User login
American Society of Hematology (ASH): ASH 2015
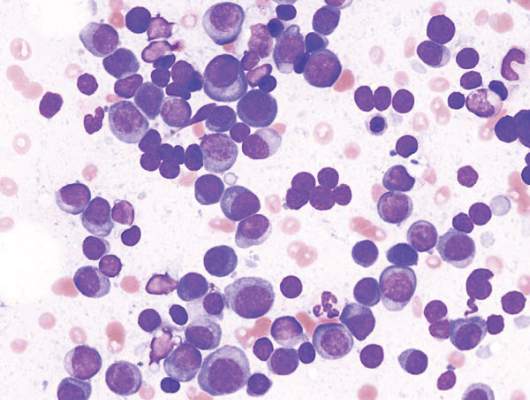
ASH: Genes tag increased risk for avascular necrosis in ALL patients under age 10
ORLANDO – Variants in mesenchymal stem cell genes that are important to bone and fat differentiation were associated with the risk of developing osteonecrosis in children under age 10 with acute lymphocytic leukemia, Dr. Seth E. Karol, of St. Jude Children’s Research Hospital in Memphis, reported at the annual meeting of the American Society of Hematology.

In a second study of children with ALL, Dr. Peter D. Cole, of Montefiore Medical Center, Albert Einstein College of Medicine, Bronx, NY, reported that patients under age 10 and homozygous for the 2R polymorphism in thymidylate synthase were at increased risk for avascular necrosis. For children over age 10 with ALL, homozygosity for the 2R polymorphism was linked with an increased risk of fracture.
The pathophysiology of bony morbidity differs depending on the patient’s age, Dr. Cole noted. “This is the first report identifying a genetic risk factor for avascular necrosis specifically among children younger than 10 years, a group where AVN is significantly less frequent.”
Bony morbidity is more common in children over age 10 with ALL, and related to exposure to crucial components of leukemia therapy including corticosteroids, asparaginase, and methotrexate. Therapy-induced osteonecrosis has become a limiting toxicity in the intensification of treatment for pediatric acute lymphoblastic leukemia (ALL), particularly among patients 10-20 years old.
However, children under 10 make up 75% of new pediatric ALL diagnoses, Dr. Karol said. So even though osteonecrosis occurs in just 3%-10% of children under age 10, those patients make up 40% of the cases.
In Dr. Cole’s study, research focused on 19 common genetic polymorphisms in peripheral blood or bone marrow collected at remission from 637 children treated for ALL.

The research targeted common variants within genes related to glucocorticoid metabolism, oxidative damage, and folate physiology. Children above and below the age of 10 years were evaluated separately. Multivariable models for bony morbidity included sex, WBC at diagnosis, race, final risk group, and asparaginase randomization. Blood was collected during treatment for analysis of serum and RBC folate.
Of the 637 patients tested, 627 achieved a complete remission and received post-induction therapy; about 10% of patients experienced avascular necrosis and 22% had at least 1 fracture. Both morbidities were significantly more common in patients aged 10 and older; 23% had avascular necrosis and 31% had a fracture. In younger children, 5.5% had avascular necrosis and 19% had fractures.
Of 626 patients tested, about 21% were homozygous for the 2R polymorphism in thymidylate synthase. In children less than age 10, this 2R/2R genotype was associated with nearly three times the rate of 5-year estimated incidence of avascular necrosis, almost 12%, as compared to the 4% rate seen in children with 2R/3R or 3R/3R genotypes.
Among children over age 10, this polymorphism was associated with an increased risk of bony fracture (multivariable HR 2.13; 95% CI 1.13-3.99; P=0.019).
Bony morbidity was not associated with the other tested polymorphisms previously linked to risk of avascular necrosis, including PAI-1 (rs6092), ABCB1 (rs1045642), and the vitamin D receptor Fok1 restriction site (rs228570). No significant association was observed between thymidylate synthase genotype and serum or RBC folate at any time point during therapy.
In the study reported by Dr. Karol, the researchers studied a discovery cohort of 82 cases of osteonecrosis and 287 controls treated on the Children’s Oncology Group (COG) NCI standard risk ALL protocol and tested for replication in 817 children less than 10 treated on the COG high risk ALL protocol.
Both discovery and replication genome-wide association studies adjusted for demographic and therapy variables known to modify the risk of osteonecrosis. Genes associated with the identified single nucleotide polymorphisms were evaluated for enrichment in biologically relevant pathways.
Within the discovery cohort and replication cohorts, top ranked variants were located near bone morphogenic protein 7 (BMP7) and PROX1-antisense RNA1. The top replicated non-synonymous SNP, rs34144324, was in a glutamate receptor gene, and the genotyping of this variant was verified in the whole exome sequencing data.
Pathway analysis of genes linked to top SNPs demonstrated enrichment in glutamate receptor signaling and adipogenesis pathways.
“Patients with osteonecrosis are 8-15 times more likely to possesses genetic variants near a gene important to bone development (BMP7) and between 3-6 times more likely to have variants near a gene important to fat levels in the blood (PROX1),” Dr. Karol reported.
Dr. Cole and Dr. Karol had no relevant financial disclosures.
On Twitter @maryjodales
ORLANDO – Variants in mesenchymal stem cell genes that are important to bone and fat differentiation were associated with the risk of developing osteonecrosis in children under age 10 with acute lymphocytic leukemia, Dr. Seth E. Karol, of St. Jude Children’s Research Hospital in Memphis, reported at the annual meeting of the American Society of Hematology.
In a second study of children with ALL, Dr. Peter D. Cole, of Montefiore Medical Center, Albert Einstein College of Medicine, Bronx, NY, reported that patients under age 10 and homozygous for the 2R polymorphism in thymidylate synthase were at increased risk for avascular necrosis. For children over age 10 with ALL, homozygosity for the 2R polymorphism was linked with an increased risk of fracture.
The pathophysiology of bony morbidity differs depending on the patient’s age, Dr. Cole noted. “This is the first report identifying a genetic risk factor for avascular necrosis specifically among children younger than 10 years, a group where AVN is significantly less frequent.”
Bony morbidity is more common in children over age 10 with ALL, and related to exposure to crucial components of leukemia therapy including corticosteroids, asparaginase, and methotrexate. Therapy-induced osteonecrosis has become a limiting toxicity in the intensification of treatment for pediatric acute lymphoblastic leukemia (ALL), particularly among patients 10-20 years old.
However, children under 10 make up 75% of new pediatric ALL diagnoses, Dr. Karol said. So even though osteonecrosis occurs in just 3%-10% of children under age 10, those patients make up 40% of the cases.
In Dr. Cole’s study, research focused on 19 common genetic polymorphisms in peripheral blood or bone marrow collected at remission from 637 children treated for ALL.
The research targeted common variants within genes related to glucocorticoid metabolism, oxidative damage, and folate physiology. Children above and below the age of 10 years were evaluated separately. Multivariable models for bony morbidity included sex, WBC at diagnosis, race, final risk group, and asparaginase randomization. Blood was collected during treatment for analysis of serum and RBC folate.
Of the 637 patients tested, 627 achieved a complete remission and received post-induction therapy; about 10% of patients experienced avascular necrosis and 22% had at least 1 fracture. Both morbidities were significantly more common in patients aged 10 and older; 23% had avascular necrosis and 31% had a fracture. In younger children, 5.5% had avascular necrosis and 19% had fractures.
Of 626 patients tested, about 21% were homozygous for the 2R polymorphism in thymidylate synthase. In children less than age 10, this 2R/2R genotype was associated with nearly three times the rate of 5-year estimated incidence of avascular necrosis, almost 12%, as compared to the 4% rate seen in children with 2R/3R or 3R/3R genotypes.
Among children over age 10, this polymorphism was associated with an increased risk of bony fracture (multivariable HR 2.13; 95% CI 1.13-3.99; P=0.019).
Bony morbidity was not associated with the other tested polymorphisms previously linked to risk of avascular necrosis, including PAI-1 (rs6092), ABCB1 (rs1045642), and the vitamin D receptor Fok1 restriction site (rs228570). No significant association was observed between thymidylate synthase genotype and serum or RBC folate at any time point during therapy.
In the study reported by Dr. Karol, the researchers studied a discovery cohort of 82 cases of osteonecrosis and 287 controls treated on the Children’s Oncology Group (COG) NCI standard risk ALL protocol and tested for replication in 817 children less than 10 treated on the COG high risk ALL protocol.
Both discovery and replication genome-wide association studies adjusted for demographic and therapy variables known to modify the risk of osteonecrosis. Genes associated with the identified single nucleotide polymorphisms were evaluated for enrichment in biologically relevant pathways.
Within the discovery cohort and replication cohorts, top ranked variants were located near bone morphogenic protein 7 (BMP7) and PROX1-antisense RNA1. The top replicated non-synonymous SNP, rs34144324, was in a glutamate receptor gene, and the genotyping of this variant was verified in the whole exome sequencing data.
Pathway analysis of genes linked to top SNPs demonstrated enrichment in glutamate receptor signaling and adipogenesis pathways.
“Patients with osteonecrosis are 8-15 times more likely to possesses genetic variants near a gene important to bone development (BMP7) and between 3-6 times more likely to have variants near a gene important to fat levels in the blood (PROX1),” Dr. Karol reported.
Dr. Cole and Dr. Karol had no relevant financial disclosures.
On Twitter @maryjodales
ORLANDO – Variants in mesenchymal stem cell genes that are important to bone and fat differentiation were associated with the risk of developing osteonecrosis in children under age 10 with acute lymphocytic leukemia, Dr. Seth E. Karol, of St. Jude Children’s Research Hospital in Memphis, reported at the annual meeting of the American Society of Hematology.
In a second study of children with ALL, Dr. Peter D. Cole, of Montefiore Medical Center, Albert Einstein College of Medicine, Bronx, NY, reported that patients under age 10 and homozygous for the 2R polymorphism in thymidylate synthase were at increased risk for avascular necrosis. For children over age 10 with ALL, homozygosity for the 2R polymorphism was linked with an increased risk of fracture.
The pathophysiology of bony morbidity differs depending on the patient’s age, Dr. Cole noted. “This is the first report identifying a genetic risk factor for avascular necrosis specifically among children younger than 10 years, a group where AVN is significantly less frequent.”
Bony morbidity is more common in children over age 10 with ALL, and related to exposure to crucial components of leukemia therapy including corticosteroids, asparaginase, and methotrexate. Therapy-induced osteonecrosis has become a limiting toxicity in the intensification of treatment for pediatric acute lymphoblastic leukemia (ALL), particularly among patients 10-20 years old.
However, children under 10 make up 75% of new pediatric ALL diagnoses, Dr. Karol said. So even though osteonecrosis occurs in just 3%-10% of children under age 10, those patients make up 40% of the cases.
In Dr. Cole’s study, research focused on 19 common genetic polymorphisms in peripheral blood or bone marrow collected at remission from 637 children treated for ALL.
The research targeted common variants within genes related to glucocorticoid metabolism, oxidative damage, and folate physiology. Children above and below the age of 10 years were evaluated separately. Multivariable models for bony morbidity included sex, WBC at diagnosis, race, final risk group, and asparaginase randomization. Blood was collected during treatment for analysis of serum and RBC folate.
Of the 637 patients tested, 627 achieved a complete remission and received post-induction therapy; about 10% of patients experienced avascular necrosis and 22% had at least 1 fracture. Both morbidities were significantly more common in patients aged 10 and older; 23% had avascular necrosis and 31% had a fracture. In younger children, 5.5% had avascular necrosis and 19% had fractures.
Of 626 patients tested, about 21% were homozygous for the 2R polymorphism in thymidylate synthase. In children less than age 10, this 2R/2R genotype was associated with nearly three times the rate of 5-year estimated incidence of avascular necrosis, almost 12%, as compared to the 4% rate seen in children with 2R/3R or 3R/3R genotypes.
Among children over age 10, this polymorphism was associated with an increased risk of bony fracture (multivariable HR 2.13; 95% CI 1.13-3.99; P=0.019).
Bony morbidity was not associated with the other tested polymorphisms previously linked to risk of avascular necrosis, including PAI-1 (rs6092), ABCB1 (rs1045642), and the vitamin D receptor Fok1 restriction site (rs228570). No significant association was observed between thymidylate synthase genotype and serum or RBC folate at any time point during therapy.
In the study reported by Dr. Karol, the researchers studied a discovery cohort of 82 cases of osteonecrosis and 287 controls treated on the Children’s Oncology Group (COG) NCI standard risk ALL protocol and tested for replication in 817 children less than 10 treated on the COG high risk ALL protocol.
Both discovery and replication genome-wide association studies adjusted for demographic and therapy variables known to modify the risk of osteonecrosis. Genes associated with the identified single nucleotide polymorphisms were evaluated for enrichment in biologically relevant pathways.
Within the discovery cohort and replication cohorts, top ranked variants were located near bone morphogenic protein 7 (BMP7) and PROX1-antisense RNA1. The top replicated non-synonymous SNP, rs34144324, was in a glutamate receptor gene, and the genotyping of this variant was verified in the whole exome sequencing data.
Pathway analysis of genes linked to top SNPs demonstrated enrichment in glutamate receptor signaling and adipogenesis pathways.
“Patients with osteonecrosis are 8-15 times more likely to possesses genetic variants near a gene important to bone development (BMP7) and between 3-6 times more likely to have variants near a gene important to fat levels in the blood (PROX1),” Dr. Karol reported.
Dr. Cole and Dr. Karol had no relevant financial disclosures.
On Twitter @maryjodales
AT ASH 2015
Key clinical point: Gene profiles can identify ALL patients under age 10 at increased risk for avascular necrosis.
Major finding: In children under 10 who were homozygous for the 2R polymorphism in thymidylate synthase the rate of 5-year estimated incidence of avascular necrosis was nearly 12%, three times higher than the 4% rate seen in children with 2R/3R or 3R/3R genotypes.
Data source: Research focused on 19 common genetic polymorphisms in peripheral blood or bone marrow collected at remission from 637 children treated for ALL.
Disclosures: Dr. Cole and Dr. Karol had no relevant financial disclosures.

