User login
Face masks can aggravate rosacea
The “maskne” phenomenon – that is, new onset or exacerbation of preexisting acne due to prolonged wearing of protective face masks – has become commonplace during the COVID-19 pandemic. Less well appreciated is that rosacea often markedly worsens, too, Giovanni Damiani, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.

“This is particularly interesting because two inflammatory dermatoses with different pathogenesis are both mechanically and microbiologically triggered by mask use,” observed Dr. Damiani, a dermatologist at the University of Milan.
He presented . These patients – 23 with papulopustular and 13 with erythematotelangiectatic rosacea – were wearing face masks for at least 6 hours per day during quarantine. Most were using what Dr. Damiani termed “community masks,” meaning they weren’t approved by the European regulatory agency as personal protective equipment.
Every yardstick Dr. Damiani and coinvestigators employed to characterize the patients’ rosacea demonstrated that the dermatosis was significantly worse during the prolonged mask-wearing period. For example, the average prequarantine score on the Global Flushing Severity Scale was 2.56, jumping to 3.97 after a month of masked quarantine. The flushing score climbed from 1.83 to 2.78 in the subgroup with papulopustular rosacea, and from 3.85 to 6.08 in patients with erythematotelangiectatic rosacea. Scores on the Clinician’s Erythema Assessment rose from 1.09 to 1.7 in the papulopustular rosacea patients, and from 2.46 to 3.54 in those with erythematotelangiectatic rosacea.
Scores on the Dermatology Life Quality Index climbed from 7.35 prequarantine to 10.65 in the subgroup with papulopustular rosacea and from 5.15 to 8.69 in patients with erythematotelangiectatic rosacea. Investigator Global Assessment and Patient’s Self-Assessment scores also deteriorated significantly after a month in masked quarantine.
Clinically, the mask-aggravated rosacea, or “maskacea,” was mainly localized to the dorsal lower third of the nose as well as the cheeks. The ocular and perioral areas and the chin were least affected.
Dr. Damiani advised his colleagues to intensify therapy promptly when patients report any worsening of their preexisting rosacea in connection with use of face masks. He has found this condition is often relatively treatment resistant so long as affected patients continue to wear face masks as an essential tool in preventing transmission of COVID-19.
The dermatologist noted that not all face masks are equal offenders when it comes to aggravating common facial dermatoses. During the spring 2020 pandemic quarantine in Milan, 11.6% of 318 mask wearers, none health care professionals, presented to Dr. Damiani and coinvestigators for treatment of facial dermatoses. The facial dermatosis rate was 5.4% among 168 users of masks bearing the European Union CE mark signifying the devices met relevant safety and performance standards, compared with 18.7% in 150 users of community masks with no CE mark. The rate of irritant contact dermatitis was zero with the CE mark masks and 4.7% with the community masks.
During quarantine, however, these patients wore their protective face masks for only a limited time, since for the most part they were restricted to home. In contrast, during the first week after the quarantine was lifted in early May and the daily hours of mask use increased, facial dermatoses were diagnosed in 8.7% of 23 users of CE-approved masks, compared with 45% of 71 wearers of community masks. Dr. Damiani and colleagues diagnosed irritant contact dermatitis in 16% of the community mask wearers post quarantine, but in not a single user of a mask bearing the CE mark.
The National Rosacea Society has issued patient guidance on avoiding rosacea flare-ups during the Covid-19 pandemic.
Dr. Damiani reported having no financial conflicts regarding his study.
The “maskne” phenomenon – that is, new onset or exacerbation of preexisting acne due to prolonged wearing of protective face masks – has become commonplace during the COVID-19 pandemic. Less well appreciated is that rosacea often markedly worsens, too, Giovanni Damiani, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“This is particularly interesting because two inflammatory dermatoses with different pathogenesis are both mechanically and microbiologically triggered by mask use,” observed Dr. Damiani, a dermatologist at the University of Milan.
He presented . These patients – 23 with papulopustular and 13 with erythematotelangiectatic rosacea – were wearing face masks for at least 6 hours per day during quarantine. Most were using what Dr. Damiani termed “community masks,” meaning they weren’t approved by the European regulatory agency as personal protective equipment.
Every yardstick Dr. Damiani and coinvestigators employed to characterize the patients’ rosacea demonstrated that the dermatosis was significantly worse during the prolonged mask-wearing period. For example, the average prequarantine score on the Global Flushing Severity Scale was 2.56, jumping to 3.97 after a month of masked quarantine. The flushing score climbed from 1.83 to 2.78 in the subgroup with papulopustular rosacea, and from 3.85 to 6.08 in patients with erythematotelangiectatic rosacea. Scores on the Clinician’s Erythema Assessment rose from 1.09 to 1.7 in the papulopustular rosacea patients, and from 2.46 to 3.54 in those with erythematotelangiectatic rosacea.
Scores on the Dermatology Life Quality Index climbed from 7.35 prequarantine to 10.65 in the subgroup with papulopustular rosacea and from 5.15 to 8.69 in patients with erythematotelangiectatic rosacea. Investigator Global Assessment and Patient’s Self-Assessment scores also deteriorated significantly after a month in masked quarantine.
Clinically, the mask-aggravated rosacea, or “maskacea,” was mainly localized to the dorsal lower third of the nose as well as the cheeks. The ocular and perioral areas and the chin were least affected.
Dr. Damiani advised his colleagues to intensify therapy promptly when patients report any worsening of their preexisting rosacea in connection with use of face masks. He has found this condition is often relatively treatment resistant so long as affected patients continue to wear face masks as an essential tool in preventing transmission of COVID-19.
The dermatologist noted that not all face masks are equal offenders when it comes to aggravating common facial dermatoses. During the spring 2020 pandemic quarantine in Milan, 11.6% of 318 mask wearers, none health care professionals, presented to Dr. Damiani and coinvestigators for treatment of facial dermatoses. The facial dermatosis rate was 5.4% among 168 users of masks bearing the European Union CE mark signifying the devices met relevant safety and performance standards, compared with 18.7% in 150 users of community masks with no CE mark. The rate of irritant contact dermatitis was zero with the CE mark masks and 4.7% with the community masks.
During quarantine, however, these patients wore their protective face masks for only a limited time, since for the most part they were restricted to home. In contrast, during the first week after the quarantine was lifted in early May and the daily hours of mask use increased, facial dermatoses were diagnosed in 8.7% of 23 users of CE-approved masks, compared with 45% of 71 wearers of community masks. Dr. Damiani and colleagues diagnosed irritant contact dermatitis in 16% of the community mask wearers post quarantine, but in not a single user of a mask bearing the CE mark.
The National Rosacea Society has issued patient guidance on avoiding rosacea flare-ups during the Covid-19 pandemic.
Dr. Damiani reported having no financial conflicts regarding his study.
The “maskne” phenomenon – that is, new onset or exacerbation of preexisting acne due to prolonged wearing of protective face masks – has become commonplace during the COVID-19 pandemic. Less well appreciated is that rosacea often markedly worsens, too, Giovanni Damiani, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“This is particularly interesting because two inflammatory dermatoses with different pathogenesis are both mechanically and microbiologically triggered by mask use,” observed Dr. Damiani, a dermatologist at the University of Milan.
He presented . These patients – 23 with papulopustular and 13 with erythematotelangiectatic rosacea – were wearing face masks for at least 6 hours per day during quarantine. Most were using what Dr. Damiani termed “community masks,” meaning they weren’t approved by the European regulatory agency as personal protective equipment.
Every yardstick Dr. Damiani and coinvestigators employed to characterize the patients’ rosacea demonstrated that the dermatosis was significantly worse during the prolonged mask-wearing period. For example, the average prequarantine score on the Global Flushing Severity Scale was 2.56, jumping to 3.97 after a month of masked quarantine. The flushing score climbed from 1.83 to 2.78 in the subgroup with papulopustular rosacea, and from 3.85 to 6.08 in patients with erythematotelangiectatic rosacea. Scores on the Clinician’s Erythema Assessment rose from 1.09 to 1.7 in the papulopustular rosacea patients, and from 2.46 to 3.54 in those with erythematotelangiectatic rosacea.
Scores on the Dermatology Life Quality Index climbed from 7.35 prequarantine to 10.65 in the subgroup with papulopustular rosacea and from 5.15 to 8.69 in patients with erythematotelangiectatic rosacea. Investigator Global Assessment and Patient’s Self-Assessment scores also deteriorated significantly after a month in masked quarantine.
Clinically, the mask-aggravated rosacea, or “maskacea,” was mainly localized to the dorsal lower third of the nose as well as the cheeks. The ocular and perioral areas and the chin were least affected.
Dr. Damiani advised his colleagues to intensify therapy promptly when patients report any worsening of their preexisting rosacea in connection with use of face masks. He has found this condition is often relatively treatment resistant so long as affected patients continue to wear face masks as an essential tool in preventing transmission of COVID-19.
The dermatologist noted that not all face masks are equal offenders when it comes to aggravating common facial dermatoses. During the spring 2020 pandemic quarantine in Milan, 11.6% of 318 mask wearers, none health care professionals, presented to Dr. Damiani and coinvestigators for treatment of facial dermatoses. The facial dermatosis rate was 5.4% among 168 users of masks bearing the European Union CE mark signifying the devices met relevant safety and performance standards, compared with 18.7% in 150 users of community masks with no CE mark. The rate of irritant contact dermatitis was zero with the CE mark masks and 4.7% with the community masks.
During quarantine, however, these patients wore their protective face masks for only a limited time, since for the most part they were restricted to home. In contrast, during the first week after the quarantine was lifted in early May and the daily hours of mask use increased, facial dermatoses were diagnosed in 8.7% of 23 users of CE-approved masks, compared with 45% of 71 wearers of community masks. Dr. Damiani and colleagues diagnosed irritant contact dermatitis in 16% of the community mask wearers post quarantine, but in not a single user of a mask bearing the CE mark.
The National Rosacea Society has issued patient guidance on avoiding rosacea flare-ups during the Covid-19 pandemic.
Dr. Damiani reported having no financial conflicts regarding his study.
FROM THE EADV CONGRESS
Expert offers clinical pearls on leg ulcer therapy
Elena Conde Montero, MD, PhD, asserted at the virtual annual congress of the European Academy of Dermatology and Venereology.
In addition to delving into the finer points of compression therapy, she offered other clinical pearls for the treatment of chronic leg ulcers. These included the use of autologous punch grafting to reduce pain as well as promote healing, when to employ adjunctive negative pressure therapy, and the benefits of liquid sevoflurane for highly effective topical analgesia during wound cleansing and debridement.
Compression therapy
“If no contraindications exist, compression therapy is the best antihypertensive and anti-inflammatory treatment for all leg ulcers, not only venous leg ulcers,” according to Dr. Conde, a dermatologist at Infanta Leonor University Hospital in Madrid.
The list of absolute contraindications to compression treatment is brief, as highlighted in a recent international consensus statement. The expert writing panel named only four: severe peripheral artery disease, the presence of an epifascial arterial bypass, severe cardiac insufficiency, and true allergy to compression material.
Compression therapy provides multiple salutary effects. These include reduced capillary filtration of fluids to tissue, decreased swelling, enhanced tissue remodeling, better lymphatic drainage, reduced inflammatory cell counts, and increased arterial flow.
“This means that people with mild arterial disease will benefit from active compression because perfusion will improve,” Dr. Conde said.
Similarly, leg ulcers secondary to pyoderma gangrenosum will benefit from the anti-inflammatory effects of compression therapy in conjunction with standard immunotherapy, added the dermatologist, who coauthored a recent publication by the European Wound Management Association entitled “Atypical Wounds: Best Clinical Practices and Challenges.”
Four broad types of compression therapy are available: compression stockings, short-stretch bandages, multicomponent bandage systems, and self-adjusting compression wrap devices. The best clinical outcomes are achieved by individualized selection of a compression method based upon patient characteristics.
Short-stretch, low-elasticity bandages – such as the classic Unna boot loaded with zinc paste and topical corticosteroids – are well suited for patients with large leg ulcers. These bandages feature high working pressures during muscle contraction. They also provide low resting pressures, which is advantageous in patients with peripheral artery disease. The major disadvantage of short-stretch bandages is the need for frequent dressing changes by a nurse or other trained professional, since the compression is quickly lost as an unwanted consequence of the welcome reduction in swelling.
Multicomponent bandage systems feature two to four layers of bandages of differing stiffness, as well as padding material and in many cases pressure indicators. These bandages can often be worn for up to a week without needing to be changed, since they maintain adequate pressure long term. “These are very easy to use by nonexperts,” Dr. Conde noted.
A caveat regarding both short-stretch bandages and the multicomponent bandage systems: before applying them, it’s important to pad at-risk areas against injury caused by high pressures. These high-risk areas include the Achilles tendon, the pretibial region, and the lateral foot.
Self-adjusting compression systems are comprised of strips of short-stretch, low-elasticity fabric, which wrap around the leg and are fixed with Velcro closures. Dr. Conde hailed these devices as “a great innovation in compression therapy, without doubt.” Their major advantage is ease of application and removal by the patient. They are best-suited for treatment of small ulcers in patients who find it difficult to use compression stockings because of obesity or osteoarthritis, in patients who can’t tolerate such stockings because they have peripheral artery disease and the stockings’ high resting pressure is uncomfortable, or in individuals ill-suited for compression bandages because they lack adequate access to nursing care for the required frequent dressing changes.
Compression stockings are a good option for small ulcers. It’s easier for patients to wear shoes with compression stockings and thereby engage in normal everyday activities than with short-stretch bandages. A tip: Many patients find it arduous to don and remove a high-compression stocking that achieves the recommended pressure of 30-40 mm Hg at the point of transition between the Achilles tendon and the calf muscle, but the same effect can be achieved by overlapping two easier-to-use lower-compression stockings.
Punch grafting
This simple, cost-effective outpatient procedure was first described as a means of enhancing wound healing 150 years ago. The method involves utilizing a scalpel, curette, or punch to obtain a series of thin split-thickness skin grafts that contain epidermis and dermis down to the superficial papillary dermis. The grafts, usually harvested from the anterior thigh, are placed on the wound. This is followed by at least 5 days of local pressure and rest to promote graft uptake.
Sequential punch grafting is an excellent option for particularly challenging chronic ulcers, including Martorell hypertensive ischemic leg ulcers and other arteriolopathic ulcers in the elderly.
“Sequential punch grafting of wounds is very common in our clinics, especially for wounds that lack perfect grafting conditions,” Dr. Conde said.
She considers Martorell hypertensive ischemic leg ulcers to be underdiagnosed and undertreated. The Martorell leg ulcer is an exceedingly painful, rapidly progressive ischemic lesion, or bilateral lesions, with inflamed irregular margins. The disorder is caused by obstruction of subcutaneous arterioles in the absence of signs of vasculitis, and generally occurs in older individuals who have had well-controlled hypertension for many years. Diabetes, obesity, dyslipidemia, and peripheral artery disease are common comorbid conditions. The most common form of treatment – bioactive dressings in a moist environment – produces unsatisfactory results because it doesn’t address the inflammatory process.
Dr. Conde and coworkers have published the full details of how they achieved complete healing of Martorell hypertensive ischemic leg ulcers 3-8 weeks after punch grafting in three affected patients, all of whom presented with pain scores of 10/10 refractory even to opioid analgesics. The punch grafting was preceded by 15 days of topical corticosteroids and low-elasticity compression bandages in order to create adequate granulation tissue in the wound bed, which had the added benefit of achieving a 2- to 3-point reduction in pain scores even before the surgical procedure.
