User login
Rowell Syndrome: Targeting a True Definition
Case Report
A 37-year-old woman was admitted to the intensive care unit secondary to the acute development of an erythematous rash with tissue sloughing that involved acral sites and mucosal surfaces. Her medical history was notable for anti-Ro/Sjögren syndrome antigen A (SS-A)–positive lupus erythematosus (LE) with a morphologic semblance to subacute cutaneous LE (SCLE). Prior treatment had included oral corticosteroids. In addition, she reported a concurrent history of acral and mucosal lesions that appeared to flare with her lupus. The nature of these lesions was not clear to the patient or her physicians. Before this particular episode, her primary care physician had attempted to wean her off of the corticosteroids. As she dropped below 20 mg of prednisone daily, new lesions developed. The patient stated that her social situation was poor and that these lesions did seem to develop more frequently during times of physical and emotional stress. She recounted her first episode developing during her second pregnancy. Oral prednisone and over-the-counter calcium with vitamin D were her only reported medications. She denied the use of any other medications, including nonsteroidal anti-inflammatory drugs, acetaminophen, and recent antibiotic therapy.
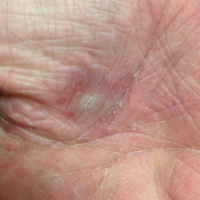
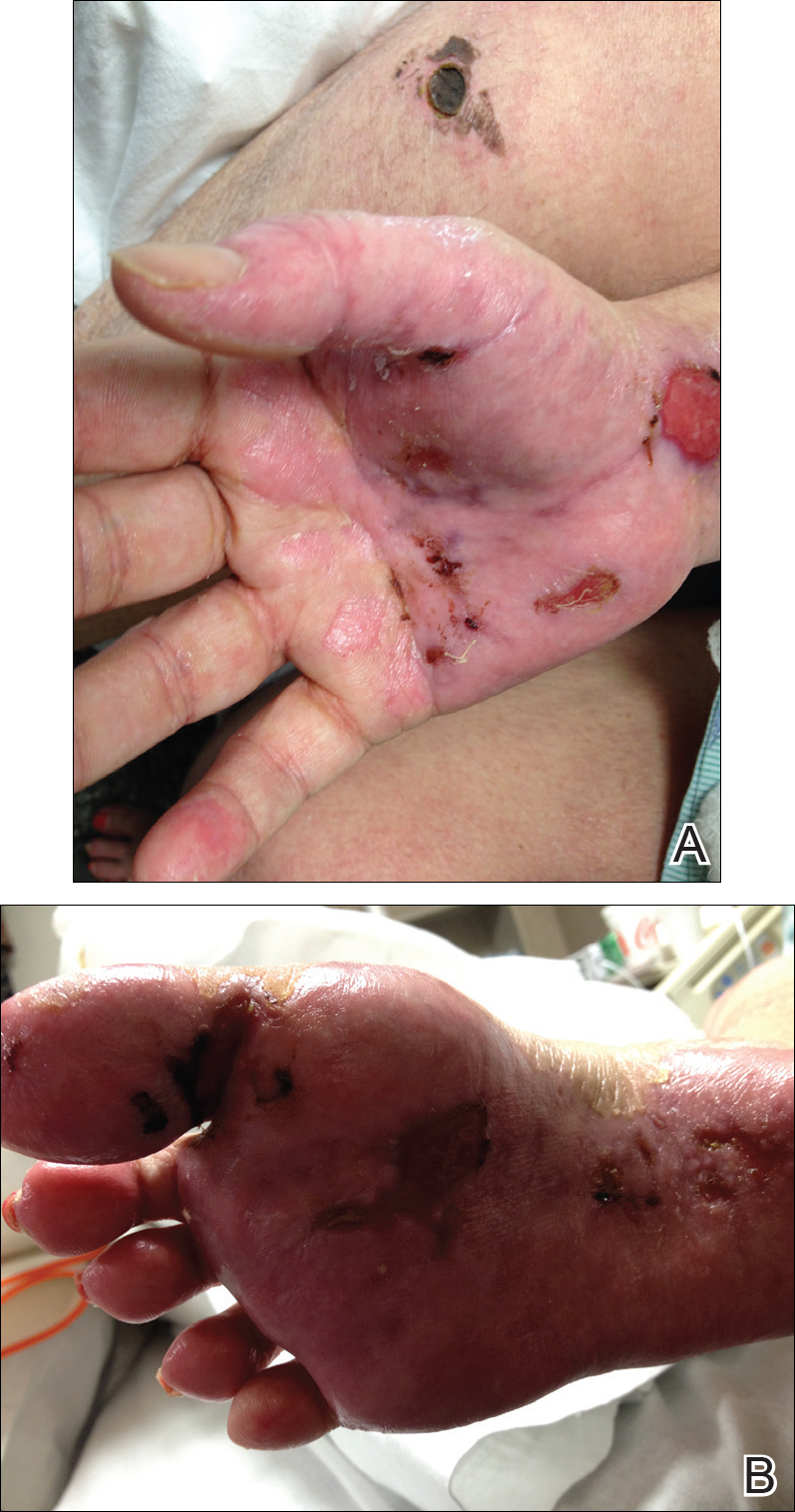
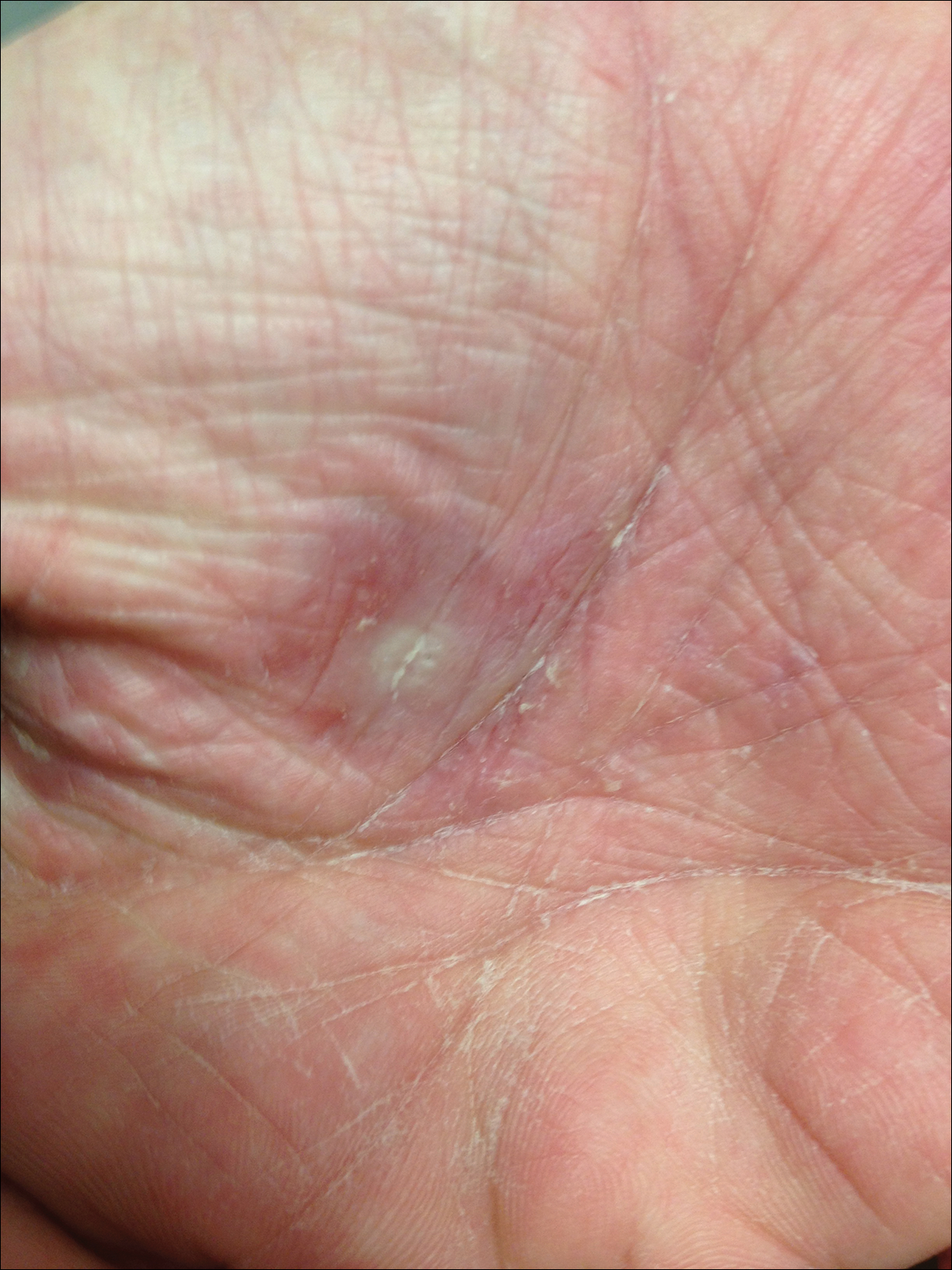
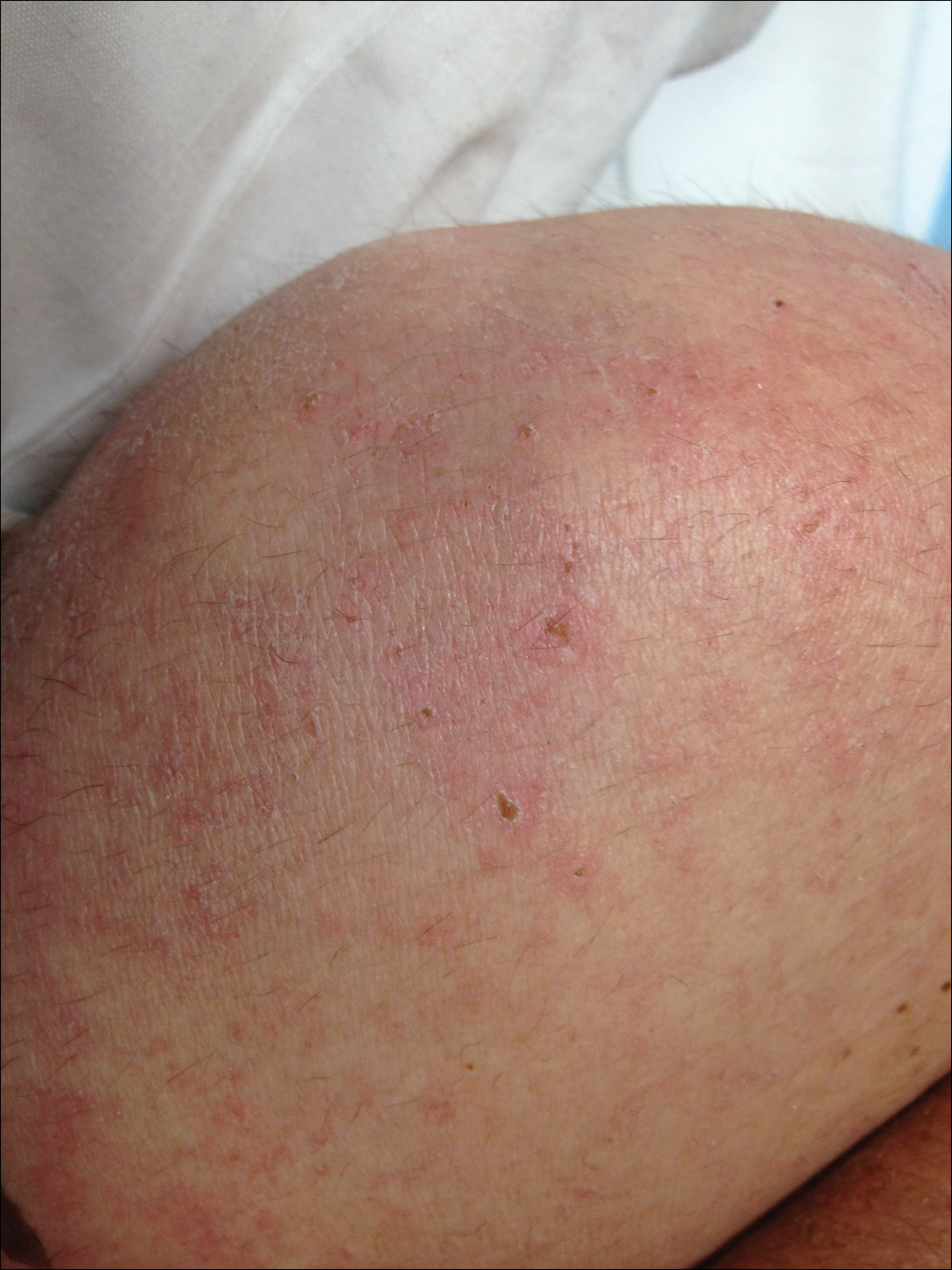
Dermatology was called in for consultation, and physical examination revealed areas of epidermal sloughing on the hands and feet. Complete clinical exposure of the underlying dermis was noted with remarkable tenderness. These lesions were noted to be in various stages of healing (Figure 1). Figure 2 displays a lesion in early development. The mucosal surfaces of the lips and eyes demonstrated hemorrhagic crusting, and some tissue sloughing was noted on the ears. A widespread erythematous exanthema with fine scaling was noted on the face, neck, chest, back, abdomen, arms, and legs (Figure 3).
