User login
Primary Cutaneous Apocrine Carcinoma Arising Within a Nevus Sebaceus
Nevus sebaceus (NS) is a benign hair follicle neoplasm present in approximately 1.3% of the population, typically involving the scalp, neck, or face.1 These lesions usually are present at birth or identified soon after, during the first year. They present as a yellowish hairless patch or plaque but can develop a more papillomatous appearance, especially after puberty. Historically, the concern with NS was its tendency to transform into basal cell carcinoma (BCC), which prompted surgical excision of the lesion during childhood. This theory has been discounted more recently, as further research has suggested that what was once thought to be BCC may have been confused with the similarly appearing trichoblastoma; however, malignant transformation of NS does still occur, with BCC still being the most common.2 We present the case of a long-standing NS with rare transformation to apocrine carcinoma.
Case Report
A 76-year-old woman presented with several new lesions within a previously diagnosed NS. She reported having the large plaque for as long as she could recall but reported that several new growths developed within the plaque over the last 2 months, slowly increasing in size. She reported a prior biopsy within the growth several years prior, which she described as an irritated seborrheic keratosis.
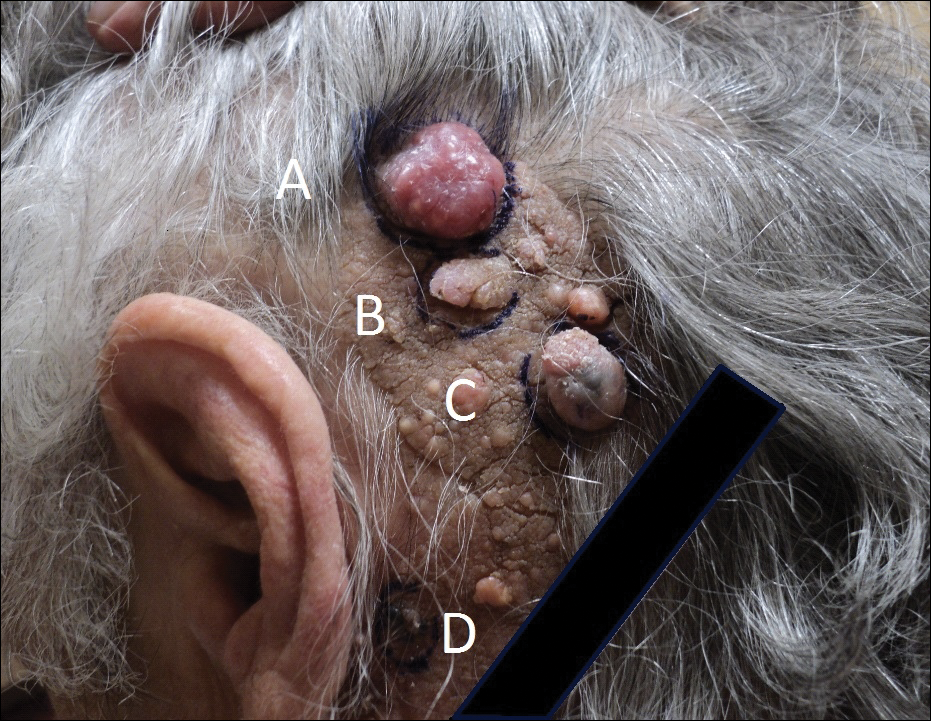
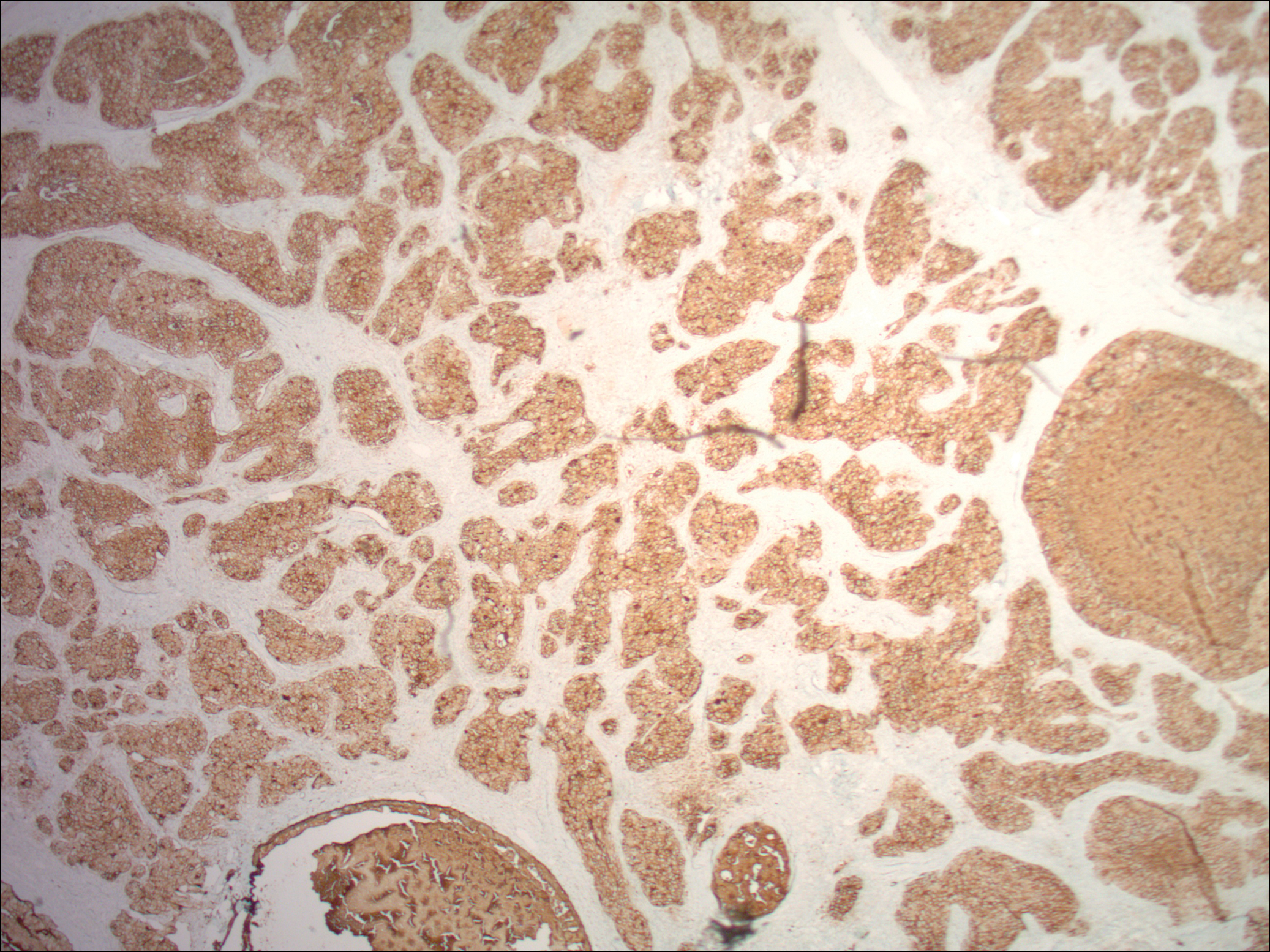
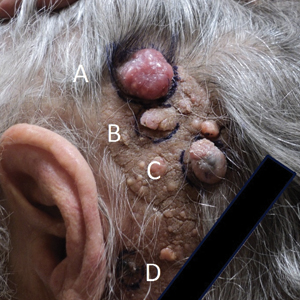
Physical examination demonstrated 4 distinct lesions within the flesh-colored, verrucous plaque located on the left side of the temporal scalp (Figure 1). The first lesion was a 2.5-cm pearly, pink, exophytic tumor (labeled as A in Figure 1). The next 2 lesions were brown, pedunculated, verrucous papules (labeled as B and C in Figure 1). The last lesion was a purple papule (labeled as D in Figure 1). Four shave biopsies were performed for histologic analysis of the lesions. Lesions B, C, and D were consistent with trichoblastomas, as pathology showed basaloid epithelial tumors that displayed primitive follicular structures, areas of stromal induction, and some pigmentation. Lesion A, originally thought to be suspicious for a BCC, was determined to be a primary cutaneous apocrine adenocarcinoma upon pathologic review. The pathology showed a dermal tumor displaying solid and tubular areas with decapitation secretion. Nuclear pleomorphism and mitoses were present (Figure 2), and staining for carcinoembryonic antigen was positive (Figure 3). Immunoreactivity with epithelial membrane antigen and cytokeratin 7 was noted as well as focal positivity for mammaglobin. Primary apocrine carcinoma was favored over metastatic carcinoma due to the location of the lesion within an NS along with a negative history of internal malignancy. Dermatopathology recommended complete removal of all lesions within the NS.

Upon discussing biopsy results and recommendations with our patient, she agreed to undergo excision with intraoperative pathology by a plastic surgeon within our practice to ensure clear margins. The surgical defect following excision was sizeable and closed utilizing a rhomboid flap, full-thickness skin graft, and a split-thickness skin graft. At surgical follow-up, she was doing well and there have been no signs of local recurrence for 10 months since excision.