The pain-reducing effect of punch grafting isn’t as well appreciated as the wound-healing effect. Dr. Conde was first author of a recent study in which investigators systematically measured pain reduction in 136 patients with hard-to-heal leg ulcers of various etiologies treated with punch grafting. Nearly three-quarters of those who presented with painful ulcers were pain free after punch grafting, and the rest experienced greater than 70% pain reduction.
Pain suppression wasn’t dependent upon the percentage of graft uptake in this study. That’s because, as long as the wound isn’t overcleaned during dressing changes, even grafts that haven’t attached to the wound will release growth factors that promote wound healing, Dr. Conde explained.
Adjunctive negative pressure therapy
Portable vacuum-based negative pressure therapy devices are easy to use as a means to promote punch graft uptake. Negative pressure is best employed as an adjunct to punch grafting in suboptimal wound beds, longstanding ulcers, in patients with previous graft failure, or in challenging anatomic locations, such as the Achilles tendon or ankle. Dr. Conde has found the combination of punch grafting and negative pressure therapy especially helpful in patients with clinically inactive pyoderma gangrenosum.
Topical sevoflurane for analgesia
Most of the literature on topical sevoflurane for ulcer care has been published by Spanish researchers, but this form of analgesia deserves much more widespread use, according to Dr. Conde.
Sevoflurane is most often used as a gas in general anesthesia. In liquid form, however, it not only has a rapid, long-lasting analgesic effect when applied to painful leg ulcers, it also promotes healing because it is both antibacterial and a vasodilator. So before performing a potentially painful ulcer or wound cleaning, Dr. Conde recommended protecting perilesional skin with petroleum jelly, then irrigating the ulcer site with liquid sevoflurane. After that, it’s advisable to wait just 5-10 minutes before proceeding.
“It takes effect in much less time than EMLA cream,” she noted.
In one study of 30 adults aged over age 65 years with painful chronic venous ulcers refractory to conventional analgesics who underwent ulcer cleaning supported by topical sevoflurane at a dose of roughly 1 mL/cm2 of ulcer area every 2 days for a month, Spanish investigators documented onset of analgesic effect in 2-7 minutes, with a duration of 8-18 hours. The researchers found that the use of backup conventional analgesics ranging from acetaminophen to opioids was diminished. Side effects were limited to mild, transient itching and redness.
Dr. Conde reported having no financial conflicts of interest regarding her presentation.
Elena Conde Montero, MD, PhD, asserted at the virtual annual congress of the European Academy of Dermatology and Venereology.
In addition to delving into the finer points of compression therapy, she offered other clinical pearls for the treatment of chronic leg ulcers. These included the use of autologous punch grafting to reduce pain as well as promote healing, when to employ adjunctive negative pressure therapy, and the benefits of liquid sevoflurane for highly effective topical analgesia during wound cleansing and debridement.
Compression therapy
“If no contraindications exist, compression therapy is the best antihypertensive and anti-inflammatory treatment for all leg ulcers, not only venous leg ulcers,” according to Dr. Conde, a dermatologist at Infanta Leonor University Hospital in Madrid.
The list of absolute contraindications to compression treatment is brief, as highlighted in a recent international consensus statement. The expert writing panel named only four: severe peripheral artery disease, the presence of an epifascial arterial bypass, severe cardiac insufficiency, and true allergy to compression material.
Compression therapy provides multiple salutary effects. These include reduced capillary filtration of fluids to tissue, decreased swelling, enhanced tissue remodeling, better lymphatic drainage, reduced inflammatory cell counts, and increased arterial flow.
“This means that people with mild arterial disease will benefit from active compression because perfusion will improve,” Dr. Conde said.
Similarly, leg ulcers secondary to pyoderma gangrenosum will benefit from the anti-inflammatory effects of compression therapy in conjunction with standard immunotherapy, added the dermatologist, who coauthored a recent publication by the European Wound Management Association entitled “Atypical Wounds: Best Clinical Practices and Challenges.”
Four broad types of compression therapy are available: compression stockings, short-stretch bandages, multicomponent bandage systems, and self-adjusting compression wrap devices. The best clinical outcomes are achieved by individualized selection of a compression method based upon patient characteristics.
Short-stretch, low-elasticity bandages – such as the classic Unna boot loaded with zinc paste and topical corticosteroids – are well suited for patients with large leg ulcers. These bandages feature high working pressures during muscle contraction. They also provide low resting pressures, which is advantageous in patients with peripheral artery disease. The major disadvantage of short-stretch bandages is the need for frequent dressing changes by a nurse or other trained professional, since the compression is quickly lost as an unwanted consequence of the welcome reduction in swelling.
Multicomponent bandage systems feature two to four layers of bandages of differing stiffness, as well as padding material and in many cases pressure indicators. These bandages can often be worn for up to a week without needing to be changed, since they maintain adequate pressure long term. “These are very easy to use by nonexperts,” Dr. Conde noted.
A caveat regarding both short-stretch bandages and the multicomponent bandage systems: before applying them, it’s important to pad at-risk areas against injury caused by high pressures. These high-risk areas include the Achilles tendon, the pretibial region, and the lateral foot.
Self-adjusting compression systems are comprised of strips of short-stretch, low-elasticity fabric, which wrap around the leg and are fixed with Velcro closures. Dr. Conde hailed these devices as “a great innovation in compression therapy, without doubt.” Their major advantage is ease of application and removal by the patient. They are best-suited for treatment of small ulcers in patients who find it difficult to use compression stockings because of obesity or osteoarthritis, in patients who can’t tolerate such stockings because they have peripheral artery disease and the stockings’ high resting pressure is uncomfortable, or in individuals ill-suited for compression bandages because they lack adequate access to nursing care for the required frequent dressing changes.
Compression stockings are a good option for small ulcers. It’s easier for patients to wear shoes with compression stockings and thereby engage in normal everyday activities than with short-stretch bandages. A tip: Many patients find it arduous to don and remove a high-compression stocking that achieves the recommended pressure of 30-40 mm Hg at the point of transition between the Achilles tendon and the calf muscle, but the same effect can be achieved by overlapping two easier-to-use lower-compression stockings.
Punch grafting
This simple, cost-effective outpatient procedure was first described as a means of enhancing wound healing 150 years ago. The method involves utilizing a scalpel, curette, or punch to obtain a series of thin split-thickness skin grafts that contain epidermis and dermis down to the superficial papillary dermis. The grafts, usually harvested from the anterior thigh, are placed on the wound. This is followed by at least 5 days of local pressure and rest to promote graft uptake.
Sequential punch grafting is an excellent option for particularly challenging chronic ulcers, including Martorell hypertensive ischemic leg ulcers and other arteriolopathic ulcers in the elderly.
“Sequential punch grafting of wounds is very common in our clinics, especially for wounds that lack perfect grafting conditions,” Dr. Conde said.
She considers Martorell hypertensive ischemic leg ulcers to be underdiagnosed and undertreated. The Martorell leg ulcer is an exceedingly painful, rapidly progressive ischemic lesion, or bilateral lesions, with inflamed irregular margins. The disorder is caused by obstruction of subcutaneous arterioles in the absence of signs of vasculitis, and generally occurs in older individuals who have had well-controlled hypertension for many years. Diabetes, obesity, dyslipidemia, and peripheral artery disease are common comorbid conditions. The most common form of treatment – bioactive dressings in a moist environment – produces unsatisfactory results because it doesn’t address the inflammatory process.
Dr. Conde and coworkers have published the full details of how they achieved complete healing of Martorell hypertensive ischemic leg ulcers 3-8 weeks after punch grafting in three affected patients, all of whom presented with pain scores of 10/10 refractory even to opioid analgesics. The punch grafting was preceded by 15 days of topical corticosteroids and low-elasticity compression bandages in order to create adequate granulation tissue in the wound bed, which had the added benefit of achieving a 2- to 3-point reduction in pain scores even before the surgical procedure.
The pain-reducing effect of punch grafting isn’t as well appreciated as the wound-healing effect. Dr. Conde was first author of a recent study in which investigators systematically measured pain reduction in 136 patients with hard-to-heal leg ulcers of various etiologies treated with punch grafting. Nearly three-quarters of those who presented with painful ulcers were pain free after punch grafting, and the rest experienced greater than 70% pain reduction.
Pain suppression wasn’t dependent upon the percentage of graft uptake in this study. That’s because, as long as the wound isn’t overcleaned during dressing changes, even grafts that haven’t attached to the wound will release growth factors that promote wound healing, Dr. Conde explained.
Adjunctive negative pressure therapy
Portable vacuum-based negative pressure therapy devices are easy to use as a means to promote punch graft uptake. Negative pressure is best employed as an adjunct to punch grafting in suboptimal wound beds, longstanding ulcers, in patients with previous graft failure, or in challenging anatomic locations, such as the Achilles tendon or ankle. Dr. Conde has found the combination of punch grafting and negative pressure therapy especially helpful in patients with clinically inactive pyoderma gangrenosum.
Topical sevoflurane for analgesia
Most of the literature on topical sevoflurane for ulcer care has been published by Spanish researchers, but this form of analgesia deserves much more widespread use, according to Dr. Conde.
Sevoflurane is most often used as a gas in general anesthesia. In liquid form, however, it not only has a rapid, long-lasting analgesic effect when applied to painful leg ulcers, it also promotes healing because it is both antibacterial and a vasodilator. So before performing a potentially painful ulcer or wound cleaning, Dr. Conde recommended protecting perilesional skin with petroleum jelly, then irrigating the ulcer site with liquid sevoflurane. After that, it’s advisable to wait just 5-10 minutes before proceeding.
“It takes effect in much less time than EMLA cream,” she noted.
In one study of 30 adults aged over age 65 years with painful chronic venous ulcers refractory to conventional analgesics who underwent ulcer cleaning supported by topical sevoflurane at a dose of roughly 1 mL/cm2 of ulcer area every 2 days for a month, Spanish investigators documented onset of analgesic effect in 2-7 minutes, with a duration of 8-18 hours. The researchers found that the use of backup conventional analgesics ranging from acetaminophen to opioids was diminished. Side effects were limited to mild, transient itching and redness.
Dr. Conde reported having no financial conflicts of interest regarding her presentation.
Elena Conde Montero, MD, PhD, asserted at the virtual annual congress of the European Academy of Dermatology and Venereology.
In addition to delving into the finer points of compression therapy, she offered other clinical pearls for the treatment of chronic leg ulcers. These included the use of autologous punch grafting to reduce pain as well as promote healing, when to employ adjunctive negative pressure therapy, and the benefits of liquid sevoflurane for highly effective topical analgesia during wound cleansing and debridement.
Compression therapy
“If no contraindications exist, compression therapy is the best antihypertensive and anti-inflammatory treatment for all leg ulcers, not only venous leg ulcers,” according to Dr. Conde, a dermatologist at Infanta Leonor University Hospital in Madrid.
The list of absolute contraindications to compression treatment is brief, as highlighted in a recent international consensus statement. The expert writing panel named only four: severe peripheral artery disease, the presence of an epifascial arterial bypass, severe cardiac insufficiency, and true allergy to compression material.
Compression therapy provides multiple salutary effects. These include reduced capillary filtration of fluids to tissue, decreased swelling, enhanced tissue remodeling, better lymphatic drainage, reduced inflammatory cell counts, and increased arterial flow.
“This means that people with mild arterial disease will benefit from active compression because perfusion will improve,” Dr. Conde said.
Similarly, leg ulcers secondary to pyoderma gangrenosum will benefit from the anti-inflammatory effects of compression therapy in conjunction with standard immunotherapy, added the dermatologist, who coauthored a recent publication by the European Wound Management Association entitled “Atypical Wounds: Best Clinical Practices and Challenges.”
Four broad types of compression therapy are available: compression stockings, short-stretch bandages, multicomponent bandage systems, and self-adjusting compression wrap devices. The best clinical outcomes are achieved by individualized selection of a compression method based upon patient characteristics.
Short-stretch, low-elasticity bandages – such as the classic Unna boot loaded with zinc paste and topical corticosteroids – are well suited for patients with large leg ulcers. These bandages feature high working pressures during muscle contraction. They also provide low resting pressures, which is advantageous in patients with peripheral artery disease. The major disadvantage of short-stretch bandages is the need for frequent dressing changes by a nurse or other trained professional, since the compression is quickly lost as an unwanted consequence of the welcome reduction in swelling.
Multicomponent bandage systems feature two to four layers of bandages of differing stiffness, as well as padding material and in many cases pressure indicators. These bandages can often be worn for up to a week without needing to be changed, since they maintain adequate pressure long term. “These are very easy to use by nonexperts,” Dr. Conde noted.
A caveat regarding both short-stretch bandages and the multicomponent bandage systems: before applying them, it’s important to pad at-risk areas against injury caused by high pressures. These high-risk areas include the Achilles tendon, the pretibial region, and the lateral foot.
Self-adjusting compression systems are comprised of strips of short-stretch, low-elasticity fabric, which wrap around the leg and are fixed with Velcro closures. Dr. Conde hailed these devices as “a great innovation in compression therapy, without doubt.” Their major advantage is ease of application and removal by the patient. They are best-suited for treatment of small ulcers in patients who find it difficult to use compression stockings because of obesity or osteoarthritis, in patients who can’t tolerate such stockings because they have peripheral artery disease and the stockings’ high resting pressure is uncomfortable, or in individuals ill-suited for compression bandages because they lack adequate access to nursing care for the required frequent dressing changes.
Compression stockings are a good option for small ulcers. It’s easier for patients to wear shoes with compression stockings and thereby engage in normal everyday activities than with short-stretch bandages. A tip: Many patients find it arduous to don and remove a high-compression stocking that achieves the recommended pressure of 30-40 mm Hg at the point of transition between the Achilles tendon and the calf muscle, but the same effect can be achieved by overlapping two easier-to-use lower-compression stockings.
Punch grafting
This simple, cost-effective outpatient procedure was first described as a means of enhancing wound healing 150 years ago. The method involves utilizing a scalpel, curette, or punch to obtain a series of thin split-thickness skin grafts that contain epidermis and dermis down to the superficial papillary dermis. The grafts, usually harvested from the anterior thigh, are placed on the wound. This is followed by at least 5 days of local pressure and rest to promote graft uptake.
Sequential punch grafting is an excellent option for particularly challenging chronic ulcers, including Martorell hypertensive ischemic leg ulcers and other arteriolopathic ulcers in the elderly.
“Sequential punch grafting of wounds is very common in our clinics, especially for wounds that lack perfect grafting conditions,” Dr. Conde said.
She considers Martorell hypertensive ischemic leg ulcers to be underdiagnosed and undertreated. The Martorell leg ulcer is an exceedingly painful, rapidly progressive ischemic lesion, or bilateral lesions, with inflamed irregular margins. The disorder is caused by obstruction of subcutaneous arterioles in the absence of signs of vasculitis, and generally occurs in older individuals who have had well-controlled hypertension for many years. Diabetes, obesity, dyslipidemia, and peripheral artery disease are common comorbid conditions. The most common form of treatment – bioactive dressings in a moist environment – produces unsatisfactory results because it doesn’t address the inflammatory process.
Dr. Conde and coworkers have published the full details of how they achieved complete healing of Martorell hypertensive ischemic leg ulcers 3-8 weeks after punch grafting in three affected patients, all of whom presented with pain scores of 10/10 refractory even to opioid analgesics. The punch grafting was preceded by 15 days of topical corticosteroids and low-elasticity compression bandages in order to create adequate granulation tissue in the wound bed, which had the added benefit of achieving a 2- to 3-point reduction in pain scores even before the surgical procedure.
