User login
The Diagnosis: Interstitial Granulomatous Dermatitis
Interstitial granulomatous dermatitis (IGD) is rare, and the exact incidence is unknown, with only a few cases reported in the literature annually.1 Although IGD may arise in both children and adults, it occurs more commonly in adults, with an age of onset of 52 to 58.5 years. Interstitial granulomatous dermatitis also shows a female predominance.1
Interstitial granulomatous dermatitis may present as annular flesh-colored or erythematous to violaceous papules and plaques, or less commonly erythematous linear cordlike subcutaneous nodules (called the rope sign).1 Lesions often are asymptomatic but may be pruritic or tender. Interstitial granulomatous dermatitis has been associated with autoimmune conditions such as rheumatoid arthritis, systemic lupus erythematosus, and primary biliary cholangitis, and rarely malignancy.2 Interstitial granulomatous drug reactions can occur months to years after initiation of therapy with offending agents, and common causes include calcium channel blockers, statins, and tumor necrosis factor α inhibitors.3
Interstitial granulomatous dermatitis and palisaded neutrophilic and granulomatous dermatitis (PNGD) demonstrate overlapping clinical features and are thought to be part of the same spectrum of granulomatous dermatitis.4 Both IGD and PNGD may present with symmetric flesh-colored to erythematous papules or erythematous annular or linear plaques.5 Interstitial granulomatous dermatitis and PNGD may be differentiated through histopathologic examination.
