer Society: Cancer Facts and Figures 2018. Atlanta, Ga: American Cancer Society, 2018. Available online. Last accessed January 5, 2018.
- Wibmer C, Leithner A, Zielonke N, et al.: Increasing incidence rates of soft tissue sarcomas? A population-based epidemiologic study and literature review. Ann Oncol 21 (5): 1106-11, 2010. [PUBMED Abstract]
- Singer S, Nielsen T, Antonescu CR: Molecular biology of soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1522-32.
- Singer S, Maki RG, O'Sullivan B: Soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1533-77.
- Malawer MM, Helman LJ, O'Sullivan B: Sarcomas of bone. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1578-1609.
- Gustafson P, Dreinhöfer KE, Rydholm A: Soft tissue sarcoma should be treated at a tumor center. A comparison of quality of surgery in 375 patients. Acta Orthop Scand 65 (1): 47-50, 1994. [PUBMED Abstract]
- Coindre JM, Terrier P, Guillou L, et al.: Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 91 (10): 1914-26, 2001. [PUBMED Abstract]
- Kasper B, Ouali M, van Glabbeke M, et al.: Prognostic factors in adolescents and young adults (AYA) with high risk soft tissue sarcoma (STS) treated by adjuvant chemotherapy: a study based on pooled European Organisation for Research and Treatment of Cancer (EORTC) clinical trials 62771 and 62931. Eur J Cancer 49 (2): 449-56, 2013. [PUBMED Abstract]
- Vraa S, Keller J, Nielsen OS, et al.: Prognostic factors in soft tissue sarcomas: the Aarhus experience. Eur J Cancer 34 (12): 1876-82, 1998. [PUBMED Abstract]
- Trovik LH, Ovrebo K, Almquist M, et al.: Adjuvant radiotherapy in retroperitoneal sarcomas. A Scandinavian Sarcoma Group study of 97 patients. Acta Oncol 53 (9): 1165-72, 2014. [PUBMED Abstract]
- Rothermundt C, Whelan JS, Dileo P, et al.: What is the role of routine follow-up for localised limb soft tissue sarcomas? A retrospective analysis of 174 patients. Br J Cancer 110 (10): 2420-6, 2014. [PUBMED Abstract]
Cellular Classification of Adult Soft Tissue Sarcoma
Soft tissue sarcomas are classified histologically according to the soft tissue cell of origin. Additional studies, including electron microscopy, specialized immunohistochemistry, flow cytometry, cytogenetics, and tissue culture studies may allow identification of particular subtypes within the major histologic categories. For example, S100 antigen suggests neural sheath origin, cytokeratin suggests epithelioid or synovial cell origin, and factor VIII-related antigen suggests endothelial origin. Likewise, some subtypes of sarcomas have characteristic genetic markers, but these markers are not generally used in the routine clinical setting (e.g., translocation t(X;18)(p11;q11) in synovial sarcomas and translocation t(12;16)(q13;p11) in myxoid and round-cell sarcomas).[1-3]
The histologic grade reflects the metastatic potential of these tumors more accurately than the classic cellular classification listed below. Pathologists assign a grade based on the number of mitoses per high-powered field, the presence of necrosis, cellular and nuclear morphology, and the degree of cellularity; discordance among expert pathologists regarding tumor grade, and even histologic subtype, can be substantial.[4]
The World Health Organization lists the following cell types in its classification of soft tissue sarcomas:[5,6]
- Adipocytic tumors.
- Dedifferentiated liposarcoma.*
- Myxoid/round cell liposarcoma.
- Pleomorphic liposarcoma.
- Fibroblastic/myofibroblastic tumors.
- Fibrosarcoma.**
- Myxofibrosarcoma, low grade.
- Low-grade fibromyxoid sarcoma.
- Sclerosing epithelioid fibrosarcoma.
- So-called fibrohistiocytic tumors.
- Undifferentiated pleomorphic sarcoma/malignant fibrous histiocytoma (including pleomorphic, giant cell, myxoid/high-grade myxofibrosarcoma, and inflammatory forms).
- Smooth muscle tumors.
- Leiomyosarcoma.
- Skeletal muscle tumors.
- Rhabdomyosarcoma (embryonal, alveolar, and pleomorphic forms).
- Vascular tumors.
- Epithelioid hemangioendothelioma.
- Angiosarcoma, deep.***
- Tumors of peripheral nerves.
- Malignant peripheral nerve sheath tumor.
- Chondro-osseous tumors.
- Extraskeletal chondrosarcoma (mesenchymal and other variants).
- Extraskeletal osteosarcoma.
- Tumors of uncertain differentiation.
- Synovial sarcoma.
- Epithelioid sarcoma.
- Alveolar soft part sarcoma.
- Clear cell sarcoma of soft tissue.
- Extraskeletal myxoid chondrosarcoma.
- Primitive neuroectodermal tumor/extraskeletal Ewing tumor.
- Desmoplastic small round cell tumor.
- Extrarenal rhabdoid tumor.
- Undifferentiated sarcoma; sarcoma, not otherwise specified.
[Note: *It is recognized that dedifferentiated liposarcoma primarily arises in the context of deep atypical lipomatous tumor/well-differentiated liposarcoma, a sarcoma of intermediate malignancy because of the lack of metastatic capacity. **The category of fibrosarcoma can be inclusive of fibrosarcomatous differentiation in dermatofibrosarcoma protuberans. ***Cutaneous angiosarcoma may be difficult to stage using the American Joint Committee on Cancer system. (Refer to the PDQ summary on Gastrointestinal Stromal Tumors for more information.)]
References
- Singer S, Nielsen T, Antonescu CR: Molecular biology of soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1522-32.
- Singer S, Maki RG, O'Sullivan B: Soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1533-77.
- Malawer MM, Helman LJ, O'Sullivan B: Sarcomas of bone. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1578-1609.
- Alvegård TA, Berg NO: Histopathology peer review of high-grade soft tissue sarcoma: the Scandinavian Sarcoma Group experience. J Clin Oncol 7 (12): 1845-51, 1989. [PUBMED Abstract]
- Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-6.
- Brodowicz T, Schwameis E, Widder J, et al.: Intensified Adjuvant IFADIC Chemotherapy for Adult Soft Tissue Sarcoma: A Prospective Randomized Feasibility Trial. Sarcoma 4 (4): 151-60, 2000. [PUBMED Abstract]
Stage Information for Adult Soft Tissue Sarcoma
Note: The American Joint Committee on Cancer (AJCC) has published the 8th edition of the AJCC Cancer Staging Manual, which includes revisions to the staging for this disease. Implementation of the 8th edition began in January 2018. The PDQ Adult Treatment Editorial Board, which maintains this summary, is reviewing the revised staging and will make appropriate changes as needed.
Staging has an important role in determining the most effective treatment for soft tissue sarcoma. Clinical staging involves magnetic resonance imaging (MRI) or computed tomography (CT) of the primary tumor area and a chest CT to look for metastasis to the lung (the most common site of distant spread). An abdominal CT scan is done in the case of retroperitoneal sarcomas because the liver may be the site of initial clinical metastasis for these tumors.
The stage is determined by the size of the tumor, the histologic grade, and whether there is spread to lymph nodes or distant sites. Intracompartmental or extracompartmental extension of extremity sarcomas is also important for surgical decision making. For complete staging, a thorough review of all biopsy specimens (including those from the primary tumor, lymph nodes, or other suspicious lesions) is essential. CT scan of the chest is recommended for sarcomas larger than 5 cm (T2) or with moderate to poor differentiation (grades 2–4). Nodal involvement is rare, occurring in fewer than 3% of patients with sarcoma.[1]
Lymph node involvement in soft tissue sarcomas of adulthood is rare but is somewhat more frequent in some subtypes (e.g., rhabdomyosarcoma, vascular sarcomas, clear cell sarcomas, and epithelioid sarcomas) when they are high grade.[2] Because treatment decisions are predicated on pathology staging, patients should be staged before, and again after, any neoadjuvant therapy. The assessment of tumor grade can be affected in either direction, but more frequently decreased because of differential cellular loss related to the neoadjuvant chemotherapy or radiation.[3] Grade, which is based on cellular differentiation, mitotic rate, and extent of necrosis, should be recorded for all soft tissue sarcomas. A three-grade system (G1–G3) is preferred. (See below.)
The AJCC has designated staging by the four criteria of tumor size, nodal status, metastasis, and grade (TNMG).[3] The characteristic molecular markers of some sarcomas are not formally incorporated in the staging system pending further evaluation of their impact on prognosis. Recurrent sarcomas are restaged using the same system as for primary tumors with the specification that the tumor is recurrent.
Definitions of TNM and Grade
| aReprinted with permission from AJCC: Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-8. | |
| bSuperficial tumor is located exclusively above the superficial fascia without invasion of the fascia; deep tumor is located either exclusively beneath the superficial fascia, superficial to the fascia with invasion of or through the fascia, or both superficial yet beneath the fascia. | |
| TX | Primary tumor cannot be assessed. |
| T0 | No evidence of primary tumor. |
| T1 | Tumor ≤5 cm in greatest dimension. (Size should be regarded as a continuous variable, and the measurement should be provided.) |
| T1a | Superficial tumor.b |
| T1b | Deep tumor.b |
| T2 | Tumor >5 cm in greatest dimension.b |
| T2a | Superficial tumor.b |
| T2b | Deep tumor. |
| aReprinted with permission from AJCC: Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-8. | |
| bPresence of positive nodes (N1) in M0 tumors is considered Stage III. | |
| NX | Regional lymph nodes cannot be assessed. |
| N0 | No regional lymph node metastasis. |
| N1b | Regional lymph node metastasis. |
| aReprinted with permission from AJCC: Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-8. | |
| M0 | No distant metastasis. |
| M1 | Distant metastasis. |
| aReprinted with permission from AJCC: Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-8. | ||||
| Stage IA | T1a | N0 | M0 | G1, GX |
| T1b | N0 | M0 | G1, GX | |
| Stage IB | T2a | N0 | M0 | G1, GX |
| T2b | N0 | M0 | G1, GX | |
| Stage IIA | T1a | N0 | M0 | G2, G3 |
| T1b | N0 | M0 | G2, G3 | |
| Stage IIB | T2a | N0 | M0 | G2 |
| T2b | N0 | M0 | G2 | |
| Stage III | T2a, T2b | N0 | M0 | G3 |
| Any T | N1 | M0 | Any G | |
| Stage IV | Any T | Any N | M1 | Any G |
Neurovascular and bone invasion are indicators of poor prognosis, but they are not incorporated into the formal staging system.
References
- Fong Y, Coit DG, Woodruff JM, et al.: Lymph node metastasis from soft tissue sarcoma in adults. Analysis of data from a prospective database of 1772 sarcoma patients. Ann Surg 217 (1): 72-7, 1993. [PUBMED Abstract]
- Mazeron JJ, Suit HD: Lymph nodes as sites of metastases from sarcomas of soft tissue. Cancer 60 (8): 1800-8, 1987. [PUBMED Abstract]
- Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-6.
Treatment Option Overview
Multimodality Approach
In most cases, a combined modality approach of preoperative radiation therapy (preRT) or postoperative radiation therapy (PORT) is used, rather than the radical surgical procedures, such as amputation, that were used in the past. It may even be possible to use surgery without PORT in selected cases. For example, a case series was reported from a specialized sarcoma treatment referral center in which 74 selected patients with primary extremity and trunk tumors 5 cm or smaller were found to have no histologic involvement of the surgical margins. The patients were observed without radiation therapy, and the estimated local recurrence rate after 10 years was 11%.[1][Level of evidence: 3iiiDiv] The role of chemotherapy is not as well defined as is the role for radiation therapy. Because of the evolving nature of the treatment options for this disease, patients should be considered when available. Information about ongoing clinical trials is available from the NCI website.
Role of Surgery
Surgical resection is the mainstay of therapy for soft tissue sarcomas. When feasible, wide-margin function–sparing surgical excision is the cornerstone of effective treatment for extremity tumors. This may be facilitated by soft tissue reconstructive surgery, which generally permits wider margins than those obtained when the surgical plan involves direct closure of the excision site.[2] Cutting into the tumor mass or shelling out the gross tumor along the plane of the pseudocapsule of compressed tumor cells and reactive tissue that often surrounds soft tissue sarcomas are associated with an elevated risk of local recurrence. Even high-grade, soft tissue sarcomas of the extremities can usually be effectively treated while preserving the limb with combined-modality treatment consisting of preRT or PORT to reduce local recurrence. (Refer to the Role of Radiation Therapy section of this summary for more information.)
Only one small, single-institution, randomized trial has directly compared amputation to limb-sparing surgery for soft tissue sarcomas of the extremities.[3] In a 2:1 randomization ratio, 27 patients with high-grade extremity sarcomas were assigned to a wide excision plus PORT (45 Gy–50 Gy to the wide local excision area, and a total of 60 Gy–70 Gy to the tumor bed over 6–7 weeks), and 16 were assigned to amputation at or above the joint proximal to the tumor. Both groups received adjuvant chemotherapy (i.e., doxorubicin, cyclophosphamide, and high-dose methotrexate). At 63 months, with a median follow-up of 56 months, there were four local recurrences in the 27 patients who underwent limb-sparing surgery and no recurrences in the 16 patients who underwent amputation P2 = .12. Overall survival (OS) rates were not statistically significantly different (actuarial 5-year survival rate, 83% vs. 88%, P2 = .99).[3][Level of evidence: 1iiA]
Local control of high-grade soft tissue sarcomas of the trunk and the head and neck can be achieved with surgery in combination with radiation therapy.[4] It may be possible to use surgery without PORT in selected cases. For example, a case series was reported from a specialized sarcoma treatment referral center in which 74 selected patients with primary extremity and trunk tumors 5 cm or smaller were found to have no histologic involvement of the surgical margins.[1] They were observed without radiation therapy, and the estimated local recurrence rate after 10 years was 11%.[1][Level of evidence: 3iiiDiv] The role of chemotherapy is not as well defined as is the role of radiation therapy. Because of the evolving nature of the treatment options for this disease, patients should be offered the option of clinical trials when available.
Effective treatment of retroperitoneal sarcomas requires removal of all gross disease while sparing adjacent viscera not invaded by tumor. The prognosis for patients with high-grade retroperitoneal sarcomas is less favorable than for patients with tumors at other sites, partly because of the difficulty in completely resecting these tumors and the dose-limiting toxicity of high-dose radiation therapy on visceral organs.[5-8]
In the setting of distant metastasis, surgery may be associated with long-term, disease-free survival in patients with pulmonary metastasis and optimal underlying disease biology (i.e., patients with a limited number of metastases and slow nodule growth) who have undergone or are undergoing complete resection of the primary tumor.[9-11] It is not clear to what degree the favorable outcomes are attributable to the efficacy of surgery or the careful selection of patients based on factors that are associated with less-virulent disease.
Role of Radiation Therapy
Radiation plays an important role in limb-sparing therapy. Pre- and postoperative external-beam radiation therapies (EBRT), as well as brachytherapy, have been shown to decrease the risk of local recurrence. They have not been shown to increase OS but are used to avoid amputation for all but the most locally advanced tumors or for limbs seriously compromised by vascular disease, where acceptable functional preservation is not possible. In the case of EBRT, irradiation of the entire limb circumference is avoided to preserve vascular and nerve structures that are critical to function and preservation of the limb.
PORT
PORT has been tested in a single-institution, randomized trial of 141 patients with extremity sarcomas who were treated with limb-sparing surgery. Patients with high-grade tumors (n = 91) also received adjuvant chemotherapy (i.e., five 28-day cycles of doxorubicin and cyclophosphamide). All patients were randomly assigned to receive radiation (45 Gy to a wide field, plus a tumor-bed boost of 18 Gy over 6–7 weeks), concurrent with chemotherapy in the case of high-grade tumors versus no radiation.[12] At up to 12 years of follow-up, there was one local recurrence in the 70 patients randomly assigned to receive radiation versus 17 recurrences in the 71 control patients (P = .0001), with similar reduction in risk of local recurrence for both high- and low-grade tumors. However, there was no difference in OS between the radiation and control groups.[12][Level of evidence: 1iiDiii] Global quality of life was similar in the two groups, but the radiation therapy group had substantially worse functional deficits resulting from reduced strength and joint motion as well as increased edema.
To limit acute toxicity with preRT, smaller fields and lower doses are generally given than is the case with PORT. PreRT has been directly compared with PORT for extremity soft tissue sarcomas in a multicenter randomized trial.[13-15] Designed to include 266 patients, the trial was stopped early after 190 patients had been accrued because of an increase in wound complications in the preRT group. The scheduled radiation in the preRT group was a wide field of 50 Gy in 2-Gy fractions (first phase of the trial) with an additional 16 Gy to 20 Gy to the tumor bed and a 2-cm margin (second phase of the trial) only if tumor cells were found at the surgical margins.
Patients in the PORT group were scheduled to receive radiation during both phases of the trial. The wound-complication rates were 35% versus 17% in the preRT and PORT groups, respectively (P = .01). In addition, limb function at 6 weeks after surgery was worse in the preRT group (P = .01).[13] At 5 years, the two groups had similar local control rates (93% vs. 92%) and OS (73% vs. 67%, P = .48).[14] Of the 129 patients evaluated for limb function at 21 to 27 months after surgery (n = 73 for preRT and n = 56 for PORT), limb function was similar in both groups, but there was a statistical trend for less fibrosis in the preRT group (P = .07).[15]
Brachytherapy
Brachytherapy has also been investigated as an adjuvant therapy for soft tissue sarcomas. Although it has possible advantages of convenience and less radiation to normal surrounding tissue relative to EBRT, the two treatment strategies have not been directly compared in terms of efficacy or morbidity. However, adjuvant brachytherapy has been compared with surgery without radiation. The time interval between preRT and surgical excision in extremity soft tissue sarcoma had minimal influence on the development of wound complications. Four- or 5-week intervals showed equivalent complication rates between patients who did or did not develop wound complications, suggesting an optimal interval to reduce potential complications.[16]
In a single-institution trial, 164 patients with sarcomas of the extremity or superficial trunk were randomly assigned during surgery, if all gross tumor could be excised, to receive an iridium Ir 192 implant (delivering 42 Gy–45 Gy over 4–6 days; 78 patients) or to a control arm of no radiation (86 patients).[17,18] Some of the patients with high-grade tumors received adjuvant doxorubicin-based chemotherapy if they were thought to be at a high risk for metastasis (34 patients in each study arm). With a median follow-up of 76 months, the 5-year actuarial local recurrence rates were 18% and 31% in the brachytherapy and control arms, respectively (P = .04). This difference was limited to patients with high-grade tumors. There was no discernible difference in sarcoma-specific survival rates between the brachytherapy and control arms (84% and 81%, respectively; P = .65), and there was no difference in the high tumor-grade group.[17][Level of evidence: 1iiDiii] The rates of clinically important wound complications (e.g., need for operative revision or repeated seroma drainage, wound separation, large hematomas, or purulent infection) were 24% and 14% in the radiation and control arms, respectively (P = .13); wound reoperation rates were 10% and 0%, respectively (P = .006).[18]
Intensity-modulated radiation therapy
Intensity-modulated radiation therapy (IMRT) has been used to deliver preRT or PORT to patients with extremity soft tissue sarcomas in an effort to spare the femur, joints, and selected other normal tissues from the full prescription dose and to maintain local control while potentially reducing radiation therapy-related morbidity. Initial single-institution reports suggest that high rates of local control with some reduction in morbidity are possible with this technique.[19,20] Retrospective comparison of IMRT compared with 3-dimensional, conformal radiation therapy demonstrates that local recurrence for primary soft tissue sarcomas of the extremity was worse in the non-IMRT group.[21][Level of evidence: 3iiiDiv]
Surgery and radiation therapy
In some tumors of the extremities or trunk, surgery alone can be performed without the use of radiation. Evidence for this approach is limited to single-institution, relatively small, case series [1,22,23] or analysis of outcomes in the National Cancer Institute's Surveillance, Epidemiology, and End Results (SEER) tumor registry.[24] However, these comparisons suffer from low statistical power and differential evaluability rates that could have introduced bias.[1] Patient selection factors may vary among surgeons. In general, this approach is considered in patients with low-grade tumors of the extremity or superficial trunk that are 5 cm or smaller in diameter (T1) and have microscopically negative surgical margins; long-term local tumor control is about 90% in such patients.[25]
A patterns-of-care study using SEER data was queried to identify patients undergoing surgery for truncal and extremity soft tissue sarcomas from 2004 to 2009.[26] Of 5,075 patients, 50% received radiation therapy. Radiation was considered to be underused in a significant portion of patients undergoing treatment for soft tissue sarcoma in the United States. Although routine radiation therapy is not recommended for stage I patients, 25% of them still underwent radiation. Even though routine radiation therapy is recommended for patients with stage II and III tumors, only 60% of them underwent radiation. On multivariate analysis, predictors of radiation therapy included age younger than 50 years (odds ratio [OR], 1.57; 95% confidence interval [CI], 1.28–1.91), malignant fibrous histiocytoma histology (OR, 1.47; 95% CI, 1.3–1.92), T2 classification (OR, 1.88; 95% CI, 1.60–2.20), and G3 (OR, 6.27; 95% CI, 5.10–7.72). Patients with stage III soft tissue sarcoma who received radiation therapy showed improved disease-specific survival at 5 years compared with those who did not (68% vs. 46%, P < .001).[26][Level of evidence: 3iDii]
On occasion, surgical excision cannot be performed in the initial management of soft tissue sarcomas because the morbidity would be unacceptable or nearby critical organs make complete resection impossible. In such circumstances, radiation has been used as the primary therapy.[27] However, this must be considered a treatment of last resort. Experience is limited to retrospective case series from single centers.[27][Level of evidence: 3iiiDiv]
Role of Adjuvant or Neoadjuvant Chemotherapy for Clinically Localized Tumors
The role of adjuvant chemotherapy is not completely clear. The investigation of its use falls into two categories or generations—pre- and post-ifosfamide regimens. In discussions with a patient, any potential benefits should be considered in the context of the short- and long-term toxicities of the chemotherapy.
First-generation trials (preifosfamide)
Several prospective, randomized trials were unable to determine conclusively whether doxorubicin-based adjuvant chemotherapy benefits adults with resectable soft tissue sarcomas. The majority of these studies accrued small numbers of patients and did not demonstrate a metastasis-free survival or an OS benefit for adjuvant chemotherapy.[4] A small study of adjuvant chemotherapy showed a positive effect on both disease-free survival (DFS) and OS in patients treated with postoperative chemotherapy.[28] There was wide interstudy variability among the reported trials, including differences in therapeutic regimens, drug doses, sample size, tumor site, and histologic grade.
A quantitative meta-analysis of updated data from 1,568 individual patients in 14 trials of doxorubicin-based adjuvant therapy showed an absolute benefit from adjuvant therapy of 6% for a local relapse-free interval (95% CI, 1%–10%), 10% for a distant relapse-free interval (95% CI, 5%–15%), and 10% for recurrence-free survival (95% CI, 5%–15%). A statistically significant OS benefit at 10 years was not detected: absolute difference 4% (95% CI, -1%–+9%).[29,30][Level of evidence: 1iiDii] However, only a small proportion of patients in this meta-analysis were treated with ifosfamide, an agent with demonstrated activity against soft tissue sarcoma. In addition, a subset analysis suggested that patients with sarcomas of the extremities may have benefited from adjuvant chemotherapy (hazard ratio [HR] for death, 0.8, P = .029), but there was no clear evidence that patients with extremity sarcomas had outcomes that were statistically significantly different from the outcomes of patients with tumors at other sites (P = .58).[30]
Second-generation trials (postifosfamide)
Subsequent chemotherapy trials were performed using anthracycline and ifosfamide combinations in patients who primarily had extremity or truncal soft tissue sarcomas. The data are conflicting, and the issue is still not settled. In a small feasibility study, 59 patients with high-risk, soft tissue sarcomas, 58 of whom had an extremity or the trunk as the primary site, underwent primary resection plus PORT and were randomly assigned to observation versus a dose-dense regimen of six 14-day courses of ifosfamide, dacarbazine (DTIC), and doxorubicin (IFADIC regimen) with granulocyte colony-stimulating factor (G-CSF) bone marrow support and mesna uroprotection.[31] There were no statistically significant differences in OS or relapse-free survival (RFS), but the study was severely underpowered.
In a second trial performed by the Italian National Council for Research, high-risk patients were treated with local therapy (i.e., wide resection plus preRT or PORT, or amputation as clinically necessary) and were then randomly assigned to observation versus five 21-day cycles of 4-epidoxorubicin (epirubicin) plus ifosfamide (with mesna and G-CSF).[28,32] Based on power calculations, the planned study size was 190 patients, but the trial was stopped after 104 patients had been entered because an interim analysis revealed a statistically significant (P = .001) difference in DFS favoring the chemotherapy arm. By the time of the initial peer-reviewed report of the study, the DFS still favored the chemotherapy group (median DFS of 48 months vs. 16 months), but the P value had risen to .04.[28]
Although there was no difference in metastasis-free survival at the time of the report, there was an improvement in median OS (75 months vs. 46 months, P = .03). However, at the follow-up report (at a median of 89.6 months in a range of 56–119 months), OS differences were no longer statistically significant (58.5% vs. 43.1% [P = .07]). The DFS difference had also lost statistical significance (47.2% vs. 16.0% [P = .09]).[32] In summary, the trial was underpowered because it was stopped early, and the early promising results that led to stopping the trial diminished as the trial matured.
In a third, underpowered, single-center trial, 88 patients with high-risk, soft tissue sarcomas (64 of whom had extremity or truncal primary tumors) underwent surgery (with or without radiation) and were then randomly assigned to receive four 21-day cycles of chemotherapy (epirubicin [n = 26] or epirubicin plus ifosfamide [n = 19]) versus no adjuvant chemotherapy (n = 43).[33] The trial was closed prematurely because of a slow accrual rate. After a median follow-up of 94 months, the 5-year DFS in the chemotherapy and control arms was 69% versus 44%, respectively (P = .01); the 5-year OS rates were 72% versus 47% (P = .06). All of the benefit associated with chemotherapy appeared restricted to the 19 patients who received epirubicin plus ifosfamide.
In yet another underpowered trial, 137 patients with high-risk, soft tissue sarcomas (93% with extremity or truncal primary tumors) who met the eligibility criteria were randomly assigned to undergo surgical resection (with or without radiation) or to receive three preoperative 21-day cycles of doxorubicin plus ifosfamide.[34] This multicenter European Organization for Research and Treatment of Cancer trial (EORTC-62874) was closed because of slow accrual and results that were not promising enough to continue. With a median follow-up of 7.3 years, the 5-year DFS in the surgery alone and chemotherapy plus surgery arms was 52% and 56%, respectively (P = .35); and OS was 64% and 65%, respectively (P = .22).
These last four trials have been combined with the 14 first-generation trials in a trial-level meta-analysis.[35] Of the 18 randomized trials of patients with resectable soft tissue sarcomas, five trials used a combination of doxorubicin (50–90 mg/m2 per cycle) plus ifosfamide (1,500–5,000 mg/m2 per cycle). The remaining 13 trials used doxorubicin (50–70 mg/m2 per cycle) alone or with other drugs. The absolute risk reduction in local recurrence rates associated with any chemotherapy added to local therapy was 4 percentage points (95% CI, 0%–7%), and it was 5 percentage points (95% CI, 1%–12%) when ifosfamide was combined with doxorubicin. The absolute reduction in overall mortality was 6 percentage points with any chemotherapy (95% CI, 2%–11%; [i.e., a reduction from 46%–40%]), 11 percentage points for doxorubicin plus ifosfamide (95% CI, 3%–19%; [i.e., a reduction from 41%–30%]), and 5 percentage points for doxorubicin without ifosfamide.[35][Level of evidence: 1iiA]
An additional multicenter randomized trial (EORTC-62931 [NCT00002641]), the largest trial reported to date using adjuvant doxorubicin (75 mg/m2) plus ifosfamide (5,000 mg/m2), was subsequently published in abstract form and was not included in the above meta-analysis.[36] The results differed from those reported in the meta-analysis.[35] After local therapy, 351 patients were randomly assigned to five 21-day cycles of adjuvant therapy versus observation. The trial was stopped for futility because the 5-year RFS was 52% in both arms. OS was 64% in the chemotherapy arm versus 69% in the observation arm. In a subsequent abstract, the EORTC investigators reported a combined analysis of this trial and their previous trial (EORTC-62771) [37] of adjuvant cyclophosphamide plus doxorubicin plus DTIC (CYVADIC), representing the two largest trials of adjuvant therapy for adult soft tissue sarcoma in the literature.[38] The combined analysis showed no improvement in either RFS or OS associated with adjuvant chemotherapy.[38][Level of evidence: 1iiA]
In summary, the impact of adjuvant chemotherapy on survival is not clear but is likely to be small in absolute magnitude. Therefore, in discussions with a patient, any potential benefits should be considered in the context of the short- and long-term toxicities of the chemotherapy.
Role of regional hyperthermia
The use of regional hyperthermia to enhance the local effects of systemic chemotherapy in the neoadjuvant and adjuvant setting is under investigation. In a multicenter phase III trial, 341 patients with high-risk (tumor ≥5 cm, grade 2–3, and deep to fascia), soft tissue sarcomas (149 extremity tumors and 192 nonextremity tumors) were randomly allocated to receive four 21-day cycles of chemotherapy (etoposide 125 mg/m2 on days 1 and 4; ifosfamide 1,500 mg/m2 on days 1–4; doxorubicin 50 mg/m2 on day 1) with or without regional hyperthermia both before and after local therapy.[39] Approximately 11% of the patients were being treated for recurrent tumors. The regional hyperthermia was designed to produce tumor temperatures of 42°C for 60 minutes and was given on days 1 and 4 of each chemotherapy cycle. After the first four cycles of chemotherapy, definitive surgical excision of the tumor was performed, if possible, followed by radiation therapy, if indicated (i.e., a 52.7 Gy median dose delivered), and then the last four cycles of chemotherapy plus or minus hyperthermia. Three of the nine treatment centers with particular expertise in hyperthermia treated 91% of the patients in the trial.
The median duration of follow-up was 34 months. Local progression occurred in 56 patients in the hyperthermia group and 76 patients in the control group. The relative HR for local progression or death was 0.58 (95% CI, 0.41–0.84), with an absolute difference at 2 years of 15% (76% vs. 61%; 95% CI of the difference 6–26). The decreased risk of local progression or death was seen in both extremity and nonextremity tumors. However, hyperthermia had no effect on distant failure rates nor was there a statistically significant effect on OS (HR, .88, 95% CI, 0.64–1.21; P = .43).[39][Level of evidence: 1iiDiii] There was a higher rate of grade 3 to 4 leucopenia in the hyperthermia group: 77.6% versus 63.5% (P = .005). Since a large proportion of the patients were treated at centers with special expertise, there is no certainty that the finding can be generalized to apply to other settings.
Role of isolated limb perfusion
Isolated limb perfusion is under investigation as a means to deliver high doses of chemotherapy and permit limb salvage in unresectable primary or recurrent extremity soft tissue sarcomas that would otherwise require amputation, in the opinion of the surgeon.[40,41] Common drugs used in the procedure are TNF-alpha, melphalan, and interferon-gamma. Experience is limited to case series with response rates and reported avoidance of amputation as the outcome.[40,41][Level of evidence: 3iiiDiv] The technique requires specialized expertise to avoid severe local and systemic toxicity including systemic effects of TNF-alpha. The technique has not been directly compared with standard approaches using combined systemic and local therapy.
Role of chemotherapy for advanced disease
Doxorubicin is a mainstay of systemic therapy in the management of locally advanced and metastatic soft tissue sarcoma. Pegylated liposomal encapsulated doxorubicin is a formulation of doxorubicin designed to prolong the half-life of circulating doxorubicin and slow the release of active drugs.[42] The changed pharmacokinetics result in less myelosuppression and possibly less cardiotoxic effects, but there is a substantial incidence of hypersensitivity-like reactions and hand-foot syndrome. Its clinical activity relative to unencapsulated doxorubicin is not clear.[42][Level of evidence: 3iiiDiv] Other drugs that are thought to have clinical activity as single agents are ifosfamide, epirubicin, gemcitabine, and paclitaxel.[43-46][Level of Evidence: 3iiiDiv] Their clinical activity relative to single-agent doxorubicin is not clear, and they are not known to have superior activity.
There is controversy about the clinical benefit of adding other drugs to doxorubicin as a single agent. A systematic evidence review and meta-analysis conducted by the Cochrane Collaboration summarized the eight randomized trials reported from 1976 to 1995.[47] No additional randomized trials had been reported or were known to be in progress between 1995 and the 2002 literature search. Single-agent doxorubicin had been compared with a variety of doxorubicin-containing combinations that included vincristine, vindesine, cyclophosphamide, streptozotocin, mitomycin-C, cisplatin, and/or ifosfamide. Combination regimens consistently caused more nausea and hematologic toxicity. However, the better response rates associated with combination therapy were marginal and depended on the statistical model used (fixed effects model ORresponse = 1.29; 95% CI, 1.03–1.60, P = .03; random effects model ORresp = 1.26; 95% CI, 0.96–1.67, P = .10) There was no statistically significant difference in the 1- (ORmortality = 0.87; 95% CI, 0.73–1.05, P = .14) or 2-year mortality rates (ORmortality = 0.84; 95% CI, 0.67–1.06, P = .13).
These results were very similar even when the analyses were restricted to the four trials that used DTIC and/or ifosfamide as part of the combination regimen with doxorubicin agents that were postulated to have greater activity than the others tested. A subsequent meta-analysis of all three published randomized trials of chemotherapy regimens that contained ifosfamide versus those that did not came to similar conclusions: tumor response rates were better when the regimen included ifosfamide (RRresponse = 1.52; 95% CI, 1.11–2.08), but mortality at 1 year was not (RRmortality = 0.98; 95% CI, 0.85–1.13).[48][Level of evidence: 1iiDiv]. Therefore, response rate was a poor surrogate for OS. Quality-of-life outcomes were not reported in any of the above-mentioned randomized trials, but toxicity was worse when agents were added to doxorubicin.
References
- Pisters PW, Pollock RE, Lewis VO, et al.: Long-term results of prospective trial of surgery alone with selective use of radiation for patients with T1 extremity and trunk soft tissue sarcomas. Ann Surg 246 (4): 675-81; discussion 681-2, 2007. [PUBMED Abstract]
- Lohman RF, Nabawi AS, Reece GP, et al.: Soft tissue sarcoma of the upper extremity: a 5-year experience at two institutions emphasizing the role of soft tissue flap reconstruction. Cancer 94 (8): 2256-64, 2002. [PUBMED Abstract]
- Rosenberg SA, Tepper J, Glatstein E, et al.: The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg 196 (3): 305-15, 1982. [PUBMED Abstract]
- O'Byrne K, Steward WP: The role of adjuvant chemotherapy in the treatment of adult soft tissue sarcomas. Crit Rev Oncol Hematol 27 (3): 221-7, 1998. [PUBMED Abstract]
- Singer S, Nielsen T, Antonescu CR: Molecular biology of soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1522-32.
- Singer S, Maki RG, O'Sullivan B: Soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1533-77.
- Malawer MM, Helman LJ, O'Sullivan B: Sarcomas of bone. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1578-1609.
- Lewis JJ, Leung D, Woodruff JM, et al.: Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg 228 (3): 355-65, 1998. [PUBMED Abstract]
- van Geel AN, Pastorino U, Jauch KW, et al.: Surgical treatment of lung metastases: The European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group study of 255 patients. Cancer 77 (4): 675-82, 1996. [PUBMED Abstract]
- Casson AG, Putnam JB, Natarajan G, et al.: Five-year survival after pulmonary metastasectomy for adult soft tissue sarcoma. Cancer 69 (3): 662-8, 1992. [PUBMED Abstract]
- Putnam JB Jr, Roth JA: Surgical treatment for pulmonary metastases from sarcoma. Hematol Oncol Clin North Am 9 (4): 869-87, 1995. [PUBMED Abstract]
- Yang JC, Chang AE, Baker AR, et al.: Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 16 (1): 197-203, 1998. [PUBMED Abstract]
- O'Sullivan B, Davis AM, Turcotte R, et al.: Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 359 (9325): 2235-41, 2002. [PUBMED Abstract]
- O'Sullivan B, Davis A, Turcotte R, et al.: Five-year results of a randomized phase III trial of pre-operative vs post-operative radiotherapy in extremity soft tissue sarcoma. [Abstract] J Clin Oncol 22 (Suppl 14): A-9007, 819s, 2004.
- Davis AM, O'Sullivan B, Turcotte R, et al.: Late radiation morbidity following randomization to preoperative versus postoperative radiotherapy in extremity soft tissue sarcoma. Radiother Oncol 75 (1): 48-53, 2005. [PUBMED Abstract]
- Griffin AM, Dickie CI, Catton CN, et al.: The influence of time interval between preoperative radiation and surgical resection on the development of wound healing complications in extremity soft tissue sarcoma. Ann Surg Oncol 22 (9): 2824-30, 2015. [PUBMED Abstract]
- Pisters PW, Harrison LB, Leung DH, et al.: Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol 14 (3): 859-68, 1996. [PUBMED Abstract]
- Alektiar KM, Zelefsky MJ, Brennan MF: Morbidity of adjuvant brachytherapy in soft tissue sarcoma of the extremity and superficial trunk. Int J Radiat Oncol Biol Phys 47 (5): 1273-9, 2000. [PUBMED Abstract]
- Alektiar KM, Brennan MF, Healey JH, et al.: Impact of intensity-modulated radiation therapy on local control in primary soft-tissue sarcoma of the extremity. J Clin Oncol 26 (20): 3440-4, 2008. [PUBMED Abstract]
- Alektiar KM, Brennan MF, Singer S: Local control comparison of adjuvant brachytherapy to intensity-modulated radiotherapy in primary high-grade sarcoma of the extremity. Cancer 117 (14): 3229-34, 2011. [PUBMED Abstract]
- Folkert MR, Singer S, Brennan MF, et al.: Comparison of local recurrence with conventional and intensity-modulated radiation therapy for primary soft-tissue sarcomas of the extremity. J Clin Oncol 32 (29): 3236-41, 2014. [PUBMED Abstract]
- Fabrizio PL, Stafford SL, Pritchard DJ: Extremity soft-tissue sarcomas selectively treated with surgery alone. Int J Radiat Oncol Biol Phys 48 (1): 227-32, 2000. [PUBMED Abstract]
- Rydholm A, Gustafson P, Rööser B, et al.: Limb-sparing surgery without radiotherapy based on anatomic location of soft tissue sarcoma. J Clin Oncol 9 (10): 1757-65, 1991. [PUBMED Abstract]
- Al-Refaie WB, Habermann EB, Jensen EH, et al.: Surgery alone is adequate treatment for early stage soft tissue sarcoma of the extremity. Br J Surg 97 (5): 707-13, 2010. [PUBMED Abstract]
- Rydholm A: Surgery without radiotherapy in soft tissue sarcoma. Acta Orthop Scand Suppl 273: 117-9, 1997. [PUBMED Abstract]
- Bagaria SP, Ashman JB, Daugherty LC, et al.: Compliance with National Comprehensive Cancer Network guidelines in the use of radiation therapy for extremity and superficial trunk soft tissue sarcoma in the United States. J Surg Oncol 109 (7): 633-8, 2014. [PUBMED Abstract]
- Kepka L, DeLaney TF, Suit HD, et al.: Results of radiation therapy for unresected soft-tissue sarcomas. Int J Radiat Oncol Biol Phys 63 (3): 852-9, 2005. [PUBMED Abstract]
- Frustaci S, Gherlinzoni F, De Paoli A, et al.: Adjuvant chemotherapy for adult soft tissue sarcomas of the extremities and girdles: results of the Italian randomized cooperative trial. J Clin Oncol 19 (5): 1238-47, 2001. [PUBMED Abstract]
- Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet 350 (9092): 1647-54, 1997. [PUBMED Abstract]
- Sarcoma Meta-analysis Collaboration (SMAC): Adjuvant chemotherapy for localised resectable soft tissue sarcoma in adults. Cochrane Database Syst Rev (4): CD001419, 2000. [PUBMED Abstract]
- Brodowicz T, Schwameis E, Widder J, et al.: Intensified Adjuvant IFADIC Chemotherapy for Adult Soft Tissue Sarcoma: A Prospective Randomized Feasibility Trial. Sarcoma 4 (4): 151-60, 2000. [PUBMED Abstract]
- Frustaci S, De Paoli A, Bidoli E, et al.: Ifosfamide in the adjuvant therapy of soft tissue sarcomas. Oncology 65 (Suppl 2): 80-4, 2003. [PUBMED Abstract]
- Petrioli R, Coratti A, Correale P, et al.: Adjuvant epirubicin with or without Ifosfamide for adult soft-tissue sarcoma. Am J Clin Oncol 25 (5): 468-73, 2002. [PUBMED Abstract]
- Gortzak E, Azzarelli A, Buesa J, et al.: A randomised phase II study on neo-adjuvant chemotherapy for 'high-risk' adult soft-tissue sarcoma. Eur J Cancer 37 (9): 1096-103, 2001. [PUBMED Abstract]
- Pervaiz N, Colterjohn N, Farrokhyar F, et al.: A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 113 (3): 573-81, 2008. [PUBMED Abstract]
- Woll PJ, van Glabbeke M, Hohenberger P, et al.: Adjuvant chemotherapy (CT) with doxorubicin and ifosfamide in resected soft tissue sarcoma (STS): Interim analysis of a randomised phase III trial. [Abstract] J Clin Oncol 25 (Suppl 18): A-10008, 2007.
- Bramwell V, Rouesse J, Steward W, et al.: Adjuvant CYVADIC chemotherapy for adult soft tissue sarcoma--reduced local recurrence but no improvement in survival: a study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol 12 (6): 1137-49, 1994. [PUBMED Abstract]
- Le Cesne A, Van Glabbeke M, Woll PJ, et al.: The end of adjuvant chemotherapy (adCT) era with doxorubicin-based regimen in resected high-grade soft tissue sarcoma (STS): pooled analysis of the two STBSG-EORTC phase III clinical trials. [Abstract] J Clin Oncol 26 (Suppl 15): A-10525, 2008.
- Issels RD, Lindner LH, Verweij J, et al.: Neo-adjuvant chemotherapy alone or with regional hyperthermia for localised high-risk soft-tissue sarcoma: a randomised phase 3 multicentre study. Lancet Oncol 11 (6): 561-70, 2010. [PUBMED Abstract]
- Eggermont AM, de Wilt JH, ten Hagen TL: Current uses of isolated limb perfusion in the clinic and a model system for new strategies. Lancet Oncol 4 (7): 429-37, 2003. [PUBMED Abstract]
- Bonvalot S, Laplanche A, Lejeune F, et al.: Limb salvage with isolated perfusion for soft tissue sarcoma: could less TNF-alpha be better? Ann Oncol 16 (7): 1061-8, 2005. [PUBMED Abstract]
- Grenader T, Goldberg A, Hadas-Halperin I, et al.: Long-term response to pegylated liposomal doxorubicin in patients with metastatic soft tissue sarcomas. Anticancer Drugs 20 (1): 15-20, 2009. [PUBMED Abstract]
- Lorigan P, Verweij J, Papai Z, et al.: Phase III trial of two investigational schedules of ifosfamide compared with standard-dose doxorubicin in advanced or metastatic soft tissue sarcoma: a European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol 25 (21): 3144-50, 2007. [PUBMED Abstract]
- Nielsen OS, Dombernowsky P, Mouridsen H, et al.: High-dose epirubicin is not an alternative to standard-dose doxorubicin in the treatment of advanced soft tissue sarcomas. A study of the EORTC soft tissue and bone sarcoma group. Br J Cancer 78 (12): 1634-9, 1998. [PUBMED Abstract]
- Maki RG, Wathen JK, Patel SR, et al.: Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 25 (19): 2755-63, 2007. [PUBMED Abstract]
- Okuno S, Ryan LM, Edmonson JH, et al.: Phase II trial of gemcitabine in patients with advanced sarcomas (E1797): a trial of the Eastern Cooperative Oncology Group. Cancer 97 (8): 1969-73, 2003. [PUBMED Abstract]
- Bramwell VH, Anderson D, Charette ML, et al.: Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev (3): CD003293, 2003. [PUBMED Abstract]
- Verma S, Younus J, Stys-Norman D, et al.: Meta-analysis of ifosfamide-based combination chemotherapy in advanced soft tissue sarcoma. Cancer Treat Rev 34 (4): 339-47, 2008. [PUBMED Abstract]
Stage I Adult Soft Tissue Sarcoma
Refer to the Treatment Option Overview section of this summary for a more detailed discussion of the roles of surgery and radiation therapy.
Low-grade soft tissue sarcomas have little metastatic potential, but they have a propensity to recur locally. Accordingly, surgical excision with negative tissue margins of 1 cm to 2 cm or larger in all directions is the treatment of choice for patients with these early-stage sarcomas.[1-3] The Mohs surgical technique may be considered as an alternative to wide surgical excision for the very rare, small, well-differentiated primary sarcomas of the skin when cosmetic results are considered to be important, as margins can be assured with minimal normal tissue removal.[4]
Carefully executed high-dose radiation therapy using a shrinking-field technique may be beneficial for unresectable tumors or for resectable tumors in which a high likelihood of residual disease is thought to be present when margins are judged to be inadequate, and when wider resection would require either an amputation or the removal of a vital organ.[5] Because of the low metastatic potential of these tumors, chemotherapy is usually not given.[6,7]
Standard treatment options:
- Surgical excision of tumors 5 cm or smaller in diameter with negative tissue margins in all directions.[8-12]
- Surgical excision with preoperative radiation therapy (preRT) or postoperative radiation therapy (PORT). Radiation decreases the risk of local recurrence but has not been shown to increase overall survival.[13-16]
- If the tumor is unresectable, high-dose preRT may be used.[17]
- For tumors of the retroperitoneum, trunk, and head and neck, the following are options:
- Surgical resection with the option of PORT if negative margins cannot be obtained. Wide margins are unusual in these sites, and radiation therapy is usually advocated for trunk and head and neck primary sites.[18]
- PreRT followed by maximal surgical resection. Radiation therapy may be used in sarcomas of the trunk and head and neck to maximize local control because of the inability to obtain wide surgical margins.[19]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Singer S, Nielsen T, Antonescu CR: Molecular biology of soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1522-32.
- Singer S, Maki RG, O'Sullivan B: Soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1533-77.
- Malawer MM, Helman LJ, O'Sullivan B: Sarcomas of bone. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1578-1609.
- Fish FS: Soft tissue sarcomas in dermatology. Dermatol Surg 22 (3): 268-73, 1996. [PUBMED Abstract]
- Temple WJ, Temple CL, Arthur K, et al.: Prospective cohort study of neoadjuvant treatment in conservative surgery of soft tissue sarcomas. Ann Surg Oncol 4 (7): 586-90, 1997 Oct-Nov. [PUBMED Abstract]
- Sarcoma Meta-analysis Collaboration (SMAC): Adjuvant chemotherapy for localised resectable soft tissue sarcoma in adults. Cochrane Database Syst Rev (4): CD001419, 2000. [PUBMED Abstract]
- Pervaiz N, Colterjohn N, Farrokhyar F, et al.: A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 113 (3): 573-81, 2008. [PUBMED Abstract]
- Al-Refaie WB, Habermann EB, Jensen EH, et al.: Surgery alone is adequate treatment for early stage soft tissue sarcoma of the extremity. Br J Surg 97 (5): 707-13, 2010. [PUBMED Abstract]
- Pisters PW, Pollock RE, Lewis VO, et al.: Long-term results of prospective trial of surgery alone with selective use of radiation for patients with T1 extremity and trunk soft tissue sarcomas. Ann Surg 246 (4): 675-81; discussion 681-2, 2007. [PUBMED Abstract]
- Fabrizio PL, Stafford SL, Pritchard DJ: Extremity soft-tissue sarcomas selectively treated with surgery alone. Int J Radiat Oncol Biol Phys 48 (1): 227-32, 2000. [PUBMED Abstract]
- Rydholm A, Gustafson P, Rööser B, et al.: Limb-sparing surgery without radiotherapy based on anatomic location of soft tissue sarcoma. J Clin Oncol 9 (10): 1757-65, 1991. [PUBMED Abstract]
- Rydholm A: Surgery without radiotherapy in soft tissue sarcoma. Acta Orthop Scand Suppl 273: 117-9, 1997. [PUBMED Abstract]
- Yang JC, Chang AE, Baker AR, et al.: Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 16 (1): 197-203, 1998. [PUBMED Abstract]
- O'Sullivan B, Davis AM, Turcotte R, et al.: Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 359 (9325): 2235-41, 2002. [PUBMED Abstract]
- O'Sullivan B, Davis A, Turcotte R, et al.: Five-year results of a randomized phase III trial of pre-operative vs post-operative radiotherapy in extremity soft tissue sarcoma. [Abstract] J Clin Oncol 22 (Suppl 14): A-9007, 819s, 2004.
- Davis AM, O'Sullivan B, Turcotte R, et al.: Late radiation morbidity following randomization to preoperative versus postoperative radiotherapy in extremity soft tissue sarcoma. Radiother Oncol 75 (1): 48-53, 2005. [PUBMED Abstract]
- Kepka L, DeLaney TF, Suit HD, et al.: Results of radiation therapy for unresected soft-tissue sarcomas. Int J Radiat Oncol Biol Phys 63 (3): 852-9, 2005. [PUBMED Abstract]
- Brennan MF, Singer S, Maki RG: Sarcomas of the soft tissue and bone. In: DeVita VT Jr, Hellman S, Rosenberg SA, eds.: Cancer: Principles and Practice of Oncology. Vols. 1 & 2. 8th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2008, pp 1741-1833.
- Baldini EH, Wang D, Haas RL, et al.: Treatment Guidelines for Preoperative Radiation Therapy for Retroperitoneal Sarcoma: Preliminary Consensus of an International Expert Panel. Int J Radiat Oncol Biol Phys 92 (3): 602-12, 2015. [PUBMED Abstract]
Stage II and Node-Negative Stage III Adult Soft Tissue Sarcoma
Refer to the Treatment Option Overview section of this summary for a more detailed discussion of the roles of surgery, radiation therapy, and chemotherapy.
High-grade localized soft tissue sarcomas have an increased potential for local recurrence and metastasis. For sarcomas of the extremities, local control comparable to that obtained with amputation may be achieved with limb-sparing surgery that involves wide local excision in combination with preoperative radiation therapy (preRT) or postoperative radiation therapy (PORT).
Complete surgical resection is often difficult for sarcomas of the retroperitoneum because of their large size before detection and anatomical location.[1,2] As opposed to soft tissue sarcomas of the extremities, local recurrence is the most common cause of death in patients with retroperitoneal soft tissue sarcomas. Complete surgical resection (i.e., removal of the entire gross tumor) is the most important factor in preventing local recurrence and, in many instances, requires resection of adjacent viscera. For retroperitoneal sarcomas, retrospective comparison of surgery alone versus preRT review suggests that preRT is associated with improved local recurrence-free survival, but not disease-free survival.[3]
Standard treatment options:
- Surgical excision with preRT or PORT. Radiation decreases the risk of local recurrence but has not been shown to increase overall survival.[4-8]
- Surgical excision with negative tissue margins in all directions. This approach is generally restricted to low-grade tumors ( ≤5 cm in diameter) of the extremities or superficial trunk with microscopically negative surgical tumor margins.[9-13]
- If the tumor is unresectable, high-dose radiation therapy may be used, but poor local control is likely to result.[14]
- In some situations, radiation therapy and/or chemotherapy may be used before surgery in an attempt to convert a marginally resectable tumor to one that can be adequately resected with limb preservation; this treatment may be followed by PORT.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Heslin MJ, Lewis JJ, Nadler E, et al.: Prognostic factors associated with long-term survival for retroperitoneal sarcoma: implications for management. J Clin Oncol 15 (8): 2832-9, 1997. [PUBMED Abstract]
- Jaques DP, Coit DG, Hajdu SI, et al.: Management of primary and recurrent soft-tissue sarcoma of the retroperitoneum. Ann Surg 212 (1): 51-9, 1990. [PUBMED Abstract]
- Kelly KJ, Yoon SS, Kuk D, et al.: Comparison of Perioperative Radiation Therapy and Surgery Versus Surgery Alone in 204 Patients With Primary Retroperitoneal Sarcoma: A Retrospective 2-Institution Study. Ann Surg 262 (1): 156-62, 2015. [PUBMED Abstract]
- Yang JC, Chang AE, Baker AR, et al.: Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 16 (1): 197-203, 1998. [PUBMED Abstract]
- Rosenberg SA, Tepper J, Glatstein E, et al.: The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg 196 (3): 305-15, 1982. [PUBMED Abstract]
- O'Sullivan B, Davis AM, Turcotte R, et al.: Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 359 (9325): 2235-41, 2002. [PUBMED Abstract]
- O'Sullivan B, Davis A, Turcotte R, et al.: Five-year results of a randomized phase III trial of pre-operative vs post-operative radiotherapy in extremity soft tissue sarcoma. [Abstract] J Clin Oncol 22 (Suppl 14): A-9007, 819s, 2004.
- Davis AM, O'Sullivan B, Turcotte R, et al.: Late radiation morbidity following randomization to preoperative versus postoperative radiotherapy in extremity soft tissue sarcoma. Radiother Oncol 75 (1): 48-53, 2005. [PUBMED Abstract]
- Al-Refaie WB, Habermann EB, Jensen EH, et al.: Surgery alone is adequate treatment for early stage soft tissue sarcoma of the extremity. Br J Surg 97 (5): 707-13, 2010. [PUBMED Abstract]
- Pisters PW, Pollock RE, Lewis VO, et al.: Long-term results of prospective trial of surgery alone with selective use of radiation for patients with T1 extremity and trunk soft tissue sarcomas. Ann Surg 246 (4): 675-81; discussion 681-2, 2007. [PUBMED Abstract]
- Fabrizio PL, Stafford SL, Pritchard DJ: Extremity soft-tissue sarcomas selectively treated with surgery alone. Int J Radiat Oncol Biol Phys 48 (1): 227-32, 2000. [PUBMED Abstract]
- Rydholm A, Gustafson P, Rööser B, et al.: Limb-sparing surgery without radiotherapy based on anatomic location of soft tissue sarcoma. J Clin Oncol 9 (10): 1757-65, 1991. [PUBMED Abstract]
- Rydholm A: Surgery without radiotherapy in soft tissue sarcoma. Acta Orthop Scand Suppl 273: 117-9, 1997. [PUBMED Abstract]
- Kepka L, DeLaney TF, Suit HD, et al.: Results of radiation therapy for unresected soft-tissue sarcomas. Int J Radiat Oncol Biol Phys 63 (3): 852-9, 2005. [PUBMED Abstract]
Advanced Stage III (N1) Adult Soft Tissue Sarcoma
Refer to the Treatment Option Overview section of this summary for a more detailed discussion of the roles of surgery, radiation therapy, and chemotherapy.
Regional lymph node involvement by soft tissue sarcomas of adulthood is very infrequent. However, sarcoma types that more commonly spread to lymph nodes include high-grade rhabdomyosarcoma, vascular sarcomas, and epithelioid sarcomas.[1]
Standard treatment options:
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Mazeron JJ, Suit HD: Lymph nodes as sites of metastases from sarcomas of soft tissue. Cancer 60 (8): 1800-8, 1987. [PUBMED Abstract]
- Watson DI, Coventry BJ, Langlois SL, et al.: Soft-tissue sarcoma of the extremity. Experience with limb-sparing surgery. Med J Aust 160 (7): 412-6, 1994. [PUBMED Abstract]
- Cormier JN, Huang X, Xing Y, et al.: Cohort analysis of patients with localized, high-risk, extremity soft tissue sarcoma treated at two cancer centers: chemotherapy-associated outcomes. J Clin Oncol 22 (22): 4567-74, 2004. [PUBMED Abstract]
- O'Byrne K, Steward WP: The role of adjuvant chemotherapy in the treatment of adult soft tissue sarcomas. Crit Rev Oncol Hematol 27 (3): 221-7, 1998. [PUBMED Abstract]
- Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet 350 (9092): 1647-54, 1997. [PUBMED Abstract]
Stage IV Adult Soft Tissue Sarcoma
Refer to the Treatment Option Overview section of this summary for a more detailed discussion of the roles of surgery, radiation therapy, and chemotherapy.
In the setting of lung metastasis, resection of metastatic tumors may be associated with long-term disease-free survival in patients selected for optimal underlying disease biology (i.e., patients with a limited number of metastases and slow tumor growth).[1-3] It is not clear to what degree the favorable outcomes are attributable to the efficacy of surgery or to careful selection of patients based upon factors that are associated with less-virulent disease.[1-3] The value of resection of hepatic metastases is unclear.
As noted in the Treatment Option Overview section above, doxorubicin is the standard systemic therapy in the management of metastatic sarcomas.[4,5] Other drugs that may have clinical activity as single agents are ifosfamide, epirubicin, gemcitabine, and paclitaxel.[6-9] Their clinical activity relative to single-agent doxorubicin is not clear, and they are not known to have superior activity. There is controversy about whether adding drugs to doxorubicin offers clinical benefit beyond what is achieved by doxorubicin as a single agent. To avoid severe toxicity in older patients, sequential use of single agents may be the preferred strategy for palliation.
A randomized study assessed whether dose intensification of doxorubicin with ifosfamide improved the survival of patients with advanced soft-tissue sarcoma compared with doxorubicin alone.[10] Two hundred twenty-eight patients were randomly assigned to receive doxorubicin, and 227 patients were randomly assigned to receive doxorubicin and ifosfamide. Median follow-up was 56 months (interquartile range [IQR], 31–77) in the doxorubicin-only group and 59 months (IQR, 36–72) in the combination group.
There was no significant difference in overall survival (OS) between groups (median OS, 12.8 months; 95.5% confidence interval [CI], 10.5–14.3 in the doxorubicin-alone group vs. 14.3 months; range, 12.5–16.5 months in the doxorubicin and ifosfamide group; hazard ratio [HR], 0.83; 95.5% CI 0.67–1.03; stratified log-rank test P = .076). Median progression-free survival was significantly higher for the doxorubicin and ifosfamide group (7.4 months; 95% CI, 6.6-8.3) than for the doxorubicin-alone group (4.6 months; range, 2.9–5.6 months; HR, 0.74; 95% CI, 0.60–0.90; stratified log-rank test P = .003). More patients in the doxorubicin and ifosfamide group than in the doxorubicin-alone group had an overall response (60 [26%] of 227 patients vs. 31 [14%] of 228; P < .0006). The most common grade 3 and 4 toxic effects, which were all more common with doxorubicin and ifosfamide than with doxorubicin alone, were leucopenia (97 [43%] of 224 patients vs. 40 [18%] of 223 patients), neutropenia (93 [42%] vs. 83 [37%]), febrile neutropenia (103 (46%) vs. 30 [13%]), anemia (78 [35%] vs. 10 [5%]), and thrombocytopenia (75 [33%]) vs. 1 [<1%]).[10][Level of evidence: 1iiA] Treatment intensification with doxorubicin and ifosfamide for palliation of advanced soft tissue sarcoma is not indicated.
Standard treatment options
- Chemotherapy.
- Single-agent chemotherapy, with subsequent single agents for disease regrowth.[4-6,8,9,11] Doxorubicin is generally the first-line agent. Ifosfamide also has substantial single-agent activity.
- Doxorubicin-based combination chemotherapy. A variety of regimens have been used, but none has been proven to increase OS compared with doxorubicin alone.[4,5] There is some evidence that the addition of ifosfamide increases response rates (but not survival). Toxicity is increased with the addition of drugs to doxorubicin. No quality-of-life studies have been reported in comparisons of single-agent therapy versus combination therapy.
- Resection of pulmonary lesions may be performed if the primary tumor is under control.[1-3]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- van Geel AN, Pastorino U, Jauch KW, et al.: Surgical treatment of lung metastases: The European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group study of 255 patients. Cancer 77 (4): 675-82, 1996. [PUBMED Abstract]
- Casson AG, Putnam JB, Natarajan G, et al.: Five-year survival after pulmonary metastasectomy for adult soft tissue sarcoma. Cancer 69 (3): 662-8, 1992. [PUBMED Abstract]
- Putnam JB Jr, Roth JA: Surgical treatment for pulmonary metastases from sarcoma. Hematol Oncol Clin North Am 9 (4): 869-87, 1995. [PUBMED Abstract]
- Bramwell VH, Anderson D, Charette ML, et al.: Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev (3): CD003293, 2003. [PUBMED Abstract]
- Verma S, Younus J, Stys-Norman D, et al.: Meta-analysis of ifosfamide-based combination chemotherapy in advanced soft tissue sarcoma. Cancer Treat Rev 34 (4): 339-47, 2008. [PUBMED Abstract]
- Lorigan P, Verweij J, Papai Z, et al.: Phase III trial of two investigational schedules of ifosfamide compared with standard-dose doxorubicin in advanced or metastatic soft tissue sarcoma: a European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol 25 (21): 3144-50, 2007. [PUBMED Abstract]
- Nielsen OS, Dombernowsky P, Mouridsen H, et al.: High-dose epirubicin is not an alternative to standard-dose doxorubicin in the treatment of advanced soft tissue sarcomas. A study of the EORTC soft tissue and bone sarcoma group. Br J Cancer 78 (12): 1634-9, 1998. [PUBMED Abstract]
- Maki RG, Wathen JK, Patel SR, et al.: Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 25 (19): 2755-63, 2007. [PUBMED Abstract]
- Okuno S, Ryan LM, Edmonson JH, et al.: Phase II trial of gemcitabine in patients with advanced sarcomas (E1797): a trial of the Eastern Cooperative Oncology Group. Cancer 97 (8): 1969-73, 2003. [PUBMED Abstract]
- Judson I, Verweij J, Gelderblom H, et al.: Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol 15 (4): 415-23, 2014. [PUBMED Abstract]
- Grenader T, Goldberg A, Hadas-Halperin I, et al.: Long-term response to pegylated liposomal doxorubicin in patients with metastatic soft tissue sarcomas. Anticancer Drugs 20 (1): 15-20, 2009. [PUBMED Abstract]
Recurrent Adult Soft Tissue Sarcoma
Treatment of patients with recurrent soft tissue sarcoma depends on the type of initial presentation and treatment. Patients who develop a local recurrence often can be treated by local therapy: surgical excision plus radiation therapy after previous minimal therapy or amputation after previous aggressive treatment.[1-7] Resection of limited pulmonary metastases may be associated with favorable disease-free survival.[8-10][Level of evidence: 3iiiDiv] However, the contribution of selection factors, such as low tumor burden, slow tumor growth, and long disease-free interval, to these favorable outcomes is not known.
There is no standard chemotherapy for recurrent soft tissue sarcomas that have progressed after doxorubicin as a single agent or in combination with other agents that have clinical activity, such as ifosfamide, epirubicin, gemcitabine, and paclitaxel. Any of these agents not previously administered to the patient may be used sequentially at the time of recurrence or progression.[11-14][Level of Evidence: 3iiiDiv] None of these agents has been shown to increase overall survival in this setting, therefore, clinical trials are an appropriate option.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Singer S, Nielsen T, Antonescu CR: Molecular biology of soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1522-32.
- Singer S, Maki RG, O'Sullivan B: Soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1533-77.
- Malawer MM, Helman LJ, O'Sullivan B: Sarcomas of bone. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1578-1609.
- Midis GP, Pollock RE, Chen NP, et al.: Locally recurrent soft tissue sarcoma of the extremities. Surgery 123 (6): 666-71, 1998. [PUBMED Abstract]
- Essner R, Selch M, Eilber FR: Reirradiation for extremity soft tissue sarcomas. Local control and complications. Cancer 67 (11): 2813-7, 1991. [PUBMED Abstract]
- Singer S, Antman K, Corson JM, et al.: Long-term salvageability for patients with locally recurrent soft-tissue sarcomas. Arch Surg 127 (5): 548-53; discussion 553-4, 1992. [PUBMED Abstract]
- Lewis JJ, Leung D, Heslin M, et al.: Association of local recurrence with subsequent survival in extremity soft tissue sarcoma. J Clin Oncol 15 (2): 646-52, 1997. [PUBMED Abstract]
- van Geel AN, Pastorino U, Jauch KW, et al.: Surgical treatment of lung metastases: The European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group study of 255 patients. Cancer 77 (4): 675-82, 1996. [PUBMED Abstract]
- Casson AG, Putnam JB, Natarajan G, et al.: Five-year survival after pulmonary metastasectomy for adult soft tissue sarcoma. Cancer 69 (3): 662-8, 1992. [PUBMED Abstract]
- Putnam JB Jr, Roth JA: Surgical treatment for pulmonary metastases from sarcoma. Hematol Oncol Clin North Am 9 (4): 869-87, 1995. [PUBMED Abstract]
- Lorigan P, Verweij J, Papai Z, et al.: Phase III trial of two investigational schedules of ifosfamide compared with standard-dose doxorubicin in advanced or metastatic soft tissue sarcoma: a European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol 25 (21): 3144-50, 2007. [PUBMED Abstract]
- Nielsen OS, Dombernowsky P, Mouridsen H, et al.: High-dose epirubicin is not an alternative to standard-dose doxorubicin in the treatment of advanced soft tissue sarcomas. A study of the EORTC soft tissue and bone sarcoma group. Br J Cancer 78 (12): 1634-9, 1998. [PUBMED Abstract]
- Maki RG, Wathen JK, Patel SR, et al.: Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 25 (19): 2755-63, 2007. [PUBMED Abstract]
- Okuno S, Ryan LM, Edmonson JH, et al.: Phase II trial of gemcitabine in patients with advanced sarcomas (E1797): a trial of the Eastern Cooperative Oncology Group. Cancer 97 (8): 1969-73, 2003. [PUBMED Abstract]
Changes to This Summary (02/01/2018)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
General Information About Adult Soft Tissue Sarcoma
Updated statistics with estimated new cases and deaths for 2018 (cited American Cancer Society as reference 1).
Stage Information for Adult Soft Tissue Sarcoma
An editorial change was made to this section.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of adult soft tissue sarcoma. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Adult Soft Tissue Sarcoma Treatment are:
- Russell S. Berman, MD (New York University School of Medicine)
- Minh Tam Truong, MD (Boston University Medical Center)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Adult Soft Tissue Sarcoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/soft-tissue-sarcoma/hp/adult-soft-tissue-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389481]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Updated: February 1, 2018
Source URL: https://www.cancer.gov/publishedcontent/syndication/2127.htm
Source Agency: National Cancer Institute (NCI)
Captured Date: 2013-09-14 09:00:45.0
Adult Soft Tissue Sarcoma Treatment (PDQ®)–Health Professional Version
General Information About Adult Soft Tissue Sarcoma
Incidence and Mortality
Estimated new cases and deaths from soft tissue sarcoma in the United States in 2018:[1]
- New cases: 13,040.
- Deaths: 5,150.
Soft tissue sarcomas are malignant tumors that arise in any of the mesodermal tissues of the extremities (50%), trunk and retroperitoneum (40%), or head and neck (10%). The reported international incidence rates range from 1.8 to 5 per 100,000 individuals per year.[2]
Risk Factors and Genetic Factors
The risk of sporadic soft tissue sarcomas is increased by previous radiation therapy and, in the case of lymphangiosarcoma, by chronic lymphedema. The chemicals Thorotrast (thorium dioxide), vinyl chloride, and arsenic are also established carcinogens for hepatic angiosarcomas.[3-5]
Soft tissue sarcomas occur with greater frequency in patients with the following inherited syndromes:[3-5]
- Nevoid basal cell carcinoma syndrome (Gorlin syndrome: PTC gene mutation).
- Gardner syndrome (APC mutation).
- Li-Fraumeni syndrome (p53 mutation).
- Tuberous sclerosis (Bourneville disease: TSC1 or TSC2 mutation).
- von Recklinghausen disease (neurofibromatosis type 1: NF1 mutation).
- Werner syndrome (adult progeria: WRN mutation).
Diagnosis
Soft tissue sarcomas may be heterogeneous, so adequate tissue should be obtained via either core-needle or incisional biopsy for microscopic examination to determine histologic type and tumor grade. Careful planning of the initial biopsy is important to avoid compromising subsequent curative resection. Since the selection of treatment is determined by the grade of the tumor, it is essential to have a careful review of the biopsy tissue by a pathologist who is experienced in diagnosing sarcomas. Complete staging and treatment planning by a multidisciplinary team of cancer specialists is required to determine the optimal treatment for patients with this disease.
There is evidence that at least some favorable clinical outcomes may be associated with referral to a specialized sarcoma treatment center. In a population-based consecutive series of 375 soft tissue sarcoma patients in Sweden, local recurrence rates of resected tumors were higher in patients who were not referred to the specialized center: in 35 of 78 (45%) patients not referred; in 24 of 102 (24%) patients referred after initial surgery or incisional biopsy; and in 36 of 195 (18%) patients referred before any surgical procedure (P = .0001 for the difference between those never referred vs. those referred before any surgical procedure).[6][Level of evidence: 3iDii] However, there were no statistically significant differences in death from sarcoma between the groups of patients.
Prognostic Factors
The prognosis for patients with adult soft tissue sarcomas depends on several factors, including:[3-5,7,8]
- Patient’s age.
- Size, sarcoma subtype, histologic grade, mitotic activity, and stage of the tumor.
Factors associated with a poorer prognosis include the following:[9]
- Age older than 60 years.
- Tumors larger than 5 cm in greatest dimension.
- High-grade histology with high mitotic activity.
- Positive margins after resection.[10]
Although low-grade tumors are usually curable by surgery alone, higher-grade sarcomas (as determined by the mitotic index and by the presence of hemorrhage and necrosis) are associated with higher local-treatment failure rates and increased metastatic potential.
Surveillance for Relapse
A retrospective review included 174 consecutive patients with a soft tissue sarcoma of the limb who underwent follow-up by oncologists at a single center from 2003 to 2009.[11] The rate and site of recurrence and mode of detection were analyzed. Eighty-two patients (47%) experienced relapse. Isolated local recurrences occurred in 26 patients and local relapse with synchronous pulmonary metastases occurred in 5 patients. Local recurrences were detected clinically in 30 of the 31 patients; magnetic resonance imaging identified only one local recurrence. Twenty-eight patients developed isolated lung metastases; in 9 patients, the lung metastases were amenable to resections, 7 of whom were free of disease after treatment. Lung metastases were detected by chest x-ray in 19 patients, by computed tomography scanning in 3 patients, and clinically in 11 patients. Twenty-three patients developed nonpulmonary metastases. More than 80% of the relapses occurred in the first 2 years of follow-up; however, later recurrences were also observed.[11][Level of evidence: 3iiDi] This study supports imaging surveillance for detection of lung metastases, whereas local recurrences at the primary site were usually detected by clinical examination. The impact of picking up metastases from overall survival or quality-of-life data is unknown.
Related Summaries
Other PDQ summaries containing information about soft tissue sarcoma include:
References
- American Cancer Society: Cancer Facts and Figures 2018. Atlanta, Ga: American Cancer Society, 2018. Available online. Last accessed January 5, 2018.
- Wibmer C, Leithner A, Zielonke N, et al.: Increasing incidence rates of soft tissue sarcomas? A population-based epidemiologic study and literature review. Ann Oncol 21 (5): 1106-11, 2010. [PUBMED Abstract]
- Singer S, Nielsen T, Antonescu CR: Molecular biology of soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1522-32.
- Singer S, Maki RG, O'Sullivan B: Soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1533-77.
- Malawer MM, Helman LJ, O'Sullivan B: Sarcomas of bone. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1578-1609.
- Gustafson P, Dreinhöfer KE, Rydholm A: Soft tissue sarcoma should be treated at a tumor center. A comparison of quality of surgery in 375 patients. Acta Orthop Scand 65 (1): 47-50, 1994. [PUBMED Abstract]
- Coindre JM, Terrier P, Guillou L, et al.: Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 91 (10): 1914-26, 2001. [PUBMED Abstract]
- Kasper B, Ouali M, van Glabbeke M, et al.: Prognostic factors in adolescents and young adults (AYA) with high risk soft tissue sarcoma (STS) treated by adjuvant chemotherapy: a study based on pooled European Organisation for Research and Treatment of Cancer (EORTC) clinical trials 62771 and 62931. Eur J Cancer 49 (2): 449-56, 2013. [PUBMED Abstract]
- Vraa S, Keller J, Nielsen OS, et al.: Prognostic factors in soft tissue sarcomas: the Aarhus experience. Eur J Cancer 34 (12): 1876-82, 1998. [PUBMED Abstract]
- Trovik LH, Ovrebo K, Almquist M, et al.: Adjuvant radiotherapy in retroperitoneal sarcomas. A Scandinavian Sarcoma Group study of 97 patients. Acta Oncol 53 (9): 1165-72, 2014. [PUBMED Abstract]
- Rothermundt C, Whelan JS, Dileo P, et al.: What is the role of routine follow-up for localised limb soft tissue sarcomas? A retrospective analysis of 174 patients. Br J Cancer 110 (10): 2420-6, 2014. [PUBMED Abstract]
Cellular Classification of Adult Soft Tissue Sarcoma
Soft tissue sarcomas are classified histologically according to the soft tissue cell of origin. Additional studies, including electron microscopy, specialized immunohistochemistry, flow cytometry, cytogenetics, and tissue culture studies may allow identification of particular subtypes within the major histologic categories. For example, S100 antigen suggests neural sheath origin, cytokeratin suggests epithelioid or synovial cell origin, and factor VIII-related antigen suggests endothelial origin. Likewise, some subtypes of sarcomas have characteristic genetic markers, but these markers are not generally used in the routine clinical setting (e.g., translocation t(X;18)(p11;q11) in synovial sarcomas and translocation t(12;16)(q13;p11) in myxoid and round-cell sarcomas).[1-3]
The histologic grade reflects the metastatic potential of these tumors more accurately than the classic cellular classification listed below. Pathologists assign a grade based on the number of mitoses per high-powered field, the presence of necrosis, cellular and nuclear morphology, and the degree of cellularity; discordance among expert pathologists regarding tumor grade, and even histologic subtype, can be substantial.[4]
The World Health Organization lists the following cell types in its classification of soft tissue sarcomas:[5,6]
- Adipocytic tumors.
- Dedifferentiated liposarcoma.*
- Myxoid/round cell liposarcoma.
- Pleomorphic liposarcoma.
- Fibroblastic/myofibroblastic tumors.
- Fibrosarcoma.**
- Myxofibrosarcoma, low grade.
- Low-grade fibromyxoid sarcoma.
- Sclerosing epithelioid fibrosarcoma.
- So-called fibrohistiocytic tumors.
- Undifferentiated pleomorphic sarcoma/malignant fibrous histiocytoma (including pleomorphic, giant cell, myxoid/high-grade myxofibrosarcoma, and inflammatory forms).
- Smooth muscle tumors.
- Leiomyosarcoma.
- Skeletal muscle tumors.
- Rhabdomyosarcoma (embryonal, alveolar, and pleomorphic forms).
- Vascular tumors.
- Epithelioid hemangioendothelioma.
- Angiosarcoma, deep.***
- Tumors of peripheral nerves.
- Malignant peripheral nerve sheath tumor.
- Chondro-osseous tumors.
- Extraskeletal chondrosarcoma (mesenchymal and other variants).
- Extraskeletal osteosarcoma.
- Tumors of uncertain differentiation.
- Synovial sarcoma.
- Epithelioid sarcoma.
- Alveolar soft part sarcoma.
- Clear cell sarcoma of soft tissue.
- Extraskeletal myxoid chondrosarcoma.
- Primitive neuroectodermal tumor/extraskeletal Ewing tumor.
- Desmoplastic small round cell tumor.
- Extrarenal rhabdoid tumor.
- Undifferentiated sarcoma; sarcoma, not otherwise specified.
[Note: *It is recognized that dedifferentiated liposarcoma primarily arises in the context of deep atypical lipomatous tumor/well-differentiated liposarcoma, a sarcoma of intermediate malignancy because of the lack of metastatic capacity. **The category of fibrosarcoma can be inclusive of fibrosarcomatous differentiation in dermatofibrosarcoma protuberans. ***Cutaneous angiosarcoma may be difficult to stage using the American Joint Committee on Cancer system. (Refer to the PDQ summary on Gastrointestinal Stromal Tumors for more information.)]
References
- Singer S, Nielsen T, Antonescu CR: Molecular biology of soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1522-32.
- Singer S, Maki RG, O'Sullivan B: Soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1533-77.
- Malawer MM, Helman LJ, O'Sullivan B: Sarcomas of bone. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1578-1609.
- Alvegård TA, Berg NO: Histopathology peer review of high-grade soft tissue sarcoma: the Scandinavian Sarcoma Group experience. J Clin Oncol 7 (12): 1845-51, 1989. [PUBMED Abstract]
- Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-6.
- Brodowicz T, Schwameis E, Widder J, et al.: Intensified Adjuvant IFADIC Chemotherapy for Adult Soft Tissue Sarcoma: A Prospective Randomized Feasibility Trial. Sarcoma 4 (4): 151-60, 2000. [PUBMED Abstract]
Stage Information for Adult Soft Tissue Sarcoma
Note: The American Joint Committee on Cancer (AJCC) has published the 8th edition of the AJCC Cancer Staging Manual, which includes revisions to the staging for this disease. Implementation of the 8th edition began in January 2018. The PDQ Adult Treatment Editorial Board, which maintains this summary, is reviewing the revised staging and will make appropriate changes as needed.
Staging has an important role in determining the most effective treatment for soft tissue sarcoma. Clinical staging involves magnetic resonance imaging (MRI) or computed tomography (CT) of the primary tumor area and a chest CT to look for metastasis to the lung (the most common site of distant spread). An abdominal CT scan is done in the case of retroperitoneal sarcomas because the liver may be the site of initial clinical metastasis for these tumors.
The stage is determined by the size of the tumor, the histologic grade, and whether there is spread to lymph nodes or distant sites. Intracompartmental or extracompartmental extension of extremity sarcomas is also important for surgical decision making. For complete staging, a thorough review of all biopsy specimens (including those from the primary tumor, lymph nodes, or other suspicious lesions) is essential. CT scan of the chest is recommended for sarcomas larger than 5 cm (T2) or with moderate to poor differentiation (grades 2–4). Nodal involvement is rare, occurring in fewer than 3% of patients with sarcoma.[1]
Lymph node involvement in soft tissue sarcomas of adulthood is rare but is somewhat more frequent in some subtypes (e.g., rhabdomyosarcoma, vascular sarcomas, clear cell sarcomas, and epithelioid sarcomas) when they are high grade.[2] Because treatment decisions are predicated on pathology staging, patients should be staged before, and again after, any neoadjuvant therapy. The assessment of tumor grade can be affected in either direction, but more frequently decreased because of differential cellular loss related to the neoadjuvant chemotherapy or radiation.[3] Grade, which is based on cellular differentiation, mitotic rate, and extent of necrosis, should be recorded for all soft tissue sarcomas. A three-grade system (G1–G3) is preferred. (See below.)
The AJCC has designated staging by the four criteria of tumor size, nodal status, metastasis, and grade (TNMG).[3] The characteristic molecular markers of some sarcomas are not formally incorporated in the staging system pending further evaluation of their impact on prognosis. Recurrent sarcomas are restaged using the same system as for primary tumors with the specification that the tumor is recurrent.
Definitions of TNM and Grade
| aReprinted with permission from AJCC: Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-8. | |
| bSuperficial tumor is located exclusively above the superficial fascia without invasion of the fascia; deep tumor is located either exclusively beneath the superficial fascia, superficial to the fascia with invasion of or through the fascia, or both superficial yet beneath the fascia. | |
| TX | Primary tumor cannot be assessed. |
| T0 | No evidence of primary tumor. |
| T1 | Tumor ≤5 cm in greatest dimension. (Size should be regarded as a continuous variable, and the measurement should be provided.) |
| T1a | Superficial tumor.b |
| T1b | Deep tumor.b |
| T2 | Tumor >5 cm in greatest dimension.b |
| T2a | Superficial tumor.b |
| T2b | Deep tumor. |
| aReprinted with permission from AJCC: Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-8. | |
| bPresence of positive nodes (N1) in M0 tumors is considered Stage III. | |
| NX | Regional lymph nodes cannot be assessed. |
| N0 | No regional lymph node metastasis. |
| N1b | Regional lymph node metastasis. |
| aReprinted with permission from AJCC: Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-8. | |
| M0 | No distant metastasis. |
| M1 | Distant metastasis. |
| aReprinted with permission from AJCC: Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-8. | ||||
| Stage IA | T1a | N0 | M0 | G1, GX |
| T1b | N0 | M0 | G1, GX | |
| Stage IB | T2a | N0 | M0 | G1, GX |
| T2b | N0 | M0 | G1, GX | |
| Stage IIA | T1a | N0 | M0 | G2, G3 |
| T1b | N0 | M0 | G2, G3 | |
| Stage IIB | T2a | N0 | M0 | G2 |
| T2b | N0 | M0 | G2 | |
| Stage III | T2a, T2b | N0 | M0 | G3 |
| Any T | N1 | M0 | Any G | |
| Stage IV | Any T | Any N | M1 | Any G |
Neurovascular and bone invasion are indicators of poor prognosis, but they are not incorporated into the formal staging system.
References
- Fong Y, Coit DG, Woodruff JM, et al.: Lymph node metastasis from soft tissue sarcoma in adults. Analysis of data from a prospective database of 1772 sarcoma patients. Ann Surg 217 (1): 72-7, 1993. [PUBMED Abstract]
- Mazeron JJ, Suit HD: Lymph nodes as sites of metastases from sarcomas of soft tissue. Cancer 60 (8): 1800-8, 1987. [PUBMED Abstract]
- Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-6.
Treatment Option Overview
Multimodality Approach
In most cases, a combined modality approach of preoperative radiation therapy (preRT) or postoperative radiation therapy (PORT) is used, rather than the radical surgical procedures, such as amputation, that were used in the past. It may even be possible to use surgery without PORT in selected cases. For example, a case series was reported from a specialized sarcoma treatment referral center in which 74 selected patients with primary extremity and trunk tumors 5 cm or smaller were found to have no histologic involvement of the surgical margins. The patients were observed without radiation therapy, and the estimated local recurrence rate after 10 years was 11%.[1][Level of evidence: 3iiiDiv] The role of chemotherapy is not as well defined as is the role for radiation therapy. Because of the evolving nature of the treatment options for this disease, patients should be considered when available. Information about ongoing clinical trials is available from the NCI website.
Role of Surgery
Surgical resection is the mainstay of therapy for soft tissue sarcomas. When feasible, wide-margin function–sparing surgical excision is the cornerstone of effective treatment for extremity tumors. This may be facilitated by soft tissue reconstructive surgery, which generally permits wider margins than those obtained when the surgical plan involves direct closure of the excision site.[2] Cutting into the tumor mass or shelling out the gross tumor along the plane of the pseudocapsule of compressed tumor cells and reactive tissue that often surrounds soft tissue sarcomas are associated with an elevated risk of local recurrence. Even high-grade, soft tissue sarcomas of the extremities can usually be effectively treated while preserving the limb with combined-modality treatment consisting of preRT or PORT to reduce local recurrence. (Refer to the Role of Radiation Therapy section of this summary for more information.)
Only one small, single-institution, randomized trial has directly compared amputation to limb-sparing surgery for soft tissue sarcomas of the extremities.[3] In a 2:1 randomization ratio, 27 patients with high-grade extremity sarcomas were assigned to a wide excision plus PORT (45 Gy–50 Gy to the wide local excision area, and a total of 60 Gy–70 Gy to the tumor bed over 6–7 weeks), and 16 were assigned to amputation at or above the joint proximal to the tumor. Both groups received adjuvant chemotherapy (i.e., doxorubicin, cyclophosphamide, and high-dose methotrexate). At 63 months, with a median follow-up of 56 months, there were four local recurrences in the 27 patients who underwent limb-sparing surgery and no recurrences in the 16 patients who underwent amputation P2 = .12. Overall survival (OS) rates were not statistically significantly different (actuarial 5-year survival rate, 83% vs. 88%, P2 = .99).[3][Level of evidence: 1iiA]
Local control of high-grade soft tissue sarcomas of the trunk and the head and neck can be achieved with surgery in combination with radiation therapy.[4] It may be possible to use surgery without PORT in selected cases. For example, a case series was reported from a specialized sarcoma treatment referral center in which 74 selected patients with primary extremity and trunk tumors 5 cm or smaller were found to have no histologic involvement of the surgical margins.[1] They were observed without radiation therapy, and the estimated local recurrence rate after 10 years was 11%.[1][Level of evidence: 3iiiDiv] The role of chemotherapy is not as well defined as is the role of radiation therapy. Because of the evolving nature of the treatment options for this disease, patients should be offered the option of clinical trials when available.
Effective treatment of retroperitoneal sarcomas requires removal of all gross disease while sparing adjacent viscera not invaded by tumor. The prognosis for patients with high-grade retroperitoneal sarcomas is less favorable than for patients with tumors at other sites, partly because of the difficulty in completely resecting these tumors and the dose-limiting toxicity of high-dose radiation therapy on visceral organs.[5-8]
In the setting of distant metastasis, surgery may be associated with long-term, disease-free survival in patients with pulmonary metastasis and optimal underlying disease biology (i.e., patients with a limited number of metastases and slow nodule growth) who have undergone or are undergoing complete resection of the primary tumor.[9-11] It is not clear to what degree the favorable outcomes are attributable to the efficacy of surgery or the careful selection of patients based on factors that are associated with less-virulent disease.
Role of Radiation Therapy
Radiation plays an important role in limb-sparing therapy. Pre- and postoperative external-beam radiation therapies (EBRT), as well as brachytherapy, have been shown to decrease the risk of local recurrence. They have not been shown to increase OS but are used to avoid amputation for all but the most locally advanced tumors or for limbs seriously compromised by vascular disease, where acceptable functional preservation is not possible. In the case of EBRT, irradiation of the entire limb circumference is avoided to preserve vascular and nerve structures that are critical to function and preservation of the limb.
PORT
PORT has been tested in a single-institution, randomized trial of 141 patients with extremity sarcomas who were treated with limb-sparing surgery. Patients with high-grade tumors (n = 91) also received adjuvant chemotherapy (i.e., five 28-day cycles of doxorubicin and cyclophosphamide). All patients were randomly assigned to receive radiation (45 Gy to a wide field, plus a tumor-bed boost of 18 Gy over 6–7 weeks), concurrent with chemotherapy in the case of high-grade tumors versus no radiation.[12] At up to 12 years of follow-up, there was one local recurrence in the 70 patients randomly assigned to receive radiation versus 17 recurrences in the 71 control patients (P = .0001), with similar reduction in risk of local recurrence for both high- and low-grade tumors. However, there was no difference in OS between the radiation and control groups.[12][Level of evidence: 1iiDiii] Global quality of life was similar in the two groups, but the radiation therapy group had substantially worse functional deficits resulting from reduced strength and joint motion as well as increased edema.
To limit acute toxicity with preRT, smaller fields and lower doses are generally given than is the case with PORT. PreRT has been directly compared with PORT for extremity soft tissue sarcomas in a multicenter randomized trial.[13-15] Designed to include 266 patients, the trial was stopped early after 190 patients had been accrued because of an increase in wound complications in the preRT group. The scheduled radiation in the preRT group was a wide field of 50 Gy in 2-Gy fractions (first phase of the trial) with an additional 16 Gy to 20 Gy to the tumor bed and a 2-cm margin (second phase of the trial) only if tumor cells were found at the surgical margins.
Patients in the PORT group were scheduled to receive radiation during both phases of the trial. The wound-complication rates were 35% versus 17% in the preRT and PORT groups, respectively (P = .01). In addition, limb function at 6 weeks after surgery was worse in the preRT group (P = .01).[13] At 5 years, the two groups had similar local control rates (93% vs. 92%) and OS (73% vs. 67%, P = .48).[14] Of the 129 patients evaluated for limb function at 21 to 27 months after surgery (n = 73 for preRT and n = 56 for PORT), limb function was similar in both groups, but there was a statistical trend for less fibrosis in the preRT group (P = .07).[15]
Brachytherapy
Brachytherapy has also been investigated as an adjuvant therapy for soft tissue sarcomas. Although it has possible advantages of convenience and less radiation to normal surrounding tissue relative to EBRT, the two treatment strategies have not been directly compared in terms of efficacy or morbidity. However, adjuvant brachytherapy has been compared with surgery without radiation. The time interval between preRT and surgical excision in extremity soft tissue sarcoma had minimal influence on the development of wound complications. Four- or 5-week intervals showed equivalent complication rates between patients who did or did not develop wound complications, suggesting an optimal interval to reduce potential complications.[16]
In a single-institution trial, 164 patients with sarcomas of the extremity or superficial trunk were randomly assigned during surgery, if all gross tumor could be excised, to receive an iridium Ir 192 implant (delivering 42 Gy–45 Gy over 4–6 days; 78 patients) or to a control arm of no radiation (86 patients).[17,18] Some of the patients with high-grade tumors received adjuvant doxorubicin-based chemotherapy if they were thought to be at a high risk for metastasis (34 patients in each study arm). With a median follow-up of 76 months, the 5-year actuarial local recurrence rates were 18% and 31% in the brachytherapy and control arms, respectively (P = .04). This difference was limited to patients with high-grade tumors. There was no discernible difference in sarcoma-specific survival rates between the brachytherapy and control arms (84% and 81%, respectively; P = .65), and there was no difference in the high tumor-grade group.[17][Level of evidence: 1iiDiii] The rates of clinically important wound complications (e.g., need for operative revision or repeated seroma drainage, wound separation, large hematomas, or purulent infection) were 24% and 14% in the radiation and control arms, respectively (P = .13); wound reoperation rates were 10% and 0%, respectively (P = .006).[18]
Intensity-modulated radiation therapy
Intensity-modulated radiation therapy (IMRT) has been used to deliver preRT or PORT to patients with extremity soft tissue sarcomas in an effort to spare the femur, joints, and selected other normal tissues from the full prescription dose and to maintain local control while potentially reducing radiation therapy-related morbidity. Initial single-institution reports suggest that high rates of local control with some reduction in morbidity are possible with this technique.[19,20] Retrospective comparison of IMRT compared with 3-dimensional, conformal radiation therapy demonstrates that local recurrence for primary soft tissue sarcomas of the extremity was worse in the non-IMRT group.[21][Level of evidence: 3iiiDiv]
Surgery and radiation therapy
In some tumors of the extremities or trunk, surgery alone can be performed without the use of radiation. Evidence for this approach is limited to single-institution, relatively small, case series [1,22,23] or analysis of outcomes in the National Cancer Institute's Surveillance, Epidemiology, and End Results (SEER) tumor registry.[24] However, these comparisons suffer from low statistical power and differential evaluability rates that could have introduced bias.[1] Patient selection factors may vary among surgeons. In general, this approach is considered in patients with low-grade tumors of the extremity or superficial trunk that are 5 cm or smaller in diameter (T1) and have microscopically negative surgical margins; long-term local tumor control is about 90% in such patients.[25]
A patterns-of-care study using SEER data was queried to identify patients undergoing surgery for truncal and extremity soft tissue sarcomas from 2004 to 2009.[26] Of 5,075 patients, 50% received radiation therapy. Radiation was considered to be underused in a significant portion of patients undergoing treatment for soft tissue sarcoma in the United States. Although routine radiation therapy is not recommended for stage I patients, 25% of them still underwent radiation. Even though routine radiation therapy is recommended for patients with stage II and III tumors, only 60% of them underwent radiation. On multivariate analysis, predictors of radiation therapy included age younger than 50 years (odds ratio [OR], 1.57; 95% confidence interval [CI], 1.28–1.91), malignant fibrous histiocytoma histology (OR, 1.47; 95% CI, 1.3–1.92), T2 classification (OR, 1.88; 95% CI, 1.60–2.20), and G3 (OR, 6.27; 95% CI, 5.10–7.72). Patients with stage III soft tissue sarcoma who received radiation therapy showed improved disease-specific survival at 5 years compared with those who did not (68% vs. 46%, P < .001).[26][Level of evidence: 3iDii]
On occasion, surgical excision cannot be performed in the initial management of soft tissue sarcomas because the morbidity would be unacceptable or nearby critical organs make complete resection impossible. In such circumstances, radiation has been used as the primary therapy.[27] However, this must be considered a treatment of last resort. Experience is limited to retrospective case series from single centers.[27][Level of evidence: 3iiiDiv]
Role of Adjuvant or Neoadjuvant Chemotherapy for Clinically Localized Tumors
The role of adjuvant chemotherapy is not completely clear. The investigation of its use falls into two categories or generations—pre- and post-ifosfamide regimens. In discussions with a patient, any potential benefits should be considered in the context of the short- and long-term toxicities of the chemotherapy.
First-generation trials (preifosfamide)
Several prospective, randomized trials were unable to determine conclusively whether doxorubicin-based adjuvant chemotherapy benefits adults with resectable soft tissue sarcomas. The majority of these studies accrued small numbers of patients and did not demonstrate a metastasis-free survival or an OS benefit for adjuvant chemotherapy.[4] A small study of adjuvant chemotherapy showed a positive effect on both disease-free survival (DFS) and OS in patients treated with postoperative chemotherapy.[28] There was wide interstudy variability among the reported trials, including differences in therapeutic regimens, drug doses, sample size, tumor site, and histologic grade.
A quantitative meta-analysis of updated data from 1,568 individual patients in 14 trials of doxorubicin-based adjuvant therapy showed an absolute benefit from adjuvant therapy of 6% for a local relapse-free interval (95% CI, 1%–10%), 10% for a distant relapse-free interval (95% CI, 5%–15%), and 10% for recurrence-free survival (95% CI, 5%–15%). A statistically significant OS benefit at 10 years was not detected: absolute difference 4% (95% CI, -1%–+9%).[29,30][Level of evidence: 1iiDii] However, only a small proportion of patients in this meta-analysis were treated with ifosfamide, an agent with demonstrated activity against soft tissue sarcoma. In addition, a subset analysis suggested that patients with sarcomas of the extremities may have benefited from adjuvant chemotherapy (hazard ratio [HR] for death, 0.8, P = .029), but there was no clear evidence that patients with extremity sarcomas had outcomes that were statistically significantly different from the outcomes of patients with tumors at other sites (P = .58).[30]
Second-generation trials (postifosfamide)
Subsequent chemotherapy trials were performed using anthracycline and ifosfamide combinations in patients who primarily had extremity or truncal soft tissue sarcomas. The data are conflicting, and the issue is still not settled. In a small feasibility study, 59 patients with high-risk, soft tissue sarcomas, 58 of whom had an extremity or the trunk as the primary site, underwent primary resection plus PORT and were randomly assigned to observation versus a dose-dense regimen of six 14-day courses of ifosfamide, dacarbazine (DTIC), and doxorubicin (IFADIC regimen) with granulocyte colony-stimulating factor (G-CSF) bone marrow support and mesna uroprotection.[31] There were no statistically significant differences in OS or relapse-free survival (RFS), but the study was severely underpowered.
In a second trial performed by the Italian National Council for Research, high-risk patients were treated with local therapy (i.e., wide resection plus preRT or PORT, or amputation as clinically necessary) and were then randomly assigned to observation versus five 21-day cycles of 4-epidoxorubicin (epirubicin) plus ifosfamide (with mesna and G-CSF).[28,32] Based on power calculations, the planned study size was 190 patients, but the trial was stopped after 104 patients had been entered because an interim analysis revealed a statistically significant (P = .001) difference in DFS favoring the chemotherapy arm. By the time of the initial peer-reviewed report of the study, the DFS still favored the chemotherapy group (median DFS of 48 months vs. 16 months), but the P value had risen to .04.[28]
Although there was no difference in metastasis-free survival at the time of the report, there was an improvement in median OS (75 months vs. 46 months, P = .03). However, at the follow-up report (at a median of 89.6 months in a range of 56–119 months), OS differences were no longer statistically significant (58.5% vs. 43.1% [P = .07]). The DFS difference had also lost statistical significance (47.2% vs. 16.0% [P = .09]).[32] In summary, the trial was underpowered because it was stopped early, and the early promising results that led to stopping the trial diminished as the trial matured.
In a third, underpowered, single-center trial, 88 patients with high-risk, soft tissue sarcomas (64 of whom had extremity or truncal primary tumors) underwent surgery (with or without radiation) and were then randomly assigned to receive four 21-day cycles of chemotherapy (epirubicin [n = 26] or epirubicin plus ifosfamide [n = 19]) versus no adjuvant chemotherapy (n = 43).[33] The trial was closed prematurely because of a slow accrual rate. After a median follow-up of 94 months, the 5-year DFS in the chemotherapy and control arms was 69% versus 44%, respectively (P = .01); the 5-year OS rates were 72% versus 47% (P = .06). All of the benefit associated with chemotherapy appeared restricted to the 19 patients who received epirubicin plus ifosfamide.
In yet another underpowered trial, 137 patients with high-risk, soft tissue sarcomas (93% with extremity or truncal primary tumors) who met the eligibility criteria were randomly assigned to undergo surgical resection (with or without radiation) or to receive three preoperative 21-day cycles of doxorubicin plus ifosfamide.[34] This multicenter European Organization for Research and Treatment of Cancer trial (EORTC-62874) was closed because of slow accrual and results that were not promising enough to continue. With a median follow-up of 7.3 years, the 5-year DFS in the surgery alone and chemotherapy plus surgery arms was 52% and 56%, respectively (P = .35); and OS was 64% and 65%, respectively (P = .22).
These last four trials have been combined with the 14 first-generation trials in a trial-level meta-analysis.[35] Of the 18 randomized trials of patients with resectable soft tissue sarcomas, five trials used a combination of doxorubicin (50–90 mg/m2 per cycle) plus ifosfamide (1,500–5,000 mg/m2 per cycle). The remaining 13 trials used doxorubicin (50–70 mg/m2 per cycle) alone or with other drugs. The absolute risk reduction in local recurrence rates associated with any chemotherapy added to local therapy was 4 percentage points (95% CI, 0%–7%), and it was 5 percentage points (95% CI, 1%–12%) when ifosfamide was combined with doxorubicin. The absolute reduction in overall mortality was 6 percentage points with any chemotherapy (95% CI, 2%–11%; [i.e., a reduction from 46%–40%]), 11 percentage points for doxorubicin plus ifosfamide (95% CI, 3%–19%; [i.e., a reduction from 41%–30%]), and 5 percentage points for doxorubicin without ifosfamide.[35][Level of evidence: 1iiA]
An additional multicenter randomized trial (EORTC-62931 [NCT00002641]), the largest trial reported to date using adjuvant doxorubicin (75 mg/m2) plus ifosfamide (5,000 mg/m2), was subsequently published in abstract form and was not included in the above meta-analysis.[36] The results differed from those reported in the meta-analysis.[35] After local therapy, 351 patients were randomly assigned to five 21-day cycles of adjuvant therapy versus observation. The trial was stopped for futility because the 5-year RFS was 52% in both arms. OS was 64% in the chemotherapy arm versus 69% in the observation arm. In a subsequent abstract, the EORTC investigators reported a combined analysis of this trial and their previous trial (EORTC-62771) [37] of adjuvant cyclophosphamide plus doxorubicin plus DTIC (CYVADIC), representing the two largest trials of adjuvant therapy for adult soft tissue sarcoma in the literature.[38] The combined analysis showed no improvement in either RFS or OS associated with adjuvant chemotherapy.[38][Level of evidence: 1iiA]
In summary, the impact of adjuvant chemotherapy on survival is not clear but is likely to be small in absolute magnitude. Therefore, in discussions with a patient, any potential benefits should be considered in the context of the short- and long-term toxicities of the chemotherapy.
Role of regional hyperthermia
The use of regional hyperthermia to enhance the local effects of systemic chemotherapy in the neoadjuvant and adjuvant setting is under investigation. In a multicenter phase III trial, 341 patients with high-risk (tumor ≥5 cm, grade 2–3, and deep to fascia), soft tissue sarcomas (149 extremity tumors and 192 nonextremity tumors) were randomly allocated to receive four 21-day cycles of chemotherapy (etoposide 125 mg/m2 on days 1 and 4; ifosfamide 1,500 mg/m2 on days 1–4; doxorubicin 50 mg/m2 on day 1) with or without regional hyperthermia both before and after local therapy.[39] Approximately 11% of the patients were being treated for recurrent tumors. The regional hyperthermia was designed to produce tumor temperatures of 42°C for 60 minutes and was given on days 1 and 4 of each chemotherapy cycle. After the first four cycles of chemotherapy, definitive surgical excision of the tumor was performed, if possible, followed by radiation therapy, if indicated (i.e., a 52.7 Gy median dose delivered), and then the last four cycles of chemotherapy plus or minus hyperthermia. Three of the nine treatment centers with particular expertise in hyperthermia treated 91% of the patients in the trial.
The median duration of follow-up was 34 months. Local progression occurred in 56 patients in the hyperthermia group and 76 patients in the control group. The relative HR for local progression or death was 0.58 (95% CI, 0.41–0.84), with an absolute difference at 2 years of 15% (76% vs. 61%; 95% CI of the difference 6–26). The decreased risk of local progression or death was seen in both extremity and nonextremity tumors. However, hyperthermia had no effect on distant failure rates nor was there a statistically significant effect on OS (HR, .88, 95% CI, 0.64–1.21; P = .43).[39][Level of evidence: 1iiDiii] There was a higher rate of grade 3 to 4 leucopenia in the hyperthermia group: 77.6% versus 63.5% (P = .005). Since a large proportion of the patients were treated at centers with special expertise, there is no certainty that the finding can be generalized to apply to other settings.
Role of isolated limb perfusion
Isolated limb perfusion is under investigation as a means to deliver high doses of chemotherapy and permit limb salvage in unresectable primary or recurrent extremity soft tissue sarcomas that would otherwise require amputation, in the opinion of the surgeon.[40,41] Common drugs used in the procedure are TNF-alpha, melphalan, and interferon-gamma. Experience is limited to case series with response rates and reported avoidance of amputation as the outcome.[40,41][Level of evidence: 3iiiDiv] The technique requires specialized expertise to avoid severe local and systemic toxicity including systemic effects of TNF-alpha. The technique has not been directly compared with standard approaches using combined systemic and local therapy.
Role of chemotherapy for advanced disease
Doxorubicin is a mainstay of systemic therapy in the management of locally advanced and metastatic soft tissue sarcoma. Pegylated liposomal encapsulated doxorubicin is a formulation of doxorubicin designed to prolong the half-life of circulating doxorubicin and slow the release of active drugs.[42] The changed pharmacokinetics result in less myelosuppression and possibly less cardiotoxic effects, but there is a substantial incidence of hypersensitivity-like reactions and hand-foot syndrome. Its clinical activity relative to unencapsulated doxorubicin is not clear.[42][Level of evidence: 3iiiDiv] Other drugs that are thought to have clinical activity as single agents are ifosfamide, epirubicin, gemcitabine, and paclitaxel.[43-46][Level of Evidence: 3iiiDiv] Their clinical activity relative to single-agent doxorubicin is not clear, and they are not known to have superior activity.
There is controversy about the clinical benefit of adding other drugs to doxorubicin as a single agent. A systematic evidence review and meta-analysis conducted by the Cochrane Collaboration summarized the eight randomized trials reported from 1976 to 1995.[47] No additional randomized trials had been reported or were known to be in progress between 1995 and the 2002 literature search. Single-agent doxorubicin had been compared with a variety of doxorubicin-containing combinations that included vincristine, vindesine, cyclophosphamide, streptozotocin, mitomycin-C, cisplatin, and/or ifosfamide. Combination regimens consistently caused more nausea and hematologic toxicity. However, the better response rates associated with combination therapy were marginal and depended on the statistical model used (fixed effects model ORresponse = 1.29; 95% CI, 1.03–1.60, P = .03; random effects model ORresp = 1.26; 95% CI, 0.96–1.67, P = .10) There was no statistically significant difference in the 1- (ORmortality = 0.87; 95% CI, 0.73–1.05, P = .14) or 2-year mortality rates (ORmortality = 0.84; 95% CI, 0.67–1.06, P = .13).
These results were very similar even when the analyses were restricted to the four trials that used DTIC and/or ifosfamide as part of the combination regimen with doxorubicin agents that were postulated to have greater activity than the others tested. A subsequent meta-analysis of all three published randomized trials of chemotherapy regimens that contained ifosfamide versus those that did not came to similar conclusions: tumor response rates were better when the regimen included ifosfamide (RRresponse = 1.52; 95% CI, 1.11–2.08), but mortality at 1 year was not (RRmortality = 0.98; 95% CI, 0.85–1.13).[48][Level of evidence: 1iiDiv]. Therefore, response rate was a poor surrogate for OS. Quality-of-life outcomes were not reported in any of the above-mentioned randomized trials, but toxicity was worse when agents were added to doxorubicin.
References
- Pisters PW, Pollock RE, Lewis VO, et al.: Long-term results of prospective trial of surgery alone with selective use of radiation for patients with T1 extremity and trunk soft tissue sarcomas. Ann Surg 246 (4): 675-81; discussion 681-2, 2007. [PUBMED Abstract]
- Lohman RF, Nabawi AS, Reece GP, et al.: Soft tissue sarcoma of the upper extremity: a 5-year experience at two institutions emphasizing the role of soft tissue flap reconstruction. Cancer 94 (8): 2256-64, 2002. [PUBMED Abstract]
- Rosenberg SA, Tepper J, Glatstein E, et al.: The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg 196 (3): 305-15, 1982. [PUBMED Abstract]
- O'Byrne K, Steward WP: The role of adjuvant chemotherapy in the treatment of adult soft tissue sarcomas. Crit Rev Oncol Hematol 27 (3): 221-7, 1998. [PUBMED Abstract]
- Singer S, Nielsen T, Antonescu CR: Molecular biology of soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1522-32.
- Singer S, Maki RG, O'Sullivan B: Soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1533-77.
- Malawer MM, Helman LJ, O'Sullivan B: Sarcomas of bone. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1578-1609.
- Lewis JJ, Leung D, Woodruff JM, et al.: Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg 228 (3): 355-65, 1998. [PUBMED Abstract]
- van Geel AN, Pastorino U, Jauch KW, et al.: Surgical treatment of lung metastases: The European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group study of 255 patients. Cancer 77 (4): 675-82, 1996. [PUBMED Abstract]
- Casson AG, Putnam JB, Natarajan G, et al.: Five-year survival after pulmonary metastasectomy for adult soft tissue sarcoma. Cancer 69 (3): 662-8, 1992. [PUBMED Abstract]
- Putnam JB Jr, Roth JA: Surgical treatment for pulmonary metastases from sarcoma. Hematol Oncol Clin North Am 9 (4): 869-87, 1995. [PUBMED Abstract]
- Yang JC, Chang AE, Baker AR, et al.: Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 16 (1): 197-203, 1998. [PUBMED Abstract]
- O'Sullivan B, Davis AM, Turcotte R, et al.: Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 359 (9325): 2235-41, 2002. [PUBMED Abstract]
- O'Sullivan B, Davis A, Turcotte R, et al.: Five-year results of a randomized phase III trial of pre-operative vs post-operative radiotherapy in extremity soft tissue sarcoma. [Abstract] J Clin Oncol 22 (Suppl 14): A-9007, 819s, 2004.
- Davis AM, O'Sullivan B, Turcotte R, et al.: Late radiation morbidity following randomization to preoperative versus postoperative radiotherapy in extremity soft tissue sarcoma. Radiother Oncol 75 (1): 48-53, 2005. [PUBMED Abstract]
- Griffin AM, Dickie CI, Catton CN, et al.: The influence of time interval between preoperative radiation and surgical resection on the development of wound healing complications in extremity soft tissue sarcoma. Ann Surg Oncol 22 (9): 2824-30, 2015. [PUBMED Abstract]
- Pisters PW, Harrison LB, Leung DH, et al.: Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol 14 (3): 859-68, 1996. [PUBMED Abstract]
- Alektiar KM, Zelefsky MJ, Brennan MF: Morbidity of adjuvant brachytherapy in soft tissue sarcoma of the extremity and superficial trunk. Int J Radiat Oncol Biol Phys 47 (5): 1273-9, 2000. [PUBMED Abstract]
- Alektiar KM, Brennan MF, Healey JH, et al.: Impact of intensity-modulated radiation therapy on local control in primary soft-tissue sarcoma of the extremity. J Clin Oncol 26 (20): 3440-4, 2008. [PUBMED Abstract]
- Alektiar KM, Brennan MF, Singer S: Local control comparison of adjuvant brachytherapy to intensity-modulated radiotherapy in primary high-grade sarcoma of the extremity. Cancer 117 (14): 3229-34, 2011. [PUBMED Abstract]
- Folkert MR, Singer S, Brennan MF, et al.: Comparison of local recurrence with conventional and intensity-modulated radiation therapy for primary soft-tissue sarcomas of the extremity. J Clin Oncol 32 (29): 3236-41, 2014. [PUBMED Abstract]
- Fabrizio PL, Stafford SL, Pritchard DJ: Extremity soft-tissue sarcomas selectively treated with surgery alone. Int J Radiat Oncol Biol Phys 48 (1): 227-32, 2000. [PUBMED Abstract]
- Rydholm A, Gustafson P, Rööser B, et al.: Limb-sparing surgery without radiotherapy based on anatomic location of soft tissue sarcoma. J Clin Oncol 9 (10): 1757-65, 1991. [PUBMED Abstract]
- Al-Refaie WB, Habermann EB, Jensen EH, et al.: Surgery alone is adequate treatment for early stage soft tissue sarcoma of the extremity. Br J Surg 97 (5): 707-13, 2010. [PUBMED Abstract]
- Rydholm A: Surgery without radiotherapy in soft tissue sarcoma. Acta Orthop Scand Suppl 273: 117-9, 1997. [PUBMED Abstract]
- Bagaria SP, Ashman JB, Daugherty LC, et al.: Compliance with National Comprehensive Cancer Network guidelines in the use of radiation therapy for extremity and superficial trunk soft tissue sarcoma in the United States. J Surg Oncol 109 (7): 633-8, 2014. [PUBMED Abstract]
- Kepka L, DeLaney TF, Suit HD, et al.: Results of radiation therapy for unresected soft-tissue sarcomas. Int J Radiat Oncol Biol Phys 63 (3): 852-9, 2005. [PUBMED Abstract]
- Frustaci S, Gherlinzoni F, De Paoli A, et al.: Adjuvant chemotherapy for adult soft tissue sarcomas of the extremities and girdles: results of the Italian randomized cooperative trial. J Clin Oncol 19 (5): 1238-47, 2001. [PUBMED Abstract]
- Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet 350 (9092): 1647-54, 1997. [PUBMED Abstract]
- Sarcoma Meta-analysis Collaboration (SMAC): Adjuvant chemotherapy for localised resectable soft tissue sarcoma in adults. Cochrane Database Syst Rev (4): CD001419, 2000. [PUBMED Abstract]
- Brodowicz T, Schwameis E, Widder J, et al.: Intensified Adjuvant IFADIC Chemotherapy for Adult Soft Tissue Sarcoma: A Prospective Randomized Feasibility Trial. Sarcoma 4 (4): 151-60, 2000. [PUBMED Abstract]
- Frustaci S, De Paoli A, Bidoli E, et al.: Ifosfamide in the adjuvant therapy of soft tissue sarcomas. Oncology 65 (Suppl 2): 80-4, 2003. [PUBMED Abstract]
- Petrioli R, Coratti A, Correale P, et al.: Adjuvant epirubicin with or without Ifosfamide for adult soft-tissue sarcoma. Am J Clin Oncol 25 (5): 468-73, 2002. [PUBMED Abstract]
- Gortzak E, Azzarelli A, Buesa J, et al.: A randomised phase II study on neo-adjuvant chemotherapy for 'high-risk' adult soft-tissue sarcoma. Eur J Cancer 37 (9): 1096-103, 2001. [PUBMED Abstract]
- Pervaiz N, Colterjohn N, Farrokhyar F, et al.: A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 113 (3): 573-81, 2008. [PUBMED Abstract]
- Woll PJ, van Glabbeke M, Hohenberger P, et al.: Adjuvant chemotherapy (CT) with doxorubicin and ifosfamide in resected soft tissue sarcoma (STS): Interim analysis of a randomised phase III trial. [Abstract] J Clin Oncol 25 (Suppl 18): A-10008, 2007.
- Bramwell V, Rouesse J, Steward W, et al.: Adjuvant CYVADIC chemotherapy for adult soft tissue sarcoma--reduced local recurrence but no improvement in survival: a study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol 12 (6): 1137-49, 1994. [PUBMED Abstract]
- Le Cesne A, Van Glabbeke M, Woll PJ, et al.: The end of adjuvant chemotherapy (adCT) era with doxorubicin-based regimen in resected high-grade soft tissue sarcoma (STS): pooled analysis of the two STBSG-EORTC phase III clinical trials. [Abstract] J Clin Oncol 26 (Suppl 15): A-10525, 2008.
- Issels RD, Lindner LH, Verweij J, et al.: Neo-adjuvant chemotherapy alone or with regional hyperthermia for localised high-risk soft-tissue sarcoma: a randomised phase 3 multicentre study. Lancet Oncol 11 (6): 561-70, 2010. [PUBMED Abstract]
- Eggermont AM, de Wilt JH, ten Hagen TL: Current uses of isolated limb perfusion in the clinic and a model system for new strategies. Lancet Oncol 4 (7): 429-37, 2003. [PUBMED Abstract]
- Bonvalot S, Laplanche A, Lejeune F, et al.: Limb salvage with isolated perfusion for soft tissue sarcoma: could less TNF-alpha be better? Ann Oncol 16 (7): 1061-8, 2005. [PUBMED Abstract]
- Grenader T, Goldberg A, Hadas-Halperin I, et al.: Long-term response to pegylated liposomal doxorubicin in patients with metastatic soft tissue sarcomas. Anticancer Drugs 20 (1): 15-20, 2009. [PUBMED Abstract]
- Lorigan P, Verweij J, Papai Z, et al.: Phase III trial of two investigational schedules of ifosfamide compared with standard-dose doxorubicin in advanced or metastatic soft tissue sarcoma: a European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol 25 (21): 3144-50, 2007. [PUBMED Abstract]
- Nielsen OS, Dombernowsky P, Mouridsen H, et al.: High-dose epirubicin is not an alternative to standard-dose doxorubicin in the treatment of advanced soft tissue sarcomas. A study of the EORTC soft tissue and bone sarcoma group. Br J Cancer 78 (12): 1634-9, 1998. [PUBMED Abstract]
- Maki RG, Wathen JK, Patel SR, et al.: Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 25 (19): 2755-63, 2007. [PUBMED Abstract]
- Okuno S, Ryan LM, Edmonson JH, et al.: Phase II trial of gemcitabine in patients with advanced sarcomas (E1797): a trial of the Eastern Cooperative Oncology Group. Cancer 97 (8): 1969-73, 2003. [PUBMED Abstract]
- Bramwell VH, Anderson D, Charette ML, et al.: Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev (3): CD003293, 2003. [PUBMED Abstract]
- Verma S, Younus J, Stys-Norman D, et al.: Meta-analysis of ifosfamide-based combination chemotherapy in advanced soft tissue sarcoma. Cancer Treat Rev 34 (4): 339-47, 2008. [PUBMED Abstract]
Stage I Adult Soft Tissue Sarcoma
Refer to the Treatment Option Overview section of this summary for a more detailed discussion of the roles of surgery and radiation therapy.
Low-grade soft tissue sarcomas have little metastatic potential, but they have a propensity to recur locally. Accordingly, surgical excision with negative tissue margins of 1 cm to 2 cm or larger in all directions is the treatment of choice for patients with these early-stage sarcomas.[1-3] The Mohs surgical technique may be considered as an alternative to wide surgical excision for the very rare, small, well-differentiated primary sarcomas of the skin when cosmetic results are considered to be important, as margins can be assured with minimal normal tissue removal.[4]
Carefully executed high-dose radiation therapy using a shrinking-field technique may be beneficial for unresectable tumors or for resectable tumors in which a high likelihood of residual disease is thought to be present when margins are judged to be inadequate, and when wider resection would require either an amputation or the removal of a vital organ.[5] Because of the low metastatic potential of these tumors, chemotherapy is usually not given.[6,7]
Standard treatment options:
- Surgical excision of tumors 5 cm or smaller in diameter with negative tissue margins in all directions.[8-12]
- Surgical excision with preoperative radiation therapy (preRT) or postoperative radiation therapy (PORT). Radiation decreases the risk of local recurrence but has not been shown to increase overall survival.[13-16]
- If the tumor is unresectable, high-dose preRT may be used.[17]
- For tumors of the retroperitoneum, trunk, and head and neck, the following are options:
- Surgical resection with the option of PORT if negative margins cannot be obtained. Wide margins are unusual in these sites, and radiation therapy is usually advocated for trunk and head and neck primary sites.[18]
- PreRT followed by maximal surgical resection. Radiation therapy may be used in sarcomas of the trunk and head and neck to maximize local control because of the inability to obtain wide surgical margins.[19]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Singer S, Nielsen T, Antonescu CR: Molecular biology of soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1522-32.
- Singer S, Maki RG, O'Sullivan B: Soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1533-77.
- Malawer MM, Helman LJ, O'Sullivan B: Sarcomas of bone. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1578-1609.
- Fish FS: Soft tissue sarcomas in dermatology. Dermatol Surg 22 (3): 268-73, 1996. [PUBMED Abstract]
- Temple WJ, Temple CL, Arthur K, et al.: Prospective cohort study of neoadjuvant treatment in conservative surgery of soft tissue sarcomas. Ann Surg Oncol 4 (7): 586-90, 1997 Oct-Nov. [PUBMED Abstract]
- Sarcoma Meta-analysis Collaboration (SMAC): Adjuvant chemotherapy for localised resectable soft tissue sarcoma in adults. Cochrane Database Syst Rev (4): CD001419, 2000. [PUBMED Abstract]
- Pervaiz N, Colterjohn N, Farrokhyar F, et al.: A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 113 (3): 573-81, 2008. [PUBMED Abstract]
- Al-Refaie WB, Habermann EB, Jensen EH, et al.: Surgery alone is adequate treatment for early stage soft tissue sarcoma of the extremity. Br J Surg 97 (5): 707-13, 2010. [PUBMED Abstract]
- Pisters PW, Pollock RE, Lewis VO, et al.: Long-term results of prospective trial of surgery alone with selective use of radiation for patients with T1 extremity and trunk soft tissue sarcomas. Ann Surg 246 (4): 675-81; discussion 681-2, 2007. [PUBMED Abstract]
- Fabrizio PL, Stafford SL, Pritchard DJ: Extremity soft-tissue sarcomas selectively treated with surgery alone. Int J Radiat Oncol Biol Phys 48 (1): 227-32, 2000. [PUBMED Abstract]
- Rydholm A, Gustafson P, Rööser B, et al.: Limb-sparing surgery without radiotherapy based on anatomic location of soft tissue sarcoma. J Clin Oncol 9 (10): 1757-65, 1991. [PUBMED Abstract]
- Rydholm A: Surgery without radiotherapy in soft tissue sarcoma. Acta Orthop Scand Suppl 273: 117-9, 1997. [PUBMED Abstract]
- Yang JC, Chang AE, Baker AR, et al.: Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 16 (1): 197-203, 1998. [PUBMED Abstract]
- O'Sullivan B, Davis AM, Turcotte R, et al.: Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 359 (9325): 2235-41, 2002. [PUBMED Abstract]
- O'Sullivan B, Davis A, Turcotte R, et al.: Five-year results of a randomized phase III trial of pre-operative vs post-operative radiotherapy in extremity soft tissue sarcoma. [Abstract] J Clin Oncol 22 (Suppl 14): A-9007, 819s, 2004.
- Davis AM, O'Sullivan B, Turcotte R, et al.: Late radiation morbidity following randomization to preoperative versus postoperative radiotherapy in extremity soft tissue sarcoma. Radiother Oncol 75 (1): 48-53, 2005. [PUBMED Abstract]
- Kepka L, DeLaney TF, Suit HD, et al.: Results of radiation therapy for unresected soft-tissue sarcomas. Int J Radiat Oncol Biol Phys 63 (3): 852-9, 2005. [PUBMED Abstract]
- Brennan MF, Singer S, Maki RG: Sarcomas of the soft tissue and bone. In: DeVita VT Jr, Hellman S, Rosenberg SA, eds.: Cancer: Principles and Practice of Oncology. Vols. 1 & 2. 8th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2008, pp 1741-1833.
- Baldini EH, Wang D, Haas RL, et al.: Treatment Guidelines for Preoperative Radiation Therapy for Retroperitoneal Sarcoma: Preliminary Consensus of an International Expert Panel. Int J Radiat Oncol Biol Phys 92 (3): 602-12, 2015. [PUBMED Abstract]
Stage II and Node-Negative Stage III Adult Soft Tissue Sarcoma
Refer to the Treatment Option Overview section of this summary for a more detailed discussion of the roles of surgery, radiation therapy, and chemotherapy.
High-grade localized soft tissue sarcomas have an increased potential for local recurrence and metastasis. For sarcomas of the extremities, local control comparable to that obtained with amputation may be achieved with limb-sparing surgery that involves wide local excision in combination with preoperative radiation therapy (preRT) or postoperative radiation therapy (PORT).
Complete surgical resection is often difficult for sarcomas of the retroperitoneum because of their large size before detection and anatomical location.[1,2] As opposed to soft tissue sarcomas of the extremities, local recurrence is the most common cause of death in patients with retroperitoneal soft tissue sarcomas. Complete surgical resection (i.e., removal of the entire gross tumor) is the most important factor in preventing local recurrence and, in many instances, requires resection of adjacent viscera. For retroperitoneal sarcomas, retrospective comparison of surgery alone versus preRT review suggests that preRT is associated with improved local recurrence-free survival, but not disease-free survival.[3]
Standard treatment options:
- Surgical excision with preRT or PORT. Radiation decreases the risk of local recurrence but has not been shown to increase overall survival.[4-8]
- Surgical excision with negative tissue margins in all directions. This approach is generally restricted to low-grade tumors ( ≤5 cm in diameter) of the extremities or superficial trunk with microscopically negative surgical tumor margins.[9-13]
- If the tumor is unresectable, high-dose radiation therapy may be used, but poor local control is likely to result.[14]
- In some situations, radiation therapy and/or chemotherapy may be used before surgery in an attempt to convert a marginally resectable tumor to one that can be adequately resected with limb preservation; this treatment may be followed by PORT.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Heslin MJ, Lewis JJ, Nadler E, et al.: Prognostic factors associated with long-term survival for retroperitoneal sarcoma: implications for management. J Clin Oncol 15 (8): 2832-9, 1997. [PUBMED Abstract]
- Jaques DP, Coit DG, Hajdu SI, et al.: Management of primary and recurrent soft-tissue sarcoma of the retroperitoneum. Ann Surg 212 (1): 51-9, 1990. [PUBMED Abstract]
- Kelly KJ, Yoon SS, Kuk D, et al.: Comparison of Perioperative Radiation Therapy and Surgery Versus Surgery Alone in 204 Patients With Primary Retroperitoneal Sarcoma: A Retrospective 2-Institution Study. Ann Surg 262 (1): 156-62, 2015. [PUBMED Abstract]
- Yang JC, Chang AE, Baker AR, et al.: Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 16 (1): 197-203, 1998. [PUBMED Abstract]
- Rosenberg SA, Tepper J, Glatstein E, et al.: The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg 196 (3): 305-15, 1982. [PUBMED Abstract]
- O'Sullivan B, Davis AM, Turcotte R, et al.: Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 359 (9325): 2235-41, 2002. [PUBMED Abstract]
- O'Sullivan B, Davis A, Turcotte R, et al.: Five-year results of a randomized phase III trial of pre-operative vs post-operative radiotherapy in extremity soft tissue sarcoma. [Abstract] J Clin Oncol 22 (Suppl 14): A-9007, 819s, 2004.
- Davis AM, O'Sullivan B, Turcotte R, et al.: Late radiation morbidity following randomization to preoperative versus postoperative radiotherapy in extremity soft tissue sarcoma. Radiother Oncol 75 (1): 48-53, 2005. [PUBMED Abstract]
- Al-Refaie WB, Habermann EB, Jensen EH, et al.: Surgery alone is adequate treatment for early stage soft tissue sarcoma of the extremity. Br J Surg 97 (5): 707-13, 2010. [PUBMED Abstract]
- Pisters PW, Pollock RE, Lewis VO, et al.: Long-term results of prospective trial of surgery alone with selective use of radiation for patients with T1 extremity and trunk soft tissue sarcomas. Ann Surg 246 (4): 675-81; discussion 681-2, 2007. [PUBMED Abstract]
- Fabrizio PL, Stafford SL, Pritchard DJ: Extremity soft-tissue sarcomas selectively treated with surgery alone. Int J Radiat Oncol Biol Phys 48 (1): 227-32, 2000. [PUBMED Abstract]
- Rydholm A, Gustafson P, Rööser B, et al.: Limb-sparing surgery without radiotherapy based on anatomic location of soft tissue sarcoma. J Clin Oncol 9 (10): 1757-65, 1991. [PUBMED Abstract]
- Rydholm A: Surgery without radiotherapy in soft tissue sarcoma. Acta Orthop Scand Suppl 273: 117-9, 1997. [PUBMED Abstract]
- Kepka L, DeLaney TF, Suit HD, et al.: Results of radiation therapy for unresected soft-tissue sarcomas. Int J Radiat Oncol Biol Phys 63 (3): 852-9, 2005. [PUBMED Abstract]
Advanced Stage III (N1) Adult Soft Tissue Sarcoma
Refer to the Treatment Option Overview section of this summary for a more detailed discussion of the roles of surgery, radiation therapy, and chemotherapy.
Regional lymph node involvement by soft tissue sarcomas of adulthood is very infrequent. However, sarcoma types that more commonly spread to lymph nodes include high-grade rhabdomyosarcoma, vascular sarcomas, and epithelioid sarcomas.[1]
Standard treatment options:
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Mazeron JJ, Suit HD: Lymph nodes as sites of metastases from sarcomas of soft tissue. Cancer 60 (8): 1800-8, 1987. [PUBMED Abstract]
- Watson DI, Coventry BJ, Langlois SL, et al.: Soft-tissue sarcoma of the extremity. Experience with limb-sparing surgery. Med J Aust 160 (7): 412-6, 1994. [PUBMED Abstract]
- Cormier JN, Huang X, Xing Y, et al.: Cohort analysis of patients with localized, high-risk, extremity soft tissue sarcoma treated at two cancer centers: chemotherapy-associated outcomes. J Clin Oncol 22 (22): 4567-74, 2004. [PUBMED Abstract]
- O'Byrne K, Steward WP: The role of adjuvant chemotherapy in the treatment of adult soft tissue sarcomas. Crit Rev Oncol Hematol 27 (3): 221-7, 1998. [PUBMED Abstract]
- Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet 350 (9092): 1647-54, 1997. [PUBMED Abstract]
Stage IV Adult Soft Tissue Sarcoma
Refer to the Treatment Option Overview section of this summary for a more detailed discussion of the roles of surgery, radiation therapy, and chemotherapy.
In the setting of lung metastasis, resection of metastatic tumors may be associated with long-term disease-free survival in patients selected for optimal underlying disease biology (i.e., patients with a limited number of metastases and slow tumor growth).[1-3] It is not clear to what degree the favorable outcomes are attributable to the efficacy of surgery or to careful selection of patients based upon factors that are associated with less-virulent disease.[1-3] The value of resection of hepatic metastases is unclear.
As noted in the Treatment Option Overview section above, doxorubicin is the standard systemic therapy in the management of metastatic sarcomas.[4,5] Other drugs that may have clinical activity as single agents are ifosfamide, epirubicin, gemcitabine, and paclitaxel.[6-9] Their clinical activity relative to single-agent doxorubicin is not clear, and they are not known to have superior activity. There is controversy about whether adding drugs to doxorubicin offers clinical benefit beyond what is achieved by doxorubicin as a single agent. To avoid severe toxicity in older patients, sequential use of single agents may be the preferred strategy for palliation.
A randomized study assessed whether dose intensification of doxorubicin with ifosfamide improved the survival of patients with advanced soft-tissue sarcoma compared with doxorubicin alone.[10] Two hundred twenty-eight patients were randomly assigned to receive doxorubicin, and 227 patients were randomly assigned to receive doxorubicin and ifosfamide. Median follow-up was 56 months (interquartile range [IQR], 31–77) in the doxorubicin-only group and 59 months (IQR, 36–72) in the combination group.
There was no significant difference in overall survival (OS) between groups (median OS, 12.8 months; 95.5% confidence interval [CI], 10.5–14.3 in the doxorubicin-alone group vs. 14.3 months; range, 12.5–16.5 months in the doxorubicin and ifosfamide group; hazard ratio [HR], 0.83; 95.5% CI 0.67–1.03; stratified log-rank test P = .076). Median progression-free survival was significantly higher for the doxorubicin and ifosfamide group (7.4 months; 95% CI, 6.6-8.3) than for the doxorubicin-alone group (4.6 months; range, 2.9–5.6 months; HR, 0.74; 95% CI, 0.60–0.90; stratified log-rank test P = .003). More patients in the doxorubicin and ifosfamide group than in the doxorubicin-alone group had an overall response (60 [26%] of 227 patients vs. 31 [14%] of 228; P < .0006). The most common grade 3 and 4 toxic effects, which were all more common with doxorubicin and ifosfamide than with doxorubicin alone, were leucopenia (97 [43%] of 224 patients vs. 40 [18%] of 223 patients), neutropenia (93 [42%] vs. 83 [37%]), febrile neutropenia (103 (46%) vs. 30 [13%]), anemia (78 [35%] vs. 10 [5%]), and thrombocytopenia (75 [33%]) vs. 1 [<1%]).[10][Level of evidence: 1iiA] Treatment intensification with doxorubicin and ifosfamide for palliation of advanced soft tissue sarcoma is not indicated.
Standard treatment options
- Chemotherapy.
- Single-agent chemotherapy, with subsequent single agents for disease regrowth.[4-6,8,9,11] Doxorubicin is generally the first-line agent. Ifosfamide also has substantial single-agent activity.
- Doxorubicin-based combination chemotherapy. A variety of regimens have been used, but none has been proven to increase OS compared with doxorubicin alone.[4,5] There is some evidence that the addition of ifosfamide increases response rates (but not survival). Toxicity is increased with the addition of drugs to doxorubicin. No quality-of-life studies have been reported in comparisons of single-agent therapy versus combination therapy.
- Resection of pulmonary lesions may be performed if the primary tumor is under control.[1-3]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- van Geel AN, Pastorino U, Jauch KW, et al.: Surgical treatment of lung metastases: The European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group study of 255 patients. Cancer 77 (4): 675-82, 1996. [PUBMED Abstract]
- Casson AG, Putnam JB, Natarajan G, et al.: Five-year survival after pulmonary metastasectomy for adult soft tissue sarcoma. Cancer 69 (3): 662-8, 1992. [PUBMED Abstract]
- Putnam JB Jr, Roth JA: Surgical treatment for pulmonary metastases from sarcoma. Hematol Oncol Clin North Am 9 (4): 869-87, 1995. [PUBMED Abstract]
- Bramwell VH, Anderson D, Charette ML, et al.: Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev (3): CD003293, 2003. [PUBMED Abstract]
- Verma S, Younus J, Stys-Norman D, et al.: Meta-analysis of ifosfamide-based combination chemotherapy in advanced soft tissue sarcoma. Cancer Treat Rev 34 (4): 339-47, 2008. [PUBMED Abstract]
- Lorigan P, Verweij J, Papai Z, et al.: Phase III trial of two investigational schedules of ifosfamide compared with standard-dose doxorubicin in advanced or metastatic soft tissue sarcoma: a European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol 25 (21): 3144-50, 2007. [PUBMED Abstract]
- Nielsen OS, Dombernowsky P, Mouridsen H, et al.: High-dose epirubicin is not an alternative to standard-dose doxorubicin in the treatment of advanced soft tissue sarcomas. A study of the EORTC soft tissue and bone sarcoma group. Br J Cancer 78 (12): 1634-9, 1998. [PUBMED Abstract]
- Maki RG, Wathen JK, Patel SR, et al.: Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 25 (19): 2755-63, 2007. [PUBMED Abstract]
- Okuno S, Ryan LM, Edmonson JH, et al.: Phase II trial of gemcitabine in patients with advanced sarcomas (E1797): a trial of the Eastern Cooperative Oncology Group. Cancer 97 (8): 1969-73, 2003. [PUBMED Abstract]
- Judson I, Verweij J, Gelderblom H, et al.: Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol 15 (4): 415-23, 2014. [PUBMED Abstract]
- Grenader T, Goldberg A, Hadas-Halperin I, et al.: Long-term response to pegylated liposomal doxorubicin in patients with metastatic soft tissue sarcomas. Anticancer Drugs 20 (1): 15-20, 2009. [PUBMED Abstract]
Recurrent Adult Soft Tissue Sarcoma
Treatment of patients with recurrent soft tissue sarcoma depends on the type of initial presentation and treatment. Patients who develop a local recurrence often can be treated by local therapy: surgical excision plus radiation therapy after previous minimal therapy or amputation after previous aggressive treatment.[1-7] Resection of limited pulmonary metastases may be associated with favorable disease-free survival.[8-10][Level of evidence: 3iiiDiv] However, the contribution of selection factors, such as low tumor burden, slow tumor growth, and long disease-free interval, to these favorable outcomes is not known.
There is no standard chemotherapy for recurrent soft tissue sarcomas that have progressed after doxorubicin as a single agent or in combination with other agents that have clinical activity, such as ifosfamide, epirubicin, gemcitabine, and paclitaxel. Any of these agents not previously administered to the patient may be used sequentially at the time of recurrence or progression.[11-14][Level of Evidence: 3iiiDiv] None of these agents has been shown to increase overall survival in this setting, therefore, clinical trials are an appropriate option.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Singer S, Nielsen T, Antonescu CR: Molecular biology of soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1522-32.
- Singer S, Maki RG, O'Sullivan B: Soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1533-77.
- Malawer MM, Helman LJ, O'Sullivan B: Sarcomas of bone. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1578-1609.
- Midis GP, Pollock RE, Chen NP, et al.: Locally recurrent soft tissue sarcoma of the extremities. Surgery 123 (6): 666-71, 1998. [PUBMED Abstract]
- Essner R, Selch M, Eilber FR: Reirradiation for extremity soft tissue sarcomas. Local control and complications. Cancer 67 (11): 2813-7, 1991. [PUBMED Abstract]
- Singer S, Antman K, Corson JM, et al.: Long-term salvageability for patients with locally recurrent soft-tissue sarcomas. Arch Surg 127 (5): 548-53; discussion 553-4, 1992. [PUBMED Abstract]
- Lewis JJ, Leung D, Heslin M, et al.: Association of local recurrence with subsequent survival in extremity soft tissue sarcoma. J Clin Oncol 15 (2): 646-52, 1997. [PUBMED Abstract]
- van Geel AN, Pastorino U, Jauch KW, et al.: Surgical treatment of lung metastases: The European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group study of 255 patients. Cancer 77 (4): 675-82, 1996. [PUBMED Abstract]
- Casson AG, Putnam JB, Natarajan G, et al.: Five-year survival after pulmonary metastasectomy for adult soft tissue sarcoma. Cancer 69 (3): 662-8, 1992. [PUBMED Abstract]
- Putnam JB Jr, Roth JA: Surgical treatment for pulmonary metastases from sarcoma. Hematol Oncol Clin North Am 9 (4): 869-87, 1995. [PUBMED Abstract]
- Lorigan P, Verweij J, Papai Z, et al.: Phase III trial of two investigational schedules of ifosfamide compared with standard-dose doxorubicin in advanced or metastatic soft tissue sarcoma: a European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol 25 (21): 3144-50, 2007. [PUBMED Abstract]
- Nielsen OS, Dombernowsky P, Mouridsen H, et al.: High-dose epirubicin is not an alternative to standard-dose doxorubicin in the treatment of advanced soft tissue sarcomas. A study of the EORTC soft tissue and bone sarcoma group. Br J Cancer 78 (12): 1634-9, 1998. [PUBMED Abstract]
- Maki RG, Wathen JK, Patel SR, et al.: Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 25 (19): 2755-63, 2007. [PUBMED Abstract]
- Okuno S, Ryan LM, Edmonson JH, et al.: Phase II trial of gemcitabine in patients with advanced sarcomas (E1797): a trial of the Eastern Cooperative Oncology Group. Cancer 97 (8): 1969-73, 2003. [PUBMED Abstract]
Changes to This Summary (02/01/2018)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
General Information About Adult Soft Tissue Sarcoma
Updated statistics with estimated new cases and deaths for 2018 (cited American Cancer Society as reference 1).
Stage Information for Adult Soft Tissue Sarcoma
An editorial change was made to this section.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of adult soft tissue sarcoma. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Adult Soft Tissue Sarcoma Treatment are:
- Russell S. Berman, MD (New York University School of Medicine)
- Minh Tam Truong, MD (Boston University Medical Center)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Adult Soft Tissue Sarcoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/soft-tissue-sarcoma/hp/adult-soft-tissue-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389481]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Updated: February 1, 2018
Source URL: https://www.cancer.gov/publishedcontent/syndication/2127.htm
Source Agency: National Cancer Institute (NCI)
Captured Date: 2013-09-14 09:00:45.0
Adult Soft Tissue Sarcoma Treatment (PDQ®)–Health Professional Version
General Information About Adult Soft Tissue Sarcoma
Incidence and Mortality
Estimated new cases and deaths from soft tissue sarcoma in the United States in 2018:[1]
- New cases: 13,040.
- Deaths: 5,150.
Soft tissue sarcomas are malignant tumors that arise in any of the mesodermal tissues of the extremities (50%), trunk and retroperitoneum (40%), or head and neck (10%). The reported international incidence rates range from 1.8 to 5 per 100,000 individuals per year.[2]
Risk Factors and Genetic Factors
The risk of sporadic soft tissue sarcomas is increased by previous radiation therapy and, in the case of lymphangiosarcoma, by chronic lymphedema. The chemicals Thorotrast (thorium dioxide), vinyl chloride, and arsenic are also established carcinogens for hepatic angiosarcomas.[3-5]
Soft tissue sarcomas occur with greater frequency in patients with the following inherited syndromes:[3-5]
- Nevoid basal cell carcinoma syndrome (Gorlin syndrome: PTC gene mutation).
- Gardner syndrome (APC mutation).
- Li-Fraumeni syndrome (p53 mutation).
- Tuberous sclerosis (Bourneville disease: TSC1 or TSC2 mutation).
- von Recklinghausen disease (neurofibromatosis type 1: NF1 mutation).
- Werner syndrome (adult progeria: WRN mutation).
Diagnosis
Soft tissue sarcomas may be heterogeneous, so adequate tissue should be obtained via either core-needle or incisional biopsy for microscopic examination to determine histologic type and tumor grade. Careful planning of the initial biopsy is important to avoid compromising subsequent curative resection. Since the selection of treatment is determined by the grade of the tumor, it is essential to have a careful review of the biopsy tissue by a pathologist who is experienced in diagnosing sarcomas. Complete staging and treatment planning by a multidisciplinary team of cancer specialists is required to determine the optimal treatment for patients with this disease.
There is evidence that at least some favorable clinical outcomes may be associated with referral to a specialized sarcoma treatment center. In a population-based consecutive series of 375 soft tissue sarcoma patients in Sweden, local recurrence rates of resected tumors were higher in patients who were not referred to the specialized center: in 35 of 78 (45%) patients not referred; in 24 of 102 (24%) patients referred after initial surgery or incisional biopsy; and in 36 of 195 (18%) patients referred before any surgical procedure (P = .0001 for the difference between those never referred vs. those referred before any surgical procedure).[6][Level of evidence: 3iDii] However, there were no statistically significant differences in death from sarcoma between the groups of patients.
Prognostic Factors
The prognosis for patients with adult soft tissue sarcomas depends on several factors, including:[3-5,7,8]
- Patient’s age.
- Size, sarcoma subtype, histologic grade, mitotic activity, and stage of the tumor.
Factors associated with a poorer prognosis include the following:[9]
- Age older than 60 years.
- Tumors larger than 5 cm in greatest dimension.
- High-grade histology with high mitotic activity.
- Positive margins after resection.[10]
Although low-grade tumors are usually curable by surgery alone, higher-grade sarcomas (as determined by the mitotic index and by the presence of hemorrhage and necrosis) are associated with higher local-treatment failure rates and increased metastatic potential.
Surveillance for Relapse
A retrospective review included 174 consecutive patients with a soft tissue sarcoma of the limb who underwent follow-up by oncologists at a single center from 2003 to 2009.[11] The rate and site of recurrence and mode of detection were analyzed. Eighty-two patients (47%) experienced relapse. Isolated local recurrences occurred in 26 patients and local relapse with synchronous pulmonary metastases occurred in 5 patients. Local recurrences were detected clinically in 30 of the 31 patients; magnetic resonance imaging identified only one local recurrence. Twenty-eight patients developed isolated lung metastases; in 9 patients, the lung metastases were amenable to resections, 7 of whom were free of disease after treatment. Lung metastases were detected by chest x-ray in 19 patients, by computed tomography scanning in 3 patients, and clinically in 11 patients. Twenty-three patients developed nonpulmonary metastases. More than 80% of the relapses occurred in the first 2 years of follow-up; however, later recurrences were also observed.[11][Level of evidence: 3iiDi] This study supports imaging surveillance for detection of lung metastases, whereas local recurrences at the primary site were usually detected by clinical examination. The impact of picking up metastases from overall survival or quality-of-life data is unknown.
Related Summaries
Other PDQ summaries containing information about soft tissue sarcoma include:
References
- American Cancer Society: Cancer Facts and Figures 2018. Atlanta, Ga: American Cancer Society, 2018. Available online. Last accessed January 5, 2018.
- Wibmer C, Leithner A, Zielonke N, et al.: Increasing incidence rates of soft tissue sarcomas? A population-based epidemiologic study and literature review. Ann Oncol 21 (5): 1106-11, 2010. [PUBMED Abstract]
- Singer S, Nielsen T, Antonescu CR: Molecular biology of soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1522-32.
- Singer S, Maki RG, O'Sullivan B: Soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1533-77.
- Malawer MM, Helman LJ, O'Sullivan B: Sarcomas of bone. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1578-1609.
- Gustafson P, Dreinhöfer KE, Rydholm A: Soft tissue sarcoma should be treated at a tumor center. A comparison of quality of surgery in 375 patients. Acta Orthop Scand 65 (1): 47-50, 1994. [PUBMED Abstract]
- Coindre JM, Terrier P, Guillou L, et al.: Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 91 (10): 1914-26, 2001. [PUBMED Abstract]
- Kasper B, Ouali M, van Glabbeke M, et al.: Prognostic factors in adolescents and young adults (AYA) with high risk soft tissue sarcoma (STS) treated by adjuvant chemotherapy: a study based on pooled European Organisation for Research and Treatment of Cancer (EORTC) clinical trials 62771 and 62931. Eur J Cancer 49 (2): 449-56, 2013. [PUBMED Abstract]
- Vraa S, Keller J, Nielsen OS, et al.: Prognostic factors in soft tissue sarcomas: the Aarhus experience. Eur J Cancer 34 (12): 1876-82, 1998. [PUBMED Abstract]
- Trovik LH, Ovrebo K, Almquist M, et al.: Adjuvant radiotherapy in retroperitoneal sarcomas. A Scandinavian Sarcoma Group study of 97 patients. Acta Oncol 53 (9): 1165-72, 2014. [PUBMED Abstract]
- Rothermundt C, Whelan JS, Dileo P, et al.: What is the role of routine follow-up for localised limb soft tissue sarcomas? A retrospective analysis of 174 patients. Br J Cancer 110 (10): 2420-6, 2014. [PUBMED Abstract]
Cellular Classification of Adult Soft Tissue Sarcoma
Soft tissue sarcomas are classified histologically according to the soft tissue cell of origin. Additional studies, including electron microscopy, specialized immunohistochemistry, flow cytometry, cytogenetics, and tissue culture studies may allow identification of particular subtypes within the major histologic categories. For example, S100 antigen suggests neural sheath origin, cytokeratin suggests epithelioid or synovial cell origin, and factor VIII-related antigen suggests endothelial origin. Likewise, some subtypes of sarcomas have characteristic genetic markers, but these markers are not generally used in the routine clinical setting (e.g., translocation t(X;18)(p11;q11) in synovial sarcomas and translocation t(12;16)(q13;p11) in myxoid and round-cell sarcomas).[1-3]
The histologic grade reflects the metastatic potential of these tumors more accurately than the classic cellular classification listed below. Pathologists assign a grade based on the number of mitoses per high-powered field, the presence of necrosis, cellular and nuclear morphology, and the degree of cellularity; discordance among expert pathologists regarding tumor grade, and even histologic subtype, can be substantial.[4]
The World Health Organization lists the following cell types in its classification of soft tissue sarcomas:[5,6]
- Adipocytic tumors.
- Dedifferentiated liposarcoma.*
- Myxoid/round cell liposarcoma.
- Pleomorphic liposarcoma.
- Fibroblastic/myofibroblastic tumors.
- Fibrosarcoma.**
- Myxofibrosarcoma, low grade.
- Low-grade fibromyxoid sarcoma.
- Sclerosing epithelioid fibrosarcoma.
- So-called fibrohistiocytic tumors.
- Undifferentiated pleomorphic sarcoma/malignant fibrous histiocytoma (including pleomorphic, giant cell, myxoid/high-grade myxofibrosarcoma, and inflammatory forms).
- Smooth muscle tumors.
- Leiomyosarcoma.
- Skeletal muscle tumors.
- Rhabdomyosarcoma (embryonal, alveolar, and pleomorphic forms).
- Vascular tumors.
- Epithelioid hemangioendothelioma.
- Angiosarcoma, deep.***
- Tumors of peripheral nerves.
- Malignant peripheral nerve sheath tumor.
- Chondro-osseous tumors.
- Extraskeletal chondrosarcoma (mesenchymal and other variants).
- Extraskeletal osteosarcoma.
- Tumors of uncertain differentiation.
- Synovial sarcoma.
- Epithelioid sarcoma.
- Alveolar soft part sarcoma.
- Clear cell sarcoma of soft tissue.
- Extraskeletal myxoid chondrosarcoma.
- Primitive neuroectodermal tumor/extraskeletal Ewing tumor.
- Desmoplastic small round cell tumor.
- Extrarenal rhabdoid tumor.
- Undifferentiated sarcoma; sarcoma, not otherwise specified.
[Note: *It is recognized that dedifferentiated liposarcoma primarily arises in the context of deep atypical lipomatous tumor/well-differentiated liposarcoma, a sarcoma of intermediate malignancy because of the lack of metastatic capacity. **The category of fibrosarcoma can be inclusive of fibrosarcomatous differentiation in dermatofibrosarcoma protuberans. ***Cutaneous angiosarcoma may be difficult to stage using the American Joint Committee on Cancer system. (Refer to the PDQ summary on Gastrointestinal Stromal Tumors for more information.)]
References
- Singer S, Nielsen T, Antonescu CR: Molecular biology of soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1522-32.
- Singer S, Maki RG, O'Sullivan B: Soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1533-77.
- Malawer MM, Helman LJ, O'Sullivan B: Sarcomas of bone. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1578-1609.
- Alvegård TA, Berg NO: Histopathology peer review of high-grade soft tissue sarcoma: the Scandinavian Sarcoma Group experience. J Clin Oncol 7 (12): 1845-51, 1989. [PUBMED Abstract]
- Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-6.
- Brodowicz T, Schwameis E, Widder J, et al.: Intensified Adjuvant IFADIC Chemotherapy for Adult Soft Tissue Sarcoma: A Prospective Randomized Feasibility Trial. Sarcoma 4 (4): 151-60, 2000. [PUBMED Abstract]
Stage Information for Adult Soft Tissue Sarcoma
Note: The American Joint Committee on Cancer (AJCC) has published the 8th edition of the AJCC Cancer Staging Manual, which includes revisions to the staging for this disease. Implementation of the 8th edition began in January 2018. The PDQ Adult Treatment Editorial Board, which maintains this summary, is reviewing the revised staging and will make appropriate changes as needed.
Staging has an important role in determining the most effective treatment for soft tissue sarcoma. Clinical staging involves magnetic resonance imaging (MRI) or computed tomography (CT) of the primary tumor area and a chest CT to look for metastasis to the lung (the most common site of distant spread). An abdominal CT scan is done in the case of retroperitoneal sarcomas because the liver may be the site of initial clinical metastasis for these tumors.
The stage is determined by the size of the tumor, the histologic grade, and whether there is spread to lymph nodes or distant sites. Intracompartmental or extracompartmental extension of extremity sarcomas is also important for surgical decision making. For complete staging, a thorough review of all biopsy specimens (including those from the primary tumor, lymph nodes, or other suspicious lesions) is essential. CT scan of the chest is recommended for sarcomas larger than 5 cm (T2) or with moderate to poor differentiation (grades 2–4). Nodal involvement is rare, occurring in fewer than 3% of patients with sarcoma.[1]
Lymph node involvement in soft tissue sarcomas of adulthood is rare but is somewhat more frequent in some subtypes (e.g., rhabdomyosarcoma, vascular sarcomas, clear cell sarcomas, and epithelioid sarcomas) when they are high grade.[2] Because treatment decisions are predicated on pathology staging, patients should be staged before, and again after, any neoadjuvant therapy. The assessment of tumor grade can be affected in either direction, but more frequently decreased because of differential cellular loss related to the neoadjuvant chemotherapy or radiation.[3] Grade, which is based on cellular differentiation, mitotic rate, and extent of necrosis, should be recorded for all soft tissue sarcomas. A three-grade system (G1–G3) is preferred. (See below.)
The AJCC has designated staging by the four criteria of tumor size, nodal status, metastasis, and grade (TNMG).[3] The characteristic molecular markers of some sarcomas are not formally incorporated in the staging system pending further evaluation of their impact on prognosis. Recurrent sarcomas are restaged using the same system as for primary tumors with the specification that the tumor is recurrent.
Definitions of TNM and Grade
| aReprinted with permission from AJCC: Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-8. | |
| bSuperficial tumor is located exclusively above the superficial fascia without invasion of the fascia; deep tumor is located either exclusively beneath the superficial fascia, superficial to the fascia with invasion of or through the fascia, or both superficial yet beneath the fascia. | |
| TX | Primary tumor cannot be assessed. |
| T0 | No evidence of primary tumor. |
| T1 | Tumor ≤5 cm in greatest dimension. (Size should be regarded as a continuous variable, and the measurement should be provided.) |
| T1a | Superficial tumor.b |
| T1b | Deep tumor.b |
| T2 | Tumor >5 cm in greatest dimension.b |
| T2a | Superficial tumor.b |
| T2b | Deep tumor. |
| aReprinted with permission from AJCC: Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-8. | |
| bPresence of positive nodes (N1) in M0 tumors is considered Stage III. | |
| NX | Regional lymph nodes cannot be assessed. |
| N0 | No regional lymph node metastasis. |
| N1b | Regional lymph node metastasis. |
| aReprinted with permission from AJCC: Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-8. | |
| M0 | No distant metastasis. |
| M1 | Distant metastasis. |
| aReprinted with permission from AJCC: Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-8. | ||||
| Stage IA | T1a | N0 | M0 | G1, GX |
| T1b | N0 | M0 | G1, GX | |
| Stage IB | T2a | N0 | M0 | G1, GX |
| T2b | N0 | M0 | G1, GX | |
| Stage IIA | T1a | N0 | M0 | G2, G3 |
| T1b | N0 | M0 | G2, G3 | |
| Stage IIB | T2a | N0 | M0 | G2 |
| T2b | N0 | M0 | G2 | |
| Stage III | T2a, T2b | N0 | M0 | G3 |
| Any T | N1 | M0 | Any G | |
| Stage IV | Any T | Any N | M1 | Any G |
Neurovascular and bone invasion are indicators of poor prognosis, but they are not incorporated into the formal staging system.
References
- Fong Y, Coit DG, Woodruff JM, et al.: Lymph node metastasis from soft tissue sarcoma in adults. Analysis of data from a prospective database of 1772 sarcoma patients. Ann Surg 217 (1): 72-7, 1993. [PUBMED Abstract]
- Mazeron JJ, Suit HD: Lymph nodes as sites of metastases from sarcomas of soft tissue. Cancer 60 (8): 1800-8, 1987. [PUBMED Abstract]
- Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-6.
Treatment Option Overview
Multimodality Approach
In most cases, a combined modality approach of preoperative radiation therapy (preRT) or postoperative radiation therapy (PORT) is used, rather than the radical surgical procedures, such as amputation, that were used in the past. It may even be possible to use surgery without PORT in selected cases. For example, a case series was reported from a specialized sarcoma treatment referral center in which 74 selected patients with primary extremity and trunk tumors 5 cm or smaller were found to have no histologic involvement of the surgical margins. The patients were observed without radiation therapy, and the estimated local recurrence rate after 10 years was 11%.[1][Level of evidence: 3iiiDiv] The role of chemotherapy is not as well defined as is the role for radiation therapy. Because of the evolving nature of the treatment options for this disease, patients should be considered when available. Information about ongoing clinical trials is available from the NCI website.
Role of Surgery
Surgical resection is the mainstay of therapy for soft tissue sarcomas. When feasible, wide-margin function–sparing surgical excision is the cornerstone of effective treatment for extremity tumors. This may be facilitated by soft tissue reconstructive surgery, which generally permits wider margins than those obtained when the surgical plan involves direct closure of the excision site.[2] Cutting into the tumor mass or shelling out the gross tumor along the plane of the pseudocapsule of compressed tumor cells and reactive tissue that often surrounds soft tissue sarcomas are associated with an elevated risk of local recurrence. Even high-grade, soft tissue sarcomas of the extremities can usually be effectively treated while preserving the limb with combined-modality treatment consisting of preRT or PORT to reduce local recurrence. (Refer to the Role of Radiation Therapy section of this summary for more information.)
Only one small, single-institution, randomized trial has directly compared amputation to limb-sparing surgery for soft tissue sarcomas of the extremities.[3] In a 2:1 randomization ratio, 27 patients with high-grade extremity sarcomas were assigned to a wide excision plus PORT (45 Gy–50 Gy to the wide local excision area, and a total of 60 Gy–70 Gy to the tumor bed over 6–7 weeks), and 16 were assigned to amputation at or above the joint proximal to the tumor. Both groups received adjuvant chemotherapy (i.e., doxorubicin, cyclophosphamide, and high-dose methotrexate). At 63 months, with a median follow-up of 56 months, there were four local recurrences in the 27 patients who underwent limb-sparing surgery and no recurrences in the 16 patients who underwent amputation P2 = .12. Overall survival (OS) rates were not statistically significantly different (actuarial 5-year survival rate, 83% vs. 88%, P2 = .99).[3][Level of evidence: 1iiA]
Local control of high-grade soft tissue sarcomas of the trunk and the head and neck can be achieved with surgery in combination with radiation therapy.[4] It may be possible to use surgery without PORT in selected cases. For example, a case series was reported from a specialized sarcoma treatment referral center in which 74 selected patients with primary extremity and trunk tumors 5 cm or smaller were found to have no histologic involvement of the surgical margins.[1] They were observed without radiation therapy, and the estimated local recurrence rate after 10 years was 11%.[1][Level of evidence: 3iiiDiv] The role of chemotherapy is not as well defined as is the role of radiation therapy. Because of the evolving nature of the treatment options for this disease, patients should be offered the option of clinical trials when available.
Effective treatment of retroperitoneal sarcomas requires removal of all gross disease while sparing adjacent viscera not invaded by tumor. The prognosis for patients with high-grade retroperitoneal sarcomas is less favorable than for patients with tumors at other sites, partly because of the difficulty in completely resecting these tumors and the dose-limiting toxicity of high-dose radiation therapy on visceral organs.[5-8]
In the setting of distant metastasis, surgery may be associated with long-term, disease-free survival in patients with pulmonary metastasis and optimal underlying disease biology (i.e., patients with a limited number of metastases and slow nodule growth) who have undergone or are undergoing complete resection of the primary tumor.[9-11] It is not clear to what degree the favorable outcomes are attributable to the efficacy of surgery or the careful selection of patients based on factors that are associated with less-virulent disease.
Role of Radiation Therapy
Radiation plays an important role in limb-sparing therapy. Pre- and postoperative external-beam radiation therapies (EBRT), as well as brachytherapy, have been shown to decrease the risk of local recurrence. They have not been shown to increase OS but are used to avoid amputation for all but the most locally advanced tumors or for limbs seriously compromised by vascular disease, where acceptable functional preservation is not possible. In the case of EBRT, irradiation of the entire limb circumference is avoided to preserve vascular and nerve structures that are critical to function and preservation of the limb.
PORT
PORT has been tested in a single-institution, randomized trial of 141 patients with extremity sarcomas who were treated with limb-sparing surgery. Patients with high-grade tumors (n = 91) also received adjuvant chemotherapy (i.e., five 28-day cycles of doxorubicin and cyclophosphamide). All patients were randomly assigned to receive radiation (45 Gy to a wide field, plus a tumor-bed boost of 18 Gy over 6–7 weeks), concurrent with chemotherapy in the case of high-grade tumors versus no radiation.[12] At up to 12 years of follow-up, there was one local recurrence in the 70 patients randomly assigned to receive radiation versus 17 recurrences in the 71 control patients (P = .0001), with similar reduction in risk of local recurrence for both high- and low-grade tumors. However, there was no difference in OS between the radiation and control groups.[12][Level of evidence: 1iiDiii] Global quality of life was similar in the two groups, but the radiation therapy group had substantially worse functional deficits resulting from reduced strength and joint motion as well as increased edema.
To limit acute toxicity with preRT, smaller fields and lower doses are generally given than is the case with PORT. PreRT has been directly compared with PORT for extremity soft tissue sarcomas in a multicenter randomized trial.[13-15] Designed to include 266 patients, the trial was stopped early after 190 patients had been accrued because of an increase in wound complications in the preRT group. The scheduled radiation in the preRT group was a wide field of 50 Gy in 2-Gy fractions (first phase of the trial) with an additional 16 Gy to 20 Gy to the tumor bed and a 2-cm margin (second phase of the trial) only if tumor cells were found at the surgical margins.
Patients in the PORT group were scheduled to receive radiation during both phases of the trial. The wound-complication rates were 35% versus 17% in the preRT and PORT groups, respectively (P = .01). In addition, limb function at 6 weeks after surgery was worse in the preRT group (P = .01).[13] At 5 years, the two groups had similar local control rates (93% vs. 92%) and OS (73% vs. 67%, P = .48).[14] Of the 129 patients evaluated for limb function at 21 to 27 months after surgery (n = 73 for preRT and n = 56 for PORT), limb function was similar in both groups, but there was a statistical trend for less fibrosis in the preRT group (P = .07).[15]
Brachytherapy
Brachytherapy has also been investigated as an adjuvant therapy for soft tissue sarcomas. Although it has possible advantages of convenience and less radiation to normal surrounding tissue relative to EBRT, the two treatment strategies have not been directly compared in terms of efficacy or morbidity. However, adjuvant brachytherapy has been compared with surgery without radiation. The time interval between preRT and surgical excision in extremity soft tissue sarcoma had minimal influence on the development of wound complications. Four- or 5-week intervals showed equivalent complication rates between patients who did or did not develop wound complications, suggesting an optimal interval to reduce potential complications.[16]
In a single-institution trial, 164 patients with sarcomas of the extremity or superficial trunk were randomly assigned during surgery, if all gross tumor could be excised, to receive an iridium Ir 192 implant (delivering 42 Gy–45 Gy over 4–6 days; 78 patients) or to a control arm of no radiation (86 patients).[17,18] Some of the patients with high-grade tumors received adjuvant doxorubicin-based chemotherapy if they were thought to be at a high risk for metastasis (34 patients in each study arm). With a median follow-up of 76 months, the 5-year actuarial local recurrence rates were 18% and 31% in the brachytherapy and control arms, respectively (P = .04). This difference was limited to patients with high-grade tumors. There was no discernible difference in sarcoma-specific survival rates between the brachytherapy and control arms (84% and 81%, respectively; P = .65), and there was no difference in the high tumor-grade group.[17][Level of evidence: 1iiDiii] The rates of clinically important wound complications (e.g., need for operative revision or repeated seroma drainage, wound separation, large hematomas, or purulent infection) were 24% and 14% in the radiation and control arms, respectively (P = .13); wound reoperation rates were 10% and 0%, respectively (P = .006).[18]
Intensity-modulated radiation therapy
Intensity-modulated radiation therapy (IMRT) has been used to deliver preRT or PORT to patients with extremity soft tissue sarcomas in an effort to spare the femur, joints, and selected other normal tissues from the full prescription dose and to maintain local control while potentially reducing radiation therapy-related morbidity. Initial single-institution reports suggest that high rates of local control with some reduction in morbidity are possible with this technique.[19,20] Retrospective comparison of IMRT compared with 3-dimensional, conformal radiation therapy demonstrates that local recurrence for primary soft tissue sarcomas of the extremity was worse in the non-IMRT group.[21][Level of evidence: 3iiiDiv]
Surgery and radiation therapy
In some tumors of the extremities or trunk, surgery alone can be performed without the use of radiation. Evidence for this approach is limited to single-institution, relatively small, case series [1,22,23] or analysis of outcomes in the National Cancer Institute's Surveillance, Epidemiology, and End Results (SEER) tumor registry.[24] However, these comparisons suffer from low statistical power and differential evaluability rates that could have introduced bias.[1] Patient selection factors may vary among surgeons. In general, this approach is considered in patients with low-grade tumors of the extremity or superficial trunk that are 5 cm or smaller in diameter (T1) and have microscopically negative surgical margins; long-term local tumor control is about 90% in such patients.[25]
A patterns-of-care study using SEER data was queried to identify patients undergoing surgery for truncal and extremity soft tissue sarcomas from 2004 to 2009.[26] Of 5,075 patients, 50% received radiation therapy. Radiation was considered to be underused in a significant portion of patients undergoing treatment for soft tissue sarcoma in the United States. Although routine radiation therapy is not recommended for stage I patients, 25% of them still underwent radiation. Even though routine radiation therapy is recommended for patients with stage II and III tumors, only 60% of them underwent radiation. On multivariate analysis, predictors of radiation therapy included age younger than 50 years (odds ratio [OR], 1.57; 95% confidence interval [CI], 1.28–1.91), malignant fibrous histiocytoma histology (OR, 1.47; 95% CI, 1.3–1.92), T2 classification (OR, 1.88; 95% CI, 1.60–2.20), and G3 (OR, 6.27; 95% CI, 5.10–7.72). Patients with stage III soft tissue sarcoma who received radiation therapy showed improved disease-specific survival at 5 years compared with those who did not (68% vs. 46%, P < .001).[26][Level of evidence: 3iDii]
On occasion, surgical excision cannot be performed in the initial management of soft tissue sarcomas because the morbidity would be unacceptable or nearby critical organs make complete resection impossible. In such circumstances, radiation has been used as the primary therapy.[27] However, this must be considered a treatment of last resort. Experience is limited to retrospective case series from single centers.[27][Level of evidence: 3iiiDiv]
Role of Adjuvant or Neoadjuvant Chemotherapy for Clinically Localized Tumors
The role of adjuvant chemotherapy is not completely clear. The investigation of its use falls into two categories or generations—pre- and post-ifosfamide regimens. In discussions with a patient, any potential benefits should be considered in the context of the short- and long-term toxicities of the chemotherapy.
First-generation trials (preifosfamide)
Several prospective, randomized trials were unable to determine conclusively whether doxorubicin-based adjuvant chemotherapy benefits adults with resectable soft tissue sarcomas. The majority of these studies accrued small numbers of patients and did not demonstrate a metastasis-free survival or an OS benefit for adjuvant chemotherapy.[4] A small study of adjuvant chemotherapy showed a positive effect on both disease-free survival (DFS) and OS in patients treated with postoperative chemotherapy.[28] There was wide interstudy variability among the reported trials, including differences in therapeutic regimens, drug doses, sample size, tumor site, and histologic grade.
A quantitative meta-analysis of updated data from 1,568 individual patients in 14 trials of doxorubicin-based adjuvant therapy showed an absolute benefit from adjuvant therapy of 6% for a local relapse-free interval (95% CI, 1%–10%), 10% for a distant relapse-free interval (95% CI, 5%–15%), and 10% for recurrence-free survival (95% CI, 5%–15%). A statistically significant OS benefit at 10 years was not detected: absolute difference 4% (95% CI, -1%–+9%).[29,30][Level of evidence: 1iiDii] However, only a small proportion of patients in this meta-analysis were treated with ifosfamide, an agent with demonstrated activity against soft tissue sarcoma. In addition, a subset analysis suggested that patients with sarcomas of the extremities may have benefited from adjuvant chemotherapy (hazard ratio [HR] for death, 0.8, P = .029), but there was no clear evidence that patients with extremity sarcomas had outcomes that were statistically significantly different from the outcomes of patients with tumors at other sites (P = .58).[30]
Second-generation trials (postifosfamide)
Subsequent chemotherapy trials were performed using anthracycline and ifosfamide combinations in patients who primarily had extremity or truncal soft tissue sarcomas. The data are conflicting, and the issue is still not settled. In a small feasibility study, 59 patients with high-risk, soft tissue sarcomas, 58 of whom had an extremity or the trunk as the primary site, underwent primary resection plus PORT and were randomly assigned to observation versus a dose-dense regimen of six 14-day courses of ifosfamide, dacarbazine (DTIC), and doxorubicin (IFADIC regimen) with granulocyte colony-stimulating factor (G-CSF) bone marrow support and mesna uroprotection.[31] There were no statistically significant differences in OS or relapse-free survival (RFS), but the study was severely underpowered.
In a second trial performed by the Italian National Council for Research, high-risk patients were treated with local therapy (i.e., wide resection plus preRT or PORT, or amputation as clinically necessary) and were then randomly assigned to observation versus five 21-day cycles of 4-epidoxorubicin (epirubicin) plus ifosfamide (with mesna and G-CSF).[28,32] Based on power calculations, the planned study size was 190 patients, but the trial was stopped after 104 patients had been entered because an interim analysis revealed a statistically significant (P = .001) difference in DFS favoring the chemotherapy arm. By the time of the initial peer-reviewed report of the study, the DFS still favored the chemotherapy group (median DFS of 48 months vs. 16 months), but the P value had risen to .04.[28]
Although there was no difference in metastasis-free survival at the time of the report, there was an improvement in median OS (75 months vs. 46 months, P = .03). However, at the follow-up report (at a median of 89.6 months in a range of 56–119 months), OS differences were no longer statistically significant (58.5% vs. 43.1% [P = .07]). The DFS difference had also lost statistical significance (47.2% vs. 16.0% [P = .09]).[32] In summary, the trial was underpowered because it was stopped early, and the early promising results that led to stopping the trial diminished as the trial matured.
In a third, underpowered, single-center trial, 88 patients with high-risk, soft tissue sarcomas (64 of whom had extremity or truncal primary tumors) underwent surgery (with or without radiation) and were then randomly assigned to receive four 21-day cycles of chemotherapy (epirubicin [n = 26] or epirubicin plus ifosfamide [n = 19]) versus no adjuvant chemotherapy (n = 43).[33] The trial was closed prematurely because of a slow accrual rate. After a median follow-up of 94 months, the 5-year DFS in the chemotherapy and control arms was 69% versus 44%, respectively (P = .01); the 5-year OS rates were 72% versus 47% (P = .06). All of the benefit associated with chemotherapy appeared restricted to the 19 patients who received epirubicin plus ifosfamide.
In yet another underpowered trial, 137 patients with high-risk, soft tissue sarcomas (93% with extremity or truncal primary tumors) who met the eligibility criteria were randomly assigned to undergo surgical resection (with or without radiation) or to receive three preoperative 21-day cycles of doxorubicin plus ifosfamide.[34] This multicenter European Organization for Research and Treatment of Cancer trial (EORTC-62874) was closed because of slow accrual and results that were not promising enough to continue. With a median follow-up of 7.3 years, the 5-year DFS in the surgery alone and chemotherapy plus surgery arms was 52% and 56%, respectively (P = .35); and OS was 64% and 65%, respectively (P = .22).
These last four trials have been combined with the 14 first-generation trials in a trial-level meta-analysis.[35] Of the 18 randomized trials of patients with resectable soft tissue sarcomas, five trials used a combination of doxorubicin (50–90 mg/m2 per cycle) plus ifosfamide (1,500–5,000 mg/m2 per cycle). The remaining 13 trials used doxorubicin (50–70 mg/m2 per cycle) alone or with other drugs. The absolute risk reduction in local recurrence rates associated with any chemotherapy added to local therapy was 4 percentage points (95% CI, 0%–7%), and it was 5 percentage points (95% CI, 1%–12%) when ifosfamide was combined with doxorubicin. The absolute reduction in overall mortality was 6 percentage points with any chemotherapy (95% CI, 2%–11%; [i.e., a reduction from 46%–40%]), 11 percentage points for doxorubicin plus ifosfamide (95% CI, 3%–19%; [i.e., a reduction from 41%–30%]), and 5 percentage points for doxorubicin without ifosfamide.[35][Level of evidence: 1iiA]
An additional multicenter randomized trial (EORTC-62931 [NCT00002641]), the largest trial reported to date using adjuvant doxorubicin (75 mg/m2) plus ifosfamide (5,000 mg/m2), was subsequently published in abstract form and was not included in the above meta-analysis.[36] The results differed from those reported in the meta-analysis.[35] After local therapy, 351 patients were randomly assigned to five 21-day cycles of adjuvant therapy versus observation. The trial was stopped for futility because the 5-year RFS was 52% in both arms. OS was 64% in the chemotherapy arm versus 69% in the observation arm. In a subsequent abstract, the EORTC investigators reported a combined analysis of this trial and their previous trial (EORTC-62771) [37] of adjuvant cyclophosphamide plus doxorubicin plus DTIC (CYVADIC), representing the two largest trials of adjuvant therapy for adult soft tissue sarcoma in the literature.[38] The combined analysis showed no improvement in either RFS or OS associated with adjuvant chemotherapy.[38][Level of evidence: 1iiA]
In summary, the impact of adjuvant chemotherapy on survival is not clear but is likely to be small in absolute magnitude. Therefore, in discussions with a patient, any potential benefits should be considered in the context of the short- and long-term toxicities of the chemotherapy.
Role of regional hyperthermia
The use of regional hyperthermia to enhance the local effects of systemic chemotherapy in the neoadjuvant and adjuvant setting is under investigation. In a multicenter phase III trial, 341 patients with high-risk (tumor ≥5 cm, grade 2–3, and deep to fascia), soft tissue sarcomas (149 extremity tumors and 192 nonextremity tumors) were randomly allocated to receive four 21-day cycles of chemotherapy (etoposide 125 mg/m2 on days 1 and 4; ifosfamide 1,500 mg/m2 on days 1–4; doxorubicin 50 mg/m2 on day 1) with or without regional hyperthermia both before and after local therapy.[39] Approximately 11% of the patients were being treated for recurrent tumors. The regional hyperthermia was designed to produce tumor temperatures of 42°C for 60 minutes and was given on days 1 and 4 of each chemotherapy cycle. After the first four cycles of chemotherapy, definitive surgical excision of the tumor was performed, if possible, followed by radiation therapy, if indicated (i.e., a 52.7 Gy median dose delivered), and then the last four cycles of chemotherapy plus or minus hyperthermia. Three of the nine treatment centers with particular expertise in hyperthermia treated 91% of the patients in the trial.
The median duration of follow-up was 34 months. Local progression occurred in 56 patients in the hyperthermia group and 76 patients in the control group. The relative HR for local progression or death was 0.58 (95% CI, 0.41–0.84), with an absolute difference at 2 years of 15% (76% vs. 61%; 95% CI of the difference 6–26). The decreased risk of local progression or death was seen in both extremity and nonextremity tumors. However, hyperthermia had no effect on distant failure rates nor was there a statistically significant effect on OS (HR, .88, 95% CI, 0.64–1.21; P = .43).[39][Level of evidence: 1iiDiii] There was a higher rate of grade 3 to 4 leucopenia in the hyperthermia group: 77.6% versus 63.5% (P = .005). Since a large proportion of the patients were treated at centers with special expertise, there is no certainty that the finding can be generalized to apply to other settings.
Role of isolated limb perfusion
Isolated limb perfusion is under investigation as a means to deliver high doses of chemotherapy and permit limb salvage in unresectable primary or recurrent extremity soft tissue sarcomas that would otherwise require amputation, in the opinion of the surgeon.[40,41] Common drugs used in the procedure are TNF-alpha, melphalan, and interferon-gamma. Experience is limited to case series with response rates and reported avoidance of amputation as the outcome.[40,41][Level of evidence: 3iiiDiv] The technique requires specialized expertise to avoid severe local and systemic toxicity including systemic effects of TNF-alpha. The technique has not been directly compared with standard approaches using combined systemic and local therapy.
Role of chemotherapy for advanced disease
Doxorubicin is a mainstay of systemic therapy in the management of locally advanced and metastatic soft tissue sarcoma. Pegylated liposomal encapsulated doxorubicin is a formulation of doxorubicin designed to prolong the half-life of circulating doxorubicin and slow the release of active drugs.[42] The changed pharmacokinetics result in less myelosuppression and possibly less cardiotoxic effects, but there is a substantial incidence of hypersensitivity-like reactions and hand-foot syndrome. Its clinical activity relative to unencapsulated doxorubicin is not clear.[42][Level of evidence: 3iiiDiv] Other drugs that are thought to have clinical activity as single agents are ifosfamide, epirubicin, gemcitabine, and paclitaxel.[43-46][Level of Evidence: 3iiiDiv] Their clinical activity relative to single-agent doxorubicin is not clear, and they are not known to have superior activity.
There is controversy about the clinical benefit of adding other drugs to doxorubicin as a single agent. A systematic evidence review and meta-analysis conducted by the Cochrane Collaboration summarized the eight randomized trials reported from 1976 to 1995.[47] No additional randomized trials had been reported or were known to be in progress between 1995 and the 2002 literature search. Single-agent doxorubicin had been compared with a variety of doxorubicin-containing combinations that included vincristine, vindesine, cyclophosphamide, streptozotocin, mitomycin-C, cisplatin, and/or ifosfamide. Combination regimens consistently caused more nausea and hematologic toxicity. However, the better response rates associated with combination therapy were marginal and depended on the statistical model used (fixed effects model ORresponse = 1.29; 95% CI, 1.03–1.60, P = .03; random effects model ORresp = 1.26; 95% CI, 0.96–1.67, P = .10) There was no statistically significant difference in the 1- (ORmortality = 0.87; 95% CI, 0.73–1.05, P = .14) or 2-year mortality rates (ORmortality = 0.84; 95% CI, 0.67–1.06, P = .13).
These results were very similar even when the analyses were restricted to the four trials that used DTIC and/or ifosfamide as part of the combination regimen with doxorubicin agents that were postulated to have greater activity than the others tested. A subsequent meta-analysis of all three published randomized trials of chemotherapy regimens that contained ifosfamide versus those that did not came to similar conclusions: tumor response rates were better when the regimen included ifosfamide (RRresponse = 1.52; 95% CI, 1.11–2.08), but mortality at 1 year was not (RRmortality = 0.98; 95% CI, 0.85–1.13).[48][Level of evidence: 1iiDiv]. Therefore, response rate was a poor surrogate for OS. Quality-of-life outcomes were not reported in any of the above-mentioned randomized trials, but toxicity was worse when agents were added to doxorubicin.
References
- Pisters PW, Pollock RE, Lewis VO, et al.: Long-term results of prospective trial of surgery alone with selective use of radiation for patients with T1 extremity and trunk soft tissue sarcomas. Ann Surg 246 (4): 675-81; discussion 681-2, 2007. [PUBMED Abstract]
- Lohman RF, Nabawi AS, Reece GP, et al.: Soft tissue sarcoma of the upper extremity: a 5-year experience at two institutions emphasizing the role of soft tissue flap reconstruction. Cancer 94 (8): 2256-64, 2002. [PUBMED Abstract]
- Rosenberg SA, Tepper J, Glatstein E, et al.: The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg 196 (3): 305-15, 1982. [PUBMED Abstract]
- O'Byrne K, Steward WP: The role of adjuvant chemotherapy in the treatment of adult soft tissue sarcomas. Crit Rev Oncol Hematol 27 (3): 221-7, 1998. [PUBMED Abstract]
- Singer S, Nielsen T, Antonescu CR: Molecular biology of soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1522-32.
- Singer S, Maki RG, O'Sullivan B: Soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1533-77.
- Malawer MM, Helman LJ, O'Sullivan B: Sarcomas of bone. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1578-1609.
- Lewis JJ, Leung D, Woodruff JM, et al.: Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg 228 (3): 355-65, 1998. [PUBMED Abstract]
- van Geel AN, Pastorino U, Jauch KW, et al.: Surgical treatment of lung metastases: The European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group study of 255 patients. Cancer 77 (4): 675-82, 1996. [PUBMED Abstract]
- Casson AG, Putnam JB, Natarajan G, et al.: Five-year survival after pulmonary metastasectomy for adult soft tissue sarcoma. Cancer 69 (3): 662-8, 1992. [PUBMED Abstract]
- Putnam JB Jr, Roth JA: Surgical treatment for pulmonary metastases from sarcoma. Hematol Oncol Clin North Am 9 (4): 869-87, 1995. [PUBMED Abstract]
- Yang JC, Chang AE, Baker AR, et al.: Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 16 (1): 197-203, 1998. [PUBMED Abstract]
- O'Sullivan B, Davis AM, Turcotte R, et al.: Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 359 (9325): 2235-41, 2002. [PUBMED Abstract]
- O'Sullivan B, Davis A, Turcotte R, et al.: Five-year results of a randomized phase III trial of pre-operative vs post-operative radiotherapy in extremity soft tissue sarcoma. [Abstract] J Clin Oncol 22 (Suppl 14): A-9007, 819s, 2004.