VIDEO: Gene therapy repairs immune function in older patients with SCID-XI
ORLANDO – Older patients with x-linked severe combined immunodeficiency syndrome (SCID-X1) treated initially with haploidentical stem cell transplantation without conditioning have impaired immunity and experience severe, life-long morbidities. Dr. Harry L. Malech, chief of the Laboratory of Host Defenses and Chief, Genetic Immunotherapy Section, U.S. National Institute of Allergy and Infectious Diseases, Bethesda, Md., discusses how investigators at the U.S. NAID are treating autologous CD34 cells from patients with SCID-X1 with a lentiviral vector that transduced the cells with normal copies of the gene which in its mutated form causes SCID-X1. In early trials, they have been able to significantly improve immune function in some patients without an apparent increase in risk for malignancies, a common problem with earlier gene therapies.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ORLANDO – Older patients with x-linked severe combined immunodeficiency syndrome (SCID-X1) treated initially with haploidentical stem cell transplantation without conditioning have impaired immunity and experience severe, life-long morbidities. Dr. Harry L. Malech, chief of the Laboratory of Host Defenses and Chief, Genetic Immunotherapy Section, U.S. National Institute of Allergy and Infectious Diseases, Bethesda, Md., discusses how investigators at the U.S. NAID are treating autologous CD34 cells from patients with SCID-X1 with a lentiviral vector that transduced the cells with normal copies of the gene which in its mutated form causes SCID-X1. In early trials, they have been able to significantly improve immune function in some patients without an apparent increase in risk for malignancies, a common problem with earlier gene therapies.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ORLANDO – Older patients with x-linked severe combined immunodeficiency syndrome (SCID-X1) treated initially with haploidentical stem cell transplantation without conditioning have impaired immunity and experience severe, life-long morbidities. Dr. Harry L. Malech, chief of the Laboratory of Host Defenses and Chief, Genetic Immunotherapy Section, U.S. National Institute of Allergy and Infectious Diseases, Bethesda, Md., discusses how investigators at the U.S. NAID are treating autologous CD34 cells from patients with SCID-X1 with a lentiviral vector that transduced the cells with normal copies of the gene which in its mutated form causes SCID-X1. In early trials, they have been able to significantly improve immune function in some patients without an apparent increase in risk for malignancies, a common problem with earlier gene therapies.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT ASH 2015

ASH: Pill bottles flag noncompliance in patients on ALL maintenance
ORLANDO – Pill containers are not known for eloquence, but they tell a disturbing tale of patient over-reporting of adherence to essential leukemia maintenance therapy.
Among 416 children with acute lymphoblastic leukemia (ALL) in first remission, the number of days patients or their guardians reported that the child took his/her daily oral 6-mercaptopurine (6MP) for maintenance was significantly higher than the number of days the pill bottles were opened, as reported by an electronic medication management system.
“Over-reporting was more likely in patients who were non-adherent, older, of non-white race, and came from households with lower paternal education,” said Dr. Wendy Landier from the Institute for Cancer Outcomes and Survivorship at the University of Alabama at Birmingham.

The results suggest that patient self-reports of adherence to 6MP oral maintenance therapy may be unreliable, and should be taken with a grain of salt, Dr. Landier said in a briefing at the American Society of Hematology annual meeting.
The investigators had previously reported that poor adherence to oral 6MP maintenance therapy was associated with a nealy four-fold increased risk for relapse (JCO 30[17]:2094-101, Blood 124[15]:2345-53).
To better understand why some patients are poorly adherent to 6MP maintenance therapy, the investigator provided 416 children with ALL in first remission with pill containers equipped with a Medication Event Management System (MEMS) that electronically recorded the dates and times that each pill bottle was opened over a 16-week period. Patients age 12 and older or the parents/guardians of younger children were also asked to report the date and time of each daily oral dose at the end of each 28-day study month.
The authors collected and evaluated a total of 1344 patient-months of self-report and MEMS data, compared the records, and stratified patients as either “perfect reporters”, whose self-reports matched the objective MEMS data; “over-reporters”, who self-report exceeded the MEMS data on 5 or more days per month for more than half of study months, and “others.”The median patient age at study entry was 6 years (range 2 to 20 years).
The authors reported in their study that 40.4% of patients were not adherent to oral 6MP therapy, as evidence by a mismatch between self-report and objective (MEMS) reporting.
The overall adjusted mean number of self-reported days per month that patients took their pills ranged from a low of 25.8+5.3 to a high of 26.1+4.5. The pill bottles, however, told a different tale, reporting that they had been opened from a low of 22.8 ± 6.4 to a high of 25.4 ± 4.5 days per month.
Month-by-month correlations between self-report and MEMS 0.36 to 0.58, and were all statistically significant.
In logistic regression models adjusted for thiopurine methyltransferase (TMPT) genotype, 6MP dose intensity, and 6-thiogaunine levels in red cells, significant predictors for over-reporting included older age, with an odds ratio (OR) of 1.07 for every 1-year increase in age (P = .04); Hispanic origin (OR 2.4, P = .02); Asian origin, (OR 3.1 P = .02; African-American origin (OR 5.3, P < .001); paternal education level lower than college (OR 2.1, P = .02); and 6MP non-adherence (OR 8.6, P < .0001).
Dr. Landier noted that 78.6% of over-reporters were non-adherent, compared with only 2% of perfect reporters.

“The real importance of this paper from my perspective is this: there is a famous quote that if the patient doesn’t take the medication it can’t work and in an era where we’re trying to improve the care of patients, one of the things we oftentimes forget that if the medication isn’t being taken, the patient can’t get better from it,” said Dr. Mark Crowther, Professor and Chair in the Department of Pathology and Molecular Medicine of McMaster University in Hamilton, Ontario, Canada. Dr. Crowther moderated a briefing where Dr. Cserti-Gazdewich presented the data.
The study was supported by grants from the National Institutes of Health and from St. Baldrick’s Foundation. Dr. Landier and Dr. Crowther reported no relevant conflicts of interest.
ORLANDO – Pill containers are not known for eloquence, but they tell a disturbing tale of patient over-reporting of adherence to essential leukemia maintenance therapy.
Among 416 children with acute lymphoblastic leukemia (ALL) in first remission, the number of days patients or their guardians reported that the child took his/her daily oral 6-mercaptopurine (6MP) for maintenance was significantly higher than the number of days the pill bottles were opened, as reported by an electronic medication management system.
“Over-reporting was more likely in patients who were non-adherent, older, of non-white race, and came from households with lower paternal education,” said Dr. Wendy Landier from the Institute for Cancer Outcomes and Survivorship at the University of Alabama at Birmingham.
The results suggest that patient self-reports of adherence to 6MP oral maintenance therapy may be unreliable, and should be taken with a grain of salt, Dr. Landier said in a briefing at the American Society of Hematology annual meeting.
The investigators had previously reported that poor adherence to oral 6MP maintenance therapy was associated with a nealy four-fold increased risk for relapse (JCO 30[17]:2094-101, Blood 124[15]:2345-53).
To better understand why some patients are poorly adherent to 6MP maintenance therapy, the investigator provided 416 children with ALL in first remission with pill containers equipped with a Medication Event Management System (MEMS) that electronically recorded the dates and times that each pill bottle was opened over a 16-week period. Patients age 12 and older or the parents/guardians of younger children were also asked to report the date and time of each daily oral dose at the end of each 28-day study month.
The authors collected and evaluated a total of 1344 patient-months of self-report and MEMS data, compared the records, and stratified patients as either “perfect reporters”, whose self-reports matched the objective MEMS data; “over-reporters”, who self-report exceeded the MEMS data on 5 or more days per month for more than half of study months, and “others.”The median patient age at study entry was 6 years (range 2 to 20 years).
The authors reported in their study that 40.4% of patients were not adherent to oral 6MP therapy, as evidence by a mismatch between self-report and objective (MEMS) reporting.
The overall adjusted mean number of self-reported days per month that patients took their pills ranged from a low of 25.8+5.3 to a high of 26.1+4.5. The pill bottles, however, told a different tale, reporting that they had been opened from a low of 22.8 ± 6.4 to a high of 25.4 ± 4.5 days per month.
Month-by-month correlations between self-report and MEMS 0.36 to 0.58, and were all statistically significant.
In logistic regression models adjusted for thiopurine methyltransferase (TMPT) genotype, 6MP dose intensity, and 6-thiogaunine levels in red cells, significant predictors for over-reporting included older age, with an odds ratio (OR) of 1.07 for every 1-year increase in age (P = .04); Hispanic origin (OR 2.4, P = .02); Asian origin, (OR 3.1 P = .02; African-American origin (OR 5.3, P < .001); paternal education level lower than college (OR 2.1, P = .02); and 6MP non-adherence (OR 8.6, P < .0001).
Dr. Landier noted that 78.6% of over-reporters were non-adherent, compared with only 2% of perfect reporters.
“The real importance of this paper from my perspective is this: there is a famous quote that if the patient doesn’t take the medication it can’t work and in an era where we’re trying to improve the care of patients, one of the things we oftentimes forget that if the medication isn’t being taken, the patient can’t get better from it,” said Dr. Mark Crowther, Professor and Chair in the Department of Pathology and Molecular Medicine of McMaster University in Hamilton, Ontario, Canada. Dr. Crowther moderated a briefing where Dr. Cserti-Gazdewich presented the data.
The study was supported by grants from the National Institutes of Health and from St. Baldrick’s Foundation. Dr. Landier and Dr. Crowther reported no relevant conflicts of interest.
ORLANDO – Pill containers are not known for eloquence, but they tell a disturbing tale of patient over-reporting of adherence to essential leukemia maintenance therapy.
Among 416 children with acute lymphoblastic leukemia (ALL) in first remission, the number of days patients or their guardians reported that the child took his/her daily oral 6-mercaptopurine (6MP) for maintenance was significantly higher than the number of days the pill bottles were opened, as reported by an electronic medication management system.
“Over-reporting was more likely in patients who were non-adherent, older, of non-white race, and came from households with lower paternal education,” said Dr. Wendy Landier from the Institute for Cancer Outcomes and Survivorship at the University of Alabama at Birmingham.
The results suggest that patient self-reports of adherence to 6MP oral maintenance therapy may be unreliable, and should be taken with a grain of salt, Dr. Landier said in a briefing at the American Society of Hematology annual meeting.
The investigators had previously reported that poor adherence to oral 6MP maintenance therapy was associated with a nealy four-fold increased risk for relapse (JCO 30[17]:2094-101, Blood 124[15]:2345-53).
To better understand why some patients are poorly adherent to 6MP maintenance therapy, the investigator provided 416 children with ALL in first remission with pill containers equipped with a Medication Event Management System (MEMS) that electronically recorded the dates and times that each pill bottle was opened over a 16-week period. Patients age 12 and older or the parents/guardians of younger children were also asked to report the date and time of each daily oral dose at the end of each 28-day study month.
The authors collected and evaluated a total of 1344 patient-months of self-report and MEMS data, compared the records, and stratified patients as either “perfect reporters”, whose self-reports matched the objective MEMS data; “over-reporters”, who self-report exceeded the MEMS data on 5 or more days per month for more than half of study months, and “others.”The median patient age at study entry was 6 years (range 2 to 20 years).
The authors reported in their study that 40.4% of patients were not adherent to oral 6MP therapy, as evidence by a mismatch between self-report and objective (MEMS) reporting.
The overall adjusted mean number of self-reported days per month that patients took their pills ranged from a low of 25.8+5.3 to a high of 26.1+4.5. The pill bottles, however, told a different tale, reporting that they had been opened from a low of 22.8 ± 6.4 to a high of 25.4 ± 4.5 days per month.
Month-by-month correlations between self-report and MEMS 0.36 to 0.58, and were all statistically significant.
In logistic regression models adjusted for thiopurine methyltransferase (TMPT) genotype, 6MP dose intensity, and 6-thiogaunine levels in red cells, significant predictors for over-reporting included older age, with an odds ratio (OR) of 1.07 for every 1-year increase in age (P = .04); Hispanic origin (OR 2.4, P = .02); Asian origin, (OR 3.1 P = .02; African-American origin (OR 5.3, P < .001); paternal education level lower than college (OR 2.1, P = .02); and 6MP non-adherence (OR 8.6, P < .0001).
Dr. Landier noted that 78.6% of over-reporters were non-adherent, compared with only 2% of perfect reporters.
“The real importance of this paper from my perspective is this: there is a famous quote that if the patient doesn’t take the medication it can’t work and in an era where we’re trying to improve the care of patients, one of the things we oftentimes forget that if the medication isn’t being taken, the patient can’t get better from it,” said Dr. Mark Crowther, Professor and Chair in the Department of Pathology and Molecular Medicine of McMaster University in Hamilton, Ontario, Canada. Dr. Crowther moderated a briefing where Dr. Cserti-Gazdewich presented the data.
The study was supported by grants from the National Institutes of Health and from St. Baldrick’s Foundation. Dr. Landier and Dr. Crowther reported no relevant conflicts of interest.
AT ASH 2015
Key clinical point: Patients in first ALL remission may not be as adherent to 6MP maintenance therapy as they claim.
Major finding: In all, 40.4% of patients were not adherent to oral 6MP maintenance therapy.
Data source: Observational study comparing patient reported and electronically monitored dosing in 416 children/young adults with ALL in first remission.
Disclosures: The study was supported by grants from the National Institutes of Health and from St. Baldrick’s Foundation. Dr. Landier and Dr. Crowther reported no relevant conflicts of interest.