Laboratory evaluation revealed positive antinuclear antibodies (ANAs), anti-Ro/SS-A antibodies, anti-La/Sjögren syndrome antigen B (SS-B) antibodies, and anti–double-stranded DNA. The hemoglobin level was 9.4 g/dL (reference range, 12–15 g/dL) and hematocrit was 28.8% (reference range, 36%–47%). The mean corpuscular hemoglobin level was 32 pg/cell (reference range, 27–31 pg/cell), and the mean corpuscular hemoglobin concentration was 32.5 g/dL (reference range, 30–35 g/dL). Rheumatoid factor (RF) and herpes simplex virus types 1 and 2 IgM were all found to be negative.
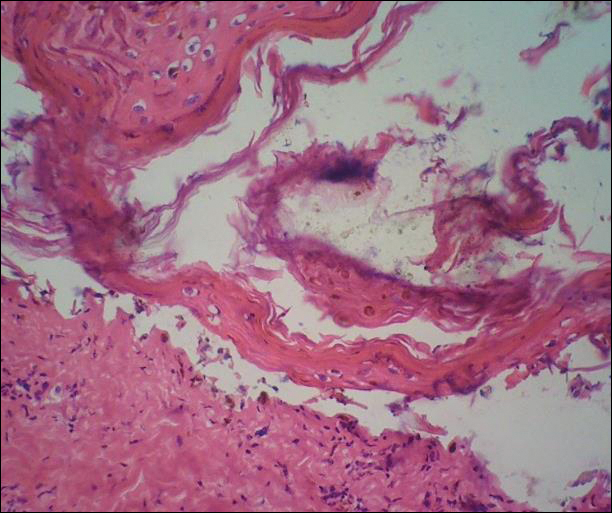
A deep shave biopsy obtained from the patient’s right knee revealed an atrophic interface dermatitis associated with a lymphocytic eccrine hidradenitis accompanied by abundant mesenchymal mucin deposition (Figure 4). Direct immunofluorescence (DIF) from the same area demonstrated IgG and IgM along the dermoepidermal junction with some granular deposition. Frozen sections performed on acral lesions demonstrated epidermal necrosis (Figure 5). Direct immunofluorescence of acral lesions was negative. In light of these findings, a diagnosis of Rowell syndrome (RS) was suspected to be the most likely explanation for the presentation.
