User login
Traumatic Ulcerative Granuloma With Stromal Eosinophilia: A Malignant-Appearing Benign Lesion
Traumatic ulcerative granuloma with stromal eosinophilia (TUGSE) is an uncommon, benign, self-limited condition that is restricted to the oral mucosa, most commonly seen in the fifth to seventh decades of life.1-3 The pathogenesis of TUGSE is unknown, but current theory suggests trauma is the instigating factor. The presence of CD30+ mononuclear cells within TUGSE raises the possibility of a CD30+ lymphoproliferative disorder in some cases.4 However, because CD30+ cells are not uncommon in other benign reactive processes, they may simply represent a reactive phenomenon.3
Traumatic ulcerative granuloma with stromal eosinophilia traverses multiple disciplines, including dermatology, oral surgery, dentistry, and pathology, resulting in a diverse nomenclature including traumatic granuloma of the tongue, traumatic eosinophilic granuloma of the oral mucosa, ulcerated granuloma eosinophilicum diutinum, and eosinophilic ulcer of the oral mucosa.1,4-6 It is important to differentiate eosinophilic granuloma of the oral mucosa from the eosinophilic granuloma that is associated with Langerhans cell histiocytosis. Although both may present with oral ulceration, Langerhans cell–associated eosinophilic granuloma typically develops from underlying bone, whereas eosinophilic granuloma of the oral mucosa (TUGSE) is described as nonosseous.7,8 Furthermore, the gingiva is the most common oral site in Langerhans cell–associated eosinophilic granuloma, whereas the tongue is most commonly involved in TUGSE.8 Shapiro and Juhlin9 clearly distinguished TUGSE from Langerhans cell–associated eosinophilic granuloma in 1970. Histologically, the 2 conditions are completely different.
When ulcerative granulomas develop in the pediatric population, usually in children younger than 2 years, it is termed Riga-Fede disease.10 These children were typically breastfeeding, suckling, or teething, suggesting trauma as a triggering event. In 1961, Hjorting-Hansen and Schmidt5 described 3 separate lesions similar to Riga-Fede disease in an adult patient. Subsequently, Riga-Fede disease was grouped under TUGSE.3
Histologically, TUGSE shows an ulcerated epithelium with a polymorphic inflammatory cell infiltrate that has a large predominance of eosinophils. The infiltrate affects the superficial and deep layers of the muscle tissue and penetrates into the salivary glands. Large atypical mononuclear cells with an ovoid and pale-appearing nucleus often are present. These cells may be mitotically active and stain positively for CD30.1,4,11 CD68+ macrophages, T lymphocytes, and factor XIIIa–positive dendritic cells commonly are present.12
Given the presence of large atypical CD30+ cells in many lesions, the possibility of a CD30+ lymphoproliferative disorder has been postulated by some authors. Indeed, lymphomatoid papulosis (LyP) has been documented to involve the oral mucosa.2,4
Case Report
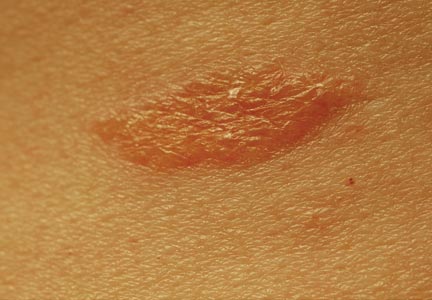
An 81-year-old man presented with a rapidly enlarging, 1.7×1.3-cm, vascular-appearing nodule with a collarette of mucosal epithelium on the left side of the dorsal surface of the tongue of 2 weeks’ duration (Figure 1). He denied any history of trauma, tobacco chewing, weight change, fever, or fatigue; however, he did report a 30 pack-year smoking history. There was no other pertinent medical history to include medications or allergies.

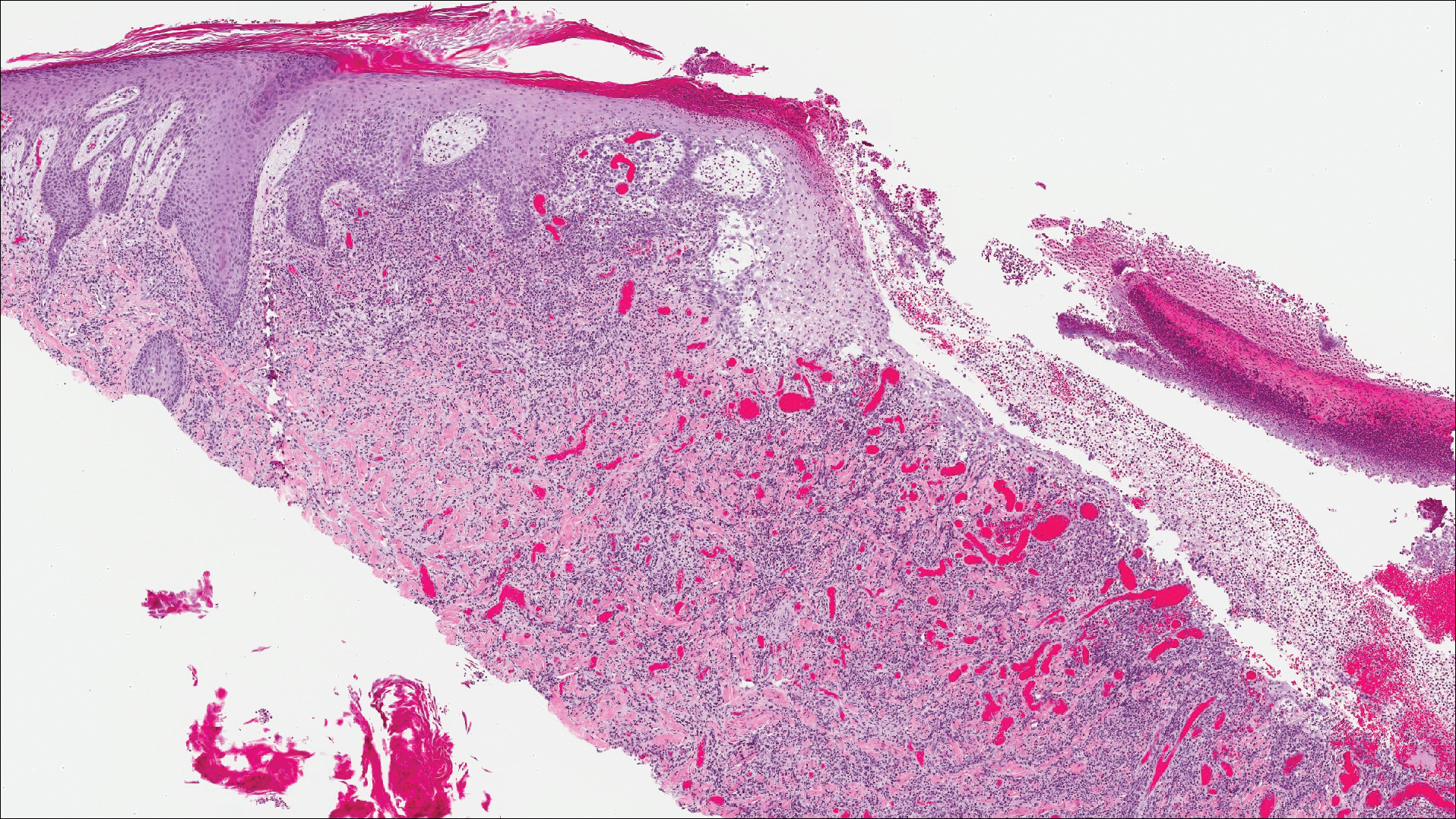
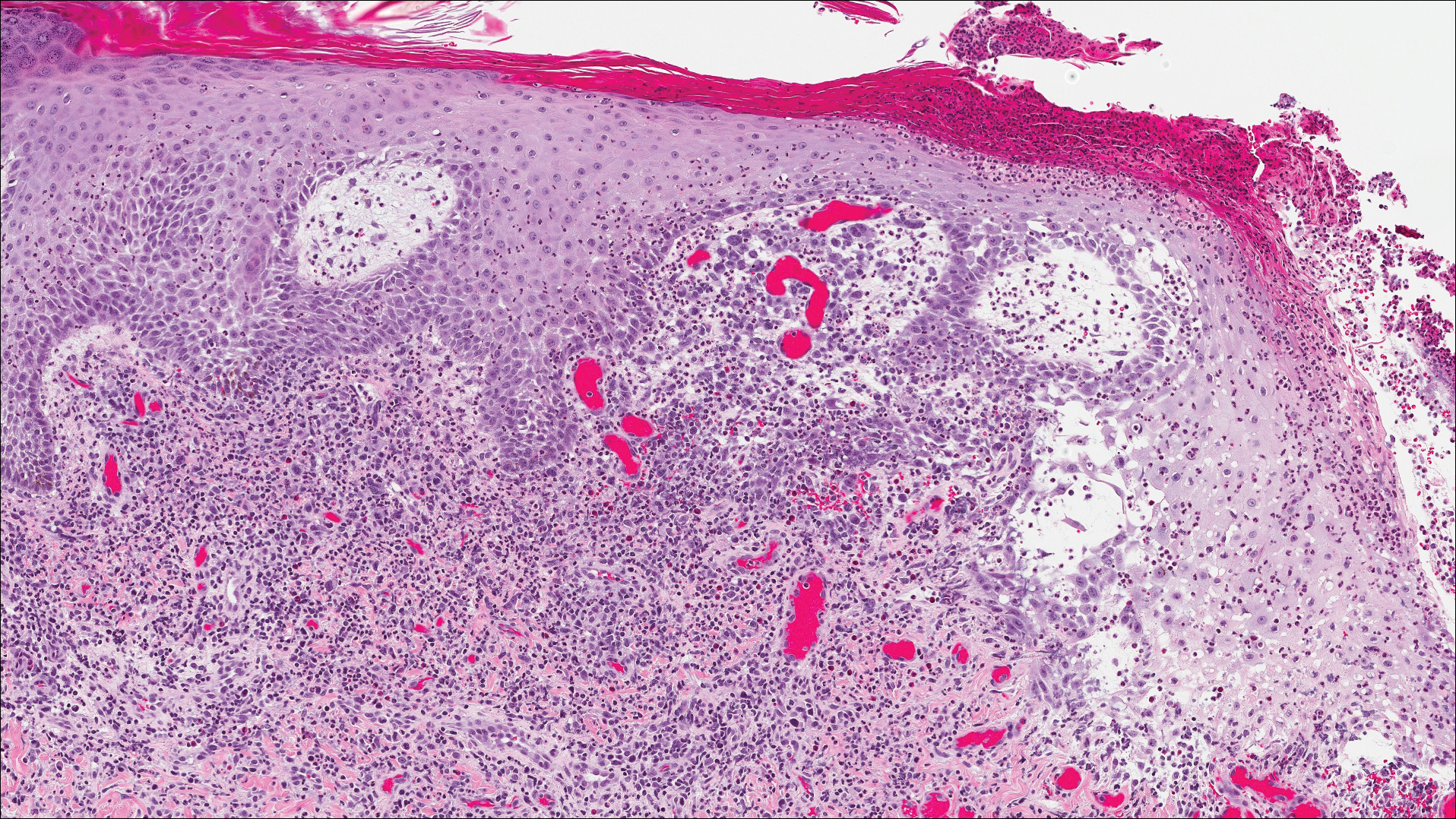
The differential diagnosis included pyogenic granuloma, granular cell tumor, squamous cell carcinoma, other neoplasms (eg, oral lymphoma, salivary gland tumors), and a traumatic blood blister from tongue biting. The patient was referred to the oral maxillofacial surgery department for an excisional biopsy, which showed a solitary ulcerated nodule with associated granulation tissue, thrombus, and fibrinoid debris (Figure 2). A surrounding dense mixed inflammatory cell infiltrate composed of lymphocytes, histiocytes, and numerous eosinophils was noted extending through the submucosal tissue and underlying striated muscle fibers (Figure 3). The adjacent mucosal epithelium appeared normal. CD30 staining showed only rare positive cells. These findings were consistent with TUGSE.


Due to the benign nature of TUGSE, the patient was released with symptomatic care and instructed to return for any new growth. The growth spontaneously resolved over 1 month and no recurrence or new lesions were reported 1 year later.
Comment
Despite encompassing multiple disciplines of medicine, TUGSE has minimal exposure in the dermatologic literature. It is an important clinical and histologic diagnosis that will provide reassurance to the patient when accurately identified and reduce potentially harmful treatments.
Clinical Presentation
Typically, TUGSE presents as a painful solitary nodule with a central ulcer and yellow fibrinous base. The margins of the ulcer typically have an indurated and rolled appearance.1,4 More than 50% of the lesions develop on the tongue, specifically the dorsal or lateral surfaces, but they may present anywhere in the oral mucosa.7 Traumatic ulcerative granuloma with stromal eosinophilia is a fast-growing lesion, typically developing in days to weeks. Although it spontaneously regresses, the lesion may take weeks or months to resolve. In one case, it resolved 1 year later.1 Traumatic ulcerative granuloma with stromal eosinophilia has a bimodal age distribution, generally appearing in the first 2 years of life and later in the fifth through seventh decades. The male-to-female predominance is equal.1,7,11 Reoccurrence is rare, but some reports have shown patients with multiple episodes of TUGSE.13,14
Differential Diagnosis
The clinical differential diagnosis for TUGSE includes squamous cell carcinoma, pyogenic granuloma, lymphoproliferative disorder, traumatic neuroma, Langerhans cell histiocytosis, granulomatous disorders, and oral lymphoma. Inflammatory disorders such as syphilis, Behçet’s disease, herpes, histoplasmosis, Wegener granulomatosis, and others also should be considered.
Immunohistochemistry
Immunohistochemical analysis of TUGSE lesions recently has revealed the presence of CD30+ cells. These cells are associated with cutaneous lymphoproliferative disorders including LyP, anaplastic large cell lymphoma (ALCL), and borderline CD30+ lesions, among others. Systemic diseases with CD30+ cells include mycosis fungoides, other T-cell lymphomas, and Hodgkin lymphoma.15,16 Once CD30+ cells were recognized, multiple authors began speculating there was a correlation between TUGSE and the CD30+ lymphoproliferative disorders.1,2,13 Anaplastic large cell lymphoma and LyP of the oral mucosa have been reported in several cases.17-20 One report described 2 cases of ulcerated CD30+ T-cell non-Hodgkin lymphoma of the oral mucosa, one of which showed eosinophilic infiltrates and was initially thought to be TUGSE. Based on these overlapping clinical and histologic features, the authors hypothesized there was a correlation between oral ALCL, LyP, and TUGSE.17 In one report, a patient developed multiple TUGSE lesions throughout his life, suggesting a pathologic process similar to LyP. The lesion biopsied showed that 70% of the T cells expressed CD30 (Ki-1) antigen.13
Underlying Causes
In support of an underlying immunologic process that augments the growth of these lesions, 2 separate case reports of TUGSE in the presence of human T-lymphotropic virus 1 (HTLV-1) and Epstein-Barr virus have been documented.2,21 Concurrent presentation of TUGSE and HTLV-1 in one report demonstrated eosinophilia in both the oral lesion and peripheral blood, suggesting an immunologic relationship. Furthermore, the authors postulated that local trauma initiated the development of TUGSE, providing the catalyst for the HTLV-1 carrier to develop peripheral eosinophilia.21
In the second case, a 12-year-old boy developed TUGSE in the presence of Epstein-Barr virus.2 Immunologically, this virus can be reactivated from its latent stage during immunosuppression. Epstein-Barr virus has been implicated in lymphoproliferative diseases of both B- and T-cell origin, including CD30+ ALCL and LyP.22,23 The authors in this report again hypothesized there was a correlation between lymphoproliferative disorders and TUGSE lesions.2,24
Alternatively, TUGSE may simply be a reactive process to trauma or another underlying trigger. It has been speculated that the presence of eosinophils correlates with antigen insertion into the oral mucosa, whereas other ulcers of the oral mucosa are devoid of eosinophils.1 These antigens may include microorganisms, endogenous degradation products, or foreign proteins.7,25 Additionally, the presence of CD30+ lymphocytes is not isolated to lymphoproliferative disorders. CD30+ cells have been documented in arthropod bite reactions, atopic dermatitis, drug reactions, molluscum contagiosum, and scabies, among others.1,26
Healing and Management
The length of healing in TUGSE ulcers has substantial variability, from days to up to 1 year in an isolated case.1,24 Sequential expression of transforming growth factor (TGF) α and TGF-β expressed by tissue eosinophils may be underlying factors associated with a quicker healing response as demonstrated by similar ulcers in hamsters.27 Chronic nonhealing oral ulcers, particularly TUGSE lesions that demonstrated the typical increase in eosinophils in 11 of 12 cases, showed minimal TGF-α or TGF-β expression by eosinophils, perhaps indicating a possible mechanism leading to delayed wound healing in some cases. Interestingly, incisional biopsies often led to rapid wound healing, suggesting that the biopsy itself allowed for a transition back to the regular wound-healing processes.28
Traumatic ulcerative granuloma with stromal eosinophilia spontaneously resolves on its own in most cases; however, because of the concern for malignancy, it has the potential to be overtreated.26 Symptomatic treatment only is the mainstay of therapy. The patient should be instructed to avoid trauma, and referral to a dental professional is indicated when associated with dentures or other periprosthetic devices. Diet should consist of soft foods while avoiding spicy foods. Topical or oral analgesics may be necessary if substantial pain is associated with the lesion.2 Oral prednisolone was used in a patient with concurrent HTLV-1 and TUGSE to treat peripheral eosinophilia.21 The patient’s peripheral eosinophils dropped to 1% in 1 day, and the patient’s oral lesion began to improve at day 3 and disappeared by day 10. Although TUGSE may spontaneously resolve within a 10-day period without steroids, it may be a reasonable treatment to improve healing time in an otherwise healthy individual.21,26 If there is concern for malignancy, the patient should have the lesion biopsied to provide reassurance and for the added benefit of a transition to normal healing response and decreased healing time.28
Clinical Recognition
The clinician should be aware of the possibility of a CD30+ lymphoproliferative disorder, which has been associated with TUGSE in some cases, or may simulate TUGSE both clinically and histologically. Further studies are needed to clarify the relationship between these 2 entities. Whether it is a true relationship, simple coincidence, or simply overlapping clinical and histologic features remains to be determined.
- Hirshberg A, Amariglio N, Akrish S, et al. Traumatic ulcerative granuloma with stromal eosinophilia: reactive lesion of the oral mucosa. Am J Clin Pathol. 2006;126:522-529.
- Abdel-Naser MB, Tsatsou F, Hippe S, et al. Oral eosinophilic ulcer, an Epstein-Barr virus-associated CD30+ lymphoproliferation? [published online April 5, 2011]. Dermatology. 2011;222:113-118.
- Fonseca FP, Benevenuto de Andrade BA, Coletta RD, et al. Clinicopathological and immunohistochemical analysis of 19 cases of oral eosinophilic ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;115:532-540.
