User login
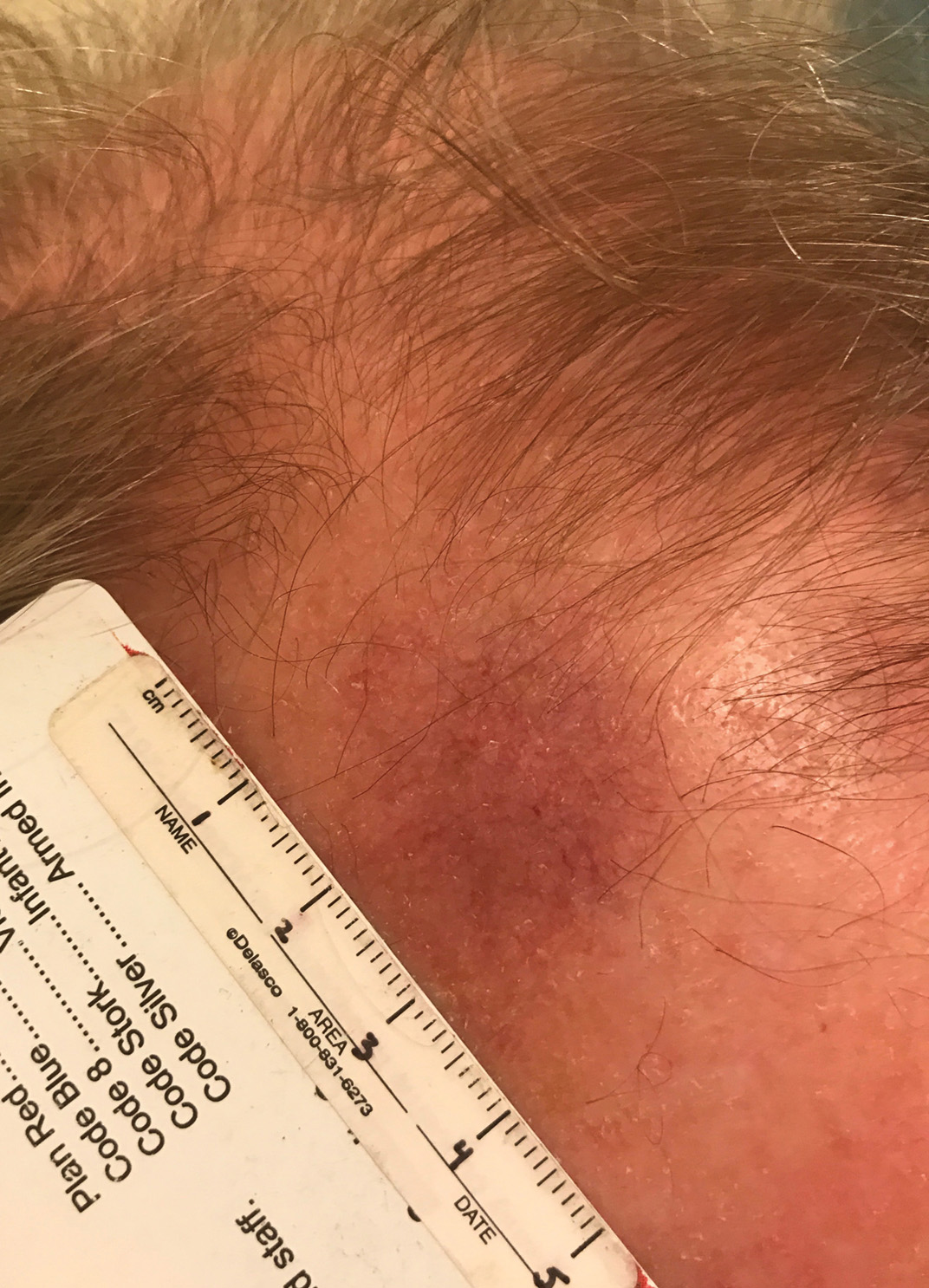
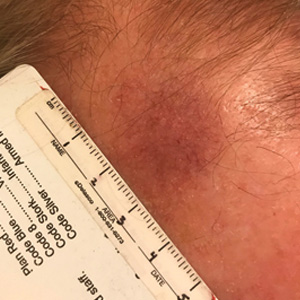
Telangiectatic Patch on the Forehead
The Diagnosis: Cutaneous B-cell Lymphoma
Histopathology was suggestive of cutaneous B-cell lymphoma (Figure). Further immunohistochemical studies including Bcl-6 positivity and Bcl-2 negativity in the large atypical cells supported a diagnosis of primary cutaneous follicle center lymphoma (PCFCL). The designation of primary cutaneous B-cell lymphoma includes several different types of lymphoma, including marginal zone lymphoma, diffuse large B-cell lymphoma, and intravascular lymphoma. To be considered a primary cutaneous lymphoma, there must be evidence of the lymphoma in the skin without concomitant evidence of systemic involvement, as determined through a full staging workup. Primary cutaneous follicle center lymphoma is an indolent lymphoma that most commonly presents as solitary or grouped, pink to plum-colored papules, plaques, nodules, and tumors on the scalp, forehead, or back.1 The lesions often are biopsied as suspected basal cell carcinomas or Merkel cell carcinomas (MCCs). Lesions on the face or scalp may easily evade diagnosis, as they initially may mimic rosacea or insect bites. Less common presentations include infiltrative lesions that cause rhinophymatous changes or scarring alopecia. Multifocal or disseminated lesions rarely can be observed. This case presentation is unique in its patchy appearance that clinically resembled angiosarcoma.2 When identified and treated, the disease-specific 5-year survival rate for PCFCL is greater than 95%.3
Merkel cell carcinoma was first described in 1972 and has been diagnosed with increasing frequency each year.4 It generally presents as an erythematous or violaceous, tender, indurated nodule on sun-exposed skin of the head or neck in elderly White men. However, other presentations have been reported, including papules, plaques, cystlike structures, pruritic tumors, pedunculated lesions, subcutaneous masses, and telangiectatic papules.5 Histopathologically, MCC is characterized by dermal nests and sheets of basaloid cells with finely granular salt and pepper-like chromatin. The histologic features can resemble other small blue cell tumors; therefore, the differential diagnosis can be broad.5 Immunohistochemistry that can confirm the diagnosis of MCC generally will be positive for cytokeratin 20 and neuroendocrine markers but negative for cytokeratin 7 and thyroid transcription factor 1. Merkel cell carcinoma is an aggressive tumor with a high risk for local recurrence and distant metastasis that carries a generally poor prognosis, especially when there is evidence of metastatic disease at presentation.5,6
Rosacea can appear as telangiectatic patches, though generally not as one discrete patch limited to the forehead, as in our patient. Histologic features vary based on the age of the lesion and clinical variant. In early lesions there is a mild perivascular lymphoplasmacytic infiltrate within the dermis, while older lesions can have a mixed infiltrate crowded around vessels and adnexal structures. Granulomas often are seen near hair follicles and interspersed throughout the dermis with ectatic vessels and dermal edema.7
Angiosarcoma is divided into 3 clinicopathological subtypes: idiopathic angiosarcoma of the head and neck, angiosarcoma in the setting of lymphedema, and postirradiation angiosarcoma.7 Idiopathic angiosarcoma most closely mimics PCFCL, as it can present as single or multifocal nodules, plaques, or patches. Histologically, the 3 groups appear similar with poorly circumscribed, infiltrative, dermal tumors. The neoplastic endothelial cells have large hyperchromatic nuclei that protrude into vascular lumens. The prognosis for idiopathic angiosarcoma of the head and neck is poor, with a 5-year survival rate of 15% to 34%, which often is due to delayed diagnosis.7
Pigmented purpuric dermatoses (PPDs) are chronic skin disorders characterized by purpura due to extravasation of blood from capillaries; the resulting hemosiderin deposition leads to pigmentation.7 There are various forms of PPD, which are classified into groups based on clinical appearance including Schamberg disease, purpura annularis telangiectodes of Majocchi, pigmented purpuric lichenoid dermatosis of Gougerot and Blum, lichen aureus, and others including eczematid and itching variants, which some consider to be distinct entities. Purpura annularis telangiectodes of Majocchi is the specific PPD that should be included in the clinical differential for PCFCL because it presents as annular patches with telangiectasias. Histologically, PPDs are characterized by a CD4+ lymphocytic infiltrate in the upper dermis with extravasated red blood cells and the presence of hemosiderin mostly within macrophages and a lack of true vasculitis. Clonality of the T cells has been shown, and there is some evidence that PPD may overlap with mycosis fungoides. However, this overlap mainly has been seen in patients with widespread lesions and would not apply to this case. In general, patients with PPD can be reassured of the benign process. In cases of widespread PPD, patients should be followed clinically to assess for progression to mycosis fungoides, though the likelihood is low.7
Our patient underwent a full staging workup, which confirmed the diagnosis of PCFCL. He was treated with radiation to the forehead that resulted in clearance of the lesion. Approximately 2 years after the initial diagnosis, the patient was alive and well with no evidence of recurrence of PCFCL.
