User login
Derm diagnoses you can’t afford to miss
- Management of hereditary angioedema should include fresh frozen plasma containing C1 inhibitor (C1-INH), whenever possible; if C1-INH-containing plasma is unavailable, fresh frozen plasma can be used instead (SOR: A).
- Do not give neomycin to patients with suspected cellulitis; the drug may promote antibiotic resistance in Staphylococcus aureus, a pathogen often associated with this condition (SOR: A).
- Whenever a patient presents with erythematous skin lesions and a recent history of receiving penicillin or a cephalosporin antibiotic, a sulfa derivative, or an anticonvulsant, the suspected medication should be stopped until Stevens-Johnson syndrome is ruled out (SOR: A).
Strength of recommendation (SOR)
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
Skin eruptions are a common reason for visits to primary care physicians. While most are innocuous, some are associated with—or are early warning signs of—severe allergic reactions or other emergent conditions. A 9-year-old patient I’ll call Julie is a case in point.
The first time Julie’s parents brought her to our clinic, she’d been complaining of a sore throat and had a fever that hovered between 102° and 103°F for several days. The physician who examined Julie found mild maxillary tenderness. A rapid streptococcal throat swab was negative; her doctor prescribed a 10-day course of trimethoprim/sulfamethoxazole (TMP/SMX) for presumed acute sinusitis.
Thirteen days later (3 days after the patient completed the course of antibiotics), Julie’s parents brought her back to the clinic. Her throat still hurt, and she had erythematous oval lesions on her trunk and upper extremities. Her physician diagnosed scarlet fever and wrote a prescription for penicillin.
The following day, Julie was taken to the emergency department (ED) with bilateral conjunctival hyperemia and diffuse, confluent erythematous macules throughout her body. The ED physician who examined Julie found a 2.5 × 2.0 cm targetoid lesion with a necrotic, purpuric center on her lower back—a diagnostic clue to the cause of her signs and symptoms.
If you had been Julie’s physician, would you have been alert to that clue?
For family physicians accustomed to seeing relatively mild skin disorders, recognizing and responding to dermatologic conditions with potentially dire outcomes can be challenging. This review, and the images that accompany it, will help you sharpen your dermatologic diagnostic and treatment skills, both for benign disorders and those that are less common and more severe. We’ll also tell you more about Julie and her diagnosis.
Urticaria: A simple case of hives?
This common allergic reaction affects close to 10% of the population at some point in their lives. The affected areas are itchy and have raised, circumscribed red welts with surrounding erythema.1 Urticaria can occur throughout the body, with new lesions often erupting as the old ones disappear.2,3
Despite the persistent itchiness that patients typically complain of, however, urticaria is usually self-limiting, and rarely life-threatening. Acute urticaria normally resolves within 2 to 6 weeks.2,4
In most cases, urticaria arises secondary to exposure to an allergenic substance, chemical, or emotional stress.4,5 In rare instances, systemic diseases, such as hematologic malignancies, can also cause urticarial lesions to erupt throughout the body.4
Body piercing, cosmetics, latex exposure, Helicobacter pylori, insects, and angiotensin-converting enzyme (ACE) inhibitors have been identified as common triggers of urticaria, as have nonsteroidal anti-inflammatory drugs (NSAIDs) and antibiotics, animal dander, and foods such as shellfish, nuts, and dairy products.4,6
Treatment of all forms of urticaria should be based on identification and strict avoidance of the causative agent, if it’s known.7 Following withdrawal of the specific agent, symptomatic treatment with medications such as histamine antagonists and corticosteroids remains the mainstay of therapy.4,8 A daily dose of 40 to 60 mg prednisone for 5 days is a reasonable therapeutic regimen for adults; a 5-day course of 1 mg/kg per day is suitable for pediatric patients.4,8,9
In the event that topical or oral therapy is ineffective in mild cases of urticaria, intravenous (IV) diphenhydramine (50 mg) can be administered every 6 to 8 hours.4,8 IV diphenhydramine typically takes 30 minutes to work, while corticosteroids take at least 2 hours to reach full effect.4,8
In an emergency setting, subcutaneous epinephrine (0.3-0.5 mg) can be useful in treating severe urticaria.4,8 And recent clinical trials have demonstrated complete clearance of urticaria with leukotriene inhibitors, such as montelukast (10 mg).10
Chronic urticaria, trigger unknown
Although acute urticaria is responsive to treatment, chronic urticaria—lesions that do not resolve after 6 weeks—poses a greater challenge. In up to 80% of cases of chronic urticaria, no identifiable trigger is found.3,8 Long-term treatment of patients with this chronic condition, including lifestyle changes (to avoid environmental or dietary triggers) and a medication regimen for 6 months or more, leads to complete resolution of symptoms in most cases.7,8
Angioedema: Less common, more dangerous
Angioedema is part of the same disease spectrum as urticaria, but it affects the deeper tissues—involving mucosal and submucosal swelling. Angioedema affects just 0.1% to 0.2% of the general population, but up to 15% of patients with urticaria.8
The risk increases with the use of ACE inhibitors; for every 1000 patients taking ACE inhibitors, 0.4 to 3.5 develop angioedema.11 A recent double-blind study involving 25,642 patients being treated with ACE inhibitors revealed that those taking a combination of ACE inhibitors were more likely to develop angioedema than those on monotherapy.12
Angioedema is characterized by the sudden appearance of painful, localized erythematous wheals with central blanching.13 It results from increased vascular permeability in capillaries of the dermis, which leads to fluid leakage.13,14 Increased accumulation of fluids from the vessels of the skin results in rapidly developing nonpitting edema that most often affects the hands, face (and lips), neck, and oropharynx (FIGURE 1).14 Although the head and neck are the most commonly involved areas, angioedema can also affect the digestive tract, leading to nausea, vomiting, diarrhea, and abdominal pain secondary to bowel edema.13
FIGURE 1
Angioedema: A look at the most commonly affected areas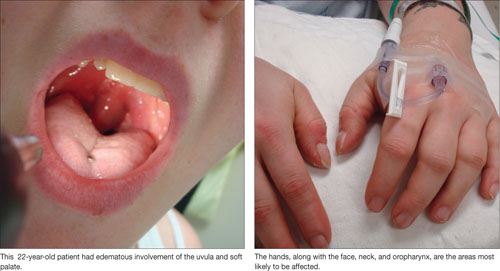
Airway management is imperative
In severe cases of angioedema, involvement of the oral mucosa results in stridor, followed by upper airway obstruction. To avoid hypoxemia and death,13,14 rapid preparation for emergency intervention to maintain the airway is critical; mortality can be as high as 30% in patients with airway compromise.13,15 In severe cases in which edema engulfs the oropharynx, oral intubation is often impossible, and nasotracheal intubation may be warranted.15,16 A failure to maintain adequate airway function via nasotracheal intubation may signal the need for an emergency tracheotomy.15,16
Is the angioedema hereditary or acquired?
Hereditary and acquired angioedema are treated differently. After ensuring that the patient has a patent airway, distinguishing between them is critical. Hereditary angioedema is the result of an inherited deficiency of plasma protein C1 inhibitor (C1-INH). Acquired angioedema is typically caused by enhanced consumption of endogenous C1-INH,13,17 leading to a net deficit of circulating C1-INH, and can be triggered by food, pharmacologic agents, and, occasionally, by systemic disorders.13 African Americans and patients with renal impairment appear to be at increased risk for acquired angioedema.13 In both hereditary and acquired angioiedema, decreased levels of C1-INH lead to disruption of the complement pathway.
Although the clinical presentation of acquired and hereditary angioedema is similar, a focused patient history can be used to distinguish between them. Age, health status, and medication history are the key considerations. Hereditary angioedema most commonly occurs—in recurrent attacks after minor trauma—in children with no underlying disease, with worsening symptoms during puberty. Acquired angioedema is generally seen in adult and elderly patients with malignancies or other underlying disorders.
Mild to moderate cases of acquired angioedema respond well to oral corticosteroids and antihistamines. Severe cases often require administration of subcutaneous epinephrine, followed by IV steroids. Management of hereditary angioedema should include C1-INH-containing fresh frozen plasma,13,16,18 although fresh frozen plasma can be used if plasma with C1-INH is not available.13
While oral corticosteroids and anti-histamines may be effective adjunctive therapy for hereditary angioedema, they are not likely to reverse acute attacks in this patient population. IV diphenhydramine (50-100 mg) or IV cimetidine (300 mg) every 6 to 8 hours is a reasonable therapeutic regimen for acute attacks of hereditary angioedema.
A recent randomized double-blind trial involving 40 patients with hereditary angioedema studied the administration of ecallantide, a kallikrein inhibitor for which US Food and Drug Administration approval is pending.19 Nearly 3 out of 4 of those who received ecallantide (72.5%) for acute attacks of angioedema showed significant improvement within 4 hours.20 The use of kallikrein inhibitors, which target inflammatory blood components, is not widespread, but may hold promise for the treatment of hereditary angioedema.20
Cellulitis: On the lookout for infiltration
Cellulitis is a bacterial infection of the skin that affects approximately 24.6 in 1000 people and is rarely associated with death.21 It occurs when bacteria enter through disrupted areas in the skin, particularly when skin integrity is compromised by recent surgery, piercing, wounds, athlete’s foot, or even dermatitis.22,23Streptococcus and Staphylococcus are the 2 most common infectious agents, and methicillin-resistant Staphylococcus aureus (MRSA) is increasingly common.22,23
Although cellulitis is primarily superficial in nature, it may progress to a serious condition by infiltrating underlying tissues and spreading to nearby lymphatic tissue and the bloodstream to cause lymphadenitis or bacteremia.21,23 In instances of cellulitis-induced bacteremia, mortality rates increase if prompt, targeted treatment is not provided.23
Raised erythematous plaques are the cardinal features of cellulitis, with the affected areas warm to the touch, red, and tender.21,23 As the condition progresses, the affected area tends to enlarge and expand (FIGURE 2),24 and the patient often becomes febrile.22,24
The risk of developing cellulitis increases with age, compromised immune status, diabetes, obesity, IV drug use, lymphedema, and chronic corticosteroid use.22,24
Cellulitis is often diagnosed solely on the basis of clinical presentation, although aspiration of purulent discharge from the wound and a gram stain of the culture can confirm the diagnosis.25,26 (See “Is it cellulitis or stasis dermatitis?”.)
