User login
Cannabis and schizophrenia: A complex relationship
Approximately 1 in 200 individuals will be diagnosed with schizophrenia in their lifetime.1 DSM-5 criteria for the diagnosis of schizophrenia require the presence of ≥2 of 5 symptoms: delusions, hallucinations, disordered speech, grossly disorganized (or catatonic) behavior, and negative symptoms such as flat affect or avolition.2 Multiple studies have found increased rates of cannabis use among patients with schizophrenia. Because cognitive deficits are the chief predictor of clinical outcomes and quality of life in individuals with schizophrenia, the cognitive effects of cannabis use among these patients are of clinical significance.3 As legislation increasingly allows for the sale, possession, and consumption of cannabis, it is crucial to provide clinicians with evidence-based recommendations for treating patients who regularly use cannabis (approximately 8% of the adult population3). In this article, we analyze several peer-reviewed studies to investigate the impact of cannabis use on the onset and development of schizophrenia.
A look at substance-induced psychosis
Schizophrenia is associated with several structural brain changes, and some of these changes may be influenced by cannabis use (Box4). The biochemical etiology of schizophrenia is poorly understood but thought to involve dopamine, glutamate, serotonin, and gamma-aminobutyric acid. Certain positive symptoms, such as hallucinations, are uniquely human and difficult to study in animal models.5 Psychoactive substance use, especially cannabis, is frequently comorbid with schizophrenia. Additionally, certain individuals may be more predisposed to substance-induced psychosis than others based on genetic variation and underlying brain structure changes.4 Substance-induced psychosis is a psychotic state following the ingestion of a psychoactive substance or drug withdrawal lasting ≥48 hours.6 The psychoactive effects of cannabis have been associated with an exacerbation of existing schizophrenia symptoms.7 In 1998, Hall7 proposed 2 hypotheses to explain the relationship between cannabis and psychosis. The first was that heavy consumption of cannabis triggers a specific type of cannabis psychosis.7 The second was that cannabis use exacerbates existing schizophrenia, making the symptoms worse.7 Hall7 concluded that there was a complicated interaction among an individual’s vulnerability to their stressors, environment, and genetics.
Box
Schizophrenia is associated with several structural changes in the brain, including lateral ventriculomegaly, reduced prefrontal cortex volume, and generalized atrophy. These changes may precede illness and act as a risk marker.4 A multivariate regression analysis that compared patients with schizophrenia who were cannabis users vs patients with schizophrenia who were nonusers found that those with high-level cannabis use had relatively higher left and right lateral ventricle volume (r = 0.208, P = .13, and r = 0.226, P = .007, respectively) as well as increased third ventricle volume (r = 0.271, P = .001).4 These changes were dose-dependent and may lead to worse disease outcomes.4
Cannabis, COMT, and homocysteine
Great advances have been made in our ability to examine the association between genetics and metabolism. One example of this is the interaction between the catechol-O-methyltransferase (COMT) gene and the active component of cannabis, delta-9-tetrahydrocannabinol (THC). COMT codes for an enzyme that degrades cortical dopamine. The Val158Met polymorphism of this gene increases COMT activity, leading to increased dopamine catabolism, and thus decreased levels of extracellular dopamine, which induces an increase in mesolimbic dopaminergic activity, thereby increasing susceptibility to psychosis.3
In a study that genotyped 135 patients with schizophrenia, the Val158Met polymorphism was present in 29.63% of participants.3 Because THC can induce episodes of psychosis, individuals with this polymorphism may be at a higher risk of developing schizophrenia. Compared to Met carrier control participants with similar histories of cannabis consumption, those with the Val158Met polymorphism demonstrated markedly worse performance on tests of verbal fluency and processing speed.3 This is clinically significant because cognitive impairments are a major prognostic factor in schizophrenia, and identifying patients with this polymorphism could help personalize interventions for those who consume cannabis and are at risk of developing schizophrenia.
A study that evaluated 56 patients with first-episode schizophrenia found that having a history of cannabis abuse was associated with significantly higher levels of homocysteine as well as lower levels of high-density lipoprotein and vitamin B12.8 Homocysteine is an agonist at the glutamate binding site and a partial antagonist at the glycine co-agonist site in the N-methyl-
The C677T polymorphism in MTHFR may predict the risk of developing metabolic syndrome in patients taking second-generation antipsychotics.8 Elevations in homocysteine by as little as 5 μmol/L may increase schizophrenia risk by 70% compared to controls, possibly due to homocysteine initiating neuronal apoptosis, catalyzing dysfunction of the mitochondria, or increasing oxidative stress.8 There is a positive correlation between homocysteine levels and severity of negative symptoms (P = .006) and general psychopathology (P = .008) of schizophrenia when analyzed using the Positive and Negative Syndrome Scale.8 Negative symptoms such as blunted affect, apathy, anhedonia, and loss of motivation significantly impact the social and economic outcomes of patients diagnosed with schizophrenia.
Research paints a mixed picture
A Danish study analyzed the rates of conversion to schizophrenia or bipolar disorder (BD) among 6,788 individuals who received a diagnosis of substance-induced psychosis from 1994 to 2014.6 Ten comparison participants were selected for each case participant, matched on sex and year/month of birth. Participants were followed until the first occurrence of schizophrenia or BD, death, or emigration from Denmark. Substances implicated in the initial psychotic episode included cannabis, alcohol, opioids, sedatives, cocaine, amphetamines, hallucinogens, and combinations of substances.
Continue to: The overall conversion rate...
The overall conversion rate over 20 years was 32.2% (95% CI, 29.7 to 34.9), with 26.0% developing schizophrenia vs 8.4% developing BD.6 Of the substances involved, cannabis was the most common, implicated in 41.2% (95% CI, 36.6 to 46.2) of cases.6 One-half of male patients converted within 2.0 years and one-half of female patients converted within 4.4 years after a cannabis-induced psychosis.6
This study had several limitations. It could not account for any short-term psychotic symptoms experienced by the general population, especially after cannabis use. Such patients might not seek treatment. Thus, the results might not be generalizable to the general population. The study did not evaluate if conversion rates differed based on continued substance use following the psychosis episode, or the amount of each substance taken prior to the episode. Dose-dependence was not well elucidated, and this study only looked at patients from Denmark and did not account for socioeconomic status.6
Another Danish study looked at the influences of gender and cannabis use in the early course of the disease in 133 patients with schizophrenia.9 These researchers found that male gender was a significant predictor of earlier onset of dysfunction socially and in the workplace, as well as a higher risk of developing negative symptoms. However, compared to gender, cannabis use was a stronger predictor of age at first psychotic episode. For cannabis users, the median age of onset of negative symptoms was 23.7, compared to 38.4 for nonusers (P < .001).9
Cannabis use is significantly elevated among individuals with psychosis, with a 12-month prevalence of 29.2% compared to 4.0% among the general population of the United States.10 In a study that assessed 229 patients with a schizophrenia spectrum disorder during their first hospitalization and 6 months, 2 years, 4 years, and 10 years later, Foti et al10 found that the lifetime rate of cannabis use was 66.2%. Survival analysis found cannabis use doubled the risk of the onset of psychosis compared to nonusers of the same age (hazard ratio [HR] = 1.97; 95% CI, 1.48 to 2.62, P < .001), even after adjusting for socioeconomic status, age, and gender (HR = 1.34; 95% CI, 1.01 to 1.77, P < .05).10 Additionally, Foti et al10 found significant positive correlations between psychotic symptoms and cannabis use in patients with schizophrenia over the course of 10 years. An increase in symptoms was associated with a higher likelihood of cannabis use, and a decrease in symptoms was correlated with a lower likelihood of use (adjusted odds ratio = 1.64; 95% CI, 1.12 to 2.43, P < .0125).10
Ortiz-Medina et al11 conducted a meta-analysis of 22 studies of 15 cohorts from healthy populations and 12 other cohort follow-up studies that evaluated the onset of psychotic symptoms in individuals who used cannabis. Most studies found associations between cannabis use and the onset of symptoms of schizophrenia, and most determined cannabis was also a major risk factor for other psychotic disorders. Analyses of dose-dependence indicated that repeated cannabis use increased the risk of developing psychotic symptoms. This risk is increased when an individual starts using cannabis before age 15.11 Age seemed to be a stronger predictor of onset and outcome than sex, with no significant differences between men and women. One study in this review found that approximately 8% to 13% cases of schizophrenia may have been solely due to cannabis.11 The most significant limitation to the studies analyzed in this review is that retrospective studies utilize self-reported questionnaires.