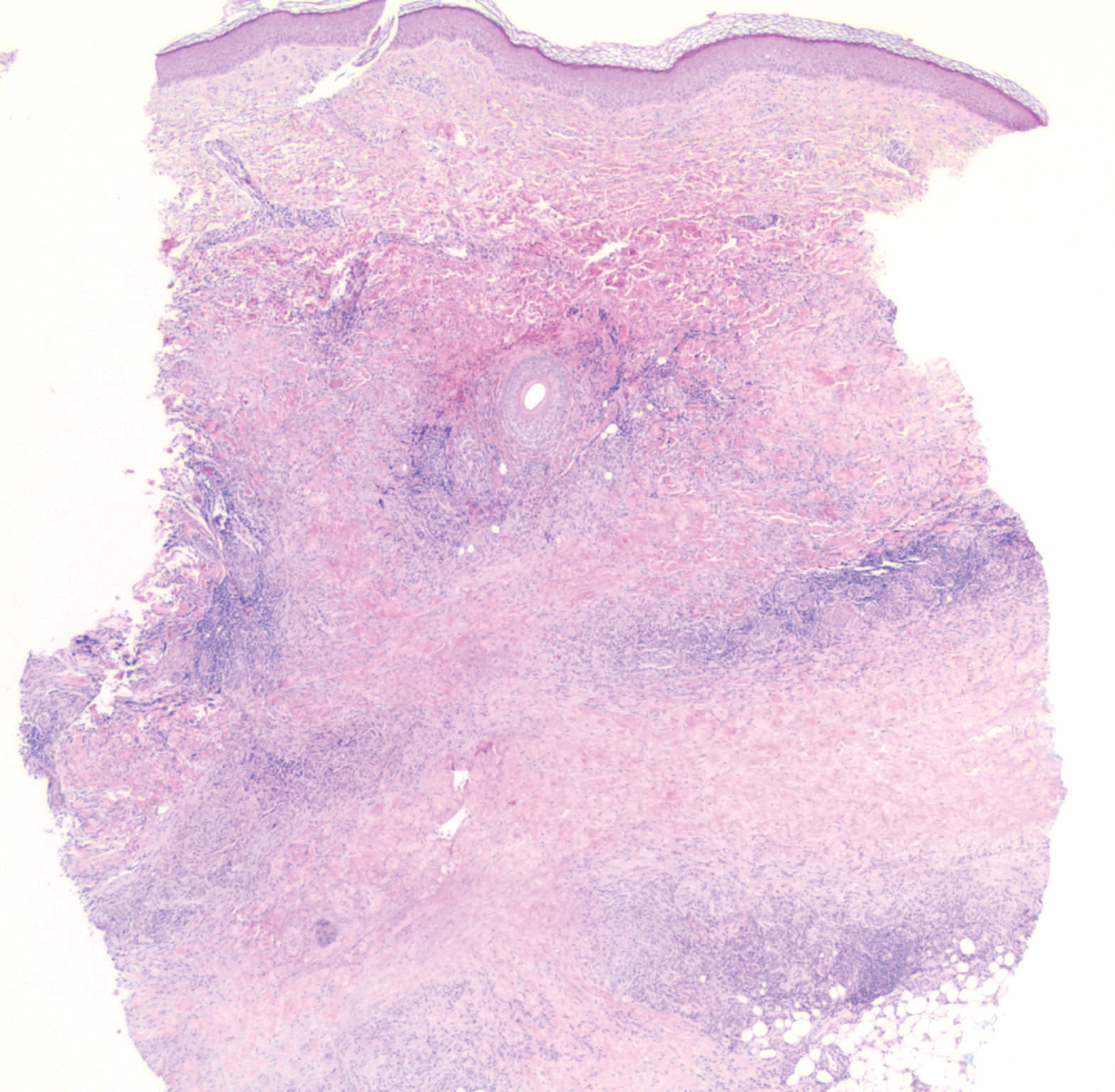
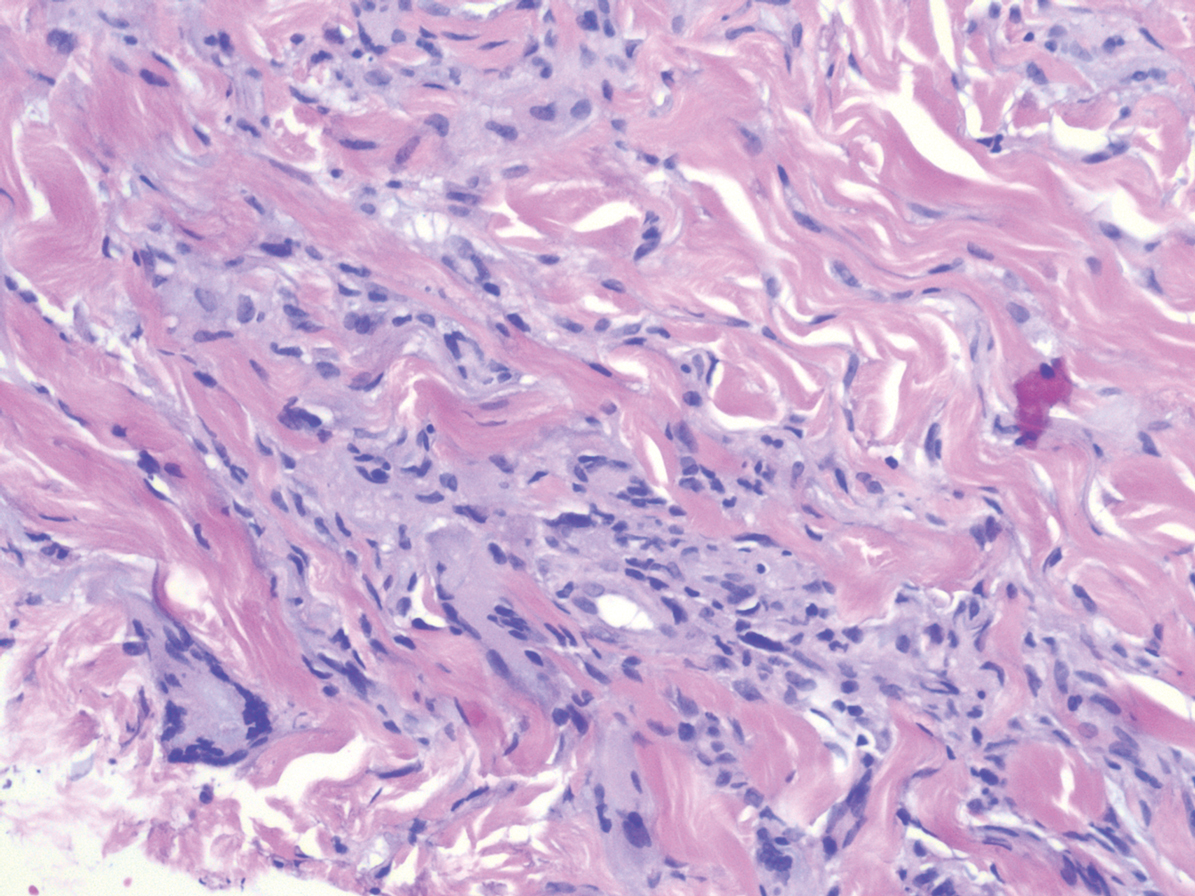
Histopathology of IGD shows an interstitial infiltrate of epithelioid histiocytes in the dermis, often surrounding foci of degenerated collagen resembling palisading granulomas (quiz images).1 Perivascular and interstitial lymphocytic infiltrates also are present in most cases. Epidermal changes are minimal in IGD but can be associated with interstitial granulomatous drug reactions.1 There usually is no vasculitis, and mucin typically is absent, unlike granuloma annulare (GA).3,6 In comparison, histopathologic examination of PNGD shows basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris with focal areas of leukocytoclastic vasculitis and rare mucin.5
No specific treatment is recommended, and lesions may resolve without any therapy. Reported treatments include topical, intralesional, or systemic steroids; nonsteroidal anti-inflammatory drugs; methotrexate; hydroxychloroquine; and cyclosporine.6 Due to the strong association with systemic diseases, it is important to evaluate patients with IGD for autoimmune diseases and conduct age-appropriate cancer screening. Furthermore, a review of medications is warranted to assess the possibility of interstitial granulomatous drug reactions.6 In our patient, rheumatologic workup and age-appropriate cancer screenings were negative, and the rash spontaneously resolved without treatment.
