User login
<p>For ClinicalEdge use only</p>
FDA Boxed Warning Updates
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential bene t from the drug that it is essential that it be considered in assessing the risks and bene ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted.
PRANDIMET (METFORMIN HYDROCHLORIDE; REPAGLINIDE):
- Edited boxed warning April 7, 2017
Post-marketing cases of metforminassociated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin-associated lactic acidosis is often subtle, accompanied only by nonspecific symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin-associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/pyruvate ratio; and metformin plasma levels generally > 5 mcg/mL.
Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, carbonic anyhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Steps to reduce the risk of and manage metformin-associated lactic acidosis in these high risk groups are provided in the full prescribing information.
If metformin-associated lactic acidosis is suspected, immediately discontinue PrandiMet and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
AVANDAMET (METFORMIN HYDROCHLORIDE; ROSIGLITAZONE MALEATE):
- Edited and updated boxed warning April 7, 2017
WARNING: CONGESTIVE HEART FAILURE and LACTIC ACIDOSIS
Rosiglitazone maleate: CONGESTIVE HEART FAILURE
- Thiazolidinediones, including rosiglitazone, cause or exacerbate congestive heart failure in some patients. After initiation of Avandamet, and after dose increases, observe patients carefully for signs and symptoms of heart failure (including excessive, rapid weight gain, dyspnea, and/or edema). If these signs and symptoms develop, the heart failure should be managed according to current standards of care. Furthermore, discontinuation or dose reduction of Avandamet must be considered.
- Avandamet is not recommended in patients with symptomatic heart failure. Initiation of Avandamet in patients with established NYHA Class III or IV heart failure is contraindicated.
Metformin hydrochloride: LACTIC ACIDOSIS
- Postmarketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin- associated lactic acidosis often subtle, accompanied only by nonspeci c symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin- associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/L), anion gap acidosis (without evidence of ketonuria or ketonemia), and increased lactate/pyruvate ratio; and metformin plasma levels generally > 5 mcg/mL.
- Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, carbonic anhydrase
inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment. Steps to reduce the risk of and manage metformin-associated lactic acidosis in these highrisk groups are provided in the Full Prescribing Information. - If metformin-associated lactic acidosis is suspected, immediately discontinue Avandamet and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
OPTIRAY (160, 240, 300, 320, AND 350):
- Edited boxed warning April 7, 2017
PLR conversion, addition of the following:
WARNING: NOT FOR INTRATHECAL USE
Inadvertent intrathecal administration may cause death, convulsions, cerebral hemorrhage, coma, paralysis, arachnoiditis, acute renal failure, cardiac arrest, seizures, rhabdomyolysis, hyperthermia, and brain edema.
GLUMETZA (METFORMIN HYDROCHLORIDE):
- Edited boxed warning April 7, 2017
LACTIC ACIDOSIS
Postmarketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metforminassociated lactic acidosis is often subtle, accompanied only by nonspeci c symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin-associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/pyruvate ratio, and metformin plasma levels generally > 5 mcg/mL.
Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, carbonic anhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Steps to reduce the risk of and manage metformin-associated lactic acidosis in these high risk groups are provided in the full prescribing information.
If metformin-associated lactic acidosis is suspected, immediately discontinue Glumetza and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
SABRIL (VIGABATRIN):
- Edited boxed warning April 7, 2017
WARNING: PERMANENT VISION LOSS
Because of the risk of permanent vision loss, Sabril is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the Vigabatrin REMS Program. Further information is available at www.vigabatrinREMS.com or 1-866-244-8175.
VALCYTE (VALGANCICLOVIR HYDROCHLORIDE):
- Edited boxed warning April 7, 2017
WARNING: HEMATOLOGIC TOXICITY, IMPAIRMENT OF FERTILITY, FETAL TOXICITY, MUTAGENESIS AND CARCINOGENESIS
- Hematologic Toxicity: Severe leukopenia, neutropenia, anemia, thrombocytopenia, pancytopenia, and bone marrow failure including aplastic anemia have been reported in patients treated with Valcyte.
- Impairment of Fertility: Based on animal data, Valcyte may cause temporary or permanent inhibition of spermatogenesis in males and suppression of fertility in females.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential bene t from the drug that it is essential that it be considered in assessing the risks and bene ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted.
PRANDIMET (METFORMIN HYDROCHLORIDE; REPAGLINIDE):
- Edited boxed warning April 7, 2017
Post-marketing cases of metforminassociated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin-associated lactic acidosis is often subtle, accompanied only by nonspecific symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin-associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/pyruvate ratio; and metformin plasma levels generally > 5 mcg/mL.
Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, carbonic anyhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Steps to reduce the risk of and manage metformin-associated lactic acidosis in these high risk groups are provided in the full prescribing information.
If metformin-associated lactic acidosis is suspected, immediately discontinue PrandiMet and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
AVANDAMET (METFORMIN HYDROCHLORIDE; ROSIGLITAZONE MALEATE):
- Edited and updated boxed warning April 7, 2017
WARNING: CONGESTIVE HEART FAILURE and LACTIC ACIDOSIS
Rosiglitazone maleate: CONGESTIVE HEART FAILURE
- Thiazolidinediones, including rosiglitazone, cause or exacerbate congestive heart failure in some patients. After initiation of Avandamet, and after dose increases, observe patients carefully for signs and symptoms of heart failure (including excessive, rapid weight gain, dyspnea, and/or edema). If these signs and symptoms develop, the heart failure should be managed according to current standards of care. Furthermore, discontinuation or dose reduction of Avandamet must be considered.
- Avandamet is not recommended in patients with symptomatic heart failure. Initiation of Avandamet in patients with established NYHA Class III or IV heart failure is contraindicated.
Metformin hydrochloride: LACTIC ACIDOSIS
- Postmarketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin- associated lactic acidosis often subtle, accompanied only by nonspeci c symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin- associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/L), anion gap acidosis (without evidence of ketonuria or ketonemia), and increased lactate/pyruvate ratio; and metformin plasma levels generally > 5 mcg/mL.
- Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, carbonic anhydrase
inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment. Steps to reduce the risk of and manage metformin-associated lactic acidosis in these highrisk groups are provided in the Full Prescribing Information. - If metformin-associated lactic acidosis is suspected, immediately discontinue Avandamet and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
OPTIRAY (160, 240, 300, 320, AND 350):
- Edited boxed warning April 7, 2017
PLR conversion, addition of the following:
WARNING: NOT FOR INTRATHECAL USE
Inadvertent intrathecal administration may cause death, convulsions, cerebral hemorrhage, coma, paralysis, arachnoiditis, acute renal failure, cardiac arrest, seizures, rhabdomyolysis, hyperthermia, and brain edema.
GLUMETZA (METFORMIN HYDROCHLORIDE):
- Edited boxed warning April 7, 2017
LACTIC ACIDOSIS
Postmarketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metforminassociated lactic acidosis is often subtle, accompanied only by nonspeci c symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin-associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/pyruvate ratio, and metformin plasma levels generally > 5 mcg/mL.
Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, carbonic anhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Steps to reduce the risk of and manage metformin-associated lactic acidosis in these high risk groups are provided in the full prescribing information.
If metformin-associated lactic acidosis is suspected, immediately discontinue Glumetza and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
SABRIL (VIGABATRIN):
- Edited boxed warning April 7, 2017
WARNING: PERMANENT VISION LOSS
Because of the risk of permanent vision loss, Sabril is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the Vigabatrin REMS Program. Further information is available at www.vigabatrinREMS.com or 1-866-244-8175.
VALCYTE (VALGANCICLOVIR HYDROCHLORIDE):
- Edited boxed warning April 7, 2017
WARNING: HEMATOLOGIC TOXICITY, IMPAIRMENT OF FERTILITY, FETAL TOXICITY, MUTAGENESIS AND CARCINOGENESIS
- Hematologic Toxicity: Severe leukopenia, neutropenia, anemia, thrombocytopenia, pancytopenia, and bone marrow failure including aplastic anemia have been reported in patients treated with Valcyte.
- Impairment of Fertility: Based on animal data, Valcyte may cause temporary or permanent inhibition of spermatogenesis in males and suppression of fertility in females.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential bene t from the drug that it is essential that it be considered in assessing the risks and bene ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted.
PRANDIMET (METFORMIN HYDROCHLORIDE; REPAGLINIDE):
- Edited boxed warning April 7, 2017
Post-marketing cases of metforminassociated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin-associated lactic acidosis is often subtle, accompanied only by nonspecific symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin-associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/pyruvate ratio; and metformin plasma levels generally > 5 mcg/mL.
Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, carbonic anyhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Steps to reduce the risk of and manage metformin-associated lactic acidosis in these high risk groups are provided in the full prescribing information.
