User login
Chronic lymphocytic leukemia (CLL) is a common hematological malignancy in the U.S. with 15,000 new patients diagnosed each year.1 This leukemia is frequently diagnosed in veterans since it is more commonly seen in an elderly male population. The disease is characterized by a slow accumulation of mature B cells that are functionally incompetent and resist apoptosis. CLL has an indolent clinical course, but about 60% to 70% of patients require treatment. The disease also runs a variable course, and a number of genetic abnormalities and prognostic markers have been defined to subclassify CLL patients and prognosticate.2-4 This article reviews important CLL signaling pathways and novel therapeutic agents in this leukemia.
Signaling Pathways
B-Cell Receptor Signaling
The B-cell receptor (BCR) signaling is the major signaling pathway in CLL, because it defines clinical, biologic, and prognostic characteristics of the disease.5 The BCR is composed of a surface transmembrane immunoglobulin that binds the antigen with CD79 alpha and beta chains. The activation of BCR results in the formation of a signaling complex or signalosome, which includes Lyn, Syk, BTK, and ZAP-70, among other components that assemble with other adaptor proteins (Figure). This assembly of proteins occurs on the cytoplasmic tails of immunoglobulin chains on regions called immunoreceptor tyrosine-based motifs (ITAMs).
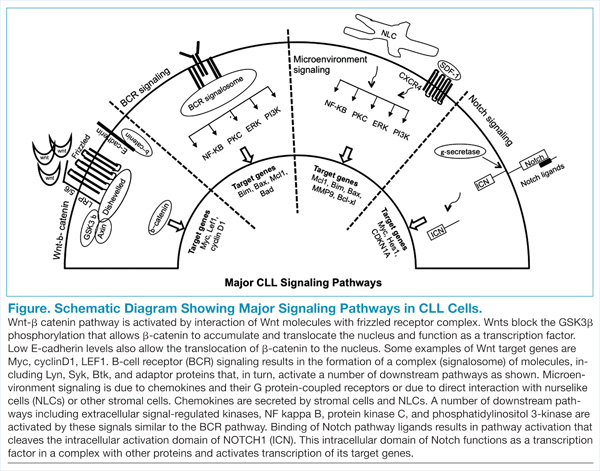
With the assembly of this signaling complex, BCR stimulates a number of downstream pathways, such as phosphatidylinositol 3-kinase (PI3K), protein kinase B (Akt), protein kinase C, nuclear factor-κB (NFκB), and extracellular signal-regulated kinases (ERKs) (Figure). Activation of these pathways results in cell proliferation, resistance to apoptosis, increased cell motility and migration. Recent studies have identified additional novel components of this signaling complex, including a guanine nucleotide exchange factor (GEF) RASGRF1. This GEF is activated by BCR signaling and, in turn, stimulates the ERK pathway by increasing the production of active GTP-bound Ras.6
The ability of BCR to activate a number of downstream signaling pathways makes it a highly relevant and investigated pathway in this leukemia. Inhibitors have been developed and/or identified against a number of signalosome components to block the BCR signaling.7 Syk and Lyn are Src kinases, and their phosphorylation is one of the initial events of BCR signaling. Syk is overexpressed in CLL specimens, and Syk inhibitors (R406 and P505-15, also known as PRT062607) have shown activity in CLL.8,9 Dasatinib is a Src inhibitor that also shows activity in CLL specimens and is being studied in combination with chemotherapy drugs in refractory CLL patients.10
BTK, a component of the BCR signalosome, is required for BCR function, and loss of its function is seen in X-linked agammaglobulinemia. PCI-32765 (ibrutinib) is an oral BTK inhibitor that irreversibly inactivates this kinase and has been approved for clinical use in CLL patients.11,12 Another signaling pathway activated by BCR is the PI3K, and a promising inhibitor (CAL-101) blocks its activity in CLL specimens.13 Investigative work has identified that the delta isoform of PI3K p110 is highly expressed in B cells and lymphocytes.14 This is a catalytic subunit of a class I PI3K with a role in BCR signaling. A selective inhibitor GS-1101 (CAL-101) is able to block PI3K signaling in CLL specimens and inhibits Akt phosphorylation and other downstream effectors along with induction of apoptosis.15 The clinical data with BTK and PI3K inhibitors will be discussed later in this review.
CLL and the Microenvironment
Interactions between CLL cells and the microenvironment allow CLL cells to thrive in certain niche environments.16,17 Interaction mainly occurs via bone marrow stromal cells and nurselike cells (NLCs), which evolve from monocytes (Figure). These interactions can be divided into 2 groups. First, CLL cell growth is supported by a number of chemokine receptor-ligand interactions. CXCR4 is the receptor for CXCL12 (SDF-1) that stimulates chemotaxis and tissue homing. Another chemokine is CXCL13, which acts via its receptor CXCR5 and is involved in chemotaxis and activation of other kinases. Second, NLCs also support CLL cells by expressing TNF family members BAFF and APRIL, which interact with their receptors and activate the NFκB pathway.
Leukemic cells also express VLA-4 integrins, which further their support adhesion to the stromal cells and predict for an aggressive phenotype. Specific inhibitors that block the stimulation by chemokines and cytokines are not yet available; however, one can envision that this class of inhibitors will decrease the chemoresistance of leukemic cells and will be used in conjunction with other chemotherapy agents. Interestingly, inhibitors that block BCR-mediated signaling (BTK and PI3K inhibitors) also inhibit signaling via the microenvironment and chemokines.
Wnt-β-catenin Pathway
Wnt signaling affects developmental pathways, and its aberrant activation has major oncogenic effects as well. This pathway is activated in CLL as these leukemic cells express high levels of Wnt and frizzled along with epigenetic downregulation of Wnt pathway antagonist genes, including secreted frizzled-related protein (SFRP) family members and WIF1 (Figure).18-20 The binding of Wnts to their cognate receptors results in inhibition of GSK3β phosphorylation and stabilization of β-catenin, which then translocates to the nucleus and interacts with lymphoid-enhancing (LEF) and T-cell transcription factors to activate transcription of Wnt-target genes. Lack of E-cadherin expression in CLL cells also results in an increase in translocation of β-catenin and upregulation of the Wnt pathway.20
Wnt-target genes include Myc, LEF, cyclinD1, COX-2, and MMP. Gene expression profiling from our laboratory and other groups have identified the overexpression of these wnt-target genes and support this pathway activation in CLL cells.20 This is a promising signalling pathway and an active area of research for developing inhibitors that will have a growth inhibitory effect on CLL leukemic cells. GSK3b inhibitors and other drugs that re-express epigenetically silenced Wnt antagonist genes have been shown to inhibit this pathway activity in CLL cells in vitro.