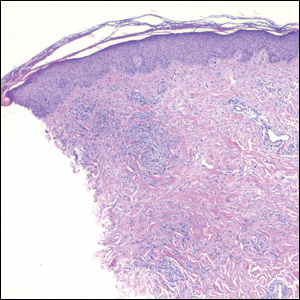
Granuloma annulare presents with asymptomatic flesh-colored to erythematous papules and plaques in an annular configuration. In the localized variant of GA, plaques frequently localize to the distal extremities, especially the dorsal hands, as in our patient. Other variants include generalized GA, subcutaneous GA, and perforating GA. Mucin and a palisading or interstitial pattern of granulomatous inflammation are key features on histopathology in all subtypes of GA (Figure 1).7 Patch GA is a rare variant that presents with asymptomatic erythematous to brown patches, is associated with interstitial-type inflammation on histopathology, and can be difficult to distinguish from IGD.8 Granuloma annulare with interstitial inflammation on histology can be differentiated from IGD by the comparative lack of mucin in IGD.7
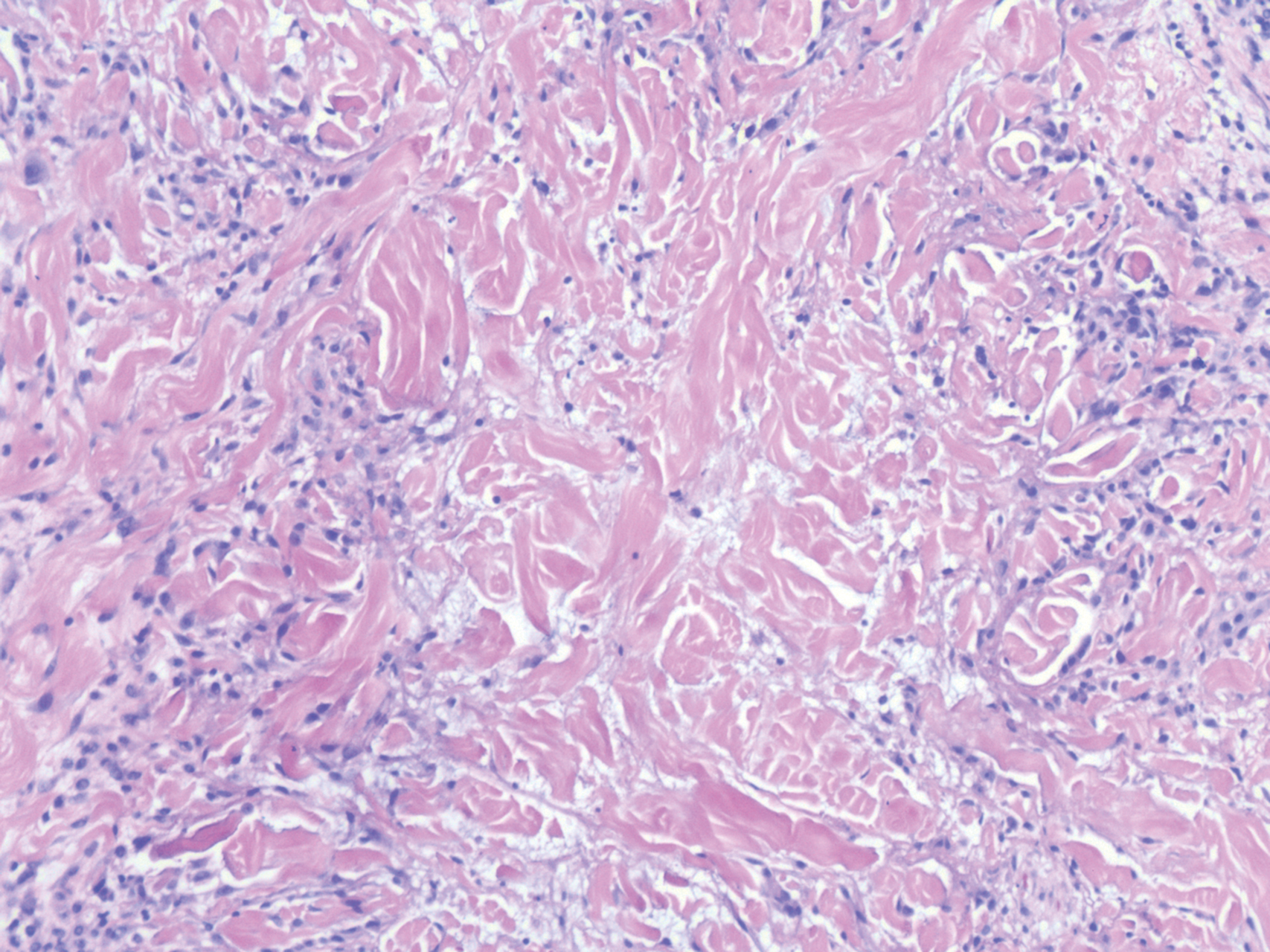
Sweet syndrome (SS) is characterized by sudden-onset, painful, erythematous plaques and/or nodules, commonly associated with fever and leukocytosis. Clinical variants of SS include pustular and bullous SS; giant cellulitis-like SS; necrotizing SS; and neutrophilic dermatosis of the dorsal hands presenting with hemorrhagic bullae, plaques, and pustules.7-9 Histopathologic examination shows dense nodular or perivascular neutrophilic infiltrate in the dermis without evidence of vasculitis (Figure 2).10 Histopathologic variants include histiocytoid, lymphocytic, subcutaneous, and cryptococcoid.9 The classic variant of SS has a bandlike, predominantly neutrophilic infiltrate with marked leukocytoclasia, which can be differentiated from the histiocytoid infiltrate of IGD.11 It has been shown that the infiltrate of the histiocytoid variant of SS is composed of myeloperoxidase-positive, immature myeloid cells rather than true histiocytes, and therefore can be differentiated from IGD.12 Lastly, all variants of SS have dermal edema, which typically is absent in IGD, and SS has no evidence of necrobiosis.
