User login
Spontaneous Regression of Merkel Cell Carcinoma
Merkel cell carcinoma (MCC) is a rare, rapidly growing, aggressive neoplasm with a generally poor prognosis. The cells of origin are highly anaplastic and share structural and immunohistochemical features with various neuroectodermally derived cells. Although Merkel cells, which are slow-acting cutaneous mechanoreceptors located in the basal layer of the epidermis, and MCC share immunohistochemical and ultrastructural features, there is limited evidence of a direct histogenetic relationship between the two.1,2 Additionally, some extracutaneous neuroendocrine tumors have features similar to MCC; therefore, although it may be more accurate and perhaps more practical to describe these lesions as primary neuroendocrine carcinomas of the skin, the term MCC is more commonly used both in the literature and in clinical practice.1,2
Merkel cell carcinoma typically presents in the head and neck region in white patients older than 70 years of age and in the immunocompromised population.3-6 The mean age of diagnosis is 76 years for women and 74 years for men.7 The incidence of MCC in the United States tripled over a 15-year period, and there are approximately 1500 new cases of MCC diagnosed each year, making it about 40 times less common than melanoma.8 The 5-year survival rate for patients without lymph node involvement is 75%, whereas the 5-year survival rate for patients with distant metastases is 25%.9
Merkel cell carcinoma is thought to develop through 1 of 2 distinct pathways. In a virally mediated pathway, which represents at least 80% of cases, the Merkel cell polyomavirus (MCV) monoclonally integrates into the host genome and promotes oncogenesis via altered p53 and retinoblastoma protein expression.10-12 The remainder of cases are believed to develop via a nonvirally mediated pathway in which genetic anomalies, immune status, and environmental factors influence oncogenesis.10-13
Due to the similarity between MCC and metastatic neuroendocrine neoplasms, especially small-cell lung carcinomas, immunohistochemistry is important in making the diagnosis. Cytokeratin 20 and neuron-specific enolase positivity and thyroid transcription factor 1 negativity are the most useful markers in identifying MCC.
Regression of MCC is a very rare and poorly understood event. A 2010 review of the literature described 22 cases of spontaneous regression.14 We report a rare case of rapid and complete regression of MCC following punch biopsy in a 96-year-old woman.
Case Report

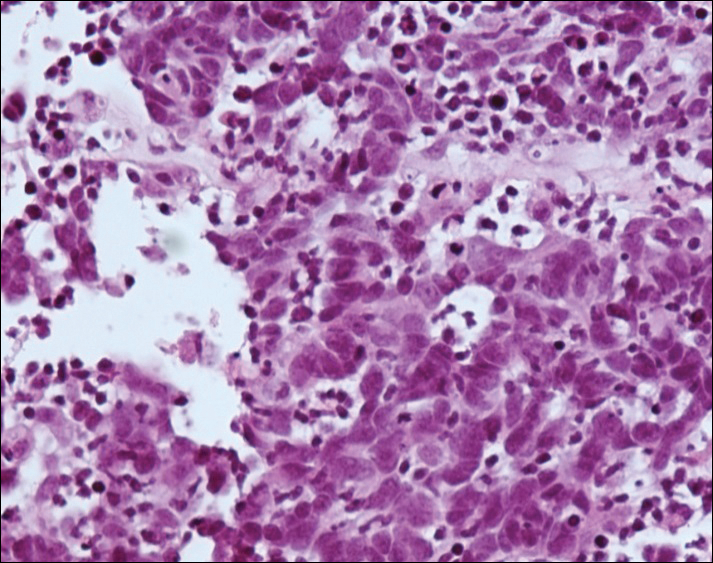
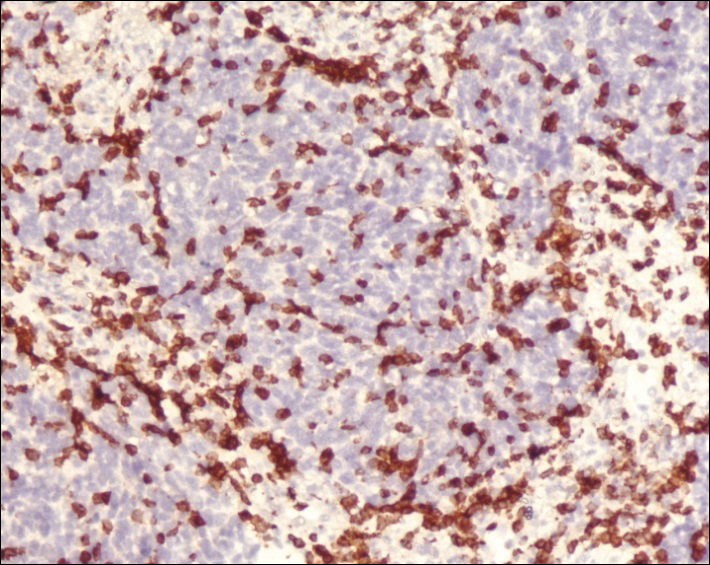
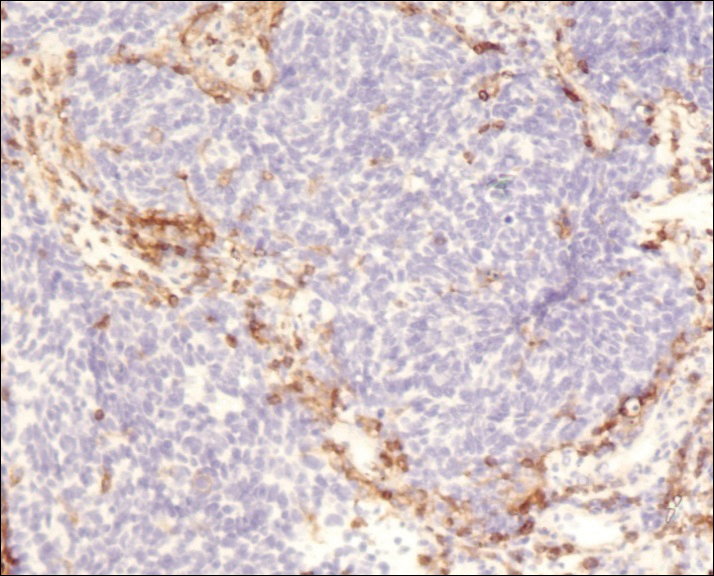
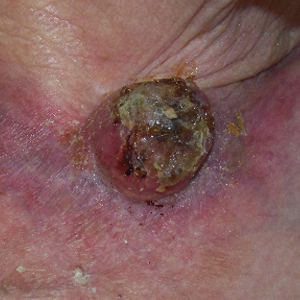
A 4-mm punch biopsy was obtained at a follow-up visit 4 weeks later (12 weeks after the reported onset of the lesion). Hematoxylin and eosin staining showed a small-cell neoplasm with stippled nuclei and scant cytoplasm forming a nested and somewhat trabecular pattern. Mitotic activity, apoptosis, and nuclear molding also were present (Figure 2). The tumor cells were positive for cytokeratin 20 with a dotlike, paranuclear pattern (Figure 3). Staining for CAM 5.2 also was positive. Cytokeratin 5/6, human melanoma black 45, and leukocyte common antigen were negative. The immunophenotyping of the lymphocytic response to the tumor showed that the majority of intratumoral lymphocytes were CD8 positive (Figure 4). CD4-positive lymphocytes were predominantly seen at the periphery of the tumor nests without tumor infiltration (Figure 5). Based on these findings, a diagnosis of MCC was made. The patient’s family declined treatment based on her advanced age and current health status, which included advanced dementia.

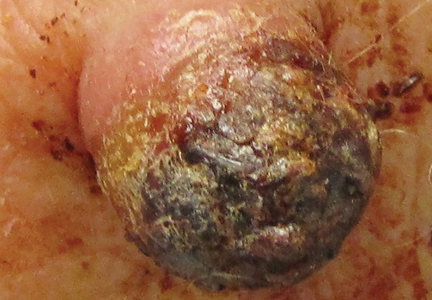
Two weeks after the punch biopsy, the lesion had noticeably decreased in size and lost its dome-shaped appearance. Within 8 weeks after biopsy (20 weeks since the lesion first appeared), the lesion had completely resolved (Figure 6). The patient was lost to follow-up months later, but no recurrence of the lesion was reported.

Comment
Spontaneous regression is not unique to MCC, as this phenomenon also has been reported in keratoacanthoma, lymphoma, basal cell carcinoma, and melanoma.15 Complete spontaneous regression is defined as occurring in the absence of therapy that is intended to have a treatment effect.15,16 Spontaneous regression is estimated to occur in malignant neoplasms at a rate of 1 case per 60,000 to 100,000 (approximately 0.0013% of all malignant neoplasms).17 Considering the reported prevalence of MCC and the number of cases that have been known to regress, the estimated incidence of complete spontaneous regression may be as high as 1.5%.14 Though spontaneous regression of MCC is more prevalent than expected, it still is considered a rare phenomenon. A 2010 review of the literature yielded 22 cases of complete spontaneous regression of MCC.14 No recurrences have been observed; however, follow-up was relatively short in some cases.
In a unique report by Bertolotti et al,18 a patient with MCC on the nasal tip presented 4 weeks after biopsy with complete spontaneous regression of the tumor, which was associated with bilateral cervical lymph node involvement as noted by hypermetabolic uptake on positron emission tomography scanning. The patient underwent radiation therapy and was disease free at 12 months’ follow-up.18
Complete spontaneous regression has been described in MCC patients with local disease, regional recurrences, and metastatic disease.19 In
The histopathologic features observed in our case, specifically intratumoral CD8-positive cytotoxic lymphocytes and peritumoral CD4-positive cells, were similar to the findings in other reported cases. In one series of 2 cases, the one case showed scar tissue with a moderate, predominantly T-lymphocytic infiltrate and no tumor cells, and the second showed cellular proliferation in the deep dermis with dense lymphocytic infiltrates primarily composed of CD3-positive T cells.14 Other studies of regression of both localized and metastatic MCC demonstrated infiltration by CD4-positive, CD8-positive, and CD3-positive lymphocytes and foamy macrophages.21-23
The discovery of the MCV was one of the most important advances in elucidating the pathogenesis of MCC.10,24-26 Merkel cell polyomavirus DNA has been detected in a majority of MCC cases.25,27 Viral integration has been shown to take place early, prior to tumor clonal expansion.10 Importantly, not all cases of MCC show MCV infection, and MCV infection is not exclusive to MCC.28 Merkel cell polyomavirus is considered to be part of the normal human flora, and asymptomatic infection is quite common.29 It has been identified in 80% of adults older than 50 years of age and, interestingly, in 35% of children by 13 years of age or younger.30,31 It remains unclear what role the presence of MCV plays in determining MCC prognosis. Several reports have demonstrated lower disease-specific mortality associated with MCV-positive MCC.32-35 In contrast, Schrama et al36 correlated the MCV status of 174 MCC tumors and found no difference in clinical behavior or prognosis between MCV-positive and MCV-negative MCCs.
Immunosuppression also may play a role in the development of MCC.5,25 There is increased prevalence of MCC in the human immunodeficiency virus–positive population, as well as in organ-transplant recipients and patients with leukemia. Chronic lymphocytic leukemia seems to be the most frequent neoplasia associated with development of MCC.37
The mechanism of MCC regression remains unclear, but many investigators emphasize the importance of T-cell–mediated immunity.16,21-23,38,39 Apoptosis also has been shown to play an important role.40 Our case showed tumor-infiltrating CD8-positive lymphocytes and CD4-positive lymphocytes present predominantly at the periphery of the tumor, with close proximity to the tumor nests but with no tumor infiltration (Figure 3). This distribution was consistently present in multiple sections of the tumor. These findings are consistent with prior reports of both CD4-positive and CD8-positive T lymphocytes associated with MCC regression. Our findings confirm that immune response may play an important role in spontaneous regression of MCC.
There is much speculation regarding the initial biopsy of an MCC lesion (or other traumatic event) and its role in tumor regression. Koba et al41 examined the effect of biopsy on CD8-positive lymphocytic infiltration of MCC tumor cells and found that biopsy does not commonly alter intratumoral CD8-positive infiltration. These findings suggest trauma does not directly induce immunologic recognition of this cancer.
Conclusion
We report a case of complete spontaneous regression of a localized MCC following a punch biopsy. The histopathology showed a brisk T-lymphocyte response with intratumoral CD8-positive cytotoxic lymphocytes and peritumoral CD4-positive cells. The age and clinical profile of our patient as well as the clinicopathologic characteristics of the tumor regression are similar to other reported cases. Further research is needed to elucidate the mechanism of MCC regression, and a better understanding of this fascinating phenomenon could help in development of new immunotherapeutic approaches.
- Sibley RK, Dehner LP, Rosai J. Primary neuroendocrine (Merkel cell?) carcinoma of the skin. I. a clinicopathologic and ultrastructural study of 43 cases. Am J Surg Pathol. 1985;9:95-108.
- Sibley RK, Dahl D. Primary neuroendocrine (Merkel cell?) carcinoma of the skin. II. an immunocytochemical study of 21 cases. Am J Surg Pathol. 1985;9:109-116.
- Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375-381.
- Penn I, First MR. Merkel’s cell carcinoma in organ recipients: report of 41 cases. Transplantation. 1999;68:1717-1721.
- Gooptu C, Woolloons A, Ross J, et al. Merkel cell carcinoma arising after therapeutic immunosuppression. Br J Dermatol. 1997;137:637-641.
- Plunkett TA, Harris AJ, Ogg CS, et al. The treatment of Merkel cell carcinoma and its association with immunosuppression. Br J Dermatol. 1998;139:345-346.
- Calder KB, Smoller BR. New insights into Merkel cell carcinoma. Adv Anat Pathol. 2010;17:155-161.
- Hodgson NC. Merkel cell carcinoma: changing incidence trends. J Surg Oncol. 2005;89:1-4.
- Agelli M, Clegg LX. Epidemiology of primary Merkel cell carcinoma in the United States. J Am Acad Dermatol. 2003;49:832-841.
- Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
- Amber K, McLeod MP, Nouri K. The Merkel cell polyomavirus and its involvement in Merkel cell carcinoma. Dermatol Surg. 2013;39:232-238.
- Decaprio JA. Does detection of Merkel cell polyomavirus in Merkel cell carcinoma provide prognostic information? J Natl Cancer Inst. 2009;101:905-907.
- Popp S, Waltering S, Herbst C, et al. UV-B-type mutations and chromosomal imbalances indicate common pathways for the development of Merkel and skin squamous cell carcinomas. Int J Cancer. 2002;99:352-360.
- Ciudad C, Avilés JA, Alfageme F, et al. Spontaneous regression in Merkel cell carcinoma: report of two cases with description of dermoscopic features and review of literature. Dermatol Surg. 2010;36:687-693.
- O’Rourke MGE, Bell JR. Merkel cell tumor with spontaneous regression. J Dermatol Surg Oncol. 1986;12:994-997.
- Connelly TJ, Cribier B, Brown TJ, et al. Complete spontaneous regression of Merkel cell carcinoma: a review of 10 reported cases. Dermatol Surg. 2000;26:853-856.
- Cole WH. Efforts to explain spontaneous regression of cancer. J Surg Oncol. 1981;17:201-209.
- Bertolotti A, Conte H, Francois L, et al. Merkel cell carcinoma: complete clinical remission associated with disease progression. JAMA Dermatol. 2013;149:501-502.
- Pang C, Sharma D, Sankar T. Spontaneous regression of Merkel cell carcinoma: a case report and review of the literature [published online November 13, 2014]. Int J Surg Case Rep. 2015;7C:104-108.
- Richetta AG, Mancini M, Torroni A, et al. Total spontaneous regression of advanced Merkel cell carcinoma after biopsy: review and a new case. Dermatol Surg. 2008;34:815-822.
- Vesely MJ, Murray DJ, Neligan PC, et al. Complete spontaneous regression in Merkel cell carcinoma. J Plast Reconstr Aesthet Surg. 2008;61:165-171.
- Kayashima K, Ono T, Johno M, et al. Spontaneous regression in Merkel cell (neuroendocrine) carcinoma of the skin. Arch Dermatol. 1991;127:550-553.
- Maruo K, Kayashima KI, Ono T. Regressing Merkel cell carcinoma-a case showing replacement of tumour cells by foamy cells. Br J Dermatol. 2000;142:1184-1189.
- Duncavage E, Zehnbauer B, Pfeifer J. Prevalence of Merkel cell polyomavirus in Merkel cell carcinoma. Mod Pathol. 2009;22:516-521.
- Kassem A, Schopflin A, Diaz C, et al. Frequent detection of Merkel cell polyomavirus in human Merkel cell carcinomas and identification of unique deletion in the VP1 gene. Cancer Res. 2008;68:5009-5013.
- Becker J, Schrama D, Houben R. Merkel cell carcinoma. Cell Mol Life Sci. 2009;66:1-8.
- Haitz KA, Rady PL, Nguyen HP, et al. Merkel cell polyomavirus DNA detection in a patient with Merkel cell carcinoma and multiple other skin cancers. Int J Dermatol. 2012;51:442-444.
- Andres C, Puchta U, Sander CA, et al. Prevalence of Merkel cell polyomavirus DNA in cutaneous lymphomas, pseudolymphomas, and inflammatory skin diseases. Am J Dermatopathol. 2010;32:593-598.
- Showalter RM, Pastrana DV, Pumphrey KA, et al. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe. 2010;7:509-515.
- Tolstov YL, Pastrana DV, Feng H, et al. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int J Cancer. 2009;125:1250-1256.
- Chen T, Hedman L, Mattila PS, et al. Serological evidence of Merkel cell polyomavirus primary infections in childhood. J Clin Virol. 2011;50:125-129.
- Laude HC, Jonchère B, Maubec E, et al. Distinct Merkel cell polyomavirus molecular features in tumour and non tumour specimens from patients with Merkel cell carcinoma. PLoS Pathog. 2010;6:e1001076.
- Waltari M, Sihto H, Kukko H, et al. Association of Merkel cell polyomavirus infection with tumor p53, KIT, stem cell factor, PDGFR-alpha and survival in Merkel cell carcinoma. Int J Cancer. 2011;129:619-628.
- Sihto H, Kukko H, Koljonen V, et al. Clinical factors associated with Merkel cell polyomavirus infection in Merkel cell carcinoma. J Natl Cancer Inst. 2009;101:938-945.
- Paulson KG, Lemos BD, Feng B, et al. Array-CGH reveals recurrent genomic changes in Merkel cell carcinoma including amplification of L-Myc. J Invest Dermatol. 2009;129:1547-1555.
- Schrama D, Peitsch WK, Zapatka M, et al. Merkel cell polyomavirus status is not associated with clinical course of Merkel cell carcinoma. J Invest Dermatol. 2011;131:1631-1638.
- Tadmor T, Aviv A, Polliack A. Merkel cell carcinoma, chronic lymphocytic leukemia and other lymphoproliferative disorders: an old bond with possible new viral ties. Ann Oncol. 2011;22:250-256.
- Wooff J, Trites JR, Walsh NM, et al. Complete spontaneous regression of metastatic Merkel cell carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:614-617.
- Turk TO, Smoljan I, Nacinovic A, et al. Spontaneous regression of Merkel cell carcinoma in a patient with chronic lymphocytic leukemia: a case report. J Med Case Rep. 2009;3:7270.
- Mori Y, Tanaka K, Cui CY, et al. A study of apoptosis in Merkel cell carcinoma. an immunohistochemical, ultrasctructural, DNA ladder and TUNEL labeling study. Am J Dermatopathol. 2001;23:16-23.