In conclusion, it is imperative to identify unusual, macular, vascular-appearing patches, especially on the head and neck in older individuals. Because the clinical presentations of PCFCL, angiosarcoma, rosacea, MCC, and PPD can overlap with one another as well as with other entities, it is necessary to have a high level of suspicion and low threshold to biopsy these types of lesions, as outcomes can be drastically different.
- Goyal A, LeBlanc RE, Carter JB. Cutaneous B-cell lymphoma. Hematol Oncol Clin North Am. 2019;33:149-161.
- Massone C, Fink-Puches R, Cerroni L. Atypical clinical presentation of primary and secondary cutaneous follicle center lymphoma (FCL) on the head characterized by macular lesions. J Am Acad Dermatol. 2016;75:1000-1006.
- Wilcox RA. Cutaneous B-cell lymphomas: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:1052-1055.
- Conic RRZ, Ko J, Saridakis S, et al. Sentinel lymph node biopsy in Merkel cell carcinoma: predictors of sentinel lymph node positivity and association with overall survival. J Am Acad Dermatol. 2019;81:364-372
- Coggshall K, Tello TL, North JP, et al. Merkel cell carcinoma: an update and review: pathogenesis, diagnosis, and staging. J Am Acad Dermatol. 2018;78:433-442.
- Tello TL, Coggshall K, Yom SS, et al. Merkel cell carcinoma: an update and review: current and future therapy. J Am Acad Dermatol. 2018;78:445-454.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
The Diagnosis: Cutaneous B-cell Lymphoma
Histopathology was suggestive of cutaneous B-cell lymphoma (Figure). Further immunohistochemical studies including Bcl-6 positivity and Bcl-2 negativity in the large atypical cells supported a diagnosis of primary cutaneous follicle center lymphoma (PCFCL). The designation of primary cutaneous B-cell lymphoma includes several different types of lymphoma, including marginal zone lymphoma, diffuse large B-cell lymphoma, and intravascular lymphoma. To be considered a primary cutaneous lymphoma, there must be evidence of the lymphoma in the skin without concomitant evidence of systemic involvement, as determined through a full staging workup. Primary cutaneous follicle center lymphoma is an indolent lymphoma that most commonly presents as solitary or grouped, pink to plum-colored papules, plaques, nodules, and tumors on the scalp, forehead, or back.1 The lesions often are biopsied as suspected basal cell carcinomas or Merkel cell carcinomas (MCCs). Lesions on the face or scalp may easily evade diagnosis, as they initially may mimic rosacea or insect bites. Less common presentations include infiltrative lesions that cause rhinophymatous changes or scarring alopecia. Multifocal or disseminated lesions rarely can be observed. This case presentation is unique in its patchy appearance that clinically resembled angiosarcoma.2 When identified and treated, the disease-specific 5-year survival rate for PCFCL is greater than 95%.3
Merkel cell carcinoma was first described in 1972 and has been diagnosed with increasing frequency each year.4 It generally presents as an erythematous or violaceous, tender, indurated nodule on sun-exposed skin of the head or neck in elderly White men. However, other presentations have been reported, including papules, plaques, cystlike structures, pruritic tumors, pedunculated lesions, subcutaneous masses, and telangiectatic papules.5 Histopathologically, MCC is characterized by dermal nests and sheets of basaloid cells with finely granular salt and pepper-like chromatin. The histologic features can resemble other small blue cell tumors; therefore, the differential diagnosis can be broad.5 Immunohistochemistry that can confirm the diagnosis of MCC generally will be positive for cytokeratin 20 and neuroendocrine markers but negative for cytokeratin 7 and thyroid transcription factor 1. Merkel cell carcinoma is an aggressive tumor with a high risk for local recurrence and distant metastasis that carries a generally poor prognosis, especially when there is evidence of metastatic disease at presentation.5,6
Rosacea can appear as telangiectatic patches, though generally not as one discrete patch limited to the forehead, as in our patient. Histologic features vary based on the age of the lesion and clinical variant. In early lesions there is a mild perivascular lymphoplasmacytic infiltrate within the dermis, while older lesions can have a mixed infiltrate crowded around vessels and adnexal structures. Granulomas often are seen near hair follicles and interspersed throughout the dermis with ectatic vessels and dermal edema.7
Angiosarcoma is divided into 3 clinicopathological subtypes: idiopathic angiosarcoma of the head and neck, angiosarcoma in the setting of lymphedema, and postirradiation angiosarcoma.7 Idiopathic angiosarcoma most closely mimics PCFCL, as it can present as single or multifocal nodules, plaques, or patches. Histologically, the 3 groups appear similar with poorly circumscribed, infiltrative, dermal tumors. The neoplastic endothelial cells have large hyperchromatic nuclei that protrude into vascular lumens. The prognosis for idiopathic angiosarcoma of the head and neck is poor, with a 5-year survival rate of 15% to 34%, which often is due to delayed diagnosis.7
Pigmented purpuric dermatoses (PPDs) are chronic skin disorders characterized by purpura due to extravasation of blood from capillaries; the resulting hemosiderin deposition leads to pigmentation.7 There are various forms of PPD, which are classified into groups based on clinical appearance including Schamberg disease, purpura annularis telangiectodes of Majocchi, pigmented purpuric lichenoid dermatosis of Gougerot and Blum, lichen aureus, and others including eczematid and itching variants, which some consider to be distinct entities. Purpura annularis telangiectodes of Majocchi is the specific PPD that should be included in the clinical differential for PCFCL because it presents as annular patches with telangiectasias. Histologically, PPDs are characterized by a CD4+ lymphocytic infiltrate in the upper dermis with extravasated red blood cells and the presence of hemosiderin mostly within macrophages and a lack of true vasculitis. Clonality of the T cells has been shown, and there is some evidence that PPD may overlap with mycosis fungoides. However, this overlap mainly has been seen in patients with widespread lesions and would not apply to this case. In general, patients with PPD can be reassured of the benign process. In cases of widespread PPD, patients should be followed clinically to assess for progression to mycosis fungoides, though the likelihood is low.7
Our patient underwent a full staging workup, which confirmed the diagnosis of PCFCL. He was treated with radiation to the forehead that resulted in clearance of the lesion. Approximately 2 years after the initial diagnosis, the patient was alive and well with no evidence of recurrence of PCFCL.
In conclusion, it is imperative to identify unusual, macular, vascular-appearing patches, especially on the head and neck in older individuals. Because the clinical presentations of PCFCL, angiosarcoma, rosacea, MCC, and PPD can overlap with one another as well as with other entities, it is necessary to have a high level of suspicion and low threshold to biopsy these types of lesions, as outcomes can be drastically different.
The Diagnosis: Cutaneous B-cell Lymphoma
Histopathology was suggestive of cutaneous B-cell lymphoma (Figure). Further immunohistochemical studies including Bcl-6 positivity and Bcl-2 negativity in the large atypical cells supported a diagnosis of primary cutaneous follicle center lymphoma (PCFCL). The designation of primary cutaneous B-cell lymphoma includes several different types of lymphoma, including marginal zone lymphoma, diffuse large B-cell lymphoma, and intravascular lymphoma. To be considered a primary cutaneous lymphoma, there must be evidence of the lymphoma in the skin without concomitant evidence of systemic involvement, as determined through a full staging workup. Primary cutaneous follicle center lymphoma is an indolent lymphoma that most commonly presents as solitary or grouped, pink to plum-colored papules, plaques, nodules, and tumors on the scalp, forehead, or back.1 The lesions often are biopsied as suspected basal cell carcinomas or Merkel cell carcinomas (MCCs). Lesions on the face or scalp may easily evade diagnosis, as they initially may mimic rosacea or insect bites. Less common presentations include infiltrative lesions that cause rhinophymatous changes or scarring alopecia. Multifocal or disseminated lesions rarely can be observed. This case presentation is unique in its patchy appearance that clinically resembled angiosarcoma.2 When identified and treated, the disease-specific 5-year survival rate for PCFCL is greater than 95%.3
Merkel cell carcinoma was first described in 1972 and has been diagnosed with increasing frequency each year.4 It generally presents as an erythematous or violaceous, tender, indurated nodule on sun-exposed skin of the head or neck in elderly White men. However, other presentations have been reported, including papules, plaques, cystlike structures, pruritic tumors, pedunculated lesions, subcutaneous masses, and telangiectatic papules.5 Histopathologically, MCC is characterized by dermal nests and sheets of basaloid cells with finely granular salt and pepper-like chromatin. The histologic features can resemble other small blue cell tumors; therefore, the differential diagnosis can be broad.5 Immunohistochemistry that can confirm the diagnosis of MCC generally will be positive for cytokeratin 20 and neuroendocrine markers but negative for cytokeratin 7 and thyroid transcription factor 1. Merkel cell carcinoma is an aggressive tumor with a high risk for local recurrence and distant metastasis that carries a generally poor prognosis, especially when there is evidence of metastatic disease at presentation.5,6
Rosacea can appear as telangiectatic patches, though generally not as one discrete patch limited to the forehead, as in our patient. Histologic features vary based on the age of the lesion and clinical variant. In early lesions there is a mild perivascular lymphoplasmacytic infiltrate within the dermis, while older lesions can have a mixed infiltrate crowded around vessels and adnexal structures. Granulomas often are seen near hair follicles and interspersed throughout the dermis with ectatic vessels and dermal edema.7
Angiosarcoma is divided into 3 clinicopathological subtypes: idiopathic angiosarcoma of the head and neck, angiosarcoma in the setting of lymphedema, and postirradiation angiosarcoma.7 Idiopathic angiosarcoma most closely mimics PCFCL, as it can present as single or multifocal nodules, plaques, or patches. Histologically, the 3 groups appear similar with poorly circumscribed, infiltrative, dermal tumors. The neoplastic endothelial cells have large hyperchromatic nuclei that protrude into vascular lumens. The prognosis for idiopathic angiosarcoma of the head and neck is poor, with a 5-year survival rate of 15% to 34%, which often is due to delayed diagnosis.7
Pigmented purpuric dermatoses (PPDs) are chronic skin disorders characterized by purpura due to extravasation of blood from capillaries; the resulting hemosiderin deposition leads to pigmentation.7 There are various forms of PPD, which are classified into groups based on clinical appearance including Schamberg disease, purpura annularis telangiectodes of Majocchi, pigmented purpuric lichenoid dermatosis of Gougerot and Blum, lichen aureus, and others including eczematid and itching variants, which some consider to be distinct entities. Purpura annularis telangiectodes of Majocchi is the specific PPD that should be included in the clinical differential for PCFCL because it presents as annular patches with telangiectasias. Histologically, PPDs are characterized by a CD4+ lymphocytic infiltrate in the upper dermis with extravasated red blood cells and the presence of hemosiderin mostly within macrophages and a lack of true vasculitis. Clonality of the T cells has been shown, and there is some evidence that PPD may overlap with mycosis fungoides. However, this overlap mainly has been seen in patients with widespread lesions and would not apply to this case. In general, patients with PPD can be reassured of the benign process. In cases of widespread PPD, patients should be followed clinically to assess for progression to mycosis fungoides, though the likelihood is low.7
Our patient underwent a full staging workup, which confirmed the diagnosis of PCFCL. He was treated with radiation to the forehead that resulted in clearance of the lesion. Approximately 2 years after the initial diagnosis, the patient was alive and well with no evidence of recurrence of PCFCL.