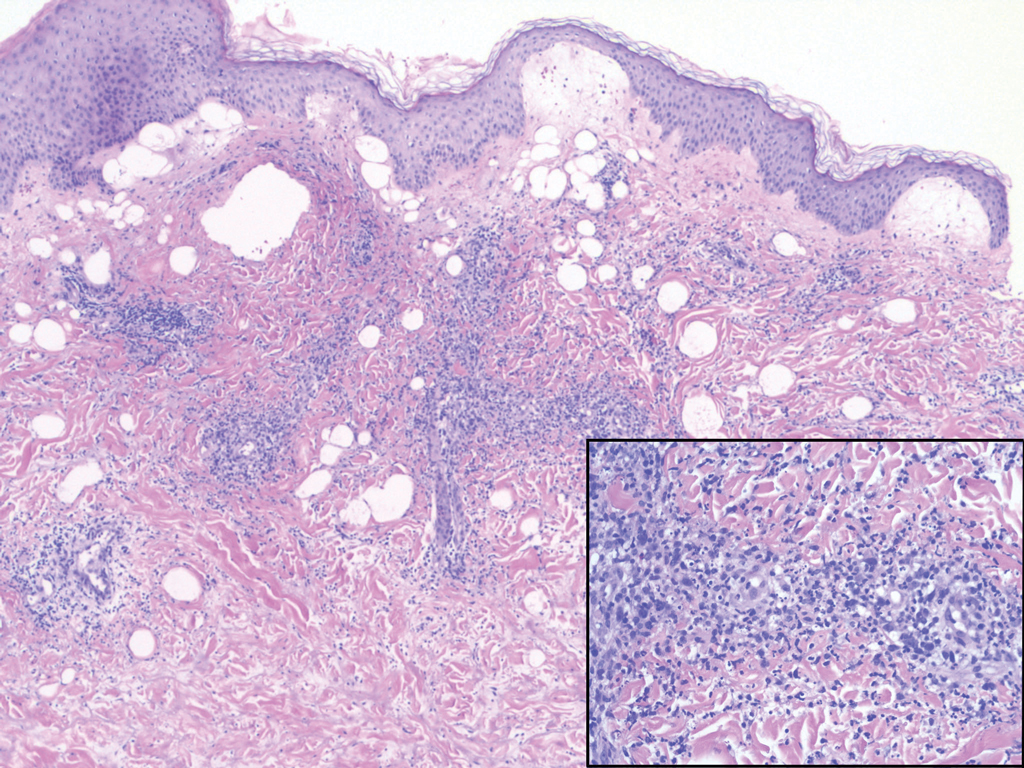
Erythema elevatum diutinum (EED) is a rare disease that presents with bilateral violaceous or erythematous to brown papules, plaques, or nodules. Lesions frequently localize to extensor surfaces, including the hands and fingers, and may be asymptomatic or associated with pruritus, burning, or tingling.13 Early EED lesions are characterized by leukocytoclastic vasculitis of the papillary and mid-dermal vessels with a perivascular neutrophilic infiltrate and perivascular fibrinoid necrosis. With older EED lesions, dermal and perivascular onion skin-like fibrosis become more prominent (Figure 3).14 The neutrophilic infiltrate, dermal fibrosis, and chronic vasculitic changes distinguish EED from IGD.
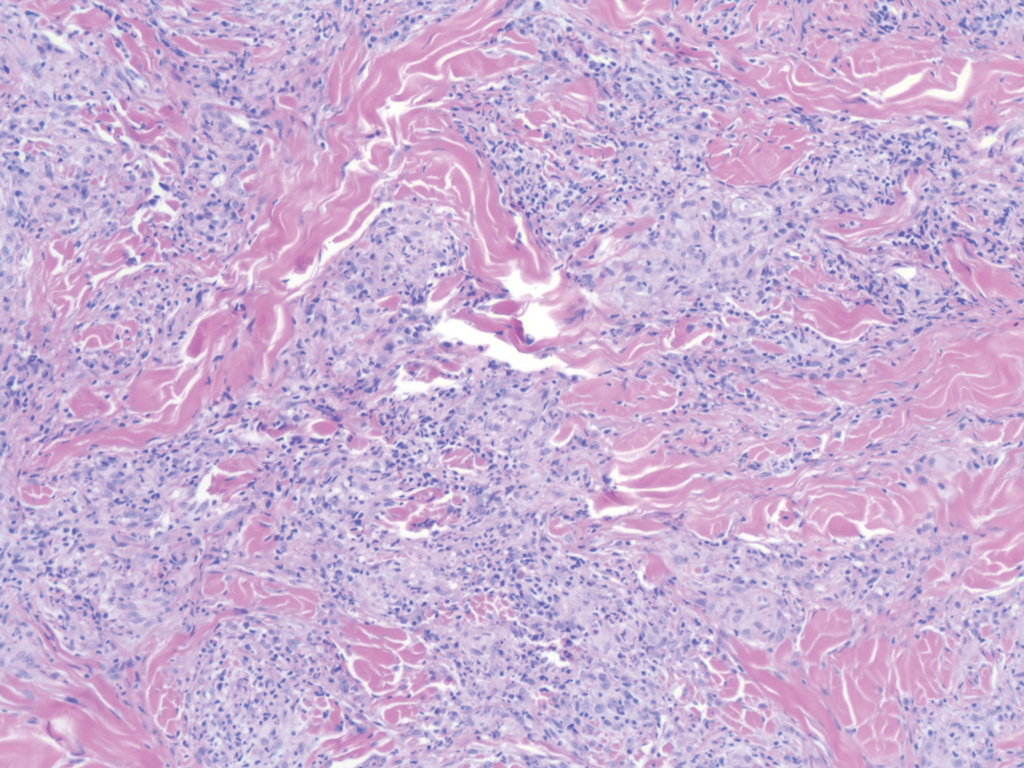
Necrobiosis lipoidica (NL) is a rare disease that presents with well-demarcated, yellow to red-brown papules and nodules most commonly localized to the bilateral lower extremities on the pretibial area. Papules and nodules evolve into plaques over time, and ulceration is common.15 On histopathology, NL primarily exhibits granulomatous inflammation with parallel palisading (Figure 4). The hallmark feature is necrobiosis--or degeneration--of collagen; the alternation of necrobiotic collagen and inflammatory infiltrate creates a layered cake-like appearance on low power.16 The clinical presentation as well as the dermal necrobiotic granuloma consisting of a large confluent area of necrobiosis centered in the superficial dermis and subcutaneous tissue of NL distinguishes it from the histiocytic infiltrate of IGD.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Terziroli Beretta-Piccoli B, Mainetti C, Peeters MA, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1644-1663.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Huizenga T, Kado JA, Pellicane B, et al. Interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis. Cutis. 2018;101:E19-E21.
