User login
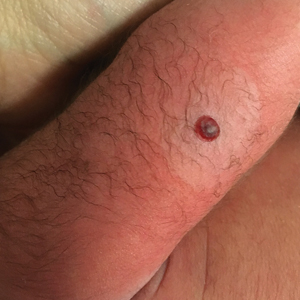
Hemorrhagic Crusted Papule on the Arm
The Diagnosis: Self-healing Langerhans Cell Histiocytosis
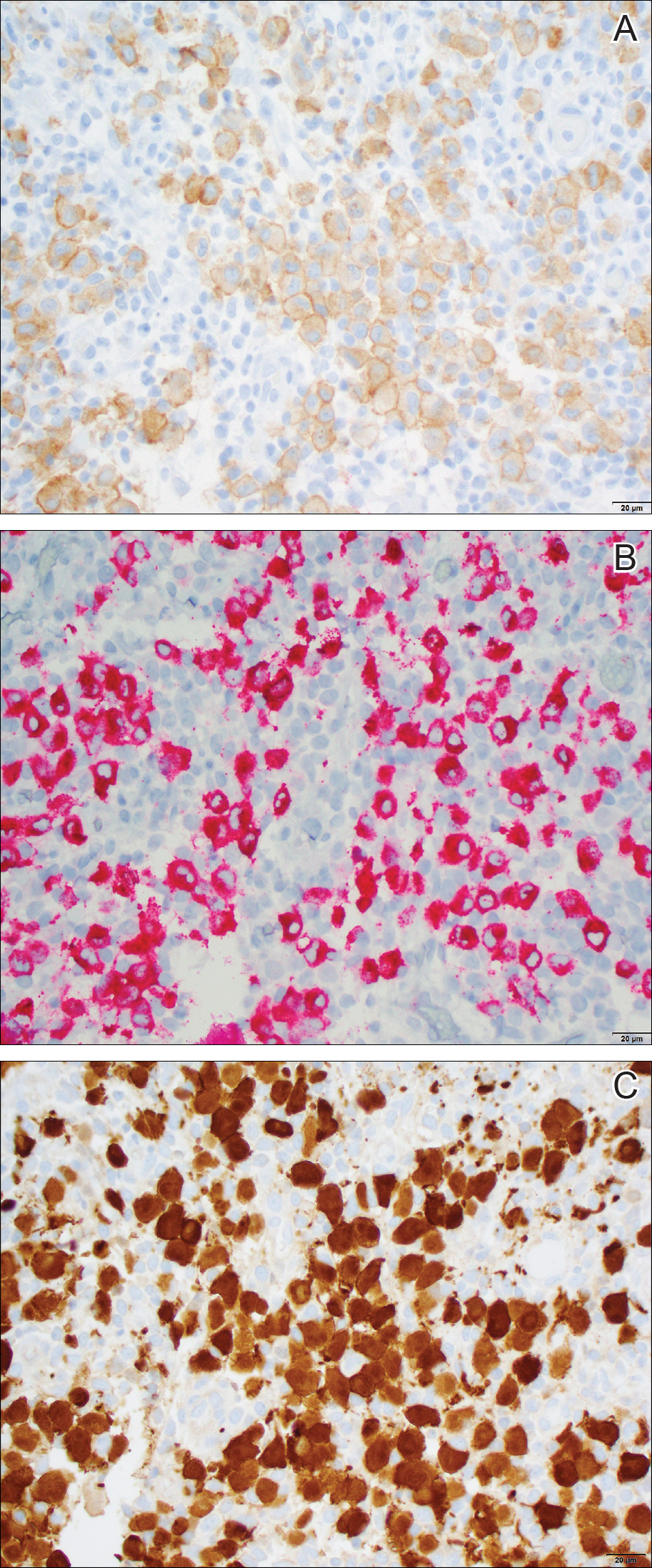
Histopathologic examination showed an infiltrate of mononuclear cells with indented nuclei admixed with a variable dermal inflammatory infiltrate. Immunohistochemistry demonstrated cells that were strongly positive for CD1a (Figure, A) and langerin (Figure, B) antigens as well as S-100 protein (Figure, C), which was consistent with Langerhans cell histiocytosis (LCH).
