User login
Tumor Necrosis Factor α Inhibitor–Induced Lupuslike Syndrome in a Patient Prescribed Certolizumab Pegol
To the Editor:
Tumor necrosis factor α (TNF-α) inhibitor–induced lupuslike syndrome (TAILS) is a newly described entity that refers to the onset of subacute cutaneous lupus erythematosus (SCLE) during drug therapy with TNF-α antagonists. The condition is unique because it is thought to occur via a separate pathophysiologic mechanism than all other agents implicated in the development of drug-induced lupus erythematosus (DILE). Infliximab and etanercept are the 2 most common TNF-α antagonists associated with TAILS. Although rare, adalimumab, golimumab, and certolizumab pegol have been reported to induce this state of autoimmunity. We report an uncommon presentation of TAILS in a patient taking certolizumab pegol with a brief discussion of the pathogenesis underlying TAILS.

A 71-year-old woman presented to the dermatology clinic with a rash located on the arms, face, and trunk that she reported as having been present for months. She had a medical history of rheumatoid arthritis and currently was receiving certolizumab pegol injections. Physical examination revealed erythematous patches and plaques with overlying scaling and evidence of atrophic scarring on sun-exposed areas of the body. The lesions predominantly were in a symmetrical distribution across the extensor surfaces of both outer arms as well as the posterior superior thoracic region extending anteriorly along the bilateral supraclavicular area (Figures 1 and 2). A 4-mm punch biopsy was obtained and sent for histologic analysis, along with a sample of the patient’s serum for antinuclear antibody (ANA) testing.
.")
Hematoxylin and eosin–stained tissue sections of the right superior thoracic lesions revealed epidermal atrophy, hyperkeratosis, and vacuolar alteration of the basal layer with apoptosis, consistent with a lichenoid tissue reaction. In addition, both superficial and deep perivascular and periadnexal lymphocytic infiltrates were observed as well as increased dermal mucin. Serologic testing was performed with a comprehensive ANA panel of the patient’s serum (Table). Of note, there was a speckled ANA pattern (1:1280), with elevated anti–double-stranded DNA (anti-dsDNA) and anti–Sjögren syndrome–related antigen A (anti-SSA)(also called anti-Ro antibodies) levels. The patient’s rheumatologist was consulted; certolizumab pegol was removed from the current drug regimen and switched to a daily regimen of hydroxychloroquine and prednisone. Seven weeks after discontinuation of certolizumab pegol, the patient was symptom free and without any cutaneous involvement. Based on the histologic analysis, presence of anti-SSA (Ro) autoantibodies, and the resolution of symptoms following withdrawal of anti–TNF-α therapy, a diagnosis of TAILS was made.

Subacute cutaneous lupus erythematosus, the most common subset of DILE, typically presents with annular polycyclic or papulosquamous skin eruptions on the legs; patients often test positive for anti-SSA/Ro and/or anti–Sjögren syndrome–related antigen B (also called anti-La) antibodies. Pharmaceutical agents linked to the development of SCLE are calcium channel blockers, angiotensin-converting enzyme inhibitors, thiazide diuretics, terbinafine, the chemotherapeutic agent gemcitabine, and TNF-α antagonists.1,2 Tumor necrosis factor α antagonists are biologic agents that commonly are used in the management of systemic inflammatory diseases such as ulcerative colitis, Crohn disease, seronegative spondyloarthropathies, and rheumatoid arthritis. Among this family of therapeutics includes adalimumab (humanized monoclonal antibody), infliximab (chimeric monoclonal TNF-α antagonist), etanercept (soluble receptor fusion protein), certolizumab pegol (Fab fraction of a human IgG monoclonal antibody), and golimumab (humanized monoclonal antibody).
Tumor necrosis factor α inhibitor–induced lupuslike syndrome most commonly occurs in women in the fifth decade of life, and it is seen more often in those using infliximab or entanercept.3 Although reports do exist, TAILS rarely complicates treatment with adalimumab, golimumab, or certolizumab.4,5 Due to the lack of reports, there are no diagnostic criteria nor an acceptable theory regarding the pathogenesis. In one study in France, the estimated incidence was thought to be 0.19% for infliximab and 0.18% for etanercept.6 Tumor necrosis factor α inhibitor–induced lupuslike syndrome is unique in that it is thought to occur by a different mechanism than that of other known offending agents in the development of DILE. Molecular mimicry, direct cytotoxicity, altered T-cell gene expression, and disruption of central immune tolerance have all been hypothesized to cause drug-induced systemic lupus erythematosus, SCLE, and chronic cutaneous lupus erythematosus. Tumor necrosis factor α inhibitors, are postulated to cause the induction of SCLE via an independent route separate from not only other drugs that cause SCLE but also all forms of DILE as a whole, making it a distinctive player within the realm of agents known to cause a lupuslike syndrome. The following hypotheses may explain this occurrence:
1. Increased humoral autoimmunity: Under normal circumstances, TNF-α activation leads to upregulation in the production of cytotoxic CD8+ T lymphocytes. The upregulation of CD8+ T lymphocytes concurrently leads to a simultaneous suppression of B lymphocytes. Inhibiting the effects of TNF-α on the other hand promotes cytotoxic T-lymphocyte suppression, leading to an increased synthesis of B cells and subsequently a state of increased humoral autoimmunity.7
2. Infection: The immunosuppressive effects of TNF-α inhibitors are well known, and the propensity to develop microbial infections, such as tuberculosis, is markedly increased on the use of these agents. Infections brought on by TNF-α inhibitor usage are hypothesized to induce a widespread activation of polyclonal B lymphocytes, eventually leading to the formation of antibodies against these polyclonal B lymphocytes and subsequently SCLE.8
3. Helper T cell (TH2) response: The inhibition of TH1 CD4+ lymphocytes by TNF-α inversely leads to an increased production of TH2 CD4+ lymphocytes. This increase in the levels of circulating TH2 CD4+ lymphocytes brought on by the action of anti–TNF-α agents is thought to promote the development of SCLE.9,10
4. Apoptosis theory: Molecules of TNF-α inhibitors are capable of binding to TNF-α receptors on the cell surface. In doing so, cellular apoptosis is triggered, resulting in the release of nucleosomal autoantigens from the apoptotic cells. In susceptible individuals, autoantibodies then begin to form against the nucleosomal autoantigens, leading to an autoimmune reaction that is characterized by SCLE.11,12
Major histone compatibility (MHC) antigen testing performed by Sontheimer et al12 established the presence of the HLA class I, HLA-B8, and/or HLA-DR3 haplotypes in patients with SCLE.13,14 Furthermore, there is a well-known association between the antinuclear profile of known SCLE patients and the presence of anti-SSA (Ro) antibodies.13 Therefore, we propose that in susceptible individuals, such as those with the HLA class I, HLA-B8, or HLA-DR3 haplotypes, the initiation of a TNF-α inhibitor causes cellular apoptosis with the subsequent release of nucleosomal and cytoplasmic components (namely that of the Ro autoantigens), inducing a state of autoimmunity. An ensuing immunogenic response is then initiated in predisposed individuals for which anti-SSA (Ro) autoantibodies are produced against these previously mentioned autoantigens.
Drug-induced SCLE is most common in females (71%), with a median age of 58 years. The most common site of cutaneous manifestations is the legs.15 Although our patient was in the eighth decade of life with predominant cutaneous involvement of the upper extremity, the erythematous plaques with a symmetric, annular, polycyclic appearance in photosensitive regions raised a heightened suspicion for lupus erythematosus. Histology classically involves an interface dermatitis with vacuolar or hydropic change and lymphocytic infiltrates,16 consistent with the analysis of tissue sections from our patient. Moreover, the speckled ANA profile with positive anti-dsDNA and anti-SSA (Ro) antibodies in the absence of a negative rheumatoid factor and anticyclic citrullinated peptide antibodies strongly favored the diagnosis of SCLE over alternative diagnoses.2
The supraclavicular rash in our patient raises clinical suspicion for the shawl sign of dermatomyositis, which also is associated with musculoskeletal pain and photosensitivity. In addition, skin biopsy revealed vacuolar alteration of the basement membrane zoneand dermal mucin in both lupus erythematosus and dermatomyositis; therefore, skin biopsy is of little use in distinguishing the 2 conditions, and antibody testing must be performed. Although anti-SSA (Ro) antibodies commonly are associated with SCLE, there are reports involving positivity for the extractable nuclear antigen in cases of dermatomyositis.17 Based on our patient’s current drug regimen, including that of a known offending agent for SCLE, a presumptive diagnosis of TAILS was made. Following withdrawal of certolizumab pegol injections and subsequent resolution of the skin lesions, our patient was given a definitive diagnosis of TAILS based on clinical and pathological assessments.
The clinical diagnosis of TAILS should be made according to the triad of at least 1 serologic and 1 nonserologic American College of Rheumatology criteria, such as anti-SSA (Ro) antibodies and a photosensitive rash, respectively, as well as a relationship between the onset of symptoms and TNF-α inhibitor therapy.18 Both the definitive diagnosis and the treatment of TAILS can be made via withdrawal of the TNF-α inhibitor, which was true in our case whereby chronologically the onset of use with a TNF-α inhibitor was associated with disease onset. Furthermore, withdrawal led to complete improvement of all signs and symptoms, collectively supporting a diagnosis of TAILS. Notably, switching to a different TNF-α inhibitor has been shown to be safe and effective.19
- Marzano AV, Vezzoli P, Crosti C. Drug-induced lupus: an update on its dermatological aspects. Lupus. 2009;18:935-940.
- Wiznia LE, Subtil A, Choi JN. Subacute cutaneous lupus erythematosus induced by chemotherapy: gemcitabine as a causative agent. JAMA Dermatol. 2013;149:1071-1075.
- Williams VL, Cohen PR. TNF alpha antagonist-induced lupus-like syndrome: report and review of the literature with implications for treatment with alternative TNF alpha antagonists. Int J Dermatol. 2011;50:619-625.
- Pasut G. Pegylation of biological molecules and potential benefits: pharmacological properties of certolizumab pegol. Bio Drugs. 2014;28(suppl 1):15-23.
