User login
Telehealth finds acceptance among patients with CF, clinicians
(CF) and the physicians who treat them, according to three new studies. The surveys examined attitudes during the COVID-19 pandemic, which complicates interpretation of the survey, but the results nevertheless bode well for telehealth’s future in the management of CF.

“Patients could be responding positively just because they could have a visit during the pandemic,” said Andrew NeSmith, during a presentation of a survey of adults with CF at the virtual North American Cystic Fibrosis Conference. Mr. NeSmith is the clinical data coordinator at the University of Alabama at Birmingham Cystic Fibrosis Center.
Other posters at the conference examined attitudes among pediatric populations and treating physicians, with generally positive results, which has generated optimism that telehealth could become an important element of care after the pandemic fades. “This data suggests that telehealth could be integrated into routine follow-up care in the CF chronic care model,” said Mr. NeSmith.
His team collected responses from 119 individuals at the University of Alabama at Birmingham; Boston Children’s Hospital; Brigham and Women’s Hospital, Boston; Virginia Commonwealth University, Richmond; and West Virginia University, Morgantown. A total of 28% had conducted a prior telehealth visit before the study; 92% of visits were conducted with a medical doctor. Only 13% reported experiencing difficulties with their first telehealth visit. Eighty-five percent rated convenience, and 77% rated their satisfaction with telehealth as “high.” Most (92%) said they were able to see their desired disciplines, 95% felt all of their issues had been addressed, and 83% strongly agreed that telehealth visits were of adequate length.
Not everything was rosy. A total of 48% of participants expressed at least moderate concern over a lack of pulmonary function test or throat/sputum culture. There were much fewer concerns over missing vital signs or weight measurements.
The overall results weren’t surprising to Robert Giusti, MD, clinical professor of pediatrics at New York University and director of the Pediatric Cystic Fibrosis Center, New York, who was not involved in the study. “I was expecting that patients were going to like it. It makes their life easier,” he said in an interview.
A survey of families of pediatric individuals with CF at seven centers found similar levels of satisfaction. A total of 23% had used telehealth previously; 96% rated convenience, and 93% rated satisfaction as “high.” Almost all (99%) felt that all concerns were met, 98% said that sessions were adequately long, and 87% had no trouble connecting to the visit.
Some participants in this survey had concerns about what might be missing with a televisit. Half (52%) had at least a moderate concern over lack of pulmonary function tests, 45% over lack of vital signs, 29% about lack of weight measurements, and 64% about the need for throat/sputum culture. Despite those issues, 69% preferred that “some” and 22% preferred that “most” future visits be conducted by telehealth.
A survey of physicians who used telehealth with CF patients also found broad support. They reported some challenges, with 70% saying they experienced technical difficulty, and 77% saying it “took time” to resolve a visit with only 18% reporting that visits were “quickly resolved.” Most (86%) said they were satisfied with telehealth for care delivery, and 78% said it was appropriate for most patients. Most said telehealth improved the patient-physician relationship, and they believed visits were more efficient when conducted via telehealth than in person. A majority (81%) endorsed using telehealth for some visits, and 12% for most visits.
A key question will be how telehealth affects patient outcomes, according to Ryan Perkins, MD, who was a coauthor of the survey of physicians. “If they’re not doing as well from an outcomes perspective, that would be a huge limitation to our patients,” said Dr. Perkins, who is a pediatric and adult pulmonary fellow at Boston Children’s Hospital and Brigham and Women’s Hospital.
Although the study examined only models of care that were entirely virtual, Dr. Perkins noted that hybrid in-person/virtual care models are also possible. “Do we have better outcomes doing it that way? Is there higher patient satisfaction? I’m sure that will be a hot topic moving forward.”
Dr. Perkins noted that patients expressed concern about not being able to get sputum cultures and spirometry recordings during telehealth sessions. “That’s not really surprising to me, but I think it raises the question as we’re imagining care models for the future – how can we implement those components into future care delivery?”
Another hurdle will be insurance coverage. “My fear is that insurance companies are going to cut down the amount of reimbursement for telehealth visits in the future and just going to make it more complicated,” said Dr. Giusti. “Certainly, though, I think telehealth is an important outreach that we’d like to continue with our patients.”
Mr. NeSmith, Dr. Giusti, and Dr. Perkins reported no relevant financial disclosures.
SOURCE: NeSmith A et al. NACFC 2020, Abstracts 797, 799, 810.
(CF) and the physicians who treat them, according to three new studies. The surveys examined attitudes during the COVID-19 pandemic, which complicates interpretation of the survey, but the results nevertheless bode well for telehealth’s future in the management of CF.
“Patients could be responding positively just because they could have a visit during the pandemic,” said Andrew NeSmith, during a presentation of a survey of adults with CF at the virtual North American Cystic Fibrosis Conference. Mr. NeSmith is the clinical data coordinator at the University of Alabama at Birmingham Cystic Fibrosis Center.
Other posters at the conference examined attitudes among pediatric populations and treating physicians, with generally positive results, which has generated optimism that telehealth could become an important element of care after the pandemic fades. “This data suggests that telehealth could be integrated into routine follow-up care in the CF chronic care model,” said Mr. NeSmith.
His team collected responses from 119 individuals at the University of Alabama at Birmingham; Boston Children’s Hospital; Brigham and Women’s Hospital, Boston; Virginia Commonwealth University, Richmond; and West Virginia University, Morgantown. A total of 28% had conducted a prior telehealth visit before the study; 92% of visits were conducted with a medical doctor. Only 13% reported experiencing difficulties with their first telehealth visit. Eighty-five percent rated convenience, and 77% rated their satisfaction with telehealth as “high.” Most (92%) said they were able to see their desired disciplines, 95% felt all of their issues had been addressed, and 83% strongly agreed that telehealth visits were of adequate length.
Not everything was rosy. A total of 48% of participants expressed at least moderate concern over a lack of pulmonary function test or throat/sputum culture. There were much fewer concerns over missing vital signs or weight measurements.
The overall results weren’t surprising to Robert Giusti, MD, clinical professor of pediatrics at New York University and director of the Pediatric Cystic Fibrosis Center, New York, who was not involved in the study. “I was expecting that patients were going to like it. It makes their life easier,” he said in an interview.
A survey of families of pediatric individuals with CF at seven centers found similar levels of satisfaction. A total of 23% had used telehealth previously; 96% rated convenience, and 93% rated satisfaction as “high.” Almost all (99%) felt that all concerns were met, 98% said that sessions were adequately long, and 87% had no trouble connecting to the visit.
Some participants in this survey had concerns about what might be missing with a televisit. Half (52%) had at least a moderate concern over lack of pulmonary function tests, 45% over lack of vital signs, 29% about lack of weight measurements, and 64% about the need for throat/sputum culture. Despite those issues, 69% preferred that “some” and 22% preferred that “most” future visits be conducted by telehealth.
A survey of physicians who used telehealth with CF patients also found broad support. They reported some challenges, with 70% saying they experienced technical difficulty, and 77% saying it “took time” to resolve a visit with only 18% reporting that visits were “quickly resolved.” Most (86%) said they were satisfied with telehealth for care delivery, and 78% said it was appropriate for most patients. Most said telehealth improved the patient-physician relationship, and they believed visits were more efficient when conducted via telehealth than in person. A majority (81%) endorsed using telehealth for some visits, and 12% for most visits.
A key question will be how telehealth affects patient outcomes, according to Ryan Perkins, MD, who was a coauthor of the survey of physicians. “If they’re not doing as well from an outcomes perspective, that would be a huge limitation to our patients,” said Dr. Perkins, who is a pediatric and adult pulmonary fellow at Boston Children’s Hospital and Brigham and Women’s Hospital.
Although the study examined only models of care that were entirely virtual, Dr. Perkins noted that hybrid in-person/virtual care models are also possible. “Do we have better outcomes doing it that way? Is there higher patient satisfaction? I’m sure that will be a hot topic moving forward.”
Dr. Perkins noted that patients expressed concern about not being able to get sputum cultures and spirometry recordings during telehealth sessions. “That’s not really surprising to me, but I think it raises the question as we’re imagining care models for the future – how can we implement those components into future care delivery?”
Another hurdle will be insurance coverage. “My fear is that insurance companies are going to cut down the amount of reimbursement for telehealth visits in the future and just going to make it more complicated,” said Dr. Giusti. “Certainly, though, I think telehealth is an important outreach that we’d like to continue with our patients.”
Mr. NeSmith, Dr. Giusti, and Dr. Perkins reported no relevant financial disclosures.
SOURCE: NeSmith A et al. NACFC 2020, Abstracts 797, 799, 810.
(CF) and the physicians who treat them, according to three new studies. The surveys examined attitudes during the COVID-19 pandemic, which complicates interpretation of the survey, but the results nevertheless bode well for telehealth’s future in the management of CF.
“Patients could be responding positively just because they could have a visit during the pandemic,” said Andrew NeSmith, during a presentation of a survey of adults with CF at the virtual North American Cystic Fibrosis Conference. Mr. NeSmith is the clinical data coordinator at the University of Alabama at Birmingham Cystic Fibrosis Center.