In conclusion, it is imperative to identify unusual, macular, vascular-appearing patches, especially on the head and neck in older individuals. Because the clinical presentations of PCFCL, angiosarcoma, rosacea, MCC, and PPD can overlap with one another as well as with other entities, it is necessary to have a high level of suspicion and low threshold to biopsy these types of lesions, as outcomes can be drastically different.
- Goyal A, LeBlanc RE, Carter JB. Cutaneous B-cell lymphoma. Hematol Oncol Clin North Am. 2019;33:149-161.
- Massone C, Fink-Puches R, Cerroni L. Atypical clinical presentation of primary and secondary cutaneous follicle center lymphoma (FCL) on the head characterized by macular lesions. J Am Acad Dermatol. 2016;75:1000-1006.
- Wilcox RA. Cutaneous B-cell lymphomas: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:1052-1055.
- Conic RRZ, Ko J, Saridakis S, et al. Sentinel lymph node biopsy in Merkel cell carcinoma: predictors of sentinel lymph node positivity and association with overall survival. J Am Acad Dermatol. 2019;81:364-372
- Coggshall K, Tello TL, North JP, et al. Merkel cell carcinoma: an update and review: pathogenesis, diagnosis, and staging. J Am Acad Dermatol. 2018;78:433-442.
- Tello TL, Coggshall K, Yom SS, et al. Merkel cell carcinoma: an update and review: current and future therapy. J Am Acad Dermatol. 2018;78:445-454.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
- Goyal A, LeBlanc RE, Carter JB. Cutaneous B-cell lymphoma. Hematol Oncol Clin North Am. 2019;33:149-161.
- Massone C, Fink-Puches R, Cerroni L. Atypical clinical presentation of primary and secondary cutaneous follicle center lymphoma (FCL) on the head characterized by macular lesions. J Am Acad Dermatol. 2016;75:1000-1006.
- Wilcox RA. Cutaneous B-cell lymphomas: 2016 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91:1052-1055.
- Conic RRZ, Ko J, Saridakis S, et al. Sentinel lymph node biopsy in Merkel cell carcinoma: predictors of sentinel lymph node positivity and association with overall survival. J Am Acad Dermatol. 2019;81:364-372
- Coggshall K, Tello TL, North JP, et al. Merkel cell carcinoma: an update and review: pathogenesis, diagnosis, and staging. J Am Acad Dermatol. 2018;78:433-442.
- Tello TL, Coggshall K, Yom SS, et al. Merkel cell carcinoma: an update and review: current and future therapy. J Am Acad Dermatol. 2018;78:445-454.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016.
Asymptomatic Nodule on the Back
The Diagnosis: Primary Cutaneous Perivascular Epithelioid Cell Tumor
Perivascular epithelioid cell tumors (PEComas) were first described in 1996.1 They comprise a family of rare mesenchymal neoplasms that have a unique characteristic of staining positive for melanocytic and smooth muscle markers on immunohistochemistry.2 These neoplasms have been described in many areas of the body including the uterus, bladder, heart, pancreas, and prostate. The majority of PEComas are extracutaneous, with only 8% of reported cases originating on the skin.3 A case of primary cutaneous PEComa (pcPEComa) was described in 2003.4 The primary cutaneous form is extremely rare.3,5-7
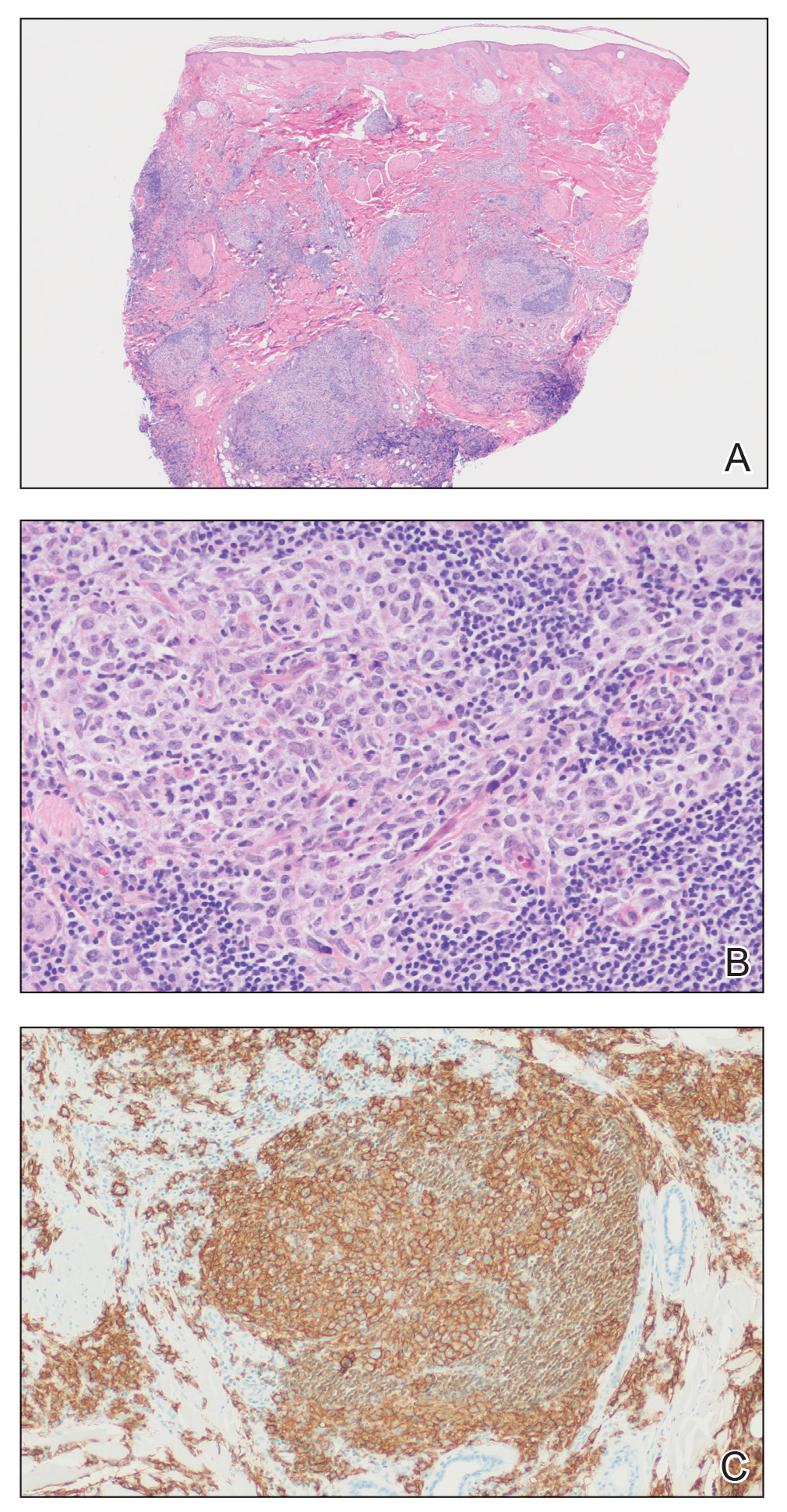
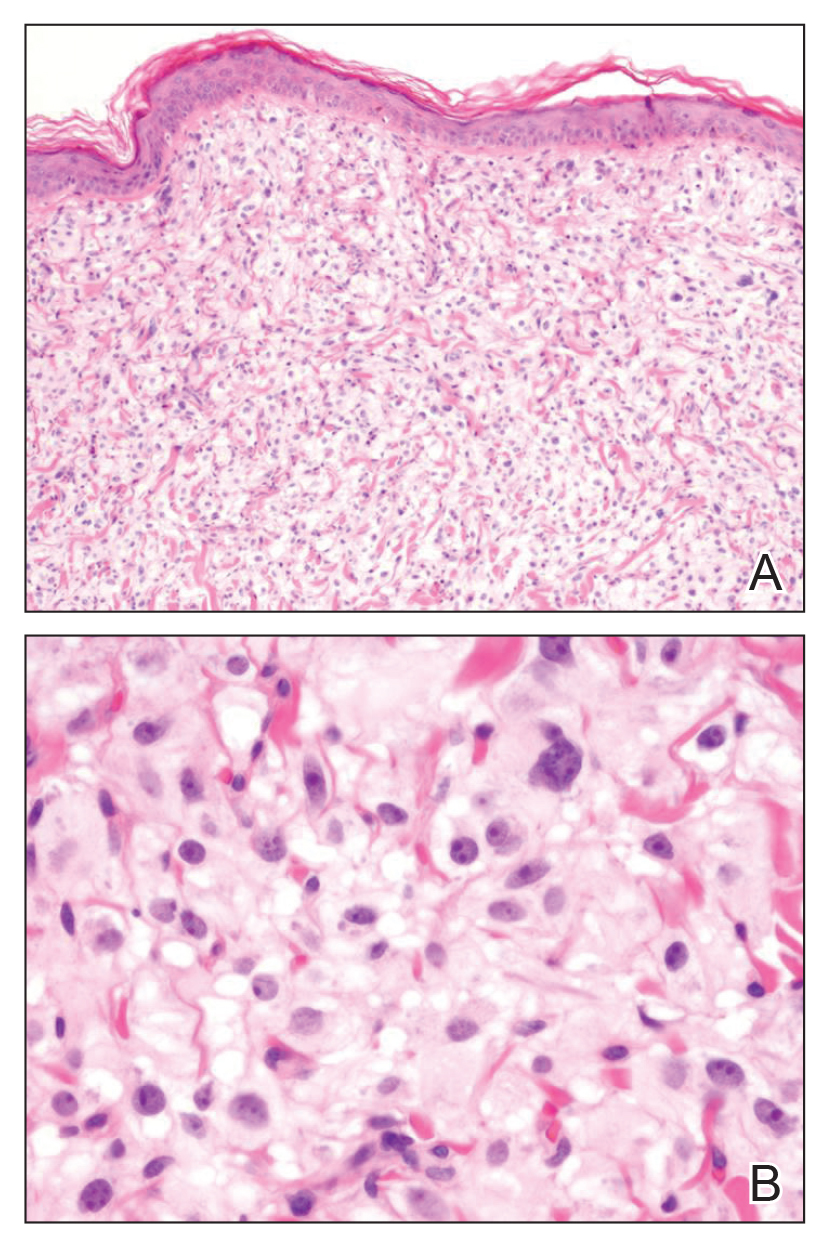
A broad deep shave biopsy was performed in our patient in an attempt to sample the entire lesion. Histopathologic examination of the nodule demonstrated a dermal neoplasm comprised of a diffuse proliferation of large polygonal cells with abundant clear cytoplasm, fine chromatin, and prominent nucleoli (Figure 1A). Higher-power magnification showed moderate nuclear pleomorphism and only rare mitotic figures (Figure 1B).
Immunohistochemical staining revealed positivity for myomelanocytic markers with positivity for human melanoma black 45 (HMB-45)(Figure 2) and desmin (not shown). Additionally, the tumor was positive for CD163 and negative for smooth muscle actin, cytokeratin, and S-100 protein.