Intravenous corticosteroids and antibiotics were administered, and over a 2-week hospitalization, the lesions on the feet and hands slowly reepithelialized. Physical therapy was required to aid in ambulation. The patient was discharged on a tapering course of oral prednisone and hydroxychloroquine. After 6 months of therapy with hydroxychloroquine 200 mg twice daily, the patient continued to experience recurrent bouts of acral lesions, and pulse doses of oral prednisone were required. The lesions currently are controlled with azathioprine 50 mg twice daily and prednisone 10 mg by mouth daily.

Comment
The 4 prototypical patients identified by Rowell et al1 in 1963 in the first account of the eponymous syndrome were all females with discoid lupus erythematosus (DLE) and perniosis. In addition, they all displayed positive RF and saline extract of human tissue antibodies (analogous to anti-Ro/SS-A and anti-La/SS-B).2 Since then, at least 132 patients with clinical symptoms suspicious of RS have been identified with variations on these original criteria.3 The reported permutations of the lupus component of the disease include cutaneous LE (CLE), bullous systemic LE, necrotic lesions associated with antiphospholipid syndrome, annular/polycyclic SCLE, systemic LE (SLE) without CLE, SLE with lupus nephritis, SLE with pericarditis, SLE with systemic vasculitis, Sjögren syndrome, rheumatoid arthritis, and necrotizing lymphadenitis.2 In addition, variations of the erythema multiforme (EM)–like lesions found in reported cases include changes to their gross appearance (flat vs raised), location (acral or mucosal involvement), and resemblance to other conditions (Stevens-Johnson syndrome or toxic epidermal necrolysis).2,3 From this information alone, it is clear that, as further cases have been chronicled, defining exact criteria for the disease has been challenging.
The essential question concerning the existence of RS hinges on the strength of its distinctiveness: Is it a unique disorder or merely another variant of lupus? Antiga et al2 concluded that it should be characterized as a variant of SCLE. Lee at al4 agreed, stating that “[i]n view of the lack of specific features that distinguish RS from LE, Kuhn et al5 suggested that [RS] is probably not a distinct entity and is now widely considered to be a variant of SCLE.” One of the primary contributors to this conclusion is that the laboratory findings of reported patients with SCLE have more closely mirrored the original cases from Rowell et al’s1 report than those of typical LE. Patients with SCLE have demonstrated positive ANA antibodies in 60% to 80% of cases, positive anti-Ro/SS-A antibodies in 40% to 100% of cases, positive anti-La/SS-B antibodies in 12% to 42% of cases, positive anti–double-stranded DNA in 1.2% to 10% of cases, and positive RF antibodies in 33% of cases.2 An argument could certainly be made to ascribe our patient’s condition to an SCLE variant, as 4 of 5 preceding laboratory findings were found to be positive; however, the majority of reported cases of SCLE have been linked to drugs (ie, hydrochlorothiazide, angiotensin-converting enzyme inhibitors, calcium channel blockers, terbinafine),2 which has not commonly been the attributable etiology of other cases of RS, including the 4 cases reported by Rowell et al.1
In a review of the literature on RS since 2010 in addition to their report of 132 new cases, Torchia et al3 outlined a set of diagnostic standards for the condition consisting of major and minor criteria. According to the authors, if all 4 major and 1 minor criteria are met, the patient meets the standards for true RS. The major criteria include the following: (1) presence of chronic CLE [DLE and/or chilblain]; (2) presence of EM-like lesions [typical or atypical targets]; (3) at least 1 positivity among speckled ANA, anti-Ro/SS-A, and anti-La/SS-B antibodies; and (4) negative DIF on lesional EM-like targetoid lesions. The minor criteria include the following: (1) absence of infectious or pharmacologic triggers; (2) absence of typical EM location (acral and mucosal); and (3) presence of at least 1 additional American College of Rheumatology criterion for diagnosis of SLE8 besides discoid rash and positive ANA antibodies and excluding photosensitivity, malar rash, and oral ulcers. Using these criteria, the patient in our case met the standards for diagnosis of RS.
One area of disagreement that has been encountered in the literature is the exact histologic determination of true RS, specifically related to the microscopic findings of the EM-like lesions. Two cases presented by Modi et al6 were interpreted under the stipulation that true RS must contain histologic LE and histologic EM. Because the EM-appearing lesions revealed LE histology, the cases were concluded to be variants of LE. These cases are similar to our case in that the EM-like lesions in our patient demonstrated LE pathology. Torchia et al,3 as demonstrated in the above criteria, seemed to be less concerned about the histology of the EM-like lesions, only requiring them to show negative DIF.
Conclusion
In the search for answers concerning RS, many unanswered questions remain: Where should the line be drawn in the inclusion of so many variations of both the LE and EM components of the condition? Also, should these elements even be approached as distinct components in the first place? Viewing the majority of RS cases as simply simultaneous LE and EM, Shteyngarts et al7 concluded that “the concomitant occurrence of EM with LE did not change the course, therapy, or prognosis of either disease. SLE and DLE can coexist with EM, but the coexistence does not impart any unusual characteristic to either illness. Rowell’s syndrome is not reproducible, and the immunologic disturbances in such patients are probably coincidental.”
