User login
Rare Cutaneous Presentation of Burkitt Lymphoma
To the Editor:
A 73-year-old man was admitted to the hospital with progressive abdominal and hip pain of several weeks’ duration that was accompanied by unilateral swelling of the left leg. He had a medical history of hypertension, hyperlipidemia, and prediabetes. Computed tomography (CT) showed extensive intra-abdominal, retroperitoneal, and pelvic lymphadenopathy in addition to poorly defined hepatic lesions.
A CT-guided core biopsy of a left inguinal lymph node showed Burkitt lymphoma. Fluorescence in situ hybridization was positive for oncogene c-MYC rearrangement on chromosome 8q24 and negative for B-cell lymphoma 2 (BCL2) and B-cell lymphoma 6 (BCL6) gene rearrangements. Flow cytometry demonstrated an aberrant population of κ light chain-restricted CD5−CD10+ B lymphocytes.
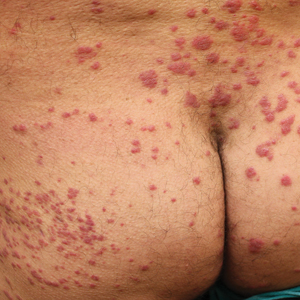
The patient’s overall disease burden was consistent with stage IV Burkitt lymphoma. R-miniCHOP chemotherapy—rituximab plus a reduced dose of cyclophosphamide, doxorubicin, vincristine sulfate, and prednisone—was initiated. Approximately 2 weeks after chemotherapy was initiated, the patient developed a firm erythematous eruption on the left hip (Figure 1A). His regimen was then switched to R-EPOCH—rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, and doxorubicin—at the time of discharge, and he was referred to dermatology due to an initial concern of an adverse reaction to R-EPOCH chemotherapy. The patient denied any pain, pruritus, or irritation. Physical examination showed multifocal, subcutaneous, indurated, erythematous and violaceous nodules without epidermal changes. Some nodules on the lateral aspect of the hip coalesced to form firm plaques.

A punch biopsy specimen showed markedly atypical lymphocytes with enlarged nuclei and scant cytoplasm present throughout the dermis (Figures 2A and 2B). Numerous apoptotic cells and cellular debris were seen. Immunohistochemical staining demonstrated that the lymphocytic infiltrate comprised CD79a+ B cells that were positive for Bcl-6 and CD10 and negative for Bcl-2 (Figures 2C and 2D). There also was diminished focal expression of CD20. Ki-67 protein staining was intensely positive and demonstrated a very high proliferative index.

Taken together, these findings were consistent with a diagnosis of cutaneous metastasis of Burkitt lymphoma. The patient’s cutaneous lesions improved after continued aggressive chemotherapy. At follow-up 2 weeks after biopsy, he was receiving his second round of R-EPOCH chemotherapy with appreciable regression of skin lesions (Figure 1B). However, he then developed right-side double vision, ptosis, and right-side facial paresthesia. Although magnetic resonance imaging of the brain and lumbar puncture did not show evidence of central nervous system involvement, the chemotherapy regimen was switched to dose-adjusted CVAD-R—hypercyclophosphamide, vincristine, doxorubicin hydrochloride, and dexamethasone plus rituximab—for empiric treatment of central nervous system disease. Although treatment was complicated by sepsis with extended-spectrum β-lactamase-producing Enterobacter cloacae, Burkitt lymphoma was found to be in remission after 3 cycles of CVAD-R and 5 months of chemotherapy.
Burkitt lymphoma is a B-cell non-Hodgkin malignancy caused by translocation of chromosome 8 and chromosome 14, leading to overexpression of c-MYC and subsequent hyperproliferation of B lymphocytes.1,2 The disease is divided into 3 major categories: sporadic, endemic, and immunodeficiency related.3 The endemic variant is the most prevalent subtype in Africa and is associated with Plasmodium falciparum malaria; the sporadic variant is the most common subtype in the rest of the world.4
Burkitt lymphoma is highly aggressive and is characterized by unusually high rates of mitosis and apoptosis that result in abundant cellular debris and a distinctive starry-sky pattern on histopathology.5,6 Extranodal metastasis is common,7 but cutaneous involvement is exceedingly rare, with only a few cases having been reported.8-14 Cutaneous metastasis of Burkitt lymphoma often is associated with a high overall disease burden and poor prognosis.8,11
Immunodeficiency-related Burkitt lymphoma is particularly aggressive. Notably, 3 of 7 (42.9%) reported cases of cutaneous Burkitt lymphoma occurred in HIV-positive patients.11,13 In one case, cutaneous involvement was the first sign of relapsed disease that had been in remission.12
Although c-MYC rearrangement is required to make a diagnosis of Burkitt lymphoma, the disease also is present in a minority of cases of diffuse large B-cell lymphoma (DLBCL)(6%).15 Although DLBCL typically can be differentiated from Burkitt lymphoma by the large nuclear size and characteristic vesicular nuclei of B cells, few cases of DLBCL with c-MYC rearrangement histologically mimic Burkitt lymphoma. However, key features such as immunohistochemical staining for Bcl-2 and CD10 can be used to distinguish these 2 entities.16 Bcl-2 negativity and CD10 positivity, as seen in our patient, is considered more characteristic of Burkitt lymphoma. This staining pattern in combination with a high Ki-67 fraction (>95%) and the presence of monomorphic medium-sized cells is more consistent with a diagnosis of Burkitt lymphoma than of DLBCL.17
Earlier case reports have documented that cutaneous lesions of Burkitt lymphoma can occur in a variety of ways. Hematogenous spread is the likely route of metastasis for lesions distant to the primary site or those that have widespread distribution.18 Alternatively, other reports have suggested that cutaneous metastases can occur from local invasion and subcutaneous extension of malignant cells after a surgical procedure.10,19 For example, cutaneous Burkitt lymphoma has been reported in the setting of celioscopy, occurring directly at the surgical site.19 In our patient, we believe that the route of metastatic spread likely was through subcutaneous invasion secondary to CT-guided core biopsy, which was supported by the observation that the onset of cutaneous manifestations was temporally related to the procedure and that the lesions occurred on the skin directly overlying the biopsy site.
In conclusion, we describe an exceedingly rare presentation of cutaneous Burkitt lymphoma in which a surgical procedure likely served as an inciting event that triggered seeding of malignant cells to the skin. Cutaneous spread of Burkitt lymphoma is infrequently reported; all such reports that provide long-term follow-up data have described it in association with high disease burden and often a lethal outcome.8,11,12 Our patient had complete resolution of cutaneous lesions with chemotherapy. It is unclear if the presence of cutaneous lesions can serve as a prognostic indicator and requires further investigation. However, our case provides preliminary evidence to suggest that cutaneous metastases do not always represent aggressive disease and that cutaneous lesions may respond well to chemotherapy.
- Kalisz K, Alessandrino F, Beck R, et al. An update on Burkitt lymphoma: a review of pathogenesis and multimodality imaging assessment of disease presentation, treatment response, and recurrence. Insights Imaging. 2019;10:56. doi:10.1186/s13244-019-0733-7
- Dunleavy K, Gross TG. Management of aggressive B-cell NHLs in the AYA population: an adult vs pediatric perspective. Blood. 2018;132:369-375. doi:10.1182/blood-2018-02-778480
- Noy A. Burkitt lymphoma—subtypes, pathogenesis, and treatment strategies. Clin Lymphoma Myeloma Leuk. 2020;20(Suppl 1):S37-S38. doi:10.1016/S2152-2650(20)30455-9
- Lenze D, Leoncini L, Hummel M, et al. The different epidemiologic subtypes of Burkitt lymphoma share a homogenous micro RNA profile distinct from diffuse large B-cell lymphoma. Leukemia. 2011;25:1869-1876. doi:10.1038/leu.2011.156
- Bellan C, Lazzi S, De Falco G, et al. Burkitt’s lymphoma: new insights into molecular pathogenesis. J Clin Pathol. 2003;56:188-192. doi:10.1136/jcp.56.3.188
- Chuang S-S, Ye H, Du M-Q, et al. Histopathology and immunohistochemistry in distinguishing Burkitt lymphoma from diffuse large B-cell lymphoma with very high proliferation index and with or without a starry-sky pattern: a comparative study with EBER and FISH. Am J Clin Pathol. 2007;128:558-564. doi:10.1309/EQJR3D3V0CCQGP04
- Baker PS, Gold KG, Lane KA, et al. Orbital burkitt lymphoma in immunocompetent patients: a report of 3 cases and a review of the literature. Ophthalmic Plast Reconstr Surg. 2009;25:464-468. doi:10.1097/IOP.0b013e3181b80fde
- Fuhrmann TL, Ignatovich YV, Pentland A. Cutaneous metastatic disease: Burkitt lymphoma. J Am Acad Dermatol. 2011;64:1196-1197. doi:10.1016/j.jaad.2009.08.033
- Burns CA, Scott GA, Miller CC. Leukemia cutis at the site of trauma in a patient with Burkitt leukemia. Cutis. 2005;75:54-56.
- Jacobson MA, Hutcheson ACS, Hurray DH, et al. Cutaneous involvement by Burkitt lymphoma. J Am Acad Dermatol. 2006;54:1111-1113. doi:10.1016/j.jaad.2006.02.030
- Berk DR, Cheng A, Lind AC, et al. Burkitt lymphoma with cutaneous involvement. Dermatol Online J. 2008;14:14.