- Koba S, Paulson KG, Nagase K, et al. Diagnostic biopsy does not commonly induce intratumoral CD8 T cell infiltration in Merkel cell carcinoma. PLoS ONE. 2012;7:e41465.
Merkel cell carcinoma (MCC) is a rare, rapidly growing, aggressive neoplasm with a generally poor prognosis. The cells of origin are highly anaplastic and share structural and immunohistochemical features with various neuroectodermally derived cells. Although Merkel cells, which are slow-acting cutaneous mechanoreceptors located in the basal layer of the epidermis, and MCC share immunohistochemical and ultrastructural features, there is limited evidence of a direct histogenetic relationship between the two.1,2 Additionally, some extracutaneous neuroendocrine tumors have features similar to MCC; therefore, although it may be more accurate and perhaps more practical to describe these lesions as primary neuroendocrine carcinomas of the skin, the term MCC is more commonly used both in the literature and in clinical practice.1,2
Merkel cell carcinoma typically presents in the head and neck region in white patients older than 70 years of age and in the immunocompromised population.3-6 The mean age of diagnosis is 76 years for women and 74 years for men.7 The incidence of MCC in the United States tripled over a 15-year period, and there are approximately 1500 new cases of MCC diagnosed each year, making it about 40 times less common than melanoma.8 The 5-year survival rate for patients without lymph node involvement is 75%, whereas the 5-year survival rate for patients with distant metastases is 25%.9
Merkel cell carcinoma is thought to develop through 1 of 2 distinct pathways. In a virally mediated pathway, which represents at least 80% of cases, the Merkel cell polyomavirus (MCV) monoclonally integrates into the host genome and promotes oncogenesis via altered p53 and retinoblastoma protein expression.10-12 The remainder of cases are believed to develop via a nonvirally mediated pathway in which genetic anomalies, immune status, and environmental factors influence oncogenesis.10-13
Due to the similarity between MCC and metastatic neuroendocrine neoplasms, especially small-cell lung carcinomas, immunohistochemistry is important in making the diagnosis. Cytokeratin 20 and neuron-specific enolase positivity and thyroid transcription factor 1 negativity are the most useful markers in identifying MCC.
Regression of MCC is a very rare and poorly understood event. A 2010 review of the literature described 22 cases of spontaneous regression.14 We report a rare case of rapid and complete regression of MCC following punch biopsy in a 96-year-old woman.
Case Report
A 4-mm punch biopsy was obtained at a follow-up visit 4 weeks later (12 weeks after the reported onset of the lesion). Hematoxylin and eosin staining showed a small-cell neoplasm with stippled nuclei and scant cytoplasm forming a nested and somewhat trabecular pattern. Mitotic activity, apoptosis, and nuclear molding also were present (Figure 2). The tumor cells were positive for cytokeratin 20 with a dotlike, paranuclear pattern (Figure 3). Staining for CAM 5.2 also was positive. Cytokeratin 5/6, human melanoma black 45, and leukocyte common antigen were negative. The immunophenotyping of the lymphocytic response to the tumor showed that the majority of intratumoral lymphocytes were CD8 positive (Figure 4). CD4-positive lymphocytes were predominantly seen at the periphery of the tumor nests without tumor infiltration (Figure 5). Based on these findings, a diagnosis of MCC was made. The patient’s family declined treatment based on her advanced age and current health status, which included advanced dementia.
Two weeks after the punch biopsy, the lesion had noticeably decreased in size and lost its dome-shaped appearance. Within 8 weeks after biopsy (20 weeks since the lesion first appeared), the lesion had completely resolved (Figure 6). The patient was lost to follow-up months later, but no recurrence of the lesion was reported.
Comment
Spontaneous regression is not unique to MCC, as this phenomenon also has been reported in keratoacanthoma, lymphoma, basal cell carcinoma, and melanoma.15 Complete spontaneous regression is defined as occurring in the absence of therapy that is intended to have a treatment effect.15,16 Spontaneous regression is estimated to occur in malignant neoplasms at a rate of 1 case per 60,000 to 100,000 (approximately 0.0013% of all malignant neoplasms).17 Considering the reported prevalence of MCC and the number of cases that have been known to regress, the estimated incidence of complete spontaneous regression may be as high as 1.5%.14 Though spontaneous regression of MCC is more prevalent than expected, it still is considered a rare phenomenon. A 2010 review of the literature yielded 22 cases of complete spontaneous regression of MCC.14 No recurrences have been observed; however, follow-up was relatively short in some cases.
In a unique report by Bertolotti et al,18 a patient with MCC on the nasal tip presented 4 weeks after biopsy with complete spontaneous regression of the tumor, which was associated with bilateral cervical lymph node involvement as noted by hypermetabolic uptake on positron emission tomography scanning. The patient underwent radiation therapy and was disease free at 12 months’ follow-up.18
Complete spontaneous regression has been described in MCC patients with local disease, regional recurrences, and metastatic disease.19 In
The histopathologic features observed in our case, specifically intratumoral CD8-positive cytotoxic lymphocytes and peritumoral CD4-positive cells, were similar to the findings in other reported cases. In one series of 2 cases, the one case showed scar tissue with a moderate, predominantly T-lymphocytic infiltrate and no tumor cells, and the second showed cellular proliferation in the deep dermis with dense lymphocytic infiltrates primarily composed of CD3-positive T cells.14 Other studies of regression of both localized and metastatic MCC demonstrated infiltration by CD4-positive, CD8-positive, and CD3-positive lymphocytes and foamy macrophages.21-23
The discovery of the MCV was one of the most important advances in elucidating the pathogenesis of MCC.10,24-26 Merkel cell polyomavirus DNA has been detected in a majority of MCC cases.25,27 Viral integration has been shown to take place early, prior to tumor clonal expansion.10 Importantly, not all cases of MCC show MCV infection, and MCV infection is not exclusive to MCC.28 Merkel cell polyomavirus is considered to be part of the normal human flora, and asymptomatic infection is quite common.29 It has been identified in 80% of adults older than 50 years of age and, interestingly, in 35% of children by 13 years of age or younger.30,31 It remains unclear what role the presence of MCV plays in determining MCC prognosis. Several reports have demonstrated lower disease-specific mortality associated with MCV-positive MCC.32-35 In contrast, Schrama et al36 correlated the MCV status of 174 MCC tumors and found no difference in clinical behavior or prognosis between MCV-positive and MCV-negative MCCs.
Immunosuppression also may play a role in the development of MCC.5,25 There is increased prevalence of MCC in the human immunodeficiency virus–positive population, as well as in organ-transplant recipients and patients with leukemia. Chronic lymphocytic leukemia seems to be the most frequent neoplasia associated with development of MCC.37
The mechanism of MCC regression remains unclear, but many investigators emphasize the importance of T-cell–mediated immunity.16,21-23,38,39 Apoptosis also has been shown to play an important role.40 Our case showed tumor-infiltrating CD8-positive lymphocytes and CD4-positive lymphocytes present predominantly at the periphery of the tumor, with close proximity to the tumor nests but with no tumor infiltration (Figure 3). This distribution was consistently present in multiple sections of the tumor. These findings are consistent with prior reports of both CD4-positive and CD8-positive T lymphocytes associated with MCC regression. Our findings confirm that immune response may play an important role in spontaneous regression of MCC.
There is much speculation regarding the initial biopsy of an MCC lesion (or other traumatic event) and its role in tumor regression. Koba et al41 examined the effect of biopsy on CD8-positive lymphocytic infiltration of MCC tumor cells and found that biopsy does not commonly alter intratumoral CD8-positive infiltration. These findings suggest trauma does not directly induce immunologic recognition of this cancer.
Conclusion
We report a case of complete spontaneous regression of a localized MCC following a punch biopsy. The histopathology showed a brisk T-lymphocyte response with intratumoral CD8-positive cytotoxic lymphocytes and peritumoral CD4-positive cells. The age and clinical profile of our patient as well as the clinicopathologic characteristics of the tumor regression are similar to other reported cases. Further research is needed to elucidate the mechanism of MCC regression, and a better understanding of this fascinating phenomenon could help in development of new immunotherapeutic approaches.
Merkel cell carcinoma (MCC) is a rare, rapidly growing, aggressive neoplasm with a generally poor prognosis. The cells of origin are highly anaplastic and share structural and immunohistochemical features with various neuroectodermally derived cells. Although Merkel cells, which are slow-acting cutaneous mechanoreceptors located in the basal layer of the epidermis, and MCC share immunohistochemical and ultrastructural features, there is limited evidence of a direct histogenetic relationship between the two.1,2 Additionally, some extracutaneous neuroendocrine tumors have features similar to MCC; therefore, although it may be more accurate and perhaps more practical to describe these lesions as primary neuroendocrine carcinomas of the skin, the term MCC is more commonly used both in the literature and in clinical practice.1,2
Merkel cell carcinoma typically presents in the head and neck region in white patients older than 70 years of age and in the immunocompromised population.3-6 The mean age of diagnosis is 76 years for women and 74 years for men.7 The incidence of MCC in the United States tripled over a 15-year period, and there are approximately 1500 new cases of MCC diagnosed each year, making it about 40 times less common than melanoma.8 The 5-year survival rate for patients without lymph node involvement is 75%, whereas the 5-year survival rate for patients with distant metastases is 25%.9
Merkel cell carcinoma is thought to develop through 1 of 2 distinct pathways. In a virally mediated pathway, which represents at least 80% of cases, the Merkel cell polyomavirus (MCV) monoclonally integrates into the host genome and promotes oncogenesis via altered p53 and retinoblastoma protein expression.10-12 The remainder of cases are believed to develop via a nonvirally mediated pathway in which genetic anomalies, immune status, and environmental factors influence oncogenesis.10-13
Due to the similarity between MCC and metastatic neuroendocrine neoplasms, especially small-cell lung carcinomas, immunohistochemistry is important in making the diagnosis. Cytokeratin 20 and neuron-specific enolase positivity and thyroid transcription factor 1 negativity are the most useful markers in identifying MCC.
Regression of MCC is a very rare and poorly understood event. A 2010 review of the literature described 22 cases of spontaneous regression.14 We report a rare case of rapid and complete regression of MCC following punch biopsy in a 96-year-old woman.
Case Report
A 4-mm punch biopsy was obtained at a follow-up visit 4 weeks later (12 weeks after the reported onset of the lesion). Hematoxylin and eosin staining showed a small-cell neoplasm with stippled nuclei and scant cytoplasm forming a nested and somewhat trabecular pattern. Mitotic activity, apoptosis, and nuclear molding also were present (Figure 2). The tumor cells were positive for cytokeratin 20 with a dotlike, paranuclear pattern (Figure 3). Staining for CAM 5.2 also was positive. Cytokeratin 5/6, human melanoma black 45, and leukocyte common antigen were negative. The immunophenotyping of the lymphocytic response to the tumor showed that the majority of intratumoral lymphocytes were CD8 positive (Figure 4). CD4-positive lymphocytes were predominantly seen at the periphery of the tumor nests without tumor infiltration (Figure 5). Based on these findings, a diagnosis of MCC was made. The patient’s family declined treatment based on her advanced age and current health status, which included advanced dementia.
Two weeks after the punch biopsy, the lesion had noticeably decreased in size and lost its dome-shaped appearance. Within 8 weeks after biopsy (20 weeks since the lesion first appeared), the lesion had completely resolved (Figure 6). The patient was lost to follow-up months later, but no recurrence of the lesion was reported.
Comment
Spontaneous regression is not unique to MCC, as this phenomenon also has been reported in keratoacanthoma, lymphoma, basal cell carcinoma, and melanoma.15 Complete spontaneous regression is defined as occurring in the absence of therapy that is intended to have a treatment effect.15,16 Spontaneous regression is estimated to occur in malignant neoplasms at a rate of 1 case per 60,000 to 100,000 (approximately 0.0013% of all malignant neoplasms).17 Considering the reported prevalence of MCC and the number of cases that have been known to regress, the estimated incidence of complete spontaneous regression may be as high as 1.5%.14 Though spontaneous regression of MCC is more prevalent than expected, it still is considered a rare phenomenon. A 2010 review of the literature yielded 22 cases of complete spontaneous regression of MCC.14 No recurrences have been observed; however, follow-up was relatively short in some cases.
In a unique report by Bertolotti et al,18 a patient with MCC on the nasal tip presented 4 weeks after biopsy with complete spontaneous regression of the tumor, which was associated with bilateral cervical lymph node involvement as noted by hypermetabolic uptake on positron emission tomography scanning. The patient underwent radiation therapy and was disease free at 12 months’ follow-up.18
Complete spontaneous regression has been described in MCC patients with local disease, regional recurrences, and metastatic disease.19 In
The histopathologic features observed in our case, specifically intratumoral CD8-positive cytotoxic lymphocytes and peritumoral CD4-positive cells, were similar to the findings in other reported cases. In one series of 2 cases, the one case showed scar tissue with a moderate, predominantly T-lymphocytic infiltrate and no tumor cells, and the second showed cellular proliferation in the deep dermis with dense lymphocytic infiltrates primarily composed of CD3-positive T cells.14 Other studies of regression of both localized and metastatic MCC demonstrated infiltration by CD4-positive, CD8-positive, and CD3-positive lymphocytes and foamy macrophages.21-23
The discovery of the MCV was one of the most important advances in elucidating the pathogenesis of MCC.10,24-26 Merkel cell polyomavirus DNA has been detected in a majority of MCC cases.25,27 Viral integration has been shown to take place early, prior to tumor clonal expansion.10 Importantly, not all cases of MCC show MCV infection, and MCV infection is not exclusive to MCC.28 Merkel cell polyomavirus is considered to be part of the normal human flora, and asymptomatic infection is quite common.29 It has been identified in 80% of adults older than 50 years of age and, interestingly, in 35% of children by 13 years of age or younger.30,31 It remains unclear what role the presence of MCV plays in determining MCC prognosis. Several reports have demonstrated lower disease-specific mortality associated with MCV-positive MCC.32-35 In contrast, Schrama et al36 correlated the MCV status of 174 MCC tumors and found no difference in clinical behavior or prognosis between MCV-positive and MCV-negative MCCs.
Immunosuppression also may play a role in the development of MCC.5,25 There is increased prevalence of MCC in the human immunodeficiency virus–positive population, as well as in organ-transplant recipients and patients with leukemia. Chronic lymphocytic leukemia seems to be the most frequent neoplasia associated with development of MCC.37
The mechanism of MCC regression remains unclear, but many investigators emphasize the importance of T-cell–mediated immunity.16,21-23,38,39 Apoptosis also has been shown to play an important role.40 Our case showed tumor-infiltrating CD8-positive lymphocytes and CD4-positive lymphocytes present predominantly at the periphery of the tumor, with close proximity to the tumor nests but with no tumor infiltration (Figure 3). This distribution was consistently present in multiple sections of the tumor. These findings are consistent with prior reports of both CD4-positive and CD8-positive T lymphocytes associated with MCC regression. Our findings confirm that immune response may play an important role in spontaneous regression of MCC.
There is much speculation regarding the initial biopsy of an MCC lesion (or other traumatic event) and its role in tumor regression. Koba et al41 examined the effect of biopsy on CD8-positive lymphocytic infiltration of MCC tumor cells and found that biopsy does not commonly alter intratumoral CD8-positive infiltration. These findings suggest trauma does not directly induce immunologic recognition of this cancer.
Conclusion
We report a case of complete spontaneous regression of a localized MCC following a punch biopsy. The histopathology showed a brisk T-lymphocyte response with intratumoral CD8-positive cytotoxic lymphocytes and peritumoral CD4-positive cells. The age and clinical profile of our patient as well as the clinicopathologic characteristics of the tumor regression are similar to other reported cases. Further research is needed to elucidate the mechanism of MCC regression, and a better understanding of this fascinating phenomenon could help in development of new immunotherapeutic approaches.
- Sibley RK, Dehner LP, Rosai J. Primary neuroendocrine (Merkel cell?) carcinoma of the skin. I. a clinicopathologic and ultrastructural study of 43 cases. Am J Surg Pathol. 1985;9:95-108.
- Sibley RK, Dahl D. Primary neuroendocrine (Merkel cell?) carcinoma of the skin. II. an immunocytochemical study of 21 cases. Am J Surg Pathol. 1985;9:109-116.
- Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375-381.
- Penn I, First MR. Merkel’s cell carcinoma in organ recipients: report of 41 cases. Transplantation. 1999;68:1717-1721.
- Gooptu C, Woolloons A, Ross J, et al. Merkel cell carcinoma arising after therapeutic immunosuppression. Br J Dermatol. 1997;137:637-641.
- Plunkett TA, Harris AJ, Ogg CS, et al. The treatment of Merkel cell carcinoma and its association with immunosuppression. Br J Dermatol. 1998;139:345-346.
- Calder KB, Smoller BR. New insights into Merkel cell carcinoma. Adv Anat Pathol. 2010;17:155-161.
- Hodgson NC. Merkel cell carcinoma: changing incidence trends. J Surg Oncol. 2005;89:1-4.
- Agelli M, Clegg LX. Epidemiology of primary Merkel cell carcinoma in the United States. J Am Acad Dermatol. 2003;49:832-841.
- Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
- Amber K, McLeod MP, Nouri K. The Merkel cell polyomavirus and its involvement in Merkel cell carcinoma. Dermatol Surg. 2013;39:232-238.
- Decaprio JA. Does detection of Merkel cell polyomavirus in Merkel cell carcinoma provide prognostic information? J Natl Cancer Inst. 2009;101:905-907.
- Popp S, Waltering S, Herbst C, et al. UV-B-type mutations and chromosomal imbalances indicate common pathways for the development of Merkel and skin squamous cell carcinomas. Int J Cancer. 2002;99:352-360.
- Ciudad C, Avilés JA, Alfageme F, et al. Spontaneous regression in Merkel cell carcinoma: report of two cases with description of dermoscopic features and review of literature. Dermatol Surg. 2010;36:687-693.
- O’Rourke MGE, Bell JR. Merkel cell tumor with spontaneous regression. J Dermatol Surg Oncol. 1986;12:994-997.
- Connelly TJ, Cribier B, Brown TJ, et al. Complete spontaneous regression of Merkel cell carcinoma: a review of 10 reported cases. Dermatol Surg. 2000;26:853-856.
- Cole WH. Efforts to explain spontaneous regression of cancer. J Surg Oncol. 1981;17:201-209.
- Bertolotti A, Conte H, Francois L, et al. Merkel cell carcinoma: complete clinical remission associated with disease progression. JAMA Dermatol. 2013;149:501-502.
- Pang C, Sharma D, Sankar T. Spontaneous regression of Merkel cell carcinoma: a case report and review of the literature [published online November 13, 2014]. Int J Surg Case Rep. 2015;7C:104-108.
- Richetta AG, Mancini M, Torroni A, et al. Total spontaneous regression of advanced Merkel cell carcinoma after biopsy: review and a new case. Dermatol Surg. 2008;34:815-822.
- Vesely MJ, Murray DJ, Neligan PC, et al. Complete spontaneous regression in Merkel cell carcinoma. J Plast Reconstr Aesthet Surg. 2008;61:165-171.
- Kayashima K, Ono T, Johno M, et al. Spontaneous regression in Merkel cell (neuroendocrine) carcinoma of the skin. Arch Dermatol. 1991;127:550-553.
- Maruo K, Kayashima KI, Ono T. Regressing Merkel cell carcinoma-a case showing replacement of tumour cells by foamy cells. Br J Dermatol. 2000;142:1184-1189.
- Duncavage E, Zehnbauer B, Pfeifer J. Prevalence of Merkel cell polyomavirus in Merkel cell carcinoma. Mod Pathol. 2009;22:516-521.
- Kassem A, Schopflin A, Diaz C, et al. Frequent detection of Merkel cell polyomavirus in human Merkel cell carcinomas and identification of unique deletion in the VP1 gene. Cancer Res. 2008;68:5009-5013.