If the condition is a genuine pathological individuality, should we not view the seemingly separate LE and EM as the product of a single underlying biochemical process? These questions and others in the search for a true definition of the disease should continue to be debated. It is clear that further investigation is warranted in the understanding of the underlying mechanism of the pathology.
- Rowell NR, Beck JS, Anderson JR. Lupus erythematosus and erythema multiforme-like lesions: a syndrome with characteristic immunological abnormalities. Arch Dermatol. 1963;88:176-180.
- Antiga E, Caproni M, Bonciani D, et al. The last word on the so-called ‘Rowell’s syndrome’? Lupus. 2012;21:577-585.
- Torchia D, Romanelli P, Kerdel FA. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol. 2012;67:417-421.
- Lee A, Batra P, Furer V, et al. Rowell syndrome (systemic lupus erythematosus + erythema multiforme). Dermatol Online J. 2009;15:1.
- Kuhn A, Sticherling M, Bonsmann G. Clinical manifestations of cutaneous lupus erythematosus. J Dtsch Dermatol Ges. 2007;5:1124-1140.
- Modi GM, Shen A, Mazloom A, et al. Lupus erythematosus masquerading as erythema multiforme: does Rowell syndrome really exist? Dermatol Online J. 2009;15:5.
- Shteyngarts AR, Warner MR, Camisa C. Lupus erythematosus associated with erythema multiforme: does Rowell’s syndrome exist? J Am Acad Dermatol. 1999;40(5 pt 1):773-777.
- Lupus diagnosis. Lupus Research Alliance website. http://lupusresearchinstitute.org/lupus-facts/lupus-diagnosis. Accessed July 11, 2017.
Case Report
A 37-year-old woman was admitted to the intensive care unit secondary to the acute development of an erythematous rash with tissue sloughing that involved acral sites and mucosal surfaces. Her medical history was notable for anti-Ro/Sjögren syndrome antigen A (SS-A)–positive lupus erythematosus (LE) with a morphologic semblance to subacute cutaneous LE (SCLE). Prior treatment had included oral corticosteroids. In addition, she reported a concurrent history of acral and mucosal lesions that appeared to flare with her lupus. The nature of these lesions was not clear to the patient or her physicians. Before this particular episode, her primary care physician had attempted to wean her off of the corticosteroids. As she dropped below 20 mg of prednisone daily, new lesions developed. The patient stated that her social situation was poor and that these lesions did seem to develop more frequently during times of physical and emotional stress. She recounted her first episode developing during her second pregnancy. Oral prednisone and over-the-counter calcium with vitamin D were her only reported medications. She denied the use of any other medications, including nonsteroidal anti-inflammatory drugs, acetaminophen, and recent antibiotic therapy.
Dermatology was called in for consultation, and physical examination revealed areas of epidermal sloughing on the hands and feet. Complete clinical exposure of the underlying dermis was noted with remarkable tenderness. These lesions were noted to be in various stages of healing (Figure 1). Figure 2 displays a lesion in early development. The mucosal surfaces of the lips and eyes demonstrated hemorrhagic crusting, and some tissue sloughing was noted on the ears. A widespread erythematous exanthema with fine scaling was noted on the face, neck, chest, back, abdomen, arms, and legs (Figure 3).
Laboratory evaluation revealed positive antinuclear antibodies (ANAs), anti-Ro/SS-A antibodies, anti-La/Sjögren syndrome antigen B (SS-B) antibodies, and anti–double-stranded DNA. The hemoglobin level was 9.4 g/dL (reference range, 12–15 g/dL) and hematocrit was 28.8% (reference range, 36%–47%). The mean corpuscular hemoglobin level was 32 pg/cell (reference range, 27–31 pg/cell), and the mean corpuscular hemoglobin concentration was 32.5 g/dL (reference range, 30–35 g/dL). Rheumatoid factor (RF) and herpes simplex virus types 1 and 2 IgM were all found to be negative.
A deep shave biopsy obtained from the patient’s right knee revealed an atrophic interface dermatitis associated with a lymphocytic eccrine hidradenitis accompanied by abundant mesenchymal mucin deposition (Figure 4). Direct immunofluorescence (DIF) from the same area demonstrated IgG and IgM along the dermoepidermal junction with some granular deposition. Frozen sections performed on acral lesions demonstrated epidermal necrosis (Figure 5). Direct immunofluorescence of acral lesions was negative. In light of these findings, a diagnosis of Rowell syndrome (RS) was suspected to be the most likely explanation for the presentation.
Intravenous corticosteroids and antibiotics were administered, and over a 2-week hospitalization, the lesions on the feet and hands slowly reepithelialized. Physical therapy was required to aid in ambulation. The patient was discharged on a tapering course of oral prednisone and hydroxychloroquine. After 6 months of therapy with hydroxychloroquine 200 mg twice daily, the patient continued to experience recurrent bouts of acral lesions, and pulse doses of oral prednisone were required. The lesions currently are controlled with azathioprine 50 mg twice daily and prednisone 10 mg by mouth daily.