Notch Pathway Activation
High-throughput exome sequencing has identified recurring mutations in a number of genes, including NOTCH1.21 Analysis of additional CLL patients confirmed activating NOTCH1 mutations in 10% to 15% of CLL patients and were also associated with poor outcome.22 This pathway is activated by ligands such as Jagged and Delta-like, which interact with the Notch receptor, which is then cleaved by γ-secretases. The cleaved intracellular domain of the NOTCH1 receptor in combination with other factors activates transcription of target genes, including Myc and HES1 (Figure). Besides the mutations that generate a truncated protein or may stabilize the pathway, the Notch pathway is also constitutively active in CLL specimens.23 Notch stimulation increases activity of prosurvival pathways and genes such as NFκB that resist apoptotic signals. The pathway can be inhibited by γ-secretase inhibitors (GSIs), which reduce the levels of cleaved NOTCH1 protein and downregulated Notch target genes. This pathway is also able to modulate the microenvironment stimuli as the GSIs inhibit responses to chemokines such as CXCL12 and inhibit migration and invasion.24
Newer Theraputic Agents
Work on signaling mechanisms paid dividends in CLL with the recent development of 2 inhibitors. Ibrutinib (BTK inhibitor) and idelalisib (PI3K inhibitor) are being studied in clinical trials, and both drugs block the BCR and microenvironment signaling pathways, thereby inhibiting the growth of CLL cells.
BTK Inhibitor: Ibrutinib
The activity of BTK is critical for a number of CLL signaling pathways, and it is a component of the initial signaling complex or signalosome that is formed with BCR signaling. Studies have shown that inhibiting this kinase blocks a number of pathways, including ERK, NFκB, and others. The drug ibrutinib blocks this kinase by forming a covalent bond and inhibiting its enzyme activity. This orally bioavailable drug showed activity in phase 1 trials in different B-cell malignancies.25 In a phase 2 study, high-risk CLL patients were given 2 different doses of this inhibitor, and the overall response rate was 71% with an overall survival at 26 months of 83%.11 Responses were seen in all patients irrespective of clinical and genetic risk factors. Based on these findings, the drug was approved for clinical use in patients with relapsed or refractory disease. Recently, there are data on the use of this drug as frontline therapy in elderly patients, and the drug was well tolerated.26 There are additional ongoing trials to compare this drug with other agents, including chlorambucil (in chemotherapy-naïve patients) and ofatumumab (in relapsed or refractory patients).
PI3 Kinase p110 Delta Inhibitor: Idelalisib
The crucial finding for the development of this inhibitor was the over-expression of the delta isoform of PI3K p110 in B-cell malignancies.14 The drug CAL-101 selectively inhibits this constitutively active isoform and induces apoptosis in a number of B-cell malignancies.15,27 In the phase 1 trial, this inhibitor was evaluated in relapsed/refractory patients at multiple dose levels.28 There was inhibition of PI3K signaling with an overall response rate of 72%, and a partial response rate of 39% was observed in CLL patients. This was followed by a randomized, placebo-controlled phase 3 study in which patients with myelosuppression, decreased renal function, or other illnesses were treated with either rituximab alone or with rituximab and idelalisib.29
At the time of reporting, the median progression-free survival (PFS) was 5.5 months in the placebo arm and was not reached in the idelalisib arm. Overall response rates were higher in the idelalisib group (81% vs 13%) with similar toxicity profiles in the 2 groups. This drug is now being extensively studied in combination with bendamustine and other anti-CD20 antibodies in clinical trials.
A unique toxicity observed with both these inhibitors is the initial lymphocytosis. In the case of ibrutinib, this was seen in a majority of patients (77%) and at the same time there was a response in the nodal disease, implying a redistribution of leukemic cells from the tissues to the peripheral blood.30
A potential explanation is that these drugs inhibit signaling via chemokines and other components of the microenvironment and by inhibiting the homing signals, allows leukemic cells to move out of their niche areas. This was analyzed in a recent study that compared clinical and biochemical parameters of patients who had a complete or partial response with ibrutinib compared with a “partial response except for lymphocytosis.”30 Patients with “partial response except for lymphocytosis” were found to have favorable prognostic factors, and the persisting leukemic cells were not clonally different from the original cells. The progression free survival of patients with “partial response except for lymphocytosis” was also similar to the subgroup with no prolonged lymphocytosis.
Discussion
Several therapeutic agents with novel mechanisms of action are effective in killing the CLL leukemic cells, and a number of targeted agents are currently in the pipeline. The next challenge for treating CLL will be the proper integration of these novel targeted agents with the traditional chemotherapy and chemoimmunotherapy approaches. Let us consider CLL patients in different clinical settings. First, a patient aged 60 years who is otherwise healthy will be treated with possibly all the available chemotherapy and chemoimmunotherapy options, as well as the newer targeted agents. In this clinical setting sequencing of therapy is not a major concern. On the other hand, a patient aged 70 years who is already refractory to multiple lines of therapy is a good candidate for these newer drugs.
The more controversial use of these targeted agents will be in an older patient with some comorbidities and newly diagnosed CLL. In this clinical setting, should one go with traditional chemotherapy/chemoimmunotherapy approaches or consider newer targeted agents? These issues are now being addressed in clinical trials, and with acceptable toxicity profiles these newer drugs will move to the frontline setting.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect an endorsement by or opinion of Federal Practitioner, Frontline Medical Communications, the U.S. Air Force, the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drug combinations–including indications, contraindications, warnings, and adverse effects–before administering pharmacologic therapy to patients.
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9-29.
2. Döhner H, Stilgenbauer S, Döhner K, Bentz M, Lichter P. Chromosome aberrations in B-cell chronic lymphocytic leukemia: reassessment based on molecular cytogenetic analysis. J Mol Med. 1999;77(2):266-281.
3. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848-1854.
4. Chen L, Widhopf G, Huynh L, et al. Expression of ZAP-70 is associated with increased B-cell receptor signaling in chronic lymphocytic leukemia. Blood. 2002;100(13):4609-4614.
5. Wickremasinghe RG, Prentice AG, Steele AJ. Aberrantly activated anti-apoptotic signalling mechanisms in chronic lymphocytic leukaemia cells: Clues to the identification of novel therapeutic targets. Br J Haematol. 2011;153(5):545-556.
6. Liao W, Jordaan G, Coriaty N, Sharma S. Amplification of B cell receptor-Erk signaling by Rasgrf-1 overexpression in chronic lymphocytic leukemia [published online ahead of print April 2, 2014]. Leuk Lymphoma. doi: 10.3109/10428194.2014898759.
7. Burger JA. Inhibiting B-cell receptor signaling pathways in chronic lymphocytic leukemia. Curr Hematol Malig Rep. 2012;7(1):26-33.
8. Buchner M, Fuchs S, Prinz G, et al. Spleen tyrosine kinase is overexpressed and represents a potential therapeutic target in chronic lymphocytic leukemia. Cancer Res. 2009;69(13):5424-5432.
9. Spurgeon SE, Coffey G, Fletcher LB, et al. The selective SYK inhibitor P505-15 (PRT062607) inhibits B cell signaling and function in vitro and in vivo and augments the activity of fludarabine in chronic lymphocytic leukemia. J Pharmacol Exp Ther. 2013;344(2):378-387.
10. Veldurthy A, Patz M, Hagist S, et al. The kinase inhibitor dasatinib induces apoptosis in chronic lymphocytic leukemia cells in vitro with preference for a subgroup of patients with unmutated IgVH genes. Blood. 2008;112(4):1443-1452.
11. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia [published correction appears in N Engl J Med. 2014;370(8):786]. N Engl J Med. 2013;369(1):32-42.
12. Cheng S, Ma J, Guo A, et al. BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia. 2014;28(3):649-657.
13. Lannutti BJ, Meadows SA, Herman SE, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591-594.
14. Chantry D, Vojtek A, Kashishian A, et al. p110delta, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J Biol Chem. 1997;272(31):19236-19241.
15. Hoellenriegel J, Meadows SA, Sivina M, et al. The phosphoinositide 3’-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011;118(13):3603-3612.
16. Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F. The microenvironment in mature B-cell malignancies: A target for new treatment strategies. Blood. 2009;114(16):3367-3375.
17. ten Hacken E, Burger JA. Molecular pathways: targeting the microenvironment in chronic lymphocytic leukemia—focus on the B-cell receptor. Clin Cancer Res. 2014;20(3):548-556.
18. Gandhirajan RK, Poll-Wolbeck SJ, Gehrke I, Kreuzer KA. Wnt/b-catenin/LEF-1 signaling in chronic lymphocytic leukemia (CLL): a target for current and potential therapeutic options. Curr Cancer Drug Targets. 2010;10(7):716-727.
19. Gutierrez A, Jr, Tschumper RC, Wu X, et al. LEF-1 is a prosurvival factor in chronic lymphocytic leukemia and is expressed in the preleukemic state of monoclonal B-cell lymphocytosis. Blood. 2010;116(16):2975-2983.
20. Jordaan G, Liao W, Sharma S. E-cadherin gene re-expression in chronic lymphocytic leukemia cells by HDAC inhibitors. BMC Cancer. 2013;13:88.
21. Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101-105.
22. Fabbri G, Rasi S, Rossi D, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208(7):1389-1401.
23. Rosati E, Sabatini R, Rampino G, et al. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood. 2009;113(4):856-865.
24. López-Guerra M, Xargay-Torrent S, Rosich L, et al. The g-secretase inhibitor PF-03084014 combined with fludarabine antagonizes migration, invasion and angiogenesis in NOTCH1-mutated CLL cells [published online ahead of print April 30, 2014]. Leukemia. doi: 10.1038/leu.2014.143.
25. Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31(1):88-94.
26. O’Brien S, Furman RR, Coutre SE, et al. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: An open-label, multicentre, phase 1b/2 trial. Lancet Oncol. 2014;15(1):48-58.
27. Brown JR, Byrd JC, Coutre SE, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110∂, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;123(22):3390-3397.
28. Brown JR, Furman RR, Flinn I, et al. Final results of a phase I study of idelalisib (GS-1101) a selective inhibitor of PI3K∂, in patients with relapsed or refractory CLL. J Clin Oncol. 2013;31:Absract 7003.
29. Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997-1007.
30. Woyach JA, Smucker K, Smith LL, et al. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate a suboptimal response to therapy. Blood. 2013;123(12):1810-1817.
Chronic lymphocytic leukemia (CLL) is a common hematological malignancy in the U.S. with 15,000 new patients diagnosed each year.1 This leukemia is frequently diagnosed in veterans since it is more commonly seen in an elderly male population. The disease is characterized by a slow accumulation of mature B cells that are functionally incompetent and resist apoptosis. CLL has an indolent clinical course, but about 60% to 70% of patients require treatment. The disease also runs a variable course, and a number of genetic abnormalities and prognostic markers have been defined to subclassify CLL patients and prognosticate.2-4 This article reviews important CLL signaling pathways and novel therapeutic agents in this leukemia.
Signaling Pathways
B-Cell Receptor Signaling
The B-cell receptor (BCR) signaling is the major signaling pathway in CLL, because it defines clinical, biologic, and prognostic characteristics of the disease.5 The BCR is composed of a surface transmembrane immunoglobulin that binds the antigen with CD79 alpha and beta chains. The activation of BCR results in the formation of a signaling complex or signalosome, which includes Lyn, Syk, BTK, and ZAP-70, among other components that assemble with other adaptor proteins (Figure). This assembly of proteins occurs on the cytoplasmic tails of immunoglobulin chains on regions called immunoreceptor tyrosine-based motifs (ITAMs).
