User login
Personalizing guideline-driven cancer screening
Reports of cancer date back thousands of years to Egyptian texts. Its existence baffled scientists until the 1950s, when Watson, Crick, and Franklin discovered the structure of DNA, laying the groundwork for identifying the genetic pathways leading to cancer. Currently, cancer is a leading global cause of death and the second leading cause of death in the United States.1,2
In an effort to curtail cancer and its related morbidity and mortality, population-based screening programs have been implemented with tests that identify precancerous lesions and, preferably, early-stage rather than late-stage cancer.
Screening for cancer can lead to early diagnosis and prevent death from cancer, but the topic continues to provoke controversy.
VALUE OF SCREENING QUESTIONED
In a commentary in the March 2019 Cleveland Clinic Journal of Medicine, Kim et al3 argued that cancer screening is not very effective and that we need to find the balance between the potential benefit and harm.
Using data from the US Preventive Services Task Force (USPSTF) and various studies, the authors showed that although screening can prevent some deaths from breast, colon, prostate, and lung cancer, at least 3 times as many people who are screened still die of those diseases. Given that screening does not eliminate all cancer deaths, has not been definitely shown to decrease the all-cause mortality rate, and has the potential to harm through false-positive results, overdiagnosis, and overtreatment, the authors questioned the utility of screening and encouraged us to discuss the benefits and harms with our patients.
In view of the apparently meager benefit, the USPSTF has relaxed its recommendations for screening for breast and prostate cancer in average-risk populations in recent years, a move that has evoked strong reactions from some clinicians. Proponents of screening argue that preventing late-stage cancers can save money, as the direct and indirect costs of morbidity associated with late-stage cancers are substantial, and that patients prefer screening when a test is available. Current models of screening efficacy do not take these factors into account.4
Kim et al, in defending the USPSTF’s position, suggested that the motivation for aggressive testing may be a belief that no harm is greater than the benefit of saving a life. They illustrated this through a Swiftian “modest proposal,” ie, universal prophylactic organectomy to prevent cancer. This hypothetical extreme measure would nearly eliminate the risk of cancer in the removed organs and prevent overdiagnosis and overtreatment of malignancies, but at substantial harm and cost.
In response to this proposal, we would like to point out the alternative extreme: stop all cancer screening programs. The pendulum would swing from what was previously considered a benefit—cancer prevention—to a harm, ie, cancer.
IN DEFENSE OF CANCER SCREENING
Observational studies, systematic reviews, meta-analyses, and modeling studies show that screening for cervical, colorectal, breast, and prostate cancer decreases disease-specific mortality.5–11
For example, in lung cancer, the National Lung Screening Trial demonstrated reductions in disease-specific and overall mortality in patients at high risk who underwent low-dose screening computed tomography.12
In breast cancer, a systematic review demonstrated decreased disease-specific mortality for women ages 50 through 79 who underwent screening mammography.13
In cervical cancer, lower rates of cancer-related death and invasive cancer have also been shown with screening.14
In colorectal cancer, great strides have been made in reducing both the incidence of and mortality from this disease over the past 30 years through fecal occult blood testing. Early detection shifts the 5-year survival rate—14% for late-stage cancer—to over 90%.15 Colorectal cancer screening has also been shown to be cost-effective, with savings in excess of $30,000 per life-year gained from screening.16
Moreover, recent data from the Prostate, Lung, Colorectal, and Ovarian Cancer (PLCO) screening trial17 demonstrated a 2-fold higher overall non-cancer-related mortality rate in participants who did not adhere to screening compared with those who were fully adherent to all sex-specific PLCO screening tests when adjusted for age, sex, and ethnicity. Although a possible explanation is that people who adhere to screening recommendations are also likely to have a healthier lifestyle overall, the association persisted (although it was slightly attenuated) even after adjusting for medical risk and behavioral factors.
ON THIS WE CAN AGREE
Like Kim et al, we also believe an informed discussion of screening should occur with each patient—and challenge Kim et al to design an efficient and practical approach to allow providers to do so in a busy office visit aimed to address and manage other competing diseases.
In addition, medical science needs to improve. Methods to increase the efficacy of screening and decrease risks should be explored; these include improving test and operator performance, reducing nonadherence to screening, investigating novel biomarkers or precursors of cancer and pathways that escape current detection, and devising better risk-stratification tools.
Bodies such as the USPSTF should use models that account for factors not considered previously but important when informing patients of potential benefits and harm. Examples include varying sensitivities and specificities at different rounds of testing and accounting for the variability in risk or efficacy affected by race, ethnicity, sex, and patient preferences.
We practice in the era of evidence-based medicine. Guidelines and recommendations are based on the available evidence. As more studies are published, disease mechanisms are better understood, and the effects of previous recommendations are evaluated, cancer screening programs will be further refined or replaced. The balance between benefit and harm will be further delineated.
Kim et al knocked on the door of personalized medicine, where individual screening will be based on individual risk. Until that door is opened, screening should be personalized through the risk-benefit discussions we have with our patients. Ultimately, the choice to undergo screening is the patient’s.
- Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends—an update. Cancer Epidemiol Biomarkers Prev 2016; 25(1):16–27. doi:10.1158/1055-9965.EPI-15-0578
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018; 68(1):7–30. doi:10.3322/caac.21442
- Kim MS, Nishikawa G, Prasad V. Cancer screening: a modest proposal for prevention. Cleve Clin J Med 2019; 86(3):157–160. doi:10.3949/ccjm.86a.18092
- Knudsen AB, Zauber AG, Rutter CM, et al. Estimation of benefits, burden, and harms of colorectal cancer screening strategies: modeling study for the US Preventive Services Task Force. JAMA 2016; 315(23):2595–2609. doi:10.1001/jama.2016.6828
- Peirson L, Fitzpatrick-Lewis D, Ciliska D, Warren R. Screening for cervical cancer: a systematic review and meta-analysis. Syst Rev 2013; 2:35. doi:10.1186/2046-4053-2-35
- Whitlock EP, Vesco KK, Eder M, Lin JS, Senger CA, Burda BU. Liquid-based cytology and human papillomavirus testing to screen for cervical cancer: a systematic review for the U.S. Preventive Services Task Force. Ann Intern Med 2011; 155(10):687–697. doi:10.7326/0003-4819-155-10-201111150-00376
- Yang DX, Gross CP, Soulos PR, Yu JB. Estimating the magnitude of colorectal cancers prevented during the era of screening: 1976 to 2009. Cancer 2014; 120:2893–2901. doi:10.1002/cncr.28794
- Edwards BK, Ward E, Kohler BA, et al. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer 2010; 116(3):544–573. doi:10.1002/cncr.24760
- Myers ER, Moorman P, Gierisch JM, et al. Benefits and harms of breast cancer screening: a systematic review. JAMA 2015; 314(15):1615–1634. doi:10.1001/jama.2015.13183
- Independent UK Panel on Breast Cancer Screening. The benefits and harms of breast cancer screening: an independent review. Lancet 2012; 380(9855):1778–1786. doi:10.1016/S0140-6736(12)61611-0
- Etzioni R, Tsodikov A, Mariotto A, et al. Quantifying the role of PSA screening in the US prostate cancer mortality decline. Cancer Causes Control 2008; 19(2):175–181. doi:10.1007/s10552-007-9083-8
- National Lung Screening Trial Research Team, Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365(5):395–409. doi:10.1056/NEJMoa1102873
- Nelson HD, Fu R, Cantor A, et al. Effectiveness of breast cancer screening: systematic review and meta-analysis to update the 2009 U.S. Preventive Services Task Force recommendation. Ann Intern Med 2016; 164(4):244–255. doi:10.7326/M15-0969
- US Preventive Services Task Force, Curry SJ, Krist AH, Owens DK, et al. Screening for cervical cancer: US Preventive Services Task Force recommendation statement. JAMA 2018; 320(7):674–686. doi:10.1001/jama.2018.10897
- Kopetz S, Chang GJ, Overman MJ, et al. Improved survival in metastatic colorectal cancer is associated with adoption of hepatic resection and improved chemotherapy. J Clin Oncol 2009; 27(22):3677–3683. doi:10.1200/JCO.2008.20.5278
- Patel S, Kilgore M. Cost effectiveness of colorectal cancer screening strategies. Cancer Control 2015; 22(2):248–258. doi:10.1177/107327481502200219
- Pierre-Victor D, Pinsky PF. Association of nonadherence to cancer screening examinations with mortality from unrelated causes: a secondary analysis of the PLCO cancer screening trial. JAMA Intern Med 2019; 179(2):196–203. doi:10.1001/jamainternmed.2018.5982
Reports of cancer date back thousands of years to Egyptian texts. Its existence baffled scientists until the 1950s, when Watson, Crick, and Franklin discovered the structure of DNA, laying the groundwork for identifying the genetic pathways leading to cancer. Currently, cancer is a leading global cause of death and the second leading cause of death in the United States.1,2
In an effort to curtail cancer and its related morbidity and mortality, population-based screening programs have been implemented with tests that identify precancerous lesions and, preferably, early-stage rather than late-stage cancer.
Screening for cancer can lead to early diagnosis and prevent death from cancer, but the topic continues to provoke controversy.
VALUE OF SCREENING QUESTIONED
In a commentary in the March 2019 Cleveland Clinic Journal of Medicine, Kim et al3 argued that cancer screening is not very effective and that we need to find the balance between the potential benefit and harm.
Using data from the US Preventive Services Task Force (USPSTF) and various studies, the authors showed that although screening can prevent some deaths from breast, colon, prostate, and lung cancer, at least 3 times as many people who are screened still die of those diseases. Given that screening does not eliminate all cancer deaths, has not been definitely shown to decrease the all-cause mortality rate, and has the potential to harm through false-positive results, overdiagnosis, and overtreatment, the authors questioned the utility of screening and encouraged us to discuss the benefits and harms with our patients.
In view of the apparently meager benefit, the USPSTF has relaxed its recommendations for screening for breast and prostate cancer in average-risk populations in recent years, a move that has evoked strong reactions from some clinicians. Proponents of screening argue that preventing late-stage cancers can save money, as the direct and indirect costs of morbidity associated with late-stage cancers are substantial, and that patients prefer screening when a test is available. Current models of screening efficacy do not take these factors into account.4
Kim et al, in defending the USPSTF’s position, suggested that the motivation for aggressive testing may be a belief that no harm is greater than the benefit of saving a life. They illustrated this through a Swiftian “modest proposal,” ie, universal prophylactic organectomy to prevent cancer. This hypothetical extreme measure would nearly eliminate the risk of cancer in the removed organs and prevent overdiagnosis and overtreatment of malignancies, but at substantial harm and cost.
In response to this proposal, we would like to point out the alternative extreme: stop all cancer screening programs. The pendulum would swing from what was previously considered a benefit—cancer prevention—to a harm, ie, cancer.
IN DEFENSE OF CANCER SCREENING
Observational studies, systematic reviews, meta-analyses, and modeling studies show that screening for cervical, colorectal, breast, and prostate cancer decreases disease-specific mortality.5–11
For example, in lung cancer, the National Lung Screening Trial demonstrated reductions in disease-specific and overall mortality in patients at high risk who underwent low-dose screening computed tomography.12
In breast cancer, a systematic review demonstrated decreased disease-specific mortality for women ages 50 through 79 who underwent screening mammography.13
In cervical cancer, lower rates of cancer-related death and invasive cancer have also been shown with screening.14
In colorectal cancer, great strides have been made in reducing both the incidence of and mortality from this disease over the past 30 years through fecal occult blood testing. Early detection shifts the 5-year survival rate—14% for late-stage cancer—to over 90%.15 Colorectal cancer screening has also been shown to be cost-effective, with savings in excess of $30,000 per life-year gained from screening.16
Moreover, recent data from the Prostate, Lung, Colorectal, and Ovarian Cancer (PLCO) screening trial17 demonstrated a 2-fold higher overall non-cancer-related mortality rate in participants who did not adhere to screening compared with those who were fully adherent to all sex-specific PLCO screening tests when adjusted for age, sex, and ethnicity. Although a possible explanation is that people who adhere to screening recommendations are also likely to have a healthier lifestyle overall, the association persisted (although it was slightly attenuated) even after adjusting for medical risk and behavioral factors.
ON THIS WE CAN AGREE
Like Kim et al, we also believe an informed discussion of screening should occur with each patient—and challenge Kim et al to design an efficient and practical approach to allow providers to do so in a busy office visit aimed to address and manage other competing diseases.
In addition, medical science needs to improve. Methods to increase the efficacy of screening and decrease risks should be explored; these include improving test and operator performance, reducing nonadherence to screening, investigating novel biomarkers or precursors of cancer and pathways that escape current detection, and devising better risk-stratification tools.
Bodies such as the USPSTF should use models that account for factors not considered previously but important when informing patients of potential benefits and harm. Examples include varying sensitivities and specificities at different rounds of testing and accounting for the variability in risk or efficacy affected by race, ethnicity, sex, and patient preferences.
We practice in the era of evidence-based medicine. Guidelines and recommendations are based on the available evidence. As more studies are published, disease mechanisms are better understood, and the effects of previous recommendations are evaluated, cancer screening programs will be further refined or replaced. The balance between benefit and harm will be further delineated.
Kim et al knocked on the door of personalized medicine, where individual screening will be based on individual risk. Until that door is opened, screening should be personalized through the risk-benefit discussions we have with our patients. Ultimately, the choice to undergo screening is the patient’s.
Reports of cancer date back thousands of years to Egyptian texts. Its existence baffled scientists until the 1950s, when Watson, Crick, and Franklin discovered the structure of DNA, laying the groundwork for identifying the genetic pathways leading to cancer. Currently, cancer is a leading global cause of death and the second leading cause of death in the United States.1,2
In an effort to curtail cancer and its related morbidity and mortality, population-based screening programs have been implemented with tests that identify precancerous lesions and, preferably, early-stage rather than late-stage cancer.
Screening for cancer can lead to early diagnosis and prevent death from cancer, but the topic continues to provoke controversy.
VALUE OF SCREENING QUESTIONED
In a commentary in the March 2019 Cleveland Clinic Journal of Medicine, Kim et al3 argued that cancer screening is not very effective and that we need to find the balance between the potential benefit and harm.
Using data from the US Preventive Services Task Force (USPSTF) and various studies, the authors showed that although screening can prevent some deaths from breast, colon, prostate, and lung cancer, at least 3 times as many people who are screened still die of those diseases. Given that screening does not eliminate all cancer deaths, has not been definitely shown to decrease the all-cause mortality rate, and has the potential to harm through false-positive results, overdiagnosis, and overtreatment, the authors questioned the utility of screening and encouraged us to discuss the benefits and harms with our patients.
In view of the apparently meager benefit, the USPSTF has relaxed its recommendations for screening for breast and prostate cancer in average-risk populations in recent years, a move that has evoked strong reactions from some clinicians. Proponents of screening argue that preventing late-stage cancers can save money, as the direct and indirect costs of morbidity associated with late-stage cancers are substantial, and that patients prefer screening when a test is available. Current models of screening efficacy do not take these factors into account.4
Kim et al, in defending the USPSTF’s position, suggested that the motivation for aggressive testing may be a belief that no harm is greater than the benefit of saving a life. They illustrated this through a Swiftian “modest proposal,” ie, universal prophylactic organectomy to prevent cancer. This hypothetical extreme measure would nearly eliminate the risk of cancer in the removed organs and prevent overdiagnosis and overtreatment of malignancies, but at substantial harm and cost.
In response to this proposal, we would like to point out the alternative extreme: stop all cancer screening programs. The pendulum would swing from what was previously considered a benefit—cancer prevention—to a harm, ie, cancer.
IN DEFENSE OF CANCER SCREENING
Observational studies, systematic reviews, meta-analyses, and modeling studies show that screening for cervical, colorectal, breast, and prostate cancer decreases disease-specific mortality.5–11
For example, in lung cancer, the National Lung Screening Trial demonstrated reductions in disease-specific and overall mortality in patients at high risk who underwent low-dose screening computed tomography.12
In breast cancer, a systematic review demonstrated decreased disease-specific mortality for women ages 50 through 79 who underwent screening mammography.13
In cervical cancer, lower rates of cancer-related death and invasive cancer have also been shown with screening.14
In colorectal cancer, great strides have been made in reducing both the incidence of and mortality from this disease over the past 30 years through fecal occult blood testing. Early detection shifts the 5-year survival rate—14% for late-stage cancer—to over 90%.15 Colorectal cancer screening has also been shown to be cost-effective, with savings in excess of $30,000 per life-year gained from screening.16
Moreover, recent data from the Prostate, Lung, Colorectal, and Ovarian Cancer (PLCO) screening trial17 demonstrated a 2-fold higher overall non-cancer-related mortality rate in participants who did not adhere to screening compared with those who were fully adherent to all sex-specific PLCO screening tests when adjusted for age, sex, and ethnicity. Although a possible explanation is that people who adhere to screening recommendations are also likely to have a healthier lifestyle overall, the association persisted (although it was slightly attenuated) even after adjusting for medical risk and behavioral factors.
ON THIS WE CAN AGREE
Like Kim et al, we also believe an informed discussion of screening should occur with each patient—and challenge Kim et al to design an efficient and practical approach to allow providers to do so in a busy office visit aimed to address and manage other competing diseases.
In addition, medical science needs to improve. Methods to increase the efficacy of screening and decrease risks should be explored; these include improving test and operator performance, reducing nonadherence to screening, investigating novel biomarkers or precursors of cancer and pathways that escape current detection, and devising better risk-stratification tools.
Bodies such as the USPSTF should use models that account for factors not considered previously but important when informing patients of potential benefits and harm. Examples include varying sensitivities and specificities at different rounds of testing and accounting for the variability in risk or efficacy affected by race, ethnicity, sex, and patient preferences.
We practice in the era of evidence-based medicine. Guidelines and recommendations are based on the available evidence. As more studies are published, disease mechanisms are better understood, and the effects of previous recommendations are evaluated, cancer screening programs will be further refined or replaced. The balance between benefit and harm will be further delineated.
Kim et al knocked on the door of personalized medicine, where individual screening will be based on individual risk. Until that door is opened, screening should be personalized through the risk-benefit discussions we have with our patients. Ultimately, the choice to undergo screening is the patient’s.
- Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends—an update. Cancer Epidemiol Biomarkers Prev 2016; 25(1):16–27. doi:10.1158/1055-9965.EPI-15-0578
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018; 68(1):7–30. doi:10.3322/caac.21442
- Kim MS, Nishikawa G, Prasad V. Cancer screening: a modest proposal for prevention. Cleve Clin J Med 2019; 86(3):157–160. doi:10.3949/ccjm.86a.18092
- Knudsen AB, Zauber AG, Rutter CM, et al. Estimation of benefits, burden, and harms of colorectal cancer screening strategies: modeling study for the US Preventive Services Task Force. JAMA 2016; 315(23):2595–2609. doi:10.1001/jama.2016.6828
- Peirson L, Fitzpatrick-Lewis D, Ciliska D, Warren R. Screening for cervical cancer: a systematic review and meta-analysis. Syst Rev 2013; 2:35. doi:10.1186/2046-4053-2-35
- Whitlock EP, Vesco KK, Eder M, Lin JS, Senger CA, Burda BU. Liquid-based cytology and human papillomavirus testing to screen for cervical cancer: a systematic review for the U.S. Preventive Services Task Force. Ann Intern Med 2011; 155(10):687–697. doi:10.7326/0003-4819-155-10-201111150-00376
- Yang DX, Gross CP, Soulos PR, Yu JB. Estimating the magnitude of colorectal cancers prevented during the era of screening: 1976 to 2009. Cancer 2014; 120:2893–2901. doi:10.1002/cncr.28794
- Edwards BK, Ward E, Kohler BA, et al. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer 2010; 116(3):544–573. doi:10.1002/cncr.24760
- Myers ER, Moorman P, Gierisch JM, et al. Benefits and harms of breast cancer screening: a systematic review. JAMA 2015; 314(15):1615–1634. doi:10.1001/jama.2015.13183
- Independent UK Panel on Breast Cancer Screening. The benefits and harms of breast cancer screening: an independent review. Lancet 2012; 380(9855):1778–1786. doi:10.1016/S0140-6736(12)61611-0
- Etzioni R, Tsodikov A, Mariotto A, et al. Quantifying the role of PSA screening in the US prostate cancer mortality decline. Cancer Causes Control 2008; 19(2):175–181. doi:10.1007/s10552-007-9083-8
- National Lung Screening Trial Research Team, Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365(5):395–409. doi:10.1056/NEJMoa1102873
- Nelson HD, Fu R, Cantor A, et al. Effectiveness of breast cancer screening: systematic review and meta-analysis to update the 2009 U.S. Preventive Services Task Force recommendation. Ann Intern Med 2016; 164(4):244–255. doi:10.7326/M15-0969
- US Preventive Services Task Force, Curry SJ, Krist AH, Owens DK, et al. Screening for cervical cancer: US Preventive Services Task Force recommendation statement. JAMA 2018; 320(7):674–686. doi:10.1001/jama.2018.10897
- Kopetz S, Chang GJ, Overman MJ, et al. Improved survival in metastatic colorectal cancer is associated with adoption of hepatic resection and improved chemotherapy. J Clin Oncol 2009; 27(22):3677–3683. doi:10.1200/JCO.2008.20.5278
- Patel S, Kilgore M. Cost effectiveness of colorectal cancer screening strategies. Cancer Control 2015; 22(2):248–258. doi:10.1177/107327481502200219
- Pierre-Victor D, Pinsky PF. Association of nonadherence to cancer screening examinations with mortality from unrelated causes: a secondary analysis of the PLCO cancer screening trial. JAMA Intern Med 2019; 179(2):196–203. doi:10.1001/jamainternmed.2018.5982
- Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends—an update. Cancer Epidemiol Biomarkers Prev 2016; 25(1):16–27. doi:10.1158/1055-9965.EPI-15-0578
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018; 68(1):7–30. doi:10.3322/caac.21442
- Kim MS, Nishikawa G, Prasad V. Cancer screening: a modest proposal for prevention. Cleve Clin J Med 2019; 86(3):157–160. doi:10.3949/ccjm.86a.18092
- Knudsen AB, Zauber AG, Rutter CM, et al. Estimation of benefits, burden, and harms of colorectal cancer screening strategies: modeling study for the US Preventive Services Task Force. JAMA 2016; 315(23):2595–2609. doi:10.1001/jama.2016.6828
- Peirson L, Fitzpatrick-Lewis D, Ciliska D, Warren R. Screening for cervical cancer: a systematic review and meta-analysis. Syst Rev 2013; 2:35. doi:10.1186/2046-4053-2-35
- Whitlock EP, Vesco KK, Eder M, Lin JS, Senger CA, Burda BU. Liquid-based cytology and human papillomavirus testing to screen for cervical cancer: a systematic review for the U.S. Preventive Services Task Force. Ann Intern Med 2011; 155(10):687–697. doi:10.7326/0003-4819-155-10-201111150-00376
- Yang DX, Gross CP, Soulos PR, Yu JB. Estimating the magnitude of colorectal cancers prevented during the era of screening: 1976 to 2009. Cancer 2014; 120:2893–2901. doi:10.1002/cncr.28794
- Edwards BK, Ward E, Kohler BA, et al. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer 2010; 116(3):544–573. doi:10.1002/cncr.24760
- Myers ER, Moorman P, Gierisch JM, et al. Benefits and harms of breast cancer screening: a systematic review. JAMA 2015; 314(15):1615–1634. doi:10.1001/jama.2015.13183
- Independent UK Panel on Breast Cancer Screening. The benefits and harms of breast cancer screening: an independent review. Lancet 2012; 380(9855):1778–1786. doi:10.1016/S0140-6736(12)61611-0
- Etzioni R, Tsodikov A, Mariotto A, et al. Quantifying the role of PSA screening in the US prostate cancer mortality decline. Cancer Causes Control 2008; 19(2):175–181. doi:10.1007/s10552-007-9083-8
- National Lung Screening Trial Research Team, Aberle DR, Adams AM, Berg CD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011; 365(5):395–409. doi:10.1056/NEJMoa1102873
- Nelson HD, Fu R, Cantor A, et al. Effectiveness of breast cancer screening: systematic review and meta-analysis to update the 2009 U.S. Preventive Services Task Force recommendation. Ann Intern Med 2016; 164(4):244–255. doi:10.7326/M15-0969
- US Preventive Services Task Force, Curry SJ, Krist AH, Owens DK, et al. Screening for cervical cancer: US Preventive Services Task Force recommendation statement. JAMA 2018; 320(7):674–686. doi:10.1001/jama.2018.10897
- Kopetz S, Chang GJ, Overman MJ, et al. Improved survival in metastatic colorectal cancer is associated with adoption of hepatic resection and improved chemotherapy. J Clin Oncol 2009; 27(22):3677–3683. doi:10.1200/JCO.2008.20.5278
- Patel S, Kilgore M. Cost effectiveness of colorectal cancer screening strategies. Cancer Control 2015; 22(2):248–258. doi:10.1177/107327481502200219
- Pierre-Victor D, Pinsky PF. Association of nonadherence to cancer screening examinations with mortality from unrelated causes: a secondary analysis of the PLCO cancer screening trial. JAMA Intern Med 2019; 179(2):196–203. doi:10.1001/jamainternmed.2018.5982

Sessile serrated polyps: Cancer risk and appropriate surveillance
Sessile serrated polyps are a type of polyp recently recognized to be a precursor of colorectal cancer. They arise from a pathway of genetic alterations different from the pathway that causes the more common and well-understood conventional adenomas (also called tubular adenomas, tubulovillous adenomas, and villous adenomas).
We do not yet know enough about the lifetime colorectal cancer risk for individuals with sessile serrated polyps, nor do we know the optimal surveillance interval for patients who have these polyps on colonoscopy. It is believed that sessile serrated polyps may be the cause of a substantial number of “interval” colorectal cancers—ie, cancers that occur after colonoscopy but before the next scheduled examination.
Serrated polyps get their name from their jagged appearance on microscopy. In the past, all serrated colorectal lesions were called hyperplastic polyps. But with the advent of molecular and genetic diagnostics and with the ability to recognize the subtle morphologic differences of serrated lesions, they have been reclassified into those without malignant potential (hyperplastic polyps) and those that are neoplastic (sessile serrated polyps and traditional serrated adenomas) (Table 1).
In this article, we discuss the evolving understanding of the different types of serrated polyps, and we offer our thoughts on a reasonable postpolypectomy surveillance plan in patients with these lesions. We focus on sessile serrated polyps, the most common form of serrated polyp with cancerous potential, since it may be one of our greatest challenges in optimal colorectal cancer prevention.
CLINICAL SCENARIO
A 65-year-old woman with no family history of colorectal cancer undergoes screening colonoscopy, during which three polyps are found and removed—a 3-mm tubular adenoma in the sigmoid colon, an 8-mm sessile serrated polyp at the hepatic flexure, and a 2-mm hyperplastic polyp in the rectum. When should she undergo follow-up colonoscopy?
Based on the number, size, and pathologic makeup of the polyps in this patient, we would recommend follow-up surveillance colonoscopy in 5 years.