- Davis AM, O'Sullivan B, Turcotte R, et al.: Late radiation morbidity following randomization to preoperative versus postoperative radiotherapy in extremity soft tissue sarcoma. Radiother Oncol 75 (1): 48-53, 2005. [PUBMED Abstract]
- Griffin AM, Dickie CI, Catton CN, et al.: The influence of time interval between preoperative radiation and surgical resection on the development of wound healing complications in extremity soft tissue sarcoma. Ann Surg Oncol 22 (9): 2824-30, 2015. [PUBMED Abstract]
- Pisters PW, Harrison LB, Leung DH, et al.: Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol 14 (3): 859-68, 1996. [PUBMED Abstract]
- Alektiar KM, Zelefsky MJ, Brennan MF: Morbidity of adjuvant brachytherapy in soft tissue sarcoma of the extremity and superficial trunk. Int J Radiat Oncol Biol Phys 47 (5): 1273-9, 2000. [PUBMED Abstract]
- Alektiar KM, Brennan MF, Healey JH, et al.: Impact of intensity-modulated radiation therapy on local control in primary soft-tissue sarcoma of the extremity. J Clin Oncol 26 (20): 3440-4, 2008. [PUBMED Abstract]
- Alektiar KM, Brennan MF, Singer S: Local control comparison of adjuvant brachytherapy to intensity-modulated radiotherapy in primary high-grade sarcoma of the extremity. Cancer 117 (14): 3229-34, 2011. [PUBMED Abstract]
- Folkert MR, Singer S, Brennan MF, et al.: Comparison of local recurrence with conventional and intensity-modulated radiation therapy for primary soft-tissue sarcomas of the extremity. J Clin Oncol 32 (29): 3236-41, 2014. [PUBMED Abstract]
- Fabrizio PL, Stafford SL, Pritchard DJ: Extremity soft-tissue sarcomas selectively treated with surgery alone. Int J Radiat Oncol Biol Phys 48 (1): 227-32, 2000. [PUBMED Abstract]
- Rydholm A, Gustafson P, Rööser B, et al.: Limb-sparing surgery without radiotherapy based on anatomic location of soft tissue sarcoma. J Clin Oncol 9 (10): 1757-65, 1991. [PUBMED Abstract]
- Al-Refaie WB, Habermann EB, Jensen EH, et al.: Surgery alone is adequate treatment for early stage soft tissue sarcoma of the extremity. Br J Surg 97 (5): 707-13, 2010. [PUBMED Abstract]
- Rydholm A: Surgery without radiotherapy in soft tissue sarcoma. Acta Orthop Scand Suppl 273: 117-9, 1997. [PUBMED Abstract]
- Bagaria SP, Ashman JB, Daugherty LC, et al.: Compliance with National Comprehensive Cancer Network guidelines in the use of radiation therapy for extremity and superficial trunk soft tissue sarcoma in the United States. J Surg Oncol 109 (7): 633-8, 2014. [PUBMED Abstract]
- Kepka L, DeLaney TF, Suit HD, et al.: Results of radiation therapy for unresected soft-tissue sarcomas. Int J Radiat Oncol Biol Phys 63 (3): 852-9, 2005. [PUBMED Abstract]
- Frustaci S, Gherlinzoni F, De Paoli A, et al.: Adjuvant chemotherapy for adult soft tissue sarcomas of the extremities and girdles: results of the Italian randomized cooperative trial. J Clin Oncol 19 (5): 1238-47, 2001. [PUBMED Abstract]
- Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet 350 (9092): 1647-54, 1997. [PUBMED Abstract]
- Sarcoma Meta-analysis Collaboration (SMAC): Adjuvant chemotherapy for localised resectable soft tissue sarcoma in adults. Cochrane Database Syst Rev (4): CD001419, 2000. [PUBMED Abstract]
- Brodowicz T, Schwameis E, Widder J, et al.: Intensified Adjuvant IFADIC Chemotherapy for Adult Soft Tissue Sarcoma: A Prospective Randomized Feasibility Trial. Sarcoma 4 (4): 151-60, 2000. [PUBMED Abstract]
- Frustaci S, De Paoli A, Bidoli E, et al.: Ifosfamide in the adjuvant therapy of soft tissue sarcomas. Oncology 65 (Suppl 2): 80-4, 2003. [PUBMED Abstract]
- Petrioli R, Coratti A, Correale P, et al.: Adjuvant epirubicin with or without Ifosfamide for adult soft-tissue sarcoma. Am J Clin Oncol 25 (5): 468-73, 2002. [PUBMED Abstract]
- Gortzak E, Azzarelli A, Buesa J, et al.: A randomised phase II study on neo-adjuvant chemotherapy for 'high-risk' adult soft-tissue sarcoma. Eur J Cancer 37 (9): 1096-103, 2001. [PUBMED Abstract]
- Pervaiz N, Colterjohn N, Farrokhyar F, et al.: A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 113 (3): 573-81, 2008. [PUBMED Abstract]
- Woll PJ, van Glabbeke M, Hohenberger P, et al.: Adjuvant chemotherapy (CT) with doxorubicin and ifosfamide in resected soft tissue sarcoma (STS): Interim analysis of a randomised phase III trial. [Abstract] J Clin Oncol 25 (Suppl 18): A-10008, 2007.
- Bramwell V, Rouesse J, Steward W, et al.: Adjuvant CYVADIC chemotherapy for adult soft tissue sarcoma--reduced local recurrence but no improvement in survival: a study of the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. J Clin Oncol 12 (6): 1137-49, 1994. [PUBMED Abstract]
- Le Cesne A, Van Glabbeke M, Woll PJ, et al.: The end of adjuvant chemotherapy (adCT) era with doxorubicin-based regimen in resected high-grade soft tissue sarcoma (STS): pooled analysis of the two STBSG-EORTC phase III clinical trials. [Abstract] J Clin Oncol 26 (Suppl 15): A-10525, 2008.
- Issels RD, Lindner LH, Verweij J, et al.: Neo-adjuvant chemotherapy alone or with regional hyperthermia for localised high-risk soft-tissue sarcoma: a randomised phase 3 multicentre study. Lancet Oncol 11 (6): 561-70, 2010. [PUBMED Abstract]
- Eggermont AM, de Wilt JH, ten Hagen TL: Current uses of isolated limb perfusion in the clinic and a model system for new strategies. Lancet Oncol 4 (7): 429-37, 2003. [PUBMED Abstract]
- Bonvalot S, Laplanche A, Lejeune F, et al.: Limb salvage with isolated perfusion for soft tissue sarcoma: could less TNF-alpha be better? Ann Oncol 16 (7): 1061-8, 2005. [PUBMED Abstract]
- Grenader T, Goldberg A, Hadas-Halperin I, et al.: Long-term response to pegylated liposomal doxorubicin in patients with metastatic soft tissue sarcomas. Anticancer Drugs 20 (1): 15-20, 2009. [PUBMED Abstract]
- Lorigan P, Verweij J, Papai Z, et al.: Phase III trial of two investigational schedules of ifosfamide compared with standard-dose doxorubicin in advanced or metastatic soft tissue sarcoma: a European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol 25 (21): 3144-50, 2007. [PUBMED Abstract]
- Nielsen OS, Dombernowsky P, Mouridsen H, et al.: High-dose epirubicin is not an alternative to standard-dose doxorubicin in the treatment of advanced soft tissue sarcomas. A study of the EORTC soft tissue and bone sarcoma group. Br J Cancer 78 (12): 1634-9, 1998. [PUBMED Abstract]
- Maki RG, Wathen JK, Patel SR, et al.: Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 25 (19): 2755-63, 2007. [PUBMED Abstract]
- Okuno S, Ryan LM, Edmonson JH, et al.: Phase II trial of gemcitabine in patients with advanced sarcomas (E1797): a trial of the Eastern Cooperative Oncology Group. Cancer 97 (8): 1969-73, 2003. [PUBMED Abstract]
- Bramwell VH, Anderson D, Charette ML, et al.: Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev (3): CD003293, 2003. [PUBMED Abstract]
- Verma S, Younus J, Stys-Norman D, et al.: Meta-analysis of ifosfamide-based combination chemotherapy in advanced soft tissue sarcoma. Cancer Treat Rev 34 (4): 339-47, 2008. [PUBMED Abstract]
Stage I Adult Soft Tissue Sarcoma
Refer to the Treatment Option Overview section of this summary for a more detailed discussion of the roles of surgery and radiation therapy.
Low-grade soft tissue sarcomas have little metastatic potential, but they have a propensity to recur locally. Accordingly, surgical excision with negative tissue margins of 1 cm to 2 cm or larger in all directions is the treatment of choice for patients with these early-stage sarcomas.[1-3] The Mohs surgical technique may be considered as an alternative to wide surgical excision for the very rare, small, well-differentiated primary sarcomas of the skin when cosmetic results are considered to be important, as margins can be assured with minimal normal tissue removal.[4]
Carefully executed high-dose radiation therapy using a shrinking-field technique may be beneficial for unresectable tumors or for resectable tumors in which a high likelihood of residual disease is thought to be present when margins are judged to be inadequate, and when wider resection would require either an amputation or the removal of a vital organ.[5] Because of the low metastatic potential of these tumors, chemotherapy is usually not given.[6,7]
Standard treatment options:
- Surgical excision of tumors 5 cm or smaller in diameter with negative tissue margins in all directions.[8-12]
- Surgical excision with preoperative radiation therapy (preRT) or postoperative radiation therapy (PORT). Radiation decreases the risk of local recurrence but has not been shown to increase overall survival.[13-16]
- If the tumor is unresectable, high-dose preRT may be used.[17]
- For tumors of the retroperitoneum, trunk, and head and neck, the following are options:
- Surgical resection with the option of PORT if negative margins cannot be obtained. Wide margins are unusual in these sites, and radiation therapy is usually advocated for trunk and head and neck primary sites.[18]
- PreRT followed by maximal surgical resection. Radiation therapy may be used in sarcomas of the trunk and head and neck to maximize local control because of the inability to obtain wide surgical margins.[19]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Singer S, Nielsen T, Antonescu CR: Molecular biology of soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1522-32.
- Singer S, Maki RG, O'Sullivan B: Soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1533-77.
- Malawer MM, Helman LJ, O'Sullivan B: Sarcomas of bone. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1578-1609.
- Fish FS: Soft tissue sarcomas in dermatology. Dermatol Surg 22 (3): 268-73, 1996. [PUBMED Abstract]
- Temple WJ, Temple CL, Arthur K, et al.: Prospective cohort study of neoadjuvant treatment in conservative surgery of soft tissue sarcomas. Ann Surg Oncol 4 (7): 586-90, 1997 Oct-Nov. [PUBMED Abstract]
- Sarcoma Meta-analysis Collaboration (SMAC): Adjuvant chemotherapy for localised resectable soft tissue sarcoma in adults. Cochrane Database Syst Rev (4): CD001419, 2000. [PUBMED Abstract]
- Pervaiz N, Colterjohn N, Farrokhyar F, et al.: A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer 113 (3): 573-81, 2008. [PUBMED Abstract]
- Al-Refaie WB, Habermann EB, Jensen EH, et al.: Surgery alone is adequate treatment for early stage soft tissue sarcoma of the extremity. Br J Surg 97 (5): 707-13, 2010. [PUBMED Abstract]
- Pisters PW, Pollock RE, Lewis VO, et al.: Long-term results of prospective trial of surgery alone with selective use of radiation for patients with T1 extremity and trunk soft tissue sarcomas. Ann Surg 246 (4): 675-81; discussion 681-2, 2007. [PUBMED Abstract]
- Fabrizio PL, Stafford SL, Pritchard DJ: Extremity soft-tissue sarcomas selectively treated with surgery alone. Int J Radiat Oncol Biol Phys 48 (1): 227-32, 2000. [PUBMED Abstract]
- Rydholm A, Gustafson P, Rööser B, et al.: Limb-sparing surgery without radiotherapy based on anatomic location of soft tissue sarcoma. J Clin Oncol 9 (10): 1757-65, 1991. [PUBMED Abstract]
- Rydholm A: Surgery without radiotherapy in soft tissue sarcoma. Acta Orthop Scand Suppl 273: 117-9, 1997. [PUBMED Abstract]
- Yang JC, Chang AE, Baker AR, et al.: Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 16 (1): 197-203, 1998. [PUBMED Abstract]
- O'Sullivan B, Davis AM, Turcotte R, et al.: Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 359 (9325): 2235-41, 2002. [PUBMED Abstract]
- O'Sullivan B, Davis A, Turcotte R, et al.: Five-year results of a randomized phase III trial of pre-operative vs post-operative radiotherapy in extremity soft tissue sarcoma. [Abstract] J Clin Oncol 22 (Suppl 14): A-9007, 819s, 2004.
- Davis AM, O'Sullivan B, Turcotte R, et al.: Late radiation morbidity following randomization to preoperative versus postoperative radiotherapy in extremity soft tissue sarcoma. Radiother Oncol 75 (1): 48-53, 2005. [PUBMED Abstract]
- Kepka L, DeLaney TF, Suit HD, et al.: Results of radiation therapy for unresected soft-tissue sarcomas. Int J Radiat Oncol Biol Phys 63 (3): 852-9, 2005. [PUBMED Abstract]
- Brennan MF, Singer S, Maki RG: Sarcomas of the soft tissue and bone. In: DeVita VT Jr, Hellman S, Rosenberg SA, eds.: Cancer: Principles and Practice of Oncology. Vols. 1 & 2. 8th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2008, pp 1741-1833.
- Baldini EH, Wang D, Haas RL, et al.: Treatment Guidelines for Preoperative Radiation Therapy for Retroperitoneal Sarcoma: Preliminary Consensus of an International Expert Panel. Int J Radiat Oncol Biol Phys 92 (3): 602-12, 2015. [PUBMED Abstract]
Stage II and Node-Negative Stage III Adult Soft Tissue Sarcoma
Refer to the Treatment Option Overview section of this summary for a more detailed discussion of the roles of surgery, radiation therapy, and chemotherapy.
High-grade localized soft tissue sarcomas have an increased potential for local recurrence and metastasis. For sarcomas of the extremities, local control comparable to that obtained with amputation may be achieved with limb-sparing surgery that involves wide local excision in combination with preoperative radiation therapy (preRT) or postoperative radiation therapy (PORT).
Complete surgical resection is often difficult for sarcomas of the retroperitoneum because of their large size before detection and anatomical location.[1,2] As opposed to soft tissue sarcomas of the extremities, local recurrence is the most common cause of death in patients with retroperitoneal soft tissue sarcomas. Complete surgical resection (i.e., removal of the entire gross tumor) is the most important factor in preventing local recurrence and, in many instances, requires resection of adjacent viscera. For retroperitoneal sarcomas, retrospective comparison of surgery alone versus preRT review suggests that preRT is associated with improved local recurrence-free survival, but not disease-free survival.[3]
Standard treatment options:
- Surgical excision with preRT or PORT. Radiation decreases the risk of local recurrence but has not been shown to increase overall survival.[4-8]
- Surgical excision with negative tissue margins in all directions. This approach is generally restricted to low-grade tumors ( ≤5 cm in diameter) of the extremities or superficial trunk with microscopically negative surgical tumor margins.[9-13]
- If the tumor is unresectable, high-dose radiation therapy may be used, but poor local control is likely to result.[14]
- In some situations, radiation therapy and/or chemotherapy may be used before surgery in an attempt to convert a marginally resectable tumor to one that can be adequately resected with limb preservation; this treatment may be followed by PORT.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Heslin MJ, Lewis JJ, Nadler E, et al.: Prognostic factors associated with long-term survival for retroperitoneal sarcoma: implications for management. J Clin Oncol 15 (8): 2832-9, 1997. [PUBMED Abstract]
- Jaques DP, Coit DG, Hajdu SI, et al.: Management of primary and recurrent soft-tissue sarcoma of the retroperitoneum. Ann Surg 212 (1): 51-9, 1990. [PUBMED Abstract]
- Kelly KJ, Yoon SS, Kuk D, et al.: Comparison of Perioperative Radiation Therapy and Surgery Versus Surgery Alone in 204 Patients With Primary Retroperitoneal Sarcoma: A Retrospective 2-Institution Study. Ann Surg 262 (1): 156-62, 2015. [PUBMED Abstract]
- Yang JC, Chang AE, Baker AR, et al.: Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol 16 (1): 197-203, 1998. [PUBMED Abstract]
- Rosenberg SA, Tepper J, Glatstein E, et al.: The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg 196 (3): 305-15, 1982. [PUBMED Abstract]
- O'Sullivan B, Davis AM, Turcotte R, et al.: Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 359 (9325): 2235-41, 2002. [PUBMED Abstract]
- O'Sullivan B, Davis A, Turcotte R, et al.: Five-year results of a randomized phase III trial of pre-operative vs post-operative radiotherapy in extremity soft tissue sarcoma. [Abstract] J Clin Oncol 22 (Suppl 14): A-9007, 819s, 2004.
- Davis AM, O'Sullivan B, Turcotte R, et al.: Late radiation morbidity following randomization to preoperative versus postoperative radiotherapy in extremity soft tissue sarcoma. Radiother Oncol 75 (1): 48-53, 2005. [PUBMED Abstract]
- Al-Refaie WB, Habermann EB, Jensen EH, et al.: Surgery alone is adequate treatment for early stage soft tissue sarcoma of the extremity. Br J Surg 97 (5): 707-13, 2010. [PUBMED Abstract]
- Pisters PW, Pollock RE, Lewis VO, et al.: Long-term results of prospective trial of surgery alone with selective use of radiation for patients with T1 extremity and trunk soft tissue sarcomas. Ann Surg 246 (4): 675-81; discussion 681-2, 2007. [PUBMED Abstract]
- Fabrizio PL, Stafford SL, Pritchard DJ: Extremity soft-tissue sarcomas selectively treated with surgery alone. Int J Radiat Oncol Biol Phys 48 (1): 227-32, 2000. [PUBMED Abstract]
- Rydholm A, Gustafson P, Rööser B, et al.: Limb-sparing surgery without radiotherapy based on anatomic location of soft tissue sarcoma. J Clin Oncol 9 (10): 1757-65, 1991. [PUBMED Abstract]
- Rydholm A: Surgery without radiotherapy in soft tissue sarcoma. Acta Orthop Scand Suppl 273: 117-9, 1997. [PUBMED Abstract]
- Kepka L, DeLaney TF, Suit HD, et al.: Results of radiation therapy for unresected soft-tissue sarcomas. Int J Radiat Oncol Biol Phys 63 (3): 852-9, 2005. [PUBMED Abstract]
Advanced Stage III (N1) Adult Soft Tissue Sarcoma
Refer to the Treatment Option Overview section of this summary for a more detailed discussion of the roles of surgery, radiation therapy, and chemotherapy.
Regional lymph node involvement by soft tissue sarcomas of adulthood is very infrequent. However, sarcoma types that more commonly spread to lymph nodes include high-grade rhabdomyosarcoma, vascular sarcomas, and epithelioid sarcomas.[1]
Standard treatment options:
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Mazeron JJ, Suit HD: Lymph nodes as sites of metastases from sarcomas of soft tissue. Cancer 60 (8): 1800-8, 1987. [PUBMED Abstract]
- Watson DI, Coventry BJ, Langlois SL, et al.: Soft-tissue sarcoma of the extremity. Experience with limb-sparing surgery. Med J Aust 160 (7): 412-6, 1994. [PUBMED Abstract]
- Cormier JN, Huang X, Xing Y, et al.: Cohort analysis of patients with localized, high-risk, extremity soft tissue sarcoma treated at two cancer centers: chemotherapy-associated outcomes. J Clin Oncol 22 (22): 4567-74, 2004. [PUBMED Abstract]
- O'Byrne K, Steward WP: The role of adjuvant chemotherapy in the treatment of adult soft tissue sarcomas. Crit Rev Oncol Hematol 27 (3): 221-7, 1998. [PUBMED Abstract]
- Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet 350 (9092): 1647-54, 1997. [PUBMED Abstract]
Stage IV Adult Soft Tissue Sarcoma
Refer to the Treatment Option Overview section of this summary for a more detailed discussion of the roles of surgery, radiation therapy, and chemotherapy.
In the setting of lung metastasis, resection of metastatic tumors may be associated with long-term disease-free survival in patients selected for optimal underlying disease biology (i.e., patients with a limited number of metastases and slow tumor growth).[1-3] It is not clear to what degree the favorable outcomes are attributable to the efficacy of surgery or to careful selection of patients based upon factors that are associated with less-virulent disease.[1-3] The value of resection of hepatic metastases is unclear.
As noted in the Treatment Option Overview section above, doxorubicin is the standard systemic therapy in the management of metastatic sarcomas.[4,5] Other drugs that may have clinical activity as single agents are ifosfamide, epirubicin, gemcitabine, and paclitaxel.[6-9] Their clinical activity relative to single-agent doxorubicin is not clear, and they are not known to have superior activity. There is controversy about whether adding drugs to doxorubicin offers clinical benefit beyond what is achieved by doxorubicin as a single agent. To avoid severe toxicity in older patients, sequential use of single agents may be the preferred strategy for palliation.
A randomized study assessed whether dose intensification of doxorubicin with ifosfamide improved the survival of patients with advanced soft-tissue sarcoma compared with doxorubicin alone.[10] Two hundred twenty-eight patients were randomly assigned to receive doxorubicin, and 227 patients were randomly assigned to receive doxorubicin and ifosfamide. Median follow-up was 56 months (interquartile range [IQR], 31–77) in the doxorubicin-only group and 59 months (IQR, 36–72) in the combination group.
There was no significant difference in overall survival (OS) between groups (median OS, 12.8 months; 95.5% confidence interval [CI], 10.5–14.3 in the doxorubicin-alone group vs. 14.3 months; range, 12.5–16.5 months in the doxorubicin and ifosfamide group; hazard ratio [HR], 0.83; 95.5% CI 0.67–1.03; stratified log-rank test P = .076). Median progression-free survival was significantly higher for the doxorubicin and ifosfamide group (7.4 months; 95% CI, 6.6-8.3) than for the doxorubicin-alone group (4.6 months; range, 2.9–5.6 months; HR, 0.74; 95% CI, 0.60–0.90; stratified log-rank test P = .003). More patients in the doxorubicin and ifosfamide group than in the doxorubicin-alone group had an overall response (60 [26%] of 227 patients vs. 31 [14%] of 228; P < .0006). The most common grade 3 and 4 toxic effects, which were all more common with doxorubicin and ifosfamide than with doxorubicin alone, were leucopenia (97 [43%] of 224 patients vs. 40 [18%] of 223 patients), neutropenia (93 [42%] vs. 83 [37%]), febrile neutropenia (103 (46%) vs. 30 [13%]), anemia (78 [35%] vs. 10 [5%]), and thrombocytopenia (75 [33%]) vs. 1 [<1%]).[10][Level of evidence: 1iiA] Treatment intensification with doxorubicin and ifosfamide for palliation of advanced soft tissue sarcoma is not indicated.
Standard treatment options
- Chemotherapy.
- Single-agent chemotherapy, with subsequent single agents for disease regrowth.[4-6,8,9,11] Doxorubicin is generally the first-line agent. Ifosfamide also has substantial single-agent activity.
- Doxorubicin-based combination chemotherapy. A variety of regimens have been used, but none has been proven to increase OS compared with doxorubicin alone.[4,5] There is some evidence that the addition of ifosfamide increases response rates (but not survival). Toxicity is increased with the addition of drugs to doxorubicin. No quality-of-life studies have been reported in comparisons of single-agent therapy versus combination therapy.
- Resection of pulmonary lesions may be performed if the primary tumor is under control.[1-3]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- van Geel AN, Pastorino U, Jauch KW, et al.: Surgical treatment of lung metastases: The European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group study of 255 patients. Cancer 77 (4): 675-82, 1996. [PUBMED Abstract]
- Casson AG, Putnam JB, Natarajan G, et al.: Five-year survival after pulmonary metastasectomy for adult soft tissue sarcoma. Cancer 69 (3): 662-8, 1992. [PUBMED Abstract]
- Putnam JB Jr, Roth JA: Surgical treatment for pulmonary metastases from sarcoma. Hematol Oncol Clin North Am 9 (4): 869-87, 1995. [PUBMED Abstract]
- Bramwell VH, Anderson D, Charette ML, et al.: Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft tissue sarcoma. Cochrane Database Syst Rev (3): CD003293, 2003. [PUBMED Abstract]
- Verma S, Younus J, Stys-Norman D, et al.: Meta-analysis of ifosfamide-based combination chemotherapy in advanced soft tissue sarcoma. Cancer Treat Rev 34 (4): 339-47, 2008. [PUBMED Abstract]
- Lorigan P, Verweij J, Papai Z, et al.: Phase III trial of two investigational schedules of ifosfamide compared with standard-dose doxorubicin in advanced or metastatic soft tissue sarcoma: a European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol 25 (21): 3144-50, 2007. [PUBMED Abstract]
- Nielsen OS, Dombernowsky P, Mouridsen H, et al.: High-dose epirubicin is not an alternative to standard-dose doxorubicin in the treatment of advanced soft tissue sarcomas. A study of the EORTC soft tissue and bone sarcoma group. Br J Cancer 78 (12): 1634-9, 1998. [PUBMED Abstract]
- Maki RG, Wathen JK, Patel SR, et al.: Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 25 (19): 2755-63, 2007. [PUBMED Abstract]
- Okuno S, Ryan LM, Edmonson JH, et al.: Phase II trial of gemcitabine in patients with advanced sarcomas (E1797): a trial of the Eastern Cooperative Oncology Group. Cancer 97 (8): 1969-73, 2003. [PUBMED Abstract]
- Judson I, Verweij J, Gelderblom H, et al.: Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol 15 (4): 415-23, 2014. [PUBMED Abstract]
- Grenader T, Goldberg A, Hadas-Halperin I, et al.: Long-term response to pegylated liposomal doxorubicin in patients with metastatic soft tissue sarcomas. Anticancer Drugs 20 (1): 15-20, 2009. [PUBMED Abstract]
Recurrent Adult Soft Tissue Sarcoma
Treatment of patients with recurrent soft tissue sarcoma depends on the type of initial presentation and treatment. Patients who develop a local recurrence often can be treated by local therapy: surgical excision plus radiation therapy after previous minimal therapy or amputation after previous aggressive treatment.[1-7] Resection of limited pulmonary metastases may be associated with favorable disease-free survival.[8-10][Level of evidence: 3iiiDiv] However, the contribution of selection factors, such as low tumor burden, slow tumor growth, and long disease-free interval, to these favorable outcomes is not known.
There is no standard chemotherapy for recurrent soft tissue sarcomas that have progressed after doxorubicin as a single agent or in combination with other agents that have clinical activity, such as ifosfamide, epirubicin, gemcitabine, and paclitaxel. Any of these agents not previously administered to the patient may be used sequentially at the time of recurrence or progression.[11-14][Level of Evidence: 3iiiDiv] None of these agents has been shown to increase overall survival in this setting, therefore, clinical trials are an appropriate option.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Singer S, Nielsen T, Antonescu CR: Molecular biology of soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1522-32.
- Singer S, Maki RG, O'Sullivan B: Soft tissue sarcoma. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1533-77.
- Malawer MM, Helman LJ, O'Sullivan B: Sarcomas of bone. In: DeVita VT Jr, Lawrence TS, Rosenberg SA: Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia, Pa: Lippincott Williams & Wilkins, 2011, pp 1578-1609.
- Midis GP, Pollock RE, Chen NP, et al.: Locally recurrent soft tissue sarcoma of the extremities. Surgery 123 (6): 666-71, 1998. [PUBMED Abstract]
- Essner R, Selch M, Eilber FR: Reirradiation for extremity soft tissue sarcomas. Local control and complications. Cancer 67 (11): 2813-7, 1991. [PUBMED Abstract]
- Singer S, Antman K, Corson JM, et al.: Long-term salvageability for patients with locally recurrent soft-tissue sarcomas. Arch Surg 127 (5): 548-53; discussion 553-4, 1992. [PUBMED Abstract]
- Lewis JJ, Leung D, Heslin M, et al.: Association of local recurrence with subsequent survival in extremity soft tissue sarcoma. J Clin Oncol 15 (2): 646-52, 1997. [PUBMED Abstract]
- van Geel AN, Pastorino U, Jauch KW, et al.: Surgical treatment of lung metastases: The European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group study of 255 patients. Cancer 77 (4): 675-82, 1996. [PUBMED Abstract]
- Casson AG, Putnam JB, Natarajan G, et al.: Five-year survival after pulmonary metastasectomy for adult soft tissue sarcoma. Cancer 69 (3): 662-8, 1992. [PUBMED Abstract]
- Putnam JB Jr, Roth JA: Surgical treatment for pulmonary metastases from sarcoma. Hematol Oncol Clin North Am 9 (4): 869-87, 1995. [PUBMED Abstract]
- Lorigan P, Verweij J, Papai Z, et al.: Phase III trial of two investigational schedules of ifosfamide compared with standard-dose doxorubicin in advanced or metastatic soft tissue sarcoma: a European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol 25 (21): 3144-50, 2007. [PUBMED Abstract]
- Nielsen OS, Dombernowsky P, Mouridsen H, et al.: High-dose epirubicin is not an alternative to standard-dose doxorubicin in the treatment of advanced soft tissue sarcomas. A study of the EORTC soft tissue and bone sarcoma group. Br J Cancer 78 (12): 1634-9, 1998. [PUBMED Abstract]
- Maki RG, Wathen JK, Patel SR, et al.: Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 25 (19): 2755-63, 2007. [PUBMED Abstract]
- Okuno S, Ryan LM, Edmonson JH, et al.: Phase II trial of gemcitabine in patients with advanced sarcomas (E1797): a trial of the Eastern Cooperative Oncology Group. Cancer 97 (8): 1969-73, 2003. [PUBMED Abstract]
Changes to This Summary (02/01/2018)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
General Information About Adult Soft Tissue Sarcoma
Updated statistics with estimated new cases and deaths for 2018 (cited American Cancer Society as reference 1).
Stage Information for Adult Soft Tissue Sarcoma
An editorial change was made to this section.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of adult soft tissue sarcoma. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Adult Soft Tissue Sarcoma Treatment are:
- Russell S. Berman, MD (New York University School of Medicine)
- Minh Tam Truong, MD (Boston University Medical Center)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Adult Soft Tissue Sarcoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/soft-tissue-sarcoma/hp/adult-soft-tissue-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389481]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Updated: February 1, 2018
Source URL: https://www.cancer.gov/publishedcontent/syndication/2127.htm
Source Agency: National Cancer Institute (NCI)
Captured Date: 2013-09-14 09:00:45.0
Childhood soft tissue sarcoma: Professional resources from the National Cancer Institute
Childhood Soft Tissue Sarcoma Treatment (PDQ®)–Health Professional Version
General Information About Childhood Soft Tissue Sarcoma
Dramatic improvements in survival have been achieved for children and adolescents with cancer. Between 1975 and 2010, childhood cancer mortality decreased by more than 50%.[1] Childhood and adolescent cancer survivors require close monitoring because cancer therapy side effects may persist or develop months or years after treatment. (Refer to the PDQ summary on Late Effects of Treatment for Childhood Cancer for specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors.)
Rhabdomyosarcoma, a tumor of striated muscle, is the most common soft tissue sarcoma in children aged 0 to 14 years and accounts for 50% of tumors in this age group.[2] (Refer to the PDQ summary on Childhood Rhabdomyosarcoma Treatment for more information.) In pediatrics, the remaining soft tissue sarcomas are commonly referred to as nonrhabdomyosarcomatous soft tissue sarcomas and account for approximately 3% of all childhood tumors.[3] This heterogeneous group of tumors includes the following neoplasms:[4]
- Connective tissue (e.g., desmoid-type fibromatosis).
- Peripheral nervous system (e.g., malignant peripheral nerve sheath tumor).
- Smooth muscle (e.g., leiomyosarcoma).
- Vascular tissue (blood and lymphatic vessels, e.g., angiosarcoma). (Refer to the PDQ summary on Childhood Vascular Tumors Treatment for more information about childhood vascular tumors.)
Distribution of Soft Tissue Sarcoma by Age and Histology
Pediatric soft tissue sarcomas are a heterogenous group of malignant tumors that originate from primitive mesenchymal tissue and account for 7% of all childhood tumors.[5]
The distribution of soft tissue sarcomas by histology and age, based on the Surveillance, Epidemiology, and End Results (SEER) information from 1975 to 2012, is depicted in Table 1. The distribution of histologic subtypes by age is also shown in Figure 2.
| Age <5 y | Age 5–9 y | Age 10–14 y | Age 15–19 y | % of the Total Number of STS Cases <20 y | |||
|---|---|---|---|---|---|---|---|
| pPNET = peripheral primitive neuroectodermal tumors; SEER = Surveillance, Epidemiology, and End Results; STS = soft tissue sarcoma. | |||||||
| aSEER data is available at http://seer.cancer.gov. | |||||||
| bDermatofibrosarcoma accounts for 75% of these cases. | |||||||
| All soft tissue and other extraosseous sarcomas | 923 | 631 | 946 | 1,267 | 100 | ||
| Rhabdomyosarcomas | 551 | 348 | 312 | 270 | 39 | ||
| Fibrosarcomas, peripheral nerve, and other fibrous neoplasms | 116 | 50 | 88 | 141 | 10 | ||
| Fibroblastic and myofibroblastic tumors | 97 | 24 | 31 | 62 | 6 | ||
| Nerve sheath tumors | 19 | 26 | 56 | 77 | 5 | ||
| Other fibromatous neoplasms | 0 | 0 | 1 | 2 | 0.1 | ||
| Kaposi sarcoma | 2 | 1 | 1 | 9 | 0.3 | ||
| Other specified soft tissue sarcomas | 194 | 190 | 424 | 708 | 40 | ||
| Ewing tumor and Askin tumor of soft tissue | 27 | 30 | 62 | 92 | 6 | ||
| pPNET of soft tissue | 21 | 18 | 36 | 46 | 3.2 | ||
| Extrarenal rhabdoid tumor | 61 | 3 | 7 | 3 | 2 | ||
| Liposarcomas | 3 | 5 | 22 | 57 | 2.3 | ||
| Fibrohistiocytic tumors b | 34 | 54 | 108 | 188 | 10 | ||
| Leiomyosarcomas | 9 | 14 | 15 | 36 | 2 | ||
| Synovial sarcomas | 10 | 34 | 111 | 175 | 9 | ||
| Blood vessel tumors | 11 | 7 | 8 | 25 | 1.4 | ||
| Osseous and chondromatous neoplasms of soft tissue | 1 | 6 | 13 | 10 | 0.8 | ||
| Alveolar soft parts sarcoma | 4 | 3 | 16 | 29 | 1.4 | ||
| Miscellaneous soft tissue sarcomas | 13 | 16 | 36 | 47 | 3 | ||
| Unspecified soft tissue sarcomas | 60 | 40 | 111 | 139 | 9.3 | ||
Nonrhabdomyosarcomatous soft tissue sarcomas are more common in adolescents and adults,[4] and most of the information regarding treatment and natural history of the disease in younger patients has been based on adult studies. The distributions of these tumors by age according to stage, histologic subtype, and tumor site are shown in Figures 1, 2, and 3, respectively.[6]



Risk Factors
Some genetic and environmental factors have been associated with the development of nonrhabdomyosarcomatous soft tissue sarcoma, including the following:
- Genetic factors:
- Li-Fraumeni syndrome: Patients with Li-Fraumeni syndrome (usually due to heritable cancer-associated changes of the TP53 tumor suppressor gene) have an increased risk of developing soft tissue tumors (mostly nonrhabdomyosarcomatous soft tissue sarcomas), bone sarcomas, breast cancer, brain tumors, and acute leukemia.[7,8]
- Familial adenomatous polyposis: Patients with familial adenomatous polyposis are at increased risk of developing desmoid-type fibromatosis.[9]
- Retinoblastoma (RB1) gene: Germline mutations of the retinoblastoma gene have been associated with an increased risk of developing soft tissue sarcomas, particularly leiomyosarcoma.[10]
- SMARCB1 gene: Germline mutations or deletions of the SMARCB1 (INI1) gene are associated with an increased risk of developing extrarenal rhabdoid tumors.[11]
- Neurofibromatosis type 1: Approximately 4% of patients with neurofibromatosis type 1 develop malignant peripheral nerve sheath tumors, which usually develop after a long latency; some patients develop multiple lesions.[12-14]
- Werner syndrome: Werner syndrome is characterized by spontaneous chromosomal instability, resulting in increased susceptibility to cancer and premature aging. An excess of soft tissue sarcomas has been reported in patients with Werner syndrome.[15]
- Environmental factors:
- Radiation: Some nonrhabdomyosarcomatous soft tissue sarcomas (particularly malignant fibrous histiocytoma) can develop within a previously irradiated site.[3,16]
- Epstein-Barr virus infection in patients with AIDS: Some nonrhabdomyosarcomatous soft tissue sarcomas (e.g., leiomyosarcoma) have been linked to Epstein-Barr virus infection in patients with AIDS.[3,17]
Clinical Presentation
Although nonrhabdomyosarcomatous soft tissue sarcomas can develop in any part of the body, they arise most commonly in the trunk and extremities.[18-20] These neoplasms can present initially as an asymptomatic solid mass, or they may be symptomatic because of local invasion of adjacent anatomical structures. Although rare, these tumors can arise primarily in brain tissue and are treated according to the histotype.[21]
Systemic symptoms (e.g., fever, weight loss, and night sweats) are rare. Hypoglycemia and hypophosphatemic rickets have been reported in cases of hemangiopericytoma, whereas hyperglycemia has been noted in patients with fibrosarcoma of the lung.[22]
Diagnostic and Staging Evaluation
When a suspicious lesion is identified, it is crucial that a complete workup, followed by adequate biopsy be performed. It is best to image the lesion using the following procedures before initiating any intervention:
- Plain films. Plain films can be used to rule out bone involvement and detect calcifications that may be seen in soft tissue tumors such as extraskeletal osteosarcoma or synovial sarcoma.
- Chest computed tomography (CT). Chest CT is essential to assess the presence of metastases.
- Abdominal CT or magnetic resonance imaging (MRI). Abdominal CT or MRI can be used to image intra-abdominal tumors, such as liposarcoma.
- Extremity MRI. MRI is essential for extremity lesions.
- Positron emission tomography (PET) scan and bone scan. In children with rhabdomyosarcoma, PET-CT performed better than conventional imaging in identifying nodal, bone, bone marrow, and soft tissue disease. The authors of an imaging comparison study suggest that bone scans with technetium Tc 99m might be eliminated as a staging procedure.[23] The use of this modality in pediatric nonrhabdomyosarcomatous soft tissue sarcoma has not been studied extensively. However, a small study of nine patients with nonrhabdomyosarcomatous soft tissue sarcoma suggests that PET-CT is more accurate and cost effective than either modality alone in identifying distant metastatic disease.[24]
The imaging characteristics of some tumors can be highly suggestive of this diagnosis. For example, the imaging characteristics of pediatric low-grade fibromyxoid sarcoma and alveolar soft part sarcoma have been described and can aid in the diagnosis of these rare neoplasms.[25]
Biopsy strategies
Although nonrhabdomyosarcomatous soft tissue tumors are fairly readily distinguished pathologically from rhabdomyosarcoma and Ewing sarcoma, the classification of childhood nonrhabdomyosarcomatous soft tissue sarcoma type is often difficult. Core-needle biopsy, incisional biopsy, or excisional biopsy can be used to diagnose a nonrhabdomyosarcomatous soft tissue sarcoma. If possible, the surgeon who will perform the definitive resection needs to be involved in the biopsy decision. Poorly placed incisional or needle biopsies may adversely affect the performance of the primary resection.
Considerations related to the selection of a biopsy procedure are as follows:
- Given the diagnostic importance of translocations, a core-needle biopsy or small incisional biopsy that obtains adequate tumor tissue is crucial to allow for conventional histology, immunocytochemical analysis, and other studies such as light and electron microscopy, cytogenetics, fluorescence in situ hybridization, and molecular pathology.[26,27] Core-needle biopsy for a deep-seated tumor can lead to formation of a hematoma, which affects subsequent resection and/or radiation; in these cases, incisional biopsy is the preferred procedure.
- Fine-needle biopsy is usually not recommended because it is difficult to determine the accurate histologic diagnosis and grade of the tumor in this heterogeneous group of tumors.
- Image guidance using ultrasound, CT scan, or MRI may be necessary to ensure a representative biopsy.[28]
- Needle biopsy techniques must ensure adequate tissue sampling. The acquisition of multiple cores of tissue may be required.
- Incisional biopsies must not compromise subsequent wide local excision.
- Excisional biopsy of the lesion is only appropriate for small superficial lesions (<3 cm in size) and are discouraged.[29,30] If an excisional biopsy is contemplated, then MRI of the area is recommended to define the area of involvement as subsequent surgery or radiation therapy is likely.
- Various institutional series have demonstrated the feasibility and effectiveness of sentinel node biopsy as a staging procedure in pediatric patients with soft tissue sarcomas.[31-36]
- Transverse extremity incisions are avoided to reduce skin loss and because they require a greater cross-sectional volume of tissue to be covered in the radiation field. Other extensive surgical procedures are also avoided before definitive diagnosis. For these reasons, open biopsy or multiple core-needle biopsies are strongly encouraged so that adequate tumor tissue can be obtained to allow crucial studies to be performed and to avoid limiting future treatment options.
Unplanned resection
In children with unplanned resection of nonrhabdomyosarcomatous soft tissue sarcomas, primary re-excision is frequently recommended because many patients will have tumor present in the re-excision specimen.[37,38] A single-institution analysis of adolescents and adults compared patients with unplanned excision of soft tissue sarcoma to stage-matched controls. In this retrospective analysis, unplanned initial excision of soft tissue sarcoma resulted in increased risk of local recurrence, metastasis, and death; this increase was greatest for high-grade tumors.[39][Level of evidence: 3iiA]
Chromosomal abnormalities
Many nonrhabdomyosarcomatous soft tissue sarcomas are characterized by chromosomal abnormalities. Some of these chromosomal translocations lead to a fusion of two disparate genes. The resulting fusion transcript can be readily detected by using polymerase chain reaction-based techniques, thus facilitating the diagnosis of those neoplasms that have translocations.
Some of the most frequent aberrations seen in nonrhabdomyosarcomatous soft tissue tumors are listed in Table 2.
| Histology | Chromosomal Aberrations | Genes Involved |
|---|---|---|
| aAdapted from Sandberg,[40] Slater et al.,[41] Mertens et al.,[42] and Romeo.[43] | ||
| Alveolar soft part sarcoma | t(x;17)(p11.2;q25) | ASPL/TFE3 [44-46] |
| Angiomatoid fibrous histiocytoma | t(12;16)(q13;p11), t(2;22)(q33;q12), t(12;22)(q13;q12) | FUS/ATF1, EWSR1/CREB1,[47] EWS/ATF1 |
| Clear cell sarcoma | t(12;22)(q13;q12), t(2;22)(q33;q12) | ATF1/EWS, EWSR1/CREB1 |
| Congenital (infantile) fibrosarcoma/mesoblastic nephroma | t(12;15)(p13;q25) | ETV-NTRK3 |
| Dermatofibrosarcoma protuberans | t(17;22)(q22;q13) | COL1A1/PDGFB |
| Desmoid fibromatosis | Trisomy 8 or 20, loss of 5q21 | CTNNB1 or APC mutations |
| Desmoplastic small round cell tumors | t(11;22)(p13;q12) | EWS/WT1 [48,49] |
| Epithelioid hemangioendothelioma | t(1;3)(p36;q25) [50] | WWTR1/CAMTA1 |
| Epithelioid sarcoma | Inactivation SMARCB1 | SMARCB1 |
| Extraskeletal myxoid chondrosarcoma | t(9;22)(q22;q12), t(9;17)(q22;q11), t(9;15)(q22;q21), t(3;9)(q11;q22) | EWSR1/NR4A3, TAF2N/NR4A3, TCF12/NR4A3, TGF/NR4A3 |
| Hemangiopericytoma | t(12;19)(q13;q13.3) and t(13;22)(q22;q13.3) | |
| Infantile fibrosarcoma | t(12;15)(p13;q25) | ETV6/NTRK3 |
| Inflammatory myofibroblastic tumor | t(1;2)(q23;q23), t(2;19)(q23;q13), t(2;17)(q23;q23), t(2;2)(p23;q13), t(2;11)(p23;p15) [51] | TPM3/ALK, TPM4/ALK, CLTC/ALK, RANBP2/ALK, CARS/ALK, RAS |
| Low-grade fibromyxoid sarcoma | t(7;16)(q33;p11), t(11;16)(p11;p11) | FUS/CREB3L2, FUS/CREB3L1 |
| Malignant peripheral nerve sheath tumor | 17q11.2, loss or rearrangement 10p, 11q, 17q, 22q | NF1 |
| Mesenchymal chondrosarcoma | Del(8)(q13.3q21.1) | HEY1/NCOA2 |
| Myoepithelioma | t(19;22)(q13;q12), t(1;22)(q23;q12), t(6;22)(p21;q12) | EWSR/ZNF44, EWSR/PBX1, EWSR/POU5F1 |
| Myxoid/round cell liposarcoma | t(12;16)(q13;p11), t(12;22)(q13;q12) | FUS/DD1T3, EWSR/DD1T3 |
| Rhabdoid tumor | Inactivation SMARCB1 | SMARCB1 |
| Solitary fibrous tumor | Inv(12)(q13q13) | NAB2/STAT6 |
| Synovial sarcoma | t(x;18)(p11.2;q11.2) | SYT/SSX |
| Tenosynovial giant cell tumor | t(1;2)(p13;q35) | COL6A3/CSF1 |
Prognosis
The prognosis of nonrhabdomyosarcomatous soft tissue sarcoma varies greatly depending on the following factors:[52-54]
- Site of the primary tumor.
- Tumor size.
- Tumor grade. (Refer to the Prognostic Significance of Tumor Grading section of this summary for more information.)
- Tumor histology.
- Depth of tumor invasion.
- Presence of metastases.
- Resectability of the tumor.
- Use of radiation therapy.
Several adult and pediatric series have shown that patients with large or invasive tumors have a significantly worse prognosis than do those with small, noninvasive tumors. A retrospective review of soft tissue sarcomas in children and adolescents suggests that the 5 cm cutoff used for adults with soft tissue sarcoma may not be ideal for smaller children, especially infants. The review identified an interaction between tumor diameter and body surface area.[55] This relationship requires further study to determine the therapeutic implications of the observation.
In a review of a large adult series of nonrhabdomyosarcomatous soft tissue sarcomas, superficial extremity sarcomas had a better prognosis than did deep tumors. Thus, in addition to grade and size, the depth of invasion of the tumor should be considered.[56]
Some pediatric nonrhabdomyosarcomatous soft tissue sarcomas are associated with a better outcome. For instance, infantile fibrosarcoma, presenting in infants and children younger than 5 years, has an excellent prognosis given that surgery alone can cure a significant number of these patients and the tumor is highly chemosensitive.[3]
Soft tissue sarcomas in older children and adolescents often behave similarly to those in adult patients.[3,26] A large, prospective, multinational Children's Oncology Group study (ARST0332 [NCT00346164]) enrolled newly diagnosed patients younger than 30 years. Patients were assigned to treatment on the basis of their risk group (refer to Figure 4).[57][Level of evidence: 2A]

- Arm A (grossly excised low-grade tumor and ≤5 cm widely excised high-grade tumor): Surgery only.
- Arm B (≤5 cm marginally resected high-grade tumor): 55.8 Gy of radiation therapy.
- Arm C (>5 cm grossly resected tumor ± metastases): Ifosfamide/doxorubicin chemotherapy and 55.8 Gy of radiation therapy.
- Arm D (>5 cm unresected tumor ± metastases): Preoperative ifosfamide/doxorubicin chemotherapy and 45 Gy of radiation therapy, and then surgery and a radiation boost that was based on margins.
Of 551 patients enrolled, at a median follow-up of 2.6 years, the preliminary analysis estimated the following 3-year survival rates:[57]
- Arm A: 91% event-free survival (EFS); 99% overall survival (OS).
- Arm B: 79% EFS; 100% OS.
- Arm C: 68% EFS; 81% OS.
- Arm D: 52% EFS; 66% OS.
Pediatric patients with unresected localized nonrhabdomyosarcomatous soft tissue sarcomas have a poor outcome. Only about one-third of patients treated with multimodality therapy remain disease free.[52,58]; [59,60][Level of evidence: 3iiiA] In a review of 30 Italian patients with nonrhabdomyosarcomatous soft tissue sarcoma at visceral sites, only ten patients survived at 5 years. Unfavorable prognostic factors included inability to achieve complete resection, large tumor size, tumor invasion, histologic subtype, and lung-pleura sites.[61][Level of evidence: 3iiB]
In a pooled analysis from U.S. and European pediatric centers, outcome was better for patients whose tumor removal procedure was deemed complete than for patients whose tumor removal was incomplete. Outcome was better for patients who received radiation therapy than for patients who did not.[59][Level of evidence: 3iiiA]
Because long-term related morbidity must be minimized while disease-free survival is maximized, the ideal therapy for each patient must be carefully and individually determined utilizing these prognostic factors before initiating therapy.[19,62-66]
Related Summaries
Refer to the following PDQ summaries for information about other types of sarcoma:
- Childhood Rhabdomyosarcoma Treatment.
- Childhood Vascular Tumors Treatment.
- Ewing Sarcoma Treatment (extraosseous Ewing, peripheral neuroepithelioma, and Askin tumors).
- Unusual Cancers of Childhood Treatment (gastrointestinal stromal tumors).
- Adult Soft Tissue Sarcoma Treatment.
References
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014. [PUBMED Abstract]
- Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. Bethesda, Md: National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649. Also available online. Last accessed January 24, 2018.
- Spunt SL, Million L, Coffin C: The nonrhabdomyosarcoma soft tissue sarcoma. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 827-54.
- Weiss SW, Goldblum JR: General considerations. In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. St. Louis, Mo: Mosby, 2008, pp 1-14.
- Pappo AS, Pratt CB: Soft tissue sarcomas in children. Cancer Treat Res 91: 205-22, 1997. [PUBMED Abstract]
- Ferrari A, Sultan I, Huang TT, et al.: Soft tissue sarcoma across the age spectrum: a population-based study from the Surveillance Epidemiology and End Results database. Pediatr Blood Cancer 57 (6): 943-9, 2011. [PUBMED Abstract]
- Chang F, Syrjänen S, Syrjänen K: Implications of the p53 tumor-suppressor gene in clinical oncology. J Clin Oncol 13 (4): 1009-22, 1995. [PUBMED Abstract]
- Plon SE, Malkin D: Childhood cancer and hereditary. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 13-31.
- Groen EJ, Roos A, Muntinghe FL, et al.: Extra-intestinal manifestations of familial adenomatous polyposis. Ann Surg Oncol 15 (9): 2439-50, 2008. [PUBMED Abstract]
- Kleinerman RA, Tucker MA, Abramson DH, et al.: Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst 99 (1): 24-31, 2007. [PUBMED Abstract]
- Eaton KW, Tooke LS, Wainwright LM, et al.: Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56 (1): 7-15, 2011. [PUBMED Abstract]
- Weiss SW, Goldblum JR: Benign tumors of peripheral nerves. In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. St. Louis, Mo: Mosby, 2008, pp 825-901.
- deCou JM, Rao BN, Parham DM, et al.: Malignant peripheral nerve sheath tumors: the St. Jude Children's Research Hospital experience. Ann Surg Oncol 2 (6): 524-9, 1995. [PUBMED Abstract]
- Stark AM, Buhl R, Hugo HH, et al.: Malignant peripheral nerve sheath tumours--report of 8 cases and review of the literature. Acta Neurochir (Wien) 143 (4): 357-63; discussion 363-4, 2001. [PUBMED Abstract]
- Goto M, Miller RW, Ishikawa Y, et al.: Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol Biomarkers Prev 5 (4): 239-46, 1996. [PUBMED Abstract]
- Weiss SW, Goldblum JR: Malignant fibrous histiocytoma (pleomorphic undifferentiated sarcoma). In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. St. Louis, Mo: Mosby, 2008, pp 403-27.
- McClain KL, Leach CT, Jenson HB, et al.: Association of Epstein-Barr virus with leiomyosarcomas in children with AIDS. N Engl J Med 332 (1): 12-8, 1995. [PUBMED Abstract]
- Dillon P, Maurer H, Jenkins J, et al.: A prospective study of nonrhabdomyosarcoma soft tissue sarcomas in the pediatric age group. J Pediatr Surg 27 (2): 241-4; discussion 244-5, 1992. [PUBMED Abstract]
- Rao BN: Nonrhabdomyosarcoma in children: prognostic factors influencing survival. Semin Surg Oncol 9 (6): 524-31, 1993 Nov-Dec. [PUBMED Abstract]
- Zeytoonjian T, Mankin HJ, Gebhardt MC, et al.: Distal lower extremity sarcomas: frequency of occurrence and patient survival rate. Foot Ankle Int 25 (5): 325-30, 2004. [PUBMED Abstract]
- Benesch M, von Bueren AO, Dantonello T, et al.: Primary intracranial soft tissue sarcoma in children and adolescents: a cooperative analysis of the European CWS and HIT study groups. J Neurooncol 111 (3): 337-45, 2013. [PUBMED Abstract]
- Weiss SW, Goldblum JR: Miscellaneous tumors of intermediate malignancy. In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. St. Louis, Mo: Mosby, 2008, pp 1093-1160.
- Federico SM, Spunt SL, Krasin MJ, et al.: Comparison of PET-CT and conventional imaging in staging pediatric rhabdomyosarcoma. Pediatr Blood Cancer 60 (7): 1128-34, 2013. [PUBMED Abstract]
- Tateishi U, Hosono A, Makimoto A, et al.: Accuracy of 18F fluorodeoxyglucose positron emission tomography/computed tomography in staging of pediatric sarcomas. J Pediatr Hematol Oncol 29 (9): 608-12, 2007. [PUBMED Abstract]
- Sargar K, Kao SC, Spunt SL, et al.: MRI and CT of Low-Grade Fibromyxoid Sarcoma in Children: A Report From Children's Oncology Group Study ARST0332. AJR Am J Roentgenol 205 (2): 414-20, 2015. [PUBMED Abstract]
- Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 4th ed. St. Louis, Mo: Mosby, 2001.
- Recommendations for the reporting of soft tissue sarcomas. Association of Directors of Anatomic and Surgical Pathology. Mod Pathol 11 (12): 1257-61, 1998. [PUBMED Abstract]
- Chowdhury T, Barnacle A, Haque S, et al.: Ultrasound-guided core needle biopsy for the diagnosis of rhabdomyosarcoma in childhood. Pediatr Blood Cancer 53 (3): 356-60, 2009. [PUBMED Abstract]
- Coffin CM, Dehner LP, O'Shea PA: Pediatric Soft Tissue Tumors: A Clinical, Pathological, and Therapeutic Approach. Baltimore, Md: Williams and Wilkins, 1997.
- Smith LM, Watterson J, Scott SM: Medical and surgical management of pediatric soft tissue tumors. In: Coffin CM, Dehner LP, O'Shea PA: Pediatric Soft Tissue Tumors: A Clinical, Pathological, and Therapeutic Approach. Baltimore, Md: Williams and Wilkins, 1997, pp 360-71.
- Neville HL, Andrassy RJ, Lally KP, et al.: Lymphatic mapping with sentinel node biopsy in pediatric patients. J Pediatr Surg 35 (6): 961-4, 2000. [PUBMED Abstract]
- Neville HL, Raney RB, Andrassy RJ, et al.: Multidisciplinary management of pediatric soft-tissue sarcoma. Oncology (Huntingt) 14 (10): 1471-81; discussion 1482-6, 1489-90, 2000. [PUBMED Abstract]
- Kayton ML, Delgado R, Busam K, et al.: Experience with 31 sentinel lymph node biopsies for sarcomas and carcinomas in pediatric patients. Cancer 112 (9): 2052-9, 2008. [PUBMED Abstract]
- Dall'Igna P, De Corti F, Alaggio R, et al.: Sentinel node biopsy in pediatric patients: the experience in a single institution. Eur J Pediatr Surg 24 (6): 482-7, 2014. [PUBMED Abstract]
- Parida L, Morrisson GT, Shammas A, et al.: Role of lymphoscintigraphy and sentinel lymph node biopsy in the management of pediatric melanoma and sarcoma. Pediatr Surg Int 28 (6): 571-8, 2012. [PUBMED Abstract]
- Alcorn KM, Deans KJ, Congeni A, et al.: Sentinel lymph node biopsy in pediatric soft tissue sarcoma patients: utility and concordance with imaging. J Pediatr Surg 48 (9): 1903-6, 2013. [PUBMED Abstract]
- Chui CH, Spunt SL, Liu T, et al.: Is reexcision in pediatric nonrhabdomyosarcoma soft tissue sarcoma necessary after an initial unplanned resection? J Pediatr Surg 37 (10): 1424-9, 2002. [PUBMED Abstract]
- Cecchetto G, Guglielmi M, Inserra A, et al.: Primary re-excision: the Italian experience in patients with localized soft-tissue sarcomas. Pediatr Surg Int 17 (7): 532-4, 2001. [PUBMED Abstract]
- Qureshi YA, Huddy JR, Miller JD, et al.: Unplanned excision of soft tissue sarcoma results in increased rates of local recurrence despite full further oncological treatment. Ann Surg Oncol 19 (3): 871-7, 2012. [PUBMED Abstract]
- Sandberg AA: Translocations in malignant tumors. Am J Pathol 159 (6): 1979-80, 2001. [PUBMED Abstract]
- Slater O, Shipley J: Clinical relevance of molecular genetics to paediatric sarcomas. J Clin Pathol 60 (11): 1187-94, 2007. [PUBMED Abstract]
- Mertens F, Antonescu CR, Hohenberger P, et al.: Translocation-related sarcomas. Semin Oncol 36 (4): 312-23, 2009. [PUBMED Abstract]
- Romeo S, Dei Tos AP: Clinical application of molecular pathology in sarcomas. Curr Opin Oncol 23 (4): 379-84, 2011. [PUBMED Abstract]
- Ladanyi M, Lui MY, Antonescu CR, et al.: The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene 20 (1): 48-57, 2001. [PUBMED Abstract]
- Ladanyi M: The emerging molecular genetics of sarcoma translocations. Diagn Mol Pathol 4 (3): 162-73, 1995. [PUBMED Abstract]
- Williams A, Bartle G, Sumathi VP, et al.: Detection of ASPL/TFE3 fusion transcripts and the TFE3 antigen in formalin-fixed, paraffin-embedded tissue in a series of 18 cases of alveolar soft part sarcoma: useful diagnostic tools in cases with unusual histological features. Virchows Arch 458 (3): 291-300, 2011. [PUBMED Abstract]
- Antonescu CR, Dal Cin P, Nafa K, et al.: EWSR1-CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer 46 (12): 1051-60, 2007. [PUBMED Abstract]
- Barnoud R, Sabourin JC, Pasquier D, et al.: Immunohistochemical expression of WT1 by desmoplastic small round cell tumor: a comparative study with other small round cell tumors. Am J Surg Pathol 24 (6): 830-6, 2000. [PUBMED Abstract]
- Wang LL, Perlman EJ, Vujanic GM, et al.: Desmoplastic small round cell tumor of the kidney in childhood. Am J Surg Pathol 31 (4): 576-84, 2007. [PUBMED Abstract]
- Errani C, Zhang L, Sung YS, et al.: A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer 50 (8): 644-53, 2011. [PUBMED Abstract]
- Jain S, Xu R, Prieto VG, et al.: Molecular classification of soft tissue sarcomas and its clinical applications. Int J Clin Exp Pathol 3 (4): 416-28, 2010. [PUBMED Abstract]
- Spunt SL, Hill DA, Motosue AM, et al.: Clinical features and outcome of initially unresected nonmetastatic pediatric nonrhabdomyosarcoma soft tissue sarcoma. J Clin Oncol 20 (15): 3225-35, 2002. [PUBMED Abstract]
- Spunt SL, Poquette CA, Hurt YS, et al.: Prognostic factors for children and adolescents with surgically resected nonrhabdomyosarcoma soft tissue sarcoma: an analysis of 121 patients treated at St Jude Children's Research Hospital. J Clin Oncol 17 (12): 3697-705, 1999. [PUBMED Abstract]
- Ferrari A, Casanova M, Collini P, et al.: Adult-type soft tissue sarcomas in pediatric-age patients: experience at the Istituto Nazionale Tumori in Milan. J Clin Oncol 23 (18): 4021-30, 2005. [PUBMED Abstract]
- Ferrari A, Miceli R, Meazza C, et al.: Soft tissue sarcomas of childhood and adolescence: the prognostic role of tumor size in relation to patient body size. J Clin Oncol 27 (3): 371-6, 2009. [PUBMED Abstract]
- Brooks AD, Heslin MJ, Leung DH, et al.: Superficial extremity soft tissue sarcoma: an analysis of prognostic factors. Ann Surg Oncol 5 (1): 41-7, 1998 Jan-Feb. [PUBMED Abstract]
- Spunt SL, Million L, Anderson JR, et al.: Risk-based treatment for nonrhabdomyosarcoma soft tissue sarcomas (NRSTS) in patients under 30 years of age: Children’s Oncology Group study ARST0332. [Abstract] J Clin Oncol 32 (Suppl 15): A-10008, 2014. Also available online. Last accessed April 02, 2018.
- O'Sullivan B, Davis AM, Turcotte R, et al.: Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 359 (9325): 2235-41, 2002. [PUBMED Abstract]
- Ferrari A, Miceli R, Rey A, et al.: Non-metastatic unresected paediatric non-rhabdomyosarcoma soft tissue sarcomas: results of a pooled analysis from United States and European groups. Eur J Cancer 47 (5): 724-31, 2011. [PUBMED Abstract]
- Smith KB, Indelicato DJ, Knapik JA, et al.: Definitive radiotherapy for unresectable pediatric and young adult nonrhabdomyosarcoma soft tissue sarcoma. Pediatr Blood Cancer 57 (2): 247-51, 2011. [PUBMED Abstract]
- Ferrari A, Magni C, Bergamaschi L, et al.: Pediatric nonrhabdomyosarcoma soft tissue sarcomas arising at visceral sites. Pediatr Blood Cancer 64 (9): , 2017. [PUBMED Abstract]
- Dillon PW, Whalen TV, Azizkhan RG, et al.: Neonatal soft tissue sarcomas: the influence of pathology on treatment and survival. Children's Cancer Group Surgical Committee. J Pediatr Surg 30 (7): 1038-41, 1995. [PUBMED Abstract]
- Pappo AS, Fontanesi J, Luo X, et al.: Synovial sarcoma in children and adolescents: the St Jude Children's Research Hospital experience. J Clin Oncol 12 (11): 2360-6, 1994. [PUBMED Abstract]
- Marcus KC, Grier HE, Shamberger RC, et al.: Childhood soft tissue sarcoma: a 20-year experience. J Pediatr 131 (4): 603-7, 1997. [PUBMED Abstract]
- Pratt CB, Pappo AS, Gieser P, et al.: Role of adjuvant chemotherapy in the treatment of surgically resected pediatric nonrhabdomyosarcomatous soft tissue sarcomas: A Pediatric Oncology Group Study. J Clin Oncol 17 (4): 1219, 1999. [PUBMED Abstract]
- Pratt CB, Maurer HM, Gieser P, et al.: Treatment of unresectable or metastatic pediatric soft tissue sarcomas with surgery, irradiation, and chemotherapy: a Pediatric Oncology Group study. Med Pediatr Oncol 30 (4): 201-9, 1998. [PUBMED Abstract]
Histopathological Classification of Childhood Soft Tissue Sarcoma
World Health Organization (WHO) Classification of Soft Tissue Sarcomas
The WHO lists the following cell types in its classification of soft tissue sarcomas:[1,2]
- Adipocytic tumors.
- Intermediate (locally aggressive).
- Malignant.
- Chondro-osseous tumors.
- Extraskeletal mesenchymal chondrosarcoma.[3]
- Extraskeletal osteosarcoma.
- Soft tissue chondroma.
- Fibroblastic/myofibroblastic tumors.
- Intermediate-grade (locally aggressive).
- Desmoid-type fibromatosis (previously called desmoid tumor or aggressive fibromatoses).
- Giant cell fibroblastoma.
- Lipofibromatosis.
- Palmar/plantar fibromatosis.
- Intermediate-grade (rarely metastasizing).
- Dermatofibrosarcoma protuberans.
- Infantile fibrosarcoma.[4]
- Inflammatory myofibroblastic tumor.
- Low-grade myofibroblastic tumor.
- Myxoinflammatory fibroblastic sarcoma.
- Solitary fibrous tumor.
- Malignant.
- Intermediate-grade (locally aggressive).
- Skeletal muscle tumors.
- Rhabdomyosarcoma (embryonal, alveolar, and pleomorphic forms). (Refer to the PDQ summary on Childhood Rhabdomyosarcoma Treatment for more information.)
- Smooth muscle tumors.
- So-called fibrohistiocytic tumors (intermediate, rarely metastasizing).
- Giant cell tumors of soft tissue.
- Plexiform fibrohistiocytic tumor.
- Tumors of peripheral nerves.
- Pericytic (perivascular) tumors.
- Malignant glomus tumor and variants.
- Myopericytoma.
- Angioleiomyoma.
- Myofibroma.
- Infantile myofibroma (previously called hemangiopericytoma [infantile]).
- Myofibromatosis.
- Infantile myofibromatosis.
- Tumors of uncertain differentiation.
- Alveolar soft part sarcoma.
- Clear cell sarcoma of soft tissue.
- Desmoplastic small round cell tumor.
- Epithelioid sarcoma.
- Extrarenal rhabdoid tumor.
- Extraskeletal myxoid chondrosarcoma.
- Neoplasms with perivascular epithelioid cell differentiation (PEComa NOS, malignant).
- Primitive neuroectodermal tumor/extraskeletal Ewing tumor.
- Synovial sarcoma (NOS, spindle cell, and biphasic varieties).
- Undifferentiated/unclassified sarcomas.
- Undifferentiated epithelial sarcoma.
- Undifferentiated pleomorphic sarcoma.
- Undifferentiated round cell sarcoma.
- Undifferentiated sarcoma; sarcoma, NOS.[6]
- Undifferentiated spindle cell sarcoma.
- Vascular tumors.
References
- Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-6.
- Brodowicz T, Schwameis E, Widder J, et al.: Intensified Adjuvant IFADIC Chemotherapy for Adult Soft Tissue Sarcoma: A Prospective Randomized Feasibility Trial. Sarcoma 4 (4): 151-60, 2000. [PUBMED Abstract]
- Dantonello TM, Int-Veen C, Leuschner I, et al.: Mesenchymal chondrosarcoma of soft tissues and bone in children, adolescents, and young adults: experiences of the CWS and COSS study groups. Cancer 112 (11): 2424-31, 2008. [PUBMED Abstract]
- Steelman C, Katzenstein H, Parham D, et al.: Unusual presentation of congenital infantile fibrosarcoma in seven infants with molecular-genetic analysis. Fetal Pediatr Pathol 30 (5): 329-37, 2011. [PUBMED Abstract]
- Evans HL: Low-grade fibromyxoid sarcoma: a clinicopathologic study of 33 cases with long-term follow-up. Am J Surg Pathol 35 (10): 1450-62, 2011. [PUBMED Abstract]
- Alaggio R, Collini P, Randall RL, et al.: Undifferentiated high-grade pleomorphic sarcomas in children: a clinicopathologic study of 10 cases and review of literature. Pediatr Dev Pathol 13 (3): 209-17, 2010 May-Jun. [PUBMED Abstract]
Staging and Grading Systems for Childhood Soft Tissue Sarcoma
Clinical staging has an important role in predicting the clinical outcome and determining the most effective therapy for pediatric soft tissue sarcomas. As yet, there is no well-accepted staging system that is applicable to all childhood sarcomas. The system from the American Joint Committee on Cancer (AJCC) that is used for adults has not been validated in pediatric studies. Although a standardized staging system for pediatric nonrhabdomyosarcomatous soft tissue sarcoma does not exist, two systems are currently in use for staging pediatric nonrhabdomyosarcomatous soft tissue sarcoma.[1]
- Surgico-pathologic staging system: The surgico-pathologic staging system used by the Intergroup Rhabdomyosarcoma Study (see below) is based on the amount, or extent, of tumor that remains after initial surgery and whether the disease has metastasized. This staging system was used in early pediatric trials.[2]
- TNM staging system: The TNM staging system is a collaborative effort between the AJCC (United States) and the International Union Against Cancer (worldwide). Staging is based on the extent of the tumor (T), the extent of spread to the lymph nodes (N), and the presence of metastasis (M). Refer to Tables 3, 4, 5, and 6 for the staging of soft tissue sarcoma from the eighth edition of the AJCC Cancer Staging Manual.[3-7] The last Children's Oncology Group trial used the sixth edition AJCC Cancer Staging Manual for soft tissue sarcoma (with central pathology review).[1] A review of children with non-rhabdomyosarcoma soft tissue sarcomas was performed with data from the Surveillance, Epidemiology, and End Results (SEER) program and identified 941 patients between 1988 and 2007.[8] The COG risk stratification was validated in this cohort.
Intergroup Rhabdomyosarcoma Study Staging System
Nonmetastatic disease
- Group I: Localized tumor completely resected with histologically negative margins.
- Group II: Grossly resected tumor with microscopic residual tumor at the margin(s) and/or extension into regional lymph nodes.
- IIA: Localized, grossly resected tumor with microscopic residual disease.
- IIB: Regional disease with involved nodes completely resected with no microscopic disease. The most proximal (to the patient, most distal to the tumor) regional lymph node must be negative.
- IIC: Regional disease with involved nodes grossly resected but with evidence of residual microscopic disease at the primary site and/or histologic involvement of the most proximal regional lymph node in the dissection.
- Group III: Localized tumor, incompletely resected, or biopsy only, with gross residual tumor.
Metastatic disease
- Group IV: Any localized or regional tumor with distant metastases present at the time of diagnosis. This includes the presence of malignant cells in effusions (pleural, peritoneal) and/or cerebrospinal fluid (rare).
Recurrent/progressive disease
- Any soft tissue sarcoma that recurs after initial treatment or progresses after radiation therapy, chemotherapy, or initial surgery.
TNM Staging System
The eighth edition of the AJCC Cancer Staging Manual has designated staging by the four criteria of tumor size, nodal status, histologic grade, and metastasis and by anatomic primary tumor site (head and neck; trunk and extremities; abdomen and thoracic visceral organs; retroperitoneum; and unusual histologies and sites) (refer to Tables 3, 4, 5, and 6).[3-7] For information on unusual histologies and sites, refer to the AJCC Cancer Staging Manual.[7]
| T Category | Soft Tissue Sarcoma of the Trunk, Extremities, and Retroperitoneum | Soft Tissue Sarcoma of the Head and Neck | Soft Tissue Sarcoma of the Abdomen and Thoracic Visceral Organs |
|---|---|---|---|
| aAdapted from O'Sullivan et al.,[3] Yoon et al.,[4] Raut et al.,[5] and Pollock et al.[6] | |||
| TX | Primary tumor cannot be assessed. | Primary tumor cannot be assessed. | Primary tumor cannot be assessed. |
| T0 | No evidence of primary tumor. | ||
| T1 | Tumor ≤5 cm in greatest dimension. | Tumor ≤2 cm. | Organ confined. |
| T2 | Tumor >5 cm and ≤10 cm in greatest dimension. | Tumor >2 to ≤4 cm. | Tumor extension into tissue beyond organ. |
| T2a | Invades serosa or visceral peritoneum. | ||
| T2b | Extension beyond serosa (mesentery). | ||
| T3 | Tumor >10 cm and ≤15 cm in greatest dimension. | Tumor >4 cm. | Invades another organ. |
| T4 | Tumor >15 cm in greatest dimension. | Tumor with invasion of adjoining structures. | Multifocal involvement. |
| T4a | Tumor with orbital invasion, skull base/dural invasion, invasion of central compartment viscera, involvement of facial skeleton, or invasion of pterygoid muscles. | Multifocal (2 sites). | |
| T4b | Tumor with brain parenchymal invasion, carotid artery encasement, prevertebral muscle invasion, or central nervous system involvement via perineural spread. | Multifocal (3–5 sites). | |
| T4c | Multifocal (>5 sites). | ||
| aAdapted from O'Sullivan et al.,[3] Yoon et al.,[4] Raut et al.,[5] and Pollock et al.[6] | |
| bFor soft tissue sarcoma of the abdomen and thoracic visceral organs, N0 = no lymph node involvement or unknown lymph node status and N1 = lymph node involvement present. | |
| N0 | No regional lymph node metastasis or unknown lymph node status.b |
| N1 | Regional lymph node metastasis.b |
| aAdapted from O'Sullivan et al.,[3] Yoon et al.,[4] Raut et al.,[5] and Pollock et al.[6] | |
| bFor soft tissue sarcoma of the abdomen and thoracic visceral organs, M0 = no metastases and M1 = metastases present. | |
| M0 | No distant metastasis.b |
| M1 | Distant metastasis.b |
| Stage | T | N | M | Grade |
|---|---|---|---|---|
| aAdapted from Yoon et al. [4] and Pollock et al.[6] | ||||
| bStage IIIB for soft tissue sarcoma of the retroperitoneum; stage IV for soft tissue sarcoma of the trunk and extremities. | ||||
| IA | T1 | N0 | M0 | G1, GX |
| IB | T2, T3, T4 | N0 | M0 | G1, GX |
| II | T1 | N0 | M0 | G2, G3 |
| IIIA | T2 | N0 | M0 | G2, G3 |
| IIIB | T3, T4 | N0 | M0 | G2, G3 |
| IIIB/IVb | Any T | N1 | M0 | Any G |
| IV | Any T | Any N | M1 | Any G |
Soft Tissue Sarcoma Tumor Pathological Grading System
In most cases, accurate histopathologic classification alone of soft tissue sarcomas does not yield optimal information about their clinical behavior. Therefore, several histologic parameters are evaluated in the grading process, including the following:
- Degree of cellularity.
- Cellular pleomorphism.
- Mitotic activity.
- Degree of necrosis.
- Invasive growth.
This process is used to improve the correlation between histologic findings and clinical outcome.[9] In children, grading of soft tissue sarcoma is compromised by the good prognosis of certain tumors, such as infantile fibrosarcoma and hemangiopericytoma, which have a good prognosis in children younger than 4 years, and also angiomatoid fibrous histiocytoma and dermatofibrosarcoma protuberans, which may recur locally if incompletely excised, but usually do not metastasize.
Testing the validity of a grading system within the pediatric population is difficult because of the rarity of these neoplasms. In March 1986, the Pediatric Oncology Group (POG) conducted a prospective study on pediatric soft tissue sarcomas other than rhabdomyosarcoma and devised the POG grading system. Analysis of outcome for patients with localized soft tissue sarcomas other than rhabdomyosarcoma demonstrated that patients with grade 3 tumors fared significantly worse than those with grade 1 or grade 2 lesions. This finding suggests that this system can accurately predict the clinical behavior of nonrhabdomyosarcomatous soft tissue sarcoma.[9-11]
The grading systems developed by the POG and the French Federation of Comprehensive Cancer Centers (Fédération Nationale des Centres de Lutte Contre Le Cancer [FNCLCC]) Sarcoma Group are described below. These grading systems are being compared by the central review pathologists on the COG-ARST0332 study. The study has closed and results are pending.
POG grading system
The POG grading system is described below.[9] It is an older grading system of historical value that is no longer being used for treatment.
Grade I
Grade I lesions are based on histologic type, well-differentiated cytohistologic features, and/or age of the patient.
- Angiomatoid fibrous histiocytoma.
- Dermatofibrosarcoma protuberans.
- Liposarcoma–myxoid or well-differentiated.
- Myxoid chondrosarcoma.
- Well-differentiated malignant peripheral nerve sheath tumor.
- Well-differentiated or infantile (aged ≤4 years) fibrosarcoma.
- Well-differentiated or infantile (aged ≤4 years) hemangiopericytoma.
Grade II
Grade II lesions are soft tissue sarcomas not included in grade I or III by histologic diagnosis (with <5 mitoses/10 high-power fields or <15% necrosis):
- 15% or less of the surface area shows necrosis (primary criteria).
- The mitotic count is <5 mitotic figures per 10 high-power fields (40X objective) (primary criteria).
- Nuclear atypia is not marked (secondary criteria).
- The tumor is not markedly cellular (secondary criteria).
Grade III
Grade III lesions are similar to grade II lesions and include certain tumors known to be clinically aggressive by virtue of histologic diagnosis and non-grade I tumors (with >4 mitoses per 10 high-power fields or >15% necrosis):
- Alveolar soft part sarcoma.
- Extraskeletal osteogenic sarcoma.
- Malignant triton tumor.
- Mesenchymal chondrosarcoma.
- Pleomorphic or round-cell liposarcoma.
- Any other sarcoma not in grade I with >15% necrosis and/or ≥5 mitotic figures per 10 high-power fields (40X objective). Marked atypia and cellularity are less predictive but may assist in placing tumors in this category.
FNCLCC grading system
The FNCLCC histologic grading system was developed for adults with soft tissue sarcoma. The purpose of the grading system is to predict which patients will develop metastasis and subsequently benefit from postoperative chemotherapy.[12,13] The system is described in Table 7 and Table 8.
| FNCLCC = Fédération Nationale des Centres de Lutte Contre Le Cancer; HPF = high-power field. | |
| Tumor Differentiation | |
| Score 1 | Sarcoma closely resembling normal adult mesenchymal tissue (e.g., well-differentiated liposarcoma) |
| Score 2 | Sarcomas for which histologic typing is certain (e.g., myxoid liposarcoma) |
| Score 3 | Embryonal and undifferentiated sarcomas, sarcomas of doubtful type, and synovial sarcomas |
| Mitotic Count | |
| Score 1 | 0–9 mitoses per 10 HPF |
| Score 2 | 10–19 mitoses per 10 HPF |
| Score 3 | ≥20 mitoses per 10 HPF |
| Tumor Necrosis | |
| Score 0 | No necrosis |
| Score 1 | <50% tumor necrosis |
| Score 2 | ≥50% tumor necrosis |
| Total Score | Histologic Grade |
|---|---|
| 2–3 | Grade I |
| 4–5 | Grade II |
| 6–8 | Grade III |
Prognostic Significance of Tumor Grading
The POG and FNCLCC grading systems have proven to be of prognostic value in pediatric and adult nonrhabdomyosarcomatous soft tissue sarcomas.[14-18] In a study of 130 tumors from children and adolescents with nonrhabdomyosarcomatous soft tissue sarcoma enrolled in three prospective clinical trials, a correlation was found between the POG-assigned grade and the FNCLCC-assigned grade. However, grading did not correlate in all cases; 44 patients whose tumors received discrepant grades (POG grade 3, FNCLCC grade 1 or 2) had outcomes between concurrent grade 3 and grades 1 and 2. A mitotic index of 10 or greater emerged as an important prognostic factor.[19] The recently completed COG-ARST0332 trial will analyze data comparing the POG and FNCLCC pathologic grading systems to determine which system better correlates with clinical outcomes. The current open trial (ARST1321 [NCT02180867]) uses the FNCLCC system to assign histological grade.
References
- American Joint Committee on Cancer: AJCC Cancer Staging Manual. 6th ed. New York, NY: Springer, 2002.
- Maurer HM, Beltangady M, Gehan EA, et al.: The Intergroup Rhabdomyosarcoma Study-I. A final report. Cancer 61 (2): 209-20, 1988. [PUBMED Abstract]
- O'Sullivan B, Maki RG, Agulnik M, et al.: Soft tissue sarcoma of the head and neck. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 499-505.
- Yoon SS, Maki RG, Asare EA, et al.: Soft tissue sarcoma of the trunk and extremities. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 507-15.
- Raut CP, Maki RG, Baldini EH, et al.: Soft tissue sarcoma of the abdomen and thoracic visceral organs. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 517-21.
- Pollock RE, Maki RG, Baldini EH, et al.: Soft tissue sarcoma of the retroperitoneum. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 531-7.
- Maki RG, Folpe AL, Guadagnolo BA, et al.: Soft tissue sarcoma - unusual histologies and sites. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 539-45.
- Waxweiler TV, Rusthoven CG, Proper MS, et al.: Non-Rhabdomyosarcoma Soft Tissue Sarcomas in Children: A Surveillance, Epidemiology, and End Results Analysis Validating COG Risk Stratifications. Int J Radiat Oncol Biol Phys 92 (2): 339-48, 2015. [PUBMED Abstract]
- Parham DM, Webber BL, Jenkins JJ 3rd, et al.: Nonrhabdomyosarcomatous soft tissue sarcomas of childhood: formulation of a simplified system for grading. Mod Pathol 8 (7): 705-10, 1995. [PUBMED Abstract]
- Recommendations for the reporting of soft tissue sarcomas. Association of Directors of Anatomic and Surgical Pathology. Mod Pathol 11 (12): 1257-61, 1998. [PUBMED Abstract]
- Skytting B, Meis-Kindblom JM, Larsson O, et al.: Synovial sarcoma--identification of favorable and unfavorable histologic types: a Scandinavian sarcoma group study of 104 cases. Acta Orthop Scand 70 (6): 543-54, 1999. [PUBMED Abstract]
- Coindre JM, Terrier P, Guillou L, et al.: Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 91 (10): 1914-26, 2001. [PUBMED Abstract]
- Guillou L, Coindre JM, Bonichon F, et al.: Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 15 (1): 350-62, 1997. [PUBMED Abstract]
- Rao BN: Nonrhabdomyosarcoma in children: prognostic factors influencing survival. Semin Surg Oncol 9 (6): 524-31, 1993 Nov-Dec. [PUBMED Abstract]
- Pisters PW, Leung DH, Woodruff J, et al.: Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 14 (5): 1679-89, 1996. [PUBMED Abstract]
- Coindre JM, Terrier P, Bui NB, et al.: Prognostic factors in adult patients with locally controlled soft tissue sarcoma. A study of 546 patients from the French Federation of Cancer Centers Sarcoma Group. J Clin Oncol 14 (3): 869-77, 1996. [PUBMED Abstract]
- Pappo AS, Fontanesi J, Luo X, et al.: Synovial sarcoma in children and adolescents: the St Jude Children's Research Hospital experience. J Clin Oncol 12 (11): 2360-6, 1994. [PUBMED Abstract]
- Pratt CB, Maurer HM, Gieser P, et al.: Treatment of unresectable or metastatic pediatric soft tissue sarcomas with surgery, irradiation, and chemotherapy: a Pediatric Oncology Group study. Med Pediatr Oncol 30 (4): 201-9, 1998. [PUBMED Abstract]
- Khoury JD, Coffin CM, Spunt SL, et al.: Grading of nonrhabdomyosarcoma soft tissue sarcoma in children and adolescents: a comparison of parameters used for the Fédération Nationale des Centers de Lutte Contre le Cancer and Pediatric Oncology Group Systems. Cancer 116 (9): 2266-74, 2010. [PUBMED Abstract]
Treatment Option Overview for Childhood Soft Tissue Sarcoma
Because of the rarity of pediatric nonrhabdomyosarcomatous soft tissue sarcomas, coordination of treatment by a multidisciplinary team comprising oncologists (pediatric or medical), pathologists, surgeons, and radiation oncologists should be considered for all children, adolescents, and young adults with these tumors. In addition, to better define the tumors' natural history and response to therapy, entry into national or institutional treatment protocols should be considered for children with rare neoplasms. Information about ongoing clinical trials is available from the NCI website.
Surgery
After an appropriate biopsy and pathologic diagnosis, every attempt is made to resect the primary tumor with negative margins before or after chemotherapy and/or radiation therapy. Involvement of a surgeon with special expertise in the resection of soft tissue sarcomas in the decision is highly desirable.
The timing of surgery depends on an assessment of the feasibility and morbidity of surgery. If the initial operation fails to achieve pathologically negative tissue margins or if the initial surgery was done without the knowledge that cancer was present, a re-excision of the affected area is performed to obtain clear, but not necessarily wide, margins.[1-4] This surgical tenet is true even if no mass is detected by magnetic resonance imaging after initial surgery.[5]; [6][Level of evidence: 3iiA]
Regional lymph node metastases at diagnosis are unusual and are most often seen in patients with epithelioid and clear cell sarcomas.[7,8] Various institutional series have demonstrated the feasibility and effectiveness of sentinel node biopsy as a staging procedure in pediatric patients with soft tissue sarcomas.[9-14]
Radiation Therapy
Considerations for radiation therapy are based on the potential for surgery, with or without chemotherapy, to obtain local control without loss of critical organs or significant functional, cosmetic, or psychological impairment. This will vary according to the following:
- Patient variables (e.g., age and sex).
- Tumor variables (e.g., histopathology, site, size, and grade).
- Surgical margin status.
- Expectations for radiation-induced morbidities (e.g., impaired bone or muscle development, organ damage, or second malignancy).
Radiation therapy can be given preoperatively. Radiation field size and dose will be based on patient and tumor variables and the operability of the tumor. Preoperative radiation therapy has been associated with excellent local control rates.[15,16] This approach has the advantage of treating smaller tissue volumes because it does not necessitate treating a postsurgical bed; it also has the advantage of somewhat lower radiation doses because relative hypoxia from surgical disruption of vasculature and scarring is not present. Preoperative radiation therapy has been associated with an increased rate of wound complications in adults, primarily in lower extremity tumors, but the degree of this is questionable.[17] Conversely, preoperative radiation therapy may lead to less fibrosis than with postoperative approaches, perhaps due to the smaller treatment volume and dose.[18]
Retroperitoneal sarcomas are unique in that radiosensitivity of the bowel to injury makes postoperative radiation therapy less desirable.[19,20] Postoperative adhesions and bowel immobility can increase the risk of damage from any given radiation dose. This contrasts with the preoperative approach in which the tumor often displaces bowel outside of the radiation field, and any exposed bowel is more mobile, which decreases exposure to specific bowel segments.
Radiation therapy can also be given postoperatively. In general, radiation is indicated for patients with inadequate surgical margins and for larger, high-grade tumors.[21,22] This is particularly important in high-grade tumors with tumor margins smaller than 1 cm.[23,24]; [25][Level of evidence: 3iiDiv] With combined surgery and radiation therapy, local control of the primary tumor can be achieved in more than 80% of patients.[26,27]
Brachytherapy and intraoperative radiation may be applicable in select situations.[27-29]; [30][Level of evidence: 3iiiDii]
Radiation volume and dose depend on the patient, tumor, and surgical variables noted above, as well as the following:
- Patient age and growth potential.
- Ability to avoid critical organs, epiphyseal plates, and lymphatics (but not the neurovascular bundles that are relatively radiation tolerant).
- Functional/cosmetic outcome.
Radiation doses are typically 45 Gy to 50 Gy preoperatively, with consideration for postoperative boost of 10 Gy to 20 Gy if resection margins are microscopically or grossly positive, or planned brachytherapy if the resection is predicted to be subtotal. However, data documenting the efficacy of a postoperative boost are lacking.[31] The postoperative radiation dose is 55 Gy to 60 Gy, or rarely, higher when unresectable gross residual disease exists.
Radiation margins are typically 2 cm to 4 cm longitudinally and encompass fascial planes axially.[32,33]
Chemotherapy
The role of postoperative chemotherapy remains unclear as evidenced by the following studies:[34]
- A meta-analysis of data from all randomized trials of adults with soft tissue sarcoma concluded that recurrence-free survival was better with postoperative chemotherapy for patients with high-grade tumors larger than 5 cm.[35]
- In a European trial, adults with completely resected soft tissue sarcoma were randomly assigned to observation or postoperative chemotherapy with ifosfamide and doxorubicin. Postoperative chemotherapy was not associated with improved event-free survival (EFS) or overall survival (OS). It is difficult to extrapolate this trial to pediatric patients because the trial included 1) a wide variety of histologies; 2) a relatively low dose of ifosfamide; 3) patients assigned to chemotherapy had definitive radiation delayed until completion of chemotherapy; and 4) almost one-half of the patients in the trial had intermediate-grade tumors. In the discussion section, the authors merged their patients with previously published series, including those from the European meta-analysis, and concluded that the results suggested a benefit for postoperative chemotherapy.[36][Level of evidence: 1iiA]
- The largest prospective pediatric trial failed to demonstrate any benefit with postoperative vincristine, dactinomycin, cyclophosphamide, and doxorubicin.[26]
- Doxorubicin and ifosfamide were used in the risk-based COG ARST0332 (NCT00346164) trial. Although this was not a randomized study, results at 2.6 years show that patients with high-risk (>5 cm and high grade), grossly resected, nonmetastatic tumors who were treated with radiation therapy and postoperative doxorubicin and ifosfamide had a 3-year EFS of 68% and OS of 81%. In patients with metastatic disease treated with preoperative chemotherapy and radiation therapy, the estimated 3-year failure-free survival was 52% and OS was 66%.[37][Level of evidence: 3iiiA]
Targeted Therapy
The use of angiogenesis and mammalian target of rapamycin (mTOR) inhibitors has been explored in the treatment of adult soft tissue sarcomas but not in pediatrics.
- In a trial of 711 randomly assigned adult patients who achieved a response or stable disease after chemotherapy, the administration of ridaforolimus was associated with a 3-week improvement in progression-free survival (PFS) when compared with placebo.[38]
- In another trial of 371 randomly assigned adult patients with metastatic soft tissue sarcoma that progressed after chemotherapy, pazopanib was compared with placebo. The median PFS for the pazopanib arm was 4.6 months compared with 1.6 months for the placebo arm. OS was not different between the two arms.[39]
- In a randomized study of 182 previously treated adult patients with recurrent liposarcoma, leiomyosarcoma, synovial sarcoma, and other sarcomas, patients with nonadipocytic tumors who were treated with regorafenib had significant improvements in progression-free survival when compared with patients who were treated with placebo.[40]
Special Considerations for the Treatment of Children With Soft Tissue Sarcoma
Cancer in children and adolescents is rare, although the overall incidence of childhood cancer has been slowly increasing since 1975.[41] Children and adolescents with cancer should be referred to medical centers that have a multidisciplinary team of cancer specialists with experience treating the cancers that occur during childhood and adolescence. This multidisciplinary team approach incorporates the skills of the following health care professionals and others to ensure that children receive treatment, supportive care, and rehabilitation that will achieve optimal survival and quality of life:
- Primary care physicians.
- Pediatric surgical specialists.
- Pediatric radiation oncologists.
- Pediatric medical oncologists/hematologists.
- Rehabilitation specialists.
- Pediatric nurse specialists.
- Social workers.
- Child life professionals.
- Psychologists.
(Refer to the PDQ Supportive and Palliative Care summaries for specific information about supportive care for children and adolescents with cancer.)
Guidelines for pediatric cancer centers and their role in the treatment of pediatric patients with cancer have been outlined by the American Academy of Pediatrics.[42] At these pediatric cancer centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate in these trials is offered to most patients/families. Multidisciplinary evaluation in pediatric cancer centers that have surgical and radiotherapeutic expertise is of critical importance to ensure the best clinical outcome for these patients. Although surgery with or without radiation therapy can be curative for a significant proportion of patients, the addition of chemotherapy might benefit subsets of children with the disease; therefore, enrollment into clinical trials is encouraged. Clinical trials for children and adolescents with cancer are generally designed to compare potentially better therapy with therapy that is currently accepted as standard. Most of the progress made in identifying curative therapies for childhood cancers has been achieved through clinical trials. Information about ongoing clinical trials is available from the NCI website.
Many therapeutic strategies for children and adolescents with soft tissue tumors are similar to those for adult patients, although there are important differences. For example, the biology of the neoplasm in pediatric patients may differ dramatically from that of the adult lesion. Additionally, limb-sparing procedures are more difficult to perform in pediatric patients. The morbidity associated with radiation therapy, particularly in infants and young children, may be much greater than that observed in adults.[43]
Improved outcomes with multimodality therapy in adults and children with soft tissue sarcomas over the past 20 years has caused increasing concern about the potential long-term side effects of this therapy in children, especially when considering the expected longer life span of children versus adults. Therefore, to maximize tumor control and minimize long-term morbidity, treatment must be individualized for children and adolescents with nonrhabdomyosarcomatous soft tissue sarcoma. These patients should be enrolled in prospective studies that accurately assess any potential complications.[44]
References
- Sugiura H, Takahashi M, Katagiri H, et al.: Additional wide resection of malignant soft tissue tumors. Clin Orthop (394): 201-10, 2002. [PUBMED Abstract]
- Cecchetto G, Guglielmi M, Inserra A, et al.: Primary re-excision: the Italian experience in patients with localized soft-tissue sarcomas. Pediatr Surg Int 17 (7): 532-4, 2001. [PUBMED Abstract]
- Chui CH, Spunt SL, Liu T, et al.: Is reexcision in pediatric nonrhabdomyosarcoma soft tissue sarcoma necessary after an initial unplanned resection? J Pediatr Surg 37 (10): 1424-9, 2002. [PUBMED Abstract]
- Paulino AC, Ritchie J, Wen BC: The value of postoperative radiotherapy in childhood nonrhabdomyosarcoma soft tissue sarcoma. Pediatr Blood Cancer 43 (5): 587-93, 2004. [PUBMED Abstract]
- Kaste SC, Hill A, Conley L, et al.: Magnetic resonance imaging after incomplete resection of soft tissue sarcoma. Clin Orthop (397): 204-11, 2002. [PUBMED Abstract]
- Chandrasekar CR, Wafa H, Grimer RJ, et al.: The effect of an unplanned excision of a soft-tissue sarcoma on prognosis. J Bone Joint Surg Br 90 (2): 203-8, 2008. [PUBMED Abstract]
- Daigeler A, Kuhnen C, Moritz R, et al.: Lymph node metastases in soft tissue sarcomas: a single center analysis of 1,597 patients. Langenbecks Arch Surg 394 (2): 321-9, 2009. [PUBMED Abstract]
- Mazeron JJ, Suit HD: Lymph nodes as sites of metastases from sarcomas of soft tissue. Cancer 60 (8): 1800-8, 1987. [PUBMED Abstract]
- Neville HL, Andrassy RJ, Lally KP, et al.: Lymphatic mapping with sentinel node biopsy in pediatric patients. J Pediatr Surg 35 (6): 961-4, 2000. [PUBMED Abstract]
- Neville HL, Raney RB, Andrassy RJ, et al.: Multidisciplinary management of pediatric soft-tissue sarcoma. Oncology (Huntingt) 14 (10): 1471-81; discussion 1482-6, 1489-90, 2000. [PUBMED Abstract]
- Kayton ML, Delgado R, Busam K, et al.: Experience with 31 sentinel lymph node biopsies for sarcomas and carcinomas in pediatric patients. Cancer 112 (9): 2052-9, 2008. [PUBMED Abstract]
- Dall'Igna P, De Corti F, Alaggio R, et al.: Sentinel node biopsy in pediatric patients: the experience in a single institution. Eur J Pediatr Surg 24 (6): 482-7, 2014. [PUBMED Abstract]
- Parida L, Morrisson GT, Shammas A, et al.: Role of lymphoscintigraphy and sentinel lymph node biopsy in the management of pediatric melanoma and sarcoma. Pediatr Surg Int 28 (6): 571-8, 2012. [PUBMED Abstract]
- Alcorn KM, Deans KJ, Congeni A, et al.: Sentinel lymph node biopsy in pediatric soft tissue sarcoma patients: utility and concordance with imaging. J Pediatr Surg 48 (9): 1903-6, 2013. [PUBMED Abstract]
- Virkus WW, Mollabashy A, Reith JD, et al.: Preoperative radiotherapy in the treatment of soft tissue sarcomas. Clin Orthop (397): 177-89, 2002. [PUBMED Abstract]
- Zagars GK, Ballo MT, Pisters PW, et al.: Preoperative vs. postoperative radiation therapy for soft tissue sarcoma: a retrospective comparative evaluation of disease outcome. Int J Radiat Oncol Biol Phys 56 (2): 482-8, 2003. [PUBMED Abstract]
- O'Sullivan B, Davis AM, Turcotte R, et al.: Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 359 (9325): 2235-41, 2002. [PUBMED Abstract]
- Davis AM, O'Sullivan B, Turcotte R, et al.: Late radiation morbidity following randomization to preoperative versus postoperative radiotherapy in extremity soft tissue sarcoma. Radiother Oncol 75 (1): 48-53, 2005. [PUBMED Abstract]
- Baldini EH, Wang D, Haas RL, et al.: Treatment Guidelines for Preoperative Radiation Therapy for Retroperitoneal Sarcoma: Preliminary Consensus of an International Expert Panel. Int J Radiat Oncol Biol Phys 92 (3): 602-12, 2015. [PUBMED Abstract]
- Bishop AJ, Zagars GK, Torres KE, et al.: Combined Modality Management of Retroperitoneal Sarcomas: A Single-Institution Series of 121 Patients. Int J Radiat Oncol Biol Phys 93 (1): 158-65, 2015. [PUBMED Abstract]
- Marcus KC, Grier HE, Shamberger RC, et al.: Childhood soft tissue sarcoma: a 20-year experience. J Pediatr 131 (4): 603-7, 1997. [PUBMED Abstract]
- Delaney TF, Kepka L, Goldberg SI, et al.: Radiation therapy for control of soft-tissue sarcomas resected with positive margins. Int J Radiat Oncol Biol Phys 67 (5): 1460-9, 2007. [PUBMED Abstract]
- Blakely ML, Spurbeck WW, Pappo AS, et al.: The impact of margin of resection on outcome in pediatric nonrhabdomyosarcoma soft tissue sarcoma. J Pediatr Surg 34 (5): 672-5, 1999. [PUBMED Abstract]
- Skytting B: Synovial sarcoma. A Scandinavian Sarcoma Group project. Acta Orthop Scand Suppl 291: 1-28, 2000. [PUBMED Abstract]
- Hua C, Gray JM, Merchant TE, et al.: Treatment planning and delivery of external beam radiotherapy for pediatric sarcoma: the St. Jude Children's Research Hospital experience. Int J Radiat Oncol Biol Phys 70 (5): 1598-606, 2008. [PUBMED Abstract]
- Pratt CB, Pappo AS, Gieser P, et al.: Role of adjuvant chemotherapy in the treatment of surgically resected pediatric nonrhabdomyosarcomatous soft tissue sarcomas: A Pediatric Oncology Group Study. J Clin Oncol 17 (4): 1219, 1999. [PUBMED Abstract]
- Merchant TE, Parsh N, del Valle PL, et al.: Brachytherapy for pediatric soft-tissue sarcoma. Int J Radiat Oncol Biol Phys 46 (2): 427-32, 2000. [PUBMED Abstract]
- Schomberg PJ, Gunderson LL, Moir CR, et al.: Intraoperative electron irradiation in the management of pediatric malignancies. Cancer 79 (11): 2251-6, 1997. [PUBMED Abstract]
- Nag S, Shasha D, Janjan N, et al.: The American Brachytherapy Society recommendations for brachytherapy of soft tissue sarcomas. Int J Radiat Oncol Biol Phys 49 (4): 1033-43, 2001. [PUBMED Abstract]
- Viani GA, Novaes PE, Jacinto AA, et al.: High-dose-rate brachytherapy for soft tissue sarcoma in children: a single institution experience. Radiat Oncol 3: 9, 2008. [PUBMED Abstract]
- Al Yami A, Griffin AM, Ferguson PC, et al.: Positive surgical margins in soft tissue sarcoma treated with preoperative radiation: is a postoperative boost necessary? Int J Radiat Oncol Biol Phys 77 (4): 1191-7, 2010. [PUBMED Abstract]
- Wang D, Bosch W, Kirsch DG, et al.: Variation in the gross tumor volume and clinical target volume for preoperative radiotherapy of primary large high-grade soft tissue sarcoma of the extremity among RTOG sarcoma radiation oncologists. Int J Radiat Oncol Biol Phys 81 (5): e775-80, 2011. [PUBMED Abstract]
- Bahig H, Roberge D, Bosch W, et al.: Agreement among RTOG sarcoma radiation oncologists in contouring suspicious peritumoral edema for preoperative radiation therapy of soft tissue sarcoma of the extremity. Int J Radiat Oncol Biol Phys 86 (2): 298-303, 2013. [PUBMED Abstract]
- Ferrari A: Role of chemotherapy in pediatric nonrhabdomyosarcoma soft-tissue sarcomas. Expert Rev Anticancer Ther 8 (6): 929-38, 2008. [PUBMED Abstract]
- Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet 350 (9092): 1647-54, 1997. [PUBMED Abstract]
- Woll PJ, Reichardt P, Le Cesne A, et al.: Adjuvant chemotherapy with doxorubicin, ifosfamide, and lenograstim for resected soft-tissue sarcoma (EORTC 62931): a multicentre randomised controlled trial. Lancet Oncol 13 (10): 1045-54, 2012. [PUBMED Abstract]
- Spunt SL, Million L, Anderson JR, et al.: Risk-based treatment for nonrhabdomyosarcoma soft tissue sarcomas (NRSTS) in patients under 30 years of age: Children’s Oncology Group study ARST0332. [Abstract] J Clin Oncol 32 (Suppl 15): A-10008, 2014. Also available online. Last accessed April 02, 2018.
- Demetri GD, Chawla SP, Ray-Coquard I, et al.: Results of an international randomized phase III trial of the mammalian target of rapamycin inhibitor ridaforolimus versus placebo to control metastatic sarcomas in patients after benefit from prior chemotherapy. J Clin Oncol 31 (19): 2485-92, 2013. [PUBMED Abstract]
- van der Graaf WT, Blay JY, Chawla SP, et al.: Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 379 (9829): 1879-86, 2012. [PUBMED Abstract]
- Mir O, Brodowicz T, Italiano A, et al.: Safety and efficacy of regorafenib in patients with advanced soft tissue sarcoma (REGOSARC): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol 17 (12): 1732-1742, 2016. [PUBMED Abstract]
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014. [PUBMED Abstract]
- Corrigan JJ, Feig SA; American Academy of Pediatrics: Guidelines for pediatric cancer centers. Pediatrics 113 (6): 1833-5, 2004. [PUBMED Abstract]
- Suit H, Spiro I: Radiation as a therapeutic modality in sarcomas of the soft tissue. Hematol Oncol Clin North Am 9 (4): 733-46, 1995. [PUBMED Abstract]
- Spunt SL, Million L, Coffin C: The nonrhabdomyosarcoma soft tissue sarcoma. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 827-54.
Treatment of Newly Diagnosed Childhood Soft Tissue Sarcoma
Adipocytic Tumors
Liposarcoma
Liposarcoma accounts for 3% of soft tissue sarcoma in patients younger than 20 years (refer to Table 1).
Liposarcoma is rare in the pediatric population. In a review of 182 pediatric patients with adult-type sarcomas, only 14 had a diagnosis of liposarcoma.[1] One retrospective study identified 34 patients younger than 22 years from 1960 to 2011.[2] There were roughly equal numbers of male and female patients and the median age was 18 years. In an international clinicopathological review, the characteristics of 82 cases of pediatric liposarcoma were reported. The median age was 15.5 years and females were more commonly affected.[3] In both reports, the great majority of patients had myxoid liposarcoma.
Histopathologic classification
The World Health Organization (WHO) classification for liposarcoma is as follows:
- Intermediate grade (rarely metastasizing).
- Atypical lipomatous neoplasm/well-differentiated liposarcoma. These tumors do not metastasize unless they undergo dedifferentiation.
- Malignant.
- Liposarcoma, not otherwise specified (NOS).
- Myxoid liposarcoma. Pure myxoid liposarcomas are characterized by a t(12;16)(q13;p11) translocation and can metastasize but usually have an excellent outcome in the absence of a round cell component.[4]
- Dedifferentiated liposarcoma.
- Pleomorphic liposarcoma.
Clinical presentation
The majority of liposarcomas in the pediatric and adolescent age range are low grade and located subcutaneously. Metastasis to lymph nodes is very uncommon, and the great majority of metastases are pulmonary. Tumors arising in the periphery are more likely to be low grade and myxoid. Tumors arising centrally are more likely to be high grade, pleomorphic, and present with metastasis or recur with metastasis.
Prognosis
Higher grade or central tumors are associated with a significantly higher risk of death. In a retrospective review, 5-year survival for central tumors was 42%. In the international review, seven of ten patients with pleomorphic myxoid liposarcoma died because of their disease.[3] In a retrospective study of 14 patients, 5-year survival was 78% and tumor grade, histologic subtype, and primary location correlated with survival.[2]
Treatment
Treatment options for liposarcoma include the following:
Surgery is the most important treatment for liposarcoma. After surgical resection of myxoid liposarcoma, event-free survival (EFS) and overall survival (OS) are roughly 90%. If initial surgery is incomplete, re-excision should be performed to achieve a wide margin of resection. Local recurrences have been seen and are controlled with a second resection of the tumor.
There are reports of the use of chemotherapy to decrease the size of liposarcoma before surgery to facilitate complete resection, particularly in central tumors.[10,11] The role of postoperative chemotherapy for liposarcoma is poorly defined. There does not appear to be a need for any postoperative therapy for completely resected myxoid liposarcoma. Even with the use of postoperative chemotherapy, the survival of pleomorphic liposarcoma remains poor.[12]
Trabectedin has produced encouraging responses in adults with advanced myxoid liposarcoma.[13] In one study, adult patients with recurrent liposarcoma and leiomyosarcoma were randomly assigned to treatment with either trabectedin or dacarbazine. Patients treated with trabectedin had a 45% reduction in disease progression.[14][Level of evidence: 1iiDiii] There are very limited data to support the use of trabectedin in pediatric patients.[15]
Treatment options under clinical evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma, excluding myxoid liposarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with liposarcoma are eligible for this trial.
Chondro-osseous Tumors
Chondro-osseous tumors include the following tumor subtypes:
- Extraskeletal mesenchymal chondrosarcoma.
- Extraskeletal osteosarcoma.
- Soft tissue chondroma.
Extraskeletal mesenchymal chondrosarcoma
Osseous and chondromatous neoplasms account for 0.8% of soft tissue sarcoma in patients younger than 20 years (refer to Table 1).
Histopathology and molecular features
Mesenchymal chondrosarcoma is a rare tumor characterized by small round cells and hyaline cartilage that more commonly affects young adults and has a predilection for involving the head and neck region.
Mesenchymal chondrosarcoma has been associated with consistent chromosomal rearrangement. A retrospective analysis of cases of mesenchymal chondrosarcoma identified a HEY1-NCOA2 fusion in 10 of 15 tested specimens.[16] This gene fusion was not associated with chromosomal changes that could be detected by karyotyping. In one instance, translocation t(1;5)(q42;q32) was identified in a case of mesenchymal chondrosarcoma and shown to be associated with a novel IRF2BP-CDX1 fusion gene.[17]
Prognosis
A retrospective survey of European institutions identified 113 children and adults with mesenchymal chondrosarcoma. Factors associated with better outcome included the following:[18][Level of evidence: 3iiiA]
- Lack of metastatic disease at initial presentation.
- Clear resection margins.
- Administration of postoperative chemotherapy following resection for patients with initially localized disease.
Treatment
Treatment options for extraskeletal mesenchymal chondrosarcoma include the following:
A review of 15 patients younger than 26 years from the German Cooperative Soft Tissue Sarcoma Study Group (11 with soft-tissue lesions) and the German-Austrian-Swiss Cooperative Osteosarcoma Study Group (four with primary bone lesions) protocols suggests that complete surgical removal, or incomplete resection followed by radiation therapy, is necessary for local control.[19][Level of evidence: 3iiA]
A single-institution, retrospective review identified 12 pediatric patients with mesenchymal chondrosarcoma.[20] The presence of the NCOA2 rearrangement in tumors was documented in these patients. It was also confirmed that surgical resection is necessary for cure. Eleven patients presented with localized disease and one presented with pulmonary nodules. All patients received chemotherapy—six patients before and after surgical resection and six patients only after resection. All patients received postoperative chemotherapy (most commonly ifosfamide/doxorubicin) with or without radiation therapy (median dose, 59.4 Gy). At a median follow-up of 4.8 years, 5-year disease-free survival (DFS) was 68.2% (95% CI, 39.8%–96.6%) and OS was 88.9% (95% CI, 66.9%–100%).
Extraskeletal osteosarcoma
Osseous and chondromatous neoplasms account for 0.8% of soft tissue sarcomas in patients younger than 20 years (refer to Table 1).
Extraskeletal osteosarcoma is extremely rare in the pediatric and adolescent age range. A 2003 review identified only ten case reports in the medical literature.[21]
Prognosis
Extraskeletal osteosarcoma is associated with a high risk of local recurrence and pulmonary metastasis.[22]
Treatment
Treatment options for extraskeletal osteosarcoma include the following:
- Surgery followed by chemotherapy.
(Refer to the PDQ summary on Osteosarcoma and Malignant Fibrous Histiocytoma of Bone Treatment for more information.)
Treatment options under clinical evaluation
Information about National Cancer Institute NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with extraskeletal mesenchymal chondrosarcoma and extraskeletal osteosarcoma are eligible for this trial.
Fibroblastic/Myofibroblastic Tumors
Fibroblastic/myofibroblastic tumors include the following tumor subtypes:
- Fibroblastic/myofibroblastic tumors.
- Intermediate grade (locally aggressive).
- Desmoid-type fibromatosis (previously called desmoid tumor or aggressive fibromatoses).
- Giant cell fibroblastoma.
- Lipofibromatosis.
- Palmar/plantar fibromatosis.
- Intermediate grade (rarely metastasizing).
- Dermatofibrosarcoma protuberans.
- Infantile fibrosarcoma.
- Inflammatory myofibroblastic tumor.
- Low-grade myofibroblastic sarcoma.
- Myxoinflammatory fibroblastic sarcoma.
- Solitary fibrous tumor.
- Malignant.
- Intermediate grade (locally aggressive).
Desmoid-type fibromatosis
Desmoid-type fibromatosis has previously been called desmoid tumors or aggressive fibromatoses.
Risk factors
A small number of desmoid-type fibromatosis tumors may occur in association with a mutation in the adenomatous polyposis coli (APC) gene (associated with intestinal polyps and a high incidence of colon cancer). In a study of 519 patients older than 10 years with a diagnosis of desmoid-type fibromatosis, 39 (7.5%, a possible underestimation) were found to have familial adenomatous polyposis (FAP).[23] The patients with FAP and desmoid-type fibromatosis were younger, more often male, and had more abdominal wall or mesenteric tumors than did patients with desmoid-type fibromatosis without FAP.
A family history of colon cancer, the presence of congenital hyperplasia of the retinal pigment epithelium,[24,25] or location of the desmoid-type fibromatosis in the abdomen or abdominal wall [23] should prompt referral to a genetic counselor. Currently, there are no general recommendations for genetic testing in children with desmoid-type fibromatosis. Pathology and molecular characteristics of the tumor only provide guidance for screening. If the tumor has a somatic CTNNB1 mutation, screening is not necessary, because the APC gene mutation has not been described in this setting. If a CTNNB1 mutation is not identified, screening for the APC mutation may be warranted.[26,27] (Refer to the Familial Adenomatous Polyposis (FAP) section of the PDQ summary on Genetics of Colorectal Cancer for more information.)
Prognosis
Desmoid-type fibromatosis has an extremely low potential to metastasize. The tumors are locally infiltrating, and surgical control can be difficult because of the need to preserve normal structures.
These tumors have a high potential for local recurrence. Desmoid-type fibromatosis has a highly variable natural history, including well documented examples of spontaneous regression.[28] Mutations in exon 3 of the beta-catenin gene are seen in over 80% of desmoid-type fibromatosis and the mutation 45F has been associated with an increased risk of disease recurrence.[29] Repeated surgical resection can sometimes bring recurrent lesions under control.[30]
Treatment
Evaluation of the benefit of interventions for treatment of desmoid-type fibromatosis has been extremely difficult, because desmoid-type fibromatosis has a highly variable natural history. Large adult series and smaller pediatric series have reported long periods of disease stabilization and even regression without systemic therapy.[30,31]; [32][Level of evidence: 3iiiDi]
Treatment options for desmoid-type fibromatosis include the following:
- Surgery.
- Observation, for tumors that are incompletely resected or recurrent that do not pose a danger to vital organs, if other treatment options are not available.[30,33-39] Whenever possible, however, the treatment of choice is complete resection.
- Chemotherapy, for unresectable or recurrent tumors.
- Other drug therapy, such as nonsteroidal anti-inflammatory drugs (NSAIDs) or antiestrogen therapy.
- Surgery preceded or followed by radiation therapy, for incompletely resected tumors or to avoid recurrence and subsequent surgery that may result in functional or cosmetic compromise.
- Radiation therapy alone, for unresectable tumors.
The treatment of choice is resection to achieve clear margins. However, a retrospective review of children who underwent surgery for desmoid-type fibromatosis at the St. Jude Children’s Research Hospital (SJCRH) reported no correlation between surgical margins and risk of recurrence.[39]
When the diagnosis is known and complete surgical excision is not feasible, and if the tumor poses significant potential for mortality or morbidity, preoperative strategies may include the following:[40,41]
- Observation.
- Chemotherapy.
- Anti-estrogen therapy.
- NSAID therapy.
- External-beam radiation therapy.
Desmoid-type fibromatosis often behaves in a nonaggressive manner. In a study that included mostly adults with extra-abdominal primary fibromatosis, nonsurgical approaches (medical and observation) had similar 3-year EFS compared with surgery.[34] In a subsequent study of adolescents and adults with abdominal wall aggressive fibromatosis, 102 patients were treated with a watch and wait approach, of which 65 patients required no further treatment at 3 years. Approximately one-third of patients had regression of the tumor.[33]
Chemotherapy regimens may include the following:
- Combination chemotherapy using vinblastine and methotrexate produced objective responses in about one-third of patients with unresectable or recurrent desmoid-type fibromatosis.[40]
- A series of mainly adult patients with FAP and unresectable desmoid-type fibromatosis that were unresponsive to hormone therapy showed that doxorubicin plus dacarbazine followed by meloxicam (an NSAID) can be safely administered and can induce responses.[42]
- Pegylated liposomal doxorubicin has been used with some responses.[43] In a series of five patients, a median progression-free interval of 29 months was reported.[44]
- Tyrosine kinase inhibitors: A small retrospective study of adults with desmoid-type fibromatosis showed objective responses to the multi-targeted kinase inhibitor sorafenib.[45][Level of evidence: 3iiiDiv] Previous studies with imatinib did not support its use.[46,47] A small series reported symptomatic improvement and stable disease in seven patients with desmoid-type fibromatosis who were treated with pazopanib.[48]
- The NOTCH pathway has been implicated in the development of desmoid tumors.[49] Partial responses to the gamma secretase inhibitor PF-03084014 have been noted in adults with desmoid-type fibromatosis.[50][Level of evidence: 3iiiDiv]
- Hydroxyurea has been used successfully to treat a few patients after other treatments, but more data are needed.[51-53]
Other drug therapy may include the following:
- NSAIDs such as sulindac have been used in single cases for desmoid-type fibromatosis; the responses seen were usually disease stabilization.[54]
- Antiestrogen treatment, usually tamoxifen, plus sulindac has also resulted in disease stabilization.[55] A prospective trial of the combination of tamoxifen and sulindac reported few side effects, although asymptomatic ovarian cysts were common in girls. This combination showed relatively little activity, as measured by rates of response and progression-free survival (PFS).[56][Level of evidence: 2Diii]
Postoperative radiation therapy is a consideration when progression would entail additional surgery that might cause functional or cosmetic compromise and if radiation is considered acceptable in terms of morbidities.
Radiation has been used for unresectable desmoid-type fibromatosis or postoperatively for tumors with inadequate resections. The potential long-term complications of radiation therapy, especially subsequent neoplasms, make using this modality less appealing in a young population.[57]
Dermatofibrosarcoma protuberans
Dermatofibrosarcoma is a rare tumor that can be present in all age groups, but many of the reported cases arise in children.[58-60] A review of 451 cases in children younger than 20 years in the SEER database found that the incidence was 1 case per 1 million, highest among black patients aged 15 to 19 years. The most common sites were trunk and extremities, which is similar to what is found in adults. Ninety-five percent of patients underwent surgery. OS was 100% at 5 years, 98% at 15 years, and 97% at 30 years. Males had decreased survival compared with females (P < .05).[61][Level of evidence: 3iA]
Molecular features
The tumor has a consistent chromosomal translocation t(17;22)(q22;q13) that juxtaposes the COL1A1 gene with the PDGF-beta gene.
Treatment
Treatment of dermatofibrosarcoma protuberans includes the following:
- Surgery.
- Surgery preceded or followed by radiation therapy.
- Radiation therapy and imatinib therapy, for unresectable or recurrent tumors.
Most dermatofibrosarcoma tumors can be cured by complete surgical resection. Wide excision with negative margins or Mohs or modified Mohs surgery will prevent most tumors from recurring.[62] Despite the locally aggressive behavior of the tumor, lymph node or visceral metastasis rarely occurs.
In retrospective reviews, postoperative radiation therapy after incomplete excision may have decreased the likelihood of recurrence.[63,64]
When surgical resection cannot be accomplished or the tumor is recurrent, treatment with imatinib has been effective.[65-67] Because metastatic disease is more likely after multiple recurrences, radiation or other adjuvant therapy should be considered in patients with recurrence that cannot be managed surgically.[59,61]
Guidelines for workup and management of dermatofibrosarcoma protuberans have been published.[68]
Infantile fibrosarcoma
There are two distinct types of fibrosarcoma in children and adolescents: infantile fibrosarcoma (also called congenital fibrosarcoma) and fibrosarcoma that is indistinguishable from fibrosarcoma seen in adults. These are two distinct pathologic diagnoses and require different treatments. Adult-type fibrosarcoma is addressed below.
Infantile fibrosarcoma usually occurs in children younger than 1 year. It occasionally occurs in children up to age 4 years. A tumor with similar morphology has been identified in older children; in these older children, the tumors do not have the t(12;15)(ETV-NTRK3) translocation that is characteristic of the younger patients.[69] In several of these patients, BRAF gene fusions have been identified.
Clinical presentation
Infantile fibrosarcoma usually presents with a rapidly growing mass, often noted at birth or even seen in prenatal ultrasound. The tumors are often quite large at the time of presentation.[70]
Molecular features
The tumor usually has a characteristic cytogenetic translocation t(12;15)(ETV-NTRK3). Infantile fibrosarcoma shares this translocation and a virtually identical histologic appearance with mesoblastic nephroma.
Prognosis
These tumors have a low incidence of metastases at diagnosis.
Treatment
Treatment options for infantile fibrosarcoma include the following:
- Surgery followed by observation.
- Surgery followed by chemotherapy.
- Chemotherapy followed by surgery.
Complete resection is curative in the majority of patients with infantile fibrosarcoma. However, the large size of the lesion frequently makes resection without major functional consequences impossible (for instance, tumors of the extremities often require amputation for complete excision). The European pediatric group has reported that observation may also be an option in patients with group II disease after surgery.[71] Twelve patients with group II disease received no further therapy and two patients relapsed. One patient obtained a complete remission after chemotherapy. Postoperative chemotherapy was administered to patients with higher group disease and those who progressed. In a subsequent study, only one of seven patients with group II disease progressed during observation; that patient achieved complete remission with chemotherapy.[72][Level of evidence: 3iiA]
Preoperative chemotherapy has made a more conservative surgical approach possible; agents active in this setting include vincristine, dactinomycin, cyclophosphamide, and ifosfamide.[73,74]; [72,75][Level of evidence: 3iiA]; [76][Level of evidence: 3iiB]
Three studies of patients with infantile fibrosarcoma suggest that an alkylator-free regimen is effective and should be used as the first treatment choice in patients with macroscopic disease.[71,72,77] Two cases with variant LMNA/NTRK1 fusions responded to crizotinib.[78,79]
A pediatric patient (aged 16 months) with refractory infantile fibrosarcoma with constitutive activation of the tropomyosin-related kinase signaling pathway from an ETS variant gene 6–neurotrophin 3 receptor gene fusion (ETV6-NTRK3) responded to LOXO-101, with a 90% reduction in tumor size after 2 months of treatment.[80]
A patient aged 2 months with infantile fibrosarcoma was initially treated with chemotherapy. At disease progression, a response was seen with pazopanib.[81]
A rare case of spontaneous regression without treatment has been reported.[82][Level of evidence: 3iiiDiv]
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- LOXO-TRK-15003 (NCT02637687) (Oral TRK Inhibitor LOXO-101 for Treatment of Advanced Pediatric Solid or Primary Central Nervous System [CNS] Tumors): A phase I trial of the pan-TRK inhibitor LOXO-101 is being conducted for children with solid tumors or brain tumors whose disease has progressed or was nonresponsive to available therapies, and for which no standard or available curative therapy exists. LOXO-101 is a highly selective inhibitor of all three TRK family kinases.
- RXDX-101-03 (NCT02650401) (Study of RXDX-101 in Children With Recurrent or Refractory Solid Tumors and Primary CNS Tumors): This is a four-part, open-label, phase I/Ib, dose-escalation study in pediatric patients with: 1) relapsed or refractory solid tumors; 2) primary CNS tumors; 3) neuroblastoma; and 4) non-neuroblastoma, extracranial solid tumors with NTRK1/2/3, ROS1 or ALK gene rearrangements. The study is designed to explore the safety, maximum tolerated dose or recommended phase II dose, pharmacokinetics, and antitumor activity of entrectinib (RXDX-101).
Inflammatory myofibroblastic tumor
Inflammatory myofibroblastic tumor is a rare mesenchymal tumor that has a predilection for children and adolescents.[83-85]
Clinical presentation
Inflammatory myofibroblastic tumors are rare tumors that affect soft tissues and visceral organs of children and young adults.[86] They rarely metastasize but tend to be locally invasive. Usual anatomical sites of disease include soft tissue, lungs, spleen, colon, and breast.[83] A review of 42 cases of pediatric inflammatory myofibroblastic tumor of the bladder was published in 2015.[87]
Molecular features
Roughly half of inflammatory myofibroblastic tumors exhibit a clonal mutation that activates the anaplastic lymphoma kinase (ALK)-receptor tyrosine kinase gene at chromosome 2p23.[88] ROS1 and PDGFR-beta kinase fusions have been identified in 8 of 11 cases (73%) who are negative for ALK by immunohistochemistry.[89][Level of evidence: 3iiiDiv]
Prognosis
Inflammatory myofibroblastic tumor recurs frequently but is rarely metastatic.[83-85]
Treatment
Treatment options for inflammatory myofibroblastic tumor include the following:
- Surgery.
- Chemotherapy.
- Steroid therapy.
- NSAID therapy.
- Targeted therapy (ALK inhibitors).
Complete surgical removal, when feasible, is the mainstay of therapy.[90] In a series of nine patients, four patients achieved continuous remission after complete resection, three patients with residual disease recurred but later achieved continuous remission, and one patient with metastatic disease responded to multiagent chemotherapy.[91][Level of evidence: 3iiA] The benefit of chemotherapy has been noted in case reports.[92] There are case reports of response to either steroids or NSAIDs.[93,94] A series of 32 patients aged 18 years and younger found that complete excision was the mainstay of therapy, although some patients were treated with steroids or cytotoxic chemotherapy. OS was 94%; three patients relapsed and two of them died of the disease. With complete excision, with or without other treatments such as steroids, there was a high survival rate for patients with this disease.[95][Level of evidence: 3iiA]
Inflammatory myofibroblastic tumors respond to crizotinib. Two adults with ALK-rearranged inflammatory myofibroblastic tumor achieved partial response with crizotinib.[96][Level of evidence: 3iiiDiv] For pediatric patients with measurable disease, the use of crizotinib achieved partial tumor responses in three of six patients with ALK-translocated inflammatory myofibroblastic tumors.[97] A case report of a patient aged 16 years with metastatic/multifocal ALK-positive inflammatory myofibroblastic tumor demonstrated a complete response and a 3-year disease-free interval with crizotinib therapy.[98] In a phase I trial of ceritinib for adult patients previously treated with ALK inhibitors, one patient with inflammatory myofibroblastic tumor had a partial response.[99] Finally, one study included 14 patients with inflammatory myofibroblastic tumor who were treated with crizotinib. With crizotinib therapy, five patients had a complete response, seven had a partial response, and the remaining two had stable disease; no patient had relapsed at the time the article was published.[100][Level of evidence: 3iiDiv]
Adult-type fibrosarcoma
These tumors lack the translocation seen in infantile fibrosarcomas. They present like the great majority of nonrhabdomyosarcomas and the management approach is similar.
Low-grade fibromyxoid sarcoma
Low-grade fibromyxoid sarcoma is a histologically deceptive soft tissue neoplasm that most commonly affects young and middle-aged adults, is commonly located deep within the extremities, and is characterized by a FUS/CREB3L3 translocation.[101,102]
Prognosis
In a review of 33 patients (three were younger than 18 years) with low grade fibromyxoid sarcoma, 21 of 33 patients developed a local recurrence after intervals of up to 15 years (median, 3.5 years) and 15 developed metastases up to 45 years (median, 5 years) from diagnosis, most commonly to the lungs and pleura, emphasizing the need for continued follow-up of these patients.[101] Even after metastases occur, the course may be indolent.[103]
In another report, 14 of 73 cases were younger than 18 years of age. In this series with a relatively short follow up (median of 24 months), only 8 of 54 patients with adequate follow up developed local (9%) or distant (6%) recurrence. This report suggests that the behavior of this tumor might be significantly better than previously reported.[104] However, because of the occurrence of late metastases, careful monitoring of these patients is warranted.
The most recent Children's Oncology Group (COG) trial (ARST0332 [NCT00346164]) enrolled 11 patients with this tumor entity. The median age at diagnosis was 13 years and males were more commonly affected. The most common sites were the lower and upper extremity (n = 9) and none of the patients had developed local or distant disease recurrence at a median follow up of 2.7 years.[105]
Treatment
Treatment options for low-grade fibromyxoid sarcoma include the following:
- Surgery.
The limited treatment information for low-grade fibromyxoid sarcoma suggest that surgery is the treatment of choice as the tumor is not very chemosensitive.[103] There are little data regarding the use of chemotherapy and/or radiation therapy in this disease. One report suggests that trabectedin may be effective in the treatment of low-grade fibromyxoid sarcoma.[106]
Myxofibrosarcoma
Myxofibrosarcoma is a rare lesion, especially in childhood. It is typically treated with complete surgical resection.
Sclerosing epithelioid fibrosarcoma
Sclerosing epithelioid fibrosarcoma is a rare malignant sarcoma that commonly harbors EWSR1 gene rearrangements and has an aggressive clinical course.[107] It is typically treated with complete surgical excision. Long-term follow-up is recommended because local recurrence and metastases can occur late.
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with infantile fibrosarcoma, inflammatory myofibroblastic tumor, low-grade myofibroblastic tumor, myxoinflammatory fibroblastic sarcoma, solitary fibrous tumor, adult-type fibrosarcoma, low-grade fibromyxoid sarcoma, myxofibrosarcoma, and sclerosing epithelioid fibrosarcoma are eligible for this trial.
Skeletal Muscle Tumors
Rhabdomyosarcoma
Refer to the PDQ summary on Childhood Rhabdomyosarcoma Treatment for more information.
Smooth Muscle Tumors
Leiomyosarcoma
Leiomyosarcoma accounts for 2% of soft tissue sarcoma in patients younger than 20 years (refer to Table 1).
Risk factors
Among 43 children with HIV/AIDS who developed tumors, eight developed Epstein-Barr virus–associated leiomyosarcoma.[108] Survivors of hereditary retinoblastoma have a statistically significant increased risk of developing leiomyosarcoma and 78% of these were diagnosed 30 or more years after the initial diagnosis of retinoblastoma.[109]
Treatment
Treatment options for leiomyosarcoma include the following:
- Chemotherapy (trabectedin).
In an open-label study of trabectedin in adult patients with recurrent sarcomas, the best overall response rate (complete remission and partial remission) was seen in patients with leiomyosarcoma (7.5%).[110] The clinical benefit rate (includes stable disease) for leiomyosarcoma was 54%. In another adult study, patients with recurrent liposarcoma and leiomyosarcoma were randomly assigned to receive treatment with either trabectedin or dacarbazine. Patients treated with trabectedin had a 45% reduction in disease progression.[14] There are no data to support the use of trabectedin in pediatric patients.
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with leiomyosarcoma are eligible for this trial.
So-called Fibrohistiocytic Tumors
So-called fibrohistiocytic tumors include the following tumor subtypes:
- Giant cell tumor of soft tissue.
- Plexiform fibrohistiocytic tumor.
Plexiform fibrohistiocytic tumor
Plexiform histiocytic tumor is a rare, low- to intermediate-grade tumor that most commonly affects children and young adults. Depending on the series, the median age at presentation ranges from 8 to 14.5 years; however, the tumor has been described in patients as young as 3 months.[111,112]
Clinical presentation
The tumor commonly arises as a painless mass in the skin or subcutaneous tissue and most often involves the upper extremities, including the fingers, hand, and wrist.[113-115] There are rare reports of spread to regional lymph nodes or the lungs.[111,115,116]
Molecular features
No consistent chromosomal anomalies have been detected but a t(4;15)(q21;q15) translocation has been reported.[117]
Prognosis
Plexiform fibrohistiocytic tumor is an intermediate-grade tumor that rarely metastasizes.
Treatment
Surgery is the treatment of choice but local recurrence has been reported in 12% to 50% of cases.[118]
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with giant cell tumors of soft tissue and plexiform fibrohistiocytic tumor are eligible for this trial.
Tumors of Peripheral Nerves
Ectomesenchymoma
Ectomesenchymoma is a rare nerve sheath tumor that mainly occurs in children. It is a biphenotypic soft tissue sarcoma with both mesenchymal and ectodermal components. Elements similar to rhabdomyosarcoma have been identified.
The German Soft Tissue Sarcoma Group (Cooperative Weichteilsarkom Studiengruppe [CWS]) reported on six patients (ages 0.2–13.5 years) registered over 14 years.[119][Level of evidence: 3iiA] The tumors were located in various sites including the extremities, abdomen, and orbit. All six patients were treated with surgery and chemotherapy directed at rhabdomyosarcoma. Two patients received radiation therapy. Three patients recurred with rhabdomyosarcoma features. Although data are scant, it appears that the tumor may respond to chemotherapy.[119]
Malignant peripheral nerve sheath tumor
Malignant peripheral nerve sheath tumors account for 5% of soft tissue sarcoma in patients younger than 20 years (refer to Table 1).
Risk factors
Malignant peripheral nerve sheath tumor can arise sporadically and in children with type 1 neurofibromatosis (NF1).[120]
Molecular features
Inactivating mutations of SUZ12 have been described in these tumors and are absent in neurofibromas.[121]
Prognosis
Features associated with a favorable prognosis include the following:[120,122-124]
- Smaller tumor size. In a multivariate analysis, only tumor size and nuclear p53 expression were found to be independent predictors of disease-specific survival.[123]
- Male sex and non-Hispanic white race.[125]
- No metastasis at presentation. A retrospective review of 140 patients with malignant peripheral nerve sheath tumor from the MD Anderson Cancer Center included children and adolescents. The disease-specific survival at 10 years was 32%. In this series, presence of metastatic disease was associated with a much worse prognosis.[123]
- Lower stage.
- Lower histologic grade.
- Extremity as the primary site.
Features associated with an unfavorable prognosis include the following:[126]
- High grade.
- Deep tumor location.
- Locally advanced stage at diagnosis.
- Macroscopically incomplete resection (R2).
For patients with localized disease in the MD Anderson Cancer Center study, there was no significant difference in outcome between patients with and without NF1.[123] In other studies, it was not clear whether the absence of NF1 is a favorable prognostic factor as it has been associated with both favorable [122] and unfavorable outcomes.[120,122,124] In the French Sarcoma Group study, NF1 was associated with other adverse prognostic features, but was not an independent predictor of poor outcome.[126] The Italian Sarcoma Group reported on outcomes after recurrence in 73 children and adolescents with malignant peripheral nerve sheath tumor.[127][Level of evidence: 3iiiA] The median overall survival after first relapse was 11 months, and the survival rates were 39.2% at 1 year and 15.8% at 5 years. The factors associated with a better prognosis for these patients who relapsed were less initial tumor invasiveness, longer time to relapse, and the achievement of a secondary complete remission (which was related to the feasibility of radical surgery).
Treatment
Treatment options for malignant peripheral nerve sheath tumor include the following:
Complete surgical removal of the tumor, whenever possible, is the mainstay of treatment.
The role of radiation therapy is difficult to assess, but durable local control of known postoperative microscopic residual tumor is not assured after radiation therapy.
Chemotherapy has achieved objective responses in childhood malignant peripheral nerve sheath tumor. A large retrospective analysis of the German and Italian experience with malignant peripheral nerve sheath tumor reported that 65% of measurable tumors had objective responses to ifosfamide-containing chemotherapy regimens, but the analysis did not conclusively demonstrate improved survival for chemotherapy.[120] This retrospective analysis also noted a trend toward improved outcome with postoperative radiation therapy.[120] A series of 37 young patients with malignant peripheral nerve sheath tumor and NF1 showed that most patients had large invasive tumors that were poorly responsive to chemotherapy; PFS was 19% and 5-year OS was 28%.[128]
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with malignant peripheral nerve sheath tumor are eligible for this trial.
- SARC023 (NCT02008877) (Ganetespib and Sirolimus in Patients With Malignant Peripheral Nerve Sheath Tumors): This trial is testing the combination of ganetespib, the heat shock protein inhibitor, and sirolimus, the mammalian target of rapamycin (mTOR) inhibitor, for the treatment of patients with unresectable or metastatic malignant peripheral nerve sheath tumors. Patients with unresectable soft tissue or bone sarcomas are eligible for phase I of the trial. Patients with unresectable malignant peripheral nerve sheath tumors are eligible for phase II of the trial. Eligibility is restricted to patients aged 18 years and older.
- NCT02601937 (A Phase 1 Study of the EZH2 Inhibitor Tazemetostat in Pediatric Subjects With Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma): Patients with INI1-negative tumors are eligible for targeted treatment with an EZH2 inhibitor. This is a phase I, open-label, dose-escalation, and dose-expansion study with a twice-daily oral dose of tazemetostat.
- ADVL1522 (NCT02452554) (Lorvotuzumab Mertansine in Treating Younger Patients with Relapsed or Refractory Wilms Tumor, Rhabdomyosarcoma, Neuroblastoma, Pleuropulmonary Blastoma, Malignant Peripheral Nerve Sheath Tumor, or Synovial Sarcoma): This is a phase II study of IMGN901 (lorvotuzumab mertansine) in children with relapsed or refractory Wilms tumor, rhabdomyosarcoma, neuroblastoma, pleuropulmonary blastoma, malignant peripheral nerve sheath tumor, and synovial sarcoma. This trial is studying the effects of IMGN901, an antibody-drug conjugate that links a potent antimitotic to antibodies that target CD56.
Malignant triton tumor
Malignant triton tumors are a variant of malignant peripheral nerve sheath tumors. They occur most often in patients with neurofibromatosis type I and consist of neurogenic and rhabdomyoblastic components. Malignant triton tumors are high-grade malignancies. They usually occur before age 35 years and are very rare in children (case reports only).[129]
Malignant triton tumors are not usually responsive to chemotherapy and radiation therapy but have been treated with rhabdomyosarcoma therapy.[129][Level of evidence: 3iiiA] (Refer to the PDQ summary on Childhood Rhabdomyosarcoma Treatment for more information.)
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with malignant triton tumor are eligible for this trial.
Pericytic (Perivascular) Tumors
Myopericytoma
Infantile hemangiopericytoma is a subtype of myopericytoma.
Hemangiopericytoma is a highly vascularized tumor of uncertain origin.
Histology
Histologically, hemangiopericytomas are composed of packed round or fusiform cells that are arranged around a complex vasculature, forming many branch-like structures. Hyalinization is often present. Infantile hemangiopericytomas have similar histology but many are multilobular with vasculature outside the tumor mass.[130]
Treatment and outcome
Treatment of infantile hemangiopericytomas includes the following:
- Chemotherapy.
In a series of 17 children, the differences in metastatic potential and response to treatment were clearly demonstrated for adult and infantile hemangiopericytomas.[131] Eleven children were older than 1 year. Several of these patients had disease in the lymph nodes or lungs. Six patients with stage II or III disease progressed and died. Three patients with stage I disease survived, although one had recurrence in the lungs. Six patients had infantile hemangiopericytoma, most were greater than stage I (5 of 6). All six patients survived and three had good responses to vincristine, actinomycin, and cyclophosphamide. Hemangiopericytoma in children younger than 1 year seems to have a better prognosis than in children older than 1 year.[132-134]
Infantile myofibromatosis
This entity is a fibrous tumor of infancy and childhood that most commonly presents in the first 2 years of life.[135] The lesion can present as a single subcutaneous nodule (myofibroma) most commonly involving the head and neck region or lesions can affect multiple skin areas, muscle, and bone (myofibromatosis).[136-139]
An autosomal dominant form of the disease has been described and it is associated with germline mutations of the PDGFRB gene.[140]
Treatment
These lesions have an excellent prognosis and can regress spontaneously.
About one-third of cases with multicentric involvement will also have visceral involvement, and the prognosis for these patients is poor.[138,139,141] The use of combination therapy with vincristine/dactinomycin and vinblastine/methotrexate have proven effective in cases of multicentric disease with visceral involvement and in cases in which the disease has progressed and has threatened the life of the patient (e.g., upper airway obstruction).[138,139,142]
Tumors of Uncertain Differentiation
Tumors of uncertain differentiation include the following tumor subtypes:
- Alveolar soft part sarcoma.
- Clear cell sarcoma of soft tissue.
- Desmoplastic small round cell tumor.
- Epithelioid sarcoma.
- Extrarenal rhabdoid tumor.
- Extraskeletal myxoid chondrosarcoma.
- Neoplasms with perivascular epithelioid cell differentiation (PEComa NOS, malignant)
- Primitive neuroectodermal tumor (PNET)/extraskeletal Ewing tumor.
- Synovial sarcoma (NOS, spindle cell, and biphasic varieties).
Alveolar soft part sarcoma
Alveolar soft parts sarcomas account for 1.4% of soft tissue sarcomas in patients younger than 20 years (refer to Table 1).
Clinical presentation
The median age at presentation is 25 years, and alveolar soft part sarcoma most commonly arises in the extremities but can occur in the oral and maxillofacial region.[143-145] Alveolar soft part sarcoma in children can present with evidence of metastatic disease.[146]
Molecular features
This tumor of uncertain histogenesis is characterized by a consistent chromosomal translocation t(X;17)(p11.2;q25) that fuses the ASPSCR1 gene with the TFE3 gene.[147,148]
Prognosis
Alveolar soft part sarcoma in children may have an indolent course.[146] Patients with alveolar soft part sarcoma may relapse several years after a prolonged period of apparent remission.[149] Because these tumors are rare, all children with alveolar soft part sarcoma should be considered for enrollment in prospective clinical trials.
In a series of 19 treated patients, one group reported a 5-year OS rate of 80%, a 91% OS rate for patients with localized disease, a 100% OS rate for patients with tumors 5 cm or smaller, and a 31% OS rate for patients with tumors larger than 5 cm.[150] In another series of 33 patients, OS was 68% at 5 years from diagnosis and 53% at 10 years from diagnosis. Survival was better for smaller tumors (≤5 cm) and completely resected tumors.[151][Level of evidence: 3iiA] Delayed metastases to the brain and lung are uncommon.[143] A retrospective review of children and young adults younger than 30 years (median age, 17 years; range, 1.5–30 years) from four institutions identified 69 patients treated primarily with surgery between 1980 and 2014.[152][Level of evidence: 3iiA] The ASPL-TFE3 translocation was present in all 26 patients tested. There were 19 patients with Intergroup Rhabdomyosarcoma Study (IRS) postsurgical staging group I tumors (28%), 7 patients with IRS group II tumors (10%), 5 patients with IRS group III tumors (7%), and 38 patients with IRS group IV tumors (55%). The 5-year EFS was 80% and the OS was 87% for the 31 patients with localized tumors (IRS postsurgical groups I, II, and III). The 5-year EFS was 7% and the OS was 61% for the 38 patients with metastatic tumors (IRS postsurgical group IV).
Treatment
Treatment options for alveolar soft part sarcoma include the following:
The standard approach is complete resection of the primary lesion.[150] If complete excision is not feasible, radiation therapy should be administered. A study from China reported on 18 patients with alveolar soft part sarcoma of the oral and maxillofacial region; 15 patients were younger than 30 years.[145][Level of evidence: 3iiDii] Surgical removal with negative margins was the primary treatment. All patients survived, and only one patient had metastatic disease recurrence.
A series of 51 pediatric patients aged 0 to 21 years with alveolar soft part sarcoma found an OS rate at 10 years of 78% and an EFS rate of about 63%. Patients with localized disease (n = 37) had a 10-year OS of 87%, and the 14 patients with metastases at diagnosis had a 10-year OS of 44%, partly resulting from surgical removal of primary tumor and lung metastases in some patients. Only 3 of 18 patients (17%) with measurable disease had a response to conventional antisarcoma chemotherapy, but two of four patients treated with sunitinib had a partial response.[143][Level of evidence: 3iiiA] There have been sporadic reports of objective responses to interferon-alpha and bevacizumab.[143,153,154]
A small retrospective study of nine adult patients with metastatic alveolar soft part sarcoma treated with sunitinib reported partial response in five patients and stable disease in two patients.[155][Level of evidence: 3iiiDiv] In a phase II trial of cediranib, an inhibitor of all three known vascular epidermal growth factor receptors, 15 of 43 adult patients (35%) with metastatic alveolar soft part sarcoma had a partial response.[156][Level of evidence: 3iiDiv]
There have been no open trials for patients with metastatic alveolar soft part sarcoma.
Treatment options under clinical evaluation for alveolar soft part sarcoma
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- NCT00942877 (Phase II Study of Cediranib [AZD2171] in Patients With Alveolar Soft Part Sarcoma): A phase II study of cediranib in patients with alveolar soft part sarcoma is being conducted in patients younger than 16 years at the Clinical Center of the National Institutes of Health.
- NCT01391962 (Sunitinib or Cediranib for Alveolar Soft Part Sarcoma): A phase II trial in which patients with metastatic alveolar soft part sarcoma are randomly assigned to either sunitinib or cediranib monotherapy, with crossover at disease progression. Patients aged 16 years and older are eligible. This study is being conducted at the Clinical Center of the National Institutes of Health.
Clear cell sarcoma of soft tissue
Clear cell sarcoma (formerly and inappropriately called malignant melanoma of soft parts) is a rare soft tissue sarcoma that typically involves the deep soft tissues of the extremities. It is also called clear cell sarcoma of tendons and aponeuroses. The tumor often affects adolescents and young adults.
Patients who have small, localized tumors with low mitotic rate and intermediate histologic grade fare best.[157]
Clinical presentation
The tumor most commonly affects the lower extremity, particularly the foot, heel, and ankle.[158,159] It has a high propensity for nodal dissemination, especially metastases to regional lymph nodes (12%–43%).[159,160] The tumor typically has an indolent clinical course.
Molecular features
Clear cell sarcoma of soft tissue is characterized by an EWS-ATF1 fusion.[161]
Treatment
Treatment options for clear cell sarcoma of soft tissue include the following:
In a series of 28 pediatric patients reported by the Italian and German Soft Tissue Cooperative Studies, the median age at diagnosis was 14 years and the lower extremity was the most common primary site (50%). Surgery with or without radiotherapy is the treatment of choice and offers the best chance for cure. In this series, 12 of 13 patients with completely resected tumors were cured. For patients with more advanced disease the outcome is poor and chemotherapy is rarely effective.[162]; [163][Level of evidence: 3iiDii]
Desmoplastic small round cell tumor
Desmoplastic small round cell tumor is a rare primitive sarcoma.
Clinical presentation
Desmoplastic small round cell tumor most frequently involves the abdomen, pelvis, or tissues around the testes, but it may occur in the kidney.[164-167] The tumor occurs more commonly in males and may spread to the lungs and elsewhere. Peritoneal and pelvic lesions frequently have widespread peritoneal implants.[168]
In a large, single-institution series of 65 patients, a correlation was made between computed tomography (CT) scans in most patients and positron-emission tomography (PET)/CT scans in 11 patients. PET/CT scans had very few false-negative results and detected metastatic sites missed on conventional CT scans.[168]
Molecular features
Cytogenetic studies of these tumors have demonstrated the recurrent translocation t(11;22)(p13;q12), which has been characterized as a fusion of the WT1 and EWS genes.[167,169] The WT1-EWS fusion confirms the diagnosis of desmoplastic small round cell tumor.
Prognosis
The overall prognosis for desmoplastic small round cell tumor remains extremely poor, with reported rates of death at 90%. Greater than 90% tumor resection either at presentation or after preoperative chemotherapy may be a favorable prognostic factor for OS.[170,171]; [172][Level of evidence: 3iiiA]
Treatment
There is no standard approach to the treatment of desmoplastic small round cell tumor.
Treatment options for desmoplastic small round cell tumor include the following:
- Surgery.
- Chemotherapy followed by surgery.
- Radiation therapy.
Complete surgical resections are rare, and the overall prognosis for desmoplastic small round cell tumor remains extremely poor, with reported rates of death at 90%. Treatment may include chemotherapy, surgery, and radiation therapy. Multiagent chemotherapy analogous to that used for sarcomas has been used, as well as total abdominal radiation therapy.[164,165,170,173-176]
A single-institution study reported that five of five patients with recurrent desmoplastic small round cell tumor had partial responses to treatment with the combination of vinorelbine, cyclophosphamide, and temsirolimus.[177]
The Center for International Blood and Marrow Transplant Research (CIBMTR) analyzed patients with desmoplastic small round cell tumor in their registry who received consolidation with high dose chemotherapy and autologous stem cell reconstitution.[178] While this retrospective registry analysis suggested some benefit for this approach, other investigators have abandoned the approach because of excessive toxicity and lack of efficacy.[170]
Epithelioid sarcoma
Epithelioid sarcoma is a rare mesenchymal tumor of uncertain histogenesis which displays multilineage differentiation.[179]
Clinical presentation
Epithelioid sarcoma commonly presents as a slowly growing firm nodule based in the deep soft tissue; the proximal type predominantly affects adults and involves the axial skeleton and proximal sites. The tumor is highly aggressive and has a propensity for lymph node metastases.
Molecular features
Epithelioid sarcoma is characterized by inactivation of the SMARCB1 gene, which is present in both conventional and proximal types of epithelioid sarcoma.[180] This abnormality leads to increased dependence on EZH2 and tumor formation.[181]
Treatment
Treatment options for epithelioid sarcoma include the following
- Chemotherapy.
- Surgery.
- Surgery preceded or followed by radiation therapy.
Patients should be carefully evaluated for the presence of involved lymph nodes; suspicious lymph nodes should be biopsied. Surgical removal of primary and recurrent tumor(s) is the most effective treatment.[182][Level of evidence: 3iiiA]
In a review of 30 pediatric patients with epithelioid sarcoma (median age at presentation, 12 years), responses to chemotherapy were reported in 40% of patients using sarcoma-based regimens, and 60% of patients were alive at 5 years after initial diagnosis.[183] A single-institution retrospective review of 20 patients, including children and adults (median age, 27.3 years), found no difference in the probability of recurrence between patients who received chemotherapy and those who did not receive chemotherapy and suggested that radiation therapy may be useful.[182]
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- NCT02601937 (A Phase 1 Study of the EZH2 Inhibitor Tazemetostat in Pediatric Subjects With Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma): Patients with INI1-negative tumors are eligible for targeted treatment with an EZH2 inhibitor. This is a phase I, open-label, dose-escalation, and dose-expansion study with a twice-daily oral dose of tazemetostat.
Extrarenal (extracranial) rhabdoid tumor
Malignant rhabdoid tumors were first described in children with renal tumors in 1981 (refer to the PDQ summary on Wilms Tumor and Other Childhood Kidney Tumors Treatment for more information) and were later found in a variety of extrarenal sites. These tumors are uncommon and highly malignant, especially in children younger than 2 years.
Extrarenal (extracranial) rhabdoid tumors account for 2% of soft tissue sarcoma in patients younger than 20 years (refer to Table 1).
Molecular features
The first sizeable series of 26 children with extrarenal extracranial malignant rhabdoid tumor of soft tissues came from patients enrolled on the Intergroup Rhabdomyosarcoma Studies I through III during a review of pathology material. Only five patients (19%) were alive without disease.[184] Later, investigation of children with atypical teratoid/rhabdoid tumors of the brain, as well as those with renal and extrarenal malignant rhabdoid tumors, found germline and acquired mutations of the SMARCB1 gene in all 29 tumors tested.[185] Rhabdoid tumors may be associated with germline mutations of the SMARCB1 gene and may be inherited from an apparently unaffected parent.[186] This observation was extended to 32 malignant rhabdoid tumors at all sites in patients whose mean age at diagnosis was 12 months.[187]
Prognosis
In a Surveillance, Epidemiology, and End Results (SEER) study of 229 patients with renal, central nervous system, and extrarenal malignant rhabdoid tumor, patients aged 2 to 18 years, limited extent of tumor, and delivery of radiation therapy were shown to affect the outcome favorably compared with other patients (P < .002 for each comparison). Site of the primary tumor was not prognostically significant. OS at 5 years was 33%.[188]
Treatment
Treatment includes surgical removal when possible, chemotherapy as used for soft tissue sarcomas (but no single regimen is currently accepted as best), and radiation therapy.[189][Level of evidence: 3iA]; [190,191][Level of evidence: 3iiiB]
Responses to alisertib have been documented in four patients with central nervous system (CNS) atypical teratoid/rhabdoid tumors.[192] (Refer to the PDQ summary on Childhood Central Nervous System Atypical Teratoid/Rhabdoid Tumor Treatment summary for more information about CNS atypical teratoid/rhabdoid tumors.)
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- NCT02601937 (A Phase 1 Study of the EZH2 Inhibitor Tazemetostat in Pediatric Subjects With Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma): Patients with INI1-negative tumors are eligible for targeted treatment with an EZH2 inhibitor. This is a phase I, open-label, dose-escalation, and dose-expansion study with a twice-daily oral dose of tazemetostat.
Extraskeletal myxoid chondrosarcoma
Extraskeletal myxoid chondrosarcoma is relatively rare among soft tissue sarcomas, representing only 2.3% of all soft tissue sarcoma.[193] It has been reported in children and adolescents.[194]
Molecular features
Extraskeletal myxoid chondrosarcoma is a multinodular neoplasm. The rounded cells are arranged in cords and strands in a chondroitin sulfate myxoid background. Several cytogenetic abnormalities have been identified (refer to Table 2), with the most frequent being the translocation t(9;22)(q22;q12), involving the EWSR1/NR4A3 genes.[195]
Prognosis
The tumor has traditionally been considered of low-grade malignant potential.[196] However, recent reports from large institutions showed that extraskeletal myxoid chondrosarcoma has significant malignant potential, especially if patients are followed for a long time.[197,198] Patients tend to have slow protracted courses. Nodal involvement has been well described. Local recurrence (57%) and metastatic spread to lungs (26%) have been reported.[198]
Treatment
Treatment options for extraskeletal myxoid chondrosarcoma include the following:
- Surgery.
- Radiation therapy.
The therapeutic benefit of chemotherapy has not been established. Aggressive local control and resection of metastases led to OS of 87% at 5 years and 63% at 10 years. Tumors were relatively resistant to radiation therapy.[197]
There may be potential genetic targets for small molecules, but these should be studied as part of a clinical trial. In an adult study, six of ten patients who received sunitinib achieved a partial response.[199]
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- NCT02601937 (A Phase 1 Study of the EZH2 Inhibitor Tazemetostat in Pediatric Subjects With Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma): Patients with INI1-negative tumors are eligible for targeted treatment with an EZH2 inhibitor. This is a phase I, open-label, dose-escalation, and dose-expansion study with a twice-daily oral dose of tazemetostat.
Neoplasms with perivascular epithelioid cell differentiation (PEComas)
Risk factors and molecular features
Benign PEComas are common in tuberous sclerosis, an autosomal dominant syndrome that also predisposes to renal cell cancer and brain tumors. Tuberous sclerosis is caused by germline inactivation of either TSC1 (9q34) or TSC2 (16p13.3), and the same tumor suppressor genes are inactivated somatically in sporadic PEComas.[200] Inactivation of either gene results in stimulation of the mTOR pathway, providing the basis for the treatment of nonsurgically curable PEComas with mTOR inhibitors.[201,202] A small proportion of PEComas have TFE3 rearrangements with fusions involving various genes including SFPQ/PSF and RAD51B.[203]
Clinical presentation
PEComas occur in various rare gastrointestinal, pulmonary, gynecologic, and genitourinary sites. Soft tissue, visceral, and gynecologic PEComas are more commonly seen in middle-aged female patients and are usually not associated with the tuberous sclerosis complex.[204] The disease course may be indolent.
Prognosis
Most PEComas have a benign clinical course, but malignant behavior has been reported and can be predicted based on the size of the tumor, mitotic rate, and presence of necrosis.[205]
Treatment
Treatment options have not been defined. Treatment may include surgery or observation followed by surgery when the tumor is large.[206]
Clinical activity with mTOR inhibitors, such as sirolimus, in tumors with evidence of mTORC1 activation and TSC loss has been well documented.[207]
Primitive neuroectodermal tumor (PNET)/extraskeletal Ewing tumor
(Refer to the PDQ summary on Ewing Sarcoma Treatment for more information.)
Synovial sarcoma
Synovial sarcoma accounts for 9% of soft tissue sarcomas in patients younger than 20 years (refer to Table 1).
Synovial sarcoma is one of the most common nonrhabdomyosarcomatous soft tissue sarcomas in children and adolescents. In a 1973 to 2005 SEER review, 1,268 patients with synovial sarcoma were identified. Approximately 17% of these patients were children and adolescents and the median age at diagnosis was 34 years.[208]
Histologic classification
Synovial sarcoma can be subclassified as the following types:
- Synovial sarcoma, NOS.
- Synovial sarcoma, spindle cell.
- Synovial sarcoma, biphasic.
Clinical presentation
The most common tumor location is the extremities, followed by trunk and head and neck.[208] Rarely, a synovial sarcoma may arise in the heart or pericardium.[209]
The most common site of metastasis is the lung.[210,211] The risk of metastases is highly influenced by tumor size; it is estimated that patients with tumors that are larger than 5 cm have a 32-fold risk of developing metastases when compared with other patients.
Diagnostic evaluation
The diagnosis of synovial sarcoma is made by immunohistochemical analysis, ultrastructural findings, and demonstration of the specific chromosomal translocation t(x;18)(p11.2;q11.2). This abnormality is specific for synovial sarcoma and is found in all morphologic subtypes. Synovial sarcoma results in rearrangement of the SYT gene on chromosome 18 with one of the subtypes (1, 2, or 4) of the SSX gene on chromosome X.[212,213] It is thought that the SYT/SSX18 transcript promotes epigenetic silencing of key tumor suppressor genes.[214]
In one report, reduced INI1 nuclear reactivity on immunohistochemical staining was seen in 49 cases of synovial sarcoma, suggesting that this pattern may help distinguish synovial sarcoma from other histologies.[215]
Prognosis
Patients younger than 10 years have more favorable outcomes and clinical features, including extremity primaries, smaller tumors, and localized disease, than do older patients.[208,216] A meta-analysis also suggested that response to chemotherapy was correlated with improved survival.[217]
The following studies have reported multiple factors associated with unfavorable outcomes:
- In a retrospective analysis of synovial sarcoma in children and adolescents who were treated in Germany and Italy, tumor size (>5 cm or ≤5 cm in greatest dimension) was an important predictor of EFS.[218] In this analysis, local invasiveness conferred an inferior probability of EFS, but surgical margins were not associated with clinical outcome.
- In a single-institution retrospective analysis of 111 patients with synovial sarcoma who were younger than 22 years at diagnosis, larger tumor size, greater depth in tissue, greater local invasiveness, and more proximal tumor location were associated with poorer OS.[219][Level of evidence: 3iiA]
- A multicenter analysis of 219 children from various treating centers including Germany, SJCRH, Instituto Tumori, and MD Anderson Cancer Center reported an estimated 5-year OS of 80% and EFS rate of 72%.[217] In this analysis, an interaction between tumor size and invasiveness was observed; in multivariate analysis, patients with large or invasive tumors or with Intergroup Rhabdomyosarcoma Study Clinical Group III disease (localized, incompletely resected or with biopsy only) and IV (metastases at diagnosis) had decreased OS. Treatment with radiation therapy was related to improved OS (hazard ratio, 0.4; 95% confidence interval, 0.2–0.7). In Intergroup Rhabdomyosarcoma Study Group III patients, objective response to chemotherapy (18 of 30 [60%]) correlated with improved survival. In adults, factors such as International Union Against Cancer/American Joint Committee on Cancer stage III and stage IVA, tumor necrosis, truncal location, elevated mitotic rate, age, and histologic grade have been associated with a worse prognosis.[220-222]
- Expression and genomic index prognostic signatures have been studied in synovial sarcoma. Complex genomic profiles, with greater rearrangement of the genome, are more common in adults than in younger patients with synovial sarcoma and are associated with a higher risk of metastasis.[223]
- A review of 84 patients with localized synovial sarcoma who had information on fusion status (SYT-SSX) and histologic grading found no difference in OS according to these criteria. However, for tumor size at diagnosis, the study showed that patients with tumors between 5 cm and 10 cm had a worse prognosis than those with smaller tumors (P = .02), and patients with tumors larger than 10 cm had even worse OS (P = .0003).[224][Level of evidence: 3iiiA]
- The German CWS group reviewed 27 evaluable patients younger than 21 years with pulmonary metastases among 296 patients with synovial sarcoma. Metastases involved the lungs in all patients. The 5-year EFS rate was 26%, and the OS rate was 30%. The most important prognostic factor at presentation was that the metastases were limited to one lesion in one lung or one lesion in both lungs (a group they termed oligometastatic). Treatment elements associated with superior survival were adequate local therapy of the primary tumor and, if feasible, for the metastases. The use of whole-lung irradiation did not correlate with better outcomes.[225][Level of evidence: 3iiA]
Survival after relapse is poor (30% at 5 years). Factors associated with outcome after relapse include duration of first remission (> or ≤ 18 months) and lack of a second remission.[226]
Treatment
Treatment options for synovial sarcoma include the following:
The COG and the European Pediatric Soft Tissue Sarcoma Study Group reported a combined analysis of 60 patients younger than 21 years with localized synovial sarcoma prospectively assigned to surgery without adjuvant radiation therapy or chemotherapy.[227] Enrollment was limited to patients with initial complete resection with histologically free margins, with a grade 2 tumor of any size or a grade 3 tumor 5 cm or smaller. The 3-year EFS was 90% (median follow-up, 5.2 years; range, 1.9–9.1). All eight events were local tumor recurrence; no metastatic recurrences were seen. All patients with recurrent disease were effectively treated with salvage therapy, resulting in 100% OS.
Synovial sarcoma appears to be more sensitive to chemotherapy than many other soft tissue sarcomas, and children with synovial sarcoma seem to have a better prognosis when compared with adults.[11,211,222,228-232] The most commonly used regimens for the treatment of synovial sarcoma incorporate ifosfamide and doxorubicin.[217,231,233] Response rates to the ifosfamide and doxorubicin regimen are higher than in other nonrhabdomyosarcomatous soft tissue sarcomas.[234]
Several studies have reported the following chemotherapy-associated treatment findings:
- Several treatment centers advocate postoperative chemotherapy after resection and radiation therapy of synovial sarcoma in children and young adults.[217,218,235-237]
- The International Society of Pediatric Oncology-Malignant Mesenchymal Tumors studies showed that select patients (young age, <5 cm resected tumors) with nonmetastatic synovial sarcoma can have excellent outcome in the absence of radiation, but it is still unclear whether that approach obviates an advantage of radiation for local or regional control.[236]
- A German trial suggested a benefit for postoperative chemotherapy in children with synovial sarcoma.[237]
- A meta-analysis also suggested that chemotherapy may provide benefit.[217]
- In the most recent COG ARST0332 (NCT00346164) study, 129 patients with synovial sarcoma were prospectively treated using a risk-based therapy program (as detailed in the prognosis section), of which 43 were categorized as low risk, 66 as intermediate risk, and 20 as high risk. At a median follow-up of 2.6 years, 3-year EFS for low-, intermediate-, and high-risk groups were 83%, 79%, and 16%, respectively. The use of risk factor–directed therapy accurately predicted outcomes.[238]
- The European Pediatric Soft Tissue Sarcoma Study Group performed a prospective study of patients younger than 21 years with synovial sarcoma (CCLG-EPSSG-NRSTS-2005 [NCT00334854]).[239][Level of evidence: 3iiA] Patients were stratified into the following three risks groups and nonrandomly assigned to treatment by risk group:
- Low-risk patients had Intergroup Rhabdomyosarcoma Study (IRS) group I tumors less than 5 cm in size and nonaxial primary tumors.
- Intermediate-risk patients had no axial primary tumors and IRS group I tumors greater than 5 cm or IRS group II tumors.
- High-risk patients included all patients with axial primary sites (head and neck, lung and pleura, trunk, retroperitoneal), IRS group III tumors, or N1 tumors.
Outcomes for patients treated on the CCLG-EPSSG-NRSTS-2005 trial are described in Table 9.
Table 9. Event-Free Survival (EFS) and Overall Survival (OS) in Patients With Low-, Intermediate-, and High-Risk Synovial Sarcoma Treated on the CCLG-EPSSG-NRSTS-2005 Trial Risk Group Treatment 3-Year EFS (%) 3-Year OS (%) IRS = Intergroup Rhabdomyosarcoma Study; RT = radiation therapy. aChemotherapy was ifosfamide/doxorubicin, with doxorubicin omitted during radiation therapy. b59.4 Gy in cases without the option of secondary resection; 50.4 Gy as preoperative radiation therapy; 50.4, 54, and 59.4 Gy as postoperative radiation therapy, in the case of R0, R1, and R2 resections, respectively (no additional radiation therapy in the case of secondary complete resections with free margins, in children younger than 6 years). Low Surgery alone 92 100 Intermediate Surgery, 3–6 cycles chemotherapya ± RTb 91 100 High (IRS group III) 3 cycles of chemotherapya surgery, 3 additional cycles of chemotherapy, ± RTb 77 94 High (axial primary sites) Surgery, 6 cycles of chemotherapya, RTb 78 100
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- ADP 04511 (NCT01343043) (A Pilot Study of Genetically Engineered NY-ESO-1 Specific [c259] T Cells in HLA-A2+ Patients With Synovial Sarcoma): Patients with unresectable, metastatic, or recurrent synovial sarcoma undergo apheresis. Cells are shipped to a central laboratory where they undergo NY-ESO-1 transduction, expansion, and cryopreservation. Patients undergo lymphodepletion with fludarabine and cyclophosphamide, followed by an infusion of autologous transfected cells. Eligibility is restricted to patients with HLA type A2+, age older than 4 years, and weight greater than 18 kg.
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with alveolar soft part sarcoma, clear cell sarcoma of soft tissue, epithelioid sarcoma, extraskeletal myxoid chondrosarcoma, PEComa, and synovial sarcoma are eligible for this trial.
- NCT02601937 (A Phase 1 Study of the EZH2 Inhibitor Tazemetostat in Pediatric Subjects With Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma): Patients with INI1-negative tumors are eligible for targeted treatment with an EZH2 inhibitor. This is a phase I, open-label, dose-escalation, and dose-expansion study with a twice-daily oral dose of tazemetostat.
- ADVL1522 (NCT02452554) (Lorvotuzumab Mertansine in Treating Younger Patients with Relapsed or Refractory Wilms Tumor, Rhabdomyosarcoma, Neuroblastoma, Pleuropulmonary Blastoma, Malignant Peripheral Nerve Sheath Tumor, or Synovial Sarcoma): This is a phase II study of IMGN901 (lorvotuzumab mertansine) in children with relapsed or refractory Wilms tumor, rhabdomyosarcoma, neuroblastoma, pleuropulmonary blastoma, malignant peripheral nerve sheath tumor, and synovial sarcoma. This trial is studying the effects of IMGN901, an antibody-drug conjugate that links a potent antimitotic to antibodies that target CD56.
Undifferentiated/unclassified sarcoma
Patients with undifferentiated soft tissue sarcoma had been eligible for participation in rhabdomyosarcoma trials coordinated by the Intergroup Rhabdomyosarcoma Study Group and the COG from 1972 to 2006. The rationale was the observation that patients with undifferentiated soft tissue sarcoma had similar sites of disease and outcome as those with alveolar rhabdomyosarcoma. Therapeutic trials for adults with soft tissue sarcoma include patients with undifferentiated soft tissue sarcoma and other histologies, which are treated similarly, using ifosfamide and doxorubicin, and sometimes with other chemotherapy agents, surgery, and radiation therapy.
In the COG ARST0332 (NCT00346164) trial, patients with high-grade undifferentiated sarcoma were treated with an ifosfamide and doxorubicin-based regimen and were treated with rhabdomyosarcoma-directed therapies in previous Intergroup Rhabdomyosarcoma Study Group studies with a 5-year survival estimate for nonmetastatic patients of 72%.[240][Level of evidence: 3iiA] Currently, these patients are eligible for the COG open ARST1321 (NCT02180867) trial for patients with nonrhabdomyosarcomatous soft tissue sarcoma.
Undifferentiated pleomorphic sarcoma/malignant fibrous histiocytoma (high-grade)
At one time, malignant fibrous histiocytoma was the single most common histotype among adults with soft tissue sarcomas. Since it was first recognized in the early 1960s, malignant fibrous histiocytoma has been plagued by controversy in terms of both its histogenesis and its validity as a clinicopathologic entity. The latest WHO classification no longer includes malignant fibrous histiocytoma as a distinct diagnostic category but rather as a subtype of an undifferentiated pleomorphic sarcoma.[241]
This entity accounts for 2% to 6% of all childhood soft tissue sarcomas.[242] These tumors can arise in previously irradiated sites or as a second malignancy in patients with retinoblastoma.
These tumors occur mainly in the second decade of life. In a series of ten patients, the median age was 10 years and the tumor was most commonly located in the extremities. In this series, all tumors were localized and five of nine (for whom follow-up was available) were alive and in first remission.[242] In another series of 17 pediatric patients with malignant fibrous histiocytoma, the median age at diagnosis was 5 years and the extremities were involved in eight cases.[243] All patients with metastatic disease died and two patients experienced a clinical response to a doxorubicin-based regimen.
(Refer to the PDQ summary on Osteosarcoma and Malignant Fibrous Histiocytoma of Bone Treatment for more information about the treatment of malignant fibrous histiocytoma of bone.)
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with undifferentiated epithelial sarcoma, undifferentiated pleomorphic sarcoma, undifferentiated round cell sarcoma, and undifferentiated spindle cell sarcoma are eligible for this trial.
Vascular Tumors
Vascular tumors vary from hemangiomas, which are always considered benign, to angiosarcomas, which are highly malignant.[244] Vascular tumors include the following tumor subtypes:
Angiosarcoma of the soft tissue
Incidence
Angiosarcoma is a rare (accounting for 2% of sarcomas), aggressive, vascular tumor that can arise in any part of the body, but is more common in the soft tissue. Angiosarcoma has an estimated incidence of 2 cases per 1 million; in the United States, it annually affects approximately 600 people who are typically aged 60 to 70 years.[245]
Angiosarcomas are extremely rare in children and it is unclear if the pathophysiology of this tumor is different in the pediatric population. Cases have been reported in neonates and toddlers, with presentation of multiple cutaneous lesions and liver lesions, some of which are GLUT1 positive.[246-249] Most angiosarcomas involve the skin and superficial soft tissue, although the liver, spleen, and lung can be affected; bone is rarely affected.
Risk factors
Established risk factors include vinyl chloride exposure, radiation exposure, and chronic lymphedema from any cause, including Stewart-Treves syndrome.[250]
Pathology and biology
Angiosarcomas are largely aneuploid tumors. The rare cases of angiosarcoma that arise from benign lesions such as hemangiomas have a distinct pathway that needs to be investigated. MYC amplification is seen in radiation-induced angiosarcoma. KDR-VEGFR2 mutations and FLT4-VEGFR3 amplifications have been seen with a frequency of less than 50%.[250]
Histopathologic diagnosis can be very difficult because there can be areas of varied atypia. The common feature is an irregular network of channels in a dissective pattern along dermal collagen bundles. There is varied cellular shape, size, mitosis, endothelial multilayering, and papillary formation. Epithelioid cells can also be present. Necrosis and hemorrhage are common. Tumors stain for factor VIII, CD31, and CD34. Some liver lesions can mimic infantile hemangiomas and have focal GLUT1 positivity. Nomenclature of these liver lesions has been difficult and confusing with use of terminology from 1971 (e.g., type I hemangioendothelioma: infantile hemangioma; type II hemangioendothelioma: low-grade angiosarcoma; type III hemangioendothelioma: high-grade angiosarcoma).[247]
Treatment of angiosarcoma of the soft tissue
Treatment options for angiosarcoma of the soft tissue include the following:
- Surgery (localized disease).
- Radiation therapy (localized cutaneous disease in adults).
- Surgery, chemotherapy, and radiation therapy (metastatic disease).
Localized disease is cured by aggressive surgery. Complete surgical excision appears to be crucial for angiosarcomas and lymphangiosarcomas despite evidence of tumor shrinkage in some patients who were treated with local or systemic therapy.[248,251-253] A review of 222 patients (median age, 62 years; range, age 15–90 years) showed an overall disease-specific survival (DSS) rate of 38% at 5 years. Five-year DSS was 44% in 138 patients with localized, resected tumors but only 16% in 43 patients with metastases at diagnosis.[253] Data on liver transplantation for localized angiosarcoma are limited.[254][Level of evidence: 3iiA]
Localized disease, especially cutaneous angiosarcoma, can be treated with radiation therapy. Most of these reported cases are in adults.[255]
Multimodal treatment with surgery, systemic chemotherapy, and radiation therapy is used for metastatic disease, although it is rarely curative.[256] Disease control is the objective in metastatic angiosarcoma, with published progression-free survival rates between 3 months and 7 months [257] and a median overall survival (OS) rate of 14 months to 18 months.[258] In both adults and children, 5-year OS rates between 20% and 35% are reported.[248,249,259]
In a child diagnosed with angiosarcoma secondary to malignant transformation from infantile hemangioma, response to treatment with bevacizumab, a monoclonal antibody against vascular endothelial growth factor, combined with systemic chemotherapy, has been reported.[246,256] A report of eight cases of liver angiosarcoma in children highlighted the misuse of the term hemangioendothelioma and the importance of early diagnosis and treatment of these tumors.[260]
Biologic agents that inhibit angiogenesis have shown activity in adults with angiosarcoma.[247,259]
Treatment options under clinical evaluation for angiosarcoma of the soft tissue
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery [PAZNTIS]): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with angiosarcoma of the soft tissue are eligible for this trial.
- NCT01532687 (Gemcitabine Hydrochloride With or Without Pazopanib Hydrochloride in Treating Patients With Refractory Soft Tissue Sarcoma): This randomized phase II trial studies how well gemcitabine hydrochloride works with or without pazopanib hydrochloride in treating patients with refractory soft tissue sarcoma.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
Epithelioid hemangioendothelioma
Incidence and outcome
This tumor was first described in soft tissue by Weiss and Enzinger in 1982. Epithelioid hemangioendotheliomas can occur at younger ages, but the peak incidence is in the fourth and fifth decades of life. The tumors can have an indolent or very aggressive course, with overall survival of 73% at 5 years. There are case reports of patients with untreated multiple lesions who have a very benign course compared with other patients who have a very aggressive course. Some pathologists have tried to stratify patients to evaluate risks and adjust treatment, but more research is needed.[261-267]
The presence of effusions, tumor size larger than 3 cm, and a high mitotic index (>3 mitoses/50 high-power fields) have been associated with unfavorable outcomes.[263]
Pathology and biology
A WWTR1-CAMTA1 gene fusion has been found in a large percentage of patients; less commonly, a YAP1-TFE3 gene fusion has been reported.[261] These fusions are not directly targetable with current medicines. Monoclonality has been described in multiple liver lesions, suggesting a metastatic process.
Histologically, these lesions are characterized as epithelioid lesions arranged in nests, strands, and trabecular patterns, with infrequent vascular spaces. Features that may be associated with aggressive clinical behavior include cellular atypia, one or more mitoses per 10 high-power fields, an increased proportion of spindled cells, focal necrosis, and metaplastic bone formation.[263]
The number of pediatric patients reported in the literature is limited.
Clinical presentation and diagnostic evaluation
Common sites of involvement are liver alone (21%), liver plus lung (18%), lung alone (12%), and bone alone (14%).[263,268,269] Clinical presentation depends on site of involvement, as follows:
- Liver: Hepatic nodules have central vascularity on ultrasound, contrast-enhancing lesions by computed tomography, and low T1 signal and moderate T2 signal on magnetic resonance imaging.
- Lung: Pulmonary epithelioid hemangioendothelioma may be an asymptomatic finding on chest x-ray or be associated with pleuritic pain, hemoptysis, anemia, and fibrosis.
- Bone: Bone metastasis may be associated with pathologic fracture. On x-rays, they are well-defined osteolytic lesions and can be multiple or solitary.
- Soft tissue: Thirty percent of soft tissue cases are associated with metastases, and when present, can have a very aggressive course, with limited response to chemotherapy.
- Skin: Cutaneous lesions can be raised and nodular or can be warm red-brown plaques.
Treatment of epithelioid hemangioendothelioma
Treatment options for epithelioid hemangioendothelioma include the following:
- Observation.
- Surgery.
- Immunotherapy.
- Targeted therapy.
- Chemotherapy.
For indolent cases, observation is warranted. For more aggressive cases, multiple medications have been used, including interferon, thalidomide, sorafenib, pazopanib, and sirolimus.[270] The most aggressive cases are treated with angiosarcoma-type chemotherapy. Surgery is used when possible. Liver transplantation has been used with aggressive liver lesions, both with and without metastases.[263,271-274]
Treatment options under clinical evaluation for epithelioid hemangioendothelioma
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- NCT03148275 (Trametinib in Treating Patients with Epithelioid Hemangioendothelioma That Is Metastatic, Locally Advanced, or Cannot Be Removed by Surgery): This is a phase II trial assessing the efficacy of trametinib, with patient-reported outcomes as secondary aims.
- NCT01532687 (Gemcitabine Hydrochloride With or Without Pazopanib Hydrochloride in Treating Patients With Refractory Soft Tissue Sarcoma): This randomized phase II trial studies how well gemcitabine hydrochloride works with or without pazopanib hydrochloride in treating patients with refractory soft tissue sarcoma.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Ferrari A, Casanova M, Collini P, et al.: Adult-type soft tissue sarcomas in pediatric-age patients: experience at the Istituto Nazionale Tumori in Milan. J Clin Oncol 23 (18): 4021-30, 2005. [PUBMED Abstract]
- Stanelle EJ, Christison-Lagay ER, Sidebotham EL, et al.: Prognostic factors and survival in pediatric and adolescent liposarcoma. Sarcoma 2012: 870910, 2012. [PUBMED Abstract]
- Alaggio R, Coffin CM, Weiss SW, et al.: Liposarcomas in young patients: a study of 82 cases occurring in patients younger than 22 years of age. Am J Surg Pathol 33 (5): 645-58, 2009. [PUBMED Abstract]
- Sreekantaiah C, Karakousis CP, Leong SP, et al.: Cytogenetic findings in liposarcoma correlate with histopathologic subtypes. Cancer 69 (10): 2484-95, 1992. [PUBMED Abstract]
- Sugiura H, Takahashi M, Katagiri H, et al.: Additional wide resection of malignant soft tissue tumors. Clin Orthop (394): 201-10, 2002. [PUBMED Abstract]
- Cecchetto G, Guglielmi M, Inserra A, et al.: Primary re-excision: the Italian experience in patients with localized soft-tissue sarcomas. Pediatr Surg Int 17 (7): 532-4, 2001. [PUBMED Abstract]
- Chui CH, Spunt SL, Liu T, et al.: Is reexcision in pediatric nonrhabdomyosarcoma soft tissue sarcoma necessary after an initial unplanned resection? J Pediatr Surg 37 (10): 1424-9, 2002. [PUBMED Abstract]
- Bahig H, Roberge D, Bosch W, et al.: Agreement among RTOG sarcoma radiation oncologists in contouring suspicious peritumoral edema for preoperative radiation therapy of soft tissue sarcoma of the extremity. Int J Radiat Oncol Biol Phys 86 (2): 298-303, 2013. [PUBMED Abstract]
- Baldini EH, Wang D, Haas RL, et al.: Treatment Guidelines for Preoperative Radiation Therapy for Retroperitoneal Sarcoma: Preliminary Consensus of an International Expert Panel. Int J Radiat Oncol Biol Phys 92 (3): 602-12, 2015. [PUBMED Abstract]
- Ferrari A, Casanova M, Spreafico F, et al.: Childhood liposarcoma: a single-institutional twenty-year experience. Pediatr Hematol Oncol 16 (5): 415-21, 1999 Sep-Oct. [PUBMED Abstract]
- Cecchetto G, Alaggio R, Dall'Igna P, et al.: Localized unresectable non-rhabdo soft tissue sarcomas of the extremities in pediatric age: results from the Italian studies. Cancer 104 (9): 2006-12, 2005. [PUBMED Abstract]
- Huh WW, Yuen C, Munsell M, et al.: Liposarcoma in children and young adults: a multi-institutional experience. Pediatr Blood Cancer 57 (7): 1142-6, 2011. [PUBMED Abstract]
- Gronchi A, Bui BN, Bonvalot S, et al.: Phase II clinical trial of neoadjuvant trabectedin in patients with advanced localized myxoid liposarcoma. Ann Oncol 23 (3): 771-6, 2012. [PUBMED Abstract]
- Demetri GD, von Mehren M, Jones RL, et al.: Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. J Clin Oncol 34 (8): 786-93, 2016. [PUBMED Abstract]
- Baruchel S, Pappo A, Krailo M, et al.: A phase 2 trial of trabectedin in children with recurrent rhabdomyosarcoma, Ewing sarcoma and non-rhabdomyosarcoma soft tissue sarcomas: a report from the Children's Oncology Group. Eur J Cancer 48 (4): 579-85, 2012. [PUBMED Abstract]
- Wang L, Motoi T, Khanin R, et al.: Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer 51 (2): 127-39, 2012. [PUBMED Abstract]
- Nyquist KB, Panagopoulos I, Thorsen J, et al.: Whole-transcriptome sequencing identifies novel IRF2BP2-CDX1 fusion gene brought about by translocation t(1;5)(q42;q32) in mesenchymal chondrosarcoma. PLoS One 7 (11): e49705, 2012. [PUBMED Abstract]
- Frezza AM, Cesari M, Baumhoer D, et al.: Mesenchymal chondrosarcoma: prognostic factors and outcome in 113 patients. A European Musculoskeletal Oncology Society study. Eur J Cancer 51 (3): 374-81, 2015. [PUBMED Abstract]
- Dantonello TM, Int-Veen C, Leuschner I, et al.: Mesenchymal chondrosarcoma of soft tissues and bone in children, adolescents, and young adults: experiences of the CWS and COSS study groups. Cancer 112 (11): 2424-31, 2008. [PUBMED Abstract]
- Bishop MW, Somerville JM, Bahrami A, et al.: Mesenchymal Chondrosarcoma in Children and Young Adults: A Single Institution Retrospective Review. Sarcoma 2015: 608279, 2015. [PUBMED Abstract]
- Wodowski K, Hill DA, Pappo AS, et al.: A chemosensitive pediatric extraosseous osteosarcoma: case report and review of the literature. J Pediatr Hematol Oncol 25 (1): 73-7, 2003. [PUBMED Abstract]
- Sordillo PP, Hajdu SI, Magill GB, et al.: Extraosseous osteogenic sarcoma. A review of 48 patients. Cancer 51 (4): 727-34, 1983. [PUBMED Abstract]
- Nieuwenhuis MH, Casparie M, Mathus-Vliegen LM, et al.: A nation-wide study comparing sporadic and familial adenomatous polyposis-related desmoid-type fibromatoses. Int J Cancer 129 (1): 256-61, 2011. [PUBMED Abstract]
- Rossato M, Rigotti M, Grazia M, et al.: Congenital hypertrophy of the retinal pigment epithelium (CHRPE) and familial adenomatous polyposis (FAP). Acta Ophthalmol Scand 74 (4): 338-42, 1996. [PUBMED Abstract]
- Baker RH, Heinemann MH, Miller HH, et al.: Hyperpigmented lesions of the retinal pigment epithelium in familial adenomatous polyposis. Am J Med Genet 31 (2): 427-35, 1988. [PUBMED Abstract]
- Kattentidt Mouravieva AA, Geurts-Giele IR, de Krijger RR, et al.: Identification of Familial Adenomatous Polyposis carriers among children with desmoid tumours. Eur J Cancer 48 (12): 1867-74, 2012. [PUBMED Abstract]
- Wang WL, Nero C, Pappo A, et al.: CTNNB1 genotyping and APC screening in pediatric desmoid tumors: a proposed algorithm. Pediatr Dev Pathol 15 (5): 361-7, 2012 Sep-Oct. [PUBMED Abstract]
- Lewis JJ, Boland PJ, Leung DH, et al.: The enigma of desmoid tumors. Ann Surg 229 (6): 866-72; discussion 872-3, 1999. [PUBMED Abstract]
- Lazar AJ, Tuvin D, Hajibashi S, et al.: Specific mutations in the beta-catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am J Pathol 173 (5): 1518-27, 2008. [PUBMED Abstract]
- Faulkner LB, Hajdu SI, Kher U, et al.: Pediatric desmoid tumor: retrospective analysis of 63 cases. J Clin Oncol 13 (11): 2813-8, 1995. [PUBMED Abstract]
- Merchant NB, Lewis JJ, Woodruff JM, et al.: Extremity and trunk desmoid tumors: a multifactorial analysis of outcome. Cancer 86 (10): 2045-52, 1999. [PUBMED Abstract]
- Honeyman JN, Theilen TM, Knowles MA, et al.: Desmoid fibromatosis in children and adolescents: a conservative approach to management. J Pediatr Surg 48 (1): 62-6, 2013. [PUBMED Abstract]
- Bonvalot S, Ternès N, Fiore M, et al.: Spontaneous regression of primary abdominal wall desmoid tumors: more common than previously thought. Ann Surg Oncol 20 (13): 4096-102, 2013. [PUBMED Abstract]
- Bonvalot S, Eldweny H, Haddad V, et al.: Extra-abdominal primary fibromatosis: Aggressive management could be avoided in a subgroup of patients. Eur J Surg Oncol 34 (4): 462-8, 2008. [PUBMED Abstract]
- Merchant TE, Nguyen D, Walter AW, et al.: Long-term results with radiation therapy for pediatric desmoid tumors. Int J Radiat Oncol Biol Phys 47 (5): 1267-71, 2000. [PUBMED Abstract]
- Zelefsky MJ, Harrison LB, Shiu MH, et al.: Combined surgical resection and iridium 192 implantation for locally advanced and recurrent desmoid tumors. Cancer 67 (2): 380-4, 1991. [PUBMED Abstract]
- Weiss AJ, Lackman RD: Low-dose chemotherapy of desmoid tumors. Cancer 64 (6): 1192-4, 1989. [PUBMED Abstract]
- Klein WA, Miller HH, Anderson M, et al.: The use of indomethacin, sulindac, and tamoxifen for the treatment of desmoid tumors associated with familial polyposis. Cancer 60 (12): 2863-8, 1987. [PUBMED Abstract]
- Soto-Miranda MA, Sandoval JA, Rao B, et al.: Surgical treatment of pediatric desmoid tumors. A 12-year, single-center experience. Ann Surg Oncol 20 (11): 3384-90, 2013. [PUBMED Abstract]
- Skapek SX, Ferguson WS, Granowetter L, et al.: Vinblastine and methotrexate for desmoid fibromatosis in children: results of a Pediatric Oncology Group Phase II Trial. J Clin Oncol 25 (5): 501-6, 2007. [PUBMED Abstract]
- Gandhi MM, Nathan PC, Weitzman S, et al.: Successful treatment of life-threatening generalized infantile myofibromatosis using low-dose chemotherapy. J Pediatr Hematol Oncol 25 (9): 750-4, 2003. [PUBMED Abstract]
- Gega M, Yanagi H, Yoshikawa R, et al.: Successful chemotherapeutic modality of doxorubicin plus dacarbazine for the treatment of desmoid tumors in association with familial adenomatous polyposis. J Clin Oncol 24 (1): 102-5, 2006. [PUBMED Abstract]
- Constantinidou A, Jones RL, Scurr M, et al.: Pegylated liposomal doxorubicin, an effective, well-tolerated treatment for refractory aggressive fibromatosis. Eur J Cancer 45 (17): 2930-4, 2009. [PUBMED Abstract]
- Ananth P, Werger A, Voss S, et al.: Liposomal doxorubicin: Effective treatment for pediatric desmoid fibromatosis. Pediatr Blood Cancer 64 (7): , 2017. [PUBMED Abstract]
- Gounder MM, Lefkowitz RA, Keohan ML, et al.: Activity of Sorafenib against desmoid tumor/deep fibromatosis. Clin Cancer Res 17 (12): 4082-90, 2011. [PUBMED Abstract]
- Heinrich MC, McArthur GA, Demetri GD, et al.: Clinical and molecular studies of the effect of imatinib on advanced aggressive fibromatosis (desmoid tumor). J Clin Oncol 24 (7): 1195-203, 2006. [PUBMED Abstract]
- Chugh R, Wathen JK, Patel SR, et al.: Efficacy of imatinib in aggressive fibromatosis: Results of a phase II multicenter Sarcoma Alliance for Research through Collaboration (SARC) trial. Clin Cancer Res 16 (19): 4884-91, 2010. [PUBMED Abstract]
- Agresta L, Kim H, Turpin BK, et al.: Pazopanib therapy for desmoid tumors in adolescent and young adult patients. Pediatr Blood Cancer : , 2018. [PUBMED Abstract]
- Shang H, Braggio D, Lee YJ, et al.: Targeting the Notch pathway: A potential therapeutic approach for desmoid tumors. Cancer 121 (22): 4088-96, 2015. [PUBMED Abstract]
- Messersmith WA, Shapiro GI, Cleary JM, et al.: A Phase I, dose-finding study in patients with advanced solid malignancies of the oral γ-secretase inhibitor PF-03084014. Clin Cancer Res 21 (1): 60-7, 2015. [PUBMED Abstract]
- Bisogno G, Tagarelli A, Stramare R, et al.: Hydroxyurea treatment can avoid the need for aggressive surgery in pediatric fibromatosis. J Pediatr Hematol Oncol 35 (4): e171-3, 2013. [PUBMED Abstract]
- Meazza C, Casanova M, Trecate G, et al.: Objective response to hydroxyurea in a patient with heavily pre-treated aggressive fibromatosis. Pediatr Blood Cancer 55 (3): 587-8, 2010. [PUBMED Abstract]
- Balamuth NJ, Womer RB: Successful treatment of fibromatosis with hydroxyurea: Analysis of 20 pediatric cases. [Abstract] The Connective Tissue Oncology Society (CTOS) 14th Annual Meeting, 13–15 November 2008, London, United Kingdom A-34852, 2008. Also available online. Last accessed April 02, 2018.