Other posters at the conference examined attitudes among pediatric populations and treating physicians, with generally positive results, which has generated optimism that telehealth could become an important element of care after the pandemic fades. “This data suggests that telehealth could be integrated into routine follow-up care in the CF chronic care model,” said Mr. NeSmith.
His team collected responses from 119 individuals at the University of Alabama at Birmingham; Boston Children’s Hospital; Brigham and Women’s Hospital, Boston; Virginia Commonwealth University, Richmond; and West Virginia University, Morgantown. A total of 28% had conducted a prior telehealth visit before the study; 92% of visits were conducted with a medical doctor. Only 13% reported experiencing difficulties with their first telehealth visit. Eighty-five percent rated convenience, and 77% rated their satisfaction with telehealth as “high.” Most (92%) said they were able to see their desired disciplines, 95% felt all of their issues had been addressed, and 83% strongly agreed that telehealth visits were of adequate length.
Not everything was rosy. A total of 48% of participants expressed at least moderate concern over a lack of pulmonary function test or throat/sputum culture. There were much fewer concerns over missing vital signs or weight measurements.
The overall results weren’t surprising to Robert Giusti, MD, clinical professor of pediatrics at New York University and director of the Pediatric Cystic Fibrosis Center, New York, who was not involved in the study. “I was expecting that patients were going to like it. It makes their life easier,” he said in an interview.
A survey of families of pediatric individuals with CF at seven centers found similar levels of satisfaction. A total of 23% had used telehealth previously; 96% rated convenience, and 93% rated satisfaction as “high.” Almost all (99%) felt that all concerns were met, 98% said that sessions were adequately long, and 87% had no trouble connecting to the visit.
Some participants in this survey had concerns about what might be missing with a televisit. Half (52%) had at least a moderate concern over lack of pulmonary function tests, 45% over lack of vital signs, 29% about lack of weight measurements, and 64% about the need for throat/sputum culture. Despite those issues, 69% preferred that “some” and 22% preferred that “most” future visits be conducted by telehealth.
A survey of physicians who used telehealth with CF patients also found broad support. They reported some challenges, with 70% saying they experienced technical difficulty, and 77% saying it “took time” to resolve a visit with only 18% reporting that visits were “quickly resolved.” Most (86%) said they were satisfied with telehealth for care delivery, and 78% said it was appropriate for most patients. Most said telehealth improved the patient-physician relationship, and they believed visits were more efficient when conducted via telehealth than in person. A majority (81%) endorsed using telehealth for some visits, and 12% for most visits.
A key question will be how telehealth affects patient outcomes, according to Ryan Perkins, MD, who was a coauthor of the survey of physicians. “If they’re not doing as well from an outcomes perspective, that would be a huge limitation to our patients,” said Dr. Perkins, who is a pediatric and adult pulmonary fellow at Boston Children’s Hospital and Brigham and Women’s Hospital.
Although the study examined only models of care that were entirely virtual, Dr. Perkins noted that hybrid in-person/virtual care models are also possible. “Do we have better outcomes doing it that way? Is there higher patient satisfaction? I’m sure that will be a hot topic moving forward.”
Dr. Perkins noted that patients expressed concern about not being able to get sputum cultures and spirometry recordings during telehealth sessions. “That’s not really surprising to me, but I think it raises the question as we’re imagining care models for the future – how can we implement those components into future care delivery?”
Another hurdle will be insurance coverage. “My fear is that insurance companies are going to cut down the amount of reimbursement for telehealth visits in the future and just going to make it more complicated,” said Dr. Giusti. “Certainly, though, I think telehealth is an important outreach that we’d like to continue with our patients.”
Mr. NeSmith, Dr. Giusti, and Dr. Perkins reported no relevant financial disclosures.
SOURCE: NeSmith A et al. NACFC 2020, Abstracts 797, 799, 810.
FROM NACFC 2020
Poverty raises depression risk in patients with cystic fibrosis
Poor people with chronic illness have greater difficulty managing their disease than do their better-off counterparts, and a new study confirms this reality for patients with cystic fibrosis.

and anxiety symptoms, according to a new cross-sectional study. The data were drawn from the Cystic Fibrosis Foundation’s Success with Therapies Research Consortium.
“Assessing the special challenges that individuals with lower SES face, including financial barriers, is essential to understand how we can address the unique combinations of adherence barriers. In other chronic disorders, financial barriers or lower socioeconomic status is associated with nonadherence, but this relationship has not been well established in cystic fibrosis,” said Kimberly Dickinson, MD, MPH, of Johns Hopkins University, Baltimore, during her presentation of the results at the virtual North American Cystic Fibrosis Conference.
“I’ve always thought that my patients in the poorer population were doing worse, and I think this demonstrates that that’s true,” said Robert Giusti, MD, in an interview. Dr. Giusti is a clinical professor of pediatrics at the New York University and director of the Pediatric Cystic Fibrosis Center in New York. He was not involved in the study.
“These are very pertinent issues, especially if you think about the pandemic, and some of the issues related to mental health. It just highlights the importance of socioeconomic status and screening for some of the known risk factors so that we can develop interventions or programs to provide equitable care to all of our cystic fibrosis patients,” said Ryan Perkins, MD, who moderated the session where the study was presented. He is a pediatric and adult pulmonary fellow at Boston Children’s Hospital and Brigham and Women’s Hospital, also in Boston.
The researchers looked retrospectively at 1 year’s worth of pharmacy refill receipts and number of times prescriptions were refilled versus the number of times prescribed, then calculated medicinal possession ratios. This was cross-referenced with annual household income and insurance status of patients with CF at 12 pediatric and 9 adult CF care centers, for a total of 376 patients (128 pediatric and 248 adult).
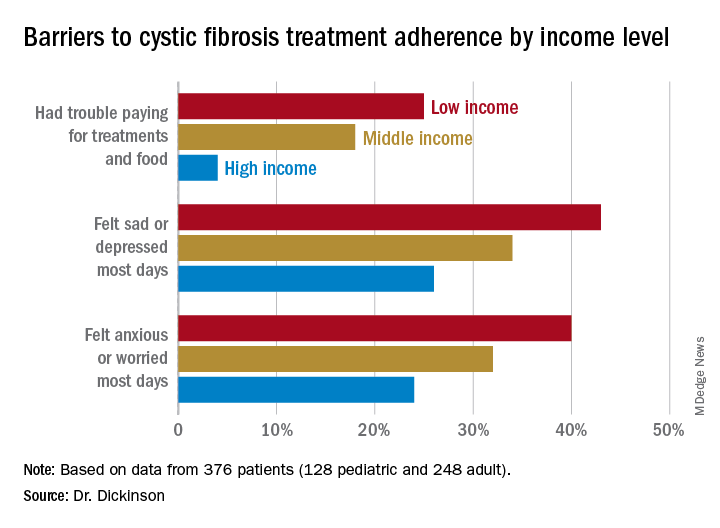
In this population, 32% of participants had public or no insurance, 68% had private or military insurance. The public/no insurance group was more likely than the private/military insurance group to report having trouble paying for treatments, food, or critical expenses related to CF care (23.3% vs. 12.1%, respectively); feeling symptoms on most days of depression (42.5% vs. 31.3%) or anxiety (40.0% vs. 28.5%); and experiencing conflict or stress with loved ones over treatments (30.0% vs. 20.3%) (P < .05 for all).
In all, 35% had a household income less than $40,000 per year, 33% between $44,000 and $100,000, and 32% higher than $100,000. The low-income group had a lower composite medication possession ratio (0.41) than the middle- (0.44) or high-income (0.52) groups, were more likely to have trouble paying for treatments, food, or treatment-related expenses (25%, 18%, 4%, respectively); were more likely most days to report symptoms of depression (43%, 34%, 26%) or anxiety (40%, 32%, 24%), and to have concerns about whether treatments were effective (42%, 27%, 29%). They were more likely to not be able to maintain a daily schedule or routine for treatments (28%, 22%, 14%).
The study showed that adherence barriers and suboptimal adherence are issues that cross all socioeconomic categories, though they were more problematic in the lowest bracket. Greater anxiety and depression among lower income individuals and those with private or no insurance was a key finding, according to Dr. Dickinson. “It highlights the importance of screening for mental health comorbidities that may impact non-adherence,” she said.
The study received funding from the Cystic Fibrosis Foundation. Dr. Dickinson, Dr. Giusti, and Dr. Perkins have no relevant financial disclosures.
Poor people with chronic illness have greater difficulty managing their disease than do their better-off counterparts, and a new study confirms this reality for patients with cystic fibrosis.
and anxiety symptoms, according to a new cross-sectional study. The data were drawn from the Cystic Fibrosis Foundation’s Success with Therapies Research Consortium.