- Alobeid B, Pan LX, Milligan L, et al. Eosinophil-rich CD30+ lymphoproliferative disorder of the oral mucosa. Am J Clin Pathol. 2004;121:43-50.
- Hjorting-Hansen E, Schmidt H. Ulcerated granuloma eosinophilicum diutinum of the tongue. report of a case. Acta Derm Venereol. 1961;41:235-239.
- Velez A, Alamillos FJ, Dean A, et al. Eosinophilic ulcer of the oral mucosa: report of a recurrent case on the tongue. Clin Exp Dermatol. 1997;22:154-156.
- Elzay RP. Traumatic ulcerative granuloma with stromal eosinophilia (Riga-Fede’s disease and traumatic eosinophilic granuloma). Oral Surg Oral Med Oral Pathol. 1983;55:497-506.
- Val-Bernal JF, Gonzalez-Vela MC, Sanchez-Santolino S, et al. Localized eosinophilic (Langerhans’ cell) granuloma of the lower lip. a lesion that may cause diagnostic error. J Cutan Pathol. 2009;36:1109-1113.
- Shapiro L, Juhlin EA. Eosinophilic ulcer of the tongue report of two cases and review of the literature. Dermatologica. 1970;140:242-250.
- Amberg S. Sublingual growth in infants. Am J Med Sci. 1902;126:257-269.
- EI-Mofty SK, Swanson PE, Wick MR, et al. Eosinophilic ulcer of the oral mucosa: report of 38 new cases with immunohistochemical observations. Oral Surg Oral Med Oral Pathol. 1993;75:716-722.
- Regezi JA, Zarbo RJ, Daniels TE, et al. Oral traumatic granuloma: characterization of the cellular infiltrate. Oral Surg Oral Med Oral Pathol. 1993;75:723-727.
- Ficarra G, Prignano F, Romagnoli P. Traumatic eosinophilic granuloma of the oral mucosa: a CD30+ (Ki-1) lymphoproliferative disorder? Oral Oncol. 1997;33:375-379.
- Doyle JL, Geary W, Baden E. Eosinophilic ulcer. J Oral Maxillofac Surg. 1989;47:349-352.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Stein H, Mason DY, Gerdes J, et al. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood. 1985;66:848-858.
- Rosenberg A, Biesma DH, Sie-Go DMDS, et al. Primary extranodal CD30-positive T-cell non-Hodgkin’s lymphoma of the oral mucosa. report of two cases. Int J Oral Maxillofac Surg. 1996;25:57-59.
- Kato N, Tomita Y, Yoshida K, et al. Involvement of the tongue by lymphomatoid papulosis. Am J Dermatopathol. 1998;20:522-526.
- Savarrio L, Gibson J, Dunlop DJ, et al. Spontaneous regression of an anaplastic large cell lymphoma in the oral cavity: first reported case and review of the literature. Oral Oncol. 1999;35:609-613.
- Sciubba J, Said-Al-Naief N, Fantasia J. Critical review of lymphomatoid papulosis of the oral cavity with case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:195-204.
- Yamazaki H, Shirasugi Y, Kajiwara H, et al. Concurrent onset of eosinophilic ulcer of the oral mucosa with peripheral eosinophilia in a human T-cell leukemia virus type I carrier. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;114:E43-E48.
- Dojcinov SD, Venkataram G, Raffeld M, et al. EBV positive mucocutaneous ulcer—a study of 26 cases associated with various sources of immunosuppression. Am J Surg Pathol. 2010;34:405-417.
- Kim YC, Yang WI, Lee MG, et al. Epstein-Barr virus in CD30 anaplastic large cell lymphoma involving the skin and lymphomatoid papulosis in South Korea. Int J Dermatol. 2006;45:1312-1316.
- Pietersma F, Piriou E, van Baarle D. Immune surveillance of EBV-infected B cells and the development of non-Hodgkin lymphomas in immunocompromised patients. Leuk Lymphoma. 2008;49:1028-1041.
- Salisbury CL, Budnick SD, Li S. T cell receptor gene rearrangement and CD 30 immunoreactivity in traumatic ulcerative granuloma with stromal eosinophilia of oral cavity. Am J Clin Pathol. 2009;132:722-727.
- Marszalek A, Neska-Dlugosz I. Traumatic ulcerative granuloma with stromal eosinophilia. a case report and short literature review. Pol J Pathol. 2011;3:172-175.
- Wong DT, Donoff RB, Yang J, et al. Sequential expression of transforming growth factors alpha and beta 1 by eosinophils during cutaneous wound healing in the hamster. Am J Pathol. 1993;143:130-142.
- Elovic AE, Gallagher GT, Kabani S, et al. Lack of TGF-alpha and TGF-beta synthesis by human eosinophils in chronic oral ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:672-681.
Traumatic ulcerative granuloma with stromal eosinophilia (TUGSE) is an uncommon, benign, self-limited condition that is restricted to the oral mucosa, most commonly seen in the fifth to seventh decades of life.1-3 The pathogenesis of TUGSE is unknown, but current theory suggests trauma is the instigating factor. The presence of CD30+ mononuclear cells within TUGSE raises the possibility of a CD30+ lymphoproliferative disorder in some cases.4 However, because CD30+ cells are not uncommon in other benign reactive processes, they may simply represent a reactive phenomenon.3
Traumatic ulcerative granuloma with stromal eosinophilia traverses multiple disciplines, including dermatology, oral surgery, dentistry, and pathology, resulting in a diverse nomenclature including traumatic granuloma of the tongue, traumatic eosinophilic granuloma of the oral mucosa, ulcerated granuloma eosinophilicum diutinum, and eosinophilic ulcer of the oral mucosa.1,4-6 It is important to differentiate eosinophilic granuloma of the oral mucosa from the eosinophilic granuloma that is associated with Langerhans cell histiocytosis. Although both may present with oral ulceration, Langerhans cell–associated eosinophilic granuloma typically develops from underlying bone, whereas eosinophilic granuloma of the oral mucosa (TUGSE) is described as nonosseous.7,8 Furthermore, the gingiva is the most common oral site in Langerhans cell–associated eosinophilic granuloma, whereas the tongue is most commonly involved in TUGSE.8 Shapiro and Juhlin9 clearly distinguished TUGSE from Langerhans cell–associated eosinophilic granuloma in 1970. Histologically, the 2 conditions are completely different.
When ulcerative granulomas develop in the pediatric population, usually in children younger than 2 years, it is termed Riga-Fede disease.10 These children were typically breastfeeding, suckling, or teething, suggesting trauma as a triggering event. In 1961, Hjorting-Hansen and Schmidt5 described 3 separate lesions similar to Riga-Fede disease in an adult patient. Subsequently, Riga-Fede disease was grouped under TUGSE.3
Histologically, TUGSE shows an ulcerated epithelium with a polymorphic inflammatory cell infiltrate that has a large predominance of eosinophils. The infiltrate affects the superficial and deep layers of the muscle tissue and penetrates into the salivary glands. Large atypical mononuclear cells with an ovoid and pale-appearing nucleus often are present. These cells may be mitotically active and stain positively for CD30.1,4,11 CD68+ macrophages, T lymphocytes, and factor XIIIa–positive dendritic cells commonly are present.12
Given the presence of large atypical CD30+ cells in many lesions, the possibility of a CD30+ lymphoproliferative disorder has been postulated by some authors. Indeed, lymphomatoid papulosis (LyP) has been documented to involve the oral mucosa.2,4
Case Report
An 81-year-old man presented with a rapidly enlarging, 1.7×1.3-cm, vascular-appearing nodule with a collarette of mucosal epithelium on the left side of the dorsal surface of the tongue of 2 weeks’ duration (Figure 1). He denied any history of trauma, tobacco chewing, weight change, fever, or fatigue; however, he did report a 30 pack-year smoking history. There was no other pertinent medical history to include medications or allergies.
The differential diagnosis included pyogenic granuloma, granular cell tumor, squamous cell carcinoma, other neoplasms (eg, oral lymphoma, salivary gland tumors), and a traumatic blood blister from tongue biting. The patient was referred to the oral maxillofacial surgery department for an excisional biopsy, which showed a solitary ulcerated nodule with associated granulation tissue, thrombus, and fibrinoid debris (Figure 2). A surrounding dense mixed inflammatory cell infiltrate composed of lymphocytes, histiocytes, and numerous eosinophils was noted extending through the submucosal tissue and underlying striated muscle fibers (Figure 3). The adjacent mucosal epithelium appeared normal. CD30 staining showed only rare positive cells. These findings were consistent with TUGSE.
Due to the benign nature of TUGSE, the patient was released with symptomatic care and instructed to return for any new growth. The growth spontaneously resolved over 1 month and no recurrence or new lesions were reported 1 year later.
Comment
Despite encompassing multiple disciplines of medicine, TUGSE has minimal exposure in the dermatologic literature. It is an important clinical and histologic diagnosis that will provide reassurance to the patient when accurately identified and reduce potentially harmful treatments.
Clinical Presentation
Typically, TUGSE presents as a painful solitary nodule with a central ulcer and yellow fibrinous base. The margins of the ulcer typically have an indurated and rolled appearance.1,4 More than 50% of the lesions develop on the tongue, specifically the dorsal or lateral surfaces, but they may present anywhere in the oral mucosa.7 Traumatic ulcerative granuloma with stromal eosinophilia is a fast-growing lesion, typically developing in days to weeks. Although it spontaneously regresses, the lesion may take weeks or months to resolve. In one case, it resolved 1 year later.1 Traumatic ulcerative granuloma with stromal eosinophilia has a bimodal age distribution, generally appearing in the first 2 years of life and later in the fifth through seventh decades. The male-to-female predominance is equal.1,7,11 Reoccurrence is rare, but some reports have shown patients with multiple episodes of TUGSE.13,14
Differential Diagnosis
The clinical differential diagnosis for TUGSE includes squamous cell carcinoma, pyogenic granuloma, lymphoproliferative disorder, traumatic neuroma, Langerhans cell histiocytosis, granulomatous disorders, and oral lymphoma. Inflammatory disorders such as syphilis, Behçet’s disease, herpes, histoplasmosis, Wegener granulomatosis, and others also should be considered.
Immunohistochemistry
Immunohistochemical analysis of TUGSE lesions recently has revealed the presence of CD30+ cells. These cells are associated with cutaneous lymphoproliferative disorders including LyP, anaplastic large cell lymphoma (ALCL), and borderline CD30+ lesions, among others. Systemic diseases with CD30+ cells include mycosis fungoides, other T-cell lymphomas, and Hodgkin lymphoma.15,16 Once CD30+ cells were recognized, multiple authors began speculating there was a correlation between TUGSE and the CD30+ lymphoproliferative disorders.1,2,13 Anaplastic large cell lymphoma and LyP of the oral mucosa have been reported in several cases.17-20 One report described 2 cases of ulcerated CD30+ T-cell non-Hodgkin lymphoma of the oral mucosa, one of which showed eosinophilic infiltrates and was initially thought to be TUGSE. Based on these overlapping clinical and histologic features, the authors hypothesized there was a correlation between oral ALCL, LyP, and TUGSE.17 In one report, a patient developed multiple TUGSE lesions throughout his life, suggesting a pathologic process similar to LyP. The lesion biopsied showed that 70% of the T cells expressed CD30 (Ki-1) antigen.13
Underlying Causes
In support of an underlying immunologic process that augments the growth of these lesions, 2 separate case reports of TUGSE in the presence of human T-lymphotropic virus 1 (HTLV-1) and Epstein-Barr virus have been documented.2,21 Concurrent presentation of TUGSE and HTLV-1 in one report demonstrated eosinophilia in both the oral lesion and peripheral blood, suggesting an immunologic relationship. Furthermore, the authors postulated that local trauma initiated the development of TUGSE, providing the catalyst for the HTLV-1 carrier to develop peripheral eosinophilia.21
In the second case, a 12-year-old boy developed TUGSE in the presence of Epstein-Barr virus.2 Immunologically, this virus can be reactivated from its latent stage during immunosuppression. Epstein-Barr virus has been implicated in lymphoproliferative diseases of both B- and T-cell origin, including CD30+ ALCL and LyP.22,23 The authors in this report again hypothesized there was a correlation between lymphoproliferative disorders and TUGSE lesions.2,24
Alternatively, TUGSE may simply be a reactive process to trauma or another underlying trigger. It has been speculated that the presence of eosinophils correlates with antigen insertion into the oral mucosa, whereas other ulcers of the oral mucosa are devoid of eosinophils.1 These antigens may include microorganisms, endogenous degradation products, or foreign proteins.7,25 Additionally, the presence of CD30+ lymphocytes is not isolated to lymphoproliferative disorders. CD30+ cells have been documented in arthropod bite reactions, atopic dermatitis, drug reactions, molluscum contagiosum, and scabies, among others.1,26
Healing and Management
The length of healing in TUGSE ulcers has substantial variability, from days to up to 1 year in an isolated case.1,24 Sequential expression of transforming growth factor (TGF) α and TGF-β expressed by tissue eosinophils may be underlying factors associated with a quicker healing response as demonstrated by similar ulcers in hamsters.27 Chronic nonhealing oral ulcers, particularly TUGSE lesions that demonstrated the typical increase in eosinophils in 11 of 12 cases, showed minimal TGF-α or TGF-β expression by eosinophils, perhaps indicating a possible mechanism leading to delayed wound healing in some cases. Interestingly, incisional biopsies often led to rapid wound healing, suggesting that the biopsy itself allowed for a transition back to the regular wound-healing processes.28
Traumatic ulcerative granuloma with stromal eosinophilia spontaneously resolves on its own in most cases; however, because of the concern for malignancy, it has the potential to be overtreated.26 Symptomatic treatment only is the mainstay of therapy. The patient should be instructed to avoid trauma, and referral to a dental professional is indicated when associated with dentures or other periprosthetic devices. Diet should consist of soft foods while avoiding spicy foods. Topical or oral analgesics may be necessary if substantial pain is associated with the lesion.2 Oral prednisolone was used in a patient with concurrent HTLV-1 and TUGSE to treat peripheral eosinophilia.21 The patient’s peripheral eosinophils dropped to 1% in 1 day, and the patient’s oral lesion began to improve at day 3 and disappeared by day 10. Although TUGSE may spontaneously resolve within a 10-day period without steroids, it may be a reasonable treatment to improve healing time in an otherwise healthy individual.21,26 If there is concern for malignancy, the patient should have the lesion biopsied to provide reassurance and for the added benefit of a transition to normal healing response and decreased healing time.28
Clinical Recognition
The clinician should be aware of the possibility of a CD30+ lymphoproliferative disorder, which has been associated with TUGSE in some cases, or may simulate TUGSE both clinically and histologically. Further studies are needed to clarify the relationship between these 2 entities. Whether it is a true relationship, simple coincidence, or simply overlapping clinical and histologic features remains to be determined.
Traumatic ulcerative granuloma with stromal eosinophilia (TUGSE) is an uncommon, benign, self-limited condition that is restricted to the oral mucosa, most commonly seen in the fifth to seventh decades of life.1-3 The pathogenesis of TUGSE is unknown, but current theory suggests trauma is the instigating factor. The presence of CD30+ mononuclear cells within TUGSE raises the possibility of a CD30+ lymphoproliferative disorder in some cases.4 However, because CD30+ cells are not uncommon in other benign reactive processes, they may simply represent a reactive phenomenon.3
Traumatic ulcerative granuloma with stromal eosinophilia traverses multiple disciplines, including dermatology, oral surgery, dentistry, and pathology, resulting in a diverse nomenclature including traumatic granuloma of the tongue, traumatic eosinophilic granuloma of the oral mucosa, ulcerated granuloma eosinophilicum diutinum, and eosinophilic ulcer of the oral mucosa.1,4-6 It is important to differentiate eosinophilic granuloma of the oral mucosa from the eosinophilic granuloma that is associated with Langerhans cell histiocytosis. Although both may present with oral ulceration, Langerhans cell–associated eosinophilic granuloma typically develops from underlying bone, whereas eosinophilic granuloma of the oral mucosa (TUGSE) is described as nonosseous.7,8 Furthermore, the gingiva is the most common oral site in Langerhans cell–associated eosinophilic granuloma, whereas the tongue is most commonly involved in TUGSE.8 Shapiro and Juhlin9 clearly distinguished TUGSE from Langerhans cell–associated eosinophilic granuloma in 1970. Histologically, the 2 conditions are completely different.
When ulcerative granulomas develop in the pediatric population, usually in children younger than 2 years, it is termed Riga-Fede disease.10 These children were typically breastfeeding, suckling, or teething, suggesting trauma as a triggering event. In 1961, Hjorting-Hansen and Schmidt5 described 3 separate lesions similar to Riga-Fede disease in an adult patient. Subsequently, Riga-Fede disease was grouped under TUGSE.3
Histologically, TUGSE shows an ulcerated epithelium with a polymorphic inflammatory cell infiltrate that has a large predominance of eosinophils. The infiltrate affects the superficial and deep layers of the muscle tissue and penetrates into the salivary glands. Large atypical mononuclear cells with an ovoid and pale-appearing nucleus often are present. These cells may be mitotically active and stain positively for CD30.1,4,11 CD68+ macrophages, T lymphocytes, and factor XIIIa–positive dendritic cells commonly are present.12
Given the presence of large atypical CD30+ cells in many lesions, the possibility of a CD30+ lymphoproliferative disorder has been postulated by some authors. Indeed, lymphomatoid papulosis (LyP) has been documented to involve the oral mucosa.2,4
Case Report
An 81-year-old man presented with a rapidly enlarging, 1.7×1.3-cm, vascular-appearing nodule with a collarette of mucosal epithelium on the left side of the dorsal surface of the tongue of 2 weeks’ duration (Figure 1). He denied any history of trauma, tobacco chewing, weight change, fever, or fatigue; however, he did report a 30 pack-year smoking history. There was no other pertinent medical history to include medications or allergies.
The differential diagnosis included pyogenic granuloma, granular cell tumor, squamous cell carcinoma, other neoplasms (eg, oral lymphoma, salivary gland tumors), and a traumatic blood blister from tongue biting. The patient was referred to the oral maxillofacial surgery department for an excisional biopsy, which showed a solitary ulcerated nodule with associated granulation tissue, thrombus, and fibrinoid debris (Figure 2). A surrounding dense mixed inflammatory cell infiltrate composed of lymphocytes, histiocytes, and numerous eosinophils was noted extending through the submucosal tissue and underlying striated muscle fibers (Figure 3). The adjacent mucosal epithelium appeared normal. CD30 staining showed only rare positive cells. These findings were consistent with TUGSE.
Due to the benign nature of TUGSE, the patient was released with symptomatic care and instructed to return for any new growth. The growth spontaneously resolved over 1 month and no recurrence or new lesions were reported 1 year later.
Comment
Despite encompassing multiple disciplines of medicine, TUGSE has minimal exposure in the dermatologic literature. It is an important clinical and histologic diagnosis that will provide reassurance to the patient when accurately identified and reduce potentially harmful treatments.
