User login
Recognizing and treating cutaneous signs of liver disease
Dysfunction in the body’s second largest organ, the liver, often yields changes in the body’s largest organ, the skin. If we can recognize these manifestations early, we are better able to promptly diagnose and treat the underlying liver disease, as well as the skin lesions.
The liver has many jobs: synthesizing proteins such as clotting factors, complements, and albumin; neutralizing toxins; and metabolizing lipids and carbohydrates. Insults to the liver can compromise any of these functions, affecting visceral organs, joints, gastrointestinal tissues, and the skin. Dermatologic signs of specific liver diseases include alopecia and vitiligo associated with autoimmune hepatitis, and xanthelasma in chronic cholestatic liver disease.
This article reviews the important cutaneous manifestations of specific liver diseases. We focus first on skin conditions that may represent liver disease, and then we discuss several major liver diseases and their typical cutaneous manifestations.
JAUNDICE AND HYPERBILIRUBINEMIA
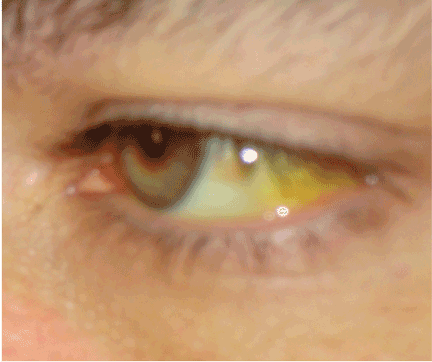
Establishing whether the excess bilirubin is conjugated or unconjugated gives a clue as to whether the cause is prehepatic, intrahepatic, or posthepatic.3–8 One of the liver’s main functions is to conjugate bilirubin into a secretable form. Prehepatic causes of jaundice include hemolysis and ineffective erythropoiesis, both of which lead to higher levels of circulating unconjugated bilirubin.4 Intrahepatic causes of jaundice can lead to both unconjugated and conjugated hyperbilirubinemia.4,8 Posthepatic causes such as bile duct obstruction primarily result in conjugated hyperbilirubinemia.4
PRURITUS AND PRURIGO NODULARIS
Pruritus can be multifactorial or the result of a specific dermatologic or systemic condition.9 A thorough history and physical examination are warranted to rule out hepatic or systemic causes of itching.10
The liver neutralizes toxins and filters bile salts. If its function is impaired, these materials can accumulate in the body, and deposition in the skin causes irritation and itching.11,12 In cholestatic liver disorders such as primary sclerosing cholangitis and obstructive gallstone disease, pruritus tends to be generalized, but worse on the hands and feet.13
Although the severity of pruritus is not directly associated with the level of bile salts and toxic substances, lowering bile salt levels can mitigate symptoms.11
Treatment. Pruritus due to liver disease is particularly resistant to therapy.
In a strategy described by Mela et al for managing pruritus in chronic liver disease,14 the initial treatment is the anion exchange resin cholestyramine (Questran) at a starting dose of 4 g/day, gradually increased to 24 g/day in two doses at mealtimes.
If the pruritus does not respond adequately to cholestyramine or the patient cannot tolerate the drug, then the antituberculosis drug rifampin (Rifadin) can be tried. Rifampin promotes metabolism of endogenous pruritogens and has been effective against cholestatic pruritus when started at 150 mg/day and increased up to 600 mg/day, depending on the clinical response.14
Third-line drug therapies include opioid antagonists such as naltrexone (ReVia) and nalmefene (Revex).14,15
Plasmapheresis can be considered if drug therapy fails.16 Experimental therapies include albumin dialysis using the molecular adsorbent recirculating system (a form of artificial liver support), antioxidant treatment, and bright-light therapy.15 Liver transplantation, when appropriate, also resolves cholestatic pruritus.14
Prurigo nodularis
Prurigo nodularis, distinguished by firm, crusty nodules, is associated with viral infections (eg, hepatitis C, human immunodeficiency virus), bacterial infections, and kidney dysfunction.17,18 The lesions are intensely pruritic and often lead to persistent scratching, excoriation, and, ultimately, diffuse scarring.19
Treatment. Although the exact cause of prurigo nodularis is not known and no cure exists, corticosteroid or antihistamine ointments control the symptoms in most patients with hepatitis.19 Low doses of thalidomide (Thalomid), a tumor necrosis factor antagonist, have also been used safely and effectively.18,19
SUPERFICIAL VASCULAR SIGNS
Spider angiomas
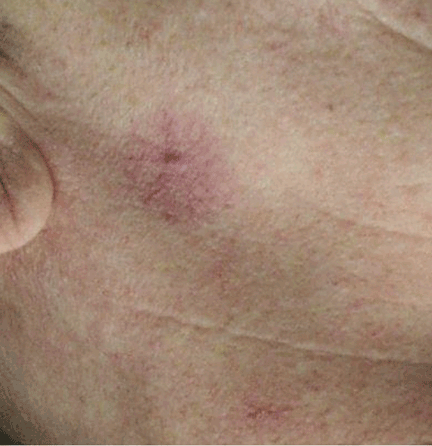
Spider angiomas can occur anywhere on the body, but they occur most often on the face and the trunk.21,23 A key feature is that they disappear when pressure is applied and reappear when pressure is removed.23,24 Biopsy is rarely necessary for diagnosis.
These lesions occur with elevated estrogen levels, such as in cirrhosis, during estrogen therapy, or during pregnancy.25–28 Although spider angiomas are common in pregnant women and in children, adults with spider angiomas deserve a workup for liver dysfunction.29
Given their innocuous nature and asymptomatic course, spider angiomas themselves require no medical treatment.
Bier spots
Bier spots are small, irregularly shaped, hypopigmented patches on the arms and legs. They are likely due to venous stasis associated with functional damage to the small vessels of the skin.30
Since Bier spots are a sign of liver disease, they must be distinguished from true pigmentation disorders. A key distinguishing feature is that Bier spots disappear when pressure is applied. Also, raising the affected limb from a dependent position causes the hypopigmented macules of Bier spots to disappear, which is not the case in true pigmentation disorders.10,30
Paper-money skin
Paper-money skin (or “dollar-paper” markings) describes the condition in which the upper trunk is covered with many randomly scattered, needle-thin superficial capillaries. It often occurs in association with spider angiomas. The name comes from the resemblance the thread-like capillaries have to the finely chopped silk threads in American dollar bills.10,31 The condition is commonly seen in patients with alcoholic cirrhosis and may improve with hemodialysis.31
PALMAR ERYTHEMA
Palmar erythema is a florid, crimson coloration of the palms of the hands and the fingertips. It can occur anywhere on the palm and fingers but is most common on the hypothenar eminence. It can occur in a number of liver conditions but most often with cirrhosis.32 Hepatic compromise, as seen in alcoholic liver disease, disrupts the body’s androgen balance, causing local vasodilation and erythema.32,33 Although the exact mechanism remains unknown, research suggests that prostacyclins and nitric oxide play a role, as both are increased in liver disease.32,33
XANTHELASMA
Xanthelasma—a localized cholesterol deposit beneath the skin and especially beneath the eyelids—is a common manifestation of hypercholesterolemia. Xanthelasma often presents as a painless, yellowish, soft plaque with well-defined borders,34 which may enlarge over the course of weeks.
Several liver diseases can lead to various forms of secondary dyslipoproteinemia.35 The most common dyslipoproteinemias in liver disease are hypertriglyceridemia and low levels of high-density lipoprotein cholesterol, and both of these often accompany fatty liver disease.36 Hypercholesterolemia is a common feature of primary biliary cirrhosis and other forms of cholestatic liver disease.37 Studies suggest that the total plasma cholesterol level is elevated in as many as 50% of patients with compromised liver function.38
Treatment. The underlying hyperlipidemia is treated with cholesterol-lowering drugs. Laser treatment and surgical excision have proven efficacious in treating the lesions.39
OTHER CUTANEOUS FINDINGS IN LIVER DISEASE
Bleeding and bruising. Liver disease can cause hypersplenism and thrombocytopenia, in addition to a decrease in clotting factors. These may present with a myriad of cutaneous symptoms, including purpura, bleeding gums, and easy bruising and bleeding, even from minor trauma.40–42
Hyperpigmentation of the skin may accompany hemochromatosis, alcoholic liver disease, and cirrhosis.43–45
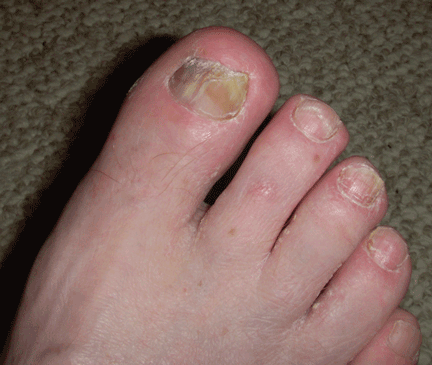
“Terry’s nails,” in which the proximal two-thirds of the nail plate turns powdery white with a ground-glass opacity, may develop in patients with advanced cirrhosis.48
ALCOHOLIC CIRRHOSIS AND THE SKIN
The cutaneous changes associated with alcoholic cirrhosis are more widely recognized than those due to other forms of liver dysfunction. In the United States, approximately 3 million people have alcoholic cirrhosis, the second-leading reason for liver transplantation.49,50
As the body’s main site of alcohol metabolism, the liver is the organ most affected by excessive alcohol intake, which can lead to end-stage liver disease secondary to alcoholic cirrhosis.41,51 The characteristic feature of cirrhosis is advanced fibrous scarring of parenchymal tissue and the formation of regenerative nodules with increased resistance to blood flow throughout the organ.41,52 The insufficient blood flow damages vital structures in the liver and compromises liver function. For example, liver cirrhosis leads to defective hepatic synthesis of clotting factors and results in bleeding disorders.
Cutaneous lesions often accompany alcoholic cirrhosis and have been detected in up to 43% of people with chronic alcoholism.53 Skin changes in alcoholic cirrhosis can be of great diagnostic value. The combined prevalence of spider angiomas, palmar erythema, and Dupuytren contracture in alcoholic cirrhosis was found to be 72%. Paper-money skin and Dupuytren contracture are more distinct lesions for alcoholic cirrhosis.31 Recognizing these skin changes contributes to the diagnosis and staging of liver cirrhosis.51,52
Dupuytren contracture
Dupuytren contracture is characterized by progressive fibrosis and thickening of tendons in the palmar fascia, the connective tissue that lies beneath the skin of the palms.54 Over time, as fibrotic involvement expands across the fascia, rampant stiffness of the joints ensues, sometimes to a point where the fingers cannot fully flex or extend.54
Although the exact cause of Dupuytren contracture is unknown, it appears to be associated with excess alcohol consumption and can be found in patients with alcoholic cirrhosis.54,55 These patients often present with painless stiffness of the fingers, curling of fingers, and loss of motion in involved fingers.54 Surgery in the form of limited fasciectomy has been curative in such patients.54
Disseminated superficial porokeratosis
Porokeratosis is a keratinization disorder of clonal origin that presents as a linear configuration of white scaly papules that coalesce into plaques throughout the body.56 Although it most commonly afflicts fair-skinned people, patients with alcoholic cirrhosis have a much greater susceptibility than the general population.57,58
A recent study58 documented that the lesions completely resolved when liver function improved, thus underlining the relationship between the two conditions. Since immunosuppression has been linked to eruption of the lesion, the fact that both humoral and cell-mediated immune responses are impaired in alcoholic liver disease provides another dimension to the association between porokeratosis and alcoholic cirrhosis.58
These lesions can transform into squamous cell carcinoma.59 The risk of widespread metastases in squamous cell carcinoma highlights the importance of dermatologic consultation in such patients.59
HEPATITIS C AND THE SKIN
Extrahepatic manifestations have been documented in up to 74% of people with hepatitis C virus infection.60 In addition to parasthesias, arthralgias, and myalgias, hepatitis C has a significant association with porphyria cutanea tarda, lichen planus, vitiligo, sialadenitis, urticarial vasculitis, corneal ulcers, xerosis, pruritus, and prurigo nodularis.60–64 Although the primary causative agents of sialadenitis are bacteria, viruses such as hepatitis C have been implicated as a cause of chronic sialadenitis with associated xerostomia.65
Patients with hepatitis C being treated with interferons also present with cutaneous manifestations such as hyperkeratosis and vasculitis.63
Porphyria cutanea tarda
Porphyria cutanea tarda is the most common of the porphyrias, disorders distinguished by deficiencies or defects in one or more of the enzymes responsible for hepatic production of heme.66 If these enzymes are impaired, heme precursors such as porphyrins accumulate.66
Porphyria cutanea tarda results from a deficiency of the hepatic enzyme uroporphyrin decarboxylase. In the absence of this enzyme, shortwave visible light activates uroporphyrin deposited in the skin, resulting in a photochemical reaction that generates reactive oxygen species that lead to the characteristic skin blistering.
Although porphyria cutanea tarda is associated with liver disease in general, recent studies confirm that patients with hepatitis C are at particularly high risk.67 Those with the disorder often present with skin photosensitivity. 68 Many develop blisters on sun-exposed skin, including the dorsal aspects of the hands and forearms and on the neck and face. Chronic porphyria cutanea tarda can lead to scarring, alopecia, and skin ulceration.69 As the blisters heal, keratin-filled milial cysts may develop in the areas of ulceration.
The condition is also commonly associated with melasma-like hyperpigmentation and hypertrichosis in sun-exposed areas of the head and neck. People of Northern European ancestry may be more at risk than the general population because of a presumed genetic susceptibility.70
Treatment. Because many patients with porphyria cutanea tarda have iron overload, they need to restrict foods rich in iron and to avoid alcohol.71,72 Severe cases may necessitate iron removal via phlebotomy or antimalarial therapy. Patients with porphyria cutanea tarda induced by hepatitis C should have their bodily iron stores depleted before starting antiviral therapy.60
Lichen planus
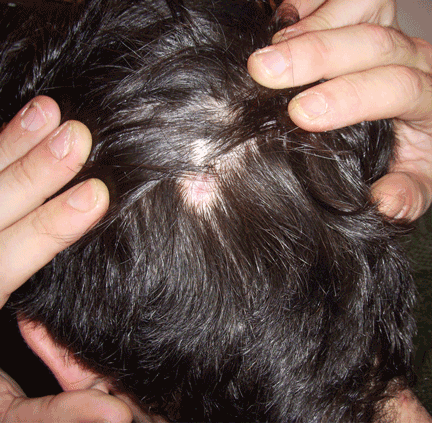
Lichen planopilaris is a subset of lichen planus that causes scaling and atrophy of the scalp and permanent hair loss (Figure 4).73
Interferon-induced vitiligo
Vitiligo is an autoimmune disease in which melanocytes in the skin are destroyed, with resulting depigmentation in affected areas.75 Although it has no specific association with liver disease, it has been linked to treatments for hepatitis C such as interferons.76 Interferon-induced vitiligo often completely resolves when interferon is stopped.77
Typical findings include aggregations of irregularly shaped white patches in a focal or segmental pattern.78 The diagnosis is based on the medical history, physical examination, and sometimes skin biopsy.
HEMOCHROMATOSIS
Hemochromatosis or “bronze diabetes” is a devastating multisystem disease with a relentless course. It is among the most common genetic disorders of metabolism, and results in deposition of iron in tissues and organs throughout the body, including the liver, usually in patients ages 30 to 40.
As iron stores increase in tissues and organs, multiorgan failure and associated complications may ensue. In addition, surplus iron stores can also result in widespread bronze discoloration of skin exposed to the sun. Hemochromatosis also results in loss of body hair, ichthyosiform alterations, and koilonychia.79
Treatments that lower serum iron levels can reverse the cutaneous manifestations of the disorder and minimize the risk of organ failure.
Since the condition is inherited in an autosomal-recessive pattern, family members of patients should consider being screened.80
Hyperpigmentation in hemochromatosis
Hyperpigmentation is an early sign of hemochromatosis, affecting up to 90% of patients. Usually, sun-exposed areas of the body are the most prone and take on a grayish or brownish-bronze hue.81 Cutaneous iron deposits injure vital skin structures, initiating a process that culminates in enhanced melanin production by melanocytes.82 Exposure to ultraviolet light may have synergistic effects with iron, hastening the process of hyperpigmentation. As a result of this synergistic effect, many patients with hemochromatosis notice tanning with minimal sun exposure.
Although organ function can improve immediately with phlebotomy to reduce iron stores, skin hyperpigmentation does not immediately resolve.81,82
Ichthyosiform alterations in hemochromatosis
Ichthyosiform changes, in which the skin takes on the appearance of fish scales,83 can be seen in patients with hemochromatosis.80 Affected areas typically become extremely dry. Treatment includes topical hydrating creams and ointments. Avoiding sunlight is paramount, as sunlight exposure may exacerbate the condition.83
- Morioka D, Togo S, Kumamoto T, et al. Six consecutive cases of successful adult ABO-incompatible living donor liver transplantation: a proposal for grading the severity of antibody-mediated rejection. Transplantation 2008; 85:171–178.
- Bertini G, Rubaltelli FF. Non-invasive bilirubinometry in neonatal jaundice. Semin Neonatol 2002; 7:129–133.
- Clementi M, Di Gianantonio E, Fabris L, et al. Inheritance of hyperbilirubinemia: evidence for a major autosomal recessive gene. Dig Liver Dis 2007; 39:351–355.
- Roche SP, Kobos R. Jaundice in the adult patient. Am Fam Physician 2004; 69:299–304.
- Odemis B, Parlak E, Basar O, Yüksel O, Sahin B. Biliary tract obstruction secondary to malignant lymphoma: experience at a referral center. Dig Dis Sci 2007; 52:2323–2332.
- Caddy GR, Tham TC. Gallstone disease: symptoms, diagnosis and endoscopic management of common bile duct stones. Best Pract Res Clin Gastroenterol 2006; 20:1085–1101.
- Heathcote EJ. Diagnosis and management of cholestatic liver disease. Clin Gastroenterol Hepatol 2007; 5:776–782.
- Faust TW, Reddy KR. Postoperative jaundice. Clin Liver Dis 2004; 8:151–166.
- Maticic M, Poljak M, Lunder T, Rener-Sitar K, Stojanovic L. Lichen planus and other cutaneous manifestations in chronic hepatitis C: pre- and post-interferon-based treatment prevalence vary in a cohort of patients from low hepatitis C virus endemic area. J Eur Acad Dermatol Venereol 2008; 22:779–788.
- Polat M, Oztas P, Ilhan MN, Yalçin B, Alli N. Generalized pruritus: a prospective study concerning etiology. Am J Clin Dermatol 2008; 9:39–44.
- Gaspari R, Avolio AW, Zileri Dal Verme L, et al. Molecular adsorbent recirculating system in liver transplantation: safety and efficacy. Transplant Proc 2006; 38:3544–3551.
- Dasgupta R, Saha I, Pal S, et al. Immunosuppression, hepatotoxicity and depression of antioxidant status by arecoline in albino mice. Toxicology 2006; 227:94–104.
- Mayo MJ, Handem I, Saldana S, Jacobe H, Getachew Y, Rush AJ. Sertraline as a first-line treatment for cholestatic pruritus. Hepatology 2007; 45:666–674.
- Mela M, Mancuso A, Burroughs AK. Review article: pruritus in cholestatic and other liver diseases. Aliment Pharmacol Ther 2003; 17:857–870.
- Montero JL, Pozo JC, Barrera P, et al. Treatment of refractory cholestatic pruritus with molecular adsorbent recirculating system (MARS). Transplant Proc 2006; 38:2511–2513.
- Neff GW, O'Brien CB, Reddy KR, et al. Preliminary observation with dronabinol in patients with intractable pruritus secondary to cholestatic liver disease. Am J Gastroenterol 2002; 97:2117–2119.
- Neri S, Raciti C, D’Angelo G, Ierna D, Bruno CM. Hyde’s prurigo nodularis and chronic HCV hepatitis. J Hepatol 1998; 28:161–164.
- Brown MA, George CR, Dunstan CR, Kalowski S, Corrigan AB. Prurigo nodularis and aluminium overload in maintenance haemodialysis. Lancet 1992; 340:48.
- Stander S, Luger T, Metze D. Treatment of prurigo nodularis with topical capsaicin. J Am Acad Dermatol 2001; 44:471–478.
- Requena L, Sangueza OP. Cutaneous vascular anomalies. Part I. Hamartomas, malformations, and dilation of preexisting vessels. J Am Acad Dermatol 1997; 37:523–549.
- Khasnis A, Gokula RM. Spider nevus. J Postgrad Med 2002; 48:307–309.
- Kaul V, Friedenberg FK, Braitman LE, et al. Development and validation of a model to diagnose cirrhosis in patients with hepatitis C. Am J Gastroenterol 2002; 97:2623–2628.
- Banyai AL. There is more than surface appearance to skin spiders. Chest 1971; 60:48.
- Errickson CV, Matus NR. Skin disorders of pregnancy. Am Fam Physician 1994; 49:605–610.
- Li CP, Lee FY, Hwang SJ, et al. Spider angiomas in patients with liver cirrhosis: role of alcoholism and impaired liver function. Scand J Gastroenterol 1999; 34:520–523.
- Li CP, Lee FY, Hwang SJ, et al. Role of substance P in the pathogenesis of spider angiomas in patients with nonalcoholic liver cirrhosis. Am J Gastroenterol 1999; 94:502–507.