“Assessing the special challenges that individuals with lower SES face, including financial barriers, is essential to understand how we can address the unique combinations of adherence barriers. In other chronic disorders, financial barriers or lower socioeconomic status is associated with nonadherence, but this relationship has not been well established in cystic fibrosis,” said Kimberly Dickinson, MD, MPH, of Johns Hopkins University, Baltimore, during her presentation of the results at the virtual North American Cystic Fibrosis Conference.
“I’ve always thought that my patients in the poorer population were doing worse, and I think this demonstrates that that’s true,” said Robert Giusti, MD, in an interview. Dr. Giusti is a clinical professor of pediatrics at the New York University and director of the Pediatric Cystic Fibrosis Center in New York. He was not involved in the study.
“These are very pertinent issues, especially if you think about the pandemic, and some of the issues related to mental health. It just highlights the importance of socioeconomic status and screening for some of the known risk factors so that we can develop interventions or programs to provide equitable care to all of our cystic fibrosis patients,” said Ryan Perkins, MD, who moderated the session where the study was presented. He is a pediatric and adult pulmonary fellow at Boston Children’s Hospital and Brigham and Women’s Hospital, also in Boston.
The researchers looked retrospectively at 1 year’s worth of pharmacy refill receipts and number of times prescriptions were refilled versus the number of times prescribed, then calculated medicinal possession ratios. This was cross-referenced with annual household income and insurance status of patients with CF at 12 pediatric and 9 adult CF care centers, for a total of 376 patients (128 pediatric and 248 adult).
In this population, 32% of participants had public or no insurance, 68% had private or military insurance. The public/no insurance group was more likely than the private/military insurance group to report having trouble paying for treatments, food, or critical expenses related to CF care (23.3% vs. 12.1%, respectively); feeling symptoms on most days of depression (42.5% vs. 31.3%) or anxiety (40.0% vs. 28.5%); and experiencing conflict or stress with loved ones over treatments (30.0% vs. 20.3%) (P < .05 for all).
In all, 35% had a household income less than $40,000 per year, 33% between $44,000 and $100,000, and 32% higher than $100,000. The low-income group had a lower composite medication possession ratio (0.41) than the middle- (0.44) or high-income (0.52) groups, were more likely to have trouble paying for treatments, food, or treatment-related expenses (25%, 18%, 4%, respectively); were more likely most days to report symptoms of depression (43%, 34%, 26%) or anxiety (40%, 32%, 24%), and to have concerns about whether treatments were effective (42%, 27%, 29%). They were more likely to not be able to maintain a daily schedule or routine for treatments (28%, 22%, 14%).
The study showed that adherence barriers and suboptimal adherence are issues that cross all socioeconomic categories, though they were more problematic in the lowest bracket. Greater anxiety and depression among lower income individuals and those with private or no insurance was a key finding, according to Dr. Dickinson. “It highlights the importance of screening for mental health comorbidities that may impact non-adherence,” she said.
The study received funding from the Cystic Fibrosis Foundation. Dr. Dickinson, Dr. Giusti, and Dr. Perkins have no relevant financial disclosures.
Poor people with chronic illness have greater difficulty managing their disease than do their better-off counterparts, and a new study confirms this reality for patients with cystic fibrosis.
and anxiety symptoms, according to a new cross-sectional study. The data were drawn from the Cystic Fibrosis Foundation’s Success with Therapies Research Consortium.
“Assessing the special challenges that individuals with lower SES face, including financial barriers, is essential to understand how we can address the unique combinations of adherence barriers. In other chronic disorders, financial barriers or lower socioeconomic status is associated with nonadherence, but this relationship has not been well established in cystic fibrosis,” said Kimberly Dickinson, MD, MPH, of Johns Hopkins University, Baltimore, during her presentation of the results at the virtual North American Cystic Fibrosis Conference.
“I’ve always thought that my patients in the poorer population were doing worse, and I think this demonstrates that that’s true,” said Robert Giusti, MD, in an interview. Dr. Giusti is a clinical professor of pediatrics at the New York University and director of the Pediatric Cystic Fibrosis Center in New York. He was not involved in the study.
“These are very pertinent issues, especially if you think about the pandemic, and some of the issues related to mental health. It just highlights the importance of socioeconomic status and screening for some of the known risk factors so that we can develop interventions or programs to provide equitable care to all of our cystic fibrosis patients,” said Ryan Perkins, MD, who moderated the session where the study was presented. He is a pediatric and adult pulmonary fellow at Boston Children’s Hospital and Brigham and Women’s Hospital, also in Boston.
The researchers looked retrospectively at 1 year’s worth of pharmacy refill receipts and number of times prescriptions were refilled versus the number of times prescribed, then calculated medicinal possession ratios. This was cross-referenced with annual household income and insurance status of patients with CF at 12 pediatric and 9 adult CF care centers, for a total of 376 patients (128 pediatric and 248 adult).
In this population, 32% of participants had public or no insurance, 68% had private or military insurance. The public/no insurance group was more likely than the private/military insurance group to report having trouble paying for treatments, food, or critical expenses related to CF care (23.3% vs. 12.1%, respectively); feeling symptoms on most days of depression (42.5% vs. 31.3%) or anxiety (40.0% vs. 28.5%); and experiencing conflict or stress with loved ones over treatments (30.0% vs. 20.3%) (P < .05 for all).
In all, 35% had a household income less than $40,000 per year, 33% between $44,000 and $100,000, and 32% higher than $100,000. The low-income group had a lower composite medication possession ratio (0.41) than the middle- (0.44) or high-income (0.52) groups, were more likely to have trouble paying for treatments, food, or treatment-related expenses (25%, 18%, 4%, respectively); were more likely most days to report symptoms of depression (43%, 34%, 26%) or anxiety (40%, 32%, 24%), and to have concerns about whether treatments were effective (42%, 27%, 29%). They were more likely to not be able to maintain a daily schedule or routine for treatments (28%, 22%, 14%).
The study showed that adherence barriers and suboptimal adherence are issues that cross all socioeconomic categories, though they were more problematic in the lowest bracket. Greater anxiety and depression among lower income individuals and those with private or no insurance was a key finding, according to Dr. Dickinson. “It highlights the importance of screening for mental health comorbidities that may impact non-adherence,” she said.
The study received funding from the Cystic Fibrosis Foundation. Dr. Dickinson, Dr. Giusti, and Dr. Perkins have no relevant financial disclosures.
FROM NACFC 2020
.
Triple combination therapy for cystic fibrosis linked to plunging hospitalizations
.
The triple combination therapy elexacaftor/tezacaftor/ivacaftor was associated with a near elimination of hospital stays in one hospital in Oregon, according to a new report. The hospital savings still weren’t nearly enough to pay for the cost of therapy, but the study underscores what many institutions have observed and adds a new layer to the view of quality of life improvements that the new therapy brings.
“After we started prescribing it, we noticed pretty quickly that hospitalizations appeared to be declining after patients started triple combination therapy, and we were hearing [similar reports] from other centers as well. We wanted to quantify this,” Eric C. Walter, MD, a pulmonologist at the Kaiser Permanente Cystic Fibrosis Clinic in Portland, Ore., said during a presentation of the results at the virtual North American Cystic Fibrosis Conference.
“We’re seeing that across the board in real practice, the number of cystic fibrosis patients that have to be hospitalized since starting this triple combination has gone down,” Robert Giusti, MD, said in an interview. “When they’ve had pulmonary exacerbations in the past, it was frequently because they failed outpatient antibiotics, but I think with triple combination therapy, if they do get sick, the likelihood is they will respond to oral antibiotics, so they may not need that prolonged IV course in the hospital.” Dr. Giusti is clinical professor of pediatrics at New York University and director of the Pediatric Cystic Fibrosis Center. He was not involved in the study.
The therapy gained Food and Drug Administration approval in 2019 for the treatment of individuals with CF who are aged 12 years and older, and who have at least one copy of the F508del mutation. Its cost is about $317,000 per year within the Kaiser Permanente system, according to Dr. Walter. His group compared hospitalization days for CF-related diagnoses from Jan. 1 through Aug. 31, 2020, before and after initiation of triple combination therapy.
Of 47 eligible patients, 32 initiated therapy during the study period; 38% had severe lung disease, defined by forced expiratory volume in 1 second (FEV1) value less than 40%. In 2020, before initiation of therapy, there were an average of 27 hospital days per month, all among patients with severe lung disease.
Among the therapy group, there were no hospitalizations after initiation of therapy through Aug. 31. Dr. Walter noted that the first hospitalization of a patient on triple combination therapy didn’t occur until early October.
At an average daily cost of $6,700, the researchers calculated that triple combination therapy saved about $189,000 per month in this group of patients. Comparing numbers to previous years, in which some patients with FEV1 greater than 40% were hospitalized, the researchers calculated that the therapy saved about $151,000 per month among individuals with severe lung disease: Patients with severe lung disease contributed about 80% to total hospital costs.
The drug itself for the whole group cost $845,000, dwarfing the $189,000 savings overall. But among patients with severe disease, hospitalization savings were about $151,000 per month, while the drug cost in this group was $316,800 per month.