Comment
Presentation
Nevus sebaceus is the most common adnexal tumor and is classified as a benign congenital hair follicle tumor that is located most commonly on the scalp but also occurs on the face and neck.1 The lesions usually are present at birth but also can develop during the first year of life.2 Diagnosis may be later, during adolescence, when patients seek medical attention during the lesion’s rapid growth phase.1 Nevus sebaceus also is known as an organoid nevus because it may contain all components of the skin. It was originally identified by Jadassohn in 1895.3 It presents as a yellowish, smooth, hairless patch or plaque in prepubertal patients. During adolescence, the lesion typically becomes more yellowish, as well as papillomatous, scaly, or warty. The reported incidence of NS is 0.05% to 1% in dermatology patients.2
Differential
Nevus sebaceus also is a component of several syndromes that should be kept in mind, including Schimmelpenning-Feuerstein-Mims syndrome, which presents with neurologic, skeletal, genitourinary, cardiovascular, and ophthalmic disorders, in addition to cutaneous features. Others include phacomatosis pigmentokeratotica, didmyosis aplasticosebacea, SCALP syndrome (sebaceus nevus, central nervous system malformations, aplasia cutis congenita, limbal dermoid, and pigmented nevus), and more.4,5
Etiology
The etiology of NS has not been completely determined. One study that evaluated 44 NS tissue samples suggested the presence of human papillomavirus (HPV) in NS formation, finding that 82% of NS lesions studied contained HPV DNA. From these results, Carlson et al6 suggested a possible maternal transmission of HPV and infection of ectodermal cells as a potential cause of NS; however, this hypothesis was soon challenged by a study that showed a complete absence of HPV in 16 samples via histological evaluation and polymerase chain reaction for a broad range of HPV types.7 There were investigations into a patched (PTCH) deletion as the cause of NS and thus explained the historically high rate of secondary BCC.8 Further studies showed no mutations at the PTCH locus in trichoblastomas or other tumors arising from NS.9,10
More recent studies have recognized HRAS and KRAS mutations as a causative factor in NS.11 Nevus sebaceus belongs to a group of syndromes resulting from lethal mutations that survive via mosaicism. Nevus sebaceus is caused by postzygotic HRAS or KRAS mutations and is known as a mosaic RASopathy.12 In fact, there is growing evidence to suggest that other nevoid proliferations including keratinocytic epidermal nevi and melanocytic nevi also fall into the spectrum of mosaic RASopathies.13
Staging
There are 3 clinical stages of NS, originally described by Mehregan and Pinkus.14 In stage I (historically known as the infantile stage), the lesion presents as a yellow to pink, smooth, hairless patch. Histologic features include immature hair follicles and hypoplastic sebaceous glands. In stage II (also known as the puberty stage), the lesion becomes more pronounced. Firmer plaques can develop with hyperkeratosis. Hormonal changes cause sebaceous glands to develop, accompanied by epidermal hyperplasia and maturation of apocrine glands. Stage III (the tumoral stage) is a period that various neoplasms have the highest likelihood of occurring. Nevus sebaceus in an adolescent or adult demonstrates mature adnexal structures and greater epidermal hyperplasia.2,4,15
Malignancy
By virtue of these stages of NS development, malignant transformation is expected most often during stage III. However, cases have been reported of malignant tumor development in NS in children before puberty. Two case reports described a 7-year-old boy and a 10-year-old boy diagnosed with a BCC arising from an NS.16,17 However, secondary BCC formation before 16 years of age is rare. Basal cell carcinoma arising from an NS has been commonly reported and is the most common malignant neoplasm in NS (1.1%).2,3 However, the most common neoplasm overall is trichoblastoma (7.4%). The second most common tumor was syringocystadenoma papilliferum, occurring in approximately 5.2% of NS cases. The neoplasm rate in NS was found to be proportional to the patient age.2,18 Multiple studies have shown the overall rate of secondary neoplasms in NS to be 13% to 21.4%, with malignant tumors composing 0.8% to 2.5%.2,15,19 Other neoplasms that have been reported include keratoacanthoma, trichilemmoma, sebaceoma, nevocellular nevus, squamous cell carcinoma, adnexal carcinoma, apocrine adenocarcinoma, and malignant melanoma.19-21
It is argued that the reported rate of BCC formation is overestimated, as prior studies incorrectly labeled trichoblastomas as BCCs. In fact, the largest studies of NS from the 1990s revealed lower rates of malignant secondary tumors than previously determined.4
The identification of apocrine adenocarcinoma tumors arising from NS is exceedingly rare. A study performed by Cribier et al19 in 2000 retrospect
Histopathology
Histopathologic examination reveals considerable variation in morphology, and an underlying pattern has been difficult to recognize. Unfortunately, some authors have concluded that the diagnosis of apocrine carcinoma is relatively subjective.26 Robson et al26 identified 3 general architectural patterns: tubular, tubulopapillary, and solid. Tubular structures consisted of glands and ducts lined by a single or multilayered epithelium. Tubulopapillary architecture was characterized by epithelium forming papillary folds without a fibrovascular core. The solid morphology showed sheets of cells with limited ductal or tubular formation.26 The most specific criteria of these apocrine carcinomas are identification of decapitation secretion, periodic acid–Schiff–positive diastase-resistant material present in the cells or lumen, and positive immunostaining for gross cystic disease fluid protein-15.27
Robson et al26 reported estrogen receptor positivity and androgen receptor positivity in 62% and 64% of 24 primary apocrine carcinoma cases, respectively. However, whether these markers are as common in NS-related apocrine carcinomas has yet to be noted in the literature. One study reports a case of apocrine carcinoma from NS with positive staining for human epidermal growth factor-2, a cell membrane receptor tyrosine kinase commonly investigated in breast cancers and extramammary Paget disease.22
These apocrine carcinomas do have the potential for lymphatic metastasis, as seen with multiple studies. Domingo and Helwig21 identified regional lymph node metastasis in 2 of its 4 apocrine carcinoma patients. Robson et al26 reported lymphovascular invasion in 4 cases and perineural invasion in 2 of 24 patients studied. However, even in the context of recurrence and regional metastasis, the prognosis was good and seldom fatal.26
Treatment
The most effective treatment of NS is excision of dermal and epidermal components. Excision should be completed with a minimum of 2- to 3-mm margins and full thickness down to the underlying supporting fat.28 Historically, the practice of prophylactic excision of NS was supported by the potential for malignant transformation; however, early excision of NS may be less reasonable in light of these more recent studies showing lower incidence of BCC (0.8%), replaced by benign trichoblastomas.19 In the case of apocrine carcinoma development, excision is undoubtedly recommended, with unclear recommendations regarding further evaluation for metastasis.
Excision also may be favored for cosmetic purposes, given the visible regions where NS tends to develop. Chepla and Gosain29 argued that surgical intervention should be based on other factors such as location on the scalp, alopecia, and other issues affecting appearance and monitoring rather than incidence of malignant transformation. Close monitoring and biopsy of suspicious areas is a more conservative option.
Other therapies include CO2 laser, as demonstrated by Kiedrowicz et al,30 on linear NS in a patient with Schimmelpenning-Feuerstein-Mims syndrome.31 However, this approach is palliative and not effective in removing the entire lesion. Electrodesiccation and curettage and dermabrasion also are not good options for the same reason.4
Occurrence in Children
Nevus sebaceus in children, accompanied by other findings suggestive of epidermal nevus syndromes, should prompt further investigation. Schimmelpenning-Feuerstein-Mims syndrome includes major neurological abnormalities including hemimegalencephaly and seizures.32
Conclusion
Apocrine carcinomas are malignant neoplasms that may rarely arise within an NS. Their clinical identification is difficult and requires histopathologic evaluation. Upon recognition, prompt excision with tumor-free margins is recommended. As a rare entity, little data is available regarding its metastatic potential or overall survival rates. Further investigation is clearly necessary as new cases arise.
- Kamyab-Hesari K, Balochi K, Afshar N, et al. Clinicopathological study of 1016 consecutive adnexal skin tumors. Acta Med Iran. 2013;51:879-885.
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337.
- Ball EA, Hussain M, Moss AL. Squamous cell carcinoma and basal cell carcinoma arising in a naevus sebaceous of Jadassohn: case report and literature review. Clin Exp Dermatol. 2005;30:259-260.
- Moody MN, Landau JM, Goldberg LH. Nevus sebaceous revisited. Pediatr Dermatol. 2012;29:15-23.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22; quiz 23-24.
- Carlson JA, Cribier B, Nuovo G, et al. Epidermodysplasia verruciformis-associated and genital-mucosal high-risk human papillomavirus DNA are prevalent in nevus sebaceus of Jadassohn. J Am Acad Dermatol. 2008;59:279-294.
- Kim D, Benjamin LT, Sahoo MK, et al. Human papilloma virus is not prevalent in nevus sebaceus [published online November 14, 2013]. Pediatr Dermatol. 2014;31:326-330.
- Xin H, Matt D, Qin JZ, et al. The sebaceous nevus: a nevus with deletions of the PTCH gene. Cancer Res. 1999;59:1834-1836.
- Hafner C, Schmiemann V, Ruetten A, et al. PTCH mutations are not mainly involved in the pathogenesis of sporadic trichoblastomas. Hum Pathol. 2007;38:1496-1500.
- Takata M, Tojo M, Hatta N, et al. No evidence of deregulated patched-hedgehog signaling pathway in trichoblastomas and other tumors arising within nevus sebaceous. J Invest Dermatol. 2001;117:1666-1670.
- Levinsohn JL, Tian LC, Boyden LM, et al. Whole-exome sequencing reveals somatic mutations in HRAS and KRAS, which cause nevus sebaceus [published online October 25, 2012]. J Invest Dermatol. 2013;133:827-830.
- Happle R. Nevus sebaceus is a mosaic RASopathy. J Invest Dermatol. 2013;133:597-600.
- Luo S, Tsao H. Epidermal, sebaceous, and melanocytic nevoid proliferations are spectrums of mosaic RASopathies. J Invest Dermatol. 2014;134:2493-2496.
- Mehregan AH, Pinkus H. Life history of organoid nevi. special reference to nevus sebaceus of Jadassohn. Arch Dermatol. 1965;91:574-588.
- Muñoz-Pérez MA, García-Hernandez MJ, Ríos JJ, et al. Sebaceus naevi: a clinicopathologic study. J Eur Acad Dermatol Venereol. 2002;16:319-324.
- Altaykan A, Ersoy-Evans S, Erkin G, et al. Basal cell carcinoma arising in nevus sebaceous during childhood. Pediatr Dermatol. 2008;25:616-619.
- Turner CD, Shea CR, Rosoff PM. Basal cell carcinoma originating from a nevus sebaceus on the scalp of a 7-year-old boy. J Pediatr Hematol Oncol. 2001;23:247-249.
- Jaqueti G, Requena L, Sánchez Yus E. Trichoblastoma is the most common neoplasm developed in nevus sebaceus of Jadassohn: a clinicopathologic study of a series of 155 cases. Am J Dermatopathol. 2000;22:108-118.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2, pt 1):263-268.
- Paudel U, Jha A, Pokhrel DB, et al. Apocrine carcinoma developing in a naevus sebaceous of scalp. Kathmandu Univ Med J (KUMJ). 2012;10:103-105.
- Domingo J, Helwig EB. Malignant neoplasms associated with nevus sebaceus of Jadassohn. J Am Acad Dermatol. 1979;1:545-556.
- Tanese K, Wakabayashi A, Suzuki T, et al. Immunoexpression of human epidermal growth factor receptor-2 in apocrine carcinoma arising in naevus sebaceous, case report [published online August 23, 2009]. J Eur Acad Dermatol Venereol. 2010;24:360-362.
- Dalle S, Skowron F, Balme B, et al. Apocrine carcinoma developed in nevus sebaceus of Jadassohn. Eur J Dermatol. 2003;13:487-489.
- Jacyk WK, Requena L, Sánchez Yus E, et al. Tubular apocrine carcinoma arising in a nevus sebaceus of Jadassohn. Am J Dermatopathol. 1998;20:389-392.
- Ansai S, Koseki S, Hashimoto H, et al. A case of ductal sweat gland carcinoma connected to syringocystadenoma papilliferum arising in nevus sebaceus. J Cutan Pathol. 1994;21:557-563.