If metformin-associated lactic acidosis is suspected, immediately discontinue PrandiMet and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
AVANDAMET (METFORMIN HYDROCHLORIDE; ROSIGLITAZONE MALEATE):
- Edited and updated boxed warning April 7, 2017
WARNING: CONGESTIVE HEART FAILURE and LACTIC ACIDOSIS
Rosiglitazone maleate: CONGESTIVE HEART FAILURE
- Thiazolidinediones, including rosiglitazone, cause or exacerbate congestive heart failure in some patients. After initiation of Avandamet, and after dose increases, observe patients carefully for signs and symptoms of heart failure (including excessive, rapid weight gain, dyspnea, and/or edema). If these signs and symptoms develop, the heart failure should be managed according to current standards of care. Furthermore, discontinuation or dose reduction of Avandamet must be considered.
- Avandamet is not recommended in patients with symptomatic heart failure. Initiation of Avandamet in patients with established NYHA Class III or IV heart failure is contraindicated.
Metformin hydrochloride: LACTIC ACIDOSIS
- Postmarketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin- associated lactic acidosis often subtle, accompanied only by nonspeci c symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin- associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/L), anion gap acidosis (without evidence of ketonuria or ketonemia), and increased lactate/pyruvate ratio; and metformin plasma levels generally > 5 mcg/mL.
- Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, carbonic anhydrase
inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment. Steps to reduce the risk of and manage metformin-associated lactic acidosis in these highrisk groups are provided in the Full Prescribing Information. - If metformin-associated lactic acidosis is suspected, immediately discontinue Avandamet and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
OPTIRAY (160, 240, 300, 320, AND 350):
- Edited boxed warning April 7, 2017
PLR conversion, addition of the following:
WARNING: NOT FOR INTRATHECAL USE
Inadvertent intrathecal administration may cause death, convulsions, cerebral hemorrhage, coma, paralysis, arachnoiditis, acute renal failure, cardiac arrest, seizures, rhabdomyolysis, hyperthermia, and brain edema.
GLUMETZA (METFORMIN HYDROCHLORIDE):
- Edited boxed warning April 7, 2017
LACTIC ACIDOSIS
Postmarketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metforminassociated lactic acidosis is often subtle, accompanied only by nonspeci c symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metformin-associated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/pyruvate ratio, and metformin plasma levels generally > 5 mcg/mL.
Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (eg, carbonic anhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (eg, acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Steps to reduce the risk of and manage metformin-associated lactic acidosis in these high risk groups are provided in the full prescribing information.
If metformin-associated lactic acidosis is suspected, immediately discontinue Glumetza and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
SABRIL (VIGABATRIN):
- Edited boxed warning April 7, 2017
WARNING: PERMANENT VISION LOSS
Because of the risk of permanent vision loss, Sabril is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the Vigabatrin REMS Program. Further information is available at www.vigabatrinREMS.com or 1-866-244-8175.
VALCYTE (VALGANCICLOVIR HYDROCHLORIDE):
- Edited boxed warning April 7, 2017
WARNING: HEMATOLOGIC TOXICITY, IMPAIRMENT OF FERTILITY, FETAL TOXICITY, MUTAGENESIS AND CARCINOGENESIS
- Hematologic Toxicity: Severe leukopenia, neutropenia, anemia, thrombocytopenia, pancytopenia, and bone marrow failure including aplastic anemia have been reported in patients treated with Valcyte.
- Impairment of Fertility: Based on animal data, Valcyte may cause temporary or permanent inhibition of spermatogenesis in males and suppression of fertility in females.
Improved Access to Drug Safety Labeling Changes Information
The FDA has made it easier and faster for health care professionals (HCPs) to get up-to-date drug safety information for the more than 18,000 approved drugs via its Drug Safety Labeling Changes (SLCs) database. The FDA Center for Drug Evaluation and Research recently launched a new searchable and downloadable database for SLCs information (http://www.fda.gov/slc). In most cases, the improved website provides supplemental labeling information within days of a safety label change. Now when a physician or other HCP prescribes a medicine using an e-prescribing system, the updated drug safety information displays much faster than it did with the previous safety labeling changes system. Here’s how.
Shortly after FDA approval of the new drug safety information for an existing drug, the information is entered into the safety labeling changes database. Health information technology (IT) vendors that provide clinical and drug information support for hospitals and pharmacies are then alerted to integrate the updated data into their systems as well. Instead of waiting weeks for the monthly release of all safety labeling updates, this information now is accessible within days.
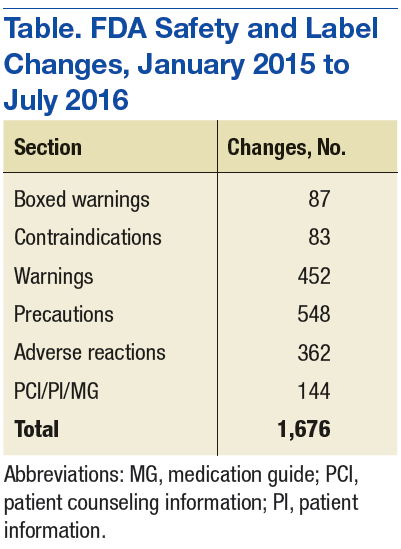
Although SLCs have been available online for many years, previously they were aggregated and posted only monthly. This time frame meant that if a new safety concern was reflected in an approved labeling change early in a month, then the information was not publicly posted until the following month—4 to 5 weeks later. The FDA recognized the need to apply new digital functionalities to shorten the time between an SLC approval and the public availability of the safety information. Between January 2015 and July 2016, FDA made more than 1,500 SLCs (Table).
As health care professionals know, the “labeling” of a medicine includes detailed information provided in the package insert that accompanies the drug whether it’s on the box, inside the product box, or folded and glued to the lid of a bottle. The product labeling includes a summary for the safe and effective use of the drug and is generally intended for use by prescribers and pharmacists.
However, when a drug is approved, not every safety concern or risk potential can be identified or known. Safety information can change multiple times over the lifetime of a drug as the FDA learns about new risks, interactions with other medications, and adverse effects.
After the FDA becomes aware of new safety information, changes to the product labeling may be required. That’s why postmarketing safety oversight is essential to learn more about the effects of medicines when they are used by a large number of people over a long period. If new safety concerns emerge after a medicine is used in a real-world setting, the FDA may require a “Safety Labeling Change.” The FDA’s new, faster connection between updated safety information and safety alerts on the pharmacy computer system can help build improved confidence into each drug prescription.
The new SLCs website contains a database of changed safety information from all sections of the label that addresses a drug’s safety, including:
- Boxed warning
- Contraindications
- Warnings and precautions
- Adverse reactions
- Drug interactions
- Use in specific populations
- Patient counseling information/patient information/medication guide
Health care providers, health IT vendors, and the public now have access to critical safety data that can impact the health of a patient faster than before.
Providing drug safety labeling changes quickly to health care vendors facilitates having the data further integrated into systems frequently accessed by HCPs. It also carries SLC data downstream for integration into drug information systems and other electronic venues, such as social media, news feeds, and websites, with vast reach among health care professionals, patients, and consumers. Some of these include WebMD, Medscape, American Society of Health-System Pharmacists, PDR.net, Epocrates, First Databank, and Yahoo Health.
The data files are downloadable in a comma-separated values format—a feature that allows information to be gathered faster. There also are hyperlinks to the labeling revisions at Drugs@FDA, and notifications are sent to subscribers via an RSS feed.
The FDA continues to pursue and provide innovative ways to rapidly access important information that protects and advances public health and will work to better identify class labeling changes. The FDA’s primary goal for the redesigned SLC Internet interface is to deliver drug safety labeling changes as quickly and efficiently as possible, to help create and promote better patient health.
The FDA has made it easier and faster for health care professionals (HCPs) to get up-to-date drug safety information for the more than 18,000 approved drugs via its Drug Safety Labeling Changes (SLCs) database. The FDA Center for Drug Evaluation and Research recently launched a new searchable and downloadable database for SLCs information (http://www.fda.gov/slc). In most cases, the improved website provides supplemental labeling information within days of a safety label change. Now when a physician or other HCP prescribes a medicine using an e-prescribing system, the updated drug safety information displays much faster than it did with the previous safety labeling changes system. Here’s how.
Shortly after FDA approval of the new drug safety information for an existing drug, the information is entered into the safety labeling changes database. Health information technology (IT) vendors that provide clinical and drug information support for hospitals and pharmacies are then alerted to integrate the updated data into their systems as well. Instead of waiting weeks for the monthly release of all safety labeling updates, this information now is accessible within days.
Although SLCs have been available online for many years, previously they were aggregated and posted only monthly. This time frame meant that if a new safety concern was reflected in an approved labeling change early in a month, then the information was not publicly posted until the following month—4 to 5 weeks later. The FDA recognized the need to apply new digital functionalities to shorten the time between an SLC approval and the public availability of the safety information. Between January 2015 and July 2016, FDA made more than 1,500 SLCs (Table).