Clinical Presentation
Typically, TUGSE presents as a painful solitary nodule with a central ulcer and yellow fibrinous base. The margins of the ulcer typically have an indurated and rolled appearance.1,4 More than 50% of the lesions develop on the tongue, specifically the dorsal or lateral surfaces, but they may present anywhere in the oral mucosa.7 Traumatic ulcerative granuloma with stromal eosinophilia is a fast-growing lesion, typically developing in days to weeks. Although it spontaneously regresses, the lesion may take weeks or months to resolve. In one case, it resolved 1 year later.1 Traumatic ulcerative granuloma with stromal eosinophilia has a bimodal age distribution, generally appearing in the first 2 years of life and later in the fifth through seventh decades. The male-to-female predominance is equal.1,7,11 Reoccurrence is rare, but some reports have shown patients with multiple episodes of TUGSE.13,14
Differential Diagnosis
The clinical differential diagnosis for TUGSE includes squamous cell carcinoma, pyogenic granuloma, lymphoproliferative disorder, traumatic neuroma, Langerhans cell histiocytosis, granulomatous disorders, and oral lymphoma. Inflammatory disorders such as syphilis, Behçet’s disease, herpes, histoplasmosis, Wegener granulomatosis, and others also should be considered.
Immunohistochemistry
Immunohistochemical analysis of TUGSE lesions recently has revealed the presence of CD30+ cells. These cells are associated with cutaneous lymphoproliferative disorders including LyP, anaplastic large cell lymphoma (ALCL), and borderline CD30+ lesions, among others. Systemic diseases with CD30+ cells include mycosis fungoides, other T-cell lymphomas, and Hodgkin lymphoma.15,16 Once CD30+ cells were recognized, multiple authors began speculating there was a correlation between TUGSE and the CD30+ lymphoproliferative disorders.1,2,13 Anaplastic large cell lymphoma and LyP of the oral mucosa have been reported in several cases.17-20 One report described 2 cases of ulcerated CD30+ T-cell non-Hodgkin lymphoma of the oral mucosa, one of which showed eosinophilic infiltrates and was initially thought to be TUGSE. Based on these overlapping clinical and histologic features, the authors hypothesized there was a correlation between oral ALCL, LyP, and TUGSE.17 In one report, a patient developed multiple TUGSE lesions throughout his life, suggesting a pathologic process similar to LyP. The lesion biopsied showed that 70% of the T cells expressed CD30 (Ki-1) antigen.13
Underlying Causes
In support of an underlying immunologic process that augments the growth of these lesions, 2 separate case reports of TUGSE in the presence of human T-lymphotropic virus 1 (HTLV-1) and Epstein-Barr virus have been documented.2,21 Concurrent presentation of TUGSE and HTLV-1 in one report demonstrated eosinophilia in both the oral lesion and peripheral blood, suggesting an immunologic relationship. Furthermore, the authors postulated that local trauma initiated the development of TUGSE, providing the catalyst for the HTLV-1 carrier to develop peripheral eosinophilia.21
In the second case, a 12-year-old boy developed TUGSE in the presence of Epstein-Barr virus.2 Immunologically, this virus can be reactivated from its latent stage during immunosuppression. Epstein-Barr virus has been implicated in lymphoproliferative diseases of both B- and T-cell origin, including CD30+ ALCL and LyP.22,23 The authors in this report again hypothesized there was a correlation between lymphoproliferative disorders and TUGSE lesions.2,24
Alternatively, TUGSE may simply be a reactive process to trauma or another underlying trigger. It has been speculated that the presence of eosinophils correlates with antigen insertion into the oral mucosa, whereas other ulcers of the oral mucosa are devoid of eosinophils.1 These antigens may include microorganisms, endogenous degradation products, or foreign proteins.7,25 Additionally, the presence of CD30+ lymphocytes is not isolated to lymphoproliferative disorders. CD30+ cells have been documented in arthropod bite reactions, atopic dermatitis, drug reactions, molluscum contagiosum, and scabies, among others.1,26
Healing and Management
The length of healing in TUGSE ulcers has substantial variability, from days to up to 1 year in an isolated case.1,24 Sequential expression of transforming growth factor (TGF) α and TGF-β expressed by tissue eosinophils may be underlying factors associated with a quicker healing response as demonstrated by similar ulcers in hamsters.27 Chronic nonhealing oral ulcers, particularly TUGSE lesions that demonstrated the typical increase in eosinophils in 11 of 12 cases, showed minimal TGF-α or TGF-β expression by eosinophils, perhaps indicating a possible mechanism leading to delayed wound healing in some cases. Interestingly, incisional biopsies often led to rapid wound healing, suggesting that the biopsy itself allowed for a transition back to the regular wound-healing processes.28
Traumatic ulcerative granuloma with stromal eosinophilia spontaneously resolves on its own in most cases; however, because of the concern for malignancy, it has the potential to be overtreated.26 Symptomatic treatment only is the mainstay of therapy. The patient should be instructed to avoid trauma, and referral to a dental professional is indicated when associated with dentures or other periprosthetic devices. Diet should consist of soft foods while avoiding spicy foods. Topical or oral analgesics may be necessary if substantial pain is associated with the lesion.2 Oral prednisolone was used in a patient with concurrent HTLV-1 and TUGSE to treat peripheral eosinophilia.21 The patient’s peripheral eosinophils dropped to 1% in 1 day, and the patient’s oral lesion began to improve at day 3 and disappeared by day 10. Although TUGSE may spontaneously resolve within a 10-day period without steroids, it may be a reasonable treatment to improve healing time in an otherwise healthy individual.21,26 If there is concern for malignancy, the patient should have the lesion biopsied to provide reassurance and for the added benefit of a transition to normal healing response and decreased healing time.28
Clinical Recognition
The clinician should be aware of the possibility of a CD30+ lymphoproliferative disorder, which has been associated with TUGSE in some cases, or may simulate TUGSE both clinically and histologically. Further studies are needed to clarify the relationship between these 2 entities. Whether it is a true relationship, simple coincidence, or simply overlapping clinical and histologic features remains to be determined.
- Hirshberg A, Amariglio N, Akrish S, et al. Traumatic ulcerative granuloma with stromal eosinophilia: reactive lesion of the oral mucosa. Am J Clin Pathol. 2006;126:522-529.
- Abdel-Naser MB, Tsatsou F, Hippe S, et al. Oral eosinophilic ulcer, an Epstein-Barr virus-associated CD30+ lymphoproliferation? [published online April 5, 2011]. Dermatology. 2011;222:113-118.
- Fonseca FP, Benevenuto de Andrade BA, Coletta RD, et al. Clinicopathological and immunohistochemical analysis of 19 cases of oral eosinophilic ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;115:532-540.
- Alobeid B, Pan LX, Milligan L, et al. Eosinophil-rich CD30+ lymphoproliferative disorder of the oral mucosa. Am J Clin Pathol. 2004;121:43-50.
- Hjorting-Hansen E, Schmidt H. Ulcerated granuloma eosinophilicum diutinum of the tongue. report of a case. Acta Derm Venereol. 1961;41:235-239.
- Velez A, Alamillos FJ, Dean A, et al. Eosinophilic ulcer of the oral mucosa: report of a recurrent case on the tongue. Clin Exp Dermatol. 1997;22:154-156.
- Elzay RP. Traumatic ulcerative granuloma with stromal eosinophilia (Riga-Fede’s disease and traumatic eosinophilic granuloma). Oral Surg Oral Med Oral Pathol. 1983;55:497-506.
- Val-Bernal JF, Gonzalez-Vela MC, Sanchez-Santolino S, et al. Localized eosinophilic (Langerhans’ cell) granuloma of the lower lip. a lesion that may cause diagnostic error. J Cutan Pathol. 2009;36:1109-1113.
- Shapiro L, Juhlin EA. Eosinophilic ulcer of the tongue report of two cases and review of the literature. Dermatologica. 1970;140:242-250.
- Amberg S. Sublingual growth in infants. Am J Med Sci. 1902;126:257-269.
- EI-Mofty SK, Swanson PE, Wick MR, et al. Eosinophilic ulcer of the oral mucosa: report of 38 new cases with immunohistochemical observations. Oral Surg Oral Med Oral Pathol. 1993;75:716-722.
- Regezi JA, Zarbo RJ, Daniels TE, et al. Oral traumatic granuloma: characterization of the cellular infiltrate. Oral Surg Oral Med Oral Pathol. 1993;75:723-727.
- Ficarra G, Prignano F, Romagnoli P. Traumatic eosinophilic granuloma of the oral mucosa: a CD30+ (Ki-1) lymphoproliferative disorder? Oral Oncol. 1997;33:375-379.
- Doyle JL, Geary W, Baden E. Eosinophilic ulcer. J Oral Maxillofac Surg. 1989;47:349-352.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Stein H, Mason DY, Gerdes J, et al. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood. 1985;66:848-858.
- Rosenberg A, Biesma DH, Sie-Go DMDS, et al. Primary extranodal CD30-positive T-cell non-Hodgkin’s lymphoma of the oral mucosa. report of two cases. Int J Oral Maxillofac Surg. 1996;25:57-59.
- Kato N, Tomita Y, Yoshida K, et al. Involvement of the tongue by lymphomatoid papulosis. Am J Dermatopathol. 1998;20:522-526.
- Savarrio L, Gibson J, Dunlop DJ, et al. Spontaneous regression of an anaplastic large cell lymphoma in the oral cavity: first reported case and review of the literature. Oral Oncol. 1999;35:609-613.
- Sciubba J, Said-Al-Naief N, Fantasia J. Critical review of lymphomatoid papulosis of the oral cavity with case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:195-204.
- Yamazaki H, Shirasugi Y, Kajiwara H, et al. Concurrent onset of eosinophilic ulcer of the oral mucosa with peripheral eosinophilia in a human T-cell leukemia virus type I carrier. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;114:E43-E48.
- Dojcinov SD, Venkataram G, Raffeld M, et al. EBV positive mucocutaneous ulcer—a study of 26 cases associated with various sources of immunosuppression. Am J Surg Pathol. 2010;34:405-417.
- Kim YC, Yang WI, Lee MG, et al. Epstein-Barr virus in CD30 anaplastic large cell lymphoma involving the skin and lymphomatoid papulosis in South Korea. Int J Dermatol. 2006;45:1312-1316.
- Pietersma F, Piriou E, van Baarle D. Immune surveillance of EBV-infected B cells and the development of non-Hodgkin lymphomas in immunocompromised patients. Leuk Lymphoma. 2008;49:1028-1041.
- Salisbury CL, Budnick SD, Li S. T cell receptor gene rearrangement and CD 30 immunoreactivity in traumatic ulcerative granuloma with stromal eosinophilia of oral cavity. Am J Clin Pathol. 2009;132:722-727.
- Marszalek A, Neska-Dlugosz I. Traumatic ulcerative granuloma with stromal eosinophilia. a case report and short literature review. Pol J Pathol. 2011;3:172-175.
- Wong DT, Donoff RB, Yang J, et al. Sequential expression of transforming growth factors alpha and beta 1 by eosinophils during cutaneous wound healing in the hamster. Am J Pathol. 1993;143:130-142.
- Elovic AE, Gallagher GT, Kabani S, et al. Lack of TGF-alpha and TGF-beta synthesis by human eosinophils in chronic oral ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:672-681.
- Hirshberg A, Amariglio N, Akrish S, et al. Traumatic ulcerative granuloma with stromal eosinophilia: reactive lesion of the oral mucosa. Am J Clin Pathol. 2006;126:522-529.
- Abdel-Naser MB, Tsatsou F, Hippe S, et al. Oral eosinophilic ulcer, an Epstein-Barr virus-associated CD30+ lymphoproliferation? [published online April 5, 2011]. Dermatology. 2011;222:113-118.
- Fonseca FP, Benevenuto de Andrade BA, Coletta RD, et al. Clinicopathological and immunohistochemical analysis of 19 cases of oral eosinophilic ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;115:532-540.
- Alobeid B, Pan LX, Milligan L, et al. Eosinophil-rich CD30+ lymphoproliferative disorder of the oral mucosa. Am J Clin Pathol. 2004;121:43-50.
- Hjorting-Hansen E, Schmidt H. Ulcerated granuloma eosinophilicum diutinum of the tongue. report of a case. Acta Derm Venereol. 1961;41:235-239.
- Velez A, Alamillos FJ, Dean A, et al. Eosinophilic ulcer of the oral mucosa: report of a recurrent case on the tongue. Clin Exp Dermatol. 1997;22:154-156.
- Elzay RP. Traumatic ulcerative granuloma with stromal eosinophilia (Riga-Fede’s disease and traumatic eosinophilic granuloma). Oral Surg Oral Med Oral Pathol. 1983;55:497-506.
- Val-Bernal JF, Gonzalez-Vela MC, Sanchez-Santolino S, et al. Localized eosinophilic (Langerhans’ cell) granuloma of the lower lip. a lesion that may cause diagnostic error. J Cutan Pathol. 2009;36:1109-1113.
- Shapiro L, Juhlin EA. Eosinophilic ulcer of the tongue report of two cases and review of the literature. Dermatologica. 1970;140:242-250.
- Amberg S. Sublingual growth in infants. Am J Med Sci. 1902;126:257-269.
- EI-Mofty SK, Swanson PE, Wick MR, et al. Eosinophilic ulcer of the oral mucosa: report of 38 new cases with immunohistochemical observations. Oral Surg Oral Med Oral Pathol. 1993;75:716-722.
- Regezi JA, Zarbo RJ, Daniels TE, et al. Oral traumatic granuloma: characterization of the cellular infiltrate. Oral Surg Oral Med Oral Pathol. 1993;75:723-727.
- Ficarra G, Prignano F, Romagnoli P. Traumatic eosinophilic granuloma of the oral mucosa: a CD30+ (Ki-1) lymphoproliferative disorder? Oral Oncol. 1997;33:375-379.
- Doyle JL, Geary W, Baden E. Eosinophilic ulcer. J Oral Maxillofac Surg. 1989;47:349-352.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Stein H, Mason DY, Gerdes J, et al. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood. 1985;66:848-858.
- Rosenberg A, Biesma DH, Sie-Go DMDS, et al. Primary extranodal CD30-positive T-cell non-Hodgkin’s lymphoma of the oral mucosa. report of two cases. Int J Oral Maxillofac Surg. 1996;25:57-59.
- Kato N, Tomita Y, Yoshida K, et al. Involvement of the tongue by lymphomatoid papulosis. Am J Dermatopathol. 1998;20:522-526.
- Savarrio L, Gibson J, Dunlop DJ, et al. Spontaneous regression of an anaplastic large cell lymphoma in the oral cavity: first reported case and review of the literature. Oral Oncol. 1999;35:609-613.
- Sciubba J, Said-Al-Naief N, Fantasia J. Critical review of lymphomatoid papulosis of the oral cavity with case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:195-204.
- Yamazaki H, Shirasugi Y, Kajiwara H, et al. Concurrent onset of eosinophilic ulcer of the oral mucosa with peripheral eosinophilia in a human T-cell leukemia virus type I carrier. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;114:E43-E48.
- Dojcinov SD, Venkataram G, Raffeld M, et al. EBV positive mucocutaneous ulcer—a study of 26 cases associated with various sources of immunosuppression. Am J Surg Pathol. 2010;34:405-417.
- Kim YC, Yang WI, Lee MG, et al. Epstein-Barr virus in CD30 anaplastic large cell lymphoma involving the skin and lymphomatoid papulosis in South Korea. Int J Dermatol. 2006;45:1312-1316.
- Pietersma F, Piriou E, van Baarle D. Immune surveillance of EBV-infected B cells and the development of non-Hodgkin lymphomas in immunocompromised patients. Leuk Lymphoma. 2008;49:1028-1041.
- Salisbury CL, Budnick SD, Li S. T cell receptor gene rearrangement and CD 30 immunoreactivity in traumatic ulcerative granuloma with stromal eosinophilia of oral cavity. Am J Clin Pathol. 2009;132:722-727.
- Marszalek A, Neska-Dlugosz I. Traumatic ulcerative granuloma with stromal eosinophilia. a case report and short literature review. Pol J Pathol. 2011;3:172-175.
- Wong DT, Donoff RB, Yang J, et al. Sequential expression of transforming growth factors alpha and beta 1 by eosinophils during cutaneous wound healing in the hamster. Am J Pathol. 1993;143:130-142.
- Elovic AE, Gallagher GT, Kabani S, et al. Lack of TGF-alpha and TGF-beta synthesis by human eosinophils in chronic oral ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:672-681.
Practice Points
- Immunohistochemical staining of traumatic ulcerative granuloma with stromal eosinophilia (TUGSE) may suggest an underlying lymphoproliferative disorder.
- Early recognition of TUGSE, which often is malignant appearing, is key, with watchful waiting as the mainstay therapy.
- Adjunctive therapy for TUGSE includes prednisolone and oral analgesics.
Chronic Diffuse Erythematous Papulonodules
The Diagnosis: Lymphomatoid Papulosis
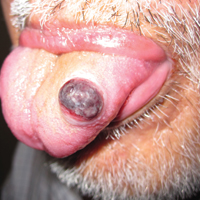
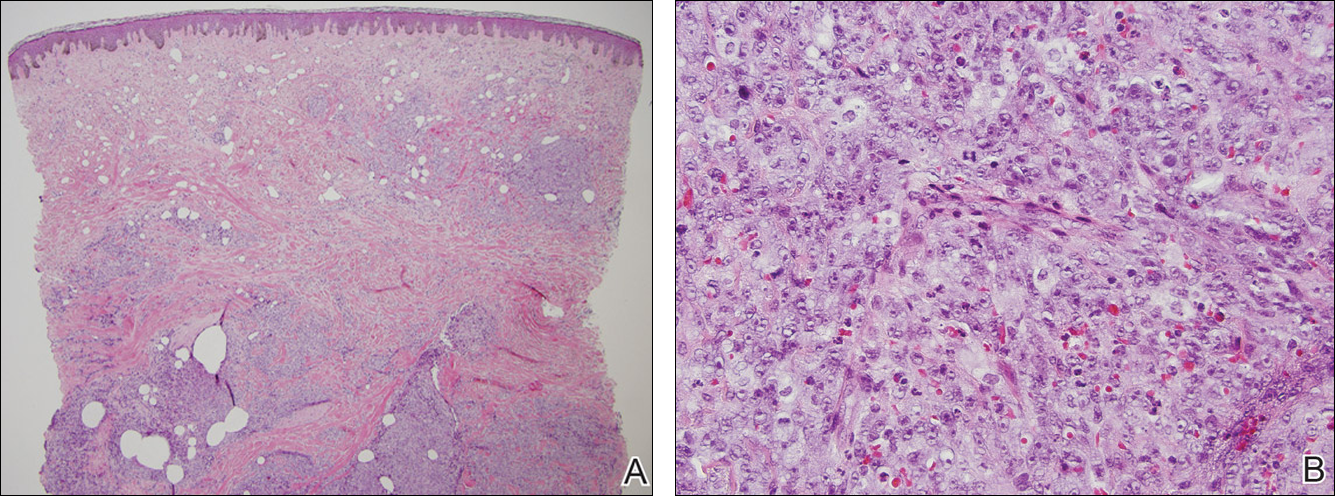
A shave biopsy of an established lesion on the volar aspect of the left wrist was performed (Figure 1). The biopsy showed an ulcerated nodular lesion characterized by a dense mixed inflammatory cell infiltrate in the dermis composed of lymphocytes, histiocytes, scattered neutrophils, and numerous eosinophils (Figure 2). Notably there was a minor population of large atypical cells with immunoblastic and anaplastic morphology present individually and in small clusters most prominently within the upper dermis (Figures 3 and 4). Immunohistochemistry of the anaplastic cells revealed a CD30+, CD3−, CD4+, CD5−, CD8−, CD2−, CD7−, CD56−, ALK1− (anaplastic lymphoma kinase-1), PAX5− (paired box protein-5), CD20−, and CD15− phenotype. These morphologic and immunohistochemical features suggested a CD30+ cutaneous lymphoproliferative disorder. The clinical history of recurrent self-healing papulonodules in an otherwise-healthy patient established the diagnosis of lymphomatoid papulosis (LyP).