- Robson A, Lazar AJ, Ben Nagi J, et al. Primary cutaneous apocrine carcinoma: a clinico-pathologic analysis of 24 cases. Am J Surg Pathol. 2008;32:682-690.
- Paties C, Taccagni GL, Papotti M, et al. Apocrine carcinoma of the skin. a clinicopathologic, immunocytochemical, and ultrastructural study. Cancer. 1993;71:375-381.
- Davison SP, Khachemoune A, Yu D, et al. Nevus sebaceus of Jadassohn revisited with reconstruction options. Int J Dermatol. 2005;44:145-150.
- Chepla KJ, Gosain AK. Giant nevus sebaceus: definition, surgical techniques, and rationale for treatment. Plast Reconstr Surg. 2012;130:296E-304E.
- Kiedrowicz M, Kacalak-Rzepka A, Królicki A et al. Therapeutic effects of CO2 laser therapy of linear nevus sebaceous in the course of the Schimmelpenning-Feuerstein-Mims syndrome. Postepy Dermatol Allergol. 2013;30:320-323.
- Ashinoff R. Linear nevus sebaceus of Jadassohn treated with the carbon dioxide laser. Pediatr Dermatol. 1993;10:189-191.
- van de Warrenburg BP, van Gulik S, Renier WO, et al. The linear naevus sebaceus syndrome. Clin Neurol Neurosurg. 1998;100:126-132.
Nevus sebaceus (NS) is a benign hair follicle neoplasm present in approximately 1.3% of the population, typically involving the scalp, neck, or face.1 These lesions usually are present at birth or identified soon after, during the first year. They present as a yellowish hairless patch or plaque but can develop a more papillomatous appearance, especially after puberty. Historically, the concern with NS was its tendency to transform into basal cell carcinoma (BCC), which prompted surgical excision of the lesion during childhood. This theory has been discounted more recently, as further research has suggested that what was once thought to be BCC may have been confused with the similarly appearing trichoblastoma; however, malignant transformation of NS does still occur, with BCC still being the most common.2 We present the case of a long-standing NS with rare transformation to apocrine carcinoma.
Case Report
A 76-year-old woman presented with several new lesions within a previously diagnosed NS. She reported having the large plaque for as long as she could recall but reported that several new growths developed within the plaque over the last 2 months, slowly increasing in size. She reported a prior biopsy within the growth several years prior, which she described as an irritated seborrheic keratosis.
Physical examination demonstrated 4 distinct lesions within the flesh-colored, verrucous plaque located on the left side of the temporal scalp (Figure 1). The first lesion was a 2.5-cm pearly, pink, exophytic tumor (labeled as A in Figure 1). The next 2 lesions were brown, pedunculated, verrucous papules (labeled as B and C in Figure 1). The last lesion was a purple papule (labeled as D in Figure 1). Four shave biopsies were performed for histologic analysis of the lesions. Lesions B, C, and D were consistent with trichoblastomas, as pathology showed basaloid epithelial tumors that displayed primitive follicular structures, areas of stromal induction, and some pigmentation. Lesion A, originally thought to be suspicious for a BCC, was determined to be a primary cutaneous apocrine adenocarcinoma upon pathologic review. The pathology showed a dermal tumor displaying solid and tubular areas with decapitation secretion. Nuclear pleomorphism and mitoses were present (Figure 2), and staining for carcinoembryonic antigen was positive (Figure 3). Immunoreactivity with epithelial membrane antigen and cytokeratin 7 was noted as well as focal positivity for mammaglobin. Primary apocrine carcinoma was favored over metastatic carcinoma due to the location of the lesion within an NS along with a negative history of internal malignancy. Dermatopathology recommended complete removal of all lesions within the NS.
Upon discussing biopsy results and recommendations with our patient, she agreed to undergo excision with intraoperative pathology by a plastic surgeon within our practice to ensure clear margins. The surgical defect following excision was sizeable and closed utilizing a rhomboid flap, full-thickness skin graft, and a split-thickness skin graft. At surgical follow-up, she was doing well and there have been no signs of local recurrence for 10 months since excision.
Comment
Presentation
Nevus sebaceus is the most common adnexal tumor and is classified as a benign congenital hair follicle tumor that is located most commonly on the scalp but also occurs on the face and neck.1 The lesions usually are present at birth but also can develop during the first year of life.2 Diagnosis may be later, during adolescence, when patients seek medical attention during the lesion’s rapid growth phase.1 Nevus sebaceus also is known as an organoid nevus because it may contain all components of the skin. It was originally identified by Jadassohn in 1895.3 It presents as a yellowish, smooth, hairless patch or plaque in prepubertal patients. During adolescence, the lesion typically becomes more yellowish, as well as papillomatous, scaly, or warty. The reported incidence of NS is 0.05% to 1% in dermatology patients.2
Differential
Nevus sebaceus also is a component of several syndromes that should be kept in mind, including Schimmelpenning-Feuerstein-Mims syndrome, which presents with neurologic, skeletal, genitourinary, cardiovascular, and ophthalmic disorders, in addition to cutaneous features. Others include phacomatosis pigmentokeratotica, didmyosis aplasticosebacea, SCALP syndrome (sebaceus nevus, central nervous system malformations, aplasia cutis congenita, limbal dermoid, and pigmented nevus), and more.4,5
Etiology
The etiology of NS has not been completely determined. One study that evaluated 44 NS tissue samples suggested the presence of human papillomavirus (HPV) in NS formation, finding that 82% of NS lesions studied contained HPV DNA. From these results, Carlson et al6 suggested a possible maternal transmission of HPV and infection of ectodermal cells as a potential cause of NS; however, this hypothesis was soon challenged by a study that showed a complete absence of HPV in 16 samples via histological evaluation and polymerase chain reaction for a broad range of HPV types.7 There were investigations into a patched (PTCH) deletion as the cause of NS and thus explained the historically high rate of secondary BCC.8 Further studies showed no mutations at the PTCH locus in trichoblastomas or other tumors arising from NS.9,10
More recent studies have recognized HRAS and KRAS mutations as a causative factor in NS.11 Nevus sebaceus belongs to a group of syndromes resulting from lethal mutations that survive via mosaicism. Nevus sebaceus is caused by postzygotic HRAS or KRAS mutations and is known as a mosaic RASopathy.12 In fact, there is growing evidence to suggest that other nevoid proliferations including keratinocytic epidermal nevi and melanocytic nevi also fall into the spectrum of mosaic RASopathies.13
Staging
There are 3 clinical stages of NS, originally described by Mehregan and Pinkus.14 In stage I (historically known as the infantile stage), the lesion presents as a yellow to pink, smooth, hairless patch. Histologic features include immature hair follicles and hypoplastic sebaceous glands. In stage II (also known as the puberty stage), the lesion becomes more pronounced. Firmer plaques can develop with hyperkeratosis. Hormonal changes cause sebaceous glands to develop, accompanied by epidermal hyperplasia and maturation of apocrine glands. Stage III (the tumoral stage) is a period that various neoplasms have the highest likelihood of occurring. Nevus sebaceus in an adolescent or adult demonstrates mature adnexal structures and greater epidermal hyperplasia.2,4,15
Malignancy
By virtue of these stages of NS development, malignant transformation is expected most often during stage III. However, cases have been reported of malignant tumor development in NS in children before puberty. Two case reports described a 7-year-old boy and a 10-year-old boy diagnosed with a BCC arising from an NS.16,17 However, secondary BCC formation before 16 years of age is rare. Basal cell carcinoma arising from an NS has been commonly reported and is the most common malignant neoplasm in NS (1.1%).2,3 However, the most common neoplasm overall is trichoblastoma (7.4%). The second most common tumor was syringocystadenoma papilliferum, occurring in approximately 5.2% of NS cases. The neoplasm rate in NS was found to be proportional to the patient age.2,18 Multiple studies have shown the overall rate of secondary neoplasms in NS to be 13% to 21.4%, with malignant tumors composing 0.8% to 2.5%.2,15,19 Other neoplasms that have been reported include keratoacanthoma, trichilemmoma, sebaceoma, nevocellular nevus, squamous cell carcinoma, adnexal carcinoma, apocrine adenocarcinoma, and malignant melanoma.19-21
It is argued that the reported rate of BCC formation is overestimated, as prior studies incorrectly labeled trichoblastomas as BCCs. In fact, the largest studies of NS from the 1990s revealed lower rates of malignant secondary tumors than previously determined.4
The identification of apocrine adenocarcinoma tumors arising from NS is exceedingly rare. A study performed by Cribier et al19 in 2000 retrospect
Histopathology
Histopathologic examination reveals considerable variation in morphology, and an underlying pattern has been difficult to recognize. Unfortunately, some authors have concluded that the diagnosis of apocrine carcinoma is relatively subjective.26 Robson et al26 identified 3 general architectural patterns: tubular, tubulopapillary, and solid. Tubular structures consisted of glands and ducts lined by a single or multilayered epithelium. Tubulopapillary architecture was characterized by epithelium forming papillary folds without a fibrovascular core. The solid morphology showed sheets of cells with limited ductal or tubular formation.26 The most specific criteria of these apocrine carcinomas are identification of decapitation secretion, periodic acid–Schiff–positive diastase-resistant material present in the cells or lumen, and positive immunostaining for gross cystic disease fluid protein-15.27
Robson et al26 reported estrogen receptor positivity and androgen receptor positivity in 62% and 64% of 24 primary apocrine carcinoma cases, respectively. However, whether these markers are as common in NS-related apocrine carcinomas has yet to be noted in the literature. One study reports a case of apocrine carcinoma from NS with positive staining for human epidermal growth factor-2, a cell membrane receptor tyrosine kinase commonly investigated in breast cancers and extramammary Paget disease.22
These apocrine carcinomas do have the potential for lymphatic metastasis, as seen with multiple studies. Domingo and Helwig21 identified regional lymph node metastasis in 2 of its 4 apocrine carcinoma patients. Robson et al26 reported lymphovascular invasion in 4 cases and perineural invasion in 2 of 24 patients studied. However, even in the context of recurrence and regional metastasis, the prognosis was good and seldom fatal.26
Treatment
The most effective treatment of NS is excision of dermal and epidermal components. Excision should be completed with a minimum of 2- to 3-mm margins and full thickness down to the underlying supporting fat.28 Historically, the practice of prophylactic excision of NS was supported by the potential for malignant transformation; however, early excision of NS may be less reasonable in light of these more recent studies showing lower incidence of BCC (0.8%), replaced by benign trichoblastomas.19 In the case of apocrine carcinoma development, excision is undoubtedly recommended, with unclear recommendations regarding further evaluation for metastasis.