- Rosenbach M, English JC 3rd. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Mutasim DF, Bridges AG. Patch granuloma annulare: clinicopathologic study of 6 patients. J Am Acad Dermatol. 2000;42:417-421.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006.
- Dabade TS, Davis MD. Diagnosis and treatment of the neutrophilic dermatoses (pyoderma gangrenosum, Sweet's syndrome). Dermatol Ther. 2011;24:273-284.
- Davis M, Moschella L. Neutrophilic dermatoses. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:2102-2112.
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Sardiña LA, Jour G, Piliang MP, et al. Erythema elevatum diutinum a rare and poorly understood cutaneous vasculitis: a single institution experience. J Cutan Pathol. 2019;46:97-101.
- Reid SD, Ladizinski B, Lee K, et al. Update on necrobiosis lipoidica: a review of etiology, diagnosis, and treatment options. J Am Acad Dermatol. 2013;69:783-791.
- Sibbald C, Reid S, Alavi A. Necrobiosis lipoidica. Dermatol Clin. 2015;33:343-360.
The Diagnosis: Interstitial Granulomatous Dermatitis
Interstitial granulomatous dermatitis (IGD) is rare, and the exact incidence is unknown, with only a few cases reported in the literature annually.1 Although IGD may arise in both children and adults, it occurs more commonly in adults, with an age of onset of 52 to 58.5 years. Interstitial granulomatous dermatitis also shows a female predominance.1
Interstitial granulomatous dermatitis may present as annular flesh-colored or erythematous to violaceous papules and plaques, or less commonly erythematous linear cordlike subcutaneous nodules (called the rope sign).1 Lesions often are asymptomatic but may be pruritic or tender. Interstitial granulomatous dermatitis has been associated with autoimmune conditions such as rheumatoid arthritis, systemic lupus erythematosus, and primary biliary cholangitis, and rarely malignancy.2 Interstitial granulomatous drug reactions can occur months to years after initiation of therapy with offending agents, and common causes include calcium channel blockers, statins, and tumor necrosis factor α inhibitors.3
Interstitial granulomatous dermatitis and palisaded neutrophilic and granulomatous dermatitis (PNGD) demonstrate overlapping clinical features and are thought to be part of the same spectrum of granulomatous dermatitis.4 Both IGD and PNGD may present with symmetric flesh-colored to erythematous papules or erythematous annular or linear plaques.5 Interstitial granulomatous dermatitis and PNGD may be differentiated through histopathologic examination.
Histopathology of IGD shows an interstitial infiltrate of epithelioid histiocytes in the dermis, often surrounding foci of degenerated collagen resembling palisading granulomas (quiz images).1 Perivascular and interstitial lymphocytic infiltrates also are present in most cases. Epidermal changes are minimal in IGD but can be associated with interstitial granulomatous drug reactions.1 There usually is no vasculitis, and mucin typically is absent, unlike granuloma annulare (GA).3,6 In comparison, histopathologic examination of PNGD shows basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris with focal areas of leukocytoclastic vasculitis and rare mucin.5
No specific treatment is recommended, and lesions may resolve without any therapy. Reported treatments include topical, intralesional, or systemic steroids; nonsteroidal anti-inflammatory drugs; methotrexate; hydroxychloroquine; and cyclosporine.6 Due to the strong association with systemic diseases, it is important to evaluate patients with IGD for autoimmune diseases and conduct age-appropriate cancer screening. Furthermore, a review of medications is warranted to assess the possibility of interstitial granulomatous drug reactions.6 In our patient, rheumatologic workup and age-appropriate cancer screenings were negative, and the rash spontaneously resolved without treatment.