The pain-reducing effect of punch grafting isn’t as well appreciated as the wound-healing effect. Dr. Conde was first author of a recent study in which investigators systematically measured pain reduction in 136 patients with hard-to-heal leg ulcers of various etiologies treated with punch grafting. Nearly three-quarters of those who presented with painful ulcers were pain free after punch grafting, and the rest experienced greater than 70% pain reduction.
Pain suppression wasn’t dependent upon the percentage of graft uptake in this study. That’s because, as long as the wound isn’t overcleaned during dressing changes, even grafts that haven’t attached to the wound will release growth factors that promote wound healing, Dr. Conde explained.
Adjunctive negative pressure therapy
Portable vacuum-based negative pressure therapy devices are easy to use as a means to promote punch graft uptake. Negative pressure is best employed as an adjunct to punch grafting in suboptimal wound beds, longstanding ulcers, in patients with previous graft failure, or in challenging anatomic locations, such as the Achilles tendon or ankle. Dr. Conde has found the combination of punch grafting and negative pressure therapy especially helpful in patients with clinically inactive pyoderma gangrenosum.
Topical sevoflurane for analgesia
Most of the literature on topical sevoflurane for ulcer care has been published by Spanish researchers, but this form of analgesia deserves much more widespread use, according to Dr. Conde.
Sevoflurane is most often used as a gas in general anesthesia. In liquid form, however, it not only has a rapid, long-lasting analgesic effect when applied to painful leg ulcers, it also promotes healing because it is both antibacterial and a vasodilator. So before performing a potentially painful ulcer or wound cleaning, Dr. Conde recommended protecting perilesional skin with petroleum jelly, then irrigating the ulcer site with liquid sevoflurane. After that, it’s advisable to wait just 5-10 minutes before proceeding.
“It takes effect in much less time than EMLA cream,” she noted.
In one study of 30 adults aged over age 65 years with painful chronic venous ulcers refractory to conventional analgesics who underwent ulcer cleaning supported by topical sevoflurane at a dose of roughly 1 mL/cm2 of ulcer area every 2 days for a month, Spanish investigators documented onset of analgesic effect in 2-7 minutes, with a duration of 8-18 hours. The researchers found that the use of backup conventional analgesics ranging from acetaminophen to opioids was diminished. Side effects were limited to mild, transient itching and redness.
Dr. Conde reported having no financial conflicts of interest regarding her presentation.
FROM THE EADV CONGRESS
Oral JAK inhibitor for alopecia areata advances to phase 3
An of a trio of 24-week, phase 2 randomized trials, James V. Cassella, PhD, reported at the virtual annual congress of the European Academy of Dermatology and Venereology.
“So far, I’d say that the results are very encouraging from this study, as we see good safety and continued stability of the hair regrowth in the open-label extension,” said Dr. Cassella, chief development officer at Concert Pharmaceuticals of Lexington, Mass.
The study drug, known for now as CTP-543, is a deuterium-modified form of the selective JAK1/2 inhibitor ruxolitinib. CTP-543 was expressly designed for treatment of alopecia areata, a chronic autoimmune disease for which no Food and Drug Administration–approved therapy exists. Dr. Cassella characterized alopecia areata as a “devastating and poorly treated” autoimmune disease which affects men, women, and children of all ages and has a lifetime risk of about 2%.
Participants in the open-label extension were typically in their late 30s, with an average disease duration of 3-4 years. Two-thirds were women, and more than three-quarters were White. Their baseline Severity of Alopecia Areata Tool (SALT) score was in the high 80s, indicative of moderate to severe disease.
About 92% of eligible participants from the three phase 2 trials elected to enroll in the long-term extension, a remarkably high participation rate reflective of the major unmet need for effective treatments for this chronic disease.
Efficacy
Of the 152 patients who enrolled in an ongoing open-label extension, which had been going for 108 weeks at the time of the presentation, 130 who completed at least 1 year on CTP-543 formed the focus of Dr. Cassella’s presentation. The great majority of participants were on 12 mg twice daily. By week 24 in the original phase 2 trials, SALT scores decreased by more than 50%, compared with baseline, with an average score of about 40. This level of efficacy was maintained through the first 6 months of the extension study – meaning a total of at least 1 year on treatment – in roughly two-thirds of participants, while one-third saw further progressive improvement during the first 6 months of the open-label extension, with SALT scores dropping into the 20-30 range.
“You see remarkable stability of hair regrowth beyond 6 months,” Dr. Cassella commented. Only 2 patients experienced a clear loss of response during the open-label extension.
Safety and tolerability
About 15% of patients have dropped out of the long-term study, which in only three cases was because of adverse events. Treatment-emergent adverse events, which occurred in 13% of participants, were rated as mild in 76% and moderate in 21%. The most common were nasopharyngitis, acne, and elevated creatine phosphokinase. Two severe adverse events were deemed “possibly related” to treatment. No new types of side effects have emerged during the long-term extension study. Laboratory monitoring of hemoglobin, neutrophils, and platelets has shown stable levels over time.
An audience member asked how quickly hair loss occurs upon stopping therapy.
“Surprisingly, in the few dose interruptions we’ve had – for changes in hematologic parameters, for example – we have not seen any hair loss in up to a 3-week time period. It’s a very interesting question that we will continue to study,” Dr. Cassella replied. He added: “The general thinking in the community has been, at some point with JAK inhibitors, you will lose your hair if you stop treatment.”
Dr. Cassella is an employee of Concert Pharmaceuticals, and has received company stock.
An of a trio of 24-week, phase 2 randomized trials, James V. Cassella, PhD, reported at the virtual annual congress of the European Academy of Dermatology and Venereology.
“So far, I’d say that the results are very encouraging from this study, as we see good safety and continued stability of the hair regrowth in the open-label extension,” said Dr. Cassella, chief development officer at Concert Pharmaceuticals of Lexington, Mass.
The study drug, known for now as CTP-543, is a deuterium-modified form of the selective JAK1/2 inhibitor ruxolitinib. CTP-543 was expressly designed for treatment of alopecia areata, a chronic autoimmune disease for which no Food and Drug Administration–approved therapy exists. Dr. Cassella characterized alopecia areata as a “devastating and poorly treated” autoimmune disease which affects men, women, and children of all ages and has a lifetime risk of about 2%.
Participants in the open-label extension were typically in their late 30s, with an average disease duration of 3-4 years. Two-thirds were women, and more than three-quarters were White. Their baseline Severity of Alopecia Areata Tool (SALT) score was in the high 80s, indicative of moderate to severe disease.
About 92% of eligible participants from the three phase 2 trials elected to enroll in the long-term extension, a remarkably high participation rate reflective of the major unmet need for effective treatments for this chronic disease.
Efficacy
Of the 152 patients who enrolled in an ongoing open-label extension, which had been going for 108 weeks at the time of the presentation, 130 who completed at least 1 year on CTP-543 formed the focus of Dr. Cassella’s presentation. The great majority of participants were on 12 mg twice daily. By week 24 in the original phase 2 trials, SALT scores decreased by more than 50%, compared with baseline, with an average score of about 40. This level of efficacy was maintained through the first 6 months of the extension study – meaning a total of at least 1 year on treatment – in roughly two-thirds of participants, while one-third saw further progressive improvement during the first 6 months of the open-label extension, with SALT scores dropping into the 20-30 range.
“You see remarkable stability of hair regrowth beyond 6 months,” Dr. Cassella commented. Only 2 patients experienced a clear loss of response during the open-label extension.
Safety and tolerability
About 15% of patients have dropped out of the long-term study, which in only three cases was because of adverse events. Treatment-emergent adverse events, which occurred in 13% of participants, were rated as mild in 76% and moderate in 21%. The most common were nasopharyngitis, acne, and elevated creatine phosphokinase. Two severe adverse events were deemed “possibly related” to treatment. No new types of side effects have emerged during the long-term extension study. Laboratory monitoring of hemoglobin, neutrophils, and platelets has shown stable levels over time.
An audience member asked how quickly hair loss occurs upon stopping therapy.
“Surprisingly, in the few dose interruptions we’ve had – for changes in hematologic parameters, for example – we have not seen any hair loss in up to a 3-week time period. It’s a very interesting question that we will continue to study,” Dr. Cassella replied. He added: “The general thinking in the community has been, at some point with JAK inhibitors, you will lose your hair if you stop treatment.”
Dr. Cassella is an employee of Concert Pharmaceuticals, and has received company stock.
An of a trio of 24-week, phase 2 randomized trials, James V. Cassella, PhD, reported at the virtual annual congress of the European Academy of Dermatology and Venereology.
“So far, I’d say that the results are very encouraging from this study, as we see good safety and continued stability of the hair regrowth in the open-label extension,” said Dr. Cassella, chief development officer at Concert Pharmaceuticals of Lexington, Mass.
The study drug, known for now as CTP-543, is a deuterium-modified form of the selective JAK1/2 inhibitor ruxolitinib. CTP-543 was expressly designed for treatment of alopecia areata, a chronic autoimmune disease for which no Food and Drug Administration–approved therapy exists. Dr. Cassella characterized alopecia areata as a “devastating and poorly treated” autoimmune disease which affects men, women, and children of all ages and has a lifetime risk of about 2%.
Participants in the open-label extension were typically in their late 30s, with an average disease duration of 3-4 years. Two-thirds were women, and more than three-quarters were White. Their baseline Severity of Alopecia Areata Tool (SALT) score was in the high 80s, indicative of moderate to severe disease.
About 92% of eligible participants from the three phase 2 trials elected to enroll in the long-term extension, a remarkably high participation rate reflective of the major unmet need for effective treatments for this chronic disease.
Efficacy
Of the 152 patients who enrolled in an ongoing open-label extension, which had been going for 108 weeks at the time of the presentation, 130 who completed at least 1 year on CTP-543 formed the focus of Dr. Cassella’s presentation. The great majority of participants were on 12 mg twice daily. By week 24 in the original phase 2 trials, SALT scores decreased by more than 50%, compared with baseline, with an average score of about 40. This level of efficacy was maintained through the first 6 months of the extension study – meaning a total of at least 1 year on treatment – in roughly two-thirds of participants, while one-third saw further progressive improvement during the first 6 months of the open-label extension, with SALT scores dropping into the 20-30 range.
“You see remarkable stability of hair regrowth beyond 6 months,” Dr. Cassella commented. Only 2 patients experienced a clear loss of response during the open-label extension.
Safety and tolerability
About 15% of patients have dropped out of the long-term study, which in only three cases was because of adverse events. Treatment-emergent adverse events, which occurred in 13% of participants, were rated as mild in 76% and moderate in 21%. The most common were nasopharyngitis, acne, and elevated creatine phosphokinase. Two severe adverse events were deemed “possibly related” to treatment. No new types of side effects have emerged during the long-term extension study. Laboratory monitoring of hemoglobin, neutrophils, and platelets has shown stable levels over time.
An audience member asked how quickly hair loss occurs upon stopping therapy.
“Surprisingly, in the few dose interruptions we’ve had – for changes in hematologic parameters, for example – we have not seen any hair loss in up to a 3-week time period. It’s a very interesting question that we will continue to study,” Dr. Cassella replied. He added: “The general thinking in the community has been, at some point with JAK inhibitors, you will lose your hair if you stop treatment.”
Dr. Cassella is an employee of Concert Pharmaceuticals, and has received company stock.
FROM THE EADV CONGRESS
Improvements in chronic hand eczema seen with oral gusacitinib in phase 2 study
in a phase 2b, randomized trial, Howard Sofen, MD, reported at the virtual annual congress of the European Academy of Dermatology and Venereology.
The once-daily drug proved effective for this challenging condition, regardless of whether an individual’s chronic hand eczema was driven chiefly by irritant contact dermatitis, allergic contact dermatitis, or atopic dermatitis, added Dr. Sofen, medical director of Dermatology Research Associates, Los Angeles, and chief of the dermatology division at LA County/Olive View Medical Center.
Gusacitinib is a once-daily oral inhibitor of Janus kinase 1, 2, and 3, tyrosine kinase 2, and spleen tyrosine kinase (SYK). As such, it targets the Th1, Th2, Th17, and Th22 cytokine pathways, as well as SYK-mediated interleukin-17 signaling of keratinocyte proliferation and differentiation. Thus, its spectrum of activity makes it a candidate for the treatment of a variety of other inflammatory dermatologic diseases, although chronic hand eczema alone affects an estimated 7 million Americans, the dermatologist noted.
The phase 2b, double-blind, 16-week, multicenter, randomized trial included 97 patients who were randomized to oral gusacitinib as monotherapy at 40 or 80 mg once daily or placebo. All participants had chronic hand eczema of more than 6 months duration that was refractory to potent or superpotent topical and/or systemic steroids. Participants were split 60/40 between those with severe chronic hand eczema, defined by a baseline score on the 0-4 Physician’s Global Assessment scale, and moderate disease, with a PGA of 3.
The primary endpoint was the percent improvement in modified total lesion severity score (mTLSS) at week 16 from a mean baseline of 13.2. A clearcut dose response was evident: Gusacitinib at 80 mg/day achieved a 69.5% decrease, while 40 mg brought a 40% reduction, which wasn’t significantly better than the 33.5% decrease in placebo-treated controls.
The rapidity of response was noteworthy in these steroid-refractory patients. The 80-mg group showed significant separation from placebo by 2 weeks, with a mean 40.1% reduction in mTLSS versus 13.6% with placebo.
The secondary endpoint was achievement of a PGA score of 0 or 1 – that is, clear or almost clear – with a 2-grade improvement over placebo. This was achieved in 31.3% of patients assigned to the higher dose of gusacitinib at week 16, a success rate fivefold higher than the 6.3% rate in controls. The two groups separated on this endpoint at week 2, the first assessment. At week 8 there was an eightfold difference in response: 25% in patients on gusacitinib at 80 mg, 3.1% with placebo.
The other secondary endpoint was improvement in itch as measured by the mTLSS pruritus 0-3 subscore. As for the other outcomes, the improvement in itch was rapid. At week 2, patients on gusacitinib at 80 mg averaged a 43.1% reduction from their baseline pruritus score, compared with 4.6% with placebo. At week 16, the reductions were 65.7% and 29.8%, respectively.
Both doses of gusacitinib were well tolerated, according to Dr. Sofer. No thromboembolic events, major adverse cardiovascular events, or opportunistic infections occurred during the short 16-week study. The drug’s safety profile was consistent with what’s been seen in a collective gusacitinib clinical trial experience totaling more than 350 patients: mild to moderate nasopharyngitis, headache, asymptomatic elevations in creatine phosphokinase, and a slight increase in HDL cholesterol accompanied by a small reduction in LDL cholesterol.
Dr. Sofen reported receiving research funding from and serving as a consultant to Asana BioSciences, the study sponsor, as well as more than half a dozen other pharmaceutical companies.
in a phase 2b, randomized trial, Howard Sofen, MD, reported at the virtual annual congress of the European Academy of Dermatology and Venereology.
The once-daily drug proved effective for this challenging condition, regardless of whether an individual’s chronic hand eczema was driven chiefly by irritant contact dermatitis, allergic contact dermatitis, or atopic dermatitis, added Dr. Sofen, medical director of Dermatology Research Associates, Los Angeles, and chief of the dermatology division at LA County/Olive View Medical Center.