Comment
The 4 prototypical patients identified by Rowell et al1 in 1963 in the first account of the eponymous syndrome were all females with discoid lupus erythematosus (DLE) and perniosis. In addition, they all displayed positive RF and saline extract of human tissue antibodies (analogous to anti-Ro/SS-A and anti-La/SS-B).2 Since then, at least 132 patients with clinical symptoms suspicious of RS have been identified with variations on these original criteria.3 The reported permutations of the lupus component of the disease include cutaneous LE (CLE), bullous systemic LE, necrotic lesions associated with antiphospholipid syndrome, annular/polycyclic SCLE, systemic LE (SLE) without CLE, SLE with lupus nephritis, SLE with pericarditis, SLE with systemic vasculitis, Sjögren syndrome, rheumatoid arthritis, and necrotizing lymphadenitis.2 In addition, variations of the erythema multiforme (EM)–like lesions found in reported cases include changes to their gross appearance (flat vs raised), location (acral or mucosal involvement), and resemblance to other conditions (Stevens-Johnson syndrome or toxic epidermal necrolysis).2,3 From this information alone, it is clear that, as further cases have been chronicled, defining exact criteria for the disease has been challenging.
The essential question concerning the existence of RS hinges on the strength of its distinctiveness: Is it a unique disorder or merely another variant of lupus? Antiga et al2 concluded that it should be characterized as a variant of SCLE. Lee at al4 agreed, stating that “[i]n view of the lack of specific features that distinguish RS from LE, Kuhn et al5 suggested that [RS] is probably not a distinct entity and is now widely considered to be a variant of SCLE.” One of the primary contributors to this conclusion is that the laboratory findings of reported patients with SCLE have more closely mirrored the original cases from Rowell et al’s1 report than those of typical LE. Patients with SCLE have demonstrated positive ANA antibodies in 60% to 80% of cases, positive anti-Ro/SS-A antibodies in 40% to 100% of cases, positive anti-La/SS-B antibodies in 12% to 42% of cases, positive anti–double-stranded DNA in 1.2% to 10% of cases, and positive RF antibodies in 33% of cases.2 An argument could certainly be made to ascribe our patient’s condition to an SCLE variant, as 4 of 5 preceding laboratory findings were found to be positive; however, the majority of reported cases of SCLE have been linked to drugs (ie, hydrochlorothiazide, angiotensin-converting enzyme inhibitors, calcium channel blockers, terbinafine),2 which has not commonly been the attributable etiology of other cases of RS, including the 4 cases reported by Rowell et al.1
In a review of the literature on RS since 2010 in addition to their report of 132 new cases, Torchia et al3 outlined a set of diagnostic standards for the condition consisting of major and minor criteria. According to the authors, if all 4 major and 1 minor criteria are met, the patient meets the standards for true RS. The major criteria include the following: (1) presence of chronic CLE [DLE and/or chilblain]; (2) presence of EM-like lesions [typical or atypical targets]; (3) at least 1 positivity among speckled ANA, anti-Ro/SS-A, and anti-La/SS-B antibodies; and (4) negative DIF on lesional EM-like targetoid lesions. The minor criteria include the following: (1) absence of infectious or pharmacologic triggers; (2) absence of typical EM location (acral and mucosal); and (3) presence of at least 1 additional American College of Rheumatology criterion for diagnosis of SLE8 besides discoid rash and positive ANA antibodies and excluding photosensitivity, malar rash, and oral ulcers. Using these criteria, the patient in our case met the standards for diagnosis of RS.
One area of disagreement that has been encountered in the literature is the exact histologic determination of true RS, specifically related to the microscopic findings of the EM-like lesions. Two cases presented by Modi et al6 were interpreted under the stipulation that true RS must contain histologic LE and histologic EM. Because the EM-appearing lesions revealed LE histology, the cases were concluded to be variants of LE. These cases are similar to our case in that the EM-like lesions in our patient demonstrated LE pathology. Torchia et al,3 as demonstrated in the above criteria, seemed to be less concerned about the histology of the EM-like lesions, only requiring them to show negative DIF.
Conclusion
In the search for answers concerning RS, many unanswered questions remain: Where should the line be drawn in the inclusion of so many variations of both the LE and EM components of the condition? Also, should these elements even be approached as distinct components in the first place? Viewing the majority of RS cases as simply simultaneous LE and EM, Shteyngarts et al7 concluded that “the concomitant occurrence of EM with LE did not change the course, therapy, or prognosis of either disease. SLE and DLE can coexist with EM, but the coexistence does not impart any unusual characteristic to either illness. Rowell’s syndrome is not reproducible, and the immunologic disturbances in such patients are probably coincidental.”
If the condition is a genuine pathological individuality, should we not view the seemingly separate LE and EM as the product of a single underlying biochemical process? These questions and others in the search for a true definition of the disease should continue to be debated. It is clear that further investigation is warranted in the understanding of the underlying mechanism of the pathology.
Case Report
A 37-year-old woman was admitted to the intensive care unit secondary to the acute development of an erythematous rash with tissue sloughing that involved acral sites and mucosal surfaces. Her medical history was notable for anti-Ro/Sjögren syndrome antigen A (SS-A)–positive lupus erythematosus (LE) with a morphologic semblance to subacute cutaneous LE (SCLE). Prior treatment had included oral corticosteroids. In addition, she reported a concurrent history of acral and mucosal lesions that appeared to flare with her lupus. The nature of these lesions was not clear to the patient or her physicians. Before this particular episode, her primary care physician had attempted to wean her off of the corticosteroids. As she dropped below 20 mg of prednisone daily, new lesions developed. The patient stated that her social situation was poor and that these lesions did seem to develop more frequently during times of physical and emotional stress. She recounted her first episode developing during her second pregnancy. Oral prednisone and over-the-counter calcium with vitamin D were her only reported medications. She denied the use of any other medications, including nonsteroidal anti-inflammatory drugs, acetaminophen, and recent antibiotic therapy.