- Becker J, Schrama D, Houben R. Merkel cell carcinoma. Cell Mol Life Sci. 2009;66:1-8.
- Haitz KA, Rady PL, Nguyen HP, et al. Merkel cell polyomavirus DNA detection in a patient with Merkel cell carcinoma and multiple other skin cancers. Int J Dermatol. 2012;51:442-444.
- Andres C, Puchta U, Sander CA, et al. Prevalence of Merkel cell polyomavirus DNA in cutaneous lymphomas, pseudolymphomas, and inflammatory skin diseases. Am J Dermatopathol. 2010;32:593-598.
- Showalter RM, Pastrana DV, Pumphrey KA, et al. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe. 2010;7:509-515.
- Tolstov YL, Pastrana DV, Feng H, et al. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int J Cancer. 2009;125:1250-1256.
- Chen T, Hedman L, Mattila PS, et al. Serological evidence of Merkel cell polyomavirus primary infections in childhood. J Clin Virol. 2011;50:125-129.
- Laude HC, Jonchère B, Maubec E, et al. Distinct Merkel cell polyomavirus molecular features in tumour and non tumour specimens from patients with Merkel cell carcinoma. PLoS Pathog. 2010;6:e1001076.
- Waltari M, Sihto H, Kukko H, et al. Association of Merkel cell polyomavirus infection with tumor p53, KIT, stem cell factor, PDGFR-alpha and survival in Merkel cell carcinoma. Int J Cancer. 2011;129:619-628.
- Sihto H, Kukko H, Koljonen V, et al. Clinical factors associated with Merkel cell polyomavirus infection in Merkel cell carcinoma. J Natl Cancer Inst. 2009;101:938-945.
- Paulson KG, Lemos BD, Feng B, et al. Array-CGH reveals recurrent genomic changes in Merkel cell carcinoma including amplification of L-Myc. J Invest Dermatol. 2009;129:1547-1555.
- Schrama D, Peitsch WK, Zapatka M, et al. Merkel cell polyomavirus status is not associated with clinical course of Merkel cell carcinoma. J Invest Dermatol. 2011;131:1631-1638.
- Tadmor T, Aviv A, Polliack A. Merkel cell carcinoma, chronic lymphocytic leukemia and other lymphoproliferative disorders: an old bond with possible new viral ties. Ann Oncol. 2011;22:250-256.
- Wooff J, Trites JR, Walsh NM, et al. Complete spontaneous regression of metastatic Merkel cell carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:614-617.
- Turk TO, Smoljan I, Nacinovic A, et al. Spontaneous regression of Merkel cell carcinoma in a patient with chronic lymphocytic leukemia: a case report. J Med Case Rep. 2009;3:7270.
- Mori Y, Tanaka K, Cui CY, et al. A study of apoptosis in Merkel cell carcinoma. an immunohistochemical, ultrasctructural, DNA ladder and TUNEL labeling study. Am J Dermatopathol. 2001;23:16-23.
- Koba S, Paulson KG, Nagase K, et al. Diagnostic biopsy does not commonly induce intratumoral CD8 T cell infiltration in Merkel cell carcinoma. PLoS ONE. 2012;7:e41465.
- Sibley RK, Dehner LP, Rosai J. Primary neuroendocrine (Merkel cell?) carcinoma of the skin. I. a clinicopathologic and ultrastructural study of 43 cases. Am J Surg Pathol. 1985;9:95-108.
- Sibley RK, Dahl D. Primary neuroendocrine (Merkel cell?) carcinoma of the skin. II. an immunocytochemical study of 21 cases. Am J Surg Pathol. 1985;9:109-116.
- Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375-381.
- Penn I, First MR. Merkel’s cell carcinoma in organ recipients: report of 41 cases. Transplantation. 1999;68:1717-1721.
- Gooptu C, Woolloons A, Ross J, et al. Merkel cell carcinoma arising after therapeutic immunosuppression. Br J Dermatol. 1997;137:637-641.
- Plunkett TA, Harris AJ, Ogg CS, et al. The treatment of Merkel cell carcinoma and its association with immunosuppression. Br J Dermatol. 1998;139:345-346.
- Calder KB, Smoller BR. New insights into Merkel cell carcinoma. Adv Anat Pathol. 2010;17:155-161.
- Hodgson NC. Merkel cell carcinoma: changing incidence trends. J Surg Oncol. 2005;89:1-4.
- Agelli M, Clegg LX. Epidemiology of primary Merkel cell carcinoma in the United States. J Am Acad Dermatol. 2003;49:832-841.
- Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
- Amber K, McLeod MP, Nouri K. The Merkel cell polyomavirus and its involvement in Merkel cell carcinoma. Dermatol Surg. 2013;39:232-238.
- Decaprio JA. Does detection of Merkel cell polyomavirus in Merkel cell carcinoma provide prognostic information? J Natl Cancer Inst. 2009;101:905-907.
- Popp S, Waltering S, Herbst C, et al. UV-B-type mutations and chromosomal imbalances indicate common pathways for the development of Merkel and skin squamous cell carcinomas. Int J Cancer. 2002;99:352-360.
- Ciudad C, Avilés JA, Alfageme F, et al. Spontaneous regression in Merkel cell carcinoma: report of two cases with description of dermoscopic features and review of literature. Dermatol Surg. 2010;36:687-693.
- O’Rourke MGE, Bell JR. Merkel cell tumor with spontaneous regression. J Dermatol Surg Oncol. 1986;12:994-997.
- Connelly TJ, Cribier B, Brown TJ, et al. Complete spontaneous regression of Merkel cell carcinoma: a review of 10 reported cases. Dermatol Surg. 2000;26:853-856.
- Cole WH. Efforts to explain spontaneous regression of cancer. J Surg Oncol. 1981;17:201-209.
- Bertolotti A, Conte H, Francois L, et al. Merkel cell carcinoma: complete clinical remission associated with disease progression. JAMA Dermatol. 2013;149:501-502.
- Pang C, Sharma D, Sankar T. Spontaneous regression of Merkel cell carcinoma: a case report and review of the literature [published online November 13, 2014]. Int J Surg Case Rep. 2015;7C:104-108.
- Richetta AG, Mancini M, Torroni A, et al. Total spontaneous regression of advanced Merkel cell carcinoma after biopsy: review and a new case. Dermatol Surg. 2008;34:815-822.
- Vesely MJ, Murray DJ, Neligan PC, et al. Complete spontaneous regression in Merkel cell carcinoma. J Plast Reconstr Aesthet Surg. 2008;61:165-171.
- Kayashima K, Ono T, Johno M, et al. Spontaneous regression in Merkel cell (neuroendocrine) carcinoma of the skin. Arch Dermatol. 1991;127:550-553.
- Maruo K, Kayashima KI, Ono T. Regressing Merkel cell carcinoma-a case showing replacement of tumour cells by foamy cells. Br J Dermatol. 2000;142:1184-1189.
- Duncavage E, Zehnbauer B, Pfeifer J. Prevalence of Merkel cell polyomavirus in Merkel cell carcinoma. Mod Pathol. 2009;22:516-521.
- Kassem A, Schopflin A, Diaz C, et al. Frequent detection of Merkel cell polyomavirus in human Merkel cell carcinomas and identification of unique deletion in the VP1 gene. Cancer Res. 2008;68:5009-5013.
- Becker J, Schrama D, Houben R. Merkel cell carcinoma. Cell Mol Life Sci. 2009;66:1-8.
- Haitz KA, Rady PL, Nguyen HP, et al. Merkel cell polyomavirus DNA detection in a patient with Merkel cell carcinoma and multiple other skin cancers. Int J Dermatol. 2012;51:442-444.
- Andres C, Puchta U, Sander CA, et al. Prevalence of Merkel cell polyomavirus DNA in cutaneous lymphomas, pseudolymphomas, and inflammatory skin diseases. Am J Dermatopathol. 2010;32:593-598.
- Showalter RM, Pastrana DV, Pumphrey KA, et al. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe. 2010;7:509-515.
- Tolstov YL, Pastrana DV, Feng H, et al. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int J Cancer. 2009;125:1250-1256.
- Chen T, Hedman L, Mattila PS, et al. Serological evidence of Merkel cell polyomavirus primary infections in childhood. J Clin Virol. 2011;50:125-129.
- Laude HC, Jonchère B, Maubec E, et al. Distinct Merkel cell polyomavirus molecular features in tumour and non tumour specimens from patients with Merkel cell carcinoma. PLoS Pathog. 2010;6:e1001076.
- Waltari M, Sihto H, Kukko H, et al. Association of Merkel cell polyomavirus infection with tumor p53, KIT, stem cell factor, PDGFR-alpha and survival in Merkel cell carcinoma. Int J Cancer. 2011;129:619-628.
- Sihto H, Kukko H, Koljonen V, et al. Clinical factors associated with Merkel cell polyomavirus infection in Merkel cell carcinoma. J Natl Cancer Inst. 2009;101:938-945.
- Paulson KG, Lemos BD, Feng B, et al. Array-CGH reveals recurrent genomic changes in Merkel cell carcinoma including amplification of L-Myc. J Invest Dermatol. 2009;129:1547-1555.
- Schrama D, Peitsch WK, Zapatka M, et al. Merkel cell polyomavirus status is not associated with clinical course of Merkel cell carcinoma. J Invest Dermatol. 2011;131:1631-1638.
- Tadmor T, Aviv A, Polliack A. Merkel cell carcinoma, chronic lymphocytic leukemia and other lymphoproliferative disorders: an old bond with possible new viral ties. Ann Oncol. 2011;22:250-256.
- Wooff J, Trites JR, Walsh NM, et al. Complete spontaneous regression of metastatic Merkel cell carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:614-617.
- Turk TO, Smoljan I, Nacinovic A, et al. Spontaneous regression of Merkel cell carcinoma in a patient with chronic lymphocytic leukemia: a case report. J Med Case Rep. 2009;3:7270.
- Mori Y, Tanaka K, Cui CY, et al. A study of apoptosis in Merkel cell carcinoma. an immunohistochemical, ultrasctructural, DNA ladder and TUNEL labeling study. Am J Dermatopathol. 2001;23:16-23.
- Koba S, Paulson KG, Nagase K, et al. Diagnostic biopsy does not commonly induce intratumoral CD8 T cell infiltration in Merkel cell carcinoma. PLoS ONE. 2012;7:e41465.
Practice Points
- Merkel cell carcinoma (MCC) is a rare malignancy with a high rate of metastasis and poor prognosis.
- T-cell mediated immunity appears to play an important role in tumor regression in MCC.
- Merkel cell polyomavirus appears to play a role in the pathogenesis of MCC and may be associated with a better prognosis.
- A better understanding of spontaneous regression of MCC could help in the development of new immunotherapeutic approaches to this malignancy.
Photosensitive Atopic Dermatitis Exacerbated by UVB Exposure
Atopic dermatitis (AD) is the most common inflammatory skin condition, affecting approximately 15% to 20% of the global population.1,2 Atopic dermatitis is characterized by a chronic relapsing dermatitis with pruritus, often beginning in infancy or childhood. Atopic dermatitis is caused by a defect in epidermal barrier function, which results in increased transepidermal water loss.1 The criteria for AD include a pruritic skin condition plus 3 or more of the following: history of involvement of the skin creases, history of asthma or hay fever, history of AD in a first-degree relative (in children), 1-year history of generally dry skin, visible flexural eczema, and an age of onset of less than 2 years. Adults with AD frequently present with hand or facial dermatitis.1
UV light therapies including narrowband UVB (NB-UVB), UVA1, and psoralen plus UVA (PUVA) have all been used as effective treatments of AD.3,4 UV light is beneficial for AD patients due to its immunomodulatory effects, thickening of the stratum corneum, and the reduction of Staphylococcus aureus in the skin.2 Most patients with AD improve with light therapy; however, it is estimated that 1% to 3% of patients with AD will experience a paradoxical worsening of their AD after exposure to UV light.2,5 This condition is referred to as photosensitive AD and is characterized by a photodistributed rash in patients who fulfill the criteria of AD. Photosensitive AD has a female predominance and generally affects patients with late-onset disease with development of AD after puberty.2,5 The pathogenesis for the development of photosensitivity in patients with AD who previously tolerated exposure to sunlight is unknown.5 We describe a case of photosensitive AD exacerbated by UVB exposure.
Case Report
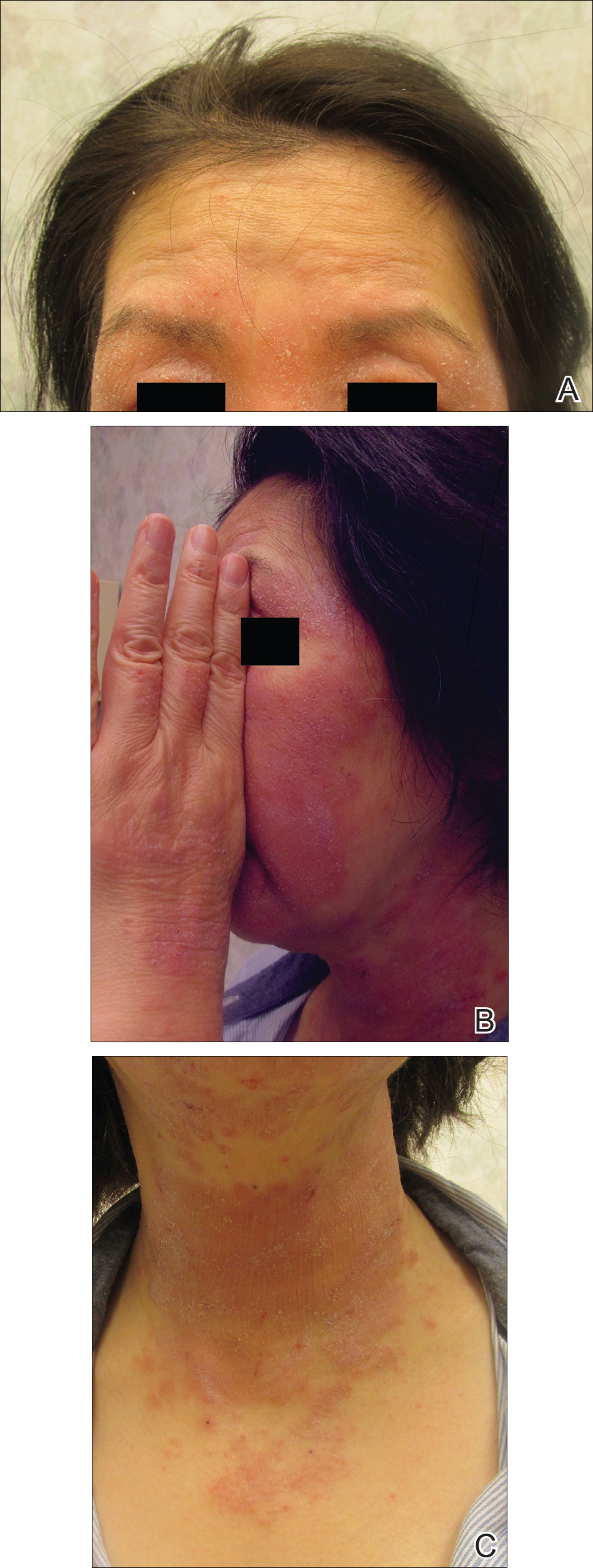
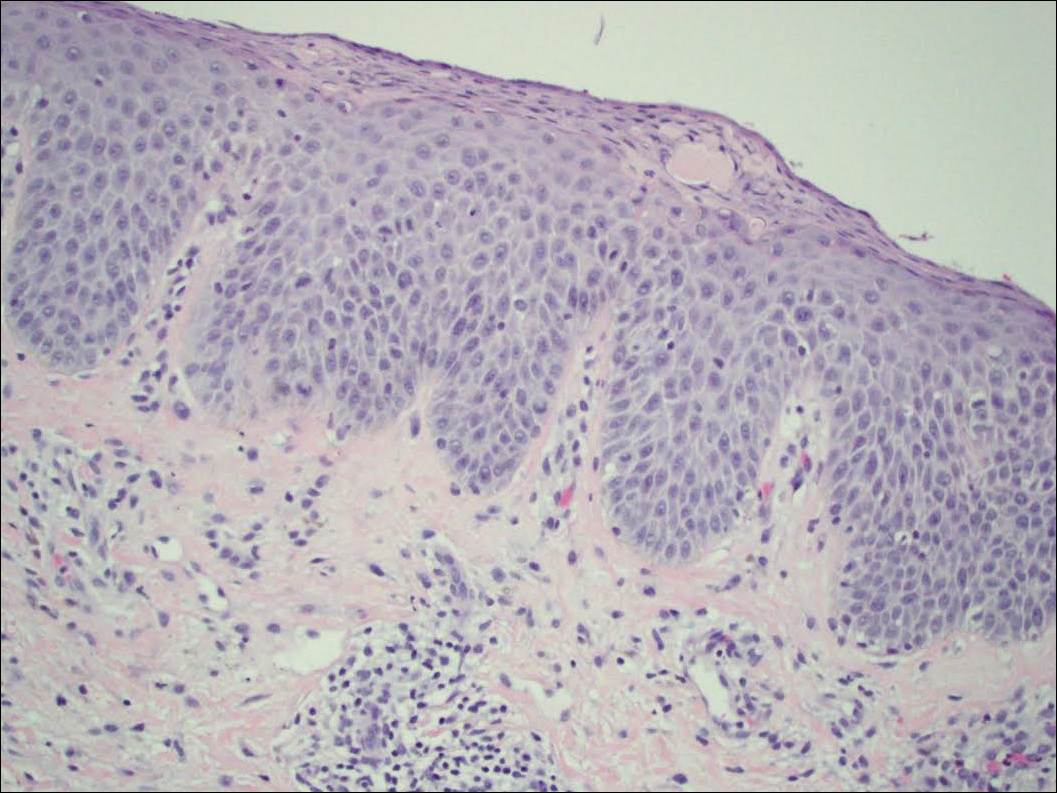
On physical examination the patient had thin, well-demarcated, erythematous papules and plaques with scaling, primarily on sun-exposed skin on the forehead (Figure 1A), cheeks (Figure 1B), eyelids, upper lip, neck (Figures 1B and 1C), upper chest (Figure 1C), and dorsal aspect of the hands, with excoriated pink papules on the forearms, shoulders, and back. A punch biopsy of the right neck showed spongiotic dermatitis with a perivascular lymphohistiocytic infiltrate (Figure 2). Further workup was pursued including complete blood cell count, comprehensive metabolic profile, liver function panel, Sjögren syndrome antigen A/Sjögren syndrome antigen B test, antinuclear antibody test, human immunodeficiency virus 1/2 antigen/antibody test, hepatitis panel, and mycobacterium tuberculosis test, which were all within reference range. Photodermatosis was suspected and she underwent phototesting including UVA, NB-UVB, and visible light. Phototesting confirmed she had a UVB photosensitivity with a markedly decreased minimal erythema dose (MED) to NB-UVB. The MED to NB-UVB was positive at 24 hours to all tested sites, the lowest of which was 0.135 J/cm2. Eczematous changes began to develop at day 6 at doses of 0.945 and 1.080 J/cm2. The patient also underwent visible light testing, which was negative. The patient was patch tested for multiple standardized agents as well as personal products, all of which were negative. Subsequent photopatch testing revealed a slightly positive reaction to benzophenone 4, a common ingredient in sunscreens.
The patient was then started on mycophenolate mofetil and prednisone. Repeat MED testing to NB-UVB was performed. Her repeat MED to NB-UVB was determined to be 0.405 J/cm2, and hardening commenced at 3 times per week at 70% of the MED (0.2835 J/cm2). She began to flare and develop an eczematous reaction, thus the dose was decreased to 50% of the MED (0.2025 J/cm2), which she tolerated.