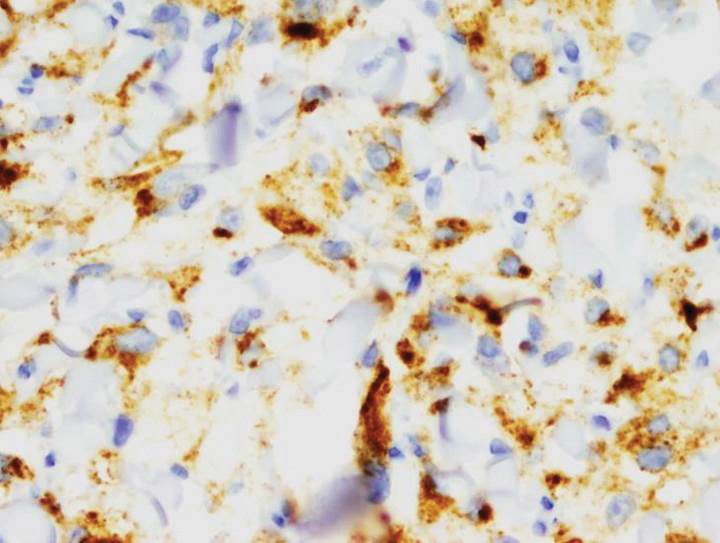
Perivascular epithelioid cell tumors are characterized histologically as mesenchymal neoplasms containing large epithelioid to spindled cells with a slightly granular, vacuolated cytoplasm. These cells often are found in close proximity to vascular structures.3,5,8 The hallmark of PEComas is the expression of both melanocytic and muscle markers.3,8 A review of staining patterns of pcPEComas emphasized that immunophenotypes between visceral and primary cutaneous forms may vary considerably.3,5,8 The most consistent and sensitive melanocytic marker is HMB-45 (88%-92% positive).3,8 Positive Melan-A staining varies in the literature from 0% to 50% of cases.3 Our patient's neoplasm expressed the characteristic myomelanocytic immunophenotype with both HMB-45 and desmin positivity.
Given the histologic characteristics, these lesions can be mistaken for melanocytic and other nonmelanocytic tumors with a clear cell morphology such as balloon cell nevus, hypomelanotic blue nevus, and melanoma.2,3 A pigmented case of pcPEComa was reported in 2015 and was originally diagnosed as metastatic melanoma.6 Unlike pcPEComa, melanoma usually stains positive with S-100 protein in up to 99% of cases8 and is negative for muscle markers; however, a case series reported S-100 protein positivity in 38% of pcPEComas.3 Nonmelanocytic neoplasms in the histologic differential diagnosis include clear cell sarcoma and clear cell renal cell carcinoma, both of which show immunoreactivity for cytokeratin.9
Histologic criteria exist for establishing malignancy potential for visceral PEComas but not for pcPEComas, though it has been suggested that the same malignancy criteria should be applied to pcPEComas.3,9 Features associated with malignancy include size greater than 8 cm, mitotic activity greater than 1 mitosis per 50 high-power fields, infiltrative growth pattern, high nuclear grade, necrosis, and vascular invasion. Based on these criteria, fulfilling 2 or more features technically classifies the lesion as malignant, 1 feature classifies it as uncertain malignant potential, and a lack of these features renders the lesion benign.9
The overwhelming majority of pcPEComas are considered benign. One case of pcPEComa was considered malignant with a high mitotic rate (5 mitoses per 10 high-power fields) and nuclear atypia.10 Further workup with thoracic computed tomography and positron emission tomography-computed tomography was negative for metastasis. Treatment with wide excision and radiotherapy was performed with no sign of recurrence at 24-month follow-up.10
Although pcPEComas arising from the dermis seem to be benign overall, PEComas originating from the subcutaneous tissue may have greater malignancy potential. Two cases of subcutaneous PEComas presenting as nodules resulted in metastasis; one case had local nodal metastasis and another developed metastasis to the lungs months later.10,11
- Zamboni G, Pea M, Martignoni G, et al. Clear cell “sugar” tumorof the pancreas. a novel member of the family of lesions characterizedby the presence of perivascular epithelioid cells. Am J Surg Pathol.1996;20:722-730.
- Folpe AK, Wiatkowski D. Perivascular epithelioid cell neoplasms: pathology and pathogenesis. Hum Pathol. 2010;41:1-15.
- Charli-Joseph Y, Saggini A, Vemula S, et al. Primary cutaneous perivascularepithelioid cell tumor: a clinicopathological and molecular reappraisal. J Am Acad Dermatol. 2014;71:1127-1136.
- Crowson AN, Taylor JR, Magro CM. Cutaneous clear cell myomelanocytictumor-perivascular epithelioid cell tumor: first reported case. Mod Pathol. 2003;16:90A.
- Chaplin A, Conrad D, Tatlidil C, et al. Primary cutaneous PEComa. Am J Dermatopathol. 2010;32:310-312.
- Navale P, Asgari M, Chen S. Pigmented perivascular epithelioid cell tumor of the skin. Am J Dermatopathol. 2015;37:866-869.