ASH: Genes, induction response identify high risk childhood B-lymphoblastic leukemia patients with good outcomes
ORLANDO – Cytogenetic profile and response to initial induction therapy can identify a subgroup of National Cancer Institute (NCI) high risk childhood B-lymphoblastic leukemia patients who have good outcomes, Dr. Elizabeth Raetz said, reporting results for her colleagues in the Children’s Oncology Group (COG).
Outcomes were excellent for patients with favorable cytogenetic features and rapid minimal residual disease responses during induction therapy, said Dr. Raetz, of Huntsman Cancer Institute and Primary Children’s Hospital, University of Utah, Salt Lake City. Notably, 5-year overall survival (OS) was over 98% for those with favorable cytogenetic subsets, nearly half of all patients, in a combined analysis of standard risk and high risk patients.

The findings suggest that these patients would not benefit from further intensification of their chemotherapy, sparing them possible toxicity and late effects.
The COG used clinical, biologic, and early disease response measures to study 11,144 patients, aged 1-30 years, enrolled on the COG AALL03B1 classification study. Patients began either a standard risk 3-drug or high risk 4-drug induction therapy based on NCI risk group, she said at the annual meeting of the American Society of Hematology.
After induction therapy, patients were classified into low (29%), standard (33%), high (34%), or very high (4%) risk groups for treatment allocation. Variables used for risk classification included age, initial WBC, extramedullary disease status, blast cytogenetics, early treatment response based on bone marrow morphology and minimal residual disease in marrow at day 29.
Rapid early response was defined as having less than 5% blasts in bone marrow by day 15 plus flow cytometry-based minimal residual disease less than 0.1% on day 29 of induction. Those with either 5% or more blasts in their marrow at day 15 or minimal residual disease of 0.1% or more at day 29 were deemed slow early responders.
Of the 96% of patients evaluable for post-induction treatment assignment, 5104 (65%) were treated for NCI standard risk and 2791 were treated for NCI high-risk B-ALL.
Rapid early responses were seen in 84% and slow early responses in 16%. For those with rapid early responses, the 5-year event-free survival rates was 89% and the overall survival rate was 95%. For those with slow early responses, EFS was 68% and OS was 84%.
The standard risk and high risk groups were then combined and analyzed based on cytogenetic subtype. The favorable gene ETV6-RUNX1 was seen in 26% (1,928 patients) and associated with an EFS of 93% as compared to an EFS of 84% for the 5578 patients without ETV6-RUNX1. OS was 98% for positive patients and 92% for negative patients. In the 21% (1,483 patients) with the triple trisomy 4/10/17, EFS was 95% as compared to an EFS of 84% for the 5,603 patients without the trisomy. OS was 99% and 92%, respectively.
In a subsequent analysis using current COG minimum residual disease response measures – with rapid early response defined as day 8 blood minimal residual disease less than 1% and day 29 marrow minimal residual disease less than 0.01% – the 243 high risk patients who had favorable cytogenetics and no detectable leukemia cells in their CSF had 5 years EFS of 95% and OS of 98%.
Dr. Raetz had no relevant financial disclosures.
Abstract 807: Genetic and Response-Based Risk Classification Identifies a Subgroup of NCI High Risk Childhood B-Lymphoblastic Leukemia (HR B-ALL) with Outstanding Outcomes: A Report from the Children’s Oncology Group (COG).
On Twitter @maryjodales
ORLANDO – Cytogenetic profile and response to initial induction therapy can identify a subgroup of National Cancer Institute (NCI) high risk childhood B-lymphoblastic leukemia patients who have good outcomes, Dr. Elizabeth Raetz said, reporting results for her colleagues in the Children’s Oncology Group (COG).
Outcomes were excellent for patients with favorable cytogenetic features and rapid minimal residual disease responses during induction therapy, said Dr. Raetz, of Huntsman Cancer Institute and Primary Children’s Hospital, University of Utah, Salt Lake City. Notably, 5-year overall survival (OS) was over 98% for those with favorable cytogenetic subsets, nearly half of all patients, in a combined analysis of standard risk and high risk patients.
The findings suggest that these patients would not benefit from further intensification of their chemotherapy, sparing them possible toxicity and late effects.
The COG used clinical, biologic, and early disease response measures to study 11,144 patients, aged 1-30 years, enrolled on the COG AALL03B1 classification study. Patients began either a standard risk 3-drug or high risk 4-drug induction therapy based on NCI risk group, she said at the annual meeting of the American Society of Hematology.
After induction therapy, patients were classified into low (29%), standard (33%), high (34%), or very high (4%) risk groups for treatment allocation. Variables used for risk classification included age, initial WBC, extramedullary disease status, blast cytogenetics, early treatment response based on bone marrow morphology and minimal residual disease in marrow at day 29.
Rapid early response was defined as having less than 5% blasts in bone marrow by day 15 plus flow cytometry-based minimal residual disease less than 0.1% on day 29 of induction. Those with either 5% or more blasts in their marrow at day 15 or minimal residual disease of 0.1% or more at day 29 were deemed slow early responders.
Of the 96% of patients evaluable for post-induction treatment assignment, 5104 (65%) were treated for NCI standard risk and 2791 were treated for NCI high-risk B-ALL.
Rapid early responses were seen in 84% and slow early responses in 16%. For those with rapid early responses, the 5-year event-free survival rates was 89% and the overall survival rate was 95%. For those with slow early responses, EFS was 68% and OS was 84%.
The standard risk and high risk groups were then combined and analyzed based on cytogenetic subtype. The favorable gene ETV6-RUNX1 was seen in 26% (1,928 patients) and associated with an EFS of 93% as compared to an EFS of 84% for the 5578 patients without ETV6-RUNX1. OS was 98% for positive patients and 92% for negative patients. In the 21% (1,483 patients) with the triple trisomy 4/10/17, EFS was 95% as compared to an EFS of 84% for the 5,603 patients without the trisomy. OS was 99% and 92%, respectively.
In a subsequent analysis using current COG minimum residual disease response measures – with rapid early response defined as day 8 blood minimal residual disease less than 1% and day 29 marrow minimal residual disease less than 0.01% – the 243 high risk patients who had favorable cytogenetics and no detectable leukemia cells in their CSF had 5 years EFS of 95% and OS of 98%.
Dr. Raetz had no relevant financial disclosures.
Abstract 807: Genetic and Response-Based Risk Classification Identifies a Subgroup of NCI High Risk Childhood B-Lymphoblastic Leukemia (HR B-ALL) with Outstanding Outcomes: A Report from the Children’s Oncology Group (COG).
On Twitter @maryjodales
ORLANDO – Cytogenetic profile and response to initial induction therapy can identify a subgroup of National Cancer Institute (NCI) high risk childhood B-lymphoblastic leukemia patients who have good outcomes, Dr. Elizabeth Raetz said, reporting results for her colleagues in the Children’s Oncology Group (COG).
Outcomes were excellent for patients with favorable cytogenetic features and rapid minimal residual disease responses during induction therapy, said Dr. Raetz, of Huntsman Cancer Institute and Primary Children’s Hospital, University of Utah, Salt Lake City. Notably, 5-year overall survival (OS) was over 98% for those with favorable cytogenetic subsets, nearly half of all patients, in a combined analysis of standard risk and high risk patients.
The findings suggest that these patients would not benefit from further intensification of their chemotherapy, sparing them possible toxicity and late effects.
The COG used clinical, biologic, and early disease response measures to study 11,144 patients, aged 1-30 years, enrolled on the COG AALL03B1 classification study. Patients began either a standard risk 3-drug or high risk 4-drug induction therapy based on NCI risk group, she said at the annual meeting of the American Society of Hematology.
After induction therapy, patients were classified into low (29%), standard (33%), high (34%), or very high (4%) risk groups for treatment allocation. Variables used for risk classification included age, initial WBC, extramedullary disease status, blast cytogenetics, early treatment response based on bone marrow morphology and minimal residual disease in marrow at day 29.
Rapid early response was defined as having less than 5% blasts in bone marrow by day 15 plus flow cytometry-based minimal residual disease less than 0.1% on day 29 of induction. Those with either 5% or more blasts in their marrow at day 15 or minimal residual disease of 0.1% or more at day 29 were deemed slow early responders.
Of the 96% of patients evaluable for post-induction treatment assignment, 5104 (65%) were treated for NCI standard risk and 2791 were treated for NCI high-risk B-ALL.
Rapid early responses were seen in 84% and slow early responses in 16%. For those with rapid early responses, the 5-year event-free survival rates was 89% and the overall survival rate was 95%. For those with slow early responses, EFS was 68% and OS was 84%.
The standard risk and high risk groups were then combined and analyzed based on cytogenetic subtype. The favorable gene ETV6-RUNX1 was seen in 26% (1,928 patients) and associated with an EFS of 93% as compared to an EFS of 84% for the 5578 patients without ETV6-RUNX1. OS was 98% for positive patients and 92% for negative patients. In the 21% (1,483 patients) with the triple trisomy 4/10/17, EFS was 95% as compared to an EFS of 84% for the 5,603 patients without the trisomy. OS was 99% and 92%, respectively.
In a subsequent analysis using current COG minimum residual disease response measures – with rapid early response defined as day 8 blood minimal residual disease less than 1% and day 29 marrow minimal residual disease less than 0.01% – the 243 high risk patients who had favorable cytogenetics and no detectable leukemia cells in their CSF had 5 years EFS of 95% and OS of 98%.
Dr. Raetz had no relevant financial disclosures.
Abstract 807: Genetic and Response-Based Risk Classification Identifies a Subgroup of NCI High Risk Childhood B-Lymphoblastic Leukemia (HR B-ALL) with Outstanding Outcomes: A Report from the Children’s Oncology Group (COG).
On Twitter @maryjodales
AT ASH 2015
Key clinical point: Knowing which patients would not benefit from further intensification of their chemotherapy might spare them possible toxicity and late effects.
Major finding: The 243 high risk patients who had favorable cytogenetics and no detectable leukemia cells in their CSF had 5 year EFS of 94% and OS of 98%.
Data source: 11,144 patients, aged 1-30 years, enrolled on the COG AALL03B1 classification study.
Disclosures: Dr. Raetz had no relevant financial disclosures.