Gusacitinib is a once-daily oral inhibitor of Janus kinase 1, 2, and 3, tyrosine kinase 2, and spleen tyrosine kinase (SYK). As such, it targets the Th1, Th2, Th17, and Th22 cytokine pathways, as well as SYK-mediated interleukin-17 signaling of keratinocyte proliferation and differentiation. Thus, its spectrum of activity makes it a candidate for the treatment of a variety of other inflammatory dermatologic diseases, although chronic hand eczema alone affects an estimated 7 million Americans, the dermatologist noted.
The phase 2b, double-blind, 16-week, multicenter, randomized trial included 97 patients who were randomized to oral gusacitinib as monotherapy at 40 or 80 mg once daily or placebo. All participants had chronic hand eczema of more than 6 months duration that was refractory to potent or superpotent topical and/or systemic steroids. Participants were split 60/40 between those with severe chronic hand eczema, defined by a baseline score on the 0-4 Physician’s Global Assessment scale, and moderate disease, with a PGA of 3.
The primary endpoint was the percent improvement in modified total lesion severity score (mTLSS) at week 16 from a mean baseline of 13.2. A clearcut dose response was evident: Gusacitinib at 80 mg/day achieved a 69.5% decrease, while 40 mg brought a 40% reduction, which wasn’t significantly better than the 33.5% decrease in placebo-treated controls.
The rapidity of response was noteworthy in these steroid-refractory patients. The 80-mg group showed significant separation from placebo by 2 weeks, with a mean 40.1% reduction in mTLSS versus 13.6% with placebo.
The secondary endpoint was achievement of a PGA score of 0 or 1 – that is, clear or almost clear – with a 2-grade improvement over placebo. This was achieved in 31.3% of patients assigned to the higher dose of gusacitinib at week 16, a success rate fivefold higher than the 6.3% rate in controls. The two groups separated on this endpoint at week 2, the first assessment. At week 8 there was an eightfold difference in response: 25% in patients on gusacitinib at 80 mg, 3.1% with placebo.
The other secondary endpoint was improvement in itch as measured by the mTLSS pruritus 0-3 subscore. As for the other outcomes, the improvement in itch was rapid. At week 2, patients on gusacitinib at 80 mg averaged a 43.1% reduction from their baseline pruritus score, compared with 4.6% with placebo. At week 16, the reductions were 65.7% and 29.8%, respectively.
Both doses of gusacitinib were well tolerated, according to Dr. Sofer. No thromboembolic events, major adverse cardiovascular events, or opportunistic infections occurred during the short 16-week study. The drug’s safety profile was consistent with what’s been seen in a collective gusacitinib clinical trial experience totaling more than 350 patients: mild to moderate nasopharyngitis, headache, asymptomatic elevations in creatine phosphokinase, and a slight increase in HDL cholesterol accompanied by a small reduction in LDL cholesterol.
Dr. Sofen reported receiving research funding from and serving as a consultant to Asana BioSciences, the study sponsor, as well as more than half a dozen other pharmaceutical companies.
in a phase 2b, randomized trial, Howard Sofen, MD, reported at the virtual annual congress of the European Academy of Dermatology and Venereology.
The once-daily drug proved effective for this challenging condition, regardless of whether an individual’s chronic hand eczema was driven chiefly by irritant contact dermatitis, allergic contact dermatitis, or atopic dermatitis, added Dr. Sofen, medical director of Dermatology Research Associates, Los Angeles, and chief of the dermatology division at LA County/Olive View Medical Center.
Gusacitinib is a once-daily oral inhibitor of Janus kinase 1, 2, and 3, tyrosine kinase 2, and spleen tyrosine kinase (SYK). As such, it targets the Th1, Th2, Th17, and Th22 cytokine pathways, as well as SYK-mediated interleukin-17 signaling of keratinocyte proliferation and differentiation. Thus, its spectrum of activity makes it a candidate for the treatment of a variety of other inflammatory dermatologic diseases, although chronic hand eczema alone affects an estimated 7 million Americans, the dermatologist noted.
The phase 2b, double-blind, 16-week, multicenter, randomized trial included 97 patients who were randomized to oral gusacitinib as monotherapy at 40 or 80 mg once daily or placebo. All participants had chronic hand eczema of more than 6 months duration that was refractory to potent or superpotent topical and/or systemic steroids. Participants were split 60/40 between those with severe chronic hand eczema, defined by a baseline score on the 0-4 Physician’s Global Assessment scale, and moderate disease, with a PGA of 3.
The primary endpoint was the percent improvement in modified total lesion severity score (mTLSS) at week 16 from a mean baseline of 13.2. A clearcut dose response was evident: Gusacitinib at 80 mg/day achieved a 69.5% decrease, while 40 mg brought a 40% reduction, which wasn’t significantly better than the 33.5% decrease in placebo-treated controls.
The rapidity of response was noteworthy in these steroid-refractory patients. The 80-mg group showed significant separation from placebo by 2 weeks, with a mean 40.1% reduction in mTLSS versus 13.6% with placebo.
The secondary endpoint was achievement of a PGA score of 0 or 1 – that is, clear or almost clear – with a 2-grade improvement over placebo. This was achieved in 31.3% of patients assigned to the higher dose of gusacitinib at week 16, a success rate fivefold higher than the 6.3% rate in controls. The two groups separated on this endpoint at week 2, the first assessment. At week 8 there was an eightfold difference in response: 25% in patients on gusacitinib at 80 mg, 3.1% with placebo.
The other secondary endpoint was improvement in itch as measured by the mTLSS pruritus 0-3 subscore. As for the other outcomes, the improvement in itch was rapid. At week 2, patients on gusacitinib at 80 mg averaged a 43.1% reduction from their baseline pruritus score, compared with 4.6% with placebo. At week 16, the reductions were 65.7% and 29.8%, respectively.
Both doses of gusacitinib were well tolerated, according to Dr. Sofer. No thromboembolic events, major adverse cardiovascular events, or opportunistic infections occurred during the short 16-week study. The drug’s safety profile was consistent with what’s been seen in a collective gusacitinib clinical trial experience totaling more than 350 patients: mild to moderate nasopharyngitis, headache, asymptomatic elevations in creatine phosphokinase, and a slight increase in HDL cholesterol accompanied by a small reduction in LDL cholesterol.
Dr. Sofen reported receiving research funding from and serving as a consultant to Asana BioSciences, the study sponsor, as well as more than half a dozen other pharmaceutical companies.
FROM THE EADV CONGRESS
Tildrakizumab for psoriasis shows durable efficacy over 5 years
The full during more than 5,400 patient-years of prospective follow-up, Diamont Thaçi, MD, PhD, reported at the virtual annual congress of the European Academy of Dermatology and Venereology.
For example, 89% of patients who had a PASI-75 response on the 100-mg dose of tildrakizumab (Ilumya) – the dose approved in the United States – at week 28 in the parent reSURFACE 1 and reSURFACE 2 trials maintained their PASI-75 response throughout the next 4½ years in the long-term extension study, as did 93% of those with a week 28 PASI-75 response on 200 mg, a dose approved elsewhere, said Dr. Thaçi, professor of dermatology and director of the Comprehensive Center for Inflammation Medicine at Lübeck (Germany) University.
The same held true for PASI-90, a response achieved by 71% of participants on 100 mg of tildrakizumab at week 28 and 66% at week 244, and by 73% of those on the 200-mg dose at week 28 and 70% at 5 years. A PASI-100 response was documented at week 28 in 29% of patients on the lower dose and 37% of those on 200 mg, with week 244 PASI-100 rates of 33% and 41%, respectively.
The long-term extension study enrolled 622 patients with moderate to severe chronic plaque psoriasis with at least a PASI-75 response to 100 mg or 200 mg of the humanized monoclonal antibody interleukin-23p19 inhibitor at week 28 in reSURFACE 1 or 2, or who were partial or nonresponders to etanercept in reSURFACE 2 and were then switched to tildrakizumab at 200 mg. Five hundred and forty-five of the 622 patients (88%) completed the full 5 years of the extension study.
Very few patients left the study because of loss of efficacy or adverse events. Indeed, the exposure-adjusted rate of drug-related serious adverse events was 0.8 cases per 100 patient-years at tildrakizumab 100 mg and 0.5 per 100 patient-years at 200 mg. Moreover, the rates of drug-related serious adverse events leading to treatment continuation were 0.3 and 0.2 per 100 patient-years at the 100-mg and 200-mg doses. Rates of treatment-emergent severe infection were 1.2 and 1.3 per 100 patient-years on the lower and higher doses. Major adverse cardiovascular events occurred at rates of 0.5 and 0.7 cases per 100 patient-years.
“I think the adverse events are generally similar to what has been seen with other biologics, but slightly less with tildrakizumab. Registries will provide a clearer picture. What’s interesting is that even if you double the dosage you don’t see an increase in side effects,” Dr. Thaçi said.
Asked what happens when a tildrakizumab responder stops taking the monoclonal antibody, he replied, “This is something very interesting we see with the IL-23 inhibitors: The disease comes back very slowly. It takes months, and sometimes years, for the patient to lose the PASI-75 or even the PASI-90 response. But we still consider that continuous treatment is probably the better way to go because we cannot be sure who will lose or regain response. At the moment we don’t have a biomarker to tell us what we should do in our daily practice.”
Dr. Thaçi reported serving as an adviser to and paid investigator for Almirall, the study sponsor, and approximately 20 other pharmaceutical companies.
The full during more than 5,400 patient-years of prospective follow-up, Diamont Thaçi, MD, PhD, reported at the virtual annual congress of the European Academy of Dermatology and Venereology.
For example, 89% of patients who had a PASI-75 response on the 100-mg dose of tildrakizumab (Ilumya) – the dose approved in the United States – at week 28 in the parent reSURFACE 1 and reSURFACE 2 trials maintained their PASI-75 response throughout the next 4½ years in the long-term extension study, as did 93% of those with a week 28 PASI-75 response on 200 mg, a dose approved elsewhere, said Dr. Thaçi, professor of dermatology and director of the Comprehensive Center for Inflammation Medicine at Lübeck (Germany) University.
The same held true for PASI-90, a response achieved by 71% of participants on 100 mg of tildrakizumab at week 28 and 66% at week 244, and by 73% of those on the 200-mg dose at week 28 and 70% at 5 years. A PASI-100 response was documented at week 28 in 29% of patients on the lower dose and 37% of those on 200 mg, with week 244 PASI-100 rates of 33% and 41%, respectively.
The long-term extension study enrolled 622 patients with moderate to severe chronic plaque psoriasis with at least a PASI-75 response to 100 mg or 200 mg of the humanized monoclonal antibody interleukin-23p19 inhibitor at week 28 in reSURFACE 1 or 2, or who were partial or nonresponders to etanercept in reSURFACE 2 and were then switched to tildrakizumab at 200 mg. Five hundred and forty-five of the 622 patients (88%) completed the full 5 years of the extension study.
Very few patients left the study because of loss of efficacy or adverse events. Indeed, the exposure-adjusted rate of drug-related serious adverse events was 0.8 cases per 100 patient-years at tildrakizumab 100 mg and 0.5 per 100 patient-years at 200 mg. Moreover, the rates of drug-related serious adverse events leading to treatment continuation were 0.3 and 0.2 per 100 patient-years at the 100-mg and 200-mg doses. Rates of treatment-emergent severe infection were 1.2 and 1.3 per 100 patient-years on the lower and higher doses. Major adverse cardiovascular events occurred at rates of 0.5 and 0.7 cases per 100 patient-years.
“I think the adverse events are generally similar to what has been seen with other biologics, but slightly less with tildrakizumab. Registries will provide a clearer picture. What’s interesting is that even if you double the dosage you don’t see an increase in side effects,” Dr. Thaçi said.
Asked what happens when a tildrakizumab responder stops taking the monoclonal antibody, he replied, “This is something very interesting we see with the IL-23 inhibitors: The disease comes back very slowly. It takes months, and sometimes years, for the patient to lose the PASI-75 or even the PASI-90 response. But we still consider that continuous treatment is probably the better way to go because we cannot be sure who will lose or regain response. At the moment we don’t have a biomarker to tell us what we should do in our daily practice.”
Dr. Thaçi reported serving as an adviser to and paid investigator for Almirall, the study sponsor, and approximately 20 other pharmaceutical companies.
The full during more than 5,400 patient-years of prospective follow-up, Diamont Thaçi, MD, PhD, reported at the virtual annual congress of the European Academy of Dermatology and Venereology.
For example, 89% of patients who had a PASI-75 response on the 100-mg dose of tildrakizumab (Ilumya) – the dose approved in the United States – at week 28 in the parent reSURFACE 1 and reSURFACE 2 trials maintained their PASI-75 response throughout the next 4½ years in the long-term extension study, as did 93% of those with a week 28 PASI-75 response on 200 mg, a dose approved elsewhere, said Dr. Thaçi, professor of dermatology and director of the Comprehensive Center for Inflammation Medicine at Lübeck (Germany) University.
The same held true for PASI-90, a response achieved by 71% of participants on 100 mg of tildrakizumab at week 28 and 66% at week 244, and by 73% of those on the 200-mg dose at week 28 and 70% at 5 years. A PASI-100 response was documented at week 28 in 29% of patients on the lower dose and 37% of those on 200 mg, with week 244 PASI-100 rates of 33% and 41%, respectively.
The long-term extension study enrolled 622 patients with moderate to severe chronic plaque psoriasis with at least a PASI-75 response to 100 mg or 200 mg of the humanized monoclonal antibody interleukin-23p19 inhibitor at week 28 in reSURFACE 1 or 2, or who were partial or nonresponders to etanercept in reSURFACE 2 and were then switched to tildrakizumab at 200 mg. Five hundred and forty-five of the 622 patients (88%) completed the full 5 years of the extension study.
Very few patients left the study because of loss of efficacy or adverse events. Indeed, the exposure-adjusted rate of drug-related serious adverse events was 0.8 cases per 100 patient-years at tildrakizumab 100 mg and 0.5 per 100 patient-years at 200 mg. Moreover, the rates of drug-related serious adverse events leading to treatment continuation were 0.3 and 0.2 per 100 patient-years at the 100-mg and 200-mg doses. Rates of treatment-emergent severe infection were 1.2 and 1.3 per 100 patient-years on the lower and higher doses. Major adverse cardiovascular events occurred at rates of 0.5 and 0.7 cases per 100 patient-years.
“I think the adverse events are generally similar to what has been seen with other biologics, but slightly less with tildrakizumab. Registries will provide a clearer picture. What’s interesting is that even if you double the dosage you don’t see an increase in side effects,” Dr. Thaçi said.
Asked what happens when a tildrakizumab responder stops taking the monoclonal antibody, he replied, “This is something very interesting we see with the IL-23 inhibitors: The disease comes back very slowly. It takes months, and sometimes years, for the patient to lose the PASI-75 or even the PASI-90 response. But we still consider that continuous treatment is probably the better way to go because we cannot be sure who will lose or regain response. At the moment we don’t have a biomarker to tell us what we should do in our daily practice.”
Dr. Thaçi reported serving as an adviser to and paid investigator for Almirall, the study sponsor, and approximately 20 other pharmaceutical companies.
FROM THE EADV CONGRESS
Abrocitinib highly effective as long-term monotherapy in AD
through 48 weeks of follow-up in the JADE EXTEND study, Kristian Reich, MD, reported at the virtual annual congress of the European Academy of Dermatology and Venereology.