- Ieremia E, Robson A. Cutaneous PEComa. Am J Dermatopathol. 2014;36:E198-E201.
- Calder K, Schlauder S, Morgan M. Malignant perivascularepithelioid cell tumor (‘PEComa’): a case report and literature review of cutaneous/subcutaneous presentations. J Cutan Pathol. 2008;35:499-503.
- Folpe A, Mentzel T, Lehr H, et al. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: a clinicopathologic study of 26 cases and review of the literature. Am J Dermatopathol. 2005; 29:1558-1575.
- Greveling K, Winnepenninckx V, Nagtzaam I, et al. Malignant perivascular epithelioid cell tumor: a case report of a cutaneous tumor on the cheek of a male patient. J Am Acad Dermatol. 2013;69:E262-E264.
- Shon W, Kim J, Sukov W, et al. Malignant TFE3-rearranged perivascular epithelioid cell neoplasm (PEComa) presenting as a subcutaneous mass. Br J Dermatol. 2015;174:617-620.
The Diagnosis: Primary Cutaneous Perivascular Epithelioid Cell Tumor
Perivascular epithelioid cell tumors (PEComas) were first described in 1996.1 They comprise a family of rare mesenchymal neoplasms that have a unique characteristic of staining positive for melanocytic and smooth muscle markers on immunohistochemistry.2 These neoplasms have been described in many areas of the body including the uterus, bladder, heart, pancreas, and prostate. The majority of PEComas are extracutaneous, with only 8% of reported cases originating on the skin.3 A case of primary cutaneous PEComa (pcPEComa) was described in 2003.4 The primary cutaneous form is extremely rare.3,5-7
A broad deep shave biopsy was performed in our patient in an attempt to sample the entire lesion. Histopathologic examination of the nodule demonstrated a dermal neoplasm comprised of a diffuse proliferation of large polygonal cells with abundant clear cytoplasm, fine chromatin, and prominent nucleoli (Figure 1A). Higher-power magnification showed moderate nuclear pleomorphism and only rare mitotic figures (Figure 1B).
Immunohistochemical staining revealed positivity for myomelanocytic markers with positivity for human melanoma black 45 (HMB-45)(Figure 2) and desmin (not shown). Additionally, the tumor was positive for CD163 and negative for smooth muscle actin, cytokeratin, and S-100 protein.
Perivascular epithelioid cell tumors are characterized histologically as mesenchymal neoplasms containing large epithelioid to spindled cells with a slightly granular, vacuolated cytoplasm. These cells often are found in close proximity to vascular structures.3,5,8 The hallmark of PEComas is the expression of both melanocytic and muscle markers.3,8 A review of staining patterns of pcPEComas emphasized that immunophenotypes between visceral and primary cutaneous forms may vary considerably.3,5,8 The most consistent and sensitive melanocytic marker is HMB-45 (88%-92% positive).3,8 Positive Melan-A staining varies in the literature from 0% to 50% of cases.3 Our patient's neoplasm expressed the characteristic myomelanocytic immunophenotype with both HMB-45 and desmin positivity.
Given the histologic characteristics, these lesions can be mistaken for melanocytic and other nonmelanocytic tumors with a clear cell morphology such as balloon cell nevus, hypomelanotic blue nevus, and melanoma.2,3 A pigmented case of pcPEComa was reported in 2015 and was originally diagnosed as metastatic melanoma.6 Unlike pcPEComa, melanoma usually stains positive with S-100 protein in up to 99% of cases8 and is negative for muscle markers; however, a case series reported S-100 protein positivity in 38% of pcPEComas.3 Nonmelanocytic neoplasms in the histologic differential diagnosis include clear cell sarcoma and clear cell renal cell carcinoma, both of which show immunoreactivity for cytokeratin.9
Histologic criteria exist for establishing malignancy potential for visceral PEComas but not for pcPEComas, though it has been suggested that the same malignancy criteria should be applied to pcPEComas.3,9 Features associated with malignancy include size greater than 8 cm, mitotic activity greater than 1 mitosis per 50 high-power fields, infiltrative growth pattern, high nuclear grade, necrosis, and vascular invasion. Based on these criteria, fulfilling 2 or more features technically classifies the lesion as malignant, 1 feature classifies it as uncertain malignant potential, and a lack of these features renders the lesion benign.9
The overwhelming majority of pcPEComas are considered benign. One case of pcPEComa was considered malignant with a high mitotic rate (5 mitoses per 10 high-power fields) and nuclear atypia.10 Further workup with thoracic computed tomography and positron emission tomography-computed tomography was negative for metastasis. Treatment with wide excision and radiotherapy was performed with no sign of recurrence at 24-month follow-up.10
Although pcPEComas arising from the dermis seem to be benign overall, PEComas originating from the subcutaneous tissue may have greater malignancy potential. Two cases of subcutaneous PEComas presenting as nodules resulted in metastasis; one case had local nodal metastasis and another developed metastasis to the lungs months later.10,11
The Diagnosis: Primary Cutaneous Perivascular Epithelioid Cell Tumor
Perivascular epithelioid cell tumors (PEComas) were first described in 1996.1 They comprise a family of rare mesenchymal neoplasms that have a unique characteristic of staining positive for melanocytic and smooth muscle markers on immunohistochemistry.2 These neoplasms have been described in many areas of the body including the uterus, bladder, heart, pancreas, and prostate. The majority of PEComas are extracutaneous, with only 8% of reported cases originating on the skin.3 A case of primary cutaneous PEComa (pcPEComa) was described in 2003.4 The primary cutaneous form is extremely rare.3,5-7
A broad deep shave biopsy was performed in our patient in an attempt to sample the entire lesion. Histopathologic examination of the nodule demonstrated a dermal neoplasm comprised of a diffuse proliferation of large polygonal cells with abundant clear cytoplasm, fine chromatin, and prominent nucleoli (Figure 1A). Higher-power magnification showed moderate nuclear pleomorphism and only rare mitotic figures (Figure 1B).
Immunohistochemical staining revealed positivity for myomelanocytic markers with positivity for human melanoma black 45 (HMB-45)(Figure 2) and desmin (not shown). Additionally, the tumor was positive for CD163 and negative for smooth muscle actin, cytokeratin, and S-100 protein.
Perivascular epithelioid cell tumors are characterized histologically as mesenchymal neoplasms containing large epithelioid to spindled cells with a slightly granular, vacuolated cytoplasm. These cells often are found in close proximity to vascular structures.3,5,8 The hallmark of PEComas is the expression of both melanocytic and muscle markers.3,8 A review of staining patterns of pcPEComas emphasized that immunophenotypes between visceral and primary cutaneous forms may vary considerably.3,5,8 The most consistent and sensitive melanocytic marker is HMB-45 (88%-92% positive).3,8 Positive Melan-A staining varies in the literature from 0% to 50% of cases.3 Our patient's neoplasm expressed the characteristic myomelanocytic immunophenotype with both HMB-45 and desmin positivity.
Given the histologic characteristics, these lesions can be mistaken for melanocytic and other nonmelanocytic tumors with a clear cell morphology such as balloon cell nevus, hypomelanotic blue nevus, and melanoma.2,3 A pigmented case of pcPEComa was reported in 2015 and was originally diagnosed as metastatic melanoma.6 Unlike pcPEComa, melanoma usually stains positive with S-100 protein in up to 99% of cases8 and is negative for muscle markers; however, a case series reported S-100 protein positivity in 38% of pcPEComas.3 Nonmelanocytic neoplasms in the histologic differential diagnosis include clear cell sarcoma and clear cell renal cell carcinoma, both of which show immunoreactivity for cytokeratin.9
Histologic criteria exist for establishing malignancy potential for visceral PEComas but not for pcPEComas, though it has been suggested that the same malignancy criteria should be applied to pcPEComas.3,9 Features associated with malignancy include size greater than 8 cm, mitotic activity greater than 1 mitosis per 50 high-power fields, infiltrative growth pattern, high nuclear grade, necrosis, and vascular invasion. Based on these criteria, fulfilling 2 or more features technically classifies the lesion as malignant, 1 feature classifies it as uncertain malignant potential, and a lack of these features renders the lesion benign.9
The overwhelming majority of pcPEComas are considered benign. One case of pcPEComa was considered malignant with a high mitotic rate (5 mitoses per 10 high-power fields) and nuclear atypia.10 Further workup with thoracic computed tomography and positron emission tomography-computed tomography was negative for metastasis. Treatment with wide excision and radiotherapy was performed with no sign of recurrence at 24-month follow-up.10
Although pcPEComas arising from the dermis seem to be benign overall, PEComas originating from the subcutaneous tissue may have greater malignancy potential. Two cases of subcutaneous PEComas presenting as nodules resulted in metastasis; one case had local nodal metastasis and another developed metastasis to the lungs months later.10,11
- Zamboni G, Pea M, Martignoni G, et al. Clear cell “sugar” tumorof the pancreas. a novel member of the family of lesions characterizedby the presence of perivascular epithelioid cells. Am J Surg Pathol.1996;20:722-730.