ASH: Longer-stored RBCs equivalent to shorter for children with severe anemia
ORLANDO – Stored red blood cells continue to do their primary job of tissue reoxygenation for more than a month after being collected, a finding that has profound positive implications for countries where blood products are in perennially short supply and demand is high, said investigators from Africa and North America.
In a randomized controlled clinical trial, red blood cells (RBCs) stored for 25 to 35 days were not-inferior to RBCs stored for 1 to 10 days for tissue reox-ygenation in African children with severe anemia with lactic acidosis.
“We found no justification to shorten the current storage duration for RBCs judged by their fundamental role to deliver oxygen,” said Dr. Christine M. Cserti-Gazdewich from the University of Toronto, Ontario, Canada.
Their findings suggest that stored RBCs should be evaluated by their ability to effectively deliver oxygen to tissues rather than by the cell survival measures and in vitro markers of hemolysis used as the current standard for viability by regulatory agencies, Dr. Cserti-Gazdewich said at the American Society of Hematology annual meeting.
The study clinicaltrials.gov ID NCT01586923 is also published online in JAMA.
“This study won’t change our clinical practice, but it will substantiate what we have been doing,” said co-author Dr. Walter H “Sunny” Dzik from Mas-sachusetts General Hospital in Boston, in an interview.
“We didn’t go into this business to harm people, yet there’s a lot of lab data that has been pointing fingers at us, suggesting rather forcefully that by try-ing to manage blood inventories using older blood that we’re harming people,” he said.
Concerns about the shelf life of RBCs derive from evidence that the cells undergo cumulative changes in structure, biochemistry, and enzymatic active that ultimately could degrade their ability to deliver oxygen to target tissues. But those studies were based largely on laboratory findings and not on clinical practice, she noted.
To see whether RBCs stored for up to 5 weeks could be as effective at tissue oxygenation as more recently packaged cells, the investigators conducted a randomized clinical trial at a university hospital urgent-care facility in Kampala, Uganda, where diseases such as malaria and sickle-cell anemia, as well as malnutrion, cause severe anemia on a scale seldom seen in the developed world.
They enrolled 290 children from the ages of 6 to 60 months who presented with lactic acidosis due to severe anemia in the absence of shock, trauma, impaired cardiac function, refractory hypoxia, liver disease, or tissue injury. The patients all had hemoglobin levels of 5 g/dL or5 less, and serum lactate levels of 5 mM or greater.
The patients were randomly assigned, 145 to each study arm, to receive leukoreduced RBCs stored for either 1-10 days or from 25 to 35 days. All patients received 10 mL/kg of RBCs over 2 hours at the start of the study and, if indicated per protocol, an additional 10 mL/kg during hours 4-6. Blood lactate levels were measured at baseline and at 2, 4, 6, 8 and 24 hours.
The mean hemoglobin level at presentation was 3.7 ±1.3 g/dL and mean lactate was 9.3 ±3.4 mM.
The investigators found that the proportion of patients achieving a lactate ≤ 3 mM at 8 hours was 58% among patients who received the short-storage RBCs, compared with 61% among patients who received long-storage cells (P = 0.72). This result met the primary endpoint of non-inferiority for long-er-stored RBCs.
In fact, there were no significant differences between the groups in mean lactate levels at any of the time points beyond baseline.
There were no significant differences in lactate clearance, and there were similar degrees of improvement in clinical assessments, serial measurements of hemoglobin concentration, cerebral tissue oxygen saturation, and electrolyte abnormalities following RBC transfusions.
Adverse events, 30-day recovery, and survival were also comparable between the groups.
The authors also found in a pre-specified sub-group analysis that there were no significant between group differences in those patients who received a total of 20 mL/kg RBCs, Dr. Cserti-Gazdewich said.
“This is a crushingly urgent problem, in that there have been belief systems developed over the last couple of years that older blood is not as good as newer blood, without any real evidence to support that. The evidence that Christine is [presenting], I think, is pretty compelling evidence that that hy-pothesis is incorrect,” commented Dr. Mark Crowther, Professor and Chair in the Department of Pathology and Molecular Medicine of McMaster Univer-sity in Hamilton, Ontario, Canada. Dr. Crowther moderated a briefing where Dr. Cserti-Gazdewich presented the data.
The study was funded by the National Institutes of Health. Dr. Cserti-Gazdewich, Dr. Dzik, and Dr. Crowther reported having no relevant conflicts of interest.
ORLANDO – Stored red blood cells continue to do their primary job of tissue reoxygenation for more than a month after being collected, a finding that has profound positive implications for countries where blood products are in perennially short supply and demand is high, said investigators from Africa and North America.
In a randomized controlled clinical trial, red blood cells (RBCs) stored for 25 to 35 days were not-inferior to RBCs stored for 1 to 10 days for tissue reox-ygenation in African children with severe anemia with lactic acidosis.
“We found no justification to shorten the current storage duration for RBCs judged by their fundamental role to deliver oxygen,” said Dr. Christine M. Cserti-Gazdewich from the University of Toronto, Ontario, Canada.
Their findings suggest that stored RBCs should be evaluated by their ability to effectively deliver oxygen to tissues rather than by the cell survival measures and in vitro markers of hemolysis used as the current standard for viability by regulatory agencies, Dr. Cserti-Gazdewich said at the American Society of Hematology annual meeting.
The study clinicaltrials.gov ID NCT01586923 is also published online in JAMA.
“This study won’t change our clinical practice, but it will substantiate what we have been doing,” said co-author Dr. Walter H “Sunny” Dzik from Mas-sachusetts General Hospital in Boston, in an interview.
“We didn’t go into this business to harm people, yet there’s a lot of lab data that has been pointing fingers at us, suggesting rather forcefully that by try-ing to manage blood inventories using older blood that we’re harming people,” he said.
Concerns about the shelf life of RBCs derive from evidence that the cells undergo cumulative changes in structure, biochemistry, and enzymatic active that ultimately could degrade their ability to deliver oxygen to target tissues. But those studies were based largely on laboratory findings and not on clinical practice, she noted.
To see whether RBCs stored for up to 5 weeks could be as effective at tissue oxygenation as more recently packaged cells, the investigators conducted a randomized clinical trial at a university hospital urgent-care facility in Kampala, Uganda, where diseases such as malaria and sickle-cell anemia, as well as malnutrion, cause severe anemia on a scale seldom seen in the developed world.
They enrolled 290 children from the ages of 6 to 60 months who presented with lactic acidosis due to severe anemia in the absence of shock, trauma, impaired cardiac function, refractory hypoxia, liver disease, or tissue injury. The patients all had hemoglobin levels of 5 g/dL or5 less, and serum lactate levels of 5 mM or greater.
The patients were randomly assigned, 145 to each study arm, to receive leukoreduced RBCs stored for either 1-10 days or from 25 to 35 days. All patients received 10 mL/kg of RBCs over 2 hours at the start of the study and, if indicated per protocol, an additional 10 mL/kg during hours 4-6. Blood lactate levels were measured at baseline and at 2, 4, 6, 8 and 24 hours.
The mean hemoglobin level at presentation was 3.7 ±1.3 g/dL and mean lactate was 9.3 ±3.4 mM.
The investigators found that the proportion of patients achieving a lactate ≤ 3 mM at 8 hours was 58% among patients who received the short-storage RBCs, compared with 61% among patients who received long-storage cells (P = 0.72). This result met the primary endpoint of non-inferiority for long-er-stored RBCs.
In fact, there were no significant differences between the groups in mean lactate levels at any of the time points beyond baseline.
There were no significant differences in lactate clearance, and there were similar degrees of improvement in clinical assessments, serial measurements of hemoglobin concentration, cerebral tissue oxygen saturation, and electrolyte abnormalities following RBC transfusions.
Adverse events, 30-day recovery, and survival were also comparable between the groups.
The authors also found in a pre-specified sub-group analysis that there were no significant between group differences in those patients who received a total of 20 mL/kg RBCs, Dr. Cserti-Gazdewich said.
“This is a crushingly urgent problem, in that there have been belief systems developed over the last couple of years that older blood is not as good as newer blood, without any real evidence to support that. The evidence that Christine is [presenting], I think, is pretty compelling evidence that that hy-pothesis is incorrect,” commented Dr. Mark Crowther, Professor and Chair in the Department of Pathology and Molecular Medicine of McMaster Univer-sity in Hamilton, Ontario, Canada. Dr. Crowther moderated a briefing where Dr. Cserti-Gazdewich presented the data.
The study was funded by the National Institutes of Health. Dr. Cserti-Gazdewich, Dr. Dzik, and Dr. Crowther reported having no relevant conflicts of interest.
ORLANDO – Stored red blood cells continue to do their primary job of tissue reoxygenation for more than a month after being collected, a finding that has profound positive implications for countries where blood products are in perennially short supply and demand is high, said investigators from Africa and North America.
In a randomized controlled clinical trial, red blood cells (RBCs) stored for 25 to 35 days were not-inferior to RBCs stored for 1 to 10 days for tissue reox-ygenation in African children with severe anemia with lactic acidosis.
“We found no justification to shorten the current storage duration for RBCs judged by their fundamental role to deliver oxygen,” said Dr. Christine M. Cserti-Gazdewich from the University of Toronto, Ontario, Canada.
Their findings suggest that stored RBCs should be evaluated by their ability to effectively deliver oxygen to tissues rather than by the cell survival measures and in vitro markers of hemolysis used as the current standard for viability by regulatory agencies, Dr. Cserti-Gazdewich said at the American Society of Hematology annual meeting.
The study clinicaltrials.gov ID NCT01586923 is also published online in JAMA.
“This study won’t change our clinical practice, but it will substantiate what we have been doing,” said co-author Dr. Walter H “Sunny” Dzik from Mas-sachusetts General Hospital in Boston, in an interview.
“We didn’t go into this business to harm people, yet there’s a lot of lab data that has been pointing fingers at us, suggesting rather forcefully that by try-ing to manage blood inventories using older blood that we’re harming people,” he said.
Concerns about the shelf life of RBCs derive from evidence that the cells undergo cumulative changes in structure, biochemistry, and enzymatic active that ultimately could degrade their ability to deliver oxygen to target tissues. But those studies were based largely on laboratory findings and not on clinical practice, she noted.
To see whether RBCs stored for up to 5 weeks could be as effective at tissue oxygenation as more recently packaged cells, the investigators conducted a randomized clinical trial at a university hospital urgent-care facility in Kampala, Uganda, where diseases such as malaria and sickle-cell anemia, as well as malnutrion, cause severe anemia on a scale seldom seen in the developed world.
They enrolled 290 children from the ages of 6 to 60 months who presented with lactic acidosis due to severe anemia in the absence of shock, trauma, impaired cardiac function, refractory hypoxia, liver disease, or tissue injury. The patients all had hemoglobin levels of 5 g/dL or5 less, and serum lactate levels of 5 mM or greater.
The patients were randomly assigned, 145 to each study arm, to receive leukoreduced RBCs stored for either 1-10 days or from 25 to 35 days. All patients received 10 mL/kg of RBCs over 2 hours at the start of the study and, if indicated per protocol, an additional 10 mL/kg during hours 4-6. Blood lactate levels were measured at baseline and at 2, 4, 6, 8 and 24 hours.
The mean hemoglobin level at presentation was 3.7 ±1.3 g/dL and mean lactate was 9.3 ±3.4 mM.
The investigators found that the proportion of patients achieving a lactate ≤ 3 mM at 8 hours was 58% among patients who received the short-storage RBCs, compared with 61% among patients who received long-storage cells (P = 0.72). This result met the primary endpoint of non-inferiority for long-er-stored RBCs.
In fact, there were no significant differences between the groups in mean lactate levels at any of the time points beyond baseline.
There were no significant differences in lactate clearance, and there were similar degrees of improvement in clinical assessments, serial measurements of hemoglobin concentration, cerebral tissue oxygen saturation, and electrolyte abnormalities following RBC transfusions.
Adverse events, 30-day recovery, and survival were also comparable between the groups.
The authors also found in a pre-specified sub-group analysis that there were no significant between group differences in those patients who received a total of 20 mL/kg RBCs, Dr. Cserti-Gazdewich said.
“This is a crushingly urgent problem, in that there have been belief systems developed over the last couple of years that older blood is not as good as newer blood, without any real evidence to support that. The evidence that Christine is [presenting], I think, is pretty compelling evidence that that hy-pothesis is incorrect,” commented Dr. Mark Crowther, Professor and Chair in the Department of Pathology and Molecular Medicine of McMaster Univer-sity in Hamilton, Ontario, Canada. Dr. Crowther moderated a briefing where Dr. Cserti-Gazdewich presented the data.
The study was funded by the National Institutes of Health. Dr. Cserti-Gazdewich, Dr. Dzik, and Dr. Crowther reported having no relevant conflicts of interest.
AT ASH 2015
Key clinical point: Red blood cells stored for up to 35 days were not inferior to more recently collected RBCs for tissue reoxygenation.
Major finding: The proportion of children with severe anemia achieving a lactate ≤ 3 mM at 8 hours was 58% among patients who received the short-storage RBCs, compared with 61% among patients who received long-storage cells.
Data source: Randomized controlled clinical trial in 290 children with lactic acidosis due to severe anemia.
Disclosures: The study was funded by the National Institutes of Health. Dr. Cserti-Gazdewich, Dr. Dzik, and Dr. Crowther reported having no relevant conflicts of interest.
ASH: Gene therapy restores immune function to older children with SCID-X1
ORLANDO – – Gene therapy can help restore immune function to older children and young adults with X-linked severe combined immunodeficiency, a team of US investigators reports.
“This is the first demonstration of the use of gene therapy to salvage failed allogeneic hematopoietic stem cell transplants in older SCID-X1 patients,” said Dr. Suk See De Ravin from the Laboratory of Host Defenses at the National Institutes of Health in Bethesda, Maryland.
Although allogeneic hematopoietic stem cell transplantation (HSCT) from a matched sibling donor can be curative, transplants using parental bone marrow or genetically modified autologous transplants without myeloconditiong restore T-cell-mediated immunity but not humoral immunity, Dr. De Ravin said at the American Society of Hematology annual meeting.