- Sadick NS, Niedt GW. A study of estrogen and progesterone receptors in spider telangiectasias of the lower extremities. J Dermatol Surg Oncol 1990; 16:620–623.
- Henry F, Quatresooz P, Valverde-Lopez JC, Pierard GE. Blood vessel changes during pregnancy: a review. Am J Clin Dermatol 2006; 7:65–69.
- Finn SM, Rowland M, Lawlor F, et al. The significance of cutaneous spider naevi in children. Arch Dis Child 2006; 91:604–605.
- Peyrot I, Boulinguez S, Sparsa A, Le Meur Y, Bonnetblanc JM, Bedane C. Bier’s white spots associated with scleroderma renal crisis. Clin Exp Dermatol 2007; 32:165–167.
- Satoh T, Yokozeki H, Nishioka K. Vascular spiders and paper money skin improved by hemodialysis. Dermatology 2002; 205:73–74.
- Serrao R, Zirwas M, English JC. Palmar erythema. Am J Clin Dermatol 2007; 8:347–356.
- Matsumoto M, Ohki K, Nagai I, Oshibuchi T. Lung traction causes an increase in plasma prostacyclin concentration and decrease in mean arterial blood pressure. Anesth Analg 1992; 75:773–776.
- Otto AI, Horvath I, Feldmann J. Multiple firm, painless erythematous papules with a yellowish hue. Arch Dermatol 2005; 141:1595–1600.
- Gandelman G, Aronow WS, Weiss MB. Resolving hyperlipidemia after liver transplantation in a patient with primary sclerosing cholangitis. Am J Ther 2006; 13:171–174.
- Assy N, Kaita K, Mymin D, Levy C, Rosser B, Minuk G. Fatty infiltration of liver in hyperlipidemic patients. Dig Dis Sci 2000; 45:1929–1934.
- Allocca M, Crosignani A, Gritti A, et al. Hypercholesterolaemia is not associated with early atherosclerotic lesions in primary biliary cirrhosis. Gut 2006; 55:1795–1800.
- Dickson E, Fleming C, Ludwig J. Primary biliary cirrhosis. In:Popper H, Schaffner F, editors. Progress in Liver Diseases. New York: Grune and Stratton; 1978:487.
- Elner VM, Mintz R, Demirci H, Hassan AS. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Trans Am Ophthalmol Soc 2005; 103:69–73.
- Craxi A, Camma C, Giunta M. Clinical aspects of bleeding complications in cirrhotic patients. Blood Coagul Fibrinolysis 2000; 11( suppl 1):S75–579.
- Kajihara M, Okazaki Y, Kato S, et al. Evaluation of platelet kinetics in patients with liver cirrhosis: similarity to idiopathic thrombocytopenic purpura. J Gastroenterol Hepatol 2007; 22:112–118.
- Levine N. Patient reports six-month history of minimally pruritic purple dots on legs. Non-blanching macules developed over six months. Geriatrics 2006; 61:22.
- Barton JC, Rao SV, Pereira NM, et al. Juvenile hemochromatosis in the southeastern United States: a report of seven cases in two kinships. Blood Cells Mol Dis 2002; 29:104–115.
- Smith AG, Shuster S, Bomford A, Williams R. Plasma immunoreactive beta-melanocyte-stimulating hormone in chronic liver disease and fulminant hepatic failure. J Invest Dermatol 1978; 70:326–327.
- Barton JC, McDonnell SM, Adams PC, et al. Management of hemochromatosis. Hemochromatosis Management Working Group. Ann Intern Med 1998; 129:932–939.
- Bahnsen M, Gluud C, Johnsen SG, et al. Pituitary-testicular function in patients with alcoholic cirrhosis of the liver. Eur J Clin Invest 1981; 11:473–479.
- Kumar N, Aggarwal SR, Anand BS. Comparison of truncal hair distribution in alcoholic liver disease and alcohol-related chronic pancreatitis. J Gastroenterol Hepatol 2001; 16:855–856.
- Holzberg M, Walker HK. Terry's nails: revised definition and new correlations. Lancet 1984; 1:896–899.
- Mandayam S, Jamal MM, Morgan TR. Epidemiology of alcoholic liver disease. Semin Liver Dis 2004; 24:217–232.
- Belle SH, Beringer KC, Detre KM. Liver transplantation in the United States: results from the National Pitt-UNOS Liver Transplant Registry. United Network for Organ Sharing Clin Transpl 1994:19–35.
- Dunn W, Xu R, Schwimmer JB. Modest wine drinking and decreased prevalence of suspected nonalcoholic fatty liver disease. Hepatology 2008; 47:1947–1954.
- Afford SC, Fisher NC, Neil DA, et al. Distinct patterns of chemokine expression are associated with leukocyte recruitment in alcoholic hepatitis and alcoholic cirrhosis. J Pathol 1998; 186:82–89.
- Evstaf’ev VV, Levin MM. Dermatologic pathology in chronic alcoholics. Vestn Dermatol Venerol 1989; 8:72–74.
- Jerosch-Herold C, Shepstone L, Chojnowski AJ, Larson D. Splinting after contracture release for Dupuytren's contracture (SCoRD): protocol of a pragmatic, multi-centre, randomized controlled trial. BMC Musculoskelet Disord 2008; 9:62.
- Houghton S, Holdstock G, Cockerell R, Wright R. Dupuytren’s contracture, chronic liver disease and IgA immune complexes. Liver 1983; 3:220–224.
- Ibbotson SH. Disseminated superficial porokeratosis: what is the association with ultraviolet radiation? Clin Exp Dermatol 1996; 21:48–50.
- Kono T, Kobayashi H, Ishii M, Nishiguchi S, Taniguchi S. Synchronous development of disseminated superficial porokeratosis and hepatitis C virus-related hepatocellular carcinoma. J Am Acad Dermatol 2000; 43:966–968.
- Park BS, Moon SE, Kim JA. Disseminated superficial porokeratosis in a patient with chronic liver disease. J Dermatol 1997; 24:485–487.
- Murata Y, Kumano K, Takai T. Type 2 segmental manifestation of disseminated superficial porokeratosis showing a systematized pattern of involvement and pronounced cancer proneness. Eur J Dermatol 2001; 11:191–194.
- Galossi A, Guarisco R, Bellis L, Puoti C. Extrahepatic manifestations of chronic HCV infection. J Gastrointestin Liver Dis 2007; 16:65–73.
- El-Serag HB, Hampel H, Yeh C, Rabeneck L. Extrahepatic manifestations of hepatitis C among United States male veterans. Hepatology 2002; 36:1439–1445.
- Stefanova-Petrova DV, Tzvetanska AH, Naumova EJ, et al. Chronic hepatitis C virus infection: prevalence of extrahepatic manifestations and association with cryoglobulinemia in Bulgarian patients. World J Gastroenterol 2007; 13:6518–6528.
- Vassilopoulos D, Calabrese LH. Extrahepatic immunological complications of hepatitis C virus infection. AIDS 2005; 19( suppl 3):S123–S127.
- Hsing AW, Zhang M, Rashid A, et al. Hepatitis B and C virus infection and the risk of biliary tract cancer: a population-based study in China. Int J Cancer 2008; 122:1849–1853.
- Madrid C, Courtois B, Duran D. Chronic sialadenitis revealing hepatitis C: a case report. Med Oral 2004; 9:328–332.
- Lançoni G, Ravinal RC, Costa RS, Roselino AM. Mast cells and transforming growth factor-beta expression: a possible relationship in the development of porphyria cutanea tarda skin lesions. Int J Dermatol 2008; 47:575–581.
- Toll A, Celis R, Ozalla MD, Bruguera M, Herrero C, Ercilla MG. The prevalence of HFE C282Y gene mutation is increased in Spanish patients with porphyria cutanea tarda without hepatitis C virus infection. J Eur Acad Dermatol Venereol 2006; 20:1201–1206.
- Badminton MN, Elder GH. Management of acute and cutaneous porphyrias. Int J Clin Pract 2002; 56:272–278.
- Jackson JM, Callen JP. Scarring alopecia and sclerodermatous changes of the scalp in a patient with hepatitis C infection. J Am Acad Dermatol 1998; 39:824–826.
- Mortimore M, Merryweather-Clarke AT, Robson KJ, Powell LW. The haemochromatosis gene: a global perspective and implications for the Asia-Pacific region. J Gastroenterol Hepatol 1999; 14:838–843.
- Shehan JM, Huerter CJ. Porphyria cutanea tarda associated with an acute gastrointestinal bleed: the roles of supplemental iron and blood transfusion. Cutis 2001; 68:147–150.
- Lambrecht RW, Thapar M, Bonkovsky HL. Genetic aspects of porphyria cutanea tarda. Semin Liver Dis 2007; 27:99–108.
- d’Ovidio R, Sgarra C, Conserva A, Angelotti UF, Erriquez R, Foti C. Alterated integrin expression in lichen planopilaris. Head Face Med 2007; 3:11.
- Chuang TY, Stitle L, Brashear R, Lewis C. Hepatitis C virus and lichen planus: a case-control study of 340 patients. J Am Acad Dermatol 1999; 41:787–789.
- Kemp EH, Gavalas NG, Gawkrodger DJ, Weetman AP. Autoantibody responses to melanocytes in the depigmenting skin disease vitiligo. Autoimmun Rev 2007; 6:138–142.
- Tomasiewicz K, Modrzewska R, Semczuk G. Vitiligo associated with pegylated interferon and ribavirin treatment of patients with chronic hepatitis C: a case report. Adv Ther 2006; 23:139–142.
- Simsek H, Savas C, Akkiz H, Telatar H. Interferon-induced vitiligo in a patient with chronic viral hepatitis C infection. Dermatology 1996; 193:65–66.
- Mulekar SV, Al Issa A, Asaad M, Ghwish B, Al Eisa A. Mixed vitiligo. J Cutan Med Surg. 2006; 10:104–107.
- Waalen J, Felitti V, Gelbart T, Ho NJ, Beutler E. Prevalence of hemochromatosis-related symptoms among individuals with mutations in the HFE gene. Mayo Clin Proc 2002; 77:522–530.
- Walker AP, Tucker DC, Hall MA, et al. Results communication and patient education after screening for possible hemochromatosis and iron overload: experience from the HEIRS study of a large ethnically and linguistically diverse group. Genet Med 2007; 9:778–791.
- Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, cafe au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician 2003; 68:1955–1960.
- Tsuji T. Experimental hemosiderosis: relationship between skin pigmentation and hemosiderin. Acta Derm Venereol 1980; 60:109–114.
- Oji V, Traupe H. Ichthyoses: differential diagnosis and molecular genetics. Eur J Dermatol 2006; 16:349–359.
Dysfunction in the body’s second largest organ, the liver, often yields changes in the body’s largest organ, the skin. If we can recognize these manifestations early, we are better able to promptly diagnose and treat the underlying liver disease, as well as the skin lesions.
The liver has many jobs: synthesizing proteins such as clotting factors, complements, and albumin; neutralizing toxins; and metabolizing lipids and carbohydrates. Insults to the liver can compromise any of these functions, affecting visceral organs, joints, gastrointestinal tissues, and the skin. Dermatologic signs of specific liver diseases include alopecia and vitiligo associated with autoimmune hepatitis, and xanthelasma in chronic cholestatic liver disease.
This article reviews the important cutaneous manifestations of specific liver diseases. We focus first on skin conditions that may represent liver disease, and then we discuss several major liver diseases and their typical cutaneous manifestations.
JAUNDICE AND HYPERBILIRUBINEMIA
Establishing whether the excess bilirubin is conjugated or unconjugated gives a clue as to whether the cause is prehepatic, intrahepatic, or posthepatic.3–8 One of the liver’s main functions is to conjugate bilirubin into a secretable form. Prehepatic causes of jaundice include hemolysis and ineffective erythropoiesis, both of which lead to higher levels of circulating unconjugated bilirubin.4 Intrahepatic causes of jaundice can lead to both unconjugated and conjugated hyperbilirubinemia.4,8 Posthepatic causes such as bile duct obstruction primarily result in conjugated hyperbilirubinemia.4
PRURITUS AND PRURIGO NODULARIS
Pruritus can be multifactorial or the result of a specific dermatologic or systemic condition.9 A thorough history and physical examination are warranted to rule out hepatic or systemic causes of itching.10
The liver neutralizes toxins and filters bile salts. If its function is impaired, these materials can accumulate in the body, and deposition in the skin causes irritation and itching.11,12 In cholestatic liver disorders such as primary sclerosing cholangitis and obstructive gallstone disease, pruritus tends to be generalized, but worse on the hands and feet.13
Although the severity of pruritus is not directly associated with the level of bile salts and toxic substances, lowering bile salt levels can mitigate symptoms.11
Treatment. Pruritus due to liver disease is particularly resistant to therapy.
In a strategy described by Mela et al for managing pruritus in chronic liver disease,14 the initial treatment is the anion exchange resin cholestyramine (Questran) at a starting dose of 4 g/day, gradually increased to 24 g/day in two doses at mealtimes.
If the pruritus does not respond adequately to cholestyramine or the patient cannot tolerate the drug, then the antituberculosis drug rifampin (Rifadin) can be tried. Rifampin promotes metabolism of endogenous pruritogens and has been effective against cholestatic pruritus when started at 150 mg/day and increased up to 600 mg/day, depending on the clinical response.14
Third-line drug therapies include opioid antagonists such as naltrexone (ReVia) and nalmefene (Revex).14,15
Plasmapheresis can be considered if drug therapy fails.16 Experimental therapies include albumin dialysis using the molecular adsorbent recirculating system (a form of artificial liver support), antioxidant treatment, and bright-light therapy.15 Liver transplantation, when appropriate, also resolves cholestatic pruritus.14
Prurigo nodularis
Prurigo nodularis, distinguished by firm, crusty nodules, is associated with viral infections (eg, hepatitis C, human immunodeficiency virus), bacterial infections, and kidney dysfunction.17,18 The lesions are intensely pruritic and often lead to persistent scratching, excoriation, and, ultimately, diffuse scarring.19
Treatment. Although the exact cause of prurigo nodularis is not known and no cure exists, corticosteroid or antihistamine ointments control the symptoms in most patients with hepatitis.19 Low doses of thalidomide (Thalomid), a tumor necrosis factor antagonist, have also been used safely and effectively.18,19
SUPERFICIAL VASCULAR SIGNS
Spider angiomas
Spider angiomas can occur anywhere on the body, but they occur most often on the face and the trunk.21,23 A key feature is that they disappear when pressure is applied and reappear when pressure is removed.23,24 Biopsy is rarely necessary for diagnosis.
These lesions occur with elevated estrogen levels, such as in cirrhosis, during estrogen therapy, or during pregnancy.25–28 Although spider angiomas are common in pregnant women and in children, adults with spider angiomas deserve a workup for liver dysfunction.29
Given their innocuous nature and asymptomatic course, spider angiomas themselves require no medical treatment.
Bier spots
Bier spots are small, irregularly shaped, hypopigmented patches on the arms and legs. They are likely due to venous stasis associated with functional damage to the small vessels of the skin.30
Since Bier spots are a sign of liver disease, they must be distinguished from true pigmentation disorders. A key distinguishing feature is that Bier spots disappear when pressure is applied. Also, raising the affected limb from a dependent position causes the hypopigmented macules of Bier spots to disappear, which is not the case in true pigmentation disorders.10,30
Paper-money skin
Paper-money skin (or “dollar-paper” markings) describes the condition in which the upper trunk is covered with many randomly scattered, needle-thin superficial capillaries. It often occurs in association with spider angiomas. The name comes from the resemblance the thread-like capillaries have to the finely chopped silk threads in American dollar bills.10,31 The condition is commonly seen in patients with alcoholic cirrhosis and may improve with hemodialysis.31
PALMAR ERYTHEMA
Palmar erythema is a florid, crimson coloration of the palms of the hands and the fingertips. It can occur anywhere on the palm and fingers but is most common on the hypothenar eminence. It can occur in a number of liver conditions but most often with cirrhosis.32 Hepatic compromise, as seen in alcoholic liver disease, disrupts the body’s androgen balance, causing local vasodilation and erythema.32,33 Although the exact mechanism remains unknown, research suggests that prostacyclins and nitric oxide play a role, as both are increased in liver disease.32,33
XANTHELASMA
Xanthelasma—a localized cholesterol deposit beneath the skin and especially beneath the eyelids—is a common manifestation of hypercholesterolemia. Xanthelasma often presents as a painless, yellowish, soft plaque with well-defined borders,34 which may enlarge over the course of weeks.
Several liver diseases can lead to various forms of secondary dyslipoproteinemia.35 The most common dyslipoproteinemias in liver disease are hypertriglyceridemia and low levels of high-density lipoprotein cholesterol, and both of these often accompany fatty liver disease.36 Hypercholesterolemia is a common feature of primary biliary cirrhosis and other forms of cholestatic liver disease.37 Studies suggest that the total plasma cholesterol level is elevated in as many as 50% of patients with compromised liver function.38
Treatment. The underlying hyperlipidemia is treated with cholesterol-lowering drugs. Laser treatment and surgical excision have proven efficacious in treating the lesions.39
OTHER CUTANEOUS FINDINGS IN LIVER DISEASE
Bleeding and bruising. Liver disease can cause hypersplenism and thrombocytopenia, in addition to a decrease in clotting factors. These may present with a myriad of cutaneous symptoms, including purpura, bleeding gums, and easy bruising and bleeding, even from minor trauma.40–42
Hyperpigmentation of the skin may accompany hemochromatosis, alcoholic liver disease, and cirrhosis.43–45
“Terry’s nails,” in which the proximal two-thirds of the nail plate turns powdery white with a ground-glass opacity, may develop in patients with advanced cirrhosis.48
ALCOHOLIC CIRRHOSIS AND THE SKIN
The cutaneous changes associated with alcoholic cirrhosis are more widely recognized than those due to other forms of liver dysfunction. In the United States, approximately 3 million people have alcoholic cirrhosis, the second-leading reason for liver transplantation.49,50
As the body’s main site of alcohol metabolism, the liver is the organ most affected by excessive alcohol intake, which can lead to end-stage liver disease secondary to alcoholic cirrhosis.41,51 The characteristic feature of cirrhosis is advanced fibrous scarring of parenchymal tissue and the formation of regenerative nodules with increased resistance to blood flow throughout the organ.41,52 The insufficient blood flow damages vital structures in the liver and compromises liver function. For example, liver cirrhosis leads to defective hepatic synthesis of clotting factors and results in bleeding disorders.
Cutaneous lesions often accompany alcoholic cirrhosis and have been detected in up to 43% of people with chronic alcoholism.53 Skin changes in alcoholic cirrhosis can be of great diagnostic value. The combined prevalence of spider angiomas, palmar erythema, and Dupuytren contracture in alcoholic cirrhosis was found to be 72%. Paper-money skin and Dupuytren contracture are more distinct lesions for alcoholic cirrhosis.31 Recognizing these skin changes contributes to the diagnosis and staging of liver cirrhosis.51,52
Dupuytren contracture
Dupuytren contracture is characterized by progressive fibrosis and thickening of tendons in the palmar fascia, the connective tissue that lies beneath the skin of the palms.54 Over time, as fibrotic involvement expands across the fascia, rampant stiffness of the joints ensues, sometimes to a point where the fingers cannot fully flex or extend.54
Although the exact cause of Dupuytren contracture is unknown, it appears to be associated with excess alcohol consumption and can be found in patients with alcoholic cirrhosis.54,55 These patients often present with painless stiffness of the fingers, curling of fingers, and loss of motion in involved fingers.54 Surgery in the form of limited fasciectomy has been curative in such patients.54
Disseminated superficial porokeratosis
Porokeratosis is a keratinization disorder of clonal origin that presents as a linear configuration of white scaly papules that coalesce into plaques throughout the body.56 Although it most commonly afflicts fair-skinned people, patients with alcoholic cirrhosis have a much greater susceptibility than the general population.57,58
A recent study58 documented that the lesions completely resolved when liver function improved, thus underlining the relationship between the two conditions. Since immunosuppression has been linked to eruption of the lesion, the fact that both humoral and cell-mediated immune responses are impaired in alcoholic liver disease provides another dimension to the association between porokeratosis and alcoholic cirrhosis.58
These lesions can transform into squamous cell carcinoma.59 The risk of widespread metastases in squamous cell carcinoma highlights the importance of dermatologic consultation in such patients.59
HEPATITIS C AND THE SKIN
Extrahepatic manifestations have been documented in up to 74% of people with hepatitis C virus infection.60 In addition to parasthesias, arthralgias, and myalgias, hepatitis C has a significant association with porphyria cutanea tarda, lichen planus, vitiligo, sialadenitis, urticarial vasculitis, corneal ulcers, xerosis, pruritus, and prurigo nodularis.60–64 Although the primary causative agents of sialadenitis are bacteria, viruses such as hepatitis C have been implicated as a cause of chronic sialadenitis with associated xerostomia.65
Patients with hepatitis C being treated with interferons also present with cutaneous manifestations such as hyperkeratosis and vasculitis.63
Porphyria cutanea tarda
Porphyria cutanea tarda is the most common of the porphyrias, disorders distinguished by deficiencies or defects in one or more of the enzymes responsible for hepatic production of heme.66 If these enzymes are impaired, heme precursors such as porphyrins accumulate.66
Porphyria cutanea tarda results from a deficiency of the hepatic enzyme uroporphyrin decarboxylase. In the absence of this enzyme, shortwave visible light activates uroporphyrin deposited in the skin, resulting in a photochemical reaction that generates reactive oxygen species that lead to the characteristic skin blistering.