As health care professionals know, the “labeling” of a medicine includes detailed information provided in the package insert that accompanies the drug whether it’s on the box, inside the product box, or folded and glued to the lid of a bottle. The product labeling includes a summary for the safe and effective use of the drug and is generally intended for use by prescribers and pharmacists.
However, when a drug is approved, not every safety concern or risk potential can be identified or known. Safety information can change multiple times over the lifetime of a drug as the FDA learns about new risks, interactions with other medications, and adverse effects.
After the FDA becomes aware of new safety information, changes to the product labeling may be required. That’s why postmarketing safety oversight is essential to learn more about the effects of medicines when they are used by a large number of people over a long period. If new safety concerns emerge after a medicine is used in a real-world setting, the FDA may require a “Safety Labeling Change.” The FDA’s new, faster connection between updated safety information and safety alerts on the pharmacy computer system can help build improved confidence into each drug prescription.
The new SLCs website contains a database of changed safety information from all sections of the label that addresses a drug’s safety, including:
- Boxed warning
- Contraindications
- Warnings and precautions
- Adverse reactions
- Drug interactions
- Use in specific populations
- Patient counseling information/patient information/medication guide
Health care providers, health IT vendors, and the public now have access to critical safety data that can impact the health of a patient faster than before.
Providing drug safety labeling changes quickly to health care vendors facilitates having the data further integrated into systems frequently accessed by HCPs. It also carries SLC data downstream for integration into drug information systems and other electronic venues, such as social media, news feeds, and websites, with vast reach among health care professionals, patients, and consumers. Some of these include WebMD, Medscape, American Society of Health-System Pharmacists, PDR.net, Epocrates, First Databank, and Yahoo Health.
The data files are downloadable in a comma-separated values format—a feature that allows information to be gathered faster. There also are hyperlinks to the labeling revisions at Drugs@FDA, and notifications are sent to subscribers via an RSS feed.
The FDA continues to pursue and provide innovative ways to rapidly access important information that protects and advances public health and will work to better identify class labeling changes. The FDA’s primary goal for the redesigned SLC Internet interface is to deliver drug safety labeling changes as quickly and efficiently as possible, to help create and promote better patient health.
The FDA has made it easier and faster for health care professionals (HCPs) to get up-to-date drug safety information for the more than 18,000 approved drugs via its Drug Safety Labeling Changes (SLCs) database. The FDA Center for Drug Evaluation and Research recently launched a new searchable and downloadable database for SLCs information (http://www.fda.gov/slc). In most cases, the improved website provides supplemental labeling information within days of a safety label change. Now when a physician or other HCP prescribes a medicine using an e-prescribing system, the updated drug safety information displays much faster than it did with the previous safety labeling changes system. Here’s how.
Shortly after FDA approval of the new drug safety information for an existing drug, the information is entered into the safety labeling changes database. Health information technology (IT) vendors that provide clinical and drug information support for hospitals and pharmacies are then alerted to integrate the updated data into their systems as well. Instead of waiting weeks for the monthly release of all safety labeling updates, this information now is accessible within days.
Although SLCs have been available online for many years, previously they were aggregated and posted only monthly. This time frame meant that if a new safety concern was reflected in an approved labeling change early in a month, then the information was not publicly posted until the following month—4 to 5 weeks later. The FDA recognized the need to apply new digital functionalities to shorten the time between an SLC approval and the public availability of the safety information. Between January 2015 and July 2016, FDA made more than 1,500 SLCs (Table).
As health care professionals know, the “labeling” of a medicine includes detailed information provided in the package insert that accompanies the drug whether it’s on the box, inside the product box, or folded and glued to the lid of a bottle. The product labeling includes a summary for the safe and effective use of the drug and is generally intended for use by prescribers and pharmacists.
However, when a drug is approved, not every safety concern or risk potential can be identified or known. Safety information can change multiple times over the lifetime of a drug as the FDA learns about new risks, interactions with other medications, and adverse effects.
After the FDA becomes aware of new safety information, changes to the product labeling may be required. That’s why postmarketing safety oversight is essential to learn more about the effects of medicines when they are used by a large number of people over a long period. If new safety concerns emerge after a medicine is used in a real-world setting, the FDA may require a “Safety Labeling Change.” The FDA’s new, faster connection between updated safety information and safety alerts on the pharmacy computer system can help build improved confidence into each drug prescription.
The new SLCs website contains a database of changed safety information from all sections of the label that addresses a drug’s safety, including:
- Boxed warning
- Contraindications
- Warnings and precautions
- Adverse reactions
- Drug interactions
- Use in specific populations
- Patient counseling information/patient information/medication guide
Health care providers, health IT vendors, and the public now have access to critical safety data that can impact the health of a patient faster than before.
Providing drug safety labeling changes quickly to health care vendors facilitates having the data further integrated into systems frequently accessed by HCPs. It also carries SLC data downstream for integration into drug information systems and other electronic venues, such as social media, news feeds, and websites, with vast reach among health care professionals, patients, and consumers. Some of these include WebMD, Medscape, American Society of Health-System Pharmacists, PDR.net, Epocrates, First Databank, and Yahoo Health.
The data files are downloadable in a comma-separated values format—a feature that allows information to be gathered faster. There also are hyperlinks to the labeling revisions at Drugs@FDA, and notifications are sent to subscribers via an RSS feed.
The FDA continues to pursue and provide innovative ways to rapidly access important information that protects and advances public health and will work to better identify class labeling changes. The FDA’s primary goal for the redesigned SLC Internet interface is to deliver drug safety labeling changes as quickly and efficiently as possible, to help create and promote better patient health.
New and Updated FDA Boxed Warnings
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential bene t from the drug that it is essential that it be considered in assessing the risks and bene ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted.
IMODIUM (LOPERAMIDE HYDROCHLORIDE):
- New warning December 2016
WARNING: TORSADES DE POINTES AND SUDDEN DEATH
Cases of Torsades de Pointes, cardiac arrest, and death have been reported with the use of a higher than recommended dosages of Imodium (see WARNINGS and OVERDOSAGE).
Imodium is contraindicated in pediatric patients less than 2 years of age (see CONTRANIDICATIONS).
Avoid Imodium dosages higher than recommended in adults and pediatric patients 2 years of age and older due to the risk of serious cardiac adverse reactions (see DOSAGE AND ADMINISTRATION).
AUBAGIO (TERIFLUNOMIDE) TABLETS:
- Edited and updated warning December 2016
Risk of Teratogenicity
Aubagio is contraindicated for use in pregnant women and in women of reproductive potential who are not using effective contraception because of the potential for fetal harm. Teratogenicity and embryolethality occurred in animals at plasma teriflunomide exposures lower than that in humans. Exclude pregnancy before the start of treatment with Aubagio in females of reproductive potential. Advise females of reproductive potential to use effective contraception during Aubagio treatment and during an accelerated drug elimination procedure after Aubagio treatment. Stop Aubagio and use an accelerated drug elimination procedure if the patient becomes pregnant.
PROMACTA (ELTROMBOPAG) TABLETS, FOR ORAL USE AND ORAL SUSPENSION:
- Edited and updated warning December 2016
Chronic Hepatitis C
Promacta may increase the risk of severe and potentially lifethreatening hepatotoxicity. Monitor hepatic function and discontinue dosing as recommended.
ICLUSIG (PONATINIB HYDROCHLORIDE):
- Edited and updated warning December 2016
WARNING: ARTERIAL OCCLUSION, VENOUS THROMBOEMBOLISM, HEART FAILURE, and HEPATOTOXICITY
Arterial Occlusion
Arterial occlusions have occurred in at least 35% of Iclusig-treated patients. Some patients experienced more than 1 type of event. Events observed included fatal myocardial infarction, stroke, stenosis of large arterial vessels of the brain, severe peripheral vascular disease, and the need for urgent revascularization procedures. Patients with and without cardiovascular risk factors, including patients age 50 years or younger, experienced these events. Monitor for evidence of arterial occlusion. Interrupt or stop Iclusig immediately for arterial occlusion.
Venous Thromboembolism
Venous occlusive events have occurred in 6% of Iclusig-treated patients. Monitor for evidence of venous thromboembolism. Consider dose modification or discontinuation of Iclusig in patients who develop serious venous thromboembolism.
Heart Failure
Heart failure, including fatalities, occurred in 9% of Iclusig-treated patients.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential bene t from the drug that it is essential that it be considered in assessing the risks and bene ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted.
IMODIUM (LOPERAMIDE HYDROCHLORIDE):
- New warning December 2016
WARNING: TORSADES DE POINTES AND SUDDEN DEATH
Cases of Torsades de Pointes, cardiac arrest, and death have been reported with the use of a higher than recommended dosages of Imodium (see WARNINGS and OVERDOSAGE).
Imodium is contraindicated in pediatric patients less than 2 years of age (see CONTRANIDICATIONS).