THE SERRATED POLYP PATHWAY: A DIFFERENT PATH TO COLORECTAL CANCER
Colorectal cancer is the third most common cancer in the United States.1 From 70% to 80% of these cancers arise from adenomatous polyps via the adenoma-carcinoma pathway. This molecular pathway develops through chromosomal instability (CIN) and involves the loss of heterozygosity (the loss of function of one allele). This leads to the progressive accumulation of mutations in tumor-suppressor genes such as adenomatous polyposis coli (APC) and p53, and oncogenes such as KRAS. The result of these mutations is the development of adenomatous polyps that lead to microsatellite-stable colorectal cancers (Figure 1).2
More recently, studies have shown that the other 20% to 30% of colorectal cancers likely arise through a separate pathway, called the serrated polyp pathway or serrated neoplasia pathway. In contrast to CIN, this pathway is characterized by methylation of CpG islands (CIMP–CpG island methylation phenotype, CIMP) in the promoter regions of specific genes.3 Central to the serrated polyp pathway is progressive methylation in colonic mucosa; mutation in the BRAF oncogene, activating cell proliferation leading to a sessile serrated polyp; and epigenetic silencing of the DNA mismatch repair gene hMLH1, which is a key step in the progression to a sessile serrated polyp with dysplasia, which may rapidly become a microsatellite-unstable colorectal cancer.4
Histologically, serrated polyps have a serrated or sawtooth appearance from the folding in of the crypt epithelium, and they include hyperplastic polyps, traditional serrated adenomas, and sessile serrated polyps (sessile serrated adenomas).
Sessile serrated polyps and traditional serrated adenomas (which are rare) are thought to be precancerous, whereas hyperplastic polyps do not have malignant potential.
COMMON, BUT PREVALENCE IS NOT CLEARLY ESTABLISHED
The histologic criteria for sessile serrated polyps and traditional serrated adenomas have been elucidated,4–7 but the epidemiology of these serrated polyps is not clear. Small studies have shown that sessile serrated polyps account for 2% to 9% of all polyps removed at colonoscopy8–10; however, larger studies are needed to determine the prevalence because detection by an endoscopist and pathologic diagnosis of these polyps are both operator-dependent.
Traditional serrated adenomas are the least common type of serrated polyp, with a reported prevalence of 0.3%.7 Hyperplastic polyps are by far the most common, accounting for 20% to 30% of all polyps removed at colonoscopy.9,11 Sessile serrated polyps have a predilection for the proximal colon and are associated with female sex and with smoking, 12,13 but no consistent effect of other factors on their formation has been reported. In contrast, Wallace et al13 found that obesity, cigarette smoking, dietary fat intake, total caloric intake, and the consumption of red meat were associated with an increased risk of distal (but not proximal) serrated polyps, including hyperplastic polyps, sessile serrated polyps, and traditional serrated adenomas.
HYPERPLASTIC POLYPS
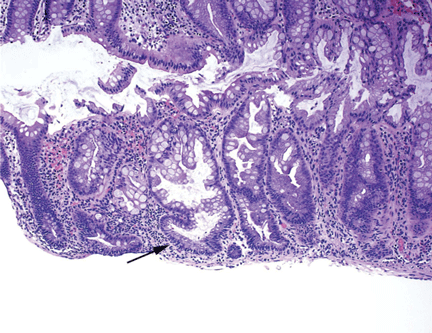
Hyperplastic polyps usually occur in the rectosigmoid colon. They appear as slightly elevated, whitish lesions with a diameter less than 5 mm (Figure 2). Microscopically, the serrated architecture is present in the upper half of their crypts (Figure 3). The proliferative zone is more or less normally located in the basal half of the crypt (the nonserrated portion), with nuclei that are small, uniform, and basally located.14 The bases of the crypts have a rounded contour and do not grow laterally along the muscularis mucosae.
SESSILE SERRATED POLYPS
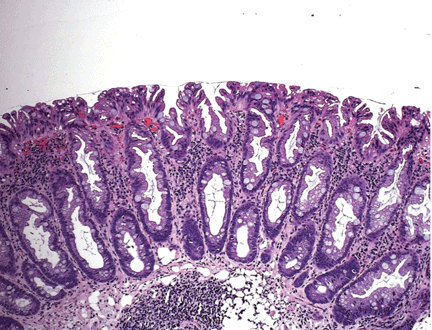
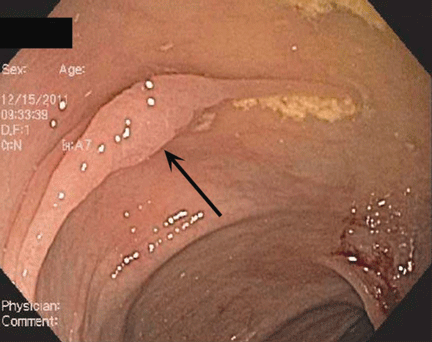
Endoscopically, sessile serrated polyps are often subtle, appear flat or slightly elevated, and can be covered by yellow mucus (Figure 4). They are typically found in the proximal colon and are usually larger than typical adenomas, with 50% being larger than 10 mm.10
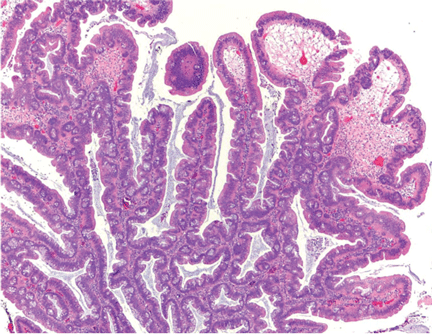
Histologically, the serrations are more prominent than those of hyperplastic polyps and involve the entire length of the crypt (Figure 5). The crypt bases are often dilated and display lateral growth along the lamina muscularis mucosae, resembling a letter t or l. The lamina muscularis mucosae is often thinner than normal. Crypts from sessile serrated polyps are occasionally found beneath the muscularis mucosae, a condition called pseudoinvasion.7
TRADITIONAL SERRATED ADENOMAS
Traditional serrated adenomas are usually left-sided. In contrast to the other types of serrated polyps, they are histologically often villiform and are lined by cells with elongated nuclei and abundant eosinophilic cytoplasm (Figure 6). Unlike those in sessile serrated polyps, the crypt bases do not display an abnormal architecture; rather, traditional serrated adenomas have abundant ectopic crypts (“budding crypts”) in the long, slender villi.7
Traditional serrated adenomas also appear to be genetically distinct from sessile serrated polyps. They are most often characterized by a KRAS (or less commonly, BRAF) mutation and commonly have methylation of the DNA repair gene MGMT (O-6-methylguanine-DNA methyltransferase) rather than hMLH1.
CHALLENGES TO EFFECTIVE COLONOSCOPY
Colonoscopic polypectomy of adenomatous polyps reduces the incidence of colorectal cancer and the rate of death from it.15,16 However, recent data show that colonoscopy may not be as effective as once thought. As many as 9% of patients with colorectal cancer have had a “normal” colonoscopic examination in the preceding 3 years.17,18 In addition, the reduction in incidence and mortality rates was less for cancers in the proximal colon than for cancers in the distal colon.19,20
Possible explanations for this discrepancy include the skill of the endoscopist, technical limitations of the examination, incomplete removal of polyps, and inadequate bowel preparation. Several studies have shown that interval colorectal cancers are more likely to be found in the proximal colon and to have the same molecular characteristics as sessile serrated polyps and the serrated colorectal cancer pathway (CIMP-high and MSI-H).21,22 Therefore, it is now thought that sessile serrated polyps may account for a substantial portion of “postcolonoscopy cancers” (ie, interval cancers) that arise in the proximal colon.
Two large studies of screening colonoscopy confirmed that the ability to detect sessile serrated polyps depends greatly on the skill of the endoscopist. Hetzel et al9 studied the differences in the rates of polyp detection among endoscopists performing more than 7,000 colonoscopies. Detection rates varied significantly for adenomas, hyperplastic polyps, and sessile serrated polyps, with the greatest variability noted in the detection of sessile serrated polyps. Significant variability was also noted in the ability of the pathologist to diagnose sessile serrated polyps.9
In the other study, a strong correlation was found between physicians who are “high detectors” of adenomas and their detection rates for proximal serrated polyps.23 There is widespread acceptance that screening colonoscopy in average-risk patients age 50 and older should detect adenomas in more than 25% of men and more than 15% of women. There is no current minimum recommended detection rate for sessile serrated polyps, but some have suggested 1.5%.8
POLYPS AS PREDICTORS OF CANCER RISK
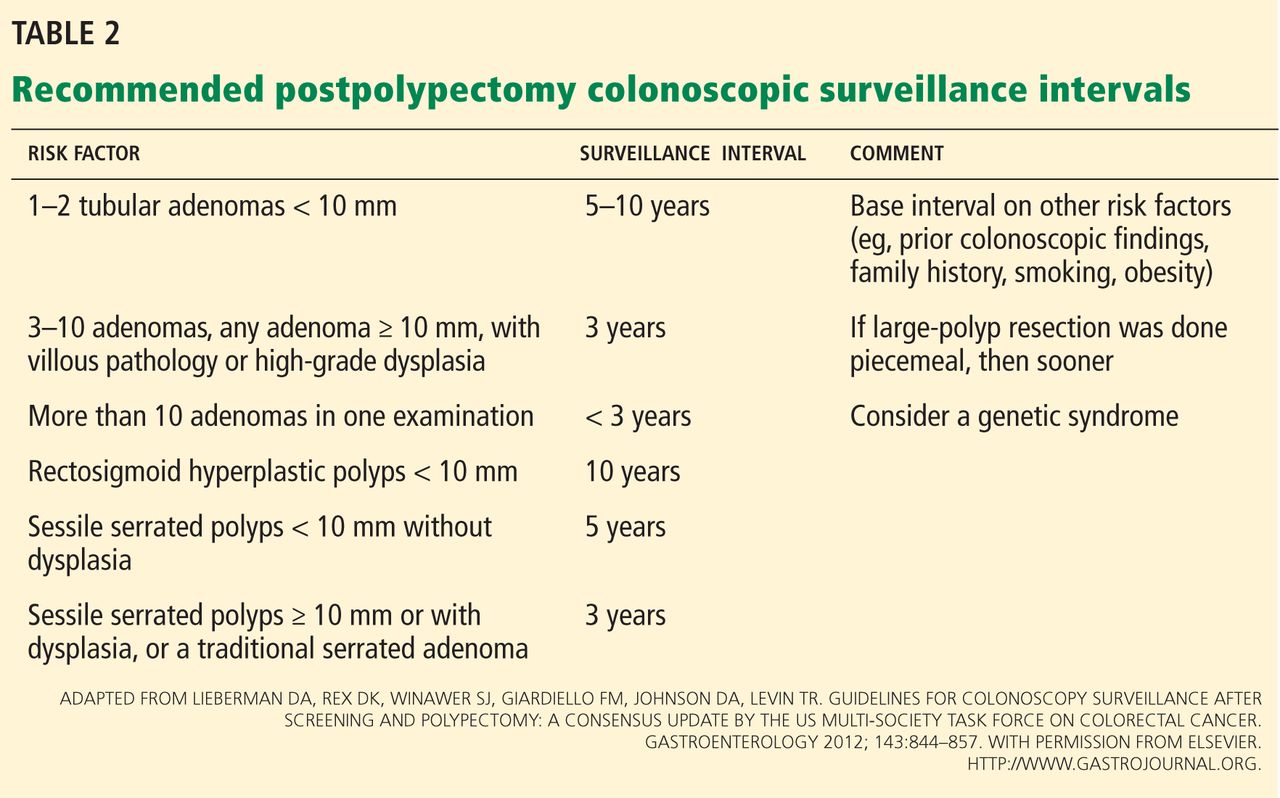
Certain polyp characteristics predict the risk of metachronous, advanced neoplasia. Advanced neoplasms are defined as invasive carcinomas, adenomas 10 mm or larger, or adenomas with any villous histology or high-grade dysplasia. Patients with one or two small tubular adenomas have a much lower risk of metachronous advanced neoplasia than do patients with more than two adenomas or advanced neoplasms.24 Current recommended surveillance intervals vary on that basis (Table 2).25
People who harbor serrated neoplasms are at high risk of synchronous serrated polyps and advanced adenomatous neoplasia. Pai et al26 found that patients with one sessile serrated polyp were four times more likely to have additional serrated polyps at the same time than an unselected population. The authors suggested that this indicates a strong colonic mucosal-field defect in patients with sessile serrated polyps, thereby predisposing them to the development of synchronous serrated polyps.
Li et al27 found that large serrated polyps (ie, > 10 mm) are associated with a risk of synchronous advanced neoplasia that is three times higher than in patients without adenomas. Schreiner et al28 determined that patients with either a proximal or a large serrated polyp were at higher risk of synchronous advanced neoplasia compared with patients who did not have those lesions. Vu et al29 found that patients who have both sessile serrated polyps and conventional adenomas have significantly larger and more numerous lesions of both types.29 In addition, these lesions are more likely to be pathologically advanced when compared with people with only one or the other type.
In the only study of the risk of advanced neoplasia on follow-up colonoscopy,28 patients with advanced neoplasia and proximal serrated polyps at baseline examination were twice as likely to have advanced neoplasia during subsequent surveillance than those with only advanced neoplasia at baseline examination.28
Therefore, it seems clear that the presence of large or proximal serrated polyps or serrated neoplasms predicts the presence of synchronous and likely metachronous advanced neoplasms.
Guidelines for postpolypectomy surveillance for individuals with serrated lesions of the colon have recently been published.25 Patients with large serrated lesions (≥ 10 mm) or an advanced serrated lesion (a sessile serrated polyp with or without cytologic dysplasia or a traditional serrated adenoma) should be followed closely. Patients with small (< 10-mm) rectosigmoid hyperplastic polyps should be followed as average-risk patients. If a patient with a sessile serrated polyp also has adenomas, the surveillance interval should be the shortest interval recommended for either lesion.29
SURVEILLANCE FOR OUR PATIENT
In our patient, given the number, size, and histologic features of the polyps found, surveillance colonoscopy should be considered in 5 years. Although the clinical significance of the serrated pathway to colorectal cancer cannot be argued, further study is required to understand the lifetime risk to patients with serrated neoplasms and the optimal surveillance interval.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62:10–29.
- Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology 2010; 138;2059–2072.
- Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology 2010; 138:2088–2100.
- Snover DC. Update on the serrated pathway to colorectal carcinoma. Hum Pathol 2011; 42:1–10.
- O’Brien MJ, Yang S, Mack C, et al. Comparison of microsatellite instability, CpG island methylation phenotype, BRAF and KRAS status in serrated polyps and traditional adenomas indicates separate pathways to distinct colorectal carcinoma end points. Am J Surg Pathol 2006; 30:1491–1501.
- Torlakovic E, Skovlund E, Snover DC, Torlakovic G, Nesland JM. Morphologic reappraisal of serrated colorectal polyps. Am J Surg Pathol 2003; 27:65–81.
- Torlakovic EE, Gomez JD, Driman DK, et al. Sessile serrated adenoma (SSA) vs traditional serrated adenoma (TSA). Am J Surg Pathol 2008; 32:21–29.
- Sanaka MR, Gohel T, Podugu A, et al. Quality indicators to enhance adenoma detection rate: should there be reconsideration of the current standard? Gastrointest Endosc 2011; 73:AB138.
- Hetzel JT, Huang CS, Coukos JA, et al. Variation in the detection of serrated polyps in an average risk colorectal cancer screening cohort. Am J Gastroenterol 2010; 105:2656–2664.
- Spring KJ, Zhao ZZ, Karamatic R, et al. High prevalence of sessile serrated adenomas with BRAF mutations: a prospective study of patients undergoing colonoscopy. Gastroenterology 2006; 131:1400–1407.
- Higuchi T, Sugihara K, Jass JR. Demographic and pathological characteristics of serrated polyps of colorectum. Histopathology 2005; 47:32–40.
- Lieberman DA, Prindiville S, Weiss DG, Willett W; VA Cooperative Study Group 380. Risk factors for advanced colonic neoplasia and hyperplastic polyps in asymptomatic individuals. JAMA 2003; 290:2959–2967.
- Wallace K, Grau MV, Ahnen D, et al. The association of lifestyle and dietary factors with the risk for serrated polyps of the colorectum. Cancer Epidemiol Biomarkers Prev 2009; 18:2310–2317.
- Rex DK, Ahnen DJ, Baron JA, Batts KP, Burke CA, et al. Serrated lesions of the colorectum: review and recommendations from an expert panel. Am J Gastroenterol 2012; 107:1315–1329.
- Winawer SJ, Zauber AG, Ho MN, et al. Prevention of colorectal cancer by colonoscopic polypectomy. The National Polyp Study Workgroup. N Engl J Med 1993; 329:1977–1981.
- Zauber AG, Winawer SJ, O’Brien MJ, et al. Colonoscopic polypectomy and long-term prevention of colorectal-cancer deaths. N Engl J Med 2012; 366:687–696.
- Sawhney MS, Farrar WD, Gudiseva S, et al. Microsatellite instability in interval colon cancers. Gastroenterology 2006; 131:1700–1705.
- Baxter NN, Sutradhar R, Forbes SS, Paszat lF, Saskin R, Rabeneck l. Analysis of administrative data finds endoscopist quality measures associated with postcolonoscopy colorectal cancer. Gastroenterology 2011; 140:65–72.
- Singh H, Nugent Z, Demers AA, Kliewer EV, Mahmud SM, Bernstein CN. The reduction in colorectal cancer mortality after colonoscopy varies by site of the cancer. Gastroenterology 2010; 139:1128–1137.
- Baxter NN, Goldwasser MA, Paszat lF, Saskin R, Urbach DR, Rabeneck l. Association of colonoscopy and death from colorectal cancer. Ann Intern Med 2009; 150:1–8.
- Arain MA, Sawhney M, Sheikh S, et al. CIMP status of interval colon cancers: another piece to the puzzle. Am J Gastroenterol 2010; 105:1189–1195.
- Farrar WD, Sawhney MS, Nelson DB, Lederle FA, Bond JH. Colorectal cancers found after a complete colonoscopy. Clin Gastroenterol Hepatol 2006; 4:1259–1264.
- Kahi CJ, Hewett DG, Norton Dl, Eckert GJ, Rex DK. Prevalence and variable detection of proximal colon serrated polyps during screening colonoscopy. Clin Gastroenterol Hepatol 2011; 9:42–46.
- Martínez ME, Baron JA, Lieberman DA, et al. A pooled analysis of advanced colorectal neoplasia diagnoses after colonoscopic polypectomy. Gastroenterology 2009; 136:832–841.
- Lieberman DA, Rex DK, Winawer SJ, Giardiello FM, Johnson DA, Levin TR. Guidelines for colonoscopy surveillance after screening and polypectomy: a consensus update by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 2012; 143:844–857.
- Pai RK, Hart J, Noffsinger AE. Sessile serrated adenomas strongly predispose to synchronous serrated polyps in nonsyndromic patients. Histopathology 2010; 56:581–588.
- Li D, Jin C, McCulloch C, et al. Association of large serrated polyps with synchronous advanced colorectal neoplasia. Am J Gastroenterol 2009; 104:695–702.
- Schreiner MA, Weiss DG, Lieberman DA. Proximal and large hyperplastic and nondysplastic serrated polyps detected by colonoscopy are associated with neoplasia. Gastroenterology 2010; 139:1497–1502.
- Vu HT, Lopez R, Bennett A, Burke CA. Individuals with sessile serrated polyps express an aggressive colorectal phenotype. Dis Colon Rectum 2011; 54:1216–1223.
Sessile serrated polyps are a type of polyp recently recognized to be a precursor of colorectal cancer. They arise from a pathway of genetic alterations different from the pathway that causes the more common and well-understood conventional adenomas (also called tubular adenomas, tubulovillous adenomas, and villous adenomas).
We do not yet know enough about the lifetime colorectal cancer risk for individuals with sessile serrated polyps, nor do we know the optimal surveillance interval for patients who have these polyps on colonoscopy. It is believed that sessile serrated polyps may be the cause of a substantial number of “interval” colorectal cancers—ie, cancers that occur after colonoscopy but before the next scheduled examination.
Serrated polyps get their name from their jagged appearance on microscopy. In the past, all serrated colorectal lesions were called hyperplastic polyps. But with the advent of molecular and genetic diagnostics and with the ability to recognize the subtle morphologic differences of serrated lesions, they have been reclassified into those without malignant potential (hyperplastic polyps) and those that are neoplastic (sessile serrated polyps and traditional serrated adenomas) (Table 1).
In this article, we discuss the evolving understanding of the different types of serrated polyps, and we offer our thoughts on a reasonable postpolypectomy surveillance plan in patients with these lesions. We focus on sessile serrated polyps, the most common form of serrated polyp with cancerous potential, since it may be one of our greatest challenges in optimal colorectal cancer prevention.
CLINICAL SCENARIO
A 65-year-old woman with no family history of colorectal cancer undergoes screening colonoscopy, during which three polyps are found and removed—a 3-mm tubular adenoma in the sigmoid colon, an 8-mm sessile serrated polyp at the hepatic flexure, and a 2-mm hyperplastic polyp in the rectum. When should she undergo follow-up colonoscopy?
Based on the number, size, and pathologic makeup of the polyps in this patient, we would recommend follow-up surveillance colonoscopy in 5 years.
THE SERRATED POLYP PATHWAY: A DIFFERENT PATH TO COLORECTAL CANCER
Colorectal cancer is the third most common cancer in the United States.1 From 70% to 80% of these cancers arise from adenomatous polyps via the adenoma-carcinoma pathway. This molecular pathway develops through chromosomal instability (CIN) and involves the loss of heterozygosity (the loss of function of one allele). This leads to the progressive accumulation of mutations in tumor-suppressor genes such as adenomatous polyposis coli (APC) and p53, and oncogenes such as KRAS. The result of these mutations is the development of adenomatous polyps that lead to microsatellite-stable colorectal cancers (Figure 1).2
More recently, studies have shown that the other 20% to 30% of colorectal cancers likely arise through a separate pathway, called the serrated polyp pathway or serrated neoplasia pathway. In contrast to CIN, this pathway is characterized by methylation of CpG islands (CIMP–CpG island methylation phenotype, CIMP) in the promoter regions of specific genes.3 Central to the serrated polyp pathway is progressive methylation in colonic mucosa; mutation in the BRAF oncogene, activating cell proliferation leading to a sessile serrated polyp; and epigenetic silencing of the DNA mismatch repair gene hMLH1, which is a key step in the progression to a sessile serrated polyp with dysplasia, which may rapidly become a microsatellite-unstable colorectal cancer.4
Histologically, serrated polyps have a serrated or sawtooth appearance from the folding in of the crypt epithelium, and they include hyperplastic polyps, traditional serrated adenomas, and sessile serrated polyps (sessile serrated adenomas).
Sessile serrated polyps and traditional serrated adenomas (which are rare) are thought to be precancerous, whereas hyperplastic polyps do not have malignant potential.
COMMON, BUT PREVALENCE IS NOT CLEARLY ESTABLISHED
The histologic criteria for sessile serrated polyps and traditional serrated adenomas have been elucidated,4–7 but the epidemiology of these serrated polyps is not clear. Small studies have shown that sessile serrated polyps account for 2% to 9% of all polyps removed at colonoscopy8–10; however, larger studies are needed to determine the prevalence because detection by an endoscopist and pathologic diagnosis of these polyps are both operator-dependent.
Traditional serrated adenomas are the least common type of serrated polyp, with a reported prevalence of 0.3%.7 Hyperplastic polyps are by far the most common, accounting for 20% to 30% of all polyps removed at colonoscopy.9,11 Sessile serrated polyps have a predilection for the proximal colon and are associated with female sex and with smoking, 12,13 but no consistent effect of other factors on their formation has been reported. In contrast, Wallace et al13 found that obesity, cigarette smoking, dietary fat intake, total caloric intake, and the consumption of red meat were associated with an increased risk of distal (but not proximal) serrated polyps, including hyperplastic polyps, sessile serrated polyps, and traditional serrated adenomas.
HYPERPLASTIC POLYPS
Hyperplastic polyps usually occur in the rectosigmoid colon. They appear as slightly elevated, whitish lesions with a diameter less than 5 mm (Figure 2). Microscopically, the serrated architecture is present in the upper half of their crypts (Figure 3). The proliferative zone is more or less normally located in the basal half of the crypt (the nonserrated portion), with nuclei that are small, uniform, and basally located.14 The bases of the crypts have a rounded contour and do not grow laterally along the muscularis mucosae.
SESSILE SERRATED POLYPS
Endoscopically, sessile serrated polyps are often subtle, appear flat or slightly elevated, and can be covered by yellow mucus (Figure 4). They are typically found in the proximal colon and are usually larger than typical adenomas, with 50% being larger than 10 mm.10
Histologically, the serrations are more prominent than those of hyperplastic polyps and involve the entire length of the crypt (Figure 5). The crypt bases are often dilated and display lateral growth along the lamina muscularis mucosae, resembling a letter t or l. The lamina muscularis mucosae is often thinner than normal. Crypts from sessile serrated polyps are occasionally found beneath the muscularis mucosae, a condition called pseudoinvasion.7
TRADITIONAL SERRATED ADENOMAS
Traditional serrated adenomas are usually left-sided. In contrast to the other types of serrated polyps, they are histologically often villiform and are lined by cells with elongated nuclei and abundant eosinophilic cytoplasm (Figure 6). Unlike those in sessile serrated polyps, the crypt bases do not display an abnormal architecture; rather, traditional serrated adenomas have abundant ectopic crypts (“budding crypts”) in the long, slender villi.7
Traditional serrated adenomas also appear to be genetically distinct from sessile serrated polyps. They are most often characterized by a KRAS (or less commonly, BRAF) mutation and commonly have methylation of the DNA repair gene MGMT (O-6-methylguanine-DNA methyltransferase) rather than hMLH1.
CHALLENGES TO EFFECTIVE COLONOSCOPY
Colonoscopic polypectomy of adenomatous polyps reduces the incidence of colorectal cancer and the rate of death from it.15,16 However, recent data show that colonoscopy may not be as effective as once thought. As many as 9% of patients with colorectal cancer have had a “normal” colonoscopic examination in the preceding 3 years.17,18 In addition, the reduction in incidence and mortality rates was less for cancers in the proximal colon than for cancers in the distal colon.19,20
Possible explanations for this discrepancy include the skill of the endoscopist, technical limitations of the examination, incomplete removal of polyps, and inadequate bowel preparation. Several studies have shown that interval colorectal cancers are more likely to be found in the proximal colon and to have the same molecular characteristics as sessile serrated polyps and the serrated colorectal cancer pathway (CIMP-high and MSI-H).21,22 Therefore, it is now thought that sessile serrated polyps may account for a substantial portion of “postcolonoscopy cancers” (ie, interval cancers) that arise in the proximal colon.
Two large studies of screening colonoscopy confirmed that the ability to detect sessile serrated polyps depends greatly on the skill of the endoscopist. Hetzel et al9 studied the differences in the rates of polyp detection among endoscopists performing more than 7,000 colonoscopies. Detection rates varied significantly for adenomas, hyperplastic polyps, and sessile serrated polyps, with the greatest variability noted in the detection of sessile serrated polyps. Significant variability was also noted in the ability of the pathologist to diagnose sessile serrated polyps.9
In the other study, a strong correlation was found between physicians who are “high detectors” of adenomas and their detection rates for proximal serrated polyps.23 There is widespread acceptance that screening colonoscopy in average-risk patients age 50 and older should detect adenomas in more than 25% of men and more than 15% of women. There is no current minimum recommended detection rate for sessile serrated polyps, but some have suggested 1.5%.8
POLYPS AS PREDICTORS OF CANCER RISK
Certain polyp characteristics predict the risk of metachronous, advanced neoplasia. Advanced neoplasms are defined as invasive carcinomas, adenomas 10 mm or larger, or adenomas with any villous histology or high-grade dysplasia. Patients with one or two small tubular adenomas have a much lower risk of metachronous advanced neoplasia than do patients with more than two adenomas or advanced neoplasms.24 Current recommended surveillance intervals vary on that basis (Table 2).25
People who harbor serrated neoplasms are at high risk of synchronous serrated polyps and advanced adenomatous neoplasia. Pai et al26 found that patients with one sessile serrated polyp were four times more likely to have additional serrated polyps at the same time than an unselected population. The authors suggested that this indicates a strong colonic mucosal-field defect in patients with sessile serrated polyps, thereby predisposing them to the development of synchronous serrated polyps.
