User login
Solitary Tender Nodule on the Back
The Diagnosis: Solitary Fibrous Tumor
Solitary fibrous tumors (SFTs), as first described by Klemperer and Rabin1 in 1931, are relatively uncommon mesenchymal neoplasms that occur primarily in the pleura. This lesion is now known to affect many other extrathoracic sites, such as the liver, kidney, adrenal glands, thyroid, central nervous system, and soft tissue, with rare examples originating from the skin.2 Okamura et al3 reported the first known case of cutaneous SFT in 1997, with most of the literature limited to case reports. Erdag et al2 described one of the largest case series of primary cutaneous SFTs. These lesions can occur across a wide age range but tend to primarily affect middle-aged adults. Solitary fibrous tumors have been known to have no sex predilection; however, Erdag et al2 found a male predominance with a male to female ratio of 4 to 1.
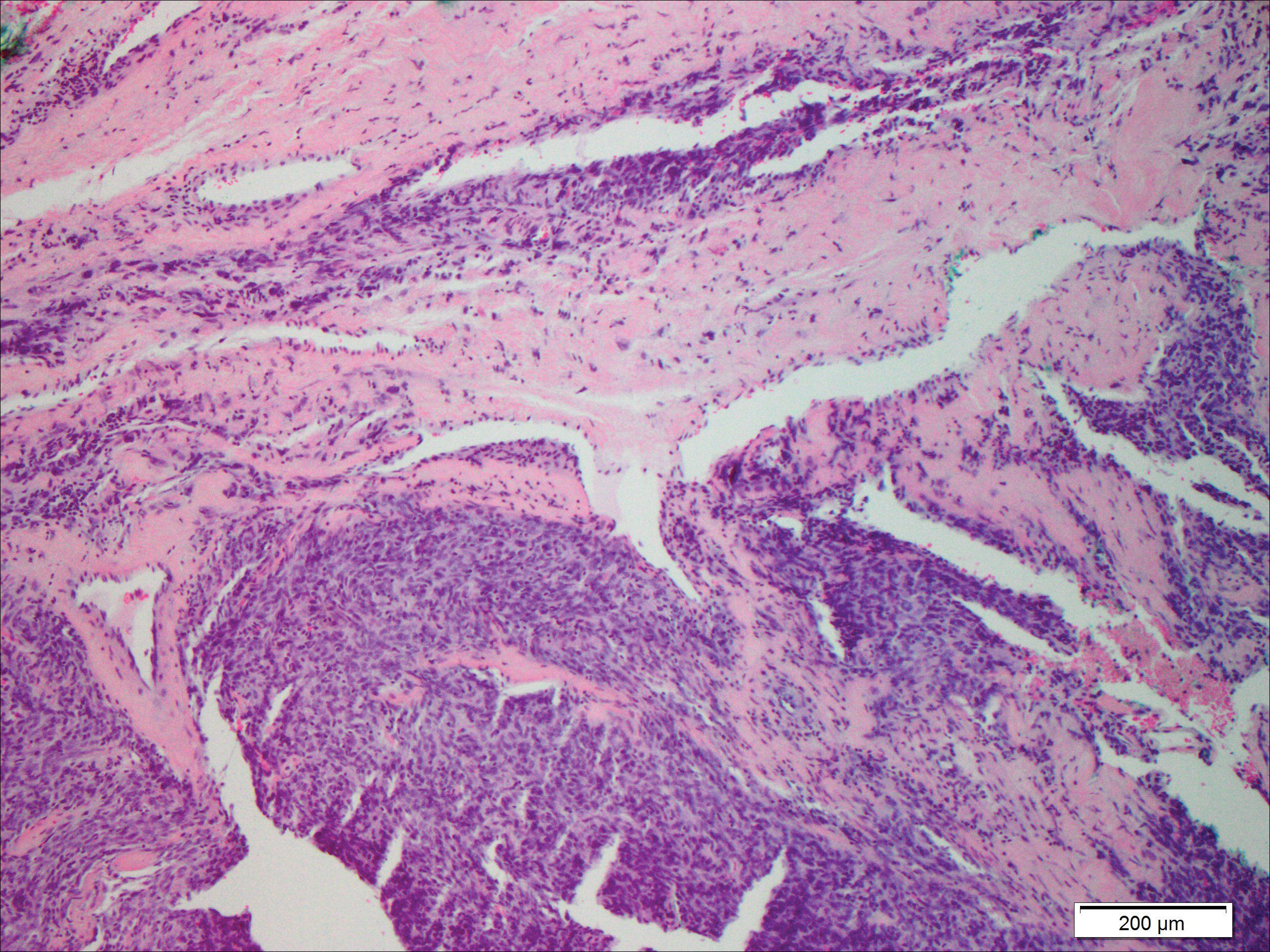
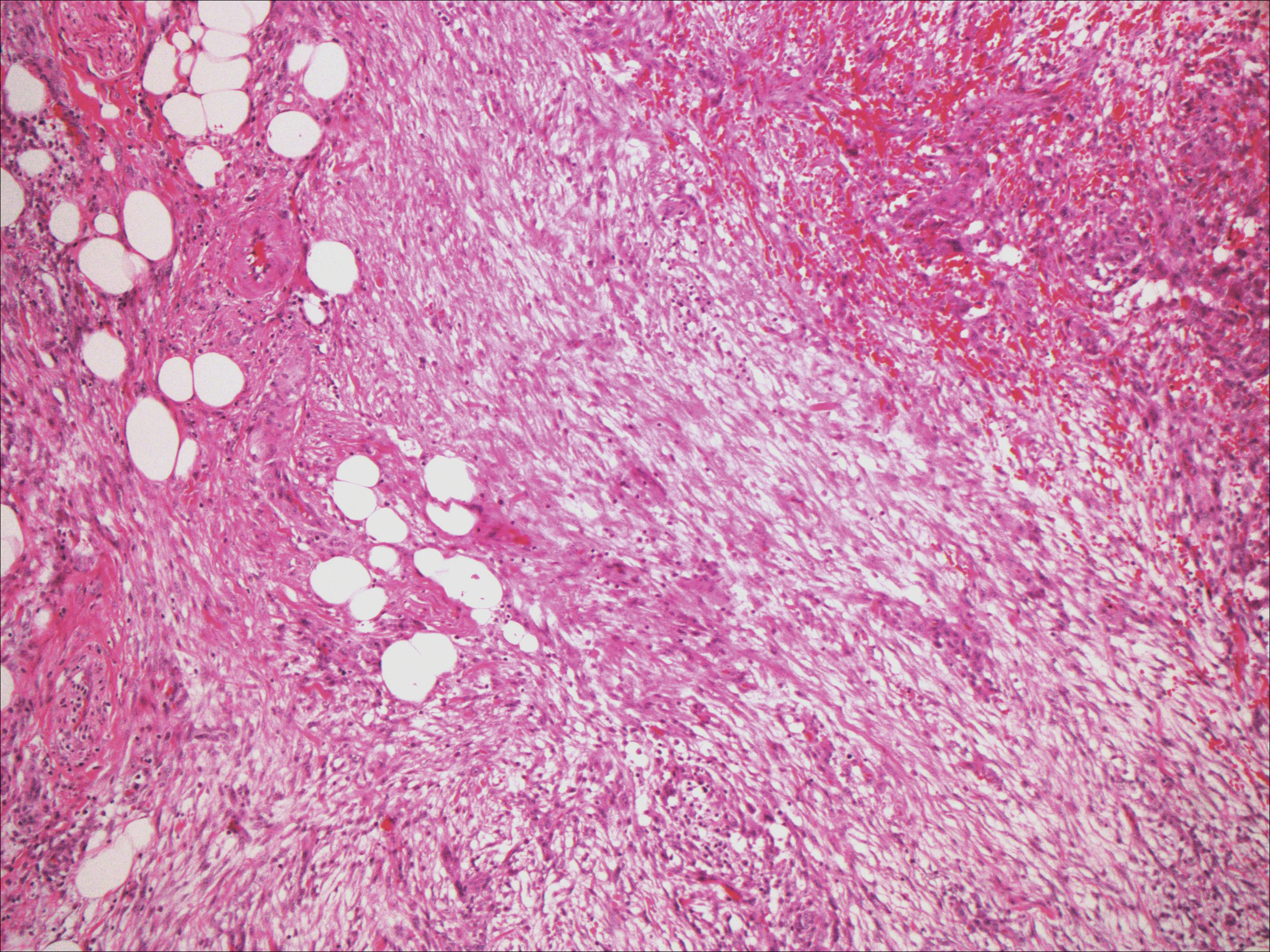
Histopathologically, a cutaneous SFT is known to appear as a well-circumscribed nodular spindle cell proliferation arranged in interlacing fascicles with an abundant hyalinized collagen stroma (quiz image). Alternating hypocellular and hypercellular areas can be seen. Supporting vasculature often is relatively prominent, represented by angulated and branching staghorn blood vessels (Figure 1).2 A common histopathologic finding of SFTs is a patternless pattern, which suggests that the tumor can have a variety of morphologic appearances (eg, storiform, fascicular, neural, herringbone growth patterns), making histologic diagnosis difficult (quiz image).4 Therefore, immunohistochemistry plays a large role in the diagnosis of this tumor. The most important positive markers include CD34, CD99, B-cell lymphoma 2 (BCL-2), and signal transducer and activator of transcription 6 (STAT6).5 Nuclear STAT6 staining is an immunomarker for NGFI-A binding protein 2 (NAB2)-STAT6 gene fusion, which is specific for SFT.5,6 Vivero et al7 also reported glutamate receptor, inotropic, AMPA 2 (GRIA2) as a useful immunostain in SFT, though it is also expressed in dermatofibrosarcoma protuberans (DFSP). In this case, the clinical and histopathologic findings best supported a diagnosis of SFT. Some consider hemangiopericytomas to be examples of SFTs; however, true hemangiopericytomas lack the thick hyalinized collagen and hypercellular areas seen in SFT.