Although porphyria cutanea tarda is associated with liver disease in general, recent studies confirm that patients with hepatitis C are at particularly high risk.67 Those with the disorder often present with skin photosensitivity. 68 Many develop blisters on sun-exposed skin, including the dorsal aspects of the hands and forearms and on the neck and face. Chronic porphyria cutanea tarda can lead to scarring, alopecia, and skin ulceration.69 As the blisters heal, keratin-filled milial cysts may develop in the areas of ulceration.
The condition is also commonly associated with melasma-like hyperpigmentation and hypertrichosis in sun-exposed areas of the head and neck. People of Northern European ancestry may be more at risk than the general population because of a presumed genetic susceptibility.70
Treatment. Because many patients with porphyria cutanea tarda have iron overload, they need to restrict foods rich in iron and to avoid alcohol.71,72 Severe cases may necessitate iron removal via phlebotomy or antimalarial therapy. Patients with porphyria cutanea tarda induced by hepatitis C should have their bodily iron stores depleted before starting antiviral therapy.60
Lichen planus
Lichen planopilaris is a subset of lichen planus that causes scaling and atrophy of the scalp and permanent hair loss (Figure 4).73
Interferon-induced vitiligo
Vitiligo is an autoimmune disease in which melanocytes in the skin are destroyed, with resulting depigmentation in affected areas.75 Although it has no specific association with liver disease, it has been linked to treatments for hepatitis C such as interferons.76 Interferon-induced vitiligo often completely resolves when interferon is stopped.77
Typical findings include aggregations of irregularly shaped white patches in a focal or segmental pattern.78 The diagnosis is based on the medical history, physical examination, and sometimes skin biopsy.
HEMOCHROMATOSIS
Hemochromatosis or “bronze diabetes” is a devastating multisystem disease with a relentless course. It is among the most common genetic disorders of metabolism, and results in deposition of iron in tissues and organs throughout the body, including the liver, usually in patients ages 30 to 40.
As iron stores increase in tissues and organs, multiorgan failure and associated complications may ensue. In addition, surplus iron stores can also result in widespread bronze discoloration of skin exposed to the sun. Hemochromatosis also results in loss of body hair, ichthyosiform alterations, and koilonychia.79
Treatments that lower serum iron levels can reverse the cutaneous manifestations of the disorder and minimize the risk of organ failure.
Since the condition is inherited in an autosomal-recessive pattern, family members of patients should consider being screened.80
Hyperpigmentation in hemochromatosis
Hyperpigmentation is an early sign of hemochromatosis, affecting up to 90% of patients. Usually, sun-exposed areas of the body are the most prone and take on a grayish or brownish-bronze hue.81 Cutaneous iron deposits injure vital skin structures, initiating a process that culminates in enhanced melanin production by melanocytes.82 Exposure to ultraviolet light may have synergistic effects with iron, hastening the process of hyperpigmentation. As a result of this synergistic effect, many patients with hemochromatosis notice tanning with minimal sun exposure.
Although organ function can improve immediately with phlebotomy to reduce iron stores, skin hyperpigmentation does not immediately resolve.81,82
Ichthyosiform alterations in hemochromatosis
Ichthyosiform changes, in which the skin takes on the appearance of fish scales,83 can be seen in patients with hemochromatosis.80 Affected areas typically become extremely dry. Treatment includes topical hydrating creams and ointments. Avoiding sunlight is paramount, as sunlight exposure may exacerbate the condition.83
Dysfunction in the body’s second largest organ, the liver, often yields changes in the body’s largest organ, the skin. If we can recognize these manifestations early, we are better able to promptly diagnose and treat the underlying liver disease, as well as the skin lesions.
The liver has many jobs: synthesizing proteins such as clotting factors, complements, and albumin; neutralizing toxins; and metabolizing lipids and carbohydrates. Insults to the liver can compromise any of these functions, affecting visceral organs, joints, gastrointestinal tissues, and the skin. Dermatologic signs of specific liver diseases include alopecia and vitiligo associated with autoimmune hepatitis, and xanthelasma in chronic cholestatic liver disease.
This article reviews the important cutaneous manifestations of specific liver diseases. We focus first on skin conditions that may represent liver disease, and then we discuss several major liver diseases and their typical cutaneous manifestations.
JAUNDICE AND HYPERBILIRUBINEMIA
Establishing whether the excess bilirubin is conjugated or unconjugated gives a clue as to whether the cause is prehepatic, intrahepatic, or posthepatic.3–8 One of the liver’s main functions is to conjugate bilirubin into a secretable form. Prehepatic causes of jaundice include hemolysis and ineffective erythropoiesis, both of which lead to higher levels of circulating unconjugated bilirubin.4 Intrahepatic causes of jaundice can lead to both unconjugated and conjugated hyperbilirubinemia.4,8 Posthepatic causes such as bile duct obstruction primarily result in conjugated hyperbilirubinemia.4
PRURITUS AND PRURIGO NODULARIS
Pruritus can be multifactorial or the result of a specific dermatologic or systemic condition.9 A thorough history and physical examination are warranted to rule out hepatic or systemic causes of itching.10
The liver neutralizes toxins and filters bile salts. If its function is impaired, these materials can accumulate in the body, and deposition in the skin causes irritation and itching.11,12 In cholestatic liver disorders such as primary sclerosing cholangitis and obstructive gallstone disease, pruritus tends to be generalized, but worse on the hands and feet.13
Although the severity of pruritus is not directly associated with the level of bile salts and toxic substances, lowering bile salt levels can mitigate symptoms.11
Treatment. Pruritus due to liver disease is particularly resistant to therapy.
In a strategy described by Mela et al for managing pruritus in chronic liver disease,14 the initial treatment is the anion exchange resin cholestyramine (Questran) at a starting dose of 4 g/day, gradually increased to 24 g/day in two doses at mealtimes.
If the pruritus does not respond adequately to cholestyramine or the patient cannot tolerate the drug, then the antituberculosis drug rifampin (Rifadin) can be tried. Rifampin promotes metabolism of endogenous pruritogens and has been effective against cholestatic pruritus when started at 150 mg/day and increased up to 600 mg/day, depending on the clinical response.14
Third-line drug therapies include opioid antagonists such as naltrexone (ReVia) and nalmefene (Revex).14,15
Plasmapheresis can be considered if drug therapy fails.16 Experimental therapies include albumin dialysis using the molecular adsorbent recirculating system (a form of artificial liver support), antioxidant treatment, and bright-light therapy.15 Liver transplantation, when appropriate, also resolves cholestatic pruritus.14
Prurigo nodularis
Prurigo nodularis, distinguished by firm, crusty nodules, is associated with viral infections (eg, hepatitis C, human immunodeficiency virus), bacterial infections, and kidney dysfunction.17,18 The lesions are intensely pruritic and often lead to persistent scratching, excoriation, and, ultimately, diffuse scarring.19
Treatment. Although the exact cause of prurigo nodularis is not known and no cure exists, corticosteroid or antihistamine ointments control the symptoms in most patients with hepatitis.19 Low doses of thalidomide (Thalomid), a tumor necrosis factor antagonist, have also been used safely and effectively.18,19
SUPERFICIAL VASCULAR SIGNS
Spider angiomas
Spider angiomas can occur anywhere on the body, but they occur most often on the face and the trunk.21,23 A key feature is that they disappear when pressure is applied and reappear when pressure is removed.23,24 Biopsy is rarely necessary for diagnosis.
These lesions occur with elevated estrogen levels, such as in cirrhosis, during estrogen therapy, or during pregnancy.25–28 Although spider angiomas are common in pregnant women and in children, adults with spider angiomas deserve a workup for liver dysfunction.29
Given their innocuous nature and asymptomatic course, spider angiomas themselves require no medical treatment.
Bier spots
Bier spots are small, irregularly shaped, hypopigmented patches on the arms and legs. They are likely due to venous stasis associated with functional damage to the small vessels of the skin.30
Since Bier spots are a sign of liver disease, they must be distinguished from true pigmentation disorders. A key distinguishing feature is that Bier spots disappear when pressure is applied. Also, raising the affected limb from a dependent position causes the hypopigmented macules of Bier spots to disappear, which is not the case in true pigmentation disorders.10,30
Paper-money skin
Paper-money skin (or “dollar-paper” markings) describes the condition in which the upper trunk is covered with many randomly scattered, needle-thin superficial capillaries. It often occurs in association with spider angiomas. The name comes from the resemblance the thread-like capillaries have to the finely chopped silk threads in American dollar bills.10,31 The condition is commonly seen in patients with alcoholic cirrhosis and may improve with hemodialysis.31
PALMAR ERYTHEMA
Palmar erythema is a florid, crimson coloration of the palms of the hands and the fingertips. It can occur anywhere on the palm and fingers but is most common on the hypothenar eminence. It can occur in a number of liver conditions but most often with cirrhosis.32 Hepatic compromise, as seen in alcoholic liver disease, disrupts the body’s androgen balance, causing local vasodilation and erythema.32,33 Although the exact mechanism remains unknown, research suggests that prostacyclins and nitric oxide play a role, as both are increased in liver disease.32,33
XANTHELASMA
Xanthelasma—a localized cholesterol deposit beneath the skin and especially beneath the eyelids—is a common manifestation of hypercholesterolemia. Xanthelasma often presents as a painless, yellowish, soft plaque with well-defined borders,34 which may enlarge over the course of weeks.
Several liver diseases can lead to various forms of secondary dyslipoproteinemia.35 The most common dyslipoproteinemias in liver disease are hypertriglyceridemia and low levels of high-density lipoprotein cholesterol, and both of these often accompany fatty liver disease.36 Hypercholesterolemia is a common feature of primary biliary cirrhosis and other forms of cholestatic liver disease.37 Studies suggest that the total plasma cholesterol level is elevated in as many as 50% of patients with compromised liver function.38
Treatment. The underlying hyperlipidemia is treated with cholesterol-lowering drugs. Laser treatment and surgical excision have proven efficacious in treating the lesions.39
OTHER CUTANEOUS FINDINGS IN LIVER DISEASE
Bleeding and bruising. Liver disease can cause hypersplenism and thrombocytopenia, in addition to a decrease in clotting factors. These may present with a myriad of cutaneous symptoms, including purpura, bleeding gums, and easy bruising and bleeding, even from minor trauma.40–42
Hyperpigmentation of the skin may accompany hemochromatosis, alcoholic liver disease, and cirrhosis.43–45
“Terry’s nails,” in which the proximal two-thirds of the nail plate turns powdery white with a ground-glass opacity, may develop in patients with advanced cirrhosis.48
ALCOHOLIC CIRRHOSIS AND THE SKIN
The cutaneous changes associated with alcoholic cirrhosis are more widely recognized than those due to other forms of liver dysfunction. In the United States, approximately 3 million people have alcoholic cirrhosis, the second-leading reason for liver transplantation.49,50
As the body’s main site of alcohol metabolism, the liver is the organ most affected by excessive alcohol intake, which can lead to end-stage liver disease secondary to alcoholic cirrhosis.41,51 The characteristic feature of cirrhosis is advanced fibrous scarring of parenchymal tissue and the formation of regenerative nodules with increased resistance to blood flow throughout the organ.41,52 The insufficient blood flow damages vital structures in the liver and compromises liver function. For example, liver cirrhosis leads to defective hepatic synthesis of clotting factors and results in bleeding disorders.
Cutaneous lesions often accompany alcoholic cirrhosis and have been detected in up to 43% of people with chronic alcoholism.53 Skin changes in alcoholic cirrhosis can be of great diagnostic value. The combined prevalence of spider angiomas, palmar erythema, and Dupuytren contracture in alcoholic cirrhosis was found to be 72%. Paper-money skin and Dupuytren contracture are more distinct lesions for alcoholic cirrhosis.31 Recognizing these skin changes contributes to the diagnosis and staging of liver cirrhosis.51,52
Dupuytren contracture
Dupuytren contracture is characterized by progressive fibrosis and thickening of tendons in the palmar fascia, the connective tissue that lies beneath the skin of the palms.54 Over time, as fibrotic involvement expands across the fascia, rampant stiffness of the joints ensues, sometimes to a point where the fingers cannot fully flex or extend.54
Although the exact cause of Dupuytren contracture is unknown, it appears to be associated with excess alcohol consumption and can be found in patients with alcoholic cirrhosis.54,55 These patients often present with painless stiffness of the fingers, curling of fingers, and loss of motion in involved fingers.54 Surgery in the form of limited fasciectomy has been curative in such patients.54
Disseminated superficial porokeratosis
Porokeratosis is a keratinization disorder of clonal origin that presents as a linear configuration of white scaly papules that coalesce into plaques throughout the body.56 Although it most commonly afflicts fair-skinned people, patients with alcoholic cirrhosis have a much greater susceptibility than the general population.57,58
A recent study58 documented that the lesions completely resolved when liver function improved, thus underlining the relationship between the two conditions. Since immunosuppression has been linked to eruption of the lesion, the fact that both humoral and cell-mediated immune responses are impaired in alcoholic liver disease provides another dimension to the association between porokeratosis and alcoholic cirrhosis.58
These lesions can transform into squamous cell carcinoma.59 The risk of widespread metastases in squamous cell carcinoma highlights the importance of dermatologic consultation in such patients.59
HEPATITIS C AND THE SKIN
Extrahepatic manifestations have been documented in up to 74% of people with hepatitis C virus infection.60 In addition to parasthesias, arthralgias, and myalgias, hepatitis C has a significant association with porphyria cutanea tarda, lichen planus, vitiligo, sialadenitis, urticarial vasculitis, corneal ulcers, xerosis, pruritus, and prurigo nodularis.60–64 Although the primary causative agents of sialadenitis are bacteria, viruses such as hepatitis C have been implicated as a cause of chronic sialadenitis with associated xerostomia.65
Patients with hepatitis C being treated with interferons also present with cutaneous manifestations such as hyperkeratosis and vasculitis.63
Porphyria cutanea tarda
Porphyria cutanea tarda is the most common of the porphyrias, disorders distinguished by deficiencies or defects in one or more of the enzymes responsible for hepatic production of heme.66 If these enzymes are impaired, heme precursors such as porphyrins accumulate.66
Porphyria cutanea tarda results from a deficiency of the hepatic enzyme uroporphyrin decarboxylase. In the absence of this enzyme, shortwave visible light activates uroporphyrin deposited in the skin, resulting in a photochemical reaction that generates reactive oxygen species that lead to the characteristic skin blistering.
Although porphyria cutanea tarda is associated with liver disease in general, recent studies confirm that patients with hepatitis C are at particularly high risk.67 Those with the disorder often present with skin photosensitivity. 68 Many develop blisters on sun-exposed skin, including the dorsal aspects of the hands and forearms and on the neck and face. Chronic porphyria cutanea tarda can lead to scarring, alopecia, and skin ulceration.69 As the blisters heal, keratin-filled milial cysts may develop in the areas of ulceration.
The condition is also commonly associated with melasma-like hyperpigmentation and hypertrichosis in sun-exposed areas of the head and neck. People of Northern European ancestry may be more at risk than the general population because of a presumed genetic susceptibility.70
Treatment. Because many patients with porphyria cutanea tarda have iron overload, they need to restrict foods rich in iron and to avoid alcohol.71,72 Severe cases may necessitate iron removal via phlebotomy or antimalarial therapy. Patients with porphyria cutanea tarda induced by hepatitis C should have their bodily iron stores depleted before starting antiviral therapy.60
Lichen planus
Lichen planopilaris is a subset of lichen planus that causes scaling and atrophy of the scalp and permanent hair loss (Figure 4).73
Interferon-induced vitiligo
Vitiligo is an autoimmune disease in which melanocytes in the skin are destroyed, with resulting depigmentation in affected areas.75 Although it has no specific association with liver disease, it has been linked to treatments for hepatitis C such as interferons.76 Interferon-induced vitiligo often completely resolves when interferon is stopped.77
Typical findings include aggregations of irregularly shaped white patches in a focal or segmental pattern.78 The diagnosis is based on the medical history, physical examination, and sometimes skin biopsy.
HEMOCHROMATOSIS
Hemochromatosis or “bronze diabetes” is a devastating multisystem disease with a relentless course. It is among the most common genetic disorders of metabolism, and results in deposition of iron in tissues and organs throughout the body, including the liver, usually in patients ages 30 to 40.
As iron stores increase in tissues and organs, multiorgan failure and associated complications may ensue. In addition, surplus iron stores can also result in widespread bronze discoloration of skin exposed to the sun. Hemochromatosis also results in loss of body hair, ichthyosiform alterations, and koilonychia.79
Treatments that lower serum iron levels can reverse the cutaneous manifestations of the disorder and minimize the risk of organ failure.
Since the condition is inherited in an autosomal-recessive pattern, family members of patients should consider being screened.80
Hyperpigmentation in hemochromatosis
Hyperpigmentation is an early sign of hemochromatosis, affecting up to 90% of patients. Usually, sun-exposed areas of the body are the most prone and take on a grayish or brownish-bronze hue.81 Cutaneous iron deposits injure vital skin structures, initiating a process that culminates in enhanced melanin production by melanocytes.82 Exposure to ultraviolet light may have synergistic effects with iron, hastening the process of hyperpigmentation. As a result of this synergistic effect, many patients with hemochromatosis notice tanning with minimal sun exposure.
Although organ function can improve immediately with phlebotomy to reduce iron stores, skin hyperpigmentation does not immediately resolve.81,82
Ichthyosiform alterations in hemochromatosis
Ichthyosiform changes, in which the skin takes on the appearance of fish scales,83 can be seen in patients with hemochromatosis.80 Affected areas typically become extremely dry. Treatment includes topical hydrating creams and ointments. Avoiding sunlight is paramount, as sunlight exposure may exacerbate the condition.83
- Morioka D, Togo S, Kumamoto T, et al. Six consecutive cases of successful adult ABO-incompatible living donor liver transplantation: a proposal for grading the severity of antibody-mediated rejection. Transplantation 2008; 85:171–178.
- Bertini G, Rubaltelli FF. Non-invasive bilirubinometry in neonatal jaundice. Semin Neonatol 2002; 7:129–133.
- Clementi M, Di Gianantonio E, Fabris L, et al. Inheritance of hyperbilirubinemia: evidence for a major autosomal recessive gene. Dig Liver Dis 2007; 39:351–355.
- Roche SP, Kobos R. Jaundice in the adult patient. Am Fam Physician 2004; 69:299–304.
- Odemis B, Parlak E, Basar O, Yüksel O, Sahin B. Biliary tract obstruction secondary to malignant lymphoma: experience at a referral center. Dig Dis Sci 2007; 52:2323–2332.
- Caddy GR, Tham TC. Gallstone disease: symptoms, diagnosis and endoscopic management of common bile duct stones. Best Pract Res Clin Gastroenterol 2006; 20:1085–1101.
- Heathcote EJ. Diagnosis and management of cholestatic liver disease. Clin Gastroenterol Hepatol 2007; 5:776–782.
- Faust TW, Reddy KR. Postoperative jaundice. Clin Liver Dis 2004; 8:151–166.
- Maticic M, Poljak M, Lunder T, Rener-Sitar K, Stojanovic L. Lichen planus and other cutaneous manifestations in chronic hepatitis C: pre- and post-interferon-based treatment prevalence vary in a cohort of patients from low hepatitis C virus endemic area. J Eur Acad Dermatol Venereol 2008; 22:779–788.
- Polat M, Oztas P, Ilhan MN, Yalçin B, Alli N. Generalized pruritus: a prospective study concerning etiology. Am J Clin Dermatol 2008; 9:39–44.
- Gaspari R, Avolio AW, Zileri Dal Verme L, et al. Molecular adsorbent recirculating system in liver transplantation: safety and efficacy. Transplant Proc 2006; 38:3544–3551.
- Dasgupta R, Saha I, Pal S, et al. Immunosuppression, hepatotoxicity and depression of antioxidant status by arecoline in albino mice. Toxicology 2006; 227:94–104.
- Mayo MJ, Handem I, Saldana S, Jacobe H, Getachew Y, Rush AJ. Sertraline as a first-line treatment for cholestatic pruritus. Hepatology 2007; 45:666–674.
- Mela M, Mancuso A, Burroughs AK. Review article: pruritus in cholestatic and other liver diseases. Aliment Pharmacol Ther 2003; 17:857–870.
- Montero JL, Pozo JC, Barrera P, et al. Treatment of refractory cholestatic pruritus with molecular adsorbent recirculating system (MARS). Transplant Proc 2006; 38:2511–2513.
- Neff GW, O'Brien CB, Reddy KR, et al. Preliminary observation with dronabinol in patients with intractable pruritus secondary to cholestatic liver disease. Am J Gastroenterol 2002; 97:2117–2119.
- Neri S, Raciti C, D’Angelo G, Ierna D, Bruno CM. Hyde’s prurigo nodularis and chronic HCV hepatitis. J Hepatol 1998; 28:161–164.