Direct immunofluorescence can be used when cultures are difficult to obtain, but this technique is seldom necessary.22 If infiltration of underlying soft tissues is suspected based on clinical findings, magnetic resonance imaging can be a useful tool in evaluating the extent of the infection and in directing appropriate debridement and drainage of affected areas.22,27
Patients with venous insufficiency may present with stasis dermatitis, which often results in breakdown of the skin and ulceration that bears a striking resemblance to cellulitis. Thus, these conditions can be easily confused, and may lead to unnecessary antibiotic use and, possibly, hospitalization in patients with venous insufficiency.26,28
Despite the similarities of these conditions, a focused patient history and physical exam can prevent such confusion. Stasis dermatitis arises as a result of venous insufficiency, so it is likely to be accompanied by pitting edema that responds to leg raising and to the use of elastic compression stockings—interventions that are seldom effective for cellulitis.26 In addition, cellulitis tends to be unilateral, while stasis dermatitis often has bilateral involvement.
FIGURE 2
Diagnosing cellulitis based on clinical presentation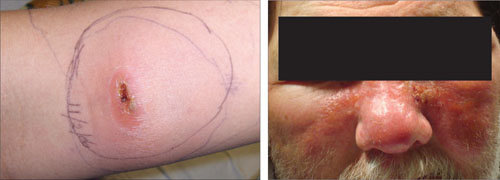
The raised erythematous lesions that are a hallmark of cellulitis, shown here on the arm and face, are warm to the touch, red, and tender.
Treatment: Targeted antibiotics and preventive measures
Because of the likelihood of recurrence with cellulitis, treating the condition involves both preventive and curative measures. Mild cases can be treated in an outpatient setting with a 7- to 10-day course of oral cephalosporins or antibiotics with similar coverage.22,25,26 A recent randomized study involving 391 patients found that cure rates for cellulitis treated with cephalexin were between 83% and 92%, depending on the pathogen involved.26
For severe cases of cellulitis, patients who are immunocompromised, and cases that are refractory to oral medications, hospital admission is recommended, and use of IV antibiotics is routinely required.22,25,26 For patients with MRSA, a drug such as vancomycin IV may be warranted; a reasonable dose would be 15 mg/kg every 12 hours.22,26,27,29
A recent randomized, multicenter study demonstrated that vancomycin effectively treated approximately 67% of cases of MRSA-induced cellulitis.26 Neomycin should be strictly avoided whenever cellulitis is suspected, because of its propensity to promote antibiotic resistance to S. aureus.29
Patient education emphasizing preventive measures is critical for minimizing recurrence of cellulitis.22,25,26 Encourage patients to wash with antibacterial soap and water daily, apply topical antibiotic ointment, and keep the wound completely covered at all times. Advise them to change bandages and wash their hands frequently.27,29 Patients with diabetes and others with decreased circulation in the extremities need to take further precautions, such as moisturizing the skin regularly in order to prevent cuts in their skin.22,29
SJS: Triggered by drugs, and infections, too
Stevens-Johnson syndrome (SJS), also known as erythema multiforme major, is an often-debilitating and possibly fatal adverse reaction, typically (but not exclusively) to a drug. It manifests as full-thickness epidermal necrosis of the mucous membranes.30,31 SJS occurs at a rate of about 1 to 7 cases per million people per year, and has a mortality rate of approximately 5%.30-32 The types of medication that most commonly precipitate SJS are anticonvulsants, sulfa drugs, penicillin-related and cephalosporin antibiotics, anti-inflammatory agents, and certain neoplastic drugs.30,32 SJS can develop in response to infections and neoplasms (TABLE) as well, and in many cases a cause is never found.
Patients with widespread involvement often complain of a burning sensation, particularly around the mouth.32-36 In some cases, this is the presenting sign, because the oral mucosa tends to be among the first mucous membranes involved.
TABLE
Stevens-Johnson syndrome: Pinpointing the cause32
| MORE FREQUENT ETIOLOGY |
| Drugs: Allopurinol, anticonvulsants, antiparasitics, barbiturates, NSAIDs, penicillin-related and cephalosporin antibiotics, sulfas, tetracyclines |
| LESS FREQUENT ETIOLOGY |
| Bacterial: Diphtheria, group A Streptococcus, Mycoplasma pneumoniae, tularemia, typhoid |
| Fungal: Coccidiomycosis, dermatophytosis, histoplasmosis, |
| Protozoan: Plasmodium, trichomoniasis |
| Viral: AIDS, Coxsackie, Epstein-Barr, HSV, influenza |
| AIDS, acquired immune deficiency syndrome; HSV, herpes-simplex virus; NSAIDs, nonsteroidal anti-inflammatory drugs. |
Targetoid lesions, Nikolsky sign are diagnostic clues
The characteristic skin lesions seen with SJS consist of initially erythematous macules that rapidly develop central necrosis to form vesiculation, as well as other variable areas of denudation (FIGURE 3). These vesicles tend to demonstrate confluence and often show a positive Nikolsky sign—epidermal detachment of superficial layers of the skin when slight pressure is applied. The lesions typically take on a targetoid appearance. If left untreated, the blisters often result in ulceration and hemorrhagic crusting of affected areas.
Like most skin disorders, SJS is initially diagnosed on the basis of clinical presentation. However, it is a rare disorder, and commonly misdiagnosed. Among the disorders SJS has been mistaken for are staphylococcal scalded skin syndrome, toxic shock syndrome, exfoliative dermatitis, scarlet fever, erythema multiforme, and iatrogenic chemical burns.32,37 (Erythema multiforme, SJS, and toxic epidermal necrolysis [TEN] are considered part of the same disease spectrum; erythema multiforme typically presents with few random lesions and no mucosal involvement, SJS with mucosal involvement on up to 30% of the body surface, and TEN with >30%.)32,37 Skin biopsies and immunofluorescence studies are recommended to confirm the diagnosis.
During the course of SJS, the mucous membranes of the oropharynx, ocular cavity, gastrointestinal system, nasal cavity, genitourinary system, and lower respiratory tracts are typically affected.32-35 As the condition progresses, increased epidermal erosion can lead to the sloughing off of up to 100% of the epidermis, resulting in considerable fluid loss.32,34
Corneal ulceration, anterior uveitis, panophthalmitis, polyarthritis, hematuria, and acute tubular necrosis leading to renal failure may also occur.32,35,37 Scarring within vital ocular structures can result in corneal opacity and lead to significant visual impairment. In severe cases, blood loss and fluid loss increase the risk of bacterial superinfection and sepsis.35,37
FIGURE 3
Targetoid lesions are characteristic of SJS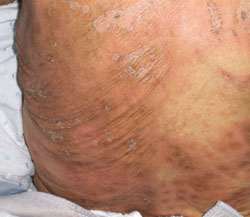
Characteristic diffuse erythematous macules with necrotic centers and overlying blistering on the back of a patient with Stevens-Johnson syndrome.
Supportive therapy, wound care are key components of treatment
Rapid cessation of the offending agent with targeted dermatologic management can reduce morbidity by promoting rapid re-epithelialization of affected skin. (See “SJS is diagnosed, but not quickly,” [Verdicts] on page 332, for a discussion of the dangers of delayed diagnosis and failure to promptly stop the drug causing the acute reaction.)
Closely monitoring the patient for fluid and electrolyte abnormalities is also crucial. Corticosteroids and IV immunoglobulins have been suggested for early severe cases of SJS, but their efficacy in treating this condition has yet to be established by prospective double-blind studies.32-37
The skin lesions associated with SJS should be treated in the same way you would treat thermal burns, with local wound care, warm compresses, and topical anesthetics for pain reduction.36,37 Oral lesions are managed with diphenhydramine or sodium bicarbonate mouthwashes and glycerin swabs.
An ophthalmologic consultation is mandatory because of the risk of vision loss associated with corneal scarring.
And now, a return to our 9-year-old patient
When we left off our discussion of Julie, the ED physician who examined her had detected a targetoid lesion with a necrotic, purpuric center—a finding that we described as a diagnostic clue. The second diagnostic clue? The presence of the Nikolsky sign, which the doctor detected by applying slight pressure to the lesion. Julie was admitted to the hospital with a presumed diagnosis of SJS, which skin biopsies and immunofluorescence studies later confirmed.
It wasn’t clear whether Julie had had a reaction to the sulfa (which she’d completed) or to the penicillin (which she’d just begun taking), or whether she had a synergistic reaction to both. Although the exact cause remained uncertain, as it often does, the penicillin was stopped immediately. She received dermatologic treatment without delay and was monitored closely for fluid and electrolyte status. Since Julie had signs of ocular involvement, daily erythromycin and corticosteroid eyedrops were administered to minimize the risk of infection and reduce local inflammation. Given the risk of long-term ocular complications in patients with SJS, we recommended continued ophthalmologic care.
Nine days after she was admitted, Julie’s symptoms resolved, with the exception of persistent complaints of dry eye. At discharge, Julie was given artificial tears to minimize ocular irritation. We suspected that she had dry eyes because of SJS-induced corneal scarring, but we were unable to confirm our suspicion because our patient failed to return for scheduled ophthalmologic appointments. She was subsequently lost to follow up.
Acknowledgement
The authors wish to thank Azita Hamedani, MD, MPH, FACEP, for her critical editing of the text.