- Mudduluru BM, Shah S, Shamah S. et al. TNF-alpha antagonist induced lupus on three different agents. Postgrad Med. 2017;129:304-306.
- De Bandt M. Anti-TNF-alpha-induced lupus. Arthritis Res Ther. 2019;21:235.
- Costa MF, Said NR, Zimmermann B. Drug-induced lupus due to anti-tumor necrosis factor alfa agents. Semin Arthritis Rheum. 2008;37:381-387.
- Caramaschi P, Biasi D, Colombatti M. Anti-TNF alpha therapy in rheumatoid arthritis and autoimmunity. Rheumatol Int. 2006;26:209-214.
- Yung RL, Quddus J, Chrisp CE, et al. Mechanism of drug-induced lupus. I. cloned Th2 cells modified with DNA methylation inhibitors in vitro cause autoimmunity in vivo. J Immunol. 1995;154:3025-3035.
- Yung R, Powers D, Johnson K, et al. Mechanisms of drug-induced lupus. II. T cells overexpressing lymphocyte function-associated antigen 1 become autoreactive and cause a lupuslike disease in syngeneic mice. J Clin Invest. 1996;97:2866-2871.
- Sontheimer RD, Stastny P, Gilliam JN. Human histocompatibility antigen associations in subacute cutaneous lupus erythematosus. J Clin Invest. 1981;67:312-316.
- Sontheimer RD, Maddison PJ, Reichlin M, et al. Serologic and HLA associations in subacute cutaneous lupus erythematosus, a clinical subset of lupus erythematosus. Ann Intern Med. 1982;97:664-671.
- Lee LA, Roberts CM, Frank MB, et al. The autoantibody response to Ro/SSA in cutaneous lupus erythematosus. Arch Dermatol. 1994;130:1262-1268.
- Deutscher SL, Harley JB, Keene JD. Molecular analysis of the 60-kDa human Ro ribonucleoprotein. Proc Natl Acad Sci. 1988;85:9479-9483.
- DalleVedove C, Simon JC, Girolomoni G. Drug-induced lupus erythematosus with emphasis on skin manifestations and the role of anti-TNFα agents [article in German]. J Dtsch Dermatol Ges. 2012;10:889-897.
- Okon LG, Werth VP. Cutaneous lupus erythematosus: diagnosis and treatment. Best Pract Res Clin Rheumatol. 2013;27:391-404.
- Schulte-Pelkum J, Fritzler M, Mahler M. Latest update on the Ro/SS-A autoantibody system. Autoimmun Rev. 2009;8:632-637.
- De Bandt M, Sibilia J, Le Loët X, et al. Systemic lupus erythematosus induced by anti-tumour necrosis factor alpha therapy: a French national survey. Arthritis Res Ther. 2005;7:R545-R551.
- Lupu A, Tieranu C, Constantinescu CL, et al. TNFα inhibitor induced lupus-like syndrome (TAILS) in a patient with IBD. Current Health Sci J. 2014;40:285-288.
To the Editor:
Tumor necrosis factor α (TNF-α) inhibitor–induced lupuslike syndrome (TAILS) is a newly described entity that refers to the onset of subacute cutaneous lupus erythematosus (SCLE) during drug therapy with TNF-α antagonists. The condition is unique because it is thought to occur via a separate pathophysiologic mechanism than all other agents implicated in the development of drug-induced lupus erythematosus (DILE). Infliximab and etanercept are the 2 most common TNF-α antagonists associated with TAILS. Although rare, adalimumab, golimumab, and certolizumab pegol have been reported to induce this state of autoimmunity. We report an uncommon presentation of TAILS in a patient taking certolizumab pegol with a brief discussion of the pathogenesis underlying TAILS.
A 71-year-old woman presented to the dermatology clinic with a rash located on the arms, face, and trunk that she reported as having been present for months. She had a medical history of rheumatoid arthritis and currently was receiving certolizumab pegol injections. Physical examination revealed erythematous patches and plaques with overlying scaling and evidence of atrophic scarring on sun-exposed areas of the body. The lesions predominantly were in a symmetrical distribution across the extensor surfaces of both outer arms as well as the posterior superior thoracic region extending anteriorly along the bilateral supraclavicular area (Figures 1 and 2). A 4-mm punch biopsy was obtained and sent for histologic analysis, along with a sample of the patient’s serum for antinuclear antibody (ANA) testing.
Hematoxylin and eosin–stained tissue sections of the right superior thoracic lesions revealed epidermal atrophy, hyperkeratosis, and vacuolar alteration of the basal layer with apoptosis, consistent with a lichenoid tissue reaction. In addition, both superficial and deep perivascular and periadnexal lymphocytic infiltrates were observed as well as increased dermal mucin. Serologic testing was performed with a comprehensive ANA panel of the patient’s serum (Table). Of note, there was a speckled ANA pattern (1:1280), with elevated anti–double-stranded DNA (anti-dsDNA) and anti–Sjögren syndrome–related antigen A (anti-SSA)(also called anti-Ro antibodies) levels. The patient’s rheumatologist was consulted; certolizumab pegol was removed from the current drug regimen and switched to a daily regimen of hydroxychloroquine and prednisone. Seven weeks after discontinuation of certolizumab pegol, the patient was symptom free and without any cutaneous involvement. Based on the histologic analysis, presence of anti-SSA (Ro) autoantibodies, and the resolution of symptoms following withdrawal of anti–TNF-α therapy, a diagnosis of TAILS was made.
Subacute cutaneous lupus erythematosus, the most common subset of DILE, typically presents with annular polycyclic or papulosquamous skin eruptions on the legs; patients often test positive for anti-SSA/Ro and/or anti–Sjögren syndrome–related antigen B (also called anti-La) antibodies. Pharmaceutical agents linked to the development of SCLE are calcium channel blockers, angiotensin-converting enzyme inhibitors, thiazide diuretics, terbinafine, the chemotherapeutic agent gemcitabine, and TNF-α antagonists.1,2 Tumor necrosis factor α antagonists are biologic agents that commonly are used in the management of systemic inflammatory diseases such as ulcerative colitis, Crohn disease, seronegative spondyloarthropathies, and rheumatoid arthritis. Among this family of therapeutics includes adalimumab (humanized monoclonal antibody), infliximab (chimeric monoclonal TNF-α antagonist), etanercept (soluble receptor fusion protein), certolizumab pegol (Fab fraction of a human IgG monoclonal antibody), and golimumab (humanized monoclonal antibody).
Tumor necrosis factor α inhibitor–induced lupuslike syndrome most commonly occurs in women in the fifth decade of life, and it is seen more often in those using infliximab or entanercept.3 Although reports do exist, TAILS rarely complicates treatment with adalimumab, golimumab, or certolizumab.4,5 Due to the lack of reports, there are no diagnostic criteria nor an acceptable theory regarding the pathogenesis. In one study in France, the estimated incidence was thought to be 0.19% for infliximab and 0.18% for etanercept.6 Tumor necrosis factor α inhibitor–induced lupuslike syndrome is unique in that it is thought to occur by a different mechanism than that of other known offending agents in the development of DILE. Molecular mimicry, direct cytotoxicity, altered T-cell gene expression, and disruption of central immune tolerance have all been hypothesized to cause drug-induced systemic lupus erythematosus, SCLE, and chronic cutaneous lupus erythematosus. Tumor necrosis factor α inhibitors, are postulated to cause the induction of SCLE via an independent route separate from not only other drugs that cause SCLE but also all forms of DILE as a whole, making it a distinctive player within the realm of agents known to cause a lupuslike syndrome. The following hypotheses may explain this occurrence:
1. Increased humoral autoimmunity: Under normal circumstances, TNF-α activation leads to upregulation in the production of cytotoxic CD8+ T lymphocytes. The upregulation of CD8+ T lymphocytes concurrently leads to a simultaneous suppression of B lymphocytes. Inhibiting the effects of TNF-α on the other hand promotes cytotoxic T-lymphocyte suppression, leading to an increased synthesis of B cells and subsequently a state of increased humoral autoimmunity.7
2. Infection: The immunosuppressive effects of TNF-α inhibitors are well known, and the propensity to develop microbial infections, such as tuberculosis, is markedly increased on the use of these agents. Infections brought on by TNF-α inhibitor usage are hypothesized to induce a widespread activation of polyclonal B lymphocytes, eventually leading to the formation of antibodies against these polyclonal B lymphocytes and subsequently SCLE.8
3. Helper T cell (TH2) response: The inhibition of TH1 CD4+ lymphocytes by TNF-α inversely leads to an increased production of TH2 CD4+ lymphocytes. This increase in the levels of circulating TH2 CD4+ lymphocytes brought on by the action of anti–TNF-α agents is thought to promote the development of SCLE.9,10
4. Apoptosis theory: Molecules of TNF-α inhibitors are capable of binding to TNF-α receptors on the cell surface. In doing so, cellular apoptosis is triggered, resulting in the release of nucleosomal autoantigens from the apoptotic cells. In susceptible individuals, autoantibodies then begin to form against the nucleosomal autoantigens, leading to an autoimmune reaction that is characterized by SCLE.11,12
Major histone compatibility (MHC) antigen testing performed by Sontheimer et al12 established the presence of the HLA class I, HLA-B8, and/or HLA-DR3 haplotypes in patients with SCLE.13,14 Furthermore, there is a well-known association between the antinuclear profile of known SCLE patients and the presence of anti-SSA (Ro) antibodies.13 Therefore, we propose that in susceptible individuals, such as those with the HLA class I, HLA-B8, or HLA-DR3 haplotypes, the initiation of a TNF-α inhibitor causes cellular apoptosis with the subsequent release of nucleosomal and cytoplasmic components (namely that of the Ro autoantigens), inducing a state of autoimmunity. An ensuing immunogenic response is then initiated in predisposed individuals for which anti-SSA (Ro) autoantibodies are produced against these previously mentioned autoantigens.