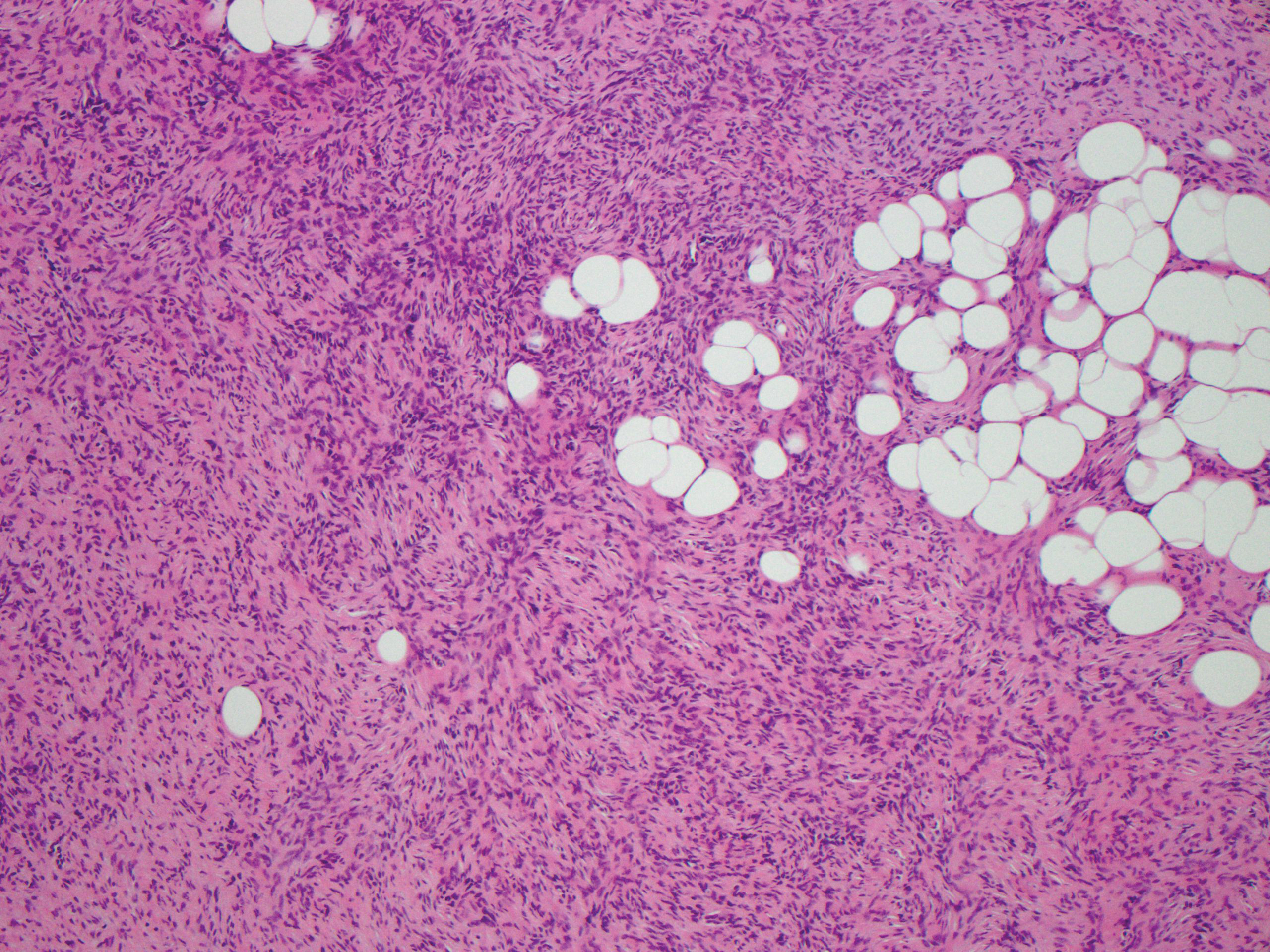
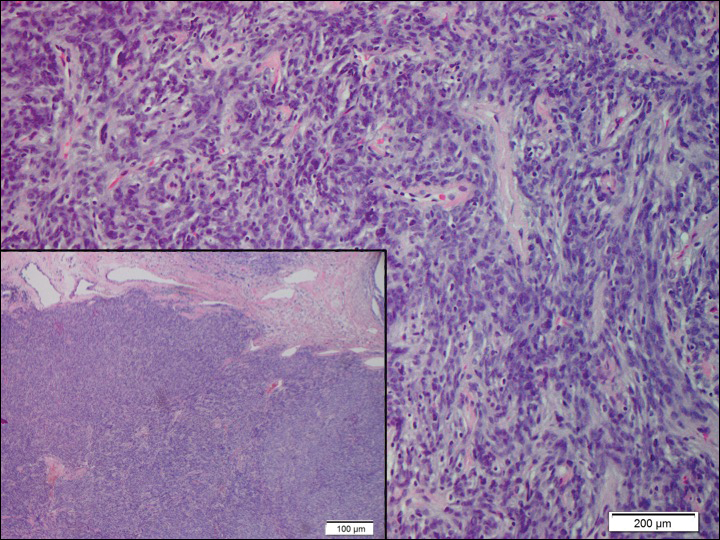
A cellular dermatofibroma generally presents as a single round, reddish brown papule or nodule approximately 0.5 to 1 cm in diameter that is firm to palpation with a central depression or dimple created over the lesion from the lateral pressure. Cellular dermatofibromas mostly occur in middle-aged adults, with the most common locations on the legs and on the sides of the trunk. They are thought to arise after injuries to the skin. On histopathologic examination, cellular dermatofibromas typically exhibit a proliferation of fibrohistiocytic cells with collagen trapping, often at the periphery of the tumor (Figure 2). Although cellular dermatofibromas appear clinically different than SFTs, they often mimic SFTs histopathologically. Immunostaining also can be helpful in differentiating cellular dermatofibromas in which cells stain positive for factor XIIIa. CD34 staining is negative.

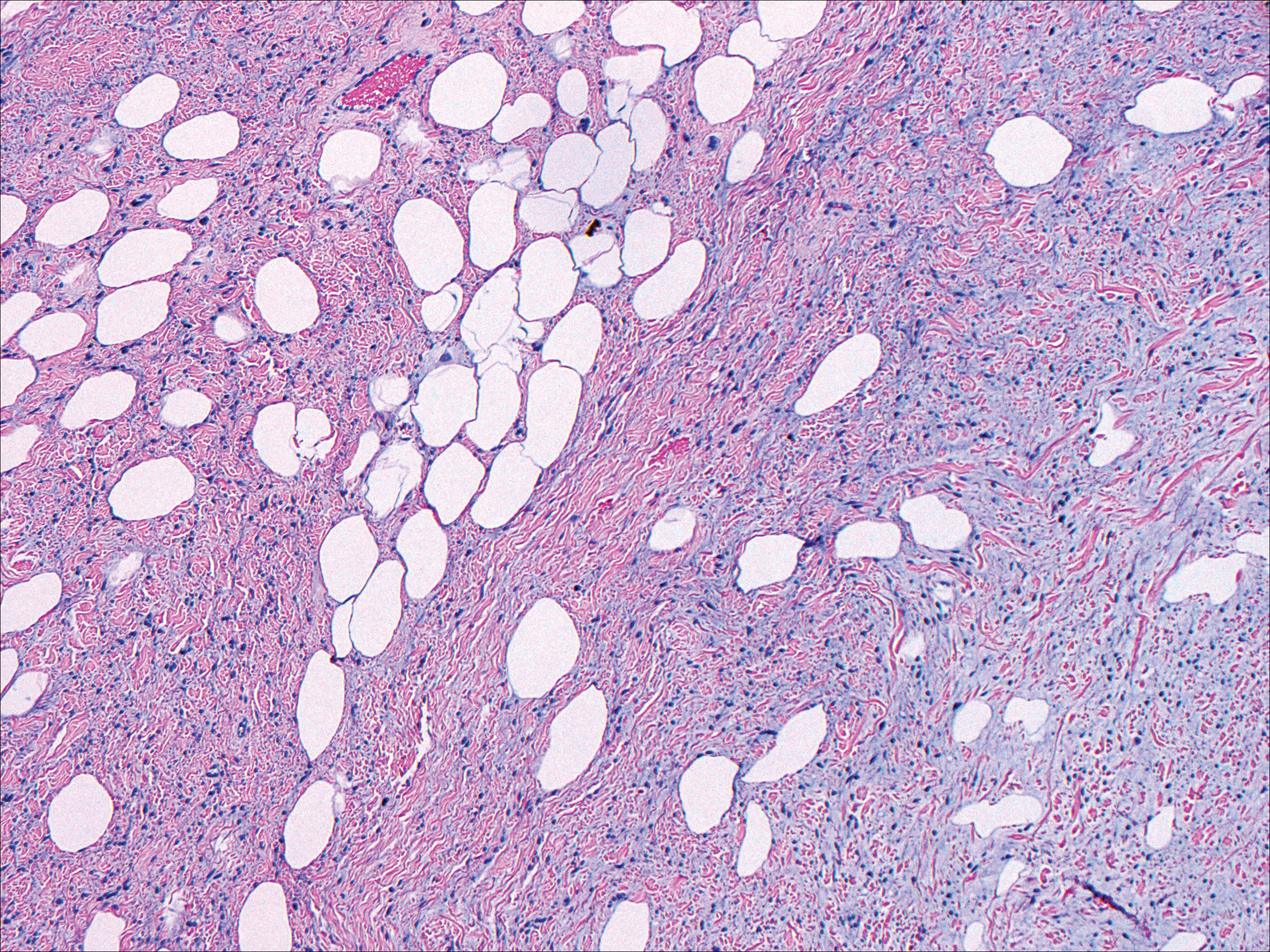
Dermatofibrosarcoma protuberans usually appears as one or multiple firm, red to violaceous nodules or plaques. They most often occur on the trunk in middle-aged adults. Histopathologically, DFSP presents with a dense, hypercellular, spindle cell proliferation that demonstrates a typical storiform pattern. The tumor generally infiltrates into the deep dermis and subcutaneous adipose layer with characteristic adipocyte entrapment (Figure 3). Positive CD34 and negative factor XIIIa staining helps to differentiate DFSP from a cellular dermatofibroma. Immunohistochemically, it is more difficult to distinguish DFSP from SFT, as both are CD34+ spindle cell neoplasms that also stain positive for CD99 and BCL-2.2 GRIA2 positivity also is seen in both SFT and DFSP.7 However, differentiation can be made on morphologic grounds alone, as DFSP has ill-defined tumor borders with adnexal and fat entrapment and SFT tends to be more circumscribed with prominent arborizing hyalinized vessels.8
Spindle cell lipoma (SCL) is an asymptomatic subcutaneous tumor commonly located on the back, neck, and shoulders in older patients, typically men. It often presents as a solitary lesion, though multiple lesions may occur. It is a well-circumscribed tumor of mature adipose tissue with areas of spindle cell proliferation and ropey collagen bundles (Figure 4). In early lesions, the spindle cell areas are myxoid with the presence of many mast cells.9 The spindle cells stain positive for CD34. Although spindle cell lipoma would be included in both the clinical and histopathologic differential diagnosis for SFT, its histopathologic features often are enough to differentiate SCL, which is highlighted by the aforementioned features as well as a relatively low cellularity and lack of ectatic vessels.8 However, discerning tumor variants, such as low-fat pseudoangiomatous SCL and lipomatous or myxoid SFT, might prove more challenging.
Nodular fasciitis typically presents as a rapidly growing subcutaneous nodule that may be tender. It is a benign reactive process usually affecting the arms and trunk of young to middle-aged adults, though it commonly involves the head and neck region in children.10 The tumor histopathologically appears as a well-circumscribed subcutaneous or fascial nodule with an angulated appearance. Spindle-shaped and stellate fibroblasts are loosely arranged in an edematous myxomatous stroma with a feathered appearance (Figure 5). Extravasated erythrocytes often are present. With time, collagen bundles become thicker and hyalinized. Immunohistochemical studies demonstrate positivity for vimentin, calponin, muscle-specific actin, and smooth muscle actin. Desmin, CD34, cytokeratin, and S-100 typically are negative.10-12 Therefore, CD34 staining is one of the main differentiating factors between nodular fasciitis and SFTs.
- Klemperer P, Rabin CB. Primary neoplasms of the pleura: a report of five cases. Arch Pathol. 1931;11:385-412.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
- Okamura JM, Barr RJ, Battifora H. Solitary fibrous tumor of the skin. Am J Dermatopathol. 1997;19:515-518.
- Lee JY, Park SE, Shin SJ, et al. Solitary fibrous tumor with myxoid stromal change. Am J Dermatopathol. 2015;37:570-573.
- Geramizadeh B, Marzban M, Churg A. Role of immunohistochemistry in the diagnosis of solitary fibrous tumor, a review. Iran J Pathol. 2016;11:195-293.
- Creytens D, Ferdinande L, Dorpe JV. Histopathologically malignant solitary fibrous tumor of the skin: a report of an unusual case. J Cutan Pathol. 2016;43:629-631.
- Vivero M, Doyle LA, Fletcher CD, et al. GRIA2 is a novel diagnostic marker for solitary fibrous tumour identified through gene expression profiling. Histopathology. 2014;65:71-80.
- Wood L, Fountaine TJ, Rosamilia L, et al. Cutaneous CD34 spindle cell neoplasms: histopathologic features distinguish spindle cell lipoma, solitary fibrous tumor, and dermatofibrosarcoma protuberans. Am J Dermatopathol. 2010;32:764-768.
- Khatib Y, Khade AL, Shah VB, et al. Cytohistological features of spindle cell lipoma--a case report with differential diagnosis. J Clin Diagn Res. 2017;11:10-11.
- Kumar E, Patel NR, Demicco EG, et al. Cutaneous nodular fasciitis with genetic analysis: a case series. J Cutan Pathol. 2016;43:1143-1149.
- Bracey TS, Wharton S, Smith ME. Nodular 'fasciitis' presenting as a cutaneous polyp. J Cutan Pathol. 2009;36:980-982.
- Perez-Montiel MD, Plaza JA, Dominguez-Malagon H, et al. Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. Am J Dermatopathol. 2006;28:105-111.