- Bachmeyer C, Bazarbachi A, Rio B, et al. Specific cutaneous involvement indicating relapse of Burkitt’s lymphoma. Am J Hematol. 1997;54:176. doi:10.1002/(sici)1096-8652(199702)54:2<176::aid-ajh20>3.0.co;2-c
- Rogers A, Graves M, Toscano M, et al. A unique cutaneous presentation of Burkitt lymphoma. Am J Dermatopathol. 2014;36:997-1001. doi:10.1097/DAD.0000000000000004
- Thakkar D, Lipi L, Misra R, et al. Skin involvement in Burkitt’s lymphoma. Hematol Oncol Stem Cell Ther. 2018;11:251-252. doi:10.1016/j.hemonc.2018.01.002
- Akasaka T, Akasaka H, Ueda C, et al. Molecular and clinical features of non-Burkitt’s, diffuse large-cell lymphoma of B-cell type associated with the c-MYC/immunoglobulin heavy-chain fusion gene. J Clin Oncol. 2000;18:510-518. doi:10.1200/JCO.2000.18.3.510
- Nakamura N, Nakamine H, Tamaru J-I, et al. The distinction between Burkitt lymphoma and diffuse large B-cell lymphoma with c-myc rearrangement. Mod Pathol. 2002;15:771-776. doi:10.1097/01.MP.0000019577.73786.64
- Bellan C, Stefano L, Giulia de F, et al. Burkitt lymphoma versus diffuse large B-cell lymphoma: a practical approach. Hematol Oncol. 2010;28:53-56. doi:10.1002/hon.916
- Amonchaisakda N, Aiempanakit K, Apinantriyo B. Burkitt lymphoma initially mimicking varicella zoster infection. IDCases. 2020;21:E00818. doi:10.1016/j.idcr.2020.e00818
- Aractingi S, Marolleau JP, Daniel MT, et al. Subcutaneous localizations of Burkitt lymphoma after celioscopy. Am J Hematol. 1993;42:408. doi:10.1002/ajh.2830420421
To the Editor:
A 73-year-old man was admitted to the hospital with progressive abdominal and hip pain of several weeks’ duration that was accompanied by unilateral swelling of the left leg. He had a medical history of hypertension, hyperlipidemia, and prediabetes. Computed tomography (CT) showed extensive intra-abdominal, retroperitoneal, and pelvic lymphadenopathy in addition to poorly defined hepatic lesions.
A CT-guided core biopsy of a left inguinal lymph node showed Burkitt lymphoma. Fluorescence in situ hybridization was positive for oncogene c-MYC rearrangement on chromosome 8q24 and negative for B-cell lymphoma 2 (BCL2) and B-cell lymphoma 6 (BCL6) gene rearrangements. Flow cytometry demonstrated an aberrant population of κ light chain-restricted CD5−CD10+ B lymphocytes.
The patient’s overall disease burden was consistent with stage IV Burkitt lymphoma. R-miniCHOP chemotherapy—rituximab plus a reduced dose of cyclophosphamide, doxorubicin, vincristine sulfate, and prednisone—was initiated. Approximately 2 weeks after chemotherapy was initiated, the patient developed a firm erythematous eruption on the left hip (Figure 1A). His regimen was then switched to R-EPOCH—rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, and doxorubicin—at the time of discharge, and he was referred to dermatology due to an initial concern of an adverse reaction to R-EPOCH chemotherapy. The patient denied any pain, pruritus, or irritation. Physical examination showed multifocal, subcutaneous, indurated, erythematous and violaceous nodules without epidermal changes. Some nodules on the lateral aspect of the hip coalesced to form firm plaques.
A punch biopsy specimen showed markedly atypical lymphocytes with enlarged nuclei and scant cytoplasm present throughout the dermis (Figures 2A and 2B). Numerous apoptotic cells and cellular debris were seen. Immunohistochemical staining demonstrated that the lymphocytic infiltrate comprised CD79a+ B cells that were positive for Bcl-6 and CD10 and negative for Bcl-2 (Figures 2C and 2D). There also was diminished focal expression of CD20. Ki-67 protein staining was intensely positive and demonstrated a very high proliferative index.
Taken together, these findings were consistent with a diagnosis of cutaneous metastasis of Burkitt lymphoma. The patient’s cutaneous lesions improved after continued aggressive chemotherapy. At follow-up 2 weeks after biopsy, he was receiving his second round of R-EPOCH chemotherapy with appreciable regression of skin lesions (Figure 1B). However, he then developed right-side double vision, ptosis, and right-side facial paresthesia. Although magnetic resonance imaging of the brain and lumbar puncture did not show evidence of central nervous system involvement, the chemotherapy regimen was switched to dose-adjusted CVAD-R—hypercyclophosphamide, vincristine, doxorubicin hydrochloride, and dexamethasone plus rituximab—for empiric treatment of central nervous system disease. Although treatment was complicated by sepsis with extended-spectrum β-lactamase-producing Enterobacter cloacae, Burkitt lymphoma was found to be in remission after 3 cycles of CVAD-R and 5 months of chemotherapy.
Burkitt lymphoma is a B-cell non-Hodgkin malignancy caused by translocation of chromosome 8 and chromosome 14, leading to overexpression of c-MYC and subsequent hyperproliferation of B lymphocytes.1,2 The disease is divided into 3 major categories: sporadic, endemic, and immunodeficiency related.3 The endemic variant is the most prevalent subtype in Africa and is associated with Plasmodium falciparum malaria; the sporadic variant is the most common subtype in the rest of the world.4
Burkitt lymphoma is highly aggressive and is characterized by unusually high rates of mitosis and apoptosis that result in abundant cellular debris and a distinctive starry-sky pattern on histopathology.5,6 Extranodal metastasis is common,7 but cutaneous involvement is exceedingly rare, with only a few cases having been reported.8-14 Cutaneous metastasis of Burkitt lymphoma often is associated with a high overall disease burden and poor prognosis.8,11
Immunodeficiency-related Burkitt lymphoma is particularly aggressive. Notably, 3 of 7 (42.9%) reported cases of cutaneous Burkitt lymphoma occurred in HIV-positive patients.11,13 In one case, cutaneous involvement was the first sign of relapsed disease that had been in remission.12
Although c-MYC rearrangement is required to make a diagnosis of Burkitt lymphoma, the disease also is present in a minority of cases of diffuse large B-cell lymphoma (DLBCL)(6%).15 Although DLBCL typically can be differentiated from Burkitt lymphoma by the large nuclear size and characteristic vesicular nuclei of B cells, few cases of DLBCL with c-MYC rearrangement histologically mimic Burkitt lymphoma. However, key features such as immunohistochemical staining for Bcl-2 and CD10 can be used to distinguish these 2 entities.16 Bcl-2 negativity and CD10 positivity, as seen in our patient, is considered more characteristic of Burkitt lymphoma. This staining pattern in combination with a high Ki-67 fraction (>95%) and the presence of monomorphic medium-sized cells is more consistent with a diagnosis of Burkitt lymphoma than of DLBCL.17
Earlier case reports have documented that cutaneous lesions of Burkitt lymphoma can occur in a variety of ways. Hematogenous spread is the likely route of metastasis for lesions distant to the primary site or those that have widespread distribution.18 Alternatively, other reports have suggested that cutaneous metastases can occur from local invasion and subcutaneous extension of malignant cells after a surgical procedure.10,19 For example, cutaneous Burkitt lymphoma has been reported in the setting of celioscopy, occurring directly at the surgical site.19 In our patient, we believe that the route of metastatic spread likely was through subcutaneous invasion secondary to CT-guided core biopsy, which was supported by the observation that the onset of cutaneous manifestations was temporally related to the procedure and that the lesions occurred on the skin directly overlying the biopsy site.
In conclusion, we describe an exceedingly rare presentation of cutaneous Burkitt lymphoma in which a surgical procedure likely served as an inciting event that triggered seeding of malignant cells to the skin. Cutaneous spread of Burkitt lymphoma is infrequently reported; all such reports that provide long-term follow-up data have described it in association with high disease burden and often a lethal outcome.8,11,12 Our patient had complete resolution of cutaneous lesions with chemotherapy. It is unclear if the presence of cutaneous lesions can serve as a prognostic indicator and requires further investigation. However, our case provides preliminary evidence to suggest that cutaneous metastases do not always represent aggressive disease and that cutaneous lesions may respond well to chemotherapy.
To the Editor:
A 73-year-old man was admitted to the hospital with progressive abdominal and hip pain of several weeks’ duration that was accompanied by unilateral swelling of the left leg. He had a medical history of hypertension, hyperlipidemia, and prediabetes. Computed tomography (CT) showed extensive intra-abdominal, retroperitoneal, and pelvic lymphadenopathy in addition to poorly defined hepatic lesions.
A CT-guided core biopsy of a left inguinal lymph node showed Burkitt lymphoma. Fluorescence in situ hybridization was positive for oncogene c-MYC rearrangement on chromosome 8q24 and negative for B-cell lymphoma 2 (BCL2) and B-cell lymphoma 6 (BCL6) gene rearrangements. Flow cytometry demonstrated an aberrant population of κ light chain-restricted CD5−CD10+ B lymphocytes.
The patient’s overall disease burden was consistent with stage IV Burkitt lymphoma. R-miniCHOP chemotherapy—rituximab plus a reduced dose of cyclophosphamide, doxorubicin, vincristine sulfate, and prednisone—was initiated. Approximately 2 weeks after chemotherapy was initiated, the patient developed a firm erythematous eruption on the left hip (Figure 1A). His regimen was then switched to R-EPOCH—rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, and doxorubicin—at the time of discharge, and he was referred to dermatology due to an initial concern of an adverse reaction to R-EPOCH chemotherapy. The patient denied any pain, pruritus, or irritation. Physical examination showed multifocal, subcutaneous, indurated, erythematous and violaceous nodules without epidermal changes. Some nodules on the lateral aspect of the hip coalesced to form firm plaques.