Comment
Classification and Clinical Presentation
The literature on photosensitive AD is scant, and this disease entity is rare. Alternative names include photoaggravated AD, photosensitive eczema, and light-exacerbated eczema.5 Two main studies have been conducted in recent years that were intended to characterize photosensitive AD. ten Berge et al5 conducted a retrospective study of 145 patients with AD that were phototested in 2009. They found that 3% of their total AD patient population had photosensitive AD.5 In 2016, Ellenbogen et al2 performed a similar single-center retrospective analysis of 17 patients with long-standing AD who suddenly developed photosensitivity.
Patients with photosensitive AD typically present with lesions on sun-exposed skin with coexisting eczematous lesions in sites with a predilection for AD.2 In the study conducted by ten Berge et al,5 2 main reaction patterns were observed: erythematous papules with pruritus and an eczematous reaction.
Histopathology
The histopathologic findings of photosensitive AD are nonspecific but are characterized by spongiotic dermatitis with a perivascular lymphohistiocytic infiltrate.2
Diagnosis With Phototesting
Phototesting of patients with AD should be considered if there is a suspicion for photosensitivity based on persistent disease despite use of photoprotection and local treatment.5-7 Patients may not notice a correlation of skin exacerbations with UV exposure, especially if they are only sensitive to UVA, as it is still present on cloudy days and can penetrate glass windows.8 Phototesting evaluates the degree of sensitivity to UV light and the specific wavelength eliciting the cutaneous response. Phototesting consists of determining the MED to UVA and UVB, the minimal phototoxic dose for PUVA, and visible light exposure. Further evaluation may include photoprovocation testing or photopatch testing, as these patients can have coexisting photocontact allergies.
The MED is defined as the minimal dose of UV light needed to induce perceptible erythema in exposed skin.5 It is dependent on the light source and patient’s skin type, and individual units may vary. To determine the MED to UVA or UVB, 2×2-cm skin fields are irradiated with increasing cumulative UVA/UVB. The dose varies by skin type and it is then read at 24 hours. The majority of patients with photosensitive AD are reported to have a normal MED; however, some studies have reported the MED to be decreased.5,7-9 ten Berge et al5 found 7% of their study participants exhibited a lower MED, as seen in our patient.
The minimal phototoxic dose for PUVA is defined as the least exposure dose of UVA 1 hour after ingestion of 0.4 mg/kg of methoxsalen that produces pink erythema with 4 distinct borders at 48, 72, or 96 hours after ingestion.10 Visible light exposure is tested using a slide projector as the light source to an approximately 10×5-cm area of skin for 45 minutes. Any immediate or delayed reaction is abnormal and considered positive.10
Photoprovocation testing has been performed in several studies.2,5 It consists of exposing an 8-cm area of skin to 80 J/cm2 UVA and 10 mJ/cm2 UVB, which is read at 24, 48, or 72 hours. A papular or eczematous reaction is considered positive.2,11
The results of phototesting have varied between studies. ten Berge et al5 phototested 107 patients with AD and photosensitivity and 17% were found to be solely sensitive to UVA whereas 67% were found to be sensitive to UVA and UVB. In contrast, Ellenbogen et al2 only tested 17 patients with AD and photosensitivity and they found that 56% (9/16) were sensitive to UVA alone while only 44% (7/16) were sensitive to UVA and UVB.
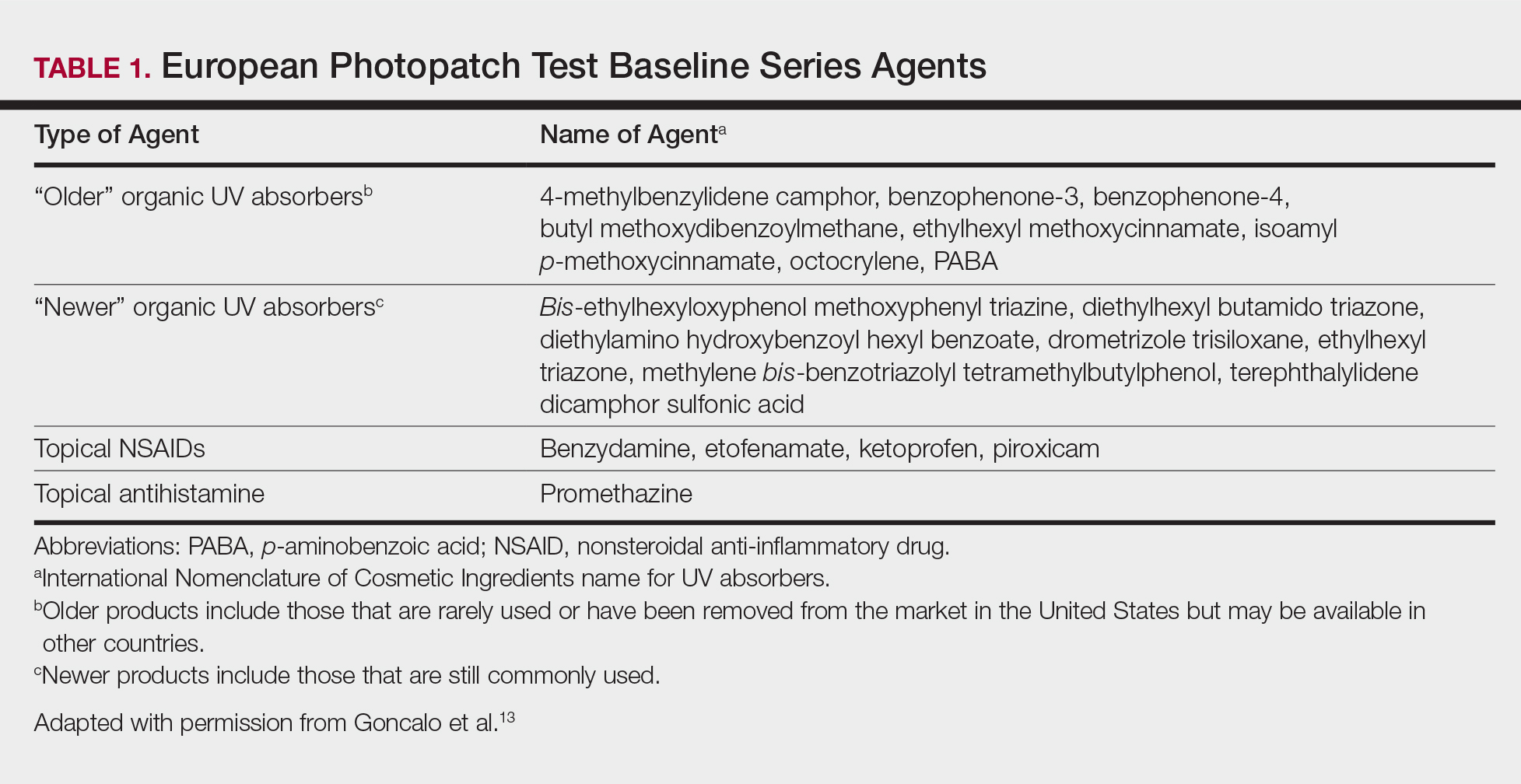
Photopatch testing can help to rule out photosensitivity due to a substance in the presence of UV light. In studies of patients with photosensitive AD (N=125), photocontact reactions occurred in 23% and were predominantly associated with sunscreens, skin care products, and fragrances.5,12 Photopatch testing is done by placing duplicate sets of patches on nonlesional skin using the Finn Chamber technique. A published list of allergens, which were agreed upon by the European Society of Contact Dermatitis and the European Society for Photodermatology in 2000 are seen in Table 1.13 The list contains mainly UV filters and drugs. The patients’ own products also should be tested in addition to the published list of allergens, but a maximum of 30 patches should be placed at one time. The patches are removed at either 24 or 48 hours; some researchers have found greater sensitivity with the 48-hour time period, while others have not found a significant difference.10 One set of skin fields then is covered with an impermeable occlusive dressing as a control while the other is irradiated with 5 J/cm2 of a broad-spectrum UVA light source. UVA fluorescent lamps are the light source of choice because of their widespread availability, reproducible broad spectrum, and beam uniformity.10 In the study conducted by ten Berge et al,5 photopatch testing was performed on 125 patients, and 29 patients were found to be positive to one or more substances. Ellenbogen et al2 photopatch tested 5 patients with photosensitive AD and a clinical suspicion of photoallergy; however, all 5 were negative. Our patient underwent traditional patch testing due to clinical suspicion of a coexisting contact allergy, which was negative.
Differential Diagnosis
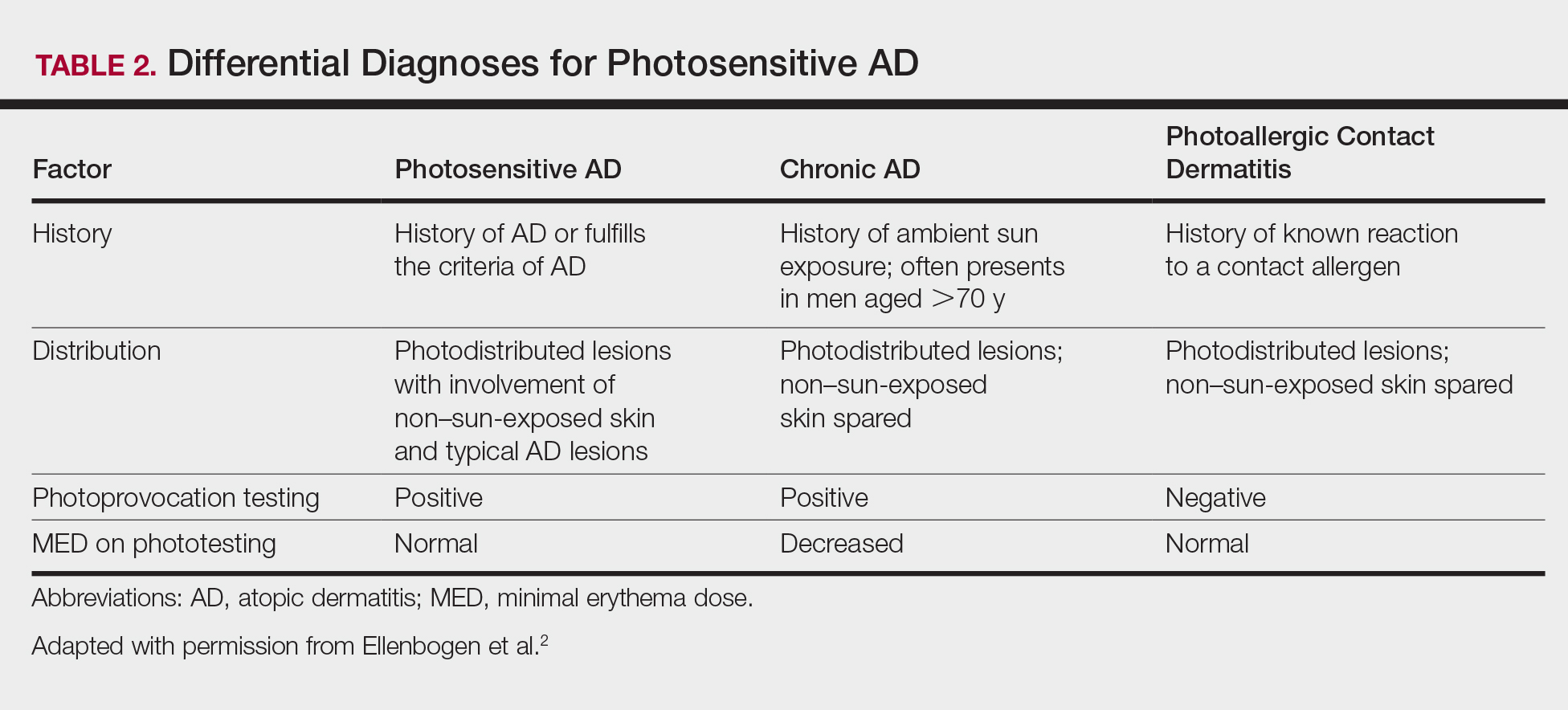
The differential diagnosis for photosensitive AD includes PMLE with coexisting AD, chronic AD, and photoallergic contact dermatitis. Photosensitive AD worsens with increasing exposure to uncontrolled sunlight, in contrast to patients with PMLE who experience UV radiation (UVR) hardening with increasing UV exposure during the summer months, resulting in improvement of skin lesions. Patients with chronic AD generally report a history of chronic ambient sun exposure and exhibit well-demarcated eczematous lesions in a photodistributed pattern with sparing of sun-protected skin.2 In contrast, photosensitive AD involves both sun-exposed and covered areas of the body. Chronic AD will have a positive photoprovocation test with a decreased MED (Table 2). Photoallergic contact dermatitis also will have photodistributed eczematous lesions with relative sparing of non–sun-exposed skin; however, these patients generally have negative photoprovocation testing with a normal MED.2 These patients may or may not have a history of reaction to a known allergen, but they likely will have a positive photopatch test.
Treatment
The treatment of photosensitive AD is based on the severity of the photosensitivity. Treatment for mild disease is limited to sun protection in addition to topical corticosteroids or topical calcineurin inhibitors. For moderate disease and unsatisfactory relief with proper sun protection, UVR hardening is recommended. If severe disease is present, immunosuppression with medications such as corticosteroids, cyclosporine, and mycophenolate mofetil is suggested to prevent flaring of disease during UVR hardening.2,5,8,14
Conclusion
Photosensitive AD is a rare entity characterized by a photodistributed rash and involvement of non–sun-exposed skin. Patients will either have a history of AD or fulfill the criteria of AD. They have positive photoprovocation testing and generally have a normal MED. They may have positive photopatch testing with coexisting photoallergies. Histopathology is nonspecific but shows spongiotic dermatitis with perivascular lymphohistiocytic infiltrate. Diagnosis is essential, as this disease can be life altering and affect quality of life. Effective treatment options are available, and the therapeutic ladder is based on severity of disease.2,5
- Bieber T, Bussmann C. Atopic dermatitis. In: Bolognia JL, Jorizzo J, Rapini R, eds. Dermatology. 3rd ed. New York, NY: Elsevier; 2012:203-230.
- Ellenbogen E, Wesselmann U, Hofmann SC, et al. Photosensitive atopic dermatitis—a neglected subset: clinical, laboratory, histological and photobiological workup. J Eur Acad Dermatol Venereol. 2016;30:270-275.
- Yule S, Dawe RS, Cameron H, et al. Does narrow-band ultraviolet B phototherapy work in atopic dermatitis through a local or a systemic effect? Photodermatol Photoimmunol Photomed. 2005;21:333-335.
- Sidbury R, Davis DM, Cohen DE, et al. Guidelines of care for the management of atopic dermatitis. section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71:327-349.
- ten Berge O, van Weelden H, Bruijnzeel-Koomen CA, et al. Throwing a light on photosensitivity in atopic dermatitis: a retrospective study. Am J Clin Dermatol. 2009;10:119-123.
- O’Gorman SM, Murphy GM. Photoaggravated disorders. Dermatol Clin. 2014;32:385-398.
- Crouch RB, Foley PA, Baker CS. Analysis of patients with suspected photosensitivity referred for investigation to an Australian photodermatology clinic. J Am Acad Dermatol. 2003;48:714-720.
- Russell SC, Dawes RS, Collins P, et al. The photosensitivity dermatitis and actinic reticuloid syndrome (chronic actinic dermatitis) occurring in seven young atopic dermatitis patients. Br J Dermatol. 1998;138:496-501.
- Tajima T, Ibe M, Matsushita T, et al. A variety of skin responses to ultraviolet irradiation in patients with atopic dermatitis. J Dermatol Sci. 1998;17:101-107.
- Faurschou A, Wulf HC. European Dermatology Guideline for the photodermatoses: phototesting. European Dermatology Forum website. http://www.euroderm.org/edf/index.php/edf-guidelines/category/3-guidelines-on-photodermatoses. Accessed August 21, 2017.
- Keong CH, Kurumaji Y, Miyamoto C, et al. Photosensitivity in atopic dermatitis: demonstration of abnormal response to UVB. J Dermatol. 1992;19:342-347.
- Lee PA, Freeman S. Photosensitivity: the 9-year experience at a Sydney contact dermatitis clinic. Australas J Dermatol. 2002;43:289-292.
- Goncalo M, Ferguson J, Bonevalle A, et al. Photopatch testing: recommendations for a European photopatch test baseline series. Contact Dermatitis. 2013;68:239-243.
- Amon U, Mangalo S, Roth A. Clinical relevance of increased UV-sensitivity in patients with atopic dermatitis. J Allergy Clin Immunol. 2011;127:AB39.
Atopic dermatitis (AD) is the most common inflammatory skin condition, affecting approximately 15% to 20% of the global population.1,2 Atopic dermatitis is characterized by a chronic relapsing dermatitis with pruritus, often beginning in infancy or childhood. Atopic dermatitis is caused by a defect in epidermal barrier function, which results in increased transepidermal water loss.1 The criteria for AD include a pruritic skin condition plus 3 or more of the following: history of involvement of the skin creases, history of asthma or hay fever, history of AD in a first-degree relative (in children), 1-year history of generally dry skin, visible flexural eczema, and an age of onset of less than 2 years. Adults with AD frequently present with hand or facial dermatitis.1
UV light therapies including narrowband UVB (NB-UVB), UVA1, and psoralen plus UVA (PUVA) have all been used as effective treatments of AD.3,4 UV light is beneficial for AD patients due to its immunomodulatory effects, thickening of the stratum corneum, and the reduction of Staphylococcus aureus in the skin.2 Most patients with AD improve with light therapy; however, it is estimated that 1% to 3% of patients with AD will experience a paradoxical worsening of their AD after exposure to UV light.2,5 This condition is referred to as photosensitive AD and is characterized by a photodistributed rash in patients who fulfill the criteria of AD. Photosensitive AD has a female predominance and generally affects patients with late-onset disease with development of AD after puberty.2,5 The pathogenesis for the development of photosensitivity in patients with AD who previously tolerated exposure to sunlight is unknown.5 We describe a case of photosensitive AD exacerbated by UVB exposure.
Case Report
On physical examination the patient had thin, well-demarcated, erythematous papules and plaques with scaling, primarily on sun-exposed skin on the forehead (Figure 1A), cheeks (Figure 1B), eyelids, upper lip, neck (Figures 1B and 1C), upper chest (Figure 1C), and dorsal aspect of the hands, with excoriated pink papules on the forearms, shoulders, and back. A punch biopsy of the right neck showed spongiotic dermatitis with a perivascular lymphohistiocytic infiltrate (Figure 2). Further workup was pursued including complete blood cell count, comprehensive metabolic profile, liver function panel, Sjögren syndrome antigen A/Sjögren syndrome antigen B test, antinuclear antibody test, human immunodeficiency virus 1/2 antigen/antibody test, hepatitis panel, and mycobacterium tuberculosis test, which were all within reference range. Photodermatosis was suspected and she underwent phototesting including UVA, NB-UVB, and visible light. Phototesting confirmed she had a UVB photosensitivity with a markedly decreased minimal erythema dose (MED) to NB-UVB. The MED to NB-UVB was positive at 24 hours to all tested sites, the lowest of which was 0.135 J/cm2. Eczematous changes began to develop at day 6 at doses of 0.945 and 1.080 J/cm2. The patient also underwent visible light testing, which was negative. The patient was patch tested for multiple standardized agents as well as personal products, all of which were negative. Subsequent photopatch testing revealed a slightly positive reaction to benzophenone 4, a common ingredient in sunscreens.