Histiocytoses are a heterogeneous group of disorders in which the infiltrating cells belong to the mononuclear phagocyte system.1,2 Langerhans cell histiocytosis is the most common dendritic cell-related histiocytosis, occurring in approximately 5 per 1 million children annually, giving it an incidence comparable to pediatric Hodgkin lymphoma and acute myeloid leukemia.1,2
Historically, there has been much debate about the pathogenesis of the disease.2 Until recently it was unknown whether LCH was primarily a neoplastic or an inflammatory disorder. Although the condition initially was thought to have a reactive etiology,1 more recent evidence suggests a clonal neoplastic process. Langerhans cell histiocytosis lesions are clonal and display malignancy-associated mechanisms such as immune evasion. Genome sequencing has revealed several mutations in precursor myeloid cells that result in the common downstream hyperactivation of the mitogen-activated protein kinase signaling pathway that regulates cell proliferation and differentiation.1
Langerhans cell histiocytosis displays a wide spectrum of clinical phenotypes, which historically were subclassified as eosinophilic granulomas (localized lesions in bone), Hand-Schüller-Christian disease (multiple organ involvement with the classic triad of skull defects, diabetes insipidus, and exophthalmos), and Letterer-Siwe disease (visceral lesions involving multiple organs).3 However, in 1997 the Reclassification Working Group of the Histiocyte Society redefined LCH as single-system single site (SS-s) LCH, single-system multisite LCH, and multisystem LCH.4
In SS-s LCH, the most common site is bone (82%), followed by the skin (12%).5 Skin SS-s LCH classically presents as multiple skin lesions at birth without systemic manifestations; the lesions spontaneously involute within a few months.6 Less commonly, skin SS-s LCH can present as a single lesion. Berger et al7 described 4 neonates with unilesional skin SS-s LCH. Since then, more than 30 cases have been reported in the literature,8 and we report herein another unilesional self-healing LCH.
The morphology of skin lesions in self-healing LCH is highly variable, with the most common being multiple erythematous crusted papules (50%), followed by eczematous scaly lesions resembling seborrheic dermatitis in intertriginous areas (37.5%).3,6 Unilesional self-healing LCH typically presents as an ulcerated or crusted nodule or papule on the trunk. This variability results in a large differential diagnosis. Self-healing LCH is easily mistaken for infectious processes including neonatal herpes simplex and varicella-zoster virus infection.9 Often, the dermatology department is consulted to rule out LCH when the asymptomatic neonate does not respond to parenteral acyclovir.
Less commonly, the magenta-colored papulonodules of self-healing LCH can mimic blueberry muffin rash and mandate a workup for intrauterine infections, especially cytomegalovirus, rubella, and blood dyscrasia.10 Other noninfectious processes in the differential of self-healing LCH include congenital infantile hemangioma, neonatal lupus erythematosus, seborrheic dermatitis (cradle cap), pyogenic granuloma, and psoriasis.3,10 Definitive diagnosis requires histopathology.
Because unilesional self-healing LCH has an excellent prognosis and usually resolves on its own, therapy is unnecessary.3,8 One large retrospective study (N=146) found that of all patients with skin lesions, 56% were managed with biopsy only.5 Other options include watchful waiting and topical corticosteroids. If the skin lesions are large, ulcerated, and/or painful, alkylating antitumor agents have been used. For extensive cutaneous disease, systemic corticosteroids combined with chemotherapy and psoralen plus UVA can be effective.6
The primary concern in the management of self-healing LCH is that the solitary skin lesion may be the harbinger of an aggressive disorder that can progress to systemic disease.5 Moreover, recurrent visceral or disseminated disease may occur months to years after resolution of solitary skin lesions.9 Studies have shown that localized and disseminated disease cannot be differentiated on the basis of clinical findings, histology, immunohistochemistry, or biomarkers.3,11 As a result, an evaluation for systemic disease should be performed at the time of diagnosis for cutaneous LCH.3,9 Minimum baseline studies recommended by the Writing Group of the Histiocyte Society include a complete blood cell count, liver function tests, coagulation studies, chest radiography, skeletal surveys, and urine osmolality testing.12 Periodic clinical follow-up is recommended for all variants of LCH.9
Our case was diagnosed as self-healing LCH based on histologic findings. No treatment was required, and at 3-month follow-up the infant was asymptomatic without recurrence and was meeting all developmental milestones.