The head-turning outcomes achieved at the higher studied dose of 200 mg once daily as monotherapy – namely, 87% of patients had an EASI-75 response, defined as at least a 75% reduction from baseline in Eczema Area and Severity Index score, and 62% had an EASI-90 response – herald a new era in the management of atopic dermatitis, predicted Dr. Reich, of the Center for Translational Research in Inflammatory Skin Diseases at the University Medical Center Hamburg-Eppendorf (Germany).
“I think we will see an evolution in the treatment goals in atopic dermatitis. It’s really good to see nearly 90% of the patients achieved EASI-75 over time. I am completely convinced that if you ultimately want to have a happy patient, you will see treatment goals moving up. We have already seen this in psoriasis. I want to see drugs that give the majority of my patients an EASI-75. And ultimately I want to see EASI-90 for my patients,” he said.
Concurrent with his presentation at the EADV congress, Pfizer announced it has filed for marketing approval of abrocitinib at 100 mg and 200 mg once daily for the treatment of moderate to severe AD. The Food and Drug Administration has granted the application priority review status, with a decision due next April. The company has also filed for marketing approval with the European Medicines Agency.
The JADE EXTEND study is an ongoing extension of the previously reported phase 3, randomized, double-blind, placebo-controlled, 12-week JADE MONO-1 and JADE MONO-2 trials. The two trials included a total of 309 patients on abrocitinib at 200 mg/day and 314 on the selective Janus kinase (JAK) 1 inhibitor at 100 mg/day, 519 of whom subsequently entered the long-term extension study on their same dose. The 70% who required no supplemental topical therapy through 48 weeks were the focus of the analysis presented by Dr. Reich.
The proportion of strong responders increased up until the week 24 or 36 assessments, then remained steady until week 48. For example, the EASI-75 rate in patients on abrocitinib at 200 mg/day rose from 82.5% at week 16, to 86.2% at week 24, 90.1% at week 36, and reached 87.2% at week 48. The EASI-90 rates at the same time points were 56.7%, 64.5%, 65.5%, and 61.6%, respectively. And the EASI-100 rates were 24%, 31.6%, 29.6%, and 24%, respectively.
Not surprisingly, the EASI-75 rates in patients on abrocitinib at 100 mg/day were less robust: 64.4% at week 16, 75.5% at week 24, 74.5% at week 36, and 68% at week 48.
An Investigator’s Global Assessment score of 0 or 1 – that is, clear or almost clear – was achieved at week 16 in 55% of patients on 200 mg/day, 64.5% at week 24, 66% at week 36, and 60.5% at week 48. In patients on the 100-mg dose, the corresponding figures were 36.5%, 46.6%, 53.3%, and 45.2%.
A hallmark of all of the JAK inhibitors under study for AD is what Dr. Reich characterized as “an amazingly fast reduction of itch,” the dominant symptom of the disease. A clinically meaningful reduction of at least 4 points in the Peak Pruritus Numerical Rating Scale – a response of 4 or greater is considered clinically important – from the mean baseline score of 7.1 was present at week 12 in 56.3% of patients on abrocitinib at 200 mg, in 74.3% at week 16, and in 72.5% at week 48. The proportion of patients achieving this endpoint on 100 mg was 41.6% at week 12, 49.4% at week 16, and 52% at week 48.
Serious treatment-emergent adverse events occurred in 6.1% of JADE EXTEND participants on abrocitinib at 100 mg and 12.8% of those on 200 mg. These events included oral herpes and elevated creatine phosphokinase levels. The sole case of pulmonary embolism that occurred during the study was deemed unrelated to treatment.
“What this is telling me here is there are no signals that we haven’t seen earlier with this drug and with other JAK inhibitors before,” the dermatologist observed. “But I want to see more data. I want to see the overall safety, not just for a year, but for 2, 3, 4, and 5 years.”
Asked by an audience member if nonresponsiveness to one JAK inhibitor predicts nonresponse to others, Dr. Reich speculated that it’s likely to be so. He noted that all three of the JAK inhibitors furthest along in the developmental pipeline for atopic dermatitis – abrocitinib, baricitinib, and upadacitinib – are inhibitors of JAK 1, although baricitinib also targets JAK 2.
“I would think that if you really are a nonresponder to any of these that it will be hard to get a good response with the others. We’re not talking about antibodies here, where there may be different epitopes. The affinity is different, and we have seen that if you have no response to a weak TNF [tumor necrosis factor] inhibitor, you can still have a response to a strong TNF inhibitor. I don’t expect the same here,” according to Dr. Reich.
He reported serving as an adviser to and paid clinical research for Pfizer, which sponsored JADE EXTEND, as well as more than two dozen other pharmaceutical companies.
through 48 weeks of follow-up in the JADE EXTEND study, Kristian Reich, MD, reported at the virtual annual congress of the European Academy of Dermatology and Venereology.
The head-turning outcomes achieved at the higher studied dose of 200 mg once daily as monotherapy – namely, 87% of patients had an EASI-75 response, defined as at least a 75% reduction from baseline in Eczema Area and Severity Index score, and 62% had an EASI-90 response – herald a new era in the management of atopic dermatitis, predicted Dr. Reich, of the Center for Translational Research in Inflammatory Skin Diseases at the University Medical Center Hamburg-Eppendorf (Germany).
“I think we will see an evolution in the treatment goals in atopic dermatitis. It’s really good to see nearly 90% of the patients achieved EASI-75 over time. I am completely convinced that if you ultimately want to have a happy patient, you will see treatment goals moving up. We have already seen this in psoriasis. I want to see drugs that give the majority of my patients an EASI-75. And ultimately I want to see EASI-90 for my patients,” he said.
Concurrent with his presentation at the EADV congress, Pfizer announced it has filed for marketing approval of abrocitinib at 100 mg and 200 mg once daily for the treatment of moderate to severe AD. The Food and Drug Administration has granted the application priority review status, with a decision due next April. The company has also filed for marketing approval with the European Medicines Agency.
The JADE EXTEND study is an ongoing extension of the previously reported phase 3, randomized, double-blind, placebo-controlled, 12-week JADE MONO-1 and JADE MONO-2 trials. The two trials included a total of 309 patients on abrocitinib at 200 mg/day and 314 on the selective Janus kinase (JAK) 1 inhibitor at 100 mg/day, 519 of whom subsequently entered the long-term extension study on their same dose. The 70% who required no supplemental topical therapy through 48 weeks were the focus of the analysis presented by Dr. Reich.
The proportion of strong responders increased up until the week 24 or 36 assessments, then remained steady until week 48. For example, the EASI-75 rate in patients on abrocitinib at 200 mg/day rose from 82.5% at week 16, to 86.2% at week 24, 90.1% at week 36, and reached 87.2% at week 48. The EASI-90 rates at the same time points were 56.7%, 64.5%, 65.5%, and 61.6%, respectively. And the EASI-100 rates were 24%, 31.6%, 29.6%, and 24%, respectively.
Not surprisingly, the EASI-75 rates in patients on abrocitinib at 100 mg/day were less robust: 64.4% at week 16, 75.5% at week 24, 74.5% at week 36, and 68% at week 48.
An Investigator’s Global Assessment score of 0 or 1 – that is, clear or almost clear – was achieved at week 16 in 55% of patients on 200 mg/day, 64.5% at week 24, 66% at week 36, and 60.5% at week 48. In patients on the 100-mg dose, the corresponding figures were 36.5%, 46.6%, 53.3%, and 45.2%.
A hallmark of all of the JAK inhibitors under study for AD is what Dr. Reich characterized as “an amazingly fast reduction of itch,” the dominant symptom of the disease. A clinically meaningful reduction of at least 4 points in the Peak Pruritus Numerical Rating Scale – a response of 4 or greater is considered clinically important – from the mean baseline score of 7.1 was present at week 12 in 56.3% of patients on abrocitinib at 200 mg, in 74.3% at week 16, and in 72.5% at week 48. The proportion of patients achieving this endpoint on 100 mg was 41.6% at week 12, 49.4% at week 16, and 52% at week 48.
Serious treatment-emergent adverse events occurred in 6.1% of JADE EXTEND participants on abrocitinib at 100 mg and 12.8% of those on 200 mg. These events included oral herpes and elevated creatine phosphokinase levels. The sole case of pulmonary embolism that occurred during the study was deemed unrelated to treatment.
“What this is telling me here is there are no signals that we haven’t seen earlier with this drug and with other JAK inhibitors before,” the dermatologist observed. “But I want to see more data. I want to see the overall safety, not just for a year, but for 2, 3, 4, and 5 years.”
Asked by an audience member if nonresponsiveness to one JAK inhibitor predicts nonresponse to others, Dr. Reich speculated that it’s likely to be so. He noted that all three of the JAK inhibitors furthest along in the developmental pipeline for atopic dermatitis – abrocitinib, baricitinib, and upadacitinib – are inhibitors of JAK 1, although baricitinib also targets JAK 2.
“I would think that if you really are a nonresponder to any of these that it will be hard to get a good response with the others. We’re not talking about antibodies here, where there may be different epitopes. The affinity is different, and we have seen that if you have no response to a weak TNF [tumor necrosis factor] inhibitor, you can still have a response to a strong TNF inhibitor. I don’t expect the same here,” according to Dr. Reich.
He reported serving as an adviser to and paid clinical research for Pfizer, which sponsored JADE EXTEND, as well as more than two dozen other pharmaceutical companies.
through 48 weeks of follow-up in the JADE EXTEND study, Kristian Reich, MD, reported at the virtual annual congress of the European Academy of Dermatology and Venereology.
The head-turning outcomes achieved at the higher studied dose of 200 mg once daily as monotherapy – namely, 87% of patients had an EASI-75 response, defined as at least a 75% reduction from baseline in Eczema Area and Severity Index score, and 62% had an EASI-90 response – herald a new era in the management of atopic dermatitis, predicted Dr. Reich, of the Center for Translational Research in Inflammatory Skin Diseases at the University Medical Center Hamburg-Eppendorf (Germany).
“I think we will see an evolution in the treatment goals in atopic dermatitis. It’s really good to see nearly 90% of the patients achieved EASI-75 over time. I am completely convinced that if you ultimately want to have a happy patient, you will see treatment goals moving up. We have already seen this in psoriasis. I want to see drugs that give the majority of my patients an EASI-75. And ultimately I want to see EASI-90 for my patients,” he said.
Concurrent with his presentation at the EADV congress, Pfizer announced it has filed for marketing approval of abrocitinib at 100 mg and 200 mg once daily for the treatment of moderate to severe AD. The Food and Drug Administration has granted the application priority review status, with a decision due next April. The company has also filed for marketing approval with the European Medicines Agency.
The JADE EXTEND study is an ongoing extension of the previously reported phase 3, randomized, double-blind, placebo-controlled, 12-week JADE MONO-1 and JADE MONO-2 trials. The two trials included a total of 309 patients on abrocitinib at 200 mg/day and 314 on the selective Janus kinase (JAK) 1 inhibitor at 100 mg/day, 519 of whom subsequently entered the long-term extension study on their same dose. The 70% who required no supplemental topical therapy through 48 weeks were the focus of the analysis presented by Dr. Reich.
The proportion of strong responders increased up until the week 24 or 36 assessments, then remained steady until week 48. For example, the EASI-75 rate in patients on abrocitinib at 200 mg/day rose from 82.5% at week 16, to 86.2% at week 24, 90.1% at week 36, and reached 87.2% at week 48. The EASI-90 rates at the same time points were 56.7%, 64.5%, 65.5%, and 61.6%, respectively. And the EASI-100 rates were 24%, 31.6%, 29.6%, and 24%, respectively.
Not surprisingly, the EASI-75 rates in patients on abrocitinib at 100 mg/day were less robust: 64.4% at week 16, 75.5% at week 24, 74.5% at week 36, and 68% at week 48.
An Investigator’s Global Assessment score of 0 or 1 – that is, clear or almost clear – was achieved at week 16 in 55% of patients on 200 mg/day, 64.5% at week 24, 66% at week 36, and 60.5% at week 48. In patients on the 100-mg dose, the corresponding figures were 36.5%, 46.6%, 53.3%, and 45.2%.
A hallmark of all of the JAK inhibitors under study for AD is what Dr. Reich characterized as “an amazingly fast reduction of itch,” the dominant symptom of the disease. A clinically meaningful reduction of at least 4 points in the Peak Pruritus Numerical Rating Scale – a response of 4 or greater is considered clinically important – from the mean baseline score of 7.1 was present at week 12 in 56.3% of patients on abrocitinib at 200 mg, in 74.3% at week 16, and in 72.5% at week 48. The proportion of patients achieving this endpoint on 100 mg was 41.6% at week 12, 49.4% at week 16, and 52% at week 48.
Serious treatment-emergent adverse events occurred in 6.1% of JADE EXTEND participants on abrocitinib at 100 mg and 12.8% of those on 200 mg. These events included oral herpes and elevated creatine phosphokinase levels. The sole case of pulmonary embolism that occurred during the study was deemed unrelated to treatment.
“What this is telling me here is there are no signals that we haven’t seen earlier with this drug and with other JAK inhibitors before,” the dermatologist observed. “But I want to see more data. I want to see the overall safety, not just for a year, but for 2, 3, 4, and 5 years.”
Asked by an audience member if nonresponsiveness to one JAK inhibitor predicts nonresponse to others, Dr. Reich speculated that it’s likely to be so. He noted that all three of the JAK inhibitors furthest along in the developmental pipeline for atopic dermatitis – abrocitinib, baricitinib, and upadacitinib – are inhibitors of JAK 1, although baricitinib also targets JAK 2.
“I would think that if you really are a nonresponder to any of these that it will be hard to get a good response with the others. We’re not talking about antibodies here, where there may be different epitopes. The affinity is different, and we have seen that if you have no response to a weak TNF [tumor necrosis factor] inhibitor, you can still have a response to a strong TNF inhibitor. I don’t expect the same here,” according to Dr. Reich.
He reported serving as an adviser to and paid clinical research for Pfizer, which sponsored JADE EXTEND, as well as more than two dozen other pharmaceutical companies.
FROM THE EADV CONGRESS
Topical tapinarof effective in pivotal psoriasis trials
in two identical pivotal phase 3, randomized trials, Mark G. Lebwohl, MD, reported at the virtual annual congress of the European Academy of Dermatology and Venereology.

“Tapinarof cream has the potential to be a first-in-class topical therapeutic aryl hydrocarbon receptor modulating agent and will provide physicians and patients with a novel nonsteroidal topical treatment option that’s effective and well tolerated,” predicted Dr. Lebwohl, professor and chair of the department of dermatology at the Icahn School of Medicine at Mount Sinai, New York.
Dermavant Sciences, the company developing topical tapinarof for treatment of atopic dermatitis as well as psoriasis, announced that upon completion of an ongoing long-term extension study the company plans to file for approval of the drug for psoriasis in 2021.
The two pivotal phase 3 trials, PSOARING 1 and PSOARING 2, randomized a total of 1,025 patients with plaque psoriasis to once-daily tapinarof cream 1% or its vehicle. “This was a fairly difficult group of patients,” Dr. Lebwohl said. Roughly 80% had moderate psoriasis as defined by a baseline Physician Global Assessment (PGA) score of 3, with the remainder split evenly between mild and severe disease. Participants averaged 8% body surface area involvement. Body mass index was on average greater than 31 kg/m2.
The primary efficacy endpoint was a PGA score of 0 or 1 – that is, clear or almost clear – plus at least a 2-grade improvement in PGA from baseline at week 12. This was achieved in 35.4% of patients on tapinarof cream once daily in PSOARING 1 and 40.2% in PSOARING 2, compared with 6.0% and 6.3% of vehicle-treated controls, a highly significant difference (both P < .0001).
