User login
The Medical Roundtable: Type II Pulmonary Hypertension in the Setting of Heart Failure with Preserved Ejection Fraction
DR. GIVERTZ: I’m Michael Givertz. I am the Medical Director of the Heart Transplant and Mechanical Circulatory Support Program at the Brigham and Women’s Hospital in Boston, Massachusetts. I am interested in the management of patients with advanced heart disease and those with heart failure with preserved ejection fraction (HFpEF) as well as those with reduced ejection fraction (EF). In particular, I focus on patients with comorbidities, including pulmonary hypertension (PH) and chronic kidney disease.
DR. LEWIS: I’m Greg Lewis from Massachusetts General Hospital (MGH), Boston. I am a member of the Heart Failure and Transplant Unit, and I direct the cardiopulmonary exercise labs at MGH. Like Dr. Givertz, I am interested in heart failure (HF) with both preserved EF and reduced EF and in patients who have PH complicating both the abovementioned conditions.
DR. PRITZKER: I’m Marc Pritzker. I’m a professor of Cardiovascular Medicine and Surgery at the University of Minnesota and a part of the Advanced Heart Failure Group here. I’m also the Director of the Pulmonary Hypertension Center, and I have a special interest in diastolic dysfunction, which is going to be the leading cause of PH in Western society,1 if it isn’t already.
DR. FRANCIS: Let’s begin with a case and then proceed from there. It’s a short vignette.
An 83-year-old woman is suffering from long-standing diabetes, coronary disease, and hypertension. She’s admitted to the hospital with acute HF. An echocardiogram was performed, which indicates a normal EF of 65%. She has left ventricular (LV) hypertrophy, a small LV cavity, and a large left atrium.
Doppler studies indicate that the left atrial pressure is probably slightly elevated. There is clear diastolic dysfunction, as shown by echocardiography, and enough tricuspid regurgitation to suggest that the pulmonary artery (PA) pressure is approximately 70/30 mm Hg.
She has been given intravenous furosemide, her condition improves, and she is subsequently discharged. She then visits the outpatient clinic where follow-up echocardiogray indicates the persistence of PH. Her estimated PA pressure by echocardiogram is slightly lower (60/30 mm Hg), although her LV filling pressures are probably lower.
Although I’m not exactly sure of the situation at the Brigham or MGH, I suspect that it’s similar to the situation here and around the country: We are now seeing many such patients. They’re often elderly women with many comorbid conditions, and they’re sometimes admitted with atrial fibrillation, which makes the estimation of diastolic function problematic. They’re treated conventionally with diuretics and antihypertensive therapies, but they tend to fare poorly. Often, they return to the hospital and pose a recurrent problem.
Dr. Pritzker, let’s start with you. The patient’s PA pressure is quite high, at least based on the estimation by an echocardiogram. It’s fairly common for these patients to have PH, but this patient’s PA pressure is probably disproportionately high relative to her slightly elevated left atrial pressure.
Granted that we don’t have the hemodynamic data from the catheterization lab; nonetheless, am I right? Are we seeing more of these patients with so-called reactive or disproportionate PH or hypertension that is higher than what you would expect, given the clinical context?
DR. PRITZKER: I agree. We probably encounter 3 or 4 new patients a week who are referred to us because of certain observations during echocardiographic screening. In reality, these patients have diastolic dysfunction, and probably half of the patients in our clinic have PH that appears to be higher than the classical expectation that the PA pressure follows the left atrial pressure.
This situation could be viewed in 2 ways. One is the eyeball method, which many people do and just say, “Well, that seems high to me compared to what the wedge pressure is.” The transpulmonary gradient, which is the mean PA pressure minus the wedge pressure, or, sometimes, the pulmonary diastolic pressure minus the wedge pressure, is above 15 mm Hg.
Based on the information given in this case, the patient’s mean PA pressure is above 40 mm Hg. I’m assuming that modestly elevated pulmonary capillary wedge pressure is approximately 15 to 20 mm Hg, which indicates an elevated transpulmonary gradient by any measurement.
DR. FRANCIS: Dr. Givertz, what is the scope of this problem in your hospital?
DR. GIVERTZ: As Dr. Pritzker was alluding to, we are seeing many such patients. I’m not sure if we’re actually looking for them or if we are looking more closely at the estimates of PA pressure by echocardiogram, which we may have been less attuned to previously.
The problem of HFpEF, which is a broad definition for this patient’s condition, is that it’s not something new: It’s something that we’ve been struggling with for many years, particularly in older patients. As Dr. Francis mentioned, these patients are typically women with a history of hypertension and often, other comorbidities.
This case would seem unusual, mostly due to the estimated PA pressure. I think that’s probably what caught the eye of the clinicians here.
We probably wouldn’t have given it a second thought if the estimated PA pressure was in the 40s or, maybe, 50s. It is only once the pressure increases above 60 mm Hg that we begin to realize that some other factor could be involved or that something was overlooked, which we now need to look at a little more closely.
Since you didn’t mention anything about the mitral valve, let’s assume that there isn’t any mitral valve disease here and we’re not missing significant mitral regurgitation at rest or ischemic mitral regurgitation, which is something you would want to rule out. This is a common problem, and I think we’re seeing it more often because we’re more closely looking for it.
DR. FRANCIS: I agree, Dr. Givertz. But, it turns out that this patient has minimal mitral regurgitation.
Dr. Lewis, as you have a focused laboratory interest in this problem, what do you think of this case, particularly with regard to the scope of the problem?
DR. LEWIS: I think that it is clearly a major problem. I would like to add that these patients can come to subspecialty care through different routes. It’s not uncommon for a patient like the one in question to be hospitalized through the general medical service or to be assessed by a pulmonary specialist or general cardiologist.
Unlike patients who have advanced LV systolic dysfunction and are often routed into the well-oiled machine of an advanced cardiomyopathy clinic, these patients are identified through providers with different scopes of practice. For example, I believe that these patients are often initially referred to a pulmonary physician.
This makes the problem more challenging because there’s a less-inherent ownership within a predefined group of clinicians. Additionally, this case has features that are clearly suggestive of a pre-capillary PH as well as provide evidence of increased left-sided filling pressures. These patients may be directed through different routes of care in the hospital.
It does present a slight problem, particularly when the house staff is presenting the case and people have their own ideas about what we should call it. Dr. Givertz, what should we call this syndrome? What do you refer to it as at the Brigham?
DR. GIVERTZ: That’s an excellent question. We have moved away from the older terminology of diastolic HF, as probably, most have, although there are some strong proponents of this terminology, which assumes that there is some abnormality in diastolic function.
In an 83-year-old woman with long-standing hypertension, I think LV hypertrophy, a small cavity on her echocardiogram report, and a large left atrium are certainly markers of diastolic dysfunction. So, calling this diastolic HF might be justified.
We probably overstep that terminology in patients who have the clinical syndrome of HF, as this patient does, with dyspnea on exertion, volume overload, and a normal or near-normal EF. So, this meets the basic definition of HFpEF, and there are echocardiographic indicators of abnormal diastolic function. So, you could call it diastolic HF.
The key indicator here is the breadth. Although it is just a descriptive term, I would prefer using a term like HFpEF, and we generally use this term on rounds in the hospital or in an ambulatory setting.
The terminology is important because we don’t assume that this is just a problem of diastole or relaxation. There may be other comorbidities and aspects related to the pathophysiology,2 which may have a larger role to play than only causing a simple abnormality in diastolic function.
DR. FRANCIS: I agree, Dr. Givertz, although I have found that there are certain experts and authors who are accustomed to a specific terminology, which is unfortunate because some patients don’t really have diastolic dysfunction, and I don’t think we understand the underlying pathophysiology very well.
DR. PRITZKER: Drs. Givertz and Lewis, is there a certain trigger in echocardiogram reports such that when an elevation in PA pressure is evident, as in this case, the examiners make the diastology a little bit more manifest or look at tricuspid annular plane systolic excursion or PA acceleration time?
I think we all have difficulties with that. I have looked at reports from several different hospitals, and nobody provides a consistent report once an increase in PA pressure has been identified.
DR. GIVERTZ: I think that’s an excellent point. We get comprehensive echocardiogram reports; it’s the way that our laboratory runs. But we don’t have a trigger that leads to the reporting of additional indices of diastolic function. With high PA pressures, a focus on the right ventricle (RV) (in terms of tricuspid annular plane systolic excursion measurements or RV dimensions) would be particularly helpful in terms of having additional information about the etiology of PH.
DR. LEWIS: In our institution, there is no dedicated report type for this constellation of findings. The conclusion of the interpretation would indicate that findings were consistent with diastolic dysfunction.
Dr. Francis, you mentioned in the description that there was some indication of left atrial pressure elevation in the Doppler studies. I know our echocardiographers at MGH generally avoid reporting Doppler measurements such as E/A ratios or estimates of left atrial pressure based on E/E' values.Ultimately, for patient management, it will be important to establish widely accepted echocardiographic criteria that differentiate precapillary PH, postcapillary PH, and mixed PH.
DR. FRANCIS: That’s a good point. As you know, some hospitals, like my former institution the Cleveland Clinic, did attempt to carry out the calculations, but in most places, these calculations are really not done. I personally assessed the echocardiogram as a consequence of my obsessive behavior. I have also talked to the echocardiographers about it and gathered an intuitive sense of impaired filling.
If you were to guess, you would say that the left atrial pressure is somewhat higher than normal. This is a really subjective phrase. It wasn’t actually measured, of course, and it wasn’t calculated.
DR. PRITZKER: I think it would be helpful if each institution could, at least, have a trigger, so that when the patient goes back to a family practitioner, a pulmonologist or another specialist, there is some comprehensive information that can give the practitioner some direction.
As echocardiograms become more capable of calculating these derived values (E/E prime, E to A, etc.), even general cardiologists begin feeling confused. So I like Dr. Lewis’ idea about inserting a comment at the end to at least point people in some direction.
DR. FRANCIS: Let’s focus a little on what’s different about this particular patient, which is something that I’m personally interested in. Dr. Lewis, why is it that some of these patients have higher PA pressures than what you would expect, given the clinical context? Is there something unusual about them? Is there something relevant that we can discuss, which might help identify such a patient?
DR. LEWIS: I think that’s a key question regarding the evaluation of patients like this one. For me, the first thing to do when I see a patient like this is to make sure that there aren’t other factors, other than left heart disease, that may be mediating the PH.
Even if there is a suggestion of elevated left atrial pressure, it’s important to think beyond the left heart, in terms of additional direct insults that may have been sustained by the pulmonary circulation. For example, does the patient have chronic obstructive pulmonary disease that may be contributing to the high pulmonary arterial pressures? Does the patient have sleep apnea or a history of chronic thromboembolic disease in the lung? Even an overlapping syndrome such as scleroderma or sarcoidosis can cause LV dysfunction as well as pulmonary arterial hypertension but would be unusual to detect at an advanced age in terms of relevance to this case.
Epidemiologic studies can also provide clues regarding the type of patients who tend to have higher pulmonary arterial pressures (eg, older individuals or people with higher systolic blood pressure).
I have been surprised by our relative lack of ability to predict who’s going to have a higher transpulmonary gradient, based on the clinical features alone. For a given left atrial pressure, why do some patients have higher transpulmonary gradients than others?
Elevation of transpulmonary gradients is not simply related to degree of elevation in left-sided pressure alone. That is clear from the relatively weak correlation between PA systolic pressure and left-sided filling pressure.3 I don’t think it is related to the duration of HF alone either.
In our patient population, we’ve recorded the time when patients were diagnosed with HF because I commonly hear, “Well, if the patient has HF for a sufficient amount of time, he/she will gradually develop increases in transpulmonary gradient because of high pressure on the left side of the heart.” The duration of HF was not significantly related to the transpulmonary gradient. Therefore, I believe we need to look at new ways of understanding which patients will develop high transpulmonary gradients.
There’s some work being conducted in this area. Investigators at Dr. Francis’s former institution recently published a study about arginine metabolism dysregulation and some circulating markers that were high in patients who had a high burden of precapillary PH and HF.4
Dr. Givertz has been involved in studies evaluating endothelin levels or cyclic guanosine monophosphate-adenosine monophosphate release through the pulmonary circulation as potential indicators of dysregulation in signaling systems within pulmonary circulation. There may also be permissive genotypes that predispose patients to develop precapillary PH in the setting of left heart dysfunction.
So, I think that the question is a good one, but for me, it raises many more questions about why these patients are different. I don’t think we can explain it on the basis of clinical characteristics alone.
DR. FRANCIS: Dr. Lewis has had extensive experience in studying these patients in the laboratory and performing exercise tests on them. Dr. Lewis, from a clinical rather than research standpoint, when would you actually consider conducting more invasive studies like you are currently doing in the research laboratory?
DR. LEWIS: We haven’t talked about invasive phenotyping of patients yet, but to address Dr. Givertz’s point, the question pertains to when we need to carefully study the conditions in the pulmonary circulation. I believe that knowing the left atrial pressure is important for truly understanding the transpulmonary gradient, and I’d like to know whether it’s greater than 15 mm Hg, as Dr. Pritzker indicated; this level serves as a trigger for looking carefully into the pulmonary circulation.
Dr. Francis, I find the exercise status to be particularly informative in patients who exhibit shortness of breath with exertion. However, it’s not entirely clear whether their symptoms are being mediated by left-heart disease or a primary insult to the pulmonary circulation.
Often, even when patients have a resting right-heart catheterization, we see borderline elevations in pulmonary arterial pressure, say, a mean pressure of 20 to 25 mm Hg, and a pulmonary capillary wedge pressure of 10 to 15 mm Hg, but symptoms are present.
In such cases, instead of looking at hemodynamic measurements during a single moment in time, I find it quite helpful to make serial measurements under different loading conditions that are introduced by the very highly relevant physiologic state of exercise. We often encounter patients who have shortness of breath with exertion, which is not entirely explained by the initial diagnostic testing results.
I find it particularly useful to characterize the changes in the pulmonary capillary wedge pressure and transpulmonary gradient when patients are subjected to exercise. We can begin to classify patients based on whether their burden of precapillary PH is high or there is truly a left-sided predominance to their symptoms.
DR. FRANCIS: This gives rise to question of whether therapeutic targets exist. We all know that they exist, but what is the therapy? Perhaps, we’re not so sure about that.
DR. PRITZKER: We’re predominantly using sildenafil because it’s easily available in the hospital and doesn’t require cumbersome pre-approval for acute use like in the case of endothelin antagonists. We have a good experience of using it, and we follow it up with 6-min walks, which perhaps, in this population, is slightly easier to do.
DR. FRANCIS: Dr. Pritzker, what is your threshold for administering sildenafil? Is it lower?
DR. PRITZKER: Yes, because I think there’s more relevant evidence, including the provocative work of an Italian group and Dr. Lewis’ work. We don’t necessarily know why we’re making them better yet, but clearly, people are having fewer symptoms. This is more anecdotal than longitudinal, but patients have fewer symptoms, feel better, and seem to have better function. We also see fewer hospital admissions. I think a previous paper published at the end of last year presented similar findings.
DR. FRANCIS: Dr. Givertz, can you comment about the therapy?
DR. GIVERTZ: I think we’re all gaining experience with using sildenafil or longer-acting phosphodiesterase type 5 (PDE5) inhibitors in this patient population, but that might not be the first thing I would reach for.
This patient was admitted with acute HF. Presumably, there was a fluid problem here as well. I might be initially inclined to make sure I have optimized diuretic therapy. If this patient wasn’t already on nitrates, given the underlying coronary disease and hypertension, I think administering nitrates would be a reasonable next step.
Although there are certainly patients who cannot tolerate nitrate therapy due to its side effects such as headache or light-headedness, I might try administering nitrates before opting for a PDE5 inhibitor.
DR. PRITZKER: I assumed that the patient had been stabilized, and we still had a therapeutic gap after conventional therapy.
DR. FRANCIS: That’s a good point because it is related to the timing of initiation of such therapy; I am quite conservative about it. I don’t have as much experience as the other physicians on the panel, but my tendency would be to go with conventional medical therapy, as bad as it is, and if the patient didn’t improve thereafter, I would think about sending the patient to Dr. Pritzker or beginning sildenafil treatment myself.
Dr. Lewis, you’re actually involved in a National Institute of Health study, a consortium study. Is that right?
DR. LEWIS: That’s right; it’s the Phosphodiesterase-5 Inhibition to Improve Clinical Status and Exercise Capacity in Diastolic Heart Failure (RELAX)trial.5 Michael and Maggie Redfield from the Mayo Clinic are involved as well, and Dr. Redfield is the principal architect of the trial. But, we are adjudicating the primary end point here, which is a cardiopulmonary exercise endpoint.
Dr. Francis, I agree with Drs. Pritzker and Givertz in terms of the therapeutic approach. There certainly have been some cautionary accounts from other pulmonary vasodilators that have been tried in patients who have left heart disease, predominantly those with systolic dysfunction and have not resulted in improved outcomes.6 Therefore, I tend to refrain from using endothelin antagonists as well as the prostacyclins.
There has been some promising work done with PDE5 inhibitors in left-heart disease, and we will know the results from the RELAX trial soon. We completed enrollment of just over 200 patients, and we’re trying to determine whether we can improve exercise capacity over a period of 24 weeks with sildenafil in patients with HFpEF.
The results of PDE5 inhibition in HF with preserved LVEF from the Guazzi group demonstrated that patients with a significant burden of right-heart disease may derive significant improvement in precapillary PH with sildenafil.7 I believe if there is right-heart dysfunction that is largely attributed to high pulmonary vascular resistance or a high transpulmonary gradient, then pulmonary vasodilators like PDE5 inhibitors will have a promising role to play. Potentially, there are also some novel therapies involving the use of some of the soluble guanylate cyclase stimulators and activators in trials.
If patients have a predominant left-heart dysfunction with a modest transpulmonary gradient, I think they are much better served by trying to optimize the diuretic therapy and treat the systemic hypertension.
DR. FRANCIS: Thank you, Dr. Lewis. I think we agree that this is clearly an emerging problem, and it’s prevalence is increasing, partly because of the aging population. It is true that these patients are often admitted to general medicine wards and are sometimes transferred to the cardiology groups when they are faring poorly, and I don’t know what we can do about that.