Cost savings are important, but the improvement in quality of life for a patient – avoiding hospitalization, fewer impacts on work and education – should not be overlooked, according to Ryan Perkins, MD, a pediatric and adult pulmonary fellow at Boston Children’s Hospital and Brigham and Women’s Hospital, who moderated the session. “Some of these aren’t things people typically quantify and assign a price tag to,” Dr. Perkins said in an interview.
A big limitation of the work is that it was conducted during the COVID-19 pandemic, which may have reduced hospitalizations. “We did have patients that called in, told us they were sick, that they needed to be treated for an exacerbation but didn’t want to go to the hospital,” said Dr. Walter. To help adjust for this, Dr. Walter’s team plans to compare intravenous antibiotic exposure before and after triple combination therapy, reasoning that it could help clarify the pandemic’s impact on hospitalizations.
Dr. Walter, Dr. Giusti, and Dr. Perkins have no relevant financial disclosures.
SOURCE: Walter E et al. NACFC 2020. Abstract 795.
.
The triple combination therapy elexacaftor/tezacaftor/ivacaftor was associated with a near elimination of hospital stays in one hospital in Oregon, according to a new report. The hospital savings still weren’t nearly enough to pay for the cost of therapy, but the study underscores what many institutions have observed and adds a new layer to the view of quality of life improvements that the new therapy brings.
“After we started prescribing it, we noticed pretty quickly that hospitalizations appeared to be declining after patients started triple combination therapy, and we were hearing [similar reports] from other centers as well. We wanted to quantify this,” Eric C. Walter, MD, a pulmonologist at the Kaiser Permanente Cystic Fibrosis Clinic in Portland, Ore., said during a presentation of the results at the virtual North American Cystic Fibrosis Conference.
“We’re seeing that across the board in real practice, the number of cystic fibrosis patients that have to be hospitalized since starting this triple combination has gone down,” Robert Giusti, MD, said in an interview. “When they’ve had pulmonary exacerbations in the past, it was frequently because they failed outpatient antibiotics, but I think with triple combination therapy, if they do get sick, the likelihood is they will respond to oral antibiotics, so they may not need that prolonged IV course in the hospital.” Dr. Giusti is clinical professor of pediatrics at New York University and director of the Pediatric Cystic Fibrosis Center. He was not involved in the study.
The therapy gained Food and Drug Administration approval in 2019 for the treatment of individuals with CF who are aged 12 years and older, and who have at least one copy of the F508del mutation. Its cost is about $317,000 per year within the Kaiser Permanente system, according to Dr. Walter. His group compared hospitalization days for CF-related diagnoses from Jan. 1 through Aug. 31, 2020, before and after initiation of triple combination therapy.
Of 47 eligible patients, 32 initiated therapy during the study period; 38% had severe lung disease, defined by forced expiratory volume in 1 second (FEV1) value less than 40%. In 2020, before initiation of therapy, there were an average of 27 hospital days per month, all among patients with severe lung disease.
Among the therapy group, there were no hospitalizations after initiation of therapy through Aug. 31. Dr. Walter noted that the first hospitalization of a patient on triple combination therapy didn’t occur until early October.
At an average daily cost of $6,700, the researchers calculated that triple combination therapy saved about $189,000 per month in this group of patients. Comparing numbers to previous years, in which some patients with FEV1 greater than 40% were hospitalized, the researchers calculated that the therapy saved about $151,000 per month among individuals with severe lung disease: Patients with severe lung disease contributed about 80% to total hospital costs.
The drug itself for the whole group cost $845,000, dwarfing the $189,000 savings overall. But among patients with severe disease, hospitalization savings were about $151,000 per month, while the drug cost in this group was $316,800 per month.
Cost savings are important, but the improvement in quality of life for a patient – avoiding hospitalization, fewer impacts on work and education – should not be overlooked, according to Ryan Perkins, MD, a pediatric and adult pulmonary fellow at Boston Children’s Hospital and Brigham and Women’s Hospital, who moderated the session. “Some of these aren’t things people typically quantify and assign a price tag to,” Dr. Perkins said in an interview.
A big limitation of the work is that it was conducted during the COVID-19 pandemic, which may have reduced hospitalizations. “We did have patients that called in, told us they were sick, that they needed to be treated for an exacerbation but didn’t want to go to the hospital,” said Dr. Walter. To help adjust for this, Dr. Walter’s team plans to compare intravenous antibiotic exposure before and after triple combination therapy, reasoning that it could help clarify the pandemic’s impact on hospitalizations.
Dr. Walter, Dr. Giusti, and Dr. Perkins have no relevant financial disclosures.
SOURCE: Walter E et al. NACFC 2020. Abstract 795.
.
The triple combination therapy elexacaftor/tezacaftor/ivacaftor was associated with a near elimination of hospital stays in one hospital in Oregon, according to a new report. The hospital savings still weren’t nearly enough to pay for the cost of therapy, but the study underscores what many institutions have observed and adds a new layer to the view of quality of life improvements that the new therapy brings.
“After we started prescribing it, we noticed pretty quickly that hospitalizations appeared to be declining after patients started triple combination therapy, and we were hearing [similar reports] from other centers as well. We wanted to quantify this,” Eric C. Walter, MD, a pulmonologist at the Kaiser Permanente Cystic Fibrosis Clinic in Portland, Ore., said during a presentation of the results at the virtual North American Cystic Fibrosis Conference.
“We’re seeing that across the board in real practice, the number of cystic fibrosis patients that have to be hospitalized since starting this triple combination has gone down,” Robert Giusti, MD, said in an interview. “When they’ve had pulmonary exacerbations in the past, it was frequently because they failed outpatient antibiotics, but I think with triple combination therapy, if they do get sick, the likelihood is they will respond to oral antibiotics, so they may not need that prolonged IV course in the hospital.” Dr. Giusti is clinical professor of pediatrics at New York University and director of the Pediatric Cystic Fibrosis Center. He was not involved in the study.
The therapy gained Food and Drug Administration approval in 2019 for the treatment of individuals with CF who are aged 12 years and older, and who have at least one copy of the F508del mutation. Its cost is about $317,000 per year within the Kaiser Permanente system, according to Dr. Walter. His group compared hospitalization days for CF-related diagnoses from Jan. 1 through Aug. 31, 2020, before and after initiation of triple combination therapy.
Of 47 eligible patients, 32 initiated therapy during the study period; 38% had severe lung disease, defined by forced expiratory volume in 1 second (FEV1) value less than 40%. In 2020, before initiation of therapy, there were an average of 27 hospital days per month, all among patients with severe lung disease.
Among the therapy group, there were no hospitalizations after initiation of therapy through Aug. 31. Dr. Walter noted that the first hospitalization of a patient on triple combination therapy didn’t occur until early October.
At an average daily cost of $6,700, the researchers calculated that triple combination therapy saved about $189,000 per month in this group of patients. Comparing numbers to previous years, in which some patients with FEV1 greater than 40% were hospitalized, the researchers calculated that the therapy saved about $151,000 per month among individuals with severe lung disease: Patients with severe lung disease contributed about 80% to total hospital costs.
The drug itself for the whole group cost $845,000, dwarfing the $189,000 savings overall. But among patients with severe disease, hospitalization savings were about $151,000 per month, while the drug cost in this group was $316,800 per month.
Cost savings are important, but the improvement in quality of life for a patient – avoiding hospitalization, fewer impacts on work and education – should not be overlooked, according to Ryan Perkins, MD, a pediatric and adult pulmonary fellow at Boston Children’s Hospital and Brigham and Women’s Hospital, who moderated the session. “Some of these aren’t things people typically quantify and assign a price tag to,” Dr. Perkins said in an interview.
A big limitation of the work is that it was conducted during the COVID-19 pandemic, which may have reduced hospitalizations. “We did have patients that called in, told us they were sick, that they needed to be treated for an exacerbation but didn’t want to go to the hospital,” said Dr. Walter. To help adjust for this, Dr. Walter’s team plans to compare intravenous antibiotic exposure before and after triple combination therapy, reasoning that it could help clarify the pandemic’s impact on hospitalizations.
Dr. Walter, Dr. Giusti, and Dr. Perkins have no relevant financial disclosures.
SOURCE: Walter E et al. NACFC 2020. Abstract 795.
FROM NACFC 2020
Cystic fibrosis patients’ vulnerability to COVID-19 infection: Preliminary data ease fears
But early results suggest that social distance measures and perhaps the younger average age of individuals with CF have prevented a severe impact on this patient population.
Not all of the news is good. Some research suggests that posttransplant individuals may be at greater risk of severe outcomes. However, researchers warned that the data are too sparse to draw firm conclusions, and ongoing analyses of patient registries and other sources should lend greater insight into the burden of COVID-19 among individuals with CF. Those were some of the conclusions presented at a session of the virtual North American Cystic Fibrosis Conference.