Granuloma annulare presents with asymptomatic flesh-colored to erythematous papules and plaques in an annular configuration. In the localized variant of GA, plaques frequently localize to the distal extremities, especially the dorsal hands, as in our patient. Other variants include generalized GA, subcutaneous GA, and perforating GA. Mucin and a palisading or interstitial pattern of granulomatous inflammation are key features on histopathology in all subtypes of GA (Figure 1).7 Patch GA is a rare variant that presents with asymptomatic erythematous to brown patches, is associated with interstitial-type inflammation on histopathology, and can be difficult to distinguish from IGD.8 Granuloma annulare with interstitial inflammation on histology can be differentiated from IGD by the comparative lack of mucin in IGD.7
Sweet syndrome (SS) is characterized by sudden-onset, painful, erythematous plaques and/or nodules, commonly associated with fever and leukocytosis. Clinical variants of SS include pustular and bullous SS; giant cellulitis-like SS; necrotizing SS; and neutrophilic dermatosis of the dorsal hands presenting with hemorrhagic bullae, plaques, and pustules.7-9 Histopathologic examination shows dense nodular or perivascular neutrophilic infiltrate in the dermis without evidence of vasculitis (Figure 2).10 Histopathologic variants include histiocytoid, lymphocytic, subcutaneous, and cryptococcoid.9 The classic variant of SS has a bandlike, predominantly neutrophilic infiltrate with marked leukocytoclasia, which can be differentiated from the histiocytoid infiltrate of IGD.11 It has been shown that the infiltrate of the histiocytoid variant of SS is composed of myeloperoxidase-positive, immature myeloid cells rather than true histiocytes, and therefore can be differentiated from IGD.12 Lastly, all variants of SS have dermal edema, which typically is absent in IGD, and SS has no evidence of necrobiosis.
Erythema elevatum diutinum (EED) is a rare disease that presents with bilateral violaceous or erythematous to brown papules, plaques, or nodules. Lesions frequently localize to extensor surfaces, including the hands and fingers, and may be asymptomatic or associated with pruritus, burning, or tingling.13 Early EED lesions are characterized by leukocytoclastic vasculitis of the papillary and mid-dermal vessels with a perivascular neutrophilic infiltrate and perivascular fibrinoid necrosis. With older EED lesions, dermal and perivascular onion skin-like fibrosis become more prominent (Figure 3).14 The neutrophilic infiltrate, dermal fibrosis, and chronic vasculitic changes distinguish EED from IGD.
Necrobiosis lipoidica (NL) is a rare disease that presents with well-demarcated, yellow to red-brown papules and nodules most commonly localized to the bilateral lower extremities on the pretibial area. Papules and nodules evolve into plaques over time, and ulceration is common.15 On histopathology, NL primarily exhibits granulomatous inflammation with parallel palisading (Figure 4). The hallmark feature is necrobiosis--or degeneration--of collagen; the alternation of necrobiotic collagen and inflammatory infiltrate creates a layered cake-like appearance on low power.16 The clinical presentation as well as the dermal necrobiotic granuloma consisting of a large confluent area of necrobiosis centered in the superficial dermis and subcutaneous tissue of NL distinguishes it from the histiocytic infiltrate of IGD.
The Diagnosis: Interstitial Granulomatous Dermatitis
Interstitial granulomatous dermatitis (IGD) is rare, and the exact incidence is unknown, with only a few cases reported in the literature annually.1 Although IGD may arise in both children and adults, it occurs more commonly in adults, with an age of onset of 52 to 58.5 years. Interstitial granulomatous dermatitis also shows a female predominance.1
Interstitial granulomatous dermatitis may present as annular flesh-colored or erythematous to violaceous papules and plaques, or less commonly erythematous linear cordlike subcutaneous nodules (called the rope sign).1 Lesions often are asymptomatic but may be pruritic or tender. Interstitial granulomatous dermatitis has been associated with autoimmune conditions such as rheumatoid arthritis, systemic lupus erythematosus, and primary biliary cholangitis, and rarely malignancy.2 Interstitial granulomatous drug reactions can occur months to years after initiation of therapy with offending agents, and common causes include calcium channel blockers, statins, and tumor necrosis factor α inhibitors.3
Interstitial granulomatous dermatitis and palisaded neutrophilic and granulomatous dermatitis (PNGD) demonstrate overlapping clinical features and are thought to be part of the same spectrum of granulomatous dermatitis.4 Both IGD and PNGD may present with symmetric flesh-colored to erythematous papules or erythematous annular or linear plaques.5 Interstitial granulomatous dermatitis and PNGD may be differentiated through histopathologic examination.
Histopathology of IGD shows an interstitial infiltrate of epithelioid histiocytes in the dermis, often surrounding foci of degenerated collagen resembling palisading granulomas (quiz images).1 Perivascular and interstitial lymphocytic infiltrates also are present in most cases. Epidermal changes are minimal in IGD but can be associated with interstitial granulomatous drug reactions.1 There usually is no vasculitis, and mucin typically is absent, unlike granuloma annulare (GA).3,6 In comparison, histopathologic examination of PNGD shows basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris with focal areas of leukocytoclastic vasculitis and rare mucin.5
No specific treatment is recommended, and lesions may resolve without any therapy. Reported treatments include topical, intralesional, or systemic steroids; nonsteroidal anti-inflammatory drugs; methotrexate; hydroxychloroquine; and cyclosporine.6 Due to the strong association with systemic diseases, it is important to evaluate patients with IGD for autoimmune diseases and conduct age-appropriate cancer screening. Furthermore, a review of medications is warranted to assess the possibility of interstitial granulomatous drug reactions.6 In our patient, rheumatologic workup and age-appropriate cancer screenings were negative, and the rash spontaneously resolved without treatment.