With the assembly of this signaling complex, BCR stimulates a number of downstream pathways, such as phosphatidylinositol 3-kinase (PI3K), protein kinase B (Akt), protein kinase C, nuclear factor-κB (NFκB), and extracellular signal-regulated kinases (ERKs) (Figure). Activation of these pathways results in cell proliferation, resistance to apoptosis, increased cell motility and migration. Recent studies have identified additional novel components of this signaling complex, including a guanine nucleotide exchange factor (GEF) RASGRF1. This GEF is activated by BCR signaling and, in turn, stimulates the ERK pathway by increasing the production of active GTP-bound Ras.6
The ability of BCR to activate a number of downstream signaling pathways makes it a highly relevant and investigated pathway in this leukemia. Inhibitors have been developed and/or identified against a number of signalosome components to block the BCR signaling.7 Syk and Lyn are Src kinases, and their phosphorylation is one of the initial events of BCR signaling. Syk is overexpressed in CLL specimens, and Syk inhibitors (R406 and P505-15, also known as PRT062607) have shown activity in CLL.8,9 Dasatinib is a Src inhibitor that also shows activity in CLL specimens and is being studied in combination with chemotherapy drugs in refractory CLL patients.10
BTK, a component of the BCR signalosome, is required for BCR function, and loss of its function is seen in X-linked agammaglobulinemia. PCI-32765 (ibrutinib) is an oral BTK inhibitor that irreversibly inactivates this kinase and has been approved for clinical use in CLL patients.11,12 Another signaling pathway activated by BCR is the PI3K, and a promising inhibitor (CAL-101) blocks its activity in CLL specimens.13 Investigative work has identified that the delta isoform of PI3K p110 is highly expressed in B cells and lymphocytes.14 This is a catalytic subunit of a class I PI3K with a role in BCR signaling. A selective inhibitor GS-1101 (CAL-101) is able to block PI3K signaling in CLL specimens and inhibits Akt phosphorylation and other downstream effectors along with induction of apoptosis.15 The clinical data with BTK and PI3K inhibitors will be discussed later in this review.
CLL and the Microenvironment
Interactions between CLL cells and the microenvironment allow CLL cells to thrive in certain niche environments.16,17 Interaction mainly occurs via bone marrow stromal cells and nurselike cells (NLCs), which evolve from monocytes (Figure). These interactions can be divided into 2 groups. First, CLL cell growth is supported by a number of chemokine receptor-ligand interactions. CXCR4 is the receptor for CXCL12 (SDF-1) that stimulates chemotaxis and tissue homing. Another chemokine is CXCL13, which acts via its receptor CXCR5 and is involved in chemotaxis and activation of other kinases. Second, NLCs also support CLL cells by expressing TNF family members BAFF and APRIL, which interact with their receptors and activate the NFκB pathway.
Leukemic cells also express VLA-4 integrins, which further their support adhesion to the stromal cells and predict for an aggressive phenotype. Specific inhibitors that block the stimulation by chemokines and cytokines are not yet available; however, one can envision that this class of inhibitors will decrease the chemoresistance of leukemic cells and will be used in conjunction with other chemotherapy agents. Interestingly, inhibitors that block BCR-mediated signaling (BTK and PI3K inhibitors) also inhibit signaling via the microenvironment and chemokines.
Wnt-β-catenin Pathway
Wnt signaling affects developmental pathways, and its aberrant activation has major oncogenic effects as well. This pathway is activated in CLL as these leukemic cells express high levels of Wnt and frizzled along with epigenetic downregulation of Wnt pathway antagonist genes, including secreted frizzled-related protein (SFRP) family members and WIF1 (Figure).18-20 The binding of Wnts to their cognate receptors results in inhibition of GSK3β phosphorylation and stabilization of β-catenin, which then translocates to the nucleus and interacts with lymphoid-enhancing (LEF) and T-cell transcription factors to activate transcription of Wnt-target genes. Lack of E-cadherin expression in CLL cells also results in an increase in translocation of β-catenin and upregulation of the Wnt pathway.20
Wnt-target genes include Myc, LEF, cyclinD1, COX-2, and MMP. Gene expression profiling from our laboratory and other groups have identified the overexpression of these wnt-target genes and support this pathway activation in CLL cells.20 This is a promising signalling pathway and an active area of research for developing inhibitors that will have a growth inhibitory effect on CLL leukemic cells. GSK3b inhibitors and other drugs that re-express epigenetically silenced Wnt antagonist genes have been shown to inhibit this pathway activity in CLL cells in vitro.
Notch Pathway Activation
High-throughput exome sequencing has identified recurring mutations in a number of genes, including NOTCH1.21 Analysis of additional CLL patients confirmed activating NOTCH1 mutations in 10% to 15% of CLL patients and were also associated with poor outcome.22 This pathway is activated by ligands such as Jagged and Delta-like, which interact with the Notch receptor, which is then cleaved by γ-secretases. The cleaved intracellular domain of the NOTCH1 receptor in combination with other factors activates transcription of target genes, including Myc and HES1 (Figure). Besides the mutations that generate a truncated protein or may stabilize the pathway, the Notch pathway is also constitutively active in CLL specimens.23 Notch stimulation increases activity of prosurvival pathways and genes such as NFκB that resist apoptotic signals. The pathway can be inhibited by γ-secretase inhibitors (GSIs), which reduce the levels of cleaved NOTCH1 protein and downregulated Notch target genes. This pathway is also able to modulate the microenvironment stimuli as the GSIs inhibit responses to chemokines such as CXCL12 and inhibit migration and invasion.24
Newer Theraputic Agents
Work on signaling mechanisms paid dividends in CLL with the recent development of 2 inhibitors. Ibrutinib (BTK inhibitor) and idelalisib (PI3K inhibitor) are being studied in clinical trials, and both drugs block the BCR and microenvironment signaling pathways, thereby inhibiting the growth of CLL cells.