- Brown MA, George CR, Dunstan CR, Kalowski S, Corrigan AB. Prurigo nodularis and aluminium overload in maintenance haemodialysis. Lancet 1992; 340:48.
- Stander S, Luger T, Metze D. Treatment of prurigo nodularis with topical capsaicin. J Am Acad Dermatol 2001; 44:471–478.
- Requena L, Sangueza OP. Cutaneous vascular anomalies. Part I. Hamartomas, malformations, and dilation of preexisting vessels. J Am Acad Dermatol 1997; 37:523–549.
- Khasnis A, Gokula RM. Spider nevus. J Postgrad Med 2002; 48:307–309.
- Kaul V, Friedenberg FK, Braitman LE, et al. Development and validation of a model to diagnose cirrhosis in patients with hepatitis C. Am J Gastroenterol 2002; 97:2623–2628.
- Banyai AL. There is more than surface appearance to skin spiders. Chest 1971; 60:48.
- Errickson CV, Matus NR. Skin disorders of pregnancy. Am Fam Physician 1994; 49:605–610.
- Li CP, Lee FY, Hwang SJ, et al. Spider angiomas in patients with liver cirrhosis: role of alcoholism and impaired liver function. Scand J Gastroenterol 1999; 34:520–523.
- Li CP, Lee FY, Hwang SJ, et al. Role of substance P in the pathogenesis of spider angiomas in patients with nonalcoholic liver cirrhosis. Am J Gastroenterol 1999; 94:502–507.
- Sadick NS, Niedt GW. A study of estrogen and progesterone receptors in spider telangiectasias of the lower extremities. J Dermatol Surg Oncol 1990; 16:620–623.
- Henry F, Quatresooz P, Valverde-Lopez JC, Pierard GE. Blood vessel changes during pregnancy: a review. Am J Clin Dermatol 2006; 7:65–69.
- Finn SM, Rowland M, Lawlor F, et al. The significance of cutaneous spider naevi in children. Arch Dis Child 2006; 91:604–605.
- Peyrot I, Boulinguez S, Sparsa A, Le Meur Y, Bonnetblanc JM, Bedane C. Bier’s white spots associated with scleroderma renal crisis. Clin Exp Dermatol 2007; 32:165–167.
- Satoh T, Yokozeki H, Nishioka K. Vascular spiders and paper money skin improved by hemodialysis. Dermatology 2002; 205:73–74.
- Serrao R, Zirwas M, English JC. Palmar erythema. Am J Clin Dermatol 2007; 8:347–356.
- Matsumoto M, Ohki K, Nagai I, Oshibuchi T. Lung traction causes an increase in plasma prostacyclin concentration and decrease in mean arterial blood pressure. Anesth Analg 1992; 75:773–776.
- Otto AI, Horvath I, Feldmann J. Multiple firm, painless erythematous papules with a yellowish hue. Arch Dermatol 2005; 141:1595–1600.
- Gandelman G, Aronow WS, Weiss MB. Resolving hyperlipidemia after liver transplantation in a patient with primary sclerosing cholangitis. Am J Ther 2006; 13:171–174.
- Assy N, Kaita K, Mymin D, Levy C, Rosser B, Minuk G. Fatty infiltration of liver in hyperlipidemic patients. Dig Dis Sci 2000; 45:1929–1934.
- Allocca M, Crosignani A, Gritti A, et al. Hypercholesterolaemia is not associated with early atherosclerotic lesions in primary biliary cirrhosis. Gut 2006; 55:1795–1800.
- Dickson E, Fleming C, Ludwig J. Primary biliary cirrhosis. In:Popper H, Schaffner F, editors. Progress in Liver Diseases. New York: Grune and Stratton; 1978:487.
- Elner VM, Mintz R, Demirci H, Hassan AS. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Trans Am Ophthalmol Soc 2005; 103:69–73.
- Craxi A, Camma C, Giunta M. Clinical aspects of bleeding complications in cirrhotic patients. Blood Coagul Fibrinolysis 2000; 11( suppl 1):S75–579.
- Kajihara M, Okazaki Y, Kato S, et al. Evaluation of platelet kinetics in patients with liver cirrhosis: similarity to idiopathic thrombocytopenic purpura. J Gastroenterol Hepatol 2007; 22:112–118.
- Levine N. Patient reports six-month history of minimally pruritic purple dots on legs. Non-blanching macules developed over six months. Geriatrics 2006; 61:22.
- Barton JC, Rao SV, Pereira NM, et al. Juvenile hemochromatosis in the southeastern United States: a report of seven cases in two kinships. Blood Cells Mol Dis 2002; 29:104–115.
- Smith AG, Shuster S, Bomford A, Williams R. Plasma immunoreactive beta-melanocyte-stimulating hormone in chronic liver disease and fulminant hepatic failure. J Invest Dermatol 1978; 70:326–327.
- Barton JC, McDonnell SM, Adams PC, et al. Management of hemochromatosis. Hemochromatosis Management Working Group. Ann Intern Med 1998; 129:932–939.
- Bahnsen M, Gluud C, Johnsen SG, et al. Pituitary-testicular function in patients with alcoholic cirrhosis of the liver. Eur J Clin Invest 1981; 11:473–479.
- Kumar N, Aggarwal SR, Anand BS. Comparison of truncal hair distribution in alcoholic liver disease and alcohol-related chronic pancreatitis. J Gastroenterol Hepatol 2001; 16:855–856.
- Holzberg M, Walker HK. Terry's nails: revised definition and new correlations. Lancet 1984; 1:896–899.
- Mandayam S, Jamal MM, Morgan TR. Epidemiology of alcoholic liver disease. Semin Liver Dis 2004; 24:217–232.
- Belle SH, Beringer KC, Detre KM. Liver transplantation in the United States: results from the National Pitt-UNOS Liver Transplant Registry. United Network for Organ Sharing Clin Transpl 1994:19–35.
- Dunn W, Xu R, Schwimmer JB. Modest wine drinking and decreased prevalence of suspected nonalcoholic fatty liver disease. Hepatology 2008; 47:1947–1954.
- Afford SC, Fisher NC, Neil DA, et al. Distinct patterns of chemokine expression are associated with leukocyte recruitment in alcoholic hepatitis and alcoholic cirrhosis. J Pathol 1998; 186:82–89.
- Evstaf’ev VV, Levin MM. Dermatologic pathology in chronic alcoholics. Vestn Dermatol Venerol 1989; 8:72–74.
- Jerosch-Herold C, Shepstone L, Chojnowski AJ, Larson D. Splinting after contracture release for Dupuytren's contracture (SCoRD): protocol of a pragmatic, multi-centre, randomized controlled trial. BMC Musculoskelet Disord 2008; 9:62.
- Houghton S, Holdstock G, Cockerell R, Wright R. Dupuytren’s contracture, chronic liver disease and IgA immune complexes. Liver 1983; 3:220–224.
- Ibbotson SH. Disseminated superficial porokeratosis: what is the association with ultraviolet radiation? Clin Exp Dermatol 1996; 21:48–50.
- Kono T, Kobayashi H, Ishii M, Nishiguchi S, Taniguchi S. Synchronous development of disseminated superficial porokeratosis and hepatitis C virus-related hepatocellular carcinoma. J Am Acad Dermatol 2000; 43:966–968.
- Park BS, Moon SE, Kim JA. Disseminated superficial porokeratosis in a patient with chronic liver disease. J Dermatol 1997; 24:485–487.
- Murata Y, Kumano K, Takai T. Type 2 segmental manifestation of disseminated superficial porokeratosis showing a systematized pattern of involvement and pronounced cancer proneness. Eur J Dermatol 2001; 11:191–194.
- Galossi A, Guarisco R, Bellis L, Puoti C. Extrahepatic manifestations of chronic HCV infection. J Gastrointestin Liver Dis 2007; 16:65–73.
- El-Serag HB, Hampel H, Yeh C, Rabeneck L. Extrahepatic manifestations of hepatitis C among United States male veterans. Hepatology 2002; 36:1439–1445.
- Stefanova-Petrova DV, Tzvetanska AH, Naumova EJ, et al. Chronic hepatitis C virus infection: prevalence of extrahepatic manifestations and association with cryoglobulinemia in Bulgarian patients. World J Gastroenterol 2007; 13:6518–6528.
- Vassilopoulos D, Calabrese LH. Extrahepatic immunological complications of hepatitis C virus infection. AIDS 2005; 19( suppl 3):S123–S127.
- Hsing AW, Zhang M, Rashid A, et al. Hepatitis B and C virus infection and the risk of biliary tract cancer: a population-based study in China. Int J Cancer 2008; 122:1849–1853.
- Madrid C, Courtois B, Duran D. Chronic sialadenitis revealing hepatitis C: a case report. Med Oral 2004; 9:328–332.
- Lançoni G, Ravinal RC, Costa RS, Roselino AM. Mast cells and transforming growth factor-beta expression: a possible relationship in the development of porphyria cutanea tarda skin lesions. Int J Dermatol 2008; 47:575–581.
- Toll A, Celis R, Ozalla MD, Bruguera M, Herrero C, Ercilla MG. The prevalence of HFE C282Y gene mutation is increased in Spanish patients with porphyria cutanea tarda without hepatitis C virus infection. J Eur Acad Dermatol Venereol 2006; 20:1201–1206.
- Badminton MN, Elder GH. Management of acute and cutaneous porphyrias. Int J Clin Pract 2002; 56:272–278.
- Jackson JM, Callen JP. Scarring alopecia and sclerodermatous changes of the scalp in a patient with hepatitis C infection. J Am Acad Dermatol 1998; 39:824–826.
- Mortimore M, Merryweather-Clarke AT, Robson KJ, Powell LW. The haemochromatosis gene: a global perspective and implications for the Asia-Pacific region. J Gastroenterol Hepatol 1999; 14:838–843.
- Shehan JM, Huerter CJ. Porphyria cutanea tarda associated with an acute gastrointestinal bleed: the roles of supplemental iron and blood transfusion. Cutis 2001; 68:147–150.
- Lambrecht RW, Thapar M, Bonkovsky HL. Genetic aspects of porphyria cutanea tarda. Semin Liver Dis 2007; 27:99–108.
- d’Ovidio R, Sgarra C, Conserva A, Angelotti UF, Erriquez R, Foti C. Alterated integrin expression in lichen planopilaris. Head Face Med 2007; 3:11.
- Chuang TY, Stitle L, Brashear R, Lewis C. Hepatitis C virus and lichen planus: a case-control study of 340 patients. J Am Acad Dermatol 1999; 41:787–789.
- Kemp EH, Gavalas NG, Gawkrodger DJ, Weetman AP. Autoantibody responses to melanocytes in the depigmenting skin disease vitiligo. Autoimmun Rev 2007; 6:138–142.
- Tomasiewicz K, Modrzewska R, Semczuk G. Vitiligo associated with pegylated interferon and ribavirin treatment of patients with chronic hepatitis C: a case report. Adv Ther 2006; 23:139–142.
- Simsek H, Savas C, Akkiz H, Telatar H. Interferon-induced vitiligo in a patient with chronic viral hepatitis C infection. Dermatology 1996; 193:65–66.
- Mulekar SV, Al Issa A, Asaad M, Ghwish B, Al Eisa A. Mixed vitiligo. J Cutan Med Surg. 2006; 10:104–107.
- Waalen J, Felitti V, Gelbart T, Ho NJ, Beutler E. Prevalence of hemochromatosis-related symptoms among individuals with mutations in the HFE gene. Mayo Clin Proc 2002; 77:522–530.
- Walker AP, Tucker DC, Hall MA, et al. Results communication and patient education after screening for possible hemochromatosis and iron overload: experience from the HEIRS study of a large ethnically and linguistically diverse group. Genet Med 2007; 9:778–791.
- Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, cafe au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician 2003; 68:1955–1960.
- Tsuji T. Experimental hemosiderosis: relationship between skin pigmentation and hemosiderin. Acta Derm Venereol 1980; 60:109–114.
- Oji V, Traupe H. Ichthyoses: differential diagnosis and molecular genetics. Eur J Dermatol 2006; 16:349–359.
- Morioka D, Togo S, Kumamoto T, et al. Six consecutive cases of successful adult ABO-incompatible living donor liver transplantation: a proposal for grading the severity of antibody-mediated rejection. Transplantation 2008; 85:171–178.
- Bertini G, Rubaltelli FF. Non-invasive bilirubinometry in neonatal jaundice. Semin Neonatol 2002; 7:129–133.
- Clementi M, Di Gianantonio E, Fabris L, et al. Inheritance of hyperbilirubinemia: evidence for a major autosomal recessive gene. Dig Liver Dis 2007; 39:351–355.
- Roche SP, Kobos R. Jaundice in the adult patient. Am Fam Physician 2004; 69:299–304.
- Odemis B, Parlak E, Basar O, Yüksel O, Sahin B. Biliary tract obstruction secondary to malignant lymphoma: experience at a referral center. Dig Dis Sci 2007; 52:2323–2332.
- Caddy GR, Tham TC. Gallstone disease: symptoms, diagnosis and endoscopic management of common bile duct stones. Best Pract Res Clin Gastroenterol 2006; 20:1085–1101.
- Heathcote EJ. Diagnosis and management of cholestatic liver disease. Clin Gastroenterol Hepatol 2007; 5:776–782.
- Faust TW, Reddy KR. Postoperative jaundice. Clin Liver Dis 2004; 8:151–166.
- Maticic M, Poljak M, Lunder T, Rener-Sitar K, Stojanovic L. Lichen planus and other cutaneous manifestations in chronic hepatitis C: pre- and post-interferon-based treatment prevalence vary in a cohort of patients from low hepatitis C virus endemic area. J Eur Acad Dermatol Venereol 2008; 22:779–788.
- Polat M, Oztas P, Ilhan MN, Yalçin B, Alli N. Generalized pruritus: a prospective study concerning etiology. Am J Clin Dermatol 2008; 9:39–44.
- Gaspari R, Avolio AW, Zileri Dal Verme L, et al. Molecular adsorbent recirculating system in liver transplantation: safety and efficacy. Transplant Proc 2006; 38:3544–3551.
- Dasgupta R, Saha I, Pal S, et al. Immunosuppression, hepatotoxicity and depression of antioxidant status by arecoline in albino mice. Toxicology 2006; 227:94–104.
- Mayo MJ, Handem I, Saldana S, Jacobe H, Getachew Y, Rush AJ. Sertraline as a first-line treatment for cholestatic pruritus. Hepatology 2007; 45:666–674.
- Mela M, Mancuso A, Burroughs AK. Review article: pruritus in cholestatic and other liver diseases. Aliment Pharmacol Ther 2003; 17:857–870.
- Montero JL, Pozo JC, Barrera P, et al. Treatment of refractory cholestatic pruritus with molecular adsorbent recirculating system (MARS). Transplant Proc 2006; 38:2511–2513.
- Neff GW, O'Brien CB, Reddy KR, et al. Preliminary observation with dronabinol in patients with intractable pruritus secondary to cholestatic liver disease. Am J Gastroenterol 2002; 97:2117–2119.
- Neri S, Raciti C, D’Angelo G, Ierna D, Bruno CM. Hyde’s prurigo nodularis and chronic HCV hepatitis. J Hepatol 1998; 28:161–164.
- Brown MA, George CR, Dunstan CR, Kalowski S, Corrigan AB. Prurigo nodularis and aluminium overload in maintenance haemodialysis. Lancet 1992; 340:48.
- Stander S, Luger T, Metze D. Treatment of prurigo nodularis with topical capsaicin. J Am Acad Dermatol 2001; 44:471–478.
- Requena L, Sangueza OP. Cutaneous vascular anomalies. Part I. Hamartomas, malformations, and dilation of preexisting vessels. J Am Acad Dermatol 1997; 37:523–549.
- Khasnis A, Gokula RM. Spider nevus. J Postgrad Med 2002; 48:307–309.
- Kaul V, Friedenberg FK, Braitman LE, et al. Development and validation of a model to diagnose cirrhosis in patients with hepatitis C. Am J Gastroenterol 2002; 97:2623–2628.
- Banyai AL. There is more than surface appearance to skin spiders. Chest 1971; 60:48.
- Errickson CV, Matus NR. Skin disorders of pregnancy. Am Fam Physician 1994; 49:605–610.
- Li CP, Lee FY, Hwang SJ, et al. Spider angiomas in patients with liver cirrhosis: role of alcoholism and impaired liver function. Scand J Gastroenterol 1999; 34:520–523.
- Li CP, Lee FY, Hwang SJ, et al. Role of substance P in the pathogenesis of spider angiomas in patients with nonalcoholic liver cirrhosis. Am J Gastroenterol 1999; 94:502–507.
- Sadick NS, Niedt GW. A study of estrogen and progesterone receptors in spider telangiectasias of the lower extremities. J Dermatol Surg Oncol 1990; 16:620–623.
- Henry F, Quatresooz P, Valverde-Lopez JC, Pierard GE. Blood vessel changes during pregnancy: a review. Am J Clin Dermatol 2006; 7:65–69.
- Finn SM, Rowland M, Lawlor F, et al. The significance of cutaneous spider naevi in children. Arch Dis Child 2006; 91:604–605.
- Peyrot I, Boulinguez S, Sparsa A, Le Meur Y, Bonnetblanc JM, Bedane C. Bier’s white spots associated with scleroderma renal crisis. Clin Exp Dermatol 2007; 32:165–167.
- Satoh T, Yokozeki H, Nishioka K. Vascular spiders and paper money skin improved by hemodialysis. Dermatology 2002; 205:73–74.
- Serrao R, Zirwas M, English JC. Palmar erythema. Am J Clin Dermatol 2007; 8:347–356.
- Matsumoto M, Ohki K, Nagai I, Oshibuchi T. Lung traction causes an increase in plasma prostacyclin concentration and decrease in mean arterial blood pressure. Anesth Analg 1992; 75:773–776.
- Otto AI, Horvath I, Feldmann J. Multiple firm, painless erythematous papules with a yellowish hue. Arch Dermatol 2005; 141:1595–1600.
- Gandelman G, Aronow WS, Weiss MB. Resolving hyperlipidemia after liver transplantation in a patient with primary sclerosing cholangitis. Am J Ther 2006; 13:171–174.
- Assy N, Kaita K, Mymin D, Levy C, Rosser B, Minuk G. Fatty infiltration of liver in hyperlipidemic patients. Dig Dis Sci 2000; 45:1929–1934.
- Allocca M, Crosignani A, Gritti A, et al. Hypercholesterolaemia is not associated with early atherosclerotic lesions in primary biliary cirrhosis. Gut 2006; 55:1795–1800.
- Dickson E, Fleming C, Ludwig J. Primary biliary cirrhosis. In:Popper H, Schaffner F, editors. Progress in Liver Diseases. New York: Grune and Stratton; 1978:487.
- Elner VM, Mintz R, Demirci H, Hassan AS. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Trans Am Ophthalmol Soc 2005; 103:69–73.
- Craxi A, Camma C, Giunta M. Clinical aspects of bleeding complications in cirrhotic patients. Blood Coagul Fibrinolysis 2000; 11( suppl 1):S75–579.
- Kajihara M, Okazaki Y, Kato S, et al. Evaluation of platelet kinetics in patients with liver cirrhosis: similarity to idiopathic thrombocytopenic purpura. J Gastroenterol Hepatol 2007; 22:112–118.
- Levine N. Patient reports six-month history of minimally pruritic purple dots on legs. Non-blanching macules developed over six months. Geriatrics 2006; 61:22.
- Barton JC, Rao SV, Pereira NM, et al. Juvenile hemochromatosis in the southeastern United States: a report of seven cases in two kinships. Blood Cells Mol Dis 2002; 29:104–115.
- Smith AG, Shuster S, Bomford A, Williams R. Plasma immunoreactive beta-melanocyte-stimulating hormone in chronic liver disease and fulminant hepatic failure. J Invest Dermatol 1978; 70:326–327.
- Barton JC, McDonnell SM, Adams PC, et al. Management of hemochromatosis. Hemochromatosis Management Working Group. Ann Intern Med 1998; 129:932–939.
- Bahnsen M, Gluud C, Johnsen SG, et al. Pituitary-testicular function in patients with alcoholic cirrhosis of the liver. Eur J Clin Invest 1981; 11:473–479.
- Kumar N, Aggarwal SR, Anand BS. Comparison of truncal hair distribution in alcoholic liver disease and alcohol-related chronic pancreatitis. J Gastroenterol Hepatol 2001; 16:855–856.
- Holzberg M, Walker HK. Terry's nails: revised definition and new correlations. Lancet 1984; 1:896–899.
- Mandayam S, Jamal MM, Morgan TR. Epidemiology of alcoholic liver disease. Semin Liver Dis 2004; 24:217–232.
- Belle SH, Beringer KC, Detre KM. Liver transplantation in the United States: results from the National Pitt-UNOS Liver Transplant Registry. United Network for Organ Sharing Clin Transpl 1994:19–35.
- Dunn W, Xu R, Schwimmer JB. Modest wine drinking and decreased prevalence of suspected nonalcoholic fatty liver disease. Hepatology 2008; 47:1947–1954.
- Afford SC, Fisher NC, Neil DA, et al. Distinct patterns of chemokine expression are associated with leukocyte recruitment in alcoholic hepatitis and alcoholic cirrhosis. J Pathol 1998; 186:82–89.