A punch biopsy specimen showed markedly atypical lymphocytes with enlarged nuclei and scant cytoplasm present throughout the dermis (Figures 2A and 2B). Numerous apoptotic cells and cellular debris were seen. Immunohistochemical staining demonstrated that the lymphocytic infiltrate comprised CD79a+ B cells that were positive for Bcl-6 and CD10 and negative for Bcl-2 (Figures 2C and 2D). There also was diminished focal expression of CD20. Ki-67 protein staining was intensely positive and demonstrated a very high proliferative index.
Taken together, these findings were consistent with a diagnosis of cutaneous metastasis of Burkitt lymphoma. The patient’s cutaneous lesions improved after continued aggressive chemotherapy. At follow-up 2 weeks after biopsy, he was receiving his second round of R-EPOCH chemotherapy with appreciable regression of skin lesions (Figure 1B). However, he then developed right-side double vision, ptosis, and right-side facial paresthesia. Although magnetic resonance imaging of the brain and lumbar puncture did not show evidence of central nervous system involvement, the chemotherapy regimen was switched to dose-adjusted CVAD-R—hypercyclophosphamide, vincristine, doxorubicin hydrochloride, and dexamethasone plus rituximab—for empiric treatment of central nervous system disease. Although treatment was complicated by sepsis with extended-spectrum β-lactamase-producing Enterobacter cloacae, Burkitt lymphoma was found to be in remission after 3 cycles of CVAD-R and 5 months of chemotherapy.
Burkitt lymphoma is a B-cell non-Hodgkin malignancy caused by translocation of chromosome 8 and chromosome 14, leading to overexpression of c-MYC and subsequent hyperproliferation of B lymphocytes.1,2 The disease is divided into 3 major categories: sporadic, endemic, and immunodeficiency related.3 The endemic variant is the most prevalent subtype in Africa and is associated with Plasmodium falciparum malaria; the sporadic variant is the most common subtype in the rest of the world.4
Burkitt lymphoma is highly aggressive and is characterized by unusually high rates of mitosis and apoptosis that result in abundant cellular debris and a distinctive starry-sky pattern on histopathology.5,6 Extranodal metastasis is common,7 but cutaneous involvement is exceedingly rare, with only a few cases having been reported.8-14 Cutaneous metastasis of Burkitt lymphoma often is associated with a high overall disease burden and poor prognosis.8,11
Immunodeficiency-related Burkitt lymphoma is particularly aggressive. Notably, 3 of 7 (42.9%) reported cases of cutaneous Burkitt lymphoma occurred in HIV-positive patients.11,13 In one case, cutaneous involvement was the first sign of relapsed disease that had been in remission.12
Although c-MYC rearrangement is required to make a diagnosis of Burkitt lymphoma, the disease also is present in a minority of cases of diffuse large B-cell lymphoma (DLBCL)(6%).15 Although DLBCL typically can be differentiated from Burkitt lymphoma by the large nuclear size and characteristic vesicular nuclei of B cells, few cases of DLBCL with c-MYC rearrangement histologically mimic Burkitt lymphoma. However, key features such as immunohistochemical staining for Bcl-2 and CD10 can be used to distinguish these 2 entities.16 Bcl-2 negativity and CD10 positivity, as seen in our patient, is considered more characteristic of Burkitt lymphoma. This staining pattern in combination with a high Ki-67 fraction (>95%) and the presence of monomorphic medium-sized cells is more consistent with a diagnosis of Burkitt lymphoma than of DLBCL.17
Earlier case reports have documented that cutaneous lesions of Burkitt lymphoma can occur in a variety of ways. Hematogenous spread is the likely route of metastasis for lesions distant to the primary site or those that have widespread distribution.18 Alternatively, other reports have suggested that cutaneous metastases can occur from local invasion and subcutaneous extension of malignant cells after a surgical procedure.10,19 For example, cutaneous Burkitt lymphoma has been reported in the setting of celioscopy, occurring directly at the surgical site.19 In our patient, we believe that the route of metastatic spread likely was through subcutaneous invasion secondary to CT-guided core biopsy, which was supported by the observation that the onset of cutaneous manifestations was temporally related to the procedure and that the lesions occurred on the skin directly overlying the biopsy site.
In conclusion, we describe an exceedingly rare presentation of cutaneous Burkitt lymphoma in which a surgical procedure likely served as an inciting event that triggered seeding of malignant cells to the skin. Cutaneous spread of Burkitt lymphoma is infrequently reported; all such reports that provide long-term follow-up data have described it in association with high disease burden and often a lethal outcome.8,11,12 Our patient had complete resolution of cutaneous lesions with chemotherapy. It is unclear if the presence of cutaneous lesions can serve as a prognostic indicator and requires further investigation. However, our case provides preliminary evidence to suggest that cutaneous metastases do not always represent aggressive disease and that cutaneous lesions may respond well to chemotherapy.
- Kalisz K, Alessandrino F, Beck R, et al. An update on Burkitt lymphoma: a review of pathogenesis and multimodality imaging assessment of disease presentation, treatment response, and recurrence. Insights Imaging. 2019;10:56. doi:10.1186/s13244-019-0733-7
- Dunleavy K, Gross TG. Management of aggressive B-cell NHLs in the AYA population: an adult vs pediatric perspective. Blood. 2018;132:369-375. doi:10.1182/blood-2018-02-778480
- Noy A. Burkitt lymphoma—subtypes, pathogenesis, and treatment strategies. Clin Lymphoma Myeloma Leuk. 2020;20(Suppl 1):S37-S38. doi:10.1016/S2152-2650(20)30455-9
- Lenze D, Leoncini L, Hummel M, et al. The different epidemiologic subtypes of Burkitt lymphoma share a homogenous micro RNA profile distinct from diffuse large B-cell lymphoma. Leukemia. 2011;25:1869-1876. doi:10.1038/leu.2011.156
- Bellan C, Lazzi S, De Falco G, et al. Burkitt’s lymphoma: new insights into molecular pathogenesis. J Clin Pathol. 2003;56:188-192. doi:10.1136/jcp.56.3.188
- Chuang S-S, Ye H, Du M-Q, et al. Histopathology and immunohistochemistry in distinguishing Burkitt lymphoma from diffuse large B-cell lymphoma with very high proliferation index and with or without a starry-sky pattern: a comparative study with EBER and FISH. Am J Clin Pathol. 2007;128:558-564. doi:10.1309/EQJR3D3V0CCQGP04
- Baker PS, Gold KG, Lane KA, et al. Orbital burkitt lymphoma in immunocompetent patients: a report of 3 cases and a review of the literature. Ophthalmic Plast Reconstr Surg. 2009;25:464-468. doi:10.1097/IOP.0b013e3181b80fde
- Fuhrmann TL, Ignatovich YV, Pentland A. Cutaneous metastatic disease: Burkitt lymphoma. J Am Acad Dermatol. 2011;64:1196-1197. doi:10.1016/j.jaad.2009.08.033
- Burns CA, Scott GA, Miller CC. Leukemia cutis at the site of trauma in a patient with Burkitt leukemia. Cutis. 2005;75:54-56.
- Jacobson MA, Hutcheson ACS, Hurray DH, et al. Cutaneous involvement by Burkitt lymphoma. J Am Acad Dermatol. 2006;54:1111-1113. doi:10.1016/j.jaad.2006.02.030
- Berk DR, Cheng A, Lind AC, et al. Burkitt lymphoma with cutaneous involvement. Dermatol Online J. 2008;14:14.