Correspondence
Ribhi Hazin, MD, 29 Garden Street, Suite 214, Cambridge, MA 02138; Ribhi.hazin@gmail.com
1. Noe R, Cohen AL, Lederman E, et al. Skin disorders among construction workers following Hurricane Katrina and Hurricane Rita: an outbreak investigation in New Orleans, Louisiana. Arch Dermatol. 2007;143:1393-1398.
2. Kulthanan K, Jiamton S, Thumpimukvatana N, et al. Chronic idiopathic urticaria: prevalence and clinical course. J Dermatol. 2007;34:294-301.
3. Stanway AD, Cohen SN, Chen C, et al. H1-antihistamines for chronic urticaria. Cochrane Database Syst Rev. 2008(2):CD006137.-
4. Grattan CE, Humphreys F. British Association of Dermatologists Therapy Guidelines and Audit Subcommittee. Guidelines for evaluation and management of urticaria in adults and children. Br J Dermatol. 2007;157:1116-1123.
5. Kozel MM, Mekkes JR, Bossuyt PM, et al. Natural course of physical and chronic urticaria and angioedema in 220 patients. J Am Acad Dermatol. 2001;45:387-391.
6. Nettis E, Colanardi MC, Soccio AL, et al. Double-blind, placebo-controlled study of sublingual immunotherapy in patients with latex-induced urticaria: a 12-month study. Br J Dermatol. 2007;156:674-681.
7. Lee EE, Maibach HI. Treatment of urticaria. An evidence-based evaluation of antihistamines. Am J Clin Dermatol. 2001;2:27-32.
8. Humphreys F, Hunter JA. The characteristics of urticaria in 390 patients. Br J Dermatol. 1998;138:635-638.
9. Serhat Inaloz H, Ozturk S, Akcali C, et al. Low-dose and short-term cyclosporine treatment in patients with chronic idiopathic urticaria: A clinical and immunological evaluation. J Dermatol. 2008;35:276-282.
10. Erbagci Z. The leukotriene receptor antagonist montelukast in the treatment of chronic idiopathic urticaria. A single-blind, placebo-controlled, crossover clinical study. J Allergy Clin Immunol. 2002;110:484-488.
11. Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med. 2005;165:1637-1642.
12. Yusuf S, Teo KK, Pogue J, et al. ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008;358:1547-1559.
13. Joint Task Force on Practice Parameters; American Academy of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. The diagnosis and management of anaphylaxis: an updated practice parameter. J Allergy Clin Immunol. 2005;115(3 Suppl 2):s483-s523.
14. Bas M, Kirchhartz N, Hochfeld J, et al. Potential role of vasomotor effects of fibrinogen in bradykinin-induced angioedema. J Allergy Clin Immunol. 2008;121:969e2-975e2.
15. Sica DA, Black HR. Angioedema in heart failure: occurrence with ACE inhibitors and safety of angiotensin receptor blocker therapy. Congest Heart Fail. 2002;8:334-341.
16. Banerji A, Clark S, Blanda M, et al. Multicenter study of patients with angiotensin-converting enzyme inhibitor-induced angioedema who present to the emergency department. Ann Allergy Asthma Immunol. 2008;100:327-332.
17. Cicardi M, Zingale LC, Zanichelli A, et al. The use of plasma-derived C1 inhibitor in the treatment of hereditary angioedema. Expert Opin Pharmacother. 2007;8:3173-3181.
18. Bork K, Bygum A, Hardt J. Benefits and risks of danazol in hereditary angioedema: a long-term survey of 118 patients. Ann Allergy Asthma Immunol. 2008;100:153-161.
19. US Food and Drug Administration. Advisory Committee Briefing Document. Kalbitor (ecallantide) for acute attacks of hereditary angioedema. Available at: http://www.fda.gov/ohrms/dockets/AC/09/briefing/2009-4413b1-03-Dyax.pdf. Accessed May 5, 2009.
20. Schneider L, Lumry W, Vegh A, et al. Critical role of kallikrein in hereditary angioedema pathogenesis: a clinical trial of ecallantide, a novel kallikrein inhibitor. J Allergy Clin Immunol. 2007;120:416-422.
21. Ellis Simonsen SM, van Orman ER, Hatch BE, et al. Cellulitis incidence in a defined population. Epidemiol Infect. 2006;134:293-299.
22. Stevens DL, Bisno AL, Chambers HF, et al. Infectious Diseases Society of America. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin Infect Dis. 2005;41:1373-1406.
23. Morris A. What are the benefits of treatments? Cellulitis and erysipelas. BMJ Clin Evid. 2005;13:2066-2069.
24. Murray H, Stiell I, Wells G. Treatment failure in emergency department patients with cellulitis. CJEM. 2005;7:228-234.
25. Meier DE, Nkor SK, Aasa D, et al. Prospective randomized comparison of two preoperative skin preparation techniques in a developing world country. World J Surg. 2001;25:441-443.
26. Giordano PA, Elston D, Akinlade BK, et al. Cefdinir vs. cephalexin for mild to moderate uncomplicated skin and skin structure infections in adolescents and adults. Curr Med Res Opin. 2006;22:2419-2428.
27. Halpern J, Holder R, Langford NJ. Ethnicity and other risk factors for acute lower limb cellulitis: a U.K.-based prospective case-control study. Br J Dermatol. 2008;158:1288-1292.
28. Lin YT, Lu PW. Retrospective study of pediatric facial cellulitis of odontogenic origin. Pediatr Infect Dis J. 2006;25:339-342.
29. Rajendran PM, Young D, Maurer T, et al. Randomized, double-blind, placebo-controlled trial of cephalexin for treatment of uncomplicated skin abscesses in a population at risk for community-acquired methicillin-resistant Staphylococcus aureus infection. Antimicrob Agents Chemo. 2007;51:4044-4048.
30. Schneck J, Fagot JP, Sekula P, et al. Effects of treatments on the mortality of Stevens-Johnson syndrome and toxic epidermal necrolysis: a retrospective study on patients included in the prospective EuroSCAR Study. J Am Acad Dermatol. 2008;58:33-40.
31. Chan HL, Stern RS, Arndt KA, et al. The incidence of erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis. A population-based study with particular reference to reactions caused by drugs among outpatients. Arch Dermatol. 1990;126:43-47.
32. Hazin R, Ibrahimi OA, Hazin MI, et al. Stevens-Johnson syndrome: pathogenesis, diagnosis, and management. Ann Med, 2008;40:129-138.
33. Chung WH, Hung SI, Hong HS, et al. Medical genetics: a marker for Stevens-Johnson syndrome. Nature. 2004;428:486.-
34. Jette N, Hemming K, Hutton JL, et al. Topiramate add-on for drug-resistant partial epilepsy. Cochrane Database Syst Rev. 2008;(3):DC001417.-
35. Gürcan HM, Ahmed AR. Efficacy of various intravenous immunoglobulin therapy protocols in auto-immune and chronic inflammatory disorders. Ann Pharmacother. 2007;41:812-523.
36. Strom BL, Carson JL, Halpern AC, et al. A population-based study of Stevens-Johnson syndrome. Incidence and antecedent drug exposures. Arch Dermatol. 1991;127:831-838.
37. Gravante G, Delogu D, Marianetti M, et al. Toxic epidermal necrolysis and Steven-Johnson syndrome: 11-years experience and outcome. Eur Rev Med Pharmacol Sci. 2007;11:119-127.
- Management of hereditary angioedema should include fresh frozen plasma containing C1 inhibitor (C1-INH), whenever possible; if C1-INH-containing plasma is unavailable, fresh frozen plasma can be used instead (SOR: A).
- Do not give neomycin to patients with suspected cellulitis; the drug may promote antibiotic resistance in Staphylococcus aureus, a pathogen often associated with this condition (SOR: A).
- Whenever a patient presents with erythematous skin lesions and a recent history of receiving penicillin or a cephalosporin antibiotic, a sulfa derivative, or an anticonvulsant, the suspected medication should be stopped until Stevens-Johnson syndrome is ruled out (SOR: A).
Strength of recommendation (SOR)
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
Skin eruptions are a common reason for visits to primary care physicians. While most are innocuous, some are associated with—or are early warning signs of—severe allergic reactions or other emergent conditions. A 9-year-old patient I’ll call Julie is a case in point.
The first time Julie’s parents brought her to our clinic, she’d been complaining of a sore throat and had a fever that hovered between 102° and 103°F for several days. The physician who examined Julie found mild maxillary tenderness. A rapid streptococcal throat swab was negative; her doctor prescribed a 10-day course of trimethoprim/sulfamethoxazole (TMP/SMX) for presumed acute sinusitis.
Thirteen days later (3 days after the patient completed the course of antibiotics), Julie’s parents brought her back to the clinic. Her throat still hurt, and she had erythematous oval lesions on her trunk and upper extremities. Her physician diagnosed scarlet fever and wrote a prescription for penicillin.
The following day, Julie was taken to the emergency department (ED) with bilateral conjunctival hyperemia and diffuse, confluent erythematous macules throughout her body. The ED physician who examined Julie found a 2.5 × 2.0 cm targetoid lesion with a necrotic, purpuric center on her lower back—a diagnostic clue to the cause of her signs and symptoms.
If you had been Julie’s physician, would you have been alert to that clue?
For family physicians accustomed to seeing relatively mild skin disorders, recognizing and responding to dermatologic conditions with potentially dire outcomes can be challenging. This review, and the images that accompany it, will help you sharpen your dermatologic diagnostic and treatment skills, both for benign disorders and those that are less common and more severe. We’ll also tell you more about Julie and her diagnosis.
Urticaria: A simple case of hives?
