User login
Systemic Targeted Treatments for Basal Cell Carcinoma
Basal cell carcinoma (BCC) is the most common keratinocyte carcinoma and affects more than 3 million individuals per year in the United States.1 Approximately 40% of patients diagnosed with BCC will develop another BCC within 5 years of the initial diagnosis.2 Most cases are successfully treated with surgical excision and occasionally topical therapy or radiotherapy. Despite the high cure rate with conventional treatments, BCC can recur and can cause substantial destruction of the surrounding tissue if left untreated.3-5 In some instances, BCC can even metastasize and lead to death.6 For patients who are poor candidates for surgical or topical treatment modalities because of locally advanced BCC (laBCC) or metastatic BCC (mBCC), systemic treatment may be indicated. Vismodegib, sonidegib, and cemiplimab are the only systemic medications approved by the US Food and Drug Administration (FDA) for the treatment of laBCC and/or mBCC. Vismodegib and sonidegib target the sonic hedgehog (SHH) signaling pathway that is abnormally activated in more than 90% of BCCs.7 Cemiplimab is an immune checkpoint inhibitor (ICI) that targets the programmed cell death protein 1 (PD-1) receptor.8 Herein, we review the clinical utility of these medications and their evolving roles in the treatment of BCC.
SHH Pathway Inhibitors
The SHH pathway is a key regulator of cell proliferation and differentiation during embryogenesis.7 During adulthood, SHH signaling decreases but still plays an important role in stem cell activation and in regulation of the hair follicle growth cycle.9,10 However, de novo mutations in the genes that comprise the SHH pathway can result in aberrant constitutive activation, leading to unrestricted cell proliferation. Genetic mutations resulting in activation of Smoothened (SMO), a G-protein–coupled receptor involved in the signal transduction and propagation of the SHH pathway, have been implicated in the pathogenesis of BCC. Inactivating mutations also are commonly observed in patched homolog 1, an upstream cell-surface protein that inhibits SMO.7 The mechanism by which vismodegib and sonidegib, 2 of the FDA-approved oral medications for the treatment of advanced BCC, block the SHH pathway is through the selective inhibition of SMO.7,11
Vismodegib first received FDA approval in 2012 for the treatment of laBCC and mBCC after initial results from the pivotal ERIVANCE phase 2 trial demonstrated an objective response rate (ORR) of 43% (27/63) and 30% (10/33) in patients with locally advanced and metastatic disease, respectively. In this single-arm study, all enrolled patients (63 with laBCC and 33 with mBCC) received 150 mg of oral vismodegib daily.12 Updated results at 39 months demonstrated improved ORRs of 60% (38/63) and 48% (16/33) for the laBCC and mBCC groups, respectively. A complete response (CR) and partial response (PR) were observed in 32% (n=20) and 29% (n=18) of patients with laBCC, respectively.13 These results have been confirmed in subsequent studies, including the large international open-label trial known as STEVIE, with ORRs of 68.5% for 1119 cases of laBCC and 37% for 96 cases of mBCC.14-17 The CR and PR rates were 33% and 35%, respectively, for the laBCC group. The CR and PR rates for the mBCC group were 5% and 32%, respectively.14
The FDA approval of sonidegib for laBCC—but not mBCC—occurred in 2015 after the pivotal BOLT randomized phase 2 trial demonstrated an initial ORR of 43% (18/42) for laBCC and 15% (2/13) for mBCC after administration of 200 mg of sonidegib daily.18 A final follow-up analysis at 42 months resulted in ORRs of 56% (37/66) and 8% (1/13) for the laBCC and mBCC groups, respectively.19 Additionally, improved efficacy was not observed in the 151 patients who were randomized to receive treatment with the higher 800-mg dose; however, they did experience a higher incidence of adverse events.18,19
Currently, the true clinical differences between vismodegib and sonidegib remain uncertain, as no head-to-head trials have been conducted. Moreover, direct comparison of the data from the ERIVANCE and BOLT trials is challenging owing to fundamental differences in methodologic design, including the criteria used to assess BCC severity. The ERIVANCE trial utilized the conventional Response Evaluation Criteria in Solid Tumors (RECIST), while BOLT used the rigorous modified RECIST. However, an expert consensus study attempted to compare the 2 trials by modifying the outcomes from BOLT with the former RECIST criteria. The expert group found that the 2 SHH inhibitors had comparable efficacy and adverse event profiles.20 Nevertheless, a recent meta-analysis found that although ORRs for laBCC were similar between the 2 drugs, the CR rate for vismodegib was 31% compared with 3% for sonidegib. Additionally, for mBCC, they reported the ORR of vismodegib to be 2.7 times higher than that of sonidegib (39% vs 15%).21
Immune Checkpoint Inhibitors
Immune checkpoint inhibitors have successfully been utilized in the treatment of cutaneous squamous cell carcinoma (cSCC); however, their use for treating BCC has been limited until recently.22-25 In February 2021, cemiplimab became the first and only ICI approved for the treatment of laBCC and mBCC in patients who did not respond to or were intolerant to prior SHH inhibitor therapy.26 Cemiplimab—a human monoclonal antibody against the PD-1 receptor expressed on T cells—blocks its interaction with programmed cell death ligand 1 and programmed cell death ligand 2 present on tumor cells. The blockade of the PD-1 pathway releases the inhibition of the antitumor immune response and enables appropriate cytotoxic T-cell activity to occur.8
The FDA approval of cemiplimab for the treatment of advanced BCC was based on an open-label, multicenter, single-arm phase 2 trial (NCT03132636) evaluating 84 patients with laBCC refractory or intolerant to SHH inhibitor therapy.26 Patients received an intravenous infusion of cemiplimab 350 mg every 3 weeks for up to 93 weeks or until disease progression or unacceptable toxicity. An ORR of 31% (26/84) was observed with a CR and PR of 6% (5/84) and 25% (21/84), respectively. The median duration of follow-up was 15 months.26 Given the clinically meaningful results of this trial, investigating the efficacy of other PD-1 inhibitors, such as pembrolizumab and nivolumab, for treatment of advanced BCC may prove worthwhile.
Adverse Effects of Systemic Treatments
The 2 approved SHH inhibitors—vismodegib and sonidegib—appear to have similar side-effect profiles, with the most common adverse effects being muscle spasms, dysgeusia, alopecia, nausea, vomiting, diarrhea, weight loss, and fatigue.20,21,27 These side effects occur at high frequencies (>40%) for both SHH inhibitors and often lead to discontinuation of the medication.21 Rates of treatment discontinuation range from 15% to 50% on average.12-14,18 Fortunately, the majority of these adverse effects do not appear to increase in severity or frequency with prolonged use of these medications.14,16,28
Various conservative and pharmacologic measures can be implemented to help manage side effects. For muscle spasms, which are the most commonly reported adverse effect, supplementation with magnesium, transcutaneous electrical nerve stimulation, acupuncture, massages, stretching, and thermal compresses can potentially be beneficial.29 Calcium channel blockers also may be effective, as one small prospective cohort study reported a reduction in the frequency of muscle cramps with amlodipine 10 mg daily.30 For alopecia, which typically is reversible and caused by SHH inhibition of the normal hair cycle, minoxidil theoretically can help, as it reduces telogen arrest and extends the anagen growth phase.31,32 Although usually mild and self-limiting, management of dysgeusia, weight loss, and gastrointestinal upset often can be managed with dietary changes, such as smaller, more frequent meals.33,34 Finally, alternative dosing strategies and drug holidays have been employed to mitigate these side effects and increase drug tolerability.35,36 These are discussed in the Alternative Dosing section.
Given the essential role of the SHH pathway in embryologic development, SHH inhibitors carry a black box warning of embryofetal teratogenicity and are contraindicated in females who are pregnant or breastfeeding. For females of reproductive potential, verification of pregnancy status should be performed prior to initiating treatment with an SHH inhibitor. These patients should be counseled on the use of contraception during treatment and for at least 24 months and 20 months after cessation of vismodegib and sonidegib, respectively.27,37,38 Male patients, even after a vasectomy, should use barrier contraception during treatment and for at least 3 months and 8 months after the final dose of vismodegib and sonidegib, respectively.37,38
Laboratory abnormalities commonly associated with SHH inhibitors include elevated hepatic enzymes, particularly with vismodegib, and elevated creatine kinase levels, particularly with sonidegib.28,39 Other laboratory abnormalities that can occur include hypercholesterolemia, hypercreatininemia, hyperglycemia, and increased serum lipase levels.19,28 Although these laboratory abnormalities usually are asymptomatic and self-limiting, regular monitoring should be performed.
There also is concern that SHH inhibitors may induce the development of cSCC. A case-control study of 55 cases and 125 control patients found an increased risk for cSCC in those previously treated with vismodegib, with a hazard ratio of 8.12.40 However, a subsequent retrospective cohort study of 1675 patients with BCC failed to find any association with cSCC among those treated with vismodegib compared to those who received standard surgical therapy.41 Clinical data for sonidegib are lacking, but the BOLT trial found that cSCC occurred in 3 patients receiving treatment with the SHH inhibitor.18 Thus, further studies are needed to more thoroughly assess this concern. Close monitoring for cSCC may be warranted in patients prescribed SHH inhibitors at this time.
Cemiplimab has demonstrated an acceptable safety profile and is generally well tolerated. In the phase 2 trial of cemiplimab for cSCC, approximately 5% of patients discontinued treatment because of adverse effects. The most commonly reported side effects of cemiplimab were diarrhea (27%), fatigue (24%), nausea (17%), constipation (15%), and rash (15%).23 In the phase 2 trial for laBCC, grade 3 or 4 adverse events occurred in 48% of patients, with hypertension (5%) being the most common.26 Although rare, immune-mediated adverse reactions also can occur, given the mechanism of action of ICIs. These side effects, ranging in severity from mild to fatal, include pneumonitis, colitis, hepatitis, nephritis, myocarditis, and hypophysitis. Therefore, close monitoring for these immune-mediated reactions is critical, but most can be managed with corticosteroids or treatment interruption if they occur.42,43
No absolute contraindications exist for cemiplimab; however, extreme caution should be taken in immunosuppressed individuals, such as solid organ transplant recipients and those with chronic lymphocytic leukemia (CLL), as safety data are limited in these patients.44,45 Although small retrospective studies have reported reasonable tolerability in solid organ transplant recipients treated with ICIs, an allograft rejection rate of 41% was found in a meta-analysis of 64 patients.46-48 In CLL patients with keratinocyte carcinomas, ICIs have been safely used and have even demonstrated efficacy for CLL in some cases.49-52
Alternative Dosing
The side effects of SHH inhibitors have led to alternative dosing strategies to prevent medication discontinuation and improve adherence. In patients with basal cell nevus syndrome, multimonth drug holidays have been shown to increase drug tolerability without compromising efficacy.35,36 Weekly intermittent dosing regimens of vismodegib ranging from 1 week on followed by 1 to 3 weeks off demonstrated efficacy in a retrospective study of 7 patients with advanced BCC.53 All 7 patients experienced improvement in their BCCs, with 3 patients experiencing CR. Importantly, treatment-related adverse effects were mild and well tolerated, with no patients terminating the medication.53 Two other retrospective case series of patients with advanced BCC treated with vismodegib reported similar findings for those placed on an intermittent dosing schedule ranging from once every other day to once per week.54,55
In the large phase 2 randomized trial known as MIKIE, 2 different intermittent dosing regimens of 150 mg vismodegib daily for patients with multiple BCCs were found to have good activity and tolerability.56 The first group (n=116) received vismodegib for 12 weeks, then 3 rounds of 8 weeks of placebo, followed by 12 weeks of vismodegib; there was a 63% reduction in clinically evident BCCs after 73 weeks. The second group (n=113) received the medication for 24 weeks, then 3 rounds of 8 weeks of placebo, followed by 8 weeks of vismodegib; there was a 54% reduction at the end of 73 weeks.56 Subsequent analyses found improvements in health-related quality-of-life outcomes that were similar for both groups.57
Consequently, alternative dosing schedules appear to be a viable option for patients at risk of discontinuing treatment because of adverse effects, and current data support the recently approved recommendations of dose interruptions of up to 8 weeks to manage adverse effects in patients with laBCC or mBCC.58 Nevertheless, further clinical studies are required to determine the optimal intermittent dosing regimen for patients treated with SHH inhibitors.
Neoadjuvant Administration
Recently, vismodegib has been studied as a neoadjuvant therapy for BCC with promising results. Several small retrospective studies and case reports have documented successful treatment of both operable and inoperable periocular laBCC, with preservation of the eye in all patients.59-61 An open-label trial of 15 patients with advanced BCC who received neoadjuvant vismodegib for 3 to 6 months prior to surgical excision reported a mean reduction of 35% in the final surgical defect size, with no recurrence at 22 months.62,63 The latest and largest study performed was a phase 2 open-label trial known as VISMONEO, where 44 of 55 laBCC patients (80%) receiving neoadjuvant vismodegib for a mean duration of 6 months (range, 4–10 months) achieved the primary end point of tumor surgical downstaging.64 Of the 44 patients who had tumor downstaging, 27 (61%) experienced histologically proven CRs. Additionally, a 66% reduction in the average target lesion size was reported in this group compared to29% in the 11 patients who did not have tumor downstaging (P=.0002).64 Thus, SHH inhibitors may hold an important neoadjuvant role in the treatment of BCC by decreasing surgical defect size and allowing for surgical management of previously inoperable cases.
Synergism With Radiation
Preliminary data suggest SHH inhibitors may help potentiate the effects of radiation therapy for the treatment of BCC. Currently, the evidence primarily is limited to case studies, with several reports describing complete remission in patients with advanced BCCs who were considered unsuitable candidates for surgery. In these cases, vismodegib was administered either prior to or concurrently with radiation treatment.65-69 An in vitro study also documented the radiation-sensitizing effects of vismodegib in a BCC cell line.70 Recently, a phase 2 trial (ClinicalTrials.gov identifier NCT01835626) evaluating the concurrent use of vismodegib and radiotherapy for patients with advanced BCC was completed, but data has yet to be published.
Synergism With and Benefit of Antifungal Therapy
The antifungal drug itraconazole is a potent inhibitor of the SHH pathway and may have an adjunctive role in the treatment of BCC. Similar to vismodegib and sonidegib, itraconazole acts as a direct antagonist of SMO. However, it is thought to bind to a distinct site on SMO.71,72 An open-label, exploratory phase 2 trial of 19 patients with BCC found that oral itraconazole 200 to 400 mg daily decreased tumor proliferative index by 45% (P=.04), as measured by Ki-67; SHH activity by 65% (P=.03), as measured by GLI1 messenger RNA; and mean tumor area by 24%.73 In a case series of 5 patients with mBCC refractory to conventional SHH inhibitor therapy, combined treatment with itraconazole and arsenic trioxide resulted in stable disease and a 75% reduction in SHH activity (P<.001).74 One case report documented tumor regression leading to stable disease for 15 months in a patient with laBCC treated with itraconazole monotherapy due to being unable to afford vismodegib or sonidegib. However, within 2 months of treatment discontinuation, the lesion progressed considerably.75 The efficacy of a topical formulation of itraconazole also has been tested in an open-label, placebo-controlled phase 2 trial, but no benefit was observed.76
Posaconazole is a second-generation antifungal agent that may serve as a potential alternative to itraconazole.77 Although clinical data are lacking, a basic science study found that posaconazole could inhibit the growth of SHH-dependent BCC in vivo (in mice).78 Furthermore, posaconazole has demonstrated a better safety profile with fewer and more mild side effects than itraconazole and does not require dose adjustment for those with hepatic or renal failure.79,80 Thus, posaconazole may be a safer alternative to itraconazole for the treatment of BCC. Further clinical studies are needed to elucidate the potential synergistic effects of these antifungal agents with the 2 currently approved SHH inhibitors for the treatment of advanced BCC.
Drug Resistance
Treatment resistance to SHH inhibitors, though uncommon, is a growing concern. Acquired mutations in the SMO binding site or downstream mediators of the SHH pathway have been shown to confer resistance to vismodegib and sonidegib.72,81-83 In addition, it appears that there may be shared resistance among the drugs in this class. One study assessing the efficacy of sonidegib in 9 patients with laBCC resistant to vismodegib found that these patients also did not respond to sonidegib.84 Interestingly, 1 case report documented tumor regression of an intracranial BCC in a patient treated with sonidegib and itraconazole after failure with vismodegib.85 An in vitro study also found that itraconazole maintained SHH inhibitory activity for all drug-resistant SMO mutations that have been reported.72 Therefore, itraconazole monotherapy or combination therapy with a canonical SHH inhibitor may be considered for patients with recalcitrant BCC and warrants further investigation.
Taladegib is a newly developed SMO inhibitor that may serve as another promising alternative for patients who develop resistance to vismodegib or sonidegib. A phase 1 trial of taladegib for advanced BCC found an ORR of 69% (11/16) in the SHH inhibitor–naïve group and an ORR of 36% (11/32) in the group previously treated with a SHH inhibitor.86 Additionally, the safety profile and frequency of adverse effects appear to be similar to those associated with vismodegib and sonidegib.86,87 Unfortunately, no clinical trials evaluating taladegib for BCC are ongoing or in development at this time.
Recurrence
There appears to be a relatively high rate of recurrence for BCC patients who achieve a CR to SHH inhibitors. In a retrospective study of 116 laBCC patients who experienced a CR after vismodegib therapy, 54 patients (47%) relapsed at 36 months. Among the 54 patients that relapsed, 27 were re-treated with vismodegib, which resulted in an ORR of 85% (23/27), a CR rate of 37% (10/27), and a PR rate of 48% (13/27).88 Another retrospective study of 35 laBCC patients who relapsed after vismodegib treatment reported a 31% (11/35) clinical recurrence rate at 6-month follow-up.89 An observational retrospective study also assessed the efficacy of SHH inhibitor maintenance therapy for advanced BCC patients who achieved a CR.90 In the study, 27 (64%) patients received a maintenance dose of 150 mg vismodegib once per week for 1 year, while 15 (36%) patients decided not to take a maintenance dose following CR of their BCC. All patients who took the maintenance therapy did not experience clinical recurrence at 1-year follow-up, whereas 26% of patients not on the maintenance dose relapsed.90 Consequently, these results indicate that BCC recurrence is frequent after SHH inhibitor therapy and highlights the importance of close surveillance after CR is attained. Nevertheless, most patients still respond to treatment with SHH inhibitors after relapsing, and intermittent maintenance doses may be an effective means to reduce risk of recurrence.
Conclusion
Vismodegib and sonidegib are SHH inhibitors approved for the treatment of laBCC and mBCC. Cemiplimab is now also approved for patients who do not respond to SHH inhibitors or for whom SHH inhibitors are not tolerable. Although these systemic targeted therapies can lead to notable tumor shrinkage and even complete regression in some patients, recurrence is common, and adverse effects may limit their use. Drug resistance is an emerging issue that requires additional examination. Further clinical studies are needed to determine which patients are likely to respond to these targeted treatments.
Various intermittent and maintenance drug regimens should be evaluated for their potential to mitigate adverse effects and reduce risk of recurrence, respectively. The synergistic effects of these medications with other therapies as well as their neoadjuvant and adjuvant roles should be further investigated. For example, administration of an SHH inhibitor prior to surgical excision of a BCC may allow for a smaller surgical defect size, thereby improving cosmetic and functional outcomes. Moreover, these systemic targeted medications may allow for previously inoperable tumors to become amenable to surgical treatment.
Although SHH inhibitors and PD-1 inhibitors represent a major advancement in the field of oncodermatology, real-world efficacy and safety data in the upcoming years will be important for elucidating their true benefit for patients with BCC.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the U.S. population, 2012. JAMA Dermatol. 2015;151:1081-1086.
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: epidemiology; pathophysiology; clinical and histological subtypes; and disease associations. J Am Acad Dermatol. 2019;80:303-317.
- Rees JR, Zens MS, Celaya MO, et al. Survival after squamous cell and basal cell carcinoma of the skin: a retrospective cohort analysis. Int J Cancer. 2015;137:878-884.
- Kasumagic-Halilovic E, Hasic M, Ovcina-Kurtovic N. A clinical study of basal cell carcinoma. Med Arch. 2019;73:394-398.
- Goldenberg G, Karagiannis T, Palmer JB, et al. Incidence and prevalence of basal cell carcinoma (BCC) and locally advanced BCC (LABCC) in a large commercially insured population in the United States: a retrospective cohort study. J Am Acad Dermatol. 2016;75:957.e952-966.e952.
- Laga AC, Schaefer IM, Sholl LM, et al. Metastatic basal cell carcinoma. Am J Clin Pathol. 2019;152:706-717.
- Rimkus TK, Carpenter RL, Qasem S, et al. Targeting the sonic hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancers (Basel). 2016;8:22.
- Burova E, Hermann A, Waite J, et al. Characterization of the anti–PD-1 antibody REGN2810 and its antitumor activity in human PD-1 knock-in mice. Mol Cancer Ther. 2017;16:861-870.
- Bhardwaj G, Murdoch B, Wu D, et al. Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nat Immunol. 2001;2:172-180.
- Paladini RD, Saleh J, Qian C, et al. Modulation of hair growth with small molecule agonists of the hedgehog signaling pathway. J Invest Dermatol. 2005;125:638-646.
- Von Hoff DD, LoRusso PM, Rudin CM, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361:1164-1172.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Sekulic A, Migden MR, Basset-Seguin N, et al. Long-term safety and efficacy of vismodegib in patients with advanced basal cell carcinoma: final update of the pivotal ERIVANCE BCC study. BMC Cancer. 2017;17:332.
- Basset-Séguin N, Hauschild A, Kunstfeld R, et al. Vismodegib in patients with advanced basal cell carcinoma: primary analysis of STEVIE, an international, open-label trial. Eur J Cancer. 2017;86:334-348.
- Cozzani R, Del Aguila R, Carrizo M, et al. Efficacy and safety profile of vismodegib in a real-world setting cohort of patients with advanced basal cell carcinoma in Argentina. Int J Dermatol. 2020;59:627-632.
- Spallone G, Sollena P, Ventura A, et al. Efficacy and safety of vismodegib treatment in patients with advanced basal cell carcinoma and multiple comorbidities. Dermatol Ther. 2019;32:E13108.
- Fosko SW, Chu MB, Armbrecht E, et al. Efficacy, rate of tumor response, and safety of a short course (12-24 weeks) of oral vismodegib in various histologic subtypes (infiltrative, nodular, and superficial) of high-risk or locally advanced basal cell carcinoma, in an open-label, prospective case series clinical trial. J Am Acad Dermatol. 2020;82:946-954.
- Migden MR, Guminski A, Gutzmer R, et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): a multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 2015;16:716-728.
- Dummer R, Guminksi A, Gutzmer R, et al. Long-term efficacy and safety of sonidegib in patients with advanced basal cell carcinoma: 42-month analysis of the phase II randomized, double-blind BOLT study. Br J Dermatol. 2020;182:1369-1378.
- Dummer R, Ascierto PA, Basset-Seguin N, et al. Sonidegib and vismodegib in the treatment of patients with locally advanced basal cell carcinoma: a joint expert opinion. J Eur Acad Dermatol Venereol. 2020;34:1944-1956.
- Xie P, Lefrançois P. Efficacy, safety, and comparison of sonic hedgehog inhibitors in basal cell carcinomas: a systematic review and meta-analysis. J Am Acad Dermatol. 2018;79:1089-1100.e1017.
- Gentzler R, Hall R, Kunk PR, et al. Beyond melanoma: inhibiting the PD-1/PD-L1 pathway in solid tumors. Immunotherapy. 2016;8:583-600.
- Guminski AD, Lim AML, Khushalani NI, et al. Phase 2 study of cemiplimab, a human monoclonal anti-PD-1, in patients (pts) with metastatic cutaneous squamous cell carcinoma (mCSCC; group 1): 12-month follow-up [abstract]. J Clin Oncol. 2019;37(15 suppl):9526.
- Grob JJ, Gonzalez Mendoza R, Basset-Seguin N, et al. Pembrolizumab for recurrent/metastatic cutaneous squamous cell carcinoma (cSCC): efficacy and safety results from the phase II KEYNOTE-629 study [abstract]. Ann Oncol. 2019;30 (suppl 5):v908.
- Maubec E, Boubaya M, Petrow P, et al. Pembrolizumab as first-line therapy in patients with unresectable cutaneous squamous cell carcinoma (cSCC): phase 2 results from CARSKIN [abstract]. J Clin Oncol. 2019;37(15 suppl):9547.
- Stratigos AJ, Sekulic A, Peris K, et al. Cemiplimab in locally advanced basal cell carcinoma after hedgehog inhibitor therapy: an open-label, multi-centre, single-arm, phase 2 trial. Lancet Oncol. 2021;22:848-857.
- Carpenter RL, Ray H. Safety and tolerability of sonic hedgehog pathway inhibitors in cancer. Drug Saf. 2019;42:263-279.
- Villani A, Fabbrocini G, Costa C, et al. Sonidegib: safety and efficacy in treatment of advanced basal cell carcinoma. Dermatol Ther (Heidelb). 2020;10:401-412.
- Wright A, Sluka KA. Nonpharmacological treatments for musculoskeletal pain. Clin J Pain. 2001;17:33-46.
- Ally MS, Tang JY, Lindgren J, et al. Effect of calcium channel blockade on vismodegib-induced muscle cramps. JAMA Dermatol. 2015;151:1132-1134.
- Yang X, Thai K-E. Treatment of permanent chemotherapy-induced alopecia with low dose oral minoxidil. Australas J Dermatol. 2016;57:e130-e132.
- Ferguson JS, Hannam S, Toholka R, et al. Hair loss and hedgehog inhibitors: a class effect? Br J Dermatol. 2015;173:262-264.
- Kumbargere Nagraj S, George RP, Shetty N, et al. Interventions for managing taste disturbances. Cochrane Database Syst Rev. 2017;12:CD010470.
- Jacobsen AA, Kydd AR, Strasswimmer J. Practical management of the adverse effects of hedgehog pathway inhibitor therapy for basal cell carcinoma. J Am Acad Dermatol. 2017;76:767-768.
- Ally MS, Tang JY, Joseph T, et al. The use of vismodegib to shrink keratocystic odontogenic tumors in patients with basal cell nevus syndrome. JAMA Dermatol. 2014;150:542-545.
- Yang X, Dinehart SM. Intermittent vismodegib therapy in basal cell nevus syndrome. JAMA Dermatol. 2016;152:223-224.
- Erivedge. Prescribing information. Genentech; 2015.
- Odomzo. Prescribing information. Novartis; 2015.
- Ventarola DJ, Silverstein DI. Vismodegib-associated hepatotoxicity: a potential side effect detected in postmarketing surveillance. J Am Acad Dermatol. 2014;71:397-398.
- Mohan SV, Chang J, Li S, et al. Increased risk of cutaneous squamous cell carcinoma after vismodegib therapy for basal cell carcinoma. JAMA Dermatol. 2016;152:527-532.
- Bhutani T, Abrouk M, Sima CS, et al. Risk of cutaneous squamous cell carcinoma after treatment of basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2017;77:713-718.
- Morgado M, Plácido A, Morgado S, et al. Management of the adverse effects of immune checkpoint inhibitors. Vaccines (Basel). 2020;8:575.
- Martins F, Sofiya L, Sykiotis GP, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16:563-580.
- Ntsethe A, Dludla PV, Nyambuya TM, et al. The impact of immune checkpoint inhibitors in patients with chronic lymphocytic leukemia (CLL): a protocol for a systematic review and meta-analysis of randomized controlled trials. Medicine (Baltimore). 2020;99:E21167.
- Johnson DB, Sullivan RJ, Menzies AM. Immune checkpoint inhibitors in challenging populations. Cancer. 2017;123:1904-1911.
- Tsung I, Worden FP, Fontana RJ. Safety and efficacy of checkpoint inhibitors in solid organ transplant recipients with cutaneous squamous cell carcinoma [abstract]. J Clin Oncol. 2020;38(15 suppl):E22014.
- Owoyemi I, Vaughan LE, Costello CM, et al. Clinical outcomes of solid organ transplant recipients with metastatic cancers who are treated with immune checkpoint inhibitors: a single-center analysis. Cancer. 2020;126:4780-4787.
- Kumar V, Shinagare AB, Rennke HG, et al. The safety and efficacy of checkpoint inhibitors in transplant recipients: a case series and systematic review of literature. Oncologist. 2020;25:505-514.
- Arenbergerova M, Fialova A, Arenberger P, et al. Killing two birds with one stone: response to pembrolizumab in a patient with metastatic melanoma and B-cell chronic lymphocytic leukaemia. J Eur Acad Dermatol Venereol. 2018;32:E72-E74.
- Archibald WJ, Meacham PJ, Williams AM, et al. Management of melanoma in patients with chronic lymphocytic leukemia. Leuk Res. 2018;71:43-46.
- Ding W, LaPlant BR, Call TG, et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood. 2017;129:3419-3427.
- Leiter U, Loquai C, Reinhardt L, et al. Immune checkpoint inhibition therapy for advanced skin cancer in patients with concomitant hematological malignancy: a retrospective multicenter DeCOG study of 84 patients. J Immunother Cancer. 2020;8:E000897.
- Becker LR, Aakhus AE, Reich HC, et al. A novel alternate dosing of vismodegib for treatment of patients with advanced basal cell carcinomas. JAMA Dermatol. 2017;153:321-322.
- Woltsche N, Pichler N, Wolf I, et al. Managing adverse effects by dose reduction during routine treatment of locally advanced basal cell carcinoma with the hedgehog inhibitor vismodegib: a single centre experience. J Eur Acad Dermatol Venereol. 2019;33:E144-E145.
- Wong C, Poblete-Lopez C, Vidimos A. Comparison of daily dosing versus Monday through Friday dosing of vismodegib for locally advanced basal cell carcinoma and basal cell nevus syndrome: a retrospective case series. J Am Acad Dermatol. 2020;82:1539-1542.
- Dréno B, Kunstfeld R, Hauschild A, et al. Two intermittent vismodegib dosing regimens in patients with multiple basal-cell carcinomas (MIKIE): a randomised, regimen-controlled, double-blind, phase 2 trial. Lancet Oncol. 2017;18:404-412.
- Schadendorf D, Hauschild A, Fosko S, et al. Quality-of-life analysis with intermittent vismodegib regimens in patients with multiple basal cell carcinomas: patient-reported outcomes from the MIKIE study. J Eur Acad Dermatol Venereol. 2020;34:E526-E529.
- Chanu P, Musib L, Wang X, et al. Vismodegib efficacy in advanced basal cell carcinoma maintained with 8-week dose interruptions: a model-based evaluation. J Invest Dermatol. 2021;141:930-933.
- Su MG, Potts LB, Tsai JH. Treatment of periocular basal cell carcinoma with neoadjuvant vismodegib. Am J Ophthalmol Case Rep. 2020;19:100755.
- González AR, Etchichury D, Gil ME, et al. Neoadjuvant vismodegib and Mohs micrographic surgery for locally advanced periocular basal cell carcinoma. Ophthalmic Plast Reconstr Surg. 2019;35:56-61.
- Sagiv O, Nagarajan P, Ferrarotto R, et al. Ocular preservation with neoadjuvant vismodegib in patients with locally advanced periocular basal cell carcinoma. Br J Ophthalmol. 2019;103:775-780.
- Ally MS, Aasi S, Wysong A, et al. An investigator-initiated open-label clinical trial of vismodegib as a neoadjuvant to surgery for high-risk basal cell carcinoma. J Am Acad Dermatol. 2014;71:904-911.e1.
- Kwon GP, Ally MS, Bailey-Healy I, et al. Update to an open-label clinical trial of vismodegib as neoadjuvant before surgery for high-risk basal cell carcinoma (BCC). J Am Acad Dermatol. 2016;75:213-215.