Avoid Imodium dosages higher than recommended in adults and pediatric patients 2 years of age and older due to the risk of serious cardiac adverse reactions (see DOSAGE AND ADMINISTRATION).
AUBAGIO (TERIFLUNOMIDE) TABLETS:
- Edited and updated warning December 2016
Risk of Teratogenicity
Aubagio is contraindicated for use in pregnant women and in women of reproductive potential who are not using effective contraception because of the potential for fetal harm. Teratogenicity and embryolethality occurred in animals at plasma teriflunomide exposures lower than that in humans. Exclude pregnancy before the start of treatment with Aubagio in females of reproductive potential. Advise females of reproductive potential to use effective contraception during Aubagio treatment and during an accelerated drug elimination procedure after Aubagio treatment. Stop Aubagio and use an accelerated drug elimination procedure if the patient becomes pregnant.
PROMACTA (ELTROMBOPAG) TABLETS, FOR ORAL USE AND ORAL SUSPENSION:
- Edited and updated warning December 2016
Chronic Hepatitis C
Promacta may increase the risk of severe and potentially lifethreatening hepatotoxicity. Monitor hepatic function and discontinue dosing as recommended.
ICLUSIG (PONATINIB HYDROCHLORIDE):
- Edited and updated warning December 2016
WARNING: ARTERIAL OCCLUSION, VENOUS THROMBOEMBOLISM, HEART FAILURE, and HEPATOTOXICITY
Arterial Occlusion
Arterial occlusions have occurred in at least 35% of Iclusig-treated patients. Some patients experienced more than 1 type of event. Events observed included fatal myocardial infarction, stroke, stenosis of large arterial vessels of the brain, severe peripheral vascular disease, and the need for urgent revascularization procedures. Patients with and without cardiovascular risk factors, including patients age 50 years or younger, experienced these events. Monitor for evidence of arterial occlusion. Interrupt or stop Iclusig immediately for arterial occlusion.
Venous Thromboembolism
Venous occlusive events have occurred in 6% of Iclusig-treated patients. Monitor for evidence of venous thromboembolism. Consider dose modification or discontinuation of Iclusig in patients who develop serious venous thromboembolism.
Heart Failure
Heart failure, including fatalities, occurred in 9% of Iclusig-treated patients.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential bene t from the drug that it is essential that it be considered in assessing the risks and bene ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted.
IMODIUM (LOPERAMIDE HYDROCHLORIDE):
- New warning December 2016
WARNING: TORSADES DE POINTES AND SUDDEN DEATH
Cases of Torsades de Pointes, cardiac arrest, and death have been reported with the use of a higher than recommended dosages of Imodium (see WARNINGS and OVERDOSAGE).
Imodium is contraindicated in pediatric patients less than 2 years of age (see CONTRANIDICATIONS).
Avoid Imodium dosages higher than recommended in adults and pediatric patients 2 years of age and older due to the risk of serious cardiac adverse reactions (see DOSAGE AND ADMINISTRATION).
AUBAGIO (TERIFLUNOMIDE) TABLETS:
- Edited and updated warning December 2016
Risk of Teratogenicity
Aubagio is contraindicated for use in pregnant women and in women of reproductive potential who are not using effective contraception because of the potential for fetal harm. Teratogenicity and embryolethality occurred in animals at plasma teriflunomide exposures lower than that in humans. Exclude pregnancy before the start of treatment with Aubagio in females of reproductive potential. Advise females of reproductive potential to use effective contraception during Aubagio treatment and during an accelerated drug elimination procedure after Aubagio treatment. Stop Aubagio and use an accelerated drug elimination procedure if the patient becomes pregnant.
PROMACTA (ELTROMBOPAG) TABLETS, FOR ORAL USE AND ORAL SUSPENSION:
- Edited and updated warning December 2016
Chronic Hepatitis C
Promacta may increase the risk of severe and potentially lifethreatening hepatotoxicity. Monitor hepatic function and discontinue dosing as recommended.
ICLUSIG (PONATINIB HYDROCHLORIDE):
- Edited and updated warning December 2016
WARNING: ARTERIAL OCCLUSION, VENOUS THROMBOEMBOLISM, HEART FAILURE, and HEPATOTOXICITY
Arterial Occlusion
Arterial occlusions have occurred in at least 35% of Iclusig-treated patients. Some patients experienced more than 1 type of event. Events observed included fatal myocardial infarction, stroke, stenosis of large arterial vessels of the brain, severe peripheral vascular disease, and the need for urgent revascularization procedures. Patients with and without cardiovascular risk factors, including patients age 50 years or younger, experienced these events. Monitor for evidence of arterial occlusion. Interrupt or stop Iclusig immediately for arterial occlusion.
Venous Thromboembolism
Venous occlusive events have occurred in 6% of Iclusig-treated patients. Monitor for evidence of venous thromboembolism. Consider dose modification or discontinuation of Iclusig in patients who develop serious venous thromboembolism.
Heart Failure
Heart failure, including fatalities, occurred in 9% of Iclusig-treated patients.
Recent FDA Boxed Warnings
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. You can search these and other label changes in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential bene t from the drug that it is essential that it be considered in assessing the risks and bene ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted.
QUINOLONE:
- Edited and updated warning September 2016
WARNING: SERIOUS ADVERSE REACTIONS INCLUDING TENDINITIS, TENDON RUPTURE, PERIPHERAL NEUROPATHY, CENTRAL NERVOUS SYSTEM EFFECTS AND EXACERBATION OF MYASTHENIA GRAVIS
Fluoroquinolones have been associated with disabling and potentially irreversible serious adverse reactions that have occurred together including:
- Tendinitis and tendon rupture
- Peripheral neuropathy
- Central nervous system effects
Discontinue immediately and avoid the use of fluoroquinolones in patients who experience any of these serious adverse reactions. Fluoroquinolones may exacerbate muscle weakness in patients with myasthenia gravis. Avoid quinolones in patients with known history of myasthenia gravis. Because fluoroquinolones
have been associated with serious adverse reactions, reserve quinolones for use in patients who have no alternative treatment options for the following
indications:
Avelox (moxifloxacin hydrochloride): Avelox in sodium chloride 0.8% in plastic container; moxifloxacin hydrochloride; Cipro in dextrose 5% in plastic container):
Acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis.
Cipro (ciprofloxacin; ciprofloxacin hydrochloride): Acute exacerbation of chronic bronchitis, acute uncomplicated cystitis, and acute sinusitis.
Cipro XR; Noroxin (norfloxacin): Uncomplicated urinary tract infections.
Factive (gemifloxacin mesylate): Acute bacterial exacerbation of chronic bronchitis.
Levaquin (levofloxacin): Uncomplicated urinary tract infection, acute bacterial exacerbation of chronic bronchitis, and acute bacterial sinusitis.
KRYSTEXXA (PEGLOTICASE):
- Added section to warning September 2016
WARNING: ANAPHYLAXIS AND INFUSION REACTIONS; G6PD DEFICIENCY ASSOCIATED HEMOLYSIS AND METHEMOGLOBINEMIA (Title Updated)
Addition of: Screen patients at risk for G6PD deficiency prior to starting Krystexxa. Hemolysis and methemoglobinemia have been reported with Krystexxa in patients with G6PD deficiency. Do not administer Krystexxa to patients with G6PD deficiency.
PLAVIX (CLOPIDOGREL BISULFATE):
- Edited and updated warning September 2016
WARNING: DIMINISHED ANTIPLATELET EFFECT IN PATIENTS WITH TWO LOSS-OF-FUNCTION ALLELES OF THE CYP2C19 GENE
The effectiveness of Plavix results from its antiplatelet activity, which is dependent on its conversion to an active metabolite by the cytochrome P450 (CYP) system, principally CYP2C19. Plavix at recommended doses forms less of the active metabolite and so has a reduced effect on platelet activity in patients who are homozygous for nonfunctional alleles of the CYP2C19 gene, (termed “CYP2C19 poor metabolizers”). Tests are available to identify patients who are CYP2C19 poor metabolizers. Consider use of another platelet P2Y12 inhibitor in patients identified as CYP2C19 poor metabolizers.
SYNJARDY (EMPAGLIFLOZIN; METFORMIN HYDROCHLORIDE):
- Edited and updated warning September 2016
Postmarketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin-associated lactic acidosis is often subtle, accompanied only by nonspecific symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metforminassociated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/pyruvate ratio; and metformin plasma levels generally > 5 mcg/mL.
Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (e.g., carbonic anhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (e.g., acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Steps to reduce the risk of and manage metformin-associated lactic acidosis in these high-risk groups are provided in the full prescribing information.
If metformin-associated lactic acidosis is suspected, immediately discontinue Synjardy and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
ZYDELIG (IDELALISIB)
- Edited and updated warning September 2016
WARNING: FATAL AND SERIOUS TOXICITIES: HEPATIC, SEVERE DIARRHEA, COLITIS, PNEUMONITIS, INFECTIONS, AND INTESTINAL PERFORATION
- Fatal and/or serious hepatotoxicity occurred in 11 % to 18% of Zydelig-treated patients. Monitor hepatic function prior to and during treatment. Interrupt and then reduce or discontinue Zydelig as recommended.