- Bachmeyer C, Bazarbachi A, Rio B, et al. Specific cutaneous involvement indicating relapse of Burkitt’s lymphoma. Am J Hematol. 1997;54:176. doi:10.1002/(sici)1096-8652(199702)54:2<176::aid-ajh20>3.0.co;2-c
- Rogers A, Graves M, Toscano M, et al. A unique cutaneous presentation of Burkitt lymphoma. Am J Dermatopathol. 2014;36:997-1001. doi:10.1097/DAD.0000000000000004
- Thakkar D, Lipi L, Misra R, et al. Skin involvement in Burkitt’s lymphoma. Hematol Oncol Stem Cell Ther. 2018;11:251-252. doi:10.1016/j.hemonc.2018.01.002
- Akasaka T, Akasaka H, Ueda C, et al. Molecular and clinical features of non-Burkitt’s, diffuse large-cell lymphoma of B-cell type associated with the c-MYC/immunoglobulin heavy-chain fusion gene. J Clin Oncol. 2000;18:510-518. doi:10.1200/JCO.2000.18.3.510
- Nakamura N, Nakamine H, Tamaru J-I, et al. The distinction between Burkitt lymphoma and diffuse large B-cell lymphoma with c-myc rearrangement. Mod Pathol. 2002;15:771-776. doi:10.1097/01.MP.0000019577.73786.64
- Bellan C, Stefano L, Giulia de F, et al. Burkitt lymphoma versus diffuse large B-cell lymphoma: a practical approach. Hematol Oncol. 2010;28:53-56. doi:10.1002/hon.916
- Amonchaisakda N, Aiempanakit K, Apinantriyo B. Burkitt lymphoma initially mimicking varicella zoster infection. IDCases. 2020;21:E00818. doi:10.1016/j.idcr.2020.e00818
- Aractingi S, Marolleau JP, Daniel MT, et al. Subcutaneous localizations of Burkitt lymphoma after celioscopy. Am J Hematol. 1993;42:408. doi:10.1002/ajh.2830420421
- Kalisz K, Alessandrino F, Beck R, et al. An update on Burkitt lymphoma: a review of pathogenesis and multimodality imaging assessment of disease presentation, treatment response, and recurrence. Insights Imaging. 2019;10:56. doi:10.1186/s13244-019-0733-7
- Dunleavy K, Gross TG. Management of aggressive B-cell NHLs in the AYA population: an adult vs pediatric perspective. Blood. 2018;132:369-375. doi:10.1182/blood-2018-02-778480
- Noy A. Burkitt lymphoma—subtypes, pathogenesis, and treatment strategies. Clin Lymphoma Myeloma Leuk. 2020;20(Suppl 1):S37-S38. doi:10.1016/S2152-2650(20)30455-9
- Lenze D, Leoncini L, Hummel M, et al. The different epidemiologic subtypes of Burkitt lymphoma share a homogenous micro RNA profile distinct from diffuse large B-cell lymphoma. Leukemia. 2011;25:1869-1876. doi:10.1038/leu.2011.156
- Bellan C, Lazzi S, De Falco G, et al. Burkitt’s lymphoma: new insights into molecular pathogenesis. J Clin Pathol. 2003;56:188-192. doi:10.1136/jcp.56.3.188
- Chuang S-S, Ye H, Du M-Q, et al. Histopathology and immunohistochemistry in distinguishing Burkitt lymphoma from diffuse large B-cell lymphoma with very high proliferation index and with or without a starry-sky pattern: a comparative study with EBER and FISH. Am J Clin Pathol. 2007;128:558-564. doi:10.1309/EQJR3D3V0CCQGP04
- Baker PS, Gold KG, Lane KA, et al. Orbital burkitt lymphoma in immunocompetent patients: a report of 3 cases and a review of the literature. Ophthalmic Plast Reconstr Surg. 2009;25:464-468. doi:10.1097/IOP.0b013e3181b80fde
- Fuhrmann TL, Ignatovich YV, Pentland A. Cutaneous metastatic disease: Burkitt lymphoma. J Am Acad Dermatol. 2011;64:1196-1197. doi:10.1016/j.jaad.2009.08.033
- Burns CA, Scott GA, Miller CC. Leukemia cutis at the site of trauma in a patient with Burkitt leukemia. Cutis. 2005;75:54-56.
- Jacobson MA, Hutcheson ACS, Hurray DH, et al. Cutaneous involvement by Burkitt lymphoma. J Am Acad Dermatol. 2006;54:1111-1113. doi:10.1016/j.jaad.2006.02.030
- Berk DR, Cheng A, Lind AC, et al. Burkitt lymphoma with cutaneous involvement. Dermatol Online J. 2008;14:14.
- Bachmeyer C, Bazarbachi A, Rio B, et al. Specific cutaneous involvement indicating relapse of Burkitt’s lymphoma. Am J Hematol. 1997;54:176. doi:10.1002/(sici)1096-8652(199702)54:2<176::aid-ajh20>3.0.co;2-c
- Rogers A, Graves M, Toscano M, et al. A unique cutaneous presentation of Burkitt lymphoma. Am J Dermatopathol. 2014;36:997-1001. doi:10.1097/DAD.0000000000000004
- Thakkar D, Lipi L, Misra R, et al. Skin involvement in Burkitt’s lymphoma. Hematol Oncol Stem Cell Ther. 2018;11:251-252. doi:10.1016/j.hemonc.2018.01.002
- Akasaka T, Akasaka H, Ueda C, et al. Molecular and clinical features of non-Burkitt’s, diffuse large-cell lymphoma of B-cell type associated with the c-MYC/immunoglobulin heavy-chain fusion gene. J Clin Oncol. 2000;18:510-518. doi:10.1200/JCO.2000.18.3.510
- Nakamura N, Nakamine H, Tamaru J-I, et al. The distinction between Burkitt lymphoma and diffuse large B-cell lymphoma with c-myc rearrangement. Mod Pathol. 2002;15:771-776. doi:10.1097/01.MP.0000019577.73786.64
- Bellan C, Stefano L, Giulia de F, et al. Burkitt lymphoma versus diffuse large B-cell lymphoma: a practical approach. Hematol Oncol. 2010;28:53-56. doi:10.1002/hon.916
- Amonchaisakda N, Aiempanakit K, Apinantriyo B. Burkitt lymphoma initially mimicking varicella zoster infection. IDCases. 2020;21:E00818. doi:10.1016/j.idcr.2020.e00818
- Aractingi S, Marolleau JP, Daniel MT, et al. Subcutaneous localizations of Burkitt lymphoma after celioscopy. Am J Hematol. 1993;42:408. doi:10.1002/ajh.2830420421
Practice Points
- Cutaneous metastasis is exceedingly rare in Burkitt lymphoma. When cutaneous involvement does occur, it can represent an uncommon consequence of a surgical procedure, serving as the inciting event for hematogenous spread and local tumor extension into the skin.
- Although cutaneous metasis of Burkitt lymphoma typically is associated with high disease burden and mortality, our case demonstrated that cutaneous spread can be present even in a patient who has a positive outcome. Our patient was able to achieve disease remission and complete resolution of cutaneous lesions with continued chemotherapy, suggesting that cutaneous metastasis does not always portend a poor prognosis.

Testosterone Pellet–Induced Generalized Drug Eruption
To the Editor:
Testosterone-replacement therapy (TRT) is indicated for hypogonadism. The benefits of TRT are well documented, with multiple options available for delivery. Testosterone pellet implantation (TPI) is an effective treatment option for hypogonadism with minimal adverse reactions. Availability of TRT is increasing, as facilities are offering off-label applications. Although TPI generally is well tolerated, cutaneous reactions have been documented. We present a patient with drug-induced dermatitis following TPI.
A 51-year-old man with hypogonadism presented with an extremely pruritic rash that began on the left buttock 3 days after receiving his fourth TPI. The patient had received subcutaneous insertions of 8 testosterone pellets (75 mg per pellet every 6 months) to the left buttock. He denied any history of a similar rash. His medical history was remarkable for hyperlipidemia, which was controlled with niacin and omega-3 fatty acids (fish oil). Other medications included glucosamine. Before presenting to our clinic, he was given a 40-mg intramuscular injection of triamcinolone acetonide and trimethoprim-sulfamethoxazole twice daily for 7 days, a methylprednisolone dose pack, and triamcinolone ointment 0.1% twice daily by his primary care physician, all without improvement of the rash.
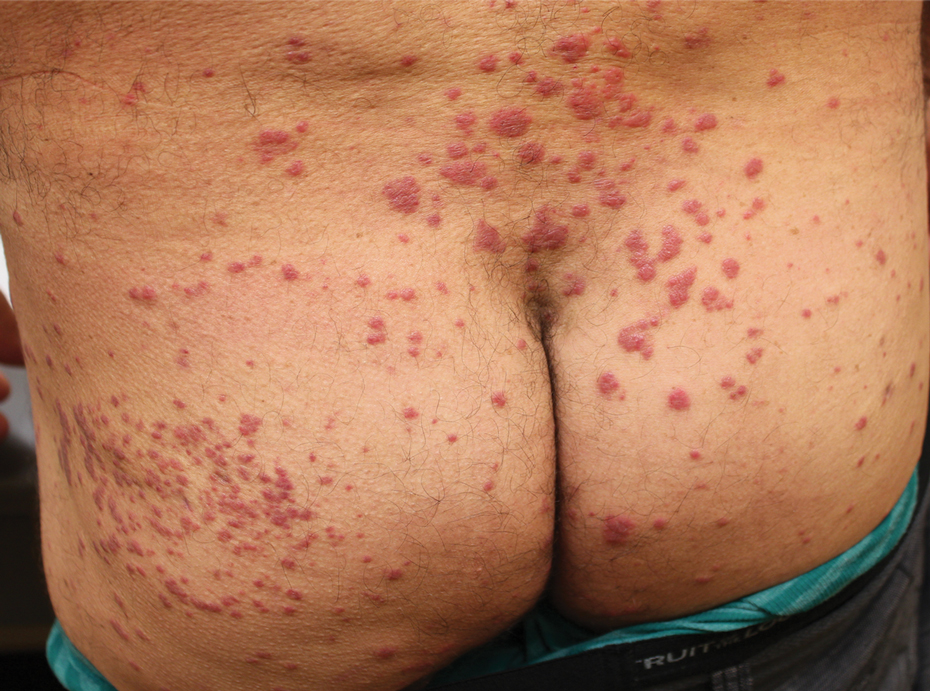
Physical examination revealed multiple well-circumscribed, coalescing clusters of darkly erythematous papules and dermal plaques of varying size on the buttocks with extension to the lower back, abdomen, and thighs (Figure 1). The differential diagnosis included lichenoid eruption, pseudolymphoma, sarcoidosis, and granuloma annulare.