The prespecified secondary endpoint was a 75% improvement in Psoriasis Area and Severity Index (PASI) score from baseline to week 12. The PASI 75 rates were 36.1% and 47.6% with tapinarof, significantly better than the 10.2% and 6.9% rates in controls.
The most common adverse event associated with tapinarof was folliculitis, which occurred in 20.6% of treated patients in PSOARING 1 and in 15.7% in PSOARING 2. More than 98% of cases were mild or moderate. The folliculitis led to study discontinuation in only 1.8% and 0.9% of subjects in the two trials.
The other noteworthy adverse event was contact dermatitis. It occurred in 3.8% and 4.7% of tapinarof-treated patients, again with low study discontinuation rates of 1.5% and 2.2%.
During the audience discussion, Linda Stein Gold, MD, lead investigator for PSOARING 2, was asked about this folliculitis. She said the mechanism is unclear, as is the best management. She encountered it in patients, didn’t treat it, and it went away on its own. It’s not a bacterial folliculitis; when cultured it invariably proved culture negative, she noted.
The comparative efficacy of tapinarof cream versus the potent and superpotent topical corticosteroids commonly used in the treatment of psoriasis hasn’t been evaluated in head-to-head studies. Her experience and that of the other investigators has been that tapinarof’s efficacy is comparably strong, “but we don’t have the steroid side effects,” said Dr. Stein Gold, director of dermatology clinical research at Henry Ford Health System in Detroit.
In an interview, Dr. Lebwohl said tapinarof, if approved, could help meet a major unmet need for new and better topical therapies for psoriasis.
“You keep hearing about all these biologic agents and small-molecule pills coming out, but the majority of patients still only need topical therapy,” he observed.
Moreover, even when patients with more severe disease achieve a PASI 75 or PASI 90 response with systemic therapy, they usually still need supplemental topical therapy to get them closer to the goal of clear skin.
The superpotent steroids that are the current mainstay of topical therapy come with predictable side effects that dictate a 2- to 4-week limit on their approved use. Also, they’re not supposed to be applied to the face or to intertriginous sites, including the groin, axillae, and under the breasts. In contrast, tapinarof has proved safe and effective in these sensitive areas.
Asked to predict how tapinarof is likely to be used in clinical practice, Dr. Lebwohl replied: “The efficacy was equivalent to strong topical steroids, so I think it could be used first line in place of topical steroids. And in particular, in patients with psoriasis at facial and intertriginous sites, I think an argument can be made for insisting that it be first line.”
He also expects that physicians will end up utilizing tapinarof for a varied group of steroid-responsive dermatoses beyond psoriasis and atopic dermatitis.
“It clearly reduces inflammation, which is why I would expect it would work well for those,” the dermatologist said.
The mechanism of action of tapinarof has been worked out. The drug enters the cell and binds to the aryl hydrocarbon receptor, forming a complex that enters the nucleus. There it joins with the aryl hydrocarbon receptor nuclear translocator, which regulates gene expression so as to reduce production of inflammatory cytokines while promoting an increase in skin barrier proteins, which is why tapinarof is also being developed as an atopic dermatitis therapy.
Dr. Lebwohl and Dr. Stein Gold reported receiving research funds from and serving as consultants to Dermavant Sciences as well as numerous other pharmaceutical companies.
in two identical pivotal phase 3, randomized trials, Mark G. Lebwohl, MD, reported at the virtual annual congress of the European Academy of Dermatology and Venereology.
“Tapinarof cream has the potential to be a first-in-class topical therapeutic aryl hydrocarbon receptor modulating agent and will provide physicians and patients with a novel nonsteroidal topical treatment option that’s effective and well tolerated,” predicted Dr. Lebwohl, professor and chair of the department of dermatology at the Icahn School of Medicine at Mount Sinai, New York.
Dermavant Sciences, the company developing topical tapinarof for treatment of atopic dermatitis as well as psoriasis, announced that upon completion of an ongoing long-term extension study the company plans to file for approval of the drug for psoriasis in 2021.
The two pivotal phase 3 trials, PSOARING 1 and PSOARING 2, randomized a total of 1,025 patients with plaque psoriasis to once-daily tapinarof cream 1% or its vehicle. “This was a fairly difficult group of patients,” Dr. Lebwohl said. Roughly 80% had moderate psoriasis as defined by a baseline Physician Global Assessment (PGA) score of 3, with the remainder split evenly between mild and severe disease. Participants averaged 8% body surface area involvement. Body mass index was on average greater than 31 kg/m2.
The primary efficacy endpoint was a PGA score of 0 or 1 – that is, clear or almost clear – plus at least a 2-grade improvement in PGA from baseline at week 12. This was achieved in 35.4% of patients on tapinarof cream once daily in PSOARING 1 and 40.2% in PSOARING 2, compared with 6.0% and 6.3% of vehicle-treated controls, a highly significant difference (both P < .0001).
The prespecified secondary endpoint was a 75% improvement in Psoriasis Area and Severity Index (PASI) score from baseline to week 12. The PASI 75 rates were 36.1% and 47.6% with tapinarof, significantly better than the 10.2% and 6.9% rates in controls.
The most common adverse event associated with tapinarof was folliculitis, which occurred in 20.6% of treated patients in PSOARING 1 and in 15.7% in PSOARING 2. More than 98% of cases were mild or moderate. The folliculitis led to study discontinuation in only 1.8% and 0.9% of subjects in the two trials.
The other noteworthy adverse event was contact dermatitis. It occurred in 3.8% and 4.7% of tapinarof-treated patients, again with low study discontinuation rates of 1.5% and 2.2%.
During the audience discussion, Linda Stein Gold, MD, lead investigator for PSOARING 2, was asked about this folliculitis. She said the mechanism is unclear, as is the best management. She encountered it in patients, didn’t treat it, and it went away on its own. It’s not a bacterial folliculitis; when cultured it invariably proved culture negative, she noted.
The comparative efficacy of tapinarof cream versus the potent and superpotent topical corticosteroids commonly used in the treatment of psoriasis hasn’t been evaluated in head-to-head studies. Her experience and that of the other investigators has been that tapinarof’s efficacy is comparably strong, “but we don’t have the steroid side effects,” said Dr. Stein Gold, director of dermatology clinical research at Henry Ford Health System in Detroit.
In an interview, Dr. Lebwohl said tapinarof, if approved, could help meet a major unmet need for new and better topical therapies for psoriasis.
“You keep hearing about all these biologic agents and small-molecule pills coming out, but the majority of patients still only need topical therapy,” he observed.
Moreover, even when patients with more severe disease achieve a PASI 75 or PASI 90 response with systemic therapy, they usually still need supplemental topical therapy to get them closer to the goal of clear skin.
The superpotent steroids that are the current mainstay of topical therapy come with predictable side effects that dictate a 2- to 4-week limit on their approved use. Also, they’re not supposed to be applied to the face or to intertriginous sites, including the groin, axillae, and under the breasts. In contrast, tapinarof has proved safe and effective in these sensitive areas.
Asked to predict how tapinarof is likely to be used in clinical practice, Dr. Lebwohl replied: “The efficacy was equivalent to strong topical steroids, so I think it could be used first line in place of topical steroids. And in particular, in patients with psoriasis at facial and intertriginous sites, I think an argument can be made for insisting that it be first line.”
He also expects that physicians will end up utilizing tapinarof for a varied group of steroid-responsive dermatoses beyond psoriasis and atopic dermatitis.
“It clearly reduces inflammation, which is why I would expect it would work well for those,” the dermatologist said.
The mechanism of action of tapinarof has been worked out. The drug enters the cell and binds to the aryl hydrocarbon receptor, forming a complex that enters the nucleus. There it joins with the aryl hydrocarbon receptor nuclear translocator, which regulates gene expression so as to reduce production of inflammatory cytokines while promoting an increase in skin barrier proteins, which is why tapinarof is also being developed as an atopic dermatitis therapy.
Dr. Lebwohl and Dr. Stein Gold reported receiving research funds from and serving as consultants to Dermavant Sciences as well as numerous other pharmaceutical companies.
in two identical pivotal phase 3, randomized trials, Mark G. Lebwohl, MD, reported at the virtual annual congress of the European Academy of Dermatology and Venereology.
“Tapinarof cream has the potential to be a first-in-class topical therapeutic aryl hydrocarbon receptor modulating agent and will provide physicians and patients with a novel nonsteroidal topical treatment option that’s effective and well tolerated,” predicted Dr. Lebwohl, professor and chair of the department of dermatology at the Icahn School of Medicine at Mount Sinai, New York.
Dermavant Sciences, the company developing topical tapinarof for treatment of atopic dermatitis as well as psoriasis, announced that upon completion of an ongoing long-term extension study the company plans to file for approval of the drug for psoriasis in 2021.
The two pivotal phase 3 trials, PSOARING 1 and PSOARING 2, randomized a total of 1,025 patients with plaque psoriasis to once-daily tapinarof cream 1% or its vehicle. “This was a fairly difficult group of patients,” Dr. Lebwohl said. Roughly 80% had moderate psoriasis as defined by a baseline Physician Global Assessment (PGA) score of 3, with the remainder split evenly between mild and severe disease. Participants averaged 8% body surface area involvement. Body mass index was on average greater than 31 kg/m2.
The primary efficacy endpoint was a PGA score of 0 or 1 – that is, clear or almost clear – plus at least a 2-grade improvement in PGA from baseline at week 12. This was achieved in 35.4% of patients on tapinarof cream once daily in PSOARING 1 and 40.2% in PSOARING 2, compared with 6.0% and 6.3% of vehicle-treated controls, a highly significant difference (both P < .0001).
The prespecified secondary endpoint was a 75% improvement in Psoriasis Area and Severity Index (PASI) score from baseline to week 12. The PASI 75 rates were 36.1% and 47.6% with tapinarof, significantly better than the 10.2% and 6.9% rates in controls.
The most common adverse event associated with tapinarof was folliculitis, which occurred in 20.6% of treated patients in PSOARING 1 and in 15.7% in PSOARING 2. More than 98% of cases were mild or moderate. The folliculitis led to study discontinuation in only 1.8% and 0.9% of subjects in the two trials.
The other noteworthy adverse event was contact dermatitis. It occurred in 3.8% and 4.7% of tapinarof-treated patients, again with low study discontinuation rates of 1.5% and 2.2%.
During the audience discussion, Linda Stein Gold, MD, lead investigator for PSOARING 2, was asked about this folliculitis. She said the mechanism is unclear, as is the best management. She encountered it in patients, didn’t treat it, and it went away on its own. It’s not a bacterial folliculitis; when cultured it invariably proved culture negative, she noted.
The comparative efficacy of tapinarof cream versus the potent and superpotent topical corticosteroids commonly used in the treatment of psoriasis hasn’t been evaluated in head-to-head studies. Her experience and that of the other investigators has been that tapinarof’s efficacy is comparably strong, “but we don’t have the steroid side effects,” said Dr. Stein Gold, director of dermatology clinical research at Henry Ford Health System in Detroit.
In an interview, Dr. Lebwohl said tapinarof, if approved, could help meet a major unmet need for new and better topical therapies for psoriasis.
“You keep hearing about all these biologic agents and small-molecule pills coming out, but the majority of patients still only need topical therapy,” he observed.
Moreover, even when patients with more severe disease achieve a PASI 75 or PASI 90 response with systemic therapy, they usually still need supplemental topical therapy to get them closer to the goal of clear skin.
The superpotent steroids that are the current mainstay of topical therapy come with predictable side effects that dictate a 2- to 4-week limit on their approved use. Also, they’re not supposed to be applied to the face or to intertriginous sites, including the groin, axillae, and under the breasts. In contrast, tapinarof has proved safe and effective in these sensitive areas.
Asked to predict how tapinarof is likely to be used in clinical practice, Dr. Lebwohl replied: “The efficacy was equivalent to strong topical steroids, so I think it could be used first line in place of topical steroids. And in particular, in patients with psoriasis at facial and intertriginous sites, I think an argument can be made for insisting that it be first line.”
He also expects that physicians will end up utilizing tapinarof for a varied group of steroid-responsive dermatoses beyond psoriasis and atopic dermatitis.
“It clearly reduces inflammation, which is why I would expect it would work well for those,” the dermatologist said.
The mechanism of action of tapinarof has been worked out. The drug enters the cell and binds to the aryl hydrocarbon receptor, forming a complex that enters the nucleus. There it joins with the aryl hydrocarbon receptor nuclear translocator, which regulates gene expression so as to reduce production of inflammatory cytokines while promoting an increase in skin barrier proteins, which is why tapinarof is also being developed as an atopic dermatitis therapy.
Dr. Lebwohl and Dr. Stein Gold reported receiving research funds from and serving as consultants to Dermavant Sciences as well as numerous other pharmaceutical companies.
FROM THE EADV CONGRESS

Who’s at risk for depression on isotretinoin?
A Sanaa Butt, MD, reported at the virtual annual congress of the European Academy of Dermatology and Venereology.
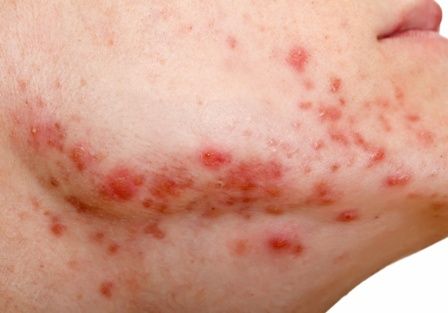
This was, however, the sole identifiable risk factor for treatment-limiting depressive symptoms in acne patients on isotretinoin in the study of 3,151 consecutive acne patients taking isotretinoin. There was no significant difference between those who did or did not develop depression on the oral retinoid in terms of age, gender, or daily dose of the drug at the time it was discontinued.
“Depressive symptoms occurred at any time from the date of initiation of isotretinoin up to 6 months into therapy, with no identifiable peak time period,” said Dr. Butt, a dermatologist with the U.K. National Health Service Tayside district at Ninewells Hospital, Dundee, Scotland. “Lower doses appear not to be protective,” she added.
The Tayside district has a catchment of roughly 450,000 people. The local population tends to stay put because Tayside is an economically disadvantaged and remote part of Scotland. There are very few private practice dermatologists in the area, so Dr. Butt and coinvestigators are confident their observational study of NHS patients captured the great majority of isotretinoin users in northern Scotland.
The investigators utilized software to analyze the contents of more than 8,000 digitized letters exchanged between NHS Tayside dermatologists and general practitioners during 2005-2018, zeroing in on 3,151 consecutive patients on isotretinoin for acne and 158 on the drug for other conditions, most often rosacea or folliculitis. They then drilled down further through the letters, electronically searching for key words such as suicide, depression, and anxiety. In this way, they ultimately identified 30 patients who discontinued the drug because they developed depressive symptoms. All 30 were on the drug for acne.
The annual incidence of treatment-limiting depressive mood changes was 0.96%, a figure that remained steady over the 13-year study period, even though prescribing of isotretinoin increased over time. This flat incidence rate effectively rules out the potential for confounding because of assessor bias, especially since many different NHS dermatologists were prescribing the drug, Dr. Butt said.
Half of acne patients prescribed isotretinoin were female and 50% were male. And 15 cases of treatment discontinuation caused by development of depressive symptoms occurred in females, 15 in males. A history of past depressive illness was present in 9.3% of females who started on isotretinoin and in 4.5% of the males. The relative risk of treatment-limiting depressive mood changes was increased 790% among females with a prior history of depressive illness and 440% in males with such a history.