- Fatal and/or serious and severe diarrhea or colitis occurred in 14% to 19% of Zydelig-treated patients. Monitor for the development of severe diarrhea or colitis. Interrupt and then reduce or discontinue Zydelig as recommended.
- Fatal and/or serious pneumonitis occurred in 4% of Zydelig-treated patients. Monitor for pulmonary symptoms and bilateral interstitial infiltrates. Interrupt or discontinue Zydelig as recommended.
- Fatal and/or serious infections occurred in 21% to 36% of Zydelig-treated patients. Monitor for signs and symptoms of infection. Interrupt Zydelig if infection is suspected.
- Fatal and serious intestinal perforation can occur in Zydelig-treated patients across clinical trials. Discontinue Zydelig for intestinal perforation.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. You can search these and other label changes in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential bene t from the drug that it is essential that it be considered in assessing the risks and bene ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted.
QUINOLONE:
- Edited and updated warning September 2016
WARNING: SERIOUS ADVERSE REACTIONS INCLUDING TENDINITIS, TENDON RUPTURE, PERIPHERAL NEUROPATHY, CENTRAL NERVOUS SYSTEM EFFECTS AND EXACERBATION OF MYASTHENIA GRAVIS
Fluoroquinolones have been associated with disabling and potentially irreversible serious adverse reactions that have occurred together including:
- Tendinitis and tendon rupture
- Peripheral neuropathy
- Central nervous system effects
Discontinue immediately and avoid the use of fluoroquinolones in patients who experience any of these serious adverse reactions. Fluoroquinolones may exacerbate muscle weakness in patients with myasthenia gravis. Avoid quinolones in patients with known history of myasthenia gravis. Because fluoroquinolones
have been associated with serious adverse reactions, reserve quinolones for use in patients who have no alternative treatment options for the following
indications:
Avelox (moxifloxacin hydrochloride): Avelox in sodium chloride 0.8% in plastic container; moxifloxacin hydrochloride; Cipro in dextrose 5% in plastic container):
Acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis.
Cipro (ciprofloxacin; ciprofloxacin hydrochloride): Acute exacerbation of chronic bronchitis, acute uncomplicated cystitis, and acute sinusitis.
Cipro XR; Noroxin (norfloxacin): Uncomplicated urinary tract infections.
Factive (gemifloxacin mesylate): Acute bacterial exacerbation of chronic bronchitis.
Levaquin (levofloxacin): Uncomplicated urinary tract infection, acute bacterial exacerbation of chronic bronchitis, and acute bacterial sinusitis.
KRYSTEXXA (PEGLOTICASE):
- Added section to warning September 2016
WARNING: ANAPHYLAXIS AND INFUSION REACTIONS; G6PD DEFICIENCY ASSOCIATED HEMOLYSIS AND METHEMOGLOBINEMIA (Title Updated)
Addition of: Screen patients at risk for G6PD deficiency prior to starting Krystexxa. Hemolysis and methemoglobinemia have been reported with Krystexxa in patients with G6PD deficiency. Do not administer Krystexxa to patients with G6PD deficiency.
PLAVIX (CLOPIDOGREL BISULFATE):
- Edited and updated warning September 2016
WARNING: DIMINISHED ANTIPLATELET EFFECT IN PATIENTS WITH TWO LOSS-OF-FUNCTION ALLELES OF THE CYP2C19 GENE
The effectiveness of Plavix results from its antiplatelet activity, which is dependent on its conversion to an active metabolite by the cytochrome P450 (CYP) system, principally CYP2C19. Plavix at recommended doses forms less of the active metabolite and so has a reduced effect on platelet activity in patients who are homozygous for nonfunctional alleles of the CYP2C19 gene, (termed “CYP2C19 poor metabolizers”). Tests are available to identify patients who are CYP2C19 poor metabolizers. Consider use of another platelet P2Y12 inhibitor in patients identified as CYP2C19 poor metabolizers.
SYNJARDY (EMPAGLIFLOZIN; METFORMIN HYDROCHLORIDE):
- Edited and updated warning September 2016
Postmarketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin-associated lactic acidosis is often subtle, accompanied only by nonspecific symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metforminassociated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/pyruvate ratio; and metformin plasma levels generally > 5 mcg/mL.
Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (e.g., carbonic anhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (e.g., acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Steps to reduce the risk of and manage metformin-associated lactic acidosis in these high-risk groups are provided in the full prescribing information.
If metformin-associated lactic acidosis is suspected, immediately discontinue Synjardy and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
ZYDELIG (IDELALISIB)
- Edited and updated warning September 2016
WARNING: FATAL AND SERIOUS TOXICITIES: HEPATIC, SEVERE DIARRHEA, COLITIS, PNEUMONITIS, INFECTIONS, AND INTESTINAL PERFORATION
- Fatal and/or serious hepatotoxicity occurred in 11 % to 18% of Zydelig-treated patients. Monitor hepatic function prior to and during treatment. Interrupt and then reduce or discontinue Zydelig as recommended.
- Fatal and/or serious and severe diarrhea or colitis occurred in 14% to 19% of Zydelig-treated patients. Monitor for the development of severe diarrhea or colitis. Interrupt and then reduce or discontinue Zydelig as recommended.
- Fatal and/or serious pneumonitis occurred in 4% of Zydelig-treated patients. Monitor for pulmonary symptoms and bilateral interstitial infiltrates. Interrupt or discontinue Zydelig as recommended.
- Fatal and/or serious infections occurred in 21% to 36% of Zydelig-treated patients. Monitor for signs and symptoms of infection. Interrupt Zydelig if infection is suspected.
- Fatal and serious intestinal perforation can occur in Zydelig-treated patients across clinical trials. Discontinue Zydelig for intestinal perforation.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. You can search these and other label changes in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential bene t from the drug that it is essential that it be considered in assessing the risks and bene ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted.
QUINOLONE:
- Edited and updated warning September 2016
WARNING: SERIOUS ADVERSE REACTIONS INCLUDING TENDINITIS, TENDON RUPTURE, PERIPHERAL NEUROPATHY, CENTRAL NERVOUS SYSTEM EFFECTS AND EXACERBATION OF MYASTHENIA GRAVIS
Fluoroquinolones have been associated with disabling and potentially irreversible serious adverse reactions that have occurred together including:
- Tendinitis and tendon rupture
- Peripheral neuropathy
- Central nervous system effects
Discontinue immediately and avoid the use of fluoroquinolones in patients who experience any of these serious adverse reactions. Fluoroquinolones may exacerbate muscle weakness in patients with myasthenia gravis. Avoid quinolones in patients with known history of myasthenia gravis. Because fluoroquinolones
have been associated with serious adverse reactions, reserve quinolones for use in patients who have no alternative treatment options for the following
indications:
Avelox (moxifloxacin hydrochloride): Avelox in sodium chloride 0.8% in plastic container; moxifloxacin hydrochloride; Cipro in dextrose 5% in plastic container):
Acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis.
Cipro (ciprofloxacin; ciprofloxacin hydrochloride): Acute exacerbation of chronic bronchitis, acute uncomplicated cystitis, and acute sinusitis.
Cipro XR; Noroxin (norfloxacin): Uncomplicated urinary tract infections.
Factive (gemifloxacin mesylate): Acute bacterial exacerbation of chronic bronchitis.
Levaquin (levofloxacin): Uncomplicated urinary tract infection, acute bacterial exacerbation of chronic bronchitis, and acute bacterial sinusitis.
KRYSTEXXA (PEGLOTICASE):
- Added section to warning September 2016
WARNING: ANAPHYLAXIS AND INFUSION REACTIONS; G6PD DEFICIENCY ASSOCIATED HEMOLYSIS AND METHEMOGLOBINEMIA (Title Updated)
Addition of: Screen patients at risk for G6PD deficiency prior to starting Krystexxa. Hemolysis and methemoglobinemia have been reported with Krystexxa in patients with G6PD deficiency. Do not administer Krystexxa to patients with G6PD deficiency.
PLAVIX (CLOPIDOGREL BISULFATE):
- Edited and updated warning September 2016
WARNING: DIMINISHED ANTIPLATELET EFFECT IN PATIENTS WITH TWO LOSS-OF-FUNCTION ALLELES OF THE CYP2C19 GENE
The effectiveness of Plavix results from its antiplatelet activity, which is dependent on its conversion to an active metabolite by the cytochrome P450 (CYP) system, principally CYP2C19. Plavix at recommended doses forms less of the active metabolite and so has a reduced effect on platelet activity in patients who are homozygous for nonfunctional alleles of the CYP2C19 gene, (termed “CYP2C19 poor metabolizers”). Tests are available to identify patients who are CYP2C19 poor metabolizers. Consider use of another platelet P2Y12 inhibitor in patients identified as CYP2C19 poor metabolizers.