- Mortier L, Bertrand N, Basset-Seguin N, et al. Vismodegib in neoadjuvant treatment of locally advanced basal cell carcinoma: first results of a multicenter, open-label, phase 2 trial (VISMONEO study) [abstract]. J Clin Oncol. 2018;36(15 suppl):9509.
- Strasswimmer JM. Potential synergy of radiation therapy with vismodegib for basal cell carcinoma. JAMA Dermatol. 2015;151:925-926.
- Gathings RM, Orscheln CS, Huang WW. Compassionate use of vismodegib and adjuvant radiotherapy in the treatment of multiple locally advanced and inoperable basal cell carcinomas and squamous cell carcinomas of the skin. J Am Acad Dermatol. 2014;70:E88-E89.
- Franco AI, Eastwick G, Farah R, et al. Upfront radiotherapy with concurrent and adjuvant vismodegib is effective and well-tolerated in a patient with advanced, multifocal basal cell carcinoma. Case Rep Dermatol Med. 2018;2018:2354146.
- Pollom EL, Bui TT, Chang AL, et al. Concurrent vismodegib and radiotherapy for recurrent, advanced basal cell carcinoma. JAMA Dermatol. 2015;151:998-1001.
- Janela-Lapert R, Dubray B, Duval-Modeste A, et al. Treatment of advanced basal cell carcinoma with vismodegib followed by radiotherapy [in French]. Ann Dermatol Venereol. 2020;147:780-782.
- Hehlgans S, Booms P, Güllülü Ö, et al. Radiation sensitization of basal cell and head and neck squamous cell carcinoma by the hedgehog pathway inhibitor vismodegib. Int J Mol Sci. 2018;19:2485.
- Kim J, Tang JY, Gong R, et al. Itraconazole, a commonly used antifungal that inhibits hedgehog pathway activity and cancer growth. Cancer Cell. 2010;17:388-399.
- Kim J, Aftab BT, Tang JY, et al. Itraconazole and arsenic trioxide inhibit hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell. 2013;23:23-34.
- Kim DJ, Kim J, Spaunhurst K, et al. Open-label, exploratory phase II trial of oral itraconazole for the treatment of basal cell carcinoma. J Clin Oncol. 2014;32:745-751.
- Ally MS, Ransohoff K, Sarin K, et al. Effects of combined treatment with arsenic trioxide and itraconazole in patients with refractory metastatic basal cell carcinoma. JAMA Dermatol. 2016;152:452-456.
- Cia˛z˙yn´yska M, Narbutt J, Skibin´ska M, et al. Itraconazole—a new player in the therapy of advanced basal cell carcinoma: a case report. JCO Oncol Pract. 2020;16:837-838.
- Sohn GK, Kwon GP, Bailey-Healy I, et al. Topical itraconazole for the treatment of basal cell carcinoma in patients with basal cell nevus syndrome or high-frequency basal cell carcinomas: a phase 2, open-label, placebo-controlled trial. JAMA Dermatol. 2019;155:1078-1080.
- Lass-Flörl C. Triazole antifungal agents in invasive fungal infections: a comparative review. Drugs. 2011;71:2405-2419.
- Chen B, Trang V, Lee A, et al. Posaconazole, a second-generation triazole antifungal drug, inhibits the hedgehog signaling pathway and progression of basal cell carcinoma. Mol Cancer Ther. 2016;15:866-876.
- Katragkou A, Tsikopoulou F, Roilides E, et al. Posaconazole: when and how? the clinician’s view. Mycoses. 2012;55:110-122.
- Raad II, Graybill JR, Bustamante AB, et al. Safety of long-term oral posaconazole use in the treatment of refractory invasive fungal infections. Clin Infect Dis. 2006;42:1726-1734.
- Atwood SX, Sarin KY, Whitson RJ, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342-353.
- Sun Q, Atzmony L, Zaki T, et al. Clues to primary vismodegib resistance lie in histology and genetics. J Clin Pathol. 2020;73:678-680.
- Verkouteren BJA, Wakkee M, van Geel M, et al. Molecular testing in metastatic basal cell carcinoma. J Am Acad Dermatol. 2021;85:1135-1142.
- Danial C, Sarin KY, Oro AE, et al. An investigator-initiated open-label trial of sonidegib in advanced basal cell carcinoma patients resistant to vismodegib. Clin Cancer Res. 2016;22:1325-1329.
- Yoon J, Apicelli AJ 3rd, Pavlopoulos TV. Intracranial regression of an advanced basal cell carcinoma using sonidegib and itraconazole after failure with vismodegib. JAAD Case Rep. 2017;4:10-12.
- Bendell J, Andre V, Ho A, et al. Phase I study of LY2940680, a Smo antagonist, in patients with advanced cancer including treatment-naïve and previously treated basal cell carcinoma. Clin Cancer Res. 2018;24:2082-2091.
- Ueno H, Kondo S, Yoshikawa S, et al. A phase I and pharmacokinetic study of taladegib, a Smoothened inhibitor, in Japanese patients with advanced solid tumors. Invest New Drugs. 2018;36:647-656.
- Herms F, Lambert J, Grob JJ, et al. Follow-up of patients with complete remission of locally advanced basal cell carcinoma after vismodegib discontinuation: a multicenter French study of 116 patients. J Clin Oncol. 2019;37:3275-3282.
- Villani A, Megna M, Fabbrocini G, et al. Long-term efficacy of vismodegib after its withdrawal and patients’ health-related quality of life using the Dermatology Life Quality Index (DLQI). Dermatol Ther (Heidelb). 2019;9:719-724.
- Scalvenzi M, Cappello M, Costa C, et al. Low-dose vismodegib as maintenance therapy after locally advanced basal cell carcinoma complete remission: high efficacy with minimal toxicity. Dermatol Ther (Heidelb). 2020;10:465-468.
Basal cell carcinoma (BCC) is the most common keratinocyte carcinoma and affects more than 3 million individuals per year in the United States.1 Approximately 40% of patients diagnosed with BCC will develop another BCC within 5 years of the initial diagnosis.2 Most cases are successfully treated with surgical excision and occasionally topical therapy or radiotherapy. Despite the high cure rate with conventional treatments, BCC can recur and can cause substantial destruction of the surrounding tissue if left untreated.3-5 In some instances, BCC can even metastasize and lead to death.6 For patients who are poor candidates for surgical or topical treatment modalities because of locally advanced BCC (laBCC) or metastatic BCC (mBCC), systemic treatment may be indicated. Vismodegib, sonidegib, and cemiplimab are the only systemic medications approved by the US Food and Drug Administration (FDA) for the treatment of laBCC and/or mBCC. Vismodegib and sonidegib target the sonic hedgehog (SHH) signaling pathway that is abnormally activated in more than 90% of BCCs.7 Cemiplimab is an immune checkpoint inhibitor (ICI) that targets the programmed cell death protein 1 (PD-1) receptor.8 Herein, we review the clinical utility of these medications and their evolving roles in the treatment of BCC.
SHH Pathway Inhibitors
The SHH pathway is a key regulator of cell proliferation and differentiation during embryogenesis.7 During adulthood, SHH signaling decreases but still plays an important role in stem cell activation and in regulation of the hair follicle growth cycle.9,10 However, de novo mutations in the genes that comprise the SHH pathway can result in aberrant constitutive activation, leading to unrestricted cell proliferation. Genetic mutations resulting in activation of Smoothened (SMO), a G-protein–coupled receptor involved in the signal transduction and propagation of the SHH pathway, have been implicated in the pathogenesis of BCC. Inactivating mutations also are commonly observed in patched homolog 1, an upstream cell-surface protein that inhibits SMO.7 The mechanism by which vismodegib and sonidegib, 2 of the FDA-approved oral medications for the treatment of advanced BCC, block the SHH pathway is through the selective inhibition of SMO.7,11
Vismodegib first received FDA approval in 2012 for the treatment of laBCC and mBCC after initial results from the pivotal ERIVANCE phase 2 trial demonstrated an objective response rate (ORR) of 43% (27/63) and 30% (10/33) in patients with locally advanced and metastatic disease, respectively. In this single-arm study, all enrolled patients (63 with laBCC and 33 with mBCC) received 150 mg of oral vismodegib daily.12 Updated results at 39 months demonstrated improved ORRs of 60% (38/63) and 48% (16/33) for the laBCC and mBCC groups, respectively. A complete response (CR) and partial response (PR) were observed in 32% (n=20) and 29% (n=18) of patients with laBCC, respectively.13 These results have been confirmed in subsequent studies, including the large international open-label trial known as STEVIE, with ORRs of 68.5% for 1119 cases of laBCC and 37% for 96 cases of mBCC.14-17 The CR and PR rates were 33% and 35%, respectively, for the laBCC group. The CR and PR rates for the mBCC group were 5% and 32%, respectively.14
The FDA approval of sonidegib for laBCC—but not mBCC—occurred in 2015 after the pivotal BOLT randomized phase 2 trial demonstrated an initial ORR of 43% (18/42) for laBCC and 15% (2/13) for mBCC after administration of 200 mg of sonidegib daily.18 A final follow-up analysis at 42 months resulted in ORRs of 56% (37/66) and 8% (1/13) for the laBCC and mBCC groups, respectively.19 Additionally, improved efficacy was not observed in the 151 patients who were randomized to receive treatment with the higher 800-mg dose; however, they did experience a higher incidence of adverse events.18,19
Currently, the true clinical differences between vismodegib and sonidegib remain uncertain, as no head-to-head trials have been conducted. Moreover, direct comparison of the data from the ERIVANCE and BOLT trials is challenging owing to fundamental differences in methodologic design, including the criteria used to assess BCC severity. The ERIVANCE trial utilized the conventional Response Evaluation Criteria in Solid Tumors (RECIST), while BOLT used the rigorous modified RECIST. However, an expert consensus study attempted to compare the 2 trials by modifying the outcomes from BOLT with the former RECIST criteria. The expert group found that the 2 SHH inhibitors had comparable efficacy and adverse event profiles.20 Nevertheless, a recent meta-analysis found that although ORRs for laBCC were similar between the 2 drugs, the CR rate for vismodegib was 31% compared with 3% for sonidegib. Additionally, for mBCC, they reported the ORR of vismodegib to be 2.7 times higher than that of sonidegib (39% vs 15%).21
Immune Checkpoint Inhibitors
Immune checkpoint inhibitors have successfully been utilized in the treatment of cutaneous squamous cell carcinoma (cSCC); however, their use for treating BCC has been limited until recently.22-25 In February 2021, cemiplimab became the first and only ICI approved for the treatment of laBCC and mBCC in patients who did not respond to or were intolerant to prior SHH inhibitor therapy.26 Cemiplimab—a human monoclonal antibody against the PD-1 receptor expressed on T cells—blocks its interaction with programmed cell death ligand 1 and programmed cell death ligand 2 present on tumor cells. The blockade of the PD-1 pathway releases the inhibition of the antitumor immune response and enables appropriate cytotoxic T-cell activity to occur.8
The FDA approval of cemiplimab for the treatment of advanced BCC was based on an open-label, multicenter, single-arm phase 2 trial (NCT03132636) evaluating 84 patients with laBCC refractory or intolerant to SHH inhibitor therapy.26 Patients received an intravenous infusion of cemiplimab 350 mg every 3 weeks for up to 93 weeks or until disease progression or unacceptable toxicity. An ORR of 31% (26/84) was observed with a CR and PR of 6% (5/84) and 25% (21/84), respectively. The median duration of follow-up was 15 months.26 Given the clinically meaningful results of this trial, investigating the efficacy of other PD-1 inhibitors, such as pembrolizumab and nivolumab, for treatment of advanced BCC may prove worthwhile.
Adverse Effects of Systemic Treatments
The 2 approved SHH inhibitors—vismodegib and sonidegib—appear to have similar side-effect profiles, with the most common adverse effects being muscle spasms, dysgeusia, alopecia, nausea, vomiting, diarrhea, weight loss, and fatigue.20,21,27 These side effects occur at high frequencies (>40%) for both SHH inhibitors and often lead to discontinuation of the medication.21 Rates of treatment discontinuation range from 15% to 50% on average.12-14,18 Fortunately, the majority of these adverse effects do not appear to increase in severity or frequency with prolonged use of these medications.14,16,28
Various conservative and pharmacologic measures can be implemented to help manage side effects. For muscle spasms, which are the most commonly reported adverse effect, supplementation with magnesium, transcutaneous electrical nerve stimulation, acupuncture, massages, stretching, and thermal compresses can potentially be beneficial.29 Calcium channel blockers also may be effective, as one small prospective cohort study reported a reduction in the frequency of muscle cramps with amlodipine 10 mg daily.30 For alopecia, which typically is reversible and caused by SHH inhibition of the normal hair cycle, minoxidil theoretically can help, as it reduces telogen arrest and extends the anagen growth phase.31,32 Although usually mild and self-limiting, management of dysgeusia, weight loss, and gastrointestinal upset often can be managed with dietary changes, such as smaller, more frequent meals.33,34 Finally, alternative dosing strategies and drug holidays have been employed to mitigate these side effects and increase drug tolerability.35,36 These are discussed in the Alternative Dosing section.
Given the essential role of the SHH pathway in embryologic development, SHH inhibitors carry a black box warning of embryofetal teratogenicity and are contraindicated in females who are pregnant or breastfeeding. For females of reproductive potential, verification of pregnancy status should be performed prior to initiating treatment with an SHH inhibitor. These patients should be counseled on the use of contraception during treatment and for at least 24 months and 20 months after cessation of vismodegib and sonidegib, respectively.27,37,38 Male patients, even after a vasectomy, should use barrier contraception during treatment and for at least 3 months and 8 months after the final dose of vismodegib and sonidegib, respectively.37,38
Laboratory abnormalities commonly associated with SHH inhibitors include elevated hepatic enzymes, particularly with vismodegib, and elevated creatine kinase levels, particularly with sonidegib.28,39 Other laboratory abnormalities that can occur include hypercholesterolemia, hypercreatininemia, hyperglycemia, and increased serum lipase levels.19,28 Although these laboratory abnormalities usually are asymptomatic and self-limiting, regular monitoring should be performed.
There also is concern that SHH inhibitors may induce the development of cSCC. A case-control study of 55 cases and 125 control patients found an increased risk for cSCC in those previously treated with vismodegib, with a hazard ratio of 8.12.40 However, a subsequent retrospective cohort study of 1675 patients with BCC failed to find any association with cSCC among those treated with vismodegib compared to those who received standard surgical therapy.41 Clinical data for sonidegib are lacking, but the BOLT trial found that cSCC occurred in 3 patients receiving treatment with the SHH inhibitor.18 Thus, further studies are needed to more thoroughly assess this concern. Close monitoring for cSCC may be warranted in patients prescribed SHH inhibitors at this time.
Cemiplimab has demonstrated an acceptable safety profile and is generally well tolerated. In the phase 2 trial of cemiplimab for cSCC, approximately 5% of patients discontinued treatment because of adverse effects. The most commonly reported side effects of cemiplimab were diarrhea (27%), fatigue (24%), nausea (17%), constipation (15%), and rash (15%).23 In the phase 2 trial for laBCC, grade 3 or 4 adverse events occurred in 48% of patients, with hypertension (5%) being the most common.26 Although rare, immune-mediated adverse reactions also can occur, given the mechanism of action of ICIs. These side effects, ranging in severity from mild to fatal, include pneumonitis, colitis, hepatitis, nephritis, myocarditis, and hypophysitis. Therefore, close monitoring for these immune-mediated reactions is critical, but most can be managed with corticosteroids or treatment interruption if they occur.42,43
No absolute contraindications exist for cemiplimab; however, extreme caution should be taken in immunosuppressed individuals, such as solid organ transplant recipients and those with chronic lymphocytic leukemia (CLL), as safety data are limited in these patients.44,45 Although small retrospective studies have reported reasonable tolerability in solid organ transplant recipients treated with ICIs, an allograft rejection rate of 41% was found in a meta-analysis of 64 patients.46-48 In CLL patients with keratinocyte carcinomas, ICIs have been safely used and have even demonstrated efficacy for CLL in some cases.49-52
Alternative Dosing
The side effects of SHH inhibitors have led to alternative dosing strategies to prevent medication discontinuation and improve adherence. In patients with basal cell nevus syndrome, multimonth drug holidays have been shown to increase drug tolerability without compromising efficacy.35,36 Weekly intermittent dosing regimens of vismodegib ranging from 1 week on followed by 1 to 3 weeks off demonstrated efficacy in a retrospective study of 7 patients with advanced BCC.53 All 7 patients experienced improvement in their BCCs, with 3 patients experiencing CR. Importantly, treatment-related adverse effects were mild and well tolerated, with no patients terminating the medication.53 Two other retrospective case series of patients with advanced BCC treated with vismodegib reported similar findings for those placed on an intermittent dosing schedule ranging from once every other day to once per week.54,55
In the large phase 2 randomized trial known as MIKIE, 2 different intermittent dosing regimens of 150 mg vismodegib daily for patients with multiple BCCs were found to have good activity and tolerability.56 The first group (n=116) received vismodegib for 12 weeks, then 3 rounds of 8 weeks of placebo, followed by 12 weeks of vismodegib; there was a 63% reduction in clinically evident BCCs after 73 weeks. The second group (n=113) received the medication for 24 weeks, then 3 rounds of 8 weeks of placebo, followed by 8 weeks of vismodegib; there was a 54% reduction at the end of 73 weeks.56 Subsequent analyses found improvements in health-related quality-of-life outcomes that were similar for both groups.57
Consequently, alternative dosing schedules appear to be a viable option for patients at risk of discontinuing treatment because of adverse effects, and current data support the recently approved recommendations of dose interruptions of up to 8 weeks to manage adverse effects in patients with laBCC or mBCC.58 Nevertheless, further clinical studies are required to determine the optimal intermittent dosing regimen for patients treated with SHH inhibitors.
Neoadjuvant Administration
Recently, vismodegib has been studied as a neoadjuvant therapy for BCC with promising results. Several small retrospective studies and case reports have documented successful treatment of both operable and inoperable periocular laBCC, with preservation of the eye in all patients.59-61 An open-label trial of 15 patients with advanced BCC who received neoadjuvant vismodegib for 3 to 6 months prior to surgical excision reported a mean reduction of 35% in the final surgical defect size, with no recurrence at 22 months.62,63 The latest and largest study performed was a phase 2 open-label trial known as VISMONEO, where 44 of 55 laBCC patients (80%) receiving neoadjuvant vismodegib for a mean duration of 6 months (range, 4–10 months) achieved the primary end point of tumor surgical downstaging.64 Of the 44 patients who had tumor downstaging, 27 (61%) experienced histologically proven CRs. Additionally, a 66% reduction in the average target lesion size was reported in this group compared to29% in the 11 patients who did not have tumor downstaging (P=.0002).64 Thus, SHH inhibitors may hold an important neoadjuvant role in the treatment of BCC by decreasing surgical defect size and allowing for surgical management of previously inoperable cases.
Synergism With Radiation
Preliminary data suggest SHH inhibitors may help potentiate the effects of radiation therapy for the treatment of BCC. Currently, the evidence primarily is limited to case studies, with several reports describing complete remission in patients with advanced BCCs who were considered unsuitable candidates for surgery. In these cases, vismodegib was administered either prior to or concurrently with radiation treatment.65-69 An in vitro study also documented the radiation-sensitizing effects of vismodegib in a BCC cell line.70 Recently, a phase 2 trial (ClinicalTrials.gov identifier NCT01835626) evaluating the concurrent use of vismodegib and radiotherapy for patients with advanced BCC was completed, but data has yet to be published.
Synergism With and Benefit of Antifungal Therapy
The antifungal drug itraconazole is a potent inhibitor of the SHH pathway and may have an adjunctive role in the treatment of BCC. Similar to vismodegib and sonidegib, itraconazole acts as a direct antagonist of SMO. However, it is thought to bind to a distinct site on SMO.71,72 An open-label, exploratory phase 2 trial of 19 patients with BCC found that oral itraconazole 200 to 400 mg daily decreased tumor proliferative index by 45% (P=.04), as measured by Ki-67; SHH activity by 65% (P=.03), as measured by GLI1 messenger RNA; and mean tumor area by 24%.73 In a case series of 5 patients with mBCC refractory to conventional SHH inhibitor therapy, combined treatment with itraconazole and arsenic trioxide resulted in stable disease and a 75% reduction in SHH activity (P<.001).74 One case report documented tumor regression leading to stable disease for 15 months in a patient with laBCC treated with itraconazole monotherapy due to being unable to afford vismodegib or sonidegib. However, within 2 months of treatment discontinuation, the lesion progressed considerably.75 The efficacy of a topical formulation of itraconazole also has been tested in an open-label, placebo-controlled phase 2 trial, but no benefit was observed.76
Posaconazole is a second-generation antifungal agent that may serve as a potential alternative to itraconazole.77 Although clinical data are lacking, a basic science study found that posaconazole could inhibit the growth of SHH-dependent BCC in vivo (in mice).78 Furthermore, posaconazole has demonstrated a better safety profile with fewer and more mild side effects than itraconazole and does not require dose adjustment for those with hepatic or renal failure.79,80 Thus, posaconazole may be a safer alternative to itraconazole for the treatment of BCC. Further clinical studies are needed to elucidate the potential synergistic effects of these antifungal agents with the 2 currently approved SHH inhibitors for the treatment of advanced BCC.
Drug Resistance
Treatment resistance to SHH inhibitors, though uncommon, is a growing concern. Acquired mutations in the SMO binding site or downstream mediators of the SHH pathway have been shown to confer resistance to vismodegib and sonidegib.72,81-83 In addition, it appears that there may be shared resistance among the drugs in this class. One study assessing the efficacy of sonidegib in 9 patients with laBCC resistant to vismodegib found that these patients also did not respond to sonidegib.84 Interestingly, 1 case report documented tumor regression of an intracranial BCC in a patient treated with sonidegib and itraconazole after failure with vismodegib.85 An in vitro study also found that itraconazole maintained SHH inhibitory activity for all drug-resistant SMO mutations that have been reported.72 Therefore, itraconazole monotherapy or combination therapy with a canonical SHH inhibitor may be considered for patients with recalcitrant BCC and warrants further investigation.
Taladegib is a newly developed SMO inhibitor that may serve as another promising alternative for patients who develop resistance to vismodegib or sonidegib. A phase 1 trial of taladegib for advanced BCC found an ORR of 69% (11/16) in the SHH inhibitor–naïve group and an ORR of 36% (11/32) in the group previously treated with a SHH inhibitor.86 Additionally, the safety profile and frequency of adverse effects appear to be similar to those associated with vismodegib and sonidegib.86,87 Unfortunately, no clinical trials evaluating taladegib for BCC are ongoing or in development at this time.
Recurrence
There appears to be a relatively high rate of recurrence for BCC patients who achieve a CR to SHH inhibitors. In a retrospective study of 116 laBCC patients who experienced a CR after vismodegib therapy, 54 patients (47%) relapsed at 36 months. Among the 54 patients that relapsed, 27 were re-treated with vismodegib, which resulted in an ORR of 85% (23/27), a CR rate of 37% (10/27), and a PR rate of 48% (13/27).88 Another retrospective study of 35 laBCC patients who relapsed after vismodegib treatment reported a 31% (11/35) clinical recurrence rate at 6-month follow-up.89 An observational retrospective study also assessed the efficacy of SHH inhibitor maintenance therapy for advanced BCC patients who achieved a CR.90 In the study, 27 (64%) patients received a maintenance dose of 150 mg vismodegib once per week for 1 year, while 15 (36%) patients decided not to take a maintenance dose following CR of their BCC. All patients who took the maintenance therapy did not experience clinical recurrence at 1-year follow-up, whereas 26% of patients not on the maintenance dose relapsed.90 Consequently, these results indicate that BCC recurrence is frequent after SHH inhibitor therapy and highlights the importance of close surveillance after CR is attained. Nevertheless, most patients still respond to treatment with SHH inhibitors after relapsing, and intermittent maintenance doses may be an effective means to reduce risk of recurrence.
Conclusion
Vismodegib and sonidegib are SHH inhibitors approved for the treatment of laBCC and mBCC. Cemiplimab is now also approved for patients who do not respond to SHH inhibitors or for whom SHH inhibitors are not tolerable. Although these systemic targeted therapies can lead to notable tumor shrinkage and even complete regression in some patients, recurrence is common, and adverse effects may limit their use. Drug resistance is an emerging issue that requires additional examination. Further clinical studies are needed to determine which patients are likely to respond to these targeted treatments.
Various intermittent and maintenance drug regimens should be evaluated for their potential to mitigate adverse effects and reduce risk of recurrence, respectively. The synergistic effects of these medications with other therapies as well as their neoadjuvant and adjuvant roles should be further investigated. For example, administration of an SHH inhibitor prior to surgical excision of a BCC may allow for a smaller surgical defect size, thereby improving cosmetic and functional outcomes. Moreover, these systemic targeted medications may allow for previously inoperable tumors to become amenable to surgical treatment.
Although SHH inhibitors and PD-1 inhibitors represent a major advancement in the field of oncodermatology, real-world efficacy and safety data in the upcoming years will be important for elucidating their true benefit for patients with BCC.
Basal cell carcinoma (BCC) is the most common keratinocyte carcinoma and affects more than 3 million individuals per year in the United States.1 Approximately 40% of patients diagnosed with BCC will develop another BCC within 5 years of the initial diagnosis.2 Most cases are successfully treated with surgical excision and occasionally topical therapy or radiotherapy. Despite the high cure rate with conventional treatments, BCC can recur and can cause substantial destruction of the surrounding tissue if left untreated.3-5 In some instances, BCC can even metastasize and lead to death.6 For patients who are poor candidates for surgical or topical treatment modalities because of locally advanced BCC (laBCC) or metastatic BCC (mBCC), systemic treatment may be indicated. Vismodegib, sonidegib, and cemiplimab are the only systemic medications approved by the US Food and Drug Administration (FDA) for the treatment of laBCC and/or mBCC. Vismodegib and sonidegib target the sonic hedgehog (SHH) signaling pathway that is abnormally activated in more than 90% of BCCs.7 Cemiplimab is an immune checkpoint inhibitor (ICI) that targets the programmed cell death protein 1 (PD-1) receptor.8 Herein, we review the clinical utility of these medications and their evolving roles in the treatment of BCC.
SHH Pathway Inhibitors
The SHH pathway is a key regulator of cell proliferation and differentiation during embryogenesis.7 During adulthood, SHH signaling decreases but still plays an important role in stem cell activation and in regulation of the hair follicle growth cycle.9,10 However, de novo mutations in the genes that comprise the SHH pathway can result in aberrant constitutive activation, leading to unrestricted cell proliferation. Genetic mutations resulting in activation of Smoothened (SMO), a G-protein–coupled receptor involved in the signal transduction and propagation of the SHH pathway, have been implicated in the pathogenesis of BCC. Inactivating mutations also are commonly observed in patched homolog 1, an upstream cell-surface protein that inhibits SMO.7 The mechanism by which vismodegib and sonidegib, 2 of the FDA-approved oral medications for the treatment of advanced BCC, block the SHH pathway is through the selective inhibition of SMO.7,11
Vismodegib first received FDA approval in 2012 for the treatment of laBCC and mBCC after initial results from the pivotal ERIVANCE phase 2 trial demonstrated an objective response rate (ORR) of 43% (27/63) and 30% (10/33) in patients with locally advanced and metastatic disease, respectively. In this single-arm study, all enrolled patients (63 with laBCC and 33 with mBCC) received 150 mg of oral vismodegib daily.12 Updated results at 39 months demonstrated improved ORRs of 60% (38/63) and 48% (16/33) for the laBCC and mBCC groups, respectively. A complete response (CR) and partial response (PR) were observed in 32% (n=20) and 29% (n=18) of patients with laBCC, respectively.13 These results have been confirmed in subsequent studies, including the large international open-label trial known as STEVIE, with ORRs of 68.5% for 1119 cases of laBCC and 37% for 96 cases of mBCC.14-17 The CR and PR rates were 33% and 35%, respectively, for the laBCC group. The CR and PR rates for the mBCC group were 5% and 32%, respectively.14
The FDA approval of sonidegib for laBCC—but not mBCC—occurred in 2015 after the pivotal BOLT randomized phase 2 trial demonstrated an initial ORR of 43% (18/42) for laBCC and 15% (2/13) for mBCC after administration of 200 mg of sonidegib daily.18 A final follow-up analysis at 42 months resulted in ORRs of 56% (37/66) and 8% (1/13) for the laBCC and mBCC groups, respectively.19 Additionally, improved efficacy was not observed in the 151 patients who were randomized to receive treatment with the higher 800-mg dose; however, they did experience a higher incidence of adverse events.18,19
Currently, the true clinical differences between vismodegib and sonidegib remain uncertain, as no head-to-head trials have been conducted. Moreover, direct comparison of the data from the ERIVANCE and BOLT trials is challenging owing to fundamental differences in methodologic design, including the criteria used to assess BCC severity. The ERIVANCE trial utilized the conventional Response Evaluation Criteria in Solid Tumors (RECIST), while BOLT used the rigorous modified RECIST. However, an expert consensus study attempted to compare the 2 trials by modifying the outcomes from BOLT with the former RECIST criteria. The expert group found that the 2 SHH inhibitors had comparable efficacy and adverse event profiles.20 Nevertheless, a recent meta-analysis found that although ORRs for laBCC were similar between the 2 drugs, the CR rate for vismodegib was 31% compared with 3% for sonidegib. Additionally, for mBCC, they reported the ORR of vismodegib to be 2.7 times higher than that of sonidegib (39% vs 15%).21
Immune Checkpoint Inhibitors
Immune checkpoint inhibitors have successfully been utilized in the treatment of cutaneous squamous cell carcinoma (cSCC); however, their use for treating BCC has been limited until recently.22-25 In February 2021, cemiplimab became the first and only ICI approved for the treatment of laBCC and mBCC in patients who did not respond to or were intolerant to prior SHH inhibitor therapy.26 Cemiplimab—a human monoclonal antibody against the PD-1 receptor expressed on T cells—blocks its interaction with programmed cell death ligand 1 and programmed cell death ligand 2 present on tumor cells. The blockade of the PD-1 pathway releases the inhibition of the antitumor immune response and enables appropriate cytotoxic T-cell activity to occur.8
The FDA approval of cemiplimab for the treatment of advanced BCC was based on an open-label, multicenter, single-arm phase 2 trial (NCT03132636) evaluating 84 patients with laBCC refractory or intolerant to SHH inhibitor therapy.26 Patients received an intravenous infusion of cemiplimab 350 mg every 3 weeks for up to 93 weeks or until disease progression or unacceptable toxicity. An ORR of 31% (26/84) was observed with a CR and PR of 6% (5/84) and 25% (21/84), respectively. The median duration of follow-up was 15 months.26 Given the clinically meaningful results of this trial, investigating the efficacy of other PD-1 inhibitors, such as pembrolizumab and nivolumab, for treatment of advanced BCC may prove worthwhile.
Adverse Effects of Systemic Treatments
The 2 approved SHH inhibitors—vismodegib and sonidegib—appear to have similar side-effect profiles, with the most common adverse effects being muscle spasms, dysgeusia, alopecia, nausea, vomiting, diarrhea, weight loss, and fatigue.20,21,27 These side effects occur at high frequencies (>40%) for both SHH inhibitors and often lead to discontinuation of the medication.21 Rates of treatment discontinuation range from 15% to 50% on average.12-14,18 Fortunately, the majority of these adverse effects do not appear to increase in severity or frequency with prolonged use of these medications.14,16,28
Various conservative and pharmacologic measures can be implemented to help manage side effects. For muscle spasms, which are the most commonly reported adverse effect, supplementation with magnesium, transcutaneous electrical nerve stimulation, acupuncture, massages, stretching, and thermal compresses can potentially be beneficial.29 Calcium channel blockers also may be effective, as one small prospective cohort study reported a reduction in the frequency of muscle cramps with amlodipine 10 mg daily.30 For alopecia, which typically is reversible and caused by SHH inhibition of the normal hair cycle, minoxidil theoretically can help, as it reduces telogen arrest and extends the anagen growth phase.31,32 Although usually mild and self-limiting, management of dysgeusia, weight loss, and gastrointestinal upset often can be managed with dietary changes, such as smaller, more frequent meals.33,34 Finally, alternative dosing strategies and drug holidays have been employed to mitigate these side effects and increase drug tolerability.35,36 These are discussed in the Alternative Dosing section.