Continue to: Other researchers have found...
Other researchers have found it would take a relatively high number of individuals to stop using cannabis to prevent 1 case of schizophrenia. In a study of data from England and Wales, Hickman et al12 evaluated the best available estimates of the incidence of schizophrenia, rates of heavy and light cannabis use, and risk that cannabis causes schizophrenia to determine the number needed to prevent (NNP) 1 case of schizophrenia. They estimated that it would require approximately 2,800 men age 20 to 24 (90% CI, 2,018 to 4,530) and 4,700 men age 35 to 39 (90% CI, 3,114 to 8,416) who heavily used cannabis to stop their consumption to prevent 1 case of schizophrenia.12 For women with heavy cannabis use, the mean NNP was 5,470 for women age 25 to 29 (90% CI, 3,640 to 9,839) and 10,870 for women age 35 to 39 (90% CI, 6,786 to 22,732).12 For light cannabis users, the NNP was 4 to 5 times higher than the NNP for heavy cannabis users. This suggests that clinical interventions aimed at preventing dependence on cannabis would be more effective than interventions aimed at eliminating cannabis use.
Medical cannabis and increased potency
In recent years, the use of medical cannabis, which is used to address adverse effects of chemotherapy as well as neuropathic pain, Parkinson’s disease, and epilepsy, has been increasing.13 However, there is a lack of well-conducted randomized clinical trials evaluating medical cannabis’ efficacy and safety. As medical cannabis continues to gain public acceptance and more states permit its legal use, patients and physicians should be fully informed of the known adverse effects, including impaired attention, learning, and motivation.13
Several studies have drawn attention to the dose-dependence of many of cannabis’ effects. Since at least the 1960s, the concentration of THC in cannabis has increased substantially, thus increasing its potency. Based on 66,747 samples across 8 studies, 1 meta-analysis estimated that THC concentrations in herbal cannabis increased by 0.29% (P < .001) each year between 1970 and 2017.14 Similarly, THC concentrations in cannabis resins were found to have increased by 0.57% (P = .017) each year between 1975 and 2017.14 Cannabis products with high concentrations of THC carry an increased risk of addiction and mental health disorders.14
Identifying those at highest risk
Despite ongoing research, scientific consensus on the relationship of cannabis to schizophrenia and psychosis has yet to be reached. The disparity between the relatively high prevalence of regular adult use of cannabis (8%7)and the low incidence of cannabis-induced psychosis suggests that cannabis use alone is unlikely to lead to episodes of psychosis in individuals who are not predisposed to such episodes. Sarrazin et al15 evaluated 170 patients with schizophrenia, 31 of whom had cannabis use disorder. They found no significant difference in lifetime symptom dimensions between groups, and proposed that cannabis-associated schizophrenia should not be categorized as a distinct clinical entity of schizophrenia with specific features.15
Policies that encourage follow-up of patients after episodes of drug-induced psychosis may mitigate the adverse social and economic effects of schizophrenia. Currently, these policies are not widely implemented in health care institutions, possibly because psychotic symptoms may fade after the drug’s effects have dissipated. Despite this, these patients are at high risk of developing schizophrenia and self-harm. New-onset schizophrenia should be promptly identified because delayed diagnosis is associated with worse prognosis.6 Additionally, identifying genetic susceptibilities to schizophrenia—such as the Val158Met polymorphisms—in individuals who use cannabis could help clinicians manage or slow the onset or progression of schizophrenia.3 Motivational interviewing strategies should be used to minimize or eliminate cannabis use in individuals with active schizophrenia or psychosis, thus preventing worse outcomes.
Bottom Line
Identifying susceptibilities to schizophrenia may guide interventions in patients who use cannabis. Several large studies have suggested that cannabis use may exacerbate symptoms and worsen the prognosis of schizophrenia. Motivational interviewing strategies aimed at minimizing cannabis use may improve outcomes in patients with schizophrenia.
Related Resources
- Khokhar JY, Dwiel LL, Henricks AM, et al. The link between schizophrenia and substance use disorder: a unifying hypothesis. Schizophr Res. 2018;194:78-85. doi:10.1016/j. schres.2017.04.016
- Otite ES, Solanky A, Doumas S. Adolescents, THC, and the risk of psychosis. Current Psychiatry. 2021;20(12):e1-e2. doi:10.12788/cp.0197
1. Simeone JC, Ward AJ, Rotella P, et al. An evaluation of variation in published estimates of schizophrenia prevalence from 1990-2013: a systematic literature review. BMC Psychiatry. 2015;15(1):193. doi:10.1186/s12888-015-0578-7
2. Tandon R, Gaebel W, Barch DM, et al. Definition and description of schizophrenia in the DSM-5. Schizophr Res. 2013;150(1):3-10. doi:10.1016/j.schres.2013.05.028
3. Bosia M, Buonocore M, Bechi M, et al. Schizophrenia, cannabis use and catechol-O-methyltransferase (COMT): modeling the interplay on cognition. Prog Neuropsychopharmacol Biol Psychiatry. 2019;92:363-368. doi:10.1016/j.pnpbp.2019.02.009
4. Welch KA, McIntosh AM, Job DE, et al. The impact of substance use on brain structure in people at high risk of developing schizophrenia. Schizophr Bull. 2011;37(5):1066-1076. doi:10.1093/schbul/sbq013
5. Winship IR, Dursun SM, Baker GB, et al. An overview of animal models related to schizophrenia. Can J Psychiatry. 2019;64(1):5-17. doi:10.1177/0706743718773728
6. Starzer MSK, Nordentoft M, Hjorthøj C. Rates and predictors of conversion to schizophrenia or bipolar disorder following substance-induced psychosis. Am J Psychiatry. 2018;175(4):343-350. doi:10.1176/appi.ajp.2017.17020223
7. Hall W. Cannabis use and psychosis. Drug Alcohol Rev. 1998;17(4):433-444. doi:10.1080/09595239800187271
8. Misiak B, Frydecka D, Slezak R, et al. Elevated homocysteine level in first-episode schizophrenia patients—the relevance of family history of schizophrenia and lifetime diagnosis of cannabis abuse. Metab Brain Dis. 2014;29(3):661-670. doi:10.1007/s11011-014-9534-3
9. Veen ND, Selten J, van der Tweel I, et al. Cannabis use and age at onset of schizophrenia. Am J Psychiatry. 2004;161(3):501-506. doi:10.1176/appi.ajp.161.3.501
10. Foti DJ, Kotov R, Guey LT, et al. Cannabis use and the course of schizophrenia: 10-year follow-up after first hospitalization. Am J Psychiatry. 2010;167(8):987-993. doi:10.1176/appi.ajp.2010.09020189
11. Ortiz-Medina MB, Perea M, Torales J, et al. Cannabis consumption and psychosis or schizophrenia development. Int J Soc Psychiatry. 2018;64(7):690-704. doi:10.1177/0020764018801690
12. Hickman M, Vickerman P, Macleod J, et al. If cannabis caused schizophrenia—how many cannabis users may need to be prevented in order to prevent one case of schizophrenia? England and Wales calculations. Addiction. 2009;104(11):1856-1861. doi:10.1111/j.1360-0443.2009.02736.x
13. Gupta S, Phalen T, Gupta S. Medical marijuana: do the benefits outweigh the risks? Current Psychiatry. 2018;17(1):34-41.
14. Freeman TP, Craft S, Wilson J, et al. Changes in delta-9-tetrahydrocannabinol (THC) and cannabidiol (CBD) concentrations in cannabis over time: systematic review and meta-analysis. Addiction. 2021;116(5):1000-1010. doi:10.1111/add.15253
15. Sarrazin S, Louppe F, Doukhan R, et al. A clinical comparison of schizophrenia with and without pre-onset cannabis use disorder: a retrospective cohort study using categorical and dimensional approaches. Ann Gen Psychiatry. 2015;14:44. doi:10.1186/s12991-015-0083-x
Approximately 1 in 200 individuals will be diagnosed with schizophrenia in their lifetime.1 DSM-5 criteria for the diagnosis of schizophrenia require the presence of ≥2 of 5 symptoms: delusions, hallucinations, disordered speech, grossly disorganized (or catatonic) behavior, and negative symptoms such as flat affect or avolition.2 Multiple studies have found increased rates of cannabis use among patients with schizophrenia. Because cognitive deficits are the chief predictor of clinical outcomes and quality of life in individuals with schizophrenia, the cognitive effects of cannabis use among these patients are of clinical significance.3 As legislation increasingly allows for the sale, possession, and consumption of cannabis, it is crucial to provide clinicians with evidence-based recommendations for treating patients who regularly use cannabis (approximately 8% of the adult population3). In this article, we analyze several peer-reviewed studies to investigate the impact of cannabis use on the onset and development of schizophrenia.