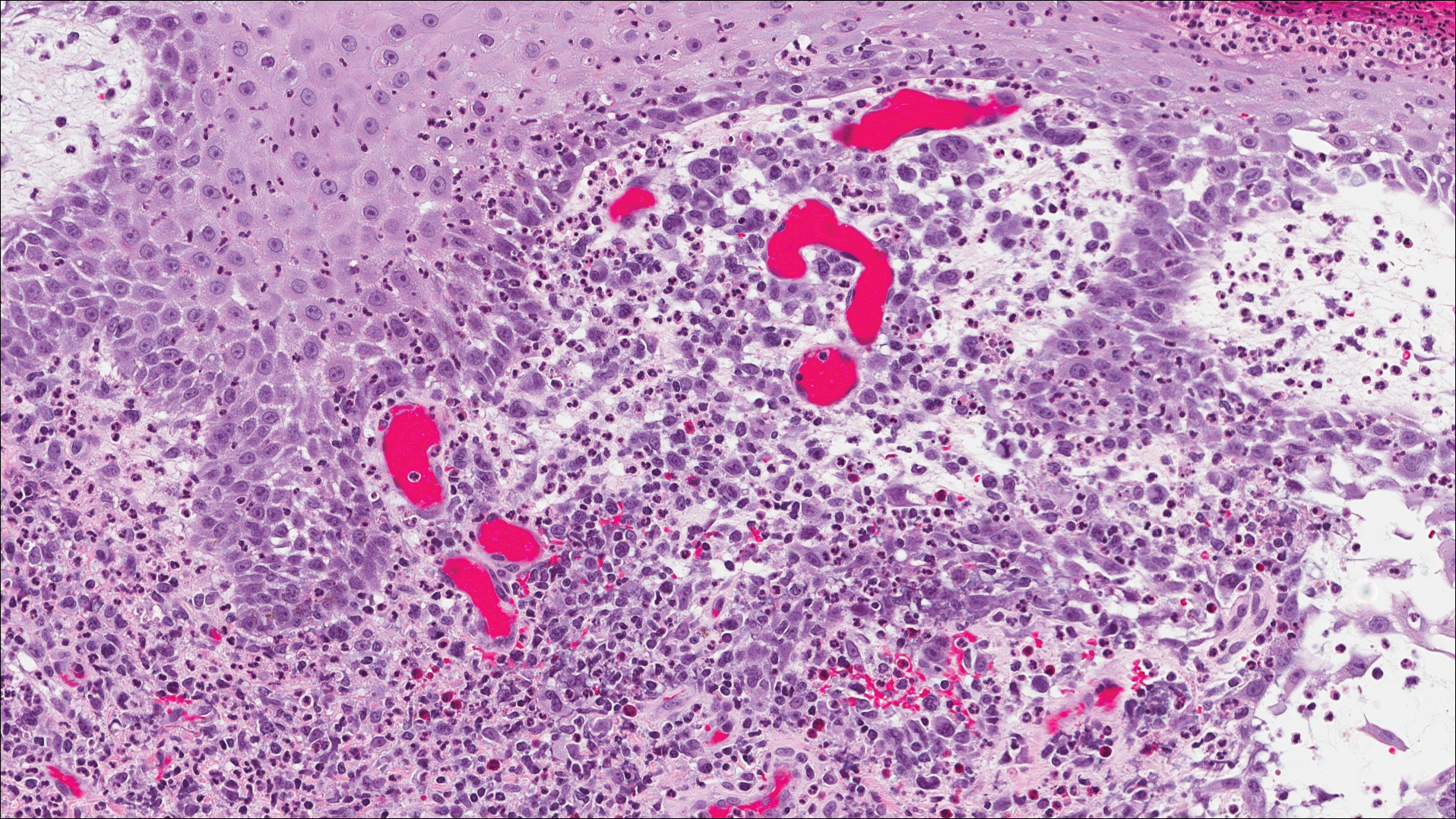
Lymphomatoid papulosis is a lymphoproliferative disorder characterized by recurrent crops of self-resolving eruptive papulonodular skin lesions that may show a variety of histologic features including a CD30+ malignant T-cell lymphoma.1 Lymphomatoid papulosis was first described in 19681 but debate continues whether the condition should be considered malignant or benign.2 Although the prognosis is excellent, LyP is characterized by a protracted course, often lasting many years. Additionally, these patients have a lifelong increased risk for development of a second cutaneous or systemic lymphoma such as mycosis fungoides (MF), cutaneous or nodal anaplastic large cell lymphoma (ALCL), or Hodgkin lymphoma, among others.
Lymphomatoid papulosis is a rare disease occurring in all ethnic groups and at any age, though most commonly presenting in the fifth decade of life. Finding large atypical T cells expressing CD30 in recurring skin lesions is highly suggestive of LyP; however, large CD30+ cells also can be seen in numerous benign reactive processes such as arthropod assault, drug eruption, viral skin infections, and other dermatoses, thus clinical correlation is always paramount. The cause of LyP is largely unknown; however, spontaneous regression may be explained by CD30-CD30 ligand interaction3 as well as an increased proapoptotic milieu.4 Specific translocations such as interferon regulatory factor-4 have been hypothesized as a risk factor for malignant progression.5-7 Additionally, an inactivating gene mutation resulting in loss of transforming growth factor β1 receptor expression and subsequent unresponsiveness to the growth inhibitory effect of transforming growth factor β may play a role in progression of LyP to ALCL.8
Clinically, LyP consists of red-brown papules and nodules generally smaller than 2 cm, often with central hemorrhage, necrosis, and crusting. Lesions are at different stages of eruption and resolution. They are often grouped but may be disseminated. Spontaneous regression typically occurs within 3 to 8 weeks. Pruritus or mild tenderness may occur as well as residual hyperpigmentation or scarring. Systemic symptoms are notably absent.
The histologic features of LyP vary according to the age of the lesion and subtype.2 Early lesions may only show a few inflammatory cells, but as lesions evolve, larger immunoblastlike CD30+ atypical cells accumulate that may resemble the Reed-Sternberg cells of Hodgkin lymphoma. Of the 5 subtypes, the most common is type A. It is characterized by a wedge-shaped infiltrate with a mixed population of scattered or clustered, large, atypical CD30+ cells, lymphocytes, neutrophils, eosinophils, and histiocytes.9 Frequent mitoses often are seen. Type B appears similar to MF due to a predominantly epidermotropic infiltrate of CD3+ and often CD30− atypical cells. Spontaneously regressing papules favor LyP, whereas persistent patches or plaques favor MF. Type C appears identical to ALCL with diffuse sheets of large atypical CD30+ cells and relatively few inflammatory cells, but spontaneously regressing lesions again favor LyP, whereas persistent tumors favor ALCL. Type D appears similar to primary cutaneous aggressive epidermotropic CD8+ cytotoxic T cell lymphoma due to a markedly epidermotropic infiltrate of small atypical CD8+ and CD30+ lymphocytes, often TIA-1+ (T-cell intracytoplasmic antigen-1) or granzyme B+, but CD30 positivity and self-resolving lesions favor LyP. Type E mimics extranodal natural killer/T cell lymphoma (nasal type) due to angioinvasive CD30+ and beta F1+ T lymphocytes, often CD8+ and/or TIA-1+, but self-resolving lesions again favor LyP, as well as absence of Epstein-Barr virus and CD56−.9
The most common therapeutic approaches to LyP include topical steroids, phototherapy, and low-dose methotrexate.10 However, treatment does not change overall disease course or reduce the future risk for developing an associated lymphoma. Accordingly, abstaining from active therapeutic intervention is reasonable, especially in patients with only a few asymptomatic lesions.
- Macaulay WL. Lymphomatoid papulosis: a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Slater DN. The new World Health Organization-European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas: a practical marriage of two giants. Br J Dermatol. 2005;153:874-880.
- Mori M, Manuelli C, Pimpinelli N, et al. CD30-CD30 ligand interaction in primary cutaneous CD30(+) T-cell lymphomas: a clue to the pathophysiology of clinical regression. Blood. 1999;94:3077-3083.
- Greisser J, Doebbeling U, Roos M, et al. Apoptosis in CD30-positive lymphoproliferative disorders of the skin. Exp Dermatol. 2005;14:380-385.
- Kiran T, Demirkesen C, Eker C, et al. The significance of MUM1/IRF4 protein expression and IRF4 translocation of CD30(+) cutaneous T-cell lymphoproliferative disorders: a study of 53 cases. Leuk Res. 2013;37:396-400.
- Wada DA, Law ME, Hsi ED, et al. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: a multicenter study of 204 skin biopsies. Mod Pathol. 2011;24:596-605.
- Pham-Ledard A, Prochazkova-Carlotti M, Laharanne E, et al. IRF4 gene rearrangements define a subgroup of CD30-positive cutaneous T-cell lymphoma: a study of 54 cases. J Invest Dermatol. 2010;130:816-825.
- Schiemann WP, Pfeifer WM, Levi E, et al. A deletion in the gene for transforming growth factor β type I receptor abolishes growth regulation by transforming growth factor β in a cutaneous T-cell lymphoma. Blood. 1999;94:2854-2861.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
The Diagnosis: Lymphomatoid Papulosis
A shave biopsy of an established lesion on the volar aspect of the left wrist was performed (Figure 1). The biopsy showed an ulcerated nodular lesion characterized by a dense mixed inflammatory cell infiltrate in the dermis composed of lymphocytes, histiocytes, scattered neutrophils, and numerous eosinophils (Figure 2). Notably there was a minor population of large atypical cells with immunoblastic and anaplastic morphology present individually and in small clusters most prominently within the upper dermis (Figures 3 and 4). Immunohistochemistry of the anaplastic cells revealed a CD30+, CD3−, CD4+, CD5−, CD8−, CD2−, CD7−, CD56−, ALK1− (anaplastic lymphoma kinase-1), PAX5− (paired box protein-5), CD20−, and CD15− phenotype. These morphologic and immunohistochemical features suggested a CD30+ cutaneous lymphoproliferative disorder. The clinical history of recurrent self-healing papulonodules in an otherwise-healthy patient established the diagnosis of lymphomatoid papulosis (LyP).
Lymphomatoid papulosis is a lymphoproliferative disorder characterized by recurrent crops of self-resolving eruptive papulonodular skin lesions that may show a variety of histologic features including a CD30+ malignant T-cell lymphoma.1 Lymphomatoid papulosis was first described in 19681 but debate continues whether the condition should be considered malignant or benign.2 Although the prognosis is excellent, LyP is characterized by a protracted course, often lasting many years. Additionally, these patients have a lifelong increased risk for development of a second cutaneous or systemic lymphoma such as mycosis fungoides (MF), cutaneous or nodal anaplastic large cell lymphoma (ALCL), or Hodgkin lymphoma, among others.
Lymphomatoid papulosis is a rare disease occurring in all ethnic groups and at any age, though most commonly presenting in the fifth decade of life. Finding large atypical T cells expressing CD30 in recurring skin lesions is highly suggestive of LyP; however, large CD30+ cells also can be seen in numerous benign reactive processes such as arthropod assault, drug eruption, viral skin infections, and other dermatoses, thus clinical correlation is always paramount. The cause of LyP is largely unknown; however, spontaneous regression may be explained by CD30-CD30 ligand interaction3 as well as an increased proapoptotic milieu.4 Specific translocations such as interferon regulatory factor-4 have been hypothesized as a risk factor for malignant progression.5-7 Additionally, an inactivating gene mutation resulting in loss of transforming growth factor β1 receptor expression and subsequent unresponsiveness to the growth inhibitory effect of transforming growth factor β may play a role in progression of LyP to ALCL.8
Clinically, LyP consists of red-brown papules and nodules generally smaller than 2 cm, often with central hemorrhage, necrosis, and crusting. Lesions are at different stages of eruption and resolution. They are often grouped but may be disseminated. Spontaneous regression typically occurs within 3 to 8 weeks. Pruritus or mild tenderness may occur as well as residual hyperpigmentation or scarring. Systemic symptoms are notably absent.
The histologic features of LyP vary according to the age of the lesion and subtype.2 Early lesions may only show a few inflammatory cells, but as lesions evolve, larger immunoblastlike CD30+ atypical cells accumulate that may resemble the Reed-Sternberg cells of Hodgkin lymphoma. Of the 5 subtypes, the most common is type A. It is characterized by a wedge-shaped infiltrate with a mixed population of scattered or clustered, large, atypical CD30+ cells, lymphocytes, neutrophils, eosinophils, and histiocytes.9 Frequent mitoses often are seen. Type B appears similar to MF due to a predominantly epidermotropic infiltrate of CD3+ and often CD30− atypical cells. Spontaneously regressing papules favor LyP, whereas persistent patches or plaques favor MF. Type C appears identical to ALCL with diffuse sheets of large atypical CD30+ cells and relatively few inflammatory cells, but spontaneously regressing lesions again favor LyP, whereas persistent tumors favor ALCL. Type D appears similar to primary cutaneous aggressive epidermotropic CD8+ cytotoxic T cell lymphoma due to a markedly epidermotropic infiltrate of small atypical CD8+ and CD30+ lymphocytes, often TIA-1+ (T-cell intracytoplasmic antigen-1) or granzyme B+, but CD30 positivity and self-resolving lesions favor LyP. Type E mimics extranodal natural killer/T cell lymphoma (nasal type) due to angioinvasive CD30+ and beta F1+ T lymphocytes, often CD8+ and/or TIA-1+, but self-resolving lesions again favor LyP, as well as absence of Epstein-Barr virus and CD56−.9
The most common therapeutic approaches to LyP include topical steroids, phototherapy, and low-dose methotrexate.10 However, treatment does not change overall disease course or reduce the future risk for developing an associated lymphoma. Accordingly, abstaining from active therapeutic intervention is reasonable, especially in patients with only a few asymptomatic lesions.
The Diagnosis: Lymphomatoid Papulosis
A shave biopsy of an established lesion on the volar aspect of the left wrist was performed (Figure 1). The biopsy showed an ulcerated nodular lesion characterized by a dense mixed inflammatory cell infiltrate in the dermis composed of lymphocytes, histiocytes, scattered neutrophils, and numerous eosinophils (Figure 2). Notably there was a minor population of large atypical cells with immunoblastic and anaplastic morphology present individually and in small clusters most prominently within the upper dermis (Figures 3 and 4). Immunohistochemistry of the anaplastic cells revealed a CD30+, CD3−, CD4+, CD5−, CD8−, CD2−, CD7−, CD56−, ALK1− (anaplastic lymphoma kinase-1), PAX5− (paired box protein-5), CD20−, and CD15− phenotype. These morphologic and immunohistochemical features suggested a CD30+ cutaneous lymphoproliferative disorder. The clinical history of recurrent self-healing papulonodules in an otherwise-healthy patient established the diagnosis of lymphomatoid papulosis (LyP).
Lymphomatoid papulosis is a lymphoproliferative disorder characterized by recurrent crops of self-resolving eruptive papulonodular skin lesions that may show a variety of histologic features including a CD30+ malignant T-cell lymphoma.1 Lymphomatoid papulosis was first described in 19681 but debate continues whether the condition should be considered malignant or benign.2 Although the prognosis is excellent, LyP is characterized by a protracted course, often lasting many years. Additionally, these patients have a lifelong increased risk for development of a second cutaneous or systemic lymphoma such as mycosis fungoides (MF), cutaneous or nodal anaplastic large cell lymphoma (ALCL), or Hodgkin lymphoma, among others.
Lymphomatoid papulosis is a rare disease occurring in all ethnic groups and at any age, though most commonly presenting in the fifth decade of life. Finding large atypical T cells expressing CD30 in recurring skin lesions is highly suggestive of LyP; however, large CD30+ cells also can be seen in numerous benign reactive processes such as arthropod assault, drug eruption, viral skin infections, and other dermatoses, thus clinical correlation is always paramount. The cause of LyP is largely unknown; however, spontaneous regression may be explained by CD30-CD30 ligand interaction3 as well as an increased proapoptotic milieu.4 Specific translocations such as interferon regulatory factor-4 have been hypothesized as a risk factor for malignant progression.5-7 Additionally, an inactivating gene mutation resulting in loss of transforming growth factor β1 receptor expression and subsequent unresponsiveness to the growth inhibitory effect of transforming growth factor β may play a role in progression of LyP to ALCL.8
Clinically, LyP consists of red-brown papules and nodules generally smaller than 2 cm, often with central hemorrhage, necrosis, and crusting. Lesions are at different stages of eruption and resolution. They are often grouped but may be disseminated. Spontaneous regression typically occurs within 3 to 8 weeks. Pruritus or mild tenderness may occur as well as residual hyperpigmentation or scarring. Systemic symptoms are notably absent.
The histologic features of LyP vary according to the age of the lesion and subtype.2 Early lesions may only show a few inflammatory cells, but as lesions evolve, larger immunoblastlike CD30+ atypical cells accumulate that may resemble the Reed-Sternberg cells of Hodgkin lymphoma. Of the 5 subtypes, the most common is type A. It is characterized by a wedge-shaped infiltrate with a mixed population of scattered or clustered, large, atypical CD30+ cells, lymphocytes, neutrophils, eosinophils, and histiocytes.9 Frequent mitoses often are seen. Type B appears similar to MF due to a predominantly epidermotropic infiltrate of CD3+ and often CD30− atypical cells. Spontaneously regressing papules favor LyP, whereas persistent patches or plaques favor MF. Type C appears identical to ALCL with diffuse sheets of large atypical CD30+ cells and relatively few inflammatory cells, but spontaneously regressing lesions again favor LyP, whereas persistent tumors favor ALCL. Type D appears similar to primary cutaneous aggressive epidermotropic CD8+ cytotoxic T cell lymphoma due to a markedly epidermotropic infiltrate of small atypical CD8+ and CD30+ lymphocytes, often TIA-1+ (T-cell intracytoplasmic antigen-1) or granzyme B+, but CD30 positivity and self-resolving lesions favor LyP. Type E mimics extranodal natural killer/T cell lymphoma (nasal type) due to angioinvasive CD30+ and beta F1+ T lymphocytes, often CD8+ and/or TIA-1+, but self-resolving lesions again favor LyP, as well as absence of Epstein-Barr virus and CD56−.9
The most common therapeutic approaches to LyP include topical steroids, phototherapy, and low-dose methotrexate.10 However, treatment does not change overall disease course or reduce the future risk for developing an associated lymphoma. Accordingly, abstaining from active therapeutic intervention is reasonable, especially in patients with only a few asymptomatic lesions.