Given the essential role of the SHH pathway in embryologic development, SHH inhibitors carry a black box warning of embryofetal teratogenicity and are contraindicated in females who are pregnant or breastfeeding. For females of reproductive potential, verification of pregnancy status should be performed prior to initiating treatment with an SHH inhibitor. These patients should be counseled on the use of contraception during treatment and for at least 24 months and 20 months after cessation of vismodegib and sonidegib, respectively.27,37,38 Male patients, even after a vasectomy, should use barrier contraception during treatment and for at least 3 months and 8 months after the final dose of vismodegib and sonidegib, respectively.37,38
Laboratory abnormalities commonly associated with SHH inhibitors include elevated hepatic enzymes, particularly with vismodegib, and elevated creatine kinase levels, particularly with sonidegib.28,39 Other laboratory abnormalities that can occur include hypercholesterolemia, hypercreatininemia, hyperglycemia, and increased serum lipase levels.19,28 Although these laboratory abnormalities usually are asymptomatic and self-limiting, regular monitoring should be performed.
There also is concern that SHH inhibitors may induce the development of cSCC. A case-control study of 55 cases and 125 control patients found an increased risk for cSCC in those previously treated with vismodegib, with a hazard ratio of 8.12.40 However, a subsequent retrospective cohort study of 1675 patients with BCC failed to find any association with cSCC among those treated with vismodegib compared to those who received standard surgical therapy.41 Clinical data for sonidegib are lacking, but the BOLT trial found that cSCC occurred in 3 patients receiving treatment with the SHH inhibitor.18 Thus, further studies are needed to more thoroughly assess this concern. Close monitoring for cSCC may be warranted in patients prescribed SHH inhibitors at this time.
Cemiplimab has demonstrated an acceptable safety profile and is generally well tolerated. In the phase 2 trial of cemiplimab for cSCC, approximately 5% of patients discontinued treatment because of adverse effects. The most commonly reported side effects of cemiplimab were diarrhea (27%), fatigue (24%), nausea (17%), constipation (15%), and rash (15%).23 In the phase 2 trial for laBCC, grade 3 or 4 adverse events occurred in 48% of patients, with hypertension (5%) being the most common.26 Although rare, immune-mediated adverse reactions also can occur, given the mechanism of action of ICIs. These side effects, ranging in severity from mild to fatal, include pneumonitis, colitis, hepatitis, nephritis, myocarditis, and hypophysitis. Therefore, close monitoring for these immune-mediated reactions is critical, but most can be managed with corticosteroids or treatment interruption if they occur.42,43
No absolute contraindications exist for cemiplimab; however, extreme caution should be taken in immunosuppressed individuals, such as solid organ transplant recipients and those with chronic lymphocytic leukemia (CLL), as safety data are limited in these patients.44,45 Although small retrospective studies have reported reasonable tolerability in solid organ transplant recipients treated with ICIs, an allograft rejection rate of 41% was found in a meta-analysis of 64 patients.46-48 In CLL patients with keratinocyte carcinomas, ICIs have been safely used and have even demonstrated efficacy for CLL in some cases.49-52
Alternative Dosing
The side effects of SHH inhibitors have led to alternative dosing strategies to prevent medication discontinuation and improve adherence. In patients with basal cell nevus syndrome, multimonth drug holidays have been shown to increase drug tolerability without compromising efficacy.35,36 Weekly intermittent dosing regimens of vismodegib ranging from 1 week on followed by 1 to 3 weeks off demonstrated efficacy in a retrospective study of 7 patients with advanced BCC.53 All 7 patients experienced improvement in their BCCs, with 3 patients experiencing CR. Importantly, treatment-related adverse effects were mild and well tolerated, with no patients terminating the medication.53 Two other retrospective case series of patients with advanced BCC treated with vismodegib reported similar findings for those placed on an intermittent dosing schedule ranging from once every other day to once per week.54,55
In the large phase 2 randomized trial known as MIKIE, 2 different intermittent dosing regimens of 150 mg vismodegib daily for patients with multiple BCCs were found to have good activity and tolerability.56 The first group (n=116) received vismodegib for 12 weeks, then 3 rounds of 8 weeks of placebo, followed by 12 weeks of vismodegib; there was a 63% reduction in clinically evident BCCs after 73 weeks. The second group (n=113) received the medication for 24 weeks, then 3 rounds of 8 weeks of placebo, followed by 8 weeks of vismodegib; there was a 54% reduction at the end of 73 weeks.56 Subsequent analyses found improvements in health-related quality-of-life outcomes that were similar for both groups.57
Consequently, alternative dosing schedules appear to be a viable option for patients at risk of discontinuing treatment because of adverse effects, and current data support the recently approved recommendations of dose interruptions of up to 8 weeks to manage adverse effects in patients with laBCC or mBCC.58 Nevertheless, further clinical studies are required to determine the optimal intermittent dosing regimen for patients treated with SHH inhibitors.
Neoadjuvant Administration
Recently, vismodegib has been studied as a neoadjuvant therapy for BCC with promising results. Several small retrospective studies and case reports have documented successful treatment of both operable and inoperable periocular laBCC, with preservation of the eye in all patients.59-61 An open-label trial of 15 patients with advanced BCC who received neoadjuvant vismodegib for 3 to 6 months prior to surgical excision reported a mean reduction of 35% in the final surgical defect size, with no recurrence at 22 months.62,63 The latest and largest study performed was a phase 2 open-label trial known as VISMONEO, where 44 of 55 laBCC patients (80%) receiving neoadjuvant vismodegib for a mean duration of 6 months (range, 4–10 months) achieved the primary end point of tumor surgical downstaging.64 Of the 44 patients who had tumor downstaging, 27 (61%) experienced histologically proven CRs. Additionally, a 66% reduction in the average target lesion size was reported in this group compared to29% in the 11 patients who did not have tumor downstaging (P=.0002).64 Thus, SHH inhibitors may hold an important neoadjuvant role in the treatment of BCC by decreasing surgical defect size and allowing for surgical management of previously inoperable cases.
Synergism With Radiation
Preliminary data suggest SHH inhibitors may help potentiate the effects of radiation therapy for the treatment of BCC. Currently, the evidence primarily is limited to case studies, with several reports describing complete remission in patients with advanced BCCs who were considered unsuitable candidates for surgery. In these cases, vismodegib was administered either prior to or concurrently with radiation treatment.65-69 An in vitro study also documented the radiation-sensitizing effects of vismodegib in a BCC cell line.70 Recently, a phase 2 trial (ClinicalTrials.gov identifier NCT01835626) evaluating the concurrent use of vismodegib and radiotherapy for patients with advanced BCC was completed, but data has yet to be published.
Synergism With and Benefit of Antifungal Therapy
The antifungal drug itraconazole is a potent inhibitor of the SHH pathway and may have an adjunctive role in the treatment of BCC. Similar to vismodegib and sonidegib, itraconazole acts as a direct antagonist of SMO. However, it is thought to bind to a distinct site on SMO.71,72 An open-label, exploratory phase 2 trial of 19 patients with BCC found that oral itraconazole 200 to 400 mg daily decreased tumor proliferative index by 45% (P=.04), as measured by Ki-67; SHH activity by 65% (P=.03), as measured by GLI1 messenger RNA; and mean tumor area by 24%.73 In a case series of 5 patients with mBCC refractory to conventional SHH inhibitor therapy, combined treatment with itraconazole and arsenic trioxide resulted in stable disease and a 75% reduction in SHH activity (P<.001).74 One case report documented tumor regression leading to stable disease for 15 months in a patient with laBCC treated with itraconazole monotherapy due to being unable to afford vismodegib or sonidegib. However, within 2 months of treatment discontinuation, the lesion progressed considerably.75 The efficacy of a topical formulation of itraconazole also has been tested in an open-label, placebo-controlled phase 2 trial, but no benefit was observed.76
Posaconazole is a second-generation antifungal agent that may serve as a potential alternative to itraconazole.77 Although clinical data are lacking, a basic science study found that posaconazole could inhibit the growth of SHH-dependent BCC in vivo (in mice).78 Furthermore, posaconazole has demonstrated a better safety profile with fewer and more mild side effects than itraconazole and does not require dose adjustment for those with hepatic or renal failure.79,80 Thus, posaconazole may be a safer alternative to itraconazole for the treatment of BCC. Further clinical studies are needed to elucidate the potential synergistic effects of these antifungal agents with the 2 currently approved SHH inhibitors for the treatment of advanced BCC.
Drug Resistance
Treatment resistance to SHH inhibitors, though uncommon, is a growing concern. Acquired mutations in the SMO binding site or downstream mediators of the SHH pathway have been shown to confer resistance to vismodegib and sonidegib.72,81-83 In addition, it appears that there may be shared resistance among the drugs in this class. One study assessing the efficacy of sonidegib in 9 patients with laBCC resistant to vismodegib found that these patients also did not respond to sonidegib.84 Interestingly, 1 case report documented tumor regression of an intracranial BCC in a patient treated with sonidegib and itraconazole after failure with vismodegib.85 An in vitro study also found that itraconazole maintained SHH inhibitory activity for all drug-resistant SMO mutations that have been reported.72 Therefore, itraconazole monotherapy or combination therapy with a canonical SHH inhibitor may be considered for patients with recalcitrant BCC and warrants further investigation.
Taladegib is a newly developed SMO inhibitor that may serve as another promising alternative for patients who develop resistance to vismodegib or sonidegib. A phase 1 trial of taladegib for advanced BCC found an ORR of 69% (11/16) in the SHH inhibitor–naïve group and an ORR of 36% (11/32) in the group previously treated with a SHH inhibitor.86 Additionally, the safety profile and frequency of adverse effects appear to be similar to those associated with vismodegib and sonidegib.86,87 Unfortunately, no clinical trials evaluating taladegib for BCC are ongoing or in development at this time.
Recurrence
There appears to be a relatively high rate of recurrence for BCC patients who achieve a CR to SHH inhibitors. In a retrospective study of 116 laBCC patients who experienced a CR after vismodegib therapy, 54 patients (47%) relapsed at 36 months. Among the 54 patients that relapsed, 27 were re-treated with vismodegib, which resulted in an ORR of 85% (23/27), a CR rate of 37% (10/27), and a PR rate of 48% (13/27).88 Another retrospective study of 35 laBCC patients who relapsed after vismodegib treatment reported a 31% (11/35) clinical recurrence rate at 6-month follow-up.89 An observational retrospective study also assessed the efficacy of SHH inhibitor maintenance therapy for advanced BCC patients who achieved a CR.90 In the study, 27 (64%) patients received a maintenance dose of 150 mg vismodegib once per week for 1 year, while 15 (36%) patients decided not to take a maintenance dose following CR of their BCC. All patients who took the maintenance therapy did not experience clinical recurrence at 1-year follow-up, whereas 26% of patients not on the maintenance dose relapsed.90 Consequently, these results indicate that BCC recurrence is frequent after SHH inhibitor therapy and highlights the importance of close surveillance after CR is attained. Nevertheless, most patients still respond to treatment with SHH inhibitors after relapsing, and intermittent maintenance doses may be an effective means to reduce risk of recurrence.
Conclusion
Vismodegib and sonidegib are SHH inhibitors approved for the treatment of laBCC and mBCC. Cemiplimab is now also approved for patients who do not respond to SHH inhibitors or for whom SHH inhibitors are not tolerable. Although these systemic targeted therapies can lead to notable tumor shrinkage and even complete regression in some patients, recurrence is common, and adverse effects may limit their use. Drug resistance is an emerging issue that requires additional examination. Further clinical studies are needed to determine which patients are likely to respond to these targeted treatments.
Various intermittent and maintenance drug regimens should be evaluated for their potential to mitigate adverse effects and reduce risk of recurrence, respectively. The synergistic effects of these medications with other therapies as well as their neoadjuvant and adjuvant roles should be further investigated. For example, administration of an SHH inhibitor prior to surgical excision of a BCC may allow for a smaller surgical defect size, thereby improving cosmetic and functional outcomes. Moreover, these systemic targeted medications may allow for previously inoperable tumors to become amenable to surgical treatment.
Although SHH inhibitors and PD-1 inhibitors represent a major advancement in the field of oncodermatology, real-world efficacy and safety data in the upcoming years will be important for elucidating their true benefit for patients with BCC.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the U.S. population, 2012. JAMA Dermatol. 2015;151:1081-1086.
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: epidemiology; pathophysiology; clinical and histological subtypes; and disease associations. J Am Acad Dermatol. 2019;80:303-317.
- Rees JR, Zens MS, Celaya MO, et al. Survival after squamous cell and basal cell carcinoma of the skin: a retrospective cohort analysis. Int J Cancer. 2015;137:878-884.
- Kasumagic-Halilovic E, Hasic M, Ovcina-Kurtovic N. A clinical study of basal cell carcinoma. Med Arch. 2019;73:394-398.
- Goldenberg G, Karagiannis T, Palmer JB, et al. Incidence and prevalence of basal cell carcinoma (BCC) and locally advanced BCC (LABCC) in a large commercially insured population in the United States: a retrospective cohort study. J Am Acad Dermatol. 2016;75:957.e952-966.e952.
- Laga AC, Schaefer IM, Sholl LM, et al. Metastatic basal cell carcinoma. Am J Clin Pathol. 2019;152:706-717.
- Rimkus TK, Carpenter RL, Qasem S, et al. Targeting the sonic hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancers (Basel). 2016;8:22.
- Burova E, Hermann A, Waite J, et al. Characterization of the anti–PD-1 antibody REGN2810 and its antitumor activity in human PD-1 knock-in mice. Mol Cancer Ther. 2017;16:861-870.
- Bhardwaj G, Murdoch B, Wu D, et al. Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nat Immunol. 2001;2:172-180.
- Paladini RD, Saleh J, Qian C, et al. Modulation of hair growth with small molecule agonists of the hedgehog signaling pathway. J Invest Dermatol. 2005;125:638-646.
- Von Hoff DD, LoRusso PM, Rudin CM, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361:1164-1172.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Sekulic A, Migden MR, Basset-Seguin N, et al. Long-term safety and efficacy of vismodegib in patients with advanced basal cell carcinoma: final update of the pivotal ERIVANCE BCC study. BMC Cancer. 2017;17:332.
- Basset-Séguin N, Hauschild A, Kunstfeld R, et al. Vismodegib in patients with advanced basal cell carcinoma: primary analysis of STEVIE, an international, open-label trial. Eur J Cancer. 2017;86:334-348.
- Cozzani R, Del Aguila R, Carrizo M, et al. Efficacy and safety profile of vismodegib in a real-world setting cohort of patients with advanced basal cell carcinoma in Argentina. Int J Dermatol. 2020;59:627-632.
- Spallone G, Sollena P, Ventura A, et al. Efficacy and safety of vismodegib treatment in patients with advanced basal cell carcinoma and multiple comorbidities. Dermatol Ther. 2019;32:E13108.
- Fosko SW, Chu MB, Armbrecht E, et al. Efficacy, rate of tumor response, and safety of a short course (12-24 weeks) of oral vismodegib in various histologic subtypes (infiltrative, nodular, and superficial) of high-risk or locally advanced basal cell carcinoma, in an open-label, prospective case series clinical trial. J Am Acad Dermatol. 2020;82:946-954.
- Migden MR, Guminski A, Gutzmer R, et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): a multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 2015;16:716-728.
- Dummer R, Guminksi A, Gutzmer R, et al. Long-term efficacy and safety of sonidegib in patients with advanced basal cell carcinoma: 42-month analysis of the phase II randomized, double-blind BOLT study. Br J Dermatol. 2020;182:1369-1378.
- Dummer R, Ascierto PA, Basset-Seguin N, et al. Sonidegib and vismodegib in the treatment of patients with locally advanced basal cell carcinoma: a joint expert opinion. J Eur Acad Dermatol Venereol. 2020;34:1944-1956.
- Xie P, Lefrançois P. Efficacy, safety, and comparison of sonic hedgehog inhibitors in basal cell carcinomas: a systematic review and meta-analysis. J Am Acad Dermatol. 2018;79:1089-1100.e1017.
- Gentzler R, Hall R, Kunk PR, et al. Beyond melanoma: inhibiting the PD-1/PD-L1 pathway in solid tumors. Immunotherapy. 2016;8:583-600.
- Guminski AD, Lim AML, Khushalani NI, et al. Phase 2 study of cemiplimab, a human monoclonal anti-PD-1, in patients (pts) with metastatic cutaneous squamous cell carcinoma (mCSCC; group 1): 12-month follow-up [abstract]. J Clin Oncol. 2019;37(15 suppl):9526.
- Grob JJ, Gonzalez Mendoza R, Basset-Seguin N, et al. Pembrolizumab for recurrent/metastatic cutaneous squamous cell carcinoma (cSCC): efficacy and safety results from the phase II KEYNOTE-629 study [abstract]. Ann Oncol. 2019;30 (suppl 5):v908.
- Maubec E, Boubaya M, Petrow P, et al. Pembrolizumab as first-line therapy in patients with unresectable cutaneous squamous cell carcinoma (cSCC): phase 2 results from CARSKIN [abstract]. J Clin Oncol. 2019;37(15 suppl):9547.
- Stratigos AJ, Sekulic A, Peris K, et al. Cemiplimab in locally advanced basal cell carcinoma after hedgehog inhibitor therapy: an open-label, multi-centre, single-arm, phase 2 trial. Lancet Oncol. 2021;22:848-857.
- Carpenter RL, Ray H. Safety and tolerability of sonic hedgehog pathway inhibitors in cancer. Drug Saf. 2019;42:263-279.
- Villani A, Fabbrocini G, Costa C, et al. Sonidegib: safety and efficacy in treatment of advanced basal cell carcinoma. Dermatol Ther (Heidelb). 2020;10:401-412.
- Wright A, Sluka KA. Nonpharmacological treatments for musculoskeletal pain. Clin J Pain. 2001;17:33-46.
- Ally MS, Tang JY, Lindgren J, et al. Effect of calcium channel blockade on vismodegib-induced muscle cramps. JAMA Dermatol. 2015;151:1132-1134.
- Yang X, Thai K-E. Treatment of permanent chemotherapy-induced alopecia with low dose oral minoxidil. Australas J Dermatol. 2016;57:e130-e132.
- Ferguson JS, Hannam S, Toholka R, et al. Hair loss and hedgehog inhibitors: a class effect? Br J Dermatol. 2015;173:262-264.
- Kumbargere Nagraj S, George RP, Shetty N, et al. Interventions for managing taste disturbances. Cochrane Database Syst Rev. 2017;12:CD010470.
- Jacobsen AA, Kydd AR, Strasswimmer J. Practical management of the adverse effects of hedgehog pathway inhibitor therapy for basal cell carcinoma. J Am Acad Dermatol. 2017;76:767-768.
- Ally MS, Tang JY, Joseph T, et al. The use of vismodegib to shrink keratocystic odontogenic tumors in patients with basal cell nevus syndrome. JAMA Dermatol. 2014;150:542-545.
- Yang X, Dinehart SM. Intermittent vismodegib therapy in basal cell nevus syndrome. JAMA Dermatol. 2016;152:223-224.
- Erivedge. Prescribing information. Genentech; 2015.
- Odomzo. Prescribing information. Novartis; 2015.
- Ventarola DJ, Silverstein DI. Vismodegib-associated hepatotoxicity: a potential side effect detected in postmarketing surveillance. J Am Acad Dermatol. 2014;71:397-398.
- Mohan SV, Chang J, Li S, et al. Increased risk of cutaneous squamous cell carcinoma after vismodegib therapy for basal cell carcinoma. JAMA Dermatol. 2016;152:527-532.
- Bhutani T, Abrouk M, Sima CS, et al. Risk of cutaneous squamous cell carcinoma after treatment of basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2017;77:713-718.
- Morgado M, Plácido A, Morgado S, et al. Management of the adverse effects of immune checkpoint inhibitors. Vaccines (Basel). 2020;8:575.
- Martins F, Sofiya L, Sykiotis GP, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16:563-580.
- Ntsethe A, Dludla PV, Nyambuya TM, et al. The impact of immune checkpoint inhibitors in patients with chronic lymphocytic leukemia (CLL): a protocol for a systematic review and meta-analysis of randomized controlled trials. Medicine (Baltimore). 2020;99:E21167.
- Johnson DB, Sullivan RJ, Menzies AM. Immune checkpoint inhibitors in challenging populations. Cancer. 2017;123:1904-1911.
- Tsung I, Worden FP, Fontana RJ. Safety and efficacy of checkpoint inhibitors in solid organ transplant recipients with cutaneous squamous cell carcinoma [abstract]. J Clin Oncol. 2020;38(15 suppl):E22014.
- Owoyemi I, Vaughan LE, Costello CM, et al. Clinical outcomes of solid organ transplant recipients with metastatic cancers who are treated with immune checkpoint inhibitors: a single-center analysis. Cancer. 2020;126:4780-4787.
- Kumar V, Shinagare AB, Rennke HG, et al. The safety and efficacy of checkpoint inhibitors in transplant recipients: a case series and systematic review of literature. Oncologist. 2020;25:505-514.
- Arenbergerova M, Fialova A, Arenberger P, et al. Killing two birds with one stone: response to pembrolizumab in a patient with metastatic melanoma and B-cell chronic lymphocytic leukaemia. J Eur Acad Dermatol Venereol. 2018;32:E72-E74.
- Archibald WJ, Meacham PJ, Williams AM, et al. Management of melanoma in patients with chronic lymphocytic leukemia. Leuk Res. 2018;71:43-46.
- Ding W, LaPlant BR, Call TG, et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood. 2017;129:3419-3427.
- Leiter U, Loquai C, Reinhardt L, et al. Immune checkpoint inhibition therapy for advanced skin cancer in patients with concomitant hematological malignancy: a retrospective multicenter DeCOG study of 84 patients. J Immunother Cancer. 2020;8:E000897.
- Becker LR, Aakhus AE, Reich HC, et al. A novel alternate dosing of vismodegib for treatment of patients with advanced basal cell carcinomas. JAMA Dermatol. 2017;153:321-322.
- Woltsche N, Pichler N, Wolf I, et al. Managing adverse effects by dose reduction during routine treatment of locally advanced basal cell carcinoma with the hedgehog inhibitor vismodegib: a single centre experience. J Eur Acad Dermatol Venereol. 2019;33:E144-E145.
- Wong C, Poblete-Lopez C, Vidimos A. Comparison of daily dosing versus Monday through Friday dosing of vismodegib for locally advanced basal cell carcinoma and basal cell nevus syndrome: a retrospective case series. J Am Acad Dermatol. 2020;82:1539-1542.
- Dréno B, Kunstfeld R, Hauschild A, et al. Two intermittent vismodegib dosing regimens in patients with multiple basal-cell carcinomas (MIKIE): a randomised, regimen-controlled, double-blind, phase 2 trial. Lancet Oncol. 2017;18:404-412.
- Schadendorf D, Hauschild A, Fosko S, et al. Quality-of-life analysis with intermittent vismodegib regimens in patients with multiple basal cell carcinomas: patient-reported outcomes from the MIKIE study. J Eur Acad Dermatol Venereol. 2020;34:E526-E529.
- Chanu P, Musib L, Wang X, et al. Vismodegib efficacy in advanced basal cell carcinoma maintained with 8-week dose interruptions: a model-based evaluation. J Invest Dermatol. 2021;141:930-933.
- Su MG, Potts LB, Tsai JH. Treatment of periocular basal cell carcinoma with neoadjuvant vismodegib. Am J Ophthalmol Case Rep. 2020;19:100755.
- González AR, Etchichury D, Gil ME, et al. Neoadjuvant vismodegib and Mohs micrographic surgery for locally advanced periocular basal cell carcinoma. Ophthalmic Plast Reconstr Surg. 2019;35:56-61.
- Sagiv O, Nagarajan P, Ferrarotto R, et al. Ocular preservation with neoadjuvant vismodegib in patients with locally advanced periocular basal cell carcinoma. Br J Ophthalmol. 2019;103:775-780.
- Ally MS, Aasi S, Wysong A, et al. An investigator-initiated open-label clinical trial of vismodegib as a neoadjuvant to surgery for high-risk basal cell carcinoma. J Am Acad Dermatol. 2014;71:904-911.e1.
- Kwon GP, Ally MS, Bailey-Healy I, et al. Update to an open-label clinical trial of vismodegib as neoadjuvant before surgery for high-risk basal cell carcinoma (BCC). J Am Acad Dermatol. 2016;75:213-215.
- Mortier L, Bertrand N, Basset-Seguin N, et al. Vismodegib in neoadjuvant treatment of locally advanced basal cell carcinoma: first results of a multicenter, open-label, phase 2 trial (VISMONEO study) [abstract]. J Clin Oncol. 2018;36(15 suppl):9509.
- Strasswimmer JM. Potential synergy of radiation therapy with vismodegib for basal cell carcinoma. JAMA Dermatol. 2015;151:925-926.
- Gathings RM, Orscheln CS, Huang WW. Compassionate use of vismodegib and adjuvant radiotherapy in the treatment of multiple locally advanced and inoperable basal cell carcinomas and squamous cell carcinomas of the skin. J Am Acad Dermatol. 2014;70:E88-E89.
- Franco AI, Eastwick G, Farah R, et al. Upfront radiotherapy with concurrent and adjuvant vismodegib is effective and well-tolerated in a patient with advanced, multifocal basal cell carcinoma. Case Rep Dermatol Med. 2018;2018:2354146.
- Pollom EL, Bui TT, Chang AL, et al. Concurrent vismodegib and radiotherapy for recurrent, advanced basal cell carcinoma. JAMA Dermatol. 2015;151:998-1001.
- Janela-Lapert R, Dubray B, Duval-Modeste A, et al. Treatment of advanced basal cell carcinoma with vismodegib followed by radiotherapy [in French]. Ann Dermatol Venereol. 2020;147:780-782.
- Hehlgans S, Booms P, Güllülü Ö, et al. Radiation sensitization of basal cell and head and neck squamous cell carcinoma by the hedgehog pathway inhibitor vismodegib. Int J Mol Sci. 2018;19:2485.
- Kim J, Tang JY, Gong R, et al. Itraconazole, a commonly used antifungal that inhibits hedgehog pathway activity and cancer growth. Cancer Cell. 2010;17:388-399.
- Kim J, Aftab BT, Tang JY, et al. Itraconazole and arsenic trioxide inhibit hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell. 2013;23:23-34.
- Kim DJ, Kim J, Spaunhurst K, et al. Open-label, exploratory phase II trial of oral itraconazole for the treatment of basal cell carcinoma. J Clin Oncol. 2014;32:745-751.
- Ally MS, Ransohoff K, Sarin K, et al. Effects of combined treatment with arsenic trioxide and itraconazole in patients with refractory metastatic basal cell carcinoma. JAMA Dermatol. 2016;152:452-456.
- Cia˛z˙yn´yska M, Narbutt J, Skibin´ska M, et al. Itraconazole—a new player in the therapy of advanced basal cell carcinoma: a case report. JCO Oncol Pract. 2020;16:837-838.
- Sohn GK, Kwon GP, Bailey-Healy I, et al. Topical itraconazole for the treatment of basal cell carcinoma in patients with basal cell nevus syndrome or high-frequency basal cell carcinomas: a phase 2, open-label, placebo-controlled trial. JAMA Dermatol. 2019;155:1078-1080.
- Lass-Flörl C. Triazole antifungal agents in invasive fungal infections: a comparative review. Drugs. 2011;71:2405-2419.
- Chen B, Trang V, Lee A, et al. Posaconazole, a second-generation triazole antifungal drug, inhibits the hedgehog signaling pathway and progression of basal cell carcinoma. Mol Cancer Ther. 2016;15:866-876.
- Katragkou A, Tsikopoulou F, Roilides E, et al. Posaconazole: when and how? the clinician’s view. Mycoses. 2012;55:110-122.
- Raad II, Graybill JR, Bustamante AB, et al. Safety of long-term oral posaconazole use in the treatment of refractory invasive fungal infections. Clin Infect Dis. 2006;42:1726-1734.
- Atwood SX, Sarin KY, Whitson RJ, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342-353.
- Sun Q, Atzmony L, Zaki T, et al. Clues to primary vismodegib resistance lie in histology and genetics. J Clin Pathol. 2020;73:678-680.
- Verkouteren BJA, Wakkee M, van Geel M, et al. Molecular testing in metastatic basal cell carcinoma. J Am Acad Dermatol. 2021;85:1135-1142.
- Danial C, Sarin KY, Oro AE, et al. An investigator-initiated open-label trial of sonidegib in advanced basal cell carcinoma patients resistant to vismodegib. Clin Cancer Res. 2016;22:1325-1329.
- Yoon J, Apicelli AJ 3rd, Pavlopoulos TV. Intracranial regression of an advanced basal cell carcinoma using sonidegib and itraconazole after failure with vismodegib. JAAD Case Rep. 2017;4:10-12.
- Bendell J, Andre V, Ho A, et al. Phase I study of LY2940680, a Smo antagonist, in patients with advanced cancer including treatment-naïve and previously treated basal cell carcinoma. Clin Cancer Res. 2018;24:2082-2091.
- Ueno H, Kondo S, Yoshikawa S, et al. A phase I and pharmacokinetic study of taladegib, a Smoothened inhibitor, in Japanese patients with advanced solid tumors. Invest New Drugs. 2018;36:647-656.
- Herms F, Lambert J, Grob JJ, et al. Follow-up of patients with complete remission of locally advanced basal cell carcinoma after vismodegib discontinuation: a multicenter French study of 116 patients. J Clin Oncol. 2019;37:3275-3282.
- Villani A, Megna M, Fabbrocini G, et al. Long-term efficacy of vismodegib after its withdrawal and patients’ health-related quality of life using the Dermatology Life Quality Index (DLQI). Dermatol Ther (Heidelb). 2019;9:719-724.
- Scalvenzi M, Cappello M, Costa C, et al. Low-dose vismodegib as maintenance therapy after locally advanced basal cell carcinoma complete remission: high efficacy with minimal toxicity. Dermatol Ther (Heidelb). 2020;10:465-468.
- Rogers HW, Weinstock MA, Feldman SR, et al. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the U.S. population, 2012. JAMA Dermatol. 2015;151:1081-1086.
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: epidemiology; pathophysiology; clinical and histological subtypes; and disease associations. J Am Acad Dermatol. 2019;80:303-317.
- Rees JR, Zens MS, Celaya MO, et al. Survival after squamous cell and basal cell carcinoma of the skin: a retrospective cohort analysis. Int J Cancer. 2015;137:878-884.
- Kasumagic-Halilovic E, Hasic M, Ovcina-Kurtovic N. A clinical study of basal cell carcinoma. Med Arch. 2019;73:394-398.
- Goldenberg G, Karagiannis T, Palmer JB, et al. Incidence and prevalence of basal cell carcinoma (BCC) and locally advanced BCC (LABCC) in a large commercially insured population in the United States: a retrospective cohort study. J Am Acad Dermatol. 2016;75:957.e952-966.e952.
- Laga AC, Schaefer IM, Sholl LM, et al. Metastatic basal cell carcinoma. Am J Clin Pathol. 2019;152:706-717.
- Rimkus TK, Carpenter RL, Qasem S, et al. Targeting the sonic hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancers (Basel). 2016;8:22.