Excision also may be favored for cosmetic purposes, given the visible regions where NS tends to develop. Chepla and Gosain29 argued that surgical intervention should be based on other factors such as location on the scalp, alopecia, and other issues affecting appearance and monitoring rather than incidence of malignant transformation. Close monitoring and biopsy of suspicious areas is a more conservative option.
Other therapies include CO2 laser, as demonstrated by Kiedrowicz et al,30 on linear NS in a patient with Schimmelpenning-Feuerstein-Mims syndrome.31 However, this approach is palliative and not effective in removing the entire lesion. Electrodesiccation and curettage and dermabrasion also are not good options for the same reason.4
Occurrence in Children
Nevus sebaceus in children, accompanied by other findings suggestive of epidermal nevus syndromes, should prompt further investigation. Schimmelpenning-Feuerstein-Mims syndrome includes major neurological abnormalities including hemimegalencephaly and seizures.32
Conclusion
Apocrine carcinomas are malignant neoplasms that may rarely arise within an NS. Their clinical identification is difficult and requires histopathologic evaluation. Upon recognition, prompt excision with tumor-free margins is recommended. As a rare entity, little data is available regarding its metastatic potential or overall survival rates. Further investigation is clearly necessary as new cases arise.
Nevus sebaceus (NS) is a benign hair follicle neoplasm present in approximately 1.3% of the population, typically involving the scalp, neck, or face.1 These lesions usually are present at birth or identified soon after, during the first year. They present as a yellowish hairless patch or plaque but can develop a more papillomatous appearance, especially after puberty. Historically, the concern with NS was its tendency to transform into basal cell carcinoma (BCC), which prompted surgical excision of the lesion during childhood. This theory has been discounted more recently, as further research has suggested that what was once thought to be BCC may have been confused with the similarly appearing trichoblastoma; however, malignant transformation of NS does still occur, with BCC still being the most common.2 We present the case of a long-standing NS with rare transformation to apocrine carcinoma.
Case Report
A 76-year-old woman presented with several new lesions within a previously diagnosed NS. She reported having the large plaque for as long as she could recall but reported that several new growths developed within the plaque over the last 2 months, slowly increasing in size. She reported a prior biopsy within the growth several years prior, which she described as an irritated seborrheic keratosis.
Physical examination demonstrated 4 distinct lesions within the flesh-colored, verrucous plaque located on the left side of the temporal scalp (Figure 1). The first lesion was a 2.5-cm pearly, pink, exophytic tumor (labeled as A in Figure 1). The next 2 lesions were brown, pedunculated, verrucous papules (labeled as B and C in Figure 1). The last lesion was a purple papule (labeled as D in Figure 1). Four shave biopsies were performed for histologic analysis of the lesions. Lesions B, C, and D were consistent with trichoblastomas, as pathology showed basaloid epithelial tumors that displayed primitive follicular structures, areas of stromal induction, and some pigmentation. Lesion A, originally thought to be suspicious for a BCC, was determined to be a primary cutaneous apocrine adenocarcinoma upon pathologic review. The pathology showed a dermal tumor displaying solid and tubular areas with decapitation secretion. Nuclear pleomorphism and mitoses were present (Figure 2), and staining for carcinoembryonic antigen was positive (Figure 3). Immunoreactivity with epithelial membrane antigen and cytokeratin 7 was noted as well as focal positivity for mammaglobin. Primary apocrine carcinoma was favored over metastatic carcinoma due to the location of the lesion within an NS along with a negative history of internal malignancy. Dermatopathology recommended complete removal of all lesions within the NS.
Upon discussing biopsy results and recommendations with our patient, she agreed to undergo excision with intraoperative pathology by a plastic surgeon within our practice to ensure clear margins. The surgical defect following excision was sizeable and closed utilizing a rhomboid flap, full-thickness skin graft, and a split-thickness skin graft. At surgical follow-up, she was doing well and there have been no signs of local recurrence for 10 months since excision.
Comment
Presentation
Nevus sebaceus is the most common adnexal tumor and is classified as a benign congenital hair follicle tumor that is located most commonly on the scalp but also occurs on the face and neck.1 The lesions usually are present at birth but also can develop during the first year of life.2 Diagnosis may be later, during adolescence, when patients seek medical attention during the lesion’s rapid growth phase.1 Nevus sebaceus also is known as an organoid nevus because it may contain all components of the skin. It was originally identified by Jadassohn in 1895.3 It presents as a yellowish, smooth, hairless patch or plaque in prepubertal patients. During adolescence, the lesion typically becomes more yellowish, as well as papillomatous, scaly, or warty. The reported incidence of NS is 0.05% to 1% in dermatology patients.2
Differential
Nevus sebaceus also is a component of several syndromes that should be kept in mind, including Schimmelpenning-Feuerstein-Mims syndrome, which presents with neurologic, skeletal, genitourinary, cardiovascular, and ophthalmic disorders, in addition to cutaneous features. Others include phacomatosis pigmentokeratotica, didmyosis aplasticosebacea, SCALP syndrome (sebaceus nevus, central nervous system malformations, aplasia cutis congenita, limbal dermoid, and pigmented nevus), and more.4,5
Etiology
The etiology of NS has not been completely determined. One study that evaluated 44 NS tissue samples suggested the presence of human papillomavirus (HPV) in NS formation, finding that 82% of NS lesions studied contained HPV DNA. From these results, Carlson et al6 suggested a possible maternal transmission of HPV and infection of ectodermal cells as a potential cause of NS; however, this hypothesis was soon challenged by a study that showed a complete absence of HPV in 16 samples via histological evaluation and polymerase chain reaction for a broad range of HPV types.7 There were investigations into a patched (PTCH) deletion as the cause of NS and thus explained the historically high rate of secondary BCC.8 Further studies showed no mutations at the PTCH locus in trichoblastomas or other tumors arising from NS.9,10
More recent studies have recognized HRAS and KRAS mutations as a causative factor in NS.11 Nevus sebaceus belongs to a group of syndromes resulting from lethal mutations that survive via mosaicism. Nevus sebaceus is caused by postzygotic HRAS or KRAS mutations and is known as a mosaic RASopathy.12 In fact, there is growing evidence to suggest that other nevoid proliferations including keratinocytic epidermal nevi and melanocytic nevi also fall into the spectrum of mosaic RASopathies.13
Staging
There are 3 clinical stages of NS, originally described by Mehregan and Pinkus.14 In stage I (historically known as the infantile stage), the lesion presents as a yellow to pink, smooth, hairless patch. Histologic features include immature hair follicles and hypoplastic sebaceous glands. In stage II (also known as the puberty stage), the lesion becomes more pronounced. Firmer plaques can develop with hyperkeratosis. Hormonal changes cause sebaceous glands to develop, accompanied by epidermal hyperplasia and maturation of apocrine glands. Stage III (the tumoral stage) is a period that various neoplasms have the highest likelihood of occurring. Nevus sebaceus in an adolescent or adult demonstrates mature adnexal structures and greater epidermal hyperplasia.2,4,15
Malignancy
By virtue of these stages of NS development, malignant transformation is expected most often during stage III. However, cases have been reported of malignant tumor development in NS in children before puberty. Two case reports described a 7-year-old boy and a 10-year-old boy diagnosed with a BCC arising from an NS.16,17 However, secondary BCC formation before 16 years of age is rare. Basal cell carcinoma arising from an NS has been commonly reported and is the most common malignant neoplasm in NS (1.1%).2,3 However, the most common neoplasm overall is trichoblastoma (7.4%). The second most common tumor was syringocystadenoma papilliferum, occurring in approximately 5.2% of NS cases. The neoplasm rate in NS was found to be proportional to the patient age.2,18 Multiple studies have shown the overall rate of secondary neoplasms in NS to be 13% to 21.4%, with malignant tumors composing 0.8% to 2.5%.2,15,19 Other neoplasms that have been reported include keratoacanthoma, trichilemmoma, sebaceoma, nevocellular nevus, squamous cell carcinoma, adnexal carcinoma, apocrine adenocarcinoma, and malignant melanoma.19-21
It is argued that the reported rate of BCC formation is overestimated, as prior studies incorrectly labeled trichoblastomas as BCCs. In fact, the largest studies of NS from the 1990s revealed lower rates of malignant secondary tumors than previously determined.4
The identification of apocrine adenocarcinoma tumors arising from NS is exceedingly rare. A study performed by Cribier et al19 in 2000 retrospect
Histopathology
Histopathologic examination reveals considerable variation in morphology, and an underlying pattern has been difficult to recognize. Unfortunately, some authors have concluded that the diagnosis of apocrine carcinoma is relatively subjective.26 Robson et al26 identified 3 general architectural patterns: tubular, tubulopapillary, and solid. Tubular structures consisted of glands and ducts lined by a single or multilayered epithelium. Tubulopapillary architecture was characterized by epithelium forming papillary folds without a fibrovascular core. The solid morphology showed sheets of cells with limited ductal or tubular formation.26 The most specific criteria of these apocrine carcinomas are identification of decapitation secretion, periodic acid–Schiff–positive diastase-resistant material present in the cells or lumen, and positive immunostaining for gross cystic disease fluid protein-15.27
Robson et al26 reported estrogen receptor positivity and androgen receptor positivity in 62% and 64% of 24 primary apocrine carcinoma cases, respectively. However, whether these markers are as common in NS-related apocrine carcinomas has yet to be noted in the literature. One study reports a case of apocrine carcinoma from NS with positive staining for human epidermal growth factor-2, a cell membrane receptor tyrosine kinase commonly investigated in breast cancers and extramammary Paget disease.22
These apocrine carcinomas do have the potential for lymphatic metastasis, as seen with multiple studies. Domingo and Helwig21 identified regional lymph node metastasis in 2 of its 4 apocrine carcinoma patients. Robson et al26 reported lymphovascular invasion in 4 cases and perineural invasion in 2 of 24 patients studied. However, even in the context of recurrence and regional metastasis, the prognosis was good and seldom fatal.26
Treatment
The most effective treatment of NS is excision of dermal and epidermal components. Excision should be completed with a minimum of 2- to 3-mm margins and full thickness down to the underlying supporting fat.28 Historically, the practice of prophylactic excision of NS was supported by the potential for malignant transformation; however, early excision of NS may be less reasonable in light of these more recent studies showing lower incidence of BCC (0.8%), replaced by benign trichoblastomas.19 In the case of apocrine carcinoma development, excision is undoubtedly recommended, with unclear recommendations regarding further evaluation for metastasis.
Excision also may be favored for cosmetic purposes, given the visible regions where NS tends to develop. Chepla and Gosain29 argued that surgical intervention should be based on other factors such as location on the scalp, alopecia, and other issues affecting appearance and monitoring rather than incidence of malignant transformation. Close monitoring and biopsy of suspicious areas is a more conservative option.