- Macaulay WL. Lymphomatoid papulosis: a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Slater DN. The new World Health Organization-European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas: a practical marriage of two giants. Br J Dermatol. 2005;153:874-880.
- Mori M, Manuelli C, Pimpinelli N, et al. CD30-CD30 ligand interaction in primary cutaneous CD30(+) T-cell lymphomas: a clue to the pathophysiology of clinical regression. Blood. 1999;94:3077-3083.
- Greisser J, Doebbeling U, Roos M, et al. Apoptosis in CD30-positive lymphoproliferative disorders of the skin. Exp Dermatol. 2005;14:380-385.
- Kiran T, Demirkesen C, Eker C, et al. The significance of MUM1/IRF4 protein expression and IRF4 translocation of CD30(+) cutaneous T-cell lymphoproliferative disorders: a study of 53 cases. Leuk Res. 2013;37:396-400.
- Wada DA, Law ME, Hsi ED, et al. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: a multicenter study of 204 skin biopsies. Mod Pathol. 2011;24:596-605.
- Pham-Ledard A, Prochazkova-Carlotti M, Laharanne E, et al. IRF4 gene rearrangements define a subgroup of CD30-positive cutaneous T-cell lymphoma: a study of 54 cases. J Invest Dermatol. 2010;130:816-825.
- Schiemann WP, Pfeifer WM, Levi E, et al. A deletion in the gene for transforming growth factor β type I receptor abolishes growth regulation by transforming growth factor β in a cutaneous T-cell lymphoma. Blood. 1999;94:2854-2861.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
- Macaulay WL. Lymphomatoid papulosis: a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Slater DN. The new World Health Organization-European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas: a practical marriage of two giants. Br J Dermatol. 2005;153:874-880.
- Mori M, Manuelli C, Pimpinelli N, et al. CD30-CD30 ligand interaction in primary cutaneous CD30(+) T-cell lymphomas: a clue to the pathophysiology of clinical regression. Blood. 1999;94:3077-3083.
- Greisser J, Doebbeling U, Roos M, et al. Apoptosis in CD30-positive lymphoproliferative disorders of the skin. Exp Dermatol. 2005;14:380-385.
- Kiran T, Demirkesen C, Eker C, et al. The significance of MUM1/IRF4 protein expression and IRF4 translocation of CD30(+) cutaneous T-cell lymphoproliferative disorders: a study of 53 cases. Leuk Res. 2013;37:396-400.
- Wada DA, Law ME, Hsi ED, et al. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: a multicenter study of 204 skin biopsies. Mod Pathol. 2011;24:596-605.
- Pham-Ledard A, Prochazkova-Carlotti M, Laharanne E, et al. IRF4 gene rearrangements define a subgroup of CD30-positive cutaneous T-cell lymphoma: a study of 54 cases. J Invest Dermatol. 2010;130:816-825.
- Schiemann WP, Pfeifer WM, Levi E, et al. A deletion in the gene for transforming growth factor β type I receptor abolishes growth regulation by transforming growth factor β in a cutaneous T-cell lymphoma. Blood. 1999;94:2854-2861.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
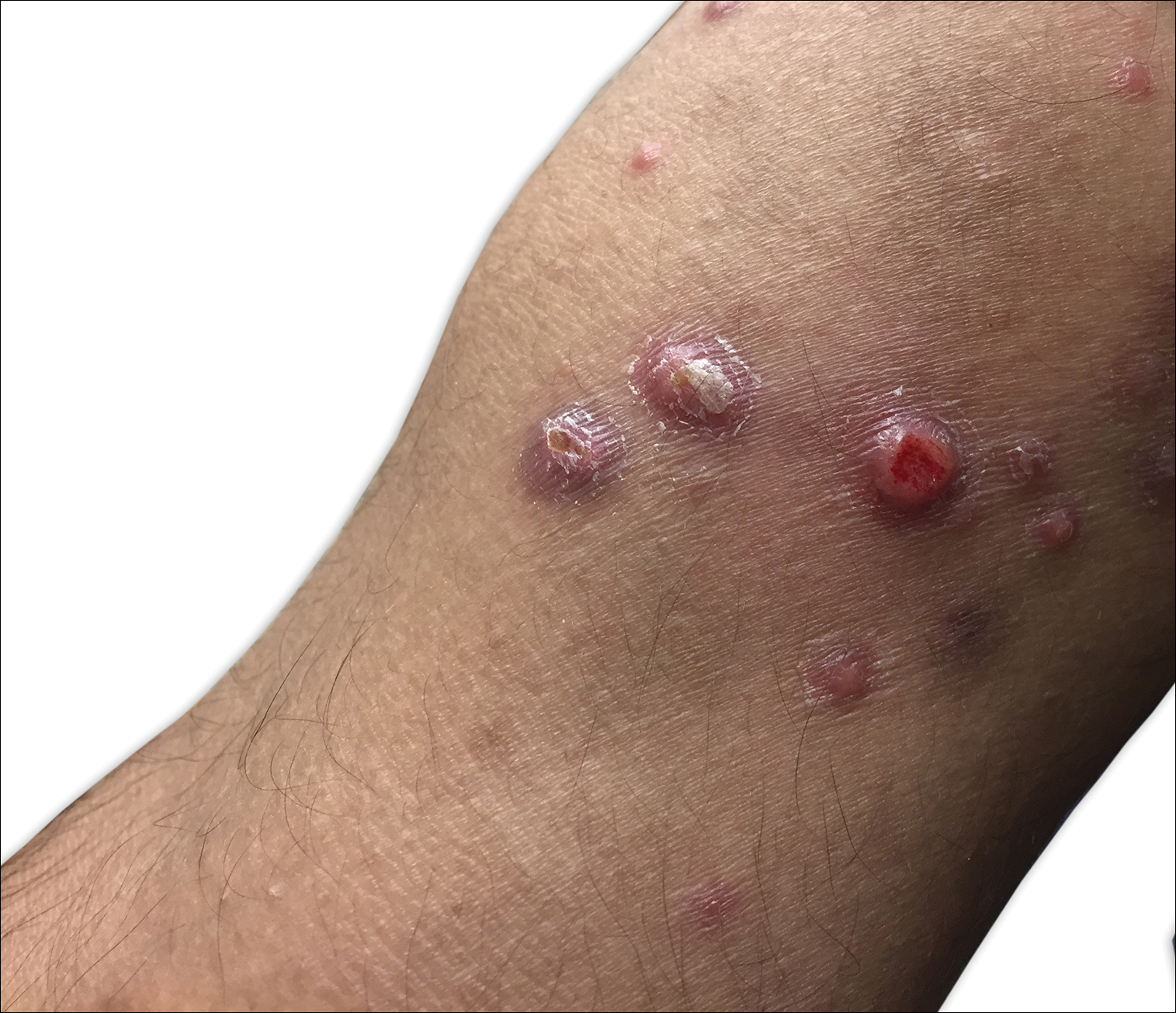
A 29-year-old man from Saudi Arabia presented with slightly tender skin lesions occurring in crops every few months over the last 7 years. The lesions typically would occur on the inguinal area, lower abdomen, buttocks, thighs, or arms, resolving within a few weeks despite no treatment. The patient denied having systemic symptoms such as fevers, chills, sweats, chest pain, shortness of breath, or unexpected weight loss. Physical examination revealed multiple erythematous papulonodules, some ulcerated with a superficial crust, grouped predominantly on the medial aspect of the right upper arm and left lower inguinal region. Isolated lesions also were present on the forearms, dorsal aspects of the hands, abdomen, and thighs. The grouped papulonodules were intermixed with faint hyperpigmented macules indicative of prior lesions. No oral lesions were noted, and there was no marked axillary or inguinal lymphadenopathy.
Enlarging Breast Lesion
The Diagnosis: Radiation-Associated Angiosarcoma
At the time of presentation, a 4-mm lesional punch biopsy was obtained (Figure), which revealed an epithelioid neoplasm within the dermis expressing CD31 and CD34, and staining negatively for S-100, CD45, and estrogen and progesterone receptors. The histologic and immunophenotypic findings were compatible with the diagnosis of angiosarcoma. Given the patient’s history of radiation for breast carcinoma several years ago, this tumor was consistent with radiation-associated angiosarcoma (RAAS).
Development of secondary angiosarcoma has been linked to both prior radiation (RAAS) and chronic lymphedema (Stewart-Treves syndrome).1 Radiation-associated angiosarcoma is defined as a “pathologically confirmed breast or chest wall angiosarcoma arising within a previously irradiated field.”2 The incidence of RAAS is estimated to be 0.9 per 1000 individuals following radiation treatment of breast cancer over the subsequent 15 years and a mean time from radiation to development of 7 years.1 Incidence is expected to increase in the future due to improved likelihood of surviving early-stage breast carcinoma and the increased use of external beam radiation therapy for management of breast cancer.
Differentiating between primary and secondary angiosarcoma of the breast is important. Although primary breast angiosarcoma usually arises in women aged 30 to 40 years, RAAS tends to arise in older women (mean age, 68 years) and is seen only in those women with prior radiation.2 Additionally, high-level amplification of MYC, a known photo-oncogene, on chromosome 8 is a key genetic alteration of RAAS that helps to distinguish it from primary angiosarcoma, though this variance may be present in only half of RAAS cases.3 Immunohistochemical analysis of tumor cells for MYC expression correlates well with this amplification and also is helpful in distinguishing atypical vascular lesions from RAAS.4 Atypical vascular lesions, similar to RAAS, occur years after radiation exposure and may have a similar clinical presentation. Atypical vascular lesions do not progress to angiosarcoma in reported cases, but clinical and histologic overlap with RAAS make the diagnosis difficult.5 In these cases, analysis with fluorescence in situ hybridization or immunohistochemistry for the MYC amplification is important to differentiate these tumors.6
At the time of presentation, the majority of patients with RAAS of the breast have localized disease, often with a variable presentation. In all known cases, there have been skin changes present, emphasizing the importance of both patient and clinician vigilance on a regular basis in at-risk individuals. In one study, the most common presentation was breast ecchymosis, which was observed in 55% of patients.7 These lesions involve the dermis and are commonly mistaken for benign conditions such as infection or hemorrhage.2 In 2 other studies, RAAS most often manifested as a skin nodule or apparent tumor, closely followed by either a rash or bruiselike presentation.1,2
The overall recommendation for management of patients with ecchymotic skin lesions in previously irradiated regions is to obtain a biopsy specimen for tissue diagnosis. Although there is no standard of care for the management of RAAS, a multidisciplinary approach involving specialists from oncology, surgical oncology, and radiation oncology is recommended. Most often, radical surgery encompassing both the breast parenchyma and the at-risk radiated skin is performed. Extensive surgery has demonstrated the best survival benefits compared to mastectomy alone.7 Chemotherapeutics also may be used as adjuncts to surgery, which have been determined to decrease local recurrence rates but have no proven survival benefits.2 Adverse prognostic factors for survival are tumor size greater than 10 cm and development of local and/or distant metastases.2 Following the diagnosis of RAAS, our patient underwent radical mastectomy with adjuvant chemotherapy and remained disease free 6 months after surgery.
In summary, RAAS is a well-known, albeit relatively uncommon, consequence of radiation therapy. Dermatologists, oncologists, and primary care providers play an important role in recognizing this entity when evaluating patients with ecchymotic lesions as well as nodules or tumors within an irradiated field. Biopsy should be obtained promptly to prevent delay in diagnosis and to expedite referral to appropriate specialists for further evaluation and treatment.
- Seinen JM, Emelie S, Verstappen V, et al. Radiation-associated angiosarcoma after breast cancer: high recurrence rate and poor survival despite surgical treatment with R0 resection. Ann Surg Oncol. 2012;19:2700-2706.
- Torres KE, Ravi V, Kin K, et al. Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radiotherapy for breast cancer. Ann Surg Oncol. 2013;20:1267-1274.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Ginter PS, Mosquera JM, MacDonald TY, et al. Diagnostic utility of MYC amplification and anti-MYC immunohistochemistry in atypical vascular lesions, primary or radiation-induced mammary angiosarcomas, and primary angiosarcomas of other sites. Hum Pathol. 2014;45:709-716.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological immunohistochemical and molecular analysis of 66 cases. Mod Pathol. 2012;25:75-85.
- Fernandez AP, Sun Y, Tubbs RR, et al. FISH for MYC amplification and anti-MYC immunohistochemistry: useful diagnostic tools in the assessment of secondary angiosarcoma and atypical vascular proliferations. J Cutan Pathol. 2012;39:234-242.
- Morgan EA, Kozono DE, Wang Q, et al. Cutaneous radiation-associated angiosarcoma of the breast: poor prognosis in a rare secondary malignancy. Ann Surg Oncol. 2012;19:3801-3808.
The Diagnosis: Radiation-Associated Angiosarcoma
At the time of presentation, a 4-mm lesional punch biopsy was obtained (Figure), which revealed an epithelioid neoplasm within the dermis expressing CD31 and CD34, and staining negatively for S-100, CD45, and estrogen and progesterone receptors. The histologic and immunophenotypic findings were compatible with the diagnosis of angiosarcoma. Given the patient’s history of radiation for breast carcinoma several years ago, this tumor was consistent with radiation-associated angiosarcoma (RAAS).
Development of secondary angiosarcoma has been linked to both prior radiation (RAAS) and chronic lymphedema (Stewart-Treves syndrome).1 Radiation-associated angiosarcoma is defined as a “pathologically confirmed breast or chest wall angiosarcoma arising within a previously irradiated field.”2 The incidence of RAAS is estimated to be 0.9 per 1000 individuals following radiation treatment of breast cancer over the subsequent 15 years and a mean time from radiation to development of 7 years.1 Incidence is expected to increase in the future due to improved likelihood of surviving early-stage breast carcinoma and the increased use of external beam radiation therapy for management of breast cancer.
Differentiating between primary and secondary angiosarcoma of the breast is important. Although primary breast angiosarcoma usually arises in women aged 30 to 40 years, RAAS tends to arise in older women (mean age, 68 years) and is seen only in those women with prior radiation.2 Additionally, high-level amplification of MYC, a known photo-oncogene, on chromosome 8 is a key genetic alteration of RAAS that helps to distinguish it from primary angiosarcoma, though this variance may be present in only half of RAAS cases.3 Immunohistochemical analysis of tumor cells for MYC expression correlates well with this amplification and also is helpful in distinguishing atypical vascular lesions from RAAS.4 Atypical vascular lesions, similar to RAAS, occur years after radiation exposure and may have a similar clinical presentation. Atypical vascular lesions do not progress to angiosarcoma in reported cases, but clinical and histologic overlap with RAAS make the diagnosis difficult.5 In these cases, analysis with fluorescence in situ hybridization or immunohistochemistry for the MYC amplification is important to differentiate these tumors.6
At the time of presentation, the majority of patients with RAAS of the breast have localized disease, often with a variable presentation. In all known cases, there have been skin changes present, emphasizing the importance of both patient and clinician vigilance on a regular basis in at-risk individuals. In one study, the most common presentation was breast ecchymosis, which was observed in 55% of patients.7 These lesions involve the dermis and are commonly mistaken for benign conditions such as infection or hemorrhage.2 In 2 other studies, RAAS most often manifested as a skin nodule or apparent tumor, closely followed by either a rash or bruiselike presentation.1,2
The overall recommendation for management of patients with ecchymotic skin lesions in previously irradiated regions is to obtain a biopsy specimen for tissue diagnosis. Although there is no standard of care for the management of RAAS, a multidisciplinary approach involving specialists from oncology, surgical oncology, and radiation oncology is recommended. Most often, radical surgery encompassing both the breast parenchyma and the at-risk radiated skin is performed. Extensive surgery has demonstrated the best survival benefits compared to mastectomy alone.7 Chemotherapeutics also may be used as adjuncts to surgery, which have been determined to decrease local recurrence rates but have no proven survival benefits.2 Adverse prognostic factors for survival are tumor size greater than 10 cm and development of local and/or distant metastases.2 Following the diagnosis of RAAS, our patient underwent radical mastectomy with adjuvant chemotherapy and remained disease free 6 months after surgery.
In summary, RAAS is a well-known, albeit relatively uncommon, consequence of radiation therapy. Dermatologists, oncologists, and primary care providers play an important role in recognizing this entity when evaluating patients with ecchymotic lesions as well as nodules or tumors within an irradiated field. Biopsy should be obtained promptly to prevent delay in diagnosis and to expedite referral to appropriate specialists for further evaluation and treatment.
The Diagnosis: Radiation-Associated Angiosarcoma
At the time of presentation, a 4-mm lesional punch biopsy was obtained (Figure), which revealed an epithelioid neoplasm within the dermis expressing CD31 and CD34, and staining negatively for S-100, CD45, and estrogen and progesterone receptors. The histologic and immunophenotypic findings were compatible with the diagnosis of angiosarcoma. Given the patient’s history of radiation for breast carcinoma several years ago, this tumor was consistent with radiation-associated angiosarcoma (RAAS).
Development of secondary angiosarcoma has been linked to both prior radiation (RAAS) and chronic lymphedema (Stewart-Treves syndrome).1 Radiation-associated angiosarcoma is defined as a “pathologically confirmed breast or chest wall angiosarcoma arising within a previously irradiated field.”2 The incidence of RAAS is estimated to be 0.9 per 1000 individuals following radiation treatment of breast cancer over the subsequent 15 years and a mean time from radiation to development of 7 years.1 Incidence is expected to increase in the future due to improved likelihood of surviving early-stage breast carcinoma and the increased use of external beam radiation therapy for management of breast cancer.