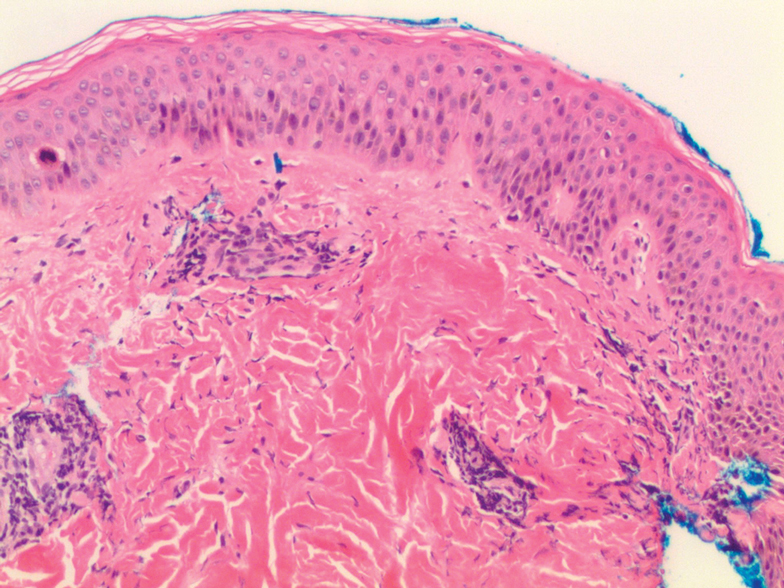
Histologic examination of a punch biopsy revealed an epidermis with a normal stratum corneum and subtle cell-poor vacuolar interface dermatitis with rare necrotic keratinocytes. There was a mild perivascular lymphocytic infiltrate with slight edema within the dermis without notable eosinophils or findings indicative of a vasculitic process (Figure 2).
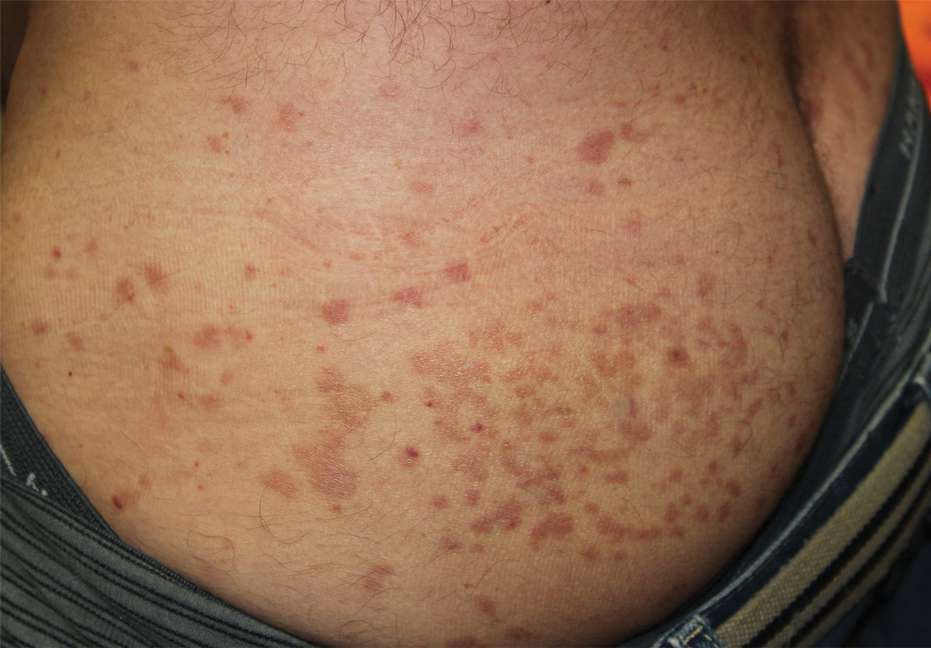
Oral prednisone 60 mg daily and betamethasone ointment 0.05% applied twice daily were started, with notable improvement of the rash in 1 week (Figure 3). Given the temporal relationship of the TPI, histologic findings suggestive of drug eruption, and resolution of symptoms shortly after treatment, a diagnosis of testosterone pellet–induced generalized dermatitis was established.
Testosterone-replacement therapy is the principal treatment of male pathologic hypoandrogenism, but off-label prescription frequently occurs for age-related hypogonadism and hypoactive sexual desire disorder.1 Testosterone-replacement therapy also can enhance sexual desire and function and improve mood in premenopausal and postmenopausal women with testosterone deficiency.2 Delivery options include topicals, intramuscular injections, oral formulations, transdermal patches and gels, and subcutaneous placement of testosterone pellets (TPI).Cutaneous reactions to TPI are rare. Hirsutism, male-pattern hair loss, and acne are possible cutaneous adverse reactions.3 In addition, a localized erythematous pruritic eruption at the implantation site and an immunologic foreign-body reaction to testosterone pellets have been reported.4
In one case report, a man developed recurrent ill-defined, erythematous, scaly plaques and patches over the buttocks and thighs, consistent with testosterone-induced eczematous dermatitis, subsequent to his second TPI. The patient presented with the eruption within 4 weeks after the most recent implantation, similar to our case, but differed temporally in initial presentation, presenting after the second implantation.5 Our case differed in morphologic presentation (dermal plaques as opposed to eczematous change) and refractoriness to triamcinolone injection.
Testosterone-replacement therapy is becoming more widely available. Lack of regulation of proper marketing by such facilities as medical spas that offer TPI for off-label applications has led to a rampant increase in TRT prescribing, possibly foreshadowing an increase in adverse cutaneous reactions to TRT.6
Our case of histologically consistent testosterone pellet–induced dermatitis highlights a rare cutaneous adverse reaction that can occur subsequent to TPI and illustrates the efficacy of high-dose oral steroids as a treatment option. With increased use of TRT, physicians should be cognizant of the potential adverse cutaneous effects related to this treatment and counsel patients appropriately prior to initiating treatment.
Acknowledgment
We thank the patient for granting permission to publish this case.
- Clayton AH, Kingsberg SA, Goldstein I. Evaluation and management of hypoactive sexual desire disorder. Sex Med. 2018;6:59-74.
- Glaser R, Dimitrakakis C. Testosterone therapy in women: myths and misconceptions. Maturitas. 2013;74:230-234.
- Testopel (testosterone pellet) [package insert]. Endo Pharmaceuticals, Inc; 2016. Accessed December 16, 2020. https://dailymed.nlm.nih.gov/dailymed/fda/fdaDrugXsl.cfm?setid=a1741a0b-3d4c-42dc-880d-a06e96cce9ef&type=display
- Cavender RK, Fairall M. Subcutaneous testosterone pellet implant (Testopel) therapy for men with testosterone deficiency syndrome: a single-site retrospective safety analysis. J Sex Med. 2009;6:3177-3192.
- Heldt Manica LA, Cohen PR. Testosterone pellet associated dermatitis: report and review of Testopel-related cutaneous adverse effects. Cureus. 2017;9:e1560.
- Mintzes B. The marketing of testosterone treatments for age-related low testosterone or ‘Low T’. Curr Opin Endocrinol Diabetes Obes. 2018;25:224-230.
To the Editor:
Testosterone-replacement therapy (TRT) is indicated for hypogonadism. The benefits of TRT are well documented, with multiple options available for delivery. Testosterone pellet implantation (TPI) is an effective treatment option for hypogonadism with minimal adverse reactions. Availability of TRT is increasing, as facilities are offering off-label applications. Although TPI generally is well tolerated, cutaneous reactions have been documented. We present a patient with drug-induced dermatitis following TPI.
A 51-year-old man with hypogonadism presented with an extremely pruritic rash that began on the left buttock 3 days after receiving his fourth TPI. The patient had received subcutaneous insertions of 8 testosterone pellets (75 mg per pellet every 6 months) to the left buttock. He denied any history of a similar rash. His medical history was remarkable for hyperlipidemia, which was controlled with niacin and omega-3 fatty acids (fish oil). Other medications included glucosamine. Before presenting to our clinic, he was given a 40-mg intramuscular injection of triamcinolone acetonide and trimethoprim-sulfamethoxazole twice daily for 7 days, a methylprednisolone dose pack, and triamcinolone ointment 0.1% twice daily by his primary care physician, all without improvement of the rash.
Physical examination revealed multiple well-circumscribed, coalescing clusters of darkly erythematous papules and dermal plaques of varying size on the buttocks with extension to the lower back, abdomen, and thighs (Figure 1). The differential diagnosis included lichenoid eruption, pseudolymphoma, sarcoidosis, and granuloma annulare.
Histologic examination of a punch biopsy revealed an epidermis with a normal stratum corneum and subtle cell-poor vacuolar interface dermatitis with rare necrotic keratinocytes. There was a mild perivascular lymphocytic infiltrate with slight edema within the dermis without notable eosinophils or findings indicative of a vasculitic process (Figure 2).
Oral prednisone 60 mg daily and betamethasone ointment 0.05% applied twice daily were started, with notable improvement of the rash in 1 week (Figure 3). Given the temporal relationship of the TPI, histologic findings suggestive of drug eruption, and resolution of symptoms shortly after treatment, a diagnosis of testosterone pellet–induced generalized dermatitis was established.
Testosterone-replacement therapy is the principal treatment of male pathologic hypoandrogenism, but off-label prescription frequently occurs for age-related hypogonadism and hypoactive sexual desire disorder.1 Testosterone-replacement therapy also can enhance sexual desire and function and improve mood in premenopausal and postmenopausal women with testosterone deficiency.2 Delivery options include topicals, intramuscular injections, oral formulations, transdermal patches and gels, and subcutaneous placement of testosterone pellets (TPI).Cutaneous reactions to TPI are rare. Hirsutism, male-pattern hair loss, and acne are possible cutaneous adverse reactions.3 In addition, a localized erythematous pruritic eruption at the implantation site and an immunologic foreign-body reaction to testosterone pellets have been reported.4
In one case report, a man developed recurrent ill-defined, erythematous, scaly plaques and patches over the buttocks and thighs, consistent with testosterone-induced eczematous dermatitis, subsequent to his second TPI. The patient presented with the eruption within 4 weeks after the most recent implantation, similar to our case, but differed temporally in initial presentation, presenting after the second implantation.5 Our case differed in morphologic presentation (dermal plaques as opposed to eczematous change) and refractoriness to triamcinolone injection.