- Burova E, Hermann A, Waite J, et al. Characterization of the anti–PD-1 antibody REGN2810 and its antitumor activity in human PD-1 knock-in mice. Mol Cancer Ther. 2017;16:861-870.
- Bhardwaj G, Murdoch B, Wu D, et al. Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nat Immunol. 2001;2:172-180.
- Paladini RD, Saleh J, Qian C, et al. Modulation of hair growth with small molecule agonists of the hedgehog signaling pathway. J Invest Dermatol. 2005;125:638-646.
- Von Hoff DD, LoRusso PM, Rudin CM, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361:1164-1172.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Sekulic A, Migden MR, Basset-Seguin N, et al. Long-term safety and efficacy of vismodegib in patients with advanced basal cell carcinoma: final update of the pivotal ERIVANCE BCC study. BMC Cancer. 2017;17:332.
- Basset-Séguin N, Hauschild A, Kunstfeld R, et al. Vismodegib in patients with advanced basal cell carcinoma: primary analysis of STEVIE, an international, open-label trial. Eur J Cancer. 2017;86:334-348.
- Cozzani R, Del Aguila R, Carrizo M, et al. Efficacy and safety profile of vismodegib in a real-world setting cohort of patients with advanced basal cell carcinoma in Argentina. Int J Dermatol. 2020;59:627-632.
- Spallone G, Sollena P, Ventura A, et al. Efficacy and safety of vismodegib treatment in patients with advanced basal cell carcinoma and multiple comorbidities. Dermatol Ther. 2019;32:E13108.
- Fosko SW, Chu MB, Armbrecht E, et al. Efficacy, rate of tumor response, and safety of a short course (12-24 weeks) of oral vismodegib in various histologic subtypes (infiltrative, nodular, and superficial) of high-risk or locally advanced basal cell carcinoma, in an open-label, prospective case series clinical trial. J Am Acad Dermatol. 2020;82:946-954.
- Migden MR, Guminski A, Gutzmer R, et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): a multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 2015;16:716-728.
- Dummer R, Guminksi A, Gutzmer R, et al. Long-term efficacy and safety of sonidegib in patients with advanced basal cell carcinoma: 42-month analysis of the phase II randomized, double-blind BOLT study. Br J Dermatol. 2020;182:1369-1378.
- Dummer R, Ascierto PA, Basset-Seguin N, et al. Sonidegib and vismodegib in the treatment of patients with locally advanced basal cell carcinoma: a joint expert opinion. J Eur Acad Dermatol Venereol. 2020;34:1944-1956.
- Xie P, Lefrançois P. Efficacy, safety, and comparison of sonic hedgehog inhibitors in basal cell carcinomas: a systematic review and meta-analysis. J Am Acad Dermatol. 2018;79:1089-1100.e1017.
- Gentzler R, Hall R, Kunk PR, et al. Beyond melanoma: inhibiting the PD-1/PD-L1 pathway in solid tumors. Immunotherapy. 2016;8:583-600.
- Guminski AD, Lim AML, Khushalani NI, et al. Phase 2 study of cemiplimab, a human monoclonal anti-PD-1, in patients (pts) with metastatic cutaneous squamous cell carcinoma (mCSCC; group 1): 12-month follow-up [abstract]. J Clin Oncol. 2019;37(15 suppl):9526.
- Grob JJ, Gonzalez Mendoza R, Basset-Seguin N, et al. Pembrolizumab for recurrent/metastatic cutaneous squamous cell carcinoma (cSCC): efficacy and safety results from the phase II KEYNOTE-629 study [abstract]. Ann Oncol. 2019;30 (suppl 5):v908.
- Maubec E, Boubaya M, Petrow P, et al. Pembrolizumab as first-line therapy in patients with unresectable cutaneous squamous cell carcinoma (cSCC): phase 2 results from CARSKIN [abstract]. J Clin Oncol. 2019;37(15 suppl):9547.
- Stratigos AJ, Sekulic A, Peris K, et al. Cemiplimab in locally advanced basal cell carcinoma after hedgehog inhibitor therapy: an open-label, multi-centre, single-arm, phase 2 trial. Lancet Oncol. 2021;22:848-857.
- Carpenter RL, Ray H. Safety and tolerability of sonic hedgehog pathway inhibitors in cancer. Drug Saf. 2019;42:263-279.
- Villani A, Fabbrocini G, Costa C, et al. Sonidegib: safety and efficacy in treatment of advanced basal cell carcinoma. Dermatol Ther (Heidelb). 2020;10:401-412.
- Wright A, Sluka KA. Nonpharmacological treatments for musculoskeletal pain. Clin J Pain. 2001;17:33-46.
- Ally MS, Tang JY, Lindgren J, et al. Effect of calcium channel blockade on vismodegib-induced muscle cramps. JAMA Dermatol. 2015;151:1132-1134.
- Yang X, Thai K-E. Treatment of permanent chemotherapy-induced alopecia with low dose oral minoxidil. Australas J Dermatol. 2016;57:e130-e132.
- Ferguson JS, Hannam S, Toholka R, et al. Hair loss and hedgehog inhibitors: a class effect? Br J Dermatol. 2015;173:262-264.
- Kumbargere Nagraj S, George RP, Shetty N, et al. Interventions for managing taste disturbances. Cochrane Database Syst Rev. 2017;12:CD010470.
- Jacobsen AA, Kydd AR, Strasswimmer J. Practical management of the adverse effects of hedgehog pathway inhibitor therapy for basal cell carcinoma. J Am Acad Dermatol. 2017;76:767-768.
- Ally MS, Tang JY, Joseph T, et al. The use of vismodegib to shrink keratocystic odontogenic tumors in patients with basal cell nevus syndrome. JAMA Dermatol. 2014;150:542-545.
- Yang X, Dinehart SM. Intermittent vismodegib therapy in basal cell nevus syndrome. JAMA Dermatol. 2016;152:223-224.
- Erivedge. Prescribing information. Genentech; 2015.
- Odomzo. Prescribing information. Novartis; 2015.
- Ventarola DJ, Silverstein DI. Vismodegib-associated hepatotoxicity: a potential side effect detected in postmarketing surveillance. J Am Acad Dermatol. 2014;71:397-398.
- Mohan SV, Chang J, Li S, et al. Increased risk of cutaneous squamous cell carcinoma after vismodegib therapy for basal cell carcinoma. JAMA Dermatol. 2016;152:527-532.
- Bhutani T, Abrouk M, Sima CS, et al. Risk of cutaneous squamous cell carcinoma after treatment of basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2017;77:713-718.
- Morgado M, Plácido A, Morgado S, et al. Management of the adverse effects of immune checkpoint inhibitors. Vaccines (Basel). 2020;8:575.
- Martins F, Sofiya L, Sykiotis GP, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16:563-580.
- Ntsethe A, Dludla PV, Nyambuya TM, et al. The impact of immune checkpoint inhibitors in patients with chronic lymphocytic leukemia (CLL): a protocol for a systematic review and meta-analysis of randomized controlled trials. Medicine (Baltimore). 2020;99:E21167.
- Johnson DB, Sullivan RJ, Menzies AM. Immune checkpoint inhibitors in challenging populations. Cancer. 2017;123:1904-1911.
- Tsung I, Worden FP, Fontana RJ. Safety and efficacy of checkpoint inhibitors in solid organ transplant recipients with cutaneous squamous cell carcinoma [abstract]. J Clin Oncol. 2020;38(15 suppl):E22014.
- Owoyemi I, Vaughan LE, Costello CM, et al. Clinical outcomes of solid organ transplant recipients with metastatic cancers who are treated with immune checkpoint inhibitors: a single-center analysis. Cancer. 2020;126:4780-4787.
- Kumar V, Shinagare AB, Rennke HG, et al. The safety and efficacy of checkpoint inhibitors in transplant recipients: a case series and systematic review of literature. Oncologist. 2020;25:505-514.
- Arenbergerova M, Fialova A, Arenberger P, et al. Killing two birds with one stone: response to pembrolizumab in a patient with metastatic melanoma and B-cell chronic lymphocytic leukaemia. J Eur Acad Dermatol Venereol. 2018;32:E72-E74.
- Archibald WJ, Meacham PJ, Williams AM, et al. Management of melanoma in patients with chronic lymphocytic leukemia. Leuk Res. 2018;71:43-46.
- Ding W, LaPlant BR, Call TG, et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood. 2017;129:3419-3427.
- Leiter U, Loquai C, Reinhardt L, et al. Immune checkpoint inhibition therapy for advanced skin cancer in patients with concomitant hematological malignancy: a retrospective multicenter DeCOG study of 84 patients. J Immunother Cancer. 2020;8:E000897.
- Becker LR, Aakhus AE, Reich HC, et al. A novel alternate dosing of vismodegib for treatment of patients with advanced basal cell carcinomas. JAMA Dermatol. 2017;153:321-322.
- Woltsche N, Pichler N, Wolf I, et al. Managing adverse effects by dose reduction during routine treatment of locally advanced basal cell carcinoma with the hedgehog inhibitor vismodegib: a single centre experience. J Eur Acad Dermatol Venereol. 2019;33:E144-E145.
- Wong C, Poblete-Lopez C, Vidimos A. Comparison of daily dosing versus Monday through Friday dosing of vismodegib for locally advanced basal cell carcinoma and basal cell nevus syndrome: a retrospective case series. J Am Acad Dermatol. 2020;82:1539-1542.
- Dréno B, Kunstfeld R, Hauschild A, et al. Two intermittent vismodegib dosing regimens in patients with multiple basal-cell carcinomas (MIKIE): a randomised, regimen-controlled, double-blind, phase 2 trial. Lancet Oncol. 2017;18:404-412.
- Schadendorf D, Hauschild A, Fosko S, et al. Quality-of-life analysis with intermittent vismodegib regimens in patients with multiple basal cell carcinomas: patient-reported outcomes from the MIKIE study. J Eur Acad Dermatol Venereol. 2020;34:E526-E529.
- Chanu P, Musib L, Wang X, et al. Vismodegib efficacy in advanced basal cell carcinoma maintained with 8-week dose interruptions: a model-based evaluation. J Invest Dermatol. 2021;141:930-933.
- Su MG, Potts LB, Tsai JH. Treatment of periocular basal cell carcinoma with neoadjuvant vismodegib. Am J Ophthalmol Case Rep. 2020;19:100755.
- González AR, Etchichury D, Gil ME, et al. Neoadjuvant vismodegib and Mohs micrographic surgery for locally advanced periocular basal cell carcinoma. Ophthalmic Plast Reconstr Surg. 2019;35:56-61.
- Sagiv O, Nagarajan P, Ferrarotto R, et al. Ocular preservation with neoadjuvant vismodegib in patients with locally advanced periocular basal cell carcinoma. Br J Ophthalmol. 2019;103:775-780.
- Ally MS, Aasi S, Wysong A, et al. An investigator-initiated open-label clinical trial of vismodegib as a neoadjuvant to surgery for high-risk basal cell carcinoma. J Am Acad Dermatol. 2014;71:904-911.e1.
- Kwon GP, Ally MS, Bailey-Healy I, et al. Update to an open-label clinical trial of vismodegib as neoadjuvant before surgery for high-risk basal cell carcinoma (BCC). J Am Acad Dermatol. 2016;75:213-215.
- Mortier L, Bertrand N, Basset-Seguin N, et al. Vismodegib in neoadjuvant treatment of locally advanced basal cell carcinoma: first results of a multicenter, open-label, phase 2 trial (VISMONEO study) [abstract]. J Clin Oncol. 2018;36(15 suppl):9509.
- Strasswimmer JM. Potential synergy of radiation therapy with vismodegib for basal cell carcinoma. JAMA Dermatol. 2015;151:925-926.
- Gathings RM, Orscheln CS, Huang WW. Compassionate use of vismodegib and adjuvant radiotherapy in the treatment of multiple locally advanced and inoperable basal cell carcinomas and squamous cell carcinomas of the skin. J Am Acad Dermatol. 2014;70:E88-E89.
- Franco AI, Eastwick G, Farah R, et al. Upfront radiotherapy with concurrent and adjuvant vismodegib is effective and well-tolerated in a patient with advanced, multifocal basal cell carcinoma. Case Rep Dermatol Med. 2018;2018:2354146.
- Pollom EL, Bui TT, Chang AL, et al. Concurrent vismodegib and radiotherapy for recurrent, advanced basal cell carcinoma. JAMA Dermatol. 2015;151:998-1001.
- Janela-Lapert R, Dubray B, Duval-Modeste A, et al. Treatment of advanced basal cell carcinoma with vismodegib followed by radiotherapy [in French]. Ann Dermatol Venereol. 2020;147:780-782.
- Hehlgans S, Booms P, Güllülü Ö, et al. Radiation sensitization of basal cell and head and neck squamous cell carcinoma by the hedgehog pathway inhibitor vismodegib. Int J Mol Sci. 2018;19:2485.
- Kim J, Tang JY, Gong R, et al. Itraconazole, a commonly used antifungal that inhibits hedgehog pathway activity and cancer growth. Cancer Cell. 2010;17:388-399.
- Kim J, Aftab BT, Tang JY, et al. Itraconazole and arsenic trioxide inhibit hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell. 2013;23:23-34.
- Kim DJ, Kim J, Spaunhurst K, et al. Open-label, exploratory phase II trial of oral itraconazole for the treatment of basal cell carcinoma. J Clin Oncol. 2014;32:745-751.
- Ally MS, Ransohoff K, Sarin K, et al. Effects of combined treatment with arsenic trioxide and itraconazole in patients with refractory metastatic basal cell carcinoma. JAMA Dermatol. 2016;152:452-456.
- Cia˛z˙yn´yska M, Narbutt J, Skibin´ska M, et al. Itraconazole—a new player in the therapy of advanced basal cell carcinoma: a case report. JCO Oncol Pract. 2020;16:837-838.
- Sohn GK, Kwon GP, Bailey-Healy I, et al. Topical itraconazole for the treatment of basal cell carcinoma in patients with basal cell nevus syndrome or high-frequency basal cell carcinomas: a phase 2, open-label, placebo-controlled trial. JAMA Dermatol. 2019;155:1078-1080.
- Lass-Flörl C. Triazole antifungal agents in invasive fungal infections: a comparative review. Drugs. 2011;71:2405-2419.
- Chen B, Trang V, Lee A, et al. Posaconazole, a second-generation triazole antifungal drug, inhibits the hedgehog signaling pathway and progression of basal cell carcinoma. Mol Cancer Ther. 2016;15:866-876.
- Katragkou A, Tsikopoulou F, Roilides E, et al. Posaconazole: when and how? the clinician’s view. Mycoses. 2012;55:110-122.
- Raad II, Graybill JR, Bustamante AB, et al. Safety of long-term oral posaconazole use in the treatment of refractory invasive fungal infections. Clin Infect Dis. 2006;42:1726-1734.
- Atwood SX, Sarin KY, Whitson RJ, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342-353.
- Sun Q, Atzmony L, Zaki T, et al. Clues to primary vismodegib resistance lie in histology and genetics. J Clin Pathol. 2020;73:678-680.
- Verkouteren BJA, Wakkee M, van Geel M, et al. Molecular testing in metastatic basal cell carcinoma. J Am Acad Dermatol. 2021;85:1135-1142.
- Danial C, Sarin KY, Oro AE, et al. An investigator-initiated open-label trial of sonidegib in advanced basal cell carcinoma patients resistant to vismodegib. Clin Cancer Res. 2016;22:1325-1329.
- Yoon J, Apicelli AJ 3rd, Pavlopoulos TV. Intracranial regression of an advanced basal cell carcinoma using sonidegib and itraconazole after failure with vismodegib. JAAD Case Rep. 2017;4:10-12.
- Bendell J, Andre V, Ho A, et al. Phase I study of LY2940680, a Smo antagonist, in patients with advanced cancer including treatment-naïve and previously treated basal cell carcinoma. Clin Cancer Res. 2018;24:2082-2091.
- Ueno H, Kondo S, Yoshikawa S, et al. A phase I and pharmacokinetic study of taladegib, a Smoothened inhibitor, in Japanese patients with advanced solid tumors. Invest New Drugs. 2018;36:647-656.
- Herms F, Lambert J, Grob JJ, et al. Follow-up of patients with complete remission of locally advanced basal cell carcinoma after vismodegib discontinuation: a multicenter French study of 116 patients. J Clin Oncol. 2019;37:3275-3282.
- Villani A, Megna M, Fabbrocini G, et al. Long-term efficacy of vismodegib after its withdrawal and patients’ health-related quality of life using the Dermatology Life Quality Index (DLQI). Dermatol Ther (Heidelb). 2019;9:719-724.
- Scalvenzi M, Cappello M, Costa C, et al. Low-dose vismodegib as maintenance therapy after locally advanced basal cell carcinoma complete remission: high efficacy with minimal toxicity. Dermatol Ther (Heidelb). 2020;10:465-468.
Practice Points
- The sonic hedgehog (SHH) inhibitors vismodegib and sonidegib currently are the only 2 oral medications approved by the US Food and Drug Administration for the first-line treatment of locally advanced basal cell carcinoma (BCC). Vismodegib also is approved for metastatic BCC.
- Cemiplimab, a programmed cell death protein 1 inhibitor, is now an approved treatment for patients with advanced BCC refractory or intolerant to SHH inhibitor therapy.
- Adverse effects of SHH inhibitors, most commonly muscle spasms, often lead to treatment discontinuation, but intermittent dosing regimens can be used to increase tolerability and adherence.
- Combining SHH inhibitors with radiotherapy or antifungal therapy as well as maintenance dosing strategies may help reduce the risk of recurrence.
- Neoadjuvant administration of a SHH inhibitor may enable surgical excision of previously inoperable cases through tumor shrinkage.
Squamoid Eccrine Ductal Carcinoma
Squamoid eccrine ductal carcinoma (SEDC) is an aggressive underrecognized cutaneous malignancy of unknown etiology.1 It is most likely to occur in sun-exposed areas of the body, most commonly the head and neck. Risk factors include male sex, increased age, and chronic immunosuppression.1-4 Current reports suggest that SEDC is likely a high-grade subtype of squamous cell carcinoma (SCC) with a high risk for local recurrence (25%) and metastasis (13%).1,3,5,6 There are as few as 56 cases of SEDC reported in the literature; however, the number of cases may be closer to 100 due to SEDC being classified as either adenosquamous carcinoma of the skin or ductal eccrine carcinoma with squamous differentiation.1
Clinically, SEDC mimics keratinocyte carcinomas. Histologically, SEDC is biphasic, with a superficial portion resembling well-differentiated SCC and a deeply invasive portion having infiltrative irregular cords with ductal differentiation. Perineural invasion (PNI) frequently is present. Multiple connections to the overlying epidermis also can be seen, serving as a subtle clue to the diagnosis on broad superficial specimens.1-3 Due to superficial sampling, approximately 50% of reported cases are misdiagnosed as SCC during the initial biopsy.4 The diagnosis of SEDC often is made during complete excision when deeper tissue is sampled. Establishing an accurate diagnosis is important given the more aggressive nature of SEDC compared with SCC and its proclivity for PNI.1,3,6 The purpose of this review is to increase awareness of this underrecognized entity and describe the histologic findings that help distinguish SEDC from SCC.
Patient Chart Review
We reviewed chart notes as well as frozen and formalin-fixed paraffin-embedded tissue sections from all 5 patients diagnosed with SEDC at a single institution between November 2018 and May 2020. The mean age of patients was 81 years, and 4 were male. Four of the patients presented for MMS with a preoperative diagnosis of SCC per the original biopsy results. Only 1 patient had a preoperative diagnosis of SEDC. The details of each case are recorded in the Table. All tumors were greater than 2 cm in diameter on initial presentation, were located on the head, and clinically resembled keratinocyte carcinoma with either a nodular or plaquelike appearance (Figure 1).
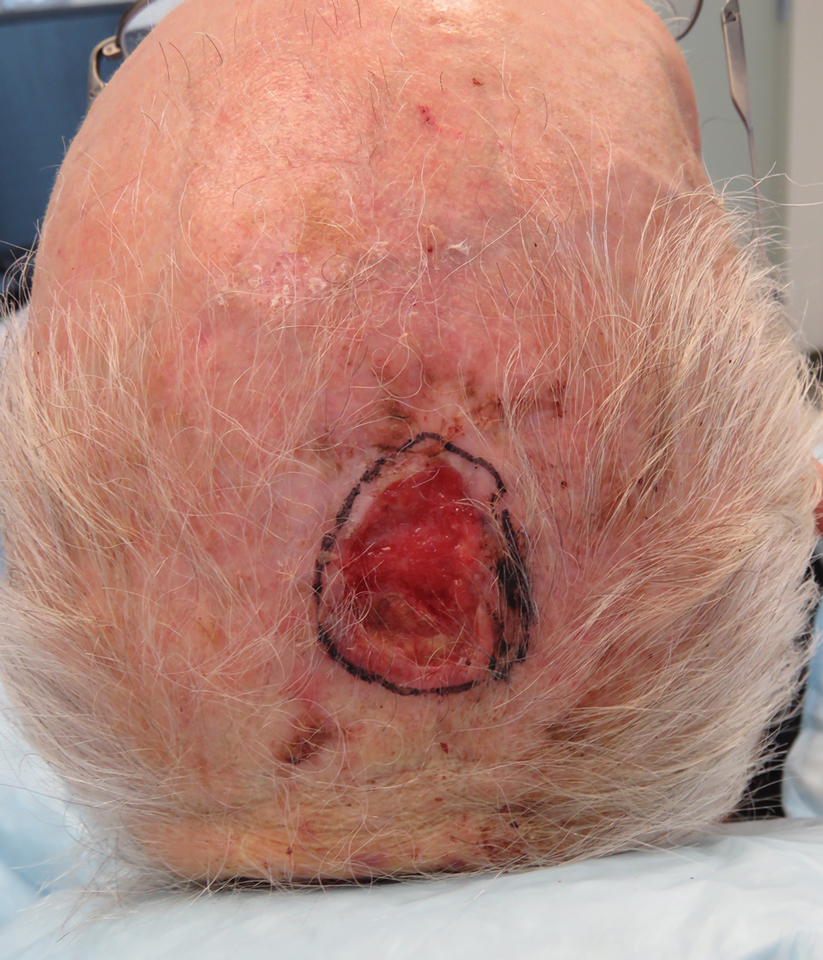
Intraoperative histologic examination of the excised tissue revealed a biphasic pattern consisting of superficial SCC features overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation in all 5 patients (Figures 2–4). Immunohistochemical staining with cytokeratin AE1/AE3 revealed thin strands of carcinoma in the mid to deeper dermis with squamous differentiation and eccrine ductal differentiation (Figure 5), thus confirming the diagnosis in all 5 patients.
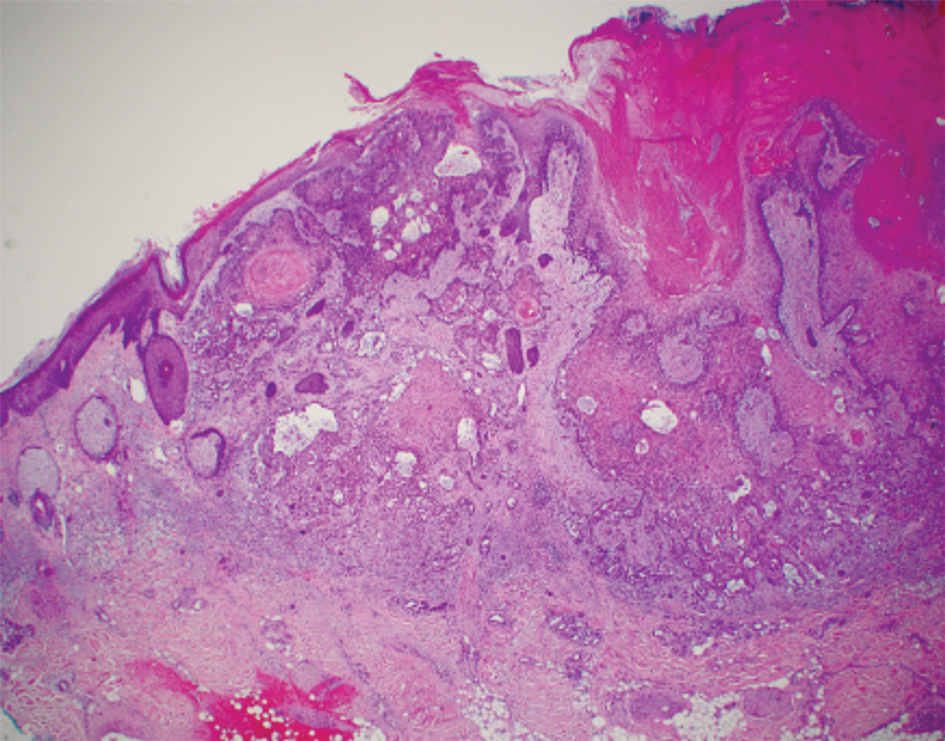


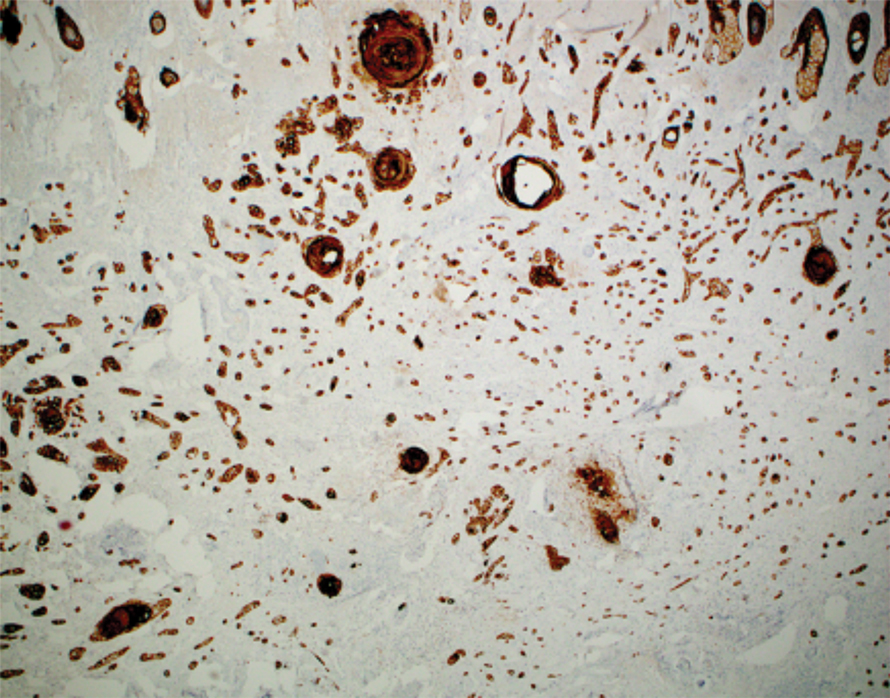
The median depth of tumor invasion was 4.1 mm (range, 2.2–5.45 mm). Ulceration was seen in 3 of the patients, and PNI of large-caliber nerves was observed in all 5 patients. A connection with the overlying epidermis was present in all 5 patients. All 5 patients required more than 1 Mohs stage for complete tumor clearance (Table).
In 4 of the patients, nodal imaging performed at the time of diagnosis revealed no evidence of metastasis. Two patients received adjuvant radiation therapy, and none demonstrated evidence of recurrence. The mean follow-up time was 11 months (range, 6.5–18 months) for the 4 cases with available follow-up data (Table).
Literature Review
A PubMed review of the literature using the search term squamoid eccrine ductal carcinoma resulted in 28 articles, 19 of which were included in the review based on inclusion criteria (original articles available in English, in full text, and pertained to SEDC). Our review yielded 56 cases of SEDC.1-19 The mean age of patients with SEDC was 72 years. The number of male and female cases was 52% (29/56) and 48% (27/56), respectively. The most common location of SEDC was on the head or neck (71% [40/56]), followed by the extremities (19% [11/56]). Immunosuppression was noted in 9% (5/56) of cases. Wide local excision was the most commonly employed treatment modality (91% [51/56]), with MMS being used in 4 patients (7%). Adjuvant radiation was reported in 5% (3/56) of cases. Perineural invasion was reported in 34% (19/56) of cases. Recurrence was seen in 23% (13/56) of cases, with a mean time to recurrence of 10.4 months. Metastasis to regional lymph nodes was observed in 13% (7/56) of cases, with 7% (4/56) of those cases having distant metastases.
Comment
Squamoid eccrine ductal carcinoma was successfully treated with MMS in all 5 of the patients we reviewed. Recognition of a distinct biphasic pattern consisting of squamous differentiation superficially with epidermal connection overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation should lead to consideration of this diagnosis. A thorough inspection for PNI also should be performed, as this finding was present in all of 5 cases and in 34% of reported cases in our literature review.
The differential diagnosis for SEDC includes SCC, metastatic adenocarcinoma with squamoid features, and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma (MAC), and porocarcinoma with squamous differentiation. The combination of histologic features with the immunoexpression profile of carcinoembryonic antigen (CEA), epithelial membrane antigen (EMA), cytokeratin (CK) 5/6, and p63 can effectively exclude the other entities in the differential and confirm the diagnosis of SEDC.1,3,4 While the diagnosis of SEDC relies on the specific histologic features of multiple surface attachments and superficial squamoid changes with deep ductular elements, immunohistochemistry can nonetheless be adjunctive in difficult cases. Positive immunohistochemical staining for CEA and EMA can help to highlight and delineate true glandular elements, whereas CK5/6 highlights the overall contour of the tumor, displaying more clearly the multiple epidermal attachments and the subtle infiltrative nature of the deeper components of invasive cords and ducts. In addition, the combination of CK5/6 and p63 positivity supports the primary cutaneous nature of the lesion rather than metastatic adenocarcinoma.13,20 Other markers of eccrine secretory coils, such as CK7, CAM5.2, and S100, also are sometimes used for confirmation, some of which can aid in distinction from noneccrine sweat gland differentiation, as CK7 and CAM5.2 are negative in both luminal and basal cells of the dermal duct while being positive within the secretory coil, and S100 protein is expressed within eccrine secretory coil but negative within the apocrine sweat glands.2,4,21
The clinical findings from our chart review corroborated those reported in the literature. The mean age of SEDC in the 5 patients we reviewed was 81 years, and all cases presented on the head, consistent with the findings observed in the literature. Although 4 of our cases were male, there may not be a difference in risk based on sex as previously thought.1 Our literature review revealed an almost equivalent percentage of male and female cases, with 52% being male.
Immunosuppression has been associated with an increased risk for SEDC. Our literature review revealed that approximately 9% (5/56) of cases occurred in immunosuppressed individuals. Two of these reported cases were in the setting of underlying chronic lymphocytic leukemia, 2 in individuals with a history of organ transplant, and 1 treated with azathioprine for myasthenia gravis.2,4,10,12,13 Our chart review supported this correlation, as all 5 patients had a medical history potentially consistent with being in an immunocompromised state (Table). Notably, patient 5 represents a unique case of SEDC occurring in the setting of HIV. The patient had HIV for 33 years, with his most recent CD4+ count of 794 mm3 and HIV-1 RNA load of 35 copies/mL. Given that HIV-positive individuals may have more than a 2-fold increased risk of SCC, a greater degree of suspicion for SEDC should be maintained for these patients.22,23
The etiology of SEDC is controversial but is thought to be either an SCC arising from eccrine glands or a variant of eccrine carcinoma with extensive squamoid differentiation.4,6,13,14,17,24 While SEDC certainly appears to share the proclivity for PNI with the malignant eccrine tumor MAC, it is simultaneously quite distinct, demonstrating nuclear pleomorphism and mitotic activity, both of which are lacking in the bland nature of MACs.12,25
The exact prevalence of SEDC is difficult to ascertain because of its frequent misdiagnosis and variable nomenclature used within the literature. Most reported cases of SEDC are mistakenly diagnosed as SCC on the initial shave or punch biopsy because of superficial sampling. This also was the case in 4 of the patients we reviewed. In addition, there are reported cases of SEDC that were referred to by the investigators as cutaneous adenosquamous carcinoma (cASC), among other descriptors, such as ductal eccrine carcinoma with squamous differentiation, adnexal carcinoma with squamous and ductal differentiation, and syringoid eccrine carcinoma.26-32 While the World Health Organization classifies SEDC as a distinct variant of cASC, which is a rare variant of SCC in itself, the 2 can be differentiated. Despite the similar clinical and histologic features shared between cASC and SEDC, the neoplastic aggregates in SEDC exhibit ductal differentiation containing lumina positive for CEA and EMA.4 Overall, we favor the term squamoid eccrine ductal carcinoma, as there has recently been more uniformity for the designation of this disease entity as such.