Differentiating between primary and secondary angiosarcoma of the breast is important. Although primary breast angiosarcoma usually arises in women aged 30 to 40 years, RAAS tends to arise in older women (mean age, 68 years) and is seen only in those women with prior radiation.2 Additionally, high-level amplification of MYC, a known photo-oncogene, on chromosome 8 is a key genetic alteration of RAAS that helps to distinguish it from primary angiosarcoma, though this variance may be present in only half of RAAS cases.3 Immunohistochemical analysis of tumor cells for MYC expression correlates well with this amplification and also is helpful in distinguishing atypical vascular lesions from RAAS.4 Atypical vascular lesions, similar to RAAS, occur years after radiation exposure and may have a similar clinical presentation. Atypical vascular lesions do not progress to angiosarcoma in reported cases, but clinical and histologic overlap with RAAS make the diagnosis difficult.5 In these cases, analysis with fluorescence in situ hybridization or immunohistochemistry for the MYC amplification is important to differentiate these tumors.6
At the time of presentation, the majority of patients with RAAS of the breast have localized disease, often with a variable presentation. In all known cases, there have been skin changes present, emphasizing the importance of both patient and clinician vigilance on a regular basis in at-risk individuals. In one study, the most common presentation was breast ecchymosis, which was observed in 55% of patients.7 These lesions involve the dermis and are commonly mistaken for benign conditions such as infection or hemorrhage.2 In 2 other studies, RAAS most often manifested as a skin nodule or apparent tumor, closely followed by either a rash or bruiselike presentation.1,2
The overall recommendation for management of patients with ecchymotic skin lesions in previously irradiated regions is to obtain a biopsy specimen for tissue diagnosis. Although there is no standard of care for the management of RAAS, a multidisciplinary approach involving specialists from oncology, surgical oncology, and radiation oncology is recommended. Most often, radical surgery encompassing both the breast parenchyma and the at-risk radiated skin is performed. Extensive surgery has demonstrated the best survival benefits compared to mastectomy alone.7 Chemotherapeutics also may be used as adjuncts to surgery, which have been determined to decrease local recurrence rates but have no proven survival benefits.2 Adverse prognostic factors for survival are tumor size greater than 10 cm and development of local and/or distant metastases.2 Following the diagnosis of RAAS, our patient underwent radical mastectomy with adjuvant chemotherapy and remained disease free 6 months after surgery.
In summary, RAAS is a well-known, albeit relatively uncommon, consequence of radiation therapy. Dermatologists, oncologists, and primary care providers play an important role in recognizing this entity when evaluating patients with ecchymotic lesions as well as nodules or tumors within an irradiated field. Biopsy should be obtained promptly to prevent delay in diagnosis and to expedite referral to appropriate specialists for further evaluation and treatment.
- Seinen JM, Emelie S, Verstappen V, et al. Radiation-associated angiosarcoma after breast cancer: high recurrence rate and poor survival despite surgical treatment with R0 resection. Ann Surg Oncol. 2012;19:2700-2706.
- Torres KE, Ravi V, Kin K, et al. Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radiotherapy for breast cancer. Ann Surg Oncol. 2013;20:1267-1274.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Ginter PS, Mosquera JM, MacDonald TY, et al. Diagnostic utility of MYC amplification and anti-MYC immunohistochemistry in atypical vascular lesions, primary or radiation-induced mammary angiosarcomas, and primary angiosarcomas of other sites. Hum Pathol. 2014;45:709-716.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological immunohistochemical and molecular analysis of 66 cases. Mod Pathol. 2012;25:75-85.
- Fernandez AP, Sun Y, Tubbs RR, et al. FISH for MYC amplification and anti-MYC immunohistochemistry: useful diagnostic tools in the assessment of secondary angiosarcoma and atypical vascular proliferations. J Cutan Pathol. 2012;39:234-242.
- Morgan EA, Kozono DE, Wang Q, et al. Cutaneous radiation-associated angiosarcoma of the breast: poor prognosis in a rare secondary malignancy. Ann Surg Oncol. 2012;19:3801-3808.
- Seinen JM, Emelie S, Verstappen V, et al. Radiation-associated angiosarcoma after breast cancer: high recurrence rate and poor survival despite surgical treatment with R0 resection. Ann Surg Oncol. 2012;19:2700-2706.
- Torres KE, Ravi V, Kin K, et al. Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radiotherapy for breast cancer. Ann Surg Oncol. 2013;20:1267-1274.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Ginter PS, Mosquera JM, MacDonald TY, et al. Diagnostic utility of MYC amplification and anti-MYC immunohistochemistry in atypical vascular lesions, primary or radiation-induced mammary angiosarcomas, and primary angiosarcomas of other sites. Hum Pathol. 2014;45:709-716.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological immunohistochemical and molecular analysis of 66 cases. Mod Pathol. 2012;25:75-85.
- Fernandez AP, Sun Y, Tubbs RR, et al. FISH for MYC amplification and anti-MYC immunohistochemistry: useful diagnostic tools in the assessment of secondary angiosarcoma and atypical vascular proliferations. J Cutan Pathol. 2012;39:234-242.
- Morgan EA, Kozono DE, Wang Q, et al. Cutaneous radiation-associated angiosarcoma of the breast: poor prognosis in a rare secondary malignancy. Ann Surg Oncol. 2012;19:3801-3808.

A 75-year-old woman with a history of stage II invasive ductal carcinoma of the right breast presented to the dermatology clinic with an enlarging, indurated, ecchymotic plaque on the inferior aspect of the right breast of 2 months’ duration. The patient underwent a lumpectomy, radiation, and adjuvant chemotherapy 13 years prior to presentation. Review of systems was otherwise noncontributory.
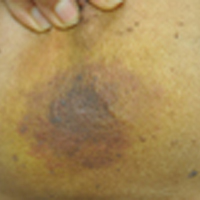
Diagnosis and Treatment of Leprosy Type 1 (Reversal) Reaction
Leprosy is a chronic granulomatous infection caused by the organism Mycobacterium leprae that primarily affects the skin and peripheral nerves.1 The organism is thought to be transmitted from person to person via the nasal secretions of infected individuals and is known to have a long incubation period, lasting 2 to 6 years.2 Leprosy has several distinct clinical presentations depending on the host immune response to the infection.3 Treatment typically involves antimicrobials (eg, clofazimine, dapsone, rifampin). Once treatment has begun, an important aspect of patient care is the recognition and treatment of leprosy reactions. Leprosy reactions are acute inflammatory complications that typically occur during the treatment course but also may occur in untreated disease. Type 1 (reversal) and type 2 (erythema nodosum leprosum) reactions are the 2 main types of leprosy reactions, which may affect 30% to 50% of all leprosy patients combined.4 Vasculonecrotic reactions (Lucio leprosy phenomenon) in leprosy are much less common.
We report a case of a 44-year-old man who repeatedly developed physical findings consistent with type 1 reactions after undergoing multiple treatments for leprosy. A discussion of leprosy, as well as its clinical manifestations, treatment options, and management of reversal reactions, also will be provided.
Case Report
A healthy 44-year-old man presented with a several month history of elevated, erythematous to yellow, anesthetic papules and plaques on the trunk (Figure 1). No other systemic symptoms were noted. Biopsies of multiple skin lesions showed noncaseating granulomas with preferential extension in a perineural pattern and tracking along the arrector pili muscle (Figure 2). The cutaneous nerves appeared to be slightly enlarged. The patient reported a history of living in Louisiana and growing up with armadillos in the backyard, often filling the holes that they dug, but he denied having direct contact with or eating armadillos. In childhood, the patient traveled across the border to Mexico a few times but only for the day. He spent several months in the Middle East (ie, Diego Garcia, Saudi Arabia) more than 10 years prior to presentation, and he spent 2 weeks in Korea approximately 2 years prior to presentation but did not travel off the US air base. He had never traveled to South America or Africa. The clinical and histopathologic findings were consistent with Hansen disease (leprosy) and were determined to be tuberculoid in type given the limited clinical presentation, tuberculoid granulomas on biopsy, and no visible organisms on histopathologic analysis.
|
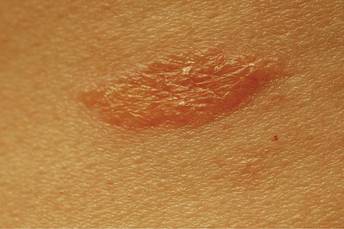
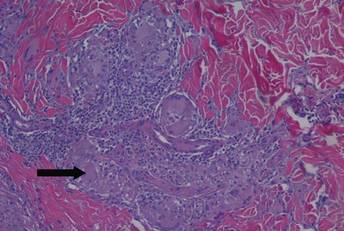
The patient initially was started on rifampin but was unable to tolerate treatment due to subsequent hepatotoxicity. He then was transitioned to a dual regimen of clofazimine and dapsone, which he tolerated well for the full 12-month treatment course. The cutaneous lesions quickly resolved after starting treatment, leaving a fine “cigarette paper–like” atrophy of the skin. After 12 months, it was subsequently presumed that the patient’s disease had been cured and treatment was stopped.
Nine months later, the patient noted new papules and plaques beginning to reappear in the truncal region. He was seen in clinic and a repeat biopsy was conducted, revealing perineural inflammation and noncaseating granulomas that were similar to the initial biopsies. Fite staining showed no acid-fast bacilli. Polymerase chain reaction was negative for M leprae. Nevertheless, a diagnosis of recurrent leprosy was made based on the patient’s clinical manifestations. He initially was started on dapsone, minocycline, and levofloxacin but was unable to tolerate the minocycline due to subsequent vertigo. After 1 month of treatment with dapsone and levofloxacin, the patient was clinically clear of all skin lesions and a repeat 12-month course of treatment was completed.
One year after completing the second 12-month treatment course, the patient again developed recurrent, indurated, erythematous papules and plaques on the trunk. Expert consultation from the National Hansen’s Disease (Leprosy) Program determined that the patient was experiencing a type 1 (reversal) reaction, not recurrent disease. Intralesional triamcinolone acetonide (10 mg/cc) was subsequently administered within the individual lesions. After a few treatments, the patient experienced notable regression of the lesions and has since been free of recurrent reactions (Figures 3 and 4).
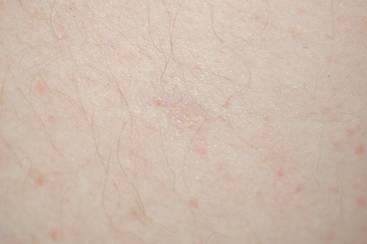
|
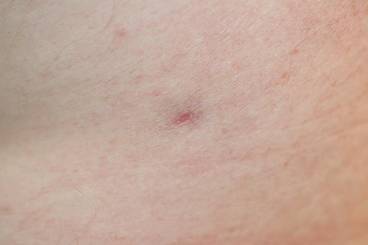
Comment
Mycobacterium leprae
Mycobacterium leprae is an obligate intracellular bacillus that is confined to humans, armadillos of specific locales, and sphagnum moss. It is an acid-fast bacillus that is microscopically indistinguishable from other mycobacteria and is best detected on Fite staining of tissue sections.5
Mycobacterium leprae has 2 unique properties. It is thermolabile, growing best at 27°C to 30°C. Given its thermal sensitivity, M leprae has a preference for peripheral tissues including the skin, peripheral nerves, and the mucosa of the upper airways. It also may affect other tissues such as the bones and some viscera.2 The other unique quality of M leprae is its slow replication, with a generation time of 12 to 14 days. Because of the slow growth of M leprae, the incubation period in humans typically ranges from 2 to 6 years, with the minimal incubation period being 2 to 3 years and the maximum incubation period being as long as 40 years.6
Perhaps the greatest challenge to investigators is the fact that M leprae cannot be grown via normal laboratory culture methods. A possible explanation is reductive evolution, which may have led to a number of inactivated (pseudogenes) in the genome of this organism. In fact, close genetic examination of this organism has led to the conclusion that only half of the genome of M leprae is actually functional. This gene decay may explain the specific host tropism of M leprae as well as the inability to culture this organism in a laboratory setting.5,7
Incidence
Leprosy is primarily a disease of developing countries. More than 80% of the world’s cases of leprosy occur in India, China, Myanmar, Indonesia, Brazil, Nigeria, Madagascar, and Nepal. Although Africa has the highest prevalence, Asia is known to have had the most cases.5 In contrast, leprosy is largely absent from Europe, Canada, and the United States, except as imported cases or scattered cases along the southern border of the United States. In the United States, for example, fewer than 100 cases of leprosy are diagnosed each year, with almost all cases identified in immigrants from endemic areas.6
The global burden of leprosy, defined as the number of new cases detected annually, is stabilizing, which can be attributed in large part to the World Health Organization’s commitment in 1991 to eliminate leprosy as a public health concern by the year 2000 by implementing worldwide treatment regimes. Elimination was defined as a prevalence of less than 1 case per 10,000 persons.8 By 2012, only 3 of 122 countries had not achieved this standard, which is evidence of the program’s success.9
Disease Transmission
There is still some uncertainty involving the mode by which leprosy is transmitted. The most widely held view is that M leprae infection occurs primarily via nasal secretions.10 Transmission is thought to be respiratory, as large numbers of bacilli typically are found in the nasal secretions of untreated patients with multibacillary disease.6 Although nasal secretions often are regarded as the most common mode of M leprae transmission, other possible modes of transmission also may be important, including direct dermal inoculation and vector transmission, though neither has been proven.10 Finally, studies involving patients with confirmed exposure to armadillos have demonstrated a 2-fold increase in the incidence of leprosy versus the general population.11 Because this topic remains controversial, additional studies are needed to ascertain the mechanism of transmission of leprosy between humans and armadillos to confirm the evidence of this study.
Classification
Clinical manifestations of leprosy vary in accordance with the immune response of the host, with the more severe forms of the disease presenting in patients with the least immunity to M leprae.12 Traditionally, patient disease is classified using the Ridley-Jopling scale, which includes tuberculoid, borderline tuberculoid, borderline, borderline lepromatous, and lepromatous types of leprosy.
Tuberculoid leprosy, as noted in our patient, is characterized by a high degree of cellular immunity, a low antigen load, a small number or absence of acid-fast bacilli in skin lesions, and a predominance of helper T cells. Skin lesions in tuberculoid leprosy usually consist of 1 to 2 large hypopigmented or erythematous anesthetic lesions with raised margins and possible overlying scale.13 In tuberculoid leprosy, neural involvement often is asymmetrical and localized and may be the sole clinical finding.10
In stark contrast, lepromatous leprosy is characterized by low cellular immunity, a large antigen load, numerous acid-fast bacilli in tissues, and a predominance of suppressor T cells. Patients with lepromatous leprosy develop widespread disease that includes cutaneous findings of diffuse erythematous macules, nodules, and papules. Disease also can be demonstrated in the upper respiratory tract, anterior chambers of the eyes, testes, lymph nodes, periosteum, and superficial sensory and motor nerves of patients with lepromatous leprosy.12 Neural involvement typically is more symmetrical and diffuse than in patients with tuberculoid leprosy.10
The spectrum of disease between tuberculoid leprosy and lepromatous leprosy includes borderline tuberculoid leprosy, borderline leprosy, and borderline lepromatous leprosy.14 The clinical presentation of borderline leprosy also varies according to the patient’s immune response. Skin lesions vary in number and usually are associated with loss of sensation. Bacilli spreading throughout the bloodstream can lead to more diffuse systemic involvement. Clinical improvement of borderline leprosy to the tuberculoid type often is seen with treatment. Disease progression or deterioration to the lepromatous type can occur with immune system compromise.14
Treatment Options
Treatment of leprosy typically involves multidrug therapy. There are several effective chemotherapeutic agents against M leprae, including dapsone, clofazimine, ofloxacin, and minocycline.15 The World Health Organization recommendations for treatment are based on the classification of patient disease as either multibacillary or paucibacillary.16 Currently, patients are classified as multibacillary if they have 6 or more skin lesions and paucibacillary if they have fewer than 6 lesions.5 World Health Organization recommendations for paucibacillary leprosy include monthly doses of rifampin along with daily doses of dapsone for 6 months. Multibacillary patients usually are treated with a combination of rifampin, dapsone, and clofazimine for 12 months.1
Management of Reversal Reactions
Leprosy reactions can occur in all leprosy patients most commonly during multidrug therapy and represent a delayed hypersensitivity response to M leprae antigens.17 Type 1 and 2 reactions together affect 40% to 50% of all patients at least once during their disease course. Type 1 reactions occur in patients in the tuberculoid and borderline portion of the spectrum. These reactions manifest as erythema and induration of existing plaques. Frequently, progressive neuritis leads to sensory and motor neuropathy. These type 1 reactions typically develop gradually and may last for several weeks.4 Type 2 reactions occur in patients with borderline lepromatous leprosy and lepromatous leprosy and are characterized by the appearance of tender, erythematous, subcutaneous nodules. They are often accompanied by systemic symptoms such as malaise, fever, edema, arthralgia, and weight loss. Organ systems including the joints, eyes, testes, and nervous system also may be affected.18 The natural course of a type 2 reaction is 1 to 2 weeks, but many patients experience multiple recurrences over several months.
All leprosy reactions are believed to be immunologically mediated; however, the mechanism responsible for each reaction type remains poorly defined. The histology of type 1 reactions is that of a delayed-type hypersensitivity response with CD4+ T cells, macrophages, and expression of IL-2 in lesions. In type 1 reactions, increases in cytokines including IL-1, IL-2, IL-12, IFN-γ, and tumor necrosis factor a have been documented both locally within the skin and systemically in the serum. However, studies have not been able to differentiate if this enhanced TH1 response is related to an immunological versus an inflammatory process.19
Type 2 leprosy reactions occur in patients with poor cellular immunity to M leprae. The acute lesions typically are characterized by a neutrophilic infiltrate superimposed on a chronic lepromatous pattern, and there is a systemic inflammatory response to immune complex deposition. It has been proposed that type 2 leprosy reactions are a type of Arthus reaction characterized by deposition of an immunoglobulin-antigen complex in vascular endothelium with subsequent complement activation. Both immunoglobulin and complement have been demonstrated in the reactive nodules of type 2 reactions, and serum complement is decreased in these patients, supporting this pathogenic process.4 Other studies have identified possible immune cell activation in type 2 reactions, including increases in TH2-related cytokines.19
These immunologic reactions can ultimately lead to impaired motor, sensory, and autonomic nerve function if allowed to progress.20 As a result, anesthetic limbs are subjected to repeated trauma, infection, and pressure necrosis that may lead to limb deformity. Autonomic nerve dysfunction may lead to loss of the corneal reflex, which can result in blindness. Common motor findings include wrist and foot drop as well as clawing of the hand from damage to the nerves of the upper extremity.20
Treatment of both type 1 and 2 leprosy reactions is imperative, as these inflammatory reactions are responsible for a great deal of the permanent nerve damage, deformity, and disability that is associated with leprosy.21 Oral and intralesional corticosteroids typically are highly effective for the clinical treatment of type 1 and 2 leprosy reactions given their anti-inflammatory properties. Our patient’s type 1 leprosy reaction responded well to intralesional corticosteroid injections. Thalidomide also has proven to be highly effective in treating type 2 reactions and was used frequently prior to realization of its teratogenic effects. It is now prohibited for use in women of childbearing age but is still routinely used in many countries for the treatment of type 2 reactions in men and postmenopausal women. Other therapies for type 2 reactions that have been used with some success include cyclosporine, azathioprine, and pentoxifylline.4
Conclusion
In summary, we present a unique case of multiple cutaneous reversal reactions in a patient with leprosy years after successful antimicrobial therapy. Proper recognition of this phenomenon is important to avoid overtreatment for mistaken recurrent disease. Although rare in the United States, leprosy should be considered in the differential diagnosis of patients presenting with hypoesthetic or anesthetic skin lesions, chronic annular dermatitis, papular or nodular granulomatous skin lesions, diffuse cutaneous infiltrative disease, peripheral neuropathy, and a history of travel to regions where the disease is known to be endemic. Additionally, if left untreated, M leprae infection and subsequent type 1 or type 2 reactions can lead to devastating neurologic and cutaneous sequelae. Prompt recognition and treatment of these reactions is imperative to prevent these long-term complications.