- Berres ML, Merad M, Allen CE. Progress in understanding the pathogenesis of Langerhans cell histiocytosis: back to histiocytosis X? Br J Haematol. 2015;169:3-13.
- Jordan MB, Filipovich AH. Histiocytic disorders. In: Hoffman R, Benz EJ Jr, Silberstein LE, eds. Hematology: Basic Principles and Practice. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:686-700.
- Stein SL, Paller AS, Haut PR, et al. Langerhans cell histiocytosis presenting in the neonatal period: a retrospective case series. Arch Pediatr Adolesc Med. 2001;155:778-783.
- Favara BE, Feller AC, Pauli M, et al. Contemporary classification of histiocytic disorders. Pediatr Blood Cancer. 1997;29:157-166.
- Morimoto A, Ishida Y, Suzuki N, et al. Nationwide survey of single-system single site Langerhans cell histiocytosis in Japan. Pediatr Blood Cancer. 2010;54:98-102.
- Morren MA, Broecke KV, Vangeebergen L, et al. Diverse cutaneous presentations of Langerhans cell histiocytosis in children: a retrospective cohort study. Pediatr Blood Cancer. 2016;63:486-492.
- Berger TG, Lane AT, Headington JT, et al. A solitary variant of congenital self-healing reticulohistiocytosis: solitary Hashimoto-Pritzker disease. Pediatr Dermatol. 1986;3:230.
- Wheller L, Carman N, Butler G. Unilesional self-limited Langerhans cell histiocytosis: a case report and review of the literature. J Cutan Pathol. 2013;40:595-599.
- Battistella M, Fraitag S, Teillac DH, et al. Neonatal and early infantile cutaneous Langerhans cell histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol. 2010;146:149-156.
- Mehta V, Balachandran C, Lonikar V. Blueberry muffin baby: a pictoral differential diagnosis. Dermatol Online J. 2008;14:8.
- Kapur P, Erickson C, Rakheja D, et al. Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease): ten-year experience at Dallas Children's Medical Center. J Am Acad Dermatol. 2007;56:290-294.
- Writing Group of the Histiocyte Society. Histiocytosis syndromes in children. Lancet. 1987;24:208-209.
The Diagnosis: Self-healing Langerhans Cell Histiocytosis
Histopathologic examination showed an infiltrate of mononuclear cells with indented nuclei admixed with a variable dermal inflammatory infiltrate. Immunohistochemistry demonstrated cells that were strongly positive for CD1a (Figure, A) and langerin (Figure, B) antigens as well as S-100 protein (Figure, C), which was consistent with Langerhans cell histiocytosis (LCH).
Histiocytoses are a heterogeneous group of disorders in which the infiltrating cells belong to the mononuclear phagocyte system.1,2 Langerhans cell histiocytosis is the most common dendritic cell-related histiocytosis, occurring in approximately 5 per 1 million children annually, giving it an incidence comparable to pediatric Hodgkin lymphoma and acute myeloid leukemia.1,2
Historically, there has been much debate about the pathogenesis of the disease.2 Until recently it was unknown whether LCH was primarily a neoplastic or an inflammatory disorder. Although the condition initially was thought to have a reactive etiology,1 more recent evidence suggests a clonal neoplastic process. Langerhans cell histiocytosis lesions are clonal and display malignancy-associated mechanisms such as immune evasion. Genome sequencing has revealed several mutations in precursor myeloid cells that result in the common downstream hyperactivation of the mitogen-activated protein kinase signaling pathway that regulates cell proliferation and differentiation.1
Langerhans cell histiocytosis displays a wide spectrum of clinical phenotypes, which historically were subclassified as eosinophilic granulomas (localized lesions in bone), Hand-Schüller-Christian disease (multiple organ involvement with the classic triad of skull defects, diabetes insipidus, and exophthalmos), and Letterer-Siwe disease (visceral lesions involving multiple organs).3 However, in 1997 the Reclassification Working Group of the Histiocyte Society redefined LCH as single-system single site (SS-s) LCH, single-system multisite LCH, and multisystem LCH.4
In SS-s LCH, the most common site is bone (82%), followed by the skin (12%).5 Skin SS-s LCH classically presents as multiple skin lesions at birth without systemic manifestations; the lesions spontaneously involute within a few months.6 Less commonly, skin SS-s LCH can present as a single lesion. Berger et al7 described 4 neonates with unilesional skin SS-s LCH. Since then, more than 30 cases have been reported in the literature,8 and we report herein another unilesional self-healing LCH.