Granuloma annulare presents with asymptomatic flesh-colored to erythematous papules and plaques in an annular configuration. In the localized variant of GA, plaques frequently localize to the distal extremities, especially the dorsal hands, as in our patient. Other variants include generalized GA, subcutaneous GA, and perforating GA. Mucin and a palisading or interstitial pattern of granulomatous inflammation are key features on histopathology in all subtypes of GA (Figure 1).7 Patch GA is a rare variant that presents with asymptomatic erythematous to brown patches, is associated with interstitial-type inflammation on histopathology, and can be difficult to distinguish from IGD.8 Granuloma annulare with interstitial inflammation on histology can be differentiated from IGD by the comparative lack of mucin in IGD.7
Sweet syndrome (SS) is characterized by sudden-onset, painful, erythematous plaques and/or nodules, commonly associated with fever and leukocytosis. Clinical variants of SS include pustular and bullous SS; giant cellulitis-like SS; necrotizing SS; and neutrophilic dermatosis of the dorsal hands presenting with hemorrhagic bullae, plaques, and pustules.7-9 Histopathologic examination shows dense nodular or perivascular neutrophilic infiltrate in the dermis without evidence of vasculitis (Figure 2).10 Histopathologic variants include histiocytoid, lymphocytic, subcutaneous, and cryptococcoid.9 The classic variant of SS has a bandlike, predominantly neutrophilic infiltrate with marked leukocytoclasia, which can be differentiated from the histiocytoid infiltrate of IGD.11 It has been shown that the infiltrate of the histiocytoid variant of SS is composed of myeloperoxidase-positive, immature myeloid cells rather than true histiocytes, and therefore can be differentiated from IGD.12 Lastly, all variants of SS have dermal edema, which typically is absent in IGD, and SS has no evidence of necrobiosis.
Erythema elevatum diutinum (EED) is a rare disease that presents with bilateral violaceous or erythematous to brown papules, plaques, or nodules. Lesions frequently localize to extensor surfaces, including the hands and fingers, and may be asymptomatic or associated with pruritus, burning, or tingling.13 Early EED lesions are characterized by leukocytoclastic vasculitis of the papillary and mid-dermal vessels with a perivascular neutrophilic infiltrate and perivascular fibrinoid necrosis. With older EED lesions, dermal and perivascular onion skin-like fibrosis become more prominent (Figure 3).14 The neutrophilic infiltrate, dermal fibrosis, and chronic vasculitic changes distinguish EED from IGD.
Necrobiosis lipoidica (NL) is a rare disease that presents with well-demarcated, yellow to red-brown papules and nodules most commonly localized to the bilateral lower extremities on the pretibial area. Papules and nodules evolve into plaques over time, and ulceration is common.15 On histopathology, NL primarily exhibits granulomatous inflammation with parallel palisading (Figure 4). The hallmark feature is necrobiosis--or degeneration--of collagen; the alternation of necrobiotic collagen and inflammatory infiltrate creates a layered cake-like appearance on low power.16 The clinical presentation as well as the dermal necrobiotic granuloma consisting of a large confluent area of necrobiosis centered in the superficial dermis and subcutaneous tissue of NL distinguishes it from the histiocytic infiltrate of IGD.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Terziroli Beretta-Piccoli B, Mainetti C, Peeters MA, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1644-1663.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Huizenga T, Kado JA, Pellicane B, et al. Interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis. Cutis. 2018;101:E19-E21.
- Rosenbach M, English JC 3rd. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Mutasim DF, Bridges AG. Patch granuloma annulare: clinicopathologic study of 6 patients. J Am Acad Dermatol. 2000;42:417-421.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006.
- Dabade TS, Davis MD. Diagnosis and treatment of the neutrophilic dermatoses (pyoderma gangrenosum, Sweet's syndrome). Dermatol Ther. 2011;24:273-284.
- Davis M, Moschella L. Neutrophilic dermatoses. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:2102-2112.
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Sardiña LA, Jour G, Piliang MP, et al. Erythema elevatum diutinum a rare and poorly understood cutaneous vasculitis: a single institution experience. J Cutan Pathol. 2019;46:97-101.
- Reid SD, Ladizinski B, Lee K, et al. Update on necrobiosis lipoidica: a review of etiology, diagnosis, and treatment options. J Am Acad Dermatol. 2013;69:783-791.
- Sibbald C, Reid S, Alavi A. Necrobiosis lipoidica. Dermatol Clin. 2015;33:343-360.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Terziroli Beretta-Piccoli B, Mainetti C, Peeters MA, et al. Cutaneous granulomatosis: a comprehensive review. Clin Rev Allergy Immunol. 2018;54:131-146.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1644-1663.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Huizenga T, Kado JA, Pellicane B, et al. Interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis. Cutis. 2018;101:E19-E21.