Additionally, transplants in older children with SCID-X1 who have persistent immune system defects – despite having received a transplant from a haploidentical donor in infancy – leave the patients with serious medical problems, including the life-long need for immunoglobulin G (IgG) supplementation, recurrent and chronic infections, warts, malnutrition, growth failure, and progressive diseases of the gut and lung.
Previous attempts at using gene therapy to correct mutations in IL2RG, the cause of SCID-X1, used mouse retroviral vectors to insert normal IL2RG into autologous hematopoietic stem cells without chemotherapy conditioning. This treatment restored T-cell immunity, but not B-cell- or natural killer (NK)-cell immunity
Of even greater concern is the fact that among infants with SCID-X1 who received autologous stem cells transduced with murine gamma retrovirus carrying the common gamma chain, 25% developed vector-associated leukemia.
To overcome these problems, investigators in the current study used a lentiviral vector containing an insulator fragment from a chicken beta-globin gene. The insulator fragment allows expression of the gamma chain complementary DNA while protecting against up-regulation of neighboring oncogenes.
Dr. De Ravin reported data on five patients from the ages of 7 to 24 years who had worsening immune dysfunction and complex medical problems, including dependence on immunoglobulin G (IgG) supplementations. All of the patients had previously undergone one or more HSCT from haploidentical donors.
The patients were treated with granulocyte-colony stimulating factor and plerixafor to mobilize peripheral blood cells, and then underwent apheresis to isolate CD34 cells. The cells were transduced in vitro with a lentiviral vector, and reinfused into patients after conditioning with low-dose busulfan (6 mg/kg). The vector was developed by researchers at St Jude Children’s Hospital in Memphis, Tennessee.
At the most recent follow-up, the first two patients treated with the protocol had stable engraftment of gamma-chain expressing cells gene with enhanced expression of B, T, and NK cells, with the cells continuing to show improvements, Early data on the remaining four patients indicates a similar positive trend.
Chimerism studies of the patients’ T-cells showed that the host cells were continuing to increase their contribution, suggesting gradual replacement over time of the T-cell graft, Dr. De Ravin said.
In the second patient treated, an increase in NK cells corresponded with an improvement in chronic warts. In addition, the first two patients began to produce both IgG and antigen-specific responses to vaccination, clearance of chronic norovirus infections, and resolution of their protein-losing enteropathy.
“Gene therapy does not appear to reverse prior organ damage, supporting early intervention to improve immunity in these patients before such damage occurs,” Dr. De Ravin said.
The study was supported by the National Institutes of Health. The viral vector was developed at St. Jude Children’s Research Hospital in Memphis, Tennessee. The authors reported no relevant conflicts of interest.
ORLANDO – – Gene therapy can help restore immune function to older children and young adults with X-linked severe combined immunodeficiency, a team of US investigators reports.
“This is the first demonstration of the use of gene therapy to salvage failed allogeneic hematopoietic stem cell transplants in older SCID-X1 patients,” said Dr. Suk See De Ravin from the Laboratory of Host Defenses at the National Institutes of Health in Bethesda, Maryland.
Although allogeneic hematopoietic stem cell transplantation (HSCT) from a matched sibling donor can be curative, transplants using parental bone marrow or genetically modified autologous transplants without myeloconditiong restore T-cell-mediated immunity but not humoral immunity, Dr. De Ravin said at the American Society of Hematology annual meeting.
Additionally, transplants in older children with SCID-X1 who have persistent immune system defects – despite having received a transplant from a haploidentical donor in infancy – leave the patients with serious medical problems, including the life-long need for immunoglobulin G (IgG) supplementation, recurrent and chronic infections, warts, malnutrition, growth failure, and progressive diseases of the gut and lung.
Previous attempts at using gene therapy to correct mutations in IL2RG, the cause of SCID-X1, used mouse retroviral vectors to insert normal IL2RG into autologous hematopoietic stem cells without chemotherapy conditioning. This treatment restored T-cell immunity, but not B-cell- or natural killer (NK)-cell immunity
Of even greater concern is the fact that among infants with SCID-X1 who received autologous stem cells transduced with murine gamma retrovirus carrying the common gamma chain, 25% developed vector-associated leukemia.
To overcome these problems, investigators in the current study used a lentiviral vector containing an insulator fragment from a chicken beta-globin gene. The insulator fragment allows expression of the gamma chain complementary DNA while protecting against up-regulation of neighboring oncogenes.
Dr. De Ravin reported data on five patients from the ages of 7 to 24 years who had worsening immune dysfunction and complex medical problems, including dependence on immunoglobulin G (IgG) supplementations. All of the patients had previously undergone one or more HSCT from haploidentical donors.
The patients were treated with granulocyte-colony stimulating factor and plerixafor to mobilize peripheral blood cells, and then underwent apheresis to isolate CD34 cells. The cells were transduced in vitro with a lentiviral vector, and reinfused into patients after conditioning with low-dose busulfan (6 mg/kg). The vector was developed by researchers at St Jude Children’s Hospital in Memphis, Tennessee.
At the most recent follow-up, the first two patients treated with the protocol had stable engraftment of gamma-chain expressing cells gene with enhanced expression of B, T, and NK cells, with the cells continuing to show improvements, Early data on the remaining four patients indicates a similar positive trend.
Chimerism studies of the patients’ T-cells showed that the host cells were continuing to increase their contribution, suggesting gradual replacement over time of the T-cell graft, Dr. De Ravin said.
In the second patient treated, an increase in NK cells corresponded with an improvement in chronic warts. In addition, the first two patients began to produce both IgG and antigen-specific responses to vaccination, clearance of chronic norovirus infections, and resolution of their protein-losing enteropathy.
“Gene therapy does not appear to reverse prior organ damage, supporting early intervention to improve immunity in these patients before such damage occurs,” Dr. De Ravin said.
The study was supported by the National Institutes of Health. The viral vector was developed at St. Jude Children’s Research Hospital in Memphis, Tennessee. The authors reported no relevant conflicts of interest.
ORLANDO – – Gene therapy can help restore immune function to older children and young adults with X-linked severe combined immunodeficiency, a team of US investigators reports.
“This is the first demonstration of the use of gene therapy to salvage failed allogeneic hematopoietic stem cell transplants in older SCID-X1 patients,” said Dr. Suk See De Ravin from the Laboratory of Host Defenses at the National Institutes of Health in Bethesda, Maryland.
Although allogeneic hematopoietic stem cell transplantation (HSCT) from a matched sibling donor can be curative, transplants using parental bone marrow or genetically modified autologous transplants without myeloconditiong restore T-cell-mediated immunity but not humoral immunity, Dr. De Ravin said at the American Society of Hematology annual meeting.
Additionally, transplants in older children with SCID-X1 who have persistent immune system defects – despite having received a transplant from a haploidentical donor in infancy – leave the patients with serious medical problems, including the life-long need for immunoglobulin G (IgG) supplementation, recurrent and chronic infections, warts, malnutrition, growth failure, and progressive diseases of the gut and lung.
Previous attempts at using gene therapy to correct mutations in IL2RG, the cause of SCID-X1, used mouse retroviral vectors to insert normal IL2RG into autologous hematopoietic stem cells without chemotherapy conditioning. This treatment restored T-cell immunity, but not B-cell- or natural killer (NK)-cell immunity
Of even greater concern is the fact that among infants with SCID-X1 who received autologous stem cells transduced with murine gamma retrovirus carrying the common gamma chain, 25% developed vector-associated leukemia.
To overcome these problems, investigators in the current study used a lentiviral vector containing an insulator fragment from a chicken beta-globin gene. The insulator fragment allows expression of the gamma chain complementary DNA while protecting against up-regulation of neighboring oncogenes.
Dr. De Ravin reported data on five patients from the ages of 7 to 24 years who had worsening immune dysfunction and complex medical problems, including dependence on immunoglobulin G (IgG) supplementations. All of the patients had previously undergone one or more HSCT from haploidentical donors.
The patients were treated with granulocyte-colony stimulating factor and plerixafor to mobilize peripheral blood cells, and then underwent apheresis to isolate CD34 cells. The cells were transduced in vitro with a lentiviral vector, and reinfused into patients after conditioning with low-dose busulfan (6 mg/kg). The vector was developed by researchers at St Jude Children’s Hospital in Memphis, Tennessee.
At the most recent follow-up, the first two patients treated with the protocol had stable engraftment of gamma-chain expressing cells gene with enhanced expression of B, T, and NK cells, with the cells continuing to show improvements, Early data on the remaining four patients indicates a similar positive trend.
Chimerism studies of the patients’ T-cells showed that the host cells were continuing to increase their contribution, suggesting gradual replacement over time of the T-cell graft, Dr. De Ravin said.
In the second patient treated, an increase in NK cells corresponded with an improvement in chronic warts. In addition, the first two patients began to produce both IgG and antigen-specific responses to vaccination, clearance of chronic norovirus infections, and resolution of their protein-losing enteropathy.
“Gene therapy does not appear to reverse prior organ damage, supporting early intervention to improve immunity in these patients before such damage occurs,” Dr. De Ravin said.
The study was supported by the National Institutes of Health. The viral vector was developed at St. Jude Children’s Research Hospital in Memphis, Tennessee. The authors reported no relevant conflicts of interest.
AT ASH 2015
Key clinical point: Gene therapy can correct B, T, and NK cell immunity in older patients with SCID-X1.
Major finding: The first two patients treated with the protocol had stable engraftment of gamma-chain expressing cells gene with enhanced expression of B, T, and NK cells, with the cells continuing to show improvements.
Data source: Clinical study of 5 patients with X-linked severe combined immunodeficiency syndrome (SCID-X1).
Disclosures: The study was supported by the National Institutes of Health. The viral vector was developed at St. Jude Children’s Research Hospital in Memphis, Tennessee. The authors reported no relevant conflicts of interest.

ASH: Donor CAR-T cells elicit responses in mixture of progressive B-cell cancers
ORLANDO – A single infusion of donor-derived chimeric antigen receptor (CAR)-modified T cells targeting CD19 achieved remission in 9 of 20 patients with B-cell malignancies that progressed after allogeneic stem cell transplant, a study shows.
The seven complete remissions and two partial remissions occurred without any chemotherapy and without causing acute graft-versus-host disease (GVHD).

The experimental anti-CD 19 CAR T-cells seem particularly effective against acute lymphoid leukemia (ALL) and chronic lymphocytic leukemia (CLL), but responses also occurred in lymphoma, Dr. James Kochenderfer of the Center for Cancer Research, National Cancer Institute, in Bethesda, Md., reported at the annual meeting of the American Society of Hematology.
B-cell malignancies that persist after transplantation are often treated with unmanipulated donor lymphocytes, but these infusions are often ineffective and associated with significant morbidity and mortality from GVHD.
To improve on this approach, 20 patients were infused with T cells obtained from the original stem cell donor and transduced with a CD19-directed CAR that was encoded by a gamma-retroviral vector and included a CD28 co-stimulatory domain. The highest dose reached in the phase I study was 107 total cell/kg. Production of the anti-CD19 CAR T cells took only eight days for each patient, Dr. Kochenderfer said at a press briefing.
The patients had received at least one standard donor-leukocyte infusion, had to have minimal or no GVHD, and could not be receiving systemic immunosuppressive drugs.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The highest response rates were in ALL, where four of five patients obtained complete remission (CR) with no detectable minimal residual disease by multi-color flow cytometry, Dr. Kochenderfer said. Two of these patients later relapsed, one is in ongoing CR at 18 months, and one went on to a second allogeneic transplant and continues in complete remission.
The longest ongoing CR at 36 months occurred in a patient treated for CLL. Another patient achieved a partial remission (PR) ongoing at 18 months, two patients progressed, and one has stable disease.
In five patients treated for Mantle cell lymphoma, there is one CR ongoing at 31 months, one PR, and three stable diseases.
Three of the five patients treated for diffuse large B-cell lymphoma experienced stable disease, one progressive disease, and one obtained a CR, but is no longer evaluable because she received other therapies for chronic GVHD. Dr. Kochenderfer went on describe an impressive response in this patient, who had large lymphoma masses at the back of her head and in her eye socket before infusion.
“Amazingly, the tumor masses completely disappeared within five days of CAR T-cell infusion,” he said.
Patients with high tumor burdens, however, experienced severe cytokine-release syndrome with fever, tachycardia, and hypertension that was treated with the interleukin-6 receptor antagonist tocilizumab (Actemra).
Only one case of mild aphasia occurred, which contrasts with other CAR T-cell therapies where neurotoxicity is common, Dr. Kochenderfer said.
One patient had continued worsening of pre-existing chronic GVHD after CAR T-cell therapy, and one patient developed very mild chronic eye GVHD more than a year after infusion.
The press corps was not fully convinced by the findings, however, asking Dr. Kochenderfer why they should be excited by the 40% remission rate when other CAR T-cell therapies have yielded remission rates as high as 90%.
Dr. Kochenderfer pointed out that four of the five ALL patients (80%) achieved a MRD-negative complete response, which compares favorably with other protocols. The remaining patients had far more advanced, treatment-resident disease of varying histologies than evaluated in other trials and, unlike most trials, all patients had received an allogeneic transplant. Further, the investigators used no chemotherapy whatsoever, whereas other CAR T-cells trials have used chemotherapy, sometimes in huge does, he said.
ORLANDO – A single infusion of donor-derived chimeric antigen receptor (CAR)-modified T cells targeting CD19 achieved remission in 9 of 20 patients with B-cell malignancies that progressed after allogeneic stem cell transplant, a study shows.
The seven complete remissions and two partial remissions occurred without any chemotherapy and without causing acute graft-versus-host disease (GVHD).
The experimental anti-CD 19 CAR T-cells seem particularly effective against acute lymphoid leukemia (ALL) and chronic lymphocytic leukemia (CLL), but responses also occurred in lymphoma, Dr. James Kochenderfer of the Center for Cancer Research, National Cancer Institute, in Bethesda, Md., reported at the annual meeting of the American Society of Hematology.
B-cell malignancies that persist after transplantation are often treated with unmanipulated donor lymphocytes, but these infusions are often ineffective and associated with significant morbidity and mortality from GVHD.
To improve on this approach, 20 patients were infused with T cells obtained from the original stem cell donor and transduced with a CD19-directed CAR that was encoded by a gamma-retroviral vector and included a CD28 co-stimulatory domain. The highest dose reached in the phase I study was 107 total cell/kg. Production of the anti-CD19 CAR T cells took only eight days for each patient, Dr. Kochenderfer said at a press briefing.
The patients had received at least one standard donor-leukocyte infusion, had to have minimal or no GVHD, and could not be receiving systemic immunosuppressive drugs.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The highest response rates were in ALL, where four of five patients obtained complete remission (CR) with no detectable minimal residual disease by multi-color flow cytometry, Dr. Kochenderfer said. Two of these patients later relapsed, one is in ongoing CR at 18 months, and one went on to a second allogeneic transplant and continues in complete remission.
The longest ongoing CR at 36 months occurred in a patient treated for CLL. Another patient achieved a partial remission (PR) ongoing at 18 months, two patients progressed, and one has stable disease.
In five patients treated for Mantle cell lymphoma, there is one CR ongoing at 31 months, one PR, and three stable diseases.
Three of the five patients treated for diffuse large B-cell lymphoma experienced stable disease, one progressive disease, and one obtained a CR, but is no longer evaluable because she received other therapies for chronic GVHD. Dr. Kochenderfer went on describe an impressive response in this patient, who had large lymphoma masses at the back of her head and in her eye socket before infusion.
“Amazingly, the tumor masses completely disappeared within five days of CAR T-cell infusion,” he said.
Patients with high tumor burdens, however, experienced severe cytokine-release syndrome with fever, tachycardia, and hypertension that was treated with the interleukin-6 receptor antagonist tocilizumab (Actemra).
Only one case of mild aphasia occurred, which contrasts with other CAR T-cell therapies where neurotoxicity is common, Dr. Kochenderfer said.
One patient had continued worsening of pre-existing chronic GVHD after CAR T-cell therapy, and one patient developed very mild chronic eye GVHD more than a year after infusion.
The press corps was not fully convinced by the findings, however, asking Dr. Kochenderfer why they should be excited by the 40% remission rate when other CAR T-cell therapies have yielded remission rates as high as 90%.
Dr. Kochenderfer pointed out that four of the five ALL patients (80%) achieved a MRD-negative complete response, which compares favorably with other protocols. The remaining patients had far more advanced, treatment-resident disease of varying histologies than evaluated in other trials and, unlike most trials, all patients had received an allogeneic transplant. Further, the investigators used no chemotherapy whatsoever, whereas other CAR T-cells trials have used chemotherapy, sometimes in huge does, he said.
ORLANDO – A single infusion of donor-derived chimeric antigen receptor (CAR)-modified T cells targeting CD19 achieved remission in 9 of 20 patients with B-cell malignancies that progressed after allogeneic stem cell transplant, a study shows.
The seven complete remissions and two partial remissions occurred without any chemotherapy and without causing acute graft-versus-host disease (GVHD).
The experimental anti-CD 19 CAR T-cells seem particularly effective against acute lymphoid leukemia (ALL) and chronic lymphocytic leukemia (CLL), but responses also occurred in lymphoma, Dr. James Kochenderfer of the Center for Cancer Research, National Cancer Institute, in Bethesda, Md., reported at the annual meeting of the American Society of Hematology.
B-cell malignancies that persist after transplantation are often treated with unmanipulated donor lymphocytes, but these infusions are often ineffective and associated with significant morbidity and mortality from GVHD.
To improve on this approach, 20 patients were infused with T cells obtained from the original stem cell donor and transduced with a CD19-directed CAR that was encoded by a gamma-retroviral vector and included a CD28 co-stimulatory domain. The highest dose reached in the phase I study was 107 total cell/kg. Production of the anti-CD19 CAR T cells took only eight days for each patient, Dr. Kochenderfer said at a press briefing.
The patients had received at least one standard donor-leukocyte infusion, had to have minimal or no GVHD, and could not be receiving systemic immunosuppressive drugs.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The highest response rates were in ALL, where four of five patients obtained complete remission (CR) with no detectable minimal residual disease by multi-color flow cytometry, Dr. Kochenderfer said. Two of these patients later relapsed, one is in ongoing CR at 18 months, and one went on to a second allogeneic transplant and continues in complete remission.
The longest ongoing CR at 36 months occurred in a patient treated for CLL. Another patient achieved a partial remission (PR) ongoing at 18 months, two patients progressed, and one has stable disease.
In five patients treated for Mantle cell lymphoma, there is one CR ongoing at 31 months, one PR, and three stable diseases.
Three of the five patients treated for diffuse large B-cell lymphoma experienced stable disease, one progressive disease, and one obtained a CR, but is no longer evaluable because she received other therapies for chronic GVHD. Dr. Kochenderfer went on describe an impressive response in this patient, who had large lymphoma masses at the back of her head and in her eye socket before infusion.
“Amazingly, the tumor masses completely disappeared within five days of CAR T-cell infusion,” he said.
Patients with high tumor burdens, however, experienced severe cytokine-release syndrome with fever, tachycardia, and hypertension that was treated with the interleukin-6 receptor antagonist tocilizumab (Actemra).
Only one case of mild aphasia occurred, which contrasts with other CAR T-cell therapies where neurotoxicity is common, Dr. Kochenderfer said.
One patient had continued worsening of pre-existing chronic GVHD after CAR T-cell therapy, and one patient developed very mild chronic eye GVHD more than a year after infusion.
The press corps was not fully convinced by the findings, however, asking Dr. Kochenderfer why they should be excited by the 40% remission rate when other CAR T-cell therapies have yielded remission rates as high as 90%.
Dr. Kochenderfer pointed out that four of the five ALL patients (80%) achieved a MRD-negative complete response, which compares favorably with other protocols. The remaining patients had far more advanced, treatment-resident disease of varying histologies than evaluated in other trials and, unlike most trials, all patients had received an allogeneic transplant. Further, the investigators used no chemotherapy whatsoever, whereas other CAR T-cells trials have used chemotherapy, sometimes in huge does, he said.
AT ASH 2015
Key clinical point: Allogeneic anti-CD19 CAR T-cell therapy showed promise in a treatment approach for B-cell malignancies persisting after allogeneic transplantation.
Major finding: Nine of 20 patients achieved remission with anti-CD19 CAR T-cell therapy.
Data source: Phase I study in 20 patients with CD19-positive B-cell malignancies progressing after allogeneic transplant.
Disclosures: Dr. Kochenderfer reported research funding from Bluebird bio, the study sponsor.