The patient was then started on mycophenolate mofetil and prednisone. Repeat MED testing to NB-UVB was performed. Her repeat MED to NB-UVB was determined to be 0.405 J/cm2, and hardening commenced at 3 times per week at 70% of the MED (0.2835 J/cm2). She began to flare and develop an eczematous reaction, thus the dose was decreased to 50% of the MED (0.2025 J/cm2), which she tolerated.
Comment
Classification and Clinical Presentation
The literature on photosensitive AD is scant, and this disease entity is rare. Alternative names include photoaggravated AD, photosensitive eczema, and light-exacerbated eczema.5 Two main studies have been conducted in recent years that were intended to characterize photosensitive AD. ten Berge et al5 conducted a retrospective study of 145 patients with AD that were phototested in 2009. They found that 3% of their total AD patient population had photosensitive AD.5 In 2016, Ellenbogen et al2 performed a similar single-center retrospective analysis of 17 patients with long-standing AD who suddenly developed photosensitivity.
Patients with photosensitive AD typically present with lesions on sun-exposed skin with coexisting eczematous lesions in sites with a predilection for AD.2 In the study conducted by ten Berge et al,5 2 main reaction patterns were observed: erythematous papules with pruritus and an eczematous reaction.
Histopathology
The histopathologic findings of photosensitive AD are nonspecific but are characterized by spongiotic dermatitis with a perivascular lymphohistiocytic infiltrate.2
Diagnosis With Phototesting
Phototesting of patients with AD should be considered if there is a suspicion for photosensitivity based on persistent disease despite use of photoprotection and local treatment.5-7 Patients may not notice a correlation of skin exacerbations with UV exposure, especially if they are only sensitive to UVA, as it is still present on cloudy days and can penetrate glass windows.8 Phototesting evaluates the degree of sensitivity to UV light and the specific wavelength eliciting the cutaneous response. Phototesting consists of determining the MED to UVA and UVB, the minimal phototoxic dose for PUVA, and visible light exposure. Further evaluation may include photoprovocation testing or photopatch testing, as these patients can have coexisting photocontact allergies.
The MED is defined as the minimal dose of UV light needed to induce perceptible erythema in exposed skin.5 It is dependent on the light source and patient’s skin type, and individual units may vary. To determine the MED to UVA or UVB, 2×2-cm skin fields are irradiated with increasing cumulative UVA/UVB. The dose varies by skin type and it is then read at 24 hours. The majority of patients with photosensitive AD are reported to have a normal MED; however, some studies have reported the MED to be decreased.5,7-9 ten Berge et al5 found 7% of their study participants exhibited a lower MED, as seen in our patient.
The minimal phototoxic dose for PUVA is defined as the least exposure dose of UVA 1 hour after ingestion of 0.4 mg/kg of methoxsalen that produces pink erythema with 4 distinct borders at 48, 72, or 96 hours after ingestion.10 Visible light exposure is tested using a slide projector as the light source to an approximately 10×5-cm area of skin for 45 minutes. Any immediate or delayed reaction is abnormal and considered positive.10
Photoprovocation testing has been performed in several studies.2,5 It consists of exposing an 8-cm area of skin to 80 J/cm2 UVA and 10 mJ/cm2 UVB, which is read at 24, 48, or 72 hours. A papular or eczematous reaction is considered positive.2,11
The results of phototesting have varied between studies. ten Berge et al5 phototested 107 patients with AD and photosensitivity and 17% were found to be solely sensitive to UVA whereas 67% were found to be sensitive to UVA and UVB. In contrast, Ellenbogen et al2 only tested 17 patients with AD and photosensitivity and they found that 56% (9/16) were sensitive to UVA alone while only 44% (7/16) were sensitive to UVA and UVB.
Photopatch testing can help to rule out photosensitivity due to a substance in the presence of UV light. In studies of patients with photosensitive AD (N=125), photocontact reactions occurred in 23% and were predominantly associated with sunscreens, skin care products, and fragrances.5,12 Photopatch testing is done by placing duplicate sets of patches on nonlesional skin using the Finn Chamber technique. A published list of allergens, which were agreed upon by the European Society of Contact Dermatitis and the European Society for Photodermatology in 2000 are seen in Table 1.13 The list contains mainly UV filters and drugs. The patients’ own products also should be tested in addition to the published list of allergens, but a maximum of 30 patches should be placed at one time. The patches are removed at either 24 or 48 hours; some researchers have found greater sensitivity with the 48-hour time period, while others have not found a significant difference.10 One set of skin fields then is covered with an impermeable occlusive dressing as a control while the other is irradiated with 5 J/cm2 of a broad-spectrum UVA light source. UVA fluorescent lamps are the light source of choice because of their widespread availability, reproducible broad spectrum, and beam uniformity.10 In the study conducted by ten Berge et al,5 photopatch testing was performed on 125 patients, and 29 patients were found to be positive to one or more substances. Ellenbogen et al2 photopatch tested 5 patients with photosensitive AD and a clinical suspicion of photoallergy; however, all 5 were negative. Our patient underwent traditional patch testing due to clinical suspicion of a coexisting contact allergy, which was negative.
Differential Diagnosis
The differential diagnosis for photosensitive AD includes PMLE with coexisting AD, chronic AD, and photoallergic contact dermatitis. Photosensitive AD worsens with increasing exposure to uncontrolled sunlight, in contrast to patients with PMLE who experience UV radiation (UVR) hardening with increasing UV exposure during the summer months, resulting in improvement of skin lesions. Patients with chronic AD generally report a history of chronic ambient sun exposure and exhibit well-demarcated eczematous lesions in a photodistributed pattern with sparing of sun-protected skin.2 In contrast, photosensitive AD involves both sun-exposed and covered areas of the body. Chronic AD will have a positive photoprovocation test with a decreased MED (Table 2). Photoallergic contact dermatitis also will have photodistributed eczematous lesions with relative sparing of non–sun-exposed skin; however, these patients generally have negative photoprovocation testing with a normal MED.2 These patients may or may not have a history of reaction to a known allergen, but they likely will have a positive photopatch test.
Treatment
The treatment of photosensitive AD is based on the severity of the photosensitivity. Treatment for mild disease is limited to sun protection in addition to topical corticosteroids or topical calcineurin inhibitors. For moderate disease and unsatisfactory relief with proper sun protection, UVR hardening is recommended. If severe disease is present, immunosuppression with medications such as corticosteroids, cyclosporine, and mycophenolate mofetil is suggested to prevent flaring of disease during UVR hardening.2,5,8,14
Conclusion
Photosensitive AD is a rare entity characterized by a photodistributed rash and involvement of non–sun-exposed skin. Patients will either have a history of AD or fulfill the criteria of AD. They have positive photoprovocation testing and generally have a normal MED. They may have positive photopatch testing with coexisting photoallergies. Histopathology is nonspecific but shows spongiotic dermatitis with perivascular lymphohistiocytic infiltrate. Diagnosis is essential, as this disease can be life altering and affect quality of life. Effective treatment options are available, and the therapeutic ladder is based on severity of disease.2,5
Atopic dermatitis (AD) is the most common inflammatory skin condition, affecting approximately 15% to 20% of the global population.1,2 Atopic dermatitis is characterized by a chronic relapsing dermatitis with pruritus, often beginning in infancy or childhood. Atopic dermatitis is caused by a defect in epidermal barrier function, which results in increased transepidermal water loss.1 The criteria for AD include a pruritic skin condition plus 3 or more of the following: history of involvement of the skin creases, history of asthma or hay fever, history of AD in a first-degree relative (in children), 1-year history of generally dry skin, visible flexural eczema, and an age of onset of less than 2 years. Adults with AD frequently present with hand or facial dermatitis.1
UV light therapies including narrowband UVB (NB-UVB), UVA1, and psoralen plus UVA (PUVA) have all been used as effective treatments of AD.3,4 UV light is beneficial for AD patients due to its immunomodulatory effects, thickening of the stratum corneum, and the reduction of Staphylococcus aureus in the skin.2 Most patients with AD improve with light therapy; however, it is estimated that 1% to 3% of patients with AD will experience a paradoxical worsening of their AD after exposure to UV light.2,5 This condition is referred to as photosensitive AD and is characterized by a photodistributed rash in patients who fulfill the criteria of AD. Photosensitive AD has a female predominance and generally affects patients with late-onset disease with development of AD after puberty.2,5 The pathogenesis for the development of photosensitivity in patients with AD who previously tolerated exposure to sunlight is unknown.5 We describe a case of photosensitive AD exacerbated by UVB exposure.
Case Report
On physical examination the patient had thin, well-demarcated, erythematous papules and plaques with scaling, primarily on sun-exposed skin on the forehead (Figure 1A), cheeks (Figure 1B), eyelids, upper lip, neck (Figures 1B and 1C), upper chest (Figure 1C), and dorsal aspect of the hands, with excoriated pink papules on the forearms, shoulders, and back. A punch biopsy of the right neck showed spongiotic dermatitis with a perivascular lymphohistiocytic infiltrate (Figure 2). Further workup was pursued including complete blood cell count, comprehensive metabolic profile, liver function panel, Sjögren syndrome antigen A/Sjögren syndrome antigen B test, antinuclear antibody test, human immunodeficiency virus 1/2 antigen/antibody test, hepatitis panel, and mycobacterium tuberculosis test, which were all within reference range. Photodermatosis was suspected and she underwent phototesting including UVA, NB-UVB, and visible light. Phototesting confirmed she had a UVB photosensitivity with a markedly decreased minimal erythema dose (MED) to NB-UVB. The MED to NB-UVB was positive at 24 hours to all tested sites, the lowest of which was 0.135 J/cm2. Eczematous changes began to develop at day 6 at doses of 0.945 and 1.080 J/cm2. The patient also underwent visible light testing, which was negative. The patient was patch tested for multiple standardized agents as well as personal products, all of which were negative. Subsequent photopatch testing revealed a slightly positive reaction to benzophenone 4, a common ingredient in sunscreens.
The patient was then started on mycophenolate mofetil and prednisone. Repeat MED testing to NB-UVB was performed. Her repeat MED to NB-UVB was determined to be 0.405 J/cm2, and hardening commenced at 3 times per week at 70% of the MED (0.2835 J/cm2). She began to flare and develop an eczematous reaction, thus the dose was decreased to 50% of the MED (0.2025 J/cm2), which she tolerated.
Comment
Classification and Clinical Presentation
The literature on photosensitive AD is scant, and this disease entity is rare. Alternative names include photoaggravated AD, photosensitive eczema, and light-exacerbated eczema.5 Two main studies have been conducted in recent years that were intended to characterize photosensitive AD. ten Berge et al5 conducted a retrospective study of 145 patients with AD that were phototested in 2009. They found that 3% of their total AD patient population had photosensitive AD.5 In 2016, Ellenbogen et al2 performed a similar single-center retrospective analysis of 17 patients with long-standing AD who suddenly developed photosensitivity.
Patients with photosensitive AD typically present with lesions on sun-exposed skin with coexisting eczematous lesions in sites with a predilection for AD.2 In the study conducted by ten Berge et al,5 2 main reaction patterns were observed: erythematous papules with pruritus and an eczematous reaction.
Histopathology
The histopathologic findings of photosensitive AD are nonspecific but are characterized by spongiotic dermatitis with a perivascular lymphohistiocytic infiltrate.2
Diagnosis With Phototesting
Phototesting of patients with AD should be considered if there is a suspicion for photosensitivity based on persistent disease despite use of photoprotection and local treatment.5-7 Patients may not notice a correlation of skin exacerbations with UV exposure, especially if they are only sensitive to UVA, as it is still present on cloudy days and can penetrate glass windows.8 Phototesting evaluates the degree of sensitivity to UV light and the specific wavelength eliciting the cutaneous response. Phototesting consists of determining the MED to UVA and UVB, the minimal phototoxic dose for PUVA, and visible light exposure. Further evaluation may include photoprovocation testing or photopatch testing, as these patients can have coexisting photocontact allergies.
The MED is defined as the minimal dose of UV light needed to induce perceptible erythema in exposed skin.5 It is dependent on the light source and patient’s skin type, and individual units may vary. To determine the MED to UVA or UVB, 2×2-cm skin fields are irradiated with increasing cumulative UVA/UVB. The dose varies by skin type and it is then read at 24 hours. The majority of patients with photosensitive AD are reported to have a normal MED; however, some studies have reported the MED to be decreased.5,7-9 ten Berge et al5 found 7% of their study participants exhibited a lower MED, as seen in our patient.
The minimal phototoxic dose for PUVA is defined as the least exposure dose of UVA 1 hour after ingestion of 0.4 mg/kg of methoxsalen that produces pink erythema with 4 distinct borders at 48, 72, or 96 hours after ingestion.10 Visible light exposure is tested using a slide projector as the light source to an approximately 10×5-cm area of skin for 45 minutes. Any immediate or delayed reaction is abnormal and considered positive.10
Photoprovocation testing has been performed in several studies.2,5 It consists of exposing an 8-cm area of skin to 80 J/cm2 UVA and 10 mJ/cm2 UVB, which is read at 24, 48, or 72 hours. A papular or eczematous reaction is considered positive.2,11
The results of phototesting have varied between studies. ten Berge et al5 phototested 107 patients with AD and photosensitivity and 17% were found to be solely sensitive to UVA whereas 67% were found to be sensitive to UVA and UVB. In contrast, Ellenbogen et al2 only tested 17 patients with AD and photosensitivity and they found that 56% (9/16) were sensitive to UVA alone while only 44% (7/16) were sensitive to UVA and UVB.
Photopatch testing can help to rule out photosensitivity due to a substance in the presence of UV light. In studies of patients with photosensitive AD (N=125), photocontact reactions occurred in 23% and were predominantly associated with sunscreens, skin care products, and fragrances.5,12 Photopatch testing is done by placing duplicate sets of patches on nonlesional skin using the Finn Chamber technique. A published list of allergens, which were agreed upon by the European Society of Contact Dermatitis and the European Society for Photodermatology in 2000 are seen in Table 1.13 The list contains mainly UV filters and drugs. The patients’ own products also should be tested in addition to the published list of allergens, but a maximum of 30 patches should be placed at one time. The patches are removed at either 24 or 48 hours; some researchers have found greater sensitivity with the 48-hour time period, while others have not found a significant difference.10 One set of skin fields then is covered with an impermeable occlusive dressing as a control while the other is irradiated with 5 J/cm2 of a broad-spectrum UVA light source. UVA fluorescent lamps are the light source of choice because of their widespread availability, reproducible broad spectrum, and beam uniformity.10 In the study conducted by ten Berge et al,5 photopatch testing was performed on 125 patients, and 29 patients were found to be positive to one or more substances. Ellenbogen et al2 photopatch tested 5 patients with photosensitive AD and a clinical suspicion of photoallergy; however, all 5 were negative. Our patient underwent traditional patch testing due to clinical suspicion of a coexisting contact allergy, which was negative.
Differential Diagnosis
The differential diagnosis for photosensitive AD includes PMLE with coexisting AD, chronic AD, and photoallergic contact dermatitis. Photosensitive AD worsens with increasing exposure to uncontrolled sunlight, in contrast to patients with PMLE who experience UV radiation (UVR) hardening with increasing UV exposure during the summer months, resulting in improvement of skin lesions. Patients with chronic AD generally report a history of chronic ambient sun exposure and exhibit well-demarcated eczematous lesions in a photodistributed pattern with sparing of sun-protected skin.2 In contrast, photosensitive AD involves both sun-exposed and covered areas of the body. Chronic AD will have a positive photoprovocation test with a decreased MED (Table 2). Photoallergic contact dermatitis also will have photodistributed eczematous lesions with relative sparing of non–sun-exposed skin; however, these patients generally have negative photoprovocation testing with a normal MED.2 These patients may or may not have a history of reaction to a known allergen, but they likely will have a positive photopatch test.
Treatment
The treatment of photosensitive AD is based on the severity of the photosensitivity. Treatment for mild disease is limited to sun protection in addition to topical corticosteroids or topical calcineurin inhibitors. For moderate disease and unsatisfactory relief with proper sun protection, UVR hardening is recommended. If severe disease is present, immunosuppression with medications such as corticosteroids, cyclosporine, and mycophenolate mofetil is suggested to prevent flaring of disease during UVR hardening.2,5,8,14
Conclusion
Photosensitive AD is a rare entity characterized by a photodistributed rash and involvement of non–sun-exposed skin. Patients will either have a history of AD or fulfill the criteria of AD. They have positive photoprovocation testing and generally have a normal MED. They may have positive photopatch testing with coexisting photoallergies. Histopathology is nonspecific but shows spongiotic dermatitis with perivascular lymphohistiocytic infiltrate. Diagnosis is essential, as this disease can be life altering and affect quality of life. Effective treatment options are available, and the therapeutic ladder is based on severity of disease.2,5
- Bieber T, Bussmann C. Atopic dermatitis. In: Bolognia JL, Jorizzo J, Rapini R, eds. Dermatology. 3rd ed. New York, NY: Elsevier; 2012:203-230.
- Ellenbogen E, Wesselmann U, Hofmann SC, et al. Photosensitive atopic dermatitis—a neglected subset: clinical, laboratory, histological and photobiological workup. J Eur Acad Dermatol Venereol. 2016;30:270-275.
- Yule S, Dawe RS, Cameron H, et al. Does narrow-band ultraviolet B phototherapy work in atopic dermatitis through a local or a systemic effect? Photodermatol Photoimmunol Photomed. 2005;21:333-335.
- Sidbury R, Davis DM, Cohen DE, et al. Guidelines of care for the management of atopic dermatitis. section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71:327-349.
- ten Berge O, van Weelden H, Bruijnzeel-Koomen CA, et al. Throwing a light on photosensitivity in atopic dermatitis: a retrospective study. Am J Clin Dermatol. 2009;10:119-123.
- O’Gorman SM, Murphy GM. Photoaggravated disorders. Dermatol Clin. 2014;32:385-398.
- Crouch RB, Foley PA, Baker CS. Analysis of patients with suspected photosensitivity referred for investigation to an Australian photodermatology clinic. J Am Acad Dermatol. 2003;48:714-720.
- Russell SC, Dawes RS, Collins P, et al. The photosensitivity dermatitis and actinic reticuloid syndrome (chronic actinic dermatitis) occurring in seven young atopic dermatitis patients. Br J Dermatol. 1998;138:496-501.
- Tajima T, Ibe M, Matsushita T, et al. A variety of skin responses to ultraviolet irradiation in patients with atopic dermatitis. J Dermatol Sci. 1998;17:101-107.
- Faurschou A, Wulf HC. European Dermatology Guideline for the photodermatoses: phototesting. European Dermatology Forum website. http://www.euroderm.org/edf/index.php/edf-guidelines/category/3-guidelines-on-photodermatoses. Accessed August 21, 2017.
- Keong CH, Kurumaji Y, Miyamoto C, et al. Photosensitivity in atopic dermatitis: demonstration of abnormal response to UVB. J Dermatol. 1992;19:342-347.
- Lee PA, Freeman S. Photosensitivity: the 9-year experience at a Sydney contact dermatitis clinic. Australas J Dermatol. 2002;43:289-292.
- Goncalo M, Ferguson J, Bonevalle A, et al. Photopatch testing: recommendations for a European photopatch test baseline series. Contact Dermatitis. 2013;68:239-243.
- Amon U, Mangalo S, Roth A. Clinical relevance of increased UV-sensitivity in patients with atopic dermatitis. J Allergy Clin Immunol. 2011;127:AB39.
- Bieber T, Bussmann C. Atopic dermatitis. In: Bolognia JL, Jorizzo J, Rapini R, eds. Dermatology. 3rd ed. New York, NY: Elsevier; 2012:203-230.