- Rosenbach M, English JC 3rd. Reactive granulomatous dermatitis: a review of palisaded neutrophilic and granulomatous dermatitis, interstitial granulomatous dermatitis, interstitial granulomatous drug reaction, and a proposed reclassification. Dermatol Clin. 2015;33:373-387.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Mutasim DF, Bridges AG. Patch granuloma annulare: clinicopathologic study of 6 patients. J Am Acad Dermatol. 2000;42:417-421.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses: pathogenesis, Sweet syndrome, neutrophilic eccrine hidradenitis, and Behçet disease. J Am Acad Dermatol. 2018;79:987-1006.
- Dabade TS, Davis MD. Diagnosis and treatment of the neutrophilic dermatoses (pyoderma gangrenosum, Sweet's syndrome). Dermatol Ther. 2011;24:273-284.
- Davis M, Moschella L. Neutrophilic dermatoses. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:2102-2112.
- Requena L, Kutzner H, Palmedo G, et al. Histiocytoid Sweet syndrome: a dermal infiltration of immature neutrophilic granulocytes. Arch Dermatol. 2005;141:834-842.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Sardiña LA, Jour G, Piliang MP, et al. Erythema elevatum diutinum a rare and poorly understood cutaneous vasculitis: a single institution experience. J Cutan Pathol. 2019;46:97-101.
- Reid SD, Ladizinski B, Lee K, et al. Update on necrobiosis lipoidica: a review of etiology, diagnosis, and treatment options. J Am Acad Dermatol. 2013;69:783-791.
- Sibbald C, Reid S, Alavi A. Necrobiosis lipoidica. Dermatol Clin. 2015;33:343-360.
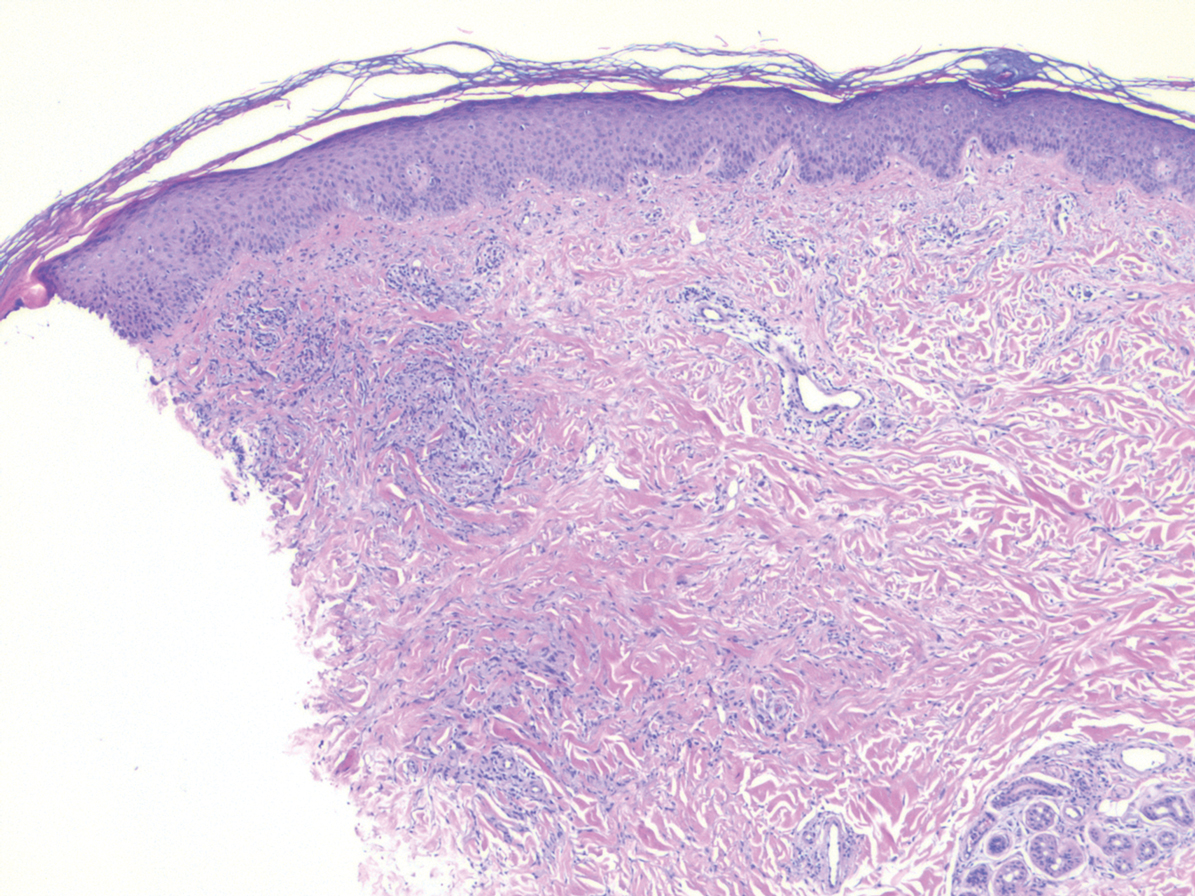
A 58-year-old woman with a medical history of asthma, hypertension, hypothyroidism, and hyperlipidemia presented with a painful rash of 10 days' duration. The rash was associated with fever at home (temperature, 38.5.2 °C), and a review of systems was positive for joint pain. Physical examination revealed numerous 8- to 10-mm, erythematous, discus-shaped papules on the bilateral dorsal hands, bilateral palms, right knee, and right dorsal foot with slight tenderness to palpation. A papule on the right dorsal hand was biopsied.