Li et al27 found that large serrated polyps (ie, > 10 mm) are associated with a risk of synchronous advanced neoplasia that is three times higher than in patients without adenomas. Schreiner et al28 determined that patients with either a proximal or a large serrated polyp were at higher risk of synchronous advanced neoplasia compared with patients who did not have those lesions. Vu et al29 found that patients who have both sessile serrated polyps and conventional adenomas have significantly larger and more numerous lesions of both types.29 In addition, these lesions are more likely to be pathologically advanced when compared with people with only one or the other type.
In the only study of the risk of advanced neoplasia on follow-up colonoscopy,28 patients with advanced neoplasia and proximal serrated polyps at baseline examination were twice as likely to have advanced neoplasia during subsequent surveillance than those with only advanced neoplasia at baseline examination.28
Therefore, it seems clear that the presence of large or proximal serrated polyps or serrated neoplasms predicts the presence of synchronous and likely metachronous advanced neoplasms.
Guidelines for postpolypectomy surveillance for individuals with serrated lesions of the colon have recently been published.25 Patients with large serrated lesions (≥ 10 mm) or an advanced serrated lesion (a sessile serrated polyp with or without cytologic dysplasia or a traditional serrated adenoma) should be followed closely. Patients with small (< 10-mm) rectosigmoid hyperplastic polyps should be followed as average-risk patients. If a patient with a sessile serrated polyp also has adenomas, the surveillance interval should be the shortest interval recommended for either lesion.29
SURVEILLANCE FOR OUR PATIENT
In our patient, given the number, size, and histologic features of the polyps found, surveillance colonoscopy should be considered in 5 years. Although the clinical significance of the serrated pathway to colorectal cancer cannot be argued, further study is required to understand the lifetime risk to patients with serrated neoplasms and the optimal surveillance interval.
Sessile serrated polyps are a type of polyp recently recognized to be a precursor of colorectal cancer. They arise from a pathway of genetic alterations different from the pathway that causes the more common and well-understood conventional adenomas (also called tubular adenomas, tubulovillous adenomas, and villous adenomas).
We do not yet know enough about the lifetime colorectal cancer risk for individuals with sessile serrated polyps, nor do we know the optimal surveillance interval for patients who have these polyps on colonoscopy. It is believed that sessile serrated polyps may be the cause of a substantial number of “interval” colorectal cancers—ie, cancers that occur after colonoscopy but before the next scheduled examination.
Serrated polyps get their name from their jagged appearance on microscopy. In the past, all serrated colorectal lesions were called hyperplastic polyps. But with the advent of molecular and genetic diagnostics and with the ability to recognize the subtle morphologic differences of serrated lesions, they have been reclassified into those without malignant potential (hyperplastic polyps) and those that are neoplastic (sessile serrated polyps and traditional serrated adenomas) (Table 1).
In this article, we discuss the evolving understanding of the different types of serrated polyps, and we offer our thoughts on a reasonable postpolypectomy surveillance plan in patients with these lesions. We focus on sessile serrated polyps, the most common form of serrated polyp with cancerous potential, since it may be one of our greatest challenges in optimal colorectal cancer prevention.
CLINICAL SCENARIO
A 65-year-old woman with no family history of colorectal cancer undergoes screening colonoscopy, during which three polyps are found and removed—a 3-mm tubular adenoma in the sigmoid colon, an 8-mm sessile serrated polyp at the hepatic flexure, and a 2-mm hyperplastic polyp in the rectum. When should she undergo follow-up colonoscopy?
Based on the number, size, and pathologic makeup of the polyps in this patient, we would recommend follow-up surveillance colonoscopy in 5 years.
THE SERRATED POLYP PATHWAY: A DIFFERENT PATH TO COLORECTAL CANCER
Colorectal cancer is the third most common cancer in the United States.1 From 70% to 80% of these cancers arise from adenomatous polyps via the adenoma-carcinoma pathway. This molecular pathway develops through chromosomal instability (CIN) and involves the loss of heterozygosity (the loss of function of one allele). This leads to the progressive accumulation of mutations in tumor-suppressor genes such as adenomatous polyposis coli (APC) and p53, and oncogenes such as KRAS. The result of these mutations is the development of adenomatous polyps that lead to microsatellite-stable colorectal cancers (Figure 1).2
More recently, studies have shown that the other 20% to 30% of colorectal cancers likely arise through a separate pathway, called the serrated polyp pathway or serrated neoplasia pathway. In contrast to CIN, this pathway is characterized by methylation of CpG islands (CIMP–CpG island methylation phenotype, CIMP) in the promoter regions of specific genes.3 Central to the serrated polyp pathway is progressive methylation in colonic mucosa; mutation in the BRAF oncogene, activating cell proliferation leading to a sessile serrated polyp; and epigenetic silencing of the DNA mismatch repair gene hMLH1, which is a key step in the progression to a sessile serrated polyp with dysplasia, which may rapidly become a microsatellite-unstable colorectal cancer.4
Histologically, serrated polyps have a serrated or sawtooth appearance from the folding in of the crypt epithelium, and they include hyperplastic polyps, traditional serrated adenomas, and sessile serrated polyps (sessile serrated adenomas).
Sessile serrated polyps and traditional serrated adenomas (which are rare) are thought to be precancerous, whereas hyperplastic polyps do not have malignant potential.
COMMON, BUT PREVALENCE IS NOT CLEARLY ESTABLISHED
The histologic criteria for sessile serrated polyps and traditional serrated adenomas have been elucidated,4–7 but the epidemiology of these serrated polyps is not clear. Small studies have shown that sessile serrated polyps account for 2% to 9% of all polyps removed at colonoscopy8–10; however, larger studies are needed to determine the prevalence because detection by an endoscopist and pathologic diagnosis of these polyps are both operator-dependent.
Traditional serrated adenomas are the least common type of serrated polyp, with a reported prevalence of 0.3%.7 Hyperplastic polyps are by far the most common, accounting for 20% to 30% of all polyps removed at colonoscopy.9,11 Sessile serrated polyps have a predilection for the proximal colon and are associated with female sex and with smoking, 12,13 but no consistent effect of other factors on their formation has been reported. In contrast, Wallace et al13 found that obesity, cigarette smoking, dietary fat intake, total caloric intake, and the consumption of red meat were associated with an increased risk of distal (but not proximal) serrated polyps, including hyperplastic polyps, sessile serrated polyps, and traditional serrated adenomas.
HYPERPLASTIC POLYPS
Hyperplastic polyps usually occur in the rectosigmoid colon. They appear as slightly elevated, whitish lesions with a diameter less than 5 mm (Figure 2). Microscopically, the serrated architecture is present in the upper half of their crypts (Figure 3). The proliferative zone is more or less normally located in the basal half of the crypt (the nonserrated portion), with nuclei that are small, uniform, and basally located.14 The bases of the crypts have a rounded contour and do not grow laterally along the muscularis mucosae.
SESSILE SERRATED POLYPS
Endoscopically, sessile serrated polyps are often subtle, appear flat or slightly elevated, and can be covered by yellow mucus (Figure 4). They are typically found in the proximal colon and are usually larger than typical adenomas, with 50% being larger than 10 mm.10
Histologically, the serrations are more prominent than those of hyperplastic polyps and involve the entire length of the crypt (Figure 5). The crypt bases are often dilated and display lateral growth along the lamina muscularis mucosae, resembling a letter t or l. The lamina muscularis mucosae is often thinner than normal. Crypts from sessile serrated polyps are occasionally found beneath the muscularis mucosae, a condition called pseudoinvasion.7
TRADITIONAL SERRATED ADENOMAS
Traditional serrated adenomas are usually left-sided. In contrast to the other types of serrated polyps, they are histologically often villiform and are lined by cells with elongated nuclei and abundant eosinophilic cytoplasm (Figure 6). Unlike those in sessile serrated polyps, the crypt bases do not display an abnormal architecture; rather, traditional serrated adenomas have abundant ectopic crypts (“budding crypts”) in the long, slender villi.7
Traditional serrated adenomas also appear to be genetically distinct from sessile serrated polyps. They are most often characterized by a KRAS (or less commonly, BRAF) mutation and commonly have methylation of the DNA repair gene MGMT (O-6-methylguanine-DNA methyltransferase) rather than hMLH1.
CHALLENGES TO EFFECTIVE COLONOSCOPY
Colonoscopic polypectomy of adenomatous polyps reduces the incidence of colorectal cancer and the rate of death from it.15,16 However, recent data show that colonoscopy may not be as effective as once thought. As many as 9% of patients with colorectal cancer have had a “normal” colonoscopic examination in the preceding 3 years.17,18 In addition, the reduction in incidence and mortality rates was less for cancers in the proximal colon than for cancers in the distal colon.19,20
Possible explanations for this discrepancy include the skill of the endoscopist, technical limitations of the examination, incomplete removal of polyps, and inadequate bowel preparation. Several studies have shown that interval colorectal cancers are more likely to be found in the proximal colon and to have the same molecular characteristics as sessile serrated polyps and the serrated colorectal cancer pathway (CIMP-high and MSI-H).21,22 Therefore, it is now thought that sessile serrated polyps may account for a substantial portion of “postcolonoscopy cancers” (ie, interval cancers) that arise in the proximal colon.
Two large studies of screening colonoscopy confirmed that the ability to detect sessile serrated polyps depends greatly on the skill of the endoscopist. Hetzel et al9 studied the differences in the rates of polyp detection among endoscopists performing more than 7,000 colonoscopies. Detection rates varied significantly for adenomas, hyperplastic polyps, and sessile serrated polyps, with the greatest variability noted in the detection of sessile serrated polyps. Significant variability was also noted in the ability of the pathologist to diagnose sessile serrated polyps.9
In the other study, a strong correlation was found between physicians who are “high detectors” of adenomas and their detection rates for proximal serrated polyps.23 There is widespread acceptance that screening colonoscopy in average-risk patients age 50 and older should detect adenomas in more than 25% of men and more than 15% of women. There is no current minimum recommended detection rate for sessile serrated polyps, but some have suggested 1.5%.8
POLYPS AS PREDICTORS OF CANCER RISK
Certain polyp characteristics predict the risk of metachronous, advanced neoplasia. Advanced neoplasms are defined as invasive carcinomas, adenomas 10 mm or larger, or adenomas with any villous histology or high-grade dysplasia. Patients with one or two small tubular adenomas have a much lower risk of metachronous advanced neoplasia than do patients with more than two adenomas or advanced neoplasms.24 Current recommended surveillance intervals vary on that basis (Table 2).25
People who harbor serrated neoplasms are at high risk of synchronous serrated polyps and advanced adenomatous neoplasia. Pai et al26 found that patients with one sessile serrated polyp were four times more likely to have additional serrated polyps at the same time than an unselected population. The authors suggested that this indicates a strong colonic mucosal-field defect in patients with sessile serrated polyps, thereby predisposing them to the development of synchronous serrated polyps.
Li et al27 found that large serrated polyps (ie, > 10 mm) are associated with a risk of synchronous advanced neoplasia that is three times higher than in patients without adenomas. Schreiner et al28 determined that patients with either a proximal or a large serrated polyp were at higher risk of synchronous advanced neoplasia compared with patients who did not have those lesions. Vu et al29 found that patients who have both sessile serrated polyps and conventional adenomas have significantly larger and more numerous lesions of both types.29 In addition, these lesions are more likely to be pathologically advanced when compared with people with only one or the other type.
In the only study of the risk of advanced neoplasia on follow-up colonoscopy,28 patients with advanced neoplasia and proximal serrated polyps at baseline examination were twice as likely to have advanced neoplasia during subsequent surveillance than those with only advanced neoplasia at baseline examination.28
Therefore, it seems clear that the presence of large or proximal serrated polyps or serrated neoplasms predicts the presence of synchronous and likely metachronous advanced neoplasms.
Guidelines for postpolypectomy surveillance for individuals with serrated lesions of the colon have recently been published.25 Patients with large serrated lesions (≥ 10 mm) or an advanced serrated lesion (a sessile serrated polyp with or without cytologic dysplasia or a traditional serrated adenoma) should be followed closely. Patients with small (< 10-mm) rectosigmoid hyperplastic polyps should be followed as average-risk patients. If a patient with a sessile serrated polyp also has adenomas, the surveillance interval should be the shortest interval recommended for either lesion.29
SURVEILLANCE FOR OUR PATIENT
In our patient, given the number, size, and histologic features of the polyps found, surveillance colonoscopy should be considered in 5 years. Although the clinical significance of the serrated pathway to colorectal cancer cannot be argued, further study is required to understand the lifetime risk to patients with serrated neoplasms and the optimal surveillance interval.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62:10–29.
- Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology 2010; 138;2059–2072.
- Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology 2010; 138:2088–2100.
- Snover DC. Update on the serrated pathway to colorectal carcinoma. Hum Pathol 2011; 42:1–10.
- O’Brien MJ, Yang S, Mack C, et al. Comparison of microsatellite instability, CpG island methylation phenotype, BRAF and KRAS status in serrated polyps and traditional adenomas indicates separate pathways to distinct colorectal carcinoma end points. Am J Surg Pathol 2006; 30:1491–1501.
- Torlakovic E, Skovlund E, Snover DC, Torlakovic G, Nesland JM. Morphologic reappraisal of serrated colorectal polyps. Am J Surg Pathol 2003; 27:65–81.
- Torlakovic EE, Gomez JD, Driman DK, et al. Sessile serrated adenoma (SSA) vs traditional serrated adenoma (TSA). Am J Surg Pathol 2008; 32:21–29.
- Sanaka MR, Gohel T, Podugu A, et al. Quality indicators to enhance adenoma detection rate: should there be reconsideration of the current standard? Gastrointest Endosc 2011; 73:AB138.
- Hetzel JT, Huang CS, Coukos JA, et al. Variation in the detection of serrated polyps in an average risk colorectal cancer screening cohort. Am J Gastroenterol 2010; 105:2656–2664.
- Spring KJ, Zhao ZZ, Karamatic R, et al. High prevalence of sessile serrated adenomas with BRAF mutations: a prospective study of patients undergoing colonoscopy. Gastroenterology 2006; 131:1400–1407.
- Higuchi T, Sugihara K, Jass JR. Demographic and pathological characteristics of serrated polyps of colorectum. Histopathology 2005; 47:32–40.
- Lieberman DA, Prindiville S, Weiss DG, Willett W; VA Cooperative Study Group 380. Risk factors for advanced colonic neoplasia and hyperplastic polyps in asymptomatic individuals. JAMA 2003; 290:2959–2967.
- Wallace K, Grau MV, Ahnen D, et al. The association of lifestyle and dietary factors with the risk for serrated polyps of the colorectum. Cancer Epidemiol Biomarkers Prev 2009; 18:2310–2317.
- Rex DK, Ahnen DJ, Baron JA, Batts KP, Burke CA, et al. Serrated lesions of the colorectum: review and recommendations from an expert panel. Am J Gastroenterol 2012; 107:1315–1329.
- Winawer SJ, Zauber AG, Ho MN, et al. Prevention of colorectal cancer by colonoscopic polypectomy. The National Polyp Study Workgroup. N Engl J Med 1993; 329:1977–1981.
- Zauber AG, Winawer SJ, O’Brien MJ, et al. Colonoscopic polypectomy and long-term prevention of colorectal-cancer deaths. N Engl J Med 2012; 366:687–696.
- Sawhney MS, Farrar WD, Gudiseva S, et al. Microsatellite instability in interval colon cancers. Gastroenterology 2006; 131:1700–1705.
- Baxter NN, Sutradhar R, Forbes SS, Paszat lF, Saskin R, Rabeneck l. Analysis of administrative data finds endoscopist quality measures associated with postcolonoscopy colorectal cancer. Gastroenterology 2011; 140:65–72.
- Singh H, Nugent Z, Demers AA, Kliewer EV, Mahmud SM, Bernstein CN. The reduction in colorectal cancer mortality after colonoscopy varies by site of the cancer. Gastroenterology 2010; 139:1128–1137.
- Baxter NN, Goldwasser MA, Paszat lF, Saskin R, Urbach DR, Rabeneck l. Association of colonoscopy and death from colorectal cancer. Ann Intern Med 2009; 150:1–8.
- Arain MA, Sawhney M, Sheikh S, et al. CIMP status of interval colon cancers: another piece to the puzzle. Am J Gastroenterol 2010; 105:1189–1195.
- Farrar WD, Sawhney MS, Nelson DB, Lederle FA, Bond JH. Colorectal cancers found after a complete colonoscopy. Clin Gastroenterol Hepatol 2006; 4:1259–1264.
- Kahi CJ, Hewett DG, Norton Dl, Eckert GJ, Rex DK. Prevalence and variable detection of proximal colon serrated polyps during screening colonoscopy. Clin Gastroenterol Hepatol 2011; 9:42–46.
- Martínez ME, Baron JA, Lieberman DA, et al. A pooled analysis of advanced colorectal neoplasia diagnoses after colonoscopic polypectomy. Gastroenterology 2009; 136:832–841.
- Lieberman DA, Rex DK, Winawer SJ, Giardiello FM, Johnson DA, Levin TR. Guidelines for colonoscopy surveillance after screening and polypectomy: a consensus update by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 2012; 143:844–857.
- Pai RK, Hart J, Noffsinger AE. Sessile serrated adenomas strongly predispose to synchronous serrated polyps in nonsyndromic patients. Histopathology 2010; 56:581–588.
- Li D, Jin C, McCulloch C, et al. Association of large serrated polyps with synchronous advanced colorectal neoplasia. Am J Gastroenterol 2009; 104:695–702.
- Schreiner MA, Weiss DG, Lieberman DA. Proximal and large hyperplastic and nondysplastic serrated polyps detected by colonoscopy are associated with neoplasia. Gastroenterology 2010; 139:1497–1502.
- Vu HT, Lopez R, Bennett A, Burke CA. Individuals with sessile serrated polyps express an aggressive colorectal phenotype. Dis Colon Rectum 2011; 54:1216–1223.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62:10–29.
- Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology 2010; 138;2059–2072.
- Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology 2010; 138:2088–2100.
- Snover DC. Update on the serrated pathway to colorectal carcinoma. Hum Pathol 2011; 42:1–10.
- O’Brien MJ, Yang S, Mack C, et al. Comparison of microsatellite instability, CpG island methylation phenotype, BRAF and KRAS status in serrated polyps and traditional adenomas indicates separate pathways to distinct colorectal carcinoma end points. Am J Surg Pathol 2006; 30:1491–1501.
- Torlakovic E, Skovlund E, Snover DC, Torlakovic G, Nesland JM. Morphologic reappraisal of serrated colorectal polyps. Am J Surg Pathol 2003; 27:65–81.
- Torlakovic EE, Gomez JD, Driman DK, et al. Sessile serrated adenoma (SSA) vs traditional serrated adenoma (TSA). Am J Surg Pathol 2008; 32:21–29.
- Sanaka MR, Gohel T, Podugu A, et al. Quality indicators to enhance adenoma detection rate: should there be reconsideration of the current standard? Gastrointest Endosc 2011; 73:AB138.
- Hetzel JT, Huang CS, Coukos JA, et al. Variation in the detection of serrated polyps in an average risk colorectal cancer screening cohort. Am J Gastroenterol 2010; 105:2656–2664.
- Spring KJ, Zhao ZZ, Karamatic R, et al. High prevalence of sessile serrated adenomas with BRAF mutations: a prospective study of patients undergoing colonoscopy. Gastroenterology 2006; 131:1400–1407.
- Higuchi T, Sugihara K, Jass JR. Demographic and pathological characteristics of serrated polyps of colorectum. Histopathology 2005; 47:32–40.
- Lieberman DA, Prindiville S, Weiss DG, Willett W; VA Cooperative Study Group 380. Risk factors for advanced colonic neoplasia and hyperplastic polyps in asymptomatic individuals. JAMA 2003; 290:2959–2967.
- Wallace K, Grau MV, Ahnen D, et al. The association of lifestyle and dietary factors with the risk for serrated polyps of the colorectum. Cancer Epidemiol Biomarkers Prev 2009; 18:2310–2317.
- Rex DK, Ahnen DJ, Baron JA, Batts KP, Burke CA, et al. Serrated lesions of the colorectum: review and recommendations from an expert panel. Am J Gastroenterol 2012; 107:1315–1329.
- Winawer SJ, Zauber AG, Ho MN, et al. Prevention of colorectal cancer by colonoscopic polypectomy. The National Polyp Study Workgroup. N Engl J Med 1993; 329:1977–1981.
- Zauber AG, Winawer SJ, O’Brien MJ, et al. Colonoscopic polypectomy and long-term prevention of colorectal-cancer deaths. N Engl J Med 2012; 366:687–696.
- Sawhney MS, Farrar WD, Gudiseva S, et al. Microsatellite instability in interval colon cancers. Gastroenterology 2006; 131:1700–1705.
- Baxter NN, Sutradhar R, Forbes SS, Paszat lF, Saskin R, Rabeneck l. Analysis of administrative data finds endoscopist quality measures associated with postcolonoscopy colorectal cancer. Gastroenterology 2011; 140:65–72.
- Singh H, Nugent Z, Demers AA, Kliewer EV, Mahmud SM, Bernstein CN. The reduction in colorectal cancer mortality after colonoscopy varies by site of the cancer. Gastroenterology 2010; 139:1128–1137.
- Baxter NN, Goldwasser MA, Paszat lF, Saskin R, Urbach DR, Rabeneck l. Association of colonoscopy and death from colorectal cancer. Ann Intern Med 2009; 150:1–8.
- Arain MA, Sawhney M, Sheikh S, et al. CIMP status of interval colon cancers: another piece to the puzzle. Am J Gastroenterol 2010; 105:1189–1195.
- Farrar WD, Sawhney MS, Nelson DB, Lederle FA, Bond JH. Colorectal cancers found after a complete colonoscopy. Clin Gastroenterol Hepatol 2006; 4:1259–1264.
- Kahi CJ, Hewett DG, Norton Dl, Eckert GJ, Rex DK. Prevalence and variable detection of proximal colon serrated polyps during screening colonoscopy. Clin Gastroenterol Hepatol 2011; 9:42–46.
- Martínez ME, Baron JA, Lieberman DA, et al. A pooled analysis of advanced colorectal neoplasia diagnoses after colonoscopic polypectomy. Gastroenterology 2009; 136:832–841.
- Lieberman DA, Rex DK, Winawer SJ, Giardiello FM, Johnson DA, Levin TR. Guidelines for colonoscopy surveillance after screening and polypectomy: a consensus update by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 2012; 143:844–857.
- Pai RK, Hart J, Noffsinger AE. Sessile serrated adenomas strongly predispose to synchronous serrated polyps in nonsyndromic patients. Histopathology 2010; 56:581–588.
- Li D, Jin C, McCulloch C, et al. Association of large serrated polyps with synchronous advanced colorectal neoplasia. Am J Gastroenterol 2009; 104:695–702.
- Schreiner MA, Weiss DG, Lieberman DA. Proximal and large hyperplastic and nondysplastic serrated polyps detected by colonoscopy are associated with neoplasia. Gastroenterology 2010; 139:1497–1502.
- Vu HT, Lopez R, Bennett A, Burke CA. Individuals with sessile serrated polyps express an aggressive colorectal phenotype. Dis Colon Rectum 2011; 54:1216–1223.
KEY POINTS
- From 20% to 30% of colorectal cancers arise through the serrated polyp pathway (the serrated neoplasia pathway.)
- Histologically, serrated polyps have a serrated or sawtooth appearance from the folding in of the crypt epithelium. Types of serrated polyps include hyperplastic polyps, traditional serrated adenomas, and sessile serrated polyps (also known as sessile serrated adenomas).
- Guidelines for surveillance after polypectomy of serrated lesions recommend that patients with a large (≥ 10-mm) or a sessile serrated polyp with cytologic dysplasia or a traditional serrated adenoma be followed more closely than patients with a sessile serrated polyp smaller than 10 mm. Patients with small rectosigmoid hyperplastic polyps should be followed the same as people at average risk.
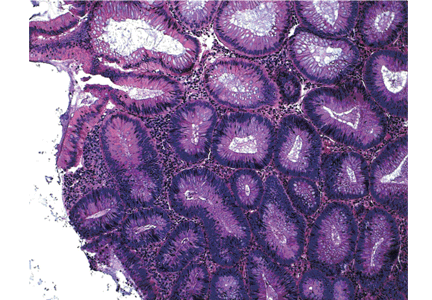
Detecting and managing hereditary colorectal cancer syndromes in your practice
Hereditary colorectal cancer syndromes account for 5% to 10% of cases of colorectal cancer.
Identifying these patients in clinical practice begins by assessing a patient’s personal and family health history. An accurate and comprehensive family history should cover three generations and include ethnic background, ages and causes of death of relatives, and any diagnosis of cancer, including age at onset and history of polyps.

Red flags for a hereditary colorectal cancer syndrome in the personal or family history are:
- Early age of onset of cancer (eg, colorectal cancer before age 50)
- More than 10 colorectal adenomas
- Synchronous (ie, occurring at the same time) or metachronous (occurring at different times) primary cancers
- Multiple relatives in successive generations with the same or related cancers (eg, colon or endometrial cancer)
- A family member with a known hereditary colorectal cancer syndrome (Table 1).
Any of these red flags should prompt a referral for genetic counseling.
SYNDROMES ARE CLASSIFIED AS WITH OR WITHOUT POLYPOSIS

Many hereditary syndromes are associated with a higher risk of colorectal cancer. Generally, they can be divided into two categories (Table 2): polyposis syndromes (in which patients have numerous colorectal polyps) and nonpolyposis syndromes (with few or no polyps).
These two main types are subclassified on the basis of the histology of most of the polyps detected: adenomatous, hamartomatous, serrated, or mixed types.
In this review, we will address the three most common of these syndromes: Lynch syndrome (hereditary nonpolyposis colorectal cancer), familial adenomatous polyposis, and MYH-associated polyposis. However, as noted in Table 2, other hereditary colorectal cancer syndromes exist, and suspicion of these conditions should prompt a referral for further evaluation.
LYNCH SYNDROME (HEREDITARY NONPOLYPOSIS COLORECTAL CANCER)
Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer, predisposes people to a variety of cancers.
Colorectal cancer is the most common type of cancer associated with Lynch syndrome. Recent research suggests that the cumulative risk of developing colorectal cancer by age 80 is 42% for all patients with Lynch syndrome.1 The median age at onset is 45 years.1 For patients who undergo segmental resection of their initial cancer, the cumulative risk of metachronous colorectal cancer (ie, a new tumor arising later) is 16% at 10 years, 41% at 20 years, and up to 62% after 30 years.2
Endometrial cancer occurs in 17% to 57% of women with Lynch syndrome by age 70, with a median age at onset of 49 years.1
Other extracolonic cancers in Lynch syndrome include cancers of the:
- Stomach (1%–10% risk by age 70 years)
- Ovaries (1%–20% risk)
- Hepatobiliary tract (1%–2% risk)
- Urinary tract (1%–12% risk)
- Small bowel (1%–2% risk)
- Brain (1%–8% risk)
- Skin (sebaceous adenomas, adenocarcinomas, and keratoacanthomas).1,3,4
Earlier studies reported higher rates of associated cancer than those shown here. However, their data were largely derived from registries and may be overestimates. The numbers shown above are from population-based studies.