The morphology of skin lesions in self-healing LCH is highly variable, with the most common being multiple erythematous crusted papules (50%), followed by eczematous scaly lesions resembling seborrheic dermatitis in intertriginous areas (37.5%).3,6 Unilesional self-healing LCH typically presents as an ulcerated or crusted nodule or papule on the trunk. This variability results in a large differential diagnosis. Self-healing LCH is easily mistaken for infectious processes including neonatal herpes simplex and varicella-zoster virus infection.9 Often, the dermatology department is consulted to rule out LCH when the asymptomatic neonate does not respond to parenteral acyclovir.
Less commonly, the magenta-colored papulonodules of self-healing LCH can mimic blueberry muffin rash and mandate a workup for intrauterine infections, especially cytomegalovirus, rubella, and blood dyscrasia.10 Other noninfectious processes in the differential of self-healing LCH include congenital infantile hemangioma, neonatal lupus erythematosus, seborrheic dermatitis (cradle cap), pyogenic granuloma, and psoriasis.3,10 Definitive diagnosis requires histopathology.
Because unilesional self-healing LCH has an excellent prognosis and usually resolves on its own, therapy is unnecessary.3,8 One large retrospective study (N=146) found that of all patients with skin lesions, 56% were managed with biopsy only.5 Other options include watchful waiting and topical corticosteroids. If the skin lesions are large, ulcerated, and/or painful, alkylating antitumor agents have been used. For extensive cutaneous disease, systemic corticosteroids combined with chemotherapy and psoralen plus UVA can be effective.6
The primary concern in the management of self-healing LCH is that the solitary skin lesion may be the harbinger of an aggressive disorder that can progress to systemic disease.5 Moreover, recurrent visceral or disseminated disease may occur months to years after resolution of solitary skin lesions.9 Studies have shown that localized and disseminated disease cannot be differentiated on the basis of clinical findings, histology, immunohistochemistry, or biomarkers.3,11 As a result, an evaluation for systemic disease should be performed at the time of diagnosis for cutaneous LCH.3,9 Minimum baseline studies recommended by the Writing Group of the Histiocyte Society include a complete blood cell count, liver function tests, coagulation studies, chest radiography, skeletal surveys, and urine osmolality testing.12 Periodic clinical follow-up is recommended for all variants of LCH.9
Our case was diagnosed as self-healing LCH based on histologic findings. No treatment was required, and at 3-month follow-up the infant was asymptomatic without recurrence and was meeting all developmental milestones.
The Diagnosis: Self-healing Langerhans Cell Histiocytosis
Histopathologic examination showed an infiltrate of mononuclear cells with indented nuclei admixed with a variable dermal inflammatory infiltrate. Immunohistochemistry demonstrated cells that were strongly positive for CD1a (Figure, A) and langerin (Figure, B) antigens as well as S-100 protein (Figure, C), which was consistent with Langerhans cell histiocytosis (LCH).