Dr. Butt reported having no financial conflicts regarding her NHS-funded study.
A Sanaa Butt, MD, reported at the virtual annual congress of the European Academy of Dermatology and Venereology.
This was, however, the sole identifiable risk factor for treatment-limiting depressive symptoms in acne patients on isotretinoin in the study of 3,151 consecutive acne patients taking isotretinoin. There was no significant difference between those who did or did not develop depression on the oral retinoid in terms of age, gender, or daily dose of the drug at the time it was discontinued.
“Depressive symptoms occurred at any time from the date of initiation of isotretinoin up to 6 months into therapy, with no identifiable peak time period,” said Dr. Butt, a dermatologist with the U.K. National Health Service Tayside district at Ninewells Hospital, Dundee, Scotland. “Lower doses appear not to be protective,” she added.
The Tayside district has a catchment of roughly 450,000 people. The local population tends to stay put because Tayside is an economically disadvantaged and remote part of Scotland. There are very few private practice dermatologists in the area, so Dr. Butt and coinvestigators are confident their observational study of NHS patients captured the great majority of isotretinoin users in northern Scotland.
The investigators utilized software to analyze the contents of more than 8,000 digitized letters exchanged between NHS Tayside dermatologists and general practitioners during 2005-2018, zeroing in on 3,151 consecutive patients on isotretinoin for acne and 158 on the drug for other conditions, most often rosacea or folliculitis. They then drilled down further through the letters, electronically searching for key words such as suicide, depression, and anxiety. In this way, they ultimately identified 30 patients who discontinued the drug because they developed depressive symptoms. All 30 were on the drug for acne.
The annual incidence of treatment-limiting depressive mood changes was 0.96%, a figure that remained steady over the 13-year study period, even though prescribing of isotretinoin increased over time. This flat incidence rate effectively rules out the potential for confounding because of assessor bias, especially since many different NHS dermatologists were prescribing the drug, Dr. Butt said.
Half of acne patients prescribed isotretinoin were female and 50% were male. And 15 cases of treatment discontinuation caused by development of depressive symptoms occurred in females, 15 in males. A history of past depressive illness was present in 9.3% of females who started on isotretinoin and in 4.5% of the males. The relative risk of treatment-limiting depressive mood changes was increased 790% among females with a prior history of depressive illness and 440% in males with such a history.
Dr. Butt reported having no financial conflicts regarding her NHS-funded study.
A Sanaa Butt, MD, reported at the virtual annual congress of the European Academy of Dermatology and Venereology.
This was, however, the sole identifiable risk factor for treatment-limiting depressive symptoms in acne patients on isotretinoin in the study of 3,151 consecutive acne patients taking isotretinoin. There was no significant difference between those who did or did not develop depression on the oral retinoid in terms of age, gender, or daily dose of the drug at the time it was discontinued.
“Depressive symptoms occurred at any time from the date of initiation of isotretinoin up to 6 months into therapy, with no identifiable peak time period,” said Dr. Butt, a dermatologist with the U.K. National Health Service Tayside district at Ninewells Hospital, Dundee, Scotland. “Lower doses appear not to be protective,” she added.
The Tayside district has a catchment of roughly 450,000 people. The local population tends to stay put because Tayside is an economically disadvantaged and remote part of Scotland. There are very few private practice dermatologists in the area, so Dr. Butt and coinvestigators are confident their observational study of NHS patients captured the great majority of isotretinoin users in northern Scotland.
The investigators utilized software to analyze the contents of more than 8,000 digitized letters exchanged between NHS Tayside dermatologists and general practitioners during 2005-2018, zeroing in on 3,151 consecutive patients on isotretinoin for acne and 158 on the drug for other conditions, most often rosacea or folliculitis. They then drilled down further through the letters, electronically searching for key words such as suicide, depression, and anxiety. In this way, they ultimately identified 30 patients who discontinued the drug because they developed depressive symptoms. All 30 were on the drug for acne.
The annual incidence of treatment-limiting depressive mood changes was 0.96%, a figure that remained steady over the 13-year study period, even though prescribing of isotretinoin increased over time. This flat incidence rate effectively rules out the potential for confounding because of assessor bias, especially since many different NHS dermatologists were prescribing the drug, Dr. Butt said.
Half of acne patients prescribed isotretinoin were female and 50% were male. And 15 cases of treatment discontinuation caused by development of depressive symptoms occurred in females, 15 in males. A history of past depressive illness was present in 9.3% of females who started on isotretinoin and in 4.5% of the males. The relative risk of treatment-limiting depressive mood changes was increased 790% among females with a prior history of depressive illness and 440% in males with such a history.
Dr. Butt reported having no financial conflicts regarding her NHS-funded study.
FROM THE EADV CONGRESS
Survey finds European dermatologists unhappy with pandemic teledermatology experience
intensely, according to the findings of a survey presented at the virtual annual congress of the European Academy of Dermatology and Venereology.
“The results of our survey clearly show 7 out of 10 participating dermatologists declared that they were not happy with teledermatology, and most of them declared that they were not at all happy,” according to Mariano Suppa, MD, PhD, of the department of dermatology and venereology, Free University of Brussels.
“It was very interesting: it was not just about the lack of a good quality of consultation, but was also related to some extent to a lack of respect from some patients, and also a lack of empathy. The majority of survey respondents felt [attacked] by their own patients because they were proposing teledermatology. So, yes, we were forced to go to teledermatology, and I think we will be again to some extent, but clearly we’re not happy about it,” he elaborated in response to a question from session chair Brigitte Dreno, MD, professor of dermatology and vice dean of the faculty of medicine at the University of Nantes (France).
The survey, conducted by the EADV communication committee, assessed the pandemic’s impact on European dermatologists’ professional practices and personal lives through 30 brief questions, with space at the end for additional open-ended comments. In the comments section, many dermatologists vented about their income loss, the disorganized response to round one of the pandemic, and most of all about teledermatology. Common complaints were that teledermatology required a huge consumption of energy and constituted a major intrusion upon the physicians’ personal lives. And then there was the common theme of unkind treatment by some patients.
The survey was sent twice in June 2020 to more than 4,800 EADV members. It was completed by 490 dermatologists from 39 countries. Dr. Suppa attributed the low response rate to physician weariness of the topic due to saturation news media coverage of the pandemic.
Sixty-nine percent of responding dermatologists were women. Fifty-two percent of participants were over age 50, 81% lived in a city, and 53% worked in a university or public hospital or clinic. Twelve percent lived alone.
Impact on professional practice
Many European dermatologists were on the front lines in dealing with the first wave of COVID-19. Twenty-eight percent worked in a COVID-19 unit. Forty-eight percent of dermatologists performed COVID-19 tests, and those who didn’t either had no patient contact or couldn’t get test kits. Thirty-five percent of dermatologists saw patients who presented with skin signs of COVID-19. Four percent of survey respondents became infected.
Seventy percent rescheduled or canceled all or most patient appointments. Clinical care was prioritized: during the peak of the pandemic, 76% of dermatologists saw only urgent cases – mostly potentially serious rashes – and dermato-oncology patients. Seventy-six percent of dermatologists performed teledermatology, although by June 60% of respondents reported seeing at least three-quarters of their patients face-to-face.
Twenty-three percent of dermatologists reported having lost most or all of their income during March through June, and another 26% lost about half.
Impact on dermatologists’ personal lives
About half of survey respondents reported feeling stressed, and a similar percentage checked the box marked ‘anxiety.’ Nine percent reported depressive symptoms, 15% mentioned feeling anger, 17% uselessness, and 2% admitted suicidal ideation. But 30% of dermatologists reported experiencing no negative psychological effects whatsoever stemming from the pandemic.
Sixteen percent of dermatologists reported drinking more alcohol during sequestration.
But respondents cited positive effects as well: a renewed appreciation of the importance of time, and enjoyment of the additional time spent with family and alone. Many dermatologists relished the opportunity to spend more time cooking, reading literature, doing research, listening to or playing music, and practicing yoga or meditation. And dermatologists took solace and pride in being members of the vital medical community.
Dr. Dreno asked if the survey revealed evidence of underdiagnosis and undertreatment of dermatologic diseases during the pandemic. Dr. Suppa replied that the survey didn’t address that issue, but it’s his personal opinion that this was no doubt the case. Roughly one-quarter of dermatologists canceled all appointments, and when dermatology clinics became filled beginning in June, he and his colleagues saw a number of cases of delayed-diagnosis advanced skin cancer.
“I think that the diseases that were really penalized were the chronic inflammatory diseases, such as psoriasis, hidradenitis suppurativa, and also atopic dermatitis. We were doing a lot of telephone consultations for those patients at that time, and we saw in June that for those particular patients there was an unmet need in the pandemic because some of them really needed to have been seen. I think this is a lesson we should learn for the second wave that we’re unfortunately facing right now: We need to adopt restrictive measures to avoid spreading the pandemic, yes, for sure, but we need to keep in mind that there is not just COVID-19, but also other important diseases,” Dr. Suppa said.
A second EADV survey will be performed during the fall/winter wave of the pandemic.
Dr. Suppa reported having no financial conflicts regarding the EADV-funded survey.
SOURCE: Suppa M. EADV 2020. Presentation D3T03.4D
intensely, according to the findings of a survey presented at the virtual annual congress of the European Academy of Dermatology and Venereology.
“The results of our survey clearly show 7 out of 10 participating dermatologists declared that they were not happy with teledermatology, and most of them declared that they were not at all happy,” according to Mariano Suppa, MD, PhD, of the department of dermatology and venereology, Free University of Brussels.
“It was very interesting: it was not just about the lack of a good quality of consultation, but was also related to some extent to a lack of respect from some patients, and also a lack of empathy. The majority of survey respondents felt [attacked] by their own patients because they were proposing teledermatology. So, yes, we were forced to go to teledermatology, and I think we will be again to some extent, but clearly we’re not happy about it,” he elaborated in response to a question from session chair Brigitte Dreno, MD, professor of dermatology and vice dean of the faculty of medicine at the University of Nantes (France).
The survey, conducted by the EADV communication committee, assessed the pandemic’s impact on European dermatologists’ professional practices and personal lives through 30 brief questions, with space at the end for additional open-ended comments. In the comments section, many dermatologists vented about their income loss, the disorganized response to round one of the pandemic, and most of all about teledermatology. Common complaints were that teledermatology required a huge consumption of energy and constituted a major intrusion upon the physicians’ personal lives. And then there was the common theme of unkind treatment by some patients.
The survey was sent twice in June 2020 to more than 4,800 EADV members. It was completed by 490 dermatologists from 39 countries. Dr. Suppa attributed the low response rate to physician weariness of the topic due to saturation news media coverage of the pandemic.
Sixty-nine percent of responding dermatologists were women. Fifty-two percent of participants were over age 50, 81% lived in a city, and 53% worked in a university or public hospital or clinic. Twelve percent lived alone.
Impact on professional practice
Many European dermatologists were on the front lines in dealing with the first wave of COVID-19. Twenty-eight percent worked in a COVID-19 unit. Forty-eight percent of dermatologists performed COVID-19 tests, and those who didn’t either had no patient contact or couldn’t get test kits. Thirty-five percent of dermatologists saw patients who presented with skin signs of COVID-19. Four percent of survey respondents became infected.
Seventy percent rescheduled or canceled all or most patient appointments. Clinical care was prioritized: during the peak of the pandemic, 76% of dermatologists saw only urgent cases – mostly potentially serious rashes – and dermato-oncology patients. Seventy-six percent of dermatologists performed teledermatology, although by June 60% of respondents reported seeing at least three-quarters of their patients face-to-face.
Twenty-three percent of dermatologists reported having lost most or all of their income during March through June, and another 26% lost about half.
Impact on dermatologists’ personal lives
About half of survey respondents reported feeling stressed, and a similar percentage checked the box marked ‘anxiety.’ Nine percent reported depressive symptoms, 15% mentioned feeling anger, 17% uselessness, and 2% admitted suicidal ideation. But 30% of dermatologists reported experiencing no negative psychological effects whatsoever stemming from the pandemic.
Sixteen percent of dermatologists reported drinking more alcohol during sequestration.
But respondents cited positive effects as well: a renewed appreciation of the importance of time, and enjoyment of the additional time spent with family and alone. Many dermatologists relished the opportunity to spend more time cooking, reading literature, doing research, listening to or playing music, and practicing yoga or meditation. And dermatologists took solace and pride in being members of the vital medical community.
Dr. Dreno asked if the survey revealed evidence of underdiagnosis and undertreatment of dermatologic diseases during the pandemic. Dr. Suppa replied that the survey didn’t address that issue, but it’s his personal opinion that this was no doubt the case. Roughly one-quarter of dermatologists canceled all appointments, and when dermatology clinics became filled beginning in June, he and his colleagues saw a number of cases of delayed-diagnosis advanced skin cancer.
“I think that the diseases that were really penalized were the chronic inflammatory diseases, such as psoriasis, hidradenitis suppurativa, and also atopic dermatitis. We were doing a lot of telephone consultations for those patients at that time, and we saw in June that for those particular patients there was an unmet need in the pandemic because some of them really needed to have been seen. I think this is a lesson we should learn for the second wave that we’re unfortunately facing right now: We need to adopt restrictive measures to avoid spreading the pandemic, yes, for sure, but we need to keep in mind that there is not just COVID-19, but also other important diseases,” Dr. Suppa said.
A second EADV survey will be performed during the fall/winter wave of the pandemic.
Dr. Suppa reported having no financial conflicts regarding the EADV-funded survey.
SOURCE: Suppa M. EADV 2020. Presentation D3T03.4D
intensely, according to the findings of a survey presented at the virtual annual congress of the European Academy of Dermatology and Venereology.
“The results of our survey clearly show 7 out of 10 participating dermatologists declared that they were not happy with teledermatology, and most of them declared that they were not at all happy,” according to Mariano Suppa, MD, PhD, of the department of dermatology and venereology, Free University of Brussels.
“It was very interesting: it was not just about the lack of a good quality of consultation, but was also related to some extent to a lack of respect from some patients, and also a lack of empathy. The majority of survey respondents felt [attacked] by their own patients because they were proposing teledermatology. So, yes, we were forced to go to teledermatology, and I think we will be again to some extent, but clearly we’re not happy about it,” he elaborated in response to a question from session chair Brigitte Dreno, MD, professor of dermatology and vice dean of the faculty of medicine at the University of Nantes (France).
The survey, conducted by the EADV communication committee, assessed the pandemic’s impact on European dermatologists’ professional practices and personal lives through 30 brief questions, with space at the end for additional open-ended comments. In the comments section, many dermatologists vented about their income loss, the disorganized response to round one of the pandemic, and most of all about teledermatology. Common complaints were that teledermatology required a huge consumption of energy and constituted a major intrusion upon the physicians’ personal lives. And then there was the common theme of unkind treatment by some patients.
The survey was sent twice in June 2020 to more than 4,800 EADV members. It was completed by 490 dermatologists from 39 countries. Dr. Suppa attributed the low response rate to physician weariness of the topic due to saturation news media coverage of the pandemic.
Sixty-nine percent of responding dermatologists were women. Fifty-two percent of participants were over age 50, 81% lived in a city, and 53% worked in a university or public hospital or clinic. Twelve percent lived alone.
Impact on professional practice
Many European dermatologists were on the front lines in dealing with the first wave of COVID-19. Twenty-eight percent worked in a COVID-19 unit. Forty-eight percent of dermatologists performed COVID-19 tests, and those who didn’t either had no patient contact or couldn’t get test kits. Thirty-five percent of dermatologists saw patients who presented with skin signs of COVID-19. Four percent of survey respondents became infected.