BTK Inhibitor: Ibrutinib
The activity of BTK is critical for a number of CLL signaling pathways, and it is a component of the initial signaling complex or signalosome that is formed with BCR signaling. Studies have shown that inhibiting this kinase blocks a number of pathways, including ERK, NFκB, and others. The drug ibrutinib blocks this kinase by forming a covalent bond and inhibiting its enzyme activity. This orally bioavailable drug showed activity in phase 1 trials in different B-cell malignancies.25 In a phase 2 study, high-risk CLL patients were given 2 different doses of this inhibitor, and the overall response rate was 71% with an overall survival at 26 months of 83%.11 Responses were seen in all patients irrespective of clinical and genetic risk factors. Based on these findings, the drug was approved for clinical use in patients with relapsed or refractory disease. Recently, there are data on the use of this drug as frontline therapy in elderly patients, and the drug was well tolerated.26 There are additional ongoing trials to compare this drug with other agents, including chlorambucil (in chemotherapy-naïve patients) and ofatumumab (in relapsed or refractory patients).
PI3 Kinase p110 Delta Inhibitor: Idelalisib
The crucial finding for the development of this inhibitor was the over-expression of the delta isoform of PI3K p110 in B-cell malignancies.14 The drug CAL-101 selectively inhibits this constitutively active isoform and induces apoptosis in a number of B-cell malignancies.15,27 In the phase 1 trial, this inhibitor was evaluated in relapsed/refractory patients at multiple dose levels.28 There was inhibition of PI3K signaling with an overall response rate of 72%, and a partial response rate of 39% was observed in CLL patients. This was followed by a randomized, placebo-controlled phase 3 study in which patients with myelosuppression, decreased renal function, or other illnesses were treated with either rituximab alone or with rituximab and idelalisib.29
At the time of reporting, the median progression-free survival (PFS) was 5.5 months in the placebo arm and was not reached in the idelalisib arm. Overall response rates were higher in the idelalisib group (81% vs 13%) with similar toxicity profiles in the 2 groups. This drug is now being extensively studied in combination with bendamustine and other anti-CD20 antibodies in clinical trials.
A unique toxicity observed with both these inhibitors is the initial lymphocytosis. In the case of ibrutinib, this was seen in a majority of patients (77%) and at the same time there was a response in the nodal disease, implying a redistribution of leukemic cells from the tissues to the peripheral blood.30
A potential explanation is that these drugs inhibit signaling via chemokines and other components of the microenvironment and by inhibiting the homing signals, allows leukemic cells to move out of their niche areas. This was analyzed in a recent study that compared clinical and biochemical parameters of patients who had a complete or partial response with ibrutinib compared with a “partial response except for lymphocytosis.”30 Patients with “partial response except for lymphocytosis” were found to have favorable prognostic factors, and the persisting leukemic cells were not clonally different from the original cells. The progression free survival of patients with “partial response except for lymphocytosis” was also similar to the subgroup with no prolonged lymphocytosis.
Discussion
Several therapeutic agents with novel mechanisms of action are effective in killing the CLL leukemic cells, and a number of targeted agents are currently in the pipeline. The next challenge for treating CLL will be the proper integration of these novel targeted agents with the traditional chemotherapy and chemoimmunotherapy approaches. Let us consider CLL patients in different clinical settings. First, a patient aged 60 years who is otherwise healthy will be treated with possibly all the available chemotherapy and chemoimmunotherapy options, as well as the newer targeted agents. In this clinical setting sequencing of therapy is not a major concern. On the other hand, a patient aged 70 years who is already refractory to multiple lines of therapy is a good candidate for these newer drugs.
The more controversial use of these targeted agents will be in an older patient with some comorbidities and newly diagnosed CLL. In this clinical setting, should one go with traditional chemotherapy/chemoimmunotherapy approaches or consider newer targeted agents? These issues are now being addressed in clinical trials, and with acceptable toxicity profiles these newer drugs will move to the frontline setting.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect an endorsement by or opinion of Federal Practitioner, Frontline Medical Communications, the U.S. Air Force, the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drug combinations–including indications, contraindications, warnings, and adverse effects–before administering pharmacologic therapy to patients.
Chronic lymphocytic leukemia (CLL) is a common hematological malignancy in the U.S. with 15,000 new patients diagnosed each year.1 This leukemia is frequently diagnosed in veterans since it is more commonly seen in an elderly male population. The disease is characterized by a slow accumulation of mature B cells that are functionally incompetent and resist apoptosis. CLL has an indolent clinical course, but about 60% to 70% of patients require treatment. The disease also runs a variable course, and a number of genetic abnormalities and prognostic markers have been defined to subclassify CLL patients and prognosticate.2-4 This article reviews important CLL signaling pathways and novel therapeutic agents in this leukemia.
Signaling Pathways
B-Cell Receptor Signaling
The B-cell receptor (BCR) signaling is the major signaling pathway in CLL, because it defines clinical, biologic, and prognostic characteristics of the disease.5 The BCR is composed of a surface transmembrane immunoglobulin that binds the antigen with CD79 alpha and beta chains. The activation of BCR results in the formation of a signaling complex or signalosome, which includes Lyn, Syk, BTK, and ZAP-70, among other components that assemble with other adaptor proteins (Figure). This assembly of proteins occurs on the cytoplasmic tails of immunoglobulin chains on regions called immunoreceptor tyrosine-based motifs (ITAMs).
With the assembly of this signaling complex, BCR stimulates a number of downstream pathways, such as phosphatidylinositol 3-kinase (PI3K), protein kinase B (Akt), protein kinase C, nuclear factor-κB (NFκB), and extracellular signal-regulated kinases (ERKs) (Figure). Activation of these pathways results in cell proliferation, resistance to apoptosis, increased cell motility and migration. Recent studies have identified additional novel components of this signaling complex, including a guanine nucleotide exchange factor (GEF) RASGRF1. This GEF is activated by BCR signaling and, in turn, stimulates the ERK pathway by increasing the production of active GTP-bound Ras.6
The ability of BCR to activate a number of downstream signaling pathways makes it a highly relevant and investigated pathway in this leukemia. Inhibitors have been developed and/or identified against a number of signalosome components to block the BCR signaling.7 Syk and Lyn are Src kinases, and their phosphorylation is one of the initial events of BCR signaling. Syk is overexpressed in CLL specimens, and Syk inhibitors (R406 and P505-15, also known as PRT062607) have shown activity in CLL.8,9 Dasatinib is a Src inhibitor that also shows activity in CLL specimens and is being studied in combination with chemotherapy drugs in refractory CLL patients.10
BTK, a component of the BCR signalosome, is required for BCR function, and loss of its function is seen in X-linked agammaglobulinemia. PCI-32765 (ibrutinib) is an oral BTK inhibitor that irreversibly inactivates this kinase and has been approved for clinical use in CLL patients.11,12 Another signaling pathway activated by BCR is the PI3K, and a promising inhibitor (CAL-101) blocks its activity in CLL specimens.13 Investigative work has identified that the delta isoform of PI3K p110 is highly expressed in B cells and lymphocytes.14 This is a catalytic subunit of a class I PI3K with a role in BCR signaling. A selective inhibitor GS-1101 (CAL-101) is able to block PI3K signaling in CLL specimens and inhibits Akt phosphorylation and other downstream effectors along with induction of apoptosis.15 The clinical data with BTK and PI3K inhibitors will be discussed later in this review.