There does seem to be a subset of patients with disproportionate PH, and there’s a lot of interest in that particular subset. I don’t know if they’re clearly identifiable from the phenoclinical phenotype, but I think that they pose more difficult problems.
Lastly, I’m delighted to know that the RELAX trial is on the verge of completion, and we will probably have some results soon. Management of patients with HFpEF and PH has been a problem for all of us, and as yet, we do not have good, randomized, controlled trials to help us understand how to manage them.
On that note, I’d like to thank the discussants for their participation.
FoxP2 Media LLC is the publisher of The Medical Roundtable.
DR. GIVERTZ: I’m Michael Givertz. I am the Medical Director of the Heart Transplant and Mechanical Circulatory Support Program at the Brigham and Women’s Hospital in Boston, Massachusetts. I am interested in the management of patients with advanced heart disease and those with heart failure with preserved ejection fraction (HFpEF) as well as those with reduced ejection fraction (EF). In particular, I focus on patients with comorbidities, including pulmonary hypertension (PH) and chronic kidney disease.
DR. LEWIS: I’m Greg Lewis from Massachusetts General Hospital (MGH), Boston. I am a member of the Heart Failure and Transplant Unit, and I direct the cardiopulmonary exercise labs at MGH. Like Dr. Givertz, I am interested in heart failure (HF) with both preserved EF and reduced EF and in patients who have PH complicating both the abovementioned conditions.
DR. PRITZKER: I’m Marc Pritzker. I’m a professor of Cardiovascular Medicine and Surgery at the University of Minnesota and a part of the Advanced Heart Failure Group here. I’m also the Director of the Pulmonary Hypertension Center, and I have a special interest in diastolic dysfunction, which is going to be the leading cause of PH in Western society,1 if it isn’t already.
DR. FRANCIS: Let’s begin with a case and then proceed from there. It’s a short vignette.
An 83-year-old woman is suffering from long-standing diabetes, coronary disease, and hypertension. She’s admitted to the hospital with acute HF. An echocardiogram was performed, which indicates a normal EF of 65%. She has left ventricular (LV) hypertrophy, a small LV cavity, and a large left atrium.
Doppler studies indicate that the left atrial pressure is probably slightly elevated. There is clear diastolic dysfunction, as shown by echocardiography, and enough tricuspid regurgitation to suggest that the pulmonary artery (PA) pressure is approximately 70/30 mm Hg.
She has been given intravenous furosemide, her condition improves, and she is subsequently discharged. She then visits the outpatient clinic where follow-up echocardiogray indicates the persistence of PH. Her estimated PA pressure by echocardiogram is slightly lower (60/30 mm Hg), although her LV filling pressures are probably lower.
Although I’m not exactly sure of the situation at the Brigham or MGH, I suspect that it’s similar to the situation here and around the country: We are now seeing many such patients. They’re often elderly women with many comorbid conditions, and they’re sometimes admitted with atrial fibrillation, which makes the estimation of diastolic function problematic. They’re treated conventionally with diuretics and antihypertensive therapies, but they tend to fare poorly. Often, they return to the hospital and pose a recurrent problem.
Dr. Pritzker, let’s start with you. The patient’s PA pressure is quite high, at least based on the estimation by an echocardiogram. It’s fairly common for these patients to have PH, but this patient’s PA pressure is probably disproportionately high relative to her slightly elevated left atrial pressure.
Granted that we don’t have the hemodynamic data from the catheterization lab; nonetheless, am I right? Are we seeing more of these patients with so-called reactive or disproportionate PH or hypertension that is higher than what you would expect, given the clinical context?
DR. PRITZKER: I agree. We probably encounter 3 or 4 new patients a week who are referred to us because of certain observations during echocardiographic screening. In reality, these patients have diastolic dysfunction, and probably half of the patients in our clinic have PH that appears to be higher than the classical expectation that the PA pressure follows the left atrial pressure.
This situation could be viewed in 2 ways. One is the eyeball method, which many people do and just say, “Well, that seems high to me compared to what the wedge pressure is.” The transpulmonary gradient, which is the mean PA pressure minus the wedge pressure, or, sometimes, the pulmonary diastolic pressure minus the wedge pressure, is above 15 mm Hg.
Based on the information given in this case, the patient’s mean PA pressure is above 40 mm Hg. I’m assuming that modestly elevated pulmonary capillary wedge pressure is approximately 15 to 20 mm Hg, which indicates an elevated transpulmonary gradient by any measurement.
DR. FRANCIS: Dr. Givertz, what is the scope of this problem in your hospital?
DR. GIVERTZ: As Dr. Pritzker was alluding to, we are seeing many such patients. I’m not sure if we’re actually looking for them or if we are looking more closely at the estimates of PA pressure by echocardiogram, which we may have been less attuned to previously.
The problem of HFpEF, which is a broad definition for this patient’s condition, is that it’s not something new: It’s something that we’ve been struggling with for many years, particularly in older patients. As Dr. Francis mentioned, these patients are typically women with a history of hypertension and often, other comorbidities.
This case would seem unusual, mostly due to the estimated PA pressure. I think that’s probably what caught the eye of the clinicians here.
We probably wouldn’t have given it a second thought if the estimated PA pressure was in the 40s or, maybe, 50s. It is only once the pressure increases above 60 mm Hg that we begin to realize that some other factor could be involved or that something was overlooked, which we now need to look at a little more closely.
Since you didn’t mention anything about the mitral valve, let’s assume that there isn’t any mitral valve disease here and we’re not missing significant mitral regurgitation at rest or ischemic mitral regurgitation, which is something you would want to rule out. This is a common problem, and I think we’re seeing it more often because we’re more closely looking for it.
DR. FRANCIS: I agree, Dr. Givertz. But, it turns out that this patient has minimal mitral regurgitation.
Dr. Lewis, as you have a focused laboratory interest in this problem, what do you think of this case, particularly with regard to the scope of the problem?
DR. LEWIS: I think that it is clearly a major problem. I would like to add that these patients can come to subspecialty care through different routes. It’s not uncommon for a patient like the one in question to be hospitalized through the general medical service or to be assessed by a pulmonary specialist or general cardiologist.
Unlike patients who have advanced LV systolic dysfunction and are often routed into the well-oiled machine of an advanced cardiomyopathy clinic, these patients are identified through providers with different scopes of practice. For example, I believe that these patients are often initially referred to a pulmonary physician.
This makes the problem more challenging because there’s a less-inherent ownership within a predefined group of clinicians. Additionally, this case has features that are clearly suggestive of a pre-capillary PH as well as provide evidence of increased left-sided filling pressures. These patients may be directed through different routes of care in the hospital.
It does present a slight problem, particularly when the house staff is presenting the case and people have their own ideas about what we should call it. Dr. Givertz, what should we call this syndrome? What do you refer to it as at the Brigham?
DR. GIVERTZ: That’s an excellent question. We have moved away from the older terminology of diastolic HF, as probably, most have, although there are some strong proponents of this terminology, which assumes that there is some abnormality in diastolic function.
In an 83-year-old woman with long-standing hypertension, I think LV hypertrophy, a small cavity on her echocardiogram report, and a large left atrium are certainly markers of diastolic dysfunction. So, calling this diastolic HF might be justified.
We probably overstep that terminology in patients who have the clinical syndrome of HF, as this patient does, with dyspnea on exertion, volume overload, and a normal or near-normal EF. So, this meets the basic definition of HFpEF, and there are echocardiographic indicators of abnormal diastolic function. So, you could call it diastolic HF.
The key indicator here is the breadth. Although it is just a descriptive term, I would prefer using a term like HFpEF, and we generally use this term on rounds in the hospital or in an ambulatory setting.
The terminology is important because we don’t assume that this is just a problem of diastole or relaxation. There may be other comorbidities and aspects related to the pathophysiology,2 which may have a larger role to play than only causing a simple abnormality in diastolic function.
DR. FRANCIS: I agree, Dr. Givertz, although I have found that there are certain experts and authors who are accustomed to a specific terminology, which is unfortunate because some patients don’t really have diastolic dysfunction, and I don’t think we understand the underlying pathophysiology very well.
DR. PRITZKER: Drs. Givertz and Lewis, is there a certain trigger in echocardiogram reports such that when an elevation in PA pressure is evident, as in this case, the examiners make the diastology a little bit more manifest or look at tricuspid annular plane systolic excursion or PA acceleration time?
I think we all have difficulties with that. I have looked at reports from several different hospitals, and nobody provides a consistent report once an increase in PA pressure has been identified.
DR. GIVERTZ: I think that’s an excellent point. We get comprehensive echocardiogram reports; it’s the way that our laboratory runs. But we don’t have a trigger that leads to the reporting of additional indices of diastolic function. With high PA pressures, a focus on the right ventricle (RV) (in terms of tricuspid annular plane systolic excursion measurements or RV dimensions) would be particularly helpful in terms of having additional information about the etiology of PH.
DR. LEWIS: In our institution, there is no dedicated report type for this constellation of findings. The conclusion of the interpretation would indicate that findings were consistent with diastolic dysfunction.
Dr. Francis, you mentioned in the description that there was some indication of left atrial pressure elevation in the Doppler studies. I know our echocardiographers at MGH generally avoid reporting Doppler measurements such as E/A ratios or estimates of left atrial pressure based on E/E' values.Ultimately, for patient management, it will be important to establish widely accepted echocardiographic criteria that differentiate precapillary PH, postcapillary PH, and mixed PH.
DR. FRANCIS: That’s a good point. As you know, some hospitals, like my former institution the Cleveland Clinic, did attempt to carry out the calculations, but in most places, these calculations are really not done. I personally assessed the echocardiogram as a consequence of my obsessive behavior. I have also talked to the echocardiographers about it and gathered an intuitive sense of impaired filling.
If you were to guess, you would say that the left atrial pressure is somewhat higher than normal. This is a really subjective phrase. It wasn’t actually measured, of course, and it wasn’t calculated.
DR. PRITZKER: I think it would be helpful if each institution could, at least, have a trigger, so that when the patient goes back to a family practitioner, a pulmonologist or another specialist, there is some comprehensive information that can give the practitioner some direction.
As echocardiograms become more capable of calculating these derived values (E/E prime, E to A, etc.), even general cardiologists begin feeling confused. So I like Dr. Lewis’ idea about inserting a comment at the end to at least point people in some direction.
DR. FRANCIS: Let’s focus a little on what’s different about this particular patient, which is something that I’m personally interested in. Dr. Lewis, why is it that some of these patients have higher PA pressures than what you would expect, given the clinical context? Is there something unusual about them? Is there something relevant that we can discuss, which might help identify such a patient?
DR. LEWIS: I think that’s a key question regarding the evaluation of patients like this one. For me, the first thing to do when I see a patient like this is to make sure that there aren’t other factors, other than left heart disease, that may be mediating the PH.
Even if there is a suggestion of elevated left atrial pressure, it’s important to think beyond the left heart, in terms of additional direct insults that may have been sustained by the pulmonary circulation. For example, does the patient have chronic obstructive pulmonary disease that may be contributing to the high pulmonary arterial pressures? Does the patient have sleep apnea or a history of chronic thromboembolic disease in the lung? Even an overlapping syndrome such as scleroderma or sarcoidosis can cause LV dysfunction as well as pulmonary arterial hypertension but would be unusual to detect at an advanced age in terms of relevance to this case.
Epidemiologic studies can also provide clues regarding the type of patients who tend to have higher pulmonary arterial pressures (eg, older individuals or people with higher systolic blood pressure).
I have been surprised by our relative lack of ability to predict who’s going to have a higher transpulmonary gradient, based on the clinical features alone. For a given left atrial pressure, why do some patients have higher transpulmonary gradients than others?
Elevation of transpulmonary gradients is not simply related to degree of elevation in left-sided pressure alone. That is clear from the relatively weak correlation between PA systolic pressure and left-sided filling pressure.3 I don’t think it is related to the duration of HF alone either.
In our patient population, we’ve recorded the time when patients were diagnosed with HF because I commonly hear, “Well, if the patient has HF for a sufficient amount of time, he/she will gradually develop increases in transpulmonary gradient because of high pressure on the left side of the heart.” The duration of HF was not significantly related to the transpulmonary gradient. Therefore, I believe we need to look at new ways of understanding which patients will develop high transpulmonary gradients.
There’s some work being conducted in this area. Investigators at Dr. Francis’s former institution recently published a study about arginine metabolism dysregulation and some circulating markers that were high in patients who had a high burden of precapillary PH and HF.4
Dr. Givertz has been involved in studies evaluating endothelin levels or cyclic guanosine monophosphate-adenosine monophosphate release through the pulmonary circulation as potential indicators of dysregulation in signaling systems within pulmonary circulation. There may also be permissive genotypes that predispose patients to develop precapillary PH in the setting of left heart dysfunction.
So, I think that the question is a good one, but for me, it raises many more questions about why these patients are different. I don’t think we can explain it on the basis of clinical characteristics alone.
DR. FRANCIS: Dr. Lewis has had extensive experience in studying these patients in the laboratory and performing exercise tests on them. Dr. Lewis, from a clinical rather than research standpoint, when would you actually consider conducting more invasive studies like you are currently doing in the research laboratory?
DR. LEWIS: We haven’t talked about invasive phenotyping of patients yet, but to address Dr. Givertz’s point, the question pertains to when we need to carefully study the conditions in the pulmonary circulation. I believe that knowing the left atrial pressure is important for truly understanding the transpulmonary gradient, and I’d like to know whether it’s greater than 15 mm Hg, as Dr. Pritzker indicated; this level serves as a trigger for looking carefully into the pulmonary circulation.
Dr. Francis, I find the exercise status to be particularly informative in patients who exhibit shortness of breath with exertion. However, it’s not entirely clear whether their symptoms are being mediated by left-heart disease or a primary insult to the pulmonary circulation.
Often, even when patients have a resting right-heart catheterization, we see borderline elevations in pulmonary arterial pressure, say, a mean pressure of 20 to 25 mm Hg, and a pulmonary capillary wedge pressure of 10 to 15 mm Hg, but symptoms are present.
In such cases, instead of looking at hemodynamic measurements during a single moment in time, I find it quite helpful to make serial measurements under different loading conditions that are introduced by the very highly relevant physiologic state of exercise. We often encounter patients who have shortness of breath with exertion, which is not entirely explained by the initial diagnostic testing results.
I find it particularly useful to characterize the changes in the pulmonary capillary wedge pressure and transpulmonary gradient when patients are subjected to exercise. We can begin to classify patients based on whether their burden of precapillary PH is high or there is truly a left-sided predominance to their symptoms.
DR. FRANCIS: This gives rise to question of whether therapeutic targets exist. We all know that they exist, but what is the therapy? Perhaps, we’re not so sure about that.
DR. PRITZKER: We’re predominantly using sildenafil because it’s easily available in the hospital and doesn’t require cumbersome pre-approval for acute use like in the case of endothelin antagonists. We have a good experience of using it, and we follow it up with 6-min walks, which perhaps, in this population, is slightly easier to do.
DR. FRANCIS: Dr. Pritzker, what is your threshold for administering sildenafil? Is it lower?
DR. PRITZKER: Yes, because I think there’s more relevant evidence, including the provocative work of an Italian group and Dr. Lewis’ work. We don’t necessarily know why we’re making them better yet, but clearly, people are having fewer symptoms. This is more anecdotal than longitudinal, but patients have fewer symptoms, feel better, and seem to have better function. We also see fewer hospital admissions. I think a previous paper published at the end of last year presented similar findings.
DR. FRANCIS: Dr. Givertz, can you comment about the therapy?
DR. GIVERTZ: I think we’re all gaining experience with using sildenafil or longer-acting phosphodiesterase type 5 (PDE5) inhibitors in this patient population, but that might not be the first thing I would reach for.
This patient was admitted with acute HF. Presumably, there was a fluid problem here as well. I might be initially inclined to make sure I have optimized diuretic therapy. If this patient wasn’t already on nitrates, given the underlying coronary disease and hypertension, I think administering nitrates would be a reasonable next step.
Although there are certainly patients who cannot tolerate nitrate therapy due to its side effects such as headache or light-headedness, I might try administering nitrates before opting for a PDE5 inhibitor.
DR. PRITZKER: I assumed that the patient had been stabilized, and we still had a therapeutic gap after conventional therapy.
DR. FRANCIS: That’s a good point because it is related to the timing of initiation of such therapy; I am quite conservative about it. I don’t have as much experience as the other physicians on the panel, but my tendency would be to go with conventional medical therapy, as bad as it is, and if the patient didn’t improve thereafter, I would think about sending the patient to Dr. Pritzker or beginning sildenafil treatment myself.
Dr. Lewis, you’re actually involved in a National Institute of Health study, a consortium study. Is that right?
DR. LEWIS: That’s right; it’s the Phosphodiesterase-5 Inhibition to Improve Clinical Status and Exercise Capacity in Diastolic Heart Failure (RELAX)trial.5 Michael and Maggie Redfield from the Mayo Clinic are involved as well, and Dr. Redfield is the principal architect of the trial. But, we are adjudicating the primary end point here, which is a cardiopulmonary exercise endpoint.
Dr. Francis, I agree with Drs. Pritzker and Givertz in terms of the therapeutic approach. There certainly have been some cautionary accounts from other pulmonary vasodilators that have been tried in patients who have left heart disease, predominantly those with systolic dysfunction and have not resulted in improved outcomes.6 Therefore, I tend to refrain from using endothelin antagonists as well as the prostacyclins.
There has been some promising work done with PDE5 inhibitors in left-heart disease, and we will know the results from the RELAX trial soon. We completed enrollment of just over 200 patients, and we’re trying to determine whether we can improve exercise capacity over a period of 24 weeks with sildenafil in patients with HFpEF.
The results of PDE5 inhibition in HF with preserved LVEF from the Guazzi group demonstrated that patients with a significant burden of right-heart disease may derive significant improvement in precapillary PH with sildenafil.7 I believe if there is right-heart dysfunction that is largely attributed to high pulmonary vascular resistance or a high transpulmonary gradient, then pulmonary vasodilators like PDE5 inhibitors will have a promising role to play. Potentially, there are also some novel therapies involving the use of some of the soluble guanylate cyclase stimulators and activators in trials.