- Meazza C, Bisogno G, Gronchi A, et al.: Aggressive fibromatosis in children and adolescents: the Italian experience. Cancer 116 (1): 233-40, 2010. [PUBMED Abstract]
- Hansmann A, Adolph C, Vogel T, et al.: High-dose tamoxifen and sulindac as first-line treatment for desmoid tumors. Cancer 100 (3): 612-20, 2004. [PUBMED Abstract]
- Skapek SX, Anderson JR, Hill DA, et al.: Safety and efficacy of high-dose tamoxifen and sulindac for desmoid tumor in children: results of a Children's Oncology Group (COG) phase II study. Pediatr Blood Cancer 60 (7): 1108-12, 2013. [PUBMED Abstract]
- Rutenberg MS, Indelicato DJ, Knapik JA, et al.: External-beam radiotherapy for pediatric and young adult desmoid tumors. Pediatr Blood Cancer 57 (3): 435-42, 2011. [PUBMED Abstract]
- Buckley PG, Mantripragada KK, Benetkiewicz M, et al.: A full-coverage, high-resolution human chromosome 22 genomic microarray for clinical and research applications. Hum Mol Genet 11 (25): 3221-9, 2002. [PUBMED Abstract]
- Valdivielso-Ramos M, Torrelo A, Campos M, et al.: Pediatric dermatofibrosarcoma protuberans in Madrid, Spain: multi-institutional outcomes. Pediatr Dermatol 31 (6): 676-82, 2014 Nov-Dec. [PUBMED Abstract]
- Gooskens SL, Oranje AP, van Adrichem LN, et al.: Imatinib mesylate for children with dermatofibrosarcoma protuberans (DFSP). Pediatr Blood Cancer 55 (2): 369-73, 2010. [PUBMED Abstract]
- Rubio GA, Alvarado A, Gerth DJ, et al.: Incidence and Outcomes of Dermatofibrosarcoma Protuberans in the US Pediatric Population. J Craniofac Surg 28 (1): 182-184, 2017. [PUBMED Abstract]
- Meguerditchian AN, Wang J, Lema B, et al.: Wide excision or Mohs micrographic surgery for the treatment of primary dermatofibrosarcoma protuberans. Am J Clin Oncol 33 (3): 300-3, 2010. [PUBMED Abstract]
- Dagan R, Morris CG, Zlotecki RA, et al.: Radiotherapy in the treatment of dermatofibrosarcoma protuberans. Am J Clin Oncol 28 (6): 537-9, 2005. [PUBMED Abstract]
- Sun LM, Wang CJ, Huang CC, et al.: Dermatofibrosarcoma protuberans: treatment results of 35 cases. Radiother Oncol 57 (2): 175-81, 2000. [PUBMED Abstract]
- Price VE, Fletcher JA, Zielenska M, et al.: Imatinib mesylate: an attractive alternative in young children with large, surgically challenging dermatofibrosarcoma protuberans. Pediatr Blood Cancer 44 (5): 511-5, 2005. [PUBMED Abstract]
- McArthur GA, Demetri GD, van Oosterom A, et al.: Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J Clin Oncol 23 (4): 866-73, 2005. [PUBMED Abstract]
- Rutkowski P, Van Glabbeke M, Rankin CJ, et al.: Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials. J Clin Oncol 28 (10): 1772-9, 2010. [PUBMED Abstract]
- Miller SJ, Alam M, Andersen JS, et al.: Dermatofibrosarcoma protuberans. J Natl Compr Canc Netw 10 (3): 312-8, 2012. [PUBMED Abstract]
- Kao YC, Fletcher CDM, Alaggio R, et al.: Recurrent BRAF Gene Fusions in a Subset of Pediatric Spindle Cell Sarcomas: Expanding the Genetic Spectrum of Tumors With Overlapping Features With Infantile Fibrosarcoma. Am J Surg Pathol 42 (1): 28-38, 2018. [PUBMED Abstract]
- Sulkowski JP, Raval MV, Browne M: Margin status and multimodal therapy in infantile fibrosarcoma. Pediatr Surg Int 29 (8): 771-6, 2013. [PUBMED Abstract]
- Orbach D, Rey A, Cecchetto G, et al.: Infantile fibrosarcoma: management based on the European experience. J Clin Oncol 28 (2): 318-23, 2010. [PUBMED Abstract]
- Orbach D, Brennan B, De Paoli A, et al.: Conservative strategy in infantile fibrosarcoma is possible: The European paediatric Soft tissue sarcoma Study Group experience. Eur J Cancer 57: 1-9, 2016. [PUBMED Abstract]
- Spunt SL, Million L, Coffin C: The nonrhabdomyosarcoma soft tissue sarcoma. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 827-54.
- Loh ML, Ahn P, Perez-Atayde AR, et al.: Treatment of infantile fibrosarcoma with chemotherapy and surgery: results from the Dana-Farber Cancer Institute and Children's Hospital, Boston. J Pediatr Hematol Oncol 24 (9): 722-6, 2002. [PUBMED Abstract]
- Akyüz C, Küpeli S, Varan A, et al.: Infantile fibrosarcoma: retrospective analysis of eleven patients. Tumori 97 (2): 166-9, 2011 Mar-Apr. [PUBMED Abstract]
- Gallego S, Pericas N, Barber I, et al.: Infantile fibrosarcoma of the retroperitoneum: a site of unfavorable prognosis? Pediatr Hematol Oncol 28 (5): 451-3, 2011. [PUBMED Abstract]
- Parida L, Fernandez-Pineda I, Uffman JK, et al.: Clinical management of infantile fibrosarcoma: a retrospective single-institution review. Pediatr Surg Int 29 (7): 703-8, 2013. [PUBMED Abstract]
- Mody RJ, Wu YM, Lonigro RJ, et al.: Integrative Clinical Sequencing in the Management of Refractory or Relapsed Cancer in Youth. JAMA 314 (9): 913-25, 2015. [PUBMED Abstract]
- Wong V, Pavlick D, Brennan T, et al.: Evaluation of a Congenital Infantile Fibrosarcoma by Comprehensive Genomic Profiling Reveals an LMNA-NTRK1 Gene Fusion Responsive to Crizotinib. J Natl Cancer Inst 108 (1): , 2016. [PUBMED Abstract]
- Nagasubramanian R, Wei J, Gordon P, et al.: Infantile Fibrosarcoma With NTRK3-ETV6 Fusion Successfully Treated With the Tropomyosin-Related Kinase Inhibitor LOXO-101. Pediatr Blood Cancer 63 (8): 1468-70, 2016. [PUBMED Abstract]
- Yanagisawa R, Noguchi M, Fujita K, et al.: Preoperative Treatment With Pazopanib in a Case of Chemotherapy-Resistant Infantile Fibrosarcoma. Pediatr Blood Cancer 63 (2): 348-51, 2016. [PUBMED Abstract]
- Madden NP, Spicer RD, Allibone EB, et al.: Spontaneous regression of neonatal fibrosarcoma. Br J Cancer Suppl 18: S72-5, 1992. [PUBMED Abstract]
- Kovach SJ, Fischer AC, Katzman PJ, et al.: Inflammatory myofibroblastic tumors. J Surg Oncol 94 (5): 385-91, 2006. [PUBMED Abstract]
- Brodlie M, Barwick SC, Wood KM, et al.: Inflammatory myofibroblastic tumours of the respiratory tract: paediatric case series with varying clinical presentations. J Laryngol Otol 125 (8): 865-8, 2011. [PUBMED Abstract]
- Xiao Y, Zhou S, Ma C, et al.: Radiological and histopathological features of hepatic inflammatory myofibroblastic tumour: analysis of 10 cases. Clin Radiol 68 (11): 1114-20, 2013. [PUBMED Abstract]
- Karnak I, Senocak ME, Ciftci AO, et al.: Inflammatory myofibroblastic tumor in children: diagnosis and treatment. J Pediatr Surg 36 (6): 908-12, 2001. [PUBMED Abstract]
- Collin M, Charles A, Barker A, et al.: Inflammatory myofibroblastic tumour of the bladder in children: a review. J Pediatr Urol 11 (5): 239-45, 2015. [PUBMED Abstract]
- Coffin CM, Hornick JL, Fletcher CD: Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol 31 (4): 509-20, 2007. [PUBMED Abstract]
- Lovly CM, Gupta A, Lipson D, et al.: Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov 4 (8): 889-95, 2014. [PUBMED Abstract]
- Devaney KO, Lafeir DJ, Triantafyllou A, et al.: Inflammatory myofibroblastic tumors of the head and neck: evaluation of clinicopathologic and prognostic features. Eur Arch Otorhinolaryngol 269 (12): 2461-5, 2012. [PUBMED Abstract]
- Mehta B, Mascarenhas L, Zhou S, et al.: Inflammatory myofibroblastic tumors in childhood. Pediatr Hematol Oncol 30 (7): 640-5, 2013. [PUBMED Abstract]
- Favini F, Resti AG, Collini P, et al.: Inflammatory myofibroblastic tumor of the conjunctiva: response to chemotherapy with low-dose methotrexate and vinorelbine. Pediatr Blood Cancer 54 (3): 483-5, 2010. [PUBMED Abstract]
- Doski JJ, Priebe CJ Jr, Driessnack M, et al.: Corticosteroids in the management of unresected plasma cell granuloma (inflammatory pseudotumor) of the lung. J Pediatr Surg 26 (9): 1064-6, 1991. [PUBMED Abstract]
- Diop B, Konate I, Ka S, et al.: Mesenteric myofibroblastic tumor: NSAID therapy after incomplete resection. J Visc Surg 148 (4): e311-4, 2011. [PUBMED Abstract]
- Dalton BG, Thomas PG, Sharp NE, et al.: Inflammatory myofibroblastic tumors in children. J Pediatr Surg 51 (4): 541-4, 2016. [PUBMED Abstract]
- Butrynski JE, D'Adamo DR, Hornick JL, et al.: Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med 363 (18): 1727-33, 2010. [PUBMED Abstract]
- Mossé YP, Lim MS, Voss SD, et al.: Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children's Oncology Group phase 1 consortium study. Lancet Oncol 14 (6): 472-80, 2013. [PUBMED Abstract]
- Gaudichon J, Jeanne-Pasquier C, Deparis M, et al.: Complete and Repeated Response of a Metastatic ALK-rearranged Inflammatory Myofibroblastic Tumor to Crizotinib in a Teenage Girl. J Pediatr Hematol Oncol 38 (4): 308-11, 2016. [PUBMED Abstract]
- Nishio M, Murakami H, Horiike A, et al.: Phase I Study of Ceritinib (LDK378) in Japanese Patients with Advanced, Anaplastic Lymphoma Kinase-Rearranged Non-Small-Cell Lung Cancer or Other Tumors. J Thorac Oncol 10 (7): 1058-66, 2015. [PUBMED Abstract]
- Mossé YP, Voss SD, Lim MS, et al.: Targeting ALK With Crizotinib in Pediatric Anaplastic Large Cell Lymphoma and Inflammatory Myofibroblastic Tumor: A Children's Oncology Group Study. J Clin Oncol 35 (28): 3215-3221, 2017. [PUBMED Abstract]
- Evans HL: Low-grade fibromyxoid sarcoma: a clinicopathologic study of 33 cases with long-term follow-up. Am J Surg Pathol 35 (10): 1450-62, 2011. [PUBMED Abstract]
- Guillou L, Benhattar J, Gengler C, et al.: Translocation-positive low-grade fibromyxoid sarcoma: clinicopathologic and molecular analysis of a series expanding the morphologic spectrum and suggesting potential relationship to sclerosing epithelioid fibrosarcoma: a study from the French Sarcoma Group. Am J Surg Pathol 31 (9): 1387-402, 2007. [PUBMED Abstract]
- O'Sullivan MJ, Sirgi KE, Dehner LP: Low-grade fibrosarcoma (hyalinizing spindle cell tumor with giant rosettes) with pulmonary metastases at presentation: case report and review of the literature. Int J Surg Pathol 10 (3): 211-6, 2002. [PUBMED Abstract]
- Folpe AL, Lane KL, Paull G, et al.: Low-grade fibromyxoid sarcoma and hyalinizing spindle cell tumor with giant rosettes: a clinicopathologic study of 73 cases supporting their identity and assessing the impact of high-grade areas. Am J Surg Pathol 24 (10): 1353-60, 2000. [PUBMED Abstract]
- Sargar K, Kao SC, Spunt SL, et al.: MRI and CT of Low-Grade Fibromyxoid Sarcoma in Children: A Report From Children's Oncology Group Study ARST0332. AJR Am J Roentgenol 205 (2): 414-20, 2015. [PUBMED Abstract]
- Maretty-Nielsen K, Baerentzen S, Keller J, et al.: Low-Grade Fibromyxoid Sarcoma: Incidence, Treatment Strategy of Metastases, and Clinical Significance of the FUS Gene. Sarcoma 2013: 256280, 2013. [PUBMED Abstract]
- Prieto-Granada C, Zhang L, Chen HW, et al.: A genetic dichotomy between pure sclerosing epithelioid fibrosarcoma (SEF) and hybrid SEF/low-grade fibromyxoid sarcoma: a pathologic and molecular study of 18 cases. Genes Chromosomes Cancer 54 (1): 28-38, 2015. [PUBMED Abstract]
- Pollock BH, Jenson HB, Leach CT, et al.: Risk factors for pediatric human immunodeficiency virus-related malignancy. JAMA 289 (18): 2393-9, 2003. [PUBMED Abstract]
- Kleinerman RA, Tucker MA, Abramson DH, et al.: Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst 99 (1): 24-31, 2007. [PUBMED Abstract]
- Samuels BL, Chawla S, Patel S, et al.: Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol 24 (6): 1703-9, 2013. [PUBMED Abstract]
- Enzinger FM, Zhang RY: Plexiform fibrohistiocytic tumor presenting in children and young adults. An analysis of 65 cases. Am J Surg Pathol 12 (11): 818-26, 1988. [PUBMED Abstract]
- Black J, Coffin CM, Dehner LP: Fibrohistiocytic tumors and related neoplasms in children and adolescents. Pediatr Dev Pathol 15 (1 Suppl): 181-210, 2012. [PUBMED Abstract]
- Moosavi C, Jha P, Fanburg-Smith JC: An update on plexiform fibrohistiocytic tumor and addition of 66 new cases from the Armed Forces Institute of Pathology, in honor of Franz M. Enzinger, MD. Ann Diagn Pathol 11 (5): 313-9, 2007. [PUBMED Abstract]
- Billings SD, Folpe AL: Cutaneous and subcutaneous fibrohistiocytic tumors of intermediate malignancy: an update. Am J Dermatopathol 26 (2): 141-55, 2004. [PUBMED Abstract]
- Remstein ED, Arndt CA, Nascimento AG: Plexiform fibrohistiocytic tumor: clinicopathologic analysis of 22 cases. Am J Surg Pathol 23 (6): 662-70, 1999. [PUBMED Abstract]
- Salomao DR, Nascimento AG: Plexiform fibrohistiocytic tumor with systemic metastases: a case report. Am J Surg Pathol 21 (4): 469-76, 1997. [PUBMED Abstract]
- Redlich GC, Montgomery KD, Allgood GA, et al.: Plexiform fibrohistiocytic tumor with a clonal cytogenetic anomaly. Cancer Genet Cytogenet 108 (2): 141-3, 1999. [PUBMED Abstract]
- Luzar B, Calonje E: Cutaneous fibrohistiocytic tumours - an update. Histopathology 56 (1): 148-65, 2010. [PUBMED Abstract]
- Dantonello TM, Leuschner I, Vokuhl C, et al.: Malignant ectomesenchymoma in children and adolescents: report from the Cooperative Weichteilsarkom Studiengruppe (CWS). Pediatr Blood Cancer 60 (2): 224-9, 2013. [PUBMED Abstract]
- Carli M, Ferrari A, Mattke A, et al.: Pediatric malignant peripheral nerve sheath tumor: the Italian and German soft tissue sarcoma cooperative group. J Clin Oncol 23 (33): 8422-30, 2005. [PUBMED Abstract]
- Zhang M, Wang Y, Jones S, et al.: Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet 46 (11): 1170-2, 2014. [PUBMED Abstract]
- Hagel C, Zils U, Peiper M, et al.: Histopathology and clinical outcome of NF1-associated vs. sporadic malignant peripheral nerve sheath tumors. J Neurooncol 82 (2): 187-92, 2007. [PUBMED Abstract]
- Zou C, Smith KD, Liu J, et al.: Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg 249 (6): 1014-22, 2009. [PUBMED Abstract]
- Okada K, Hasegawa T, Tajino T, et al.: Clinical relevance of pathological grades of malignant peripheral nerve sheath tumor: a multi-institution TMTS study of 56 cases in Northern Japan. Ann Surg Oncol 14 (2): 597-604, 2007. [PUBMED Abstract]
- Amirian ES, Goodman JC, New P, et al.: Pediatric and adult malignant peripheral nerve sheath tumors: an analysis of data from the surveillance, epidemiology, and end results program. J Neurooncol 116 (3): 609-16, 2014. [PUBMED Abstract]
- Valentin T, Le Cesne A, Ray-Coquard I, et al.: Management and prognosis of malignant peripheral nerve sheath tumors: The experience of the French Sarcoma Group (GSF-GETO). Eur J Cancer 56: 77-84, 2016. [PUBMED Abstract]
- Bergamaschi L, Bisogno G, Manzitti C, et al.: Salvage rates and prognostic factors after relapse in children and adolescents with malignant peripheral nerve sheath tumors. Pediatr Blood Cancer 65 (2): , 2018. [PUBMED Abstract]
- Ferrari A, Bisogno G, Macaluso A, et al.: Soft-tissue sarcomas in children and adolescents with neurofibromatosis type 1. Cancer 109 (7): 1406-12, 2007. [PUBMED Abstract]
- Okur FV, Oguz A, Karadeniz C, et al.: Malignant triton tumor of the pelvis in a 2-year-old boy. J Pediatr Hematol Oncol 28 (3): 173-6, 2006. [PUBMED Abstract]
- Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 4th ed. St. Louis, Mo: Mosby, 2001.
- Fernandez-Pineda I, Parida L, Jenkins JJ, et al.: Childhood hemangiopericytoma: review of St Jude Children's Research Hospital. J Pediatr Hematol Oncol 33 (5): 356-9, 2011. [PUBMED Abstract]
- Rodriguez-Galindo C, Ramsey K, Jenkins JJ, et al.: Hemangiopericytoma in children and infants. Cancer 88 (1): 198-204, 2000. [PUBMED Abstract]
- Ferrari A, Casanova M, Bisogno G, et al.: Hemangiopericytoma in pediatric ages: a report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Cancer 92 (10): 2692-8, 2001. [PUBMED Abstract]
- Bien E, Stachowicz-Stencel T, Godzinski J, et al.: Retrospective multi-institutional study on hemangiopericytoma in Polish children. Pediatr Int 51 (1): 19-24, 2009. [PUBMED Abstract]
- Wiswell TE, Davis J, Cunningham BE, et al.: Infantile myofibromatosis: the most common fibrous tumor of infancy. J Pediatr Surg 23 (4): 315-8, 1988. [PUBMED Abstract]
- Chung EB, Enzinger FM: Infantile myofibromatosis. Cancer 48 (8): 1807-18, 1981. [PUBMED Abstract]
- Modi N: Congenital generalised fibromatosis. Arch Dis Child 57 (11): 881-2, 1982. [PUBMED Abstract]
- Levine E, Fréneaux P, Schleiermacher G, et al.: Risk-adapted therapy for infantile myofibromatosis in children. Pediatr Blood Cancer 59 (1): 115-20, 2012. [PUBMED Abstract]
- Larralde M, Hoffner MV, Boggio P, et al.: Infantile myofibromatosis: report of nine patients. Pediatr Dermatol 27 (1): 29-33, 2010 Jan-Feb. [PUBMED Abstract]
- Cheung YH, Gayden T, Campeau PM, et al.: A recurrent PDGFRB mutation causes familial infantile myofibromatosis. Am J Hum Genet 92 (6): 996-1000, 2013. [PUBMED Abstract]
- Gopal M, Chahal G, Al-Rifai Z, et al.: Infantile myofibromatosis. Pediatr Surg Int 24 (3): 287-91, 2008. [PUBMED Abstract]
- Weaver MS, Navid F, Huppmann A, et al.: Vincristine and Dactinomycin in Infantile Myofibromatosis With a Review of Treatment Options. J Pediatr Hematol Oncol 37 (3): 237-41, 2015. [PUBMED Abstract]
- Orbach D, Brennan B, Casanova M, et al.: Paediatric and adolescent alveolar soft part sarcoma: A joint series from European cooperative groups. Pediatr Blood Cancer 60 (11): 1826-32, 2013. [PUBMED Abstract]
- Ferrari A, Sultan I, Huang TT, et al.: Soft tissue sarcoma across the age spectrum: a population-based study from the Surveillance Epidemiology and End Results database. Pediatr Blood Cancer 57 (6): 943-9, 2011. [PUBMED Abstract]
- Wang HW, Qin XJ, Yang WJ, et al.: Alveolar soft part sarcoma of the oral and maxillofacial region: clinical analysis in a series of 18 patients. Oral Surg Oral Med Oral Pathol Oral Radiol 119 (4): 396-401, 2015. [PUBMED Abstract]
- Kayton ML, Meyers P, Wexler LH, et al.: Clinical presentation, treatment, and outcome of alveolar soft part sarcoma in children, adolescents, and young adults. J Pediatr Surg 41 (1): 187-93, 2006. [PUBMED Abstract]
- Ladanyi M, Lui MY, Antonescu CR, et al.: The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene 20 (1): 48-57, 2001. [PUBMED Abstract]
- Williams A, Bartle G, Sumathi VP, et al.: Detection of ASPL/TFE3 fusion transcripts and the TFE3 antigen in formalin-fixed, paraffin-embedded tissue in a series of 18 cases of alveolar soft part sarcoma: useful diagnostic tools in cases with unusual histological features. Virchows Arch 458 (3): 291-300, 2011. [PUBMED Abstract]
- Lieberman PH, Brennan MF, Kimmel M, et al.: Alveolar soft-part sarcoma. A clinico-pathologic study of half a century. Cancer 63 (1): 1-13, 1989. [PUBMED Abstract]
- Casanova M, Ferrari A, Bisogno G, et al.: Alveolar soft part sarcoma in children and adolescents: A report from the Soft-Tissue Sarcoma Italian Cooperative Group. Ann Oncol 11 (11): 1445-9, 2000. [PUBMED Abstract]
- Pennacchioli E, Fiore M, Collini P, et al.: Alveolar soft part sarcoma: clinical presentation, treatment, and outcome in a series of 33 patients at a single institution. Ann Surg Oncol 17 (12): 3229-33, 2010. [PUBMED Abstract]
- Flores RJ, Harrison DJ, Federman NC, et al.: Alveolar soft part sarcoma in children and young adults: A report of 69 cases. Pediatr Blood Cancer : , 2018. [PUBMED Abstract]
- Roozendaal KJ, de Valk B, ten Velden JJ, et al.: Alveolar soft-part sarcoma responding to interferon alpha-2b. Br J Cancer 89 (2): 243-5, 2003. [PUBMED Abstract]
- Conde N, Cruz O, Albert A, et al.: Antiangiogenic treatment as a pre-operative management of alveolar soft-part sarcoma. Pediatr Blood Cancer 57 (6): 1071-3, 2011. [PUBMED Abstract]
- Stacchiotti S, Negri T, Zaffaroni N, et al.: Sunitinib in advanced alveolar soft part sarcoma: evidence of a direct antitumor effect. Ann Oncol 22 (7): 1682-90, 2011. [PUBMED Abstract]
- Kummar S, Allen D, Monks A, et al.: Cediranib for metastatic alveolar soft part sarcoma. J Clin Oncol 31 (18): 2296-302, 2013. [PUBMED Abstract]
- Coindre JM, Hostein I, Terrier P, et al.: Diagnosis of clear cell sarcoma by real-time reverse transcriptase-polymerase chain reaction analysis of paraffin embedded tissues: clinicopathologic and molecular analysis of 44 patients from the French sarcoma group. Cancer 107 (5): 1055-64, 2006. [PUBMED Abstract]
- Meis-Kindblom JM: Clear cell sarcoma of tendons and aponeuroses: a historical perspective and tribute to the man behind the entity. Adv Anat Pathol 13 (6): 286-92, 2006. [PUBMED Abstract]
- Dim DC, Cooley LD, Miranda RN: Clear cell sarcoma of tendons and aponeuroses: a review. Arch Pathol Lab Med 131 (1): 152-6, 2007. [PUBMED Abstract]
- Blazer DG 3rd, Lazar AJ, Xing Y, et al.: Clinical outcomes of molecularly confirmed clear cell sarcoma from a single institution and in comparison with data from the Surveillance, Epidemiology, and End Results registry. Cancer 115 (13): 2971-9, 2009. [PUBMED Abstract]
- Fujimura Y, Siddique H, Lee L, et al.: EWS-ATF-1 chimeric protein in soft tissue clear cell sarcoma associates with CREB-binding protein and interferes with p53-mediated trans-activation function. Oncogene 20 (46): 6653-9, 2001. [PUBMED Abstract]
- Ferrari A, Casanova M, Bisogno G, et al.: Clear cell sarcoma of tendons and aponeuroses in pediatric patients: a report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Cancer 94 (12): 3269-76, 2002. [PUBMED Abstract]
- Karita M, Tsuchiya H, Yamamoto N, et al.: Caffeine-potentiated chemotherapy for clear cell sarcoma: a report of five cases. Int J Clin Oncol 18 (1): 33-7, 2013. [PUBMED Abstract]
- Leuschner I, Radig K, Harms D: Desmoplastic small round cell tumor. Semin Diagn Pathol 13 (3): 204-12, 1996. [PUBMED Abstract]
- Kushner BH, LaQuaglia MP, Wollner N, et al.: Desmoplastic small round-cell tumor: prolonged progression-free survival with aggressive multimodality therapy. J Clin Oncol 14 (5): 1526-31, 1996. [PUBMED Abstract]
- Saab R, Khoury JD, Krasin M, et al.: Desmoplastic small round cell tumor in childhood: the St. Jude Children's Research Hospital experience. Pediatr Blood Cancer 49 (3): 274-9, 2007. [PUBMED Abstract]
- Wang LL, Perlman EJ, Vujanic GM, et al.: Desmoplastic small round cell tumor of the kidney in childhood. Am J Surg Pathol 31 (4): 576-84, 2007. [PUBMED Abstract]
- Arora VC, Price AP, Fleming S, et al.: Characteristic imaging features of desmoplastic small round cell tumour. Pediatr Radiol 43 (1): 93-102, 2013. [PUBMED Abstract]
- Gerald WL, Ladanyi M, de Alava E, et al.: Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): desmoplastic small round-cell tumor and its variants. J Clin Oncol 16 (9): 3028-36, 1998. [PUBMED Abstract]
- Lal DR, Su WT, Wolden SL, et al.: Results of multimodal treatment for desmoplastic small round cell tumors. J Pediatr Surg 40 (1): 251-5, 2005. [PUBMED Abstract]
- Philippe-Chomette P, Kabbara N, Andre N, et al.: Desmoplastic small round cell tumors with EWS-WT1 fusion transcript in children and young adults. Pediatr Blood Cancer 58 (6): 891-7, 2012. [PUBMED Abstract]
- Sedig L, Geiger J, Mody R, et al.: Paratesticular desmoplastic small round cell tumors: A case report and review of the literature. Pediatr Blood Cancer 64 (12): , 2017. [PUBMED Abstract]
- Schwarz RE, Gerald WL, Kushner BH, et al.: Desmoplastic small round cell tumors: prognostic indicators and results of surgical management. Ann Surg Oncol 5 (5): 416-22, 1998 Jul-Aug. [PUBMED Abstract]
- Goodman KA, Wolden SL, La Quaglia MP, et al.: Whole abdominopelvic radiotherapy for desmoplastic small round-cell tumor. Int J Radiat Oncol Biol Phys 54 (1): 170-6, 2002. [PUBMED Abstract]
- Osborne EM, Briere TM, Hayes-Jordan A, et al.: Survival and toxicity following sequential multimodality treatment including whole abdominopelvic radiotherapy for patients with desmoplastic small round cell tumor. Radiother Oncol 119 (1): 40-4, 2016. [PUBMED Abstract]
- Atallah V, Honore C, Orbach D, et al.: Role of Adjuvant Radiation Therapy After Surgery for Abdominal Desmoplastic Small Round Cell Tumors. Int J Radiat Oncol Biol Phys 95 (4): 1244-53, 2016. [PUBMED Abstract]
- Tarek N, Hayes-Jordan A, Salvador L, et al.: Recurrent desmoplastic small round cell tumor responding to an mTOR inhibitor containing regimen. Pediatr Blood Cancer 65 (1): , 2018. [PUBMED Abstract]
- Cook RJ, Wang Z, Arora M, et al.: Clinical outcomes of patients with desmoplastic small round cell tumor of the peritoneum undergoing autologous HCT: a CIBMTR retrospective analysis. Bone Marrow Transplant 47 (11): 1455-8, 2012. [PUBMED Abstract]
- Chbani L, Guillou L, Terrier P, et al.: Epithelioid sarcoma: a clinicopathologic and immunohistochemical analysis of 106 cases from the French sarcoma group. Am J Clin Pathol 131 (2): 222-7, 2009. [PUBMED Abstract]
- Hornick JL, Dal Cin P, Fletcher CD: Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol 33 (4): 542-50, 2009. [PUBMED Abstract]
- Knutson SK, Warholic NM, Wigle TJ, et al.: Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 110 (19): 7922-7, 2013. [PUBMED Abstract]
- Guzzetta AA, Montgomery EA, Lyu H, et al.: Epithelioid sarcoma: one institution's experience with a rare sarcoma. J Surg Res 177 (1): 116-22, 2012. [PUBMED Abstract]
- Casanova M, Ferrari A, Collini P, et al.: Epithelioid sarcoma in children and adolescents: a report from the Italian Soft Tissue Sarcoma Committee. Cancer 106 (3): 708-17, 2006. [PUBMED Abstract]
- Kodet R, Newton WA Jr, Sachs N, et al.: Rhabdoid tumors of soft tissues: a clinicopathologic study of 26 cases enrolled on the Intergroup Rhabdomyosarcoma Study. Hum Pathol 22 (7): 674-84, 1991. [PUBMED Abstract]
- Biegel JA, Zhou JY, Rorke LB, et al.: Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 59 (1): 74-9, 1999. [PUBMED Abstract]
- Eaton KW, Tooke LS, Wainwright LM, et al.: Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56 (1): 7-15, 2011. [PUBMED Abstract]
- Lee RS, Stewart C, Carter SL, et al.: A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest 122 (8): 2983-8, 2012. [PUBMED Abstract]
- Sultan I, Qaddoumi I, Rodríguez-Galindo C, et al.: Age, stage, and radiotherapy, but not primary tumor site, affects the outcome of patients with malignant rhabdoid tumors. Pediatr Blood Cancer 54 (1): 35-40, 2010. [PUBMED Abstract]
- Puri DR, Meyers PA, Kraus DH, et al.: Radiotherapy in the multimodal treatment of extrarenal extracranial malignant rhabdoid tumors. Pediatr Blood Cancer 50 (1): 167-9, 2008. [PUBMED Abstract]
- Madigan CE, Armenian SH, Malogolowkin MH, et al.: Extracranial malignant rhabdoid tumors in childhood: the Childrens Hospital Los Angeles experience. Cancer 110 (9): 2061-6, 2007. [PUBMED Abstract]
- Bourdeaut F, Fréneaux P, Thuille B, et al.: Extra-renal non-cerebral rhabdoid tumours. Pediatr Blood Cancer 51 (3): 363-8, 2008. [PUBMED Abstract]
- Wetmore C, Boyett J, Li S, et al.: Alisertib is active as single agent in recurrent atypical teratoid rhabdoid tumors in 4 children. Neuro Oncol 17 (6): 882-8, 2015. [PUBMED Abstract]
- Tsuneyoshi M, Enjoji M, Iwasaki H, et al.: Extraskeletal myxoid chondrosarcoma--a clinicopathologic and electron microscopic study. Acta Pathol Jpn 31 (3): 439-47, 1981. [PUBMED Abstract]
- Hachitanda Y, Tsuneyoshi M, Daimaru Y, et al.: Extraskeletal myxoid chondrosarcoma in young children. Cancer 61 (12): 2521-6, 1988. [PUBMED Abstract]
- Hisaoka M, Ishida T, Imamura T, et al.: TFG is a novel fusion partner of NOR1 in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer 40 (4): 325-8, 2004. [PUBMED Abstract]
- Enzinger FM, Shiraki M: Extraskeletal myxoid chondrosarcoma. An analysis of 34 cases. Hum Pathol 3 (3): 421-35, 1972. [PUBMED Abstract]
- McGrory JE, Rock MG, Nascimento AG, et al.: Extraskeletal myxoid chondrosarcoma. Clin Orthop Relat Res (382): 185-90, 2001. [PUBMED Abstract]
- Drilon AD, Popat S, Bhuchar G, et al.: Extraskeletal myxoid chondrosarcoma: a retrospective review from 2 referral centers emphasizing long-term outcomes with surgery and chemotherapy. Cancer 113 (12): 3364-71, 2008. [PUBMED Abstract]
- Stacchiotti S, Pantaleo MA, Astolfi A, et al.: Activity of sunitinib in extraskeletal myxoid chondrosarcoma. Eur J Cancer 50 (9): 1657-64, 2014. [PUBMED Abstract]
- Martignoni G, Pea M, Reghellin D, et al.: Molecular pathology of lymphangioleiomyomatosis and other perivascular epithelioid cell tumors. Arch Pathol Lab Med 134 (1): 33-40, 2010. [PUBMED Abstract]
- Bissler JJ, McCormack FX, Young LR, et al.: Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med 358 (2): 140-51, 2008. [PUBMED Abstract]
- Davies DM, Johnson SR, Tattersfield AE, et al.: Sirolimus therapy in tuberous sclerosis or sporadic lymphangioleiomyomatosis. N Engl J Med 358 (2): 200-3, 2008. [PUBMED Abstract]
- Agaram NP, Sung YS, Zhang L, et al.: Dichotomy of Genetic Abnormalities in PEComas With Therapeutic Implications. Am J Surg Pathol 39 (6): 813-25, 2015. [PUBMED Abstract]
- Folpe A, Inwards C, eds.: Bone and Soft Tissue Pathology: A Volume in the Foundations in Diagnostic Pathology. Philadelphia, Pa: WB Saunders Co, 2010.
- Armah HB, Parwani AV: Perivascular epithelioid cell tumor. Arch Pathol Lab Med 133 (4): 648-54, 2009. [PUBMED Abstract]
- Alaggio R, Cecchetto G, Martignoni G, et al.: Malignant perivascular epithelioid cell tumor in children: description of a case and review of the literature. J Pediatr Surg 47 (6): e31-40, 2012. [PUBMED Abstract]
- Wagner AJ, Malinowska-Kolodziej I, Morgan JA, et al.: Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: targeting the pathogenic activation of mTORC1 in tumors. J Clin Oncol 28 (5): 835-40, 2010. [PUBMED Abstract]
- Sultan I, Rodriguez-Galindo C, Saab R, et al.: Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005: an analysis of 1268 patients. Cancer 115 (15): 3537-47, 2009. [PUBMED Abstract]
- Wang JG, Li NN: Primary cardiac synovial sarcoma. Ann Thorac Surg 95 (6): 2202-9, 2013. [PUBMED Abstract]
- Pappo AS, Fontanesi J, Luo X, et al.: Synovial sarcoma in children and adolescents: the St Jude Children's Research Hospital experience. J Clin Oncol 12 (11): 2360-6, 1994. [PUBMED Abstract]
- Ferrari A, De Salvo GL, Oberlin O, et al.: Synovial sarcoma in children and adolescents: a critical reappraisal of staging investigations in relation to the rate of metastatic involvement at diagnosis. Eur J Cancer 48 (9): 1370-5, 2012. [PUBMED Abstract]
- van de Rijn M, Barr FG, Collins MH, et al.: Absence of SYT-SSX fusion products in soft tissue tumors other than synovial sarcoma. Am J Clin Pathol 112 (1): 43-9, 1999. [PUBMED Abstract]
- Krsková L, Sumerauer D, Stejskalová E, et al.: A novel variant of SYT-SSX1 fusion gene in a case of spindle cell synovial sarcoma. Diagn Mol Pathol 16 (3): 179-83, 2007. [PUBMED Abstract]
- Su L, Sampaio AV, Jones KB, et al.: Deconstruction of the SS18-SSX fusion oncoprotein complex: insights into disease etiology and therapeutics. Cancer Cell 21 (3): 333-47, 2012. [PUBMED Abstract]
- Arnold MA, Arnold CA, Li G, et al.: A unique pattern of INI1 immunohistochemistry distinguishes synovial sarcoma from its histologic mimics. Hum Pathol 44 (5): 881-7, 2013. [PUBMED Abstract]
- Vlenterie M, Ho VK, Kaal SE, et al.: Age as an independent prognostic factor for survival of localised synovial sarcoma patients. Br J Cancer 113 (11): 1602-6, 2015. [PUBMED Abstract]
- Okcu MF, Munsell M, Treuner J, et al.: Synovial sarcoma of childhood and adolescence: a multicenter, multivariate analysis of outcome. J Clin Oncol 21 (8): 1602-11, 2003. [PUBMED Abstract]
- Brecht IB, Ferrari A, Int-Veen C, et al.: Grossly-resected synovial sarcoma treated by the German and Italian Pediatric Soft Tissue Sarcoma Cooperative Groups: discussion on the role of adjuvant therapies. Pediatr Blood Cancer 46 (1): 11-7, 2006. [PUBMED Abstract]
- Stanelle EJ, Christison-Lagay ER, Healey JH, et al.: Pediatric and adolescent synovial sarcoma: multivariate analysis of prognostic factors and survival outcomes. Ann Surg Oncol 20 (1): 73-9, 2013. [PUBMED Abstract]
- Trassard M, Le Doussal V, Hacène K, et al.: Prognostic factors in localized primary synovial sarcoma: a multicenter study of 128 adult patients. J Clin Oncol 19 (2): 525-34, 2001. [PUBMED Abstract]
- Guillou L, Benhattar J, Bonichon F, et al.: Histologic grade, but not SYT-SSX fusion type, is an important prognostic factor in patients with synovial sarcoma: a multicenter, retrospective analysis. J Clin Oncol 22 (20): 4040-50, 2004. [PUBMED Abstract]
- Ferrari A, Gronchi A, Casanova M, et al.: Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer 101 (3): 627-34, 2004. [PUBMED Abstract]
- Lagarde P, Przybyl J, Brulard C, et al.: Chromosome instability accounts for reverse metastatic outcomes of pediatric and adult synovial sarcomas. J Clin Oncol 31 (5): 608-15, 2013. [PUBMED Abstract]
- Stegmaier S, Leuschner I, Poremba C, et al.: The prognostic impact of SYT-SSX fusion type and histological grade in pediatric patients with synovial sarcoma treated according to the CWS (Cooperative Weichteilsarkom Studie) trials. Pediatr Blood Cancer 64 (1): 89-95, 2017. [PUBMED Abstract]
- Scheer M, Dantonello T, Hallmen E, et al.: Primary Metastatic Synovial Sarcoma: Experience of the CWS Study Group. Pediatr Blood Cancer 63 (7): 1198-206, 2016. [PUBMED Abstract]
- Ferrari A, De Salvo GL, Dall'Igna P, et al.: Salvage rates and prognostic factors after relapse in children and adolescents with initially localised synovial sarcoma. Eur J Cancer 48 (18): 3448-55, 2012. [PUBMED Abstract]
- Ferrari A, Chi YY, De Salvo GL, et al.: Surgery alone is sufficient therapy for children and adolescents with low-risk synovial sarcoma: A joint analysis from the European paediatric soft tissue sarcoma Study Group and the Children's Oncology Group. Eur J Cancer 78: 1-6, 2017. [PUBMED Abstract]
- McGrory JE, Pritchard DJ, Arndt CA, et al.: Nonrhabdomyosarcoma soft tissue sarcomas in children. The Mayo Clinic experience. Clin Orthop (374): 247-58, 2000. [PUBMED Abstract]
- Van Glabbeke M, van Oosterom AT, Oosterhuis JW, et al.: Prognostic factors for the outcome of chemotherapy in advanced soft tissue sarcoma: an analysis of 2,185 patients treated with anthracycline-containing first-line regimens--a European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol 17 (1): 150-7, 1999. [PUBMED Abstract]
- Koscielniak E, Harms D, Henze G, et al.: Results of treatment for soft tissue sarcoma in childhood and adolescence: a final report of the German Cooperative Soft Tissue Sarcoma Study CWS-86. J Clin Oncol 17 (12): 3706-19, 1999. [PUBMED Abstract]
- Pappo AS, Devidas M, Jenkins J, et al.: Phase II trial of neoadjuvant vincristine, ifosfamide, and doxorubicin with granulocyte colony-stimulating factor support in children and adolescents with advanced-stage nonrhabdomyosarcomatous soft tissue sarcomas: a Pediatric Oncology Group Study. J Clin Oncol 23 (18): 4031-8, 2005. [PUBMED Abstract]
- Pappo AS, Rao BN, Jenkins JJ, et al.: Metastatic nonrhabdomyosarcomatous soft-tissue sarcomas in children and adolescents: the St. Jude Children's Research Hospital experience. Med Pediatr Oncol 33 (2): 76-82, 1999. [PUBMED Abstract]
- Brennan B, Stevens M, Kelsey A, et al.: Synovial sarcoma in childhood and adolescence: a retrospective series of 77 patients registered by the Children's Cancer and Leukaemia Group between 1991 and 2006. Pediatr Blood Cancer 55 (1): 85-90, 2010. [PUBMED Abstract]
- Ferrari A, Miceli R, Rey A, et al.: Non-metastatic unresected paediatric non-rhabdomyosarcoma soft tissue sarcomas: results of a pooled analysis from United States and European groups. Eur J Cancer 47 (5): 724-31, 2011. [PUBMED Abstract]
- Raney RB: Synovial sarcoma in young people: background, prognostic factors, and therapeutic questions. J Pediatr Hematol Oncol 27 (4): 207-11, 2005. [PUBMED Abstract]
- Orbach D, Mc Dowell H, Rey A, et al.: Sparing strategy does not compromise prognosis in pediatric localized synovial sarcoma: experience of the International Society of Pediatric Oncology, Malignant Mesenchymal Tumors (SIOP-MMT) Working Group. Pediatr Blood Cancer 57 (7): 1130-6, 2011. [PUBMED Abstract]
- Ladenstein R, Treuner J, Koscielniak E, et al.: Synovial sarcoma of childhood and adolescence. Report of the German CWS-81 study. Cancer 71 (11): 3647-55, 1993. [PUBMED Abstract]
- Venkatramani R, Anderson JR, Million L, et al.: Risk-based treatment for synovial sarcoma in patients under 30 years of age: Children’s Oncology Group study ARST0332. [Abstract] J Clin Oncol 33 (15 Suppl): A-10012, 2015. Also available online. Last accessed April 02, 2018.
- Ferrari A, De Salvo GL, Brennan B, et al.: Synovial sarcoma in children and adolescents: the European Pediatric Soft Tissue Sarcoma Study Group prospective trial (EpSSG NRSTS 2005). Ann Oncol 26 (3): 567-72, 2015. [PUBMED Abstract]
- Spunt SL, Million L, Anderson JR, et al.: Risk-based treatment for nonrhabdomyosarcoma soft tissue sarcomas (NRSTS) in patients under 30 years of age: Children’s Oncology Group study ARST0332. [Abstract] J Clin Oncol 32 (Suppl 15): A-10008, 2014. Also available online. Last accessed April 02, 2018.
- Randall RL, Albritton KH, Ferney BJ, et al.: Malignant fibrous histiocytoma of soft tissue: an abandoned diagnosis. Am J Orthop 33 (12): 602-8, 2004. [PUBMED Abstract]
- Alaggio R, Collini P, Randall RL, et al.: Undifferentiated high-grade pleomorphic sarcomas in children: a clinicopathologic study of 10 cases and review of literature. Pediatr Dev Pathol 13 (3): 209-17, 2010 May-Jun. [PUBMED Abstract]
- Daw NC, Billups CA, Pappo AS, et al.: Malignant fibrous histiocytoma and other fibrohistiocytic tumors in pediatric patients: the St. Jude Children's Research Hospital experience. Cancer 97 (11): 2839-47, 2003. [PUBMED Abstract]
- Coffin CM, Dehner LP: Vascular tumors in children and adolescents: a clinicopathologic study of 228 tumors in 222 patients. Pathol Annu 28 Pt 1: 97-120, 1993. [PUBMED Abstract]
- Cioffi A, Reichert S, Antonescu CR, et al.: Angiosarcomas and other sarcomas of endothelial origin. Hematol Oncol Clin North Am 27 (5): 975-88, 2013. [PUBMED Abstract]
- Jeng MR, Fuh B, Blatt J, et al.: Malignant transformation of infantile hemangioma to angiosarcoma: response to chemotherapy with bevacizumab. Pediatr Blood Cancer 61 (11): 2115-7, 2014. [PUBMED Abstract]
- Dehner LP, Ishak KG: Vascular tumors of the liver in infants and children. A study of 30 cases and review of the literature. Arch Pathol 92 (2): 101-11, 1971. [PUBMED Abstract]
- Ferrari A, Casanova M, Bisogno G, et al.: Malignant vascular tumors in children and adolescents: a report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Med Pediatr Oncol 39 (2): 109-14, 2002. [PUBMED Abstract]
- Deyrup AT, Miettinen M, North PE, et al.: Pediatric cutaneous angiosarcomas: a clinicopathologic study of 10 cases. Am J Surg Pathol 35 (1): 70-5, 2011. [PUBMED Abstract]
- Elliott P, Kleinschmidt I: Angiosarcoma of the liver in Great Britain in proximity to vinyl chloride sites. Occup Environ Med 54 (1): 14-8, 1997. [PUBMED Abstract]
- Lezama-del Valle P, Gerald WL, Tsai J, et al.: Malignant vascular tumors in young patients. Cancer 83 (8): 1634-9, 1998. [PUBMED Abstract]
- Fata F, O'Reilly E, Ilson D, et al.: Paclitaxel in the treatment of patients with angiosarcoma of the scalp or face. Cancer 86 (10): 2034-7, 1999. [PUBMED Abstract]
- Lahat G, Dhuka AR, Hallevi H, et al.: Angiosarcoma: clinical and molecular insights. Ann Surg 251 (6): 1098-106, 2010. [PUBMED Abstract]
- Orlando G, Adam R, Mirza D, et al.: Hepatic hemangiosarcoma: an absolute contraindication to liver transplantation--the European Liver Transplant Registry experience. Transplantation 95 (6): 872-7, 2013. [PUBMED Abstract]
- Sanada T, Nakayama H, Irisawa R, et al.: Clinical outcome and dose volume evaluation in patients who undergo brachytherapy for angiosarcoma of the scalp and face. Mol Clin Oncol 6 (3): 334-340, 2017. [PUBMED Abstract]
- Dickson MA, D'Adamo DR, Keohan ML, et al.: Phase II Trial of Gemcitabine and Docetaxel with Bevacizumab in Soft Tissue Sarcoma. Sarcoma 2015: 532478, 2015. [PUBMED Abstract]
- North PE, Waner M, Mizeracki A, et al.: A unique microvascular phenotype shared by juvenile hemangiomas and human placenta. Arch Dermatol 137 (5): 559-70, 2001. [PUBMED Abstract]
- Boye E, Yu Y, Paranya G, et al.: Clonality and altered behavior of endothelial cells from hemangiomas. J Clin Invest 107 (6): 745-52, 2001. [PUBMED Abstract]
- Ravi V, Patel S: Vascular sarcomas. Curr Oncol Rep 15 (4): 347-55, 2013. [PUBMED Abstract]
- Grassia KL, Peterman CM, Iacobas I, et al.: Clinical case series of pediatric hepatic angiosarcoma. Pediatr Blood Cancer 64 (11): , 2017. [PUBMED Abstract]
- Mehrabi A, Kashfi A, Fonouni H, et al.: Primary malignant hepatic epithelioid hemangioendothelioma: a comprehensive review of the literature with emphasis on the surgical therapy. Cancer 107 (9): 2108-21, 2006. [PUBMED Abstract]
- Haro A, Saitoh G, Tamiya S, et al.: Four-year natural clinical course of pulmonary epithelioid hemangioendothelioma without therapy. Thorac Cancer 6 (4): 544-7, 2015. [PUBMED Abstract]
- Sardaro A, Bardoscia L, Petruzzelli MF, et al.: Epithelioid hemangioendothelioma: an overview and update on a rare vascular tumor. Oncol Rev 8 (2): 259, 2014. [PUBMED Abstract]
- Dong K, Wang XX, Feng JL, et al.: Pathological characteristics of liver biopsies in eight patients with hepatic epithelioid hemangioendothelioma. Int J Clin Exp Pathol 8 (9): 11015-23, 2015. [PUBMED Abstract]
- Adams DM, Hammill A: Other vascular tumors. Semin Pediatr Surg 23 (4): 173-7, 2014. [PUBMED Abstract]
- Xiao Y, Wang C, Song Y, et al.: Primary epithelioid hemangioendothelioma of the kidney: the first case report in a child and literature review. Urology 82 (4): 925-7, 2013. [PUBMED Abstract]
- Reich S, Ringe H, Uhlenberg B, et al.: Epithelioid hemangioendothelioma of the lung presenting with pneumonia and heart rhythm disturbances in a teenage girl. J Pediatr Hematol Oncol 32 (4): 274-6, 2010. [PUBMED Abstract]
- Daller JA, Bueno J, Gutierrez J, et al.: Hepatic hemangioendothelioma: clinical experience and management strategy. J Pediatr Surg 34 (1): 98-105; discussion 105-6, 1999. [PUBMED Abstract]
- Ackermann O, Fabre M, Franchi S, et al.: Widening spectrum of liver angiosarcoma in children. J Pediatr Gastroenterol Nutr 53 (6): 615-9, 2011. [PUBMED Abstract]
- Stacchiotti S, Provenzano S, Dagrada G, et al.: Sirolimus in Advanced Epithelioid Hemangioendothelioma: A Retrospective Case-Series Analysis from the Italian Rare Cancer Network Database. Ann Surg Oncol 23 (9): 2735-44, 2016. [PUBMED Abstract]
- Semenisty V, Naroditsky I, Keidar Z, et al.: Pazopanib for metastatic pulmonary epithelioid hemangioendothelioma-a suitable treatment option: case report and review of anti-angiogenic treatment options. BMC Cancer 15: 402, 2015. [PUBMED Abstract]
- Raheja A, Suri A, Singh S, et al.: Multimodality management of a giant skull base hemangioendothelioma of the sphenopetroclival region. J Clin Neurosci 22 (9): 1495-8, 2015. [PUBMED Abstract]
- Ahmad N, Adams DM, Wang J, et al.: Hepatic epithelioid hemangioendothelioma in a patient with hemochromatosis. J Natl Compr Canc Netw 12 (9): 1203-7, 2014. [PUBMED Abstract]
- Otte JB, Zimmerman A: The role of liver transplantation for pediatric epithelioid hemangioendothelioma. Pediatr Transplant 14 (3): 295-7, 2010. [PUBMED Abstract]
Treatment of Metastatic Childhood Soft Tissue Sarcoma
Standard treatment options for metastatic childhood soft tissue sarcoma include the following:
- Combination therapy using chemotherapy, radiation therapy, and surgical resection of pulmonary metastases.
For treatment options, refer to the individual tumor type sections of the summary.
The prognosis for children with metastatic soft tissue sarcomas is poor,[1-6] and these children should receive combined treatment with chemotherapy, radiation therapy, and surgical resection of pulmonary metastases. In a prospective randomized trial, chemotherapy with vincristine, dactinomycin, doxorubicin, and cyclophosphamide, with or without dacarbazine, led to tumor responses in one-third of patients with unresectable or metastatic disease. The estimated 4-year survival rate, however, was poor, with fewer than one-third of children surviving.[6-8]
Pulmonary Metastases
Generally, children with isolated pulmonary metastases should be considered for a surgical procedure in an attempt to resect all gross disease.[9] For patients with multiple or recurrent pulmonary metastases, additional surgical procedures can be performed if the morbidity is deemed acceptable. In a retrospective review, patients with synovial sarcoma and pulmonary metastases for whom it was possible to completely resect all metastatic lung lesions had better survival than did patients for whom it was not possible to achieve complete resections.[9][Level of evidence: 3iiiA] Formal segmentectomy, lobectomy, and mediastinal lymph node dissection are unnecessary.[10]
An alternative approach is focused radiation therapy (fractionated stereotactic radiation therapy), which has been successfully used in adults to control lesions. The estimated 5-year survival rate after thoracotomy for pulmonary metastasectomy has ranged from 10% to 58% in adult studies. Emerging data suggest a similar outcome after the administration of focused radiation therapy.[11]
References
- Demetri GD, Elias AD: Results of single-agent and combination chemotherapy for advanced soft tissue sarcomas. Implications for decision making in the clinic. Hematol Oncol Clin North Am 9 (4): 765-85, 1995. [PUBMED Abstract]
- Elias A, Ryan L, Sulkes A, et al.: Response to mesna, doxorubicin, ifosfamide, and dacarbazine in 108 patients with metastatic or unresectable sarcoma and no prior chemotherapy. J Clin Oncol 7 (9): 1208-16, 1989. [PUBMED Abstract]
- Edmonson JH, Ryan LM, Blum RH, et al.: Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol 11 (7): 1269-75, 1993. [PUBMED Abstract]
- Rao BN: Nonrhabdomyosarcoma in children: prognostic factors influencing survival. Semin Surg Oncol 9 (6): 524-31, 1993 Nov-Dec. [PUBMED Abstract]
- deCou JM, Rao BN, Parham DM, et al.: Malignant peripheral nerve sheath tumors: the St. Jude Children's Research Hospital experience. Ann Surg Oncol 2 (6): 524-9, 1995. [PUBMED Abstract]
- Pappo AS, Rao BN, Jenkins JJ, et al.: Metastatic nonrhabdomyosarcomatous soft-tissue sarcomas in children and adolescents: the St. Jude Children's Research Hospital experience. Med Pediatr Oncol 33 (2): 76-82, 1999. [PUBMED Abstract]
- Pratt CB, Pappo AS, Gieser P, et al.: Role of adjuvant chemotherapy in the treatment of surgically resected pediatric nonrhabdomyosarcomatous soft tissue sarcomas: A Pediatric Oncology Group Study. J Clin Oncol 17 (4): 1219, 1999. [PUBMED Abstract]
- Pratt CB, Maurer HM, Gieser P, et al.: Treatment of unresectable or metastatic pediatric soft tissue sarcomas with surgery, irradiation, and chemotherapy: a Pediatric Oncology Group study. Med Pediatr Oncol 30 (4): 201-9, 1998. [PUBMED Abstract]
- Stanelle EJ, Christison-Lagay ER, Wolden SL, et al.: Pulmonary metastasectomy in pediatric/adolescent patients with synovial sarcoma: an institutional review. J Pediatr Surg 48 (4): 757-63, 2013. [PUBMED Abstract]
- Putnam JB Jr, Roth JA: Surgical treatment for pulmonary metastases from sarcoma. Hematol Oncol Clin North Am 9 (4): 869-87, 1995. [PUBMED Abstract]
- Dhakal S, Corbin KS, Milano MT, et al.: Stereotactic body radiotherapy for pulmonary metastases from soft-tissue sarcomas: excellent local lesion control and improved patient survival. Int J Radiat Oncol Biol Phys 82 (2): 940-5, 2012. [PUBMED Abstract]
Treatment of Progressive/Recurrent Childhood Soft Tissue Sarcoma
With the possible exception of infants with infantile fibrosarcoma, the prognosis for patients with recurrent or progressive disease is poor. No prospective trial has been able to prove that enhanced local control of pediatric soft tissue sarcomas will ultimately improve survival. Therefore, treatment should be individualized for the site of recurrence, biologic characteristics of the tumor (e.g., grade, invasiveness, and size), previous therapies, and individual patient considerations.
Treatment options for recurrent or progressive disease include the following:
- Surgical excision of local recurrence or isolated pulmonary recurrence.
- An Italian review of 73 patients with recurrent malignant peripheral nerve sheath tumors found that most relapses were local. Multivariate analysis showed that the factors associated with improved survival were no tumor invasiveness at initial diagnosis (T1), time of recurrence more than 12 months after initial diagnosis, and achievement of a second complete response with surgical removal of the recurrence(s). Only 15.8% of patients who had complete surgical excisions of local recurrence(s) were alive at 5 years.[1][Level of evidence: 3iiiA]
- Surgical excision of local recurrence followed by radiation therapy or brachytherapy (if no previous radiation therapy was given).
- Limb amputation (only for some children with extremity sarcomas that have already received radiation therapy).
- Gemcitabine and docetaxel.[2]
- Trabectedin.[3-5]
- Pazopanib. A phase I trial of pazopanib reported one partial response in a patient with desmoplastic small round cell tumor and prolonged disease stabilization in eight patients with recurrent sarcoma.[6][Level of evidence: 2Diii] Pazopanib has been approved for use in recurrent soft tissue sarcoma. The clinical trial that was used to obtain approval was limited to adults and demonstrated disease stabilization and prolonged time to progression; it did not demonstrate improved overall survival.[7] One 13-year-old boy and one 14-year-old girl with multiply recurrent synovial sarcoma and lung metastases had responses to pazopanib for 14 and 15 months, respectively.[8][Level of evidence: 3iiDi]
- A clinical trial of new chemotherapeutic regimens.
Resection is the standard treatment for recurrent pediatric nonrhabdomyosarcomatous soft tissue sarcomas. If the patient has not yet received radiation therapy, postoperative radiation should be considered after local excision of the recurrent tumor. Limb-sparing procedures with postoperative brachytherapy have been evaluated in adults but have not been studied extensively in children. For some children with extremity sarcomas who have received previous radiation therapy, amputation may be the only therapeutic option.
Pulmonary metastasectomy may achieve prolonged disease control for some patients.[9] A large, retrospective analysis of patients with recurrent soft tissue sarcoma showed that isolated local relapse had a better prognosis and that resection of pulmonary metastases improved the probability of survival.[10] In 31 children and adolescents younger than 23 years with pulmonary metastases from synovial sarcoma, complete resection of lung metastases appeared to prolong survival when compared with ten other patients who were not considered candidates for metastasectomy.[11][Level of evidence: 3iiiA] All patients with recurrent tumors should be considered for current clinical trials.
Published results of two studies addressed the outcomes for children with relapsed synovial sarcoma. Most patients in one study had distant relapse (29 of 44 patients),[12] while most patients in the second study had local relapse (27 of 37 patients).[13] Distant recurrence was a poor prognostic variable, while tumor resectability at relapse (as manifested by extremity recurrence) was associated with a better outcome in both studies.
Treatment Options Under Clinical Evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- APEC1621 (NCT03155620) (Pediatric MATCH: Targeted Therapy Directed by Genetic Testing in Treating Pediatric Patients with Relapsed or Refractory Advanced Solid Tumors, Non-Hodgkin Lymphomas, or Histiocytic Disorders): NCI-COG Pediatric Molecular Analysis for Therapeutic Choice (MATCH), referred to as Pediatric MATCH, will match targeted agents with specific molecular changes identified using a next-generation sequencing targeted assay of more than 3,000 different mutations across more than 160 genes in refractory and recurrent solid tumors. Children and adolescents aged 1 to 21 years are eligible for the trial.
Tumor tissue from progressive or recurrent disease must be available for molecular characterization. Patients with tumors that have molecular variants addressed by treatment arms included in the trial will be offered treatment on Pediatric MATCH. Additional information can be obtained on the ClinicalTrials.gov website for APEC1621 (NCT03155620).
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Bergamaschi L, Bisogno G, Manzitti C, et al.: Salvage rates and prognostic factors after relapse in children and adolescents with malignant peripheral nerve sheath tumors. Pediatr Blood Cancer 65 (2): , 2018. [PUBMED Abstract]
- Maki RG, Wathen JK, Patel SR, et al.: Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 25 (19): 2755-63, 2007. [PUBMED Abstract]
- Le Cesne A, Cresta S, Maki RG, et al.: A retrospective analysis of antitumour activity with trabectedin in translocation-related sarcomas. Eur J Cancer 48 (16): 3036-44, 2012. [PUBMED Abstract]
- Garcia-Carbonero R, Supko JG, Maki RG, et al.: Ecteinascidin-743 (ET-743) for chemotherapy-naive patients with advanced soft tissue sarcomas: multicenter phase II and pharmacokinetic study. J Clin Oncol 23 (24): 5484-92, 2005. [PUBMED Abstract]
- Garcia-Carbonero R, Supko JG, Manola J, et al.: Phase II and pharmacokinetic study of ecteinascidin 743 in patients with progressive sarcomas of soft tissues refractory to chemotherapy. J Clin Oncol 22 (8): 1480-90, 2004. [PUBMED Abstract]
- Glade Bender JL, Lee A, Reid JM, et al.: Phase I pharmacokinetic and pharmacodynamic study of pazopanib in children with soft tissue sarcoma and other refractory solid tumors: a children's oncology group phase I consortium report. J Clin Oncol 31 (24): 3034-43, 2013. [PUBMED Abstract]
- van der Graaf WT, Blay JY, Chawla SP, et al.: Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 379 (9829): 1879-86, 2012. [PUBMED Abstract]
- Casanova M, Basso E, Magni C, et al.: Response to pazopanib in two pediatric patients with pretreated relapsing synovial sarcoma. Tumori 103 (1): e1-e3, 2017. [PUBMED Abstract]
- Belal A, Salah E, Hajjar W, et al.: Pulmonary metastatectomy for soft tissue sarcomas: is it valuable? J Cardiovasc Surg (Torino) 42 (6): 835-40, 2001. [PUBMED Abstract]
- Zagars GK, Ballo MT, Pisters PW, et al.: Prognostic factors for disease-specific survival after first relapse of soft-tissue sarcoma: analysis of 402 patients with disease relapse after initial conservative surgery and radiotherapy. Int J Radiat Oncol Biol Phys 57 (3): 739-47, 2003. [PUBMED Abstract]
- Stanelle EJ, Christison-Lagay ER, Wolden SL, et al.: Pulmonary metastasectomy in pediatric/adolescent patients with synovial sarcoma: an institutional review. J Pediatr Surg 48 (4): 757-63, 2013. [PUBMED Abstract]
- Ferrari A, De Salvo GL, Dall'Igna P, et al.: Salvage rates and prognostic factors after relapse in children and adolescents with initially localised synovial sarcoma. Eur J Cancer 48 (18): 3448-55, 2012. [PUBMED Abstract]
- Soole F, Maupain C, Defachelles AS, et al.: Synovial sarcoma relapses in children and adolescents: prognostic factors, treatment, and outcome. Pediatr Blood Cancer 61 (8): 1387-93, 2014. [PUBMED Abstract]
Changes to This Summary (04/02/2018)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Treatment of Newly Diagnosed Childhood Soft Tissue Sarcoma
Added text to state that a small series reported symptomatic improvement and stable disease in seven patients with desmoid-type fibromatosis who were treated with pazopanib (cited Agresta et al. as reference 48).
Added text to state that a tumor with morphology similar to that of infantile fibrosarcoma has been identified in older children; in these older children, the tumors do not have the t(12;15)(ETV-NTRK3) translocation that is characteristic of the younger patients. In several of these patients, BRAF gene fusions have been identified (cited Kao et al. as reference 69).
Added text about the outcome results of 73 children and adolescents with recurrent malignant peripheral nerve sheath tumor reported by the Italian Sarcoma Group (cited Bergamaschi et al. as reference 127 and level of evidence 3iiiA).
Added text about the patient characteristics and results of a retrospective review of children and young adults younger than 30 years from four institutions, which identified 69 patients with alveolar soft part sarcoma treated primarily with surgery between 1980 and 2014 (cited Flores et al. as reference 152 and level of evidence 3iiA).
Added Sedig et al. as reference 172 and level of evidence 3iiiA.
Treatment of Progressive/Recurrent Childhood Soft Tissue Sarcoma
Added text about the prognostic factors and outcome results reported in an Italian review of 73 children and adolescents with recurrent malignant peripheral nerve sheath tumor (cited Bergamaschi et al. as reference 1 and level of evidence 3iiiA).
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of childhood soft tissue sarcoma. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Childhood Soft Tissue Sarcoma Treatment are:
- Denise Adams, MD (Children's Hospital Boston)
- Louis S. Constine, MD (James P. Wilmot Cancer Center at University of Rochester Medical Center)
- Holcombe Edwin Grier, MD (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Andrea A. Hayes-Jordan, MD, FACS, FAAP (M.D. Anderson Cancer Center)
- Paul A. Meyers, MD (Memorial Sloan-Kettering Cancer Center)
- Thomas A. Olson, MD (Aflac Cancer and Blood Disorders Center of Children's Healthcare of Atlanta - Egleston Campus)
- Alberto S. Pappo, MD (St. Jude Children's Research Hospital)
- R Beverly Raney, MD (Consultant)
- Stephen J. Shochat, MD (St. Jude Children's Research Hospital)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Childhood Soft Tissue Sarcoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/soft-tissue-sarcoma/hp/child-soft-tissue-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389361]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Updated: April 2, 2018
Source URL: https://www.cancer.gov/publishedcontent/syndication/3899.htm
Source Agency: National Cancer Institute (NCI)
Captured Date: 2013-09-14 09:01:57.0
Childhood Soft Tissue Sarcoma Treatment (PDQ®)–Health Professional Version
General Information About Childhood Soft Tissue Sarcoma
Dramatic improvements in survival have been achieved for children and adolescents with cancer. Between 1975 and 2010, childhood cancer mortality decreased by more than 50%.[1] Childhood and adolescent cancer survivors require close monitoring because cancer therapy side effects may persist or develop months or years after treatment. (Refer to the PDQ summary on Late Effects of Treatment for Childhood Cancer for specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors.)
Rhabdomyosarcoma, a tumor of striated muscle, is the most common soft tissue sarcoma in children aged 0 to 14 years and accounts for 50% of tumors in this age group.[2] (Refer to the PDQ summary on Childhood Rhabdomyosarcoma Treatment for more information.) In pediatrics, the remaining soft tissue sarcomas are commonly referred to as nonrhabdomyosarcomatous soft tissue sarcomas and account for approximately 3% of all childhood tumors.[3] This heterogeneous group of tumors includes the following neoplasms:[4]
- Connective tissue (e.g., desmoid-type fibromatosis).
- Peripheral nervous system (e.g., malignant peripheral nerve sheath tumor).
- Smooth muscle (e.g., leiomyosarcoma).
- Vascular tissue (blood and lymphatic vessels, e.g., angiosarcoma). (Refer to the PDQ summary on Childhood Vascular Tumors Treatment for more information about childhood vascular tumors.)
Distribution of Soft Tissue Sarcoma by Age and Histology
Pediatric soft tissue sarcomas are a heterogenous group of malignant tumors that originate from primitive mesenchymal tissue and account for 7% of all childhood tumors.[5]
The distribution of soft tissue sarcomas by histology and age, based on the Surveillance, Epidemiology, and End Results (SEER) information from 1975 to 2012, is depicted in Table 1. The distribution of histologic subtypes by age is also shown in Figure 2.
| Age <5 y | Age 5–9 y | Age 10–14 y | Age 15–19 y | % of the Total Number of STS Cases <20 y | |||
|---|---|---|---|---|---|---|---|
| pPNET = peripheral primitive neuroectodermal tumors; SEER = Surveillance, Epidemiology, and End Results; STS = soft tissue sarcoma. | |||||||
| aSEER data is available at http://seer.cancer.gov. | |||||||
| bDermatofibrosarcoma accounts for 75% of these cases. | |||||||
| All soft tissue and other extraosseous sarcomas | 923 | 631 | 946 | 1,267 | 100 | ||
| Rhabdomyosarcomas | 551 | 348 | 312 | 270 | 39 | ||
| Fibrosarcomas, peripheral nerve, and other fibrous neoplasms | 116 | 50 | 88 | 141 | 10 | ||
| Fibroblastic and myofibroblastic tumors | 97 | 24 | 31 | 62 | 6 | ||
| Nerve sheath tumors | 19 | 26 | 56 | 77 | 5 | ||
| Other fibromatous neoplasms | 0 | 0 | 1 | 2 | 0.1 | ||
| Kaposi sarcoma | 2 | 1 | 1 | 9 | 0.3 | ||
| Other specified soft tissue sarcomas | 194 | 190 | 424 | 708 | 40 | ||
| Ewing tumor and Askin tumor of soft tissue | 27 | 30 | 62 | 92 | 6 | ||
| pPNET of soft tissue | 21 | 18 | 36 | 46 | 3.2 | ||
| Extrarenal rhabdoid tumor | 61 | 3 | 7 | 3 | 2 | ||
| Liposarcomas | 3 | 5 | 22 | 57 | 2.3 | ||
| Fibrohistiocytic tumors b | 34 | 54 | 108 | 188 | 10 | ||
| Leiomyosarcomas | 9 | 14 | 15 | 36 | 2 | ||
| Synovial sarcomas | 10 | 34 | 111 | 175 | 9 | ||
| Blood vessel tumors | 11 | 7 | 8 | 25 | 1.4 | ||
| Osseous and chondromatous neoplasms of soft tissue | 1 | 6 | 13 | 10 | 0.8 | ||
| Alveolar soft parts sarcoma | 4 | 3 | 16 | 29 | 1.4 | ||
| Miscellaneous soft tissue sarcomas | 13 | 16 | 36 | 47 | 3 | ||
| Unspecified soft tissue sarcomas | 60 | 40 | 111 | 139 | 9.3 | ||
Nonrhabdomyosarcomatous soft tissue sarcomas are more common in adolescents and adults,[4] and most of the information regarding treatment and natural history of the disease in younger patients has been based on adult studies. The distributions of these tumors by age according to stage, histologic subtype, and tumor site are shown in Figures 1, 2, and 3, respectively.[6]
Risk Factors
Some genetic and environmental factors have been associated with the development of nonrhabdomyosarcomatous soft tissue sarcoma, including the following:
- Genetic factors:
- Li-Fraumeni syndrome: Patients with Li-Fraumeni syndrome (usually due to heritable cancer-associated changes of the TP53 tumor suppressor gene) have an increased risk of developing soft tissue tumors (mostly nonrhabdomyosarcomatous soft tissue sarcomas), bone sarcomas, breast cancer, brain tumors, and acute leukemia.[7,8]
- Familial adenomatous polyposis: Patients with familial adenomatous polyposis are at increased risk of developing desmoid-type fibromatosis.[9]
- Retinoblastoma (RB1) gene: Germline mutations of the retinoblastoma gene have been associated with an increased risk of developing soft tissue sarcomas, particularly leiomyosarcoma.[10]
- SMARCB1 gene: Germline mutations or deletions of the SMARCB1 (INI1) gene are associated with an increased risk of developing extrarenal rhabdoid tumors.[11]
- Neurofibromatosis type 1: Approximately 4% of patients with neurofibromatosis type 1 develop malignant peripheral nerve sheath tumors, which usually develop after a long latency; some patients develop multiple lesions.[12-14]
- Werner syndrome: Werner syndrome is characterized by spontaneous chromosomal instability, resulting in increased susceptibility to cancer and premature aging. An excess of soft tissue sarcomas has been reported in patients with Werner syndrome.[15]
- Environmental factors:
- Radiation: Some nonrhabdomyosarcomatous soft tissue sarcomas (particularly malignant fibrous histiocytoma) can develop within a previously irradiated site.[3,16]
- Epstein-Barr virus infection in patients with AIDS: Some nonrhabdomyosarcomatous soft tissue sarcomas (e.g., leiomyosarcoma) have been linked to Epstein-Barr virus infection in patients with AIDS.[3,17]
Clinical Presentation
Although nonrhabdomyosarcomatous soft tissue sarcomas can develop in any part of the body, they arise most commonly in the trunk and extremities.[18-20] These neoplasms can present initially as an asymptomatic solid mass, or they may be symptomatic because of local invasion of adjacent anatomical structures. Although rare, these tumors can arise primarily in brain tissue and are treated according to the histotype.[21]
Systemic symptoms (e.g., fever, weight loss, and night sweats) are rare. Hypoglycemia and hypophosphatemic rickets have been reported in cases of hemangiopericytoma, whereas hyperglycemia has been noted in patients with fibrosarcoma of the lung.[22]
Diagnostic and Staging Evaluation
When a suspicious lesion is identified, it is crucial that a complete workup, followed by adequate biopsy be performed. It is best to image the lesion using the following procedures before initiating any intervention:
- Plain films. Plain films can be used to rule out bone involvement and detect calcifications that may be seen in soft tissue tumors such as extraskeletal osteosarcoma or synovial sarcoma.
- Chest computed tomography (CT). Chest CT is essential to assess the presence of metastases.
- Abdominal CT or magnetic resonance imaging (MRI). Abdominal CT or MRI can be used to image intra-abdominal tumors, such as liposarcoma.
- Extremity MRI. MRI is essential for extremity lesions.
- Positron emission tomography (PET) scan and bone scan. In children with rhabdomyosarcoma, PET-CT performed better than conventional imaging in identifying nodal, bone, bone marrow, and soft tissue disease. The authors of an imaging comparison study suggest that bone scans with technetium Tc 99m might be eliminated as a staging procedure.[23] The use of this modality in pediatric nonrhabdomyosarcomatous soft tissue sarcoma has not been studied extensively. However, a small study of nine patients with nonrhabdomyosarcomatous soft tissue sarcoma suggests that PET-CT is more accurate and cost effective than either modality alone in identifying distant metastatic disease.[24]
The imaging characteristics of some tumors can be highly suggestive of this diagnosis. For example, the imaging characteristics of pediatric low-grade fibromyxoid sarcoma and alveolar soft part sarcoma have been described and can aid in the diagnosis of these rare neoplasms.[25]
Biopsy strategies
Although nonrhabdomyosarcomatous soft tissue tumors are fairly readily distinguished pathologically from rhabdomyosarcoma and Ewing sarcoma, the classification of childhood nonrhabdomyosarcomatous soft tissue sarcoma type is often difficult. Core-needle biopsy, incisional biopsy, or excisional biopsy can be used to diagnose a nonrhabdomyosarcomatous soft tissue sarcoma. If possible, the surgeon who will perform the definitive resection needs to be involved in the biopsy decision. Poorly placed incisional or needle biopsies may adversely affect the performance of the primary resection.
Considerations related to the selection of a biopsy procedure are as follows:
- Given the diagnostic importance of translocations, a core-needle biopsy or small incisional biopsy that obtains adequate tumor tissue is crucial to allow for conventional histology, immunocytochemical analysis, and other studies such as light and electron microscopy, cytogenetics, fluorescence in situ hybridization, and molecular pathology.[26,27] Core-needle biopsy for a deep-seated tumor can lead to formation of a hematoma, which affects subsequent resection and/or radiation; in these cases, incisional biopsy is the preferred procedure.
- Fine-needle biopsy is usually not recommended because it is difficult to determine the accurate histologic diagnosis and grade of the tumor in this heterogeneous group of tumors.
- Image guidance using ultrasound, CT scan, or MRI may be necessary to ensure a representative biopsy.[28]
- Needle biopsy techniques must ensure adequate tissue sampling. The acquisition of multiple cores of tissue may be required.
- Incisional biopsies must not compromise subsequent wide local excision.
- Excisional biopsy of the lesion is only appropriate for small superficial lesions (<3 cm in size) and are discouraged.[29,30] If an excisional biopsy is contemplated, then MRI of the area is recommended to define the area of involvement as subsequent surgery or radiation therapy is likely.
- Various institutional series have demonstrated the feasibility and effectiveness of sentinel node biopsy as a staging procedure in pediatric patients with soft tissue sarcomas.[31-36]
- Transverse extremity incisions are avoided to reduce skin loss and because they require a greater cross-sectional volume of tissue to be covered in the radiation field. Other extensive surgical procedures are also avoided before definitive diagnosis. For these reasons, open biopsy or multiple core-needle biopsies are strongly encouraged so that adequate tumor tissue can be obtained to allow crucial studies to be performed and to avoid limiting future treatment options.
Unplanned resection
In children with unplanned resection of nonrhabdomyosarcomatous soft tissue sarcomas, primary re-excision is frequently recommended because many patients will have tumor present in the re-excision specimen.[37,38] A single-institution analysis of adolescents and adults compared patients with unplanned excision of soft tissue sarcoma to stage-matched controls. In this retrospective analysis, unplanned initial excision of soft tissue sarcoma resulted in increased risk of local recurrence, metastasis, and death; this increase was greatest for high-grade tumors.[39][Level of evidence: 3iiA]
Chromosomal abnormalities
Many nonrhabdomyosarcomatous soft tissue sarcomas are characterized by chromosomal abnormalities. Some of these chromosomal translocations lead to a fusion of two disparate genes. The resulting fusion transcript can be readily detected by using polymerase chain reaction-based techniques, thus facilitating the diagnosis of those neoplasms that have translocations.
Some of the most frequent aberrations seen in nonrhabdomyosarcomatous soft tissue tumors are listed in Table 2.
| Histology | Chromosomal Aberrations | Genes Involved |
|---|---|---|
| aAdapted from Sandberg,[40] Slater et al.,[41] Mertens et al.,[42] and Romeo.[43] | ||
| Alveolar soft part sarcoma | t(x;17)(p11.2;q25) | ASPL/TFE3 [44-46] |
| Angiomatoid fibrous histiocytoma | t(12;16)(q13;p11), t(2;22)(q33;q12), t(12;22)(q13;q12) | FUS/ATF1, EWSR1/CREB1,[47] EWS/ATF1 |
| Clear cell sarcoma | t(12;22)(q13;q12), t(2;22)(q33;q12) | ATF1/EWS, EWSR1/CREB1 |
| Congenital (infantile) fibrosarcoma/mesoblastic nephroma | t(12;15)(p13;q25) | ETV-NTRK3 |
| Dermatofibrosarcoma protuberans | t(17;22)(q22;q13) | COL1A1/PDGFB |
| Desmoid fibromatosis | Trisomy 8 or 20, loss of 5q21 | CTNNB1 or APC mutations |
| Desmoplastic small round cell tumors | t(11;22)(p13;q12) | EWS/WT1 [48,49] |
| Epithelioid hemangioendothelioma | t(1;3)(p36;q25) [50] | WWTR1/CAMTA1 |
| Epithelioid sarcoma | Inactivation SMARCB1 | SMARCB1 |
| Extraskeletal myxoid chondrosarcoma | t(9;22)(q22;q12), t(9;17)(q22;q11), t(9;15)(q22;q21), t(3;9)(q11;q22) | EWSR1/NR4A3, TAF2N/NR4A3, TCF12/NR4A3, TGF/NR4A3 |
| Hemangiopericytoma | t(12;19)(q13;q13.3) and t(13;22)(q22;q13.3) | |
| Infantile fibrosarcoma | t(12;15)(p13;q25) | ETV6/NTRK3 |
| Inflammatory myofibroblastic tumor | t(1;2)(q23;q23), t(2;19)(q23;q13), t(2;17)(q23;q23), t(2;2)(p23;q13), t(2;11)(p23;p15) [51] | TPM3/ALK, TPM4/ALK, CLTC/ALK, RANBP2/ALK, CARS/ALK, RAS |
| Low-grade fibromyxoid sarcoma | t(7;16)(q33;p11), t(11;16)(p11;p11) | FUS/CREB3L2, FUS/CREB3L1 |
| Malignant peripheral nerve sheath tumor | 17q11.2, loss or rearrangement 10p, 11q, 17q, 22q | NF1 |
| Mesenchymal chondrosarcoma | Del(8)(q13.3q21.1) | HEY1/NCOA2 |
| Myoepithelioma | t(19;22)(q13;q12), t(1;22)(q23;q12), t(6;22)(p21;q12) | EWSR/ZNF44, EWSR/PBX1, EWSR/POU5F1 |
| Myxoid/round cell liposarcoma | t(12;16)(q13;p11), t(12;22)(q13;q12) | FUS/DD1T3, EWSR/DD1T3 |
| Rhabdoid tumor | Inactivation SMARCB1 | SMARCB1 |
| Solitary fibrous tumor | Inv(12)(q13q13) | NAB2/STAT6 |
| Synovial sarcoma | t(x;18)(p11.2;q11.2) | SYT/SSX |
| Tenosynovial giant cell tumor | t(1;2)(p13;q35) | COL6A3/CSF1 |
Prognosis
The prognosis of nonrhabdomyosarcomatous soft tissue sarcoma varies greatly depending on the following factors:[52-54]
- Site of the primary tumor.
- Tumor size.
- Tumor grade. (Refer to the Prognostic Significance of Tumor Grading section of this summary for more information.)
- Tumor histology.
- Depth of tumor invasion.
- Presence of metastases.
- Resectability of the tumor.
- Use of radiation therapy.
Several adult and pediatric series have shown that patients with large or invasive tumors have a significantly worse prognosis than do those with small, noninvasive tumors. A retrospective review of soft tissue sarcomas in children and adolescents suggests that the 5 cm cutoff used for adults with soft tissue sarcoma may not be ideal for smaller children, especially infants. The review identified an interaction between tumor diameter and body surface area.[55] This relationship requires further study to determine the therapeutic implications of the observation.
In a review of a large adult series of nonrhabdomyosarcomatous soft tissue sarcomas, superficial extremity sarcomas had a better prognosis than did deep tumors. Thus, in addition to grade and size, the depth of invasion of the tumor should be considered.[56]
Some pediatric nonrhabdomyosarcomatous soft tissue sarcomas are associated with a better outcome. For instance, infantile fibrosarcoma, presenting in infants and children younger than 5 years, has an excellent prognosis given that surgery alone can cure a significant number of these patients and the tumor is highly chemosensitive.[3]
Soft tissue sarcomas in older children and adolescents often behave similarly to those in adult patients.[3,26] A large, prospective, multinational Children's Oncology Group study (ARST0332 [NCT00346164]) enrolled newly diagnosed patients younger than 30 years. Patients were assigned to treatment on the basis of their risk group (refer to Figure 4).[57][Level of evidence: 2A]
- Arm A (grossly excised low-grade tumor and ≤5 cm widely excised high-grade tumor): Surgery only.
- Arm B (≤5 cm marginally resected high-grade tumor): 55.8 Gy of radiation therapy.
- Arm C (>5 cm grossly resected tumor ± metastases): Ifosfamide/doxorubicin chemotherapy and 55.8 Gy of radiation therapy.
- Arm D (>5 cm unresected tumor ± metastases): Preoperative ifosfamide/doxorubicin chemotherapy and 45 Gy of radiation therapy, and then surgery and a radiation boost that was based on margins.
Of 551 patients enrolled, at a median follow-up of 2.6 years, the preliminary analysis estimated the following 3-year survival rates:[57]
- Arm A: 91% event-free survival (EFS); 99% overall survival (OS).
- Arm B: 79% EFS; 100% OS.
- Arm C: 68% EFS; 81% OS.
- Arm D: 52% EFS; 66% OS.
Pediatric patients with unresected localized nonrhabdomyosarcomatous soft tissue sarcomas have a poor outcome. Only about one-third of patients treated with multimodality therapy remain disease free.[52,58]; [59,60][Level of evidence: 3iiiA] In a review of 30 Italian patients with nonrhabdomyosarcomatous soft tissue sarcoma at visceral sites, only ten patients survived at 5 years. Unfavorable prognostic factors included inability to achieve complete resection, large tumor size, tumor invasion, histologic subtype, and lung-pleura sites.[61][Level of evidence: 3iiB]
In a pooled analysis from U.S. and European pediatric centers, outcome was better for patients whose tumor removal procedure was deemed complete than for patients whose tumor removal was incomplete. Outcome was better for patients who received radiation therapy than for patients who did not.[59][Level of evidence: 3iiiA]
Because long-term related morbidity must be minimized while disease-free survival is maximized, the ideal therapy for each patient must be carefully and individually determined utilizing these prognostic factors before initiating therapy.[19,62-66]
Related Summaries
Refer to the following PDQ summaries for information about other types of sarcoma:
- Childhood Rhabdomyosarcoma Treatment.
- Childhood Vascular Tumors Treatment.
- Ewing Sarcoma Treatment (extraosseous Ewing, peripheral neuroepithelioma, and Askin tumors).
- Unusual Cancers of Childhood Treatment (gastrointestinal stromal tumors).
- Adult Soft Tissue Sarcoma Treatment.
References
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014. [PUBMED Abstract]
- Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. Bethesda, Md: National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649. Also available online. Last accessed January 24, 2018.
- Spunt SL, Million L, Coffin C: The nonrhabdomyosarcoma soft tissue sarcoma. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 827-54.
- Weiss SW, Goldblum JR: General considerations. In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. St. Louis, Mo: Mosby, 2008, pp 1-14.
- Pappo AS, Pratt CB: Soft tissue sarcomas in children. Cancer Treat Res 91: 205-22, 1997. [PUBMED Abstract]
- Ferrari A, Sultan I, Huang TT, et al.: Soft tissue sarcoma across the age spectrum: a population-based study from the Surveillance Epidemiology and End Results database. Pediatr Blood Cancer 57 (6): 943-9, 2011. [PUBMED Abstract]
- Chang F, Syrjänen S, Syrjänen K: Implications of the p53 tumor-suppressor gene in clinical oncology. J Clin Oncol 13 (4): 1009-22, 1995. [PUBMED Abstract]
- Plon SE, Malkin D: Childhood cancer and hereditary. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 13-31.
- Groen EJ, Roos A, Muntinghe FL, et al.: Extra-intestinal manifestations of familial adenomatous polyposis. Ann Surg Oncol 15 (9): 2439-50, 2008. [PUBMED Abstract]
- Kleinerman RA, Tucker MA, Abramson DH, et al.: Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst 99 (1): 24-31, 2007. [PUBMED Abstract]
- Eaton KW, Tooke LS, Wainwright LM, et al.: Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56 (1): 7-15, 2011. [PUBMED Abstract]
- Weiss SW, Goldblum JR: Benign tumors of peripheral nerves. In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. St. Louis, Mo: Mosby, 2008, pp 825-901.
- deCou JM, Rao BN, Parham DM, et al.: Malignant peripheral nerve sheath tumors: the St. Jude Children's Research Hospital experience. Ann Surg Oncol 2 (6): 524-9, 1995. [PUBMED Abstract]
- Stark AM, Buhl R, Hugo HH, et al.: Malignant peripheral nerve sheath tumours--report of 8 cases and review of the literature. Acta Neurochir (Wien) 143 (4): 357-63; discussion 363-4, 2001. [PUBMED Abstract]
- Goto M, Miller RW, Ishikawa Y, et al.: Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol Biomarkers Prev 5 (4): 239-46, 1996. [PUBMED Abstract]
- Weiss SW, Goldblum JR: Malignant fibrous histiocytoma (pleomorphic undifferentiated sarcoma). In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. St. Louis, Mo: Mosby, 2008, pp 403-27.
- McClain KL, Leach CT, Jenson HB, et al.: Association of Epstein-Barr virus with leiomyosarcomas in children with AIDS. N Engl J Med 332 (1): 12-8, 1995. [PUBMED Abstract]
- Dillon P, Maurer H, Jenkins J, et al.: A prospective study of nonrhabdomyosarcoma soft tissue sarcomas in the pediatric age group. J Pediatr Surg 27 (2): 241-4; discussion 244-5, 1992. [PUBMED Abstract]
- Rao BN: Nonrhabdomyosarcoma in children: prognostic factors influencing survival. Semin Surg Oncol 9 (6): 524-31, 1993 Nov-Dec. [PUBMED Abstract]
- Zeytoonjian T, Mankin HJ, Gebhardt MC, et al.: Distal lower extremity sarcomas: frequency of occurrence and patient survival rate. Foot Ankle Int 25 (5): 325-30, 2004. [PUBMED Abstract]
- Benesch M, von Bueren AO, Dantonello T, et al.: Primary intracranial soft tissue sarcoma in children and adolescents: a cooperative analysis of the European CWS and HIT study groups. J Neurooncol 111 (3): 337-45, 2013. [PUBMED Abstract]
- Weiss SW, Goldblum JR: Miscellaneous tumors of intermediate malignancy. In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. St. Louis, Mo: Mosby, 2008, pp 1093-1160.
- Federico SM, Spunt SL, Krasin MJ, et al.: Comparison of PET-CT and conventional imaging in staging pediatric rhabdomyosarcoma. Pediatr Blood Cancer 60 (7): 1128-34, 2013. [PUBMED Abstract]
- Tateishi U, Hosono A, Makimoto A, et al.: Accuracy of 18F fluorodeoxyglucose positron emission tomography/computed tomography in staging of pediatric sarcomas. J Pediatr Hematol Oncol 29 (9): 608-12, 2007. [PUBMED Abstract]
- Sargar K, Kao SC, Spunt SL, et al.: MRI and CT of Low-Grade Fibromyxoid Sarcoma in Children: A Report From Children's Oncology Group Study ARST0332. AJR Am J Roentgenol 205 (2): 414-20, 2015. [PUBMED Abstract]
- Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 4th ed. St. Louis, Mo: Mosby, 2001.
- Recommendations for the reporting of soft tissue sarcomas. Association of Directors of Anatomic and Surgical Pathology. Mod Pathol 11 (12): 1257-61, 1998. [PUBMED Abstract]
- Chowdhury T, Barnacle A, Haque S, et al.: Ultrasound-guided core needle biopsy for the diagnosis of rhabdomyosarcoma in childhood. Pediatr Blood Cancer 53 (3): 356-60, 2009. [PUBMED Abstract]
- Coffin CM, Dehner LP, O'Shea PA: Pediatric Soft Tissue Tumors: A Clinical, Pathological, and Therapeutic Approach. Baltimore, Md: Williams and Wilkins, 1997.
- Smith LM, Watterson J, Scott SM: Medical and surgical management of pediatric soft tissue tumors. In: Coffin CM, Dehner LP, O'Shea PA: Pediatric Soft Tissue Tumors: A Clinical, Pathological, and Therapeutic Approach. Baltimore, Md: Williams and Wilkins, 1997, pp 360-71.
- Neville HL, Andrassy RJ, Lally KP, et al.: Lymphatic mapping with sentinel node biopsy in pediatric patients. J Pediatr Surg 35 (6): 961-4, 2000. [PUBMED Abstract]
- Neville HL, Raney RB, Andrassy RJ, et al.: Multidisciplinary management of pediatric soft-tissue sarcoma. Oncology (Huntingt) 14 (10): 1471-81; discussion 1482-6, 1489-90, 2000. [PUBMED Abstract]
- Kayton ML, Delgado R, Busam K, et al.: Experience with 31 sentinel lymph node biopsies for sarcomas and carcinomas in pediatric patients. Cancer 112 (9): 2052-9, 2008. [PUBMED Abstract]
- Dall'Igna P, De Corti F, Alaggio R, et al.: Sentinel node biopsy in pediatric patients: the experience in a single institution. Eur J Pediatr Surg 24 (6): 482-7, 2014. [PUBMED Abstract]
- Parida L, Morrisson GT, Shammas A, et al.: Role of lymphoscintigraphy and sentinel lymph node biopsy in the management of pediatric melanoma and sarcoma. Pediatr Surg Int 28 (6): 571-8, 2012. [PUBMED Abstract]
- Alcorn KM, Deans KJ, Congeni A, et al.: Sentinel lymph node biopsy in pediatric soft tissue sarcoma patients: utility and concordance with imaging. J Pediatr Surg 48 (9): 1903-6, 2013. [PUBMED Abstract]
- Chui CH, Spunt SL, Liu T, et al.: Is reexcision in pediatric nonrhabdomyosarcoma soft tissue sarcoma necessary after an initial unplanned resection? J Pediatr Surg 37 (10): 1424-9, 2002. [PUBMED Abstract]
- Cecchetto G, Guglielmi M, Inserra A, et al.: Primary re-excision: the Italian experience in patients with localized soft-tissue sarcomas. Pediatr Surg Int 17 (7): 532-4, 2001. [PUBMED Abstract]
- Qureshi YA, Huddy JR, Miller JD, et al.: Unplanned excision of soft tissue sarcoma results in increased rates of local recurrence despite full further oncological treatment. Ann Surg Oncol 19 (3): 871-7, 2012. [PUBMED Abstract]
- Sandberg AA: Translocations in malignant tumors. Am J Pathol 159 (6): 1979-80, 2001. [PUBMED Abstract]
- Slater O, Shipley J: Clinical relevance of molecular genetics to paediatric sarcomas. J Clin Pathol 60 (11): 1187-94, 2007. [PUBMED Abstract]
- Mertens F, Antonescu CR, Hohenberger P, et al.: Translocation-related sarcomas. Semin Oncol 36 (4): 312-23, 2009. [PUBMED Abstract]
- Romeo S, Dei Tos AP: Clinical application of molecular pathology in sarcomas. Curr Opin Oncol 23 (4): 379-84, 2011. [PUBMED Abstract]
- Ladanyi M, Lui MY, Antonescu CR, et al.: The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene 20 (1): 48-57, 2001. [PUBMED Abstract]
- Ladanyi M: The emerging molecular genetics of sarcoma translocations. Diagn Mol Pathol 4 (3): 162-73, 1995. [PUBMED Abstract]
- Williams A, Bartle G, Sumathi VP, et al.: Detection of ASPL/TFE3 fusion transcripts and the TFE3 antigen in formalin-fixed, paraffin-embedded tissue in a series of 18 cases of alveolar soft part sarcoma: useful diagnostic tools in cases with unusual histological features. Virchows Arch 458 (3): 291-300, 2011. [PUBMED Abstract]
- Antonescu CR, Dal Cin P, Nafa K, et al.: EWSR1-CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer 46 (12): 1051-60, 2007. [PUBMED Abstract]
- Barnoud R, Sabourin JC, Pasquier D, et al.: Immunohistochemical expression of WT1 by desmoplastic small round cell tumor: a comparative study with other small round cell tumors. Am J Surg Pathol 24 (6): 830-6, 2000. [PUBMED Abstract]
- Wang LL, Perlman EJ, Vujanic GM, et al.: Desmoplastic small round cell tumor of the kidney in childhood. Am J Surg Pathol 31 (4): 576-84, 2007. [PUBMED Abstract]
- Errani C, Zhang L, Sung YS, et al.: A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer 50 (8): 644-53, 2011. [PUBMED Abstract]
- Jain S, Xu R, Prieto VG, et al.: Molecular classification of soft tissue sarcomas and its clinical applications. Int J Clin Exp Pathol 3 (4): 416-28, 2010. [PUBMED Abstract]
- Spunt SL, Hill DA, Motosue AM, et al.: Clinical features and outcome of initially unresected nonmetastatic pediatric nonrhabdomyosarcoma soft tissue sarcoma. J Clin Oncol 20 (15): 3225-35, 2002. [PUBMED Abstract]
- Spunt SL, Poquette CA, Hurt YS, et al.: Prognostic factors for children and adolescents with surgically resected nonrhabdomyosarcoma soft tissue sarcoma: an analysis of 121 patients treated at St Jude Children's Research Hospital. J Clin Oncol 17 (12): 3697-705, 1999. [PUBMED Abstract]
- Ferrari A, Casanova M, Collini P, et al.: Adult-type soft tissue sarcomas in pediatric-age patients: experience at the Istituto Nazionale Tumori in Milan. J Clin Oncol 23 (18): 4021-30, 2005. [PUBMED Abstract]
- Ferrari A, Miceli R, Meazza C, et al.: Soft tissue sarcomas of childhood and adolescence: the prognostic role of tumor size in relation to patient body size. J Clin Oncol 27 (3): 371-6, 2009. [PUBMED Abstract]
- Brooks AD, Heslin MJ, Leung DH, et al.: Superficial extremity soft tissue sarcoma: an analysis of prognostic factors. Ann Surg Oncol 5 (1): 41-7, 1998 Jan-Feb. [PUBMED Abstract]
- Spunt SL, Million L, Anderson JR, et al.: Risk-based treatment for nonrhabdomyosarcoma soft tissue sarcomas (NRSTS) in patients under 30 years of age: Children’s Oncology Group study ARST0332. [Abstract] J Clin Oncol 32 (Suppl 15): A-10008, 2014. Also available online. Last accessed April 02, 2018.
- O'Sullivan B, Davis AM, Turcotte R, et al.: Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 359 (9325): 2235-41, 2002. [PUBMED Abstract]
- Ferrari A, Miceli R, Rey A, et al.: Non-metastatic unresected paediatric non-rhabdomyosarcoma soft tissue sarcomas: results of a pooled analysis from United States and European groups. Eur J Cancer 47 (5): 724-31, 2011. [PUBMED Abstract]
- Smith KB, Indelicato DJ, Knapik JA, et al.: Definitive radiotherapy for unresectable pediatric and young adult nonrhabdomyosarcoma soft tissue sarcoma. Pediatr Blood Cancer 57 (2): 247-51, 2011. [PUBMED Abstract]
- Ferrari A, Magni C, Bergamaschi L, et al.: Pediatric nonrhabdomyosarcoma soft tissue sarcomas arising at visceral sites. Pediatr Blood Cancer 64 (9): , 2017. [PUBMED Abstract]
- Dillon PW, Whalen TV, Azizkhan RG, et al.: Neonatal soft tissue sarcomas: the influence of pathology on treatment and survival. Children's Cancer Group Surgical Committee. J Pediatr Surg 30 (7): 1038-41, 1995. [PUBMED Abstract]
- Pappo AS, Fontanesi J, Luo X, et al.: Synovial sarcoma in children and adolescents: the St Jude Children's Research Hospital experience. J Clin Oncol 12 (11): 2360-6, 1994. [PUBMED Abstract]
- Marcus KC, Grier HE, Shamberger RC, et al.: Childhood soft tissue sarcoma: a 20-year experience. J Pediatr 131 (4): 603-7, 1997. [PUBMED Abstract]
- Pratt CB, Pappo AS, Gieser P, et al.: Role of adjuvant chemotherapy in the treatment of surgically resected pediatric nonrhabdomyosarcomatous soft tissue sarcomas: A Pediatric Oncology Group Study. J Clin Oncol 17 (4): 1219, 1999. [PUBMED Abstract]
- Pratt CB, Maurer HM, Gieser P, et al.: Treatment of unresectable or metastatic pediatric soft tissue sarcomas with surgery, irradiation, and chemotherapy: a Pediatric Oncology Group study. Med Pediatr Oncol 30 (4): 201-9, 1998. [PUBMED Abstract]
Histopathological Classification of Childhood Soft Tissue Sarcoma
World Health Organization (WHO) Classification of Soft Tissue Sarcomas
The WHO lists the following cell types in its classification of soft tissue sarcomas:[1,2]
- Adipocytic tumors.
- Intermediate (locally aggressive).
- Malignant.
- Chondro-osseous tumors.
- Extraskeletal mesenchymal chondrosarcoma.[3]
- Extraskeletal osteosarcoma.
- Soft tissue chondroma.
- Fibroblastic/myofibroblastic tumors.
- Intermediate-grade (locally aggressive).
- Desmoid-type fibromatosis (previously called desmoid tumor or aggressive fibromatoses).
- Giant cell fibroblastoma.
- Lipofibromatosis.
- Palmar/plantar fibromatosis.
- Intermediate-grade (rarely metastasizing).
- Dermatofibrosarcoma protuberans.
- Infantile fibrosarcoma.[4]
- Inflammatory myofibroblastic tumor.
- Low-grade myofibroblastic tumor.
- Myxoinflammatory fibroblastic sarcoma.
- Solitary fibrous tumor.
- Malignant.
- Intermediate-grade (locally aggressive).
- Skeletal muscle tumors.
- Rhabdomyosarcoma (embryonal, alveolar, and pleomorphic forms). (Refer to the PDQ summary on Childhood Rhabdomyosarcoma Treatment for more information.)
- Smooth muscle tumors.
- So-called fibrohistiocytic tumors (intermediate, rarely metastasizing).
- Giant cell tumors of soft tissue.
- Plexiform fibrohistiocytic tumor.
- Tumors of peripheral nerves.
- Pericytic (perivascular) tumors.
- Malignant glomus tumor and variants.
- Myopericytoma.
- Angioleiomyoma.
- Myofibroma.
- Infantile myofibroma (previously called hemangiopericytoma [infantile]).
- Myofibromatosis.
- Infantile myofibromatosis.
- Tumors of uncertain differentiation.
- Alveolar soft part sarcoma.
- Clear cell sarcoma of soft tissue.
- Desmoplastic small round cell tumor.
- Epithelioid sarcoma.
- Extrarenal rhabdoid tumor.
- Extraskeletal myxoid chondrosarcoma.
- Neoplasms with perivascular epithelioid cell differentiation (PEComa NOS, malignant).
- Primitive neuroectodermal tumor/extraskeletal Ewing tumor.
- Synovial sarcoma (NOS, spindle cell, and biphasic varieties).
- Undifferentiated/unclassified sarcomas.
- Undifferentiated epithelial sarcoma.
- Undifferentiated pleomorphic sarcoma.
- Undifferentiated round cell sarcoma.
- Undifferentiated sarcoma; sarcoma, NOS.[6]
- Undifferentiated spindle cell sarcoma.
- Vascular tumors.
References
- Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-6.
- Brodowicz T, Schwameis E, Widder J, et al.: Intensified Adjuvant IFADIC Chemotherapy for Adult Soft Tissue Sarcoma: A Prospective Randomized Feasibility Trial. Sarcoma 4 (4): 151-60, 2000. [PUBMED Abstract]
- Dantonello TM, Int-Veen C, Leuschner I, et al.: Mesenchymal chondrosarcoma of soft tissues and bone in children, adolescents, and young adults: experiences of the CWS and COSS study groups. Cancer 112 (11): 2424-31, 2008. [PUBMED Abstract]
- Steelman C, Katzenstein H, Parham D, et al.: Unusual presentation of congenital infantile fibrosarcoma in seven infants with molecular-genetic analysis. Fetal Pediatr Pathol 30 (5): 329-37, 2011. [PUBMED Abstract]
- Evans HL: Low-grade fibromyxoid sarcoma: a clinicopathologic study of 33 cases with long-term follow-up. Am J Surg Pathol 35 (10): 1450-62, 2011. [PUBMED Abstract]
- Alaggio R, Collini P, Randall RL, et al.: Undifferentiated high-grade pleomorphic sarcomas in children: a clinicopathologic study of 10 cases and review of literature. Pediatr Dev Pathol 13 (3): 209-17, 2010 May-Jun. [PUBMED Abstract]
Staging and Grading Systems for Childhood Soft Tissue Sarcoma
Clinical staging has an important role in predicting the clinical outcome and determining the most effective therapy for pediatric soft tissue sarcomas. As yet, there is no well-accepted staging system that is applicable to all childhood sarcomas. The system from the American Joint Committee on Cancer (AJCC) that is used for adults has not been validated in pediatric studies. Although a standardized staging system for pediatric nonrhabdomyosarcomatous soft tissue sarcoma does not exist, two systems are currently in use for staging pediatric nonrhabdomyosarcomatous soft tissue sarcoma.[1]
- Surgico-pathologic staging system: The surgico-pathologic staging system used by the Intergroup Rhabdomyosarcoma Study (see below) is based on the amount, or extent, of tumor that remains after initial surgery and whether the disease has metastasized. This staging system was used in early pediatric trials.[2]
- TNM staging system: The TNM staging system is a collaborative effort between the AJCC (United States) and the International Union Against Cancer (worldwide). Staging is based on the extent of the tumor (T), the extent of spread to the lymph nodes (N), and the presence of metastasis (M). Refer to Tables 3, 4, 5, and 6 for the staging of soft tissue sarcoma from the eighth edition of the AJCC Cancer Staging Manual.[3-7] The last Children's Oncology Group trial used the sixth edition AJCC Cancer Staging Manual for soft tissue sarcoma (with central pathology review).[1] A review of children with non-rhabdomyosarcoma soft tissue sarcomas was performed with data from the Surveillance, Epidemiology, and End Results (SEER) program and identified 941 patients between 1988 and 2007.[8] The COG risk stratification was validated in this cohort.
Intergroup Rhabdomyosarcoma Study Staging System
Nonmetastatic disease
- Group I: Localized tumor completely resected with histologically negative margins.
- Group II: Grossly resected tumor with microscopic residual tumor at the margin(s) and/or extension into regional lymph nodes.
- IIA: Localized, grossly resected tumor with microscopic residual disease.
- IIB: Regional disease with involved nodes completely resected with no microscopic disease. The most proximal (to the patient, most distal to the tumor) regional lymph node must be negative.
- IIC: Regional disease with involved nodes grossly resected but with evidence of residual microscopic disease at the primary site and/or histologic involvement of the most proximal regional lymph node in the dissection.
- Group III: Localized tumor, incompletely resected, or biopsy only, with gross residual tumor.
Metastatic disease
- Group IV: Any localized or regional tumor with distant metastases present at the time of diagnosis. This includes the presence of malignant cells in effusions (pleural, peritoneal) and/or cerebrospinal fluid (rare).
Recurrent/progressive disease
- Any soft tissue sarcoma that recurs after initial treatment or progresses after radiation therapy, chemotherapy, or initial surgery.
TNM Staging System
The eighth edition of the AJCC Cancer Staging Manual has designated staging by the four criteria of tumor size, nodal status, histologic grade, and metastasis and by anatomic primary tumor site (head and neck; trunk and extremities; abdomen and thoracic visceral organs; retroperitoneum; and unusual histologies and sites) (refer to Tables 3, 4, 5, and 6).[3-7] For information on unusual histologies and sites, refer to the AJCC Cancer Staging Manual.[7]
| T Category | Soft Tissue Sarcoma of the Trunk, Extremities, and Retroperitoneum | Soft Tissue Sarcoma of the Head and Neck | Soft Tissue Sarcoma of the Abdomen and Thoracic Visceral Organs |
|---|---|---|---|
| aAdapted from O'Sullivan et al.,[3] Yoon et al.,[4] Raut et al.,[5] and Pollock et al.[6] | |||
| TX | Primary tumor cannot be assessed. | Primary tumor cannot be assessed. | Primary tumor cannot be assessed. |
| T0 | No evidence of primary tumor. | ||
| T1 | Tumor ≤5 cm in greatest dimension. | Tumor ≤2 cm. | Organ confined. |
| T2 | Tumor >5 cm and ≤10 cm in greatest dimension. | Tumor >2 to ≤4 cm. | Tumor extension into tissue beyond organ. |
| T2a | Invades serosa or visceral peritoneum. | ||
| T2b | Extension beyond serosa (mesentery). | ||
| T3 | Tumor >10 cm and ≤15 cm in greatest dimension. | Tumor >4 cm. | Invades another organ. |
| T4 | Tumor >15 cm in greatest dimension. | Tumor with invasion of adjoining structures. | Multifocal involvement. |
| T4a | Tumor with orbital invasion, skull base/dural invasion, invasion of central compartment viscera, involvement of facial skeleton, or invasion of pterygoid muscles. | Multifocal (2 sites). | |
| T4b | Tumor with brain parenchymal invasion, carotid artery encasement, prevertebral muscle invasion, or central nervous system involvement via perineural spread. | Multifocal (3–5 sites). | |
| T4c | Multifocal (>5 sites). | ||
| aAdapted from O'Sullivan et al.,[3] Yoon et al.,[4] Raut et al.,[5] and Pollock et al.[6] | |
| bFor soft tissue sarcoma of the abdomen and thoracic visceral organs, N0 = no lymph node involvement or unknown lymph node status and N1 = lymph node involvement present. | |
| N0 | No regional lymph node metastasis or unknown lymph node status.b |
| N1 | Regional lymph node metastasis.b |
| aAdapted from O'Sullivan et al.,[3] Yoon et al.,[4] Raut et al.,[5] and Pollock et al.[6] | |
| bFor soft tissue sarcoma of the abdomen and thoracic visceral organs, M0 = no metastases and M1 = metastases present. | |
| M0 | No distant metastasis.b |
| M1 | Distant metastasis.b |
| Stage | T | N | M | Grade |
|---|---|---|---|---|
| aAdapted from Yoon et al. [4] and Pollock et al.[6] | ||||
| bStage IIIB for soft tissue sarcoma of the retroperitoneum; stage IV for soft tissue sarcoma of the trunk and extremities. | ||||
| IA | T1 | N0 | M0 | G1, GX |
| IB | T2, T3, T4 | N0 | M0 | G1, GX |
| II | T1 | N0 | M0 | G2, G3 |
| IIIA | T2 | N0 | M0 | G2, G3 |
| IIIB | T3, T4 | N0 | M0 | G2, G3 |
| IIIB/IVb | Any T | N1 | M0 | Any G |
| IV | Any T | Any N | M1 | Any G |
Soft Tissue Sarcoma Tumor Pathological Grading System
In most cases, accurate histopathologic classification alone of soft tissue sarcomas does not yield optimal information about their clinical behavior. Therefore, several histologic parameters are evaluated in the grading process, including the following:
- Degree of cellularity.
- Cellular pleomorphism.
- Mitotic activity.
- Degree of necrosis.
- Invasive growth.
This process is used to improve the correlation between histologic findings and clinical outcome.[9] In children, grading of soft tissue sarcoma is compromised by the good prognosis of certain tumors, such as infantile fibrosarcoma and hemangiopericytoma, which have a good prognosis in children younger than 4 years, and also angiomatoid fibrous histiocytoma and dermatofibrosarcoma protuberans, which may recur locally if incompletely excised, but usually do not metastasize.
Testing the validity of a grading system within the pediatric population is difficult because of the rarity of these neoplasms. In March 1986, the Pediatric Oncology Group (POG) conducted a prospective study on pediatric soft tissue sarcomas other than rhabdomyosarcoma and devised the POG grading system. Analysis of outcome for patients with localized soft tissue sarcomas other than rhabdomyosarcoma demonstrated that patients with grade 3 tumors fared significantly worse than those with grade 1 or grade 2 lesions. This finding suggests that this system can accurately predict the clinical behavior of nonrhabdomyosarcomatous soft tissue sarcoma.[9-11]
The grading systems developed by the POG and the French Federation of Comprehensive Cancer Centers (Fédération Nationale des Centres de Lutte Contre Le Cancer [FNCLCC]) Sarcoma Group are described below. These grading systems are being compared by the central review pathologists on the COG-ARST0332 study. The study has closed and results are pending.
POG grading system
The POG grading system is described below.[9] It is an older grading system of historical value that is no longer being used for treatment.
Grade I
Grade I lesions are based on histologic type, well-differentiated cytohistologic features, and/or age of the patient.
- Angiomatoid fibrous histiocytoma.
- Dermatofibrosarcoma protuberans.
- Liposarcoma–myxoid or well-differentiated.
- Myxoid chondrosarcoma.
- Well-differentiated malignant peripheral nerve sheath tumor.
- Well-differentiated or infantile (aged ≤4 years) fibrosarcoma.
- Well-differentiated or infantile (aged ≤4 years) hemangiopericytoma.
Grade II
Grade II lesions are soft tissue sarcomas not included in grade I or III by histologic diagnosis (with <5 mitoses/10 high-power fields or <15% necrosis):
- 15% or less of the surface area shows necrosis (primary criteria).
- The mitotic count is <5 mitotic figures per 10 high-power fields (40X objective) (primary criteria).
- Nuclear atypia is not marked (secondary criteria).
- The tumor is not markedly cellular (secondary criteria).
Grade III
Grade III lesions are similar to grade II lesions and include certain tumors known to be clinically aggressive by virtue of histologic diagnosis and non-grade I tumors (with >4 mitoses per 10 high-power fields or >15% necrosis):
- Alveolar soft part sarcoma.
- Extraskeletal osteogenic sarcoma.
- Malignant triton tumor.
- Mesenchymal chondrosarcoma.
- Pleomorphic or round-cell liposarcoma.
- Any other sarcoma not in grade I with >15% necrosis and/or ≥5 mitotic figures per 10 high-power fields (40X objective). Marked atypia and cellularity are less predictive but may assist in placing tumors in this category.
FNCLCC grading system
The FNCLCC histologic grading system was developed for adults with soft tissue sarcoma. The purpose of the grading system is to predict which patients will develop metastasis and subsequently benefit from postoperative chemotherapy.[12,13] The system is described in Table 7 and Table 8.
| FNCLCC = Fédération Nationale des Centres de Lutte Contre Le Cancer; HPF = high-power field. | |
| Tumor Differentiation | |
| Score 1 | Sarcoma closely resembling normal adult mesenchymal tissue (e.g., well-differentiated liposarcoma) |
| Score 2 | Sarcomas for which histologic typing is certain (e.g., myxoid liposarcoma) |
| Score 3 | Embryonal and undifferentiated sarcomas, sarcomas of doubtful type, and synovial sarcomas |
| Mitotic Count | |
| Score 1 | 0–9 mitoses per 10 HPF |
| Score 2 | 10–19 mitoses per 10 HPF |
| Score 3 | ≥20 mitoses per 10 HPF |
| Tumor Necrosis | |
| Score 0 | No necrosis |
| Score 1 | <50% tumor necrosis |
| Score 2 | ≥50% tumor necrosis |
| Total Score | Histologic Grade |
|---|---|
| 2–3 | Grade I |
| 4–5 | Grade II |
| 6–8 | Grade III |
Prognostic Significance of Tumor Grading
The POG and FNCLCC grading systems have proven to be of prognostic value in pediatric and adult nonrhabdomyosarcomatous soft tissue sarcomas.[14-18] In a study of 130 tumors from children and adolescents with nonrhabdomyosarcomatous soft tissue sarcoma enrolled in three prospective clinical trials, a correlation was found between the POG-assigned grade and the FNCLCC-assigned grade. However, grading did not correlate in all cases; 44 patients whose tumors received discrepant grades (POG grade 3, FNCLCC grade 1 or 2) had outcomes between concurrent grade 3 and grades 1 and 2. A mitotic index of 10 or greater emerged as an important prognostic factor.[19] The recently completed COG-ARST0332 trial will analyze data comparing the POG and FNCLCC pathologic grading systems to determine which system better correlates with clinical outcomes. The current open trial (ARST1321 [NCT02180867]) uses the FNCLCC system to assign histological grade.
References
- American Joint Committee on Cancer: AJCC Cancer Staging Manual. 6th ed. New York, NY: Springer, 2002.
- Maurer HM, Beltangady M, Gehan EA, et al.: The Intergroup Rhabdomyosarcoma Study-I. A final report. Cancer 61 (2): 209-20, 1988. [PUBMED Abstract]
- O'Sullivan B, Maki RG, Agulnik M, et al.: Soft tissue sarcoma of the head and neck. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 499-505.
- Yoon SS, Maki RG, Asare EA, et al.: Soft tissue sarcoma of the trunk and extremities. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 507-15.
- Raut CP, Maki RG, Baldini EH, et al.: Soft tissue sarcoma of the abdomen and thoracic visceral organs. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 517-21.
- Pollock RE, Maki RG, Baldini EH, et al.: Soft tissue sarcoma of the retroperitoneum. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 531-7.
- Maki RG, Folpe AL, Guadagnolo BA, et al.: Soft tissue sarcoma - unusual histologies and sites. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 539-45.
- Waxweiler TV, Rusthoven CG, Proper MS, et al.: Non-Rhabdomyosarcoma Soft Tissue Sarcomas in Children: A Surveillance, Epidemiology, and End Results Analysis Validating COG Risk Stratifications. Int J Radiat Oncol Biol Phys 92 (2): 339-48, 2015. [PUBMED Abstract]
- Parham DM, Webber BL, Jenkins JJ 3rd, et al.: Nonrhabdomyosarcomatous soft tissue sarcomas of childhood: formulation of a simplified system for grading. Mod Pathol 8 (7): 705-10, 1995. [PUBMED Abstract]
- Recommendations for the reporting of soft tissue sarcomas. Association of Directors of Anatomic and Surgical Pathology. Mod Pathol 11 (12): 1257-61, 1998. [PUBMED Abstract]
- Skytting B, Meis-Kindblom JM, Larsson O, et al.: Synovial sarcoma--identification of favorable and unfavorable histologic types: a Scandinavian sarcoma group study of 104 cases. Acta Orthop Scand 70 (6): 543-54, 1999. [PUBMED Abstract]
- Coindre JM, Terrier P, Guillou L, et al.: Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 91 (10): 1914-26, 2001. [PUBMED Abstract]
- Guillou L, Coindre JM, Bonichon F, et al.: Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 15 (1): 350-62, 1997. [PUBMED Abstract]
- Rao BN: Nonrhabdomyosarcoma in children: prognostic factors influencing survival. Semin Surg Oncol 9 (6): 524-31, 1993 Nov-Dec. [PUBMED Abstract]
- Pisters PW, Leung DH, Woodruff J, et al.: Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 14 (5): 1679-89, 1996. [PUBMED Abstract]
- Coindre JM, Terrier P, Bui NB, et al.: Prognostic factors in adult patients with locally controlled soft tissue sarcoma. A study of 546 patients from the French Federation of Cancer Centers Sarcoma Group. J Clin Oncol 14 (3): 869-77, 1996. [PUBMED Abstract]
- Pappo AS, Fontanesi J, Luo X, et al.: Synovial sarcoma in children and adolescents: the St Jude Children's Research Hospital experience. J Clin Oncol 12 (11): 2360-6, 1994. [PUBMED Abstract]
- Pratt CB, Maurer HM, Gieser P, et al.: Treatment of unresectable or metastatic pediatric soft tissue sarcomas with surgery, irradiation, and chemotherapy: a Pediatric Oncology Group study. Med Pediatr Oncol 30 (4): 201-9, 1998. [PUBMED Abstract]
- Khoury JD, Coffin CM, Spunt SL, et al.: Grading of nonrhabdomyosarcoma soft tissue sarcoma in children and adolescents: a comparison of parameters used for the Fédération Nationale des Centers de Lutte Contre le Cancer and Pediatric Oncology Group Systems. Cancer 116 (9): 2266-74, 2010. [PUBMED Abstract]
Treatment Option Overview for Childhood Soft Tissue Sarcoma
Because of the rarity of pediatric nonrhabdomyosarcomatous soft tissue sarcomas, coordination of treatment by a multidisciplinary team comprising oncologists (pediatric or medical), pathologists, surgeons, and radiation oncologists should be considered for all children, adolescents, and young adults with these tumors. In addition, to better define the tumors' natural history and response to therapy, entry into national or institutional treatment protocols should be considered for children with rare neoplasms. Information about ongoing clinical trials is available from the NCI website.
Surgery
After an appropriate biopsy and pathologic diagnosis, every attempt is made to resect the primary tumor with negative margins before or after chemotherapy and/or radiation therapy. Involvement of a surgeon with special expertise in the resection of soft tissue sarcomas in the decision is highly desirable.
The timing of surgery depends on an assessment of the feasibility and morbidity of surgery. If the initial operation fails to achieve pathologically negative tissue margins or if the initial surgery was done without the knowledge that cancer was present, a re-excision of the affected area is performed to obtain clear, but not necessarily wide, margins.[1-4] This surgical tenet is true even if no mass is detected by magnetic resonance imaging after initial surgery.[5]; [6][Level of evidence: 3iiA]
Regional lymph node metastases at diagnosis are unusual and are most often seen in patients with epithelioid and clear cell sarcomas.[7,8] Various institutional series have demonstrated the feasibility and effectiveness of sentinel node biopsy as a staging procedure in pediatric patients with soft tissue sarcomas.[9-14]
Radiation Therapy
Considerations for radiation therapy are based on the potential for surgery, with or without chemotherapy, to obtain local control without loss of critical organs or significant functional, cosmetic, or psychological impairment. This will vary according to the following:
- Patient variables (e.g., age and sex).
- Tumor variables (e.g., histopathology, site, size, and grade).
- Surgical margin status.
- Expectations for radiation-induced morbidities (e.g., impaired bone or muscle development, organ damage, or second malignancy).
Radiation therapy can be given preoperatively. Radiation field size and dose will be based on patient and tumor variables and the operability of the tumor. Preoperative radiation therapy has been associated with excellent local control rates.[15,16] This approach has the advantage of treating smaller tissue volumes because it does not necessitate treating a postsurgical bed; it also has the advantage of somewhat lower radiation doses because relative hypoxia from surgical disruption of vasculature and scarring is not present. Preoperative radiation therapy has been associated with an increased rate of wound complications in adults, primarily in lower extremity tumors, but the degree of this is questionable.[17] Conversely, preoperative radiation therapy may lead to less fibrosis than with postoperative approaches, perhaps due to the smaller treatment volume and dose.[18]
Retroperitoneal sarcomas are unique in that radiosensitivity of the bowel to injury makes postoperative radiation therapy less desirable.[19,20] Postoperative adhesions and bowel immobility can increase the risk of damage from any given radiation dose. This contrasts with the preoperative approach in which the tumor often displaces bowel outside of the radiation field, and any exposed bowel is more mobile, which decreases exposure to specific bowel segments.
Radiation therapy can also be given postoperatively. In general, radiation is indicated for patients with inadequate surgical margins and for larger, high-grade tumors.[21,22] This is particularly important in high-grade tumors with tumor margins smaller than 1 cm.[23,24]; [25][Level of evidence: 3iiDiv] With combined surgery and radiation therapy, local control of the primary tumor can be achieved in more than 80% of patients.[26,27]
Brachytherapy and intraoperative radiation may be applicable in select situations.[27-29]; [30][Level of evidence: 3iiiDii]
Radiation volume and dose depend on the patient, tumor, and surgical variables noted above, as well as the following:
- Patient age and growth potential.
- Ability to avoid critical organs, epiphyseal plates, and lymphatics (but not the neurovascular bundles that are relatively radiation tolerant).
- Functional/cosmetic outcome.
Radiation doses are typically 45 Gy to 50 Gy preoperatively, with consideration for postoperative boost of 10 Gy to 20 Gy if resection margins are microscopically or grossly positive, or planned brachytherapy if the resection is predicted to be subtotal. However, data documenting the efficacy of a postoperative boost are lacking.[31] The postoperative radiation dose is 55 Gy to 60 Gy, or rarely, higher when unresectable gross residual disease exists.
Radiation margins are typically 2 cm to 4 cm longitudinally and encompass fascial planes axially.[32,33]
Chemotherapy
The role of postoperative chemotherapy remains unclear as evidenced by the following studies:[34]
- A meta-analysis of data from all randomized trials of adults with soft tissue sarcoma concluded that recurrence-free survival was better with postoperative chemotherapy for patients with high-grade tumors larger than 5 cm.[35]
- In a European trial, adults with completely resected soft tissue sarcoma were randomly assigned to observation or postoperative chemotherapy with ifosfamide and doxorubicin. Postoperative chemotherapy was not associated with improved event-free survival (EFS) or overall survival (OS). It is difficult to extrapolate this trial to pediatric patients because the trial included 1) a wide variety of histologies; 2) a relatively low dose of ifosfamide; 3) patients assigned to chemotherapy had definitive radiation delayed until completion of chemotherapy; and 4) almost one-half of the patients in the trial had intermediate-grade tumors. In the discussion section, the authors merged their patients with previously published series, including those from the European meta-analysis, and concluded that the results suggested a benefit for postoperative chemotherapy.[36][Level of evidence: 1iiA]
- The largest prospective pediatric trial failed to demonstrate any benefit with postoperative vincristine, dactinomycin, cyclophosphamide, and doxorubicin.[26]
- Doxorubicin and ifosfamide were used in the risk-based COG ARST0332 (NCT00346164) trial. Although this was not a randomized study, results at 2.6 years show that patients with high-risk (>5 cm and high grade), grossly resected, nonmetastatic tumors who were treated with radiation therapy and postoperative doxorubicin and ifosfamide had a 3-year EFS of 68% and OS of 81%. In patients with metastatic disease treated with preoperative chemotherapy and radiation therapy, the estimated 3-year failure-free survival was 52% and OS was 66%.[37][Level of evidence: 3iiiA]
Targeted Therapy
The use of angiogenesis and mammalian target of rapamycin (mTOR) inhibitors has been explored in the treatment of adult soft tissue sarcomas but not in pediatrics.
- In a trial of 711 randomly assigned adult patients who achieved a response or stable disease after chemotherapy, the administration of ridaforolimus was associated with a 3-week improvement in progression-free survival (PFS) when compared with placebo.[38]
- In another trial of 371 randomly assigned adult patients with metastatic soft tissue sarcoma that progressed after chemotherapy, pazopanib was compared with placebo. The median PFS for the pazopanib arm was 4.6 months compared with 1.6 months for the placebo arm. OS was not different between the two arms.[39]
- In a randomized study of 182 previously treated adult patients with recurrent liposarcoma, leiomyosarcoma, synovial sarcoma, and other sarcomas, patients with nonadipocytic tumors who were treated with regorafenib had significant improvements in progression-free survival when compared with patients who were treated with placebo.[40]
Special Considerations for the Treatment of Children With Soft Tissue Sarcoma
Cancer in children and adolescents is rare, although the overall incidence of childhood cancer has been slowly increasing since 1975.[41] Children and adolescents with cancer should be referred to medical centers that have a multidisciplinary team of cancer specialists with experience treating the cancers that occur during childhood and adolescence. This multidisciplinary team approach incorporates the skills of the following health care professionals and others to ensure that children receive treatment, supportive care, and rehabilitation that will achieve optimal survival and quality of life:
- Primary care physicians.
- Pediatric surgical specialists.
- Pediatric radiation oncologists.
- Pediatric medical oncologists/hematologists.
- Rehabilitation specialists.
- Pediatric nurse specialists.
- Social workers.
- Child life professionals.
- Psychologists.
(Refer to the PDQ Supportive and Palliative Care summaries for specific information about supportive care for children and adolescents with cancer.)
Guidelines for pediatric cancer centers and their role in the treatment of pediatric patients with cancer have been outlined by the American Academy of Pediatrics.[42] At these pediatric cancer centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate in these trials is offered to most patients/families. Multidisciplinary evaluation in pediatric cancer centers that have surgical and radiotherapeutic expertise is of critical importance to ensure the best clinical outcome for these patients. Although surgery with or without radiation therapy can be curative for a significant proportion of patients, the addition of chemotherapy might benefit subsets of children with the disease; therefore, enrollment into clinical trials is encouraged. Clinical trials for children and adolescents with cancer are generally designed to compare potentially better therapy with therapy that is currently accepted as standard. Most of the progress made in identifying curative therapies for childhood cancers has been achieved through clinical trials. Information about ongoing clinical trials is available from the NCI website.
Many therapeutic strategies for children and adolescents with soft tissue tumors are similar to those for adult patients, although there are important differences. For example, the biology of the neoplasm in pediatric patients may differ dramatically from that of the adult lesion. Additionally, limb-sparing procedures are more difficult to perform in pediatric patients. The morbidity associated with radiation therapy, particularly in infants and young children, may be much greater than that observed in adults.[43]
Improved outcomes with multimodality therapy in adults and children with soft tissue sarcomas over the past 20 years has caused increasing concern about the potential long-term side effects of this therapy in children, especially when considering the expected longer life span of children versus adults. Therefore, to maximize tumor control and minimize long-term morbidity, treatment must be individualized for children and adolescents with nonrhabdomyosarcomatous soft tissue sarcoma. These patients should be enrolled in prospective studies that accurately assess any potential complications.[44]
References
- Sugiura H, Takahashi M, Katagiri H, et al.: Additional wide resection of malignant soft tissue tumors. Clin Orthop (394): 201-10, 2002. [PUBMED Abstract]
- Cecchetto G, Guglielmi M, Inserra A, et al.: Primary re-excision: the Italian experience in patients with localized soft-tissue sarcomas. Pediatr Surg Int 17 (7): 532-4, 2001. [PUBMED Abstract]
- Chui CH, Spunt SL, Liu T, et al.: Is reexcision in pediatric nonrhabdomyosarcoma soft tissue sarcoma necessary after an initial unplanned resection? J Pediatr Surg 37 (10): 1424-9, 2002. [PUBMED Abstract]
- Paulino AC, Ritchie J, Wen BC: The value of postoperative radiotherapy in childhood nonrhabdomyosarcoma soft tissue sarcoma. Pediatr Blood Cancer 43 (5): 587-93, 2004. [PUBMED Abstract]
- Kaste SC, Hill A, Conley L, et al.: Magnetic resonance imaging after incomplete resection of soft tissue sarcoma. Clin Orthop (397): 204-11, 2002. [PUBMED Abstract]
- Chandrasekar CR, Wafa H, Grimer RJ, et al.: The effect of an unplanned excision of a soft-tissue sarcoma on prognosis. J Bone Joint Surg Br 90 (2): 203-8, 2008. [PUBMED Abstract]
- Daigeler A, Kuhnen C, Moritz R, et al.: Lymph node metastases in soft tissue sarcomas: a single center analysis of 1,597 patients. Langenbecks Arch Surg 394 (2): 321-9, 2009. [PUBMED Abstract]
- Mazeron JJ, Suit HD: Lymph nodes as sites of metastases from sarcomas of soft tissue. Cancer 60 (8): 1800-8, 1987. [PUBMED Abstract]
- Neville HL, Andrassy RJ, Lally KP, et al.: Lymphatic mapping with sentinel node biopsy in pediatric patients. J Pediatr Surg 35 (6): 961-4, 2000. [PUBMED Abstract]
- Neville HL, Raney RB, Andrassy RJ, et al.: Multidisciplinary management of pediatric soft-tissue sarcoma. Oncology (Huntingt) 14 (10): 1471-81; discussion 1482-6, 1489-90, 2000. [PUBMED Abstract]
- Kayton ML, Delgado R, Busam K, et al.: Experience with 31 sentinel lymph node biopsies for sarcomas and carcinomas in pediatric patients. Cancer 112 (9): 2052-9, 2008. [PUBMED Abstract]
- Dall'Igna P, De Corti F, Alaggio R, et al.: Sentinel node biopsy in pediatric patients: the experience in a single institution. Eur J Pediatr Surg 24 (6): 482-7, 2014. [PUBMED Abstract]
- Parida L, Morrisson GT, Shammas A, et al.: Role of lymphoscintigraphy and sentinel lymph node biopsy in the management of pediatric melanoma and sarcoma. Pediatr Surg Int 28 (6): 571-8, 2012. [PUBMED Abstract]
- Alcorn KM, Deans KJ, Congeni A, et al.: Sentinel lymph node biopsy in pediatric soft tissue sarcoma patients: utility and concordance with imaging. J Pediatr Surg 48 (9): 1903-6, 2013. [PUBMED Abstract]
- Virkus WW, Mollabashy A, Reith JD, et al.: Preoperative radiotherapy in the treatment of soft tissue sarcomas. Clin Orthop (397): 177-89, 2002. [PUBMED Abstract]
- Zagars GK, Ballo MT, Pisters PW, et al.: Preoperative vs. postoperative radiation therapy for soft tissue sarcoma: a retrospective comparative evaluation of disease outcome. Int J Radiat Oncol Biol Phys 56 (2): 482-8, 2003. [PUBMED Abstract]
- O'Sullivan B, Davis AM, Turcotte R, et al.: Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 359 (9325): 2235-41, 2002. [PUBMED Abstract]
- Davis AM, O'Sullivan B, Turcotte R, et al.: Late radiation morbidity following randomization to preoperative versus postoperative radiotherapy in extremity soft tissue sarcoma. Radiother Oncol 75 (1): 48-53, 2005. [PUBMED Abstract]
- Baldini EH, Wang D, Haas RL, et al.: Treatment Guidelines for Preoperative Radiation Therapy for Retroperitoneal Sarcoma: Preliminary Consensus of an International Expert Panel. Int J Radiat Oncol Biol Phys 92 (3): 602-12, 2015. [PUBMED Abstract]
- Bishop AJ, Zagars GK, Torres KE, et al.: Combined Modality Management of Retroperitoneal Sarcomas: A Single-Institution Series of 121 Patients. Int J Radiat Oncol Biol Phys 93 (1): 158-65, 2015. [PUBMED Abstract]
- Marcus KC, Grier HE, Shamberger RC, et al.: Childhood soft tissue sarcoma: a 20-year experience. J Pediatr 131 (4): 603-7, 1997. [PUBMED Abstract]
- Delaney TF, Kepka L, Goldberg SI, et al.: Radiation therapy for control of soft-tissue sarcomas resected with positive margins. Int J Radiat Oncol Biol Phys 67 (5): 1460-9, 2007. [PUBMED Abstract]
- Blakely ML, Spurbeck WW, Pappo AS, et al.: The impact of margin of resection on outcome in pediatric nonrhabdomyosarcoma soft tissue sarcoma. J Pediatr Surg 34 (5): 672-5, 1999. [PUBMED Abstract]
- Skytting B: Synovial sarcoma. A Scandinavian Sarcoma Group project. Acta Orthop Scand Suppl 291: 1-28, 2000. [PUBMED Abstract]
- Hua C, Gray JM, Merchant TE, et al.: Treatment planning and delivery of external beam radiotherapy for pediatric sarcoma: the St. Jude Children's Research Hospital experience. Int J Radiat Oncol Biol Phys 70 (5): 1598-606, 2008. [PUBMED Abstract]
- Pratt CB, Pappo AS, Gieser P, et al.: Role of adjuvant chemotherapy in the treatment of surgically resected pediatric nonrhabdomyosarcomatous soft tissue sarcomas: A Pediatric Oncology Group Study. J Clin Oncol 17 (4): 1219, 1999. [PUBMED Abstract]
- Merchant TE, Parsh N, del Valle PL, et al.: Brachytherapy for pediatric soft-tissue sarcoma. Int J Radiat Oncol Biol Phys 46 (2): 427-32, 2000. [PUBMED Abstract]
- Schomberg PJ, Gunderson LL, Moir CR, et al.: Intraoperative electron irradiation in the management of pediatric malignancies. Cancer 79 (11): 2251-6, 1997. [PUBMED Abstract]
- Nag S, Shasha D, Janjan N, et al.: The American Brachytherapy Society recommendations for brachytherapy of soft tissue sarcomas. Int J Radiat Oncol Biol Phys 49 (4): 1033-43, 2001. [PUBMED Abstract]
- Viani GA, Novaes PE, Jacinto AA, et al.: High-dose-rate brachytherapy for soft tissue sarcoma in children: a single institution experience. Radiat Oncol 3: 9, 2008. [PUBMED Abstract]
- Al Yami A, Griffin AM, Ferguson PC, et al.: Positive surgical margins in soft tissue sarcoma treated with preoperative radiation: is a postoperative boost necessary? Int J Radiat Oncol Biol Phys 77 (4): 1191-7, 2010. [PUBMED Abstract]
- Wang D, Bosch W, Kirsch DG, et al.: Variation in the gross tumor volume and clinical target volume for preoperative radiotherapy of primary large high-grade soft tissue sarcoma of the extremity among RTOG sarcoma radiation oncologists. Int J Radiat Oncol Biol Phys 81 (5): e775-80, 2011. [PUBMED Abstract]
- Bahig H, Roberge D, Bosch W, et al.: Agreement among RTOG sarcoma radiation oncologists in contouring suspicious peritumoral edema for preoperative radiation therapy of soft tissue sarcoma of the extremity. Int J Radiat Oncol Biol Phys 86 (2): 298-303, 2013. [PUBMED Abstract]
- Ferrari A: Role of chemotherapy in pediatric nonrhabdomyosarcoma soft-tissue sarcomas. Expert Rev Anticancer Ther 8 (6): 929-38, 2008. [PUBMED Abstract]
- Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet 350 (9092): 1647-54, 1997. [PUBMED Abstract]
- Woll PJ, Reichardt P, Le Cesne A, et al.: Adjuvant chemotherapy with doxorubicin, ifosfamide, and lenograstim for resected soft-tissue sarcoma (EORTC 62931): a multicentre randomised controlled trial. Lancet Oncol 13 (10): 1045-54, 2012. [PUBMED Abstract]
- Spunt SL, Million L, Anderson JR, et al.: Risk-based treatment for nonrhabdomyosarcoma soft tissue sarcomas (NRSTS) in patients under 30 years of age: Children’s Oncology Group study ARST0332. [Abstract] J Clin Oncol 32 (Suppl 15): A-10008, 2014. Also available online. Last accessed April 02, 2018.
- Demetri GD, Chawla SP, Ray-Coquard I, et al.: Results of an international randomized phase III trial of the mammalian target of rapamycin inhibitor ridaforolimus versus placebo to control metastatic sarcomas in patients after benefit from prior chemotherapy. J Clin Oncol 31 (19): 2485-92, 2013. [PUBMED Abstract]
- van der Graaf WT, Blay JY, Chawla SP, et al.: Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 379 (9829): 1879-86, 2012. [PUBMED Abstract]
- Mir O, Brodowicz T, Italiano A, et al.: Safety and efficacy of regorafenib in patients with advanced soft tissue sarcoma (REGOSARC): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol 17 (12): 1732-1742, 2016. [PUBMED Abstract]
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014. [PUBMED Abstract]
- Corrigan JJ, Feig SA; American Academy of Pediatrics: Guidelines for pediatric cancer centers. Pediatrics 113 (6): 1833-5, 2004. [PUBMED Abstract]
- Suit H, Spiro I: Radiation as a therapeutic modality in sarcomas of the soft tissue. Hematol Oncol Clin North Am 9 (4): 733-46, 1995. [PUBMED Abstract]
- Spunt SL, Million L, Coffin C: The nonrhabdomyosarcoma soft tissue sarcoma. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 827-54.
Treatment of Newly Diagnosed Childhood Soft Tissue Sarcoma
Adipocytic Tumors
Liposarcoma
Liposarcoma accounts for 3% of soft tissue sarcoma in patients younger than 20 years (refer to Table 1).
Liposarcoma is rare in the pediatric population. In a review of 182 pediatric patients with adult-type sarcomas, only 14 had a diagnosis of liposarcoma.[1] One retrospective study identified 34 patients younger than 22 years from 1960 to 2011.[2] There were roughly equal numbers of male and female patients and the median age was 18 years. In an international clinicopathological review, the characteristics of 82 cases of pediatric liposarcoma were reported. The median age was 15.5 years and females were more commonly affected.[3] In both reports, the great majority of patients had myxoid liposarcoma.
Histopathologic classification
The World Health Organization (WHO) classification for liposarcoma is as follows:
- Intermediate grade (rarely metastasizing).
- Atypical lipomatous neoplasm/well-differentiated liposarcoma. These tumors do not metastasize unless they undergo dedifferentiation.
- Malignant.
- Liposarcoma, not otherwise specified (NOS).
- Myxoid liposarcoma. Pure myxoid liposarcomas are characterized by a t(12;16)(q13;p11) translocation and can metastasize but usually have an excellent outcome in the absence of a round cell component.[4]
- Dedifferentiated liposarcoma.
- Pleomorphic liposarcoma.
Clinical presentation
The majority of liposarcomas in the pediatric and adolescent age range are low grade and located subcutaneously. Metastasis to lymph nodes is very uncommon, and the great majority of metastases are pulmonary. Tumors arising in the periphery are more likely to be low grade and myxoid. Tumors arising centrally are more likely to be high grade, pleomorphic, and present with metastasis or recur with metastasis.
Prognosis
Higher grade or central tumors are associated with a significantly higher risk of death. In a retrospective review, 5-year survival for central tumors was 42%. In the international review, seven of ten patients with pleomorphic myxoid liposarcoma died because of their disease.[3] In a retrospective study of 14 patients, 5-year survival was 78% and tumor grade, histologic subtype, and primary location correlated with survival.[2]
Treatment
Treatment options for liposarcoma include the following:
Surgery is the most important treatment for liposarcoma. After surgical resection of myxoid liposarcoma, event-free survival (EFS) and overall survival (OS) are roughly 90%. If initial surgery is incomplete, re-excision should be performed to achieve a wide margin of resection. Local recurrences have been seen and are controlled with a second resection of the tumor.
There are reports of the use of chemotherapy to decrease the size of liposarcoma before surgery to facilitate complete resection, particularly in central tumors.[10,11] The role of postoperative chemotherapy for liposarcoma is poorly defined. There does not appear to be a need for any postoperative therapy for completely resected myxoid liposarcoma. Even with the use of postoperative chemotherapy, the survival of pleomorphic liposarcoma remains poor.[12]
Trabectedin has produced encouraging responses in adults with advanced myxoid liposarcoma.[13] In one study, adult patients with recurrent liposarcoma and leiomyosarcoma were randomly assigned to treatment with either trabectedin or dacarbazine. Patients treated with trabectedin had a 45% reduction in disease progression.[14][Level of evidence: 1iiDiii] There are very limited data to support the use of trabectedin in pediatric patients.[15]
Treatment options under clinical evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma, excluding myxoid liposarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with liposarcoma are eligible for this trial.
Chondro-osseous Tumors
Chondro-osseous tumors include the following tumor subtypes:
- Extraskeletal mesenchymal chondrosarcoma.
- Extraskeletal osteosarcoma.
- Soft tissue chondroma.
Extraskeletal mesenchymal chondrosarcoma
Osseous and chondromatous neoplasms account for 0.8% of soft tissue sarcoma in patients younger than 20 years (refer to Table 1).
Histopathology and molecular features
Mesenchymal chondrosarcoma is a rare tumor characterized by small round cells and hyaline cartilage that more commonly affects young adults and has a predilection for involving the head and neck region.
Mesenchymal chondrosarcoma has been associated with consistent chromosomal rearrangement. A retrospective analysis of cases of mesenchymal chondrosarcoma identified a HEY1-NCOA2 fusion in 10 of 15 tested specimens.[16] This gene fusion was not associated with chromosomal changes that could be detected by karyotyping. In one instance, translocation t(1;5)(q42;q32) was identified in a case of mesenchymal chondrosarcoma and shown to be associated with a novel IRF2BP-CDX1 fusion gene.[17]
Prognosis
A retrospective survey of European institutions identified 113 children and adults with mesenchymal chondrosarcoma. Factors associated with better outcome included the following:[18][Level of evidence: 3iiiA]
- Lack of metastatic disease at initial presentation.
- Clear resection margins.
- Administration of postoperative chemotherapy following resection for patients with initially localized disease.
Treatment
Treatment options for extraskeletal mesenchymal chondrosarcoma include the following:
A review of 15 patients younger than 26 years from the German Cooperative Soft Tissue Sarcoma Study Group (11 with soft-tissue lesions) and the German-Austrian-Swiss Cooperative Osteosarcoma Study Group (four with primary bone lesions) protocols suggests that complete surgical removal, or incomplete resection followed by radiation therapy, is necessary for local control.[19][Level of evidence: 3iiA]
A single-institution, retrospective review identified 12 pediatric patients with mesenchymal chondrosarcoma.[20] The presence of the NCOA2 rearrangement in tumors was documented in these patients. It was also confirmed that surgical resection is necessary for cure. Eleven patients presented with localized disease and one presented with pulmonary nodules. All patients received chemotherapy—six patients before and after surgical resection and six patients only after resection. All patients received postoperative chemotherapy (most commonly ifosfamide/doxorubicin) with or without radiation therapy (median dose, 59.4 Gy). At a median follow-up of 4.8 years, 5-year disease-free survival (DFS) was 68.2% (95% CI, 39.8%–96.6%) and OS was 88.9% (95% CI, 66.9%–100%).
Extraskeletal osteosarcoma
Osseous and chondromatous neoplasms account for 0.8% of soft tissue sarcomas in patients younger than 20 years (refer to Table 1).
Extraskeletal osteosarcoma is extremely rare in the pediatric and adolescent age range. A 2003 review identified only ten case reports in the medical literature.[21]
Prognosis
Extraskeletal osteosarcoma is associated with a high risk of local recurrence and pulmonary metastasis.[22]
Treatment
Treatment options for extraskeletal osteosarcoma include the following:
- Surgery followed by chemotherapy.
(Refer to the PDQ summary on Osteosarcoma and Malignant Fibrous Histiocytoma of Bone Treatment for more information.)
Treatment options under clinical evaluation
Information about National Cancer Institute NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with extraskeletal mesenchymal chondrosarcoma and extraskeletal osteosarcoma are eligible for this trial.
Fibroblastic/Myofibroblastic Tumors
Fibroblastic/myofibroblastic tumors include the following tumor subtypes:
- Fibroblastic/myofibroblastic tumors.
- Intermediate grade (locally aggressive).
- Desmoid-type fibromatosis (previously called desmoid tumor or aggressive fibromatoses).
- Giant cell fibroblastoma.
- Lipofibromatosis.
- Palmar/plantar fibromatosis.
- Intermediate grade (rarely metastasizing).
- Dermatofibrosarcoma protuberans.
- Infantile fibrosarcoma.
- Inflammatory myofibroblastic tumor.
- Low-grade myofibroblastic sarcoma.
- Myxoinflammatory fibroblastic sarcoma.
- Solitary fibrous tumor.
- Malignant.
- Intermediate grade (locally aggressive).
Desmoid-type fibromatosis
Desmoid-type fibromatosis has previously been called desmoid tumors or aggressive fibromatoses.
Risk factors
A small number of desmoid-type fibromatosis tumors may occur in association with a mutation in the adenomatous polyposis coli (APC) gene (associated with intestinal polyps and a high incidence of colon cancer). In a study of 519 patients older than 10 years with a diagnosis of desmoid-type fibromatosis, 39 (7.5%, a possible underestimation) were found to have familial adenomatous polyposis (FAP).[23] The patients with FAP and desmoid-type fibromatosis were younger, more often male, and had more abdominal wall or mesenteric tumors than did patients with desmoid-type fibromatosis without FAP.
A family history of colon cancer, the presence of congenital hyperplasia of the retinal pigment epithelium,[24,25] or location of the desmoid-type fibromatosis in the abdomen or abdominal wall [23] should prompt referral to a genetic counselor. Currently, there are no general recommendations for genetic testing in children with desmoid-type fibromatosis. Pathology and molecular characteristics of the tumor only provide guidance for screening. If the tumor has a somatic CTNNB1 mutation, screening is not necessary, because the APC gene mutation has not been described in this setting. If a CTNNB1 mutation is not identified, screening for the APC mutation may be warranted.[26,27] (Refer to the Familial Adenomatous Polyposis (FAP) section of the PDQ summary on Genetics of Colorectal Cancer for more information.)
Prognosis
Desmoid-type fibromatosis has an extremely low potential to metastasize. The tumors are locally infiltrating, and surgical control can be difficult because of the need to preserve normal structures.
These tumors have a high potential for local recurrence. Desmoid-type fibromatosis has a highly variable natural history, including well documented examples of spontaneous regression.[28] Mutations in exon 3 of the beta-catenin gene are seen in over 80% of desmoid-type fibromatosis and the mutation 45F has been associated with an increased risk of disease recurrence.[29] Repeated surgical resection can sometimes bring recurrent lesions under control.[30]
Treatment
Evaluation of the benefit of interventions for treatment of desmoid-type fibromatosis has been extremely difficult, because desmoid-type fibromatosis has a highly variable natural history. Large adult series and smaller pediatric series have reported long periods of disease stabilization and even regression without systemic therapy.[30,31]; [32][Level of evidence: 3iiiDi]
Treatment options for desmoid-type fibromatosis include the following:
- Surgery.
- Observation, for tumors that are incompletely resected or recurrent that do not pose a danger to vital organs, if other treatment options are not available.[30,33-39] Whenever possible, however, the treatment of choice is complete resection.
- Chemotherapy, for unresectable or recurrent tumors.
- Other drug therapy, such as nonsteroidal anti-inflammatory drugs (NSAIDs) or antiestrogen therapy.
- Surgery preceded or followed by radiation therapy, for incompletely resected tumors or to avoid recurrence and subsequent surgery that may result in functional or cosmetic compromise.
- Radiation therapy alone, for unresectable tumors.
The treatment of choice is resection to achieve clear margins. However, a retrospective review of children who underwent surgery for desmoid-type fibromatosis at the St. Jude Children’s Research Hospital (SJCRH) reported no correlation between surgical margins and risk of recurrence.[39]
When the diagnosis is known and complete surgical excision is not feasible, and if the tumor poses significant potential for mortality or morbidity, preoperative strategies may include the following:[40,41]
- Observation.
- Chemotherapy.
- Anti-estrogen therapy.
- NSAID therapy.
- External-beam radiation therapy.
Desmoid-type fibromatosis often behaves in a nonaggressive manner. In a study that included mostly adults with extra-abdominal primary fibromatosis, nonsurgical approaches (medical and observation) had similar 3-year EFS compared with surgery.[34] In a subsequent study of adolescents and adults with abdominal wall aggressive fibromatosis, 102 patients were treated with a watch and wait approach, of which 65 patients required no further treatment at 3 years. Approximately one-third of patients had regression of the tumor.[33]
Chemotherapy regimens may include the following:
- Combination chemotherapy using vinblastine and methotrexate produced objective responses in about one-third of patients with unresectable or recurrent desmoid-type fibromatosis.[40]
- A series of mainly adult patients with FAP and unresectable desmoid-type fibromatosis that were unresponsive to hormone therapy showed that doxorubicin plus dacarbazine followed by meloxicam (an NSAID) can be safely administered and can induce responses.[42]
- Pegylated liposomal doxorubicin has been used with some responses.[43] In a series of five patients, a median progression-free interval of 29 months was reported.[44]
- Tyrosine kinase inhibitors: A small retrospective study of adults with desmoid-type fibromatosis showed objective responses to the multi-targeted kinase inhibitor sorafenib.[45][Level of evidence: 3iiiDiv] Previous studies with imatinib did not support its use.[46,47] A small series reported symptomatic improvement and stable disease in seven patients with desmoid-type fibromatosis who were treated with pazopanib.[48]
- The NOTCH pathway has been implicated in the development of desmoid tumors.[49] Partial responses to the gamma secretase inhibitor PF-03084014 have been noted in adults with desmoid-type fibromatosis.[50][Level of evidence: 3iiiDiv]
- Hydroxyurea has been used successfully to treat a few patients after other treatments, but more data are needed.[51-53]
Other drug therapy may include the following:
- NSAIDs such as sulindac have been used in single cases for desmoid-type fibromatosis; the responses seen were usually disease stabilization.[54]
- Antiestrogen treatment, usually tamoxifen, plus sulindac has also resulted in disease stabilization.[55] A prospective trial of the combination of tamoxifen and sulindac reported few side effects, although asymptomatic ovarian cysts were common in girls. This combination showed relatively little activity, as measured by rates of response and progression-free survival (PFS).[56][Level of evidence: 2Diii]
Postoperative radiation therapy is a consideration when progression would entail additional surgery that might cause functional or cosmetic compromise and if radiation is considered acceptable in terms of morbidities.
Radiation has been used for unresectable desmoid-type fibromatosis or postoperatively for tumors with inadequate resections. The potential long-term complications of radiation therapy, especially subsequent neoplasms, make using this modality less appealing in a young population.[57]
Dermatofibrosarcoma protuberans
Dermatofibrosarcoma is a rare tumor that can be present in all age groups, but many of the reported cases arise in children.[58-60] A review of 451 cases in children younger than 20 years in the SEER database found that the incidence was 1 case per 1 million, highest among black patients aged 15 to 19 years. The most common sites were trunk and extremities, which is similar to what is found in adults. Ninety-five percent of patients underwent surgery. OS was 100% at 5 years, 98% at 15 years, and 97% at 30 years. Males had decreased survival compared with females (P < .05).[61][Level of evidence: 3iA]
Molecular features
The tumor has a consistent chromosomal translocation t(17;22)(q22;q13) that juxtaposes the COL1A1 gene with the PDGF-beta gene.
Treatment
Treatment of dermatofibrosarcoma protuberans includes the following:
- Surgery.
- Surgery preceded or followed by radiation therapy.
- Radiation therapy and imatinib therapy, for unresectable or recurrent tumors.
Most dermatofibrosarcoma tumors can be cured by complete surgical resection. Wide excision with negative margins or Mohs or modified Mohs surgery will prevent most tumors from recurring.[62] Despite the locally aggressive behavior of the tumor, lymph node or visceral metastasis rarely occurs.
In retrospective reviews, postoperative radiation therapy after incomplete excision may have decreased the likelihood of recurrence.[63,64]
When surgical resection cannot be accomplished or the tumor is recurrent, treatment with imatinib has been effective.[65-67] Because metastatic disease is more likely after multiple recurrences, radiation or other adjuvant therapy should be considered in patients with recurrence that cannot be managed surgically.[59,61]
Guidelines for workup and management of dermatofibrosarcoma protuberans have been published.[68]
Infantile fibrosarcoma
There are two distinct types of fibrosarcoma in children and adolescents: infantile fibrosarcoma (also called congenital fibrosarcoma) and fibrosarcoma that is indistinguishable from fibrosarcoma seen in adults. These are two distinct pathologic diagnoses and require different treatments. Adult-type fibrosarcoma is addressed below.
Infantile fibrosarcoma usually occurs in children younger than 1 year. It occasionally occurs in children up to age 4 years. A tumor with similar morphology has been identified in older children; in these older children, the tumors do not have the t(12;15)(ETV-NTRK3) translocation that is characteristic of the younger patients.[69] In several of these patients, BRAF gene fusions have been identified.
Clinical presentation
Infantile fibrosarcoma usually presents with a rapidly growing mass, often noted at birth or even seen in prenatal ultrasound. The tumors are often quite large at the time of presentation.[70]
Molecular features
The tumor usually has a characteristic cytogenetic translocation t(12;15)(ETV-NTRK3). Infantile fibrosarcoma shares this translocation and a virtually identical histologic appearance with mesoblastic nephroma.
Prognosis
These tumors have a low incidence of metastases at diagnosis.
Treatment
Treatment options for infantile fibrosarcoma include the following:
- Surgery followed by observation.
- Surgery followed by chemotherapy.
- Chemotherapy followed by surgery.
Complete resection is curative in the majority of patients with infantile fibrosarcoma. However, the large size of the lesion frequently makes resection without major functional consequences impossible (for instance, tumors of the extremities often require amputation for complete excision). The European pediatric group has reported that observation may also be an option in patients with group II disease after surgery.[71] Twelve patients with group II disease received no further therapy and two patients relapsed. One patient obtained a complete remission after chemotherapy. Postoperative chemotherapy was administered to patients with higher group disease and those who progressed. In a subsequent study, only one of seven patients with group II disease progressed during observation; that patient achieved complete remission with chemotherapy.[72][Level of evidence: 3iiA]
Preoperative chemotherapy has made a more conservative surgical approach possible; agents active in this setting include vincristine, dactinomycin, cyclophosphamide, and ifosfamide.[73,74]; [72,75][Level of evidence: 3iiA]; [76][Level of evidence: 3iiB]
Three studies of patients with infantile fibrosarcoma suggest that an alkylator-free regimen is effective and should be used as the first treatment choice in patients with macroscopic disease.[71,72,77] Two cases with variant LMNA/NTRK1 fusions responded to crizotinib.[78,79]
A pediatric patient (aged 16 months) with refractory infantile fibrosarcoma with constitutive activation of the tropomyosin-related kinase signaling pathway from an ETS variant gene 6–neurotrophin 3 receptor gene fusion (ETV6-NTRK3) responded to LOXO-101, with a 90% reduction in tumor size after 2 months of treatment.[80]
A patient aged 2 months with infantile fibrosarcoma was initially treated with chemotherapy. At disease progression, a response was seen with pazopanib.[81]
A rare case of spontaneous regression without treatment has been reported.[82][Level of evidence: 3iiiDiv]
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- LOXO-TRK-15003 (NCT02637687) (Oral TRK Inhibitor LOXO-101 for Treatment of Advanced Pediatric Solid or Primary Central Nervous System [CNS] Tumors): A phase I trial of the pan-TRK inhibitor LOXO-101 is being conducted for children with solid tumors or brain tumors whose disease has progressed or was nonresponsive to available therapies, and for which no standard or available curative therapy exists. LOXO-101 is a highly selective inhibitor of all three TRK family kinases.
- RXDX-101-03 (NCT02650401) (Study of RXDX-101 in Children With Recurrent or Refractory Solid Tumors and Primary CNS Tumors): This is a four-part, open-label, phase I/Ib, dose-escalation study in pediatric patients with: 1) relapsed or refractory solid tumors; 2) primary CNS tumors; 3) neuroblastoma; and 4) non-neuroblastoma, extracranial solid tumors with NTRK1/2/3, ROS1 or ALK gene rearrangements. The study is designed to explore the safety, maximum tolerated dose or recommended phase II dose, pharmacokinetics, and antitumor activity of entrectinib (RXDX-101).
Inflammatory myofibroblastic tumor
Inflammatory myofibroblastic tumor is a rare mesenchymal tumor that has a predilection for children and adolescents.[83-85]
Clinical presentation
Inflammatory myofibroblastic tumors are rare tumors that affect soft tissues and visceral organs of children and young adults.[86] They rarely metastasize but tend to be locally invasive. Usual anatomical sites of disease include soft tissue, lungs, spleen, colon, and breast.[83] A review of 42 cases of pediatric inflammatory myofibroblastic tumor of the bladder was published in 2015.[87]
Molecular features
Roughly half of inflammatory myofibroblastic tumors exhibit a clonal mutation that activates the anaplastic lymphoma kinase (ALK)-receptor tyrosine kinase gene at chromosome 2p23.[88] ROS1 and PDGFR-beta kinase fusions have been identified in 8 of 11 cases (73%) who are negative for ALK by immunohistochemistry.[89][Level of evidence: 3iiiDiv]
Prognosis
Inflammatory myofibroblastic tumor recurs frequently but is rarely metastatic.[83-85]
Treatment
Treatment options for inflammatory myofibroblastic tumor include the following:
- Surgery.
- Chemotherapy.
- Steroid therapy.
- NSAID therapy.
- Targeted therapy (ALK inhibitors).
Complete surgical removal, when feasible, is the mainstay of therapy.[90] In a series of nine patients, four patients achieved continuous remission after complete resection, three patients with residual disease recurred but later achieved continuous remission, and one patient with metastatic disease responded to multiagent chemotherapy.[91][Level of evidence: 3iiA] The benefit of chemotherapy has been noted in case reports.[92] There are case reports of response to either steroids or NSAIDs.[93,94] A series of 32 patients aged 18 years and younger found that complete excision was the mainstay of therapy, although some patients were treated with steroids or cytotoxic chemotherapy. OS was 94%; three patients relapsed and two of them died of the disease. With complete excision, with or without other treatments such as steroids, there was a high survival rate for patients with this disease.[95][Level of evidence: 3iiA]
Inflammatory myofibroblastic tumors respond to crizotinib. Two adults with ALK-rearranged inflammatory myofibroblastic tumor achieved partial response with crizotinib.[96][Level of evidence: 3iiiDiv] For pediatric patients with measurable disease, the use of crizotinib achieved partial tumor responses in three of six patients with ALK-translocated inflammatory myofibroblastic tumors.[97] A case report of a patient aged 16 years with metastatic/multifocal ALK-positive inflammatory myofibroblastic tumor demonstrated a complete response and a 3-year disease-free interval with crizotinib therapy.[98] In a phase I trial of ceritinib for adult patients previously treated with ALK inhibitors, one patient with inflammatory myofibroblastic tumor had a partial response.[99] Finally, one study included 14 patients with inflammatory myofibroblastic tumor who were treated with crizotinib. With crizotinib therapy, five patients had a complete response, seven had a partial response, and the remaining two had stable disease; no patient had relapsed at the time the article was published.[100][Level of evidence: 3iiDiv]
Adult-type fibrosarcoma
These tumors lack the translocation seen in infantile fibrosarcomas. They present like the great majority of nonrhabdomyosarcomas and the management approach is similar.
Low-grade fibromyxoid sarcoma
Low-grade fibromyxoid sarcoma is a histologically deceptive soft tissue neoplasm that most commonly affects young and middle-aged adults, is commonly located deep within the extremities, and is characterized by a FUS/CREB3L3 translocation.[101,102]
Prognosis
In a review of 33 patients (three were younger than 18 years) with low grade fibromyxoid sarcoma, 21 of 33 patients developed a local recurrence after intervals of up to 15 years (median, 3.5 years) and 15 developed metastases up to 45 years (median, 5 years) from diagnosis, most commonly to the lungs and pleura, emphasizing the need for continued follow-up of these patients.[101] Even after metastases occur, the course may be indolent.[103]
In another report, 14 of 73 cases were younger than 18 years of age. In this series with a relatively short follow up (median of 24 months), only 8 of 54 patients with adequate follow up developed local (9%) or distant (6%) recurrence. This report suggests that the behavior of this tumor might be significantly better than previously reported.[104] However, because of the occurrence of late metastases, careful monitoring of these patients is warranted.
The most recent Children's Oncology Group (COG) trial (ARST0332 [NCT00346164]) enrolled 11 patients with this tumor entity. The median age at diagnosis was 13 years and males were more commonly affected. The most common sites were the lower and upper extremity (n = 9) and none of the patients had developed local or distant disease recurrence at a median follow up of 2.7 years.[105]
Treatment
Treatment options for low-grade fibromyxoid sarcoma include the following:
- Surgery.
The limited treatment information for low-grade fibromyxoid sarcoma suggest that surgery is the treatment of choice as the tumor is not very chemosensitive.[103] There are little data regarding the use of chemotherapy and/or radiation therapy in this disease. One report suggests that trabectedin may be effective in the treatment of low-grade fibromyxoid sarcoma.[106]
Myxofibrosarcoma
Myxofibrosarcoma is a rare lesion, especially in childhood. It is typically treated with complete surgical resection.
Sclerosing epithelioid fibrosarcoma
Sclerosing epithelioid fibrosarcoma is a rare malignant sarcoma that commonly harbors EWSR1 gene rearrangements and has an aggressive clinical course.[107] It is typically treated with complete surgical excision. Long-term follow-up is recommended because local recurrence and metastases can occur late.
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with infantile fibrosarcoma, inflammatory myofibroblastic tumor, low-grade myofibroblastic tumor, myxoinflammatory fibroblastic sarcoma, solitary fibrous tumor, adult-type fibrosarcoma, low-grade fibromyxoid sarcoma, myxofibrosarcoma, and sclerosing epithelioid fibrosarcoma are eligible for this trial.
Skeletal Muscle Tumors
Rhabdomyosarcoma
Refer to the PDQ summary on Childhood Rhabdomyosarcoma Treatment for more information.
Smooth Muscle Tumors
Leiomyosarcoma
Leiomyosarcoma accounts for 2% of soft tissue sarcoma in patients younger than 20 years (refer to Table 1).
Risk factors
Among 43 children with HIV/AIDS who developed tumors, eight developed Epstein-Barr virus–associated leiomyosarcoma.[108] Survivors of hereditary retinoblastoma have a statistically significant increased risk of developing leiomyosarcoma and 78% of these were diagnosed 30 or more years after the initial diagnosis of retinoblastoma.[109]
Treatment
Treatment options for leiomyosarcoma include the following:
- Chemotherapy (trabectedin).
In an open-label study of trabectedin in adult patients with recurrent sarcomas, the best overall response rate (complete remission and partial remission) was seen in patients with leiomyosarcoma (7.5%).[110] The clinical benefit rate (includes stable disease) for leiomyosarcoma was 54%. In another adult study, patients with recurrent liposarcoma and leiomyosarcoma were randomly assigned to receive treatment with either trabectedin or dacarbazine. Patients treated with trabectedin had a 45% reduction in disease progression.[14] There are no data to support the use of trabectedin in pediatric patients.
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with leiomyosarcoma are eligible for this trial.
So-called Fibrohistiocytic Tumors
So-called fibrohistiocytic tumors include the following tumor subtypes:
- Giant cell tumor of soft tissue.
- Plexiform fibrohistiocytic tumor.
Plexiform fibrohistiocytic tumor
Plexiform histiocytic tumor is a rare, low- to intermediate-grade tumor that most commonly affects children and young adults. Depending on the series, the median age at presentation ranges from 8 to 14.5 years; however, the tumor has been described in patients as young as 3 months.[111,112]
Clinical presentation
The tumor commonly arises as a painless mass in the skin or subcutaneous tissue and most often involves the upper extremities, including the fingers, hand, and wrist.[113-115] There are rare reports of spread to regional lymph nodes or the lungs.[111,115,116]
Molecular features
No consistent chromosomal anomalies have been detected but a t(4;15)(q21;q15) translocation has been reported.[117]
Prognosis
Plexiform fibrohistiocytic tumor is an intermediate-grade tumor that rarely metastasizes.
Treatment
Surgery is the treatment of choice but local recurrence has been reported in 12% to 50% of cases.[118]
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with giant cell tumors of soft tissue and plexiform fibrohistiocytic tumor are eligible for this trial.
Tumors of Peripheral Nerves
Ectomesenchymoma
Ectomesenchymoma is a rare nerve sheath tumor that mainly occurs in children. It is a biphenotypic soft tissue sarcoma with both mesenchymal and ectodermal components. Elements similar to rhabdomyosarcoma have been identified.
The German Soft Tissue Sarcoma Group (Cooperative Weichteilsarkom Studiengruppe [CWS]) reported on six patients (ages 0.2–13.5 years) registered over 14 years.[119][Level of evidence: 3iiA] The tumors were located in various sites including the extremities, abdomen, and orbit. All six patients were treated with surgery and chemotherapy directed at rhabdomyosarcoma. Two patients received radiation therapy. Three patients recurred with rhabdomyosarcoma features. Although data are scant, it appears that the tumor may respond to chemotherapy.[119]
Malignant peripheral nerve sheath tumor
Malignant peripheral nerve sheath tumors account for 5% of soft tissue sarcoma in patients younger than 20 years (refer to Table 1).
Risk factors
Malignant peripheral nerve sheath tumor can arise sporadically and in children with type 1 neurofibromatosis (NF1).[120]
Molecular features
Inactivating mutations of SUZ12 have been described in these tumors and are absent in neurofibromas.[121]
Prognosis
Features associated with a favorable prognosis include the following:[120,122-124]
- Smaller tumor size. In a multivariate analysis, only tumor size and nuclear p53 expression were found to be independent predictors of disease-specific survival.[123]
- Male sex and non-Hispanic white race.[125]
- No metastasis at presentation. A retrospective review of 140 patients with malignant peripheral nerve sheath tumor from the MD Anderson Cancer Center included children and adolescents. The disease-specific survival at 10 years was 32%. In this series, presence of metastatic disease was associated with a much worse prognosis.[123]
- Lower stage.
- Lower histologic grade.
- Extremity as the primary site.
Features associated with an unfavorable prognosis include the following:[126]
- High grade.
- Deep tumor location.
- Locally advanced stage at diagnosis.
- Macroscopically incomplete resection (R2).
For patients with localized disease in the MD Anderson Cancer Center study, there was no significant difference in outcome between patients with and without NF1.[123] In other studies, it was not clear whether the absence of NF1 is a favorable prognostic factor as it has been associated with both favorable [122] and unfavorable outcomes.[120,122,124] In the French Sarcoma Group study, NF1 was associated with other adverse prognostic features, but was not an independent predictor of poor outcome.[126] The Italian Sarcoma Group reported on outcomes after recurrence in 73 children and adolescents with malignant peripheral nerve sheath tumor.[127][Level of evidence: 3iiiA] The median overall survival after first relapse was 11 months, and the survival rates were 39.2% at 1 year and 15.8% at 5 years. The factors associated with a better prognosis for these patients who relapsed were less initial tumor invasiveness, longer time to relapse, and the achievement of a secondary complete remission (which was related to the feasibility of radical surgery).
Treatment
Treatment options for malignant peripheral nerve sheath tumor include the following:
Complete surgical removal of the tumor, whenever possible, is the mainstay of treatment.
The role of radiation therapy is difficult to assess, but durable local control of known postoperative microscopic residual tumor is not assured after radiation therapy.
Chemotherapy has achieved objective responses in childhood malignant peripheral nerve sheath tumor. A large retrospective analysis of the German and Italian experience with malignant peripheral nerve sheath tumor reported that 65% of measurable tumors had objective responses to ifosfamide-containing chemotherapy regimens, but the analysis did not conclusively demonstrate improved survival for chemotherapy.[120] This retrospective analysis also noted a trend toward improved outcome with postoperative radiation therapy.[120] A series of 37 young patients with malignant peripheral nerve sheath tumor and NF1 showed that most patients had large invasive tumors that were poorly responsive to chemotherapy; PFS was 19% and 5-year OS was 28%.[128]
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with malignant peripheral nerve sheath tumor are eligible for this trial.
- SARC023 (NCT02008877) (Ganetespib and Sirolimus in Patients With Malignant Peripheral Nerve Sheath Tumors): This trial is testing the combination of ganetespib, the heat shock protein inhibitor, and sirolimus, the mammalian target of rapamycin (mTOR) inhibitor, for the treatment of patients with unresectable or metastatic malignant peripheral nerve sheath tumors. Patients with unresectable soft tissue or bone sarcomas are eligible for phase I of the trial. Patients with unresectable malignant peripheral nerve sheath tumors are eligible for phase II of the trial. Eligibility is restricted to patients aged 18 years and older.
- NCT02601937 (A Phase 1 Study of the EZH2 Inhibitor Tazemetostat in Pediatric Subjects With Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma): Patients with INI1-negative tumors are eligible for targeted treatment with an EZH2 inhibitor. This is a phase I, open-label, dose-escalation, and dose-expansion study with a twice-daily oral dose of tazemetostat.
- ADVL1522 (NCT02452554) (Lorvotuzumab Mertansine in Treating Younger Patients with Relapsed or Refractory Wilms Tumor, Rhabdomyosarcoma, Neuroblastoma, Pleuropulmonary Blastoma, Malignant Peripheral Nerve Sheath Tumor, or Synovial Sarcoma): This is a phase II study of IMGN901 (lorvotuzumab mertansine) in children with relapsed or refractory Wilms tumor, rhabdomyosarcoma, neuroblastoma, pleuropulmonary blastoma, malignant peripheral nerve sheath tumor, and synovial sarcoma. This trial is studying the effects of IMGN901, an antibody-drug conjugate that links a potent antimitotic to antibodies that target CD56.
Malignant triton tumor
Malignant triton tumors are a variant of malignant peripheral nerve sheath tumors. They occur most often in patients with neurofibromatosis type I and consist of neurogenic and rhabdomyoblastic components. Malignant triton tumors are high-grade malignancies. They usually occur before age 35 years and are very rare in children (case reports only).[129]
Malignant triton tumors are not usually responsive to chemotherapy and radiation therapy but have been treated with rhabdomyosarcoma therapy.[129][Level of evidence: 3iiiA] (Refer to the PDQ summary on Childhood Rhabdomyosarcoma Treatment for more information.)
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with malignant triton tumor are eligible for this trial.
Pericytic (Perivascular) Tumors
Myopericytoma
Infantile hemangiopericytoma is a subtype of myopericytoma.
Hemangiopericytoma is a highly vascularized tumor of uncertain origin.
Histology
Histologically, hemangiopericytomas are composed of packed round or fusiform cells that are arranged around a complex vasculature, forming many branch-like structures. Hyalinization is often present. Infantile hemangiopericytomas have similar histology but many are multilobular with vasculature outside the tumor mass.[130]
Treatment and outcome
Treatment of infantile hemangiopericytomas includes the following:
- Chemotherapy.
In a series of 17 children, the differences in metastatic potential and response to treatment were clearly demonstrated for adult and infantile hemangiopericytomas.[131] Eleven children were older than 1 year. Several of these patients had disease in the lymph nodes or lungs. Six patients with stage II or III disease progressed and died. Three patients with stage I disease survived, although one had recurrence in the lungs. Six patients had infantile hemangiopericytoma, most were greater than stage I (5 of 6). All six patients survived and three had good responses to vincristine, actinomycin, and cyclophosphamide. Hemangiopericytoma in children younger than 1 year seems to have a better prognosis than in children older than 1 year.[132-134]
Infantile myofibromatosis
This entity is a fibrous tumor of infancy and childhood that most commonly presents in the first 2 years of life.[135] The lesion can present as a single subcutaneous nodule (myofibroma) most commonly involving the head and neck region or lesions can affect multiple skin areas, muscle, and bone (myofibromatosis).[136-139]
An autosomal dominant form of the disease has been described and it is associated with germline mutations of the PDGFRB gene.[140]
Treatment
These lesions have an excellent prognosis and can regress spontaneously.
About one-third of cases with multicentric involvement will also have visceral involvement, and the prognosis for these patients is poor.[138,139,141] The use of combination therapy with vincristine/dactinomycin and vinblastine/methotrexate have proven effective in cases of multicentric disease with visceral involvement and in cases in which the disease has progressed and has threatened the life of the patient (e.g., upper airway obstruction).[138,139,142]
Tumors of Uncertain Differentiation
Tumors of uncertain differentiation include the following tumor subtypes:
- Alveolar soft part sarcoma.
- Clear cell sarcoma of soft tissue.
- Desmoplastic small round cell tumor.
- Epithelioid sarcoma.
- Extrarenal rhabdoid tumor.
- Extraskeletal myxoid chondrosarcoma.
- Neoplasms with perivascular epithelioid cell differentiation (PEComa NOS, malignant)
- Primitive neuroectodermal tumor (PNET)/extraskeletal Ewing tumor.
- Synovial sarcoma (NOS, spindle cell, and biphasic varieties).
Alveolar soft part sarcoma
Alveolar soft parts sarcomas account for 1.4% of soft tissue sarcomas in patients younger than 20 years (refer to Table 1).
Clinical presentation
The median age at presentation is 25 years, and alveolar soft part sarcoma most commonly arises in the extremities but can occur in the oral and maxillofacial region.[143-145] Alveolar soft part sarcoma in children can present with evidence of metastatic disease.[146]
Molecular features
This tumor of uncertain histogenesis is characterized by a consistent chromosomal translocation t(X;17)(p11.2;q25) that fuses the ASPSCR1 gene with the TFE3 gene.[147,148]
Prognosis
Alveolar soft part sarcoma in children may have an indolent course.[146] Patients with alveolar soft part sarcoma may relapse several years after a prolonged period of apparent remission.[149] Because these tumors are rare, all children with alveolar soft part sarcoma should be considered for enrollment in prospective clinical trials.