D.B. Sanders, MD, who is a pediatric pulmonologist at Riley Hospital for Children and the Indiana University, both in Indianapolis, presented data from the Cystic Fibrosis Foundation’s Patient Registry, which includes patients in the United States. As in other populations, he showed that health care use has gone down among individuals with CF. From April to September 2019, 81% of clinical encounters were in the clinic and 12% in the hospital. Over the same period in 2020, those numbers dropped to 35% and 4%, respectively, with 30% by phone or computer. In-person health care use rebounded somewhat between July 1 and Sept. 16, with 53% of encounters at the clinic, 5% at the hospital, and 28% conducted virtually. There were also dips in forced expiratory volume in one second (FEV1) and microbiology testing, from about 90% occurring during health encounters at the end of 2019 to fewer than 10% of encounters by April.
As of Aug. 17, Dr. Sanders reported that 3,048 individuals with CF had been tested for COVID-19, with 174 positive results.
Racial and ethnic disparities in positive test results seen in other populations were also observable among individuals with CF. Several groups made up a higher proportion of COVID-19–positive CF patients than the general CF population, including Hispanics (18% vs. 9%), Blacks (7% vs. 5%), and individuals with FEV1 value less than 40% predicted (14% vs. 8%).
As of Sept. 17, there had been 51 hospitalizations and two deaths in the United States among 212 individuals with CF who tested positive for COVID-19, with increasing numbers that mirror trends in the U.S. population. One death occurred in a patient with advanced lung disease, the other in a post–lung transplant patient. “Thankfully [the numbers are] not higher, but this is being followed very closely,” said Dr. Sanders during his presentation.
One encouraging bit of news was that hospitalizations among individuals with CF have dropped since the start of the pandemic. “I think this shows how good our families are at socially distancing, wearing masks, and now that they not being exposed to viruses, I think we’re seeing the fruits of this with fewer hospitalizations,” said Dr. Sanders. He noted that it’s possible some of the decline could have been to reluctance to go to the hospital, and the introduction of triple combination cystic fibrosis transmembrane conductance regulator modulator therapy has also likely contributed. “We were already seeing fewer hospitalizations even before the pandemic hit,” he said.
At the session, Rebecca Cosgriff, director of data and quality improvement at the Cystic Fibrosis Trust in the United Kingdom, presented an international perspective on COVID-19 cases among individuals with CF. At the beginning of the pandemic, the Cystic Fibrosis Global Registry Harmonization Group recruited country coordinators to collect anonymized data on infections, hospitalizations, and other outcomes. In April, the group published its initial findings from 40 cases in eight countries, which concluded that these cases generally resembled the broader population in clinical course, which assuaged initial fears.
Ms. Cosgriff reported on results from a second round of data collection with a cutoff date of June 19, which expanded to 19 countries and included many from South America and more in Europe. The network encompassed about 85,000 individuals with CF, and tallied 181 cases of COVID-19. A total of 149 cases were nontransplant, and 32 were posttransplant (28 lung only). Fully 15% of the nontransplant group were over age 40 years, compared with 41% in the transplant group. Homozygous F508del mutations were more common in the posttransplant group (59% vs. 36%). However, lung function, as estimated by the best FEV1 measured in the previous year prior to infection, differed between the nontransplant (73%) and posttransplant (80%) COVID-19 patients.
Across all age groups, hospitalizations were more common in patients with best FEV1 percentage predicted values less than 70% (P = .001). Ms. Cosgriff also expressed concern about the posttransplant group. “Across all outcomes that might be indicative of infection severity – hospitalization, ICU admission, new supplementary oxygen, and non-invasive ventilation – the proportion of the posttransplant group was higher across the board,” she said during her presentation.
There were seven deaths. Ms. Cosgriff noted that there were too few deaths to analyze trends, but she presented a slide showing characteristics of deceased patients. “Factors like being post–lung transplant, being male, having less FEV1 than predicted, being over 40, or having CF-related diabetes, all appear pretty frequently amongst the cohort of people who died,” she said.
Overall, the results of these surveys are encouraging, suggesting that early fears that COVID-19 cases could be more severe among individuals with CF may not have been borne out so far. Dr. Sanders noted in his talk that there aren’t enough cases in the U.S. cohort to show links to risk factors with statistical significance. “But thankfully we’re not seeing a host of negative outcomes,” he said.
Dr. Sanders and Ms Cosgriff have no relevant financial disclosures.
But early results suggest that social distance measures and perhaps the younger average age of individuals with CF have prevented a severe impact on this patient population.
Not all of the news is good. Some research suggests that posttransplant individuals may be at greater risk of severe outcomes. However, researchers warned that the data are too sparse to draw firm conclusions, and ongoing analyses of patient registries and other sources should lend greater insight into the burden of COVID-19 among individuals with CF. Those were some of the conclusions presented at a session of the virtual North American Cystic Fibrosis Conference.
D.B. Sanders, MD, who is a pediatric pulmonologist at Riley Hospital for Children and the Indiana University, both in Indianapolis, presented data from the Cystic Fibrosis Foundation’s Patient Registry, which includes patients in the United States. As in other populations, he showed that health care use has gone down among individuals with CF. From April to September 2019, 81% of clinical encounters were in the clinic and 12% in the hospital. Over the same period in 2020, those numbers dropped to 35% and 4%, respectively, with 30% by phone or computer. In-person health care use rebounded somewhat between July 1 and Sept. 16, with 53% of encounters at the clinic, 5% at the hospital, and 28% conducted virtually. There were also dips in forced expiratory volume in one second (FEV1) and microbiology testing, from about 90% occurring during health encounters at the end of 2019 to fewer than 10% of encounters by April.
As of Aug. 17, Dr. Sanders reported that 3,048 individuals with CF had been tested for COVID-19, with 174 positive results.
Racial and ethnic disparities in positive test results seen in other populations were also observable among individuals with CF. Several groups made up a higher proportion of COVID-19–positive CF patients than the general CF population, including Hispanics (18% vs. 9%), Blacks (7% vs. 5%), and individuals with FEV1 value less than 40% predicted (14% vs. 8%).
As of Sept. 17, there had been 51 hospitalizations and two deaths in the United States among 212 individuals with CF who tested positive for COVID-19, with increasing numbers that mirror trends in the U.S. population. One death occurred in a patient with advanced lung disease, the other in a post–lung transplant patient. “Thankfully [the numbers are] not higher, but this is being followed very closely,” said Dr. Sanders during his presentation.
One encouraging bit of news was that hospitalizations among individuals with CF have dropped since the start of the pandemic. “I think this shows how good our families are at socially distancing, wearing masks, and now that they not being exposed to viruses, I think we’re seeing the fruits of this with fewer hospitalizations,” said Dr. Sanders. He noted that it’s possible some of the decline could have been to reluctance to go to the hospital, and the introduction of triple combination cystic fibrosis transmembrane conductance regulator modulator therapy has also likely contributed. “We were already seeing fewer hospitalizations even before the pandemic hit,” he said.
At the session, Rebecca Cosgriff, director of data and quality improvement at the Cystic Fibrosis Trust in the United Kingdom, presented an international perspective on COVID-19 cases among individuals with CF. At the beginning of the pandemic, the Cystic Fibrosis Global Registry Harmonization Group recruited country coordinators to collect anonymized data on infections, hospitalizations, and other outcomes. In April, the group published its initial findings from 40 cases in eight countries, which concluded that these cases generally resembled the broader population in clinical course, which assuaged initial fears.
Ms. Cosgriff reported on results from a second round of data collection with a cutoff date of June 19, which expanded to 19 countries and included many from South America and more in Europe. The network encompassed about 85,000 individuals with CF, and tallied 181 cases of COVID-19. A total of 149 cases were nontransplant, and 32 were posttransplant (28 lung only). Fully 15% of the nontransplant group were over age 40 years, compared with 41% in the transplant group. Homozygous F508del mutations were more common in the posttransplant group (59% vs. 36%). However, lung function, as estimated by the best FEV1 measured in the previous year prior to infection, differed between the nontransplant (73%) and posttransplant (80%) COVID-19 patients.
Across all age groups, hospitalizations were more common in patients with best FEV1 percentage predicted values less than 70% (P = .001). Ms. Cosgriff also expressed concern about the posttransplant group. “Across all outcomes that might be indicative of infection severity – hospitalization, ICU admission, new supplementary oxygen, and non-invasive ventilation – the proportion of the posttransplant group was higher across the board,” she said during her presentation.
There were seven deaths. Ms. Cosgriff noted that there were too few deaths to analyze trends, but she presented a slide showing characteristics of deceased patients. “Factors like being post–lung transplant, being male, having less FEV1 than predicted, being over 40, or having CF-related diabetes, all appear pretty frequently amongst the cohort of people who died,” she said.
Overall, the results of these surveys are encouraging, suggesting that early fears that COVID-19 cases could be more severe among individuals with CF may not have been borne out so far. Dr. Sanders noted in his talk that there aren’t enough cases in the U.S. cohort to show links to risk factors with statistical significance. “But thankfully we’re not seeing a host of negative outcomes,” he said.
Dr. Sanders and Ms Cosgriff have no relevant financial disclosures.