Histiocytoses are a heterogeneous group of disorders in which the infiltrating cells belong to the mononuclear phagocyte system.1,2 Langerhans cell histiocytosis is the most common dendritic cell-related histiocytosis, occurring in approximately 5 per 1 million children annually, giving it an incidence comparable to pediatric Hodgkin lymphoma and acute myeloid leukemia.1,2
Historically, there has been much debate about the pathogenesis of the disease.2 Until recently it was unknown whether LCH was primarily a neoplastic or an inflammatory disorder. Although the condition initially was thought to have a reactive etiology,1 more recent evidence suggests a clonal neoplastic process. Langerhans cell histiocytosis lesions are clonal and display malignancy-associated mechanisms such as immune evasion. Genome sequencing has revealed several mutations in precursor myeloid cells that result in the common downstream hyperactivation of the mitogen-activated protein kinase signaling pathway that regulates cell proliferation and differentiation.1
Langerhans cell histiocytosis displays a wide spectrum of clinical phenotypes, which historically were subclassified as eosinophilic granulomas (localized lesions in bone), Hand-Schüller-Christian disease (multiple organ involvement with the classic triad of skull defects, diabetes insipidus, and exophthalmos), and Letterer-Siwe disease (visceral lesions involving multiple organs).3 However, in 1997 the Reclassification Working Group of the Histiocyte Society redefined LCH as single-system single site (SS-s) LCH, single-system multisite LCH, and multisystem LCH.4
In SS-s LCH, the most common site is bone (82%), followed by the skin (12%).5 Skin SS-s LCH classically presents as multiple skin lesions at birth without systemic manifestations; the lesions spontaneously involute within a few months.6 Less commonly, skin SS-s LCH can present as a single lesion. Berger et al7 described 4 neonates with unilesional skin SS-s LCH. Since then, more than 30 cases have been reported in the literature,8 and we report herein another unilesional self-healing LCH.
The morphology of skin lesions in self-healing LCH is highly variable, with the most common being multiple erythematous crusted papules (50%), followed by eczematous scaly lesions resembling seborrheic dermatitis in intertriginous areas (37.5%).3,6 Unilesional self-healing LCH typically presents as an ulcerated or crusted nodule or papule on the trunk. This variability results in a large differential diagnosis. Self-healing LCH is easily mistaken for infectious processes including neonatal herpes simplex and varicella-zoster virus infection.9 Often, the dermatology department is consulted to rule out LCH when the asymptomatic neonate does not respond to parenteral acyclovir.
Less commonly, the magenta-colored papulonodules of self-healing LCH can mimic blueberry muffin rash and mandate a workup for intrauterine infections, especially cytomegalovirus, rubella, and blood dyscrasia.10 Other noninfectious processes in the differential of self-healing LCH include congenital infantile hemangioma, neonatal lupus erythematosus, seborrheic dermatitis (cradle cap), pyogenic granuloma, and psoriasis.3,10 Definitive diagnosis requires histopathology.
Because unilesional self-healing LCH has an excellent prognosis and usually resolves on its own, therapy is unnecessary.3,8 One large retrospective study (N=146) found that of all patients with skin lesions, 56% were managed with biopsy only.5 Other options include watchful waiting and topical corticosteroids. If the skin lesions are large, ulcerated, and/or painful, alkylating antitumor agents have been used. For extensive cutaneous disease, systemic corticosteroids combined with chemotherapy and psoralen plus UVA can be effective.6
The primary concern in the management of self-healing LCH is that the solitary skin lesion may be the harbinger of an aggressive disorder that can progress to systemic disease.5 Moreover, recurrent visceral or disseminated disease may occur months to years after resolution of solitary skin lesions.9 Studies have shown that localized and disseminated disease cannot be differentiated on the basis of clinical findings, histology, immunohistochemistry, or biomarkers.3,11 As a result, an evaluation for systemic disease should be performed at the time of diagnosis for cutaneous LCH.3,9 Minimum baseline studies recommended by the Writing Group of the Histiocyte Society include a complete blood cell count, liver function tests, coagulation studies, chest radiography, skeletal surveys, and urine osmolality testing.12 Periodic clinical follow-up is recommended for all variants of LCH.9
Our case was diagnosed as self-healing LCH based on histologic findings. No treatment was required, and at 3-month follow-up the infant was asymptomatic without recurrence and was meeting all developmental milestones.
- Berres ML, Merad M, Allen CE. Progress in understanding the pathogenesis of Langerhans cell histiocytosis: back to histiocytosis X? Br J Haematol. 2015;169:3-13.
- Jordan MB, Filipovich AH. Histiocytic disorders. In: Hoffman R, Benz EJ Jr, Silberstein LE, eds. Hematology: Basic Principles and Practice. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:686-700.