It is unclear whether the high incidence of local recurrence (23% [13/56]) of SEDC reported in the literature is related to the treatment modality employed (ie, wide local excision) or due to the innate aggressiveness of SEDC.1,3,5 The literature has shown that MMS has lower recurrence rates than other treatments at 5-year follow-up for SCC (3.1%–5%) and eccrine carcinomas (0%–5%).33,34 Although studies assessing tumor behavior or comparing treatment modalities are limited because of the rarity and underrecognition of SEDC, MMS has been used several times for SEDC with only 1 recurrence reported.4,13,17,24 Given that all 5 of the patients we reviewed required more than 1 Mohs stage for complete tumor clearance and none demonstrated evidence of recurrence or metastasis (Table), we recommend MMS as the treatment of choice for SEDC.
Conclusion
Squamoid eccrine ductal carcinoma is a rare but likely underdiagnosed cutaneous tumor of uncertain etiology. Because of its propensity for recurrence and metastasis, excision of SEDC with complete circumferential peripheral and deep margin assessment with close follow-up is recommended.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Jacob J, Kugelman L. Squamoid eccrine ductal carcinoma. Cutis. 2018;101:378-380, 385.
- Yim S, Lee YH, Chae SW, et al. Squamoid eccrine ductal carcinoma of the ear helix. Clin Case Rep. 2019;7:1409-1411.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Jung YH, Jo HJ, Kang MS. Squamoid eccrine ductal carcinoma of the scalp. Korean J Pathol. 2012;46:278-281.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;91:799-802.
- Phan K, Kim L, Lim P, et al. A case report of temple squamoid eccrine ductal carcinoma: a diagnostic challenge beneath the tip of the iceberg. Dermatol Ther. 2020;33:E13213.
- McKissack SS, Wohltmann W, Dalton SR, et al. Squamoid eccrine ductal carcinoma: an aggressive mimicker of squamous cell carcinoma. Am J Dermatopathol. 2019;41:140-143.
- Lobo-Jardim MM, Souza BdCE, Kakizaki P, et al. Dermoscopy of squamoid eccrine ductal carcinoma: an aid for early diagnosis. An Bras Dermatol. 2018;93:893-895.
- Chan H, Howard V, Moir D, et al. Squamoid eccrine ductal carcinoma of the scalp. Aust J Dermatol. 2016;57:E117-E119.
- Wang B, Jarell AD, Bingham JL, et al. PET/CT imaging of squamoid eccrine ductal carcinoma. Clin Nucl Med. 2015;40:322-324.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma). J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. J Clin Aesthet Dermatol. 2013;6:33-36.
- Pusiol T, Morichetti D, Zorzi MG, et al. Squamoid eccrine ductal carcinoma: inappropriate diagnosis. Dermatol Surg. 2011;37:1819-1820.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Wasserman DI, Sack J, Gonzalez-Serva A, et al. Sentinel lymph node biopsy for a squamoid eccrine carcinoma with lymphatic invasion. Dermatol Surg. 2007;33:1126-1129.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Herrero J, Monteagudo C, Jorda E, et al. Squamoid eccrine ductal carcinoma. Histopathology. 1998;32:478-480.
- Wong TY, Suster S, Mihm MC. Squamoid eccrine ductal carcinoma. Histopathology. 1997;30:288-293.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Dabbs DJ. Diagnostic Immunohistochemistry: Theranostic and Genomic Applications. 4th ed. Elsevier/Saunders; 2014.
- Silverberg MJ, Leyden W, Warton EM, et al. HIV infection status, immunodeficiency, and the incidence of non-melanoma skin cancer. J Natl Cancer Inst. 2013;105:350-360.
- Asgari MM, Ray GT, Quesenberry CP Jr, et al. Association of multiple primary skin cancers with human immunodeficiency virus infection, CD4 count, and viral load. JAMA Dermatol. 2017;153:892-896.
- Tolkachjov SN. Adnexal carcinomas treated with Mohs micrographic surgery: a comprehensive review. Dermatol Surg. 2017;43:1199-1207.
- Kazakov DV. Cutaneous Adnexal Tumors. Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2012.
- Weidner N, Foucar E. Adenosquamous carcinoma of the skin. an aggressive mucin- and gland-forming squamous carcinoma. Arch Dermatol. 1985;121:775-779.
- Banks ER, Cooper PH. Adenosquamous carcinoma of the skin: a report of 10 cases. J Cutan Pathol. 1991;18:227-234.
- Ko CJ, Leffell DJ, McNiff JM. Adenosquamous carcinoma: a report of nine cases with p63 and cytokeratin 5/6 staining. J Cutan Pathol. 2009;36:448-452.
- Patel V, Squires SM, Liu DY, et al. Cutaneous adenosquamous carcinoma: a rare neoplasm with biphasic differentiation. Cutis. 2014;94:231-233.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
- Azorín D, López-Ríos F, Ballestín C, et al. Primary cutaneous adenosquamous carcinoma: a case report and review of the literature. J Cutan Pathol. 2001;28:542-545.
- Wildemore JK, Lee JB, Humphreys TR. Mohs surgery for malignant eccrine neoplasms. Dermatol Surg. 2004;30(12 pt 2):1574-1579.
- Garcia-Zuazaga J, Olbricht SM. Cutaneous squamous cell carcinoma. Adv Dermatol. 2008;24:33-57.
Squamoid eccrine ductal carcinoma (SEDC) is an aggressive underrecognized cutaneous malignancy of unknown etiology.1 It is most likely to occur in sun-exposed areas of the body, most commonly the head and neck. Risk factors include male sex, increased age, and chronic immunosuppression.1-4 Current reports suggest that SEDC is likely a high-grade subtype of squamous cell carcinoma (SCC) with a high risk for local recurrence (25%) and metastasis (13%).1,3,5,6 There are as few as 56 cases of SEDC reported in the literature; however, the number of cases may be closer to 100 due to SEDC being classified as either adenosquamous carcinoma of the skin or ductal eccrine carcinoma with squamous differentiation.1
Clinically, SEDC mimics keratinocyte carcinomas. Histologically, SEDC is biphasic, with a superficial portion resembling well-differentiated SCC and a deeply invasive portion having infiltrative irregular cords with ductal differentiation. Perineural invasion (PNI) frequently is present. Multiple connections to the overlying epidermis also can be seen, serving as a subtle clue to the diagnosis on broad superficial specimens.1-3 Due to superficial sampling, approximately 50% of reported cases are misdiagnosed as SCC during the initial biopsy.4 The diagnosis of SEDC often is made during complete excision when deeper tissue is sampled. Establishing an accurate diagnosis is important given the more aggressive nature of SEDC compared with SCC and its proclivity for PNI.1,3,6 The purpose of this review is to increase awareness of this underrecognized entity and describe the histologic findings that help distinguish SEDC from SCC.
Patient Chart Review
We reviewed chart notes as well as frozen and formalin-fixed paraffin-embedded tissue sections from all 5 patients diagnosed with SEDC at a single institution between November 2018 and May 2020. The mean age of patients was 81 years, and 4 were male. Four of the patients presented for MMS with a preoperative diagnosis of SCC per the original biopsy results. Only 1 patient had a preoperative diagnosis of SEDC. The details of each case are recorded in the Table. All tumors were greater than 2 cm in diameter on initial presentation, were located on the head, and clinically resembled keratinocyte carcinoma with either a nodular or plaquelike appearance (Figure 1).
Intraoperative histologic examination of the excised tissue revealed a biphasic pattern consisting of superficial SCC features overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation in all 5 patients (Figures 2–4). Immunohistochemical staining with cytokeratin AE1/AE3 revealed thin strands of carcinoma in the mid to deeper dermis with squamous differentiation and eccrine ductal differentiation (Figure 5), thus confirming the diagnosis in all 5 patients.
The median depth of tumor invasion was 4.1 mm (range, 2.2–5.45 mm). Ulceration was seen in 3 of the patients, and PNI of large-caliber nerves was observed in all 5 patients. A connection with the overlying epidermis was present in all 5 patients. All 5 patients required more than 1 Mohs stage for complete tumor clearance (Table).
In 4 of the patients, nodal imaging performed at the time of diagnosis revealed no evidence of metastasis. Two patients received adjuvant radiation therapy, and none demonstrated evidence of recurrence. The mean follow-up time was 11 months (range, 6.5–18 months) for the 4 cases with available follow-up data (Table).
Literature Review
A PubMed review of the literature using the search term squamoid eccrine ductal carcinoma resulted in 28 articles, 19 of which were included in the review based on inclusion criteria (original articles available in English, in full text, and pertained to SEDC). Our review yielded 56 cases of SEDC.1-19 The mean age of patients with SEDC was 72 years. The number of male and female cases was 52% (29/56) and 48% (27/56), respectively. The most common location of SEDC was on the head or neck (71% [40/56]), followed by the extremities (19% [11/56]). Immunosuppression was noted in 9% (5/56) of cases. Wide local excision was the most commonly employed treatment modality (91% [51/56]), with MMS being used in 4 patients (7%). Adjuvant radiation was reported in 5% (3/56) of cases. Perineural invasion was reported in 34% (19/56) of cases. Recurrence was seen in 23% (13/56) of cases, with a mean time to recurrence of 10.4 months. Metastasis to regional lymph nodes was observed in 13% (7/56) of cases, with 7% (4/56) of those cases having distant metastases.
Comment
Squamoid eccrine ductal carcinoma was successfully treated with MMS in all 5 of the patients we reviewed. Recognition of a distinct biphasic pattern consisting of squamous differentiation superficially with epidermal connection overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation should lead to consideration of this diagnosis. A thorough inspection for PNI also should be performed, as this finding was present in all of 5 cases and in 34% of reported cases in our literature review.
The differential diagnosis for SEDC includes SCC, metastatic adenocarcinoma with squamoid features, and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma (MAC), and porocarcinoma with squamous differentiation. The combination of histologic features with the immunoexpression profile of carcinoembryonic antigen (CEA), epithelial membrane antigen (EMA), cytokeratin (CK) 5/6, and p63 can effectively exclude the other entities in the differential and confirm the diagnosis of SEDC.1,3,4 While the diagnosis of SEDC relies on the specific histologic features of multiple surface attachments and superficial squamoid changes with deep ductular elements, immunohistochemistry can nonetheless be adjunctive in difficult cases. Positive immunohistochemical staining for CEA and EMA can help to highlight and delineate true glandular elements, whereas CK5/6 highlights the overall contour of the tumor, displaying more clearly the multiple epidermal attachments and the subtle infiltrative nature of the deeper components of invasive cords and ducts. In addition, the combination of CK5/6 and p63 positivity supports the primary cutaneous nature of the lesion rather than metastatic adenocarcinoma.13,20 Other markers of eccrine secretory coils, such as CK7, CAM5.2, and S100, also are sometimes used for confirmation, some of which can aid in distinction from noneccrine sweat gland differentiation, as CK7 and CAM5.2 are negative in both luminal and basal cells of the dermal duct while being positive within the secretory coil, and S100 protein is expressed within eccrine secretory coil but negative within the apocrine sweat glands.2,4,21
The clinical findings from our chart review corroborated those reported in the literature. The mean age of SEDC in the 5 patients we reviewed was 81 years, and all cases presented on the head, consistent with the findings observed in the literature. Although 4 of our cases were male, there may not be a difference in risk based on sex as previously thought.1 Our literature review revealed an almost equivalent percentage of male and female cases, with 52% being male.
Immunosuppression has been associated with an increased risk for SEDC. Our literature review revealed that approximately 9% (5/56) of cases occurred in immunosuppressed individuals. Two of these reported cases were in the setting of underlying chronic lymphocytic leukemia, 2 in individuals with a history of organ transplant, and 1 treated with azathioprine for myasthenia gravis.2,4,10,12,13 Our chart review supported this correlation, as all 5 patients had a medical history potentially consistent with being in an immunocompromised state (Table). Notably, patient 5 represents a unique case of SEDC occurring in the setting of HIV. The patient had HIV for 33 years, with his most recent CD4+ count of 794 mm3 and HIV-1 RNA load of 35 copies/mL. Given that HIV-positive individuals may have more than a 2-fold increased risk of SCC, a greater degree of suspicion for SEDC should be maintained for these patients.22,23
The etiology of SEDC is controversial but is thought to be either an SCC arising from eccrine glands or a variant of eccrine carcinoma with extensive squamoid differentiation.4,6,13,14,17,24 While SEDC certainly appears to share the proclivity for PNI with the malignant eccrine tumor MAC, it is simultaneously quite distinct, demonstrating nuclear pleomorphism and mitotic activity, both of which are lacking in the bland nature of MACs.12,25
The exact prevalence of SEDC is difficult to ascertain because of its frequent misdiagnosis and variable nomenclature used within the literature. Most reported cases of SEDC are mistakenly diagnosed as SCC on the initial shave or punch biopsy because of superficial sampling. This also was the case in 4 of the patients we reviewed. In addition, there are reported cases of SEDC that were referred to by the investigators as cutaneous adenosquamous carcinoma (cASC), among other descriptors, such as ductal eccrine carcinoma with squamous differentiation, adnexal carcinoma with squamous and ductal differentiation, and syringoid eccrine carcinoma.26-32 While the World Health Organization classifies SEDC as a distinct variant of cASC, which is a rare variant of SCC in itself, the 2 can be differentiated. Despite the similar clinical and histologic features shared between cASC and SEDC, the neoplastic aggregates in SEDC exhibit ductal differentiation containing lumina positive for CEA and EMA.4 Overall, we favor the term squamoid eccrine ductal carcinoma, as there has recently been more uniformity for the designation of this disease entity as such.
It is unclear whether the high incidence of local recurrence (23% [13/56]) of SEDC reported in the literature is related to the treatment modality employed (ie, wide local excision) or due to the innate aggressiveness of SEDC.1,3,5 The literature has shown that MMS has lower recurrence rates than other treatments at 5-year follow-up for SCC (3.1%–5%) and eccrine carcinomas (0%–5%).33,34 Although studies assessing tumor behavior or comparing treatment modalities are limited because of the rarity and underrecognition of SEDC, MMS has been used several times for SEDC with only 1 recurrence reported.4,13,17,24 Given that all 5 of the patients we reviewed required more than 1 Mohs stage for complete tumor clearance and none demonstrated evidence of recurrence or metastasis (Table), we recommend MMS as the treatment of choice for SEDC.
Conclusion
Squamoid eccrine ductal carcinoma is a rare but likely underdiagnosed cutaneous tumor of uncertain etiology. Because of its propensity for recurrence and metastasis, excision of SEDC with complete circumferential peripheral and deep margin assessment with close follow-up is recommended.
Squamoid eccrine ductal carcinoma (SEDC) is an aggressive underrecognized cutaneous malignancy of unknown etiology.1 It is most likely to occur in sun-exposed areas of the body, most commonly the head and neck. Risk factors include male sex, increased age, and chronic immunosuppression.1-4 Current reports suggest that SEDC is likely a high-grade subtype of squamous cell carcinoma (SCC) with a high risk for local recurrence (25%) and metastasis (13%).1,3,5,6 There are as few as 56 cases of SEDC reported in the literature; however, the number of cases may be closer to 100 due to SEDC being classified as either adenosquamous carcinoma of the skin or ductal eccrine carcinoma with squamous differentiation.1
Clinically, SEDC mimics keratinocyte carcinomas. Histologically, SEDC is biphasic, with a superficial portion resembling well-differentiated SCC and a deeply invasive portion having infiltrative irregular cords with ductal differentiation. Perineural invasion (PNI) frequently is present. Multiple connections to the overlying epidermis also can be seen, serving as a subtle clue to the diagnosis on broad superficial specimens.1-3 Due to superficial sampling, approximately 50% of reported cases are misdiagnosed as SCC during the initial biopsy.4 The diagnosis of SEDC often is made during complete excision when deeper tissue is sampled. Establishing an accurate diagnosis is important given the more aggressive nature of SEDC compared with SCC and its proclivity for PNI.1,3,6 The purpose of this review is to increase awareness of this underrecognized entity and describe the histologic findings that help distinguish SEDC from SCC.
Patient Chart Review
We reviewed chart notes as well as frozen and formalin-fixed paraffin-embedded tissue sections from all 5 patients diagnosed with SEDC at a single institution between November 2018 and May 2020. The mean age of patients was 81 years, and 4 were male. Four of the patients presented for MMS with a preoperative diagnosis of SCC per the original biopsy results. Only 1 patient had a preoperative diagnosis of SEDC. The details of each case are recorded in the Table. All tumors were greater than 2 cm in diameter on initial presentation, were located on the head, and clinically resembled keratinocyte carcinoma with either a nodular or plaquelike appearance (Figure 1).
Intraoperative histologic examination of the excised tissue revealed a biphasic pattern consisting of superficial SCC features overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation in all 5 patients (Figures 2–4). Immunohistochemical staining with cytokeratin AE1/AE3 revealed thin strands of carcinoma in the mid to deeper dermis with squamous differentiation and eccrine ductal differentiation (Figure 5), thus confirming the diagnosis in all 5 patients.
The median depth of tumor invasion was 4.1 mm (range, 2.2–5.45 mm). Ulceration was seen in 3 of the patients, and PNI of large-caliber nerves was observed in all 5 patients. A connection with the overlying epidermis was present in all 5 patients. All 5 patients required more than 1 Mohs stage for complete tumor clearance (Table).
In 4 of the patients, nodal imaging performed at the time of diagnosis revealed no evidence of metastasis. Two patients received adjuvant radiation therapy, and none demonstrated evidence of recurrence. The mean follow-up time was 11 months (range, 6.5–18 months) for the 4 cases with available follow-up data (Table).
Literature Review
A PubMed review of the literature using the search term squamoid eccrine ductal carcinoma resulted in 28 articles, 19 of which were included in the review based on inclusion criteria (original articles available in English, in full text, and pertained to SEDC). Our review yielded 56 cases of SEDC.1-19 The mean age of patients with SEDC was 72 years. The number of male and female cases was 52% (29/56) and 48% (27/56), respectively. The most common location of SEDC was on the head or neck (71% [40/56]), followed by the extremities (19% [11/56]). Immunosuppression was noted in 9% (5/56) of cases. Wide local excision was the most commonly employed treatment modality (91% [51/56]), with MMS being used in 4 patients (7%). Adjuvant radiation was reported in 5% (3/56) of cases. Perineural invasion was reported in 34% (19/56) of cases. Recurrence was seen in 23% (13/56) of cases, with a mean time to recurrence of 10.4 months. Metastasis to regional lymph nodes was observed in 13% (7/56) of cases, with 7% (4/56) of those cases having distant metastases.
Comment
Squamoid eccrine ductal carcinoma was successfully treated with MMS in all 5 of the patients we reviewed. Recognition of a distinct biphasic pattern consisting of squamous differentiation superficially with epidermal connection overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation should lead to consideration of this diagnosis. A thorough inspection for PNI also should be performed, as this finding was present in all of 5 cases and in 34% of reported cases in our literature review.
The differential diagnosis for SEDC includes SCC, metastatic adenocarcinoma with squamoid features, and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma (MAC), and porocarcinoma with squamous differentiation. The combination of histologic features with the immunoexpression profile of carcinoembryonic antigen (CEA), epithelial membrane antigen (EMA), cytokeratin (CK) 5/6, and p63 can effectively exclude the other entities in the differential and confirm the diagnosis of SEDC.1,3,4 While the diagnosis of SEDC relies on the specific histologic features of multiple surface attachments and superficial squamoid changes with deep ductular elements, immunohistochemistry can nonetheless be adjunctive in difficult cases. Positive immunohistochemical staining for CEA and EMA can help to highlight and delineate true glandular elements, whereas CK5/6 highlights the overall contour of the tumor, displaying more clearly the multiple epidermal attachments and the subtle infiltrative nature of the deeper components of invasive cords and ducts. In addition, the combination of CK5/6 and p63 positivity supports the primary cutaneous nature of the lesion rather than metastatic adenocarcinoma.13,20 Other markers of eccrine secretory coils, such as CK7, CAM5.2, and S100, also are sometimes used for confirmation, some of which can aid in distinction from noneccrine sweat gland differentiation, as CK7 and CAM5.2 are negative in both luminal and basal cells of the dermal duct while being positive within the secretory coil, and S100 protein is expressed within eccrine secretory coil but negative within the apocrine sweat glands.2,4,21
The clinical findings from our chart review corroborated those reported in the literature. The mean age of SEDC in the 5 patients we reviewed was 81 years, and all cases presented on the head, consistent with the findings observed in the literature. Although 4 of our cases were male, there may not be a difference in risk based on sex as previously thought.1 Our literature review revealed an almost equivalent percentage of male and female cases, with 52% being male.
Immunosuppression has been associated with an increased risk for SEDC. Our literature review revealed that approximately 9% (5/56) of cases occurred in immunosuppressed individuals. Two of these reported cases were in the setting of underlying chronic lymphocytic leukemia, 2 in individuals with a history of organ transplant, and 1 treated with azathioprine for myasthenia gravis.2,4,10,12,13 Our chart review supported this correlation, as all 5 patients had a medical history potentially consistent with being in an immunocompromised state (Table). Notably, patient 5 represents a unique case of SEDC occurring in the setting of HIV. The patient had HIV for 33 years, with his most recent CD4+ count of 794 mm3 and HIV-1 RNA load of 35 copies/mL. Given that HIV-positive individuals may have more than a 2-fold increased risk of SCC, a greater degree of suspicion for SEDC should be maintained for these patients.22,23
The etiology of SEDC is controversial but is thought to be either an SCC arising from eccrine glands or a variant of eccrine carcinoma with extensive squamoid differentiation.4,6,13,14,17,24 While SEDC certainly appears to share the proclivity for PNI with the malignant eccrine tumor MAC, it is simultaneously quite distinct, demonstrating nuclear pleomorphism and mitotic activity, both of which are lacking in the bland nature of MACs.12,25
The exact prevalence of SEDC is difficult to ascertain because of its frequent misdiagnosis and variable nomenclature used within the literature. Most reported cases of SEDC are mistakenly diagnosed as SCC on the initial shave or punch biopsy because of superficial sampling. This also was the case in 4 of the patients we reviewed. In addition, there are reported cases of SEDC that were referred to by the investigators as cutaneous adenosquamous carcinoma (cASC), among other descriptors, such as ductal eccrine carcinoma with squamous differentiation, adnexal carcinoma with squamous and ductal differentiation, and syringoid eccrine carcinoma.26-32 While the World Health Organization classifies SEDC as a distinct variant of cASC, which is a rare variant of SCC in itself, the 2 can be differentiated. Despite the similar clinical and histologic features shared between cASC and SEDC, the neoplastic aggregates in SEDC exhibit ductal differentiation containing lumina positive for CEA and EMA.4 Overall, we favor the term squamoid eccrine ductal carcinoma, as there has recently been more uniformity for the designation of this disease entity as such.
It is unclear whether the high incidence of local recurrence (23% [13/56]) of SEDC reported in the literature is related to the treatment modality employed (ie, wide local excision) or due to the innate aggressiveness of SEDC.1,3,5 The literature has shown that MMS has lower recurrence rates than other treatments at 5-year follow-up for SCC (3.1%–5%) and eccrine carcinomas (0%–5%).33,34 Although studies assessing tumor behavior or comparing treatment modalities are limited because of the rarity and underrecognition of SEDC, MMS has been used several times for SEDC with only 1 recurrence reported.4,13,17,24 Given that all 5 of the patients we reviewed required more than 1 Mohs stage for complete tumor clearance and none demonstrated evidence of recurrence or metastasis (Table), we recommend MMS as the treatment of choice for SEDC.
Conclusion
Squamoid eccrine ductal carcinoma is a rare but likely underdiagnosed cutaneous tumor of uncertain etiology. Because of its propensity for recurrence and metastasis, excision of SEDC with complete circumferential peripheral and deep margin assessment with close follow-up is recommended.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Jacob J, Kugelman L. Squamoid eccrine ductal carcinoma. Cutis. 2018;101:378-380, 385.
- Yim S, Lee YH, Chae SW, et al. Squamoid eccrine ductal carcinoma of the ear helix. Clin Case Rep. 2019;7:1409-1411.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Jung YH, Jo HJ, Kang MS. Squamoid eccrine ductal carcinoma of the scalp. Korean J Pathol. 2012;46:278-281.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;91:799-802.
- Phan K, Kim L, Lim P, et al. A case report of temple squamoid eccrine ductal carcinoma: a diagnostic challenge beneath the tip of the iceberg. Dermatol Ther. 2020;33:E13213.
- McKissack SS, Wohltmann W, Dalton SR, et al. Squamoid eccrine ductal carcinoma: an aggressive mimicker of squamous cell carcinoma. Am J Dermatopathol. 2019;41:140-143.
- Lobo-Jardim MM, Souza BdCE, Kakizaki P, et al. Dermoscopy of squamoid eccrine ductal carcinoma: an aid for early diagnosis. An Bras Dermatol. 2018;93:893-895.
- Chan H, Howard V, Moir D, et al. Squamoid eccrine ductal carcinoma of the scalp. Aust J Dermatol. 2016;57:E117-E119.
- Wang B, Jarell AD, Bingham JL, et al. PET/CT imaging of squamoid eccrine ductal carcinoma. Clin Nucl Med. 2015;40:322-324.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma). J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. J Clin Aesthet Dermatol. 2013;6:33-36.
- Pusiol T, Morichetti D, Zorzi MG, et al. Squamoid eccrine ductal carcinoma: inappropriate diagnosis. Dermatol Surg. 2011;37:1819-1820.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Wasserman DI, Sack J, Gonzalez-Serva A, et al. Sentinel lymph node biopsy for a squamoid eccrine carcinoma with lymphatic invasion. Dermatol Surg. 2007;33:1126-1129.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Herrero J, Monteagudo C, Jorda E, et al. Squamoid eccrine ductal carcinoma. Histopathology. 1998;32:478-480.
- Wong TY, Suster S, Mihm MC. Squamoid eccrine ductal carcinoma. Histopathology. 1997;30:288-293.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Dabbs DJ. Diagnostic Immunohistochemistry: Theranostic and Genomic Applications. 4th ed. Elsevier/Saunders; 2014.
- Silverberg MJ, Leyden W, Warton EM, et al. HIV infection status, immunodeficiency, and the incidence of non-melanoma skin cancer. J Natl Cancer Inst. 2013;105:350-360.
- Asgari MM, Ray GT, Quesenberry CP Jr, et al. Association of multiple primary skin cancers with human immunodeficiency virus infection, CD4 count, and viral load. JAMA Dermatol. 2017;153:892-896.
- Tolkachjov SN. Adnexal carcinomas treated with Mohs micrographic surgery: a comprehensive review. Dermatol Surg. 2017;43:1199-1207.
- Kazakov DV. Cutaneous Adnexal Tumors. Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2012.
- Weidner N, Foucar E. Adenosquamous carcinoma of the skin. an aggressive mucin- and gland-forming squamous carcinoma. Arch Dermatol. 1985;121:775-779.
- Banks ER, Cooper PH. Adenosquamous carcinoma of the skin: a report of 10 cases. J Cutan Pathol. 1991;18:227-234.
- Ko CJ, Leffell DJ, McNiff JM. Adenosquamous carcinoma: a report of nine cases with p63 and cytokeratin 5/6 staining. J Cutan Pathol. 2009;36:448-452.
- Patel V, Squires SM, Liu DY, et al. Cutaneous adenosquamous carcinoma: a rare neoplasm with biphasic differentiation. Cutis. 2014;94:231-233.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
- Azorín D, López-Ríos F, Ballestín C, et al. Primary cutaneous adenosquamous carcinoma: a case report and review of the literature. J Cutan Pathol. 2001;28:542-545.
- Wildemore JK, Lee JB, Humphreys TR. Mohs surgery for malignant eccrine neoplasms. Dermatol Surg. 2004;30(12 pt 2):1574-1579.
- Garcia-Zuazaga J, Olbricht SM. Cutaneous squamous cell carcinoma. Adv Dermatol. 2008;24:33-57.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Jacob J, Kugelman L. Squamoid eccrine ductal carcinoma. Cutis. 2018;101:378-380, 385.
- Yim S, Lee YH, Chae SW, et al. Squamoid eccrine ductal carcinoma of the ear helix. Clin Case Rep. 2019;7:1409-1411.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Jung YH, Jo HJ, Kang MS. Squamoid eccrine ductal carcinoma of the scalp. Korean J Pathol. 2012;46:278-281.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;91:799-802.
- Phan K, Kim L, Lim P, et al. A case report of temple squamoid eccrine ductal carcinoma: a diagnostic challenge beneath the tip of the iceberg. Dermatol Ther. 2020;33:E13213.
- McKissack SS, Wohltmann W, Dalton SR, et al. Squamoid eccrine ductal carcinoma: an aggressive mimicker of squamous cell carcinoma. Am J Dermatopathol. 2019;41:140-143.
- Lobo-Jardim MM, Souza BdCE, Kakizaki P, et al. Dermoscopy of squamoid eccrine ductal carcinoma: an aid for early diagnosis. An Bras Dermatol. 2018;93:893-895.
- Chan H, Howard V, Moir D, et al. Squamoid eccrine ductal carcinoma of the scalp. Aust J Dermatol. 2016;57:E117-E119.
- Wang B, Jarell AD, Bingham JL, et al. PET/CT imaging of squamoid eccrine ductal carcinoma. Clin Nucl Med. 2015;40:322-324.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma). J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. J Clin Aesthet Dermatol. 2013;6:33-36.
- Pusiol T, Morichetti D, Zorzi MG, et al. Squamoid eccrine ductal carcinoma: inappropriate diagnosis. Dermatol Surg. 2011;37:1819-1820.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Wasserman DI, Sack J, Gonzalez-Serva A, et al. Sentinel lymph node biopsy for a squamoid eccrine carcinoma with lymphatic invasion. Dermatol Surg. 2007;33:1126-1129.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Herrero J, Monteagudo C, Jorda E, et al. Squamoid eccrine ductal carcinoma. Histopathology. 1998;32:478-480.
- Wong TY, Suster S, Mihm MC. Squamoid eccrine ductal carcinoma. Histopathology. 1997;30:288-293.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Dabbs DJ. Diagnostic Immunohistochemistry: Theranostic and Genomic Applications. 4th ed. Elsevier/Saunders; 2014.
- Silverberg MJ, Leyden W, Warton EM, et al. HIV infection status, immunodeficiency, and the incidence of non-melanoma skin cancer. J Natl Cancer Inst. 2013;105:350-360.
- Asgari MM, Ray GT, Quesenberry CP Jr, et al. Association of multiple primary skin cancers with human immunodeficiency virus infection, CD4 count, and viral load. JAMA Dermatol. 2017;153:892-896.
- Tolkachjov SN. Adnexal carcinomas treated with Mohs micrographic surgery: a comprehensive review. Dermatol Surg. 2017;43:1199-1207.