- Folpe AK, Wiatkowski D. Perivascular epithelioid cell neoplasms: pathology and pathogenesis. Hum Pathol. 2010;41:1-15.
- Charli-Joseph Y, Saggini A, Vemula S, et al. Primary cutaneous perivascularepithelioid cell tumor: a clinicopathological and molecular reappraisal. J Am Acad Dermatol. 2014;71:1127-1136.
- Crowson AN, Taylor JR, Magro CM. Cutaneous clear cell myomelanocytictumor-perivascular epithelioid cell tumor: first reported case. Mod Pathol. 2003;16:90A.
- Chaplin A, Conrad D, Tatlidil C, et al. Primary cutaneous PEComa. Am J Dermatopathol. 2010;32:310-312.
- Navale P, Asgari M, Chen S. Pigmented perivascular epithelioid cell tumor of the skin. Am J Dermatopathol. 2015;37:866-869.
- Ieremia E, Robson A. Cutaneous PEComa. Am J Dermatopathol. 2014;36:E198-E201.
- Calder K, Schlauder S, Morgan M. Malignant perivascularepithelioid cell tumor (‘PEComa’): a case report and literature review of cutaneous/subcutaneous presentations. J Cutan Pathol. 2008;35:499-503.
- Folpe A, Mentzel T, Lehr H, et al. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: a clinicopathologic study of 26 cases and review of the literature. Am J Dermatopathol. 2005; 29:1558-1575.
- Greveling K, Winnepenninckx V, Nagtzaam I, et al. Malignant perivascular epithelioid cell tumor: a case report of a cutaneous tumor on the cheek of a male patient. J Am Acad Dermatol. 2013;69:E262-E264.
- Shon W, Kim J, Sukov W, et al. Malignant TFE3-rearranged perivascular epithelioid cell neoplasm (PEComa) presenting as a subcutaneous mass. Br J Dermatol. 2015;174:617-620.
- Zamboni G, Pea M, Martignoni G, et al. Clear cell “sugar” tumorof the pancreas. a novel member of the family of lesions characterizedby the presence of perivascular epithelioid cells. Am J Surg Pathol.1996;20:722-730.
- Folpe AK, Wiatkowski D. Perivascular epithelioid cell neoplasms: pathology and pathogenesis. Hum Pathol. 2010;41:1-15.
- Charli-Joseph Y, Saggini A, Vemula S, et al. Primary cutaneous perivascularepithelioid cell tumor: a clinicopathological and molecular reappraisal. J Am Acad Dermatol. 2014;71:1127-1136.
- Crowson AN, Taylor JR, Magro CM. Cutaneous clear cell myomelanocytictumor-perivascular epithelioid cell tumor: first reported case. Mod Pathol. 2003;16:90A.
- Chaplin A, Conrad D, Tatlidil C, et al. Primary cutaneous PEComa. Am J Dermatopathol. 2010;32:310-312.
- Navale P, Asgari M, Chen S. Pigmented perivascular epithelioid cell tumor of the skin. Am J Dermatopathol. 2015;37:866-869.
- Ieremia E, Robson A. Cutaneous PEComa. Am J Dermatopathol. 2014;36:E198-E201.
- Calder K, Schlauder S, Morgan M. Malignant perivascularepithelioid cell tumor (‘PEComa’): a case report and literature review of cutaneous/subcutaneous presentations. J Cutan Pathol. 2008;35:499-503.
- Folpe A, Mentzel T, Lehr H, et al. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: a clinicopathologic study of 26 cases and review of the literature. Am J Dermatopathol. 2005; 29:1558-1575.
- Greveling K, Winnepenninckx V, Nagtzaam I, et al. Malignant perivascular epithelioid cell tumor: a case report of a cutaneous tumor on the cheek of a male patient. J Am Acad Dermatol. 2013;69:E262-E264.
- Shon W, Kim J, Sukov W, et al. Malignant TFE3-rearranged perivascular epithelioid cell neoplasm (PEComa) presenting as a subcutaneous mass. Br J Dermatol. 2015;174:617-620.
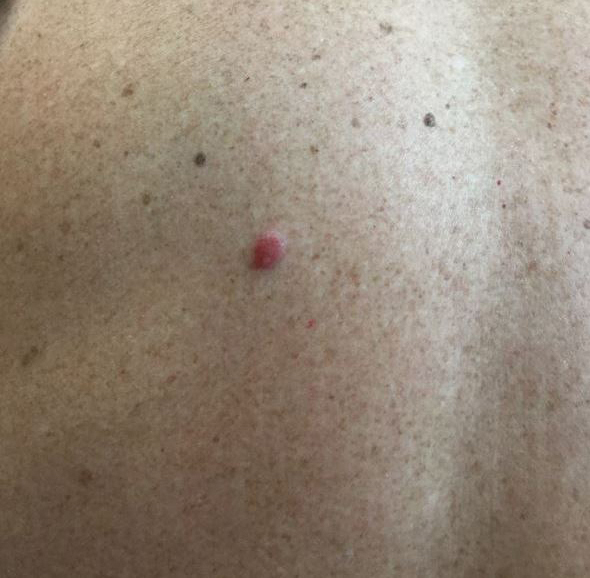
A 54-year-old man presented with an asymptomatic nodule on the left side of the mid back that had been slowly growing in size over the last 12 months. The patient had 2 other lesions on the nasal supratip and left upper arm that were concerning for basal cell carcinoma. The patient’s medical history was notable for stage IV mantle cell lymphoma diagnosed 8 years prior by lymph node biopsy. He completed multiple rounds of methotrexate and CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy over 2 years and later received a stem cell transplant; he had been in clinical remission for the last 6 years. On review of symptoms he denied any fevers, chills, fatigue, night sweats, or constitutional symptoms. The remainder of the review of symptoms was negative. Physical examination showed a 1.5×1.0-cm pink, firm, nontender nodule on the left side of the mid back.
Sunburn Purpura
To the Editor:
Chronic UV exposure has been linked to increased skin fragility and the development of purpuric lesions, a benign condition known as actinic purpura and commonly seen in elderly patients. Petechial skin changes acutely following intense sun exposure is a rare phenomenon referred to as sunburn purpura, photolocalized purpura, or solar purpura.
A 19-year-old woman presented with red and purple spots on the pretibial region of both legs extending to the thigh. One week prior to presentation she had a severe sunburn affecting most of the body, which resolved without blistering. Two days later, the spots appeared within the most severely sunburned areas of both legs. The patient reported that the lesions were mildly painful to palpation, but she was more concerned about the appearance. She denied any history of similar skin changes associated with sun exposure. The patient was otherwise healthy and denied any recent illnesses. She noted a history of mild bruising and bleeding with a resulting unremarkable workup by her primary care physician. The only medication taken was etonogestrel-ethinyl estradiol vaginal ring.
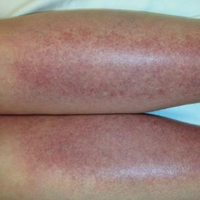
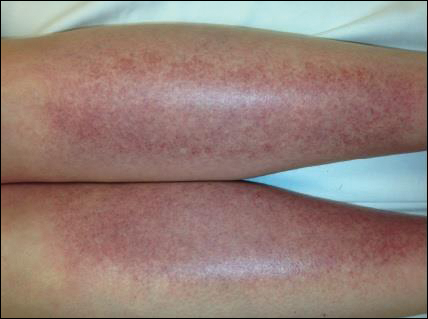
The scalp, face, arms, trunk, and legs were examined, and nonpalpable petechial changes were noted on the anterior aspect of the legs (Figure 1), with changes more prominent on the distal aspect of the legs. Mild superficial epidermal exfoliation was noted on both anterior thighs. The area of the lesions was not warm. The lesions were mildly tender to palpation. The remainder of the physical examination was unremarkable.