This common allergic reaction affects close to 10% of the population at some point in their lives. The affected areas are itchy and have raised, circumscribed red welts with surrounding erythema.1 Urticaria can occur throughout the body, with new lesions often erupting as the old ones disappear.2,3
Despite the persistent itchiness that patients typically complain of, however, urticaria is usually self-limiting, and rarely life-threatening. Acute urticaria normally resolves within 2 to 6 weeks.2,4
In most cases, urticaria arises secondary to exposure to an allergenic substance, chemical, or emotional stress.4,5 In rare instances, systemic diseases, such as hematologic malignancies, can also cause urticarial lesions to erupt throughout the body.4
Body piercing, cosmetics, latex exposure, Helicobacter pylori, insects, and angiotensin-converting enzyme (ACE) inhibitors have been identified as common triggers of urticaria, as have nonsteroidal anti-inflammatory drugs (NSAIDs) and antibiotics, animal dander, and foods such as shellfish, nuts, and dairy products.4,6
Treatment of all forms of urticaria should be based on identification and strict avoidance of the causative agent, if it’s known.7 Following withdrawal of the specific agent, symptomatic treatment with medications such as histamine antagonists and corticosteroids remains the mainstay of therapy.4,8 A daily dose of 40 to 60 mg prednisone for 5 days is a reasonable therapeutic regimen for adults; a 5-day course of 1 mg/kg per day is suitable for pediatric patients.4,8,9
In the event that topical or oral therapy is ineffective in mild cases of urticaria, intravenous (IV) diphenhydramine (50 mg) can be administered every 6 to 8 hours.4,8 IV diphenhydramine typically takes 30 minutes to work, while corticosteroids take at least 2 hours to reach full effect.4,8
In an emergency setting, subcutaneous epinephrine (0.3-0.5 mg) can be useful in treating severe urticaria.4,8 And recent clinical trials have demonstrated complete clearance of urticaria with leukotriene inhibitors, such as montelukast (10 mg).10
Chronic urticaria, trigger unknown
Although acute urticaria is responsive to treatment, chronic urticaria—lesions that do not resolve after 6 weeks—poses a greater challenge. In up to 80% of cases of chronic urticaria, no identifiable trigger is found.3,8 Long-term treatment of patients with this chronic condition, including lifestyle changes (to avoid environmental or dietary triggers) and a medication regimen for 6 months or more, leads to complete resolution of symptoms in most cases.7,8
Angioedema: Less common, more dangerous
Angioedema is part of the same disease spectrum as urticaria, but it affects the deeper tissues—involving mucosal and submucosal swelling. Angioedema affects just 0.1% to 0.2% of the general population, but up to 15% of patients with urticaria.8
The risk increases with the use of ACE inhibitors; for every 1000 patients taking ACE inhibitors, 0.4 to 3.5 develop angioedema.11 A recent double-blind study involving 25,642 patients being treated with ACE inhibitors revealed that those taking a combination of ACE inhibitors were more likely to develop angioedema than those on monotherapy.12
Angioedema is characterized by the sudden appearance of painful, localized erythematous wheals with central blanching.13 It results from increased vascular permeability in capillaries of the dermis, which leads to fluid leakage.13,14 Increased accumulation of fluids from the vessels of the skin results in rapidly developing nonpitting edema that most often affects the hands, face (and lips), neck, and oropharynx (FIGURE 1).14 Although the head and neck are the most commonly involved areas, angioedema can also affect the digestive tract, leading to nausea, vomiting, diarrhea, and abdominal pain secondary to bowel edema.13
FIGURE 1
Angioedema: A look at the most commonly affected areas
Airway management is imperative
In severe cases of angioedema, involvement of the oral mucosa results in stridor, followed by upper airway obstruction. To avoid hypoxemia and death,13,14 rapid preparation for emergency intervention to maintain the airway is critical; mortality can be as high as 30% in patients with airway compromise.13,15 In severe cases in which edema engulfs the oropharynx, oral intubation is often impossible, and nasotracheal intubation may be warranted.15,16 A failure to maintain adequate airway function via nasotracheal intubation may signal the need for an emergency tracheotomy.15,16
Is the angioedema hereditary or acquired?
Hereditary and acquired angioedema are treated differently. After ensuring that the patient has a patent airway, distinguishing between them is critical. Hereditary angioedema is the result of an inherited deficiency of plasma protein C1 inhibitor (C1-INH). Acquired angioedema is typically caused by enhanced consumption of endogenous C1-INH,13,17 leading to a net deficit of circulating C1-INH, and can be triggered by food, pharmacologic agents, and, occasionally, by systemic disorders.13 African Americans and patients with renal impairment appear to be at increased risk for acquired angioedema.13 In both hereditary and acquired angioiedema, decreased levels of C1-INH lead to disruption of the complement pathway.
Although the clinical presentation of acquired and hereditary angioedema is similar, a focused patient history can be used to distinguish between them. Age, health status, and medication history are the key considerations. Hereditary angioedema most commonly occurs—in recurrent attacks after minor trauma—in children with no underlying disease, with worsening symptoms during puberty. Acquired angioedema is generally seen in adult and elderly patients with malignancies or other underlying disorders.
Mild to moderate cases of acquired angioedema respond well to oral corticosteroids and antihistamines. Severe cases often require administration of subcutaneous epinephrine, followed by IV steroids. Management of hereditary angioedema should include C1-INH-containing fresh frozen plasma,13,16,18 although fresh frozen plasma can be used if plasma with C1-INH is not available.13
While oral corticosteroids and anti-histamines may be effective adjunctive therapy for hereditary angioedema, they are not likely to reverse acute attacks in this patient population. IV diphenhydramine (50-100 mg) or IV cimetidine (300 mg) every 6 to 8 hours is a reasonable therapeutic regimen for acute attacks of hereditary angioedema.
A recent randomized double-blind trial involving 40 patients with hereditary angioedema studied the administration of ecallantide, a kallikrein inhibitor for which US Food and Drug Administration approval is pending.19 Nearly 3 out of 4 of those who received ecallantide (72.5%) for acute attacks of angioedema showed significant improvement within 4 hours.20 The use of kallikrein inhibitors, which target inflammatory blood components, is not widespread, but may hold promise for the treatment of hereditary angioedema.20
Cellulitis: On the lookout for infiltration
Cellulitis is a bacterial infection of the skin that affects approximately 24.6 in 1000 people and is rarely associated with death.21 It occurs when bacteria enter through disrupted areas in the skin, particularly when skin integrity is compromised by recent surgery, piercing, wounds, athlete’s foot, or even dermatitis.22,23Streptococcus and Staphylococcus are the 2 most common infectious agents, and methicillin-resistant Staphylococcus aureus (MRSA) is increasingly common.22,23
Although cellulitis is primarily superficial in nature, it may progress to a serious condition by infiltrating underlying tissues and spreading to nearby lymphatic tissue and the bloodstream to cause lymphadenitis or bacteremia.21,23 In instances of cellulitis-induced bacteremia, mortality rates increase if prompt, targeted treatment is not provided.23
Raised erythematous plaques are the cardinal features of cellulitis, with the affected areas warm to the touch, red, and tender.21,23 As the condition progresses, the affected area tends to enlarge and expand (FIGURE 2),24 and the patient often becomes febrile.22,24
The risk of developing cellulitis increases with age, compromised immune status, diabetes, obesity, IV drug use, lymphedema, and chronic corticosteroid use.22,24
Cellulitis is often diagnosed solely on the basis of clinical presentation, although aspiration of purulent discharge from the wound and a gram stain of the culture can confirm the diagnosis.25,26 (See “Is it cellulitis or stasis dermatitis?”.)
Direct immunofluorescence can be used when cultures are difficult to obtain, but this technique is seldom necessary.22 If infiltration of underlying soft tissues is suspected based on clinical findings, magnetic resonance imaging can be a useful tool in evaluating the extent of the infection and in directing appropriate debridement and drainage of affected areas.22,27
Patients with venous insufficiency may present with stasis dermatitis, which often results in breakdown of the skin and ulceration that bears a striking resemblance to cellulitis. Thus, these conditions can be easily confused, and may lead to unnecessary antibiotic use and, possibly, hospitalization in patients with venous insufficiency.26,28
Despite the similarities of these conditions, a focused patient history and physical exam can prevent such confusion. Stasis dermatitis arises as a result of venous insufficiency, so it is likely to be accompanied by pitting edema that responds to leg raising and to the use of elastic compression stockings—interventions that are seldom effective for cellulitis.26 In addition, cellulitis tends to be unilateral, while stasis dermatitis often has bilateral involvement.
FIGURE 2
Diagnosing cellulitis based on clinical presentation
The raised erythematous lesions that are a hallmark of cellulitis, shown here on the arm and face, are warm to the touch, red, and tender.
Treatment: Targeted antibiotics and preventive measures
Because of the likelihood of recurrence with cellulitis, treating the condition involves both preventive and curative measures. Mild cases can be treated in an outpatient setting with a 7- to 10-day course of oral cephalosporins or antibiotics with similar coverage.22,25,26 A recent randomized study involving 391 patients found that cure rates for cellulitis treated with cephalexin were between 83% and 92%, depending on the pathogen involved.26
For severe cases of cellulitis, patients who are immunocompromised, and cases that are refractory to oral medications, hospital admission is recommended, and use of IV antibiotics is routinely required.22,25,26 For patients with MRSA, a drug such as vancomycin IV may be warranted; a reasonable dose would be 15 mg/kg every 12 hours.22,26,27,29
A recent randomized, multicenter study demonstrated that vancomycin effectively treated approximately 67% of cases of MRSA-induced cellulitis.26 Neomycin should be strictly avoided whenever cellulitis is suspected, because of its propensity to promote antibiotic resistance to S. aureus.29
Patient education emphasizing preventive measures is critical for minimizing recurrence of cellulitis.22,25,26 Encourage patients to wash with antibacterial soap and water daily, apply topical antibiotic ointment, and keep the wound completely covered at all times. Advise them to change bandages and wash their hands frequently.27,29 Patients with diabetes and others with decreased circulation in the extremities need to take further precautions, such as moisturizing the skin regularly in order to prevent cuts in their skin.22,29
SJS: Triggered by drugs, and infections, too
Stevens-Johnson syndrome (SJS), also known as erythema multiforme major, is an often-debilitating and possibly fatal adverse reaction, typically (but not exclusively) to a drug. It manifests as full-thickness epidermal necrosis of the mucous membranes.30,31 SJS occurs at a rate of about 1 to 7 cases per million people per year, and has a mortality rate of approximately 5%.30-32 The types of medication that most commonly precipitate SJS are anticonvulsants, sulfa drugs, penicillin-related and cephalosporin antibiotics, anti-inflammatory agents, and certain neoplastic drugs.30,32 SJS can develop in response to infections and neoplasms (TABLE) as well, and in many cases a cause is never found.