The Diagnosis: Solitary Fibrous Tumor
Solitary fibrous tumors (SFTs), as first described by Klemperer and Rabin1 in 1931, are relatively uncommon mesenchymal neoplasms that occur primarily in the pleura. This lesion is now known to affect many other extrathoracic sites, such as the liver, kidney, adrenal glands, thyroid, central nervous system, and soft tissue, with rare examples originating from the skin.2 Okamura et al3 reported the first known case of cutaneous SFT in 1997, with most of the literature limited to case reports. Erdag et al2 described one of the largest case series of primary cutaneous SFTs. These lesions can occur across a wide age range but tend to primarily affect middle-aged adults. Solitary fibrous tumors have been known to have no sex predilection; however, Erdag et al2 found a male predominance with a male to female ratio of 4 to 1.
Histopathologically, a cutaneous SFT is known to appear as a well-circumscribed nodular spindle cell proliferation arranged in interlacing fascicles with an abundant hyalinized collagen stroma (quiz image). Alternating hypocellular and hypercellular areas can be seen. Supporting vasculature often is relatively prominent, represented by angulated and branching staghorn blood vessels (Figure 1).2 A common histopathologic finding of SFTs is a patternless pattern, which suggests that the tumor can have a variety of morphologic appearances (eg, storiform, fascicular, neural, herringbone growth patterns), making histologic diagnosis difficult (quiz image).4 Therefore, immunohistochemistry plays a large role in the diagnosis of this tumor. The most important positive markers include CD34, CD99, B-cell lymphoma 2 (BCL-2), and signal transducer and activator of transcription 6 (STAT6).5 Nuclear STAT6 staining is an immunomarker for NGFI-A binding protein 2 (NAB2)-STAT6 gene fusion, which is specific for SFT.5,6 Vivero et al7 also reported glutamate receptor, inotropic, AMPA 2 (GRIA2) as a useful immunostain in SFT, though it is also expressed in dermatofibrosarcoma protuberans (DFSP). In this case, the clinical and histopathologic findings best supported a diagnosis of SFT. Some consider hemangiopericytomas to be examples of SFTs; however, true hemangiopericytomas lack the thick hyalinized collagen and hypercellular areas seen in SFT.
A cellular dermatofibroma generally presents as a single round, reddish brown papule or nodule approximately 0.5 to 1 cm in diameter that is firm to palpation with a central depression or dimple created over the lesion from the lateral pressure. Cellular dermatofibromas mostly occur in middle-aged adults, with the most common locations on the legs and on the sides of the trunk. They are thought to arise after injuries to the skin. On histopathologic examination, cellular dermatofibromas typically exhibit a proliferation of fibrohistiocytic cells with collagen trapping, often at the periphery of the tumor (Figure 2). Although cellular dermatofibromas appear clinically different than SFTs, they often mimic SFTs histopathologically. Immunostaining also can be helpful in differentiating cellular dermatofibromas in which cells stain positive for factor XIIIa. CD34 staining is negative.
Dermatofibrosarcoma protuberans usually appears as one or multiple firm, red to violaceous nodules or plaques. They most often occur on the trunk in middle-aged adults. Histopathologically, DFSP presents with a dense, hypercellular, spindle cell proliferation that demonstrates a typical storiform pattern. The tumor generally infiltrates into the deep dermis and subcutaneous adipose layer with characteristic adipocyte entrapment (Figure 3). Positive CD34 and negative factor XIIIa staining helps to differentiate DFSP from a cellular dermatofibroma. Immunohistochemically, it is more difficult to distinguish DFSP from SFT, as both are CD34+ spindle cell neoplasms that also stain positive for CD99 and BCL-2.2 GRIA2 positivity also is seen in both SFT and DFSP.7 However, differentiation can be made on morphologic grounds alone, as DFSP has ill-defined tumor borders with adnexal and fat entrapment and SFT tends to be more circumscribed with prominent arborizing hyalinized vessels.8
Spindle cell lipoma (SCL) is an asymptomatic subcutaneous tumor commonly located on the back, neck, and shoulders in older patients, typically men. It often presents as a solitary lesion, though multiple lesions may occur. It is a well-circumscribed tumor of mature adipose tissue with areas of spindle cell proliferation and ropey collagen bundles (Figure 4). In early lesions, the spindle cell areas are myxoid with the presence of many mast cells.9 The spindle cells stain positive for CD34. Although spindle cell lipoma would be included in both the clinical and histopathologic differential diagnosis for SFT, its histopathologic features often are enough to differentiate SCL, which is highlighted by the aforementioned features as well as a relatively low cellularity and lack of ectatic vessels.8 However, discerning tumor variants, such as low-fat pseudoangiomatous SCL and lipomatous or myxoid SFT, might prove more challenging.
Nodular fasciitis typically presents as a rapidly growing subcutaneous nodule that may be tender. It is a benign reactive process usually affecting the arms and trunk of young to middle-aged adults, though it commonly involves the head and neck region in children.10 The tumor histopathologically appears as a well-circumscribed subcutaneous or fascial nodule with an angulated appearance. Spindle-shaped and stellate fibroblasts are loosely arranged in an edematous myxomatous stroma with a feathered appearance (Figure 5). Extravasated erythrocytes often are present. With time, collagen bundles become thicker and hyalinized. Immunohistochemical studies demonstrate positivity for vimentin, calponin, muscle-specific actin, and smooth muscle actin. Desmin, CD34, cytokeratin, and S-100 typically are negative.10-12 Therefore, CD34 staining is one of the main differentiating factors between nodular fasciitis and SFTs.
The Diagnosis: Solitary Fibrous Tumor
Solitary fibrous tumors (SFTs), as first described by Klemperer and Rabin1 in 1931, are relatively uncommon mesenchymal neoplasms that occur primarily in the pleura. This lesion is now known to affect many other extrathoracic sites, such as the liver, kidney, adrenal glands, thyroid, central nervous system, and soft tissue, with rare examples originating from the skin.2 Okamura et al3 reported the first known case of cutaneous SFT in 1997, with most of the literature limited to case reports. Erdag et al2 described one of the largest case series of primary cutaneous SFTs. These lesions can occur across a wide age range but tend to primarily affect middle-aged adults. Solitary fibrous tumors have been known to have no sex predilection; however, Erdag et al2 found a male predominance with a male to female ratio of 4 to 1.
Histopathologically, a cutaneous SFT is known to appear as a well-circumscribed nodular spindle cell proliferation arranged in interlacing fascicles with an abundant hyalinized collagen stroma (quiz image). Alternating hypocellular and hypercellular areas can be seen. Supporting vasculature often is relatively prominent, represented by angulated and branching staghorn blood vessels (Figure 1).2 A common histopathologic finding of SFTs is a patternless pattern, which suggests that the tumor can have a variety of morphologic appearances (eg, storiform, fascicular, neural, herringbone growth patterns), making histologic diagnosis difficult (quiz image).4 Therefore, immunohistochemistry plays a large role in the diagnosis of this tumor. The most important positive markers include CD34, CD99, B-cell lymphoma 2 (BCL-2), and signal transducer and activator of transcription 6 (STAT6).5 Nuclear STAT6 staining is an immunomarker for NGFI-A binding protein 2 (NAB2)-STAT6 gene fusion, which is specific for SFT.5,6 Vivero et al7 also reported glutamate receptor, inotropic, AMPA 2 (GRIA2) as a useful immunostain in SFT, though it is also expressed in dermatofibrosarcoma protuberans (DFSP). In this case, the clinical and histopathologic findings best supported a diagnosis of SFT. Some consider hemangiopericytomas to be examples of SFTs; however, true hemangiopericytomas lack the thick hyalinized collagen and hypercellular areas seen in SFT.
A cellular dermatofibroma generally presents as a single round, reddish brown papule or nodule approximately 0.5 to 1 cm in diameter that is firm to palpation with a central depression or dimple created over the lesion from the lateral pressure. Cellular dermatofibromas mostly occur in middle-aged adults, with the most common locations on the legs and on the sides of the trunk. They are thought to arise after injuries to the skin. On histopathologic examination, cellular dermatofibromas typically exhibit a proliferation of fibrohistiocytic cells with collagen trapping, often at the periphery of the tumor (Figure 2). Although cellular dermatofibromas appear clinically different than SFTs, they often mimic SFTs histopathologically. Immunostaining also can be helpful in differentiating cellular dermatofibromas in which cells stain positive for factor XIIIa. CD34 staining is negative.
Dermatofibrosarcoma protuberans usually appears as one or multiple firm, red to violaceous nodules or plaques. They most often occur on the trunk in middle-aged adults. Histopathologically, DFSP presents with a dense, hypercellular, spindle cell proliferation that demonstrates a typical storiform pattern. The tumor generally infiltrates into the deep dermis and subcutaneous adipose layer with characteristic adipocyte entrapment (Figure 3). Positive CD34 and negative factor XIIIa staining helps to differentiate DFSP from a cellular dermatofibroma. Immunohistochemically, it is more difficult to distinguish DFSP from SFT, as both are CD34+ spindle cell neoplasms that also stain positive for CD99 and BCL-2.2 GRIA2 positivity also is seen in both SFT and DFSP.7 However, differentiation can be made on morphologic grounds alone, as DFSP has ill-defined tumor borders with adnexal and fat entrapment and SFT tends to be more circumscribed with prominent arborizing hyalinized vessels.8
Spindle cell lipoma (SCL) is an asymptomatic subcutaneous tumor commonly located on the back, neck, and shoulders in older patients, typically men. It often presents as a solitary lesion, though multiple lesions may occur. It is a well-circumscribed tumor of mature adipose tissue with areas of spindle cell proliferation and ropey collagen bundles (Figure 4). In early lesions, the spindle cell areas are myxoid with the presence of many mast cells.9 The spindle cells stain positive for CD34. Although spindle cell lipoma would be included in both the clinical and histopathologic differential diagnosis for SFT, its histopathologic features often are enough to differentiate SCL, which is highlighted by the aforementioned features as well as a relatively low cellularity and lack of ectatic vessels.8 However, discerning tumor variants, such as low-fat pseudoangiomatous SCL and lipomatous or myxoid SFT, might prove more challenging.
Nodular fasciitis typically presents as a rapidly growing subcutaneous nodule that may be tender. It is a benign reactive process usually affecting the arms and trunk of young to middle-aged adults, though it commonly involves the head and neck region in children.10 The tumor histopathologically appears as a well-circumscribed subcutaneous or fascial nodule with an angulated appearance. Spindle-shaped and stellate fibroblasts are loosely arranged in an edematous myxomatous stroma with a feathered appearance (Figure 5). Extravasated erythrocytes often are present. With time, collagen bundles become thicker and hyalinized. Immunohistochemical studies demonstrate positivity for vimentin, calponin, muscle-specific actin, and smooth muscle actin. Desmin, CD34, cytokeratin, and S-100 typically are negative.10-12 Therefore, CD34 staining is one of the main differentiating factors between nodular fasciitis and SFTs.
- Klemperer P, Rabin CB. Primary neoplasms of the pleura: a report of five cases. Arch Pathol. 1931;11:385-412.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
- Okamura JM, Barr RJ, Battifora H. Solitary fibrous tumor of the skin. Am J Dermatopathol. 1997;19:515-518.
- Lee JY, Park SE, Shin SJ, et al. Solitary fibrous tumor with myxoid stromal change. Am J Dermatopathol. 2015;37:570-573.
- Geramizadeh B, Marzban M, Churg A. Role of immunohistochemistry in the diagnosis of solitary fibrous tumor, a review. Iran J Pathol. 2016;11:195-293.
- Creytens D, Ferdinande L, Dorpe JV. Histopathologically malignant solitary fibrous tumor of the skin: a report of an unusual case. J Cutan Pathol. 2016;43:629-631.
- Vivero M, Doyle LA, Fletcher CD, et al. GRIA2 is a novel diagnostic marker for solitary fibrous tumour identified through gene expression profiling. Histopathology. 2014;65:71-80.
- Wood L, Fountaine TJ, Rosamilia L, et al. Cutaneous CD34 spindle cell neoplasms: histopathologic features distinguish spindle cell lipoma, solitary fibrous tumor, and dermatofibrosarcoma protuberans. Am J Dermatopathol. 2010;32:764-768.