Other therapies include CO2 laser, as demonstrated by Kiedrowicz et al,30 on linear NS in a patient with Schimmelpenning-Feuerstein-Mims syndrome.31 However, this approach is palliative and not effective in removing the entire lesion. Electrodesiccation and curettage and dermabrasion also are not good options for the same reason.4
Occurrence in Children
Nevus sebaceus in children, accompanied by other findings suggestive of epidermal nevus syndromes, should prompt further investigation. Schimmelpenning-Feuerstein-Mims syndrome includes major neurological abnormalities including hemimegalencephaly and seizures.32
Conclusion
Apocrine carcinomas are malignant neoplasms that may rarely arise within an NS. Their clinical identification is difficult and requires histopathologic evaluation. Upon recognition, prompt excision with tumor-free margins is recommended. As a rare entity, little data is available regarding its metastatic potential or overall survival rates. Further investigation is clearly necessary as new cases arise.
- Kamyab-Hesari K, Balochi K, Afshar N, et al. Clinicopathological study of 1016 consecutive adnexal skin tumors. Acta Med Iran. 2013;51:879-885.
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337.
- Ball EA, Hussain M, Moss AL. Squamous cell carcinoma and basal cell carcinoma arising in a naevus sebaceous of Jadassohn: case report and literature review. Clin Exp Dermatol. 2005;30:259-260.
- Moody MN, Landau JM, Goldberg LH. Nevus sebaceous revisited. Pediatr Dermatol. 2012;29:15-23.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22; quiz 23-24.
- Carlson JA, Cribier B, Nuovo G, et al. Epidermodysplasia verruciformis-associated and genital-mucosal high-risk human papillomavirus DNA are prevalent in nevus sebaceus of Jadassohn. J Am Acad Dermatol. 2008;59:279-294.
- Kim D, Benjamin LT, Sahoo MK, et al. Human papilloma virus is not prevalent in nevus sebaceus [published online November 14, 2013]. Pediatr Dermatol. 2014;31:326-330.
- Xin H, Matt D, Qin JZ, et al. The sebaceous nevus: a nevus with deletions of the PTCH gene. Cancer Res. 1999;59:1834-1836.
- Hafner C, Schmiemann V, Ruetten A, et al. PTCH mutations are not mainly involved in the pathogenesis of sporadic trichoblastomas. Hum Pathol. 2007;38:1496-1500.
- Takata M, Tojo M, Hatta N, et al. No evidence of deregulated patched-hedgehog signaling pathway in trichoblastomas and other tumors arising within nevus sebaceous. J Invest Dermatol. 2001;117:1666-1670.
- Levinsohn JL, Tian LC, Boyden LM, et al. Whole-exome sequencing reveals somatic mutations in HRAS and KRAS, which cause nevus sebaceus [published online October 25, 2012]. J Invest Dermatol. 2013;133:827-830.
- Happle R. Nevus sebaceus is a mosaic RASopathy. J Invest Dermatol. 2013;133:597-600.
- Luo S, Tsao H. Epidermal, sebaceous, and melanocytic nevoid proliferations are spectrums of mosaic RASopathies. J Invest Dermatol. 2014;134:2493-2496.
- Mehregan AH, Pinkus H. Life history of organoid nevi. special reference to nevus sebaceus of Jadassohn. Arch Dermatol. 1965;91:574-588.
- Muñoz-Pérez MA, García-Hernandez MJ, Ríos JJ, et al. Sebaceus naevi: a clinicopathologic study. J Eur Acad Dermatol Venereol. 2002;16:319-324.
- Altaykan A, Ersoy-Evans S, Erkin G, et al. Basal cell carcinoma arising in nevus sebaceous during childhood. Pediatr Dermatol. 2008;25:616-619.
- Turner CD, Shea CR, Rosoff PM. Basal cell carcinoma originating from a nevus sebaceus on the scalp of a 7-year-old boy. J Pediatr Hematol Oncol. 2001;23:247-249.
- Jaqueti G, Requena L, Sánchez Yus E. Trichoblastoma is the most common neoplasm developed in nevus sebaceus of Jadassohn: a clinicopathologic study of a series of 155 cases. Am J Dermatopathol. 2000;22:108-118.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2, pt 1):263-268.
- Paudel U, Jha A, Pokhrel DB, et al. Apocrine carcinoma developing in a naevus sebaceous of scalp. Kathmandu Univ Med J (KUMJ). 2012;10:103-105.
- Domingo J, Helwig EB. Malignant neoplasms associated with nevus sebaceus of Jadassohn. J Am Acad Dermatol. 1979;1:545-556.
- Tanese K, Wakabayashi A, Suzuki T, et al. Immunoexpression of human epidermal growth factor receptor-2 in apocrine carcinoma arising in naevus sebaceous, case report [published online August 23, 2009]. J Eur Acad Dermatol Venereol. 2010;24:360-362.
- Dalle S, Skowron F, Balme B, et al. Apocrine carcinoma developed in nevus sebaceus of Jadassohn. Eur J Dermatol. 2003;13:487-489.
- Jacyk WK, Requena L, Sánchez Yus E, et al. Tubular apocrine carcinoma arising in a nevus sebaceus of Jadassohn. Am J Dermatopathol. 1998;20:389-392.
- Ansai S, Koseki S, Hashimoto H, et al. A case of ductal sweat gland carcinoma connected to syringocystadenoma papilliferum arising in nevus sebaceus. J Cutan Pathol. 1994;21:557-563.
- Robson A, Lazar AJ, Ben Nagi J, et al. Primary cutaneous apocrine carcinoma: a clinico-pathologic analysis of 24 cases. Am J Surg Pathol. 2008;32:682-690.
- Paties C, Taccagni GL, Papotti M, et al. Apocrine carcinoma of the skin. a clinicopathologic, immunocytochemical, and ultrastructural study. Cancer. 1993;71:375-381.
- Davison SP, Khachemoune A, Yu D, et al. Nevus sebaceus of Jadassohn revisited with reconstruction options. Int J Dermatol. 2005;44:145-150.
- Chepla KJ, Gosain AK. Giant nevus sebaceus: definition, surgical techniques, and rationale for treatment. Plast Reconstr Surg. 2012;130:296E-304E.
- Kiedrowicz M, Kacalak-Rzepka A, Królicki A et al. Therapeutic effects of CO2 laser therapy of linear nevus sebaceous in the course of the Schimmelpenning-Feuerstein-Mims syndrome. Postepy Dermatol Allergol. 2013;30:320-323.
- Ashinoff R. Linear nevus sebaceus of Jadassohn treated with the carbon dioxide laser. Pediatr Dermatol. 1993;10:189-191.
- van de Warrenburg BP, van Gulik S, Renier WO, et al. The linear naevus sebaceus syndrome. Clin Neurol Neurosurg. 1998;100:126-132.
- Kamyab-Hesari K, Balochi K, Afshar N, et al. Clinicopathological study of 1016 consecutive adnexal skin tumors. Acta Med Iran. 2013;51:879-885.
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337.
- Ball EA, Hussain M, Moss AL. Squamous cell carcinoma and basal cell carcinoma arising in a naevus sebaceous of Jadassohn: case report and literature review. Clin Exp Dermatol. 2005;30:259-260.
- Moody MN, Landau JM, Goldberg LH. Nevus sebaceous revisited. Pediatr Dermatol. 2012;29:15-23.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22; quiz 23-24.
- Carlson JA, Cribier B, Nuovo G, et al. Epidermodysplasia verruciformis-associated and genital-mucosal high-risk human papillomavirus DNA are prevalent in nevus sebaceus of Jadassohn. J Am Acad Dermatol. 2008;59:279-294.
- Kim D, Benjamin LT, Sahoo MK, et al. Human papilloma virus is not prevalent in nevus sebaceus [published online November 14, 2013]. Pediatr Dermatol. 2014;31:326-330.
- Xin H, Matt D, Qin JZ, et al. The sebaceous nevus: a nevus with deletions of the PTCH gene. Cancer Res. 1999;59:1834-1836.
- Hafner C, Schmiemann V, Ruetten A, et al. PTCH mutations are not mainly involved in the pathogenesis of sporadic trichoblastomas. Hum Pathol. 2007;38:1496-1500.
- Takata M, Tojo M, Hatta N, et al. No evidence of deregulated patched-hedgehog signaling pathway in trichoblastomas and other tumors arising within nevus sebaceous. J Invest Dermatol. 2001;117:1666-1670.
- Levinsohn JL, Tian LC, Boyden LM, et al. Whole-exome sequencing reveals somatic mutations in HRAS and KRAS, which cause nevus sebaceus [published online October 25, 2012]. J Invest Dermatol. 2013;133:827-830.
- Happle R. Nevus sebaceus is a mosaic RASopathy. J Invest Dermatol. 2013;133:597-600.
- Luo S, Tsao H. Epidermal, sebaceous, and melanocytic nevoid proliferations are spectrums of mosaic RASopathies. J Invest Dermatol. 2014;134:2493-2496.
- Mehregan AH, Pinkus H. Life history of organoid nevi. special reference to nevus sebaceus of Jadassohn. Arch Dermatol. 1965;91:574-588.
- Muñoz-Pérez MA, García-Hernandez MJ, Ríos JJ, et al. Sebaceus naevi: a clinicopathologic study. J Eur Acad Dermatol Venereol. 2002;16:319-324.
- Altaykan A, Ersoy-Evans S, Erkin G, et al. Basal cell carcinoma arising in nevus sebaceous during childhood. Pediatr Dermatol. 2008;25:616-619.
- Turner CD, Shea CR, Rosoff PM. Basal cell carcinoma originating from a nevus sebaceus on the scalp of a 7-year-old boy. J Pediatr Hematol Oncol. 2001;23:247-249.
- Jaqueti G, Requena L, Sánchez Yus E. Trichoblastoma is the most common neoplasm developed in nevus sebaceus of Jadassohn: a clinicopathologic study of a series of 155 cases. Am J Dermatopathol. 2000;22:108-118.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2, pt 1):263-268.
- Paudel U, Jha A, Pokhrel DB, et al. Apocrine carcinoma developing in a naevus sebaceous of scalp. Kathmandu Univ Med J (KUMJ). 2012;10:103-105.
- Domingo J, Helwig EB. Malignant neoplasms associated with nevus sebaceus of Jadassohn. J Am Acad Dermatol. 1979;1:545-556.
- Tanese K, Wakabayashi A, Suzuki T, et al. Immunoexpression of human epidermal growth factor receptor-2 in apocrine carcinoma arising in naevus sebaceous, case report [published online August 23, 2009]. J Eur Acad Dermatol Venereol. 2010;24:360-362.