A look at substance-induced psychosis
Schizophrenia is associated with several structural brain changes, and some of these changes may be influenced by cannabis use (Box4). The biochemical etiology of schizophrenia is poorly understood but thought to involve dopamine, glutamate, serotonin, and gamma-aminobutyric acid. Certain positive symptoms, such as hallucinations, are uniquely human and difficult to study in animal models.5 Psychoactive substance use, especially cannabis, is frequently comorbid with schizophrenia. Additionally, certain individuals may be more predisposed to substance-induced psychosis than others based on genetic variation and underlying brain structure changes.4 Substance-induced psychosis is a psychotic state following the ingestion of a psychoactive substance or drug withdrawal lasting ≥48 hours.6 The psychoactive effects of cannabis have been associated with an exacerbation of existing schizophrenia symptoms.7 In 1998, Hall7 proposed 2 hypotheses to explain the relationship between cannabis and psychosis. The first was that heavy consumption of cannabis triggers a specific type of cannabis psychosis.7 The second was that cannabis use exacerbates existing schizophrenia, making the symptoms worse.7 Hall7 concluded that there was a complicated interaction among an individual’s vulnerability to their stressors, environment, and genetics.
Box
Schizophrenia is associated with several structural changes in the brain, including lateral ventriculomegaly, reduced prefrontal cortex volume, and generalized atrophy. These changes may precede illness and act as a risk marker.4 A multivariate regression analysis that compared patients with schizophrenia who were cannabis users vs patients with schizophrenia who were nonusers found that those with high-level cannabis use had relatively higher left and right lateral ventricle volume (r = 0.208, P = .13, and r = 0.226, P = .007, respectively) as well as increased third ventricle volume (r = 0.271, P = .001).4 These changes were dose-dependent and may lead to worse disease outcomes.4
Cannabis, COMT, and homocysteine
Great advances have been made in our ability to examine the association between genetics and metabolism. One example of this is the interaction between the catechol-O-methyltransferase (COMT) gene and the active component of cannabis, delta-9-tetrahydrocannabinol (THC). COMT codes for an enzyme that degrades cortical dopamine. The Val158Met polymorphism of this gene increases COMT activity, leading to increased dopamine catabolism, and thus decreased levels of extracellular dopamine, which induces an increase in mesolimbic dopaminergic activity, thereby increasing susceptibility to psychosis.3
In a study that genotyped 135 patients with schizophrenia, the Val158Met polymorphism was present in 29.63% of participants.3 Because THC can induce episodes of psychosis, individuals with this polymorphism may be at a higher risk of developing schizophrenia. Compared to Met carrier control participants with similar histories of cannabis consumption, those with the Val158Met polymorphism demonstrated markedly worse performance on tests of verbal fluency and processing speed.3 This is clinically significant because cognitive impairments are a major prognostic factor in schizophrenia, and identifying patients with this polymorphism could help personalize interventions for those who consume cannabis and are at risk of developing schizophrenia.
A study that evaluated 56 patients with first-episode schizophrenia found that having a history of cannabis abuse was associated with significantly higher levels of homocysteine as well as lower levels of high-density lipoprotein and vitamin B12.8 Homocysteine is an agonist at the glutamate binding site and a partial antagonist at the glycine co-agonist site in the N-methyl-
The C677T polymorphism in MTHFR may predict the risk of developing metabolic syndrome in patients taking second-generation antipsychotics.8 Elevations in homocysteine by as little as 5 μmol/L may increase schizophrenia risk by 70% compared to controls, possibly due to homocysteine initiating neuronal apoptosis, catalyzing dysfunction of the mitochondria, or increasing oxidative stress.8 There is a positive correlation between homocysteine levels and severity of negative symptoms (P = .006) and general psychopathology (P = .008) of schizophrenia when analyzed using the Positive and Negative Syndrome Scale.8 Negative symptoms such as blunted affect, apathy, anhedonia, and loss of motivation significantly impact the social and economic outcomes of patients diagnosed with schizophrenia.
Research paints a mixed picture
A Danish study analyzed the rates of conversion to schizophrenia or bipolar disorder (BD) among 6,788 individuals who received a diagnosis of substance-induced psychosis from 1994 to 2014.6 Ten comparison participants were selected for each case participant, matched on sex and year/month of birth. Participants were followed until the first occurrence of schizophrenia or BD, death, or emigration from Denmark. Substances implicated in the initial psychotic episode included cannabis, alcohol, opioids, sedatives, cocaine, amphetamines, hallucinogens, and combinations of substances.
Continue to: The overall conversion rate...
The overall conversion rate over 20 years was 32.2% (95% CI, 29.7 to 34.9), with 26.0% developing schizophrenia vs 8.4% developing BD.6 Of the substances involved, cannabis was the most common, implicated in 41.2% (95% CI, 36.6 to 46.2) of cases.6 One-half of male patients converted within 2.0 years and one-half of female patients converted within 4.4 years after a cannabis-induced psychosis.6
This study had several limitations. It could not account for any short-term psychotic symptoms experienced by the general population, especially after cannabis use. Such patients might not seek treatment. Thus, the results might not be generalizable to the general population. The study did not evaluate if conversion rates differed based on continued substance use following the psychosis episode, or the amount of each substance taken prior to the episode. Dose-dependence was not well elucidated, and this study only looked at patients from Denmark and did not account for socioeconomic status.6
Another Danish study looked at the influences of gender and cannabis use in the early course of the disease in 133 patients with schizophrenia.9 These researchers found that male gender was a significant predictor of earlier onset of dysfunction socially and in the workplace, as well as a higher risk of developing negative symptoms. However, compared to gender, cannabis use was a stronger predictor of age at first psychotic episode. For cannabis users, the median age of onset of negative symptoms was 23.7, compared to 38.4 for nonusers (P < .001).9
Cannabis use is significantly elevated among individuals with psychosis, with a 12-month prevalence of 29.2% compared to 4.0% among the general population of the United States.10 In a study that assessed 229 patients with a schizophrenia spectrum disorder during their first hospitalization and 6 months, 2 years, 4 years, and 10 years later, Foti et al10 found that the lifetime rate of cannabis use was 66.2%. Survival analysis found cannabis use doubled the risk of the onset of psychosis compared to nonusers of the same age (hazard ratio [HR] = 1.97; 95% CI, 1.48 to 2.62, P < .001), even after adjusting for socioeconomic status, age, and gender (HR = 1.34; 95% CI, 1.01 to 1.77, P < .05).10 Additionally, Foti et al10 found significant positive correlations between psychotic symptoms and cannabis use in patients with schizophrenia over the course of 10 years. An increase in symptoms was associated with a higher likelihood of cannabis use, and a decrease in symptoms was correlated with a lower likelihood of use (adjusted odds ratio = 1.64; 95% CI, 1.12 to 2.43, P < .0125).10
Ortiz-Medina et al11 conducted a meta-analysis of 22 studies of 15 cohorts from healthy populations and 12 other cohort follow-up studies that evaluated the onset of psychotic symptoms in individuals who used cannabis. Most studies found associations between cannabis use and the onset of symptoms of schizophrenia, and most determined cannabis was also a major risk factor for other psychotic disorders. Analyses of dose-dependence indicated that repeated cannabis use increased the risk of developing psychotic symptoms. This risk is increased when an individual starts using cannabis before age 15.11 Age seemed to be a stronger predictor of onset and outcome than sex, with no significant differences between men and women. One study in this review found that approximately 8% to 13% cases of schizophrenia may have been solely due to cannabis.11 The most significant limitation to the studies analyzed in this review is that retrospective studies utilize self-reported questionnaires.
Continue to: Other researchers have found...