Patients with widespread involvement often complain of a burning sensation, particularly around the mouth.32-36 In some cases, this is the presenting sign, because the oral mucosa tends to be among the first mucous membranes involved.
TABLE
Stevens-Johnson syndrome: Pinpointing the cause32
| MORE FREQUENT ETIOLOGY |
| Drugs: Allopurinol, anticonvulsants, antiparasitics, barbiturates, NSAIDs, penicillin-related and cephalosporin antibiotics, sulfas, tetracyclines |
| LESS FREQUENT ETIOLOGY |
| Bacterial: Diphtheria, group A Streptococcus, Mycoplasma pneumoniae, tularemia, typhoid |
| Fungal: Coccidiomycosis, dermatophytosis, histoplasmosis, |
| Protozoan: Plasmodium, trichomoniasis |
| Viral: AIDS, Coxsackie, Epstein-Barr, HSV, influenza |
| AIDS, acquired immune deficiency syndrome; HSV, herpes-simplex virus; NSAIDs, nonsteroidal anti-inflammatory drugs. |
Targetoid lesions, Nikolsky sign are diagnostic clues
The characteristic skin lesions seen with SJS consist of initially erythematous macules that rapidly develop central necrosis to form vesiculation, as well as other variable areas of denudation (FIGURE 3). These vesicles tend to demonstrate confluence and often show a positive Nikolsky sign—epidermal detachment of superficial layers of the skin when slight pressure is applied. The lesions typically take on a targetoid appearance. If left untreated, the blisters often result in ulceration and hemorrhagic crusting of affected areas.
Like most skin disorders, SJS is initially diagnosed on the basis of clinical presentation. However, it is a rare disorder, and commonly misdiagnosed. Among the disorders SJS has been mistaken for are staphylococcal scalded skin syndrome, toxic shock syndrome, exfoliative dermatitis, scarlet fever, erythema multiforme, and iatrogenic chemical burns.32,37 (Erythema multiforme, SJS, and toxic epidermal necrolysis [TEN] are considered part of the same disease spectrum; erythema multiforme typically presents with few random lesions and no mucosal involvement, SJS with mucosal involvement on up to 30% of the body surface, and TEN with >30%.)32,37 Skin biopsies and immunofluorescence studies are recommended to confirm the diagnosis.
During the course of SJS, the mucous membranes of the oropharynx, ocular cavity, gastrointestinal system, nasal cavity, genitourinary system, and lower respiratory tracts are typically affected.32-35 As the condition progresses, increased epidermal erosion can lead to the sloughing off of up to 100% of the epidermis, resulting in considerable fluid loss.32,34
Corneal ulceration, anterior uveitis, panophthalmitis, polyarthritis, hematuria, and acute tubular necrosis leading to renal failure may also occur.32,35,37 Scarring within vital ocular structures can result in corneal opacity and lead to significant visual impairment. In severe cases, blood loss and fluid loss increase the risk of bacterial superinfection and sepsis.35,37
FIGURE 3
Targetoid lesions are characteristic of SJS
Characteristic diffuse erythematous macules with necrotic centers and overlying blistering on the back of a patient with Stevens-Johnson syndrome.
Supportive therapy, wound care are key components of treatment
Rapid cessation of the offending agent with targeted dermatologic management can reduce morbidity by promoting rapid re-epithelialization of affected skin. (See “SJS is diagnosed, but not quickly,” [Verdicts] on page 332, for a discussion of the dangers of delayed diagnosis and failure to promptly stop the drug causing the acute reaction.)
Closely monitoring the patient for fluid and electrolyte abnormalities is also crucial. Corticosteroids and IV immunoglobulins have been suggested for early severe cases of SJS, but their efficacy in treating this condition has yet to be established by prospective double-blind studies.32-37
The skin lesions associated with SJS should be treated in the same way you would treat thermal burns, with local wound care, warm compresses, and topical anesthetics for pain reduction.36,37 Oral lesions are managed with diphenhydramine or sodium bicarbonate mouthwashes and glycerin swabs.
An ophthalmologic consultation is mandatory because of the risk of vision loss associated with corneal scarring.
And now, a return to our 9-year-old patient
When we left off our discussion of Julie, the ED physician who examined her had detected a targetoid lesion with a necrotic, purpuric center—a finding that we described as a diagnostic clue. The second diagnostic clue? The presence of the Nikolsky sign, which the doctor detected by applying slight pressure to the lesion. Julie was admitted to the hospital with a presumed diagnosis of SJS, which skin biopsies and immunofluorescence studies later confirmed.
It wasn’t clear whether Julie had had a reaction to the sulfa (which she’d completed) or to the penicillin (which she’d just begun taking), or whether she had a synergistic reaction to both. Although the exact cause remained uncertain, as it often does, the penicillin was stopped immediately. She received dermatologic treatment without delay and was monitored closely for fluid and electrolyte status. Since Julie had signs of ocular involvement, daily erythromycin and corticosteroid eyedrops were administered to minimize the risk of infection and reduce local inflammation. Given the risk of long-term ocular complications in patients with SJS, we recommended continued ophthalmologic care.
Nine days after she was admitted, Julie’s symptoms resolved, with the exception of persistent complaints of dry eye. At discharge, Julie was given artificial tears to minimize ocular irritation. We suspected that she had dry eyes because of SJS-induced corneal scarring, but we were unable to confirm our suspicion because our patient failed to return for scheduled ophthalmologic appointments. She was subsequently lost to follow up.
Acknowledgement
The authors wish to thank Azita Hamedani, MD, MPH, FACEP, for her critical editing of the text.
Correspondence
Ribhi Hazin, MD, 29 Garden Street, Suite 214, Cambridge, MA 02138; Ribhi.hazin@gmail.com
- Management of hereditary angioedema should include fresh frozen plasma containing C1 inhibitor (C1-INH), whenever possible; if C1-INH-containing plasma is unavailable, fresh frozen plasma can be used instead (SOR: A).
- Do not give neomycin to patients with suspected cellulitis; the drug may promote antibiotic resistance in Staphylococcus aureus, a pathogen often associated with this condition (SOR: A).
- Whenever a patient presents with erythematous skin lesions and a recent history of receiving penicillin or a cephalosporin antibiotic, a sulfa derivative, or an anticonvulsant, the suspected medication should be stopped until Stevens-Johnson syndrome is ruled out (SOR: A).
Strength of recommendation (SOR)
- Good-quality patient-oriented evidence
- Inconsistent or limited-quality patient-oriented evidence
- Consensus, usual practice, opinion, disease-oriented evidence, case series
Skin eruptions are a common reason for visits to primary care physicians. While most are innocuous, some are associated with—or are early warning signs of—severe allergic reactions or other emergent conditions. A 9-year-old patient I’ll call Julie is a case in point.
The first time Julie’s parents brought her to our clinic, she’d been complaining of a sore throat and had a fever that hovered between 102° and 103°F for several days. The physician who examined Julie found mild maxillary tenderness. A rapid streptococcal throat swab was negative; her doctor prescribed a 10-day course of trimethoprim/sulfamethoxazole (TMP/SMX) for presumed acute sinusitis.
Thirteen days later (3 days after the patient completed the course of antibiotics), Julie’s parents brought her back to the clinic. Her throat still hurt, and she had erythematous oval lesions on her trunk and upper extremities. Her physician diagnosed scarlet fever and wrote a prescription for penicillin.
The following day, Julie was taken to the emergency department (ED) with bilateral conjunctival hyperemia and diffuse, confluent erythematous macules throughout her body. The ED physician who examined Julie found a 2.5 × 2.0 cm targetoid lesion with a necrotic, purpuric center on her lower back—a diagnostic clue to the cause of her signs and symptoms.
If you had been Julie’s physician, would you have been alert to that clue?
For family physicians accustomed to seeing relatively mild skin disorders, recognizing and responding to dermatologic conditions with potentially dire outcomes can be challenging. This review, and the images that accompany it, will help you sharpen your dermatologic diagnostic and treatment skills, both for benign disorders and those that are less common and more severe. We’ll also tell you more about Julie and her diagnosis.
Urticaria: A simple case of hives?