Dermatology was called in for consultation, and physical examination revealed areas of epidermal sloughing on the hands and feet. Complete clinical exposure of the underlying dermis was noted with remarkable tenderness. These lesions were noted to be in various stages of healing (Figure 1). Figure 2 displays a lesion in early development. The mucosal surfaces of the lips and eyes demonstrated hemorrhagic crusting, and some tissue sloughing was noted on the ears. A widespread erythematous exanthema with fine scaling was noted on the face, neck, chest, back, abdomen, arms, and legs (Figure 3).
Laboratory evaluation revealed positive antinuclear antibodies (ANAs), anti-Ro/SS-A antibodies, anti-La/Sjögren syndrome antigen B (SS-B) antibodies, and anti–double-stranded DNA. The hemoglobin level was 9.4 g/dL (reference range, 12–15 g/dL) and hematocrit was 28.8% (reference range, 36%–47%). The mean corpuscular hemoglobin level was 32 pg/cell (reference range, 27–31 pg/cell), and the mean corpuscular hemoglobin concentration was 32.5 g/dL (reference range, 30–35 g/dL). Rheumatoid factor (RF) and herpes simplex virus types 1 and 2 IgM were all found to be negative.
A deep shave biopsy obtained from the patient’s right knee revealed an atrophic interface dermatitis associated with a lymphocytic eccrine hidradenitis accompanied by abundant mesenchymal mucin deposition (Figure 4). Direct immunofluorescence (DIF) from the same area demonstrated IgG and IgM along the dermoepidermal junction with some granular deposition. Frozen sections performed on acral lesions demonstrated epidermal necrosis (Figure 5). Direct immunofluorescence of acral lesions was negative. In light of these findings, a diagnosis of Rowell syndrome (RS) was suspected to be the most likely explanation for the presentation.
Intravenous corticosteroids and antibiotics were administered, and over a 2-week hospitalization, the lesions on the feet and hands slowly reepithelialized. Physical therapy was required to aid in ambulation. The patient was discharged on a tapering course of oral prednisone and hydroxychloroquine. After 6 months of therapy with hydroxychloroquine 200 mg twice daily, the patient continued to experience recurrent bouts of acral lesions, and pulse doses of oral prednisone were required. The lesions currently are controlled with azathioprine 50 mg twice daily and prednisone 10 mg by mouth daily.
Comment
The 4 prototypical patients identified by Rowell et al1 in 1963 in the first account of the eponymous syndrome were all females with discoid lupus erythematosus (DLE) and perniosis. In addition, they all displayed positive RF and saline extract of human tissue antibodies (analogous to anti-Ro/SS-A and anti-La/SS-B).2 Since then, at least 132 patients with clinical symptoms suspicious of RS have been identified with variations on these original criteria.3 The reported permutations of the lupus component of the disease include cutaneous LE (CLE), bullous systemic LE, necrotic lesions associated with antiphospholipid syndrome, annular/polycyclic SCLE, systemic LE (SLE) without CLE, SLE with lupus nephritis, SLE with pericarditis, SLE with systemic vasculitis, Sjögren syndrome, rheumatoid arthritis, and necrotizing lymphadenitis.2 In addition, variations of the erythema multiforme (EM)–like lesions found in reported cases include changes to their gross appearance (flat vs raised), location (acral or mucosal involvement), and resemblance to other conditions (Stevens-Johnson syndrome or toxic epidermal necrolysis).2,3 From this information alone, it is clear that, as further cases have been chronicled, defining exact criteria for the disease has been challenging.
The essential question concerning the existence of RS hinges on the strength of its distinctiveness: Is it a unique disorder or merely another variant of lupus? Antiga et al2 concluded that it should be characterized as a variant of SCLE. Lee at al4 agreed, stating that “[i]n view of the lack of specific features that distinguish RS from LE, Kuhn et al5 suggested that [RS] is probably not a distinct entity and is now widely considered to be a variant of SCLE.” One of the primary contributors to this conclusion is that the laboratory findings of reported patients with SCLE have more closely mirrored the original cases from Rowell et al’s1 report than those of typical LE. Patients with SCLE have demonstrated positive ANA antibodies in 60% to 80% of cases, positive anti-Ro/SS-A antibodies in 40% to 100% of cases, positive anti-La/SS-B antibodies in 12% to 42% of cases, positive anti–double-stranded DNA in 1.2% to 10% of cases, and positive RF antibodies in 33% of cases.2 An argument could certainly be made to ascribe our patient’s condition to an SCLE variant, as 4 of 5 preceding laboratory findings were found to be positive; however, the majority of reported cases of SCLE have been linked to drugs (ie, hydrochlorothiazide, angiotensin-converting enzyme inhibitors, calcium channel blockers, terbinafine),2 which has not commonly been the attributable etiology of other cases of RS, including the 4 cases reported by Rowell et al.1
In a review of the literature on RS since 2010 in addition to their report of 132 new cases, Torchia et al3 outlined a set of diagnostic standards for the condition consisting of major and minor criteria. According to the authors, if all 4 major and 1 minor criteria are met, the patient meets the standards for true RS. The major criteria include the following: (1) presence of chronic CLE [DLE and/or chilblain]; (2) presence of EM-like lesions [typical or atypical targets]; (3) at least 1 positivity among speckled ANA, anti-Ro/SS-A, and anti-La/SS-B antibodies; and (4) negative DIF on lesional EM-like targetoid lesions. The minor criteria include the following: (1) absence of infectious or pharmacologic triggers; (2) absence of typical EM location (acral and mucosal); and (3) presence of at least 1 additional American College of Rheumatology criterion for diagnosis of SLE8 besides discoid rash and positive ANA antibodies and excluding photosensitivity, malar rash, and oral ulcers. Using these criteria, the patient in our case met the standards for diagnosis of RS.