Other researchers have found it would take a relatively high number of individuals to stop using cannabis to prevent 1 case of schizophrenia. In a study of data from England and Wales, Hickman et al12 evaluated the best available estimates of the incidence of schizophrenia, rates of heavy and light cannabis use, and risk that cannabis causes schizophrenia to determine the number needed to prevent (NNP) 1 case of schizophrenia. They estimated that it would require approximately 2,800 men age 20 to 24 (90% CI, 2,018 to 4,530) and 4,700 men age 35 to 39 (90% CI, 3,114 to 8,416) who heavily used cannabis to stop their consumption to prevent 1 case of schizophrenia.12 For women with heavy cannabis use, the mean NNP was 5,470 for women age 25 to 29 (90% CI, 3,640 to 9,839) and 10,870 for women age 35 to 39 (90% CI, 6,786 to 22,732).12 For light cannabis users, the NNP was 4 to 5 times higher than the NNP for heavy cannabis users. This suggests that clinical interventions aimed at preventing dependence on cannabis would be more effective than interventions aimed at eliminating cannabis use.
Medical cannabis and increased potency
In recent years, the use of medical cannabis, which is used to address adverse effects of chemotherapy as well as neuropathic pain, Parkinson’s disease, and epilepsy, has been increasing.13 However, there is a lack of well-conducted randomized clinical trials evaluating medical cannabis’ efficacy and safety. As medical cannabis continues to gain public acceptance and more states permit its legal use, patients and physicians should be fully informed of the known adverse effects, including impaired attention, learning, and motivation.13
Several studies have drawn attention to the dose-dependence of many of cannabis’ effects. Since at least the 1960s, the concentration of THC in cannabis has increased substantially, thus increasing its potency. Based on 66,747 samples across 8 studies, 1 meta-analysis estimated that THC concentrations in herbal cannabis increased by 0.29% (P < .001) each year between 1970 and 2017.14 Similarly, THC concentrations in cannabis resins were found to have increased by 0.57% (P = .017) each year between 1975 and 2017.14 Cannabis products with high concentrations of THC carry an increased risk of addiction and mental health disorders.14
Identifying those at highest risk
Despite ongoing research, scientific consensus on the relationship of cannabis to schizophrenia and psychosis has yet to be reached. The disparity between the relatively high prevalence of regular adult use of cannabis (8%7)and the low incidence of cannabis-induced psychosis suggests that cannabis use alone is unlikely to lead to episodes of psychosis in individuals who are not predisposed to such episodes. Sarrazin et al15 evaluated 170 patients with schizophrenia, 31 of whom had cannabis use disorder. They found no significant difference in lifetime symptom dimensions between groups, and proposed that cannabis-associated schizophrenia should not be categorized as a distinct clinical entity of schizophrenia with specific features.15
Policies that encourage follow-up of patients after episodes of drug-induced psychosis may mitigate the adverse social and economic effects of schizophrenia. Currently, these policies are not widely implemented in health care institutions, possibly because psychotic symptoms may fade after the drug’s effects have dissipated. Despite this, these patients are at high risk of developing schizophrenia and self-harm. New-onset schizophrenia should be promptly identified because delayed diagnosis is associated with worse prognosis.6 Additionally, identifying genetic susceptibilities to schizophrenia—such as the Val158Met polymorphisms—in individuals who use cannabis could help clinicians manage or slow the onset or progression of schizophrenia.3 Motivational interviewing strategies should be used to minimize or eliminate cannabis use in individuals with active schizophrenia or psychosis, thus preventing worse outcomes.
Bottom Line
Identifying susceptibilities to schizophrenia may guide interventions in patients who use cannabis. Several large studies have suggested that cannabis use may exacerbate symptoms and worsen the prognosis of schizophrenia. Motivational interviewing strategies aimed at minimizing cannabis use may improve outcomes in patients with schizophrenia.
Related Resources
- Khokhar JY, Dwiel LL, Henricks AM, et al. The link between schizophrenia and substance use disorder: a unifying hypothesis. Schizophr Res. 2018;194:78-85. doi:10.1016/j. schres.2017.04.016
- Otite ES, Solanky A, Doumas S. Adolescents, THC, and the risk of psychosis. Current Psychiatry. 2021;20(12):e1-e2. doi:10.12788/cp.0197
Approximately 1 in 200 individuals will be diagnosed with schizophrenia in their lifetime.1 DSM-5 criteria for the diagnosis of schizophrenia require the presence of ≥2 of 5 symptoms: delusions, hallucinations, disordered speech, grossly disorganized (or catatonic) behavior, and negative symptoms such as flat affect or avolition.2 Multiple studies have found increased rates of cannabis use among patients with schizophrenia. Because cognitive deficits are the chief predictor of clinical outcomes and quality of life in individuals with schizophrenia, the cognitive effects of cannabis use among these patients are of clinical significance.3 As legislation increasingly allows for the sale, possession, and consumption of cannabis, it is crucial to provide clinicians with evidence-based recommendations for treating patients who regularly use cannabis (approximately 8% of the adult population3). In this article, we analyze several peer-reviewed studies to investigate the impact of cannabis use on the onset and development of schizophrenia.
A look at substance-induced psychosis
Schizophrenia is associated with several structural brain changes, and some of these changes may be influenced by cannabis use (Box4). The biochemical etiology of schizophrenia is poorly understood but thought to involve dopamine, glutamate, serotonin, and gamma-aminobutyric acid. Certain positive symptoms, such as hallucinations, are uniquely human and difficult to study in animal models.5 Psychoactive substance use, especially cannabis, is frequently comorbid with schizophrenia. Additionally, certain individuals may be more predisposed to substance-induced psychosis than others based on genetic variation and underlying brain structure changes.4 Substance-induced psychosis is a psychotic state following the ingestion of a psychoactive substance or drug withdrawal lasting ≥48 hours.6 The psychoactive effects of cannabis have been associated with an exacerbation of existing schizophrenia symptoms.7 In 1998, Hall7 proposed 2 hypotheses to explain the relationship between cannabis and psychosis. The first was that heavy consumption of cannabis triggers a specific type of cannabis psychosis.7 The second was that cannabis use exacerbates existing schizophrenia, making the symptoms worse.7 Hall7 concluded that there was a complicated interaction among an individual’s vulnerability to their stressors, environment, and genetics.
Box
Schizophrenia is associated with several structural changes in the brain, including lateral ventriculomegaly, reduced prefrontal cortex volume, and generalized atrophy. These changes may precede illness and act as a risk marker.4 A multivariate regression analysis that compared patients with schizophrenia who were cannabis users vs patients with schizophrenia who were nonusers found that those with high-level cannabis use had relatively higher left and right lateral ventricle volume (r = 0.208, P = .13, and r = 0.226, P = .007, respectively) as well as increased third ventricle volume (r = 0.271, P = .001).4 These changes were dose-dependent and may lead to worse disease outcomes.4
Cannabis, COMT, and homocysteine
Great advances have been made in our ability to examine the association between genetics and metabolism. One example of this is the interaction between the catechol-O-methyltransferase (COMT) gene and the active component of cannabis, delta-9-tetrahydrocannabinol (THC). COMT codes for an enzyme that degrades cortical dopamine. The Val158Met polymorphism of this gene increases COMT activity, leading to increased dopamine catabolism, and thus decreased levels of extracellular dopamine, which induces an increase in mesolimbic dopaminergic activity, thereby increasing susceptibility to psychosis.3
In a study that genotyped 135 patients with schizophrenia, the Val158Met polymorphism was present in 29.63% of participants.3 Because THC can induce episodes of psychosis, individuals with this polymorphism may be at a higher risk of developing schizophrenia. Compared to Met carrier control participants with similar histories of cannabis consumption, those with the Val158Met polymorphism demonstrated markedly worse performance on tests of verbal fluency and processing speed.3 This is clinically significant because cognitive impairments are a major prognostic factor in schizophrenia, and identifying patients with this polymorphism could help personalize interventions for those who consume cannabis and are at risk of developing schizophrenia.
A study that evaluated 56 patients with first-episode schizophrenia found that having a history of cannabis abuse was associated with significantly higher levels of homocysteine as well as lower levels of high-density lipoprotein and vitamin B12.8 Homocysteine is an agonist at the glutamate binding site and a partial antagonist at the glycine co-agonist site in the N-methyl-
The C677T polymorphism in MTHFR may predict the risk of developing metabolic syndrome in patients taking second-generation antipsychotics.8 Elevations in homocysteine by as little as 5 μmol/L may increase schizophrenia risk by 70% compared to controls, possibly due to homocysteine initiating neuronal apoptosis, catalyzing dysfunction of the mitochondria, or increasing oxidative stress.8 There is a positive correlation between homocysteine levels and severity of negative symptoms (P = .006) and general psychopathology (P = .008) of schizophrenia when analyzed using the Positive and Negative Syndrome Scale.8 Negative symptoms such as blunted affect, apathy, anhedonia, and loss of motivation significantly impact the social and economic outcomes of patients diagnosed with schizophrenia.
Research paints a mixed picture
A Danish study analyzed the rates of conversion to schizophrenia or bipolar disorder (BD) among 6,788 individuals who received a diagnosis of substance-induced psychosis from 1994 to 2014.6 Ten comparison participants were selected for each case participant, matched on sex and year/month of birth. Participants were followed until the first occurrence of schizophrenia or BD, death, or emigration from Denmark. Substances implicated in the initial psychotic episode included cannabis, alcohol, opioids, sedatives, cocaine, amphetamines, hallucinogens, and combinations of substances.
Continue to: The overall conversion rate...
The overall conversion rate over 20 years was 32.2% (95% CI, 29.7 to 34.9), with 26.0% developing schizophrenia vs 8.4% developing BD.6 Of the substances involved, cannabis was the most common, implicated in 41.2% (95% CI, 36.6 to 46.2) of cases.6 One-half of male patients converted within 2.0 years and one-half of female patients converted within 4.4 years after a cannabis-induced psychosis.6
This study had several limitations. It could not account for any short-term psychotic symptoms experienced by the general population, especially after cannabis use. Such patients might not seek treatment. Thus, the results might not be generalizable to the general population. The study did not evaluate if conversion rates differed based on continued substance use following the psychosis episode, or the amount of each substance taken prior to the episode. Dose-dependence was not well elucidated, and this study only looked at patients from Denmark and did not account for socioeconomic status.6
Another Danish study looked at the influences of gender and cannabis use in the early course of the disease in 133 patients with schizophrenia.9 These researchers found that male gender was a significant predictor of earlier onset of dysfunction socially and in the workplace, as well as a higher risk of developing negative symptoms. However, compared to gender, cannabis use was a stronger predictor of age at first psychotic episode. For cannabis users, the median age of onset of negative symptoms was 23.7, compared to 38.4 for nonusers (P < .001).9
Cannabis use is significantly elevated among individuals with psychosis, with a 12-month prevalence of 29.2% compared to 4.0% among the general population of the United States.10 In a study that assessed 229 patients with a schizophrenia spectrum disorder during their first hospitalization and 6 months, 2 years, 4 years, and 10 years later, Foti et al10 found that the lifetime rate of cannabis use was 66.2%. Survival analysis found cannabis use doubled the risk of the onset of psychosis compared to nonusers of the same age (hazard ratio [HR] = 1.97; 95% CI, 1.48 to 2.62, P < .001), even after adjusting for socioeconomic status, age, and gender (HR = 1.34; 95% CI, 1.01 to 1.77, P < .05).10 Additionally, Foti et al10 found significant positive correlations between psychotic symptoms and cannabis use in patients with schizophrenia over the course of 10 years. An increase in symptoms was associated with a higher likelihood of cannabis use, and a decrease in symptoms was correlated with a lower likelihood of use (adjusted odds ratio = 1.64; 95% CI, 1.12 to 2.43, P < .0125).10
Ortiz-Medina et al11 conducted a meta-analysis of 22 studies of 15 cohorts from healthy populations and 12 other cohort follow-up studies that evaluated the onset of psychotic symptoms in individuals who used cannabis. Most studies found associations between cannabis use and the onset of symptoms of schizophrenia, and most determined cannabis was also a major risk factor for other psychotic disorders. Analyses of dose-dependence indicated that repeated cannabis use increased the risk of developing psychotic symptoms. This risk is increased when an individual starts using cannabis before age 15.11 Age seemed to be a stronger predictor of onset and outcome than sex, with no significant differences between men and women. One study in this review found that approximately 8% to 13% cases of schizophrenia may have been solely due to cannabis.11 The most significant limitation to the studies analyzed in this review is that retrospective studies utilize self-reported questionnaires.
Continue to: Other researchers have found...
Other researchers have found it would take a relatively high number of individuals to stop using cannabis to prevent 1 case of schizophrenia. In a study of data from England and Wales, Hickman et al12 evaluated the best available estimates of the incidence of schizophrenia, rates of heavy and light cannabis use, and risk that cannabis causes schizophrenia to determine the number needed to prevent (NNP) 1 case of schizophrenia. They estimated that it would require approximately 2,800 men age 20 to 24 (90% CI, 2,018 to 4,530) and 4,700 men age 35 to 39 (90% CI, 3,114 to 8,416) who heavily used cannabis to stop their consumption to prevent 1 case of schizophrenia.12 For women with heavy cannabis use, the mean NNP was 5,470 for women age 25 to 29 (90% CI, 3,640 to 9,839) and 10,870 for women age 35 to 39 (90% CI, 6,786 to 22,732).12 For light cannabis users, the NNP was 4 to 5 times higher than the NNP for heavy cannabis users. This suggests that clinical interventions aimed at preventing dependence on cannabis would be more effective than interventions aimed at eliminating cannabis use.
Medical cannabis and increased potency
In recent years, the use of medical cannabis, which is used to address adverse effects of chemotherapy as well as neuropathic pain, Parkinson’s disease, and epilepsy, has been increasing.13 However, there is a lack of well-conducted randomized clinical trials evaluating medical cannabis’ efficacy and safety. As medical cannabis continues to gain public acceptance and more states permit its legal use, patients and physicians should be fully informed of the known adverse effects, including impaired attention, learning, and motivation.13
Several studies have drawn attention to the dose-dependence of many of cannabis’ effects. Since at least the 1960s, the concentration of THC in cannabis has increased substantially, thus increasing its potency. Based on 66,747 samples across 8 studies, 1 meta-analysis estimated that THC concentrations in herbal cannabis increased by 0.29% (P < .001) each year between 1970 and 2017.14 Similarly, THC concentrations in cannabis resins were found to have increased by 0.57% (P = .017) each year between 1975 and 2017.14 Cannabis products with high concentrations of THC carry an increased risk of addiction and mental health disorders.14
Identifying those at highest risk
Despite ongoing research, scientific consensus on the relationship of cannabis to schizophrenia and psychosis has yet to be reached. The disparity between the relatively high prevalence of regular adult use of cannabis (8%7)and the low incidence of cannabis-induced psychosis suggests that cannabis use alone is unlikely to lead to episodes of psychosis in individuals who are not predisposed to such episodes. Sarrazin et al15 evaluated 170 patients with schizophrenia, 31 of whom had cannabis use disorder. They found no significant difference in lifetime symptom dimensions between groups, and proposed that cannabis-associated schizophrenia should not be categorized as a distinct clinical entity of schizophrenia with specific features.15
Policies that encourage follow-up of patients after episodes of drug-induced psychosis may mitigate the adverse social and economic effects of schizophrenia. Currently, these policies are not widely implemented in health care institutions, possibly because psychotic symptoms may fade after the drug’s effects have dissipated. Despite this, these patients are at high risk of developing schizophrenia and self-harm. New-onset schizophrenia should be promptly identified because delayed diagnosis is associated with worse prognosis.6 Additionally, identifying genetic susceptibilities to schizophrenia—such as the Val158Met polymorphisms—in individuals who use cannabis could help clinicians manage or slow the onset or progression of schizophrenia.3 Motivational interviewing strategies should be used to minimize or eliminate cannabis use in individuals with active schizophrenia or psychosis, thus preventing worse outcomes.
Bottom Line
Identifying susceptibilities to schizophrenia may guide interventions in patients who use cannabis. Several large studies have suggested that cannabis use may exacerbate symptoms and worsen the prognosis of schizophrenia. Motivational interviewing strategies aimed at minimizing cannabis use may improve outcomes in patients with schizophrenia.
Related Resources
- Khokhar JY, Dwiel LL, Henricks AM, et al. The link between schizophrenia and substance use disorder: a unifying hypothesis. Schizophr Res. 2018;194:78-85. doi:10.1016/j. schres.2017.04.016
- Otite ES, Solanky A, Doumas S. Adolescents, THC, and the risk of psychosis. Current Psychiatry. 2021;20(12):e1-e2. doi:10.12788/cp.0197
1. Simeone JC, Ward AJ, Rotella P, et al. An evaluation of variation in published estimates of schizophrenia prevalence from 1990-2013: a systematic literature review. BMC Psychiatry. 2015;15(1):193. doi:10.1186/s12888-015-0578-7
2. Tandon R, Gaebel W, Barch DM, et al. Definition and description of schizophrenia in the DSM-5. Schizophr Res. 2013;150(1):3-10. doi:10.1016/j.schres.2013.05.028
3. Bosia M, Buonocore M, Bechi M, et al. Schizophrenia, cannabis use and catechol-O-methyltransferase (COMT): modeling the interplay on cognition. Prog Neuropsychopharmacol Biol Psychiatry. 2019;92:363-368. doi:10.1016/j.pnpbp.2019.02.009
4. Welch KA, McIntosh AM, Job DE, et al. The impact of substance use on brain structure in people at high risk of developing schizophrenia. Schizophr Bull. 2011;37(5):1066-1076. doi:10.1093/schbul/sbq013
5. Winship IR, Dursun SM, Baker GB, et al. An overview of animal models related to schizophrenia. Can J Psychiatry. 2019;64(1):5-17. doi:10.1177/0706743718773728
6. Starzer MSK, Nordentoft M, Hjorthøj C. Rates and predictors of conversion to schizophrenia or bipolar disorder following substance-induced psychosis. Am J Psychiatry. 2018;175(4):343-350. doi:10.1176/appi.ajp.2017.17020223
7. Hall W. Cannabis use and psychosis. Drug Alcohol Rev. 1998;17(4):433-444. doi:10.1080/09595239800187271
8. Misiak B, Frydecka D, Slezak R, et al. Elevated homocysteine level in first-episode schizophrenia patients—the relevance of family history of schizophrenia and lifetime diagnosis of cannabis abuse. Metab Brain Dis. 2014;29(3):661-670. doi:10.1007/s11011-014-9534-3
9. Veen ND, Selten J, van der Tweel I, et al. Cannabis use and age at onset of schizophrenia. Am J Psychiatry. 2004;161(3):501-506. doi:10.1176/appi.ajp.161.3.501
10. Foti DJ, Kotov R, Guey LT, et al. Cannabis use and the course of schizophrenia: 10-year follow-up after first hospitalization. Am J Psychiatry. 2010;167(8):987-993. doi:10.1176/appi.ajp.2010.09020189
11. Ortiz-Medina MB, Perea M, Torales J, et al. Cannabis consumption and psychosis or schizophrenia development. Int J Soc Psychiatry. 2018;64(7):690-704. doi:10.1177/0020764018801690
12. Hickman M, Vickerman P, Macleod J, et al. If cannabis caused schizophrenia—how many cannabis users may need to be prevented in order to prevent one case of schizophrenia? England and Wales calculations. Addiction. 2009;104(11):1856-1861. doi:10.1111/j.1360-0443.2009.02736.x
13. Gupta S, Phalen T, Gupta S. Medical marijuana: do the benefits outweigh the risks? Current Psychiatry. 2018;17(1):34-41.
14. Freeman TP, Craft S, Wilson J, et al. Changes in delta-9-tetrahydrocannabinol (THC) and cannabidiol (CBD) concentrations in cannabis over time: systematic review and meta-analysis. Addiction. 2021;116(5):1000-1010. doi:10.1111/add.15253
15. Sarrazin S, Louppe F, Doukhan R, et al. A clinical comparison of schizophrenia with and without pre-onset cannabis use disorder: a retrospective cohort study using categorical and dimensional approaches. Ann Gen Psychiatry. 2015;14:44. doi:10.1186/s12991-015-0083-x
1. Simeone JC, Ward AJ, Rotella P, et al. An evaluation of variation in published estimates of schizophrenia prevalence from 1990-2013: a systematic literature review. BMC Psychiatry. 2015;15(1):193. doi:10.1186/s12888-015-0578-7
2. Tandon R, Gaebel W, Barch DM, et al. Definition and description of schizophrenia in the DSM-5. Schizophr Res. 2013;150(1):3-10. doi:10.1016/j.schres.2013.05.028
3. Bosia M, Buonocore M, Bechi M, et al. Schizophrenia, cannabis use and catechol-O-methyltransferase (COMT): modeling the interplay on cognition. Prog Neuropsychopharmacol Biol Psychiatry. 2019;92:363-368. doi:10.1016/j.pnpbp.2019.02.009
4. Welch KA, McIntosh AM, Job DE, et al. The impact of substance use on brain structure in people at high risk of developing schizophrenia. Schizophr Bull. 2011;37(5):1066-1076. doi:10.1093/schbul/sbq013
5. Winship IR, Dursun SM, Baker GB, et al. An overview of animal models related to schizophrenia. Can J Psychiatry. 2019;64(1):5-17. doi:10.1177/0706743718773728
6. Starzer MSK, Nordentoft M, Hjorthøj C. Rates and predictors of conversion to schizophrenia or bipolar disorder following substance-induced psychosis. Am J Psychiatry. 2018;175(4):343-350. doi:10.1176/appi.ajp.2017.17020223
7. Hall W. Cannabis use and psychosis. Drug Alcohol Rev. 1998;17(4):433-444. doi:10.1080/09595239800187271
8. Misiak B, Frydecka D, Slezak R, et al. Elevated homocysteine level in first-episode schizophrenia patients—the relevance of family history of schizophrenia and lifetime diagnosis of cannabis abuse. Metab Brain Dis. 2014;29(3):661-670. doi:10.1007/s11011-014-9534-3
9. Veen ND, Selten J, van der Tweel I, et al. Cannabis use and age at onset of schizophrenia. Am J Psychiatry. 2004;161(3):501-506. doi:10.1176/appi.ajp.161.3.501
10. Foti DJ, Kotov R, Guey LT, et al. Cannabis use and the course of schizophrenia: 10-year follow-up after first hospitalization. Am J Psychiatry. 2010;167(8):987-993. doi:10.1176/appi.ajp.2010.09020189
11. Ortiz-Medina MB, Perea M, Torales J, et al. Cannabis consumption and psychosis or schizophrenia development. Int J Soc Psychiatry. 2018;64(7):690-704. doi:10.1177/0020764018801690
12. Hickman M, Vickerman P, Macleod J, et al. If cannabis caused schizophrenia—how many cannabis users may need to be prevented in order to prevent one case of schizophrenia? England and Wales calculations. Addiction. 2009;104(11):1856-1861. doi:10.1111/j.1360-0443.2009.02736.x
13. Gupta S, Phalen T, Gupta S. Medical marijuana: do the benefits outweigh the risks? Current Psychiatry. 2018;17(1):34-41.
14. Freeman TP, Craft S, Wilson J, et al. Changes in delta-9-tetrahydrocannabinol (THC) and cannabidiol (CBD) concentrations in cannabis over time: systematic review and meta-analysis. Addiction. 2021;116(5):1000-1010. doi:10.1111/add.15253
15. Sarrazin S, Louppe F, Doukhan R, et al. A clinical comparison of schizophrenia with and without pre-onset cannabis use disorder: a retrospective cohort study using categorical and dimensional approaches. Ann Gen Psychiatry. 2015;14:44. doi:10.1186/s12991-015-0083-x

Nutraceuticals for traumatic brain injury: Should you recommend their use?
Traumatic brain injury (TBI) affects more than 2 million people in the United States each year.1 TBI can trigger a cascade of secondary injury mechanisms, such as inflammation, hypoxic/ischemic injury, excitotoxicity, and oxidative stress,2 that could contribute to cognitive and behavioral changes. Although neuropsychiatric symptoms might not be obvious after a TBI, they have a high prevalence in these patients, can last long term, and may be difficult to treat.3 Despite research advances in understanding the biological basis of TBI and identifying potential therapeutic targets, treatment options for individuals with TBI remain limited.
As a result, clinicians have turned to alternative treatments for TBI, including nutraceuticals. In this article, we will:
- provide an overview of nutraceuticals used in treating TBI, first exploring outcomes soon after TBI, then concentrating on neuropsychiatric outcomes
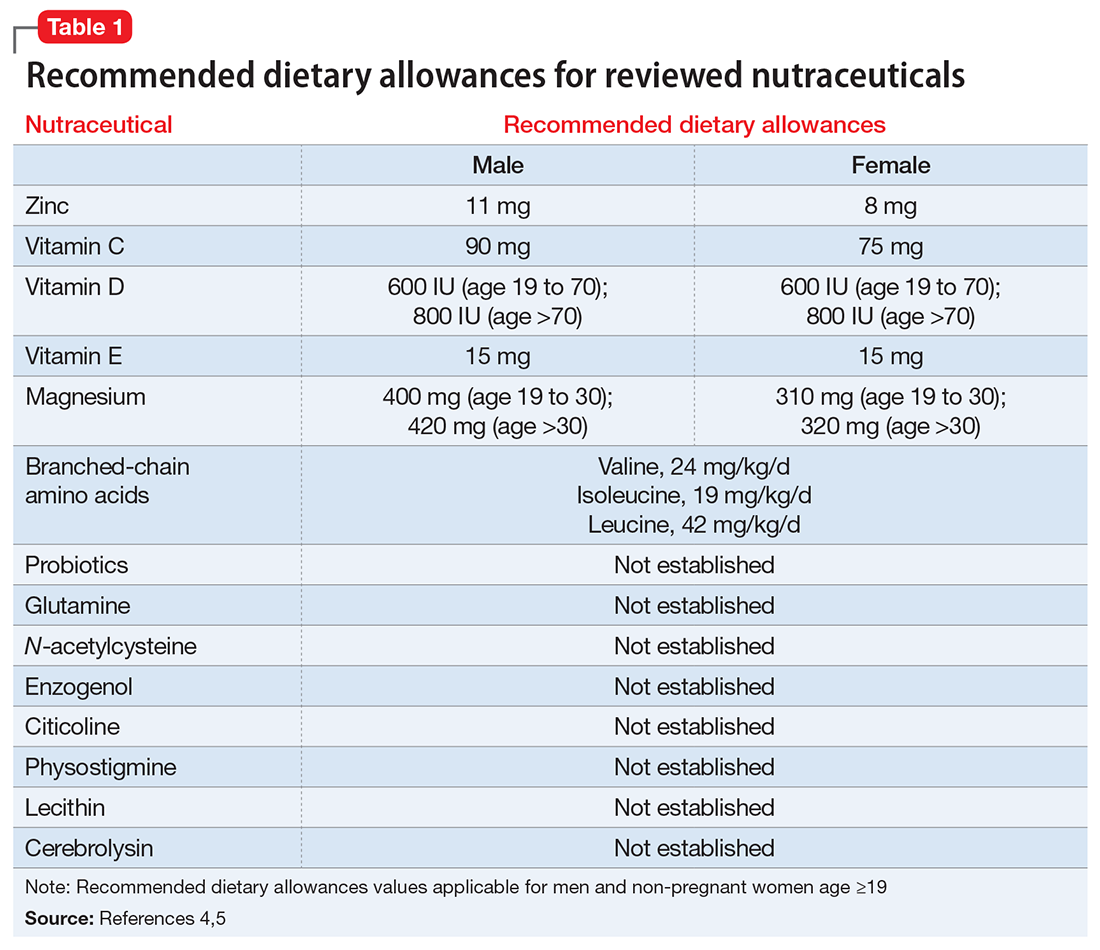
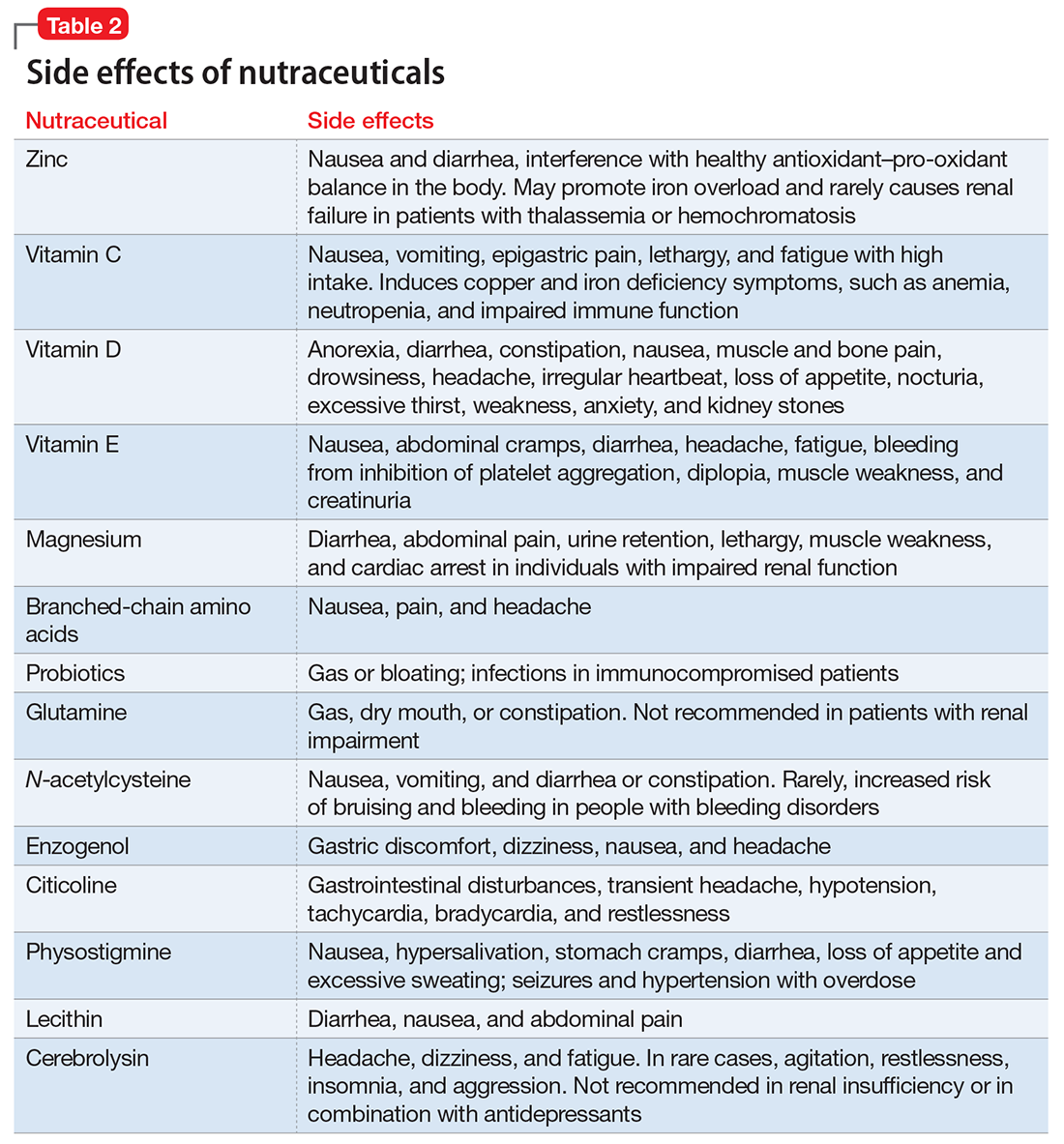
- evaluate the existing evidence, including recommended dietary allowances (Table 1)4,5 and side effects (Table 2)
- review recommendations for their clinical use.
Pharmacologic approaches are limited
Nutraceuticals have gained attention for managing TBI-associated neuropsychiatric disorders because of the limited evidence supporting current approaches. Existing strategies encompass pharmacologic and non-pharmacologic interventions, psychoeducation, supportive and behavioral psychotherapies, and cognitive rehabilitation.6
Many pharmacologic options exist for specific neurobehavioral symptoms, but the evidence for their use is based on small studies, case reports, and knowledge extrapolated from their use in idiopathic psychiatric disorders.7,8 No FDA-approved drugs have been effective for treating neuropsychiatric disturbances after a TBI. Off-label use of antidepressants, anticonvulsants, dopaminergic agents, and cholinesterase inhibitors in TBI has been associated with inadequate clinical response and/or intolerable side effects.9,10
What are nutraceuticals?
DeFelice11 introduced the term “nutraceutical” to refer to “any substance that is a food or part of a food and provides medical or health benefits, including the prevention and treatment of disease.” The term has been expanded to include dietary supplements, such as vitamins, minerals, amino acids, herbal or other botanicals, and food products that provide health benefits beyond what they normally provide in food form. The FDA does not regulate the marketing or manufacturing of nutraceuticals; therefore, their bioavailability and metabolism can vary.