SYNJARDY (EMPAGLIFLOZIN; METFORMIN HYDROCHLORIDE):
- Edited and updated warning September 2016
Postmarketing cases of metformin-associated lactic acidosis have resulted in death, hypothermia, hypotension, and resistant bradyarrhythmias. The onset of metformin-associated lactic acidosis is often subtle, accompanied only by nonspecific symptoms such as malaise, myalgias, respiratory distress, somnolence, and abdominal pain. Metforminassociated lactic acidosis was characterized by elevated blood lactate levels (> 5 mmol/Liter), anion gap acidosis (without evidence of ketonuria or ketonemia), an increased lactate/pyruvate ratio; and metformin plasma levels generally > 5 mcg/mL.
Risk factors for metformin-associated lactic acidosis include renal impairment, concomitant use of certain drugs (e.g., carbonic anhydrase inhibitors such as topiramate), age 65 years old or greater, having a radiological study with contrast, surgery and other procedures, hypoxic states (e.g., acute congestive heart failure), excessive alcohol intake, and hepatic impairment.
Steps to reduce the risk of and manage metformin-associated lactic acidosis in these high-risk groups are provided in the full prescribing information.
If metformin-associated lactic acidosis is suspected, immediately discontinue Synjardy and institute general supportive measures in a hospital setting. Prompt hemodialysis is recommended.
ZYDELIG (IDELALISIB)
- Edited and updated warning September 2016
WARNING: FATAL AND SERIOUS TOXICITIES: HEPATIC, SEVERE DIARRHEA, COLITIS, PNEUMONITIS, INFECTIONS, AND INTESTINAL PERFORATION
- Fatal and/or serious hepatotoxicity occurred in 11 % to 18% of Zydelig-treated patients. Monitor hepatic function prior to and during treatment. Interrupt and then reduce or discontinue Zydelig as recommended.
- Fatal and/or serious and severe diarrhea or colitis occurred in 14% to 19% of Zydelig-treated patients. Monitor for the development of severe diarrhea or colitis. Interrupt and then reduce or discontinue Zydelig as recommended.
- Fatal and/or serious pneumonitis occurred in 4% of Zydelig-treated patients. Monitor for pulmonary symptoms and bilateral interstitial infiltrates. Interrupt or discontinue Zydelig as recommended.
- Fatal and/or serious infections occurred in 21% to 36% of Zydelig-treated patients. Monitor for signs and symptoms of infection. Interrupt Zydelig if infection is suspected.
- Fatal and serious intestinal perforation can occur in Zydelig-treated patients across clinical trials. Discontinue Zydelig for intestinal perforation.
FDA approves topical oxymetazoline for rosacea
A topical cream containing the vasoconstrictor oxymetazoline has been approved by the Food and Drug Administration to treat symptoms of rosacea, its manufacturer announced.
Oxymetazoline hydrochloride cream 1%, which will be marketed as Rhofade by Allergan, is indicated for the treatment of “persistent facial erythema associated with rosacea in adults.” While nasal sprays containing a lower concentration of oxymetazoline HCl, an alpha1A-adrenoceptor agonist, have been used off label for a decade, this is the first time this ingredient has been harnessed to formulate an approved rosacea treatment.
![]()
Safety results from three pooled trials showed 2% of patients in the active treatment arms (489 people) had treatment-site dermatitis, and 1% had worsening of rosacea symptoms, pruritus, or pain. The vehicle cream groups (483 people) experienced similar rates of pruritus but negligible rates of other adverse effects, according to the prescribing information.
Brimonidine (Mirvaso) is another topical treatment approved by the FDA for treating rosacea, and its active ingredient is also an alpha-adrenergic agonist that works on the cutaneous microvasculature. However, there are differences in the two agents’ activity. Oxymetazoline acts on alpha1A receptors and brimonidine on alpha2 receptors. There have been reports of rebound erythema more severe than at baseline with brimonidine, and its manufacturer, Galderma, acknowledges the phenomenon in patient labeling.
When Allergan announced the FDA application for oxymetazoline in May 2016, it issued a press statement, describing oxymetazoline as a “sympathomimetic agonist that is selective for the alpha1A adrenoceptor or over other alpha1 adrenoceptors and nonselective for the alpha2 adrenoceptors.”In a 1-year open label trial of oxymetazoline (440 people), 3% of patients had worsening inflammatory lesions of rosacea, according to the prescribing information for oxymetazoline HCl 1%.
A topical cream containing the vasoconstrictor oxymetazoline has been approved by the Food and Drug Administration to treat symptoms of rosacea, its manufacturer announced.
Oxymetazoline hydrochloride cream 1%, which will be marketed as Rhofade by Allergan, is indicated for the treatment of “persistent facial erythema associated with rosacea in adults.” While nasal sprays containing a lower concentration of oxymetazoline HCl, an alpha1A-adrenoceptor agonist, have been used off label for a decade, this is the first time this ingredient has been harnessed to formulate an approved rosacea treatment.
![]()
Safety results from three pooled trials showed 2% of patients in the active treatment arms (489 people) had treatment-site dermatitis, and 1% had worsening of rosacea symptoms, pruritus, or pain. The vehicle cream groups (483 people) experienced similar rates of pruritus but negligible rates of other adverse effects, according to the prescribing information.
Brimonidine (Mirvaso) is another topical treatment approved by the FDA for treating rosacea, and its active ingredient is also an alpha-adrenergic agonist that works on the cutaneous microvasculature. However, there are differences in the two agents’ activity. Oxymetazoline acts on alpha1A receptors and brimonidine on alpha2 receptors. There have been reports of rebound erythema more severe than at baseline with brimonidine, and its manufacturer, Galderma, acknowledges the phenomenon in patient labeling.
When Allergan announced the FDA application for oxymetazoline in May 2016, it issued a press statement, describing oxymetazoline as a “sympathomimetic agonist that is selective for the alpha1A adrenoceptor or over other alpha1 adrenoceptors and nonselective for the alpha2 adrenoceptors.”In a 1-year open label trial of oxymetazoline (440 people), 3% of patients had worsening inflammatory lesions of rosacea, according to the prescribing information for oxymetazoline HCl 1%.
A topical cream containing the vasoconstrictor oxymetazoline has been approved by the Food and Drug Administration to treat symptoms of rosacea, its manufacturer announced.
Oxymetazoline hydrochloride cream 1%, which will be marketed as Rhofade by Allergan, is indicated for the treatment of “persistent facial erythema associated with rosacea in adults.” While nasal sprays containing a lower concentration of oxymetazoline HCl, an alpha1A-adrenoceptor agonist, have been used off label for a decade, this is the first time this ingredient has been harnessed to formulate an approved rosacea treatment.
![]()
Safety results from three pooled trials showed 2% of patients in the active treatment arms (489 people) had treatment-site dermatitis, and 1% had worsening of rosacea symptoms, pruritus, or pain. The vehicle cream groups (483 people) experienced similar rates of pruritus but negligible rates of other adverse effects, according to the prescribing information.
Brimonidine (Mirvaso) is another topical treatment approved by the FDA for treating rosacea, and its active ingredient is also an alpha-adrenergic agonist that works on the cutaneous microvasculature. However, there are differences in the two agents’ activity. Oxymetazoline acts on alpha1A receptors and brimonidine on alpha2 receptors. There have been reports of rebound erythema more severe than at baseline with brimonidine, and its manufacturer, Galderma, acknowledges the phenomenon in patient labeling.
When Allergan announced the FDA application for oxymetazoline in May 2016, it issued a press statement, describing oxymetazoline as a “sympathomimetic agonist that is selective for the alpha1A adrenoceptor or over other alpha1 adrenoceptors and nonselective for the alpha2 adrenoceptors.”In a 1-year open label trial of oxymetazoline (440 people), 3% of patients had worsening inflammatory lesions of rosacea, according to the prescribing information for oxymetazoline HCl 1%.
FDA, EPA clarify which fish pregnant women and young children should eat
The Food and Drug Administration and the Environmental Protection Agency have issued updated guidance about fish consumption for pregnant women and young children, clarifying which types of fish are recommended and what types of fish to avoid.
In guidance issued Jan. 18, the agencies sort 62 types of fish into three categories based on mercury level: best choices, good choices, and fish to avoid. They recommend that women who are pregnant, women who may become pregnant, breastfeeding mothers, and young children eat two to three servings of fish in the “best choices” category per week. Women and young children are advised to eat one serving per week of fish in the “good choices” category, according to the announcement. Fish in the “best choices” category make up nearly 90% of fish eaten in the United States, according to the FDA.