In a series of 19 treated patients, one group reported a 5-year OS rate of 80%, a 91% OS rate for patients with localized disease, a 100% OS rate for patients with tumors 5 cm or smaller, and a 31% OS rate for patients with tumors larger than 5 cm.[150] In another series of 33 patients, OS was 68% at 5 years from diagnosis and 53% at 10 years from diagnosis. Survival was better for smaller tumors (≤5 cm) and completely resected tumors.[151][Level of evidence: 3iiA] Delayed metastases to the brain and lung are uncommon.[143] A retrospective review of children and young adults younger than 30 years (median age, 17 years; range, 1.5–30 years) from four institutions identified 69 patients treated primarily with surgery between 1980 and 2014.[152][Level of evidence: 3iiA] The ASPL-TFE3 translocation was present in all 26 patients tested. There were 19 patients with Intergroup Rhabdomyosarcoma Study (IRS) postsurgical staging group I tumors (28%), 7 patients with IRS group II tumors (10%), 5 patients with IRS group III tumors (7%), and 38 patients with IRS group IV tumors (55%). The 5-year EFS was 80% and the OS was 87% for the 31 patients with localized tumors (IRS postsurgical groups I, II, and III). The 5-year EFS was 7% and the OS was 61% for the 38 patients with metastatic tumors (IRS postsurgical group IV).
Treatment
Treatment options for alveolar soft part sarcoma include the following:
The standard approach is complete resection of the primary lesion.[150] If complete excision is not feasible, radiation therapy should be administered. A study from China reported on 18 patients with alveolar soft part sarcoma of the oral and maxillofacial region; 15 patients were younger than 30 years.[145][Level of evidence: 3iiDii] Surgical removal with negative margins was the primary treatment. All patients survived, and only one patient had metastatic disease recurrence.
A series of 51 pediatric patients aged 0 to 21 years with alveolar soft part sarcoma found an OS rate at 10 years of 78% and an EFS rate of about 63%. Patients with localized disease (n = 37) had a 10-year OS of 87%, and the 14 patients with metastases at diagnosis had a 10-year OS of 44%, partly resulting from surgical removal of primary tumor and lung metastases in some patients. Only 3 of 18 patients (17%) with measurable disease had a response to conventional antisarcoma chemotherapy, but two of four patients treated with sunitinib had a partial response.[143][Level of evidence: 3iiiA] There have been sporadic reports of objective responses to interferon-alpha and bevacizumab.[143,153,154]
A small retrospective study of nine adult patients with metastatic alveolar soft part sarcoma treated with sunitinib reported partial response in five patients and stable disease in two patients.[155][Level of evidence: 3iiiDiv] In a phase II trial of cediranib, an inhibitor of all three known vascular epidermal growth factor receptors, 15 of 43 adult patients (35%) with metastatic alveolar soft part sarcoma had a partial response.[156][Level of evidence: 3iiDiv]
There have been no open trials for patients with metastatic alveolar soft part sarcoma.
Treatment options under clinical evaluation for alveolar soft part sarcoma
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- NCT00942877 (Phase II Study of Cediranib [AZD2171] in Patients With Alveolar Soft Part Sarcoma): A phase II study of cediranib in patients with alveolar soft part sarcoma is being conducted in patients younger than 16 years at the Clinical Center of the National Institutes of Health.
- NCT01391962 (Sunitinib or Cediranib for Alveolar Soft Part Sarcoma): A phase II trial in which patients with metastatic alveolar soft part sarcoma are randomly assigned to either sunitinib or cediranib monotherapy, with crossover at disease progression. Patients aged 16 years and older are eligible. This study is being conducted at the Clinical Center of the National Institutes of Health.
Clear cell sarcoma of soft tissue
Clear cell sarcoma (formerly and inappropriately called malignant melanoma of soft parts) is a rare soft tissue sarcoma that typically involves the deep soft tissues of the extremities. It is also called clear cell sarcoma of tendons and aponeuroses. The tumor often affects adolescents and young adults.
Patients who have small, localized tumors with low mitotic rate and intermediate histologic grade fare best.[157]
Clinical presentation
The tumor most commonly affects the lower extremity, particularly the foot, heel, and ankle.[158,159] It has a high propensity for nodal dissemination, especially metastases to regional lymph nodes (12%–43%).[159,160] The tumor typically has an indolent clinical course.
Molecular features
Clear cell sarcoma of soft tissue is characterized by an EWS-ATF1 fusion.[161]
Treatment
Treatment options for clear cell sarcoma of soft tissue include the following:
In a series of 28 pediatric patients reported by the Italian and German Soft Tissue Cooperative Studies, the median age at diagnosis was 14 years and the lower extremity was the most common primary site (50%). Surgery with or without radiotherapy is the treatment of choice and offers the best chance for cure. In this series, 12 of 13 patients with completely resected tumors were cured. For patients with more advanced disease the outcome is poor and chemotherapy is rarely effective.[162]; [163][Level of evidence: 3iiDii]
Desmoplastic small round cell tumor
Desmoplastic small round cell tumor is a rare primitive sarcoma.
Clinical presentation
Desmoplastic small round cell tumor most frequently involves the abdomen, pelvis, or tissues around the testes, but it may occur in the kidney.[164-167] The tumor occurs more commonly in males and may spread to the lungs and elsewhere. Peritoneal and pelvic lesions frequently have widespread peritoneal implants.[168]
In a large, single-institution series of 65 patients, a correlation was made between computed tomography (CT) scans in most patients and positron-emission tomography (PET)/CT scans in 11 patients. PET/CT scans had very few false-negative results and detected metastatic sites missed on conventional CT scans.[168]
Molecular features
Cytogenetic studies of these tumors have demonstrated the recurrent translocation t(11;22)(p13;q12), which has been characterized as a fusion of the WT1 and EWS genes.[167,169] The WT1-EWS fusion confirms the diagnosis of desmoplastic small round cell tumor.
Prognosis
The overall prognosis for desmoplastic small round cell tumor remains extremely poor, with reported rates of death at 90%. Greater than 90% tumor resection either at presentation or after preoperative chemotherapy may be a favorable prognostic factor for OS.[170,171]; [172][Level of evidence: 3iiiA]
Treatment
There is no standard approach to the treatment of desmoplastic small round cell tumor.
Treatment options for desmoplastic small round cell tumor include the following:
- Surgery.
- Chemotherapy followed by surgery.
- Radiation therapy.
Complete surgical resections are rare, and the overall prognosis for desmoplastic small round cell tumor remains extremely poor, with reported rates of death at 90%. Treatment may include chemotherapy, surgery, and radiation therapy. Multiagent chemotherapy analogous to that used for sarcomas has been used, as well as total abdominal radiation therapy.[164,165,170,173-176]
A single-institution study reported that five of five patients with recurrent desmoplastic small round cell tumor had partial responses to treatment with the combination of vinorelbine, cyclophosphamide, and temsirolimus.[177]
The Center for International Blood and Marrow Transplant Research (CIBMTR) analyzed patients with desmoplastic small round cell tumor in their registry who received consolidation with high dose chemotherapy and autologous stem cell reconstitution.[178] While this retrospective registry analysis suggested some benefit for this approach, other investigators have abandoned the approach because of excessive toxicity and lack of efficacy.[170]
Epithelioid sarcoma
Epithelioid sarcoma is a rare mesenchymal tumor of uncertain histogenesis which displays multilineage differentiation.[179]
Clinical presentation
Epithelioid sarcoma commonly presents as a slowly growing firm nodule based in the deep soft tissue; the proximal type predominantly affects adults and involves the axial skeleton and proximal sites. The tumor is highly aggressive and has a propensity for lymph node metastases.
Molecular features
Epithelioid sarcoma is characterized by inactivation of the SMARCB1 gene, which is present in both conventional and proximal types of epithelioid sarcoma.[180] This abnormality leads to increased dependence on EZH2 and tumor formation.[181]
Treatment
Treatment options for epithelioid sarcoma include the following
- Chemotherapy.
- Surgery.
- Surgery preceded or followed by radiation therapy.
Patients should be carefully evaluated for the presence of involved lymph nodes; suspicious lymph nodes should be biopsied. Surgical removal of primary and recurrent tumor(s) is the most effective treatment.[182][Level of evidence: 3iiiA]
In a review of 30 pediatric patients with epithelioid sarcoma (median age at presentation, 12 years), responses to chemotherapy were reported in 40% of patients using sarcoma-based regimens, and 60% of patients were alive at 5 years after initial diagnosis.[183] A single-institution retrospective review of 20 patients, including children and adults (median age, 27.3 years), found no difference in the probability of recurrence between patients who received chemotherapy and those who did not receive chemotherapy and suggested that radiation therapy may be useful.[182]
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- NCT02601937 (A Phase 1 Study of the EZH2 Inhibitor Tazemetostat in Pediatric Subjects With Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma): Patients with INI1-negative tumors are eligible for targeted treatment with an EZH2 inhibitor. This is a phase I, open-label, dose-escalation, and dose-expansion study with a twice-daily oral dose of tazemetostat.
Extrarenal (extracranial) rhabdoid tumor
Malignant rhabdoid tumors were first described in children with renal tumors in 1981 (refer to the PDQ summary on Wilms Tumor and Other Childhood Kidney Tumors Treatment for more information) and were later found in a variety of extrarenal sites. These tumors are uncommon and highly malignant, especially in children younger than 2 years.
Extrarenal (extracranial) rhabdoid tumors account for 2% of soft tissue sarcoma in patients younger than 20 years (refer to Table 1).
Molecular features
The first sizeable series of 26 children with extrarenal extracranial malignant rhabdoid tumor of soft tissues came from patients enrolled on the Intergroup Rhabdomyosarcoma Studies I through III during a review of pathology material. Only five patients (19%) were alive without disease.[184] Later, investigation of children with atypical teratoid/rhabdoid tumors of the brain, as well as those with renal and extrarenal malignant rhabdoid tumors, found germline and acquired mutations of the SMARCB1 gene in all 29 tumors tested.[185] Rhabdoid tumors may be associated with germline mutations of the SMARCB1 gene and may be inherited from an apparently unaffected parent.[186] This observation was extended to 32 malignant rhabdoid tumors at all sites in patients whose mean age at diagnosis was 12 months.[187]
Prognosis
In a Surveillance, Epidemiology, and End Results (SEER) study of 229 patients with renal, central nervous system, and extrarenal malignant rhabdoid tumor, patients aged 2 to 18 years, limited extent of tumor, and delivery of radiation therapy were shown to affect the outcome favorably compared with other patients (P < .002 for each comparison). Site of the primary tumor was not prognostically significant. OS at 5 years was 33%.[188]
Treatment
Treatment includes surgical removal when possible, chemotherapy as used for soft tissue sarcomas (but no single regimen is currently accepted as best), and radiation therapy.[189][Level of evidence: 3iA]; [190,191][Level of evidence: 3iiiB]
Responses to alisertib have been documented in four patients with central nervous system (CNS) atypical teratoid/rhabdoid tumors.[192] (Refer to the PDQ summary on Childhood Central Nervous System Atypical Teratoid/Rhabdoid Tumor Treatment summary for more information about CNS atypical teratoid/rhabdoid tumors.)
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- NCT02601937 (A Phase 1 Study of the EZH2 Inhibitor Tazemetostat in Pediatric Subjects With Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma): Patients with INI1-negative tumors are eligible for targeted treatment with an EZH2 inhibitor. This is a phase I, open-label, dose-escalation, and dose-expansion study with a twice-daily oral dose of tazemetostat.
Extraskeletal myxoid chondrosarcoma
Extraskeletal myxoid chondrosarcoma is relatively rare among soft tissue sarcomas, representing only 2.3% of all soft tissue sarcoma.[193] It has been reported in children and adolescents.[194]
Molecular features
Extraskeletal myxoid chondrosarcoma is a multinodular neoplasm. The rounded cells are arranged in cords and strands in a chondroitin sulfate myxoid background. Several cytogenetic abnormalities have been identified (refer to Table 2), with the most frequent being the translocation t(9;22)(q22;q12), involving the EWSR1/NR4A3 genes.[195]
Prognosis
The tumor has traditionally been considered of low-grade malignant potential.[196] However, recent reports from large institutions showed that extraskeletal myxoid chondrosarcoma has significant malignant potential, especially if patients are followed for a long time.[197,198] Patients tend to have slow protracted courses. Nodal involvement has been well described. Local recurrence (57%) and metastatic spread to lungs (26%) have been reported.[198]
Treatment
Treatment options for extraskeletal myxoid chondrosarcoma include the following:
- Surgery.
- Radiation therapy.
The therapeutic benefit of chemotherapy has not been established. Aggressive local control and resection of metastases led to OS of 87% at 5 years and 63% at 10 years. Tumors were relatively resistant to radiation therapy.[197]
There may be potential genetic targets for small molecules, but these should be studied as part of a clinical trial. In an adult study, six of ten patients who received sunitinib achieved a partial response.[199]
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- NCT02601937 (A Phase 1 Study of the EZH2 Inhibitor Tazemetostat in Pediatric Subjects With Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma): Patients with INI1-negative tumors are eligible for targeted treatment with an EZH2 inhibitor. This is a phase I, open-label, dose-escalation, and dose-expansion study with a twice-daily oral dose of tazemetostat.
Neoplasms with perivascular epithelioid cell differentiation (PEComas)
Risk factors and molecular features
Benign PEComas are common in tuberous sclerosis, an autosomal dominant syndrome that also predisposes to renal cell cancer and brain tumors. Tuberous sclerosis is caused by germline inactivation of either TSC1 (9q34) or TSC2 (16p13.3), and the same tumor suppressor genes are inactivated somatically in sporadic PEComas.[200] Inactivation of either gene results in stimulation of the mTOR pathway, providing the basis for the treatment of nonsurgically curable PEComas with mTOR inhibitors.[201,202] A small proportion of PEComas have TFE3 rearrangements with fusions involving various genes including SFPQ/PSF and RAD51B.[203]
Clinical presentation
PEComas occur in various rare gastrointestinal, pulmonary, gynecologic, and genitourinary sites. Soft tissue, visceral, and gynecologic PEComas are more commonly seen in middle-aged female patients and are usually not associated with the tuberous sclerosis complex.[204] The disease course may be indolent.
Prognosis
Most PEComas have a benign clinical course, but malignant behavior has been reported and can be predicted based on the size of the tumor, mitotic rate, and presence of necrosis.[205]
Treatment
Treatment options have not been defined. Treatment may include surgery or observation followed by surgery when the tumor is large.[206]
Clinical activity with mTOR inhibitors, such as sirolimus, in tumors with evidence of mTORC1 activation and TSC loss has been well documented.[207]
Primitive neuroectodermal tumor (PNET)/extraskeletal Ewing tumor
(Refer to the PDQ summary on Ewing Sarcoma Treatment for more information.)
Synovial sarcoma
Synovial sarcoma accounts for 9% of soft tissue sarcomas in patients younger than 20 years (refer to Table 1).
Synovial sarcoma is one of the most common nonrhabdomyosarcomatous soft tissue sarcomas in children and adolescents. In a 1973 to 2005 SEER review, 1,268 patients with synovial sarcoma were identified. Approximately 17% of these patients were children and adolescents and the median age at diagnosis was 34 years.[208]
Histologic classification
Synovial sarcoma can be subclassified as the following types:
- Synovial sarcoma, NOS.
- Synovial sarcoma, spindle cell.
- Synovial sarcoma, biphasic.
Clinical presentation
The most common tumor location is the extremities, followed by trunk and head and neck.[208] Rarely, a synovial sarcoma may arise in the heart or pericardium.[209]
The most common site of metastasis is the lung.[210,211] The risk of metastases is highly influenced by tumor size; it is estimated that patients with tumors that are larger than 5 cm have a 32-fold risk of developing metastases when compared with other patients.
Diagnostic evaluation
The diagnosis of synovial sarcoma is made by immunohistochemical analysis, ultrastructural findings, and demonstration of the specific chromosomal translocation t(x;18)(p11.2;q11.2). This abnormality is specific for synovial sarcoma and is found in all morphologic subtypes. Synovial sarcoma results in rearrangement of the SYT gene on chromosome 18 with one of the subtypes (1, 2, or 4) of the SSX gene on chromosome X.[212,213] It is thought that the SYT/SSX18 transcript promotes epigenetic silencing of key tumor suppressor genes.[214]
In one report, reduced INI1 nuclear reactivity on immunohistochemical staining was seen in 49 cases of synovial sarcoma, suggesting that this pattern may help distinguish synovial sarcoma from other histologies.[215]
Prognosis
Patients younger than 10 years have more favorable outcomes and clinical features, including extremity primaries, smaller tumors, and localized disease, than do older patients.[208,216] A meta-analysis also suggested that response to chemotherapy was correlated with improved survival.[217]
The following studies have reported multiple factors associated with unfavorable outcomes:
- In a retrospective analysis of synovial sarcoma in children and adolescents who were treated in Germany and Italy, tumor size (>5 cm or ≤5 cm in greatest dimension) was an important predictor of EFS.[218] In this analysis, local invasiveness conferred an inferior probability of EFS, but surgical margins were not associated with clinical outcome.
- In a single-institution retrospective analysis of 111 patients with synovial sarcoma who were younger than 22 years at diagnosis, larger tumor size, greater depth in tissue, greater local invasiveness, and more proximal tumor location were associated with poorer OS.[219][Level of evidence: 3iiA]
- A multicenter analysis of 219 children from various treating centers including Germany, SJCRH, Instituto Tumori, and MD Anderson Cancer Center reported an estimated 5-year OS of 80% and EFS rate of 72%.[217] In this analysis, an interaction between tumor size and invasiveness was observed; in multivariate analysis, patients with large or invasive tumors or with Intergroup Rhabdomyosarcoma Study Clinical Group III disease (localized, incompletely resected or with biopsy only) and IV (metastases at diagnosis) had decreased OS. Treatment with radiation therapy was related to improved OS (hazard ratio, 0.4; 95% confidence interval, 0.2–0.7). In Intergroup Rhabdomyosarcoma Study Group III patients, objective response to chemotherapy (18 of 30 [60%]) correlated with improved survival. In adults, factors such as International Union Against Cancer/American Joint Committee on Cancer stage III and stage IVA, tumor necrosis, truncal location, elevated mitotic rate, age, and histologic grade have been associated with a worse prognosis.[220-222]
- Expression and genomic index prognostic signatures have been studied in synovial sarcoma. Complex genomic profiles, with greater rearrangement of the genome, are more common in adults than in younger patients with synovial sarcoma and are associated with a higher risk of metastasis.[223]
- A review of 84 patients with localized synovial sarcoma who had information on fusion status (SYT-SSX) and histologic grading found no difference in OS according to these criteria. However, for tumor size at diagnosis, the study showed that patients with tumors between 5 cm and 10 cm had a worse prognosis than those with smaller tumors (P = .02), and patients with tumors larger than 10 cm had even worse OS (P = .0003).[224][Level of evidence: 3iiiA]
- The German CWS group reviewed 27 evaluable patients younger than 21 years with pulmonary metastases among 296 patients with synovial sarcoma. Metastases involved the lungs in all patients. The 5-year EFS rate was 26%, and the OS rate was 30%. The most important prognostic factor at presentation was that the metastases were limited to one lesion in one lung or one lesion in both lungs (a group they termed oligometastatic). Treatment elements associated with superior survival were adequate local therapy of the primary tumor and, if feasible, for the metastases. The use of whole-lung irradiation did not correlate with better outcomes.[225][Level of evidence: 3iiA]
Survival after relapse is poor (30% at 5 years). Factors associated with outcome after relapse include duration of first remission (> or ≤ 18 months) and lack of a second remission.[226]
Treatment
Treatment options for synovial sarcoma include the following:
The COG and the European Pediatric Soft Tissue Sarcoma Study Group reported a combined analysis of 60 patients younger than 21 years with localized synovial sarcoma prospectively assigned to surgery without adjuvant radiation therapy or chemotherapy.[227] Enrollment was limited to patients with initial complete resection with histologically free margins, with a grade 2 tumor of any size or a grade 3 tumor 5 cm or smaller. The 3-year EFS was 90% (median follow-up, 5.2 years; range, 1.9–9.1). All eight events were local tumor recurrence; no metastatic recurrences were seen. All patients with recurrent disease were effectively treated with salvage therapy, resulting in 100% OS.
Synovial sarcoma appears to be more sensitive to chemotherapy than many other soft tissue sarcomas, and children with synovial sarcoma seem to have a better prognosis when compared with adults.[11,211,222,228-232] The most commonly used regimens for the treatment of synovial sarcoma incorporate ifosfamide and doxorubicin.[217,231,233] Response rates to the ifosfamide and doxorubicin regimen are higher than in other nonrhabdomyosarcomatous soft tissue sarcomas.[234]
Several studies have reported the following chemotherapy-associated treatment findings:
- Several treatment centers advocate postoperative chemotherapy after resection and radiation therapy of synovial sarcoma in children and young adults.[217,218,235-237]
- The International Society of Pediatric Oncology-Malignant Mesenchymal Tumors studies showed that select patients (young age, <5 cm resected tumors) with nonmetastatic synovial sarcoma can have excellent outcome in the absence of radiation, but it is still unclear whether that approach obviates an advantage of radiation for local or regional control.[236]
- A German trial suggested a benefit for postoperative chemotherapy in children with synovial sarcoma.[237]
- A meta-analysis also suggested that chemotherapy may provide benefit.[217]
- In the most recent COG ARST0332 (NCT00346164) study, 129 patients with synovial sarcoma were prospectively treated using a risk-based therapy program (as detailed in the prognosis section), of which 43 were categorized as low risk, 66 as intermediate risk, and 20 as high risk. At a median follow-up of 2.6 years, 3-year EFS for low-, intermediate-, and high-risk groups were 83%, 79%, and 16%, respectively. The use of risk factor–directed therapy accurately predicted outcomes.[238]
- The European Pediatric Soft Tissue Sarcoma Study Group performed a prospective study of patients younger than 21 years with synovial sarcoma (CCLG-EPSSG-NRSTS-2005 [NCT00334854]).[239][Level of evidence: 3iiA] Patients were stratified into the following three risks groups and nonrandomly assigned to treatment by risk group:
- Low-risk patients had Intergroup Rhabdomyosarcoma Study (IRS) group I tumors less than 5 cm in size and nonaxial primary tumors.
- Intermediate-risk patients had no axial primary tumors and IRS group I tumors greater than 5 cm or IRS group II tumors.
- High-risk patients included all patients with axial primary sites (head and neck, lung and pleura, trunk, retroperitoneal), IRS group III tumors, or N1 tumors.
Outcomes for patients treated on the CCLG-EPSSG-NRSTS-2005 trial are described in Table 9.
Table 9. Event-Free Survival (EFS) and Overall Survival (OS) in Patients With Low-, Intermediate-, and High-Risk Synovial Sarcoma Treated on the CCLG-EPSSG-NRSTS-2005 Trial Risk Group Treatment 3-Year EFS (%) 3-Year OS (%) IRS = Intergroup Rhabdomyosarcoma Study; RT = radiation therapy. aChemotherapy was ifosfamide/doxorubicin, with doxorubicin omitted during radiation therapy. b59.4 Gy in cases without the option of secondary resection; 50.4 Gy as preoperative radiation therapy; 50.4, 54, and 59.4 Gy as postoperative radiation therapy, in the case of R0, R1, and R2 resections, respectively (no additional radiation therapy in the case of secondary complete resections with free margins, in children younger than 6 years). Low Surgery alone 92 100 Intermediate Surgery, 3–6 cycles chemotherapya ± RTb 91 100 High (IRS group III) 3 cycles of chemotherapya surgery, 3 additional cycles of chemotherapy, ± RTb 77 94 High (axial primary sites) Surgery, 6 cycles of chemotherapya, RTb 78 100
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- ADP 04511 (NCT01343043) (A Pilot Study of Genetically Engineered NY-ESO-1 Specific [c259] T Cells in HLA-A2+ Patients With Synovial Sarcoma): Patients with unresectable, metastatic, or recurrent synovial sarcoma undergo apheresis. Cells are shipped to a central laboratory where they undergo NY-ESO-1 transduction, expansion, and cryopreservation. Patients undergo lymphodepletion with fludarabine and cyclophosphamide, followed by an infusion of autologous transfected cells. Eligibility is restricted to patients with HLA type A2+, age older than 4 years, and weight greater than 18 kg.
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with alveolar soft part sarcoma, clear cell sarcoma of soft tissue, epithelioid sarcoma, extraskeletal myxoid chondrosarcoma, PEComa, and synovial sarcoma are eligible for this trial.
- NCT02601937 (A Phase 1 Study of the EZH2 Inhibitor Tazemetostat in Pediatric Subjects With Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma): Patients with INI1-negative tumors are eligible for targeted treatment with an EZH2 inhibitor. This is a phase I, open-label, dose-escalation, and dose-expansion study with a twice-daily oral dose of tazemetostat.
- ADVL1522 (NCT02452554) (Lorvotuzumab Mertansine in Treating Younger Patients with Relapsed or Refractory Wilms Tumor, Rhabdomyosarcoma, Neuroblastoma, Pleuropulmonary Blastoma, Malignant Peripheral Nerve Sheath Tumor, or Synovial Sarcoma): This is a phase II study of IMGN901 (lorvotuzumab mertansine) in children with relapsed or refractory Wilms tumor, rhabdomyosarcoma, neuroblastoma, pleuropulmonary blastoma, malignant peripheral nerve sheath tumor, and synovial sarcoma. This trial is studying the effects of IMGN901, an antibody-drug conjugate that links a potent antimitotic to antibodies that target CD56.
Undifferentiated/unclassified sarcoma
Patients with undifferentiated soft tissue sarcoma had been eligible for participation in rhabdomyosarcoma trials coordinated by the Intergroup Rhabdomyosarcoma Study Group and the COG from 1972 to 2006. The rationale was the observation that patients with undifferentiated soft tissue sarcoma had similar sites of disease and outcome as those with alveolar rhabdomyosarcoma. Therapeutic trials for adults with soft tissue sarcoma include patients with undifferentiated soft tissue sarcoma and other histologies, which are treated similarly, using ifosfamide and doxorubicin, and sometimes with other chemotherapy agents, surgery, and radiation therapy.
In the COG ARST0332 (NCT00346164) trial, patients with high-grade undifferentiated sarcoma were treated with an ifosfamide and doxorubicin-based regimen and were treated with rhabdomyosarcoma-directed therapies in previous Intergroup Rhabdomyosarcoma Study Group studies with a 5-year survival estimate for nonmetastatic patients of 72%.[240][Level of evidence: 3iiA] Currently, these patients are eligible for the COG open ARST1321 (NCT02180867) trial for patients with nonrhabdomyosarcomatous soft tissue sarcoma.
Undifferentiated pleomorphic sarcoma/malignant fibrous histiocytoma (high-grade)
At one time, malignant fibrous histiocytoma was the single most common histotype among adults with soft tissue sarcomas. Since it was first recognized in the early 1960s, malignant fibrous histiocytoma has been plagued by controversy in terms of both its histogenesis and its validity as a clinicopathologic entity. The latest WHO classification no longer includes malignant fibrous histiocytoma as a distinct diagnostic category but rather as a subtype of an undifferentiated pleomorphic sarcoma.[241]
This entity accounts for 2% to 6% of all childhood soft tissue sarcomas.[242] These tumors can arise in previously irradiated sites or as a second malignancy in patients with retinoblastoma.
These tumors occur mainly in the second decade of life. In a series of ten patients, the median age was 10 years and the tumor was most commonly located in the extremities. In this series, all tumors were localized and five of nine (for whom follow-up was available) were alive and in first remission.[242] In another series of 17 pediatric patients with malignant fibrous histiocytoma, the median age at diagnosis was 5 years and the extremities were involved in eight cases.[243] All patients with metastatic disease died and two patients experienced a clinical response to a doxorubicin-based regimen.
(Refer to the PDQ summary on Osteosarcoma and Malignant Fibrous Histiocytoma of Bone Treatment for more information about the treatment of malignant fibrous histiocytoma of bone.)
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with undifferentiated epithelial sarcoma, undifferentiated pleomorphic sarcoma, undifferentiated round cell sarcoma, and undifferentiated spindle cell sarcoma are eligible for this trial.
Vascular Tumors
Vascular tumors vary from hemangiomas, which are always considered benign, to angiosarcomas, which are highly malignant.[244] Vascular tumors include the following tumor subtypes:
Angiosarcoma of the soft tissue
Incidence
Angiosarcoma is a rare (accounting for 2% of sarcomas), aggressive, vascular tumor that can arise in any part of the body, but is more common in the soft tissue. Angiosarcoma has an estimated incidence of 2 cases per 1 million; in the United States, it annually affects approximately 600 people who are typically aged 60 to 70 years.[245]
Angiosarcomas are extremely rare in children and it is unclear if the pathophysiology of this tumor is different in the pediatric population. Cases have been reported in neonates and toddlers, with presentation of multiple cutaneous lesions and liver lesions, some of which are GLUT1 positive.[246-249] Most angiosarcomas involve the skin and superficial soft tissue, although the liver, spleen, and lung can be affected; bone is rarely affected.
Risk factors
Established risk factors include vinyl chloride exposure, radiation exposure, and chronic lymphedema from any cause, including Stewart-Treves syndrome.[250]
Pathology and biology
Angiosarcomas are largely aneuploid tumors. The rare cases of angiosarcoma that arise from benign lesions such as hemangiomas have a distinct pathway that needs to be investigated. MYC amplification is seen in radiation-induced angiosarcoma. KDR-VEGFR2 mutations and FLT4-VEGFR3 amplifications have been seen with a frequency of less than 50%.[250]
Histopathologic diagnosis can be very difficult because there can be areas of varied atypia. The common feature is an irregular network of channels in a dissective pattern along dermal collagen bundles. There is varied cellular shape, size, mitosis, endothelial multilayering, and papillary formation. Epithelioid cells can also be present. Necrosis and hemorrhage are common. Tumors stain for factor VIII, CD31, and CD34. Some liver lesions can mimic infantile hemangiomas and have focal GLUT1 positivity. Nomenclature of these liver lesions has been difficult and confusing with use of terminology from 1971 (e.g., type I hemangioendothelioma: infantile hemangioma; type II hemangioendothelioma: low-grade angiosarcoma; type III hemangioendothelioma: high-grade angiosarcoma).[247]
Treatment of angiosarcoma of the soft tissue
Treatment options for angiosarcoma of the soft tissue include the following:
- Surgery (localized disease).
- Radiation therapy (localized cutaneous disease in adults).
- Surgery, chemotherapy, and radiation therapy (metastatic disease).
Localized disease is cured by aggressive surgery. Complete surgical excision appears to be crucial for angiosarcomas and lymphangiosarcomas despite evidence of tumor shrinkage in some patients who were treated with local or systemic therapy.[248,251-253] A review of 222 patients (median age, 62 years; range, age 15–90 years) showed an overall disease-specific survival (DSS) rate of 38% at 5 years. Five-year DSS was 44% in 138 patients with localized, resected tumors but only 16% in 43 patients with metastases at diagnosis.[253] Data on liver transplantation for localized angiosarcoma are limited.[254][Level of evidence: 3iiA]
Localized disease, especially cutaneous angiosarcoma, can be treated with radiation therapy. Most of these reported cases are in adults.[255]
Multimodal treatment with surgery, systemic chemotherapy, and radiation therapy is used for metastatic disease, although it is rarely curative.[256] Disease control is the objective in metastatic angiosarcoma, with published progression-free survival rates between 3 months and 7 months [257] and a median overall survival (OS) rate of 14 months to 18 months.[258] In both adults and children, 5-year OS rates between 20% and 35% are reported.[248,249,259]
In a child diagnosed with angiosarcoma secondary to malignant transformation from infantile hemangioma, response to treatment with bevacizumab, a monoclonal antibody against vascular endothelial growth factor, combined with systemic chemotherapy, has been reported.[246,256] A report of eight cases of liver angiosarcoma in children highlighted the misuse of the term hemangioendothelioma and the importance of early diagnosis and treatment of these tumors.[260]
Biologic agents that inhibit angiogenesis have shown activity in adults with angiosarcoma.[247,259]
Treatment options under clinical evaluation for angiosarcoma of the soft tissue
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery [PAZNTIS]): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with angiosarcoma of the soft tissue are eligible for this trial.
- NCT01532687 (Gemcitabine Hydrochloride With or Without Pazopanib Hydrochloride in Treating Patients With Refractory Soft Tissue Sarcoma): This randomized phase II trial studies how well gemcitabine hydrochloride works with or without pazopanib hydrochloride in treating patients with refractory soft tissue sarcoma.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
Epithelioid hemangioendothelioma
Incidence and outcome
This tumor was first described in soft tissue by Weiss and Enzinger in 1982. Epithelioid hemangioendotheliomas can occur at younger ages, but the peak incidence is in the fourth and fifth decades of life. The tumors can have an indolent or very aggressive course, with overall survival of 73% at 5 years. There are case reports of patients with untreated multiple lesions who have a very benign course compared with other patients who have a very aggressive course. Some pathologists have tried to stratify patients to evaluate risks and adjust treatment, but more research is needed.[261-267]
The presence of effusions, tumor size larger than 3 cm, and a high mitotic index (>3 mitoses/50 high-power fields) have been associated with unfavorable outcomes.[263]
Pathology and biology
A WWTR1-CAMTA1 gene fusion has been found in a large percentage of patients; less commonly, a YAP1-TFE3 gene fusion has been reported.[261] These fusions are not directly targetable with current medicines. Monoclonality has been described in multiple liver lesions, suggesting a metastatic process.
Histologically, these lesions are characterized as epithelioid lesions arranged in nests, strands, and trabecular patterns, with infrequent vascular spaces. Features that may be associated with aggressive clinical behavior include cellular atypia, one or more mitoses per 10 high-power fields, an increased proportion of spindled cells, focal necrosis, and metaplastic bone formation.[263]
The number of pediatric patients reported in the literature is limited.
Clinical presentation and diagnostic evaluation
Common sites of involvement are liver alone (21%), liver plus lung (18%), lung alone (12%), and bone alone (14%).[263,268,269] Clinical presentation depends on site of involvement, as follows:
- Liver: Hepatic nodules have central vascularity on ultrasound, contrast-enhancing lesions by computed tomography, and low T1 signal and moderate T2 signal on magnetic resonance imaging.
- Lung: Pulmonary epithelioid hemangioendothelioma may be an asymptomatic finding on chest x-ray or be associated with pleuritic pain, hemoptysis, anemia, and fibrosis.
- Bone: Bone metastasis may be associated with pathologic fracture. On x-rays, they are well-defined osteolytic lesions and can be multiple or solitary.
- Soft tissue: Thirty percent of soft tissue cases are associated with metastases, and when present, can have a very aggressive course, with limited response to chemotherapy.
- Skin: Cutaneous lesions can be raised and nodular or can be warm red-brown plaques.
Treatment of epithelioid hemangioendothelioma
Treatment options for epithelioid hemangioendothelioma include the following:
- Observation.
- Surgery.
- Immunotherapy.
- Targeted therapy.
- Chemotherapy.
For indolent cases, observation is warranted. For more aggressive cases, multiple medications have been used, including interferon, thalidomide, sorafenib, pazopanib, and sirolimus.[270] The most aggressive cases are treated with angiosarcoma-type chemotherapy. Surgery is used when possible. Liver transplantation has been used with aggressive liver lesions, both with and without metastases.[263,271-274]
Treatment options under clinical evaluation for epithelioid hemangioendothelioma
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- NCT03148275 (Trametinib in Treating Patients with Epithelioid Hemangioendothelioma That Is Metastatic, Locally Advanced, or Cannot Be Removed by Surgery): This is a phase II trial assessing the efficacy of trametinib, with patient-reported outcomes as secondary aims.
- NCT01532687 (Gemcitabine Hydrochloride With or Without Pazopanib Hydrochloride in Treating Patients With Refractory Soft Tissue Sarcoma): This randomized phase II trial studies how well gemcitabine hydrochloride works with or without pazopanib hydrochloride in treating patients with refractory soft tissue sarcoma.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Ferrari A, Casanova M, Collini P, et al.: Adult-type soft tissue sarcomas in pediatric-age patients: experience at the Istituto Nazionale Tumori in Milan. J Clin Oncol 23 (18): 4021-30, 2005. [PUBMED Abstract]
- Stanelle EJ, Christison-Lagay ER, Sidebotham EL, et al.: Prognostic factors and survival in pediatric and adolescent liposarcoma. Sarcoma 2012: 870910, 2012. [PUBMED Abstract]
- Alaggio R, Coffin CM, Weiss SW, et al.: Liposarcomas in young patients: a study of 82 cases occurring in patients younger than 22 years of age. Am J Surg Pathol 33 (5): 645-58, 2009. [PUBMED Abstract]
- Sreekantaiah C, Karakousis CP, Leong SP, et al.: Cytogenetic findings in liposarcoma correlate with histopathologic subtypes. Cancer 69 (10): 2484-95, 1992. [PUBMED Abstract]
- Sugiura H, Takahashi M, Katagiri H, et al.: Additional wide resection of malignant soft tissue tumors. Clin Orthop (394): 201-10, 2002. [PUBMED Abstract]
- Cecchetto G, Guglielmi M, Inserra A, et al.: Primary re-excision: the Italian experience in patients with localized soft-tissue sarcomas. Pediatr Surg Int 17 (7): 532-4, 2001. [PUBMED Abstract]
- Chui CH, Spunt SL, Liu T, et al.: Is reexcision in pediatric nonrhabdomyosarcoma soft tissue sarcoma necessary after an initial unplanned resection? J Pediatr Surg 37 (10): 1424-9, 2002. [PUBMED Abstract]
- Bahig H, Roberge D, Bosch W, et al.: Agreement among RTOG sarcoma radiation oncologists in contouring suspicious peritumoral edema for preoperative radiation therapy of soft tissue sarcoma of the extremity. Int J Radiat Oncol Biol Phys 86 (2): 298-303, 2013. [PUBMED Abstract]
- Baldini EH, Wang D, Haas RL, et al.: Treatment Guidelines for Preoperative Radiation Therapy for Retroperitoneal Sarcoma: Preliminary Consensus of an International Expert Panel. Int J Radiat Oncol Biol Phys 92 (3): 602-12, 2015. [PUBMED Abstract]
- Ferrari A, Casanova M, Spreafico F, et al.: Childhood liposarcoma: a single-institutional twenty-year experience. Pediatr Hematol Oncol 16 (5): 415-21, 1999 Sep-Oct. [PUBMED Abstract]
- Cecchetto G, Alaggio R, Dall'Igna P, et al.: Localized unresectable non-rhabdo soft tissue sarcomas of the extremities in pediatric age: results from the Italian studies. Cancer 104 (9): 2006-12, 2005. [PUBMED Abstract]
- Huh WW, Yuen C, Munsell M, et al.: Liposarcoma in children and young adults: a multi-institutional experience. Pediatr Blood Cancer 57 (7): 1142-6, 2011. [PUBMED Abstract]
- Gronchi A, Bui BN, Bonvalot S, et al.: Phase II clinical trial of neoadjuvant trabectedin in patients with advanced localized myxoid liposarcoma. Ann Oncol 23 (3): 771-6, 2012. [PUBMED Abstract]
- Demetri GD, von Mehren M, Jones RL, et al.: Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. J Clin Oncol 34 (8): 786-93, 2016. [PUBMED Abstract]
- Baruchel S, Pappo A, Krailo M, et al.: A phase 2 trial of trabectedin in children with recurrent rhabdomyosarcoma, Ewing sarcoma and non-rhabdomyosarcoma soft tissue sarcomas: a report from the Children's Oncology Group. Eur J Cancer 48 (4): 579-85, 2012. [PUBMED Abstract]
- Wang L, Motoi T, Khanin R, et al.: Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer 51 (2): 127-39, 2012. [PUBMED Abstract]
- Nyquist KB, Panagopoulos I, Thorsen J, et al.: Whole-transcriptome sequencing identifies novel IRF2BP2-CDX1 fusion gene brought about by translocation t(1;5)(q42;q32) in mesenchymal chondrosarcoma. PLoS One 7 (11): e49705, 2012. [PUBMED Abstract]
- Frezza AM, Cesari M, Baumhoer D, et al.: Mesenchymal chondrosarcoma: prognostic factors and outcome in 113 patients. A European Musculoskeletal Oncology Society study. Eur J Cancer 51 (3): 374-81, 2015. [PUBMED Abstract]
- Dantonello TM, Int-Veen C, Leuschner I, et al.: Mesenchymal chondrosarcoma of soft tissues and bone in children, adolescents, and young adults: experiences of the CWS and COSS study groups. Cancer 112 (11): 2424-31, 2008. [PUBMED Abstract]
- Bishop MW, Somerville JM, Bahrami A, et al.: Mesenchymal Chondrosarcoma in Children and Young Adults: A Single Institution Retrospective Review. Sarcoma 2015: 608279, 2015. [PUBMED Abstract]
- Wodowski K, Hill DA, Pappo AS, et al.: A chemosensitive pediatric extraosseous osteosarcoma: case report and review of the literature. J Pediatr Hematol Oncol 25 (1): 73-7, 2003. [PUBMED Abstract]
- Sordillo PP, Hajdu SI, Magill GB, et al.: Extraosseous osteogenic sarcoma. A review of 48 patients. Cancer 51 (4): 727-34, 1983. [PUBMED Abstract]
- Nieuwenhuis MH, Casparie M, Mathus-Vliegen LM, et al.: A nation-wide study comparing sporadic and familial adenomatous polyposis-related desmoid-type fibromatoses. Int J Cancer 129 (1): 256-61, 2011. [PUBMED Abstract]
- Rossato M, Rigotti M, Grazia M, et al.: Congenital hypertrophy of the retinal pigment epithelium (CHRPE) and familial adenomatous polyposis (FAP). Acta Ophthalmol Scand 74 (4): 338-42, 1996. [PUBMED Abstract]
- Baker RH, Heinemann MH, Miller HH, et al.: Hyperpigmented lesions of the retinal pigment epithelium in familial adenomatous polyposis. Am J Med Genet 31 (2): 427-35, 1988. [PUBMED Abstract]
- Kattentidt Mouravieva AA, Geurts-Giele IR, de Krijger RR, et al.: Identification of Familial Adenomatous Polyposis carriers among children with desmoid tumours. Eur J Cancer 48 (12): 1867-74, 2012. [PUBMED Abstract]
- Wang WL, Nero C, Pappo A, et al.: CTNNB1 genotyping and APC screening in pediatric desmoid tumors: a proposed algorithm. Pediatr Dev Pathol 15 (5): 361-7, 2012 Sep-Oct. [PUBMED Abstract]
- Lewis JJ, Boland PJ, Leung DH, et al.: The enigma of desmoid tumors. Ann Surg 229 (6): 866-72; discussion 872-3, 1999. [PUBMED Abstract]
- Lazar AJ, Tuvin D, Hajibashi S, et al.: Specific mutations in the beta-catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am J Pathol 173 (5): 1518-27, 2008. [PUBMED Abstract]
- Faulkner LB, Hajdu SI, Kher U, et al.: Pediatric desmoid tumor: retrospective analysis of 63 cases. J Clin Oncol 13 (11): 2813-8, 1995. [PUBMED Abstract]
- Merchant NB, Lewis JJ, Woodruff JM, et al.: Extremity and trunk desmoid tumors: a multifactorial analysis of outcome. Cancer 86 (10): 2045-52, 1999. [PUBMED Abstract]
- Honeyman JN, Theilen TM, Knowles MA, et al.: Desmoid fibromatosis in children and adolescents: a conservative approach to management. J Pediatr Surg 48 (1): 62-6, 2013. [PUBMED Abstract]
- Bonvalot S, Ternès N, Fiore M, et al.: Spontaneous regression of primary abdominal wall desmoid tumors: more common than previously thought. Ann Surg Oncol 20 (13): 4096-102, 2013. [PUBMED Abstract]
- Bonvalot S, Eldweny H, Haddad V, et al.: Extra-abdominal primary fibromatosis: Aggressive management could be avoided in a subgroup of patients. Eur J Surg Oncol 34 (4): 462-8, 2008. [PUBMED Abstract]
- Merchant TE, Nguyen D, Walter AW, et al.: Long-term results with radiation therapy for pediatric desmoid tumors. Int J Radiat Oncol Biol Phys 47 (5): 1267-71, 2000. [PUBMED Abstract]
- Zelefsky MJ, Harrison LB, Shiu MH, et al.: Combined surgical resection and iridium 192 implantation for locally advanced and recurrent desmoid tumors. Cancer 67 (2): 380-4, 1991. [PUBMED Abstract]
- Weiss AJ, Lackman RD: Low-dose chemotherapy of desmoid tumors. Cancer 64 (6): 1192-4, 1989. [PUBMED Abstract]
- Klein WA, Miller HH, Anderson M, et al.: The use of indomethacin, sulindac, and tamoxifen for the treatment of desmoid tumors associated with familial polyposis. Cancer 60 (12): 2863-8, 1987. [PUBMED Abstract]
- Soto-Miranda MA, Sandoval JA, Rao B, et al.: Surgical treatment of pediatric desmoid tumors. A 12-year, single-center experience. Ann Surg Oncol 20 (11): 3384-90, 2013. [PUBMED Abstract]
- Skapek SX, Ferguson WS, Granowetter L, et al.: Vinblastine and methotrexate for desmoid fibromatosis in children: results of a Pediatric Oncology Group Phase II Trial. J Clin Oncol 25 (5): 501-6, 2007. [PUBMED Abstract]
- Gandhi MM, Nathan PC, Weitzman S, et al.: Successful treatment of life-threatening generalized infantile myofibromatosis using low-dose chemotherapy. J Pediatr Hematol Oncol 25 (9): 750-4, 2003. [PUBMED Abstract]
- Gega M, Yanagi H, Yoshikawa R, et al.: Successful chemotherapeutic modality of doxorubicin plus dacarbazine for the treatment of desmoid tumors in association with familial adenomatous polyposis. J Clin Oncol 24 (1): 102-5, 2006. [PUBMED Abstract]
- Constantinidou A, Jones RL, Scurr M, et al.: Pegylated liposomal doxorubicin, an effective, well-tolerated treatment for refractory aggressive fibromatosis. Eur J Cancer 45 (17): 2930-4, 2009. [PUBMED Abstract]
- Ananth P, Werger A, Voss S, et al.: Liposomal doxorubicin: Effective treatment for pediatric desmoid fibromatosis. Pediatr Blood Cancer 64 (7): , 2017. [PUBMED Abstract]
- Gounder MM, Lefkowitz RA, Keohan ML, et al.: Activity of Sorafenib against desmoid tumor/deep fibromatosis. Clin Cancer Res 17 (12): 4082-90, 2011. [PUBMED Abstract]
- Heinrich MC, McArthur GA, Demetri GD, et al.: Clinical and molecular studies of the effect of imatinib on advanced aggressive fibromatosis (desmoid tumor). J Clin Oncol 24 (7): 1195-203, 2006. [PUBMED Abstract]
- Chugh R, Wathen JK, Patel SR, et al.: Efficacy of imatinib in aggressive fibromatosis: Results of a phase II multicenter Sarcoma Alliance for Research through Collaboration (SARC) trial. Clin Cancer Res 16 (19): 4884-91, 2010. [PUBMED Abstract]
- Agresta L, Kim H, Turpin BK, et al.: Pazopanib therapy for desmoid tumors in adolescent and young adult patients. Pediatr Blood Cancer : , 2018. [PUBMED Abstract]
- Shang H, Braggio D, Lee YJ, et al.: Targeting the Notch pathway: A potential therapeutic approach for desmoid tumors. Cancer 121 (22): 4088-96, 2015. [PUBMED Abstract]
- Messersmith WA, Shapiro GI, Cleary JM, et al.: A Phase I, dose-finding study in patients with advanced solid malignancies of the oral γ-secretase inhibitor PF-03084014. Clin Cancer Res 21 (1): 60-7, 2015. [PUBMED Abstract]
- Bisogno G, Tagarelli A, Stramare R, et al.: Hydroxyurea treatment can avoid the need for aggressive surgery in pediatric fibromatosis. J Pediatr Hematol Oncol 35 (4): e171-3, 2013. [PUBMED Abstract]
- Meazza C, Casanova M, Trecate G, et al.: Objective response to hydroxyurea in a patient with heavily pre-treated aggressive fibromatosis. Pediatr Blood Cancer 55 (3): 587-8, 2010. [PUBMED Abstract]
- Balamuth NJ, Womer RB: Successful treatment of fibromatosis with hydroxyurea: Analysis of 20 pediatric cases. [Abstract] The Connective Tissue Oncology Society (CTOS) 14th Annual Meeting, 13–15 November 2008, London, United Kingdom A-34852, 2008. Also available online. Last accessed April 02, 2018.
- Meazza C, Bisogno G, Gronchi A, et al.: Aggressive fibromatosis in children and adolescents: the Italian experience. Cancer 116 (1): 233-40, 2010. [PUBMED Abstract]
- Hansmann A, Adolph C, Vogel T, et al.: High-dose tamoxifen and sulindac as first-line treatment for desmoid tumors. Cancer 100 (3): 612-20, 2004. [PUBMED Abstract]
- Skapek SX, Anderson JR, Hill DA, et al.: Safety and efficacy of high-dose tamoxifen and sulindac for desmoid tumor in children: results of a Children's Oncology Group (COG) phase II study. Pediatr Blood Cancer 60 (7): 1108-12, 2013. [PUBMED Abstract]
- Rutenberg MS, Indelicato DJ, Knapik JA, et al.: External-beam radiotherapy for pediatric and young adult desmoid tumors. Pediatr Blood Cancer 57 (3): 435-42, 2011. [PUBMED Abstract]
- Buckley PG, Mantripragada KK, Benetkiewicz M, et al.: A full-coverage, high-resolution human chromosome 22 genomic microarray for clinical and research applications. Hum Mol Genet 11 (25): 3221-9, 2002. [PUBMED Abstract]
- Valdivielso-Ramos M, Torrelo A, Campos M, et al.: Pediatric dermatofibrosarcoma protuberans in Madrid, Spain: multi-institutional outcomes. Pediatr Dermatol 31 (6): 676-82, 2014 Nov-Dec. [PUBMED Abstract]
- Gooskens SL, Oranje AP, van Adrichem LN, et al.: Imatinib mesylate for children with dermatofibrosarcoma protuberans (DFSP). Pediatr Blood Cancer 55 (2): 369-73, 2010. [PUBMED Abstract]
- Rubio GA, Alvarado A, Gerth DJ, et al.: Incidence and Outcomes of Dermatofibrosarcoma Protuberans in the US Pediatric Population. J Craniofac Surg 28 (1): 182-184, 2017. [PUBMED Abstract]
- Meguerditchian AN, Wang J, Lema B, et al.: Wide excision or Mohs micrographic surgery for the treatment of primary dermatofibrosarcoma protuberans. Am J Clin Oncol 33 (3): 300-3, 2010. [PUBMED Abstract]
- Dagan R, Morris CG, Zlotecki RA, et al.: Radiotherapy in the treatment of dermatofibrosarcoma protuberans. Am J Clin Oncol 28 (6): 537-9, 2005. [PUBMED Abstract]
- Sun LM, Wang CJ, Huang CC, et al.: Dermatofibrosarcoma protuberans: treatment results of 35 cases. Radiother Oncol 57 (2): 175-81, 2000. [PUBMED Abstract]
- Price VE, Fletcher JA, Zielenska M, et al.: Imatinib mesylate: an attractive alternative in young children with large, surgically challenging dermatofibrosarcoma protuberans. Pediatr Blood Cancer 44 (5): 511-5, 2005. [PUBMED Abstract]
- McArthur GA, Demetri GD, van Oosterom A, et al.: Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J Clin Oncol 23 (4): 866-73, 2005. [PUBMED Abstract]
- Rutkowski P, Van Glabbeke M, Rankin CJ, et al.: Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials. J Clin Oncol 28 (10): 1772-9, 2010. [PUBMED Abstract]
- Miller SJ, Alam M, Andersen JS, et al.: Dermatofibrosarcoma protuberans. J Natl Compr Canc Netw 10 (3): 312-8, 2012. [PUBMED Abstract]
- Kao YC, Fletcher CDM, Alaggio R, et al.: Recurrent BRAF Gene Fusions in a Subset of Pediatric Spindle Cell Sarcomas: Expanding the Genetic Spectrum of Tumors With Overlapping Features With Infantile Fibrosarcoma. Am J Surg Pathol 42 (1): 28-38, 2018. [PUBMED Abstract]
- Sulkowski JP, Raval MV, Browne M: Margin status and multimodal therapy in infantile fibrosarcoma. Pediatr Surg Int 29 (8): 771-6, 2013. [PUBMED Abstract]
- Orbach D, Rey A, Cecchetto G, et al.: Infantile fibrosarcoma: management based on the European experience. J Clin Oncol 28 (2): 318-23, 2010. [PUBMED Abstract]
- Orbach D, Brennan B, De Paoli A, et al.: Conservative strategy in infantile fibrosarcoma is possible: The European paediatric Soft tissue sarcoma Study Group experience. Eur J Cancer 57: 1-9, 2016. [PUBMED Abstract]
- Spunt SL, Million L, Coffin C: The nonrhabdomyosarcoma soft tissue sarcoma. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 827-54.
- Loh ML, Ahn P, Perez-Atayde AR, et al.: Treatment of infantile fibrosarcoma with chemotherapy and surgery: results from the Dana-Farber Cancer Institute and Children's Hospital, Boston. J Pediatr Hematol Oncol 24 (9): 722-6, 2002. [PUBMED Abstract]
- Akyüz C, Küpeli S, Varan A, et al.: Infantile fibrosarcoma: retrospective analysis of eleven patients. Tumori 97 (2): 166-9, 2011 Mar-Apr. [PUBMED Abstract]
- Gallego S, Pericas N, Barber I, et al.: Infantile fibrosarcoma of the retroperitoneum: a site of unfavorable prognosis? Pediatr Hematol Oncol 28 (5): 451-3, 2011. [PUBMED Abstract]
- Parida L, Fernandez-Pineda I, Uffman JK, et al.: Clinical management of infantile fibrosarcoma: a retrospective single-institution review. Pediatr Surg Int 29 (7): 703-8, 2013. [PUBMED Abstract]
- Mody RJ, Wu YM, Lonigro RJ, et al.: Integrative Clinical Sequencing in the Management of Refractory or Relapsed Cancer in Youth. JAMA 314 (9): 913-25, 2015. [PUBMED Abstract]
- Wong V, Pavlick D, Brennan T, et al.: Evaluation of a Congenital Infantile Fibrosarcoma by Comprehensive Genomic Profiling Reveals an LMNA-NTRK1 Gene Fusion Responsive to Crizotinib. J Natl Cancer Inst 108 (1): , 2016. [PUBMED Abstract]
- Nagasubramanian R, Wei J, Gordon P, et al.: Infantile Fibrosarcoma With NTRK3-ETV6 Fusion Successfully Treated With the Tropomyosin-Related Kinase Inhibitor LOXO-101. Pediatr Blood Cancer 63 (8): 1468-70, 2016. [PUBMED Abstract]
- Yanagisawa R, Noguchi M, Fujita K, et al.: Preoperative Treatment With Pazopanib in a Case of Chemotherapy-Resistant Infantile Fibrosarcoma. Pediatr Blood Cancer 63 (2): 348-51, 2016. [PUBMED Abstract]
- Madden NP, Spicer RD, Allibone EB, et al.: Spontaneous regression of neonatal fibrosarcoma. Br J Cancer Suppl 18: S72-5, 1992. [PUBMED Abstract]
- Kovach SJ, Fischer AC, Katzman PJ, et al.: Inflammatory myofibroblastic tumors. J Surg Oncol 94 (5): 385-91, 2006. [PUBMED Abstract]
- Brodlie M, Barwick SC, Wood KM, et al.: Inflammatory myofibroblastic tumours of the respiratory tract: paediatric case series with varying clinical presentations. J Laryngol Otol 125 (8): 865-8, 2011. [PUBMED Abstract]
- Xiao Y, Zhou S, Ma C, et al.: Radiological and histopathological features of hepatic inflammatory myofibroblastic tumour: analysis of 10 cases. Clin Radiol 68 (11): 1114-20, 2013. [PUBMED Abstract]
- Karnak I, Senocak ME, Ciftci AO, et al.: Inflammatory myofibroblastic tumor in children: diagnosis and treatment. J Pediatr Surg 36 (6): 908-12, 2001. [PUBMED Abstract]
- Collin M, Charles A, Barker A, et al.: Inflammatory myofibroblastic tumour of the bladder in children: a review. J Pediatr Urol 11 (5): 239-45, 2015. [PUBMED Abstract]
- Coffin CM, Hornick JL, Fletcher CD: Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol 31 (4): 509-20, 2007. [PUBMED Abstract]
- Lovly CM, Gupta A, Lipson D, et al.: Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov 4 (8): 889-95, 2014. [PUBMED Abstract]
- Devaney KO, Lafeir DJ, Triantafyllou A, et al.: Inflammatory myofibroblastic tumors of the head and neck: evaluation of clinicopathologic and prognostic features. Eur Arch Otorhinolaryngol 269 (12): 2461-5, 2012. [PUBMED Abstract]
- Mehta B, Mascarenhas L, Zhou S, et al.: Inflammatory myofibroblastic tumors in childhood. Pediatr Hematol Oncol 30 (7): 640-5, 2013. [PUBMED Abstract]
- Favini F, Resti AG, Collini P, et al.: Inflammatory myofibroblastic tumor of the conjunctiva: response to chemotherapy with low-dose methotrexate and vinorelbine. Pediatr Blood Cancer 54 (3): 483-5, 2010. [PUBMED Abstract]
- Doski JJ, Priebe CJ Jr, Driessnack M, et al.: Corticosteroids in the management of unresected plasma cell granuloma (inflammatory pseudotumor) of the lung. J Pediatr Surg 26 (9): 1064-6, 1991. [PUBMED Abstract]
- Diop B, Konate I, Ka S, et al.: Mesenteric myofibroblastic tumor: NSAID therapy after incomplete resection. J Visc Surg 148 (4): e311-4, 2011. [PUBMED Abstract]
- Dalton BG, Thomas PG, Sharp NE, et al.: Inflammatory myofibroblastic tumors in children. J Pediatr Surg 51 (4): 541-4, 2016. [PUBMED Abstract]
- Butrynski JE, D'Adamo DR, Hornick JL, et al.: Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med 363 (18): 1727-33, 2010. [PUBMED Abstract]
- Mossé YP, Lim MS, Voss SD, et al.: Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children's Oncology Group phase 1 consortium study. Lancet Oncol 14 (6): 472-80, 2013. [PUBMED Abstract]
- Gaudichon J, Jeanne-Pasquier C, Deparis M, et al.: Complete and Repeated Response of a Metastatic ALK-rearranged Inflammatory Myofibroblastic Tumor to Crizotinib in a Teenage Girl. J Pediatr Hematol Oncol 38 (4): 308-11, 2016. [PUBMED Abstract]
- Nishio M, Murakami H, Horiike A, et al.: Phase I Study of Ceritinib (LDK378) in Japanese Patients with Advanced, Anaplastic Lymphoma Kinase-Rearranged Non-Small-Cell Lung Cancer or Other Tumors. J Thorac Oncol 10 (7): 1058-66, 2015. [PUBMED Abstract]
- Mossé YP, Voss SD, Lim MS, et al.: Targeting ALK With Crizotinib in Pediatric Anaplastic Large Cell Lymphoma and Inflammatory Myofibroblastic Tumor: A Children's Oncology Group Study. J Clin Oncol 35 (28): 3215-3221, 2017. [PUBMED Abstract]
- Evans HL: Low-grade fibromyxoid sarcoma: a clinicopathologic study of 33 cases with long-term follow-up. Am J Surg Pathol 35 (10): 1450-62, 2011. [PUBMED Abstract]
- Guillou L, Benhattar J, Gengler C, et al.: Translocation-positive low-grade fibromyxoid sarcoma: clinicopathologic and molecular analysis of a series expanding the morphologic spectrum and suggesting potential relationship to sclerosing epithelioid fibrosarcoma: a study from the French Sarcoma Group. Am J Surg Pathol 31 (9): 1387-402, 2007. [PUBMED Abstract]
- O'Sullivan MJ, Sirgi KE, Dehner LP: Low-grade fibrosarcoma (hyalinizing spindle cell tumor with giant rosettes) with pulmonary metastases at presentation: case report and review of the literature. Int J Surg Pathol 10 (3): 211-6, 2002. [PUBMED Abstract]
- Folpe AL, Lane KL, Paull G, et al.: Low-grade fibromyxoid sarcoma and hyalinizing spindle cell tumor with giant rosettes: a clinicopathologic study of 73 cases supporting their identity and assessing the impact of high-grade areas. Am J Surg Pathol 24 (10): 1353-60, 2000. [PUBMED Abstract]
- Sargar K, Kao SC, Spunt SL, et al.: MRI and CT of Low-Grade Fibromyxoid Sarcoma in Children: A Report From Children's Oncology Group Study ARST0332. AJR Am J Roentgenol 205 (2): 414-20, 2015. [PUBMED Abstract]
- Maretty-Nielsen K, Baerentzen S, Keller J, et al.: Low-Grade Fibromyxoid Sarcoma: Incidence, Treatment Strategy of Metastases, and Clinical Significance of the FUS Gene. Sarcoma 2013: 256280, 2013. [PUBMED Abstract]
- Prieto-Granada C, Zhang L, Chen HW, et al.: A genetic dichotomy between pure sclerosing epithelioid fibrosarcoma (SEF) and hybrid SEF/low-grade fibromyxoid sarcoma: a pathologic and molecular study of 18 cases. Genes Chromosomes Cancer 54 (1): 28-38, 2015. [PUBMED Abstract]
- Pollock BH, Jenson HB, Leach CT, et al.: Risk factors for pediatric human immunodeficiency virus-related malignancy. JAMA 289 (18): 2393-9, 2003. [PUBMED Abstract]
- Kleinerman RA, Tucker MA, Abramson DH, et al.: Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst 99 (1): 24-31, 2007. [PUBMED Abstract]
- Samuels BL, Chawla S, Patel S, et al.: Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol 24 (6): 1703-9, 2013. [PUBMED Abstract]
- Enzinger FM, Zhang RY: Plexiform fibrohistiocytic tumor presenting in children and young adults. An analysis of 65 cases. Am J Surg Pathol 12 (11): 818-26, 1988. [PUBMED Abstract]
- Black J, Coffin CM, Dehner LP: Fibrohistiocytic tumors and related neoplasms in children and adolescents. Pediatr Dev Pathol 15 (1 Suppl): 181-210, 2012. [PUBMED Abstract]
- Moosavi C, Jha P, Fanburg-Smith JC: An update on plexiform fibrohistiocytic tumor and addition of 66 new cases from the Armed Forces Institute of Pathology, in honor of Franz M. Enzinger, MD. Ann Diagn Pathol 11 (5): 313-9, 2007. [PUBMED Abstract]
- Billings SD, Folpe AL: Cutaneous and subcutaneous fibrohistiocytic tumors of intermediate malignancy: an update. Am J Dermatopathol 26 (2): 141-55, 2004. [PUBMED Abstract]
- Remstein ED, Arndt CA, Nascimento AG: Plexiform fibrohistiocytic tumor: clinicopathologic analysis of 22 cases. Am J Surg Pathol 23 (6): 662-70, 1999. [PUBMED Abstract]
- Salomao DR, Nascimento AG: Plexiform fibrohistiocytic tumor with systemic metastases: a case report. Am J Surg Pathol 21 (4): 469-76, 1997. [PUBMED Abstract]
- Redlich GC, Montgomery KD, Allgood GA, et al.: Plexiform fibrohistiocytic tumor with a clonal cytogenetic anomaly. Cancer Genet Cytogenet 108 (2): 141-3, 1999. [PUBMED Abstract]
- Luzar B, Calonje E: Cutaneous fibrohistiocytic tumours - an update. Histopathology 56 (1): 148-65, 2010. [PUBMED Abstract]
- Dantonello TM, Leuschner I, Vokuhl C, et al.: Malignant ectomesenchymoma in children and adolescents: report from the Cooperative Weichteilsarkom Studiengruppe (CWS). Pediatr Blood Cancer 60 (2): 224-9, 2013. [PUBMED Abstract]
- Carli M, Ferrari A, Mattke A, et al.: Pediatric malignant peripheral nerve sheath tumor: the Italian and German soft tissue sarcoma cooperative group. J Clin Oncol 23 (33): 8422-30, 2005. [PUBMED Abstract]
- Zhang M, Wang Y, Jones S, et al.: Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet 46 (11): 1170-2, 2014. [PUBMED Abstract]
- Hagel C, Zils U, Peiper M, et al.: Histopathology and clinical outcome of NF1-associated vs. sporadic malignant peripheral nerve sheath tumors. J Neurooncol 82 (2): 187-92, 2007. [PUBMED Abstract]
- Zou C, Smith KD, Liu J, et al.: Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg 249 (6): 1014-22, 2009. [PUBMED Abstract]
- Okada K, Hasegawa T, Tajino T, et al.: Clinical relevance of pathological grades of malignant peripheral nerve sheath tumor: a multi-institution TMTS study of 56 cases in Northern Japan. Ann Surg Oncol 14 (2): 597-604, 2007. [PUBMED Abstract]
- Amirian ES, Goodman JC, New P, et al.: Pediatric and adult malignant peripheral nerve sheath tumors: an analysis of data from the surveillance, epidemiology, and end results program. J Neurooncol 116 (3): 609-16, 2014. [PUBMED Abstract]
- Valentin T, Le Cesne A, Ray-Coquard I, et al.: Management and prognosis of malignant peripheral nerve sheath tumors: The experience of the French Sarcoma Group (GSF-GETO). Eur J Cancer 56: 77-84, 2016. [PUBMED Abstract]
- Bergamaschi L, Bisogno G, Manzitti C, et al.: Salvage rates and prognostic factors after relapse in children and adolescents with malignant peripheral nerve sheath tumors. Pediatr Blood Cancer 65 (2): , 2018. [PUBMED Abstract]
- Ferrari A, Bisogno G, Macaluso A, et al.: Soft-tissue sarcomas in children and adolescents with neurofibromatosis type 1. Cancer 109 (7): 1406-12, 2007. [PUBMED Abstract]
- Okur FV, Oguz A, Karadeniz C, et al.: Malignant triton tumor of the pelvis in a 2-year-old boy. J Pediatr Hematol Oncol 28 (3): 173-6, 2006. [PUBMED Abstract]
- Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 4th ed. St. Louis, Mo: Mosby, 2001.
- Fernandez-Pineda I, Parida L, Jenkins JJ, et al.: Childhood hemangiopericytoma: review of St Jude Children's Research Hospital. J Pediatr Hematol Oncol 33 (5): 356-9, 2011. [PUBMED Abstract]
- Rodriguez-Galindo C, Ramsey K, Jenkins JJ, et al.: Hemangiopericytoma in children and infants. Cancer 88 (1): 198-204, 2000. [PUBMED Abstract]
- Ferrari A, Casanova M, Bisogno G, et al.: Hemangiopericytoma in pediatric ages: a report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Cancer 92 (10): 2692-8, 2001. [PUBMED Abstract]
- Bien E, Stachowicz-Stencel T, Godzinski J, et al.: Retrospective multi-institutional study on hemangiopericytoma in Polish children. Pediatr Int 51 (1): 19-24, 2009. [PUBMED Abstract]
- Wiswell TE, Davis J, Cunningham BE, et al.: Infantile myofibromatosis: the most common fibrous tumor of infancy. J Pediatr Surg 23 (4): 315-8, 1988. [PUBMED Abstract]
- Chung EB, Enzinger FM: Infantile myofibromatosis. Cancer 48 (8): 1807-18, 1981. [PUBMED Abstract]
- Modi N: Congenital generalised fibromatosis. Arch Dis Child 57 (11): 881-2, 1982. [PUBMED Abstract]
- Levine E, Fréneaux P, Schleiermacher G, et al.: Risk-adapted therapy for infantile myofibromatosis in children. Pediatr Blood Cancer 59 (1): 115-20, 2012. [PUBMED Abstract]
- Larralde M, Hoffner MV, Boggio P, et al.: Infantile myofibromatosis: report of nine patients. Pediatr Dermatol 27 (1): 29-33, 2010 Jan-Feb. [PUBMED Abstract]
- Cheung YH, Gayden T, Campeau PM, et al.: A recurrent PDGFRB mutation causes familial infantile myofibromatosis. Am J Hum Genet 92 (6): 996-1000, 2013. [PUBMED Abstract]
- Gopal M, Chahal G, Al-Rifai Z, et al.: Infantile myofibromatosis. Pediatr Surg Int 24 (3): 287-91, 2008. [PUBMED Abstract]
- Weaver MS, Navid F, Huppmann A, et al.: Vincristine and Dactinomycin in Infantile Myofibromatosis With a Review of Treatment Options. J Pediatr Hematol Oncol 37 (3): 237-41, 2015. [PUBMED Abstract]
- Orbach D, Brennan B, Casanova M, et al.: Paediatric and adolescent alveolar soft part sarcoma: A joint series from European cooperative groups. Pediatr Blood Cancer 60 (11): 1826-32, 2013. [PUBMED Abstract]
- Ferrari A, Sultan I, Huang TT, et al.: Soft tissue sarcoma across the age spectrum: a population-based study from the Surveillance Epidemiology and End Results database. Pediatr Blood Cancer 57 (6): 943-9, 2011. [PUBMED Abstract]
- Wang HW, Qin XJ, Yang WJ, et al.: Alveolar soft part sarcoma of the oral and maxillofacial region: clinical analysis in a series of 18 patients. Oral Surg Oral Med Oral Pathol Oral Radiol 119 (4): 396-401, 2015. [PUBMED Abstract]
- Kayton ML, Meyers P, Wexler LH, et al.: Clinical presentation, treatment, and outcome of alveolar soft part sarcoma in children, adolescents, and young adults. J Pediatr Surg 41 (1): 187-93, 2006. [PUBMED Abstract]
- Ladanyi M, Lui MY, Antonescu CR, et al.: The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene 20 (1): 48-57, 2001. [PUBMED Abstract]
- Williams A, Bartle G, Sumathi VP, et al.: Detection of ASPL/TFE3 fusion transcripts and the TFE3 antigen in formalin-fixed, paraffin-embedded tissue in a series of 18 cases of alveolar soft part sarcoma: useful diagnostic tools in cases with unusual histological features. Virchows Arch 458 (3): 291-300, 2011. [PUBMED Abstract]
- Lieberman PH, Brennan MF, Kimmel M, et al.: Alveolar soft-part sarcoma. A clinico-pathologic study of half a century. Cancer 63 (1): 1-13, 1989. [PUBMED Abstract]
- Casanova M, Ferrari A, Bisogno G, et al.: Alveolar soft part sarcoma in children and adolescents: A report from the Soft-Tissue Sarcoma Italian Cooperative Group. Ann Oncol 11 (11): 1445-9, 2000. [PUBMED Abstract]
- Pennacchioli E, Fiore M, Collini P, et al.: Alveolar soft part sarcoma: clinical presentation, treatment, and outcome in a series of 33 patients at a single institution. Ann Surg Oncol 17 (12): 3229-33, 2010. [PUBMED Abstract]
- Flores RJ, Harrison DJ, Federman NC, et al.: Alveolar soft part sarcoma in children and young adults: A report of 69 cases. Pediatr Blood Cancer : , 2018. [PUBMED Abstract]
- Roozendaal KJ, de Valk B, ten Velden JJ, et al.: Alveolar soft-part sarcoma responding to interferon alpha-2b. Br J Cancer 89 (2): 243-5, 2003. [PUBMED Abstract]
- Conde N, Cruz O, Albert A, et al.: Antiangiogenic treatment as a pre-operative management of alveolar soft-part sarcoma. Pediatr Blood Cancer 57 (6): 1071-3, 2011. [PUBMED Abstract]
- Stacchiotti S, Negri T, Zaffaroni N, et al.: Sunitinib in advanced alveolar soft part sarcoma: evidence of a direct antitumor effect. Ann Oncol 22 (7): 1682-90, 2011. [PUBMED Abstract]
- Kummar S, Allen D, Monks A, et al.: Cediranib for metastatic alveolar soft part sarcoma. J Clin Oncol 31 (18): 2296-302, 2013. [PUBMED Abstract]
- Coindre JM, Hostein I, Terrier P, et al.: Diagnosis of clear cell sarcoma by real-time reverse transcriptase-polymerase chain reaction analysis of paraffin embedded tissues: clinicopathologic and molecular analysis of 44 patients from the French sarcoma group. Cancer 107 (5): 1055-64, 2006. [PUBMED Abstract]
- Meis-Kindblom JM: Clear cell sarcoma of tendons and aponeuroses: a historical perspective and tribute to the man behind the entity. Adv Anat Pathol 13 (6): 286-92, 2006. [PUBMED Abstract]
- Dim DC, Cooley LD, Miranda RN: Clear cell sarcoma of tendons and aponeuroses: a review. Arch Pathol Lab Med 131 (1): 152-6, 2007. [PUBMED Abstract]
- Blazer DG 3rd, Lazar AJ, Xing Y, et al.: Clinical outcomes of molecularly confirmed clear cell sarcoma from a single institution and in comparison with data from the Surveillance, Epidemiology, and End Results registry. Cancer 115 (13): 2971-9, 2009. [PUBMED Abstract]
- Fujimura Y, Siddique H, Lee L, et al.: EWS-ATF-1 chimeric protein in soft tissue clear cell sarcoma associates with CREB-binding protein and interferes with p53-mediated trans-activation function. Oncogene 20 (46): 6653-9, 2001. [PUBMED Abstract]
- Ferrari A, Casanova M, Bisogno G, et al.: Clear cell sarcoma of tendons and aponeuroses in pediatric patients: a report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Cancer 94 (12): 3269-76, 2002. [PUBMED Abstract]
- Karita M, Tsuchiya H, Yamamoto N, et al.: Caffeine-potentiated chemotherapy for clear cell sarcoma: a report of five cases. Int J Clin Oncol 18 (1): 33-7, 2013. [PUBMED Abstract]
- Leuschner I, Radig K, Harms D: Desmoplastic small round cell tumor. Semin Diagn Pathol 13 (3): 204-12, 1996. [PUBMED Abstract]
- Kushner BH, LaQuaglia MP, Wollner N, et al.: Desmoplastic small round-cell tumor: prolonged progression-free survival with aggressive multimodality therapy. J Clin Oncol 14 (5): 1526-31, 1996. [PUBMED Abstract]
- Saab R, Khoury JD, Krasin M, et al.: Desmoplastic small round cell tumor in childhood: the St. Jude Children's Research Hospital experience. Pediatr Blood Cancer 49 (3): 274-9, 2007. [PUBMED Abstract]
- Wang LL, Perlman EJ, Vujanic GM, et al.: Desmoplastic small round cell tumor of the kidney in childhood. Am J Surg Pathol 31 (4): 576-84, 2007. [PUBMED Abstract]
- Arora VC, Price AP, Fleming S, et al.: Characteristic imaging features of desmoplastic small round cell tumour. Pediatr Radiol 43 (1): 93-102, 2013. [PUBMED Abstract]
- Gerald WL, Ladanyi M, de Alava E, et al.: Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): desmoplastic small round-cell tumor and its variants. J Clin Oncol 16 (9): 3028-36, 1998. [PUBMED Abstract]
- Lal DR, Su WT, Wolden SL, et al.: Results of multimodal treatment for desmoplastic small round cell tumors. J Pediatr Surg 40 (1): 251-5, 2005. [PUBMED Abstract]
- Philippe-Chomette P, Kabbara N, Andre N, et al.: Desmoplastic small round cell tumors with EWS-WT1 fusion transcript in children and young adults. Pediatr Blood Cancer 58 (6): 891-7, 2012. [PUBMED Abstract]
- Sedig L, Geiger J, Mody R, et al.: Paratesticular desmoplastic small round cell tumors: A case report and review of the literature. Pediatr Blood Cancer 64 (12): , 2017. [PUBMED Abstract]
- Schwarz RE, Gerald WL, Kushner BH, et al.: Desmoplastic small round cell tumors: prognostic indicators and results of surgical management. Ann Surg Oncol 5 (5): 416-22, 1998 Jul-Aug. [PUBMED Abstract]
- Goodman KA, Wolden SL, La Quaglia MP, et al.: Whole abdominopelvic radiotherapy for desmoplastic small round-cell tumor. Int J Radiat Oncol Biol Phys 54 (1): 170-6, 2002. [PUBMED Abstract]
- Osborne EM, Briere TM, Hayes-Jordan A, et al.: Survival and toxicity following sequential multimodality treatment including whole abdominopelvic radiotherapy for patients with desmoplastic small round cell tumor. Radiother Oncol 119 (1): 40-4, 2016. [PUBMED Abstract]
- Atallah V, Honore C, Orbach D, et al.: Role of Adjuvant Radiation Therapy After Surgery for Abdominal Desmoplastic Small Round Cell Tumors. Int J Radiat Oncol Biol Phys 95 (4): 1244-53, 2016. [PUBMED Abstract]
- Tarek N, Hayes-Jordan A, Salvador L, et al.: Recurrent desmoplastic small round cell tumor responding to an mTOR inhibitor containing regimen. Pediatr Blood Cancer 65 (1): , 2018. [PUBMED Abstract]
- Cook RJ, Wang Z, Arora M, et al.: Clinical outcomes of patients with desmoplastic small round cell tumor of the peritoneum undergoing autologous HCT: a CIBMTR retrospective analysis. Bone Marrow Transplant 47 (11): 1455-8, 2012. [PUBMED Abstract]
- Chbani L, Guillou L, Terrier P, et al.: Epithelioid sarcoma: a clinicopathologic and immunohistochemical analysis of 106 cases from the French sarcoma group. Am J Clin Pathol 131 (2): 222-7, 2009. [PUBMED Abstract]
- Hornick JL, Dal Cin P, Fletcher CD: Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol 33 (4): 542-50, 2009. [PUBMED Abstract]
- Knutson SK, Warholic NM, Wigle TJ, et al.: Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 110 (19): 7922-7, 2013. [PUBMED Abstract]
- Guzzetta AA, Montgomery EA, Lyu H, et al.: Epithelioid sarcoma: one institution's experience with a rare sarcoma. J Surg Res 177 (1): 116-22, 2012. [PUBMED Abstract]
- Casanova M, Ferrari A, Collini P, et al.: Epithelioid sarcoma in children and adolescents: a report from the Italian Soft Tissue Sarcoma Committee. Cancer 106 (3): 708-17, 2006. [PUBMED Abstract]
- Kodet R, Newton WA Jr, Sachs N, et al.: Rhabdoid tumors of soft tissues: a clinicopathologic study of 26 cases enrolled on the Intergroup Rhabdomyosarcoma Study. Hum Pathol 22 (7): 674-84, 1991. [PUBMED Abstract]
- Biegel JA, Zhou JY, Rorke LB, et al.: Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 59 (1): 74-9, 1999. [PUBMED Abstract]
- Eaton KW, Tooke LS, Wainwright LM, et al.: Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56 (1): 7-15, 2011. [PUBMED Abstract]
- Lee RS, Stewart C, Carter SL, et al.: A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest 122 (8): 2983-8, 2012. [PUBMED Abstract]
- Sultan I, Qaddoumi I, Rodríguez-Galindo C, et al.: Age, stage, and radiotherapy, but not primary tumor site, affects the outcome of patients with malignant rhabdoid tumors. Pediatr Blood Cancer 54 (1): 35-40, 2010. [PUBMED Abstract]
- Puri DR, Meyers PA, Kraus DH, et al.: Radiotherapy in the multimodal treatment of extrarenal extracranial malignant rhabdoid tumors. Pediatr Blood Cancer 50 (1): 167-9, 2008. [PUBMED Abstract]
- Madigan CE, Armenian SH, Malogolowkin MH, et al.: Extracranial malignant rhabdoid tumors in childhood: the Childrens Hospital Los Angeles experience. Cancer 110 (9): 2061-6, 2007. [PUBMED Abstract]
- Bourdeaut F, Fréneaux P, Thuille B, et al.: Extra-renal non-cerebral rhabdoid tumours. Pediatr Blood Cancer 51 (3): 363-8, 2008. [PUBMED Abstract]
- Wetmore C, Boyett J, Li S, et al.: Alisertib is active as single agent in recurrent atypical teratoid rhabdoid tumors in 4 children. Neuro Oncol 17 (6): 882-8, 2015. [PUBMED Abstract]
- Tsuneyoshi M, Enjoji M, Iwasaki H, et al.: Extraskeletal myxoid chondrosarcoma--a clinicopathologic and electron microscopic study. Acta Pathol Jpn 31 (3): 439-47, 1981. [PUBMED Abstract]
- Hachitanda Y, Tsuneyoshi M, Daimaru Y, et al.: Extraskeletal myxoid chondrosarcoma in young children. Cancer 61 (12): 2521-6, 1988. [PUBMED Abstract]
- Hisaoka M, Ishida T, Imamura T, et al.: TFG is a novel fusion partner of NOR1 in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer 40 (4): 325-8, 2004. [PUBMED Abstract]
- Enzinger FM, Shiraki M: Extraskeletal myxoid chondrosarcoma. An analysis of 34 cases. Hum Pathol 3 (3): 421-35, 1972. [PUBMED Abstract]
- McGrory JE, Rock MG, Nascimento AG, et al.: Extraskeletal myxoid chondrosarcoma. Clin Orthop Relat Res (382): 185-90, 2001. [PUBMED Abstract]
- Drilon AD, Popat S, Bhuchar G, et al.: Extraskeletal myxoid chondrosarcoma: a retrospective review from 2 referral centers emphasizing long-term outcomes with surgery and chemotherapy. Cancer 113 (12): 3364-71, 2008. [PUBMED Abstract]
- Stacchiotti S, Pantaleo MA, Astolfi A, et al.: Activity of sunitinib in extraskeletal myxoid chondrosarcoma. Eur J Cancer 50 (9): 1657-64, 2014. [PUBMED Abstract]
- Martignoni G, Pea M, Reghellin D, et al.: Molecular pathology of lymphangioleiomyomatosis and other perivascular epithelioid cell tumors. Arch Pathol Lab Med 134 (1): 33-40, 2010. [PUBMED Abstract]
- Bissler JJ, McCormack FX, Young LR, et al.: Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med 358 (2): 140-51, 2008. [PUBMED Abstract]
- Davies DM, Johnson SR, Tattersfield AE, et al.: Sirolimus therapy in tuberous sclerosis or sporadic lymphangioleiomyomatosis. N Engl J Med 358 (2): 200-3, 2008. [PUBMED Abstract]
- Agaram NP, Sung YS, Zhang L, et al.: Dichotomy of Genetic Abnormalities in PEComas With Therapeutic Implications. Am J Surg Pathol 39 (6): 813-25, 2015. [PUBMED Abstract]
- Folpe A, Inwards C, eds.: Bone and Soft Tissue Pathology: A Volume in the Foundations in Diagnostic Pathology. Philadelphia, Pa: WB Saunders Co, 2010.
- Armah HB, Parwani AV: Perivascular epithelioid cell tumor. Arch Pathol Lab Med 133 (4): 648-54, 2009. [PUBMED Abstract]
- Alaggio R, Cecchetto G, Martignoni G, et al.: Malignant perivascular epithelioid cell tumor in children: description of a case and review of the literature. J Pediatr Surg 47 (6): e31-40, 2012. [PUBMED Abstract]
- Wagner AJ, Malinowska-Kolodziej I, Morgan JA, et al.: Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: targeting the pathogenic activation of mTORC1 in tumors. J Clin Oncol 28 (5): 835-40, 2010. [PUBMED Abstract]
- Sultan I, Rodriguez-Galindo C, Saab R, et al.: Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005: an analysis of 1268 patients. Cancer 115 (15): 3537-47, 2009. [PUBMED Abstract]
- Wang JG, Li NN: Primary cardiac synovial sarcoma. Ann Thorac Surg 95 (6): 2202-9, 2013. [PUBMED Abstract]
- Pappo AS, Fontanesi J, Luo X, et al.: Synovial sarcoma in children and adolescents: the St Jude Children's Research Hospital experience. J Clin Oncol 12 (11): 2360-6, 1994. [PUBMED Abstract]
- Ferrari A, De Salvo GL, Oberlin O, et al.: Synovial sarcoma in children and adolescents: a critical reappraisal of staging investigations in relation to the rate of metastatic involvement at diagnosis. Eur J Cancer 48 (9): 1370-5, 2012. [PUBMED Abstract]
- van de Rijn M, Barr FG, Collins MH, et al.: Absence of SYT-SSX fusion products in soft tissue tumors other than synovial sarcoma. Am J Clin Pathol 112 (1): 43-9, 1999. [PUBMED Abstract]
- Krsková L, Sumerauer D, Stejskalová E, et al.: A novel variant of SYT-SSX1 fusion gene in a case of spindle cell synovial sarcoma. Diagn Mol Pathol 16 (3): 179-83, 2007. [PUBMED Abstract]
- Su L, Sampaio AV, Jones KB, et al.: Deconstruction of the SS18-SSX fusion oncoprotein complex: insights into disease etiology and therapeutics. Cancer Cell 21 (3): 333-47, 2012. [PUBMED Abstract]
- Arnold MA, Arnold CA, Li G, et al.: A unique pattern of INI1 immunohistochemistry distinguishes synovial sarcoma from its histologic mimics. Hum Pathol 44 (5): 881-7, 2013. [PUBMED Abstract]
- Vlenterie M, Ho VK, Kaal SE, et al.: Age as an independent prognostic factor for survival of localised synovial sarcoma patients. Br J Cancer 113 (11): 1602-6, 2015. [PUBMED Abstract]
- Okcu MF, Munsell M, Treuner J, et al.: Synovial sarcoma of childhood and adolescence: a multicenter, multivariate analysis of outcome. J Clin Oncol 21 (8): 1602-11, 2003. [PUBMED Abstract]
- Brecht IB, Ferrari A, Int-Veen C, et al.: Grossly-resected synovial sarcoma treated by the German and Italian Pediatric Soft Tissue Sarcoma Cooperative Groups: discussion on the role of adjuvant therapies. Pediatr Blood Cancer 46 (1): 11-7, 2006. [PUBMED Abstract]
- Stanelle EJ, Christison-Lagay ER, Healey JH, et al.: Pediatric and adolescent synovial sarcoma: multivariate analysis of prognostic factors and survival outcomes. Ann Surg Oncol 20 (1): 73-9, 2013. [PUBMED Abstract]
- Trassard M, Le Doussal V, Hacène K, et al.: Prognostic factors in localized primary synovial sarcoma: a multicenter study of 128 adult patients. J Clin Oncol 19 (2): 525-34, 2001. [PUBMED Abstract]
- Guillou L, Benhattar J, Bonichon F, et al.: Histologic grade, but not SYT-SSX fusion type, is an important prognostic factor in patients with synovial sarcoma: a multicenter, retrospective analysis. J Clin Oncol 22 (20): 4040-50, 2004. [PUBMED Abstract]
- Ferrari A, Gronchi A, Casanova M, et al.: Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer 101 (3): 627-34, 2004. [PUBMED Abstract]
- Lagarde P, Przybyl J, Brulard C, et al.: Chromosome instability accounts for reverse metastatic outcomes of pediatric and adult synovial sarcomas. J Clin Oncol 31 (5): 608-15, 2013. [PUBMED Abstract]
- Stegmaier S, Leuschner I, Poremba C, et al.: The prognostic impact of SYT-SSX fusion type and histological grade in pediatric patients with synovial sarcoma treated according to the CWS (Cooperative Weichteilsarkom Studie) trials. Pediatr Blood Cancer 64 (1): 89-95, 2017. [PUBMED Abstract]
- Scheer M, Dantonello T, Hallmen E, et al.: Primary Metastatic Synovial Sarcoma: Experience of the CWS Study Group. Pediatr Blood Cancer 63 (7): 1198-206, 2016. [PUBMED Abstract]
- Ferrari A, De Salvo GL, Dall'Igna P, et al.: Salvage rates and prognostic factors after relapse in children and adolescents with initially localised synovial sarcoma. Eur J Cancer 48 (18): 3448-55, 2012. [PUBMED Abstract]
- Ferrari A, Chi YY, De Salvo GL, et al.: Surgery alone is sufficient therapy for children and adolescents with low-risk synovial sarcoma: A joint analysis from the European paediatric soft tissue sarcoma Study Group and the Children's Oncology Group. Eur J Cancer 78: 1-6, 2017. [PUBMED Abstract]
- McGrory JE, Pritchard DJ, Arndt CA, et al.: Nonrhabdomyosarcoma soft tissue sarcomas in children. The Mayo Clinic experience. Clin Orthop (374): 247-58, 2000. [PUBMED Abstract]
- Van Glabbeke M, van Oosterom AT, Oosterhuis JW, et al.: Prognostic factors for the outcome of chemotherapy in advanced soft tissue sarcoma: an analysis of 2,185 patients treated with anthracycline-containing first-line regimens--a European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol 17 (1): 150-7, 1999. [PUBMED Abstract]
- Koscielniak E, Harms D, Henze G, et al.: Results of treatment for soft tissue sarcoma in childhood and adolescence: a final report of the German Cooperative Soft Tissue Sarcoma Study CWS-86. J Clin Oncol 17 (12): 3706-19, 1999. [PUBMED Abstract]
- Pappo AS, Devidas M, Jenkins J, et al.: Phase II trial of neoadjuvant vincristine, ifosfamide, and doxorubicin with granulocyte colony-stimulating factor support in children and adolescents with advanced-stage nonrhabdomyosarcomatous soft tissue sarcomas: a Pediatric Oncology Group Study. J Clin Oncol 23 (18): 4031-8, 2005. [PUBMED Abstract]
- Pappo AS, Rao BN, Jenkins JJ, et al.: Metastatic nonrhabdomyosarcomatous soft-tissue sarcomas in children and adolescents: the St. Jude Children's Research Hospital experience. Med Pediatr Oncol 33 (2): 76-82, 1999. [PUBMED Abstract]
- Brennan B, Stevens M, Kelsey A, et al.: Synovial sarcoma in childhood and adolescence: a retrospective series of 77 patients registered by the Children's Cancer and Leukaemia Group between 1991 and 2006. Pediatr Blood Cancer 55 (1): 85-90, 2010. [PUBMED Abstract]
- Ferrari A, Miceli R, Rey A, et al.: Non-metastatic unresected paediatric non-rhabdomyosarcoma soft tissue sarcomas: results of a pooled analysis from United States and European groups. Eur J Cancer 47 (5): 724-31, 2011. [PUBMED Abstract]
- Raney RB: Synovial sarcoma in young people: background, prognostic factors, and therapeutic questions. J Pediatr Hematol Oncol 27 (4): 207-11, 2005. [PUBMED Abstract]
- Orbach D, Mc Dowell H, Rey A, et al.: Sparing strategy does not compromise prognosis in pediatric localized synovial sarcoma: experience of the International Society of Pediatric Oncology, Malignant Mesenchymal Tumors (SIOP-MMT) Working Group. Pediatr Blood Cancer 57 (7): 1130-6, 2011. [PUBMED Abstract]
- Ladenstein R, Treuner J, Koscielniak E, et al.: Synovial sarcoma of childhood and adolescence. Report of the German CWS-81 study. Cancer 71 (11): 3647-55, 1993. [PUBMED Abstract]
- Venkatramani R, Anderson JR, Million L, et al.: Risk-based treatment for synovial sarcoma in patients under 30 years of age: Children’s Oncology Group study ARST0332. [Abstract] J Clin Oncol 33 (15 Suppl): A-10012, 2015. Also available online. Last accessed April 02, 2018.
- Ferrari A, De Salvo GL, Brennan B, et al.: Synovial sarcoma in children and adolescents: the European Pediatric Soft Tissue Sarcoma Study Group prospective trial (EpSSG NRSTS 2005). Ann Oncol 26 (3): 567-72, 2015. [PUBMED Abstract]
- Spunt SL, Million L, Anderson JR, et al.: Risk-based treatment for nonrhabdomyosarcoma soft tissue sarcomas (NRSTS) in patients under 30 years of age: Children’s Oncology Group study ARST0332. [Abstract] J Clin Oncol 32 (Suppl 15): A-10008, 2014. Also available online. Last accessed April 02, 2018.
- Randall RL, Albritton KH, Ferney BJ, et al.: Malignant fibrous histiocytoma of soft tissue: an abandoned diagnosis. Am J Orthop 33 (12): 602-8, 2004. [PUBMED Abstract]
- Alaggio R, Collini P, Randall RL, et al.: Undifferentiated high-grade pleomorphic sarcomas in children: a clinicopathologic study of 10 cases and review of literature. Pediatr Dev Pathol 13 (3): 209-17, 2010 May-Jun. [PUBMED Abstract]
- Daw NC, Billups CA, Pappo AS, et al.: Malignant fibrous histiocytoma and other fibrohistiocytic tumors in pediatric patients: the St. Jude Children's Research Hospital experience. Cancer 97 (11): 2839-47, 2003. [PUBMED Abstract]
- Coffin CM, Dehner LP: Vascular tumors in children and adolescents: a clinicopathologic study of 228 tumors in 222 patients. Pathol Annu 28 Pt 1: 97-120, 1993. [PUBMED Abstract]
- Cioffi A, Reichert S, Antonescu CR, et al.: Angiosarcomas and other sarcomas of endothelial origin. Hematol Oncol Clin North Am 27 (5): 975-88, 2013. [PUBMED Abstract]
- Jeng MR, Fuh B, Blatt J, et al.: Malignant transformation of infantile hemangioma to angiosarcoma: response to chemotherapy with bevacizumab. Pediatr Blood Cancer 61 (11): 2115-7, 2014. [PUBMED Abstract]
- Dehner LP, Ishak KG: Vascular tumors of the liver in infants and children. A study of 30 cases and review of the literature. Arch Pathol 92 (2): 101-11, 1971. [PUBMED Abstract]
- Ferrari A, Casanova M, Bisogno G, et al.: Malignant vascular tumors in children and adolescents: a report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Med Pediatr Oncol 39 (2): 109-14, 2002. [PUBMED Abstract]
- Deyrup AT, Miettinen M, North PE, et al.: Pediatric cutaneous angiosarcomas: a clinicopathologic study of 10 cases. Am J Surg Pathol 35 (1): 70-5, 2011. [PUBMED Abstract]
- Elliott P, Kleinschmidt I: Angiosarcoma of the liver in Great Britain in proximity to vinyl chloride sites. Occup Environ Med 54 (1): 14-8, 1997. [PUBMED Abstract]
- Lezama-del Valle P, Gerald WL, Tsai J, et al.: Malignant vascular tumors in young patients. Cancer 83 (8): 1634-9, 1998. [PUBMED Abstract]
- Fata F, O'Reilly E, Ilson D, et al.: Paclitaxel in the treatment of patients with angiosarcoma of the scalp or face. Cancer 86 (10): 2034-7, 1999. [PUBMED Abstract]
- Lahat G, Dhuka AR, Hallevi H, et al.: Angiosarcoma: clinical and molecular insights. Ann Surg 251 (6): 1098-106, 2010. [PUBMED Abstract]
- Orlando G, Adam R, Mirza D, et al.: Hepatic hemangiosarcoma: an absolute contraindication to liver transplantation--the European Liver Transplant Registry experience. Transplantation 95 (6): 872-7, 2013. [PUBMED Abstract]
- Sanada T, Nakayama H, Irisawa R, et al.: Clinical outcome and dose volume evaluation in patients who undergo brachytherapy for angiosarcoma of the scalp and face. Mol Clin Oncol 6 (3): 334-340, 2017. [PUBMED Abstract]
- Dickson MA, D'Adamo DR, Keohan ML, et al.: Phase II Trial of Gemcitabine and Docetaxel with Bevacizumab in Soft Tissue Sarcoma. Sarcoma 2015: 532478, 2015. [PUBMED Abstract]
- North PE, Waner M, Mizeracki A, et al.: A unique microvascular phenotype shared by juvenile hemangiomas and human placenta. Arch Dermatol 137 (5): 559-70, 2001. [PUBMED Abstract]
- Boye E, Yu Y, Paranya G, et al.: Clonality and altered behavior of endothelial cells from hemangiomas. J Clin Invest 107 (6): 745-52, 2001. [PUBMED Abstract]
- Ravi V, Patel S: Vascular sarcomas. Curr Oncol Rep 15 (4): 347-55, 2013. [PUBMED Abstract]
- Grassia KL, Peterman CM, Iacobas I, et al.: Clinical case series of pediatric hepatic angiosarcoma. Pediatr Blood Cancer 64 (11): , 2017. [PUBMED Abstract]
- Mehrabi A, Kashfi A, Fonouni H, et al.: Primary malignant hepatic epithelioid hemangioendothelioma: a comprehensive review of the literature with emphasis on the surgical therapy. Cancer 107 (9): 2108-21, 2006. [PUBMED Abstract]
- Haro A, Saitoh G, Tamiya S, et al.: Four-year natural clinical course of pulmonary epithelioid hemangioendothelioma without therapy. Thorac Cancer 6 (4): 544-7, 2015. [PUBMED Abstract]
- Sardaro A, Bardoscia L, Petruzzelli MF, et al.: Epithelioid hemangioendothelioma: an overview and update on a rare vascular tumor. Oncol Rev 8 (2): 259, 2014. [PUBMED Abstract]
- Dong K, Wang XX, Feng JL, et al.: Pathological characteristics of liver biopsies in eight patients with hepatic epithelioid hemangioendothelioma. Int J Clin Exp Pathol 8 (9): 11015-23, 2015. [PUBMED Abstract]
- Adams DM, Hammill A: Other vascular tumors. Semin Pediatr Surg 23 (4): 173-7, 2014. [PUBMED Abstract]
- Xiao Y, Wang C, Song Y, et al.: Primary epithelioid hemangioendothelioma of the kidney: the first case report in a child and literature review. Urology 82 (4): 925-7, 2013. [PUBMED Abstract]
- Reich S, Ringe H, Uhlenberg B, et al.: Epithelioid hemangioendothelioma of the lung presenting with pneumonia and heart rhythm disturbances in a teenage girl. J Pediatr Hematol Oncol 32 (4): 274-6, 2010. [PUBMED Abstract]
- Daller JA, Bueno J, Gutierrez J, et al.: Hepatic hemangioendothelioma: clinical experience and management strategy. J Pediatr Surg 34 (1): 98-105; discussion 105-6, 1999. [PUBMED Abstract]
- Ackermann O, Fabre M, Franchi S, et al.: Widening spectrum of liver angiosarcoma in children. J Pediatr Gastroenterol Nutr 53 (6): 615-9, 2011. [PUBMED Abstract]
- Stacchiotti S, Provenzano S, Dagrada G, et al.: Sirolimus in Advanced Epithelioid Hemangioendothelioma: A Retrospective Case-Series Analysis from the Italian Rare Cancer Network Database. Ann Surg Oncol 23 (9): 2735-44, 2016. [PUBMED Abstract]
- Semenisty V, Naroditsky I, Keidar Z, et al.: Pazopanib for metastatic pulmonary epithelioid hemangioendothelioma-a suitable treatment option: case report and review of anti-angiogenic treatment options. BMC Cancer 15: 402, 2015. [PUBMED Abstract]
- Raheja A, Suri A, Singh S, et al.: Multimodality management of a giant skull base hemangioendothelioma of the sphenopetroclival region. J Clin Neurosci 22 (9): 1495-8, 2015. [PUBMED Abstract]
- Ahmad N, Adams DM, Wang J, et al.: Hepatic epithelioid hemangioendothelioma in a patient with hemochromatosis. J Natl Compr Canc Netw 12 (9): 1203-7, 2014. [PUBMED Abstract]
- Otte JB, Zimmerman A: The role of liver transplantation for pediatric epithelioid hemangioendothelioma. Pediatr Transplant 14 (3): 295-7, 2010. [PUBMED Abstract]
Treatment of Metastatic Childhood Soft Tissue Sarcoma
Standard treatment options for metastatic childhood soft tissue sarcoma include the following:
- Combination therapy using chemotherapy, radiation therapy, and surgical resection of pulmonary metastases.
For treatment options, refer to the individual tumor type sections of the summary.
The prognosis for children with metastatic soft tissue sarcomas is poor,[1-6] and these children should receive combined treatment with chemotherapy, radiation therapy, and surgical resection of pulmonary metastases. In a prospective randomized trial, chemotherapy with vincristine, dactinomycin, doxorubicin, and cyclophosphamide, with or without dacarbazine, led to tumor responses in one-third of patients with unresectable or metastatic disease. The estimated 4-year survival rate, however, was poor, with fewer than one-third of children surviving.[6-8]
Pulmonary Metastases
Generally, children with isolated pulmonary metastases should be considered for a surgical procedure in an attempt to resect all gross disease.[9] For patients with multiple or recurrent pulmonary metastases, additional surgical procedures can be performed if the morbidity is deemed acceptable. In a retrospective review, patients with synovial sarcoma and pulmonary metastases for whom it was possible to completely resect all metastatic lung lesions had better survival than did patients for whom it was not possible to achieve complete resections.[9][Level of evidence: 3iiiA] Formal segmentectomy, lobectomy, and mediastinal lymph node dissection are unnecessary.[10]
An alternative approach is focused radiation therapy (fractionated stereotactic radiation therapy), which has been successfully used in adults to control lesions. The estimated 5-year survival rate after thoracotomy for pulmonary metastasectomy has ranged from 10% to 58% in adult studies. Emerging data suggest a similar outcome after the administration of focused radiation therapy.[11]
References
- Demetri GD, Elias AD: Results of single-agent and combination chemotherapy for advanced soft tissue sarcomas. Implications for decision making in the clinic. Hematol Oncol Clin North Am 9 (4): 765-85, 1995. [PUBMED Abstract]
- Elias A, Ryan L, Sulkes A, et al.: Response to mesna, doxorubicin, ifosfamide, and dacarbazine in 108 patients with metastatic or unresectable sarcoma and no prior chemotherapy. J Clin Oncol 7 (9): 1208-16, 1989. [PUBMED Abstract]
- Edmonson JH, Ryan LM, Blum RH, et al.: Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol 11 (7): 1269-75, 1993. [PUBMED Abstract]
- Rao BN: Nonrhabdomyosarcoma in children: prognostic factors influencing survival. Semin Surg Oncol 9 (6): 524-31, 1993 Nov-Dec. [PUBMED Abstract]
- deCou JM, Rao BN, Parham DM, et al.: Malignant peripheral nerve sheath tumors: the St. Jude Children's Research Hospital experience. Ann Surg Oncol 2 (6): 524-9, 1995. [PUBMED Abstract]
- Pappo AS, Rao BN, Jenkins JJ, et al.: Metastatic nonrhabdomyosarcomatous soft-tissue sarcomas in children and adolescents: the St. Jude Children's Research Hospital experience. Med Pediatr Oncol 33 (2): 76-82, 1999. [PUBMED Abstract]
- Pratt CB, Pappo AS, Gieser P, et al.: Role of adjuvant chemotherapy in the treatment of surgically resected pediatric nonrhabdomyosarcomatous soft tissue sarcomas: A Pediatric Oncology Group Study. J Clin Oncol 17 (4): 1219, 1999. [PUBMED Abstract]
- Pratt CB, Maurer HM, Gieser P, et al.: Treatment of unresectable or metastatic pediatric soft tissue sarcomas with surgery, irradiation, and chemotherapy: a Pediatric Oncology Group study. Med Pediatr Oncol 30 (4): 201-9, 1998. [PUBMED Abstract]
- Stanelle EJ, Christison-Lagay ER, Wolden SL, et al.: Pulmonary metastasectomy in pediatric/adolescent patients with synovial sarcoma: an institutional review. J Pediatr Surg 48 (4): 757-63, 2013. [PUBMED Abstract]
- Putnam JB Jr, Roth JA: Surgical treatment for pulmonary metastases from sarcoma. Hematol Oncol Clin North Am 9 (4): 869-87, 1995. [PUBMED Abstract]
- Dhakal S, Corbin KS, Milano MT, et al.: Stereotactic body radiotherapy for pulmonary metastases from soft-tissue sarcomas: excellent local lesion control and improved patient survival. Int J Radiat Oncol Biol Phys 82 (2): 940-5, 2012. [PUBMED Abstract]
Treatment of Progressive/Recurrent Childhood Soft Tissue Sarcoma
With the possible exception of infants with infantile fibrosarcoma, the prognosis for patients with recurrent or progressive disease is poor. No prospective trial has been able to prove that enhanced local control of pediatric soft tissue sarcomas will ultimately improve survival. Therefore, treatment should be individualized for the site of recurrence, biologic characteristics of the tumor (e.g., grade, invasiveness, and size), previous therapies, and individual patient considerations.
Treatment options for recurrent or progressive disease include the following:
- Surgical excision of local recurrence or isolated pulmonary recurrence.
- An Italian review of 73 patients with recurrent malignant peripheral nerve sheath tumors found that most relapses were local. Multivariate analysis showed that the factors associated with improved survival were no tumor invasiveness at initial diagnosis (T1), time of recurrence more than 12 months after initial diagnosis, and achievement of a second complete response with surgical removal of the recurrence(s). Only 15.8% of patients who had complete surgical excisions of local recurrence(s) were alive at 5 years.[1][Level of evidence: 3iiiA]
- Surgical excision of local recurrence followed by radiation therapy or brachytherapy (if no previous radiation therapy was given).
- Limb amputation (only for some children with extremity sarcomas that have already received radiation therapy).
- Gemcitabine and docetaxel.[2]
- Trabectedin.[3-5]
- Pazopanib. A phase I trial of pazopanib reported one partial response in a patient with desmoplastic small round cell tumor and prolonged disease stabilization in eight patients with recurrent sarcoma.[6][Level of evidence: 2Diii] Pazopanib has been approved for use in recurrent soft tissue sarcoma. The clinical trial that was used to obtain approval was limited to adults and demonstrated disease stabilization and prolonged time to progression; it did not demonstrate improved overall survival.[7] One 13-year-old boy and one 14-year-old girl with multiply recurrent synovial sarcoma and lung metastases had responses to pazopanib for 14 and 15 months, respectively.[8][Level of evidence: 3iiDi]
- A clinical trial of new chemotherapeutic regimens.
Resection is the standard treatment for recurrent pediatric nonrhabdomyosarcomatous soft tissue sarcomas. If the patient has not yet received radiation therapy, postoperative radiation should be considered after local excision of the recurrent tumor. Limb-sparing procedures with postoperative brachytherapy have been evaluated in adults but have not been studied extensively in children. For some children with extremity sarcomas who have received previous radiation therapy, amputation may be the only therapeutic option.
Pulmonary metastasectomy may achieve prolonged disease control for some patients.[9] A large, retrospective analysis of patients with recurrent soft tissue sarcoma showed that isolated local relapse had a better prognosis and that resection of pulmonary metastases improved the probability of survival.[10] In 31 children and adolescents younger than 23 years with pulmonary metastases from synovial sarcoma, complete resection of lung metastases appeared to prolong survival when compared with ten other patients who were not considered candidates for metastasectomy.[11][Level of evidence: 3iiiA] All patients with recurrent tumors should be considered for current clinical trials.
Published results of two studies addressed the outcomes for children with relapsed synovial sarcoma. Most patients in one study had distant relapse (29 of 44 patients),[12] while most patients in the second study had local relapse (27 of 37 patients).[13] Distant recurrence was a poor prognostic variable, while tumor resectability at relapse (as manifested by extremity recurrence) was associated with a better outcome in both studies.
Treatment Options Under Clinical Evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- APEC1621 (NCT03155620) (Pediatric MATCH: Targeted Therapy Directed by Genetic Testing in Treating Pediatric Patients with Relapsed or Refractory Advanced Solid Tumors, Non-Hodgkin Lymphomas, or Histiocytic Disorders): NCI-COG Pediatric Molecular Analysis for Therapeutic Choice (MATCH), referred to as Pediatric MATCH, will match targeted agents with specific molecular changes identified using a next-generation sequencing targeted assay of more than 3,000 different mutations across more than 160 genes in refractory and recurrent solid tumors. Children and adolescents aged 1 to 21 years are eligible for the trial.
Tumor tissue from progressive or recurrent disease must be available for molecular characterization. Patients with tumors that have molecular variants addressed by treatment arms included in the trial will be offered treatment on Pediatric MATCH. Additional information can be obtained on the ClinicalTrials.gov website for APEC1621 (NCT03155620).
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Bergamaschi L, Bisogno G, Manzitti C, et al.: Salvage rates and prognostic factors after relapse in children and adolescents with malignant peripheral nerve sheath tumors. Pediatr Blood Cancer 65 (2): , 2018. [PUBMED Abstract]
- Maki RG, Wathen JK, Patel SR, et al.: Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 25 (19): 2755-63, 2007. [PUBMED Abstract]
- Le Cesne A, Cresta S, Maki RG, et al.: A retrospective analysis of antitumour activity with trabectedin in translocation-related sarcomas. Eur J Cancer 48 (16): 3036-44, 2012. [PUBMED Abstract]
- Garcia-Carbonero R, Supko JG, Maki RG, et al.: Ecteinascidin-743 (ET-743) for chemotherapy-naive patients with advanced soft tissue sarcomas: multicenter phase II and pharmacokinetic study. J Clin Oncol 23 (24): 5484-92, 2005. [PUBMED Abstract]
- Garcia-Carbonero R, Supko JG, Manola J, et al.: Phase II and pharmacokinetic study of ecteinascidin 743 in patients with progressive sarcomas of soft tissues refractory to chemotherapy. J Clin Oncol 22 (8): 1480-90, 2004. [PUBMED Abstract]
- Glade Bender JL, Lee A, Reid JM, et al.: Phase I pharmacokinetic and pharmacodynamic study of pazopanib in children with soft tissue sarcoma and other refractory solid tumors: a children's oncology group phase I consortium report. J Clin Oncol 31 (24): 3034-43, 2013. [PUBMED Abstract]
- van der Graaf WT, Blay JY, Chawla SP, et al.: Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 379 (9829): 1879-86, 2012. [PUBMED Abstract]
- Casanova M, Basso E, Magni C, et al.: Response to pazopanib in two pediatric patients with pretreated relapsing synovial sarcoma. Tumori 103 (1): e1-e3, 2017. [PUBMED Abstract]
- Belal A, Salah E, Hajjar W, et al.: Pulmonary metastatectomy for soft tissue sarcomas: is it valuable? J Cardiovasc Surg (Torino) 42 (6): 835-40, 2001. [PUBMED Abstract]
- Zagars GK, Ballo MT, Pisters PW, et al.: Prognostic factors for disease-specific survival after first relapse of soft-tissue sarcoma: analysis of 402 patients with disease relapse after initial conservative surgery and radiotherapy. Int J Radiat Oncol Biol Phys 57 (3): 739-47, 2003. [PUBMED Abstract]
- Stanelle EJ, Christison-Lagay ER, Wolden SL, et al.: Pulmonary metastasectomy in pediatric/adolescent patients with synovial sarcoma: an institutional review. J Pediatr Surg 48 (4): 757-63, 2013. [PUBMED Abstract]
- Ferrari A, De Salvo GL, Dall'Igna P, et al.: Salvage rates and prognostic factors after relapse in children and adolescents with initially localised synovial sarcoma. Eur J Cancer 48 (18): 3448-55, 2012. [PUBMED Abstract]
- Soole F, Maupain C, Defachelles AS, et al.: Synovial sarcoma relapses in children and adolescents: prognostic factors, treatment, and outcome. Pediatr Blood Cancer 61 (8): 1387-93, 2014. [PUBMED Abstract]
Changes to This Summary (04/02/2018)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Treatment of Newly Diagnosed Childhood Soft Tissue Sarcoma
Added text to state that a small series reported symptomatic improvement and stable disease in seven patients with desmoid-type fibromatosis who were treated with pazopanib (cited Agresta et al. as reference 48).
Added text to state that a tumor with morphology similar to that of infantile fibrosarcoma has been identified in older children; in these older children, the tumors do not have the t(12;15)(ETV-NTRK3) translocation that is characteristic of the younger patients. In several of these patients, BRAF gene fusions have been identified (cited Kao et al. as reference 69).
Added text about the outcome results of 73 children and adolescents with recurrent malignant peripheral nerve sheath tumor reported by the Italian Sarcoma Group (cited Bergamaschi et al. as reference 127 and level of evidence 3iiiA).
Added text about the patient characteristics and results of a retrospective review of children and young adults younger than 30 years from four institutions, which identified 69 patients with alveolar soft part sarcoma treated primarily with surgery between 1980 and 2014 (cited Flores et al. as reference 152 and level of evidence 3iiA).
Added Sedig et al. as reference 172 and level of evidence 3iiiA.
Treatment of Progressive/Recurrent Childhood Soft Tissue Sarcoma
Added text about the prognostic factors and outcome results reported in an Italian review of 73 children and adolescents with recurrent malignant peripheral nerve sheath tumor (cited Bergamaschi et al. as reference 1 and level of evidence 3iiiA).
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of childhood soft tissue sarcoma. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Childhood Soft Tissue Sarcoma Treatment are:
- Denise Adams, MD (Children's Hospital Boston)
- Louis S. Constine, MD (James P. Wilmot Cancer Center at University of Rochester Medical Center)
- Holcombe Edwin Grier, MD (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Andrea A. Hayes-Jordan, MD, FACS, FAAP (M.D. Anderson Cancer Center)
- Paul A. Meyers, MD (Memorial Sloan-Kettering Cancer Center)
- Thomas A. Olson, MD (Aflac Cancer and Blood Disorders Center of Children's Healthcare of Atlanta - Egleston Campus)
- Alberto S. Pappo, MD (St. Jude Children's Research Hospital)
- R Beverly Raney, MD (Consultant)
- Stephen J. Shochat, MD (St. Jude Children's Research Hospital)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Childhood Soft Tissue Sarcoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/soft-tissue-sarcoma/hp/child-soft-tissue-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389361]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Updated: April 2, 2018
Source URL: https://www.cancer.gov/publishedcontent/syndication/3899.htm
Source Agency: National Cancer Institute (NCI)
Captured Date: 2013-09-14 09:01:57.0
Childhood Soft Tissue Sarcoma Treatment (PDQ®)–Health Professional Version
General Information About Childhood Soft Tissue Sarcoma
Dramatic improvements in survival have been achieved for children and adolescents with cancer. Between 1975 and 2010, childhood cancer mortality decreased by more than 50%.[1] Childhood and adolescent cancer survivors require close monitoring because cancer therapy side effects may persist or develop months or years after treatment. (Refer to the PDQ summary on Late Effects of Treatment for Childhood Cancer for specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors.)
Rhabdomyosarcoma, a tumor of striated muscle, is the most common soft tissue sarcoma in children aged 0 to 14 years and accounts for 50% of tumors in this age group.[2] (Refer to the PDQ summary on Childhood Rhabdomyosarcoma Treatment for more information.) In pediatrics, the remaining soft tissue sarcomas are commonly referred to as nonrhabdomyosarcomatous soft tissue sarcomas and account for approximately 3% of all childhood tumors.[3] This heterogeneous group of tumors includes the following neoplasms:[4]
- Connective tissue (e.g., desmoid-type fibromatosis).
- Peripheral nervous system (e.g., malignant peripheral nerve sheath tumor).
- Smooth muscle (e.g., leiomyosarcoma).
- Vascular tissue (blood and lymphatic vessels, e.g., angiosarcoma). (Refer to the PDQ summary on Childhood Vascular Tumors Treatment for more information about childhood vascular tumors.)
Distribution of Soft Tissue Sarcoma by Age and Histology
Pediatric soft tissue sarcomas are a heterogenous group of malignant tumors that originate from primitive mesenchymal tissue and account for 7% of all childhood tumors.[5]
The distribution of soft tissue sarcomas by histology and age, based on the Surveillance, Epidemiology, and End Results (SEER) information from 1975 to 2012, is depicted in Table 1. The distribution of histologic subtypes by age is also shown in Figure 2.
| Age <5 y | Age 5–9 y | Age 10–14 y | Age 15–19 y | % of the Total Number of STS Cases <20 y | |||
|---|---|---|---|---|---|---|---|
| pPNET = peripheral primitive neuroectodermal tumors; SEER = Surveillance, Epidemiology, and End Results; STS = soft tissue sarcoma. | |||||||
| aSEER data is available at http://seer.cancer.gov. | |||||||
| bDermatofibrosarcoma accounts for 75% of these cases. | |||||||
| All soft tissue and other extraosseous sarcomas | 923 | 631 | 946 | 1,267 | 100 | ||
| Rhabdomyosarcomas | 551 | 348 | 312 | 270 | 39 | ||
| Fibrosarcomas, peripheral nerve, and other fibrous neoplasms | 116 | 50 | 88 | 141 | 10 | ||
| Fibroblastic and myofibroblastic tumors | 97 | 24 | 31 | 62 | 6 | ||
| Nerve sheath tumors | 19 | 26 | 56 | 77 | 5 | ||
| Other fibromatous neoplasms | 0 | 0 | 1 | 2 | 0.1 | ||
| Kaposi sarcoma | 2 | 1 | 1 | 9 | 0.3 | ||
| Other specified soft tissue sarcomas | 194 | 190 | 424 | 708 | 40 | ||
| Ewing tumor and Askin tumor of soft tissue | 27 | 30 | 62 | 92 | 6 | ||
| pPNET of soft tissue | 21 | 18 | 36 | 46 | 3.2 | ||
| Extrarenal rhabdoid tumor | 61 | 3 | 7 | 3 | 2 | ||
| Liposarcomas | 3 | 5 | 22 | 57 | 2.3 | ||
| Fibrohistiocytic tumors b | 34 | 54 | 108 | 188 | 10 | ||
| Leiomyosarcomas | 9 | 14 | 15 | 36 | 2 | ||
| Synovial sarcomas | 10 | 34 | 111 | 175 | 9 | ||
| Blood vessel tumors | 11 | 7 | 8 | 25 | 1.4 | ||
| Osseous and chondromatous neoplasms of soft tissue | 1 | 6 | 13 | 10 | 0.8 | ||
| Alveolar soft parts sarcoma | 4 | 3 | 16 | 29 | 1.4 | ||
| Miscellaneous soft tissue sarcomas | 13 | 16 | 36 | 47 | 3 | ||
| Unspecified soft tissue sarcomas | 60 | 40 | 111 | 139 | 9.3 | ||
Nonrhabdomyosarcomatous soft tissue sarcomas are more common in adolescents and adults,[4] and most of the information regarding treatment and natural history of the disease in younger patients has been based on adult studies. The distributions of these tumors by age according to stage, histologic subtype, and tumor site are shown in Figures 1, 2, and 3, respectively.[6]
Risk Factors
Some genetic and environmental factors have been associated with the development of nonrhabdomyosarcomatous soft tissue sarcoma, including the following:
- Genetic factors:
- Li-Fraumeni syndrome: Patients with Li-Fraumeni syndrome (usually due to heritable cancer-associated changes of the TP53 tumor suppressor gene) have an increased risk of developing soft tissue tumors (mostly nonrhabdomyosarcomatous soft tissue sarcomas), bone sarcomas, breast cancer, brain tumors, and acute leukemia.[7,8]
- Familial adenomatous polyposis: Patients with familial adenomatous polyposis are at increased risk of developing desmoid-type fibromatosis.[9]
- Retinoblastoma (RB1) gene: Germline mutations of the retinoblastoma gene have been associated with an increased risk of developing soft tissue sarcomas, particularly leiomyosarcoma.[10]
- SMARCB1 gene: Germline mutations or deletions of the SMARCB1 (INI1) gene are associated with an increased risk of developing extrarenal rhabdoid tumors.[11]
- Neurofibromatosis type 1: Approximately 4% of patients with neurofibromatosis type 1 develop malignant peripheral nerve sheath tumors, which usually develop after a long latency; some patients develop multiple lesions.[12-14]
- Werner syndrome: Werner syndrome is characterized by spontaneous chromosomal instability, resulting in increased susceptibility to cancer and premature aging. An excess of soft tissue sarcomas has been reported in patients with Werner syndrome.[15]
- Environmental factors:
- Radiation: Some nonrhabdomyosarcomatous soft tissue sarcomas (particularly malignant fibrous histiocytoma) can develop within a previously irradiated site.[3,16]
- Epstein-Barr virus infection in patients with AIDS: Some nonrhabdomyosarcomatous soft tissue sarcomas (e.g., leiomyosarcoma) have been linked to Epstein-Barr virus infection in patients with AIDS.[3,17]
Clinical Presentation
Although nonrhabdomyosarcomatous soft tissue sarcomas can develop in any part of the body, they arise most commonly in the trunk and extremities.[18-20] These neoplasms can present initially as an asymptomatic solid mass, or they may be symptomatic because of local invasion of adjacent anatomical structures. Although rare, these tumors can arise primarily in brain tissue and are treated according to the histotype.[21]
Systemic symptoms (e.g., fever, weight loss, and night sweats) are rare. Hypoglycemia and hypophosphatemic rickets have been reported in cases of hemangiopericytoma, whereas hyperglycemia has been noted in patients with fibrosarcoma of the lung.[22]
Diagnostic and Staging Evaluation
When a suspicious lesion is identified, it is crucial that a complete workup, followed by adequate biopsy be performed. It is best to image the lesion using the following procedures before initiating any intervention:
- Plain films. Plain films can be used to rule out bone involvement and detect calcifications that may be seen in soft tissue tumors such as extraskeletal osteosarcoma or synovial sarcoma.
- Chest computed tomography (CT). Chest CT is essential to assess the presence of metastases.
- Abdominal CT or magnetic resonance imaging (MRI). Abdominal CT or MRI can be used to image intra-abdominal tumors, such as liposarcoma.
- Extremity MRI. MRI is essential for extremity lesions.
- Positron emission tomography (PET) scan and bone scan. In children with rhabdomyosarcoma, PET-CT performed better than conventional imaging in identifying nodal, bone, bone marrow, and soft tissue disease. The authors of an imaging comparison study suggest that bone scans with technetium Tc 99m might be eliminated as a staging procedure.[23] The use of this modality in pediatric nonrhabdomyosarcomatous soft tissue sarcoma has not been studied extensively. However, a small study of nine patients with nonrhabdomyosarcomatous soft tissue sarcoma suggests that PET-CT is more accurate and cost effective than either modality alone in identifying distant metastatic disease.[24]
The imaging characteristics of some tumors can be highly suggestive of this diagnosis. For example, the imaging characteristics of pediatric low-grade fibromyxoid sarcoma and alveolar soft part sarcoma have been described and can aid in the diagnosis of these rare neoplasms.[25]
Biopsy strategies
Although nonrhabdomyosarcomatous soft tissue tumors are fairly readily distinguished pathologically from rhabdomyosarcoma and Ewing sarcoma, the classification of childhood nonrhabdomyosarcomatous soft tissue sarcoma type is often difficult. Core-needle biopsy, incisional biopsy, or excisional biopsy can be used to diagnose a nonrhabdomyosarcomatous soft tissue sarcoma. If possible, the surgeon who will perform the definitive resection needs to be involved in the biopsy decision. Poorly placed incisional or needle biopsies may adversely affect the performance of the primary resection.
Considerations related to the selection of a biopsy procedure are as follows:
- Given the diagnostic importance of translocations, a core-needle biopsy or small incisional biopsy that obtains adequate tumor tissue is crucial to allow for conventional histology, immunocytochemical analysis, and other studies such as light and electron microscopy, cytogenetics, fluorescence in situ hybridization, and molecular pathology.[26,27] Core-needle biopsy for a deep-seated tumor can lead to formation of a hematoma, which affects subsequent resection and/or radiation; in these cases, incisional biopsy is the preferred procedure.
- Fine-needle biopsy is usually not recommended because it is difficult to determine the accurate histologic diagnosis and grade of the tumor in this heterogeneous group of tumors.
- Image guidance using ultrasound, CT scan, or MRI may be necessary to ensure a representative biopsy.[28]
- Needle biopsy techniques must ensure adequate tissue sampling. The acquisition of multiple cores of tissue may be required.
- Incisional biopsies must not compromise subsequent wide local excision.
- Excisional biopsy of the lesion is only appropriate for small superficial lesions (<3 cm in size) and are discouraged.[29,30] If an excisional biopsy is contemplated, then MRI of the area is recommended to define the area of involvement as subsequent surgery or radiation therapy is likely.
- Various institutional series have demonstrated the feasibility and effectiveness of sentinel node biopsy as a staging procedure in pediatric patients with soft tissue sarcomas.[31-36]
- Transverse extremity incisions are avoided to reduce skin loss and because they require a greater cross-sectional volume of tissue to be covered in the radiation field. Other extensive surgical procedures are also avoided before definitive diagnosis. For these reasons, open biopsy or multiple core-needle biopsies are strongly encouraged so that adequate tumor tissue can be obtained to allow crucial studies to be performed and to avoid limiting future treatment options.
Unplanned resection
In children with unplanned resection of nonrhabdomyosarcomatous soft tissue sarcomas, primary re-excision is frequently recommended because many patients will have tumor present in the re-excision specimen.[37,38] A single-institution analysis of adolescents and adults compared patients with unplanned excision of soft tissue sarcoma to stage-matched controls. In this retrospective analysis, unplanned initial excision of soft tissue sarcoma resulted in increased risk of local recurrence, metastasis, and death; this increase was greatest for high-grade tumors.[39][Level of evidence: 3iiA]
Chromosomal abnormalities
Many nonrhabdomyosarcomatous soft tissue sarcomas are characterized by chromosomal abnormalities. Some of these chromosomal translocations lead to a fusion of two disparate genes. The resulting fusion transcript can be readily detected by using polymerase chain reaction-based techniques, thus facilitating the diagnosis of those neoplasms that have translocations.
Some of the most frequent aberrations seen in nonrhabdomyosarcomatous soft tissue tumors are listed in Table 2.
| Histology | Chromosomal Aberrations | Genes Involved |
|---|---|---|
| aAdapted from Sandberg,[40] Slater et al.,[41] Mertens et al.,[42] and Romeo.[43] | ||
| Alveolar soft part sarcoma | t(x;17)(p11.2;q25) | ASPL/TFE3 [44-46] |
| Angiomatoid fibrous histiocytoma | t(12;16)(q13;p11), t(2;22)(q33;q12), t(12;22)(q13;q12) | FUS/ATF1, EWSR1/CREB1,[47] EWS/ATF1 |
| Clear cell sarcoma | t(12;22)(q13;q12), t(2;22)(q33;q12) | ATF1/EWS, EWSR1/CREB1 |
| Congenital (infantile) fibrosarcoma/mesoblastic nephroma | t(12;15)(p13;q25) | ETV-NTRK3 |
| Dermatofibrosarcoma protuberans | t(17;22)(q22;q13) | COL1A1/PDGFB |
| Desmoid fibromatosis | Trisomy 8 or 20, loss of 5q21 | CTNNB1 or APC mutations |
| Desmoplastic small round cell tumors | t(11;22)(p13;q12) | EWS/WT1 [48,49] |
| Epithelioid hemangioendothelioma | t(1;3)(p36;q25) [50] | WWTR1/CAMTA1 |
| Epithelioid sarcoma | Inactivation SMARCB1 | SMARCB1 |
| Extraskeletal myxoid chondrosarcoma | t(9;22)(q22;q12), t(9;17)(q22;q11), t(9;15)(q22;q21), t(3;9)(q11;q22) | EWSR1/NR4A3, TAF2N/NR4A3, TCF12/NR4A3, TGF/NR4A3 |
| Hemangiopericytoma | t(12;19)(q13;q13.3) and t(13;22)(q22;q13.3) | |
| Infantile fibrosarcoma | t(12;15)(p13;q25) | ETV6/NTRK3 |
| Inflammatory myofibroblastic tumor | t(1;2)(q23;q23), t(2;19)(q23;q13), t(2;17)(q23;q23), t(2;2)(p23;q13), t(2;11)(p23;p15) [51] | TPM3/ALK, TPM4/ALK, CLTC/ALK, RANBP2/ALK, CARS/ALK, RAS |
| Low-grade fibromyxoid sarcoma | t(7;16)(q33;p11), t(11;16)(p11;p11) | FUS/CREB3L2, FUS/CREB3L1 |
| Malignant peripheral nerve sheath tumor | 17q11.2, loss or rearrangement 10p, 11q, 17q, 22q | NF1 |
| Mesenchymal chondrosarcoma | Del(8)(q13.3q21.1) | HEY1/NCOA2 |
| Myoepithelioma | t(19;22)(q13;q12), t(1;22)(q23;q12), t(6;22)(p21;q12) | EWSR/ZNF44, EWSR/PBX1, EWSR/POU5F1 |
| Myxoid/round cell liposarcoma | t(12;16)(q13;p11), t(12;22)(q13;q12) | FUS/DD1T3, EWSR/DD1T3 |
| Rhabdoid tumor | Inactivation SMARCB1 | SMARCB1 |
| Solitary fibrous tumor | Inv(12)(q13q13) | NAB2/STAT6 |
| Synovial sarcoma | t(x;18)(p11.2;q11.2) | SYT/SSX |
| Tenosynovial giant cell tumor | t(1;2)(p13;q35) | COL6A3/CSF1 |
Prognosis
The prognosis of nonrhabdomyosarcomatous soft tissue sarcoma varies greatly depending on the following factors:[52-54]
- Site of the primary tumor.
- Tumor size.
- Tumor grade. (Refer to the Prognostic Significance of Tumor Grading section of this summary for more information.)
- Tumor histology.
- Depth of tumor invasion.
- Presence of metastases.
- Resectability of the tumor.
- Use of radiation therapy.
Several adult and pediatric series have shown that patients with large or invasive tumors have a significantly worse prognosis than do those with small, noninvasive tumors. A retrospective review of soft tissue sarcomas in children and adolescents suggests that the 5 cm cutoff used for adults with soft tissue sarcoma may not be ideal for smaller children, especially infants. The review identified an interaction between tumor diameter and body surface area.[55] This relationship requires further study to determine the therapeutic implications of the observation.
In a review of a large adult series of nonrhabdomyosarcomatous soft tissue sarcomas, superficial extremity sarcomas had a better prognosis than did deep tumors. Thus, in addition to grade and size, the depth of invasion of the tumor should be considered.[56]
Some pediatric nonrhabdomyosarcomatous soft tissue sarcomas are associated with a better outcome. For instance, infantile fibrosarcoma, presenting in infants and children younger than 5 years, has an excellent prognosis given that surgery alone can cure a significant number of these patients and the tumor is highly chemosensitive.[3]
Soft tissue sarcomas in older children and adolescents often behave similarly to those in adult patients.[3,26] A large, prospective, multinational Children's Oncology Group study (ARST0332 [NCT00346164]) enrolled newly diagnosed patients younger than 30 years. Patients were assigned to treatment on the basis of their risk group (refer to Figure 4).[57][Level of evidence: 2A]
- Arm A (grossly excised low-grade tumor and ≤5 cm widely excised high-grade tumor): Surgery only.
- Arm B (≤5 cm marginally resected high-grade tumor): 55.8 Gy of radiation therapy.
- Arm C (>5 cm grossly resected tumor ± metastases): Ifosfamide/doxorubicin chemotherapy and 55.8 Gy of radiation therapy.
- Arm D (>5 cm unresected tumor ± metastases): Preoperative ifosfamide/doxorubicin chemotherapy and 45 Gy of radiation therapy, and then surgery and a radiation boost that was based on margins.
Of 551 patients enrolled, at a median follow-up of 2.6 years, the preliminary analysis estimated the following 3-year survival rates:[57]
- Arm A: 91% event-free survival (EFS); 99% overall survival (OS).
- Arm B: 79% EFS; 100% OS.
- Arm C: 68% EFS; 81% OS.
- Arm D: 52% EFS; 66% OS.
Pediatric patients with unresected localized nonrhabdomyosarcomatous soft tissue sarcomas have a poor outcome. Only about one-third of patients treated with multimodality therapy remain disease free.[52,58]; [59,60][Level of evidence: 3iiiA] In a review of 30 Italian patients with nonrhabdomyosarcomatous soft tissue sarcoma at visceral sites, only ten patients survived at 5 years. Unfavorable prognostic factors included inability to achieve complete resection, large tumor size, tumor invasion, histologic subtype, and lung-pleura sites.[61][Level of evidence: 3iiB]
In a pooled analysis from U.S. and European pediatric centers, outcome was better for patients whose tumor removal procedure was deemed complete than for patients whose tumor removal was incomplete. Outcome was better for patients who received radiation therapy than for patients who did not.[59][Level of evidence: 3iiiA]
Because long-term related morbidity must be minimized while disease-free survival is maximized, the ideal therapy for each patient must be carefully and individually determined utilizing these prognostic factors before initiating therapy.[19,62-66]
Related Summaries
Refer to the following PDQ summaries for information about other types of sarcoma:
- Childhood Rhabdomyosarcoma Treatment.
- Childhood Vascular Tumors Treatment.
- Ewing Sarcoma Treatment (extraosseous Ewing, peripheral neuroepithelioma, and Askin tumors).
- Unusual Cancers of Childhood Treatment (gastrointestinal stromal tumors).
- Adult Soft Tissue Sarcoma Treatment.
References
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014. [PUBMED Abstract]
- Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. Bethesda, Md: National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649. Also available online. Last accessed January 24, 2018.
- Spunt SL, Million L, Coffin C: The nonrhabdomyosarcoma soft tissue sarcoma. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 827-54.
- Weiss SW, Goldblum JR: General considerations. In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. St. Louis, Mo: Mosby, 2008, pp 1-14.
- Pappo AS, Pratt CB: Soft tissue sarcomas in children. Cancer Treat Res 91: 205-22, 1997. [PUBMED Abstract]
- Ferrari A, Sultan I, Huang TT, et al.: Soft tissue sarcoma across the age spectrum: a population-based study from the Surveillance Epidemiology and End Results database. Pediatr Blood Cancer 57 (6): 943-9, 2011. [PUBMED Abstract]
- Chang F, Syrjänen S, Syrjänen K: Implications of the p53 tumor-suppressor gene in clinical oncology. J Clin Oncol 13 (4): 1009-22, 1995. [PUBMED Abstract]
- Plon SE, Malkin D: Childhood cancer and hereditary. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 13-31.
- Groen EJ, Roos A, Muntinghe FL, et al.: Extra-intestinal manifestations of familial adenomatous polyposis. Ann Surg Oncol 15 (9): 2439-50, 2008. [PUBMED Abstract]
- Kleinerman RA, Tucker MA, Abramson DH, et al.: Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst 99 (1): 24-31, 2007. [PUBMED Abstract]
- Eaton KW, Tooke LS, Wainwright LM, et al.: Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56 (1): 7-15, 2011. [PUBMED Abstract]
- Weiss SW, Goldblum JR: Benign tumors of peripheral nerves. In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. St. Louis, Mo: Mosby, 2008, pp 825-901.
- deCou JM, Rao BN, Parham DM, et al.: Malignant peripheral nerve sheath tumors: the St. Jude Children's Research Hospital experience. Ann Surg Oncol 2 (6): 524-9, 1995. [PUBMED Abstract]
- Stark AM, Buhl R, Hugo HH, et al.: Malignant peripheral nerve sheath tumours--report of 8 cases and review of the literature. Acta Neurochir (Wien) 143 (4): 357-63; discussion 363-4, 2001. [PUBMED Abstract]
- Goto M, Miller RW, Ishikawa Y, et al.: Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol Biomarkers Prev 5 (4): 239-46, 1996. [PUBMED Abstract]
- Weiss SW, Goldblum JR: Malignant fibrous histiocytoma (pleomorphic undifferentiated sarcoma). In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. St. Louis, Mo: Mosby, 2008, pp 403-27.
- McClain KL, Leach CT, Jenson HB, et al.: Association of Epstein-Barr virus with leiomyosarcomas in children with AIDS. N Engl J Med 332 (1): 12-8, 1995. [PUBMED Abstract]
- Dillon P, Maurer H, Jenkins J, et al.: A prospective study of nonrhabdomyosarcoma soft tissue sarcomas in the pediatric age group. J Pediatr Surg 27 (2): 241-4; discussion 244-5, 1992. [PUBMED Abstract]
- Rao BN: Nonrhabdomyosarcoma in children: prognostic factors influencing survival. Semin Surg Oncol 9 (6): 524-31, 1993 Nov-Dec. [PUBMED Abstract]
- Zeytoonjian T, Mankin HJ, Gebhardt MC, et al.: Distal lower extremity sarcomas: frequency of occurrence and patient survival rate. Foot Ankle Int 25 (5): 325-30, 2004. [PUBMED Abstract]
- Benesch M, von Bueren AO, Dantonello T, et al.: Primary intracranial soft tissue sarcoma in children and adolescents: a cooperative analysis of the European CWS and HIT study groups. J Neurooncol 111 (3): 337-45, 2013. [PUBMED Abstract]
- Weiss SW, Goldblum JR: Miscellaneous tumors of intermediate malignancy. In: Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 5th ed. St. Louis, Mo: Mosby, 2008, pp 1093-1160.
- Federico SM, Spunt SL, Krasin MJ, et al.: Comparison of PET-CT and conventional imaging in staging pediatric rhabdomyosarcoma. Pediatr Blood Cancer 60 (7): 1128-34, 2013. [PUBMED Abstract]
- Tateishi U, Hosono A, Makimoto A, et al.: Accuracy of 18F fluorodeoxyglucose positron emission tomography/computed tomography in staging of pediatric sarcomas. J Pediatr Hematol Oncol 29 (9): 608-12, 2007. [PUBMED Abstract]
- Sargar K, Kao SC, Spunt SL, et al.: MRI and CT of Low-Grade Fibromyxoid Sarcoma in Children: A Report From Children's Oncology Group Study ARST0332. AJR Am J Roentgenol 205 (2): 414-20, 2015. [PUBMED Abstract]
- Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 4th ed. St. Louis, Mo: Mosby, 2001.
- Recommendations for the reporting of soft tissue sarcomas. Association of Directors of Anatomic and Surgical Pathology. Mod Pathol 11 (12): 1257-61, 1998. [PUBMED Abstract]
- Chowdhury T, Barnacle A, Haque S, et al.: Ultrasound-guided core needle biopsy for the diagnosis of rhabdomyosarcoma in childhood. Pediatr Blood Cancer 53 (3): 356-60, 2009. [PUBMED Abstract]
- Coffin CM, Dehner LP, O'Shea PA: Pediatric Soft Tissue Tumors: A Clinical, Pathological, and Therapeutic Approach. Baltimore, Md: Williams and Wilkins, 1997.
- Smith LM, Watterson J, Scott SM: Medical and surgical management of pediatric soft tissue tumors. In: Coffin CM, Dehner LP, O'Shea PA: Pediatric Soft Tissue Tumors: A Clinical, Pathological, and Therapeutic Approach. Baltimore, Md: Williams and Wilkins, 1997, pp 360-71.
- Neville HL, Andrassy RJ, Lally KP, et al.: Lymphatic mapping with sentinel node biopsy in pediatric patients. J Pediatr Surg 35 (6): 961-4, 2000. [PUBMED Abstract]
- Neville HL, Raney RB, Andrassy RJ, et al.: Multidisciplinary management of pediatric soft-tissue sarcoma. Oncology (Huntingt) 14 (10): 1471-81; discussion 1482-6, 1489-90, 2000. [PUBMED Abstract]
- Kayton ML, Delgado R, Busam K, et al.: Experience with 31 sentinel lymph node biopsies for sarcomas and carcinomas in pediatric patients. Cancer 112 (9): 2052-9, 2008. [PUBMED Abstract]
- Dall'Igna P, De Corti F, Alaggio R, et al.: Sentinel node biopsy in pediatric patients: the experience in a single institution. Eur J Pediatr Surg 24 (6): 482-7, 2014. [PUBMED Abstract]
- Parida L, Morrisson GT, Shammas A, et al.: Role of lymphoscintigraphy and sentinel lymph node biopsy in the management of pediatric melanoma and sarcoma. Pediatr Surg Int 28 (6): 571-8, 2012. [PUBMED Abstract]
- Alcorn KM, Deans KJ, Congeni A, et al.: Sentinel lymph node biopsy in pediatric soft tissue sarcoma patients: utility and concordance with imaging. J Pediatr Surg 48 (9): 1903-6, 2013. [PUBMED Abstract]
- Chui CH, Spunt SL, Liu T, et al.: Is reexcision in pediatric nonrhabdomyosarcoma soft tissue sarcoma necessary after an initial unplanned resection? J Pediatr Surg 37 (10): 1424-9, 2002. [PUBMED Abstract]
- Cecchetto G, Guglielmi M, Inserra A, et al.: Primary re-excision: the Italian experience in patients with localized soft-tissue sarcomas. Pediatr Surg Int 17 (7): 532-4, 2001. [PUBMED Abstract]
- Qureshi YA, Huddy JR, Miller JD, et al.: Unplanned excision of soft tissue sarcoma results in increased rates of local recurrence despite full further oncological treatment. Ann Surg Oncol 19 (3): 871-7, 2012. [PUBMED Abstract]
- Sandberg AA: Translocations in malignant tumors. Am J Pathol 159 (6): 1979-80, 2001. [PUBMED Abstract]
- Slater O, Shipley J: Clinical relevance of molecular genetics to paediatric sarcomas. J Clin Pathol 60 (11): 1187-94, 2007. [PUBMED Abstract]
- Mertens F, Antonescu CR, Hohenberger P, et al.: Translocation-related sarcomas. Semin Oncol 36 (4): 312-23, 2009. [PUBMED Abstract]
- Romeo S, Dei Tos AP: Clinical application of molecular pathology in sarcomas. Curr Opin Oncol 23 (4): 379-84, 2011. [PUBMED Abstract]
- Ladanyi M, Lui MY, Antonescu CR, et al.: The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene 20 (1): 48-57, 2001. [PUBMED Abstract]
- Ladanyi M: The emerging molecular genetics of sarcoma translocations. Diagn Mol Pathol 4 (3): 162-73, 1995. [PUBMED Abstract]
- Williams A, Bartle G, Sumathi VP, et al.: Detection of ASPL/TFE3 fusion transcripts and the TFE3 antigen in formalin-fixed, paraffin-embedded tissue in a series of 18 cases of alveolar soft part sarcoma: useful diagnostic tools in cases with unusual histological features. Virchows Arch 458 (3): 291-300, 2011. [PUBMED Abstract]
- Antonescu CR, Dal Cin P, Nafa K, et al.: EWSR1-CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer 46 (12): 1051-60, 2007. [PUBMED Abstract]
- Barnoud R, Sabourin JC, Pasquier D, et al.: Immunohistochemical expression of WT1 by desmoplastic small round cell tumor: a comparative study with other small round cell tumors. Am J Surg Pathol 24 (6): 830-6, 2000. [PUBMED Abstract]
- Wang LL, Perlman EJ, Vujanic GM, et al.: Desmoplastic small round cell tumor of the kidney in childhood. Am J Surg Pathol 31 (4): 576-84, 2007. [PUBMED Abstract]
- Errani C, Zhang L, Sung YS, et al.: A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer 50 (8): 644-53, 2011. [PUBMED Abstract]
- Jain S, Xu R, Prieto VG, et al.: Molecular classification of soft tissue sarcomas and its clinical applications. Int J Clin Exp Pathol 3 (4): 416-28, 2010. [PUBMED Abstract]
- Spunt SL, Hill DA, Motosue AM, et al.: Clinical features and outcome of initially unresected nonmetastatic pediatric nonrhabdomyosarcoma soft tissue sarcoma. J Clin Oncol 20 (15): 3225-35, 2002. [PUBMED Abstract]
- Spunt SL, Poquette CA, Hurt YS, et al.: Prognostic factors for children and adolescents with surgically resected nonrhabdomyosarcoma soft tissue sarcoma: an analysis of 121 patients treated at St Jude Children's Research Hospital. J Clin Oncol 17 (12): 3697-705, 1999. [PUBMED Abstract]
- Ferrari A, Casanova M, Collini P, et al.: Adult-type soft tissue sarcomas in pediatric-age patients: experience at the Istituto Nazionale Tumori in Milan. J Clin Oncol 23 (18): 4021-30, 2005. [PUBMED Abstract]
- Ferrari A, Miceli R, Meazza C, et al.: Soft tissue sarcomas of childhood and adolescence: the prognostic role of tumor size in relation to patient body size. J Clin Oncol 27 (3): 371-6, 2009. [PUBMED Abstract]
- Brooks AD, Heslin MJ, Leung DH, et al.: Superficial extremity soft tissue sarcoma: an analysis of prognostic factors. Ann Surg Oncol 5 (1): 41-7, 1998 Jan-Feb. [PUBMED Abstract]
- Spunt SL, Million L, Anderson JR, et al.: Risk-based treatment for nonrhabdomyosarcoma soft tissue sarcomas (NRSTS) in patients under 30 years of age: Children’s Oncology Group study ARST0332. [Abstract] J Clin Oncol 32 (Suppl 15): A-10008, 2014. Also available online. Last accessed April 02, 2018.
- O'Sullivan B, Davis AM, Turcotte R, et al.: Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 359 (9325): 2235-41, 2002. [PUBMED Abstract]
- Ferrari A, Miceli R, Rey A, et al.: Non-metastatic unresected paediatric non-rhabdomyosarcoma soft tissue sarcomas: results of a pooled analysis from United States and European groups. Eur J Cancer 47 (5): 724-31, 2011. [PUBMED Abstract]
- Smith KB, Indelicato DJ, Knapik JA, et al.: Definitive radiotherapy for unresectable pediatric and young adult nonrhabdomyosarcoma soft tissue sarcoma. Pediatr Blood Cancer 57 (2): 247-51, 2011. [PUBMED Abstract]
- Ferrari A, Magni C, Bergamaschi L, et al.: Pediatric nonrhabdomyosarcoma soft tissue sarcomas arising at visceral sites. Pediatr Blood Cancer 64 (9): , 2017. [PUBMED Abstract]
- Dillon PW, Whalen TV, Azizkhan RG, et al.: Neonatal soft tissue sarcomas: the influence of pathology on treatment and survival. Children's Cancer Group Surgical Committee. J Pediatr Surg 30 (7): 1038-41, 1995. [PUBMED Abstract]
- Pappo AS, Fontanesi J, Luo X, et al.: Synovial sarcoma in children and adolescents: the St Jude Children's Research Hospital experience. J Clin Oncol 12 (11): 2360-6, 1994. [PUBMED Abstract]
- Marcus KC, Grier HE, Shamberger RC, et al.: Childhood soft tissue sarcoma: a 20-year experience. J Pediatr 131 (4): 603-7, 1997. [PUBMED Abstract]
- Pratt CB, Pappo AS, Gieser P, et al.: Role of adjuvant chemotherapy in the treatment of surgically resected pediatric nonrhabdomyosarcomatous soft tissue sarcomas: A Pediatric Oncology Group Study. J Clin Oncol 17 (4): 1219, 1999. [PUBMED Abstract]
- Pratt CB, Maurer HM, Gieser P, et al.: Treatment of unresectable or metastatic pediatric soft tissue sarcomas with surgery, irradiation, and chemotherapy: a Pediatric Oncology Group study. Med Pediatr Oncol 30 (4): 201-9, 1998. [PUBMED Abstract]
Histopathological Classification of Childhood Soft Tissue Sarcoma
World Health Organization (WHO) Classification of Soft Tissue Sarcomas
The WHO lists the following cell types in its classification of soft tissue sarcomas:[1,2]
- Adipocytic tumors.
- Intermediate (locally aggressive).
- Malignant.
- Chondro-osseous tumors.
- Extraskeletal mesenchymal chondrosarcoma.[3]
- Extraskeletal osteosarcoma.
- Soft tissue chondroma.
- Fibroblastic/myofibroblastic tumors.
- Intermediate-grade (locally aggressive).
- Desmoid-type fibromatosis (previously called desmoid tumor or aggressive fibromatoses).
- Giant cell fibroblastoma.
- Lipofibromatosis.
- Palmar/plantar fibromatosis.
- Intermediate-grade (rarely metastasizing).
- Dermatofibrosarcoma protuberans.
- Infantile fibrosarcoma.[4]
- Inflammatory myofibroblastic tumor.
- Low-grade myofibroblastic tumor.
- Myxoinflammatory fibroblastic sarcoma.
- Solitary fibrous tumor.
- Malignant.
- Intermediate-grade (locally aggressive).
- Skeletal muscle tumors.
- Rhabdomyosarcoma (embryonal, alveolar, and pleomorphic forms). (Refer to the PDQ summary on Childhood Rhabdomyosarcoma Treatment for more information.)
- Smooth muscle tumors.
- So-called fibrohistiocytic tumors (intermediate, rarely metastasizing).
- Giant cell tumors of soft tissue.
- Plexiform fibrohistiocytic tumor.
- Tumors of peripheral nerves.
- Pericytic (perivascular) tumors.
- Malignant glomus tumor and variants.
- Myopericytoma.
- Angioleiomyoma.
- Myofibroma.
- Infantile myofibroma (previously called hemangiopericytoma [infantile]).
- Myofibromatosis.
- Infantile myofibromatosis.
- Tumors of uncertain differentiation.
- Alveolar soft part sarcoma.
- Clear cell sarcoma of soft tissue.
- Desmoplastic small round cell tumor.
- Epithelioid sarcoma.
- Extrarenal rhabdoid tumor.
- Extraskeletal myxoid chondrosarcoma.
- Neoplasms with perivascular epithelioid cell differentiation (PEComa NOS, malignant).
- Primitive neuroectodermal tumor/extraskeletal Ewing tumor.
- Synovial sarcoma (NOS, spindle cell, and biphasic varieties).
- Undifferentiated/unclassified sarcomas.
- Undifferentiated epithelial sarcoma.
- Undifferentiated pleomorphic sarcoma.
- Undifferentiated round cell sarcoma.
- Undifferentiated sarcoma; sarcoma, NOS.[6]
- Undifferentiated spindle cell sarcoma.
- Vascular tumors.
References
- Soft tissue sarcoma. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 291-6.
- Brodowicz T, Schwameis E, Widder J, et al.: Intensified Adjuvant IFADIC Chemotherapy for Adult Soft Tissue Sarcoma: A Prospective Randomized Feasibility Trial. Sarcoma 4 (4): 151-60, 2000. [PUBMED Abstract]
- Dantonello TM, Int-Veen C, Leuschner I, et al.: Mesenchymal chondrosarcoma of soft tissues and bone in children, adolescents, and young adults: experiences of the CWS and COSS study groups. Cancer 112 (11): 2424-31, 2008. [PUBMED Abstract]
- Steelman C, Katzenstein H, Parham D, et al.: Unusual presentation of congenital infantile fibrosarcoma in seven infants with molecular-genetic analysis. Fetal Pediatr Pathol 30 (5): 329-37, 2011. [PUBMED Abstract]
- Evans HL: Low-grade fibromyxoid sarcoma: a clinicopathologic study of 33 cases with long-term follow-up. Am J Surg Pathol 35 (10): 1450-62, 2011. [PUBMED Abstract]
- Alaggio R, Collini P, Randall RL, et al.: Undifferentiated high-grade pleomorphic sarcomas in children: a clinicopathologic study of 10 cases and review of literature. Pediatr Dev Pathol 13 (3): 209-17, 2010 May-Jun. [PUBMED Abstract]
Staging and Grading Systems for Childhood Soft Tissue Sarcoma
Clinical staging has an important role in predicting the clinical outcome and determining the most effective therapy for pediatric soft tissue sarcomas. As yet, there is no well-accepted staging system that is applicable to all childhood sarcomas. The system from the American Joint Committee on Cancer (AJCC) that is used for adults has not been validated in pediatric studies. Although a standardized staging system for pediatric nonrhabdomyosarcomatous soft tissue sarcoma does not exist, two systems are currently in use for staging pediatric nonrhabdomyosarcomatous soft tissue sarcoma.[1]
- Surgico-pathologic staging system: The surgico-pathologic staging system used by the Intergroup Rhabdomyosarcoma Study (see below) is based on the amount, or extent, of tumor that remains after initial surgery and whether the disease has metastasized. This staging system was used in early pediatric trials.[2]
- TNM staging system: The TNM staging system is a collaborative effort between the AJCC (United States) and the International Union Against Cancer (worldwide). Staging is based on the extent of the tumor (T), the extent of spread to the lymph nodes (N), and the presence of metastasis (M). Refer to Tables 3, 4, 5, and 6 for the staging of soft tissue sarcoma from the eighth edition of the AJCC Cancer Staging Manual.[3-7] The last Children's Oncology Group trial used the sixth edition AJCC Cancer Staging Manual for soft tissue sarcoma (with central pathology review).[1] A review of children with non-rhabdomyosarcoma soft tissue sarcomas was performed with data from the Surveillance, Epidemiology, and End Results (SEER) program and identified 941 patients between 1988 and 2007.[8] The COG risk stratification was validated in this cohort.
Intergroup Rhabdomyosarcoma Study Staging System
Nonmetastatic disease
- Group I: Localized tumor completely resected with histologically negative margins.
- Group II: Grossly resected tumor with microscopic residual tumor at the margin(s) and/or extension into regional lymph nodes.
- IIA: Localized, grossly resected tumor with microscopic residual disease.
- IIB: Regional disease with involved nodes completely resected with no microscopic disease. The most proximal (to the patient, most distal to the tumor) regional lymph node must be negative.
- IIC: Regional disease with involved nodes grossly resected but with evidence of residual microscopic disease at the primary site and/or histologic involvement of the most proximal regional lymph node in the dissection.
- Group III: Localized tumor, incompletely resected, or biopsy only, with gross residual tumor.
Metastatic disease
- Group IV: Any localized or regional tumor with distant metastases present at the time of diagnosis. This includes the presence of malignant cells in effusions (pleural, peritoneal) and/or cerebrospinal fluid (rare).
Recurrent/progressive disease
- Any soft tissue sarcoma that recurs after initial treatment or progresses after radiation therapy, chemotherapy, or initial surgery.
TNM Staging System
The eighth edition of the AJCC Cancer Staging Manual has designated staging by the four criteria of tumor size, nodal status, histologic grade, and metastasis and by anatomic primary tumor site (head and neck; trunk and extremities; abdomen and thoracic visceral organs; retroperitoneum; and unusual histologies and sites) (refer to Tables 3, 4, 5, and 6).[3-7] For information on unusual histologies and sites, refer to the AJCC Cancer Staging Manual.[7]
| T Category | Soft Tissue Sarcoma of the Trunk, Extremities, and Retroperitoneum | Soft Tissue Sarcoma of the Head and Neck | Soft Tissue Sarcoma of the Abdomen and Thoracic Visceral Organs |
|---|---|---|---|
| aAdapted from O'Sullivan et al.,[3] Yoon et al.,[4] Raut et al.,[5] and Pollock et al.[6] | |||
| TX | Primary tumor cannot be assessed. | Primary tumor cannot be assessed. | Primary tumor cannot be assessed. |
| T0 | No evidence of primary tumor. | ||
| T1 | Tumor ≤5 cm in greatest dimension. | Tumor ≤2 cm. | Organ confined. |
| T2 | Tumor >5 cm and ≤10 cm in greatest dimension. | Tumor >2 to ≤4 cm. | Tumor extension into tissue beyond organ. |
| T2a | Invades serosa or visceral peritoneum. | ||
| T2b | Extension beyond serosa (mesentery). | ||
| T3 | Tumor >10 cm and ≤15 cm in greatest dimension. | Tumor >4 cm. | Invades another organ. |
| T4 | Tumor >15 cm in greatest dimension. | Tumor with invasion of adjoining structures. | Multifocal involvement. |
| T4a | Tumor with orbital invasion, skull base/dural invasion, invasion of central compartment viscera, involvement of facial skeleton, or invasion of pterygoid muscles. | Multifocal (2 sites). | |
| T4b | Tumor with brain parenchymal invasion, carotid artery encasement, prevertebral muscle invasion, or central nervous system involvement via perineural spread. | Multifocal (3–5 sites). | |
| T4c | Multifocal (>5 sites). | ||
| aAdapted from O'Sullivan et al.,[3] Yoon et al.,[4] Raut et al.,[5] and Pollock et al.[6] | |
| bFor soft tissue sarcoma of the abdomen and thoracic visceral organs, N0 = no lymph node involvement or unknown lymph node status and N1 = lymph node involvement present. | |
| N0 | No regional lymph node metastasis or unknown lymph node status.b |
| N1 | Regional lymph node metastasis.b |
| aAdapted from O'Sullivan et al.,[3] Yoon et al.,[4] Raut et al.,[5] and Pollock et al.[6] | |
| bFor soft tissue sarcoma of the abdomen and thoracic visceral organs, M0 = no metastases and M1 = metastases present. | |
| M0 | No distant metastasis.b |
| M1 | Distant metastasis.b |
| Stage | T | N | M | Grade |
|---|---|---|---|---|
| aAdapted from Yoon et al. [4] and Pollock et al.[6] | ||||
| bStage IIIB for soft tissue sarcoma of the retroperitoneum; stage IV for soft tissue sarcoma of the trunk and extremities. | ||||
| IA | T1 | N0 | M0 | G1, GX |
| IB | T2, T3, T4 | N0 | M0 | G1, GX |
| II | T1 | N0 | M0 | G2, G3 |
| IIIA | T2 | N0 | M0 | G2, G3 |
| IIIB | T3, T4 | N0 | M0 | G2, G3 |
| IIIB/IVb | Any T | N1 | M0 | Any G |
| IV | Any T | Any N | M1 | Any G |
Soft Tissue Sarcoma Tumor Pathological Grading System
In most cases, accurate histopathologic classification alone of soft tissue sarcomas does not yield optimal information about their clinical behavior. Therefore, several histologic parameters are evaluated in the grading process, including the following:
- Degree of cellularity.
- Cellular pleomorphism.
- Mitotic activity.
- Degree of necrosis.
- Invasive growth.
This process is used to improve the correlation between histologic findings and clinical outcome.[9] In children, grading of soft tissue sarcoma is compromised by the good prognosis of certain tumors, such as infantile fibrosarcoma and hemangiopericytoma, which have a good prognosis in children younger than 4 years, and also angiomatoid fibrous histiocytoma and dermatofibrosarcoma protuberans, which may recur locally if incompletely excised, but usually do not metastasize.
Testing the validity of a grading system within the pediatric population is difficult because of the rarity of these neoplasms. In March 1986, the Pediatric Oncology Group (POG) conducted a prospective study on pediatric soft tissue sarcomas other than rhabdomyosarcoma and devised the POG grading system. Analysis of outcome for patients with localized soft tissue sarcomas other than rhabdomyosarcoma demonstrated that patients with grade 3 tumors fared significantly worse than those with grade 1 or grade 2 lesions. This finding suggests that this system can accurately predict the clinical behavior of nonrhabdomyosarcomatous soft tissue sarcoma.[9-11]
The grading systems developed by the POG and the French Federation of Comprehensive Cancer Centers (Fédération Nationale des Centres de Lutte Contre Le Cancer [FNCLCC]) Sarcoma Group are described below. These grading systems are being compared by the central review pathologists on the COG-ARST0332 study. The study has closed and results are pending.
POG grading system
The POG grading system is described below.[9] It is an older grading system of historical value that is no longer being used for treatment.
Grade I
Grade I lesions are based on histologic type, well-differentiated cytohistologic features, and/or age of the patient.
- Angiomatoid fibrous histiocytoma.
- Dermatofibrosarcoma protuberans.
- Liposarcoma–myxoid or well-differentiated.
- Myxoid chondrosarcoma.
- Well-differentiated malignant peripheral nerve sheath tumor.
- Well-differentiated or infantile (aged ≤4 years) fibrosarcoma.
- Well-differentiated or infantile (aged ≤4 years) hemangiopericytoma.
Grade II
Grade II lesions are soft tissue sarcomas not included in grade I or III by histologic diagnosis (with <5 mitoses/10 high-power fields or <15% necrosis):
- 15% or less of the surface area shows necrosis (primary criteria).
- The mitotic count is <5 mitotic figures per 10 high-power fields (40X objective) (primary criteria).
- Nuclear atypia is not marked (secondary criteria).
- The tumor is not markedly cellular (secondary criteria).
Grade III
Grade III lesions are similar to grade II lesions and include certain tumors known to be clinically aggressive by virtue of histologic diagnosis and non-grade I tumors (with >4 mitoses per 10 high-power fields or >15% necrosis):
- Alveolar soft part sarcoma.
- Extraskeletal osteogenic sarcoma.
- Malignant triton tumor.
- Mesenchymal chondrosarcoma.
- Pleomorphic or round-cell liposarcoma.
- Any other sarcoma not in grade I with >15% necrosis and/or ≥5 mitotic figures per 10 high-power fields (40X objective). Marked atypia and cellularity are less predictive but may assist in placing tumors in this category.
FNCLCC grading system
The FNCLCC histologic grading system was developed for adults with soft tissue sarcoma. The purpose of the grading system is to predict which patients will develop metastasis and subsequently benefit from postoperative chemotherapy.[12,13] The system is described in Table 7 and Table 8.
| FNCLCC = Fédération Nationale des Centres de Lutte Contre Le Cancer; HPF = high-power field. | |
| Tumor Differentiation | |
| Score 1 | Sarcoma closely resembling normal adult mesenchymal tissue (e.g., well-differentiated liposarcoma) |
| Score 2 | Sarcomas for which histologic typing is certain (e.g., myxoid liposarcoma) |
| Score 3 | Embryonal and undifferentiated sarcomas, sarcomas of doubtful type, and synovial sarcomas |
| Mitotic Count | |
| Score 1 | 0–9 mitoses per 10 HPF |
| Score 2 | 10–19 mitoses per 10 HPF |
| Score 3 | ≥20 mitoses per 10 HPF |
| Tumor Necrosis | |
| Score 0 | No necrosis |
| Score 1 | <50% tumor necrosis |
| Score 2 | ≥50% tumor necrosis |
| Total Score | Histologic Grade |
|---|---|
| 2–3 | Grade I |
| 4–5 | Grade II |
| 6–8 | Grade III |
Prognostic Significance of Tumor Grading
The POG and FNCLCC grading systems have proven to be of prognostic value in pediatric and adult nonrhabdomyosarcomatous soft tissue sarcomas.[14-18] In a study of 130 tumors from children and adolescents with nonrhabdomyosarcomatous soft tissue sarcoma enrolled in three prospective clinical trials, a correlation was found between the POG-assigned grade and the FNCLCC-assigned grade. However, grading did not correlate in all cases; 44 patients whose tumors received discrepant grades (POG grade 3, FNCLCC grade 1 or 2) had outcomes between concurrent grade 3 and grades 1 and 2. A mitotic index of 10 or greater emerged as an important prognostic factor.[19] The recently completed COG-ARST0332 trial will analyze data comparing the POG and FNCLCC pathologic grading systems to determine which system better correlates with clinical outcomes. The current open trial (ARST1321 [NCT02180867]) uses the FNCLCC system to assign histological grade.
References
- American Joint Committee on Cancer: AJCC Cancer Staging Manual. 6th ed. New York, NY: Springer, 2002.
- Maurer HM, Beltangady M, Gehan EA, et al.: The Intergroup Rhabdomyosarcoma Study-I. A final report. Cancer 61 (2): 209-20, 1988. [PUBMED Abstract]
- O'Sullivan B, Maki RG, Agulnik M, et al.: Soft tissue sarcoma of the head and neck. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 499-505.
- Yoon SS, Maki RG, Asare EA, et al.: Soft tissue sarcoma of the trunk and extremities. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 507-15.
- Raut CP, Maki RG, Baldini EH, et al.: Soft tissue sarcoma of the abdomen and thoracic visceral organs. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 517-21.
- Pollock RE, Maki RG, Baldini EH, et al.: Soft tissue sarcoma of the retroperitoneum. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 531-7.
- Maki RG, Folpe AL, Guadagnolo BA, et al.: Soft tissue sarcoma - unusual histologies and sites. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. New York, NY: Springer, 2017, pp 539-45.
- Waxweiler TV, Rusthoven CG, Proper MS, et al.: Non-Rhabdomyosarcoma Soft Tissue Sarcomas in Children: A Surveillance, Epidemiology, and End Results Analysis Validating COG Risk Stratifications. Int J Radiat Oncol Biol Phys 92 (2): 339-48, 2015. [PUBMED Abstract]
- Parham DM, Webber BL, Jenkins JJ 3rd, et al.: Nonrhabdomyosarcomatous soft tissue sarcomas of childhood: formulation of a simplified system for grading. Mod Pathol 8 (7): 705-10, 1995. [PUBMED Abstract]
- Recommendations for the reporting of soft tissue sarcomas. Association of Directors of Anatomic and Surgical Pathology. Mod Pathol 11 (12): 1257-61, 1998. [PUBMED Abstract]
- Skytting B, Meis-Kindblom JM, Larsson O, et al.: Synovial sarcoma--identification of favorable and unfavorable histologic types: a Scandinavian sarcoma group study of 104 cases. Acta Orthop Scand 70 (6): 543-54, 1999. [PUBMED Abstract]
- Coindre JM, Terrier P, Guillou L, et al.: Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 91 (10): 1914-26, 2001. [PUBMED Abstract]
- Guillou L, Coindre JM, Bonichon F, et al.: Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 15 (1): 350-62, 1997. [PUBMED Abstract]
- Rao BN: Nonrhabdomyosarcoma in children: prognostic factors influencing survival. Semin Surg Oncol 9 (6): 524-31, 1993 Nov-Dec. [PUBMED Abstract]
- Pisters PW, Leung DH, Woodruff J, et al.: Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 14 (5): 1679-89, 1996. [PUBMED Abstract]
- Coindre JM, Terrier P, Bui NB, et al.: Prognostic factors in adult patients with locally controlled soft tissue sarcoma. A study of 546 patients from the French Federation of Cancer Centers Sarcoma Group. J Clin Oncol 14 (3): 869-77, 1996. [PUBMED Abstract]
- Pappo AS, Fontanesi J, Luo X, et al.: Synovial sarcoma in children and adolescents: the St Jude Children's Research Hospital experience. J Clin Oncol 12 (11): 2360-6, 1994. [PUBMED Abstract]
- Pratt CB, Maurer HM, Gieser P, et al.: Treatment of unresectable or metastatic pediatric soft tissue sarcomas with surgery, irradiation, and chemotherapy: a Pediatric Oncology Group study. Med Pediatr Oncol 30 (4): 201-9, 1998. [PUBMED Abstract]
- Khoury JD, Coffin CM, Spunt SL, et al.: Grading of nonrhabdomyosarcoma soft tissue sarcoma in children and adolescents: a comparison of parameters used for the Fédération Nationale des Centers de Lutte Contre le Cancer and Pediatric Oncology Group Systems. Cancer 116 (9): 2266-74, 2010. [PUBMED Abstract]
Treatment Option Overview for Childhood Soft Tissue Sarcoma
Because of the rarity of pediatric nonrhabdomyosarcomatous soft tissue sarcomas, coordination of treatment by a multidisciplinary team comprising oncologists (pediatric or medical), pathologists, surgeons, and radiation oncologists should be considered for all children, adolescents, and young adults with these tumors. In addition, to better define the tumors' natural history and response to therapy, entry into national or institutional treatment protocols should be considered for children with rare neoplasms. Information about ongoing clinical trials is available from the NCI website.
Surgery
After an appropriate biopsy and pathologic diagnosis, every attempt is made to resect the primary tumor with negative margins before or after chemotherapy and/or radiation therapy. Involvement of a surgeon with special expertise in the resection of soft tissue sarcomas in the decision is highly desirable.
The timing of surgery depends on an assessment of the feasibility and morbidity of surgery. If the initial operation fails to achieve pathologically negative tissue margins or if the initial surgery was done without the knowledge that cancer was present, a re-excision of the affected area is performed to obtain clear, but not necessarily wide, margins.[1-4] This surgical tenet is true even if no mass is detected by magnetic resonance imaging after initial surgery.[5]; [6][Level of evidence: 3iiA]
Regional lymph node metastases at diagnosis are unusual and are most often seen in patients with epithelioid and clear cell sarcomas.[7,8] Various institutional series have demonstrated the feasibility and effectiveness of sentinel node biopsy as a staging procedure in pediatric patients with soft tissue sarcomas.[9-14]
Radiation Therapy
Considerations for radiation therapy are based on the potential for surgery, with or without chemotherapy, to obtain local control without loss of critical organs or significant functional, cosmetic, or psychological impairment. This will vary according to the following:
- Patient variables (e.g., age and sex).
- Tumor variables (e.g., histopathology, site, size, and grade).
- Surgical margin status.
- Expectations for radiation-induced morbidities (e.g., impaired bone or muscle development, organ damage, or second malignancy).
Radiation therapy can be given preoperatively. Radiation field size and dose will be based on patient and tumor variables and the operability of the tumor. Preoperative radiation therapy has been associated with excellent local control rates.[15,16] This approach has the advantage of treating smaller tissue volumes because it does not necessitate treating a postsurgical bed; it also has the advantage of somewhat lower radiation doses because relative hypoxia from surgical disruption of vasculature and scarring is not present. Preoperative radiation therapy has been associated with an increased rate of wound complications in adults, primarily in lower extremity tumors, but the degree of this is questionable.[17] Conversely, preoperative radiation therapy may lead to less fibrosis than with postoperative approaches, perhaps due to the smaller treatment volume and dose.[18]
Retroperitoneal sarcomas are unique in that radiosensitivity of the bowel to injury makes postoperative radiation therapy less desirable.[19,20] Postoperative adhesions and bowel immobility can increase the risk of damage from any given radiation dose. This contrasts with the preoperative approach in which the tumor often displaces bowel outside of the radiation field, and any exposed bowel is more mobile, which decreases exposure to specific bowel segments.
Radiation therapy can also be given postoperatively. In general, radiation is indicated for patients with inadequate surgical margins and for larger, high-grade tumors.[21,22] This is particularly important in high-grade tumors with tumor margins smaller than 1 cm.[23,24]; [25][Level of evidence: 3iiDiv] With combined surgery and radiation therapy, local control of the primary tumor can be achieved in more than 80% of patients.[26,27]
Brachytherapy and intraoperative radiation may be applicable in select situations.[27-29]; [30][Level of evidence: 3iiiDii]
Radiation volume and dose depend on the patient, tumor, and surgical variables noted above, as well as the following:
- Patient age and growth potential.
- Ability to avoid critical organs, epiphyseal plates, and lymphatics (but not the neurovascular bundles that are relatively radiation tolerant).
- Functional/cosmetic outcome.
Radiation doses are typically 45 Gy to 50 Gy preoperatively, with consideration for postoperative boost of 10 Gy to 20 Gy if resection margins are microscopically or grossly positive, or planned brachytherapy if the resection is predicted to be subtotal. However, data documenting the efficacy of a postoperative boost are lacking.[31] The postoperative radiation dose is 55 Gy to 60 Gy, or rarely, higher when unresectable gross residual disease exists.
Radiation margins are typically 2 cm to 4 cm longitudinally and encompass fascial planes axially.[32,33]
Chemotherapy
The role of postoperative chemotherapy remains unclear as evidenced by the following studies:[34]
- A meta-analysis of data from all randomized trials of adults with soft tissue sarcoma concluded that recurrence-free survival was better with postoperative chemotherapy for patients with high-grade tumors larger than 5 cm.[35]
- In a European trial, adults with completely resected soft tissue sarcoma were randomly assigned to observation or postoperative chemotherapy with ifosfamide and doxorubicin. Postoperative chemotherapy was not associated with improved event-free survival (EFS) or overall survival (OS). It is difficult to extrapolate this trial to pediatric patients because the trial included 1) a wide variety of histologies; 2) a relatively low dose of ifosfamide; 3) patients assigned to chemotherapy had definitive radiation delayed until completion of chemotherapy; and 4) almost one-half of the patients in the trial had intermediate-grade tumors. In the discussion section, the authors merged their patients with previously published series, including those from the European meta-analysis, and concluded that the results suggested a benefit for postoperative chemotherapy.[36][Level of evidence: 1iiA]
- The largest prospective pediatric trial failed to demonstrate any benefit with postoperative vincristine, dactinomycin, cyclophosphamide, and doxorubicin.[26]
- Doxorubicin and ifosfamide were used in the risk-based COG ARST0332 (NCT00346164) trial. Although this was not a randomized study, results at 2.6 years show that patients with high-risk (>5 cm and high grade), grossly resected, nonmetastatic tumors who were treated with radiation therapy and postoperative doxorubicin and ifosfamide had a 3-year EFS of 68% and OS of 81%. In patients with metastatic disease treated with preoperative chemotherapy and radiation therapy, the estimated 3-year failure-free survival was 52% and OS was 66%.[37][Level of evidence: 3iiiA]
Targeted Therapy
The use of angiogenesis and mammalian target of rapamycin (mTOR) inhibitors has been explored in the treatment of adult soft tissue sarcomas but not in pediatrics.
- In a trial of 711 randomly assigned adult patients who achieved a response or stable disease after chemotherapy, the administration of ridaforolimus was associated with a 3-week improvement in progression-free survival (PFS) when compared with placebo.[38]
- In another trial of 371 randomly assigned adult patients with metastatic soft tissue sarcoma that progressed after chemotherapy, pazopanib was compared with placebo. The median PFS for the pazopanib arm was 4.6 months compared with 1.6 months for the placebo arm. OS was not different between the two arms.[39]
- In a randomized study of 182 previously treated adult patients with recurrent liposarcoma, leiomyosarcoma, synovial sarcoma, and other sarcomas, patients with nonadipocytic tumors who were treated with regorafenib had significant improvements in progression-free survival when compared with patients who were treated with placebo.[40]
Special Considerations for the Treatment of Children With Soft Tissue Sarcoma
Cancer in children and adolescents is rare, although the overall incidence of childhood cancer has been slowly increasing since 1975.[41] Children and adolescents with cancer should be referred to medical centers that have a multidisciplinary team of cancer specialists with experience treating the cancers that occur during childhood and adolescence. This multidisciplinary team approach incorporates the skills of the following health care professionals and others to ensure that children receive treatment, supportive care, and rehabilitation that will achieve optimal survival and quality of life:
- Primary care physicians.
- Pediatric surgical specialists.
- Pediatric radiation oncologists.
- Pediatric medical oncologists/hematologists.
- Rehabilitation specialists.
- Pediatric nurse specialists.
- Social workers.
- Child life professionals.
- Psychologists.
(Refer to the PDQ Supportive and Palliative Care summaries for specific information about supportive care for children and adolescents with cancer.)
Guidelines for pediatric cancer centers and their role in the treatment of pediatric patients with cancer have been outlined by the American Academy of Pediatrics.[42] At these pediatric cancer centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate in these trials is offered to most patients/families. Multidisciplinary evaluation in pediatric cancer centers that have surgical and radiotherapeutic expertise is of critical importance to ensure the best clinical outcome for these patients. Although surgery with or without radiation therapy can be curative for a significant proportion of patients, the addition of chemotherapy might benefit subsets of children with the disease; therefore, enrollment into clinical trials is encouraged. Clinical trials for children and adolescents with cancer are generally designed to compare potentially better therapy with therapy that is currently accepted as standard. Most of the progress made in identifying curative therapies for childhood cancers has been achieved through clinical trials. Information about ongoing clinical trials is available from the NCI website.
Many therapeutic strategies for children and adolescents with soft tissue tumors are similar to those for adult patients, although there are important differences. For example, the biology of the neoplasm in pediatric patients may differ dramatically from that of the adult lesion. Additionally, limb-sparing procedures are more difficult to perform in pediatric patients. The morbidity associated with radiation therapy, particularly in infants and young children, may be much greater than that observed in adults.[43]
Improved outcomes with multimodality therapy in adults and children with soft tissue sarcomas over the past 20 years has caused increasing concern about the potential long-term side effects of this therapy in children, especially when considering the expected longer life span of children versus adults. Therefore, to maximize tumor control and minimize long-term morbidity, treatment must be individualized for children and adolescents with nonrhabdomyosarcomatous soft tissue sarcoma. These patients should be enrolled in prospective studies that accurately assess any potential complications.[44]
References
- Sugiura H, Takahashi M, Katagiri H, et al.: Additional wide resection of malignant soft tissue tumors. Clin Orthop (394): 201-10, 2002. [PUBMED Abstract]
- Cecchetto G, Guglielmi M, Inserra A, et al.: Primary re-excision: the Italian experience in patients with localized soft-tissue sarcomas. Pediatr Surg Int 17 (7): 532-4, 2001. [PUBMED Abstract]
- Chui CH, Spunt SL, Liu T, et al.: Is reexcision in pediatric nonrhabdomyosarcoma soft tissue sarcoma necessary after an initial unplanned resection? J Pediatr Surg 37 (10): 1424-9, 2002. [PUBMED Abstract]
- Paulino AC, Ritchie J, Wen BC: The value of postoperative radiotherapy in childhood nonrhabdomyosarcoma soft tissue sarcoma. Pediatr Blood Cancer 43 (5): 587-93, 2004. [PUBMED Abstract]
- Kaste SC, Hill A, Conley L, et al.: Magnetic resonance imaging after incomplete resection of soft tissue sarcoma. Clin Orthop (397): 204-11, 2002. [PUBMED Abstract]
- Chandrasekar CR, Wafa H, Grimer RJ, et al.: The effect of an unplanned excision of a soft-tissue sarcoma on prognosis. J Bone Joint Surg Br 90 (2): 203-8, 2008. [PUBMED Abstract]
- Daigeler A, Kuhnen C, Moritz R, et al.: Lymph node metastases in soft tissue sarcomas: a single center analysis of 1,597 patients. Langenbecks Arch Surg 394 (2): 321-9, 2009. [PUBMED Abstract]
- Mazeron JJ, Suit HD: Lymph nodes as sites of metastases from sarcomas of soft tissue. Cancer 60 (8): 1800-8, 1987. [PUBMED Abstract]
- Neville HL, Andrassy RJ, Lally KP, et al.: Lymphatic mapping with sentinel node biopsy in pediatric patients. J Pediatr Surg 35 (6): 961-4, 2000. [PUBMED Abstract]
- Neville HL, Raney RB, Andrassy RJ, et al.: Multidisciplinary management of pediatric soft-tissue sarcoma. Oncology (Huntingt) 14 (10): 1471-81; discussion 1482-6, 1489-90, 2000. [PUBMED Abstract]
- Kayton ML, Delgado R, Busam K, et al.: Experience with 31 sentinel lymph node biopsies for sarcomas and carcinomas in pediatric patients. Cancer 112 (9): 2052-9, 2008. [PUBMED Abstract]
- Dall'Igna P, De Corti F, Alaggio R, et al.: Sentinel node biopsy in pediatric patients: the experience in a single institution. Eur J Pediatr Surg 24 (6): 482-7, 2014. [PUBMED Abstract]
- Parida L, Morrisson GT, Shammas A, et al.: Role of lymphoscintigraphy and sentinel lymph node biopsy in the management of pediatric melanoma and sarcoma. Pediatr Surg Int 28 (6): 571-8, 2012. [PUBMED Abstract]
- Alcorn KM, Deans KJ, Congeni A, et al.: Sentinel lymph node biopsy in pediatric soft tissue sarcoma patients: utility and concordance with imaging. J Pediatr Surg 48 (9): 1903-6, 2013. [PUBMED Abstract]
- Virkus WW, Mollabashy A, Reith JD, et al.: Preoperative radiotherapy in the treatment of soft tissue sarcomas. Clin Orthop (397): 177-89, 2002. [PUBMED Abstract]
- Zagars GK, Ballo MT, Pisters PW, et al.: Preoperative vs. postoperative radiation therapy for soft tissue sarcoma: a retrospective comparative evaluation of disease outcome. Int J Radiat Oncol Biol Phys 56 (2): 482-8, 2003. [PUBMED Abstract]
- O'Sullivan B, Davis AM, Turcotte R, et al.: Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet 359 (9325): 2235-41, 2002. [PUBMED Abstract]
- Davis AM, O'Sullivan B, Turcotte R, et al.: Late radiation morbidity following randomization to preoperative versus postoperative radiotherapy in extremity soft tissue sarcoma. Radiother Oncol 75 (1): 48-53, 2005. [PUBMED Abstract]
- Baldini EH, Wang D, Haas RL, et al.: Treatment Guidelines for Preoperative Radiation Therapy for Retroperitoneal Sarcoma: Preliminary Consensus of an International Expert Panel. Int J Radiat Oncol Biol Phys 92 (3): 602-12, 2015. [PUBMED Abstract]
- Bishop AJ, Zagars GK, Torres KE, et al.: Combined Modality Management of Retroperitoneal Sarcomas: A Single-Institution Series of 121 Patients. Int J Radiat Oncol Biol Phys 93 (1): 158-65, 2015. [PUBMED Abstract]
- Marcus KC, Grier HE, Shamberger RC, et al.: Childhood soft tissue sarcoma: a 20-year experience. J Pediatr 131 (4): 603-7, 1997. [PUBMED Abstract]
- Delaney TF, Kepka L, Goldberg SI, et al.: Radiation therapy for control of soft-tissue sarcomas resected with positive margins. Int J Radiat Oncol Biol Phys 67 (5): 1460-9, 2007. [PUBMED Abstract]
- Blakely ML, Spurbeck WW, Pappo AS, et al.: The impact of margin of resection on outcome in pediatric nonrhabdomyosarcoma soft tissue sarcoma. J Pediatr Surg 34 (5): 672-5, 1999. [PUBMED Abstract]
- Skytting B: Synovial sarcoma. A Scandinavian Sarcoma Group project. Acta Orthop Scand Suppl 291: 1-28, 2000. [PUBMED Abstract]
- Hua C, Gray JM, Merchant TE, et al.: Treatment planning and delivery of external beam radiotherapy for pediatric sarcoma: the St. Jude Children's Research Hospital experience. Int J Radiat Oncol Biol Phys 70 (5): 1598-606, 2008. [PUBMED Abstract]
- Pratt CB, Pappo AS, Gieser P, et al.: Role of adjuvant chemotherapy in the treatment of surgically resected pediatric nonrhabdomyosarcomatous soft tissue sarcomas: A Pediatric Oncology Group Study. J Clin Oncol 17 (4): 1219, 1999. [PUBMED Abstract]
- Merchant TE, Parsh N, del Valle PL, et al.: Brachytherapy for pediatric soft-tissue sarcoma. Int J Radiat Oncol Biol Phys 46 (2): 427-32, 2000. [PUBMED Abstract]
- Schomberg PJ, Gunderson LL, Moir CR, et al.: Intraoperative electron irradiation in the management of pediatric malignancies. Cancer 79 (11): 2251-6, 1997. [PUBMED Abstract]
- Nag S, Shasha D, Janjan N, et al.: The American Brachytherapy Society recommendations for brachytherapy of soft tissue sarcomas. Int J Radiat Oncol Biol Phys 49 (4): 1033-43, 2001. [PUBMED Abstract]
- Viani GA, Novaes PE, Jacinto AA, et al.: High-dose-rate brachytherapy for soft tissue sarcoma in children: a single institution experience. Radiat Oncol 3: 9, 2008. [PUBMED Abstract]
- Al Yami A, Griffin AM, Ferguson PC, et al.: Positive surgical margins in soft tissue sarcoma treated with preoperative radiation: is a postoperative boost necessary? Int J Radiat Oncol Biol Phys 77 (4): 1191-7, 2010. [PUBMED Abstract]
- Wang D, Bosch W, Kirsch DG, et al.: Variation in the gross tumor volume and clinical target volume for preoperative radiotherapy of primary large high-grade soft tissue sarcoma of the extremity among RTOG sarcoma radiation oncologists. Int J Radiat Oncol Biol Phys 81 (5): e775-80, 2011. [PUBMED Abstract]
- Bahig H, Roberge D, Bosch W, et al.: Agreement among RTOG sarcoma radiation oncologists in contouring suspicious peritumoral edema for preoperative radiation therapy of soft tissue sarcoma of the extremity. Int J Radiat Oncol Biol Phys 86 (2): 298-303, 2013. [PUBMED Abstract]
- Ferrari A: Role of chemotherapy in pediatric nonrhabdomyosarcoma soft-tissue sarcomas. Expert Rev Anticancer Ther 8 (6): 929-38, 2008. [PUBMED Abstract]
- Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet 350 (9092): 1647-54, 1997. [PUBMED Abstract]
- Woll PJ, Reichardt P, Le Cesne A, et al.: Adjuvant chemotherapy with doxorubicin, ifosfamide, and lenograstim for resected soft-tissue sarcoma (EORTC 62931): a multicentre randomised controlled trial. Lancet Oncol 13 (10): 1045-54, 2012. [PUBMED Abstract]
- Spunt SL, Million L, Anderson JR, et al.: Risk-based treatment for nonrhabdomyosarcoma soft tissue sarcomas (NRSTS) in patients under 30 years of age: Children’s Oncology Group study ARST0332. [Abstract] J Clin Oncol 32 (Suppl 15): A-10008, 2014. Also available online. Last accessed April 02, 2018.