- Kazakov DV. Cutaneous Adnexal Tumors. Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2012.
- Weidner N, Foucar E. Adenosquamous carcinoma of the skin. an aggressive mucin- and gland-forming squamous carcinoma. Arch Dermatol. 1985;121:775-779.
- Banks ER, Cooper PH. Adenosquamous carcinoma of the skin: a report of 10 cases. J Cutan Pathol. 1991;18:227-234.
- Ko CJ, Leffell DJ, McNiff JM. Adenosquamous carcinoma: a report of nine cases with p63 and cytokeratin 5/6 staining. J Cutan Pathol. 2009;36:448-452.
- Patel V, Squires SM, Liu DY, et al. Cutaneous adenosquamous carcinoma: a rare neoplasm with biphasic differentiation. Cutis. 2014;94:231-233.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
- Azorín D, López-Ríos F, Ballestín C, et al. Primary cutaneous adenosquamous carcinoma: a case report and review of the literature. J Cutan Pathol. 2001;28:542-545.
- Wildemore JK, Lee JB, Humphreys TR. Mohs surgery for malignant eccrine neoplasms. Dermatol Surg. 2004;30(12 pt 2):1574-1579.
- Garcia-Zuazaga J, Olbricht SM. Cutaneous squamous cell carcinoma. Adv Dermatol. 2008;24:33-57.
PRACTICE POINTS
- Squamoid eccrine ductal carcinoma is an aggressive underrecognized cutaneous malignancy that often is misdiagnosed as squamous cell carcinoma (SCC) during initial biopsy.
- Squamoid eccrine ductal carcinoma has a biphasic histologic appearance with a superficial portion resembling well-differentiated SCC and a deeply invasive portion comprised of infiltrative irregular cords with ductal differentiation.
- Excision with complete circumferential peripheral and deep margin assessment with close follow-up is recommended for these patients because of the high risk for recurrence and metastasis.
Ulcerated Nodule on the Scalp
The Diagnosis: Proliferating Pilar Tumor
Proliferating pilar tumor (PPT), or cyst, is a neoplasm of trichilemmal keratinization first described by Wilson-Jones1 in 1966. Proliferating pilar tumors lie on a spectrum with malignant PPT, which is a rare adnexal neoplasm first described by Saida et al2 in 1983. The incidence of PPT is unknown given the paucity of cases and the possible misdiagnosis as squamous cell carcinoma (SCC). Proliferating pilar tumors tend to present on the head and neck of older females as a multilobular and sometimes ulcerating nodule.3 Although PPT can occur de novo, the majority of cases are thought to develop progressively from a benign pilar cyst. Histopathologically, PPT is characterized by cords and nests of squamous cells that display trichilemmal keratinization (quiz images).
Classification of PPT as benign or malignant is challenging, though criteria have been proposed.3-7 Lesions with minimal infiltration into the surrounding dermis and scant mitosis typically behave in a benign manner, while lesions showing nuclear atypia, atypical mitosis, and irregular infiltration into the surrounding dermis can have up to a 50% locoregional recurrence rate.3 In addition, distinguishing a PPT from an SCC or trichilemmal carcinoma also can be difficult; however, SCC is favored when there is a lack of trichilemmal keratinization or when squamous atypia is present in the adjacent epidermis.8 Trichilemmal carcinoma is a rare tumor that has been questioned as a distinct entity.9-12
Pilomatricoma, also known as calcifying epithelioma of Malherbe, is a benign pilar tumor that presents as a slowly growing nodule on the head or neck area or arms.13,14 Most pilomatricomas develop by the second decade of life. Multiple lesions may be present in association with myotonic dystrophy or Gardner syndrome among other syndromes.15-17 Similar to PPT, pilomatricomas present as large dermal nodules; however, they tend to be circumscribed and have a trabecular network that consists of basophilic cells and eosinophilic keratinized shadow cells (Figure 1).18 Calcification may be seen and bone formation subsequently may occur.19
Most sources now consider keratoacanthoma (KA) as a well-differentiated SCC.20 The typical presentation consists of a rapidly growing erythematous to flesh-colored nodule with a central keratinous plug that develops over a period of weeks. If untreated, KAs may resolve over a period of months and leave a depressed scar. Local destruction can result from KAs, and they have the potential to transform into a more aggressive SCC. Accordingly, most clinicians use tissue destructive methods, excision, or Mohs micrographic surgery for treatment based on location. Histologically, a well-circumscribed proliferation of glassy cytoplasm is noted. A depressed keratin-filled center is surrounded by a lip of epithelium extending over the lesion (Figure 2).20,21 Pseudoepitheliomatous hyperplasia accompanied by hypergranulosis is seen in the center of KAs rather than at the periphery, which is typical of non-KA SCCs. Typical KAs lack acantholysis, a feature suggesting a non-KA type of SCC. Neutrophilic microabscesses and eosinophils commonly are seen in KAs.20,21
Inverted follicular keratosis is a benign tumor that gained traction as its own entity in the 1960s.22 These lesions typically develop from the follicular infundibulum, but some consider them a version of a wart or seborrheic keratosis.23 They generally are flesh-colored nodules on the upper cutaneous lip or face. Treatment usually consists of complete excision. There are many different growth patterns described, but they typically are endophytic tumors with eosinophilic squamous cells in the center and more basophilic cells at the periphery (Figure 3).24 Characteristically, there are squamous eddies throughout the tumor (Figure 3 [inset]). There also may be a scant lymphohistiocytic infiltrate within the dermis surrounding the lesion. 
Trichilemmomas are flesh-colored adnexal neoplasms that may present as a solitary lesion or in clusters on the face. They have been reported to occur on all nonglabrous skin sites.25 Multiple lesions may occur in association with Cowden syndrome or with nevus sebaceous.26 A desmoplastic variant of trichilemmomas has been reported.27 Desmoplastic trichilemmomas appear as well-circumscribed tumors of outer root sheath differentiation with lobules extending down into the dermis.28 Vacuolated glycogen-filled keratinocytes are scattered throughout the lesion but are most prominent at the base. At the periphery of the lobules, peripheral palisading of basaloid cells is accompanied by a thickened eosinophilic basement membrane that is periodic acid-Schiff positive. Typical trichilemmomas also can display these features; however, the main differentiating feature of a desmoplastic trichilemmoma is the pink hyalinized stroma separating small islets of basophilic cells (Figure 4). Differentiation from an invasive malignant carcinoma sometimes can be challenging without a focus of typical trichilemmoma or if the biopsy specimen is too superficial.29

Pilar cysts are common tumors that typically arise on the scalp and sometimes are proliferating. Proliferating pilar tumor should be kept on the differential when secondary changes such as ulceration occur in the primary lesion of the scalp. Microscopically, and sometimes clinically, PPT can be difficult to differentiate from other mimickers.
- Wilson-Jones E. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Saida T, Oohara K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatologica. 1983;166:203-208.
- Ye J, Nappi O, Swanson PE, et al. Proliferating pilar tumours: a clinicopathological study of 76 cases with a proposal for definition of benign and malignant variants. Am J Clin Pathol. 2004;122:566-574.
- Garg PK, Dangi A, Khurana N, et al. Malignant proliferating trichilemmal cyst: a case report with review of literature. Malaysian J Pathol. 2009;31:71-76.
- Herrero J, Monteagudo C, Ruiz A, et al. Malignant proliferating trichilemmal tumors: a histopathological and immunohistochemical study of three cases with DNA ploidy and morphometric evaluation. Histopathology. 1998;33:542-546.
- Haas N, Audring H, Sterry W. Carcinoma arising in a proliferating trichilemmal cyst expresses fetal and trichilemmal hair phenotype. Am J Dermatopathol. 2002;24:340-344.
- Rutty GN, Richman PI, Laing JH. Malignant change in trichilemmal cysts: a study of cell proliferation and DNA content. Histopathology. 1992;21:465-468.
- Brownstein MH, Arluk DJ. Proliferating trichilemmal cyst: a simulant of squamous cell carcinoma. Cancer. 1981;48:1207-1214.
- Misago N, Ackerman AB. Tricholemmal carcinoma? Dermatopathol Pract Concept. 1999;5:205-206.
- Misago N, Narisawa Y. Tricholemmal carcinoma in continuity with trichoblastoma within nevus sebaceous. Am J Dermatopathol. 2002;24:149-155.
- Liang H, Wu H, Giorgadze TA, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
- Swanson PE, Marrogi AJ, Williams DJ, et al. Trichilemmal carcinoma: clinicopathologic study of 10 cases. J Cutan Pathol. 1992;19:100-109.
- Mehregan AH. Hair follicle tumors of the skin. J Cutan Pathol. 1985;12:189-195.
- Julian CG, Bowers PW. A clinical review of 209 pilomatricomas. J Am Acad Dermatol. 1998;39:191-195.
- Marrogi AJ, Wick MR, Dehner LP. Pilomatrical neoplasms in children and young adults. Am J Dermatopathol. 1992;14:87-94.
- Berberian BJ, Colonna TM, Battaglia M, et al. Multiple pilomatricomas in association with myotonic dystrophy and a family history of melanoma. J Am Acad Dermatol. 1997;37:268-269.
- Cooper PH, Fechner RE. Pilomatricoma-like changes in the epidermal cysts of Gardner's syndrome. J Am Acad Dermatol. 1983;8:639-644.
- Kaddu S, Soyer HP, Cerroni L, et al. Clinical and histopathologic spectrum of pilomatricomas in adults. Int J Dermatol. 1994;33:705-708.
- Sano Y, Mihara M, Miyamoto T, et al. Simultaneous occurrence of calcification and amyloid deposit in pilomatricoma. Acta Derm Venereol. 1990;70:256-259.
- Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30:1-19.
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233.
- Mehregan AH. Inverted follicular keratosis. Arch Dermatol. 1964;89:117-123.
- Spielvogel RL, Austin C, Ackerman AB. Inverted follicular keratosis is not a specific keratosis but a verruca vulgaris (or seborrheic keratosis) with squamous eddies. Am J Dermatopathol. 1983;5:427-445.
- Mehregan AH. Inverted follicular keratosis is a distinct follicular tumor. Am J Dermatopathol. 1983;5:467-470.
- Brownstein MH. Trichilemmoma. benign follicular tumor or viral wart? Am J Dermatopathol. 1980;2:229-231.
- Brownstein MH. Multiple trichilemmomas in Cowden's syndrome. Arch Dermatol. 1979;115:111.
- Roson E, Gomez Centeno P, Sanchez Aguilar D, et al. Desmoplastic trichilemmoma arising within a nevus sebaceous. Am J Dermatopathol. 1998;20:495-497.
- Tellechea O, Reis JP, Baptista AP. Desmoplastic trichilemmoma. Am J Dermatopathol. 1992;14:107-114.
- Sharma R, Sirohi D, Sengupta P, et al. Desmoplastic trichilemmoma of the facial region mimicking invasive carcinoma. J Maxillofac Oral Surg. 2010;10:71-73.
The Diagnosis: Proliferating Pilar Tumor
Proliferating pilar tumor (PPT), or cyst, is a neoplasm of trichilemmal keratinization first described by Wilson-Jones1 in 1966. Proliferating pilar tumors lie on a spectrum with malignant PPT, which is a rare adnexal neoplasm first described by Saida et al2 in 1983. The incidence of PPT is unknown given the paucity of cases and the possible misdiagnosis as squamous cell carcinoma (SCC). Proliferating pilar tumors tend to present on the head and neck of older females as a multilobular and sometimes ulcerating nodule.3 Although PPT can occur de novo, the majority of cases are thought to develop progressively from a benign pilar cyst. Histopathologically, PPT is characterized by cords and nests of squamous cells that display trichilemmal keratinization (quiz images).
Classification of PPT as benign or malignant is challenging, though criteria have been proposed.3-7 Lesions with minimal infiltration into the surrounding dermis and scant mitosis typically behave in a benign manner, while lesions showing nuclear atypia, atypical mitosis, and irregular infiltration into the surrounding dermis can have up to a 50% locoregional recurrence rate.3 In addition, distinguishing a PPT from an SCC or trichilemmal carcinoma also can be difficult; however, SCC is favored when there is a lack of trichilemmal keratinization or when squamous atypia is present in the adjacent epidermis.8 Trichilemmal carcinoma is a rare tumor that has been questioned as a distinct entity.9-12
Pilomatricoma, also known as calcifying epithelioma of Malherbe, is a benign pilar tumor that presents as a slowly growing nodule on the head or neck area or arms.13,14 Most pilomatricomas develop by the second decade of life. Multiple lesions may be present in association with myotonic dystrophy or Gardner syndrome among other syndromes.15-17 Similar to PPT, pilomatricomas present as large dermal nodules; however, they tend to be circumscribed and have a trabecular network that consists of basophilic cells and eosinophilic keratinized shadow cells (Figure 1).18 Calcification may be seen and bone formation subsequently may occur.19
Most sources now consider keratoacanthoma (KA) as a well-differentiated SCC.20 The typical presentation consists of a rapidly growing erythematous to flesh-colored nodule with a central keratinous plug that develops over a period of weeks. If untreated, KAs may resolve over a period of months and leave a depressed scar. Local destruction can result from KAs, and they have the potential to transform into a more aggressive SCC. Accordingly, most clinicians use tissue destructive methods, excision, or Mohs micrographic surgery for treatment based on location. Histologically, a well-circumscribed proliferation of glassy cytoplasm is noted. A depressed keratin-filled center is surrounded by a lip of epithelium extending over the lesion (Figure 2).20,21 Pseudoepitheliomatous hyperplasia accompanied by hypergranulosis is seen in the center of KAs rather than at the periphery, which is typical of non-KA SCCs. Typical KAs lack acantholysis, a feature suggesting a non-KA type of SCC. Neutrophilic microabscesses and eosinophils commonly are seen in KAs.20,21
Inverted follicular keratosis is a benign tumor that gained traction as its own entity in the 1960s.22 These lesions typically develop from the follicular infundibulum, but some consider them a version of a wart or seborrheic keratosis.23 They generally are flesh-colored nodules on the upper cutaneous lip or face. Treatment usually consists of complete excision. There are many different growth patterns described, but they typically are endophytic tumors with eosinophilic squamous cells in the center and more basophilic cells at the periphery (Figure 3).24 Characteristically, there are squamous eddies throughout the tumor (Figure 3 [inset]). There also may be a scant lymphohistiocytic infiltrate within the dermis surrounding the lesion.
Trichilemmomas are flesh-colored adnexal neoplasms that may present as a solitary lesion or in clusters on the face. They have been reported to occur on all nonglabrous skin sites.25 Multiple lesions may occur in association with Cowden syndrome or with nevus sebaceous.26 A desmoplastic variant of trichilemmomas has been reported.27 Desmoplastic trichilemmomas appear as well-circumscribed tumors of outer root sheath differentiation with lobules extending down into the dermis.28 Vacuolated glycogen-filled keratinocytes are scattered throughout the lesion but are most prominent at the base. At the periphery of the lobules, peripheral palisading of basaloid cells is accompanied by a thickened eosinophilic basement membrane that is periodic acid-Schiff positive. Typical trichilemmomas also can display these features; however, the main differentiating feature of a desmoplastic trichilemmoma is the pink hyalinized stroma separating small islets of basophilic cells (Figure 4). Differentiation from an invasive malignant carcinoma sometimes can be challenging without a focus of typical trichilemmoma or if the biopsy specimen is too superficial.29
Pilar cysts are common tumors that typically arise on the scalp and sometimes are proliferating. Proliferating pilar tumor should be kept on the differential when secondary changes such as ulceration occur in the primary lesion of the scalp. Microscopically, and sometimes clinically, PPT can be difficult to differentiate from other mimickers.
The Diagnosis: Proliferating Pilar Tumor
Proliferating pilar tumor (PPT), or cyst, is a neoplasm of trichilemmal keratinization first described by Wilson-Jones1 in 1966. Proliferating pilar tumors lie on a spectrum with malignant PPT, which is a rare adnexal neoplasm first described by Saida et al2 in 1983. The incidence of PPT is unknown given the paucity of cases and the possible misdiagnosis as squamous cell carcinoma (SCC). Proliferating pilar tumors tend to present on the head and neck of older females as a multilobular and sometimes ulcerating nodule.3 Although PPT can occur de novo, the majority of cases are thought to develop progressively from a benign pilar cyst. Histopathologically, PPT is characterized by cords and nests of squamous cells that display trichilemmal keratinization (quiz images).
Classification of PPT as benign or malignant is challenging, though criteria have been proposed.3-7 Lesions with minimal infiltration into the surrounding dermis and scant mitosis typically behave in a benign manner, while lesions showing nuclear atypia, atypical mitosis, and irregular infiltration into the surrounding dermis can have up to a 50% locoregional recurrence rate.3 In addition, distinguishing a PPT from an SCC or trichilemmal carcinoma also can be difficult; however, SCC is favored when there is a lack of trichilemmal keratinization or when squamous atypia is present in the adjacent epidermis.8 Trichilemmal carcinoma is a rare tumor that has been questioned as a distinct entity.9-12
Pilomatricoma, also known as calcifying epithelioma of Malherbe, is a benign pilar tumor that presents as a slowly growing nodule on the head or neck area or arms.13,14 Most pilomatricomas develop by the second decade of life. Multiple lesions may be present in association with myotonic dystrophy or Gardner syndrome among other syndromes.15-17 Similar to PPT, pilomatricomas present as large dermal nodules; however, they tend to be circumscribed and have a trabecular network that consists of basophilic cells and eosinophilic keratinized shadow cells (Figure 1).18 Calcification may be seen and bone formation subsequently may occur.19
Most sources now consider keratoacanthoma (KA) as a well-differentiated SCC.20 The typical presentation consists of a rapidly growing erythematous to flesh-colored nodule with a central keratinous plug that develops over a period of weeks. If untreated, KAs may resolve over a period of months and leave a depressed scar. Local destruction can result from KAs, and they have the potential to transform into a more aggressive SCC. Accordingly, most clinicians use tissue destructive methods, excision, or Mohs micrographic surgery for treatment based on location. Histologically, a well-circumscribed proliferation of glassy cytoplasm is noted. A depressed keratin-filled center is surrounded by a lip of epithelium extending over the lesion (Figure 2).20,21 Pseudoepitheliomatous hyperplasia accompanied by hypergranulosis is seen in the center of KAs rather than at the periphery, which is typical of non-KA SCCs. Typical KAs lack acantholysis, a feature suggesting a non-KA type of SCC. Neutrophilic microabscesses and eosinophils commonly are seen in KAs.20,21
Inverted follicular keratosis is a benign tumor that gained traction as its own entity in the 1960s.22 These lesions typically develop from the follicular infundibulum, but some consider them a version of a wart or seborrheic keratosis.23 They generally are flesh-colored nodules on the upper cutaneous lip or face. Treatment usually consists of complete excision. There are many different growth patterns described, but they typically are endophytic tumors with eosinophilic squamous cells in the center and more basophilic cells at the periphery (Figure 3).24 Characteristically, there are squamous eddies throughout the tumor (Figure 3 [inset]). There also may be a scant lymphohistiocytic infiltrate within the dermis surrounding the lesion.
Trichilemmomas are flesh-colored adnexal neoplasms that may present as a solitary lesion or in clusters on the face. They have been reported to occur on all nonglabrous skin sites.25 Multiple lesions may occur in association with Cowden syndrome or with nevus sebaceous.26 A desmoplastic variant of trichilemmomas has been reported.27 Desmoplastic trichilemmomas appear as well-circumscribed tumors of outer root sheath differentiation with lobules extending down into the dermis.28 Vacuolated glycogen-filled keratinocytes are scattered throughout the lesion but are most prominent at the base. At the periphery of the lobules, peripheral palisading of basaloid cells is accompanied by a thickened eosinophilic basement membrane that is periodic acid-Schiff positive. Typical trichilemmomas also can display these features; however, the main differentiating feature of a desmoplastic trichilemmoma is the pink hyalinized stroma separating small islets of basophilic cells (Figure 4). Differentiation from an invasive malignant carcinoma sometimes can be challenging without a focus of typical trichilemmoma or if the biopsy specimen is too superficial.29
Pilar cysts are common tumors that typically arise on the scalp and sometimes are proliferating. Proliferating pilar tumor should be kept on the differential when secondary changes such as ulceration occur in the primary lesion of the scalp. Microscopically, and sometimes clinically, PPT can be difficult to differentiate from other mimickers.
- Wilson-Jones E. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Saida T, Oohara K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatologica. 1983;166:203-208.
- Ye J, Nappi O, Swanson PE, et al. Proliferating pilar tumours: a clinicopathological study of 76 cases with a proposal for definition of benign and malignant variants. Am J Clin Pathol. 2004;122:566-574.
- Garg PK, Dangi A, Khurana N, et al. Malignant proliferating trichilemmal cyst: a case report with review of literature. Malaysian J Pathol. 2009;31:71-76.
- Herrero J, Monteagudo C, Ruiz A, et al. Malignant proliferating trichilemmal tumors: a histopathological and immunohistochemical study of three cases with DNA ploidy and morphometric evaluation. Histopathology. 1998;33:542-546.
- Haas N, Audring H, Sterry W. Carcinoma arising in a proliferating trichilemmal cyst expresses fetal and trichilemmal hair phenotype. Am J Dermatopathol. 2002;24:340-344.
- Rutty GN, Richman PI, Laing JH. Malignant change in trichilemmal cysts: a study of cell proliferation and DNA content. Histopathology. 1992;21:465-468.
- Brownstein MH, Arluk DJ. Proliferating trichilemmal cyst: a simulant of squamous cell carcinoma. Cancer. 1981;48:1207-1214.
- Misago N, Ackerman AB. Tricholemmal carcinoma? Dermatopathol Pract Concept. 1999;5:205-206.
- Misago N, Narisawa Y. Tricholemmal carcinoma in continuity with trichoblastoma within nevus sebaceous. Am J Dermatopathol. 2002;24:149-155.
- Liang H, Wu H, Giorgadze TA, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
- Swanson PE, Marrogi AJ, Williams DJ, et al. Trichilemmal carcinoma: clinicopathologic study of 10 cases. J Cutan Pathol. 1992;19:100-109.
- Mehregan AH. Hair follicle tumors of the skin. J Cutan Pathol. 1985;12:189-195.
- Julian CG, Bowers PW. A clinical review of 209 pilomatricomas. J Am Acad Dermatol. 1998;39:191-195.
- Marrogi AJ, Wick MR, Dehner LP. Pilomatrical neoplasms in children and young adults. Am J Dermatopathol. 1992;14:87-94.
- Berberian BJ, Colonna TM, Battaglia M, et al. Multiple pilomatricomas in association with myotonic dystrophy and a family history of melanoma. J Am Acad Dermatol. 1997;37:268-269.
- Cooper PH, Fechner RE. Pilomatricoma-like changes in the epidermal cysts of Gardner's syndrome. J Am Acad Dermatol. 1983;8:639-644.
- Kaddu S, Soyer HP, Cerroni L, et al. Clinical and histopathologic spectrum of pilomatricomas in adults. Int J Dermatol. 1994;33:705-708.
- Sano Y, Mihara M, Miyamoto T, et al. Simultaneous occurrence of calcification and amyloid deposit in pilomatricoma. Acta Derm Venereol. 1990;70:256-259.
- Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30:1-19.
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233.
- Mehregan AH. Inverted follicular keratosis. Arch Dermatol. 1964;89:117-123.
- Spielvogel RL, Austin C, Ackerman AB. Inverted follicular keratosis is not a specific keratosis but a verruca vulgaris (or seborrheic keratosis) with squamous eddies. Am J Dermatopathol. 1983;5:427-445.
- Mehregan AH. Inverted follicular keratosis is a distinct follicular tumor. Am J Dermatopathol. 1983;5:467-470.
- Brownstein MH. Trichilemmoma. benign follicular tumor or viral wart? Am J Dermatopathol. 1980;2:229-231.
- Brownstein MH. Multiple trichilemmomas in Cowden's syndrome. Arch Dermatol. 1979;115:111.
- Roson E, Gomez Centeno P, Sanchez Aguilar D, et al. Desmoplastic trichilemmoma arising within a nevus sebaceous. Am J Dermatopathol. 1998;20:495-497.
- Tellechea O, Reis JP, Baptista AP. Desmoplastic trichilemmoma. Am J Dermatopathol. 1992;14:107-114.
- Sharma R, Sirohi D, Sengupta P, et al. Desmoplastic trichilemmoma of the facial region mimicking invasive carcinoma. J Maxillofac Oral Surg. 2010;10:71-73.
- Wilson-Jones E. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Saida T, Oohara K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatologica. 1983;166:203-208.
- Ye J, Nappi O, Swanson PE, et al. Proliferating pilar tumours: a clinicopathological study of 76 cases with a proposal for definition of benign and malignant variants. Am J Clin Pathol. 2004;122:566-574.
- Garg PK, Dangi A, Khurana N, et al. Malignant proliferating trichilemmal cyst: a case report with review of literature. Malaysian J Pathol. 2009;31:71-76.
- Herrero J, Monteagudo C, Ruiz A, et al. Malignant proliferating trichilemmal tumors: a histopathological and immunohistochemical study of three cases with DNA ploidy and morphometric evaluation. Histopathology. 1998;33:542-546.
- Haas N, Audring H, Sterry W. Carcinoma arising in a proliferating trichilemmal cyst expresses fetal and trichilemmal hair phenotype. Am J Dermatopathol. 2002;24:340-344.
- Rutty GN, Richman PI, Laing JH. Malignant change in trichilemmal cysts: a study of cell proliferation and DNA content. Histopathology. 1992;21:465-468.
- Brownstein MH, Arluk DJ. Proliferating trichilemmal cyst: a simulant of squamous cell carcinoma. Cancer. 1981;48:1207-1214.
- Misago N, Ackerman AB. Tricholemmal carcinoma? Dermatopathol Pract Concept. 1999;5:205-206.
- Misago N, Narisawa Y. Tricholemmal carcinoma in continuity with trichoblastoma within nevus sebaceous. Am J Dermatopathol. 2002;24:149-155.
- Liang H, Wu H, Giorgadze TA, et al. Podoplanin is a highly sensitive and specific marker to distinguish primary skin adnexal carcinomas from adenocarcinomas metastatic to skin. Am J Surg Pathol. 2007;31:304-310.
- Swanson PE, Marrogi AJ, Williams DJ, et al. Trichilemmal carcinoma: clinicopathologic study of 10 cases. J Cutan Pathol. 1992;19:100-109.
- Mehregan AH. Hair follicle tumors of the skin. J Cutan Pathol. 1985;12:189-195.
- Julian CG, Bowers PW. A clinical review of 209 pilomatricomas. J Am Acad Dermatol. 1998;39:191-195.
- Marrogi AJ, Wick MR, Dehner LP. Pilomatrical neoplasms in children and young adults. Am J Dermatopathol. 1992;14:87-94.
- Berberian BJ, Colonna TM, Battaglia M, et al. Multiple pilomatricomas in association with myotonic dystrophy and a family history of melanoma. J Am Acad Dermatol. 1997;37:268-269.
- Cooper PH, Fechner RE. Pilomatricoma-like changes in the epidermal cysts of Gardner's syndrome. J Am Acad Dermatol. 1983;8:639-644.
- Kaddu S, Soyer HP, Cerroni L, et al. Clinical and histopathologic spectrum of pilomatricomas in adults. Int J Dermatol. 1994;33:705-708.
- Sano Y, Mihara M, Miyamoto T, et al. Simultaneous occurrence of calcification and amyloid deposit in pilomatricoma. Acta Derm Venereol. 1990;70:256-259.
- Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30:1-19.
- Kwiek B, Schwartz RA. Keratoacanthoma (KA): an update and review. J Am Acad Dermatol. 2016;74:1220-1233.
- Mehregan AH. Inverted follicular keratosis. Arch Dermatol. 1964;89:117-123.
- Spielvogel RL, Austin C, Ackerman AB. Inverted follicular keratosis is not a specific keratosis but a verruca vulgaris (or seborrheic keratosis) with squamous eddies. Am J Dermatopathol. 1983;5:427-445.
- Mehregan AH. Inverted follicular keratosis is a distinct follicular tumor. Am J Dermatopathol. 1983;5:467-470.
- Brownstein MH. Trichilemmoma. benign follicular tumor or viral wart? Am J Dermatopathol. 1980;2:229-231.
- Brownstein MH. Multiple trichilemmomas in Cowden's syndrome. Arch Dermatol. 1979;115:111.
- Roson E, Gomez Centeno P, Sanchez Aguilar D, et al. Desmoplastic trichilemmoma arising within a nevus sebaceous. Am J Dermatopathol. 1998;20:495-497.
- Tellechea O, Reis JP, Baptista AP. Desmoplastic trichilemmoma. Am J Dermatopathol. 1992;14:107-114.
- Sharma R, Sirohi D, Sengupta P, et al. Desmoplastic trichilemmoma of the facial region mimicking invasive carcinoma. J Maxillofac Oral Surg. 2010;10:71-73.
A 66-year-old woman presented to the dermatology clinic with a rapidly enlarging, draining lesion on the scalp. The lesion seemed to enlarge over the last 3 months from a lesion that had been there for years. Physical examination revealed a 2.2-cm ulcerated nodule on the right parietal scalp. A shave biopsy was obtained.
Systemic Medications Linked to an Increased Risk for Skin Malignancy
Dermatologists are increasingly called on to evaluate patients with complex medical problems who are often taking many medications. Over the last several decades, many new drugs that target molecular pathways in carcinogenesis and the inflammatory immune system have been developed. Increased skin cancer risk has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, Janus kinase (JAK) inhibitors, and phosphodiesterase 5 (PDE-5) inhibitors. We review the literature and data regarding the significance and strength of these associations and the molecular pathways by which these medications promote cutaneous tumorigenesis. The association of skin cancer with drugs that either induce photosensitivity—nonsteroidal anti-inflammatory drugs, antibiotics (eg, tetracyclines, fluoroquinolones, trimethoprim-sulfamethoxazole), voriconazole, thiazides—or suppress the immune system—certain biologics (eg, anti–tumor necrosis factor agents), calcineurin inhibitors, thiopurines, methotrexate, cyclosporine—is well known and is therefore not reviewed in this discussion.
BRAF Inhibitors
The mitogen-activated protein kinase (MAPK) pathway (also known as the RAS/RAF/MAPK signaling pathway) is important in growth factor–receptor signaling and plays a key role in cell differentiation, survival, and proliferation. Activating mutations in this pathway allow cells to grow and proliferate in a growth factor–independent manner. Twenty percent of human cancers harbor a mutation in the RAS oncogene, an upstream mediator of the pathway.1 Activating mutations in BRAF, a serine/threonine kinase, predominate in cutaneous melanoma and also have been found in 40% to 70% of papillary thyroid malignancies, 10% to 20% of cholangiocarcinomas, and 5% to 20% of colorectal carcinomas. The most common BRAF mutation in cutaneous melanoma is V600E, which involves a glutamic acid for valine substitution at codon 600. This mutation activates BRAF 500-fold and is present in approximately 50% of melanomas.1,2
Vemurafenib, a selective BRAF inhibitor, was approved by the US Food and Drug Administration (FDA) for the treatment of metastatic melanoma in the United States in 2011. Phase 3 trial data demonstrated that vemurafenib resulted in improved survival and decreased risk for disease progression compared to dacarbazine, the former best treatment.3 During phase 1 testing, it became apparent that vemurafenib treatment was associated with a 31% increased risk for squamous cell carcinoma (SCC), most commonly well-differentiated SCC, and keratoacanthomas (KAs).4 This association was confirmed in phase 2 and 3 studies, though the incidence was lower. McArthur et al5 reported a 19% incidence of cutaneous SCC with extended follow-up analysis of the phase 3 trial. Dabrafenib, another BRAF inhibitor, has been similarly associated with increasing the risk for SCC and KA.