Genetics of Lynch syndrome
Lynch syndrome is caused by a germline mutation in the MLH1, MSH2, MSH6, PMS2, or EPCAM genes.5 These genes code for proteins that are responsible DNA mismatch repair—one of the cell’s proofreading mechanisms during DNA replication.
These mutations are inherited in an autosomal dominant manner. Though de novo mutations in these genes have been reported, they are rare and the exact frequency with which they occur is unknown.6
In whom should Lynch syndrome be suspected?
Lynch syndrome can be suspected on the basis of family history and clinical criteria.
In 1991, the same group of experts who coined the term “hereditary nonpolyposis colorectal cancer” developed family history criteria for it1:
- At least three relatives with histologically confirmed colorectal cancer, one of whom is a first-degree relative of the other two
- At least two successive generations involved
- At least one of the cancers diagnosed before age 50
- Familial adenomatous polyposis is excluded.

Known as the Amsterdam criteria, these were to be used in collaborative studies of families with hereditary colorectal cancer.7 In 1999, these criteria were broadened to include extracolonic cancers and became known as the Amsterdam II criteria (Table 3).8
Patients whose families meet the Amsterdam II criteria or who have molecular pathologic evidence of Lynch syndrome (see below) are appropriate candidates for genetic counseling and testing.
Diagnosis of Lynch syndrome
The diagnosis of Lynch syndrome is based on molecular pathologic analysis (performed on tumor samples) and confirmed by genetic testing.
Molecular pathologic evidence of Lynch syndrome includes microsatellite instability and loss of expression of one or more of the DNA mismatch repair proteins (detected using immunohistochemistry) (more on these below). The revised Bethesda guidelines (TABLE 3) were intended to identify individuals whose tumors should be tested for one or both of these phenomena.9
In 2009, the Evaluation of Genomic Applications in Practice and Prevention working group recommended that all patients with newly diagnosed colorectal cancer undergo microsatellite instability analysis, immunohistochemistry testing, or both, regardless of whether they meet the Amsterdam II or the Bethesda guideline criteria.10
Microsatellite instability analysis. Microsatellites are short sequences of repeated DNA. The tumor cells of patients who carry defective mismatch repair genes have microsatellites that are longer or shorter than in normal cells, a condition called microsatellite instability (ie, “MSI-high”).
Microsatellite instability testing, using a standardized panel of five DNA markers, is performed on normal and tumor tissue. If more than two of the five microsatellite markers in the tumor show instability, the lesion is considered to have a high level of microsatellite instability. About 15% of colorectal cancers have this high level, although most are not associated with Lynch syndrome and lose MLH1 expression by promoter methylation.11,12
While only 2% of patients with colorectal cancer have Lynch syndrome, from 90% to 95% of colorectal cancers from patients with Lynch syndrome have high levels of microsatellite instability.10 The presence of MLH1 promoter hypermethylation, the BRAF mutation V600E, or both within the tumor suggests that the cancer is not associated with Lynch syndrome.
Some families that meet the Amsterdam I criteria have microsatellite-stable tumors: their condition has been called familial colorectal cancer type X.13 This condition is associated with a higher risk of colorectal cancer but not the other malignancies observed in Lynch syndrome.
Immunohistochemistry is performed to assess for expression of the mismatch repair proteins MSH2, MSH6, MLH1, and PMS2. Absence of expression of the specific protein within tumor cells compared with normal cells within the specimen suggests dysfunction of the specific gene and guides germline mutation testing (Figure 1). For example, a patient who lacks expression of the MSH2 protein in his or her colon cancer most likely has a mutation in the MSH2 gene. Therefore, germ-line genetic testing should initially target the MSH2 gene. Approximately 88% of Lynch syndrome-associated colorectal cancers have abnormal immunohistochemical staining.10
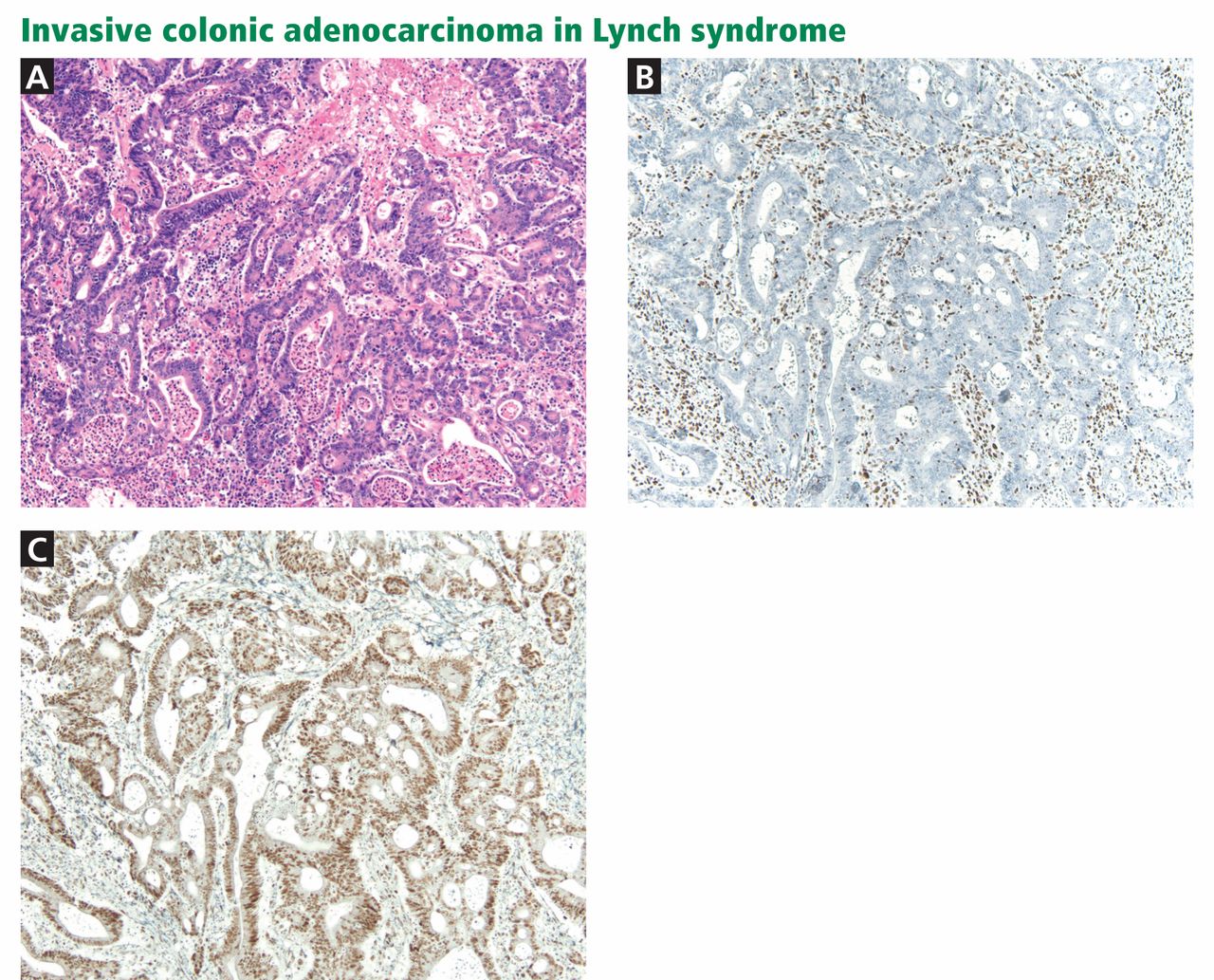
Testing for microsatellite instability and mismatch repair gene expression ideally precedes germline genetic testing and helps to guide which gene or genes should be tested.9,14
Genetic testing for Lynch syndrome is routinely performed on a blood or saliva sample, using DNA from white blood cells and sequencing the gene or genes involved to look for mutations. Positive results from a germline genetic test confirm the diagnosis of Lynch syndrome and allow for predictive testing for relatives at risk. The term Lynch syndrome is used exclusively to describe individuals with evidence of a mutation in one of the mismatch repair genes.15
If a patient’s results are positive, genetic counseling and genetic testing should be offered to at-risk relatives age 18 and over.
Management of Lynch syndrome
Aggressive cancer surveillance is essential for people with Lynch syndrome and for those who are considered at risk but have not pursued genetic testing, such as a sibling of a person with Lynch syndrome.
Colorectal cancer. Colonoscopy is recommended every 1 to 2 years beginning at the age of 20 to 25 years, or 2 to 5 years earlier than the age of the youngest relative affected with colorectal cancer if the initial diagnosis was before age 25. When patients turn 40 years old, colonoscopy is done annually.16–18 A significant reduction in cancer incidence and in the mortality rate has been shown with colonoscopic surveillance.19–21
Chemoprevention may also have a role. Patients with Lynch syndrome who took aspirin 600 mg per day for an average of 25 months had a significantly lower incidence of colorectal cancer during a 55-month follow-up period compared with patients randomized to placebo.22
For patients with Lynch syndrome who are diagnosed with colorectal cancer, the high risk of metachronous cancers after standard segmental colectomy calls for a more extended resection. Retrospective analysis of 382 Lynch syndrome patients found that none of the 50 who underwent total or subtotal colectomy were diagnosed with metachronous colorectal cancer, whereas a metachronous cancer developed in 74 (22%) of the 332 patients who had had segmented resection.2 Annual surveillance of the remaining colon, rectum, or both is indicated postoperatively.
Gynecologic cancers. Women with Lynch syndrome should also consider gynecologic surveillance and risk-reducing surgery. This includes annual gynecologic examination, transvaginal ultrasonography, and endometrial aspiration, beginning at age 30 to 35 years. Although this surveillance does detect premalignant lesions and early symptomatic cancers, its effect on the mortality rate is unknown. Hysterectomy with bilateral salpingo-oophorectomy has been shown to significantly reduce endometrial and ovarian cancers in women with Lynch syndrome.23,24
Urothelial cancers. Carriers of MSH2 mutations have a significantly higher risk of urothelial cancers.4 Therefore, MSH2 carriers should consider ultrasonography of the urinary tract, urinary cytology, and urinalysis every 1 to 2 years beginning at age 40.4
Other extracolonic cancers. Poor evidence exists for systematic screening for the other extracolonic tumors associated with Lynch syndrome. However, the National Comprehensive Cancer Network advises considering esophagogastroduodenoscopy with extended duodenoscopy as well as capsule endoscopy every 2 to 3 years beginning at age 30 to 35.14
ADENOMATOUS POLYPOSIS SYNDROMES
Familial adenomatous polyposis and MYH-associated polyposis are the next most common hereditary colorectal cancer syndromes. Each of these accounts for about 1% of cases of colorectal cancer. Clinically, these two syndromes can be challenging to distinguish because they overlap phenotypically to a significant degree.
FAMILIAL ADENOMATOUS POLYPOSIS
Familial adenomatous polyposis is caused by mutations in the APC gene. Its prevalence is 2.29 to 3.2 per 100,000 individuals.25,26
Genetics of familial adenomatous polyposis
APC is the only gene known to cause familial adenomatous polyposis. Mutations in APC are inherited in an autosomal dominant manner. Approximately 25% of cases of familial adenomatous polyposis are due to a de novo mutation in APC.27
Clinical presentation of familial adenomatous polyposis
Familial adenomatous polyposis is classified by the burden of colorectal adenomas.
Patients who have fewer than 100 adenomas have an attenuated form of the disease. In this group, polyps usually begin to form in the late teenage years or early 20s and tend to develop in the proximal colon. The attenuated form is associated with an approximately 70% lifetime risk of colorectal cancer.28
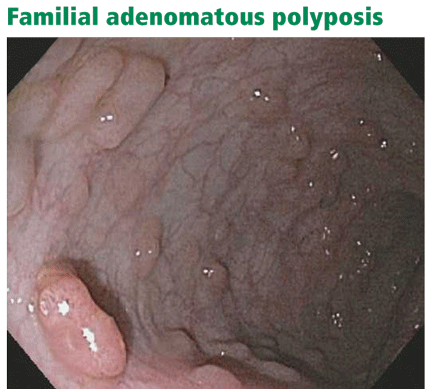
Patients who have more than 100 polyps are considered to have the classic form of the disease, and those with more than 1,000 polyps have profuse familial adenomatous polyposis (Figure 2). In these groups, polyps typically begin to develop in the preteenage to mid-teenage years. Without surgery, there is nearly a 100% risk of colorectal cancer. The average age at diagnosis of colorectal cancer is 39 years for patients with classic disease.
Upper gastrointestinal polyps are common in familial adenomatous polyposis. Nearly 90% of patients develop duodenal adenomas by a mean age of 44, with a cumulative lifetime risk of nearly 100%.29 Fundic gland polyposis occurs in nearly 90% of patients,30 while gastric adenomas are reported in fewer than 15% of patients.
Duodenal and periampullary cancer is the second most common malignancy in familial adenomatous polyposis. The lifetime risk ranges from 2% to 36%, depending on the Spigelman stage. People with Spigelman stage I, II, or III have a 2.5% risk of duodenal cancer, while those with stage IV disease have up to a 36% lifetime risk.
Gastric cancer, arising from fundic gland polyps, has been reported but is rare in Western populations.
In familial adenomatous polyposis, the incidence of jejunal adenomas and cancer is less than 10%, and the risk of ileal adenomas and cancer is less than 1%.31
Familial adenomatous polyposis is also associated with a higher risk of other malignancies, including:
- Pancreatic cancer (2% lifetime risk)
- Thyroid cancer (2% to 3% lifetime risk, typically papillary carcinoma)32
- Hepatoblastoma (1% to 2% lifetime risk)
- Brain tumors (< 1% lifetime risk)
- Biliary cancer (higher risk than in the general population).33
Benign extracolonic manifestations that have been observed include osteomas, dental abnormalities (supernumerary teeth, unerupted or absent teeth, odontomas), congenital hypertrophy of the retinal pigment epithelium, benign cutaneous lesions (epidermoid cysts and fibromas), and desmoid tumors.33 The term “Gardner syndrome” has been used to describe patients who have familial adenomatous polyposis but also have osteomas and soft-tissue tumors.34 These patients carry the same risk of colorectal cancer as other patients with familial adenomatous polyposis.
Diagnosing familial adenomatous polyposis
The diagnosis of familial adenomatous polyposis is suspected when a patient has more than 10 adenomatous polyps.
Seventy-five percent of patients with familial adenomatous polyposis have a family history of the condition. Therefore, most cases are identified at a young age on screening sigmoidoscopy or colonoscopy or by predictive gene testing. Patients rarely have cancer at the time of diagnosis.
The other 25% of patients typically are diagnosed when symptoms develop from the polyps or cancer. Over 50% of these symptomatic patients have cancer at the time of diagnosis.
It is recommended that people who have more than 10 adenomas detected on a single colonoscopy or who are first-degree relatives of patients with familial adenomatous polyposis undergo a genetic evaluation and testing for mutations in the APC gene.14 Once an APC mutation is identified in the family, at-risk relatives should be offered testing around age 10 years for families with classic familial adenomatous polyposis or in the mid to late teenage years for those with the attenuated form. It also appropriate to refer patients with desmoid tumors, duodenal adenomas, and bilateral or multifocal congenital hypertrophy of the retinal pigment epithelium for a genetic evaluation.
Management of familial adenomatous polyposis
Flexible sigmoidoscopy every 1 to 2 years beginning at age 10 to 12 years is recommended for individuals and families who have been phenotypically or genetically diagnosed with familial adenomatous polyposis.35–37 If colorectal adenomas are found, surgical options should be discussed and annual colonoscopic surveillance should commence.
For people with the attenuated form, because of the later age of disease onset and the tendency for right-sided disease, colonoscopy every 1 to 2 years should commence at about age 18.35–37 If polyps are found, colonoscopy should be performed every year.
The decision of when to offer colectomy is based on polyp burden (taking into account the number, pathologic appearance, and size of the polyps) and psychosocial factors such as patient maturity. Surgical options include total colectomy and ileorectal anastomosis or total proctocolectomy and ileal pouch anal anastomosis.38 Colonic and extracolonic phenotype as well as genotype should factor into the type of operation recommended. After colectomy, annual endoscopic surveillance of the rectum or ileal pouch is indicated to screen for recurrent polyposis and cancer.
Chemoprevention with sulindac (Clinoril) 150 mg or celecoxib (Celebrex) 400 mg twice a day causes regression of colorectal adenomas in familial adenomatous polyposis and may be useful as an adjunct to endoscopy in managing the colorectal polyp burden.39,40
Forward and side-viewing upper endoscopy should commence at age 20. This should include visualization and biopsy of the papilla and periampulllary region.29 The frequency of endoscopic surveillance depends on the Spigelman stage, which reflects the duodenal polyp burden. It is recommended that patients with Spigelman stage IV duodenal polyposis be seen in consultation with an experienced gastrointestinal surgeon for consideration of a prophylactic, pylorus-preserving, pancreas-sparing duodenectomy. This procedure has been shown to be more effective in polyp control and cancer prevention than endoscopic polyp ablation and local surgical resection.41
Some evidence for the utility of celecoxib 400 mg twice daily for the regression of duodenal polyposis was noted in a 6-month placebo-controlled trial.42 Some experts recommend removal of large duodenal adenomas, with adjunctive celecoxib therapy to control polyposis burden.30
People with familial adenomatous polyposis have been shown to have a 2.6% risk of thyroid cancer, and ultrasonography of the neck with attention to the thyroid is recommended for them.32
MYH-ASSOCIATED POLYPOSIS
Biallelic mutations in the MYH gene result in an adenomatous polyposis syndrome that may be indistinguishable from the attenuated or classic forms of familial adenomatous polyposis. A characteristic autosomal recessive pattern of inheritance in the family can be useful for identifying these patients in the clinic.
Genetics of MYH-associated polyposis
MYH-associated polyposis is the only known autosomal recessive hereditary colorectal cancer syndrome. In white populations, the most commonly reported mutations in MYH are Y179C (previously called Y165C) and G396D (previously called G382D), which account for up to 80% of cases.43 These two mutations are estimated to occur in 1% to 2% of the general population.44
Clinical presentation of MYH-associated polyposis
MYH-associated polyposis typically presents as multiple adenomatous polyps and is diagnosed at a mean age of 47 years. Eleven percent to 42% of affected individuals are reported to have fewer than 100 adenomas, while a minority (7.5% to 29%) of patients present with classic polyposis.45–47 In one study, an estimated 19% of patients presented with colorectal cancer and reported no history of colorectal polyps.48 Synchronous colorectal cancer is seen in more than 60% of patients with biallelic MYH mutations.49 Patients with monoallelic (heterozygous) MYH mutations appear to have the same risk of developing colorectal adenomas and cancer as the general population.49
Upper-gastrointestinal polyps have been reported in MYH-associated polyposis; as many as 17% to 25% of patients have duodenal adenomas.50,51
Diagnosis of MYH-associated polyposis
Genetic testing for biallelic MYH mutations should be performed in patients who test negative for an APC mutation but who have clinical features of familial adenomatous polyposis, a personal history of more than 10 colorectal adenomas, or a recessive family history of polyposis. 14 It has been shown that up to 29% of patients with familial adenomatous polyposis who are APC-negative will have biallelic mutations in the MYH gene.52 The siblings of a patient with biallelic MYH mutations should be offered genetic counseling and testing in their late teens or early 20s. All children of an individual with MYH-associated polyposis will carry one MYH mutation and are only at risk of having the syndrome if the other parent is also a MYH carrier and passed on his or her mutation.
Management of MYH-associated polyposis
The management of patients with MYH-associated polyposis is similar to that recommended for attenuated and classic familial adenomatous polyposis.14 Genetic counseling and testing and colonic and extracolonic surveillance are warranted. There are no data on the use of chemoprevention in MYH-associated polyposis. Surgery should be considered early because of the high risk of colorectal cancer, even in individuals with very few adenomas. Patients with monoallelic MYH mutations should follow the general population screening guidelines for colorectal cancer.49
GENETIC COUNSELING AND GENETIC TESTING
The American College of Gastroenterology advises that patients suspected of having hereditary colorectal cancer syndromes be advised to pursue genetic counseling and, if appropriate, genetic testing.16 They further recommend genetic counseling and informed consent before genetic testing.16
Genetic counseling is a process of working with patients and families whereby:
- A detailed medical and family history is obtained
- A formal risk assessment is performed
- Education about the disease in question and about genetic testing is provided
- Psychosocial concerns are assessed
- Informed consent is obtained when genetic testing is recommended.53
This process is important for helping patients better understand their cancer risks, the benefits and limitations of genetic testing, and the protections that are in place for people who undergo genetic testing, including the Genetic Information Non-Discrimination Act.
In 1996 the American Society of Clinical Oncology issued a policy statement highlighting the essential elements of informed consent for genetic testing for cancer susceptibility, and this was updated in 2003.54 In particular, it notes that patients should be informed of the implications of positive and negative results and of the possibility that the test may be uninformative.
When a hereditary colorectal cancer syndrome is suspected, a positive genetic test result confirms the diagnosis and allows for predictive testing of the patient’s relatives. However, no genetic test for a hereditary colorectal cancer syndrome is 100% sensitive. Therefore, a negative result does not rule out the syndrome in question.
Further, all cancer susceptibility genes have variants of uncertain significance, which are genetic alterations for which there are insufficient data to determine if the mutation is disease-causing or polymorphic (benign). Both negative and uninformative results can be confusing for patients and providers and can lead to false reassurance or undue worry when patients are not properly educated about these potential outcomes of testing.
Genetic testing is an evolving field, and with additional research and improved testing technologies, appropriate diagnoses can be made over time. That is why it is important for the genetic counseling relationship to continue over time.
- Bonadona V, Bonaïti B, Olschwang S, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011; 305:2304–2310.
- Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011; 60:950–957.
- Barrow E, Robinson L, Alduaij W, et al. Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: a report of 121 families with proven mutations. Clin Genet 2009; 75:141–149.
- van der Post RS, Kiemeney LA, Ligtenberg MJ, et al. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J Med Genet 2010; 47:464–470.
- Wijnen JT, Vasen HF, Khan PM, et al. Clinical findings with implications for genetic testing in families with clustering of colorectal cancer. N Engl J Med 1998; 339:511–518.
- Bisgaard ML, Bernstein I. HNPCC mutation rate. Familial Cancer 2003; 2.
- Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum 1991; 34:424–425.
- Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999; 116:1453–1456.
- Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96:261–268.
- Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP Working Group: can UGT1A1 genotyping reduce morbidity and mortality in patients with metastatic colorectal cancer treated with irinotecan? Genet Med 2009; 11:15–20.
- Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993; 260:812–816.
- Kim H, Jen J, Vogelstein B, Hamilton SR. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol 1994; 145:148–156.
- Lindor NM, Rabe K, Petersen GM, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA 2005; 293:1979–1985.
- National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology (NCCN guidelines) colorectal cancer screening version 2.2011. www.nccn.org. Accessed October 2, 2012.
- Jass JR. Hereditary non-polyposis colorectal cancer: the rise and fall of a confusing term. World J Gastroenterol 2006; 12:4943–4950.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 [corrected]. Am J Gastroenterol 2009; 104:739–750.
- Winawer S, Fletcher R, Rex D, et al; Gastrointestinal Consortium Panel. Colorectal cancer screening and surveillance: clinical guidelines and rationale-update based on new evidence. Gastroenterology 2003; 124:544–560.
- Lindor NM, Petersen GM, Hadley DW, et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA 2006; 296:1507–1517.
- de Jong AE, Hendriks YM, Kleibeuker JH, et al. Decrease in mortality in Lynch syndrome families because of surveillance. Gastroenterology 2006; 130:665–671.
- Mecklin JP, Aarnio M, Läärä E, et al. Development of colorectal tumors in colonoscopic surveillance in Lynch syndrome. Gastroenterology 2007; 133:1093–1098.
- Engel C, Rahner N, Schulmann K, et al; German HNPCC Consortium. Efficacy of annual colonoscopic surveillance in individuals with hereditary nonpolyposis colorectal cancer. Clin Gastroenterol Hepatol 2010; 8:174–182.
- Burn J, Gerdes AM, Macrae F, et al; CAPP2 Investigators. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 2011; 378:2081–2087.
- Vasen HF, Möslein G, Alonso A, et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J Med Genet 2007; 44:353–362.
- Manchanda R, Menon U, Michaelson-Cohen R, Beller U, Jacobs I. Hereditary non-polyposis colorectal cancer or Lynch syndrome: the gynaecological perspective. Curr Opin Obstet Gynecol 2009; 21:31–38.
- Burn J, Chapman P, Delhanty J, et al. The UK Northern region genetic register for familial adenomatous polyposis coli: use of age of onset, congenital hypertrophy of the retinal pigment epithelium, and DNA markers in risk calculations. J Med Genet 1991; 28:289–296.
- Järvinen HJ. Epidemiology of familial adenomatous polyposis in Finland: impact of family screening on the colorectal cancer rate and survival. Gut 1992; 33:357–360.
- Bisgaard ML, Fenger K, Bülow S, Niebuhr E, Mohr J. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat 1994; 3:121–125.
- Neklason DW, Stevens J, Boucher KM, et al. American founder mutation for attenuated familial adenomatous polyposis. Clin Gastroenterol Hepatol 2008; 6:46–52.
- Burke CA, Beck GJ, Church JM, van Stolk RU. The natural history of untreated duodenal and ampullary adenomas in patients with familial adenomatous polyposis followed in an endoscopic surveillance program. Gastrointest Endosc 1999; 49:358–364.
- Bianchi LK, Burke CA, Bennett AE, Lopez R, Hasson H, Church JM. Fundic gland polyp dysplasia is common in familial adenomatous polyposis. Clin Gastroenterol Hepatol 2008; 6:180–185.
- Kadmon M, Tandara A, Herfarth C. Duodenal adenomatosis in familial adenomatous polyposis coli. A review of the literature and results from the Heidelberg Polyposis Register. Int J Colorectal Dis 2001; 16:63–75.
- Jarrar AM, Milas M, Mitchell J, et al. Screening for thyroid cancer in patients with familial adenomatous polyposis. Ann Surg 2011; 253:515–521.
- Jasperson KW, Burt RW. APC-associated polyposis conditions. In:Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews (Internet). Seattle, WA: University of Washington; 2011.
- Gardner EJ, Richards RC. Multiple cutaneous and subcutaneous lesions occurring simultaneously with hereditary polyposis and osteomatosis. Am J Hum Genet 1953; 5:139–147.
- Dunlop MG; British Society for Gastroenterology. Guidance on gastrointestinal surveillance for hereditary non-polyposis colorectal cancer, familial adenomatous polyposis, juvenile polyposis, and Peutz-Jeghers syndrome. Gut 2002; 51(suppl 5):V21–V27.
- Burke W, Petersen G, Lynch P, et al. Recommendations for follow-up care of individuals with an inherited predisposition to cancer. I. Hereditary nonpolyposis colon cancer. Cancer Genetics Studies Consortium. JAMA 1997; 277:915–919.
- Vasen HF, Möslein G, Alonso A, et al. Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut 2008; 57:704–713.
- Church J. Familial adenomatous polyposis. Surg Oncol Clin N Am 2009; 18:585–598.
- Giardiello FM, Hamilton SR, Krush AJ, et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med 1993; 328:1313–1316.
- Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 2000; 342:1946–1952.
- Johnson MD, Mackey R, Brown N, Church J, Burke C, Walsh RM. Outcome based on management for duodenal adenomas: sporadic versus familial disease. J Gastrointest Surg 2010; 14:229–235.