But early results suggest that social distance measures and perhaps the younger average age of individuals with CF have prevented a severe impact on this patient population.
Not all of the news is good. Some research suggests that posttransplant individuals may be at greater risk of severe outcomes. However, researchers warned that the data are too sparse to draw firm conclusions, and ongoing analyses of patient registries and other sources should lend greater insight into the burden of COVID-19 among individuals with CF. Those were some of the conclusions presented at a session of the virtual North American Cystic Fibrosis Conference.
D.B. Sanders, MD, who is a pediatric pulmonologist at Riley Hospital for Children and the Indiana University, both in Indianapolis, presented data from the Cystic Fibrosis Foundation’s Patient Registry, which includes patients in the United States. As in other populations, he showed that health care use has gone down among individuals with CF. From April to September 2019, 81% of clinical encounters were in the clinic and 12% in the hospital. Over the same period in 2020, those numbers dropped to 35% and 4%, respectively, with 30% by phone or computer. In-person health care use rebounded somewhat between July 1 and Sept. 16, with 53% of encounters at the clinic, 5% at the hospital, and 28% conducted virtually. There were also dips in forced expiratory volume in one second (FEV1) and microbiology testing, from about 90% occurring during health encounters at the end of 2019 to fewer than 10% of encounters by April.
As of Aug. 17, Dr. Sanders reported that 3,048 individuals with CF had been tested for COVID-19, with 174 positive results.
Racial and ethnic disparities in positive test results seen in other populations were also observable among individuals with CF. Several groups made up a higher proportion of COVID-19–positive CF patients than the general CF population, including Hispanics (18% vs. 9%), Blacks (7% vs. 5%), and individuals with FEV1 value less than 40% predicted (14% vs. 8%).
As of Sept. 17, there had been 51 hospitalizations and two deaths in the United States among 212 individuals with CF who tested positive for COVID-19, with increasing numbers that mirror trends in the U.S. population. One death occurred in a patient with advanced lung disease, the other in a post–lung transplant patient. “Thankfully [the numbers are] not higher, but this is being followed very closely,” said Dr. Sanders during his presentation.
One encouraging bit of news was that hospitalizations among individuals with CF have dropped since the start of the pandemic. “I think this shows how good our families are at socially distancing, wearing masks, and now that they not being exposed to viruses, I think we’re seeing the fruits of this with fewer hospitalizations,” said Dr. Sanders. He noted that it’s possible some of the decline could have been to reluctance to go to the hospital, and the introduction of triple combination cystic fibrosis transmembrane conductance regulator modulator therapy has also likely contributed. “We were already seeing fewer hospitalizations even before the pandemic hit,” he said.
At the session, Rebecca Cosgriff, director of data and quality improvement at the Cystic Fibrosis Trust in the United Kingdom, presented an international perspective on COVID-19 cases among individuals with CF. At the beginning of the pandemic, the Cystic Fibrosis Global Registry Harmonization Group recruited country coordinators to collect anonymized data on infections, hospitalizations, and other outcomes. In April, the group published its initial findings from 40 cases in eight countries, which concluded that these cases generally resembled the broader population in clinical course, which assuaged initial fears.
Ms. Cosgriff reported on results from a second round of data collection with a cutoff date of June 19, which expanded to 19 countries and included many from South America and more in Europe. The network encompassed about 85,000 individuals with CF, and tallied 181 cases of COVID-19. A total of 149 cases were nontransplant, and 32 were posttransplant (28 lung only). Fully 15% of the nontransplant group were over age 40 years, compared with 41% in the transplant group. Homozygous F508del mutations were more common in the posttransplant group (59% vs. 36%). However, lung function, as estimated by the best FEV1 measured in the previous year prior to infection, differed between the nontransplant (73%) and posttransplant (80%) COVID-19 patients.
Across all age groups, hospitalizations were more common in patients with best FEV1 percentage predicted values less than 70% (P = .001). Ms. Cosgriff also expressed concern about the posttransplant group. “Across all outcomes that might be indicative of infection severity – hospitalization, ICU admission, new supplementary oxygen, and non-invasive ventilation – the proportion of the posttransplant group was higher across the board,” she said during her presentation.
There were seven deaths. Ms. Cosgriff noted that there were too few deaths to analyze trends, but she presented a slide showing characteristics of deceased patients. “Factors like being post–lung transplant, being male, having less FEV1 than predicted, being over 40, or having CF-related diabetes, all appear pretty frequently amongst the cohort of people who died,” she said.
Overall, the results of these surveys are encouraging, suggesting that early fears that COVID-19 cases could be more severe among individuals with CF may not have been borne out so far. Dr. Sanders noted in his talk that there aren’t enough cases in the U.S. cohort to show links to risk factors with statistical significance. “But thankfully we’re not seeing a host of negative outcomes,” he said.
Dr. Sanders and Ms Cosgriff have no relevant financial disclosures.
FROM NACFC 2020
Home spirometry improved monitoring of cystic fibrosis patients during COVID-19 pandemic
Home spirometry has become increasingly used among cystic fibrosis patients during the COVID-19 pandemic, and new research suggests that home devices perform reasonably well. Forced expiratory volume in 1 second (FEV1) values were a bit lower than values seen in clinical spirometry performed in the same patient at a nearby time point, but the procedure reliably picked up decreases in FEV1, potentially helping patients and clinicians spot exacerbations early.
“Home spirometry was sort of a curiosity that was slowly working its way into cystic fibrosis research in 2019, and then all of a sudden in 2020 it became front and center as the only way to continue with clinical monitoring and research in many cases,” Alexander Paynter, MS, a biostatistician at the Cystic Fibrosis Foundation’s Therapeutic Development Network Coordinating Center, said during a talk at the virtual North American Cystic Fibrosis Conference.
To better determine how closely home spirometry matches clinical spirometry, Mr. Paynter and his colleagues analyzed data from the eICE study, which included 267 cystic fibrosis patients aged 14 and over at 14 cystic fibrosis centers. They were randomized to use home spirometry as an early intervention to detect exacerbations, or to continue usual clinic care with visits to the clinic every 3 months. The dataset includes twice-weekly home spirometry values, with a full-year of follow-up data. The researchers compared the home spirometry data to the clinical data closest in time to it. Clinic spirometry data with no corresponding home data within 7 days were discarded.
There was an estimated difference of –2.01 mL between home and clinic tests, with home spirometry producing lower values (95% confidence interval, –3.56 to –0.45). “There is actually a bias in home spirometry as compared to clinic spirometry,” concluded Mr. Paynter.
One explanation for lower values in home spirometry is that users are inexperienced with the device. If that’s true, then agreement should improve over time, but the researchers didn’t see strong evidence of that. Among 44 patients who completed five clinical visits, there was a difference of –2.97 (standard deviation [SD], 10.51) at baseline, –1.66 at 3 months (SD, 13.49), –3.7 at 6 months (SD, 12.44), –0.86 at 9 months (SD, 13.73), and –0.53 at 12 months (SD, 13.35). Though there was improvement over time, “we don’t find a lot of evidence that this bias completely resolves,” said Mr. Paynter.
In fact, a more likely explanation is the presence of coaching by a technician during clinical spirometry, according to Robert J. Giusti, MD, clinical professor of pediatrics and director of the Pediatric Cystic Fibrosis Center at New York University. “When they’re doing it at home, they don’t do it with the same effort, so I think that coaching through telemedicine during the home spirometry would make that difference disappear,” he said when asked to comment on the study.
The researchers found that change-based endpoints were similar between clinic and at-home spirometry. Compared to baseline, the two showed similar declines over time. “The clinic and home observations tend to track each other pretty well. At 6 months, for instance, it’s about a change of three points decrease (in both). But the bad news is that the variability is much greater in home devices,” said Mr. Paynter, noting larger confidence intervals and standard deviation values associated with home spirometry. That could influence future clinical designs that may rely on home spirometry, since a larger confidence interval means reduced power, which could double or even quadruple the number of participants needed to achieve the required power, he said.
But from a clinical standpoint, the ability of home spirometry to consistently detect a change from baseline could be quite valuable to future patient management, according to Dr. Giusti. “It looks like home spirometry will show that kind of a decrease, so that it’s still sensitive to pick up the concern that a patient is getting worse at home,” he said.
That could be useful even after the COVID-19 pandemic passes, as patients continue to embrace home monitoring. Physicians could keep track of patients and keep them focused on their care and treatment through frequent telemedicine visits combined with home spirometry. “I really think home spirometry will keep us more focused on how the patients are doing and make for better outcomes,” said Dr. Giusti.
Mr. Paynter and Dr. Giusti have no relevant financial disclosures.
SOURCE: Alex Paynter et al. NACFC 2020. Poster 643.
Home spirometry has become increasingly used among cystic fibrosis patients during the COVID-19 pandemic, and new research suggests that home devices perform reasonably well. Forced expiratory volume in 1 second (FEV1) values were a bit lower than values seen in clinical spirometry performed in the same patient at a nearby time point, but the procedure reliably picked up decreases in FEV1, potentially helping patients and clinicians spot exacerbations early.