- Evstaf’ev VV, Levin MM. Dermatologic pathology in chronic alcoholics. Vestn Dermatol Venerol 1989; 8:72–74.
- Jerosch-Herold C, Shepstone L, Chojnowski AJ, Larson D. Splinting after contracture release for Dupuytren's contracture (SCoRD): protocol of a pragmatic, multi-centre, randomized controlled trial. BMC Musculoskelet Disord 2008; 9:62.
- Houghton S, Holdstock G, Cockerell R, Wright R. Dupuytren’s contracture, chronic liver disease and IgA immune complexes. Liver 1983; 3:220–224.
- Ibbotson SH. Disseminated superficial porokeratosis: what is the association with ultraviolet radiation? Clin Exp Dermatol 1996; 21:48–50.
- Kono T, Kobayashi H, Ishii M, Nishiguchi S, Taniguchi S. Synchronous development of disseminated superficial porokeratosis and hepatitis C virus-related hepatocellular carcinoma. J Am Acad Dermatol 2000; 43:966–968.
- Park BS, Moon SE, Kim JA. Disseminated superficial porokeratosis in a patient with chronic liver disease. J Dermatol 1997; 24:485–487.
- Murata Y, Kumano K, Takai T. Type 2 segmental manifestation of disseminated superficial porokeratosis showing a systematized pattern of involvement and pronounced cancer proneness. Eur J Dermatol 2001; 11:191–194.
- Galossi A, Guarisco R, Bellis L, Puoti C. Extrahepatic manifestations of chronic HCV infection. J Gastrointestin Liver Dis 2007; 16:65–73.
- El-Serag HB, Hampel H, Yeh C, Rabeneck L. Extrahepatic manifestations of hepatitis C among United States male veterans. Hepatology 2002; 36:1439–1445.
- Stefanova-Petrova DV, Tzvetanska AH, Naumova EJ, et al. Chronic hepatitis C virus infection: prevalence of extrahepatic manifestations and association with cryoglobulinemia in Bulgarian patients. World J Gastroenterol 2007; 13:6518–6528.
- Vassilopoulos D, Calabrese LH. Extrahepatic immunological complications of hepatitis C virus infection. AIDS 2005; 19( suppl 3):S123–S127.
- Hsing AW, Zhang M, Rashid A, et al. Hepatitis B and C virus infection and the risk of biliary tract cancer: a population-based study in China. Int J Cancer 2008; 122:1849–1853.
- Madrid C, Courtois B, Duran D. Chronic sialadenitis revealing hepatitis C: a case report. Med Oral 2004; 9:328–332.
- Lançoni G, Ravinal RC, Costa RS, Roselino AM. Mast cells and transforming growth factor-beta expression: a possible relationship in the development of porphyria cutanea tarda skin lesions. Int J Dermatol 2008; 47:575–581.
- Toll A, Celis R, Ozalla MD, Bruguera M, Herrero C, Ercilla MG. The prevalence of HFE C282Y gene mutation is increased in Spanish patients with porphyria cutanea tarda without hepatitis C virus infection. J Eur Acad Dermatol Venereol 2006; 20:1201–1206.
- Badminton MN, Elder GH. Management of acute and cutaneous porphyrias. Int J Clin Pract 2002; 56:272–278.
- Jackson JM, Callen JP. Scarring alopecia and sclerodermatous changes of the scalp in a patient with hepatitis C infection. J Am Acad Dermatol 1998; 39:824–826.
- Mortimore M, Merryweather-Clarke AT, Robson KJ, Powell LW. The haemochromatosis gene: a global perspective and implications for the Asia-Pacific region. J Gastroenterol Hepatol 1999; 14:838–843.
- Shehan JM, Huerter CJ. Porphyria cutanea tarda associated with an acute gastrointestinal bleed: the roles of supplemental iron and blood transfusion. Cutis 2001; 68:147–150.
- Lambrecht RW, Thapar M, Bonkovsky HL. Genetic aspects of porphyria cutanea tarda. Semin Liver Dis 2007; 27:99–108.
- d’Ovidio R, Sgarra C, Conserva A, Angelotti UF, Erriquez R, Foti C. Alterated integrin expression in lichen planopilaris. Head Face Med 2007; 3:11.
- Chuang TY, Stitle L, Brashear R, Lewis C. Hepatitis C virus and lichen planus: a case-control study of 340 patients. J Am Acad Dermatol 1999; 41:787–789.
- Kemp EH, Gavalas NG, Gawkrodger DJ, Weetman AP. Autoantibody responses to melanocytes in the depigmenting skin disease vitiligo. Autoimmun Rev 2007; 6:138–142.
- Tomasiewicz K, Modrzewska R, Semczuk G. Vitiligo associated with pegylated interferon and ribavirin treatment of patients with chronic hepatitis C: a case report. Adv Ther 2006; 23:139–142.
- Simsek H, Savas C, Akkiz H, Telatar H. Interferon-induced vitiligo in a patient with chronic viral hepatitis C infection. Dermatology 1996; 193:65–66.
- Mulekar SV, Al Issa A, Asaad M, Ghwish B, Al Eisa A. Mixed vitiligo. J Cutan Med Surg. 2006; 10:104–107.
- Waalen J, Felitti V, Gelbart T, Ho NJ, Beutler E. Prevalence of hemochromatosis-related symptoms among individuals with mutations in the HFE gene. Mayo Clin Proc 2002; 77:522–530.
- Walker AP, Tucker DC, Hall MA, et al. Results communication and patient education after screening for possible hemochromatosis and iron overload: experience from the HEIRS study of a large ethnically and linguistically diverse group. Genet Med 2007; 9:778–791.
- Stulberg DL, Clark N, Tovey D. Common hyperpigmentation disorders in adults: Part I. Diagnostic approach, cafe au lait macules, diffuse hyperpigmentation, sun exposure, and phototoxic reactions. Am Fam Physician 2003; 68:1955–1960.
- Tsuji T. Experimental hemosiderosis: relationship between skin pigmentation and hemosiderin. Acta Derm Venereol 1980; 60:109–114.
- Oji V, Traupe H. Ichthyoses: differential diagnosis and molecular genetics. Eur J Dermatol 2006; 16:349–359.
KEY POINTS
- Pruritus due to liver disease is particularly resistant to therapy. Cholestyramine (Questran) 4 g/day, gradually increased to 24 g/day, is one option. If the pruritus does not respond or the patient cannot tolerate cholestyramine, rifampin (Rifadin) can be tried.
- Spider angiomas, Bier spots, and “paper-money” skin are all superficial vascular problems that may be related to liver disease.
- Cutaneous lesions often accompany alcoholic cirrhosis and have been detected in up to 43% of people with chronic alcoholism. The combination of spider angiomas, palmar erythema, and Dupuytren contracture is common in alcoholic cirrhosis.
- Although porphyria cutanea tarda is associated with liver disease in general, recent studies show that patients with hepatitis C are at particularly high risk.
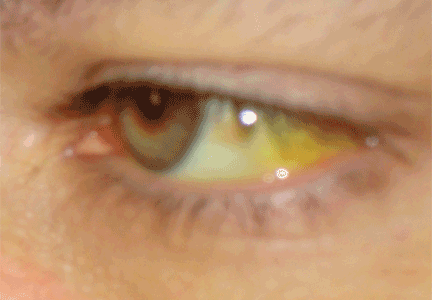
Derm diagnoses you can’t afford to miss
- Management of hereditary angioedema should include fresh frozen plasma containing C1 inhibitor (C1-INH), whenever possible; if C1-INH-containing plasma is unavailable, fresh frozen plasma can be used instead (SOR: A).
- Do not give neomycin to patients with suspected cellulitis; the drug may promote antibiotic resistance in Staphylococcus aureus, a pathogen often associated with this condition (SOR: A).
- Whenever a patient presents with erythematous skin lesions and a recent history of receiving penicillin or a cephalosporin antibiotic, a sulfa derivative, or an anticonvulsant, the suspected medication should be stopped until Stevens-Johnson syndrome is ruled out (SOR: A).
Strength of recommendation (SOR)
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
Skin eruptions are a common reason for visits to primary care physicians. While most are innocuous, some are associated with—or are early warning signs of—severe allergic reactions or other emergent conditions. A 9-year-old patient I’ll call Julie is a case in point.
The first time Julie’s parents brought her to our clinic, she’d been complaining of a sore throat and had a fever that hovered between 102° and 103°F for several days. The physician who examined Julie found mild maxillary tenderness. A rapid streptococcal throat swab was negative; her doctor prescribed a 10-day course of trimethoprim/sulfamethoxazole (TMP/SMX) for presumed acute sinusitis.
Thirteen days later (3 days after the patient completed the course of antibiotics), Julie’s parents brought her back to the clinic. Her throat still hurt, and she had erythematous oval lesions on her trunk and upper extremities. Her physician diagnosed scarlet fever and wrote a prescription for penicillin.
The following day, Julie was taken to the emergency department (ED) with bilateral conjunctival hyperemia and diffuse, confluent erythematous macules throughout her body. The ED physician who examined Julie found a 2.5 × 2.0 cm targetoid lesion with a necrotic, purpuric center on her lower back—a diagnostic clue to the cause of her signs and symptoms.
If you had been Julie’s physician, would you have been alert to that clue?
For family physicians accustomed to seeing relatively mild skin disorders, recognizing and responding to dermatologic conditions with potentially dire outcomes can be challenging. This review, and the images that accompany it, will help you sharpen your dermatologic diagnostic and treatment skills, both for benign disorders and those that are less common and more severe. We’ll also tell you more about Julie and her diagnosis.
Urticaria: A simple case of hives?
This common allergic reaction affects close to 10% of the population at some point in their lives. The affected areas are itchy and have raised, circumscribed red welts with surrounding erythema.1 Urticaria can occur throughout the body, with new lesions often erupting as the old ones disappear.2,3
Despite the persistent itchiness that patients typically complain of, however, urticaria is usually self-limiting, and rarely life-threatening. Acute urticaria normally resolves within 2 to 6 weeks.2,4
In most cases, urticaria arises secondary to exposure to an allergenic substance, chemical, or emotional stress.4,5 In rare instances, systemic diseases, such as hematologic malignancies, can also cause urticarial lesions to erupt throughout the body.4
Body piercing, cosmetics, latex exposure, Helicobacter pylori, insects, and angiotensin-converting enzyme (ACE) inhibitors have been identified as common triggers of urticaria, as have nonsteroidal anti-inflammatory drugs (NSAIDs) and antibiotics, animal dander, and foods such as shellfish, nuts, and dairy products.4,6
Treatment of all forms of urticaria should be based on identification and strict avoidance of the causative agent, if it’s known.7 Following withdrawal of the specific agent, symptomatic treatment with medications such as histamine antagonists and corticosteroids remains the mainstay of therapy.4,8 A daily dose of 40 to 60 mg prednisone for 5 days is a reasonable therapeutic regimen for adults; a 5-day course of 1 mg/kg per day is suitable for pediatric patients.4,8,9
In the event that topical or oral therapy is ineffective in mild cases of urticaria, intravenous (IV) diphenhydramine (50 mg) can be administered every 6 to 8 hours.4,8 IV diphenhydramine typically takes 30 minutes to work, while corticosteroids take at least 2 hours to reach full effect.4,8
In an emergency setting, subcutaneous epinephrine (0.3-0.5 mg) can be useful in treating severe urticaria.4,8 And recent clinical trials have demonstrated complete clearance of urticaria with leukotriene inhibitors, such as montelukast (10 mg).10
Chronic urticaria, trigger unknown
Although acute urticaria is responsive to treatment, chronic urticaria—lesions that do not resolve after 6 weeks—poses a greater challenge. In up to 80% of cases of chronic urticaria, no identifiable trigger is found.3,8 Long-term treatment of patients with this chronic condition, including lifestyle changes (to avoid environmental or dietary triggers) and a medication regimen for 6 months or more, leads to complete resolution of symptoms in most cases.7,8
Angioedema: Less common, more dangerous
Angioedema is part of the same disease spectrum as urticaria, but it affects the deeper tissues—involving mucosal and submucosal swelling. Angioedema affects just 0.1% to 0.2% of the general population, but up to 15% of patients with urticaria.8
The risk increases with the use of ACE inhibitors; for every 1000 patients taking ACE inhibitors, 0.4 to 3.5 develop angioedema.11 A recent double-blind study involving 25,642 patients being treated with ACE inhibitors revealed that those taking a combination of ACE inhibitors were more likely to develop angioedema than those on monotherapy.12
Angioedema is characterized by the sudden appearance of painful, localized erythematous wheals with central blanching.13 It results from increased vascular permeability in capillaries of the dermis, which leads to fluid leakage.13,14 Increased accumulation of fluids from the vessels of the skin results in rapidly developing nonpitting edema that most often affects the hands, face (and lips), neck, and oropharynx (FIGURE 1).14 Although the head and neck are the most commonly involved areas, angioedema can also affect the digestive tract, leading to nausea, vomiting, diarrhea, and abdominal pain secondary to bowel edema.13
FIGURE 1
Angioedema: A look at the most commonly affected areas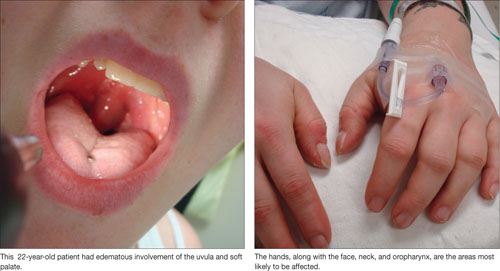
Airway management is imperative
In severe cases of angioedema, involvement of the oral mucosa results in stridor, followed by upper airway obstruction. To avoid hypoxemia and death,13,14 rapid preparation for emergency intervention to maintain the airway is critical; mortality can be as high as 30% in patients with airway compromise.13,15 In severe cases in which edema engulfs the oropharynx, oral intubation is often impossible, and nasotracheal intubation may be warranted.15,16 A failure to maintain adequate airway function via nasotracheal intubation may signal the need for an emergency tracheotomy.15,16
Is the angioedema hereditary or acquired?
Hereditary and acquired angioedema are treated differently. After ensuring that the patient has a patent airway, distinguishing between them is critical. Hereditary angioedema is the result of an inherited deficiency of plasma protein C1 inhibitor (C1-INH). Acquired angioedema is typically caused by enhanced consumption of endogenous C1-INH,13,17 leading to a net deficit of circulating C1-INH, and can be triggered by food, pharmacologic agents, and, occasionally, by systemic disorders.13 African Americans and patients with renal impairment appear to be at increased risk for acquired angioedema.13 In both hereditary and acquired angioiedema, decreased levels of C1-INH lead to disruption of the complement pathway.
Although the clinical presentation of acquired and hereditary angioedema is similar, a focused patient history can be used to distinguish between them. Age, health status, and medication history are the key considerations. Hereditary angioedema most commonly occurs—in recurrent attacks after minor trauma—in children with no underlying disease, with worsening symptoms during puberty. Acquired angioedema is generally seen in adult and elderly patients with malignancies or other underlying disorders.
Mild to moderate cases of acquired angioedema respond well to oral corticosteroids and antihistamines. Severe cases often require administration of subcutaneous epinephrine, followed by IV steroids. Management of hereditary angioedema should include C1-INH-containing fresh frozen plasma,13,16,18 although fresh frozen plasma can be used if plasma with C1-INH is not available.13
While oral corticosteroids and anti-histamines may be effective adjunctive therapy for hereditary angioedema, they are not likely to reverse acute attacks in this patient population. IV diphenhydramine (50-100 mg) or IV cimetidine (300 mg) every 6 to 8 hours is a reasonable therapeutic regimen for acute attacks of hereditary angioedema.
A recent randomized double-blind trial involving 40 patients with hereditary angioedema studied the administration of ecallantide, a kallikrein inhibitor for which US Food and Drug Administration approval is pending.19 Nearly 3 out of 4 of those who received ecallantide (72.5%) for acute attacks of angioedema showed significant improvement within 4 hours.20 The use of kallikrein inhibitors, which target inflammatory blood components, is not widespread, but may hold promise for the treatment of hereditary angioedema.20
Cellulitis: On the lookout for infiltration
Cellulitis is a bacterial infection of the skin that affects approximately 24.6 in 1000 people and is rarely associated with death.21 It occurs when bacteria enter through disrupted areas in the skin, particularly when skin integrity is compromised by recent surgery, piercing, wounds, athlete’s foot, or even dermatitis.22,23Streptococcus and Staphylococcus are the 2 most common infectious agents, and methicillin-resistant Staphylococcus aureus (MRSA) is increasingly common.22,23
Although cellulitis is primarily superficial in nature, it may progress to a serious condition by infiltrating underlying tissues and spreading to nearby lymphatic tissue and the bloodstream to cause lymphadenitis or bacteremia.21,23 In instances of cellulitis-induced bacteremia, mortality rates increase if prompt, targeted treatment is not provided.23
Raised erythematous plaques are the cardinal features of cellulitis, with the affected areas warm to the touch, red, and tender.21,23 As the condition progresses, the affected area tends to enlarge and expand (FIGURE 2),24 and the patient often becomes febrile.22,24
The risk of developing cellulitis increases with age, compromised immune status, diabetes, obesity, IV drug use, lymphedema, and chronic corticosteroid use.22,24
Cellulitis is often diagnosed solely on the basis of clinical presentation, although aspiration of purulent discharge from the wound and a gram stain of the culture can confirm the diagnosis.25,26 (See “Is it cellulitis or stasis dermatitis?”.)
Direct immunofluorescence can be used when cultures are difficult to obtain, but this technique is seldom necessary.22 If infiltration of underlying soft tissues is suspected based on clinical findings, magnetic resonance imaging can be a useful tool in evaluating the extent of the infection and in directing appropriate debridement and drainage of affected areas.22,27
Patients with venous insufficiency may present with stasis dermatitis, which often results in breakdown of the skin and ulceration that bears a striking resemblance to cellulitis. Thus, these conditions can be easily confused, and may lead to unnecessary antibiotic use and, possibly, hospitalization in patients with venous insufficiency.26,28
Despite the similarities of these conditions, a focused patient history and physical exam can prevent such confusion. Stasis dermatitis arises as a result of venous insufficiency, so it is likely to be accompanied by pitting edema that responds to leg raising and to the use of elastic compression stockings—interventions that are seldom effective for cellulitis.26 In addition, cellulitis tends to be unilateral, while stasis dermatitis often has bilateral involvement.
FIGURE 2
Diagnosing cellulitis based on clinical presentation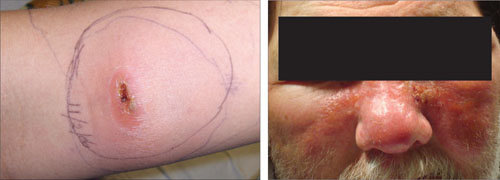
The raised erythematous lesions that are a hallmark of cellulitis, shown here on the arm and face, are warm to the touch, red, and tender.
Treatment: Targeted antibiotics and preventive measures
Because of the likelihood of recurrence with cellulitis, treating the condition involves both preventive and curative measures. Mild cases can be treated in an outpatient setting with a 7- to 10-day course of oral cephalosporins or antibiotics with similar coverage.22,25,26 A recent randomized study involving 391 patients found that cure rates for cellulitis treated with cephalexin were between 83% and 92%, depending on the pathogen involved.26
For severe cases of cellulitis, patients who are immunocompromised, and cases that are refractory to oral medications, hospital admission is recommended, and use of IV antibiotics is routinely required.22,25,26 For patients with MRSA, a drug such as vancomycin IV may be warranted; a reasonable dose would be 15 mg/kg every 12 hours.22,26,27,29
A recent randomized, multicenter study demonstrated that vancomycin effectively treated approximately 67% of cases of MRSA-induced cellulitis.26 Neomycin should be strictly avoided whenever cellulitis is suspected, because of its propensity to promote antibiotic resistance to S. aureus.29
Patient education emphasizing preventive measures is critical for minimizing recurrence of cellulitis.22,25,26 Encourage patients to wash with antibacterial soap and water daily, apply topical antibiotic ointment, and keep the wound completely covered at all times. Advise them to change bandages and wash their hands frequently.27,29 Patients with diabetes and others with decreased circulation in the extremities need to take further precautions, such as moisturizing the skin regularly in order to prevent cuts in their skin.22,29
SJS: Triggered by drugs, and infections, too
Stevens-Johnson syndrome (SJS), also known as erythema multiforme major, is an often-debilitating and possibly fatal adverse reaction, typically (but not exclusively) to a drug. It manifests as full-thickness epidermal necrosis of the mucous membranes.30,31 SJS occurs at a rate of about 1 to 7 cases per million people per year, and has a mortality rate of approximately 5%.30-32 The types of medication that most commonly precipitate SJS are anticonvulsants, sulfa drugs, penicillin-related and cephalosporin antibiotics, anti-inflammatory agents, and certain neoplastic drugs.30,32 SJS can develop in response to infections and neoplasms (TABLE) as well, and in many cases a cause is never found.
Patients with widespread involvement often complain of a burning sensation, particularly around the mouth.32-36 In some cases, this is the presenting sign, because the oral mucosa tends to be among the first mucous membranes involved.