- Stein SL, Paller AS, Haut PR, et al. Langerhans cell histiocytosis presenting in the neonatal period: a retrospective case series. Arch Pediatr Adolesc Med. 2001;155:778-783.
- Favara BE, Feller AC, Pauli M, et al. Contemporary classification of histiocytic disorders. Pediatr Blood Cancer. 1997;29:157-166.
- Morimoto A, Ishida Y, Suzuki N, et al. Nationwide survey of single-system single site Langerhans cell histiocytosis in Japan. Pediatr Blood Cancer. 2010;54:98-102.
- Morren MA, Broecke KV, Vangeebergen L, et al. Diverse cutaneous presentations of Langerhans cell histiocytosis in children: a retrospective cohort study. Pediatr Blood Cancer. 2016;63:486-492.
- Berger TG, Lane AT, Headington JT, et al. A solitary variant of congenital self-healing reticulohistiocytosis: solitary Hashimoto-Pritzker disease. Pediatr Dermatol. 1986;3:230.
- Wheller L, Carman N, Butler G. Unilesional self-limited Langerhans cell histiocytosis: a case report and review of the literature. J Cutan Pathol. 2013;40:595-599.
- Battistella M, Fraitag S, Teillac DH, et al. Neonatal and early infantile cutaneous Langerhans cell histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol. 2010;146:149-156.
- Mehta V, Balachandran C, Lonikar V. Blueberry muffin baby: a pictoral differential diagnosis. Dermatol Online J. 2008;14:8.
- Kapur P, Erickson C, Rakheja D, et al. Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease): ten-year experience at Dallas Children's Medical Center. J Am Acad Dermatol. 2007;56:290-294.
- Writing Group of the Histiocyte Society. Histiocytosis syndromes in children. Lancet. 1987;24:208-209.
- Berres ML, Merad M, Allen CE. Progress in understanding the pathogenesis of Langerhans cell histiocytosis: back to histiocytosis X? Br J Haematol. 2015;169:3-13.
- Jordan MB, Filipovich AH. Histiocytic disorders. In: Hoffman R, Benz EJ Jr, Silberstein LE, eds. Hematology: Basic Principles and Practice. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:686-700.
- Stein SL, Paller AS, Haut PR, et al. Langerhans cell histiocytosis presenting in the neonatal period: a retrospective case series. Arch Pediatr Adolesc Med. 2001;155:778-783.
- Favara BE, Feller AC, Pauli M, et al. Contemporary classification of histiocytic disorders. Pediatr Blood Cancer. 1997;29:157-166.
- Morimoto A, Ishida Y, Suzuki N, et al. Nationwide survey of single-system single site Langerhans cell histiocytosis in Japan. Pediatr Blood Cancer. 2010;54:98-102.
- Morren MA, Broecke KV, Vangeebergen L, et al. Diverse cutaneous presentations of Langerhans cell histiocytosis in children: a retrospective cohort study. Pediatr Blood Cancer. 2016;63:486-492.
- Berger TG, Lane AT, Headington JT, et al. A solitary variant of congenital self-healing reticulohistiocytosis: solitary Hashimoto-Pritzker disease. Pediatr Dermatol. 1986;3:230.
- Wheller L, Carman N, Butler G. Unilesional self-limited Langerhans cell histiocytosis: a case report and review of the literature. J Cutan Pathol. 2013;40:595-599.
- Battistella M, Fraitag S, Teillac DH, et al. Neonatal and early infantile cutaneous Langerhans cell histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol. 2010;146:149-156.
- Mehta V, Balachandran C, Lonikar V. Blueberry muffin baby: a pictoral differential diagnosis. Dermatol Online J. 2008;14:8.
- Kapur P, Erickson C, Rakheja D, et al. Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease): ten-year experience at Dallas Children's Medical Center. J Am Acad Dermatol. 2007;56:290-294.
- Writing Group of the Histiocyte Society. Histiocytosis syndromes in children. Lancet. 1987;24:208-209.