- Demetri GD, Chawla SP, Ray-Coquard I, et al.: Results of an international randomized phase III trial of the mammalian target of rapamycin inhibitor ridaforolimus versus placebo to control metastatic sarcomas in patients after benefit from prior chemotherapy. J Clin Oncol 31 (19): 2485-92, 2013. [PUBMED Abstract]
- van der Graaf WT, Blay JY, Chawla SP, et al.: Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 379 (9829): 1879-86, 2012. [PUBMED Abstract]
- Mir O, Brodowicz T, Italiano A, et al.: Safety and efficacy of regorafenib in patients with advanced soft tissue sarcoma (REGOSARC): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol 17 (12): 1732-1742, 2016. [PUBMED Abstract]
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014. [PUBMED Abstract]
- Corrigan JJ, Feig SA; American Academy of Pediatrics: Guidelines for pediatric cancer centers. Pediatrics 113 (6): 1833-5, 2004. [PUBMED Abstract]
- Suit H, Spiro I: Radiation as a therapeutic modality in sarcomas of the soft tissue. Hematol Oncol Clin North Am 9 (4): 733-46, 1995. [PUBMED Abstract]
- Spunt SL, Million L, Coffin C: The nonrhabdomyosarcoma soft tissue sarcoma. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 827-54.
Treatment of Newly Diagnosed Childhood Soft Tissue Sarcoma
Adipocytic Tumors
Liposarcoma
Liposarcoma accounts for 3% of soft tissue sarcoma in patients younger than 20 years (refer to Table 1).
Liposarcoma is rare in the pediatric population. In a review of 182 pediatric patients with adult-type sarcomas, only 14 had a diagnosis of liposarcoma.[1] One retrospective study identified 34 patients younger than 22 years from 1960 to 2011.[2] There were roughly equal numbers of male and female patients and the median age was 18 years. In an international clinicopathological review, the characteristics of 82 cases of pediatric liposarcoma were reported. The median age was 15.5 years and females were more commonly affected.[3] In both reports, the great majority of patients had myxoid liposarcoma.
Histopathologic classification
The World Health Organization (WHO) classification for liposarcoma is as follows:
- Intermediate grade (rarely metastasizing).
- Atypical lipomatous neoplasm/well-differentiated liposarcoma. These tumors do not metastasize unless they undergo dedifferentiation.
- Malignant.
- Liposarcoma, not otherwise specified (NOS).
- Myxoid liposarcoma. Pure myxoid liposarcomas are characterized by a t(12;16)(q13;p11) translocation and can metastasize but usually have an excellent outcome in the absence of a round cell component.[4]
- Dedifferentiated liposarcoma.
- Pleomorphic liposarcoma.
Clinical presentation
The majority of liposarcomas in the pediatric and adolescent age range are low grade and located subcutaneously. Metastasis to lymph nodes is very uncommon, and the great majority of metastases are pulmonary. Tumors arising in the periphery are more likely to be low grade and myxoid. Tumors arising centrally are more likely to be high grade, pleomorphic, and present with metastasis or recur with metastasis.
Prognosis
Higher grade or central tumors are associated with a significantly higher risk of death. In a retrospective review, 5-year survival for central tumors was 42%. In the international review, seven of ten patients with pleomorphic myxoid liposarcoma died because of their disease.[3] In a retrospective study of 14 patients, 5-year survival was 78% and tumor grade, histologic subtype, and primary location correlated with survival.[2]
Treatment
Treatment options for liposarcoma include the following:
Surgery is the most important treatment for liposarcoma. After surgical resection of myxoid liposarcoma, event-free survival (EFS) and overall survival (OS) are roughly 90%. If initial surgery is incomplete, re-excision should be performed to achieve a wide margin of resection. Local recurrences have been seen and are controlled with a second resection of the tumor.
There are reports of the use of chemotherapy to decrease the size of liposarcoma before surgery to facilitate complete resection, particularly in central tumors.[10,11] The role of postoperative chemotherapy for liposarcoma is poorly defined. There does not appear to be a need for any postoperative therapy for completely resected myxoid liposarcoma. Even with the use of postoperative chemotherapy, the survival of pleomorphic liposarcoma remains poor.[12]
Trabectedin has produced encouraging responses in adults with advanced myxoid liposarcoma.[13] In one study, adult patients with recurrent liposarcoma and leiomyosarcoma were randomly assigned to treatment with either trabectedin or dacarbazine. Patients treated with trabectedin had a 45% reduction in disease progression.[14][Level of evidence: 1iiDiii] There are very limited data to support the use of trabectedin in pediatric patients.[15]
Treatment options under clinical evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma, excluding myxoid liposarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with liposarcoma are eligible for this trial.
Chondro-osseous Tumors
Chondro-osseous tumors include the following tumor subtypes:
- Extraskeletal mesenchymal chondrosarcoma.
- Extraskeletal osteosarcoma.
- Soft tissue chondroma.
Extraskeletal mesenchymal chondrosarcoma
Osseous and chondromatous neoplasms account for 0.8% of soft tissue sarcoma in patients younger than 20 years (refer to Table 1).
Histopathology and molecular features
Mesenchymal chondrosarcoma is a rare tumor characterized by small round cells and hyaline cartilage that more commonly affects young adults and has a predilection for involving the head and neck region.
Mesenchymal chondrosarcoma has been associated with consistent chromosomal rearrangement. A retrospective analysis of cases of mesenchymal chondrosarcoma identified a HEY1-NCOA2 fusion in 10 of 15 tested specimens.[16] This gene fusion was not associated with chromosomal changes that could be detected by karyotyping. In one instance, translocation t(1;5)(q42;q32) was identified in a case of mesenchymal chondrosarcoma and shown to be associated with a novel IRF2BP-CDX1 fusion gene.[17]
Prognosis
A retrospective survey of European institutions identified 113 children and adults with mesenchymal chondrosarcoma. Factors associated with better outcome included the following:[18][Level of evidence: 3iiiA]
- Lack of metastatic disease at initial presentation.
- Clear resection margins.
- Administration of postoperative chemotherapy following resection for patients with initially localized disease.
Treatment
Treatment options for extraskeletal mesenchymal chondrosarcoma include the following:
A review of 15 patients younger than 26 years from the German Cooperative Soft Tissue Sarcoma Study Group (11 with soft-tissue lesions) and the German-Austrian-Swiss Cooperative Osteosarcoma Study Group (four with primary bone lesions) protocols suggests that complete surgical removal, or incomplete resection followed by radiation therapy, is necessary for local control.[19][Level of evidence: 3iiA]
A single-institution, retrospective review identified 12 pediatric patients with mesenchymal chondrosarcoma.[20] The presence of the NCOA2 rearrangement in tumors was documented in these patients. It was also confirmed that surgical resection is necessary for cure. Eleven patients presented with localized disease and one presented with pulmonary nodules. All patients received chemotherapy—six patients before and after surgical resection and six patients only after resection. All patients received postoperative chemotherapy (most commonly ifosfamide/doxorubicin) with or without radiation therapy (median dose, 59.4 Gy). At a median follow-up of 4.8 years, 5-year disease-free survival (DFS) was 68.2% (95% CI, 39.8%–96.6%) and OS was 88.9% (95% CI, 66.9%–100%).
Extraskeletal osteosarcoma
Osseous and chondromatous neoplasms account for 0.8% of soft tissue sarcomas in patients younger than 20 years (refer to Table 1).
Extraskeletal osteosarcoma is extremely rare in the pediatric and adolescent age range. A 2003 review identified only ten case reports in the medical literature.[21]
Prognosis
Extraskeletal osteosarcoma is associated with a high risk of local recurrence and pulmonary metastasis.[22]
Treatment
Treatment options for extraskeletal osteosarcoma include the following:
- Surgery followed by chemotherapy.
(Refer to the PDQ summary on Osteosarcoma and Malignant Fibrous Histiocytoma of Bone Treatment for more information.)
Treatment options under clinical evaluation
Information about National Cancer Institute NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with extraskeletal mesenchymal chondrosarcoma and extraskeletal osteosarcoma are eligible for this trial.
Fibroblastic/Myofibroblastic Tumors
Fibroblastic/myofibroblastic tumors include the following tumor subtypes:
- Fibroblastic/myofibroblastic tumors.
- Intermediate grade (locally aggressive).
- Desmoid-type fibromatosis (previously called desmoid tumor or aggressive fibromatoses).
- Giant cell fibroblastoma.
- Lipofibromatosis.
- Palmar/plantar fibromatosis.
- Intermediate grade (rarely metastasizing).
- Dermatofibrosarcoma protuberans.
- Infantile fibrosarcoma.
- Inflammatory myofibroblastic tumor.
- Low-grade myofibroblastic sarcoma.
- Myxoinflammatory fibroblastic sarcoma.
- Solitary fibrous tumor.
- Malignant.
- Intermediate grade (locally aggressive).
Desmoid-type fibromatosis
Desmoid-type fibromatosis has previously been called desmoid tumors or aggressive fibromatoses.
Risk factors
A small number of desmoid-type fibromatosis tumors may occur in association with a mutation in the adenomatous polyposis coli (APC) gene (associated with intestinal polyps and a high incidence of colon cancer). In a study of 519 patients older than 10 years with a diagnosis of desmoid-type fibromatosis, 39 (7.5%, a possible underestimation) were found to have familial adenomatous polyposis (FAP).[23] The patients with FAP and desmoid-type fibromatosis were younger, more often male, and had more abdominal wall or mesenteric tumors than did patients with desmoid-type fibromatosis without FAP.
A family history of colon cancer, the presence of congenital hyperplasia of the retinal pigment epithelium,[24,25] or location of the desmoid-type fibromatosis in the abdomen or abdominal wall [23] should prompt referral to a genetic counselor. Currently, there are no general recommendations for genetic testing in children with desmoid-type fibromatosis. Pathology and molecular characteristics of the tumor only provide guidance for screening. If the tumor has a somatic CTNNB1 mutation, screening is not necessary, because the APC gene mutation has not been described in this setting. If a CTNNB1 mutation is not identified, screening for the APC mutation may be warranted.[26,27] (Refer to the Familial Adenomatous Polyposis (FAP) section of the PDQ summary on Genetics of Colorectal Cancer for more information.)
Prognosis
Desmoid-type fibromatosis has an extremely low potential to metastasize. The tumors are locally infiltrating, and surgical control can be difficult because of the need to preserve normal structures.
These tumors have a high potential for local recurrence. Desmoid-type fibromatosis has a highly variable natural history, including well documented examples of spontaneous regression.[28] Mutations in exon 3 of the beta-catenin gene are seen in over 80% of desmoid-type fibromatosis and the mutation 45F has been associated with an increased risk of disease recurrence.[29] Repeated surgical resection can sometimes bring recurrent lesions under control.[30]
Treatment
Evaluation of the benefit of interventions for treatment of desmoid-type fibromatosis has been extremely difficult, because desmoid-type fibromatosis has a highly variable natural history. Large adult series and smaller pediatric series have reported long periods of disease stabilization and even regression without systemic therapy.[30,31]; [32][Level of evidence: 3iiiDi]
Treatment options for desmoid-type fibromatosis include the following:
- Surgery.
- Observation, for tumors that are incompletely resected or recurrent that do not pose a danger to vital organs, if other treatment options are not available.[30,33-39] Whenever possible, however, the treatment of choice is complete resection.
- Chemotherapy, for unresectable or recurrent tumors.
- Other drug therapy, such as nonsteroidal anti-inflammatory drugs (NSAIDs) or antiestrogen therapy.
- Surgery preceded or followed by radiation therapy, for incompletely resected tumors or to avoid recurrence and subsequent surgery that may result in functional or cosmetic compromise.
- Radiation therapy alone, for unresectable tumors.
The treatment of choice is resection to achieve clear margins. However, a retrospective review of children who underwent surgery for desmoid-type fibromatosis at the St. Jude Children’s Research Hospital (SJCRH) reported no correlation between surgical margins and risk of recurrence.[39]
When the diagnosis is known and complete surgical excision is not feasible, and if the tumor poses significant potential for mortality or morbidity, preoperative strategies may include the following:[40,41]
- Observation.
- Chemotherapy.
- Anti-estrogen therapy.
- NSAID therapy.
- External-beam radiation therapy.
Desmoid-type fibromatosis often behaves in a nonaggressive manner. In a study that included mostly adults with extra-abdominal primary fibromatosis, nonsurgical approaches (medical and observation) had similar 3-year EFS compared with surgery.[34] In a subsequent study of adolescents and adults with abdominal wall aggressive fibromatosis, 102 patients were treated with a watch and wait approach, of which 65 patients required no further treatment at 3 years. Approximately one-third of patients had regression of the tumor.[33]
Chemotherapy regimens may include the following:
- Combination chemotherapy using vinblastine and methotrexate produced objective responses in about one-third of patients with unresectable or recurrent desmoid-type fibromatosis.[40]
- A series of mainly adult patients with FAP and unresectable desmoid-type fibromatosis that were unresponsive to hormone therapy showed that doxorubicin plus dacarbazine followed by meloxicam (an NSAID) can be safely administered and can induce responses.[42]
- Pegylated liposomal doxorubicin has been used with some responses.[43] In a series of five patients, a median progression-free interval of 29 months was reported.[44]
- Tyrosine kinase inhibitors: A small retrospective study of adults with desmoid-type fibromatosis showed objective responses to the multi-targeted kinase inhibitor sorafenib.[45][Level of evidence: 3iiiDiv] Previous studies with imatinib did not support its use.[46,47] A small series reported symptomatic improvement and stable disease in seven patients with desmoid-type fibromatosis who were treated with pazopanib.[48]
- The NOTCH pathway has been implicated in the development of desmoid tumors.[49] Partial responses to the gamma secretase inhibitor PF-03084014 have been noted in adults with desmoid-type fibromatosis.[50][Level of evidence: 3iiiDiv]
- Hydroxyurea has been used successfully to treat a few patients after other treatments, but more data are needed.[51-53]
Other drug therapy may include the following:
- NSAIDs such as sulindac have been used in single cases for desmoid-type fibromatosis; the responses seen were usually disease stabilization.[54]
- Antiestrogen treatment, usually tamoxifen, plus sulindac has also resulted in disease stabilization.[55] A prospective trial of the combination of tamoxifen and sulindac reported few side effects, although asymptomatic ovarian cysts were common in girls. This combination showed relatively little activity, as measured by rates of response and progression-free survival (PFS).[56][Level of evidence: 2Diii]
Postoperative radiation therapy is a consideration when progression would entail additional surgery that might cause functional or cosmetic compromise and if radiation is considered acceptable in terms of morbidities.
Radiation has been used for unresectable desmoid-type fibromatosis or postoperatively for tumors with inadequate resections. The potential long-term complications of radiation therapy, especially subsequent neoplasms, make using this modality less appealing in a young population.[57]
Dermatofibrosarcoma protuberans
Dermatofibrosarcoma is a rare tumor that can be present in all age groups, but many of the reported cases arise in children.[58-60] A review of 451 cases in children younger than 20 years in the SEER database found that the incidence was 1 case per 1 million, highest among black patients aged 15 to 19 years. The most common sites were trunk and extremities, which is similar to what is found in adults. Ninety-five percent of patients underwent surgery. OS was 100% at 5 years, 98% at 15 years, and 97% at 30 years. Males had decreased survival compared with females (P < .05).[61][Level of evidence: 3iA]
Molecular features
The tumor has a consistent chromosomal translocation t(17;22)(q22;q13) that juxtaposes the COL1A1 gene with the PDGF-beta gene.
Treatment
Treatment of dermatofibrosarcoma protuberans includes the following:
- Surgery.
- Surgery preceded or followed by radiation therapy.
- Radiation therapy and imatinib therapy, for unresectable or recurrent tumors.
Most dermatofibrosarcoma tumors can be cured by complete surgical resection. Wide excision with negative margins or Mohs or modified Mohs surgery will prevent most tumors from recurring.[62] Despite the locally aggressive behavior of the tumor, lymph node or visceral metastasis rarely occurs.
In retrospective reviews, postoperative radiation therapy after incomplete excision may have decreased the likelihood of recurrence.[63,64]
When surgical resection cannot be accomplished or the tumor is recurrent, treatment with imatinib has been effective.[65-67] Because metastatic disease is more likely after multiple recurrences, radiation or other adjuvant therapy should be considered in patients with recurrence that cannot be managed surgically.[59,61]
Guidelines for workup and management of dermatofibrosarcoma protuberans have been published.[68]
Infantile fibrosarcoma
There are two distinct types of fibrosarcoma in children and adolescents: infantile fibrosarcoma (also called congenital fibrosarcoma) and fibrosarcoma that is indistinguishable from fibrosarcoma seen in adults. These are two distinct pathologic diagnoses and require different treatments. Adult-type fibrosarcoma is addressed below.
Infantile fibrosarcoma usually occurs in children younger than 1 year. It occasionally occurs in children up to age 4 years. A tumor with similar morphology has been identified in older children; in these older children, the tumors do not have the t(12;15)(ETV-NTRK3) translocation that is characteristic of the younger patients.[69] In several of these patients, BRAF gene fusions have been identified.
Clinical presentation
Infantile fibrosarcoma usually presents with a rapidly growing mass, often noted at birth or even seen in prenatal ultrasound. The tumors are often quite large at the time of presentation.[70]
Molecular features
The tumor usually has a characteristic cytogenetic translocation t(12;15)(ETV-NTRK3). Infantile fibrosarcoma shares this translocation and a virtually identical histologic appearance with mesoblastic nephroma.
Prognosis
These tumors have a low incidence of metastases at diagnosis.
Treatment
Treatment options for infantile fibrosarcoma include the following:
- Surgery followed by observation.
- Surgery followed by chemotherapy.
- Chemotherapy followed by surgery.
Complete resection is curative in the majority of patients with infantile fibrosarcoma. However, the large size of the lesion frequently makes resection without major functional consequences impossible (for instance, tumors of the extremities often require amputation for complete excision). The European pediatric group has reported that observation may also be an option in patients with group II disease after surgery.[71] Twelve patients with group II disease received no further therapy and two patients relapsed. One patient obtained a complete remission after chemotherapy. Postoperative chemotherapy was administered to patients with higher group disease and those who progressed. In a subsequent study, only one of seven patients with group II disease progressed during observation; that patient achieved complete remission with chemotherapy.[72][Level of evidence: 3iiA]
Preoperative chemotherapy has made a more conservative surgical approach possible; agents active in this setting include vincristine, dactinomycin, cyclophosphamide, and ifosfamide.[73,74]; [72,75][Level of evidence: 3iiA]; [76][Level of evidence: 3iiB]
Three studies of patients with infantile fibrosarcoma suggest that an alkylator-free regimen is effective and should be used as the first treatment choice in patients with macroscopic disease.[71,72,77] Two cases with variant LMNA/NTRK1 fusions responded to crizotinib.[78,79]
A pediatric patient (aged 16 months) with refractory infantile fibrosarcoma with constitutive activation of the tropomyosin-related kinase signaling pathway from an ETS variant gene 6–neurotrophin 3 receptor gene fusion (ETV6-NTRK3) responded to LOXO-101, with a 90% reduction in tumor size after 2 months of treatment.[80]
A patient aged 2 months with infantile fibrosarcoma was initially treated with chemotherapy. At disease progression, a response was seen with pazopanib.[81]
A rare case of spontaneous regression without treatment has been reported.[82][Level of evidence: 3iiiDiv]
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- LOXO-TRK-15003 (NCT02637687) (Oral TRK Inhibitor LOXO-101 for Treatment of Advanced Pediatric Solid or Primary Central Nervous System [CNS] Tumors): A phase I trial of the pan-TRK inhibitor LOXO-101 is being conducted for children with solid tumors or brain tumors whose disease has progressed or was nonresponsive to available therapies, and for which no standard or available curative therapy exists. LOXO-101 is a highly selective inhibitor of all three TRK family kinases.
- RXDX-101-03 (NCT02650401) (Study of RXDX-101 in Children With Recurrent or Refractory Solid Tumors and Primary CNS Tumors): This is a four-part, open-label, phase I/Ib, dose-escalation study in pediatric patients with: 1) relapsed or refractory solid tumors; 2) primary CNS tumors; 3) neuroblastoma; and 4) non-neuroblastoma, extracranial solid tumors with NTRK1/2/3, ROS1 or ALK gene rearrangements. The study is designed to explore the safety, maximum tolerated dose or recommended phase II dose, pharmacokinetics, and antitumor activity of entrectinib (RXDX-101).
Inflammatory myofibroblastic tumor
Inflammatory myofibroblastic tumor is a rare mesenchymal tumor that has a predilection for children and adolescents.[83-85]
Clinical presentation
Inflammatory myofibroblastic tumors are rare tumors that affect soft tissues and visceral organs of children and young adults.[86] They rarely metastasize but tend to be locally invasive. Usual anatomical sites of disease include soft tissue, lungs, spleen, colon, and breast.[83] A review of 42 cases of pediatric inflammatory myofibroblastic tumor of the bladder was published in 2015.[87]
Molecular features
Roughly half of inflammatory myofibroblastic tumors exhibit a clonal mutation that activates the anaplastic lymphoma kinase (ALK)-receptor tyrosine kinase gene at chromosome 2p23.[88] ROS1 and PDGFR-beta kinase fusions have been identified in 8 of 11 cases (73%) who are negative for ALK by immunohistochemistry.[89][Level of evidence: 3iiiDiv]
Prognosis
Inflammatory myofibroblastic tumor recurs frequently but is rarely metastatic.[83-85]
Treatment
Treatment options for inflammatory myofibroblastic tumor include the following:
- Surgery.
- Chemotherapy.
- Steroid therapy.
- NSAID therapy.
- Targeted therapy (ALK inhibitors).
Complete surgical removal, when feasible, is the mainstay of therapy.[90] In a series of nine patients, four patients achieved continuous remission after complete resection, three patients with residual disease recurred but later achieved continuous remission, and one patient with metastatic disease responded to multiagent chemotherapy.[91][Level of evidence: 3iiA] The benefit of chemotherapy has been noted in case reports.[92] There are case reports of response to either steroids or NSAIDs.[93,94] A series of 32 patients aged 18 years and younger found that complete excision was the mainstay of therapy, although some patients were treated with steroids or cytotoxic chemotherapy. OS was 94%; three patients relapsed and two of them died of the disease. With complete excision, with or without other treatments such as steroids, there was a high survival rate for patients with this disease.[95][Level of evidence: 3iiA]
Inflammatory myofibroblastic tumors respond to crizotinib. Two adults with ALK-rearranged inflammatory myofibroblastic tumor achieved partial response with crizotinib.[96][Level of evidence: 3iiiDiv] For pediatric patients with measurable disease, the use of crizotinib achieved partial tumor responses in three of six patients with ALK-translocated inflammatory myofibroblastic tumors.[97] A case report of a patient aged 16 years with metastatic/multifocal ALK-positive inflammatory myofibroblastic tumor demonstrated a complete response and a 3-year disease-free interval with crizotinib therapy.[98] In a phase I trial of ceritinib for adult patients previously treated with ALK inhibitors, one patient with inflammatory myofibroblastic tumor had a partial response.[99] Finally, one study included 14 patients with inflammatory myofibroblastic tumor who were treated with crizotinib. With crizotinib therapy, five patients had a complete response, seven had a partial response, and the remaining two had stable disease; no patient had relapsed at the time the article was published.[100][Level of evidence: 3iiDiv]
Adult-type fibrosarcoma
These tumors lack the translocation seen in infantile fibrosarcomas. They present like the great majority of nonrhabdomyosarcomas and the management approach is similar.
Low-grade fibromyxoid sarcoma
Low-grade fibromyxoid sarcoma is a histologically deceptive soft tissue neoplasm that most commonly affects young and middle-aged adults, is commonly located deep within the extremities, and is characterized by a FUS/CREB3L3 translocation.[101,102]
Prognosis
In a review of 33 patients (three were younger than 18 years) with low grade fibromyxoid sarcoma, 21 of 33 patients developed a local recurrence after intervals of up to 15 years (median, 3.5 years) and 15 developed metastases up to 45 years (median, 5 years) from diagnosis, most commonly to the lungs and pleura, emphasizing the need for continued follow-up of these patients.[101] Even after metastases occur, the course may be indolent.[103]
In another report, 14 of 73 cases were younger than 18 years of age. In this series with a relatively short follow up (median of 24 months), only 8 of 54 patients with adequate follow up developed local (9%) or distant (6%) recurrence. This report suggests that the behavior of this tumor might be significantly better than previously reported.[104] However, because of the occurrence of late metastases, careful monitoring of these patients is warranted.
The most recent Children's Oncology Group (COG) trial (ARST0332 [NCT00346164]) enrolled 11 patients with this tumor entity. The median age at diagnosis was 13 years and males were more commonly affected. The most common sites were the lower and upper extremity (n = 9) and none of the patients had developed local or distant disease recurrence at a median follow up of 2.7 years.[105]
Treatment
Treatment options for low-grade fibromyxoid sarcoma include the following:
- Surgery.
The limited treatment information for low-grade fibromyxoid sarcoma suggest that surgery is the treatment of choice as the tumor is not very chemosensitive.[103] There are little data regarding the use of chemotherapy and/or radiation therapy in this disease. One report suggests that trabectedin may be effective in the treatment of low-grade fibromyxoid sarcoma.[106]
Myxofibrosarcoma
Myxofibrosarcoma is a rare lesion, especially in childhood. It is typically treated with complete surgical resection.
Sclerosing epithelioid fibrosarcoma
Sclerosing epithelioid fibrosarcoma is a rare malignant sarcoma that commonly harbors EWSR1 gene rearrangements and has an aggressive clinical course.[107] It is typically treated with complete surgical excision. Long-term follow-up is recommended because local recurrence and metastases can occur late.
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with infantile fibrosarcoma, inflammatory myofibroblastic tumor, low-grade myofibroblastic tumor, myxoinflammatory fibroblastic sarcoma, solitary fibrous tumor, adult-type fibrosarcoma, low-grade fibromyxoid sarcoma, myxofibrosarcoma, and sclerosing epithelioid fibrosarcoma are eligible for this trial.
Skeletal Muscle Tumors
Rhabdomyosarcoma
Refer to the PDQ summary on Childhood Rhabdomyosarcoma Treatment for more information.
Smooth Muscle Tumors
Leiomyosarcoma
Leiomyosarcoma accounts for 2% of soft tissue sarcoma in patients younger than 20 years (refer to Table 1).
Risk factors
Among 43 children with HIV/AIDS who developed tumors, eight developed Epstein-Barr virus–associated leiomyosarcoma.[108] Survivors of hereditary retinoblastoma have a statistically significant increased risk of developing leiomyosarcoma and 78% of these were diagnosed 30 or more years after the initial diagnosis of retinoblastoma.[109]
Treatment
Treatment options for leiomyosarcoma include the following:
- Chemotherapy (trabectedin).
In an open-label study of trabectedin in adult patients with recurrent sarcomas, the best overall response rate (complete remission and partial remission) was seen in patients with leiomyosarcoma (7.5%).[110] The clinical benefit rate (includes stable disease) for leiomyosarcoma was 54%. In another adult study, patients with recurrent liposarcoma and leiomyosarcoma were randomly assigned to receive treatment with either trabectedin or dacarbazine. Patients treated with trabectedin had a 45% reduction in disease progression.[14] There are no data to support the use of trabectedin in pediatric patients.
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with leiomyosarcoma are eligible for this trial.
So-called Fibrohistiocytic Tumors
So-called fibrohistiocytic tumors include the following tumor subtypes:
- Giant cell tumor of soft tissue.
- Plexiform fibrohistiocytic tumor.
Plexiform fibrohistiocytic tumor
Plexiform histiocytic tumor is a rare, low- to intermediate-grade tumor that most commonly affects children and young adults. Depending on the series, the median age at presentation ranges from 8 to 14.5 years; however, the tumor has been described in patients as young as 3 months.[111,112]
Clinical presentation
The tumor commonly arises as a painless mass in the skin or subcutaneous tissue and most often involves the upper extremities, including the fingers, hand, and wrist.[113-115] There are rare reports of spread to regional lymph nodes or the lungs.[111,115,116]
Molecular features
No consistent chromosomal anomalies have been detected but a t(4;15)(q21;q15) translocation has been reported.[117]
Prognosis
Plexiform fibrohistiocytic tumor is an intermediate-grade tumor that rarely metastasizes.
Treatment
Surgery is the treatment of choice but local recurrence has been reported in 12% to 50% of cases.[118]
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with giant cell tumors of soft tissue and plexiform fibrohistiocytic tumor are eligible for this trial.
Tumors of Peripheral Nerves
Ectomesenchymoma
Ectomesenchymoma is a rare nerve sheath tumor that mainly occurs in children. It is a biphenotypic soft tissue sarcoma with both mesenchymal and ectodermal components. Elements similar to rhabdomyosarcoma have been identified.
The German Soft Tissue Sarcoma Group (Cooperative Weichteilsarkom Studiengruppe [CWS]) reported on six patients (ages 0.2–13.5 years) registered over 14 years.[119][Level of evidence: 3iiA] The tumors were located in various sites including the extremities, abdomen, and orbit. All six patients were treated with surgery and chemotherapy directed at rhabdomyosarcoma. Two patients received radiation therapy. Three patients recurred with rhabdomyosarcoma features. Although data are scant, it appears that the tumor may respond to chemotherapy.[119]
Malignant peripheral nerve sheath tumor
Malignant peripheral nerve sheath tumors account for 5% of soft tissue sarcoma in patients younger than 20 years (refer to Table 1).
Risk factors
Malignant peripheral nerve sheath tumor can arise sporadically and in children with type 1 neurofibromatosis (NF1).[120]
Molecular features
Inactivating mutations of SUZ12 have been described in these tumors and are absent in neurofibromas.[121]
Prognosis
Features associated with a favorable prognosis include the following:[120,122-124]
- Smaller tumor size. In a multivariate analysis, only tumor size and nuclear p53 expression were found to be independent predictors of disease-specific survival.[123]
- Male sex and non-Hispanic white race.[125]
- No metastasis at presentation. A retrospective review of 140 patients with malignant peripheral nerve sheath tumor from the MD Anderson Cancer Center included children and adolescents. The disease-specific survival at 10 years was 32%. In this series, presence of metastatic disease was associated with a much worse prognosis.[123]
- Lower stage.
- Lower histologic grade.
- Extremity as the primary site.
Features associated with an unfavorable prognosis include the following:[126]
- High grade.
- Deep tumor location.
- Locally advanced stage at diagnosis.
- Macroscopically incomplete resection (R2).
For patients with localized disease in the MD Anderson Cancer Center study, there was no significant difference in outcome between patients with and without NF1.[123] In other studies, it was not clear whether the absence of NF1 is a favorable prognostic factor as it has been associated with both favorable [122] and unfavorable outcomes.[120,122,124] In the French Sarcoma Group study, NF1 was associated with other adverse prognostic features, but was not an independent predictor of poor outcome.[126] The Italian Sarcoma Group reported on outcomes after recurrence in 73 children and adolescents with malignant peripheral nerve sheath tumor.[127][Level of evidence: 3iiiA] The median overall survival after first relapse was 11 months, and the survival rates were 39.2% at 1 year and 15.8% at 5 years. The factors associated with a better prognosis for these patients who relapsed were less initial tumor invasiveness, longer time to relapse, and the achievement of a secondary complete remission (which was related to the feasibility of radical surgery).
Treatment
Treatment options for malignant peripheral nerve sheath tumor include the following:
Complete surgical removal of the tumor, whenever possible, is the mainstay of treatment.
The role of radiation therapy is difficult to assess, but durable local control of known postoperative microscopic residual tumor is not assured after radiation therapy.
Chemotherapy has achieved objective responses in childhood malignant peripheral nerve sheath tumor. A large retrospective analysis of the German and Italian experience with malignant peripheral nerve sheath tumor reported that 65% of measurable tumors had objective responses to ifosfamide-containing chemotherapy regimens, but the analysis did not conclusively demonstrate improved survival for chemotherapy.[120] This retrospective analysis also noted a trend toward improved outcome with postoperative radiation therapy.[120] A series of 37 young patients with malignant peripheral nerve sheath tumor and NF1 showed that most patients had large invasive tumors that were poorly responsive to chemotherapy; PFS was 19% and 5-year OS was 28%.[128]
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with malignant peripheral nerve sheath tumor are eligible for this trial.
- SARC023 (NCT02008877) (Ganetespib and Sirolimus in Patients With Malignant Peripheral Nerve Sheath Tumors): This trial is testing the combination of ganetespib, the heat shock protein inhibitor, and sirolimus, the mammalian target of rapamycin (mTOR) inhibitor, for the treatment of patients with unresectable or metastatic malignant peripheral nerve sheath tumors. Patients with unresectable soft tissue or bone sarcomas are eligible for phase I of the trial. Patients with unresectable malignant peripheral nerve sheath tumors are eligible for phase II of the trial. Eligibility is restricted to patients aged 18 years and older.
- NCT02601937 (A Phase 1 Study of the EZH2 Inhibitor Tazemetostat in Pediatric Subjects With Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma): Patients with INI1-negative tumors are eligible for targeted treatment with an EZH2 inhibitor. This is a phase I, open-label, dose-escalation, and dose-expansion study with a twice-daily oral dose of tazemetostat.
- ADVL1522 (NCT02452554) (Lorvotuzumab Mertansine in Treating Younger Patients with Relapsed or Refractory Wilms Tumor, Rhabdomyosarcoma, Neuroblastoma, Pleuropulmonary Blastoma, Malignant Peripheral Nerve Sheath Tumor, or Synovial Sarcoma): This is a phase II study of IMGN901 (lorvotuzumab mertansine) in children with relapsed or refractory Wilms tumor, rhabdomyosarcoma, neuroblastoma, pleuropulmonary blastoma, malignant peripheral nerve sheath tumor, and synovial sarcoma. This trial is studying the effects of IMGN901, an antibody-drug conjugate that links a potent antimitotic to antibodies that target CD56.
Malignant triton tumor
Malignant triton tumors are a variant of malignant peripheral nerve sheath tumors. They occur most often in patients with neurofibromatosis type I and consist of neurogenic and rhabdomyoblastic components. Malignant triton tumors are high-grade malignancies. They usually occur before age 35 years and are very rare in children (case reports only).[129]
Malignant triton tumors are not usually responsive to chemotherapy and radiation therapy but have been treated with rhabdomyosarcoma therapy.[129][Level of evidence: 3iiiA] (Refer to the PDQ summary on Childhood Rhabdomyosarcoma Treatment for more information.)
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with malignant triton tumor are eligible for this trial.
Pericytic (Perivascular) Tumors
Myopericytoma
Infantile hemangiopericytoma is a subtype of myopericytoma.
Hemangiopericytoma is a highly vascularized tumor of uncertain origin.
Histology
Histologically, hemangiopericytomas are composed of packed round or fusiform cells that are arranged around a complex vasculature, forming many branch-like structures. Hyalinization is often present. Infantile hemangiopericytomas have similar histology but many are multilobular with vasculature outside the tumor mass.[130]
Treatment and outcome
Treatment of infantile hemangiopericytomas includes the following:
- Chemotherapy.
In a series of 17 children, the differences in metastatic potential and response to treatment were clearly demonstrated for adult and infantile hemangiopericytomas.[131] Eleven children were older than 1 year. Several of these patients had disease in the lymph nodes or lungs. Six patients with stage II or III disease progressed and died. Three patients with stage I disease survived, although one had recurrence in the lungs. Six patients had infantile hemangiopericytoma, most were greater than stage I (5 of 6). All six patients survived and three had good responses to vincristine, actinomycin, and cyclophosphamide. Hemangiopericytoma in children younger than 1 year seems to have a better prognosis than in children older than 1 year.[132-134]
Infantile myofibromatosis
This entity is a fibrous tumor of infancy and childhood that most commonly presents in the first 2 years of life.[135] The lesion can present as a single subcutaneous nodule (myofibroma) most commonly involving the head and neck region or lesions can affect multiple skin areas, muscle, and bone (myofibromatosis).[136-139]
An autosomal dominant form of the disease has been described and it is associated with germline mutations of the PDGFRB gene.[140]
Treatment
These lesions have an excellent prognosis and can regress spontaneously.
About one-third of cases with multicentric involvement will also have visceral involvement, and the prognosis for these patients is poor.[138,139,141] The use of combination therapy with vincristine/dactinomycin and vinblastine/methotrexate have proven effective in cases of multicentric disease with visceral involvement and in cases in which the disease has progressed and has threatened the life of the patient (e.g., upper airway obstruction).[138,139,142]
Tumors of Uncertain Differentiation
Tumors of uncertain differentiation include the following tumor subtypes:
- Alveolar soft part sarcoma.
- Clear cell sarcoma of soft tissue.
- Desmoplastic small round cell tumor.
- Epithelioid sarcoma.
- Extrarenal rhabdoid tumor.
- Extraskeletal myxoid chondrosarcoma.
- Neoplasms with perivascular epithelioid cell differentiation (PEComa NOS, malignant)
- Primitive neuroectodermal tumor (PNET)/extraskeletal Ewing tumor.
- Synovial sarcoma (NOS, spindle cell, and biphasic varieties).
Alveolar soft part sarcoma
Alveolar soft parts sarcomas account for 1.4% of soft tissue sarcomas in patients younger than 20 years (refer to Table 1).
Clinical presentation
The median age at presentation is 25 years, and alveolar soft part sarcoma most commonly arises in the extremities but can occur in the oral and maxillofacial region.[143-145] Alveolar soft part sarcoma in children can present with evidence of metastatic disease.[146]
Molecular features
This tumor of uncertain histogenesis is characterized by a consistent chromosomal translocation t(X;17)(p11.2;q25) that fuses the ASPSCR1 gene with the TFE3 gene.[147,148]
Prognosis
Alveolar soft part sarcoma in children may have an indolent course.[146] Patients with alveolar soft part sarcoma may relapse several years after a prolonged period of apparent remission.[149] Because these tumors are rare, all children with alveolar soft part sarcoma should be considered for enrollment in prospective clinical trials.
In a series of 19 treated patients, one group reported a 5-year OS rate of 80%, a 91% OS rate for patients with localized disease, a 100% OS rate for patients with tumors 5 cm or smaller, and a 31% OS rate for patients with tumors larger than 5 cm.[150] In another series of 33 patients, OS was 68% at 5 years from diagnosis and 53% at 10 years from diagnosis. Survival was better for smaller tumors (≤5 cm) and completely resected tumors.[151][Level of evidence: 3iiA] Delayed metastases to the brain and lung are uncommon.[143] A retrospective review of children and young adults younger than 30 years (median age, 17 years; range, 1.5–30 years) from four institutions identified 69 patients treated primarily with surgery between 1980 and 2014.[152][Level of evidence: 3iiA] The ASPL-TFE3 translocation was present in all 26 patients tested. There were 19 patients with Intergroup Rhabdomyosarcoma Study (IRS) postsurgical staging group I tumors (28%), 7 patients with IRS group II tumors (10%), 5 patients with IRS group III tumors (7%), and 38 patients with IRS group IV tumors (55%). The 5-year EFS was 80% and the OS was 87% for the 31 patients with localized tumors (IRS postsurgical groups I, II, and III). The 5-year EFS was 7% and the OS was 61% for the 38 patients with metastatic tumors (IRS postsurgical group IV).
Treatment
Treatment options for alveolar soft part sarcoma include the following:
The standard approach is complete resection of the primary lesion.[150] If complete excision is not feasible, radiation therapy should be administered. A study from China reported on 18 patients with alveolar soft part sarcoma of the oral and maxillofacial region; 15 patients were younger than 30 years.[145][Level of evidence: 3iiDii] Surgical removal with negative margins was the primary treatment. All patients survived, and only one patient had metastatic disease recurrence.
A series of 51 pediatric patients aged 0 to 21 years with alveolar soft part sarcoma found an OS rate at 10 years of 78% and an EFS rate of about 63%. Patients with localized disease (n = 37) had a 10-year OS of 87%, and the 14 patients with metastases at diagnosis had a 10-year OS of 44%, partly resulting from surgical removal of primary tumor and lung metastases in some patients. Only 3 of 18 patients (17%) with measurable disease had a response to conventional antisarcoma chemotherapy, but two of four patients treated with sunitinib had a partial response.[143][Level of evidence: 3iiiA] There have been sporadic reports of objective responses to interferon-alpha and bevacizumab.[143,153,154]
A small retrospective study of nine adult patients with metastatic alveolar soft part sarcoma treated with sunitinib reported partial response in five patients and stable disease in two patients.[155][Level of evidence: 3iiiDiv] In a phase II trial of cediranib, an inhibitor of all three known vascular epidermal growth factor receptors, 15 of 43 adult patients (35%) with metastatic alveolar soft part sarcoma had a partial response.[156][Level of evidence: 3iiDiv]
There have been no open trials for patients with metastatic alveolar soft part sarcoma.
Treatment options under clinical evaluation for alveolar soft part sarcoma
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- NCT00942877 (Phase II Study of Cediranib [AZD2171] in Patients With Alveolar Soft Part Sarcoma): A phase II study of cediranib in patients with alveolar soft part sarcoma is being conducted in patients younger than 16 years at the Clinical Center of the National Institutes of Health.
- NCT01391962 (Sunitinib or Cediranib for Alveolar Soft Part Sarcoma): A phase II trial in which patients with metastatic alveolar soft part sarcoma are randomly assigned to either sunitinib or cediranib monotherapy, with crossover at disease progression. Patients aged 16 years and older are eligible. This study is being conducted at the Clinical Center of the National Institutes of Health.
Clear cell sarcoma of soft tissue
Clear cell sarcoma (formerly and inappropriately called malignant melanoma of soft parts) is a rare soft tissue sarcoma that typically involves the deep soft tissues of the extremities. It is also called clear cell sarcoma of tendons and aponeuroses. The tumor often affects adolescents and young adults.
Patients who have small, localized tumors with low mitotic rate and intermediate histologic grade fare best.[157]
Clinical presentation
The tumor most commonly affects the lower extremity, particularly the foot, heel, and ankle.[158,159] It has a high propensity for nodal dissemination, especially metastases to regional lymph nodes (12%–43%).[159,160] The tumor typically has an indolent clinical course.
Molecular features
Clear cell sarcoma of soft tissue is characterized by an EWS-ATF1 fusion.[161]
Treatment
Treatment options for clear cell sarcoma of soft tissue include the following:
In a series of 28 pediatric patients reported by the Italian and German Soft Tissue Cooperative Studies, the median age at diagnosis was 14 years and the lower extremity was the most common primary site (50%). Surgery with or without radiotherapy is the treatment of choice and offers the best chance for cure. In this series, 12 of 13 patients with completely resected tumors were cured. For patients with more advanced disease the outcome is poor and chemotherapy is rarely effective.[162]; [163][Level of evidence: 3iiDii]
Desmoplastic small round cell tumor
Desmoplastic small round cell tumor is a rare primitive sarcoma.
Clinical presentation
Desmoplastic small round cell tumor most frequently involves the abdomen, pelvis, or tissues around the testes, but it may occur in the kidney.[164-167] The tumor occurs more commonly in males and may spread to the lungs and elsewhere. Peritoneal and pelvic lesions frequently have widespread peritoneal implants.[168]
In a large, single-institution series of 65 patients, a correlation was made between computed tomography (CT) scans in most patients and positron-emission tomography (PET)/CT scans in 11 patients. PET/CT scans had very few false-negative results and detected metastatic sites missed on conventional CT scans.[168]
Molecular features
Cytogenetic studies of these tumors have demonstrated the recurrent translocation t(11;22)(p13;q12), which has been characterized as a fusion of the WT1 and EWS genes.[167,169] The WT1-EWS fusion confirms the diagnosis of desmoplastic small round cell tumor.
Prognosis
The overall prognosis for desmoplastic small round cell tumor remains extremely poor, with reported rates of death at 90%. Greater than 90% tumor resection either at presentation or after preoperative chemotherapy may be a favorable prognostic factor for OS.[170,171]; [172][Level of evidence: 3iiiA]
Treatment
There is no standard approach to the treatment of desmoplastic small round cell tumor.
Treatment options for desmoplastic small round cell tumor include the following:
- Surgery.
- Chemotherapy followed by surgery.
- Radiation therapy.
Complete surgical resections are rare, and the overall prognosis for desmoplastic small round cell tumor remains extremely poor, with reported rates of death at 90%. Treatment may include chemotherapy, surgery, and radiation therapy. Multiagent chemotherapy analogous to that used for sarcomas has been used, as well as total abdominal radiation therapy.[164,165,170,173-176]
A single-institution study reported that five of five patients with recurrent desmoplastic small round cell tumor had partial responses to treatment with the combination of vinorelbine, cyclophosphamide, and temsirolimus.[177]
The Center for International Blood and Marrow Transplant Research (CIBMTR) analyzed patients with desmoplastic small round cell tumor in their registry who received consolidation with high dose chemotherapy and autologous stem cell reconstitution.[178] While this retrospective registry analysis suggested some benefit for this approach, other investigators have abandoned the approach because of excessive toxicity and lack of efficacy.[170]
Epithelioid sarcoma
Epithelioid sarcoma is a rare mesenchymal tumor of uncertain histogenesis which displays multilineage differentiation.[179]
Clinical presentation
Epithelioid sarcoma commonly presents as a slowly growing firm nodule based in the deep soft tissue; the proximal type predominantly affects adults and involves the axial skeleton and proximal sites. The tumor is highly aggressive and has a propensity for lymph node metastases.
Molecular features
Epithelioid sarcoma is characterized by inactivation of the SMARCB1 gene, which is present in both conventional and proximal types of epithelioid sarcoma.[180] This abnormality leads to increased dependence on EZH2 and tumor formation.[181]
Treatment
Treatment options for epithelioid sarcoma include the following
- Chemotherapy.
- Surgery.
- Surgery preceded or followed by radiation therapy.
Patients should be carefully evaluated for the presence of involved lymph nodes; suspicious lymph nodes should be biopsied. Surgical removal of primary and recurrent tumor(s) is the most effective treatment.[182][Level of evidence: 3iiiA]
In a review of 30 pediatric patients with epithelioid sarcoma (median age at presentation, 12 years), responses to chemotherapy were reported in 40% of patients using sarcoma-based regimens, and 60% of patients were alive at 5 years after initial diagnosis.[183] A single-institution retrospective review of 20 patients, including children and adults (median age, 27.3 years), found no difference in the probability of recurrence between patients who received chemotherapy and those who did not receive chemotherapy and suggested that radiation therapy may be useful.[182]
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- NCT02601937 (A Phase 1 Study of the EZH2 Inhibitor Tazemetostat in Pediatric Subjects With Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma): Patients with INI1-negative tumors are eligible for targeted treatment with an EZH2 inhibitor. This is a phase I, open-label, dose-escalation, and dose-expansion study with a twice-daily oral dose of tazemetostat.
Extrarenal (extracranial) rhabdoid tumor
Malignant rhabdoid tumors were first described in children with renal tumors in 1981 (refer to the PDQ summary on Wilms Tumor and Other Childhood Kidney Tumors Treatment for more information) and were later found in a variety of extrarenal sites. These tumors are uncommon and highly malignant, especially in children younger than 2 years.
Extrarenal (extracranial) rhabdoid tumors account for 2% of soft tissue sarcoma in patients younger than 20 years (refer to Table 1).
Molecular features
The first sizeable series of 26 children with extrarenal extracranial malignant rhabdoid tumor of soft tissues came from patients enrolled on the Intergroup Rhabdomyosarcoma Studies I through III during a review of pathology material. Only five patients (19%) were alive without disease.[184] Later, investigation of children with atypical teratoid/rhabdoid tumors of the brain, as well as those with renal and extrarenal malignant rhabdoid tumors, found germline and acquired mutations of the SMARCB1 gene in all 29 tumors tested.[185] Rhabdoid tumors may be associated with germline mutations of the SMARCB1 gene and may be inherited from an apparently unaffected parent.[186] This observation was extended to 32 malignant rhabdoid tumors at all sites in patients whose mean age at diagnosis was 12 months.[187]
Prognosis
In a Surveillance, Epidemiology, and End Results (SEER) study of 229 patients with renal, central nervous system, and extrarenal malignant rhabdoid tumor, patients aged 2 to 18 years, limited extent of tumor, and delivery of radiation therapy were shown to affect the outcome favorably compared with other patients (P < .002 for each comparison). Site of the primary tumor was not prognostically significant. OS at 5 years was 33%.[188]
Treatment
Treatment includes surgical removal when possible, chemotherapy as used for soft tissue sarcomas (but no single regimen is currently accepted as best), and radiation therapy.[189][Level of evidence: 3iA]; [190,191][Level of evidence: 3iiiB]
Responses to alisertib have been documented in four patients with central nervous system (CNS) atypical teratoid/rhabdoid tumors.[192] (Refer to the PDQ summary on Childhood Central Nervous System Atypical Teratoid/Rhabdoid Tumor Treatment summary for more information about CNS atypical teratoid/rhabdoid tumors.)
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- NCT02601937 (A Phase 1 Study of the EZH2 Inhibitor Tazemetostat in Pediatric Subjects With Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma): Patients with INI1-negative tumors are eligible for targeted treatment with an EZH2 inhibitor. This is a phase I, open-label, dose-escalation, and dose-expansion study with a twice-daily oral dose of tazemetostat.
Extraskeletal myxoid chondrosarcoma
Extraskeletal myxoid chondrosarcoma is relatively rare among soft tissue sarcomas, representing only 2.3% of all soft tissue sarcoma.[193] It has been reported in children and adolescents.[194]
Molecular features
Extraskeletal myxoid chondrosarcoma is a multinodular neoplasm. The rounded cells are arranged in cords and strands in a chondroitin sulfate myxoid background. Several cytogenetic abnormalities have been identified (refer to Table 2), with the most frequent being the translocation t(9;22)(q22;q12), involving the EWSR1/NR4A3 genes.[195]
Prognosis
The tumor has traditionally been considered of low-grade malignant potential.[196] However, recent reports from large institutions showed that extraskeletal myxoid chondrosarcoma has significant malignant potential, especially if patients are followed for a long time.[197,198] Patients tend to have slow protracted courses. Nodal involvement has been well described. Local recurrence (57%) and metastatic spread to lungs (26%) have been reported.[198]
Treatment
Treatment options for extraskeletal myxoid chondrosarcoma include the following:
- Surgery.
- Radiation therapy.
The therapeutic benefit of chemotherapy has not been established. Aggressive local control and resection of metastases led to OS of 87% at 5 years and 63% at 10 years. Tumors were relatively resistant to radiation therapy.[197]
There may be potential genetic targets for small molecules, but these should be studied as part of a clinical trial. In an adult study, six of ten patients who received sunitinib achieved a partial response.[199]
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- NCT02601937 (A Phase 1 Study of the EZH2 Inhibitor Tazemetostat in Pediatric Subjects With Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma): Patients with INI1-negative tumors are eligible for targeted treatment with an EZH2 inhibitor. This is a phase I, open-label, dose-escalation, and dose-expansion study with a twice-daily oral dose of tazemetostat.
Neoplasms with perivascular epithelioid cell differentiation (PEComas)
Risk factors and molecular features
Benign PEComas are common in tuberous sclerosis, an autosomal dominant syndrome that also predisposes to renal cell cancer and brain tumors. Tuberous sclerosis is caused by germline inactivation of either TSC1 (9q34) or TSC2 (16p13.3), and the same tumor suppressor genes are inactivated somatically in sporadic PEComas.[200] Inactivation of either gene results in stimulation of the mTOR pathway, providing the basis for the treatment of nonsurgically curable PEComas with mTOR inhibitors.[201,202] A small proportion of PEComas have TFE3 rearrangements with fusions involving various genes including SFPQ/PSF and RAD51B.[203]
Clinical presentation
PEComas occur in various rare gastrointestinal, pulmonary, gynecologic, and genitourinary sites. Soft tissue, visceral, and gynecologic PEComas are more commonly seen in middle-aged female patients and are usually not associated with the tuberous sclerosis complex.[204] The disease course may be indolent.
Prognosis
Most PEComas have a benign clinical course, but malignant behavior has been reported and can be predicted based on the size of the tumor, mitotic rate, and presence of necrosis.[205]
Treatment
Treatment options have not been defined. Treatment may include surgery or observation followed by surgery when the tumor is large.[206]
Clinical activity with mTOR inhibitors, such as sirolimus, in tumors with evidence of mTORC1 activation and TSC loss has been well documented.[207]
Primitive neuroectodermal tumor (PNET)/extraskeletal Ewing tumor
(Refer to the PDQ summary on Ewing Sarcoma Treatment for more information.)
Synovial sarcoma
Synovial sarcoma accounts for 9% of soft tissue sarcomas in patients younger than 20 years (refer to Table 1).
Synovial sarcoma is one of the most common nonrhabdomyosarcomatous soft tissue sarcomas in children and adolescents. In a 1973 to 2005 SEER review, 1,268 patients with synovial sarcoma were identified. Approximately 17% of these patients were children and adolescents and the median age at diagnosis was 34 years.[208]
Histologic classification
Synovial sarcoma can be subclassified as the following types:
- Synovial sarcoma, NOS.
- Synovial sarcoma, spindle cell.
- Synovial sarcoma, biphasic.
Clinical presentation
The most common tumor location is the extremities, followed by trunk and head and neck.[208] Rarely, a synovial sarcoma may arise in the heart or pericardium.[209]
The most common site of metastasis is the lung.[210,211] The risk of metastases is highly influenced by tumor size; it is estimated that patients with tumors that are larger than 5 cm have a 32-fold risk of developing metastases when compared with other patients.
Diagnostic evaluation
The diagnosis of synovial sarcoma is made by immunohistochemical analysis, ultrastructural findings, and demonstration of the specific chromosomal translocation t(x;18)(p11.2;q11.2). This abnormality is specific for synovial sarcoma and is found in all morphologic subtypes. Synovial sarcoma results in rearrangement of the SYT gene on chromosome 18 with one of the subtypes (1, 2, or 4) of the SSX gene on chromosome X.[212,213] It is thought that the SYT/SSX18 transcript promotes epigenetic silencing of key tumor suppressor genes.[214]
In one report, reduced INI1 nuclear reactivity on immunohistochemical staining was seen in 49 cases of synovial sarcoma, suggesting that this pattern may help distinguish synovial sarcoma from other histologies.[215]
Prognosis
Patients younger than 10 years have more favorable outcomes and clinical features, including extremity primaries, smaller tumors, and localized disease, than do older patients.[208,216] A meta-analysis also suggested that response to chemotherapy was correlated with improved survival.[217]
The following studies have reported multiple factors associated with unfavorable outcomes:
- In a retrospective analysis of synovial sarcoma in children and adolescents who were treated in Germany and Italy, tumor size (>5 cm or ≤5 cm in greatest dimension) was an important predictor of EFS.[218] In this analysis, local invasiveness conferred an inferior probability of EFS, but surgical margins were not associated with clinical outcome.
- In a single-institution retrospective analysis of 111 patients with synovial sarcoma who were younger than 22 years at diagnosis, larger tumor size, greater depth in tissue, greater local invasiveness, and more proximal tumor location were associated with poorer OS.[219][Level of evidence: 3iiA]
- A multicenter analysis of 219 children from various treating centers including Germany, SJCRH, Instituto Tumori, and MD Anderson Cancer Center reported an estimated 5-year OS of 80% and EFS rate of 72%.[217] In this analysis, an interaction between tumor size and invasiveness was observed; in multivariate analysis, patients with large or invasive tumors or with Intergroup Rhabdomyosarcoma Study Clinical Group III disease (localized, incompletely resected or with biopsy only) and IV (metastases at diagnosis) had decreased OS. Treatment with radiation therapy was related to improved OS (hazard ratio, 0.4; 95% confidence interval, 0.2–0.7). In Intergroup Rhabdomyosarcoma Study Group III patients, objective response to chemotherapy (18 of 30 [60%]) correlated with improved survival. In adults, factors such as International Union Against Cancer/American Joint Committee on Cancer stage III and stage IVA, tumor necrosis, truncal location, elevated mitotic rate, age, and histologic grade have been associated with a worse prognosis.[220-222]
- Expression and genomic index prognostic signatures have been studied in synovial sarcoma. Complex genomic profiles, with greater rearrangement of the genome, are more common in adults than in younger patients with synovial sarcoma and are associated with a higher risk of metastasis.[223]
- A review of 84 patients with localized synovial sarcoma who had information on fusion status (SYT-SSX) and histologic grading found no difference in OS according to these criteria. However, for tumor size at diagnosis, the study showed that patients with tumors between 5 cm and 10 cm had a worse prognosis than those with smaller tumors (P = .02), and patients with tumors larger than 10 cm had even worse OS (P = .0003).[224][Level of evidence: 3iiiA]
- The German CWS group reviewed 27 evaluable patients younger than 21 years with pulmonary metastases among 296 patients with synovial sarcoma. Metastases involved the lungs in all patients. The 5-year EFS rate was 26%, and the OS rate was 30%. The most important prognostic factor at presentation was that the metastases were limited to one lesion in one lung or one lesion in both lungs (a group they termed oligometastatic). Treatment elements associated with superior survival were adequate local therapy of the primary tumor and, if feasible, for the metastases. The use of whole-lung irradiation did not correlate with better outcomes.[225][Level of evidence: 3iiA]
Survival after relapse is poor (30% at 5 years). Factors associated with outcome after relapse include duration of first remission (> or ≤ 18 months) and lack of a second remission.[226]
Treatment
Treatment options for synovial sarcoma include the following:
The COG and the European Pediatric Soft Tissue Sarcoma Study Group reported a combined analysis of 60 patients younger than 21 years with localized synovial sarcoma prospectively assigned to surgery without adjuvant radiation therapy or chemotherapy.[227] Enrollment was limited to patients with initial complete resection with histologically free margins, with a grade 2 tumor of any size or a grade 3 tumor 5 cm or smaller. The 3-year EFS was 90% (median follow-up, 5.2 years; range, 1.9–9.1). All eight events were local tumor recurrence; no metastatic recurrences were seen. All patients with recurrent disease were effectively treated with salvage therapy, resulting in 100% OS.
Synovial sarcoma appears to be more sensitive to chemotherapy than many other soft tissue sarcomas, and children with synovial sarcoma seem to have a better prognosis when compared with adults.[11,211,222,228-232] The most commonly used regimens for the treatment of synovial sarcoma incorporate ifosfamide and doxorubicin.[217,231,233] Response rates to the ifosfamide and doxorubicin regimen are higher than in other nonrhabdomyosarcomatous soft tissue sarcomas.[234]
Several studies have reported the following chemotherapy-associated treatment findings:
- Several treatment centers advocate postoperative chemotherapy after resection and radiation therapy of synovial sarcoma in children and young adults.[217,218,235-237]
- The International Society of Pediatric Oncology-Malignant Mesenchymal Tumors studies showed that select patients (young age, <5 cm resected tumors) with nonmetastatic synovial sarcoma can have excellent outcome in the absence of radiation, but it is still unclear whether that approach obviates an advantage of radiation for local or regional control.[236]
- A German trial suggested a benefit for postoperative chemotherapy in children with synovial sarcoma.[237]
- A meta-analysis also suggested that chemotherapy may provide benefit.[217]
- In the most recent COG ARST0332 (NCT00346164) study, 129 patients with synovial sarcoma were prospectively treated using a risk-based therapy program (as detailed in the prognosis section), of which 43 were categorized as low risk, 66 as intermediate risk, and 20 as high risk. At a median follow-up of 2.6 years, 3-year EFS for low-, intermediate-, and high-risk groups were 83%, 79%, and 16%, respectively. The use of risk factor–directed therapy accurately predicted outcomes.[238]
- The European Pediatric Soft Tissue Sarcoma Study Group performed a prospective study of patients younger than 21 years with synovial sarcoma (CCLG-EPSSG-NRSTS-2005 [NCT00334854]).[239][Level of evidence: 3iiA] Patients were stratified into the following three risks groups and nonrandomly assigned to treatment by risk group:
- Low-risk patients had Intergroup Rhabdomyosarcoma Study (IRS) group I tumors less than 5 cm in size and nonaxial primary tumors.
- Intermediate-risk patients had no axial primary tumors and IRS group I tumors greater than 5 cm or IRS group II tumors.
- High-risk patients included all patients with axial primary sites (head and neck, lung and pleura, trunk, retroperitoneal), IRS group III tumors, or N1 tumors.
Outcomes for patients treated on the CCLG-EPSSG-NRSTS-2005 trial are described in Table 9.
Table 9. Event-Free Survival (EFS) and Overall Survival (OS) in Patients With Low-, Intermediate-, and High-Risk Synovial Sarcoma Treated on the CCLG-EPSSG-NRSTS-2005 Trial Risk Group Treatment 3-Year EFS (%) 3-Year OS (%) IRS = Intergroup Rhabdomyosarcoma Study; RT = radiation therapy. aChemotherapy was ifosfamide/doxorubicin, with doxorubicin omitted during radiation therapy. b59.4 Gy in cases without the option of secondary resection; 50.4 Gy as preoperative radiation therapy; 50.4, 54, and 59.4 Gy as postoperative radiation therapy, in the case of R0, R1, and R2 resections, respectively (no additional radiation therapy in the case of secondary complete resections with free margins, in children younger than 6 years). Low Surgery alone 92 100 Intermediate Surgery, 3–6 cycles chemotherapya ± RTb 91 100 High (IRS group III) 3 cycles of chemotherapya surgery, 3 additional cycles of chemotherapy, ± RTb 77 94 High (axial primary sites) Surgery, 6 cycles of chemotherapya, RTb 78 100
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- ADP 04511 (NCT01343043) (A Pilot Study of Genetically Engineered NY-ESO-1 Specific [c259] T Cells in HLA-A2+ Patients With Synovial Sarcoma): Patients with unresectable, metastatic, or recurrent synovial sarcoma undergo apheresis. Cells are shipped to a central laboratory where they undergo NY-ESO-1 transduction, expansion, and cryopreservation. Patients undergo lymphodepletion with fludarabine and cyclophosphamide, followed by an infusion of autologous transfected cells. Eligibility is restricted to patients with HLA type A2+, age older than 4 years, and weight greater than 18 kg.
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with alveolar soft part sarcoma, clear cell sarcoma of soft tissue, epithelioid sarcoma, extraskeletal myxoid chondrosarcoma, PEComa, and synovial sarcoma are eligible for this trial.
- NCT02601937 (A Phase 1 Study of the EZH2 Inhibitor Tazemetostat in Pediatric Subjects With Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma): Patients with INI1-negative tumors are eligible for targeted treatment with an EZH2 inhibitor. This is a phase I, open-label, dose-escalation, and dose-expansion study with a twice-daily oral dose of tazemetostat.
- ADVL1522 (NCT02452554) (Lorvotuzumab Mertansine in Treating Younger Patients with Relapsed or Refractory Wilms Tumor, Rhabdomyosarcoma, Neuroblastoma, Pleuropulmonary Blastoma, Malignant Peripheral Nerve Sheath Tumor, or Synovial Sarcoma): This is a phase II study of IMGN901 (lorvotuzumab mertansine) in children with relapsed or refractory Wilms tumor, rhabdomyosarcoma, neuroblastoma, pleuropulmonary blastoma, malignant peripheral nerve sheath tumor, and synovial sarcoma. This trial is studying the effects of IMGN901, an antibody-drug conjugate that links a potent antimitotic to antibodies that target CD56.
Undifferentiated/unclassified sarcoma
Patients with undifferentiated soft tissue sarcoma had been eligible for participation in rhabdomyosarcoma trials coordinated by the Intergroup Rhabdomyosarcoma Study Group and the COG from 1972 to 2006. The rationale was the observation that patients with undifferentiated soft tissue sarcoma had similar sites of disease and outcome as those with alveolar rhabdomyosarcoma. Therapeutic trials for adults with soft tissue sarcoma include patients with undifferentiated soft tissue sarcoma and other histologies, which are treated similarly, using ifosfamide and doxorubicin, and sometimes with other chemotherapy agents, surgery, and radiation therapy.
In the COG ARST0332 (NCT00346164) trial, patients with high-grade undifferentiated sarcoma were treated with an ifosfamide and doxorubicin-based regimen and were treated with rhabdomyosarcoma-directed therapies in previous Intergroup Rhabdomyosarcoma Study Group studies with a 5-year survival estimate for nonmetastatic patients of 72%.[240][Level of evidence: 3iiA] Currently, these patients are eligible for the COG open ARST1321 (NCT02180867) trial for patients with nonrhabdomyosarcomatous soft tissue sarcoma.
Undifferentiated pleomorphic sarcoma/malignant fibrous histiocytoma (high-grade)
At one time, malignant fibrous histiocytoma was the single most common histotype among adults with soft tissue sarcomas. Since it was first recognized in the early 1960s, malignant fibrous histiocytoma has been plagued by controversy in terms of both its histogenesis and its validity as a clinicopathologic entity. The latest WHO classification no longer includes malignant fibrous histiocytoma as a distinct diagnostic category but rather as a subtype of an undifferentiated pleomorphic sarcoma.[241]
This entity accounts for 2% to 6% of all childhood soft tissue sarcomas.[242] These tumors can arise in previously irradiated sites or as a second malignancy in patients with retinoblastoma.
These tumors occur mainly in the second decade of life. In a series of ten patients, the median age was 10 years and the tumor was most commonly located in the extremities. In this series, all tumors were localized and five of nine (for whom follow-up was available) were alive and in first remission.[242] In another series of 17 pediatric patients with malignant fibrous histiocytoma, the median age at diagnosis was 5 years and the extremities were involved in eight cases.[243] All patients with metastatic disease died and two patients experienced a clinical response to a doxorubicin-based regimen.
(Refer to the PDQ summary on Osteosarcoma and Malignant Fibrous Histiocytoma of Bone Treatment for more information about the treatment of malignant fibrous histiocytoma of bone.)
Treatment options under clinical evaluation
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable, intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with undifferentiated epithelial sarcoma, undifferentiated pleomorphic sarcoma, undifferentiated round cell sarcoma, and undifferentiated spindle cell sarcoma are eligible for this trial.
Vascular Tumors
Vascular tumors vary from hemangiomas, which are always considered benign, to angiosarcomas, which are highly malignant.[244] Vascular tumors include the following tumor subtypes:
Angiosarcoma of the soft tissue
Incidence
Angiosarcoma is a rare (accounting for 2% of sarcomas), aggressive, vascular tumor that can arise in any part of the body, but is more common in the soft tissue. Angiosarcoma has an estimated incidence of 2 cases per 1 million; in the United States, it annually affects approximately 600 people who are typically aged 60 to 70 years.[245]
Angiosarcomas are extremely rare in children and it is unclear if the pathophysiology of this tumor is different in the pediatric population. Cases have been reported in neonates and toddlers, with presentation of multiple cutaneous lesions and liver lesions, some of which are GLUT1 positive.[246-249] Most angiosarcomas involve the skin and superficial soft tissue, although the liver, spleen, and lung can be affected; bone is rarely affected.
Risk factors
Established risk factors include vinyl chloride exposure, radiation exposure, and chronic lymphedema from any cause, including Stewart-Treves syndrome.[250]
Pathology and biology
Angiosarcomas are largely aneuploid tumors. The rare cases of angiosarcoma that arise from benign lesions such as hemangiomas have a distinct pathway that needs to be investigated. MYC amplification is seen in radiation-induced angiosarcoma. KDR-VEGFR2 mutations and FLT4-VEGFR3 amplifications have been seen with a frequency of less than 50%.[250]
Histopathologic diagnosis can be very difficult because there can be areas of varied atypia. The common feature is an irregular network of channels in a dissective pattern along dermal collagen bundles. There is varied cellular shape, size, mitosis, endothelial multilayering, and papillary formation. Epithelioid cells can also be present. Necrosis and hemorrhage are common. Tumors stain for factor VIII, CD31, and CD34. Some liver lesions can mimic infantile hemangiomas and have focal GLUT1 positivity. Nomenclature of these liver lesions has been difficult and confusing with use of terminology from 1971 (e.g., type I hemangioendothelioma: infantile hemangioma; type II hemangioendothelioma: low-grade angiosarcoma; type III hemangioendothelioma: high-grade angiosarcoma).[247]
Treatment of angiosarcoma of the soft tissue
Treatment options for angiosarcoma of the soft tissue include the following:
- Surgery (localized disease).
- Radiation therapy (localized cutaneous disease in adults).
- Surgery, chemotherapy, and radiation therapy (metastatic disease).
Localized disease is cured by aggressive surgery. Complete surgical excision appears to be crucial for angiosarcomas and lymphangiosarcomas despite evidence of tumor shrinkage in some patients who were treated with local or systemic therapy.[248,251-253] A review of 222 patients (median age, 62 years; range, age 15–90 years) showed an overall disease-specific survival (DSS) rate of 38% at 5 years. Five-year DSS was 44% in 138 patients with localized, resected tumors but only 16% in 43 patients with metastases at diagnosis.[253] Data on liver transplantation for localized angiosarcoma are limited.[254][Level of evidence: 3iiA]
Localized disease, especially cutaneous angiosarcoma, can be treated with radiation therapy. Most of these reported cases are in adults.[255]
Multimodal treatment with surgery, systemic chemotherapy, and radiation therapy is used for metastatic disease, although it is rarely curative.[256] Disease control is the objective in metastatic angiosarcoma, with published progression-free survival rates between 3 months and 7 months [257] and a median overall survival (OS) rate of 14 months to 18 months.[258] In both adults and children, 5-year OS rates between 20% and 35% are reported.[248,249,259]
In a child diagnosed with angiosarcoma secondary to malignant transformation from infantile hemangioma, response to treatment with bevacizumab, a monoclonal antibody against vascular endothelial growth factor, combined with systemic chemotherapy, has been reported.[246,256] A report of eight cases of liver angiosarcoma in children highlighted the misuse of the term hemangioendothelioma and the importance of early diagnosis and treatment of these tumors.[260]
Biologic agents that inhibit angiogenesis have shown activity in adults with angiosarcoma.[247,259]
Treatment options under clinical evaluation for angiosarcoma of the soft tissue
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- ARST1321 (NCT02180867) (Radiation Therapy With or Without Combination Chemotherapy or Pazopanib Hydrochloride Before Surgery in Treating Patients With Newly Diagnosed Nonrhabdomyosarcoma Soft Tissue Sarcomas That Can be Removed by Surgery [PAZNTIS]): This study will first determine the feasibility of adding a tyrosine kinase inhibitor in combination with radiation therapy or chemotherapy (ifosfamide/etoposide) and radiation therapy in pediatric and adult patients newly diagnosed with unresected intermediate-risk and high-risk nonrhabdomyosarcomatous soft tissue sarcoma. Subsequently, this trial will compare the rates of near-complete pathologic response (>90% necrosis) of: 1) preoperative pazopanib plus chemoradiation therapy versus preoperative chemoradiation therapy alone for potentially resectable (>5 cm), grade 3 intermediate-risk to high-risk chemotherapy-sensitive adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma; and 2) pazopanib plus preoperative radiation therapy versus preoperative radiation therapy alone for potentially resectable intermediate-risk to high-risk adult and pediatric nonrhabdomyosarcomatous soft tissue sarcoma. Patients with angiosarcoma of the soft tissue are eligible for this trial.
- NCT01532687 (Gemcitabine Hydrochloride With or Without Pazopanib Hydrochloride in Treating Patients With Refractory Soft Tissue Sarcoma): This randomized phase II trial studies how well gemcitabine hydrochloride works with or without pazopanib hydrochloride in treating patients with refractory soft tissue sarcoma.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
Epithelioid hemangioendothelioma
Incidence and outcome
This tumor was first described in soft tissue by Weiss and Enzinger in 1982. Epithelioid hemangioendotheliomas can occur at younger ages, but the peak incidence is in the fourth and fifth decades of life. The tumors can have an indolent or very aggressive course, with overall survival of 73% at 5 years. There are case reports of patients with untreated multiple lesions who have a very benign course compared with other patients who have a very aggressive course. Some pathologists have tried to stratify patients to evaluate risks and adjust treatment, but more research is needed.[261-267]
The presence of effusions, tumor size larger than 3 cm, and a high mitotic index (>3 mitoses/50 high-power fields) have been associated with unfavorable outcomes.[263]
Pathology and biology
A WWTR1-CAMTA1 gene fusion has been found in a large percentage of patients; less commonly, a YAP1-TFE3 gene fusion has been reported.[261] These fusions are not directly targetable with current medicines. Monoclonality has been described in multiple liver lesions, suggesting a metastatic process.
Histologically, these lesions are characterized as epithelioid lesions arranged in nests, strands, and trabecular patterns, with infrequent vascular spaces. Features that may be associated with aggressive clinical behavior include cellular atypia, one or more mitoses per 10 high-power fields, an increased proportion of spindled cells, focal necrosis, and metaplastic bone formation.[263]
The number of pediatric patients reported in the literature is limited.
Clinical presentation and diagnostic evaluation
Common sites of involvement are liver alone (21%), liver plus lung (18%), lung alone (12%), and bone alone (14%).[263,268,269] Clinical presentation depends on site of involvement, as follows:
- Liver: Hepatic nodules have central vascularity on ultrasound, contrast-enhancing lesions by computed tomography, and low T1 signal and moderate T2 signal on magnetic resonance imaging.
- Lung: Pulmonary epithelioid hemangioendothelioma may be an asymptomatic finding on chest x-ray or be associated with pleuritic pain, hemoptysis, anemia, and fibrosis.
- Bone: Bone metastasis may be associated with pathologic fracture. On x-rays, they are well-defined osteolytic lesions and can be multiple or solitary.
- Soft tissue: Thirty percent of soft tissue cases are associated with metastases, and when present, can have a very aggressive course, with limited response to chemotherapy.
- Skin: Cutaneous lesions can be raised and nodular or can be warm red-brown plaques.
Treatment of epithelioid hemangioendothelioma
Treatment options for epithelioid hemangioendothelioma include the following:
- Observation.
- Surgery.
- Immunotherapy.
- Targeted therapy.
- Chemotherapy.
For indolent cases, observation is warranted. For more aggressive cases, multiple medications have been used, including interferon, thalidomide, sorafenib, pazopanib, and sirolimus.[270] The most aggressive cases are treated with angiosarcoma-type chemotherapy. Surgery is used when possible. Liver transplantation has been used with aggressive liver lesions, both with and without metastases.[263,271-274]
Treatment options under clinical evaluation for epithelioid hemangioendothelioma
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- NCT03148275 (Trametinib in Treating Patients with Epithelioid Hemangioendothelioma That Is Metastatic, Locally Advanced, or Cannot Be Removed by Surgery): This is a phase II trial assessing the efficacy of trametinib, with patient-reported outcomes as secondary aims.
- NCT01532687 (Gemcitabine Hydrochloride With or Without Pazopanib Hydrochloride in Treating Patients With Refractory Soft Tissue Sarcoma): This randomized phase II trial studies how well gemcitabine hydrochloride works with or without pazopanib hydrochloride in treating patients with refractory soft tissue sarcoma.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Ferrari A, Casanova M, Collini P, et al.: Adult-type soft tissue sarcomas in pediatric-age patients: experience at the Istituto Nazionale Tumori in Milan. J Clin Oncol 23 (18): 4021-30, 2005. [PUBMED Abstract]
- Stanelle EJ, Christison-Lagay ER, Sidebotham EL, et al.: Prognostic factors and survival in pediatric and adolescent liposarcoma. Sarcoma 2012: 870910, 2012. [PUBMED Abstract]
- Alaggio R, Coffin CM, Weiss SW, et al.: Liposarcomas in young patients: a study of 82 cases occurring in patients younger than 22 years of age. Am J Surg Pathol 33 (5): 645-58, 2009. [PUBMED Abstract]
- Sreekantaiah C, Karakousis CP, Leong SP, et al.: Cytogenetic findings in liposarcoma correlate with histopathologic subtypes. Cancer 69 (10): 2484-95, 1992. [PUBMED Abstract]
- Sugiura H, Takahashi M, Katagiri H, et al.: Additional wide resection of malignant soft tissue tumors. Clin Orthop (394): 201-10, 2002. [PUBMED Abstract]
- Cecchetto G, Guglielmi M, Inserra A, et al.: Primary re-excision: the Italian experience in patients with localized soft-tissue sarcomas. Pediatr Surg Int 17 (7): 532-4, 2001. [PUBMED Abstract]
- Chui CH, Spunt SL, Liu T, et al.: Is reexcision in pediatric nonrhabdomyosarcoma soft tissue sarcoma necessary after an initial unplanned resection? J Pediatr Surg 37 (10): 1424-9, 2002. [PUBMED Abstract]
- Bahig H, Roberge D, Bosch W, et al.: Agreement among RTOG sarcoma radiation oncologists in contouring suspicious peritumoral edema for preoperative radiation therapy of soft tissue sarcoma of the extremity. Int J Radiat Oncol Biol Phys 86 (2): 298-303, 2013. [PUBMED Abstract]
- Baldini EH, Wang D, Haas RL, et al.: Treatment Guidelines for Preoperative Radiation Therapy for Retroperitoneal Sarcoma: Preliminary Consensus of an International Expert Panel. Int J Radiat Oncol Biol Phys 92 (3): 602-12, 2015. [PUBMED Abstract]
- Ferrari A, Casanova M, Spreafico F, et al.: Childhood liposarcoma: a single-institutional twenty-year experience. Pediatr Hematol Oncol 16 (5): 415-21, 1999 Sep-Oct. [PUBMED Abstract]
- Cecchetto G, Alaggio R, Dall'Igna P, et al.: Localized unresectable non-rhabdo soft tissue sarcomas of the extremities in pediatric age: results from the Italian studies. Cancer 104 (9): 2006-12, 2005. [PUBMED Abstract]
- Huh WW, Yuen C, Munsell M, et al.: Liposarcoma in children and young adults: a multi-institutional experience. Pediatr Blood Cancer 57 (7): 1142-6, 2011. [PUBMED Abstract]
- Gronchi A, Bui BN, Bonvalot S, et al.: Phase II clinical trial of neoadjuvant trabectedin in patients with advanced localized myxoid liposarcoma. Ann Oncol 23 (3): 771-6, 2012. [PUBMED Abstract]
- Demetri GD, von Mehren M, Jones RL, et al.: Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. J Clin Oncol 34 (8): 786-93, 2016. [PUBMED Abstract]
- Baruchel S, Pappo A, Krailo M, et al.: A phase 2 trial of trabectedin in children with recurrent rhabdomyosarcoma, Ewing sarcoma and non-rhabdomyosarcoma soft tissue sarcomas: a report from the Children's Oncology Group. Eur J Cancer 48 (4): 579-85, 2012. [PUBMED Abstract]
- Wang L, Motoi T, Khanin R, et al.: Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer 51 (2): 127-39, 2012. [PUBMED Abstract]
- Nyquist KB, Panagopoulos I, Thorsen J, et al.: Whole-transcriptome sequencing identifies novel IRF2BP2-CDX1 fusion gene brought about by translocation t(1;5)(q42;q32) in mesenchymal chondrosarcoma. PLoS One 7 (11): e49705, 2012. [PUBMED Abstract]
- Frezza AM, Cesari M, Baumhoer D, et al.: Mesenchymal chondrosarcoma: prognostic factors and outcome in 113 patients. A European Musculoskeletal Oncology Society study. Eur J Cancer 51 (3): 374-81, 2015. [PUBMED Abstract]
- Dantonello TM, Int-Veen C, Leuschner I, et al.: Mesenchymal chondrosarcoma of soft tissues and bone in children, adolescents, and young adults: experiences of the CWS and COSS study groups. Cancer 112 (11): 2424-31, 2008. [PUBMED Abstract]
- Bishop MW, Somerville JM, Bahrami A, et al.: Mesenchymal Chondrosarcoma in Children and Young Adults: A Single Institution Retrospective Review. Sarcoma 2015: 608279, 2015. [PUBMED Abstract]
- Wodowski K, Hill DA, Pappo AS, et al.: A chemosensitive pediatric extraosseous osteosarcoma: case report and review of the literature. J Pediatr Hematol Oncol 25 (1): 73-7, 2003. [PUBMED Abstract]
- Sordillo PP, Hajdu SI, Magill GB, et al.: Extraosseous osteogenic sarcoma. A review of 48 patients. Cancer 51 (4): 727-34, 1983. [PUBMED Abstract]
- Nieuwenhuis MH, Casparie M, Mathus-Vliegen LM, et al.: A nation-wide study comparing sporadic and familial adenomatous polyposis-related desmoid-type fibromatoses. Int J Cancer 129 (1): 256-61, 2011. [PUBMED Abstract]
- Rossato M, Rigotti M, Grazia M, et al.: Congenital hypertrophy of the retinal pigment epithelium (CHRPE) and familial adenomatous polyposis (FAP). Acta Ophthalmol Scand 74 (4): 338-42, 1996. [PUBMED Abstract]
- Baker RH, Heinemann MH, Miller HH, et al.: Hyperpigmented lesions of the retinal pigment epithelium in familial adenomatous polyposis. Am J Med Genet 31 (2): 427-35, 1988. [PUBMED Abstract]
- Kattentidt Mouravieva AA, Geurts-Giele IR, de Krijger RR, et al.: Identification of Familial Adenomatous Polyposis carriers among children with desmoid tumours. Eur J Cancer 48 (12): 1867-74, 2012. [PUBMED Abstract]
- Wang WL, Nero C, Pappo A, et al.: CTNNB1 genotyping and APC screening in pediatric desmoid tumors: a proposed algorithm. Pediatr Dev Pathol 15 (5): 361-7, 2012 Sep-Oct. [PUBMED Abstract]
- Lewis JJ, Boland PJ, Leung DH, et al.: The enigma of desmoid tumors. Ann Surg 229 (6): 866-72; discussion 872-3, 1999. [PUBMED Abstract]
- Lazar AJ, Tuvin D, Hajibashi S, et al.: Specific mutations in the beta-catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am J Pathol 173 (5): 1518-27, 2008. [PUBMED Abstract]
- Faulkner LB, Hajdu SI, Kher U, et al.: Pediatric desmoid tumor: retrospective analysis of 63 cases. J Clin Oncol 13 (11): 2813-8, 1995. [PUBMED Abstract]
- Merchant NB, Lewis JJ, Woodruff JM, et al.: Extremity and trunk desmoid tumors: a multifactorial analysis of outcome. Cancer 86 (10): 2045-52, 1999. [PUBMED Abstract]
- Honeyman JN, Theilen TM, Knowles MA, et al.: Desmoid fibromatosis in children and adolescents: a conservative approach to management. J Pediatr Surg 48 (1): 62-6, 2013. [PUBMED Abstract]
- Bonvalot S, Ternès N, Fiore M, et al.: Spontaneous regression of primary abdominal wall desmoid tumors: more common than previously thought. Ann Surg Oncol 20 (13): 4096-102, 2013. [PUBMED Abstract]
- Bonvalot S, Eldweny H, Haddad V, et al.: Extra-abdominal primary fibromatosis: Aggressive management could be avoided in a subgroup of patients. Eur J Surg Oncol 34 (4): 462-8, 2008. [PUBMED Abstract]
- Merchant TE, Nguyen D, Walter AW, et al.: Long-term results with radiation therapy for pediatric desmoid tumors. Int J Radiat Oncol Biol Phys 47 (5): 1267-71, 2000. [PUBMED Abstract]
- Zelefsky MJ, Harrison LB, Shiu MH, et al.: Combined surgical resection and iridium 192 implantation for locally advanced and recurrent desmoid tumors. Cancer 67 (2): 380-4, 1991. [PUBMED Abstract]
- Weiss AJ, Lackman RD: Low-dose chemotherapy of desmoid tumors. Cancer 64 (6): 1192-4, 1989. [PUBMED Abstract]
- Klein WA, Miller HH, Anderson M, et al.: The use of indomethacin, sulindac, and tamoxifen for the treatment of desmoid tumors associated with familial polyposis. Cancer 60 (12): 2863-8, 1987. [PUBMED Abstract]
- Soto-Miranda MA, Sandoval JA, Rao B, et al.: Surgical treatment of pediatric desmoid tumors. A 12-year, single-center experience. Ann Surg Oncol 20 (11): 3384-90, 2013. [PUBMED Abstract]
- Skapek SX, Ferguson WS, Granowetter L, et al.: Vinblastine and methotrexate for desmoid fibromatosis in children: results of a Pediatric Oncology Group Phase II Trial. J Clin Oncol 25 (5): 501-6, 2007. [PUBMED Abstract]
- Gandhi MM, Nathan PC, Weitzman S, et al.: Successful treatment of life-threatening generalized infantile myofibromatosis using low-dose chemotherapy. J Pediatr Hematol Oncol 25 (9): 750-4, 2003. [PUBMED Abstract]
- Gega M, Yanagi H, Yoshikawa R, et al.: Successful chemotherapeutic modality of doxorubicin plus dacarbazine for the treatment of desmoid tumors in association with familial adenomatous polyposis. J Clin Oncol 24 (1): 102-5, 2006. [PUBMED Abstract]
- Constantinidou A, Jones RL, Scurr M, et al.: Pegylated liposomal doxorubicin, an effective, well-tolerated treatment for refractory aggressive fibromatosis. Eur J Cancer 45 (17): 2930-4, 2009. [PUBMED Abstract]
- Ananth P, Werger A, Voss S, et al.: Liposomal doxorubicin: Effective treatment for pediatric desmoid fibromatosis. Pediatr Blood Cancer 64 (7): , 2017. [PUBMED Abstract]
- Gounder MM, Lefkowitz RA, Keohan ML, et al.: Activity of Sorafenib against desmoid tumor/deep fibromatosis. Clin Cancer Res 17 (12): 4082-90, 2011. [PUBMED Abstract]
- Heinrich MC, McArthur GA, Demetri GD, et al.: Clinical and molecular studies of the effect of imatinib on advanced aggressive fibromatosis (desmoid tumor). J Clin Oncol 24 (7): 1195-203, 2006. [PUBMED Abstract]
- Chugh R, Wathen JK, Patel SR, et al.: Efficacy of imatinib in aggressive fibromatosis: Results of a phase II multicenter Sarcoma Alliance for Research through Collaboration (SARC) trial. Clin Cancer Res 16 (19): 4884-91, 2010. [PUBMED Abstract]
- Agresta L, Kim H, Turpin BK, et al.: Pazopanib therapy for desmoid tumors in adolescent and young adult patients. Pediatr Blood Cancer : , 2018. [PUBMED Abstract]
- Shang H, Braggio D, Lee YJ, et al.: Targeting the Notch pathway: A potential therapeutic approach for desmoid tumors. Cancer 121 (22): 4088-96, 2015. [PUBMED Abstract]
- Messersmith WA, Shapiro GI, Cleary JM, et al.: A Phase I, dose-finding study in patients with advanced solid malignancies of the oral γ-secretase inhibitor PF-03084014. Clin Cancer Res 21 (1): 60-7, 2015. [PUBMED Abstract]
- Bisogno G, Tagarelli A, Stramare R, et al.: Hydroxyurea treatment can avoid the need for aggressive surgery in pediatric fibromatosis. J Pediatr Hematol Oncol 35 (4): e171-3, 2013. [PUBMED Abstract]
- Meazza C, Casanova M, Trecate G, et al.: Objective response to hydroxyurea in a patient with heavily pre-treated aggressive fibromatosis. Pediatr Blood Cancer 55 (3): 587-8, 2010. [PUBMED Abstract]
- Balamuth NJ, Womer RB: Successful treatment of fibromatosis with hydroxyurea: Analysis of 20 pediatric cases. [Abstract] The Connective Tissue Oncology Society (CTOS) 14th Annual Meeting, 13–15 November 2008, London, United Kingdom A-34852, 2008. Also available online. Last accessed April 02, 2018.
- Meazza C, Bisogno G, Gronchi A, et al.: Aggressive fibromatosis in children and adolescents: the Italian experience. Cancer 116 (1): 233-40, 2010. [PUBMED Abstract]
- Hansmann A, Adolph C, Vogel T, et al.: High-dose tamoxifen and sulindac as first-line treatment for desmoid tumors. Cancer 100 (3): 612-20, 2004. [PUBMED Abstract]
- Skapek SX, Anderson JR, Hill DA, et al.: Safety and efficacy of high-dose tamoxifen and sulindac for desmoid tumor in children: results of a Children's Oncology Group (COG) phase II study. Pediatr Blood Cancer 60 (7): 1108-12, 2013. [PUBMED Abstract]
- Rutenberg MS, Indelicato DJ, Knapik JA, et al.: External-beam radiotherapy for pediatric and young adult desmoid tumors. Pediatr Blood Cancer 57 (3): 435-42, 2011. [PUBMED Abstract]
- Buckley PG, Mantripragada KK, Benetkiewicz M, et al.: A full-coverage, high-resolution human chromosome 22 genomic microarray for clinical and research applications. Hum Mol Genet 11 (25): 3221-9, 2002. [PUBMED Abstract]
- Valdivielso-Ramos M, Torrelo A, Campos M, et al.: Pediatric dermatofibrosarcoma protuberans in Madrid, Spain: multi-institutional outcomes. Pediatr Dermatol 31 (6): 676-82, 2014 Nov-Dec. [PUBMED Abstract]
- Gooskens SL, Oranje AP, van Adrichem LN, et al.: Imatinib mesylate for children with dermatofibrosarcoma protuberans (DFSP). Pediatr Blood Cancer 55 (2): 369-73, 2010. [PUBMED Abstract]
- Rubio GA, Alvarado A, Gerth DJ, et al.: Incidence and Outcomes of Dermatofibrosarcoma Protuberans in the US Pediatric Population. J Craniofac Surg 28 (1): 182-184, 2017. [PUBMED Abstract]
- Meguerditchian AN, Wang J, Lema B, et al.: Wide excision or Mohs micrographic surgery for the treatment of primary dermatofibrosarcoma protuberans. Am J Clin Oncol 33 (3): 300-3, 2010. [PUBMED Abstract]
- Dagan R, Morris CG, Zlotecki RA, et al.: Radiotherapy in the treatment of dermatofibrosarcoma protuberans. Am J Clin Oncol 28 (6): 537-9, 2005. [PUBMED Abstract]
- Sun LM, Wang CJ, Huang CC, et al.: Dermatofibrosarcoma protuberans: treatment results of 35 cases. Radiother Oncol 57 (2): 175-81, 2000. [PUBMED Abstract]
- Price VE, Fletcher JA, Zielenska M, et al.: Imatinib mesylate: an attractive alternative in young children with large, surgically challenging dermatofibrosarcoma protuberans. Pediatr Blood Cancer 44 (5): 511-5, 2005. [PUBMED Abstract]
- McArthur GA, Demetri GD, van Oosterom A, et al.: Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J Clin Oncol 23 (4): 866-73, 2005. [PUBMED Abstract]
- Rutkowski P, Van Glabbeke M, Rankin CJ, et al.: Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials. J Clin Oncol 28 (10): 1772-9, 2010. [PUBMED Abstract]
- Miller SJ, Alam M, Andersen JS, et al.: Dermatofibrosarcoma protuberans. J Natl Compr Canc Netw 10 (3): 312-8, 2012. [PUBMED Abstract]
- Kao YC, Fletcher CDM, Alaggio R, et al.: Recurrent BRAF Gene Fusions in a Subset of Pediatric Spindle Cell Sarcomas: Expanding the Genetic Spectrum of Tumors With Overlapping Features With Infantile Fibrosarcoma. Am J Surg Pathol 42 (1): 28-38, 2018. [PUBMED Abstract]
- Sulkowski JP, Raval MV, Browne M: Margin status and multimodal therapy in infantile fibrosarcoma. Pediatr Surg Int 29 (8): 771-6, 2013. [PUBMED Abstract]
- Orbach D, Rey A, Cecchetto G, et al.: Infantile fibrosarcoma: management based on the European experience. J Clin Oncol 28 (2): 318-23, 2010. [PUBMED Abstract]
- Orbach D, Brennan B, De Paoli A, et al.: Conservative strategy in infantile fibrosarcoma is possible: The European paediatric Soft tissue sarcoma Study Group experience. Eur J Cancer 57: 1-9, 2016. [PUBMED Abstract]
- Spunt SL, Million L, Coffin C: The nonrhabdomyosarcoma soft tissue sarcoma. In: Pizzo PA, Poplack DG, eds.: Principles and Practice of Pediatric Oncology. 7th ed. Philadelphia, Pa: Lippincott Williams and Wilkins, 2015, pp 827-54.
- Loh ML, Ahn P, Perez-Atayde AR, et al.: Treatment of infantile fibrosarcoma with chemotherapy and surgery: results from the Dana-Farber Cancer Institute and Children's Hospital, Boston. J Pediatr Hematol Oncol 24 (9): 722-6, 2002. [PUBMED Abstract]
- Akyüz C, Küpeli S, Varan A, et al.: Infantile fibrosarcoma: retrospective analysis of eleven patients. Tumori 97 (2): 166-9, 2011 Mar-Apr. [PUBMED Abstract]
- Gallego S, Pericas N, Barber I, et al.: Infantile fibrosarcoma of the retroperitoneum: a site of unfavorable prognosis? Pediatr Hematol Oncol 28 (5): 451-3, 2011. [PUBMED Abstract]
- Parida L, Fernandez-Pineda I, Uffman JK, et al.: Clinical management of infantile fibrosarcoma: a retrospective single-institution review. Pediatr Surg Int 29 (7): 703-8, 2013. [PUBMED Abstract]
- Mody RJ, Wu YM, Lonigro RJ, et al.: Integrative Clinical Sequencing in the Management of Refractory or Relapsed Cancer in Youth. JAMA 314 (9): 913-25, 2015. [PUBMED Abstract]
- Wong V, Pavlick D, Brennan T, et al.: Evaluation of a Congenital Infantile Fibrosarcoma by Comprehensive Genomic Profiling Reveals an LMNA-NTRK1 Gene Fusion Responsive to Crizotinib. J Natl Cancer Inst 108 (1): , 2016. [PUBMED Abstract]
- Nagasubramanian R, Wei J, Gordon P, et al.: Infantile Fibrosarcoma With NTRK3-ETV6 Fusion Successfully Treated With the Tropomyosin-Related Kinase Inhibitor LOXO-101. Pediatr Blood Cancer 63 (8): 1468-70, 2016. [PUBMED Abstract]
- Yanagisawa R, Noguchi M, Fujita K, et al.: Preoperative Treatment With Pazopanib in a Case of Chemotherapy-Resistant Infantile Fibrosarcoma. Pediatr Blood Cancer 63 (2): 348-51, 2016. [PUBMED Abstract]
- Madden NP, Spicer RD, Allibone EB, et al.: Spontaneous regression of neonatal fibrosarcoma. Br J Cancer Suppl 18: S72-5, 1992. [PUBMED Abstract]
- Kovach SJ, Fischer AC, Katzman PJ, et al.: Inflammatory myofibroblastic tumors. J Surg Oncol 94 (5): 385-91, 2006. [PUBMED Abstract]
- Brodlie M, Barwick SC, Wood KM, et al.: Inflammatory myofibroblastic tumours of the respiratory tract: paediatric case series with varying clinical presentations. J Laryngol Otol 125 (8): 865-8, 2011. [PUBMED Abstract]
- Xiao Y, Zhou S, Ma C, et al.: Radiological and histopathological features of hepatic inflammatory myofibroblastic tumour: analysis of 10 cases. Clin Radiol 68 (11): 1114-20, 2013. [PUBMED Abstract]
- Karnak I, Senocak ME, Ciftci AO, et al.: Inflammatory myofibroblastic tumor in children: diagnosis and treatment. J Pediatr Surg 36 (6): 908-12, 2001. [PUBMED Abstract]
- Collin M, Charles A, Barker A, et al.: Inflammatory myofibroblastic tumour of the bladder in children: a review. J Pediatr Urol 11 (5): 239-45, 2015. [PUBMED Abstract]
- Coffin CM, Hornick JL, Fletcher CD: Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol 31 (4): 509-20, 2007. [PUBMED Abstract]
- Lovly CM, Gupta A, Lipson D, et al.: Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov 4 (8): 889-95, 2014. [PUBMED Abstract]
- Devaney KO, Lafeir DJ, Triantafyllou A, et al.: Inflammatory myofibroblastic tumors of the head and neck: evaluation of clinicopathologic and prognostic features. Eur Arch Otorhinolaryngol 269 (12): 2461-5, 2012. [PUBMED Abstract]
- Mehta B, Mascarenhas L, Zhou S, et al.: Inflammatory myofibroblastic tumors in childhood. Pediatr Hematol Oncol 30 (7): 640-5, 2013. [PUBMED Abstract]
- Favini F, Resti AG, Collini P, et al.: Inflammatory myofibroblastic tumor of the conjunctiva: response to chemotherapy with low-dose methotrexate and vinorelbine. Pediatr Blood Cancer 54 (3): 483-5, 2010. [PUBMED Abstract]
- Doski JJ, Priebe CJ Jr, Driessnack M, et al.: Corticosteroids in the management of unresected plasma cell granuloma (inflammatory pseudotumor) of the lung. J Pediatr Surg 26 (9): 1064-6, 1991. [PUBMED Abstract]
- Diop B, Konate I, Ka S, et al.: Mesenteric myofibroblastic tumor: NSAID therapy after incomplete resection. J Visc Surg 148 (4): e311-4, 2011. [PUBMED Abstract]
- Dalton BG, Thomas PG, Sharp NE, et al.: Inflammatory myofibroblastic tumors in children. J Pediatr Surg 51 (4): 541-4, 2016. [PUBMED Abstract]
- Butrynski JE, D'Adamo DR, Hornick JL, et al.: Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med 363 (18): 1727-33, 2010. [PUBMED Abstract]
- Mossé YP, Lim MS, Voss SD, et al.: Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children's Oncology Group phase 1 consortium study. Lancet Oncol 14 (6): 472-80, 2013. [PUBMED Abstract]
- Gaudichon J, Jeanne-Pasquier C, Deparis M, et al.: Complete and Repeated Response of a Metastatic ALK-rearranged Inflammatory Myofibroblastic Tumor to Crizotinib in a Teenage Girl. J Pediatr Hematol Oncol 38 (4): 308-11, 2016. [PUBMED Abstract]
- Nishio M, Murakami H, Horiike A, et al.: Phase I Study of Ceritinib (LDK378) in Japanese Patients with Advanced, Anaplastic Lymphoma Kinase-Rearranged Non-Small-Cell Lung Cancer or Other Tumors. J Thorac Oncol 10 (7): 1058-66, 2015. [PUBMED Abstract]
- Mossé YP, Voss SD, Lim MS, et al.: Targeting ALK With Crizotinib in Pediatric Anaplastic Large Cell Lymphoma and Inflammatory Myofibroblastic Tumor: A Children's Oncology Group Study. J Clin Oncol 35 (28): 3215-3221, 2017. [PUBMED Abstract]
- Evans HL: Low-grade fibromyxoid sarcoma: a clinicopathologic study of 33 cases with long-term follow-up. Am J Surg Pathol 35 (10): 1450-62, 2011. [PUBMED Abstract]
- Guillou L, Benhattar J, Gengler C, et al.: Translocation-positive low-grade fibromyxoid sarcoma: clinicopathologic and molecular analysis of a series expanding the morphologic spectrum and suggesting potential relationship to sclerosing epithelioid fibrosarcoma: a study from the French Sarcoma Group. Am J Surg Pathol 31 (9): 1387-402, 2007. [PUBMED Abstract]
- O'Sullivan MJ, Sirgi KE, Dehner LP: Low-grade fibrosarcoma (hyalinizing spindle cell tumor with giant rosettes) with pulmonary metastases at presentation: case report and review of the literature. Int J Surg Pathol 10 (3): 211-6, 2002. [PUBMED Abstract]
- Folpe AL, Lane KL, Paull G, et al.: Low-grade fibromyxoid sarcoma and hyalinizing spindle cell tumor with giant rosettes: a clinicopathologic study of 73 cases supporting their identity and assessing the impact of high-grade areas. Am J Surg Pathol 24 (10): 1353-60, 2000. [PUBMED Abstract]
- Sargar K, Kao SC, Spunt SL, et al.: MRI and CT of Low-Grade Fibromyxoid Sarcoma in Children: A Report From Children's Oncology Group Study ARST0332. AJR Am J Roentgenol 205 (2): 414-20, 2015. [PUBMED Abstract]
- Maretty-Nielsen K, Baerentzen S, Keller J, et al.: Low-Grade Fibromyxoid Sarcoma: Incidence, Treatment Strategy of Metastases, and Clinical Significance of the FUS Gene. Sarcoma 2013: 256280, 2013. [PUBMED Abstract]
- Prieto-Granada C, Zhang L, Chen HW, et al.: A genetic dichotomy between pure sclerosing epithelioid fibrosarcoma (SEF) and hybrid SEF/low-grade fibromyxoid sarcoma: a pathologic and molecular study of 18 cases. Genes Chromosomes Cancer 54 (1): 28-38, 2015. [PUBMED Abstract]
- Pollock BH, Jenson HB, Leach CT, et al.: Risk factors for pediatric human immunodeficiency virus-related malignancy. JAMA 289 (18): 2393-9, 2003. [PUBMED Abstract]
- Kleinerman RA, Tucker MA, Abramson DH, et al.: Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst 99 (1): 24-31, 2007. [PUBMED Abstract]
- Samuels BL, Chawla S, Patel S, et al.: Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol 24 (6): 1703-9, 2013. [PUBMED Abstract]
- Enzinger FM, Zhang RY: Plexiform fibrohistiocytic tumor presenting in children and young adults. An analysis of 65 cases. Am J Surg Pathol 12 (11): 818-26, 1988. [PUBMED Abstract]
- Black J, Coffin CM, Dehner LP: Fibrohistiocytic tumors and related neoplasms in children and adolescents. Pediatr Dev Pathol 15 (1 Suppl): 181-210, 2012. [PUBMED Abstract]
- Moosavi C, Jha P, Fanburg-Smith JC: An update on plexiform fibrohistiocytic tumor and addition of 66 new cases from the Armed Forces Institute of Pathology, in honor of Franz M. Enzinger, MD. Ann Diagn Pathol 11 (5): 313-9, 2007. [PUBMED Abstract]
- Billings SD, Folpe AL: Cutaneous and subcutaneous fibrohistiocytic tumors of intermediate malignancy: an update. Am J Dermatopathol 26 (2): 141-55, 2004. [PUBMED Abstract]
- Remstein ED, Arndt CA, Nascimento AG: Plexiform fibrohistiocytic tumor: clinicopathologic analysis of 22 cases. Am J Surg Pathol 23 (6): 662-70, 1999. [PUBMED Abstract]
- Salomao DR, Nascimento AG: Plexiform fibrohistiocytic tumor with systemic metastases: a case report. Am J Surg Pathol 21 (4): 469-76, 1997. [PUBMED Abstract]
- Redlich GC, Montgomery KD, Allgood GA, et al.: Plexiform fibrohistiocytic tumor with a clonal cytogenetic anomaly. Cancer Genet Cytogenet 108 (2): 141-3, 1999. [PUBMED Abstract]
- Luzar B, Calonje E: Cutaneous fibrohistiocytic tumours - an update. Histopathology 56 (1): 148-65, 2010. [PUBMED Abstract]
- Dantonello TM, Leuschner I, Vokuhl C, et al.: Malignant ectomesenchymoma in children and adolescents: report from the Cooperative Weichteilsarkom Studiengruppe (CWS). Pediatr Blood Cancer 60 (2): 224-9, 2013. [PUBMED Abstract]
- Carli M, Ferrari A, Mattke A, et al.: Pediatric malignant peripheral nerve sheath tumor: the Italian and German soft tissue sarcoma cooperative group. J Clin Oncol 23 (33): 8422-30, 2005. [PUBMED Abstract]
- Zhang M, Wang Y, Jones S, et al.: Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet 46 (11): 1170-2, 2014. [PUBMED Abstract]
- Hagel C, Zils U, Peiper M, et al.: Histopathology and clinical outcome of NF1-associated vs. sporadic malignant peripheral nerve sheath tumors. J Neurooncol 82 (2): 187-92, 2007. [PUBMED Abstract]
- Zou C, Smith KD, Liu J, et al.: Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg 249 (6): 1014-22, 2009. [PUBMED Abstract]
- Okada K, Hasegawa T, Tajino T, et al.: Clinical relevance of pathological grades of malignant peripheral nerve sheath tumor: a multi-institution TMTS study of 56 cases in Northern Japan. Ann Surg Oncol 14 (2): 597-604, 2007. [PUBMED Abstract]
- Amirian ES, Goodman JC, New P, et al.: Pediatric and adult malignant peripheral nerve sheath tumors: an analysis of data from the surveillance, epidemiology, and end results program. J Neurooncol 116 (3): 609-16, 2014. [PUBMED Abstract]
- Valentin T, Le Cesne A, Ray-Coquard I, et al.: Management and prognosis of malignant peripheral nerve sheath tumors: The experience of the French Sarcoma Group (GSF-GETO). Eur J Cancer 56: 77-84, 2016. [PUBMED Abstract]
- Bergamaschi L, Bisogno G, Manzitti C, et al.: Salvage rates and prognostic factors after relapse in children and adolescents with malignant peripheral nerve sheath tumors. Pediatr Blood Cancer 65 (2): , 2018. [PUBMED Abstract]
- Ferrari A, Bisogno G, Macaluso A, et al.: Soft-tissue sarcomas in children and adolescents with neurofibromatosis type 1. Cancer 109 (7): 1406-12, 2007. [PUBMED Abstract]
- Okur FV, Oguz A, Karadeniz C, et al.: Malignant triton tumor of the pelvis in a 2-year-old boy. J Pediatr Hematol Oncol 28 (3): 173-6, 2006. [PUBMED Abstract]
- Weiss SW, Goldblum JR: Enzinger and Weiss's Soft Tissue Tumors. 4th ed. St. Louis, Mo: Mosby, 2001.
- Fernandez-Pineda I, Parida L, Jenkins JJ, et al.: Childhood hemangiopericytoma: review of St Jude Children's Research Hospital. J Pediatr Hematol Oncol 33 (5): 356-9, 2011. [PUBMED Abstract]
- Rodriguez-Galindo C, Ramsey K, Jenkins JJ, et al.: Hemangiopericytoma in children and infants. Cancer 88 (1): 198-204, 2000. [PUBMED Abstract]
- Ferrari A, Casanova M, Bisogno G, et al.: Hemangiopericytoma in pediatric ages: a report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Cancer 92 (10): 2692-8, 2001. [PUBMED Abstract]
- Bien E, Stachowicz-Stencel T, Godzinski J, et al.: Retrospective multi-institutional study on hemangiopericytoma in Polish children. Pediatr Int 51 (1): 19-24, 2009. [PUBMED Abstract]
- Wiswell TE, Davis J, Cunningham BE, et al.: Infantile myofibromatosis: the most common fibrous tumor of infancy. J Pediatr Surg 23 (4): 315-8, 1988. [PUBMED Abstract]
- Chung EB, Enzinger FM: Infantile myofibromatosis. Cancer 48 (8): 1807-18, 1981. [PUBMED Abstract]
- Modi N: Congenital generalised fibromatosis. Arch Dis Child 57 (11): 881-2, 1982. [PUBMED Abstract]
- Levine E, Fréneaux P, Schleiermacher G, et al.: Risk-adapted therapy for infantile myofibromatosis in children. Pediatr Blood Cancer 59 (1): 115-20, 2012. [PUBMED Abstract]
- Larralde M, Hoffner MV, Boggio P, et al.: Infantile myofibromatosis: report of nine patients. Pediatr Dermatol 27 (1): 29-33, 2010 Jan-Feb. [PUBMED Abstract]
- Cheung YH, Gayden T, Campeau PM, et al.: A recurrent PDGFRB mutation causes familial infantile myofibromatosis. Am J Hum Genet 92 (6): 996-1000, 2013. [PUBMED Abstract]
- Gopal M, Chahal G, Al-Rifai Z, et al.: Infantile myofibromatosis. Pediatr Surg Int 24 (3): 287-91, 2008. [PUBMED Abstract]
- Weaver MS, Navid F, Huppmann A, et al.: Vincristine and Dactinomycin in Infantile Myofibromatosis With a Review of Treatment Options. J Pediatr Hematol Oncol 37 (3): 237-41, 2015. [PUBMED Abstract]
- Orbach D, Brennan B, Casanova M, et al.: Paediatric and adolescent alveolar soft part sarcoma: A joint series from European cooperative groups. Pediatr Blood Cancer 60 (11): 1826-32, 2013. [PUBMED Abstract]
- Ferrari A, Sultan I, Huang TT, et al.: Soft tissue sarcoma across the age spectrum: a population-based study from the Surveillance Epidemiology and End Results database. Pediatr Blood Cancer 57 (6): 943-9, 2011. [PUBMED Abstract]
- Wang HW, Qin XJ, Yang WJ, et al.: Alveolar soft part sarcoma of the oral and maxillofacial region: clinical analysis in a series of 18 patients. Oral Surg Oral Med Oral Pathol Oral Radiol 119 (4): 396-401, 2015. [PUBMED Abstract]
- Kayton ML, Meyers P, Wexler LH, et al.: Clinical presentation, treatment, and outcome of alveolar soft part sarcoma in children, adolescents, and young adults. J Pediatr Surg 41 (1): 187-93, 2006. [PUBMED Abstract]
- Ladanyi M, Lui MY, Antonescu CR, et al.: The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene 20 (1): 48-57, 2001. [PUBMED Abstract]
- Williams A, Bartle G, Sumathi VP, et al.: Detection of ASPL/TFE3 fusion transcripts and the TFE3 antigen in formalin-fixed, paraffin-embedded tissue in a series of 18 cases of alveolar soft part sarcoma: useful diagnostic tools in cases with unusual histological features. Virchows Arch 458 (3): 291-300, 2011. [PUBMED Abstract]
- Lieberman PH, Brennan MF, Kimmel M, et al.: Alveolar soft-part sarcoma. A clinico-pathologic study of half a century. Cancer 63 (1): 1-13, 1989. [PUBMED Abstract]
- Casanova M, Ferrari A, Bisogno G, et al.: Alveolar soft part sarcoma in children and adolescents: A report from the Soft-Tissue Sarcoma Italian Cooperative Group. Ann Oncol 11 (11): 1445-9, 2000. [PUBMED Abstract]
- Pennacchioli E, Fiore M, Collini P, et al.: Alveolar soft part sarcoma: clinical presentation, treatment, and outcome in a series of 33 patients at a single institution. Ann Surg Oncol 17 (12): 3229-33, 2010. [PUBMED Abstract]
- Flores RJ, Harrison DJ, Federman NC, et al.: Alveolar soft part sarcoma in children and young adults: A report of 69 cases. Pediatr Blood Cancer : , 2018. [PUBMED Abstract]
- Roozendaal KJ, de Valk B, ten Velden JJ, et al.: Alveolar soft-part sarcoma responding to interferon alpha-2b. Br J Cancer 89 (2): 243-5, 2003. [PUBMED Abstract]
- Conde N, Cruz O, Albert A, et al.: Antiangiogenic treatment as a pre-operative management of alveolar soft-part sarcoma. Pediatr Blood Cancer 57 (6): 1071-3, 2011. [PUBMED Abstract]
- Stacchiotti S, Negri T, Zaffaroni N, et al.: Sunitinib in advanced alveolar soft part sarcoma: evidence of a direct antitumor effect. Ann Oncol 22 (7): 1682-90, 2011. [PUBMED Abstract]
- Kummar S, Allen D, Monks A, et al.: Cediranib for metastatic alveolar soft part sarcoma. J Clin Oncol 31 (18): 2296-302, 2013. [PUBMED Abstract]
- Coindre JM, Hostein I, Terrier P, et al.: Diagnosis of clear cell sarcoma by real-time reverse transcriptase-polymerase chain reaction analysis of paraffin embedded tissues: clinicopathologic and molecular analysis of 44 patients from the French sarcoma group. Cancer 107 (5): 1055-64, 2006. [PUBMED Abstract]
- Meis-Kindblom JM: Clear cell sarcoma of tendons and aponeuroses: a historical perspective and tribute to the man behind the entity. Adv Anat Pathol 13 (6): 286-92, 2006. [PUBMED Abstract]
- Dim DC, Cooley LD, Miranda RN: Clear cell sarcoma of tendons and aponeuroses: a review. Arch Pathol Lab Med 131 (1): 152-6, 2007. [PUBMED Abstract]
- Blazer DG 3rd, Lazar AJ, Xing Y, et al.: Clinical outcomes of molecularly confirmed clear cell sarcoma from a single institution and in comparison with data from the Surveillance, Epidemiology, and End Results registry. Cancer 115 (13): 2971-9, 2009. [PUBMED Abstract]
- Fujimura Y, Siddique H, Lee L, et al.: EWS-ATF-1 chimeric protein in soft tissue clear cell sarcoma associates with CREB-binding protein and interferes with p53-mediated trans-activation function. Oncogene 20 (46): 6653-9, 2001. [PUBMED Abstract]
- Ferrari A, Casanova M, Bisogno G, et al.: Clear cell sarcoma of tendons and aponeuroses in pediatric patients: a report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Cancer 94 (12): 3269-76, 2002. [PUBMED Abstract]
- Karita M, Tsuchiya H, Yamamoto N, et al.: Caffeine-potentiated chemotherapy for clear cell sarcoma: a report of five cases. Int J Clin Oncol 18 (1): 33-7, 2013. [PUBMED Abstract]
- Leuschner I, Radig K, Harms D: Desmoplastic small round cell tumor. Semin Diagn Pathol 13 (3): 204-12, 1996. [PUBMED Abstract]
- Kushner BH, LaQuaglia MP, Wollner N, et al.: Desmoplastic small round-cell tumor: prolonged progression-free survival with aggressive multimodality therapy. J Clin Oncol 14 (5): 1526-31, 1996. [PUBMED Abstract]
- Saab R, Khoury JD, Krasin M, et al.: Desmoplastic small round cell tumor in childhood: the St. Jude Children's Research Hospital experience. Pediatr Blood Cancer 49 (3): 274-9, 2007. [PUBMED Abstract]
- Wang LL, Perlman EJ, Vujanic GM, et al.: Desmoplastic small round cell tumor of the kidney in childhood. Am J Surg Pathol 31 (4): 576-84, 2007. [PUBMED Abstract]
- Arora VC, Price AP, Fleming S, et al.: Characteristic imaging features of desmoplastic small round cell tumour. Pediatr Radiol 43 (1): 93-102, 2013. [PUBMED Abstract]
- Gerald WL, Ladanyi M, de Alava E, et al.: Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): desmoplastic small round-cell tumor and its variants. J Clin Oncol 16 (9): 3028-36, 1998. [PUBMED Abstract]
- Lal DR, Su WT, Wolden SL, et al.: Results of multimodal treatment for desmoplastic small round cell tumors. J Pediatr Surg 40 (1): 251-5, 2005. [PUBMED Abstract]
- Philippe-Chomette P, Kabbara N, Andre N, et al.: Desmoplastic small round cell tumors with EWS-WT1 fusion transcript in children and young adults. Pediatr Blood Cancer 58 (6): 891-7, 2012. [PUBMED Abstract]
- Sedig L, Geiger J, Mody R, et al.: Paratesticular desmoplastic small round cell tumors: A case report and review of the literature. Pediatr Blood Cancer 64 (12): , 2017. [PUBMED Abstract]
- Schwarz RE, Gerald WL, Kushner BH, et al.: Desmoplastic small round cell tumors: prognostic indicators and results of surgical management. Ann Surg Oncol 5 (5): 416-22, 1998 Jul-Aug. [PUBMED Abstract]
- Goodman KA, Wolden SL, La Quaglia MP, et al.: Whole abdominopelvic radiotherapy for desmoplastic small round-cell tumor. Int J Radiat Oncol Biol Phys 54 (1): 170-6, 2002. [PUBMED Abstract]
- Osborne EM, Briere TM, Hayes-Jordan A, et al.: Survival and toxicity following sequential multimodality treatment including whole abdominopelvic radiotherapy for patients with desmoplastic small round cell tumor. Radiother Oncol 119 (1): 40-4, 2016. [PUBMED Abstract]
- Atallah V, Honore C, Orbach D, et al.: Role of Adjuvant Radiation Therapy After Surgery for Abdominal Desmoplastic Small Round Cell Tumors. Int J Radiat Oncol Biol Phys 95 (4): 1244-53, 2016. [PUBMED Abstract]
- Tarek N, Hayes-Jordan A, Salvador L, et al.: Recurrent desmoplastic small round cell tumor responding to an mTOR inhibitor containing regimen. Pediatr Blood Cancer 65 (1): , 2018. [PUBMED Abstract]
- Cook RJ, Wang Z, Arora M, et al.: Clinical outcomes of patients with desmoplastic small round cell tumor of the peritoneum undergoing autologous HCT: a CIBMTR retrospective analysis. Bone Marrow Transplant 47 (11): 1455-8, 2012. [PUBMED Abstract]
- Chbani L, Guillou L, Terrier P, et al.: Epithelioid sarcoma: a clinicopathologic and immunohistochemical analysis of 106 cases from the French sarcoma group. Am J Clin Pathol 131 (2): 222-7, 2009. [PUBMED Abstract]
- Hornick JL, Dal Cin P, Fletcher CD: Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol 33 (4): 542-50, 2009. [PUBMED Abstract]
- Knutson SK, Warholic NM, Wigle TJ, et al.: Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 110 (19): 7922-7, 2013. [PUBMED Abstract]
- Guzzetta AA, Montgomery EA, Lyu H, et al.: Epithelioid sarcoma: one institution's experience with a rare sarcoma. J Surg Res 177 (1): 116-22, 2012. [PUBMED Abstract]
- Casanova M, Ferrari A, Collini P, et al.: Epithelioid sarcoma in children and adolescents: a report from the Italian Soft Tissue Sarcoma Committee. Cancer 106 (3): 708-17, 2006. [PUBMED Abstract]
- Kodet R, Newton WA Jr, Sachs N, et al.: Rhabdoid tumors of soft tissues: a clinicopathologic study of 26 cases enrolled on the Intergroup Rhabdomyosarcoma Study. Hum Pathol 22 (7): 674-84, 1991. [PUBMED Abstract]
- Biegel JA, Zhou JY, Rorke LB, et al.: Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer Res 59 (1): 74-9, 1999. [PUBMED Abstract]
- Eaton KW, Tooke LS, Wainwright LM, et al.: Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56 (1): 7-15, 2011. [PUBMED Abstract]
- Lee RS, Stewart C, Carter SL, et al.: A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest 122 (8): 2983-8, 2012. [PUBMED Abstract]
- Sultan I, Qaddoumi I, Rodríguez-Galindo C, et al.: Age, stage, and radiotherapy, but not primary tumor site, affects the outcome of patients with malignant rhabdoid tumors. Pediatr Blood Cancer 54 (1): 35-40, 2010. [PUBMED Abstract]
- Puri DR, Meyers PA, Kraus DH, et al.: Radiotherapy in the multimodal treatment of extrarenal extracranial malignant rhabdoid tumors. Pediatr Blood Cancer 50 (1): 167-9, 2008. [PUBMED Abstract]
- Madigan CE, Armenian SH, Malogolowkin MH, et al.: Extracranial malignant rhabdoid tumors in childhood: the Childrens Hospital Los Angeles experience. Cancer 110 (9): 2061-6, 2007. [PUBMED Abstract]
- Bourdeaut F, Fréneaux P, Thuille B, et al.: Extra-renal non-cerebral rhabdoid tumours. Pediatr Blood Cancer 51 (3): 363-8, 2008. [PUBMED Abstract]
- Wetmore C, Boyett J, Li S, et al.: Alisertib is active as single agent in recurrent atypical teratoid rhabdoid tumors in 4 children. Neuro Oncol 17 (6): 882-8, 2015. [PUBMED Abstract]
- Tsuneyoshi M, Enjoji M, Iwasaki H, et al.: Extraskeletal myxoid chondrosarcoma--a clinicopathologic and electron microscopic study. Acta Pathol Jpn 31 (3): 439-47, 1981. [PUBMED Abstract]
- Hachitanda Y, Tsuneyoshi M, Daimaru Y, et al.: Extraskeletal myxoid chondrosarcoma in young children. Cancer 61 (12): 2521-6, 1988. [PUBMED Abstract]
- Hisaoka M, Ishida T, Imamura T, et al.: TFG is a novel fusion partner of NOR1 in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer 40 (4): 325-8, 2004. [PUBMED Abstract]
- Enzinger FM, Shiraki M: Extraskeletal myxoid chondrosarcoma. An analysis of 34 cases. Hum Pathol 3 (3): 421-35, 1972. [PUBMED Abstract]
- McGrory JE, Rock MG, Nascimento AG, et al.: Extraskeletal myxoid chondrosarcoma. Clin Orthop Relat Res (382): 185-90, 2001. [PUBMED Abstract]
- Drilon AD, Popat S, Bhuchar G, et al.: Extraskeletal myxoid chondrosarcoma: a retrospective review from 2 referral centers emphasizing long-term outcomes with surgery and chemotherapy. Cancer 113 (12): 3364-71, 2008. [PUBMED Abstract]
- Stacchiotti S, Pantaleo MA, Astolfi A, et al.: Activity of sunitinib in extraskeletal myxoid chondrosarcoma. Eur J Cancer 50 (9): 1657-64, 2014. [PUBMED Abstract]
- Martignoni G, Pea M, Reghellin D, et al.: Molecular pathology of lymphangioleiomyomatosis and other perivascular epithelioid cell tumors. Arch Pathol Lab Med 134 (1): 33-40, 2010. [PUBMED Abstract]
- Bissler JJ, McCormack FX, Young LR, et al.: Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med 358 (2): 140-51, 2008. [PUBMED Abstract]
- Davies DM, Johnson SR, Tattersfield AE, et al.: Sirolimus therapy in tuberous sclerosis or sporadic lymphangioleiomyomatosis. N Engl J Med 358 (2): 200-3, 2008. [PUBMED Abstract]
- Agaram NP, Sung YS, Zhang L, et al.: Dichotomy of Genetic Abnormalities in PEComas With Therapeutic Implications. Am J Surg Pathol 39 (6): 813-25, 2015. [PUBMED Abstract]
- Folpe A, Inwards C, eds.: Bone and Soft Tissue Pathology: A Volume in the Foundations in Diagnostic Pathology. Philadelphia, Pa: WB Saunders Co, 2010.
- Armah HB, Parwani AV: Perivascular epithelioid cell tumor. Arch Pathol Lab Med 133 (4): 648-54, 2009. [PUBMED Abstract]
- Alaggio R, Cecchetto G, Martignoni G, et al.: Malignant perivascular epithelioid cell tumor in children: description of a case and review of the literature. J Pediatr Surg 47 (6): e31-40, 2012. [PUBMED Abstract]
- Wagner AJ, Malinowska-Kolodziej I, Morgan JA, et al.: Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: targeting the pathogenic activation of mTORC1 in tumors. J Clin Oncol 28 (5): 835-40, 2010. [PUBMED Abstract]
- Sultan I, Rodriguez-Galindo C, Saab R, et al.: Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005: an analysis of 1268 patients. Cancer 115 (15): 3537-47, 2009. [PUBMED Abstract]
- Wang JG, Li NN: Primary cardiac synovial sarcoma. Ann Thorac Surg 95 (6): 2202-9, 2013. [PUBMED Abstract]
- Pappo AS, Fontanesi J, Luo X, et al.: Synovial sarcoma in children and adolescents: the St Jude Children's Research Hospital experience. J Clin Oncol 12 (11): 2360-6, 1994. [PUBMED Abstract]
- Ferrari A, De Salvo GL, Oberlin O, et al.: Synovial sarcoma in children and adolescents: a critical reappraisal of staging investigations in relation to the rate of metastatic involvement at diagnosis. Eur J Cancer 48 (9): 1370-5, 2012. [PUBMED Abstract]
- van de Rijn M, Barr FG, Collins MH, et al.: Absence of SYT-SSX fusion products in soft tissue tumors other than synovial sarcoma. Am J Clin Pathol 112 (1): 43-9, 1999. [PUBMED Abstract]
- Krsková L, Sumerauer D, Stejskalová E, et al.: A novel variant of SYT-SSX1 fusion gene in a case of spindle cell synovial sarcoma. Diagn Mol Pathol 16 (3): 179-83, 2007. [PUBMED Abstract]
- Su L, Sampaio AV, Jones KB, et al.: Deconstruction of the SS18-SSX fusion oncoprotein complex: insights into disease etiology and therapeutics. Cancer Cell 21 (3): 333-47, 2012. [PUBMED Abstract]
- Arnold MA, Arnold CA, Li G, et al.: A unique pattern of INI1 immunohistochemistry distinguishes synovial sarcoma from its histologic mimics. Hum Pathol 44 (5): 881-7, 2013. [PUBMED Abstract]
- Vlenterie M, Ho VK, Kaal SE, et al.: Age as an independent prognostic factor for survival of localised synovial sarcoma patients. Br J Cancer 113 (11): 1602-6, 2015. [PUBMED Abstract]
- Okcu MF, Munsell M, Treuner J, et al.: Synovial sarcoma of childhood and adolescence: a multicenter, multivariate analysis of outcome. J Clin Oncol 21 (8): 1602-11, 2003. [PUBMED Abstract]
- Brecht IB, Ferrari A, Int-Veen C, et al.: Grossly-resected synovial sarcoma treated by the German and Italian Pediatric Soft Tissue Sarcoma Cooperative Groups: discussion on the role of adjuvant therapies. Pediatr Blood Cancer 46 (1): 11-7, 2006. [PUBMED Abstract]
- Stanelle EJ, Christison-Lagay ER, Healey JH, et al.: Pediatric and adolescent synovial sarcoma: multivariate analysis of prognostic factors and survival outcomes. Ann Surg Oncol 20 (1): 73-9, 2013. [PUBMED Abstract]
- Trassard M, Le Doussal V, Hacène K, et al.: Prognostic factors in localized primary synovial sarcoma: a multicenter study of 128 adult patients. J Clin Oncol 19 (2): 525-34, 2001. [PUBMED Abstract]
- Guillou L, Benhattar J, Bonichon F, et al.: Histologic grade, but not SYT-SSX fusion type, is an important prognostic factor in patients with synovial sarcoma: a multicenter, retrospective analysis. J Clin Oncol 22 (20): 4040-50, 2004. [PUBMED Abstract]
- Ferrari A, Gronchi A, Casanova M, et al.: Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer 101 (3): 627-34, 2004. [PUBMED Abstract]
- Lagarde P, Przybyl J, Brulard C, et al.: Chromosome instability accounts for reverse metastatic outcomes of pediatric and adult synovial sarcomas. J Clin Oncol 31 (5): 608-15, 2013. [PUBMED Abstract]
- Stegmaier S, Leuschner I, Poremba C, et al.: The prognostic impact of SYT-SSX fusion type and histological grade in pediatric patients with synovial sarcoma treated according to the CWS (Cooperative Weichteilsarkom Studie) trials. Pediatr Blood Cancer 64 (1): 89-95, 2017. [PUBMED Abstract]
- Scheer M, Dantonello T, Hallmen E, et al.: Primary Metastatic Synovial Sarcoma: Experience of the CWS Study Group. Pediatr Blood Cancer 63 (7): 1198-206, 2016. [PUBMED Abstract]
- Ferrari A, De Salvo GL, Dall'Igna P, et al.: Salvage rates and prognostic factors after relapse in children and adolescents with initially localised synovial sarcoma. Eur J Cancer 48 (18): 3448-55, 2012. [PUBMED Abstract]
- Ferrari A, Chi YY, De Salvo GL, et al.: Surgery alone is sufficient therapy for children and adolescents with low-risk synovial sarcoma: A joint analysis from the European paediatric soft tissue sarcoma Study Group and the Children's Oncology Group. Eur J Cancer 78: 1-6, 2017. [PUBMED Abstract]
- McGrory JE, Pritchard DJ, Arndt CA, et al.: Nonrhabdomyosarcoma soft tissue sarcomas in children. The Mayo Clinic experience. Clin Orthop (374): 247-58, 2000. [PUBMED Abstract]
- Van Glabbeke M, van Oosterom AT, Oosterhuis JW, et al.: Prognostic factors for the outcome of chemotherapy in advanced soft tissue sarcoma: an analysis of 2,185 patients treated with anthracycline-containing first-line regimens--a European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol 17 (1): 150-7, 1999. [PUBMED Abstract]
- Koscielniak E, Harms D, Henze G, et al.: Results of treatment for soft tissue sarcoma in childhood and adolescence: a final report of the German Cooperative Soft Tissue Sarcoma Study CWS-86. J Clin Oncol 17 (12): 3706-19, 1999. [PUBMED Abstract]
- Pappo AS, Devidas M, Jenkins J, et al.: Phase II trial of neoadjuvant vincristine, ifosfamide, and doxorubicin with granulocyte colony-stimulating factor support in children and adolescents with advanced-stage nonrhabdomyosarcomatous soft tissue sarcomas: a Pediatric Oncology Group Study. J Clin Oncol 23 (18): 4031-8, 2005. [PUBMED Abstract]
- Pappo AS, Rao BN, Jenkins JJ, et al.: Metastatic nonrhabdomyosarcomatous soft-tissue sarcomas in children and adolescents: the St. Jude Children's Research Hospital experience. Med Pediatr Oncol 33 (2): 76-82, 1999. [PUBMED Abstract]
- Brennan B, Stevens M, Kelsey A, et al.: Synovial sarcoma in childhood and adolescence: a retrospective series of 77 patients registered by the Children's Cancer and Leukaemia Group between 1991 and 2006. Pediatr Blood Cancer 55 (1): 85-90, 2010. [PUBMED Abstract]
- Ferrari A, Miceli R, Rey A, et al.: Non-metastatic unresected paediatric non-rhabdomyosarcoma soft tissue sarcomas: results of a pooled analysis from United States and European groups. Eur J Cancer 47 (5): 724-31, 2011. [PUBMED Abstract]
- Raney RB: Synovial sarcoma in young people: background, prognostic factors, and therapeutic questions. J Pediatr Hematol Oncol 27 (4): 207-11, 2005. [PUBMED Abstract]
- Orbach D, Mc Dowell H, Rey A, et al.: Sparing strategy does not compromise prognosis in pediatric localized synovial sarcoma: experience of the International Society of Pediatric Oncology, Malignant Mesenchymal Tumors (SIOP-MMT) Working Group. Pediatr Blood Cancer 57 (7): 1130-6, 2011. [PUBMED Abstract]
- Ladenstein R, Treuner J, Koscielniak E, et al.: Synovial sarcoma of childhood and adolescence. Report of the German CWS-81 study. Cancer 71 (11): 3647-55, 1993. [PUBMED Abstract]
- Venkatramani R, Anderson JR, Million L, et al.: Risk-based treatment for synovial sarcoma in patients under 30 years of age: Children’s Oncology Group study ARST0332. [Abstract] J Clin Oncol 33 (15 Suppl): A-10012, 2015. Also available online. Last accessed April 02, 2018.
- Ferrari A, De Salvo GL, Brennan B, et al.: Synovial sarcoma in children and adolescents: the European Pediatric Soft Tissue Sarcoma Study Group prospective trial (EpSSG NRSTS 2005). Ann Oncol 26 (3): 567-72, 2015. [PUBMED Abstract]
- Spunt SL, Million L, Anderson JR, et al.: Risk-based treatment for nonrhabdomyosarcoma soft tissue sarcomas (NRSTS) in patients under 30 years of age: Children’s Oncology Group study ARST0332. [Abstract] J Clin Oncol 32 (Suppl 15): A-10008, 2014. Also available online. Last accessed April 02, 2018.
- Randall RL, Albritton KH, Ferney BJ, et al.: Malignant fibrous histiocytoma of soft tissue: an abandoned diagnosis. Am J Orthop 33 (12): 602-8, 2004. [PUBMED Abstract]
- Alaggio R, Collini P, Randall RL, et al.: Undifferentiated high-grade pleomorphic sarcomas in children: a clinicopathologic study of 10 cases and review of literature. Pediatr Dev Pathol 13 (3): 209-17, 2010 May-Jun. [PUBMED Abstract]
- Daw NC, Billups CA, Pappo AS, et al.: Malignant fibrous histiocytoma and other fibrohistiocytic tumors in pediatric patients: the St. Jude Children's Research Hospital experience. Cancer 97 (11): 2839-47, 2003. [PUBMED Abstract]
- Coffin CM, Dehner LP: Vascular tumors in children and adolescents: a clinicopathologic study of 228 tumors in 222 patients. Pathol Annu 28 Pt 1: 97-120, 1993. [PUBMED Abstract]
- Cioffi A, Reichert S, Antonescu CR, et al.: Angiosarcomas and other sarcomas of endothelial origin. Hematol Oncol Clin North Am 27 (5): 975-88, 2013. [PUBMED Abstract]
- Jeng MR, Fuh B, Blatt J, et al.: Malignant transformation of infantile hemangioma to angiosarcoma: response to chemotherapy with bevacizumab. Pediatr Blood Cancer 61 (11): 2115-7, 2014. [PUBMED Abstract]
- Dehner LP, Ishak KG: Vascular tumors of the liver in infants and children. A study of 30 cases and review of the literature. Arch Pathol 92 (2): 101-11, 1971. [PUBMED Abstract]
- Ferrari A, Casanova M, Bisogno G, et al.: Malignant vascular tumors in children and adolescents: a report from the Italian and German Soft Tissue Sarcoma Cooperative Group. Med Pediatr Oncol 39 (2): 109-14, 2002. [PUBMED Abstract]
- Deyrup AT, Miettinen M, North PE, et al.: Pediatric cutaneous angiosarcomas: a clinicopathologic study of 10 cases. Am J Surg Pathol 35 (1): 70-5, 2011. [PUBMED Abstract]
- Elliott P, Kleinschmidt I: Angiosarcoma of the liver in Great Britain in proximity to vinyl chloride sites. Occup Environ Med 54 (1): 14-8, 1997. [PUBMED Abstract]
- Lezama-del Valle P, Gerald WL, Tsai J, et al.: Malignant vascular tumors in young patients. Cancer 83 (8): 1634-9, 1998. [PUBMED Abstract]
- Fata F, O'Reilly E, Ilson D, et al.: Paclitaxel in the treatment of patients with angiosarcoma of the scalp or face. Cancer 86 (10): 2034-7, 1999. [PUBMED Abstract]
- Lahat G, Dhuka AR, Hallevi H, et al.: Angiosarcoma: clinical and molecular insights. Ann Surg 251 (6): 1098-106, 2010. [PUBMED Abstract]
- Orlando G, Adam R, Mirza D, et al.: Hepatic hemangiosarcoma: an absolute contraindication to liver transplantation--the European Liver Transplant Registry experience. Transplantation 95 (6): 872-7, 2013. [PUBMED Abstract]
- Sanada T, Nakayama H, Irisawa R, et al.: Clinical outcome and dose volume evaluation in patients who undergo brachytherapy for angiosarcoma of the scalp and face. Mol Clin Oncol 6 (3): 334-340, 2017. [PUBMED Abstract]
- Dickson MA, D'Adamo DR, Keohan ML, et al.: Phase II Trial of Gemcitabine and Docetaxel with Bevacizumab in Soft Tissue Sarcoma. Sarcoma 2015: 532478, 2015. [PUBMED Abstract]
- North PE, Waner M, Mizeracki A, et al.: A unique microvascular phenotype shared by juvenile hemangiomas and human placenta. Arch Dermatol 137 (5): 559-70, 2001. [PUBMED Abstract]
- Boye E, Yu Y, Paranya G, et al.: Clonality and altered behavior of endothelial cells from hemangiomas. J Clin Invest 107 (6): 745-52, 2001. [PUBMED Abstract]
- Ravi V, Patel S: Vascular sarcomas. Curr Oncol Rep 15 (4): 347-55, 2013. [PUBMED Abstract]
- Grassia KL, Peterman CM, Iacobas I, et al.: Clinical case series of pediatric hepatic angiosarcoma. Pediatr Blood Cancer 64 (11): , 2017. [PUBMED Abstract]
- Mehrabi A, Kashfi A, Fonouni H, et al.: Primary malignant hepatic epithelioid hemangioendothelioma: a comprehensive review of the literature with emphasis on the surgical therapy. Cancer 107 (9): 2108-21, 2006. [PUBMED Abstract]
- Haro A, Saitoh G, Tamiya S, et al.: Four-year natural clinical course of pulmonary epithelioid hemangioendothelioma without therapy. Thorac Cancer 6 (4): 544-7, 2015. [PUBMED Abstract]
- Sardaro A, Bardoscia L, Petruzzelli MF, et al.: Epithelioid hemangioendothelioma: an overview and update on a rare vascular tumor. Oncol Rev 8 (2): 259, 2014. [PUBMED Abstract]
- Dong K, Wang XX, Feng JL, et al.: Pathological characteristics of liver biopsies in eight patients with hepatic epithelioid hemangioendothelioma. Int J Clin Exp Pathol 8 (9): 11015-23, 2015. [PUBMED Abstract]
- Adams DM, Hammill A: Other vascular tumors. Semin Pediatr Surg 23 (4): 173-7, 2014. [PUBMED Abstract]
- Xiao Y, Wang C, Song Y, et al.: Primary epithelioid hemangioendothelioma of the kidney: the first case report in a child and literature review. Urology 82 (4): 925-7, 2013. [PUBMED Abstract]
- Reich S, Ringe H, Uhlenberg B, et al.: Epithelioid hemangioendothelioma of the lung presenting with pneumonia and heart rhythm disturbances in a teenage girl. J Pediatr Hematol Oncol 32 (4): 274-6, 2010. [PUBMED Abstract]
- Daller JA, Bueno J, Gutierrez J, et al.: Hepatic hemangioendothelioma: clinical experience and management strategy. J Pediatr Surg 34 (1): 98-105; discussion 105-6, 1999. [PUBMED Abstract]
- Ackermann O, Fabre M, Franchi S, et al.: Widening spectrum of liver angiosarcoma in children. J Pediatr Gastroenterol Nutr 53 (6): 615-9, 2011. [PUBMED Abstract]
- Stacchiotti S, Provenzano S, Dagrada G, et al.: Sirolimus in Advanced Epithelioid Hemangioendothelioma: A Retrospective Case-Series Analysis from the Italian Rare Cancer Network Database. Ann Surg Oncol 23 (9): 2735-44, 2016. [PUBMED Abstract]
- Semenisty V, Naroditsky I, Keidar Z, et al.: Pazopanib for metastatic pulmonary epithelioid hemangioendothelioma-a suitable treatment option: case report and review of anti-angiogenic treatment options. BMC Cancer 15: 402, 2015. [PUBMED Abstract]
- Raheja A, Suri A, Singh S, et al.: Multimodality management of a giant skull base hemangioendothelioma of the sphenopetroclival region. J Clin Neurosci 22 (9): 1495-8, 2015. [PUBMED Abstract]
- Ahmad N, Adams DM, Wang J, et al.: Hepatic epithelioid hemangioendothelioma in a patient with hemochromatosis. J Natl Compr Canc Netw 12 (9): 1203-7, 2014. [PUBMED Abstract]
- Otte JB, Zimmerman A: The role of liver transplantation for pediatric epithelioid hemangioendothelioma. Pediatr Transplant 14 (3): 295-7, 2010. [PUBMED Abstract]
Treatment of Metastatic Childhood Soft Tissue Sarcoma
Standard treatment options for metastatic childhood soft tissue sarcoma include the following:
- Combination therapy using chemotherapy, radiation therapy, and surgical resection of pulmonary metastases.
For treatment options, refer to the individual tumor type sections of the summary.
The prognosis for children with metastatic soft tissue sarcomas is poor,[1-6] and these children should receive combined treatment with chemotherapy, radiation therapy, and surgical resection of pulmonary metastases. In a prospective randomized trial, chemotherapy with vincristine, dactinomycin, doxorubicin, and cyclophosphamide, with or without dacarbazine, led to tumor responses in one-third of patients with unresectable or metastatic disease. The estimated 4-year survival rate, however, was poor, with fewer than one-third of children surviving.[6-8]
Pulmonary Metastases
Generally, children with isolated pulmonary metastases should be considered for a surgical procedure in an attempt to resect all gross disease.[9] For patients with multiple or recurrent pulmonary metastases, additional surgical procedures can be performed if the morbidity is deemed acceptable. In a retrospective review, patients with synovial sarcoma and pulmonary metastases for whom it was possible to completely resect all metastatic lung lesions had better survival than did patients for whom it was not possible to achieve complete resections.[9][Level of evidence: 3iiiA] Formal segmentectomy, lobectomy, and mediastinal lymph node dissection are unnecessary.[10]
An alternative approach is focused radiation therapy (fractionated stereotactic radiation therapy), which has been successfully used in adults to control lesions. The estimated 5-year survival rate after thoracotomy for pulmonary metastasectomy has ranged from 10% to 58% in adult studies. Emerging data suggest a similar outcome after the administration of focused radiation therapy.[11]
References
- Demetri GD, Elias AD: Results of single-agent and combination chemotherapy for advanced soft tissue sarcomas. Implications for decision making in the clinic. Hematol Oncol Clin North Am 9 (4): 765-85, 1995. [PUBMED Abstract]
- Elias A, Ryan L, Sulkes A, et al.: Response to mesna, doxorubicin, ifosfamide, and dacarbazine in 108 patients with metastatic or unresectable sarcoma and no prior chemotherapy. J Clin Oncol 7 (9): 1208-16, 1989. [PUBMED Abstract]
- Edmonson JH, Ryan LM, Blum RH, et al.: Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol 11 (7): 1269-75, 1993. [PUBMED Abstract]
- Rao BN: Nonrhabdomyosarcoma in children: prognostic factors influencing survival. Semin Surg Oncol 9 (6): 524-31, 1993 Nov-Dec. [PUBMED Abstract]
- deCou JM, Rao BN, Parham DM, et al.: Malignant peripheral nerve sheath tumors: the St. Jude Children's Research Hospital experience. Ann Surg Oncol 2 (6): 524-9, 1995. [PUBMED Abstract]
- Pappo AS, Rao BN, Jenkins JJ, et al.: Metastatic nonrhabdomyosarcomatous soft-tissue sarcomas in children and adolescents: the St. Jude Children's Research Hospital experience. Med Pediatr Oncol 33 (2): 76-82, 1999. [PUBMED Abstract]
- Pratt CB, Pappo AS, Gieser P, et al.: Role of adjuvant chemotherapy in the treatment of surgically resected pediatric nonrhabdomyosarcomatous soft tissue sarcomas: A Pediatric Oncology Group Study. J Clin Oncol 17 (4): 1219, 1999. [PUBMED Abstract]
- Pratt CB, Maurer HM, Gieser P, et al.: Treatment of unresectable or metastatic pediatric soft tissue sarcomas with surgery, irradiation, and chemotherapy: a Pediatric Oncology Group study. Med Pediatr Oncol 30 (4): 201-9, 1998. [PUBMED Abstract]
- Stanelle EJ, Christison-Lagay ER, Wolden SL, et al.: Pulmonary metastasectomy in pediatric/adolescent patients with synovial sarcoma: an institutional review. J Pediatr Surg 48 (4): 757-63, 2013. [PUBMED Abstract]
- Putnam JB Jr, Roth JA: Surgical treatment for pulmonary metastases from sarcoma. Hematol Oncol Clin North Am 9 (4): 869-87, 1995. [PUBMED Abstract]
- Dhakal S, Corbin KS, Milano MT, et al.: Stereotactic body radiotherapy for pulmonary metastases from soft-tissue sarcomas: excellent local lesion control and improved patient survival. Int J Radiat Oncol Biol Phys 82 (2): 940-5, 2012. [PUBMED Abstract]
Treatment of Progressive/Recurrent Childhood Soft Tissue Sarcoma
With the possible exception of infants with infantile fibrosarcoma, the prognosis for patients with recurrent or progressive disease is poor. No prospective trial has been able to prove that enhanced local control of pediatric soft tissue sarcomas will ultimately improve survival. Therefore, treatment should be individualized for the site of recurrence, biologic characteristics of the tumor (e.g., grade, invasiveness, and size), previous therapies, and individual patient considerations.
Treatment options for recurrent or progressive disease include the following:
- Surgical excision of local recurrence or isolated pulmonary recurrence.
- An Italian review of 73 patients with recurrent malignant peripheral nerve sheath tumors found that most relapses were local. Multivariate analysis showed that the factors associated with improved survival were no tumor invasiveness at initial diagnosis (T1), time of recurrence more than 12 months after initial diagnosis, and achievement of a second complete response with surgical removal of the recurrence(s). Only 15.8% of patients who had complete surgical excisions of local recurrence(s) were alive at 5 years.[1][Level of evidence: 3iiiA]
- Surgical excision of local recurrence followed by radiation therapy or brachytherapy (if no previous radiation therapy was given).
- Limb amputation (only for some children with extremity sarcomas that have already received radiation therapy).
- Gemcitabine and docetaxel.[2]
- Trabectedin.[3-5]
- Pazopanib. A phase I trial of pazopanib reported one partial response in a patient with desmoplastic small round cell tumor and prolonged disease stabilization in eight patients with recurrent sarcoma.[6][Level of evidence: 2Diii] Pazopanib has been approved for use in recurrent soft tissue sarcoma. The clinical trial that was used to obtain approval was limited to adults and demonstrated disease stabilization and prolonged time to progression; it did not demonstrate improved overall survival.[7] One 13-year-old boy and one 14-year-old girl with multiply recurrent synovial sarcoma and lung metastases had responses to pazopanib for 14 and 15 months, respectively.[8][Level of evidence: 3iiDi]
- A clinical trial of new chemotherapeutic regimens.
Resection is the standard treatment for recurrent pediatric nonrhabdomyosarcomatous soft tissue sarcomas. If the patient has not yet received radiation therapy, postoperative radiation should be considered after local excision of the recurrent tumor. Limb-sparing procedures with postoperative brachytherapy have been evaluated in adults but have not been studied extensively in children. For some children with extremity sarcomas who have received previous radiation therapy, amputation may be the only therapeutic option.
Pulmonary metastasectomy may achieve prolonged disease control for some patients.[9] A large, retrospective analysis of patients with recurrent soft tissue sarcoma showed that isolated local relapse had a better prognosis and that resection of pulmonary metastases improved the probability of survival.[10] In 31 children and adolescents younger than 23 years with pulmonary metastases from synovial sarcoma, complete resection of lung metastases appeared to prolong survival when compared with ten other patients who were not considered candidates for metastasectomy.[11][Level of evidence: 3iiiA] All patients with recurrent tumors should be considered for current clinical trials.
Published results of two studies addressed the outcomes for children with relapsed synovial sarcoma. Most patients in one study had distant relapse (29 of 44 patients),[12] while most patients in the second study had local relapse (27 of 37 patients).[13] Distant recurrence was a poor prognostic variable, while tumor resectability at relapse (as manifested by extremity recurrence) was associated with a better outcome in both studies.
Treatment Options Under Clinical Evaluation
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- APEC1621 (NCT03155620) (Pediatric MATCH: Targeted Therapy Directed by Genetic Testing in Treating Pediatric Patients with Relapsed or Refractory Advanced Solid Tumors, Non-Hodgkin Lymphomas, or Histiocytic Disorders): NCI-COG Pediatric Molecular Analysis for Therapeutic Choice (MATCH), referred to as Pediatric MATCH, will match targeted agents with specific molecular changes identified using a next-generation sequencing targeted assay of more than 3,000 different mutations across more than 160 genes in refractory and recurrent solid tumors. Children and adolescents aged 1 to 21 years are eligible for the trial.
Tumor tissue from progressive or recurrent disease must be available for molecular characterization. Patients with tumors that have molecular variants addressed by treatment arms included in the trial will be offered treatment on Pediatric MATCH. Additional information can be obtained on the ClinicalTrials.gov website for APEC1621 (NCT03155620).
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Bergamaschi L, Bisogno G, Manzitti C, et al.: Salvage rates and prognostic factors after relapse in children and adolescents with malignant peripheral nerve sheath tumors. Pediatr Blood Cancer 65 (2): , 2018. [PUBMED Abstract]
- Maki RG, Wathen JK, Patel SR, et al.: Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 25 (19): 2755-63, 2007. [PUBMED Abstract]
- Le Cesne A, Cresta S, Maki RG, et al.: A retrospective analysis of antitumour activity with trabectedin in translocation-related sarcomas. Eur J Cancer 48 (16): 3036-44, 2012. [PUBMED Abstract]
- Garcia-Carbonero R, Supko JG, Maki RG, et al.: Ecteinascidin-743 (ET-743) for chemotherapy-naive patients with advanced soft tissue sarcomas: multicenter phase II and pharmacokinetic study. J Clin Oncol 23 (24): 5484-92, 2005. [PUBMED Abstract]
- Garcia-Carbonero R, Supko JG, Manola J, et al.: Phase II and pharmacokinetic study of ecteinascidin 743 in patients with progressive sarcomas of soft tissues refractory to chemotherapy. J Clin Oncol 22 (8): 1480-90, 2004. [PUBMED Abstract]
- Glade Bender JL, Lee A, Reid JM, et al.: Phase I pharmacokinetic and pharmacodynamic study of pazopanib in children with soft tissue sarcoma and other refractory solid tumors: a children's oncology group phase I consortium report. J Clin Oncol 31 (24): 3034-43, 2013. [PUBMED Abstract]
- van der Graaf WT, Blay JY, Chawla SP, et al.: Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 379 (9829): 1879-86, 2012. [PUBMED Abstract]
- Casanova M, Basso E, Magni C, et al.: Response to pazopanib in two pediatric patients with pretreated relapsing synovial sarcoma. Tumori 103 (1): e1-e3, 2017. [PUBMED Abstract]
- Belal A, Salah E, Hajjar W, et al.: Pulmonary metastatectomy for soft tissue sarcomas: is it valuable? J Cardiovasc Surg (Torino) 42 (6): 835-40, 2001. [PUBMED Abstract]
- Zagars GK, Ballo MT, Pisters PW, et al.: Prognostic factors for disease-specific survival after first relapse of soft-tissue sarcoma: analysis of 402 patients with disease relapse after initial conservative surgery and radiotherapy. Int J Radiat Oncol Biol Phys 57 (3): 739-47, 2003. [PUBMED Abstract]
- Stanelle EJ, Christison-Lagay ER, Wolden SL, et al.: Pulmonary metastasectomy in pediatric/adolescent patients with synovial sarcoma: an institutional review. J Pediatr Surg 48 (4): 757-63, 2013. [PUBMED Abstract]
- Ferrari A, De Salvo GL, Dall'Igna P, et al.: Salvage rates and prognostic factors after relapse in children and adolescents with initially localised synovial sarcoma. Eur J Cancer 48 (18): 3448-55, 2012. [PUBMED Abstract]
- Soole F, Maupain C, Defachelles AS, et al.: Synovial sarcoma relapses in children and adolescents: prognostic factors, treatment, and outcome. Pediatr Blood Cancer 61 (8): 1387-93, 2014. [PUBMED Abstract]
Changes to This Summary (04/02/2018)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Treatment of Newly Diagnosed Childhood Soft Tissue Sarcoma
Added text to state that a small series reported symptomatic improvement and stable disease in seven patients with desmoid-type fibromatosis who were treated with pazopanib (cited Agresta et al. as reference 48).
Added text to state that a tumor with morphology similar to that of infantile fibrosarcoma has been identified in older children; in these older children, the tumors do not have the t(12;15)(ETV-NTRK3) translocation that is characteristic of the younger patients. In several of these patients, BRAF gene fusions have been identified (cited Kao et al. as reference 69).
Added text about the outcome results of 73 children and adolescents with recurrent malignant peripheral nerve sheath tumor reported by the Italian Sarcoma Group (cited Bergamaschi et al. as reference 127 and level of evidence 3iiiA).
Added text about the patient characteristics and results of a retrospective review of children and young adults younger than 30 years from four institutions, which identified 69 patients with alveolar soft part sarcoma treated primarily with surgery between 1980 and 2014 (cited Flores et al. as reference 152 and level of evidence 3iiA).
Added Sedig et al. as reference 172 and level of evidence 3iiiA.
Treatment of Progressive/Recurrent Childhood Soft Tissue Sarcoma
Added text about the prognostic factors and outcome results reported in an Italian review of 73 children and adolescents with recurrent malignant peripheral nerve sheath tumor (cited Bergamaschi et al. as reference 1 and level of evidence 3iiiA).
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of childhood soft tissue sarcoma. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Childhood Soft Tissue Sarcoma Treatment are:
- Denise Adams, MD (Children's Hospital Boston)
- Louis S. Constine, MD (James P. Wilmot Cancer Center at University of Rochester Medical Center)
- Holcombe Edwin Grier, MD (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Andrea A. Hayes-Jordan, MD, FACS, FAAP (M.D. Anderson Cancer Center)
- Paul A. Meyers, MD (Memorial Sloan-Kettering Cancer Center)
- Thomas A. Olson, MD (Aflac Cancer and Blood Disorders Center of Children's Healthcare of Atlanta - Egleston Campus)
- Alberto S. Pappo, MD (St. Jude Children's Research Hospital)
- R Beverly Raney, MD (Consultant)
- Stephen J. Shochat, MD (St. Jude Children's Research Hospital)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Childhood Soft Tissue Sarcoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/soft-tissue-sarcoma/hp/child-soft-tissue-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389361]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Updated: April 2, 2018
Source URL: https://www.cancer.gov/publishedcontent/syndication/3899.htm
Source Agency: National Cancer Institute (NCI)
Captured Date: 2013-09-14 09:01:57.0
Kaposi sarcoma: Professional resources from the National Cancer Institute
Kaposi Sarcoma Treatment (PDQ®)–Health Professional Version
General Information About Kaposi Sarcoma
Epidemiology
KS was first described in 1872 by the Hungarian dermatologist, Moritz Kaposi. From that time until the current human immunodeficiency virus (HIV) disease epidemic identified with the Acquired Immunodeficiency Syndrome (AIDS), KS remained a rare tumor. While most of the cases seen in Europe and North America have occurred in elderly men of Italian or Eastern European Jewish ancestry, the neoplasm also occurs in several other distinct populations: young black African adult males, prepubescent children, renal allograft recipients, and other patients receiving immunosuppressive therapy. The disseminated, fulminant form of KS associated with HIV disease is referred to as epidemic KS to distinguish it from the classic, African, and transplant-related varieties of the neoplasm. In addition, KS has been identified in homosexual men apart from the HIV disease epidemic.[1]
Histopathology
Although the histopathology of the different types of the Kaposi tumor is essentially identical in all of these groups, the clinical manifestations and course of the disease differ dramatically.[2] A key piece to the puzzle of KS pathogenesis was the 1994 discovery of a gamma herpes virus, human herpes virus type 8 (HHV-8), also known as Kaposi sarcoma herpes virus.[3] HHV-8 was identified in KS tissue biopsies from virtually all patients with classic, African, transplant-related, and AIDS-associated KS but was absent from noninvolved tissue.[4-7]
Classic Kaposi Sarcoma
Considered a rare disease, classic KS occurs more often in males, with a ratio of approximately 10 to 15 males to 1 female. In North Americans and Europeans, the usual age at onset is between 50 and 70 years. Classic KS tumors usually present with one or more asymptomatic red, purple, or brown patches, plaques, or nodular skin lesions. The disease is often limited to single or multiple lesions usually localized to one or both lower extremities, especially involving the ankles and soles.
Classic KS most commonly runs a relatively benign, indolent course for 10 to 15 years or more, with slow enlargement of the original tumors and the gradual development of additional lesions. Venous stasis and lymphedema of the involved lower extremity are frequent complications. In long-standing cases, systemic lesions can develop along the gastrointestinal tract, in lymph nodes, and in other organs. The visceral lesions are generally asymptomatic and are most often discovered only at autopsy, though clinically, gastrointestinal bleeding can occur. As many as 33% of the patients with classic KS develop a second primary malignancy, which is most often non-Hodgkin lymphoma.[8-10]
African Kaposi Sarcoma
In the 1950s, KS was recognized as a relatively common neoplasm endemic in native populations in equatorial Africa and comprised approximately 9% of all cancers seen in Ugandan males. African KS is seen as either an indolent neoplasm identical to the classic disease seen in Europe and North America or as an aggressive disease with fungating and exophytic tumors that may invade the subcutaneous and surrounding tissue including the underlying bone. In Africa, both the indolent and locally more aggressive forms of KS occur with a male-to-female ratio comparable to that observed with the classic KS tumor seen in North America and Europe. In general, however, patients in Africa are significantly younger than their European counterparts. A lymphadenopathic form of KS is also seen in Africa, primarily in prepubescent children (male:female ratio, 3:1). In these cases, the generalized lymphadenopathy is frequently associated with visceral organ involvement. The prognosis is very poor with a 100% fatality rate within 3 years.[11,12]
Immunosuppressive Treatment–Related Kaposi Sarcoma
In 1969, the first case of KS in association with immunosuppression in a renal transplant patient was described. Since that time, a number of renal and other organ allograft recipients who received prednisone and azathioprine developed KS shortly after the onset of immunosuppressive therapy.[13] Estimates of the incidence of KS in immunosuppressed renal transplant recipients are between 150 and 200 times the expected incidence of the tumor in the general population. The average time to develop KS after transplantation is 16 months. Although the KS tumor in iatrogenically immunosuppressed patients often remains localized to the skin, widespread dissemination with mucocutaneous or visceral organ involvement is common. In some cases, the KS tumors have regressed as a result of reduction or changes in immunosuppressive therapy. Clinical management of renal transplant patients who develop KS is difficult and requires a balance between the risk of death from generalized KS and the risk of graft rejection and complications of renal failure that may occur if the immunosuppressive therapy is discontinued.
Epidemic Kaposi Sarcoma
In 1981, a fulminant and disseminated form of KS in young homosexual or bisexual men was first reported as part of an epidemic now known as AIDS.[14] The etiology of AIDS is a T-cell lymphotropic retrovirus known as HIV. The underlying immunologic deficiency that characterizes HIV disease is an acquired profound disorder of cell-mediated immune functions. This immunologic deficiency and immune dysregulation predisposes the host to a variety of opportunistic infections and unusual neoplasms, especially KS. HIV may play an indirect role in the development of KS.[15]
Approximately 95% of all the cases of epidemic KS in the United States have been diagnosed in homosexual or bisexual men. In the past, approximately 26% of all homosexual males with HIV disease presented with, or eventually developed, KS during the course of their illness. By comparison, fewer than 3% of all heterosexual intravenous drug users with HIV disease developed KS. The proportion of HIV disease patients with KS has steadily decreased since the epidemic was first identified in 1981.[16] About 48% of AIDS patients in 1981 had KS as their presenting AIDS diagnosis. By August 1987, the cumulative proportion of AIDS patients with KS had diminished to fewer than 20%. The introduction of combined antiretroviral therapy (cART) has delayed or prevented the emergence of drug-resistant HIV strains, profoundly decreased viral load, led to increased survival, and lessened the risk of opportunistic infections.[17-19] The use of cART has been associated with a sustained and substantial decline in KS incidence in multiple large cohorts.[20-25]
The lesions that develop may involve the skin; oral mucosa; lymph nodes; and visceral organs, such as the gastrointestinal tract, lung, liver, and spleen. Most patients with HIV disease who present with the mucocutaneous lesions of KS feel healthy and are usually free of systemic symptoms, as compared with HIV patients who first develop an opportunistic infection. The sites of disease at presentation of epidemic KS are much more varied than the sites seen in other types of this neoplasm. In an early report on the clinical manifestations of the disease, 49 patients were described.[26] Of these patients, 8% had no skin involvement, 27% had localized or fewer than five skin lesions, and 63% had innumerable skin lesions widely distributed over the skin surface area. Of these patients, 61% had generalized lymphadenopathy at the time of the first examination. Four of these patients, who had generalized lymphadenopathy in the absence of skin lesions or detectable visceral organ involvement at the time of presentation, were found to have biopsy-proven KS localized to the lymph nodes. In 45% of the patients studied, KS lesions were found in one or more sites along the gastrointestinal tract. Of these patients, 29% had either unexplained fever or unexplained weight loss when first seen. While most patients present with skin disease, KS involvement of lymph nodes or the gastrointestinal tract may occasionally precede the appearance of the cutaneous lesions.
Eventually, most patients with epidemic KS develop disseminated disease. The disease often progresses in an orderly fashion from a few localized or widespread mucocutaneous lesions to more numerous lesions and generalized skin disease with lymph node, gastrointestinal tract disease, and other organ involvement. Pleuropulmonary KS is an ominous sign usually occurring late in the course of the disease, especially in those patients whose death is directly attributed to KS.[27] Most patients with epidemic KS die of one or more complicating opportunistic infections.
References
- Friedman-Kien AE, Saltzman BR, Cao YZ, et al.: Kaposi's sarcoma in HIV-negative homosexual men. Lancet 335 (8682): 168-9, 1990. [PUBMED Abstract]
- Safai B: Kaposi's sarcoma and acquired immunodeficiency syndrome. In: DeVita VT, Hellman S, Rosenberg S, eds.: AIDS: Etiology, Diagnosis, Treatment and Prevention. 4th ed. Philadelphia, Pa: Lippincott-Raven Publishers, 1997, pp 295-318.
- Chang Y, Cesarman E, Pessin MS, et al.: Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266 (5192): 1865-9, 1994. [PUBMED Abstract]
- Moore PS, Chang Y: Detection of herpesvirus-like DNA sequences in Kaposi's sarcoma in patients with and without HIV infection. N Engl J Med 332 (18): 1181-5, 1995. [PUBMED Abstract]
- Su IJ, Hsu YS, Chang YC, et al.: Herpesvirus-like DNA sequence in Kaposi's sarcoma from AIDS and non-AIDS patients in Taiwan. Lancet 345 (8951): 722-3, 1995. [PUBMED Abstract]
- Gao SJ, Kingsley L, Li M, et al.: KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi's sarcoma. Nat Med 2 (8): 925-8, 1996. [PUBMED Abstract]
- Chang Y, Ziegler J, Wabinga H, et al.: Kaposi's sarcoma-associated herpesvirus and Kaposi's sarcoma in Africa. Uganda Kaposi's Sarcoma Study Group. Arch Intern Med 156 (2): 202-4, 1996. [PUBMED Abstract]
- Safai B, Good RA: Kaposi's sarcoma: a review and recent developments. Clin Bull 10 (2): 62-9, 1980. [PUBMED Abstract]
- Reynolds WA, Winkelmann RK, Soule EH: Kaposi's sarcoma: a clinicopathologic study with particular reference to its relationship to the reticuloendothelial system. Medicine (Baltimore) 44 (5): 419-43, 1965. [PUBMED Abstract]
- Safai B, Miké V, Giraldo G, et al.: Association of Kaposi's sarcoma with second primary malignancies: possible etiopathogenic implications. Cancer 45 (6): 1472-9, 1980. [PUBMED Abstract]
- Taylor JF, Templeton AC, Vogel CL, et al.: Kaposi's sarcoma in Uganda: a clinico-pathological study. Int J Cancer 8 (1): 122-35, 1971. [PUBMED Abstract]
- Templeton AC, Bhana D: Prognosis in Kaposi's sarcoma. J Natl Cancer Inst 55 (6): 1301-4, 1975. [PUBMED Abstract]
- Penn I: Kaposi's sarcoma in organ transplant recipients: report of 20 cases. Transplantation 27 (1): 8-11, 1979. [PUBMED Abstract]
- Kaposi's sarcoma and Pneumocystis pneumonia among homosexual men--New York City and California. MMWR Morb Mortal Wkly Rep 30 (25): 305-8, 1981. [PUBMED Abstract]
- Vogel J, Hinrichs SH, Reynolds RK, et al.: The HIV tat gene induces dermal lesions resembling Kaposi's sarcoma in transgenic mice. Nature 335 (6191): 606-11, 1988. [PUBMED Abstract]
- Selik RM, Starcher ET, Curran JW: Opportunistic diseases reported in AIDS patients: frequencies, associations, and trends. AIDS 1 (3): 175-82, 1987. [PUBMED Abstract]
- Flexner C: HIV-protease inhibitors. N Engl J Med 338 (18): 1281-92, 1998. [PUBMED Abstract]
- Palella FJ Jr, Delaney KM, Moorman AC, et al.: Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med 338 (13): 853-60, 1998. [PUBMED Abstract]
- Lodi S, Guiguet M, Costagliola D, et al.: Kaposi sarcoma incidence and survival among HIV-infected homosexual men after HIV seroconversion. J Natl Cancer Inst 102 (11): 784-92, 2010. [PUBMED Abstract]
- Portsmouth S, Stebbing J, Gill J, et al.: A comparison of regimens based on non-nucleoside reverse transcriptase inhibitors or protease inhibitors in preventing Kaposi's sarcoma. AIDS 17 (11): F17-22, 2003. [PUBMED Abstract]
- International Collaboration on HIV and Cancer: Highly active antiretroviral therapy and incidence of cancer in human immunodeficiency virus-infected adults. J Natl Cancer Inst 92 (22): 1823-30, 2000. [PUBMED Abstract]
- Dupont C, Vasseur E, Beauchet A, et al.: Long-term efficacy on Kaposi's sarcoma of highly active antiretroviral therapy in a cohort of HIV-positive patients. CISIH 92. Centre d'information et de soins de l'immunodéficience humaine. AIDS 14 (8): 987-93, 2000. [PUBMED Abstract]
- Tam HK, Zhang ZF, Jacobson LP, et al.: Effect of highly active antiretroviral therapy on survival among HIV-infected men with Kaposi sarcoma or non-Hodgkin lymphoma. Int J Cancer 98 (6): 916-22, 2002. [PUBMED Abstract]
- Carrieri MP, Pradier C, Piselli P, et al.: Reduced incidence of Kaposi's sarcoma and of systemic non-hodgkin's lymphoma in HIV-infected individuals treated with highly active antiretroviral therapy. Int J Cancer 103 (1): 142-4, 2003. [PUBMED Abstract]
- Grabar S, Abraham B, Mahamat A, et al.: Differential impact of combination antiretroviral therapy in preventing Kaposi's sarcoma with and without visceral involvement. J Clin Oncol 24 (21): 3408-14, 2006. [PUBMED Abstract]
- Krigel RL, Laubenstein LJ, Muggia FM: Kaposi's sarcoma: a new staging classification. Cancer Treat Rep 67 (6): 531-4, 1983. [PUBMED Abstract]
- Gill PS, Akil B, Colletti P, et al.: Pulmonary Kaposi's sarcoma: clinical findings and results of therapy. Am J Med 87 (1): 57-61, 1989. [PUBMED Abstract]
Stage Information for Kaposi Sarcoma
The staging evaluation of patients with classic Kaposi sarcoma (KS) should be individualized. The advanced age of most of the patients, localized nature of the tumor, rarity of visceral involvement, and usually indolent course of the disease should temper the extent of the evaluation. A careful examination of the skin and lymph nodes is sufficient in most cases. For the rare patient with rapidly progressive tumor or signs or symptoms of visceral involvement, appropriate evaluation is indicated. No universally accepted classification is available for epidemic KS. Staging schemes that incorporate laboratory parameters as well as clinical features have been proposed. Since most patients with epidemic KS do not die from the disease, factors besides tumor burden are apparently involved in survival.
The conventions used to stage KS and the methods used to evaluate the benefits of KS treatment continue to evolve because of changes in the treatment of human immunodeficiency virus (HIV) and in recognition of deficiencies in standard tumor assessment. The clinical course of KS, the selection of treatment, and the response to treatment are heavily influenced by the degree of underlying immune dysfunction and opportunistic infections.
The AIDS Clinical Trials Group (ACTG) Oncology Committee has published criteria for the evaluation of epidemic KS.[1] The staging system incorporates measures of extent of disease, severity of immunodeficiency, and presence of systemic symptoms. As shown in Table 1 below, the ACTG criteria categorizes the extent of the tumor as localized or disseminated, the CD4 cell number as high or low, and a systemic illness as absent or present.
A subsequent prospective analysis of 294 patients entered on ACTG trials for KS between 1989 and 1995 showed that each of the tumor, immune system, and systemic illness variables was independently associated with survival.[2] Multivariate analysis showed that immune system impairment was the most important single predictor of survival. In patients with relatively high CD4 counts, tumor stage was predictive. A CD4 count of 150 cells/mm³ may be a better discriminator than the published cutoff of 200 cells/mm³. A study is in progress to determine if viral load adds predictive information. None of the prior studies were conducted at a time when combined antiretroviral therapy (cART) was readily available. The impact of cART on survival in KS requires continued assessment.
| Good Risk (0) | Poor Risk (1) | |
|---|---|---|
| (Any of the following) | (Any of the following) | |
| Tumor (T) | Confined to skin and/or lymph nodes and/or minimal oral disease[Note: Minimal oral disease is non-nodular KS confined to the palate.] | Tumor-associated edema or ulceration |
| Extensive oral KS | ||
| Gastrointestinal KS | ||
| KS in other non-nodal viscera | ||
| Immune system (I) | CD4 cells ≥ = 200/µL | CD4 cells <200 per cubic mm |
| Systemic illness (S) | No history of OIs or thrush[Note: OIs are opportunistic infections.] | History of OIs and/or thrush |
| No “B” symptoms[Note: “B” symptoms are unexplained fever, night sweats, >10% involuntary weight loss, or diarrhea persisting >2 weeks.] | “B” symptoms present | |
| Performance status ≥70 (Karnofsky) | Performance status <70 | |
| Other HIV-related illness (e.g., neurological disease or lymphoma) |
References
- Krown SE, Metroka C, Wernz JC: Kaposi's sarcoma in the acquired immune deficiency syndrome: a proposal for uniform evaluation, response, and staging criteria. AIDS Clinical Trials Group Oncology Committee. J Clin Oncol 7 (9): 1201-7, 1989. [PUBMED Abstract]
- Krown SE, Testa MA, Huang J: AIDS-related Kaposi's sarcoma: prospective validation of the AIDS Clinical Trials Group staging classification. AIDS Clinical Trials Group Oncology Committee. J Clin Oncol 15 (9): 3085-92, 1997. [PUBMED Abstract]
Classic Kaposi Sarcoma Treatment
Classic Kaposi sarcoma (KS) usually is limited to the skin and has an indolent course. Patients with this tumor are predisposed to the development of a second primary malignancy, and the treating physician should consider this factor when arranging a schedule of follow-up treatment for the patient.
Equivalent standard treatment options:
Solitary lesions:
- Radiation therapy: For solitary lesions or lesions of limited extent, modest doses of radiation applied to the lesions with a limited margin provide excellent control of disease in the treated area. Usually, superficial radiation beams such as electron beams are used. Some authors believe disease recurrence in adjacent, untreated skin is common if only involved-field radiation therapy is used and claim better cure rates when extended-field radiation therapy is used.[1,2]
- Low-voltage (100 kv) photon radiation: 8 Gy to 10 Gy given as a single dose or 15 Gy to 20 Gy given over 1 week because solitary lesions control nearly 100% of local disease, but recurrence in adjacent areas is common.
- Electron-beam radiation therapy (EBRT): 4 Gy given once weekly for 6 to 8 consecutive weeks with a 4-MeV to 6-MeV electron beam. Ports should include the entire skin surface 15 cm above the lesion.
- Surgical excision may be of benefit in some patients with small superficial lesions, but local recurrence is likely to be a problem. However, over the years, multiple small excisions can be performed to achieve good disease control.
Widespread skin disease:
- Radiation therapy: Modest doses are effective in controlling disease. The type of radiation (i.e., photon vs. electron) and fields used must be tailored to suit the distribution of disease in the individual patient.[2]
- Extended-field EBRT.
- For disease limited to areas distal to the knee, subtotal-skin EBRT directed to skin below the umbilicus.
- For disease that extends above the knee, total-skin EBRT.
EBRT used in this manner gave long-term results that were superior to those obtained with radiation therapy administered to successive individual lesions as they appeared.[2]
- EBRT: 4 Gy given once weekly for 6 to 8 consecutive weeks, and subtotal- or total-skin radiation therapy given for extensive disease.
- Chemotherapy: Because classic KS is such a rare disease in the United States and is usually treated initially with radiation therapy, few patients have been treated with chemotherapy, and no randomized prospective trials have compared one agent to another. Several authors have used single-agent vinblastine given as a weekly dose of approximately 0.1 mg/kg.[3-6] Almost all of the patients had good to excellent response. In most cases, patients required prolonged courses of therapy, for several years, to maintain a partial response. Doses of vinblastine were titrated in individual patients to maintain a white blood count of more than 3,000 leukocytes. Follow-up after completion of therapy was not presented. In a multicenter trial of 55 patients who were treated over a decade, a 71% overall response rate was seen using pegylated liposomal doxorubicin.[7][Level of evidence: 3iiiDiv] In addition to the positive response rates of pegylated liposomal doxorubicin and the vinca alkaloids, response rates showing a greater than 50% decrease in lesions have also been reported in small, uncontrolled series for etoposide, taxanes, gemcitabine, and interferon alfa.[8][Level of evidence: 3iiiDiv]
One patient was treated repeatedly with intralesional injections of 0.25 to 0.50 mg of vincristine, which resulted in complete disappearance of the treated lesion.[9] Multiple courses of therapy were required because of the recurrence of disease in untreated areas.
Electroporation of the skin lesions was combined with intravenous bleomycin for 19 patients with classical KS. Most patients responded after one application, the rest after two or three applications, with a median duration of response of 16 months.[10][Level of evidence: 3iiiDiv]
Lymph node and gastrointestinal tract involvement:
- Chemotherapy: Several patients who had widespread skin disease and were treated with chemotherapy also had lymph node and gastrointestinal tract involvement. The disease in these sites also responded to vinblastine. Trials are required to define therapy. One such trial, MSKCC-04055 (NCT00096538), has been completed.
- Local radiation therapy may be added to chemotherapy if individual lesions require urgent therapy.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Hamilton CR, Cummings BJ, Harwood AR: Radiotherapy of Kaposi's sarcoma. Int J Radiat Oncol Biol Phys 12 (11): 1931-5, 1986. [PUBMED Abstract]
- Nisce LZ, Safai B, Poussin-Rosillo H: Once weekly total and subtotal skin electron beam therapy for Kaposi's sarcoma. Cancer 47 (4): 640-4, 1981. [PUBMED Abstract]
- Solan AJ, Greenwald ES, Silvay O: Long-term complete remissions of Kaposi's sarcoma with vinblastine therapy. Cancer 47 (4): 637-9, 1981. [PUBMED Abstract]
- Tucker SB, Winkelmann RK: Treatment of Kaposi sarcoma with vinblastine. Arch Dermatol 112 (7): 958-61, 1976. [PUBMED Abstract]
- Scott WP, Voight JA: Kaposi's sarcoma. Management with vincaleucoblastine. Cancer 19 (4): 557-64, 1966. [PUBMED Abstract]
- Klein E, Schwartz RA, Laor Y, et al.: Treatment of Kaposi's sarcoma with vinblastine. Cancer 45 (3): 427-31, 1980. [PUBMED Abstract]
- Di Lorenzo G, Kreuter A, Di Trolio R, et al.: Activity and safety of pegylated liposomal doxorubicin as first-line therapy in the treatment of non-visceral classic Kaposi's sarcoma: a multicenter study. J Invest Dermatol 128 (6): 1578-80, 2008. [PUBMED Abstract]
- Régnier-Rosencher E, Guillot B, Dupin N: Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol 68 (2): 313-31, 2013. [PUBMED Abstract]
- Odom RB, Goette DK: Treatment of cutaneous Kaposi's sarcoma with intralesional vincristine. Arch Dermatol 114 (11): 1693-4, 1978. [PUBMED Abstract]
- Di Monta G, Caracò C, Benedetto L, et al.: Electrochemotherapy as "new standard of care" treatment for cutaneous Kaposi's sarcoma. Eur J Surg Oncol 40 (1): 61-6, 2014. [PUBMED Abstract]
Immunosuppressive Therapy–Related Kaposi Sarcoma Treatment
Some patients with Kaposi Sarcoma (KS) have noted spontaneous and lasting remissions following discontinuation of immunosuppressive therapy. In managing these patients, if immunosuppressive therapy is not critical, its discontinuation is a reasonable first step.