TABLE
Stevens-Johnson syndrome: Pinpointing the cause32
| MORE FREQUENT ETIOLOGY |
| Drugs: Allopurinol, anticonvulsants, antiparasitics, barbiturates, NSAIDs, penicillin-related and cephalosporin antibiotics, sulfas, tetracyclines |
| LESS FREQUENT ETIOLOGY |
| Bacterial: Diphtheria, group A Streptococcus, Mycoplasma pneumoniae, tularemia, typhoid |
| Fungal: Coccidiomycosis, dermatophytosis, histoplasmosis, |
| Protozoan: Plasmodium, trichomoniasis |
| Viral: AIDS, Coxsackie, Epstein-Barr, HSV, influenza |
| AIDS, acquired immune deficiency syndrome; HSV, herpes-simplex virus; NSAIDs, nonsteroidal anti-inflammatory drugs. |
Targetoid lesions, Nikolsky sign are diagnostic clues
The characteristic skin lesions seen with SJS consist of initially erythematous macules that rapidly develop central necrosis to form vesiculation, as well as other variable areas of denudation (FIGURE 3). These vesicles tend to demonstrate confluence and often show a positive Nikolsky sign—epidermal detachment of superficial layers of the skin when slight pressure is applied. The lesions typically take on a targetoid appearance. If left untreated, the blisters often result in ulceration and hemorrhagic crusting of affected areas.
Like most skin disorders, SJS is initially diagnosed on the basis of clinical presentation. However, it is a rare disorder, and commonly misdiagnosed. Among the disorders SJS has been mistaken for are staphylococcal scalded skin syndrome, toxic shock syndrome, exfoliative dermatitis, scarlet fever, erythema multiforme, and iatrogenic chemical burns.32,37 (Erythema multiforme, SJS, and toxic epidermal necrolysis [TEN] are considered part of the same disease spectrum; erythema multiforme typically presents with few random lesions and no mucosal involvement, SJS with mucosal involvement on up to 30% of the body surface, and TEN with >30%.)32,37 Skin biopsies and immunofluorescence studies are recommended to confirm the diagnosis.
During the course of SJS, the mucous membranes of the oropharynx, ocular cavity, gastrointestinal system, nasal cavity, genitourinary system, and lower respiratory tracts are typically affected.32-35 As the condition progresses, increased epidermal erosion can lead to the sloughing off of up to 100% of the epidermis, resulting in considerable fluid loss.32,34
Corneal ulceration, anterior uveitis, panophthalmitis, polyarthritis, hematuria, and acute tubular necrosis leading to renal failure may also occur.32,35,37 Scarring within vital ocular structures can result in corneal opacity and lead to significant visual impairment. In severe cases, blood loss and fluid loss increase the risk of bacterial superinfection and sepsis.35,37
FIGURE 3
Targetoid lesions are characteristic of SJS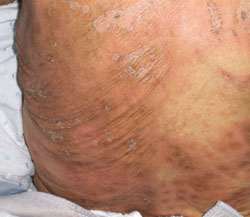
Characteristic diffuse erythematous macules with necrotic centers and overlying blistering on the back of a patient with Stevens-Johnson syndrome.
Supportive therapy, wound care are key components of treatment
Rapid cessation of the offending agent with targeted dermatologic management can reduce morbidity by promoting rapid re-epithelialization of affected skin. (See “SJS is diagnosed, but not quickly,” [Verdicts] on page 332, for a discussion of the dangers of delayed diagnosis and failure to promptly stop the drug causing the acute reaction.)
Closely monitoring the patient for fluid and electrolyte abnormalities is also crucial. Corticosteroids and IV immunoglobulins have been suggested for early severe cases of SJS, but their efficacy in treating this condition has yet to be established by prospective double-blind studies.32-37
The skin lesions associated with SJS should be treated in the same way you would treat thermal burns, with local wound care, warm compresses, and topical anesthetics for pain reduction.36,37 Oral lesions are managed with diphenhydramine or sodium bicarbonate mouthwashes and glycerin swabs.
An ophthalmologic consultation is mandatory because of the risk of vision loss associated with corneal scarring.
And now, a return to our 9-year-old patient
When we left off our discussion of Julie, the ED physician who examined her had detected a targetoid lesion with a necrotic, purpuric center—a finding that we described as a diagnostic clue. The second diagnostic clue? The presence of the Nikolsky sign, which the doctor detected by applying slight pressure to the lesion. Julie was admitted to the hospital with a presumed diagnosis of SJS, which skin biopsies and immunofluorescence studies later confirmed.
It wasn’t clear whether Julie had had a reaction to the sulfa (which she’d completed) or to the penicillin (which she’d just begun taking), or whether she had a synergistic reaction to both. Although the exact cause remained uncertain, as it often does, the penicillin was stopped immediately. She received dermatologic treatment without delay and was monitored closely for fluid and electrolyte status. Since Julie had signs of ocular involvement, daily erythromycin and corticosteroid eyedrops were administered to minimize the risk of infection and reduce local inflammation. Given the risk of long-term ocular complications in patients with SJS, we recommended continued ophthalmologic care.
Nine days after she was admitted, Julie’s symptoms resolved, with the exception of persistent complaints of dry eye. At discharge, Julie was given artificial tears to minimize ocular irritation. We suspected that she had dry eyes because of SJS-induced corneal scarring, but we were unable to confirm our suspicion because our patient failed to return for scheduled ophthalmologic appointments. She was subsequently lost to follow up.
Acknowledgement
The authors wish to thank Azita Hamedani, MD, MPH, FACEP, for her critical editing of the text.
Correspondence
Ribhi Hazin, MD, 29 Garden Street, Suite 214, Cambridge, MA 02138; Ribhi.hazin@gmail.com
1. Noe R, Cohen AL, Lederman E, et al. Skin disorders among construction workers following Hurricane Katrina and Hurricane Rita: an outbreak investigation in New Orleans, Louisiana. Arch Dermatol. 2007;143:1393-1398.
2. Kulthanan K, Jiamton S, Thumpimukvatana N, et al. Chronic idiopathic urticaria: prevalence and clinical course. J Dermatol. 2007;34:294-301.
3. Stanway AD, Cohen SN, Chen C, et al. H1-antihistamines for chronic urticaria. Cochrane Database Syst Rev. 2008(2):CD006137.-
4. Grattan CE, Humphreys F. British Association of Dermatologists Therapy Guidelines and Audit Subcommittee. Guidelines for evaluation and management of urticaria in adults and children. Br J Dermatol. 2007;157:1116-1123.
5. Kozel MM, Mekkes JR, Bossuyt PM, et al. Natural course of physical and chronic urticaria and angioedema in 220 patients. J Am Acad Dermatol. 2001;45:387-391.
6. Nettis E, Colanardi MC, Soccio AL, et al. Double-blind, placebo-controlled study of sublingual immunotherapy in patients with latex-induced urticaria: a 12-month study. Br J Dermatol. 2007;156:674-681.
7. Lee EE, Maibach HI. Treatment of urticaria. An evidence-based evaluation of antihistamines. Am J Clin Dermatol. 2001;2:27-32.
8. Humphreys F, Hunter JA. The characteristics of urticaria in 390 patients. Br J Dermatol. 1998;138:635-638.
9. Serhat Inaloz H, Ozturk S, Akcali C, et al. Low-dose and short-term cyclosporine treatment in patients with chronic idiopathic urticaria: A clinical and immunological evaluation. J Dermatol. 2008;35:276-282.
10. Erbagci Z. The leukotriene receptor antagonist montelukast in the treatment of chronic idiopathic urticaria. A single-blind, placebo-controlled, crossover clinical study. J Allergy Clin Immunol. 2002;110:484-488.
11. Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med. 2005;165:1637-1642.
12. Yusuf S, Teo KK, Pogue J, et al. ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008;358:1547-1559.
13. Joint Task Force on Practice Parameters; American Academy of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. The diagnosis and management of anaphylaxis: an updated practice parameter. J Allergy Clin Immunol. 2005;115(3 Suppl 2):s483-s523.
14. Bas M, Kirchhartz N, Hochfeld J, et al. Potential role of vasomotor effects of fibrinogen in bradykinin-induced angioedema. J Allergy Clin Immunol. 2008;121:969e2-975e2.
15. Sica DA, Black HR. Angioedema in heart failure: occurrence with ACE inhibitors and safety of angiotensin receptor blocker therapy. Congest Heart Fail. 2002;8:334-341.
16. Banerji A, Clark S, Blanda M, et al. Multicenter study of patients with angiotensin-converting enzyme inhibitor-induced angioedema who present to the emergency department. Ann Allergy Asthma Immunol. 2008;100:327-332.
17. Cicardi M, Zingale LC, Zanichelli A, et al. The use of plasma-derived C1 inhibitor in the treatment of hereditary angioedema. Expert Opin Pharmacother. 2007;8:3173-3181.
18. Bork K, Bygum A, Hardt J. Benefits and risks of danazol in hereditary angioedema: a long-term survey of 118 patients. Ann Allergy Asthma Immunol. 2008;100:153-161.
19. US Food and Drug Administration. Advisory Committee Briefing Document. Kalbitor (ecallantide) for acute attacks of hereditary angioedema. Available at: http://www.fda.gov/ohrms/dockets/AC/09/briefing/2009-4413b1-03-Dyax.pdf. Accessed May 5, 2009.
20. Schneider L, Lumry W, Vegh A, et al. Critical role of kallikrein in hereditary angioedema pathogenesis: a clinical trial of ecallantide, a novel kallikrein inhibitor. J Allergy Clin Immunol. 2007;120:416-422.
21. Ellis Simonsen SM, van Orman ER, Hatch BE, et al. Cellulitis incidence in a defined population. Epidemiol Infect. 2006;134:293-299.
22. Stevens DL, Bisno AL, Chambers HF, et al. Infectious Diseases Society of America. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin Infect Dis. 2005;41:1373-1406.
23. Morris A. What are the benefits of treatments? Cellulitis and erysipelas. BMJ Clin Evid. 2005;13:2066-2069.
24. Murray H, Stiell I, Wells G. Treatment failure in emergency department patients with cellulitis. CJEM. 2005;7:228-234.
25. Meier DE, Nkor SK, Aasa D, et al. Prospective randomized comparison of two preoperative skin preparation techniques in a developing world country. World J Surg. 2001;25:441-443.
26. Giordano PA, Elston D, Akinlade BK, et al. Cefdinir vs. cephalexin for mild to moderate uncomplicated skin and skin structure infections in adolescents and adults. Curr Med Res Opin. 2006;22:2419-2428.
27. Halpern J, Holder R, Langford NJ. Ethnicity and other risk factors for acute lower limb cellulitis: a U.K.-based prospective case-control study. Br J Dermatol. 2008;158:1288-1292.
28. Lin YT, Lu PW. Retrospective study of pediatric facial cellulitis of odontogenic origin. Pediatr Infect Dis J. 2006;25:339-342.
29. Rajendran PM, Young D, Maurer T, et al. Randomized, double-blind, placebo-controlled trial of cephalexin for treatment of uncomplicated skin abscesses in a population at risk for community-acquired methicillin-resistant Staphylococcus aureus infection. Antimicrob Agents Chemo. 2007;51:4044-4048.
30. Schneck J, Fagot JP, Sekula P, et al. Effects of treatments on the mortality of Stevens-Johnson syndrome and toxic epidermal necrolysis: a retrospective study on patients included in the prospective EuroSCAR Study. J Am Acad Dermatol. 2008;58:33-40.
31. Chan HL, Stern RS, Arndt KA, et al. The incidence of erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis. A population-based study with particular reference to reactions caused by drugs among outpatients. Arch Dermatol. 1990;126:43-47.
32. Hazin R, Ibrahimi OA, Hazin MI, et al. Stevens-Johnson syndrome: pathogenesis, diagnosis, and management. Ann Med, 2008;40:129-138.
33. Chung WH, Hung SI, Hong HS, et al. Medical genetics: a marker for Stevens-Johnson syndrome. Nature. 2004;428:486.-
34. Jette N, Hemming K, Hutton JL, et al. Topiramate add-on for drug-resistant partial epilepsy. Cochrane Database Syst Rev. 2008;(3):DC001417.-
35. Gürcan HM, Ahmed AR. Efficacy of various intravenous immunoglobulin therapy protocols in auto-immune and chronic inflammatory disorders. Ann Pharmacother. 2007;41:812-523.
36. Strom BL, Carson JL, Halpern AC, et al. A population-based study of Stevens-Johnson syndrome. Incidence and antecedent drug exposures. Arch Dermatol. 1991;127:831-838.
37. Gravante G, Delogu D, Marianetti M, et al. Toxic epidermal necrolysis and Steven-Johnson syndrome: 11-years experience and outcome. Eur Rev Med Pharmacol Sci. 2007;11:119-127.
- Management of hereditary angioedema should include fresh frozen plasma containing C1 inhibitor (C1-INH), whenever possible; if C1-INH-containing plasma is unavailable, fresh frozen plasma can be used instead (SOR: A).
- Do not give neomycin to patients with suspected cellulitis; the drug may promote antibiotic resistance in Staphylococcus aureus, a pathogen often associated with this condition (SOR: A).
- Whenever a patient presents with erythematous skin lesions and a recent history of receiving penicillin or a cephalosporin antibiotic, a sulfa derivative, or an anticonvulsant, the suspected medication should be stopped until Stevens-Johnson syndrome is ruled out (SOR: A).
Strength of recommendation (SOR)
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
Skin eruptions are a common reason for visits to primary care physicians. While most are innocuous, some are associated with—or are early warning signs of—severe allergic reactions or other emergent conditions. A 9-year-old patient I’ll call Julie is a case in point.
The first time Julie’s parents brought her to our clinic, she’d been complaining of a sore throat and had a fever that hovered between 102° and 103°F for several days. The physician who examined Julie found mild maxillary tenderness. A rapid streptococcal throat swab was negative; her doctor prescribed a 10-day course of trimethoprim/sulfamethoxazole (TMP/SMX) for presumed acute sinusitis.
Thirteen days later (3 days after the patient completed the course of antibiotics), Julie’s parents brought her back to the clinic. Her throat still hurt, and she had erythematous oval lesions on her trunk and upper extremities. Her physician diagnosed scarlet fever and wrote a prescription for penicillin.
The following day, Julie was taken to the emergency department (ED) with bilateral conjunctival hyperemia and diffuse, confluent erythematous macules throughout her body. The ED physician who examined Julie found a 2.5 × 2.0 cm targetoid lesion with a necrotic, purpuric center on her lower back—a diagnostic clue to the cause of her signs and symptoms.
If you had been Julie’s physician, would you have been alert to that clue?
For family physicians accustomed to seeing relatively mild skin disorders, recognizing and responding to dermatologic conditions with potentially dire outcomes can be challenging. This review, and the images that accompany it, will help you sharpen your dermatologic diagnostic and treatment skills, both for benign disorders and those that are less common and more severe. We’ll also tell you more about Julie and her diagnosis.
Urticaria: A simple case of hives?
This common allergic reaction affects close to 10% of the population at some point in their lives. The affected areas are itchy and have raised, circumscribed red welts with surrounding erythema.1 Urticaria can occur throughout the body, with new lesions often erupting as the old ones disappear.2,3
Despite the persistent itchiness that patients typically complain of, however, urticaria is usually self-limiting, and rarely life-threatening. Acute urticaria normally resolves within 2 to 6 weeks.2,4
In most cases, urticaria arises secondary to exposure to an allergenic substance, chemical, or emotional stress.4,5 In rare instances, systemic diseases, such as hematologic malignancies, can also cause urticarial lesions to erupt throughout the body.4
Body piercing, cosmetics, latex exposure, Helicobacter pylori, insects, and angiotensin-converting enzyme (ACE) inhibitors have been identified as common triggers of urticaria, as have nonsteroidal anti-inflammatory drugs (NSAIDs) and antibiotics, animal dander, and foods such as shellfish, nuts, and dairy products.4,6
Treatment of all forms of urticaria should be based on identification and strict avoidance of the causative agent, if it’s known.7 Following withdrawal of the specific agent, symptomatic treatment with medications such as histamine antagonists and corticosteroids remains the mainstay of therapy.4,8 A daily dose of 40 to 60 mg prednisone for 5 days is a reasonable therapeutic regimen for adults; a 5-day course of 1 mg/kg per day is suitable for pediatric patients.4,8,9
In the event that topical or oral therapy is ineffective in mild cases of urticaria, intravenous (IV) diphenhydramine (50 mg) can be administered every 6 to 8 hours.4,8 IV diphenhydramine typically takes 30 minutes to work, while corticosteroids take at least 2 hours to reach full effect.4,8
In an emergency setting, subcutaneous epinephrine (0.3-0.5 mg) can be useful in treating severe urticaria.4,8 And recent clinical trials have demonstrated complete clearance of urticaria with leukotriene inhibitors, such as montelukast (10 mg).10
Chronic urticaria, trigger unknown
Although acute urticaria is responsive to treatment, chronic urticaria—lesions that do not resolve after 6 weeks—poses a greater challenge. In up to 80% of cases of chronic urticaria, no identifiable trigger is found.3,8 Long-term treatment of patients with this chronic condition, including lifestyle changes (to avoid environmental or dietary triggers) and a medication regimen for 6 months or more, leads to complete resolution of symptoms in most cases.7,8
Angioedema: Less common, more dangerous
Angioedema is part of the same disease spectrum as urticaria, but it affects the deeper tissues—involving mucosal and submucosal swelling. Angioedema affects just 0.1% to 0.2% of the general population, but up to 15% of patients with urticaria.8
The risk increases with the use of ACE inhibitors; for every 1000 patients taking ACE inhibitors, 0.4 to 3.5 develop angioedema.11 A recent double-blind study involving 25,642 patients being treated with ACE inhibitors revealed that those taking a combination of ACE inhibitors were more likely to develop angioedema than those on monotherapy.12
Angioedema is characterized by the sudden appearance of painful, localized erythematous wheals with central blanching.13 It results from increased vascular permeability in capillaries of the dermis, which leads to fluid leakage.13,14 Increased accumulation of fluids from the vessels of the skin results in rapidly developing nonpitting edema that most often affects the hands, face (and lips), neck, and oropharynx (FIGURE 1).14 Although the head and neck are the most commonly involved areas, angioedema can also affect the digestive tract, leading to nausea, vomiting, diarrhea, and abdominal pain secondary to bowel edema.13
FIGURE 1
Angioedema: A look at the most commonly affected areas
Airway management is imperative
In severe cases of angioedema, involvement of the oral mucosa results in stridor, followed by upper airway obstruction. To avoid hypoxemia and death,13,14 rapid preparation for emergency intervention to maintain the airway is critical; mortality can be as high as 30% in patients with airway compromise.13,15 In severe cases in which edema engulfs the oropharynx, oral intubation is often impossible, and nasotracheal intubation may be warranted.15,16 A failure to maintain adequate airway function via nasotracheal intubation may signal the need for an emergency tracheotomy.15,16
Is the angioedema hereditary or acquired?
Hereditary and acquired angioedema are treated differently. After ensuring that the patient has a patent airway, distinguishing between them is critical. Hereditary angioedema is the result of an inherited deficiency of plasma protein C1 inhibitor (C1-INH). Acquired angioedema is typically caused by enhanced consumption of endogenous C1-INH,13,17 leading to a net deficit of circulating C1-INH, and can be triggered by food, pharmacologic agents, and, occasionally, by systemic disorders.13 African Americans and patients with renal impairment appear to be at increased risk for acquired angioedema.13 In both hereditary and acquired angioiedema, decreased levels of C1-INH lead to disruption of the complement pathway.
Although the clinical presentation of acquired and hereditary angioedema is similar, a focused patient history can be used to distinguish between them. Age, health status, and medication history are the key considerations. Hereditary angioedema most commonly occurs—in recurrent attacks after minor trauma—in children with no underlying disease, with worsening symptoms during puberty. Acquired angioedema is generally seen in adult and elderly patients with malignancies or other underlying disorders.
Mild to moderate cases of acquired angioedema respond well to oral corticosteroids and antihistamines. Severe cases often require administration of subcutaneous epinephrine, followed by IV steroids. Management of hereditary angioedema should include C1-INH-containing fresh frozen plasma,13,16,18 although fresh frozen plasma can be used if plasma with C1-INH is not available.13
While oral corticosteroids and anti-histamines may be effective adjunctive therapy for hereditary angioedema, they are not likely to reverse acute attacks in this patient population. IV diphenhydramine (50-100 mg) or IV cimetidine (300 mg) every 6 to 8 hours is a reasonable therapeutic regimen for acute attacks of hereditary angioedema.