- Khatib Y, Khade AL, Shah VB, et al. Cytohistological features of spindle cell lipoma--a case report with differential diagnosis. J Clin Diagn Res. 2017;11:10-11.
- Kumar E, Patel NR, Demicco EG, et al. Cutaneous nodular fasciitis with genetic analysis: a case series. J Cutan Pathol. 2016;43:1143-1149.
- Bracey TS, Wharton S, Smith ME. Nodular 'fasciitis' presenting as a cutaneous polyp. J Cutan Pathol. 2009;36:980-982.
- Perez-Montiel MD, Plaza JA, Dominguez-Malagon H, et al. Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. Am J Dermatopathol. 2006;28:105-111.
- Klemperer P, Rabin CB. Primary neoplasms of the pleura: a report of five cases. Arch Pathol. 1931;11:385-412.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
- Okamura JM, Barr RJ, Battifora H. Solitary fibrous tumor of the skin. Am J Dermatopathol. 1997;19:515-518.
- Lee JY, Park SE, Shin SJ, et al. Solitary fibrous tumor with myxoid stromal change. Am J Dermatopathol. 2015;37:570-573.
- Geramizadeh B, Marzban M, Churg A. Role of immunohistochemistry in the diagnosis of solitary fibrous tumor, a review. Iran J Pathol. 2016;11:195-293.
- Creytens D, Ferdinande L, Dorpe JV. Histopathologically malignant solitary fibrous tumor of the skin: a report of an unusual case. J Cutan Pathol. 2016;43:629-631.
- Vivero M, Doyle LA, Fletcher CD, et al. GRIA2 is a novel diagnostic marker for solitary fibrous tumour identified through gene expression profiling. Histopathology. 2014;65:71-80.
- Wood L, Fountaine TJ, Rosamilia L, et al. Cutaneous CD34 spindle cell neoplasms: histopathologic features distinguish spindle cell lipoma, solitary fibrous tumor, and dermatofibrosarcoma protuberans. Am J Dermatopathol. 2010;32:764-768.
- Khatib Y, Khade AL, Shah VB, et al. Cytohistological features of spindle cell lipoma--a case report with differential diagnosis. J Clin Diagn Res. 2017;11:10-11.
- Kumar E, Patel NR, Demicco EG, et al. Cutaneous nodular fasciitis with genetic analysis: a case series. J Cutan Pathol. 2016;43:1143-1149.
- Bracey TS, Wharton S, Smith ME. Nodular 'fasciitis' presenting as a cutaneous polyp. J Cutan Pathol. 2009;36:980-982.
- Perez-Montiel MD, Plaza JA, Dominguez-Malagon H, et al. Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. Am J Dermatopathol. 2006;28:105-111.
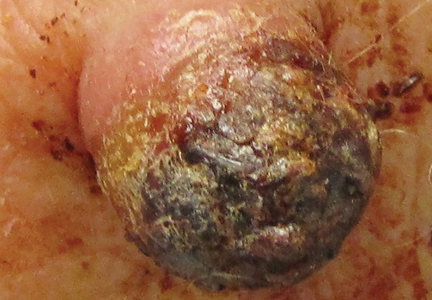
A 73-year-old man presented with a tender nodule on the back that had recently increased in size. On physical examination, a solitary 4-cm nodule was noted in the right trapezius region. The patient denied any personal or family history of similar lesions or a penchant for cysts. Due to the symptomatic nature of the lesion, surgical excision was performed.
Merkel Cell Carcinoma: A Review
Merkel cells originally were described by German histopathologist Friedrich Sigmund Merkel in 1875. These unique tactile cells were described as epidermal, nondendritic, and nonkeratinizing. Merkel cells are thought to arise from the neural crest and are believed to be primary neural cells found within the basal layer of the epidermis.1,2 They likely function primarily as slowly adapting type I mechanoreceptors. Origin from the neural crest is controversial, as other investigators have suggested derivation from epidermal keratinocytes.1,2 Tumor cells have been linked to the amine precursor uptake and decarboxylation system.3 In 1972, Toker4 described several cases of trabecular or sweat gland carcinomas of the skin. Upon further investigation, the cells that comprised these tumors were found to have dense core granules on electron microscopy, typical of Merkel cells.1,2 Other terms such as neuroendocrine carcinoma of the skin, small cell carcinoma of the skin, and anaplastic carcinoma of the skin have been used to describe Merkel cell carcinoma (MCC),1 which was suggested by De Wolf-Peeters et al5 in 1980.
Despite being a rare malignancy, MCC follows an aggressive clinical course. Upon presentation, approximately 66% of patients have local disease, 27% have nodal involvement, and 7% have distant metastasis.1 Future treatments will likely center around the novel Merkel cell polyomavirus (MCPyV) and modification of immune responses toward tumor cells. Standardization continues to be lacking in both staging and treatment of this aggressive tumor.
Epidemiology of MCC
 | ||
Figure 1. A 2.3×1.5×1.2-cm, hemorrhagic, crusted, | ||
 | ||
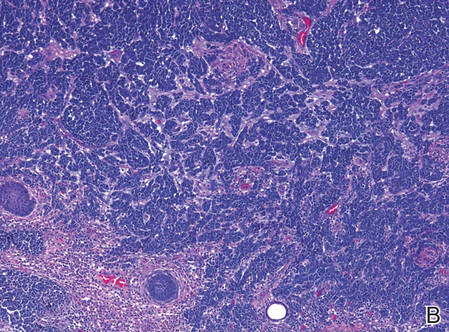 | ||
| Figure 2. Merkel cells are small- to medium-sized cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen (A)(H&E, original magnification ×40). Merkel cell carcinoma with trabecular pattern (B) (H&E, original magnification ×10). | ||
Between 1986 and 2006, the incidence of MCC grew substantially.1,2 Figures have been reported at 0.15 cases per 100,000 individuals to 0.6 cases per 100,000 individuals worldwide. In the United States, the age-adjusted incidence of MCC is estimated at 0.24 per 100,000 person-years, which is higher than the estimated 0.13 per 100,000 person-years found in Europe.3 The highest incidence worldwide has been noted in Western Australia, likely due to high levels of UV exposure.1 The incidence of MCC in psoriasis patients who are treated with oral methoxsalen (psoralen) and UVA photochemotherapy is 100 times greater than in the general population, further supporting the role of UV light in the development of MCC.1 White individuals have the highest incidence of MCC worldwide, with men being affected more frequently than women.1,3 The majority of patients with MCC are diagnosed at 70 years or older.1 Approximately 5% of reported MCC patients are diagnosed before 50 years of age.2 Immunosuppression and immunodeficiency likely play a role in the pathogenesis of MCC, and the incidence is increased in solid organ transplant recipients, most commonly renal transplant recipients,1 as well as individuals with chronic lymphocytic leukemia, human immunodeficiency virus infection, and AIDS.1,3 Patients with autoimmune diseases such as rheumatoid arthritis also are at increased risk for MCC.3 Individuals who are diagnosed with MCC are at an increased risk for development of other malignancies including nonmelanoma skin cancers, chronic lymphocytic leukemia, Hodgkin lymphoma, and non-Hodgkin lymphoma.3
Clinical Presentation of MCC
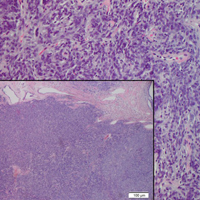
The clinical presentation of MCC can be variable. Most tumors present as firm, red to purple, nontender papules or nodules (Figure 1).1 Tumor size may range from 2 to 200 mm but is most commonly less than 20 mm.2 Growth can be rapid, and tumors are most commonly located on sun-exposed skin. The head and neck areas account for 48% of all MCC cases,1 with the eyelids being frequently involved.2 Merkel cell carcinoma also has been reported on the arms, legs, trunk, back, and buttocks.1 Non–sun-exposed areas are less commonly affected. Mucosal sites (eg, larynx, nasal cavity, pharynx, mouth) account for 5% of primary MCCs.1 Merkel cell carcinoma also has been reported to affect the vulva and penis. Subcutaneous primary MCC has presented without overlying epidermal changes.1 In a case series by Heath et al,6 14% (27/195) of MCC patients presented with nodal disease without any identifiable primary tumor, with the inguinal nodal chain being the most common for this presentation. It currently is not known whether these nodal tumors are primary tumors or metastatic disease with a regression of the primary tumor.1
|
Histopathology of MCC
 | ||
 | ||
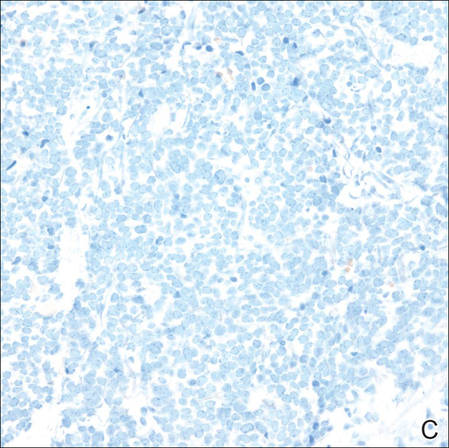 | ||
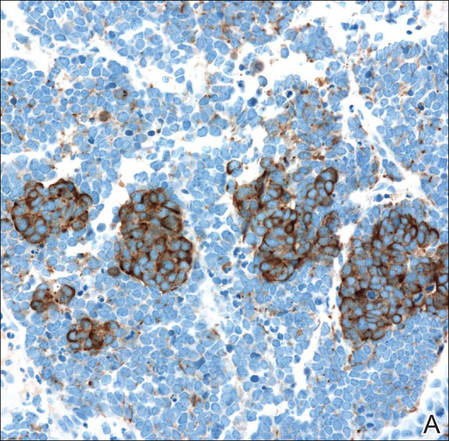
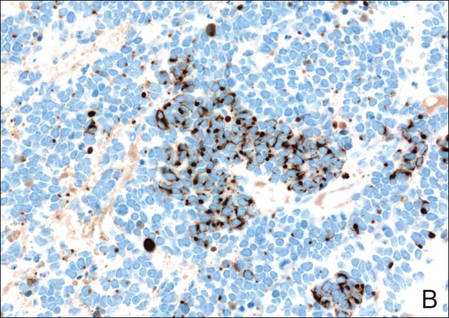
Figure 3. Positive chromogranin staining (A)(original | ||
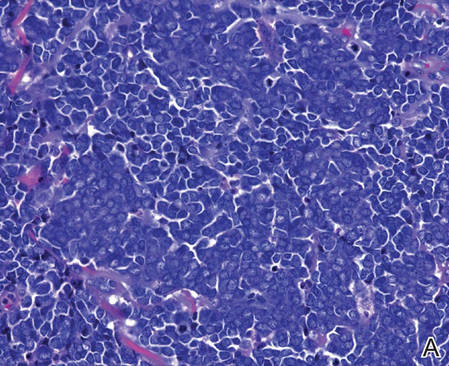
Merkel cells are small- to medium-sized basophilic cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen on histopathology (Figure 2A).1 Some tumor cells have more vesicular chromatin, multiple small nucleoli, irregular contours, and more abundant cytoplasm. In some reports, irregular contours and abundant cytoplasm were associated with no detectible MCPyV infection.1,3 Merkel cell carcinomas have a primarily nodular architecture, and classification is based on growth pattern and cell size. Three histopathologic growth patterns have been described: nodular, infiltrative, and trabecular. The trabecular pattern is composed of interconnecting strands ofcells (Figure 2B). Tumors with solely intraepidermal involvement (MCC in situ) have been described but are exceedingly rare.1 Cell types are classified according to size, with the intermediate cell type being the most common. The small cell variant may be mistaken for a lymphocytic infiltrate due to the similar size and appearance of both types of cells.1,3
Merkel cell carcinomas can have histopathological overlap with lymphomas, small cell lung cancers, carcinoid tumors, primitive neuroectodermal tumors, neuroblastomas, small cell osteosarcomas, rhabdomyosarcomas, or Ewing sarcomas.1,3 Specifically, differentiation from small cell carcinoma of the lung is of utmost importance. Merkel cell carcinoma stains positively for cytokeratins 8, 18, 19, and 20. The neuroendocrine markers chromogranin (Figure 3A), synaptophysin, and neuron-specific enolase also may show positive staining. Cytokeratin 20, low-molecular-weight cytokeratins (CAM 5.2), and neurofilament immunostains have a high sensitivity for MCC and are the most frequently used.1 Cytokeratin 20 stains in the characteristic paranuclear dot–like pattern, which is a hallmark of MCC (Figure 3B). Cytokeratin 20 positivity in conjunction with negative staining for thyroid transcription factor 1 (Figure 3C) and cytokeratin 7 can aid in differentiation from small cell carcinoma of the lung.1,3
Pathogenesis of MCC
In 2008, Feng et al7 discovered a novel polyomavirus associated with the development of MCC. This novel polyomavirus, MCPyV, is found in approximately 80% of all cases of MCC. Seventeen members of the polyomavirus family have been identified, 9 of which have been found to infect humans, including BK virus, JC virus, WU, MCPyV, human polyomavirus 6, human polyomavirus 7, trichodysplasia spinulosa–associated polyomavirus, human polyomavirus 9, and Simian virus 40.1 Merkel cell polyomavirus infection is found in approximately 60% of the general population and exposure likely occurs early in life. The virus likely is transmitted through skin shedding and nasal secretions, though it also has been found in urine specimens.3 Currently, there is no evidence to suggest vertical viral transmission from mother to fetus.