This common allergic reaction affects close to 10% of the population at some point in their lives. The affected areas are itchy and have raised, circumscribed red welts with surrounding erythema.1 Urticaria can occur throughout the body, with new lesions often erupting as the old ones disappear.2,3
Despite the persistent itchiness that patients typically complain of, however, urticaria is usually self-limiting, and rarely life-threatening. Acute urticaria normally resolves within 2 to 6 weeks.2,4
In most cases, urticaria arises secondary to exposure to an allergenic substance, chemical, or emotional stress.4,5 In rare instances, systemic diseases, such as hematologic malignancies, can also cause urticarial lesions to erupt throughout the body.4
Body piercing, cosmetics, latex exposure, Helicobacter pylori, insects, and angiotensin-converting enzyme (ACE) inhibitors have been identified as common triggers of urticaria, as have nonsteroidal anti-inflammatory drugs (NSAIDs) and antibiotics, animal dander, and foods such as shellfish, nuts, and dairy products.4,6
Treatment of all forms of urticaria should be based on identification and strict avoidance of the causative agent, if it’s known.7 Following withdrawal of the specific agent, symptomatic treatment with medications such as histamine antagonists and corticosteroids remains the mainstay of therapy.4,8 A daily dose of 40 to 60 mg prednisone for 5 days is a reasonable therapeutic regimen for adults; a 5-day course of 1 mg/kg per day is suitable for pediatric patients.4,8,9
In the event that topical or oral therapy is ineffective in mild cases of urticaria, intravenous (IV) diphenhydramine (50 mg) can be administered every 6 to 8 hours.4,8 IV diphenhydramine typically takes 30 minutes to work, while corticosteroids take at least 2 hours to reach full effect.4,8
In an emergency setting, subcutaneous epinephrine (0.3-0.5 mg) can be useful in treating severe urticaria.4,8 And recent clinical trials have demonstrated complete clearance of urticaria with leukotriene inhibitors, such as montelukast (10 mg).10
Chronic urticaria, trigger unknown
Although acute urticaria is responsive to treatment, chronic urticaria—lesions that do not resolve after 6 weeks—poses a greater challenge. In up to 80% of cases of chronic urticaria, no identifiable trigger is found.3,8 Long-term treatment of patients with this chronic condition, including lifestyle changes (to avoid environmental or dietary triggers) and a medication regimen for 6 months or more, leads to complete resolution of symptoms in most cases.7,8
Angioedema: Less common, more dangerous
Angioedema is part of the same disease spectrum as urticaria, but it affects the deeper tissues—involving mucosal and submucosal swelling. Angioedema affects just 0.1% to 0.2% of the general population, but up to 15% of patients with urticaria.8
The risk increases with the use of ACE inhibitors; for every 1000 patients taking ACE inhibitors, 0.4 to 3.5 develop angioedema.11 A recent double-blind study involving 25,642 patients being treated with ACE inhibitors revealed that those taking a combination of ACE inhibitors were more likely to develop angioedema than those on monotherapy.12
Angioedema is characterized by the sudden appearance of painful, localized erythematous wheals with central blanching.13 It results from increased vascular permeability in capillaries of the dermis, which leads to fluid leakage.13,14 Increased accumulation of fluids from the vessels of the skin results in rapidly developing nonpitting edema that most often affects the hands, face (and lips), neck, and oropharynx (FIGURE 1).14 Although the head and neck are the most commonly involved areas, angioedema can also affect the digestive tract, leading to nausea, vomiting, diarrhea, and abdominal pain secondary to bowel edema.13
FIGURE 1
Angioedema: A look at the most commonly affected areas
Airway management is imperative
In severe cases of angioedema, involvement of the oral mucosa results in stridor, followed by upper airway obstruction. To avoid hypoxemia and death,13,14 rapid preparation for emergency intervention to maintain the airway is critical; mortality can be as high as 30% in patients with airway compromise.13,15 In severe cases in which edema engulfs the oropharynx, oral intubation is often impossible, and nasotracheal intubation may be warranted.15,16 A failure to maintain adequate airway function via nasotracheal intubation may signal the need for an emergency tracheotomy.15,16
Is the angioedema hereditary or acquired?
Hereditary and acquired angioedema are treated differently. After ensuring that the patient has a patent airway, distinguishing between them is critical. Hereditary angioedema is the result of an inherited deficiency of plasma protein C1 inhibitor (C1-INH). Acquired angioedema is typically caused by enhanced consumption of endogenous C1-INH,13,17 leading to a net deficit of circulating C1-INH, and can be triggered by food, pharmacologic agents, and, occasionally, by systemic disorders.13 African Americans and patients with renal impairment appear to be at increased risk for acquired angioedema.13 In both hereditary and acquired angioiedema, decreased levels of C1-INH lead to disruption of the complement pathway.
Although the clinical presentation of acquired and hereditary angioedema is similar, a focused patient history can be used to distinguish between them. Age, health status, and medication history are the key considerations. Hereditary angioedema most commonly occurs—in recurrent attacks after minor trauma—in children with no underlying disease, with worsening symptoms during puberty. Acquired angioedema is generally seen in adult and elderly patients with malignancies or other underlying disorders.
Mild to moderate cases of acquired angioedema respond well to oral corticosteroids and antihistamines. Severe cases often require administration of subcutaneous epinephrine, followed by IV steroids. Management of hereditary angioedema should include C1-INH-containing fresh frozen plasma,13,16,18 although fresh frozen plasma can be used if plasma with C1-INH is not available.13
While oral corticosteroids and anti-histamines may be effective adjunctive therapy for hereditary angioedema, they are not likely to reverse acute attacks in this patient population. IV diphenhydramine (50-100 mg) or IV cimetidine (300 mg) every 6 to 8 hours is a reasonable therapeutic regimen for acute attacks of hereditary angioedema.
A recent randomized double-blind trial involving 40 patients with hereditary angioedema studied the administration of ecallantide, a kallikrein inhibitor for which US Food and Drug Administration approval is pending.19 Nearly 3 out of 4 of those who received ecallantide (72.5%) for acute attacks of angioedema showed significant improvement within 4 hours.20 The use of kallikrein inhibitors, which target inflammatory blood components, is not widespread, but may hold promise for the treatment of hereditary angioedema.20
Cellulitis: On the lookout for infiltration
Cellulitis is a bacterial infection of the skin that affects approximately 24.6 in 1000 people and is rarely associated with death.21 It occurs when bacteria enter through disrupted areas in the skin, particularly when skin integrity is compromised by recent surgery, piercing, wounds, athlete’s foot, or even dermatitis.22,23Streptococcus and Staphylococcus are the 2 most common infectious agents, and methicillin-resistant Staphylococcus aureus (MRSA) is increasingly common.22,23
Although cellulitis is primarily superficial in nature, it may progress to a serious condition by infiltrating underlying tissues and spreading to nearby lymphatic tissue and the bloodstream to cause lymphadenitis or bacteremia.21,23 In instances of cellulitis-induced bacteremia, mortality rates increase if prompt, targeted treatment is not provided.23
Raised erythematous plaques are the cardinal features of cellulitis, with the affected areas warm to the touch, red, and tender.21,23 As the condition progresses, the affected area tends to enlarge and expand (FIGURE 2),24 and the patient often becomes febrile.22,24
The risk of developing cellulitis increases with age, compromised immune status, diabetes, obesity, IV drug use, lymphedema, and chronic corticosteroid use.22,24
Cellulitis is often diagnosed solely on the basis of clinical presentation, although aspiration of purulent discharge from the wound and a gram stain of the culture can confirm the diagnosis.25,26 (See “Is it cellulitis or stasis dermatitis?”.)
Direct immunofluorescence can be used when cultures are difficult to obtain, but this technique is seldom necessary.22 If infiltration of underlying soft tissues is suspected based on clinical findings, magnetic resonance imaging can be a useful tool in evaluating the extent of the infection and in directing appropriate debridement and drainage of affected areas.22,27
Patients with venous insufficiency may present with stasis dermatitis, which often results in breakdown of the skin and ulceration that bears a striking resemblance to cellulitis. Thus, these conditions can be easily confused, and may lead to unnecessary antibiotic use and, possibly, hospitalization in patients with venous insufficiency.26,28
Despite the similarities of these conditions, a focused patient history and physical exam can prevent such confusion. Stasis dermatitis arises as a result of venous insufficiency, so it is likely to be accompanied by pitting edema that responds to leg raising and to the use of elastic compression stockings—interventions that are seldom effective for cellulitis.26 In addition, cellulitis tends to be unilateral, while stasis dermatitis often has bilateral involvement.
FIGURE 2
Diagnosing cellulitis based on clinical presentation
The raised erythematous lesions that are a hallmark of cellulitis, shown here on the arm and face, are warm to the touch, red, and tender.
Treatment: Targeted antibiotics and preventive measures
Because of the likelihood of recurrence with cellulitis, treating the condition involves both preventive and curative measures. Mild cases can be treated in an outpatient setting with a 7- to 10-day course of oral cephalosporins or antibiotics with similar coverage.22,25,26 A recent randomized study involving 391 patients found that cure rates for cellulitis treated with cephalexin were between 83% and 92%, depending on the pathogen involved.26
For severe cases of cellulitis, patients who are immunocompromised, and cases that are refractory to oral medications, hospital admission is recommended, and use of IV antibiotics is routinely required.22,25,26 For patients with MRSA, a drug such as vancomycin IV may be warranted; a reasonable dose would be 15 mg/kg every 12 hours.22,26,27,29
A recent randomized, multicenter study demonstrated that vancomycin effectively treated approximately 67% of cases of MRSA-induced cellulitis.26 Neomycin should be strictly avoided whenever cellulitis is suspected, because of its propensity to promote antibiotic resistance to S. aureus.29
Patient education emphasizing preventive measures is critical for minimizing recurrence of cellulitis.22,25,26 Encourage patients to wash with antibacterial soap and water daily, apply topical antibiotic ointment, and keep the wound completely covered at all times. Advise them to change bandages and wash their hands frequently.27,29 Patients with diabetes and others with decreased circulation in the extremities need to take further precautions, such as moisturizing the skin regularly in order to prevent cuts in their skin.22,29
SJS: Triggered by drugs, and infections, too
Stevens-Johnson syndrome (SJS), also known as erythema multiforme major, is an often-debilitating and possibly fatal adverse reaction, typically (but not exclusively) to a drug. It manifests as full-thickness epidermal necrosis of the mucous membranes.30,31 SJS occurs at a rate of about 1 to 7 cases per million people per year, and has a mortality rate of approximately 5%.30-32 The types of medication that most commonly precipitate SJS are anticonvulsants, sulfa drugs, penicillin-related and cephalosporin antibiotics, anti-inflammatory agents, and certain neoplastic drugs.30,32 SJS can develop in response to infections and neoplasms (TABLE) as well, and in many cases a cause is never found.
Patients with widespread involvement often complain of a burning sensation, particularly around the mouth.32-36 In some cases, this is the presenting sign, because the oral mucosa tends to be among the first mucous membranes involved.