- Phillips RK, Wallace MH, Lynch PM, et al; FAP Study Group. A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut 2002; 50:857–860.
- Tenesa A, Campbell H, Barnetson R, Porteous M, Dunlop M, Farrington SM. Association of MUTYH and colorectal cancer. Br J Cancer 2006; 95:239–242.
- Croitoru ME, Cleary SP, Di Nicola N, et al. Association between biallelic and monoallelic germline MYH gene mutations and colorectal cancer risk. J Natl Cancer Inst 2004; 96:1631–1634.
- Croitoru ME, Cleary SP, Berk T, et al. Germline MYH mutations in a clinic-based series of Canadian multiple colorectal adenoma patients. J Surg Oncol 2007; 95:499–506.
- Sampson JR, Dolwani S, Jones S, et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet 2003; 362:39–41.
- Nielsen M, Franken PF, Reinards TH, et al. Multiplicity in polyp count and extracolonic manifestations in 40 Dutch patients with MYH associated polyposis coli (MAP). J Med Genet 2005; 42:e54.
- Cleary SP, Cotterchio M, Jenkins MA, et al. Germline MutY human homologue mutations and colorectal cancer: a multisite case-control study. Gastroenterology 2009; 136:1251–1260.
- Lubbe SJ, Di Bernardo MC, Chandler IP, Houlston RS. Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J Clin Oncol 2009; 27:3975–3980.
- Aretz S, Uhlhaas S, Goergens H, et al. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006; 119:807–814.
- Vogt S, Jones N, Christian D, et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology 2009; 137:1976–1985.e1–e10.
- Gismondi V, Meta M, Bonelli L, et al. Prevalence of the Y165C, G382D and 1395delGGA germline mutations of the MYH gene in Italian patients with adenomatous polyposis coli and colorectal adenomas. Int J Cancer 2004; 109:680–684.
- Trepanier A, Ahrens M, McKinnon W, et al; National Society of Genetic Counselors. Genetic cancer risk assessment and counseling: recommendations of the national society of genetic counselors. J Genet Couns 2004; 13:83–114.
- American Society of Clinical Oncology. American Society of Clinical Oncology policy statement update: genetic testing for cancer susceptibility. J Clin Oncol 2003; 21:2397–2406.
Hereditary colorectal cancer syndromes account for 5% to 10% of cases of colorectal cancer.
Identifying these patients in clinical practice begins by assessing a patient’s personal and family health history. An accurate and comprehensive family history should cover three generations and include ethnic background, ages and causes of death of relatives, and any diagnosis of cancer, including age at onset and history of polyps.
Red flags for a hereditary colorectal cancer syndrome in the personal or family history are:
- Early age of onset of cancer (eg, colorectal cancer before age 50)
- More than 10 colorectal adenomas
- Synchronous (ie, occurring at the same time) or metachronous (occurring at different times) primary cancers
- Multiple relatives in successive generations with the same or related cancers (eg, colon or endometrial cancer)
- A family member with a known hereditary colorectal cancer syndrome (Table 1).
Any of these red flags should prompt a referral for genetic counseling.
SYNDROMES ARE CLASSIFIED AS WITH OR WITHOUT POLYPOSIS
Many hereditary syndromes are associated with a higher risk of colorectal cancer. Generally, they can be divided into two categories (Table 2): polyposis syndromes (in which patients have numerous colorectal polyps) and nonpolyposis syndromes (with few or no polyps).
These two main types are subclassified on the basis of the histology of most of the polyps detected: adenomatous, hamartomatous, serrated, or mixed types.
In this review, we will address the three most common of these syndromes: Lynch syndrome (hereditary nonpolyposis colorectal cancer), familial adenomatous polyposis, and MYH-associated polyposis. However, as noted in Table 2, other hereditary colorectal cancer syndromes exist, and suspicion of these conditions should prompt a referral for further evaluation.
LYNCH SYNDROME (HEREDITARY NONPOLYPOSIS COLORECTAL CANCER)
Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer, predisposes people to a variety of cancers.
Colorectal cancer is the most common type of cancer associated with Lynch syndrome. Recent research suggests that the cumulative risk of developing colorectal cancer by age 80 is 42% for all patients with Lynch syndrome.1 The median age at onset is 45 years.1 For patients who undergo segmental resection of their initial cancer, the cumulative risk of metachronous colorectal cancer (ie, a new tumor arising later) is 16% at 10 years, 41% at 20 years, and up to 62% after 30 years.2
Endometrial cancer occurs in 17% to 57% of women with Lynch syndrome by age 70, with a median age at onset of 49 years.1
Other extracolonic cancers in Lynch syndrome include cancers of the:
- Stomach (1%–10% risk by age 70 years)
- Ovaries (1%–20% risk)
- Hepatobiliary tract (1%–2% risk)
- Urinary tract (1%–12% risk)
- Small bowel (1%–2% risk)
- Brain (1%–8% risk)
- Skin (sebaceous adenomas, adenocarcinomas, and keratoacanthomas).1,3,4
Earlier studies reported higher rates of associated cancer than those shown here. However, their data were largely derived from registries and may be overestimates. The numbers shown above are from population-based studies.
Genetics of Lynch syndrome
Lynch syndrome is caused by a germline mutation in the MLH1, MSH2, MSH6, PMS2, or EPCAM genes.5 These genes code for proteins that are responsible DNA mismatch repair—one of the cell’s proofreading mechanisms during DNA replication.
These mutations are inherited in an autosomal dominant manner. Though de novo mutations in these genes have been reported, they are rare and the exact frequency with which they occur is unknown.6
In whom should Lynch syndrome be suspected?
Lynch syndrome can be suspected on the basis of family history and clinical criteria.
In 1991, the same group of experts who coined the term “hereditary nonpolyposis colorectal cancer” developed family history criteria for it1:
- At least three relatives with histologically confirmed colorectal cancer, one of whom is a first-degree relative of the other two
- At least two successive generations involved
- At least one of the cancers diagnosed before age 50
- Familial adenomatous polyposis is excluded.
Known as the Amsterdam criteria, these were to be used in collaborative studies of families with hereditary colorectal cancer.7 In 1999, these criteria were broadened to include extracolonic cancers and became known as the Amsterdam II criteria (Table 3).8
Patients whose families meet the Amsterdam II criteria or who have molecular pathologic evidence of Lynch syndrome (see below) are appropriate candidates for genetic counseling and testing.
Diagnosis of Lynch syndrome
The diagnosis of Lynch syndrome is based on molecular pathologic analysis (performed on tumor samples) and confirmed by genetic testing.
Molecular pathologic evidence of Lynch syndrome includes microsatellite instability and loss of expression of one or more of the DNA mismatch repair proteins (detected using immunohistochemistry) (more on these below). The revised Bethesda guidelines (TABLE 3) were intended to identify individuals whose tumors should be tested for one or both of these phenomena.9
In 2009, the Evaluation of Genomic Applications in Practice and Prevention working group recommended that all patients with newly diagnosed colorectal cancer undergo microsatellite instability analysis, immunohistochemistry testing, or both, regardless of whether they meet the Amsterdam II or the Bethesda guideline criteria.10
Microsatellite instability analysis. Microsatellites are short sequences of repeated DNA. The tumor cells of patients who carry defective mismatch repair genes have microsatellites that are longer or shorter than in normal cells, a condition called microsatellite instability (ie, “MSI-high”).
Microsatellite instability testing, using a standardized panel of five DNA markers, is performed on normal and tumor tissue. If more than two of the five microsatellite markers in the tumor show instability, the lesion is considered to have a high level of microsatellite instability. About 15% of colorectal cancers have this high level, although most are not associated with Lynch syndrome and lose MLH1 expression by promoter methylation.11,12
While only 2% of patients with colorectal cancer have Lynch syndrome, from 90% to 95% of colorectal cancers from patients with Lynch syndrome have high levels of microsatellite instability.10 The presence of MLH1 promoter hypermethylation, the BRAF mutation V600E, or both within the tumor suggests that the cancer is not associated with Lynch syndrome.
Some families that meet the Amsterdam I criteria have microsatellite-stable tumors: their condition has been called familial colorectal cancer type X.13 This condition is associated with a higher risk of colorectal cancer but not the other malignancies observed in Lynch syndrome.
Immunohistochemistry is performed to assess for expression of the mismatch repair proteins MSH2, MSH6, MLH1, and PMS2. Absence of expression of the specific protein within tumor cells compared with normal cells within the specimen suggests dysfunction of the specific gene and guides germline mutation testing (Figure 1). For example, a patient who lacks expression of the MSH2 protein in his or her colon cancer most likely has a mutation in the MSH2 gene. Therefore, germ-line genetic testing should initially target the MSH2 gene. Approximately 88% of Lynch syndrome-associated colorectal cancers have abnormal immunohistochemical staining.10
Testing for microsatellite instability and mismatch repair gene expression ideally precedes germline genetic testing and helps to guide which gene or genes should be tested.9,14
Genetic testing for Lynch syndrome is routinely performed on a blood or saliva sample, using DNA from white blood cells and sequencing the gene or genes involved to look for mutations. Positive results from a germline genetic test confirm the diagnosis of Lynch syndrome and allow for predictive testing for relatives at risk. The term Lynch syndrome is used exclusively to describe individuals with evidence of a mutation in one of the mismatch repair genes.15
If a patient’s results are positive, genetic counseling and genetic testing should be offered to at-risk relatives age 18 and over.
Management of Lynch syndrome
Aggressive cancer surveillance is essential for people with Lynch syndrome and for those who are considered at risk but have not pursued genetic testing, such as a sibling of a person with Lynch syndrome.
Colorectal cancer. Colonoscopy is recommended every 1 to 2 years beginning at the age of 20 to 25 years, or 2 to 5 years earlier than the age of the youngest relative affected with colorectal cancer if the initial diagnosis was before age 25. When patients turn 40 years old, colonoscopy is done annually.16–18 A significant reduction in cancer incidence and in the mortality rate has been shown with colonoscopic surveillance.19–21
Chemoprevention may also have a role. Patients with Lynch syndrome who took aspirin 600 mg per day for an average of 25 months had a significantly lower incidence of colorectal cancer during a 55-month follow-up period compared with patients randomized to placebo.22
For patients with Lynch syndrome who are diagnosed with colorectal cancer, the high risk of metachronous cancers after standard segmental colectomy calls for a more extended resection. Retrospective analysis of 382 Lynch syndrome patients found that none of the 50 who underwent total or subtotal colectomy were diagnosed with metachronous colorectal cancer, whereas a metachronous cancer developed in 74 (22%) of the 332 patients who had had segmented resection.2 Annual surveillance of the remaining colon, rectum, or both is indicated postoperatively.
Gynecologic cancers. Women with Lynch syndrome should also consider gynecologic surveillance and risk-reducing surgery. This includes annual gynecologic examination, transvaginal ultrasonography, and endometrial aspiration, beginning at age 30 to 35 years. Although this surveillance does detect premalignant lesions and early symptomatic cancers, its effect on the mortality rate is unknown. Hysterectomy with bilateral salpingo-oophorectomy has been shown to significantly reduce endometrial and ovarian cancers in women with Lynch syndrome.23,24
Urothelial cancers. Carriers of MSH2 mutations have a significantly higher risk of urothelial cancers.4 Therefore, MSH2 carriers should consider ultrasonography of the urinary tract, urinary cytology, and urinalysis every 1 to 2 years beginning at age 40.4
Other extracolonic cancers. Poor evidence exists for systematic screening for the other extracolonic tumors associated with Lynch syndrome. However, the National Comprehensive Cancer Network advises considering esophagogastroduodenoscopy with extended duodenoscopy as well as capsule endoscopy every 2 to 3 years beginning at age 30 to 35.14
ADENOMATOUS POLYPOSIS SYNDROMES
Familial adenomatous polyposis and MYH-associated polyposis are the next most common hereditary colorectal cancer syndromes. Each of these accounts for about 1% of cases of colorectal cancer. Clinically, these two syndromes can be challenging to distinguish because they overlap phenotypically to a significant degree.
FAMILIAL ADENOMATOUS POLYPOSIS
Familial adenomatous polyposis is caused by mutations in the APC gene. Its prevalence is 2.29 to 3.2 per 100,000 individuals.25,26
Genetics of familial adenomatous polyposis
APC is the only gene known to cause familial adenomatous polyposis. Mutations in APC are inherited in an autosomal dominant manner. Approximately 25% of cases of familial adenomatous polyposis are due to a de novo mutation in APC.27
Clinical presentation of familial adenomatous polyposis
Familial adenomatous polyposis is classified by the burden of colorectal adenomas.
Patients who have fewer than 100 adenomas have an attenuated form of the disease. In this group, polyps usually begin to form in the late teenage years or early 20s and tend to develop in the proximal colon. The attenuated form is associated with an approximately 70% lifetime risk of colorectal cancer.28
Patients who have more than 100 polyps are considered to have the classic form of the disease, and those with more than 1,000 polyps have profuse familial adenomatous polyposis (Figure 2). In these groups, polyps typically begin to develop in the preteenage to mid-teenage years. Without surgery, there is nearly a 100% risk of colorectal cancer. The average age at diagnosis of colorectal cancer is 39 years for patients with classic disease.
Upper gastrointestinal polyps are common in familial adenomatous polyposis. Nearly 90% of patients develop duodenal adenomas by a mean age of 44, with a cumulative lifetime risk of nearly 100%.29 Fundic gland polyposis occurs in nearly 90% of patients,30 while gastric adenomas are reported in fewer than 15% of patients.
Duodenal and periampullary cancer is the second most common malignancy in familial adenomatous polyposis. The lifetime risk ranges from 2% to 36%, depending on the Spigelman stage. People with Spigelman stage I, II, or III have a 2.5% risk of duodenal cancer, while those with stage IV disease have up to a 36% lifetime risk.
Gastric cancer, arising from fundic gland polyps, has been reported but is rare in Western populations.
In familial adenomatous polyposis, the incidence of jejunal adenomas and cancer is less than 10%, and the risk of ileal adenomas and cancer is less than 1%.31
Familial adenomatous polyposis is also associated with a higher risk of other malignancies, including:
- Pancreatic cancer (2% lifetime risk)
- Thyroid cancer (2% to 3% lifetime risk, typically papillary carcinoma)32
- Hepatoblastoma (1% to 2% lifetime risk)
- Brain tumors (< 1% lifetime risk)
- Biliary cancer (higher risk than in the general population).33
Benign extracolonic manifestations that have been observed include osteomas, dental abnormalities (supernumerary teeth, unerupted or absent teeth, odontomas), congenital hypertrophy of the retinal pigment epithelium, benign cutaneous lesions (epidermoid cysts and fibromas), and desmoid tumors.33 The term “Gardner syndrome” has been used to describe patients who have familial adenomatous polyposis but also have osteomas and soft-tissue tumors.34 These patients carry the same risk of colorectal cancer as other patients with familial adenomatous polyposis.
Diagnosing familial adenomatous polyposis
The diagnosis of familial adenomatous polyposis is suspected when a patient has more than 10 adenomatous polyps.
Seventy-five percent of patients with familial adenomatous polyposis have a family history of the condition. Therefore, most cases are identified at a young age on screening sigmoidoscopy or colonoscopy or by predictive gene testing. Patients rarely have cancer at the time of diagnosis.
The other 25% of patients typically are diagnosed when symptoms develop from the polyps or cancer. Over 50% of these symptomatic patients have cancer at the time of diagnosis.
It is recommended that people who have more than 10 adenomas detected on a single colonoscopy or who are first-degree relatives of patients with familial adenomatous polyposis undergo a genetic evaluation and testing for mutations in the APC gene.14 Once an APC mutation is identified in the family, at-risk relatives should be offered testing around age 10 years for families with classic familial adenomatous polyposis or in the mid to late teenage years for those with the attenuated form. It also appropriate to refer patients with desmoid tumors, duodenal adenomas, and bilateral or multifocal congenital hypertrophy of the retinal pigment epithelium for a genetic evaluation.
Management of familial adenomatous polyposis
Flexible sigmoidoscopy every 1 to 2 years beginning at age 10 to 12 years is recommended for individuals and families who have been phenotypically or genetically diagnosed with familial adenomatous polyposis.35–37 If colorectal adenomas are found, surgical options should be discussed and annual colonoscopic surveillance should commence.
For people with the attenuated form, because of the later age of disease onset and the tendency for right-sided disease, colonoscopy every 1 to 2 years should commence at about age 18.35–37 If polyps are found, colonoscopy should be performed every year.
The decision of when to offer colectomy is based on polyp burden (taking into account the number, pathologic appearance, and size of the polyps) and psychosocial factors such as patient maturity. Surgical options include total colectomy and ileorectal anastomosis or total proctocolectomy and ileal pouch anal anastomosis.38 Colonic and extracolonic phenotype as well as genotype should factor into the type of operation recommended. After colectomy, annual endoscopic surveillance of the rectum or ileal pouch is indicated to screen for recurrent polyposis and cancer.
Chemoprevention with sulindac (Clinoril) 150 mg or celecoxib (Celebrex) 400 mg twice a day causes regression of colorectal adenomas in familial adenomatous polyposis and may be useful as an adjunct to endoscopy in managing the colorectal polyp burden.39,40
Forward and side-viewing upper endoscopy should commence at age 20. This should include visualization and biopsy of the papilla and periampulllary region.29 The frequency of endoscopic surveillance depends on the Spigelman stage, which reflects the duodenal polyp burden. It is recommended that patients with Spigelman stage IV duodenal polyposis be seen in consultation with an experienced gastrointestinal surgeon for consideration of a prophylactic, pylorus-preserving, pancreas-sparing duodenectomy. This procedure has been shown to be more effective in polyp control and cancer prevention than endoscopic polyp ablation and local surgical resection.41
Some evidence for the utility of celecoxib 400 mg twice daily for the regression of duodenal polyposis was noted in a 6-month placebo-controlled trial.42 Some experts recommend removal of large duodenal adenomas, with adjunctive celecoxib therapy to control polyposis burden.30
People with familial adenomatous polyposis have been shown to have a 2.6% risk of thyroid cancer, and ultrasonography of the neck with attention to the thyroid is recommended for them.32
MYH-ASSOCIATED POLYPOSIS
Biallelic mutations in the MYH gene result in an adenomatous polyposis syndrome that may be indistinguishable from the attenuated or classic forms of familial adenomatous polyposis. A characteristic autosomal recessive pattern of inheritance in the family can be useful for identifying these patients in the clinic.
Genetics of MYH-associated polyposis
MYH-associated polyposis is the only known autosomal recessive hereditary colorectal cancer syndrome. In white populations, the most commonly reported mutations in MYH are Y179C (previously called Y165C) and G396D (previously called G382D), which account for up to 80% of cases.43 These two mutations are estimated to occur in 1% to 2% of the general population.44
Clinical presentation of MYH-associated polyposis
MYH-associated polyposis typically presents as multiple adenomatous polyps and is diagnosed at a mean age of 47 years. Eleven percent to 42% of affected individuals are reported to have fewer than 100 adenomas, while a minority (7.5% to 29%) of patients present with classic polyposis.45–47 In one study, an estimated 19% of patients presented with colorectal cancer and reported no history of colorectal polyps.48 Synchronous colorectal cancer is seen in more than 60% of patients with biallelic MYH mutations.49 Patients with monoallelic (heterozygous) MYH mutations appear to have the same risk of developing colorectal adenomas and cancer as the general population.49
Upper-gastrointestinal polyps have been reported in MYH-associated polyposis; as many as 17% to 25% of patients have duodenal adenomas.50,51
Diagnosis of MYH-associated polyposis
Genetic testing for biallelic MYH mutations should be performed in patients who test negative for an APC mutation but who have clinical features of familial adenomatous polyposis, a personal history of more than 10 colorectal adenomas, or a recessive family history of polyposis. 14 It has been shown that up to 29% of patients with familial adenomatous polyposis who are APC-negative will have biallelic mutations in the MYH gene.52 The siblings of a patient with biallelic MYH mutations should be offered genetic counseling and testing in their late teens or early 20s. All children of an individual with MYH-associated polyposis will carry one MYH mutation and are only at risk of having the syndrome if the other parent is also a MYH carrier and passed on his or her mutation.
Management of MYH-associated polyposis
The management of patients with MYH-associated polyposis is similar to that recommended for attenuated and classic familial adenomatous polyposis.14 Genetic counseling and testing and colonic and extracolonic surveillance are warranted. There are no data on the use of chemoprevention in MYH-associated polyposis. Surgery should be considered early because of the high risk of colorectal cancer, even in individuals with very few adenomas. Patients with monoallelic MYH mutations should follow the general population screening guidelines for colorectal cancer.49
GENETIC COUNSELING AND GENETIC TESTING
The American College of Gastroenterology advises that patients suspected of having hereditary colorectal cancer syndromes be advised to pursue genetic counseling and, if appropriate, genetic testing.16 They further recommend genetic counseling and informed consent before genetic testing.16
Genetic counseling is a process of working with patients and families whereby:
- A detailed medical and family history is obtained
- A formal risk assessment is performed
- Education about the disease in question and about genetic testing is provided
- Psychosocial concerns are assessed
- Informed consent is obtained when genetic testing is recommended.53
This process is important for helping patients better understand their cancer risks, the benefits and limitations of genetic testing, and the protections that are in place for people who undergo genetic testing, including the Genetic Information Non-Discrimination Act.
In 1996 the American Society of Clinical Oncology issued a policy statement highlighting the essential elements of informed consent for genetic testing for cancer susceptibility, and this was updated in 2003.54 In particular, it notes that patients should be informed of the implications of positive and negative results and of the possibility that the test may be uninformative.
When a hereditary colorectal cancer syndrome is suspected, a positive genetic test result confirms the diagnosis and allows for predictive testing of the patient’s relatives. However, no genetic test for a hereditary colorectal cancer syndrome is 100% sensitive. Therefore, a negative result does not rule out the syndrome in question.
Further, all cancer susceptibility genes have variants of uncertain significance, which are genetic alterations for which there are insufficient data to determine if the mutation is disease-causing or polymorphic (benign). Both negative and uninformative results can be confusing for patients and providers and can lead to false reassurance or undue worry when patients are not properly educated about these potential outcomes of testing.
Genetic testing is an evolving field, and with additional research and improved testing technologies, appropriate diagnoses can be made over time. That is why it is important for the genetic counseling relationship to continue over time.
Hereditary colorectal cancer syndromes account for 5% to 10% of cases of colorectal cancer.
Identifying these patients in clinical practice begins by assessing a patient’s personal and family health history. An accurate and comprehensive family history should cover three generations and include ethnic background, ages and causes of death of relatives, and any diagnosis of cancer, including age at onset and history of polyps.
Red flags for a hereditary colorectal cancer syndrome in the personal or family history are:
- Early age of onset of cancer (eg, colorectal cancer before age 50)
- More than 10 colorectal adenomas
- Synchronous (ie, occurring at the same time) or metachronous (occurring at different times) primary cancers
- Multiple relatives in successive generations with the same or related cancers (eg, colon or endometrial cancer)
- A family member with a known hereditary colorectal cancer syndrome (Table 1).
Any of these red flags should prompt a referral for genetic counseling.
SYNDROMES ARE CLASSIFIED AS WITH OR WITHOUT POLYPOSIS
Many hereditary syndromes are associated with a higher risk of colorectal cancer. Generally, they can be divided into two categories (Table 2): polyposis syndromes (in which patients have numerous colorectal polyps) and nonpolyposis syndromes (with few or no polyps).
These two main types are subclassified on the basis of the histology of most of the polyps detected: adenomatous, hamartomatous, serrated, or mixed types.
In this review, we will address the three most common of these syndromes: Lynch syndrome (hereditary nonpolyposis colorectal cancer), familial adenomatous polyposis, and MYH-associated polyposis. However, as noted in Table 2, other hereditary colorectal cancer syndromes exist, and suspicion of these conditions should prompt a referral for further evaluation.
LYNCH SYNDROME (HEREDITARY NONPOLYPOSIS COLORECTAL CANCER)
Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer, predisposes people to a variety of cancers.
Colorectal cancer is the most common type of cancer associated with Lynch syndrome. Recent research suggests that the cumulative risk of developing colorectal cancer by age 80 is 42% for all patients with Lynch syndrome.1 The median age at onset is 45 years.1 For patients who undergo segmental resection of their initial cancer, the cumulative risk of metachronous colorectal cancer (ie, a new tumor arising later) is 16% at 10 years, 41% at 20 years, and up to 62% after 30 years.2
Endometrial cancer occurs in 17% to 57% of women with Lynch syndrome by age 70, with a median age at onset of 49 years.1
Other extracolonic cancers in Lynch syndrome include cancers of the:
- Stomach (1%–10% risk by age 70 years)
- Ovaries (1%–20% risk)
- Hepatobiliary tract (1%–2% risk)
- Urinary tract (1%–12% risk)
- Small bowel (1%–2% risk)
- Brain (1%–8% risk)
- Skin (sebaceous adenomas, adenocarcinomas, and keratoacanthomas).1,3,4
Earlier studies reported higher rates of associated cancer than those shown here. However, their data were largely derived from registries and may be overestimates. The numbers shown above are from population-based studies.
Genetics of Lynch syndrome
Lynch syndrome is caused by a germline mutation in the MLH1, MSH2, MSH6, PMS2, or EPCAM genes.5 These genes code for proteins that are responsible DNA mismatch repair—one of the cell’s proofreading mechanisms during DNA replication.
These mutations are inherited in an autosomal dominant manner. Though de novo mutations in these genes have been reported, they are rare and the exact frequency with which they occur is unknown.6
In whom should Lynch syndrome be suspected?
Lynch syndrome can be suspected on the basis of family history and clinical criteria.
In 1991, the same group of experts who coined the term “hereditary nonpolyposis colorectal cancer” developed family history criteria for it1:
- At least three relatives with histologically confirmed colorectal cancer, one of whom is a first-degree relative of the other two
- At least two successive generations involved
- At least one of the cancers diagnosed before age 50
- Familial adenomatous polyposis is excluded.
Known as the Amsterdam criteria, these were to be used in collaborative studies of families with hereditary colorectal cancer.7 In 1999, these criteria were broadened to include extracolonic cancers and became known as the Amsterdam II criteria (Table 3).8
Patients whose families meet the Amsterdam II criteria or who have molecular pathologic evidence of Lynch syndrome (see below) are appropriate candidates for genetic counseling and testing.
Diagnosis of Lynch syndrome
The diagnosis of Lynch syndrome is based on molecular pathologic analysis (performed on tumor samples) and confirmed by genetic testing.
Molecular pathologic evidence of Lynch syndrome includes microsatellite instability and loss of expression of one or more of the DNA mismatch repair proteins (detected using immunohistochemistry) (more on these below). The revised Bethesda guidelines (TABLE 3) were intended to identify individuals whose tumors should be tested for one or both of these phenomena.9
In 2009, the Evaluation of Genomic Applications in Practice and Prevention working group recommended that all patients with newly diagnosed colorectal cancer undergo microsatellite instability analysis, immunohistochemistry testing, or both, regardless of whether they meet the Amsterdam II or the Bethesda guideline criteria.10
Microsatellite instability analysis. Microsatellites are short sequences of repeated DNA. The tumor cells of patients who carry defective mismatch repair genes have microsatellites that are longer or shorter than in normal cells, a condition called microsatellite instability (ie, “MSI-high”).