- Dalle S, Skowron F, Balme B, et al. Apocrine carcinoma developed in nevus sebaceus of Jadassohn. Eur J Dermatol. 2003;13:487-489.
- Jacyk WK, Requena L, Sánchez Yus E, et al. Tubular apocrine carcinoma arising in a nevus sebaceus of Jadassohn. Am J Dermatopathol. 1998;20:389-392.
- Ansai S, Koseki S, Hashimoto H, et al. A case of ductal sweat gland carcinoma connected to syringocystadenoma papilliferum arising in nevus sebaceus. J Cutan Pathol. 1994;21:557-563.
- Robson A, Lazar AJ, Ben Nagi J, et al. Primary cutaneous apocrine carcinoma: a clinico-pathologic analysis of 24 cases. Am J Surg Pathol. 2008;32:682-690.
- Paties C, Taccagni GL, Papotti M, et al. Apocrine carcinoma of the skin. a clinicopathologic, immunocytochemical, and ultrastructural study. Cancer. 1993;71:375-381.
- Davison SP, Khachemoune A, Yu D, et al. Nevus sebaceus of Jadassohn revisited with reconstruction options. Int J Dermatol. 2005;44:145-150.
- Chepla KJ, Gosain AK. Giant nevus sebaceus: definition, surgical techniques, and rationale for treatment. Plast Reconstr Surg. 2012;130:296E-304E.
- Kiedrowicz M, Kacalak-Rzepka A, Królicki A et al. Therapeutic effects of CO2 laser therapy of linear nevus sebaceous in the course of the Schimmelpenning-Feuerstein-Mims syndrome. Postepy Dermatol Allergol. 2013;30:320-323.
- Ashinoff R. Linear nevus sebaceus of Jadassohn treated with the carbon dioxide laser. Pediatr Dermatol. 1993;10:189-191.
- van de Warrenburg BP, van Gulik S, Renier WO, et al. The linear naevus sebaceus syndrome. Clin Neurol Neurosurg. 1998;100:126-132.
Practice Points
- Nevus sebaceus (NS) in the centrofacial region has been correlated with a higher risk for neurological abnormalities, including intellectual disability and seizures.
- Historically, basal cell carcinomas (BCCs) were considered a common occurrence arising from an NS, prompting prophylactic surgical excision of such lesions.
- More recently, it has been recognized that the most common tumor to arise from NS is trichoblastoma rather than BCC; in fact, BCC and other malignancies have been found to be relatively rare compared to their benign counterparts.
- In light of this discovery, observation of NS may be a more prudent course of treatment versus prophylactic surgical excision.
Imiquimod-Induced Hypopigmentation Following Treatment of Periungual Verruca Vulgaris
Imiquimod is derived from the imidazoquinoline family and works by activating both innate and adaptive immune pathways. Imiquimod binds to toll-like receptor 7 located on monocytes, macrophages, and dendritic cells,1 which allows nuclear factor κβ light chain enhancer of activated B cells to induce production of proinflammatory cytokines, including IFN-α and tumor necrosis factor α, as well as IL-1, IL-6, IL-8, IL-10, and IL-12.2 These proinflammatory cytokines play a role in the innate immunity, triggering upregulation of the adaptive immune pathway and activating type 1 helper T cells, cytotoxic T cells, and natural killer cells. These cells have antiviral and antitumoral effects that lend to their significance in coordinating innate and adaptive immune mechanisms.3 More specifically, imiquimod enhances dendritic cell migration to regional lymph nodes and induces apoptosis via activation of proapoptotic B-cell lymphoma 2 proteins.1,2 Imiquimod has been approved by the US Food and Drug Administration (FDA) to treat external genitalia and perianal condyloma acuminata, actinic keratoses (AKs), and superficial basal cell carcinoma (BCC). It often is used off label for antiviral or antitumoral therapy in Bowen disease, squamous cell carcinoma, lentigo maligna, vulvar intraepithelial neoplasia, molluscum contagiosum, common warts, and leishmaniasis.1,2 Imiquimod is generally well tolerated; erythema and irritation at the application site are the most common side effects, with pigmentary change being less common.
Case Report
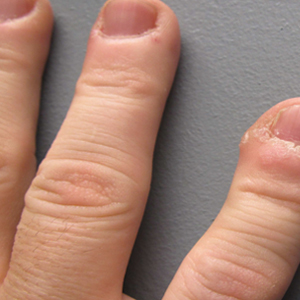
A 51-year-old man with a medical history of vitamin D deficiency, vitamin B12 deficiency, tinea pedis, and BCC presented with periungual verruca vulgaris on the right fifth digit and left thumb (Figure 1). The patient was prescribed imiquimod cream 5% to be applied 3 times weekly for 3 months. At 5-month follow-up the patient reported new-onset vitiligolike patches of depigmentation on the hands and feet that abruptly began 3 months after initiating treatment with imiquimod. On examination he had several depigmented patches with well-defined irregular borders on the bilateral dorsal hands and right foot as well as the right elbow (Figure 2). There was no personal or family history of vitiligo, thyroid disease, or autoimmune disease. Thyroid function studies and autoimmune panel were unremarkable. The patient also denied applying imiquimod to areas other than the periungual region of the right fifth digit and left thumb. He declined a biopsy of the lesions and was given a prescription for tacrolimus ointment 0.1% for twice-daily application. At 3-month follow-up the depigmented patches had spread. The patient is currently on 5-fluorouracil cream 5%. Despite loss of pigmentation, the periungual verruca vulgaris has persisted as well as depigmentation.

 and the right elbow (B).")
Comment
Imiquimod therapy is commonly used to treat conditions for which an antiviral or antitumor immune response is necessary for treatment and full resolution of skin conditions. It can yield positive results in conditions that are difficult to treat, such as periungual verruca vulgaris.4 The most common adverse effects of imiquimod include localized inflammation and application-site reactions. Pigment changes, though less common, also have been reported. From 1997 to 2003, 1257 cases of imiquimod adverse effects were reported to the FDA. There were 68 reported cases of pigmentary change, of which 51 documented vitiligo, hypopigmentation, or depigmentation. The others reported hyperpigmentation following imiquimod use.4 The imiquimod package insert lists application-site hypopigmentation as a possible adverse effect.5 Imiquimod-induced hypopigmentation and depigmentation have been reported in the peer-reviewed literature.4,6-14 Pigment loss has been reported in imiquimod treatment of condyloma acuminata, superficial BCC, nodular BCC, and extramammary Paget disease.6-8 Duration of therapy to onset of pigment loss ranged from 7 to 28 weeks.9 Imiquimod dosing varied among reported cases, ranging from 3 times weekly to daily application. Interestingly, hypopigmentation or depigmentation are not commonly associated with imiquimod use for the treatment of AKs, which Burnett and Kouba9 proposed may be due to the twice weekly imiquimod dosing regimen recommended by the FDA for the treatment of AK (below the minimum threshold for pigment loss). Our patient applied imiquimod cream 5% to periungual verruca vulgaris 3 times weekly for 3 months and may have developed vitiligolike depigmentation because he met this theoretical dosage threshold. Further research is necessary to confirm a dosage-related threshold for the development of depigmentation. Imiquimod-induced pigment loss has mainly been limited to the site of application.
Depigmentation was limited to the application site the majority of the time; however, depigmentation at adjacent sites has been reported.10 This finding was consistent with the proposed notion that cytokines induced by imiquimod have localized paracrine activity.11 Our patient was unique in that his depigmentation was present at the site of application, adjacent to the site of application, and at distant sites. He applied imiquimod only to the periungual area of the right fifth digit and left thumb but experienced depigmentation at several other sites. Although it is possible that our patient unintentionally spread imiquimod on the distant sites, it is less likely that the application would have been sufficient to cause depigmentation. Although systemic absorption of topical medications varies depending on multiple factors, the systemic absorption of imiquimod is minimal with mild systemic side effects reported, including headache, myalgia, and influenzalike symptoms.5 Thus, it is possible that our patient developed distant vitiligolike depigmentation as a systemic side effect of imiquimod therapy. Although our patient declined to have a biopsy performed, Gowda et al15 reported biopsy-proven vitiligo, demonstrating the absence of melanin and melanocytes following the use of imiquimod.
Several mechanisms have been proposed for imiquimod-induced depigmentation. For example, imiquimod may induce melanocyte apoptosis by increasing the levels of several proinflammatory and proapoptotic cytokines.16 Imiquimod-induced melanocyte apoptosis appears to involve elevated caspase-3, decreased B-cell lymphoma 2, altered mitogen-activated protein kinase expression, and ubiquitin-mediated proteolysis.13,17 Additionally, increased levels of IL-6 appear to increase melanocyte-binding molecules and increase melanocyte-leukocyte interactions. Another proposed theory targets toll-like receptor 7 on melanocytes that are acted on directly by imiquimod.11,17 In contrast, development of vitiligo following trauma (Koebner phenomenon) is not uncommon, and the immune effects induced by imiquimod may mimic those seen with trauma.14 Further research is needed to elucidate the mechanism by which imiquimod causes vitiligolike depigmentation.
Unfortunately, the depigmentation seen with imiquimod generally is permanent. Stefanaki et al10 showed repigmentation on cessation of imiquimod use. Our patient’s depigmentation remains unchanged despite treatment with tacrolimus ointment. Although it is possible for vitiligo to occur de novo without obvious inciting event or laboratory abnormality, the timeline and number of other cases in the literature make ours highly suspect for imiquimod-induced depigmentation.
Conclusion
Imiquimod is a commonly used immune-enhancing medication with an increasing list of off-label uses. Prior to prescribing imiquimod for a benign skin condition, clinicians should be cognizant of the potential for localized or possibly even distant depigmentation. We report a case of distant depigmentation following the use of imiquimod for periungual verruca vulgaris.
- Ganjian S, Ourian AJ, Shamtoub G, et al. Off-label indications for imiquimod. Dermatol Online J. 2009;15:4.
- Skinner RB Jr. Imiquimod. Dermatol Clin. 2003;21:291-300.
- Murphy K, Travers P, Walport M. Innate immunity. In: Murphy K, Travers P, Walport M, eds. Janeway’s Immunobiology. 7th ed. New York, NY: Garland Science. 2008:39-108.
- Brown T, Zirvi M, Cotsarelis G, et al. Vitiligo-like hypopigmentation associated with imiquimod treatment of genital warts. J Am Acad Dermatol. 2005;52:715-716.
- Aldara [package insert]. Bristol, TN: Graceway Pharmaceuticals, LLC; 2007.
- Kwon HH, Cho KH. Induction of vitiligo-like hypopigmentation after imiquimod treatment of extramammary Paget’s disease. Ann Dermatol. 2012;24:482-484.