“Fish are an important source of protein and other nutrients for young children and women who are or may become pregnant or are breastfeeding,” Stephen Ostroff, MD, FDA’s deputy commissioner for Foods and Veterinary Medicine, said in a statement. “This advice clearly shows the great diversity of fish in the U.S. market that they can consume safely. This new, clear and concrete advice is an excellent tool for making safe and healthy choices when buying fish.”
The updated advice cautions pregnant women and others to avoid seven types of fish that generally have higher mercury levels. This includes tilefish from the Gulf of Mexico, shark; swordfish; orange roughy, bigeye tuna; marlin, and king mackerel. Meanwhile, recommended choices lower in mercury include such fish as shrimp, pollock, salmon, canned light tuna, tilapia, catfish, and cod.
Consumers are urged to check local advisories for fish caught recreationally and gauge their fish consumption based on any local and state advisories for those waters. If no information on fishing advisories is available, the FDA recommends eating just one fish meal a week from local waters and to avoid other fish that week.
agallegos@frontlinemedcom.com
On Twitter @legal_med
The Food and Drug Administration and the Environmental Protection Agency have issued updated guidance about fish consumption for pregnant women and young children, clarifying which types of fish are recommended and what types of fish to avoid.
In guidance issued Jan. 18, the agencies sort 62 types of fish into three categories based on mercury level: best choices, good choices, and fish to avoid. They recommend that women who are pregnant, women who may become pregnant, breastfeeding mothers, and young children eat two to three servings of fish in the “best choices” category per week. Women and young children are advised to eat one serving per week of fish in the “good choices” category, according to the announcement. Fish in the “best choices” category make up nearly 90% of fish eaten in the United States, according to the FDA.
“Fish are an important source of protein and other nutrients for young children and women who are or may become pregnant or are breastfeeding,” Stephen Ostroff, MD, FDA’s deputy commissioner for Foods and Veterinary Medicine, said in a statement. “This advice clearly shows the great diversity of fish in the U.S. market that they can consume safely. This new, clear and concrete advice is an excellent tool for making safe and healthy choices when buying fish.”
The updated advice cautions pregnant women and others to avoid seven types of fish that generally have higher mercury levels. This includes tilefish from the Gulf of Mexico, shark; swordfish; orange roughy, bigeye tuna; marlin, and king mackerel. Meanwhile, recommended choices lower in mercury include such fish as shrimp, pollock, salmon, canned light tuna, tilapia, catfish, and cod.
Consumers are urged to check local advisories for fish caught recreationally and gauge their fish consumption based on any local and state advisories for those waters. If no information on fishing advisories is available, the FDA recommends eating just one fish meal a week from local waters and to avoid other fish that week.
agallegos@frontlinemedcom.com
On Twitter @legal_med
The Food and Drug Administration and the Environmental Protection Agency have issued updated guidance about fish consumption for pregnant women and young children, clarifying which types of fish are recommended and what types of fish to avoid.
In guidance issued Jan. 18, the agencies sort 62 types of fish into three categories based on mercury level: best choices, good choices, and fish to avoid. They recommend that women who are pregnant, women who may become pregnant, breastfeeding mothers, and young children eat two to three servings of fish in the “best choices” category per week. Women and young children are advised to eat one serving per week of fish in the “good choices” category, according to the announcement. Fish in the “best choices” category make up nearly 90% of fish eaten in the United States, according to the FDA.
“Fish are an important source of protein and other nutrients for young children and women who are or may become pregnant or are breastfeeding,” Stephen Ostroff, MD, FDA’s deputy commissioner for Foods and Veterinary Medicine, said in a statement. “This advice clearly shows the great diversity of fish in the U.S. market that they can consume safely. This new, clear and concrete advice is an excellent tool for making safe and healthy choices when buying fish.”
The updated advice cautions pregnant women and others to avoid seven types of fish that generally have higher mercury levels. This includes tilefish from the Gulf of Mexico, shark; swordfish; orange roughy, bigeye tuna; marlin, and king mackerel. Meanwhile, recommended choices lower in mercury include such fish as shrimp, pollock, salmon, canned light tuna, tilapia, catfish, and cod.
Consumers are urged to check local advisories for fish caught recreationally and gauge their fish consumption based on any local and state advisories for those waters. If no information on fishing advisories is available, the FDA recommends eating just one fish meal a week from local waters and to avoid other fish that week.
agallegos@frontlinemedcom.com
On Twitter @legal_med
FDA bans powdered gloves
The Food and Drug Administration has banned powdered gloves for use in health care settings, citing “numerous risks to patients and health care workers.” The ban extends to gloves currently in commercial distribution and in the hands of the ultimate user, meaning powdered gloves will have to be pulled from examination rooms and operating theaters.
“A thorough review of all currently available information supports FDA’s conclusion that powdered surgeon’s gloves, powdered patient examination gloves, and absorbable powder for lubricating a surgeon’s glove should be banned,” according to a FDA final rule available now online and scheduled for publication in the Federal Register on Dec. 19, 2016. The ban will become effective 30 days after the document’s publication in the Federal Register.
Notes the final document, “The benefits of powdered gloves appear to only include greater ease of donning and doffing, decreased tackiness, and a degree of added comfort, which FDA believes are nominal when compared to the risks posed by these devices.”
Since viable nonpowdered alternatives exist, the FDA believes that the ban would not have significant economic impact and that shortages should not affect care delivery.
Many nonpowdered gloves, said the FDA, now “have the same level of protection, dexterity, and performance” as powdered gloves.
Powder may still be used in the glove manufacturing process, but the FDA continues to recommend that no more than 2 mg of residual powder per glove remains after the manufacturing process.
Though the final document banning powdered gloves notes that the FDA received many comments asking for a ban of all natural rubber latex (NRL) gloves, the ban applied only to powdered gloves. The FDA noted that NRL gloves already must carry a statement alerting users to the risks of allergic reaction, and also noted that eliminating powder from NRL gloves reduces the risk of latex sensitization.
In explaining its analysis of the costs and benefits of the powdered glove ban, the FDA estimated that the annual net benefits would range between $26.8 million and $31.8 million.
When this ban comes into force, it will be only the second such ban; the first was the 1983 ban of prosthetic hair fibers, which were found to provide no public health benefit.
koakes@frontlinemedcom.com
On Twitter @karioakes
The Food and Drug Administration has banned powdered gloves for use in health care settings, citing “numerous risks to patients and health care workers.” The ban extends to gloves currently in commercial distribution and in the hands of the ultimate user, meaning powdered gloves will have to be pulled from examination rooms and operating theaters.
“A thorough review of all currently available information supports FDA’s conclusion that powdered surgeon’s gloves, powdered patient examination gloves, and absorbable powder for lubricating a surgeon’s glove should be banned,” according to a FDA final rule available now online and scheduled for publication in the Federal Register on Dec. 19, 2016. The ban will become effective 30 days after the document’s publication in the Federal Register.
Notes the final document, “The benefits of powdered gloves appear to only include greater ease of donning and doffing, decreased tackiness, and a degree of added comfort, which FDA believes are nominal when compared to the risks posed by these devices.”
Since viable nonpowdered alternatives exist, the FDA believes that the ban would not have significant economic impact and that shortages should not affect care delivery.
Many nonpowdered gloves, said the FDA, now “have the same level of protection, dexterity, and performance” as powdered gloves.
Powder may still be used in the glove manufacturing process, but the FDA continues to recommend that no more than 2 mg of residual powder per glove remains after the manufacturing process.
Though the final document banning powdered gloves notes that the FDA received many comments asking for a ban of all natural rubber latex (NRL) gloves, the ban applied only to powdered gloves. The FDA noted that NRL gloves already must carry a statement alerting users to the risks of allergic reaction, and also noted that eliminating powder from NRL gloves reduces the risk of latex sensitization.
In explaining its analysis of the costs and benefits of the powdered glove ban, the FDA estimated that the annual net benefits would range between $26.8 million and $31.8 million.
When this ban comes into force, it will be only the second such ban; the first was the 1983 ban of prosthetic hair fibers, which were found to provide no public health benefit.
koakes@frontlinemedcom.com
On Twitter @karioakes
The Food and Drug Administration has banned powdered gloves for use in health care settings, citing “numerous risks to patients and health care workers.” The ban extends to gloves currently in commercial distribution and in the hands of the ultimate user, meaning powdered gloves will have to be pulled from examination rooms and operating theaters.
“A thorough review of all currently available information supports FDA’s conclusion that powdered surgeon’s gloves, powdered patient examination gloves, and absorbable powder for lubricating a surgeon’s glove should be banned,” according to a FDA final rule available now online and scheduled for publication in the Federal Register on Dec. 19, 2016. The ban will become effective 30 days after the document’s publication in the Federal Register.
Notes the final document, “The benefits of powdered gloves appear to only include greater ease of donning and doffing, decreased tackiness, and a degree of added comfort, which FDA believes are nominal when compared to the risks posed by these devices.”
Since viable nonpowdered alternatives exist, the FDA believes that the ban would not have significant economic impact and that shortages should not affect care delivery.