Merkel cell polyomavirus is composed of early and late gene regions. The early gene region contains both large T antigen (LT) and small T antigen reading frames, which are necessary for viral replication.8 The late region is responsible for encoding viral proteins necessary for viral capsid assembly. Mutations found in viral protein 1 prevent formation of viral particles.9 Large T antigen is substantially overexpressed in MCC and is responsible for tumor suppression through retinoblastoma tumor suppressor protein. It also serves as a binding domain for both heat shock proteins and helicases.8,10 These domains allow the polyomaviruses to use host-cell machinery for viral genome replication while targeting tumor suppressor proteins.8 Upon viral integration into host DNA, viral replication ceases while oncogenic function persists.
The exact mechanism by which the MCPyV contributes to the development of MCC still has yet to be identified. Hypotheses suggest a combination of viral infection with external mutagens (eg, UV radiation). Experimental observations suggest viral contribution is likely due to the large percentage of MCCs that are positive for MCPyV, the identification of LT antigen expression and the role it plays in preserving cell cycle progression, and the role persistent LT antigen expression plays in continued growth of MCC cell lines in vitro.8 Two important cell line preservation mechanisms ensure continued tumor growth, including prevention of apoptosis triggered by DNA damage response mechanisms following UV damage and interaction with the retinoblastoma tumor suppressor protein allowing continued growth.8,11 Other important factors in tumor growth and survival may be the inhibition of apoptosis through the BCL2 (B-cell chronic lymphocytic leukemia/lymphoma 2) proto-oncogene and survivin (baculoviral inhibitor of apoptosis repeat-containing 5 [BIRC5]).12 Survivin has been found to play an important role in MCPyV-positive MCCs.12,13 It has been suggested that lymphangiogenesis in MCC likely is driven by vascular endothelial growth factor-C+CD68+CD163+ M2 macrophages.14 Another survival mechanism specific to polyomaviruses is their ability to interfere with the p53 tumor suppressor pathway.8 Loss of p53 expression by tumor cell nuclei has been associated with poor prognosis.15
Immune Response
Immune response as a role in tumor progression can be primarily centered on the concept of persistent antigen expression as a means of immune downregulation. Dunn et al16 suggested that cancer cells must interact through 3 consecutive phases with the host immune system (immunoediting hypothesis). In the elimination phase, the host immune system is able to recognize and destroy newly transformed cells through both the innate and adaptive immune systems. The second equilibrium phase allows the tumor to remain dormant and growth remains stagnant. Lastly, the tumor is allowed to evade the immune system through the escape phase.8
Host immune responses play an important role in both the progression and prognosis of MCC. High anti-MCPyV capsid antibody titers have been associated with better progression-free survival in some patients.8 Patients with high antibody titers (>10,000) likely have better progression-free survival than those with low antibody titers (<10,000).17 Antibody titers to the LT antigen may serve as a biomarker of MCC disease burden in the future. Rising LT antigen titers have been shown to correlate with disease progression and falling titers correlate with successful treatment.8 Tumoral infiltration of CD8+ T lymphocytes has been shown to be a predictor of survival compared to no intratumoral infiltration.6 Sihto et al18 suggested that this better prognosis from high intratumoral infiltration is not specific to MCPyV-positive MCC; however, it does highlight an important aspect of tumor evasion through the downregulation of cell surface expression of class I major histocompatibility complex antigens, which allows presentation of tumor intracellular peptides to CD8+ T lymphocytes.8 Upregulation of this specific immune response may play a role in the future treatment of MCC.
Staging and Prognosis
Due to the extremely aggressive nature of MCC, patients with local disease and tumors 2 cm or smaller in diameter have a 66% survival at 5 years.1,3 The 5-year survival rate for patients with local metastasis to regional lymph nodes ranges from 26% to 42%. Patients with distant metastasis have an 18% survival rate at 5 years.1,3 Data suggest that sentinel lymph node biopsy should be performed on all patients with MCC regardless of tumor size.1 There are no consensus guidelines to date regarding imaging for the staging of MCC patients. It is suggested that (18F)fluorodeoxyglucose positron emission tomography alone or in combination with computed tomography (CT) may be of value as a single whole-body diagnostic tool for accurate staging.10 It also has been suggested that (18F)fluorodeoxyglucose positron emission tomography and CT may offer more accurate staging than other screening modalities such as CT alone or magnetic resonance imaging.14,19
Treatment of MCC
Surgery remains the mainstay of treatment of MCC. Current National Comprehensive Cancer Network guidelines20 recommend 1- to 2-cm margins for wide local excision or treatment with Mohs micrographic surgery. Sentinel lymph node biopsy should be performed intraoperatively in patients undergoing wide local excision and preoperatively for patients undergoing Mohs micrographic surgery due to potential alterations in lymphatic drainage that may affect lymphoscintigraphy.1
Radiation may be used as primary or adjuvant therapy in patients with MCC. Radiation as primary therapy generally is reserved for patients who are not surgical candidates. It has been suggested that there was no difference in outcome in a small group of patients treated with radiation alone compared to patients who underwent surgery and radiation to the tumor bed.1 Current guidelines suggest a small group of patients may not require adjuvant therapy following adequate resection of some small tumors, and clinical observation may be appropriate.1,3 Chemotherapy may play a palliative role in patients with metastatic MCC. Merkel cell carcinoma has been shown to be chemosensitive but with a high recurrence rate.1 Because the immune system plays an important role in disease prognosis, having an intact immune system likely is paramount in the prevention of further disease progression.
Future Treatments of MCC
Future treatment of MCC may be focused on the viral etiology of most tumors and upregulation of the immune response, which may lead to the possibility of specifically interfering with virus-specific oncoproteins and stimulation of immune responses to virally infected tumor cells.8 The MCPyV large T antigen has been found to be overexpressed in some tumors and may serve as a specific target of therapy.10,21 Survivin, a key cell cycle protein encoded by LT antigen, may be an interesting target given its implication in other cancers.13 Other potential nonviral molecular target antigens include the oncoprotein H1P1 that interacts with c-KIT.8 Specific immunostimulatory cytokines that may be used to upregulate the immune response to tumoral cells may include IL-2, IL-12, IL-15, or IL-21. Therapeutic agents that may be studied in the future to target the immune exhaustion phenomenon associated with tumorigenesis include ipilimumab (cytotoxic T lymphocyte antigen 4 receptor-blocking agent) as well as programmed cell death 1 and programmed cell death 1 ligand 1 (PD-1/PD-L1).8 Neuroendocrine tumors including MCC tend to be highly vascular and express vascular endothelial growth factors and platelet-derived growth factors, which may be other potential therapeutic targets. It has been reported that approximately 95% of MCC patients have CD56+ tumors, and current clinical trials suggest a promising therapeutic response with the immunogen anti-CD56 monoclonal antibody.3
Conclusion
Merkel cell carcinoma is a rare aggressive neuroendocrine tumor that has been associated with a novel polyomavirus. Merkel cell carcinoma tends to affect elderly and immunocompromised patients as well as white individuals. Tumors are most often found in areas of high UV exposure and clinically on sun-exposed skin. Merkel cell polyomavirus is associated with approximately 80% of tumors, and tumorigenesis likely is caused by a number of sequential steps from viral integration into host DNA, mutagenic events, and specific immune responses. Currently there are no consensus guidelines for using imaging for staging of MCCs, but sentinel lymph node biopsy is recommended for all cases due to the aggressive nature of even smaller tumors. Surgery remains the mainstay of treatment, and radiation therapy may be used as a primary or adjuvant treatment. Chemotherapy usually is reserved for patients with metastatic disease purely for palliation. Future treatments of MCC likely will center on the viral etiology of MCC and upregulation of immune responses to virally infected tumor cells.
1. Han S, North J, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
2. Goessling W, McKee P, Mayer R. Merkel cell carcinoma. J Clin Oncol. 2002;20:588-598.
3. Donepudi S, DeConti R, Samlowski W. Recent advances in the understanding of the genetics, etiology, and treatment of merkel cell carcinoma. Semin Oncol. 2012;39:163-172.
4. Toker C. Trabecular carcinoma of the skin. Arch Dermatol. 1972;105:107-110.
5. De Wolff-Peeters C, Marien K, Mebis J, et al. A cutaneous APUDoma or Merkel cell tumor? a morphologically recognizable tumor with a biological and histological malignant aspect in contrast with its clinical behavior. Cancer. 1980;46:810-816.
6. Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375-381.
7. Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
8. Bhatia S, Afanasiev O, Nghiem P. Immunobiology of Merkel cell carcinoma: implications for immunotherapy of a polyomavirus-associated cancer. Curr Oncol Rep. 2011;13:488-497.
9. Amber K, McLeod M, Nouri K. The Merkel cell polyomavirus and its involvement in Merkel cell carcinoma. Dermatol Surg. 2013;39:232-238.
10. Erovic B, Al Habeeb A, Harris L, et al. Significant overexpression of the Merkel cell polyomavirus (MCPyV) large T antigen in Merkel cell carcinoma. Head Neck. 2013;35:184-189.
11. Demetriou S, Ona-Vu K, Sullivan E, et al. Defective DNA repair and cell cycle arrest in cells expressing Merkel cell polyomavirus T antigen. Int J Cancer. 2012;131:1818-1827.
12. Sahi H, Koljonen V, Kavola H, et al. Bcl-2 expression indicates better prognosis of Merkel cell carcinoma regardless of the presence of Merkel cell polyomavirus. Virchows Arch. 2012;461:553-559.
13. Arora R, Shuda M, Guastafierro A, et al. Survivin is a therapeutic target in Merkel cell carcinoma. Sci Transl Med. 2012;4:1-11.
14. Hawryluk E, O’Regan K, Sheehy N, et al. Positron emission tomography/computed tomography imaging in Merkel cell carcinoma: a study of 270 scans in 97 patients at the Dana-Farber/Brigham and Women’s Cancer Center. J Am Acad Dermatol. 2013;68:592-599.
15. Hall B, Pincus L, Yu S, et al. Immunohistochemical prognostication of Merkel cell carcinoma: p63 expression but not polyomavirus status correlates with outcome. J Cutan Pathol. 2012;39:911-917.
16. Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991-998.
17. Touze A, Le Bidre E, Laude H, et al. High levels of antibodies against Merkel cell polyomavirus identify a subset of patients with Merkel cell carcinoma with better clinical outcome. J Clin Oncol. 2011;29:1612-1619.
18. Sihto H, Bohling T, Kavola H, et al. Tumor infiltrating immune cells and outcome of Merkel cell carcinoma: a population-based study. Clin Cancer Res. 2012;18:2872-2881.
19. Colgan M, Tarantola T, Weaver A, et al. The predictive value of imaging studies in evaluating regional lymph node involvement in Merkel cell carcinoma. J Am Acad Dermatol. 2012;67:1250-1256.
20. NCCN Clinical Practice Guidelines in Oncology. National Comprehensive Cancer Network website. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#site. Accessed March 22, 2016.