Drug-induced SCLE is most common in females (71%), with a median age of 58 years. The most common site of cutaneous manifestations is the legs.15 Although our patient was in the eighth decade of life with predominant cutaneous involvement of the upper extremity, the erythematous plaques with a symmetric, annular, polycyclic appearance in photosensitive regions raised a heightened suspicion for lupus erythematosus. Histology classically involves an interface dermatitis with vacuolar or hydropic change and lymphocytic infiltrates,16 consistent with the analysis of tissue sections from our patient. Moreover, the speckled ANA profile with positive anti-dsDNA and anti-SSA (Ro) antibodies in the absence of a negative rheumatoid factor and anticyclic citrullinated peptide antibodies strongly favored the diagnosis of SCLE over alternative diagnoses.2
The supraclavicular rash in our patient raises clinical suspicion for the shawl sign of dermatomyositis, which also is associated with musculoskeletal pain and photosensitivity. In addition, skin biopsy revealed vacuolar alteration of the basement membrane zoneand dermal mucin in both lupus erythematosus and dermatomyositis; therefore, skin biopsy is of little use in distinguishing the 2 conditions, and antibody testing must be performed. Although anti-SSA (Ro) antibodies commonly are associated with SCLE, there are reports involving positivity for the extractable nuclear antigen in cases of dermatomyositis.17 Based on our patient’s current drug regimen, including that of a known offending agent for SCLE, a presumptive diagnosis of TAILS was made. Following withdrawal of certolizumab pegol injections and subsequent resolution of the skin lesions, our patient was given a definitive diagnosis of TAILS based on clinical and pathological assessments.
The clinical diagnosis of TAILS should be made according to the triad of at least 1 serologic and 1 nonserologic American College of Rheumatology criteria, such as anti-SSA (Ro) antibodies and a photosensitive rash, respectively, as well as a relationship between the onset of symptoms and TNF-α inhibitor therapy.18 Both the definitive diagnosis and the treatment of TAILS can be made via withdrawal of the TNF-α inhibitor, which was true in our case whereby chronologically the onset of use with a TNF-α inhibitor was associated with disease onset. Furthermore, withdrawal led to complete improvement of all signs and symptoms, collectively supporting a diagnosis of TAILS. Notably, switching to a different TNF-α inhibitor has been shown to be safe and effective.19
To the Editor:
Tumor necrosis factor α (TNF-α) inhibitor–induced lupuslike syndrome (TAILS) is a newly described entity that refers to the onset of subacute cutaneous lupus erythematosus (SCLE) during drug therapy with TNF-α antagonists. The condition is unique because it is thought to occur via a separate pathophysiologic mechanism than all other agents implicated in the development of drug-induced lupus erythematosus (DILE). Infliximab and etanercept are the 2 most common TNF-α antagonists associated with TAILS. Although rare, adalimumab, golimumab, and certolizumab pegol have been reported to induce this state of autoimmunity. We report an uncommon presentation of TAILS in a patient taking certolizumab pegol with a brief discussion of the pathogenesis underlying TAILS.
A 71-year-old woman presented to the dermatology clinic with a rash located on the arms, face, and trunk that she reported as having been present for months. She had a medical history of rheumatoid arthritis and currently was receiving certolizumab pegol injections. Physical examination revealed erythematous patches and plaques with overlying scaling and evidence of atrophic scarring on sun-exposed areas of the body. The lesions predominantly were in a symmetrical distribution across the extensor surfaces of both outer arms as well as the posterior superior thoracic region extending anteriorly along the bilateral supraclavicular area (Figures 1 and 2). A 4-mm punch biopsy was obtained and sent for histologic analysis, along with a sample of the patient’s serum for antinuclear antibody (ANA) testing.
Hematoxylin and eosin–stained tissue sections of the right superior thoracic lesions revealed epidermal atrophy, hyperkeratosis, and vacuolar alteration of the basal layer with apoptosis, consistent with a lichenoid tissue reaction. In addition, both superficial and deep perivascular and periadnexal lymphocytic infiltrates were observed as well as increased dermal mucin. Serologic testing was performed with a comprehensive ANA panel of the patient’s serum (Table). Of note, there was a speckled ANA pattern (1:1280), with elevated anti–double-stranded DNA (anti-dsDNA) and anti–Sjögren syndrome–related antigen A (anti-SSA)(also called anti-Ro antibodies) levels. The patient’s rheumatologist was consulted; certolizumab pegol was removed from the current drug regimen and switched to a daily regimen of hydroxychloroquine and prednisone. Seven weeks after discontinuation of certolizumab pegol, the patient was symptom free and without any cutaneous involvement. Based on the histologic analysis, presence of anti-SSA (Ro) autoantibodies, and the resolution of symptoms following withdrawal of anti–TNF-α therapy, a diagnosis of TAILS was made.
Subacute cutaneous lupus erythematosus, the most common subset of DILE, typically presents with annular polycyclic or papulosquamous skin eruptions on the legs; patients often test positive for anti-SSA/Ro and/or anti–Sjögren syndrome–related antigen B (also called anti-La) antibodies. Pharmaceutical agents linked to the development of SCLE are calcium channel blockers, angiotensin-converting enzyme inhibitors, thiazide diuretics, terbinafine, the chemotherapeutic agent gemcitabine, and TNF-α antagonists.1,2 Tumor necrosis factor α antagonists are biologic agents that commonly are used in the management of systemic inflammatory diseases such as ulcerative colitis, Crohn disease, seronegative spondyloarthropathies, and rheumatoid arthritis. Among this family of therapeutics includes adalimumab (humanized monoclonal antibody), infliximab (chimeric monoclonal TNF-α antagonist), etanercept (soluble receptor fusion protein), certolizumab pegol (Fab fraction of a human IgG monoclonal antibody), and golimumab (humanized monoclonal antibody).
Tumor necrosis factor α inhibitor–induced lupuslike syndrome most commonly occurs in women in the fifth decade of life, and it is seen more often in those using infliximab or entanercept.3 Although reports do exist, TAILS rarely complicates treatment with adalimumab, golimumab, or certolizumab.4,5 Due to the lack of reports, there are no diagnostic criteria nor an acceptable theory regarding the pathogenesis. In one study in France, the estimated incidence was thought to be 0.19% for infliximab and 0.18% for etanercept.6 Tumor necrosis factor α inhibitor–induced lupuslike syndrome is unique in that it is thought to occur by a different mechanism than that of other known offending agents in the development of DILE. Molecular mimicry, direct cytotoxicity, altered T-cell gene expression, and disruption of central immune tolerance have all been hypothesized to cause drug-induced systemic lupus erythematosus, SCLE, and chronic cutaneous lupus erythematosus. Tumor necrosis factor α inhibitors, are postulated to cause the induction of SCLE via an independent route separate from not only other drugs that cause SCLE but also all forms of DILE as a whole, making it a distinctive player within the realm of agents known to cause a lupuslike syndrome. The following hypotheses may explain this occurrence:
1. Increased humoral autoimmunity: Under normal circumstances, TNF-α activation leads to upregulation in the production of cytotoxic CD8+ T lymphocytes. The upregulation of CD8+ T lymphocytes concurrently leads to a simultaneous suppression of B lymphocytes. Inhibiting the effects of TNF-α on the other hand promotes cytotoxic T-lymphocyte suppression, leading to an increased synthesis of B cells and subsequently a state of increased humoral autoimmunity.7
2. Infection: The immunosuppressive effects of TNF-α inhibitors are well known, and the propensity to develop microbial infections, such as tuberculosis, is markedly increased on the use of these agents. Infections brought on by TNF-α inhibitor usage are hypothesized to induce a widespread activation of polyclonal B lymphocytes, eventually leading to the formation of antibodies against these polyclonal B lymphocytes and subsequently SCLE.8
3. Helper T cell (TH2) response: The inhibition of TH1 CD4+ lymphocytes by TNF-α inversely leads to an increased production of TH2 CD4+ lymphocytes. This increase in the levels of circulating TH2 CD4+ lymphocytes brought on by the action of anti–TNF-α agents is thought to promote the development of SCLE.9,10
4. Apoptosis theory: Molecules of TNF-α inhibitors are capable of binding to TNF-α receptors on the cell surface. In doing so, cellular apoptosis is triggered, resulting in the release of nucleosomal autoantigens from the apoptotic cells. In susceptible individuals, autoantibodies then begin to form against the nucleosomal autoantigens, leading to an autoimmune reaction that is characterized by SCLE.11,12
Major histone compatibility (MHC) antigen testing performed by Sontheimer et al12 established the presence of the HLA class I, HLA-B8, and/or HLA-DR3 haplotypes in patients with SCLE.13,14 Furthermore, there is a well-known association between the antinuclear profile of known SCLE patients and the presence of anti-SSA (Ro) antibodies.13 Therefore, we propose that in susceptible individuals, such as those with the HLA class I, HLA-B8, or HLA-DR3 haplotypes, the initiation of a TNF-α inhibitor causes cellular apoptosis with the subsequent release of nucleosomal and cytoplasmic components (namely that of the Ro autoantigens), inducing a state of autoimmunity. An ensuing immunogenic response is then initiated in predisposed individuals for which anti-SSA (Ro) autoantibodies are produced against these previously mentioned autoantigens.
Drug-induced SCLE is most common in females (71%), with a median age of 58 years. The most common site of cutaneous manifestations is the legs.15 Although our patient was in the eighth decade of life with predominant cutaneous involvement of the upper extremity, the erythematous plaques with a symmetric, annular, polycyclic appearance in photosensitive regions raised a heightened suspicion for lupus erythematosus. Histology classically involves an interface dermatitis with vacuolar or hydropic change and lymphocytic infiltrates,16 consistent with the analysis of tissue sections from our patient. Moreover, the speckled ANA profile with positive anti-dsDNA and anti-SSA (Ro) antibodies in the absence of a negative rheumatoid factor and anticyclic citrullinated peptide antibodies strongly favored the diagnosis of SCLE over alternative diagnoses.2
The supraclavicular rash in our patient raises clinical suspicion for the shawl sign of dermatomyositis, which also is associated with musculoskeletal pain and photosensitivity. In addition, skin biopsy revealed vacuolar alteration of the basement membrane zoneand dermal mucin in both lupus erythematosus and dermatomyositis; therefore, skin biopsy is of little use in distinguishing the 2 conditions, and antibody testing must be performed. Although anti-SSA (Ro) antibodies commonly are associated with SCLE, there are reports involving positivity for the extractable nuclear antigen in cases of dermatomyositis.17 Based on our patient’s current drug regimen, including that of a known offending agent for SCLE, a presumptive diagnosis of TAILS was made. Following withdrawal of certolizumab pegol injections and subsequent resolution of the skin lesions, our patient was given a definitive diagnosis of TAILS based on clinical and pathological assessments.