One area of disagreement that has been encountered in the literature is the exact histologic determination of true RS, specifically related to the microscopic findings of the EM-like lesions. Two cases presented by Modi et al6 were interpreted under the stipulation that true RS must contain histologic LE and histologic EM. Because the EM-appearing lesions revealed LE histology, the cases were concluded to be variants of LE. These cases are similar to our case in that the EM-like lesions in our patient demonstrated LE pathology. Torchia et al,3 as demonstrated in the above criteria, seemed to be less concerned about the histology of the EM-like lesions, only requiring them to show negative DIF.
Conclusion
In the search for answers concerning RS, many unanswered questions remain: Where should the line be drawn in the inclusion of so many variations of both the LE and EM components of the condition? Also, should these elements even be approached as distinct components in the first place? Viewing the majority of RS cases as simply simultaneous LE and EM, Shteyngarts et al7 concluded that “the concomitant occurrence of EM with LE did not change the course, therapy, or prognosis of either disease. SLE and DLE can coexist with EM, but the coexistence does not impart any unusual characteristic to either illness. Rowell’s syndrome is not reproducible, and the immunologic disturbances in such patients are probably coincidental.”
If the condition is a genuine pathological individuality, should we not view the seemingly separate LE and EM as the product of a single underlying biochemical process? These questions and others in the search for a true definition of the disease should continue to be debated. It is clear that further investigation is warranted in the understanding of the underlying mechanism of the pathology.
- Rowell NR, Beck JS, Anderson JR. Lupus erythematosus and erythema multiforme-like lesions: a syndrome with characteristic immunological abnormalities. Arch Dermatol. 1963;88:176-180.
- Antiga E, Caproni M, Bonciani D, et al. The last word on the so-called ‘Rowell’s syndrome’? Lupus. 2012;21:577-585.
- Torchia D, Romanelli P, Kerdel FA. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol. 2012;67:417-421.
- Lee A, Batra P, Furer V, et al. Rowell syndrome (systemic lupus erythematosus + erythema multiforme). Dermatol Online J. 2009;15:1.
- Kuhn A, Sticherling M, Bonsmann G. Clinical manifestations of cutaneous lupus erythematosus. J Dtsch Dermatol Ges. 2007;5:1124-1140.
- Modi GM, Shen A, Mazloom A, et al. Lupus erythematosus masquerading as erythema multiforme: does Rowell syndrome really exist? Dermatol Online J. 2009;15:5.
- Shteyngarts AR, Warner MR, Camisa C. Lupus erythematosus associated with erythema multiforme: does Rowell’s syndrome exist? J Am Acad Dermatol. 1999;40(5 pt 1):773-777.
- Lupus diagnosis. Lupus Research Alliance website. http://lupusresearchinstitute.org/lupus-facts/lupus-diagnosis. Accessed July 11, 2017.
- Rowell NR, Beck JS, Anderson JR. Lupus erythematosus and erythema multiforme-like lesions: a syndrome with characteristic immunological abnormalities. Arch Dermatol. 1963;88:176-180.
- Antiga E, Caproni M, Bonciani D, et al. The last word on the so-called ‘Rowell’s syndrome’? Lupus. 2012;21:577-585.
- Torchia D, Romanelli P, Kerdel FA. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol. 2012;67:417-421.
- Lee A, Batra P, Furer V, et al. Rowell syndrome (systemic lupus erythematosus + erythema multiforme). Dermatol Online J. 2009;15:1.
- Kuhn A, Sticherling M, Bonsmann G. Clinical manifestations of cutaneous lupus erythematosus. J Dtsch Dermatol Ges. 2007;5:1124-1140.
- Modi GM, Shen A, Mazloom A, et al. Lupus erythematosus masquerading as erythema multiforme: does Rowell syndrome really exist? Dermatol Online J. 2009;15:5.
- Shteyngarts AR, Warner MR, Camisa C. Lupus erythematosus associated with erythema multiforme: does Rowell’s syndrome exist? J Am Acad Dermatol. 1999;40(5 pt 1):773-777.
- Lupus diagnosis. Lupus Research Alliance website. http://lupusresearchinstitute.org/lupus-facts/lupus-diagnosis. Accessed July 11, 2017.
Practice Points
- Rowell syndrome (RS) is an often unrecognized unique presentation of lupus erythematosus.
- There have been a variety of historical criteria that have sought to characterize RS.