21. Angermeyer S, Hesbacher S, Becker J, et al. Merkel cell polyomavirus-positive Merkel cell carcinoma cells do not require expression of the viral small T antigen. J Invest Dermatol. 2013;133:1-6.
Merkel cells originally were described by German histopathologist Friedrich Sigmund Merkel in 1875. These unique tactile cells were described as epidermal, nondendritic, and nonkeratinizing. Merkel cells are thought to arise from the neural crest and are believed to be primary neural cells found within the basal layer of the epidermis.1,2 They likely function primarily as slowly adapting type I mechanoreceptors. Origin from the neural crest is controversial, as other investigators have suggested derivation from epidermal keratinocytes.1,2 Tumor cells have been linked to the amine precursor uptake and decarboxylation system.3 In 1972, Toker4 described several cases of trabecular or sweat gland carcinomas of the skin. Upon further investigation, the cells that comprised these tumors were found to have dense core granules on electron microscopy, typical of Merkel cells.1,2 Other terms such as neuroendocrine carcinoma of the skin, small cell carcinoma of the skin, and anaplastic carcinoma of the skin have been used to describe Merkel cell carcinoma (MCC),1 which was suggested by De Wolf-Peeters et al5 in 1980.
Despite being a rare malignancy, MCC follows an aggressive clinical course. Upon presentation, approximately 66% of patients have local disease, 27% have nodal involvement, and 7% have distant metastasis.1 Future treatments will likely center around the novel Merkel cell polyomavirus (MCPyV) and modification of immune responses toward tumor cells. Standardization continues to be lacking in both staging and treatment of this aggressive tumor.
Epidemiology of MCC
| ||
Figure 1. A 2.3×1.5×1.2-cm, hemorrhagic, crusted, | ||
| ||
| ||
| Figure 2. Merkel cells are small- to medium-sized cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen (A)(H&E, original magnification ×40). Merkel cell carcinoma with trabecular pattern (B) (H&E, original magnification ×10). | ||
Between 1986 and 2006, the incidence of MCC grew substantially.1,2 Figures have been reported at 0.15 cases per 100,000 individuals to 0.6 cases per 100,000 individuals worldwide. In the United States, the age-adjusted incidence of MCC is estimated at 0.24 per 100,000 person-years, which is higher than the estimated 0.13 per 100,000 person-years found in Europe.3 The highest incidence worldwide has been noted in Western Australia, likely due to high levels of UV exposure.1 The incidence of MCC in psoriasis patients who are treated with oral methoxsalen (psoralen) and UVA photochemotherapy is 100 times greater than in the general population, further supporting the role of UV light in the development of MCC.1 White individuals have the highest incidence of MCC worldwide, with men being affected more frequently than women.1,3 The majority of patients with MCC are diagnosed at 70 years or older.1 Approximately 5% of reported MCC patients are diagnosed before 50 years of age.2 Immunosuppression and immunodeficiency likely play a role in the pathogenesis of MCC, and the incidence is increased in solid organ transplant recipients, most commonly renal transplant recipients,1 as well as individuals with chronic lymphocytic leukemia, human immunodeficiency virus infection, and AIDS.1,3 Patients with autoimmune diseases such as rheumatoid arthritis also are at increased risk for MCC.3 Individuals who are diagnosed with MCC are at an increased risk for development of other malignancies including nonmelanoma skin cancers, chronic lymphocytic leukemia, Hodgkin lymphoma, and non-Hodgkin lymphoma.3
Clinical Presentation of MCC
The clinical presentation of MCC can be variable. Most tumors present as firm, red to purple, nontender papules or nodules (Figure 1).1 Tumor size may range from 2 to 200 mm but is most commonly less than 20 mm.2 Growth can be rapid, and tumors are most commonly located on sun-exposed skin. The head and neck areas account for 48% of all MCC cases,1 with the eyelids being frequently involved.2 Merkel cell carcinoma also has been reported on the arms, legs, trunk, back, and buttocks.1 Non–sun-exposed areas are less commonly affected. Mucosal sites (eg, larynx, nasal cavity, pharynx, mouth) account for 5% of primary MCCs.1 Merkel cell carcinoma also has been reported to affect the vulva and penis. Subcutaneous primary MCC has presented without overlying epidermal changes.1 In a case series by Heath et al,6 14% (27/195) of MCC patients presented with nodal disease without any identifiable primary tumor, with the inguinal nodal chain being the most common for this presentation. It currently is not known whether these nodal tumors are primary tumors or metastatic disease with a regression of the primary tumor.1
|
Histopathology of MCC
| ||
| ||
| ||
Figure 3. Positive chromogranin staining (A)(original | ||
Merkel cells are small- to medium-sized basophilic cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen on histopathology (Figure 2A).1 Some tumor cells have more vesicular chromatin, multiple small nucleoli, irregular contours, and more abundant cytoplasm. In some reports, irregular contours and abundant cytoplasm were associated with no detectible MCPyV infection.1,3 Merkel cell carcinomas have a primarily nodular architecture, and classification is based on growth pattern and cell size. Three histopathologic growth patterns have been described: nodular, infiltrative, and trabecular. The trabecular pattern is composed of interconnecting strands ofcells (Figure 2B). Tumors with solely intraepidermal involvement (MCC in situ) have been described but are exceedingly rare.1 Cell types are classified according to size, with the intermediate cell type being the most common. The small cell variant may be mistaken for a lymphocytic infiltrate due to the similar size and appearance of both types of cells.1,3
Merkel cell carcinomas can have histopathological overlap with lymphomas, small cell lung cancers, carcinoid tumors, primitive neuroectodermal tumors, neuroblastomas, small cell osteosarcomas, rhabdomyosarcomas, or Ewing sarcomas.1,3 Specifically, differentiation from small cell carcinoma of the lung is of utmost importance. Merkel cell carcinoma stains positively for cytokeratins 8, 18, 19, and 20. The neuroendocrine markers chromogranin (Figure 3A), synaptophysin, and neuron-specific enolase also may show positive staining. Cytokeratin 20, low-molecular-weight cytokeratins (CAM 5.2), and neurofilament immunostains have a high sensitivity for MCC and are the most frequently used.1 Cytokeratin 20 stains in the characteristic paranuclear dot–like pattern, which is a hallmark of MCC (Figure 3B). Cytokeratin 20 positivity in conjunction with negative staining for thyroid transcription factor 1 (Figure 3C) and cytokeratin 7 can aid in differentiation from small cell carcinoma of the lung.1,3
Pathogenesis of MCC
In 2008, Feng et al7 discovered a novel polyomavirus associated with the development of MCC. This novel polyomavirus, MCPyV, is found in approximately 80% of all cases of MCC. Seventeen members of the polyomavirus family have been identified, 9 of which have been found to infect humans, including BK virus, JC virus, WU, MCPyV, human polyomavirus 6, human polyomavirus 7, trichodysplasia spinulosa–associated polyomavirus, human polyomavirus 9, and Simian virus 40.1 Merkel cell polyomavirus infection is found in approximately 60% of the general population and exposure likely occurs early in life. The virus likely is transmitted through skin shedding and nasal secretions, though it also has been found in urine specimens.3 Currently, there is no evidence to suggest vertical viral transmission from mother to fetus.
Merkel cell polyomavirus is composed of early and late gene regions. The early gene region contains both large T antigen (LT) and small T antigen reading frames, which are necessary for viral replication.8 The late region is responsible for encoding viral proteins necessary for viral capsid assembly. Mutations found in viral protein 1 prevent formation of viral particles.9 Large T antigen is substantially overexpressed in MCC and is responsible for tumor suppression through retinoblastoma tumor suppressor protein. It also serves as a binding domain for both heat shock proteins and helicases.8,10 These domains allow the polyomaviruses to use host-cell machinery for viral genome replication while targeting tumor suppressor proteins.8 Upon viral integration into host DNA, viral replication ceases while oncogenic function persists.
The exact mechanism by which the MCPyV contributes to the development of MCC still has yet to be identified. Hypotheses suggest a combination of viral infection with external mutagens (eg, UV radiation). Experimental observations suggest viral contribution is likely due to the large percentage of MCCs that are positive for MCPyV, the identification of LT antigen expression and the role it plays in preserving cell cycle progression, and the role persistent LT antigen expression plays in continued growth of MCC cell lines in vitro.8 Two important cell line preservation mechanisms ensure continued tumor growth, including prevention of apoptosis triggered by DNA damage response mechanisms following UV damage and interaction with the retinoblastoma tumor suppressor protein allowing continued growth.8,11 Other important factors in tumor growth and survival may be the inhibition of apoptosis through the BCL2 (B-cell chronic lymphocytic leukemia/lymphoma 2) proto-oncogene and survivin (baculoviral inhibitor of apoptosis repeat-containing 5 [BIRC5]).12 Survivin has been found to play an important role in MCPyV-positive MCCs.12,13 It has been suggested that lymphangiogenesis in MCC likely is driven by vascular endothelial growth factor-C+CD68+CD163+ M2 macrophages.14 Another survival mechanism specific to polyomaviruses is their ability to interfere with the p53 tumor suppressor pathway.8 Loss of p53 expression by tumor cell nuclei has been associated with poor prognosis.15
Immune Response
Immune response as a role in tumor progression can be primarily centered on the concept of persistent antigen expression as a means of immune downregulation. Dunn et al16 suggested that cancer cells must interact through 3 consecutive phases with the host immune system (immunoediting hypothesis). In the elimination phase, the host immune system is able to recognize and destroy newly transformed cells through both the innate and adaptive immune systems. The second equilibrium phase allows the tumor to remain dormant and growth remains stagnant. Lastly, the tumor is allowed to evade the immune system through the escape phase.8
Host immune responses play an important role in both the progression and prognosis of MCC. High anti-MCPyV capsid antibody titers have been associated with better progression-free survival in some patients.8 Patients with high antibody titers (>10,000) likely have better progression-free survival than those with low antibody titers (<10,000).17 Antibody titers to the LT antigen may serve as a biomarker of MCC disease burden in the future. Rising LT antigen titers have been shown to correlate with disease progression and falling titers correlate with successful treatment.8 Tumoral infiltration of CD8+ T lymphocytes has been shown to be a predictor of survival compared to no intratumoral infiltration.6 Sihto et al18 suggested that this better prognosis from high intratumoral infiltration is not specific to MCPyV-positive MCC; however, it does highlight an important aspect of tumor evasion through the downregulation of cell surface expression of class I major histocompatibility complex antigens, which allows presentation of tumor intracellular peptides to CD8+ T lymphocytes.8 Upregulation of this specific immune response may play a role in the future treatment of MCC.
Staging and Prognosis
Due to the extremely aggressive nature of MCC, patients with local disease and tumors 2 cm or smaller in diameter have a 66% survival at 5 years.1,3 The 5-year survival rate for patients with local metastasis to regional lymph nodes ranges from 26% to 42%. Patients with distant metastasis have an 18% survival rate at 5 years.1,3 Data suggest that sentinel lymph node biopsy should be performed on all patients with MCC regardless of tumor size.1 There are no consensus guidelines to date regarding imaging for the staging of MCC patients. It is suggested that (18F)fluorodeoxyglucose positron emission tomography alone or in combination with computed tomography (CT) may be of value as a single whole-body diagnostic tool for accurate staging.10 It also has been suggested that (18F)fluorodeoxyglucose positron emission tomography and CT may offer more accurate staging than other screening modalities such as CT alone or magnetic resonance imaging.14,19
Treatment of MCC
Surgery remains the mainstay of treatment of MCC. Current National Comprehensive Cancer Network guidelines20 recommend 1- to 2-cm margins for wide local excision or treatment with Mohs micrographic surgery. Sentinel lymph node biopsy should be performed intraoperatively in patients undergoing wide local excision and preoperatively for patients undergoing Mohs micrographic surgery due to potential alterations in lymphatic drainage that may affect lymphoscintigraphy.1
Radiation may be used as primary or adjuvant therapy in patients with MCC. Radiation as primary therapy generally is reserved for patients who are not surgical candidates. It has been suggested that there was no difference in outcome in a small group of patients treated with radiation alone compared to patients who underwent surgery and radiation to the tumor bed.1 Current guidelines suggest a small group of patients may not require adjuvant therapy following adequate resection of some small tumors, and clinical observation may be appropriate.1,3 Chemotherapy may play a palliative role in patients with metastatic MCC. Merkel cell carcinoma has been shown to be chemosensitive but with a high recurrence rate.1 Because the immune system plays an important role in disease prognosis, having an intact immune system likely is paramount in the prevention of further disease progression.