The clinical diagnosis of TAILS should be made according to the triad of at least 1 serologic and 1 nonserologic American College of Rheumatology criteria, such as anti-SSA (Ro) antibodies and a photosensitive rash, respectively, as well as a relationship between the onset of symptoms and TNF-α inhibitor therapy.18 Both the definitive diagnosis and the treatment of TAILS can be made via withdrawal of the TNF-α inhibitor, which was true in our case whereby chronologically the onset of use with a TNF-α inhibitor was associated with disease onset. Furthermore, withdrawal led to complete improvement of all signs and symptoms, collectively supporting a diagnosis of TAILS. Notably, switching to a different TNF-α inhibitor has been shown to be safe and effective.19
- Marzano AV, Vezzoli P, Crosti C. Drug-induced lupus: an update on its dermatological aspects. Lupus. 2009;18:935-940.
- Wiznia LE, Subtil A, Choi JN. Subacute cutaneous lupus erythematosus induced by chemotherapy: gemcitabine as a causative agent. JAMA Dermatol. 2013;149:1071-1075.
- Williams VL, Cohen PR. TNF alpha antagonist-induced lupus-like syndrome: report and review of the literature with implications for treatment with alternative TNF alpha antagonists. Int J Dermatol. 2011;50:619-625.
- Pasut G. Pegylation of biological molecules and potential benefits: pharmacological properties of certolizumab pegol. Bio Drugs. 2014;28(suppl 1):15-23.
- Mudduluru BM, Shah S, Shamah S. et al. TNF-alpha antagonist induced lupus on three different agents. Postgrad Med. 2017;129:304-306.
- De Bandt M. Anti-TNF-alpha-induced lupus. Arthritis Res Ther. 2019;21:235.
- Costa MF, Said NR, Zimmermann B. Drug-induced lupus due to anti-tumor necrosis factor alfa agents. Semin Arthritis Rheum. 2008;37:381-387.
- Caramaschi P, Biasi D, Colombatti M. Anti-TNF alpha therapy in rheumatoid arthritis and autoimmunity. Rheumatol Int. 2006;26:209-214.
- Yung RL, Quddus J, Chrisp CE, et al. Mechanism of drug-induced lupus. I. cloned Th2 cells modified with DNA methylation inhibitors in vitro cause autoimmunity in vivo. J Immunol. 1995;154:3025-3035.
- Yung R, Powers D, Johnson K, et al. Mechanisms of drug-induced lupus. II. T cells overexpressing lymphocyte function-associated antigen 1 become autoreactive and cause a lupuslike disease in syngeneic mice. J Clin Invest. 1996;97:2866-2871.
- Sontheimer RD, Stastny P, Gilliam JN. Human histocompatibility antigen associations in subacute cutaneous lupus erythematosus. J Clin Invest. 1981;67:312-316.
- Sontheimer RD, Maddison PJ, Reichlin M, et al. Serologic and HLA associations in subacute cutaneous lupus erythematosus, a clinical subset of lupus erythematosus. Ann Intern Med. 1982;97:664-671.
- Lee LA, Roberts CM, Frank MB, et al. The autoantibody response to Ro/SSA in cutaneous lupus erythematosus. Arch Dermatol. 1994;130:1262-1268.
- Deutscher SL, Harley JB, Keene JD. Molecular analysis of the 60-kDa human Ro ribonucleoprotein. Proc Natl Acad Sci. 1988;85:9479-9483.
- DalleVedove C, Simon JC, Girolomoni G. Drug-induced lupus erythematosus with emphasis on skin manifestations and the role of anti-TNFα agents [article in German]. J Dtsch Dermatol Ges. 2012;10:889-897.
- Okon LG, Werth VP. Cutaneous lupus erythematosus: diagnosis and treatment. Best Pract Res Clin Rheumatol. 2013;27:391-404.
- Schulte-Pelkum J, Fritzler M, Mahler M. Latest update on the Ro/SS-A autoantibody system. Autoimmun Rev. 2009;8:632-637.
- De Bandt M, Sibilia J, Le Loët X, et al. Systemic lupus erythematosus induced by anti-tumour necrosis factor alpha therapy: a French national survey. Arthritis Res Ther. 2005;7:R545-R551.
- Lupu A, Tieranu C, Constantinescu CL, et al. TNFα inhibitor induced lupus-like syndrome (TAILS) in a patient with IBD. Current Health Sci J. 2014;40:285-288.
- Marzano AV, Vezzoli P, Crosti C. Drug-induced lupus: an update on its dermatological aspects. Lupus. 2009;18:935-940.
- Wiznia LE, Subtil A, Choi JN. Subacute cutaneous lupus erythematosus induced by chemotherapy: gemcitabine as a causative agent. JAMA Dermatol. 2013;149:1071-1075.
- Williams VL, Cohen PR. TNF alpha antagonist-induced lupus-like syndrome: report and review of the literature with implications for treatment with alternative TNF alpha antagonists. Int J Dermatol. 2011;50:619-625.
- Pasut G. Pegylation of biological molecules and potential benefits: pharmacological properties of certolizumab pegol. Bio Drugs. 2014;28(suppl 1):15-23.
- Mudduluru BM, Shah S, Shamah S. et al. TNF-alpha antagonist induced lupus on three different agents. Postgrad Med. 2017;129:304-306.
- De Bandt M. Anti-TNF-alpha-induced lupus. Arthritis Res Ther. 2019;21:235.
- Costa MF, Said NR, Zimmermann B. Drug-induced lupus due to anti-tumor necrosis factor alfa agents. Semin Arthritis Rheum. 2008;37:381-387.
- Caramaschi P, Biasi D, Colombatti M. Anti-TNF alpha therapy in rheumatoid arthritis and autoimmunity. Rheumatol Int. 2006;26:209-214.
- Yung RL, Quddus J, Chrisp CE, et al. Mechanism of drug-induced lupus. I. cloned Th2 cells modified with DNA methylation inhibitors in vitro cause autoimmunity in vivo. J Immunol. 1995;154:3025-3035.
- Yung R, Powers D, Johnson K, et al. Mechanisms of drug-induced lupus. II. T cells overexpressing lymphocyte function-associated antigen 1 become autoreactive and cause a lupuslike disease in syngeneic mice. J Clin Invest. 1996;97:2866-2871.
- Sontheimer RD, Stastny P, Gilliam JN. Human histocompatibility antigen associations in subacute cutaneous lupus erythematosus. J Clin Invest. 1981;67:312-316.
- Sontheimer RD, Maddison PJ, Reichlin M, et al. Serologic and HLA associations in subacute cutaneous lupus erythematosus, a clinical subset of lupus erythematosus. Ann Intern Med. 1982;97:664-671.
- Lee LA, Roberts CM, Frank MB, et al. The autoantibody response to Ro/SSA in cutaneous lupus erythematosus. Arch Dermatol. 1994;130:1262-1268.
- Deutscher SL, Harley JB, Keene JD. Molecular analysis of the 60-kDa human Ro ribonucleoprotein. Proc Natl Acad Sci. 1988;85:9479-9483.
- DalleVedove C, Simon JC, Girolomoni G. Drug-induced lupus erythematosus with emphasis on skin manifestations and the role of anti-TNFα agents [article in German]. J Dtsch Dermatol Ges. 2012;10:889-897.
- Okon LG, Werth VP. Cutaneous lupus erythematosus: diagnosis and treatment. Best Pract Res Clin Rheumatol. 2013;27:391-404.
- Schulte-Pelkum J, Fritzler M, Mahler M. Latest update on the Ro/SS-A autoantibody system. Autoimmun Rev. 2009;8:632-637.
- De Bandt M, Sibilia J, Le Loët X, et al. Systemic lupus erythematosus induced by anti-tumour necrosis factor alpha therapy: a French national survey. Arthritis Res Ther. 2005;7:R545-R551.
- Lupu A, Tieranu C, Constantinescu CL, et al. TNFα inhibitor induced lupus-like syndrome (TAILS) in a patient with IBD. Current Health Sci J. 2014;40:285-288.
Practice Points
- Tumor necrosis factor α (TNF-α) inhibitor–induced lupuslike syndrome (TAILS) is a form of drug-induced lupus specific to patients on anti–TNF-α therapy.
- The underlying mechanism of disease development is unique compared to other types of drug-induced lupus.
- TAILS most commonly is associated with the use of infliximab and etanercept but also has been reported with adalimumab, golimumab, and certolizumab pegol.

Rowell Syndrome: Targeting a True Definition
Case Report
A 37-year-old woman was admitted to the intensive care unit secondary to the acute development of an erythematous rash with tissue sloughing that involved acral sites and mucosal surfaces. Her medical history was notable for anti-Ro/Sjögren syndrome antigen A (SS-A)–positive lupus erythematosus (LE) with a morphologic semblance to subacute cutaneous LE (SCLE). Prior treatment had included oral corticosteroids. In addition, she reported a concurrent history of acral and mucosal lesions that appeared to flare with her lupus. The nature of these lesions was not clear to the patient or her physicians. Before this particular episode, her primary care physician had attempted to wean her off of the corticosteroids. As she dropped below 20 mg of prednisone daily, new lesions developed. The patient stated that her social situation was poor and that these lesions did seem to develop more frequently during times of physical and emotional stress. She recounted her first episode developing during her second pregnancy. Oral prednisone and over-the-counter calcium with vitamin D were her only reported medications. She denied the use of any other medications, including nonsteroidal anti-inflammatory drugs, acetaminophen, and recent antibiotic therapy.