CLL and the Microenvironment
Interactions between CLL cells and the microenvironment allow CLL cells to thrive in certain niche environments.16,17 Interaction mainly occurs via bone marrow stromal cells and nurselike cells (NLCs), which evolve from monocytes (Figure). These interactions can be divided into 2 groups. First, CLL cell growth is supported by a number of chemokine receptor-ligand interactions. CXCR4 is the receptor for CXCL12 (SDF-1) that stimulates chemotaxis and tissue homing. Another chemokine is CXCL13, which acts via its receptor CXCR5 and is involved in chemotaxis and activation of other kinases. Second, NLCs also support CLL cells by expressing TNF family members BAFF and APRIL, which interact with their receptors and activate the NFκB pathway.
Leukemic cells also express VLA-4 integrins, which further their support adhesion to the stromal cells and predict for an aggressive phenotype. Specific inhibitors that block the stimulation by chemokines and cytokines are not yet available; however, one can envision that this class of inhibitors will decrease the chemoresistance of leukemic cells and will be used in conjunction with other chemotherapy agents. Interestingly, inhibitors that block BCR-mediated signaling (BTK and PI3K inhibitors) also inhibit signaling via the microenvironment and chemokines.
Wnt-β-catenin Pathway
Wnt signaling affects developmental pathways, and its aberrant activation has major oncogenic effects as well. This pathway is activated in CLL as these leukemic cells express high levels of Wnt and frizzled along with epigenetic downregulation of Wnt pathway antagonist genes, including secreted frizzled-related protein (SFRP) family members and WIF1 (Figure).18-20 The binding of Wnts to their cognate receptors results in inhibition of GSK3β phosphorylation and stabilization of β-catenin, which then translocates to the nucleus and interacts with lymphoid-enhancing (LEF) and T-cell transcription factors to activate transcription of Wnt-target genes. Lack of E-cadherin expression in CLL cells also results in an increase in translocation of β-catenin and upregulation of the Wnt pathway.20
Wnt-target genes include Myc, LEF, cyclinD1, COX-2, and MMP. Gene expression profiling from our laboratory and other groups have identified the overexpression of these wnt-target genes and support this pathway activation in CLL cells.20 This is a promising signalling pathway and an active area of research for developing inhibitors that will have a growth inhibitory effect on CLL leukemic cells. GSK3b inhibitors and other drugs that re-express epigenetically silenced Wnt antagonist genes have been shown to inhibit this pathway activity in CLL cells in vitro.
Notch Pathway Activation
High-throughput exome sequencing has identified recurring mutations in a number of genes, including NOTCH1.21 Analysis of additional CLL patients confirmed activating NOTCH1 mutations in 10% to 15% of CLL patients and were also associated with poor outcome.22 This pathway is activated by ligands such as Jagged and Delta-like, which interact with the Notch receptor, which is then cleaved by γ-secretases. The cleaved intracellular domain of the NOTCH1 receptor in combination with other factors activates transcription of target genes, including Myc and HES1 (Figure). Besides the mutations that generate a truncated protein or may stabilize the pathway, the Notch pathway is also constitutively active in CLL specimens.23 Notch stimulation increases activity of prosurvival pathways and genes such as NFκB that resist apoptotic signals. The pathway can be inhibited by γ-secretase inhibitors (GSIs), which reduce the levels of cleaved NOTCH1 protein and downregulated Notch target genes. This pathway is also able to modulate the microenvironment stimuli as the GSIs inhibit responses to chemokines such as CXCL12 and inhibit migration and invasion.24
Newer Theraputic Agents
Work on signaling mechanisms paid dividends in CLL with the recent development of 2 inhibitors. Ibrutinib (BTK inhibitor) and idelalisib (PI3K inhibitor) are being studied in clinical trials, and both drugs block the BCR and microenvironment signaling pathways, thereby inhibiting the growth of CLL cells.
BTK Inhibitor: Ibrutinib
The activity of BTK is critical for a number of CLL signaling pathways, and it is a component of the initial signaling complex or signalosome that is formed with BCR signaling. Studies have shown that inhibiting this kinase blocks a number of pathways, including ERK, NFκB, and others. The drug ibrutinib blocks this kinase by forming a covalent bond and inhibiting its enzyme activity. This orally bioavailable drug showed activity in phase 1 trials in different B-cell malignancies.25 In a phase 2 study, high-risk CLL patients were given 2 different doses of this inhibitor, and the overall response rate was 71% with an overall survival at 26 months of 83%.11 Responses were seen in all patients irrespective of clinical and genetic risk factors. Based on these findings, the drug was approved for clinical use in patients with relapsed or refractory disease. Recently, there are data on the use of this drug as frontline therapy in elderly patients, and the drug was well tolerated.26 There are additional ongoing trials to compare this drug with other agents, including chlorambucil (in chemotherapy-naïve patients) and ofatumumab (in relapsed or refractory patients).
PI3 Kinase p110 Delta Inhibitor: Idelalisib
The crucial finding for the development of this inhibitor was the over-expression of the delta isoform of PI3K p110 in B-cell malignancies.14 The drug CAL-101 selectively inhibits this constitutively active isoform and induces apoptosis in a number of B-cell malignancies.15,27 In the phase 1 trial, this inhibitor was evaluated in relapsed/refractory patients at multiple dose levels.28 There was inhibition of PI3K signaling with an overall response rate of 72%, and a partial response rate of 39% was observed in CLL patients. This was followed by a randomized, placebo-controlled phase 3 study in which patients with myelosuppression, decreased renal function, or other illnesses were treated with either rituximab alone or with rituximab and idelalisib.29
At the time of reporting, the median progression-free survival (PFS) was 5.5 months in the placebo arm and was not reached in the idelalisib arm. Overall response rates were higher in the idelalisib group (81% vs 13%) with similar toxicity profiles in the 2 groups. This drug is now being extensively studied in combination with bendamustine and other anti-CD20 antibodies in clinical trials.