In one study, the mean time to development of SCC after initiating vemurafenib therapy was 10 weeks, with lesions reported as early as 3 weeks. Most patients had clinical signs of chronically sun damaged skin; however, a history of SCC was present in only 17%. Most lesions (63%) were characterized as KAs.6
The mechanism for BRAF inhibitor–induced squamoproliferative growth is due to paradoxical activation of the MAPK pathway in cells with wild-type BRAF that harbor upstream-activating mutations in RAS or tyrosine kinase receptors.7 In the presence of a BRAF inhibitor, inactivated BRAF forms heterodimers with wild-type CRAF (a BRAF-CRAF heterodimer). The heterodimer forms a complex with the mutant RAS that leads to transactivation of the CRAF molecule,8,9 resulting in a paradoxical increase in MAPK signaling and consequent ERK phosphorylation and activation through CRAF signaling. RAS, particularly HRAS, mutations have been found in 60% of all vemurafenib-associated SCCs and KAs. For this reason, it is thought that vemurafenib potentiates tumorigenesis in subclinical lesions harboring upstream MAPK pathway mutations as opposed to inducing de novo lesions.6
Because BRAF inhibitors are remarkably efficacious in the treatment of metastatic melanomas harboring the V600E BRAF mutation, there are no restrictions on their use, despite the known increased risk for SCC. Squamous cell carcinomas tend to be low grade, and all tumors that developed in phase 1 to 3 trials were treated with simple excision. The development of SCC did not necessitate interruption of treatment. Furthermore, the addition of MEK inhibition to BRAF inhibitor therapy reduces the risk for SCC from 19% to 7%.7,10,11
In addition to SCC, second primary melanomas (SPMs) have been reported in patients treated with BRAF inhibitors. It has been shown that these melanomas occur in melanocytes with wild-type BRAF. It has been postulated that some of these tumors occur in cells that harbor upstream mutations in RAS, whereas others might result from alternate signaling through non-RAF oncogenic pathways.9,12
Zimmer et al1 reported 12 SPMs in 11 patients treated with BRAF inhibitor therapy. They reported a median delay of 8 weeks (range, 4–27 weeks) for SPM development. Tumors were detected in early stages; 1 tumor harbored an NRAS mutation.1
Dalle et al13 reported 25 SPMs in 120 vemurafenib-treated patients. Median delay in SPM development was 14 weeks (range, 4–42 weeks). All tumors were thin, ranging from in situ to 0.45-mm thick. Wild-type BRAF was detected in the 21 melanomas sampled; 1 lesion showed mutated NRAS.13
The exact incidence of SPM in the setting of BRAF inhibition is thought to be at least 10-fold less than SCC and KA.2 Patients on BRAF inhibitor therapy should have routine full-body skin examinations, given the increased risk for SPM and SCC.
Another drug belonging to the tyrosine kinase inhibitor family, sorafenib, is used in the treatment of solid tumors, particularly hepatocellular and renal cell carcinomas, and also has been associated with development of cutaneous SCC and KAs.14 Sorafenib is a multiple tyrosine kinase inhibitor that also inhibits the RAF serine/threonine kinases. Similar to vemurafenib and dabrafenib, SCCs and KAs associated with sorafenib tend to arise in patients with chronic actinic damage during the first 2 months of treatment. It has been hypothesized that inhibition of RAF kinases is pathogenic in inducing SCCs because these lesions have not been reported with sunitinib, another multiple tyrosine kinase inhibitor that lacks the ability to inhibit serine/threonine kinases.15,16 Although SCCs and KAs associated with sorafenib tend to be low grade, it is reasonable to consider sunitinib or an alternative tyrosine kinase inhibitor in patients who develop multiple SCCs while taking sorafenib.16
Sonic Hedgehog–Inhibiting Agents
Vismodegib, the first small molecule inhibitor of the signaling protein smoothened, gained FDA approval for the treatment of metastatic or locally advanced basal cell carcinoma (BCC) in 2012. A second agent with an identical mechanism of action, sonidegib, was approved by the FDA for locally advanced BCC in 2015. Approximately 90% of BCCs contain mutations in the sonic hedgehog pathway, which lead to constitutive smoothened activation and uncontrolled cell proliferation.17 The development of smoothened inhibitors introduced a much-needed treatment for inoperable or metastatic BCC,17,18 though long-term utility is limited by drug resistance with extended use in this patient population.19,20 Several case reports have documented the emergence of KA21 and cutaneous SCC following vismodegib treatment of advanced or metastatic BCC.22-24 A larger case-control study by Mohan et al25 showed that patients with BCC treated with vismodegib had an increased risk for non-BCC malignancy (hazard ratio [HR]=6.37), most of which were cutaneous SCC (HR=8.12).
The mechanism by which selective inhibition of smoothened leads to cutaneous SCC is unclear. A study found that patients on vismodegib who developed SCC within the original BCC site had elevated ERK levels within tumor tissue, suggesting that the RAS/RAF/MAPK pathway can become upregulated during hedgehog inhibition.26 Other studies looking at hedgehog inhibition in medulloblastoma models also have shown activated RAS/RAF/MAPK pathways.25 These findings suggest that tumors under smoothened inhibition might be able to bypass the sonic hedgehog pathway and continue to grow by upregulating alternative growth pathways, such as RAS/RAF/MAPK.25,26
The incidence of cutaneous SCC following vismodegib treatment is unknown. Chang and Oro27 examined BCC tumor regrowth from secondary (acquired) resistance to vismodegib and noted that lesions recurred within 1 cm of the original tumor 21% of the time. Although none of the 12 patients whose tumors regrew during treatment were reported to have developed SCC, several demonstrated different BCC subtypes than the pretreatment specimen. The authors proposed that regrowth of BCC was due to upregulated alternative pathways allowing tumors to bypass smoothened inhibition, which is similar to the proposed mechanism for SCC development in vismodegib patients.27
Prospective studies are needed to confirm the link between vismodegib and cutaneous SCC; establish the incidence of SCC development; and identify any pretreatment factors, tumor characteristics, or treatment details (eg, dosage, duration) that might contribute to SCC development. Furthermore, because Mohan et al25 observed that vismodegib-treated patients were less likely to develop SCC in situ than controls, it is unknown if these tumors are more aggressive than traditional SCC. At this point, careful surveillance and regular full-body skin examinations are advised for patients on vismodegib for treatment of advanced BCC.
JAK Inhibitors
Another class of medications potentially associated with increased development of nonmelanoma skin cancer (NMSC) is the JAK inhibitors (also known as jakinibs). Many proinflammatory signaling pathways converge on the JAK family of enzymes—JAK1, JAK2, JAK3, and TYK2. These enzymes operate in cytokine signal transduction by phosphorylating activated cytokine receptors, which allows for recruitment and activation by means of phosphorylation of transcription factors collectively known as signal transducers and activators of transcription (STATs). Phosphorylated STATs dimerize and translocate to the nucleus, acting as direct transcription promoters. Janus kinase inhibitors modulate the immune response by reducing the effect of interleukin and interferon signaling.
Ruxolitinib, a JAK1/JAK2 inhibitor, was the first JAK inhibitor approved by the FDA and is indicated for the treatment of myelofibrosis and polycythemia vera. Additionally, oral and topical JAK inhibitors have shown efficacy in the treatment of psoriasis, rheumatoid arthritis, alopecia areata, vitiligo, and pruritus from atopic dermatitis.28
The JAK-STAT pathway is complex, and the biological activity of the pathway is both proinflammatory and pro–cell survival and proliferation. Because signaling through the pathway can increase angiogenesis and inhibit apoptosis, inhibition of this pathway has been exploited for the treatment of some tumors. However, inhibition of interferon and proinflammatory interleukin signaling also can potentially promote tumor growth by means of inhibition of downstream cytotoxic T-cell signaling, theoretically increasing the risk for NMSC. A study examining the 5-year efficacy of ruxolitinib in myelofibrosis patients (COMFORT-II trial) found that 17.1% of patients developed NMSC compared to only 2.7% of those on the best available therapy. After adjustment by patient exposure, the NMSC rate was still doubled for ruxolitinib-treated patients compared to controls (6.1/100 patient-years and 3.0/100 patient-years, respectively).29 Eighty-week follow-up of the phase 3 clinical trial of ruxolitinib for the treatment of polycythemia vera also noted an increased incidence of NMSC, albeit a more conservative increase. Patients randomized to the ruxolitinib treatment group developed NMSC at a rate of 4.4/100 patient-years, whereas the rate for controls treated with best available therapy was 2.7/100 patient-years.30 In contrast, 5-year follow-up of the COMFORT-I trial, also examining the efficacy of ruxolitinib in myelofibrosis, showed no increased risk for NMSC between ruxolitinib-treated patients and placebo (2.7/100 patient-years and 3.9/100 patient-years, respectively).31
A 2017 case series described 5 patients with myelofibrosis who developed multiple skin cancers with aggressive features while receiving ruxolitinib.32 Duration of ruxolitinib therapy ranged from 4 months to 4 years; 3 patients had a history of hydroxyurea exposure, and only 1 patient had a history of NMSC. High-risk cutaneous SCC, undifferentiated pleomorphic sarcoma, and lentigo maligna melanoma (Breslow thickness, 0.45 mm) were among the tumors reported in this series. Although no definitive conclusion can be made regarding the causality of JAK inhibitors in promoting these tumors, the association warrants further investigation. Clinicians should be aware that ruxolitinib might amplify the risk for NMSC in patients with pre-existing genetic or exposure-related susceptibility. Interruption of drug therapy may be necessary in managing patients who develop an aggressive tumor.32
In contrast, tofacitinib, which specifically inhibits JAK3, carries very low risk, if any, for NMSC when used for the treatment of psoriasis and rheumatoid arthritis. Results from 2 phase 3 trials analyzing the efficacy of tofacitinib in psoriasis demonstrated that only 2 of 1486 patients treated developed NMSC compared to none in the control group.33 Furthermore, analysis of NMSC across the tofacitinib rheumatoid arthritis clinical program, which included a total of 15,103 patient-years of exposure, demonstrated that the overall NMSC incidence was 0.55 for every 100 patient-years. Of note, the risk in patients receiving high-dose treatment (10 mg vs 5 mg) was nearly doubled in long-term follow-up studies (0.79/100 patient-years and 0.41/100 patient-years, respectively). Overall, the study concluded that treatment with tofacitinib presents no greater increased risk for NMSC than treatment with tumor necrosis factor inhibitors.33
PDE-5 Inhibitors
Phosphodiesterase 5 inhibitors, such as sildenafil citrate, have been widely prescribed for the treatment of erectile dysfunction. Studies have shown that BRAF-activated melanomas, which occur in approximately 50% to 70% of melanomas, also result in reduced PDE-5 expression.34-36 In these melanomas, downregulation of PDE-5 results in increased intracellular calcium,36 which has been shown to induce melanoma invasion.36,37 Given this similarity in molecular pathway between BRAF-activated melanomas and PDE-5 inhibitors, there has been increased concern that PDE-5 inhibitors might be associated with an increased risk for melanoma.
In 2014, Li et al38 published a retrospective analysis suggesting an association with sildenafil and an increased risk for melanoma. Their study utilized the Health Professionals Follow-up Study to identify a statistically significant elevation in the risk for invasive melanoma with both recent sildenafil use (multivariate-adjusted HR=2.24) and use at any time (HR=1.92). These results controlled for confounding variables, such as presence of major chronic disease, use of other erectile dysfunction treatments, family history of melanoma, history of sun exposure, and UV index of the patient’s residence. Notably, the study also found that sildenafil did not affect the incidence of BCC or SCC.38
In 2015, Loeb et al39 also examined the potential association between PDE-5 inhibitors and melanoma. Review of several Swedish drug and cancer registries allowed for analysis of melanoma risk and PDE-5 inhibitor use, based on number of prescriptions filled and type of PDE-5 inhibitor prescribed. Their analysis showed that men developing melanoma were more likely than nonmelanoma controls to have taken a PDE-5 inhibitor (11% vs 8%). In a subgroup analysis, however, statistical significance was shown for men with only a single prescription filled (34% of cases; P<.05), whereas the difference for men with multiple filled prescriptions did not meet statistical significance. Furthermore, the study did not find increased risk with longer-acting tadalafil and vardenafil (odds ratio [OR]=1.16) compared to sildenafil (OR=1.14). Last, use of PDE-5 inhibitors was only associated with stage 0 (OR=1.49) and stage I (OR=1.21) tumors, not with stages II to IV (OR=0.83) tumors. Although there was a statistically significant association between PDE-5 inhibitors and malignant melanoma (P<.05), the subgroup analysis findings pointed away from a causal relationship and likely toward a confounding of variable(s).39
A 2016 study by Lian et al40 looked at the risk for melanoma in a cohort of patients diagnosed with erectile dysfunction. No association between PDE-5 inhibitors and melanoma risk was shown when comparing patients who received a PDE-5 inhibitor and those who did not receive a PDE-5 inhibitor. However, secondary analysis did show that melanoma risk was increased among patients receiving more pills (34%) and prescriptions (30%). The authors concluded that there was no association between PDE-5 inhibitor use and overall increased risk for melanoma, and the increased risk associated with a greater number of pills and prescriptions would require further study.40
In contrast, a 2017 meta-analysis by Tang et al41 of 5 studies (3 of which were the aforementioned trials38-40) concluded that use of PDE-5 inhibitors was associated with a small but significantly increased risk for melanoma (OR=1.12) and BCC (OR=1.14) but not SCC. Furthermore, the study found no evidence of dosage-dependent association between PDE-5 inhibitor use and melanoma risk.41
Overall, clinical studies have been inconclusive in determining the risk for melanoma in the setting of PDE-5 inhibitor use. Studies showing an increased rate of melanoma within patient cohorts receiving PDE-5 inhibitors are limited; results might be affected by confounding variables. However, given the similarity in mechanism between PDE-5 inhibitors and HRAS-activated melanomas, it is reasonable to continue research into this potential association.
Conclusion
Since the turn of the century, drugs targeting cell-signaling pathways have been developed to treat inflammatory, oncologic, and immune conditions. The role of immunosuppressants in promoting skin cancer is well established and supported by a vast literature base. However, associations are less clear with newer immunomodulatory and antineoplastic medications. Skin cancer has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, JAK inhibitors, and PDE-5 inhibitors. In the case of JAK and PDE-5 inhibitors, the increased risk for melanoma and NMSC is somewhat inconclusive; risk is more firmly established for BRAF inhibitors and smoothened inhibitors. For the antineoplastic agents reviewed, the therapeutic effect of cancer regression is well documented, and benefits of continued therapy outweigh the increased risk for skin cancer promotion in nearly all cases. The value of early detection has been well documented for skin malignancy; therefore, increased skin surveillance and prompt management of suspicious lesions should be a priority for physicians treating patients undergoing therapy with these medications
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanoma in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
- Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA Dermatol. 2015;151:1103-1109.
- Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427-430.
- Ryan MB, Der CJ, Wang-Gillam A, et al. Targeting RAS-mutant cancers: is ERK the key? Trends Cancer. 2015;1:183-198.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
- Holderfield M, Nagel TE, Stuart DD. Mechanism and consequence of RAF kinase activation by small-molecule inhibitors. Br J Cancer. 2014;111:640-645.
- Dalle S, Poulalhon N, Debarbieux S, et al. Tracking of second primary melanomas in vemurafenib-treated patients. JAMA Dermatol. 2013;149:488-490.
- Williams VL, Cohen PR, Stewart DJ. Sorafenib-induced premalignant and malignant skin lesions. Int J Dermatol. 2011;50:396-402.
- Arnault JP, Wechsler J, Escudier B, et al. Keratoacanthomas and squamous cell carcinomas in patients receiving sorafenib. J Clin Oncol. 2009;27:e59-e61.
- Smith KJ, Haley H, Hamza S, et al. Eruptive keratoacanthoma-type squamous cell carcinomas in patients taking sorafenib for the treatment of solid tumors. Dermatol Surg. 2009;35:1766-1770.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Demirci H, Worden F, Nelson CC, et al. Efficacy of vismodegib (Erivedge) for basal cell carcinoma involving the orbit and periocular area. Ophthalmic Plast Reconstr Surg. 2015;31:463-466.
- Atwood SX, Sarin KY, Whitson RJ, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342-353.
- Ridky TW, Cotsarelis G. Vismodegib resistance in basal cell carcinoma: not a smooth fit. Cancer Cell. 2015;27:315-316.
- Aasi S, Silkiss R, Tang JY, et al. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: a report of 2 cases. JAMA Dermatol. 2013;149:242-243.
- Orouji A, Goerdt S, Utikal J, et al. Multiple highly and moderately differentiated squamous cell carcinomas of the skin during vismodegib treatment of inoperable basal cell carcinoma. Br J Dermatol. 2014;171:431-433.
- Iarrobino A, Messina JL, Kudchadkar R, et al. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2013;69:e33-e34.
- Saintes C, Saint-Jean M, Brocard A, et al. Development of squamous cell carcinoma into basal cell carcinoma under treatment with vismodegib. J Eur Acad Dermatol Venereol. 2015;29:1006-1009.
- Mohan SV, Chang J, Li S, et al. Increased risk of cutaneous squamous cell carcinoma after vismodegib therapy for basal cell carcinoma. JAMA Dermatol. 2016;152:527-532.
- Zhao X, Ponomaryov T, Ornell KJ, et al. RAS/MAPK activation drives resistance to Smo inhibition, metastasis, and tumor evolution in Shh pathway-dependent tumors. Cancer Res. 2015;75:3623-3635.
- Chang AL, Oro AE. Initial assessment of tumor regrowth after vismodegib in advanced basal cell carcinoma. Arch Dermatol. 2012;148:1324-1325.
- Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76:736-744.
- Harrison CN, Vannucchi AM, Kiladjian JJ, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30:1701-1707.
- Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica. 2016;101:821-829.
- Verstovsek S, Mesa RA, Gotlib J, et al; COMFORT-I investigators. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017;10:55.
- Blechman AB, Cabell CE, Weinberger CH, et al. Aggressive skin cancers occurring in patients treated with the Janus kinase inhibitor ruxolitinib. J Drugs Dermatol. 2017;16:508-511.
- Papp KA, Menter MA, Abe M, et al; OPT Pivotal 1 and OPT Pivotal 2 investigators. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two randomized, placebo-controlled, phase III trials. Br J Dermatol. 2015;173:949-961.
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875-885.
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851-857.
- Arozarena I, Sanchez-Laorden B, Packer L, et al. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell. 2011;19:45-57.
- Houslay MD. Hard times for oncogenic BRAF-expressing melanoma cells. Cancer Cell. 2011;19:3-4.
- Li WQ, Qureshi AA, Robinson KC, et al. Sildenafil use and increased risk of incident melanoma in US men: a prospective cohort study. JAMA Intern Med. 2014;174:964-970.
- Loeb S, Folkvaljon Y, Lambe M, et al. Use of phosphodiesterase type 5 inhibitors for erectile dysfunction and risk of malignant melanoma. JAMA. 2015;313:2449-2455.
- Lian Y, Yin H, Pollak MN, et al. Phosphodiesterase type 5 inhibitors and the risk of melanoma skin cancer. Eur Urol. 2016;70:808-815.
- Tang H, Wu W, Fu S, et al. Phosphodiesterase type 5 inhibitors and risk of melanoma: a meta-analysis. J Am Acad Dermatol. 2017;77:480.e9-488.e9.
Dermatologists are increasingly called on to evaluate patients with complex medical problems who are often taking many medications. Over the last several decades, many new drugs that target molecular pathways in carcinogenesis and the inflammatory immune system have been developed. Increased skin cancer risk has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, Janus kinase (JAK) inhibitors, and phosphodiesterase 5 (PDE-5) inhibitors. We review the literature and data regarding the significance and strength of these associations and the molecular pathways by which these medications promote cutaneous tumorigenesis. The association of skin cancer with drugs that either induce photosensitivity—nonsteroidal anti-inflammatory drugs, antibiotics (eg, tetracyclines, fluoroquinolones, trimethoprim-sulfamethoxazole), voriconazole, thiazides—or suppress the immune system—certain biologics (eg, anti–tumor necrosis factor agents), calcineurin inhibitors, thiopurines, methotrexate, cyclosporine—is well known and is therefore not reviewed in this discussion.
BRAF Inhibitors
The mitogen-activated protein kinase (MAPK) pathway (also known as the RAS/RAF/MAPK signaling pathway) is important in growth factor–receptor signaling and plays a key role in cell differentiation, survival, and proliferation. Activating mutations in this pathway allow cells to grow and proliferate in a growth factor–independent manner. Twenty percent of human cancers harbor a mutation in the RAS oncogene, an upstream mediator of the pathway.1 Activating mutations in BRAF, a serine/threonine kinase, predominate in cutaneous melanoma and also have been found in 40% to 70% of papillary thyroid malignancies, 10% to 20% of cholangiocarcinomas, and 5% to 20% of colorectal carcinomas. The most common BRAF mutation in cutaneous melanoma is V600E, which involves a glutamic acid for valine substitution at codon 600. This mutation activates BRAF 500-fold and is present in approximately 50% of melanomas.1,2
Vemurafenib, a selective BRAF inhibitor, was approved by the US Food and Drug Administration (FDA) for the treatment of metastatic melanoma in the United States in 2011. Phase 3 trial data demonstrated that vemurafenib resulted in improved survival and decreased risk for disease progression compared to dacarbazine, the former best treatment.3 During phase 1 testing, it became apparent that vemurafenib treatment was associated with a 31% increased risk for squamous cell carcinoma (SCC), most commonly well-differentiated SCC, and keratoacanthomas (KAs).4 This association was confirmed in phase 2 and 3 studies, though the incidence was lower. McArthur et al5 reported a 19% incidence of cutaneous SCC with extended follow-up analysis of the phase 3 trial. Dabrafenib, another BRAF inhibitor, has been similarly associated with increasing the risk for SCC and KA.
In one study, the mean time to development of SCC after initiating vemurafenib therapy was 10 weeks, with lesions reported as early as 3 weeks. Most patients had clinical signs of chronically sun damaged skin; however, a history of SCC was present in only 17%. Most lesions (63%) were characterized as KAs.6
The mechanism for BRAF inhibitor–induced squamoproliferative growth is due to paradoxical activation of the MAPK pathway in cells with wild-type BRAF that harbor upstream-activating mutations in RAS or tyrosine kinase receptors.7 In the presence of a BRAF inhibitor, inactivated BRAF forms heterodimers with wild-type CRAF (a BRAF-CRAF heterodimer). The heterodimer forms a complex with the mutant RAS that leads to transactivation of the CRAF molecule,8,9 resulting in a paradoxical increase in MAPK signaling and consequent ERK phosphorylation and activation through CRAF signaling. RAS, particularly HRAS, mutations have been found in 60% of all vemurafenib-associated SCCs and KAs. For this reason, it is thought that vemurafenib potentiates tumorigenesis in subclinical lesions harboring upstream MAPK pathway mutations as opposed to inducing de novo lesions.6
Because BRAF inhibitors are remarkably efficacious in the treatment of metastatic melanomas harboring the V600E BRAF mutation, there are no restrictions on their use, despite the known increased risk for SCC. Squamous cell carcinomas tend to be low grade, and all tumors that developed in phase 1 to 3 trials were treated with simple excision. The development of SCC did not necessitate interruption of treatment. Furthermore, the addition of MEK inhibition to BRAF inhibitor therapy reduces the risk for SCC from 19% to 7%.7,10,11
In addition to SCC, second primary melanomas (SPMs) have been reported in patients treated with BRAF inhibitors. It has been shown that these melanomas occur in melanocytes with wild-type BRAF. It has been postulated that some of these tumors occur in cells that harbor upstream mutations in RAS, whereas others might result from alternate signaling through non-RAF oncogenic pathways.9,12
Zimmer et al1 reported 12 SPMs in 11 patients treated with BRAF inhibitor therapy. They reported a median delay of 8 weeks (range, 4–27 weeks) for SPM development. Tumors were detected in early stages; 1 tumor harbored an NRAS mutation.1
Dalle et al13 reported 25 SPMs in 120 vemurafenib-treated patients. Median delay in SPM development was 14 weeks (range, 4–42 weeks). All tumors were thin, ranging from in situ to 0.45-mm thick. Wild-type BRAF was detected in the 21 melanomas sampled; 1 lesion showed mutated NRAS.13
The exact incidence of SPM in the setting of BRAF inhibition is thought to be at least 10-fold less than SCC and KA.2 Patients on BRAF inhibitor therapy should have routine full-body skin examinations, given the increased risk for SPM and SCC.
Another drug belonging to the tyrosine kinase inhibitor family, sorafenib, is used in the treatment of solid tumors, particularly hepatocellular and renal cell carcinomas, and also has been associated with development of cutaneous SCC and KAs.14 Sorafenib is a multiple tyrosine kinase inhibitor that also inhibits the RAF serine/threonine kinases. Similar to vemurafenib and dabrafenib, SCCs and KAs associated with sorafenib tend to arise in patients with chronic actinic damage during the first 2 months of treatment. It has been hypothesized that inhibition of RAF kinases is pathogenic in inducing SCCs because these lesions have not been reported with sunitinib, another multiple tyrosine kinase inhibitor that lacks the ability to inhibit serine/threonine kinases.15,16 Although SCCs and KAs associated with sorafenib tend to be low grade, it is reasonable to consider sunitinib or an alternative tyrosine kinase inhibitor in patients who develop multiple SCCs while taking sorafenib.16
Sonic Hedgehog–Inhibiting Agents
Vismodegib, the first small molecule inhibitor of the signaling protein smoothened, gained FDA approval for the treatment of metastatic or locally advanced basal cell carcinoma (BCC) in 2012. A second agent with an identical mechanism of action, sonidegib, was approved by the FDA for locally advanced BCC in 2015. Approximately 90% of BCCs contain mutations in the sonic hedgehog pathway, which lead to constitutive smoothened activation and uncontrolled cell proliferation.17 The development of smoothened inhibitors introduced a much-needed treatment for inoperable or metastatic BCC,17,18 though long-term utility is limited by drug resistance with extended use in this patient population.19,20 Several case reports have documented the emergence of KA21 and cutaneous SCC following vismodegib treatment of advanced or metastatic BCC.22-24 A larger case-control study by Mohan et al25 showed that patients with BCC treated with vismodegib had an increased risk for non-BCC malignancy (hazard ratio [HR]=6.37), most of which were cutaneous SCC (HR=8.12).
The mechanism by which selective inhibition of smoothened leads to cutaneous SCC is unclear. A study found that patients on vismodegib who developed SCC within the original BCC site had elevated ERK levels within tumor tissue, suggesting that the RAS/RAF/MAPK pathway can become upregulated during hedgehog inhibition.26 Other studies looking at hedgehog inhibition in medulloblastoma models also have shown activated RAS/RAF/MAPK pathways.25 These findings suggest that tumors under smoothened inhibition might be able to bypass the sonic hedgehog pathway and continue to grow by upregulating alternative growth pathways, such as RAS/RAF/MAPK.25,26
The incidence of cutaneous SCC following vismodegib treatment is unknown. Chang and Oro27 examined BCC tumor regrowth from secondary (acquired) resistance to vismodegib and noted that lesions recurred within 1 cm of the original tumor 21% of the time. Although none of the 12 patients whose tumors regrew during treatment were reported to have developed SCC, several demonstrated different BCC subtypes than the pretreatment specimen. The authors proposed that regrowth of BCC was due to upregulated alternative pathways allowing tumors to bypass smoothened inhibition, which is similar to the proposed mechanism for SCC development in vismodegib patients.27
Prospective studies are needed to confirm the link between vismodegib and cutaneous SCC; establish the incidence of SCC development; and identify any pretreatment factors, tumor characteristics, or treatment details (eg, dosage, duration) that might contribute to SCC development. Furthermore, because Mohan et al25 observed that vismodegib-treated patients were less likely to develop SCC in situ than controls, it is unknown if these tumors are more aggressive than traditional SCC. At this point, careful surveillance and regular full-body skin examinations are advised for patients on vismodegib for treatment of advanced BCC.
JAK Inhibitors
Another class of medications potentially associated with increased development of nonmelanoma skin cancer (NMSC) is the JAK inhibitors (also known as jakinibs). Many proinflammatory signaling pathways converge on the JAK family of enzymes—JAK1, JAK2, JAK3, and TYK2. These enzymes operate in cytokine signal transduction by phosphorylating activated cytokine receptors, which allows for recruitment and activation by means of phosphorylation of transcription factors collectively known as signal transducers and activators of transcription (STATs). Phosphorylated STATs dimerize and translocate to the nucleus, acting as direct transcription promoters. Janus kinase inhibitors modulate the immune response by reducing the effect of interleukin and interferon signaling.
Ruxolitinib, a JAK1/JAK2 inhibitor, was the first JAK inhibitor approved by the FDA and is indicated for the treatment of myelofibrosis and polycythemia vera. Additionally, oral and topical JAK inhibitors have shown efficacy in the treatment of psoriasis, rheumatoid arthritis, alopecia areata, vitiligo, and pruritus from atopic dermatitis.28
The JAK-STAT pathway is complex, and the biological activity of the pathway is both proinflammatory and pro–cell survival and proliferation. Because signaling through the pathway can increase angiogenesis and inhibit apoptosis, inhibition of this pathway has been exploited for the treatment of some tumors. However, inhibition of interferon and proinflammatory interleukin signaling also can potentially promote tumor growth by means of inhibition of downstream cytotoxic T-cell signaling, theoretically increasing the risk for NMSC. A study examining the 5-year efficacy of ruxolitinib in myelofibrosis patients (COMFORT-II trial) found that 17.1% of patients developed NMSC compared to only 2.7% of those on the best available therapy. After adjustment by patient exposure, the NMSC rate was still doubled for ruxolitinib-treated patients compared to controls (6.1/100 patient-years and 3.0/100 patient-years, respectively).29 Eighty-week follow-up of the phase 3 clinical trial of ruxolitinib for the treatment of polycythemia vera also noted an increased incidence of NMSC, albeit a more conservative increase. Patients randomized to the ruxolitinib treatment group developed NMSC at a rate of 4.4/100 patient-years, whereas the rate for controls treated with best available therapy was 2.7/100 patient-years.30 In contrast, 5-year follow-up of the COMFORT-I trial, also examining the efficacy of ruxolitinib in myelofibrosis, showed no increased risk for NMSC between ruxolitinib-treated patients and placebo (2.7/100 patient-years and 3.9/100 patient-years, respectively).31
A 2017 case series described 5 patients with myelofibrosis who developed multiple skin cancers with aggressive features while receiving ruxolitinib.32 Duration of ruxolitinib therapy ranged from 4 months to 4 years; 3 patients had a history of hydroxyurea exposure, and only 1 patient had a history of NMSC. High-risk cutaneous SCC, undifferentiated pleomorphic sarcoma, and lentigo maligna melanoma (Breslow thickness, 0.45 mm) were among the tumors reported in this series. Although no definitive conclusion can be made regarding the causality of JAK inhibitors in promoting these tumors, the association warrants further investigation. Clinicians should be aware that ruxolitinib might amplify the risk for NMSC in patients with pre-existing genetic or exposure-related susceptibility. Interruption of drug therapy may be necessary in managing patients who develop an aggressive tumor.32
In contrast, tofacitinib, which specifically inhibits JAK3, carries very low risk, if any, for NMSC when used for the treatment of psoriasis and rheumatoid arthritis. Results from 2 phase 3 trials analyzing the efficacy of tofacitinib in psoriasis demonstrated that only 2 of 1486 patients treated developed NMSC compared to none in the control group.33 Furthermore, analysis of NMSC across the tofacitinib rheumatoid arthritis clinical program, which included a total of 15,103 patient-years of exposure, demonstrated that the overall NMSC incidence was 0.55 for every 100 patient-years. Of note, the risk in patients receiving high-dose treatment (10 mg vs 5 mg) was nearly doubled in long-term follow-up studies (0.79/100 patient-years and 0.41/100 patient-years, respectively). Overall, the study concluded that treatment with tofacitinib presents no greater increased risk for NMSC than treatment with tumor necrosis factor inhibitors.33
PDE-5 Inhibitors
Phosphodiesterase 5 inhibitors, such as sildenafil citrate, have been widely prescribed for the treatment of erectile dysfunction. Studies have shown that BRAF-activated melanomas, which occur in approximately 50% to 70% of melanomas, also result in reduced PDE-5 expression.34-36 In these melanomas, downregulation of PDE-5 results in increased intracellular calcium,36 which has been shown to induce melanoma invasion.36,37 Given this similarity in molecular pathway between BRAF-activated melanomas and PDE-5 inhibitors, there has been increased concern that PDE-5 inhibitors might be associated with an increased risk for melanoma.