1. Kalisiak M, Yeung R, Dytoc M. Dermacase: leprosy. Can Fam Physician. 2009;55:55-56.
2. Kustner EC, Cruz MP, Dansis CP, et al. Lepromatous leprosy: a review and case report. Med Oral Patol Cir Bucal. 2006;11:474-479.
3. Nunez-Gussman J, Hwang L, Hsu S. Targetoid erythematous plaques: an unusual morphological presentation of multibacillary Hansen’s disease. Eur J Dermatol. 2001;11:65-67.
4. Scollard DM, Adams LB, Gillis TP, et al. The continuing challenges of leprosy. Clin Microbiol Rev. 2006;19:338-381.
5. Gelber RH. Leprosy (Hansen’s disease). In: Fauci AS, Kasper DL, Longo DL, et al, eds. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill; 2008:1021-1027.
6. Simon HB. Infections due to mycobacterium. In: Dale DC, Federman DD, Antman K, et al, eds. ACP Medicine. New York, NY: WebMD Professional Publishing; 2004:1703-1720.
7. Baker LP. Mycobacterium leprae interactions with the host cell: recent advances. Indian J Med Res. 2006;123:748-759.
8. Ishii N. Recent advances in the treatment of leprosy. Dermatol Online J. 2003;9:5. http://dermatology.cdlib.org/92/reviews/leprosy/ishii.html. Accessed March 16, 2015.
9. World Health Organization. Global leprosy situation, 2012. Weekly Epidemiological Rec. 2013;88:365-380. http://www.who.int/wer/2007/wer8835.pdf. Accessed March 16, 2015.
10. Gelber RH. Hansen disease. West J Med. 1993;158:583-590.
11. Deps PD, Alves L, Gripp CG, et al. Contact with armadillos increases the risk of leprosy in Brazil: a case control study. Indian J Dermatol Venereol Leprol. 2008;74:338-342.
12. Booth AV, Kovich OI. Lepromatous leprosy. Dermatol Online J. 2007;13:9.
13. Yens DA, Asters DJ, Teitel A. Subcutaneous nodules and joint deformity in leprosy. Clin Rheumatol. 2003;9:181-186.
14. Panezai S, Saleh FG. Leprosy and peripheral neuropathy. J Clin Neuromuscul Dis. 2004;5:138-145.
15. Sasaki S, Takeshita F, Okuda K, et al. Mycobacterium leprae and leprosy. Microbiol Immunol. 2001;45:729-736.
16. Kumar A, Girdhar A, Chakma J, et al. WHO Multidrug Therapy for Leprosy: Epidemiology of default in treatment in Agra District, Uttar Pradesh, India. BioMed Research International. doi:10.1155/2015/705804.
17. Fiallo P, Clapasson A, Favre A, et al. Overexpression of vascular endothelial growth factor and its endothelial cell receptor KDR in type I leprosy reaction. Am J Med Hyg. 2002;66:180-185.
18. Sales AM, de Matos HJ, Nery JAC, et al. Double-blind trial of the efficacy of pentoxifylline vs thalidomide for the treatment of type II reaction in leprosy. Braz J Med Biol Res. 2007;40:243-248.
19. Lockwood DN, Colston MJ, Khanolkar-Young SR. The detection of Mycobacterium leprae protein and carbohydrate antigens in skin and nerve from leprosy patients with type I (reversal) reactions. Am J Trop Med Hyg. 2002;66:409-415.
20. Boggild AK, Keystone JS, Kain KC. Leprosy: a primer for Canadian physicians. CMAJ. 2004;170:71-78.
21. Rook GA, Baker R. Cortisol metabolism, cortisol sensitivity and the pathogenesis of leprosy reactions. Trop Med Int Health. 1999;4:493-498.
Leprosy is a chronic granulomatous infection caused by the organism Mycobacterium leprae that primarily affects the skin and peripheral nerves.1 The organism is thought to be transmitted from person to person via the nasal secretions of infected individuals and is known to have a long incubation period, lasting 2 to 6 years.2 Leprosy has several distinct clinical presentations depending on the host immune response to the infection.3 Treatment typically involves antimicrobials (eg, clofazimine, dapsone, rifampin). Once treatment has begun, an important aspect of patient care is the recognition and treatment of leprosy reactions. Leprosy reactions are acute inflammatory complications that typically occur during the treatment course but also may occur in untreated disease. Type 1 (reversal) and type 2 (erythema nodosum leprosum) reactions are the 2 main types of leprosy reactions, which may affect 30% to 50% of all leprosy patients combined.4 Vasculonecrotic reactions (Lucio leprosy phenomenon) in leprosy are much less common.
We report a case of a 44-year-old man who repeatedly developed physical findings consistent with type 1 reactions after undergoing multiple treatments for leprosy. A discussion of leprosy, as well as its clinical manifestations, treatment options, and management of reversal reactions, also will be provided.
Case Report
A healthy 44-year-old man presented with a several month history of elevated, erythematous to yellow, anesthetic papules and plaques on the trunk (Figure 1). No other systemic symptoms were noted. Biopsies of multiple skin lesions showed noncaseating granulomas with preferential extension in a perineural pattern and tracking along the arrector pili muscle (Figure 2). The cutaneous nerves appeared to be slightly enlarged. The patient reported a history of living in Louisiana and growing up with armadillos in the backyard, often filling the holes that they dug, but he denied having direct contact with or eating armadillos. In childhood, the patient traveled across the border to Mexico a few times but only for the day. He spent several months in the Middle East (ie, Diego Garcia, Saudi Arabia) more than 10 years prior to presentation, and he spent 2 weeks in Korea approximately 2 years prior to presentation but did not travel off the US air base. He had never traveled to South America or Africa. The clinical and histopathologic findings were consistent with Hansen disease (leprosy) and were determined to be tuberculoid in type given the limited clinical presentation, tuberculoid granulomas on biopsy, and no visible organisms on histopathologic analysis.
|
The patient initially was started on rifampin but was unable to tolerate treatment due to subsequent hepatotoxicity. He then was transitioned to a dual regimen of clofazimine and dapsone, which he tolerated well for the full 12-month treatment course. The cutaneous lesions quickly resolved after starting treatment, leaving a fine “cigarette paper–like” atrophy of the skin. After 12 months, it was subsequently presumed that the patient’s disease had been cured and treatment was stopped.
Nine months later, the patient noted new papules and plaques beginning to reappear in the truncal region. He was seen in clinic and a repeat biopsy was conducted, revealing perineural inflammation and noncaseating granulomas that were similar to the initial biopsies. Fite staining showed no acid-fast bacilli. Polymerase chain reaction was negative for M leprae. Nevertheless, a diagnosis of recurrent leprosy was made based on the patient’s clinical manifestations. He initially was started on dapsone, minocycline, and levofloxacin but was unable to tolerate the minocycline due to subsequent vertigo. After 1 month of treatment with dapsone and levofloxacin, the patient was clinically clear of all skin lesions and a repeat 12-month course of treatment was completed.
One year after completing the second 12-month treatment course, the patient again developed recurrent, indurated, erythematous papules and plaques on the trunk. Expert consultation from the National Hansen’s Disease (Leprosy) Program determined that the patient was experiencing a type 1 (reversal) reaction, not recurrent disease. Intralesional triamcinolone acetonide (10 mg/cc) was subsequently administered within the individual lesions. After a few treatments, the patient experienced notable regression of the lesions and has since been free of recurrent reactions (Figures 3 and 4).
|
Comment
Mycobacterium leprae
Mycobacterium leprae is an obligate intracellular bacillus that is confined to humans, armadillos of specific locales, and sphagnum moss. It is an acid-fast bacillus that is microscopically indistinguishable from other mycobacteria and is best detected on Fite staining of tissue sections.5
Mycobacterium leprae has 2 unique properties. It is thermolabile, growing best at 27°C to 30°C. Given its thermal sensitivity, M leprae has a preference for peripheral tissues including the skin, peripheral nerves, and the mucosa of the upper airways. It also may affect other tissues such as the bones and some viscera.2 The other unique quality of M leprae is its slow replication, with a generation time of 12 to 14 days. Because of the slow growth of M leprae, the incubation period in humans typically ranges from 2 to 6 years, with the minimal incubation period being 2 to 3 years and the maximum incubation period being as long as 40 years.6
Perhaps the greatest challenge to investigators is the fact that M leprae cannot be grown via normal laboratory culture methods. A possible explanation is reductive evolution, which may have led to a number of inactivated (pseudogenes) in the genome of this organism. In fact, close genetic examination of this organism has led to the conclusion that only half of the genome of M leprae is actually functional. This gene decay may explain the specific host tropism of M leprae as well as the inability to culture this organism in a laboratory setting.5,7
Incidence
Leprosy is primarily a disease of developing countries. More than 80% of the world’s cases of leprosy occur in India, China, Myanmar, Indonesia, Brazil, Nigeria, Madagascar, and Nepal. Although Africa has the highest prevalence, Asia is known to have had the most cases.5 In contrast, leprosy is largely absent from Europe, Canada, and the United States, except as imported cases or scattered cases along the southern border of the United States. In the United States, for example, fewer than 100 cases of leprosy are diagnosed each year, with almost all cases identified in immigrants from endemic areas.6
The global burden of leprosy, defined as the number of new cases detected annually, is stabilizing, which can be attributed in large part to the World Health Organization’s commitment in 1991 to eliminate leprosy as a public health concern by the year 2000 by implementing worldwide treatment regimes. Elimination was defined as a prevalence of less than 1 case per 10,000 persons.8 By 2012, only 3 of 122 countries had not achieved this standard, which is evidence of the program’s success.9
Disease Transmission
There is still some uncertainty involving the mode by which leprosy is transmitted. The most widely held view is that M leprae infection occurs primarily via nasal secretions.10 Transmission is thought to be respiratory, as large numbers of bacilli typically are found in the nasal secretions of untreated patients with multibacillary disease.6 Although nasal secretions often are regarded as the most common mode of M leprae transmission, other possible modes of transmission also may be important, including direct dermal inoculation and vector transmission, though neither has been proven.10 Finally, studies involving patients with confirmed exposure to armadillos have demonstrated a 2-fold increase in the incidence of leprosy versus the general population.11 Because this topic remains controversial, additional studies are needed to ascertain the mechanism of transmission of leprosy between humans and armadillos to confirm the evidence of this study.
Classification
Clinical manifestations of leprosy vary in accordance with the immune response of the host, with the more severe forms of the disease presenting in patients with the least immunity to M leprae.12 Traditionally, patient disease is classified using the Ridley-Jopling scale, which includes tuberculoid, borderline tuberculoid, borderline, borderline lepromatous, and lepromatous types of leprosy.
Tuberculoid leprosy, as noted in our patient, is characterized by a high degree of cellular immunity, a low antigen load, a small number or absence of acid-fast bacilli in skin lesions, and a predominance of helper T cells. Skin lesions in tuberculoid leprosy usually consist of 1 to 2 large hypopigmented or erythematous anesthetic lesions with raised margins and possible overlying scale.13 In tuberculoid leprosy, neural involvement often is asymmetrical and localized and may be the sole clinical finding.10
In stark contrast, lepromatous leprosy is characterized by low cellular immunity, a large antigen load, numerous acid-fast bacilli in tissues, and a predominance of suppressor T cells. Patients with lepromatous leprosy develop widespread disease that includes cutaneous findings of diffuse erythematous macules, nodules, and papules. Disease also can be demonstrated in the upper respiratory tract, anterior chambers of the eyes, testes, lymph nodes, periosteum, and superficial sensory and motor nerves of patients with lepromatous leprosy.12 Neural involvement typically is more symmetrical and diffuse than in patients with tuberculoid leprosy.10
The spectrum of disease between tuberculoid leprosy and lepromatous leprosy includes borderline tuberculoid leprosy, borderline leprosy, and borderline lepromatous leprosy.14 The clinical presentation of borderline leprosy also varies according to the patient’s immune response. Skin lesions vary in number and usually are associated with loss of sensation. Bacilli spreading throughout the bloodstream can lead to more diffuse systemic involvement. Clinical improvement of borderline leprosy to the tuberculoid type often is seen with treatment. Disease progression or deterioration to the lepromatous type can occur with immune system compromise.14
Treatment Options
Treatment of leprosy typically involves multidrug therapy. There are several effective chemotherapeutic agents against M leprae, including dapsone, clofazimine, ofloxacin, and minocycline.15 The World Health Organization recommendations for treatment are based on the classification of patient disease as either multibacillary or paucibacillary.16 Currently, patients are classified as multibacillary if they have 6 or more skin lesions and paucibacillary if they have fewer than 6 lesions.5 World Health Organization recommendations for paucibacillary leprosy include monthly doses of rifampin along with daily doses of dapsone for 6 months. Multibacillary patients usually are treated with a combination of rifampin, dapsone, and clofazimine for 12 months.1
Management of Reversal Reactions
Leprosy reactions can occur in all leprosy patients most commonly during multidrug therapy and represent a delayed hypersensitivity response to M leprae antigens.17 Type 1 and 2 reactions together affect 40% to 50% of all patients at least once during their disease course. Type 1 reactions occur in patients in the tuberculoid and borderline portion of the spectrum. These reactions manifest as erythema and induration of existing plaques. Frequently, progressive neuritis leads to sensory and motor neuropathy. These type 1 reactions typically develop gradually and may last for several weeks.4 Type 2 reactions occur in patients with borderline lepromatous leprosy and lepromatous leprosy and are characterized by the appearance of tender, erythematous, subcutaneous nodules. They are often accompanied by systemic symptoms such as malaise, fever, edema, arthralgia, and weight loss. Organ systems including the joints, eyes, testes, and nervous system also may be affected.18 The natural course of a type 2 reaction is 1 to 2 weeks, but many patients experience multiple recurrences over several months.
All leprosy reactions are believed to be immunologically mediated; however, the mechanism responsible for each reaction type remains poorly defined. The histology of type 1 reactions is that of a delayed-type hypersensitivity response with CD4+ T cells, macrophages, and expression of IL-2 in lesions. In type 1 reactions, increases in cytokines including IL-1, IL-2, IL-12, IFN-γ, and tumor necrosis factor a have been documented both locally within the skin and systemically in the serum. However, studies have not been able to differentiate if this enhanced TH1 response is related to an immunological versus an inflammatory process.19
Type 2 leprosy reactions occur in patients with poor cellular immunity to M leprae. The acute lesions typically are characterized by a neutrophilic infiltrate superimposed on a chronic lepromatous pattern, and there is a systemic inflammatory response to immune complex deposition. It has been proposed that type 2 leprosy reactions are a type of Arthus reaction characterized by deposition of an immunoglobulin-antigen complex in vascular endothelium with subsequent complement activation. Both immunoglobulin and complement have been demonstrated in the reactive nodules of type 2 reactions, and serum complement is decreased in these patients, supporting this pathogenic process.4 Other studies have identified possible immune cell activation in type 2 reactions, including increases in TH2-related cytokines.19
These immunologic reactions can ultimately lead to impaired motor, sensory, and autonomic nerve function if allowed to progress.20 As a result, anesthetic limbs are subjected to repeated trauma, infection, and pressure necrosis that may lead to limb deformity. Autonomic nerve dysfunction may lead to loss of the corneal reflex, which can result in blindness. Common motor findings include wrist and foot drop as well as clawing of the hand from damage to the nerves of the upper extremity.20
Treatment of both type 1 and 2 leprosy reactions is imperative, as these inflammatory reactions are responsible for a great deal of the permanent nerve damage, deformity, and disability that is associated with leprosy.21 Oral and intralesional corticosteroids typically are highly effective for the clinical treatment of type 1 and 2 leprosy reactions given their anti-inflammatory properties. Our patient’s type 1 leprosy reaction responded well to intralesional corticosteroid injections. Thalidomide also has proven to be highly effective in treating type 2 reactions and was used frequently prior to realization of its teratogenic effects. It is now prohibited for use in women of childbearing age but is still routinely used in many countries for the treatment of type 2 reactions in men and postmenopausal women. Other therapies for type 2 reactions that have been used with some success include cyclosporine, azathioprine, and pentoxifylline.4
Conclusion
In summary, we present a unique case of multiple cutaneous reversal reactions in a patient with leprosy years after successful antimicrobial therapy. Proper recognition of this phenomenon is important to avoid overtreatment for mistaken recurrent disease. Although rare in the United States, leprosy should be considered in the differential diagnosis of patients presenting with hypoesthetic or anesthetic skin lesions, chronic annular dermatitis, papular or nodular granulomatous skin lesions, diffuse cutaneous infiltrative disease, peripheral neuropathy, and a history of travel to regions where the disease is known to be endemic. Additionally, if left untreated, M leprae infection and subsequent type 1 or type 2 reactions can lead to devastating neurologic and cutaneous sequelae. Prompt recognition and treatment of these reactions is imperative to prevent these long-term complications.
Leprosy is a chronic granulomatous infection caused by the organism Mycobacterium leprae that primarily affects the skin and peripheral nerves.1 The organism is thought to be transmitted from person to person via the nasal secretions of infected individuals and is known to have a long incubation period, lasting 2 to 6 years.2 Leprosy has several distinct clinical presentations depending on the host immune response to the infection.3 Treatment typically involves antimicrobials (eg, clofazimine, dapsone, rifampin). Once treatment has begun, an important aspect of patient care is the recognition and treatment of leprosy reactions. Leprosy reactions are acute inflammatory complications that typically occur during the treatment course but also may occur in untreated disease. Type 1 (reversal) and type 2 (erythema nodosum leprosum) reactions are the 2 main types of leprosy reactions, which may affect 30% to 50% of all leprosy patients combined.4 Vasculonecrotic reactions (Lucio leprosy phenomenon) in leprosy are much less common.
We report a case of a 44-year-old man who repeatedly developed physical findings consistent with type 1 reactions after undergoing multiple treatments for leprosy. A discussion of leprosy, as well as its clinical manifestations, treatment options, and management of reversal reactions, also will be provided.