TABLE
Stevens-Johnson syndrome: Pinpointing the cause32
| MORE FREQUENT ETIOLOGY |
| Drugs: Allopurinol, anticonvulsants, antiparasitics, barbiturates, NSAIDs, penicillin-related and cephalosporin antibiotics, sulfas, tetracyclines |
| LESS FREQUENT ETIOLOGY |
| Bacterial: Diphtheria, group A Streptococcus, Mycoplasma pneumoniae, tularemia, typhoid |
| Fungal: Coccidiomycosis, dermatophytosis, histoplasmosis, |
| Protozoan: Plasmodium, trichomoniasis |
| Viral: AIDS, Coxsackie, Epstein-Barr, HSV, influenza |
| AIDS, acquired immune deficiency syndrome; HSV, herpes-simplex virus; NSAIDs, nonsteroidal anti-inflammatory drugs. |
Targetoid lesions, Nikolsky sign are diagnostic clues
The characteristic skin lesions seen with SJS consist of initially erythematous macules that rapidly develop central necrosis to form vesiculation, as well as other variable areas of denudation (FIGURE 3). These vesicles tend to demonstrate confluence and often show a positive Nikolsky sign—epidermal detachment of superficial layers of the skin when slight pressure is applied. The lesions typically take on a targetoid appearance. If left untreated, the blisters often result in ulceration and hemorrhagic crusting of affected areas.
Like most skin disorders, SJS is initially diagnosed on the basis of clinical presentation. However, it is a rare disorder, and commonly misdiagnosed. Among the disorders SJS has been mistaken for are staphylococcal scalded skin syndrome, toxic shock syndrome, exfoliative dermatitis, scarlet fever, erythema multiforme, and iatrogenic chemical burns.32,37 (Erythema multiforme, SJS, and toxic epidermal necrolysis [TEN] are considered part of the same disease spectrum; erythema multiforme typically presents with few random lesions and no mucosal involvement, SJS with mucosal involvement on up to 30% of the body surface, and TEN with >30%.)32,37 Skin biopsies and immunofluorescence studies are recommended to confirm the diagnosis.
During the course of SJS, the mucous membranes of the oropharynx, ocular cavity, gastrointestinal system, nasal cavity, genitourinary system, and lower respiratory tracts are typically affected.32-35 As the condition progresses, increased epidermal erosion can lead to the sloughing off of up to 100% of the epidermis, resulting in considerable fluid loss.32,34
Corneal ulceration, anterior uveitis, panophthalmitis, polyarthritis, hematuria, and acute tubular necrosis leading to renal failure may also occur.32,35,37 Scarring within vital ocular structures can result in corneal opacity and lead to significant visual impairment. In severe cases, blood loss and fluid loss increase the risk of bacterial superinfection and sepsis.35,37
FIGURE 3
Targetoid lesions are characteristic of SJS
Characteristic diffuse erythematous macules with necrotic centers and overlying blistering on the back of a patient with Stevens-Johnson syndrome.
Supportive therapy, wound care are key components of treatment
Rapid cessation of the offending agent with targeted dermatologic management can reduce morbidity by promoting rapid re-epithelialization of affected skin. (See “SJS is diagnosed, but not quickly,” [Verdicts] on page 332, for a discussion of the dangers of delayed diagnosis and failure to promptly stop the drug causing the acute reaction.)
Closely monitoring the patient for fluid and electrolyte abnormalities is also crucial. Corticosteroids and IV immunoglobulins have been suggested for early severe cases of SJS, but their efficacy in treating this condition has yet to be established by prospective double-blind studies.32-37
The skin lesions associated with SJS should be treated in the same way you would treat thermal burns, with local wound care, warm compresses, and topical anesthetics for pain reduction.36,37 Oral lesions are managed with diphenhydramine or sodium bicarbonate mouthwashes and glycerin swabs.
An ophthalmologic consultation is mandatory because of the risk of vision loss associated with corneal scarring.
And now, a return to our 9-year-old patient
When we left off our discussion of Julie, the ED physician who examined her had detected a targetoid lesion with a necrotic, purpuric center—a finding that we described as a diagnostic clue. The second diagnostic clue? The presence of the Nikolsky sign, which the doctor detected by applying slight pressure to the lesion. Julie was admitted to the hospital with a presumed diagnosis of SJS, which skin biopsies and immunofluorescence studies later confirmed.
It wasn’t clear whether Julie had had a reaction to the sulfa (which she’d completed) or to the penicillin (which she’d just begun taking), or whether she had a synergistic reaction to both. Although the exact cause remained uncertain, as it often does, the penicillin was stopped immediately. She received dermatologic treatment without delay and was monitored closely for fluid and electrolyte status. Since Julie had signs of ocular involvement, daily erythromycin and corticosteroid eyedrops were administered to minimize the risk of infection and reduce local inflammation. Given the risk of long-term ocular complications in patients with SJS, we recommended continued ophthalmologic care.
Nine days after she was admitted, Julie’s symptoms resolved, with the exception of persistent complaints of dry eye. At discharge, Julie was given artificial tears to minimize ocular irritation. We suspected that she had dry eyes because of SJS-induced corneal scarring, but we were unable to confirm our suspicion because our patient failed to return for scheduled ophthalmologic appointments. She was subsequently lost to follow up.
Acknowledgement
The authors wish to thank Azita Hamedani, MD, MPH, FACEP, for her critical editing of the text.
Correspondence
Ribhi Hazin, MD, 29 Garden Street, Suite 214, Cambridge, MA 02138; Ribhi.hazin@gmail.com
1. Noe R, Cohen AL, Lederman E, et al. Skin disorders among construction workers following Hurricane Katrina and Hurricane Rita: an outbreak investigation in New Orleans, Louisiana. Arch Dermatol. 2007;143:1393-1398.
2. Kulthanan K, Jiamton S, Thumpimukvatana N, et al. Chronic idiopathic urticaria: prevalence and clinical course. J Dermatol. 2007;34:294-301.
3. Stanway AD, Cohen SN, Chen C, et al. H1-antihistamines for chronic urticaria. Cochrane Database Syst Rev. 2008(2):CD006137.-
4. Grattan CE, Humphreys F. British Association of Dermatologists Therapy Guidelines and Audit Subcommittee. Guidelines for evaluation and management of urticaria in adults and children. Br J Dermatol. 2007;157:1116-1123.
5. Kozel MM, Mekkes JR, Bossuyt PM, et al. Natural course of physical and chronic urticaria and angioedema in 220 patients. J Am Acad Dermatol. 2001;45:387-391.
6. Nettis E, Colanardi MC, Soccio AL, et al. Double-blind, placebo-controlled study of sublingual immunotherapy in patients with latex-induced urticaria: a 12-month study. Br J Dermatol. 2007;156:674-681.
7. Lee EE, Maibach HI. Treatment of urticaria. An evidence-based evaluation of antihistamines. Am J Clin Dermatol. 2001;2:27-32.
8. Humphreys F, Hunter JA. The characteristics of urticaria in 390 patients. Br J Dermatol. 1998;138:635-638.
9. Serhat Inaloz H, Ozturk S, Akcali C, et al. Low-dose and short-term cyclosporine treatment in patients with chronic idiopathic urticaria: A clinical and immunological evaluation. J Dermatol. 2008;35:276-282.
10. Erbagci Z. The leukotriene receptor antagonist montelukast in the treatment of chronic idiopathic urticaria. A single-blind, placebo-controlled, crossover clinical study. J Allergy Clin Immunol. 2002;110:484-488.
11. Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med. 2005;165:1637-1642.
12. Yusuf S, Teo KK, Pogue J, et al. ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008;358:1547-1559.
13. Joint Task Force on Practice Parameters; American Academy of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. The diagnosis and management of anaphylaxis: an updated practice parameter. J Allergy Clin Immunol. 2005;115(3 Suppl 2):s483-s523.
14. Bas M, Kirchhartz N, Hochfeld J, et al. Potential role of vasomotor effects of fibrinogen in bradykinin-induced angioedema. J Allergy Clin Immunol. 2008;121:969e2-975e2.
15. Sica DA, Black HR. Angioedema in heart failure: occurrence with ACE inhibitors and safety of angiotensin receptor blocker therapy. Congest Heart Fail. 2002;8:334-341.
16. Banerji A, Clark S, Blanda M, et al. Multicenter study of patients with angiotensin-converting enzyme inhibitor-induced angioedema who present to the emergency department. Ann Allergy Asthma Immunol. 2008;100:327-332.
17. Cicardi M, Zingale LC, Zanichelli A, et al. The use of plasma-derived C1 inhibitor in the treatment of hereditary angioedema. Expert Opin Pharmacother. 2007;8:3173-3181.
18. Bork K, Bygum A, Hardt J. Benefits and risks of danazol in hereditary angioedema: a long-term survey of 118 patients. Ann Allergy Asthma Immunol. 2008;100:153-161.
19. US Food and Drug Administration. Advisory Committee Briefing Document. Kalbitor (ecallantide) for acute attacks of hereditary angioedema. Available at: http://www.fda.gov/ohrms/dockets/AC/09/briefing/2009-4413b1-03-Dyax.pdf. Accessed May 5, 2009.
20. Schneider L, Lumry W, Vegh A, et al. Critical role of kallikrein in hereditary angioedema pathogenesis: a clinical trial of ecallantide, a novel kallikrein inhibitor. J Allergy Clin Immunol. 2007;120:416-422.
21. Ellis Simonsen SM, van Orman ER, Hatch BE, et al. Cellulitis incidence in a defined population. Epidemiol Infect. 2006;134:293-299.
22. Stevens DL, Bisno AL, Chambers HF, et al. Infectious Diseases Society of America. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin Infect Dis. 2005;41:1373-1406.
23. Morris A. What are the benefits of treatments? Cellulitis and erysipelas. BMJ Clin Evid. 2005;13:2066-2069.
24. Murray H, Stiell I, Wells G. Treatment failure in emergency department patients with cellulitis. CJEM. 2005;7:228-234.