If patients have a predominant left-heart dysfunction with a modest transpulmonary gradient, I think they are much better served by trying to optimize the diuretic therapy and treat the systemic hypertension.
DR. FRANCIS: Thank you, Dr. Lewis. I think we agree that this is clearly an emerging problem, and it’s prevalence is increasing, partly because of the aging population. It is true that these patients are often admitted to general medicine wards and are sometimes transferred to the cardiology groups when they are faring poorly, and I don’t know what we can do about that.
There does seem to be a subset of patients with disproportionate PH, and there’s a lot of interest in that particular subset. I don’t know if they’re clearly identifiable from the phenoclinical phenotype, but I think that they pose more difficult problems.
Lastly, I’m delighted to know that the RELAX trial is on the verge of completion, and we will probably have some results soon. Management of patients with HFpEF and PH has been a problem for all of us, and as yet, we do not have good, randomized, controlled trials to help us understand how to manage them.
On that note, I’d like to thank the discussants for their participation.
FoxP2 Media LLC is the publisher of The Medical Roundtable.
DR. GIVERTZ: I’m Michael Givertz. I am the Medical Director of the Heart Transplant and Mechanical Circulatory Support Program at the Brigham and Women’s Hospital in Boston, Massachusetts. I am interested in the management of patients with advanced heart disease and those with heart failure with preserved ejection fraction (HFpEF) as well as those with reduced ejection fraction (EF). In particular, I focus on patients with comorbidities, including pulmonary hypertension (PH) and chronic kidney disease.
DR. LEWIS: I’m Greg Lewis from Massachusetts General Hospital (MGH), Boston. I am a member of the Heart Failure and Transplant Unit, and I direct the cardiopulmonary exercise labs at MGH. Like Dr. Givertz, I am interested in heart failure (HF) with both preserved EF and reduced EF and in patients who have PH complicating both the abovementioned conditions.
DR. PRITZKER: I’m Marc Pritzker. I’m a professor of Cardiovascular Medicine and Surgery at the University of Minnesota and a part of the Advanced Heart Failure Group here. I’m also the Director of the Pulmonary Hypertension Center, and I have a special interest in diastolic dysfunction, which is going to be the leading cause of PH in Western society,1 if it isn’t already.
DR. FRANCIS: Let’s begin with a case and then proceed from there. It’s a short vignette.
An 83-year-old woman is suffering from long-standing diabetes, coronary disease, and hypertension. She’s admitted to the hospital with acute HF. An echocardiogram was performed, which indicates a normal EF of 65%. She has left ventricular (LV) hypertrophy, a small LV cavity, and a large left atrium.
Doppler studies indicate that the left atrial pressure is probably slightly elevated. There is clear diastolic dysfunction, as shown by echocardiography, and enough tricuspid regurgitation to suggest that the pulmonary artery (PA) pressure is approximately 70/30 mm Hg.
She has been given intravenous furosemide, her condition improves, and she is subsequently discharged. She then visits the outpatient clinic where follow-up echocardiogray indicates the persistence of PH. Her estimated PA pressure by echocardiogram is slightly lower (60/30 mm Hg), although her LV filling pressures are probably lower.
Although I’m not exactly sure of the situation at the Brigham or MGH, I suspect that it’s similar to the situation here and around the country: We are now seeing many such patients. They’re often elderly women with many comorbid conditions, and they’re sometimes admitted with atrial fibrillation, which makes the estimation of diastolic function problematic. They’re treated conventionally with diuretics and antihypertensive therapies, but they tend to fare poorly. Often, they return to the hospital and pose a recurrent problem.
Dr. Pritzker, let’s start with you. The patient’s PA pressure is quite high, at least based on the estimation by an echocardiogram. It’s fairly common for these patients to have PH, but this patient’s PA pressure is probably disproportionately high relative to her slightly elevated left atrial pressure.
Granted that we don’t have the hemodynamic data from the catheterization lab; nonetheless, am I right? Are we seeing more of these patients with so-called reactive or disproportionate PH or hypertension that is higher than what you would expect, given the clinical context?
DR. PRITZKER: I agree. We probably encounter 3 or 4 new patients a week who are referred to us because of certain observations during echocardiographic screening. In reality, these patients have diastolic dysfunction, and probably half of the patients in our clinic have PH that appears to be higher than the classical expectation that the PA pressure follows the left atrial pressure.
This situation could be viewed in 2 ways. One is the eyeball method, which many people do and just say, “Well, that seems high to me compared to what the wedge pressure is.” The transpulmonary gradient, which is the mean PA pressure minus the wedge pressure, or, sometimes, the pulmonary diastolic pressure minus the wedge pressure, is above 15 mm Hg.
Based on the information given in this case, the patient’s mean PA pressure is above 40 mm Hg. I’m assuming that modestly elevated pulmonary capillary wedge pressure is approximately 15 to 20 mm Hg, which indicates an elevated transpulmonary gradient by any measurement.
DR. FRANCIS: Dr. Givertz, what is the scope of this problem in your hospital?
DR. GIVERTZ: As Dr. Pritzker was alluding to, we are seeing many such patients. I’m not sure if we’re actually looking for them or if we are looking more closely at the estimates of PA pressure by echocardiogram, which we may have been less attuned to previously.
The problem of HFpEF, which is a broad definition for this patient’s condition, is that it’s not something new: It’s something that we’ve been struggling with for many years, particularly in older patients. As Dr. Francis mentioned, these patients are typically women with a history of hypertension and often, other comorbidities.
This case would seem unusual, mostly due to the estimated PA pressure. I think that’s probably what caught the eye of the clinicians here.
We probably wouldn’t have given it a second thought if the estimated PA pressure was in the 40s or, maybe, 50s. It is only once the pressure increases above 60 mm Hg that we begin to realize that some other factor could be involved or that something was overlooked, which we now need to look at a little more closely.
Since you didn’t mention anything about the mitral valve, let’s assume that there isn’t any mitral valve disease here and we’re not missing significant mitral regurgitation at rest or ischemic mitral regurgitation, which is something you would want to rule out. This is a common problem, and I think we’re seeing it more often because we’re more closely looking for it.
DR. FRANCIS: I agree, Dr. Givertz. But, it turns out that this patient has minimal mitral regurgitation.
Dr. Lewis, as you have a focused laboratory interest in this problem, what do you think of this case, particularly with regard to the scope of the problem?
DR. LEWIS: I think that it is clearly a major problem. I would like to add that these patients can come to subspecialty care through different routes. It’s not uncommon for a patient like the one in question to be hospitalized through the general medical service or to be assessed by a pulmonary specialist or general cardiologist.
Unlike patients who have advanced LV systolic dysfunction and are often routed into the well-oiled machine of an advanced cardiomyopathy clinic, these patients are identified through providers with different scopes of practice. For example, I believe that these patients are often initially referred to a pulmonary physician.
This makes the problem more challenging because there’s a less-inherent ownership within a predefined group of clinicians. Additionally, this case has features that are clearly suggestive of a pre-capillary PH as well as provide evidence of increased left-sided filling pressures. These patients may be directed through different routes of care in the hospital.
It does present a slight problem, particularly when the house staff is presenting the case and people have their own ideas about what we should call it. Dr. Givertz, what should we call this syndrome? What do you refer to it as at the Brigham?
DR. GIVERTZ: That’s an excellent question. We have moved away from the older terminology of diastolic HF, as probably, most have, although there are some strong proponents of this terminology, which assumes that there is some abnormality in diastolic function.
In an 83-year-old woman with long-standing hypertension, I think LV hypertrophy, a small cavity on her echocardiogram report, and a large left atrium are certainly markers of diastolic dysfunction. So, calling this diastolic HF might be justified.
We probably overstep that terminology in patients who have the clinical syndrome of HF, as this patient does, with dyspnea on exertion, volume overload, and a normal or near-normal EF. So, this meets the basic definition of HFpEF, and there are echocardiographic indicators of abnormal diastolic function. So, you could call it diastolic HF.
The key indicator here is the breadth. Although it is just a descriptive term, I would prefer using a term like HFpEF, and we generally use this term on rounds in the hospital or in an ambulatory setting.
The terminology is important because we don’t assume that this is just a problem of diastole or relaxation. There may be other comorbidities and aspects related to the pathophysiology,2 which may have a larger role to play than only causing a simple abnormality in diastolic function.
DR. FRANCIS: I agree, Dr. Givertz, although I have found that there are certain experts and authors who are accustomed to a specific terminology, which is unfortunate because some patients don’t really have diastolic dysfunction, and I don’t think we understand the underlying pathophysiology very well.
DR. PRITZKER: Drs. Givertz and Lewis, is there a certain trigger in echocardiogram reports such that when an elevation in PA pressure is evident, as in this case, the examiners make the diastology a little bit more manifest or look at tricuspid annular plane systolic excursion or PA acceleration time?
I think we all have difficulties with that. I have looked at reports from several different hospitals, and nobody provides a consistent report once an increase in PA pressure has been identified.
DR. GIVERTZ: I think that’s an excellent point. We get comprehensive echocardiogram reports; it’s the way that our laboratory runs. But we don’t have a trigger that leads to the reporting of additional indices of diastolic function. With high PA pressures, a focus on the right ventricle (RV) (in terms of tricuspid annular plane systolic excursion measurements or RV dimensions) would be particularly helpful in terms of having additional information about the etiology of PH.
DR. LEWIS: In our institution, there is no dedicated report type for this constellation of findings. The conclusion of the interpretation would indicate that findings were consistent with diastolic dysfunction.
Dr. Francis, you mentioned in the description that there was some indication of left atrial pressure elevation in the Doppler studies. I know our echocardiographers at MGH generally avoid reporting Doppler measurements such as E/A ratios or estimates of left atrial pressure based on E/E' values.Ultimately, for patient management, it will be important to establish widely accepted echocardiographic criteria that differentiate precapillary PH, postcapillary PH, and mixed PH.
DR. FRANCIS: That’s a good point. As you know, some hospitals, like my former institution the Cleveland Clinic, did attempt to carry out the calculations, but in most places, these calculations are really not done. I personally assessed the echocardiogram as a consequence of my obsessive behavior. I have also talked to the echocardiographers about it and gathered an intuitive sense of impaired filling.
If you were to guess, you would say that the left atrial pressure is somewhat higher than normal. This is a really subjective phrase. It wasn’t actually measured, of course, and it wasn’t calculated.
DR. PRITZKER: I think it would be helpful if each institution could, at least, have a trigger, so that when the patient goes back to a family practitioner, a pulmonologist or another specialist, there is some comprehensive information that can give the practitioner some direction.
As echocardiograms become more capable of calculating these derived values (E/E prime, E to A, etc.), even general cardiologists begin feeling confused. So I like Dr. Lewis’ idea about inserting a comment at the end to at least point people in some direction.
DR. FRANCIS: Let’s focus a little on what’s different about this particular patient, which is something that I’m personally interested in. Dr. Lewis, why is it that some of these patients have higher PA pressures than what you would expect, given the clinical context? Is there something unusual about them? Is there something relevant that we can discuss, which might help identify such a patient?
DR. LEWIS: I think that’s a key question regarding the evaluation of patients like this one. For me, the first thing to do when I see a patient like this is to make sure that there aren’t other factors, other than left heart disease, that may be mediating the PH.
Even if there is a suggestion of elevated left atrial pressure, it’s important to think beyond the left heart, in terms of additional direct insults that may have been sustained by the pulmonary circulation. For example, does the patient have chronic obstructive pulmonary disease that may be contributing to the high pulmonary arterial pressures? Does the patient have sleep apnea or a history of chronic thromboembolic disease in the lung? Even an overlapping syndrome such as scleroderma or sarcoidosis can cause LV dysfunction as well as pulmonary arterial hypertension but would be unusual to detect at an advanced age in terms of relevance to this case.
Epidemiologic studies can also provide clues regarding the type of patients who tend to have higher pulmonary arterial pressures (eg, older individuals or people with higher systolic blood pressure).
I have been surprised by our relative lack of ability to predict who’s going to have a higher transpulmonary gradient, based on the clinical features alone. For a given left atrial pressure, why do some patients have higher transpulmonary gradients than others?
Elevation of transpulmonary gradients is not simply related to degree of elevation in left-sided pressure alone. That is clear from the relatively weak correlation between PA systolic pressure and left-sided filling pressure.3 I don’t think it is related to the duration of HF alone either.
In our patient population, we’ve recorded the time when patients were diagnosed with HF because I commonly hear, “Well, if the patient has HF for a sufficient amount of time, he/she will gradually develop increases in transpulmonary gradient because of high pressure on the left side of the heart.” The duration of HF was not significantly related to the transpulmonary gradient. Therefore, I believe we need to look at new ways of understanding which patients will develop high transpulmonary gradients.
There’s some work being conducted in this area. Investigators at Dr. Francis’s former institution recently published a study about arginine metabolism dysregulation and some circulating markers that were high in patients who had a high burden of precapillary PH and HF.4
Dr. Givertz has been involved in studies evaluating endothelin levels or cyclic guanosine monophosphate-adenosine monophosphate release through the pulmonary circulation as potential indicators of dysregulation in signaling systems within pulmonary circulation. There may also be permissive genotypes that predispose patients to develop precapillary PH in the setting of left heart dysfunction.
So, I think that the question is a good one, but for me, it raises many more questions about why these patients are different. I don’t think we can explain it on the basis of clinical characteristics alone.
DR. FRANCIS: Dr. Lewis has had extensive experience in studying these patients in the laboratory and performing exercise tests on them. Dr. Lewis, from a clinical rather than research standpoint, when would you actually consider conducting more invasive studies like you are currently doing in the research laboratory?
DR. LEWIS: We haven’t talked about invasive phenotyping of patients yet, but to address Dr. Givertz’s point, the question pertains to when we need to carefully study the conditions in the pulmonary circulation. I believe that knowing the left atrial pressure is important for truly understanding the transpulmonary gradient, and I’d like to know whether it’s greater than 15 mm Hg, as Dr. Pritzker indicated; this level serves as a trigger for looking carefully into the pulmonary circulation.
Dr. Francis, I find the exercise status to be particularly informative in patients who exhibit shortness of breath with exertion. However, it’s not entirely clear whether their symptoms are being mediated by left-heart disease or a primary insult to the pulmonary circulation.
Often, even when patients have a resting right-heart catheterization, we see borderline elevations in pulmonary arterial pressure, say, a mean pressure of 20 to 25 mm Hg, and a pulmonary capillary wedge pressure of 10 to 15 mm Hg, but symptoms are present.
In such cases, instead of looking at hemodynamic measurements during a single moment in time, I find it quite helpful to make serial measurements under different loading conditions that are introduced by the very highly relevant physiologic state of exercise. We often encounter patients who have shortness of breath with exertion, which is not entirely explained by the initial diagnostic testing results.
I find it particularly useful to characterize the changes in the pulmonary capillary wedge pressure and transpulmonary gradient when patients are subjected to exercise. We can begin to classify patients based on whether their burden of precapillary PH is high or there is truly a left-sided predominance to their symptoms.
DR. FRANCIS: This gives rise to question of whether therapeutic targets exist. We all know that they exist, but what is the therapy? Perhaps, we’re not so sure about that.
DR. PRITZKER: We’re predominantly using sildenafil because it’s easily available in the hospital and doesn’t require cumbersome pre-approval for acute use like in the case of endothelin antagonists. We have a good experience of using it, and we follow it up with 6-min walks, which perhaps, in this population, is slightly easier to do.
DR. FRANCIS: Dr. Pritzker, what is your threshold for administering sildenafil? Is it lower?
DR. PRITZKER: Yes, because I think there’s more relevant evidence, including the provocative work of an Italian group and Dr. Lewis’ work. We don’t necessarily know why we’re making them better yet, but clearly, people are having fewer symptoms. This is more anecdotal than longitudinal, but patients have fewer symptoms, feel better, and seem to have better function. We also see fewer hospital admissions. I think a previous paper published at the end of last year presented similar findings.
DR. FRANCIS: Dr. Givertz, can you comment about the therapy?
DR. GIVERTZ: I think we’re all gaining experience with using sildenafil or longer-acting phosphodiesterase type 5 (PDE5) inhibitors in this patient population, but that might not be the first thing I would reach for.
This patient was admitted with acute HF. Presumably, there was a fluid problem here as well. I might be initially inclined to make sure I have optimized diuretic therapy. If this patient wasn’t already on nitrates, given the underlying coronary disease and hypertension, I think administering nitrates would be a reasonable next step.
Although there are certainly patients who cannot tolerate nitrate therapy due to its side effects such as headache or light-headedness, I might try administering nitrates before opting for a PDE5 inhibitor.
DR. PRITZKER: I assumed that the patient had been stabilized, and we still had a therapeutic gap after conventional therapy.
DR. FRANCIS: That’s a good point because it is related to the timing of initiation of such therapy; I am quite conservative about it. I don’t have as much experience as the other physicians on the panel, but my tendency would be to go with conventional medical therapy, as bad as it is, and if the patient didn’t improve thereafter, I would think about sending the patient to Dr. Pritzker or beginning sildenafil treatment myself.
Dr. Lewis, you’re actually involved in a National Institute of Health study, a consortium study. Is that right?
DR. LEWIS: That’s right; it’s the Phosphodiesterase-5 Inhibition to Improve Clinical Status and Exercise Capacity in Diastolic Heart Failure (RELAX)trial.5 Michael and Maggie Redfield from the Mayo Clinic are involved as well, and Dr. Redfield is the principal architect of the trial. But, we are adjudicating the primary end point here, which is a cardiopulmonary exercise endpoint.
Dr. Francis, I agree with Drs. Pritzker and Givertz in terms of the therapeutic approach. There certainly have been some cautionary accounts from other pulmonary vasodilators that have been tried in patients who have left heart disease, predominantly those with systolic dysfunction and have not resulted in improved outcomes.6 Therefore, I tend to refrain from using endothelin antagonists as well as the prostacyclins.