Case Report
A healthy 44-year-old man presented with a several month history of elevated, erythematous to yellow, anesthetic papules and plaques on the trunk (Figure 1). No other systemic symptoms were noted. Biopsies of multiple skin lesions showed noncaseating granulomas with preferential extension in a perineural pattern and tracking along the arrector pili muscle (Figure 2). The cutaneous nerves appeared to be slightly enlarged. The patient reported a history of living in Louisiana and growing up with armadillos in the backyard, often filling the holes that they dug, but he denied having direct contact with or eating armadillos. In childhood, the patient traveled across the border to Mexico a few times but only for the day. He spent several months in the Middle East (ie, Diego Garcia, Saudi Arabia) more than 10 years prior to presentation, and he spent 2 weeks in Korea approximately 2 years prior to presentation but did not travel off the US air base. He had never traveled to South America or Africa. The clinical and histopathologic findings were consistent with Hansen disease (leprosy) and were determined to be tuberculoid in type given the limited clinical presentation, tuberculoid granulomas on biopsy, and no visible organisms on histopathologic analysis.
|
The patient initially was started on rifampin but was unable to tolerate treatment due to subsequent hepatotoxicity. He then was transitioned to a dual regimen of clofazimine and dapsone, which he tolerated well for the full 12-month treatment course. The cutaneous lesions quickly resolved after starting treatment, leaving a fine “cigarette paper–like” atrophy of the skin. After 12 months, it was subsequently presumed that the patient’s disease had been cured and treatment was stopped.
Nine months later, the patient noted new papules and plaques beginning to reappear in the truncal region. He was seen in clinic and a repeat biopsy was conducted, revealing perineural inflammation and noncaseating granulomas that were similar to the initial biopsies. Fite staining showed no acid-fast bacilli. Polymerase chain reaction was negative for M leprae. Nevertheless, a diagnosis of recurrent leprosy was made based on the patient’s clinical manifestations. He initially was started on dapsone, minocycline, and levofloxacin but was unable to tolerate the minocycline due to subsequent vertigo. After 1 month of treatment with dapsone and levofloxacin, the patient was clinically clear of all skin lesions and a repeat 12-month course of treatment was completed.
One year after completing the second 12-month treatment course, the patient again developed recurrent, indurated, erythematous papules and plaques on the trunk. Expert consultation from the National Hansen’s Disease (Leprosy) Program determined that the patient was experiencing a type 1 (reversal) reaction, not recurrent disease. Intralesional triamcinolone acetonide (10 mg/cc) was subsequently administered within the individual lesions. After a few treatments, the patient experienced notable regression of the lesions and has since been free of recurrent reactions (Figures 3 and 4).
|
Comment
Mycobacterium leprae
Mycobacterium leprae is an obligate intracellular bacillus that is confined to humans, armadillos of specific locales, and sphagnum moss. It is an acid-fast bacillus that is microscopically indistinguishable from other mycobacteria and is best detected on Fite staining of tissue sections.5
Mycobacterium leprae has 2 unique properties. It is thermolabile, growing best at 27°C to 30°C. Given its thermal sensitivity, M leprae has a preference for peripheral tissues including the skin, peripheral nerves, and the mucosa of the upper airways. It also may affect other tissues such as the bones and some viscera.2 The other unique quality of M leprae is its slow replication, with a generation time of 12 to 14 days. Because of the slow growth of M leprae, the incubation period in humans typically ranges from 2 to 6 years, with the minimal incubation period being 2 to 3 years and the maximum incubation period being as long as 40 years.6
Perhaps the greatest challenge to investigators is the fact that M leprae cannot be grown via normal laboratory culture methods. A possible explanation is reductive evolution, which may have led to a number of inactivated (pseudogenes) in the genome of this organism. In fact, close genetic examination of this organism has led to the conclusion that only half of the genome of M leprae is actually functional. This gene decay may explain the specific host tropism of M leprae as well as the inability to culture this organism in a laboratory setting.5,7
Incidence
Leprosy is primarily a disease of developing countries. More than 80% of the world’s cases of leprosy occur in India, China, Myanmar, Indonesia, Brazil, Nigeria, Madagascar, and Nepal. Although Africa has the highest prevalence, Asia is known to have had the most cases.5 In contrast, leprosy is largely absent from Europe, Canada, and the United States, except as imported cases or scattered cases along the southern border of the United States. In the United States, for example, fewer than 100 cases of leprosy are diagnosed each year, with almost all cases identified in immigrants from endemic areas.6
The global burden of leprosy, defined as the number of new cases detected annually, is stabilizing, which can be attributed in large part to the World Health Organization’s commitment in 1991 to eliminate leprosy as a public health concern by the year 2000 by implementing worldwide treatment regimes. Elimination was defined as a prevalence of less than 1 case per 10,000 persons.8 By 2012, only 3 of 122 countries had not achieved this standard, which is evidence of the program’s success.9
Disease Transmission
There is still some uncertainty involving the mode by which leprosy is transmitted. The most widely held view is that M leprae infection occurs primarily via nasal secretions.10 Transmission is thought to be respiratory, as large numbers of bacilli typically are found in the nasal secretions of untreated patients with multibacillary disease.6 Although nasal secretions often are regarded as the most common mode of M leprae transmission, other possible modes of transmission also may be important, including direct dermal inoculation and vector transmission, though neither has been proven.10 Finally, studies involving patients with confirmed exposure to armadillos have demonstrated a 2-fold increase in the incidence of leprosy versus the general population.11 Because this topic remains controversial, additional studies are needed to ascertain the mechanism of transmission of leprosy between humans and armadillos to confirm the evidence of this study.
Classification
Clinical manifestations of leprosy vary in accordance with the immune response of the host, with the more severe forms of the disease presenting in patients with the least immunity to M leprae.12 Traditionally, patient disease is classified using the Ridley-Jopling scale, which includes tuberculoid, borderline tuberculoid, borderline, borderline lepromatous, and lepromatous types of leprosy.
Tuberculoid leprosy, as noted in our patient, is characterized by a high degree of cellular immunity, a low antigen load, a small number or absence of acid-fast bacilli in skin lesions, and a predominance of helper T cells. Skin lesions in tuberculoid leprosy usually consist of 1 to 2 large hypopigmented or erythematous anesthetic lesions with raised margins and possible overlying scale.13 In tuberculoid leprosy, neural involvement often is asymmetrical and localized and may be the sole clinical finding.10
In stark contrast, lepromatous leprosy is characterized by low cellular immunity, a large antigen load, numerous acid-fast bacilli in tissues, and a predominance of suppressor T cells. Patients with lepromatous leprosy develop widespread disease that includes cutaneous findings of diffuse erythematous macules, nodules, and papules. Disease also can be demonstrated in the upper respiratory tract, anterior chambers of the eyes, testes, lymph nodes, periosteum, and superficial sensory and motor nerves of patients with lepromatous leprosy.12 Neural involvement typically is more symmetrical and diffuse than in patients with tuberculoid leprosy.10
The spectrum of disease between tuberculoid leprosy and lepromatous leprosy includes borderline tuberculoid leprosy, borderline leprosy, and borderline lepromatous leprosy.14 The clinical presentation of borderline leprosy also varies according to the patient’s immune response. Skin lesions vary in number and usually are associated with loss of sensation. Bacilli spreading throughout the bloodstream can lead to more diffuse systemic involvement. Clinical improvement of borderline leprosy to the tuberculoid type often is seen with treatment. Disease progression or deterioration to the lepromatous type can occur with immune system compromise.14
Treatment Options
Treatment of leprosy typically involves multidrug therapy. There are several effective chemotherapeutic agents against M leprae, including dapsone, clofazimine, ofloxacin, and minocycline.15 The World Health Organization recommendations for treatment are based on the classification of patient disease as either multibacillary or paucibacillary.16 Currently, patients are classified as multibacillary if they have 6 or more skin lesions and paucibacillary if they have fewer than 6 lesions.5 World Health Organization recommendations for paucibacillary leprosy include monthly doses of rifampin along with daily doses of dapsone for 6 months. Multibacillary patients usually are treated with a combination of rifampin, dapsone, and clofazimine for 12 months.1
Management of Reversal Reactions
Leprosy reactions can occur in all leprosy patients most commonly during multidrug therapy and represent a delayed hypersensitivity response to M leprae antigens.17 Type 1 and 2 reactions together affect 40% to 50% of all patients at least once during their disease course. Type 1 reactions occur in patients in the tuberculoid and borderline portion of the spectrum. These reactions manifest as erythema and induration of existing plaques. Frequently, progressive neuritis leads to sensory and motor neuropathy. These type 1 reactions typically develop gradually and may last for several weeks.4 Type 2 reactions occur in patients with borderline lepromatous leprosy and lepromatous leprosy and are characterized by the appearance of tender, erythematous, subcutaneous nodules. They are often accompanied by systemic symptoms such as malaise, fever, edema, arthralgia, and weight loss. Organ systems including the joints, eyes, testes, and nervous system also may be affected.18 The natural course of a type 2 reaction is 1 to 2 weeks, but many patients experience multiple recurrences over several months.
All leprosy reactions are believed to be immunologically mediated; however, the mechanism responsible for each reaction type remains poorly defined. The histology of type 1 reactions is that of a delayed-type hypersensitivity response with CD4+ T cells, macrophages, and expression of IL-2 in lesions. In type 1 reactions, increases in cytokines including IL-1, IL-2, IL-12, IFN-γ, and tumor necrosis factor a have been documented both locally within the skin and systemically in the serum. However, studies have not been able to differentiate if this enhanced TH1 response is related to an immunological versus an inflammatory process.19
Type 2 leprosy reactions occur in patients with poor cellular immunity to M leprae. The acute lesions typically are characterized by a neutrophilic infiltrate superimposed on a chronic lepromatous pattern, and there is a systemic inflammatory response to immune complex deposition. It has been proposed that type 2 leprosy reactions are a type of Arthus reaction characterized by deposition of an immunoglobulin-antigen complex in vascular endothelium with subsequent complement activation. Both immunoglobulin and complement have been demonstrated in the reactive nodules of type 2 reactions, and serum complement is decreased in these patients, supporting this pathogenic process.4 Other studies have identified possible immune cell activation in type 2 reactions, including increases in TH2-related cytokines.19
These immunologic reactions can ultimately lead to impaired motor, sensory, and autonomic nerve function if allowed to progress.20 As a result, anesthetic limbs are subjected to repeated trauma, infection, and pressure necrosis that may lead to limb deformity. Autonomic nerve dysfunction may lead to loss of the corneal reflex, which can result in blindness. Common motor findings include wrist and foot drop as well as clawing of the hand from damage to the nerves of the upper extremity.20
Treatment of both type 1 and 2 leprosy reactions is imperative, as these inflammatory reactions are responsible for a great deal of the permanent nerve damage, deformity, and disability that is associated with leprosy.21 Oral and intralesional corticosteroids typically are highly effective for the clinical treatment of type 1 and 2 leprosy reactions given their anti-inflammatory properties. Our patient’s type 1 leprosy reaction responded well to intralesional corticosteroid injections. Thalidomide also has proven to be highly effective in treating type 2 reactions and was used frequently prior to realization of its teratogenic effects. It is now prohibited for use in women of childbearing age but is still routinely used in many countries for the treatment of type 2 reactions in men and postmenopausal women. Other therapies for type 2 reactions that have been used with some success include cyclosporine, azathioprine, and pentoxifylline.4
Conclusion
In summary, we present a unique case of multiple cutaneous reversal reactions in a patient with leprosy years after successful antimicrobial therapy. Proper recognition of this phenomenon is important to avoid overtreatment for mistaken recurrent disease. Although rare in the United States, leprosy should be considered in the differential diagnosis of patients presenting with hypoesthetic or anesthetic skin lesions, chronic annular dermatitis, papular or nodular granulomatous skin lesions, diffuse cutaneous infiltrative disease, peripheral neuropathy, and a history of travel to regions where the disease is known to be endemic. Additionally, if left untreated, M leprae infection and subsequent type 1 or type 2 reactions can lead to devastating neurologic and cutaneous sequelae. Prompt recognition and treatment of these reactions is imperative to prevent these long-term complications.
1. Kalisiak M, Yeung R, Dytoc M. Dermacase: leprosy. Can Fam Physician. 2009;55:55-56.
2. Kustner EC, Cruz MP, Dansis CP, et al. Lepromatous leprosy: a review and case report. Med Oral Patol Cir Bucal. 2006;11:474-479.
3. Nunez-Gussman J, Hwang L, Hsu S. Targetoid erythematous plaques: an unusual morphological presentation of multibacillary Hansen’s disease. Eur J Dermatol. 2001;11:65-67.
4. Scollard DM, Adams LB, Gillis TP, et al. The continuing challenges of leprosy. Clin Microbiol Rev. 2006;19:338-381.
5. Gelber RH. Leprosy (Hansen’s disease). In: Fauci AS, Kasper DL, Longo DL, et al, eds. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill; 2008:1021-1027.
6. Simon HB. Infections due to mycobacterium. In: Dale DC, Federman DD, Antman K, et al, eds. ACP Medicine. New York, NY: WebMD Professional Publishing; 2004:1703-1720.
7. Baker LP. Mycobacterium leprae interactions with the host cell: recent advances. Indian J Med Res. 2006;123:748-759.
8. Ishii N. Recent advances in the treatment of leprosy. Dermatol Online J. 2003;9:5. http://dermatology.cdlib.org/92/reviews/leprosy/ishii.html. Accessed March 16, 2015.
9. World Health Organization. Global leprosy situation, 2012. Weekly Epidemiological Rec. 2013;88:365-380. http://www.who.int/wer/2007/wer8835.pdf. Accessed March 16, 2015.
10. Gelber RH. Hansen disease. West J Med. 1993;158:583-590.
11. Deps PD, Alves L, Gripp CG, et al. Contact with armadillos increases the risk of leprosy in Brazil: a case control study. Indian J Dermatol Venereol Leprol. 2008;74:338-342.
12. Booth AV, Kovich OI. Lepromatous leprosy. Dermatol Online J. 2007;13:9.
13. Yens DA, Asters DJ, Teitel A. Subcutaneous nodules and joint deformity in leprosy. Clin Rheumatol. 2003;9:181-186.
14. Panezai S, Saleh FG. Leprosy and peripheral neuropathy. J Clin Neuromuscul Dis. 2004;5:138-145.
15. Sasaki S, Takeshita F, Okuda K, et al. Mycobacterium leprae and leprosy. Microbiol Immunol. 2001;45:729-736.
16. Kumar A, Girdhar A, Chakma J, et al. WHO Multidrug Therapy for Leprosy: Epidemiology of default in treatment in Agra District, Uttar Pradesh, India. BioMed Research International. doi:10.1155/2015/705804.
17. Fiallo P, Clapasson A, Favre A, et al. Overexpression of vascular endothelial growth factor and its endothelial cell receptor KDR in type I leprosy reaction. Am J Med Hyg. 2002;66:180-185.
18. Sales AM, de Matos HJ, Nery JAC, et al. Double-blind trial of the efficacy of pentoxifylline vs thalidomide for the treatment of type II reaction in leprosy. Braz J Med Biol Res. 2007;40:243-248.
19. Lockwood DN, Colston MJ, Khanolkar-Young SR. The detection of Mycobacterium leprae protein and carbohydrate antigens in skin and nerve from leprosy patients with type I (reversal) reactions. Am J Trop Med Hyg. 2002;66:409-415.
20. Boggild AK, Keystone JS, Kain KC. Leprosy: a primer for Canadian physicians. CMAJ. 2004;170:71-78.
21. Rook GA, Baker R. Cortisol metabolism, cortisol sensitivity and the pathogenesis of leprosy reactions. Trop Med Int Health. 1999;4:493-498.
1. Kalisiak M, Yeung R, Dytoc M. Dermacase: leprosy. Can Fam Physician. 2009;55:55-56.
2. Kustner EC, Cruz MP, Dansis CP, et al. Lepromatous leprosy: a review and case report. Med Oral Patol Cir Bucal. 2006;11:474-479.
3. Nunez-Gussman J, Hwang L, Hsu S. Targetoid erythematous plaques: an unusual morphological presentation of multibacillary Hansen’s disease. Eur J Dermatol. 2001;11:65-67.
4. Scollard DM, Adams LB, Gillis TP, et al. The continuing challenges of leprosy. Clin Microbiol Rev. 2006;19:338-381.
5. Gelber RH. Leprosy (Hansen’s disease). In: Fauci AS, Kasper DL, Longo DL, et al, eds. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill; 2008:1021-1027.
6. Simon HB. Infections due to mycobacterium. In: Dale DC, Federman DD, Antman K, et al, eds. ACP Medicine. New York, NY: WebMD Professional Publishing; 2004:1703-1720.
7. Baker LP. Mycobacterium leprae interactions with the host cell: recent advances. Indian J Med Res. 2006;123:748-759.
8. Ishii N. Recent advances in the treatment of leprosy. Dermatol Online J. 2003;9:5. http://dermatology.cdlib.org/92/reviews/leprosy/ishii.html. Accessed March 16, 2015.
9. World Health Organization. Global leprosy situation, 2012. Weekly Epidemiological Rec. 2013;88:365-380. http://www.who.int/wer/2007/wer8835.pdf. Accessed March 16, 2015.
10. Gelber RH. Hansen disease. West J Med. 1993;158:583-590.
11. Deps PD, Alves L, Gripp CG, et al. Contact with armadillos increases the risk of leprosy in Brazil: a case control study. Indian J Dermatol Venereol Leprol. 2008;74:338-342.
12. Booth AV, Kovich OI. Lepromatous leprosy. Dermatol Online J. 2007;13:9.
13. Yens DA, Asters DJ, Teitel A. Subcutaneous nodules and joint deformity in leprosy. Clin Rheumatol. 2003;9:181-186.
14. Panezai S, Saleh FG. Leprosy and peripheral neuropathy. J Clin Neuromuscul Dis. 2004;5:138-145.
15. Sasaki S, Takeshita F, Okuda K, et al. Mycobacterium leprae and leprosy. Microbiol Immunol. 2001;45:729-736.
16. Kumar A, Girdhar A, Chakma J, et al. WHO Multidrug Therapy for Leprosy: Epidemiology of default in treatment in Agra District, Uttar Pradesh, India. BioMed Research International. doi:10.1155/2015/705804.
17. Fiallo P, Clapasson A, Favre A, et al. Overexpression of vascular endothelial growth factor and its endothelial cell receptor KDR in type I leprosy reaction. Am J Med Hyg. 2002;66:180-185.
18. Sales AM, de Matos HJ, Nery JAC, et al. Double-blind trial of the efficacy of pentoxifylline vs thalidomide for the treatment of type II reaction in leprosy. Braz J Med Biol Res. 2007;40:243-248.
19. Lockwood DN, Colston MJ, Khanolkar-Young SR. The detection of Mycobacterium leprae protein and carbohydrate antigens in skin and nerve from leprosy patients with type I (reversal) reactions. Am J Trop Med Hyg. 2002;66:409-415.
20. Boggild AK, Keystone JS, Kain KC. Leprosy: a primer for Canadian physicians. CMAJ. 2004;170:71-78.
21. Rook GA, Baker R. Cortisol metabolism, cortisol sensitivity and the pathogenesis of leprosy reactions. Trop Med Int Health. 1999;4:493-498.
Practice Points
- Reversal reactions are not uncommonly witnessed in patients undergoing treatment of leprosy.
- Leprosy has several distinct clinical presentations ranging from tuberculoid leprosy to lepromatous leprosy with the extent of disease generally depending on the host’s immune response to the infection.