Testosterone-replacement therapy is becoming more widely available. Lack of regulation of proper marketing by such facilities as medical spas that offer TPI for off-label applications has led to a rampant increase in TRT prescribing, possibly foreshadowing an increase in adverse cutaneous reactions to TRT.6
Our case of histologically consistent testosterone pellet–induced dermatitis highlights a rare cutaneous adverse reaction that can occur subsequent to TPI and illustrates the efficacy of high-dose oral steroids as a treatment option. With increased use of TRT, physicians should be cognizant of the potential adverse cutaneous effects related to this treatment and counsel patients appropriately prior to initiating treatment.
Acknowledgment
We thank the patient for granting permission to publish this case.
To the Editor:
Testosterone-replacement therapy (TRT) is indicated for hypogonadism. The benefits of TRT are well documented, with multiple options available for delivery. Testosterone pellet implantation (TPI) is an effective treatment option for hypogonadism with minimal adverse reactions. Availability of TRT is increasing, as facilities are offering off-label applications. Although TPI generally is well tolerated, cutaneous reactions have been documented. We present a patient with drug-induced dermatitis following TPI.
A 51-year-old man with hypogonadism presented with an extremely pruritic rash that began on the left buttock 3 days after receiving his fourth TPI. The patient had received subcutaneous insertions of 8 testosterone pellets (75 mg per pellet every 6 months) to the left buttock. He denied any history of a similar rash. His medical history was remarkable for hyperlipidemia, which was controlled with niacin and omega-3 fatty acids (fish oil). Other medications included glucosamine. Before presenting to our clinic, he was given a 40-mg intramuscular injection of triamcinolone acetonide and trimethoprim-sulfamethoxazole twice daily for 7 days, a methylprednisolone dose pack, and triamcinolone ointment 0.1% twice daily by his primary care physician, all without improvement of the rash.
Physical examination revealed multiple well-circumscribed, coalescing clusters of darkly erythematous papules and dermal plaques of varying size on the buttocks with extension to the lower back, abdomen, and thighs (Figure 1). The differential diagnosis included lichenoid eruption, pseudolymphoma, sarcoidosis, and granuloma annulare.
Histologic examination of a punch biopsy revealed an epidermis with a normal stratum corneum and subtle cell-poor vacuolar interface dermatitis with rare necrotic keratinocytes. There was a mild perivascular lymphocytic infiltrate with slight edema within the dermis without notable eosinophils or findings indicative of a vasculitic process (Figure 2).
Oral prednisone 60 mg daily and betamethasone ointment 0.05% applied twice daily were started, with notable improvement of the rash in 1 week (Figure 3). Given the temporal relationship of the TPI, histologic findings suggestive of drug eruption, and resolution of symptoms shortly after treatment, a diagnosis of testosterone pellet–induced generalized dermatitis was established.
Testosterone-replacement therapy is the principal treatment of male pathologic hypoandrogenism, but off-label prescription frequently occurs for age-related hypogonadism and hypoactive sexual desire disorder.1 Testosterone-replacement therapy also can enhance sexual desire and function and improve mood in premenopausal and postmenopausal women with testosterone deficiency.2 Delivery options include topicals, intramuscular injections, oral formulations, transdermal patches and gels, and subcutaneous placement of testosterone pellets (TPI).Cutaneous reactions to TPI are rare. Hirsutism, male-pattern hair loss, and acne are possible cutaneous adverse reactions.3 In addition, a localized erythematous pruritic eruption at the implantation site and an immunologic foreign-body reaction to testosterone pellets have been reported.4
In one case report, a man developed recurrent ill-defined, erythematous, scaly plaques and patches over the buttocks and thighs, consistent with testosterone-induced eczematous dermatitis, subsequent to his second TPI. The patient presented with the eruption within 4 weeks after the most recent implantation, similar to our case, but differed temporally in initial presentation, presenting after the second implantation.5 Our case differed in morphologic presentation (dermal plaques as opposed to eczematous change) and refractoriness to triamcinolone injection.
Testosterone-replacement therapy is becoming more widely available. Lack of regulation of proper marketing by such facilities as medical spas that offer TPI for off-label applications has led to a rampant increase in TRT prescribing, possibly foreshadowing an increase in adverse cutaneous reactions to TRT.6
Our case of histologically consistent testosterone pellet–induced dermatitis highlights a rare cutaneous adverse reaction that can occur subsequent to TPI and illustrates the efficacy of high-dose oral steroids as a treatment option. With increased use of TRT, physicians should be cognizant of the potential adverse cutaneous effects related to this treatment and counsel patients appropriately prior to initiating treatment.
Acknowledgment
We thank the patient for granting permission to publish this case.
- Clayton AH, Kingsberg SA, Goldstein I. Evaluation and management of hypoactive sexual desire disorder. Sex Med. 2018;6:59-74.
- Glaser R, Dimitrakakis C. Testosterone therapy in women: myths and misconceptions. Maturitas. 2013;74:230-234.
- Testopel (testosterone pellet) [package insert]. Endo Pharmaceuticals, Inc; 2016. Accessed December 16, 2020. https://dailymed.nlm.nih.gov/dailymed/fda/fdaDrugXsl.cfm?setid=a1741a0b-3d4c-42dc-880d-a06e96cce9ef&type=display
- Cavender RK, Fairall M. Subcutaneous testosterone pellet implant (Testopel) therapy for men with testosterone deficiency syndrome: a single-site retrospective safety analysis. J Sex Med. 2009;6:3177-3192.
- Heldt Manica LA, Cohen PR. Testosterone pellet associated dermatitis: report and review of Testopel-related cutaneous adverse effects. Cureus. 2017;9:e1560.
- Mintzes B. The marketing of testosterone treatments for age-related low testosterone or ‘Low T’. Curr Opin Endocrinol Diabetes Obes. 2018;25:224-230.
- Clayton AH, Kingsberg SA, Goldstein I. Evaluation and management of hypoactive sexual desire disorder. Sex Med. 2018;6:59-74.
- Glaser R, Dimitrakakis C. Testosterone therapy in women: myths and misconceptions. Maturitas. 2013;74:230-234.
- Testopel (testosterone pellet) [package insert]. Endo Pharmaceuticals, Inc; 2016. Accessed December 16, 2020. https://dailymed.nlm.nih.gov/dailymed/fda/fdaDrugXsl.cfm?setid=a1741a0b-3d4c-42dc-880d-a06e96cce9ef&type=display
- Cavender RK, Fairall M. Subcutaneous testosterone pellet implant (Testopel) therapy for men with testosterone deficiency syndrome: a single-site retrospective safety analysis. J Sex Med. 2009;6:3177-3192.
- Heldt Manica LA, Cohen PR. Testosterone pellet associated dermatitis: report and review of Testopel-related cutaneous adverse effects. Cureus. 2017;9:e1560.
- Mintzes B. The marketing of testosterone treatments for age-related low testosterone or ‘Low T’. Curr Opin Endocrinol Diabetes Obes. 2018;25:224-230.
Practice Points
- Dermatologists should be aware that testosterone pellet implantation can cause dermatitis overlying the implantation site, which can generalize and differ in morphologic presentation.
- For patients presenting with a suspected case of testosterone pellet–induced dermatitis, a high-dose oral corticosteroid can be deployed as an effective therapy.
Purpura Fulminans in the Setting of Escherichia coli Septicemia
To the Editor:
Purpura fulminans is a severe and rapidly fatal thrombotic disorder that can occur in association with either hereditary or acquired deficiencies of the natural anticoagulants protein C and protein S.1 It most commonly results from the acute inflammatory response and subsequent disseminated intravascular coagulation (DIC) seen in severe bacterial septicemia. Excessive bleeding, retiform purpura, and skin necrosis may develop as a result of the coagulopathies of typical DIC.1Neisseria meningitidis, Streptococcus, and Staphylococcus frequently are implicated as pathogens, but Escherichia coli–associated purpura fulminans in adults is rare.2,3 We report a case of purpura fulminans in the setting of E coli septicemia.
A 62-year-old woman with a history of end-stage liver disease secondary to alcoholic liver cirrhosis diagnosed 13 years prior complicated by ascites and esophageal varices presented to a primary care clinic for evaluation of a recent-onset nontender lesion on the left buttock. She was hypotensive with a blood pressure of 62/48 mmHg. The patient was prescribed ciprofloxacin 250 mg twice daily and hydrocodone/acetominophen 5 mg/325 mg twice daily as needed for pain management and was discharged. Six hours later, the patient presented to the emergency department with new onset symptoms of confusion and dark-colored spots on the abdomen and lower legs, which her family members noted had developed shortly after the patient took ciprofloxacin. In the emergency department, the patient was noted to be hypotensive and febrile with a severe metabolic acidosis. She was intubated for respiratory failure and received intravenous fluid resuscitation, broad-spectrum antibiotics, and vasopressors. Blood cultures were obtained, and the dermatology department was consulted.
On physical examination, extensive purpuric, reticulated, and stellate plaques with central necrosis and hemorrhagic bullae were noted on the abdomen (Figure, A) and bilateral lower legs (Figure, B) extending onto the thighs. The patient was coagulopathic with persistent sanguineous oozing at intravenous sites and bilateral nares. A small erythematous ulcer with overlying black eschar was noted on the left medial buttock.