“Home spirometry was sort of a curiosity that was slowly working its way into cystic fibrosis research in 2019, and then all of a sudden in 2020 it became front and center as the only way to continue with clinical monitoring and research in many cases,” Alexander Paynter, MS, a biostatistician at the Cystic Fibrosis Foundation’s Therapeutic Development Network Coordinating Center, said during a talk at the virtual North American Cystic Fibrosis Conference.
To better determine how closely home spirometry matches clinical spirometry, Mr. Paynter and his colleagues analyzed data from the eICE study, which included 267 cystic fibrosis patients aged 14 and over at 14 cystic fibrosis centers. They were randomized to use home spirometry as an early intervention to detect exacerbations, or to continue usual clinic care with visits to the clinic every 3 months. The dataset includes twice-weekly home spirometry values, with a full-year of follow-up data. The researchers compared the home spirometry data to the clinical data closest in time to it. Clinic spirometry data with no corresponding home data within 7 days were discarded.
There was an estimated difference of –2.01 mL between home and clinic tests, with home spirometry producing lower values (95% confidence interval, –3.56 to –0.45). “There is actually a bias in home spirometry as compared to clinic spirometry,” concluded Mr. Paynter.
One explanation for lower values in home spirometry is that users are inexperienced with the device. If that’s true, then agreement should improve over time, but the researchers didn’t see strong evidence of that. Among 44 patients who completed five clinical visits, there was a difference of –2.97 (standard deviation [SD], 10.51) at baseline, –1.66 at 3 months (SD, 13.49), –3.7 at 6 months (SD, 12.44), –0.86 at 9 months (SD, 13.73), and –0.53 at 12 months (SD, 13.35). Though there was improvement over time, “we don’t find a lot of evidence that this bias completely resolves,” said Mr. Paynter.
In fact, a more likely explanation is the presence of coaching by a technician during clinical spirometry, according to Robert J. Giusti, MD, clinical professor of pediatrics and director of the Pediatric Cystic Fibrosis Center at New York University. “When they’re doing it at home, they don’t do it with the same effort, so I think that coaching through telemedicine during the home spirometry would make that difference disappear,” he said when asked to comment on the study.
The researchers found that change-based endpoints were similar between clinic and at-home spirometry. Compared to baseline, the two showed similar declines over time. “The clinic and home observations tend to track each other pretty well. At 6 months, for instance, it’s about a change of three points decrease (in both). But the bad news is that the variability is much greater in home devices,” said Mr. Paynter, noting larger confidence intervals and standard deviation values associated with home spirometry. That could influence future clinical designs that may rely on home spirometry, since a larger confidence interval means reduced power, which could double or even quadruple the number of participants needed to achieve the required power, he said.
But from a clinical standpoint, the ability of home spirometry to consistently detect a change from baseline could be quite valuable to future patient management, according to Dr. Giusti. “It looks like home spirometry will show that kind of a decrease, so that it’s still sensitive to pick up the concern that a patient is getting worse at home,” he said.
That could be useful even after the COVID-19 pandemic passes, as patients continue to embrace home monitoring. Physicians could keep track of patients and keep them focused on their care and treatment through frequent telemedicine visits combined with home spirometry. “I really think home spirometry will keep us more focused on how the patients are doing and make for better outcomes,” said Dr. Giusti.
Mr. Paynter and Dr. Giusti have no relevant financial disclosures.
SOURCE: Alex Paynter et al. NACFC 2020. Poster 643.
Home spirometry has become increasingly used among cystic fibrosis patients during the COVID-19 pandemic, and new research suggests that home devices perform reasonably well. Forced expiratory volume in 1 second (FEV1) values were a bit lower than values seen in clinical spirometry performed in the same patient at a nearby time point, but the procedure reliably picked up decreases in FEV1, potentially helping patients and clinicians spot exacerbations early.
“Home spirometry was sort of a curiosity that was slowly working its way into cystic fibrosis research in 2019, and then all of a sudden in 2020 it became front and center as the only way to continue with clinical monitoring and research in many cases,” Alexander Paynter, MS, a biostatistician at the Cystic Fibrosis Foundation’s Therapeutic Development Network Coordinating Center, said during a talk at the virtual North American Cystic Fibrosis Conference.
To better determine how closely home spirometry matches clinical spirometry, Mr. Paynter and his colleagues analyzed data from the eICE study, which included 267 cystic fibrosis patients aged 14 and over at 14 cystic fibrosis centers. They were randomized to use home spirometry as an early intervention to detect exacerbations, or to continue usual clinic care with visits to the clinic every 3 months. The dataset includes twice-weekly home spirometry values, with a full-year of follow-up data. The researchers compared the home spirometry data to the clinical data closest in time to it. Clinic spirometry data with no corresponding home data within 7 days were discarded.
There was an estimated difference of –2.01 mL between home and clinic tests, with home spirometry producing lower values (95% confidence interval, –3.56 to –0.45). “There is actually a bias in home spirometry as compared to clinic spirometry,” concluded Mr. Paynter.
One explanation for lower values in home spirometry is that users are inexperienced with the device. If that’s true, then agreement should improve over time, but the researchers didn’t see strong evidence of that. Among 44 patients who completed five clinical visits, there was a difference of –2.97 (standard deviation [SD], 10.51) at baseline, –1.66 at 3 months (SD, 13.49), –3.7 at 6 months (SD, 12.44), –0.86 at 9 months (SD, 13.73), and –0.53 at 12 months (SD, 13.35). Though there was improvement over time, “we don’t find a lot of evidence that this bias completely resolves,” said Mr. Paynter.
In fact, a more likely explanation is the presence of coaching by a technician during clinical spirometry, according to Robert J. Giusti, MD, clinical professor of pediatrics and director of the Pediatric Cystic Fibrosis Center at New York University. “When they’re doing it at home, they don’t do it with the same effort, so I think that coaching through telemedicine during the home spirometry would make that difference disappear,” he said when asked to comment on the study.
The researchers found that change-based endpoints were similar between clinic and at-home spirometry. Compared to baseline, the two showed similar declines over time. “The clinic and home observations tend to track each other pretty well. At 6 months, for instance, it’s about a change of three points decrease (in both). But the bad news is that the variability is much greater in home devices,” said Mr. Paynter, noting larger confidence intervals and standard deviation values associated with home spirometry. That could influence future clinical designs that may rely on home spirometry, since a larger confidence interval means reduced power, which could double or even quadruple the number of participants needed to achieve the required power, he said.
But from a clinical standpoint, the ability of home spirometry to consistently detect a change from baseline could be quite valuable to future patient management, according to Dr. Giusti. “It looks like home spirometry will show that kind of a decrease, so that it’s still sensitive to pick up the concern that a patient is getting worse at home,” he said.
That could be useful even after the COVID-19 pandemic passes, as patients continue to embrace home monitoring. Physicians could keep track of patients and keep them focused on their care and treatment through frequent telemedicine visits combined with home spirometry. “I really think home spirometry will keep us more focused on how the patients are doing and make for better outcomes,” said Dr. Giusti.
Mr. Paynter and Dr. Giusti have no relevant financial disclosures.
SOURCE: Alex Paynter et al. NACFC 2020. Poster 643.
FROM NACFC 2020
Cystic fibrosis treatment: Triple combination benefits patients with advanced disease
New CFTR [cystic fibrosis transmembrane conductance regulator] modulator therapies can offer life-altering benefits to some patients with cystic fibrosis, even those with advanced disease.
, according to a multicenter analysis of patients taking elexacaftor, tezacaftor, and ivacaftor.
The study participants had a percent predicted forced expiratory volume in 1 second (ppFEV1) of 40% or below, or other high-risk factors. Researchers compared them to control patients who were genetically ineligible for triple combination therapy.
Previous studies of such patients on individual drugs or previous combinations showed increases in lung function in patients with advanced disease, though the magnitude of improvement varied across regimens. “With this improvement, it’s unclear how CFTR modulators should affect lung transplant referral timing,” Brent Bermingham, MD, said during a presentation of the study at the virtual North American Cystic Fibrosis Conference.
“The rationale for our study was that despite patients with advanced lung disease being excluded from phase III trials (of elexacaftor, tezacaftor, and ivacaftor), they are receiving a therapy with an unknown clinical efficacy and safety profile,” said Dr. Bermingham, a pulmonary and critical care fellow at the Medical University of South Carolina, Charleston.
Lung transplant referral guidelines recommend that physicians initiate discussions about the potential benefit of lung transplant when FEV1 drops below 50% of the predicted value. Patients should be referred for a transplant when the value is below 50% and rapidly declining (>20% decline in the past 12 months), when it drops below 40% with accompanying predictors of shortened survival, or when it drops below 30%. The guidelines were published before approval of triple combination therapy.