Seventy percent rescheduled or canceled all or most patient appointments. Clinical care was prioritized: during the peak of the pandemic, 76% of dermatologists saw only urgent cases – mostly potentially serious rashes – and dermato-oncology patients. Seventy-six percent of dermatologists performed teledermatology, although by June 60% of respondents reported seeing at least three-quarters of their patients face-to-face.
Twenty-three percent of dermatologists reported having lost most or all of their income during March through June, and another 26% lost about half.
Impact on dermatologists’ personal lives
About half of survey respondents reported feeling stressed, and a similar percentage checked the box marked ‘anxiety.’ Nine percent reported depressive symptoms, 15% mentioned feeling anger, 17% uselessness, and 2% admitted suicidal ideation. But 30% of dermatologists reported experiencing no negative psychological effects whatsoever stemming from the pandemic.
Sixteen percent of dermatologists reported drinking more alcohol during sequestration.
But respondents cited positive effects as well: a renewed appreciation of the importance of time, and enjoyment of the additional time spent with family and alone. Many dermatologists relished the opportunity to spend more time cooking, reading literature, doing research, listening to or playing music, and practicing yoga or meditation. And dermatologists took solace and pride in being members of the vital medical community.
Dr. Dreno asked if the survey revealed evidence of underdiagnosis and undertreatment of dermatologic diseases during the pandemic. Dr. Suppa replied that the survey didn’t address that issue, but it’s his personal opinion that this was no doubt the case. Roughly one-quarter of dermatologists canceled all appointments, and when dermatology clinics became filled beginning in June, he and his colleagues saw a number of cases of delayed-diagnosis advanced skin cancer.
“I think that the diseases that were really penalized were the chronic inflammatory diseases, such as psoriasis, hidradenitis suppurativa, and also atopic dermatitis. We were doing a lot of telephone consultations for those patients at that time, and we saw in June that for those particular patients there was an unmet need in the pandemic because some of them really needed to have been seen. I think this is a lesson we should learn for the second wave that we’re unfortunately facing right now: We need to adopt restrictive measures to avoid spreading the pandemic, yes, for sure, but we need to keep in mind that there is not just COVID-19, but also other important diseases,” Dr. Suppa said.
A second EADV survey will be performed during the fall/winter wave of the pandemic.
Dr. Suppa reported having no financial conflicts regarding the EADV-funded survey.
SOURCE: Suppa M. EADV 2020. Presentation D3T03.4D
FROM THE EADV CONGRESS
Birch bark derivative gel found effective for EB, in phase 3 study
A . The results come from the largest double-blind, randomized trial performed in this patient population.

More than 41% of EB target wounds that were treated with Oleogel-S10 healed within 45 days, compared with about 29% of target wounds treated with placebo, in the EASE phase 3 trial, conducted at 58 sites in 28 countries.
A group of rare genetic disorders, EB “is described as the worst disease you’ve never heard of,” explained lead investigator Dedee Murrell, MD, director of dermatology, St. George Hospital at the University of New South Wales, Sydney. “It starts in children and is like having burns that heal with scars, and no treatment has been approved for it” by the Food and Drug Administration.
“This is the first large clinical trial with placebo of a topical treatment that’s worked for this terrible disease,” Dr. Murrell said in an interview. She noted that standard EB treatment currently consists of applying nonstick dressings to wounds to protect skin from trauma and infection.
Dr. Murrell, who has focused her work on EB patients since 1990, presented the findings at the virtual annual Congress of the European Academy of Dermatology and Venereology.
The trial enrolled 223 patients (average age, 12 years, but ages ranged to 81 years) with three types of EB, including dystrophic and junctional EB and Kindler syndrome. For each participant, a target wound was selected for use as the primary efficacy endpoint. Those wounds had a partial thickness of between 10 cm2 and 50 cm2 and lasted between 21 days and 9 months. Patients were stratified into groups depending on type of EB and size of target wound.
Participants were randomly assigned to receive either Oleogel-S10 (n = 109) or placebo (n = 114). All applied the blinded-study gel to all their wounds at least every 4 days at the time dressings were changed.
The primary endpoint was the percentage of patients whose target wounds completely closed within 45 days. Key secondary endpoints included time to wound healing and percentage of target wounds that healed within 90 days of treatment; incidence and severity of target wound infection; change in total body wound burden, as measured by the Epidermolysis Bullosa Disease Activity and Scarring Index skin activity subscore; change in itching, as measured by the Itch Man Scale and the Leuven Itch Scale; and adverse events.
Nearly 92% of patients who were treated with Oleogel-S10 completed the double-blind phase of the trial, compared with nearly 87% who received placebo. As noted, the primary endpoint was met, with 41.3% of Oleogel-S10 patients achieving target wound closure within 45 days, compared with 28.9% of the patients who received placebo (P = .013).
But the difference in time to wound healing by day 90 between the two patient groups was not statistically significant (P = .302), with 50.5% of Oleogel-S10 patients achieving wound closure vs. 43.9% of control patients.
Target-wound infection occurred in eight participants, including three who used Oleogel-S10 and five who received placebo; all moderate or severe infections occurred in patients who received placebo. Total wound burden was reduced to a greater extent among Oleogel-S10 patients by day 60, but there was no apparent difference at day 90.
Both treatment groups reported qualitative improvements in itch, with no significant differences between groups. The prevalence of adverse events was also similar between groups (Oleogel-S10, 81.7%; placebo, 80.7%). The most frequently reported adverse events among Oleogel-S10 patients, compared with patients who received placebo, were wound complications, pyrexia, wound infection, pruritus, and anemia; only 4.5% of adverse events were deemed severe.
Dr. Murrell said that, on the basis of the trial results, she expects the FDA to fast-track approval of Oleogel-S10, which contains triterpene extract and sunflower oil.
The gel is “a treatment patients will be able to put under their dressings, added to normal treatment, which will accelerate their wound healing, with no significant increase in any side effects,” she added.
Jemima Mellerio, MD, of St. Thomas’ Hospital in London who sees about 400 EB patients each year, agreed with Dr. Murrell that the results are “very exciting.” Dr. Mellerio was not involved in the study.
“Practicing dermatologists seeing people with EB will have something to offer that appears to speed up wound healing in chronic wounds,” Dr. Mellerio said in an interview. “It’s a positive option rather than just supportive treatment, something that makes a difference to the natural history of wounds.”
She said the trial’s biggest strength was including “such a large cohort of patients.
“It’s extremely difficult to do that kind of study, especially with a placebo-controlled arm and especially in a rare disease,” Dr. Mellerio said. “If you think about the product itself, it’s easy to apply, so it’s not particularly onerous for people to add to their daily regimen of dressings.”
The study was funded by Amryt Pharma. Dr. Murrell is an advisory board member for Amryt Pharma. Dr. Mellerio is a consultant for Amryt Pharma.
A version of this article originally appeared on Medscape.com.
A . The results come from the largest double-blind, randomized trial performed in this patient population.
More than 41% of EB target wounds that were treated with Oleogel-S10 healed within 45 days, compared with about 29% of target wounds treated with placebo, in the EASE phase 3 trial, conducted at 58 sites in 28 countries.
A group of rare genetic disorders, EB “is described as the worst disease you’ve never heard of,” explained lead investigator Dedee Murrell, MD, director of dermatology, St. George Hospital at the University of New South Wales, Sydney. “It starts in children and is like having burns that heal with scars, and no treatment has been approved for it” by the Food and Drug Administration.
“This is the first large clinical trial with placebo of a topical treatment that’s worked for this terrible disease,” Dr. Murrell said in an interview. She noted that standard EB treatment currently consists of applying nonstick dressings to wounds to protect skin from trauma and infection.
Dr. Murrell, who has focused her work on EB patients since 1990, presented the findings at the virtual annual Congress of the European Academy of Dermatology and Venereology.
The trial enrolled 223 patients (average age, 12 years, but ages ranged to 81 years) with three types of EB, including dystrophic and junctional EB and Kindler syndrome. For each participant, a target wound was selected for use as the primary efficacy endpoint. Those wounds had a partial thickness of between 10 cm2 and 50 cm2 and lasted between 21 days and 9 months. Patients were stratified into groups depending on type of EB and size of target wound.
Participants were randomly assigned to receive either Oleogel-S10 (n = 109) or placebo (n = 114). All applied the blinded-study gel to all their wounds at least every 4 days at the time dressings were changed.
The primary endpoint was the percentage of patients whose target wounds completely closed within 45 days. Key secondary endpoints included time to wound healing and percentage of target wounds that healed within 90 days of treatment; incidence and severity of target wound infection; change in total body wound burden, as measured by the Epidermolysis Bullosa Disease Activity and Scarring Index skin activity subscore; change in itching, as measured by the Itch Man Scale and the Leuven Itch Scale; and adverse events.
Nearly 92% of patients who were treated with Oleogel-S10 completed the double-blind phase of the trial, compared with nearly 87% who received placebo. As noted, the primary endpoint was met, with 41.3% of Oleogel-S10 patients achieving target wound closure within 45 days, compared with 28.9% of the patients who received placebo (P = .013).
But the difference in time to wound healing by day 90 between the two patient groups was not statistically significant (P = .302), with 50.5% of Oleogel-S10 patients achieving wound closure vs. 43.9% of control patients.
Target-wound infection occurred in eight participants, including three who used Oleogel-S10 and five who received placebo; all moderate or severe infections occurred in patients who received placebo. Total wound burden was reduced to a greater extent among Oleogel-S10 patients by day 60, but there was no apparent difference at day 90.
Both treatment groups reported qualitative improvements in itch, with no significant differences between groups. The prevalence of adverse events was also similar between groups (Oleogel-S10, 81.7%; placebo, 80.7%). The most frequently reported adverse events among Oleogel-S10 patients, compared with patients who received placebo, were wound complications, pyrexia, wound infection, pruritus, and anemia; only 4.5% of adverse events were deemed severe.
Dr. Murrell said that, on the basis of the trial results, she expects the FDA to fast-track approval of Oleogel-S10, which contains triterpene extract and sunflower oil.
The gel is “a treatment patients will be able to put under their dressings, added to normal treatment, which will accelerate their wound healing, with no significant increase in any side effects,” she added.
Jemima Mellerio, MD, of St. Thomas’ Hospital in London who sees about 400 EB patients each year, agreed with Dr. Murrell that the results are “very exciting.” Dr. Mellerio was not involved in the study.
“Practicing dermatologists seeing people with EB will have something to offer that appears to speed up wound healing in chronic wounds,” Dr. Mellerio said in an interview. “It’s a positive option rather than just supportive treatment, something that makes a difference to the natural history of wounds.”
She said the trial’s biggest strength was including “such a large cohort of patients.
“It’s extremely difficult to do that kind of study, especially with a placebo-controlled arm and especially in a rare disease,” Dr. Mellerio said. “If you think about the product itself, it’s easy to apply, so it’s not particularly onerous for people to add to their daily regimen of dressings.”
The study was funded by Amryt Pharma. Dr. Murrell is an advisory board member for Amryt Pharma. Dr. Mellerio is a consultant for Amryt Pharma.
A version of this article originally appeared on Medscape.com.
A . The results come from the largest double-blind, randomized trial performed in this patient population.
More than 41% of EB target wounds that were treated with Oleogel-S10 healed within 45 days, compared with about 29% of target wounds treated with placebo, in the EASE phase 3 trial, conducted at 58 sites in 28 countries.
A group of rare genetic disorders, EB “is described as the worst disease you’ve never heard of,” explained lead investigator Dedee Murrell, MD, director of dermatology, St. George Hospital at the University of New South Wales, Sydney. “It starts in children and is like having burns that heal with scars, and no treatment has been approved for it” by the Food and Drug Administration.
“This is the first large clinical trial with placebo of a topical treatment that’s worked for this terrible disease,” Dr. Murrell said in an interview. She noted that standard EB treatment currently consists of applying nonstick dressings to wounds to protect skin from trauma and infection.
Dr. Murrell, who has focused her work on EB patients since 1990, presented the findings at the virtual annual Congress of the European Academy of Dermatology and Venereology.
The trial enrolled 223 patients (average age, 12 years, but ages ranged to 81 years) with three types of EB, including dystrophic and junctional EB and Kindler syndrome. For each participant, a target wound was selected for use as the primary efficacy endpoint. Those wounds had a partial thickness of between 10 cm2 and 50 cm2 and lasted between 21 days and 9 months. Patients were stratified into groups depending on type of EB and size of target wound.
Participants were randomly assigned to receive either Oleogel-S10 (n = 109) or placebo (n = 114). All applied the blinded-study gel to all their wounds at least every 4 days at the time dressings were changed.
The primary endpoint was the percentage of patients whose target wounds completely closed within 45 days. Key secondary endpoints included time to wound healing and percentage of target wounds that healed within 90 days of treatment; incidence and severity of target wound infection; change in total body wound burden, as measured by the Epidermolysis Bullosa Disease Activity and Scarring Index skin activity subscore; change in itching, as measured by the Itch Man Scale and the Leuven Itch Scale; and adverse events.
Nearly 92% of patients who were treated with Oleogel-S10 completed the double-blind phase of the trial, compared with nearly 87% who received placebo. As noted, the primary endpoint was met, with 41.3% of Oleogel-S10 patients achieving target wound closure within 45 days, compared with 28.9% of the patients who received placebo (P = .013).
But the difference in time to wound healing by day 90 between the two patient groups was not statistically significant (P = .302), with 50.5% of Oleogel-S10 patients achieving wound closure vs. 43.9% of control patients.
Target-wound infection occurred in eight participants, including three who used Oleogel-S10 and five who received placebo; all moderate or severe infections occurred in patients who received placebo. Total wound burden was reduced to a greater extent among Oleogel-S10 patients by day 60, but there was no apparent difference at day 90.
Both treatment groups reported qualitative improvements in itch, with no significant differences between groups. The prevalence of adverse events was also similar between groups (Oleogel-S10, 81.7%; placebo, 80.7%). The most frequently reported adverse events among Oleogel-S10 patients, compared with patients who received placebo, were wound complications, pyrexia, wound infection, pruritus, and anemia; only 4.5% of adverse events were deemed severe.
Dr. Murrell said that, on the basis of the trial results, she expects the FDA to fast-track approval of Oleogel-S10, which contains triterpene extract and sunflower oil.
The gel is “a treatment patients will be able to put under their dressings, added to normal treatment, which will accelerate their wound healing, with no significant increase in any side effects,” she added.
Jemima Mellerio, MD, of St. Thomas’ Hospital in London who sees about 400 EB patients each year, agreed with Dr. Murrell that the results are “very exciting.” Dr. Mellerio was not involved in the study.
“Practicing dermatologists seeing people with EB will have something to offer that appears to speed up wound healing in chronic wounds,” Dr. Mellerio said in an interview. “It’s a positive option rather than just supportive treatment, something that makes a difference to the natural history of wounds.”
She said the trial’s biggest strength was including “such a large cohort of patients.
“It’s extremely difficult to do that kind of study, especially with a placebo-controlled arm and especially in a rare disease,” Dr. Mellerio said. “If you think about the product itself, it’s easy to apply, so it’s not particularly onerous for people to add to their daily regimen of dressings.”
The study was funded by Amryt Pharma. Dr. Murrell is an advisory board member for Amryt Pharma. Dr. Mellerio is a consultant for Amryt Pharma.
A version of this article originally appeared on Medscape.com.