ASH: Gene therapy reduces transfusion needs in beta-thalassemia major
ORLANDO – Lentiviral gene therapy with LentiGlobin BB305 boosts beta-globin production in patients with beta-thalassemia, but frees only some from lifelong dependence on blood transfusions, updated results of the Northstar study show.
Five patients with non-Beta-0/Beta-0 genotypes were able to stop transfusions shortly after their infusion, and remain transfusion independent for up to 16.4 months.
In four patients with the more severe form of beta-thalassemia, the Beta-0/Beta-0 genotype, red blood cell transfusion volume was reduced by 33% to 100%, with one patient stopping transfusions entirely, Dr. Mark C. Walters of the University of California-San Francisco Benioff Children’s Hospital in Oakland reported at the annual meeting of the American Society of Hematology.
Preliminary findings reported over the last two years have raised hopes that the experimental lentiviral-based therapy could be a functional cure for beta-thalassemia major and severe sickle cell disease.
Patients with beta-thalassemia major, also called Cooley’s anemia, rely on frequent blood transfusions to correct the anemia, with less than a quarter undergoing curative treatment with an allogeneic hematopoietic transplant.
In the ongoing Northstar study, 13 patients with transfusion-dependent beta-thalassemia major have been infused as of Oct. 28, 2015 with autologous CD34-positive cells transduced ex-vivo with LentiGlobin BB305 (Bluebird bio, Cambridge, Mass.), a self-inactivating, second-generation lentiviral vector containing a functioning, engineered beta-globin gene (A-T87Q). Their median age was 21 years and 11 were women.Vector-derived hemoglobin AT87Q was detectable at 6 months in 8 of 9 evaluable patients with at least six months follow-up and in 100% at 9 months, Dr. Walters reported. The median HbAT87Q level was 4.9 g/dL at 6 months, 6.5 g/dL at 9 months, and 4.2 g/dL at 12 months.
The difference in transfusion independence between genotypes is explained by endogenous non-HbAT87Q production, he said during a press briefing. While lentiglobin production was the same in patients with Beta-0/Beta-0 and non-Beta-0/Beta-0 genotypes, the Beta-0/Beta-0 patients made much smaller amounts of native hemoglobin.
Three serious post-infusion events occurred: grade 2 thrombosis, grade 3 skin infection and grade 3 veno-occlusive liver disease.
Importantly, there was no evidence of clonal dominance or replication competent lentivirus with up to 19 months follow-up.
“This is a significant advancement in the treatment of thalassemia for several reasons,” Dr. Walters told reporters. “First, compared to a bone marrow transplant, which is the only curative therapy that’s been approved, this appears to be a safer treatment in that none of these patients had a life-threatening complication. Second, because the treatment uses a thalassemia patient’s own stem cells, this bypasses the need to find a healthy bone marrow donor and thus should be more broadly available to patients affected by this disease.”
Dr. George Daley of Harvard Medical School in Boston and moderator of the press briefing, agreed that the results are an welcome advancement after decades of disappointments in the field of gene therapy including the development of insertional mutigenesis.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
In addition to the Northstar study (Ab. 201), results will be presented at the meeting from a second study examining LentiGlobin BB305 gene therapy for severe sickle cell disease and beta-thalassemia major (Ab. 202) and from the recently expanded phase I HGB-206 study in severe sickle cell disease (Ab. 3233).
Sickle cell disease represents a much larger potential market for LentiGlobin BB305, with an estimated 90,000 to 100,000 Americans affected compared with about 15,000 patients in America and Europe living with beta-thalassemia major.
ORLANDO – Lentiviral gene therapy with LentiGlobin BB305 boosts beta-globin production in patients with beta-thalassemia, but frees only some from lifelong dependence on blood transfusions, updated results of the Northstar study show.
Five patients with non-Beta-0/Beta-0 genotypes were able to stop transfusions shortly after their infusion, and remain transfusion independent for up to 16.4 months.
In four patients with the more severe form of beta-thalassemia, the Beta-0/Beta-0 genotype, red blood cell transfusion volume was reduced by 33% to 100%, with one patient stopping transfusions entirely, Dr. Mark C. Walters of the University of California-San Francisco Benioff Children’s Hospital in Oakland reported at the annual meeting of the American Society of Hematology.
Preliminary findings reported over the last two years have raised hopes that the experimental lentiviral-based therapy could be a functional cure for beta-thalassemia major and severe sickle cell disease.
Patients with beta-thalassemia major, also called Cooley’s anemia, rely on frequent blood transfusions to correct the anemia, with less than a quarter undergoing curative treatment with an allogeneic hematopoietic transplant.
In the ongoing Northstar study, 13 patients with transfusion-dependent beta-thalassemia major have been infused as of Oct. 28, 2015 with autologous CD34-positive cells transduced ex-vivo with LentiGlobin BB305 (Bluebird bio, Cambridge, Mass.), a self-inactivating, second-generation lentiviral vector containing a functioning, engineered beta-globin gene (A-T87Q). Their median age was 21 years and 11 were women.Vector-derived hemoglobin AT87Q was detectable at 6 months in 8 of 9 evaluable patients with at least six months follow-up and in 100% at 9 months, Dr. Walters reported. The median HbAT87Q level was 4.9 g/dL at 6 months, 6.5 g/dL at 9 months, and 4.2 g/dL at 12 months.
The difference in transfusion independence between genotypes is explained by endogenous non-HbAT87Q production, he said during a press briefing. While lentiglobin production was the same in patients with Beta-0/Beta-0 and non-Beta-0/Beta-0 genotypes, the Beta-0/Beta-0 patients made much smaller amounts of native hemoglobin.
Three serious post-infusion events occurred: grade 2 thrombosis, grade 3 skin infection and grade 3 veno-occlusive liver disease.
Importantly, there was no evidence of clonal dominance or replication competent lentivirus with up to 19 months follow-up.
“This is a significant advancement in the treatment of thalassemia for several reasons,” Dr. Walters told reporters. “First, compared to a bone marrow transplant, which is the only curative therapy that’s been approved, this appears to be a safer treatment in that none of these patients had a life-threatening complication. Second, because the treatment uses a thalassemia patient’s own stem cells, this bypasses the need to find a healthy bone marrow donor and thus should be more broadly available to patients affected by this disease.”
Dr. George Daley of Harvard Medical School in Boston and moderator of the press briefing, agreed that the results are an welcome advancement after decades of disappointments in the field of gene therapy including the development of insertional mutigenesis.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
In addition to the Northstar study (Ab. 201), results will be presented at the meeting from a second study examining LentiGlobin BB305 gene therapy for severe sickle cell disease and beta-thalassemia major (Ab. 202) and from the recently expanded phase I HGB-206 study in severe sickle cell disease (Ab. 3233).
Sickle cell disease represents a much larger potential market for LentiGlobin BB305, with an estimated 90,000 to 100,000 Americans affected compared with about 15,000 patients in America and Europe living with beta-thalassemia major.
ORLANDO – Lentiviral gene therapy with LentiGlobin BB305 boosts beta-globin production in patients with beta-thalassemia, but frees only some from lifelong dependence on blood transfusions, updated results of the Northstar study show.
Five patients with non-Beta-0/Beta-0 genotypes were able to stop transfusions shortly after their infusion, and remain transfusion independent for up to 16.4 months.
In four patients with the more severe form of beta-thalassemia, the Beta-0/Beta-0 genotype, red blood cell transfusion volume was reduced by 33% to 100%, with one patient stopping transfusions entirely, Dr. Mark C. Walters of the University of California-San Francisco Benioff Children’s Hospital in Oakland reported at the annual meeting of the American Society of Hematology.
Preliminary findings reported over the last two years have raised hopes that the experimental lentiviral-based therapy could be a functional cure for beta-thalassemia major and severe sickle cell disease.
Patients with beta-thalassemia major, also called Cooley’s anemia, rely on frequent blood transfusions to correct the anemia, with less than a quarter undergoing curative treatment with an allogeneic hematopoietic transplant.
In the ongoing Northstar study, 13 patients with transfusion-dependent beta-thalassemia major have been infused as of Oct. 28, 2015 with autologous CD34-positive cells transduced ex-vivo with LentiGlobin BB305 (Bluebird bio, Cambridge, Mass.), a self-inactivating, second-generation lentiviral vector containing a functioning, engineered beta-globin gene (A-T87Q). Their median age was 21 years and 11 were women.Vector-derived hemoglobin AT87Q was detectable at 6 months in 8 of 9 evaluable patients with at least six months follow-up and in 100% at 9 months, Dr. Walters reported. The median HbAT87Q level was 4.9 g/dL at 6 months, 6.5 g/dL at 9 months, and 4.2 g/dL at 12 months.
The difference in transfusion independence between genotypes is explained by endogenous non-HbAT87Q production, he said during a press briefing. While lentiglobin production was the same in patients with Beta-0/Beta-0 and non-Beta-0/Beta-0 genotypes, the Beta-0/Beta-0 patients made much smaller amounts of native hemoglobin.
Three serious post-infusion events occurred: grade 2 thrombosis, grade 3 skin infection and grade 3 veno-occlusive liver disease.
Importantly, there was no evidence of clonal dominance or replication competent lentivirus with up to 19 months follow-up.
“This is a significant advancement in the treatment of thalassemia for several reasons,” Dr. Walters told reporters. “First, compared to a bone marrow transplant, which is the only curative therapy that’s been approved, this appears to be a safer treatment in that none of these patients had a life-threatening complication. Second, because the treatment uses a thalassemia patient’s own stem cells, this bypasses the need to find a healthy bone marrow donor and thus should be more broadly available to patients affected by this disease.”
Dr. George Daley of Harvard Medical School in Boston and moderator of the press briefing, agreed that the results are an welcome advancement after decades of disappointments in the field of gene therapy including the development of insertional mutigenesis.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
In addition to the Northstar study (Ab. 201), results will be presented at the meeting from a second study examining LentiGlobin BB305 gene therapy for severe sickle cell disease and beta-thalassemia major (Ab. 202) and from the recently expanded phase I HGB-206 study in severe sickle cell disease (Ab. 3233).
Sickle cell disease represents a much larger potential market for LentiGlobin BB305, with an estimated 90,000 to 100,000 Americans affected compared with about 15,000 patients in America and Europe living with beta-thalassemia major.
AT ASH 2015
Key clinical point: Lentiviral-based gene therapy with LentiGlobin BB305 restarts hemoglobin production and leads to transfusion independence in some patients with beta-thalassemia major.
Major finding: Five patients with the non-Beta-0/Beta-0 genotype were transfusion independent post-infusion.
Data source: Phase I/II study in 13 patients with transfusion-dependent beta-thalassemia major.
Disclosures: Dr. Walters reported financial relationships with ViaCord and AllCells Inc. Several co-authors have financial relationships including employment with Bluebird bio, the study sponsor. Dr. Daley disclosed consultancy with True North Therapeutics and serving as an advisory committee member for Raze Therapeutics, Ocata Therapeutics, MPM Capital, and Solasia.