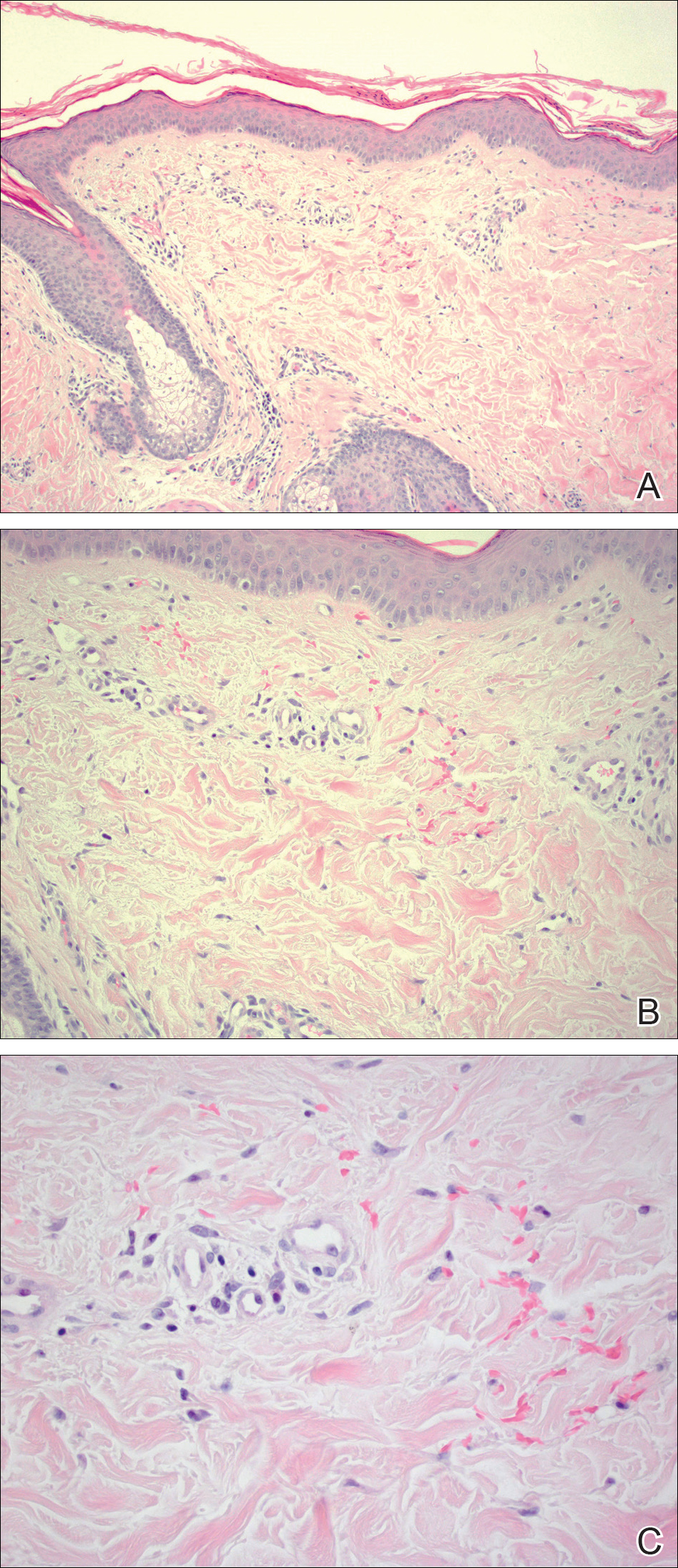
Given the timing of onset, preceding sun exposure, and the morphologic characteristics of the lesions, sunburn purpura was suspected. A punch biopsy of the anterior aspect of the left thigh was performed to rule out vasculitis. Microscopic examination revealed reactive epidermal changes with mild vascular ectasia and erythrocyte extravasation not associated with appreciable inflammation or evidence of vascular injury (Figure 2). Biopsy exposure to fluorescein-labeled antibodies directed against IgG, IgM, IgA, C3, and polyvalent immunoglobulins (IgG, IgM, and IgA) yielded no immunofluorescence. These biopsy results were consistent with sunburn purpura. Given the patient's normal platelet count, a diagnosis of idiopathic sunburn purpura was made. The patient was informed of the biopsy results and advised that the petechiae should resolve without treatment in 1 to 2 weeks, which occurred.
Sunburn purpura remains a rare phenomenon in which a petechial or purpuric rash develops acutely after intense sun exposure. We prefer the term sunburn purpura because it reflects the acuity of the phenomenon, as opposed to the previous labels solar purpura or photolocalized purpura, which also could suggest causality from chronic sun exposure. It has been proposed that sunburn purpura is a finding associated with a number of conditions rather than a unique entity.1 The following characteristics can be helpful in describing the development of sunburn purpura: delay following UV exposure, gross morphology, histologic findings, and possible associated medical conditions.1 Our case represents an important addition to the literature, as it differs from previously reported cases. Most importantly, the nonspecific biopsy findings and unremarkable laboratory findings associated with our case may represent primary or idiopathic sunburn purpura.
Previously reported cases of sunburn purpura have occurred in patients aged 10 to 66 years. It has been seen following UV exposure, vigorous exercise and high-dose aspirin, or concurrent fluoroquinolone therapy, or in the setting of erythropoietic protoporphyria, idiopathic thrombocytopenic purpura, or polymorphous light eruption.2-8 When performed, histology has revealed capillaritis, solar elastosis, perivascular infiltrate, lymphocytic perivascular infiltrate with dermal edema, or leukocytoclastic vasculitis.1,2,7-9 Our patient did not have a history of erythropoietic protoporphyria, polymorphous light eruption, or idiopathic thrombocytopenic purpura. She had not recently exercised, was not thrombocytopenic, and was not taking antiplatelet medications. She had no recent history of fluoroquinolone use. On histologic examination, our patient's biopsy demonstrated nonspecific petechial changes without signs of chronic UV exposure, dermal edema, vasculitis, lymphocytic infiltrate, or capillaritis.
Idiopathic sunburn purpura should only be diagnosed after other conditions are excluded. When evaluating a patient who presents with new-onset petechial rash following sun exposure, it is important to rule out vasculitis or thrombocytopenia as the cause, which is best achieved through skin biopsy and a platelet count, respectively. If there are no associated symptoms or thrombocytopenia and biopsy shows nonspecific vascular ectasia and erythrocyte extravasation, the physician should consider the diagnosis of idiopathic sunburn (solar or photolocalized) purpura. Along with regular UV protection, the physician should advise that the rash typically resolves without treatment in 1 to 2 weeks.
- Waters AJ, Sandhu DR, Green CM, et al. Solar capillaritis as a cause of solar purpura. Clin Exp Dermatol. 2009;34:E821-E824.
- Latenser BA, Hempstead RW. Exercise-associated solar purpura in an atypical location. Cutis. 1985;35:365-366.
- Rubegni P, Feci L, Pellegrino M, et al. Photolocalized purpura during levofloxacin therapy. Photodermatol Photoimmunol Photomed. 2012;28:105-107.
- Urbina F, Barrios M, Sudy E. Photolocalized purpura during ciprofloxacin therapy. Photodermatol Photoimmunol Photomed. 2006;22:111-112.
- Torinuki W, Miura T. Erythropoietic protoporphyria showing solar purpura. Dermatologica. 1983;167:220-222.
- Leung AK. Purpura associated with exposure to sunlight. J R Soc Med. 1986;79:423-424.
- Kalivas J, Kalivas L. Solar purpura appearing in a patient with polymorphous light eruption. Photodermatol Photoimmunol Photomed. 1995;11:31-32.
- Ros AM. Solar purpura--an unusual manifestation of polymorphous light eruption. Photodermatol. 1988;5:47-48.
- Guarrera M, Parodi A, Rebora A. Solar purpura is not related to polymorphous light eruption. Photodermatol. 1989;6:293-294.
To the Editor:
Chronic UV exposure has been linked to increased skin fragility and the development of purpuric lesions, a benign condition known as actinic purpura and commonly seen in elderly patients. Petechial skin changes acutely following intense sun exposure is a rare phenomenon referred to as sunburn purpura, photolocalized purpura, or solar purpura.
A 19-year-old woman presented with red and purple spots on the pretibial region of both legs extending to the thigh. One week prior to presentation she had a severe sunburn affecting most of the body, which resolved without blistering. Two days later, the spots appeared within the most severely sunburned areas of both legs. The patient reported that the lesions were mildly painful to palpation, but she was more concerned about the appearance. She denied any history of similar skin changes associated with sun exposure. The patient was otherwise healthy and denied any recent illnesses. She noted a history of mild bruising and bleeding with a resulting unremarkable workup by her primary care physician. The only medication taken was etonogestrel-ethinyl estradiol vaginal ring.
The scalp, face, arms, trunk, and legs were examined, and nonpalpable petechial changes were noted on the anterior aspect of the legs (Figure 1), with changes more prominent on the distal aspect of the legs. Mild superficial epidermal exfoliation was noted on both anterior thighs. The area of the lesions was not warm. The lesions were mildly tender to palpation. The remainder of the physical examination was unremarkable.
Given the timing of onset, preceding sun exposure, and the morphologic characteristics of the lesions, sunburn purpura was suspected. A punch biopsy of the anterior aspect of the left thigh was performed to rule out vasculitis. Microscopic examination revealed reactive epidermal changes with mild vascular ectasia and erythrocyte extravasation not associated with appreciable inflammation or evidence of vascular injury (Figure 2). Biopsy exposure to fluorescein-labeled antibodies directed against IgG, IgM, IgA, C3, and polyvalent immunoglobulins (IgG, IgM, and IgA) yielded no immunofluorescence. These biopsy results were consistent with sunburn purpura. Given the patient's normal platelet count, a diagnosis of idiopathic sunburn purpura was made. The patient was informed of the biopsy results and advised that the petechiae should resolve without treatment in 1 to 2 weeks, which occurred.
Sunburn purpura remains a rare phenomenon in which a petechial or purpuric rash develops acutely after intense sun exposure. We prefer the term sunburn purpura because it reflects the acuity of the phenomenon, as opposed to the previous labels solar purpura or photolocalized purpura, which also could suggest causality from chronic sun exposure. It has been proposed that sunburn purpura is a finding associated with a number of conditions rather than a unique entity.1 The following characteristics can be helpful in describing the development of sunburn purpura: delay following UV exposure, gross morphology, histologic findings, and possible associated medical conditions.1 Our case represents an important addition to the literature, as it differs from previously reported cases. Most importantly, the nonspecific biopsy findings and unremarkable laboratory findings associated with our case may represent primary or idiopathic sunburn purpura.