A unique toxicity observed with both these inhibitors is the initial lymphocytosis. In the case of ibrutinib, this was seen in a majority of patients (77%) and at the same time there was a response in the nodal disease, implying a redistribution of leukemic cells from the tissues to the peripheral blood.30
A potential explanation is that these drugs inhibit signaling via chemokines and other components of the microenvironment and by inhibiting the homing signals, allows leukemic cells to move out of their niche areas. This was analyzed in a recent study that compared clinical and biochemical parameters of patients who had a complete or partial response with ibrutinib compared with a “partial response except for lymphocytosis.”30 Patients with “partial response except for lymphocytosis” were found to have favorable prognostic factors, and the persisting leukemic cells were not clonally different from the original cells. The progression free survival of patients with “partial response except for lymphocytosis” was also similar to the subgroup with no prolonged lymphocytosis.
Discussion
Several therapeutic agents with novel mechanisms of action are effective in killing the CLL leukemic cells, and a number of targeted agents are currently in the pipeline. The next challenge for treating CLL will be the proper integration of these novel targeted agents with the traditional chemotherapy and chemoimmunotherapy approaches. Let us consider CLL patients in different clinical settings. First, a patient aged 60 years who is otherwise healthy will be treated with possibly all the available chemotherapy and chemoimmunotherapy options, as well as the newer targeted agents. In this clinical setting sequencing of therapy is not a major concern. On the other hand, a patient aged 70 years who is already refractory to multiple lines of therapy is a good candidate for these newer drugs.
The more controversial use of these targeted agents will be in an older patient with some comorbidities and newly diagnosed CLL. In this clinical setting, should one go with traditional chemotherapy/chemoimmunotherapy approaches or consider newer targeted agents? These issues are now being addressed in clinical trials, and with acceptable toxicity profiles these newer drugs will move to the frontline setting.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect an endorsement by or opinion of Federal Practitioner, Frontline Medical Communications, the U.S. Air Force, the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drug combinations–including indications, contraindications, warnings, and adverse effects–before administering pharmacologic therapy to patients.
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9-29.
2. Döhner H, Stilgenbauer S, Döhner K, Bentz M, Lichter P. Chromosome aberrations in B-cell chronic lymphocytic leukemia: reassessment based on molecular cytogenetic analysis. J Mol Med. 1999;77(2):266-281.
3. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848-1854.
4. Chen L, Widhopf G, Huynh L, et al. Expression of ZAP-70 is associated with increased B-cell receptor signaling in chronic lymphocytic leukemia. Blood. 2002;100(13):4609-4614.
5. Wickremasinghe RG, Prentice AG, Steele AJ. Aberrantly activated anti-apoptotic signalling mechanisms in chronic lymphocytic leukaemia cells: Clues to the identification of novel therapeutic targets. Br J Haematol. 2011;153(5):545-556.
6. Liao W, Jordaan G, Coriaty N, Sharma S. Amplification of B cell receptor-Erk signaling by Rasgrf-1 overexpression in chronic lymphocytic leukemia [published online ahead of print April 2, 2014]. Leuk Lymphoma. doi: 10.3109/10428194.2014898759.
7. Burger JA. Inhibiting B-cell receptor signaling pathways in chronic lymphocytic leukemia. Curr Hematol Malig Rep. 2012;7(1):26-33.
8. Buchner M, Fuchs S, Prinz G, et al. Spleen tyrosine kinase is overexpressed and represents a potential therapeutic target in chronic lymphocytic leukemia. Cancer Res. 2009;69(13):5424-5432.
9. Spurgeon SE, Coffey G, Fletcher LB, et al. The selective SYK inhibitor P505-15 (PRT062607) inhibits B cell signaling and function in vitro and in vivo and augments the activity of fludarabine in chronic lymphocytic leukemia. J Pharmacol Exp Ther. 2013;344(2):378-387.
10. Veldurthy A, Patz M, Hagist S, et al. The kinase inhibitor dasatinib induces apoptosis in chronic lymphocytic leukemia cells in vitro with preference for a subgroup of patients with unmutated IgVH genes. Blood. 2008;112(4):1443-1452.
11. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia [published correction appears in N Engl J Med. 2014;370(8):786]. N Engl J Med. 2013;369(1):32-42.
12. Cheng S, Ma J, Guo A, et al. BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia. 2014;28(3):649-657.
13. Lannutti BJ, Meadows SA, Herman SE, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591-594.
14. Chantry D, Vojtek A, Kashishian A, et al. p110delta, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J Biol Chem. 1997;272(31):19236-19241.
15. Hoellenriegel J, Meadows SA, Sivina M, et al. The phosphoinositide 3’-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011;118(13):3603-3612.
16. Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F. The microenvironment in mature B-cell malignancies: A target for new treatment strategies. Blood. 2009;114(16):3367-3375.
17. ten Hacken E, Burger JA. Molecular pathways: targeting the microenvironment in chronic lymphocytic leukemia—focus on the B-cell receptor. Clin Cancer Res. 2014;20(3):548-556.
18. Gandhirajan RK, Poll-Wolbeck SJ, Gehrke I, Kreuzer KA. Wnt/b-catenin/LEF-1 signaling in chronic lymphocytic leukemia (CLL): a target for current and potential therapeutic options. Curr Cancer Drug Targets. 2010;10(7):716-727.
19. Gutierrez A, Jr, Tschumper RC, Wu X, et al. LEF-1 is a prosurvival factor in chronic lymphocytic leukemia and is expressed in the preleukemic state of monoclonal B-cell lymphocytosis. Blood. 2010;116(16):2975-2983.
20. Jordaan G, Liao W, Sharma S. E-cadherin gene re-expression in chronic lymphocytic leukemia cells by HDAC inhibitors. BMC Cancer. 2013;13:88.
21. Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101-105.