ASH: Gene therapy eases effects of rare Wiskott-Aldrich syndrome
ORLANDO – Genetic modification of autologous stem cells provided sustained clinical benefit with good safety for children with the rare immunodeficiency disorder Wiskott-Aldrich syndrome, an international team of investigators report.
Six of eight children who received infusions of autologous stem cells that had been modified with a lentiviral vector to restore normal expression of the WAS gene had marked reductions in severe infections, fewer hospitalizations, improved hematologic parameters, and more robust immune responses than they had prior to transplant, reported Dr. Francesca Ferrua from the San Raffaele Telethon Institute for Gene Therapy in Milan, Italy.

“Importantly, with regards to safety we did not detect any serious adverse events related to gene therapy in follow up, and we did not observe any evidence of abnormal clonal proliferation after gene therapy,” she said at the American Society of Hematology annual meeting here.
The Wiskott-Aldrich syndrome is an x-linked syndrome caused by mutations in the WAS gene encoding for the WAS protein (WASP), which is involved in regulation of the cytoskeleton. The disorder, which primarily affects males, leads to immunodeficiency, microthrombocytopenia and leukocyte abnormalities. Patients develop severe eczema and other inflammatory disorders, and are at increased risk for autoimmune diseases and malignancies.
The syndrome is estimated to occur in 1-10 per 1 million males worldwide, according to The National Library of Medicine.
Allogeneic hematopoietic stem cell transplantion (HSCT) can be curative for patients with Wiskott-Aldrich syndrome, but the technique is associated with both acute transplant-related complications and long-term morbidities, particularly when there is not a perfect match between donor and recipient, Dr. Ferrua explained.Prior studies using a gamma-retroviral vector under the control of a strong viral promoter showed that gene therapy was feasible in these patients and could result in immunological improvement. The earlier attempts, however, were associated with a high risk of genotoxicity and insertional mutagenesis; seven of nine patients treated in one study developed leukemia.
In their current line of research, Dr. Ferrua and colleagues had previously reported on the use of autologous hematopoietic stem/progenitor cells modified ex vivo to correct the inherent defect in three patients with severe mutations in WAS who had no suitable stem-cell donors.
The researchers collected CD34-positive cells from each patient’s bone marrow and/or mobilized peripheral blood and transduced the cells in the laboratory with a lentivirus modified to promote normal expression of WAS. They then returned the cells to the patients after they underwent a reduced-intensity conditioning regimen using an anti-CD20 monoclonal antibody, busulfan, and fludarabine.
Long-term follow-up
At ASH 2015, Dr. Ferrua reported results on the first 8 patients treated as of October 2015. The patients were treated at a median age of 2.2 years; all are alive after a median of 3.3 years of follow up, with the longest follow up being 5.5 years
All had marked reductions in the annualized estimated rate of severe infections compared with the pre-transplant period.
Of the seven patients followed for more than 1 year, all were able to discontinue prophylaxis for infections, at a median of 13-15 months after gene therapy, and five were able to discontinue immunoglobulin supplementation.
Additionally, four of four patients had evidence of a normal immune response based on the development of specific antibodies after vaccination.
Four patients had resolution of their eczemas, and the other two with eczema had only mild cases.
At a median of 4 months after genetic therapy, none of the patients required platelet transfusions. Out to at least 1 year, there was no evidence of autoimmunity.
Among all patients, there were reductions in the frequency or severity of bleeding, no severe bleeding episodes, no hospitalizations for bleeding and a reduction in the number of hospitalizations for infections.
There were no serious adverse events related to the transplant.
The study was sponsored by IRCCS San Raffaele with support from the Fondazione Telethon and GlaxoSmithKline. Dr. Ferrua reported having no conflicts of interest.
ORLANDO – Genetic modification of autologous stem cells provided sustained clinical benefit with good safety for children with the rare immunodeficiency disorder Wiskott-Aldrich syndrome, an international team of investigators report.
Six of eight children who received infusions of autologous stem cells that had been modified with a lentiviral vector to restore normal expression of the WAS gene had marked reductions in severe infections, fewer hospitalizations, improved hematologic parameters, and more robust immune responses than they had prior to transplant, reported Dr. Francesca Ferrua from the San Raffaele Telethon Institute for Gene Therapy in Milan, Italy.
“Importantly, with regards to safety we did not detect any serious adverse events related to gene therapy in follow up, and we did not observe any evidence of abnormal clonal proliferation after gene therapy,” she said at the American Society of Hematology annual meeting here.
The Wiskott-Aldrich syndrome is an x-linked syndrome caused by mutations in the WAS gene encoding for the WAS protein (WASP), which is involved in regulation of the cytoskeleton. The disorder, which primarily affects males, leads to immunodeficiency, microthrombocytopenia and leukocyte abnormalities. Patients develop severe eczema and other inflammatory disorders, and are at increased risk for autoimmune diseases and malignancies.
The syndrome is estimated to occur in 1-10 per 1 million males worldwide, according to The National Library of Medicine.
Allogeneic hematopoietic stem cell transplantion (HSCT) can be curative for patients with Wiskott-Aldrich syndrome, but the technique is associated with both acute transplant-related complications and long-term morbidities, particularly when there is not a perfect match between donor and recipient, Dr. Ferrua explained.Prior studies using a gamma-retroviral vector under the control of a strong viral promoter showed that gene therapy was feasible in these patients and could result in immunological improvement. The earlier attempts, however, were associated with a high risk of genotoxicity and insertional mutagenesis; seven of nine patients treated in one study developed leukemia.
In their current line of research, Dr. Ferrua and colleagues had previously reported on the use of autologous hematopoietic stem/progenitor cells modified ex vivo to correct the inherent defect in three patients with severe mutations in WAS who had no suitable stem-cell donors.
The researchers collected CD34-positive cells from each patient’s bone marrow and/or mobilized peripheral blood and transduced the cells in the laboratory with a lentivirus modified to promote normal expression of WAS. They then returned the cells to the patients after they underwent a reduced-intensity conditioning regimen using an anti-CD20 monoclonal antibody, busulfan, and fludarabine.
Long-term follow-up
At ASH 2015, Dr. Ferrua reported results on the first 8 patients treated as of October 2015. The patients were treated at a median age of 2.2 years; all are alive after a median of 3.3 years of follow up, with the longest follow up being 5.5 years
All had marked reductions in the annualized estimated rate of severe infections compared with the pre-transplant period.
Of the seven patients followed for more than 1 year, all were able to discontinue prophylaxis for infections, at a median of 13-15 months after gene therapy, and five were able to discontinue immunoglobulin supplementation.
Additionally, four of four patients had evidence of a normal immune response based on the development of specific antibodies after vaccination.
Four patients had resolution of their eczemas, and the other two with eczema had only mild cases.
At a median of 4 months after genetic therapy, none of the patients required platelet transfusions. Out to at least 1 year, there was no evidence of autoimmunity.
Among all patients, there were reductions in the frequency or severity of bleeding, no severe bleeding episodes, no hospitalizations for bleeding and a reduction in the number of hospitalizations for infections.
There were no serious adverse events related to the transplant.
The study was sponsored by IRCCS San Raffaele with support from the Fondazione Telethon and GlaxoSmithKline. Dr. Ferrua reported having no conflicts of interest.
ORLANDO – Genetic modification of autologous stem cells provided sustained clinical benefit with good safety for children with the rare immunodeficiency disorder Wiskott-Aldrich syndrome, an international team of investigators report.
Six of eight children who received infusions of autologous stem cells that had been modified with a lentiviral vector to restore normal expression of the WAS gene had marked reductions in severe infections, fewer hospitalizations, improved hematologic parameters, and more robust immune responses than they had prior to transplant, reported Dr. Francesca Ferrua from the San Raffaele Telethon Institute for Gene Therapy in Milan, Italy.
“Importantly, with regards to safety we did not detect any serious adverse events related to gene therapy in follow up, and we did not observe any evidence of abnormal clonal proliferation after gene therapy,” she said at the American Society of Hematology annual meeting here.
The Wiskott-Aldrich syndrome is an x-linked syndrome caused by mutations in the WAS gene encoding for the WAS protein (WASP), which is involved in regulation of the cytoskeleton. The disorder, which primarily affects males, leads to immunodeficiency, microthrombocytopenia and leukocyte abnormalities. Patients develop severe eczema and other inflammatory disorders, and are at increased risk for autoimmune diseases and malignancies.
The syndrome is estimated to occur in 1-10 per 1 million males worldwide, according to The National Library of Medicine.
Allogeneic hematopoietic stem cell transplantion (HSCT) can be curative for patients with Wiskott-Aldrich syndrome, but the technique is associated with both acute transplant-related complications and long-term morbidities, particularly when there is not a perfect match between donor and recipient, Dr. Ferrua explained.Prior studies using a gamma-retroviral vector under the control of a strong viral promoter showed that gene therapy was feasible in these patients and could result in immunological improvement. The earlier attempts, however, were associated with a high risk of genotoxicity and insertional mutagenesis; seven of nine patients treated in one study developed leukemia.
In their current line of research, Dr. Ferrua and colleagues had previously reported on the use of autologous hematopoietic stem/progenitor cells modified ex vivo to correct the inherent defect in three patients with severe mutations in WAS who had no suitable stem-cell donors.
The researchers collected CD34-positive cells from each patient’s bone marrow and/or mobilized peripheral blood and transduced the cells in the laboratory with a lentivirus modified to promote normal expression of WAS. They then returned the cells to the patients after they underwent a reduced-intensity conditioning regimen using an anti-CD20 monoclonal antibody, busulfan, and fludarabine.
Long-term follow-up
At ASH 2015, Dr. Ferrua reported results on the first 8 patients treated as of October 2015. The patients were treated at a median age of 2.2 years; all are alive after a median of 3.3 years of follow up, with the longest follow up being 5.5 years
All had marked reductions in the annualized estimated rate of severe infections compared with the pre-transplant period.
Of the seven patients followed for more than 1 year, all were able to discontinue prophylaxis for infections, at a median of 13-15 months after gene therapy, and five were able to discontinue immunoglobulin supplementation.
Additionally, four of four patients had evidence of a normal immune response based on the development of specific antibodies after vaccination.
Four patients had resolution of their eczemas, and the other two with eczema had only mild cases.
At a median of 4 months after genetic therapy, none of the patients required platelet transfusions. Out to at least 1 year, there was no evidence of autoimmunity.
Among all patients, there were reductions in the frequency or severity of bleeding, no severe bleeding episodes, no hospitalizations for bleeding and a reduction in the number of hospitalizations for infections.
There were no serious adverse events related to the transplant.
The study was sponsored by IRCCS San Raffaele with support from the Fondazione Telethon and GlaxoSmithKline. Dr. Ferrua reported having no conflicts of interest.
AT ASH 2015
Key clinical point: Gene therapy might safely and effectively correct an inherited immunodeficiency syndrome.
Major finding: Six of eight children with Wiskott-Aldrich syndrome who received genetically modified autologous stem cells had marked clinical improvements.
Data source: International collaborative trial studying the safety and efficacy of WAS gene transfer into hematopoietic stem/progenitor cells.
Disclosures: The study was sponsored by IRCCS San Raffaele with support from the Fondazione Telethon and GlaxoSmithKline. Dr. Ferrua reported having no conflicts of interest.