- Mendonca CO, Yates VM. Permanent facial hypopigmentation following treatment with imiquimod. Clin Exp Dermatol. 2006;31:721-722.
- Zhang R, Zhu W. Genital vitiligo following use of imiquimod 5% cream. Indian J Dermatol. 2011;56:335-336.
- Burnett CT, Kouba DJ. Imiquimod-induced depigmentation: report of two cases and review of the literature. Dermatol Surg. 2012;38:1872-1875.
- Stefanaki C, Nicolaidou E, Hadjivassiliou M. Imiquimod-induced vitiligo in a patient with genital warts. J Eur Acad Dermatol Venereol. 2006;20:755-756.
- Al-Dujaili Z, Hsu S. Imiquimod-induced vitiligo. Dermatol Online J. 2007;13:10.
- Mashiah J, Brenner S. Possible mechanisms in the induction of vitiligo-like hypopigmentation by topical imiquimod. Clin Exp Dermatol. 2007;33:74-76.
- Grahovac M, Ehmann LM, Flaig M, et al. Giant basal cell carcinoma. Improvement and vitiligo-like hypopigmentation after intermittent treatment with 5% imiquimod. Acta Dermatovenerol Croat. 2012;20:275-278.
- Serrão VV, Páris FR, Feio AB. Genital vitiligo-like depigmentation following use of imiquimod 5% cream. Eur J Dermatol. 2008;18:342-343.
- Gowda S, Tillman DK, Fitzpatrick JE, et al. Imiquimod-induced vitiligo after treatment of nodular basal cell carcinoma. J Cutan Pathol. 2009;36:878-881.
- Kim CH, Ahn JH, Kang SU, et al. Imiquimod induces apoptosis of human melanocytes. Arch Dermatol Res. 2010;302:301-306.
- Eapen BR. Vitiligo, psoriasis, and imiquimod: fitting all into the same pathway. Indian J Dermatol Venereol Leprol. 2008;74:169.
Imiquimod is derived from the imidazoquinoline family and works by activating both innate and adaptive immune pathways. Imiquimod binds to toll-like receptor 7 located on monocytes, macrophages, and dendritic cells,1 which allows nuclear factor κβ light chain enhancer of activated B cells to induce production of proinflammatory cytokines, including IFN-α and tumor necrosis factor α, as well as IL-1, IL-6, IL-8, IL-10, and IL-12.2 These proinflammatory cytokines play a role in the innate immunity, triggering upregulation of the adaptive immune pathway and activating type 1 helper T cells, cytotoxic T cells, and natural killer cells. These cells have antiviral and antitumoral effects that lend to their significance in coordinating innate and adaptive immune mechanisms.3 More specifically, imiquimod enhances dendritic cell migration to regional lymph nodes and induces apoptosis via activation of proapoptotic B-cell lymphoma 2 proteins.1,2 Imiquimod has been approved by the US Food and Drug Administration (FDA) to treat external genitalia and perianal condyloma acuminata, actinic keratoses (AKs), and superficial basal cell carcinoma (BCC). It often is used off label for antiviral or antitumoral therapy in Bowen disease, squamous cell carcinoma, lentigo maligna, vulvar intraepithelial neoplasia, molluscum contagiosum, common warts, and leishmaniasis.1,2 Imiquimod is generally well tolerated; erythema and irritation at the application site are the most common side effects, with pigmentary change being less common.
Case Report
A 51-year-old man with a medical history of vitamin D deficiency, vitamin B12 deficiency, tinea pedis, and BCC presented with periungual verruca vulgaris on the right fifth digit and left thumb (Figure 1). The patient was prescribed imiquimod cream 5% to be applied 3 times weekly for 3 months. At 5-month follow-up the patient reported new-onset vitiligolike patches of depigmentation on the hands and feet that abruptly began 3 months after initiating treatment with imiquimod. On examination he had several depigmented patches with well-defined irregular borders on the bilateral dorsal hands and right foot as well as the right elbow (Figure 2). There was no personal or family history of vitiligo, thyroid disease, or autoimmune disease. Thyroid function studies and autoimmune panel were unremarkable. The patient also denied applying imiquimod to areas other than the periungual region of the right fifth digit and left thumb. He declined a biopsy of the lesions and was given a prescription for tacrolimus ointment 0.1% for twice-daily application. At 3-month follow-up the depigmented patches had spread. The patient is currently on 5-fluorouracil cream 5%. Despite loss of pigmentation, the periungual verruca vulgaris has persisted as well as depigmentation.
Comment
Imiquimod therapy is commonly used to treat conditions for which an antiviral or antitumor immune response is necessary for treatment and full resolution of skin conditions. It can yield positive results in conditions that are difficult to treat, such as periungual verruca vulgaris.4 The most common adverse effects of imiquimod include localized inflammation and application-site reactions. Pigment changes, though less common, also have been reported. From 1997 to 2003, 1257 cases of imiquimod adverse effects were reported to the FDA. There were 68 reported cases of pigmentary change, of which 51 documented vitiligo, hypopigmentation, or depigmentation. The others reported hyperpigmentation following imiquimod use.4 The imiquimod package insert lists application-site hypopigmentation as a possible adverse effect.5 Imiquimod-induced hypopigmentation and depigmentation have been reported in the peer-reviewed literature.4,6-14 Pigment loss has been reported in imiquimod treatment of condyloma acuminata, superficial BCC, nodular BCC, and extramammary Paget disease.6-8 Duration of therapy to onset of pigment loss ranged from 7 to 28 weeks.9 Imiquimod dosing varied among reported cases, ranging from 3 times weekly to daily application. Interestingly, hypopigmentation or depigmentation are not commonly associated with imiquimod use for the treatment of AKs, which Burnett and Kouba9 proposed may be due to the twice weekly imiquimod dosing regimen recommended by the FDA for the treatment of AK (below the minimum threshold for pigment loss). Our patient applied imiquimod cream 5% to periungual verruca vulgaris 3 times weekly for 3 months and may have developed vitiligolike depigmentation because he met this theoretical dosage threshold. Further research is necessary to confirm a dosage-related threshold for the development of depigmentation. Imiquimod-induced pigment loss has mainly been limited to the site of application.
Depigmentation was limited to the application site the majority of the time; however, depigmentation at adjacent sites has been reported.10 This finding was consistent with the proposed notion that cytokines induced by imiquimod have localized paracrine activity.11 Our patient was unique in that his depigmentation was present at the site of application, adjacent to the site of application, and at distant sites. He applied imiquimod only to the periungual area of the right fifth digit and left thumb but experienced depigmentation at several other sites. Although it is possible that our patient unintentionally spread imiquimod on the distant sites, it is less likely that the application would have been sufficient to cause depigmentation. Although systemic absorption of topical medications varies depending on multiple factors, the systemic absorption of imiquimod is minimal with mild systemic side effects reported, including headache, myalgia, and influenzalike symptoms.5 Thus, it is possible that our patient developed distant vitiligolike depigmentation as a systemic side effect of imiquimod therapy. Although our patient declined to have a biopsy performed, Gowda et al15 reported biopsy-proven vitiligo, demonstrating the absence of melanin and melanocytes following the use of imiquimod.
Several mechanisms have been proposed for imiquimod-induced depigmentation. For example, imiquimod may induce melanocyte apoptosis by increasing the levels of several proinflammatory and proapoptotic cytokines.16 Imiquimod-induced melanocyte apoptosis appears to involve elevated caspase-3, decreased B-cell lymphoma 2, altered mitogen-activated protein kinase expression, and ubiquitin-mediated proteolysis.13,17 Additionally, increased levels of IL-6 appear to increase melanocyte-binding molecules and increase melanocyte-leukocyte interactions. Another proposed theory targets toll-like receptor 7 on melanocytes that are acted on directly by imiquimod.11,17 In contrast, development of vitiligo following trauma (Koebner phenomenon) is not uncommon, and the immune effects induced by imiquimod may mimic those seen with trauma.14 Further research is needed to elucidate the mechanism by which imiquimod causes vitiligolike depigmentation.
Unfortunately, the depigmentation seen with imiquimod generally is permanent. Stefanaki et al10 showed repigmentation on cessation of imiquimod use. Our patient’s depigmentation remains unchanged despite treatment with tacrolimus ointment. Although it is possible for vitiligo to occur de novo without obvious inciting event or laboratory abnormality, the timeline and number of other cases in the literature make ours highly suspect for imiquimod-induced depigmentation.
Conclusion
Imiquimod is a commonly used immune-enhancing medication with an increasing list of off-label uses. Prior to prescribing imiquimod for a benign skin condition, clinicians should be cognizant of the potential for localized or possibly even distant depigmentation. We report a case of distant depigmentation following the use of imiquimod for periungual verruca vulgaris.
Imiquimod is derived from the imidazoquinoline family and works by activating both innate and adaptive immune pathways. Imiquimod binds to toll-like receptor 7 located on monocytes, macrophages, and dendritic cells,1 which allows nuclear factor κβ light chain enhancer of activated B cells to induce production of proinflammatory cytokines, including IFN-α and tumor necrosis factor α, as well as IL-1, IL-6, IL-8, IL-10, and IL-12.2 These proinflammatory cytokines play a role in the innate immunity, triggering upregulation of the adaptive immune pathway and activating type 1 helper T cells, cytotoxic T cells, and natural killer cells. These cells have antiviral and antitumoral effects that lend to their significance in coordinating innate and adaptive immune mechanisms.3 More specifically, imiquimod enhances dendritic cell migration to regional lymph nodes and induces apoptosis via activation of proapoptotic B-cell lymphoma 2 proteins.1,2 Imiquimod has been approved by the US Food and Drug Administration (FDA) to treat external genitalia and perianal condyloma acuminata, actinic keratoses (AKs), and superficial basal cell carcinoma (BCC). It often is used off label for antiviral or antitumoral therapy in Bowen disease, squamous cell carcinoma, lentigo maligna, vulvar intraepithelial neoplasia, molluscum contagiosum, common warts, and leishmaniasis.1,2 Imiquimod is generally well tolerated; erythema and irritation at the application site are the most common side effects, with pigmentary change being less common.