There has been some promising work done with PDE5 inhibitors in left-heart disease, and we will know the results from the RELAX trial soon. We completed enrollment of just over 200 patients, and we’re trying to determine whether we can improve exercise capacity over a period of 24 weeks with sildenafil in patients with HFpEF.
The results of PDE5 inhibition in HF with preserved LVEF from the Guazzi group demonstrated that patients with a significant burden of right-heart disease may derive significant improvement in precapillary PH with sildenafil.7 I believe if there is right-heart dysfunction that is largely attributed to high pulmonary vascular resistance or a high transpulmonary gradient, then pulmonary vasodilators like PDE5 inhibitors will have a promising role to play. Potentially, there are also some novel therapies involving the use of some of the soluble guanylate cyclase stimulators and activators in trials.
If patients have a predominant left-heart dysfunction with a modest transpulmonary gradient, I think they are much better served by trying to optimize the diuretic therapy and treat the systemic hypertension.
DR. FRANCIS: Thank you, Dr. Lewis. I think we agree that this is clearly an emerging problem, and it’s prevalence is increasing, partly because of the aging population. It is true that these patients are often admitted to general medicine wards and are sometimes transferred to the cardiology groups when they are faring poorly, and I don’t know what we can do about that.
There does seem to be a subset of patients with disproportionate PH, and there’s a lot of interest in that particular subset. I don’t know if they’re clearly identifiable from the phenoclinical phenotype, but I think that they pose more difficult problems.
Lastly, I’m delighted to know that the RELAX trial is on the verge of completion, and we will probably have some results soon. Management of patients with HFpEF and PH has been a problem for all of us, and as yet, we do not have good, randomized, controlled trials to help us understand how to manage them.
On that note, I’d like to thank the discussants for their participation.
FoxP2 Media LLC is the publisher of The Medical Roundtable.
FoxP2 Media LLC

Neurohormonal control of heart failure
We have known for more than 100 years that heart failure is characterized by excessive sympathetic nervous system (SNS) activity. Thanks to refinement of this concept in the 1980s and 1990s, we now have a good understanding of SNS activity in both experimental and clinical heart failure. During those two decades, we also realized the pathophysiologic importance of the renin-angiotensin-aldosterone system (RAAS) in patients with heart failure.1 By 2000, it was obvious that heart failure was inextricably intertwined with excessive neurohormonal activity.2,3 This understanding of the pathophysiology of heart failure took on greater importance with the ability to pharmacologically block these neurohormonal systems, thereby demonstrating the detrimental role of neurohormones in the onset and progression of heart failure.
This article is a brief historical and personal description of the study of neurohormonal control mechanisms as they relate to the clinical syndrome of heart failure. The article includes a personal account of how the story unfolded in the cardiology research laboratories at the University of Minnesota.
THE EARLY YEARS: NEUROHORMONAL HYPOTHESIS
A hypothesis emerged gradually in the 1980s suggesting that progression of heart failure was in part a product of excessive SNS and RAAS activity. Many believed that pharmacologic inhibition of these systems might mitigate against progressive cardiac remodeling and thereby reduce symptoms and extend life—the so called neurohormonal hypothesis.4 SNS blockers and RAAS blockers are now widely used in tandem as first-line therapy to treat patients with heart failure,5–11 but in 1980 we were just beginning to consider their therapeutic effects.
This major shift in thinking about neurohormonal systems and heart failure did not come about quickly. Early success was driven by the ability to quickly and precisely measure neurohormones in the laboratory coupled with the availability of drugs specifically designed to block the SNS and RAAS. It was also critically important to embrace the power of randomized controlled trials to test new therapies. Investigators, research nurses, and patients from many medical centers and laboratories should be credited with this astonishing success. I am proud to have been a part of this activity at the University of Minnesota.
THE COHN LABORATORY
Early work done in the 1960s by numerous investigators noted that the failing left ventricle (LV) was exquisitely sensitive to afterload conditions.12–15 John Ross and Eugene Braunwald explored this observation in patients in 1964.15 Jay Cohn, with his unique background in hypertension and hemodynamics, brought the concept back into the laboratory in the early 1970s, where he explored the mechanisms responsible for increased sensitivity to afterload in patients with heart failure.16
I had the good fortune to join Cohn’s laboratory in 1979, when this avenue of heart failure research was in full bloom. A team of investigators was gradually assembled that included Maria Teresa Olivari, who relocated from the Cardiovascular Research Institute in Milan, Italy, directed by Maurizio D. Guazzi. Also joining the group were T. Barry Levine from the University of Michigan, Ann Arbor; Steven Goldsmith from Ohio State University, Columbus; Susan Ziesche from the Minneapolis Veterans Affairs (VA) Medical Center; Thomas Rector, an expert statistician and pharmacologist at the University of Minnesota; and many research fellows, visitors, students, biochemists, statisticians, and research nurses. Joseph Franciosa joined the University of Minnesota group in 1974 and, after completing several important trials, left in 1979 to lead the cardiology group at the Philadelphia VA Medical Center.
The Cohn group developed a working hypothesis that activation of the SNS and RAAS in heart failure was most likely an adaptive mechanism intended for short-term circulatory support, such as in the setting of blood loss, dehydration, shock, volume depletion, or flight response. In patients with heart failure, according to the hypothesis, the SNS and RAAS activity persisted beyond that needed for adaptation, with chronic release of norepinephrine (NE), renin, angiotensin II, aldosterone, and other neurohormones. The neurohormones ultimately became “maladaptive.” Thanks to the assaying skills of Ada Simon, we had the early advantage of precise and rapid radioenzyme measurement of plasma norepinephrine and renin activity in the blood of patients and animals.
We believed that neurohormonal activation contributed in part to the excessive afterload conditions observed in heart failure. We also thought that excessive neurohormonal activation directly impaired cardiac systolic function. The obvious next step was to explore whether neurohormonal antagonists would improve myocardial performance.
Under the leadership of Steven Goldsmith, many studies were performed to investigate reflex control mechanisms and their pathogenic role in patients with heart failure. The accumulating data suggested that persistent, excessive neurohormonal activity was characteristic of heart failure and that it was associated with a poor prognosis.17 The precise mechanism that drives activation of the SNS remained elusive, however, and is poorly defined even today. In that era, when β-adrenergic blockers were believed to be contraindicated, we inhibited the central SNS with bromocriptine, clonidine, and guanfacine with modestly favorable responses. We inhibited circulating arginine vasopressin antibody (thanks to Prof. Alan Cowley for noting an acute favorable response).
THE PHARMACOLOGIC ERA
The 1980s and 1990s saw the availability of several pharmacologic tools for assessing the roles of the SNS and RAAS in heart failure. The hypotensive effects of angiotensin-converting enzyme (ACE) inhibitors and, later, angiotensin-receptor blockers (ARBs) were sources of concern, since many patients with advanced heart failure had low- to normal-range blood pressures before they received RAAS blockers. However, our group as well as others observed that abrupt blood pressure reduction occurred primarily in patients with very hyperreninemic responses to intravenous diuretics (ie, volume-depleted patients). Eventually, we learned that low baseline blood pressure did not adversely affect outcomes when vasodilators were used in patients with heart failure,18,19 leading us to titrate these drugs upward over days to weeks.
Several different combinations of vasodilators were used successfully to treat heart failure, including hydralazine, isosorbide dinitrate,20 ACE inhibitors,21,22 and ARBs.8,23–28 Direct-acting calcium channel blocking vasodilators, such as amlodipine, did not improve survival in patients with systolic heart failure, although they appeared to be safe in this setting.29 The aldosterone receptor blockers spironolactone30 and eplerenone31 were later demonstrated to improve survival of patients with advanced systolic heart failure when added to vasodilator therapy.
By the end of the 1990s, it was evident that drugs that blocked the SNS and RAAS were not just vasodilators or “afterload reducers,” similar to α-blockers, hydralazine, nitrates, and amlodipine. Neurohormonal blockers were doing something profoundly beneficial not observed with more direct-acting vasodilators.32–37 Simple afterload reduction was not enough in patients with systolic heart failure.
Neurohormonal antagonists were acting more directly on the myocardium. They were preventing the progression of LV remodeling and, in some cases, promoting reverse remodeling, thus improving myocardial function and favorably influencing the natural history of heart failure.31–39 We were astonished to discover that the failing, dilated heart could revert to normal size in response to neurohormone blockade with ACE inhibitors and β-adrenergic blockers; these findings were soon reported by other laboratories as well.
Contrary to our concept of heart failure in the 1970s, we now understood that the heart has inherent plasticity. It can dilate in response to abnormal loading conditions or myocardial injury, and it can restore itself to normal size when neurohormones are blocked and perverse loading conditions are improved. This reversal can occur spontaneously if an offending agent such as chronic alcohol use or inflammation is removed, but it is likely facilitated by SNS and RAAS blockers.
THE REMODELING ERA
Ken McDonald joined the University of Minnesota lab in 1989 as a research fellow. His skill in conducting both animal and clinical mechanistic studies was pivotal to our achieving our research goals. The inspired animal work by Boston-based Marc and Janice Pfeffer revealed the significance of the LV remodeling concept in the development of heart failure36: ventricular remodeling was a hallmark of systolic heart failure, and pharmacologic inhibition of LV remodeling by blocking neurohormones had profound clinical implications.
Under the direction of Wenda Carlyle, a molecular biology laboratory was established at the University of Minnesota whose work was dedicated solely to exploration of remodeling at a very basic level. Alan Hirsch was recruited from Victor Dzau’s laboratory at Brigham and Women’s Hospital in Boston to extend our efforts to understand the molecular basis of cardiac remodeling. Ken McDonald guided the use of magnetic resonance imaging to study remodeling in dogs.
The late 1970s saw the initiation and eventual execution of several important clinical trials, including the Vasodilator Heart Failure Trials (V-HeFT I and V-HeFT II)40,41 under our leadership, and Studies of Left Ventricular Dysfunction (SOLVD)5,6 under the leadership of Salim Yusuf and others at the National Heart Lung and Blood Institute (NHLBI). Many neuro hormonal and remodeling substudies sprang from these large clinical trials. Spencer Kubo joined our group from the Medical College of Cornell University in the mid-1980s, and he immediately demonstrated his prowess in clinical research. He also recruited Alan Bank to study the endothelium in both experimental and human heart failure.
Integrating the molecular, animal, and clinical laboratories allowed us to pursue many mechanistic studies. Laboratory meetings, often held on Saturday mornings, generated ideas for program projects that were subsequently funded by NHLBI. Birthday parties and other social events with laboratory staff and their families were part of our fabric. Late-night trips to the Post Office to send off abstracts for national meetings before the midnight deadline were a regular feature.
Our coordination of and participation in the large clinical trials allowed us to meet frequently in Bethesda with colleagues from other major centers, fostering many collaborations and friendships that continue to thrive. Susan Ziesche deserves much of the credit for coordinating many groups that were part of these large, complex trials. Cheryl Yano, our administrator, also played a key role. All National Institutes of Health (NIH) grants passed through Cheryl, and she worked tirelessly to ensure that the proposals were in the best possible shape before we submitted them. Inder Anand joined our group in the early 1990s and became a major analytical force. Jay Cohn was the intellectual leader of the group, as well as our soul and inspiration. People worked hard for him, and he taught us much in a setting that valued creativity and new ideas above all.
THE LATER YEARS
By 1997, the face of heart failure had changed. New treatments were effective, but there were new challenges to face. I moved that year to the Cleveland Clinic, where I spent 11 enjoyable and productive years. I returned to Minnesota in 2008 to help build a new cardiovascular division.
It is gratifying to look back and see what has become of the “neurohormonal hypothesis.” Today, nearly all major medical centers have heart failure programs, and certification in advanced heart failure/heart transplantation is a reality. Training programs in advanced heart failure and heart transplant are common. The Heart Failure Society of America sprang up in the early 1990s, dedicated to patients with heart failure. Jay Cohn founded the Journal of Cardiac Failure, which flourished under his leadership. Neurohormonal blockers are now considered standard, conventional therapy and are widely used throughout the world.
CONCLUSIONS
Still, there is much work to do. An increasing number of devices are being developed, largely for patients with more advanced heart failure, but attention is also being directed to prevention of heart failure. Identification and possible treatment of patients at risk for the development of heart failure, and identification of those who already have some early structural and functional perturbation without advanced symptoms, are critically important. Since event rates are so low in these patients, we need to create new strategies for studying interventions. In the long term, the best treatment for nearly any condition is early diagnosis and perhaps early treatment with a goal of prevention.
One consequence of our progress over the years may be that heart failure now primarily affects a more elderly group—patients who often have many associated comorbidities. The consequences include more frequent readmissions, large numbers of patients with intractable signs and symptoms, and the emergence of difficult end-of-life decisions. If we could truly prevent heart failure rather than forestall its emergence to a later point in life, perhaps we could do more good.
For me, the study of neurohormonal mechanisms in the setting of heart failure was the centerpiece of my early career. Jay Cohn had asked several of us early in our laboratory experience to choose a neurohormonal system and learn about it in great depth and detail. My assignment was the SNS. Since then, I have never tired of learning about its control mechanisms, how it achieves circulatory homeostasis, how its excess quantities can be directly toxic to the heart, and the variety of pharmacologic ways that we can control it. I am indeed fortunate to have been part of this amazing study group.
- Dzau VJ, Colucci WS, Hollenberg NK, Williams GH. Relation of the renin-angiotensin-aldosterone system to clinical state in congestive heart failure. Circulation 1981; 63:645–651.
- Francis GS, Goldsmith SR, Levine TB, Olivari MT, Cohn JN. The neurohumoral axis in congestive heart failure. Ann Intern Med 1984; 101:370–377.
- Levine TB, Francis GS, Goldsmith SR, Simon AB, Cohn JN. Activity of the sympathetic nervous system and renin-angiotensin system assessed by plasma hormone levels and their relation to hemodynamic abnormalities in congestive heart failure. Am J Cardiol 1982; 49:1659–1666.
- Packer M. The neurohormonal hypothesis: a theory to explain the mechanism of disease progression in heart failure. J Am Coll Cardiol 1992; 20:248–254.
- The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fraction and congestive heart failure. N Engl J Med 1991; 325:293–302.
- The SOLVD Investigators. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N Engl J Med 1992; 327:685–691.
- Pitt B, Zannand F, Remme WJ, et al The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med 1999; 341:709–717.
- ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008; 358:1547–1559.
- CIBIS Investigators and Committees. A randomized trial of β-blockade in heart failure: the Cardiac Insufficiency Bisoprolol Study (CIBIS). Circulation 1994; 90:1765–1773.
- Hjalmarson A, Goldstein S, Fagerberg B, et al Effects of controlled-release metoprolol on total mortality, hospitalizations, and well-being in patients with heart failure: the Metoprolol CR/XL Randomized Intervention Trial in congestive heart failure (MERITHF). JAMA 2000; 283:1295–1302.
- Packer M, Fowler MB, Roecker EB, et al Effect of carvedilol on the morbidity of patients with severe chronic heart failure: results of the carvedilol prospective randomized cumulative survival (COPERNICUS) study. Circulation 2002; 106:2194–2199.
- Imperial ES, Levy MN, Zieske H. Outflow resistance as an independent determinant of cardiac performance. Circ Res 1961; 9:1148–1155.
- Sonnenblick EH, Downing SE. Afterload as a primary determinant of ventricular performance. Am J Physiol 1963; 204:604–610.
- Wilcken DE, Charlier AA, Hoffman JI. Effects of alterations in aortic impedance on the performance of the ventricles. Circ Res 1964; 14:283–293.
- Ross J, Braunwald E. The study of left ventricular function in man by increasing resistance to ventricular ejection with angiotensin. Circulation 1964; 29:739–749.
- Cohn JN. Blood pressure and cardiac performance. Am J Med 1973; 55:351–361.
- Cohn JN, Levine TB, Olivari MT, et al Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med 1984; 311:819–823.
- Anand IS, Tam SW, Rector TS, et al Influence of blood pressure on the effectiveness of a fixed-dose combination of isosorbide dinitrate and hydralazine in the African-American Heart Failure Trial. J Am Coll Cardiol 2007; 49:32–39.
- Rouleau JL, Roecker EB, Tendra M, et al Influence of pretreatment systolic blood pressure on the effect of carvedilol in patients with severe chronic heart failure: the Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) study. J Am Coll Cardiol 2004; 43:1423–1429.
- Taylor AL, Ziesche S, Yancy C, et al Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N Engl J Med 2004; 351:2049–2057.
- Captopril Multicenter Research Group. A placebo-controlled trial of captopril in refractory chronic congestive heart failure. J Am Coll Cardiol 1983; 2:755–763.
- Pfeffer MA, Braunwald E, Moyé LA, et al Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction: results of the survival and ventricular enlargement trial—the SAVE Investigators. N Eng J Med 1992; 327:669–677.
- Curtiss C, Cohn JN, Vrobel T, Franciosa J. Role of the renin-angiotensin system in the systemic vasoconstriction of chronic congestive heart failure. Circulation 1978; 58:763–770.
- Cohn JN, Tognoni G. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl J Med 2001; 345:1667–1675.
- Young JB, Dunlap ME, Pfeffer MA, et al Mortality and morbidity reduction with Candesartan in patients with chronic heart failure and left ventricular systolic dysfunction: results of the CHARM low-left ventricular ejection trials. Circulation 2004; 110:2618–2626.
- Pfeffer MA, McMurray JJ, Velazquez EJ, et al Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 2003; 349:1893–1906.
- ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008; 358:1547–1559.
- Konstam MA, Neaton JD, Dickstein K, et al Effects of high-dose versus lose-dose losartan on clinical outcomes in patients with heart failure (HEAAL study): a randomized, double-blind trial. Lancet 2009; 374:1840–1848.
- Packer M. Prospective randomized amlodipine survival evaluation 2. Presented at: 49th American College of Cardiology meeting; March 2000; Anaheim, CA.
- Pitt B, Zannand F, Remme WJ, et al The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannand F, et al Eplerenone, a selective aldosterone blocker in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Cohn JN. Structural basis for heart failure: ventricular remodeling and its pharmacological inhibition. Circulation 1995; 91:2504–2507.
- Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling—concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. J Am Coll Cardiol 2000; 35:569–581.