Many nonpowdered gloves, said the FDA, now “have the same level of protection, dexterity, and performance” as powdered gloves.
Powder may still be used in the glove manufacturing process, but the FDA continues to recommend that no more than 2 mg of residual powder per glove remains after the manufacturing process.
Though the final document banning powdered gloves notes that the FDA received many comments asking for a ban of all natural rubber latex (NRL) gloves, the ban applied only to powdered gloves. The FDA noted that NRL gloves already must carry a statement alerting users to the risks of allergic reaction, and also noted that eliminating powder from NRL gloves reduces the risk of latex sensitization.
In explaining its analysis of the costs and benefits of the powdered glove ban, the FDA estimated that the annual net benefits would range between $26.8 million and $31.8 million.
When this ban comes into force, it will be only the second such ban; the first was the 1983 ban of prosthetic hair fibers, which were found to provide no public health benefit.
koakes@frontlinemedcom.com
On Twitter @karioakes
Absorb’s problems will revise coronary scaffold standards
One-year outcome results of the first bioresorbable coronary scaffold on the U.S. and world markets, Absorb, failed to show longer-term problems with the device that only became apparent with 3-year follow-up.
The failure of Absorb to show benefits after 3 years in the ABSORB II trial will probably not dampen enthusiasm for the concept of a bioresorbable coronary scaffold (BRS). The idea of treating coronary stenoses with a stent that disappears after a few years once it has done its job is a powerfully attractive idea, and reports from several early-stage clinical tests of new BRSs during TCT 2016 showed that many next-generation versions of these devices are in very active development.
The surprising ABSORB II results showed more than just a failure of the Absorb BRS to produce 3 years after placement the improved coronary artery vasomotion and reduced late lumen loss that were the two primary efficacy endpoints of the trial.
The results also showed troubling signs of harm from the BRS, including significantly worse late lumen loss, compared with a contemporary drug-eluting metallic stent. In addition, there was a shocking 1%/year rate of late stent thrombosis during both the second and third years following Absorb placement in coronary arteries, the period when the Absorb BRS was in the process of disappearing, and which did not occur in the study’s control patients who received a conventional, metallic drug-eluting stent.
Patrick W. Serruys, MD, lead investigator of ABSORB II, attributed these adverse outcomes to the “highly thrombogenic” proteoglycan material that formed as the Absorb BRS resorbed, and a “structural discontinuity” of the BRS as it resorbed in some patients, resulting in parts of the scaffold remnant sticking out from the coronary artery wall toward the center of the vessel.
These late flaws in the bioresorption process will now need closer scrutiny during future studies of next-generation BRSs, and will surely mean longer follow-up of pivotal trials and a shift from the 1-year follow-up data used by the Food and Drug Administration when it approved the Absorb BRS last July.
“The challenge for the field [of BRS development] is the late results, as we saw in ABSORB II,” commented David J. Cohen, MD, an interventional cardiologist at Saint Luke’s Health System in Kansas City, Mo. The ABSORB II results “will lead to reexamination of the trial design and endpoints for the next generation of BRSs,” Dr. Cohen predicted at the meeting, sponsored by the Cardiovascular Research Foundation.
“It’s not clear that BRS reduces the duration for DAPT,” Dr. Cohen noted, at least for the Absorb device, which is not full resorbed until it’s been in patients for about 3 years.
A striking property of the next-generation BRSs reported at the meeting was their use of thinner struts and faster resorption times. “These iterations hold immense promise for improving late outcomes,” commented Dean J. Kereiakes, MD, an interventional cardiologist at the Christ Hospital in Cincinnati who helped lead the large U.S. clinical trial of the Absorb BRS, ABSORB III.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
One-year outcome results of the first bioresorbable coronary scaffold on the U.S. and world markets, Absorb, failed to show longer-term problems with the device that only became apparent with 3-year follow-up.
The failure of Absorb to show benefits after 3 years in the ABSORB II trial will probably not dampen enthusiasm for the concept of a bioresorbable coronary scaffold (BRS). The idea of treating coronary stenoses with a stent that disappears after a few years once it has done its job is a powerfully attractive idea, and reports from several early-stage clinical tests of new BRSs during TCT 2016 showed that many next-generation versions of these devices are in very active development.
The surprising ABSORB II results showed more than just a failure of the Absorb BRS to produce 3 years after placement the improved coronary artery vasomotion and reduced late lumen loss that were the two primary efficacy endpoints of the trial.
The results also showed troubling signs of harm from the BRS, including significantly worse late lumen loss, compared with a contemporary drug-eluting metallic stent. In addition, there was a shocking 1%/year rate of late stent thrombosis during both the second and third years following Absorb placement in coronary arteries, the period when the Absorb BRS was in the process of disappearing, and which did not occur in the study’s control patients who received a conventional, metallic drug-eluting stent.
Patrick W. Serruys, MD, lead investigator of ABSORB II, attributed these adverse outcomes to the “highly thrombogenic” proteoglycan material that formed as the Absorb BRS resorbed, and a “structural discontinuity” of the BRS as it resorbed in some patients, resulting in parts of the scaffold remnant sticking out from the coronary artery wall toward the center of the vessel.
These late flaws in the bioresorption process will now need closer scrutiny during future studies of next-generation BRSs, and will surely mean longer follow-up of pivotal trials and a shift from the 1-year follow-up data used by the Food and Drug Administration when it approved the Absorb BRS last July.
“The challenge for the field [of BRS development] is the late results, as we saw in ABSORB II,” commented David J. Cohen, MD, an interventional cardiologist at Saint Luke’s Health System in Kansas City, Mo. The ABSORB II results “will lead to reexamination of the trial design and endpoints for the next generation of BRSs,” Dr. Cohen predicted at the meeting, sponsored by the Cardiovascular Research Foundation.
“It’s not clear that BRS reduces the duration for DAPT,” Dr. Cohen noted, at least for the Absorb device, which is not full resorbed until it’s been in patients for about 3 years.
A striking property of the next-generation BRSs reported at the meeting was their use of thinner struts and faster resorption times. “These iterations hold immense promise for improving late outcomes,” commented Dean J. Kereiakes, MD, an interventional cardiologist at the Christ Hospital in Cincinnati who helped lead the large U.S. clinical trial of the Absorb BRS, ABSORB III.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
One-year outcome results of the first bioresorbable coronary scaffold on the U.S. and world markets, Absorb, failed to show longer-term problems with the device that only became apparent with 3-year follow-up.
The failure of Absorb to show benefits after 3 years in the ABSORB II trial will probably not dampen enthusiasm for the concept of a bioresorbable coronary scaffold (BRS). The idea of treating coronary stenoses with a stent that disappears after a few years once it has done its job is a powerfully attractive idea, and reports from several early-stage clinical tests of new BRSs during TCT 2016 showed that many next-generation versions of these devices are in very active development.
The surprising ABSORB II results showed more than just a failure of the Absorb BRS to produce 3 years after placement the improved coronary artery vasomotion and reduced late lumen loss that were the two primary efficacy endpoints of the trial.
The results also showed troubling signs of harm from the BRS, including significantly worse late lumen loss, compared with a contemporary drug-eluting metallic stent. In addition, there was a shocking 1%/year rate of late stent thrombosis during both the second and third years following Absorb placement in coronary arteries, the period when the Absorb BRS was in the process of disappearing, and which did not occur in the study’s control patients who received a conventional, metallic drug-eluting stent.
Patrick W. Serruys, MD, lead investigator of ABSORB II, attributed these adverse outcomes to the “highly thrombogenic” proteoglycan material that formed as the Absorb BRS resorbed, and a “structural discontinuity” of the BRS as it resorbed in some patients, resulting in parts of the scaffold remnant sticking out from the coronary artery wall toward the center of the vessel.
These late flaws in the bioresorption process will now need closer scrutiny during future studies of next-generation BRSs, and will surely mean longer follow-up of pivotal trials and a shift from the 1-year follow-up data used by the Food and Drug Administration when it approved the Absorb BRS last July.
“The challenge for the field [of BRS development] is the late results, as we saw in ABSORB II,” commented David J. Cohen, MD, an interventional cardiologist at Saint Luke’s Health System in Kansas City, Mo. The ABSORB II results “will lead to reexamination of the trial design and endpoints for the next generation of BRSs,” Dr. Cohen predicted at the meeting, sponsored by the Cardiovascular Research Foundation.
“It’s not clear that BRS reduces the duration for DAPT,” Dr. Cohen noted, at least for the Absorb device, which is not full resorbed until it’s been in patients for about 3 years.
A striking property of the next-generation BRSs reported at the meeting was their use of thinner struts and faster resorption times. “These iterations hold immense promise for improving late outcomes,” commented Dean J. Kereiakes, MD, an interventional cardiologist at the Christ Hospital in Cincinnati who helped lead the large U.S. clinical trial of the Absorb BRS, ABSORB III.
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler