User login
To the Editor:
Palisaded neutrophilic granulomatous dermatitis (PNGD) is a rare disorder that often is associated with systemic disease. It has been shown to manifest in the presence of systemic lupus erythematosus; rheumatoid arthritis; Wegener granulomatosis; and other diseases, mainly autoimmune conditions. Interstitial granulomatous dermatitis (IGD) associated with arthritis was first described by Ackerman et al1 in 1993. In 1994, IGD was placed among the spectrum of PNGD by Chu et al.2 The disease entities included in the spectrum of PNGD of the immune complex disease are Churg-Strauss granuloma, cutaneous extravascular necrotizing granuloma, rheumatoid papules, superficial ulcerating rheumatoid necrobiosis, and IGD with arthritis.2 It has been suggested that IGD has a distinct clinical presentation with associated histopathology, while others suggest it still is part of the PNGD spectrum.2,3 We present 2 cases of granulomatous dermatitis and their findings related to IGD and PNGD.
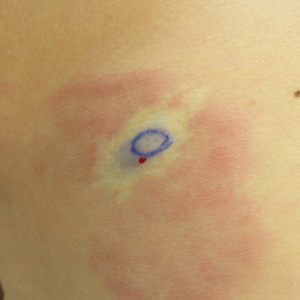
A 58-year-old woman presented with recurrent painful lesions on the trunk, arms, and legs of 2 years’ duration. The lesions spontaneously resolved without scarring or hyperpigmentation but would recur in different areas on the trunk. She was diagnosed with rheumatoid arthritis following a recent autoimmune workup. At presentation, physical examination revealed tender erythematous edematous plaques on the bilateral upper back (Figure 1) and erythematous nodules on the bilateral upper arms. The patient previously had an antinuclear antibody titer of 1:320 with a speckled pattern. A repeat antinuclear antibody titer taken 1 year later was negative. Her rheumatoid factor initially was positive and remained positive upon repeat testing. Punch biopsies were performed for histologic evaluation of the lesions and immunofluorescence. Biopsies examined with hematoxylin and eosin stain revealed perivascular and interstitial mixed (lymphocytic, neutrophilic, eosinophilic) bottom-heavy inflammation with nuclear dust and basophilic degeneration of collagen (Figure 2). Immunofluorescence studies were negative. The patient deferred treatment.

.")
A 74-year-old man presented with a rash on the flank and back with associated pruritus and occasional pain of 2 months’ duration. His primary care physician prescribed a course of cephalexin, but the rash did not improve. Review of systems was positive for intermittent swelling of the hands, feet, and lips, and negative for arthritis. His medical history included 2 episodes of rheumatic fever, one complicated by pneumonia. His medications included finasteride, simvastatin, bisoprolol-hydrochlorothiazide, aspirin, tiotropium, vitamin D, and fish oil. At presentation, physical examination revealed tender violaceous plaques with induration and central clearing distributed on the left side of the back, left side of the flank, and left axilla. The lesion on the axilla measured 30.0×3.5 cm and the lesions on the left side of the back measured 30.0×9.0 cm. The rims of the lesions were elevated and consistent with the rope sign (Figure 3). A punch biopsy of the lesion on the left axilla showed perivascular and interstitial infiltrate of lymphocytes, neutrophils, histiocytes, and eosinophils. There was no evidence of fibrin deposition in the blood vessels. Small areas of necrobiotic collagen surrounded by multinucleated giant cells and lymphocytes were noted (Figure 4). The rash improved spontaneously at the time of suture removal. No treatment was initiated.

.")
Granulomatous dermatitis in the presence of an autoimmune disorder can present as IGD or PNGD. Both forms of granulomatous dermatitis are rare conditions and considered to be part of the same clinicopathological spectrum. These conditions can be difficult to distinguish clinically but are histologically unique.
Interstitial granulomatous dermatitis and PNGD can have a variable clinical expression. Palisaded neutrophilic granulomatous dermatitis generally presents as flesh-colored to erythematous papules or plaques, most commonly located on the upper arms. The lesions may have a central umbilication with perforation and ulceration.4 Interstitial granulomatous dermatitis most commonly presents as erythematous plaques and papules. The lesions are symmetric and asymptomatic. They most commonly appear on the trunk, axillae, buttocks, thighs, and groin. Subcutaneous linear cords (the rope sign) is a characteristic associated with IGD.3,5 However, the rope sign also has been reported in a patient with PNGD with systemic lupus,6 which further demonstrates the overlapping spectrum of clinical expression seen in these 2 forms of granulomatous dermatitis. Therefore, a diagnosis cannot be made by clinical expression alone; histologic findings are needed for confirmation.
When differentiating IGD and PNGD histologically, it is important to keep in mind that these features exist on a spectrum and depend on the age of the lesion. Deposition of the immune complex around the dermal blood vessel initiates the pathogenesis. Early lesions of PNGD show a neutrophilic infiltrate, focal leukocytoclastic vasculitis, and dense nuclear dust. Developed lesions show zones of basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris.2 The histologic pattern of IGD features smaller areas of palisading histiocytes surrounding foci of degenerated collagen. Neutrophils and eosinophils are seen among the degenerated collagen. There is no evidence of vasculitis and dermal mucin usually is absent.7
Palisaded neutrophilic granulomatous dermatitis has been reported to improve with systemic steroids and dapsone.8 Th
Some authors have disputed the spectrum that Chu et al2 had determined in their study and proposed IGD is a separate entity from the PNGD spectrum. Verneuil et al9 stated that the clinical presentations in Chu et al’s2 study (symmetric papules of the extremities) had not been reported in a patient with IGD. However, in a study of IGD by Peroni et al,3 7 of 12 patients presented with symmetrical papules of the extremities. We believe that the spectrum proposed by Chu et al2 still holds true.
These 2 reports demonstrate the diverse presentation of IGD and PNGD. It is important for dermatologists to keep in mind the PNGD spectrum when a patient presents with granulomatous dermatitis in the presence of an autoimmune disorder.
- Ackerman AB, Guo Y, Vitale P. Clues to diagnosis in dermatopathology. Am Society Clin Pathol. 1993;3:309-312.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Hantash BM, Chiang D, Kohler S, et al. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58:661-664.
- Garcia-Rabasco A, Esteve-Martinez A, Zaragoza-Ninet V, et al. Interstitial granulomatous dermatitis in a patient with lupus erythematosus. Am J Dermatopathol. 2011;33:871-872.
- Gulati A, Paige D, Yaqoob M, et al. Palisaded neutrophilic granulomatous dermatitis associated with systemic lupus erythematosus presenting with the burning rope sign. J Am Acad Dermatol. 2009;61:711-714.
- Tomasini C, Pippione M. Interstitial granulomatous dermatitis with plaques. J Am Acad Dermatol. 2002;46:892-899.
- Fett N, Kovarik C, Bennett D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J Am Acad Dermatol. 2011;65:E92-E93.
- Verneuil L, Dompmartin A, Comoz F, et al. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: a disorder associated with autoantibodies. J Am Acad Dermatol. 2001;45:286-291.
To the Editor:
Palisaded neutrophilic granulomatous dermatitis (PNGD) is a rare disorder that often is associated with systemic disease. It has been shown to manifest in the presence of systemic lupus erythematosus; rheumatoid arthritis; Wegener granulomatosis; and other diseases, mainly autoimmune conditions. Interstitial granulomatous dermatitis (IGD) associated with arthritis was first described by Ackerman et al1 in 1993. In 1994, IGD was placed among the spectrum of PNGD by Chu et al.2 The disease entities included in the spectrum of PNGD of the immune complex disease are Churg-Strauss granuloma, cutaneous extravascular necrotizing granuloma, rheumatoid papules, superficial ulcerating rheumatoid necrobiosis, and IGD with arthritis.2 It has been suggested that IGD has a distinct clinical presentation with associated histopathology, while others suggest it still is part of the PNGD spectrum.2,3 We present 2 cases of granulomatous dermatitis and their findings related to IGD and PNGD.
A 58-year-old woman presented with recurrent painful lesions on the trunk, arms, and legs of 2 years’ duration. The lesions spontaneously resolved without scarring or hyperpigmentation but would recur in different areas on the trunk. She was diagnosed with rheumatoid arthritis following a recent autoimmune workup. At presentation, physical examination revealed tender erythematous edematous plaques on the bilateral upper back (Figure 1) and erythematous nodules on the bilateral upper arms. The patient previously had an antinuclear antibody titer of 1:320 with a speckled pattern. A repeat antinuclear antibody titer taken 1 year later was negative. Her rheumatoid factor initially was positive and remained positive upon repeat testing. Punch biopsies were performed for histologic evaluation of the lesions and immunofluorescence. Biopsies examined with hematoxylin and eosin stain revealed perivascular and interstitial mixed (lymphocytic, neutrophilic, eosinophilic) bottom-heavy inflammation with nuclear dust and basophilic degeneration of collagen (Figure 2). Immunofluorescence studies were negative. The patient deferred treatment.
A 74-year-old man presented with a rash on the flank and back with associated pruritus and occasional pain of 2 months’ duration. His primary care physician prescribed a course of cephalexin, but the rash did not improve. Review of systems was positive for intermittent swelling of the hands, feet, and lips, and negative for arthritis. His medical history included 2 episodes of rheumatic fever, one complicated by pneumonia. His medications included finasteride, simvastatin, bisoprolol-hydrochlorothiazide, aspirin, tiotropium, vitamin D, and fish oil. At presentation, physical examination revealed tender violaceous plaques with induration and central clearing distributed on the left side of the back, left side of the flank, and left axilla. The lesion on the axilla measured 30.0×3.5 cm and the lesions on the left side of the back measured 30.0×9.0 cm. The rims of the lesions were elevated and consistent with the rope sign (Figure 3). A punch biopsy of the lesion on the left axilla showed perivascular and interstitial infiltrate of lymphocytes, neutrophils, histiocytes, and eosinophils. There was no evidence of fibrin deposition in the blood vessels. Small areas of necrobiotic collagen surrounded by multinucleated giant cells and lymphocytes were noted (Figure 4). The rash improved spontaneously at the time of suture removal. No treatment was initiated.
Granulomatous dermatitis in the presence of an autoimmune disorder can present as IGD or PNGD. Both forms of granulomatous dermatitis are rare conditions and considered to be part of the same clinicopathological spectrum. These conditions can be difficult to distinguish clinically but are histologically unique.
Interstitial granulomatous dermatitis and PNGD can have a variable clinical expression. Palisaded neutrophilic granulomatous dermatitis generally presents as flesh-colored to erythematous papules or plaques, most commonly located on the upper arms. The lesions may have a central umbilication with perforation and ulceration.4 Interstitial granulomatous dermatitis most commonly presents as erythematous plaques and papules. The lesions are symmetric and asymptomatic. They most commonly appear on the trunk, axillae, buttocks, thighs, and groin. Subcutaneous linear cords (the rope sign) is a characteristic associated with IGD.3,5 However, the rope sign also has been reported in a patient with PNGD with systemic lupus,6 which further demonstrates the overlapping spectrum of clinical expression seen in these 2 forms of granulomatous dermatitis. Therefore, a diagnosis cannot be made by clinical expression alone; histologic findings are needed for confirmation.
When differentiating IGD and PNGD histologically, it is important to keep in mind that these features exist on a spectrum and depend on the age of the lesion. Deposition of the immune complex around the dermal blood vessel initiates the pathogenesis. Early lesions of PNGD show a neutrophilic infiltrate, focal leukocytoclastic vasculitis, and dense nuclear dust. Developed lesions show zones of basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris.2 The histologic pattern of IGD features smaller areas of palisading histiocytes surrounding foci of degenerated collagen. Neutrophils and eosinophils are seen among the degenerated collagen. There is no evidence of vasculitis and dermal mucin usually is absent.7
Palisaded neutrophilic granulomatous dermatitis has been reported to improve with systemic steroids and dapsone.8 Th
Some authors have disputed the spectrum that Chu et al2 had determined in their study and proposed IGD is a separate entity from the PNGD spectrum. Verneuil et al9 stated that the clinical presentations in Chu et al’s2 study (symmetric papules of the extremities) had not been reported in a patient with IGD. However, in a study of IGD by Peroni et al,3 7 of 12 patients presented with symmetrical papules of the extremities. We believe that the spectrum proposed by Chu et al2 still holds true.
These 2 reports demonstrate the diverse presentation of IGD and PNGD. It is important for dermatologists to keep in mind the PNGD spectrum when a patient presents with granulomatous dermatitis in the presence of an autoimmune disorder.
To the Editor:
Palisaded neutrophilic granulomatous dermatitis (PNGD) is a rare disorder that often is associated with systemic disease. It has been shown to manifest in the presence of systemic lupus erythematosus; rheumatoid arthritis; Wegener granulomatosis; and other diseases, mainly autoimmune conditions. Interstitial granulomatous dermatitis (IGD) associated with arthritis was first described by Ackerman et al1 in 1993. In 1994, IGD was placed among the spectrum of PNGD by Chu et al.2 The disease entities included in the spectrum of PNGD of the immune complex disease are Churg-Strauss granuloma, cutaneous extravascular necrotizing granuloma, rheumatoid papules, superficial ulcerating rheumatoid necrobiosis, and IGD with arthritis.2 It has been suggested that IGD has a distinct clinical presentation with associated histopathology, while others suggest it still is part of the PNGD spectrum.2,3 We present 2 cases of granulomatous dermatitis and their findings related to IGD and PNGD.
A 58-year-old woman presented with recurrent painful lesions on the trunk, arms, and legs of 2 years’ duration. The lesions spontaneously resolved without scarring or hyperpigmentation but would recur in different areas on the trunk. She was diagnosed with rheumatoid arthritis following a recent autoimmune workup. At presentation, physical examination revealed tender erythematous edematous plaques on the bilateral upper back (Figure 1) and erythematous nodules on the bilateral upper arms. The patient previously had an antinuclear antibody titer of 1:320 with a speckled pattern. A repeat antinuclear antibody titer taken 1 year later was negative. Her rheumatoid factor initially was positive and remained positive upon repeat testing. Punch biopsies were performed for histologic evaluation of the lesions and immunofluorescence. Biopsies examined with hematoxylin and eosin stain revealed perivascular and interstitial mixed (lymphocytic, neutrophilic, eosinophilic) bottom-heavy inflammation with nuclear dust and basophilic degeneration of collagen (Figure 2). Immunofluorescence studies were negative. The patient deferred treatment.
A 74-year-old man presented with a rash on the flank and back with associated pruritus and occasional pain of 2 months’ duration. His primary care physician prescribed a course of cephalexin, but the rash did not improve. Review of systems was positive for intermittent swelling of the hands, feet, and lips, and negative for arthritis. His medical history included 2 episodes of rheumatic fever, one complicated by pneumonia. His medications included finasteride, simvastatin, bisoprolol-hydrochlorothiazide, aspirin, tiotropium, vitamin D, and fish oil. At presentation, physical examination revealed tender violaceous plaques with induration and central clearing distributed on the left side of the back, left side of the flank, and left axilla. The lesion on the axilla measured 30.0×3.5 cm and the lesions on the left side of the back measured 30.0×9.0 cm. The rims of the lesions were elevated and consistent with the rope sign (Figure 3). A punch biopsy of the lesion on the left axilla showed perivascular and interstitial infiltrate of lymphocytes, neutrophils, histiocytes, and eosinophils. There was no evidence of fibrin deposition in the blood vessels. Small areas of necrobiotic collagen surrounded by multinucleated giant cells and lymphocytes were noted (Figure 4). The rash improved spontaneously at the time of suture removal. No treatment was initiated.
Granulomatous dermatitis in the presence of an autoimmune disorder can present as IGD or PNGD. Both forms of granulomatous dermatitis are rare conditions and considered to be part of the same clinicopathological spectrum. These conditions can be difficult to distinguish clinically but are histologically unique.
Interstitial granulomatous dermatitis and PNGD can have a variable clinical expression. Palisaded neutrophilic granulomatous dermatitis generally presents as flesh-colored to erythematous papules or plaques, most commonly located on the upper arms. The lesions may have a central umbilication with perforation and ulceration.4 Interstitial granulomatous dermatitis most commonly presents as erythematous plaques and papules. The lesions are symmetric and asymptomatic. They most commonly appear on the trunk, axillae, buttocks, thighs, and groin. Subcutaneous linear cords (the rope sign) is a characteristic associated with IGD.3,5 However, the rope sign also has been reported in a patient with PNGD with systemic lupus,6 which further demonstrates the overlapping spectrum of clinical expression seen in these 2 forms of granulomatous dermatitis. Therefore, a diagnosis cannot be made by clinical expression alone; histologic findings are needed for confirmation.