Future Treatments of MCC
Future treatment of MCC may be focused on the viral etiology of most tumors and upregulation of the immune response, which may lead to the possibility of specifically interfering with virus-specific oncoproteins and stimulation of immune responses to virally infected tumor cells.8 The MCPyV large T antigen has been found to be overexpressed in some tumors and may serve as a specific target of therapy.10,21 Survivin, a key cell cycle protein encoded by LT antigen, may be an interesting target given its implication in other cancers.13 Other potential nonviral molecular target antigens include the oncoprotein H1P1 that interacts with c-KIT.8 Specific immunostimulatory cytokines that may be used to upregulate the immune response to tumoral cells may include IL-2, IL-12, IL-15, or IL-21. Therapeutic agents that may be studied in the future to target the immune exhaustion phenomenon associated with tumorigenesis include ipilimumab (cytotoxic T lymphocyte antigen 4 receptor-blocking agent) as well as programmed cell death 1 and programmed cell death 1 ligand 1 (PD-1/PD-L1).8 Neuroendocrine tumors including MCC tend to be highly vascular and express vascular endothelial growth factors and platelet-derived growth factors, which may be other potential therapeutic targets. It has been reported that approximately 95% of MCC patients have CD56+ tumors, and current clinical trials suggest a promising therapeutic response with the immunogen anti-CD56 monoclonal antibody.3
Conclusion
Merkel cell carcinoma is a rare aggressive neuroendocrine tumor that has been associated with a novel polyomavirus. Merkel cell carcinoma tends to affect elderly and immunocompromised patients as well as white individuals. Tumors are most often found in areas of high UV exposure and clinically on sun-exposed skin. Merkel cell polyomavirus is associated with approximately 80% of tumors, and tumorigenesis likely is caused by a number of sequential steps from viral integration into host DNA, mutagenic events, and specific immune responses. Currently there are no consensus guidelines for using imaging for staging of MCCs, but sentinel lymph node biopsy is recommended for all cases due to the aggressive nature of even smaller tumors. Surgery remains the mainstay of treatment, and radiation therapy may be used as a primary or adjuvant treatment. Chemotherapy usually is reserved for patients with metastatic disease purely for palliation. Future treatments of MCC likely will center on the viral etiology of MCC and upregulation of immune responses to virally infected tumor cells.
Merkel cells originally were described by German histopathologist Friedrich Sigmund Merkel in 1875. These unique tactile cells were described as epidermal, nondendritic, and nonkeratinizing. Merkel cells are thought to arise from the neural crest and are believed to be primary neural cells found within the basal layer of the epidermis.1,2 They likely function primarily as slowly adapting type I mechanoreceptors. Origin from the neural crest is controversial, as other investigators have suggested derivation from epidermal keratinocytes.1,2 Tumor cells have been linked to the amine precursor uptake and decarboxylation system.3 In 1972, Toker4 described several cases of trabecular or sweat gland carcinomas of the skin. Upon further investigation, the cells that comprised these tumors were found to have dense core granules on electron microscopy, typical of Merkel cells.1,2 Other terms such as neuroendocrine carcinoma of the skin, small cell carcinoma of the skin, and anaplastic carcinoma of the skin have been used to describe Merkel cell carcinoma (MCC),1 which was suggested by De Wolf-Peeters et al5 in 1980.
Despite being a rare malignancy, MCC follows an aggressive clinical course. Upon presentation, approximately 66% of patients have local disease, 27% have nodal involvement, and 7% have distant metastasis.1 Future treatments will likely center around the novel Merkel cell polyomavirus (MCPyV) and modification of immune responses toward tumor cells. Standardization continues to be lacking in both staging and treatment of this aggressive tumor.
Epidemiology of MCC
| ||
Figure 1. A 2.3×1.5×1.2-cm, hemorrhagic, crusted, | ||
| ||
| ||
| Figure 2. Merkel cells are small- to medium-sized cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen (A)(H&E, original magnification ×40). Merkel cell carcinoma with trabecular pattern (B) (H&E, original magnification ×10). | ||
Between 1986 and 2006, the incidence of MCC grew substantially.1,2 Figures have been reported at 0.15 cases per 100,000 individuals to 0.6 cases per 100,000 individuals worldwide. In the United States, the age-adjusted incidence of MCC is estimated at 0.24 per 100,000 person-years, which is higher than the estimated 0.13 per 100,000 person-years found in Europe.3 The highest incidence worldwide has been noted in Western Australia, likely due to high levels of UV exposure.1 The incidence of MCC in psoriasis patients who are treated with oral methoxsalen (psoralen) and UVA photochemotherapy is 100 times greater than in the general population, further supporting the role of UV light in the development of MCC.1 White individuals have the highest incidence of MCC worldwide, with men being affected more frequently than women.1,3 The majority of patients with MCC are diagnosed at 70 years or older.1 Approximately 5% of reported MCC patients are diagnosed before 50 years of age.2 Immunosuppression and immunodeficiency likely play a role in the pathogenesis of MCC, and the incidence is increased in solid organ transplant recipients, most commonly renal transplant recipients,1 as well as individuals with chronic lymphocytic leukemia, human immunodeficiency virus infection, and AIDS.1,3 Patients with autoimmune diseases such as rheumatoid arthritis also are at increased risk for MCC.3 Individuals who are diagnosed with MCC are at an increased risk for development of other malignancies including nonmelanoma skin cancers, chronic lymphocytic leukemia, Hodgkin lymphoma, and non-Hodgkin lymphoma.3
Clinical Presentation of MCC
The clinical presentation of MCC can be variable. Most tumors present as firm, red to purple, nontender papules or nodules (Figure 1).1 Tumor size may range from 2 to 200 mm but is most commonly less than 20 mm.2 Growth can be rapid, and tumors are most commonly located on sun-exposed skin. The head and neck areas account for 48% of all MCC cases,1 with the eyelids being frequently involved.2 Merkel cell carcinoma also has been reported on the arms, legs, trunk, back, and buttocks.1 Non–sun-exposed areas are less commonly affected. Mucosal sites (eg, larynx, nasal cavity, pharynx, mouth) account for 5% of primary MCCs.1 Merkel cell carcinoma also has been reported to affect the vulva and penis. Subcutaneous primary MCC has presented without overlying epidermal changes.1 In a case series by Heath et al,6 14% (27/195) of MCC patients presented with nodal disease without any identifiable primary tumor, with the inguinal nodal chain being the most common for this presentation. It currently is not known whether these nodal tumors are primary tumors or metastatic disease with a regression of the primary tumor.1
|
Histopathology of MCC
| ||
| ||
| ||
Figure 3. Positive chromogranin staining (A)(original | ||
Merkel cells are small- to medium-sized basophilic cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen on histopathology (Figure 2A).1 Some tumor cells have more vesicular chromatin, multiple small nucleoli, irregular contours, and more abundant cytoplasm. In some reports, irregular contours and abundant cytoplasm were associated with no detectible MCPyV infection.1,3 Merkel cell carcinomas have a primarily nodular architecture, and classification is based on growth pattern and cell size. Three histopathologic growth patterns have been described: nodular, infiltrative, and trabecular. The trabecular pattern is composed of interconnecting strands ofcells (Figure 2B). Tumors with solely intraepidermal involvement (MCC in situ) have been described but are exceedingly rare.1 Cell types are classified according to size, with the intermediate cell type being the most common. The small cell variant may be mistaken for a lymphocytic infiltrate due to the similar size and appearance of both types of cells.1,3
Merkel cell carcinomas can have histopathological overlap with lymphomas, small cell lung cancers, carcinoid tumors, primitive neuroectodermal tumors, neuroblastomas, small cell osteosarcomas, rhabdomyosarcomas, or Ewing sarcomas.1,3 Specifically, differentiation from small cell carcinoma of the lung is of utmost importance. Merkel cell carcinoma stains positively for cytokeratins 8, 18, 19, and 20. The neuroendocrine markers chromogranin (Figure 3A), synaptophysin, and neuron-specific enolase also may show positive staining. Cytokeratin 20, low-molecular-weight cytokeratins (CAM 5.2), and neurofilament immunostains have a high sensitivity for MCC and are the most frequently used.1 Cytokeratin 20 stains in the characteristic paranuclear dot–like pattern, which is a hallmark of MCC (Figure 3B). Cytokeratin 20 positivity in conjunction with negative staining for thyroid transcription factor 1 (Figure 3C) and cytokeratin 7 can aid in differentiation from small cell carcinoma of the lung.1,3
Pathogenesis of MCC
In 2008, Feng et al7 discovered a novel polyomavirus associated with the development of MCC. This novel polyomavirus, MCPyV, is found in approximately 80% of all cases of MCC. Seventeen members of the polyomavirus family have been identified, 9 of which have been found to infect humans, including BK virus, JC virus, WU, MCPyV, human polyomavirus 6, human polyomavirus 7, trichodysplasia spinulosa–associated polyomavirus, human polyomavirus 9, and Simian virus 40.1 Merkel cell polyomavirus infection is found in approximately 60% of the general population and exposure likely occurs early in life. The virus likely is transmitted through skin shedding and nasal secretions, though it also has been found in urine specimens.3 Currently, there is no evidence to suggest vertical viral transmission from mother to fetus.
Merkel cell polyomavirus is composed of early and late gene regions. The early gene region contains both large T antigen (LT) and small T antigen reading frames, which are necessary for viral replication.8 The late region is responsible for encoding viral proteins necessary for viral capsid assembly. Mutations found in viral protein 1 prevent formation of viral particles.9 Large T antigen is substantially overexpressed in MCC and is responsible for tumor suppression through retinoblastoma tumor suppressor protein. It also serves as a binding domain for both heat shock proteins and helicases.8,10 These domains allow the polyomaviruses to use host-cell machinery for viral genome replication while targeting tumor suppressor proteins.8 Upon viral integration into host DNA, viral replication ceases while oncogenic function persists.
The exact mechanism by which the MCPyV contributes to the development of MCC still has yet to be identified. Hypotheses suggest a combination of viral infection with external mutagens (eg, UV radiation). Experimental observations suggest viral contribution is likely due to the large percentage of MCCs that are positive for MCPyV, the identification of LT antigen expression and the role it plays in preserving cell cycle progression, and the role persistent LT antigen expression plays in continued growth of MCC cell lines in vitro.8 Two important cell line preservation mechanisms ensure continued tumor growth, including prevention of apoptosis triggered by DNA damage response mechanisms following UV damage and interaction with the retinoblastoma tumor suppressor protein allowing continued growth.8,11 Other important factors in tumor growth and survival may be the inhibition of apoptosis through the BCL2 (B-cell chronic lymphocytic leukemia/lymphoma 2) proto-oncogene and survivin (baculoviral inhibitor of apoptosis repeat-containing 5 [BIRC5]).12 Survivin has been found to play an important role in MCPyV-positive MCCs.12,13 It has been suggested that lymphangiogenesis in MCC likely is driven by vascular endothelial growth factor-C+CD68+CD163+ M2 macrophages.14 Another survival mechanism specific to polyomaviruses is their ability to interfere with the p53 tumor suppressor pathway.8 Loss of p53 expression by tumor cell nuclei has been associated with poor prognosis.15
Immune Response
Immune response as a role in tumor progression can be primarily centered on the concept of persistent antigen expression as a means of immune downregulation. Dunn et al16 suggested that cancer cells must interact through 3 consecutive phases with the host immune system (immunoediting hypothesis). In the elimination phase, the host immune system is able to recognize and destroy newly transformed cells through both the innate and adaptive immune systems. The second equilibrium phase allows the tumor to remain dormant and growth remains stagnant. Lastly, the tumor is allowed to evade the immune system through the escape phase.8
Host immune responses play an important role in both the progression and prognosis of MCC. High anti-MCPyV capsid antibody titers have been associated with better progression-free survival in some patients.8 Patients with high antibody titers (>10,000) likely have better progression-free survival than those with low antibody titers (<10,000).17 Antibody titers to the LT antigen may serve as a biomarker of MCC disease burden in the future. Rising LT antigen titers have been shown to correlate with disease progression and falling titers correlate with successful treatment.8 Tumoral infiltration of CD8+ T lymphocytes has been shown to be a predictor of survival compared to no intratumoral infiltration.6 Sihto et al18 suggested that this better prognosis from high intratumoral infiltration is not specific to MCPyV-positive MCC; however, it does highlight an important aspect of tumor evasion through the downregulation of cell surface expression of class I major histocompatibility complex antigens, which allows presentation of tumor intracellular peptides to CD8+ T lymphocytes.8 Upregulation of this specific immune response may play a role in the future treatment of MCC.