- Konstam MA, Kronenberg MW, Rousseau MF, et al Effects of the angiotensin converting enzyme inhibitor enalapril on the long-term progression of left ventricular dilation in patients with asymptomatic systolic dysfunction. Circulation 1993; 88:2277–2283.
- Greenberg B, Quinones MA, Koilpillai C, et al Effects of long-term enalapril therapy on cardiac structure and function in patients with left ventricular dysfunction: results of the SOLVD echocardiography substudy. Circulation 1995; 91:2573–2581.
- Pfeffer JM, Pfeffer MA, Braunwald E. Influence of chronic captopril therapy on the infarcted left ventricle of the rat. Circ Res 1985; 57:84–95.
- Cohn JN. Structural basis for heart failure: ventricular remodeling and its pharmacological inhibition. Circulation 1995; 91:2504–2507.
- McDonald KM, Garr M, Carlyle PF, et al Relative effects of α1-adrenoceptor blockade, converting enzyme inhibitor therapy, and angiotensin II sub-type 1 receptor blockade on ventricular remodeling in the dog. Circulation 1994; 90:3034–3046.
- Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation 1990; 81:1161–1172.
- Cohn JN, Archibald DG, Ziesche S, et al Effect of vasodilator therapy on mortality in chronic congestive heart failure. N Engl J Med 1986; 314:1547–1552.
- Cohn JN, Johnson G, Ziesche S, et al A comparison of enalapril with hydralazine–isosorbide dinitrate in the treatment of chronic congestive heart failure. N Engl J Med 1991; 325:303–310.
We have known for more than 100 years that heart failure is characterized by excessive sympathetic nervous system (SNS) activity. Thanks to refinement of this concept in the 1980s and 1990s, we now have a good understanding of SNS activity in both experimental and clinical heart failure. During those two decades, we also realized the pathophysiologic importance of the renin-angiotensin-aldosterone system (RAAS) in patients with heart failure.1 By 2000, it was obvious that heart failure was inextricably intertwined with excessive neurohormonal activity.2,3 This understanding of the pathophysiology of heart failure took on greater importance with the ability to pharmacologically block these neurohormonal systems, thereby demonstrating the detrimental role of neurohormones in the onset and progression of heart failure.
This article is a brief historical and personal description of the study of neurohormonal control mechanisms as they relate to the clinical syndrome of heart failure. The article includes a personal account of how the story unfolded in the cardiology research laboratories at the University of Minnesota.
THE EARLY YEARS: NEUROHORMONAL HYPOTHESIS
A hypothesis emerged gradually in the 1980s suggesting that progression of heart failure was in part a product of excessive SNS and RAAS activity. Many believed that pharmacologic inhibition of these systems might mitigate against progressive cardiac remodeling and thereby reduce symptoms and extend life—the so called neurohormonal hypothesis.4 SNS blockers and RAAS blockers are now widely used in tandem as first-line therapy to treat patients with heart failure,5–11 but in 1980 we were just beginning to consider their therapeutic effects.
This major shift in thinking about neurohormonal systems and heart failure did not come about quickly. Early success was driven by the ability to quickly and precisely measure neurohormones in the laboratory coupled with the availability of drugs specifically designed to block the SNS and RAAS. It was also critically important to embrace the power of randomized controlled trials to test new therapies. Investigators, research nurses, and patients from many medical centers and laboratories should be credited with this astonishing success. I am proud to have been a part of this activity at the University of Minnesota.
THE COHN LABORATORY
Early work done in the 1960s by numerous investigators noted that the failing left ventricle (LV) was exquisitely sensitive to afterload conditions.12–15 John Ross and Eugene Braunwald explored this observation in patients in 1964.15 Jay Cohn, with his unique background in hypertension and hemodynamics, brought the concept back into the laboratory in the early 1970s, where he explored the mechanisms responsible for increased sensitivity to afterload in patients with heart failure.16
I had the good fortune to join Cohn’s laboratory in 1979, when this avenue of heart failure research was in full bloom. A team of investigators was gradually assembled that included Maria Teresa Olivari, who relocated from the Cardiovascular Research Institute in Milan, Italy, directed by Maurizio D. Guazzi. Also joining the group were T. Barry Levine from the University of Michigan, Ann Arbor; Steven Goldsmith from Ohio State University, Columbus; Susan Ziesche from the Minneapolis Veterans Affairs (VA) Medical Center; Thomas Rector, an expert statistician and pharmacologist at the University of Minnesota; and many research fellows, visitors, students, biochemists, statisticians, and research nurses. Joseph Franciosa joined the University of Minnesota group in 1974 and, after completing several important trials, left in 1979 to lead the cardiology group at the Philadelphia VA Medical Center.
The Cohn group developed a working hypothesis that activation of the SNS and RAAS in heart failure was most likely an adaptive mechanism intended for short-term circulatory support, such as in the setting of blood loss, dehydration, shock, volume depletion, or flight response. In patients with heart failure, according to the hypothesis, the SNS and RAAS activity persisted beyond that needed for adaptation, with chronic release of norepinephrine (NE), renin, angiotensin II, aldosterone, and other neurohormones. The neurohormones ultimately became “maladaptive.” Thanks to the assaying skills of Ada Simon, we had the early advantage of precise and rapid radioenzyme measurement of plasma norepinephrine and renin activity in the blood of patients and animals.
We believed that neurohormonal activation contributed in part to the excessive afterload conditions observed in heart failure. We also thought that excessive neurohormonal activation directly impaired cardiac systolic function. The obvious next step was to explore whether neurohormonal antagonists would improve myocardial performance.
Under the leadership of Steven Goldsmith, many studies were performed to investigate reflex control mechanisms and their pathogenic role in patients with heart failure. The accumulating data suggested that persistent, excessive neurohormonal activity was characteristic of heart failure and that it was associated with a poor prognosis.17 The precise mechanism that drives activation of the SNS remained elusive, however, and is poorly defined even today. In that era, when β-adrenergic blockers were believed to be contraindicated, we inhibited the central SNS with bromocriptine, clonidine, and guanfacine with modestly favorable responses. We inhibited circulating arginine vasopressin antibody (thanks to Prof. Alan Cowley for noting an acute favorable response).
THE PHARMACOLOGIC ERA
The 1980s and 1990s saw the availability of several pharmacologic tools for assessing the roles of the SNS and RAAS in heart failure. The hypotensive effects of angiotensin-converting enzyme (ACE) inhibitors and, later, angiotensin-receptor blockers (ARBs) were sources of concern, since many patients with advanced heart failure had low- to normal-range blood pressures before they received RAAS blockers. However, our group as well as others observed that abrupt blood pressure reduction occurred primarily in patients with very hyperreninemic responses to intravenous diuretics (ie, volume-depleted patients). Eventually, we learned that low baseline blood pressure did not adversely affect outcomes when vasodilators were used in patients with heart failure,18,19 leading us to titrate these drugs upward over days to weeks.
Several different combinations of vasodilators were used successfully to treat heart failure, including hydralazine, isosorbide dinitrate,20 ACE inhibitors,21,22 and ARBs.8,23–28 Direct-acting calcium channel blocking vasodilators, such as amlodipine, did not improve survival in patients with systolic heart failure, although they appeared to be safe in this setting.29 The aldosterone receptor blockers spironolactone30 and eplerenone31 were later demonstrated to improve survival of patients with advanced systolic heart failure when added to vasodilator therapy.
By the end of the 1990s, it was evident that drugs that blocked the SNS and RAAS were not just vasodilators or “afterload reducers,” similar to α-blockers, hydralazine, nitrates, and amlodipine. Neurohormonal blockers were doing something profoundly beneficial not observed with more direct-acting vasodilators.32–37 Simple afterload reduction was not enough in patients with systolic heart failure.
Neurohormonal antagonists were acting more directly on the myocardium. They were preventing the progression of LV remodeling and, in some cases, promoting reverse remodeling, thus improving myocardial function and favorably influencing the natural history of heart failure.31–39 We were astonished to discover that the failing, dilated heart could revert to normal size in response to neurohormone blockade with ACE inhibitors and β-adrenergic blockers; these findings were soon reported by other laboratories as well.
Contrary to our concept of heart failure in the 1970s, we now understood that the heart has inherent plasticity. It can dilate in response to abnormal loading conditions or myocardial injury, and it can restore itself to normal size when neurohormones are blocked and perverse loading conditions are improved. This reversal can occur spontaneously if an offending agent such as chronic alcohol use or inflammation is removed, but it is likely facilitated by SNS and RAAS blockers.
THE REMODELING ERA
Ken McDonald joined the University of Minnesota lab in 1989 as a research fellow. His skill in conducting both animal and clinical mechanistic studies was pivotal to our achieving our research goals. The inspired animal work by Boston-based Marc and Janice Pfeffer revealed the significance of the LV remodeling concept in the development of heart failure36: ventricular remodeling was a hallmark of systolic heart failure, and pharmacologic inhibition of LV remodeling by blocking neurohormones had profound clinical implications.
Under the direction of Wenda Carlyle, a molecular biology laboratory was established at the University of Minnesota whose work was dedicated solely to exploration of remodeling at a very basic level. Alan Hirsch was recruited from Victor Dzau’s laboratory at Brigham and Women’s Hospital in Boston to extend our efforts to understand the molecular basis of cardiac remodeling. Ken McDonald guided the use of magnetic resonance imaging to study remodeling in dogs.
The late 1970s saw the initiation and eventual execution of several important clinical trials, including the Vasodilator Heart Failure Trials (V-HeFT I and V-HeFT II)40,41 under our leadership, and Studies of Left Ventricular Dysfunction (SOLVD)5,6 under the leadership of Salim Yusuf and others at the National Heart Lung and Blood Institute (NHLBI). Many neuro hormonal and remodeling substudies sprang from these large clinical trials. Spencer Kubo joined our group from the Medical College of Cornell University in the mid-1980s, and he immediately demonstrated his prowess in clinical research. He also recruited Alan Bank to study the endothelium in both experimental and human heart failure.
Integrating the molecular, animal, and clinical laboratories allowed us to pursue many mechanistic studies. Laboratory meetings, often held on Saturday mornings, generated ideas for program projects that were subsequently funded by NHLBI. Birthday parties and other social events with laboratory staff and their families were part of our fabric. Late-night trips to the Post Office to send off abstracts for national meetings before the midnight deadline were a regular feature.
Our coordination of and participation in the large clinical trials allowed us to meet frequently in Bethesda with colleagues from other major centers, fostering many collaborations and friendships that continue to thrive. Susan Ziesche deserves much of the credit for coordinating many groups that were part of these large, complex trials. Cheryl Yano, our administrator, also played a key role. All National Institutes of Health (NIH) grants passed through Cheryl, and she worked tirelessly to ensure that the proposals were in the best possible shape before we submitted them. Inder Anand joined our group in the early 1990s and became a major analytical force. Jay Cohn was the intellectual leader of the group, as well as our soul and inspiration. People worked hard for him, and he taught us much in a setting that valued creativity and new ideas above all.
THE LATER YEARS
By 1997, the face of heart failure had changed. New treatments were effective, but there were new challenges to face. I moved that year to the Cleveland Clinic, where I spent 11 enjoyable and productive years. I returned to Minnesota in 2008 to help build a new cardiovascular division.
It is gratifying to look back and see what has become of the “neurohormonal hypothesis.” Today, nearly all major medical centers have heart failure programs, and certification in advanced heart failure/heart transplantation is a reality. Training programs in advanced heart failure and heart transplant are common. The Heart Failure Society of America sprang up in the early 1990s, dedicated to patients with heart failure. Jay Cohn founded the Journal of Cardiac Failure, which flourished under his leadership. Neurohormonal blockers are now considered standard, conventional therapy and are widely used throughout the world.
CONCLUSIONS
Still, there is much work to do. An increasing number of devices are being developed, largely for patients with more advanced heart failure, but attention is also being directed to prevention of heart failure. Identification and possible treatment of patients at risk for the development of heart failure, and identification of those who already have some early structural and functional perturbation without advanced symptoms, are critically important. Since event rates are so low in these patients, we need to create new strategies for studying interventions. In the long term, the best treatment for nearly any condition is early diagnosis and perhaps early treatment with a goal of prevention.
One consequence of our progress over the years may be that heart failure now primarily affects a more elderly group—patients who often have many associated comorbidities. The consequences include more frequent readmissions, large numbers of patients with intractable signs and symptoms, and the emergence of difficult end-of-life decisions. If we could truly prevent heart failure rather than forestall its emergence to a later point in life, perhaps we could do more good.
For me, the study of neurohormonal mechanisms in the setting of heart failure was the centerpiece of my early career. Jay Cohn had asked several of us early in our laboratory experience to choose a neurohormonal system and learn about it in great depth and detail. My assignment was the SNS. Since then, I have never tired of learning about its control mechanisms, how it achieves circulatory homeostasis, how its excess quantities can be directly toxic to the heart, and the variety of pharmacologic ways that we can control it. I am indeed fortunate to have been part of this amazing study group.
We have known for more than 100 years that heart failure is characterized by excessive sympathetic nervous system (SNS) activity. Thanks to refinement of this concept in the 1980s and 1990s, we now have a good understanding of SNS activity in both experimental and clinical heart failure. During those two decades, we also realized the pathophysiologic importance of the renin-angiotensin-aldosterone system (RAAS) in patients with heart failure.1 By 2000, it was obvious that heart failure was inextricably intertwined with excessive neurohormonal activity.2,3 This understanding of the pathophysiology of heart failure took on greater importance with the ability to pharmacologically block these neurohormonal systems, thereby demonstrating the detrimental role of neurohormones in the onset and progression of heart failure.
This article is a brief historical and personal description of the study of neurohormonal control mechanisms as they relate to the clinical syndrome of heart failure. The article includes a personal account of how the story unfolded in the cardiology research laboratories at the University of Minnesota.
THE EARLY YEARS: NEUROHORMONAL HYPOTHESIS
A hypothesis emerged gradually in the 1980s suggesting that progression of heart failure was in part a product of excessive SNS and RAAS activity. Many believed that pharmacologic inhibition of these systems might mitigate against progressive cardiac remodeling and thereby reduce symptoms and extend life—the so called neurohormonal hypothesis.4 SNS blockers and RAAS blockers are now widely used in tandem as first-line therapy to treat patients with heart failure,5–11 but in 1980 we were just beginning to consider their therapeutic effects.
This major shift in thinking about neurohormonal systems and heart failure did not come about quickly. Early success was driven by the ability to quickly and precisely measure neurohormones in the laboratory coupled with the availability of drugs specifically designed to block the SNS and RAAS. It was also critically important to embrace the power of randomized controlled trials to test new therapies. Investigators, research nurses, and patients from many medical centers and laboratories should be credited with this astonishing success. I am proud to have been a part of this activity at the University of Minnesota.
THE COHN LABORATORY
Early work done in the 1960s by numerous investigators noted that the failing left ventricle (LV) was exquisitely sensitive to afterload conditions.12–15 John Ross and Eugene Braunwald explored this observation in patients in 1964.15 Jay Cohn, with his unique background in hypertension and hemodynamics, brought the concept back into the laboratory in the early 1970s, where he explored the mechanisms responsible for increased sensitivity to afterload in patients with heart failure.16
I had the good fortune to join Cohn’s laboratory in 1979, when this avenue of heart failure research was in full bloom. A team of investigators was gradually assembled that included Maria Teresa Olivari, who relocated from the Cardiovascular Research Institute in Milan, Italy, directed by Maurizio D. Guazzi. Also joining the group were T. Barry Levine from the University of Michigan, Ann Arbor; Steven Goldsmith from Ohio State University, Columbus; Susan Ziesche from the Minneapolis Veterans Affairs (VA) Medical Center; Thomas Rector, an expert statistician and pharmacologist at the University of Minnesota; and many research fellows, visitors, students, biochemists, statisticians, and research nurses. Joseph Franciosa joined the University of Minnesota group in 1974 and, after completing several important trials, left in 1979 to lead the cardiology group at the Philadelphia VA Medical Center.
The Cohn group developed a working hypothesis that activation of the SNS and RAAS in heart failure was most likely an adaptive mechanism intended for short-term circulatory support, such as in the setting of blood loss, dehydration, shock, volume depletion, or flight response. In patients with heart failure, according to the hypothesis, the SNS and RAAS activity persisted beyond that needed for adaptation, with chronic release of norepinephrine (NE), renin, angiotensin II, aldosterone, and other neurohormones. The neurohormones ultimately became “maladaptive.” Thanks to the assaying skills of Ada Simon, we had the early advantage of precise and rapid radioenzyme measurement of plasma norepinephrine and renin activity in the blood of patients and animals.
We believed that neurohormonal activation contributed in part to the excessive afterload conditions observed in heart failure. We also thought that excessive neurohormonal activation directly impaired cardiac systolic function. The obvious next step was to explore whether neurohormonal antagonists would improve myocardial performance.
Under the leadership of Steven Goldsmith, many studies were performed to investigate reflex control mechanisms and their pathogenic role in patients with heart failure. The accumulating data suggested that persistent, excessive neurohormonal activity was characteristic of heart failure and that it was associated with a poor prognosis.17 The precise mechanism that drives activation of the SNS remained elusive, however, and is poorly defined even today. In that era, when β-adrenergic blockers were believed to be contraindicated, we inhibited the central SNS with bromocriptine, clonidine, and guanfacine with modestly favorable responses. We inhibited circulating arginine vasopressin antibody (thanks to Prof. Alan Cowley for noting an acute favorable response).
THE PHARMACOLOGIC ERA
The 1980s and 1990s saw the availability of several pharmacologic tools for assessing the roles of the SNS and RAAS in heart failure. The hypotensive effects of angiotensin-converting enzyme (ACE) inhibitors and, later, angiotensin-receptor blockers (ARBs) were sources of concern, since many patients with advanced heart failure had low- to normal-range blood pressures before they received RAAS blockers. However, our group as well as others observed that abrupt blood pressure reduction occurred primarily in patients with very hyperreninemic responses to intravenous diuretics (ie, volume-depleted patients). Eventually, we learned that low baseline blood pressure did not adversely affect outcomes when vasodilators were used in patients with heart failure,18,19 leading us to titrate these drugs upward over days to weeks.