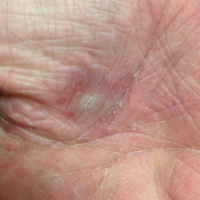
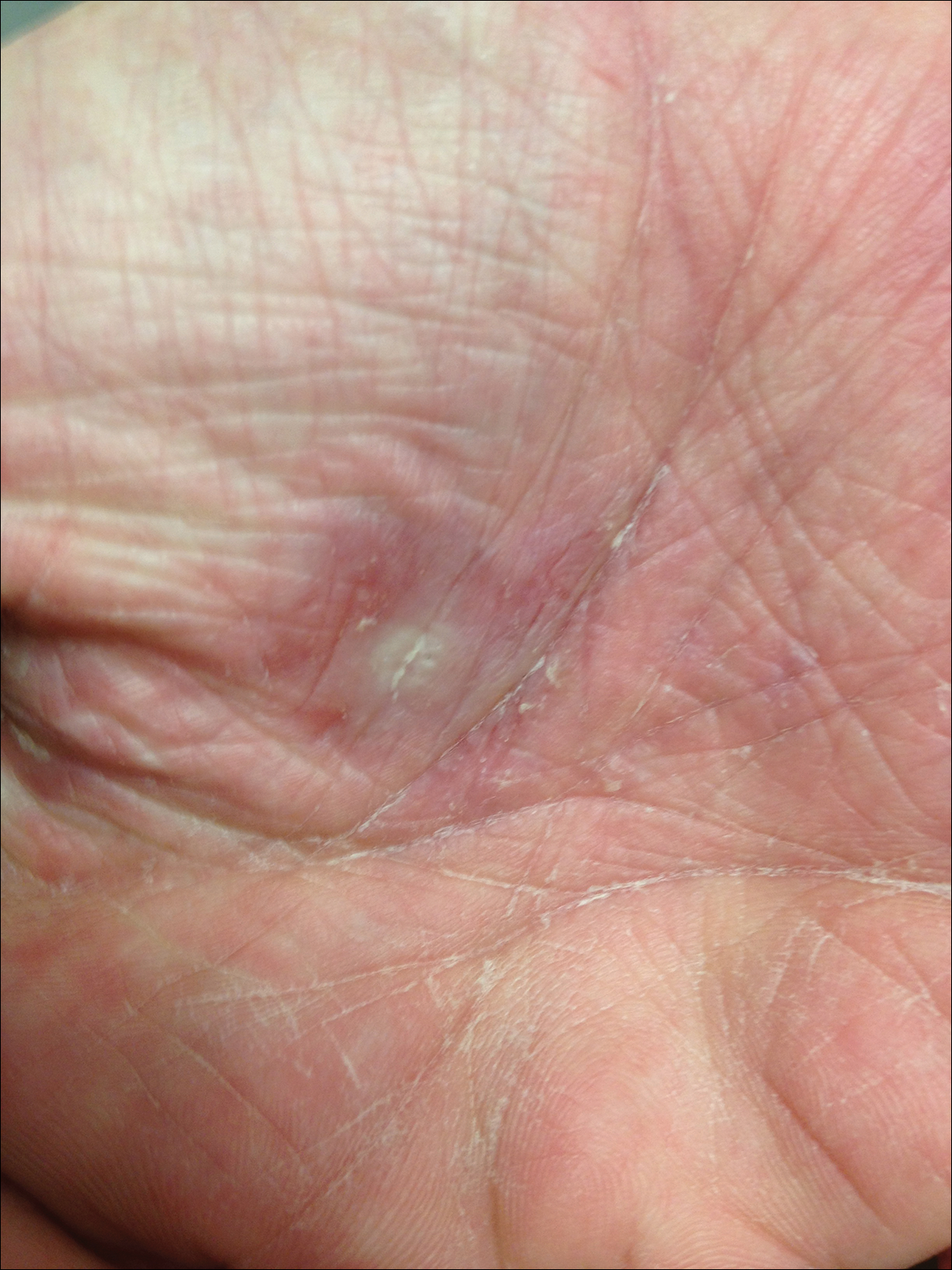
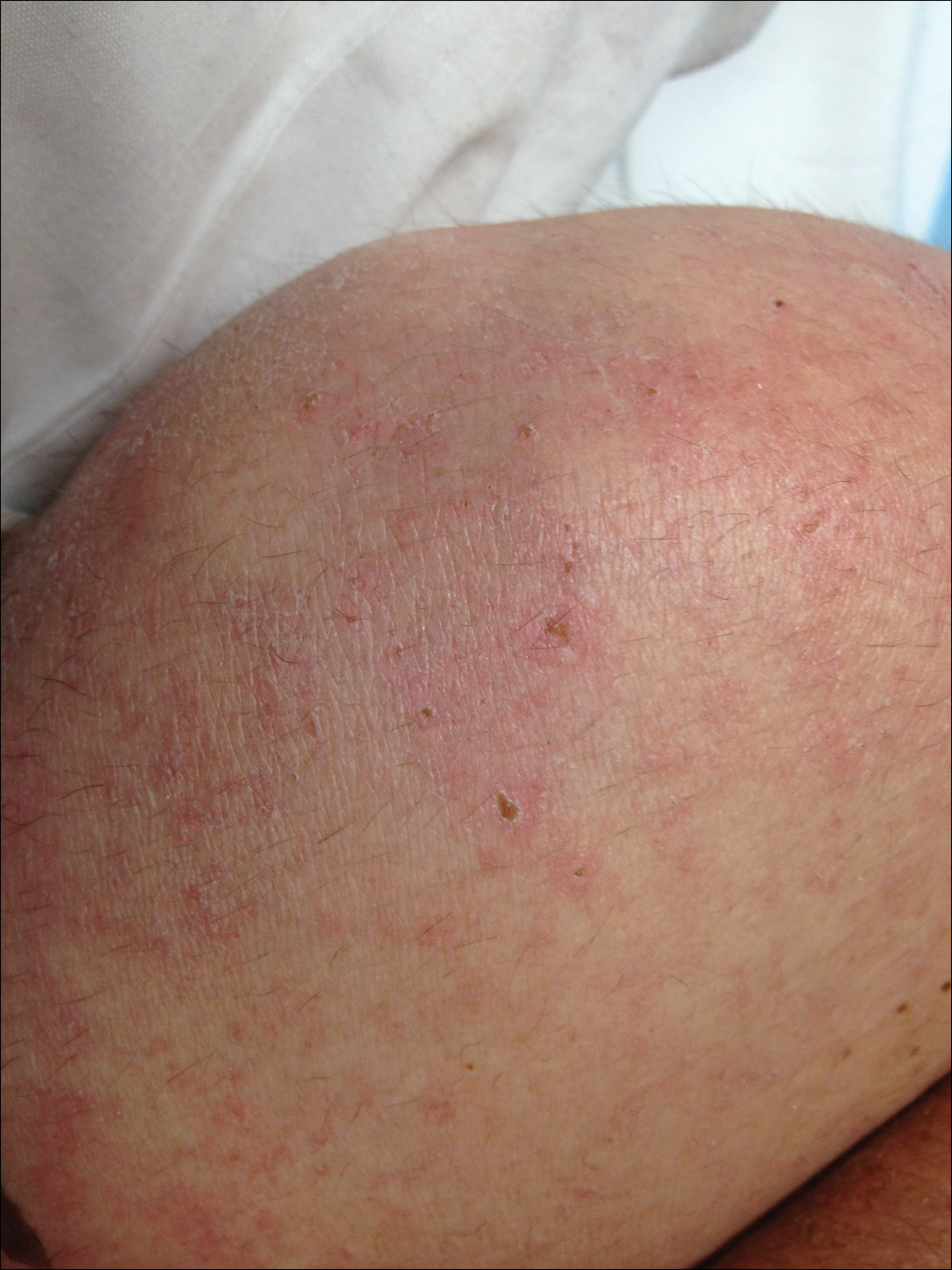
Dermatology was called in for consultation, and physical examination revealed areas of epidermal sloughing on the hands and feet. Complete clinical exposure of the underlying dermis was noted with remarkable tenderness. These lesions were noted to be in various stages of healing (Figure 1). Figure 2 displays a lesion in early development. The mucosal surfaces of the lips and eyes demonstrated hemorrhagic crusting, and some tissue sloughing was noted on the ears. A widespread erythematous exanthema with fine scaling was noted on the face, neck, chest, back, abdomen, arms, and legs (Figure 3).