Previously reported cases of sunburn purpura have occurred in patients aged 10 to 66 years. It has been seen following UV exposure, vigorous exercise and high-dose aspirin, or concurrent fluoroquinolone therapy, or in the setting of erythropoietic protoporphyria, idiopathic thrombocytopenic purpura, or polymorphous light eruption.2-8 When performed, histology has revealed capillaritis, solar elastosis, perivascular infiltrate, lymphocytic perivascular infiltrate with dermal edema, or leukocytoclastic vasculitis.1,2,7-9 Our patient did not have a history of erythropoietic protoporphyria, polymorphous light eruption, or idiopathic thrombocytopenic purpura. She had not recently exercised, was not thrombocytopenic, and was not taking antiplatelet medications. She had no recent history of fluoroquinolone use. On histologic examination, our patient's biopsy demonstrated nonspecific petechial changes without signs of chronic UV exposure, dermal edema, vasculitis, lymphocytic infiltrate, or capillaritis.
Idiopathic sunburn purpura should only be diagnosed after other conditions are excluded. When evaluating a patient who presents with new-onset petechial rash following sun exposure, it is important to rule out vasculitis or thrombocytopenia as the cause, which is best achieved through skin biopsy and a platelet count, respectively. If there are no associated symptoms or thrombocytopenia and biopsy shows nonspecific vascular ectasia and erythrocyte extravasation, the physician should consider the diagnosis of idiopathic sunburn (solar or photolocalized) purpura. Along with regular UV protection, the physician should advise that the rash typically resolves without treatment in 1 to 2 weeks.
To the Editor:
Chronic UV exposure has been linked to increased skin fragility and the development of purpuric lesions, a benign condition known as actinic purpura and commonly seen in elderly patients. Petechial skin changes acutely following intense sun exposure is a rare phenomenon referred to as sunburn purpura, photolocalized purpura, or solar purpura.
A 19-year-old woman presented with red and purple spots on the pretibial region of both legs extending to the thigh. One week prior to presentation she had a severe sunburn affecting most of the body, which resolved without blistering. Two days later, the spots appeared within the most severely sunburned areas of both legs. The patient reported that the lesions were mildly painful to palpation, but she was more concerned about the appearance. She denied any history of similar skin changes associated with sun exposure. The patient was otherwise healthy and denied any recent illnesses. She noted a history of mild bruising and bleeding with a resulting unremarkable workup by her primary care physician. The only medication taken was etonogestrel-ethinyl estradiol vaginal ring.
The scalp, face, arms, trunk, and legs were examined, and nonpalpable petechial changes were noted on the anterior aspect of the legs (Figure 1), with changes more prominent on the distal aspect of the legs. Mild superficial epidermal exfoliation was noted on both anterior thighs. The area of the lesions was not warm. The lesions were mildly tender to palpation. The remainder of the physical examination was unremarkable.
Given the timing of onset, preceding sun exposure, and the morphologic characteristics of the lesions, sunburn purpura was suspected. A punch biopsy of the anterior aspect of the left thigh was performed to rule out vasculitis. Microscopic examination revealed reactive epidermal changes with mild vascular ectasia and erythrocyte extravasation not associated with appreciable inflammation or evidence of vascular injury (Figure 2). Biopsy exposure to fluorescein-labeled antibodies directed against IgG, IgM, IgA, C3, and polyvalent immunoglobulins (IgG, IgM, and IgA) yielded no immunofluorescence. These biopsy results were consistent with sunburn purpura. Given the patient's normal platelet count, a diagnosis of idiopathic sunburn purpura was made. The patient was informed of the biopsy results and advised that the petechiae should resolve without treatment in 1 to 2 weeks, which occurred.
Sunburn purpura remains a rare phenomenon in which a petechial or purpuric rash develops acutely after intense sun exposure. We prefer the term sunburn purpura because it reflects the acuity of the phenomenon, as opposed to the previous labels solar purpura or photolocalized purpura, which also could suggest causality from chronic sun exposure. It has been proposed that sunburn purpura is a finding associated with a number of conditions rather than a unique entity.1 The following characteristics can be helpful in describing the development of sunburn purpura: delay following UV exposure, gross morphology, histologic findings, and possible associated medical conditions.1 Our case represents an important addition to the literature, as it differs from previously reported cases. Most importantly, the nonspecific biopsy findings and unremarkable laboratory findings associated with our case may represent primary or idiopathic sunburn purpura.
Previously reported cases of sunburn purpura have occurred in patients aged 10 to 66 years. It has been seen following UV exposure, vigorous exercise and high-dose aspirin, or concurrent fluoroquinolone therapy, or in the setting of erythropoietic protoporphyria, idiopathic thrombocytopenic purpura, or polymorphous light eruption.2-8 When performed, histology has revealed capillaritis, solar elastosis, perivascular infiltrate, lymphocytic perivascular infiltrate with dermal edema, or leukocytoclastic vasculitis.1,2,7-9 Our patient did not have a history of erythropoietic protoporphyria, polymorphous light eruption, or idiopathic thrombocytopenic purpura. She had not recently exercised, was not thrombocytopenic, and was not taking antiplatelet medications. She had no recent history of fluoroquinolone use. On histologic examination, our patient's biopsy demonstrated nonspecific petechial changes without signs of chronic UV exposure, dermal edema, vasculitis, lymphocytic infiltrate, or capillaritis.
Idiopathic sunburn purpura should only be diagnosed after other conditions are excluded. When evaluating a patient who presents with new-onset petechial rash following sun exposure, it is important to rule out vasculitis or thrombocytopenia as the cause, which is best achieved through skin biopsy and a platelet count, respectively. If there are no associated symptoms or thrombocytopenia and biopsy shows nonspecific vascular ectasia and erythrocyte extravasation, the physician should consider the diagnosis of idiopathic sunburn (solar or photolocalized) purpura. Along with regular UV protection, the physician should advise that the rash typically resolves without treatment in 1 to 2 weeks.
- Waters AJ, Sandhu DR, Green CM, et al. Solar capillaritis as a cause of solar purpura. Clin Exp Dermatol. 2009;34:E821-E824.
- Latenser BA, Hempstead RW. Exercise-associated solar purpura in an atypical location. Cutis. 1985;35:365-366.
- Rubegni P, Feci L, Pellegrino M, et al. Photolocalized purpura during levofloxacin therapy. Photodermatol Photoimmunol Photomed. 2012;28:105-107.
- Urbina F, Barrios M, Sudy E. Photolocalized purpura during ciprofloxacin therapy. Photodermatol Photoimmunol Photomed. 2006;22:111-112.
- Torinuki W, Miura T. Erythropoietic protoporphyria showing solar purpura. Dermatologica. 1983;167:220-222.
- Leung AK. Purpura associated with exposure to sunlight. J R Soc Med. 1986;79:423-424.
- Kalivas J, Kalivas L. Solar purpura appearing in a patient with polymorphous light eruption. Photodermatol Photoimmunol Photomed. 1995;11:31-32.
- Ros AM. Solar purpura--an unusual manifestation of polymorphous light eruption. Photodermatol. 1988;5:47-48.
- Guarrera M, Parodi A, Rebora A. Solar purpura is not related to polymorphous light eruption. Photodermatol. 1989;6:293-294.
- Waters AJ, Sandhu DR, Green CM, et al. Solar capillaritis as a cause of solar purpura. Clin Exp Dermatol. 2009;34:E821-E824.
- Latenser BA, Hempstead RW. Exercise-associated solar purpura in an atypical location. Cutis. 1985;35:365-366.
- Rubegni P, Feci L, Pellegrino M, et al. Photolocalized purpura during levofloxacin therapy. Photodermatol Photoimmunol Photomed. 2012;28:105-107.
- Urbina F, Barrios M, Sudy E. Photolocalized purpura during ciprofloxacin therapy. Photodermatol Photoimmunol Photomed. 2006;22:111-112.
- Torinuki W, Miura T. Erythropoietic protoporphyria showing solar purpura. Dermatologica. 1983;167:220-222.
- Leung AK. Purpura associated with exposure to sunlight. J R Soc Med. 1986;79:423-424.
- Kalivas J, Kalivas L. Solar purpura appearing in a patient with polymorphous light eruption. Photodermatol Photoimmunol Photomed. 1995;11:31-32.
- Ros AM. Solar purpura--an unusual manifestation of polymorphous light eruption. Photodermatol. 1988;5:47-48.
- Guarrera M, Parodi A, Rebora A. Solar purpura is not related to polymorphous light eruption. Photodermatol. 1989;6:293-294.
Practice Points
- Petechial skin changes acutely following intense sun exposure is a rare phenomenon referred to as sunburn purpura, photolocalized purpura, or solar purpura.
- Idiopathic sunburn purpura should only be diagnosed after vasculitis and/or thrombocytopenia is ruled out, which is best achieved through skin biopsy and a platelet count, respectively.
- The rash typically resolves without treatment in 1 to 2 weeks; however, a variety of UV protection modalities and education should be offered to the patient.