Microsatellite instability testing, using a standardized panel of five DNA markers, is performed on normal and tumor tissue. If more than two of the five microsatellite markers in the tumor show instability, the lesion is considered to have a high level of microsatellite instability. About 15% of colorectal cancers have this high level, although most are not associated with Lynch syndrome and lose MLH1 expression by promoter methylation.11,12
While only 2% of patients with colorectal cancer have Lynch syndrome, from 90% to 95% of colorectal cancers from patients with Lynch syndrome have high levels of microsatellite instability.10 The presence of MLH1 promoter hypermethylation, the BRAF mutation V600E, or both within the tumor suggests that the cancer is not associated with Lynch syndrome.
Some families that meet the Amsterdam I criteria have microsatellite-stable tumors: their condition has been called familial colorectal cancer type X.13 This condition is associated with a higher risk of colorectal cancer but not the other malignancies observed in Lynch syndrome.
Immunohistochemistry is performed to assess for expression of the mismatch repair proteins MSH2, MSH6, MLH1, and PMS2. Absence of expression of the specific protein within tumor cells compared with normal cells within the specimen suggests dysfunction of the specific gene and guides germline mutation testing (Figure 1). For example, a patient who lacks expression of the MSH2 protein in his or her colon cancer most likely has a mutation in the MSH2 gene. Therefore, germ-line genetic testing should initially target the MSH2 gene. Approximately 88% of Lynch syndrome-associated colorectal cancers have abnormal immunohistochemical staining.10
Testing for microsatellite instability and mismatch repair gene expression ideally precedes germline genetic testing and helps to guide which gene or genes should be tested.9,14
Genetic testing for Lynch syndrome is routinely performed on a blood or saliva sample, using DNA from white blood cells and sequencing the gene or genes involved to look for mutations. Positive results from a germline genetic test confirm the diagnosis of Lynch syndrome and allow for predictive testing for relatives at risk. The term Lynch syndrome is used exclusively to describe individuals with evidence of a mutation in one of the mismatch repair genes.15
If a patient’s results are positive, genetic counseling and genetic testing should be offered to at-risk relatives age 18 and over.
Management of Lynch syndrome
Aggressive cancer surveillance is essential for people with Lynch syndrome and for those who are considered at risk but have not pursued genetic testing, such as a sibling of a person with Lynch syndrome.
Colorectal cancer. Colonoscopy is recommended every 1 to 2 years beginning at the age of 20 to 25 years, or 2 to 5 years earlier than the age of the youngest relative affected with colorectal cancer if the initial diagnosis was before age 25. When patients turn 40 years old, colonoscopy is done annually.16–18 A significant reduction in cancer incidence and in the mortality rate has been shown with colonoscopic surveillance.19–21
Chemoprevention may also have a role. Patients with Lynch syndrome who took aspirin 600 mg per day for an average of 25 months had a significantly lower incidence of colorectal cancer during a 55-month follow-up period compared with patients randomized to placebo.22
For patients with Lynch syndrome who are diagnosed with colorectal cancer, the high risk of metachronous cancers after standard segmental colectomy calls for a more extended resection. Retrospective analysis of 382 Lynch syndrome patients found that none of the 50 who underwent total or subtotal colectomy were diagnosed with metachronous colorectal cancer, whereas a metachronous cancer developed in 74 (22%) of the 332 patients who had had segmented resection.2 Annual surveillance of the remaining colon, rectum, or both is indicated postoperatively.
Gynecologic cancers. Women with Lynch syndrome should also consider gynecologic surveillance and risk-reducing surgery. This includes annual gynecologic examination, transvaginal ultrasonography, and endometrial aspiration, beginning at age 30 to 35 years. Although this surveillance does detect premalignant lesions and early symptomatic cancers, its effect on the mortality rate is unknown. Hysterectomy with bilateral salpingo-oophorectomy has been shown to significantly reduce endometrial and ovarian cancers in women with Lynch syndrome.23,24
Urothelial cancers. Carriers of MSH2 mutations have a significantly higher risk of urothelial cancers.4 Therefore, MSH2 carriers should consider ultrasonography of the urinary tract, urinary cytology, and urinalysis every 1 to 2 years beginning at age 40.4
Other extracolonic cancers. Poor evidence exists for systematic screening for the other extracolonic tumors associated with Lynch syndrome. However, the National Comprehensive Cancer Network advises considering esophagogastroduodenoscopy with extended duodenoscopy as well as capsule endoscopy every 2 to 3 years beginning at age 30 to 35.14
ADENOMATOUS POLYPOSIS SYNDROMES
Familial adenomatous polyposis and MYH-associated polyposis are the next most common hereditary colorectal cancer syndromes. Each of these accounts for about 1% of cases of colorectal cancer. Clinically, these two syndromes can be challenging to distinguish because they overlap phenotypically to a significant degree.
FAMILIAL ADENOMATOUS POLYPOSIS
Familial adenomatous polyposis is caused by mutations in the APC gene. Its prevalence is 2.29 to 3.2 per 100,000 individuals.25,26
Genetics of familial adenomatous polyposis
APC is the only gene known to cause familial adenomatous polyposis. Mutations in APC are inherited in an autosomal dominant manner. Approximately 25% of cases of familial adenomatous polyposis are due to a de novo mutation in APC.27
Clinical presentation of familial adenomatous polyposis
Familial adenomatous polyposis is classified by the burden of colorectal adenomas.
Patients who have fewer than 100 adenomas have an attenuated form of the disease. In this group, polyps usually begin to form in the late teenage years or early 20s and tend to develop in the proximal colon. The attenuated form is associated with an approximately 70% lifetime risk of colorectal cancer.28
Patients who have more than 100 polyps are considered to have the classic form of the disease, and those with more than 1,000 polyps have profuse familial adenomatous polyposis (Figure 2). In these groups, polyps typically begin to develop in the preteenage to mid-teenage years. Without surgery, there is nearly a 100% risk of colorectal cancer. The average age at diagnosis of colorectal cancer is 39 years for patients with classic disease.
Upper gastrointestinal polyps are common in familial adenomatous polyposis. Nearly 90% of patients develop duodenal adenomas by a mean age of 44, with a cumulative lifetime risk of nearly 100%.29 Fundic gland polyposis occurs in nearly 90% of patients,30 while gastric adenomas are reported in fewer than 15% of patients.
Duodenal and periampullary cancer is the second most common malignancy in familial adenomatous polyposis. The lifetime risk ranges from 2% to 36%, depending on the Spigelman stage. People with Spigelman stage I, II, or III have a 2.5% risk of duodenal cancer, while those with stage IV disease have up to a 36% lifetime risk.
Gastric cancer, arising from fundic gland polyps, has been reported but is rare in Western populations.
In familial adenomatous polyposis, the incidence of jejunal adenomas and cancer is less than 10%, and the risk of ileal adenomas and cancer is less than 1%.31
Familial adenomatous polyposis is also associated with a higher risk of other malignancies, including:
- Pancreatic cancer (2% lifetime risk)
- Thyroid cancer (2% to 3% lifetime risk, typically papillary carcinoma)32
- Hepatoblastoma (1% to 2% lifetime risk)
- Brain tumors (< 1% lifetime risk)
- Biliary cancer (higher risk than in the general population).33
Benign extracolonic manifestations that have been observed include osteomas, dental abnormalities (supernumerary teeth, unerupted or absent teeth, odontomas), congenital hypertrophy of the retinal pigment epithelium, benign cutaneous lesions (epidermoid cysts and fibromas), and desmoid tumors.33 The term “Gardner syndrome” has been used to describe patients who have familial adenomatous polyposis but also have osteomas and soft-tissue tumors.34 These patients carry the same risk of colorectal cancer as other patients with familial adenomatous polyposis.
Diagnosing familial adenomatous polyposis
The diagnosis of familial adenomatous polyposis is suspected when a patient has more than 10 adenomatous polyps.
Seventy-five percent of patients with familial adenomatous polyposis have a family history of the condition. Therefore, most cases are identified at a young age on screening sigmoidoscopy or colonoscopy or by predictive gene testing. Patients rarely have cancer at the time of diagnosis.
The other 25% of patients typically are diagnosed when symptoms develop from the polyps or cancer. Over 50% of these symptomatic patients have cancer at the time of diagnosis.
It is recommended that people who have more than 10 adenomas detected on a single colonoscopy or who are first-degree relatives of patients with familial adenomatous polyposis undergo a genetic evaluation and testing for mutations in the APC gene.14 Once an APC mutation is identified in the family, at-risk relatives should be offered testing around age 10 years for families with classic familial adenomatous polyposis or in the mid to late teenage years for those with the attenuated form. It also appropriate to refer patients with desmoid tumors, duodenal adenomas, and bilateral or multifocal congenital hypertrophy of the retinal pigment epithelium for a genetic evaluation.
Management of familial adenomatous polyposis
Flexible sigmoidoscopy every 1 to 2 years beginning at age 10 to 12 years is recommended for individuals and families who have been phenotypically or genetically diagnosed with familial adenomatous polyposis.35–37 If colorectal adenomas are found, surgical options should be discussed and annual colonoscopic surveillance should commence.
For people with the attenuated form, because of the later age of disease onset and the tendency for right-sided disease, colonoscopy every 1 to 2 years should commence at about age 18.35–37 If polyps are found, colonoscopy should be performed every year.
The decision of when to offer colectomy is based on polyp burden (taking into account the number, pathologic appearance, and size of the polyps) and psychosocial factors such as patient maturity. Surgical options include total colectomy and ileorectal anastomosis or total proctocolectomy and ileal pouch anal anastomosis.38 Colonic and extracolonic phenotype as well as genotype should factor into the type of operation recommended. After colectomy, annual endoscopic surveillance of the rectum or ileal pouch is indicated to screen for recurrent polyposis and cancer.
Chemoprevention with sulindac (Clinoril) 150 mg or celecoxib (Celebrex) 400 mg twice a day causes regression of colorectal adenomas in familial adenomatous polyposis and may be useful as an adjunct to endoscopy in managing the colorectal polyp burden.39,40
Forward and side-viewing upper endoscopy should commence at age 20. This should include visualization and biopsy of the papilla and periampulllary region.29 The frequency of endoscopic surveillance depends on the Spigelman stage, which reflects the duodenal polyp burden. It is recommended that patients with Spigelman stage IV duodenal polyposis be seen in consultation with an experienced gastrointestinal surgeon for consideration of a prophylactic, pylorus-preserving, pancreas-sparing duodenectomy. This procedure has been shown to be more effective in polyp control and cancer prevention than endoscopic polyp ablation and local surgical resection.41
Some evidence for the utility of celecoxib 400 mg twice daily for the regression of duodenal polyposis was noted in a 6-month placebo-controlled trial.42 Some experts recommend removal of large duodenal adenomas, with adjunctive celecoxib therapy to control polyposis burden.30
People with familial adenomatous polyposis have been shown to have a 2.6% risk of thyroid cancer, and ultrasonography of the neck with attention to the thyroid is recommended for them.32
MYH-ASSOCIATED POLYPOSIS
Biallelic mutations in the MYH gene result in an adenomatous polyposis syndrome that may be indistinguishable from the attenuated or classic forms of familial adenomatous polyposis. A characteristic autosomal recessive pattern of inheritance in the family can be useful for identifying these patients in the clinic.
Genetics of MYH-associated polyposis
MYH-associated polyposis is the only known autosomal recessive hereditary colorectal cancer syndrome. In white populations, the most commonly reported mutations in MYH are Y179C (previously called Y165C) and G396D (previously called G382D), which account for up to 80% of cases.43 These two mutations are estimated to occur in 1% to 2% of the general population.44
Clinical presentation of MYH-associated polyposis
MYH-associated polyposis typically presents as multiple adenomatous polyps and is diagnosed at a mean age of 47 years. Eleven percent to 42% of affected individuals are reported to have fewer than 100 adenomas, while a minority (7.5% to 29%) of patients present with classic polyposis.45–47 In one study, an estimated 19% of patients presented with colorectal cancer and reported no history of colorectal polyps.48 Synchronous colorectal cancer is seen in more than 60% of patients with biallelic MYH mutations.49 Patients with monoallelic (heterozygous) MYH mutations appear to have the same risk of developing colorectal adenomas and cancer as the general population.49
Upper-gastrointestinal polyps have been reported in MYH-associated polyposis; as many as 17% to 25% of patients have duodenal adenomas.50,51
Diagnosis of MYH-associated polyposis
Genetic testing for biallelic MYH mutations should be performed in patients who test negative for an APC mutation but who have clinical features of familial adenomatous polyposis, a personal history of more than 10 colorectal adenomas, or a recessive family history of polyposis. 14 It has been shown that up to 29% of patients with familial adenomatous polyposis who are APC-negative will have biallelic mutations in the MYH gene.52 The siblings of a patient with biallelic MYH mutations should be offered genetic counseling and testing in their late teens or early 20s. All children of an individual with MYH-associated polyposis will carry one MYH mutation and are only at risk of having the syndrome if the other parent is also a MYH carrier and passed on his or her mutation.
Management of MYH-associated polyposis
The management of patients with MYH-associated polyposis is similar to that recommended for attenuated and classic familial adenomatous polyposis.14 Genetic counseling and testing and colonic and extracolonic surveillance are warranted. There are no data on the use of chemoprevention in MYH-associated polyposis. Surgery should be considered early because of the high risk of colorectal cancer, even in individuals with very few adenomas. Patients with monoallelic MYH mutations should follow the general population screening guidelines for colorectal cancer.49
GENETIC COUNSELING AND GENETIC TESTING
The American College of Gastroenterology advises that patients suspected of having hereditary colorectal cancer syndromes be advised to pursue genetic counseling and, if appropriate, genetic testing.16 They further recommend genetic counseling and informed consent before genetic testing.16
Genetic counseling is a process of working with patients and families whereby:
- A detailed medical and family history is obtained
- A formal risk assessment is performed
- Education about the disease in question and about genetic testing is provided
- Psychosocial concerns are assessed
- Informed consent is obtained when genetic testing is recommended.53
This process is important for helping patients better understand their cancer risks, the benefits and limitations of genetic testing, and the protections that are in place for people who undergo genetic testing, including the Genetic Information Non-Discrimination Act.
In 1996 the American Society of Clinical Oncology issued a policy statement highlighting the essential elements of informed consent for genetic testing for cancer susceptibility, and this was updated in 2003.54 In particular, it notes that patients should be informed of the implications of positive and negative results and of the possibility that the test may be uninformative.
When a hereditary colorectal cancer syndrome is suspected, a positive genetic test result confirms the diagnosis and allows for predictive testing of the patient’s relatives. However, no genetic test for a hereditary colorectal cancer syndrome is 100% sensitive. Therefore, a negative result does not rule out the syndrome in question.
Further, all cancer susceptibility genes have variants of uncertain significance, which are genetic alterations for which there are insufficient data to determine if the mutation is disease-causing or polymorphic (benign). Both negative and uninformative results can be confusing for patients and providers and can lead to false reassurance or undue worry when patients are not properly educated about these potential outcomes of testing.
Genetic testing is an evolving field, and with additional research and improved testing technologies, appropriate diagnoses can be made over time. That is why it is important for the genetic counseling relationship to continue over time.
- Bonadona V, Bonaïti B, Olschwang S, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011; 305:2304–2310.
- Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011; 60:950–957.
- Barrow E, Robinson L, Alduaij W, et al. Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: a report of 121 families with proven mutations. Clin Genet 2009; 75:141–149.
- van der Post RS, Kiemeney LA, Ligtenberg MJ, et al. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J Med Genet 2010; 47:464–470.
- Wijnen JT, Vasen HF, Khan PM, et al. Clinical findings with implications for genetic testing in families with clustering of colorectal cancer. N Engl J Med 1998; 339:511–518.
- Bisgaard ML, Bernstein I. HNPCC mutation rate. Familial Cancer 2003; 2.
- Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum 1991; 34:424–425.
- Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999; 116:1453–1456.
- Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96:261–268.
- Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP Working Group: can UGT1A1 genotyping reduce morbidity and mortality in patients with metastatic colorectal cancer treated with irinotecan? Genet Med 2009; 11:15–20.
- Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993; 260:812–816.
- Kim H, Jen J, Vogelstein B, Hamilton SR. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol 1994; 145:148–156.
- Lindor NM, Rabe K, Petersen GM, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA 2005; 293:1979–1985.
- National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology (NCCN guidelines) colorectal cancer screening version 2.2011. www.nccn.org. Accessed October 2, 2012.
- Jass JR. Hereditary non-polyposis colorectal cancer: the rise and fall of a confusing term. World J Gastroenterol 2006; 12:4943–4950.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 [corrected]. Am J Gastroenterol 2009; 104:739–750.
- Winawer S, Fletcher R, Rex D, et al; Gastrointestinal Consortium Panel. Colorectal cancer screening and surveillance: clinical guidelines and rationale-update based on new evidence. Gastroenterology 2003; 124:544–560.
- Lindor NM, Petersen GM, Hadley DW, et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA 2006; 296:1507–1517.
- de Jong AE, Hendriks YM, Kleibeuker JH, et al. Decrease in mortality in Lynch syndrome families because of surveillance. Gastroenterology 2006; 130:665–671.
- Mecklin JP, Aarnio M, Läärä E, et al. Development of colorectal tumors in colonoscopic surveillance in Lynch syndrome. Gastroenterology 2007; 133:1093–1098.
- Engel C, Rahner N, Schulmann K, et al; German HNPCC Consortium. Efficacy of annual colonoscopic surveillance in individuals with hereditary nonpolyposis colorectal cancer. Clin Gastroenterol Hepatol 2010; 8:174–182.
- Burn J, Gerdes AM, Macrae F, et al; CAPP2 Investigators. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 2011; 378:2081–2087.
- Vasen HF, Möslein G, Alonso A, et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J Med Genet 2007; 44:353–362.
- Manchanda R, Menon U, Michaelson-Cohen R, Beller U, Jacobs I. Hereditary non-polyposis colorectal cancer or Lynch syndrome: the gynaecological perspective. Curr Opin Obstet Gynecol 2009; 21:31–38.
- Burn J, Chapman P, Delhanty J, et al. The UK Northern region genetic register for familial adenomatous polyposis coli: use of age of onset, congenital hypertrophy of the retinal pigment epithelium, and DNA markers in risk calculations. J Med Genet 1991; 28:289–296.
- Järvinen HJ. Epidemiology of familial adenomatous polyposis in Finland: impact of family screening on the colorectal cancer rate and survival. Gut 1992; 33:357–360.
- Bisgaard ML, Fenger K, Bülow S, Niebuhr E, Mohr J. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat 1994; 3:121–125.
- Neklason DW, Stevens J, Boucher KM, et al. American founder mutation for attenuated familial adenomatous polyposis. Clin Gastroenterol Hepatol 2008; 6:46–52.
- Burke CA, Beck GJ, Church JM, van Stolk RU. The natural history of untreated duodenal and ampullary adenomas in patients with familial adenomatous polyposis followed in an endoscopic surveillance program. Gastrointest Endosc 1999; 49:358–364.
- Bianchi LK, Burke CA, Bennett AE, Lopez R, Hasson H, Church JM. Fundic gland polyp dysplasia is common in familial adenomatous polyposis. Clin Gastroenterol Hepatol 2008; 6:180–185.
- Kadmon M, Tandara A, Herfarth C. Duodenal adenomatosis in familial adenomatous polyposis coli. A review of the literature and results from the Heidelberg Polyposis Register. Int J Colorectal Dis 2001; 16:63–75.
- Jarrar AM, Milas M, Mitchell J, et al. Screening for thyroid cancer in patients with familial adenomatous polyposis. Ann Surg 2011; 253:515–521.
- Jasperson KW, Burt RW. APC-associated polyposis conditions. In:Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews (Internet). Seattle, WA: University of Washington; 2011.
- Gardner EJ, Richards RC. Multiple cutaneous and subcutaneous lesions occurring simultaneously with hereditary polyposis and osteomatosis. Am J Hum Genet 1953; 5:139–147.
- Dunlop MG; British Society for Gastroenterology. Guidance on gastrointestinal surveillance for hereditary non-polyposis colorectal cancer, familial adenomatous polyposis, juvenile polyposis, and Peutz-Jeghers syndrome. Gut 2002; 51(suppl 5):V21–V27.
- Burke W, Petersen G, Lynch P, et al. Recommendations for follow-up care of individuals with an inherited predisposition to cancer. I. Hereditary nonpolyposis colon cancer. Cancer Genetics Studies Consortium. JAMA 1997; 277:915–919.
- Vasen HF, Möslein G, Alonso A, et al. Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut 2008; 57:704–713.
- Church J. Familial adenomatous polyposis. Surg Oncol Clin N Am 2009; 18:585–598.
- Giardiello FM, Hamilton SR, Krush AJ, et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med 1993; 328:1313–1316.
- Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 2000; 342:1946–1952.
- Johnson MD, Mackey R, Brown N, Church J, Burke C, Walsh RM. Outcome based on management for duodenal adenomas: sporadic versus familial disease. J Gastrointest Surg 2010; 14:229–235.
- Phillips RK, Wallace MH, Lynch PM, et al; FAP Study Group. A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut 2002; 50:857–860.
- Tenesa A, Campbell H, Barnetson R, Porteous M, Dunlop M, Farrington SM. Association of MUTYH and colorectal cancer. Br J Cancer 2006; 95:239–242.
- Croitoru ME, Cleary SP, Di Nicola N, et al. Association between biallelic and monoallelic germline MYH gene mutations and colorectal cancer risk. J Natl Cancer Inst 2004; 96:1631–1634.
- Croitoru ME, Cleary SP, Berk T, et al. Germline MYH mutations in a clinic-based series of Canadian multiple colorectal adenoma patients. J Surg Oncol 2007; 95:499–506.
- Sampson JR, Dolwani S, Jones S, et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet 2003; 362:39–41.
- Nielsen M, Franken PF, Reinards TH, et al. Multiplicity in polyp count and extracolonic manifestations in 40 Dutch patients with MYH associated polyposis coli (MAP). J Med Genet 2005; 42:e54.
- Cleary SP, Cotterchio M, Jenkins MA, et al. Germline MutY human homologue mutations and colorectal cancer: a multisite case-control study. Gastroenterology 2009; 136:1251–1260.
- Lubbe SJ, Di Bernardo MC, Chandler IP, Houlston RS. Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J Clin Oncol 2009; 27:3975–3980.
- Aretz S, Uhlhaas S, Goergens H, et al. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006; 119:807–814.
- Vogt S, Jones N, Christian D, et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology 2009; 137:1976–1985.e1–e10.
- Gismondi V, Meta M, Bonelli L, et al. Prevalence of the Y165C, G382D and 1395delGGA germline mutations of the MYH gene in Italian patients with adenomatous polyposis coli and colorectal adenomas. Int J Cancer 2004; 109:680–684.
- Trepanier A, Ahrens M, McKinnon W, et al; National Society of Genetic Counselors. Genetic cancer risk assessment and counseling: recommendations of the national society of genetic counselors. J Genet Couns 2004; 13:83–114.
- American Society of Clinical Oncology. American Society of Clinical Oncology policy statement update: genetic testing for cancer susceptibility. J Clin Oncol 2003; 21:2397–2406.
- Bonadona V, Bonaïti B, Olschwang S, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011; 305:2304–2310.
- Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011; 60:950–957.
- Barrow E, Robinson L, Alduaij W, et al. Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: a report of 121 families with proven mutations. Clin Genet 2009; 75:141–149.
- van der Post RS, Kiemeney LA, Ligtenberg MJ, et al. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J Med Genet 2010; 47:464–470.
- Wijnen JT, Vasen HF, Khan PM, et al. Clinical findings with implications for genetic testing in families with clustering of colorectal cancer. N Engl J Med 1998; 339:511–518.
- Bisgaard ML, Bernstein I. HNPCC mutation rate. Familial Cancer 2003; 2.
- Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum 1991; 34:424–425.
- Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999; 116:1453–1456.
- Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96:261–268.
- Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP Working Group: can UGT1A1 genotyping reduce morbidity and mortality in patients with metastatic colorectal cancer treated with irinotecan? Genet Med 2009; 11:15–20.
- Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993; 260:812–816.
- Kim H, Jen J, Vogelstein B, Hamilton SR. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol 1994; 145:148–156.
- Lindor NM, Rabe K, Petersen GM, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA 2005; 293:1979–1985.
- National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology (NCCN guidelines) colorectal cancer screening version 2.2011. www.nccn.org. Accessed October 2, 2012.
- Jass JR. Hereditary non-polyposis colorectal cancer: the rise and fall of a confusing term. World J Gastroenterol 2006; 12:4943–4950.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 [corrected]. Am J Gastroenterol 2009; 104:739–750.
- Winawer S, Fletcher R, Rex D, et al; Gastrointestinal Consortium Panel. Colorectal cancer screening and surveillance: clinical guidelines and rationale-update based on new evidence. Gastroenterology 2003; 124:544–560.
- Lindor NM, Petersen GM, Hadley DW, et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA 2006; 296:1507–1517.
- de Jong AE, Hendriks YM, Kleibeuker JH, et al. Decrease in mortality in Lynch syndrome families because of surveillance. Gastroenterology 2006; 130:665–671.
- Mecklin JP, Aarnio M, Läärä E, et al. Development of colorectal tumors in colonoscopic surveillance in Lynch syndrome. Gastroenterology 2007; 133:1093–1098.
- Engel C, Rahner N, Schulmann K, et al; German HNPCC Consortium. Efficacy of annual colonoscopic surveillance in individuals with hereditary nonpolyposis colorectal cancer. Clin Gastroenterol Hepatol 2010; 8:174–182.
- Burn J, Gerdes AM, Macrae F, et al; CAPP2 Investigators. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 2011; 378:2081–2087.
- Vasen HF, Möslein G, Alonso A, et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J Med Genet 2007; 44:353–362.
- Manchanda R, Menon U, Michaelson-Cohen R, Beller U, Jacobs I. Hereditary non-polyposis colorectal cancer or Lynch syndrome: the gynaecological perspective. Curr Opin Obstet Gynecol 2009; 21:31–38.
- Burn J, Chapman P, Delhanty J, et al. The UK Northern region genetic register for familial adenomatous polyposis coli: use of age of onset, congenital hypertrophy of the retinal pigment epithelium, and DNA markers in risk calculations. J Med Genet 1991; 28:289–296.
- Järvinen HJ. Epidemiology of familial adenomatous polyposis in Finland: impact of family screening on the colorectal cancer rate and survival. Gut 1992; 33:357–360.
- Bisgaard ML, Fenger K, Bülow S, Niebuhr E, Mohr J. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat 1994; 3:121–125.
- Neklason DW, Stevens J, Boucher KM, et al. American founder mutation for attenuated familial adenomatous polyposis. Clin Gastroenterol Hepatol 2008; 6:46–52.
- Burke CA, Beck GJ, Church JM, van Stolk RU. The natural history of untreated duodenal and ampullary adenomas in patients with familial adenomatous polyposis followed in an endoscopic surveillance program. Gastrointest Endosc 1999; 49:358–364.
- Bianchi LK, Burke CA, Bennett AE, Lopez R, Hasson H, Church JM. Fundic gland polyp dysplasia is common in familial adenomatous polyposis. Clin Gastroenterol Hepatol 2008; 6:180–185.
- Kadmon M, Tandara A, Herfarth C. Duodenal adenomatosis in familial adenomatous polyposis coli. A review of the literature and results from the Heidelberg Polyposis Register. Int J Colorectal Dis 2001; 16:63–75.
- Jarrar AM, Milas M, Mitchell J, et al. Screening for thyroid cancer in patients with familial adenomatous polyposis. Ann Surg 2011; 253:515–521.
- Jasperson KW, Burt RW. APC-associated polyposis conditions. In:Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews (Internet). Seattle, WA: University of Washington; 2011.
- Gardner EJ, Richards RC. Multiple cutaneous and subcutaneous lesions occurring simultaneously with hereditary polyposis and osteomatosis. Am J Hum Genet 1953; 5:139–147.