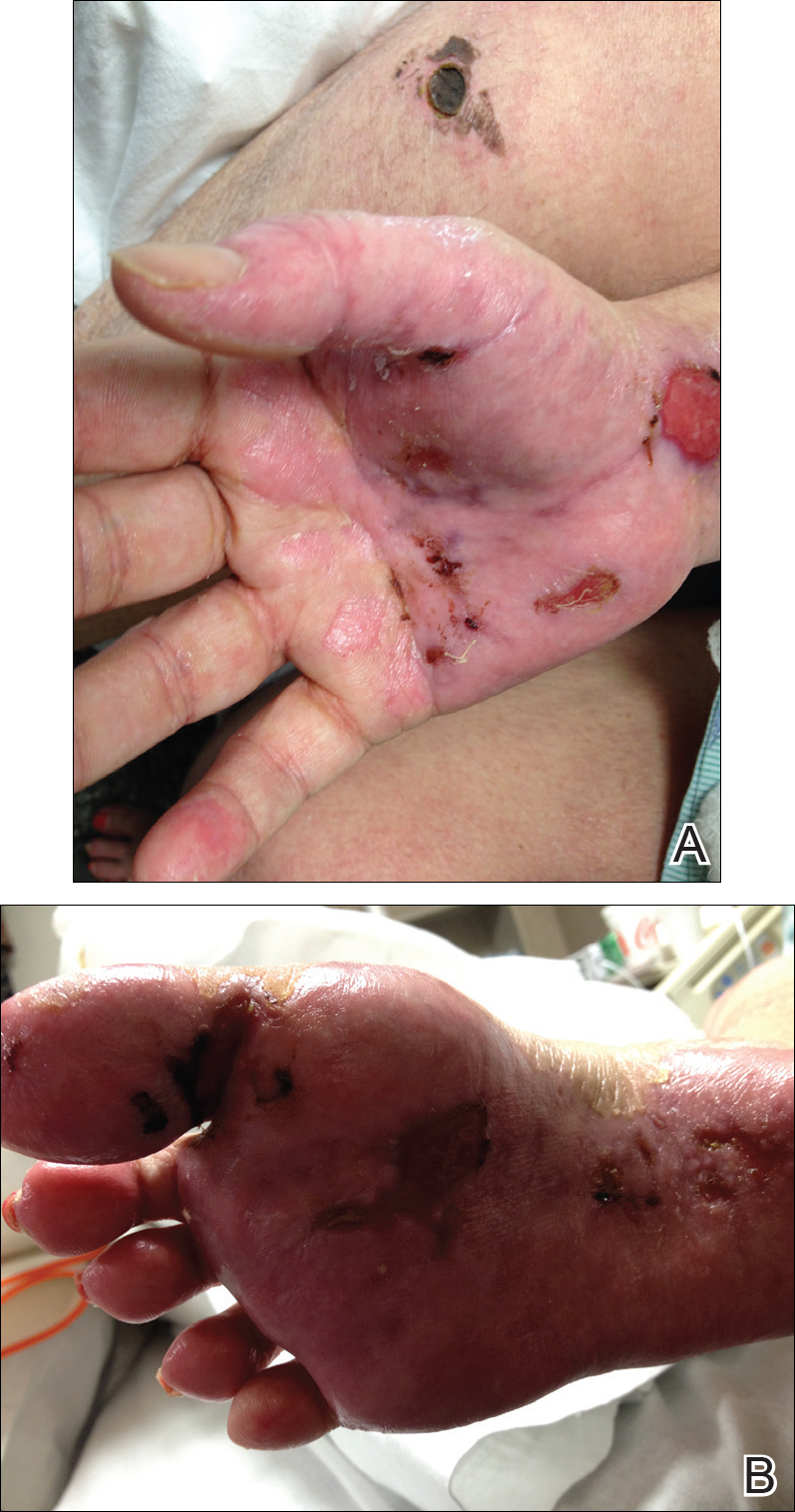
Laboratory evaluation revealed positive antinuclear antibodies (ANAs), anti-Ro/SS-A antibodies, anti-La/Sjögren syndrome antigen B (SS-B) antibodies, and anti–double-stranded DNA. The hemoglobin level was 9.4 g/dL (reference range, 12–15 g/dL) and hematocrit was 28.8% (reference range, 36%–47%). The mean corpuscular hemoglobin level was 32 pg/cell (reference range, 27–31 pg/cell), and the mean corpuscular hemoglobin concentration was 32.5 g/dL (reference range, 30–35 g/dL). Rheumatoid factor (RF) and herpes simplex virus types 1 and 2 IgM were all found to be negative.
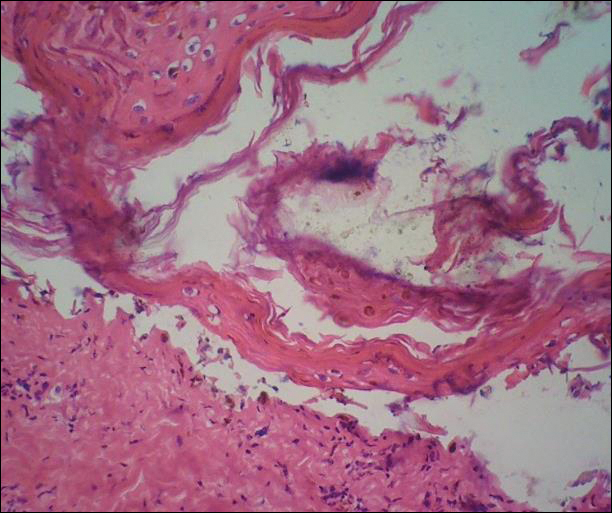
A deep shave biopsy obtained from the patient’s right knee revealed an atrophic interface dermatitis associated with a lymphocytic eccrine hidradenitis accompanied by abundant mesenchymal mucin deposition (Figure 4). Direct immunofluorescence (DIF) from the same area demonstrated IgG and IgM along the dermoepidermal junction with some granular deposition. Frozen sections performed on acral lesions demonstrated epidermal necrosis (Figure 5). Direct immunofluorescence of acral lesions was negative. In light of these findings, a diagnosis of Rowell syndrome (RS) was suspected to be the most likely explanation for the presentation.
Intravenous corticosteroids and antibiotics were administered, and over a 2-week hospitalization, the lesions on the feet and hands slowly reepithelialized. Physical therapy was required to aid in ambulation. The patient was discharged on a tapering course of oral prednisone and hydroxychloroquine. After 6 months of therapy with hydroxychloroquine 200 mg twice daily, the patient continued to experience recurrent bouts of acral lesions, and pulse doses of oral prednisone were required. The lesions currently are controlled with azathioprine 50 mg twice daily and prednisone 10 mg by mouth daily.

Comment
The 4 prototypical patients identified by Rowell et al1 in 1963 in the first account of the eponymous syndrome were all females with discoid lupus erythematosus (DLE) and perniosis. In addition, they all displayed positive RF and saline extract of human tissue antibodies (analogous to anti-Ro/SS-A and anti-La/SS-B).2 Since then, at least 132 patients with clinical symptoms suspicious of RS have been identified with variations on these original criteria.3 The reported permutations of the lupus component of the disease include cutaneous LE (CLE), bullous systemic LE, necrotic lesions associated with antiphospholipid syndrome, annular/polycyclic SCLE, systemic LE (SLE) without CLE, SLE with lupus nephritis, SLE with pericarditis, SLE with systemic vasculitis, Sjögren syndrome, rheumatoid arthritis, and necrotizing lymphadenitis.2 In addition, variations of the erythema multiforme (EM)–like lesions found in reported cases include changes to their gross appearance (flat vs raised), location (acral or mucosal involvement), and resemblance to other conditions (Stevens-Johnson syndrome or toxic epidermal necrolysis).2,3 From this information alone, it is clear that, as further cases have been chronicled, defining exact criteria for the disease has been challenging.
The essential question concerning the existence of RS hinges on the strength of its distinctiveness: Is it a unique disorder or merely another variant of lupus? Antiga et al2 concluded that it should be characterized as a variant of SCLE. Lee at al4 agreed, stating that “[i]n view of the lack of specific features that distinguish RS from LE, Kuhn et al5 suggested that [RS] is probably not a distinct entity and is now widely considered to be a variant of SCLE.” One of the primary contributors to this conclusion is that the laboratory findings of reported patients with SCLE have more closely mirrored the original cases from Rowell et al’s1 report than those of typical LE. Patients with SCLE have demonstrated positive ANA antibodies in 60% to 80% of cases, positive anti-Ro/SS-A antibodies in 40% to 100% of cases, positive anti-La/SS-B antibodies in 12% to 42% of cases, positive anti–double-stranded DNA in 1.2% to 10% of cases, and positive RF antibodies in 33% of cases.2 An argument could certainly be made to ascribe our patient’s condition to an SCLE variant, as 4 of 5 preceding laboratory findings were found to be positive; however, the majority of reported cases of SCLE have been linked to drugs (ie, hydrochlorothiazide, angiotensin-converting enzyme inhibitors, calcium channel blockers, terbinafine),2 which has not commonly been the attributable etiology of other cases of RS, including the 4 cases reported by Rowell et al.1
In a review of the literature on RS since 2010 in addition to their report of 132 new cases, Torchia et al3 outlined a set of diagnostic standards for the condition consisting of major and minor criteria. According to the authors, if all 4 major and 1 minor criteria are met, the patient meets the standards for true RS. The major criteria include the following: (1) presence of chronic CLE [DLE and/or chilblain]; (2) presence of EM-like lesions [typical or atypical targets]; (3) at least 1 positivity among speckled ANA, anti-Ro/SS-A, and anti-La/SS-B antibodies; and (4) negative DIF on lesional EM-like targetoid lesions. The minor criteria include the following: (1) absence of infectious or pharmacologic triggers; (2) absence of typical EM location (acral and mucosal); and (3) presence of at least 1 additional American College of Rheumatology criterion for diagnosis of SLE8 besides discoid rash and positive ANA antibodies and excluding photosensitivity, malar rash, and oral ulcers. Using these criteria, the patient in our case met the standards for diagnosis of RS.
One area of disagreement that has been encountered in the literature is the exact histologic determination of true RS, specifically related to the microscopic findings of the EM-like lesions. Two cases presented by Modi et al6 were interpreted under the stipulation that true RS must contain histologic LE and histologic EM. Because the EM-appearing lesions revealed LE histology, the cases were concluded to be variants of LE. These cases are similar to our case in that the EM-like lesions in our patient demonstrated LE pathology. Torchia et al,3 as demonstrated in the above criteria, seemed to be less concerned about the histology of the EM-like lesions, only requiring them to show negative DIF.
Conclusion
In the search for answers concerning RS, many unanswered questions remain: Where should the line be drawn in the inclusion of so many variations of both the LE and EM components of the condition? Also, should these elements even be approached as distinct components in the first place? Viewing the majority of RS cases as simply simultaneous LE and EM, Shteyngarts et al7 concluded that “the concomitant occurrence of EM with LE did not change the course, therapy, or prognosis of either disease. SLE and DLE can coexist with EM, but the coexistence does not impart any unusual characteristic to either illness. Rowell’s syndrome is not reproducible, and the immunologic disturbances in such patients are probably coincidental.”
If the condition is a genuine pathological individuality, should we not view the seemingly separate LE and EM as the product of a single underlying biochemical process? These questions and others in the search for a true definition of the disease should continue to be debated. It is clear that further investigation is warranted in the understanding of the underlying mechanism of the pathology.
- Rowell NR, Beck JS, Anderson JR. Lupus erythematosus and erythema multiforme-like lesions: a syndrome with characteristic immunological abnormalities. Arch Dermatol. 1963;88:176-180.
- Antiga E, Caproni M, Bonciani D, et al. The last word on the so-called ‘Rowell’s syndrome’? Lupus. 2012;21:577-585.