The researchers conducted an open-label retrospective analysis of 60 patients started on triple combination therapy between September 2019 and February 2020 at three centers in the Southeast. They compared percent predicted ppFEV1 values prior to initiation of therapy to ppFEV1 values obtained 2-12 weeks after the start of therapy. Patients on therapy were compared with 10 genetically ineligible controls. The two groups were generally similar aside from genetic status, though 100% of the therapy group had pancreatic insufficiency, compared with 90% of controls (P = .013).
The therapeutic group experienced a 7.8% increase in ppFEV1 after starting therapy (P < .001), compared with a 0.5% decrease in controls (P = .65). Before initiation of therapy, 33% of the therapy group met the criteria for initiating a transplant discussion, while 67% had been recommended for transplant. After therapy, 55% met the criteria for discussion, 33% were recommended for transplant, and 12% no longer met the criteria for discussion of transplantation. Fifty percent of controls were in discussion, and this dropped to 40%, while 50% were referred for transplantation, and this increased to 60%. On therapy, transplant referral candidates had an increase of forced vital capacity from 48.9 to 59.16 (P < .001).
Adverse events were rare, with only one discontinuation that occurred following a lung transplant and was not believed to be treatment related.
“Our study had a large number of patients taken from multiple centers, which suggests generalizabilty and real-world experience,” said Dr. Bermingham.
The results are encouraging, said Robert J. Giusti, MD, clinical professor of pediatrics at the New York University and director of the Pediatric Cystic Fibrosis Center.
“We’re all remarking how wonderful patients feel these days. It’s really a disease-altering treatment. But for the high-risk group, whose FEV1 is less than 40%, those are the patients we’re more concerned about because we thought maybe they had too much lung disease, and that they wouldn’t benefit from triple combination. But they seem to be improving, so that’s very reassuring,” said Dr. Giusti, who was not involved in the study.
The study received funding from the Cystic Fibrosis Foundation and Dartmouth College. Dr. Bermingham and Dr. Giusti have no relevant financial disclosures.
SOURCE: Bermingham B et al. NACFC 2020, Abstract 645.
New CFTR [cystic fibrosis transmembrane conductance regulator] modulator therapies can offer life-altering benefits to some patients with cystic fibrosis, even those with advanced disease.
, according to a multicenter analysis of patients taking elexacaftor, tezacaftor, and ivacaftor.
The study participants had a percent predicted forced expiratory volume in 1 second (ppFEV1) of 40% or below, or other high-risk factors. Researchers compared them to control patients who were genetically ineligible for triple combination therapy.
Previous studies of such patients on individual drugs or previous combinations showed increases in lung function in patients with advanced disease, though the magnitude of improvement varied across regimens. “With this improvement, it’s unclear how CFTR modulators should affect lung transplant referral timing,” Brent Bermingham, MD, said during a presentation of the study at the virtual North American Cystic Fibrosis Conference.
“The rationale for our study was that despite patients with advanced lung disease being excluded from phase III trials (of elexacaftor, tezacaftor, and ivacaftor), they are receiving a therapy with an unknown clinical efficacy and safety profile,” said Dr. Bermingham, a pulmonary and critical care fellow at the Medical University of South Carolina, Charleston.
Lung transplant referral guidelines recommend that physicians initiate discussions about the potential benefit of lung transplant when FEV1 drops below 50% of the predicted value. Patients should be referred for a transplant when the value is below 50% and rapidly declining (>20% decline in the past 12 months), when it drops below 40% with accompanying predictors of shortened survival, or when it drops below 30%. The guidelines were published before approval of triple combination therapy.
The researchers conducted an open-label retrospective analysis of 60 patients started on triple combination therapy between September 2019 and February 2020 at three centers in the Southeast. They compared percent predicted ppFEV1 values prior to initiation of therapy to ppFEV1 values obtained 2-12 weeks after the start of therapy. Patients on therapy were compared with 10 genetically ineligible controls. The two groups were generally similar aside from genetic status, though 100% of the therapy group had pancreatic insufficiency, compared with 90% of controls (P = .013).
The therapeutic group experienced a 7.8% increase in ppFEV1 after starting therapy (P < .001), compared with a 0.5% decrease in controls (P = .65). Before initiation of therapy, 33% of the therapy group met the criteria for initiating a transplant discussion, while 67% had been recommended for transplant. After therapy, 55% met the criteria for discussion, 33% were recommended for transplant, and 12% no longer met the criteria for discussion of transplantation. Fifty percent of controls were in discussion, and this dropped to 40%, while 50% were referred for transplantation, and this increased to 60%. On therapy, transplant referral candidates had an increase of forced vital capacity from 48.9 to 59.16 (P < .001).
Adverse events were rare, with only one discontinuation that occurred following a lung transplant and was not believed to be treatment related.
“Our study had a large number of patients taken from multiple centers, which suggests generalizabilty and real-world experience,” said Dr. Bermingham.
The results are encouraging, said Robert J. Giusti, MD, clinical professor of pediatrics at the New York University and director of the Pediatric Cystic Fibrosis Center.
“We’re all remarking how wonderful patients feel these days. It’s really a disease-altering treatment. But for the high-risk group, whose FEV1 is less than 40%, those are the patients we’re more concerned about because we thought maybe they had too much lung disease, and that they wouldn’t benefit from triple combination. But they seem to be improving, so that’s very reassuring,” said Dr. Giusti, who was not involved in the study.
The study received funding from the Cystic Fibrosis Foundation and Dartmouth College. Dr. Bermingham and Dr. Giusti have no relevant financial disclosures.
SOURCE: Bermingham B et al. NACFC 2020, Abstract 645.
New CFTR [cystic fibrosis transmembrane conductance regulator] modulator therapies can offer life-altering benefits to some patients with cystic fibrosis, even those with advanced disease.
, according to a multicenter analysis of patients taking elexacaftor, tezacaftor, and ivacaftor.
The study participants had a percent predicted forced expiratory volume in 1 second (ppFEV1) of 40% or below, or other high-risk factors. Researchers compared them to control patients who were genetically ineligible for triple combination therapy.
Previous studies of such patients on individual drugs or previous combinations showed increases in lung function in patients with advanced disease, though the magnitude of improvement varied across regimens. “With this improvement, it’s unclear how CFTR modulators should affect lung transplant referral timing,” Brent Bermingham, MD, said during a presentation of the study at the virtual North American Cystic Fibrosis Conference.
“The rationale for our study was that despite patients with advanced lung disease being excluded from phase III trials (of elexacaftor, tezacaftor, and ivacaftor), they are receiving a therapy with an unknown clinical efficacy and safety profile,” said Dr. Bermingham, a pulmonary and critical care fellow at the Medical University of South Carolina, Charleston.
Lung transplant referral guidelines recommend that physicians initiate discussions about the potential benefit of lung transplant when FEV1 drops below 50% of the predicted value. Patients should be referred for a transplant when the value is below 50% and rapidly declining (>20% decline in the past 12 months), when it drops below 40% with accompanying predictors of shortened survival, or when it drops below 30%. The guidelines were published before approval of triple combination therapy.
The researchers conducted an open-label retrospective analysis of 60 patients started on triple combination therapy between September 2019 and February 2020 at three centers in the Southeast. They compared percent predicted ppFEV1 values prior to initiation of therapy to ppFEV1 values obtained 2-12 weeks after the start of therapy. Patients on therapy were compared with 10 genetically ineligible controls. The two groups were generally similar aside from genetic status, though 100% of the therapy group had pancreatic insufficiency, compared with 90% of controls (P = .013).
The therapeutic group experienced a 7.8% increase in ppFEV1 after starting therapy (P < .001), compared with a 0.5% decrease in controls (P = .65). Before initiation of therapy, 33% of the therapy group met the criteria for initiating a transplant discussion, while 67% had been recommended for transplant. After therapy, 55% met the criteria for discussion, 33% were recommended for transplant, and 12% no longer met the criteria for discussion of transplantation. Fifty percent of controls were in discussion, and this dropped to 40%, while 50% were referred for transplantation, and this increased to 60%. On therapy, transplant referral candidates had an increase of forced vital capacity from 48.9 to 59.16 (P < .001).
Adverse events were rare, with only one discontinuation that occurred following a lung transplant and was not believed to be treatment related.
“Our study had a large number of patients taken from multiple centers, which suggests generalizabilty and real-world experience,” said Dr. Bermingham.
The results are encouraging, said Robert J. Giusti, MD, clinical professor of pediatrics at the New York University and director of the Pediatric Cystic Fibrosis Center.
“We’re all remarking how wonderful patients feel these days. It’s really a disease-altering treatment. But for the high-risk group, whose FEV1 is less than 40%, those are the patients we’re more concerned about because we thought maybe they had too much lung disease, and that they wouldn’t benefit from triple combination. But they seem to be improving, so that’s very reassuring,” said Dr. Giusti, who was not involved in the study.
The study received funding from the Cystic Fibrosis Foundation and Dartmouth College. Dr. Bermingham and Dr. Giusti have no relevant financial disclosures.
SOURCE: Bermingham B et al. NACFC 2020, Abstract 645.
FROM NACFC 2020