Standard treatment options:
- Discontinue immunosuppressive therapy (often results in tumor regression). This option is critically important in patients who are receiving immunosuppressive drugs, as in the case of certain transplant patients.
- Radiation therapy (for disease limited to skin).[1-4]
- Chemotherapy (single or multiple drug): Most systemic chemotherapy trials in KS patients have been carried out in the African and epidemic varieties. See the section on the treatment of Epidemic Kaposi Sarcoma. The applicability of the results of these trials to KS in immunosuppressed patients is unknown.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Cohen L: Dose, time, and volume parameters in irradiation therapy of Kaposi's sarcoma. Br J Radiol 35 (415): 485-488, 1962.
- Hamilton CR, Cummings BJ, Harwood AR: Radiotherapy of Kaposi's sarcoma. Int J Radiat Oncol Biol Phys 12 (11): 1931-5, 1986. [PUBMED Abstract]
- Lo TC, Salzman FA, Smedal MI, et al.: Radiotherapy for Kaposi's sarcoma. Cancer 45 (4): 684-7, 1980. [PUBMED Abstract]
- Nisce LZ, Safai B, Poussin-Rosillo H: Once weekly total and subtotal skin electron beam therapy for Kaposi's sarcoma. Cancer 47 (4): 640-4, 1981. [PUBMED Abstract]
Epidemic Kaposi Sarcoma Treatment
Treatment may result:
- In a disappearance or reduction in size of specific skin lesions, thereby alleviating the discomfort associated with the chronic edema and ulcerations that often accompany multiple skin tumors seen on the lower extremities.
- In control of symptoms associated with mucosal or visceral lesions.
No data are available, however, to show that treatment improves survival.[1] In addition to antitumor treatment, essential components of an optimal Kaposi sarcoma (KS) treatment strategy include combined antiretroviral treatment (cART), prophylaxis for opportunistic infections, and rapid recognition and treatment of intercurrent infections.
Most good-risk patients, defined by the AIDS Clinical Trials Group as T0, show tumor regression with cART alone.[2-4] Poor-risk patients, defined as T1, usually require a combination of cART and chemotherapy with discontinuation of the chemotherapy after disappearance of the skin lesion.[2-4] The combination of cART and liposomal doxorubicin resulted in a 5-year overall survival (OS) rate of 85% in 140 patients with T1 disease.[3][Level of evidence: 3iiiDiv]
Local modalities
Small localized lesions of KS may be treated by electrodesiccation and curettage, cryotherapy, or by surgical excision. KS tumors are also generally very responsive to local radiation therapy, and excellent palliation has been obtained with doses at 20 Gy or slightly higher.[5-7] One report demonstrated a response rate higher than 90%, with a median time to progression of 21 months. Although no difference in response was noted with a variety of fractionation regimens, a single fraction of 8 Gy is indicated for cutaneous lesions and is associated with significantly fewer severe reactions.[8] Radiation therapy is generally reserved to treat localized areas of the skin and oral cavity. It is less often used to control pulmonary, gastrointestinal tract, or other sites of KS lesions. Localized KS lesions have also been effectively treated with intralesional injections of vinblastine.[9] Alitretinoin 0.1% gel provided local control in a randomized prospective multicenter trial.[10][Level of evidence: 1iiDiv]
Chemotherapy
In epidemic KS, the already profoundly depressed immunologic status of the host limits the therapeutic usefulness of systemic chemotherapy. Systemic chemotherapy studies in epidemic KS have used as single agents or in combinations doxorubicin, bleomycin, vinblastine, vincristine, etoposide, paclitaxel, and docetaxel.[11-15][Level of evidence: 3iiiDiv] The combination of cART and liposomal doxorubicin resulted in a 5-year OS of 85% in 140 patients with T1 disease.[3][Level of evidence: 3iiiDiv]
Randomized multicenter trials showed an improvement in response rate (45%–60% vs. 20%–25%) and a more favorable toxic effects profile for pegylated liposomal doxorubicin or liposomal daunorubicin, compared to the combination of doxorubicin, bleomycin, and vincristine or bleomycin and vincristine.[16-18][Level of evidence: 1iiDiv] During cART, both pegylated liposomal doxorubicin and paclitaxel are active single agents with response rates close to 50%.[19][Level of evidence: 1iiDiv]
Biologic and targeted therapy
The interferon alphas have also been widely studied and show a 40% objective response rate in patients with epidemic KS.[20,21] In these reports, the responses differed significantly according to the prognostic factors of extent of disease, prior or coexistent opportunistic infections, prior treatment with chemotherapy, CD4 lymphocyte counts lower than 200 cells/mm³, the presence of circulating acid-labile interferon alpha, and an increase in beta-2-microglobulin. Several treatment studies have combined interferon alpha with other chemotherapeutic agents. Overall, these trials have shown no benefit with the interferon-chemotherapy combinations as compared to the single-agent activities.
Recombinant interferon alpha-2a and interferon alpha-2b were the first agents approved for the treatment of KS. Approval was based on single-agent studies performed in the 1980s before the advent of antiretroviral therapy. The early studies demonstrated improved efficacy at relatively high doses. High-dose monotherapy is rarely used today, and instead, interferon is given in combination with other anti-HIV drugs in doses of 4 to 18 million units. Neutropenia is dose limiting, and trials of doses of 1 to 10 million units combined with less myelosuppressive antiretrovirals are in progress. Response to interferon is slow, and the maximum effect is seen after 6 or more months. Interferon should probably not be used in the treatment of patients with rapidly progressive, symptomatic KS.
Imatinib, a c-kit/PDGF (platelet-derived growth factor) receptor inhibitor, resulted in partial responses in 10 of 30 previously treated patients (cART + chemotherapy).[22]
Bevacizumab, the humanized, antivascular, endothelial growth–factor monoclonal antibody, had a response rate in 5 of 16 patients who did not improve after the institution of cART and chemotherapy.[23][Level of evidence: 3iiiDiv]
Interleukin-12 had a response rate of 71% (95% confidence interval, 48%–89%) among 24 evaluable patients in a phase I and phase II trial.[24][Level of evidence: 3iiiDiv]
Treatment options under clinical evaluation:
- Patients with epidemic KS are appropriate candidates for clinical trials evaluating new drugs or biologicals.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Safai B: Kaposi's sarcoma and acquired immunodeficiency syndrome. In: DeVita VT, Hellman S, Rosenberg S, eds.: AIDS: Etiology, Diagnosis, Treatment and Prevention. 4th ed. Philadelphia, Pa: Lippincott-Raven Publishers, 1997, pp 295-318.
- Krown SE: Highly active antiretroviral therapy in AIDS-associated Kaposi's sarcoma: implications for the design of therapeutic trials in patients with advanced, symptomatic Kaposi's sarcoma. J Clin Oncol 22 (3): 399-402, 2004. [PUBMED Abstract]
- Bower M, Dalla Pria A, Coyle C, et al.: Prospective stage-stratified approach to AIDS-related Kaposi's sarcoma. J Clin Oncol 32 (5): 409-14, 2014. [PUBMED Abstract]
- Krell J, Stebbing J: Broader implications of a stage-guided stratified therapeutic approach for AIDS-related Kaposi's sarcoma. J Clin Oncol 32 (5): 373-5, 2014. [PUBMED Abstract]
- Cooper JS, Steinfeld AD, Lerch I: Intentions and outcomes in the radiotherapeutic management of epidemic Kaposi's sarcoma. Int J Radiat Oncol Biol Phys 20 (3): 419-22, 1991. [PUBMED Abstract]
- Nobler MP, Leddy ME, Huh SH: The impact of palliative irradiation on the management of patients with acquired immune deficiency syndrome. J Clin Oncol 5 (1): 107-12, 1987. [PUBMED Abstract]
- Singh NB, Lakier RH, Donde B: Hypofractionated radiation therapy in the treatment of epidemic Kaposi sarcoma--a prospective randomized trial. Radiother Oncol 88 (2): 211-6, 2008. [PUBMED Abstract]
- Berson AM, Quivey JM, Harris JW, et al.: Radiation therapy for AIDS-related Kaposi's Sarcoma. Int J Radiat Oncol Biol Phys 19 (3): 569-75, 1990. [PUBMED Abstract]
- Epstein JB, Lozada-Nur F, McLeod WA, et al.: Oral Kaposi's sarcoma in acquired immunodeficiency syndrome. Review of management and report of the efficacy of intralesional vinblastine. Cancer 64 (12): 2424-30, 1989. [PUBMED Abstract]
- Bodsworth NJ, Bloch M, Bower M, et al.: Phase III vehicle-controlled, multi-centered study of topical alitretinoin gel 0.1% in cutaneous AIDS-related Kaposi's sarcoma. Am J Clin Dermatol 2 (2): 77-87, 2001. [PUBMED Abstract]
- Evans SR, Krown SE, Testa MA, et al.: Phase II evaluation of low-dose oral etoposide for the treatment of relapsed or progressive AIDS-related Kaposi's sarcoma: an AIDS Clinical Trials Group clinical study. J Clin Oncol 20 (15): 3236-41, 2002. [PUBMED Abstract]
- Saville MW, Lietzau J, Pluda JM, et al.: Treatment of HIV-associated Kaposi's sarcoma with paclitaxel. Lancet 346 (8966): 26-8, 1995. [PUBMED Abstract]
- Lim ST, Tupule A, Espina BM, et al.: Weekly docetaxel is safe and effective in the treatment of advanced-stage acquired immunodeficiency syndrome-related Kaposi sarcoma. Cancer 103 (2): 417-21, 2005. [PUBMED Abstract]
- Gill PS, Tulpule A, Espina BM, et al.: Paclitaxel is safe and effective in the treatment of advanced AIDS-related Kaposi's sarcoma. J Clin Oncol 17 (6): 1876-83, 1999. [PUBMED Abstract]
- Di Lorenzo G, Konstantinopoulos PA, Pantanowitz L, et al.: Management of AIDS-related Kaposi's sarcoma. Lancet Oncol 8 (2): 167-76, 2007. [PUBMED Abstract]
- Stewart S, Jablonowski H, Goebel FD, et al.: Randomized comparative trial of pegylated liposomal doxorubicin versus bleomycin and vincristine in the treatment of AIDS-related Kaposi's sarcoma. International Pegylated Liposomal Doxorubicin Study Group. J Clin Oncol 16 (2): 683-91, 1998. [PUBMED Abstract]
- Northfelt DW, Dezube BJ, Thommes JA, et al.: Pegylated-liposomal doxorubicin versus doxorubicin, bleomycin, and vincristine in the treatment of AIDS-related Kaposi's sarcoma: results of a randomized phase III clinical trial. J Clin Oncol 16 (7): 2445-51, 1998. [PUBMED Abstract]
- Gill PS, Wernz J, Scadden DT, et al.: Randomized phase III trial of liposomal daunorubicin versus doxorubicin, bleomycin, and vincristine in AIDS-related Kaposi's sarcoma. J Clin Oncol 14 (8): 2353-64, 1996. [PUBMED Abstract]
- Cianfrocca M, Lee S, Von Roenn J, et al.: Randomized trial of paclitaxel versus pegylated liposomal doxorubicin for advanced human immunodeficiency virus-associated Kaposi sarcoma: evidence of symptom palliation from chemotherapy. Cancer 116 (16): 3969-77, 2010. [PUBMED Abstract]
- Real FX, Oettgen HF, Krown SE: Kaposi's sarcoma and the acquired immunodeficiency syndrome: treatment with high and low doses of recombinant leukocyte A interferon. J Clin Oncol 4 (4): 544-51, 1986. [PUBMED Abstract]
- Groopman JE, Gottlieb MS, Goodman J, et al.: Recombinant alpha-2 interferon therapy for Kaposi's sarcoma associated with the acquired immunodeficiency syndrome. Ann Intern Med 100 (5): 671-6, 1984. [PUBMED Abstract]
- Koon HB, Krown SE, Lee JY, et al.: Phase II trial of imatinib in AIDS-associated Kaposi's sarcoma: AIDS Malignancy Consortium Protocol 042. J Clin Oncol 32 (5): 402-8, 2014. [PUBMED Abstract]
- Uldrick TS, Wyvill KM, Kumar P, et al.: Phase II study of bevacizumab in patients with HIV-associated Kaposi's sarcoma receiving antiretroviral therapy. J Clin Oncol 30 (13): 1476-83, 2012. [PUBMED Abstract]
- Little RF, Pluda JM, Wyvill KM, et al.: Activity of subcutaneous interleukin-12 in AIDS-related Kaposi sarcoma. Blood 107 (12): 4650-7, 2006. [PUBMED Abstract]
Recurrent Kaposi Sarcoma Treatment
The prognosis for any treated Kaposi sarcoma patient with progressing, recurring, or relapsing disease is highly variable. Deciding on further treatment depends on many factors, most importantly the clinical setting (i.e., classic, immunosuppressive treatment, or AIDS) in which the tumor arises as well as individual patient considerations.
Clinical trials are appropriate and should be considered when possible.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
Changes to This Summary (01/30/2018)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Editorial changes were made to this summary.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of Kaposi sarcoma. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Kaposi Sarcoma Treatment are:
- Eric J. Seifter, MD (Johns Hopkins University)
- Minh Tam Truong, MD (Boston University Medical Center)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Kaposi Sarcoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/soft-tissue-sarcoma/hp/kaposi-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389335]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Updated: January 30, 2018
Source URL: https://www.cancer.gov/publishedcontent/syndication/3524.htm
Source Agency: National Cancer Institute (NCI)
Captured Date: 2013-09-14 09:01:43.0
Kaposi Sarcoma Treatment (PDQ®)–Health Professional Version
General Information About Kaposi Sarcoma
Epidemiology
KS was first described in 1872 by the Hungarian dermatologist, Moritz Kaposi. From that time until the current human immunodeficiency virus (HIV) disease epidemic identified with the Acquired Immunodeficiency Syndrome (AIDS), KS remained a rare tumor. While most of the cases seen in Europe and North America have occurred in elderly men of Italian or Eastern European Jewish ancestry, the neoplasm also occurs in several other distinct populations: young black African adult males, prepubescent children, renal allograft recipients, and other patients receiving immunosuppressive therapy. The disseminated, fulminant form of KS associated with HIV disease is referred to as epidemic KS to distinguish it from the classic, African, and transplant-related varieties of the neoplasm. In addition, KS has been identified in homosexual men apart from the HIV disease epidemic.[1]
Histopathology
Although the histopathology of the different types of the Kaposi tumor is essentially identical in all of these groups, the clinical manifestations and course of the disease differ dramatically.[2] A key piece to the puzzle of KS pathogenesis was the 1994 discovery of a gamma herpes virus, human herpes virus type 8 (HHV-8), also known as Kaposi sarcoma herpes virus.[3] HHV-8 was identified in KS tissue biopsies from virtually all patients with classic, African, transplant-related, and AIDS-associated KS but was absent from noninvolved tissue.[4-7]
Classic Kaposi Sarcoma
Considered a rare disease, classic KS occurs more often in males, with a ratio of approximately 10 to 15 males to 1 female. In North Americans and Europeans, the usual age at onset is between 50 and 70 years. Classic KS tumors usually present with one or more asymptomatic red, purple, or brown patches, plaques, or nodular skin lesions. The disease is often limited to single or multiple lesions usually localized to one or both lower extremities, especially involving the ankles and soles.
Classic KS most commonly runs a relatively benign, indolent course for 10 to 15 years or more, with slow enlargement of the original tumors and the gradual development of additional lesions. Venous stasis and lymphedema of the involved lower extremity are frequent complications. In long-standing cases, systemic lesions can develop along the gastrointestinal tract, in lymph nodes, and in other organs. The visceral lesions are generally asymptomatic and are most often discovered only at autopsy, though clinically, gastrointestinal bleeding can occur. As many as 33% of the patients with classic KS develop a second primary malignancy, which is most often non-Hodgkin lymphoma.[8-10]
African Kaposi Sarcoma
In the 1950s, KS was recognized as a relatively common neoplasm endemic in native populations in equatorial Africa and comprised approximately 9% of all cancers seen in Ugandan males. African KS is seen as either an indolent neoplasm identical to the classic disease seen in Europe and North America or as an aggressive disease with fungating and exophytic tumors that may invade the subcutaneous and surrounding tissue including the underlying bone. In Africa, both the indolent and locally more aggressive forms of KS occur with a male-to-female ratio comparable to that observed with the classic KS tumor seen in North America and Europe. In general, however, patients in Africa are significantly younger than their European counterparts. A lymphadenopathic form of KS is also seen in Africa, primarily in prepubescent children (male:female ratio, 3:1). In these cases, the generalized lymphadenopathy is frequently associated with visceral organ involvement. The prognosis is very poor with a 100% fatality rate within 3 years.[11,12]
Immunosuppressive Treatment–Related Kaposi Sarcoma
In 1969, the first case of KS in association with immunosuppression in a renal transplant patient was described. Since that time, a number of renal and other organ allograft recipients who received prednisone and azathioprine developed KS shortly after the onset of immunosuppressive therapy.[13] Estimates of the incidence of KS in immunosuppressed renal transplant recipients are between 150 and 200 times the expected incidence of the tumor in the general population. The average time to develop KS after transplantation is 16 months. Although the KS tumor in iatrogenically immunosuppressed patients often remains localized to the skin, widespread dissemination with mucocutaneous or visceral organ involvement is common. In some cases, the KS tumors have regressed as a result of reduction or changes in immunosuppressive therapy. Clinical management of renal transplant patients who develop KS is difficult and requires a balance between the risk of death from generalized KS and the risk of graft rejection and complications of renal failure that may occur if the immunosuppressive therapy is discontinued.
Epidemic Kaposi Sarcoma
In 1981, a fulminant and disseminated form of KS in young homosexual or bisexual men was first reported as part of an epidemic now known as AIDS.[14] The etiology of AIDS is a T-cell lymphotropic retrovirus known as HIV. The underlying immunologic deficiency that characterizes HIV disease is an acquired profound disorder of cell-mediated immune functions. This immunologic deficiency and immune dysregulation predisposes the host to a variety of opportunistic infections and unusual neoplasms, especially KS. HIV may play an indirect role in the development of KS.[15]
Approximately 95% of all the cases of epidemic KS in the United States have been diagnosed in homosexual or bisexual men. In the past, approximately 26% of all homosexual males with HIV disease presented with, or eventually developed, KS during the course of their illness. By comparison, fewer than 3% of all heterosexual intravenous drug users with HIV disease developed KS. The proportion of HIV disease patients with KS has steadily decreased since the epidemic was first identified in 1981.[16] About 48% of AIDS patients in 1981 had KS as their presenting AIDS diagnosis. By August 1987, the cumulative proportion of AIDS patients with KS had diminished to fewer than 20%. The introduction of combined antiretroviral therapy (cART) has delayed or prevented the emergence of drug-resistant HIV strains, profoundly decreased viral load, led to increased survival, and lessened the risk of opportunistic infections.[17-19] The use of cART has been associated with a sustained and substantial decline in KS incidence in multiple large cohorts.[20-25]
The lesions that develop may involve the skin; oral mucosa; lymph nodes; and visceral organs, such as the gastrointestinal tract, lung, liver, and spleen. Most patients with HIV disease who present with the mucocutaneous lesions of KS feel healthy and are usually free of systemic symptoms, as compared with HIV patients who first develop an opportunistic infection. The sites of disease at presentation of epidemic KS are much more varied than the sites seen in other types of this neoplasm. In an early report on the clinical manifestations of the disease, 49 patients were described.[26] Of these patients, 8% had no skin involvement, 27% had localized or fewer than five skin lesions, and 63% had innumerable skin lesions widely distributed over the skin surface area. Of these patients, 61% had generalized lymphadenopathy at the time of the first examination. Four of these patients, who had generalized lymphadenopathy in the absence of skin lesions or detectable visceral organ involvement at the time of presentation, were found to have biopsy-proven KS localized to the lymph nodes. In 45% of the patients studied, KS lesions were found in one or more sites along the gastrointestinal tract. Of these patients, 29% had either unexplained fever or unexplained weight loss when first seen. While most patients present with skin disease, KS involvement of lymph nodes or the gastrointestinal tract may occasionally precede the appearance of the cutaneous lesions.
Eventually, most patients with epidemic KS develop disseminated disease. The disease often progresses in an orderly fashion from a few localized or widespread mucocutaneous lesions to more numerous lesions and generalized skin disease with lymph node, gastrointestinal tract disease, and other organ involvement. Pleuropulmonary KS is an ominous sign usually occurring late in the course of the disease, especially in those patients whose death is directly attributed to KS.[27] Most patients with epidemic KS die of one or more complicating opportunistic infections.
References
- Friedman-Kien AE, Saltzman BR, Cao YZ, et al.: Kaposi's sarcoma in HIV-negative homosexual men. Lancet 335 (8682): 168-9, 1990. [PUBMED Abstract]
- Safai B: Kaposi's sarcoma and acquired immunodeficiency syndrome. In: DeVita VT, Hellman S, Rosenberg S, eds.: AIDS: Etiology, Diagnosis, Treatment and Prevention. 4th ed. Philadelphia, Pa: Lippincott-Raven Publishers, 1997, pp 295-318.
- Chang Y, Cesarman E, Pessin MS, et al.: Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266 (5192): 1865-9, 1994. [PUBMED Abstract]
- Moore PS, Chang Y: Detection of herpesvirus-like DNA sequences in Kaposi's sarcoma in patients with and without HIV infection. N Engl J Med 332 (18): 1181-5, 1995. [PUBMED Abstract]
- Su IJ, Hsu YS, Chang YC, et al.: Herpesvirus-like DNA sequence in Kaposi's sarcoma from AIDS and non-AIDS patients in Taiwan. Lancet 345 (8951): 722-3, 1995. [PUBMED Abstract]
- Gao SJ, Kingsley L, Li M, et al.: KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi's sarcoma. Nat Med 2 (8): 925-8, 1996. [PUBMED Abstract]
- Chang Y, Ziegler J, Wabinga H, et al.: Kaposi's sarcoma-associated herpesvirus and Kaposi's sarcoma in Africa. Uganda Kaposi's Sarcoma Study Group. Arch Intern Med 156 (2): 202-4, 1996. [PUBMED Abstract]
- Safai B, Good RA: Kaposi's sarcoma: a review and recent developments. Clin Bull 10 (2): 62-9, 1980. [PUBMED Abstract]
- Reynolds WA, Winkelmann RK, Soule EH: Kaposi's sarcoma: a clinicopathologic study with particular reference to its relationship to the reticuloendothelial system. Medicine (Baltimore) 44 (5): 419-43, 1965. [PUBMED Abstract]
- Safai B, Miké V, Giraldo G, et al.: Association of Kaposi's sarcoma with second primary malignancies: possible etiopathogenic implications. Cancer 45 (6): 1472-9, 1980. [PUBMED Abstract]
- Taylor JF, Templeton AC, Vogel CL, et al.: Kaposi's sarcoma in Uganda: a clinico-pathological study. Int J Cancer 8 (1): 122-35, 1971. [PUBMED Abstract]
- Templeton AC, Bhana D: Prognosis in Kaposi's sarcoma. J Natl Cancer Inst 55 (6): 1301-4, 1975. [PUBMED Abstract]
- Penn I: Kaposi's sarcoma in organ transplant recipients: report of 20 cases. Transplantation 27 (1): 8-11, 1979. [PUBMED Abstract]
- Kaposi's sarcoma and Pneumocystis pneumonia among homosexual men--New York City and California. MMWR Morb Mortal Wkly Rep 30 (25): 305-8, 1981. [PUBMED Abstract]
- Vogel J, Hinrichs SH, Reynolds RK, et al.: The HIV tat gene induces dermal lesions resembling Kaposi's sarcoma in transgenic mice. Nature 335 (6191): 606-11, 1988. [PUBMED Abstract]
- Selik RM, Starcher ET, Curran JW: Opportunistic diseases reported in AIDS patients: frequencies, associations, and trends. AIDS 1 (3): 175-82, 1987. [PUBMED Abstract]
- Flexner C: HIV-protease inhibitors. N Engl J Med 338 (18): 1281-92, 1998. [PUBMED Abstract]
- Palella FJ Jr, Delaney KM, Moorman AC, et al.: Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med 338 (13): 853-60, 1998. [PUBMED Abstract]
- Lodi S, Guiguet M, Costagliola D, et al.: Kaposi sarcoma incidence and survival among HIV-infected homosexual men after HIV seroconversion. J Natl Cancer Inst 102 (11): 784-92, 2010. [PUBMED Abstract]
- Portsmouth S, Stebbing J, Gill J, et al.: A comparison of regimens based on non-nucleoside reverse transcriptase inhibitors or protease inhibitors in preventing Kaposi's sarcoma. AIDS 17 (11): F17-22, 2003. [PUBMED Abstract]
- International Collaboration on HIV and Cancer: Highly active antiretroviral therapy and incidence of cancer in human immunodeficiency virus-infected adults. J Natl Cancer Inst 92 (22): 1823-30, 2000. [PUBMED Abstract]
- Dupont C, Vasseur E, Beauchet A, et al.: Long-term efficacy on Kaposi's sarcoma of highly active antiretroviral therapy in a cohort of HIV-positive patients. CISIH 92. Centre d'information et de soins de l'immunodéficience humaine. AIDS 14 (8): 987-93, 2000. [PUBMED Abstract]
- Tam HK, Zhang ZF, Jacobson LP, et al.: Effect of highly active antiretroviral therapy on survival among HIV-infected men with Kaposi sarcoma or non-Hodgkin lymphoma. Int J Cancer 98 (6): 916-22, 2002. [PUBMED Abstract]
- Carrieri MP, Pradier C, Piselli P, et al.: Reduced incidence of Kaposi's sarcoma and of systemic non-hodgkin's lymphoma in HIV-infected individuals treated with highly active antiretroviral therapy. Int J Cancer 103 (1): 142-4, 2003. [PUBMED Abstract]
- Grabar S, Abraham B, Mahamat A, et al.: Differential impact of combination antiretroviral therapy in preventing Kaposi's sarcoma with and without visceral involvement. J Clin Oncol 24 (21): 3408-14, 2006. [PUBMED Abstract]
- Krigel RL, Laubenstein LJ, Muggia FM: Kaposi's sarcoma: a new staging classification. Cancer Treat Rep 67 (6): 531-4, 1983. [PUBMED Abstract]
- Gill PS, Akil B, Colletti P, et al.: Pulmonary Kaposi's sarcoma: clinical findings and results of therapy. Am J Med 87 (1): 57-61, 1989. [PUBMED Abstract]
Stage Information for Kaposi Sarcoma
The staging evaluation of patients with classic Kaposi sarcoma (KS) should be individualized. The advanced age of most of the patients, localized nature of the tumor, rarity of visceral involvement, and usually indolent course of the disease should temper the extent of the evaluation. A careful examination of the skin and lymph nodes is sufficient in most cases. For the rare patient with rapidly progressive tumor or signs or symptoms of visceral involvement, appropriate evaluation is indicated. No universally accepted classification is available for epidemic KS. Staging schemes that incorporate laboratory parameters as well as clinical features have been proposed. Since most patients with epidemic KS do not die from the disease, factors besides tumor burden are apparently involved in survival.
The conventions used to stage KS and the methods used to evaluate the benefits of KS treatment continue to evolve because of changes in the treatment of human immunodeficiency virus (HIV) and in recognition of deficiencies in standard tumor assessment. The clinical course of KS, the selection of treatment, and the response to treatment are heavily influenced by the degree of underlying immune dysfunction and opportunistic infections.
The AIDS Clinical Trials Group (ACTG) Oncology Committee has published criteria for the evaluation of epidemic KS.[1] The staging system incorporates measures of extent of disease, severity of immunodeficiency, and presence of systemic symptoms. As shown in Table 1 below, the ACTG criteria categorizes the extent of the tumor as localized or disseminated, the CD4 cell number as high or low, and a systemic illness as absent or present.
A subsequent prospective analysis of 294 patients entered on ACTG trials for KS between 1989 and 1995 showed that each of the tumor, immune system, and systemic illness variables was independently associated with survival.[2] Multivariate analysis showed that immune system impairment was the most important single predictor of survival. In patients with relatively high CD4 counts, tumor stage was predictive. A CD4 count of 150 cells/mm³ may be a better discriminator than the published cutoff of 200 cells/mm³. A study is in progress to determine if viral load adds predictive information. None of the prior studies were conducted at a time when combined antiretroviral therapy (cART) was readily available. The impact of cART on survival in KS requires continued assessment.
| Good Risk (0) | Poor Risk (1) | |
|---|---|---|
| (Any of the following) | (Any of the following) | |
| Tumor (T) | Confined to skin and/or lymph nodes and/or minimal oral disease[Note: Minimal oral disease is non-nodular KS confined to the palate.] | Tumor-associated edema or ulceration |
| Extensive oral KS | ||
| Gastrointestinal KS | ||
| KS in other non-nodal viscera | ||
| Immune system (I) | CD4 cells ≥ = 200/µL | CD4 cells <200 per cubic mm |
| Systemic illness (S) | No history of OIs or thrush[Note: OIs are opportunistic infections.] | History of OIs and/or thrush |
| No “B” symptoms[Note: “B” symptoms are unexplained fever, night sweats, >10% involuntary weight loss, or diarrhea persisting >2 weeks.] | “B” symptoms present | |
| Performance status ≥70 (Karnofsky) | Performance status <70 | |
| Other HIV-related illness (e.g., neurological disease or lymphoma) |
References
- Krown SE, Metroka C, Wernz JC: Kaposi's sarcoma in the acquired immune deficiency syndrome: a proposal for uniform evaluation, response, and staging criteria. AIDS Clinical Trials Group Oncology Committee. J Clin Oncol 7 (9): 1201-7, 1989. [PUBMED Abstract]
- Krown SE, Testa MA, Huang J: AIDS-related Kaposi's sarcoma: prospective validation of the AIDS Clinical Trials Group staging classification. AIDS Clinical Trials Group Oncology Committee. J Clin Oncol 15 (9): 3085-92, 1997. [PUBMED Abstract]
Classic Kaposi Sarcoma Treatment
Classic Kaposi sarcoma (KS) usually is limited to the skin and has an indolent course. Patients with this tumor are predisposed to the development of a second primary malignancy, and the treating physician should consider this factor when arranging a schedule of follow-up treatment for the patient.
Equivalent standard treatment options:
Solitary lesions:
- Radiation therapy: For solitary lesions or lesions of limited extent, modest doses of radiation applied to the lesions with a limited margin provide excellent control of disease in the treated area. Usually, superficial radiation beams such as electron beams are used. Some authors believe disease recurrence in adjacent, untreated skin is common if only involved-field radiation therapy is used and claim better cure rates when extended-field radiation therapy is used.[1,2]
- Low-voltage (100 kv) photon radiation: 8 Gy to 10 Gy given as a single dose or 15 Gy to 20 Gy given over 1 week because solitary lesions control nearly 100% of local disease, but recurrence in adjacent areas is common.
- Electron-beam radiation therapy (EBRT): 4 Gy given once weekly for 6 to 8 consecutive weeks with a 4-MeV to 6-MeV electron beam. Ports should include the entire skin surface 15 cm above the lesion.
- Surgical excision may be of benefit in some patients with small superficial lesions, but local recurrence is likely to be a problem. However, over the years, multiple small excisions can be performed to achieve good disease control.
Widespread skin disease:
- Radiation therapy: Modest doses are effective in controlling disease. The type of radiation (i.e., photon vs. electron) and fields used must be tailored to suit the distribution of disease in the individual patient.[2]
- Extended-field EBRT.
- For disease limited to areas distal to the knee, subtotal-skin EBRT directed to skin below the umbilicus.
- For disease that extends above the knee, total-skin EBRT.
EBRT used in this manner gave long-term results that were superior to those obtained with radiation therapy administered to successive individual lesions as they appeared.[2]
- EBRT: 4 Gy given once weekly for 6 to 8 consecutive weeks, and subtotal- or total-skin radiation therapy given for extensive disease.
- Chemotherapy: Because classic KS is such a rare disease in the United States and is usually treated initially with radiation therapy, few patients have been treated with chemotherapy, and no randomized prospective trials have compared one agent to another. Several authors have used single-agent vinblastine given as a weekly dose of approximately 0.1 mg/kg.[3-6] Almost all of the patients had good to excellent response. In most cases, patients required prolonged courses of therapy, for several years, to maintain a partial response. Doses of vinblastine were titrated in individual patients to maintain a white blood count of more than 3,000 leukocytes. Follow-up after completion of therapy was not presented. In a multicenter trial of 55 patients who were treated over a decade, a 71% overall response rate was seen using pegylated liposomal doxorubicin.[7][Level of evidence: 3iiiDiv] In addition to the positive response rates of pegylated liposomal doxorubicin and the vinca alkaloids, response rates showing a greater than 50% decrease in lesions have also been reported in small, uncontrolled series for etoposide, taxanes, gemcitabine, and interferon alfa.[8][Level of evidence: 3iiiDiv]
One patient was treated repeatedly with intralesional injections of 0.25 to 0.50 mg of vincristine, which resulted in complete disappearance of the treated lesion.[9] Multiple courses of therapy were required because of the recurrence of disease in untreated areas.
Electroporation of the skin lesions was combined with intravenous bleomycin for 19 patients with classical KS. Most patients responded after one application, the rest after two or three applications, with a median duration of response of 16 months.[10][Level of evidence: 3iiiDiv]
Lymph node and gastrointestinal tract involvement:
- Chemotherapy: Several patients who had widespread skin disease and were treated with chemotherapy also had lymph node and gastrointestinal tract involvement. The disease in these sites also responded to vinblastine. Trials are required to define therapy. One such trial, MSKCC-04055 (NCT00096538), has been completed.
- Local radiation therapy may be added to chemotherapy if individual lesions require urgent therapy.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Hamilton CR, Cummings BJ, Harwood AR: Radiotherapy of Kaposi's sarcoma. Int J Radiat Oncol Biol Phys 12 (11): 1931-5, 1986. [PUBMED Abstract]
- Nisce LZ, Safai B, Poussin-Rosillo H: Once weekly total and subtotal skin electron beam therapy for Kaposi's sarcoma. Cancer 47 (4): 640-4, 1981. [PUBMED Abstract]
- Solan AJ, Greenwald ES, Silvay O: Long-term complete remissions of Kaposi's sarcoma with vinblastine therapy. Cancer 47 (4): 637-9, 1981. [PUBMED Abstract]
- Tucker SB, Winkelmann RK: Treatment of Kaposi sarcoma with vinblastine. Arch Dermatol 112 (7): 958-61, 1976. [PUBMED Abstract]
- Scott WP, Voight JA: Kaposi's sarcoma. Management with vincaleucoblastine. Cancer 19 (4): 557-64, 1966. [PUBMED Abstract]
- Klein E, Schwartz RA, Laor Y, et al.: Treatment of Kaposi's sarcoma with vinblastine. Cancer 45 (3): 427-31, 1980. [PUBMED Abstract]
- Di Lorenzo G, Kreuter A, Di Trolio R, et al.: Activity and safety of pegylated liposomal doxorubicin as first-line therapy in the treatment of non-visceral classic Kaposi's sarcoma: a multicenter study. J Invest Dermatol 128 (6): 1578-80, 2008. [PUBMED Abstract]
- Régnier-Rosencher E, Guillot B, Dupin N: Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol 68 (2): 313-31, 2013. [PUBMED Abstract]
- Odom RB, Goette DK: Treatment of cutaneous Kaposi's sarcoma with intralesional vincristine. Arch Dermatol 114 (11): 1693-4, 1978. [PUBMED Abstract]
- Di Monta G, Caracò C, Benedetto L, et al.: Electrochemotherapy as "new standard of care" treatment for cutaneous Kaposi's sarcoma. Eur J Surg Oncol 40 (1): 61-6, 2014. [PUBMED Abstract]
Immunosuppressive Therapy–Related Kaposi Sarcoma Treatment
Some patients with Kaposi Sarcoma (KS) have noted spontaneous and lasting remissions following discontinuation of immunosuppressive therapy. In managing these patients, if immunosuppressive therapy is not critical, its discontinuation is a reasonable first step.
Standard treatment options:
- Discontinue immunosuppressive therapy (often results in tumor regression). This option is critically important in patients who are receiving immunosuppressive drugs, as in the case of certain transplant patients.
- Radiation therapy (for disease limited to skin).[1-4]
- Chemotherapy (single or multiple drug): Most systemic chemotherapy trials in KS patients have been carried out in the African and epidemic varieties. See the section on the treatment of Epidemic Kaposi Sarcoma. The applicability of the results of these trials to KS in immunosuppressed patients is unknown.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Cohen L: Dose, time, and volume parameters in irradiation therapy of Kaposi's sarcoma. Br J Radiol 35 (415): 485-488, 1962.
- Hamilton CR, Cummings BJ, Harwood AR: Radiotherapy of Kaposi's sarcoma. Int J Radiat Oncol Biol Phys 12 (11): 1931-5, 1986. [PUBMED Abstract]
- Lo TC, Salzman FA, Smedal MI, et al.: Radiotherapy for Kaposi's sarcoma. Cancer 45 (4): 684-7, 1980. [PUBMED Abstract]
- Nisce LZ, Safai B, Poussin-Rosillo H: Once weekly total and subtotal skin electron beam therapy for Kaposi's sarcoma. Cancer 47 (4): 640-4, 1981. [PUBMED Abstract]
Epidemic Kaposi Sarcoma Treatment
Treatment may result:
- In a disappearance or reduction in size of specific skin lesions, thereby alleviating the discomfort associated with the chronic edema and ulcerations that often accompany multiple skin tumors seen on the lower extremities.
- In control of symptoms associated with mucosal or visceral lesions.
No data are available, however, to show that treatment improves survival.[1] In addition to antitumor treatment, essential components of an optimal Kaposi sarcoma (KS) treatment strategy include combined antiretroviral treatment (cART), prophylaxis for opportunistic infections, and rapid recognition and treatment of intercurrent infections.
Most good-risk patients, defined by the AIDS Clinical Trials Group as T0, show tumor regression with cART alone.[2-4] Poor-risk patients, defined as T1, usually require a combination of cART and chemotherapy with discontinuation of the chemotherapy after disappearance of the skin lesion.[2-4] The combination of cART and liposomal doxorubicin resulted in a 5-year overall survival (OS) rate of 85% in 140 patients with T1 disease.[3][Level of evidence: 3iiiDiv]
Local modalities
Small localized lesions of KS may be treated by electrodesiccation and curettage, cryotherapy, or by surgical excision. KS tumors are also generally very responsive to local radiation therapy, and excellent palliation has been obtained with doses at 20 Gy or slightly higher.[5-7] One report demonstrated a response rate higher than 90%, with a median time to progression of 21 months. Although no difference in response was noted with a variety of fractionation regimens, a single fraction of 8 Gy is indicated for cutaneous lesions and is associated with significantly fewer severe reactions.[8] Radiation therapy is generally reserved to treat localized areas of the skin and oral cavity. It is less often used to control pulmonary, gastrointestinal tract, or other sites of KS lesions. Localized KS lesions have also been effectively treated with intralesional injections of vinblastine.[9] Alitretinoin 0.1% gel provided local control in a randomized prospective multicenter trial.[10][Level of evidence: 1iiDiv]
Chemotherapy
In epidemic KS, the already profoundly depressed immunologic status of the host limits the therapeutic usefulness of systemic chemotherapy. Systemic chemotherapy studies in epidemic KS have used as single agents or in combinations doxorubicin, bleomycin, vinblastine, vincristine, etoposide, paclitaxel, and docetaxel.[11-15][Level of evidence: 3iiiDiv] The combination of cART and liposomal doxorubicin resulted in a 5-year OS of 85% in 140 patients with T1 disease.[3][Level of evidence: 3iiiDiv]
Randomized multicenter trials showed an improvement in response rate (45%–60% vs. 20%–25%) and a more favorable toxic effects profile for pegylated liposomal doxorubicin or liposomal daunorubicin, compared to the combination of doxorubicin, bleomycin, and vincristine or bleomycin and vincristine.[16-18][Level of evidence: 1iiDiv] During cART, both pegylated liposomal doxorubicin and paclitaxel are active single agents with response rates close to 50%.[19][Level of evidence: 1iiDiv]
Biologic and targeted therapy
The interferon alphas have also been widely studied and show a 40% objective response rate in patients with epidemic KS.[20,21] In these reports, the responses differed significantly according to the prognostic factors of extent of disease, prior or coexistent opportunistic infections, prior treatment with chemotherapy, CD4 lymphocyte counts lower than 200 cells/mm³, the presence of circulating acid-labile interferon alpha, and an increase in beta-2-microglobulin. Several treatment studies have combined interferon alpha with other chemotherapeutic agents. Overall, these trials have shown no benefit with the interferon-chemotherapy combinations as compared to the single-agent activities.
Recombinant interferon alpha-2a and interferon alpha-2b were the first agents approved for the treatment of KS. Approval was based on single-agent studies performed in the 1980s before the advent of antiretroviral therapy. The early studies demonstrated improved efficacy at relatively high doses. High-dose monotherapy is rarely used today, and instead, interferon is given in combination with other anti-HIV drugs in doses of 4 to 18 million units. Neutropenia is dose limiting, and trials of doses of 1 to 10 million units combined with less myelosuppressive antiretrovirals are in progress. Response to interferon is slow, and the maximum effect is seen after 6 or more months. Interferon should probably not be used in the treatment of patients with rapidly progressive, symptomatic KS.
Imatinib, a c-kit/PDGF (platelet-derived growth factor) receptor inhibitor, resulted in partial responses in 10 of 30 previously treated patients (cART + chemotherapy).[22]
Bevacizumab, the humanized, antivascular, endothelial growth–factor monoclonal antibody, had a response rate in 5 of 16 patients who did not improve after the institution of cART and chemotherapy.[23][Level of evidence: 3iiiDiv]
Interleukin-12 had a response rate of 71% (95% confidence interval, 48%–89%) among 24 evaluable patients in a phase I and phase II trial.[24][Level of evidence: 3iiiDiv]
Treatment options under clinical evaluation:
- Patients with epidemic KS are appropriate candidates for clinical trials evaluating new drugs or biologicals.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Safai B: Kaposi's sarcoma and acquired immunodeficiency syndrome. In: DeVita VT, Hellman S, Rosenberg S, eds.: AIDS: Etiology, Diagnosis, Treatment and Prevention. 4th ed. Philadelphia, Pa: Lippincott-Raven Publishers, 1997, pp 295-318.
- Krown SE: Highly active antiretroviral therapy in AIDS-associated Kaposi's sarcoma: implications for the design of therapeutic trials in patients with advanced, symptomatic Kaposi's sarcoma. J Clin Oncol 22 (3): 399-402, 2004. [PUBMED Abstract]
- Bower M, Dalla Pria A, Coyle C, et al.: Prospective stage-stratified approach to AIDS-related Kaposi's sarcoma. J Clin Oncol 32 (5): 409-14, 2014. [PUBMED Abstract]
- Krell J, Stebbing J: Broader implications of a stage-guided stratified therapeutic approach for AIDS-related Kaposi's sarcoma. J Clin Oncol 32 (5): 373-5, 2014. [PUBMED Abstract]
- Cooper JS, Steinfeld AD, Lerch I: Intentions and outcomes in the radiotherapeutic management of epidemic Kaposi's sarcoma. Int J Radiat Oncol Biol Phys 20 (3): 419-22, 1991. [PUBMED Abstract]
- Nobler MP, Leddy ME, Huh SH: The impact of palliative irradiation on the management of patients with acquired immune deficiency syndrome. J Clin Oncol 5 (1): 107-12, 1987. [PUBMED Abstract]
- Singh NB, Lakier RH, Donde B: Hypofractionated radiation therapy in the treatment of epidemic Kaposi sarcoma--a prospective randomized trial. Radiother Oncol 88 (2): 211-6, 2008. [PUBMED Abstract]
- Berson AM, Quivey JM, Harris JW, et al.: Radiation therapy for AIDS-related Kaposi's Sarcoma. Int J Radiat Oncol Biol Phys 19 (3): 569-75, 1990. [PUBMED Abstract]
- Epstein JB, Lozada-Nur F, McLeod WA, et al.: Oral Kaposi's sarcoma in acquired immunodeficiency syndrome. Review of management and report of the efficacy of intralesional vinblastine. Cancer 64 (12): 2424-30, 1989. [PUBMED Abstract]
- Bodsworth NJ, Bloch M, Bower M, et al.: Phase III vehicle-controlled, multi-centered study of topical alitretinoin gel 0.1% in cutaneous AIDS-related Kaposi's sarcoma. Am J Clin Dermatol 2 (2): 77-87, 2001. [PUBMED Abstract]
- Evans SR, Krown SE, Testa MA, et al.: Phase II evaluation of low-dose oral etoposide for the treatment of relapsed or progressive AIDS-related Kaposi's sarcoma: an AIDS Clinical Trials Group clinical study. J Clin Oncol 20 (15): 3236-41, 2002. [PUBMED Abstract]
- Saville MW, Lietzau J, Pluda JM, et al.: Treatment of HIV-associated Kaposi's sarcoma with paclitaxel. Lancet 346 (8966): 26-8, 1995. [PUBMED Abstract]
- Lim ST, Tupule A, Espina BM, et al.: Weekly docetaxel is safe and effective in the treatment of advanced-stage acquired immunodeficiency syndrome-related Kaposi sarcoma. Cancer 103 (2): 417-21, 2005. [PUBMED Abstract]
- Gill PS, Tulpule A, Espina BM, et al.: Paclitaxel is safe and effective in the treatment of advanced AIDS-related Kaposi's sarcoma. J Clin Oncol 17 (6): 1876-83, 1999. [PUBMED Abstract]
- Di Lorenzo G, Konstantinopoulos PA, Pantanowitz L, et al.: Management of AIDS-related Kaposi's sarcoma. Lancet Oncol 8 (2): 167-76, 2007. [PUBMED Abstract]
- Stewart S, Jablonowski H, Goebel FD, et al.: Randomized comparative trial of pegylated liposomal doxorubicin versus bleomycin and vincristine in the treatment of AIDS-related Kaposi's sarcoma. International Pegylated Liposomal Doxorubicin Study Group. J Clin Oncol 16 (2): 683-91, 1998. [PUBMED Abstract]
- Northfelt DW, Dezube BJ, Thommes JA, et al.: Pegylated-liposomal doxorubicin versus doxorubicin, bleomycin, and vincristine in the treatment of AIDS-related Kaposi's sarcoma: results of a randomized phase III clinical trial. J Clin Oncol 16 (7): 2445-51, 1998. [PUBMED Abstract]
- Gill PS, Wernz J, Scadden DT, et al.: Randomized phase III trial of liposomal daunorubicin versus doxorubicin, bleomycin, and vincristine in AIDS-related Kaposi's sarcoma. J Clin Oncol 14 (8): 2353-64, 1996. [PUBMED Abstract]
- Cianfrocca M, Lee S, Von Roenn J, et al.: Randomized trial of paclitaxel versus pegylated liposomal doxorubicin for advanced human immunodeficiency virus-associated Kaposi sarcoma: evidence of symptom palliation from chemotherapy. Cancer 116 (16): 3969-77, 2010. [PUBMED Abstract]
- Real FX, Oettgen HF, Krown SE: Kaposi's sarcoma and the acquired immunodeficiency syndrome: treatment with high and low doses of recombinant leukocyte A interferon. J Clin Oncol 4 (4): 544-51, 1986. [PUBMED Abstract]
- Groopman JE, Gottlieb MS, Goodman J, et al.: Recombinant alpha-2 interferon therapy for Kaposi's sarcoma associated with the acquired immunodeficiency syndrome. Ann Intern Med 100 (5): 671-6, 1984. [PUBMED Abstract]
- Koon HB, Krown SE, Lee JY, et al.: Phase II trial of imatinib in AIDS-associated Kaposi's sarcoma: AIDS Malignancy Consortium Protocol 042. J Clin Oncol 32 (5): 402-8, 2014. [PUBMED Abstract]
- Uldrick TS, Wyvill KM, Kumar P, et al.: Phase II study of bevacizumab in patients with HIV-associated Kaposi's sarcoma receiving antiretroviral therapy. J Clin Oncol 30 (13): 1476-83, 2012. [PUBMED Abstract]
- Little RF, Pluda JM, Wyvill KM, et al.: Activity of subcutaneous interleukin-12 in AIDS-related Kaposi sarcoma. Blood 107 (12): 4650-7, 2006. [PUBMED Abstract]
Recurrent Kaposi Sarcoma Treatment
The prognosis for any treated Kaposi sarcoma patient with progressing, recurring, or relapsing disease is highly variable. Deciding on further treatment depends on many factors, most importantly the clinical setting (i.e., classic, immunosuppressive treatment, or AIDS) in which the tumor arises as well as individual patient considerations.
Clinical trials are appropriate and should be considered when possible.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
Changes to This Summary (01/30/2018)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Editorial changes were made to this summary.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of Kaposi sarcoma. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Kaposi Sarcoma Treatment are:
- Eric J. Seifter, MD (Johns Hopkins University)
- Minh Tam Truong, MD (Boston University Medical Center)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Kaposi Sarcoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/soft-tissue-sarcoma/hp/kaposi-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389335]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Updated: January 30, 2018
Source URL: https://www.cancer.gov/publishedcontent/syndication/3524.htm
Source Agency: National Cancer Institute (NCI)
Captured Date: 2013-09-14 09:01:43.0
Kaposi Sarcoma Treatment (PDQ®)–Health Professional Version
General Information About Kaposi Sarcoma
Epidemiology
KS was first described in 1872 by the Hungarian dermatologist, Moritz Kaposi. From that time until the current human immunodeficiency virus (HIV) disease epidemic identified with the Acquired Immunodeficiency Syndrome (AIDS), KS remained a rare tumor. While most of the cases seen in Europe and North America have occurred in elderly men of Italian or Eastern European Jewish ancestry, the neoplasm also occurs in several other distinct populations: young black African adult males, prepubescent children, renal allograft recipients, and other patients receiving immunosuppressive therapy. The disseminated, fulminant form of KS associated with HIV disease is referred to as epidemic KS to distinguish it from the classic, African, and transplant-related varieties of the neoplasm. In addition, KS has been identified in homosexual men apart from the HIV disease epidemic.[1]
Histopathology
Although the histopathology of the different types of the Kaposi tumor is essentially identical in all of these groups, the clinical manifestations and course of the disease differ dramatically.[2] A key piece to the puzzle of KS pathogenesis was the 1994 discovery of a gamma herpes virus, human herpes virus type 8 (HHV-8), also known as Kaposi sarcoma herpes virus.[3] HHV-8 was identified in KS tissue biopsies from virtually all patients with classic, African, transplant-related, and AIDS-associated KS but was absent from noninvolved tissue.[4-7]
Classic Kaposi Sarcoma
Considered a rare disease, classic KS occurs more often in males, with a ratio of approximately 10 to 15 males to 1 female. In North Americans and Europeans, the usual age at onset is between 50 and 70 years. Classic KS tumors usually present with one or more asymptomatic red, purple, or brown patches, plaques, or nodular skin lesions. The disease is often limited to single or multiple lesions usually localized to one or both lower extremities, especially involving the ankles and soles.
Classic KS most commonly runs a relatively benign, indolent course for 10 to 15 years or more, with slow enlargement of the original tumors and the gradual development of additional lesions. Venous stasis and lymphedema of the involved lower extremity are frequent complications. In long-standing cases, systemic lesions can develop along the gastrointestinal tract, in lymph nodes, and in other organs. The visceral lesions are generally asymptomatic and are most often discovered only at autopsy, though clinically, gastrointestinal bleeding can occur. As many as 33% of the patients with classic KS develop a second primary malignancy, which is most often non-Hodgkin lymphoma.[8-10]
African Kaposi Sarcoma
In the 1950s, KS was recognized as a relatively common neoplasm endemic in native populations in equatorial Africa and comprised approximately 9% of all cancers seen in Ugandan males. African KS is seen as either an indolent neoplasm identical to the classic disease seen in Europe and North America or as an aggressive disease with fungating and exophytic tumors that may invade the subcutaneous and surrounding tissue including the underlying bone. In Africa, both the indolent and locally more aggressive forms of KS occur with a male-to-female ratio comparable to that observed with the classic KS tumor seen in North America and Europe. In general, however, patients in Africa are significantly younger than their European counterparts. A lymphadenopathic form of KS is also seen in Africa, primarily in prepubescent children (male:female ratio, 3:1). In these cases, the generalized lymphadenopathy is frequently associated with visceral organ involvement. The prognosis is very poor with a 100% fatality rate within 3 years.[11,12]
Immunosuppressive Treatment–Related Kaposi Sarcoma
In 1969, the first case of KS in association with immunosuppression in a renal transplant patient was described. Since that time, a number of renal and other organ allograft recipients who received prednisone and azathioprine developed KS shortly after the onset of immunosuppressive therapy.[13] Estimates of the incidence of KS in immunosuppressed renal transplant recipients are between 150 and 200 times the expected incidence of the tumor in the general population. The average time to develop KS after transplantation is 16 months. Although the KS tumor in iatrogenically immunosuppressed patients often remains localized to the skin, widespread dissemination with mucocutaneous or visceral organ involvement is common. In some cases, the KS tumors have regressed as a result of reduction or changes in immunosuppressive therapy. Clinical management of renal transplant patients who develop KS is difficult and requires a balance between the risk of death from generalized KS and the risk of graft rejection and complications of renal failure that may occur if the immunosuppressive therapy is discontinued.
Epidemic Kaposi Sarcoma
In 1981, a fulminant and disseminated form of KS in young homosexual or bisexual men was first reported as part of an epidemic now known as AIDS.[14] The etiology of AIDS is a T-cell lymphotropic retrovirus known as HIV. The underlying immunologic deficiency that characterizes HIV disease is an acquired profound disorder of cell-mediated immune functions. This immunologic deficiency and immune dysregulation predisposes the host to a variety of opportunistic infections and unusual neoplasms, especially KS. HIV may play an indirect role in the development of KS.[15]
Approximately 95% of all the cases of epidemic KS in the United States have been diagnosed in homosexual or bisexual men. In the past, approximately 26% of all homosexual males with HIV disease presented with, or eventually developed, KS during the course of their illness. By comparison, fewer than 3% of all heterosexual intravenous drug users with HIV disease developed KS. The proportion of HIV disease patients with KS has steadily decreased since the epidemic was first identified in 1981.[16] About 48% of AIDS patients in 1981 had KS as their presenting AIDS diagnosis. By August 1987, the cumulative proportion of AIDS patients with KS had diminished to fewer than 20%. The introduction of combined antiretroviral therapy (cART) has delayed or prevented the emergence of drug-resistant HIV strains, profoundly decreased viral load, led to increased survival, and lessened the risk of opportunistic infections.[17-19] The use of cART has been associated with a sustained and substantial decline in KS incidence in multiple large cohorts.[20-25]
The lesions that develop may involve the skin; oral mucosa; lymph nodes; and visceral organs, such as the gastrointestinal tract, lung, liver, and spleen. Most patients with HIV disease who present with the mucocutaneous lesions of KS feel healthy and are usually free of systemic symptoms, as compared with HIV patients who first develop an opportunistic infection. The sites of disease at presentation of epidemic KS are much more varied than the sites seen in other types of this neoplasm. In an early report on the clinical manifestations of the disease, 49 patients were described.[26] Of these patients, 8% had no skin involvement, 27% had localized or fewer than five skin lesions, and 63% had innumerable skin lesions widely distributed over the skin surface area. Of these patients, 61% had generalized lymphadenopathy at the time of the first examination. Four of these patients, who had generalized lymphadenopathy in the absence of skin lesions or detectable visceral organ involvement at the time of presentation, were found to have biopsy-proven KS localized to the lymph nodes. In 45% of the patients studied, KS lesions were found in one or more sites along the gastrointestinal tract. Of these patients, 29% had either unexplained fever or unexplained weight loss when first seen. While most patients present with skin disease, KS involvement of lymph nodes or the gastrointestinal tract may occasionally precede the appearance of the cutaneous lesions.
Eventually, most patients with epidemic KS develop disseminated disease. The disease often progresses in an orderly fashion from a few localized or widespread mucocutaneous lesions to more numerous lesions and generalized skin disease with lymph node, gastrointestinal tract disease, and other organ involvement. Pleuropulmonary KS is an ominous sign usually occurring late in the course of the disease, especially in those patients whose death is directly attributed to KS.[27] Most patients with epidemic KS die of one or more complicating opportunistic infections.
References
- Friedman-Kien AE, Saltzman BR, Cao YZ, et al.: Kaposi's sarcoma in HIV-negative homosexual men. Lancet 335 (8682): 168-9, 1990. [PUBMED Abstract]
- Safai B: Kaposi's sarcoma and acquired immunodeficiency syndrome. In: DeVita VT, Hellman S, Rosenberg S, eds.: AIDS: Etiology, Diagnosis, Treatment and Prevention. 4th ed. Philadelphia, Pa: Lippincott-Raven Publishers, 1997, pp 295-318.
- Chang Y, Cesarman E, Pessin MS, et al.: Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266 (5192): 1865-9, 1994. [PUBMED Abstract]
- Moore PS, Chang Y: Detection of herpesvirus-like DNA sequences in Kaposi's sarcoma in patients with and without HIV infection. N Engl J Med 332 (18): 1181-5, 1995. [PUBMED Abstract]
- Su IJ, Hsu YS, Chang YC, et al.: Herpesvirus-like DNA sequence in Kaposi's sarcoma from AIDS and non-AIDS patients in Taiwan. Lancet 345 (8951): 722-3, 1995. [PUBMED Abstract]
- Gao SJ, Kingsley L, Li M, et al.: KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi's sarcoma. Nat Med 2 (8): 925-8, 1996. [PUBMED Abstract]
- Chang Y, Ziegler J, Wabinga H, et al.: Kaposi's sarcoma-associated herpesvirus and Kaposi's sarcoma in Africa. Uganda Kaposi's Sarcoma Study Group. Arch Intern Med 156 (2): 202-4, 1996. [PUBMED Abstract]
- Safai B, Good RA: Kaposi's sarcoma: a review and recent developments. Clin Bull 10 (2): 62-9, 1980. [PUBMED Abstract]
- Reynolds WA, Winkelmann RK, Soule EH: Kaposi's sarcoma: a clinicopathologic study with particular reference to its relationship to the reticuloendothelial system. Medicine (Baltimore) 44 (5): 419-43, 1965. [PUBMED Abstract]
- Safai B, Miké V, Giraldo G, et al.: Association of Kaposi's sarcoma with second primary malignancies: possible etiopathogenic implications. Cancer 45 (6): 1472-9, 1980. [PUBMED Abstract]
- Taylor JF, Templeton AC, Vogel CL, et al.: Kaposi's sarcoma in Uganda: a clinico-pathological study. Int J Cancer 8 (1): 122-35, 1971. [PUBMED Abstract]
- Templeton AC, Bhana D: Prognosis in Kaposi's sarcoma. J Natl Cancer Inst 55 (6): 1301-4, 1975. [PUBMED Abstract]
- Penn I: Kaposi's sarcoma in organ transplant recipients: report of 20 cases. Transplantation 27 (1): 8-11, 1979. [PUBMED Abstract]
- Kaposi's sarcoma and Pneumocystis pneumonia among homosexual men--New York City and California. MMWR Morb Mortal Wkly Rep 30 (25): 305-8, 1981. [PUBMED Abstract]
- Vogel J, Hinrichs SH, Reynolds RK, et al.: The HIV tat gene induces dermal lesions resembling Kaposi's sarcoma in transgenic mice. Nature 335 (6191): 606-11, 1988. [PUBMED Abstract]
- Selik RM, Starcher ET, Curran JW: Opportunistic diseases reported in AIDS patients: frequencies, associations, and trends. AIDS 1 (3): 175-82, 1987. [PUBMED Abstract]
- Flexner C: HIV-protease inhibitors. N Engl J Med 338 (18): 1281-92, 1998. [PUBMED Abstract]
- Palella FJ Jr, Delaney KM, Moorman AC, et al.: Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med 338 (13): 853-60, 1998. [PUBMED Abstract]
- Lodi S, Guiguet M, Costagliola D, et al.: Kaposi sarcoma incidence and survival among HIV-infected homosexual men after HIV seroconversion. J Natl Cancer Inst 102 (11): 784-92, 2010. [PUBMED Abstract]
- Portsmouth S, Stebbing J, Gill J, et al.: A comparison of regimens based on non-nucleoside reverse transcriptase inhibitors or protease inhibitors in preventing Kaposi's sarcoma. AIDS 17 (11): F17-22, 2003. [PUBMED Abstract]
- International Collaboration on HIV and Cancer: Highly active antiretroviral therapy and incidence of cancer in human immunodeficiency virus-infected adults. J Natl Cancer Inst 92 (22): 1823-30, 2000. [PUBMED Abstract]
- Dupont C, Vasseur E, Beauchet A, et al.: Long-term efficacy on Kaposi's sarcoma of highly active antiretroviral therapy in a cohort of HIV-positive patients. CISIH 92. Centre d'information et de soins de l'immunodéficience humaine. AIDS 14 (8): 987-93, 2000. [PUBMED Abstract]
- Tam HK, Zhang ZF, Jacobson LP, et al.: Effect of highly active antiretroviral therapy on survival among HIV-infected men with Kaposi sarcoma or non-Hodgkin lymphoma. Int J Cancer 98 (6): 916-22, 2002. [PUBMED Abstract]
- Carrieri MP, Pradier C, Piselli P, et al.: Reduced incidence of Kaposi's sarcoma and of systemic non-hodgkin's lymphoma in HIV-infected individuals treated with highly active antiretroviral therapy. Int J Cancer 103 (1): 142-4, 2003. [PUBMED Abstract]
- Grabar S, Abraham B, Mahamat A, et al.: Differential impact of combination antiretroviral therapy in preventing Kaposi's sarcoma with and without visceral involvement. J Clin Oncol 24 (21): 3408-14, 2006. [PUBMED Abstract]
- Krigel RL, Laubenstein LJ, Muggia FM: Kaposi's sarcoma: a new staging classification. Cancer Treat Rep 67 (6): 531-4, 1983. [PUBMED Abstract]
- Gill PS, Akil B, Colletti P, et al.: Pulmonary Kaposi's sarcoma: clinical findings and results of therapy. Am J Med 87 (1): 57-61, 1989. [PUBMED Abstract]
Stage Information for Kaposi Sarcoma
The staging evaluation of patients with classic Kaposi sarcoma (KS) should be individualized. The advanced age of most of the patients, localized nature of the tumor, rarity of visceral involvement, and usually indolent course of the disease should temper the extent of the evaluation. A careful examination of the skin and lymph nodes is sufficient in most cases. For the rare patient with rapidly progressive tumor or signs or symptoms of visceral involvement, appropriate evaluation is indicated. No universally accepted classification is available for epidemic KS. Staging schemes that incorporate laboratory parameters as well as clinical features have been proposed. Since most patients with epidemic KS do not die from the disease, factors besides tumor burden are apparently involved in survival.
The conventions used to stage KS and the methods used to evaluate the benefits of KS treatment continue to evolve because of changes in the treatment of human immunodeficiency virus (HIV) and in recognition of deficiencies in standard tumor assessment. The clinical course of KS, the selection of treatment, and the response to treatment are heavily influenced by the degree of underlying immune dysfunction and opportunistic infections.
The AIDS Clinical Trials Group (ACTG) Oncology Committee has published criteria for the evaluation of epidemic KS.[1] The staging system incorporates measures of extent of disease, severity of immunodeficiency, and presence of systemic symptoms. As shown in Table 1 below, the ACTG criteria categorizes the extent of the tumor as localized or disseminated, the CD4 cell number as high or low, and a systemic illness as absent or present.
A subsequent prospective analysis of 294 patients entered on ACTG trials for KS between 1989 and 1995 showed that each of the tumor, immune system, and systemic illness variables was independently associated with survival.[2] Multivariate analysis showed that immune system impairment was the most important single predictor of survival. In patients with relatively high CD4 counts, tumor stage was predictive. A CD4 count of 150 cells/mm³ may be a better discriminator than the published cutoff of 200 cells/mm³. A study is in progress to determine if viral load adds predictive information. None of the prior studies were conducted at a time when combined antiretroviral therapy (cART) was readily available. The impact of cART on survival in KS requires continued assessment.
| Good Risk (0) | Poor Risk (1) | |
|---|---|---|
| (Any of the following) | (Any of the following) | |
| Tumor (T) | Confined to skin and/or lymph nodes and/or minimal oral disease[Note: Minimal oral disease is non-nodular KS confined to the palate.] | Tumor-associated edema or ulceration |
| Extensive oral KS | ||
| Gastrointestinal KS | ||
| KS in other non-nodal viscera | ||
| Immune system (I) | CD4 cells ≥ = 200/µL | CD4 cells <200 per cubic mm |
| Systemic illness (S) | No history of OIs or thrush[Note: OIs are opportunistic infections.] | History of OIs and/or thrush |
| No “B” symptoms[Note: “B” symptoms are unexplained fever, night sweats, >10% involuntary weight loss, or diarrhea persisting >2 weeks.] | “B” symptoms present | |
| Performance status ≥70 (Karnofsky) | Performance status <70 | |
| Other HIV-related illness (e.g., neurological disease or lymphoma) |
References
- Krown SE, Metroka C, Wernz JC: Kaposi's sarcoma in the acquired immune deficiency syndrome: a proposal for uniform evaluation, response, and staging criteria. AIDS Clinical Trials Group Oncology Committee. J Clin Oncol 7 (9): 1201-7, 1989. [PUBMED Abstract]
- Krown SE, Testa MA, Huang J: AIDS-related Kaposi's sarcoma: prospective validation of the AIDS Clinical Trials Group staging classification. AIDS Clinical Trials Group Oncology Committee. J Clin Oncol 15 (9): 3085-92, 1997. [PUBMED Abstract]
Classic Kaposi Sarcoma Treatment
Classic Kaposi sarcoma (KS) usually is limited to the skin and has an indolent course. Patients with this tumor are predisposed to the development of a second primary malignancy, and the treating physician should consider this factor when arranging a schedule of follow-up treatment for the patient.
Equivalent standard treatment options:
Solitary lesions:
- Radiation therapy: For solitary lesions or lesions of limited extent, modest doses of radiation applied to the lesions with a limited margin provide excellent control of disease in the treated area. Usually, superficial radiation beams such as electron beams are used. Some authors believe disease recurrence in adjacent, untreated skin is common if only involved-field radiation therapy is used and claim better cure rates when extended-field radiation therapy is used.[1,2]
- Low-voltage (100 kv) photon radiation: 8 Gy to 10 Gy given as a single dose or 15 Gy to 20 Gy given over 1 week because solitary lesions control nearly 100% of local disease, but recurrence in adjacent areas is common.
- Electron-beam radiation therapy (EBRT): 4 Gy given once weekly for 6 to 8 consecutive weeks with a 4-MeV to 6-MeV electron beam. Ports should include the entire skin surface 15 cm above the lesion.
- Surgical excision may be of benefit in some patients with small superficial lesions, but local recurrence is likely to be a problem. However, over the years, multiple small excisions can be performed to achieve good disease control.
Widespread skin disease:
- Radiation therapy: Modest doses are effective in controlling disease. The type of radiation (i.e., photon vs. electron) and fields used must be tailored to suit the distribution of disease in the individual patient.[2]
- Extended-field EBRT.
- For disease limited to areas distal to the knee, subtotal-skin EBRT directed to skin below the umbilicus.
- For disease that extends above the knee, total-skin EBRT.
EBRT used in this manner gave long-term results that were superior to those obtained with radiation therapy administered to successive individual lesions as they appeared.[2]
- EBRT: 4 Gy given once weekly for 6 to 8 consecutive weeks, and subtotal- or total-skin radiation therapy given for extensive disease.
- Chemotherapy: Because classic KS is such a rare disease in the United States and is usually treated initially with radiation therapy, few patients have been treated with chemotherapy, and no randomized prospective trials have compared one agent to another. Several authors have used single-agent vinblastine given as a weekly dose of approximately 0.1 mg/kg.[3-6] Almost all of the patients had good to excellent response. In most cases, patients required prolonged courses of therapy, for several years, to maintain a partial response. Doses of vinblastine were titrated in individual patients to maintain a white blood count of more than 3,000 leukocytes. Follow-up after completion of therapy was not presented. In a multicenter trial of 55 patients who were treated over a decade, a 71% overall response rate was seen using pegylated liposomal doxorubicin.[7][Level of evidence: 3iiiDiv] In addition to the positive response rates of pegylated liposomal doxorubicin and the vinca alkaloids, response rates showing a greater than 50% decrease in lesions have also been reported in small, uncontrolled series for etoposide, taxanes, gemcitabine, and interferon alfa.[8][Level of evidence: 3iiiDiv]
One patient was treated repeatedly with intralesional injections of 0.25 to 0.50 mg of vincristine, which resulted in complete disappearance of the treated lesion.[9] Multiple courses of therapy were required because of the recurrence of disease in untreated areas.
Electroporation of the skin lesions was combined with intravenous bleomycin for 19 patients with classical KS. Most patients responded after one application, the rest after two or three applications, with a median duration of response of 16 months.[10][Level of evidence: 3iiiDiv]
Lymph node and gastrointestinal tract involvement:
- Chemotherapy: Several patients who had widespread skin disease and were treated with chemotherapy also had lymph node and gastrointestinal tract involvement. The disease in these sites also responded to vinblastine. Trials are required to define therapy. One such trial, MSKCC-04055 (NCT00096538), has been completed.
- Local radiation therapy may be added to chemotherapy if individual lesions require urgent therapy.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Hamilton CR, Cummings BJ, Harwood AR: Radiotherapy of Kaposi's sarcoma. Int J Radiat Oncol Biol Phys 12 (11): 1931-5, 1986. [PUBMED Abstract]
- Nisce LZ, Safai B, Poussin-Rosillo H: Once weekly total and subtotal skin electron beam therapy for Kaposi's sarcoma. Cancer 47 (4): 640-4, 1981. [PUBMED Abstract]
- Solan AJ, Greenwald ES, Silvay O: Long-term complete remissions of Kaposi's sarcoma with vinblastine therapy. Cancer 47 (4): 637-9, 1981. [PUBMED Abstract]
- Tucker SB, Winkelmann RK: Treatment of Kaposi sarcoma with vinblastine. Arch Dermatol 112 (7): 958-61, 1976. [PUBMED Abstract]
- Scott WP, Voight JA: Kaposi's sarcoma. Management with vincaleucoblastine. Cancer 19 (4): 557-64, 1966. [PUBMED Abstract]
- Klein E, Schwartz RA, Laor Y, et al.: Treatment of Kaposi's sarcoma with vinblastine. Cancer 45 (3): 427-31, 1980. [PUBMED Abstract]
- Di Lorenzo G, Kreuter A, Di Trolio R, et al.: Activity and safety of pegylated liposomal doxorubicin as first-line therapy in the treatment of non-visceral classic Kaposi's sarcoma: a multicenter study. J Invest Dermatol 128 (6): 1578-80, 2008. [PUBMED Abstract]
- Régnier-Rosencher E, Guillot B, Dupin N: Treatments for classic Kaposi sarcoma: a systematic review of the literature. J Am Acad Dermatol 68 (2): 313-31, 2013. [PUBMED Abstract]
- Odom RB, Goette DK: Treatment of cutaneous Kaposi's sarcoma with intralesional vincristine. Arch Dermatol 114 (11): 1693-4, 1978. [PUBMED Abstract]
- Di Monta G, Caracò C, Benedetto L, et al.: Electrochemotherapy as "new standard of care" treatment for cutaneous Kaposi's sarcoma. Eur J Surg Oncol 40 (1): 61-6, 2014. [PUBMED Abstract]
Immunosuppressive Therapy–Related Kaposi Sarcoma Treatment
Some patients with Kaposi Sarcoma (KS) have noted spontaneous and lasting remissions following discontinuation of immunosuppressive therapy. In managing these patients, if immunosuppressive therapy is not critical, its discontinuation is a reasonable first step.
Standard treatment options:
- Discontinue immunosuppressive therapy (often results in tumor regression). This option is critically important in patients who are receiving immunosuppressive drugs, as in the case of certain transplant patients.
- Radiation therapy (for disease limited to skin).[1-4]
- Chemotherapy (single or multiple drug): Most systemic chemotherapy trials in KS patients have been carried out in the African and epidemic varieties. See the section on the treatment of Epidemic Kaposi Sarcoma. The applicability of the results of these trials to KS in immunosuppressed patients is unknown.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Cohen L: Dose, time, and volume parameters in irradiation therapy of Kaposi's sarcoma. Br J Radiol 35 (415): 485-488, 1962.
- Hamilton CR, Cummings BJ, Harwood AR: Radiotherapy of Kaposi's sarcoma. Int J Radiat Oncol Biol Phys 12 (11): 1931-5, 1986. [PUBMED Abstract]
- Lo TC, Salzman FA, Smedal MI, et al.: Radiotherapy for Kaposi's sarcoma. Cancer 45 (4): 684-7, 1980. [PUBMED Abstract]
- Nisce LZ, Safai B, Poussin-Rosillo H: Once weekly total and subtotal skin electron beam therapy for Kaposi's sarcoma. Cancer 47 (4): 640-4, 1981. [PUBMED Abstract]
Epidemic Kaposi Sarcoma Treatment
Treatment may result:
- In a disappearance or reduction in size of specific skin lesions, thereby alleviating the discomfort associated with the chronic edema and ulcerations that often accompany multiple skin tumors seen on the lower extremities.
- In control of symptoms associated with mucosal or visceral lesions.
No data are available, however, to show that treatment improves survival.[1] In addition to antitumor treatment, essential components of an optimal Kaposi sarcoma (KS) treatment strategy include combined antiretroviral treatment (cART), prophylaxis for opportunistic infections, and rapid recognition and treatment of intercurrent infections.
Most good-risk patients, defined by the AIDS Clinical Trials Group as T0, show tumor regression with cART alone.[2-4] Poor-risk patients, defined as T1, usually require a combination of cART and chemotherapy with discontinuation of the chemotherapy after disappearance of the skin lesion.[2-4] The combination of cART and liposomal doxorubicin resulted in a 5-year overall survival (OS) rate of 85% in 140 patients with T1 disease.[3][Level of evidence: 3iiiDiv]
Local modalities
Small localized lesions of KS may be treated by electrodesiccation and curettage, cryotherapy, or by surgical excision. KS tumors are also generally very responsive to local radiation therapy, and excellent palliation has been obtained with doses at 20 Gy or slightly higher.[5-7] One report demonstrated a response rate higher than 90%, with a median time to progression of 21 months. Although no difference in response was noted with a variety of fractionation regimens, a single fraction of 8 Gy is indicated for cutaneous lesions and is associated with significantly fewer severe reactions.[8] Radiation therapy is generally reserved to treat localized areas of the skin and oral cavity. It is less often used to control pulmonary, gastrointestinal tract, or other sites of KS lesions. Localized KS lesions have also been effectively treated with intralesional injections of vinblastine.[9] Alitretinoin 0.1% gel provided local control in a randomized prospective multicenter trial.[10][Level of evidence: 1iiDiv]
Chemotherapy
In epidemic KS, the already profoundly depressed immunologic status of the host limits the therapeutic usefulness of systemic chemotherapy. Systemic chemotherapy studies in epidemic KS have used as single agents or in combinations doxorubicin, bleomycin, vinblastine, vincristine, etoposide, paclitaxel, and docetaxel.[11-15][Level of evidence: 3iiiDiv] The combination of cART and liposomal doxorubicin resulted in a 5-year OS of 85% in 140 patients with T1 disease.[3][Level of evidence: 3iiiDiv]
Randomized multicenter trials showed an improvement in response rate (45%–60% vs. 20%–25%) and a more favorable toxic effects profile for pegylated liposomal doxorubicin or liposomal daunorubicin, compared to the combination of doxorubicin, bleomycin, and vincristine or bleomycin and vincristine.[16-18][Level of evidence: 1iiDiv] During cART, both pegylated liposomal doxorubicin and paclitaxel are active single agents with response rates close to 50%.[19][Level of evidence: 1iiDiv]
Biologic and targeted therapy
The interferon alphas have also been widely studied and show a 40% objective response rate in patients with epidemic KS.[20,21] In these reports, the responses differed significantly according to the prognostic factors of extent of disease, prior or coexistent opportunistic infections, prior treatment with chemotherapy, CD4 lymphocyte counts lower than 200 cells/mm³, the presence of circulating acid-labile interferon alpha, and an increase in beta-2-microglobulin. Several treatment studies have combined interferon alpha with other chemotherapeutic agents. Overall, these trials have shown no benefit with the interferon-chemotherapy combinations as compared to the single-agent activities.
Recombinant interferon alpha-2a and interferon alpha-2b were the first agents approved for the treatment of KS. Approval was based on single-agent studies performed in the 1980s before the advent of antiretroviral therapy. The early studies demonstrated improved efficacy at relatively high doses. High-dose monotherapy is rarely used today, and instead, interferon is given in combination with other anti-HIV drugs in doses of 4 to 18 million units. Neutropenia is dose limiting, and trials of doses of 1 to 10 million units combined with less myelosuppressive antiretrovirals are in progress. Response to interferon is slow, and the maximum effect is seen after 6 or more months. Interferon should probably not be used in the treatment of patients with rapidly progressive, symptomatic KS.
Imatinib, a c-kit/PDGF (platelet-derived growth factor) receptor inhibitor, resulted in partial responses in 10 of 30 previously treated patients (cART + chemotherapy).[22]
Bevacizumab, the humanized, antivascular, endothelial growth–factor monoclonal antibody, had a response rate in 5 of 16 patients who did not improve after the institution of cART and chemotherapy.[23][Level of evidence: 3iiiDiv]
Interleukin-12 had a response rate of 71% (95% confidence interval, 48%–89%) among 24 evaluable patients in a phase I and phase II trial.[24][Level of evidence: 3iiiDiv]
Treatment options under clinical evaluation:
- Patients with epidemic KS are appropriate candidates for clinical trials evaluating new drugs or biologicals.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Safai B: Kaposi's sarcoma and acquired immunodeficiency syndrome. In: DeVita VT, Hellman S, Rosenberg S, eds.: AIDS: Etiology, Diagnosis, Treatment and Prevention. 4th ed. Philadelphia, Pa: Lippincott-Raven Publishers, 1997, pp 295-318.
- Krown SE: Highly active antiretroviral therapy in AIDS-associated Kaposi's sarcoma: implications for the design of therapeutic trials in patients with advanced, symptomatic Kaposi's sarcoma. J Clin Oncol 22 (3): 399-402, 2004. [PUBMED Abstract]
- Bower M, Dalla Pria A, Coyle C, et al.: Prospective stage-stratified approach to AIDS-related Kaposi's sarcoma. J Clin Oncol 32 (5): 409-14, 2014. [PUBMED Abstract]
- Krell J, Stebbing J: Broader implications of a stage-guided stratified therapeutic approach for AIDS-related Kaposi's sarcoma. J Clin Oncol 32 (5): 373-5, 2014. [PUBMED Abstract]
- Cooper JS, Steinfeld AD, Lerch I: Intentions and outcomes in the radiotherapeutic management of epidemic Kaposi's sarcoma. Int J Radiat Oncol Biol Phys 20 (3): 419-22, 1991. [PUBMED Abstract]
- Nobler MP, Leddy ME, Huh SH: The impact of palliative irradiation on the management of patients with acquired immune deficiency syndrome. J Clin Oncol 5 (1): 107-12, 1987. [PUBMED Abstract]
- Singh NB, Lakier RH, Donde B: Hypofractionated radiation therapy in the treatment of epidemic Kaposi sarcoma--a prospective randomized trial. Radiother Oncol 88 (2): 211-6, 2008. [PUBMED Abstract]
- Berson AM, Quivey JM, Harris JW, et al.: Radiation therapy for AIDS-related Kaposi's Sarcoma. Int J Radiat Oncol Biol Phys 19 (3): 569-75, 1990. [PUBMED Abstract]
- Epstein JB, Lozada-Nur F, McLeod WA, et al.: Oral Kaposi's sarcoma in acquired immunodeficiency syndrome. Review of management and report of the efficacy of intralesional vinblastine. Cancer 64 (12): 2424-30, 1989. [PUBMED Abstract]
- Bodsworth NJ, Bloch M, Bower M, et al.: Phase III vehicle-controlled, multi-centered study of topical alitretinoin gel 0.1% in cutaneous AIDS-related Kaposi's sarcoma. Am J Clin Dermatol 2 (2): 77-87, 2001. [PUBMED Abstract]
- Evans SR, Krown SE, Testa MA, et al.: Phase II evaluation of low-dose oral etoposide for the treatment of relapsed or progressive AIDS-related Kaposi's sarcoma: an AIDS Clinical Trials Group clinical study. J Clin Oncol 20 (15): 3236-41, 2002. [PUBMED Abstract]
- Saville MW, Lietzau J, Pluda JM, et al.: Treatment of HIV-associated Kaposi's sarcoma with paclitaxel. Lancet 346 (8966): 26-8, 1995. [PUBMED Abstract]
- Lim ST, Tupule A, Espina BM, et al.: Weekly docetaxel is safe and effective in the treatment of advanced-stage acquired immunodeficiency syndrome-related Kaposi sarcoma. Cancer 103 (2): 417-21, 2005. [PUBMED Abstract]
- Gill PS, Tulpule A, Espina BM, et al.: Paclitaxel is safe and effective in the treatment of advanced AIDS-related Kaposi's sarcoma. J Clin Oncol 17 (6): 1876-83, 1999. [PUBMED Abstract]
- Di Lorenzo G, Konstantinopoulos PA, Pantanowitz L, et al.: Management of AIDS-related Kaposi's sarcoma. Lancet Oncol 8 (2): 167-76, 2007. [PUBMED Abstract]
- Stewart S, Jablonowski H, Goebel FD, et al.: Randomized comparative trial of pegylated liposomal doxorubicin versus bleomycin and vincristine in the treatment of AIDS-related Kaposi's sarcoma. International Pegylated Liposomal Doxorubicin Study Group. J Clin Oncol 16 (2): 683-91, 1998. [PUBMED Abstract]
- Northfelt DW, Dezube BJ, Thommes JA, et al.: Pegylated-liposomal doxorubicin versus doxorubicin, bleomycin, and vincristine in the treatment of AIDS-related Kaposi's sarcoma: results of a randomized phase III clinical trial. J Clin Oncol 16 (7): 2445-51, 1998. [PUBMED Abstract]
- Gill PS, Wernz J, Scadden DT, et al.: Randomized phase III trial of liposomal daunorubicin versus doxorubicin, bleomycin, and vincristine in AIDS-related Kaposi's sarcoma. J Clin Oncol 14 (8): 2353-64, 1996. [PUBMED Abstract]
- Cianfrocca M, Lee S, Von Roenn J, et al.: Randomized trial of paclitaxel versus pegylated liposomal doxorubicin for advanced human immunodeficiency virus-associated Kaposi sarcoma: evidence of symptom palliation from chemotherapy. Cancer 116 (16): 3969-77, 2010. [PUBMED Abstract]
- Real FX, Oettgen HF, Krown SE: Kaposi's sarcoma and the acquired immunodeficiency syndrome: treatment with high and low doses of recombinant leukocyte A interferon. J Clin Oncol 4 (4): 544-51, 1986. [PUBMED Abstract]
- Groopman JE, Gottlieb MS, Goodman J, et al.: Recombinant alpha-2 interferon therapy for Kaposi's sarcoma associated with the acquired immunodeficiency syndrome. Ann Intern Med 100 (5): 671-6, 1984. [PUBMED Abstract]
- Koon HB, Krown SE, Lee JY, et al.: Phase II trial of imatinib in AIDS-associated Kaposi's sarcoma: AIDS Malignancy Consortium Protocol 042. J Clin Oncol 32 (5): 402-8, 2014. [PUBMED Abstract]
- Uldrick TS, Wyvill KM, Kumar P, et al.: Phase II study of bevacizumab in patients with HIV-associated Kaposi's sarcoma receiving antiretroviral therapy. J Clin Oncol 30 (13): 1476-83, 2012. [PUBMED Abstract]
- Little RF, Pluda JM, Wyvill KM, et al.: Activity of subcutaneous interleukin-12 in AIDS-related Kaposi sarcoma. Blood 107 (12): 4650-7, 2006. [PUBMED Abstract]
Recurrent Kaposi Sarcoma Treatment
The prognosis for any treated Kaposi sarcoma patient with progressing, recurring, or relapsing disease is highly variable. Deciding on further treatment depends on many factors, most importantly the clinical setting (i.e., classic, immunosuppressive treatment, or AIDS) in which the tumor arises as well as individual patient considerations.
Clinical trials are appropriate and should be considered when possible.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
Changes to This Summary (01/30/2018)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Editorial changes were made to this summary.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of Kaposi sarcoma. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Kaposi Sarcoma Treatment are:
- Eric J. Seifter, MD (Johns Hopkins University)
- Minh Tam Truong, MD (Boston University Medical Center)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Kaposi Sarcoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/soft-tissue-sarcoma/hp/kaposi-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389335]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Updated: January 30, 2018
Source URL: https://www.cancer.gov/publishedcontent/syndication/3524.htm
Source Agency: National Cancer Institute (NCI)
Captured Date: 2013-09-14 09:01:43.0
Ewing sarcoma: Professional resources from the National Cancer Institute
Ewing Sarcoma Treatment (PDQ®)–Health Professional Version
General Information About Ewing Sarcoma
Dramatic improvements in survival have been achieved for children and adolescents with cancer.[1] Between 1975 and 2010, childhood cancer mortality decreased by more than 50%.[1] For Ewing sarcoma, the 5-year survival rate has increased over the same time from 59% to 78% for children younger than 15 years and from 20% to 60% for adolescents aged 15 to 19 years.[1]
Studies using immunohistochemical markers,[2] cytogenetics,[3,4] molecular genetics, and tissue culture [5] indicate that Ewing sarcoma is derived from a primordial bone marrow–derived mesenchymal stem cell.[6,7] Older terms such as peripheral primitive neuroectodermal tumor, Askin tumor (Ewing sarcoma of chest wall), and extraosseous Ewing sarcoma (often combined in the term Ewing sarcoma family of tumors) refer to this same tumor.
Incidence
The incidence of Ewing sarcoma has remained unchanged for 30 years.[8] The incidence for all ages is one case per 1 million people in the United States. In patients aged 10 to 19 years, the incidence is between nine and ten cases per 1 million people. The same analysis suggests that the incidence of Ewing sarcoma in the United States is nine times greater in whites than in African Americans, with an intermediate incidence in Asians.[9,10]
The relative paucity of Ewing sarcoma in people of African or Asian descent may be explained, in part, by a specific polymorphism in the EGR2 gene.
The median age of patients with Ewing sarcoma is 15 years, and more than 50% of patients are adolescents. Well-characterized cases of Ewing sarcoma in neonates and infants have been described.[11,12] Based on data from 1,426 patients entered on European Intergroup Cooperative Ewing Sarcoma Studies, 59% of patients are male and 41% are female.[13]
Clinical Presentation
Primary sites of bone disease include the following:
- Lower extremity (41%).
- Pelvis (26%).
- Chest wall (16%).
- Upper extremity (9%).
- Spine (6%).
- Hand and foot (3%).[14]
- Skull (2%).
For extraosseous primary tumors, the most common primary sites of disease include the following:[15,16]
- Trunk (32%).
- Extremity (26%).
- Head and neck (18%).
- Retroperitoneum (16%).
- Other sites (9%).
The median time from first symptom to diagnosis of Ewing sarcoma is often long, with a median interval reported from 2 to 5 months. Longer times are associated with older age and pelvic primary sites. This has not been associated with metastasis, surgical outcome, or survival.[17] Approximately 25% of patients with Ewing sarcoma have metastatic disease at the time of diagnosis.[8]
The Surveillance, Epidemiology, and End Results (SEER) database was used to compare patients younger than 40 years with Ewing sarcoma who presented with skeletal and extraosseous primary sites (refer to Table 1).[18] Patients with extraosseous Ewing sarcoma were more likely to be older, female, nonwhite, and have axial primary sites, and were less likely to have pelvic primary sites than were patients with skeletal Ewing sarcoma.
| Characteristic | Extraosseous Ewing Sarcoma | Skeletal Ewing Sarcoma | P Value |
|---|---|---|---|
| Mean age (range), years | 20 (0–39) | 16 (0–39) | <.001 |
| Male | 53% | 63% | <.001 |
| White | 85% | 93% | <.001 |
| Axial primary sites | 73% | 54% | <.001 |
| Pelvic primary sites | 20% | 27% | .001 |
Diagnostic Evaluation
The following tests and procedures may be used to diagnose or stage Ewing sarcoma:
- Physical exam and history.
- Magnetic resonance imaging (MRI).
- Computed tomography (CT) scan.
- Positron emission tomography (PET) scan.
- Bone scan.
- Bone marrow aspiration and biopsy.
- X-ray.
- Complete blood count.
- Blood chemistry studies, such as lactate dehydrogenase (LDH).
Prognostic Factors
The two major types of prognostic factors for patients with Ewing sarcoma are grouped as follows:
Pretreatment factors
- Site of tumor: Patients with Ewing sarcoma in the distal extremities have the best prognosis. Patients with Ewing sarcoma in the proximal extremities have an intermediate prognosis, followed by patients with central or pelvic sites.[19-22]
- Extraskeletal versus skeletal primary tumors: The Children's Oncology Group performed a retrospective analysis from two large cooperative trials that used similar treatment regimens.[23] They identified 213 patients with extraskeletal primary tumors and 826 patients with skeletal primary tumors. Patients with extraskeletal primary tumors were more likely to have an axial primary site, less likely to have large primary tumors, and had a statistically significant better prognosis than did patients with skeletal primary tumors.
- Tumor size or volume: Tumor size or volume has been shown to be an important prognostic factor in most studies. Cutoffs of a volume of 100 mL or 200 mL and/or single dimension greater than 8 cm are used to define larger tumors. Larger tumors tend to occur in unfavorable sites.[21,22,24]
- Age: Infants and younger patients have a better prognosis than do patients aged 15 years and older.[12,19,20,22,25,26]
In North American studies, patients younger than 10 years have a better outcome than those aged 10 to 17 years at diagnosis (relative risk [RR], 1.4). Patients older than 18 years have an inferior outcome (RR, 2.5).[27-29] A retrospective review of two consecutive German trials for Ewing sarcoma identified 47 patients older than 40 years.[30] With adequate multimodal therapy, survival was comparable to the survival observed in adolescents treated on the same trials. Review of the SEER database from 1973 to 2011 identified 1,957 patients with Ewing sarcoma.[31] Thirty-nine of these patients (2.0%) were younger than 12 months at diagnosis. Infants were less likely to receive radiation therapy and more likely to have soft tissue primary sites. Early death was more common in infants, but the overall survival (OS) did not differ significantly from that of older patients.
- Sex: Girls with Ewing sarcoma have a better prognosis than do boys with Ewing sarcoma.[9,20,22]
- Serum LDH: Increased serum LDH levels before treatment are associated with inferior prognosis. Increased LDH levels are also correlated with large primary tumors and metastatic disease.[20]
- Metastases: Any metastatic disease defined by standard imaging techniques or bone marrow aspirate/biopsy by morphology is an adverse prognostic factor. The presence or absence of metastatic disease is the single most powerful predictor of outcome. Metastases at diagnosis are detected in about 25% of patients.[8]
Patients with metastatic disease confined to the lung have a better prognosis than do patients with extrapulmonary metastatic sites.[19,21,22,32] The number of pulmonary lesions does not seem to correlate with outcome, but patients with unilateral lung involvement do better than patients with bilateral lung involvement.[33]
Patients with metastasis to only bone seem to have a better outcome than do patients with metastases to both bone and lung.[34,35]
Based on an analysis from the SEER database, regional lymph node involvement in patients is associated with an inferior overall outcome when compared with patients without regional lymph node involvement.[36]
- Previous treatment for cancer: In the SEER database, 58 patients with Ewing sarcoma who were diagnosed after treatment for a previous malignancy (2.1% of patients with Ewing sarcoma) were compared with 2,756 patients with Ewing sarcoma as a first cancer over the same period. Patients with Ewing sarcoma as a second malignant neoplasm were older (secondary Ewing sarcoma, mean age of 47.8 years; primary Ewing sarcoma, mean age of 22.5 years), more likely to have a primary tumor in an axial or extraskeletal site, and had a worse prognosis (5-year OS for patients with secondary Ewing sarcoma, 43.5%; patients with primary Ewing sarcoma, 64.2%).[37]
- Standard cytogenetics: Complex karyotype (defined as the presence of five or more independent chromosome abnormalities at diagnosis) and modal chromosome numbers lower than 50 appear to have adverse prognostic significance.[38]
- Detectable fusion transcripts in morphologically normal marrow: Reverse transcriptase polymerase chain reaction can be used to detect fusion transcripts in bone marrow. In a single retrospective study utilizing patients with normal marrow morphology and no other metastatic site, fusion transcript detection in marrow or peripheral blood was associated with an increased risk of relapse.[39]
- Other biological factors: Overexpression of the p53 protein, Ki67 expression, and loss of 16q may be adverse prognostic factors.[40-42] High expression of microsomal glutathione S-transferase, an enzyme associated with resistance to doxorubicin, is associated with inferior outcome for Ewing sarcoma.[43]
The Children's Oncology Group performed a prospective analysis of TP53 mutations and/or CDKN2A deletions in patients with Ewing sarcoma; no correlation was found with event-free survival (EFS).[44]
The following are not considered to be adverse prognostic factors for Ewing sarcoma:
- Pathologic fracture: Pathologic fractures do not appear to be a prognostic factor.[45]
- Histopathology: The degree of neural differentiation is not a prognostic factor in Ewing sarcoma.[46,47]
- Molecular pathology: The EWSR1-ETS translocation associated with Ewing sarcoma can occur at several potential breakpoints in each of the genes that join to form the novel segment of DNA. Once thought to be significant,[48] two large series have shown that the EWSR1-ETS translocation breakpoint site is not an adverse prognostic factor.[49,50]
Response to initial therapy factors
Multiple studies have shown that patients with minimal or no residual viable tumor after presurgical chemotherapy have a significantly better EFS than do patients with larger amounts of viable tumor.[51-54] Female sex and younger age predict a good histologic response to preoperative therapy.[55] For patients who receive preinduction- and postinduction-chemotherapy PET scans, decreased PET uptake after chemotherapy correlated with good histologic response and better outcome.[56-58]
Patients with poor response to presurgical chemotherapy have an increased risk for local recurrence.[59]
References
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014. [PUBMED Abstract]
- Olsen SH, Thomas DG, Lucas DR: Cluster analysis of immunohistochemical profiles in synovial sarcoma, malignant peripheral nerve sheath tumor, and Ewing sarcoma. Mod Pathol 19 (5): 659-68, 2006. [PUBMED Abstract]
- Delattre O, Zucman J, Melot T, et al.: The Ewing family of tumors--a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 331 (5): 294-9, 1994. [PUBMED Abstract]
- Dagher R, Pham TA, Sorbara L, et al.: Molecular confirmation of Ewing sarcoma. J Pediatr Hematol Oncol 23 (4): 221-4, 2001. [PUBMED Abstract]
- Llombart-Bosch A, Carda C, Peydro-Olaya A, et al.: Soft tissue Ewing's sarcoma. Characterization in established cultures and xenografts with evidence of a neuroectodermic phenotype. Cancer 66 (12): 2589-601, 1990. [PUBMED Abstract]
- Suvà ML, Riggi N, Stehle JC, et al.: Identification of cancer stem cells in Ewing's sarcoma. Cancer Res 69 (5): 1776-81, 2009. [PUBMED Abstract]
- Tirode F, Laud-Duval K, Prieur A, et al.: Mesenchymal stem cell features of Ewing tumors. Cancer Cell 11 (5): 421-9, 2007. [PUBMED Abstract]
- Esiashvili N, Goodman M, Marcus RB Jr: Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J Pediatr Hematol Oncol 30 (6): 425-30, 2008. [PUBMED Abstract]
- Jawad MU, Cheung MC, Min ES, et al.: Ewing sarcoma demonstrates racial disparities in incidence-related and sex-related differences in outcome: an analysis of 1631 cases from the SEER database, 1973-2005. Cancer 115 (15): 3526-36, 2009. [PUBMED Abstract]
- Beck R, Monument MJ, Watkins WS, et al.: EWS/FLI-responsive GGAA microsatellites exhibit polymorphic differences between European and African populations. Cancer Genet 205 (6): 304-12, 2012. [PUBMED Abstract]
- Kim SY, Tsokos M, Helman LJ: Dilemmas associated with congenital ewing sarcoma family tumors. J Pediatr Hematol Oncol 30 (1): 4-7, 2008. [PUBMED Abstract]
- van den Berg H, Dirksen U, Ranft A, et al.: Ewing tumors in infants. Pediatr Blood Cancer 50 (4): 761-4, 2008. [PUBMED Abstract]
- Paulussen M, Craft AW, Lewis I, et al.: Results of the EICESS-92 Study: two randomized trials of Ewing's sarcoma treatment--cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J Clin Oncol 26 (27): 4385-93, 2008. [PUBMED Abstract]
- Froeb D, Ranft A, Boelling T, et al.: Ewing sarcoma of the hand or foot. Klin Padiatr 224 (6): 348-52, 2012. [PUBMED Abstract]
- Raney RB, Asmar L, Newton WA Jr, et al.: Ewing's sarcoma of soft tissues in childhood: a report from the Intergroup Rhabdomyosarcoma Study, 1972 to 1991. J Clin Oncol 15 (2): 574-82, 1997. [PUBMED Abstract]
- Rowe RG, Thomas DG, Schuetze SM, et al.: Ewing sarcoma of the kidney: case series and literature review of an often overlooked entity in the diagnosis of primary renal tumors. Urology 81 (2): 347-53, 2013. [PUBMED Abstract]
- Brasme JF, Chalumeau M, Oberlin O, et al.: Time to diagnosis of Ewing tumors in children and adolescents is not associated with metastasis or survival: a prospective multicenter study of 436 patients. J Clin Oncol 32 (18): 1935-40, 2014. [PUBMED Abstract]
- Applebaum MA, Worch J, Matthay KK, et al.: Clinical features and outcomes in patients with extraskeletal Ewing sarcoma. Cancer 117 (13): 3027-32, 2011. [PUBMED Abstract]
- Cotterill SJ, Ahrens S, Paulussen M, et al.: Prognostic factors in Ewing's tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing's Sarcoma Study Group. J Clin Oncol 18 (17): 3108-14, 2000. [PUBMED Abstract]
- Bacci G, Longhi A, Ferrari S, et al.: Prognostic factors in non-metastatic Ewing's sarcoma tumor of bone: an analysis of 579 patients treated at a single institution with adjuvant or neoadjuvant chemotherapy between 1972 and 1998. Acta Oncol 45 (4): 469-75, 2006. [PUBMED Abstract]
- Rodríguez-Galindo C, Liu T, Krasin MJ, et al.: Analysis of prognostic factors in ewing sarcoma family of tumors: review of St. Jude Children's Research Hospital studies. Cancer 110 (2): 375-84, 2007. [PUBMED Abstract]
- Karski EE, McIlvaine E, Segal MR, et al.: Identification of Discrete Prognostic Groups in Ewing Sarcoma. Pediatr Blood Cancer 63 (1): 47-53, 2016. [PUBMED Abstract]
- Cash T, McIlvaine E, Krailo MD, et al.: Comparison of clinical features and outcomes in patients with extraskeletal versus skeletal localized Ewing sarcoma: A report from the Children's Oncology Group. Pediatr Blood Cancer 63 (10): 1771-9, 2016. [PUBMED Abstract]
- Ahrens S, Hoffmann C, Jabar S, et al.: Evaluation of prognostic factors in a tumor volume-adapted treatment strategy for localized Ewing sarcoma of bone: the CESS 86 experience. Cooperative Ewing Sarcoma Study. Med Pediatr Oncol 32 (3): 186-95, 1999. [PUBMED Abstract]
- De Ioris MA, Prete A, Cozza R, et al.: Ewing sarcoma of the bone in children under 6 years of age. PLoS One 8 (1): e53223, 2013. [PUBMED Abstract]
- Huh WW, Daw NC, Herzog CE, et al.: Ewing sarcoma family of tumors in children younger than 10 years of age. Pediatr Blood Cancer 64 (4): , 2017. [PUBMED Abstract]
- Grier HE, Krailo MD, Tarbell NJ, et al.: Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 348 (8): 694-701, 2003. [PUBMED Abstract]
- Granowetter L, Womer R, Devidas M, et al.: Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children's Oncology Group Study. J Clin Oncol 27 (15): 2536-41, 2009. [PUBMED Abstract]
- Womer RB, West DC, Krailo MD, et al.: Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 30 (33): 4148-54, 2012. [PUBMED Abstract]
- Pieper S, Ranft A, Braun-Munzinger G, et al.: Ewing's tumors over the age of 40: a retrospective analysis of 47 patients treated according to the International Clinical Trials EICESS 92 and EURO-E.W.I.N.G. 99. Onkologie 31 (12): 657-63, 2008. [PUBMED Abstract]
- Wong T, Goldsby RE, Wustrack R, et al.: Clinical features and outcomes of infants with Ewing sarcoma under 12 months of age. Pediatr Blood Cancer 62 (11): 1947-51, 2015. [PUBMED Abstract]
- Miser JS, Krailo MD, Tarbell NJ, et al.: Treatment of metastatic Ewing's sarcoma or primitive neuroectodermal tumor of bone: evaluation of combination ifosfamide and etoposide--a Children's Cancer Group and Pediatric Oncology Group study. J Clin Oncol 22 (14): 2873-6, 2004. [PUBMED Abstract]
- Paulussen M, Ahrens S, Craft AW, et al.: Ewing's tumors with primary lung metastases: survival analysis of 114 (European Intergroup) Cooperative Ewing's Sarcoma Studies patients. J Clin Oncol 16 (9): 3044-52, 1998. [PUBMED Abstract]
- Paulussen M, Ahrens S, Burdach S, et al.: Primary metastatic (stage IV) Ewing tumor: survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma Studies. Ann Oncol 9 (3): 275-81, 1998. [PUBMED Abstract]
- Ladenstein R, Pötschger U, Le Deley MC, et al.: Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol 28 (20): 3284-91, 2010. [PUBMED Abstract]
- Applebaum MA, Goldsby R, Neuhaus J, et al.: Clinical features and outcomes in patients with Ewing sarcoma and regional lymph node involvement. Pediatr Blood Cancer 59 (4): 617-20, 2012. [PUBMED Abstract]
- Applebaum MA, Goldsby R, Neuhaus J, et al.: Clinical features and outcomes in patients with secondary Ewing sarcoma. Pediatr Blood Cancer 60 (4): 611-5, 2013. [PUBMED Abstract]
- Roberts P, Burchill SA, Brownhill S, et al.: Ploidy and karyotype complexity are powerful prognostic indicators in the Ewing's sarcoma family of tumors: a study by the United Kingdom Cancer Cytogenetics and the Children's Cancer and Leukaemia Group. Genes Chromosomes Cancer 47 (3): 207-20, 2008. [PUBMED Abstract]
- Schleiermacher G, Peter M, Oberlin O, et al.: Increased risk of systemic relapses associated with bone marrow micrometastasis and circulating tumor cells in localized ewing tumor. J Clin Oncol 21 (1): 85-91, 2003. [PUBMED Abstract]
- Abudu A, Mangham DC, Reynolds GM, et al.: Overexpression of p53 protein in primary Ewing's sarcoma of bone: relationship to tumour stage, response and prognosis. Br J Cancer 79 (7-8): 1185-9, 1999. [PUBMED Abstract]
- López-Guerrero JA, Machado I, Scotlandi K, et al.: Clinicopathological significance of cell cycle regulation markers in a large series of genetically confirmed Ewing's sarcoma family of tumors. Int J Cancer 128 (5): 1139-50, 2011. [PUBMED Abstract]
- Ozaki T, Paulussen M, Poremba C, et al.: Genetic imbalances revealed by comparative genomic hybridization in Ewing tumors. Genes Chromosomes Cancer 32 (2): 164-71, 2001. [PUBMED Abstract]
- Scotlandi K, Remondini D, Castellani G, et al.: Overcoming resistance to conventional drugs in Ewing sarcoma and identification of molecular predictors of outcome. J Clin Oncol 27 (13): 2209-16, 2009. [PUBMED Abstract]
- Lerman DM, Monument MJ, McIlvaine E, et al.: Tumoral TP53 and/or CDKN2A alterations are not reliable prognostic biomarkers in patients with localized Ewing sarcoma: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (5): 759-65, 2015. [PUBMED Abstract]
- Bramer JA, Abudu AA, Grimer RJ, et al.: Do pathological fractures influence survival and local recurrence rate in bony sarcomas? Eur J Cancer 43 (13): 1944-51, 2007. [PUBMED Abstract]
- Parham DM, Hijazi Y, Steinberg SM, et al.: Neuroectodermal differentiation in Ewing's sarcoma family of tumors does not predict tumor behavior. Hum Pathol 30 (8): 911-8, 1999. [PUBMED Abstract]
- Luksch R, Sampietro G, Collini P, et al.: Prognostic value of clinicopathologic characteristics including neuroectodermal differentiation in osseous Ewing's sarcoma family of tumors in children. Tumori 85 (2): 101-7, 1999 Mar-Apr. [PUBMED Abstract]
- de Alava E, Kawai A, Healey JH, et al.: EWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing's sarcoma. J Clin Oncol 16 (4): 1248-55, 1998. [PUBMED Abstract]
- van Doorninck JA, Ji L, Schaub B, et al.: Current treatment protocols have eliminated the prognostic advantage of type 1 fusions in Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 28 (12): 1989-94, 2010. [PUBMED Abstract]
- Le Deley MC, Delattre O, Schaefer KL, et al.: Impact of EWS-ETS fusion type on disease progression in Ewing's sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. J Clin Oncol 28 (12): 1982-8, 2010. [PUBMED Abstract]
- Paulussen M, Ahrens S, Dunst J, et al.: Localized Ewing tumor of bone: final results of the cooperative Ewing's Sarcoma Study CESS 86. J Clin Oncol 19 (6): 1818-29, 2001. [PUBMED Abstract]
- Rosito P, Mancini AF, Rondelli R, et al.: Italian Cooperative Study for the treatment of children and young adults with localized Ewing sarcoma of bone: a preliminary report of 6 years of experience. Cancer 86 (3): 421-8, 1999. [PUBMED Abstract]
- Wunder JS, Paulian G, Huvos AG, et al.: The histological response to chemotherapy as a predictor of the oncological outcome of operative treatment of Ewing sarcoma. J Bone Joint Surg Am 80 (7): 1020-33, 1998. [PUBMED Abstract]
- Oberlin O, Deley MC, Bui BN, et al.: Prognostic factors in localized Ewing's tumours and peripheral neuroectodermal tumours: the third study of the French Society of Paediatric Oncology (EW88 study). Br J Cancer 85 (11): 1646-54, 2001. [PUBMED Abstract]
- Ferrari S, Bertoni F, Palmerini E, et al.: Predictive factors of histologic response to primary chemotherapy in patients with Ewing sarcoma. J Pediatr Hematol Oncol 29 (6): 364-8, 2007. [PUBMED Abstract]
- Hawkins DS, Schuetze SM, Butrynski JE, et al.: [18F]Fluorodeoxyglucose positron emission tomography predicts outcome for Ewing sarcoma family of tumors. J Clin Oncol 23 (34): 8828-34, 2005. [PUBMED Abstract]
- Denecke T, Hundsdörfer P, Misch D, et al.: Assessment of histological response of paediatric bone sarcomas using FDG PET in comparison to morphological volume measurement and standardized MRI parameters. Eur J Nucl Med Mol Imaging 37 (10): 1842-53, 2010. [PUBMED Abstract]
- Palmerini E, Colangeli M, Nanni C, et al.: The role of FDG PET/CT in patients treated with neoadjuvant chemotherapy for localized bone sarcomas. Eur J Nucl Med Mol Imaging 44 (2): 215-223, 2017. [PUBMED Abstract]
- Lin PP, Jaffe N, Herzog CE, et al.: Chemotherapy response is an important predictor of local recurrence in Ewing sarcoma. Cancer 109 (3): 603-11, 2007. [PUBMED Abstract]
Cellular Classification of Ewing Sarcoma
Ewing sarcoma belongs to the group of neoplasms commonly referred to as small, round, blue-cell tumors of childhood. The individual cells of Ewing sarcoma contain round-to-oval nuclei, with fine dispersed chromatin without nucleoli. Occasionally, cells with smaller, more hyperchromatic, and probably degenerative nuclei are present, giving a light cell/dark cell pattern. The cytoplasm varies in amount, but in the classic case, it is clear and contains glycogen, which can be highlighted with a periodic acid-Schiff stain. The tumor cells are tightly packed and grow in a diffuse pattern without evidence of structural organization. Tumors with the requisite translocation that show neuronal differentiation are not considered a separate entity, but rather, part of a continuum of differentiation.
The MIC2 gene product, CD99, is a surface membrane protein that is expressed in most cases of Ewing sarcoma and is useful in diagnosing these tumors when the results are interpreted in the context of clinical and pathologic parameters.[1] MIC2 positivity is not unique to Ewing sarcoma, and positivity by immunochemistry is found in several other tumors, including synovial sarcoma, non-Hodgkin lymphoma, and gastrointestinal stromal tumors.
Genomics of Ewing Sarcoma
The detection of a translocation involving the EWSR1 gene on chromosome 22 band q12 and any one of a number of partner chromosomes is the key feature in the diagnosis of Ewing sarcoma (refer to Table 2).[2] The EWSR1 gene is a member of the TET family [TLS/EWS/TAF15] of RNA-binding proteins.[3] The FLI1 gene is a member of the ETS family of DNA-binding genes. Characteristically, the amino terminus of the EWSR1 gene is juxtaposed with the carboxy terminus of the STS family gene. In most cases (90%), the carboxy terminus is provided by FLI1, a member of the family of transcription factor genes located on chromosome 11 band q24. Other family members that may combine with the EWSR1 gene are ERG, ETV1, ETV4 (also termed E1AF), and FEV.[4] Rarely, TLS, another TET family member, can substitute for EWSR1.[5] Finally, there are a few rare cases in which EWSR1 has translocated with partners that are not members of the ETS family of oncogenes. The significance of these alternate partners is not known.
Besides these consistent aberrations involving the EWSR1 gene at 22q12, additional numerical and structural aberrations have been observed in Ewing sarcoma, including gains of chromosomes 2, 5, 8, 9, 12, and 15; the nonreciprocal translocation t(1;16)(q12;q11.2); and deletions on the short arm of chromosome 6. Trisomy 20 may be associated with a more aggressive subset of Ewing sarcoma.[6]
Three papers have described the genomic landscape of Ewing sarcoma and all show that these tumors have a relatively silent genome, with a paucity of mutations in pathways that might be amenable to treatment with novel targeted therapies.[7-9] These papers also identified mutations in STAG2, a member of the cohesin complex, in about 15% to 20% of the cases, and the presence of these mutations was associated with advanced-stage disease. CDKN2A deletions were noted in 12% to 22% of cases. Finally, TP53 mutations were identified in about 6% to 7% of cases and the coexistence of STAG2 and TP53 mutations is associated with a poor clinical outcome.[7-9]
Figure 1 below from a discovery cohort (n = 99) highlights the frequency of chromosome 8 gain, the co-occurrence of chromosome 1q gain and chromosome 16q loss, the mutual exclusivity of CDKN2A deletion and STAG2 mutation, and the relative paucity of recurrent single nucleotide variants for Ewing sarcoma.[7]

Ewing sarcoma translocations can all be found with standard cytogenetic analysis. A more rapid analysis looking for a break apart of the EWS gene is now frequently done to confirm the diagnosis of Ewing sarcoma molecularly.[10] This test result must be considered with caution, however. Ewing sarcomas that utilize the TLS translocations will have negative tests because the EWSR1 gene is not translocated in those cases. In addition, other small round tumors also contain translocations of different ETS family members with EWSR1, such as desmoplastic small round cell tumor, clear cell sarcoma, extraskeletal myxoid chondrosarcoma, and myxoid liposarcoma, all of which may be positive with a EWS fluorescence in situ hybridization (FISH) break-apart probe. A detailed analysis of 85 patients with small round blue cell tumors that were negative for EWSR1 rearrangement by FISH with an EWSR1 break-apart probe identified eight patients with FUS rearrangements.[11] Four patients who had EWSR1-ERG fusions were not detected by FISH with an EWSR1 break-apart probe. The authors do not recommend relying solely on EWSR1 break-apart probes for analyzing small round blue cell tumors with strong immunohistochemical positivity for CD99.
Small round blue cell tumors of bone and soft tissue that are histologically similar to Ewing sarcoma but do not have rearrangements of the EWSR1 gene have been analyzed and translocations have been identified. These include BCOR-CCNB3, CIC-DUX4, and CIC-FOX4.[12-15] The molecular profile of these tumors is different from the profile of EWS-FLI1 translocated Ewing sarcoma, and limited evidence suggests that they have a different clinical behavior. In almost all cases, the patients were treated with therapy designed for Ewing sarcoma on the basis of the histologic and immunohistologic similarity to Ewing sarcoma. There are too few cases associated with each translocation to determine whether the prognosis for these small round blue cell tumors is distinct from the prognosis of Ewing sarcoma of similar stage and site.[12-15]
A genome-wide association study identified a region on chromosome 10q21.3 associated with an increased risk of Ewing sarcoma.[16] Deep sequencing through this region identified a polymorphism in the EGR2 gene, which appears to cooperate with the gene product of the EWSR1-FLI1 fusion that is seen in most patients with Ewing sarcoma.[17] The polymorphism associated with the increased risk is found at a much higher frequency in whites than in blacks or Asians, possibly contributing to the epidemiology of the relative infrequency of Ewing sarcoma in the latter populations.
| TET Family Partner | Fusion With ETS-like Oncogene Partner | Translocation | Comment |
|---|---|---|---|
| aThese partners are not members of the ETS family of oncogenes. | |||
| EWS | EWSR1-FLI1 | t(11;22)(q24;q12) | Most common; ~85% to 90% of cases |
| EWSR1-ERG | t(21;22)(q22;q12) | Second most common; ~10% of cases | |
| EWSR1-ETV1 | t(7;22)(p22;q12) | Rare | |
| EWSR1-ETV4 | t(17;22)(q12;q12) | Rare | |
| EWSR1-FEV | t(2;22)(q35;q12) | Rare | |
| EWSR1-NFATc2a | t(20;22)(q13;q12) | Rare | |
| EWSR1-POU5F1a | t(6;22)(p21;q12) | ||
| EWSR1-SMARCA5a | t(4;22)(q31;q12) | Rare | |
| EWSR1-ZSGa | t(6;22)(p21;q12) | ||
| EWSR1-SP3a | t(2;22)(q31;q12) | Rare | |
| TLS (also called FUS) | TLS-ERG | t(16;21)(p11;q22) | Rare |
| TLS-FEV | t(2;16)(q35;p11) | Rare | |
References
- Parham DM, Hijazi Y, Steinberg SM, et al.: Neuroectodermal differentiation in Ewing's sarcoma family of tumors does not predict tumor behavior. Hum Pathol 30 (8): 911-8, 1999. [PUBMED Abstract]
- Delattre O, Zucman J, Melot T, et al.: The Ewing family of tumors--a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 331 (5): 294-9, 1994. [PUBMED Abstract]
- Urano F, Umezawa A, Yabe H, et al.: Molecular analysis of Ewing's sarcoma: another fusion gene, EWS-E1AF, available for diagnosis. Jpn J Cancer Res 89 (7): 703-11, 1998. [PUBMED Abstract]
- Hattinger CM, Rumpler S, Strehl S, et al.: Prognostic impact of deletions at 1p36 and numerical aberrations in Ewing tumors. Genes Chromosomes Cancer 24 (3): 243-54, 1999. [PUBMED Abstract]
- Sankar S, Lessnick SL: Promiscuous partnerships in Ewing's sarcoma. Cancer Genet 204 (7): 351-65, 2011. [PUBMED Abstract]
- Roberts P, Burchill SA, Brownhill S, et al.: Ploidy and karyotype complexity are powerful prognostic indicators in the Ewing's sarcoma family of tumors: a study by the United Kingdom Cancer Cytogenetics and the Children's Cancer and Leukaemia Group. Genes Chromosomes Cancer 47 (3): 207-20, 2008. [PUBMED Abstract]
- Tirode F, Surdez D, Ma X, et al.: Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov 4 (11): 1342-53, 2014. [PUBMED Abstract]
- Crompton BD, Stewart C, Taylor-Weiner A, et al.: The genomic landscape of pediatric Ewing sarcoma. Cancer Discov 4 (11): 1326-41, 2014. [PUBMED Abstract]
- Brohl AS, Solomon DA, Chang W, et al.: The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet 10 (7): e1004475, 2014. [PUBMED Abstract]
- Monforte-Muñoz H, Lopez-Terrada D, Affendie H, et al.: Documentation of EWS gene rearrangements by fluorescence in-situ hybridization (FISH) in frozen sections of Ewing's sarcoma-peripheral primitive neuroectodermal tumor. Am J Surg Pathol 23 (3): 309-15, 1999. [PUBMED Abstract]
- Chen S, Deniz K, Sung YS, et al.: Ewing sarcoma with ERG gene rearrangements: A molecular study focusing on the prevalence of FUS-ERG and common pitfalls in detecting EWSR1-ERG fusions by FISH. Genes Chromosomes Cancer 55 (4): 340-9, 2016. [PUBMED Abstract]
- Pierron G, Tirode F, Lucchesi C, et al.: A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet 44 (4): 461-6, 2012. [PUBMED Abstract]
- Specht K, Sung YS, Zhang L, et al.: Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged Ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosomes Cancer 53 (7): 622-33, 2014. [PUBMED Abstract]
- Sugita S, Arai Y, Tonooka A, et al.: A novel CIC-FOXO4 gene fusion in undifferentiated small round cell sarcoma: a genetically distinct variant of Ewing-like sarcoma. Am J Surg Pathol 38 (11): 1571-6, 2014. [PUBMED Abstract]
- Cohen-Gogo S, Cellier C, Coindre JM, et al.: Ewing-like sarcomas with BCOR-CCNB3 fusion transcript: a clinical, radiological and pathological retrospective study from the Société Française des Cancers de L'Enfant. Pediatr Blood Cancer 61 (12): 2191-8, 2014. [PUBMED Abstract]
- Postel-Vinay S, Véron AS, Tirode F, et al.: Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat Genet 44 (3): 323-7, 2012. [PUBMED Abstract]
- Grünewald TG, Bernard V, Gilardi-Hebenstreit P, et al.: Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat Genet 47 (9): 1073-8, 2015. [PUBMED Abstract]
Stage Information for Ewing Sarcoma
Pretreatment staging studies for Ewing sarcoma may include the following:
- Magnetic resonance imaging (MRI).
- Computed tomography (CT) scan of the primary site and chest.
- Positron emission tomography using fluorine F 18-fludeoxyglucose (18F-FDG PET) or 18F-FDG PET-CT.
- Bone scan.
- Bone marrow aspiration and biopsy.
For patients with confirmed Ewing sarcoma, pretreatment staging studies include MRI and/or CT scan, depending on the primary site. Despite the fact that CT and MRI are both equivalent in terms of staging, use of both imaging modalities may help radiation therapy planning.[1] Whole-body MRI may provide additional information that could potentially alter therapy planning.[2] Additional pretreatment staging studies include bone scan and CT scan of the chest. In certain studies, determination of pretreatment tumor volume is an important variable.
Although 18F-FDG PET or 18F-FDG PET-CT are optional staging modalities, they have demonstrated high sensitivity and specificity in Ewing sarcoma and may provide additional information that alters therapy planning. In one institutional study, 18F-FDG PET had a very high correlation with bone scan; the investigators suggested that it could replace bone scan for the initial extent of disease evaluation.[3] This finding was confirmed in a single-institution retrospective review.[4] 18F-FDG PET-CT is more accurate than 18F-FDG PET alone in Ewing sarcoma.[5-7]
Bone marrow aspiration and biopsy have been considered the standard of care for Ewing sarcoma. However, two retrospective studies showed that for patients (N = 141 total) who were evaluated by bone scan and/or PET scan and lung CT without evidence of metastases, bone marrow aspirates and biopsies were negative in every case.[3,8] The need for routine use of bone marrow aspirates and biopsies in patients without bone metastases is now in question.
For Ewing sarcoma, the tumor is defined as localized when, by clinical and imaging techniques, there is no spread beyond the primary site or regional lymph node involvement. Continuous extension into adjacent soft tissue may occur. If there is a question of regional lymph node involvement, pathologic confirmation is indicated.
References
- Meyer JS, Nadel HR, Marina N, et al.: Imaging guidelines for children with Ewing sarcoma and osteosarcoma: a report from the Children's Oncology Group Bone Tumor Committee. Pediatr Blood Cancer 51 (2): 163-70, 2008. [PUBMED Abstract]
- Mentzel HJ, Kentouche K, Sauner D, et al.: Comparison of whole-body STIR-MRI and 99mTc-methylene-diphosphonate scintigraphy in children with suspected multifocal bone lesions. Eur Radiol 14 (12): 2297-302, 2004. [PUBMED Abstract]
- Newman EN, Jones RL, Hawkins DS: An evaluation of [F-18]-fluorodeoxy-D-glucose positron emission tomography, bone scan, and bone marrow aspiration/biopsy as staging investigations in Ewing sarcoma. Pediatr Blood Cancer 60 (7): 1113-7, 2013. [PUBMED Abstract]
- Ulaner GA, Magnan H, Healey JH, et al.: Is methylene diphosphonate bone scan necessary for initial staging of Ewing sarcoma if 18F-FDG PET/CT is performed? AJR Am J Roentgenol 202 (4): 859-67, 2014. [PUBMED Abstract]
- Völker T, Denecke T, Steffen I, et al.: Positron emission tomography for staging of pediatric sarcoma patients: results of a prospective multicenter trial. J Clin Oncol 25 (34): 5435-41, 2007. [PUBMED Abstract]
- Gerth HU, Juergens KU, Dirksen U, et al.: Significant benefit of multimodal imaging: PET/CT compared with PET alone in staging and follow-up of patients with Ewing tumors. J Nucl Med 48 (12): 1932-9, 2007. [PUBMED Abstract]
- Treglia G, Salsano M, Stefanelli A, et al.: Diagnostic accuracy of ¹⁸F-FDG-PET and PET/CT in patients with Ewing sarcoma family tumours: a systematic review and a meta-analysis. Skeletal Radiol 41 (3): 249-56, 2012. [PUBMED Abstract]
- Kopp LM, Hu C, Rozo B, et al.: Utility of bone marrow aspiration and biopsy in initial staging of Ewing sarcoma. Pediatr Blood Cancer 62 (1): 12-5, 2015. [PUBMED Abstract]
Treatment Option Overview for Ewing Sarcoma
It is important that patients be evaluated by specialists from the appropriate disciplines (e.g., radiologists, chemotherapists, pathologists, surgical or orthopedic oncologists, and radiation oncologists) as early as possible. Appropriate imaging studies of the site are obtained before biopsy. To ensure that the incision is placed in an acceptable location, the surgical or orthopedic oncologist who will perform the definitive surgery is involved in the decision regarding biopsy-incision placement. This is especially important if it is thought that the lesion can be totally excised or if a limb salvage procedure may be attempted. Biopsy should be from soft tissue as often as possible to avoid increasing the risk of fracture.[1] The pathologist is consulted before biopsy/surgery to ensure that the incision will not compromise the radiation port and that multiple types of adequate tissue samples are obtained. It is important to obtain fresh tissue, whenever possible, for cytogenetics and molecular pathology. A second option is to perform a needle biopsy, as long as adequate tissue is obtained for molecular biology and cytogenetics.[2]
Table 3 describes the treatment options for localized, metastatic, and recurrent Ewing sarcoma.
| Treatment Group | Standard Treatment Options | |
|---|---|---|
| Localized Ewing sarcoma | Chemotherapy | |
| Local-control measures: | ||
| Surgery | ||
| Radiation therapy | ||
| Metastatic Ewing sarcoma | Chemotherapy | |
| Surgery | ||
| Radiation therapy | ||
| Recurrent Ewing sarcoma | Chemotherapy (not considered standard treatment) | |
| Radiation therapy (not considered standard treatment) | ||
| Other therapies (not considered standard treatment) | ||
The successful treatment of patients with Ewing sarcoma requires systemic chemotherapy [3-9] in conjunction with surgery and/or radiation therapy for local tumor control.[10-14] In general, patients receive chemotherapy before instituting local-control measures. In patients who undergo surgery, surgical margins and histologic response are considered in planning postoperative therapy. Patients with metastatic disease often have a good initial response to preoperative chemotherapy, but in most cases, the disease is only partially controlled or recurs.[15-19] Patients with lung as the only metastatic site have a better prognosis than do patients with metastases to bone and/or bone marrow. Adequate local control for metastatic sites, particularly bone metastases, may be an important issue.[20]
Chemotherapy for Ewing Sarcoma
Multidrug chemotherapy for Ewing sarcoma always includes vincristine, doxorubicin, ifosfamide, and etoposide. Most protocols also use cyclophosphamide and some incorporate dactinomycin. The mode of administration and dose intensity of cyclophosphamide within courses differs markedly between protocols. A European Intergroup Cooperative Ewing Sarcoma Study (EICESS) trial suggested that 1.2 g of cyclophosphamide produced a similar event-free survival (EFS) compared with 6 g of ifosfamide in patients with lower-risk disease, and identified a trend toward better EFS for patients with localized Ewing sarcoma and higher-risk disease when treatment included etoposide (GER-GPOH-EICESS-92).[21][Level of evidence: 1iiA]
Protocols in the United States generally alternate courses of vincristine, cyclophosphamide, and doxorubicin with courses of ifosfamide/etoposide,[7] while European protocols generally combine vincristine, doxorubicin, and an alkylating agent with or without etoposide in a single treatment cycle.[9] The duration of primary chemotherapy ranges from 6 months to approximately 1 year.
Evidence (chemotherapy):
- An international consortium of European countries conducted the EURO-EWING-INTERGROUP-EE99 (NCT00020566) trial from 2000 to 2010.[22][Level of evidence: 1iiA] All patients received induction therapy with six cycles of vincristine, ifosfamide, doxorubicin, and etoposide (VIDE), followed by local control, and then one cycle of vincristine, dactinomycin, and ifosfamide (VAI). Patients were classified as standard risk if they had localized disease and good histologic response to therapy or if they had localized tumors less than 200 mL in volume at presentation; they were treated with radiation therapy alone as local treatment. Standard-risk patients (n = 856) were randomly assigned to receive either maintenance therapy with seven cycles of vincristine, dactinomycin, and cyclophosphamide (VAC) or VAI.
- There was no significant difference in EFS or overall survival (OS) between the VAC arm and the VAI arm.
- Three-year EFS for this low-risk population was 77%.
- Acute renal toxicity was lower in the VAC arm than in the VAI arm, but long-term renal function outcome and fertility analyses are still pending.
- It is difficult to compare this outcome with that of other large series because the study population excluded patients with poor response to initial therapy or patients with tumors more than 200 mL in volume who received local-control therapy with radiation alone. All other published series report results for all patients who present without clinically detectable metastasis; thus, these other series included patients with poor response and patients with larger primary tumors treated with radiation alone, all of whom were excluded from the EURO-EWING-INTERGROUP-EE99 study.
- A randomized clinical trial (COG-AEWS0031 [NCT00006734]) from the Children’s Oncology Group (COG) showed that for patients presenting without metastases, the administration of cycles of cyclophosphamide, doxorubicin, and vincristine alternating with cycles of ifosfamide and etoposide at 2-week intervals achieved superior EFS (5-year EFS, 73%) than did alternating cycles at 3-week intervals (5-year EFS, 65%).[23]
- The Brazilian Cooperative Study Group performed a multi-institutional trial that incorporated carboplatin into a risk-adapted intensive regimen in 175 children with localized or metastatic Ewing sarcoma. They found significantly increased toxicity without an improvement in outcome with the addition of carboplatin.[24][Level of evidence: 2Dii]
- The COG conducted a pilot study of the addition of cycles of cyclophosphamide and topotecan to cycles of cyclophosphamide/doxorubicin/vincristine and ifosfamide/etoposide administered in an interval-compressed (2-week instead of 3-week intervals) schedule.[25][Level of evidence: 2Di]
- Therapy was well tolerated, and the 5-year EFS for 35 patients was 80%. This pilot study became the experimental arm of COG-AEWS1031 (NCT01231906).
Local Control for Ewing Sarcoma
Treatment approaches for Ewing sarcoma titrate therapeutic aggressiveness with the goal of maximizing local control while minimizing morbidity.
Surgery is the most commonly used form of local control.[26] Radiation therapy is an effective alternative modality for local control in cases where the functional morbidity of surgery is deemed too high by experienced surgical oncologists. However, in the immature skeleton, radiation therapy can cause subsequent deformities that may be more morbid than deformities from surgery. When complete surgical resection with pathologically negative margins cannot be obtained, postoperative radiation therapy is indicated. A multidisciplinary discussion between the experienced radiation oncologist and the surgeon is necessary to determine the best treatment options for local control for a given case. For some marginally resectable lesions, a combined approach of preoperative radiation therapy followed by resection can be used.
Randomized trials that directly compare surgery and radiation therapy do not exist, and their relative roles remain controversial. Although retrospective institutional series suggest superior local control and survival with surgery than with radiation therapy, most of these studies are compromised by selection bias. An analysis using propensity scoring to adjust for clinical features that may influence the preference for surgery only, radiation only, or combined surgery and radiation demonstrated that similar EFS is achieved with each mode of local therapy after propensity adjustment.[26] Data for patients with pelvic primary Ewing sarcoma from a North American intergroup trial showed no difference in local control or survival on the basis of local-control modality—surgery alone, radiation therapy alone, or radiation plus surgery.[27]
For patients who undergo gross-total resection with microscopic residual disease, the value of adjuvant radiation therapy is controversial. Investigations addressing this issue are retrospective and nonrandomized, limiting their value.
Evidence (postoperative radiation therapy):
- Investigators from St. Jude Children’s Research Hospital reported 39 patients with localized Ewing sarcoma who received both surgery and radiation.[13]
- Local failure for patients with positive margins was 17% and OS was 71%. Local failure for patients with negative margins was 5% and OS was 94%.
- However, in a large retrospective Italian study, 45 Gy of adjuvant radiation therapy for patients with inadequate margins did not appear to improve either local control or disease-free survival.[14] It is not known whether higher doses of radiation therapy could improve outcome. These investigators concluded that patients who are anticipated to have suboptimal surgery should be considered for definitive radiation therapy.
- The EURO-EWING-INTERGROUP-EE99 (NCT00020566) study reported the outcomes of 599 patients who presented with localized disease and had surgical resection after initial chemotherapy with at least 90% necrosis of the primary tumor.[28][Level of evidence: 3iiDi] The protocol recommended postoperative radiation therapy for patients with inadequate surgical margins, vertebral primary tumors, or thoracic tumors with pleural effusion, but the decision to use postoperative radiation therapy was left to the institutional investigator.
- Patients who received postoperative radiation therapy (n = 142) had a lower risk of failure than did patients who did not receive postoperative radiation therapy, even after controlling for known prognostic factors, including age, sex, tumor site, clinical response, quality of resection, and histologic necrosis. Most of the improvement was seen in a decreased risk of local recurrence. The improvement was greater in patients who were assessed to have 100% necrosis than in patients who were assessed to have 90% to 100% necrosis.
- There is a clear interaction between systemic therapy and local-control modalities for both local control and disease-free survival. The induction regimen used in the EURO-EWING-INTERGROUP-EE99 study is less intense than the induction regimen used in contemporaneous protocols in the COG, and it is not appropriate to extrapolate the results from the EURO-EWING-INTERGROUP-EE99 study to different systemic chemotherapy regimens.
In summary, surgery is chosen as definitive local therapy for suitable patients, but radiation therapy is appropriate for patients with unresectable disease or those who would experience functional compromise by definitive surgery. The possibility of impaired function needs to be measured against the possibility of second tumors in the radiation field (refer to the Late Effects of Treatment for Ewing Sarcoma section of this summary for more information). Adjuvant radiation therapy may be considered for patients with residual microscopic disease, inadequate margins, or who have viable tumor in the resected specimen and close margins.
When preoperative assessment has suggested a high probability that surgical margins will be close or positive, preoperative radiation therapy has achieved tumor shrinkage and allowed surgical resection with clear margins.[29]
High-Dose Therapy With Stem Cell Rescue for Ewing Sarcoma
For patients with a high risk of relapse with conventional treatments, certain investigators have utilized high-dose chemotherapy with hematopoietic stem cell transplant (HSCT) as consolidation treatment, in an effort to improve outcome.[19,30-42]
Evidence (high-dose therapy with stem cell rescue):
- In a prospective study, patients with bone and/or bone marrow metastases at diagnosis were treated with aggressive chemotherapy, surgery, and/or radiation and HSCT if a good initial response was achieved.[35]
- The study showed no benefit for HSCT compared with historical controls.
- A retrospective review using international bone marrow transplant registries compared the outcomes after treatment with either reduced-intensity conditioning or high-intensity conditioning followed by allogeneic SCT for patients with Ewing sarcoma at high risk for relapse.[43][Level of evidence: 3iiiA]
- There was no difference in outcome, and the authors concluded that this suggested the absence of a clinically relevant graft-versus-tumor effect against Ewing sarcoma tumor cells with current approaches.
- Multiple small studies that report benefit for HSCT have been published but are difficult to interpret because only patients who have a good initial response to standard chemotherapy are considered for HSCT.
The role of high-dose therapy followed by stem cell rescue is being investigated in the prospective, randomized Euro-Ewing trial (EURO-EWING-INTERGROUP-EE99) for patients who present with metastases and patients with localized tumors with poor response to initial chemotherapy.
Ewing Sarcoma/Specific Sites
Multiple analyses have evaluated diagnostic findings, treatment, and outcome of patients with bone lesions at the following anatomic primary sites:
Extraosseous Ewing Sarcoma
Extraosseous Ewing sarcoma is biologically similar to Ewing sarcoma arising in bone. Historically, most children and young adults with extraosseous Ewing sarcoma were treated on protocols designed for the treatment of rhabdomyosarcoma. This is important because many of the treatment regimens for rhabdomyosarcoma do not include an anthracycline, which is a critical component of current treatment regimens for Ewing sarcoma. Currently, patients with extraosseous Ewing sarcoma are eligible for studies that include Ewing sarcoma of bone.
From 1987 to 2004, 111 patients with nonmetastatic extraosseous Ewing sarcoma were enrolled on the RMS-88 and RMS-96 protocols.[62] Patients with initial complete tumor resection received ifosfamide, vincristine, and actinomycin (IVA) while patients with residual tumor received IVA plus doxorubicin (VAIA) or IVA plus carboplatin, epirubicin, and etoposide (CEVAIE). Seventy-six percent of patients received radiation. The 5-year EFS was 59% and OS was 69%. In a multivariate analysis, independent adverse prognostic factors included axial primary, tumor size greater than 10 cm, Intergroup Rhabdomyosarcoma Studies Group III, and lack of radiation therapy.
Two hundred thirty-six patients with extraosseous Ewing sarcoma were entered on studies of the German Pediatric Oncology Group.[63] The median age at diagnosis was 15 years and 133 patients were male. Primary tumor site was either extremity (n = 62) or central site (n = 174). Sixty of the 236 patients had metastases at diagnosis. Chemotherapy consisted of vincristine, doxorubicin, cyclophosphamide, and actinomycin (VACA); CEVAIE; or VIDE. The 5-year EFS was 49% and OS was 60%. Five-year survival was 70% for patients with localized disease and 33% for patients with metastasis at diagnosis. OS in patients with localized disease did not seem related to tumor site or size. In a retrospective French study, patients with extraosseous Ewing sarcoma were treated using a rhabdomyosarcoma regimen (no anthracyclines) or a Ewing sarcoma regimen (includes anthracyclines). Patients who received the anthracycline-containing regimen had a significantly better EFS and OS than did patients who did not receive anthracyclines.[64,65] Two North American Ewing sarcoma trials have included patients with extraosseous Ewing sarcoma.[23,66] In a review of data from the POG-9354 (INT-0154) and EWS0031 (NCT00006734) studies, 213 patients with extraosseous Ewing sarcoma and 826 patients with Ewing sarcoma of bone were identified. The hazard ratio of extraosseous Ewing sarcoma was superior (0.62), and extraosseous Ewing sarcoma was a favorable risk factor, independent of age, race, and primary site.[67][Level of evidence: 3iiDi]
Cutaneous Ewing sarcoma is a soft tissue tumor in the skin or subcutaneous tissue that seems to behave as a less-aggressive tumor than primary bone or soft tissue Ewing sarcoma. Tumors can form throughout the body, although the extremity is the most common site, and they are almost always localized. In a review of 78 reported cases, some lacking molecular confirmation, the OS was 91%. Adequate local control, defined as a complete resection with negative margins, radiation therapy, or a combination, significantly reduced the incidence of relapse. Standard chemotherapy for Ewing sarcoma is often used for these patients because there are no data to suggest which patients could be treated less aggressively.[68,69] A series of 56 patients with cutaneous or subcutaneous Ewing sarcoma confirmed the excellent outcome with the use of standard systemic therapy and local control. Attempted primary definitive surgery often resulted in the need for either radiation therapy or more function-compromising surgery, supporting the recommendation of biopsy only as initial surgery, rather than upfront unplanned resection.[70][Level of evidence: 3iiD]
Special Considerations for the Treatment of Children With Cancer
Cancer in children and adolescents is rare, although the overall incidence of childhood cancer has been slowly increasing since 1975.[71] Children and adolescents with cancer should be referred to medical centers that have a multidisciplinary team of cancer specialists with experience treating the cancers that occur during childhood and adolescence. This multidisciplinary team approach incorporates the skills of the following health care professionals and others to ensure that children receive treatment, supportive care, and rehabilitation that will achieve optimal survival and quality of life:
- Primary care physicians.
- Pediatric surgeons.
- Radiation oncologists.
- Pediatric oncologists/hematologists.
- Rehabilitation specialists.
- Pediatric nurse specialists.
- Social workers.
- Child-life professionals.
- Psychologists.
(Refer to the PDQ Supportive and Palliative Care summaries for specific information about supportive care for children and adolescents with cancer.)
Guidelines for pediatric cancer centers and their role in the treatment of pediatric patients with cancer have been outlined by the American Academy of Pediatrics.[72] At these pediatric cancer centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate in these trials is offered to most patients and their families. Clinical trials for children and adolescents with cancer are generally designed to compare potentially better therapy with therapy that is currently accepted as standard. Most of the progress made in identifying curative therapies for childhood cancers has been achieved through clinical trials. Information about ongoing clinical trials is available from the NCI website.
Childhood and adolescent cancer survivors require close monitoring because cancer therapy side effects may persist or develop months or years after treatment. (Refer to the PDQ summary on Late Effects of Treatment for Childhood Cancer for specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors.)
References
- Fuchs B, Valenzuela RG, Sim FH: Pathologic fracture as a complication in the treatment of Ewing's sarcoma. Clin Orthop (415): 25-30, 2003. [PUBMED Abstract]
- Hoffer FA, Gianturco LE, Fletcher JA, et al.: Percutaneous biopsy of peripheral primitive neuroectodermal tumors and Ewing's sarcomas for cytogenetic analysis. AJR Am J Roentgenol 162 (5): 1141-2, 1994. [PUBMED Abstract]
- Craft A, Cotterill S, Malcolm A, et al.: Ifosfamide-containing chemotherapy in Ewing's sarcoma: The Second United Kingdom Children's Cancer Study Group and the Medical Research Council Ewing's Tumor Study. J Clin Oncol 16 (11): 3628-33, 1998. [PUBMED Abstract]
- Shankar AG, Pinkerton CR, Atra A, et al.: Local therapy and other factors influencing site of relapse in patients with localised Ewing's sarcoma. United Kingdom Children's Cancer Study Group (UKCCSG). Eur J Cancer 35 (12): 1698-704, 1999. [PUBMED Abstract]
- Nilbert M, Saeter G, Elomaa I, et al.: Ewing's sarcoma treatment in Scandinavia 1984-1990--ten-year results of the Scandinavian Sarcoma Group Protocol SSGIV. Acta Oncol 37 (4): 375-8, 1998. [PUBMED Abstract]
- Ferrari S, Mercuri M, Rosito P, et al.: Ifosfamide and actinomycin-D, added in the induction phase to vincristine, cyclophosphamide and doxorubicin, improve histologic response and prognosis in patients with non metastatic Ewing's sarcoma of the extremity. J Chemother 10 (6): 484-91, 1998. [PUBMED Abstract]
- Grier HE, Krailo MD, Tarbell NJ, et al.: Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 348 (8): 694-701, 2003. [PUBMED Abstract]
- Thacker MM, Temple HT, Scully SP: Current treatment for Ewing's sarcoma. Expert Rev Anticancer Ther 5 (2): 319-31, 2005. [PUBMED Abstract]
- Juergens C, Weston C, Lewis I, et al.: Safety assessment of intensive induction with vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) in the treatment of Ewing tumors in the EURO-E.W.I.N.G. 99 clinical trial. Pediatr Blood Cancer 47 (1): 22-9, 2006. [PUBMED Abstract]
- Dunst J, Schuck A: Role of radiotherapy in Ewing tumors. Pediatr Blood Cancer 42 (5): 465-70, 2004. [PUBMED Abstract]
- Donaldson SS: Ewing sarcoma: radiation dose and target volume. Pediatr Blood Cancer 42 (5): 471-6, 2004. [PUBMED Abstract]
- Bacci G, Ferrari S, Longhi A, et al.: Role of surgery in local treatment of Ewing's sarcoma of the extremities in patients undergoing adjuvant and neoadjuvant chemotherapy. Oncol Rep 11 (1): 111-20, 2004. [PUBMED Abstract]
- Krasin MJ, Rodriguez-Galindo C, Davidoff AM, et al.: Efficacy of combined surgery and irradiation for localized Ewings sarcoma family of tumors. Pediatr Blood Cancer 43 (3): 229-36, 2004. [PUBMED Abstract]
- Bacci G, Longhi A, Briccoli A, et al.: The role of surgical margins in treatment of Ewing's sarcoma family tumors: experience of a single institution with 512 patients treated with adjuvant and neoadjuvant chemotherapy. Int J Radiat Oncol Biol Phys 65 (3): 766-72, 2006. [PUBMED Abstract]
- Paulussen M, Ahrens S, Burdach S, et al.: Primary metastatic (stage IV) Ewing tumor: survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma Studies. Ann Oncol 9 (3): 275-81, 1998. [PUBMED Abstract]
- Pinkerton CR, Bataillard A, Guillo S, et al.: Treatment strategies for metastatic Ewing's sarcoma. Eur J Cancer 37 (11): 1338-44, 2001. [PUBMED Abstract]
- Miser JS, Krailo M, Meyers P, et al.: Metastatic Ewing's sarcoma(es) and primitive neuroectodermal tumor (PNET) of bone: failure of new regimens to improve outcome. [Abstract] Proceedings of the American Society of Clinical Oncology 15: A-1472, 467, 1996.
- Bernstein ML, Devidas M, Lafreniere D, et al.: Intensive therapy with growth factor support for patients with Ewing tumor metastatic at diagnosis: Pediatric Oncology Group/Children's Cancer Group Phase II Study 9457--a report from the Children's Oncology Group. J Clin Oncol 24 (1): 152-9, 2006. [PUBMED Abstract]
- Ladenstein R, Pötschger U, Le Deley MC, et al.: Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol 28 (20): 3284-91, 2010. [PUBMED Abstract]
- Haeusler J, Ranft A, Boelling T, et al.: The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES). Cancer 116 (2): 443-50, 2010. [PUBMED Abstract]
- Paulussen M, Craft AW, Lewis I, et al.: Results of the EICESS-92 Study: two randomized trials of Ewing's sarcoma treatment--cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J Clin Oncol 26 (27): 4385-93, 2008. [PUBMED Abstract]
- Le Deley MC, Paulussen M, Lewis I, et al.: Cyclophosphamide compared with ifosfamide in consolidation treatment of standard-risk Ewing sarcoma: results of the randomized noninferiority Euro-EWING99-R1 trial. J Clin Oncol 32 (23): 2440-8, 2014. [PUBMED Abstract]
- Womer RB, West DC, Krailo MD, et al.: Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 30 (33): 4148-54, 2012. [PUBMED Abstract]
- Brunetto AL, Castillo LA, Petrilli AS, et al.: Carboplatin in the treatment of Ewing sarcoma: Results of the first Brazilian collaborative study group for Ewing sarcoma family tumors-EWING1. Pediatr Blood Cancer 62 (10): 1747-53, 2015. [PUBMED Abstract]
- Mascarenhas L, Felgenhauer JL, Bond MC, et al.: Pilot Study of Adding Vincristine, Topotecan, and Cyclophosphamide to Interval-Compressed Chemotherapy in Newly Diagnosed Patients With Localized Ewing Sarcoma: A Report From the Children's Oncology Group. Pediatr Blood Cancer 63 (3): 493-8, 2016. [PUBMED Abstract]
- DuBois SG, Krailo MD, Gebhardt MC, et al.: Comparative evaluation of local control strategies in localized Ewing sarcoma of bone: a report from the Children's Oncology Group. Cancer 121 (3): 467-75, 2015. [PUBMED Abstract]
- Yock TI, Krailo M, Fryer CJ, et al.: Local control in pelvic Ewing sarcoma: analysis from INT-0091--a report from the Children's Oncology Group. J Clin Oncol 24 (24): 3838-43, 2006. [PUBMED Abstract]
- Foulon S, Brennan B, Gaspar N, et al.: Can postoperative radiotherapy be omitted in localised standard-risk Ewing sarcoma? An observational study of the Euro-E.W.I.N.G group. Eur J Cancer 61: 128-36, 2016. [PUBMED Abstract]
- Wagner TD, Kobayashi W, Dean S, et al.: Combination short-course preoperative irradiation, surgical resection, and reduced-field high-dose postoperative irradiation in the treatment of tumors involving the bone. Int J Radiat Oncol Biol Phys 73 (1): 259-66, 2009. [PUBMED Abstract]
- Kushner BH, Meyers PA: How effective is dose-intensive/myeloablative therapy against Ewing's sarcoma/primitive neuroectodermal tumor metastatic to bone or bone marrow? The Memorial Sloan-Kettering experience and a literature review. J Clin Oncol 19 (3): 870-80, 2001. [PUBMED Abstract]
- Marina N, Meyers PA: High-dose therapy and stem-cell rescue for Ewing's family of tumors in second remission. J Clin Oncol 23 (19): 4262-4, 2005. [PUBMED Abstract]
- Burdach S: Treatment of advanced Ewing tumors by combined radiochemotherapy and engineered cellular transplants. Pediatr Transplant 8 (Suppl 5): 67-82, 2004. [PUBMED Abstract]
- McTiernan A, Driver D, Michelagnoli MP, et al.: High dose chemotherapy with bone marrow or peripheral stem cell rescue is an effective treatment option for patients with relapsed or progressive Ewing's sarcoma family of tumours. Ann Oncol 17 (8): 1301-5, 2006. [PUBMED Abstract]
- Burdach S, Meyer-Bahlburg A, Laws HJ, et al.: High-dose therapy for patients with primary multifocal and early relapsed Ewing's tumors: results of two consecutive regimens assessing the role of total-body irradiation. J Clin Oncol 21 (16): 3072-8, 2003. [PUBMED Abstract]
- Meyers PA, Krailo MD, Ladanyi M, et al.: High-dose melphalan, etoposide, total-body irradiation, and autologous stem-cell reconstitution as consolidation therapy for high-risk Ewing's sarcoma does not improve prognosis. J Clin Oncol 19 (11): 2812-20, 2001. [PUBMED Abstract]
- Oberlin O, Rey A, Desfachelles AS, et al.: Impact of high-dose busulfan plus melphalan as consolidation in metastatic Ewing tumors: a study by the Société Française des Cancers de l'Enfant. J Clin Oncol 24 (24): 3997-4002, 2006. [PUBMED Abstract]
- Hawkins D, Barnett T, Bensinger W, et al.: Busulfan, melphalan, and thiotepa with or without total marrow irradiation with hematopoietic stem cell rescue for poor-risk Ewing-Sarcoma-Family tumors. Med Pediatr Oncol 34 (5): 328-37, 2000. [PUBMED Abstract]
- Rosenthal J, Bolotin E, Shakhnovits M, et al.: High-dose therapy with hematopoietic stem cell rescue in patients with poor prognosis Ewing family tumors. Bone Marrow Transplant 42 (5): 311-8, 2008. [PUBMED Abstract]
- Burdach S, Thiel U, Schöniger M, et al.: Total body MRI-governed involved compartment irradiation combined with high-dose chemotherapy and stem cell rescue improves long-term survival in Ewing tumor patients with multiple primary bone metastases. Bone Marrow Transplant 45 (3): 483-9, 2010. [PUBMED Abstract]
- Gaspar N, Rey A, Bérard PM, et al.: Risk adapted chemotherapy for localised Ewing's sarcoma of bone: the French EW93 study. Eur J Cancer 48 (9): 1376-85, 2012. [PUBMED Abstract]
- Drabko K, Raciborska A, Bilska K, et al.: Consolidation of first-line therapy with busulphan and melphalan, and autologous stem cell rescue in children with Ewing's sarcoma. Bone Marrow Transplant 47 (12): 1530-4, 2012. [PUBMED Abstract]
- Loschi S, Dufour C, Oberlin O, et al.: Tandem high-dose chemotherapy strategy as first-line treatment of primary disseminated multifocal Ewing sarcomas in children, adolescents and young adults. Bone Marrow Transplant 50 (8): 1083-8, 2015. [PUBMED Abstract]
- Thiel U, Wawer A, Wolf P, et al.: No improvement of survival with reduced- versus high-intensity conditioning for allogeneic stem cell transplants in Ewing tumor patients. Ann Oncol 22 (7): 1614-21, 2011. [PUBMED Abstract]
- Hoffmann C, Ahrens S, Dunst J, et al.: Pelvic Ewing sarcoma: a retrospective analysis of 241 cases. Cancer 85 (4): 869-77, 1999. [PUBMED Abstract]
- Sucato DJ, Rougraff B, McGrath BE, et al.: Ewing's sarcoma of the pelvis. Long-term survival and functional outcome. Clin Orthop (373): 193-201, 2000. [PUBMED Abstract]
- Bacci G, Ferrari S, Mercuri M, et al.: Multimodal therapy for the treatment of nonmetastatic Ewing sarcoma of pelvis. J Pediatr Hematol Oncol 25 (2): 118-24, 2003. [PUBMED Abstract]
- Bacci G, Ferrari S, Longhi A, et al.: Local and systemic control in Ewing's sarcoma of the femur treated with chemotherapy, and locally by radiotherapy and/or surgery. J Bone Joint Surg Br 85 (1): 107-14, 2003. [PUBMED Abstract]
- Ozaki T, Hillmann A, Hoffmann C, et al.: Ewing's sarcoma of the femur. Prognosis in 69 patients treated by the CESS group. Acta Orthop Scand 68 (1): 20-4, 1997. [PUBMED Abstract]
- Ayoub KS, Fiorenza F, Grimer RJ, et al.: Extensible endoprostheses of the humerus after resection of bone tumours. J Bone Joint Surg Br 81 (3): 495-500, 1999. [PUBMED Abstract]
- Bacci G, Palmerini E, Staals EL, et al.: Ewing's sarcoma family tumors of the humerus: outcome of patients treated with radiotherapy, surgery or surgery and adjuvant radiotherapy. Radiother Oncol 93 (2): 383-7, 2009. [PUBMED Abstract]
- Casadei R, Magnani M, Biagini R, et al.: Prognostic factors in Ewing's sarcoma of the foot. Clin Orthop (420): 230-8, 2004. [PUBMED Abstract]
- Anakwenze OA, Parker WL, Wold LE, et al.: Ewing's sarcoma of the hand. J Hand Surg Eur Vol 34 (1): 35-9, 2009. [PUBMED Abstract]
- Shamberger RC, Laquaglia MP, Krailo MD, et al.: Ewing sarcoma of the rib: results of an intergroup study with analysis of outcome by timing of resection. J Thorac Cardiovasc Surg 119 (6): 1154-61, 2000. [PUBMED Abstract]
- Sirvent N, Kanold J, Levy C, et al.: Non-metastatic Ewing's sarcoma of the ribs: the French Society of Pediatric Oncology Experience. Eur J Cancer 38 (4): 561-7, 2002. [PUBMED Abstract]
- Shamberger RC, LaQuaglia MP, Gebhardt MC, et al.: Ewing sarcoma/primitive neuroectodermal tumor of the chest wall: impact of initial versus delayed resection on tumor margins, survival, and use of radiation therapy. Ann Surg 238 (4): 563-7; discussion 567-8, 2003. [PUBMED Abstract]
- Schuck A, Ahrens S, Konarzewska A, et al.: Hemithorax irradiation for Ewing tumors of the chest wall. Int J Radiat Oncol Biol Phys 54 (3): 830-8, 2002. [PUBMED Abstract]
- Windfuhr JP: Primitive neuroectodermal tumor of the head and neck: incidence, diagnosis, and management. Ann Otol Rhinol Laryngol 113 (7): 533-43, 2004. [PUBMED Abstract]
- Venkateswaran L, Rodriguez-Galindo C, Merchant TE, et al.: Primary Ewing tumor of the vertebrae: clinical characteristics, prognostic factors, and outcome. Med Pediatr Oncol 37 (1): 30-5, 2001. [PUBMED Abstract]
- Marco RA, Gentry JB, Rhines LD, et al.: Ewing's sarcoma of the mobile spine. Spine 30 (7): 769-73, 2005. [PUBMED Abstract]
- Schuck A, Ahrens S, von Schorlemer I, et al.: Radiotherapy in Ewing tumors of the vertebrae: treatment results and local relapse analysis of the CESS 81/86 and EICESS 92 trials. Int J Radiat Oncol Biol Phys 63 (5): 1562-7, 2005. [PUBMED Abstract]
- Bacci G, Boriani S, Balladelli A, et al.: Treatment of nonmetastatic Ewing's sarcoma family tumors of the spine and sacrum: the experience from a single institution. Eur Spine J 18 (8): 1091-5, 2009. [PUBMED Abstract]
- Spiller M, Bisogno G, Ferrari A, et al.: Prognostic factors in localized extraosseus Ewing family tumors. [Abstract] Pediatr Blood Cancer 46 (10) : A-PD.024, 434, 2006.
- Ladenstein R, Pötschger U, Jürgens H, et al.: Comparison of treatment concepts for extraosseus Ewing tumors (EET) within consecutive trials of two GPOH Cooperative Study Groups. [Abstract] Pediatr Blood Cancer 45 (10) : A-P.C.004, 450, 2005.
- Castex MP, Rubie H, Stevens MC, et al.: Extraosseous localized ewing tumors: improved outcome with anthracyclines--the French society of pediatric oncology and international society of pediatric oncology. J Clin Oncol 25 (10): 1176-82, 2007. [PUBMED Abstract]
- Dantonello TM, Int-Veen C, Harms D, et al.: Cooperative trial CWS-91 for localized soft tissue sarcoma in children, adolescents, and young adults. J Clin Oncol 27 (9): 1446-55, 2009. [PUBMED Abstract]
- Granowetter L, Womer R, Devidas M, et al.: Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children's Oncology Group Study. J Clin Oncol 27 (15): 2536-41, 2009. [PUBMED Abstract]
- Cash T, McIlvaine E, Krailo MD, et al.: Comparison of clinical features and outcomes in patients with extraskeletal versus skeletal localized Ewing sarcoma: A report from the Children's Oncology Group. Pediatr Blood Cancer 63 (10): 1771-9, 2016. [PUBMED Abstract]
- Collier AB 3rd, Simpson L, Monteleone P: Cutaneous Ewing sarcoma: report of 2 cases and literature review of presentation, treatment, and outcome of 76 other reported cases. J Pediatr Hematol Oncol 33 (8): 631-4, 2011. [PUBMED Abstract]
- Terrier-Lacombe MJ, Guillou L, Chibon F, et al.: Superficial primitive Ewing's sarcoma: a clinicopathologic and molecular cytogenetic analysis of 14 cases. Mod Pathol 22 (1): 87-94, 2009. [PUBMED Abstract]
- Di Giannatale A, Frezza AM, Le Deley MC, et al.: Primary cutaneous and subcutaneous Ewing sarcoma. Pediatr Blood Cancer 62 (9): 1555-61, 2015. [PUBMED Abstract]
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014. [PUBMED Abstract]
- Corrigan JJ, Feig SA; American Academy of Pediatrics: Guidelines for pediatric cancer centers. Pediatrics 113 (6): 1833-5, 2004. [PUBMED Abstract]
Treatment of Localized Ewing Sarcoma
Standard Treatment Options for Localized Ewing Sarcoma
Standard treatment options for localized Ewing sarcoma include the following:
Because most patients with apparently localized disease at diagnosis have occult metastatic disease, multidrug chemotherapy and local disease control with surgery and/or radiation therapy is indicated in the treatment of all patients.[1-8] Current regimens for the treatment of localized Ewing sarcoma achieve event-free survival (EFS) and overall survival (OS) of approximately 70% at 5 years after diagnosis.[9]
Chemotherapy
Current standard chemotherapy in the United States includes vincristine, doxorubicin, and cyclophosphamide (VDC), alternating with ifosfamide and etoposide (IE) or VDC/IE.[9]; [10][Level of evidence: 1iiA]
Evidence (chemotherapy):
- IE has shown activity in Ewing sarcoma, and a large randomized clinical trial and a nonrandomized trial demonstrated that outcome was improved when IE was alternated with VDC.[2,9,11]
- Dactinomycin is no longer used for Ewing sarcoma in the United States but continues to be used in the Euro-Ewing studies.
- Increased dose intensity of doxorubicin during the initial months of therapy was associated with an improved outcome in a meta-analysis performed before the standard use of IE.[12]
- The use of high-dose VDC has shown promising results in small numbers of patients. A single-institution study of 44 patients treated with high-dose VDC and IE showed an 82% 4-year EFS.[13]
- However, in an intergroup trial of the Pediatric Oncology Group and the Children's Cancer Group, which compared an alkylator dose-intensified VDC/IE regimen with standard alkylator doses of the same VDC/IE regimen, no differences in outcome were observed.[14] Unlike the single-institution trial, this trial did not maintain the dose intensity of cyclophosphamide for the duration of treatment.[13]
In a Children's Oncology Group (COG) trial (COG-AEWS0031), 568 patients with newly diagnosed localized extradural Ewing sarcoma were randomly assigned to receive chemotherapy (VDC/IE) given either every 2 weeks (interval compression) or every 3 weeks (standard). Patients randomly assigned to the every 2-week interval of treatment had an improved 5-year EFS (73% vs. 65%, P = .048). There was no increase in toxicity observed with the every 2-week schedule.[10]
Local-control measures
Local control can be achieved by surgery and/or radiation therapy.
Surgery
Surgery is generally the preferred approach if the lesion is resectable.[15,16] The superiority of resection for local control has never been tested in a prospective randomized trial. The apparent superiority may represent selection bias.
- In past studies, smaller, more peripheral tumors were more likely to be treated with surgery, and larger, more central tumors were more likely to be treated with radiation therapy.[17]
- An Italian retrospective study showed that surgery improved outcome only in extremity tumors, although the number of patients with central axis Ewing sarcoma who achieved adequate margins was small.[8]
- In a series of 39 patients treated at St. Jude Children's Research Hospital who received both surgery and radiation, the 8-year local failure rate was 5% for patients with negative surgical margins and 17% for those with positive margins.[5]
- Data for patients with pelvic primary Ewing sarcoma from a North American intergroup trial showed no difference in local control or survival based on local-control modality—surgery alone, radiation therapy alone, or radiation plus surgery.[18]
Potential benefits of surgery include the following:
- If a very young child has Ewing sarcoma, surgery may be a less-morbid therapy than radiation therapy because of the retardation of bone growth caused by radiation.
- Another potential benefit for surgical resection of the primary tumor is related to the amount of necrosis in the resected tumor. Patients with residual viable tumor in the resected specimen have a worse outcome than those with complete necrosis. In a French Ewing study (EW88), EFS for patients with less than 5% viable tumor was 75%, EFS for patients with 5% to 30% viable tumor was 48%, and EFS for patients with more than 30% viable tumor was 20%.[17]
European investigators are studying whether treatment intensification (i.e., high-dose chemotherapy with stem cell rescue) will improve outcome for patients with a poor histologic response.
Radiation therapy is usually employed in the following cases:
- Patients who do not have a surgical option that preserves function.
- Patients whose tumors have been excised but with inadequate margins.
Pathologic fracture at the time of diagnosis does not preclude surgical resection and is not associated with adverse outcome.[19]
Radiation therapy
Radiation therapy is delivered in a setting in which stringent planning techniques are applied by those experienced in the treatment of Ewing sarcoma. Such an approach will result in local control of the tumor with acceptable morbidity in most patients.[1,2,20]
The radiation dose may be adjusted depending on the extent of residual disease after the initial surgical procedure. Radiation therapy is generally administered in fractionated doses totaling approximately 55.8 Gy to the prechemotherapy tumor volume. A randomized study of 40 patients with Ewing sarcoma using 55.8 Gy to the prechemotherapy tumor extent with a 2-cm margin compared with the same total-tumor dose after 39.6 Gy to the entire bone showed no difference in local control or EFS.[3] Hyperfractionated radiation therapy has not been associated with improved local control or decreased morbidity.[1]
Comparison of proton-beam radiation therapy and intensity-modulated radiation therapy (IMRT) treatment plans has shown that proton-beam radiation therapy can spare more normal tissue adjacent to Ewing sarcoma primary tumors than IMRT.[21] Follow-up remains relatively short, and there are no data available to determine whether the reduction in dose to adjacent tissue will result in improved functional outcome or reduce the risk of secondary malignancy. Because patient numbers are small and follow-up is relatively short, it is not possible to determine whether the risk of local recurrence might be increased by reducing radiation dose in tissue adjacent to the primary tumor.
Higher rates of local failure are seen in patients older than 14 years who have tumors more than 8 cm in length.[22] A retrospective analysis of patients with Ewing sarcoma of the chest wall compared patients who received hemithorax radiation therapy with those who received radiation therapy to the chest wall only. Patients with pleural invasion, pleural effusion, or intraoperative contamination were assigned to hemithorax radiation therapy. EFS was longer for patients who received hemithorax radiation, but the difference was not statistically significant. In addition, most patients with primary vertebral tumors did not receive hemithorax radiation and had a lower probability for EFS.[23]
For patients with residual disease after an attempt at surgical resection, the Intergroup Ewing Sarcoma Study (INT-0091) recommended 45 Gy to the original disease site plus a 10.8 Gy boost for patients with gross residual disease and 45 Gy plus a 5.4 Gy boost for patients with microscopic residual disease. No radiation therapy was recommended for those who have no evidence of microscopic residual disease after surgical resection.[14]
Radiation therapy is associated with the development of subsequent neoplasms. A retrospective study noted that patients who received 60 Gy or more had an incidence of second malignancy of 20%. Those who received 48 Gy to 60 Gy had an incidence of 5%, and those who received less than 48 Gy did not develop a second malignancy.[24] (Refer to the Late Effects of Treatment for Ewing Sarcoma section of this summary for more information.)
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Dunst J, Jürgens H, Sauer R, et al.: Radiation therapy in Ewing's sarcoma: an update of the CESS 86 trial. Int J Radiat Oncol Biol Phys 32 (4): 919-30, 1995. [PUBMED Abstract]
- Donaldson SS, Torrey M, Link MP, et al.: A multidisciplinary study investigating radiotherapy in Ewing's sarcoma: end results of POG #8346. Pediatric Oncology Group. Int J Radiat Oncol Biol Phys 42 (1): 125-35, 1998. [PUBMED Abstract]
- Craft A, Cotterill S, Malcolm A, et al.: Ifosfamide-containing chemotherapy in Ewing's sarcoma: The Second United Kingdom Children's Cancer Study Group and the Medical Research Council Ewing's Tumor Study. J Clin Oncol 16 (11): 3628-33, 1998. [PUBMED Abstract]
- Nilbert M, Saeter G, Elomaa I, et al.: Ewing's sarcoma treatment in Scandinavia 1984-1990--ten-year results of the Scandinavian Sarcoma Group Protocol SSGIV. Acta Oncol 37 (4): 375-8, 1998. [PUBMED Abstract]
- Krasin MJ, Davidoff AM, Rodriguez-Galindo C, et al.: Definitive surgery and multiagent systemic therapy for patients with localized Ewing sarcoma family of tumors: local outcome and prognostic factors. Cancer 104 (2): 367-73, 2005. [PUBMED Abstract]
- Bacci G, Forni C, Longhi A, et al.: Long-term outcome for patients with non-metastatic Ewing's sarcoma treated with adjuvant and neoadjuvant chemotherapies. 402 patients treated at Rizzoli between 1972 and 1992. Eur J Cancer 40 (1): 73-83, 2004. [PUBMED Abstract]
- Rosito P, Mancini AF, Rondelli R, et al.: Italian Cooperative Study for the treatment of children and young adults with localized Ewing sarcoma of bone: a preliminary report of 6 years of experience. Cancer 86 (3): 421-8, 1999. [PUBMED Abstract]
- Bacci G, Longhi A, Briccoli A, et al.: The role of surgical margins in treatment of Ewing's sarcoma family tumors: experience of a single institution with 512 patients treated with adjuvant and neoadjuvant chemotherapy. Int J Radiat Oncol Biol Phys 65 (3): 766-72, 2006. [PUBMED Abstract]
- Grier HE, Krailo MD, Tarbell NJ, et al.: Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 348 (8): 694-701, 2003. [PUBMED Abstract]
- Womer RB, West DC, Krailo MD, et al.: Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 30 (33): 4148-54, 2012. [PUBMED Abstract]
- Ferrari S, Mercuri M, Rosito P, et al.: Ifosfamide and actinomycin-D, added in the induction phase to vincristine, cyclophosphamide and doxorubicin, improve histologic response and prognosis in patients with non metastatic Ewing's sarcoma of the extremity. J Chemother 10 (6): 484-91, 1998. [PUBMED Abstract]
- Smith MA, Ungerleider RS, Horowitz ME, et al.: Influence of doxorubicin dose intensity on response and outcome for patients with osteogenic sarcoma and Ewing's sarcoma. J Natl Cancer Inst 83 (20): 1460-70, 1991. [PUBMED Abstract]
- Kolb EA, Kushner BH, Gorlick R, et al.: Long-term event-free survival after intensive chemotherapy for Ewing's family of tumors in children and young adults. J Clin Oncol 21 (18): 3423-30, 2003. [PUBMED Abstract]
- Granowetter L, Womer R, Devidas M, et al.: Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children's Oncology Group Study. J Clin Oncol 27 (15): 2536-41, 2009. [PUBMED Abstract]
- Hoffmann C, Ahrens S, Dunst J, et al.: Pelvic Ewing sarcoma: a retrospective analysis of 241 cases. Cancer 85 (4): 869-77, 1999. [PUBMED Abstract]
- Shamberger RC, Laquaglia MP, Krailo MD, et al.: Ewing sarcoma of the rib: results of an intergroup study with analysis of outcome by timing of resection. J Thorac Cardiovasc Surg 119 (6): 1154-61, 2000. [PUBMED Abstract]
- Oberlin O, Deley MC, Bui BN, et al.: Prognostic factors in localized Ewing's tumours and peripheral neuroectodermal tumours: the third study of the French Society of Paediatric Oncology (EW88 study). Br J Cancer 85 (11): 1646-54, 2001. [PUBMED Abstract]
- Yock TI, Krailo M, Fryer CJ, et al.: Local control in pelvic Ewing sarcoma: analysis from INT-0091--a report from the Children's Oncology Group. J Clin Oncol 24 (24): 3838-43, 2006. [PUBMED Abstract]
- Bramer JA, Abudu AA, Grimer RJ, et al.: Do pathological fractures influence survival and local recurrence rate in bony sarcomas? Eur J Cancer 43 (13): 1944-51, 2007. [PUBMED Abstract]
- Krasin MJ, Rodriguez-Galindo C, Billups CA, et al.: Definitive irradiation in multidisciplinary management of localized Ewing sarcoma family of tumors in pediatric patients: outcome and prognostic factors. Int J Radiat Oncol Biol Phys 60 (3): 830-8, 2004. [PUBMED Abstract]
- Rombi B, DeLaney TF, MacDonald SM, et al.: Proton radiotherapy for pediatric Ewing's sarcoma: initial clinical outcomes. Int J Radiat Oncol Biol Phys 82 (3): 1142-8, 2012. [PUBMED Abstract]
- Fuchs B, Valenzuela RG, Sim FH: Pathologic fracture as a complication in the treatment of Ewing's sarcoma. Clin Orthop (415): 25-30, 2003. [PUBMED Abstract]
- Schuck A, Ahrens S, Konarzewska A, et al.: Hemithorax irradiation for Ewing tumors of the chest wall. Int J Radiat Oncol Biol Phys 54 (3): 830-8, 2002. [PUBMED Abstract]
- Kuttesch JF Jr, Wexler LH, Marcus RB, et al.: Second malignancies after Ewing's sarcoma: radiation dose-dependency of secondary sarcomas. J Clin Oncol 14 (10): 2818-25, 1996. [PUBMED Abstract]
Treatment of Metastatic Ewing Sarcoma
Metastases at diagnosis are detected in approximately 25% of patients.[1] The prognosis of patients with metastatic disease is poor. Current therapies for patients who present with metastatic disease achieve 6-year event-free survival (EFS) of approximately 28% and overall survival (OS) of approximately 30%.[2,3] For patients with lung/pleural metastases only, 6-year EFS is approximately 40% when utilizing bilateral lung irradiation.[2,4] In contrast, patients with bone/bone marrow metastases have a 4-year EFS of approximately 28% and patients with combined lung and bone/bone marrow metastases have a 4-year EFS of approximately 14%.[4,5]
The following factors independently predict a poor outcome in patients presenting with metastatic disease:[3]
- Age older than 14 years.
- Primary tumor volume of more than 200 mL.
- More than one bone metastatic site.
- Bone marrow metastases.
- Additional lung metastases.
Standard Treatment Options for Metastatic Ewing Sarcoma
Standard treatment options for metastatic Ewing sarcoma include the following:
Chemotherapy
Standard treatment for patients with metastatic Ewing sarcoma utilizing alternating vincristine, doxorubicin, cyclophosphamide, and ifosfamide/etoposide combined with adequate local-control measures applied to both primary and metastatic sites often results in complete or partial responses; however, the overall cure rate is 20%.[5-7]
The following chemotherapy regimens have not shown benefit:
- In the Intergroup Ewing Sarcoma Study, patients with metastatic disease showed no benefit from the addition of ifosfamide and etoposide to a standard regimen of vincristine, doxorubicin, cyclophosphamide, and dactinomycin.[7]
- In another Intergroup study, increasing dose intensity of cyclophosphamide, ifosfamide, and doxorubicin did not improve outcome compared with regimens utilizing standard-dose intensity. This regimen increased toxicity and risk of second malignancy without improving EFS or OS.[2]
- Intensification of ifosfamide to 2.8 g/m2 per day for 5 days did not improve outcome when administered with standard chemotherapy in patients with newly diagnosed metastatic Ewing sarcoma.[8][Level of evidence: 3iiiDi]
Surgery and radiation therapy
Systematic use of surgery and radiation therapy for metastatic sites may improve overall outcome in patients with extrapulmonary metastases.
Evidence (surgery and radiation therapy):
- In a retrospective data analysis of 120 patients with multifocal metastatic Ewing sarcoma, patients receiving local treatment of both primary tumor and metastases had a better outcome than patients receiving local treatment of primary tumor only or with no local treatment (3-year EFS, 39% vs. 17% and 14%, P < .001).[9]
- A similar trend for better outcome with irradiation of all sites of metastatic disease was seen in three retrospective analyses of smaller groups of patients receiving radiation therapy to all tumor sites.[10-12] These results must be interpreted with caution. The patients who received local-control therapy to all known sites of metastatic disease were selected by the treating investigator, not randomly assigned. Patients with so many metastases that radiation to all sites would result in bone marrow failure were not selected to receive radiation to all sites of metastatic disease. Patients who did not achieve control of the primary tumor did not go on to have local control of all sites of metastatic disease. There was a selection bias such that while all patients in these reports had multiple sites of metastatic disease, the patients who had surgery and/or radiation therapy to all sites of clinically detectable metastatic disease had better responses to systemic therapy and fewer sites of metastasis than did patients who did not undergo similar therapy of metastatic sites.
Radiation therapy, delivered in a setting in which stringent planning techniques are applied by those experienced in the treatment of Ewing sarcoma, should be considered. Such an approach will result in local control of tumor with acceptable morbidity in most patients.[13]
The radiation dose depends on the metastatic site of disease:
- Bone and soft tissue. Stereotactic body radiation therapy has been used to treat metastatic sites in bone and soft tissue. The median total curative/definitive stereotactic body radiation therapy dose delivered was 40 Gy in five fractions (range, 30–60 Gy in 3–10 fractions). The median total palliative stereotactic body radiation therapy dose delivered was 40 Gy in five fractions (range, 16–50 Gy in 1–10 fractions). These short-course regimens with large-dose fractions are biologically equivalent to higher doses delivered with smaller-dose fractions given over longer treatment courses.[14][Level of evidence: 3iiiC]
- Pulmonary. For all patients with pulmonary metastases, whole-lung irradiation should be considered, even if complete resolution of overt pulmonary metastatic disease has been achieved with chemotherapy.[4,5,15] Radiation doses are modulated based on the amount of lung to be irradiated and on pulmonary function. Doses between 12 Gy and 15 Gy are generally used if whole lungs are treated.
Other therapies
More intensive therapies, many of which incorporate high-dose chemotherapy with or without total-body irradiation in conjunction with stem cell support, have not shown improvement in EFS rates for patients with bone and/or bone marrow metastases.[2,3,10,16-18]; [19][Level of evidence: 3iiiDi] (Refer to the High-Dose Therapy With Stem Cell Rescue for Ewing Sarcoma section of this summary for more information.)
- High-dose chemotherapy with stem cell support. One of the largest studies was the EURO-EWING-Intergroup-EE99 R3 trial that enrolled 281 patients with primary disseminated metastatic Ewing sarcoma. Patients were treated with six cycles of vincristine, ifosfamide, doxorubicin, and etoposide followed by high-dose therapy and autologous stem cell transplant and demonstrated a 3-year EFS of 27% and OS of 34%. Factors such as the presence and number of bone lesions, primary tumor volume greater than 200 mL, age older than 14 years, additional pulmonary metastases, and bone marrow involvement were identified as independent prognostic factors.[3][Level of evidence: 3iiDi] The impact of high-dose chemotherapy with peripheral blood stem cell support for patients with isolated lung metastases is unknown and is being studied in the EURO-EWING-INTERGROUP-EE99 trial, for which results are pending.[16]
- Melphalan. Melphalan, at nonmyeloablative doses, proved to be an active agent in an upfront window study for patients with metastatic disease at diagnosis; however, the cure rate remained extremely low.[20]
- Irinotecan. Irinotecan was administered as a single agent in an upfront window for newly diagnosed metastatic Ewing sarcoma patients and showed modest activity (partial response in 5 of 24 patients).[21][Level of evidence: 3iiiDiv] Further investigation is needed to determine irinotecan dosing and combinations with other agents for patients with Ewing sarcoma.
Treatment Options Under Clinical Evaluation for Metastatic Ewing Sarcoma
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- AEWS1221; NCI-2014-02380 (NCT02306161) (Combination Chemotherapy With or Without Ganitumab in Treating Patients With Newly Diagnosed Metastatic Ewing Sarcoma): This phase II study is randomly assigning newly diagnosed patients with metastatic Ewing sarcoma to multiagent chemotherapy (vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide) with or without the addition of ganitumab (AMG 479). Stereotactic body radiation therapy is being evaluated to sites of bone metastases at a dose of 40 Gy in five fractions. This is a shorter course of therapy than is the standard treatment.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Esiashvili N, Goodman M, Marcus RB Jr: Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J Pediatr Hematol Oncol 30 (6): 425-30, 2008. [PUBMED Abstract]
- Miser JS, Goldsby RE, Chen Z, et al.: Treatment of metastatic Ewing sarcoma/primitive neuroectodermal tumor of bone: evaluation of increasing the dose intensity of chemotherapy--a report from the Children's Oncology Group. Pediatr Blood Cancer 49 (7): 894-900, 2007. [PUBMED Abstract]
- Ladenstein R, Pötschger U, Le Deley MC, et al.: Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol 28 (20): 3284-91, 2010. [PUBMED Abstract]
- Paulussen M, Ahrens S, Craft AW, et al.: Ewing's tumors with primary lung metastases: survival analysis of 114 (European Intergroup) Cooperative Ewing's Sarcoma Studies patients. J Clin Oncol 16 (9): 3044-52, 1998. [PUBMED Abstract]
- Paulussen M, Ahrens S, Burdach S, et al.: Primary metastatic (stage IV) Ewing tumor: survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma Studies. Ann Oncol 9 (3): 275-81, 1998. [PUBMED Abstract]
- Pinkerton CR, Bataillard A, Guillo S, et al.: Treatment strategies for metastatic Ewing's sarcoma. Eur J Cancer 37 (11): 1338-44, 2001. [PUBMED Abstract]
- Miser JS, Krailo MD, Tarbell NJ, et al.: Treatment of metastatic Ewing's sarcoma or primitive neuroectodermal tumor of bone: evaluation of combination ifosfamide and etoposide--a Children's Cancer Group and Pediatric Oncology Group study. J Clin Oncol 22 (14): 2873-6, 2004. [PUBMED Abstract]
- Magnan H, Goodbody CM, Riedel E, et al.: Ifosfamide dose-intensification for patients with metastatic Ewing sarcoma. Pediatr Blood Cancer 62 (4): 594-7, 2015. [PUBMED Abstract]
- Haeusler J, Ranft A, Boelling T, et al.: The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES). Cancer 116 (2): 443-50, 2010. [PUBMED Abstract]
- Burdach S, Thiel U, Schöniger M, et al.: Total body MRI-governed involved compartment irradiation combined with high-dose chemotherapy and stem cell rescue improves long-term survival in Ewing tumor patients with multiple primary bone metastases. Bone Marrow Transplant 45 (3): 483-9, 2010. [PUBMED Abstract]
- Paulino AC, Mai WY, Teh BS: Radiotherapy in metastatic ewing sarcoma. Am J Clin Oncol 36 (3): 283-6, 2013. [PUBMED Abstract]
- Casey DL, Wexler LH, Meyers PA, et al.: Radiation for bone metastases in Ewing sarcoma and rhabdomyosarcoma. Pediatr Blood Cancer 62 (3): 445-9, 2015. [PUBMED Abstract]
- Donaldson SS, Torrey M, Link MP, et al.: A multidisciplinary study investigating radiotherapy in Ewing's sarcoma: end results of POG #8346. Pediatric Oncology Group. Int J Radiat Oncol Biol Phys 42 (1): 125-35, 1998. [PUBMED Abstract]
- Brown LC, Lester RA, Grams MP, et al.: Stereotactic body radiotherapy for metastatic and recurrent ewing sarcoma and osteosarcoma. Sarcoma 2014: 418270, 2014. [PUBMED Abstract]
- Spunt SL, McCarville MB, Kun LE, et al.: Selective use of whole-lung irradiation for patients with Ewing sarcoma family tumors and pulmonary metastases at the time of diagnosis. J Pediatr Hematol Oncol 23 (2): 93-8, 2001. [PUBMED Abstract]
- Meyers PA, Krailo MD, Ladanyi M, et al.: High-dose melphalan, etoposide, total-body irradiation, and autologous stem-cell reconstitution as consolidation therapy for high-risk Ewing's sarcoma does not improve prognosis. J Clin Oncol 19 (11): 2812-20, 2001. [PUBMED Abstract]
- Burdach S, Meyer-Bahlburg A, Laws HJ, et al.: High-dose therapy for patients with primary multifocal and early relapsed Ewing's tumors: results of two consecutive regimens assessing the role of total-body irradiation. J Clin Oncol 21 (16): 3072-8, 2003. [PUBMED Abstract]
- Thiel U, Wawer A, Wolf P, et al.: No improvement of survival with reduced- versus high-intensity conditioning for allogeneic stem cell transplants in Ewing tumor patients. Ann Oncol 22 (7): 1614-21, 2011. [PUBMED Abstract]
- Loschi S, Dufour C, Oberlin O, et al.: Tandem high-dose chemotherapy strategy as first-line treatment of primary disseminated multifocal Ewing sarcomas in children, adolescents and young adults. Bone Marrow Transplant 50 (8): 1083-8, 2015. [PUBMED Abstract]
- Luksch R, Grignani G, Fagioli F, et al.: Response to melphalan in up-front investigational window therapy for patients with metastatic Ewing's family tumours. Eur J Cancer 43 (5): 885-90, 2007. [PUBMED Abstract]
- Morland B, Platt K, Whelan JS: A phase II window study of irinotecan (CPT-11) in high risk Ewing sarcoma: a Euro-E.W.I.N.G. study. Pediatr Blood Cancer 61 (3): 442-5, 2014. [PUBMED Abstract]
Treatment of Recurrent Ewing Sarcoma
Recurrence of Ewing sarcoma is most common within 2 years of initial diagnosis (approximately 80%).[1,2] However, late relapses occurring more than 5 years from initial diagnosis are more common in Ewing sarcoma (13%; 95% confidence interval, 9.4–16.5) than in other pediatric solid tumors.[3] An analysis of the Surveillance, Epidemiology, and End Results database identified 1,351 patients who survived more than 60 months from diagnosis.[4] Of these patients, 209 died, with 144 of the deaths (69%) attributed to recurrent, progressive Ewing sarcoma. Black race, male sex, older age at initial diagnosis, and primary tumors of the pelvis and axial skeleton were associated with a higher risk of late death. This analysis covered the period from 1973 to 2013, and the 1,351 patients represented only 38% of the patients in the original sample, which reflects the inferior treatment outcomes from the earlier era. It is possible that patients who reach the 5-year point after more contemporary treatment may not recapitulate this experience.