In 2014, Li et al38 published a retrospective analysis suggesting an association with sildenafil and an increased risk for melanoma. Their study utilized the Health Professionals Follow-up Study to identify a statistically significant elevation in the risk for invasive melanoma with both recent sildenafil use (multivariate-adjusted HR=2.24) and use at any time (HR=1.92). These results controlled for confounding variables, such as presence of major chronic disease, use of other erectile dysfunction treatments, family history of melanoma, history of sun exposure, and UV index of the patient’s residence. Notably, the study also found that sildenafil did not affect the incidence of BCC or SCC.38
In 2015, Loeb et al39 also examined the potential association between PDE-5 inhibitors and melanoma. Review of several Swedish drug and cancer registries allowed for analysis of melanoma risk and PDE-5 inhibitor use, based on number of prescriptions filled and type of PDE-5 inhibitor prescribed. Their analysis showed that men developing melanoma were more likely than nonmelanoma controls to have taken a PDE-5 inhibitor (11% vs 8%). In a subgroup analysis, however, statistical significance was shown for men with only a single prescription filled (34% of cases; P<.05), whereas the difference for men with multiple filled prescriptions did not meet statistical significance. Furthermore, the study did not find increased risk with longer-acting tadalafil and vardenafil (odds ratio [OR]=1.16) compared to sildenafil (OR=1.14). Last, use of PDE-5 inhibitors was only associated with stage 0 (OR=1.49) and stage I (OR=1.21) tumors, not with stages II to IV (OR=0.83) tumors. Although there was a statistically significant association between PDE-5 inhibitors and malignant melanoma (P<.05), the subgroup analysis findings pointed away from a causal relationship and likely toward a confounding of variable(s).39
A 2016 study by Lian et al40 looked at the risk for melanoma in a cohort of patients diagnosed with erectile dysfunction. No association between PDE-5 inhibitors and melanoma risk was shown when comparing patients who received a PDE-5 inhibitor and those who did not receive a PDE-5 inhibitor. However, secondary analysis did show that melanoma risk was increased among patients receiving more pills (34%) and prescriptions (30%). The authors concluded that there was no association between PDE-5 inhibitor use and overall increased risk for melanoma, and the increased risk associated with a greater number of pills and prescriptions would require further study.40
In contrast, a 2017 meta-analysis by Tang et al41 of 5 studies (3 of which were the aforementioned trials38-40) concluded that use of PDE-5 inhibitors was associated with a small but significantly increased risk for melanoma (OR=1.12) and BCC (OR=1.14) but not SCC. Furthermore, the study found no evidence of dosage-dependent association between PDE-5 inhibitor use and melanoma risk.41
Overall, clinical studies have been inconclusive in determining the risk for melanoma in the setting of PDE-5 inhibitor use. Studies showing an increased rate of melanoma within patient cohorts receiving PDE-5 inhibitors are limited; results might be affected by confounding variables. However, given the similarity in mechanism between PDE-5 inhibitors and HRAS-activated melanomas, it is reasonable to continue research into this potential association.
Conclusion
Since the turn of the century, drugs targeting cell-signaling pathways have been developed to treat inflammatory, oncologic, and immune conditions. The role of immunosuppressants in promoting skin cancer is well established and supported by a vast literature base. However, associations are less clear with newer immunomodulatory and antineoplastic medications. Skin cancer has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, JAK inhibitors, and PDE-5 inhibitors. In the case of JAK and PDE-5 inhibitors, the increased risk for melanoma and NMSC is somewhat inconclusive; risk is more firmly established for BRAF inhibitors and smoothened inhibitors. For the antineoplastic agents reviewed, the therapeutic effect of cancer regression is well documented, and benefits of continued therapy outweigh the increased risk for skin cancer promotion in nearly all cases. The value of early detection has been well documented for skin malignancy; therefore, increased skin surveillance and prompt management of suspicious lesions should be a priority for physicians treating patients undergoing therapy with these medications
Dermatologists are increasingly called on to evaluate patients with complex medical problems who are often taking many medications. Over the last several decades, many new drugs that target molecular pathways in carcinogenesis and the inflammatory immune system have been developed. Increased skin cancer risk has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, Janus kinase (JAK) inhibitors, and phosphodiesterase 5 (PDE-5) inhibitors. We review the literature and data regarding the significance and strength of these associations and the molecular pathways by which these medications promote cutaneous tumorigenesis. The association of skin cancer with drugs that either induce photosensitivity—nonsteroidal anti-inflammatory drugs, antibiotics (eg, tetracyclines, fluoroquinolones, trimethoprim-sulfamethoxazole), voriconazole, thiazides—or suppress the immune system—certain biologics (eg, anti–tumor necrosis factor agents), calcineurin inhibitors, thiopurines, methotrexate, cyclosporine—is well known and is therefore not reviewed in this discussion.
BRAF Inhibitors
The mitogen-activated protein kinase (MAPK) pathway (also known as the RAS/RAF/MAPK signaling pathway) is important in growth factor–receptor signaling and plays a key role in cell differentiation, survival, and proliferation. Activating mutations in this pathway allow cells to grow and proliferate in a growth factor–independent manner. Twenty percent of human cancers harbor a mutation in the RAS oncogene, an upstream mediator of the pathway.1 Activating mutations in BRAF, a serine/threonine kinase, predominate in cutaneous melanoma and also have been found in 40% to 70% of papillary thyroid malignancies, 10% to 20% of cholangiocarcinomas, and 5% to 20% of colorectal carcinomas. The most common BRAF mutation in cutaneous melanoma is V600E, which involves a glutamic acid for valine substitution at codon 600. This mutation activates BRAF 500-fold and is present in approximately 50% of melanomas.1,2
Vemurafenib, a selective BRAF inhibitor, was approved by the US Food and Drug Administration (FDA) for the treatment of metastatic melanoma in the United States in 2011. Phase 3 trial data demonstrated that vemurafenib resulted in improved survival and decreased risk for disease progression compared to dacarbazine, the former best treatment.3 During phase 1 testing, it became apparent that vemurafenib treatment was associated with a 31% increased risk for squamous cell carcinoma (SCC), most commonly well-differentiated SCC, and keratoacanthomas (KAs).4 This association was confirmed in phase 2 and 3 studies, though the incidence was lower. McArthur et al5 reported a 19% incidence of cutaneous SCC with extended follow-up analysis of the phase 3 trial. Dabrafenib, another BRAF inhibitor, has been similarly associated with increasing the risk for SCC and KA.
In one study, the mean time to development of SCC after initiating vemurafenib therapy was 10 weeks, with lesions reported as early as 3 weeks. Most patients had clinical signs of chronically sun damaged skin; however, a history of SCC was present in only 17%. Most lesions (63%) were characterized as KAs.6
The mechanism for BRAF inhibitor–induced squamoproliferative growth is due to paradoxical activation of the MAPK pathway in cells with wild-type BRAF that harbor upstream-activating mutations in RAS or tyrosine kinase receptors.7 In the presence of a BRAF inhibitor, inactivated BRAF forms heterodimers with wild-type CRAF (a BRAF-CRAF heterodimer). The heterodimer forms a complex with the mutant RAS that leads to transactivation of the CRAF molecule,8,9 resulting in a paradoxical increase in MAPK signaling and consequent ERK phosphorylation and activation through CRAF signaling. RAS, particularly HRAS, mutations have been found in 60% of all vemurafenib-associated SCCs and KAs. For this reason, it is thought that vemurafenib potentiates tumorigenesis in subclinical lesions harboring upstream MAPK pathway mutations as opposed to inducing de novo lesions.6
Because BRAF inhibitors are remarkably efficacious in the treatment of metastatic melanomas harboring the V600E BRAF mutation, there are no restrictions on their use, despite the known increased risk for SCC. Squamous cell carcinomas tend to be low grade, and all tumors that developed in phase 1 to 3 trials were treated with simple excision. The development of SCC did not necessitate interruption of treatment. Furthermore, the addition of MEK inhibition to BRAF inhibitor therapy reduces the risk for SCC from 19% to 7%.7,10,11
In addition to SCC, second primary melanomas (SPMs) have been reported in patients treated with BRAF inhibitors. It has been shown that these melanomas occur in melanocytes with wild-type BRAF. It has been postulated that some of these tumors occur in cells that harbor upstream mutations in RAS, whereas others might result from alternate signaling through non-RAF oncogenic pathways.9,12
Zimmer et al1 reported 12 SPMs in 11 patients treated with BRAF inhibitor therapy. They reported a median delay of 8 weeks (range, 4–27 weeks) for SPM development. Tumors were detected in early stages; 1 tumor harbored an NRAS mutation.1
Dalle et al13 reported 25 SPMs in 120 vemurafenib-treated patients. Median delay in SPM development was 14 weeks (range, 4–42 weeks). All tumors were thin, ranging from in situ to 0.45-mm thick. Wild-type BRAF was detected in the 21 melanomas sampled; 1 lesion showed mutated NRAS.13
The exact incidence of SPM in the setting of BRAF inhibition is thought to be at least 10-fold less than SCC and KA.2 Patients on BRAF inhibitor therapy should have routine full-body skin examinations, given the increased risk for SPM and SCC.
Another drug belonging to the tyrosine kinase inhibitor family, sorafenib, is used in the treatment of solid tumors, particularly hepatocellular and renal cell carcinomas, and also has been associated with development of cutaneous SCC and KAs.14 Sorafenib is a multiple tyrosine kinase inhibitor that also inhibits the RAF serine/threonine kinases. Similar to vemurafenib and dabrafenib, SCCs and KAs associated with sorafenib tend to arise in patients with chronic actinic damage during the first 2 months of treatment. It has been hypothesized that inhibition of RAF kinases is pathogenic in inducing SCCs because these lesions have not been reported with sunitinib, another multiple tyrosine kinase inhibitor that lacks the ability to inhibit serine/threonine kinases.15,16 Although SCCs and KAs associated with sorafenib tend to be low grade, it is reasonable to consider sunitinib or an alternative tyrosine kinase inhibitor in patients who develop multiple SCCs while taking sorafenib.16
Sonic Hedgehog–Inhibiting Agents
Vismodegib, the first small molecule inhibitor of the signaling protein smoothened, gained FDA approval for the treatment of metastatic or locally advanced basal cell carcinoma (BCC) in 2012. A second agent with an identical mechanism of action, sonidegib, was approved by the FDA for locally advanced BCC in 2015. Approximately 90% of BCCs contain mutations in the sonic hedgehog pathway, which lead to constitutive smoothened activation and uncontrolled cell proliferation.17 The development of smoothened inhibitors introduced a much-needed treatment for inoperable or metastatic BCC,17,18 though long-term utility is limited by drug resistance with extended use in this patient population.19,20 Several case reports have documented the emergence of KA21 and cutaneous SCC following vismodegib treatment of advanced or metastatic BCC.22-24 A larger case-control study by Mohan et al25 showed that patients with BCC treated with vismodegib had an increased risk for non-BCC malignancy (hazard ratio [HR]=6.37), most of which were cutaneous SCC (HR=8.12).
The mechanism by which selective inhibition of smoothened leads to cutaneous SCC is unclear. A study found that patients on vismodegib who developed SCC within the original BCC site had elevated ERK levels within tumor tissue, suggesting that the RAS/RAF/MAPK pathway can become upregulated during hedgehog inhibition.26 Other studies looking at hedgehog inhibition in medulloblastoma models also have shown activated RAS/RAF/MAPK pathways.25 These findings suggest that tumors under smoothened inhibition might be able to bypass the sonic hedgehog pathway and continue to grow by upregulating alternative growth pathways, such as RAS/RAF/MAPK.25,26
The incidence of cutaneous SCC following vismodegib treatment is unknown. Chang and Oro27 examined BCC tumor regrowth from secondary (acquired) resistance to vismodegib and noted that lesions recurred within 1 cm of the original tumor 21% of the time. Although none of the 12 patients whose tumors regrew during treatment were reported to have developed SCC, several demonstrated different BCC subtypes than the pretreatment specimen. The authors proposed that regrowth of BCC was due to upregulated alternative pathways allowing tumors to bypass smoothened inhibition, which is similar to the proposed mechanism for SCC development in vismodegib patients.27
Prospective studies are needed to confirm the link between vismodegib and cutaneous SCC; establish the incidence of SCC development; and identify any pretreatment factors, tumor characteristics, or treatment details (eg, dosage, duration) that might contribute to SCC development. Furthermore, because Mohan et al25 observed that vismodegib-treated patients were less likely to develop SCC in situ than controls, it is unknown if these tumors are more aggressive than traditional SCC. At this point, careful surveillance and regular full-body skin examinations are advised for patients on vismodegib for treatment of advanced BCC.
JAK Inhibitors
Another class of medications potentially associated with increased development of nonmelanoma skin cancer (NMSC) is the JAK inhibitors (also known as jakinibs). Many proinflammatory signaling pathways converge on the JAK family of enzymes—JAK1, JAK2, JAK3, and TYK2. These enzymes operate in cytokine signal transduction by phosphorylating activated cytokine receptors, which allows for recruitment and activation by means of phosphorylation of transcription factors collectively known as signal transducers and activators of transcription (STATs). Phosphorylated STATs dimerize and translocate to the nucleus, acting as direct transcription promoters. Janus kinase inhibitors modulate the immune response by reducing the effect of interleukin and interferon signaling.
Ruxolitinib, a JAK1/JAK2 inhibitor, was the first JAK inhibitor approved by the FDA and is indicated for the treatment of myelofibrosis and polycythemia vera. Additionally, oral and topical JAK inhibitors have shown efficacy in the treatment of psoriasis, rheumatoid arthritis, alopecia areata, vitiligo, and pruritus from atopic dermatitis.28
The JAK-STAT pathway is complex, and the biological activity of the pathway is both proinflammatory and pro–cell survival and proliferation. Because signaling through the pathway can increase angiogenesis and inhibit apoptosis, inhibition of this pathway has been exploited for the treatment of some tumors. However, inhibition of interferon and proinflammatory interleukin signaling also can potentially promote tumor growth by means of inhibition of downstream cytotoxic T-cell signaling, theoretically increasing the risk for NMSC. A study examining the 5-year efficacy of ruxolitinib in myelofibrosis patients (COMFORT-II trial) found that 17.1% of patients developed NMSC compared to only 2.7% of those on the best available therapy. After adjustment by patient exposure, the NMSC rate was still doubled for ruxolitinib-treated patients compared to controls (6.1/100 patient-years and 3.0/100 patient-years, respectively).29 Eighty-week follow-up of the phase 3 clinical trial of ruxolitinib for the treatment of polycythemia vera also noted an increased incidence of NMSC, albeit a more conservative increase. Patients randomized to the ruxolitinib treatment group developed NMSC at a rate of 4.4/100 patient-years, whereas the rate for controls treated with best available therapy was 2.7/100 patient-years.30 In contrast, 5-year follow-up of the COMFORT-I trial, also examining the efficacy of ruxolitinib in myelofibrosis, showed no increased risk for NMSC between ruxolitinib-treated patients and placebo (2.7/100 patient-years and 3.9/100 patient-years, respectively).31
A 2017 case series described 5 patients with myelofibrosis who developed multiple skin cancers with aggressive features while receiving ruxolitinib.32 Duration of ruxolitinib therapy ranged from 4 months to 4 years; 3 patients had a history of hydroxyurea exposure, and only 1 patient had a history of NMSC. High-risk cutaneous SCC, undifferentiated pleomorphic sarcoma, and lentigo maligna melanoma (Breslow thickness, 0.45 mm) were among the tumors reported in this series. Although no definitive conclusion can be made regarding the causality of JAK inhibitors in promoting these tumors, the association warrants further investigation. Clinicians should be aware that ruxolitinib might amplify the risk for NMSC in patients with pre-existing genetic or exposure-related susceptibility. Interruption of drug therapy may be necessary in managing patients who develop an aggressive tumor.32
In contrast, tofacitinib, which specifically inhibits JAK3, carries very low risk, if any, for NMSC when used for the treatment of psoriasis and rheumatoid arthritis. Results from 2 phase 3 trials analyzing the efficacy of tofacitinib in psoriasis demonstrated that only 2 of 1486 patients treated developed NMSC compared to none in the control group.33 Furthermore, analysis of NMSC across the tofacitinib rheumatoid arthritis clinical program, which included a total of 15,103 patient-years of exposure, demonstrated that the overall NMSC incidence was 0.55 for every 100 patient-years. Of note, the risk in patients receiving high-dose treatment (10 mg vs 5 mg) was nearly doubled in long-term follow-up studies (0.79/100 patient-years and 0.41/100 patient-years, respectively). Overall, the study concluded that treatment with tofacitinib presents no greater increased risk for NMSC than treatment with tumor necrosis factor inhibitors.33
PDE-5 Inhibitors
Phosphodiesterase 5 inhibitors, such as sildenafil citrate, have been widely prescribed for the treatment of erectile dysfunction. Studies have shown that BRAF-activated melanomas, which occur in approximately 50% to 70% of melanomas, also result in reduced PDE-5 expression.34-36 In these melanomas, downregulation of PDE-5 results in increased intracellular calcium,36 which has been shown to induce melanoma invasion.36,37 Given this similarity in molecular pathway between BRAF-activated melanomas and PDE-5 inhibitors, there has been increased concern that PDE-5 inhibitors might be associated with an increased risk for melanoma.
In 2014, Li et al38 published a retrospective analysis suggesting an association with sildenafil and an increased risk for melanoma. Their study utilized the Health Professionals Follow-up Study to identify a statistically significant elevation in the risk for invasive melanoma with both recent sildenafil use (multivariate-adjusted HR=2.24) and use at any time (HR=1.92). These results controlled for confounding variables, such as presence of major chronic disease, use of other erectile dysfunction treatments, family history of melanoma, history of sun exposure, and UV index of the patient’s residence. Notably, the study also found that sildenafil did not affect the incidence of BCC or SCC.38
In 2015, Loeb et al39 also examined the potential association between PDE-5 inhibitors and melanoma. Review of several Swedish drug and cancer registries allowed for analysis of melanoma risk and PDE-5 inhibitor use, based on number of prescriptions filled and type of PDE-5 inhibitor prescribed. Their analysis showed that men developing melanoma were more likely than nonmelanoma controls to have taken a PDE-5 inhibitor (11% vs 8%). In a subgroup analysis, however, statistical significance was shown for men with only a single prescription filled (34% of cases; P<.05), whereas the difference for men with multiple filled prescriptions did not meet statistical significance. Furthermore, the study did not find increased risk with longer-acting tadalafil and vardenafil (odds ratio [OR]=1.16) compared to sildenafil (OR=1.14). Last, use of PDE-5 inhibitors was only associated with stage 0 (OR=1.49) and stage I (OR=1.21) tumors, not with stages II to IV (OR=0.83) tumors. Although there was a statistically significant association between PDE-5 inhibitors and malignant melanoma (P<.05), the subgroup analysis findings pointed away from a causal relationship and likely toward a confounding of variable(s).39
A 2016 study by Lian et al40 looked at the risk for melanoma in a cohort of patients diagnosed with erectile dysfunction. No association between PDE-5 inhibitors and melanoma risk was shown when comparing patients who received a PDE-5 inhibitor and those who did not receive a PDE-5 inhibitor. However, secondary analysis did show that melanoma risk was increased among patients receiving more pills (34%) and prescriptions (30%). The authors concluded that there was no association between PDE-5 inhibitor use and overall increased risk for melanoma, and the increased risk associated with a greater number of pills and prescriptions would require further study.40
In contrast, a 2017 meta-analysis by Tang et al41 of 5 studies (3 of which were the aforementioned trials38-40) concluded that use of PDE-5 inhibitors was associated with a small but significantly increased risk for melanoma (OR=1.12) and BCC (OR=1.14) but not SCC. Furthermore, the study found no evidence of dosage-dependent association between PDE-5 inhibitor use and melanoma risk.41
Overall, clinical studies have been inconclusive in determining the risk for melanoma in the setting of PDE-5 inhibitor use. Studies showing an increased rate of melanoma within patient cohorts receiving PDE-5 inhibitors are limited; results might be affected by confounding variables. However, given the similarity in mechanism between PDE-5 inhibitors and HRAS-activated melanomas, it is reasonable to continue research into this potential association.
Conclusion
Since the turn of the century, drugs targeting cell-signaling pathways have been developed to treat inflammatory, oncologic, and immune conditions. The role of immunosuppressants in promoting skin cancer is well established and supported by a vast literature base. However, associations are less clear with newer immunomodulatory and antineoplastic medications. Skin cancer has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, JAK inhibitors, and PDE-5 inhibitors. In the case of JAK and PDE-5 inhibitors, the increased risk for melanoma and NMSC is somewhat inconclusive; risk is more firmly established for BRAF inhibitors and smoothened inhibitors. For the antineoplastic agents reviewed, the therapeutic effect of cancer regression is well documented, and benefits of continued therapy outweigh the increased risk for skin cancer promotion in nearly all cases. The value of early detection has been well documented for skin malignancy; therefore, increased skin surveillance and prompt management of suspicious lesions should be a priority for physicians treating patients undergoing therapy with these medications
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanoma in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
- Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA Dermatol. 2015;151:1103-1109.
- Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427-430.
- Ryan MB, Der CJ, Wang-Gillam A, et al. Targeting RAS-mutant cancers: is ERK the key? Trends Cancer. 2015;1:183-198.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
- Holderfield M, Nagel TE, Stuart DD. Mechanism and consequence of RAF kinase activation by small-molecule inhibitors. Br J Cancer. 2014;111:640-645.
- Dalle S, Poulalhon N, Debarbieux S, et al. Tracking of second primary melanomas in vemurafenib-treated patients. JAMA Dermatol. 2013;149:488-490.
- Williams VL, Cohen PR, Stewart DJ. Sorafenib-induced premalignant and malignant skin lesions. Int J Dermatol. 2011;50:396-402.
- Arnault JP, Wechsler J, Escudier B, et al. Keratoacanthomas and squamous cell carcinomas in patients receiving sorafenib. J Clin Oncol. 2009;27:e59-e61.
- Smith KJ, Haley H, Hamza S, et al. Eruptive keratoacanthoma-type squamous cell carcinomas in patients taking sorafenib for the treatment of solid tumors. Dermatol Surg. 2009;35:1766-1770.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Demirci H, Worden F, Nelson CC, et al. Efficacy of vismodegib (Erivedge) for basal cell carcinoma involving the orbit and periocular area. Ophthalmic Plast Reconstr Surg. 2015;31:463-466.
- Atwood SX, Sarin KY, Whitson RJ, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342-353.
- Ridky TW, Cotsarelis G. Vismodegib resistance in basal cell carcinoma: not a smooth fit. Cancer Cell. 2015;27:315-316.
- Aasi S, Silkiss R, Tang JY, et al. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: a report of 2 cases. JAMA Dermatol. 2013;149:242-243.
- Orouji A, Goerdt S, Utikal J, et al. Multiple highly and moderately differentiated squamous cell carcinomas of the skin during vismodegib treatment of inoperable basal cell carcinoma. Br J Dermatol. 2014;171:431-433.
- Iarrobino A, Messina JL, Kudchadkar R, et al. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2013;69:e33-e34.
- Saintes C, Saint-Jean M, Brocard A, et al. Development of squamous cell carcinoma into basal cell carcinoma under treatment with vismodegib. J Eur Acad Dermatol Venereol. 2015;29:1006-1009.
- Mohan SV, Chang J, Li S, et al. Increased risk of cutaneous squamous cell carcinoma after vismodegib therapy for basal cell carcinoma. JAMA Dermatol. 2016;152:527-532.
- Zhao X, Ponomaryov T, Ornell KJ, et al. RAS/MAPK activation drives resistance to Smo inhibition, metastasis, and tumor evolution in Shh pathway-dependent tumors. Cancer Res. 2015;75:3623-3635.
- Chang AL, Oro AE. Initial assessment of tumor regrowth after vismodegib in advanced basal cell carcinoma. Arch Dermatol. 2012;148:1324-1325.
- Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76:736-744.
- Harrison CN, Vannucchi AM, Kiladjian JJ, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30:1701-1707.
- Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica. 2016;101:821-829.
- Verstovsek S, Mesa RA, Gotlib J, et al; COMFORT-I investigators. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017;10:55.
- Blechman AB, Cabell CE, Weinberger CH, et al. Aggressive skin cancers occurring in patients treated with the Janus kinase inhibitor ruxolitinib. J Drugs Dermatol. 2017;16:508-511.
- Papp KA, Menter MA, Abe M, et al; OPT Pivotal 1 and OPT Pivotal 2 investigators. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two randomized, placebo-controlled, phase III trials. Br J Dermatol. 2015;173:949-961.
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875-885.
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851-857.
- Arozarena I, Sanchez-Laorden B, Packer L, et al. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell. 2011;19:45-57.
- Houslay MD. Hard times for oncogenic BRAF-expressing melanoma cells. Cancer Cell. 2011;19:3-4.
- Li WQ, Qureshi AA, Robinson KC, et al. Sildenafil use and increased risk of incident melanoma in US men: a prospective cohort study. JAMA Intern Med. 2014;174:964-970.
- Loeb S, Folkvaljon Y, Lambe M, et al. Use of phosphodiesterase type 5 inhibitors for erectile dysfunction and risk of malignant melanoma. JAMA. 2015;313:2449-2455.
- Lian Y, Yin H, Pollak MN, et al. Phosphodiesterase type 5 inhibitors and the risk of melanoma skin cancer. Eur Urol. 2016;70:808-815.
- Tang H, Wu W, Fu S, et al. Phosphodiesterase type 5 inhibitors and risk of melanoma: a meta-analysis. J Am Acad Dermatol. 2017;77:480.e9-488.e9.
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanoma in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
- Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA Dermatol. 2015;151:1103-1109.
- Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427-430.
- Ryan MB, Der CJ, Wang-Gillam A, et al. Targeting RAS-mutant cancers: is ERK the key? Trends Cancer. 2015;1:183-198.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
- Holderfield M, Nagel TE, Stuart DD. Mechanism and consequence of RAF kinase activation by small-molecule inhibitors. Br J Cancer. 2014;111:640-645.
- Dalle S, Poulalhon N, Debarbieux S, et al. Tracking of second primary melanomas in vemurafenib-treated patients. JAMA Dermatol. 2013;149:488-490.
- Williams VL, Cohen PR, Stewart DJ. Sorafenib-induced premalignant and malignant skin lesions. Int J Dermatol. 2011;50:396-402.
- Arnault JP, Wechsler J, Escudier B, et al. Keratoacanthomas and squamous cell carcinomas in patients receiving sorafenib. J Clin Oncol. 2009;27:e59-e61.
- Smith KJ, Haley H, Hamza S, et al. Eruptive keratoacanthoma-type squamous cell carcinomas in patients taking sorafenib for the treatment of solid tumors. Dermatol Surg. 2009;35:1766-1770.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Demirci H, Worden F, Nelson CC, et al. Efficacy of vismodegib (Erivedge) for basal cell carcinoma involving the orbit and periocular area. Ophthalmic Plast Reconstr Surg. 2015;31:463-466.
- Atwood SX, Sarin KY, Whitson RJ, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342-353.
- Ridky TW, Cotsarelis G. Vismodegib resistance in basal cell carcinoma: not a smooth fit. Cancer Cell. 2015;27:315-316.
- Aasi S, Silkiss R, Tang JY, et al. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: a report of 2 cases. JAMA Dermatol. 2013;149:242-243.
- Orouji A, Goerdt S, Utikal J, et al. Multiple highly and moderately differentiated squamous cell carcinomas of the skin during vismodegib treatment of inoperable basal cell carcinoma. Br J Dermatol. 2014;171:431-433.
- Iarrobino A, Messina JL, Kudchadkar R, et al. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2013;69:e33-e34.
- Saintes C, Saint-Jean M, Brocard A, et al. Development of squamous cell carcinoma into basal cell carcinoma under treatment with vismodegib. J Eur Acad Dermatol Venereol. 2015;29:1006-1009.
- Mohan SV, Chang J, Li S, et al. Increased risk of cutaneous squamous cell carcinoma after vismodegib therapy for basal cell carcinoma. JAMA Dermatol. 2016;152:527-532.
- Zhao X, Ponomaryov T, Ornell KJ, et al. RAS/MAPK activation drives resistance to Smo inhibition, metastasis, and tumor evolution in Shh pathway-dependent tumors. Cancer Res. 2015;75:3623-3635.
- Chang AL, Oro AE. Initial assessment of tumor regrowth after vismodegib in advanced basal cell carcinoma. Arch Dermatol. 2012;148:1324-1325.
- Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76:736-744.
- Harrison CN, Vannucchi AM, Kiladjian JJ, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30:1701-1707.
- Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica. 2016;101:821-829.
- Verstovsek S, Mesa RA, Gotlib J, et al; COMFORT-I investigators. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017;10:55.
- Blechman AB, Cabell CE, Weinberger CH, et al. Aggressive skin cancers occurring in patients treated with the Janus kinase inhibitor ruxolitinib. J Drugs Dermatol. 2017;16:508-511.
- Papp KA, Menter MA, Abe M, et al; OPT Pivotal 1 and OPT Pivotal 2 investigators. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two randomized, placebo-controlled, phase III trials. Br J Dermatol. 2015;173:949-961.
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875-885.
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851-857.
- Arozarena I, Sanchez-Laorden B, Packer L, et al. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell. 2011;19:45-57.
- Houslay MD. Hard times for oncogenic BRAF-expressing melanoma cells. Cancer Cell. 2011;19:3-4.
- Li WQ, Qureshi AA, Robinson KC, et al. Sildenafil use and increased risk of incident melanoma in US men: a prospective cohort study. JAMA Intern Med. 2014;174:964-970.
- Loeb S, Folkvaljon Y, Lambe M, et al. Use of phosphodiesterase type 5 inhibitors for erectile dysfunction and risk of malignant melanoma. JAMA. 2015;313:2449-2455.
- Lian Y, Yin H, Pollak MN, et al. Phosphodiesterase type 5 inhibitors and the risk of melanoma skin cancer. Eur Urol. 2016;70:808-815.
- Tang H, Wu W, Fu S, et al. Phosphodiesterase type 5 inhibitors and risk of melanoma: a meta-analysis. J Am Acad Dermatol. 2017;77:480.e9-488.e9.
Practice Points
- Patients should be educated about the increased risk for skin malignancy while undergoing treatment with BRAF inhibitors, sonic hedgehog–inhibiting agents, Janus kinase (JAK) inhibitors, and phosphodiesterase 5 (PDE-5) inhibitors.
- For BRAF inhibitors, sonic hedgehog–inhibiting agents, and JAK inhibitors, the increased risk for skin cancer warrants regular surveillance; however, given the indications for these medications, many patients will already be receiving regular skin screenings.
- The association between PDE-5 inhibitors and melanoma as well as nonmelanoma skin cancer remains questionable, and increased skin surveillance is not recommended at this time, unless patients have other risk factors for cutaneous malignancy.