- Dunlop MG; British Society for Gastroenterology. Guidance on gastrointestinal surveillance for hereditary non-polyposis colorectal cancer, familial adenomatous polyposis, juvenile polyposis, and Peutz-Jeghers syndrome. Gut 2002; 51(suppl 5):V21–V27.
- Burke W, Petersen G, Lynch P, et al. Recommendations for follow-up care of individuals with an inherited predisposition to cancer. I. Hereditary nonpolyposis colon cancer. Cancer Genetics Studies Consortium. JAMA 1997; 277:915–919.
- Vasen HF, Möslein G, Alonso A, et al. Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut 2008; 57:704–713.
- Church J. Familial adenomatous polyposis. Surg Oncol Clin N Am 2009; 18:585–598.
- Giardiello FM, Hamilton SR, Krush AJ, et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med 1993; 328:1313–1316.
- Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 2000; 342:1946–1952.
- Johnson MD, Mackey R, Brown N, Church J, Burke C, Walsh RM. Outcome based on management for duodenal adenomas: sporadic versus familial disease. J Gastrointest Surg 2010; 14:229–235.
- Phillips RK, Wallace MH, Lynch PM, et al; FAP Study Group. A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut 2002; 50:857–860.
- Tenesa A, Campbell H, Barnetson R, Porteous M, Dunlop M, Farrington SM. Association of MUTYH and colorectal cancer. Br J Cancer 2006; 95:239–242.
- Croitoru ME, Cleary SP, Di Nicola N, et al. Association between biallelic and monoallelic germline MYH gene mutations and colorectal cancer risk. J Natl Cancer Inst 2004; 96:1631–1634.
- Croitoru ME, Cleary SP, Berk T, et al. Germline MYH mutations in a clinic-based series of Canadian multiple colorectal adenoma patients. J Surg Oncol 2007; 95:499–506.
- Sampson JR, Dolwani S, Jones S, et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet 2003; 362:39–41.
- Nielsen M, Franken PF, Reinards TH, et al. Multiplicity in polyp count and extracolonic manifestations in 40 Dutch patients with MYH associated polyposis coli (MAP). J Med Genet 2005; 42:e54.
- Cleary SP, Cotterchio M, Jenkins MA, et al. Germline MutY human homologue mutations and colorectal cancer: a multisite case-control study. Gastroenterology 2009; 136:1251–1260.
- Lubbe SJ, Di Bernardo MC, Chandler IP, Houlston RS. Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J Clin Oncol 2009; 27:3975–3980.
- Aretz S, Uhlhaas S, Goergens H, et al. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006; 119:807–814.
- Vogt S, Jones N, Christian D, et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology 2009; 137:1976–1985.e1–e10.
- Gismondi V, Meta M, Bonelli L, et al. Prevalence of the Y165C, G382D and 1395delGGA germline mutations of the MYH gene in Italian patients with adenomatous polyposis coli and colorectal adenomas. Int J Cancer 2004; 109:680–684.
- Trepanier A, Ahrens M, McKinnon W, et al; National Society of Genetic Counselors. Genetic cancer risk assessment and counseling: recommendations of the national society of genetic counselors. J Genet Couns 2004; 13:83–114.
- American Society of Clinical Oncology. American Society of Clinical Oncology policy statement update: genetic testing for cancer susceptibility. J Clin Oncol 2003; 21:2397–2406.
KEY POINTS
- Hereditary colorectal cancer syndromes carry a substantial risk of intestinal and extraintestinal tumors.
- Affected patients need increased cancer surveillance and may benefit from prophylactic surgery.
- Identifying these patients in clinical practice begins by assessing a patient’s personal and family health history.
- Patients suspected of having hereditary colorectal cancer syndromes should be referred for genetic counseling and, if appropriate, for genetic testing.

New fecal occult blood tests may improve adherence and mortality rates
New fecal occult blood tests hold promise for improving our detection of colorectal cancer and for lowering mortality rates. This is good news, because despite the proven benefit of being screened for colorectal cancer,1 only an average of 62% of eligible adults are screened,2 and colorectal cancer remains the third leading cause of cancer deaths in the United States.
Colonoscopy is often considered the gold-standard screening test for colorectal cancer. However, many patients do not undergo screening colonoscopy because it is invasive and uncomfortable, bowel preparation poses a challenge, the procedure has risks, and it is costly. Members of minority groups, people of lower socioeconomic status, and those who lack health insurance are less likely to undergo screening.
While fecal occult blood tests are cheaper and less invasive than colonoscopy, they do not allow us to prevent colorectal cancer by removing adenomatous polyps. Still, randomized controlled trials have proven that fecal occult blood testing is associated with a decrease in the rate of death from colorectal cancer,3 and it has been shown to be cost-effective.
The challenge is that all guaiac-based tests (gFOBTs), even the newest one, require strict dietary and medication restrictions to be accurate, and the difficulty of collecting stool specimens often results in either false-positive results or failure to complete the test.
The newer tests—one guaiac-based test and several fecal immunochemical tests (FITs)—are more sensitive, and the FITs are more convenient for patients to use than the older guaiac-based tests, advantages that, we hope, will increase the rates of compliance with testing.
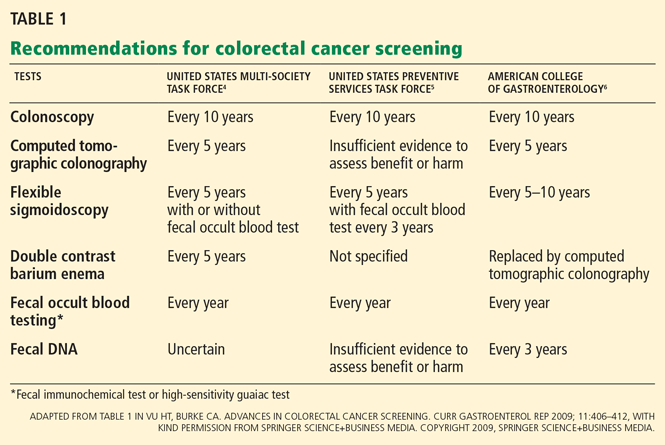
GUAIAC-BASED TESTS
Guaiac tests detect the peroxidase activity of hemoglobin. If hemoglobin is present in stool, it catalyzes the oxidation of the active compound in guaiac paper when a hydrogen peroxide developer is added. The resultant conjugated compound is blue.
The lower-sensitivity guaiac tests are commercially available as Hemoccult and Hemoccult II, and the higher-sensitivity guaiac test is Hemoccult Sensa, which has a lower threshold for detecting peroxidase. All are made by Beckman Coulter, Fullerton, CA.
Disadvantages of guaiac tests. Guaiac tests can give false-positive results by detecting pseudoperoxidases in fruits, vegetables, and nonhuman blood. In addition, they can give false-negative results in people who take excessive amounts of vitamin C, which can inhibit peroxidase activity. Therefore, patients need to follow certain dietary restrictions before testing.
Another disadvantage of guaiac tests is that they cannot differentiate between blood lost from the stomach, small bowel, or colon.
Moreover, the interpretation of guaiac tests is subject to observer variation.
Since testing involves dietary restrictions and obtaining two specimens each from three separate stools, patient compliance is poor.
Patient instructions. Patients undergoing guaiac-based fecal occult blood testing should not take nonsteroidal anti-inflammatory drugs (eg, > one adult aspirin per day) for 7 days before and during the stool collection period to avoid causing gastrointestinal bleeding. They should also not eat red meat or take vitamin C in excess of 250 mg/day for 3 days before testing and throughout the test period.
Two specimens are collected from three different stools with a wooden stick and are smeared onto the stool test card, which is then closed and returned to the physician’s office. The specimens must be collected before the stool comes into contact with the toilet water.
Efficacy of guaiac testing
Randomized, controlled trials of guaiac-based fecal occult blood testing have shown a decrease in colorectal cancer incidence.8–11
A Cochrane review12 involved more than 320,000 people in Denmark, Sweden, the United States, and the United Kingdom who underwent testing every year or every 2 years with Hemoccult or Hemoccult II. The primary analysis was by intention to treat, and it showed that participants allocated to screening had a 16% reduction in the relative risk of death from colorectal cancer, or 0.1 to 0.2 fewer colorectal cancer deaths per 1,000 patient-years. The secondary analysis was adjusted for whether the participants actually were screened; the risk reduction in death from colorectal cancer was 25% in participants who attended at least one round of screening.
FECAL IMMUNOCHEMICAL TESTS
Fecal immunochemical tests use monoclonal or polyclonal antibodies to human globin to detect human blood in stool.
Advantages of fecal immunochemical testing. The antibodies used do not cross-react with nonhuman globin or peroxidases from food sources. Therefore, these tests avoid the dietary and medication restrictions required for guaiac tests. In addition, the stool collection method is simpler, and only one stool specimen is needed instead of three. For these reasons, patient compliance may be better than with guaiac tests.
Additionally, because human globin does not survive passage through the upper gastrointestinal tract, fecal immunochemical testing is specific for bleeding from the colon and rectum.
Immunochemical tests can be read either visually or by machine. Automation allows the threshold for detection of globin to be modified to balance the test’s sensitivity and specificity for the population being served. Most studies have used a threshold of 75 ng/mL, but other studies have assessed thresholds as low as 50 ng/mL and as high as 100 ng/mL. A lower threshold of detection has been shown to increase the sensitivity and yet retain a high specificity.
The immunochemical tests are slightly more expensive than the guaiac tests. However, they are covered by insurance, including Medicare.
Disadvantages of fecal immunochemical testing. A number of tests are available; they use different antibodies and therefore differ in their sensitivity. While most screening studies used automated interpretation of the tests, some studies used visual interpretation (but trained technicians were used to decrease potential interobserver variability). Therefore, the characteristics of fecal immunochemical tests are particular to the specific test kit used.
The antibodies and their epitopes used in some fecal immunochemical tests may be unstable, so that these tests may perform poorly without refrigeration in warm climates or if there are postal delays.
Patient instructions. In some of the tests, a special wand is inserted into six different places in the stool (before the stool is in contact with toilet bowl water) and then placed in the plastic container provided. Other tests use a brush for sample collection. The container may be sent to the laboratory for automated interpretation, or, if the interpretation is performed manually, the container is shaken and a few drops of the liquid in the specimen are added to the test cassette. The interpretation is made after 5 to 10 minutes.
GUAIAC VS IMMUNOCHEMICAL TESTING IN SCREENING
Allison et al13 performed one of the earliest studies to compare the different types of fecal occult blood tests as screening tests for colorectal cancer. More than 7,500 participants in the United States who were due for screening were advised to follow the dietary restrictions for guaiac tests mentioned above for 3 to 4 days before screening and were given three specially made test cards, each of which contained three tests: Hemoccult II, Hemoccult Sensa, and the fecal immunochemical test HemeSelect (SmithKline Diagnostics, San Jose, CA), which was visually read. The authors evaluated the performance of the tests by identifying screened patients found to have colorectal cancer or an adenoma larger than 10 mm in the 2 years after screening.
Sensitivities for detecting colorectal cancer:
- 37% with Hemoccult II
- 69% with HemeSelect
- 79% with Hemoccult Sensa.
Specificities:
- 98% with Hemoccult II
- 94% with HemeSelect
- 87% with Hemoccult Sensa.
Smith et al14 evaluated the performance of two tests in a mix of a screening population and a high-risk group. More than 2,300 Australians sampled two consecutive stools for an immunochemical test, InSure (Enterix, North Ryde, NSW, Australia), and three consecutive stools for Hemoccult Sensa. They were advised to adhere to the dietary and medication restrictions listed in Beckman Coulter’s instructions for the Hemoccult Sensa test. Both tests were read visually. The sensitivity and specificity were calculated from results of colonoscopy performed in participants with a positive stool test.
InSure had a higher sensitivity than Hemoccult Sensa for colorectal cancer (87.5% vs 54.2%) and for advanced adenomas (42.6% vs 23.0%). The false-positive rate for any neoplasia was slightly higher with InSure than with Hemoccult Sensa (3.4% vs 2.5%).
Guittet et al,15 in a French study in more than 10,000 people at average risk, compared a low-sensitivity guaiac test (Hemoccult II) and an immunochemical test, Immudia/RPHA (Fujirebio, Tokyo, Japan). No dietary restrictions were required. Three stool samples were taken for the Hemoccult II and three for the immunochemical test, which was read by machine with three different thresholds for detection of globin: 20, 50, and 75 ng/mL. Positive results were followed up with colonoscopy.
The immunochemical test had a higher sensitivity for both colorectal cancer and advanced adenomas, regardless of the cutoff values of globin. At a cutoff value of 75 ng/mL, the positivity rate was similar to that of the low-sensitivity guaiac test (2.4%), and the immunochemical test offered a gain in sensitivity of 90% and a decrease in the false-positive rate of 33% for advanced neoplasia.
van Rossum et al16 performed a randomized comparison of more than 10,993 tests of Hemoccult II and the fecal immunochemical test OC-Sensor (Eiken Chemical Co., Ltd, Tokyo, Japan) in a screening population in the Netherlands. The participants were not required to follow dietary or medication restrictions. They were asked to send in cards with two samples each from three consecutive bowel movements for the Hemoccult II test and a single sample for the OC-Sensor test, for which interpretation was automated and a cutoff of 100 ng/mL or higher was considered as positive. All participants who had a positive Hemoccult II test or a positive OC-Sensor test with a globin cutoff of 50 ng/mL were advised to undergo colonoscopy.
The study found a 13% higher rate of screening participation with the immunochemical test than with the guaiac-based test, and the positivity rate was 3% higher in the immunochemical testing group (5.5%).16 Cancer was found in 11 patients with the guaiac test and in 24 patients with the immunochemical test; advanced adenomas were found in 48 patients with the guaiac test and 121 patients with the immunochemical test. The guaiac test was more specific, but the participation and detection rates for advanced adenomas and cancer were significantly higher with immunochemical testing.
Park et al17 performed a study in nearly 800 patients undergoing screening colonoscopy in South Korea. Three stool samples were collected for a low-sensitivity guaiac test (Hemoccult II) and for a fecal immunochemical test (OC-Sensor) for detecting cancer and advanced neoplasms. No dietary changes were required. At all globin thresholds between 50 and 150 ng/mL, the immunochemical test was more sensitive than the guaiac-based test, with a similar specificity.
Hundt et al18 obtained a single stool specimen from each of 1,319 German patients before they underwent scheduled screening colonoscopy. Each specimen was tested with six automated immunochemical tests with globin detection thresholds set at 10 to 50 ng/mL. In addition, participants prepared a single Hemoccult card from the same stool sample at home. They were not told to follow any dietary restrictions.
For Hemoccult, the sensitivity for advanced adenoma (1 cm or more in diameter, villous changes, or high-grade dysplasia) was 9%, and the specificity was 96%. For the immunochemical tests, the sensitivity for advanced adenoma varied from 25% to 72%, and the specificity from 70% to 97%.
The reason for the variation in performance of different fecal immunochemical tests is not clear. In some of these tests, the sensitivity can be adjusted when automated interpretation is used. It has been shown that different thresholds for the detection of globin partially explain this. Differences in collection methods also affect the result.
Itoh et al19 reported the results of a screening study done at a large Japanese corporation using a fecal immunochemical test, OC-Hemodia (Eiken Chemical Co., LTD, Tokyo, Japan). A small sample of a single stool was placed in buffer and read by machine. At a cutoff of 200 μg/mL, the sensitivity was 77.5% and the specificity was 98.9%. At a cutoff of 50 ng/mL, the sensitivity was 86.5% and the specificity was 94.9%. In this study, positive tests were followed by colonoscopy, but false-negative tests were identified from insurance claims.
Cole et al20 assessed the rates of participation in colorectal cancer screening in a study in Australia. Participants were randomized and received by mail either Hemoccult Sensa or one of two fecal immunochemical tests, Flex-Sure OBT (Beckman Coulter, Fullerton, CA) or InSure. The Hemoccult Sensa group was instructed to follow dietary and medication restrictions during stool collection, while the immunochemical test groups were not. Three stool specimens were required for the Hemoccult Sensa and FlexSure tests, while two stools were required for InSure.
The participation rate was 23.4% in the Hemoccult Sensa group, 30.5% in the Flex-Sure OBT group, and 39.6% in the InSure group (P < .001).
Hol et al21 found that the participation rate was 50% in a group asked to undergo guaiac testing requiring three samples without diet restriction and 62% in a group asked to undergo fecal immunochemical testing (OC-Sensor) requiring a single stool sample without restrictions. Higher participation rates are seen with fecal immunochemical testing than with guaiac testing and are an advantage of immunochemical testing.
CLEVELAND CLINIC SWITCHES TO FECAL IMMUNOCHEMICAL TESTING FOR COLORECTAL CANCER SCREENING
Cleveland Clinic recently switched to fecal immunochemical testing in place of Hemoccult Sensa for colorectal cancer screening. The data on fecal occult blood tests show that the sensitivities of Hemoccult Sensa and the immunochemical tests are higher than those of Hemoccult and Hemoccult II for the detection of colorectal cancer and advanced adenomas, with similar specificity. Fecal immunochemical tests have an advantage over guaiac-based tests in most screening studies by showing a superior sensitivity for advanced adenomas and colorectal cancer, as well as an increase in test adherence, likely because of the lack of dietary and medication restrictions and the lower number of stool samples required. Increased compliance should improve participation in colorectal cancer screening and positively affect colorectal cancer mortality rates.
- Edwards BK, Ward E, Kohler BA, et al. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer 2010; 116:544–573.
- Centers for Disease Control and Prevention (CDC). Vital signs: colorectal cancer screening among adults aged 50–75 years—United States, 2008. MMWR Morb Mortal Wkly Rep 2010; 59:808–812.
- Mandel JS, Church TR, Bond JH, et al. The effect of fecal occult-blood screening on the incidence of colorectal cancer. N Engl J Med 2000; 343:1603–1607.
- Levin B, Lieberman DA, McFarland B, et al; American Cancer Society Colorectal Cancer Advisory Group; US Multi-Society Task Force; American College of Radiology Colon Cancer Committee. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. Gastroenterology 2008; 134:1570–1595.
- US Preventive Services Task Force. Screening for colorectal cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 2008; 149:627–637.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 [corrected]. Am J Gastroenterol 2009; 104:739–750.
- Vu HT, Burke CA. Advances in colorectal cancer screening. Curr Gastroenterol Rep 2009; 11:406–412.
- Hardcastle JD, Chamberlain JO, Robinson MH, et al. Randomised controlled trial of faecal-occult-blood screening for colorectal cancer. Lancet 1996; 348:1472–1477.
- Kronborg O, Fenger C, Olsen J, Jørgensen OD, Søndergaard O. Randomised study of screening for colorectal cancer with faecal-occult-blood test. Lancet 1996; 348:1467–1471.
- Mandel JS, Bond JH, Church TR, et al. Reducing mortality from colorectal cancer by screening for fecal occult blood. Minnesota Colon Cancer Control Study. N Engl J Med 1993; 328:1365–1371.
- Kewenter J, Brevinge H, Engarås B, Haglind E, Ahrén C. Results of screening, rescreening, and follow-up in a prospective randomized study for detection of colorectal cancer by fecal occult blood testing. Results for 68,308 subjects. Scand J Gastroenterol 1994; 29:468–473.
- Hewitson P, Glasziou P, Irwig L, Towler B, Watson E. Screening for colorectal cancer using the faecal occult blood test, Hemoccult. Cochrane Database Syst Rev 2007;CD001216.
- Allison JE, Tekawa IS, Ransom LJ, Adrain AL. A comparison of fecal occult-blood tests for colorectal-cancer screening. N Engl J Med 1996; 334:155–159.
- Smith A, Young GP, Cole SR, Bampton P. Comparison of a brush-sampling fecal immunochemical test for hemoglobin with a sensitive guaiac-based fecal occult blood test in detection of colorectal neoplasia. Cancer 2006; 107:2152–2159.
- Guittet L, Bouvier V, Mariotte N, et al. Comparison of a guaiac based and an immunochemical faecal occult blood test in screening for colorectal cancer in a general average risk population. Gut 2007; 56:210–214.
- van Rossum LG, van Rijn AF, Laheij RJ, et al. Random comparison of guaiac and immunochemical fecal occult blood tests for colorectal cancer in a screening population. Gastroenterology 2008; 135:82–90.
- Park DI, Ryu S, Kim YH, et al. Comparison of guaiac-based and quantitative immunochemical fecal occult blood testing in a population at average risk undergoing colorectal cancer screening. Am J Gastroenterol 2010; 105:2017–2025.
- Hundt S, Haug U, Brenner H. Comparative evaluation of immunochemical fecal occult blood tests for colorectal adenoma detection. Ann Intern Med 2009; 150:162–169.
- Itoh M, Takahashi K, Nishida H, Sakagami K, Okubo T. Estimation of the optimal cut off point in a new immunological faecal occult blood test in a corporate colorectal cancer screening programme. J Med Screen 1996; 3:66–71.
- Cole SR, Young GP, Esterman A, Cadd B, Morcom J. A randomised trial of the impact of new faecal haemoglobin test technologies on population participation in screening for colorectal cancer. J Med Screen 2003; 10:117–122.
- Hol L, Wilschut JA, van Ballegooijen M, et al. Screening for colorectal cancer: random comparison of guaiac and immunochemical faecal occult blood testing at different cut-off levels. Br J Cancer 2009; 100:1103–1110.
New fecal occult blood tests hold promise for improving our detection of colorectal cancer and for lowering mortality rates. This is good news, because despite the proven benefit of being screened for colorectal cancer,1 only an average of 62% of eligible adults are screened,2 and colorectal cancer remains the third leading cause of cancer deaths in the United States.
Colonoscopy is often considered the gold-standard screening test for colorectal cancer. However, many patients do not undergo screening colonoscopy because it is invasive and uncomfortable, bowel preparation poses a challenge, the procedure has risks, and it is costly. Members of minority groups, people of lower socioeconomic status, and those who lack health insurance are less likely to undergo screening.
While fecal occult blood tests are cheaper and less invasive than colonoscopy, they do not allow us to prevent colorectal cancer by removing adenomatous polyps. Still, randomized controlled trials have proven that fecal occult blood testing is associated with a decrease in the rate of death from colorectal cancer,3 and it has been shown to be cost-effective.
The challenge is that all guaiac-based tests (gFOBTs), even the newest one, require strict dietary and medication restrictions to be accurate, and the difficulty of collecting stool specimens often results in either false-positive results or failure to complete the test.
The newer tests—one guaiac-based test and several fecal immunochemical tests (FITs)—are more sensitive, and the FITs are more convenient for patients to use than the older guaiac-based tests, advantages that, we hope, will increase the rates of compliance with testing.
GUAIAC-BASED TESTS
Guaiac tests detect the peroxidase activity of hemoglobin. If hemoglobin is present in stool, it catalyzes the oxidation of the active compound in guaiac paper when a hydrogen peroxide developer is added. The resultant conjugated compound is blue.
The lower-sensitivity guaiac tests are commercially available as Hemoccult and Hemoccult II, and the higher-sensitivity guaiac test is Hemoccult Sensa, which has a lower threshold for detecting peroxidase. All are made by Beckman Coulter, Fullerton, CA.
Disadvantages of guaiac tests. Guaiac tests can give false-positive results by detecting pseudoperoxidases in fruits, vegetables, and nonhuman blood. In addition, they can give false-negative results in people who take excessive amounts of vitamin C, which can inhibit peroxidase activity. Therefore, patients need to follow certain dietary restrictions before testing.
Another disadvantage of guaiac tests is that they cannot differentiate between blood lost from the stomach, small bowel, or colon.
Moreover, the interpretation of guaiac tests is subject to observer variation.
Since testing involves dietary restrictions and obtaining two specimens each from three separate stools, patient compliance is poor.
Patient instructions. Patients undergoing guaiac-based fecal occult blood testing should not take nonsteroidal anti-inflammatory drugs (eg, > one adult aspirin per day) for 7 days before and during the stool collection period to avoid causing gastrointestinal bleeding. They should also not eat red meat or take vitamin C in excess of 250 mg/day for 3 days before testing and throughout the test period.
Two specimens are collected from three different stools with a wooden stick and are smeared onto the stool test card, which is then closed and returned to the physician’s office. The specimens must be collected before the stool comes into contact with the toilet water.
Efficacy of guaiac testing
Randomized, controlled trials of guaiac-based fecal occult blood testing have shown a decrease in colorectal cancer incidence.8–11
A Cochrane review12 involved more than 320,000 people in Denmark, Sweden, the United States, and the United Kingdom who underwent testing every year or every 2 years with Hemoccult or Hemoccult II. The primary analysis was by intention to treat, and it showed that participants allocated to screening had a 16% reduction in the relative risk of death from colorectal cancer, or 0.1 to 0.2 fewer colorectal cancer deaths per 1,000 patient-years. The secondary analysis was adjusted for whether the participants actually were screened; the risk reduction in death from colorectal cancer was 25% in participants who attended at least one round of screening.
FECAL IMMUNOCHEMICAL TESTS
Fecal immunochemical tests use monoclonal or polyclonal antibodies to human globin to detect human blood in stool.
Advantages of fecal immunochemical testing. The antibodies used do not cross-react with nonhuman globin or peroxidases from food sources. Therefore, these tests avoid the dietary and medication restrictions required for guaiac tests. In addition, the stool collection method is simpler, and only one stool specimen is needed instead of three. For these reasons, patient compliance may be better than with guaiac tests.
Additionally, because human globin does not survive passage through the upper gastrointestinal tract, fecal immunochemical testing is specific for bleeding from the colon and rectum.
Immunochemical tests can be read either visually or by machine. Automation allows the threshold for detection of globin to be modified to balance the test’s sensitivity and specificity for the population being served. Most studies have used a threshold of 75 ng/mL, but other studies have assessed thresholds as low as 50 ng/mL and as high as 100 ng/mL. A lower threshold of detection has been shown to increase the sensitivity and yet retain a high specificity.
The immunochemical tests are slightly more expensive than the guaiac tests. However, they are covered by insurance, including Medicare.
Disadvantages of fecal immunochemical testing. A number of tests are available; they use different antibodies and therefore differ in their sensitivity. While most screening studies used automated interpretation of the tests, some studies used visual interpretation (but trained technicians were used to decrease potential interobserver variability). Therefore, the characteristics of fecal immunochemical tests are particular to the specific test kit used.
The antibodies and their epitopes used in some fecal immunochemical tests may be unstable, so that these tests may perform poorly without refrigeration in warm climates or if there are postal delays.
Patient instructions. In some of the tests, a special wand is inserted into six different places in the stool (before the stool is in contact with toilet bowl water) and then placed in the plastic container provided. Other tests use a brush for sample collection. The container may be sent to the laboratory for automated interpretation, or, if the interpretation is performed manually, the container is shaken and a few drops of the liquid in the specimen are added to the test cassette. The interpretation is made after 5 to 10 minutes.
GUAIAC VS IMMUNOCHEMICAL TESTING IN SCREENING
Allison et al13 performed one of the earliest studies to compare the different types of fecal occult blood tests as screening tests for colorectal cancer. More than 7,500 participants in the United States who were due for screening were advised to follow the dietary restrictions for guaiac tests mentioned above for 3 to 4 days before screening and were given three specially made test cards, each of which contained three tests: Hemoccult II, Hemoccult Sensa, and the fecal immunochemical test HemeSelect (SmithKline Diagnostics, San Jose, CA), which was visually read. The authors evaluated the performance of the tests by identifying screened patients found to have colorectal cancer or an adenoma larger than 10 mm in the 2 years after screening.