The overall prognosis for patients with recurrent Ewing sarcoma is poor; 5-year survival after recurrence is approximately 10% to 15%.[2,5,6]; [1][Level of evidence: 3iiA]
Prognostic factors include the following:
- Time to recurrence. Time to recurrence is the most important prognostic factor. Patients whose Ewing sarcoma recurred more than 2 years from initial diagnosis had a 5-year survival of 30% versus 7% for patients whose Ewing sarcoma recurred within 2 years.[1,2]
- Local and distant recurrence. Patients with both local recurrence and distant metastases have a worse outcome than do patients with either isolated local recurrence or metastatic recurrence alone.[1,2]
- Isolated pulmonary recurrence. Isolated pulmonary recurrence was not an important prognostic factor in a North American series.[1] In the Italian/Scandinavian experience, younger age, longer disease-free interval, and lung-only recurrence were associated with longer progression-free survival after recurrence. In this experience, patients with Ewing sarcoma that recurred after initial therapy, which included high-dose therapy with autologous stem cell rescue, were less likely to achieve a second complete remission.[7][Level of evidence: 3iiDiii]
Treatment Options for Recurrent Ewing Sarcoma
The selection of treatment for patients with recurrent disease depends on many factors, including the following:
- Site of recurrence.
- Previous treatment.
- Individual patient considerations.
There is no standardized second-line treatment for relapsed or refractory Ewing sarcoma.
Treatment options for recurrent Ewing sarcoma include the following:
Chemotherapy
Combinations of chemotherapy, such as cyclophosphamide and topotecan or irinotecan and temozolomide with or without vincristine, are active in recurrent Ewing sarcoma and can be considered for these patients.[8-13]
Evidence (chemotherapy):
- One phase II study of topotecan and cyclophosphamide showed a response in 6 of 17 patients with Ewing sarcoma; 16 of 49 patients had a clinical response in a similar trial in Germany.[8,10]
- In one retrospective series, 20 patients received temozolomide and irinotecan after recurrence. Five patients achieved a complete response and seven patients achieved a partial response.[12] A second retrospective series reported 11 of 20 objective responses in patients with recurrent Ewing sarcoma.[14][Level of evidence: 3iiDiv]
- The combination of docetaxel either with gemcitabine or irinotecan has achieved objective responses in relapsed Ewing sarcoma.[15][Level of evidence: 3iiA]; [16,17][Level of evidence: 3iiiDiv]
- High-dose ifosfamide (3 g/m2 per day for 5 days = 15 g/m2) has shown activity in patients whose Ewing sarcoma recurred after therapy that included standard ifosfamide (1.8 g/m2 per day for 5 days = 9 g/m2).[18][Level of evidence: 3iiiDiv]
Radiation therapy
Radiation therapy to bone lesions may provide palliation, although radical resection may improve outcome.[2] Patients with pulmonary metastases who have not received radiation therapy to the lungs should be considered for whole-lung irradiation.[19] Residual disease in the lung may be surgically removed.
Other therapies
Other therapies that have been studied in the treatment of recurrent Ewing sarcoma include the following:
- High-dose chemotherapy with stem cell support. Aggressive attempts to control the disease, including myeloablative regimens, have been used, but there is no evidence at this time to conclude that myeloablative therapy is superior to standard chemotherapy.[20,21]; [22][Level of evidence: 3iiA]; [23][Level of evidence: 3iiiDiii]
Most published reports about the use of high-dose therapy and stem cell support for patients with high-risk Ewing sarcoma have significant flaws in methodology. The most common error is the comparison of this high-risk group with an inappropriate control group. Patients with Ewing sarcoma at high risk of treatment failure who received high-dose therapy are compared with patients who did not receive high-dose therapy. Patients who undergo high-dose therapy must respond to systemic therapy, remain alive and respond to treatment long enough to reach the time at which stem cell therapy can be applied, be free of comorbid toxicity that precludes high-dose therapy, and have an adequate stem cell collection. Patients who undergo high-dose therapy and stem cell support are a highly selected group; comparing this patient group with all patients with high-risk Ewing sarcoma is inappropriate and leads to the erroneous conclusion that this strategy improves outcome. Surveys of patients undergoing allogeneic stem cell transplantation (SCT) for recurrent Ewing sarcoma did not show improved event-free survival when compared with autologous SCT and was associated with a higher complication rate.[20,24,25]
- Monoclonal antibody therapy. Monoclonal antibodies against the insulin-like growth factor 1 receptor (IGF1R) are reported to produce objective responses in metastatic recurrent Ewing sarcoma in roughly 10% of cases.[26-29][Level of evidence: 3iiDiv] In these studies, it was suggested that time-to-progression was prolonged compared with historical controls. Objective responses have been reported in studies combining the mTOR inhibitor temsirolimus with an IGF1R antibody. Stratification by IGF1R expression by immunohistochemistry in one of the studies did not predict clinical outcome in Ewing sarcoma patients.[30,31] Further studies are needed to identify patients who are likely to benefit from IGF1R therapy.
- Immunotherapy. Immunotherapy with antigen-specific T cells is being studied in patients with Ewing sarcoma because immune-mediated killing does not rely on pathways used by conventional therapies to which such tumors are often resistant. Several potential chimeric antigen receptors target antigens that have been identified for Ewing sarcomas. These include HER2 (human epidermal growth factor receptor 2),[32] GD2,[33] CD99 (MIC2 antigens),[34] and STEAP1 (six-transmembrane epithelial antigens of the prostate).[35] Some are in early-phase testing in sarcoma patients.[32]
Treatment Options Under Clinical Evaluation for Recurrent Ewing Sarcoma
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- APEC1621 (NCT03155620) (Pediatric MATCH: Targeted Therapy Directed by Genetic Testing in Treating Pediatric Patients with Relapsed or Refractory Advanced Solid Tumors, Non-Hodgkin Lymphomas, or Histiocytic Disorders): NCI–Children's Oncology Group Pediatric Molecular Analysis for Therapeutic Choice (MATCH), referred to as Pediatric MATCH, will match targeted agents with specific molecular changes identified using a next-generation sequencing targeted assay of more than 3,000 different mutations across more than 160 genes in refractory and recurrent solid tumors. Children and adolescents aged 1 to 21 years are eligible for the trial.
Tumor tissue from progressive or recurrent disease must be available for molecular characterization. Patients with tumors that have molecular variants addressed by treatment arms included in the trial will be offered treatment on Pediatric MATCH. Additional information can be obtained on the ClinicalTrials.gov website for APEC1621 (NCT03155620).
- ADVL1622 (NCT02867592) (Cabozantinib-S-Malate in Treating Younger Patients with Recurrent, Refractory, or Newly Diagnosed Sarcomas, Wilms Tumor, or Other Rare Tumors): This is an open-label, two-stage, phase II trial of cabozantinib in selective solid tumors, including Ewing sarcoma. Cabozantinib is an oral small molecule inhibitor of multiple tyrosine kinases, including MET, VEGFR2, and RET, which are potential therapeutic targets in many pediatric and adult solid tumors.
- SARC028; NCI-2015-00320 (NCT02301039) (A Phase II Study of the Anti-PD1 Antibody Pembrolizumab [MK-3475] in Patients With Advanced Sarcomas): The objective response rate to the anti-PD1 inhibitor pembrolizumab will be assessed in patients with refractory, recurrent, and/or metastatic high-grade soft tissue sarcomas and bone sarcomas. Patients aged 18 years and older with soft tissue sarcomas and patients aged 12 years and older with bone sarcomas are eligible.
- ADVL1412 (NCT02304458) (Nivolumab With or Without Ipilimumab in Treating Younger Patients With Recurrent or Refractory Solid Tumors or Sarcomas): Nivolumab is an anti-PD1 inhibitor that is being studied alone and in combination with ipilimumab in relapsed sarcoma patients, including patients with Ewing sarcoma.
- ADVL1411 (NCT02116777) (BMN-673 and Temozolomide in Treating Younger Patients With Refractory or Recurrent Malignancies): In this study, the PARP inhibitor BMN-673 is combined with low-dose short duration temozolomide. This is based on the in vitro and mouse human tumor xenograft models, which showed impressive activity in a broad range of pediatric cancers, including Ewing sarcoma. After identifying the recommended phase II dose, this study is open for Ewing sarcoma patients.[36]
- ADVL1615 (NCT03323034) (Pevonedistat, Irinotecan Hydrochloride, and Temozolomide in Treating Patients With Recurrent or Refractory Solid Tumors or Lymphoma): This is a phase I study of pevonedistat in combination with temozolomide and irinotecan. Pevonedistat is a novel first-in-class Nedd8 activating enzyme (NAE) inhibitor that blocks the degradation of a subset of proteins that would normally be degraded by the 26S proteasome. Pevonedistat is more specific than previous proteasome inhibitors because it blocks the degradation of cullin-RING ligases, narrowing the targets to only a handful of key regulatory proteins important in cell survival. Preclinical, antitumor activity has been observed in Ewing sarcoma.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Leavey PJ, Mascarenhas L, Marina N, et al.: Prognostic factors for patients with Ewing sarcoma (EWS) at first recurrence following multi-modality therapy: A report from the Children's Oncology Group. Pediatr Blood Cancer 51 (3): 334-8, 2008. [PUBMED Abstract]
- Stahl M, Ranft A, Paulussen M, et al.: Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatr Blood Cancer 57 (4): 549-53, 2011. [PUBMED Abstract]
- Wasilewski-Masker K, Liu Q, Yasui Y, et al.: Late recurrence in pediatric cancer: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst 101 (24): 1709-20, 2009. [PUBMED Abstract]
- Davenport JR, Vo KT, Goldsby R, et al.: Conditional Survival and Predictors of Late Death in Patients With Ewing Sarcoma. Pediatr Blood Cancer 63 (6): 1091-5, 2016. [PUBMED Abstract]
- Barker LM, Pendergrass TW, Sanders JE, et al.: Survival after recurrence of Ewing's sarcoma family of tumors. J Clin Oncol 23 (19): 4354-62, 2005. [PUBMED Abstract]
- Bacci G, Longhi A, Ferrari S, et al.: Pattern of relapse in 290 patients with nonmetastatic Ewing's sarcoma family tumors treated at a single institution with adjuvant and neoadjuvant chemotherapy between 1972 and 1999. Eur J Surg Oncol 32 (9): 974-9, 2006. [PUBMED Abstract]
- Ferrari S, Luksch R, Hall KS, et al.: Post-relapse survival in patients with Ewing sarcoma. Pediatr Blood Cancer 62 (6): 994-9, 2015. [PUBMED Abstract]
- Saylors RL 3rd, Stine KC, Sullivan J, et al.: Cyclophosphamide plus topotecan in children with recurrent or refractory solid tumors: a Pediatric Oncology Group phase II study. J Clin Oncol 19 (15): 3463-9, 2001. [PUBMED Abstract]
- McTiernan A, Driver D, Michelagnoli MP, et al.: High dose chemotherapy with bone marrow or peripheral stem cell rescue is an effective treatment option for patients with relapsed or progressive Ewing's sarcoma family of tumours. Ann Oncol 17 (8): 1301-5, 2006. [PUBMED Abstract]
- Hunold A, Weddeling N, Paulussen M, et al.: Topotecan and cyclophosphamide in patients with refractory or relapsed Ewing tumors. Pediatr Blood Cancer 47 (6): 795-800, 2006. [PUBMED Abstract]
- Wagner LM, McAllister N, Goldsby RE, et al.: Temozolomide and intravenous irinotecan for treatment of advanced Ewing sarcoma. Pediatr Blood Cancer 48 (2): 132-9, 2007. [PUBMED Abstract]
- Casey DA, Wexler LH, Merchant MS, et al.: Irinotecan and temozolomide for Ewing sarcoma: the Memorial Sloan-Kettering experience. Pediatr Blood Cancer 53 (6): 1029-34, 2009. [PUBMED Abstract]
- Raciborska A, Bilska K, Drabko K, et al.: Vincristine, irinotecan, and temozolomide in patients with relapsed and refractory Ewing sarcoma. Pediatr Blood Cancer 60 (10): 1621-5, 2013. [PUBMED Abstract]
- Kurucu N, Sari N, Ilhan IE: Irinotecan and temozolamide treatment for relapsed Ewing sarcoma: a single-center experience and review of the literature. Pediatr Hematol Oncol 32 (1): 50-9, 2015. [PUBMED Abstract]
- Fox E, Patel S, Wathen JK, et al.: Phase II study of sequential gemcitabine followed by docetaxel for recurrent Ewing sarcoma, osteosarcoma, or unresectable or locally recurrent chondrosarcoma: results of Sarcoma Alliance for Research Through Collaboration Study 003. Oncologist 17 (3): 321, 2012. [PUBMED Abstract]
- Mora J, Cruz CO, Parareda A, et al.: Treatment of relapsed/refractory pediatric sarcomas with gemcitabine and docetaxel. J Pediatr Hematol Oncol 31 (10): 723-9, 2009. [PUBMED Abstract]
- Yoon JH, Kwon MM, Park HJ, et al.: A study of docetaxel and irinotecan in children and young adults with recurrent or refractory Ewing sarcoma family of tumors. BMC Cancer 14: 622, 2014. [PUBMED Abstract]
- Ferrari S, del Prever AB, Palmerini E, et al.: Response to high-dose ifosfamide in patients with advanced/recurrent Ewing sarcoma. Pediatr Blood Cancer 52 (5): 581-4, 2009. [PUBMED Abstract]
- Rodriguez-Galindo C, Billups CA, Kun LE, et al.: Survival after recurrence of Ewing tumors: the St Jude Children's Research Hospital experience, 1979-1999. Cancer 94 (2): 561-9, 2002. [PUBMED Abstract]
- Burdach S, van Kaick B, Laws HJ, et al.: Allogeneic and autologous stem-cell transplantation in advanced Ewing tumors. An update after long-term follow-up from two centers of the European Intergroup study EICESS. Stem-Cell Transplant Programs at Düsseldorf University Medical Center, Germany and St. Anna Kinderspital, Vienna, Austria. Ann Oncol 11 (11): 1451-62, 2000. [PUBMED Abstract]
- Burdach S, Meyer-Bahlburg A, Laws HJ, et al.: High-dose therapy for patients with primary multifocal and early relapsed Ewing's tumors: results of two consecutive regimens assessing the role of total-body irradiation. J Clin Oncol 21 (16): 3072-8, 2003. [PUBMED Abstract]
- Rasper M, Jabar S, Ranft A, et al.: The value of high-dose chemotherapy in patients with first relapsed Ewing sarcoma. Pediatr Blood Cancer 61 (8): 1382-6, 2014. [PUBMED Abstract]
- Gardner SL, Carreras J, Boudreau C, et al.: Myeloablative therapy with autologous stem cell rescue for patients with Ewing sarcoma. Bone Marrow Transplant 41 (10): 867-72, 2008. [PUBMED Abstract]
- Gilman AL, Oesterheld J: Myeloablative chemotherapy with autologous stem cell rescue for Ewing sarcoma. Bone Marrow Transplant 42 (11): 761; author reply 763, 2008. [PUBMED Abstract]
- Eapen M: Response to Dr Gilman. Bone Marrow Transplant 42 (11): 763, 2008.
- Malempati S, Weigel B, Ingle AM, et al.: Phase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 30 (3): 256-62, 2012. [PUBMED Abstract]
- Juergens H, Daw NC, Geoerger B, et al.: Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J Clin Oncol 29 (34): 4534-40, 2011. [PUBMED Abstract]
- Pappo AS, Patel SR, Crowley J, et al.: R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research through Collaboration study. J Clin Oncol 29 (34): 4541-7, 2011. [PUBMED Abstract]
- Tap WD, Demetri G, Barnette P, et al.: Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J Clin Oncol 30 (15): 1849-56, 2012. [PUBMED Abstract]
- Naing A, LoRusso P, Fu S, et al.: Insulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing's sarcoma family tumors. Clin Cancer Res 18 (9): 2625-31, 2012. [PUBMED Abstract]
- Schwartz GK, Tap WD, Qin LX, et al.: Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: a multicentre, open-label, phase 2 trial. Lancet Oncol 14 (4): 371-82, 2013. [PUBMED Abstract]
- Ahmed N, Brawley VS, Hegde M, et al.: Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol 33 (15): 1688-96, 2015. [PUBMED Abstract]
- Pule MA, Savoldo B, Myers GD, et al.: Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 14 (11): 1264-70, 2008. [PUBMED Abstract]
- Scotlandi K, Baldini N, Cerisano V, et al.: CD99 engagement: an effective therapeutic strategy for Ewing tumors. Cancer Res 60 (18): 5134-42, 2000. [PUBMED Abstract]
- Grunewald TG, Diebold I, Esposito I, et al.: STEAP1 is associated with the invasive and oxidative stress phenotype of Ewing tumors. Mol Cancer Res 10 (1): 52-65, 2012. [PUBMED Abstract]
- Smith MA, Reynolds CP, Kang MH, et al.: Synergistic activity of PARP inhibition by talazoparib (BMN 673) with temozolomide in pediatric cancer models in the pediatric preclinical testing program. Clin Cancer Res 21 (4): 819-32, 2015. [PUBMED Abstract]
Late Effects of Treatment for Ewing Sarcoma
Patients treated for Ewing sarcoma have a significantly higher risk of developing subsequent neoplasms than do patients in the general population.
Treatment-related acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) have generally been reported to occur in 1% to 2% of survivors of Ewing sarcoma,[1]; [2][Level of evidence: 3iiiDi] although some dose-intensive regimens appear to be associated with a higher risk of hematological malignancy.[3,4]; [5][Level of evidence: 3ii] Treatment-related AML and MDS arise most commonly at 2 to 5 years after diagnosis.
Survivors of Ewing sarcoma remain at increased risk of developing a subsequent solid tumor throughout their lifetime. Sarcomas usually occur within the previous radiation field.[6,7] The risk of developing a sarcoma after radiation therapy is dose-dependent, with higher doses associated with an increased risk of sarcoma development.[1]; [2][Level of evidence: 3iiiDi] The cumulative incidence of subsequent neoplasms in children treated for Ewing sarcoma between 1970 and 1986 at 25 years after diagnosis was 9.0% (confidence interval, 5.8–12.2). Most of these patients received radiation therapy; comparable long-term data do not yet exist for significant numbers of patients who did not receive radiation therapy.[8]
(Refer to the PDQ summary on Late Effects of Treatment for Childhood Cancer for a full discussion of the late effects of cancer treatment in children and adolescents.)
References
- Fuchs B, Valenzuela RG, Petersen IA, et al.: Ewing's sarcoma and the development of secondary malignancies. Clin Orthop (415): 82-9, 2003. [PUBMED Abstract]
- Goldsby R, Burke C, Nagarajan R, et al.: Second solid malignancies among children, adolescents, and young adults diagnosed with malignant bone tumors after 1976: follow-up of a Children's Oncology Group cohort. Cancer 113 (9): 2597-604, 2008. [PUBMED Abstract]
- Bhatia S, Krailo MD, Chen Z, et al.: Therapy-related myelodysplasia and acute myeloid leukemia after Ewing sarcoma and primitive neuroectodermal tumor of bone: A report from the Children's Oncology Group. Blood 109 (1): 46-51, 2007. [PUBMED Abstract]
- Kushner BH, Heller G, Cheung NK, et al.: High risk of leukemia after short-term dose-intensive chemotherapy in young patients with solid tumors. J Clin Oncol 16 (9): 3016-20, 1998. [PUBMED Abstract]
- Navid F, Billups C, Liu T, et al.: Second cancers in patients with the Ewing sarcoma family of tumours. Eur J Cancer 44 (7): 983-91, 2008. [PUBMED Abstract]
- Kuttesch JF Jr, Wexler LH, Marcus RB, et al.: Second malignancies after Ewing's sarcoma: radiation dose-dependency of secondary sarcomas. J Clin Oncol 14 (10): 2818-25, 1996. [PUBMED Abstract]
- Hawkins MM, Wilson LM, Burton HS, et al.: Radiotherapy, alkylating agents, and risk of bone cancer after childhood cancer. J Natl Cancer Inst 88 (5): 270-8, 1996. [PUBMED Abstract]
- Ginsberg JP, Goodman P, Leisenring W, et al.: Long-term survivors of childhood Ewing sarcoma: report from the childhood cancer survivor study. J Natl Cancer Inst 102 (16): 1272-83, 2010. [PUBMED Abstract]
Changes to This Summary (04/04/2018)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Treatment of Recurrent Ewing Sarcoma
Added text about the ADVL1622 and ADVL1615 clinical trials as treatment options under clinical evaluation for patients with recurrent Ewing sarcoma.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of childhood Ewing sarcoma. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Ewing Sarcoma Treatment are:
- Holcombe Edwin Grier, MD (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Andrea A. Hayes-Jordan, MD, FACS, FAAP (M.D. Anderson Cancer Center)
- Karen J. Marcus, MD (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Paul A. Meyers, MD (Memorial Sloan-Kettering Cancer Center)
- Thomas A. Olson, MD (Aflac Cancer and Blood Disorders Center of Children's Healthcare of Atlanta - Egleston Campus)
- Nita Louise Seibel, MD (National Cancer Institute)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Ewing Sarcoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/bone/hp/ewing-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389480]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Updated: April 4, 2018
Source URL: https://www.cancer.gov/publishedcontent/syndication/2434.htm
Source Agency: National Cancer Institute (NCI)
Captured Date: 2013-09-14 09:00:56.0
Ewing Sarcoma Treatment (PDQ®)–Health Professional Version
General Information About Ewing Sarcoma
Dramatic improvements in survival have been achieved for children and adolescents with cancer.[1] Between 1975 and 2010, childhood cancer mortality decreased by more than 50%.[1] For Ewing sarcoma, the 5-year survival rate has increased over the same time from 59% to 78% for children younger than 15 years and from 20% to 60% for adolescents aged 15 to 19 years.[1]
Studies using immunohistochemical markers,[2] cytogenetics,[3,4] molecular genetics, and tissue culture [5] indicate that Ewing sarcoma is derived from a primordial bone marrow–derived mesenchymal stem cell.[6,7] Older terms such as peripheral primitive neuroectodermal tumor, Askin tumor (Ewing sarcoma of chest wall), and extraosseous Ewing sarcoma (often combined in the term Ewing sarcoma family of tumors) refer to this same tumor.
Incidence
The incidence of Ewing sarcoma has remained unchanged for 30 years.[8] The incidence for all ages is one case per 1 million people in the United States. In patients aged 10 to 19 years, the incidence is between nine and ten cases per 1 million people. The same analysis suggests that the incidence of Ewing sarcoma in the United States is nine times greater in whites than in African Americans, with an intermediate incidence in Asians.[9,10]
The relative paucity of Ewing sarcoma in people of African or Asian descent may be explained, in part, by a specific polymorphism in the EGR2 gene.
The median age of patients with Ewing sarcoma is 15 years, and more than 50% of patients are adolescents. Well-characterized cases of Ewing sarcoma in neonates and infants have been described.[11,12] Based on data from 1,426 patients entered on European Intergroup Cooperative Ewing Sarcoma Studies, 59% of patients are male and 41% are female.[13]
Clinical Presentation
Primary sites of bone disease include the following:
- Lower extremity (41%).
- Pelvis (26%).
- Chest wall (16%).
- Upper extremity (9%).
- Spine (6%).
- Hand and foot (3%).[14]
- Skull (2%).
For extraosseous primary tumors, the most common primary sites of disease include the following:[15,16]
- Trunk (32%).
- Extremity (26%).
- Head and neck (18%).
- Retroperitoneum (16%).
- Other sites (9%).
The median time from first symptom to diagnosis of Ewing sarcoma is often long, with a median interval reported from 2 to 5 months. Longer times are associated with older age and pelvic primary sites. This has not been associated with metastasis, surgical outcome, or survival.[17] Approximately 25% of patients with Ewing sarcoma have metastatic disease at the time of diagnosis.[8]
The Surveillance, Epidemiology, and End Results (SEER) database was used to compare patients younger than 40 years with Ewing sarcoma who presented with skeletal and extraosseous primary sites (refer to Table 1).[18] Patients with extraosseous Ewing sarcoma were more likely to be older, female, nonwhite, and have axial primary sites, and were less likely to have pelvic primary sites than were patients with skeletal Ewing sarcoma.
| Characteristic | Extraosseous Ewing Sarcoma | Skeletal Ewing Sarcoma | P Value |
|---|---|---|---|
| Mean age (range), years | 20 (0–39) | 16 (0–39) | <.001 |
| Male | 53% | 63% | <.001 |
| White | 85% | 93% | <.001 |
| Axial primary sites | 73% | 54% | <.001 |
| Pelvic primary sites | 20% | 27% | .001 |
Diagnostic Evaluation
The following tests and procedures may be used to diagnose or stage Ewing sarcoma:
- Physical exam and history.
- Magnetic resonance imaging (MRI).
- Computed tomography (CT) scan.
- Positron emission tomography (PET) scan.
- Bone scan.
- Bone marrow aspiration and biopsy.
- X-ray.
- Complete blood count.
- Blood chemistry studies, such as lactate dehydrogenase (LDH).
Prognostic Factors
The two major types of prognostic factors for patients with Ewing sarcoma are grouped as follows:
Pretreatment factors
- Site of tumor: Patients with Ewing sarcoma in the distal extremities have the best prognosis. Patients with Ewing sarcoma in the proximal extremities have an intermediate prognosis, followed by patients with central or pelvic sites.[19-22]
- Extraskeletal versus skeletal primary tumors: The Children's Oncology Group performed a retrospective analysis from two large cooperative trials that used similar treatment regimens.[23] They identified 213 patients with extraskeletal primary tumors and 826 patients with skeletal primary tumors. Patients with extraskeletal primary tumors were more likely to have an axial primary site, less likely to have large primary tumors, and had a statistically significant better prognosis than did patients with skeletal primary tumors.
- Tumor size or volume: Tumor size or volume has been shown to be an important prognostic factor in most studies. Cutoffs of a volume of 100 mL or 200 mL and/or single dimension greater than 8 cm are used to define larger tumors. Larger tumors tend to occur in unfavorable sites.[21,22,24]
- Age: Infants and younger patients have a better prognosis than do patients aged 15 years and older.[12,19,20,22,25,26]
In North American studies, patients younger than 10 years have a better outcome than those aged 10 to 17 years at diagnosis (relative risk [RR], 1.4). Patients older than 18 years have an inferior outcome (RR, 2.5).[27-29] A retrospective review of two consecutive German trials for Ewing sarcoma identified 47 patients older than 40 years.[30] With adequate multimodal therapy, survival was comparable to the survival observed in adolescents treated on the same trials. Review of the SEER database from 1973 to 2011 identified 1,957 patients with Ewing sarcoma.[31] Thirty-nine of these patients (2.0%) were younger than 12 months at diagnosis. Infants were less likely to receive radiation therapy and more likely to have soft tissue primary sites. Early death was more common in infants, but the overall survival (OS) did not differ significantly from that of older patients.
- Sex: Girls with Ewing sarcoma have a better prognosis than do boys with Ewing sarcoma.[9,20,22]
- Serum LDH: Increased serum LDH levels before treatment are associated with inferior prognosis. Increased LDH levels are also correlated with large primary tumors and metastatic disease.[20]
- Metastases: Any metastatic disease defined by standard imaging techniques or bone marrow aspirate/biopsy by morphology is an adverse prognostic factor. The presence or absence of metastatic disease is the single most powerful predictor of outcome. Metastases at diagnosis are detected in about 25% of patients.[8]
Patients with metastatic disease confined to the lung have a better prognosis than do patients with extrapulmonary metastatic sites.[19,21,22,32] The number of pulmonary lesions does not seem to correlate with outcome, but patients with unilateral lung involvement do better than patients with bilateral lung involvement.[33]
Patients with metastasis to only bone seem to have a better outcome than do patients with metastases to both bone and lung.[34,35]
Based on an analysis from the SEER database, regional lymph node involvement in patients is associated with an inferior overall outcome when compared with patients without regional lymph node involvement.[36]
- Previous treatment for cancer: In the SEER database, 58 patients with Ewing sarcoma who were diagnosed after treatment for a previous malignancy (2.1% of patients with Ewing sarcoma) were compared with 2,756 patients with Ewing sarcoma as a first cancer over the same period. Patients with Ewing sarcoma as a second malignant neoplasm were older (secondary Ewing sarcoma, mean age of 47.8 years; primary Ewing sarcoma, mean age of 22.5 years), more likely to have a primary tumor in an axial or extraskeletal site, and had a worse prognosis (5-year OS for patients with secondary Ewing sarcoma, 43.5%; patients with primary Ewing sarcoma, 64.2%).[37]
- Standard cytogenetics: Complex karyotype (defined as the presence of five or more independent chromosome abnormalities at diagnosis) and modal chromosome numbers lower than 50 appear to have adverse prognostic significance.[38]
- Detectable fusion transcripts in morphologically normal marrow: Reverse transcriptase polymerase chain reaction can be used to detect fusion transcripts in bone marrow. In a single retrospective study utilizing patients with normal marrow morphology and no other metastatic site, fusion transcript detection in marrow or peripheral blood was associated with an increased risk of relapse.[39]
- Other biological factors: Overexpression of the p53 protein, Ki67 expression, and loss of 16q may be adverse prognostic factors.[40-42] High expression of microsomal glutathione S-transferase, an enzyme associated with resistance to doxorubicin, is associated with inferior outcome for Ewing sarcoma.[43]
The Children's Oncology Group performed a prospective analysis of TP53 mutations and/or CDKN2A deletions in patients with Ewing sarcoma; no correlation was found with event-free survival (EFS).[44]
The following are not considered to be adverse prognostic factors for Ewing sarcoma:
- Pathologic fracture: Pathologic fractures do not appear to be a prognostic factor.[45]
- Histopathology: The degree of neural differentiation is not a prognostic factor in Ewing sarcoma.[46,47]
- Molecular pathology: The EWSR1-ETS translocation associated with Ewing sarcoma can occur at several potential breakpoints in each of the genes that join to form the novel segment of DNA. Once thought to be significant,[48] two large series have shown that the EWSR1-ETS translocation breakpoint site is not an adverse prognostic factor.[49,50]
Response to initial therapy factors
Multiple studies have shown that patients with minimal or no residual viable tumor after presurgical chemotherapy have a significantly better EFS than do patients with larger amounts of viable tumor.[51-54] Female sex and younger age predict a good histologic response to preoperative therapy.[55] For patients who receive preinduction- and postinduction-chemotherapy PET scans, decreased PET uptake after chemotherapy correlated with good histologic response and better outcome.[56-58]
Patients with poor response to presurgical chemotherapy have an increased risk for local recurrence.[59]
References
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014. [PUBMED Abstract]
- Olsen SH, Thomas DG, Lucas DR: Cluster analysis of immunohistochemical profiles in synovial sarcoma, malignant peripheral nerve sheath tumor, and Ewing sarcoma. Mod Pathol 19 (5): 659-68, 2006. [PUBMED Abstract]
- Delattre O, Zucman J, Melot T, et al.: The Ewing family of tumors--a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 331 (5): 294-9, 1994. [PUBMED Abstract]
- Dagher R, Pham TA, Sorbara L, et al.: Molecular confirmation of Ewing sarcoma. J Pediatr Hematol Oncol 23 (4): 221-4, 2001. [PUBMED Abstract]
- Llombart-Bosch A, Carda C, Peydro-Olaya A, et al.: Soft tissue Ewing's sarcoma. Characterization in established cultures and xenografts with evidence of a neuroectodermic phenotype. Cancer 66 (12): 2589-601, 1990. [PUBMED Abstract]
- Suvà ML, Riggi N, Stehle JC, et al.: Identification of cancer stem cells in Ewing's sarcoma. Cancer Res 69 (5): 1776-81, 2009. [PUBMED Abstract]
- Tirode F, Laud-Duval K, Prieur A, et al.: Mesenchymal stem cell features of Ewing tumors. Cancer Cell 11 (5): 421-9, 2007. [PUBMED Abstract]
- Esiashvili N, Goodman M, Marcus RB Jr: Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J Pediatr Hematol Oncol 30 (6): 425-30, 2008. [PUBMED Abstract]
- Jawad MU, Cheung MC, Min ES, et al.: Ewing sarcoma demonstrates racial disparities in incidence-related and sex-related differences in outcome: an analysis of 1631 cases from the SEER database, 1973-2005. Cancer 115 (15): 3526-36, 2009. [PUBMED Abstract]
- Beck R, Monument MJ, Watkins WS, et al.: EWS/FLI-responsive GGAA microsatellites exhibit polymorphic differences between European and African populations. Cancer Genet 205 (6): 304-12, 2012. [PUBMED Abstract]
- Kim SY, Tsokos M, Helman LJ: Dilemmas associated with congenital ewing sarcoma family tumors. J Pediatr Hematol Oncol 30 (1): 4-7, 2008. [PUBMED Abstract]
- van den Berg H, Dirksen U, Ranft A, et al.: Ewing tumors in infants. Pediatr Blood Cancer 50 (4): 761-4, 2008. [PUBMED Abstract]
- Paulussen M, Craft AW, Lewis I, et al.: Results of the EICESS-92 Study: two randomized trials of Ewing's sarcoma treatment--cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J Clin Oncol 26 (27): 4385-93, 2008. [PUBMED Abstract]
- Froeb D, Ranft A, Boelling T, et al.: Ewing sarcoma of the hand or foot. Klin Padiatr 224 (6): 348-52, 2012. [PUBMED Abstract]
- Raney RB, Asmar L, Newton WA Jr, et al.: Ewing's sarcoma of soft tissues in childhood: a report from the Intergroup Rhabdomyosarcoma Study, 1972 to 1991. J Clin Oncol 15 (2): 574-82, 1997. [PUBMED Abstract]
- Rowe RG, Thomas DG, Schuetze SM, et al.: Ewing sarcoma of the kidney: case series and literature review of an often overlooked entity in the diagnosis of primary renal tumors. Urology 81 (2): 347-53, 2013. [PUBMED Abstract]
- Brasme JF, Chalumeau M, Oberlin O, et al.: Time to diagnosis of Ewing tumors in children and adolescents is not associated with metastasis or survival: a prospective multicenter study of 436 patients. J Clin Oncol 32 (18): 1935-40, 2014. [PUBMED Abstract]
- Applebaum MA, Worch J, Matthay KK, et al.: Clinical features and outcomes in patients with extraskeletal Ewing sarcoma. Cancer 117 (13): 3027-32, 2011. [PUBMED Abstract]
- Cotterill SJ, Ahrens S, Paulussen M, et al.: Prognostic factors in Ewing's tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing's Sarcoma Study Group. J Clin Oncol 18 (17): 3108-14, 2000. [PUBMED Abstract]
- Bacci G, Longhi A, Ferrari S, et al.: Prognostic factors in non-metastatic Ewing's sarcoma tumor of bone: an analysis of 579 patients treated at a single institution with adjuvant or neoadjuvant chemotherapy between 1972 and 1998. Acta Oncol 45 (4): 469-75, 2006. [PUBMED Abstract]
- Rodríguez-Galindo C, Liu T, Krasin MJ, et al.: Analysis of prognostic factors in ewing sarcoma family of tumors: review of St. Jude Children's Research Hospital studies. Cancer 110 (2): 375-84, 2007. [PUBMED Abstract]
- Karski EE, McIlvaine E, Segal MR, et al.: Identification of Discrete Prognostic Groups in Ewing Sarcoma. Pediatr Blood Cancer 63 (1): 47-53, 2016. [PUBMED Abstract]
- Cash T, McIlvaine E, Krailo MD, et al.: Comparison of clinical features and outcomes in patients with extraskeletal versus skeletal localized Ewing sarcoma: A report from the Children's Oncology Group. Pediatr Blood Cancer 63 (10): 1771-9, 2016. [PUBMED Abstract]
- Ahrens S, Hoffmann C, Jabar S, et al.: Evaluation of prognostic factors in a tumor volume-adapted treatment strategy for localized Ewing sarcoma of bone: the CESS 86 experience. Cooperative Ewing Sarcoma Study. Med Pediatr Oncol 32 (3): 186-95, 1999. [PUBMED Abstract]
- De Ioris MA, Prete A, Cozza R, et al.: Ewing sarcoma of the bone in children under 6 years of age. PLoS One 8 (1): e53223, 2013. [PUBMED Abstract]
- Huh WW, Daw NC, Herzog CE, et al.: Ewing sarcoma family of tumors in children younger than 10 years of age. Pediatr Blood Cancer 64 (4): , 2017. [PUBMED Abstract]
- Grier HE, Krailo MD, Tarbell NJ, et al.: Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 348 (8): 694-701, 2003. [PUBMED Abstract]
- Granowetter L, Womer R, Devidas M, et al.: Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children's Oncology Group Study. J Clin Oncol 27 (15): 2536-41, 2009. [PUBMED Abstract]
- Womer RB, West DC, Krailo MD, et al.: Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 30 (33): 4148-54, 2012. [PUBMED Abstract]
- Pieper S, Ranft A, Braun-Munzinger G, et al.: Ewing's tumors over the age of 40: a retrospective analysis of 47 patients treated according to the International Clinical Trials EICESS 92 and EURO-E.W.I.N.G. 99. Onkologie 31 (12): 657-63, 2008. [PUBMED Abstract]
- Wong T, Goldsby RE, Wustrack R, et al.: Clinical features and outcomes of infants with Ewing sarcoma under 12 months of age. Pediatr Blood Cancer 62 (11): 1947-51, 2015. [PUBMED Abstract]
- Miser JS, Krailo MD, Tarbell NJ, et al.: Treatment of metastatic Ewing's sarcoma or primitive neuroectodermal tumor of bone: evaluation of combination ifosfamide and etoposide--a Children's Cancer Group and Pediatric Oncology Group study. J Clin Oncol 22 (14): 2873-6, 2004. [PUBMED Abstract]
- Paulussen M, Ahrens S, Craft AW, et al.: Ewing's tumors with primary lung metastases: survival analysis of 114 (European Intergroup) Cooperative Ewing's Sarcoma Studies patients. J Clin Oncol 16 (9): 3044-52, 1998. [PUBMED Abstract]
- Paulussen M, Ahrens S, Burdach S, et al.: Primary metastatic (stage IV) Ewing tumor: survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma Studies. Ann Oncol 9 (3): 275-81, 1998. [PUBMED Abstract]
- Ladenstein R, Pötschger U, Le Deley MC, et al.: Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol 28 (20): 3284-91, 2010. [PUBMED Abstract]
- Applebaum MA, Goldsby R, Neuhaus J, et al.: Clinical features and outcomes in patients with Ewing sarcoma and regional lymph node involvement. Pediatr Blood Cancer 59 (4): 617-20, 2012. [PUBMED Abstract]
- Applebaum MA, Goldsby R, Neuhaus J, et al.: Clinical features and outcomes in patients with secondary Ewing sarcoma. Pediatr Blood Cancer 60 (4): 611-5, 2013. [PUBMED Abstract]
- Roberts P, Burchill SA, Brownhill S, et al.: Ploidy and karyotype complexity are powerful prognostic indicators in the Ewing's sarcoma family of tumors: a study by the United Kingdom Cancer Cytogenetics and the Children's Cancer and Leukaemia Group. Genes Chromosomes Cancer 47 (3): 207-20, 2008. [PUBMED Abstract]
- Schleiermacher G, Peter M, Oberlin O, et al.: Increased risk of systemic relapses associated with bone marrow micrometastasis and circulating tumor cells in localized ewing tumor. J Clin Oncol 21 (1): 85-91, 2003. [PUBMED Abstract]
- Abudu A, Mangham DC, Reynolds GM, et al.: Overexpression of p53 protein in primary Ewing's sarcoma of bone: relationship to tumour stage, response and prognosis. Br J Cancer 79 (7-8): 1185-9, 1999. [PUBMED Abstract]
- López-Guerrero JA, Machado I, Scotlandi K, et al.: Clinicopathological significance of cell cycle regulation markers in a large series of genetically confirmed Ewing's sarcoma family of tumors. Int J Cancer 128 (5): 1139-50, 2011. [PUBMED Abstract]
- Ozaki T, Paulussen M, Poremba C, et al.: Genetic imbalances revealed by comparative genomic hybridization in Ewing tumors. Genes Chromosomes Cancer 32 (2): 164-71, 2001. [PUBMED Abstract]
- Scotlandi K, Remondini D, Castellani G, et al.: Overcoming resistance to conventional drugs in Ewing sarcoma and identification of molecular predictors of outcome. J Clin Oncol 27 (13): 2209-16, 2009. [PUBMED Abstract]
- Lerman DM, Monument MJ, McIlvaine E, et al.: Tumoral TP53 and/or CDKN2A alterations are not reliable prognostic biomarkers in patients with localized Ewing sarcoma: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (5): 759-65, 2015. [PUBMED Abstract]
- Bramer JA, Abudu AA, Grimer RJ, et al.: Do pathological fractures influence survival and local recurrence rate in bony sarcomas? Eur J Cancer 43 (13): 1944-51, 2007. [PUBMED Abstract]
- Parham DM, Hijazi Y, Steinberg SM, et al.: Neuroectodermal differentiation in Ewing's sarcoma family of tumors does not predict tumor behavior. Hum Pathol 30 (8): 911-8, 1999. [PUBMED Abstract]
- Luksch R, Sampietro G, Collini P, et al.: Prognostic value of clinicopathologic characteristics including neuroectodermal differentiation in osseous Ewing's sarcoma family of tumors in children. Tumori 85 (2): 101-7, 1999 Mar-Apr. [PUBMED Abstract]
- de Alava E, Kawai A, Healey JH, et al.: EWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing's sarcoma. J Clin Oncol 16 (4): 1248-55, 1998. [PUBMED Abstract]
- van Doorninck JA, Ji L, Schaub B, et al.: Current treatment protocols have eliminated the prognostic advantage of type 1 fusions in Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 28 (12): 1989-94, 2010. [PUBMED Abstract]
- Le Deley MC, Delattre O, Schaefer KL, et al.: Impact of EWS-ETS fusion type on disease progression in Ewing's sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. J Clin Oncol 28 (12): 1982-8, 2010. [PUBMED Abstract]
- Paulussen M, Ahrens S, Dunst J, et al.: Localized Ewing tumor of bone: final results of the cooperative Ewing's Sarcoma Study CESS 86. J Clin Oncol 19 (6): 1818-29, 2001. [PUBMED Abstract]
- Rosito P, Mancini AF, Rondelli R, et al.: Italian Cooperative Study for the treatment of children and young adults with localized Ewing sarcoma of bone: a preliminary report of 6 years of experience. Cancer 86 (3): 421-8, 1999. [PUBMED Abstract]
- Wunder JS, Paulian G, Huvos AG, et al.: The histological response to chemotherapy as a predictor of the oncological outcome of operative treatment of Ewing sarcoma. J Bone Joint Surg Am 80 (7): 1020-33, 1998. [PUBMED Abstract]
- Oberlin O, Deley MC, Bui BN, et al.: Prognostic factors in localized Ewing's tumours and peripheral neuroectodermal tumours: the third study of the French Society of Paediatric Oncology (EW88 study). Br J Cancer 85 (11): 1646-54, 2001. [PUBMED Abstract]
- Ferrari S, Bertoni F, Palmerini E, et al.: Predictive factors of histologic response to primary chemotherapy in patients with Ewing sarcoma. J Pediatr Hematol Oncol 29 (6): 364-8, 2007. [PUBMED Abstract]
- Hawkins DS, Schuetze SM, Butrynski JE, et al.: [18F]Fluorodeoxyglucose positron emission tomography predicts outcome for Ewing sarcoma family of tumors. J Clin Oncol 23 (34): 8828-34, 2005. [PUBMED Abstract]
- Denecke T, Hundsdörfer P, Misch D, et al.: Assessment of histological response of paediatric bone sarcomas using FDG PET in comparison to morphological volume measurement and standardized MRI parameters. Eur J Nucl Med Mol Imaging 37 (10): 1842-53, 2010. [PUBMED Abstract]
- Palmerini E, Colangeli M, Nanni C, et al.: The role of FDG PET/CT in patients treated with neoadjuvant chemotherapy for localized bone sarcomas. Eur J Nucl Med Mol Imaging 44 (2): 215-223, 2017. [PUBMED Abstract]
- Lin PP, Jaffe N, Herzog CE, et al.: Chemotherapy response is an important predictor of local recurrence in Ewing sarcoma. Cancer 109 (3): 603-11, 2007. [PUBMED Abstract]
Cellular Classification of Ewing Sarcoma
Ewing sarcoma belongs to the group of neoplasms commonly referred to as small, round, blue-cell tumors of childhood. The individual cells of Ewing sarcoma contain round-to-oval nuclei, with fine dispersed chromatin without nucleoli. Occasionally, cells with smaller, more hyperchromatic, and probably degenerative nuclei are present, giving a light cell/dark cell pattern. The cytoplasm varies in amount, but in the classic case, it is clear and contains glycogen, which can be highlighted with a periodic acid-Schiff stain. The tumor cells are tightly packed and grow in a diffuse pattern without evidence of structural organization. Tumors with the requisite translocation that show neuronal differentiation are not considered a separate entity, but rather, part of a continuum of differentiation.
The MIC2 gene product, CD99, is a surface membrane protein that is expressed in most cases of Ewing sarcoma and is useful in diagnosing these tumors when the results are interpreted in the context of clinical and pathologic parameters.[1] MIC2 positivity is not unique to Ewing sarcoma, and positivity by immunochemistry is found in several other tumors, including synovial sarcoma, non-Hodgkin lymphoma, and gastrointestinal stromal tumors.
Genomics of Ewing Sarcoma
The detection of a translocation involving the EWSR1 gene on chromosome 22 band q12 and any one of a number of partner chromosomes is the key feature in the diagnosis of Ewing sarcoma (refer to Table 2).[2] The EWSR1 gene is a member of the TET family [TLS/EWS/TAF15] of RNA-binding proteins.[3] The FLI1 gene is a member of the ETS family of DNA-binding genes. Characteristically, the amino terminus of the EWSR1 gene is juxtaposed with the carboxy terminus of the STS family gene. In most cases (90%), the carboxy terminus is provided by FLI1, a member of the family of transcription factor genes located on chromosome 11 band q24. Other family members that may combine with the EWSR1 gene are ERG, ETV1, ETV4 (also termed E1AF), and FEV.[4] Rarely, TLS, another TET family member, can substitute for EWSR1.[5] Finally, there are a few rare cases in which EWSR1 has translocated with partners that are not members of the ETS family of oncogenes. The significance of these alternate partners is not known.
Besides these consistent aberrations involving the EWSR1 gene at 22q12, additional numerical and structural aberrations have been observed in Ewing sarcoma, including gains of chromosomes 2, 5, 8, 9, 12, and 15; the nonreciprocal translocation t(1;16)(q12;q11.2); and deletions on the short arm of chromosome 6. Trisomy 20 may be associated with a more aggressive subset of Ewing sarcoma.[6]
Three papers have described the genomic landscape of Ewing sarcoma and all show that these tumors have a relatively silent genome, with a paucity of mutations in pathways that might be amenable to treatment with novel targeted therapies.[7-9] These papers also identified mutations in STAG2, a member of the cohesin complex, in about 15% to 20% of the cases, and the presence of these mutations was associated with advanced-stage disease. CDKN2A deletions were noted in 12% to 22% of cases. Finally, TP53 mutations were identified in about 6% to 7% of cases and the coexistence of STAG2 and TP53 mutations is associated with a poor clinical outcome.[7-9]
Figure 1 below from a discovery cohort (n = 99) highlights the frequency of chromosome 8 gain, the co-occurrence of chromosome 1q gain and chromosome 16q loss, the mutual exclusivity of CDKN2A deletion and STAG2 mutation, and the relative paucity of recurrent single nucleotide variants for Ewing sarcoma.[7]
Ewing sarcoma translocations can all be found with standard cytogenetic analysis. A more rapid analysis looking for a break apart of the EWS gene is now frequently done to confirm the diagnosis of Ewing sarcoma molecularly.[10] This test result must be considered with caution, however. Ewing sarcomas that utilize the TLS translocations will have negative tests because the EWSR1 gene is not translocated in those cases. In addition, other small round tumors also contain translocations of different ETS family members with EWSR1, such as desmoplastic small round cell tumor, clear cell sarcoma, extraskeletal myxoid chondrosarcoma, and myxoid liposarcoma, all of which may be positive with a EWS fluorescence in situ hybridization (FISH) break-apart probe. A detailed analysis of 85 patients with small round blue cell tumors that were negative for EWSR1 rearrangement by FISH with an EWSR1 break-apart probe identified eight patients with FUS rearrangements.[11] Four patients who had EWSR1-ERG fusions were not detected by FISH with an EWSR1 break-apart probe. The authors do not recommend relying solely on EWSR1 break-apart probes for analyzing small round blue cell tumors with strong immunohistochemical positivity for CD99.
Small round blue cell tumors of bone and soft tissue that are histologically similar to Ewing sarcoma but do not have rearrangements of the EWSR1 gene have been analyzed and translocations have been identified. These include BCOR-CCNB3, CIC-DUX4, and CIC-FOX4.[12-15] The molecular profile of these tumors is different from the profile of EWS-FLI1 translocated Ewing sarcoma, and limited evidence suggests that they have a different clinical behavior. In almost all cases, the patients were treated with therapy designed for Ewing sarcoma on the basis of the histologic and immunohistologic similarity to Ewing sarcoma. There are too few cases associated with each translocation to determine whether the prognosis for these small round blue cell tumors is distinct from the prognosis of Ewing sarcoma of similar stage and site.[12-15]
A genome-wide association study identified a region on chromosome 10q21.3 associated with an increased risk of Ewing sarcoma.[16] Deep sequencing through this region identified a polymorphism in the EGR2 gene, which appears to cooperate with the gene product of the EWSR1-FLI1 fusion that is seen in most patients with Ewing sarcoma.[17] The polymorphism associated with the increased risk is found at a much higher frequency in whites than in blacks or Asians, possibly contributing to the epidemiology of the relative infrequency of Ewing sarcoma in the latter populations.
| TET Family Partner | Fusion With ETS-like Oncogene Partner | Translocation | Comment |
|---|---|---|---|
| aThese partners are not members of the ETS family of oncogenes. | |||
| EWS | EWSR1-FLI1 | t(11;22)(q24;q12) | Most common; ~85% to 90% of cases |
| EWSR1-ERG | t(21;22)(q22;q12) | Second most common; ~10% of cases | |
| EWSR1-ETV1 | t(7;22)(p22;q12) | Rare | |
| EWSR1-ETV4 | t(17;22)(q12;q12) | Rare | |
| EWSR1-FEV | t(2;22)(q35;q12) | Rare | |
| EWSR1-NFATc2a | t(20;22)(q13;q12) | Rare | |
| EWSR1-POU5F1a | t(6;22)(p21;q12) | ||
| EWSR1-SMARCA5a | t(4;22)(q31;q12) | Rare | |
| EWSR1-ZSGa | t(6;22)(p21;q12) | ||
| EWSR1-SP3a | t(2;22)(q31;q12) | Rare | |
| TLS (also called FUS) | TLS-ERG | t(16;21)(p11;q22) | Rare |
| TLS-FEV | t(2;16)(q35;p11) | Rare | |
References
- Parham DM, Hijazi Y, Steinberg SM, et al.: Neuroectodermal differentiation in Ewing's sarcoma family of tumors does not predict tumor behavior. Hum Pathol 30 (8): 911-8, 1999. [PUBMED Abstract]
- Delattre O, Zucman J, Melot T, et al.: The Ewing family of tumors--a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 331 (5): 294-9, 1994. [PUBMED Abstract]
- Urano F, Umezawa A, Yabe H, et al.: Molecular analysis of Ewing's sarcoma: another fusion gene, EWS-E1AF, available for diagnosis. Jpn J Cancer Res 89 (7): 703-11, 1998. [PUBMED Abstract]
- Hattinger CM, Rumpler S, Strehl S, et al.: Prognostic impact of deletions at 1p36 and numerical aberrations in Ewing tumors. Genes Chromosomes Cancer 24 (3): 243-54, 1999. [PUBMED Abstract]
- Sankar S, Lessnick SL: Promiscuous partnerships in Ewing's sarcoma. Cancer Genet 204 (7): 351-65, 2011. [PUBMED Abstract]
- Roberts P, Burchill SA, Brownhill S, et al.: Ploidy and karyotype complexity are powerful prognostic indicators in the Ewing's sarcoma family of tumors: a study by the United Kingdom Cancer Cytogenetics and the Children's Cancer and Leukaemia Group. Genes Chromosomes Cancer 47 (3): 207-20, 2008. [PUBMED Abstract]
- Tirode F, Surdez D, Ma X, et al.: Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov 4 (11): 1342-53, 2014. [PUBMED Abstract]
- Crompton BD, Stewart C, Taylor-Weiner A, et al.: The genomic landscape of pediatric Ewing sarcoma. Cancer Discov 4 (11): 1326-41, 2014. [PUBMED Abstract]
- Brohl AS, Solomon DA, Chang W, et al.: The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet 10 (7): e1004475, 2014. [PUBMED Abstract]
- Monforte-Muñoz H, Lopez-Terrada D, Affendie H, et al.: Documentation of EWS gene rearrangements by fluorescence in-situ hybridization (FISH) in frozen sections of Ewing's sarcoma-peripheral primitive neuroectodermal tumor. Am J Surg Pathol 23 (3): 309-15, 1999. [PUBMED Abstract]
- Chen S, Deniz K, Sung YS, et al.: Ewing sarcoma with ERG gene rearrangements: A molecular study focusing on the prevalence of FUS-ERG and common pitfalls in detecting EWSR1-ERG fusions by FISH. Genes Chromosomes Cancer 55 (4): 340-9, 2016. [PUBMED Abstract]
- Pierron G, Tirode F, Lucchesi C, et al.: A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet 44 (4): 461-6, 2012. [PUBMED Abstract]
- Specht K, Sung YS, Zhang L, et al.: Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged Ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosomes Cancer 53 (7): 622-33, 2014. [PUBMED Abstract]
- Sugita S, Arai Y, Tonooka A, et al.: A novel CIC-FOXO4 gene fusion in undifferentiated small round cell sarcoma: a genetically distinct variant of Ewing-like sarcoma. Am J Surg Pathol 38 (11): 1571-6, 2014. [PUBMED Abstract]
- Cohen-Gogo S, Cellier C, Coindre JM, et al.: Ewing-like sarcomas with BCOR-CCNB3 fusion transcript: a clinical, radiological and pathological retrospective study from the Société Française des Cancers de L'Enfant. Pediatr Blood Cancer 61 (12): 2191-8, 2014. [PUBMED Abstract]
- Postel-Vinay S, Véron AS, Tirode F, et al.: Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat Genet 44 (3): 323-7, 2012. [PUBMED Abstract]
- Grünewald TG, Bernard V, Gilardi-Hebenstreit P, et al.: Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat Genet 47 (9): 1073-8, 2015. [PUBMED Abstract]
Stage Information for Ewing Sarcoma
Pretreatment staging studies for Ewing sarcoma may include the following:
- Magnetic resonance imaging (MRI).
- Computed tomography (CT) scan of the primary site and chest.
- Positron emission tomography using fluorine F 18-fludeoxyglucose (18F-FDG PET) or 18F-FDG PET-CT.
- Bone scan.
- Bone marrow aspiration and biopsy.
For patients with confirmed Ewing sarcoma, pretreatment staging studies include MRI and/or CT scan, depending on the primary site. Despite the fact that CT and MRI are both equivalent in terms of staging, use of both imaging modalities may help radiation therapy planning.[1] Whole-body MRI may provide additional information that could potentially alter therapy planning.[2] Additional pretreatment staging studies include bone scan and CT scan of the chest. In certain studies, determination of pretreatment tumor volume is an important variable.
Although 18F-FDG PET or 18F-FDG PET-CT are optional staging modalities, they have demonstrated high sensitivity and specificity in Ewing sarcoma and may provide additional information that alters therapy planning. In one institutional study, 18F-FDG PET had a very high correlation with bone scan; the investigators suggested that it could replace bone scan for the initial extent of disease evaluation.[3] This finding was confirmed in a single-institution retrospective review.[4] 18F-FDG PET-CT is more accurate than 18F-FDG PET alone in Ewing sarcoma.[5-7]
Bone marrow aspiration and biopsy have been considered the standard of care for Ewing sarcoma. However, two retrospective studies showed that for patients (N = 141 total) who were evaluated by bone scan and/or PET scan and lung CT without evidence of metastases, bone marrow aspirates and biopsies were negative in every case.[3,8] The need for routine use of bone marrow aspirates and biopsies in patients without bone metastases is now in question.
For Ewing sarcoma, the tumor is defined as localized when, by clinical and imaging techniques, there is no spread beyond the primary site or regional lymph node involvement. Continuous extension into adjacent soft tissue may occur. If there is a question of regional lymph node involvement, pathologic confirmation is indicated.
References
- Meyer JS, Nadel HR, Marina N, et al.: Imaging guidelines for children with Ewing sarcoma and osteosarcoma: a report from the Children's Oncology Group Bone Tumor Committee. Pediatr Blood Cancer 51 (2): 163-70, 2008. [PUBMED Abstract]
- Mentzel HJ, Kentouche K, Sauner D, et al.: Comparison of whole-body STIR-MRI and 99mTc-methylene-diphosphonate scintigraphy in children with suspected multifocal bone lesions. Eur Radiol 14 (12): 2297-302, 2004. [PUBMED Abstract]
- Newman EN, Jones RL, Hawkins DS: An evaluation of [F-18]-fluorodeoxy-D-glucose positron emission tomography, bone scan, and bone marrow aspiration/biopsy as staging investigations in Ewing sarcoma. Pediatr Blood Cancer 60 (7): 1113-7, 2013. [PUBMED Abstract]
- Ulaner GA, Magnan H, Healey JH, et al.: Is methylene diphosphonate bone scan necessary for initial staging of Ewing sarcoma if 18F-FDG PET/CT is performed? AJR Am J Roentgenol 202 (4): 859-67, 2014. [PUBMED Abstract]
- Völker T, Denecke T, Steffen I, et al.: Positron emission tomography for staging of pediatric sarcoma patients: results of a prospective multicenter trial. J Clin Oncol 25 (34): 5435-41, 2007. [PUBMED Abstract]
- Gerth HU, Juergens KU, Dirksen U, et al.: Significant benefit of multimodal imaging: PET/CT compared with PET alone in staging and follow-up of patients with Ewing tumors. J Nucl Med 48 (12): 1932-9, 2007. [PUBMED Abstract]
- Treglia G, Salsano M, Stefanelli A, et al.: Diagnostic accuracy of ¹⁸F-FDG-PET and PET/CT in patients with Ewing sarcoma family tumours: a systematic review and a meta-analysis. Skeletal Radiol 41 (3): 249-56, 2012. [PUBMED Abstract]
- Kopp LM, Hu C, Rozo B, et al.: Utility of bone marrow aspiration and biopsy in initial staging of Ewing sarcoma. Pediatr Blood Cancer 62 (1): 12-5, 2015. [PUBMED Abstract]
Treatment Option Overview for Ewing Sarcoma
It is important that patients be evaluated by specialists from the appropriate disciplines (e.g., radiologists, chemotherapists, pathologists, surgical or orthopedic oncologists, and radiation oncologists) as early as possible. Appropriate imaging studies of the site are obtained before biopsy. To ensure that the incision is placed in an acceptable location, the surgical or orthopedic oncologist who will perform the definitive surgery is involved in the decision regarding biopsy-incision placement. This is especially important if it is thought that the lesion can be totally excised or if a limb salvage procedure may be attempted. Biopsy should be from soft tissue as often as possible to avoid increasing the risk of fracture.[1] The pathologist is consulted before biopsy/surgery to ensure that the incision will not compromise the radiation port and that multiple types of adequate tissue samples are obtained. It is important to obtain fresh tissue, whenever possible, for cytogenetics and molecular pathology. A second option is to perform a needle biopsy, as long as adequate tissue is obtained for molecular biology and cytogenetics.[2]
Table 3 describes the treatment options for localized, metastatic, and recurrent Ewing sarcoma.
| Treatment Group | Standard Treatment Options | |
|---|---|---|
| Localized Ewing sarcoma | Chemotherapy | |
| Local-control measures: | ||
| Surgery | ||
| Radiation therapy | ||
| Metastatic Ewing sarcoma | Chemotherapy | |
| Surgery | ||
| Radiation therapy | ||
| Recurrent Ewing sarcoma | Chemotherapy (not considered standard treatment) | |
| Radiation therapy (not considered standard treatment) | ||
| Other therapies (not considered standard treatment) | ||
The successful treatment of patients with Ewing sarcoma requires systemic chemotherapy [3-9] in conjunction with surgery and/or radiation therapy for local tumor control.[10-14] In general, patients receive chemotherapy before instituting local-control measures. In patients who undergo surgery, surgical margins and histologic response are considered in planning postoperative therapy. Patients with metastatic disease often have a good initial response to preoperative chemotherapy, but in most cases, the disease is only partially controlled or recurs.[15-19] Patients with lung as the only metastatic site have a better prognosis than do patients with metastases to bone and/or bone marrow. Adequate local control for metastatic sites, particularly bone metastases, may be an important issue.[20]
Chemotherapy for Ewing Sarcoma
Multidrug chemotherapy for Ewing sarcoma always includes vincristine, doxorubicin, ifosfamide, and etoposide. Most protocols also use cyclophosphamide and some incorporate dactinomycin. The mode of administration and dose intensity of cyclophosphamide within courses differs markedly between protocols. A European Intergroup Cooperative Ewing Sarcoma Study (EICESS) trial suggested that 1.2 g of cyclophosphamide produced a similar event-free survival (EFS) compared with 6 g of ifosfamide in patients with lower-risk disease, and identified a trend toward better EFS for patients with localized Ewing sarcoma and higher-risk disease when treatment included etoposide (GER-GPOH-EICESS-92).[21][Level of evidence: 1iiA]
Protocols in the United States generally alternate courses of vincristine, cyclophosphamide, and doxorubicin with courses of ifosfamide/etoposide,[7] while European protocols generally combine vincristine, doxorubicin, and an alkylating agent with or without etoposide in a single treatment cycle.[9] The duration of primary chemotherapy ranges from 6 months to approximately 1 year.
Evidence (chemotherapy):
- An international consortium of European countries conducted the EURO-EWING-INTERGROUP-EE99 (NCT00020566) trial from 2000 to 2010.[22][Level of evidence: 1iiA] All patients received induction therapy with six cycles of vincristine, ifosfamide, doxorubicin, and etoposide (VIDE), followed by local control, and then one cycle of vincristine, dactinomycin, and ifosfamide (VAI). Patients were classified as standard risk if they had localized disease and good histologic response to therapy or if they had localized tumors less than 200 mL in volume at presentation; they were treated with radiation therapy alone as local treatment. Standard-risk patients (n = 856) were randomly assigned to receive either maintenance therapy with seven cycles of vincristine, dactinomycin, and cyclophosphamide (VAC) or VAI.
- There was no significant difference in EFS or overall survival (OS) between the VAC arm and the VAI arm.
- Three-year EFS for this low-risk population was 77%.
- Acute renal toxicity was lower in the VAC arm than in the VAI arm, but long-term renal function outcome and fertility analyses are still pending.
- It is difficult to compare this outcome with that of other large series because the study population excluded patients with poor response to initial therapy or patients with tumors more than 200 mL in volume who received local-control therapy with radiation alone. All other published series report results for all patients who present without clinically detectable metastasis; thus, these other series included patients with poor response and patients with larger primary tumors treated with radiation alone, all of whom were excluded from the EURO-EWING-INTERGROUP-EE99 study.
- A randomized clinical trial (COG-AEWS0031 [NCT00006734]) from the Children’s Oncology Group (COG) showed that for patients presenting without metastases, the administration of cycles of cyclophosphamide, doxorubicin, and vincristine alternating with cycles of ifosfamide and etoposide at 2-week intervals achieved superior EFS (5-year EFS, 73%) than did alternating cycles at 3-week intervals (5-year EFS, 65%).[23]
- The Brazilian Cooperative Study Group performed a multi-institutional trial that incorporated carboplatin into a risk-adapted intensive regimen in 175 children with localized or metastatic Ewing sarcoma. They found significantly increased toxicity without an improvement in outcome with the addition of carboplatin.[24][Level of evidence: 2Dii]
- The COG conducted a pilot study of the addition of cycles of cyclophosphamide and topotecan to cycles of cyclophosphamide/doxorubicin/vincristine and ifosfamide/etoposide administered in an interval-compressed (2-week instead of 3-week intervals) schedule.[25][Level of evidence: 2Di]
- Therapy was well tolerated, and the 5-year EFS for 35 patients was 80%. This pilot study became the experimental arm of COG-AEWS1031 (NCT01231906).
Local Control for Ewing Sarcoma
Treatment approaches for Ewing sarcoma titrate therapeutic aggressiveness with the goal of maximizing local control while minimizing morbidity.
Surgery is the most commonly used form of local control.[26] Radiation therapy is an effective alternative modality for local control in cases where the functional morbidity of surgery is deemed too high by experienced surgical oncologists. However, in the immature skeleton, radiation therapy can cause subsequent deformities that may be more morbid than deformities from surgery. When complete surgical resection with pathologically negative margins cannot be obtained, postoperative radiation therapy is indicated. A multidisciplinary discussion between the experienced radiation oncologist and the surgeon is necessary to determine the best treatment options for local control for a given case. For some marginally resectable lesions, a combined approach of preoperative radiation therapy followed by resection can be used.
Randomized trials that directly compare surgery and radiation therapy do not exist, and their relative roles remain controversial. Although retrospective institutional series suggest superior local control and survival with surgery than with radiation therapy, most of these studies are compromised by selection bias. An analysis using propensity scoring to adjust for clinical features that may influence the preference for surgery only, radiation only, or combined surgery and radiation demonstrated that similar EFS is achieved with each mode of local therapy after propensity adjustment.[26] Data for patients with pelvic primary Ewing sarcoma from a North American intergroup trial showed no difference in local control or survival on the basis of local-control modality—surgery alone, radiation therapy alone, or radiation plus surgery.[27]
For patients who undergo gross-total resection with microscopic residual disease, the value of adjuvant radiation therapy is controversial. Investigations addressing this issue are retrospective and nonrandomized, limiting their value.
Evidence (postoperative radiation therapy):
- Investigators from St. Jude Children’s Research Hospital reported 39 patients with localized Ewing sarcoma who received both surgery and radiation.[13]
- Local failure for patients with positive margins was 17% and OS was 71%. Local failure for patients with negative margins was 5% and OS was 94%.
- However, in a large retrospective Italian study, 45 Gy of adjuvant radiation therapy for patients with inadequate margins did not appear to improve either local control or disease-free survival.[14] It is not known whether higher doses of radiation therapy could improve outcome. These investigators concluded that patients who are anticipated to have suboptimal surgery should be considered for definitive radiation therapy.
- The EURO-EWING-INTERGROUP-EE99 (NCT00020566) study reported the outcomes of 599 patients who presented with localized disease and had surgical resection after initial chemotherapy with at least 90% necrosis of the primary tumor.[28][Level of evidence: 3iiDi] The protocol recommended postoperative radiation therapy for patients with inadequate surgical margins, vertebral primary tumors, or thoracic tumors with pleural effusion, but the decision to use postoperative radiation therapy was left to the institutional investigator.
- Patients who received postoperative radiation therapy (n = 142) had a lower risk of failure than did patients who did not receive postoperative radiation therapy, even after controlling for known prognostic factors, including age, sex, tumor site, clinical response, quality of resection, and histologic necrosis. Most of the improvement was seen in a decreased risk of local recurrence. The improvement was greater in patients who were assessed to have 100% necrosis than in patients who were assessed to have 90% to 100% necrosis.
- There is a clear interaction between systemic therapy and local-control modalities for both local control and disease-free survival. The induction regimen used in the EURO-EWING-INTERGROUP-EE99 study is less intense than the induction regimen used in contemporaneous protocols in the COG, and it is not appropriate to extrapolate the results from the EURO-EWING-INTERGROUP-EE99 study to different systemic chemotherapy regimens.
In summary, surgery is chosen as definitive local therapy for suitable patients, but radiation therapy is appropriate for patients with unresectable disease or those who would experience functional compromise by definitive surgery. The possibility of impaired function needs to be measured against the possibility of second tumors in the radiation field (refer to the Late Effects of Treatment for Ewing Sarcoma section of this summary for more information). Adjuvant radiation therapy may be considered for patients with residual microscopic disease, inadequate margins, or who have viable tumor in the resected specimen and close margins.
When preoperative assessment has suggested a high probability that surgical margins will be close or positive, preoperative radiation therapy has achieved tumor shrinkage and allowed surgical resection with clear margins.[29]
High-Dose Therapy With Stem Cell Rescue for Ewing Sarcoma
For patients with a high risk of relapse with conventional treatments, certain investigators have utilized high-dose chemotherapy with hematopoietic stem cell transplant (HSCT) as consolidation treatment, in an effort to improve outcome.[19,30-42]
Evidence (high-dose therapy with stem cell rescue):
- In a prospective study, patients with bone and/or bone marrow metastases at diagnosis were treated with aggressive chemotherapy, surgery, and/or radiation and HSCT if a good initial response was achieved.[35]
- The study showed no benefit for HSCT compared with historical controls.
- A retrospective review using international bone marrow transplant registries compared the outcomes after treatment with either reduced-intensity conditioning or high-intensity conditioning followed by allogeneic SCT for patients with Ewing sarcoma at high risk for relapse.[43][Level of evidence: 3iiiA]
- There was no difference in outcome, and the authors concluded that this suggested the absence of a clinically relevant graft-versus-tumor effect against Ewing sarcoma tumor cells with current approaches.
- Multiple small studies that report benefit for HSCT have been published but are difficult to interpret because only patients who have a good initial response to standard chemotherapy are considered for HSCT.
The role of high-dose therapy followed by stem cell rescue is being investigated in the prospective, randomized Euro-Ewing trial (EURO-EWING-INTERGROUP-EE99) for patients who present with metastases and patients with localized tumors with poor response to initial chemotherapy.
Ewing Sarcoma/Specific Sites
Multiple analyses have evaluated diagnostic findings, treatment, and outcome of patients with bone lesions at the following anatomic primary sites:
Extraosseous Ewing Sarcoma
Extraosseous Ewing sarcoma is biologically similar to Ewing sarcoma arising in bone. Historically, most children and young adults with extraosseous Ewing sarcoma were treated on protocols designed for the treatment of rhabdomyosarcoma. This is important because many of the treatment regimens for rhabdomyosarcoma do not include an anthracycline, which is a critical component of current treatment regimens for Ewing sarcoma. Currently, patients with extraosseous Ewing sarcoma are eligible for studies that include Ewing sarcoma of bone.
From 1987 to 2004, 111 patients with nonmetastatic extraosseous Ewing sarcoma were enrolled on the RMS-88 and RMS-96 protocols.[62] Patients with initial complete tumor resection received ifosfamide, vincristine, and actinomycin (IVA) while patients with residual tumor received IVA plus doxorubicin (VAIA) or IVA plus carboplatin, epirubicin, and etoposide (CEVAIE). Seventy-six percent of patients received radiation. The 5-year EFS was 59% and OS was 69%. In a multivariate analysis, independent adverse prognostic factors included axial primary, tumor size greater than 10 cm, Intergroup Rhabdomyosarcoma Studies Group III, and lack of radiation therapy.
Two hundred thirty-six patients with extraosseous Ewing sarcoma were entered on studies of the German Pediatric Oncology Group.[63] The median age at diagnosis was 15 years and 133 patients were male. Primary tumor site was either extremity (n = 62) or central site (n = 174). Sixty of the 236 patients had metastases at diagnosis. Chemotherapy consisted of vincristine, doxorubicin, cyclophosphamide, and actinomycin (VACA); CEVAIE; or VIDE. The 5-year EFS was 49% and OS was 60%. Five-year survival was 70% for patients with localized disease and 33% for patients with metastasis at diagnosis. OS in patients with localized disease did not seem related to tumor site or size. In a retrospective French study, patients with extraosseous Ewing sarcoma were treated using a rhabdomyosarcoma regimen (no anthracyclines) or a Ewing sarcoma regimen (includes anthracyclines). Patients who received the anthracycline-containing regimen had a significantly better EFS and OS than did patients who did not receive anthracyclines.[64,65] Two North American Ewing sarcoma trials have included patients with extraosseous Ewing sarcoma.[23,66] In a review of data from the POG-9354 (INT-0154) and EWS0031 (NCT00006734) studies, 213 patients with extraosseous Ewing sarcoma and 826 patients with Ewing sarcoma of bone were identified. The hazard ratio of extraosseous Ewing sarcoma was superior (0.62), and extraosseous Ewing sarcoma was a favorable risk factor, independent of age, race, and primary site.[67][Level of evidence: 3iiDi]
Cutaneous Ewing sarcoma is a soft tissue tumor in the skin or subcutaneous tissue that seems to behave as a less-aggressive tumor than primary bone or soft tissue Ewing sarcoma. Tumors can form throughout the body, although the extremity is the most common site, and they are almost always localized. In a review of 78 reported cases, some lacking molecular confirmation, the OS was 91%. Adequate local control, defined as a complete resection with negative margins, radiation therapy, or a combination, significantly reduced the incidence of relapse. Standard chemotherapy for Ewing sarcoma is often used for these patients because there are no data to suggest which patients could be treated less aggressively.[68,69] A series of 56 patients with cutaneous or subcutaneous Ewing sarcoma confirmed the excellent outcome with the use of standard systemic therapy and local control. Attempted primary definitive surgery often resulted in the need for either radiation therapy or more function-compromising surgery, supporting the recommendation of biopsy only as initial surgery, rather than upfront unplanned resection.[70][Level of evidence: 3iiD]
Special Considerations for the Treatment of Children With Cancer
Cancer in children and adolescents is rare, although the overall incidence of childhood cancer has been slowly increasing since 1975.[71] Children and adolescents with cancer should be referred to medical centers that have a multidisciplinary team of cancer specialists with experience treating the cancers that occur during childhood and adolescence. This multidisciplinary team approach incorporates the skills of the following health care professionals and others to ensure that children receive treatment, supportive care, and rehabilitation that will achieve optimal survival and quality of life:
- Primary care physicians.
- Pediatric surgeons.
- Radiation oncologists.
- Pediatric oncologists/hematologists.
- Rehabilitation specialists.
- Pediatric nurse specialists.
- Social workers.
- Child-life professionals.
- Psychologists.
(Refer to the PDQ Supportive and Palliative Care summaries for specific information about supportive care for children and adolescents with cancer.)
Guidelines for pediatric cancer centers and their role in the treatment of pediatric patients with cancer have been outlined by the American Academy of Pediatrics.[72] At these pediatric cancer centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate in these trials is offered to most patients and their families. Clinical trials for children and adolescents with cancer are generally designed to compare potentially better therapy with therapy that is currently accepted as standard. Most of the progress made in identifying curative therapies for childhood cancers has been achieved through clinical trials. Information about ongoing clinical trials is available from the NCI website.
Childhood and adolescent cancer survivors require close monitoring because cancer therapy side effects may persist or develop months or years after treatment. (Refer to the PDQ summary on Late Effects of Treatment for Childhood Cancer for specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors.)
References
- Fuchs B, Valenzuela RG, Sim FH: Pathologic fracture as a complication in the treatment of Ewing's sarcoma. Clin Orthop (415): 25-30, 2003. [PUBMED Abstract]
- Hoffer FA, Gianturco LE, Fletcher JA, et al.: Percutaneous biopsy of peripheral primitive neuroectodermal tumors and Ewing's sarcomas for cytogenetic analysis. AJR Am J Roentgenol 162 (5): 1141-2, 1994. [PUBMED Abstract]
- Craft A, Cotterill S, Malcolm A, et al.: Ifosfamide-containing chemotherapy in Ewing's sarcoma: The Second United Kingdom Children's Cancer Study Group and the Medical Research Council Ewing's Tumor Study. J Clin Oncol 16 (11): 3628-33, 1998. [PUBMED Abstract]
- Shankar AG, Pinkerton CR, Atra A, et al.: Local therapy and other factors influencing site of relapse in patients with localised Ewing's sarcoma. United Kingdom Children's Cancer Study Group (UKCCSG). Eur J Cancer 35 (12): 1698-704, 1999. [PUBMED Abstract]
- Nilbert M, Saeter G, Elomaa I, et al.: Ewing's sarcoma treatment in Scandinavia 1984-1990--ten-year results of the Scandinavian Sarcoma Group Protocol SSGIV. Acta Oncol 37 (4): 375-8, 1998. [PUBMED Abstract]
- Ferrari S, Mercuri M, Rosito P, et al.: Ifosfamide and actinomycin-D, added in the induction phase to vincristine, cyclophosphamide and doxorubicin, improve histologic response and prognosis in patients with non metastatic Ewing's sarcoma of the extremity. J Chemother 10 (6): 484-91, 1998. [PUBMED Abstract]
- Grier HE, Krailo MD, Tarbell NJ, et al.: Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 348 (8): 694-701, 2003. [PUBMED Abstract]
- Thacker MM, Temple HT, Scully SP: Current treatment for Ewing's sarcoma. Expert Rev Anticancer Ther 5 (2): 319-31, 2005. [PUBMED Abstract]
- Juergens C, Weston C, Lewis I, et al.: Safety assessment of intensive induction with vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) in the treatment of Ewing tumors in the EURO-E.W.I.N.G. 99 clinical trial. Pediatr Blood Cancer 47 (1): 22-9, 2006. [PUBMED Abstract]
- Dunst J, Schuck A: Role of radiotherapy in Ewing tumors. Pediatr Blood Cancer 42 (5): 465-70, 2004. [PUBMED Abstract]
- Donaldson SS: Ewing sarcoma: radiation dose and target volume. Pediatr Blood Cancer 42 (5): 471-6, 2004. [PUBMED Abstract]
- Bacci G, Ferrari S, Longhi A, et al.: Role of surgery in local treatment of Ewing's sarcoma of the extremities in patients undergoing adjuvant and neoadjuvant chemotherapy. Oncol Rep 11 (1): 111-20, 2004. [PUBMED Abstract]
- Krasin MJ, Rodriguez-Galindo C, Davidoff AM, et al.: Efficacy of combined surgery and irradiation for localized Ewings sarcoma family of tumors. Pediatr Blood Cancer 43 (3): 229-36, 2004. [PUBMED Abstract]
- Bacci G, Longhi A, Briccoli A, et al.: The role of surgical margins in treatment of Ewing's sarcoma family tumors: experience of a single institution with 512 patients treated with adjuvant and neoadjuvant chemotherapy. Int J Radiat Oncol Biol Phys 65 (3): 766-72, 2006. [PUBMED Abstract]
- Paulussen M, Ahrens S, Burdach S, et al.: Primary metastatic (stage IV) Ewing tumor: survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma Studies. Ann Oncol 9 (3): 275-81, 1998. [PUBMED Abstract]
- Pinkerton CR, Bataillard A, Guillo S, et al.: Treatment strategies for metastatic Ewing's sarcoma. Eur J Cancer 37 (11): 1338-44, 2001. [PUBMED Abstract]
- Miser JS, Krailo M, Meyers P, et al.: Metastatic Ewing's sarcoma(es) and primitive neuroectodermal tumor (PNET) of bone: failure of new regimens to improve outcome. [Abstract] Proceedings of the American Society of Clinical Oncology 15: A-1472, 467, 1996.
- Bernstein ML, Devidas M, Lafreniere D, et al.: Intensive therapy with growth factor support for patients with Ewing tumor metastatic at diagnosis: Pediatric Oncology Group/Children's Cancer Group Phase II Study 9457--a report from the Children's Oncology Group. J Clin Oncol 24 (1): 152-9, 2006. [PUBMED Abstract]
- Ladenstein R, Pötschger U, Le Deley MC, et al.: Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol 28 (20): 3284-91, 2010. [PUBMED Abstract]
- Haeusler J, Ranft A, Boelling T, et al.: The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES). Cancer 116 (2): 443-50, 2010. [PUBMED Abstract]
- Paulussen M, Craft AW, Lewis I, et al.: Results of the EICESS-92 Study: two randomized trials of Ewing's sarcoma treatment--cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J Clin Oncol 26 (27): 4385-93, 2008. [PUBMED Abstract]
- Le Deley MC, Paulussen M, Lewis I, et al.: Cyclophosphamide compared with ifosfamide in consolidation treatment of standard-risk Ewing sarcoma: results of the randomized noninferiority Euro-EWING99-R1 trial. J Clin Oncol 32 (23): 2440-8, 2014. [PUBMED Abstract]
- Womer RB, West DC, Krailo MD, et al.: Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 30 (33): 4148-54, 2012. [PUBMED Abstract]
- Brunetto AL, Castillo LA, Petrilli AS, et al.: Carboplatin in the treatment of Ewing sarcoma: Results of the first Brazilian collaborative study group for Ewing sarcoma family tumors-EWING1. Pediatr Blood Cancer 62 (10): 1747-53, 2015. [PUBMED Abstract]
- Mascarenhas L, Felgenhauer JL, Bond MC, et al.: Pilot Study of Adding Vincristine, Topotecan, and Cyclophosphamide to Interval-Compressed Chemotherapy in Newly Diagnosed Patients With Localized Ewing Sarcoma: A Report From the Children's Oncology Group. Pediatr Blood Cancer 63 (3): 493-8, 2016. [PUBMED Abstract]
- DuBois SG, Krailo MD, Gebhardt MC, et al.: Comparative evaluation of local control strategies in localized Ewing sarcoma of bone: a report from the Children's Oncology Group. Cancer 121 (3): 467-75, 2015. [PUBMED Abstract]
- Yock TI, Krailo M, Fryer CJ, et al.: Local control in pelvic Ewing sarcoma: analysis from INT-0091--a report from the Children's Oncology Group. J Clin Oncol 24 (24): 3838-43, 2006. [PUBMED Abstract]
- Foulon S, Brennan B, Gaspar N, et al.: Can postoperative radiotherapy be omitted in localised standard-risk Ewing sarcoma? An observational study of the Euro-E.W.I.N.G group. Eur J Cancer 61: 128-36, 2016. [PUBMED Abstract]
- Wagner TD, Kobayashi W, Dean S, et al.: Combination short-course preoperative irradiation, surgical resection, and reduced-field high-dose postoperative irradiation in the treatment of tumors involving the bone. Int J Radiat Oncol Biol Phys 73 (1): 259-66, 2009. [PUBMED Abstract]
- Kushner BH, Meyers PA: How effective is dose-intensive/myeloablative therapy against Ewing's sarcoma/primitive neuroectodermal tumor metastatic to bone or bone marrow? The Memorial Sloan-Kettering experience and a literature review. J Clin Oncol 19 (3): 870-80, 2001. [PUBMED Abstract]
- Marina N, Meyers PA: High-dose therapy and stem-cell rescue for Ewing's family of tumors in second remission. J Clin Oncol 23 (19): 4262-4, 2005. [PUBMED Abstract]
- Burdach S: Treatment of advanced Ewing tumors by combined radiochemotherapy and engineered cellular transplants. Pediatr Transplant 8 (Suppl 5): 67-82, 2004. [PUBMED Abstract]
- McTiernan A, Driver D, Michelagnoli MP, et al.: High dose chemotherapy with bone marrow or peripheral stem cell rescue is an effective treatment option for patients with relapsed or progressive Ewing's sarcoma family of tumours. Ann Oncol 17 (8): 1301-5, 2006. [PUBMED Abstract]
- Burdach S, Meyer-Bahlburg A, Laws HJ, et al.: High-dose therapy for patients with primary multifocal and early relapsed Ewing's tumors: results of two consecutive regimens assessing the role of total-body irradiation. J Clin Oncol 21 (16): 3072-8, 2003. [PUBMED Abstract]
- Meyers PA, Krailo MD, Ladanyi M, et al.: High-dose melphalan, etoposide, total-body irradiation, and autologous stem-cell reconstitution as consolidation therapy for high-risk Ewing's sarcoma does not improve prognosis. J Clin Oncol 19 (11): 2812-20, 2001. [PUBMED Abstract]
- Oberlin O, Rey A, Desfachelles AS, et al.: Impact of high-dose busulfan plus melphalan as consolidation in metastatic Ewing tumors: a study by the Société Française des Cancers de l'Enfant. J Clin Oncol 24 (24): 3997-4002, 2006. [PUBMED Abstract]
- Hawkins D, Barnett T, Bensinger W, et al.: Busulfan, melphalan, and thiotepa with or without total marrow irradiation with hematopoietic stem cell rescue for poor-risk Ewing-Sarcoma-Family tumors. Med Pediatr Oncol 34 (5): 328-37, 2000. [PUBMED Abstract]
- Rosenthal J, Bolotin E, Shakhnovits M, et al.: High-dose therapy with hematopoietic stem cell rescue in patients with poor prognosis Ewing family tumors. Bone Marrow Transplant 42 (5): 311-8, 2008. [PUBMED Abstract]
- Burdach S, Thiel U, Schöniger M, et al.: Total body MRI-governed involved compartment irradiation combined with high-dose chemotherapy and stem cell rescue improves long-term survival in Ewing tumor patients with multiple primary bone metastases. Bone Marrow Transplant 45 (3): 483-9, 2010. [PUBMED Abstract]
- Gaspar N, Rey A, Bérard PM, et al.: Risk adapted chemotherapy for localised Ewing's sarcoma of bone: the French EW93 study. Eur J Cancer 48 (9): 1376-85, 2012. [PUBMED Abstract]
- Drabko K, Raciborska A, Bilska K, et al.: Consolidation of first-line therapy with busulphan and melphalan, and autologous stem cell rescue in children with Ewing's sarcoma. Bone Marrow Transplant 47 (12): 1530-4, 2012. [PUBMED Abstract]
- Loschi S, Dufour C, Oberlin O, et al.: Tandem high-dose chemotherapy strategy as first-line treatment of primary disseminated multifocal Ewing sarcomas in children, adolescents and young adults. Bone Marrow Transplant 50 (8): 1083-8, 2015. [PUBMED Abstract]
- Thiel U, Wawer A, Wolf P, et al.: No improvement of survival with reduced- versus high-intensity conditioning for allogeneic stem cell transplants in Ewing tumor patients. Ann Oncol 22 (7): 1614-21, 2011. [PUBMED Abstract]
- Hoffmann C, Ahrens S, Dunst J, et al.: Pelvic Ewing sarcoma: a retrospective analysis of 241 cases. Cancer 85 (4): 869-77, 1999. [PUBMED Abstract]
- Sucato DJ, Rougraff B, McGrath BE, et al.: Ewing's sarcoma of the pelvis. Long-term survival and functional outcome. Clin Orthop (373): 193-201, 2000. [PUBMED Abstract]
- Bacci G, Ferrari S, Mercuri M, et al.: Multimodal therapy for the treatment of nonmetastatic Ewing sarcoma of pelvis. J Pediatr Hematol Oncol 25 (2): 118-24, 2003. [PUBMED Abstract]
- Bacci G, Ferrari S, Longhi A, et al.: Local and systemic control in Ewing's sarcoma of the femur treated with chemotherapy, and locally by radiotherapy and/or surgery. J Bone Joint Surg Br 85 (1): 107-14, 2003. [PUBMED Abstract]
- Ozaki T, Hillmann A, Hoffmann C, et al.: Ewing's sarcoma of the femur. Prognosis in 69 patients treated by the CESS group. Acta Orthop Scand 68 (1): 20-4, 1997. [PUBMED Abstract]
- Ayoub KS, Fiorenza F, Grimer RJ, et al.: Extensible endoprostheses of the humerus after resection of bone tumours. J Bone Joint Surg Br 81 (3): 495-500, 1999. [PUBMED Abstract]
- Bacci G, Palmerini E, Staals EL, et al.: Ewing's sarcoma family tumors of the humerus: outcome of patients treated with radiotherapy, surgery or surgery and adjuvant radiotherapy. Radiother Oncol 93 (2): 383-7, 2009. [PUBMED Abstract]
- Casadei R, Magnani M, Biagini R, et al.: Prognostic factors in Ewing's sarcoma of the foot. Clin Orthop (420): 230-8, 2004. [PUBMED Abstract]
- Anakwenze OA, Parker WL, Wold LE, et al.: Ewing's sarcoma of the hand. J Hand Surg Eur Vol 34 (1): 35-9, 2009. [PUBMED Abstract]
- Shamberger RC, Laquaglia MP, Krailo MD, et al.: Ewing sarcoma of the rib: results of an intergroup study with analysis of outcome by timing of resection. J Thorac Cardiovasc Surg 119 (6): 1154-61, 2000. [PUBMED Abstract]
- Sirvent N, Kanold J, Levy C, et al.: Non-metastatic Ewing's sarcoma of the ribs: the French Society of Pediatric Oncology Experience. Eur J Cancer 38 (4): 561-7, 2002. [PUBMED Abstract]
- Shamberger RC, LaQuaglia MP, Gebhardt MC, et al.: Ewing sarcoma/primitive neuroectodermal tumor of the chest wall: impact of initial versus delayed resection on tumor margins, survival, and use of radiation therapy. Ann Surg 238 (4): 563-7; discussion 567-8, 2003. [PUBMED Abstract]
- Schuck A, Ahrens S, Konarzewska A, et al.: Hemithorax irradiation for Ewing tumors of the chest wall. Int J Radiat Oncol Biol Phys 54 (3): 830-8, 2002. [PUBMED Abstract]
- Windfuhr JP: Primitive neuroectodermal tumor of the head and neck: incidence, diagnosis, and management. Ann Otol Rhinol Laryngol 113 (7): 533-43, 2004. [PUBMED Abstract]
- Venkateswaran L, Rodriguez-Galindo C, Merchant TE, et al.: Primary Ewing tumor of the vertebrae: clinical characteristics, prognostic factors, and outcome. Med Pediatr Oncol 37 (1): 30-5, 2001. [PUBMED Abstract]
- Marco RA, Gentry JB, Rhines LD, et al.: Ewing's sarcoma of the mobile spine. Spine 30 (7): 769-73, 2005. [PUBMED Abstract]
- Schuck A, Ahrens S, von Schorlemer I, et al.: Radiotherapy in Ewing tumors of the vertebrae: treatment results and local relapse analysis of the CESS 81/86 and EICESS 92 trials. Int J Radiat Oncol Biol Phys 63 (5): 1562-7, 2005. [PUBMED Abstract]
- Bacci G, Boriani S, Balladelli A, et al.: Treatment of nonmetastatic Ewing's sarcoma family tumors of the spine and sacrum: the experience from a single institution. Eur Spine J 18 (8): 1091-5, 2009. [PUBMED Abstract]
- Spiller M, Bisogno G, Ferrari A, et al.: Prognostic factors in localized extraosseus Ewing family tumors. [Abstract] Pediatr Blood Cancer 46 (10) : A-PD.024, 434, 2006.
- Ladenstein R, Pötschger U, Jürgens H, et al.: Comparison of treatment concepts for extraosseus Ewing tumors (EET) within consecutive trials of two GPOH Cooperative Study Groups. [Abstract] Pediatr Blood Cancer 45 (10) : A-P.C.004, 450, 2005.
- Castex MP, Rubie H, Stevens MC, et al.: Extraosseous localized ewing tumors: improved outcome with anthracyclines--the French society of pediatric oncology and international society of pediatric oncology. J Clin Oncol 25 (10): 1176-82, 2007. [PUBMED Abstract]
- Dantonello TM, Int-Veen C, Harms D, et al.: Cooperative trial CWS-91 for localized soft tissue sarcoma in children, adolescents, and young adults. J Clin Oncol 27 (9): 1446-55, 2009. [PUBMED Abstract]
- Granowetter L, Womer R, Devidas M, et al.: Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children's Oncology Group Study. J Clin Oncol 27 (15): 2536-41, 2009. [PUBMED Abstract]
- Cash T, McIlvaine E, Krailo MD, et al.: Comparison of clinical features and outcomes in patients with extraskeletal versus skeletal localized Ewing sarcoma: A report from the Children's Oncology Group. Pediatr Blood Cancer 63 (10): 1771-9, 2016. [PUBMED Abstract]
- Collier AB 3rd, Simpson L, Monteleone P: Cutaneous Ewing sarcoma: report of 2 cases and literature review of presentation, treatment, and outcome of 76 other reported cases. J Pediatr Hematol Oncol 33 (8): 631-4, 2011. [PUBMED Abstract]
- Terrier-Lacombe MJ, Guillou L, Chibon F, et al.: Superficial primitive Ewing's sarcoma: a clinicopathologic and molecular cytogenetic analysis of 14 cases. Mod Pathol 22 (1): 87-94, 2009. [PUBMED Abstract]
- Di Giannatale A, Frezza AM, Le Deley MC, et al.: Primary cutaneous and subcutaneous Ewing sarcoma. Pediatr Blood Cancer 62 (9): 1555-61, 2015. [PUBMED Abstract]
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014. [PUBMED Abstract]
- Corrigan JJ, Feig SA; American Academy of Pediatrics: Guidelines for pediatric cancer centers. Pediatrics 113 (6): 1833-5, 2004. [PUBMED Abstract]
Treatment of Localized Ewing Sarcoma
Standard Treatment Options for Localized Ewing Sarcoma
Standard treatment options for localized Ewing sarcoma include the following:
Because most patients with apparently localized disease at diagnosis have occult metastatic disease, multidrug chemotherapy and local disease control with surgery and/or radiation therapy is indicated in the treatment of all patients.[1-8] Current regimens for the treatment of localized Ewing sarcoma achieve event-free survival (EFS) and overall survival (OS) of approximately 70% at 5 years after diagnosis.[9]
Chemotherapy
Current standard chemotherapy in the United States includes vincristine, doxorubicin, and cyclophosphamide (VDC), alternating with ifosfamide and etoposide (IE) or VDC/IE.[9]; [10][Level of evidence: 1iiA]
Evidence (chemotherapy):
- IE has shown activity in Ewing sarcoma, and a large randomized clinical trial and a nonrandomized trial demonstrated that outcome was improved when IE was alternated with VDC.[2,9,11]
- Dactinomycin is no longer used for Ewing sarcoma in the United States but continues to be used in the Euro-Ewing studies.
- Increased dose intensity of doxorubicin during the initial months of therapy was associated with an improved outcome in a meta-analysis performed before the standard use of IE.[12]
- The use of high-dose VDC has shown promising results in small numbers of patients. A single-institution study of 44 patients treated with high-dose VDC and IE showed an 82% 4-year EFS.[13]
- However, in an intergroup trial of the Pediatric Oncology Group and the Children's Cancer Group, which compared an alkylator dose-intensified VDC/IE regimen with standard alkylator doses of the same VDC/IE regimen, no differences in outcome were observed.[14] Unlike the single-institution trial, this trial did not maintain the dose intensity of cyclophosphamide for the duration of treatment.[13]
In a Children's Oncology Group (COG) trial (COG-AEWS0031), 568 patients with newly diagnosed localized extradural Ewing sarcoma were randomly assigned to receive chemotherapy (VDC/IE) given either every 2 weeks (interval compression) or every 3 weeks (standard). Patients randomly assigned to the every 2-week interval of treatment had an improved 5-year EFS (73% vs. 65%, P = .048). There was no increase in toxicity observed with the every 2-week schedule.[10]
Local-control measures
Local control can be achieved by surgery and/or radiation therapy.
Surgery
Surgery is generally the preferred approach if the lesion is resectable.[15,16] The superiority of resection for local control has never been tested in a prospective randomized trial. The apparent superiority may represent selection bias.
- In past studies, smaller, more peripheral tumors were more likely to be treated with surgery, and larger, more central tumors were more likely to be treated with radiation therapy.[17]
- An Italian retrospective study showed that surgery improved outcome only in extremity tumors, although the number of patients with central axis Ewing sarcoma who achieved adequate margins was small.[8]
- In a series of 39 patients treated at St. Jude Children's Research Hospital who received both surgery and radiation, the 8-year local failure rate was 5% for patients with negative surgical margins and 17% for those with positive margins.[5]
- Data for patients with pelvic primary Ewing sarcoma from a North American intergroup trial showed no difference in local control or survival based on local-control modality—surgery alone, radiation therapy alone, or radiation plus surgery.[18]
Potential benefits of surgery include the following:
- If a very young child has Ewing sarcoma, surgery may be a less-morbid therapy than radiation therapy because of the retardation of bone growth caused by radiation.
- Another potential benefit for surgical resection of the primary tumor is related to the amount of necrosis in the resected tumor. Patients with residual viable tumor in the resected specimen have a worse outcome than those with complete necrosis. In a French Ewing study (EW88), EFS for patients with less than 5% viable tumor was 75%, EFS for patients with 5% to 30% viable tumor was 48%, and EFS for patients with more than 30% viable tumor was 20%.[17]
European investigators are studying whether treatment intensification (i.e., high-dose chemotherapy with stem cell rescue) will improve outcome for patients with a poor histologic response.
Radiation therapy is usually employed in the following cases:
- Patients who do not have a surgical option that preserves function.
- Patients whose tumors have been excised but with inadequate margins.
Pathologic fracture at the time of diagnosis does not preclude surgical resection and is not associated with adverse outcome.[19]
Radiation therapy
Radiation therapy is delivered in a setting in which stringent planning techniques are applied by those experienced in the treatment of Ewing sarcoma. Such an approach will result in local control of the tumor with acceptable morbidity in most patients.[1,2,20]
The radiation dose may be adjusted depending on the extent of residual disease after the initial surgical procedure. Radiation therapy is generally administered in fractionated doses totaling approximately 55.8 Gy to the prechemotherapy tumor volume. A randomized study of 40 patients with Ewing sarcoma using 55.8 Gy to the prechemotherapy tumor extent with a 2-cm margin compared with the same total-tumor dose after 39.6 Gy to the entire bone showed no difference in local control or EFS.[3] Hyperfractionated radiation therapy has not been associated with improved local control or decreased morbidity.[1]
Comparison of proton-beam radiation therapy and intensity-modulated radiation therapy (IMRT) treatment plans has shown that proton-beam radiation therapy can spare more normal tissue adjacent to Ewing sarcoma primary tumors than IMRT.[21] Follow-up remains relatively short, and there are no data available to determine whether the reduction in dose to adjacent tissue will result in improved functional outcome or reduce the risk of secondary malignancy. Because patient numbers are small and follow-up is relatively short, it is not possible to determine whether the risk of local recurrence might be increased by reducing radiation dose in tissue adjacent to the primary tumor.
Higher rates of local failure are seen in patients older than 14 years who have tumors more than 8 cm in length.[22] A retrospective analysis of patients with Ewing sarcoma of the chest wall compared patients who received hemithorax radiation therapy with those who received radiation therapy to the chest wall only. Patients with pleural invasion, pleural effusion, or intraoperative contamination were assigned to hemithorax radiation therapy. EFS was longer for patients who received hemithorax radiation, but the difference was not statistically significant. In addition, most patients with primary vertebral tumors did not receive hemithorax radiation and had a lower probability for EFS.[23]
For patients with residual disease after an attempt at surgical resection, the Intergroup Ewing Sarcoma Study (INT-0091) recommended 45 Gy to the original disease site plus a 10.8 Gy boost for patients with gross residual disease and 45 Gy plus a 5.4 Gy boost for patients with microscopic residual disease. No radiation therapy was recommended for those who have no evidence of microscopic residual disease after surgical resection.[14]
Radiation therapy is associated with the development of subsequent neoplasms. A retrospective study noted that patients who received 60 Gy or more had an incidence of second malignancy of 20%. Those who received 48 Gy to 60 Gy had an incidence of 5%, and those who received less than 48 Gy did not develop a second malignancy.[24] (Refer to the Late Effects of Treatment for Ewing Sarcoma section of this summary for more information.)
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Dunst J, Jürgens H, Sauer R, et al.: Radiation therapy in Ewing's sarcoma: an update of the CESS 86 trial. Int J Radiat Oncol Biol Phys 32 (4): 919-30, 1995. [PUBMED Abstract]
- Donaldson SS, Torrey M, Link MP, et al.: A multidisciplinary study investigating radiotherapy in Ewing's sarcoma: end results of POG #8346. Pediatric Oncology Group. Int J Radiat Oncol Biol Phys 42 (1): 125-35, 1998. [PUBMED Abstract]
- Craft A, Cotterill S, Malcolm A, et al.: Ifosfamide-containing chemotherapy in Ewing's sarcoma: The Second United Kingdom Children's Cancer Study Group and the Medical Research Council Ewing's Tumor Study. J Clin Oncol 16 (11): 3628-33, 1998. [PUBMED Abstract]
- Nilbert M, Saeter G, Elomaa I, et al.: Ewing's sarcoma treatment in Scandinavia 1984-1990--ten-year results of the Scandinavian Sarcoma Group Protocol SSGIV. Acta Oncol 37 (4): 375-8, 1998. [PUBMED Abstract]
- Krasin MJ, Davidoff AM, Rodriguez-Galindo C, et al.: Definitive surgery and multiagent systemic therapy for patients with localized Ewing sarcoma family of tumors: local outcome and prognostic factors. Cancer 104 (2): 367-73, 2005. [PUBMED Abstract]
- Bacci G, Forni C, Longhi A, et al.: Long-term outcome for patients with non-metastatic Ewing's sarcoma treated with adjuvant and neoadjuvant chemotherapies. 402 patients treated at Rizzoli between 1972 and 1992. Eur J Cancer 40 (1): 73-83, 2004. [PUBMED Abstract]
- Rosito P, Mancini AF, Rondelli R, et al.: Italian Cooperative Study for the treatment of children and young adults with localized Ewing sarcoma of bone: a preliminary report of 6 years of experience. Cancer 86 (3): 421-8, 1999. [PUBMED Abstract]
- Bacci G, Longhi A, Briccoli A, et al.: The role of surgical margins in treatment of Ewing's sarcoma family tumors: experience of a single institution with 512 patients treated with adjuvant and neoadjuvant chemotherapy. Int J Radiat Oncol Biol Phys 65 (3): 766-72, 2006. [PUBMED Abstract]
- Grier HE, Krailo MD, Tarbell NJ, et al.: Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 348 (8): 694-701, 2003. [PUBMED Abstract]
- Womer RB, West DC, Krailo MD, et al.: Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 30 (33): 4148-54, 2012. [PUBMED Abstract]
- Ferrari S, Mercuri M, Rosito P, et al.: Ifosfamide and actinomycin-D, added in the induction phase to vincristine, cyclophosphamide and doxorubicin, improve histologic response and prognosis in patients with non metastatic Ewing's sarcoma of the extremity. J Chemother 10 (6): 484-91, 1998. [PUBMED Abstract]
- Smith MA, Ungerleider RS, Horowitz ME, et al.: Influence of doxorubicin dose intensity on response and outcome for patients with osteogenic sarcoma and Ewing's sarcoma. J Natl Cancer Inst 83 (20): 1460-70, 1991. [PUBMED Abstract]
- Kolb EA, Kushner BH, Gorlick R, et al.: Long-term event-free survival after intensive chemotherapy for Ewing's family of tumors in children and young adults. J Clin Oncol 21 (18): 3423-30, 2003. [PUBMED Abstract]
- Granowetter L, Womer R, Devidas M, et al.: Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children's Oncology Group Study. J Clin Oncol 27 (15): 2536-41, 2009. [PUBMED Abstract]
- Hoffmann C, Ahrens S, Dunst J, et al.: Pelvic Ewing sarcoma: a retrospective analysis of 241 cases. Cancer 85 (4): 869-77, 1999. [PUBMED Abstract]
- Shamberger RC, Laquaglia MP, Krailo MD, et al.: Ewing sarcoma of the rib: results of an intergroup study with analysis of outcome by timing of resection. J Thorac Cardiovasc Surg 119 (6): 1154-61, 2000. [PUBMED Abstract]
- Oberlin O, Deley MC, Bui BN, et al.: Prognostic factors in localized Ewing's tumours and peripheral neuroectodermal tumours: the third study of the French Society of Paediatric Oncology (EW88 study). Br J Cancer 85 (11): 1646-54, 2001. [PUBMED Abstract]
- Yock TI, Krailo M, Fryer CJ, et al.: Local control in pelvic Ewing sarcoma: analysis from INT-0091--a report from the Children's Oncology Group. J Clin Oncol 24 (24): 3838-43, 2006. [PUBMED Abstract]
- Bramer JA, Abudu AA, Grimer RJ, et al.: Do pathological fractures influence survival and local recurrence rate in bony sarcomas? Eur J Cancer 43 (13): 1944-51, 2007. [PUBMED Abstract]
- Krasin MJ, Rodriguez-Galindo C, Billups CA, et al.: Definitive irradiation in multidisciplinary management of localized Ewing sarcoma family of tumors in pediatric patients: outcome and prognostic factors. Int J Radiat Oncol Biol Phys 60 (3): 830-8, 2004. [PUBMED Abstract]
- Rombi B, DeLaney TF, MacDonald SM, et al.: Proton radiotherapy for pediatric Ewing's sarcoma: initial clinical outcomes. Int J Radiat Oncol Biol Phys 82 (3): 1142-8, 2012. [PUBMED Abstract]
- Fuchs B, Valenzuela RG, Sim FH: Pathologic fracture as a complication in the treatment of Ewing's sarcoma. Clin Orthop (415): 25-30, 2003. [PUBMED Abstract]
- Schuck A, Ahrens S, Konarzewska A, et al.: Hemithorax irradiation for Ewing tumors of the chest wall. Int J Radiat Oncol Biol Phys 54 (3): 830-8, 2002. [PUBMED Abstract]
- Kuttesch JF Jr, Wexler LH, Marcus RB, et al.: Second malignancies after Ewing's sarcoma: radiation dose-dependency of secondary sarcomas. J Clin Oncol 14 (10): 2818-25, 1996. [PUBMED Abstract]
Treatment of Metastatic Ewing Sarcoma
Metastases at diagnosis are detected in approximately 25% of patients.[1] The prognosis of patients with metastatic disease is poor. Current therapies for patients who present with metastatic disease achieve 6-year event-free survival (EFS) of approximately 28% and overall survival (OS) of approximately 30%.[2,3] For patients with lung/pleural metastases only, 6-year EFS is approximately 40% when utilizing bilateral lung irradiation.[2,4] In contrast, patients with bone/bone marrow metastases have a 4-year EFS of approximately 28% and patients with combined lung and bone/bone marrow metastases have a 4-year EFS of approximately 14%.[4,5]
The following factors independently predict a poor outcome in patients presenting with metastatic disease:[3]
- Age older than 14 years.
- Primary tumor volume of more than 200 mL.
- More than one bone metastatic site.
- Bone marrow metastases.
- Additional lung metastases.
Standard Treatment Options for Metastatic Ewing Sarcoma
Standard treatment options for metastatic Ewing sarcoma include the following:
Chemotherapy
Standard treatment for patients with metastatic Ewing sarcoma utilizing alternating vincristine, doxorubicin, cyclophosphamide, and ifosfamide/etoposide combined with adequate local-control measures applied to both primary and metastatic sites often results in complete or partial responses; however, the overall cure rate is 20%.[5-7]
The following chemotherapy regimens have not shown benefit:
- In the Intergroup Ewing Sarcoma Study, patients with metastatic disease showed no benefit from the addition of ifosfamide and etoposide to a standard regimen of vincristine, doxorubicin, cyclophosphamide, and dactinomycin.[7]
- In another Intergroup study, increasing dose intensity of cyclophosphamide, ifosfamide, and doxorubicin did not improve outcome compared with regimens utilizing standard-dose intensity. This regimen increased toxicity and risk of second malignancy without improving EFS or OS.[2]
- Intensification of ifosfamide to 2.8 g/m2 per day for 5 days did not improve outcome when administered with standard chemotherapy in patients with newly diagnosed metastatic Ewing sarcoma.[8][Level of evidence: 3iiiDi]
Surgery and radiation therapy
Systematic use of surgery and radiation therapy for metastatic sites may improve overall outcome in patients with extrapulmonary metastases.
Evidence (surgery and radiation therapy):
- In a retrospective data analysis of 120 patients with multifocal metastatic Ewing sarcoma, patients receiving local treatment of both primary tumor and metastases had a better outcome than patients receiving local treatment of primary tumor only or with no local treatment (3-year EFS, 39% vs. 17% and 14%, P < .001).[9]
- A similar trend for better outcome with irradiation of all sites of metastatic disease was seen in three retrospective analyses of smaller groups of patients receiving radiation therapy to all tumor sites.[10-12] These results must be interpreted with caution. The patients who received local-control therapy to all known sites of metastatic disease were selected by the treating investigator, not randomly assigned. Patients with so many metastases that radiation to all sites would result in bone marrow failure were not selected to receive radiation to all sites of metastatic disease. Patients who did not achieve control of the primary tumor did not go on to have local control of all sites of metastatic disease. There was a selection bias such that while all patients in these reports had multiple sites of metastatic disease, the patients who had surgery and/or radiation therapy to all sites of clinically detectable metastatic disease had better responses to systemic therapy and fewer sites of metastasis than did patients who did not undergo similar therapy of metastatic sites.
Radiation therapy, delivered in a setting in which stringent planning techniques are applied by those experienced in the treatment of Ewing sarcoma, should be considered. Such an approach will result in local control of tumor with acceptable morbidity in most patients.[13]
The radiation dose depends on the metastatic site of disease:
- Bone and soft tissue. Stereotactic body radiation therapy has been used to treat metastatic sites in bone and soft tissue. The median total curative/definitive stereotactic body radiation therapy dose delivered was 40 Gy in five fractions (range, 30–60 Gy in 3–10 fractions). The median total palliative stereotactic body radiation therapy dose delivered was 40 Gy in five fractions (range, 16–50 Gy in 1–10 fractions). These short-course regimens with large-dose fractions are biologically equivalent to higher doses delivered with smaller-dose fractions given over longer treatment courses.[14][Level of evidence: 3iiiC]
- Pulmonary. For all patients with pulmonary metastases, whole-lung irradiation should be considered, even if complete resolution of overt pulmonary metastatic disease has been achieved with chemotherapy.[4,5,15] Radiation doses are modulated based on the amount of lung to be irradiated and on pulmonary function. Doses between 12 Gy and 15 Gy are generally used if whole lungs are treated.
Other therapies
More intensive therapies, many of which incorporate high-dose chemotherapy with or without total-body irradiation in conjunction with stem cell support, have not shown improvement in EFS rates for patients with bone and/or bone marrow metastases.[2,3,10,16-18]; [19][Level of evidence: 3iiiDi] (Refer to the High-Dose Therapy With Stem Cell Rescue for Ewing Sarcoma section of this summary for more information.)
- High-dose chemotherapy with stem cell support. One of the largest studies was the EURO-EWING-Intergroup-EE99 R3 trial that enrolled 281 patients with primary disseminated metastatic Ewing sarcoma. Patients were treated with six cycles of vincristine, ifosfamide, doxorubicin, and etoposide followed by high-dose therapy and autologous stem cell transplant and demonstrated a 3-year EFS of 27% and OS of 34%. Factors such as the presence and number of bone lesions, primary tumor volume greater than 200 mL, age older than 14 years, additional pulmonary metastases, and bone marrow involvement were identified as independent prognostic factors.[3][Level of evidence: 3iiDi] The impact of high-dose chemotherapy with peripheral blood stem cell support for patients with isolated lung metastases is unknown and is being studied in the EURO-EWING-INTERGROUP-EE99 trial, for which results are pending.[16]
- Melphalan. Melphalan, at nonmyeloablative doses, proved to be an active agent in an upfront window study for patients with metastatic disease at diagnosis; however, the cure rate remained extremely low.[20]
- Irinotecan. Irinotecan was administered as a single agent in an upfront window for newly diagnosed metastatic Ewing sarcoma patients and showed modest activity (partial response in 5 of 24 patients).[21][Level of evidence: 3iiiDiv] Further investigation is needed to determine irinotecan dosing and combinations with other agents for patients with Ewing sarcoma.
Treatment Options Under Clinical Evaluation for Metastatic Ewing Sarcoma
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- AEWS1221; NCI-2014-02380 (NCT02306161) (Combination Chemotherapy With or Without Ganitumab in Treating Patients With Newly Diagnosed Metastatic Ewing Sarcoma): This phase II study is randomly assigning newly diagnosed patients with metastatic Ewing sarcoma to multiagent chemotherapy (vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide) with or without the addition of ganitumab (AMG 479). Stereotactic body radiation therapy is being evaluated to sites of bone metastases at a dose of 40 Gy in five fractions. This is a shorter course of therapy than is the standard treatment.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Esiashvili N, Goodman M, Marcus RB Jr: Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J Pediatr Hematol Oncol 30 (6): 425-30, 2008. [PUBMED Abstract]
- Miser JS, Goldsby RE, Chen Z, et al.: Treatment of metastatic Ewing sarcoma/primitive neuroectodermal tumor of bone: evaluation of increasing the dose intensity of chemotherapy--a report from the Children's Oncology Group. Pediatr Blood Cancer 49 (7): 894-900, 2007. [PUBMED Abstract]
- Ladenstein R, Pötschger U, Le Deley MC, et al.: Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol 28 (20): 3284-91, 2010. [PUBMED Abstract]
- Paulussen M, Ahrens S, Craft AW, et al.: Ewing's tumors with primary lung metastases: survival analysis of 114 (European Intergroup) Cooperative Ewing's Sarcoma Studies patients. J Clin Oncol 16 (9): 3044-52, 1998. [PUBMED Abstract]
- Paulussen M, Ahrens S, Burdach S, et al.: Primary metastatic (stage IV) Ewing tumor: survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma Studies. Ann Oncol 9 (3): 275-81, 1998. [PUBMED Abstract]
- Pinkerton CR, Bataillard A, Guillo S, et al.: Treatment strategies for metastatic Ewing's sarcoma. Eur J Cancer 37 (11): 1338-44, 2001. [PUBMED Abstract]
- Miser JS, Krailo MD, Tarbell NJ, et al.: Treatment of metastatic Ewing's sarcoma or primitive neuroectodermal tumor of bone: evaluation of combination ifosfamide and etoposide--a Children's Cancer Group and Pediatric Oncology Group study. J Clin Oncol 22 (14): 2873-6, 2004. [PUBMED Abstract]
- Magnan H, Goodbody CM, Riedel E, et al.: Ifosfamide dose-intensification for patients with metastatic Ewing sarcoma. Pediatr Blood Cancer 62 (4): 594-7, 2015. [PUBMED Abstract]
- Haeusler J, Ranft A, Boelling T, et al.: The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES). Cancer 116 (2): 443-50, 2010. [PUBMED Abstract]
- Burdach S, Thiel U, Schöniger M, et al.: Total body MRI-governed involved compartment irradiation combined with high-dose chemotherapy and stem cell rescue improves long-term survival in Ewing tumor patients with multiple primary bone metastases. Bone Marrow Transplant 45 (3): 483-9, 2010. [PUBMED Abstract]
- Paulino AC, Mai WY, Teh BS: Radiotherapy in metastatic ewing sarcoma. Am J Clin Oncol 36 (3): 283-6, 2013. [PUBMED Abstract]
- Casey DL, Wexler LH, Meyers PA, et al.: Radiation for bone metastases in Ewing sarcoma and rhabdomyosarcoma. Pediatr Blood Cancer 62 (3): 445-9, 2015. [PUBMED Abstract]
- Donaldson SS, Torrey M, Link MP, et al.: A multidisciplinary study investigating radiotherapy in Ewing's sarcoma: end results of POG #8346. Pediatric Oncology Group. Int J Radiat Oncol Biol Phys 42 (1): 125-35, 1998. [PUBMED Abstract]
- Brown LC, Lester RA, Grams MP, et al.: Stereotactic body radiotherapy for metastatic and recurrent ewing sarcoma and osteosarcoma. Sarcoma 2014: 418270, 2014. [PUBMED Abstract]
- Spunt SL, McCarville MB, Kun LE, et al.: Selective use of whole-lung irradiation for patients with Ewing sarcoma family tumors and pulmonary metastases at the time of diagnosis. J Pediatr Hematol Oncol 23 (2): 93-8, 2001. [PUBMED Abstract]
- Meyers PA, Krailo MD, Ladanyi M, et al.: High-dose melphalan, etoposide, total-body irradiation, and autologous stem-cell reconstitution as consolidation therapy for high-risk Ewing's sarcoma does not improve prognosis. J Clin Oncol 19 (11): 2812-20, 2001. [PUBMED Abstract]
- Burdach S, Meyer-Bahlburg A, Laws HJ, et al.: High-dose therapy for patients with primary multifocal and early relapsed Ewing's tumors: results of two consecutive regimens assessing the role of total-body irradiation. J Clin Oncol 21 (16): 3072-8, 2003. [PUBMED Abstract]
- Thiel U, Wawer A, Wolf P, et al.: No improvement of survival with reduced- versus high-intensity conditioning for allogeneic stem cell transplants in Ewing tumor patients. Ann Oncol 22 (7): 1614-21, 2011. [PUBMED Abstract]
- Loschi S, Dufour C, Oberlin O, et al.: Tandem high-dose chemotherapy strategy as first-line treatment of primary disseminated multifocal Ewing sarcomas in children, adolescents and young adults. Bone Marrow Transplant 50 (8): 1083-8, 2015. [PUBMED Abstract]
- Luksch R, Grignani G, Fagioli F, et al.: Response to melphalan in up-front investigational window therapy for patients with metastatic Ewing's family tumours. Eur J Cancer 43 (5): 885-90, 2007. [PUBMED Abstract]
- Morland B, Platt K, Whelan JS: A phase II window study of irinotecan (CPT-11) in high risk Ewing sarcoma: a Euro-E.W.I.N.G. study. Pediatr Blood Cancer 61 (3): 442-5, 2014. [PUBMED Abstract]
Treatment of Recurrent Ewing Sarcoma
Recurrence of Ewing sarcoma is most common within 2 years of initial diagnosis (approximately 80%).[1,2] However, late relapses occurring more than 5 years from initial diagnosis are more common in Ewing sarcoma (13%; 95% confidence interval, 9.4–16.5) than in other pediatric solid tumors.[3] An analysis of the Surveillance, Epidemiology, and End Results database identified 1,351 patients who survived more than 60 months from diagnosis.[4] Of these patients, 209 died, with 144 of the deaths (69%) attributed to recurrent, progressive Ewing sarcoma. Black race, male sex, older age at initial diagnosis, and primary tumors of the pelvis and axial skeleton were associated with a higher risk of late death. This analysis covered the period from 1973 to 2013, and the 1,351 patients represented only 38% of the patients in the original sample, which reflects the inferior treatment outcomes from the earlier era. It is possible that patients who reach the 5-year point after more contemporary treatment may not recapitulate this experience.
The overall prognosis for patients with recurrent Ewing sarcoma is poor; 5-year survival after recurrence is approximately 10% to 15%.[2,5,6]; [1][Level of evidence: 3iiA]
Prognostic factors include the following:
- Time to recurrence. Time to recurrence is the most important prognostic factor. Patients whose Ewing sarcoma recurred more than 2 years from initial diagnosis had a 5-year survival of 30% versus 7% for patients whose Ewing sarcoma recurred within 2 years.[1,2]
- Local and distant recurrence. Patients with both local recurrence and distant metastases have a worse outcome than do patients with either isolated local recurrence or metastatic recurrence alone.[1,2]
- Isolated pulmonary recurrence. Isolated pulmonary recurrence was not an important prognostic factor in a North American series.[1] In the Italian/Scandinavian experience, younger age, longer disease-free interval, and lung-only recurrence were associated with longer progression-free survival after recurrence. In this experience, patients with Ewing sarcoma that recurred after initial therapy, which included high-dose therapy with autologous stem cell rescue, were less likely to achieve a second complete remission.[7][Level of evidence: 3iiDiii]
Treatment Options for Recurrent Ewing Sarcoma
The selection of treatment for patients with recurrent disease depends on many factors, including the following:
- Site of recurrence.
- Previous treatment.
- Individual patient considerations.
There is no standardized second-line treatment for relapsed or refractory Ewing sarcoma.
Treatment options for recurrent Ewing sarcoma include the following:
Chemotherapy
Combinations of chemotherapy, such as cyclophosphamide and topotecan or irinotecan and temozolomide with or without vincristine, are active in recurrent Ewing sarcoma and can be considered for these patients.[8-13]
Evidence (chemotherapy):
- One phase II study of topotecan and cyclophosphamide showed a response in 6 of 17 patients with Ewing sarcoma; 16 of 49 patients had a clinical response in a similar trial in Germany.[8,10]
- In one retrospective series, 20 patients received temozolomide and irinotecan after recurrence. Five patients achieved a complete response and seven patients achieved a partial response.[12] A second retrospective series reported 11 of 20 objective responses in patients with recurrent Ewing sarcoma.[14][Level of evidence: 3iiDiv]
- The combination of docetaxel either with gemcitabine or irinotecan has achieved objective responses in relapsed Ewing sarcoma.[15][Level of evidence: 3iiA]; [16,17][Level of evidence: 3iiiDiv]
- High-dose ifosfamide (3 g/m2 per day for 5 days = 15 g/m2) has shown activity in patients whose Ewing sarcoma recurred after therapy that included standard ifosfamide (1.8 g/m2 per day for 5 days = 9 g/m2).[18][Level of evidence: 3iiiDiv]
Radiation therapy
Radiation therapy to bone lesions may provide palliation, although radical resection may improve outcome.[2] Patients with pulmonary metastases who have not received radiation therapy to the lungs should be considered for whole-lung irradiation.[19] Residual disease in the lung may be surgically removed.
Other therapies
Other therapies that have been studied in the treatment of recurrent Ewing sarcoma include the following:
- High-dose chemotherapy with stem cell support. Aggressive attempts to control the disease, including myeloablative regimens, have been used, but there is no evidence at this time to conclude that myeloablative therapy is superior to standard chemotherapy.[20,21]; [22][Level of evidence: 3iiA]; [23][Level of evidence: 3iiiDiii]
Most published reports about the use of high-dose therapy and stem cell support for patients with high-risk Ewing sarcoma have significant flaws in methodology. The most common error is the comparison of this high-risk group with an inappropriate control group. Patients with Ewing sarcoma at high risk of treatment failure who received high-dose therapy are compared with patients who did not receive high-dose therapy. Patients who undergo high-dose therapy must respond to systemic therapy, remain alive and respond to treatment long enough to reach the time at which stem cell therapy can be applied, be free of comorbid toxicity that precludes high-dose therapy, and have an adequate stem cell collection. Patients who undergo high-dose therapy and stem cell support are a highly selected group; comparing this patient group with all patients with high-risk Ewing sarcoma is inappropriate and leads to the erroneous conclusion that this strategy improves outcome. Surveys of patients undergoing allogeneic stem cell transplantation (SCT) for recurrent Ewing sarcoma did not show improved event-free survival when compared with autologous SCT and was associated with a higher complication rate.[20,24,25]
- Monoclonal antibody therapy. Monoclonal antibodies against the insulin-like growth factor 1 receptor (IGF1R) are reported to produce objective responses in metastatic recurrent Ewing sarcoma in roughly 10% of cases.[26-29][Level of evidence: 3iiDiv] In these studies, it was suggested that time-to-progression was prolonged compared with historical controls. Objective responses have been reported in studies combining the mTOR inhibitor temsirolimus with an IGF1R antibody. Stratification by IGF1R expression by immunohistochemistry in one of the studies did not predict clinical outcome in Ewing sarcoma patients.[30,31] Further studies are needed to identify patients who are likely to benefit from IGF1R therapy.
- Immunotherapy. Immunotherapy with antigen-specific T cells is being studied in patients with Ewing sarcoma because immune-mediated killing does not rely on pathways used by conventional therapies to which such tumors are often resistant. Several potential chimeric antigen receptors target antigens that have been identified for Ewing sarcomas. These include HER2 (human epidermal growth factor receptor 2),[32] GD2,[33] CD99 (MIC2 antigens),[34] and STEAP1 (six-transmembrane epithelial antigens of the prostate).[35] Some are in early-phase testing in sarcoma patients.[32]
Treatment Options Under Clinical Evaluation for Recurrent Ewing Sarcoma
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- APEC1621 (NCT03155620) (Pediatric MATCH: Targeted Therapy Directed by Genetic Testing in Treating Pediatric Patients with Relapsed or Refractory Advanced Solid Tumors, Non-Hodgkin Lymphomas, or Histiocytic Disorders): NCI–Children's Oncology Group Pediatric Molecular Analysis for Therapeutic Choice (MATCH), referred to as Pediatric MATCH, will match targeted agents with specific molecular changes identified using a next-generation sequencing targeted assay of more than 3,000 different mutations across more than 160 genes in refractory and recurrent solid tumors. Children and adolescents aged 1 to 21 years are eligible for the trial.
Tumor tissue from progressive or recurrent disease must be available for molecular characterization. Patients with tumors that have molecular variants addressed by treatment arms included in the trial will be offered treatment on Pediatric MATCH. Additional information can be obtained on the ClinicalTrials.gov website for APEC1621 (NCT03155620).
- ADVL1622 (NCT02867592) (Cabozantinib-S-Malate in Treating Younger Patients with Recurrent, Refractory, or Newly Diagnosed Sarcomas, Wilms Tumor, or Other Rare Tumors): This is an open-label, two-stage, phase II trial of cabozantinib in selective solid tumors, including Ewing sarcoma. Cabozantinib is an oral small molecule inhibitor of multiple tyrosine kinases, including MET, VEGFR2, and RET, which are potential therapeutic targets in many pediatric and adult solid tumors.
- SARC028; NCI-2015-00320 (NCT02301039) (A Phase II Study of the Anti-PD1 Antibody Pembrolizumab [MK-3475] in Patients With Advanced Sarcomas): The objective response rate to the anti-PD1 inhibitor pembrolizumab will be assessed in patients with refractory, recurrent, and/or metastatic high-grade soft tissue sarcomas and bone sarcomas. Patients aged 18 years and older with soft tissue sarcomas and patients aged 12 years and older with bone sarcomas are eligible.
- ADVL1412 (NCT02304458) (Nivolumab With or Without Ipilimumab in Treating Younger Patients With Recurrent or Refractory Solid Tumors or Sarcomas): Nivolumab is an anti-PD1 inhibitor that is being studied alone and in combination with ipilimumab in relapsed sarcoma patients, including patients with Ewing sarcoma.
- ADVL1411 (NCT02116777) (BMN-673 and Temozolomide in Treating Younger Patients With Refractory or Recurrent Malignancies): In this study, the PARP inhibitor BMN-673 is combined with low-dose short duration temozolomide. This is based on the in vitro and mouse human tumor xenograft models, which showed impressive activity in a broad range of pediatric cancers, including Ewing sarcoma. After identifying the recommended phase II dose, this study is open for Ewing sarcoma patients.[36]
- ADVL1615 (NCT03323034) (Pevonedistat, Irinotecan Hydrochloride, and Temozolomide in Treating Patients With Recurrent or Refractory Solid Tumors or Lymphoma): This is a phase I study of pevonedistat in combination with temozolomide and irinotecan. Pevonedistat is a novel first-in-class Nedd8 activating enzyme (NAE) inhibitor that blocks the degradation of a subset of proteins that would normally be degraded by the 26S proteasome. Pevonedistat is more specific than previous proteasome inhibitors because it blocks the degradation of cullin-RING ligases, narrowing the targets to only a handful of key regulatory proteins important in cell survival. Preclinical, antitumor activity has been observed in Ewing sarcoma.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Leavey PJ, Mascarenhas L, Marina N, et al.: Prognostic factors for patients with Ewing sarcoma (EWS) at first recurrence following multi-modality therapy: A report from the Children's Oncology Group. Pediatr Blood Cancer 51 (3): 334-8, 2008. [PUBMED Abstract]
- Stahl M, Ranft A, Paulussen M, et al.: Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatr Blood Cancer 57 (4): 549-53, 2011. [PUBMED Abstract]
- Wasilewski-Masker K, Liu Q, Yasui Y, et al.: Late recurrence in pediatric cancer: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst 101 (24): 1709-20, 2009. [PUBMED Abstract]
- Davenport JR, Vo KT, Goldsby R, et al.: Conditional Survival and Predictors of Late Death in Patients With Ewing Sarcoma. Pediatr Blood Cancer 63 (6): 1091-5, 2016. [PUBMED Abstract]
- Barker LM, Pendergrass TW, Sanders JE, et al.: Survival after recurrence of Ewing's sarcoma family of tumors. J Clin Oncol 23 (19): 4354-62, 2005. [PUBMED Abstract]
- Bacci G, Longhi A, Ferrari S, et al.: Pattern of relapse in 290 patients with nonmetastatic Ewing's sarcoma family tumors treated at a single institution with adjuvant and neoadjuvant chemotherapy between 1972 and 1999. Eur J Surg Oncol 32 (9): 974-9, 2006. [PUBMED Abstract]
- Ferrari S, Luksch R, Hall KS, et al.: Post-relapse survival in patients with Ewing sarcoma. Pediatr Blood Cancer 62 (6): 994-9, 2015. [PUBMED Abstract]
- Saylors RL 3rd, Stine KC, Sullivan J, et al.: Cyclophosphamide plus topotecan in children with recurrent or refractory solid tumors: a Pediatric Oncology Group phase II study. J Clin Oncol 19 (15): 3463-9, 2001. [PUBMED Abstract]
- McTiernan A, Driver D, Michelagnoli MP, et al.: High dose chemotherapy with bone marrow or peripheral stem cell rescue is an effective treatment option for patients with relapsed or progressive Ewing's sarcoma family of tumours. Ann Oncol 17 (8): 1301-5, 2006. [PUBMED Abstract]
- Hunold A, Weddeling N, Paulussen M, et al.: Topotecan and cyclophosphamide in patients with refractory or relapsed Ewing tumors. Pediatr Blood Cancer 47 (6): 795-800, 2006. [PUBMED Abstract]
- Wagner LM, McAllister N, Goldsby RE, et al.: Temozolomide and intravenous irinotecan for treatment of advanced Ewing sarcoma. Pediatr Blood Cancer 48 (2): 132-9, 2007. [PUBMED Abstract]
- Casey DA, Wexler LH, Merchant MS, et al.: Irinotecan and temozolomide for Ewing sarcoma: the Memorial Sloan-Kettering experience. Pediatr Blood Cancer 53 (6): 1029-34, 2009. [PUBMED Abstract]
- Raciborska A, Bilska K, Drabko K, et al.: Vincristine, irinotecan, and temozolomide in patients with relapsed and refractory Ewing sarcoma. Pediatr Blood Cancer 60 (10): 1621-5, 2013. [PUBMED Abstract]
- Kurucu N, Sari N, Ilhan IE: Irinotecan and temozolamide treatment for relapsed Ewing sarcoma: a single-center experience and review of the literature. Pediatr Hematol Oncol 32 (1): 50-9, 2015. [PUBMED Abstract]
- Fox E, Patel S, Wathen JK, et al.: Phase II study of sequential gemcitabine followed by docetaxel for recurrent Ewing sarcoma, osteosarcoma, or unresectable or locally recurrent chondrosarcoma: results of Sarcoma Alliance for Research Through Collaboration Study 003. Oncologist 17 (3): 321, 2012. [PUBMED Abstract]
- Mora J, Cruz CO, Parareda A, et al.: Treatment of relapsed/refractory pediatric sarcomas with gemcitabine and docetaxel. J Pediatr Hematol Oncol 31 (10): 723-9, 2009. [PUBMED Abstract]
- Yoon JH, Kwon MM, Park HJ, et al.: A study of docetaxel and irinotecan in children and young adults with recurrent or refractory Ewing sarcoma family of tumors. BMC Cancer 14: 622, 2014. [PUBMED Abstract]
- Ferrari S, del Prever AB, Palmerini E, et al.: Response to high-dose ifosfamide in patients with advanced/recurrent Ewing sarcoma. Pediatr Blood Cancer 52 (5): 581-4, 2009. [PUBMED Abstract]
- Rodriguez-Galindo C, Billups CA, Kun LE, et al.: Survival after recurrence of Ewing tumors: the St Jude Children's Research Hospital experience, 1979-1999. Cancer 94 (2): 561-9, 2002. [PUBMED Abstract]
- Burdach S, van Kaick B, Laws HJ, et al.: Allogeneic and autologous stem-cell transplantation in advanced Ewing tumors. An update after long-term follow-up from two centers of the European Intergroup study EICESS. Stem-Cell Transplant Programs at Düsseldorf University Medical Center, Germany and St. Anna Kinderspital, Vienna, Austria. Ann Oncol 11 (11): 1451-62, 2000. [PUBMED Abstract]
- Burdach S, Meyer-Bahlburg A, Laws HJ, et al.: High-dose therapy for patients with primary multifocal and early relapsed Ewing's tumors: results of two consecutive regimens assessing the role of total-body irradiation. J Clin Oncol 21 (16): 3072-8, 2003. [PUBMED Abstract]
- Rasper M, Jabar S, Ranft A, et al.: The value of high-dose chemotherapy in patients with first relapsed Ewing sarcoma. Pediatr Blood Cancer 61 (8): 1382-6, 2014. [PUBMED Abstract]
- Gardner SL, Carreras J, Boudreau C, et al.: Myeloablative therapy with autologous stem cell rescue for patients with Ewing sarcoma. Bone Marrow Transplant 41 (10): 867-72, 2008. [PUBMED Abstract]
- Gilman AL, Oesterheld J: Myeloablative chemotherapy with autologous stem cell rescue for Ewing sarcoma. Bone Marrow Transplant 42 (11): 761; author reply 763, 2008. [PUBMED Abstract]
- Eapen M: Response to Dr Gilman. Bone Marrow Transplant 42 (11): 763, 2008.
- Malempati S, Weigel B, Ingle AM, et al.: Phase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 30 (3): 256-62, 2012. [PUBMED Abstract]
- Juergens H, Daw NC, Geoerger B, et al.: Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J Clin Oncol 29 (34): 4534-40, 2011. [PUBMED Abstract]
- Pappo AS, Patel SR, Crowley J, et al.: R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research through Collaboration study. J Clin Oncol 29 (34): 4541-7, 2011. [PUBMED Abstract]
- Tap WD, Demetri G, Barnette P, et al.: Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J Clin Oncol 30 (15): 1849-56, 2012. [PUBMED Abstract]
- Naing A, LoRusso P, Fu S, et al.: Insulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing's sarcoma family tumors. Clin Cancer Res 18 (9): 2625-31, 2012. [PUBMED Abstract]
- Schwartz GK, Tap WD, Qin LX, et al.: Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: a multicentre, open-label, phase 2 trial. Lancet Oncol 14 (4): 371-82, 2013. [PUBMED Abstract]
- Ahmed N, Brawley VS, Hegde M, et al.: Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol 33 (15): 1688-96, 2015. [PUBMED Abstract]
- Pule MA, Savoldo B, Myers GD, et al.: Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 14 (11): 1264-70, 2008. [PUBMED Abstract]
- Scotlandi K, Baldini N, Cerisano V, et al.: CD99 engagement: an effective therapeutic strategy for Ewing tumors. Cancer Res 60 (18): 5134-42, 2000. [PUBMED Abstract]
- Grunewald TG, Diebold I, Esposito I, et al.: STEAP1 is associated with the invasive and oxidative stress phenotype of Ewing tumors. Mol Cancer Res 10 (1): 52-65, 2012. [PUBMED Abstract]
- Smith MA, Reynolds CP, Kang MH, et al.: Synergistic activity of PARP inhibition by talazoparib (BMN 673) with temozolomide in pediatric cancer models in the pediatric preclinical testing program. Clin Cancer Res 21 (4): 819-32, 2015. [PUBMED Abstract]
Late Effects of Treatment for Ewing Sarcoma
Patients treated for Ewing sarcoma have a significantly higher risk of developing subsequent neoplasms than do patients in the general population.
Treatment-related acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) have generally been reported to occur in 1% to 2% of survivors of Ewing sarcoma,[1]; [2][Level of evidence: 3iiiDi] although some dose-intensive regimens appear to be associated with a higher risk of hematological malignancy.[3,4]; [5][Level of evidence: 3ii] Treatment-related AML and MDS arise most commonly at 2 to 5 years after diagnosis.
Survivors of Ewing sarcoma remain at increased risk of developing a subsequent solid tumor throughout their lifetime. Sarcomas usually occur within the previous radiation field.[6,7] The risk of developing a sarcoma after radiation therapy is dose-dependent, with higher doses associated with an increased risk of sarcoma development.[1]; [2][Level of evidence: 3iiiDi] The cumulative incidence of subsequent neoplasms in children treated for Ewing sarcoma between 1970 and 1986 at 25 years after diagnosis was 9.0% (confidence interval, 5.8–12.2). Most of these patients received radiation therapy; comparable long-term data do not yet exist for significant numbers of patients who did not receive radiation therapy.[8]
(Refer to the PDQ summary on Late Effects of Treatment for Childhood Cancer for a full discussion of the late effects of cancer treatment in children and adolescents.)
References
- Fuchs B, Valenzuela RG, Petersen IA, et al.: Ewing's sarcoma and the development of secondary malignancies. Clin Orthop (415): 82-9, 2003. [PUBMED Abstract]
- Goldsby R, Burke C, Nagarajan R, et al.: Second solid malignancies among children, adolescents, and young adults diagnosed with malignant bone tumors after 1976: follow-up of a Children's Oncology Group cohort. Cancer 113 (9): 2597-604, 2008. [PUBMED Abstract]
- Bhatia S, Krailo MD, Chen Z, et al.: Therapy-related myelodysplasia and acute myeloid leukemia after Ewing sarcoma and primitive neuroectodermal tumor of bone: A report from the Children's Oncology Group. Blood 109 (1): 46-51, 2007. [PUBMED Abstract]
- Kushner BH, Heller G, Cheung NK, et al.: High risk of leukemia after short-term dose-intensive chemotherapy in young patients with solid tumors. J Clin Oncol 16 (9): 3016-20, 1998. [PUBMED Abstract]
- Navid F, Billups C, Liu T, et al.: Second cancers in patients with the Ewing sarcoma family of tumours. Eur J Cancer 44 (7): 983-91, 2008. [PUBMED Abstract]
- Kuttesch JF Jr, Wexler LH, Marcus RB, et al.: Second malignancies after Ewing's sarcoma: radiation dose-dependency of secondary sarcomas. J Clin Oncol 14 (10): 2818-25, 1996. [PUBMED Abstract]
- Hawkins MM, Wilson LM, Burton HS, et al.: Radiotherapy, alkylating agents, and risk of bone cancer after childhood cancer. J Natl Cancer Inst 88 (5): 270-8, 1996. [PUBMED Abstract]
- Ginsberg JP, Goodman P, Leisenring W, et al.: Long-term survivors of childhood Ewing sarcoma: report from the childhood cancer survivor study. J Natl Cancer Inst 102 (16): 1272-83, 2010. [PUBMED Abstract]
Changes to This Summary (04/04/2018)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Treatment of Recurrent Ewing Sarcoma
Added text about the ADVL1622 and ADVL1615 clinical trials as treatment options under clinical evaluation for patients with recurrent Ewing sarcoma.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of childhood Ewing sarcoma. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Ewing Sarcoma Treatment are:
- Holcombe Edwin Grier, MD (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Andrea A. Hayes-Jordan, MD, FACS, FAAP (M.D. Anderson Cancer Center)
- Karen J. Marcus, MD (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Paul A. Meyers, MD (Memorial Sloan-Kettering Cancer Center)
- Thomas A. Olson, MD (Aflac Cancer and Blood Disorders Center of Children's Healthcare of Atlanta - Egleston Campus)
- Nita Louise Seibel, MD (National Cancer Institute)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Ewing Sarcoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/bone/hp/ewing-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389480]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Updated: April 4, 2018
Source URL: https://www.cancer.gov/publishedcontent/syndication/2434.htm
Source Agency: National Cancer Institute (NCI)
Captured Date: 2013-09-14 09:00:56.0
Ewing Sarcoma Treatment (PDQ®)–Health Professional Version
General Information About Ewing Sarcoma
Dramatic improvements in survival have been achieved for children and adolescents with cancer.[1] Between 1975 and 2010, childhood cancer mortality decreased by more than 50%.[1] For Ewing sarcoma, the 5-year survival rate has increased over the same time from 59% to 78% for children younger than 15 years and from 20% to 60% for adolescents aged 15 to 19 years.[1]
Studies using immunohistochemical markers,[2] cytogenetics,[3,4] molecular genetics, and tissue culture [5] indicate that Ewing sarcoma is derived from a primordial bone marrow–derived mesenchymal stem cell.[6,7] Older terms such as peripheral primitive neuroectodermal tumor, Askin tumor (Ewing sarcoma of chest wall), and extraosseous Ewing sarcoma (often combined in the term Ewing sarcoma family of tumors) refer to this same tumor.
Incidence
The incidence of Ewing sarcoma has remained unchanged for 30 years.[8] The incidence for all ages is one case per 1 million people in the United States. In patients aged 10 to 19 years, the incidence is between nine and ten cases per 1 million people. The same analysis suggests that the incidence of Ewing sarcoma in the United States is nine times greater in whites than in African Americans, with an intermediate incidence in Asians.[9,10]
The relative paucity of Ewing sarcoma in people of African or Asian descent may be explained, in part, by a specific polymorphism in the EGR2 gene.
The median age of patients with Ewing sarcoma is 15 years, and more than 50% of patients are adolescents. Well-characterized cases of Ewing sarcoma in neonates and infants have been described.[11,12] Based on data from 1,426 patients entered on European Intergroup Cooperative Ewing Sarcoma Studies, 59% of patients are male and 41% are female.[13]
Clinical Presentation
Primary sites of bone disease include the following:
- Lower extremity (41%).
- Pelvis (26%).
- Chest wall (16%).
- Upper extremity (9%).
- Spine (6%).
- Hand and foot (3%).[14]
- Skull (2%).
For extraosseous primary tumors, the most common primary sites of disease include the following:[15,16]
- Trunk (32%).
- Extremity (26%).
- Head and neck (18%).
- Retroperitoneum (16%).
- Other sites (9%).
The median time from first symptom to diagnosis of Ewing sarcoma is often long, with a median interval reported from 2 to 5 months. Longer times are associated with older age and pelvic primary sites. This has not been associated with metastasis, surgical outcome, or survival.[17] Approximately 25% of patients with Ewing sarcoma have metastatic disease at the time of diagnosis.[8]
The Surveillance, Epidemiology, and End Results (SEER) database was used to compare patients younger than 40 years with Ewing sarcoma who presented with skeletal and extraosseous primary sites (refer to Table 1).[18] Patients with extraosseous Ewing sarcoma were more likely to be older, female, nonwhite, and have axial primary sites, and were less likely to have pelvic primary sites than were patients with skeletal Ewing sarcoma.
| Characteristic | Extraosseous Ewing Sarcoma | Skeletal Ewing Sarcoma | P Value |
|---|---|---|---|
| Mean age (range), years | 20 (0–39) | 16 (0–39) | <.001 |
| Male | 53% | 63% | <.001 |
| White | 85% | 93% | <.001 |
| Axial primary sites | 73% | 54% | <.001 |
| Pelvic primary sites | 20% | 27% | .001 |
Diagnostic Evaluation
The following tests and procedures may be used to diagnose or stage Ewing sarcoma:
- Physical exam and history.
- Magnetic resonance imaging (MRI).
- Computed tomography (CT) scan.
- Positron emission tomography (PET) scan.
- Bone scan.
- Bone marrow aspiration and biopsy.
- X-ray.
- Complete blood count.
- Blood chemistry studies, such as lactate dehydrogenase (LDH).
Prognostic Factors
The two major types of prognostic factors for patients with Ewing sarcoma are grouped as follows:
Pretreatment factors
- Site of tumor: Patients with Ewing sarcoma in the distal extremities have the best prognosis. Patients with Ewing sarcoma in the proximal extremities have an intermediate prognosis, followed by patients with central or pelvic sites.[19-22]
- Extraskeletal versus skeletal primary tumors: The Children's Oncology Group performed a retrospective analysis from two large cooperative trials that used similar treatment regimens.[23] They identified 213 patients with extraskeletal primary tumors and 826 patients with skeletal primary tumors. Patients with extraskeletal primary tumors were more likely to have an axial primary site, less likely to have large primary tumors, and had a statistically significant better prognosis than did patients with skeletal primary tumors.
- Tumor size or volume: Tumor size or volume has been shown to be an important prognostic factor in most studies. Cutoffs of a volume of 100 mL or 200 mL and/or single dimension greater than 8 cm are used to define larger tumors. Larger tumors tend to occur in unfavorable sites.[21,22,24]
- Age: Infants and younger patients have a better prognosis than do patients aged 15 years and older.[12,19,20,22,25,26]
In North American studies, patients younger than 10 years have a better outcome than those aged 10 to 17 years at diagnosis (relative risk [RR], 1.4). Patients older than 18 years have an inferior outcome (RR, 2.5).[27-29] A retrospective review of two consecutive German trials for Ewing sarcoma identified 47 patients older than 40 years.[30] With adequate multimodal therapy, survival was comparable to the survival observed in adolescents treated on the same trials. Review of the SEER database from 1973 to 2011 identified 1,957 patients with Ewing sarcoma.[31] Thirty-nine of these patients (2.0%) were younger than 12 months at diagnosis. Infants were less likely to receive radiation therapy and more likely to have soft tissue primary sites. Early death was more common in infants, but the overall survival (OS) did not differ significantly from that of older patients.
- Sex: Girls with Ewing sarcoma have a better prognosis than do boys with Ewing sarcoma.[9,20,22]
- Serum LDH: Increased serum LDH levels before treatment are associated with inferior prognosis. Increased LDH levels are also correlated with large primary tumors and metastatic disease.[20]
- Metastases: Any metastatic disease defined by standard imaging techniques or bone marrow aspirate/biopsy by morphology is an adverse prognostic factor. The presence or absence of metastatic disease is the single most powerful predictor of outcome. Metastases at diagnosis are detected in about 25% of patients.[8]
Patients with metastatic disease confined to the lung have a better prognosis than do patients with extrapulmonary metastatic sites.[19,21,22,32] The number of pulmonary lesions does not seem to correlate with outcome, but patients with unilateral lung involvement do better than patients with bilateral lung involvement.[33]
Patients with metastasis to only bone seem to have a better outcome than do patients with metastases to both bone and lung.[34,35]
Based on an analysis from the SEER database, regional lymph node involvement in patients is associated with an inferior overall outcome when compared with patients without regional lymph node involvement.[36]
- Previous treatment for cancer: In the SEER database, 58 patients with Ewing sarcoma who were diagnosed after treatment for a previous malignancy (2.1% of patients with Ewing sarcoma) were compared with 2,756 patients with Ewing sarcoma as a first cancer over the same period. Patients with Ewing sarcoma as a second malignant neoplasm were older (secondary Ewing sarcoma, mean age of 47.8 years; primary Ewing sarcoma, mean age of 22.5 years), more likely to have a primary tumor in an axial or extraskeletal site, and had a worse prognosis (5-year OS for patients with secondary Ewing sarcoma, 43.5%; patients with primary Ewing sarcoma, 64.2%).[37]
- Standard cytogenetics: Complex karyotype (defined as the presence of five or more independent chromosome abnormalities at diagnosis) and modal chromosome numbers lower than 50 appear to have adverse prognostic significance.[38]
- Detectable fusion transcripts in morphologically normal marrow: Reverse transcriptase polymerase chain reaction can be used to detect fusion transcripts in bone marrow. In a single retrospective study utilizing patients with normal marrow morphology and no other metastatic site, fusion transcript detection in marrow or peripheral blood was associated with an increased risk of relapse.[39]
- Other biological factors: Overexpression of the p53 protein, Ki67 expression, and loss of 16q may be adverse prognostic factors.[40-42] High expression of microsomal glutathione S-transferase, an enzyme associated with resistance to doxorubicin, is associated with inferior outcome for Ewing sarcoma.[43]
The Children's Oncology Group performed a prospective analysis of TP53 mutations and/or CDKN2A deletions in patients with Ewing sarcoma; no correlation was found with event-free survival (EFS).[44]
The following are not considered to be adverse prognostic factors for Ewing sarcoma:
- Pathologic fracture: Pathologic fractures do not appear to be a prognostic factor.[45]
- Histopathology: The degree of neural differentiation is not a prognostic factor in Ewing sarcoma.[46,47]
- Molecular pathology: The EWSR1-ETS translocation associated with Ewing sarcoma can occur at several potential breakpoints in each of the genes that join to form the novel segment of DNA. Once thought to be significant,[48] two large series have shown that the EWSR1-ETS translocation breakpoint site is not an adverse prognostic factor.[49,50]
Response to initial therapy factors
Multiple studies have shown that patients with minimal or no residual viable tumor after presurgical chemotherapy have a significantly better EFS than do patients with larger amounts of viable tumor.[51-54] Female sex and younger age predict a good histologic response to preoperative therapy.[55] For patients who receive preinduction- and postinduction-chemotherapy PET scans, decreased PET uptake after chemotherapy correlated with good histologic response and better outcome.[56-58]
Patients with poor response to presurgical chemotherapy have an increased risk for local recurrence.[59]
References
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014. [PUBMED Abstract]
- Olsen SH, Thomas DG, Lucas DR: Cluster analysis of immunohistochemical profiles in synovial sarcoma, malignant peripheral nerve sheath tumor, and Ewing sarcoma. Mod Pathol 19 (5): 659-68, 2006. [PUBMED Abstract]
- Delattre O, Zucman J, Melot T, et al.: The Ewing family of tumors--a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 331 (5): 294-9, 1994. [PUBMED Abstract]
- Dagher R, Pham TA, Sorbara L, et al.: Molecular confirmation of Ewing sarcoma. J Pediatr Hematol Oncol 23 (4): 221-4, 2001. [PUBMED Abstract]
- Llombart-Bosch A, Carda C, Peydro-Olaya A, et al.: Soft tissue Ewing's sarcoma. Characterization in established cultures and xenografts with evidence of a neuroectodermic phenotype. Cancer 66 (12): 2589-601, 1990. [PUBMED Abstract]
- Suvà ML, Riggi N, Stehle JC, et al.: Identification of cancer stem cells in Ewing's sarcoma. Cancer Res 69 (5): 1776-81, 2009. [PUBMED Abstract]
- Tirode F, Laud-Duval K, Prieur A, et al.: Mesenchymal stem cell features of Ewing tumors. Cancer Cell 11 (5): 421-9, 2007. [PUBMED Abstract]
- Esiashvili N, Goodman M, Marcus RB Jr: Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J Pediatr Hematol Oncol 30 (6): 425-30, 2008. [PUBMED Abstract]
- Jawad MU, Cheung MC, Min ES, et al.: Ewing sarcoma demonstrates racial disparities in incidence-related and sex-related differences in outcome: an analysis of 1631 cases from the SEER database, 1973-2005. Cancer 115 (15): 3526-36, 2009. [PUBMED Abstract]
- Beck R, Monument MJ, Watkins WS, et al.: EWS/FLI-responsive GGAA microsatellites exhibit polymorphic differences between European and African populations. Cancer Genet 205 (6): 304-12, 2012. [PUBMED Abstract]
- Kim SY, Tsokos M, Helman LJ: Dilemmas associated with congenital ewing sarcoma family tumors. J Pediatr Hematol Oncol 30 (1): 4-7, 2008. [PUBMED Abstract]
- van den Berg H, Dirksen U, Ranft A, et al.: Ewing tumors in infants. Pediatr Blood Cancer 50 (4): 761-4, 2008. [PUBMED Abstract]
- Paulussen M, Craft AW, Lewis I, et al.: Results of the EICESS-92 Study: two randomized trials of Ewing's sarcoma treatment--cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J Clin Oncol 26 (27): 4385-93, 2008. [PUBMED Abstract]
- Froeb D, Ranft A, Boelling T, et al.: Ewing sarcoma of the hand or foot. Klin Padiatr 224 (6): 348-52, 2012. [PUBMED Abstract]
- Raney RB, Asmar L, Newton WA Jr, et al.: Ewing's sarcoma of soft tissues in childhood: a report from the Intergroup Rhabdomyosarcoma Study, 1972 to 1991. J Clin Oncol 15 (2): 574-82, 1997. [PUBMED Abstract]
- Rowe RG, Thomas DG, Schuetze SM, et al.: Ewing sarcoma of the kidney: case series and literature review of an often overlooked entity in the diagnosis of primary renal tumors. Urology 81 (2): 347-53, 2013. [PUBMED Abstract]
- Brasme JF, Chalumeau M, Oberlin O, et al.: Time to diagnosis of Ewing tumors in children and adolescents is not associated with metastasis or survival: a prospective multicenter study of 436 patients. J Clin Oncol 32 (18): 1935-40, 2014. [PUBMED Abstract]
- Applebaum MA, Worch J, Matthay KK, et al.: Clinical features and outcomes in patients with extraskeletal Ewing sarcoma. Cancer 117 (13): 3027-32, 2011. [PUBMED Abstract]
- Cotterill SJ, Ahrens S, Paulussen M, et al.: Prognostic factors in Ewing's tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing's Sarcoma Study Group. J Clin Oncol 18 (17): 3108-14, 2000. [PUBMED Abstract]
- Bacci G, Longhi A, Ferrari S, et al.: Prognostic factors in non-metastatic Ewing's sarcoma tumor of bone: an analysis of 579 patients treated at a single institution with adjuvant or neoadjuvant chemotherapy between 1972 and 1998. Acta Oncol 45 (4): 469-75, 2006. [PUBMED Abstract]
- Rodríguez-Galindo C, Liu T, Krasin MJ, et al.: Analysis of prognostic factors in ewing sarcoma family of tumors: review of St. Jude Children's Research Hospital studies. Cancer 110 (2): 375-84, 2007. [PUBMED Abstract]
- Karski EE, McIlvaine E, Segal MR, et al.: Identification of Discrete Prognostic Groups in Ewing Sarcoma. Pediatr Blood Cancer 63 (1): 47-53, 2016. [PUBMED Abstract]
- Cash T, McIlvaine E, Krailo MD, et al.: Comparison of clinical features and outcomes in patients with extraskeletal versus skeletal localized Ewing sarcoma: A report from the Children's Oncology Group. Pediatr Blood Cancer 63 (10): 1771-9, 2016. [PUBMED Abstract]
- Ahrens S, Hoffmann C, Jabar S, et al.: Evaluation of prognostic factors in a tumor volume-adapted treatment strategy for localized Ewing sarcoma of bone: the CESS 86 experience. Cooperative Ewing Sarcoma Study. Med Pediatr Oncol 32 (3): 186-95, 1999. [PUBMED Abstract]
- De Ioris MA, Prete A, Cozza R, et al.: Ewing sarcoma of the bone in children under 6 years of age. PLoS One 8 (1): e53223, 2013. [PUBMED Abstract]
- Huh WW, Daw NC, Herzog CE, et al.: Ewing sarcoma family of tumors in children younger than 10 years of age. Pediatr Blood Cancer 64 (4): , 2017. [PUBMED Abstract]
- Grier HE, Krailo MD, Tarbell NJ, et al.: Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 348 (8): 694-701, 2003. [PUBMED Abstract]
- Granowetter L, Womer R, Devidas M, et al.: Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children's Oncology Group Study. J Clin Oncol 27 (15): 2536-41, 2009. [PUBMED Abstract]
- Womer RB, West DC, Krailo MD, et al.: Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 30 (33): 4148-54, 2012. [PUBMED Abstract]
- Pieper S, Ranft A, Braun-Munzinger G, et al.: Ewing's tumors over the age of 40: a retrospective analysis of 47 patients treated according to the International Clinical Trials EICESS 92 and EURO-E.W.I.N.G. 99. Onkologie 31 (12): 657-63, 2008. [PUBMED Abstract]
- Wong T, Goldsby RE, Wustrack R, et al.: Clinical features and outcomes of infants with Ewing sarcoma under 12 months of age. Pediatr Blood Cancer 62 (11): 1947-51, 2015. [PUBMED Abstract]
- Miser JS, Krailo MD, Tarbell NJ, et al.: Treatment of metastatic Ewing's sarcoma or primitive neuroectodermal tumor of bone: evaluation of combination ifosfamide and etoposide--a Children's Cancer Group and Pediatric Oncology Group study. J Clin Oncol 22 (14): 2873-6, 2004. [PUBMED Abstract]
- Paulussen M, Ahrens S, Craft AW, et al.: Ewing's tumors with primary lung metastases: survival analysis of 114 (European Intergroup) Cooperative Ewing's Sarcoma Studies patients. J Clin Oncol 16 (9): 3044-52, 1998. [PUBMED Abstract]
- Paulussen M, Ahrens S, Burdach S, et al.: Primary metastatic (stage IV) Ewing tumor: survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma Studies. Ann Oncol 9 (3): 275-81, 1998. [PUBMED Abstract]
- Ladenstein R, Pötschger U, Le Deley MC, et al.: Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol 28 (20): 3284-91, 2010. [PUBMED Abstract]
- Applebaum MA, Goldsby R, Neuhaus J, et al.: Clinical features and outcomes in patients with Ewing sarcoma and regional lymph node involvement. Pediatr Blood Cancer 59 (4): 617-20, 2012. [PUBMED Abstract]
- Applebaum MA, Goldsby R, Neuhaus J, et al.: Clinical features and outcomes in patients with secondary Ewing sarcoma. Pediatr Blood Cancer 60 (4): 611-5, 2013. [PUBMED Abstract]
- Roberts P, Burchill SA, Brownhill S, et al.: Ploidy and karyotype complexity are powerful prognostic indicators in the Ewing's sarcoma family of tumors: a study by the United Kingdom Cancer Cytogenetics and the Children's Cancer and Leukaemia Group. Genes Chromosomes Cancer 47 (3): 207-20, 2008. [PUBMED Abstract]
- Schleiermacher G, Peter M, Oberlin O, et al.: Increased risk of systemic relapses associated with bone marrow micrometastasis and circulating tumor cells in localized ewing tumor. J Clin Oncol 21 (1): 85-91, 2003. [PUBMED Abstract]
- Abudu A, Mangham DC, Reynolds GM, et al.: Overexpression of p53 protein in primary Ewing's sarcoma of bone: relationship to tumour stage, response and prognosis. Br J Cancer 79 (7-8): 1185-9, 1999. [PUBMED Abstract]
- López-Guerrero JA, Machado I, Scotlandi K, et al.: Clinicopathological significance of cell cycle regulation markers in a large series of genetically confirmed Ewing's sarcoma family of tumors. Int J Cancer 128 (5): 1139-50, 2011. [PUBMED Abstract]
- Ozaki T, Paulussen M, Poremba C, et al.: Genetic imbalances revealed by comparative genomic hybridization in Ewing tumors. Genes Chromosomes Cancer 32 (2): 164-71, 2001. [PUBMED Abstract]
- Scotlandi K, Remondini D, Castellani G, et al.: Overcoming resistance to conventional drugs in Ewing sarcoma and identification of molecular predictors of outcome. J Clin Oncol 27 (13): 2209-16, 2009. [PUBMED Abstract]
- Lerman DM, Monument MJ, McIlvaine E, et al.: Tumoral TP53 and/or CDKN2A alterations are not reliable prognostic biomarkers in patients with localized Ewing sarcoma: a report from the Children's Oncology Group. Pediatr Blood Cancer 62 (5): 759-65, 2015. [PUBMED Abstract]
- Bramer JA, Abudu AA, Grimer RJ, et al.: Do pathological fractures influence survival and local recurrence rate in bony sarcomas? Eur J Cancer 43 (13): 1944-51, 2007. [PUBMED Abstract]
- Parham DM, Hijazi Y, Steinberg SM, et al.: Neuroectodermal differentiation in Ewing's sarcoma family of tumors does not predict tumor behavior. Hum Pathol 30 (8): 911-8, 1999. [PUBMED Abstract]
- Luksch R, Sampietro G, Collini P, et al.: Prognostic value of clinicopathologic characteristics including neuroectodermal differentiation in osseous Ewing's sarcoma family of tumors in children. Tumori 85 (2): 101-7, 1999 Mar-Apr. [PUBMED Abstract]
- de Alava E, Kawai A, Healey JH, et al.: EWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing's sarcoma. J Clin Oncol 16 (4): 1248-55, 1998. [PUBMED Abstract]
- van Doorninck JA, Ji L, Schaub B, et al.: Current treatment protocols have eliminated the prognostic advantage of type 1 fusions in Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 28 (12): 1989-94, 2010. [PUBMED Abstract]
- Le Deley MC, Delattre O, Schaefer KL, et al.: Impact of EWS-ETS fusion type on disease progression in Ewing's sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. J Clin Oncol 28 (12): 1982-8, 2010. [PUBMED Abstract]
- Paulussen M, Ahrens S, Dunst J, et al.: Localized Ewing tumor of bone: final results of the cooperative Ewing's Sarcoma Study CESS 86. J Clin Oncol 19 (6): 1818-29, 2001. [PUBMED Abstract]
- Rosito P, Mancini AF, Rondelli R, et al.: Italian Cooperative Study for the treatment of children and young adults with localized Ewing sarcoma of bone: a preliminary report of 6 years of experience. Cancer 86 (3): 421-8, 1999. [PUBMED Abstract]
- Wunder JS, Paulian G, Huvos AG, et al.: The histological response to chemotherapy as a predictor of the oncological outcome of operative treatment of Ewing sarcoma. J Bone Joint Surg Am 80 (7): 1020-33, 1998. [PUBMED Abstract]
- Oberlin O, Deley MC, Bui BN, et al.: Prognostic factors in localized Ewing's tumours and peripheral neuroectodermal tumours: the third study of the French Society of Paediatric Oncology (EW88 study). Br J Cancer 85 (11): 1646-54, 2001. [PUBMED Abstract]
- Ferrari S, Bertoni F, Palmerini E, et al.: Predictive factors of histologic response to primary chemotherapy in patients with Ewing sarcoma. J Pediatr Hematol Oncol 29 (6): 364-8, 2007. [PUBMED Abstract]
- Hawkins DS, Schuetze SM, Butrynski JE, et al.: [18F]Fluorodeoxyglucose positron emission tomography predicts outcome for Ewing sarcoma family of tumors. J Clin Oncol 23 (34): 8828-34, 2005. [PUBMED Abstract]
- Denecke T, Hundsdörfer P, Misch D, et al.: Assessment of histological response of paediatric bone sarcomas using FDG PET in comparison to morphological volume measurement and standardized MRI parameters. Eur J Nucl Med Mol Imaging 37 (10): 1842-53, 2010. [PUBMED Abstract]
- Palmerini E, Colangeli M, Nanni C, et al.: The role of FDG PET/CT in patients treated with neoadjuvant chemotherapy for localized bone sarcomas. Eur J Nucl Med Mol Imaging 44 (2): 215-223, 2017. [PUBMED Abstract]
- Lin PP, Jaffe N, Herzog CE, et al.: Chemotherapy response is an important predictor of local recurrence in Ewing sarcoma. Cancer 109 (3): 603-11, 2007. [PUBMED Abstract]
Cellular Classification of Ewing Sarcoma
Ewing sarcoma belongs to the group of neoplasms commonly referred to as small, round, blue-cell tumors of childhood. The individual cells of Ewing sarcoma contain round-to-oval nuclei, with fine dispersed chromatin without nucleoli. Occasionally, cells with smaller, more hyperchromatic, and probably degenerative nuclei are present, giving a light cell/dark cell pattern. The cytoplasm varies in amount, but in the classic case, it is clear and contains glycogen, which can be highlighted with a periodic acid-Schiff stain. The tumor cells are tightly packed and grow in a diffuse pattern without evidence of structural organization. Tumors with the requisite translocation that show neuronal differentiation are not considered a separate entity, but rather, part of a continuum of differentiation.
The MIC2 gene product, CD99, is a surface membrane protein that is expressed in most cases of Ewing sarcoma and is useful in diagnosing these tumors when the results are interpreted in the context of clinical and pathologic parameters.[1] MIC2 positivity is not unique to Ewing sarcoma, and positivity by immunochemistry is found in several other tumors, including synovial sarcoma, non-Hodgkin lymphoma, and gastrointestinal stromal tumors.
Genomics of Ewing Sarcoma
The detection of a translocation involving the EWSR1 gene on chromosome 22 band q12 and any one of a number of partner chromosomes is the key feature in the diagnosis of Ewing sarcoma (refer to Table 2).[2] The EWSR1 gene is a member of the TET family [TLS/EWS/TAF15] of RNA-binding proteins.[3] The FLI1 gene is a member of the ETS family of DNA-binding genes. Characteristically, the amino terminus of the EWSR1 gene is juxtaposed with the carboxy terminus of the STS family gene. In most cases (90%), the carboxy terminus is provided by FLI1, a member of the family of transcription factor genes located on chromosome 11 band q24. Other family members that may combine with the EWSR1 gene are ERG, ETV1, ETV4 (also termed E1AF), and FEV.[4] Rarely, TLS, another TET family member, can substitute for EWSR1.[5] Finally, there are a few rare cases in which EWSR1 has translocated with partners that are not members of the ETS family of oncogenes. The significance of these alternate partners is not known.
Besides these consistent aberrations involving the EWSR1 gene at 22q12, additional numerical and structural aberrations have been observed in Ewing sarcoma, including gains of chromosomes 2, 5, 8, 9, 12, and 15; the nonreciprocal translocation t(1;16)(q12;q11.2); and deletions on the short arm of chromosome 6. Trisomy 20 may be associated with a more aggressive subset of Ewing sarcoma.[6]
Three papers have described the genomic landscape of Ewing sarcoma and all show that these tumors have a relatively silent genome, with a paucity of mutations in pathways that might be amenable to treatment with novel targeted therapies.[7-9] These papers also identified mutations in STAG2, a member of the cohesin complex, in about 15% to 20% of the cases, and the presence of these mutations was associated with advanced-stage disease. CDKN2A deletions were noted in 12% to 22% of cases. Finally, TP53 mutations were identified in about 6% to 7% of cases and the coexistence of STAG2 and TP53 mutations is associated with a poor clinical outcome.[7-9]
Figure 1 below from a discovery cohort (n = 99) highlights the frequency of chromosome 8 gain, the co-occurrence of chromosome 1q gain and chromosome 16q loss, the mutual exclusivity of CDKN2A deletion and STAG2 mutation, and the relative paucity of recurrent single nucleotide variants for Ewing sarcoma.[7]
Ewing sarcoma translocations can all be found with standard cytogenetic analysis. A more rapid analysis looking for a break apart of the EWS gene is now frequently done to confirm the diagnosis of Ewing sarcoma molecularly.[10] This test result must be considered with caution, however. Ewing sarcomas that utilize the TLS translocations will have negative tests because the EWSR1 gene is not translocated in those cases. In addition, other small round tumors also contain translocations of different ETS family members with EWSR1, such as desmoplastic small round cell tumor, clear cell sarcoma, extraskeletal myxoid chondrosarcoma, and myxoid liposarcoma, all of which may be positive with a EWS fluorescence in situ hybridization (FISH) break-apart probe. A detailed analysis of 85 patients with small round blue cell tumors that were negative for EWSR1 rearrangement by FISH with an EWSR1 break-apart probe identified eight patients with FUS rearrangements.[11] Four patients who had EWSR1-ERG fusions were not detected by FISH with an EWSR1 break-apart probe. The authors do not recommend relying solely on EWSR1 break-apart probes for analyzing small round blue cell tumors with strong immunohistochemical positivity for CD99.
Small round blue cell tumors of bone and soft tissue that are histologically similar to Ewing sarcoma but do not have rearrangements of the EWSR1 gene have been analyzed and translocations have been identified. These include BCOR-CCNB3, CIC-DUX4, and CIC-FOX4.[12-15] The molecular profile of these tumors is different from the profile of EWS-FLI1 translocated Ewing sarcoma, and limited evidence suggests that they have a different clinical behavior. In almost all cases, the patients were treated with therapy designed for Ewing sarcoma on the basis of the histologic and immunohistologic similarity to Ewing sarcoma. There are too few cases associated with each translocation to determine whether the prognosis for these small round blue cell tumors is distinct from the prognosis of Ewing sarcoma of similar stage and site.[12-15]
A genome-wide association study identified a region on chromosome 10q21.3 associated with an increased risk of Ewing sarcoma.[16] Deep sequencing through this region identified a polymorphism in the EGR2 gene, which appears to cooperate with the gene product of the EWSR1-FLI1 fusion that is seen in most patients with Ewing sarcoma.[17] The polymorphism associated with the increased risk is found at a much higher frequency in whites than in blacks or Asians, possibly contributing to the epidemiology of the relative infrequency of Ewing sarcoma in the latter populations.
| TET Family Partner | Fusion With ETS-like Oncogene Partner | Translocation | Comment |
|---|---|---|---|
| aThese partners are not members of the ETS family of oncogenes. | |||
| EWS | EWSR1-FLI1 | t(11;22)(q24;q12) | Most common; ~85% to 90% of cases |
| EWSR1-ERG | t(21;22)(q22;q12) | Second most common; ~10% of cases | |
| EWSR1-ETV1 | t(7;22)(p22;q12) | Rare | |
| EWSR1-ETV4 | t(17;22)(q12;q12) | Rare | |
| EWSR1-FEV | t(2;22)(q35;q12) | Rare | |
| EWSR1-NFATc2a | t(20;22)(q13;q12) | Rare | |
| EWSR1-POU5F1a | t(6;22)(p21;q12) | ||
| EWSR1-SMARCA5a | t(4;22)(q31;q12) | Rare | |
| EWSR1-ZSGa | t(6;22)(p21;q12) | ||
| EWSR1-SP3a | t(2;22)(q31;q12) | Rare | |
| TLS (also called FUS) | TLS-ERG | t(16;21)(p11;q22) | Rare |
| TLS-FEV | t(2;16)(q35;p11) | Rare | |
References
- Parham DM, Hijazi Y, Steinberg SM, et al.: Neuroectodermal differentiation in Ewing's sarcoma family of tumors does not predict tumor behavior. Hum Pathol 30 (8): 911-8, 1999. [PUBMED Abstract]
- Delattre O, Zucman J, Melot T, et al.: The Ewing family of tumors--a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 331 (5): 294-9, 1994. [PUBMED Abstract]
- Urano F, Umezawa A, Yabe H, et al.: Molecular analysis of Ewing's sarcoma: another fusion gene, EWS-E1AF, available for diagnosis. Jpn J Cancer Res 89 (7): 703-11, 1998. [PUBMED Abstract]
- Hattinger CM, Rumpler S, Strehl S, et al.: Prognostic impact of deletions at 1p36 and numerical aberrations in Ewing tumors. Genes Chromosomes Cancer 24 (3): 243-54, 1999. [PUBMED Abstract]
- Sankar S, Lessnick SL: Promiscuous partnerships in Ewing's sarcoma. Cancer Genet 204 (7): 351-65, 2011. [PUBMED Abstract]
- Roberts P, Burchill SA, Brownhill S, et al.: Ploidy and karyotype complexity are powerful prognostic indicators in the Ewing's sarcoma family of tumors: a study by the United Kingdom Cancer Cytogenetics and the Children's Cancer and Leukaemia Group. Genes Chromosomes Cancer 47 (3): 207-20, 2008. [PUBMED Abstract]
- Tirode F, Surdez D, Ma X, et al.: Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov 4 (11): 1342-53, 2014. [PUBMED Abstract]
- Crompton BD, Stewart C, Taylor-Weiner A, et al.: The genomic landscape of pediatric Ewing sarcoma. Cancer Discov 4 (11): 1326-41, 2014. [PUBMED Abstract]
- Brohl AS, Solomon DA, Chang W, et al.: The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet 10 (7): e1004475, 2014. [PUBMED Abstract]
- Monforte-Muñoz H, Lopez-Terrada D, Affendie H, et al.: Documentation of EWS gene rearrangements by fluorescence in-situ hybridization (FISH) in frozen sections of Ewing's sarcoma-peripheral primitive neuroectodermal tumor. Am J Surg Pathol 23 (3): 309-15, 1999. [PUBMED Abstract]
- Chen S, Deniz K, Sung YS, et al.: Ewing sarcoma with ERG gene rearrangements: A molecular study focusing on the prevalence of FUS-ERG and common pitfalls in detecting EWSR1-ERG fusions by FISH. Genes Chromosomes Cancer 55 (4): 340-9, 2016. [PUBMED Abstract]
- Pierron G, Tirode F, Lucchesi C, et al.: A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet 44 (4): 461-6, 2012. [PUBMED Abstract]
- Specht K, Sung YS, Zhang L, et al.: Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged Ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosomes Cancer 53 (7): 622-33, 2014. [PUBMED Abstract]
- Sugita S, Arai Y, Tonooka A, et al.: A novel CIC-FOXO4 gene fusion in undifferentiated small round cell sarcoma: a genetically distinct variant of Ewing-like sarcoma. Am J Surg Pathol 38 (11): 1571-6, 2014. [PUBMED Abstract]
- Cohen-Gogo S, Cellier C, Coindre JM, et al.: Ewing-like sarcomas with BCOR-CCNB3 fusion transcript: a clinical, radiological and pathological retrospective study from the Société Française des Cancers de L'Enfant. Pediatr Blood Cancer 61 (12): 2191-8, 2014. [PUBMED Abstract]
- Postel-Vinay S, Véron AS, Tirode F, et al.: Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat Genet 44 (3): 323-7, 2012. [PUBMED Abstract]
- Grünewald TG, Bernard V, Gilardi-Hebenstreit P, et al.: Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat Genet 47 (9): 1073-8, 2015. [PUBMED Abstract]
Stage Information for Ewing Sarcoma
Pretreatment staging studies for Ewing sarcoma may include the following:
- Magnetic resonance imaging (MRI).
- Computed tomography (CT) scan of the primary site and chest.
- Positron emission tomography using fluorine F 18-fludeoxyglucose (18F-FDG PET) or 18F-FDG PET-CT.
- Bone scan.
- Bone marrow aspiration and biopsy.
For patients with confirmed Ewing sarcoma, pretreatment staging studies include MRI and/or CT scan, depending on the primary site. Despite the fact that CT and MRI are both equivalent in terms of staging, use of both imaging modalities may help radiation therapy planning.[1] Whole-body MRI may provide additional information that could potentially alter therapy planning.[2] Additional pretreatment staging studies include bone scan and CT scan of the chest. In certain studies, determination of pretreatment tumor volume is an important variable.
Although 18F-FDG PET or 18F-FDG PET-CT are optional staging modalities, they have demonstrated high sensitivity and specificity in Ewing sarcoma and may provide additional information that alters therapy planning. In one institutional study, 18F-FDG PET had a very high correlation with bone scan; the investigators suggested that it could replace bone scan for the initial extent of disease evaluation.[3] This finding was confirmed in a single-institution retrospective review.[4] 18F-FDG PET-CT is more accurate than 18F-FDG PET alone in Ewing sarcoma.[5-7]
Bone marrow aspiration and biopsy have been considered the standard of care for Ewing sarcoma. However, two retrospective studies showed that for patients (N = 141 total) who were evaluated by bone scan and/or PET scan and lung CT without evidence of metastases, bone marrow aspirates and biopsies were negative in every case.[3,8] The need for routine use of bone marrow aspirates and biopsies in patients without bone metastases is now in question.
For Ewing sarcoma, the tumor is defined as localized when, by clinical and imaging techniques, there is no spread beyond the primary site or regional lymph node involvement. Continuous extension into adjacent soft tissue may occur. If there is a question of regional lymph node involvement, pathologic confirmation is indicated.