- Torchia D, Romanelli P, Kerdel FA. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol. 2012;67:417-421.
- Lee A, Batra P, Furer V, et al. Rowell syndrome (systemic lupus erythematosus + erythema multiforme). Dermatol Online J. 2009;15:1.
- Kuhn A, Sticherling M, Bonsmann G. Clinical manifestations of cutaneous lupus erythematosus. J Dtsch Dermatol Ges. 2007;5:1124-1140.
- Modi GM, Shen A, Mazloom A, et al. Lupus erythematosus masquerading as erythema multiforme: does Rowell syndrome really exist? Dermatol Online J. 2009;15:5.
- Shteyngarts AR, Warner MR, Camisa C. Lupus erythematosus associated with erythema multiforme: does Rowell’s syndrome exist? J Am Acad Dermatol. 1999;40(5 pt 1):773-777.
- Lupus diagnosis. Lupus Research Alliance website. http://lupusresearchinstitute.org/lupus-facts/lupus-diagnosis. Accessed July 11, 2017.
Case Report
A 37-year-old woman was admitted to the intensive care unit secondary to the acute development of an erythematous rash with tissue sloughing that involved acral sites and mucosal surfaces. Her medical history was notable for anti-Ro/Sjögren syndrome antigen A (SS-A)–positive lupus erythematosus (LE) with a morphologic semblance to subacute cutaneous LE (SCLE). Prior treatment had included oral corticosteroids. In addition, she reported a concurrent history of acral and mucosal lesions that appeared to flare with her lupus. The nature of these lesions was not clear to the patient or her physicians. Before this particular episode, her primary care physician had attempted to wean her off of the corticosteroids. As she dropped below 20 mg of prednisone daily, new lesions developed. The patient stated that her social situation was poor and that these lesions did seem to develop more frequently during times of physical and emotional stress. She recounted her first episode developing during her second pregnancy. Oral prednisone and over-the-counter calcium with vitamin D were her only reported medications. She denied the use of any other medications, including nonsteroidal anti-inflammatory drugs, acetaminophen, and recent antibiotic therapy.
Dermatology was called in for consultation, and physical examination revealed areas of epidermal sloughing on the hands and feet. Complete clinical exposure of the underlying dermis was noted with remarkable tenderness. These lesions were noted to be in various stages of healing (Figure 1). Figure 2 displays a lesion in early development. The mucosal surfaces of the lips and eyes demonstrated hemorrhagic crusting, and some tissue sloughing was noted on the ears. A widespread erythematous exanthema with fine scaling was noted on the face, neck, chest, back, abdomen, arms, and legs (Figure 3).
Laboratory evaluation revealed positive antinuclear antibodies (ANAs), anti-Ro/SS-A antibodies, anti-La/Sjögren syndrome antigen B (SS-B) antibodies, and anti–double-stranded DNA. The hemoglobin level was 9.4 g/dL (reference range, 12–15 g/dL) and hematocrit was 28.8% (reference range, 36%–47%). The mean corpuscular hemoglobin level was 32 pg/cell (reference range, 27–31 pg/cell), and the mean corpuscular hemoglobin concentration was 32.5 g/dL (reference range, 30–35 g/dL). Rheumatoid factor (RF) and herpes simplex virus types 1 and 2 IgM were all found to be negative.
A deep shave biopsy obtained from the patient’s right knee revealed an atrophic interface dermatitis associated with a lymphocytic eccrine hidradenitis accompanied by abundant mesenchymal mucin deposition (Figure 4). Direct immunofluorescence (DIF) from the same area demonstrated IgG and IgM along the dermoepidermal junction with some granular deposition. Frozen sections performed on acral lesions demonstrated epidermal necrosis (Figure 5). Direct immunofluorescence of acral lesions was negative. In light of these findings, a diagnosis of Rowell syndrome (RS) was suspected to be the most likely explanation for the presentation.
Intravenous corticosteroids and antibiotics were administered, and over a 2-week hospitalization, the lesions on the feet and hands slowly reepithelialized. Physical therapy was required to aid in ambulation. The patient was discharged on a tapering course of oral prednisone and hydroxychloroquine. After 6 months of therapy with hydroxychloroquine 200 mg twice daily, the patient continued to experience recurrent bouts of acral lesions, and pulse doses of oral prednisone were required. The lesions currently are controlled with azathioprine 50 mg twice daily and prednisone 10 mg by mouth daily.
Comment
The 4 prototypical patients identified by Rowell et al1 in 1963 in the first account of the eponymous syndrome were all females with discoid lupus erythematosus (DLE) and perniosis. In addition, they all displayed positive RF and saline extract of human tissue antibodies (analogous to anti-Ro/SS-A and anti-La/SS-B).2 Since then, at least 132 patients with clinical symptoms suspicious of RS have been identified with variations on these original criteria.3 The reported permutations of the lupus component of the disease include cutaneous LE (CLE), bullous systemic LE, necrotic lesions associated with antiphospholipid syndrome, annular/polycyclic SCLE, systemic LE (SLE) without CLE, SLE with lupus nephritis, SLE with pericarditis, SLE with systemic vasculitis, Sjögren syndrome, rheumatoid arthritis, and necrotizing lymphadenitis.2 In addition, variations of the erythema multiforme (EM)–like lesions found in reported cases include changes to their gross appearance (flat vs raised), location (acral or mucosal involvement), and resemblance to other conditions (Stevens-Johnson syndrome or toxic epidermal necrolysis).2,3 From this information alone, it is clear that, as further cases have been chronicled, defining exact criteria for the disease has been challenging.
The essential question concerning the existence of RS hinges on the strength of its distinctiveness: Is it a unique disorder or merely another variant of lupus? Antiga et al2 concluded that it should be characterized as a variant of SCLE. Lee at al4 agreed, stating that “[i]n view of the lack of specific features that distinguish RS from LE, Kuhn et al5 suggested that [RS] is probably not a distinct entity and is now widely considered to be a variant of SCLE.” One of the primary contributors to this conclusion is that the laboratory findings of reported patients with SCLE have more closely mirrored the original cases from Rowell et al’s1 report than those of typical LE. Patients with SCLE have demonstrated positive ANA antibodies in 60% to 80% of cases, positive anti-Ro/SS-A antibodies in 40% to 100% of cases, positive anti-La/SS-B antibodies in 12% to 42% of cases, positive anti–double-stranded DNA in 1.2% to 10% of cases, and positive RF antibodies in 33% of cases.2 An argument could certainly be made to ascribe our patient’s condition to an SCLE variant, as 4 of 5 preceding laboratory findings were found to be positive; however, the majority of reported cases of SCLE have been linked to drugs (ie, hydrochlorothiazide, angiotensin-converting enzyme inhibitors, calcium channel blockers, terbinafine),2 which has not commonly been the attributable etiology of other cases of RS, including the 4 cases reported by Rowell et al.1
In a review of the literature on RS since 2010 in addition to their report of 132 new cases, Torchia et al3 outlined a set of diagnostic standards for the condition consisting of major and minor criteria. According to the authors, if all 4 major and 1 minor criteria are met, the patient meets the standards for true RS. The major criteria include the following: (1) presence of chronic CLE [DLE and/or chilblain]; (2) presence of EM-like lesions [typical or atypical targets]; (3) at least 1 positivity among speckled ANA, anti-Ro/SS-A, and anti-La/SS-B antibodies; and (4) negative DIF on lesional EM-like targetoid lesions. The minor criteria include the following: (1) absence of infectious or pharmacologic triggers; (2) absence of typical EM location (acral and mucosal); and (3) presence of at least 1 additional American College of Rheumatology criterion for diagnosis of SLE8 besides discoid rash and positive ANA antibodies and excluding photosensitivity, malar rash, and oral ulcers. Using these criteria, the patient in our case met the standards for diagnosis of RS.
One area of disagreement that has been encountered in the literature is the exact histologic determination of true RS, specifically related to the microscopic findings of the EM-like lesions. Two cases presented by Modi et al6 were interpreted under the stipulation that true RS must contain histologic LE and histologic EM. Because the EM-appearing lesions revealed LE histology, the cases were concluded to be variants of LE. These cases are similar to our case in that the EM-like lesions in our patient demonstrated LE pathology. Torchia et al,3 as demonstrated in the above criteria, seemed to be less concerned about the histology of the EM-like lesions, only requiring them to show negative DIF.
Conclusion
In the search for answers concerning RS, many unanswered questions remain: Where should the line be drawn in the inclusion of so many variations of both the LE and EM components of the condition? Also, should these elements even be approached as distinct components in the first place? Viewing the majority of RS cases as simply simultaneous LE and EM, Shteyngarts et al7 concluded that “the concomitant occurrence of EM with LE did not change the course, therapy, or prognosis of either disease. SLE and DLE can coexist with EM, but the coexistence does not impart any unusual characteristic to either illness. Rowell’s syndrome is not reproducible, and the immunologic disturbances in such patients are probably coincidental.”
If the condition is a genuine pathological individuality, should we not view the seemingly separate LE and EM as the product of a single underlying biochemical process? These questions and others in the search for a true definition of the disease should continue to be debated. It is clear that further investigation is warranted in the understanding of the underlying mechanism of the pathology.
Case Report
A 37-year-old woman was admitted to the intensive care unit secondary to the acute development of an erythematous rash with tissue sloughing that involved acral sites and mucosal surfaces. Her medical history was notable for anti-Ro/Sjögren syndrome antigen A (SS-A)–positive lupus erythematosus (LE) with a morphologic semblance to subacute cutaneous LE (SCLE). Prior treatment had included oral corticosteroids. In addition, she reported a concurrent history of acral and mucosal lesions that appeared to flare with her lupus. The nature of these lesions was not clear to the patient or her physicians. Before this particular episode, her primary care physician had attempted to wean her off of the corticosteroids. As she dropped below 20 mg of prednisone daily, new lesions developed. The patient stated that her social situation was poor and that these lesions did seem to develop more frequently during times of physical and emotional stress. She recounted her first episode developing during her second pregnancy. Oral prednisone and over-the-counter calcium with vitamin D were her only reported medications. She denied the use of any other medications, including nonsteroidal anti-inflammatory drugs, acetaminophen, and recent antibiotic therapy.