A recent randomized double-blind trial involving 40 patients with hereditary angioedema studied the administration of ecallantide, a kallikrein inhibitor for which US Food and Drug Administration approval is pending.19 Nearly 3 out of 4 of those who received ecallantide (72.5%) for acute attacks of angioedema showed significant improvement within 4 hours.20 The use of kallikrein inhibitors, which target inflammatory blood components, is not widespread, but may hold promise for the treatment of hereditary angioedema.20
Cellulitis: On the lookout for infiltration
Cellulitis is a bacterial infection of the skin that affects approximately 24.6 in 1000 people and is rarely associated with death.21 It occurs when bacteria enter through disrupted areas in the skin, particularly when skin integrity is compromised by recent surgery, piercing, wounds, athlete’s foot, or even dermatitis.22,23Streptococcus and Staphylococcus are the 2 most common infectious agents, and methicillin-resistant Staphylococcus aureus (MRSA) is increasingly common.22,23
Although cellulitis is primarily superficial in nature, it may progress to a serious condition by infiltrating underlying tissues and spreading to nearby lymphatic tissue and the bloodstream to cause lymphadenitis or bacteremia.21,23 In instances of cellulitis-induced bacteremia, mortality rates increase if prompt, targeted treatment is not provided.23
Raised erythematous plaques are the cardinal features of cellulitis, with the affected areas warm to the touch, red, and tender.21,23 As the condition progresses, the affected area tends to enlarge and expand (FIGURE 2),24 and the patient often becomes febrile.22,24
The risk of developing cellulitis increases with age, compromised immune status, diabetes, obesity, IV drug use, lymphedema, and chronic corticosteroid use.22,24
Cellulitis is often diagnosed solely on the basis of clinical presentation, although aspiration of purulent discharge from the wound and a gram stain of the culture can confirm the diagnosis.25,26 (See “Is it cellulitis or stasis dermatitis?”.)
Direct immunofluorescence can be used when cultures are difficult to obtain, but this technique is seldom necessary.22 If infiltration of underlying soft tissues is suspected based on clinical findings, magnetic resonance imaging can be a useful tool in evaluating the extent of the infection and in directing appropriate debridement and drainage of affected areas.22,27
Patients with venous insufficiency may present with stasis dermatitis, which often results in breakdown of the skin and ulceration that bears a striking resemblance to cellulitis. Thus, these conditions can be easily confused, and may lead to unnecessary antibiotic use and, possibly, hospitalization in patients with venous insufficiency.26,28
Despite the similarities of these conditions, a focused patient history and physical exam can prevent such confusion. Stasis dermatitis arises as a result of venous insufficiency, so it is likely to be accompanied by pitting edema that responds to leg raising and to the use of elastic compression stockings—interventions that are seldom effective for cellulitis.26 In addition, cellulitis tends to be unilateral, while stasis dermatitis often has bilateral involvement.
FIGURE 2
Diagnosing cellulitis based on clinical presentation
The raised erythematous lesions that are a hallmark of cellulitis, shown here on the arm and face, are warm to the touch, red, and tender.
Treatment: Targeted antibiotics and preventive measures
Because of the likelihood of recurrence with cellulitis, treating the condition involves both preventive and curative measures. Mild cases can be treated in an outpatient setting with a 7- to 10-day course of oral cephalosporins or antibiotics with similar coverage.22,25,26 A recent randomized study involving 391 patients found that cure rates for cellulitis treated with cephalexin were between 83% and 92%, depending on the pathogen involved.26
For severe cases of cellulitis, patients who are immunocompromised, and cases that are refractory to oral medications, hospital admission is recommended, and use of IV antibiotics is routinely required.22,25,26 For patients with MRSA, a drug such as vancomycin IV may be warranted; a reasonable dose would be 15 mg/kg every 12 hours.22,26,27,29
A recent randomized, multicenter study demonstrated that vancomycin effectively treated approximately 67% of cases of MRSA-induced cellulitis.26 Neomycin should be strictly avoided whenever cellulitis is suspected, because of its propensity to promote antibiotic resistance to S. aureus.29
Patient education emphasizing preventive measures is critical for minimizing recurrence of cellulitis.22,25,26 Encourage patients to wash with antibacterial soap and water daily, apply topical antibiotic ointment, and keep the wound completely covered at all times. Advise them to change bandages and wash their hands frequently.27,29 Patients with diabetes and others with decreased circulation in the extremities need to take further precautions, such as moisturizing the skin regularly in order to prevent cuts in their skin.22,29
SJS: Triggered by drugs, and infections, too
Stevens-Johnson syndrome (SJS), also known as erythema multiforme major, is an often-debilitating and possibly fatal adverse reaction, typically (but not exclusively) to a drug. It manifests as full-thickness epidermal necrosis of the mucous membranes.30,31 SJS occurs at a rate of about 1 to 7 cases per million people per year, and has a mortality rate of approximately 5%.30-32 The types of medication that most commonly precipitate SJS are anticonvulsants, sulfa drugs, penicillin-related and cephalosporin antibiotics, anti-inflammatory agents, and certain neoplastic drugs.30,32 SJS can develop in response to infections and neoplasms (TABLE) as well, and in many cases a cause is never found.
Patients with widespread involvement often complain of a burning sensation, particularly around the mouth.32-36 In some cases, this is the presenting sign, because the oral mucosa tends to be among the first mucous membranes involved.
TABLE
Stevens-Johnson syndrome: Pinpointing the cause32
| MORE FREQUENT ETIOLOGY |
| Drugs: Allopurinol, anticonvulsants, antiparasitics, barbiturates, NSAIDs, penicillin-related and cephalosporin antibiotics, sulfas, tetracyclines |
| LESS FREQUENT ETIOLOGY |
| Bacterial: Diphtheria, group A Streptococcus, Mycoplasma pneumoniae, tularemia, typhoid |
| Fungal: Coccidiomycosis, dermatophytosis, histoplasmosis, |
| Protozoan: Plasmodium, trichomoniasis |
| Viral: AIDS, Coxsackie, Epstein-Barr, HSV, influenza |
| AIDS, acquired immune deficiency syndrome; HSV, herpes-simplex virus; NSAIDs, nonsteroidal anti-inflammatory drugs. |
Targetoid lesions, Nikolsky sign are diagnostic clues
The characteristic skin lesions seen with SJS consist of initially erythematous macules that rapidly develop central necrosis to form vesiculation, as well as other variable areas of denudation (FIGURE 3). These vesicles tend to demonstrate confluence and often show a positive Nikolsky sign—epidermal detachment of superficial layers of the skin when slight pressure is applied. The lesions typically take on a targetoid appearance. If left untreated, the blisters often result in ulceration and hemorrhagic crusting of affected areas.
Like most skin disorders, SJS is initially diagnosed on the basis of clinical presentation. However, it is a rare disorder, and commonly misdiagnosed. Among the disorders SJS has been mistaken for are staphylococcal scalded skin syndrome, toxic shock syndrome, exfoliative dermatitis, scarlet fever, erythema multiforme, and iatrogenic chemical burns.32,37 (Erythema multiforme, SJS, and toxic epidermal necrolysis [TEN] are considered part of the same disease spectrum; erythema multiforme typically presents with few random lesions and no mucosal involvement, SJS with mucosal involvement on up to 30% of the body surface, and TEN with >30%.)32,37 Skin biopsies and immunofluorescence studies are recommended to confirm the diagnosis.
During the course of SJS, the mucous membranes of the oropharynx, ocular cavity, gastrointestinal system, nasal cavity, genitourinary system, and lower respiratory tracts are typically affected.32-35 As the condition progresses, increased epidermal erosion can lead to the sloughing off of up to 100% of the epidermis, resulting in considerable fluid loss.32,34
Corneal ulceration, anterior uveitis, panophthalmitis, polyarthritis, hematuria, and acute tubular necrosis leading to renal failure may also occur.32,35,37 Scarring within vital ocular structures can result in corneal opacity and lead to significant visual impairment. In severe cases, blood loss and fluid loss increase the risk of bacterial superinfection and sepsis.35,37
FIGURE 3
Targetoid lesions are characteristic of SJS
Characteristic diffuse erythematous macules with necrotic centers and overlying blistering on the back of a patient with Stevens-Johnson syndrome.
Supportive therapy, wound care are key components of treatment
Rapid cessation of the offending agent with targeted dermatologic management can reduce morbidity by promoting rapid re-epithelialization of affected skin. (See “SJS is diagnosed, but not quickly,” [Verdicts] on page 332, for a discussion of the dangers of delayed diagnosis and failure to promptly stop the drug causing the acute reaction.)
Closely monitoring the patient for fluid and electrolyte abnormalities is also crucial. Corticosteroids and IV immunoglobulins have been suggested for early severe cases of SJS, but their efficacy in treating this condition has yet to be established by prospective double-blind studies.32-37
The skin lesions associated with SJS should be treated in the same way you would treat thermal burns, with local wound care, warm compresses, and topical anesthetics for pain reduction.36,37 Oral lesions are managed with diphenhydramine or sodium bicarbonate mouthwashes and glycerin swabs.
An ophthalmologic consultation is mandatory because of the risk of vision loss associated with corneal scarring.
And now, a return to our 9-year-old patient
When we left off our discussion of Julie, the ED physician who examined her had detected a targetoid lesion with a necrotic, purpuric center—a finding that we described as a diagnostic clue. The second diagnostic clue? The presence of the Nikolsky sign, which the doctor detected by applying slight pressure to the lesion. Julie was admitted to the hospital with a presumed diagnosis of SJS, which skin biopsies and immunofluorescence studies later confirmed.
It wasn’t clear whether Julie had had a reaction to the sulfa (which she’d completed) or to the penicillin (which she’d just begun taking), or whether she had a synergistic reaction to both. Although the exact cause remained uncertain, as it often does, the penicillin was stopped immediately. She received dermatologic treatment without delay and was monitored closely for fluid and electrolyte status. Since Julie had signs of ocular involvement, daily erythromycin and corticosteroid eyedrops were administered to minimize the risk of infection and reduce local inflammation. Given the risk of long-term ocular complications in patients with SJS, we recommended continued ophthalmologic care.
Nine days after she was admitted, Julie’s symptoms resolved, with the exception of persistent complaints of dry eye. At discharge, Julie was given artificial tears to minimize ocular irritation. We suspected that she had dry eyes because of SJS-induced corneal scarring, but we were unable to confirm our suspicion because our patient failed to return for scheduled ophthalmologic appointments. She was subsequently lost to follow up.
Acknowledgement
The authors wish to thank Azita Hamedani, MD, MPH, FACEP, for her critical editing of the text.
Correspondence
Ribhi Hazin, MD, 29 Garden Street, Suite 214, Cambridge, MA 02138; Ribhi.hazin@gmail.com
- Management of hereditary angioedema should include fresh frozen plasma containing C1 inhibitor (C1-INH), whenever possible; if C1-INH-containing plasma is unavailable, fresh frozen plasma can be used instead (SOR: A).
- Do not give neomycin to patients with suspected cellulitis; the drug may promote antibiotic resistance in Staphylococcus aureus, a pathogen often associated with this condition (SOR: A).
- Whenever a patient presents with erythematous skin lesions and a recent history of receiving penicillin or a cephalosporin antibiotic, a sulfa derivative, or an anticonvulsant, the suspected medication should be stopped until Stevens-Johnson syndrome is ruled out (SOR: A).
Strength of recommendation (SOR)
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
Skin eruptions are a common reason for visits to primary care physicians. While most are innocuous, some are associated with—or are early warning signs of—severe allergic reactions or other emergent conditions. A 9-year-old patient I’ll call Julie is a case in point.
The first time Julie’s parents brought her to our clinic, she’d been complaining of a sore throat and had a fever that hovered between 102° and 103°F for several days. The physician who examined Julie found mild maxillary tenderness. A rapid streptococcal throat swab was negative; her doctor prescribed a 10-day course of trimethoprim/sulfamethoxazole (TMP/SMX) for presumed acute sinusitis.
Thirteen days later (3 days after the patient completed the course of antibiotics), Julie’s parents brought her back to the clinic. Her throat still hurt, and she had erythematous oval lesions on her trunk and upper extremities. Her physician diagnosed scarlet fever and wrote a prescription for penicillin.
The following day, Julie was taken to the emergency department (ED) with bilateral conjunctival hyperemia and diffuse, confluent erythematous macules throughout her body. The ED physician who examined Julie found a 2.5 × 2.0 cm targetoid lesion with a necrotic, purpuric center on her lower back—a diagnostic clue to the cause of her signs and symptoms.
If you had been Julie’s physician, would you have been alert to that clue?
For family physicians accustomed to seeing relatively mild skin disorders, recognizing and responding to dermatologic conditions with potentially dire outcomes can be challenging. This review, and the images that accompany it, will help you sharpen your dermatologic diagnostic and treatment skills, both for benign disorders and those that are less common and more severe. We’ll also tell you more about Julie and her diagnosis.
Urticaria: A simple case of hives?
This common allergic reaction affects close to 10% of the population at some point in their lives. The affected areas are itchy and have raised, circumscribed red welts with surrounding erythema.1 Urticaria can occur throughout the body, with new lesions often erupting as the old ones disappear.2,3
Despite the persistent itchiness that patients typically complain of, however, urticaria is usually self-limiting, and rarely life-threatening. Acute urticaria normally resolves within 2 to 6 weeks.2,4
In most cases, urticaria arises secondary to exposure to an allergenic substance, chemical, or emotional stress.4,5 In rare instances, systemic diseases, such as hematologic malignancies, can also cause urticarial lesions to erupt throughout the body.4
Body piercing, cosmetics, latex exposure, Helicobacter pylori, insects, and angiotensin-converting enzyme (ACE) inhibitors have been identified as common triggers of urticaria, as have nonsteroidal anti-inflammatory drugs (NSAIDs) and antibiotics, animal dander, and foods such as shellfish, nuts, and dairy products.4,6
Treatment of all forms of urticaria should be based on identification and strict avoidance of the causative agent, if it’s known.7 Following withdrawal of the specific agent, symptomatic treatment with medications such as histamine antagonists and corticosteroids remains the mainstay of therapy.4,8 A daily dose of 40 to 60 mg prednisone for 5 days is a reasonable therapeutic regimen for adults; a 5-day course of 1 mg/kg per day is suitable for pediatric patients.4,8,9
In the event that topical or oral therapy is ineffective in mild cases of urticaria, intravenous (IV) diphenhydramine (50 mg) can be administered every 6 to 8 hours.4,8 IV diphenhydramine typically takes 30 minutes to work, while corticosteroids take at least 2 hours to reach full effect.4,8
In an emergency setting, subcutaneous epinephrine (0.3-0.5 mg) can be useful in treating severe urticaria.4,8 And recent clinical trials have demonstrated complete clearance of urticaria with leukotriene inhibitors, such as montelukast (10 mg).10
Chronic urticaria, trigger unknown
Although acute urticaria is responsive to treatment, chronic urticaria—lesions that do not resolve after 6 weeks—poses a greater challenge. In up to 80% of cases of chronic urticaria, no identifiable trigger is found.3,8 Long-term treatment of patients with this chronic condition, including lifestyle changes (to avoid environmental or dietary triggers) and a medication regimen for 6 months or more, leads to complete resolution of symptoms in most cases.7,8
Angioedema: Less common, more dangerous
Angioedema is part of the same disease spectrum as urticaria, but it affects the deeper tissues—involving mucosal and submucosal swelling. Angioedema affects just 0.1% to 0.2% of the general population, but up to 15% of patients with urticaria.8
The risk increases with the use of ACE inhibitors; for every 1000 patients taking ACE inhibitors, 0.4 to 3.5 develop angioedema.11 A recent double-blind study involving 25,642 patients being treated with ACE inhibitors revealed that those taking a combination of ACE inhibitors were more likely to develop angioedema than those on monotherapy.12
Angioedema is characterized by the sudden appearance of painful, localized erythematous wheals with central blanching.13 It results from increased vascular permeability in capillaries of the dermis, which leads to fluid leakage.13,14 Increased accumulation of fluids from the vessels of the skin results in rapidly developing nonpitting edema that most often affects the hands, face (and lips), neck, and oropharynx (FIGURE 1).14 Although the head and neck are the most commonly involved areas, angioedema can also affect the digestive tract, leading to nausea, vomiting, diarrhea, and abdominal pain secondary to bowel edema.13
FIGURE 1
Angioedema: A look at the most commonly affected areas
Airway management is imperative
In severe cases of angioedema, involvement of the oral mucosa results in stridor, followed by upper airway obstruction. To avoid hypoxemia and death,13,14 rapid preparation for emergency intervention to maintain the airway is critical; mortality can be as high as 30% in patients with airway compromise.13,15 In severe cases in which edema engulfs the oropharynx, oral intubation is often impossible, and nasotracheal intubation may be warranted.15,16 A failure to maintain adequate airway function via nasotracheal intubation may signal the need for an emergency tracheotomy.15,16
Is the angioedema hereditary or acquired?
Hereditary and acquired angioedema are treated differently. After ensuring that the patient has a patent airway, distinguishing between them is critical. Hereditary angioedema is the result of an inherited deficiency of plasma protein C1 inhibitor (C1-INH). Acquired angioedema is typically caused by enhanced consumption of endogenous C1-INH,13,17 leading to a net deficit of circulating C1-INH, and can be triggered by food, pharmacologic agents, and, occasionally, by systemic disorders.13 African Americans and patients with renal impairment appear to be at increased risk for acquired angioedema.13 In both hereditary and acquired angioiedema, decreased levels of C1-INH lead to disruption of the complement pathway.
Although the clinical presentation of acquired and hereditary angioedema is similar, a focused patient history can be used to distinguish between them. Age, health status, and medication history are the key considerations. Hereditary angioedema most commonly occurs—in recurrent attacks after minor trauma—in children with no underlying disease, with worsening symptoms during puberty. Acquired angioedema is generally seen in adult and elderly patients with malignancies or other underlying disorders.
Mild to moderate cases of acquired angioedema respond well to oral corticosteroids and antihistamines. Severe cases often require administration of subcutaneous epinephrine, followed by IV steroids. Management of hereditary angioedema should include C1-INH-containing fresh frozen plasma,13,16,18 although fresh frozen plasma can be used if plasma with C1-INH is not available.13
While oral corticosteroids and anti-histamines may be effective adjunctive therapy for hereditary angioedema, they are not likely to reverse acute attacks in this patient population. IV diphenhydramine (50-100 mg) or IV cimetidine (300 mg) every 6 to 8 hours is a reasonable therapeutic regimen for acute attacks of hereditary angioedema.
A recent randomized double-blind trial involving 40 patients with hereditary angioedema studied the administration of ecallantide, a kallikrein inhibitor for which US Food and Drug Administration approval is pending.19 Nearly 3 out of 4 of those who received ecallantide (72.5%) for acute attacks of angioedema showed significant improvement within 4 hours.20 The use of kallikrein inhibitors, which target inflammatory blood components, is not widespread, but may hold promise for the treatment of hereditary angioedema.20
Cellulitis: On the lookout for infiltration
Cellulitis is a bacterial infection of the skin that affects approximately 24.6 in 1000 people and is rarely associated with death.21 It occurs when bacteria enter through disrupted areas in the skin, particularly when skin integrity is compromised by recent surgery, piercing, wounds, athlete’s foot, or even dermatitis.22,23Streptococcus and Staphylococcus are the 2 most common infectious agents, and methicillin-resistant Staphylococcus aureus (MRSA) is increasingly common.22,23
Although cellulitis is primarily superficial in nature, it may progress to a serious condition by infiltrating underlying tissues and spreading to nearby lymphatic tissue and the bloodstream to cause lymphadenitis or bacteremia.21,23 In instances of cellulitis-induced bacteremia, mortality rates increase if prompt, targeted treatment is not provided.23
Raised erythematous plaques are the cardinal features of cellulitis, with the affected areas warm to the touch, red, and tender.21,23 As the condition progresses, the affected area tends to enlarge and expand (FIGURE 2),24 and the patient often becomes febrile.22,24
The risk of developing cellulitis increases with age, compromised immune status, diabetes, obesity, IV drug use, lymphedema, and chronic corticosteroid use.22,24
Cellulitis is often diagnosed solely on the basis of clinical presentation, although aspiration of purulent discharge from the wound and a gram stain of the culture can confirm the diagnosis.25,26 (See “Is it cellulitis or stasis dermatitis?”.)
Direct immunofluorescence can be used when cultures are difficult to obtain, but this technique is seldom necessary.22 If infiltration of underlying soft tissues is suspected based on clinical findings, magnetic resonance imaging can be a useful tool in evaluating the extent of the infection and in directing appropriate debridement and drainage of affected areas.22,27
Patients with venous insufficiency may present with stasis dermatitis, which often results in breakdown of the skin and ulceration that bears a striking resemblance to cellulitis. Thus, these conditions can be easily confused, and may lead to unnecessary antibiotic use and, possibly, hospitalization in patients with venous insufficiency.26,28
Despite the similarities of these conditions, a focused patient history and physical exam can prevent such confusion. Stasis dermatitis arises as a result of venous insufficiency, so it is likely to be accompanied by pitting edema that responds to leg raising and to the use of elastic compression stockings—interventions that are seldom effective for cellulitis.26 In addition, cellulitis tends to be unilateral, while stasis dermatitis often has bilateral involvement.
FIGURE 2
Diagnosing cellulitis based on clinical presentation
The raised erythematous lesions that are a hallmark of cellulitis, shown here on the arm and face, are warm to the touch, red, and tender.