- Ellenbogen E, Wesselmann U, Hofmann SC, et al. Photosensitive atopic dermatitis—a neglected subset: clinical, laboratory, histological and photobiological workup. J Eur Acad Dermatol Venereol. 2016;30:270-275.
- Yule S, Dawe RS, Cameron H, et al. Does narrow-band ultraviolet B phototherapy work in atopic dermatitis through a local or a systemic effect? Photodermatol Photoimmunol Photomed. 2005;21:333-335.
- Sidbury R, Davis DM, Cohen DE, et al. Guidelines of care for the management of atopic dermatitis. section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71:327-349.
- ten Berge O, van Weelden H, Bruijnzeel-Koomen CA, et al. Throwing a light on photosensitivity in atopic dermatitis: a retrospective study. Am J Clin Dermatol. 2009;10:119-123.
- O’Gorman SM, Murphy GM. Photoaggravated disorders. Dermatol Clin. 2014;32:385-398.
- Crouch RB, Foley PA, Baker CS. Analysis of patients with suspected photosensitivity referred for investigation to an Australian photodermatology clinic. J Am Acad Dermatol. 2003;48:714-720.
- Russell SC, Dawes RS, Collins P, et al. The photosensitivity dermatitis and actinic reticuloid syndrome (chronic actinic dermatitis) occurring in seven young atopic dermatitis patients. Br J Dermatol. 1998;138:496-501.
- Tajima T, Ibe M, Matsushita T, et al. A variety of skin responses to ultraviolet irradiation in patients with atopic dermatitis. J Dermatol Sci. 1998;17:101-107.
- Faurschou A, Wulf HC. European Dermatology Guideline for the photodermatoses: phototesting. European Dermatology Forum website. http://www.euroderm.org/edf/index.php/edf-guidelines/category/3-guidelines-on-photodermatoses. Accessed August 21, 2017.
- Keong CH, Kurumaji Y, Miyamoto C, et al. Photosensitivity in atopic dermatitis: demonstration of abnormal response to UVB. J Dermatol. 1992;19:342-347.
- Lee PA, Freeman S. Photosensitivity: the 9-year experience at a Sydney contact dermatitis clinic. Australas J Dermatol. 2002;43:289-292.
- Goncalo M, Ferguson J, Bonevalle A, et al. Photopatch testing: recommendations for a European photopatch test baseline series. Contact Dermatitis. 2013;68:239-243.
- Amon U, Mangalo S, Roth A. Clinical relevance of increased UV-sensitivity in patients with atopic dermatitis. J Allergy Clin Immunol. 2011;127:AB39.
Practice Points
- Photosensitive atopic dermatitis (AD) is rare but should be considered in patients with uncontrolled AD with a rash on sun-exposed skin.
- A thorough history and physical examination of these patients can provide the necessary clues for further workup.
- Phototesting should be performed to confirm the diagnosis and evaluate the degree of sensitivity to UV light and the specific wavelength eliciting the cutaneous response.
- Photoprovocation and photopatch testing also can be useful to confirm the diagnosis.
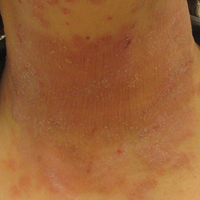
Merkel Cell Carcinoma: A Review
Merkel cells originally were described by German histopathologist Friedrich Sigmund Merkel in 1875. These unique tactile cells were described as epidermal, nondendritic, and nonkeratinizing. Merkel cells are thought to arise from the neural crest and are believed to be primary neural cells found within the basal layer of the epidermis.1,2 They likely function primarily as slowly adapting type I mechanoreceptors. Origin from the neural crest is controversial, as other investigators have suggested derivation from epidermal keratinocytes.1,2 Tumor cells have been linked to the amine precursor uptake and decarboxylation system.3 In 1972, Toker4 described several cases of trabecular or sweat gland carcinomas of the skin. Upon further investigation, the cells that comprised these tumors were found to have dense core granules on electron microscopy, typical of Merkel cells.1,2 Other terms such as neuroendocrine carcinoma of the skin, small cell carcinoma of the skin, and anaplastic carcinoma of the skin have been used to describe Merkel cell carcinoma (MCC),1 which was suggested by De Wolf-Peeters et al5 in 1980.
Despite being a rare malignancy, MCC follows an aggressive clinical course. Upon presentation, approximately 66% of patients have local disease, 27% have nodal involvement, and 7% have distant metastasis.1 Future treatments will likely center around the novel Merkel cell polyomavirus (MCPyV) and modification of immune responses toward tumor cells. Standardization continues to be lacking in both staging and treatment of this aggressive tumor.
Epidemiology of MCC
 | ||
Figure 1. A 2.3×1.5×1.2-cm, hemorrhagic, crusted, | ||
 | ||
 | ||
| Figure 2. Merkel cells are small- to medium-sized cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen (A)(H&E, original magnification ×40). Merkel cell carcinoma with trabecular pattern (B) (H&E, original magnification ×10). | ||
Between 1986 and 2006, the incidence of MCC grew substantially.1,2 Figures have been reported at 0.15 cases per 100,000 individuals to 0.6 cases per 100,000 individuals worldwide. In the United States, the age-adjusted incidence of MCC is estimated at 0.24 per 100,000 person-years, which is higher than the estimated 0.13 per 100,000 person-years found in Europe.3 The highest incidence worldwide has been noted in Western Australia, likely due to high levels of UV exposure.1 The incidence of MCC in psoriasis patients who are treated with oral methoxsalen (psoralen) and UVA photochemotherapy is 100 times greater than in the general population, further supporting the role of UV light in the development of MCC.1 White individuals have the highest incidence of MCC worldwide, with men being affected more frequently than women.1,3 The majority of patients with MCC are diagnosed at 70 years or older.1 Approximately 5% of reported MCC patients are diagnosed before 50 years of age.2 Immunosuppression and immunodeficiency likely play a role in the pathogenesis of MCC, and the incidence is increased in solid organ transplant recipients, most commonly renal transplant recipients,1 as well as individuals with chronic lymphocytic leukemia, human immunodeficiency virus infection, and AIDS.1,3 Patients with autoimmune diseases such as rheumatoid arthritis also are at increased risk for MCC.3 Individuals who are diagnosed with MCC are at an increased risk for development of other malignancies including nonmelanoma skin cancers, chronic lymphocytic leukemia, Hodgkin lymphoma, and non-Hodgkin lymphoma.3
Clinical Presentation of MCC
The clinical presentation of MCC can be variable. Most tumors present as firm, red to purple, nontender papules or nodules (Figure 1).1 Tumor size may range from 2 to 200 mm but is most commonly less than 20 mm.2 Growth can be rapid, and tumors are most commonly located on sun-exposed skin. The head and neck areas account for 48% of all MCC cases,1 with the eyelids being frequently involved.2 Merkel cell carcinoma also has been reported on the arms, legs, trunk, back, and buttocks.1 Non–sun-exposed areas are less commonly affected. Mucosal sites (eg, larynx, nasal cavity, pharynx, mouth) account for 5% of primary MCCs.1 Merkel cell carcinoma also has been reported to affect the vulva and penis. Subcutaneous primary MCC has presented without overlying epidermal changes.1 In a case series by Heath et al,6 14% (27/195) of MCC patients presented with nodal disease without any identifiable primary tumor, with the inguinal nodal chain being the most common for this presentation. It currently is not known whether these nodal tumors are primary tumors or metastatic disease with a regression of the primary tumor.1
|
Histopathology of MCC
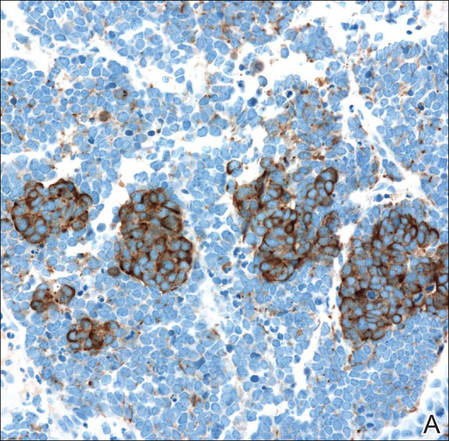 | ||
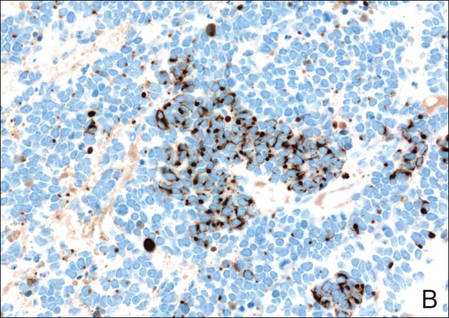 | ||
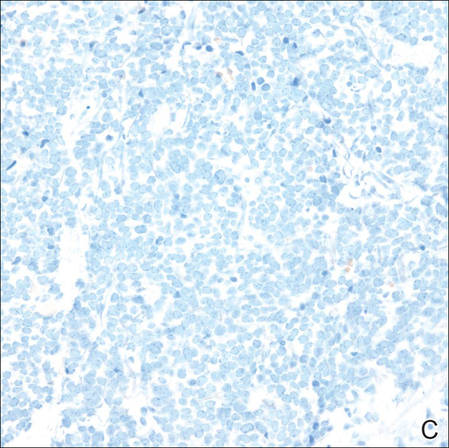 | ||
Figure 3. Positive chromogranin staining (A)(original | ||
Merkel cells are small- to medium-sized basophilic cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen on histopathology (Figure 2A).1 Some tumor cells have more vesicular chromatin, multiple small nucleoli, irregular contours, and more abundant cytoplasm. In some reports, irregular contours and abundant cytoplasm were associated with no detectible MCPyV infection.1,3 Merkel cell carcinomas have a primarily nodular architecture, and classification is based on growth pattern and cell size. Three histopathologic growth patterns have been described: nodular, infiltrative, and trabecular. The trabecular pattern is composed of interconnecting strands ofcells (Figure 2B). Tumors with solely intraepidermal involvement (MCC in situ) have been described but are exceedingly rare.1 Cell types are classified according to size, with the intermediate cell type being the most common. The small cell variant may be mistaken for a lymphocytic infiltrate due to the similar size and appearance of both types of cells.1,3
Merkel cell carcinomas can have histopathological overlap with lymphomas, small cell lung cancers, carcinoid tumors, primitive neuroectodermal tumors, neuroblastomas, small cell osteosarcomas, rhabdomyosarcomas, or Ewing sarcomas.1,3 Specifically, differentiation from small cell carcinoma of the lung is of utmost importance. Merkel cell carcinoma stains positively for cytokeratins 8, 18, 19, and 20. The neuroendocrine markers chromogranin (Figure 3A), synaptophysin, and neuron-specific enolase also may show positive staining. Cytokeratin 20, low-molecular-weight cytokeratins (CAM 5.2), and neurofilament immunostains have a high sensitivity for MCC and are the most frequently used.1 Cytokeratin 20 stains in the characteristic paranuclear dot–like pattern, which is a hallmark of MCC (Figure 3B). Cytokeratin 20 positivity in conjunction with negative staining for thyroid transcription factor 1 (Figure 3C) and cytokeratin 7 can aid in differentiation from small cell carcinoma of the lung.1,3
Pathogenesis of MCC
In 2008, Feng et al7 discovered a novel polyomavirus associated with the development of MCC. This novel polyomavirus, MCPyV, is found in approximately 80% of all cases of MCC. Seventeen members of the polyomavirus family have been identified, 9 of which have been found to infect humans, including BK virus, JC virus, WU, MCPyV, human polyomavirus 6, human polyomavirus 7, trichodysplasia spinulosa–associated polyomavirus, human polyomavirus 9, and Simian virus 40.1 Merkel cell polyomavirus infection is found in approximately 60% of the general population and exposure likely occurs early in life. The virus likely is transmitted through skin shedding and nasal secretions, though it also has been found in urine specimens.3 Currently, there is no evidence to suggest vertical viral transmission from mother to fetus.
Merkel cell polyomavirus is composed of early and late gene regions. The early gene region contains both large T antigen (LT) and small T antigen reading frames, which are necessary for viral replication.8 The late region is responsible for encoding viral proteins necessary for viral capsid assembly. Mutations found in viral protein 1 prevent formation of viral particles.9 Large T antigen is substantially overexpressed in MCC and is responsible for tumor suppression through retinoblastoma tumor suppressor protein. It also serves as a binding domain for both heat shock proteins and helicases.8,10 These domains allow the polyomaviruses to use host-cell machinery for viral genome replication while targeting tumor suppressor proteins.8 Upon viral integration into host DNA, viral replication ceases while oncogenic function persists.
The exact mechanism by which the MCPyV contributes to the development of MCC still has yet to be identified. Hypotheses suggest a combination of viral infection with external mutagens (eg, UV radiation). Experimental observations suggest viral contribution is likely due to the large percentage of MCCs that are positive for MCPyV, the identification of LT antigen expression and the role it plays in preserving cell cycle progression, and the role persistent LT antigen expression plays in continued growth of MCC cell lines in vitro.8 Two important cell line preservation mechanisms ensure continued tumor growth, including prevention of apoptosis triggered by DNA damage response mechanisms following UV damage and interaction with the retinoblastoma tumor suppressor protein allowing continued growth.8,11 Other important factors in tumor growth and survival may be the inhibition of apoptosis through the BCL2 (B-cell chronic lymphocytic leukemia/lymphoma 2) proto-oncogene and survivin (baculoviral inhibitor of apoptosis repeat-containing 5 [BIRC5]).12 Survivin has been found to play an important role in MCPyV-positive MCCs.12,13 It has been suggested that lymphangiogenesis in MCC likely is driven by vascular endothelial growth factor-C+CD68+CD163+ M2 macrophages.14 Another survival mechanism specific to polyomaviruses is their ability to interfere with the p53 tumor suppressor pathway.8 Loss of p53 expression by tumor cell nuclei has been associated with poor prognosis.15
Immune Response
Immune response as a role in tumor progression can be primarily centered on the concept of persistent antigen expression as a means of immune downregulation. Dunn et al16 suggested that cancer cells must interact through 3 consecutive phases with the host immune system (immunoediting hypothesis). In the elimination phase, the host immune system is able to recognize and destroy newly transformed cells through both the innate and adaptive immune systems. The second equilibrium phase allows the tumor to remain dormant and growth remains stagnant. Lastly, the tumor is allowed to evade the immune system through the escape phase.8
Host immune responses play an important role in both the progression and prognosis of MCC. High anti-MCPyV capsid antibody titers have been associated with better progression-free survival in some patients.8 Patients with high antibody titers (>10,000) likely have better progression-free survival than those with low antibody titers (<10,000).17 Antibody titers to the LT antigen may serve as a biomarker of MCC disease burden in the future. Rising LT antigen titers have been shown to correlate with disease progression and falling titers correlate with successful treatment.8 Tumoral infiltration of CD8+ T lymphocytes has been shown to be a predictor of survival compared to no intratumoral infiltration.6 Sihto et al18 suggested that this better prognosis from high intratumoral infiltration is not specific to MCPyV-positive MCC; however, it does highlight an important aspect of tumor evasion through the downregulation of cell surface expression of class I major histocompatibility complex antigens, which allows presentation of tumor intracellular peptides to CD8+ T lymphocytes.8 Upregulation of this specific immune response may play a role in the future treatment of MCC.
Staging and Prognosis
Due to the extremely aggressive nature of MCC, patients with local disease and tumors 2 cm or smaller in diameter have a 66% survival at 5 years.1,3 The 5-year survival rate for patients with local metastasis to regional lymph nodes ranges from 26% to 42%. Patients with distant metastasis have an 18% survival rate at 5 years.1,3 Data suggest that sentinel lymph node biopsy should be performed on all patients with MCC regardless of tumor size.1 There are no consensus guidelines to date regarding imaging for the staging of MCC patients. It is suggested that (18F)fluorodeoxyglucose positron emission tomography alone or in combination with computed tomography (CT) may be of value as a single whole-body diagnostic tool for accurate staging.10 It also has been suggested that (18F)fluorodeoxyglucose positron emission tomography and CT may offer more accurate staging than other screening modalities such as CT alone or magnetic resonance imaging.14,19
Treatment of MCC
Surgery remains the mainstay of treatment of MCC. Current National Comprehensive Cancer Network guidelines20 recommend 1- to 2-cm margins for wide local excision or treatment with Mohs micrographic surgery. Sentinel lymph node biopsy should be performed intraoperatively in patients undergoing wide local excision and preoperatively for patients undergoing Mohs micrographic surgery due to potential alterations in lymphatic drainage that may affect lymphoscintigraphy.1
Radiation may be used as primary or adjuvant therapy in patients with MCC. Radiation as primary therapy generally is reserved for patients who are not surgical candidates. It has been suggested that there was no difference in outcome in a small group of patients treated with radiation alone compared to patients who underwent surgery and radiation to the tumor bed.1 Current guidelines suggest a small group of patients may not require adjuvant therapy following adequate resection of some small tumors, and clinical observation may be appropriate.1,3 Chemotherapy may play a palliative role in patients with metastatic MCC. Merkel cell carcinoma has been shown to be chemosensitive but with a high recurrence rate.1 Because the immune system plays an important role in disease prognosis, having an intact immune system likely is paramount in the prevention of further disease progression.
Future Treatments of MCC
Future treatment of MCC may be focused on the viral etiology of most tumors and upregulation of the immune response, which may lead to the possibility of specifically interfering with virus-specific oncoproteins and stimulation of immune responses to virally infected tumor cells.8 The MCPyV large T antigen has been found to be overexpressed in some tumors and may serve as a specific target of therapy.10,21 Survivin, a key cell cycle protein encoded by LT antigen, may be an interesting target given its implication in other cancers.13 Other potential nonviral molecular target antigens include the oncoprotein H1P1 that interacts with c-KIT.8 Specific immunostimulatory cytokines that may be used to upregulate the immune response to tumoral cells may include IL-2, IL-12, IL-15, or IL-21. Therapeutic agents that may be studied in the future to target the immune exhaustion phenomenon associated with tumorigenesis include ipilimumab (cytotoxic T lymphocyte antigen 4 receptor-blocking agent) as well as programmed cell death 1 and programmed cell death 1 ligand 1 (PD-1/PD-L1).8 Neuroendocrine tumors including MCC tend to be highly vascular and express vascular endothelial growth factors and platelet-derived growth factors, which may be other potential therapeutic targets. It has been reported that approximately 95% of MCC patients have CD56+ tumors, and current clinical trials suggest a promising therapeutic response with the immunogen anti-CD56 monoclonal antibody.3
Conclusion
Merkel cell carcinoma is a rare aggressive neuroendocrine tumor that has been associated with a novel polyomavirus. Merkel cell carcinoma tends to affect elderly and immunocompromised patients as well as white individuals. Tumors are most often found in areas of high UV exposure and clinically on sun-exposed skin. Merkel cell polyomavirus is associated with approximately 80% of tumors, and tumorigenesis likely is caused by a number of sequential steps from viral integration into host DNA, mutagenic events, and specific immune responses. Currently there are no consensus guidelines for using imaging for staging of MCCs, but sentinel lymph node biopsy is recommended for all cases due to the aggressive nature of even smaller tumors. Surgery remains the mainstay of treatment, and radiation therapy may be used as a primary or adjuvant treatment. Chemotherapy usually is reserved for patients with metastatic disease purely for palliation. Future treatments of MCC likely will center on the viral etiology of MCC and upregulation of immune responses to virally infected tumor cells.
1. Han S, North J, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
2. Goessling W, McKee P, Mayer R. Merkel cell carcinoma. J Clin Oncol. 2002;20:588-598.
3. Donepudi S, DeConti R, Samlowski W. Recent advances in the understanding of the genetics, etiology, and treatment of merkel cell carcinoma. Semin Oncol. 2012;39:163-172.