References
- Meyer JS, Nadel HR, Marina N, et al.: Imaging guidelines for children with Ewing sarcoma and osteosarcoma: a report from the Children's Oncology Group Bone Tumor Committee. Pediatr Blood Cancer 51 (2): 163-70, 2008. [PUBMED Abstract]
- Mentzel HJ, Kentouche K, Sauner D, et al.: Comparison of whole-body STIR-MRI and 99mTc-methylene-diphosphonate scintigraphy in children with suspected multifocal bone lesions. Eur Radiol 14 (12): 2297-302, 2004. [PUBMED Abstract]
- Newman EN, Jones RL, Hawkins DS: An evaluation of [F-18]-fluorodeoxy-D-glucose positron emission tomography, bone scan, and bone marrow aspiration/biopsy as staging investigations in Ewing sarcoma. Pediatr Blood Cancer 60 (7): 1113-7, 2013. [PUBMED Abstract]
- Ulaner GA, Magnan H, Healey JH, et al.: Is methylene diphosphonate bone scan necessary for initial staging of Ewing sarcoma if 18F-FDG PET/CT is performed? AJR Am J Roentgenol 202 (4): 859-67, 2014. [PUBMED Abstract]
- Völker T, Denecke T, Steffen I, et al.: Positron emission tomography for staging of pediatric sarcoma patients: results of a prospective multicenter trial. J Clin Oncol 25 (34): 5435-41, 2007. [PUBMED Abstract]
- Gerth HU, Juergens KU, Dirksen U, et al.: Significant benefit of multimodal imaging: PET/CT compared with PET alone in staging and follow-up of patients with Ewing tumors. J Nucl Med 48 (12): 1932-9, 2007. [PUBMED Abstract]
- Treglia G, Salsano M, Stefanelli A, et al.: Diagnostic accuracy of ¹⁸F-FDG-PET and PET/CT in patients with Ewing sarcoma family tumours: a systematic review and a meta-analysis. Skeletal Radiol 41 (3): 249-56, 2012. [PUBMED Abstract]
- Kopp LM, Hu C, Rozo B, et al.: Utility of bone marrow aspiration and biopsy in initial staging of Ewing sarcoma. Pediatr Blood Cancer 62 (1): 12-5, 2015. [PUBMED Abstract]
Treatment Option Overview for Ewing Sarcoma
It is important that patients be evaluated by specialists from the appropriate disciplines (e.g., radiologists, chemotherapists, pathologists, surgical or orthopedic oncologists, and radiation oncologists) as early as possible. Appropriate imaging studies of the site are obtained before biopsy. To ensure that the incision is placed in an acceptable location, the surgical or orthopedic oncologist who will perform the definitive surgery is involved in the decision regarding biopsy-incision placement. This is especially important if it is thought that the lesion can be totally excised or if a limb salvage procedure may be attempted. Biopsy should be from soft tissue as often as possible to avoid increasing the risk of fracture.[1] The pathologist is consulted before biopsy/surgery to ensure that the incision will not compromise the radiation port and that multiple types of adequate tissue samples are obtained. It is important to obtain fresh tissue, whenever possible, for cytogenetics and molecular pathology. A second option is to perform a needle biopsy, as long as adequate tissue is obtained for molecular biology and cytogenetics.[2]
Table 3 describes the treatment options for localized, metastatic, and recurrent Ewing sarcoma.
| Treatment Group | Standard Treatment Options | |
|---|---|---|
| Localized Ewing sarcoma | Chemotherapy | |
| Local-control measures: | ||
| Surgery | ||
| Radiation therapy | ||
| Metastatic Ewing sarcoma | Chemotherapy | |
| Surgery | ||
| Radiation therapy | ||
| Recurrent Ewing sarcoma | Chemotherapy (not considered standard treatment) | |
| Radiation therapy (not considered standard treatment) | ||
| Other therapies (not considered standard treatment) | ||
The successful treatment of patients with Ewing sarcoma requires systemic chemotherapy [3-9] in conjunction with surgery and/or radiation therapy for local tumor control.[10-14] In general, patients receive chemotherapy before instituting local-control measures. In patients who undergo surgery, surgical margins and histologic response are considered in planning postoperative therapy. Patients with metastatic disease often have a good initial response to preoperative chemotherapy, but in most cases, the disease is only partially controlled or recurs.[15-19] Patients with lung as the only metastatic site have a better prognosis than do patients with metastases to bone and/or bone marrow. Adequate local control for metastatic sites, particularly bone metastases, may be an important issue.[20]
Chemotherapy for Ewing Sarcoma
Multidrug chemotherapy for Ewing sarcoma always includes vincristine, doxorubicin, ifosfamide, and etoposide. Most protocols also use cyclophosphamide and some incorporate dactinomycin. The mode of administration and dose intensity of cyclophosphamide within courses differs markedly between protocols. A European Intergroup Cooperative Ewing Sarcoma Study (EICESS) trial suggested that 1.2 g of cyclophosphamide produced a similar event-free survival (EFS) compared with 6 g of ifosfamide in patients with lower-risk disease, and identified a trend toward better EFS for patients with localized Ewing sarcoma and higher-risk disease when treatment included etoposide (GER-GPOH-EICESS-92).[21][Level of evidence: 1iiA]
Protocols in the United States generally alternate courses of vincristine, cyclophosphamide, and doxorubicin with courses of ifosfamide/etoposide,[7] while European protocols generally combine vincristine, doxorubicin, and an alkylating agent with or without etoposide in a single treatment cycle.[9] The duration of primary chemotherapy ranges from 6 months to approximately 1 year.
Evidence (chemotherapy):
- An international consortium of European countries conducted the EURO-EWING-INTERGROUP-EE99 (NCT00020566) trial from 2000 to 2010.[22][Level of evidence: 1iiA] All patients received induction therapy with six cycles of vincristine, ifosfamide, doxorubicin, and etoposide (VIDE), followed by local control, and then one cycle of vincristine, dactinomycin, and ifosfamide (VAI). Patients were classified as standard risk if they had localized disease and good histologic response to therapy or if they had localized tumors less than 200 mL in volume at presentation; they were treated with radiation therapy alone as local treatment. Standard-risk patients (n = 856) were randomly assigned to receive either maintenance therapy with seven cycles of vincristine, dactinomycin, and cyclophosphamide (VAC) or VAI.
- There was no significant difference in EFS or overall survival (OS) between the VAC arm and the VAI arm.
- Three-year EFS for this low-risk population was 77%.
- Acute renal toxicity was lower in the VAC arm than in the VAI arm, but long-term renal function outcome and fertility analyses are still pending.
- It is difficult to compare this outcome with that of other large series because the study population excluded patients with poor response to initial therapy or patients with tumors more than 200 mL in volume who received local-control therapy with radiation alone. All other published series report results for all patients who present without clinically detectable metastasis; thus, these other series included patients with poor response and patients with larger primary tumors treated with radiation alone, all of whom were excluded from the EURO-EWING-INTERGROUP-EE99 study.
- A randomized clinical trial (COG-AEWS0031 [NCT00006734]) from the Children’s Oncology Group (COG) showed that for patients presenting without metastases, the administration of cycles of cyclophosphamide, doxorubicin, and vincristine alternating with cycles of ifosfamide and etoposide at 2-week intervals achieved superior EFS (5-year EFS, 73%) than did alternating cycles at 3-week intervals (5-year EFS, 65%).[23]
- The Brazilian Cooperative Study Group performed a multi-institutional trial that incorporated carboplatin into a risk-adapted intensive regimen in 175 children with localized or metastatic Ewing sarcoma. They found significantly increased toxicity without an improvement in outcome with the addition of carboplatin.[24][Level of evidence: 2Dii]
- The COG conducted a pilot study of the addition of cycles of cyclophosphamide and topotecan to cycles of cyclophosphamide/doxorubicin/vincristine and ifosfamide/etoposide administered in an interval-compressed (2-week instead of 3-week intervals) schedule.[25][Level of evidence: 2Di]
- Therapy was well tolerated, and the 5-year EFS for 35 patients was 80%. This pilot study became the experimental arm of COG-AEWS1031 (NCT01231906).
Local Control for Ewing Sarcoma
Treatment approaches for Ewing sarcoma titrate therapeutic aggressiveness with the goal of maximizing local control while minimizing morbidity.
Surgery is the most commonly used form of local control.[26] Radiation therapy is an effective alternative modality for local control in cases where the functional morbidity of surgery is deemed too high by experienced surgical oncologists. However, in the immature skeleton, radiation therapy can cause subsequent deformities that may be more morbid than deformities from surgery. When complete surgical resection with pathologically negative margins cannot be obtained, postoperative radiation therapy is indicated. A multidisciplinary discussion between the experienced radiation oncologist and the surgeon is necessary to determine the best treatment options for local control for a given case. For some marginally resectable lesions, a combined approach of preoperative radiation therapy followed by resection can be used.
Randomized trials that directly compare surgery and radiation therapy do not exist, and their relative roles remain controversial. Although retrospective institutional series suggest superior local control and survival with surgery than with radiation therapy, most of these studies are compromised by selection bias. An analysis using propensity scoring to adjust for clinical features that may influence the preference for surgery only, radiation only, or combined surgery and radiation demonstrated that similar EFS is achieved with each mode of local therapy after propensity adjustment.[26] Data for patients with pelvic primary Ewing sarcoma from a North American intergroup trial showed no difference in local control or survival on the basis of local-control modality—surgery alone, radiation therapy alone, or radiation plus surgery.[27]
For patients who undergo gross-total resection with microscopic residual disease, the value of adjuvant radiation therapy is controversial. Investigations addressing this issue are retrospective and nonrandomized, limiting their value.
Evidence (postoperative radiation therapy):
- Investigators from St. Jude Children’s Research Hospital reported 39 patients with localized Ewing sarcoma who received both surgery and radiation.[13]
- Local failure for patients with positive margins was 17% and OS was 71%. Local failure for patients with negative margins was 5% and OS was 94%.
- However, in a large retrospective Italian study, 45 Gy of adjuvant radiation therapy for patients with inadequate margins did not appear to improve either local control or disease-free survival.[14] It is not known whether higher doses of radiation therapy could improve outcome. These investigators concluded that patients who are anticipated to have suboptimal surgery should be considered for definitive radiation therapy.
- The EURO-EWING-INTERGROUP-EE99 (NCT00020566) study reported the outcomes of 599 patients who presented with localized disease and had surgical resection after initial chemotherapy with at least 90% necrosis of the primary tumor.[28][Level of evidence: 3iiDi] The protocol recommended postoperative radiation therapy for patients with inadequate surgical margins, vertebral primary tumors, or thoracic tumors with pleural effusion, but the decision to use postoperative radiation therapy was left to the institutional investigator.
- Patients who received postoperative radiation therapy (n = 142) had a lower risk of failure than did patients who did not receive postoperative radiation therapy, even after controlling for known prognostic factors, including age, sex, tumor site, clinical response, quality of resection, and histologic necrosis. Most of the improvement was seen in a decreased risk of local recurrence. The improvement was greater in patients who were assessed to have 100% necrosis than in patients who were assessed to have 90% to 100% necrosis.
- There is a clear interaction between systemic therapy and local-control modalities for both local control and disease-free survival. The induction regimen used in the EURO-EWING-INTERGROUP-EE99 study is less intense than the induction regimen used in contemporaneous protocols in the COG, and it is not appropriate to extrapolate the results from the EURO-EWING-INTERGROUP-EE99 study to different systemic chemotherapy regimens.
In summary, surgery is chosen as definitive local therapy for suitable patients, but radiation therapy is appropriate for patients with unresectable disease or those who would experience functional compromise by definitive surgery. The possibility of impaired function needs to be measured against the possibility of second tumors in the radiation field (refer to the Late Effects of Treatment for Ewing Sarcoma section of this summary for more information). Adjuvant radiation therapy may be considered for patients with residual microscopic disease, inadequate margins, or who have viable tumor in the resected specimen and close margins.
When preoperative assessment has suggested a high probability that surgical margins will be close or positive, preoperative radiation therapy has achieved tumor shrinkage and allowed surgical resection with clear margins.[29]
High-Dose Therapy With Stem Cell Rescue for Ewing Sarcoma
For patients with a high risk of relapse with conventional treatments, certain investigators have utilized high-dose chemotherapy with hematopoietic stem cell transplant (HSCT) as consolidation treatment, in an effort to improve outcome.[19,30-42]
Evidence (high-dose therapy with stem cell rescue):
- In a prospective study, patients with bone and/or bone marrow metastases at diagnosis were treated with aggressive chemotherapy, surgery, and/or radiation and HSCT if a good initial response was achieved.[35]
- The study showed no benefit for HSCT compared with historical controls.
- A retrospective review using international bone marrow transplant registries compared the outcomes after treatment with either reduced-intensity conditioning or high-intensity conditioning followed by allogeneic SCT for patients with Ewing sarcoma at high risk for relapse.[43][Level of evidence: 3iiiA]
- There was no difference in outcome, and the authors concluded that this suggested the absence of a clinically relevant graft-versus-tumor effect against Ewing sarcoma tumor cells with current approaches.
- Multiple small studies that report benefit for HSCT have been published but are difficult to interpret because only patients who have a good initial response to standard chemotherapy are considered for HSCT.
The role of high-dose therapy followed by stem cell rescue is being investigated in the prospective, randomized Euro-Ewing trial (EURO-EWING-INTERGROUP-EE99) for patients who present with metastases and patients with localized tumors with poor response to initial chemotherapy.
Ewing Sarcoma/Specific Sites
Multiple analyses have evaluated diagnostic findings, treatment, and outcome of patients with bone lesions at the following anatomic primary sites:
Extraosseous Ewing Sarcoma
Extraosseous Ewing sarcoma is biologically similar to Ewing sarcoma arising in bone. Historically, most children and young adults with extraosseous Ewing sarcoma were treated on protocols designed for the treatment of rhabdomyosarcoma. This is important because many of the treatment regimens for rhabdomyosarcoma do not include an anthracycline, which is a critical component of current treatment regimens for Ewing sarcoma. Currently, patients with extraosseous Ewing sarcoma are eligible for studies that include Ewing sarcoma of bone.
From 1987 to 2004, 111 patients with nonmetastatic extraosseous Ewing sarcoma were enrolled on the RMS-88 and RMS-96 protocols.[62] Patients with initial complete tumor resection received ifosfamide, vincristine, and actinomycin (IVA) while patients with residual tumor received IVA plus doxorubicin (VAIA) or IVA plus carboplatin, epirubicin, and etoposide (CEVAIE). Seventy-six percent of patients received radiation. The 5-year EFS was 59% and OS was 69%. In a multivariate analysis, independent adverse prognostic factors included axial primary, tumor size greater than 10 cm, Intergroup Rhabdomyosarcoma Studies Group III, and lack of radiation therapy.
Two hundred thirty-six patients with extraosseous Ewing sarcoma were entered on studies of the German Pediatric Oncology Group.[63] The median age at diagnosis was 15 years and 133 patients were male. Primary tumor site was either extremity (n = 62) or central site (n = 174). Sixty of the 236 patients had metastases at diagnosis. Chemotherapy consisted of vincristine, doxorubicin, cyclophosphamide, and actinomycin (VACA); CEVAIE; or VIDE. The 5-year EFS was 49% and OS was 60%. Five-year survival was 70% for patients with localized disease and 33% for patients with metastasis at diagnosis. OS in patients with localized disease did not seem related to tumor site or size. In a retrospective French study, patients with extraosseous Ewing sarcoma were treated using a rhabdomyosarcoma regimen (no anthracyclines) or a Ewing sarcoma regimen (includes anthracyclines). Patients who received the anthracycline-containing regimen had a significantly better EFS and OS than did patients who did not receive anthracyclines.[64,65] Two North American Ewing sarcoma trials have included patients with extraosseous Ewing sarcoma.[23,66] In a review of data from the POG-9354 (INT-0154) and EWS0031 (NCT00006734) studies, 213 patients with extraosseous Ewing sarcoma and 826 patients with Ewing sarcoma of bone were identified. The hazard ratio of extraosseous Ewing sarcoma was superior (0.62), and extraosseous Ewing sarcoma was a favorable risk factor, independent of age, race, and primary site.[67][Level of evidence: 3iiDi]
Cutaneous Ewing sarcoma is a soft tissue tumor in the skin or subcutaneous tissue that seems to behave as a less-aggressive tumor than primary bone or soft tissue Ewing sarcoma. Tumors can form throughout the body, although the extremity is the most common site, and they are almost always localized. In a review of 78 reported cases, some lacking molecular confirmation, the OS was 91%. Adequate local control, defined as a complete resection with negative margins, radiation therapy, or a combination, significantly reduced the incidence of relapse. Standard chemotherapy for Ewing sarcoma is often used for these patients because there are no data to suggest which patients could be treated less aggressively.[68,69] A series of 56 patients with cutaneous or subcutaneous Ewing sarcoma confirmed the excellent outcome with the use of standard systemic therapy and local control. Attempted primary definitive surgery often resulted in the need for either radiation therapy or more function-compromising surgery, supporting the recommendation of biopsy only as initial surgery, rather than upfront unplanned resection.[70][Level of evidence: 3iiD]
Special Considerations for the Treatment of Children With Cancer
Cancer in children and adolescents is rare, although the overall incidence of childhood cancer has been slowly increasing since 1975.[71] Children and adolescents with cancer should be referred to medical centers that have a multidisciplinary team of cancer specialists with experience treating the cancers that occur during childhood and adolescence. This multidisciplinary team approach incorporates the skills of the following health care professionals and others to ensure that children receive treatment, supportive care, and rehabilitation that will achieve optimal survival and quality of life:
- Primary care physicians.
- Pediatric surgeons.
- Radiation oncologists.
- Pediatric oncologists/hematologists.
- Rehabilitation specialists.
- Pediatric nurse specialists.
- Social workers.
- Child-life professionals.
- Psychologists.
(Refer to the PDQ Supportive and Palliative Care summaries for specific information about supportive care for children and adolescents with cancer.)
Guidelines for pediatric cancer centers and their role in the treatment of pediatric patients with cancer have been outlined by the American Academy of Pediatrics.[72] At these pediatric cancer centers, clinical trials are available for most types of cancer that occur in children and adolescents, and the opportunity to participate in these trials is offered to most patients and their families. Clinical trials for children and adolescents with cancer are generally designed to compare potentially better therapy with therapy that is currently accepted as standard. Most of the progress made in identifying curative therapies for childhood cancers has been achieved through clinical trials. Information about ongoing clinical trials is available from the NCI website.
Childhood and adolescent cancer survivors require close monitoring because cancer therapy side effects may persist or develop months or years after treatment. (Refer to the PDQ summary on Late Effects of Treatment for Childhood Cancer for specific information about the incidence, type, and monitoring of late effects in childhood and adolescent cancer survivors.)
References
- Fuchs B, Valenzuela RG, Sim FH: Pathologic fracture as a complication in the treatment of Ewing's sarcoma. Clin Orthop (415): 25-30, 2003. [PUBMED Abstract]
- Hoffer FA, Gianturco LE, Fletcher JA, et al.: Percutaneous biopsy of peripheral primitive neuroectodermal tumors and Ewing's sarcomas for cytogenetic analysis. AJR Am J Roentgenol 162 (5): 1141-2, 1994. [PUBMED Abstract]
- Craft A, Cotterill S, Malcolm A, et al.: Ifosfamide-containing chemotherapy in Ewing's sarcoma: The Second United Kingdom Children's Cancer Study Group and the Medical Research Council Ewing's Tumor Study. J Clin Oncol 16 (11): 3628-33, 1998. [PUBMED Abstract]
- Shankar AG, Pinkerton CR, Atra A, et al.: Local therapy and other factors influencing site of relapse in patients with localised Ewing's sarcoma. United Kingdom Children's Cancer Study Group (UKCCSG). Eur J Cancer 35 (12): 1698-704, 1999. [PUBMED Abstract]
- Nilbert M, Saeter G, Elomaa I, et al.: Ewing's sarcoma treatment in Scandinavia 1984-1990--ten-year results of the Scandinavian Sarcoma Group Protocol SSGIV. Acta Oncol 37 (4): 375-8, 1998. [PUBMED Abstract]
- Ferrari S, Mercuri M, Rosito P, et al.: Ifosfamide and actinomycin-D, added in the induction phase to vincristine, cyclophosphamide and doxorubicin, improve histologic response and prognosis in patients with non metastatic Ewing's sarcoma of the extremity. J Chemother 10 (6): 484-91, 1998. [PUBMED Abstract]
- Grier HE, Krailo MD, Tarbell NJ, et al.: Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 348 (8): 694-701, 2003. [PUBMED Abstract]
- Thacker MM, Temple HT, Scully SP: Current treatment for Ewing's sarcoma. Expert Rev Anticancer Ther 5 (2): 319-31, 2005. [PUBMED Abstract]
- Juergens C, Weston C, Lewis I, et al.: Safety assessment of intensive induction with vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) in the treatment of Ewing tumors in the EURO-E.W.I.N.G. 99 clinical trial. Pediatr Blood Cancer 47 (1): 22-9, 2006. [PUBMED Abstract]
- Dunst J, Schuck A: Role of radiotherapy in Ewing tumors. Pediatr Blood Cancer 42 (5): 465-70, 2004. [PUBMED Abstract]
- Donaldson SS: Ewing sarcoma: radiation dose and target volume. Pediatr Blood Cancer 42 (5): 471-6, 2004. [PUBMED Abstract]
- Bacci G, Ferrari S, Longhi A, et al.: Role of surgery in local treatment of Ewing's sarcoma of the extremities in patients undergoing adjuvant and neoadjuvant chemotherapy. Oncol Rep 11 (1): 111-20, 2004. [PUBMED Abstract]
- Krasin MJ, Rodriguez-Galindo C, Davidoff AM, et al.: Efficacy of combined surgery and irradiation for localized Ewings sarcoma family of tumors. Pediatr Blood Cancer 43 (3): 229-36, 2004. [PUBMED Abstract]
- Bacci G, Longhi A, Briccoli A, et al.: The role of surgical margins in treatment of Ewing's sarcoma family tumors: experience of a single institution with 512 patients treated with adjuvant and neoadjuvant chemotherapy. Int J Radiat Oncol Biol Phys 65 (3): 766-72, 2006. [PUBMED Abstract]
- Paulussen M, Ahrens S, Burdach S, et al.: Primary metastatic (stage IV) Ewing tumor: survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma Studies. Ann Oncol 9 (3): 275-81, 1998. [PUBMED Abstract]
- Pinkerton CR, Bataillard A, Guillo S, et al.: Treatment strategies for metastatic Ewing's sarcoma. Eur J Cancer 37 (11): 1338-44, 2001. [PUBMED Abstract]
- Miser JS, Krailo M, Meyers P, et al.: Metastatic Ewing's sarcoma(es) and primitive neuroectodermal tumor (PNET) of bone: failure of new regimens to improve outcome. [Abstract] Proceedings of the American Society of Clinical Oncology 15: A-1472, 467, 1996.
- Bernstein ML, Devidas M, Lafreniere D, et al.: Intensive therapy with growth factor support for patients with Ewing tumor metastatic at diagnosis: Pediatric Oncology Group/Children's Cancer Group Phase II Study 9457--a report from the Children's Oncology Group. J Clin Oncol 24 (1): 152-9, 2006. [PUBMED Abstract]
- Ladenstein R, Pötschger U, Le Deley MC, et al.: Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol 28 (20): 3284-91, 2010. [PUBMED Abstract]
- Haeusler J, Ranft A, Boelling T, et al.: The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES). Cancer 116 (2): 443-50, 2010. [PUBMED Abstract]
- Paulussen M, Craft AW, Lewis I, et al.: Results of the EICESS-92 Study: two randomized trials of Ewing's sarcoma treatment--cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J Clin Oncol 26 (27): 4385-93, 2008. [PUBMED Abstract]
- Le Deley MC, Paulussen M, Lewis I, et al.: Cyclophosphamide compared with ifosfamide in consolidation treatment of standard-risk Ewing sarcoma: results of the randomized noninferiority Euro-EWING99-R1 trial. J Clin Oncol 32 (23): 2440-8, 2014. [PUBMED Abstract]
- Womer RB, West DC, Krailo MD, et al.: Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 30 (33): 4148-54, 2012. [PUBMED Abstract]
- Brunetto AL, Castillo LA, Petrilli AS, et al.: Carboplatin in the treatment of Ewing sarcoma: Results of the first Brazilian collaborative study group for Ewing sarcoma family tumors-EWING1. Pediatr Blood Cancer 62 (10): 1747-53, 2015. [PUBMED Abstract]
- Mascarenhas L, Felgenhauer JL, Bond MC, et al.: Pilot Study of Adding Vincristine, Topotecan, and Cyclophosphamide to Interval-Compressed Chemotherapy in Newly Diagnosed Patients With Localized Ewing Sarcoma: A Report From the Children's Oncology Group. Pediatr Blood Cancer 63 (3): 493-8, 2016. [PUBMED Abstract]
- DuBois SG, Krailo MD, Gebhardt MC, et al.: Comparative evaluation of local control strategies in localized Ewing sarcoma of bone: a report from the Children's Oncology Group. Cancer 121 (3): 467-75, 2015. [PUBMED Abstract]
- Yock TI, Krailo M, Fryer CJ, et al.: Local control in pelvic Ewing sarcoma: analysis from INT-0091--a report from the Children's Oncology Group. J Clin Oncol 24 (24): 3838-43, 2006. [PUBMED Abstract]
- Foulon S, Brennan B, Gaspar N, et al.: Can postoperative radiotherapy be omitted in localised standard-risk Ewing sarcoma? An observational study of the Euro-E.W.I.N.G group. Eur J Cancer 61: 128-36, 2016. [PUBMED Abstract]
- Wagner TD, Kobayashi W, Dean S, et al.: Combination short-course preoperative irradiation, surgical resection, and reduced-field high-dose postoperative irradiation in the treatment of tumors involving the bone. Int J Radiat Oncol Biol Phys 73 (1): 259-66, 2009. [PUBMED Abstract]
- Kushner BH, Meyers PA: How effective is dose-intensive/myeloablative therapy against Ewing's sarcoma/primitive neuroectodermal tumor metastatic to bone or bone marrow? The Memorial Sloan-Kettering experience and a literature review. J Clin Oncol 19 (3): 870-80, 2001. [PUBMED Abstract]
- Marina N, Meyers PA: High-dose therapy and stem-cell rescue for Ewing's family of tumors in second remission. J Clin Oncol 23 (19): 4262-4, 2005. [PUBMED Abstract]
- Burdach S: Treatment of advanced Ewing tumors by combined radiochemotherapy and engineered cellular transplants. Pediatr Transplant 8 (Suppl 5): 67-82, 2004. [PUBMED Abstract]
- McTiernan A, Driver D, Michelagnoli MP, et al.: High dose chemotherapy with bone marrow or peripheral stem cell rescue is an effective treatment option for patients with relapsed or progressive Ewing's sarcoma family of tumours. Ann Oncol 17 (8): 1301-5, 2006. [PUBMED Abstract]
- Burdach S, Meyer-Bahlburg A, Laws HJ, et al.: High-dose therapy for patients with primary multifocal and early relapsed Ewing's tumors: results of two consecutive regimens assessing the role of total-body irradiation. J Clin Oncol 21 (16): 3072-8, 2003. [PUBMED Abstract]
- Meyers PA, Krailo MD, Ladanyi M, et al.: High-dose melphalan, etoposide, total-body irradiation, and autologous stem-cell reconstitution as consolidation therapy for high-risk Ewing's sarcoma does not improve prognosis. J Clin Oncol 19 (11): 2812-20, 2001. [PUBMED Abstract]
- Oberlin O, Rey A, Desfachelles AS, et al.: Impact of high-dose busulfan plus melphalan as consolidation in metastatic Ewing tumors: a study by the Société Française des Cancers de l'Enfant. J Clin Oncol 24 (24): 3997-4002, 2006. [PUBMED Abstract]
- Hawkins D, Barnett T, Bensinger W, et al.: Busulfan, melphalan, and thiotepa with or without total marrow irradiation with hematopoietic stem cell rescue for poor-risk Ewing-Sarcoma-Family tumors. Med Pediatr Oncol 34 (5): 328-37, 2000. [PUBMED Abstract]
- Rosenthal J, Bolotin E, Shakhnovits M, et al.: High-dose therapy with hematopoietic stem cell rescue in patients with poor prognosis Ewing family tumors. Bone Marrow Transplant 42 (5): 311-8, 2008. [PUBMED Abstract]
- Burdach S, Thiel U, Schöniger M, et al.: Total body MRI-governed involved compartment irradiation combined with high-dose chemotherapy and stem cell rescue improves long-term survival in Ewing tumor patients with multiple primary bone metastases. Bone Marrow Transplant 45 (3): 483-9, 2010. [PUBMED Abstract]
- Gaspar N, Rey A, Bérard PM, et al.: Risk adapted chemotherapy for localised Ewing's sarcoma of bone: the French EW93 study. Eur J Cancer 48 (9): 1376-85, 2012. [PUBMED Abstract]
- Drabko K, Raciborska A, Bilska K, et al.: Consolidation of first-line therapy with busulphan and melphalan, and autologous stem cell rescue in children with Ewing's sarcoma. Bone Marrow Transplant 47 (12): 1530-4, 2012. [PUBMED Abstract]
- Loschi S, Dufour C, Oberlin O, et al.: Tandem high-dose chemotherapy strategy as first-line treatment of primary disseminated multifocal Ewing sarcomas in children, adolescents and young adults. Bone Marrow Transplant 50 (8): 1083-8, 2015. [PUBMED Abstract]
- Thiel U, Wawer A, Wolf P, et al.: No improvement of survival with reduced- versus high-intensity conditioning for allogeneic stem cell transplants in Ewing tumor patients. Ann Oncol 22 (7): 1614-21, 2011. [PUBMED Abstract]
- Hoffmann C, Ahrens S, Dunst J, et al.: Pelvic Ewing sarcoma: a retrospective analysis of 241 cases. Cancer 85 (4): 869-77, 1999. [PUBMED Abstract]
- Sucato DJ, Rougraff B, McGrath BE, et al.: Ewing's sarcoma of the pelvis. Long-term survival and functional outcome. Clin Orthop (373): 193-201, 2000. [PUBMED Abstract]
- Bacci G, Ferrari S, Mercuri M, et al.: Multimodal therapy for the treatment of nonmetastatic Ewing sarcoma of pelvis. J Pediatr Hematol Oncol 25 (2): 118-24, 2003. [PUBMED Abstract]
- Bacci G, Ferrari S, Longhi A, et al.: Local and systemic control in Ewing's sarcoma of the femur treated with chemotherapy, and locally by radiotherapy and/or surgery. J Bone Joint Surg Br 85 (1): 107-14, 2003. [PUBMED Abstract]
- Ozaki T, Hillmann A, Hoffmann C, et al.: Ewing's sarcoma of the femur. Prognosis in 69 patients treated by the CESS group. Acta Orthop Scand 68 (1): 20-4, 1997. [PUBMED Abstract]
- Ayoub KS, Fiorenza F, Grimer RJ, et al.: Extensible endoprostheses of the humerus after resection of bone tumours. J Bone Joint Surg Br 81 (3): 495-500, 1999. [PUBMED Abstract]
- Bacci G, Palmerini E, Staals EL, et al.: Ewing's sarcoma family tumors of the humerus: outcome of patients treated with radiotherapy, surgery or surgery and adjuvant radiotherapy. Radiother Oncol 93 (2): 383-7, 2009. [PUBMED Abstract]
- Casadei R, Magnani M, Biagini R, et al.: Prognostic factors in Ewing's sarcoma of the foot. Clin Orthop (420): 230-8, 2004. [PUBMED Abstract]
- Anakwenze OA, Parker WL, Wold LE, et al.: Ewing's sarcoma of the hand. J Hand Surg Eur Vol 34 (1): 35-9, 2009. [PUBMED Abstract]
- Shamberger RC, Laquaglia MP, Krailo MD, et al.: Ewing sarcoma of the rib: results of an intergroup study with analysis of outcome by timing of resection. J Thorac Cardiovasc Surg 119 (6): 1154-61, 2000. [PUBMED Abstract]
- Sirvent N, Kanold J, Levy C, et al.: Non-metastatic Ewing's sarcoma of the ribs: the French Society of Pediatric Oncology Experience. Eur J Cancer 38 (4): 561-7, 2002. [PUBMED Abstract]
- Shamberger RC, LaQuaglia MP, Gebhardt MC, et al.: Ewing sarcoma/primitive neuroectodermal tumor of the chest wall: impact of initial versus delayed resection on tumor margins, survival, and use of radiation therapy. Ann Surg 238 (4): 563-7; discussion 567-8, 2003. [PUBMED Abstract]
- Schuck A, Ahrens S, Konarzewska A, et al.: Hemithorax irradiation for Ewing tumors of the chest wall. Int J Radiat Oncol Biol Phys 54 (3): 830-8, 2002. [PUBMED Abstract]
- Windfuhr JP: Primitive neuroectodermal tumor of the head and neck: incidence, diagnosis, and management. Ann Otol Rhinol Laryngol 113 (7): 533-43, 2004. [PUBMED Abstract]
- Venkateswaran L, Rodriguez-Galindo C, Merchant TE, et al.: Primary Ewing tumor of the vertebrae: clinical characteristics, prognostic factors, and outcome. Med Pediatr Oncol 37 (1): 30-5, 2001. [PUBMED Abstract]
- Marco RA, Gentry JB, Rhines LD, et al.: Ewing's sarcoma of the mobile spine. Spine 30 (7): 769-73, 2005. [PUBMED Abstract]
- Schuck A, Ahrens S, von Schorlemer I, et al.: Radiotherapy in Ewing tumors of the vertebrae: treatment results and local relapse analysis of the CESS 81/86 and EICESS 92 trials. Int J Radiat Oncol Biol Phys 63 (5): 1562-7, 2005. [PUBMED Abstract]
- Bacci G, Boriani S, Balladelli A, et al.: Treatment of nonmetastatic Ewing's sarcoma family tumors of the spine and sacrum: the experience from a single institution. Eur Spine J 18 (8): 1091-5, 2009. [PUBMED Abstract]
- Spiller M, Bisogno G, Ferrari A, et al.: Prognostic factors in localized extraosseus Ewing family tumors. [Abstract] Pediatr Blood Cancer 46 (10) : A-PD.024, 434, 2006.
- Ladenstein R, Pötschger U, Jürgens H, et al.: Comparison of treatment concepts for extraosseus Ewing tumors (EET) within consecutive trials of two GPOH Cooperative Study Groups. [Abstract] Pediatr Blood Cancer 45 (10) : A-P.C.004, 450, 2005.
- Castex MP, Rubie H, Stevens MC, et al.: Extraosseous localized ewing tumors: improved outcome with anthracyclines--the French society of pediatric oncology and international society of pediatric oncology. J Clin Oncol 25 (10): 1176-82, 2007. [PUBMED Abstract]
- Dantonello TM, Int-Veen C, Harms D, et al.: Cooperative trial CWS-91 for localized soft tissue sarcoma in children, adolescents, and young adults. J Clin Oncol 27 (9): 1446-55, 2009. [PUBMED Abstract]
- Granowetter L, Womer R, Devidas M, et al.: Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children's Oncology Group Study. J Clin Oncol 27 (15): 2536-41, 2009. [PUBMED Abstract]
- Cash T, McIlvaine E, Krailo MD, et al.: Comparison of clinical features and outcomes in patients with extraskeletal versus skeletal localized Ewing sarcoma: A report from the Children's Oncology Group. Pediatr Blood Cancer 63 (10): 1771-9, 2016. [PUBMED Abstract]
- Collier AB 3rd, Simpson L, Monteleone P: Cutaneous Ewing sarcoma: report of 2 cases and literature review of presentation, treatment, and outcome of 76 other reported cases. J Pediatr Hematol Oncol 33 (8): 631-4, 2011. [PUBMED Abstract]
- Terrier-Lacombe MJ, Guillou L, Chibon F, et al.: Superficial primitive Ewing's sarcoma: a clinicopathologic and molecular cytogenetic analysis of 14 cases. Mod Pathol 22 (1): 87-94, 2009. [PUBMED Abstract]
- Di Giannatale A, Frezza AM, Le Deley MC, et al.: Primary cutaneous and subcutaneous Ewing sarcoma. Pediatr Blood Cancer 62 (9): 1555-61, 2015. [PUBMED Abstract]
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014. [PUBMED Abstract]
- Corrigan JJ, Feig SA; American Academy of Pediatrics: Guidelines for pediatric cancer centers. Pediatrics 113 (6): 1833-5, 2004. [PUBMED Abstract]
Treatment of Localized Ewing Sarcoma
Standard Treatment Options for Localized Ewing Sarcoma
Standard treatment options for localized Ewing sarcoma include the following:
Because most patients with apparently localized disease at diagnosis have occult metastatic disease, multidrug chemotherapy and local disease control with surgery and/or radiation therapy is indicated in the treatment of all patients.[1-8] Current regimens for the treatment of localized Ewing sarcoma achieve event-free survival (EFS) and overall survival (OS) of approximately 70% at 5 years after diagnosis.[9]
Chemotherapy
Current standard chemotherapy in the United States includes vincristine, doxorubicin, and cyclophosphamide (VDC), alternating with ifosfamide and etoposide (IE) or VDC/IE.[9]; [10][Level of evidence: 1iiA]
Evidence (chemotherapy):
- IE has shown activity in Ewing sarcoma, and a large randomized clinical trial and a nonrandomized trial demonstrated that outcome was improved when IE was alternated with VDC.[2,9,11]
- Dactinomycin is no longer used for Ewing sarcoma in the United States but continues to be used in the Euro-Ewing studies.
- Increased dose intensity of doxorubicin during the initial months of therapy was associated with an improved outcome in a meta-analysis performed before the standard use of IE.[12]
- The use of high-dose VDC has shown promising results in small numbers of patients. A single-institution study of 44 patients treated with high-dose VDC and IE showed an 82% 4-year EFS.[13]
- However, in an intergroup trial of the Pediatric Oncology Group and the Children's Cancer Group, which compared an alkylator dose-intensified VDC/IE regimen with standard alkylator doses of the same VDC/IE regimen, no differences in outcome were observed.[14] Unlike the single-institution trial, this trial did not maintain the dose intensity of cyclophosphamide for the duration of treatment.[13]
In a Children's Oncology Group (COG) trial (COG-AEWS0031), 568 patients with newly diagnosed localized extradural Ewing sarcoma were randomly assigned to receive chemotherapy (VDC/IE) given either every 2 weeks (interval compression) or every 3 weeks (standard). Patients randomly assigned to the every 2-week interval of treatment had an improved 5-year EFS (73% vs. 65%, P = .048). There was no increase in toxicity observed with the every 2-week schedule.[10]
Local-control measures
Local control can be achieved by surgery and/or radiation therapy.
Surgery
Surgery is generally the preferred approach if the lesion is resectable.[15,16] The superiority of resection for local control has never been tested in a prospective randomized trial. The apparent superiority may represent selection bias.
- In past studies, smaller, more peripheral tumors were more likely to be treated with surgery, and larger, more central tumors were more likely to be treated with radiation therapy.[17]
- An Italian retrospective study showed that surgery improved outcome only in extremity tumors, although the number of patients with central axis Ewing sarcoma who achieved adequate margins was small.[8]
- In a series of 39 patients treated at St. Jude Children's Research Hospital who received both surgery and radiation, the 8-year local failure rate was 5% for patients with negative surgical margins and 17% for those with positive margins.[5]
- Data for patients with pelvic primary Ewing sarcoma from a North American intergroup trial showed no difference in local control or survival based on local-control modality—surgery alone, radiation therapy alone, or radiation plus surgery.[18]
Potential benefits of surgery include the following:
- If a very young child has Ewing sarcoma, surgery may be a less-morbid therapy than radiation therapy because of the retardation of bone growth caused by radiation.
- Another potential benefit for surgical resection of the primary tumor is related to the amount of necrosis in the resected tumor. Patients with residual viable tumor in the resected specimen have a worse outcome than those with complete necrosis. In a French Ewing study (EW88), EFS for patients with less than 5% viable tumor was 75%, EFS for patients with 5% to 30% viable tumor was 48%, and EFS for patients with more than 30% viable tumor was 20%.[17]
European investigators are studying whether treatment intensification (i.e., high-dose chemotherapy with stem cell rescue) will improve outcome for patients with a poor histologic response.
Radiation therapy is usually employed in the following cases:
- Patients who do not have a surgical option that preserves function.
- Patients whose tumors have been excised but with inadequate margins.
Pathologic fracture at the time of diagnosis does not preclude surgical resection and is not associated with adverse outcome.[19]
Radiation therapy
Radiation therapy is delivered in a setting in which stringent planning techniques are applied by those experienced in the treatment of Ewing sarcoma. Such an approach will result in local control of the tumor with acceptable morbidity in most patients.[1,2,20]
The radiation dose may be adjusted depending on the extent of residual disease after the initial surgical procedure. Radiation therapy is generally administered in fractionated doses totaling approximately 55.8 Gy to the prechemotherapy tumor volume. A randomized study of 40 patients with Ewing sarcoma using 55.8 Gy to the prechemotherapy tumor extent with a 2-cm margin compared with the same total-tumor dose after 39.6 Gy to the entire bone showed no difference in local control or EFS.[3] Hyperfractionated radiation therapy has not been associated with improved local control or decreased morbidity.[1]
Comparison of proton-beam radiation therapy and intensity-modulated radiation therapy (IMRT) treatment plans has shown that proton-beam radiation therapy can spare more normal tissue adjacent to Ewing sarcoma primary tumors than IMRT.[21] Follow-up remains relatively short, and there are no data available to determine whether the reduction in dose to adjacent tissue will result in improved functional outcome or reduce the risk of secondary malignancy. Because patient numbers are small and follow-up is relatively short, it is not possible to determine whether the risk of local recurrence might be increased by reducing radiation dose in tissue adjacent to the primary tumor.
Higher rates of local failure are seen in patients older than 14 years who have tumors more than 8 cm in length.[22] A retrospective analysis of patients with Ewing sarcoma of the chest wall compared patients who received hemithorax radiation therapy with those who received radiation therapy to the chest wall only. Patients with pleural invasion, pleural effusion, or intraoperative contamination were assigned to hemithorax radiation therapy. EFS was longer for patients who received hemithorax radiation, but the difference was not statistically significant. In addition, most patients with primary vertebral tumors did not receive hemithorax radiation and had a lower probability for EFS.[23]
For patients with residual disease after an attempt at surgical resection, the Intergroup Ewing Sarcoma Study (INT-0091) recommended 45 Gy to the original disease site plus a 10.8 Gy boost for patients with gross residual disease and 45 Gy plus a 5.4 Gy boost for patients with microscopic residual disease. No radiation therapy was recommended for those who have no evidence of microscopic residual disease after surgical resection.[14]
Radiation therapy is associated with the development of subsequent neoplasms. A retrospective study noted that patients who received 60 Gy or more had an incidence of second malignancy of 20%. Those who received 48 Gy to 60 Gy had an incidence of 5%, and those who received less than 48 Gy did not develop a second malignancy.[24] (Refer to the Late Effects of Treatment for Ewing Sarcoma section of this summary for more information.)
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Dunst J, Jürgens H, Sauer R, et al.: Radiation therapy in Ewing's sarcoma: an update of the CESS 86 trial. Int J Radiat Oncol Biol Phys 32 (4): 919-30, 1995. [PUBMED Abstract]
- Donaldson SS, Torrey M, Link MP, et al.: A multidisciplinary study investigating radiotherapy in Ewing's sarcoma: end results of POG #8346. Pediatric Oncology Group. Int J Radiat Oncol Biol Phys 42 (1): 125-35, 1998. [PUBMED Abstract]
- Craft A, Cotterill S, Malcolm A, et al.: Ifosfamide-containing chemotherapy in Ewing's sarcoma: The Second United Kingdom Children's Cancer Study Group and the Medical Research Council Ewing's Tumor Study. J Clin Oncol 16 (11): 3628-33, 1998. [PUBMED Abstract]
- Nilbert M, Saeter G, Elomaa I, et al.: Ewing's sarcoma treatment in Scandinavia 1984-1990--ten-year results of the Scandinavian Sarcoma Group Protocol SSGIV. Acta Oncol 37 (4): 375-8, 1998. [PUBMED Abstract]
- Krasin MJ, Davidoff AM, Rodriguez-Galindo C, et al.: Definitive surgery and multiagent systemic therapy for patients with localized Ewing sarcoma family of tumors: local outcome and prognostic factors. Cancer 104 (2): 367-73, 2005. [PUBMED Abstract]
- Bacci G, Forni C, Longhi A, et al.: Long-term outcome for patients with non-metastatic Ewing's sarcoma treated with adjuvant and neoadjuvant chemotherapies. 402 patients treated at Rizzoli between 1972 and 1992. Eur J Cancer 40 (1): 73-83, 2004. [PUBMED Abstract]
- Rosito P, Mancini AF, Rondelli R, et al.: Italian Cooperative Study for the treatment of children and young adults with localized Ewing sarcoma of bone: a preliminary report of 6 years of experience. Cancer 86 (3): 421-8, 1999. [PUBMED Abstract]
- Bacci G, Longhi A, Briccoli A, et al.: The role of surgical margins in treatment of Ewing's sarcoma family tumors: experience of a single institution with 512 patients treated with adjuvant and neoadjuvant chemotherapy. Int J Radiat Oncol Biol Phys 65 (3): 766-72, 2006. [PUBMED Abstract]
- Grier HE, Krailo MD, Tarbell NJ, et al.: Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 348 (8): 694-701, 2003. [PUBMED Abstract]
- Womer RB, West DC, Krailo MD, et al.: Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 30 (33): 4148-54, 2012. [PUBMED Abstract]
- Ferrari S, Mercuri M, Rosito P, et al.: Ifosfamide and actinomycin-D, added in the induction phase to vincristine, cyclophosphamide and doxorubicin, improve histologic response and prognosis in patients with non metastatic Ewing's sarcoma of the extremity. J Chemother 10 (6): 484-91, 1998. [PUBMED Abstract]
- Smith MA, Ungerleider RS, Horowitz ME, et al.: Influence of doxorubicin dose intensity on response and outcome for patients with osteogenic sarcoma and Ewing's sarcoma. J Natl Cancer Inst 83 (20): 1460-70, 1991. [PUBMED Abstract]
- Kolb EA, Kushner BH, Gorlick R, et al.: Long-term event-free survival after intensive chemotherapy for Ewing's family of tumors in children and young adults. J Clin Oncol 21 (18): 3423-30, 2003. [PUBMED Abstract]
- Granowetter L, Womer R, Devidas M, et al.: Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children's Oncology Group Study. J Clin Oncol 27 (15): 2536-41, 2009. [PUBMED Abstract]
- Hoffmann C, Ahrens S, Dunst J, et al.: Pelvic Ewing sarcoma: a retrospective analysis of 241 cases. Cancer 85 (4): 869-77, 1999. [PUBMED Abstract]
- Shamberger RC, Laquaglia MP, Krailo MD, et al.: Ewing sarcoma of the rib: results of an intergroup study with analysis of outcome by timing of resection. J Thorac Cardiovasc Surg 119 (6): 1154-61, 2000. [PUBMED Abstract]
- Oberlin O, Deley MC, Bui BN, et al.: Prognostic factors in localized Ewing's tumours and peripheral neuroectodermal tumours: the third study of the French Society of Paediatric Oncology (EW88 study). Br J Cancer 85 (11): 1646-54, 2001. [PUBMED Abstract]
- Yock TI, Krailo M, Fryer CJ, et al.: Local control in pelvic Ewing sarcoma: analysis from INT-0091--a report from the Children's Oncology Group. J Clin Oncol 24 (24): 3838-43, 2006. [PUBMED Abstract]
- Bramer JA, Abudu AA, Grimer RJ, et al.: Do pathological fractures influence survival and local recurrence rate in bony sarcomas? Eur J Cancer 43 (13): 1944-51, 2007. [PUBMED Abstract]
- Krasin MJ, Rodriguez-Galindo C, Billups CA, et al.: Definitive irradiation in multidisciplinary management of localized Ewing sarcoma family of tumors in pediatric patients: outcome and prognostic factors. Int J Radiat Oncol Biol Phys 60 (3): 830-8, 2004. [PUBMED Abstract]
- Rombi B, DeLaney TF, MacDonald SM, et al.: Proton radiotherapy for pediatric Ewing's sarcoma: initial clinical outcomes. Int J Radiat Oncol Biol Phys 82 (3): 1142-8, 2012. [PUBMED Abstract]
- Fuchs B, Valenzuela RG, Sim FH: Pathologic fracture as a complication in the treatment of Ewing's sarcoma. Clin Orthop (415): 25-30, 2003. [PUBMED Abstract]
- Schuck A, Ahrens S, Konarzewska A, et al.: Hemithorax irradiation for Ewing tumors of the chest wall. Int J Radiat Oncol Biol Phys 54 (3): 830-8, 2002. [PUBMED Abstract]
- Kuttesch JF Jr, Wexler LH, Marcus RB, et al.: Second malignancies after Ewing's sarcoma: radiation dose-dependency of secondary sarcomas. J Clin Oncol 14 (10): 2818-25, 1996. [PUBMED Abstract]
Treatment of Metastatic Ewing Sarcoma
Metastases at diagnosis are detected in approximately 25% of patients.[1] The prognosis of patients with metastatic disease is poor. Current therapies for patients who present with metastatic disease achieve 6-year event-free survival (EFS) of approximately 28% and overall survival (OS) of approximately 30%.[2,3] For patients with lung/pleural metastases only, 6-year EFS is approximately 40% when utilizing bilateral lung irradiation.[2,4] In contrast, patients with bone/bone marrow metastases have a 4-year EFS of approximately 28% and patients with combined lung and bone/bone marrow metastases have a 4-year EFS of approximately 14%.[4,5]
The following factors independently predict a poor outcome in patients presenting with metastatic disease:[3]
- Age older than 14 years.
- Primary tumor volume of more than 200 mL.
- More than one bone metastatic site.
- Bone marrow metastases.
- Additional lung metastases.
Standard Treatment Options for Metastatic Ewing Sarcoma
Standard treatment options for metastatic Ewing sarcoma include the following:
Chemotherapy
Standard treatment for patients with metastatic Ewing sarcoma utilizing alternating vincristine, doxorubicin, cyclophosphamide, and ifosfamide/etoposide combined with adequate local-control measures applied to both primary and metastatic sites often results in complete or partial responses; however, the overall cure rate is 20%.[5-7]
The following chemotherapy regimens have not shown benefit:
- In the Intergroup Ewing Sarcoma Study, patients with metastatic disease showed no benefit from the addition of ifosfamide and etoposide to a standard regimen of vincristine, doxorubicin, cyclophosphamide, and dactinomycin.[7]
- In another Intergroup study, increasing dose intensity of cyclophosphamide, ifosfamide, and doxorubicin did not improve outcome compared with regimens utilizing standard-dose intensity. This regimen increased toxicity and risk of second malignancy without improving EFS or OS.[2]
- Intensification of ifosfamide to 2.8 g/m2 per day for 5 days did not improve outcome when administered with standard chemotherapy in patients with newly diagnosed metastatic Ewing sarcoma.[8][Level of evidence: 3iiiDi]
Surgery and radiation therapy
Systematic use of surgery and radiation therapy for metastatic sites may improve overall outcome in patients with extrapulmonary metastases.
Evidence (surgery and radiation therapy):
- In a retrospective data analysis of 120 patients with multifocal metastatic Ewing sarcoma, patients receiving local treatment of both primary tumor and metastases had a better outcome than patients receiving local treatment of primary tumor only or with no local treatment (3-year EFS, 39% vs. 17% and 14%, P < .001).[9]
- A similar trend for better outcome with irradiation of all sites of metastatic disease was seen in three retrospective analyses of smaller groups of patients receiving radiation therapy to all tumor sites.[10-12] These results must be interpreted with caution. The patients who received local-control therapy to all known sites of metastatic disease were selected by the treating investigator, not randomly assigned. Patients with so many metastases that radiation to all sites would result in bone marrow failure were not selected to receive radiation to all sites of metastatic disease. Patients who did not achieve control of the primary tumor did not go on to have local control of all sites of metastatic disease. There was a selection bias such that while all patients in these reports had multiple sites of metastatic disease, the patients who had surgery and/or radiation therapy to all sites of clinically detectable metastatic disease had better responses to systemic therapy and fewer sites of metastasis than did patients who did not undergo similar therapy of metastatic sites.
Radiation therapy, delivered in a setting in which stringent planning techniques are applied by those experienced in the treatment of Ewing sarcoma, should be considered. Such an approach will result in local control of tumor with acceptable morbidity in most patients.[13]
The radiation dose depends on the metastatic site of disease:
- Bone and soft tissue. Stereotactic body radiation therapy has been used to treat metastatic sites in bone and soft tissue. The median total curative/definitive stereotactic body radiation therapy dose delivered was 40 Gy in five fractions (range, 30–60 Gy in 3–10 fractions). The median total palliative stereotactic body radiation therapy dose delivered was 40 Gy in five fractions (range, 16–50 Gy in 1–10 fractions). These short-course regimens with large-dose fractions are biologically equivalent to higher doses delivered with smaller-dose fractions given over longer treatment courses.[14][Level of evidence: 3iiiC]
- Pulmonary. For all patients with pulmonary metastases, whole-lung irradiation should be considered, even if complete resolution of overt pulmonary metastatic disease has been achieved with chemotherapy.[4,5,15] Radiation doses are modulated based on the amount of lung to be irradiated and on pulmonary function. Doses between 12 Gy and 15 Gy are generally used if whole lungs are treated.
Other therapies
More intensive therapies, many of which incorporate high-dose chemotherapy with or without total-body irradiation in conjunction with stem cell support, have not shown improvement in EFS rates for patients with bone and/or bone marrow metastases.[2,3,10,16-18]; [19][Level of evidence: 3iiiDi] (Refer to the High-Dose Therapy With Stem Cell Rescue for Ewing Sarcoma section of this summary for more information.)
- High-dose chemotherapy with stem cell support. One of the largest studies was the EURO-EWING-Intergroup-EE99 R3 trial that enrolled 281 patients with primary disseminated metastatic Ewing sarcoma. Patients were treated with six cycles of vincristine, ifosfamide, doxorubicin, and etoposide followed by high-dose therapy and autologous stem cell transplant and demonstrated a 3-year EFS of 27% and OS of 34%. Factors such as the presence and number of bone lesions, primary tumor volume greater than 200 mL, age older than 14 years, additional pulmonary metastases, and bone marrow involvement were identified as independent prognostic factors.[3][Level of evidence: 3iiDi] The impact of high-dose chemotherapy with peripheral blood stem cell support for patients with isolated lung metastases is unknown and is being studied in the EURO-EWING-INTERGROUP-EE99 trial, for which results are pending.[16]
- Melphalan. Melphalan, at nonmyeloablative doses, proved to be an active agent in an upfront window study for patients with metastatic disease at diagnosis; however, the cure rate remained extremely low.[20]
- Irinotecan. Irinotecan was administered as a single agent in an upfront window for newly diagnosed metastatic Ewing sarcoma patients and showed modest activity (partial response in 5 of 24 patients).[21][Level of evidence: 3iiiDiv] Further investigation is needed to determine irinotecan dosing and combinations with other agents for patients with Ewing sarcoma.
Treatment Options Under Clinical Evaluation for Metastatic Ewing Sarcoma
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following is an example of a national and/or institutional clinical trial that is currently being conducted:
- AEWS1221; NCI-2014-02380 (NCT02306161) (Combination Chemotherapy With or Without Ganitumab in Treating Patients With Newly Diagnosed Metastatic Ewing Sarcoma): This phase II study is randomly assigning newly diagnosed patients with metastatic Ewing sarcoma to multiagent chemotherapy (vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide) with or without the addition of ganitumab (AMG 479). Stereotactic body radiation therapy is being evaluated to sites of bone metastases at a dose of 40 Gy in five fractions. This is a shorter course of therapy than is the standard treatment.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Esiashvili N, Goodman M, Marcus RB Jr: Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J Pediatr Hematol Oncol 30 (6): 425-30, 2008. [PUBMED Abstract]
- Miser JS, Goldsby RE, Chen Z, et al.: Treatment of metastatic Ewing sarcoma/primitive neuroectodermal tumor of bone: evaluation of increasing the dose intensity of chemotherapy--a report from the Children's Oncology Group. Pediatr Blood Cancer 49 (7): 894-900, 2007. [PUBMED Abstract]
- Ladenstein R, Pötschger U, Le Deley MC, et al.: Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol 28 (20): 3284-91, 2010. [PUBMED Abstract]
- Paulussen M, Ahrens S, Craft AW, et al.: Ewing's tumors with primary lung metastases: survival analysis of 114 (European Intergroup) Cooperative Ewing's Sarcoma Studies patients. J Clin Oncol 16 (9): 3044-52, 1998. [PUBMED Abstract]
- Paulussen M, Ahrens S, Burdach S, et al.: Primary metastatic (stage IV) Ewing tumor: survival analysis of 171 patients from the EICESS studies. European Intergroup Cooperative Ewing Sarcoma Studies. Ann Oncol 9 (3): 275-81, 1998. [PUBMED Abstract]
- Pinkerton CR, Bataillard A, Guillo S, et al.: Treatment strategies for metastatic Ewing's sarcoma. Eur J Cancer 37 (11): 1338-44, 2001. [PUBMED Abstract]
- Miser JS, Krailo MD, Tarbell NJ, et al.: Treatment of metastatic Ewing's sarcoma or primitive neuroectodermal tumor of bone: evaluation of combination ifosfamide and etoposide--a Children's Cancer Group and Pediatric Oncology Group study. J Clin Oncol 22 (14): 2873-6, 2004. [PUBMED Abstract]
- Magnan H, Goodbody CM, Riedel E, et al.: Ifosfamide dose-intensification for patients with metastatic Ewing sarcoma. Pediatr Blood Cancer 62 (4): 594-7, 2015. [PUBMED Abstract]
- Haeusler J, Ranft A, Boelling T, et al.: The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES). Cancer 116 (2): 443-50, 2010. [PUBMED Abstract]
- Burdach S, Thiel U, Schöniger M, et al.: Total body MRI-governed involved compartment irradiation combined with high-dose chemotherapy and stem cell rescue improves long-term survival in Ewing tumor patients with multiple primary bone metastases. Bone Marrow Transplant 45 (3): 483-9, 2010. [PUBMED Abstract]
- Paulino AC, Mai WY, Teh BS: Radiotherapy in metastatic ewing sarcoma. Am J Clin Oncol 36 (3): 283-6, 2013. [PUBMED Abstract]
- Casey DL, Wexler LH, Meyers PA, et al.: Radiation for bone metastases in Ewing sarcoma and rhabdomyosarcoma. Pediatr Blood Cancer 62 (3): 445-9, 2015. [PUBMED Abstract]
- Donaldson SS, Torrey M, Link MP, et al.: A multidisciplinary study investigating radiotherapy in Ewing's sarcoma: end results of POG #8346. Pediatric Oncology Group. Int J Radiat Oncol Biol Phys 42 (1): 125-35, 1998. [PUBMED Abstract]
- Brown LC, Lester RA, Grams MP, et al.: Stereotactic body radiotherapy for metastatic and recurrent ewing sarcoma and osteosarcoma. Sarcoma 2014: 418270, 2014. [PUBMED Abstract]
- Spunt SL, McCarville MB, Kun LE, et al.: Selective use of whole-lung irradiation for patients with Ewing sarcoma family tumors and pulmonary metastases at the time of diagnosis. J Pediatr Hematol Oncol 23 (2): 93-8, 2001. [PUBMED Abstract]
- Meyers PA, Krailo MD, Ladanyi M, et al.: High-dose melphalan, etoposide, total-body irradiation, and autologous stem-cell reconstitution as consolidation therapy for high-risk Ewing's sarcoma does not improve prognosis. J Clin Oncol 19 (11): 2812-20, 2001. [PUBMED Abstract]
- Burdach S, Meyer-Bahlburg A, Laws HJ, et al.: High-dose therapy for patients with primary multifocal and early relapsed Ewing's tumors: results of two consecutive regimens assessing the role of total-body irradiation. J Clin Oncol 21 (16): 3072-8, 2003. [PUBMED Abstract]
- Thiel U, Wawer A, Wolf P, et al.: No improvement of survival with reduced- versus high-intensity conditioning for allogeneic stem cell transplants in Ewing tumor patients. Ann Oncol 22 (7): 1614-21, 2011. [PUBMED Abstract]
- Loschi S, Dufour C, Oberlin O, et al.: Tandem high-dose chemotherapy strategy as first-line treatment of primary disseminated multifocal Ewing sarcomas in children, adolescents and young adults. Bone Marrow Transplant 50 (8): 1083-8, 2015. [PUBMED Abstract]
- Luksch R, Grignani G, Fagioli F, et al.: Response to melphalan in up-front investigational window therapy for patients with metastatic Ewing's family tumours. Eur J Cancer 43 (5): 885-90, 2007. [PUBMED Abstract]
- Morland B, Platt K, Whelan JS: A phase II window study of irinotecan (CPT-11) in high risk Ewing sarcoma: a Euro-E.W.I.N.G. study. Pediatr Blood Cancer 61 (3): 442-5, 2014. [PUBMED Abstract]
Treatment of Recurrent Ewing Sarcoma
Recurrence of Ewing sarcoma is most common within 2 years of initial diagnosis (approximately 80%).[1,2] However, late relapses occurring more than 5 years from initial diagnosis are more common in Ewing sarcoma (13%; 95% confidence interval, 9.4–16.5) than in other pediatric solid tumors.[3] An analysis of the Surveillance, Epidemiology, and End Results database identified 1,351 patients who survived more than 60 months from diagnosis.[4] Of these patients, 209 died, with 144 of the deaths (69%) attributed to recurrent, progressive Ewing sarcoma. Black race, male sex, older age at initial diagnosis, and primary tumors of the pelvis and axial skeleton were associated with a higher risk of late death. This analysis covered the period from 1973 to 2013, and the 1,351 patients represented only 38% of the patients in the original sample, which reflects the inferior treatment outcomes from the earlier era. It is possible that patients who reach the 5-year point after more contemporary treatment may not recapitulate this experience.
The overall prognosis for patients with recurrent Ewing sarcoma is poor; 5-year survival after recurrence is approximately 10% to 15%.[2,5,6]; [1][Level of evidence: 3iiA]
Prognostic factors include the following:
- Time to recurrence. Time to recurrence is the most important prognostic factor. Patients whose Ewing sarcoma recurred more than 2 years from initial diagnosis had a 5-year survival of 30% versus 7% for patients whose Ewing sarcoma recurred within 2 years.[1,2]
- Local and distant recurrence. Patients with both local recurrence and distant metastases have a worse outcome than do patients with either isolated local recurrence or metastatic recurrence alone.[1,2]
- Isolated pulmonary recurrence. Isolated pulmonary recurrence was not an important prognostic factor in a North American series.[1] In the Italian/Scandinavian experience, younger age, longer disease-free interval, and lung-only recurrence were associated with longer progression-free survival after recurrence. In this experience, patients with Ewing sarcoma that recurred after initial therapy, which included high-dose therapy with autologous stem cell rescue, were less likely to achieve a second complete remission.[7][Level of evidence: 3iiDiii]
Treatment Options for Recurrent Ewing Sarcoma
The selection of treatment for patients with recurrent disease depends on many factors, including the following:
- Site of recurrence.
- Previous treatment.
- Individual patient considerations.
There is no standardized second-line treatment for relapsed or refractory Ewing sarcoma.
Treatment options for recurrent Ewing sarcoma include the following:
Chemotherapy
Combinations of chemotherapy, such as cyclophosphamide and topotecan or irinotecan and temozolomide with or without vincristine, are active in recurrent Ewing sarcoma and can be considered for these patients.[8-13]
Evidence (chemotherapy):
- One phase II study of topotecan and cyclophosphamide showed a response in 6 of 17 patients with Ewing sarcoma; 16 of 49 patients had a clinical response in a similar trial in Germany.[8,10]
- In one retrospective series, 20 patients received temozolomide and irinotecan after recurrence. Five patients achieved a complete response and seven patients achieved a partial response.[12] A second retrospective series reported 11 of 20 objective responses in patients with recurrent Ewing sarcoma.[14][Level of evidence: 3iiDiv]
- The combination of docetaxel either with gemcitabine or irinotecan has achieved objective responses in relapsed Ewing sarcoma.[15][Level of evidence: 3iiA]; [16,17][Level of evidence: 3iiiDiv]
- High-dose ifosfamide (3 g/m2 per day for 5 days = 15 g/m2) has shown activity in patients whose Ewing sarcoma recurred after therapy that included standard ifosfamide (1.8 g/m2 per day for 5 days = 9 g/m2).[18][Level of evidence: 3iiiDiv]
Radiation therapy
Radiation therapy to bone lesions may provide palliation, although radical resection may improve outcome.[2] Patients with pulmonary metastases who have not received radiation therapy to the lungs should be considered for whole-lung irradiation.[19] Residual disease in the lung may be surgically removed.
Other therapies
Other therapies that have been studied in the treatment of recurrent Ewing sarcoma include the following:
- High-dose chemotherapy with stem cell support. Aggressive attempts to control the disease, including myeloablative regimens, have been used, but there is no evidence at this time to conclude that myeloablative therapy is superior to standard chemotherapy.[20,21]; [22][Level of evidence: 3iiA]; [23][Level of evidence: 3iiiDiii]
Most published reports about the use of high-dose therapy and stem cell support for patients with high-risk Ewing sarcoma have significant flaws in methodology. The most common error is the comparison of this high-risk group with an inappropriate control group. Patients with Ewing sarcoma at high risk of treatment failure who received high-dose therapy are compared with patients who did not receive high-dose therapy. Patients who undergo high-dose therapy must respond to systemic therapy, remain alive and respond to treatment long enough to reach the time at which stem cell therapy can be applied, be free of comorbid toxicity that precludes high-dose therapy, and have an adequate stem cell collection. Patients who undergo high-dose therapy and stem cell support are a highly selected group; comparing this patient group with all patients with high-risk Ewing sarcoma is inappropriate and leads to the erroneous conclusion that this strategy improves outcome. Surveys of patients undergoing allogeneic stem cell transplantation (SCT) for recurrent Ewing sarcoma did not show improved event-free survival when compared with autologous SCT and was associated with a higher complication rate.[20,24,25]
- Monoclonal antibody therapy. Monoclonal antibodies against the insulin-like growth factor 1 receptor (IGF1R) are reported to produce objective responses in metastatic recurrent Ewing sarcoma in roughly 10% of cases.[26-29][Level of evidence: 3iiDiv] In these studies, it was suggested that time-to-progression was prolonged compared with historical controls. Objective responses have been reported in studies combining the mTOR inhibitor temsirolimus with an IGF1R antibody. Stratification by IGF1R expression by immunohistochemistry in one of the studies did not predict clinical outcome in Ewing sarcoma patients.[30,31] Further studies are needed to identify patients who are likely to benefit from IGF1R therapy.
- Immunotherapy. Immunotherapy with antigen-specific T cells is being studied in patients with Ewing sarcoma because immune-mediated killing does not rely on pathways used by conventional therapies to which such tumors are often resistant. Several potential chimeric antigen receptors target antigens that have been identified for Ewing sarcomas. These include HER2 (human epidermal growth factor receptor 2),[32] GD2,[33] CD99 (MIC2 antigens),[34] and STEAP1 (six-transmembrane epithelial antigens of the prostate).[35] Some are in early-phase testing in sarcoma patients.[32]
Treatment Options Under Clinical Evaluation for Recurrent Ewing Sarcoma
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, refer to the ClinicalTrials.gov website.
The following are examples of national and/or institutional clinical trials that are currently being conducted:
- APEC1621 (NCT03155620) (Pediatric MATCH: Targeted Therapy Directed by Genetic Testing in Treating Pediatric Patients with Relapsed or Refractory Advanced Solid Tumors, Non-Hodgkin Lymphomas, or Histiocytic Disorders): NCI–Children's Oncology Group Pediatric Molecular Analysis for Therapeutic Choice (MATCH), referred to as Pediatric MATCH, will match targeted agents with specific molecular changes identified using a next-generation sequencing targeted assay of more than 3,000 different mutations across more than 160 genes in refractory and recurrent solid tumors. Children and adolescents aged 1 to 21 years are eligible for the trial.
Tumor tissue from progressive or recurrent disease must be available for molecular characterization. Patients with tumors that have molecular variants addressed by treatment arms included in the trial will be offered treatment on Pediatric MATCH. Additional information can be obtained on the ClinicalTrials.gov website for APEC1621 (NCT03155620).
- ADVL1622 (NCT02867592) (Cabozantinib-S-Malate in Treating Younger Patients with Recurrent, Refractory, or Newly Diagnosed Sarcomas, Wilms Tumor, or Other Rare Tumors): This is an open-label, two-stage, phase II trial of cabozantinib in selective solid tumors, including Ewing sarcoma. Cabozantinib is an oral small molecule inhibitor of multiple tyrosine kinases, including MET, VEGFR2, and RET, which are potential therapeutic targets in many pediatric and adult solid tumors.
- SARC028; NCI-2015-00320 (NCT02301039) (A Phase II Study of the Anti-PD1 Antibody Pembrolizumab [MK-3475] in Patients With Advanced Sarcomas): The objective response rate to the anti-PD1 inhibitor pembrolizumab will be assessed in patients with refractory, recurrent, and/or metastatic high-grade soft tissue sarcomas and bone sarcomas. Patients aged 18 years and older with soft tissue sarcomas and patients aged 12 years and older with bone sarcomas are eligible.
- ADVL1412 (NCT02304458) (Nivolumab With or Without Ipilimumab in Treating Younger Patients With Recurrent or Refractory Solid Tumors or Sarcomas): Nivolumab is an anti-PD1 inhibitor that is being studied alone and in combination with ipilimumab in relapsed sarcoma patients, including patients with Ewing sarcoma.
- ADVL1411 (NCT02116777) (BMN-673 and Temozolomide in Treating Younger Patients With Refractory or Recurrent Malignancies): In this study, the PARP inhibitor BMN-673 is combined with low-dose short duration temozolomide. This is based on the in vitro and mouse human tumor xenograft models, which showed impressive activity in a broad range of pediatric cancers, including Ewing sarcoma. After identifying the recommended phase II dose, this study is open for Ewing sarcoma patients.[36]
- ADVL1615 (NCT03323034) (Pevonedistat, Irinotecan Hydrochloride, and Temozolomide in Treating Patients With Recurrent or Refractory Solid Tumors or Lymphoma): This is a phase I study of pevonedistat in combination with temozolomide and irinotecan. Pevonedistat is a novel first-in-class Nedd8 activating enzyme (NAE) inhibitor that blocks the degradation of a subset of proteins that would normally be degraded by the 26S proteasome. Pevonedistat is more specific than previous proteasome inhibitors because it blocks the degradation of cullin-RING ligases, narrowing the targets to only a handful of key regulatory proteins important in cell survival. Preclinical, antitumor activity has been observed in Ewing sarcoma.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Leavey PJ, Mascarenhas L, Marina N, et al.: Prognostic factors for patients with Ewing sarcoma (EWS) at first recurrence following multi-modality therapy: A report from the Children's Oncology Group. Pediatr Blood Cancer 51 (3): 334-8, 2008. [PUBMED Abstract]
- Stahl M, Ranft A, Paulussen M, et al.: Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatr Blood Cancer 57 (4): 549-53, 2011. [PUBMED Abstract]
- Wasilewski-Masker K, Liu Q, Yasui Y, et al.: Late recurrence in pediatric cancer: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst 101 (24): 1709-20, 2009. [PUBMED Abstract]
- Davenport JR, Vo KT, Goldsby R, et al.: Conditional Survival and Predictors of Late Death in Patients With Ewing Sarcoma. Pediatr Blood Cancer 63 (6): 1091-5, 2016. [PUBMED Abstract]
- Barker LM, Pendergrass TW, Sanders JE, et al.: Survival after recurrence of Ewing's sarcoma family of tumors. J Clin Oncol 23 (19): 4354-62, 2005. [PUBMED Abstract]
- Bacci G, Longhi A, Ferrari S, et al.: Pattern of relapse in 290 patients with nonmetastatic Ewing's sarcoma family tumors treated at a single institution with adjuvant and neoadjuvant chemotherapy between 1972 and 1999. Eur J Surg Oncol 32 (9): 974-9, 2006. [PUBMED Abstract]
- Ferrari S, Luksch R, Hall KS, et al.: Post-relapse survival in patients with Ewing sarcoma. Pediatr Blood Cancer 62 (6): 994-9, 2015. [PUBMED Abstract]
- Saylors RL 3rd, Stine KC, Sullivan J, et al.: Cyclophosphamide plus topotecan in children with recurrent or refractory solid tumors: a Pediatric Oncology Group phase II study. J Clin Oncol 19 (15): 3463-9, 2001. [PUBMED Abstract]
- McTiernan A, Driver D, Michelagnoli MP, et al.: High dose chemotherapy with bone marrow or peripheral stem cell rescue is an effective treatment option for patients with relapsed or progressive Ewing's sarcoma family of tumours. Ann Oncol 17 (8): 1301-5, 2006. [PUBMED Abstract]
- Hunold A, Weddeling N, Paulussen M, et al.: Topotecan and cyclophosphamide in patients with refractory or relapsed Ewing tumors. Pediatr Blood Cancer 47 (6): 795-800, 2006. [PUBMED Abstract]
- Wagner LM, McAllister N, Goldsby RE, et al.: Temozolomide and intravenous irinotecan for treatment of advanced Ewing sarcoma. Pediatr Blood Cancer 48 (2): 132-9, 2007. [PUBMED Abstract]
- Casey DA, Wexler LH, Merchant MS, et al.: Irinotecan and temozolomide for Ewing sarcoma: the Memorial Sloan-Kettering experience. Pediatr Blood Cancer 53 (6): 1029-34, 2009. [PUBMED Abstract]
- Raciborska A, Bilska K, Drabko K, et al.: Vincristine, irinotecan, and temozolomide in patients with relapsed and refractory Ewing sarcoma. Pediatr Blood Cancer 60 (10): 1621-5, 2013. [PUBMED Abstract]
- Kurucu N, Sari N, Ilhan IE: Irinotecan and temozolamide treatment for relapsed Ewing sarcoma: a single-center experience and review of the literature. Pediatr Hematol Oncol 32 (1): 50-9, 2015. [PUBMED Abstract]
- Fox E, Patel S, Wathen JK, et al.: Phase II study of sequential gemcitabine followed by docetaxel for recurrent Ewing sarcoma, osteosarcoma, or unresectable or locally recurrent chondrosarcoma: results of Sarcoma Alliance for Research Through Collaboration Study 003. Oncologist 17 (3): 321, 2012. [PUBMED Abstract]
- Mora J, Cruz CO, Parareda A, et al.: Treatment of relapsed/refractory pediatric sarcomas with gemcitabine and docetaxel. J Pediatr Hematol Oncol 31 (10): 723-9, 2009. [PUBMED Abstract]
- Yoon JH, Kwon MM, Park HJ, et al.: A study of docetaxel and irinotecan in children and young adults with recurrent or refractory Ewing sarcoma family of tumors. BMC Cancer 14: 622, 2014. [PUBMED Abstract]
- Ferrari S, del Prever AB, Palmerini E, et al.: Response to high-dose ifosfamide in patients with advanced/recurrent Ewing sarcoma. Pediatr Blood Cancer 52 (5): 581-4, 2009. [PUBMED Abstract]
- Rodriguez-Galindo C, Billups CA, Kun LE, et al.: Survival after recurrence of Ewing tumors: the St Jude Children's Research Hospital experience, 1979-1999. Cancer 94 (2): 561-9, 2002. [PUBMED Abstract]
- Burdach S, van Kaick B, Laws HJ, et al.: Allogeneic and autologous stem-cell transplantation in advanced Ewing tumors. An update after long-term follow-up from two centers of the European Intergroup study EICESS. Stem-Cell Transplant Programs at Düsseldorf University Medical Center, Germany and St. Anna Kinderspital, Vienna, Austria. Ann Oncol 11 (11): 1451-62, 2000. [PUBMED Abstract]
- Burdach S, Meyer-Bahlburg A, Laws HJ, et al.: High-dose therapy for patients with primary multifocal and early relapsed Ewing's tumors: results of two consecutive regimens assessing the role of total-body irradiation. J Clin Oncol 21 (16): 3072-8, 2003. [PUBMED Abstract]
- Rasper M, Jabar S, Ranft A, et al.: The value of high-dose chemotherapy in patients with first relapsed Ewing sarcoma. Pediatr Blood Cancer 61 (8): 1382-6, 2014. [PUBMED Abstract]
- Gardner SL, Carreras J, Boudreau C, et al.: Myeloablative therapy with autologous stem cell rescue for patients with Ewing sarcoma. Bone Marrow Transplant 41 (10): 867-72, 2008. [PUBMED Abstract]
- Gilman AL, Oesterheld J: Myeloablative chemotherapy with autologous stem cell rescue for Ewing sarcoma. Bone Marrow Transplant 42 (11): 761; author reply 763, 2008. [PUBMED Abstract]
- Eapen M: Response to Dr Gilman. Bone Marrow Transplant 42 (11): 763, 2008.
- Malempati S, Weigel B, Ingle AM, et al.: Phase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 30 (3): 256-62, 2012. [PUBMED Abstract]
- Juergens H, Daw NC, Geoerger B, et al.: Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J Clin Oncol 29 (34): 4534-40, 2011. [PUBMED Abstract]
- Pappo AS, Patel SR, Crowley J, et al.: R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research through Collaboration study. J Clin Oncol 29 (34): 4541-7, 2011. [PUBMED Abstract]
- Tap WD, Demetri G, Barnette P, et al.: Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J Clin Oncol 30 (15): 1849-56, 2012. [PUBMED Abstract]
- Naing A, LoRusso P, Fu S, et al.: Insulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing's sarcoma family tumors. Clin Cancer Res 18 (9): 2625-31, 2012. [PUBMED Abstract]
- Schwartz GK, Tap WD, Qin LX, et al.: Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: a multicentre, open-label, phase 2 trial. Lancet Oncol 14 (4): 371-82, 2013. [PUBMED Abstract]
- Ahmed N, Brawley VS, Hegde M, et al.: Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol 33 (15): 1688-96, 2015. [PUBMED Abstract]
- Pule MA, Savoldo B, Myers GD, et al.: Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 14 (11): 1264-70, 2008. [PUBMED Abstract]
- Scotlandi K, Baldini N, Cerisano V, et al.: CD99 engagement: an effective therapeutic strategy for Ewing tumors. Cancer Res 60 (18): 5134-42, 2000. [PUBMED Abstract]
- Grunewald TG, Diebold I, Esposito I, et al.: STEAP1 is associated with the invasive and oxidative stress phenotype of Ewing tumors. Mol Cancer Res 10 (1): 52-65, 2012. [PUBMED Abstract]
- Smith MA, Reynolds CP, Kang MH, et al.: Synergistic activity of PARP inhibition by talazoparib (BMN 673) with temozolomide in pediatric cancer models in the pediatric preclinical testing program. Clin Cancer Res 21 (4): 819-32, 2015. [PUBMED Abstract]
Late Effects of Treatment for Ewing Sarcoma
Patients treated for Ewing sarcoma have a significantly higher risk of developing subsequent neoplasms than do patients in the general population.
Treatment-related acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) have generally been reported to occur in 1% to 2% of survivors of Ewing sarcoma,[1]; [2][Level of evidence: 3iiiDi] although some dose-intensive regimens appear to be associated with a higher risk of hematological malignancy.[3,4]; [5][Level of evidence: 3ii] Treatment-related AML and MDS arise most commonly at 2 to 5 years after diagnosis.
Survivors of Ewing sarcoma remain at increased risk of developing a subsequent solid tumor throughout their lifetime. Sarcomas usually occur within the previous radiation field.[6,7] The risk of developing a sarcoma after radiation therapy is dose-dependent, with higher doses associated with an increased risk of sarcoma development.[1]; [2][Level of evidence: 3iiiDi] The cumulative incidence of subsequent neoplasms in children treated for Ewing sarcoma between 1970 and 1986 at 25 years after diagnosis was 9.0% (confidence interval, 5.8–12.2). Most of these patients received radiation therapy; comparable long-term data do not yet exist for significant numbers of patients who did not receive radiation therapy.[8]
(Refer to the PDQ summary on Late Effects of Treatment for Childhood Cancer for a full discussion of the late effects of cancer treatment in children and adolescents.)
References
- Fuchs B, Valenzuela RG, Petersen IA, et al.: Ewing's sarcoma and the development of secondary malignancies. Clin Orthop (415): 82-9, 2003. [PUBMED Abstract]
- Goldsby R, Burke C, Nagarajan R, et al.: Second solid malignancies among children, adolescents, and young adults diagnosed with malignant bone tumors after 1976: follow-up of a Children's Oncology Group cohort. Cancer 113 (9): 2597-604, 2008. [PUBMED Abstract]
- Bhatia S, Krailo MD, Chen Z, et al.: Therapy-related myelodysplasia and acute myeloid leukemia after Ewing sarcoma and primitive neuroectodermal tumor of bone: A report from the Children's Oncology Group. Blood 109 (1): 46-51, 2007. [PUBMED Abstract]
- Kushner BH, Heller G, Cheung NK, et al.: High risk of leukemia after short-term dose-intensive chemotherapy in young patients with solid tumors. J Clin Oncol 16 (9): 3016-20, 1998. [PUBMED Abstract]
- Navid F, Billups C, Liu T, et al.: Second cancers in patients with the Ewing sarcoma family of tumours. Eur J Cancer 44 (7): 983-91, 2008. [PUBMED Abstract]
- Kuttesch JF Jr, Wexler LH, Marcus RB, et al.: Second malignancies after Ewing's sarcoma: radiation dose-dependency of secondary sarcomas. J Clin Oncol 14 (10): 2818-25, 1996. [PUBMED Abstract]
- Hawkins MM, Wilson LM, Burton HS, et al.: Radiotherapy, alkylating agents, and risk of bone cancer after childhood cancer. J Natl Cancer Inst 88 (5): 270-8, 1996. [PUBMED Abstract]
- Ginsberg JP, Goodman P, Leisenring W, et al.: Long-term survivors of childhood Ewing sarcoma: report from the childhood cancer survivor study. J Natl Cancer Inst 102 (16): 1272-83, 2010. [PUBMED Abstract]
Changes to This Summary (04/04/2018)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Treatment of Recurrent Ewing Sarcoma
Added text about the ADVL1622 and ADVL1615 clinical trials as treatment options under clinical evaluation for patients with recurrent Ewing sarcoma.
This summary is written and maintained by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of childhood Ewing sarcoma. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Pediatric Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Ewing Sarcoma Treatment are:
- Holcombe Edwin Grier, MD (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Andrea A. Hayes-Jordan, MD, FACS, FAAP (M.D. Anderson Cancer Center)
- Karen J. Marcus, MD (Dana-Farber Cancer Institute/Boston Children's Hospital)
- Paul A. Meyers, MD (Memorial Sloan-Kettering Cancer Center)
- Thomas A. Olson, MD (Aflac Cancer and Blood Disorders Center of Children's Healthcare of Atlanta - Egleston Campus)
- Nita Louise Seibel, MD (National Cancer Institute)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Pediatric Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Pediatric Treatment Editorial Board. PDQ Ewing Sarcoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/bone/hp/ewing-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389480]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Updated: April 4, 2018
Source URL: https://www.cancer.gov/publishedcontent/syndication/2434.htm
Source Agency: National Cancer Institute (NCI)
Captured Date: 2013-09-14 09:00:56.0
Uterine sarcoma: Professional resources from the National Cancer Institute
Uterine Sarcoma Treatment (PDQ®)–Health Professional Version
General Information About Uterine Sarcoma
Uterine sarcomas comprise less than 1% of gynecologic malignancies and 2% to 5% of all uterine malignancies.[1] The following tumors arise primarily from three distinct tissues:
- Carcinosarcomas arising in the endometrium, in other organs of mullerian origin, and accounting for 40% to 50% of all uterine sarcomas.
- Leiomyosarcomas arising from myometrial muscle, with a peak incidence occurring at age 50, and accounting for 30% of all uterine sarcomas.
- Sarcomas arising in the endometrial stroma, with a peak incidence occurring before menopause for the low-grade tumors and after menopause for the high-grade tumors, and accounting for 15% of all uterine sarcomas.
The three distinct entities are often grouped under uterine sarcomas; however, each type of tumor is currently being studied in separate clinical trials.
Carcinosarcomas (the preferred designation by the World Health Organization [WHO]) are also referred to as mixed mesodermal sarcomas or mullerian tumors. Controversy exists about the following issues:
- Whether they are true sarcomas.
- Whether the sarcomatous elements are actually derived from a common epithelial-cell precursor that also gives rise to the usually more abundant adenocarcinomatous elements.
The stromal components of the carcinosarcomas are further characterized by whether they contain homologous elements, such as malignant mesenchymal tissue considered possibly native to the uterus, or heterologous elements, such as striated muscle, cartilage, or bone, which are foreign to the uterus. Carcinosarcomas parallel endometrial cancer in its postmenopausal predominance and in other of its epidemiologic features; increasingly, the treatment of carcinosarcomas is becoming similar to combined modality approaches for endometrial adenocarcinomas.
Other rare forms of uterine sarcomas also fall under the WHO classification of mesenchymal and mixed tumors of the uterus. These include:[2,3]
- Mixed endometrial stromal and smooth muscle tumors.
- Adenosarcomas, in which the epithelial elements appear benign within a malignant mesenchymal background.
- Embryonal botryoides or rhabdomyosarcomas, which are found almost exclusively in infants.
- PEComa—a perivascular epithelial-cell tumor that may behave in a malignant fashion, which is the latest to be added.
(Refer to the PDQ summary on Childhood Rhabdomyosarcoma for more information.)
Risk Factors
The only documented etiologic factor in 10% to 25% of these malignancies is prior pelvic radiation therapy, which is often administered for benign uterine bleeding that began 5 to 25 years earlier. An increased incidence of uterine sarcoma has been associated with tamoxifen in the treatment of breast cancer. Subsequently, increases have also been noted when tamoxifen was given to prevent breast cancer in women at increased risk—a possible result of the estrogenic effect of tamoxifen on the uterus. Because of this increase, patients on tamoxifen should have follow-up pelvic examinations and should undergo endometrial biopsy if there is any abnormal uterine bleeding.[4-6]
Prognosis
The prognosis for women with uterine sarcoma is primarily dependent on the extent of disease at the time of diagnosis.[7] For women with carcinosarcomas, significant predictors of metastatic disease at initial surgery include:[7]
- Isthmic or cervical location.
- Lymphatic vascular space invasion.
- Serous and clear cell histology.
- Grade 2 or 3 carcinoma.
The above factors in addition to the following ones correlate with a progression-free interval:[7]
- Adnexal spread.
- Lymph node metastases.
- Tumor size.
- Peritoneal cytologic findings.
- Depth of myometrial invasion.
Factors that bear no relationship to the presence or absence of metastases at surgical exploration are:
- The presence or absence of stromal heterologous elements.
- The types of such elements.
- The grade of the stromal components.
- The mitotic activity of the stromal components.
In one study, women with a well-differentiated sarcomatous component or carcinosarcomas had significantly longer progression-free intervals than those with moderately to poorly differentiated sarcomas for the homologous and heterologous types. The recurrence rate was 44% for homologous tumors and 63% for heterologous tumors. The type of heterologous sarcoma had no effect on the progression-free interval.
For women with leiomyosarcomas, some investigators consider tumor size to be the most important prognostic factor; women with tumors greater than 5.0 cm in maximum diameter have a poor prognosis.[8] However, in a Gynecologic Oncology Group study, the mitotic index was the only factor significantly related to progression-free interval.[7] Leiomyosarcomas matched for other known prognostic factors may be more aggressive than their carcinosarcoma counterparts.[9] The 5-year survival rate for women with stage I disease, which is confined to the corpus, is approximately 50% versus 0% to 20% for the remaining stages.
Surgery alone can be curative if the malignancy is contained within the uterus. The value of pelvic radiation therapy is not established. Current studies consist primarily of phase II chemotherapy trials for patients with advanced disease. Adjuvant chemotherapy following complete resection for patients with stage I or II disease was not established to be effective in a randomized trial.[10] Yet, other nonrandomized trials have reported improved survival following adjuvant chemotherapy with or without radiation therapy.[11-13]
Related Summaries
Other PDQ summaries containing information related to uterine sarcoma include the following:
References
- Forney JP, Buschbaum HJ: Classifying, staging, and treating uterine sarcomas. Contemp Ob Gyn 18(3):47, 50, 55-56, 61-62, 64, 69, 1981.
- Gershenson D, McGuire W, Gore Martin, et al.: Gynecologic Cancer: Controversies in Management. 3rd ed. New York, NY: Churchill Livingstone, 2004.
- Tavassoéli F, Devilee P, et al.: Pathology and Genetics of Tumours of the Breast and Female Genital Organs. Lyon, France: International Agency for Research on Cancer, 2004.
- Bergman L, Beelen ML, Gallee MP, et al.: Risk and prognosis of endometrial cancer after tamoxifen for breast cancer. Comprehensive Cancer Centres' ALERT Group. Assessment of Liver and Endometrial cancer Risk following Tamoxifen. Lancet 356 (9233): 881-7, 2000. [PUBMED Abstract]
- Cohen I: Endometrial pathologies associated with postmenopausal tamoxifen treatment. Gynecol Oncol 94 (2): 256-66, 2004. [PUBMED Abstract]
- Wickerham DL, Fisher B, Wolmark N, et al.: Association of tamoxifen and uterine sarcoma. J Clin Oncol 20 (11): 2758-60, 2002. [PUBMED Abstract]
- Major FJ, Blessing JA, Silverberg SG, et al.: Prognostic factors in early-stage uterine sarcoma. A Gynecologic Oncology Group study. Cancer 71 (4 Suppl): 1702-9, 1993. [PUBMED Abstract]
- Evans HL, Chawla SP, Simpson C, et al.: Smooth muscle neoplasms of the uterus other than ordinary leiomyoma. A study of 46 cases, with emphasis on diagnostic criteria and prognostic factors. Cancer 62 (10): 2239-47, 1988. [PUBMED Abstract]
- Oláh KS, Dunn JA, Gee H: Leiomyosarcomas have a poorer prognosis than mixed mesodermal tumours when adjusting for known prognostic factors: the result of a retrospective study of 423 cases of uterine sarcoma. Br J Obstet Gynaecol 99 (7): 590-4, 1992. [PUBMED Abstract]
- Omura GA, Blessing JA, Major F, et al.: A randomized clinical trial of adjuvant adriamycin in uterine sarcomas: a Gynecologic Oncology Group Study. J Clin Oncol 3 (9): 1240-5, 1985. [PUBMED Abstract]
- Piver MS, Lele SB, Marchetti DL, et al.: Effect of adjuvant chemotherapy on time to recurrence and survival of stage I uterine sarcomas. J Surg Oncol 38 (4): 233-9, 1988. [PUBMED Abstract]
- van Nagell JR Jr, Hanson MB, Donaldson ES, et al.: Adjuvant vincristine, dactinomycin, and cyclophosphamide therapy in stage I uterine sarcomas. A pilot study. Cancer 57 (8): 1451-4, 1986. [PUBMED Abstract]
- Peters WA 3rd, Rivkin SE, Smith MR, et al.: Cisplatin and adriamycin combination chemotherapy for uterine stromal sarcomas and mixed mesodermal tumors. Gynecol Oncol 34 (3): 323-7, 1989. [PUBMED Abstract]
Cellular Classification of Uterine Sarcoma
The most common histologic types of uterine sarcomas include:
- Carcinosarcomas (mixed mesodermal sarcomas [40%–50%]).
- Leiomyosarcomas (30%).
- Endometrial stromal sarcomas (15%).
The uterine neoplasm classification of the International Society of Gynecologic Pathologists and the World Health Organization uses the term carcinosarcomas for all primary uterine neoplasms containing malignant elements of both epithelial and stromal light microscopic appearances, regardless of whether malignant heterologous elements are present.[1]
References
- Silverberg SG, Major FJ, Blessing JA, et al.: Carcinosarcoma (malignant mixed mesodermal tumor) of the uterus. A Gynecologic Oncology Group pathologic study of 203 cases. Int J Gynecol Pathol 9 (1): 1-19, 1990. [PUBMED Abstract]
Stage Information for Uterine Sarcoma
Definitions: FIGO
The Féderation Internationale de Gynécologie et d’Obstétrique (FIGO) and the American Joint Committee on Cancer (AJCC) have designated staging to define carcinoma of the corpus uteri, which applies to uterine sarcoma; the FIGO system is most commonly used.[1,2]
Uterine sarcomas include leiomyosarcomas, endometrial stromal sarcomas, and adenosarcomas.
| Stage | Description |
|---|---|
| aAdapted from FIGO Committee on Gynecologic Oncology.[1] | |
| bEither G1, G2, or G3 (G = grade). | |
| cEndocervical glandular involvement only should be considered as stage I and no longer as stage II. | |
| dPositive cytology has to be reported separately without changing the stage. | |
| Ib | Tumor confined to the corpus uteri. |
| IAb | No or less than half myometrial invasion. |
| IBb | Invasion equal to or more than half of the myometrium. |
| IIb | Tumor invades cervical stroma but does not extend beyond the uterus.c |
| IIIb | Local and/or regional spread of the tumor. |
| IIIAb | Tumor invades the serosa of the corpus uteri and/or adnexae.d |
| IIIBb | Vaginal and/or parametrial involvement.d |
| IIICb | Metastases to pelvic and/or para-aortic lymph nodes.d |
| IIIC1b | Positive pelvic nodes. |
| IIIC2b | Positive para-aortic lymph nodes with or without positive pelvic lymph nodes. |
| IVb | Tumor invades bladder and/or bowel mucosa, and/or distant metastases. |
| IVAb | Tumor invasion of bladder and/or bowel mucosa. |
| IVBb | Distant metastases, including intra-abdominal metastases and/or inguinal lymph nodes. |
References
- Pecorelli S: Revised FIGO staging for carcinoma of the vulva, cervix, and endometrium. Int J Gynaecol Obstet 105 (2): 103-4, 2009. [PUBMED Abstract]
- Corpus uteri. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 403-18.
Treatment Option Overview
Surgery is often the principal means of diagnosis and is the primary treatment for all patients with uterine sarcoma. If the diagnosis is known, the extent of surgery is planned according to the stage of the tumor. Hysterectomy is usually performed when a uterine malignancy is suspected, except for rare instances when preservation of the uterus in a young patient is deemed safe for the type of cancer (e.g., a totally confined low-grade leiomyosarcoma in a woman who desires to retain childbearing potential). Medically suitable patients with the preoperative diagnosis of uterine sarcoma are considered candidates for abdominal hysterectomy, bilateral salpingo-oophorectomy, and pelvic and periaortic selective lymphadenectomy. Cytologic washings are obtained from the pelvis and abdomen. Thorough examination of the diaphragm, omentum, and upper abdomen is performed.
There is no firm evidence from a prospective study that adjuvant chemotherapy or radiation therapy is of benefit for patients with uterine sarcoma.[1] In one Gynecologic Oncology Group (GOG) study, the use of adjuvant doxorubicin did not alter the survival rate of patients with resected stage I or stage II uterine sarcomas; however, interpretation of these results is difficult because this study included some patients who received radiation and three types of uterine sarcomas that have variable responses to doxorubicin.[1][Level of evidence: 1iiA] However, because the risk of disease recurrence is high even with localized presentations, many physicians have considered the use of adjuvant chemotherapy or radiation therapy.[2] A report of a study (GOG-0150 [NCT00002546]) that addressed radiation therapy versus adjuvant chemotherapy is awaited.[3]
References
- Omura GA, Blessing JA, Major F, et al.: A randomized clinical trial of adjuvant adriamycin in uterine sarcomas: a Gynecologic Oncology Group Study. J Clin Oncol 3 (9): 1240-5, 1985. [PUBMED Abstract]
- Kohorn EI, Schwartz PE, Chambers JT, et al.: Adjuvant therapy in mixed mullerian tumors of the uterus. Gynecol Oncol 23 (2): 212-21, 1986. [PUBMED Abstract]
- Wolfson AH, Brady MF, Mannel RS, et al.: A Gynecologic Oncology Group randomized trial of whole abdominal irradiation (WAI) vs cisplatin-ifosfamide+mesna (CIM) in optimally debulked stage I-IV carcinosarcoma (CS) of the uterus. [Abstract] J Clin Oncol 24 (Suppl 18): A-5001, 256s, 2006.
Stage I Uterine Sarcoma
Standard treatment options:
- Surgery (total abdominal hysterectomy, bilateral salpingo-oophorectomy, and pelvic and periaortic selective lymphadenectomy).
- Surgery plus pelvic radiation therapy.
- Surgery plus adjuvant chemotherapy.
- Surgery plus adjuvant radiation therapy as seen in the EORTC-55874 trial, for example.
In a nonrandomized, Gynecologic Oncology Group study in patients with stage I and II carcinosarcomas, those who had pelvic radiation therapy had a significant reduction of recurrences within the radiation treatment field but no alteration in survival.[1] A large nonrandomized study demonstrated improved survival and a lower local failure rate in patients with mixed mullerian tumors following postoperative external and intracavitary radiation therapy.[2] One nonrandomized study that predominantly included patients with carcinosarcomas appeared to show benefit for adjuvant therapy with cisplatin and doxorubicin.[3]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Hornback NB, Omura G, Major FJ: Observations on the use of adjuvant radiation therapy in patients with stage I and II uterine sarcoma. Int J Radiat Oncol Biol Phys 12 (12): 2127-30, 1986. [PUBMED Abstract]
- Larson B, Silfverswärd C, Nilsson B, et al.: Mixed müllerian tumours of the uterus--prognostic factors: a clinical and histopathologic study of 147 cases. Radiother Oncol 17 (2): 123-32, 1990. [PUBMED Abstract]
- Peters WA 3rd, Rivkin SE, Smith MR, et al.: Cisplatin and adriamycin combination chemotherapy for uterine stromal sarcomas and mixed mesodermal tumors. Gynecol Oncol 34 (3): 323-7, 1989. [PUBMED Abstract]
Stage II Uterine Sarcoma
Standard treatment options:
- Surgery (total abdominal hysterectomy, bilateral salpingo-oophorectomy, and pelvic and periaortic selective lymphadenectomy).
- Surgery plus pelvic radiation therapy.
- Surgery plus adjuvant chemotherapy.
- Surgery plus adjuvant radiation therapy (EORTC-55874).
In a nonrandomized, Gynecologic Oncology Group study in patients with stage I and II carcinosarcomas, those who had pelvic radiation therapy had a significant reduction of recurrences within the radiation treatment field but no alteration in survival.[1] One nonrandomized study that predominantly included patients with carcinosarcomas appeared to show benefit for adjuvant therapy with cisplatin and doxorubicin.[2]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Hornback NB, Omura G, Major FJ: Observations on the use of adjuvant radiation therapy in patients with stage I and II uterine sarcoma. Int J Radiat Oncol Biol Phys 12 (12): 2127-30, 1986. [PUBMED Abstract]
- Peters WA 3rd, Rivkin SE, Smith MR, et al.: Cisplatin and adriamycin combination chemotherapy for uterine stromal sarcomas and mixed mesodermal tumors. Gynecol Oncol 34 (3): 323-7, 1989. [PUBMED Abstract]
Stage III Uterine Sarcoma
Standard treatment options:
- Surgery (total abdominal hysterectomy, bilateral salpingo-oophorectomy, pelvic and periaortic selective lymphadenectomy, and resection of all gross tumor).
Treatment options under clinical evaluation:
- Surgery plus pelvic radiation therapy.
- Surgery plus adjuvant chemotherapy.
Carcinosarcomas (the preferred designation by the World Health Organization) are also referred to as mixed mesodermal or mullerian tumors. Controversy exists about the following issues:
- Whether they are true sarcomas.
- Whether the sarcomatous elements are actually derived from a common epithelial cell precursor that also gives rise to the usually more abundant adenocarcinomatous elements.
The stromal components of the carcinosarcomas are further characterized by whether they contain homologous elements (such as malignant mesenchymal tissue considered possibly native to the uterus) or heterologous elements (such as striated muscle, cartilage, or bone, which are foreign to the uterus). Carcinosarcomas parallel endometrial cancer in its postmenopausal predominance and in other of its epidemiologic features; increasingly, the treatment of carcinosarcomas is becoming similar to combined modality approaches for endometrial adenocarcinomas.
Patients who present with uterine sarcoma have been treated on a series of phase II studies by the Gynecologic Oncology Group, including the GOG-87B trial, for example.[1,2] These chemotherapy studies have documented some antitumor activity for cisplatin, doxorubicin, and ifosfamide. These studies have also documented differences in response leading to separate trials for patients with carcinosarcomas and leiomyosarcomas. As an example, in patients previously untreated with chemotherapy, ifosfamide had a 32.2% response rate in patients with carcinosarcomas [3] and a 17.2% partial response rate in patients with leiomyosarcomas.[2]
A randomized comparison that was seen in the GOG-108 trial, for example, of ifosfamide with or without cisplatin for first-line therapy for patients with measurable advanced or recurrent carcinosarcomas demonstrated a higher response rate (54% vs. 34%) and longer progression-free survival (PFS) on the combination arm (6 months vs. 4 months), but there was no significant improvement in survival (9 months vs. 8 months).[4][Level of evidence: 1iiA] The follow-up GOG-0161 [NCT00003128] study utilized 3-day ifosfamide regimens (instead of the more toxic 5-day regimen in the preceding study) for the control and for a combination with paclitaxel (with filgrastim starting on day 4).[5] The combination was superior in response rates (45% vs. 29%), PFS (8.4 months vs. 5.8 months), and overall survival (13.5 months and 8.4 months). The hazard ratio for death favored the combination 0.69 (95% confidence interval, 0.49–0.97).[5][Level of evidence: 1iiA] In this study, 52% of 179 evaluable patients had recurrent disease, 18% had stage III disease, and 30% had stage IV disease. In addition, imbalances were present in the sites of disease and in the use of prior radiation therapy, and 30 patients were excluded for wrong pathology.
A role for chemotherapy as adjuvant to surgery has not yet been established.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Thigpen JT, Blessing JA, Beecham J, et al.: Phase II trial of cisplatin as first-line chemotherapy in patients with advanced or recurrent uterine sarcomas: a Gynecologic Oncology Group study. J Clin Oncol 9 (11): 1962-6, 1991. [PUBMED Abstract]
- Sutton GP, Blessing JA, Barrett RJ, et al.: Phase II trial of ifosfamide and mesna in leiomyosarcoma of the uterus: a Gynecologic Oncology Group study. Am J Obstet Gynecol 166 (2): 556-9, 1992. [PUBMED Abstract]
- Sutton GP, Blessing JA, Rosenshein N, et al.: Phase II trial of ifosfamide and mesna in mixed mesodermal tumors of the uterus (a Gynecologic Oncology Group study). Am J Obstet Gynecol 161 (2): 309-12, 1989. [PUBMED Abstract]
- Sutton G, Brunetto VL, Kilgore L, et al.: A phase III trial of ifosfamide with or without cisplatin in carcinosarcoma of the uterus: A Gynecologic Oncology Group Study. Gynecol Oncol 79 (2): 147-53, 2000. [PUBMED Abstract]
- Homesley HD, Filiaci V, Markman M, et al.: Phase III trial of ifosfamide with or without paclitaxel in advanced uterine carcinosarcoma: a Gynecologic Oncology Group Study. J Clin Oncol 25 (5): 526-31, 2007. [PUBMED Abstract]
Stage IV Uterine Sarcoma
There is currently no standard therapy for patients with stage IV disease. These patients should be entered into an ongoing clinical trial.
Carcinosarcomas (the preferred designation by the World Health Organization) are also referred to as mixed mesodermal or mullerian tumors. Controversy exists about the following issues:
- Whether they are true sarcomas.
- Whether the sarcomatous elements are actually derived from a common epithelial cell precursor that also gives rise to the usually more abundant adenocarcinomatous elements.
The stromal components of the carcinosarcomas are further characterized by whether they contain homologous elements, such as malignant mesenchymal tissue considered possibly native to the uterus, or heterologous elements, such as striated muscle, cartilage, or bone, which is foreign to the uterus. Carcinosarcomas parallel endometrial cancer in its postmenopausal predominance and in other of its epidemiologic features; increasingly, the treatment of carcinosarcomas is becoming similar to combined modality approaches for endometrial adenocarcinomas.
Patients who present with uterine sarcoma have been treated on a series of phase II studies by the Gynecologic Oncology Group, including the GOG-87B trial, for example.[1] These chemotherapy studies have documented some antitumor activity for cisplatin, doxorubicin, and ifosfamide. These studies have also documented differences in response leading to separate trials for patients with carcinosarcomas and leiomyosarcomas. As an example, in patients previously untreated with chemotherapy, ifosfamide had a 32.2% response rate in patients with carcinosarcomas,[2] a 33% response rate in patients with endometrial stromal cell sarcomas,[3], and a 17.2% partial response rate in patients with leiomyosarcomas.[4] Doxorubicin in combination with dacarbazine or cyclophosphamide is no more active than doxorubicin alone for advanced disease.[5,6] Cisplatin has activity as first-line therapy and minimal activity as second-line therapy for patients with carcinosarcomas, but cisplatin is inactive as first- or second-line therapy for patients with leiomyosarcomas.[1,7]
A randomized comparison that was seen in the GOG-108 trial, for example, of ifosfamide with or without cisplatin for first-line therapy for patients with measurable advanced or recurrent carcinosarcomas demonstrated a higher response rate (54% vs. 34%) and longer progression-free survival (PFS) on the combination arm (6 months vs. 4 months), but there was no significant improvement in survival (9 months vs. 8 months).[8][Level of evidence: 1iiA] The follow-up GOG-0161 [NCT00003128] study utilized 3-day ifosfamide regimens (instead of the more toxic 5-day regimen in the preceding study) for the control and for a combination with paclitaxel (with filgrastim starting on day 4).[9] The combination was superior in response rates (45% vs. 29%), PFS (8.4 months vs. 5.8 months), and overall survival (13.5 months and 8.4 months). The hazard ratio for death favored the combination 0.69 (95% confidence interval, 0.49–0.97).[9][Level of evidence: 1iiA] In this study, 52% of 179 evaluable patients had recurrent disease, 18% had stage III disease, and 30% had stage IV disease. In addition, imbalances were present in the sites of disease and in the use of prior radiation therapy, and 30 patients were excluded for wrong pathology.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Thigpen JT, Blessing JA, Beecham J, et al.: Phase II trial of cisplatin as first-line chemotherapy in patients with advanced or recurrent uterine sarcomas: a Gynecologic Oncology Group study. J Clin Oncol 9 (11): 1962-6, 1991. [PUBMED Abstract]
- Sutton GP, Blessing JA, Rosenshein N, et al.: Phase II trial of ifosfamide and mesna in mixed mesodermal tumors of the uterus (a Gynecologic Oncology Group study). Am J Obstet Gynecol 161 (2): 309-12, 1989. [PUBMED Abstract]
- Sutton G, Blessing JA, Park R, et al.: Ifosfamide treatment of recurrent or metastatic endometrial stromal sarcomas previously unexposed to chemotherapy: a study of the Gynecologic Oncology Group. Obstet Gynecol 87 (5 Pt 1): 747-50, 1996. [PUBMED Abstract]
- Sutton GP, Blessing JA, Barrett RJ, et al.: Phase II trial of ifosfamide and mesna in leiomyosarcoma of the uterus: a Gynecologic Oncology Group study. Am J Obstet Gynecol 166 (2): 556-9, 1992. [PUBMED Abstract]
- Omura GA, Major FJ, Blessing JA, et al.: A randomized study of adriamycin with and without dimethyl triazenoimidazole carboxamide in advanced uterine sarcomas. Cancer 52 (4): 626-32, 1983. [PUBMED Abstract]
- Muss HB, Bundy B, DiSaia PJ, et al.: Treatment of recurrent or advanced uterine sarcoma. A randomized trial of doxorubicin versus doxorubicin and cyclophosphamide (a phase III trial of the Gynecologic Oncology Group). Cancer 55 (8): 1648-53, 1985. [PUBMED Abstract]
- Thigpen JT, Blessing JA, Wilbanks GD: Cisplatin as second-line chemotherapy in the treatment of advanced or recurrent leiomyosarcoma of the uterus. A phase II trial of the Gynecologic Oncology Group. Am J Clin Oncol 9 (1): 18-20, 1986. [PUBMED Abstract]
- Sutton G, Brunetto VL, Kilgore L, et al.: A phase III trial of ifosfamide with or without cisplatin in carcinosarcoma of the uterus: A Gynecologic Oncology Group Study. Gynecol Oncol 79 (2): 147-53, 2000. [PUBMED Abstract]
- Homesley HD, Filiaci V, Markman M, et al.: Phase III trial of ifosfamide with or without paclitaxel in advanced uterine carcinosarcoma: a Gynecologic Oncology Group Study. J Clin Oncol 25 (5): 526-31, 2007. [PUBMED Abstract]
Recurrent Uterine Sarcoma
There is currently no standard therapy for patients with recurrent disease. These patients should be entered into an ongoing clinical trial.
Patients who present with uterine sarcoma have been treated on a series of phase II studies by the Gynecologic Oncology Group, including the GOG-87B trial, for example. These chemotherapy studies have documented some antitumor activity for cisplatin, doxorubicin, and ifosfamide. These studies have also documented differences in response leading to separate trials for patients with carcinosarcomas and leiomyosarcomas. As an example, in patients previously untreated with chemotherapy, ifosfamide had a 32.2% response rate in patients with carcinosarcomas,[1] a 33% response rate in patients with endometrial stromal cell sarcomas,[2] and a 17.2% partial response rate in patients with leiomyosarcomas.[3] Doxorubicin in combination with dacarbazine or cyclophosphamide is no more active than doxorubicin alone for recurrent disease.[4,5] Cisplatin has activity as first-line therapy and minimal activity as second-line therapy for patients with carcinosarcomas, but cisplatin is inactive as first- or second-line therapy for patients with leiomyosarcomas.[6,7] A regimen of gemcitabine plus docetaxel had a 53% response rate in patients with unresectable leiomyosarcomas and is undergoing further study.[8]
A randomized comparison that was seen in the GOG-108 trial, for example, of ifosfamide with or without cisplatin for first-line therapy for patients with measurable advanced or recurrent carcinosarcomas demonstrated a higher response rate (54% vs. 34%) and longer progression-free survival (PFS) on the combination arm (6 months vs. 4 months), but there was no significant improvement in survival (9 months vs. 8 months).[9][Level of evidence: 1iiA] The follow-up GOG-0161 [NCT00003128] study utilized 3-day ifosfamide regimens (instead of the more toxic 5-day regimen in the preceding study) for the control and for a combination with paclitaxel (with filgrastim starting on day 4).[10] The combination was superior in response rates (45% vs. 29%), PFS (8.4 months vs. 5.8 months), and overall survival (13.5 months and 8.4 months). The hazard ratio for death favored the combination 0.69 (95% confidence interval, 0.49–0.97).[10][Level of evidence: 1iiA] In this study, 52% of 179 evaluable patients had recurrent disease, 18% had stage III disease, and 30% had stage IV disease. In addition, imbalances were present in the sites of disease and in the use of prior radiation therapy, and 30 patients were excluded for wrong pathology.
For patients with carcinosarcomas who have localized recurrence to the pelvis confirmed by computed tomographic scanning, radiation therapy may be effective palliation. Phase I and II clinical trials are appropriate for patients who recur with distant metastasis and are unresponsive to first-line phase II trials. High-dose progesterone hormone therapy may be of some benefit to patients with low-grade stromal sarcoma.[11]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Sutton GP, Blessing JA, Rosenshein N, et al.: Phase II trial of ifosfamide and mesna in mixed mesodermal tumors of the uterus (a Gynecologic Oncology Group study). Am J Obstet Gynecol 161 (2): 309-12, 1989. [PUBMED Abstract]
- Sutton G, Blessing JA, Park R, et al.: Ifosfamide treatment of recurrent or metastatic endometrial stromal sarcomas previously unexposed to chemotherapy: a study of the Gynecologic Oncology Group. Obstet Gynecol 87 (5 Pt 1): 747-50, 1996. [PUBMED Abstract]
- Sutton GP, Blessing JA, Barrett RJ, et al.: Phase II trial of ifosfamide and mesna in leiomyosarcoma of the uterus: a Gynecologic Oncology Group study. Am J Obstet Gynecol 166 (2): 556-9, 1992. [PUBMED Abstract]
- Omura GA, Major FJ, Blessing JA, et al.: A randomized study of adriamycin with and without dimethyl triazenoimidazole carboxamide in advanced uterine sarcomas. Cancer 52 (4): 626-32, 1983. [PUBMED Abstract]
- Muss HB, Bundy B, DiSaia PJ, et al.: Treatment of recurrent or advanced uterine sarcoma. A randomized trial of doxorubicin versus doxorubicin and cyclophosphamide (a phase III trial of the Gynecologic Oncology Group). Cancer 55 (8): 1648-53, 1985. [PUBMED Abstract]
- Thigpen JT, Blessing JA, Beecham J, et al.: Phase II trial of cisplatin as first-line chemotherapy in patients with advanced or recurrent uterine sarcomas: a Gynecologic Oncology Group study. J Clin Oncol 9 (11): 1962-6, 1991. [PUBMED Abstract]
- Thigpen JT, Blessing JA, Wilbanks GD: Cisplatin as second-line chemotherapy in the treatment of advanced or recurrent leiomyosarcoma of the uterus. A phase II trial of the Gynecologic Oncology Group. Am J Clin Oncol 9 (1): 18-20, 1986. [PUBMED Abstract]
- Hensley ML, Maki R, Venkatraman E, et al.: Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol 20 (12): 2824-31, 2002. [PUBMED Abstract]
- Sutton G, Brunetto VL, Kilgore L, et al.: A phase III trial of ifosfamide with or without cisplatin in carcinosarcoma of the uterus: A Gynecologic Oncology Group Study. Gynecol Oncol 79 (2): 147-53, 2000. [PUBMED Abstract]
- Homesley HD, Filiaci V, Markman M, et al.: Phase III trial of ifosfamide with or without paclitaxel in advanced uterine carcinosarcoma: a Gynecologic Oncology Group Study. J Clin Oncol 25 (5): 526-31, 2007. [PUBMED Abstract]
- Katz L, Merino MJ, Sakamoto H, et al.: Endometrial stromal sarcoma: a clinicopathologic study of 11 cases with determination of estrogen and progestin receptor levels in three tumors. Gynecol Oncol 26 (1): 87-97, 1987. [PUBMED Abstract]
Changes to This Summary (07/15/2015)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Editorial changes were made to this summary.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of uterine sarcoma. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Uterine Sarcoma Treatment are:
- Leslie R. Boyd, MD (New York University Medical Center)
- Franco M. Muggia, MD (New York University Medical Center)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Uterine Sarcoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/uterine/hp/uterine-sarcoma-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389327]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Uterine Sarcoma Treatment (PDQ®)–Health Professional Version
General Information About Uterine Sarcoma
Uterine sarcomas comprise less than 1% of gynecologic malignancies and 2% to 5% of all uterine malignancies.[1] The following tumors arise primarily from three distinct tissues:
- Carcinosarcomas arising in the endometrium, in other organs of mullerian origin, and accounting for 40% to 50% of all uterine sarcomas.
- Leiomyosarcomas arising from myometrial muscle, with a peak incidence occurring at age 50, and accounting for 30% of all uterine sarcomas.
- Sarcomas arising in the endometrial stroma, with a peak incidence occurring before menopause for the low-grade tumors and after menopause for the high-grade tumors, and accounting for 15% of all uterine sarcomas.
The three distinct entities are often grouped under uterine sarcomas; however, each type of tumor is currently being studied in separate clinical trials.
Carcinosarcomas (the preferred designation by the World Health Organization [WHO]) are also referred to as mixed mesodermal sarcomas or mullerian tumors. Controversy exists about the following issues:
- Whether they are true sarcomas.
- Whether the sarcomatous elements are actually derived from a common epithelial-cell precursor that also gives rise to the usually more abundant adenocarcinomatous elements.
The stromal components of the carcinosarcomas are further characterized by whether they contain homologous elements, such as malignant mesenchymal tissue considered possibly native to the uterus, or heterologous elements, such as striated muscle, cartilage, or bone, which are foreign to the uterus. Carcinosarcomas parallel endometrial cancer in its postmenopausal predominance and in other of its epidemiologic features; increasingly, the treatment of carcinosarcomas is becoming similar to combined modality approaches for endometrial adenocarcinomas.
Other rare forms of uterine sarcomas also fall under the WHO classification of mesenchymal and mixed tumors of the uterus. These include:[2,3]
- Mixed endometrial stromal and smooth muscle tumors.
- Adenosarcomas, in which the epithelial elements appear benign within a malignant mesenchymal background.
- Embryonal botryoides or rhabdomyosarcomas, which are found almost exclusively in infants.
- PEComa—a perivascular epithelial-cell tumor that may behave in a malignant fashion, which is the latest to be added.
(Refer to the PDQ summary on Childhood Rhabdomyosarcoma for more information.)
Risk Factors
The only documented etiologic factor in 10% to 25% of these malignancies is prior pelvic radiation therapy, which is often administered for benign uterine bleeding that began 5 to 25 years earlier. An increased incidence of uterine sarcoma has been associated with tamoxifen in the treatment of breast cancer. Subsequently, increases have also been noted when tamoxifen was given to prevent breast cancer in women at increased risk—a possible result of the estrogenic effect of tamoxifen on the uterus. Because of this increase, patients on tamoxifen should have follow-up pelvic examinations and should undergo endometrial biopsy if there is any abnormal uterine bleeding.[4-6]
Prognosis
The prognosis for women with uterine sarcoma is primarily dependent on the extent of disease at the time of diagnosis.[7] For women with carcinosarcomas, significant predictors of metastatic disease at initial surgery include:[7]
- Isthmic or cervical location.
- Lymphatic vascular space invasion.
- Serous and clear cell histology.
- Grade 2 or 3 carcinoma.
The above factors in addition to the following ones correlate with a progression-free interval:[7]
- Adnexal spread.
- Lymph node metastases.
- Tumor size.
- Peritoneal cytologic findings.
- Depth of myometrial invasion.
Factors that bear no relationship to the presence or absence of metastases at surgical exploration are:
- The presence or absence of stromal heterologous elements.
- The types of such elements.
- The grade of the stromal components.
- The mitotic activity of the stromal components.
In one study, women with a well-differentiated sarcomatous component or carcinosarcomas had significantly longer progression-free intervals than those with moderately to poorly differentiated sarcomas for the homologous and heterologous types. The recurrence rate was 44% for homologous tumors and 63% for heterologous tumors. The type of heterologous sarcoma had no effect on the progression-free interval.
For women with leiomyosarcomas, some investigators consider tumor size to be the most important prognostic factor; women with tumors greater than 5.0 cm in maximum diameter have a poor prognosis.[8] However, in a Gynecologic Oncology Group study, the mitotic index was the only factor significantly related to progression-free interval.[7] Leiomyosarcomas matched for other known prognostic factors may be more aggressive than their carcinosarcoma counterparts.[9] The 5-year survival rate for women with stage I disease, which is confined to the corpus, is approximately 50% versus 0% to 20% for the remaining stages.
Surgery alone can be curative if the malignancy is contained within the uterus. The value of pelvic radiation therapy is not established. Current studies consist primarily of phase II chemotherapy trials for patients with advanced disease. Adjuvant chemotherapy following complete resection for patients with stage I or II disease was not established to be effective in a randomized trial.[10] Yet, other nonrandomized trials have reported improved survival following adjuvant chemotherapy with or without radiation therapy.[11-13]
Related Summaries
Other PDQ summaries containing information related to uterine sarcoma include the following:
References
- Forney JP, Buschbaum HJ: Classifying, staging, and treating uterine sarcomas. Contemp Ob Gyn 18(3):47, 50, 55-56, 61-62, 64, 69, 1981.
- Gershenson D, McGuire W, Gore Martin, et al.: Gynecologic Cancer: Controversies in Management. 3rd ed. New York, NY: Churchill Livingstone, 2004.
- Tavassoéli F, Devilee P, et al.: Pathology and Genetics of Tumours of the Breast and Female Genital Organs. Lyon, France: International Agency for Research on Cancer, 2004.
- Bergman L, Beelen ML, Gallee MP, et al.: Risk and prognosis of endometrial cancer after tamoxifen for breast cancer. Comprehensive Cancer Centres' ALERT Group. Assessment of Liver and Endometrial cancer Risk following Tamoxifen. Lancet 356 (9233): 881-7, 2000. [PUBMED Abstract]
- Cohen I: Endometrial pathologies associated with postmenopausal tamoxifen treatment. Gynecol Oncol 94 (2): 256-66, 2004. [PUBMED Abstract]
- Wickerham DL, Fisher B, Wolmark N, et al.: Association of tamoxifen and uterine sarcoma. J Clin Oncol 20 (11): 2758-60, 2002. [PUBMED Abstract]
- Major FJ, Blessing JA, Silverberg SG, et al.: Prognostic factors in early-stage uterine sarcoma. A Gynecologic Oncology Group study. Cancer 71 (4 Suppl): 1702-9, 1993. [PUBMED Abstract]
- Evans HL, Chawla SP, Simpson C, et al.: Smooth muscle neoplasms of the uterus other than ordinary leiomyoma. A study of 46 cases, with emphasis on diagnostic criteria and prognostic factors. Cancer 62 (10): 2239-47, 1988. [PUBMED Abstract]
- Oláh KS, Dunn JA, Gee H: Leiomyosarcomas have a poorer prognosis than mixed mesodermal tumours when adjusting for known prognostic factors: the result of a retrospective study of 423 cases of uterine sarcoma. Br J Obstet Gynaecol 99 (7): 590-4, 1992. [PUBMED Abstract]
- Omura GA, Blessing JA, Major F, et al.: A randomized clinical trial of adjuvant adriamycin in uterine sarcomas: a Gynecologic Oncology Group Study. J Clin Oncol 3 (9): 1240-5, 1985. [PUBMED Abstract]
- Piver MS, Lele SB, Marchetti DL, et al.: Effect of adjuvant chemotherapy on time to recurrence and survival of stage I uterine sarcomas. J Surg Oncol 38 (4): 233-9, 1988. [PUBMED Abstract]
- van Nagell JR Jr, Hanson MB, Donaldson ES, et al.: Adjuvant vincristine, dactinomycin, and cyclophosphamide therapy in stage I uterine sarcomas. A pilot study. Cancer 57 (8): 1451-4, 1986. [PUBMED Abstract]
- Peters WA 3rd, Rivkin SE, Smith MR, et al.: Cisplatin and adriamycin combination chemotherapy for uterine stromal sarcomas and mixed mesodermal tumors. Gynecol Oncol 34 (3): 323-7, 1989. [PUBMED Abstract]
Cellular Classification of Uterine Sarcoma
The most common histologic types of uterine sarcomas include:
- Carcinosarcomas (mixed mesodermal sarcomas [40%–50%]).
- Leiomyosarcomas (30%).
- Endometrial stromal sarcomas (15%).
The uterine neoplasm classification of the International Society of Gynecologic Pathologists and the World Health Organization uses the term carcinosarcomas for all primary uterine neoplasms containing malignant elements of both epithelial and stromal light microscopic appearances, regardless of whether malignant heterologous elements are present.[1]
References
- Silverberg SG, Major FJ, Blessing JA, et al.: Carcinosarcoma (malignant mixed mesodermal tumor) of the uterus. A Gynecologic Oncology Group pathologic study of 203 cases. Int J Gynecol Pathol 9 (1): 1-19, 1990. [PUBMED Abstract]
Stage Information for Uterine Sarcoma
Definitions: FIGO
The Féderation Internationale de Gynécologie et d’Obstétrique (FIGO) and the American Joint Committee on Cancer (AJCC) have designated staging to define carcinoma of the corpus uteri, which applies to uterine sarcoma; the FIGO system is most commonly used.[1,2]
Uterine sarcomas include leiomyosarcomas, endometrial stromal sarcomas, and adenosarcomas.
| Stage | Description |
|---|---|
| aAdapted from FIGO Committee on Gynecologic Oncology.[1] | |
| bEither G1, G2, or G3 (G = grade). | |
| cEndocervical glandular involvement only should be considered as stage I and no longer as stage II. | |
| dPositive cytology has to be reported separately without changing the stage. | |
| Ib | Tumor confined to the corpus uteri. |
| IAb | No or less than half myometrial invasion. |
| IBb | Invasion equal to or more than half of the myometrium. |
| IIb | Tumor invades cervical stroma but does not extend beyond the uterus.c |
| IIIb | Local and/or regional spread of the tumor. |
| IIIAb | Tumor invades the serosa of the corpus uteri and/or adnexae.d |
| IIIBb | Vaginal and/or parametrial involvement.d |
| IIICb | Metastases to pelvic and/or para-aortic lymph nodes.d |
| IIIC1b | Positive pelvic nodes. |
| IIIC2b | Positive para-aortic lymph nodes with or without positive pelvic lymph nodes. |
| IVb | Tumor invades bladder and/or bowel mucosa, and/or distant metastases. |
| IVAb | Tumor invasion of bladder and/or bowel mucosa. |
| IVBb | Distant metastases, including intra-abdominal metastases and/or inguinal lymph nodes. |
References
- Pecorelli S: Revised FIGO staging for carcinoma of the vulva, cervix, and endometrium. Int J Gynaecol Obstet 105 (2): 103-4, 2009. [PUBMED Abstract]
- Corpus uteri. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 403-18.
Treatment Option Overview
Surgery is often the principal means of diagnosis and is the primary treatment for all patients with uterine sarcoma. If the diagnosis is known, the extent of surgery is planned according to the stage of the tumor. Hysterectomy is usually performed when a uterine malignancy is suspected, except for rare instances when preservation of the uterus in a young patient is deemed safe for the type of cancer (e.g., a totally confined low-grade leiomyosarcoma in a woman who desires to retain childbearing potential). Medically suitable patients with the preoperative diagnosis of uterine sarcoma are considered candidates for abdominal hysterectomy, bilateral salpingo-oophorectomy, and pelvic and periaortic selective lymphadenectomy. Cytologic washings are obtained from the pelvis and abdomen. Thorough examination of the diaphragm, omentum, and upper abdomen is performed.
There is no firm evidence from a prospective study that adjuvant chemotherapy or radiation therapy is of benefit for patients with uterine sarcoma.[1] In one Gynecologic Oncology Group (GOG) study, the use of adjuvant doxorubicin did not alter the survival rate of patients with resected stage I or stage II uterine sarcomas; however, interpretation of these results is difficult because this study included some patients who received radiation and three types of uterine sarcomas that have variable responses to doxorubicin.[1][Level of evidence: 1iiA] However, because the risk of disease recurrence is high even with localized presentations, many physicians have considered the use of adjuvant chemotherapy or radiation therapy.[2] A report of a study (GOG-0150 [NCT00002546]) that addressed radiation therapy versus adjuvant chemotherapy is awaited.[3]
References
- Omura GA, Blessing JA, Major F, et al.: A randomized clinical trial of adjuvant adriamycin in uterine sarcomas: a Gynecologic Oncology Group Study. J Clin Oncol 3 (9): 1240-5, 1985. [PUBMED Abstract]
- Kohorn EI, Schwartz PE, Chambers JT, et al.: Adjuvant therapy in mixed mullerian tumors of the uterus. Gynecol Oncol 23 (2): 212-21, 1986. [PUBMED Abstract]
- Wolfson AH, Brady MF, Mannel RS, et al.: A Gynecologic Oncology Group randomized trial of whole abdominal irradiation (WAI) vs cisplatin-ifosfamide+mesna (CIM) in optimally debulked stage I-IV carcinosarcoma (CS) of the uterus. [Abstract] J Clin Oncol 24 (Suppl 18): A-5001, 256s, 2006.
Stage I Uterine Sarcoma
Standard treatment options:
- Surgery (total abdominal hysterectomy, bilateral salpingo-oophorectomy, and pelvic and periaortic selective lymphadenectomy).
- Surgery plus pelvic radiation therapy.
- Surgery plus adjuvant chemotherapy.
- Surgery plus adjuvant radiation therapy as seen in the EORTC-55874 trial, for example.
In a nonrandomized, Gynecologic Oncology Group study in patients with stage I and II carcinosarcomas, those who had pelvic radiation therapy had a significant reduction of recurrences within the radiation treatment field but no alteration in survival.[1] A large nonrandomized study demonstrated improved survival and a lower local failure rate in patients with mixed mullerian tumors following postoperative external and intracavitary radiation therapy.[2] One nonrandomized study that predominantly included patients with carcinosarcomas appeared to show benefit for adjuvant therapy with cisplatin and doxorubicin.[3]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Hornback NB, Omura G, Major FJ: Observations on the use of adjuvant radiation therapy in patients with stage I and II uterine sarcoma. Int J Radiat Oncol Biol Phys 12 (12): 2127-30, 1986. [PUBMED Abstract]
- Larson B, Silfverswärd C, Nilsson B, et al.: Mixed müllerian tumours of the uterus--prognostic factors: a clinical and histopathologic study of 147 cases. Radiother Oncol 17 (2): 123-32, 1990. [PUBMED Abstract]
- Peters WA 3rd, Rivkin SE, Smith MR, et al.: Cisplatin and adriamycin combination chemotherapy for uterine stromal sarcomas and mixed mesodermal tumors. Gynecol Oncol 34 (3): 323-7, 1989. [PUBMED Abstract]
Stage II Uterine Sarcoma
Standard treatment options:
- Surgery (total abdominal hysterectomy, bilateral salpingo-oophorectomy, and pelvic and periaortic selective lymphadenectomy).
- Surgery plus pelvic radiation therapy.
- Surgery plus adjuvant chemotherapy.
- Surgery plus adjuvant radiation therapy (EORTC-55874).
In a nonrandomized, Gynecologic Oncology Group study in patients with stage I and II carcinosarcomas, those who had pelvic radiation therapy had a significant reduction of recurrences within the radiation treatment field but no alteration in survival.[1] One nonrandomized study that predominantly included patients with carcinosarcomas appeared to show benefit for adjuvant therapy with cisplatin and doxorubicin.[2]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Hornback NB, Omura G, Major FJ: Observations on the use of adjuvant radiation therapy in patients with stage I and II uterine sarcoma. Int J Radiat Oncol Biol Phys 12 (12): 2127-30, 1986. [PUBMED Abstract]
- Peters WA 3rd, Rivkin SE, Smith MR, et al.: Cisplatin and adriamycin combination chemotherapy for uterine stromal sarcomas and mixed mesodermal tumors. Gynecol Oncol 34 (3): 323-7, 1989. [PUBMED Abstract]
Stage III Uterine Sarcoma
Standard treatment options:
- Surgery (total abdominal hysterectomy, bilateral salpingo-oophorectomy, pelvic and periaortic selective lymphadenectomy, and resection of all gross tumor).
Treatment options under clinical evaluation:
- Surgery plus pelvic radiation therapy.
- Surgery plus adjuvant chemotherapy.
Carcinosarcomas (the preferred designation by the World Health Organization) are also referred to as mixed mesodermal or mullerian tumors. Controversy exists about the following issues:
- Whether they are true sarcomas.
- Whether the sarcomatous elements are actually derived from a common epithelial cell precursor that also gives rise to the usually more abundant adenocarcinomatous elements.
The stromal components of the carcinosarcomas are further characterized by whether they contain homologous elements (such as malignant mesenchymal tissue considered possibly native to the uterus) or heterologous elements (such as striated muscle, cartilage, or bone, which are foreign to the uterus). Carcinosarcomas parallel endometrial cancer in its postmenopausal predominance and in other of its epidemiologic features; increasingly, the treatment of carcinosarcomas is becoming similar to combined modality approaches for endometrial adenocarcinomas.
Patients who present with uterine sarcoma have been treated on a series of phase II studies by the Gynecologic Oncology Group, including the GOG-87B trial, for example.[1,2] These chemotherapy studies have documented some antitumor activity for cisplatin, doxorubicin, and ifosfamide. These studies have also documented differences in response leading to separate trials for patients with carcinosarcomas and leiomyosarcomas. As an example, in patients previously untreated with chemotherapy, ifosfamide had a 32.2% response rate in patients with carcinosarcomas [3] and a 17.2% partial response rate in patients with leiomyosarcomas.[2]
A randomized comparison that was seen in the GOG-108 trial, for example, of ifosfamide with or without cisplatin for first-line therapy for patients with measurable advanced or recurrent carcinosarcomas demonstrated a higher response rate (54% vs. 34%) and longer progression-free survival (PFS) on the combination arm (6 months vs. 4 months), but there was no significant improvement in survival (9 months vs. 8 months).[4][Level of evidence: 1iiA] The follow-up GOG-0161 [NCT00003128] study utilized 3-day ifosfamide regimens (instead of the more toxic 5-day regimen in the preceding study) for the control and for a combination with paclitaxel (with filgrastim starting on day 4).[5] The combination was superior in response rates (45% vs. 29%), PFS (8.4 months vs. 5.8 months), and overall survival (13.5 months and 8.4 months). The hazard ratio for death favored the combination 0.69 (95% confidence interval, 0.49–0.97).[5][Level of evidence: 1iiA] In this study, 52% of 179 evaluable patients had recurrent disease, 18% had stage III disease, and 30% had stage IV disease. In addition, imbalances were present in the sites of disease and in the use of prior radiation therapy, and 30 patients were excluded for wrong pathology.
A role for chemotherapy as adjuvant to surgery has not yet been established.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Thigpen JT, Blessing JA, Beecham J, et al.: Phase II trial of cisplatin as first-line chemotherapy in patients with advanced or recurrent uterine sarcomas: a Gynecologic Oncology Group study. J Clin Oncol 9 (11): 1962-6, 1991. [PUBMED Abstract]
- Sutton GP, Blessing JA, Barrett RJ, et al.: Phase II trial of ifosfamide and mesna in leiomyosarcoma of the uterus: a Gynecologic Oncology Group study. Am J Obstet Gynecol 166 (2): 556-9, 1992. [PUBMED Abstract]
- Sutton GP, Blessing JA, Rosenshein N, et al.: Phase II trial of ifosfamide and mesna in mixed mesodermal tumors of the uterus (a Gynecologic Oncology Group study). Am J Obstet Gynecol 161 (2): 309-12, 1989. [PUBMED Abstract]
- Sutton G, Brunetto VL, Kilgore L, et al.: A phase III trial of ifosfamide with or without cisplatin in carcinosarcoma of the uterus: A Gynecologic Oncology Group Study. Gynecol Oncol 79 (2): 147-53, 2000. [PUBMED Abstract]
- Homesley HD, Filiaci V, Markman M, et al.: Phase III trial of ifosfamide with or without paclitaxel in advanced uterine carcinosarcoma: a Gynecologic Oncology Group Study. J Clin Oncol 25 (5): 526-31, 2007. [PUBMED Abstract]
Stage IV Uterine Sarcoma
There is currently no standard therapy for patients with stage IV disease. These patients should be entered into an ongoing clinical trial.
Carcinosarcomas (the preferred designation by the World Health Organization) are also referred to as mixed mesodermal or mullerian tumors. Controversy exists about the following issues:
- Whether they are true sarcomas.
- Whether the sarcomatous elements are actually derived from a common epithelial cell precursor that also gives rise to the usually more abundant adenocarcinomatous elements.
The stromal components of the carcinosarcomas are further characterized by whether they contain homologous elements, such as malignant mesenchymal tissue considered possibly native to the uterus, or heterologous elements, such as striated muscle, cartilage, or bone, which is foreign to the uterus. Carcinosarcomas parallel endometrial cancer in its postmenopausal predominance and in other of its epidemiologic features; increasingly, the treatment of carcinosarcomas is becoming similar to combined modality approaches for endometrial adenocarcinomas.
Patients who present with uterine sarcoma have been treated on a series of phase II studies by the Gynecologic Oncology Group, including the GOG-87B trial, for example.[1] These chemotherapy studies have documented some antitumor activity for cisplatin, doxorubicin, and ifosfamide. These studies have also documented differences in response leading to separate trials for patients with carcinosarcomas and leiomyosarcomas. As an example, in patients previously untreated with chemotherapy, ifosfamide had a 32.2% response rate in patients with carcinosarcomas,[2] a 33% response rate in patients with endometrial stromal cell sarcomas,[3], and a 17.2% partial response rate in patients with leiomyosarcomas.[4] Doxorubicin in combination with dacarbazine or cyclophosphamide is no more active than doxorubicin alone for advanced disease.[5,6] Cisplatin has activity as first-line therapy and minimal activity as second-line therapy for patients with carcinosarcomas, but cisplatin is inactive as first- or second-line therapy for patients with leiomyosarcomas.[1,7]
A randomized comparison that was seen in the GOG-108 trial, for example, of ifosfamide with or without cisplatin for first-line therapy for patients with measurable advanced or recurrent carcinosarcomas demonstrated a higher response rate (54% vs. 34%) and longer progression-free survival (PFS) on the combination arm (6 months vs. 4 months), but there was no significant improvement in survival (9 months vs. 8 months).[8][Level of evidence: 1iiA] The follow-up GOG-0161 [NCT00003128] study utilized 3-day ifosfamide regimens (instead of the more toxic 5-day regimen in the preceding study) for the control and for a combination with paclitaxel (with filgrastim starting on day 4).[9] The combination was superior in response rates (45% vs. 29%), PFS (8.4 months vs. 5.8 months), and overall survival (13.5 months and 8.4 months). The hazard ratio for death favored the combination 0.69 (95% confidence interval, 0.49–0.97).[9][Level of evidence: 1iiA] In this study, 52% of 179 evaluable patients had recurrent disease, 18% had stage III disease, and 30% had stage IV disease. In addition, imbalances were present in the sites of disease and in the use of prior radiation therapy, and 30 patients were excluded for wrong pathology.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Thigpen JT, Blessing JA, Beecham J, et al.: Phase II trial of cisplatin as first-line chemotherapy in patients with advanced or recurrent uterine sarcomas: a Gynecologic Oncology Group study. J Clin Oncol 9 (11): 1962-6, 1991. [PUBMED Abstract]
- Sutton GP, Blessing JA, Rosenshein N, et al.: Phase II trial of ifosfamide and mesna in mixed mesodermal tumors of the uterus (a Gynecologic Oncology Group study). Am J Obstet Gynecol 161 (2): 309-12, 1989. [PUBMED Abstract]
- Sutton G, Blessing JA, Park R, et al.: Ifosfamide treatment of recurrent or metastatic endometrial stromal sarcomas previously unexposed to chemotherapy: a study of the Gynecologic Oncology Group. Obstet Gynecol 87 (5 Pt 1): 747-50, 1996. [PUBMED Abstract]
- Sutton GP, Blessing JA, Barrett RJ, et al.: Phase II trial of ifosfamide and mesna in leiomyosarcoma of the uterus: a Gynecologic Oncology Group study. Am J Obstet Gynecol 166 (2): 556-9, 1992. [PUBMED Abstract]
- Omura GA, Major FJ, Blessing JA, et al.: A randomized study of adriamycin with and without dimethyl triazenoimidazole carboxamide in advanced uterine sarcomas. Cancer 52 (4): 626-32, 1983. [PUBMED Abstract]
- Muss HB, Bundy B, DiSaia PJ, et al.: Treatment of recurrent or advanced uterine sarcoma. A randomized trial of doxorubicin versus doxorubicin and cyclophosphamide (a phase III trial of the Gynecologic Oncology Group). Cancer 55 (8): 1648-53, 1985. [PUBMED Abstract]
- Thigpen JT, Blessing JA, Wilbanks GD: Cisplatin as second-line chemotherapy in the treatment of advanced or recurrent leiomyosarcoma of the uterus. A phase II trial of the Gynecologic Oncology Group. Am J Clin Oncol 9 (1): 18-20, 1986. [PUBMED Abstract]
- Sutton G, Brunetto VL, Kilgore L, et al.: A phase III trial of ifosfamide with or without cisplatin in carcinosarcoma of the uterus: A Gynecologic Oncology Group Study. Gynecol Oncol 79 (2): 147-53, 2000. [PUBMED Abstract]
- Homesley HD, Filiaci V, Markman M, et al.: Phase III trial of ifosfamide with or without paclitaxel in advanced uterine carcinosarcoma: a Gynecologic Oncology Group Study. J Clin Oncol 25 (5): 526-31, 2007. [PUBMED Abstract]
Recurrent Uterine Sarcoma
There is currently no standard therapy for patients with recurrent disease. These patients should be entered into an ongoing clinical trial.
Patients who present with uterine sarcoma have been treated on a series of phase II studies by the Gynecologic Oncology Group, including the GOG-87B trial, for example. These chemotherapy studies have documented some antitumor activity for cisplatin, doxorubicin, and ifosfamide. These studies have also documented differences in response leading to separate trials for patients with carcinosarcomas and leiomyosarcomas. As an example, in patients previously untreated with chemotherapy, ifosfamide had a 32.2% response rate in patients with carcinosarcomas,[1] a 33% response rate in patients with endometrial stromal cell sarcomas,[2] and a 17.2% partial response rate in patients with leiomyosarcomas.[3] Doxorubicin in combination with dacarbazine or cyclophosphamide is no more active than doxorubicin alone for recurrent disease.[4,5] Cisplatin has activity as first-line therapy and minimal activity as second-line therapy for patients with carcinosarcomas, but cisplatin is inactive as first- or second-line therapy for patients with leiomyosarcomas.[6,7] A regimen of gemcitabine plus docetaxel had a 53% response rate in patients with unresectable leiomyosarcomas and is undergoing further study.[8]
A randomized comparison that was seen in the GOG-108 trial, for example, of ifosfamide with or without cisplatin for first-line therapy for patients with measurable advanced or recurrent carcinosarcomas demonstrated a higher response rate (54% vs. 34%) and longer progression-free survival (PFS) on the combination arm (6 months vs. 4 months), but there was no significant improvement in survival (9 months vs. 8 months).[9][Level of evidence: 1iiA] The follow-up GOG-0161 [NCT00003128] study utilized 3-day ifosfamide regimens (instead of the more toxic 5-day regimen in the preceding study) for the control and for a combination with paclitaxel (with filgrastim starting on day 4).[10] The combination was superior in response rates (45% vs. 29%), PFS (8.4 months vs. 5.8 months), and overall survival (13.5 months and 8.4 months). The hazard ratio for death favored the combination 0.69 (95% confidence interval, 0.49–0.97).[10][Level of evidence: 1iiA] In this study, 52% of 179 evaluable patients had recurrent disease, 18% had stage III disease, and 30% had stage IV disease. In addition, imbalances were present in the sites of disease and in the use of prior radiation therapy, and 30 patients were excluded for wrong pathology.
For patients with carcinosarcomas who have localized recurrence to the pelvis confirmed by computed tomographic scanning, radiation therapy may be effective palliation. Phase I and II clinical trials are appropriate for patients who recur with distant metastasis and are unresponsive to first-line phase II trials. High-dose progesterone hormone therapy may be of some benefit to patients with low-grade stromal sarcoma.[11]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Sutton GP, Blessing JA, Rosenshein N, et al.: Phase II trial of ifosfamide and mesna in mixed mesodermal tumors of the uterus (a Gynecologic Oncology Group study). Am J Obstet Gynecol 161 (2): 309-12, 1989. [PUBMED Abstract]
- Sutton G, Blessing JA, Park R, et al.: Ifosfamide treatment of recurrent or metastatic endometrial stromal sarcomas previously unexposed to chemotherapy: a study of the Gynecologic Oncology Group. Obstet Gynecol 87 (5 Pt 1): 747-50, 1996. [PUBMED Abstract]
- Sutton GP, Blessing JA, Barrett RJ, et al.: Phase II trial of ifosfamide and mesna in leiomyosarcoma of the uterus: a Gynecologic Oncology Group study. Am J Obstet Gynecol 166 (2): 556-9, 1992. [PUBMED Abstract]
- Omura GA, Major FJ, Blessing JA, et al.: A randomized study of adriamycin with and without dimethyl triazenoimidazole carboxamide in advanced uterine sarcomas. Cancer 52 (4): 626-32, 1983. [PUBMED Abstract]
- Muss HB, Bundy B, DiSaia PJ, et al.: Treatment of recurrent or advanced uterine sarcoma. A randomized trial of doxorubicin versus doxorubicin and cyclophosphamide (a phase III trial of the Gynecologic Oncology Group). Cancer 55 (8): 1648-53, 1985. [PUBMED Abstract]
- Thigpen JT, Blessing JA, Beecham J, et al.: Phase II trial of cisplatin as first-line chemotherapy in patients with advanced or recurrent uterine sarcomas: a Gynecologic Oncology Group study. J Clin Oncol 9 (11): 1962-6, 1991. [PUBMED Abstract]
- Thigpen JT, Blessing JA, Wilbanks GD: Cisplatin as second-line chemotherapy in the treatment of advanced or recurrent leiomyosarcoma of the uterus. A phase II trial of the Gynecologic Oncology Group. Am J Clin Oncol 9 (1): 18-20, 1986. [PUBMED Abstract]
- Hensley ML, Maki R, Venkatraman E, et al.: Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol 20 (12): 2824-31, 2002. [PUBMED Abstract]
- Sutton G, Brunetto VL, Kilgore L, et al.: A phase III trial of ifosfamide with or without cisplatin in carcinosarcoma of the uterus: A Gynecologic Oncology Group Study. Gynecol Oncol 79 (2): 147-53, 2000. [PUBMED Abstract]
- Homesley HD, Filiaci V, Markman M, et al.: Phase III trial of ifosfamide with or without paclitaxel in advanced uterine carcinosarcoma: a Gynecologic Oncology Group Study. J Clin Oncol 25 (5): 526-31, 2007. [PUBMED Abstract]
- Katz L, Merino MJ, Sakamoto H, et al.: Endometrial stromal sarcoma: a clinicopathologic study of 11 cases with determination of estrogen and progestin receptor levels in three tumors. Gynecol Oncol 26 (1): 87-97, 1987. [PUBMED Abstract]
Changes to This Summary (07/15/2015)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Editorial changes were made to this summary.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of uterine sarcoma. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Uterine Sarcoma Treatment are:
- Leslie R. Boyd, MD (New York University Medical Center)
- Franco M. Muggia, MD (New York University Medical Center)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Uterine Sarcoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/uterine/hp/uterine-sarcoma-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389327]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.
Uterine Sarcoma Treatment (PDQ®)–Health Professional Version
General Information About Uterine Sarcoma
Uterine sarcomas comprise less than 1% of gynecologic malignancies and 2% to 5% of all uterine malignancies.[1] The following tumors arise primarily from three distinct tissues:
- Carcinosarcomas arising in the endometrium, in other organs of mullerian origin, and accounting for 40% to 50% of all uterine sarcomas.
- Leiomyosarcomas arising from myometrial muscle, with a peak incidence occurring at age 50, and accounting for 30% of all uterine sarcomas.
- Sarcomas arising in the endometrial stroma, with a peak incidence occurring before menopause for the low-grade tumors and after menopause for the high-grade tumors, and accounting for 15% of all uterine sarcomas.
The three distinct entities are often grouped under uterine sarcomas; however, each type of tumor is currently being studied in separate clinical trials.
Carcinosarcomas (the preferred designation by the World Health Organization [WHO]) are also referred to as mixed mesodermal sarcomas or mullerian tumors. Controversy exists about the following issues:
- Whether they are true sarcomas.
- Whether the sarcomatous elements are actually derived from a common epithelial-cell precursor that also gives rise to the usually more abundant adenocarcinomatous elements.
The stromal components of the carcinosarcomas are further characterized by whether they contain homologous elements, such as malignant mesenchymal tissue considered possibly native to the uterus, or heterologous elements, such as striated muscle, cartilage, or bone, which are foreign to the uterus. Carcinosarcomas parallel endometrial cancer in its postmenopausal predominance and in other of its epidemiologic features; increasingly, the treatment of carcinosarcomas is becoming similar to combined modality approaches for endometrial adenocarcinomas.
Other rare forms of uterine sarcomas also fall under the WHO classification of mesenchymal and mixed tumors of the uterus. These include:[2,3]
- Mixed endometrial stromal and smooth muscle tumors.
- Adenosarcomas, in which the epithelial elements appear benign within a malignant mesenchymal background.
- Embryonal botryoides or rhabdomyosarcomas, which are found almost exclusively in infants.
- PEComa—a perivascular epithelial-cell tumor that may behave in a malignant fashion, which is the latest to be added.
(Refer to the PDQ summary on Childhood Rhabdomyosarcoma for more information.)
Risk Factors
The only documented etiologic factor in 10% to 25% of these malignancies is prior pelvic radiation therapy, which is often administered for benign uterine bleeding that began 5 to 25 years earlier. An increased incidence of uterine sarcoma has been associated with tamoxifen in the treatment of breast cancer. Subsequently, increases have also been noted when tamoxifen was given to prevent breast cancer in women at increased risk—a possible result of the estrogenic effect of tamoxifen on the uterus. Because of this increase, patients on tamoxifen should have follow-up pelvic examinations and should undergo endometrial biopsy if there is any abnormal uterine bleeding.[4-6]
Prognosis
The prognosis for women with uterine sarcoma is primarily dependent on the extent of disease at the time of diagnosis.[7] For women with carcinosarcomas, significant predictors of metastatic disease at initial surgery include:[7]
- Isthmic or cervical location.
- Lymphatic vascular space invasion.
- Serous and clear cell histology.
- Grade 2 or 3 carcinoma.
The above factors in addition to the following ones correlate with a progression-free interval:[7]
- Adnexal spread.
- Lymph node metastases.
- Tumor size.
- Peritoneal cytologic findings.
- Depth of myometrial invasion.
Factors that bear no relationship to the presence or absence of metastases at surgical exploration are:
- The presence or absence of stromal heterologous elements.
- The types of such elements.
- The grade of the stromal components.
- The mitotic activity of the stromal components.
In one study, women with a well-differentiated sarcomatous component or carcinosarcomas had significantly longer progression-free intervals than those with moderately to poorly differentiated sarcomas for the homologous and heterologous types. The recurrence rate was 44% for homologous tumors and 63% for heterologous tumors. The type of heterologous sarcoma had no effect on the progression-free interval.
For women with leiomyosarcomas, some investigators consider tumor size to be the most important prognostic factor; women with tumors greater than 5.0 cm in maximum diameter have a poor prognosis.[8] However, in a Gynecologic Oncology Group study, the mitotic index was the only factor significantly related to progression-free interval.[7] Leiomyosarcomas matched for other known prognostic factors may be more aggressive than their carcinosarcoma counterparts.[9] The 5-year survival rate for women with stage I disease, which is confined to the corpus, is approximately 50% versus 0% to 20% for the remaining stages.
Surgery alone can be curative if the malignancy is contained within the uterus. The value of pelvic radiation therapy is not established. Current studies consist primarily of phase II chemotherapy trials for patients with advanced disease. Adjuvant chemotherapy following complete resection for patients with stage I or II disease was not established to be effective in a randomized trial.[10] Yet, other nonrandomized trials have reported improved survival following adjuvant chemotherapy with or without radiation therapy.[11-13]
Related Summaries
Other PDQ summaries containing information related to uterine sarcoma include the following:
References
- Forney JP, Buschbaum HJ: Classifying, staging, and treating uterine sarcomas. Contemp Ob Gyn 18(3):47, 50, 55-56, 61-62, 64, 69, 1981.
- Gershenson D, McGuire W, Gore Martin, et al.: Gynecologic Cancer: Controversies in Management. 3rd ed. New York, NY: Churchill Livingstone, 2004.
- Tavassoéli F, Devilee P, et al.: Pathology and Genetics of Tumours of the Breast and Female Genital Organs. Lyon, France: International Agency for Research on Cancer, 2004.
- Bergman L, Beelen ML, Gallee MP, et al.: Risk and prognosis of endometrial cancer after tamoxifen for breast cancer. Comprehensive Cancer Centres' ALERT Group. Assessment of Liver and Endometrial cancer Risk following Tamoxifen. Lancet 356 (9233): 881-7, 2000. [PUBMED Abstract]
- Cohen I: Endometrial pathologies associated with postmenopausal tamoxifen treatment. Gynecol Oncol 94 (2): 256-66, 2004. [PUBMED Abstract]
- Wickerham DL, Fisher B, Wolmark N, et al.: Association of tamoxifen and uterine sarcoma. J Clin Oncol 20 (11): 2758-60, 2002. [PUBMED Abstract]
- Major FJ, Blessing JA, Silverberg SG, et al.: Prognostic factors in early-stage uterine sarcoma. A Gynecologic Oncology Group study. Cancer 71 (4 Suppl): 1702-9, 1993. [PUBMED Abstract]
- Evans HL, Chawla SP, Simpson C, et al.: Smooth muscle neoplasms of the uterus other than ordinary leiomyoma. A study of 46 cases, with emphasis on diagnostic criteria and prognostic factors. Cancer 62 (10): 2239-47, 1988. [PUBMED Abstract]
- Oláh KS, Dunn JA, Gee H: Leiomyosarcomas have a poorer prognosis than mixed mesodermal tumours when adjusting for known prognostic factors: the result of a retrospective study of 423 cases of uterine sarcoma. Br J Obstet Gynaecol 99 (7): 590-4, 1992. [PUBMED Abstract]
- Omura GA, Blessing JA, Major F, et al.: A randomized clinical trial of adjuvant adriamycin in uterine sarcomas: a Gynecologic Oncology Group Study. J Clin Oncol 3 (9): 1240-5, 1985. [PUBMED Abstract]
- Piver MS, Lele SB, Marchetti DL, et al.: Effect of adjuvant chemotherapy on time to recurrence and survival of stage I uterine sarcomas. J Surg Oncol 38 (4): 233-9, 1988. [PUBMED Abstract]
- van Nagell JR Jr, Hanson MB, Donaldson ES, et al.: Adjuvant vincristine, dactinomycin, and cyclophosphamide therapy in stage I uterine sarcomas. A pilot study. Cancer 57 (8): 1451-4, 1986. [PUBMED Abstract]
- Peters WA 3rd, Rivkin SE, Smith MR, et al.: Cisplatin and adriamycin combination chemotherapy for uterine stromal sarcomas and mixed mesodermal tumors. Gynecol Oncol 34 (3): 323-7, 1989. [PUBMED Abstract]
Cellular Classification of Uterine Sarcoma
The most common histologic types of uterine sarcomas include:
- Carcinosarcomas (mixed mesodermal sarcomas [40%–50%]).
- Leiomyosarcomas (30%).
- Endometrial stromal sarcomas (15%).
The uterine neoplasm classification of the International Society of Gynecologic Pathologists and the World Health Organization uses the term carcinosarcomas for all primary uterine neoplasms containing malignant elements of both epithelial and stromal light microscopic appearances, regardless of whether malignant heterologous elements are present.[1]
References
- Silverberg SG, Major FJ, Blessing JA, et al.: Carcinosarcoma (malignant mixed mesodermal tumor) of the uterus. A Gynecologic Oncology Group pathologic study of 203 cases. Int J Gynecol Pathol 9 (1): 1-19, 1990. [PUBMED Abstract]
Stage Information for Uterine Sarcoma
Definitions: FIGO
The Féderation Internationale de Gynécologie et d’Obstétrique (FIGO) and the American Joint Committee on Cancer (AJCC) have designated staging to define carcinoma of the corpus uteri, which applies to uterine sarcoma; the FIGO system is most commonly used.[1,2]
Uterine sarcomas include leiomyosarcomas, endometrial stromal sarcomas, and adenosarcomas.
| Stage | Description |
|---|---|
| aAdapted from FIGO Committee on Gynecologic Oncology.[1] | |
| bEither G1, G2, or G3 (G = grade). | |
| cEndocervical glandular involvement only should be considered as stage I and no longer as stage II. | |
| dPositive cytology has to be reported separately without changing the stage. | |
| Ib | Tumor confined to the corpus uteri. |
| IAb | No or less than half myometrial invasion. |
| IBb | Invasion equal to or more than half of the myometrium. |
| IIb | Tumor invades cervical stroma but does not extend beyond the uterus.c |
| IIIb | Local and/or regional spread of the tumor. |
| IIIAb | Tumor invades the serosa of the corpus uteri and/or adnexae.d |
| IIIBb | Vaginal and/or parametrial involvement.d |
| IIICb | Metastases to pelvic and/or para-aortic lymph nodes.d |
| IIIC1b | Positive pelvic nodes. |
| IIIC2b | Positive para-aortic lymph nodes with or without positive pelvic lymph nodes. |
| IVb | Tumor invades bladder and/or bowel mucosa, and/or distant metastases. |
| IVAb | Tumor invasion of bladder and/or bowel mucosa. |
| IVBb | Distant metastases, including intra-abdominal metastases and/or inguinal lymph nodes. |
References
- Pecorelli S: Revised FIGO staging for carcinoma of the vulva, cervix, and endometrium. Int J Gynaecol Obstet 105 (2): 103-4, 2009. [PUBMED Abstract]
- Corpus uteri. In: Edge SB, Byrd DR, Compton CC, et al., eds.: AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer, 2010, pp 403-18.
Treatment Option Overview
Surgery is often the principal means of diagnosis and is the primary treatment for all patients with uterine sarcoma. If the diagnosis is known, the extent of surgery is planned according to the stage of the tumor. Hysterectomy is usually performed when a uterine malignancy is suspected, except for rare instances when preservation of the uterus in a young patient is deemed safe for the type of cancer (e.g., a totally confined low-grade leiomyosarcoma in a woman who desires to retain childbearing potential). Medically suitable patients with the preoperative diagnosis of uterine sarcoma are considered candidates for abdominal hysterectomy, bilateral salpingo-oophorectomy, and pelvic and periaortic selective lymphadenectomy. Cytologic washings are obtained from the pelvis and abdomen. Thorough examination of the diaphragm, omentum, and upper abdomen is performed.
There is no firm evidence from a prospective study that adjuvant chemotherapy or radiation therapy is of benefit for patients with uterine sarcoma.[1] In one Gynecologic Oncology Group (GOG) study, the use of adjuvant doxorubicin did not alter the survival rate of patients with resected stage I or stage II uterine sarcomas; however, interpretation of these results is difficult because this study included some patients who received radiation and three types of uterine sarcomas that have variable responses to doxorubicin.[1][Level of evidence: 1iiA] However, because the risk of disease recurrence is high even with localized presentations, many physicians have considered the use of adjuvant chemotherapy or radiation therapy.[2] A report of a study (GOG-0150 [NCT00002546]) that addressed radiation therapy versus adjuvant chemotherapy is awaited.[3]
References
- Omura GA, Blessing JA, Major F, et al.: A randomized clinical trial of adjuvant adriamycin in uterine sarcomas: a Gynecologic Oncology Group Study. J Clin Oncol 3 (9): 1240-5, 1985. [PUBMED Abstract]
- Kohorn EI, Schwartz PE, Chambers JT, et al.: Adjuvant therapy in mixed mullerian tumors of the uterus. Gynecol Oncol 23 (2): 212-21, 1986. [PUBMED Abstract]
- Wolfson AH, Brady MF, Mannel RS, et al.: A Gynecologic Oncology Group randomized trial of whole abdominal irradiation (WAI) vs cisplatin-ifosfamide+mesna (CIM) in optimally debulked stage I-IV carcinosarcoma (CS) of the uterus. [Abstract] J Clin Oncol 24 (Suppl 18): A-5001, 256s, 2006.
Stage I Uterine Sarcoma
Standard treatment options:
- Surgery (total abdominal hysterectomy, bilateral salpingo-oophorectomy, and pelvic and periaortic selective lymphadenectomy).
- Surgery plus pelvic radiation therapy.
- Surgery plus adjuvant chemotherapy.
- Surgery plus adjuvant radiation therapy as seen in the EORTC-55874 trial, for example.
In a nonrandomized, Gynecologic Oncology Group study in patients with stage I and II carcinosarcomas, those who had pelvic radiation therapy had a significant reduction of recurrences within the radiation treatment field but no alteration in survival.[1] A large nonrandomized study demonstrated improved survival and a lower local failure rate in patients with mixed mullerian tumors following postoperative external and intracavitary radiation therapy.[2] One nonrandomized study that predominantly included patients with carcinosarcomas appeared to show benefit for adjuvant therapy with cisplatin and doxorubicin.[3]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Hornback NB, Omura G, Major FJ: Observations on the use of adjuvant radiation therapy in patients with stage I and II uterine sarcoma. Int J Radiat Oncol Biol Phys 12 (12): 2127-30, 1986. [PUBMED Abstract]
- Larson B, Silfverswärd C, Nilsson B, et al.: Mixed müllerian tumours of the uterus--prognostic factors: a clinical and histopathologic study of 147 cases. Radiother Oncol 17 (2): 123-32, 1990. [PUBMED Abstract]
- Peters WA 3rd, Rivkin SE, Smith MR, et al.: Cisplatin and adriamycin combination chemotherapy for uterine stromal sarcomas and mixed mesodermal tumors. Gynecol Oncol 34 (3): 323-7, 1989. [PUBMED Abstract]
Stage II Uterine Sarcoma
Standard treatment options:
- Surgery (total abdominal hysterectomy, bilateral salpingo-oophorectomy, and pelvic and periaortic selective lymphadenectomy).
- Surgery plus pelvic radiation therapy.
- Surgery plus adjuvant chemotherapy.
- Surgery plus adjuvant radiation therapy (EORTC-55874).
In a nonrandomized, Gynecologic Oncology Group study in patients with stage I and II carcinosarcomas, those who had pelvic radiation therapy had a significant reduction of recurrences within the radiation treatment field but no alteration in survival.[1] One nonrandomized study that predominantly included patients with carcinosarcomas appeared to show benefit for adjuvant therapy with cisplatin and doxorubicin.[2]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Hornback NB, Omura G, Major FJ: Observations on the use of adjuvant radiation therapy in patients with stage I and II uterine sarcoma. Int J Radiat Oncol Biol Phys 12 (12): 2127-30, 1986. [PUBMED Abstract]
- Peters WA 3rd, Rivkin SE, Smith MR, et al.: Cisplatin and adriamycin combination chemotherapy for uterine stromal sarcomas and mixed mesodermal tumors. Gynecol Oncol 34 (3): 323-7, 1989. [PUBMED Abstract]
Stage III Uterine Sarcoma
Standard treatment options:
- Surgery (total abdominal hysterectomy, bilateral salpingo-oophorectomy, pelvic and periaortic selective lymphadenectomy, and resection of all gross tumor).
Treatment options under clinical evaluation:
- Surgery plus pelvic radiation therapy.
- Surgery plus adjuvant chemotherapy.
Carcinosarcomas (the preferred designation by the World Health Organization) are also referred to as mixed mesodermal or mullerian tumors. Controversy exists about the following issues:
- Whether they are true sarcomas.
- Whether the sarcomatous elements are actually derived from a common epithelial cell precursor that also gives rise to the usually more abundant adenocarcinomatous elements.
The stromal components of the carcinosarcomas are further characterized by whether they contain homologous elements (such as malignant mesenchymal tissue considered possibly native to the uterus) or heterologous elements (such as striated muscle, cartilage, or bone, which are foreign to the uterus). Carcinosarcomas parallel endometrial cancer in its postmenopausal predominance and in other of its epidemiologic features; increasingly, the treatment of carcinosarcomas is becoming similar to combined modality approaches for endometrial adenocarcinomas.
Patients who present with uterine sarcoma have been treated on a series of phase II studies by the Gynecologic Oncology Group, including the GOG-87B trial, for example.[1,2] These chemotherapy studies have documented some antitumor activity for cisplatin, doxorubicin, and ifosfamide. These studies have also documented differences in response leading to separate trials for patients with carcinosarcomas and leiomyosarcomas. As an example, in patients previously untreated with chemotherapy, ifosfamide had a 32.2% response rate in patients with carcinosarcomas [3] and a 17.2% partial response rate in patients with leiomyosarcomas.[2]
A randomized comparison that was seen in the GOG-108 trial, for example, of ifosfamide with or without cisplatin for first-line therapy for patients with measurable advanced or recurrent carcinosarcomas demonstrated a higher response rate (54% vs. 34%) and longer progression-free survival (PFS) on the combination arm (6 months vs. 4 months), but there was no significant improvement in survival (9 months vs. 8 months).[4][Level of evidence: 1iiA] The follow-up GOG-0161 [NCT00003128] study utilized 3-day ifosfamide regimens (instead of the more toxic 5-day regimen in the preceding study) for the control and for a combination with paclitaxel (with filgrastim starting on day 4).[5] The combination was superior in response rates (45% vs. 29%), PFS (8.4 months vs. 5.8 months), and overall survival (13.5 months and 8.4 months). The hazard ratio for death favored the combination 0.69 (95% confidence interval, 0.49–0.97).[5][Level of evidence: 1iiA] In this study, 52% of 179 evaluable patients had recurrent disease, 18% had stage III disease, and 30% had stage IV disease. In addition, imbalances were present in the sites of disease and in the use of prior radiation therapy, and 30 patients were excluded for wrong pathology.
A role for chemotherapy as adjuvant to surgery has not yet been established.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Thigpen JT, Blessing JA, Beecham J, et al.: Phase II trial of cisplatin as first-line chemotherapy in patients with advanced or recurrent uterine sarcomas: a Gynecologic Oncology Group study. J Clin Oncol 9 (11): 1962-6, 1991. [PUBMED Abstract]
- Sutton GP, Blessing JA, Barrett RJ, et al.: Phase II trial of ifosfamide and mesna in leiomyosarcoma of the uterus: a Gynecologic Oncology Group study. Am J Obstet Gynecol 166 (2): 556-9, 1992. [PUBMED Abstract]
- Sutton GP, Blessing JA, Rosenshein N, et al.: Phase II trial of ifosfamide and mesna in mixed mesodermal tumors of the uterus (a Gynecologic Oncology Group study). Am J Obstet Gynecol 161 (2): 309-12, 1989. [PUBMED Abstract]
- Sutton G, Brunetto VL, Kilgore L, et al.: A phase III trial of ifosfamide with or without cisplatin in carcinosarcoma of the uterus: A Gynecologic Oncology Group Study. Gynecol Oncol 79 (2): 147-53, 2000. [PUBMED Abstract]
- Homesley HD, Filiaci V, Markman M, et al.: Phase III trial of ifosfamide with or without paclitaxel in advanced uterine carcinosarcoma: a Gynecologic Oncology Group Study. J Clin Oncol 25 (5): 526-31, 2007. [PUBMED Abstract]
Stage IV Uterine Sarcoma
There is currently no standard therapy for patients with stage IV disease. These patients should be entered into an ongoing clinical trial.
Carcinosarcomas (the preferred designation by the World Health Organization) are also referred to as mixed mesodermal or mullerian tumors. Controversy exists about the following issues:
- Whether they are true sarcomas.
- Whether the sarcomatous elements are actually derived from a common epithelial cell precursor that also gives rise to the usually more abundant adenocarcinomatous elements.
The stromal components of the carcinosarcomas are further characterized by whether they contain homologous elements, such as malignant mesenchymal tissue considered possibly native to the uterus, or heterologous elements, such as striated muscle, cartilage, or bone, which is foreign to the uterus. Carcinosarcomas parallel endometrial cancer in its postmenopausal predominance and in other of its epidemiologic features; increasingly, the treatment of carcinosarcomas is becoming similar to combined modality approaches for endometrial adenocarcinomas.
Patients who present with uterine sarcoma have been treated on a series of phase II studies by the Gynecologic Oncology Group, including the GOG-87B trial, for example.[1] These chemotherapy studies have documented some antitumor activity for cisplatin, doxorubicin, and ifosfamide. These studies have also documented differences in response leading to separate trials for patients with carcinosarcomas and leiomyosarcomas. As an example, in patients previously untreated with chemotherapy, ifosfamide had a 32.2% response rate in patients with carcinosarcomas,[2] a 33% response rate in patients with endometrial stromal cell sarcomas,[3], and a 17.2% partial response rate in patients with leiomyosarcomas.[4] Doxorubicin in combination with dacarbazine or cyclophosphamide is no more active than doxorubicin alone for advanced disease.[5,6] Cisplatin has activity as first-line therapy and minimal activity as second-line therapy for patients with carcinosarcomas, but cisplatin is inactive as first- or second-line therapy for patients with leiomyosarcomas.[1,7]
A randomized comparison that was seen in the GOG-108 trial, for example, of ifosfamide with or without cisplatin for first-line therapy for patients with measurable advanced or recurrent carcinosarcomas demonstrated a higher response rate (54% vs. 34%) and longer progression-free survival (PFS) on the combination arm (6 months vs. 4 months), but there was no significant improvement in survival (9 months vs. 8 months).[8][Level of evidence: 1iiA] The follow-up GOG-0161 [NCT00003128] study utilized 3-day ifosfamide regimens (instead of the more toxic 5-day regimen in the preceding study) for the control and for a combination with paclitaxel (with filgrastim starting on day 4).[9] The combination was superior in response rates (45% vs. 29%), PFS (8.4 months vs. 5.8 months), and overall survival (13.5 months and 8.4 months). The hazard ratio for death favored the combination 0.69 (95% confidence interval, 0.49–0.97).[9][Level of evidence: 1iiA] In this study, 52% of 179 evaluable patients had recurrent disease, 18% had stage III disease, and 30% had stage IV disease. In addition, imbalances were present in the sites of disease and in the use of prior radiation therapy, and 30 patients were excluded for wrong pathology.
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Thigpen JT, Blessing JA, Beecham J, et al.: Phase II trial of cisplatin as first-line chemotherapy in patients with advanced or recurrent uterine sarcomas: a Gynecologic Oncology Group study. J Clin Oncol 9 (11): 1962-6, 1991. [PUBMED Abstract]
- Sutton GP, Blessing JA, Rosenshein N, et al.: Phase II trial of ifosfamide and mesna in mixed mesodermal tumors of the uterus (a Gynecologic Oncology Group study). Am J Obstet Gynecol 161 (2): 309-12, 1989. [PUBMED Abstract]
- Sutton G, Blessing JA, Park R, et al.: Ifosfamide treatment of recurrent or metastatic endometrial stromal sarcomas previously unexposed to chemotherapy: a study of the Gynecologic Oncology Group. Obstet Gynecol 87 (5 Pt 1): 747-50, 1996. [PUBMED Abstract]
- Sutton GP, Blessing JA, Barrett RJ, et al.: Phase II trial of ifosfamide and mesna in leiomyosarcoma of the uterus: a Gynecologic Oncology Group study. Am J Obstet Gynecol 166 (2): 556-9, 1992. [PUBMED Abstract]
- Omura GA, Major FJ, Blessing JA, et al.: A randomized study of adriamycin with and without dimethyl triazenoimidazole carboxamide in advanced uterine sarcomas. Cancer 52 (4): 626-32, 1983. [PUBMED Abstract]
- Muss HB, Bundy B, DiSaia PJ, et al.: Treatment of recurrent or advanced uterine sarcoma. A randomized trial of doxorubicin versus doxorubicin and cyclophosphamide (a phase III trial of the Gynecologic Oncology Group). Cancer 55 (8): 1648-53, 1985. [PUBMED Abstract]
- Thigpen JT, Blessing JA, Wilbanks GD: Cisplatin as second-line chemotherapy in the treatment of advanced or recurrent leiomyosarcoma of the uterus. A phase II trial of the Gynecologic Oncology Group. Am J Clin Oncol 9 (1): 18-20, 1986. [PUBMED Abstract]
- Sutton G, Brunetto VL, Kilgore L, et al.: A phase III trial of ifosfamide with or without cisplatin in carcinosarcoma of the uterus: A Gynecologic Oncology Group Study. Gynecol Oncol 79 (2): 147-53, 2000. [PUBMED Abstract]
- Homesley HD, Filiaci V, Markman M, et al.: Phase III trial of ifosfamide with or without paclitaxel in advanced uterine carcinosarcoma: a Gynecologic Oncology Group Study. J Clin Oncol 25 (5): 526-31, 2007. [PUBMED Abstract]
Recurrent Uterine Sarcoma
There is currently no standard therapy for patients with recurrent disease. These patients should be entered into an ongoing clinical trial.
Patients who present with uterine sarcoma have been treated on a series of phase II studies by the Gynecologic Oncology Group, including the GOG-87B trial, for example. These chemotherapy studies have documented some antitumor activity for cisplatin, doxorubicin, and ifosfamide. These studies have also documented differences in response leading to separate trials for patients with carcinosarcomas and leiomyosarcomas. As an example, in patients previously untreated with chemotherapy, ifosfamide had a 32.2% response rate in patients with carcinosarcomas,[1] a 33% response rate in patients with endometrial stromal cell sarcomas,[2] and a 17.2% partial response rate in patients with leiomyosarcomas.[3] Doxorubicin in combination with dacarbazine or cyclophosphamide is no more active than doxorubicin alone for recurrent disease.[4,5] Cisplatin has activity as first-line therapy and minimal activity as second-line therapy for patients with carcinosarcomas, but cisplatin is inactive as first- or second-line therapy for patients with leiomyosarcomas.[6,7] A regimen of gemcitabine plus docetaxel had a 53% response rate in patients with unresectable leiomyosarcomas and is undergoing further study.[8]
A randomized comparison that was seen in the GOG-108 trial, for example, of ifosfamide with or without cisplatin for first-line therapy for patients with measurable advanced or recurrent carcinosarcomas demonstrated a higher response rate (54% vs. 34%) and longer progression-free survival (PFS) on the combination arm (6 months vs. 4 months), but there was no significant improvement in survival (9 months vs. 8 months).[9][Level of evidence: 1iiA] The follow-up GOG-0161 [NCT00003128] study utilized 3-day ifosfamide regimens (instead of the more toxic 5-day regimen in the preceding study) for the control and for a combination with paclitaxel (with filgrastim starting on day 4).[10] The combination was superior in response rates (45% vs. 29%), PFS (8.4 months vs. 5.8 months), and overall survival (13.5 months and 8.4 months). The hazard ratio for death favored the combination 0.69 (95% confidence interval, 0.49–0.97).[10][Level of evidence: 1iiA] In this study, 52% of 179 evaluable patients had recurrent disease, 18% had stage III disease, and 30% had stage IV disease. In addition, imbalances were present in the sites of disease and in the use of prior radiation therapy, and 30 patients were excluded for wrong pathology.
For patients with carcinosarcomas who have localized recurrence to the pelvis confirmed by computed tomographic scanning, radiation therapy may be effective palliation. Phase I and II clinical trials are appropriate for patients who recur with distant metastasis and are unresponsive to first-line phase II trials. High-dose progesterone hormone therapy may be of some benefit to patients with low-grade stromal sarcoma.[11]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
References
- Sutton GP, Blessing JA, Rosenshein N, et al.: Phase II trial of ifosfamide and mesna in mixed mesodermal tumors of the uterus (a Gynecologic Oncology Group study). Am J Obstet Gynecol 161 (2): 309-12, 1989. [PUBMED Abstract]
- Sutton G, Blessing JA, Park R, et al.: Ifosfamide treatment of recurrent or metastatic endometrial stromal sarcomas previously unexposed to chemotherapy: a study of the Gynecologic Oncology Group. Obstet Gynecol 87 (5 Pt 1): 747-50, 1996. [PUBMED Abstract]
- Sutton GP, Blessing JA, Barrett RJ, et al.: Phase II trial of ifosfamide and mesna in leiomyosarcoma of the uterus: a Gynecologic Oncology Group study. Am J Obstet Gynecol 166 (2): 556-9, 1992. [PUBMED Abstract]
- Omura GA, Major FJ, Blessing JA, et al.: A randomized study of adriamycin with and without dimethyl triazenoimidazole carboxamide in advanced uterine sarcomas. Cancer 52 (4): 626-32, 1983. [PUBMED Abstract]
- Muss HB, Bundy B, DiSaia PJ, et al.: Treatment of recurrent or advanced uterine sarcoma. A randomized trial of doxorubicin versus doxorubicin and cyclophosphamide (a phase III trial of the Gynecologic Oncology Group). Cancer 55 (8): 1648-53, 1985. [PUBMED Abstract]
- Thigpen JT, Blessing JA, Beecham J, et al.: Phase II trial of cisplatin as first-line chemotherapy in patients with advanced or recurrent uterine sarcomas: a Gynecologic Oncology Group study. J Clin Oncol 9 (11): 1962-6, 1991. [PUBMED Abstract]
- Thigpen JT, Blessing JA, Wilbanks GD: Cisplatin as second-line chemotherapy in the treatment of advanced or recurrent leiomyosarcoma of the uterus. A phase II trial of the Gynecologic Oncology Group. Am J Clin Oncol 9 (1): 18-20, 1986. [PUBMED Abstract]
- Hensley ML, Maki R, Venkatraman E, et al.: Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol 20 (12): 2824-31, 2002. [PUBMED Abstract]
- Sutton G, Brunetto VL, Kilgore L, et al.: A phase III trial of ifosfamide with or without cisplatin in carcinosarcoma of the uterus: A Gynecologic Oncology Group Study. Gynecol Oncol 79 (2): 147-53, 2000. [PUBMED Abstract]
- Homesley HD, Filiaci V, Markman M, et al.: Phase III trial of ifosfamide with or without paclitaxel in advanced uterine carcinosarcoma: a Gynecologic Oncology Group Study. J Clin Oncol 25 (5): 526-31, 2007. [PUBMED Abstract]
- Katz L, Merino MJ, Sakamoto H, et al.: Endometrial stromal sarcoma: a clinicopathologic study of 11 cases with determination of estrogen and progestin receptor levels in three tumors. Gynecol Oncol 26 (1): 87-97, 1987. [PUBMED Abstract]
Changes to This Summary (07/15/2015)
The PDQ cancer information summaries are reviewed regularly and updated as new information becomes available. This section describes the latest changes made to this summary as of the date above.
Editorial changes were made to this summary.
This summary is written and maintained by the PDQ Adult Treatment Editorial Board, which is editorially independent of NCI. The summary reflects an independent review of the literature and does not represent a policy statement of NCI or NIH. More information about summary policies and the role of the PDQ Editorial Boards in maintaining the PDQ summaries can be found on the About This PDQ Summary and PDQ® - NCI's Comprehensive Cancer Database pages.
About This PDQ Summary
Purpose of This Summary
This PDQ cancer information summary for health professionals provides comprehensive, peer-reviewed, evidence-based information about the treatment of uterine sarcoma. It is intended as a resource to inform and assist clinicians who care for cancer patients. It does not provide formal guidelines or recommendations for making health care decisions.
Reviewers and Updates
This summary is reviewed regularly and updated as necessary by the PDQ Adult Treatment Editorial Board, which is editorially independent of the National Cancer Institute (NCI). The summary reflects an independent review of the literature and does not represent a policy statement of NCI or the National Institutes of Health (NIH).
Board members review recently published articles each month to determine whether an article should:
- be discussed at a meeting,
- be cited with text, or
- replace or update an existing article that is already cited.
Changes to the summaries are made through a consensus process in which Board members evaluate the strength of the evidence in the published articles and determine how the article should be included in the summary.
The lead reviewers for Uterine Sarcoma Treatment are:
- Leslie R. Boyd, MD (New York University Medical Center)
- Franco M. Muggia, MD (New York University Medical Center)
Any comments or questions about the summary content should be submitted to Cancer.gov through the NCI website's Email Us. Do not contact the individual Board Members with questions or comments about the summaries. Board members will not respond to individual inquiries.
Levels of Evidence
Some of the reference citations in this summary are accompanied by a level-of-evidence designation. These designations are intended to help readers assess the strength of the evidence supporting the use of specific interventions or approaches. The PDQ Adult Treatment Editorial Board uses a formal evidence ranking system in developing its level-of-evidence designations.
Permission to Use This Summary
PDQ is a registered trademark. Although the content of PDQ documents can be used freely as text, it cannot be identified as an NCI PDQ cancer information summary unless it is presented in its entirety and is regularly updated. However, an author would be permitted to write a sentence such as “NCI’s PDQ cancer information summary about breast cancer prevention states the risks succinctly: [include excerpt from the summary].”
The preferred citation for this PDQ summary is:
PDQ® Adult Treatment Editorial Board. PDQ Uterine Sarcoma Treatment. Bethesda, MD: National Cancer Institute. Updated <MM/DD/YYYY>. Available at: https://www.cancer.gov/types/uterine/hp/uterine-sarcoma-treatment-pdq. Accessed <MM/DD/YYYY>. [PMID: 26389327]
Images in this summary are used with permission of the author(s), artist, and/or publisher for use within the PDQ summaries only. Permission to use images outside the context of PDQ information must be obtained from the owner(s) and cannot be granted by the National Cancer Institute. Information about using the illustrations in this summary, along with many other cancer-related images, is available in Visuals Online, a collection of over 2,000 scientific images.
Disclaimer
Based on the strength of the available evidence, treatment options may be described as either “standard” or “under clinical evaluation.” These classifications should not be used as a basis for insurance reimbursement determinations. More information on insurance coverage is available on Cancer.gov on the Managing Cancer Care page.
Contact Us
More information about contacting us or receiving help with the Cancer.gov website can be found on our Contact Us for Help page. Questions can also be submitted to Cancer.gov through the website’s Email Us.