22. Fabbri G, Rasi S, Rossi D, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208(7):1389-1401.
23. Rosati E, Sabatini R, Rampino G, et al. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood. 2009;113(4):856-865.
24. López-Guerra M, Xargay-Torrent S, Rosich L, et al. The g-secretase inhibitor PF-03084014 combined with fludarabine antagonizes migration, invasion and angiogenesis in NOTCH1-mutated CLL cells [published online ahead of print April 30, 2014]. Leukemia. doi: 10.1038/leu.2014.143.
25. Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31(1):88-94.
26. O’Brien S, Furman RR, Coutre SE, et al. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: An open-label, multicentre, phase 1b/2 trial. Lancet Oncol. 2014;15(1):48-58.
27. Brown JR, Byrd JC, Coutre SE, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110∂, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;123(22):3390-3397.
28. Brown JR, Furman RR, Flinn I, et al. Final results of a phase I study of idelalisib (GS-1101) a selective inhibitor of PI3K∂, in patients with relapsed or refractory CLL. J Clin Oncol. 2013;31:Absract 7003.
29. Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997-1007.
30. Woyach JA, Smucker K, Smith LL, et al. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate a suboptimal response to therapy. Blood. 2013;123(12):1810-1817.
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9-29.
2. Döhner H, Stilgenbauer S, Döhner K, Bentz M, Lichter P. Chromosome aberrations in B-cell chronic lymphocytic leukemia: reassessment based on molecular cytogenetic analysis. J Mol Med. 1999;77(2):266-281.
3. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848-1854.
4. Chen L, Widhopf G, Huynh L, et al. Expression of ZAP-70 is associated with increased B-cell receptor signaling in chronic lymphocytic leukemia. Blood. 2002;100(13):4609-4614.
5. Wickremasinghe RG, Prentice AG, Steele AJ. Aberrantly activated anti-apoptotic signalling mechanisms in chronic lymphocytic leukaemia cells: Clues to the identification of novel therapeutic targets. Br J Haematol. 2011;153(5):545-556.
6. Liao W, Jordaan G, Coriaty N, Sharma S. Amplification of B cell receptor-Erk signaling by Rasgrf-1 overexpression in chronic lymphocytic leukemia [published online ahead of print April 2, 2014]. Leuk Lymphoma. doi: 10.3109/10428194.2014898759.
7. Burger JA. Inhibiting B-cell receptor signaling pathways in chronic lymphocytic leukemia. Curr Hematol Malig Rep. 2012;7(1):26-33.
8. Buchner M, Fuchs S, Prinz G, et al. Spleen tyrosine kinase is overexpressed and represents a potential therapeutic target in chronic lymphocytic leukemia. Cancer Res. 2009;69(13):5424-5432.
9. Spurgeon SE, Coffey G, Fletcher LB, et al. The selective SYK inhibitor P505-15 (PRT062607) inhibits B cell signaling and function in vitro and in vivo and augments the activity of fludarabine in chronic lymphocytic leukemia. J Pharmacol Exp Ther. 2013;344(2):378-387.
10. Veldurthy A, Patz M, Hagist S, et al. The kinase inhibitor dasatinib induces apoptosis in chronic lymphocytic leukemia cells in vitro with preference for a subgroup of patients with unmutated IgVH genes. Blood. 2008;112(4):1443-1452.
11. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia [published correction appears in N Engl J Med. 2014;370(8):786]. N Engl J Med. 2013;369(1):32-42.
12. Cheng S, Ma J, Guo A, et al. BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia. 2014;28(3):649-657.
13. Lannutti BJ, Meadows SA, Herman SE, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591-594.
14. Chantry D, Vojtek A, Kashishian A, et al. p110delta, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J Biol Chem. 1997;272(31):19236-19241.
15. Hoellenriegel J, Meadows SA, Sivina M, et al. The phosphoinositide 3’-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011;118(13):3603-3612.
16. Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F. The microenvironment in mature B-cell malignancies: A target for new treatment strategies. Blood. 2009;114(16):3367-3375.
17. ten Hacken E, Burger JA. Molecular pathways: targeting the microenvironment in chronic lymphocytic leukemia—focus on the B-cell receptor. Clin Cancer Res. 2014;20(3):548-556.
18. Gandhirajan RK, Poll-Wolbeck SJ, Gehrke I, Kreuzer KA. Wnt/b-catenin/LEF-1 signaling in chronic lymphocytic leukemia (CLL): a target for current and potential therapeutic options. Curr Cancer Drug Targets. 2010;10(7):716-727.
19. Gutierrez A, Jr, Tschumper RC, Wu X, et al. LEF-1 is a prosurvival factor in chronic lymphocytic leukemia and is expressed in the preleukemic state of monoclonal B-cell lymphocytosis. Blood. 2010;116(16):2975-2983.
20. Jordaan G, Liao W, Sharma S. E-cadherin gene re-expression in chronic lymphocytic leukemia cells by HDAC inhibitors. BMC Cancer. 2013;13:88.
21. Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101-105.
22. Fabbri G, Rasi S, Rossi D, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208(7):1389-1401.
23. Rosati E, Sabatini R, Rampino G, et al. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood. 2009;113(4):856-865.
24. López-Guerra M, Xargay-Torrent S, Rosich L, et al. The g-secretase inhibitor PF-03084014 combined with fludarabine antagonizes migration, invasion and angiogenesis in NOTCH1-mutated CLL cells [published online ahead of print April 30, 2014]. Leukemia. doi: 10.1038/leu.2014.143.
25. Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31(1):88-94.
26. O’Brien S, Furman RR, Coutre SE, et al. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: An open-label, multicentre, phase 1b/2 trial. Lancet Oncol. 2014;15(1):48-58.
27. Brown JR, Byrd JC, Coutre SE, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110∂, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;123(22):3390-3397.
28. Brown JR, Furman RR, Flinn I, et al. Final results of a phase I study of idelalisib (GS-1101) a selective inhibitor of PI3K∂, in patients with relapsed or refractory CLL. J Clin Oncol. 2013;31:Absract 7003.
29. Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997-1007.
30. Woyach JA, Smucker K, Smith LL, et al. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate a suboptimal response to therapy. Blood. 2013;123(12):1810-1817.