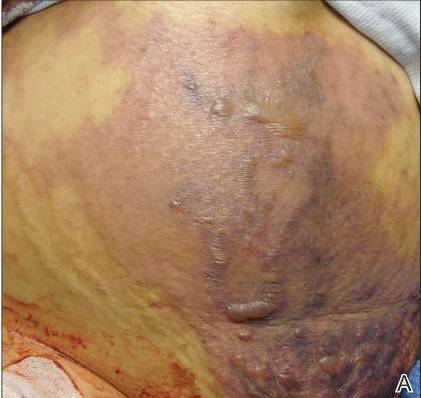
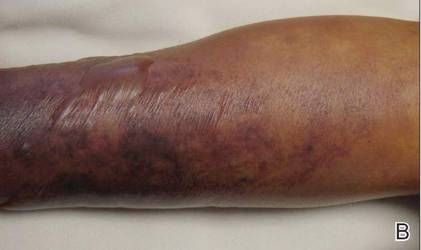
Laboratory test results showed new-onset thrombocytopenia, prolonged prothrombin time/international normalized ratio and partial thromboplastin time, and low fibrinogen levels, which confirmed a diagnosis of acute DIC. Blood cultures were positive for gram-negative rods in 4 out of 4 bottles within 12 hours of being drawn. Further testing identified the microorganism as E coli, and antibiotic susceptibility testing revealed it was sensitive to most antibiotics.
The patient was clinically diagnosed with purpura fulminans secondary to severe E coli septicemia and DIC. This life-threatening disorder is considered a medical emergency with a high mortality rate. Laboratory findings supporting DIC include the presence of schistocytes on a peripheral blood smear, thrombocytopenia, positive plasma protamine paracoagulation test, low fibrinogen levels, and positive fibrin degradation products. Reported cases of purpura fulminans in the setting of E coli septicemia are rare, and meningococcemia is the most common presentation.2,3 Bacterial components (eg, lipopolysaccharides found in the cell walls of gram-negative bacteria) may contribute to the progression of septicemia. Increased levels of endotoxin lipopolysaccharide can lead to septic shock and organ dysfunction.4 However, the release of lipooligosaccharides is associated with the development of meningococcal septicemia, and the lipopolysaccharide levels are directly correlated with prognosis in patients without meningitis.5-7
Human activated protein C concentrate (and its precursor, protein C concentrate) replacement therapy has been shown to improve outcomes in patients with meningococcemia-associated–purpura fulminans and severe sepsis, respectively.8 Heparin may be considered in the treatment of patients with purpura fulminans in addition to the replacement of any missing clotting factors or blood products.9 The international guidelines for the management of severe sepsis and septic shock include early quantitative resuscitation of the patient during the first 6 hours after recognition of sepsis, performing blood cultures before antibiotic therapy, and administering broad-spectrum antimicrobial therapy within 1 hour of recognition of septic shock.10 The elapsed time from triage to the actual administration of appropriate antimicrobials are primary determinants of patient mortality.11 Therefore, physicians must act quickly to stabilize the patient.
Gram-positive bacteria and gram-negative diplococci are common infectious agents implicated in purpura fulminans. Escherichia coli rarely has been identified as the inciting agent for purpura fulminans in adults. The increasing frequency of E coli strains that produce extended-spectrum β-lactamases—enzymes that mediate resistance to extended-spectrum (third generation) cephalosporins (eg, ceftazidime, cefotaxime, ceftriaxone) and monobactams (eg, aztreonam)—complicates matters further when deciding on appropriate antibiotics. Patients who have infections from extended-spectrum β-lactamase strains will require more potent carbapenems (eg, meropenem, imipenem) for treatment of infections. Despite undergoing treatment for septicemia, our patient went into cardiac arrest within 24 hours of presentation to the emergency department and died a few hours later. Physicians should consider E coli as an inciting agent of purpura fulminans and consider appropriate empiric antibiotics with gram-negative coverage to include E coli.
- Madden RM, Gill JC, Marlar RA. Protein C and protein S levels in two patients with acquired purpura fulminans. Br J Haematol. 1990;75:112-117.
- Nolan J, Sinclair R. Review of management of purpura fulminans and two case reports. Br J Anaesth. 2001;86:581-586.
- Huemer GM, Bonatti H, Dunst KM. Purpura fulminans due to E. coli septicemia. Wien Klin Wochenschr. 2004;116:82.
- Pugin J. Recognition of bacteria and bacterial products by host immune cells in sepsis. In: Vincent JL, ed. Yearbook of Intensive Care and Emergency Medicine. Berlin, Germany: Springer-Verlag; 1997:11-12.
- Brandtzaeg P, Oktedalen O, Kierulf P, et al. Elevated VIP and endotoxin plasma levels in human gram-negative septic shock. Regul Pept. 1989;24:37-44.
- Brandtzaeg P, Kierulf P, Gaustad P, et al. Plasma endotoxin as a predictor of multiple organ failure and death in systemic meningococcal disease. J Infect Dis. 1989;159:195-204.
- Brandtzaeg P, Ovstebøo R, Kierulf P. Compartmentalization of lipopolysaccharide production correlates with clinical presentation in meningococcal disease. J Infect Dis. 1992;166:650-652.
- Hodgson A, Ryan T, Moriarty J, et al. Plasma exchange as a source of protein C for acute onset protein C pathway failure. Br J Haematol. 2002;116:905-908.
- Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood. 1982;60:284-287.
- Dellinger RP, Levy MM, Rhodes A, et al. Surviving sepsis campaign guidelines committee including the pediatric subgroup. Crit Care Med. 2013;41:580-637.
- Gaieski DF, Mikkelsen ME, Band RA, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med. 2010;38:1045-1053.
To the Editor:
Purpura fulminans is a severe and rapidly fatal thrombotic disorder that can occur in association with either hereditary or acquired deficiencies of the natural anticoagulants protein C and protein S.1 It most commonly results from the acute inflammatory response and subsequent disseminated intravascular coagulation (DIC) seen in severe bacterial septicemia. Excessive bleeding, retiform purpura, and skin necrosis may develop as a result of the coagulopathies of typical DIC.1Neisseria meningitidis, Streptococcus, and Staphylococcus frequently are implicated as pathogens, but Escherichia coli–associated purpura fulminans in adults is rare.2,3 We report a case of purpura fulminans in the setting of E coli septicemia.
A 62-year-old woman with a history of end-stage liver disease secondary to alcoholic liver cirrhosis diagnosed 13 years prior complicated by ascites and esophageal varices presented to a primary care clinic for evaluation of a recent-onset nontender lesion on the left buttock. She was hypotensive with a blood pressure of 62/48 mmHg. The patient was prescribed ciprofloxacin 250 mg twice daily and hydrocodone/acetominophen 5 mg/325 mg twice daily as needed for pain management and was discharged. Six hours later, the patient presented to the emergency department with new onset symptoms of confusion and dark-colored spots on the abdomen and lower legs, which her family members noted had developed shortly after the patient took ciprofloxacin. In the emergency department, the patient was noted to be hypotensive and febrile with a severe metabolic acidosis. She was intubated for respiratory failure and received intravenous fluid resuscitation, broad-spectrum antibiotics, and vasopressors. Blood cultures were obtained, and the dermatology department was consulted.
On physical examination, extensive purpuric, reticulated, and stellate plaques with central necrosis and hemorrhagic bullae were noted on the abdomen (Figure, A) and bilateral lower legs (Figure, B) extending onto the thighs. The patient was coagulopathic with persistent sanguineous oozing at intravenous sites and bilateral nares. A small erythematous ulcer with overlying black eschar was noted on the left medial buttock.
Laboratory test results showed new-onset thrombocytopenia, prolonged prothrombin time/international normalized ratio and partial thromboplastin time, and low fibrinogen levels, which confirmed a diagnosis of acute DIC. Blood cultures were positive for gram-negative rods in 4 out of 4 bottles within 12 hours of being drawn. Further testing identified the microorganism as E coli, and antibiotic susceptibility testing revealed it was sensitive to most antibiotics.
The patient was clinically diagnosed with purpura fulminans secondary to severe E coli septicemia and DIC. This life-threatening disorder is considered a medical emergency with a high mortality rate. Laboratory findings supporting DIC include the presence of schistocytes on a peripheral blood smear, thrombocytopenia, positive plasma protamine paracoagulation test, low fibrinogen levels, and positive fibrin degradation products. Reported cases of purpura fulminans in the setting of E coli septicemia are rare, and meningococcemia is the most common presentation.2,3 Bacterial components (eg, lipopolysaccharides found in the cell walls of gram-negative bacteria) may contribute to the progression of septicemia. Increased levels of endotoxin lipopolysaccharide can lead to septic shock and organ dysfunction.4 However, the release of lipooligosaccharides is associated with the development of meningococcal septicemia, and the lipopolysaccharide levels are directly correlated with prognosis in patients without meningitis.5-7
Human activated protein C concentrate (and its precursor, protein C concentrate) replacement therapy has been shown to improve outcomes in patients with meningococcemia-associated–purpura fulminans and severe sepsis, respectively.8 Heparin may be considered in the treatment of patients with purpura fulminans in addition to the replacement of any missing clotting factors or blood products.9 The international guidelines for the management of severe sepsis and septic shock include early quantitative resuscitation of the patient during the first 6 hours after recognition of sepsis, performing blood cultures before antibiotic therapy, and administering broad-spectrum antimicrobial therapy within 1 hour of recognition of septic shock.10 The elapsed time from triage to the actual administration of appropriate antimicrobials are primary determinants of patient mortality.11 Therefore, physicians must act quickly to stabilize the patient.