4. Toker C. Trabecular carcinoma of the skin. Arch Dermatol. 1972;105:107-110.
5. De Wolff-Peeters C, Marien K, Mebis J, et al. A cutaneous APUDoma or Merkel cell tumor? a morphologically recognizable tumor with a biological and histological malignant aspect in contrast with its clinical behavior. Cancer. 1980;46:810-816.
6. Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375-381.
7. Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
8. Bhatia S, Afanasiev O, Nghiem P. Immunobiology of Merkel cell carcinoma: implications for immunotherapy of a polyomavirus-associated cancer. Curr Oncol Rep. 2011;13:488-497.
9. Amber K, McLeod M, Nouri K. The Merkel cell polyomavirus and its involvement in Merkel cell carcinoma. Dermatol Surg. 2013;39:232-238.
10. Erovic B, Al Habeeb A, Harris L, et al. Significant overexpression of the Merkel cell polyomavirus (MCPyV) large T antigen in Merkel cell carcinoma. Head Neck. 2013;35:184-189.
11. Demetriou S, Ona-Vu K, Sullivan E, et al. Defective DNA repair and cell cycle arrest in cells expressing Merkel cell polyomavirus T antigen. Int J Cancer. 2012;131:1818-1827.
12. Sahi H, Koljonen V, Kavola H, et al. Bcl-2 expression indicates better prognosis of Merkel cell carcinoma regardless of the presence of Merkel cell polyomavirus. Virchows Arch. 2012;461:553-559.
13. Arora R, Shuda M, Guastafierro A, et al. Survivin is a therapeutic target in Merkel cell carcinoma. Sci Transl Med. 2012;4:1-11.
14. Hawryluk E, O’Regan K, Sheehy N, et al. Positron emission tomography/computed tomography imaging in Merkel cell carcinoma: a study of 270 scans in 97 patients at the Dana-Farber/Brigham and Women’s Cancer Center. J Am Acad Dermatol. 2013;68:592-599.
15. Hall B, Pincus L, Yu S, et al. Immunohistochemical prognostication of Merkel cell carcinoma: p63 expression but not polyomavirus status correlates with outcome. J Cutan Pathol. 2012;39:911-917.
16. Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991-998.
17. Touze A, Le Bidre E, Laude H, et al. High levels of antibodies against Merkel cell polyomavirus identify a subset of patients with Merkel cell carcinoma with better clinical outcome. J Clin Oncol. 2011;29:1612-1619.
18. Sihto H, Bohling T, Kavola H, et al. Tumor infiltrating immune cells and outcome of Merkel cell carcinoma: a population-based study. Clin Cancer Res. 2012;18:2872-2881.
19. Colgan M, Tarantola T, Weaver A, et al. The predictive value of imaging studies in evaluating regional lymph node involvement in Merkel cell carcinoma. J Am Acad Dermatol. 2012;67:1250-1256.
20. NCCN Clinical Practice Guidelines in Oncology. National Comprehensive Cancer Network website. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#site. Accessed March 22, 2016.
21. Angermeyer S, Hesbacher S, Becker J, et al. Merkel cell polyomavirus-positive Merkel cell carcinoma cells do not require expression of the viral small T antigen. J Invest Dermatol. 2013;133:1-6.
Merkel cells originally were described by German histopathologist Friedrich Sigmund Merkel in 1875. These unique tactile cells were described as epidermal, nondendritic, and nonkeratinizing. Merkel cells are thought to arise from the neural crest and are believed to be primary neural cells found within the basal layer of the epidermis.1,2 They likely function primarily as slowly adapting type I mechanoreceptors. Origin from the neural crest is controversial, as other investigators have suggested derivation from epidermal keratinocytes.1,2 Tumor cells have been linked to the amine precursor uptake and decarboxylation system.3 In 1972, Toker4 described several cases of trabecular or sweat gland carcinomas of the skin. Upon further investigation, the cells that comprised these tumors were found to have dense core granules on electron microscopy, typical of Merkel cells.1,2 Other terms such as neuroendocrine carcinoma of the skin, small cell carcinoma of the skin, and anaplastic carcinoma of the skin have been used to describe Merkel cell carcinoma (MCC),1 which was suggested by De Wolf-Peeters et al5 in 1980.
Despite being a rare malignancy, MCC follows an aggressive clinical course. Upon presentation, approximately 66% of patients have local disease, 27% have nodal involvement, and 7% have distant metastasis.1 Future treatments will likely center around the novel Merkel cell polyomavirus (MCPyV) and modification of immune responses toward tumor cells. Standardization continues to be lacking in both staging and treatment of this aggressive tumor.
Epidemiology of MCC
| ||
Figure 1. A 2.3×1.5×1.2-cm, hemorrhagic, crusted, | ||
| ||
| ||
| Figure 2. Merkel cells are small- to medium-sized cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen (A)(H&E, original magnification ×40). Merkel cell carcinoma with trabecular pattern (B) (H&E, original magnification ×10). | ||
Between 1986 and 2006, the incidence of MCC grew substantially.1,2 Figures have been reported at 0.15 cases per 100,000 individuals to 0.6 cases per 100,000 individuals worldwide. In the United States, the age-adjusted incidence of MCC is estimated at 0.24 per 100,000 person-years, which is higher than the estimated 0.13 per 100,000 person-years found in Europe.3 The highest incidence worldwide has been noted in Western Australia, likely due to high levels of UV exposure.1 The incidence of MCC in psoriasis patients who are treated with oral methoxsalen (psoralen) and UVA photochemotherapy is 100 times greater than in the general population, further supporting the role of UV light in the development of MCC.1 White individuals have the highest incidence of MCC worldwide, with men being affected more frequently than women.1,3 The majority of patients with MCC are diagnosed at 70 years or older.1 Approximately 5% of reported MCC patients are diagnosed before 50 years of age.2 Immunosuppression and immunodeficiency likely play a role in the pathogenesis of MCC, and the incidence is increased in solid organ transplant recipients, most commonly renal transplant recipients,1 as well as individuals with chronic lymphocytic leukemia, human immunodeficiency virus infection, and AIDS.1,3 Patients with autoimmune diseases such as rheumatoid arthritis also are at increased risk for MCC.3 Individuals who are diagnosed with MCC are at an increased risk for development of other malignancies including nonmelanoma skin cancers, chronic lymphocytic leukemia, Hodgkin lymphoma, and non-Hodgkin lymphoma.3
Clinical Presentation of MCC
The clinical presentation of MCC can be variable. Most tumors present as firm, red to purple, nontender papules or nodules (Figure 1).1 Tumor size may range from 2 to 200 mm but is most commonly less than 20 mm.2 Growth can be rapid, and tumors are most commonly located on sun-exposed skin. The head and neck areas account for 48% of all MCC cases,1 with the eyelids being frequently involved.2 Merkel cell carcinoma also has been reported on the arms, legs, trunk, back, and buttocks.1 Non–sun-exposed areas are less commonly affected. Mucosal sites (eg, larynx, nasal cavity, pharynx, mouth) account for 5% of primary MCCs.1 Merkel cell carcinoma also has been reported to affect the vulva and penis. Subcutaneous primary MCC has presented without overlying epidermal changes.1 In a case series by Heath et al,6 14% (27/195) of MCC patients presented with nodal disease without any identifiable primary tumor, with the inguinal nodal chain being the most common for this presentation. It currently is not known whether these nodal tumors are primary tumors or metastatic disease with a regression of the primary tumor.1
|
Histopathology of MCC
| ||
| ||
| ||
Figure 3. Positive chromogranin staining (A)(original | ||
Merkel cells are small- to medium-sized basophilic cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen on histopathology (Figure 2A).1 Some tumor cells have more vesicular chromatin, multiple small nucleoli, irregular contours, and more abundant cytoplasm. In some reports, irregular contours and abundant cytoplasm were associated with no detectible MCPyV infection.1,3 Merkel cell carcinomas have a primarily nodular architecture, and classification is based on growth pattern and cell size. Three histopathologic growth patterns have been described: nodular, infiltrative, and trabecular. The trabecular pattern is composed of interconnecting strands ofcells (Figure 2B). Tumors with solely intraepidermal involvement (MCC in situ) have been described but are exceedingly rare.1 Cell types are classified according to size, with the intermediate cell type being the most common. The small cell variant may be mistaken for a lymphocytic infiltrate due to the similar size and appearance of both types of cells.1,3
Merkel cell carcinomas can have histopathological overlap with lymphomas, small cell lung cancers, carcinoid tumors, primitive neuroectodermal tumors, neuroblastomas, small cell osteosarcomas, rhabdomyosarcomas, or Ewing sarcomas.1,3 Specifically, differentiation from small cell carcinoma of the lung is of utmost importance. Merkel cell carcinoma stains positively for cytokeratins 8, 18, 19, and 20. The neuroendocrine markers chromogranin (Figure 3A), synaptophysin, and neuron-specific enolase also may show positive staining. Cytokeratin 20, low-molecular-weight cytokeratins (CAM 5.2), and neurofilament immunostains have a high sensitivity for MCC and are the most frequently used.1 Cytokeratin 20 stains in the characteristic paranuclear dot–like pattern, which is a hallmark of MCC (Figure 3B). Cytokeratin 20 positivity in conjunction with negative staining for thyroid transcription factor 1 (Figure 3C) and cytokeratin 7 can aid in differentiation from small cell carcinoma of the lung.1,3
Pathogenesis of MCC
In 2008, Feng et al7 discovered a novel polyomavirus associated with the development of MCC. This novel polyomavirus, MCPyV, is found in approximately 80% of all cases of MCC. Seventeen members of the polyomavirus family have been identified, 9 of which have been found to infect humans, including BK virus, JC virus, WU, MCPyV, human polyomavirus 6, human polyomavirus 7, trichodysplasia spinulosa–associated polyomavirus, human polyomavirus 9, and Simian virus 40.1 Merkel cell polyomavirus infection is found in approximately 60% of the general population and exposure likely occurs early in life. The virus likely is transmitted through skin shedding and nasal secretions, though it also has been found in urine specimens.3 Currently, there is no evidence to suggest vertical viral transmission from mother to fetus.
Merkel cell polyomavirus is composed of early and late gene regions. The early gene region contains both large T antigen (LT) and small T antigen reading frames, which are necessary for viral replication.8 The late region is responsible for encoding viral proteins necessary for viral capsid assembly. Mutations found in viral protein 1 prevent formation of viral particles.9 Large T antigen is substantially overexpressed in MCC and is responsible for tumor suppression through retinoblastoma tumor suppressor protein. It also serves as a binding domain for both heat shock proteins and helicases.8,10 These domains allow the polyomaviruses to use host-cell machinery for viral genome replication while targeting tumor suppressor proteins.8 Upon viral integration into host DNA, viral replication ceases while oncogenic function persists.
The exact mechanism by which the MCPyV contributes to the development of MCC still has yet to be identified. Hypotheses suggest a combination of viral infection with external mutagens (eg, UV radiation). Experimental observations suggest viral contribution is likely due to the large percentage of MCCs that are positive for MCPyV, the identification of LT antigen expression and the role it plays in preserving cell cycle progression, and the role persistent LT antigen expression plays in continued growth of MCC cell lines in vitro.8 Two important cell line preservation mechanisms ensure continued tumor growth, including prevention of apoptosis triggered by DNA damage response mechanisms following UV damage and interaction with the retinoblastoma tumor suppressor protein allowing continued growth.8,11 Other important factors in tumor growth and survival may be the inhibition of apoptosis through the BCL2 (B-cell chronic lymphocytic leukemia/lymphoma 2) proto-oncogene and survivin (baculoviral inhibitor of apoptosis repeat-containing 5 [BIRC5]).12 Survivin has been found to play an important role in MCPyV-positive MCCs.12,13 It has been suggested that lymphangiogenesis in MCC likely is driven by vascular endothelial growth factor-C+CD68+CD163+ M2 macrophages.14 Another survival mechanism specific to polyomaviruses is their ability to interfere with the p53 tumor suppressor pathway.8 Loss of p53 expression by tumor cell nuclei has been associated with poor prognosis.15
Immune Response
Immune response as a role in tumor progression can be primarily centered on the concept of persistent antigen expression as a means of immune downregulation. Dunn et al16 suggested that cancer cells must interact through 3 consecutive phases with the host immune system (immunoediting hypothesis). In the elimination phase, the host immune system is able to recognize and destroy newly transformed cells through both the innate and adaptive immune systems. The second equilibrium phase allows the tumor to remain dormant and growth remains stagnant. Lastly, the tumor is allowed to evade the immune system through the escape phase.8
Host immune responses play an important role in both the progression and prognosis of MCC. High anti-MCPyV capsid antibody titers have been associated with better progression-free survival in some patients.8 Patients with high antibody titers (>10,000) likely have better progression-free survival than those with low antibody titers (<10,000).17 Antibody titers to the LT antigen may serve as a biomarker of MCC disease burden in the future. Rising LT antigen titers have been shown to correlate with disease progression and falling titers correlate with successful treatment.8 Tumoral infiltration of CD8+ T lymphocytes has been shown to be a predictor of survival compared to no intratumoral infiltration.6 Sihto et al18 suggested that this better prognosis from high intratumoral infiltration is not specific to MCPyV-positive MCC; however, it does highlight an important aspect of tumor evasion through the downregulation of cell surface expression of class I major histocompatibility complex antigens, which allows presentation of tumor intracellular peptides to CD8+ T lymphocytes.8 Upregulation of this specific immune response may play a role in the future treatment of MCC.
Staging and Prognosis
Due to the extremely aggressive nature of MCC, patients with local disease and tumors 2 cm or smaller in diameter have a 66% survival at 5 years.1,3 The 5-year survival rate for patients with local metastasis to regional lymph nodes ranges from 26% to 42%. Patients with distant metastasis have an 18% survival rate at 5 years.1,3 Data suggest that sentinel lymph node biopsy should be performed on all patients with MCC regardless of tumor size.1 There are no consensus guidelines to date regarding imaging for the staging of MCC patients. It is suggested that (18F)fluorodeoxyglucose positron emission tomography alone or in combination with computed tomography (CT) may be of value as a single whole-body diagnostic tool for accurate staging.10 It also has been suggested that (18F)fluorodeoxyglucose positron emission tomography and CT may offer more accurate staging than other screening modalities such as CT alone or magnetic resonance imaging.14,19
Treatment of MCC
Surgery remains the mainstay of treatment of MCC. Current National Comprehensive Cancer Network guidelines20 recommend 1- to 2-cm margins for wide local excision or treatment with Mohs micrographic surgery. Sentinel lymph node biopsy should be performed intraoperatively in patients undergoing wide local excision and preoperatively for patients undergoing Mohs micrographic surgery due to potential alterations in lymphatic drainage that may affect lymphoscintigraphy.1
Radiation may be used as primary or adjuvant therapy in patients with MCC. Radiation as primary therapy generally is reserved for patients who are not surgical candidates. It has been suggested that there was no difference in outcome in a small group of patients treated with radiation alone compared to patients who underwent surgery and radiation to the tumor bed.1 Current guidelines suggest a small group of patients may not require adjuvant therapy following adequate resection of some small tumors, and clinical observation may be appropriate.1,3 Chemotherapy may play a palliative role in patients with metastatic MCC. Merkel cell carcinoma has been shown to be chemosensitive but with a high recurrence rate.1 Because the immune system plays an important role in disease prognosis, having an intact immune system likely is paramount in the prevention of further disease progression.
Future Treatments of MCC
Future treatment of MCC may be focused on the viral etiology of most tumors and upregulation of the immune response, which may lead to the possibility of specifically interfering with virus-specific oncoproteins and stimulation of immune responses to virally infected tumor cells.8 The MCPyV large T antigen has been found to be overexpressed in some tumors and may serve as a specific target of therapy.10,21 Survivin, a key cell cycle protein encoded by LT antigen, may be an interesting target given its implication in other cancers.13 Other potential nonviral molecular target antigens include the oncoprotein H1P1 that interacts with c-KIT.8 Specific immunostimulatory cytokines that may be used to upregulate the immune response to tumoral cells may include IL-2, IL-12, IL-15, or IL-21. Therapeutic agents that may be studied in the future to target the immune exhaustion phenomenon associated with tumorigenesis include ipilimumab (cytotoxic T lymphocyte antigen 4 receptor-blocking agent) as well as programmed cell death 1 and programmed cell death 1 ligand 1 (PD-1/PD-L1).8 Neuroendocrine tumors including MCC tend to be highly vascular and express vascular endothelial growth factors and platelet-derived growth factors, which may be other potential therapeutic targets. It has been reported that approximately 95% of MCC patients have CD56+ tumors, and current clinical trials suggest a promising therapeutic response with the immunogen anti-CD56 monoclonal antibody.3
Conclusion
Merkel cell carcinoma is a rare aggressive neuroendocrine tumor that has been associated with a novel polyomavirus. Merkel cell carcinoma tends to affect elderly and immunocompromised patients as well as white individuals. Tumors are most often found in areas of high UV exposure and clinically on sun-exposed skin. Merkel cell polyomavirus is associated with approximately 80% of tumors, and tumorigenesis likely is caused by a number of sequential steps from viral integration into host DNA, mutagenic events, and specific immune responses. Currently there are no consensus guidelines for using imaging for staging of MCCs, but sentinel lymph node biopsy is recommended for all cases due to the aggressive nature of even smaller tumors. Surgery remains the mainstay of treatment, and radiation therapy may be used as a primary or adjuvant treatment. Chemotherapy usually is reserved for patients with metastatic disease purely for palliation. Future treatments of MCC likely will center on the viral etiology of MCC and upregulation of immune responses to virally infected tumor cells.
Merkel cells originally were described by German histopathologist Friedrich Sigmund Merkel in 1875. These unique tactile cells were described as epidermal, nondendritic, and nonkeratinizing. Merkel cells are thought to arise from the neural crest and are believed to be primary neural cells found within the basal layer of the epidermis.1,2 They likely function primarily as slowly adapting type I mechanoreceptors. Origin from the neural crest is controversial, as other investigators have suggested derivation from epidermal keratinocytes.1,2 Tumor cells have been linked to the amine precursor uptake and decarboxylation system.3 In 1972, Toker4 described several cases of trabecular or sweat gland carcinomas of the skin. Upon further investigation, the cells that comprised these tumors were found to have dense core granules on electron microscopy, typical of Merkel cells.1,2 Other terms such as neuroendocrine carcinoma of the skin, small cell carcinoma of the skin, and anaplastic carcinoma of the skin have been used to describe Merkel cell carcinoma (MCC),1 which was suggested by De Wolf-Peeters et al5 in 1980.
Despite being a rare malignancy, MCC follows an aggressive clinical course. Upon presentation, approximately 66% of patients have local disease, 27% have nodal involvement, and 7% have distant metastasis.1 Future treatments will likely center around the novel Merkel cell polyomavirus (MCPyV) and modification of immune responses toward tumor cells. Standardization continues to be lacking in both staging and treatment of this aggressive tumor.