Sensitivities for detecting colorectal cancer:
- 37% with Hemoccult II
- 69% with HemeSelect
- 79% with Hemoccult Sensa.
Specificities:
- 98% with Hemoccult II
- 94% with HemeSelect
- 87% with Hemoccult Sensa.
Smith et al14 evaluated the performance of two tests in a mix of a screening population and a high-risk group. More than 2,300 Australians sampled two consecutive stools for an immunochemical test, InSure (Enterix, North Ryde, NSW, Australia), and three consecutive stools for Hemoccult Sensa. They were advised to adhere to the dietary and medication restrictions listed in Beckman Coulter’s instructions for the Hemoccult Sensa test. Both tests were read visually. The sensitivity and specificity were calculated from results of colonoscopy performed in participants with a positive stool test.
InSure had a higher sensitivity than Hemoccult Sensa for colorectal cancer (87.5% vs 54.2%) and for advanced adenomas (42.6% vs 23.0%). The false-positive rate for any neoplasia was slightly higher with InSure than with Hemoccult Sensa (3.4% vs 2.5%).
Guittet et al,15 in a French study in more than 10,000 people at average risk, compared a low-sensitivity guaiac test (Hemoccult II) and an immunochemical test, Immudia/RPHA (Fujirebio, Tokyo, Japan). No dietary restrictions were required. Three stool samples were taken for the Hemoccult II and three for the immunochemical test, which was read by machine with three different thresholds for detection of globin: 20, 50, and 75 ng/mL. Positive results were followed up with colonoscopy.
The immunochemical test had a higher sensitivity for both colorectal cancer and advanced adenomas, regardless of the cutoff values of globin. At a cutoff value of 75 ng/mL, the positivity rate was similar to that of the low-sensitivity guaiac test (2.4%), and the immunochemical test offered a gain in sensitivity of 90% and a decrease in the false-positive rate of 33% for advanced neoplasia.
van Rossum et al16 performed a randomized comparison of more than 10,993 tests of Hemoccult II and the fecal immunochemical test OC-Sensor (Eiken Chemical Co., Ltd, Tokyo, Japan) in a screening population in the Netherlands. The participants were not required to follow dietary or medication restrictions. They were asked to send in cards with two samples each from three consecutive bowel movements for the Hemoccult II test and a single sample for the OC-Sensor test, for which interpretation was automated and a cutoff of 100 ng/mL or higher was considered as positive. All participants who had a positive Hemoccult II test or a positive OC-Sensor test with a globin cutoff of 50 ng/mL were advised to undergo colonoscopy.
The study found a 13% higher rate of screening participation with the immunochemical test than with the guaiac-based test, and the positivity rate was 3% higher in the immunochemical testing group (5.5%).16 Cancer was found in 11 patients with the guaiac test and in 24 patients with the immunochemical test; advanced adenomas were found in 48 patients with the guaiac test and 121 patients with the immunochemical test. The guaiac test was more specific, but the participation and detection rates for advanced adenomas and cancer were significantly higher with immunochemical testing.
Park et al17 performed a study in nearly 800 patients undergoing screening colonoscopy in South Korea. Three stool samples were collected for a low-sensitivity guaiac test (Hemoccult II) and for a fecal immunochemical test (OC-Sensor) for detecting cancer and advanced neoplasms. No dietary changes were required. At all globin thresholds between 50 and 150 ng/mL, the immunochemical test was more sensitive than the guaiac-based test, with a similar specificity.
Hundt et al18 obtained a single stool specimen from each of 1,319 German patients before they underwent scheduled screening colonoscopy. Each specimen was tested with six automated immunochemical tests with globin detection thresholds set at 10 to 50 ng/mL. In addition, participants prepared a single Hemoccult card from the same stool sample at home. They were not told to follow any dietary restrictions.
For Hemoccult, the sensitivity for advanced adenoma (1 cm or more in diameter, villous changes, or high-grade dysplasia) was 9%, and the specificity was 96%. For the immunochemical tests, the sensitivity for advanced adenoma varied from 25% to 72%, and the specificity from 70% to 97%.
The reason for the variation in performance of different fecal immunochemical tests is not clear. In some of these tests, the sensitivity can be adjusted when automated interpretation is used. It has been shown that different thresholds for the detection of globin partially explain this. Differences in collection methods also affect the result.
Itoh et al19 reported the results of a screening study done at a large Japanese corporation using a fecal immunochemical test, OC-Hemodia (Eiken Chemical Co., LTD, Tokyo, Japan). A small sample of a single stool was placed in buffer and read by machine. At a cutoff of 200 μg/mL, the sensitivity was 77.5% and the specificity was 98.9%. At a cutoff of 50 ng/mL, the sensitivity was 86.5% and the specificity was 94.9%. In this study, positive tests were followed by colonoscopy, but false-negative tests were identified from insurance claims.
Cole et al20 assessed the rates of participation in colorectal cancer screening in a study in Australia. Participants were randomized and received by mail either Hemoccult Sensa or one of two fecal immunochemical tests, Flex-Sure OBT (Beckman Coulter, Fullerton, CA) or InSure. The Hemoccult Sensa group was instructed to follow dietary and medication restrictions during stool collection, while the immunochemical test groups were not. Three stool specimens were required for the Hemoccult Sensa and FlexSure tests, while two stools were required for InSure.
The participation rate was 23.4% in the Hemoccult Sensa group, 30.5% in the Flex-Sure OBT group, and 39.6% in the InSure group (P < .001).
Hol et al21 found that the participation rate was 50% in a group asked to undergo guaiac testing requiring three samples without diet restriction and 62% in a group asked to undergo fecal immunochemical testing (OC-Sensor) requiring a single stool sample without restrictions. Higher participation rates are seen with fecal immunochemical testing than with guaiac testing and are an advantage of immunochemical testing.
CLEVELAND CLINIC SWITCHES TO FECAL IMMUNOCHEMICAL TESTING FOR COLORECTAL CANCER SCREENING
Cleveland Clinic recently switched to fecal immunochemical testing in place of Hemoccult Sensa for colorectal cancer screening. The data on fecal occult blood tests show that the sensitivities of Hemoccult Sensa and the immunochemical tests are higher than those of Hemoccult and Hemoccult II for the detection of colorectal cancer and advanced adenomas, with similar specificity. Fecal immunochemical tests have an advantage over guaiac-based tests in most screening studies by showing a superior sensitivity for advanced adenomas and colorectal cancer, as well as an increase in test adherence, likely because of the lack of dietary and medication restrictions and the lower number of stool samples required. Increased compliance should improve participation in colorectal cancer screening and positively affect colorectal cancer mortality rates.
New fecal occult blood tests hold promise for improving our detection of colorectal cancer and for lowering mortality rates. This is good news, because despite the proven benefit of being screened for colorectal cancer,1 only an average of 62% of eligible adults are screened,2 and colorectal cancer remains the third leading cause of cancer deaths in the United States.
Colonoscopy is often considered the gold-standard screening test for colorectal cancer. However, many patients do not undergo screening colonoscopy because it is invasive and uncomfortable, bowel preparation poses a challenge, the procedure has risks, and it is costly. Members of minority groups, people of lower socioeconomic status, and those who lack health insurance are less likely to undergo screening.
While fecal occult blood tests are cheaper and less invasive than colonoscopy, they do not allow us to prevent colorectal cancer by removing adenomatous polyps. Still, randomized controlled trials have proven that fecal occult blood testing is associated with a decrease in the rate of death from colorectal cancer,3 and it has been shown to be cost-effective.
The challenge is that all guaiac-based tests (gFOBTs), even the newest one, require strict dietary and medication restrictions to be accurate, and the difficulty of collecting stool specimens often results in either false-positive results or failure to complete the test.
The newer tests—one guaiac-based test and several fecal immunochemical tests (FITs)—are more sensitive, and the FITs are more convenient for patients to use than the older guaiac-based tests, advantages that, we hope, will increase the rates of compliance with testing.
GUAIAC-BASED TESTS
Guaiac tests detect the peroxidase activity of hemoglobin. If hemoglobin is present in stool, it catalyzes the oxidation of the active compound in guaiac paper when a hydrogen peroxide developer is added. The resultant conjugated compound is blue.
The lower-sensitivity guaiac tests are commercially available as Hemoccult and Hemoccult II, and the higher-sensitivity guaiac test is Hemoccult Sensa, which has a lower threshold for detecting peroxidase. All are made by Beckman Coulter, Fullerton, CA.
Disadvantages of guaiac tests. Guaiac tests can give false-positive results by detecting pseudoperoxidases in fruits, vegetables, and nonhuman blood. In addition, they can give false-negative results in people who take excessive amounts of vitamin C, which can inhibit peroxidase activity. Therefore, patients need to follow certain dietary restrictions before testing.
Another disadvantage of guaiac tests is that they cannot differentiate between blood lost from the stomach, small bowel, or colon.
Moreover, the interpretation of guaiac tests is subject to observer variation.
Since testing involves dietary restrictions and obtaining two specimens each from three separate stools, patient compliance is poor.
Patient instructions. Patients undergoing guaiac-based fecal occult blood testing should not take nonsteroidal anti-inflammatory drugs (eg, > one adult aspirin per day) for 7 days before and during the stool collection period to avoid causing gastrointestinal bleeding. They should also not eat red meat or take vitamin C in excess of 250 mg/day for 3 days before testing and throughout the test period.
Two specimens are collected from three different stools with a wooden stick and are smeared onto the stool test card, which is then closed and returned to the physician’s office. The specimens must be collected before the stool comes into contact with the toilet water.
Efficacy of guaiac testing
Randomized, controlled trials of guaiac-based fecal occult blood testing have shown a decrease in colorectal cancer incidence.8–11
A Cochrane review12 involved more than 320,000 people in Denmark, Sweden, the United States, and the United Kingdom who underwent testing every year or every 2 years with Hemoccult or Hemoccult II. The primary analysis was by intention to treat, and it showed that participants allocated to screening had a 16% reduction in the relative risk of death from colorectal cancer, or 0.1 to 0.2 fewer colorectal cancer deaths per 1,000 patient-years. The secondary analysis was adjusted for whether the participants actually were screened; the risk reduction in death from colorectal cancer was 25% in participants who attended at least one round of screening.
FECAL IMMUNOCHEMICAL TESTS
Fecal immunochemical tests use monoclonal or polyclonal antibodies to human globin to detect human blood in stool.
Advantages of fecal immunochemical testing. The antibodies used do not cross-react with nonhuman globin or peroxidases from food sources. Therefore, these tests avoid the dietary and medication restrictions required for guaiac tests. In addition, the stool collection method is simpler, and only one stool specimen is needed instead of three. For these reasons, patient compliance may be better than with guaiac tests.
Additionally, because human globin does not survive passage through the upper gastrointestinal tract, fecal immunochemical testing is specific for bleeding from the colon and rectum.
Immunochemical tests can be read either visually or by machine. Automation allows the threshold for detection of globin to be modified to balance the test’s sensitivity and specificity for the population being served. Most studies have used a threshold of 75 ng/mL, but other studies have assessed thresholds as low as 50 ng/mL and as high as 100 ng/mL. A lower threshold of detection has been shown to increase the sensitivity and yet retain a high specificity.
The immunochemical tests are slightly more expensive than the guaiac tests. However, they are covered by insurance, including Medicare.
Disadvantages of fecal immunochemical testing. A number of tests are available; they use different antibodies and therefore differ in their sensitivity. While most screening studies used automated interpretation of the tests, some studies used visual interpretation (but trained technicians were used to decrease potential interobserver variability). Therefore, the characteristics of fecal immunochemical tests are particular to the specific test kit used.
The antibodies and their epitopes used in some fecal immunochemical tests may be unstable, so that these tests may perform poorly without refrigeration in warm climates or if there are postal delays.
Patient instructions. In some of the tests, a special wand is inserted into six different places in the stool (before the stool is in contact with toilet bowl water) and then placed in the plastic container provided. Other tests use a brush for sample collection. The container may be sent to the laboratory for automated interpretation, or, if the interpretation is performed manually, the container is shaken and a few drops of the liquid in the specimen are added to the test cassette. The interpretation is made after 5 to 10 minutes.
GUAIAC VS IMMUNOCHEMICAL TESTING IN SCREENING
Allison et al13 performed one of the earliest studies to compare the different types of fecal occult blood tests as screening tests for colorectal cancer. More than 7,500 participants in the United States who were due for screening were advised to follow the dietary restrictions for guaiac tests mentioned above for 3 to 4 days before screening and were given three specially made test cards, each of which contained three tests: Hemoccult II, Hemoccult Sensa, and the fecal immunochemical test HemeSelect (SmithKline Diagnostics, San Jose, CA), which was visually read. The authors evaluated the performance of the tests by identifying screened patients found to have colorectal cancer or an adenoma larger than 10 mm in the 2 years after screening.
Sensitivities for detecting colorectal cancer:
- 37% with Hemoccult II
- 69% with HemeSelect
- 79% with Hemoccult Sensa.
Specificities:
- 98% with Hemoccult II
- 94% with HemeSelect
- 87% with Hemoccult Sensa.
Smith et al14 evaluated the performance of two tests in a mix of a screening population and a high-risk group. More than 2,300 Australians sampled two consecutive stools for an immunochemical test, InSure (Enterix, North Ryde, NSW, Australia), and three consecutive stools for Hemoccult Sensa. They were advised to adhere to the dietary and medication restrictions listed in Beckman Coulter’s instructions for the Hemoccult Sensa test. Both tests were read visually. The sensitivity and specificity were calculated from results of colonoscopy performed in participants with a positive stool test.
InSure had a higher sensitivity than Hemoccult Sensa for colorectal cancer (87.5% vs 54.2%) and for advanced adenomas (42.6% vs 23.0%). The false-positive rate for any neoplasia was slightly higher with InSure than with Hemoccult Sensa (3.4% vs 2.5%).
Guittet et al,15 in a French study in more than 10,000 people at average risk, compared a low-sensitivity guaiac test (Hemoccult II) and an immunochemical test, Immudia/RPHA (Fujirebio, Tokyo, Japan). No dietary restrictions were required. Three stool samples were taken for the Hemoccult II and three for the immunochemical test, which was read by machine with three different thresholds for detection of globin: 20, 50, and 75 ng/mL. Positive results were followed up with colonoscopy.
The immunochemical test had a higher sensitivity for both colorectal cancer and advanced adenomas, regardless of the cutoff values of globin. At a cutoff value of 75 ng/mL, the positivity rate was similar to that of the low-sensitivity guaiac test (2.4%), and the immunochemical test offered a gain in sensitivity of 90% and a decrease in the false-positive rate of 33% for advanced neoplasia.
van Rossum et al16 performed a randomized comparison of more than 10,993 tests of Hemoccult II and the fecal immunochemical test OC-Sensor (Eiken Chemical Co., Ltd, Tokyo, Japan) in a screening population in the Netherlands. The participants were not required to follow dietary or medication restrictions. They were asked to send in cards with two samples each from three consecutive bowel movements for the Hemoccult II test and a single sample for the OC-Sensor test, for which interpretation was automated and a cutoff of 100 ng/mL or higher was considered as positive. All participants who had a positive Hemoccult II test or a positive OC-Sensor test with a globin cutoff of 50 ng/mL were advised to undergo colonoscopy.
The study found a 13% higher rate of screening participation with the immunochemical test than with the guaiac-based test, and the positivity rate was 3% higher in the immunochemical testing group (5.5%).16 Cancer was found in 11 patients with the guaiac test and in 24 patients with the immunochemical test; advanced adenomas were found in 48 patients with the guaiac test and 121 patients with the immunochemical test. The guaiac test was more specific, but the participation and detection rates for advanced adenomas and cancer were significantly higher with immunochemical testing.
Park et al17 performed a study in nearly 800 patients undergoing screening colonoscopy in South Korea. Three stool samples were collected for a low-sensitivity guaiac test (Hemoccult II) and for a fecal immunochemical test (OC-Sensor) for detecting cancer and advanced neoplasms. No dietary changes were required. At all globin thresholds between 50 and 150 ng/mL, the immunochemical test was more sensitive than the guaiac-based test, with a similar specificity.
Hundt et al18 obtained a single stool specimen from each of 1,319 German patients before they underwent scheduled screening colonoscopy. Each specimen was tested with six automated immunochemical tests with globin detection thresholds set at 10 to 50 ng/mL. In addition, participants prepared a single Hemoccult card from the same stool sample at home. They were not told to follow any dietary restrictions.
For Hemoccult, the sensitivity for advanced adenoma (1 cm or more in diameter, villous changes, or high-grade dysplasia) was 9%, and the specificity was 96%. For the immunochemical tests, the sensitivity for advanced adenoma varied from 25% to 72%, and the specificity from 70% to 97%.
The reason for the variation in performance of different fecal immunochemical tests is not clear. In some of these tests, the sensitivity can be adjusted when automated interpretation is used. It has been shown that different thresholds for the detection of globin partially explain this. Differences in collection methods also affect the result.
Itoh et al19 reported the results of a screening study done at a large Japanese corporation using a fecal immunochemical test, OC-Hemodia (Eiken Chemical Co., LTD, Tokyo, Japan). A small sample of a single stool was placed in buffer and read by machine. At a cutoff of 200 μg/mL, the sensitivity was 77.5% and the specificity was 98.9%. At a cutoff of 50 ng/mL, the sensitivity was 86.5% and the specificity was 94.9%. In this study, positive tests were followed by colonoscopy, but false-negative tests were identified from insurance claims.
Cole et al20 assessed the rates of participation in colorectal cancer screening in a study in Australia. Participants were randomized and received by mail either Hemoccult Sensa or one of two fecal immunochemical tests, Flex-Sure OBT (Beckman Coulter, Fullerton, CA) or InSure. The Hemoccult Sensa group was instructed to follow dietary and medication restrictions during stool collection, while the immunochemical test groups were not. Three stool specimens were required for the Hemoccult Sensa and FlexSure tests, while two stools were required for InSure.
The participation rate was 23.4% in the Hemoccult Sensa group, 30.5% in the Flex-Sure OBT group, and 39.6% in the InSure group (P < .001).
Hol et al21 found that the participation rate was 50% in a group asked to undergo guaiac testing requiring three samples without diet restriction and 62% in a group asked to undergo fecal immunochemical testing (OC-Sensor) requiring a single stool sample without restrictions. Higher participation rates are seen with fecal immunochemical testing than with guaiac testing and are an advantage of immunochemical testing.
CLEVELAND CLINIC SWITCHES TO FECAL IMMUNOCHEMICAL TESTING FOR COLORECTAL CANCER SCREENING
Cleveland Clinic recently switched to fecal immunochemical testing in place of Hemoccult Sensa for colorectal cancer screening. The data on fecal occult blood tests show that the sensitivities of Hemoccult Sensa and the immunochemical tests are higher than those of Hemoccult and Hemoccult II for the detection of colorectal cancer and advanced adenomas, with similar specificity. Fecal immunochemical tests have an advantage over guaiac-based tests in most screening studies by showing a superior sensitivity for advanced adenomas and colorectal cancer, as well as an increase in test adherence, likely because of the lack of dietary and medication restrictions and the lower number of stool samples required. Increased compliance should improve participation in colorectal cancer screening and positively affect colorectal cancer mortality rates.
- Edwards BK, Ward E, Kohler BA, et al. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer 2010; 116:544–573.
- Centers for Disease Control and Prevention (CDC). Vital signs: colorectal cancer screening among adults aged 50–75 years—United States, 2008. MMWR Morb Mortal Wkly Rep 2010; 59:808–812.
- Mandel JS, Church TR, Bond JH, et al. The effect of fecal occult-blood screening on the incidence of colorectal cancer. N Engl J Med 2000; 343:1603–1607.
- Levin B, Lieberman DA, McFarland B, et al; American Cancer Society Colorectal Cancer Advisory Group; US Multi-Society Task Force; American College of Radiology Colon Cancer Committee. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. Gastroenterology 2008; 134:1570–1595.
- US Preventive Services Task Force. Screening for colorectal cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 2008; 149:627–637.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 [corrected]. Am J Gastroenterol 2009; 104:739–750.
- Vu HT, Burke CA. Advances in colorectal cancer screening. Curr Gastroenterol Rep 2009; 11:406–412.
- Hardcastle JD, Chamberlain JO, Robinson MH, et al. Randomised controlled trial of faecal-occult-blood screening for colorectal cancer. Lancet 1996; 348:1472–1477.
- Kronborg O, Fenger C, Olsen J, Jørgensen OD, Søndergaard O. Randomised study of screening for colorectal cancer with faecal-occult-blood test. Lancet 1996; 348:1467–1471.
- Mandel JS, Bond JH, Church TR, et al. Reducing mortality from colorectal cancer by screening for fecal occult blood. Minnesota Colon Cancer Control Study. N Engl J Med 1993; 328:1365–1371.
- Kewenter J, Brevinge H, Engarås B, Haglind E, Ahrén C. Results of screening, rescreening, and follow-up in a prospective randomized study for detection of colorectal cancer by fecal occult blood testing. Results for 68,308 subjects. Scand J Gastroenterol 1994; 29:468–473.
- Hewitson P, Glasziou P, Irwig L, Towler B, Watson E. Screening for colorectal cancer using the faecal occult blood test, Hemoccult. Cochrane Database Syst Rev 2007;CD001216.
- Allison JE, Tekawa IS, Ransom LJ, Adrain AL. A comparison of fecal occult-blood tests for colorectal-cancer screening. N Engl J Med 1996; 334:155–159.
- Smith A, Young GP, Cole SR, Bampton P. Comparison of a brush-sampling fecal immunochemical test for hemoglobin with a sensitive guaiac-based fecal occult blood test in detection of colorectal neoplasia. Cancer 2006; 107:2152–2159.
- Guittet L, Bouvier V, Mariotte N, et al. Comparison of a guaiac based and an immunochemical faecal occult blood test in screening for colorectal cancer in a general average risk population. Gut 2007; 56:210–214.
- van Rossum LG, van Rijn AF, Laheij RJ, et al. Random comparison of guaiac and immunochemical fecal occult blood tests for colorectal cancer in a screening population. Gastroenterology 2008; 135:82–90.
- Park DI, Ryu S, Kim YH, et al. Comparison of guaiac-based and quantitative immunochemical fecal occult blood testing in a population at average risk undergoing colorectal cancer screening. Am J Gastroenterol 2010; 105:2017–2025.
- Hundt S, Haug U, Brenner H. Comparative evaluation of immunochemical fecal occult blood tests for colorectal adenoma detection. Ann Intern Med 2009; 150:162–169.
- Itoh M, Takahashi K, Nishida H, Sakagami K, Okubo T. Estimation of the optimal cut off point in a new immunological faecal occult blood test in a corporate colorectal cancer screening programme. J Med Screen 1996; 3:66–71.
- Cole SR, Young GP, Esterman A, Cadd B, Morcom J. A randomised trial of the impact of new faecal haemoglobin test technologies on population participation in screening for colorectal cancer. J Med Screen 2003; 10:117–122.
- Hol L, Wilschut JA, van Ballegooijen M, et al. Screening for colorectal cancer: random comparison of guaiac and immunochemical faecal occult blood testing at different cut-off levels. Br J Cancer 2009; 100:1103–1110.
- Edwards BK, Ward E, Kohler BA, et al. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer 2010; 116:544–573.
- Centers for Disease Control and Prevention (CDC). Vital signs: colorectal cancer screening among adults aged 50–75 years—United States, 2008. MMWR Morb Mortal Wkly Rep 2010; 59:808–812.
- Mandel JS, Church TR, Bond JH, et al. The effect of fecal occult-blood screening on the incidence of colorectal cancer. N Engl J Med 2000; 343:1603–1607.
- Levin B, Lieberman DA, McFarland B, et al; American Cancer Society Colorectal Cancer Advisory Group; US Multi-Society Task Force; American College of Radiology Colon Cancer Committee. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. Gastroenterology 2008; 134:1570–1595.
- US Preventive Services Task Force. Screening for colorectal cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 2008; 149:627–637.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 [corrected]. Am J Gastroenterol 2009; 104:739–750.
- Vu HT, Burke CA. Advances in colorectal cancer screening. Curr Gastroenterol Rep 2009; 11:406–412.
- Hardcastle JD, Chamberlain JO, Robinson MH, et al. Randomised controlled trial of faecal-occult-blood screening for colorectal cancer. Lancet 1996; 348:1472–1477.
- Kronborg O, Fenger C, Olsen J, Jørgensen OD, Søndergaard O. Randomised study of screening for colorectal cancer with faecal-occult-blood test. Lancet 1996; 348:1467–1471.
- Mandel JS, Bond JH, Church TR, et al. Reducing mortality from colorectal cancer by screening for fecal occult blood. Minnesota Colon Cancer Control Study. N Engl J Med 1993; 328:1365–1371.
- Kewenter J, Brevinge H, Engarås B, Haglind E, Ahrén C. Results of screening, rescreening, and follow-up in a prospective randomized study for detection of colorectal cancer by fecal occult blood testing. Results for 68,308 subjects. Scand J Gastroenterol 1994; 29:468–473.
- Hewitson P, Glasziou P, Irwig L, Towler B, Watson E. Screening for colorectal cancer using the faecal occult blood test, Hemoccult. Cochrane Database Syst Rev 2007;CD001216.
- Allison JE, Tekawa IS, Ransom LJ, Adrain AL. A comparison of fecal occult-blood tests for colorectal-cancer screening. N Engl J Med 1996; 334:155–159.
- Smith A, Young GP, Cole SR, Bampton P. Comparison of a brush-sampling fecal immunochemical test for hemoglobin with a sensitive guaiac-based fecal occult blood test in detection of colorectal neoplasia. Cancer 2006; 107:2152–2159.
- Guittet L, Bouvier V, Mariotte N, et al. Comparison of a guaiac based and an immunochemical faecal occult blood test in screening for colorectal cancer in a general average risk population. Gut 2007; 56:210–214.
- van Rossum LG, van Rijn AF, Laheij RJ, et al. Random comparison of guaiac and immunochemical fecal occult blood tests for colorectal cancer in a screening population. Gastroenterology 2008; 135:82–90.
- Park DI, Ryu S, Kim YH, et al. Comparison of guaiac-based and quantitative immunochemical fecal occult blood testing in a population at average risk undergoing colorectal cancer screening. Am J Gastroenterol 2010; 105:2017–2025.
- Hundt S, Haug U, Brenner H. Comparative evaluation of immunochemical fecal occult blood tests for colorectal adenoma detection. Ann Intern Med 2009; 150:162–169.
- Itoh M, Takahashi K, Nishida H, Sakagami K, Okubo T. Estimation of the optimal cut off point in a new immunological faecal occult blood test in a corporate colorectal cancer screening programme. J Med Screen 1996; 3:66–71.
- Cole SR, Young GP, Esterman A, Cadd B, Morcom J. A randomised trial of the impact of new faecal haemoglobin test technologies on population participation in screening for colorectal cancer. J Med Screen 2003; 10:117–122.
- Hol L, Wilschut JA, van Ballegooijen M, et al. Screening for colorectal cancer: random comparison of guaiac and immunochemical faecal occult blood testing at different cut-off levels. Br J Cancer 2009; 100:1103–1110.
KEY POINTS
- Hemoccult Sensa and several fecal immunochemical tests are more sensitive than Hemoccult and Hemoccult II for detecting colorectal cancer and advanced adenomas, with similar specificity.
- In most screening studies, fecal immunochemical tests have been more sensitive than guaiac-based tests. In addition, rates of adherence were higher, likely because dietary and medication restrictions are not needed and fewer stool samples are required.
- Better compliance should improve participation in colorectal cancer screening and reduce colorectal cancer mortality rates.