ASH: Carfilzomib doubles progression free survival in head-to-head trial with bortezomib
In a head-to-head clinical trial, carfilzomib (Kyprolis) plus dexamethasone doubled the length of progression-free survival (PFS) when compared with bortezomib (Velcade) plus dexamethasone for individuals with relapsed or refractory multiple myeloma. Response rates were also significantly higher for those receiving carfilzomib, though neither medication could overcome high-risk cytogenetic factors.
The international multi-center phase 3 open-label ENDEAVOR study (clinicaltrials.gov ID: NCT01568866) compared outcomes for a total of 929 patients who were randomized 1:1 to receive one of the treatments. Randomization was stratified to account for prior treatments, including prior proteasome treatment, as well as disease severity by International Staging System stage.
Reporting for the ENDEAVOR investigators, Dr. Meletios Dimopoulos and his co-authors published their results in The Lancet simultaneously with a presentation at the annual meeting of the American Society of Hematology [Lancet Oncol, http://dx.doi.org/10.1016/S1470-2045(15)00464-7] . Dr. Dimopoulos is a professor in the department of clinical therapeutics at the school of medicine of the National and Kapodistrian University of Athens.
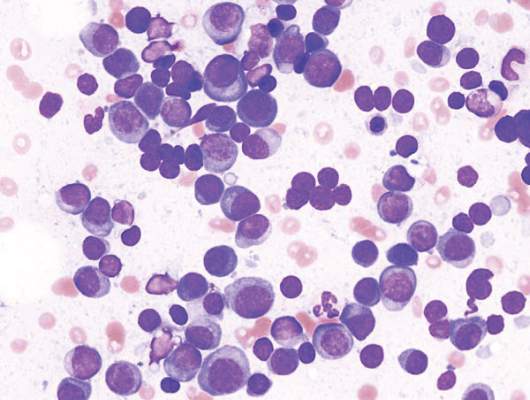
The primary endpoint, PFS in the intention-to-treat population, differed significantly between the study arms when the data were analyzed at a pre-planned interim analysis. For those receiving carfilzomib, PFS was 18.7 months, compared with 9.4 months in the bortezomib group, for a hazard ratio of 0.53 (P less than 0.0001). Data were drawn from a pre-specified interim analysis, and study participation is ongoing, with 28-day dosing cycles repeated until disease progression occurrs or the patients experience unacceptable toxicity or withdraw consent.
Investigators had pre-specified subgroup analyses to examine what populations fared better and worse in each arm. Prior bortezomib exposure did not significantly affect PFS for those receiving carfilzomib. Not enough patients had received carfilzomib prior to the study to permit analysis of this effect. Though carfilzomib arm patients with high-risk factors on cytogenetic analysis fared slightly better than those on bortezomib, “neither proteasome inhibitor appears to significantly overcome the adverse prognostic effect of high-risk cytogenetics,” wrote Dr. Dimopoulos and his co-authors.
Secondary outcome measures included the numbers of patients in each arm achieving complete response or better (with this group broken into “stringent complete response” as well as complete response). Significantly more carfilzomib than bortezomib patients reached this mark (58 patients [13%] vs 29 patients [6%], P equal to 0.001). A larger group of patients in each arm met the secondary outcome measure of experiencing very good partial response or better, with those on the carfelzomib arm faring significantly better than those receiving bortezomib (252 [54%] vs 133 [29%], P less than 0.0001).
Dr. Dimopoulos and his co-authors remarked on the importance of these findings, noting that “The finding that the proportion of patients with a complete response or better and very good partial response or better was higher in the carfilzomib group than in the bortezomib group is encouraging because studies have shown an association between depth of response and improved survival in patients with multiple myeloma.”
ENDEAVOR’s safety analyses included all patients who received at least one dose of study drug. Overall, the carfilzomib patients had a 48% rate of serious adverse events (224 of 463 patients for this analysis), compared to 36% of the bortezomib group (162 of 456 patients).
On-study death rates were similar between the two groups, with 18 of 464 patients (4%) in the carfilzomib group and 16 of 465 patients (3%) in the bortezomib group dying during the study period. Four deaths in each arm were judged related to disease progression.
A pre-planned substudy examined cardiac function for patients in both study arms, and did not find increased risk of left or right ventricular dysfunction or cumulative cardiac injury for carfilzomib when compared with bortezomib.
The open label nature of the study, an acknowledged limitation, was necessitated by the different dosing regimens of the two drugs, but the treatment allocation was hidden from the independent review committee that assessed participant disease status. The funder was also masked to per-group treatment results.
Carfilzomib’s irreversable proteosome inhibition may lead to more sustained proteasomal inhibition, Dr. Dimopoulous and his co-authors posited. Also, carfilzomib can overcome bortezomib resistance and is more potent against cell lines naive to this class of drugs in preclinical studies, they said.
“Taken together, the results from the ENDEAVOR study suggest an important role for carfilzomib-based regimens for patients with relapsed or refractory jultiple myeloma,” wrote Dr. Dimopoulos and his collaborators.
The study was supported by Onyx Pharmaceuticals, Inc, a subsidiary of Amgen. Dr. Dimopoulos reported non-financial support from Onyx Pharmaceuticals, Celgene Corporation, and Ortho-Biotech. Several co-authors reported multiple financial associations with pharmaceutical companies.
On Twitter @karioakes
In a head-to-head clinical trial, carfilzomib (Kyprolis) plus dexamethasone doubled the length of progression-free survival (PFS) when compared with bortezomib (Velcade) plus dexamethasone for individuals with relapsed or refractory multiple myeloma. Response rates were also significantly higher for those receiving carfilzomib, though neither medication could overcome high-risk cytogenetic factors.
The international multi-center phase 3 open-label ENDEAVOR study (clinicaltrials.gov ID: NCT01568866) compared outcomes for a total of 929 patients who were randomized 1:1 to receive one of the treatments. Randomization was stratified to account for prior treatments, including prior proteasome treatment, as well as disease severity by International Staging System stage.
Reporting for the ENDEAVOR investigators, Dr. Meletios Dimopoulos and his co-authors published their results in The Lancet simultaneously with a presentation at the annual meeting of the American Society of Hematology [Lancet Oncol, http://dx.doi.org/10.1016/S1470-2045(15)00464-7] . Dr. Dimopoulos is a professor in the department of clinical therapeutics at the school of medicine of the National and Kapodistrian University of Athens.
The primary endpoint, PFS in the intention-to-treat population, differed significantly between the study arms when the data were analyzed at a pre-planned interim analysis. For those receiving carfilzomib, PFS was 18.7 months, compared with 9.4 months in the bortezomib group, for a hazard ratio of 0.53 (P less than 0.0001). Data were drawn from a pre-specified interim analysis, and study participation is ongoing, with 28-day dosing cycles repeated until disease progression occurrs or the patients experience unacceptable toxicity or withdraw consent.
Investigators had pre-specified subgroup analyses to examine what populations fared better and worse in each arm. Prior bortezomib exposure did not significantly affect PFS for those receiving carfilzomib. Not enough patients had received carfilzomib prior to the study to permit analysis of this effect. Though carfilzomib arm patients with high-risk factors on cytogenetic analysis fared slightly better than those on bortezomib, “neither proteasome inhibitor appears to significantly overcome the adverse prognostic effect of high-risk cytogenetics,” wrote Dr. Dimopoulos and his co-authors.
Secondary outcome measures included the numbers of patients in each arm achieving complete response or better (with this group broken into “stringent complete response” as well as complete response). Significantly more carfilzomib than bortezomib patients reached this mark (58 patients [13%] vs 29 patients [6%], P equal to 0.001). A larger group of patients in each arm met the secondary outcome measure of experiencing very good partial response or better, with those on the carfelzomib arm faring significantly better than those receiving bortezomib (252 [54%] vs 133 [29%], P less than 0.0001).
Dr. Dimopoulos and his co-authors remarked on the importance of these findings, noting that “The finding that the proportion of patients with a complete response or better and very good partial response or better was higher in the carfilzomib group than in the bortezomib group is encouraging because studies have shown an association between depth of response and improved survival in patients with multiple myeloma.”
ENDEAVOR’s safety analyses included all patients who received at least one dose of study drug. Overall, the carfilzomib patients had a 48% rate of serious adverse events (224 of 463 patients for this analysis), compared to 36% of the bortezomib group (162 of 456 patients).
On-study death rates were similar between the two groups, with 18 of 464 patients (4%) in the carfilzomib group and 16 of 465 patients (3%) in the bortezomib group dying during the study period. Four deaths in each arm were judged related to disease progression.
A pre-planned substudy examined cardiac function for patients in both study arms, and did not find increased risk of left or right ventricular dysfunction or cumulative cardiac injury for carfilzomib when compared with bortezomib.
The open label nature of the study, an acknowledged limitation, was necessitated by the different dosing regimens of the two drugs, but the treatment allocation was hidden from the independent review committee that assessed participant disease status. The funder was also masked to per-group treatment results.
Carfilzomib’s irreversable proteosome inhibition may lead to more sustained proteasomal inhibition, Dr. Dimopoulous and his co-authors posited. Also, carfilzomib can overcome bortezomib resistance and is more potent against cell lines naive to this class of drugs in preclinical studies, they said.
“Taken together, the results from the ENDEAVOR study suggest an important role for carfilzomib-based regimens for patients with relapsed or refractory jultiple myeloma,” wrote Dr. Dimopoulos and his collaborators.
The study was supported by Onyx Pharmaceuticals, Inc, a subsidiary of Amgen. Dr. Dimopoulos reported non-financial support from Onyx Pharmaceuticals, Celgene Corporation, and Ortho-Biotech. Several co-authors reported multiple financial associations with pharmaceutical companies.
On Twitter @karioakes
In a head-to-head clinical trial, carfilzomib (Kyprolis) plus dexamethasone doubled the length of progression-free survival (PFS) when compared with bortezomib (Velcade) plus dexamethasone for individuals with relapsed or refractory multiple myeloma. Response rates were also significantly higher for those receiving carfilzomib, though neither medication could overcome high-risk cytogenetic factors.
The international multi-center phase 3 open-label ENDEAVOR study (clinicaltrials.gov ID: NCT01568866) compared outcomes for a total of 929 patients who were randomized 1:1 to receive one of the treatments. Randomization was stratified to account for prior treatments, including prior proteasome treatment, as well as disease severity by International Staging System stage.
Reporting for the ENDEAVOR investigators, Dr. Meletios Dimopoulos and his co-authors published their results in The Lancet simultaneously with a presentation at the annual meeting of the American Society of Hematology [Lancet Oncol, http://dx.doi.org/10.1016/S1470-2045(15)00464-7] . Dr. Dimopoulos is a professor in the department of clinical therapeutics at the school of medicine of the National and Kapodistrian University of Athens.
The primary endpoint, PFS in the intention-to-treat population, differed significantly between the study arms when the data were analyzed at a pre-planned interim analysis. For those receiving carfilzomib, PFS was 18.7 months, compared with 9.4 months in the bortezomib group, for a hazard ratio of 0.53 (P less than 0.0001). Data were drawn from a pre-specified interim analysis, and study participation is ongoing, with 28-day dosing cycles repeated until disease progression occurrs or the patients experience unacceptable toxicity or withdraw consent.
Investigators had pre-specified subgroup analyses to examine what populations fared better and worse in each arm. Prior bortezomib exposure did not significantly affect PFS for those receiving carfilzomib. Not enough patients had received carfilzomib prior to the study to permit analysis of this effect. Though carfilzomib arm patients with high-risk factors on cytogenetic analysis fared slightly better than those on bortezomib, “neither proteasome inhibitor appears to significantly overcome the adverse prognostic effect of high-risk cytogenetics,” wrote Dr. Dimopoulos and his co-authors.
Secondary outcome measures included the numbers of patients in each arm achieving complete response or better (with this group broken into “stringent complete response” as well as complete response). Significantly more carfilzomib than bortezomib patients reached this mark (58 patients [13%] vs 29 patients [6%], P equal to 0.001). A larger group of patients in each arm met the secondary outcome measure of experiencing very good partial response or better, with those on the carfelzomib arm faring significantly better than those receiving bortezomib (252 [54%] vs 133 [29%], P less than 0.0001).
Dr. Dimopoulos and his co-authors remarked on the importance of these findings, noting that “The finding that the proportion of patients with a complete response or better and very good partial response or better was higher in the carfilzomib group than in the bortezomib group is encouraging because studies have shown an association between depth of response and improved survival in patients with multiple myeloma.”
ENDEAVOR’s safety analyses included all patients who received at least one dose of study drug. Overall, the carfilzomib patients had a 48% rate of serious adverse events (224 of 463 patients for this analysis), compared to 36% of the bortezomib group (162 of 456 patients).
On-study death rates were similar between the two groups, with 18 of 464 patients (4%) in the carfilzomib group and 16 of 465 patients (3%) in the bortezomib group dying during the study period. Four deaths in each arm were judged related to disease progression.
A pre-planned substudy examined cardiac function for patients in both study arms, and did not find increased risk of left or right ventricular dysfunction or cumulative cardiac injury for carfilzomib when compared with bortezomib.
The open label nature of the study, an acknowledged limitation, was necessitated by the different dosing regimens of the two drugs, but the treatment allocation was hidden from the independent review committee that assessed participant disease status. The funder was also masked to per-group treatment results.
Carfilzomib’s irreversable proteosome inhibition may lead to more sustained proteasomal inhibition, Dr. Dimopoulous and his co-authors posited. Also, carfilzomib can overcome bortezomib resistance and is more potent against cell lines naive to this class of drugs in preclinical studies, they said.
“Taken together, the results from the ENDEAVOR study suggest an important role for carfilzomib-based regimens for patients with relapsed or refractory jultiple myeloma,” wrote Dr. Dimopoulos and his collaborators.
The study was supported by Onyx Pharmaceuticals, Inc, a subsidiary of Amgen. Dr. Dimopoulos reported non-financial support from Onyx Pharmaceuticals, Celgene Corporation, and Ortho-Biotech. Several co-authors reported multiple financial associations with pharmaceutical companies.
On Twitter @karioakes
FROM ASH 2015
Key clinical point: Carfilzomib nearly doubled progression-free survival (PFS) compared with bortezomib for relapsed and refractory multiple myeloma
Major finding: For those receiving carfilzomib, PFS was 18.7 months, compared with 9.4 months in the bortezomib group, for a hazard ratio of 0.53 (P less than .0001).
Data source: International randomized open-label clinical trial of 979 patients with refractory or relapsed multiple myeloma.
Disclosures: The study was supported by Onyx Pharmaceuticals, Inc, a subsidiary of Amgen. Dr. Dimopoulos reported non-financial support from Onyx Pharmaceuticals, Celgene Corporation, and Ortho-Biotech. Several co-authors reported multiple financial associations with pharmaceutical companies.