Several different combinations of vasodilators were used successfully to treat heart failure, including hydralazine, isosorbide dinitrate,20 ACE inhibitors,21,22 and ARBs.8,23–28 Direct-acting calcium channel blocking vasodilators, such as amlodipine, did not improve survival in patients with systolic heart failure, although they appeared to be safe in this setting.29 The aldosterone receptor blockers spironolactone30 and eplerenone31 were later demonstrated to improve survival of patients with advanced systolic heart failure when added to vasodilator therapy.
By the end of the 1990s, it was evident that drugs that blocked the SNS and RAAS were not just vasodilators or “afterload reducers,” similar to α-blockers, hydralazine, nitrates, and amlodipine. Neurohormonal blockers were doing something profoundly beneficial not observed with more direct-acting vasodilators.32–37 Simple afterload reduction was not enough in patients with systolic heart failure.
Neurohormonal antagonists were acting more directly on the myocardium. They were preventing the progression of LV remodeling and, in some cases, promoting reverse remodeling, thus improving myocardial function and favorably influencing the natural history of heart failure.31–39 We were astonished to discover that the failing, dilated heart could revert to normal size in response to neurohormone blockade with ACE inhibitors and β-adrenergic blockers; these findings were soon reported by other laboratories as well.
Contrary to our concept of heart failure in the 1970s, we now understood that the heart has inherent plasticity. It can dilate in response to abnormal loading conditions or myocardial injury, and it can restore itself to normal size when neurohormones are blocked and perverse loading conditions are improved. This reversal can occur spontaneously if an offending agent such as chronic alcohol use or inflammation is removed, but it is likely facilitated by SNS and RAAS blockers.
THE REMODELING ERA
Ken McDonald joined the University of Minnesota lab in 1989 as a research fellow. His skill in conducting both animal and clinical mechanistic studies was pivotal to our achieving our research goals. The inspired animal work by Boston-based Marc and Janice Pfeffer revealed the significance of the LV remodeling concept in the development of heart failure36: ventricular remodeling was a hallmark of systolic heart failure, and pharmacologic inhibition of LV remodeling by blocking neurohormones had profound clinical implications.
Under the direction of Wenda Carlyle, a molecular biology laboratory was established at the University of Minnesota whose work was dedicated solely to exploration of remodeling at a very basic level. Alan Hirsch was recruited from Victor Dzau’s laboratory at Brigham and Women’s Hospital in Boston to extend our efforts to understand the molecular basis of cardiac remodeling. Ken McDonald guided the use of magnetic resonance imaging to study remodeling in dogs.
The late 1970s saw the initiation and eventual execution of several important clinical trials, including the Vasodilator Heart Failure Trials (V-HeFT I and V-HeFT II)40,41 under our leadership, and Studies of Left Ventricular Dysfunction (SOLVD)5,6 under the leadership of Salim Yusuf and others at the National Heart Lung and Blood Institute (NHLBI). Many neuro hormonal and remodeling substudies sprang from these large clinical trials. Spencer Kubo joined our group from the Medical College of Cornell University in the mid-1980s, and he immediately demonstrated his prowess in clinical research. He also recruited Alan Bank to study the endothelium in both experimental and human heart failure.
Integrating the molecular, animal, and clinical laboratories allowed us to pursue many mechanistic studies. Laboratory meetings, often held on Saturday mornings, generated ideas for program projects that were subsequently funded by NHLBI. Birthday parties and other social events with laboratory staff and their families were part of our fabric. Late-night trips to the Post Office to send off abstracts for national meetings before the midnight deadline were a regular feature.
Our coordination of and participation in the large clinical trials allowed us to meet frequently in Bethesda with colleagues from other major centers, fostering many collaborations and friendships that continue to thrive. Susan Ziesche deserves much of the credit for coordinating many groups that were part of these large, complex trials. Cheryl Yano, our administrator, also played a key role. All National Institutes of Health (NIH) grants passed through Cheryl, and she worked tirelessly to ensure that the proposals were in the best possible shape before we submitted them. Inder Anand joined our group in the early 1990s and became a major analytical force. Jay Cohn was the intellectual leader of the group, as well as our soul and inspiration. People worked hard for him, and he taught us much in a setting that valued creativity and new ideas above all.
THE LATER YEARS
By 1997, the face of heart failure had changed. New treatments were effective, but there were new challenges to face. I moved that year to the Cleveland Clinic, where I spent 11 enjoyable and productive years. I returned to Minnesota in 2008 to help build a new cardiovascular division.
It is gratifying to look back and see what has become of the “neurohormonal hypothesis.” Today, nearly all major medical centers have heart failure programs, and certification in advanced heart failure/heart transplantation is a reality. Training programs in advanced heart failure and heart transplant are common. The Heart Failure Society of America sprang up in the early 1990s, dedicated to patients with heart failure. Jay Cohn founded the Journal of Cardiac Failure, which flourished under his leadership. Neurohormonal blockers are now considered standard, conventional therapy and are widely used throughout the world.
CONCLUSIONS
Still, there is much work to do. An increasing number of devices are being developed, largely for patients with more advanced heart failure, but attention is also being directed to prevention of heart failure. Identification and possible treatment of patients at risk for the development of heart failure, and identification of those who already have some early structural and functional perturbation without advanced symptoms, are critically important. Since event rates are so low in these patients, we need to create new strategies for studying interventions. In the long term, the best treatment for nearly any condition is early diagnosis and perhaps early treatment with a goal of prevention.
One consequence of our progress over the years may be that heart failure now primarily affects a more elderly group—patients who often have many associated comorbidities. The consequences include more frequent readmissions, large numbers of patients with intractable signs and symptoms, and the emergence of difficult end-of-life decisions. If we could truly prevent heart failure rather than forestall its emergence to a later point in life, perhaps we could do more good.
For me, the study of neurohormonal mechanisms in the setting of heart failure was the centerpiece of my early career. Jay Cohn had asked several of us early in our laboratory experience to choose a neurohormonal system and learn about it in great depth and detail. My assignment was the SNS. Since then, I have never tired of learning about its control mechanisms, how it achieves circulatory homeostasis, how its excess quantities can be directly toxic to the heart, and the variety of pharmacologic ways that we can control it. I am indeed fortunate to have been part of this amazing study group.
- Dzau VJ, Colucci WS, Hollenberg NK, Williams GH. Relation of the renin-angiotensin-aldosterone system to clinical state in congestive heart failure. Circulation 1981; 63:645–651.
- Francis GS, Goldsmith SR, Levine TB, Olivari MT, Cohn JN. The neurohumoral axis in congestive heart failure. Ann Intern Med 1984; 101:370–377.
- Levine TB, Francis GS, Goldsmith SR, Simon AB, Cohn JN. Activity of the sympathetic nervous system and renin-angiotensin system assessed by plasma hormone levels and their relation to hemodynamic abnormalities in congestive heart failure. Am J Cardiol 1982; 49:1659–1666.
- Packer M. The neurohormonal hypothesis: a theory to explain the mechanism of disease progression in heart failure. J Am Coll Cardiol 1992; 20:248–254.
- The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fraction and congestive heart failure. N Engl J Med 1991; 325:293–302.
- The SOLVD Investigators. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N Engl J Med 1992; 327:685–691.
- Pitt B, Zannand F, Remme WJ, et al The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med 1999; 341:709–717.
- ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008; 358:1547–1559.
- CIBIS Investigators and Committees. A randomized trial of β-blockade in heart failure: the Cardiac Insufficiency Bisoprolol Study (CIBIS). Circulation 1994; 90:1765–1773.
- Hjalmarson A, Goldstein S, Fagerberg B, et al Effects of controlled-release metoprolol on total mortality, hospitalizations, and well-being in patients with heart failure: the Metoprolol CR/XL Randomized Intervention Trial in congestive heart failure (MERITHF). JAMA 2000; 283:1295–1302.
- Packer M, Fowler MB, Roecker EB, et al Effect of carvedilol on the morbidity of patients with severe chronic heart failure: results of the carvedilol prospective randomized cumulative survival (COPERNICUS) study. Circulation 2002; 106:2194–2199.
- Imperial ES, Levy MN, Zieske H. Outflow resistance as an independent determinant of cardiac performance. Circ Res 1961; 9:1148–1155.
- Sonnenblick EH, Downing SE. Afterload as a primary determinant of ventricular performance. Am J Physiol 1963; 204:604–610.
- Wilcken DE, Charlier AA, Hoffman JI. Effects of alterations in aortic impedance on the performance of the ventricles. Circ Res 1964; 14:283–293.
- Ross J, Braunwald E. The study of left ventricular function in man by increasing resistance to ventricular ejection with angiotensin. Circulation 1964; 29:739–749.
- Cohn JN. Blood pressure and cardiac performance. Am J Med 1973; 55:351–361.
- Cohn JN, Levine TB, Olivari MT, et al Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med 1984; 311:819–823.
- Anand IS, Tam SW, Rector TS, et al Influence of blood pressure on the effectiveness of a fixed-dose combination of isosorbide dinitrate and hydralazine in the African-American Heart Failure Trial. J Am Coll Cardiol 2007; 49:32–39.
- Rouleau JL, Roecker EB, Tendra M, et al Influence of pretreatment systolic blood pressure on the effect of carvedilol in patients with severe chronic heart failure: the Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) study. J Am Coll Cardiol 2004; 43:1423–1429.
- Taylor AL, Ziesche S, Yancy C, et al Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N Engl J Med 2004; 351:2049–2057.
- Captopril Multicenter Research Group. A placebo-controlled trial of captopril in refractory chronic congestive heart failure. J Am Coll Cardiol 1983; 2:755–763.
- Pfeffer MA, Braunwald E, Moyé LA, et al Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction: results of the survival and ventricular enlargement trial—the SAVE Investigators. N Eng J Med 1992; 327:669–677.
- Curtiss C, Cohn JN, Vrobel T, Franciosa J. Role of the renin-angiotensin system in the systemic vasoconstriction of chronic congestive heart failure. Circulation 1978; 58:763–770.
- Cohn JN, Tognoni G. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl J Med 2001; 345:1667–1675.
- Young JB, Dunlap ME, Pfeffer MA, et al Mortality and morbidity reduction with Candesartan in patients with chronic heart failure and left ventricular systolic dysfunction: results of the CHARM low-left ventricular ejection trials. Circulation 2004; 110:2618–2626.
- Pfeffer MA, McMurray JJ, Velazquez EJ, et al Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 2003; 349:1893–1906.
- ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008; 358:1547–1559.
- Konstam MA, Neaton JD, Dickstein K, et al Effects of high-dose versus lose-dose losartan on clinical outcomes in patients with heart failure (HEAAL study): a randomized, double-blind trial. Lancet 2009; 374:1840–1848.
- Packer M. Prospective randomized amlodipine survival evaluation 2. Presented at: 49th American College of Cardiology meeting; March 2000; Anaheim, CA.
- Pitt B, Zannand F, Remme WJ, et al The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannand F, et al Eplerenone, a selective aldosterone blocker in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Cohn JN. Structural basis for heart failure: ventricular remodeling and its pharmacological inhibition. Circulation 1995; 91:2504–2507.
- Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling—concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. J Am Coll Cardiol 2000; 35:569–581.
- Konstam MA, Kronenberg MW, Rousseau MF, et al Effects of the angiotensin converting enzyme inhibitor enalapril on the long-term progression of left ventricular dilation in patients with asymptomatic systolic dysfunction. Circulation 1993; 88:2277–2283.
- Greenberg B, Quinones MA, Koilpillai C, et al Effects of long-term enalapril therapy on cardiac structure and function in patients with left ventricular dysfunction: results of the SOLVD echocardiography substudy. Circulation 1995; 91:2573–2581.
- Pfeffer JM, Pfeffer MA, Braunwald E. Influence of chronic captopril therapy on the infarcted left ventricle of the rat. Circ Res 1985; 57:84–95.
- Cohn JN. Structural basis for heart failure: ventricular remodeling and its pharmacological inhibition. Circulation 1995; 91:2504–2507.
- McDonald KM, Garr M, Carlyle PF, et al Relative effects of α1-adrenoceptor blockade, converting enzyme inhibitor therapy, and angiotensin II sub-type 1 receptor blockade on ventricular remodeling in the dog. Circulation 1994; 90:3034–3046.
- Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation 1990; 81:1161–1172.
- Cohn JN, Archibald DG, Ziesche S, et al Effect of vasodilator therapy on mortality in chronic congestive heart failure. N Engl J Med 1986; 314:1547–1552.
- Cohn JN, Johnson G, Ziesche S, et al A comparison of enalapril with hydralazine–isosorbide dinitrate in the treatment of chronic congestive heart failure. N Engl J Med 1991; 325:303–310.
- Dzau VJ, Colucci WS, Hollenberg NK, Williams GH. Relation of the renin-angiotensin-aldosterone system to clinical state in congestive heart failure. Circulation 1981; 63:645–651.
- Francis GS, Goldsmith SR, Levine TB, Olivari MT, Cohn JN. The neurohumoral axis in congestive heart failure. Ann Intern Med 1984; 101:370–377.
- Levine TB, Francis GS, Goldsmith SR, Simon AB, Cohn JN. Activity of the sympathetic nervous system and renin-angiotensin system assessed by plasma hormone levels and their relation to hemodynamic abnormalities in congestive heart failure. Am J Cardiol 1982; 49:1659–1666.
- Packer M. The neurohormonal hypothesis: a theory to explain the mechanism of disease progression in heart failure. J Am Coll Cardiol 1992; 20:248–254.
- The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fraction and congestive heart failure. N Engl J Med 1991; 325:293–302.
- The SOLVD Investigators. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N Engl J Med 1992; 327:685–691.
- Pitt B, Zannand F, Remme WJ, et al The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med 1999; 341:709–717.
- ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008; 358:1547–1559.
- CIBIS Investigators and Committees. A randomized trial of β-blockade in heart failure: the Cardiac Insufficiency Bisoprolol Study (CIBIS). Circulation 1994; 90:1765–1773.
- Hjalmarson A, Goldstein S, Fagerberg B, et al Effects of controlled-release metoprolol on total mortality, hospitalizations, and well-being in patients with heart failure: the Metoprolol CR/XL Randomized Intervention Trial in congestive heart failure (MERITHF). JAMA 2000; 283:1295–1302.
- Packer M, Fowler MB, Roecker EB, et al Effect of carvedilol on the morbidity of patients with severe chronic heart failure: results of the carvedilol prospective randomized cumulative survival (COPERNICUS) study. Circulation 2002; 106:2194–2199.
- Imperial ES, Levy MN, Zieske H. Outflow resistance as an independent determinant of cardiac performance. Circ Res 1961; 9:1148–1155.
- Sonnenblick EH, Downing SE. Afterload as a primary determinant of ventricular performance. Am J Physiol 1963; 204:604–610.
- Wilcken DE, Charlier AA, Hoffman JI. Effects of alterations in aortic impedance on the performance of the ventricles. Circ Res 1964; 14:283–293.
- Ross J, Braunwald E. The study of left ventricular function in man by increasing resistance to ventricular ejection with angiotensin. Circulation 1964; 29:739–749.
- Cohn JN. Blood pressure and cardiac performance. Am J Med 1973; 55:351–361.
- Cohn JN, Levine TB, Olivari MT, et al Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med 1984; 311:819–823.
- Anand IS, Tam SW, Rector TS, et al Influence of blood pressure on the effectiveness of a fixed-dose combination of isosorbide dinitrate and hydralazine in the African-American Heart Failure Trial. J Am Coll Cardiol 2007; 49:32–39.
- Rouleau JL, Roecker EB, Tendra M, et al Influence of pretreatment systolic blood pressure on the effect of carvedilol in patients with severe chronic heart failure: the Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) study. J Am Coll Cardiol 2004; 43:1423–1429.
- Taylor AL, Ziesche S, Yancy C, et al Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N Engl J Med 2004; 351:2049–2057.
- Captopril Multicenter Research Group. A placebo-controlled trial of captopril in refractory chronic congestive heart failure. J Am Coll Cardiol 1983; 2:755–763.
- Pfeffer MA, Braunwald E, Moyé LA, et al Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction: results of the survival and ventricular enlargement trial—the SAVE Investigators. N Eng J Med 1992; 327:669–677.
- Curtiss C, Cohn JN, Vrobel T, Franciosa J. Role of the renin-angiotensin system in the systemic vasoconstriction of chronic congestive heart failure. Circulation 1978; 58:763–770.
- Cohn JN, Tognoni G. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl J Med 2001; 345:1667–1675.
- Young JB, Dunlap ME, Pfeffer MA, et al Mortality and morbidity reduction with Candesartan in patients with chronic heart failure and left ventricular systolic dysfunction: results of the CHARM low-left ventricular ejection trials. Circulation 2004; 110:2618–2626.
- Pfeffer MA, McMurray JJ, Velazquez EJ, et al Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 2003; 349:1893–1906.
- ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 2008; 358:1547–1559.
- Konstam MA, Neaton JD, Dickstein K, et al Effects of high-dose versus lose-dose losartan on clinical outcomes in patients with heart failure (HEAAL study): a randomized, double-blind trial. Lancet 2009; 374:1840–1848.
- Packer M. Prospective randomized amlodipine survival evaluation 2. Presented at: 49th American College of Cardiology meeting; March 2000; Anaheim, CA.
- Pitt B, Zannand F, Remme WJ, et al The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannand F, et al Eplerenone, a selective aldosterone blocker in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Cohn JN. Structural basis for heart failure: ventricular remodeling and its pharmacological inhibition. Circulation 1995; 91:2504–2507.
- Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling—concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. J Am Coll Cardiol 2000; 35:569–581.
- Konstam MA, Kronenberg MW, Rousseau MF, et al Effects of the angiotensin converting enzyme inhibitor enalapril on the long-term progression of left ventricular dilation in patients with asymptomatic systolic dysfunction. Circulation 1993; 88:2277–2283.
- Greenberg B, Quinones MA, Koilpillai C, et al Effects of long-term enalapril therapy on cardiac structure and function in patients with left ventricular dysfunction: results of the SOLVD echocardiography substudy. Circulation 1995; 91:2573–2581.