Staging and Prognosis
Due to the extremely aggressive nature of MCC, patients with local disease and tumors 2 cm or smaller in diameter have a 66% survival at 5 years.1,3 The 5-year survival rate for patients with local metastasis to regional lymph nodes ranges from 26% to 42%. Patients with distant metastasis have an 18% survival rate at 5 years.1,3 Data suggest that sentinel lymph node biopsy should be performed on all patients with MCC regardless of tumor size.1 There are no consensus guidelines to date regarding imaging for the staging of MCC patients. It is suggested that (18F)fluorodeoxyglucose positron emission tomography alone or in combination with computed tomography (CT) may be of value as a single whole-body diagnostic tool for accurate staging.10 It also has been suggested that (18F)fluorodeoxyglucose positron emission tomography and CT may offer more accurate staging than other screening modalities such as CT alone or magnetic resonance imaging.14,19
Treatment of MCC
Surgery remains the mainstay of treatment of MCC. Current National Comprehensive Cancer Network guidelines20 recommend 1- to 2-cm margins for wide local excision or treatment with Mohs micrographic surgery. Sentinel lymph node biopsy should be performed intraoperatively in patients undergoing wide local excision and preoperatively for patients undergoing Mohs micrographic surgery due to potential alterations in lymphatic drainage that may affect lymphoscintigraphy.1
Radiation may be used as primary or adjuvant therapy in patients with MCC. Radiation as primary therapy generally is reserved for patients who are not surgical candidates. It has been suggested that there was no difference in outcome in a small group of patients treated with radiation alone compared to patients who underwent surgery and radiation to the tumor bed.1 Current guidelines suggest a small group of patients may not require adjuvant therapy following adequate resection of some small tumors, and clinical observation may be appropriate.1,3 Chemotherapy may play a palliative role in patients with metastatic MCC. Merkel cell carcinoma has been shown to be chemosensitive but with a high recurrence rate.1 Because the immune system plays an important role in disease prognosis, having an intact immune system likely is paramount in the prevention of further disease progression.
Future Treatments of MCC
Future treatment of MCC may be focused on the viral etiology of most tumors and upregulation of the immune response, which may lead to the possibility of specifically interfering with virus-specific oncoproteins and stimulation of immune responses to virally infected tumor cells.8 The MCPyV large T antigen has been found to be overexpressed in some tumors and may serve as a specific target of therapy.10,21 Survivin, a key cell cycle protein encoded by LT antigen, may be an interesting target given its implication in other cancers.13 Other potential nonviral molecular target antigens include the oncoprotein H1P1 that interacts with c-KIT.8 Specific immunostimulatory cytokines that may be used to upregulate the immune response to tumoral cells may include IL-2, IL-12, IL-15, or IL-21. Therapeutic agents that may be studied in the future to target the immune exhaustion phenomenon associated with tumorigenesis include ipilimumab (cytotoxic T lymphocyte antigen 4 receptor-blocking agent) as well as programmed cell death 1 and programmed cell death 1 ligand 1 (PD-1/PD-L1).8 Neuroendocrine tumors including MCC tend to be highly vascular and express vascular endothelial growth factors and platelet-derived growth factors, which may be other potential therapeutic targets. It has been reported that approximately 95% of MCC patients have CD56+ tumors, and current clinical trials suggest a promising therapeutic response with the immunogen anti-CD56 monoclonal antibody.3
Conclusion
Merkel cell carcinoma is a rare aggressive neuroendocrine tumor that has been associated with a novel polyomavirus. Merkel cell carcinoma tends to affect elderly and immunocompromised patients as well as white individuals. Tumors are most often found in areas of high UV exposure and clinically on sun-exposed skin. Merkel cell polyomavirus is associated with approximately 80% of tumors, and tumorigenesis likely is caused by a number of sequential steps from viral integration into host DNA, mutagenic events, and specific immune responses. Currently there are no consensus guidelines for using imaging for staging of MCCs, but sentinel lymph node biopsy is recommended for all cases due to the aggressive nature of even smaller tumors. Surgery remains the mainstay of treatment, and radiation therapy may be used as a primary or adjuvant treatment. Chemotherapy usually is reserved for patients with metastatic disease purely for palliation. Future treatments of MCC likely will center on the viral etiology of MCC and upregulation of immune responses to virally infected tumor cells.
1. Han S, North J, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
2. Goessling W, McKee P, Mayer R. Merkel cell carcinoma. J Clin Oncol. 2002;20:588-598.
3. Donepudi S, DeConti R, Samlowski W. Recent advances in the understanding of the genetics, etiology, and treatment of merkel cell carcinoma. Semin Oncol. 2012;39:163-172.
4. Toker C. Trabecular carcinoma of the skin. Arch Dermatol. 1972;105:107-110.
5. De Wolff-Peeters C, Marien K, Mebis J, et al. A cutaneous APUDoma or Merkel cell tumor? a morphologically recognizable tumor with a biological and histological malignant aspect in contrast with its clinical behavior. Cancer. 1980;46:810-816.
6. Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375-381.
7. Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
8. Bhatia S, Afanasiev O, Nghiem P. Immunobiology of Merkel cell carcinoma: implications for immunotherapy of a polyomavirus-associated cancer. Curr Oncol Rep. 2011;13:488-497.
9. Amber K, McLeod M, Nouri K. The Merkel cell polyomavirus and its involvement in Merkel cell carcinoma. Dermatol Surg. 2013;39:232-238.
10. Erovic B, Al Habeeb A, Harris L, et al. Significant overexpression of the Merkel cell polyomavirus (MCPyV) large T antigen in Merkel cell carcinoma. Head Neck. 2013;35:184-189.
11. Demetriou S, Ona-Vu K, Sullivan E, et al. Defective DNA repair and cell cycle arrest in cells expressing Merkel cell polyomavirus T antigen. Int J Cancer. 2012;131:1818-1827.
12. Sahi H, Koljonen V, Kavola H, et al. Bcl-2 expression indicates better prognosis of Merkel cell carcinoma regardless of the presence of Merkel cell polyomavirus. Virchows Arch. 2012;461:553-559.
13. Arora R, Shuda M, Guastafierro A, et al. Survivin is a therapeutic target in Merkel cell carcinoma. Sci Transl Med. 2012;4:1-11.
14. Hawryluk E, O’Regan K, Sheehy N, et al. Positron emission tomography/computed tomography imaging in Merkel cell carcinoma: a study of 270 scans in 97 patients at the Dana-Farber/Brigham and Women’s Cancer Center. J Am Acad Dermatol. 2013;68:592-599.
15. Hall B, Pincus L, Yu S, et al. Immunohistochemical prognostication of Merkel cell carcinoma: p63 expression but not polyomavirus status correlates with outcome. J Cutan Pathol. 2012;39:911-917.
16. Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991-998.
17. Touze A, Le Bidre E, Laude H, et al. High levels of antibodies against Merkel cell polyomavirus identify a subset of patients with Merkel cell carcinoma with better clinical outcome. J Clin Oncol. 2011;29:1612-1619.
18. Sihto H, Bohling T, Kavola H, et al. Tumor infiltrating immune cells and outcome of Merkel cell carcinoma: a population-based study. Clin Cancer Res. 2012;18:2872-2881.
19. Colgan M, Tarantola T, Weaver A, et al. The predictive value of imaging studies in evaluating regional lymph node involvement in Merkel cell carcinoma. J Am Acad Dermatol. 2012;67:1250-1256.
20. NCCN Clinical Practice Guidelines in Oncology. National Comprehensive Cancer Network website. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#site. Accessed March 22, 2016.
21. Angermeyer S, Hesbacher S, Becker J, et al. Merkel cell polyomavirus-positive Merkel cell carcinoma cells do not require expression of the viral small T antigen. J Invest Dermatol. 2013;133:1-6.
1. Han S, North J, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
2. Goessling W, McKee P, Mayer R. Merkel cell carcinoma. J Clin Oncol. 2002;20:588-598.
3. Donepudi S, DeConti R, Samlowski W. Recent advances in the understanding of the genetics, etiology, and treatment of merkel cell carcinoma. Semin Oncol. 2012;39:163-172.
4. Toker C. Trabecular carcinoma of the skin. Arch Dermatol. 1972;105:107-110.
5. De Wolff-Peeters C, Marien K, Mebis J, et al. A cutaneous APUDoma or Merkel cell tumor? a morphologically recognizable tumor with a biological and histological malignant aspect in contrast with its clinical behavior. Cancer. 1980;46:810-816.
6. Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375-381.
7. Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
8. Bhatia S, Afanasiev O, Nghiem P. Immunobiology of Merkel cell carcinoma: implications for immunotherapy of a polyomavirus-associated cancer. Curr Oncol Rep. 2011;13:488-497.
9. Amber K, McLeod M, Nouri K. The Merkel cell polyomavirus and its involvement in Merkel cell carcinoma. Dermatol Surg. 2013;39:232-238.
10. Erovic B, Al Habeeb A, Harris L, et al. Significant overexpression of the Merkel cell polyomavirus (MCPyV) large T antigen in Merkel cell carcinoma. Head Neck. 2013;35:184-189.
11. Demetriou S, Ona-Vu K, Sullivan E, et al. Defective DNA repair and cell cycle arrest in cells expressing Merkel cell polyomavirus T antigen. Int J Cancer. 2012;131:1818-1827.
12. Sahi H, Koljonen V, Kavola H, et al. Bcl-2 expression indicates better prognosis of Merkel cell carcinoma regardless of the presence of Merkel cell polyomavirus. Virchows Arch. 2012;461:553-559.
13. Arora R, Shuda M, Guastafierro A, et al. Survivin is a therapeutic target in Merkel cell carcinoma. Sci Transl Med. 2012;4:1-11.
14. Hawryluk E, O’Regan K, Sheehy N, et al. Positron emission tomography/computed tomography imaging in Merkel cell carcinoma: a study of 270 scans in 97 patients at the Dana-Farber/Brigham and Women’s Cancer Center. J Am Acad Dermatol. 2013;68:592-599.
15. Hall B, Pincus L, Yu S, et al. Immunohistochemical prognostication of Merkel cell carcinoma: p63 expression but not polyomavirus status correlates with outcome. J Cutan Pathol. 2012;39:911-917.
16. Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991-998.
17. Touze A, Le Bidre E, Laude H, et al. High levels of antibodies against Merkel cell polyomavirus identify a subset of patients with Merkel cell carcinoma with better clinical outcome. J Clin Oncol. 2011;29:1612-1619.
18. Sihto H, Bohling T, Kavola H, et al. Tumor infiltrating immune cells and outcome of Merkel cell carcinoma: a population-based study. Clin Cancer Res. 2012;18:2872-2881.
19. Colgan M, Tarantola T, Weaver A, et al. The predictive value of imaging studies in evaluating regional lymph node involvement in Merkel cell carcinoma. J Am Acad Dermatol. 2012;67:1250-1256.
20. NCCN Clinical Practice Guidelines in Oncology. National Comprehensive Cancer Network website. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#site. Accessed March 22, 2016.
21. Angermeyer S, Hesbacher S, Becker J, et al. Merkel cell polyomavirus-positive Merkel cell carcinoma cells do not require expression of the viral small T antigen. J Invest Dermatol. 2013;133:1-6.
Practice Points
- Merkel cell carcinoma has been associated with a novel polyomavirus.
- Merkel cell carcinoma follows a very aggressive course and is most likely metastatic at diagnosis.