Gram-positive bacteria and gram-negative diplococci are common infectious agents implicated in purpura fulminans. Escherichia coli rarely has been identified as the inciting agent for purpura fulminans in adults. The increasing frequency of E coli strains that produce extended-spectrum β-lactamases—enzymes that mediate resistance to extended-spectrum (third generation) cephalosporins (eg, ceftazidime, cefotaxime, ceftriaxone) and monobactams (eg, aztreonam)—complicates matters further when deciding on appropriate antibiotics. Patients who have infections from extended-spectrum β-lactamase strains will require more potent carbapenems (eg, meropenem, imipenem) for treatment of infections. Despite undergoing treatment for septicemia, our patient went into cardiac arrest within 24 hours of presentation to the emergency department and died a few hours later. Physicians should consider E coli as an inciting agent of purpura fulminans and consider appropriate empiric antibiotics with gram-negative coverage to include E coli.
To the Editor:
Purpura fulminans is a severe and rapidly fatal thrombotic disorder that can occur in association with either hereditary or acquired deficiencies of the natural anticoagulants protein C and protein S.1 It most commonly results from the acute inflammatory response and subsequent disseminated intravascular coagulation (DIC) seen in severe bacterial septicemia. Excessive bleeding, retiform purpura, and skin necrosis may develop as a result of the coagulopathies of typical DIC.1Neisseria meningitidis, Streptococcus, and Staphylococcus frequently are implicated as pathogens, but Escherichia coli–associated purpura fulminans in adults is rare.2,3 We report a case of purpura fulminans in the setting of E coli septicemia.
A 62-year-old woman with a history of end-stage liver disease secondary to alcoholic liver cirrhosis diagnosed 13 years prior complicated by ascites and esophageal varices presented to a primary care clinic for evaluation of a recent-onset nontender lesion on the left buttock. She was hypotensive with a blood pressure of 62/48 mmHg. The patient was prescribed ciprofloxacin 250 mg twice daily and hydrocodone/acetominophen 5 mg/325 mg twice daily as needed for pain management and was discharged. Six hours later, the patient presented to the emergency department with new onset symptoms of confusion and dark-colored spots on the abdomen and lower legs, which her family members noted had developed shortly after the patient took ciprofloxacin. In the emergency department, the patient was noted to be hypotensive and febrile with a severe metabolic acidosis. She was intubated for respiratory failure and received intravenous fluid resuscitation, broad-spectrum antibiotics, and vasopressors. Blood cultures were obtained, and the dermatology department was consulted.
On physical examination, extensive purpuric, reticulated, and stellate plaques with central necrosis and hemorrhagic bullae were noted on the abdomen (Figure, A) and bilateral lower legs (Figure, B) extending onto the thighs. The patient was coagulopathic with persistent sanguineous oozing at intravenous sites and bilateral nares. A small erythematous ulcer with overlying black eschar was noted on the left medial buttock.
Laboratory test results showed new-onset thrombocytopenia, prolonged prothrombin time/international normalized ratio and partial thromboplastin time, and low fibrinogen levels, which confirmed a diagnosis of acute DIC. Blood cultures were positive for gram-negative rods in 4 out of 4 bottles within 12 hours of being drawn. Further testing identified the microorganism as E coli, and antibiotic susceptibility testing revealed it was sensitive to most antibiotics.
The patient was clinically diagnosed with purpura fulminans secondary to severe E coli septicemia and DIC. This life-threatening disorder is considered a medical emergency with a high mortality rate. Laboratory findings supporting DIC include the presence of schistocytes on a peripheral blood smear, thrombocytopenia, positive plasma protamine paracoagulation test, low fibrinogen levels, and positive fibrin degradation products. Reported cases of purpura fulminans in the setting of E coli septicemia are rare, and meningococcemia is the most common presentation.2,3 Bacterial components (eg, lipopolysaccharides found in the cell walls of gram-negative bacteria) may contribute to the progression of septicemia. Increased levels of endotoxin lipopolysaccharide can lead to septic shock and organ dysfunction.4 However, the release of lipooligosaccharides is associated with the development of meningococcal septicemia, and the lipopolysaccharide levels are directly correlated with prognosis in patients without meningitis.5-7
Human activated protein C concentrate (and its precursor, protein C concentrate) replacement therapy has been shown to improve outcomes in patients with meningococcemia-associated–purpura fulminans and severe sepsis, respectively.8 Heparin may be considered in the treatment of patients with purpura fulminans in addition to the replacement of any missing clotting factors or blood products.9 The international guidelines for the management of severe sepsis and septic shock include early quantitative resuscitation of the patient during the first 6 hours after recognition of sepsis, performing blood cultures before antibiotic therapy, and administering broad-spectrum antimicrobial therapy within 1 hour of recognition of septic shock.10 The elapsed time from triage to the actual administration of appropriate antimicrobials are primary determinants of patient mortality.11 Therefore, physicians must act quickly to stabilize the patient.
Gram-positive bacteria and gram-negative diplococci are common infectious agents implicated in purpura fulminans. Escherichia coli rarely has been identified as the inciting agent for purpura fulminans in adults. The increasing frequency of E coli strains that produce extended-spectrum β-lactamases—enzymes that mediate resistance to extended-spectrum (third generation) cephalosporins (eg, ceftazidime, cefotaxime, ceftriaxone) and monobactams (eg, aztreonam)—complicates matters further when deciding on appropriate antibiotics. Patients who have infections from extended-spectrum β-lactamase strains will require more potent carbapenems (eg, meropenem, imipenem) for treatment of infections. Despite undergoing treatment for septicemia, our patient went into cardiac arrest within 24 hours of presentation to the emergency department and died a few hours later. Physicians should consider E coli as an inciting agent of purpura fulminans and consider appropriate empiric antibiotics with gram-negative coverage to include E coli.
- Madden RM, Gill JC, Marlar RA. Protein C and protein S levels in two patients with acquired purpura fulminans. Br J Haematol. 1990;75:112-117.
- Nolan J, Sinclair R. Review of management of purpura fulminans and two case reports. Br J Anaesth. 2001;86:581-586.
- Huemer GM, Bonatti H, Dunst KM. Purpura fulminans due to E. coli septicemia. Wien Klin Wochenschr. 2004;116:82.
- Pugin J. Recognition of bacteria and bacterial products by host immune cells in sepsis. In: Vincent JL, ed. Yearbook of Intensive Care and Emergency Medicine. Berlin, Germany: Springer-Verlag; 1997:11-12.
- Brandtzaeg P, Oktedalen O, Kierulf P, et al. Elevated VIP and endotoxin plasma levels in human gram-negative septic shock. Regul Pept. 1989;24:37-44.
- Brandtzaeg P, Kierulf P, Gaustad P, et al. Plasma endotoxin as a predictor of multiple organ failure and death in systemic meningococcal disease. J Infect Dis. 1989;159:195-204.
- Brandtzaeg P, Ovstebøo R, Kierulf P. Compartmentalization of lipopolysaccharide production correlates with clinical presentation in meningococcal disease. J Infect Dis. 1992;166:650-652.
- Hodgson A, Ryan T, Moriarty J, et al. Plasma exchange as a source of protein C for acute onset protein C pathway failure. Br J Haematol. 2002;116:905-908.
- Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood. 1982;60:284-287.
- Dellinger RP, Levy MM, Rhodes A, et al. Surviving sepsis campaign guidelines committee including the pediatric subgroup. Crit Care Med. 2013;41:580-637.
- Gaieski DF, Mikkelsen ME, Band RA, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med. 2010;38:1045-1053.
- Madden RM, Gill JC, Marlar RA. Protein C and protein S levels in two patients with acquired purpura fulminans. Br J Haematol. 1990;75:112-117.
- Nolan J, Sinclair R. Review of management of purpura fulminans and two case reports. Br J Anaesth. 2001;86:581-586.
- Huemer GM, Bonatti H, Dunst KM. Purpura fulminans due to E. coli septicemia. Wien Klin Wochenschr. 2004;116:82.
- Pugin J. Recognition of bacteria and bacterial products by host immune cells in sepsis. In: Vincent JL, ed. Yearbook of Intensive Care and Emergency Medicine. Berlin, Germany: Springer-Verlag; 1997:11-12.
- Brandtzaeg P, Oktedalen O, Kierulf P, et al. Elevated VIP and endotoxin plasma levels in human gram-negative septic shock. Regul Pept. 1989;24:37-44.
- Brandtzaeg P, Kierulf P, Gaustad P, et al. Plasma endotoxin as a predictor of multiple organ failure and death in systemic meningococcal disease. J Infect Dis. 1989;159:195-204.
- Brandtzaeg P, Ovstebøo R, Kierulf P. Compartmentalization of lipopolysaccharide production correlates with clinical presentation in meningococcal disease. J Infect Dis. 1992;166:650-652.
- Hodgson A, Ryan T, Moriarty J, et al. Plasma exchange as a source of protein C for acute onset protein C pathway failure. Br J Haematol. 2002;116:905-908.
- Feinstein DI. Diagnosis and management of disseminated intravascular coagulation: the role of heparin therapy. Blood. 1982;60:284-287.
- Dellinger RP, Levy MM, Rhodes A, et al. Surviving sepsis campaign guidelines committee including the pediatric subgroup. Crit Care Med. 2013;41:580-637.
- Gaieski DF, Mikkelsen ME, Band RA, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med. 2010;38:1045-1053.