- Pfeffer JM, Pfeffer MA, Braunwald E. Influence of chronic captopril therapy on the infarcted left ventricle of the rat. Circ Res 1985; 57:84–95.
- Cohn JN. Structural basis for heart failure: ventricular remodeling and its pharmacological inhibition. Circulation 1995; 91:2504–2507.
- McDonald KM, Garr M, Carlyle PF, et al Relative effects of α1-adrenoceptor blockade, converting enzyme inhibitor therapy, and angiotensin II sub-type 1 receptor blockade on ventricular remodeling in the dog. Circulation 1994; 90:3034–3046.
- Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation 1990; 81:1161–1172.
- Cohn JN, Archibald DG, Ziesche S, et al Effect of vasodilator therapy on mortality in chronic congestive heart failure. N Engl J Med 1986; 314:1547–1552.
- Cohn JN, Johnson G, Ziesche S, et al A comparison of enalapril with hydralazine–isosorbide dinitrate in the treatment of chronic congestive heart failure. N Engl J Med 1991; 325:303–310.
In reply: Acute myocardial infarction
In Reply: We thank Dr. Madias for his letter. We agree that doing a second electrocardiogram to inspect V3R, V4R, and V7 to the left of the spine and V9 to the right of the spine may provide important additional information that supports the diagnosis of acute MI. When clinical suspicion is high and the standard 12-lead electrocardiogram shows only minimal changes, then additional lead placement may be useful. Some other situations were not covered in our paper but are worthy of consideration when looking for electrocardiographic evidence of acute MI, eg:
- Patients with left main disease may demonstrate modest ST-T elevation in lead AVR with diffuse ST-T depression when having an acute MI.
- Patients with only T-wave-flattening in AVL may be having an acute MI due to isolated circumflex coronary disease.
Again, we thank Dr. Madias for his interest in our paper. We welcome his suggestion and hope that our response will be of some value to physicians responsible for making the very important decision to send a patient urgently to the cardiac catheterization laboratory.
In Reply: We thank Dr. Madias for his letter. We agree that doing a second electrocardiogram to inspect V3R, V4R, and V7 to the left of the spine and V9 to the right of the spine may provide important additional information that supports the diagnosis of acute MI. When clinical suspicion is high and the standard 12-lead electrocardiogram shows only minimal changes, then additional lead placement may be useful. Some other situations were not covered in our paper but are worthy of consideration when looking for electrocardiographic evidence of acute MI, eg:
- Patients with left main disease may demonstrate modest ST-T elevation in lead AVR with diffuse ST-T depression when having an acute MI.
- Patients with only T-wave-flattening in AVL may be having an acute MI due to isolated circumflex coronary disease.
Again, we thank Dr. Madias for his interest in our paper. We welcome his suggestion and hope that our response will be of some value to physicians responsible for making the very important decision to send a patient urgently to the cardiac catheterization laboratory.
In Reply: We thank Dr. Madias for his letter. We agree that doing a second electrocardiogram to inspect V3R, V4R, and V7 to the left of the spine and V9 to the right of the spine may provide important additional information that supports the diagnosis of acute MI. When clinical suspicion is high and the standard 12-lead electrocardiogram shows only minimal changes, then additional lead placement may be useful. Some other situations were not covered in our paper but are worthy of consideration when looking for electrocardiographic evidence of acute MI, eg:
- Patients with left main disease may demonstrate modest ST-T elevation in lead AVR with diffuse ST-T depression when having an acute MI.
- Patients with only T-wave-flattening in AVL may be having an acute MI due to isolated circumflex coronary disease.
Again, we thank Dr. Madias for his interest in our paper. We welcome his suggestion and hope that our response will be of some value to physicians responsible for making the very important decision to send a patient urgently to the cardiac catheterization laboratory.
A new, precise definition of acute myocardial infarction
Acute myocardial infarction (MI) portends important and substantial consequences. Angioplasty or fibrinolytic therapy to open the blocked coronary artery is proven to improve the patient’s chances of surviving without consequent morbidity or death. But the diagnosis is not always straightforward. The presentation of acute MI can vary widely, and a number of other conditions—many of them equally serious emergencies—can mimic its symptoms, electrocardiographic signs, and biomarker patterns.
In an attempt to improve the accuracy of the diagnosis of MI, a multinational task force met in 1999 under the auspices of the European Society of Cardiology and the American College of Cardiology. The goal was to develop a simple, clinically oriented definition of MI that could be widely adopted. A document was created and published simultaneously in 2000 in the European Heart Journal and the Journal of the American College of Cardiology.1 These organizations updated their paper in 2007 with a new definition of acute MI to account for advances in diagnosis and management.2
In this article we will review the new definition and how to make the diagnosis of acute MI today. Specifically, the updated definition includes:
- Subtypes of acute MI
- Imaging tests supporting the diagnosis
- Biomarker thresholds after percutaneous coronary intervention or bypass grafting.
TROPONIN: BETTER THAN CK, BUT NOT PERFECT
The original 2000 paper1 and the 2007 update2 featured the use of the cardiac biomarker troponin, which is considerably more sensitive and specific for heart damage than total creatine kinase (CK) or its isoform, CK-MB.
The new, more-sensitive biomarker-based definition of MI resulted in more cases of MI being diagnosed, and this has attracted the attention and scrutiny of many, especially population scientists and interventional cardiologists.3 This change has caused some controversy, especially when dealing with small rises in troponin following percutaneous coronary intervention.
In addition, some confusion over terminology remains. For example, the phrase “troponin leak” is often used to describe cases in which serum troponin levels rise but there is no MI. However, most experts believe that a rise and fall in troponin is due to true myocardial cell death. Troponin I and T are such large molecules that they cannot “leak” from a cardiac cell unless there has been irreparable cellular damage—that is, cell death.

Creatine kinase still has a role
In some cases, CK and CK-MB may be helpful in determining the acuity of myocardial necrosis, but their use will vary by institution. These biomarkers typically rise 2 to 4 hours after the initial event and fall within 24 to 48 hours, whereas troponin levels stay elevated for days or weeks. Thus, the presence of troponin without CK and CK-MB in the right clinical context may indicate a past MI that is no longer acute.
INFARCTION: CELL DEATH DUE TO ISCHEMIA
MI is myocardial cell death due to prolonged ischemia. Under the microscope, it can be categorized as coagulation necrosis in which ghost-like cell structures remain after hypoxic insult (typical of most MIs) or contraction band necrosis with amorphous cells that cannot contract anymore, the latter often a hallmark of excessive catecholamine damage or reperfusion injury. Apoptosis occurs in the heart but is technically not considered necrosis and is thought not to be associated with elevated troponin levels.6,7
In experiments in animals, cell death can occur as little as 20 minutes after coronary artery occlusion, although completion of infarction is thought to take 2 to 4 hours. The time to infarct completion may be longer in patients with collateral circulation or when the culprit coronary artery has intermittent (“stuttering”) occlusion. Preconditioning of myocardial cells with intermittent ischemia can also influence the timing of myocardial necrosis by protecting against cell death to some extent. Alteration in myocardial demand can influence the time required for completion of infarction either favorably or unfavorably; hence, reducing myocardial demand is beneficial in acute MI.
Three pathologic phases of MI
MI can be categorized pathologically as acute, healing, or healed.
Acute MI. In the first 6 hours after coronary artery occlusion, coagulation necrosis can be seen with no cellular infiltration. After 6 hours, polymorphonuclear leukocytes infiltrate the infarcted area, and this may continue for up to 7 days if coronary perfusion does not increase or myocardial demand does not decrease.
Healing MI is characterized by mononuclear cells and fibroblasts and the absence of polymorphonuclear leukocytes. The entire healing process takes 5 to 6 weeks and can be altered by coronary reperfusion.
Healed MI refers to scar tissue without cellular infiltration.
CLINICAL FEATURES VARY WIDELY
Sir William Osler said, “Variability is the law of life, and as no two faces are the same, so no two bodies are alike, and no two individuals react alike and behave alike under the abnormal conditions which we know as disease.”8
Just so, patients with acute MI display a wide variety of presentations, from no symptoms (about 25%) to severe, crushing chest pain. Discomfort may occur in the upper back, neck, jaw, teeth, arms, wrist, and epigastrium. Shortness of breath, diaphoresis, nausea, vomiting, and even syncope may occur. Unlike in acute aortic dissection, the discomfort is not usually maximal at its onset: it builds up in a crescendo manner. It is not usually changed by position, but can lessen in intensity upon standing. The discomfort in the chest is deep and visceral, and typically not well localized. A pressure sensation, air hunger, or “gas buildup” can be described. The only symptom may be shortness of breath or severe diaphoresis. The symptoms can last from minutes to hours and can be relieved by sublingual nitroglycerin. Atypical or less-prominent symptoms may make the diagnosis more difficult in the elderly, patients with diabetes mellitus, and women.
The physical examination during acute MI usually finds no clear-cut distinguishing features. The patient may appear pale and diaphoretic, and the skin cool to the touch. Heart sounds are generally soft. A fourth heart sound may be audible. Blood pressure may be low, but it can vary widely. Tachycardia, particularly sinus tachycardia, and pulmonary edema are poor prognostic signs.
In view of the wide variation in presentations, the history and physical findings can raise the suspicion of acute MI, but sequential electrocardiograms and measurements of biomarkers (troponin) are always necessary.
ELECTROCARDIOGRAPHY: NECESSARY BUT NOT SUFFICIENT
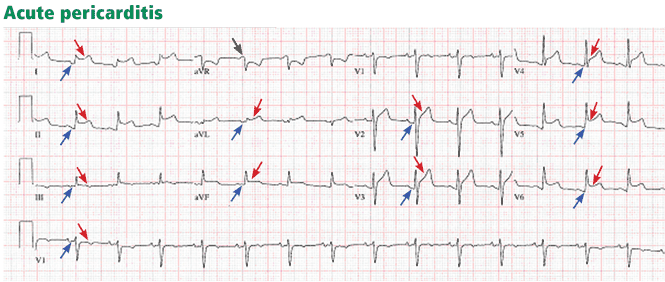

ST-elevation MI vs non-ST-elevation MI
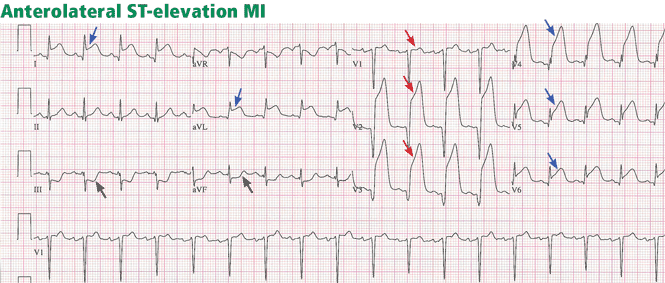
Changes in the ST segment can be very dynamic, making sequential tracings very useful. Rhythm disturbances and heart block are also more likely to be recorded when using sequential readings.
Pitfalls to electrocardiographic diagnosis
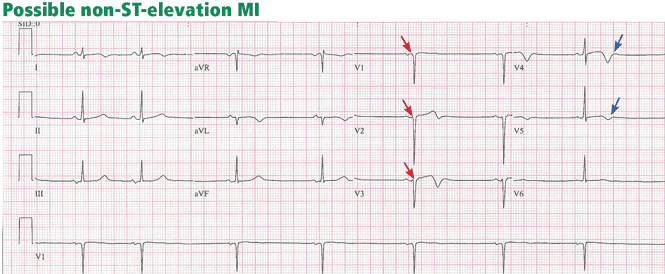
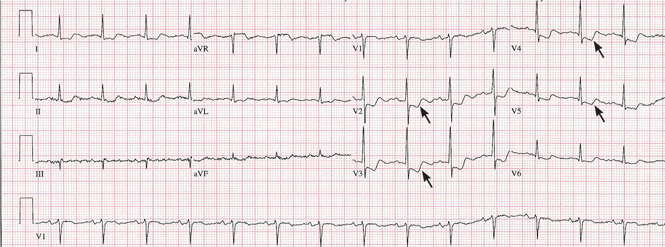
Prior MI. Q waves or QS complexes, when the Q wave is sufficiently wide (≥ 0.03 msec) or deep (≥ 1 mV), usually indicate a previous MI. However, many nuances that further raise or lower the suspicion for previous MI need to be considered. These are beyond the scope of this brief review but are available in the 2007 update.

Right ventricular infarction often requires the use of right-sided leads, which may reveal ST elevation in V4R.
ECHOCARDIOGRAPHY IF THE DIAGNOSIS IS IN DOUBT
Echocardiography is an excellent way to assess wall-motion abnormalities. In the absence of any wall-motion abnormality, a large ST-elevation MI is unlikely. A large wall-motion abnormality would verify the probability of ongoing acute MI and thus would help with rapid decision-making.
Furthermore, echocardiography can help determine the likelihood that the patient has aortic dissection or pulmonary embolism, either of which can mimic acute MI but requires very different treatment.
CLINICAL CLASSIFICATION OF ACUTE MI

The new classification scheme of the different types of MI is shown in Table 4.
The new classification scheme does not include myocardial necrosis from mechanical manipulation of the heart during open heart surgery, from cardioversion, or from toxic drugs.
As clinicians are aware, it is not unusual to see elevated biomarker levels in a host of conditions unrelated to acute myocardial ischemia or MI. The new classification of acute MI is most helpful in this regard. It will likely be even more helpful in guiding treatment and management when new ultrasensitive troponin assays are widely introduced into clinical practice.
The new classification also negotiates the controversy regarding elevated biomarker levels following percutaneous coronary intervention. In brief, elevation of biomarkers is not entirely avoidable even with a successful percutaneous coronary intervention, and furthermore, there is no scientific cutoff for biomarker elevations. So, by arbitrary convention, the troponin level must rise to more than three times the 99th percentile upper reference limit to make the diagnosis of type 4a MI. A separate type 4b MI is ascribed to angiographic or autopsy-proven stent thrombosis.
The new guidelines also suggest that troponin values be more than five times the 99th percentile of the normal reference range during the first 72 hours following coronary artery bypass graft surgery (CABG) when considering a CABG-related MI (type 5). Whenever new pathologic Q waves appear in territories other than those identified before the procedure, MI should be considered, especially if associated with elevated biomarkers, new wall-motion abnormalities, or hemodynamic instability.
Thus, the diagnosis of acute MI now has widely accepted global criteria that distinguish various types of acute MI that occur under multiple circumstances. It is expected that describing the type of acute MI according to the new criteria will further enhance our understanding of the event, its proper management, and its prognosis.
- The Joint European Society of Cardiology/American College of Cardiology Committee. Myocardial infarction redefined—a consensus document of the Joint European Society of Cardiology/American College of Cardiology Committee for the Redefinition of Myocardial Infarction. J Am Coll Cardiol 2000; 36:959–969.
- Thygesen K, Alpert JS, White HD, on behalf of the Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction. Universal definition of myocardial infarction. J Am Coll Cardiol 2007; 50:2173–2188.
- Roger VL, Killian JM, Weston SA, et al. Redefinition of myocardial infarction—prospective evaluation in the community. Circulation 2006; 114:790–797.
- Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease. J Am Coll Cardiol 2006; 48:1–11.
- French JK, White HD. Clinical implications of the new definition of myocardial infarction. Heart 2004; 90:99–106.
- James TN. The variable morphological coexistence of apoptosis and necrosis in human myocardial infarction: significance for understanding its pathogenesis, clinical course, diagnosis and prognosis. Coron Artery Dis 1998; 9:291–307.
- Sobel BE, LeWinter MM. Ingenuous interpretation of elevated blood levels of macromolecular markers of myocardial injury: a recipe for confusion. J Am Coll Cardiol 2000; 35:1355–1358.
- Osler W. Aequanimitas: With Other Addresses to Medical Students, Nurses and Practitioners of Medicine.Osler William Edition: 3, revised. Philadelphia: Blakiston’s, 1932.
- Wang F, Asinger RW, Marriott HJ. ST-segment elevation in conditions other than acute myocardial infarction. N Engl J Med 2003; 349:2128–2135.
Acute myocardial infarction (MI) portends important and substantial consequences. Angioplasty or fibrinolytic therapy to open the blocked coronary artery is proven to improve the patient’s chances of surviving without consequent morbidity or death. But the diagnosis is not always straightforward. The presentation of acute MI can vary widely, and a number of other conditions—many of them equally serious emergencies—can mimic its symptoms, electrocardiographic signs, and biomarker patterns.
In an attempt to improve the accuracy of the diagnosis of MI, a multinational task force met in 1999 under the auspices of the European Society of Cardiology and the American College of Cardiology. The goal was to develop a simple, clinically oriented definition of MI that could be widely adopted. A document was created and published simultaneously in 2000 in the European Heart Journal and the Journal of the American College of Cardiology.1 These organizations updated their paper in 2007 with a new definition of acute MI to account for advances in diagnosis and management.2
In this article we will review the new definition and how to make the diagnosis of acute MI today. Specifically, the updated definition includes:
- Subtypes of acute MI
- Imaging tests supporting the diagnosis
- Biomarker thresholds after percutaneous coronary intervention or bypass grafting.
TROPONIN: BETTER THAN CK, BUT NOT PERFECT
The original 2000 paper1 and the 2007 update2 featured the use of the cardiac biomarker troponin, which is considerably more sensitive and specific for heart damage than total creatine kinase (CK) or its isoform, CK-MB.
The new, more-sensitive biomarker-based definition of MI resulted in more cases of MI being diagnosed, and this has attracted the attention and scrutiny of many, especially population scientists and interventional cardiologists.3 This change has caused some controversy, especially when dealing with small rises in troponin following percutaneous coronary intervention.
In addition, some confusion over terminology remains. For example, the phrase “troponin leak” is often used to describe cases in which serum troponin levels rise but there is no MI. However, most experts believe that a rise and fall in troponin is due to true myocardial cell death. Troponin I and T are such large molecules that they cannot “leak” from a cardiac cell unless there has been irreparable cellular damage—that is, cell death.