Treatment: Targeted antibiotics and preventive measures
Because of the likelihood of recurrence with cellulitis, treating the condition involves both preventive and curative measures. Mild cases can be treated in an outpatient setting with a 7- to 10-day course of oral cephalosporins or antibiotics with similar coverage.22,25,26 A recent randomized study involving 391 patients found that cure rates for cellulitis treated with cephalexin were between 83% and 92%, depending on the pathogen involved.26
For severe cases of cellulitis, patients who are immunocompromised, and cases that are refractory to oral medications, hospital admission is recommended, and use of IV antibiotics is routinely required.22,25,26 For patients with MRSA, a drug such as vancomycin IV may be warranted; a reasonable dose would be 15 mg/kg every 12 hours.22,26,27,29
A recent randomized, multicenter study demonstrated that vancomycin effectively treated approximately 67% of cases of MRSA-induced cellulitis.26 Neomycin should be strictly avoided whenever cellulitis is suspected, because of its propensity to promote antibiotic resistance to S. aureus.29
Patient education emphasizing preventive measures is critical for minimizing recurrence of cellulitis.22,25,26 Encourage patients to wash with antibacterial soap and water daily, apply topical antibiotic ointment, and keep the wound completely covered at all times. Advise them to change bandages and wash their hands frequently.27,29 Patients with diabetes and others with decreased circulation in the extremities need to take further precautions, such as moisturizing the skin regularly in order to prevent cuts in their skin.22,29
SJS: Triggered by drugs, and infections, too
Stevens-Johnson syndrome (SJS), also known as erythema multiforme major, is an often-debilitating and possibly fatal adverse reaction, typically (but not exclusively) to a drug. It manifests as full-thickness epidermal necrosis of the mucous membranes.30,31 SJS occurs at a rate of about 1 to 7 cases per million people per year, and has a mortality rate of approximately 5%.30-32 The types of medication that most commonly precipitate SJS are anticonvulsants, sulfa drugs, penicillin-related and cephalosporin antibiotics, anti-inflammatory agents, and certain neoplastic drugs.30,32 SJS can develop in response to infections and neoplasms (TABLE) as well, and in many cases a cause is never found.
Patients with widespread involvement often complain of a burning sensation, particularly around the mouth.32-36 In some cases, this is the presenting sign, because the oral mucosa tends to be among the first mucous membranes involved.
TABLE
Stevens-Johnson syndrome: Pinpointing the cause32
| MORE FREQUENT ETIOLOGY |
| Drugs: Allopurinol, anticonvulsants, antiparasitics, barbiturates, NSAIDs, penicillin-related and cephalosporin antibiotics, sulfas, tetracyclines |
| LESS FREQUENT ETIOLOGY |
| Bacterial: Diphtheria, group A Streptococcus, Mycoplasma pneumoniae, tularemia, typhoid |
| Fungal: Coccidiomycosis, dermatophytosis, histoplasmosis, |
| Protozoan: Plasmodium, trichomoniasis |
| Viral: AIDS, Coxsackie, Epstein-Barr, HSV, influenza |
| AIDS, acquired immune deficiency syndrome; HSV, herpes-simplex virus; NSAIDs, nonsteroidal anti-inflammatory drugs. |
Targetoid lesions, Nikolsky sign are diagnostic clues
The characteristic skin lesions seen with SJS consist of initially erythematous macules that rapidly develop central necrosis to form vesiculation, as well as other variable areas of denudation (FIGURE 3). These vesicles tend to demonstrate confluence and often show a positive Nikolsky sign—epidermal detachment of superficial layers of the skin when slight pressure is applied. The lesions typically take on a targetoid appearance. If left untreated, the blisters often result in ulceration and hemorrhagic crusting of affected areas.
Like most skin disorders, SJS is initially diagnosed on the basis of clinical presentation. However, it is a rare disorder, and commonly misdiagnosed. Among the disorders SJS has been mistaken for are staphylococcal scalded skin syndrome, toxic shock syndrome, exfoliative dermatitis, scarlet fever, erythema multiforme, and iatrogenic chemical burns.32,37 (Erythema multiforme, SJS, and toxic epidermal necrolysis [TEN] are considered part of the same disease spectrum; erythema multiforme typically presents with few random lesions and no mucosal involvement, SJS with mucosal involvement on up to 30% of the body surface, and TEN with >30%.)32,37 Skin biopsies and immunofluorescence studies are recommended to confirm the diagnosis.
During the course of SJS, the mucous membranes of the oropharynx, ocular cavity, gastrointestinal system, nasal cavity, genitourinary system, and lower respiratory tracts are typically affected.32-35 As the condition progresses, increased epidermal erosion can lead to the sloughing off of up to 100% of the epidermis, resulting in considerable fluid loss.32,34
Corneal ulceration, anterior uveitis, panophthalmitis, polyarthritis, hematuria, and acute tubular necrosis leading to renal failure may also occur.32,35,37 Scarring within vital ocular structures can result in corneal opacity and lead to significant visual impairment. In severe cases, blood loss and fluid loss increase the risk of bacterial superinfection and sepsis.35,37
FIGURE 3
Targetoid lesions are characteristic of SJS
Characteristic diffuse erythematous macules with necrotic centers and overlying blistering on the back of a patient with Stevens-Johnson syndrome.
Supportive therapy, wound care are key components of treatment
Rapid cessation of the offending agent with targeted dermatologic management can reduce morbidity by promoting rapid re-epithelialization of affected skin. (See “SJS is diagnosed, but not quickly,” [Verdicts] on page 332, for a discussion of the dangers of delayed diagnosis and failure to promptly stop the drug causing the acute reaction.)
Closely monitoring the patient for fluid and electrolyte abnormalities is also crucial. Corticosteroids and IV immunoglobulins have been suggested for early severe cases of SJS, but their efficacy in treating this condition has yet to be established by prospective double-blind studies.32-37
The skin lesions associated with SJS should be treated in the same way you would treat thermal burns, with local wound care, warm compresses, and topical anesthetics for pain reduction.36,37 Oral lesions are managed with diphenhydramine or sodium bicarbonate mouthwashes and glycerin swabs.
An ophthalmologic consultation is mandatory because of the risk of vision loss associated with corneal scarring.
And now, a return to our 9-year-old patient
When we left off our discussion of Julie, the ED physician who examined her had detected a targetoid lesion with a necrotic, purpuric center—a finding that we described as a diagnostic clue. The second diagnostic clue? The presence of the Nikolsky sign, which the doctor detected by applying slight pressure to the lesion. Julie was admitted to the hospital with a presumed diagnosis of SJS, which skin biopsies and immunofluorescence studies later confirmed.
It wasn’t clear whether Julie had had a reaction to the sulfa (which she’d completed) or to the penicillin (which she’d just begun taking), or whether she had a synergistic reaction to both. Although the exact cause remained uncertain, as it often does, the penicillin was stopped immediately. She received dermatologic treatment without delay and was monitored closely for fluid and electrolyte status. Since Julie had signs of ocular involvement, daily erythromycin and corticosteroid eyedrops were administered to minimize the risk of infection and reduce local inflammation. Given the risk of long-term ocular complications in patients with SJS, we recommended continued ophthalmologic care.
Nine days after she was admitted, Julie’s symptoms resolved, with the exception of persistent complaints of dry eye. At discharge, Julie was given artificial tears to minimize ocular irritation. We suspected that she had dry eyes because of SJS-induced corneal scarring, but we were unable to confirm our suspicion because our patient failed to return for scheduled ophthalmologic appointments. She was subsequently lost to follow up.
Acknowledgement
The authors wish to thank Azita Hamedani, MD, MPH, FACEP, for her critical editing of the text.
Correspondence
Ribhi Hazin, MD, 29 Garden Street, Suite 214, Cambridge, MA 02138; Ribhi.hazin@gmail.com
1. Noe R, Cohen AL, Lederman E, et al. Skin disorders among construction workers following Hurricane Katrina and Hurricane Rita: an outbreak investigation in New Orleans, Louisiana. Arch Dermatol. 2007;143:1393-1398.
2. Kulthanan K, Jiamton S, Thumpimukvatana N, et al. Chronic idiopathic urticaria: prevalence and clinical course. J Dermatol. 2007;34:294-301.
3. Stanway AD, Cohen SN, Chen C, et al. H1-antihistamines for chronic urticaria. Cochrane Database Syst Rev. 2008(2):CD006137.-
4. Grattan CE, Humphreys F. British Association of Dermatologists Therapy Guidelines and Audit Subcommittee. Guidelines for evaluation and management of urticaria in adults and children. Br J Dermatol. 2007;157:1116-1123.
5. Kozel MM, Mekkes JR, Bossuyt PM, et al. Natural course of physical and chronic urticaria and angioedema in 220 patients. J Am Acad Dermatol. 2001;45:387-391.
6. Nettis E, Colanardi MC, Soccio AL, et al. Double-blind, placebo-controlled study of sublingual immunotherapy in patients with latex-induced urticaria: a 12-month study. Br J Dermatol. 2007;156:674-681.
7. Lee EE, Maibach HI. Treatment of urticaria. An evidence-based evaluation of antihistamines. Am J Clin Dermatol. 2001;2:27-32.
8. Humphreys F, Hunter JA. The characteristics of urticaria in 390 patients. Br J Dermatol. 1998;138:635-638.
9. Serhat Inaloz H, Ozturk S, Akcali C, et al. Low-dose and short-term cyclosporine treatment in patients with chronic idiopathic urticaria: A clinical and immunological evaluation. J Dermatol. 2008;35:276-282.
10. Erbagci Z. The leukotriene receptor antagonist montelukast in the treatment of chronic idiopathic urticaria. A single-blind, placebo-controlled, crossover clinical study. J Allergy Clin Immunol. 2002;110:484-488.
11. Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med. 2005;165:1637-1642.
12. Yusuf S, Teo KK, Pogue J, et al. ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008;358:1547-1559.
13. Joint Task Force on Practice Parameters; American Academy of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. The diagnosis and management of anaphylaxis: an updated practice parameter. J Allergy Clin Immunol. 2005;115(3 Suppl 2):s483-s523.
14. Bas M, Kirchhartz N, Hochfeld J, et al. Potential role of vasomotor effects of fibrinogen in bradykinin-induced angioedema. J Allergy Clin Immunol. 2008;121:969e2-975e2.
15. Sica DA, Black HR. Angioedema in heart failure: occurrence with ACE inhibitors and safety of angiotensin receptor blocker therapy. Congest Heart Fail. 2002;8:334-341.
16. Banerji A, Clark S, Blanda M, et al. Multicenter study of patients with angiotensin-converting enzyme inhibitor-induced angioedema who present to the emergency department. Ann Allergy Asthma Immunol. 2008;100:327-332.
17. Cicardi M, Zingale LC, Zanichelli A, et al. The use of plasma-derived C1 inhibitor in the treatment of hereditary angioedema. Expert Opin Pharmacother. 2007;8:3173-3181.
18. Bork K, Bygum A, Hardt J. Benefits and risks of danazol in hereditary angioedema: a long-term survey of 118 patients. Ann Allergy Asthma Immunol. 2008;100:153-161.
19. US Food and Drug Administration. Advisory Committee Briefing Document. Kalbitor (ecallantide) for acute attacks of hereditary angioedema. Available at: http://www.fda.gov/ohrms/dockets/AC/09/briefing/2009-4413b1-03-Dyax.pdf. Accessed May 5, 2009.
20. Schneider L, Lumry W, Vegh A, et al. Critical role of kallikrein in hereditary angioedema pathogenesis: a clinical trial of ecallantide, a novel kallikrein inhibitor. J Allergy Clin Immunol. 2007;120:416-422.
21. Ellis Simonsen SM, van Orman ER, Hatch BE, et al. Cellulitis incidence in a defined population. Epidemiol Infect. 2006;134:293-299.
22. Stevens DL, Bisno AL, Chambers HF, et al. Infectious Diseases Society of America. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin Infect Dis. 2005;41:1373-1406.
23. Morris A. What are the benefits of treatments? Cellulitis and erysipelas. BMJ Clin Evid. 2005;13:2066-2069.
24. Murray H, Stiell I, Wells G. Treatment failure in emergency department patients with cellulitis. CJEM. 2005;7:228-234.
25. Meier DE, Nkor SK, Aasa D, et al. Prospective randomized comparison of two preoperative skin preparation techniques in a developing world country. World J Surg. 2001;25:441-443.
26. Giordano PA, Elston D, Akinlade BK, et al. Cefdinir vs. cephalexin for mild to moderate uncomplicated skin and skin structure infections in adolescents and adults. Curr Med Res Opin. 2006;22:2419-2428.
27. Halpern J, Holder R, Langford NJ. Ethnicity and other risk factors for acute lower limb cellulitis: a U.K.-based prospective case-control study. Br J Dermatol. 2008;158:1288-1292.
28. Lin YT, Lu PW. Retrospective study of pediatric facial cellulitis of odontogenic origin. Pediatr Infect Dis J. 2006;25:339-342.
29. Rajendran PM, Young D, Maurer T, et al. Randomized, double-blind, placebo-controlled trial of cephalexin for treatment of uncomplicated skin abscesses in a population at risk for community-acquired methicillin-resistant Staphylococcus aureus infection. Antimicrob Agents Chemo. 2007;51:4044-4048.
30. Schneck J, Fagot JP, Sekula P, et al. Effects of treatments on the mortality of Stevens-Johnson syndrome and toxic epidermal necrolysis: a retrospective study on patients included in the prospective EuroSCAR Study. J Am Acad Dermatol. 2008;58:33-40.
31. Chan HL, Stern RS, Arndt KA, et al. The incidence of erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis. A population-based study with particular reference to reactions caused by drugs among outpatients. Arch Dermatol. 1990;126:43-47.
32. Hazin R, Ibrahimi OA, Hazin MI, et al. Stevens-Johnson syndrome: pathogenesis, diagnosis, and management. Ann Med, 2008;40:129-138.
33. Chung WH, Hung SI, Hong HS, et al. Medical genetics: a marker for Stevens-Johnson syndrome. Nature. 2004;428:486.-
34. Jette N, Hemming K, Hutton JL, et al. Topiramate add-on for drug-resistant partial epilepsy. Cochrane Database Syst Rev. 2008;(3):DC001417.-
35. Gürcan HM, Ahmed AR. Efficacy of various intravenous immunoglobulin therapy protocols in auto-immune and chronic inflammatory disorders. Ann Pharmacother. 2007;41:812-523.
36. Strom BL, Carson JL, Halpern AC, et al. A population-based study of Stevens-Johnson syndrome. Incidence and antecedent drug exposures. Arch Dermatol. 1991;127:831-838.
37. Gravante G, Delogu D, Marianetti M, et al. Toxic epidermal necrolysis and Steven-Johnson syndrome: 11-years experience and outcome. Eur Rev Med Pharmacol Sci. 2007;11:119-127.
1. Noe R, Cohen AL, Lederman E, et al. Skin disorders among construction workers following Hurricane Katrina and Hurricane Rita: an outbreak investigation in New Orleans, Louisiana. Arch Dermatol. 2007;143:1393-1398.
2. Kulthanan K, Jiamton S, Thumpimukvatana N, et al. Chronic idiopathic urticaria: prevalence and clinical course. J Dermatol. 2007;34:294-301.
3. Stanway AD, Cohen SN, Chen C, et al. H1-antihistamines for chronic urticaria. Cochrane Database Syst Rev. 2008(2):CD006137.-
4. Grattan CE, Humphreys F. British Association of Dermatologists Therapy Guidelines and Audit Subcommittee. Guidelines for evaluation and management of urticaria in adults and children. Br J Dermatol. 2007;157:1116-1123.
5. Kozel MM, Mekkes JR, Bossuyt PM, et al. Natural course of physical and chronic urticaria and angioedema in 220 patients. J Am Acad Dermatol. 2001;45:387-391.
6. Nettis E, Colanardi MC, Soccio AL, et al. Double-blind, placebo-controlled study of sublingual immunotherapy in patients with latex-induced urticaria: a 12-month study. Br J Dermatol. 2007;156:674-681.
7. Lee EE, Maibach HI. Treatment of urticaria. An evidence-based evaluation of antihistamines. Am J Clin Dermatol. 2001;2:27-32.
8. Humphreys F, Hunter JA. The characteristics of urticaria in 390 patients. Br J Dermatol. 1998;138:635-638.
9. Serhat Inaloz H, Ozturk S, Akcali C, et al. Low-dose and short-term cyclosporine treatment in patients with chronic idiopathic urticaria: A clinical and immunological evaluation. J Dermatol. 2008;35:276-282.
10. Erbagci Z. The leukotriene receptor antagonist montelukast in the treatment of chronic idiopathic urticaria. A single-blind, placebo-controlled, crossover clinical study. J Allergy Clin Immunol. 2002;110:484-488.
11. Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med. 2005;165:1637-1642.
12. Yusuf S, Teo KK, Pogue J, et al. ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008;358:1547-1559.
13. Joint Task Force on Practice Parameters; American Academy of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. The diagnosis and management of anaphylaxis: an updated practice parameter. J Allergy Clin Immunol. 2005;115(3 Suppl 2):s483-s523.
14. Bas M, Kirchhartz N, Hochfeld J, et al. Potential role of vasomotor effects of fibrinogen in bradykinin-induced angioedema. J Allergy Clin Immunol. 2008;121:969e2-975e2.
15. Sica DA, Black HR. Angioedema in heart failure: occurrence with ACE inhibitors and safety of angiotensin receptor blocker therapy. Congest Heart Fail. 2002;8:334-341.
16. Banerji A, Clark S, Blanda M, et al. Multicenter study of patients with angiotensin-converting enzyme inhibitor-induced angioedema who present to the emergency department. Ann Allergy Asthma Immunol. 2008;100:327-332.
17. Cicardi M, Zingale LC, Zanichelli A, et al. The use of plasma-derived C1 inhibitor in the treatment of hereditary angioedema. Expert Opin Pharmacother. 2007;8:3173-3181.
18. Bork K, Bygum A, Hardt J. Benefits and risks of danazol in hereditary angioedema: a long-term survey of 118 patients. Ann Allergy Asthma Immunol. 2008;100:153-161.
19. US Food and Drug Administration. Advisory Committee Briefing Document. Kalbitor (ecallantide) for acute attacks of hereditary angioedema. Available at: http://www.fda.gov/ohrms/dockets/AC/09/briefing/2009-4413b1-03-Dyax.pdf. Accessed May 5, 2009.
20. Schneider L, Lumry W, Vegh A, et al. Critical role of kallikrein in hereditary angioedema pathogenesis: a clinical trial of ecallantide, a novel kallikrein inhibitor. J Allergy Clin Immunol. 2007;120:416-422.
21. Ellis Simonsen SM, van Orman ER, Hatch BE, et al. Cellulitis incidence in a defined population. Epidemiol Infect. 2006;134:293-299.
22. Stevens DL, Bisno AL, Chambers HF, et al. Infectious Diseases Society of America. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin Infect Dis. 2005;41:1373-1406.
23. Morris A. What are the benefits of treatments? Cellulitis and erysipelas. BMJ Clin Evid. 2005;13:2066-2069.
24. Murray H, Stiell I, Wells G. Treatment failure in emergency department patients with cellulitis. CJEM. 2005;7:228-234.
25. Meier DE, Nkor SK, Aasa D, et al. Prospective randomized comparison of two preoperative skin preparation techniques in a developing world country. World J Surg. 2001;25:441-443.
26. Giordano PA, Elston D, Akinlade BK, et al. Cefdinir vs. cephalexin for mild to moderate uncomplicated skin and skin structure infections in adolescents and adults. Curr Med Res Opin. 2006;22:2419-2428.
27. Halpern J, Holder R, Langford NJ. Ethnicity and other risk factors for acute lower limb cellulitis: a U.K.-based prospective case-control study. Br J Dermatol. 2008;158:1288-1292.
28. Lin YT, Lu PW. Retrospective study of pediatric facial cellulitis of odontogenic origin. Pediatr Infect Dis J. 2006;25:339-342.
29. Rajendran PM, Young D, Maurer T, et al. Randomized, double-blind, placebo-controlled trial of cephalexin for treatment of uncomplicated skin abscesses in a population at risk for community-acquired methicillin-resistant Staphylococcus aureus infection. Antimicrob Agents Chemo. 2007;51:4044-4048.
30. Schneck J, Fagot JP, Sekula P, et al. Effects of treatments on the mortality of Stevens-Johnson syndrome and toxic epidermal necrolysis: a retrospective study on patients included in the prospective EuroSCAR Study. J Am Acad Dermatol. 2008;58:33-40.
31. Chan HL, Stern RS, Arndt KA, et al. The incidence of erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis. A population-based study with particular reference to reactions caused by drugs among outpatients. Arch Dermatol. 1990;126:43-47.
32. Hazin R, Ibrahimi OA, Hazin MI, et al. Stevens-Johnson syndrome: pathogenesis, diagnosis, and management. Ann Med, 2008;40:129-138.
33. Chung WH, Hung SI, Hong HS, et al. Medical genetics: a marker for Stevens-Johnson syndrome. Nature. 2004;428:486.-
34. Jette N, Hemming K, Hutton JL, et al. Topiramate add-on for drug-resistant partial epilepsy. Cochrane Database Syst Rev. 2008;(3):DC001417.-
35. Gürcan HM, Ahmed AR. Efficacy of various intravenous immunoglobulin therapy protocols in auto-immune and chronic inflammatory disorders. Ann Pharmacother. 2007;41:812-523.
36. Strom BL, Carson JL, Halpern AC, et al. A population-based study of Stevens-Johnson syndrome. Incidence and antecedent drug exposures. Arch Dermatol. 1991;127:831-838.
37. Gravante G, Delogu D, Marianetti M, et al. Toxic epidermal necrolysis and Steven-Johnson syndrome: 11-years experience and outcome. Eur Rev Med Pharmacol Sci. 2007;11:119-127.