Dermatology was called in for consultation, and physical examination revealed areas of epidermal sloughing on the hands and feet. Complete clinical exposure of the underlying dermis was noted with remarkable tenderness. These lesions were noted to be in various stages of healing (Figure 1). Figure 2 displays a lesion in early development. The mucosal surfaces of the lips and eyes demonstrated hemorrhagic crusting, and some tissue sloughing was noted on the ears. A widespread erythematous exanthema with fine scaling was noted on the face, neck, chest, back, abdomen, arms, and legs (Figure 3).
Laboratory evaluation revealed positive antinuclear antibodies (ANAs), anti-Ro/SS-A antibodies, anti-La/Sjögren syndrome antigen B (SS-B) antibodies, and anti–double-stranded DNA. The hemoglobin level was 9.4 g/dL (reference range, 12–15 g/dL) and hematocrit was 28.8% (reference range, 36%–47%). The mean corpuscular hemoglobin level was 32 pg/cell (reference range, 27–31 pg/cell), and the mean corpuscular hemoglobin concentration was 32.5 g/dL (reference range, 30–35 g/dL). Rheumatoid factor (RF) and herpes simplex virus types 1 and 2 IgM were all found to be negative.
A deep shave biopsy obtained from the patient’s right knee revealed an atrophic interface dermatitis associated with a lymphocytic eccrine hidradenitis accompanied by abundant mesenchymal mucin deposition (Figure 4). Direct immunofluorescence (DIF) from the same area demonstrated IgG and IgM along the dermoepidermal junction with some granular deposition. Frozen sections performed on acral lesions demonstrated epidermal necrosis (Figure 5). Direct immunofluorescence of acral lesions was negative. In light of these findings, a diagnosis of Rowell syndrome (RS) was suspected to be the most likely explanation for the presentation.
Intravenous corticosteroids and antibiotics were administered, and over a 2-week hospitalization, the lesions on the feet and hands slowly reepithelialized. Physical therapy was required to aid in ambulation. The patient was discharged on a tapering course of oral prednisone and hydroxychloroquine. After 6 months of therapy with hydroxychloroquine 200 mg twice daily, the patient continued to experience recurrent bouts of acral lesions, and pulse doses of oral prednisone were required. The lesions currently are controlled with azathioprine 50 mg twice daily and prednisone 10 mg by mouth daily.
Comment
The 4 prototypical patients identified by Rowell et al1 in 1963 in the first account of the eponymous syndrome were all females with discoid lupus erythematosus (DLE) and perniosis. In addition, they all displayed positive RF and saline extract of human tissue antibodies (analogous to anti-Ro/SS-A and anti-La/SS-B).2 Since then, at least 132 patients with clinical symptoms suspicious of RS have been identified with variations on these original criteria.3 The reported permutations of the lupus component of the disease include cutaneous LE (CLE), bullous systemic LE, necrotic lesions associated with antiphospholipid syndrome, annular/polycyclic SCLE, systemic LE (SLE) without CLE, SLE with lupus nephritis, SLE with pericarditis, SLE with systemic vasculitis, Sjögren syndrome, rheumatoid arthritis, and necrotizing lymphadenitis.2 In addition, variations of the erythema multiforme (EM)–like lesions found in reported cases include changes to their gross appearance (flat vs raised), location (acral or mucosal involvement), and resemblance to other conditions (Stevens-Johnson syndrome or toxic epidermal necrolysis).2,3 From this information alone, it is clear that, as further cases have been chronicled, defining exact criteria for the disease has been challenging.
The essential question concerning the existence of RS hinges on the strength of its distinctiveness: Is it a unique disorder or merely another variant of lupus? Antiga et al2 concluded that it should be characterized as a variant of SCLE. Lee at al4 agreed, stating that “[i]n view of the lack of specific features that distinguish RS from LE, Kuhn et al5 suggested that [RS] is probably not a distinct entity and is now widely considered to be a variant of SCLE.” One of the primary contributors to this conclusion is that the laboratory findings of reported patients with SCLE have more closely mirrored the original cases from Rowell et al’s1 report than those of typical LE. Patients with SCLE have demonstrated positive ANA antibodies in 60% to 80% of cases, positive anti-Ro/SS-A antibodies in 40% to 100% of cases, positive anti-La/SS-B antibodies in 12% to 42% of cases, positive anti–double-stranded DNA in 1.2% to 10% of cases, and positive RF antibodies in 33% of cases.2 An argument could certainly be made to ascribe our patient’s condition to an SCLE variant, as 4 of 5 preceding laboratory findings were found to be positive; however, the majority of reported cases of SCLE have been linked to drugs (ie, hydrochlorothiazide, angiotensin-converting enzyme inhibitors, calcium channel blockers, terbinafine),2 which has not commonly been the attributable etiology of other cases of RS, including the 4 cases reported by Rowell et al.1
In a review of the literature on RS since 2010 in addition to their report of 132 new cases, Torchia et al3 outlined a set of diagnostic standards for the condition consisting of major and minor criteria. According to the authors, if all 4 major and 1 minor criteria are met, the patient meets the standards for true RS. The major criteria include the following: (1) presence of chronic CLE [DLE and/or chilblain]; (2) presence of EM-like lesions [typical or atypical targets]; (3) at least 1 positivity among speckled ANA, anti-Ro/SS-A, and anti-La/SS-B antibodies; and (4) negative DIF on lesional EM-like targetoid lesions. The minor criteria include the following: (1) absence of infectious or pharmacologic triggers; (2) absence of typical EM location (acral and mucosal); and (3) presence of at least 1 additional American College of Rheumatology criterion for diagnosis of SLE8 besides discoid rash and positive ANA antibodies and excluding photosensitivity, malar rash, and oral ulcers. Using these criteria, the patient in our case met the standards for diagnosis of RS.
One area of disagreement that has been encountered in the literature is the exact histologic determination of true RS, specifically related to the microscopic findings of the EM-like lesions. Two cases presented by Modi et al6 were interpreted under the stipulation that true RS must contain histologic LE and histologic EM. Because the EM-appearing lesions revealed LE histology, the cases were concluded to be variants of LE. These cases are similar to our case in that the EM-like lesions in our patient demonstrated LE pathology. Torchia et al,3 as demonstrated in the above criteria, seemed to be less concerned about the histology of the EM-like lesions, only requiring them to show negative DIF.
Conclusion
In the search for answers concerning RS, many unanswered questions remain: Where should the line be drawn in the inclusion of so many variations of both the LE and EM components of the condition? Also, should these elements even be approached as distinct components in the first place? Viewing the majority of RS cases as simply simultaneous LE and EM, Shteyngarts et al7 concluded that “the concomitant occurrence of EM with LE did not change the course, therapy, or prognosis of either disease. SLE and DLE can coexist with EM, but the coexistence does not impart any unusual characteristic to either illness. Rowell’s syndrome is not reproducible, and the immunologic disturbances in such patients are probably coincidental.”
If the condition is a genuine pathological individuality, should we not view the seemingly separate LE and EM as the product of a single underlying biochemical process? These questions and others in the search for a true definition of the disease should continue to be debated. It is clear that further investigation is warranted in the understanding of the underlying mechanism of the pathology.
- Rowell NR, Beck JS, Anderson JR. Lupus erythematosus and erythema multiforme-like lesions: a syndrome with characteristic immunological abnormalities. Arch Dermatol. 1963;88:176-180.
- Antiga E, Caproni M, Bonciani D, et al. The last word on the so-called ‘Rowell’s syndrome’? Lupus. 2012;21:577-585.
- Torchia D, Romanelli P, Kerdel FA. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol. 2012;67:417-421.
- Lee A, Batra P, Furer V, et al. Rowell syndrome (systemic lupus erythematosus + erythema multiforme). Dermatol Online J. 2009;15:1.
- Kuhn A, Sticherling M, Bonsmann G. Clinical manifestations of cutaneous lupus erythematosus. J Dtsch Dermatol Ges. 2007;5:1124-1140.
- Modi GM, Shen A, Mazloom A, et al. Lupus erythematosus masquerading as erythema multiforme: does Rowell syndrome really exist? Dermatol Online J. 2009;15:5.
- Shteyngarts AR, Warner MR, Camisa C. Lupus erythematosus associated with erythema multiforme: does Rowell’s syndrome exist? J Am Acad Dermatol. 1999;40(5 pt 1):773-777.
- Lupus diagnosis. Lupus Research Alliance website. http://lupusresearchinstitute.org/lupus-facts/lupus-diagnosis. Accessed July 11, 2017.
- Rowell NR, Beck JS, Anderson JR. Lupus erythematosus and erythema multiforme-like lesions: a syndrome with characteristic immunological abnormalities. Arch Dermatol. 1963;88:176-180.
- Antiga E, Caproni M, Bonciani D, et al. The last word on the so-called ‘Rowell’s syndrome’? Lupus. 2012;21:577-585.
- Torchia D, Romanelli P, Kerdel FA. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol. 2012;67:417-421.
- Lee A, Batra P, Furer V, et al. Rowell syndrome (systemic lupus erythematosus + erythema multiforme). Dermatol Online J. 2009;15:1.
- Kuhn A, Sticherling M, Bonsmann G. Clinical manifestations of cutaneous lupus erythematosus. J Dtsch Dermatol Ges. 2007;5:1124-1140.
- Modi GM, Shen A, Mazloom A, et al. Lupus erythematosus masquerading as erythema multiforme: does Rowell syndrome really exist? Dermatol Online J. 2009;15:5.
- Shteyngarts AR, Warner MR, Camisa C. Lupus erythematosus associated with erythema multiforme: does Rowell’s syndrome exist? J Am Acad Dermatol. 1999;40(5 pt 1):773-777.
- Lupus diagnosis. Lupus Research Alliance website. http://lupusresearchinstitute.org/lupus-facts/lupus-diagnosis. Accessed July 11, 2017.
Practice Points
- Rowell syndrome (RS) is an often unrecognized unique presentation of lupus erythematosus.
- There have been a variety of historical criteria that have sought to characterize RS.