25. Meier DE, Nkor SK, Aasa D, et al. Prospective randomized comparison of two preoperative skin preparation techniques in a developing world country. World J Surg. 2001;25:441-443.
26. Giordano PA, Elston D, Akinlade BK, et al. Cefdinir vs. cephalexin for mild to moderate uncomplicated skin and skin structure infections in adolescents and adults. Curr Med Res Opin. 2006;22:2419-2428.
27. Halpern J, Holder R, Langford NJ. Ethnicity and other risk factors for acute lower limb cellulitis: a U.K.-based prospective case-control study. Br J Dermatol. 2008;158:1288-1292.
28. Lin YT, Lu PW. Retrospective study of pediatric facial cellulitis of odontogenic origin. Pediatr Infect Dis J. 2006;25:339-342.
29. Rajendran PM, Young D, Maurer T, et al. Randomized, double-blind, placebo-controlled trial of cephalexin for treatment of uncomplicated skin abscesses in a population at risk for community-acquired methicillin-resistant Staphylococcus aureus infection. Antimicrob Agents Chemo. 2007;51:4044-4048.
30. Schneck J, Fagot JP, Sekula P, et al. Effects of treatments on the mortality of Stevens-Johnson syndrome and toxic epidermal necrolysis: a retrospective study on patients included in the prospective EuroSCAR Study. J Am Acad Dermatol. 2008;58:33-40.
31. Chan HL, Stern RS, Arndt KA, et al. The incidence of erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis. A population-based study with particular reference to reactions caused by drugs among outpatients. Arch Dermatol. 1990;126:43-47.
32. Hazin R, Ibrahimi OA, Hazin MI, et al. Stevens-Johnson syndrome: pathogenesis, diagnosis, and management. Ann Med, 2008;40:129-138.
33. Chung WH, Hung SI, Hong HS, et al. Medical genetics: a marker for Stevens-Johnson syndrome. Nature. 2004;428:486.-
34. Jette N, Hemming K, Hutton JL, et al. Topiramate add-on for drug-resistant partial epilepsy. Cochrane Database Syst Rev. 2008;(3):DC001417.-
35. Gürcan HM, Ahmed AR. Efficacy of various intravenous immunoglobulin therapy protocols in auto-immune and chronic inflammatory disorders. Ann Pharmacother. 2007;41:812-523.
36. Strom BL, Carson JL, Halpern AC, et al. A population-based study of Stevens-Johnson syndrome. Incidence and antecedent drug exposures. Arch Dermatol. 1991;127:831-838.
37. Gravante G, Delogu D, Marianetti M, et al. Toxic epidermal necrolysis and Steven-Johnson syndrome: 11-years experience and outcome. Eur Rev Med Pharmacol Sci. 2007;11:119-127.
1. Noe R, Cohen AL, Lederman E, et al. Skin disorders among construction workers following Hurricane Katrina and Hurricane Rita: an outbreak investigation in New Orleans, Louisiana. Arch Dermatol. 2007;143:1393-1398.
2. Kulthanan K, Jiamton S, Thumpimukvatana N, et al. Chronic idiopathic urticaria: prevalence and clinical course. J Dermatol. 2007;34:294-301.
3. Stanway AD, Cohen SN, Chen C, et al. H1-antihistamines for chronic urticaria. Cochrane Database Syst Rev. 2008(2):CD006137.-
4. Grattan CE, Humphreys F. British Association of Dermatologists Therapy Guidelines and Audit Subcommittee. Guidelines for evaluation and management of urticaria in adults and children. Br J Dermatol. 2007;157:1116-1123.
5. Kozel MM, Mekkes JR, Bossuyt PM, et al. Natural course of physical and chronic urticaria and angioedema in 220 patients. J Am Acad Dermatol. 2001;45:387-391.
6. Nettis E, Colanardi MC, Soccio AL, et al. Double-blind, placebo-controlled study of sublingual immunotherapy in patients with latex-induced urticaria: a 12-month study. Br J Dermatol. 2007;156:674-681.
7. Lee EE, Maibach HI. Treatment of urticaria. An evidence-based evaluation of antihistamines. Am J Clin Dermatol. 2001;2:27-32.
8. Humphreys F, Hunter JA. The characteristics of urticaria in 390 patients. Br J Dermatol. 1998;138:635-638.
9. Serhat Inaloz H, Ozturk S, Akcali C, et al. Low-dose and short-term cyclosporine treatment in patients with chronic idiopathic urticaria: A clinical and immunological evaluation. J Dermatol. 2008;35:276-282.
10. Erbagci Z. The leukotriene receptor antagonist montelukast in the treatment of chronic idiopathic urticaria. A single-blind, placebo-controlled, crossover clinical study. J Allergy Clin Immunol. 2002;110:484-488.
11. Kostis JB, Kim HJ, Rusnak J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med. 2005;165:1637-1642.
12. Yusuf S, Teo KK, Pogue J, et al. ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008;358:1547-1559.
13. Joint Task Force on Practice Parameters; American Academy of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. The diagnosis and management of anaphylaxis: an updated practice parameter. J Allergy Clin Immunol. 2005;115(3 Suppl 2):s483-s523.
14. Bas M, Kirchhartz N, Hochfeld J, et al. Potential role of vasomotor effects of fibrinogen in bradykinin-induced angioedema. J Allergy Clin Immunol. 2008;121:969e2-975e2.
15. Sica DA, Black HR. Angioedema in heart failure: occurrence with ACE inhibitors and safety of angiotensin receptor blocker therapy. Congest Heart Fail. 2002;8:334-341.
16. Banerji A, Clark S, Blanda M, et al. Multicenter study of patients with angiotensin-converting enzyme inhibitor-induced angioedema who present to the emergency department. Ann Allergy Asthma Immunol. 2008;100:327-332.
17. Cicardi M, Zingale LC, Zanichelli A, et al. The use of plasma-derived C1 inhibitor in the treatment of hereditary angioedema. Expert Opin Pharmacother. 2007;8:3173-3181.
18. Bork K, Bygum A, Hardt J. Benefits and risks of danazol in hereditary angioedema: a long-term survey of 118 patients. Ann Allergy Asthma Immunol. 2008;100:153-161.
19. US Food and Drug Administration. Advisory Committee Briefing Document. Kalbitor (ecallantide) for acute attacks of hereditary angioedema. Available at: http://www.fda.gov/ohrms/dockets/AC/09/briefing/2009-4413b1-03-Dyax.pdf. Accessed May 5, 2009.
20. Schneider L, Lumry W, Vegh A, et al. Critical role of kallikrein in hereditary angioedema pathogenesis: a clinical trial of ecallantide, a novel kallikrein inhibitor. J Allergy Clin Immunol. 2007;120:416-422.
21. Ellis Simonsen SM, van Orman ER, Hatch BE, et al. Cellulitis incidence in a defined population. Epidemiol Infect. 2006;134:293-299.
22. Stevens DL, Bisno AL, Chambers HF, et al. Infectious Diseases Society of America. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin Infect Dis. 2005;41:1373-1406.
23. Morris A. What are the benefits of treatments? Cellulitis and erysipelas. BMJ Clin Evid. 2005;13:2066-2069.
24. Murray H, Stiell I, Wells G. Treatment failure in emergency department patients with cellulitis. CJEM. 2005;7:228-234.
25. Meier DE, Nkor SK, Aasa D, et al. Prospective randomized comparison of two preoperative skin preparation techniques in a developing world country. World J Surg. 2001;25:441-443.
26. Giordano PA, Elston D, Akinlade BK, et al. Cefdinir vs. cephalexin for mild to moderate uncomplicated skin and skin structure infections in adolescents and adults. Curr Med Res Opin. 2006;22:2419-2428.
27. Halpern J, Holder R, Langford NJ. Ethnicity and other risk factors for acute lower limb cellulitis: a U.K.-based prospective case-control study. Br J Dermatol. 2008;158:1288-1292.
28. Lin YT, Lu PW. Retrospective study of pediatric facial cellulitis of odontogenic origin. Pediatr Infect Dis J. 2006;25:339-342.
29. Rajendran PM, Young D, Maurer T, et al. Randomized, double-blind, placebo-controlled trial of cephalexin for treatment of uncomplicated skin abscesses in a population at risk for community-acquired methicillin-resistant Staphylococcus aureus infection. Antimicrob Agents Chemo. 2007;51:4044-4048.
30. Schneck J, Fagot JP, Sekula P, et al. Effects of treatments on the mortality of Stevens-Johnson syndrome and toxic epidermal necrolysis: a retrospective study on patients included in the prospective EuroSCAR Study. J Am Acad Dermatol. 2008;58:33-40.
31. Chan HL, Stern RS, Arndt KA, et al. The incidence of erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis. A population-based study with particular reference to reactions caused by drugs among outpatients. Arch Dermatol. 1990;126:43-47.
32. Hazin R, Ibrahimi OA, Hazin MI, et al. Stevens-Johnson syndrome: pathogenesis, diagnosis, and management. Ann Med, 2008;40:129-138.
33. Chung WH, Hung SI, Hong HS, et al. Medical genetics: a marker for Stevens-Johnson syndrome. Nature. 2004;428:486.-
34. Jette N, Hemming K, Hutton JL, et al. Topiramate add-on for drug-resistant partial epilepsy. Cochrane Database Syst Rev. 2008;(3):DC001417.-
35. Gürcan HM, Ahmed AR. Efficacy of various intravenous immunoglobulin therapy protocols in auto-immune and chronic inflammatory disorders. Ann Pharmacother. 2007;41:812-523.
36. Strom BL, Carson JL, Halpern AC, et al. A population-based study of Stevens-Johnson syndrome. Incidence and antecedent drug exposures. Arch Dermatol. 1991;127:831-838.
37. Gravante G, Delogu D, Marianetti M, et al. Toxic epidermal necrolysis and Steven-Johnson syndrome: 11-years experience and outcome. Eur Rev Med Pharmacol Sci. 2007;11:119-127.