When differentiating IGD and PNGD histologically, it is important to keep in mind that these features exist on a spectrum and depend on the age of the lesion. Deposition of the immune complex around the dermal blood vessel initiates the pathogenesis. Early lesions of PNGD show a neutrophilic infiltrate, focal leukocytoclastic vasculitis, and dense nuclear dust. Developed lesions show zones of basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris.2 The histologic pattern of IGD features smaller areas of palisading histiocytes surrounding foci of degenerated collagen. Neutrophils and eosinophils are seen among the degenerated collagen. There is no evidence of vasculitis and dermal mucin usually is absent.7
Palisaded neutrophilic granulomatous dermatitis has been reported to improve with systemic steroids and dapsone.8 Th
Some authors have disputed the spectrum that Chu et al2 had determined in their study and proposed IGD is a separate entity from the PNGD spectrum. Verneuil et al9 stated that the clinical presentations in Chu et al’s2 study (symmetric papules of the extremities) had not been reported in a patient with IGD. However, in a study of IGD by Peroni et al,3 7 of 12 patients presented with symmetrical papules of the extremities. We believe that the spectrum proposed by Chu et al2 still holds true.
These 2 reports demonstrate the diverse presentation of IGD and PNGD. It is important for dermatologists to keep in mind the PNGD spectrum when a patient presents with granulomatous dermatitis in the presence of an autoimmune disorder.
- Ackerman AB, Guo Y, Vitale P. Clues to diagnosis in dermatopathology. Am Society Clin Pathol. 1993;3:309-312.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Hantash BM, Chiang D, Kohler S, et al. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58:661-664.
- Garcia-Rabasco A, Esteve-Martinez A, Zaragoza-Ninet V, et al. Interstitial granulomatous dermatitis in a patient with lupus erythematosus. Am J Dermatopathol. 2011;33:871-872.
- Gulati A, Paige D, Yaqoob M, et al. Palisaded neutrophilic granulomatous dermatitis associated with systemic lupus erythematosus presenting with the burning rope sign. J Am Acad Dermatol. 2009;61:711-714.
- Tomasini C, Pippione M. Interstitial granulomatous dermatitis with plaques. J Am Acad Dermatol. 2002;46:892-899.
- Fett N, Kovarik C, Bennett D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J Am Acad Dermatol. 2011;65:E92-E93.
- Verneuil L, Dompmartin A, Comoz F, et al. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: a disorder associated with autoantibodies. J Am Acad Dermatol. 2001;45:286-291.
- Ackerman AB, Guo Y, Vitale P. Clues to diagnosis in dermatopathology. Am Society Clin Pathol. 1993;3:309-312.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Hantash BM, Chiang D, Kohler S, et al. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58:661-664.
- Garcia-Rabasco A, Esteve-Martinez A, Zaragoza-Ninet V, et al. Interstitial granulomatous dermatitis in a patient with lupus erythematosus. Am J Dermatopathol. 2011;33:871-872.
- Gulati A, Paige D, Yaqoob M, et al. Palisaded neutrophilic granulomatous dermatitis associated with systemic lupus erythematosus presenting with the burning rope sign. J Am Acad Dermatol. 2009;61:711-714.
- Tomasini C, Pippione M. Interstitial granulomatous dermatitis with plaques. J Am Acad Dermatol. 2002;46:892-899.
- Fett N, Kovarik C, Bennett D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J Am Acad Dermatol. 2011;65:E92-E93.
- Verneuil L, Dompmartin A, Comoz F, et al. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: a disorder associated with autoantibodies. J Am Acad Dermatol. 2001;45:286-291.
Practice Points
- The clinical features of interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis exist on a spectrum, and these is considerable overlap between the features of these 2 clinicopathologic entities.
- Interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis may respond to systemic steroids or treatment of the underlying systemic disease. Some cases spontaneously resolve.