Creatine kinase still has a role
In some cases, CK and CK-MB may be helpful in determining the acuity of myocardial necrosis, but their use will vary by institution. These biomarkers typically rise 2 to 4 hours after the initial event and fall within 24 to 48 hours, whereas troponin levels stay elevated for days or weeks. Thus, the presence of troponin without CK and CK-MB in the right clinical context may indicate a past MI that is no longer acute.
INFARCTION: CELL DEATH DUE TO ISCHEMIA
MI is myocardial cell death due to prolonged ischemia. Under the microscope, it can be categorized as coagulation necrosis in which ghost-like cell structures remain after hypoxic insult (typical of most MIs) or contraction band necrosis with amorphous cells that cannot contract anymore, the latter often a hallmark of excessive catecholamine damage or reperfusion injury. Apoptosis occurs in the heart but is technically not considered necrosis and is thought not to be associated with elevated troponin levels.6,7
In experiments in animals, cell death can occur as little as 20 minutes after coronary artery occlusion, although completion of infarction is thought to take 2 to 4 hours. The time to infarct completion may be longer in patients with collateral circulation or when the culprit coronary artery has intermittent (“stuttering”) occlusion. Preconditioning of myocardial cells with intermittent ischemia can also influence the timing of myocardial necrosis by protecting against cell death to some extent. Alteration in myocardial demand can influence the time required for completion of infarction either favorably or unfavorably; hence, reducing myocardial demand is beneficial in acute MI.
Three pathologic phases of MI
MI can be categorized pathologically as acute, healing, or healed.
Acute MI. In the first 6 hours after coronary artery occlusion, coagulation necrosis can be seen with no cellular infiltration. After 6 hours, polymorphonuclear leukocytes infiltrate the infarcted area, and this may continue for up to 7 days if coronary perfusion does not increase or myocardial demand does not decrease.
Healing MI is characterized by mononuclear cells and fibroblasts and the absence of polymorphonuclear leukocytes. The entire healing process takes 5 to 6 weeks and can be altered by coronary reperfusion.
Healed MI refers to scar tissue without cellular infiltration.
CLINICAL FEATURES VARY WIDELY
Sir William Osler said, “Variability is the law of life, and as no two faces are the same, so no two bodies are alike, and no two individuals react alike and behave alike under the abnormal conditions which we know as disease.”8
Just so, patients with acute MI display a wide variety of presentations, from no symptoms (about 25%) to severe, crushing chest pain. Discomfort may occur in the upper back, neck, jaw, teeth, arms, wrist, and epigastrium. Shortness of breath, diaphoresis, nausea, vomiting, and even syncope may occur. Unlike in acute aortic dissection, the discomfort is not usually maximal at its onset: it builds up in a crescendo manner. It is not usually changed by position, but can lessen in intensity upon standing. The discomfort in the chest is deep and visceral, and typically not well localized. A pressure sensation, air hunger, or “gas buildup” can be described. The only symptom may be shortness of breath or severe diaphoresis. The symptoms can last from minutes to hours and can be relieved by sublingual nitroglycerin. Atypical or less-prominent symptoms may make the diagnosis more difficult in the elderly, patients with diabetes mellitus, and women.
The physical examination during acute MI usually finds no clear-cut distinguishing features. The patient may appear pale and diaphoretic, and the skin cool to the touch. Heart sounds are generally soft. A fourth heart sound may be audible. Blood pressure may be low, but it can vary widely. Tachycardia, particularly sinus tachycardia, and pulmonary edema are poor prognostic signs.
In view of the wide variation in presentations, the history and physical findings can raise the suspicion of acute MI, but sequential electrocardiograms and measurements of biomarkers (troponin) are always necessary.
ELECTROCARDIOGRAPHY: NECESSARY BUT NOT SUFFICIENT
ST-elevation MI vs non-ST-elevation MI
Changes in the ST segment can be very dynamic, making sequential tracings very useful. Rhythm disturbances and heart block are also more likely to be recorded when using sequential readings.
Pitfalls to electrocardiographic diagnosis
Prior MI. Q waves or QS complexes, when the Q wave is sufficiently wide (≥ 0.03 msec) or deep (≥ 1 mV), usually indicate a previous MI. However, many nuances that further raise or lower the suspicion for previous MI need to be considered. These are beyond the scope of this brief review but are available in the 2007 update.
Right ventricular infarction often requires the use of right-sided leads, which may reveal ST elevation in V4R.
ECHOCARDIOGRAPHY IF THE DIAGNOSIS IS IN DOUBT
Echocardiography is an excellent way to assess wall-motion abnormalities. In the absence of any wall-motion abnormality, a large ST-elevation MI is unlikely. A large wall-motion abnormality would verify the probability of ongoing acute MI and thus would help with rapid decision-making.
Furthermore, echocardiography can help determine the likelihood that the patient has aortic dissection or pulmonary embolism, either of which can mimic acute MI but requires very different treatment.
CLINICAL CLASSIFICATION OF ACUTE MI
The new classification scheme of the different types of MI is shown in Table 4.
The new classification scheme does not include myocardial necrosis from mechanical manipulation of the heart during open heart surgery, from cardioversion, or from toxic drugs.
As clinicians are aware, it is not unusual to see elevated biomarker levels in a host of conditions unrelated to acute myocardial ischemia or MI. The new classification of acute MI is most helpful in this regard. It will likely be even more helpful in guiding treatment and management when new ultrasensitive troponin assays are widely introduced into clinical practice.
The new classification also negotiates the controversy regarding elevated biomarker levels following percutaneous coronary intervention. In brief, elevation of biomarkers is not entirely avoidable even with a successful percutaneous coronary intervention, and furthermore, there is no scientific cutoff for biomarker elevations. So, by arbitrary convention, the troponin level must rise to more than three times the 99th percentile upper reference limit to make the diagnosis of type 4a MI. A separate type 4b MI is ascribed to angiographic or autopsy-proven stent thrombosis.
The new guidelines also suggest that troponin values be more than five times the 99th percentile of the normal reference range during the first 72 hours following coronary artery bypass graft surgery (CABG) when considering a CABG-related MI (type 5). Whenever new pathologic Q waves appear in territories other than those identified before the procedure, MI should be considered, especially if associated with elevated biomarkers, new wall-motion abnormalities, or hemodynamic instability.
Thus, the diagnosis of acute MI now has widely accepted global criteria that distinguish various types of acute MI that occur under multiple circumstances. It is expected that describing the type of acute MI according to the new criteria will further enhance our understanding of the event, its proper management, and its prognosis.
Acute myocardial infarction (MI) portends important and substantial consequences. Angioplasty or fibrinolytic therapy to open the blocked coronary artery is proven to improve the patient’s chances of surviving without consequent morbidity or death. But the diagnosis is not always straightforward. The presentation of acute MI can vary widely, and a number of other conditions—many of them equally serious emergencies—can mimic its symptoms, electrocardiographic signs, and biomarker patterns.
In an attempt to improve the accuracy of the diagnosis of MI, a multinational task force met in 1999 under the auspices of the European Society of Cardiology and the American College of Cardiology. The goal was to develop a simple, clinically oriented definition of MI that could be widely adopted. A document was created and published simultaneously in 2000 in the European Heart Journal and the Journal of the American College of Cardiology.1 These organizations updated their paper in 2007 with a new definition of acute MI to account for advances in diagnosis and management.2
In this article we will review the new definition and how to make the diagnosis of acute MI today. Specifically, the updated definition includes:
- Subtypes of acute MI
- Imaging tests supporting the diagnosis
- Biomarker thresholds after percutaneous coronary intervention or bypass grafting.
TROPONIN: BETTER THAN CK, BUT NOT PERFECT
The original 2000 paper1 and the 2007 update2 featured the use of the cardiac biomarker troponin, which is considerably more sensitive and specific for heart damage than total creatine kinase (CK) or its isoform, CK-MB.
The new, more-sensitive biomarker-based definition of MI resulted in more cases of MI being diagnosed, and this has attracted the attention and scrutiny of many, especially population scientists and interventional cardiologists.3 This change has caused some controversy, especially when dealing with small rises in troponin following percutaneous coronary intervention.
In addition, some confusion over terminology remains. For example, the phrase “troponin leak” is often used to describe cases in which serum troponin levels rise but there is no MI. However, most experts believe that a rise and fall in troponin is due to true myocardial cell death. Troponin I and T are such large molecules that they cannot “leak” from a cardiac cell unless there has been irreparable cellular damage—that is, cell death.
Creatine kinase still has a role
In some cases, CK and CK-MB may be helpful in determining the acuity of myocardial necrosis, but their use will vary by institution. These biomarkers typically rise 2 to 4 hours after the initial event and fall within 24 to 48 hours, whereas troponin levels stay elevated for days or weeks. Thus, the presence of troponin without CK and CK-MB in the right clinical context may indicate a past MI that is no longer acute.
INFARCTION: CELL DEATH DUE TO ISCHEMIA
MI is myocardial cell death due to prolonged ischemia. Under the microscope, it can be categorized as coagulation necrosis in which ghost-like cell structures remain after hypoxic insult (typical of most MIs) or contraction band necrosis with amorphous cells that cannot contract anymore, the latter often a hallmark of excessive catecholamine damage or reperfusion injury. Apoptosis occurs in the heart but is technically not considered necrosis and is thought not to be associated with elevated troponin levels.6,7
In experiments in animals, cell death can occur as little as 20 minutes after coronary artery occlusion, although completion of infarction is thought to take 2 to 4 hours. The time to infarct completion may be longer in patients with collateral circulation or when the culprit coronary artery has intermittent (“stuttering”) occlusion. Preconditioning of myocardial cells with intermittent ischemia can also influence the timing of myocardial necrosis by protecting against cell death to some extent. Alteration in myocardial demand can influence the time required for completion of infarction either favorably or unfavorably; hence, reducing myocardial demand is beneficial in acute MI.
Three pathologic phases of MI
MI can be categorized pathologically as acute, healing, or healed.
Acute MI. In the first 6 hours after coronary artery occlusion, coagulation necrosis can be seen with no cellular infiltration. After 6 hours, polymorphonuclear leukocytes infiltrate the infarcted area, and this may continue for up to 7 days if coronary perfusion does not increase or myocardial demand does not decrease.
Healing MI is characterized by mononuclear cells and fibroblasts and the absence of polymorphonuclear leukocytes. The entire healing process takes 5 to 6 weeks and can be altered by coronary reperfusion.
Healed MI refers to scar tissue without cellular infiltration.
CLINICAL FEATURES VARY WIDELY
Sir William Osler said, “Variability is the law of life, and as no two faces are the same, so no two bodies are alike, and no two individuals react alike and behave alike under the abnormal conditions which we know as disease.”8
Just so, patients with acute MI display a wide variety of presentations, from no symptoms (about 25%) to severe, crushing chest pain. Discomfort may occur in the upper back, neck, jaw, teeth, arms, wrist, and epigastrium. Shortness of breath, diaphoresis, nausea, vomiting, and even syncope may occur. Unlike in acute aortic dissection, the discomfort is not usually maximal at its onset: it builds up in a crescendo manner. It is not usually changed by position, but can lessen in intensity upon standing. The discomfort in the chest is deep and visceral, and typically not well localized. A pressure sensation, air hunger, or “gas buildup” can be described. The only symptom may be shortness of breath or severe diaphoresis. The symptoms can last from minutes to hours and can be relieved by sublingual nitroglycerin. Atypical or less-prominent symptoms may make the diagnosis more difficult in the elderly, patients with diabetes mellitus, and women.
The physical examination during acute MI usually finds no clear-cut distinguishing features. The patient may appear pale and diaphoretic, and the skin cool to the touch. Heart sounds are generally soft. A fourth heart sound may be audible. Blood pressure may be low, but it can vary widely. Tachycardia, particularly sinus tachycardia, and pulmonary edema are poor prognostic signs.
In view of the wide variation in presentations, the history and physical findings can raise the suspicion of acute MI, but sequential electrocardiograms and measurements of biomarkers (troponin) are always necessary.
ELECTROCARDIOGRAPHY: NECESSARY BUT NOT SUFFICIENT
ST-elevation MI vs non-ST-elevation MI
Changes in the ST segment can be very dynamic, making sequential tracings very useful. Rhythm disturbances and heart block are also more likely to be recorded when using sequential readings.
Pitfalls to electrocardiographic diagnosis
Prior MI. Q waves or QS complexes, when the Q wave is sufficiently wide (≥ 0.03 msec) or deep (≥ 1 mV), usually indicate a previous MI. However, many nuances that further raise or lower the suspicion for previous MI need to be considered. These are beyond the scope of this brief review but are available in the 2007 update.
Right ventricular infarction often requires the use of right-sided leads, which may reveal ST elevation in V4R.
ECHOCARDIOGRAPHY IF THE DIAGNOSIS IS IN DOUBT
Echocardiography is an excellent way to assess wall-motion abnormalities. In the absence of any wall-motion abnormality, a large ST-elevation MI is unlikely. A large wall-motion abnormality would verify the probability of ongoing acute MI and thus would help with rapid decision-making.
Furthermore, echocardiography can help determine the likelihood that the patient has aortic dissection or pulmonary embolism, either of which can mimic acute MI but requires very different treatment.
CLINICAL CLASSIFICATION OF ACUTE MI
The new classification scheme of the different types of MI is shown in Table 4.
The new classification scheme does not include myocardial necrosis from mechanical manipulation of the heart during open heart surgery, from cardioversion, or from toxic drugs.
As clinicians are aware, it is not unusual to see elevated biomarker levels in a host of conditions unrelated to acute myocardial ischemia or MI. The new classification of acute MI is most helpful in this regard. It will likely be even more helpful in guiding treatment and management when new ultrasensitive troponin assays are widely introduced into clinical practice.
The new classification also negotiates the controversy regarding elevated biomarker levels following percutaneous coronary intervention. In brief, elevation of biomarkers is not entirely avoidable even with a successful percutaneous coronary intervention, and furthermore, there is no scientific cutoff for biomarker elevations. So, by arbitrary convention, the troponin level must rise to more than three times the 99th percentile upper reference limit to make the diagnosis of type 4a MI. A separate type 4b MI is ascribed to angiographic or autopsy-proven stent thrombosis.
The new guidelines also suggest that troponin values be more than five times the 99th percentile of the normal reference range during the first 72 hours following coronary artery bypass graft surgery (CABG) when considering a CABG-related MI (type 5). Whenever new pathologic Q waves appear in territories other than those identified before the procedure, MI should be considered, especially if associated with elevated biomarkers, new wall-motion abnormalities, or hemodynamic instability.
Thus, the diagnosis of acute MI now has widely accepted global criteria that distinguish various types of acute MI that occur under multiple circumstances. It is expected that describing the type of acute MI according to the new criteria will further enhance our understanding of the event, its proper management, and its prognosis.
- The Joint European Society of Cardiology/American College of Cardiology Committee. Myocardial infarction redefined—a consensus document of the Joint European Society of Cardiology/American College of Cardiology Committee for the Redefinition of Myocardial Infarction. J Am Coll Cardiol 2000; 36:959–969.
- Thygesen K, Alpert JS, White HD, on behalf of the Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction. Universal definition of myocardial infarction. J Am Coll Cardiol 2007; 50:2173–2188.
- Roger VL, Killian JM, Weston SA, et al. Redefinition of myocardial infarction—prospective evaluation in the community. Circulation 2006; 114:790–797.
- Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease. J Am Coll Cardiol 2006; 48:1–11.
- French JK, White HD. Clinical implications of the new definition of myocardial infarction. Heart 2004; 90:99–106.
- James TN. The variable morphological coexistence of apoptosis and necrosis in human myocardial infarction: significance for understanding its pathogenesis, clinical course, diagnosis and prognosis. Coron Artery Dis 1998; 9:291–307.
- Sobel BE, LeWinter MM. Ingenuous interpretation of elevated blood levels of macromolecular markers of myocardial injury: a recipe for confusion. J Am Coll Cardiol 2000; 35:1355–1358.
- Osler W. Aequanimitas: With Other Addresses to Medical Students, Nurses and Practitioners of Medicine.Osler William Edition: 3, revised. Philadelphia: Blakiston’s, 1932.
- Wang F, Asinger RW, Marriott HJ. ST-segment elevation in conditions other than acute myocardial infarction. N Engl J Med 2003; 349:2128–2135.
- The Joint European Society of Cardiology/American College of Cardiology Committee. Myocardial infarction redefined—a consensus document of the Joint European Society of Cardiology/American College of Cardiology Committee for the Redefinition of Myocardial Infarction. J Am Coll Cardiol 2000; 36:959–969.
- Thygesen K, Alpert JS, White HD, on behalf of the Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction. Universal definition of myocardial infarction. J Am Coll Cardiol 2007; 50:2173–2188.
- Roger VL, Killian JM, Weston SA, et al. Redefinition of myocardial infarction—prospective evaluation in the community. Circulation 2006; 114:790–797.
- Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease. J Am Coll Cardiol 2006; 48:1–11.
- French JK, White HD. Clinical implications of the new definition of myocardial infarction. Heart 2004; 90:99–106.
- James TN. The variable morphological coexistence of apoptosis and necrosis in human myocardial infarction: significance for understanding its pathogenesis, clinical course, diagnosis and prognosis. Coron Artery Dis 1998; 9:291–307.
- Sobel BE, LeWinter MM. Ingenuous interpretation of elevated blood levels of macromolecular markers of myocardial injury: a recipe for confusion. J Am Coll Cardiol 2000; 35:1355–1358.
- Osler W. Aequanimitas: With Other Addresses to Medical Students, Nurses and Practitioners of Medicine.Osler William Edition: 3, revised. Philadelphia: Blakiston’s, 1932.
- Wang F, Asinger RW, Marriott HJ. ST-segment elevation in conditions other than acute myocardial infarction. N Engl J Med 2003; 349:2128–2135.
KEY POINTS
- The clinical presentation of acute MI varies considerably from patient to patient. Therefore, one must consider the symptoms, serial electrocardiographic findings, and serial biomarker results in concert.
- Troponin I or T is now the preferred biomarker of myocardial necrosis. Still, troponin can be elevated in many conditions other than ischemic heart disease.
- Electrocardiographic signs of acute ischemia have been precisely defined, but electrocardiography can give false-positive or false-negative results in a number of conditions.
- MI is now categorized into five types depending on cause.