Case Report
A 51-year-old man with a medical history of vitamin D deficiency, vitamin B12 deficiency, tinea pedis, and BCC presented with periungual verruca vulgaris on the right fifth digit and left thumb (Figure 1). The patient was prescribed imiquimod cream 5% to be applied 3 times weekly for 3 months. At 5-month follow-up the patient reported new-onset vitiligolike patches of depigmentation on the hands and feet that abruptly began 3 months after initiating treatment with imiquimod. On examination he had several depigmented patches with well-defined irregular borders on the bilateral dorsal hands and right foot as well as the right elbow (Figure 2). There was no personal or family history of vitiligo, thyroid disease, or autoimmune disease. Thyroid function studies and autoimmune panel were unremarkable. The patient also denied applying imiquimod to areas other than the periungual region of the right fifth digit and left thumb. He declined a biopsy of the lesions and was given a prescription for tacrolimus ointment 0.1% for twice-daily application. At 3-month follow-up the depigmented patches had spread. The patient is currently on 5-fluorouracil cream 5%. Despite loss of pigmentation, the periungual verruca vulgaris has persisted as well as depigmentation.
Comment
Imiquimod therapy is commonly used to treat conditions for which an antiviral or antitumor immune response is necessary for treatment and full resolution of skin conditions. It can yield positive results in conditions that are difficult to treat, such as periungual verruca vulgaris.4 The most common adverse effects of imiquimod include localized inflammation and application-site reactions. Pigment changes, though less common, also have been reported. From 1997 to 2003, 1257 cases of imiquimod adverse effects were reported to the FDA. There were 68 reported cases of pigmentary change, of which 51 documented vitiligo, hypopigmentation, or depigmentation. The others reported hyperpigmentation following imiquimod use.4 The imiquimod package insert lists application-site hypopigmentation as a possible adverse effect.5 Imiquimod-induced hypopigmentation and depigmentation have been reported in the peer-reviewed literature.4,6-14 Pigment loss has been reported in imiquimod treatment of condyloma acuminata, superficial BCC, nodular BCC, and extramammary Paget disease.6-8 Duration of therapy to onset of pigment loss ranged from 7 to 28 weeks.9 Imiquimod dosing varied among reported cases, ranging from 3 times weekly to daily application. Interestingly, hypopigmentation or depigmentation are not commonly associated with imiquimod use for the treatment of AKs, which Burnett and Kouba9 proposed may be due to the twice weekly imiquimod dosing regimen recommended by the FDA for the treatment of AK (below the minimum threshold for pigment loss). Our patient applied imiquimod cream 5% to periungual verruca vulgaris 3 times weekly for 3 months and may have developed vitiligolike depigmentation because he met this theoretical dosage threshold. Further research is necessary to confirm a dosage-related threshold for the development of depigmentation. Imiquimod-induced pigment loss has mainly been limited to the site of application.
Depigmentation was limited to the application site the majority of the time; however, depigmentation at adjacent sites has been reported.10 This finding was consistent with the proposed notion that cytokines induced by imiquimod have localized paracrine activity.11 Our patient was unique in that his depigmentation was present at the site of application, adjacent to the site of application, and at distant sites. He applied imiquimod only to the periungual area of the right fifth digit and left thumb but experienced depigmentation at several other sites. Although it is possible that our patient unintentionally spread imiquimod on the distant sites, it is less likely that the application would have been sufficient to cause depigmentation. Although systemic absorption of topical medications varies depending on multiple factors, the systemic absorption of imiquimod is minimal with mild systemic side effects reported, including headache, myalgia, and influenzalike symptoms.5 Thus, it is possible that our patient developed distant vitiligolike depigmentation as a systemic side effect of imiquimod therapy. Although our patient declined to have a biopsy performed, Gowda et al15 reported biopsy-proven vitiligo, demonstrating the absence of melanin and melanocytes following the use of imiquimod.
Several mechanisms have been proposed for imiquimod-induced depigmentation. For example, imiquimod may induce melanocyte apoptosis by increasing the levels of several proinflammatory and proapoptotic cytokines.16 Imiquimod-induced melanocyte apoptosis appears to involve elevated caspase-3, decreased B-cell lymphoma 2, altered mitogen-activated protein kinase expression, and ubiquitin-mediated proteolysis.13,17 Additionally, increased levels of IL-6 appear to increase melanocyte-binding molecules and increase melanocyte-leukocyte interactions. Another proposed theory targets toll-like receptor 7 on melanocytes that are acted on directly by imiquimod.11,17 In contrast, development of vitiligo following trauma (Koebner phenomenon) is not uncommon, and the immune effects induced by imiquimod may mimic those seen with trauma.14 Further research is needed to elucidate the mechanism by which imiquimod causes vitiligolike depigmentation.
Unfortunately, the depigmentation seen with imiquimod generally is permanent. Stefanaki et al10 showed repigmentation on cessation of imiquimod use. Our patient’s depigmentation remains unchanged despite treatment with tacrolimus ointment. Although it is possible for vitiligo to occur de novo without obvious inciting event or laboratory abnormality, the timeline and number of other cases in the literature make ours highly suspect for imiquimod-induced depigmentation.
Conclusion
Imiquimod is a commonly used immune-enhancing medication with an increasing list of off-label uses. Prior to prescribing imiquimod for a benign skin condition, clinicians should be cognizant of the potential for localized or possibly even distant depigmentation. We report a case of distant depigmentation following the use of imiquimod for periungual verruca vulgaris.
- Ganjian S, Ourian AJ, Shamtoub G, et al. Off-label indications for imiquimod. Dermatol Online J. 2009;15:4.
- Skinner RB Jr. Imiquimod. Dermatol Clin. 2003;21:291-300.
- Murphy K, Travers P, Walport M. Innate immunity. In: Murphy K, Travers P, Walport M, eds. Janeway’s Immunobiology. 7th ed. New York, NY: Garland Science. 2008:39-108.
- Brown T, Zirvi M, Cotsarelis G, et al. Vitiligo-like hypopigmentation associated with imiquimod treatment of genital warts. J Am Acad Dermatol. 2005;52:715-716.
- Aldara [package insert]. Bristol, TN: Graceway Pharmaceuticals, LLC; 2007.
- Kwon HH, Cho KH. Induction of vitiligo-like hypopigmentation after imiquimod treatment of extramammary Paget’s disease. Ann Dermatol. 2012;24:482-484.
- Mendonca CO, Yates VM. Permanent facial hypopigmentation following treatment with imiquimod. Clin Exp Dermatol. 2006;31:721-722.
- Zhang R, Zhu W. Genital vitiligo following use of imiquimod 5% cream. Indian J Dermatol. 2011;56:335-336.
- Burnett CT, Kouba DJ. Imiquimod-induced depigmentation: report of two cases and review of the literature. Dermatol Surg. 2012;38:1872-1875.
- Stefanaki C, Nicolaidou E, Hadjivassiliou M. Imiquimod-induced vitiligo in a patient with genital warts. J Eur Acad Dermatol Venereol. 2006;20:755-756.
- Al-Dujaili Z, Hsu S. Imiquimod-induced vitiligo. Dermatol Online J. 2007;13:10.
- Mashiah J, Brenner S. Possible mechanisms in the induction of vitiligo-like hypopigmentation by topical imiquimod. Clin Exp Dermatol. 2007;33:74-76.
- Grahovac M, Ehmann LM, Flaig M, et al. Giant basal cell carcinoma. Improvement and vitiligo-like hypopigmentation after intermittent treatment with 5% imiquimod. Acta Dermatovenerol Croat. 2012;20:275-278.
- Serrão VV, Páris FR, Feio AB. Genital vitiligo-like depigmentation following use of imiquimod 5% cream. Eur J Dermatol. 2008;18:342-343.
- Gowda S, Tillman DK, Fitzpatrick JE, et al. Imiquimod-induced vitiligo after treatment of nodular basal cell carcinoma. J Cutan Pathol. 2009;36:878-881.
- Kim CH, Ahn JH, Kang SU, et al. Imiquimod induces apoptosis of human melanocytes. Arch Dermatol Res. 2010;302:301-306.
- Eapen BR. Vitiligo, psoriasis, and imiquimod: fitting all into the same pathway. Indian J Dermatol Venereol Leprol. 2008;74:169.
- Ganjian S, Ourian AJ, Shamtoub G, et al. Off-label indications for imiquimod. Dermatol Online J. 2009;15:4.
- Skinner RB Jr. Imiquimod. Dermatol Clin. 2003;21:291-300.
- Murphy K, Travers P, Walport M. Innate immunity. In: Murphy K, Travers P, Walport M, eds. Janeway’s Immunobiology. 7th ed. New York, NY: Garland Science. 2008:39-108.
- Brown T, Zirvi M, Cotsarelis G, et al. Vitiligo-like hypopigmentation associated with imiquimod treatment of genital warts. J Am Acad Dermatol. 2005;52:715-716.
- Aldara [package insert]. Bristol, TN: Graceway Pharmaceuticals, LLC; 2007.
- Kwon HH, Cho KH. Induction of vitiligo-like hypopigmentation after imiquimod treatment of extramammary Paget’s disease. Ann Dermatol. 2012;24:482-484.
- Mendonca CO, Yates VM. Permanent facial hypopigmentation following treatment with imiquimod. Clin Exp Dermatol. 2006;31:721-722.
- Zhang R, Zhu W. Genital vitiligo following use of imiquimod 5% cream. Indian J Dermatol. 2011;56:335-336.
- Burnett CT, Kouba DJ. Imiquimod-induced depigmentation: report of two cases and review of the literature. Dermatol Surg. 2012;38:1872-1875.
- Stefanaki C, Nicolaidou E, Hadjivassiliou M. Imiquimod-induced vitiligo in a patient with genital warts. J Eur Acad Dermatol Venereol. 2006;20:755-756.
- Al-Dujaili Z, Hsu S. Imiquimod-induced vitiligo. Dermatol Online J. 2007;13:10.
- Mashiah J, Brenner S. Possible mechanisms in the induction of vitiligo-like hypopigmentation by topical imiquimod. Clin Exp Dermatol. 2007;33:74-76.
- Grahovac M, Ehmann LM, Flaig M, et al. Giant basal cell carcinoma. Improvement and vitiligo-like hypopigmentation after intermittent treatment with 5% imiquimod. Acta Dermatovenerol Croat. 2012;20:275-278.
- Serrão VV, Páris FR, Feio AB. Genital vitiligo-like depigmentation following use of imiquimod 5% cream. Eur J Dermatol. 2008;18:342-343.
- Gowda S, Tillman DK, Fitzpatrick JE, et al. Imiquimod-induced vitiligo after treatment of nodular basal cell carcinoma. J Cutan Pathol. 2009;36:878-881.
- Kim CH, Ahn JH, Kang SU, et al. Imiquimod induces apoptosis of human melanocytes. Arch Dermatol Res. 2010;302:301-306.
- Eapen BR. Vitiligo, psoriasis, and imiquimod: fitting all into the same pathway. Indian J Dermatol Venereol Leprol. 2008;74:169.
Practice Points
- Imiquimod commonly is used off label to treat viral and neoplastic processes.
- Clinicians should be aware of the potential for dyspigmentation or depigmentation as a side effect from treatment.