Epidemiology of MCC
| ||
Figure 1. A 2.3×1.5×1.2-cm, hemorrhagic, crusted, | ||
| ||
| ||
| Figure 2. Merkel cells are small- to medium-sized cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen (A)(H&E, original magnification ×40). Merkel cell carcinoma with trabecular pattern (B) (H&E, original magnification ×10). | ||
Between 1986 and 2006, the incidence of MCC grew substantially.1,2 Figures have been reported at 0.15 cases per 100,000 individuals to 0.6 cases per 100,000 individuals worldwide. In the United States, the age-adjusted incidence of MCC is estimated at 0.24 per 100,000 person-years, which is higher than the estimated 0.13 per 100,000 person-years found in Europe.3 The highest incidence worldwide has been noted in Western Australia, likely due to high levels of UV exposure.1 The incidence of MCC in psoriasis patients who are treated with oral methoxsalen (psoralen) and UVA photochemotherapy is 100 times greater than in the general population, further supporting the role of UV light in the development of MCC.1 White individuals have the highest incidence of MCC worldwide, with men being affected more frequently than women.1,3 The majority of patients with MCC are diagnosed at 70 years or older.1 Approximately 5% of reported MCC patients are diagnosed before 50 years of age.2 Immunosuppression and immunodeficiency likely play a role in the pathogenesis of MCC, and the incidence is increased in solid organ transplant recipients, most commonly renal transplant recipients,1 as well as individuals with chronic lymphocytic leukemia, human immunodeficiency virus infection, and AIDS.1,3 Patients with autoimmune diseases such as rheumatoid arthritis also are at increased risk for MCC.3 Individuals who are diagnosed with MCC are at an increased risk for development of other malignancies including nonmelanoma skin cancers, chronic lymphocytic leukemia, Hodgkin lymphoma, and non-Hodgkin lymphoma.3
Clinical Presentation of MCC
The clinical presentation of MCC can be variable. Most tumors present as firm, red to purple, nontender papules or nodules (Figure 1).1 Tumor size may range from 2 to 200 mm but is most commonly less than 20 mm.2 Growth can be rapid, and tumors are most commonly located on sun-exposed skin. The head and neck areas account for 48% of all MCC cases,1 with the eyelids being frequently involved.2 Merkel cell carcinoma also has been reported on the arms, legs, trunk, back, and buttocks.1 Non–sun-exposed areas are less commonly affected. Mucosal sites (eg, larynx, nasal cavity, pharynx, mouth) account for 5% of primary MCCs.1 Merkel cell carcinoma also has been reported to affect the vulva and penis. Subcutaneous primary MCC has presented without overlying epidermal changes.1 In a case series by Heath et al,6 14% (27/195) of MCC patients presented with nodal disease without any identifiable primary tumor, with the inguinal nodal chain being the most common for this presentation. It currently is not known whether these nodal tumors are primary tumors or metastatic disease with a regression of the primary tumor.1
|
Histopathology of MCC
| ||
| ||
| ||
Figure 3. Positive chromogranin staining (A)(original | ||
Merkel cells are small- to medium-sized basophilic cells with round nuclei and scant cytoplasm. Granular or stippled chromatin can be seen on histopathology (Figure 2A).1 Some tumor cells have more vesicular chromatin, multiple small nucleoli, irregular contours, and more abundant cytoplasm. In some reports, irregular contours and abundant cytoplasm were associated with no detectible MCPyV infection.1,3 Merkel cell carcinomas have a primarily nodular architecture, and classification is based on growth pattern and cell size. Three histopathologic growth patterns have been described: nodular, infiltrative, and trabecular. The trabecular pattern is composed of interconnecting strands ofcells (Figure 2B). Tumors with solely intraepidermal involvement (MCC in situ) have been described but are exceedingly rare.1 Cell types are classified according to size, with the intermediate cell type being the most common. The small cell variant may be mistaken for a lymphocytic infiltrate due to the similar size and appearance of both types of cells.1,3
Merkel cell carcinomas can have histopathological overlap with lymphomas, small cell lung cancers, carcinoid tumors, primitive neuroectodermal tumors, neuroblastomas, small cell osteosarcomas, rhabdomyosarcomas, or Ewing sarcomas.1,3 Specifically, differentiation from small cell carcinoma of the lung is of utmost importance. Merkel cell carcinoma stains positively for cytokeratins 8, 18, 19, and 20. The neuroendocrine markers chromogranin (Figure 3A), synaptophysin, and neuron-specific enolase also may show positive staining. Cytokeratin 20, low-molecular-weight cytokeratins (CAM 5.2), and neurofilament immunostains have a high sensitivity for MCC and are the most frequently used.1 Cytokeratin 20 stains in the characteristic paranuclear dot–like pattern, which is a hallmark of MCC (Figure 3B). Cytokeratin 20 positivity in conjunction with negative staining for thyroid transcription factor 1 (Figure 3C) and cytokeratin 7 can aid in differentiation from small cell carcinoma of the lung.1,3
Pathogenesis of MCC
In 2008, Feng et al7 discovered a novel polyomavirus associated with the development of MCC. This novel polyomavirus, MCPyV, is found in approximately 80% of all cases of MCC. Seventeen members of the polyomavirus family have been identified, 9 of which have been found to infect humans, including BK virus, JC virus, WU, MCPyV, human polyomavirus 6, human polyomavirus 7, trichodysplasia spinulosa–associated polyomavirus, human polyomavirus 9, and Simian virus 40.1 Merkel cell polyomavirus infection is found in approximately 60% of the general population and exposure likely occurs early in life. The virus likely is transmitted through skin shedding and nasal secretions, though it also has been found in urine specimens.3 Currently, there is no evidence to suggest vertical viral transmission from mother to fetus.
Merkel cell polyomavirus is composed of early and late gene regions. The early gene region contains both large T antigen (LT) and small T antigen reading frames, which are necessary for viral replication.8 The late region is responsible for encoding viral proteins necessary for viral capsid assembly. Mutations found in viral protein 1 prevent formation of viral particles.9 Large T antigen is substantially overexpressed in MCC and is responsible for tumor suppression through retinoblastoma tumor suppressor protein. It also serves as a binding domain for both heat shock proteins and helicases.8,10 These domains allow the polyomaviruses to use host-cell machinery for viral genome replication while targeting tumor suppressor proteins.8 Upon viral integration into host DNA, viral replication ceases while oncogenic function persists.
The exact mechanism by which the MCPyV contributes to the development of MCC still has yet to be identified. Hypotheses suggest a combination of viral infection with external mutagens (eg, UV radiation). Experimental observations suggest viral contribution is likely due to the large percentage of MCCs that are positive for MCPyV, the identification of LT antigen expression and the role it plays in preserving cell cycle progression, and the role persistent LT antigen expression plays in continued growth of MCC cell lines in vitro.8 Two important cell line preservation mechanisms ensure continued tumor growth, including prevention of apoptosis triggered by DNA damage response mechanisms following UV damage and interaction with the retinoblastoma tumor suppressor protein allowing continued growth.8,11 Other important factors in tumor growth and survival may be the inhibition of apoptosis through the BCL2 (B-cell chronic lymphocytic leukemia/lymphoma 2) proto-oncogene and survivin (baculoviral inhibitor of apoptosis repeat-containing 5 [BIRC5]).12 Survivin has been found to play an important role in MCPyV-positive MCCs.12,13 It has been suggested that lymphangiogenesis in MCC likely is driven by vascular endothelial growth factor-C+CD68+CD163+ M2 macrophages.14 Another survival mechanism specific to polyomaviruses is their ability to interfere with the p53 tumor suppressor pathway.8 Loss of p53 expression by tumor cell nuclei has been associated with poor prognosis.15
Immune Response
Immune response as a role in tumor progression can be primarily centered on the concept of persistent antigen expression as a means of immune downregulation. Dunn et al16 suggested that cancer cells must interact through 3 consecutive phases with the host immune system (immunoediting hypothesis). In the elimination phase, the host immune system is able to recognize and destroy newly transformed cells through both the innate and adaptive immune systems. The second equilibrium phase allows the tumor to remain dormant and growth remains stagnant. Lastly, the tumor is allowed to evade the immune system through the escape phase.8
Host immune responses play an important role in both the progression and prognosis of MCC. High anti-MCPyV capsid antibody titers have been associated with better progression-free survival in some patients.8 Patients with high antibody titers (>10,000) likely have better progression-free survival than those with low antibody titers (<10,000).17 Antibody titers to the LT antigen may serve as a biomarker of MCC disease burden in the future. Rising LT antigen titers have been shown to correlate with disease progression and falling titers correlate with successful treatment.8 Tumoral infiltration of CD8+ T lymphocytes has been shown to be a predictor of survival compared to no intratumoral infiltration.6 Sihto et al18 suggested that this better prognosis from high intratumoral infiltration is not specific to MCPyV-positive MCC; however, it does highlight an important aspect of tumor evasion through the downregulation of cell surface expression of class I major histocompatibility complex antigens, which allows presentation of tumor intracellular peptides to CD8+ T lymphocytes.8 Upregulation of this specific immune response may play a role in the future treatment of MCC.
Staging and Prognosis
Due to the extremely aggressive nature of MCC, patients with local disease and tumors 2 cm or smaller in diameter have a 66% survival at 5 years.1,3 The 5-year survival rate for patients with local metastasis to regional lymph nodes ranges from 26% to 42%. Patients with distant metastasis have an 18% survival rate at 5 years.1,3 Data suggest that sentinel lymph node biopsy should be performed on all patients with MCC regardless of tumor size.1 There are no consensus guidelines to date regarding imaging for the staging of MCC patients. It is suggested that (18F)fluorodeoxyglucose positron emission tomography alone or in combination with computed tomography (CT) may be of value as a single whole-body diagnostic tool for accurate staging.10 It also has been suggested that (18F)fluorodeoxyglucose positron emission tomography and CT may offer more accurate staging than other screening modalities such as CT alone or magnetic resonance imaging.14,19
Treatment of MCC
Surgery remains the mainstay of treatment of MCC. Current National Comprehensive Cancer Network guidelines20 recommend 1- to 2-cm margins for wide local excision or treatment with Mohs micrographic surgery. Sentinel lymph node biopsy should be performed intraoperatively in patients undergoing wide local excision and preoperatively for patients undergoing Mohs micrographic surgery due to potential alterations in lymphatic drainage that may affect lymphoscintigraphy.1
Radiation may be used as primary or adjuvant therapy in patients with MCC. Radiation as primary therapy generally is reserved for patients who are not surgical candidates. It has been suggested that there was no difference in outcome in a small group of patients treated with radiation alone compared to patients who underwent surgery and radiation to the tumor bed.1 Current guidelines suggest a small group of patients may not require adjuvant therapy following adequate resection of some small tumors, and clinical observation may be appropriate.1,3 Chemotherapy may play a palliative role in patients with metastatic MCC. Merkel cell carcinoma has been shown to be chemosensitive but with a high recurrence rate.1 Because the immune system plays an important role in disease prognosis, having an intact immune system likely is paramount in the prevention of further disease progression.
Future Treatments of MCC
Future treatment of MCC may be focused on the viral etiology of most tumors and upregulation of the immune response, which may lead to the possibility of specifically interfering with virus-specific oncoproteins and stimulation of immune responses to virally infected tumor cells.8 The MCPyV large T antigen has been found to be overexpressed in some tumors and may serve as a specific target of therapy.10,21 Survivin, a key cell cycle protein encoded by LT antigen, may be an interesting target given its implication in other cancers.13 Other potential nonviral molecular target antigens include the oncoprotein H1P1 that interacts with c-KIT.8 Specific immunostimulatory cytokines that may be used to upregulate the immune response to tumoral cells may include IL-2, IL-12, IL-15, or IL-21. Therapeutic agents that may be studied in the future to target the immune exhaustion phenomenon associated with tumorigenesis include ipilimumab (cytotoxic T lymphocyte antigen 4 receptor-blocking agent) as well as programmed cell death 1 and programmed cell death 1 ligand 1 (PD-1/PD-L1).8 Neuroendocrine tumors including MCC tend to be highly vascular and express vascular endothelial growth factors and platelet-derived growth factors, which may be other potential therapeutic targets. It has been reported that approximately 95% of MCC patients have CD56+ tumors, and current clinical trials suggest a promising therapeutic response with the immunogen anti-CD56 monoclonal antibody.3
Conclusion
Merkel cell carcinoma is a rare aggressive neuroendocrine tumor that has been associated with a novel polyomavirus. Merkel cell carcinoma tends to affect elderly and immunocompromised patients as well as white individuals. Tumors are most often found in areas of high UV exposure and clinically on sun-exposed skin. Merkel cell polyomavirus is associated with approximately 80% of tumors, and tumorigenesis likely is caused by a number of sequential steps from viral integration into host DNA, mutagenic events, and specific immune responses. Currently there are no consensus guidelines for using imaging for staging of MCCs, but sentinel lymph node biopsy is recommended for all cases due to the aggressive nature of even smaller tumors. Surgery remains the mainstay of treatment, and radiation therapy may be used as a primary or adjuvant treatment. Chemotherapy usually is reserved for patients with metastatic disease purely for palliation. Future treatments of MCC likely will center on the viral etiology of MCC and upregulation of immune responses to virally infected tumor cells.
1. Han S, North J, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
2. Goessling W, McKee P, Mayer R. Merkel cell carcinoma. J Clin Oncol. 2002;20:588-598.
3. Donepudi S, DeConti R, Samlowski W. Recent advances in the understanding of the genetics, etiology, and treatment of merkel cell carcinoma. Semin Oncol. 2012;39:163-172.
4. Toker C. Trabecular carcinoma of the skin. Arch Dermatol. 1972;105:107-110.
5. De Wolff-Peeters C, Marien K, Mebis J, et al. A cutaneous APUDoma or Merkel cell tumor? a morphologically recognizable tumor with a biological and histological malignant aspect in contrast with its clinical behavior. Cancer. 1980;46:810-816.
6. Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375-381.
7. Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
8. Bhatia S, Afanasiev O, Nghiem P. Immunobiology of Merkel cell carcinoma: implications for immunotherapy of a polyomavirus-associated cancer. Curr Oncol Rep. 2011;13:488-497.
9. Amber K, McLeod M, Nouri K. The Merkel cell polyomavirus and its involvement in Merkel cell carcinoma. Dermatol Surg. 2013;39:232-238.
10. Erovic B, Al Habeeb A, Harris L, et al. Significant overexpression of the Merkel cell polyomavirus (MCPyV) large T antigen in Merkel cell carcinoma. Head Neck. 2013;35:184-189.
11. Demetriou S, Ona-Vu K, Sullivan E, et al. Defective DNA repair and cell cycle arrest in cells expressing Merkel cell polyomavirus T antigen. Int J Cancer. 2012;131:1818-1827.
12. Sahi H, Koljonen V, Kavola H, et al. Bcl-2 expression indicates better prognosis of Merkel cell carcinoma regardless of the presence of Merkel cell polyomavirus. Virchows Arch. 2012;461:553-559.
13. Arora R, Shuda M, Guastafierro A, et al. Survivin is a therapeutic target in Merkel cell carcinoma. Sci Transl Med. 2012;4:1-11.
14. Hawryluk E, O’Regan K, Sheehy N, et al. Positron emission tomography/computed tomography imaging in Merkel cell carcinoma: a study of 270 scans in 97 patients at the Dana-Farber/Brigham and Women’s Cancer Center. J Am Acad Dermatol. 2013;68:592-599.
15. Hall B, Pincus L, Yu S, et al. Immunohistochemical prognostication of Merkel cell carcinoma: p63 expression but not polyomavirus status correlates with outcome. J Cutan Pathol. 2012;39:911-917.
16. Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991-998.
17. Touze A, Le Bidre E, Laude H, et al. High levels of antibodies against Merkel cell polyomavirus identify a subset of patients with Merkel cell carcinoma with better clinical outcome. J Clin Oncol. 2011;29:1612-1619.
18. Sihto H, Bohling T, Kavola H, et al. Tumor infiltrating immune cells and outcome of Merkel cell carcinoma: a population-based study. Clin Cancer Res. 2012;18:2872-2881.
19. Colgan M, Tarantola T, Weaver A, et al. The predictive value of imaging studies in evaluating regional lymph node involvement in Merkel cell carcinoma. J Am Acad Dermatol. 2012;67:1250-1256.
20. NCCN Clinical Practice Guidelines in Oncology. National Comprehensive Cancer Network website. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#site. Accessed March 22, 2016.
21. Angermeyer S, Hesbacher S, Becker J, et al. Merkel cell polyomavirus-positive Merkel cell carcinoma cells do not require expression of the viral small T antigen. J Invest Dermatol. 2013;133:1-6.
1. Han S, North J, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
2. Goessling W, McKee P, Mayer R. Merkel cell carcinoma. J Clin Oncol. 2002;20:588-598.
3. Donepudi S, DeConti R, Samlowski W. Recent advances in the understanding of the genetics, etiology, and treatment of merkel cell carcinoma. Semin Oncol. 2012;39:163-172.
4. Toker C. Trabecular carcinoma of the skin. Arch Dermatol. 1972;105:107-110.
5. De Wolff-Peeters C, Marien K, Mebis J, et al. A cutaneous APUDoma or Merkel cell tumor? a morphologically recognizable tumor with a biological and histological malignant aspect in contrast with its clinical behavior. Cancer. 1980;46:810-816.
6. Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;58:375-381.
7. Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
8. Bhatia S, Afanasiev O, Nghiem P. Immunobiology of Merkel cell carcinoma: implications for immunotherapy of a polyomavirus-associated cancer. Curr Oncol Rep. 2011;13:488-497.
9. Amber K, McLeod M, Nouri K. The Merkel cell polyomavirus and its involvement in Merkel cell carcinoma. Dermatol Surg. 2013;39:232-238.
10. Erovic B, Al Habeeb A, Harris L, et al. Significant overexpression of the Merkel cell polyomavirus (MCPyV) large T antigen in Merkel cell carcinoma. Head Neck. 2013;35:184-189.
11. Demetriou S, Ona-Vu K, Sullivan E, et al. Defective DNA repair and cell cycle arrest in cells expressing Merkel cell polyomavirus T antigen. Int J Cancer. 2012;131:1818-1827.
12. Sahi H, Koljonen V, Kavola H, et al. Bcl-2 expression indicates better prognosis of Merkel cell carcinoma regardless of the presence of Merkel cell polyomavirus. Virchows Arch. 2012;461:553-559.
13. Arora R, Shuda M, Guastafierro A, et al. Survivin is a therapeutic target in Merkel cell carcinoma. Sci Transl Med. 2012;4:1-11.
14. Hawryluk E, O’Regan K, Sheehy N, et al. Positron emission tomography/computed tomography imaging in Merkel cell carcinoma: a study of 270 scans in 97 patients at the Dana-Farber/Brigham and Women’s Cancer Center. J Am Acad Dermatol. 2013;68:592-599.
15. Hall B, Pincus L, Yu S, et al. Immunohistochemical prognostication of Merkel cell carcinoma: p63 expression but not polyomavirus status correlates with outcome. J Cutan Pathol. 2012;39:911-917.
16. Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991-998.
17. Touze A, Le Bidre E, Laude H, et al. High levels of antibodies against Merkel cell polyomavirus identify a subset of patients with Merkel cell carcinoma with better clinical outcome. J Clin Oncol. 2011;29:1612-1619.
18. Sihto H, Bohling T, Kavola H, et al. Tumor infiltrating immune cells and outcome of Merkel cell carcinoma: a population-based study. Clin Cancer Res. 2012;18:2872-2881.
19. Colgan M, Tarantola T, Weaver A, et al. The predictive value of imaging studies in evaluating regional lymph node involvement in Merkel cell carcinoma. J Am Acad Dermatol. 2012;67:1250-1256.
20. NCCN Clinical Practice Guidelines in Oncology. National Comprehensive Cancer Network website. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#site. Accessed March 22, 2016.
21. Angermeyer S, Hesbacher S, Becker J, et al. Merkel cell polyomavirus-positive Merkel cell carcinoma cells do not require expression of the viral small T antigen. J Invest Dermatol. 2013;133:1-6.
Practice Points
- Merkel cell carcinoma has been associated with a novel polyomavirus.
- Merkel cell carcinoma follows a very aggressive course and is most likely metastatic at diagnosis.