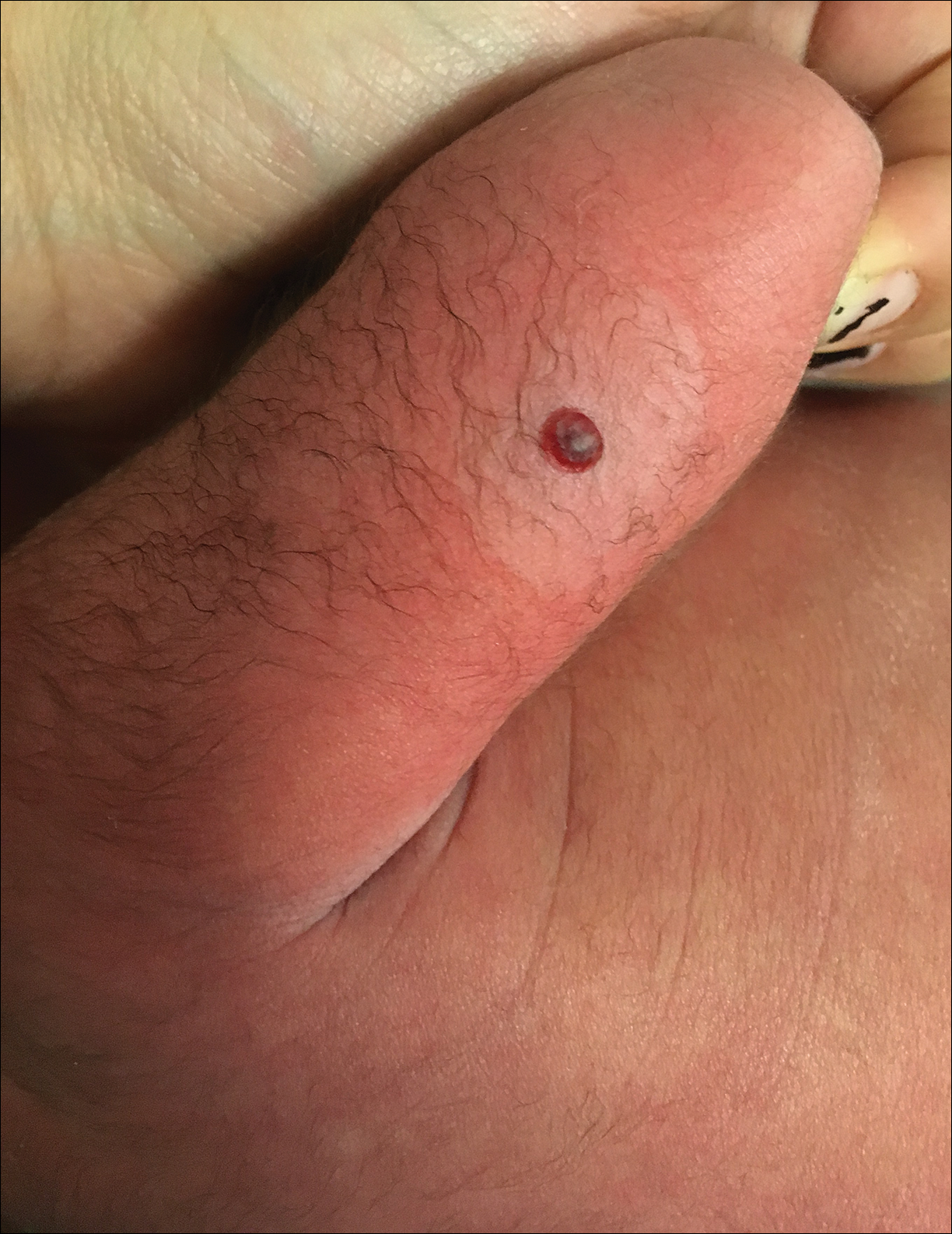
Dermatology consultation was called to the delivery room to evaluate a red, hemorrhagic, crusted, 5-mm papule on the right lateral upper arm of a preterm newborn. He appeared vigorous with an Apgar score of 7 at 1 minute and 8 at 5 minutes. Physical examination was otherwise normal. Of note, the mother presented late to prenatal care. Her herpes simplex and varicella-zoster virus status was unknown. A shave biopsy of the papule was performed at 3 days of age.