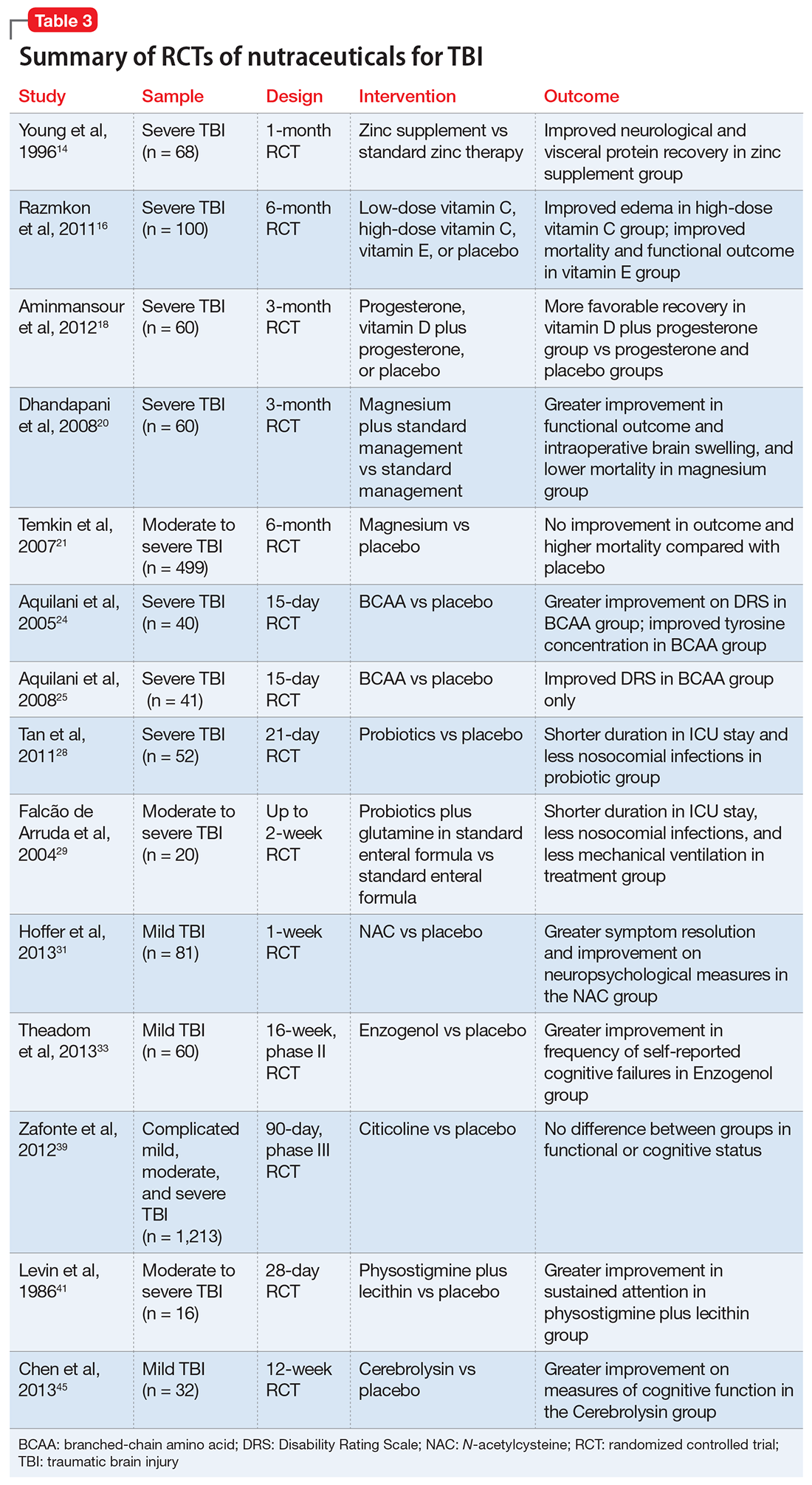
Despite their widespread use, the evidence supporting the efficacy of nutraceuticals for patients with TBI is limited. Their effects might vary by population and depend on dose, timing, TBI severity, and whether taken alone or in combination with other nutraceutical or pharmaceutical agents. Fourteen randomized controlled trials (RCTs) have addressed the use of nutraceuticals in TBI (Table 3), but further research is needed to clarify for which conditions they provide maximum benefit.
Nutraceuticals and their potential use in TBI
Zinc is considered essential for optimal CNS functioning. Patients with TBI might be at risk for zinc deficiency, which has been associated with increased cell death and behavioral deficits.12,13 A randomized, prospective, double-blinded controlled trial examined the effects of supplemental zinc administration (12 mg for 15 days) compared with standard zinc therapy (2.5 mg for 15 days) over 1 month in 68 adults with acute severe closed head injury.14 The supplemental zinc group showed improved visceral protein levels, lower mortality, and more favorable neurologic recovery based on higher adjusted mean Glasgow Coma Scale score on day 28 and mean motor score on days 15 and 21.
Rodent studies have shown that zinc supplementation could reduce deficits in spatial learning and memory and depression-like behaviors and help decrease stress and anxiety,12 although no human clinical trials have been conducted. Despite the potential neuroprotective effects of zinc supplementation, evidence exists that endogenous zinc release and accumulation following TBI can trigger cellular changes that result in neuronal death.13
Vitamins C and E. Oxidative damage is believed to play a significant role in secondary injury in TBI, so research has focused on the role of antioxidants, such as vitamins C and E, to promote post-TBI recovery.15 One RCT16 of 100 adults with acute severe head injury reported that vitamin E administration was associated with reduced mortality and lower Glasgow Outcome Scale (GOS) scores, and vitamin C was associated with stabilized or reduced perilesional edema/infarct on CT scan.
Vitamin D. An animal study reported that vitamin D supplementation can help reduce inflammation, oxidative stress, and cell death in TBI, and that vitamin D deficiency has been associated with increased inflammation and behavioral deficits.17 Further evidence suggests that vitamin D may have a synergistic effect when used in combination with the hormone progesterone. A RCT of 60 patients with severe TBI reported that 60% of those who received progesterone plus vitamin D had GOS scores of 4 (good recovery) or 5 (moderate disability) vs 45% receiving progesterone alone or 25% receiving placebo.18
Magnesium, one of the most widely used nutraceuticals, is considered essential for CNS functioning, including the regulation of N-methyl-
A RCT evaluated the safety and efficacy of magnesium supplementation in 60 patients with severe closed TBI, with one-half randomized to standard care and the other also receiving magnesium sulfate (MgSO4; initiation dose of 4 g IV and 10 g IM, continuation dose of 5 g IM every 4 hours for 24 hours).20 After 3 months, more patients in the MgSO4 group had higher GOS scores than controls (73.3% vs 40%), lower 1-month mortality rates (13.3% vs 43.3%), and lower rates of intraoperative brain swelling (29.4% vs 73.3%).
However, a larger RCT of 499 patients with moderate or severe TBI randomized to high-dose (1.25 to 2.5 mmol/L) or low-dose (1.0 to 1.85 mmol/L) IV MgSO4 or placebo provided conflicting results.21 Participants received MgSO4 8 hours after injury and continued for 5 days. After 6 months, patients in the high-dose MgSO4 and placebo groups had similar composite primary outcome measures (eg, seizures, neuropsychological measures, functional status measures), although the high-dose group had a higher mortality rate than the placebo group. Patients who received low-dose MgSO4 showed worse outcomes than those assigned to placebo.
Amino acids. Branched-chain amino acids (BCAAs), including valine, isoleucine, and leucine, are essential in protein and neurotransmitter synthesis. Reduced levels of endogenous BCAAs have been reported in patients with mild or severe TBI.22 Preclinical studies suggest that BCAAs can improve hippocampal-dependent cognitive functioning following TBI.23
Two RCTs of BCAAs have been conducted in humans. One study randomized 40 men with severe TBI to IV BCAAs or placebo.24 After 15 days, the BCAA group showed greater improvement in Disability Rating Scale scores. The study also found that supplementation increased total BCAA levels without negatively affecting plasma levels of neurotransmitter precursors tyrosine and tryptophan. A second study found that 41 patients in a vegetative or minimally conscious state who received BCAA supplementation for 15 days had higher Disability Rating Scale scores than those receiving placebo.25
Probiotics and glutamine. Probiotics are non-pathogenic microorganisms that have been shown to modulate the host’s immune system.26 TBI is associated with immunological changes, including a shift from T-helper type 1 (TH1) cells to T-helper type 2 (TH2) cells that increase susceptibility to infection.27
A RCT of 52 patients with severe TBI suggested a correlation between probiotic administration-modulated cytokine levels and TH1/TH2 balance.28 A 3-times daily probiotic mix of Bifidobacterium longum, Lactobacillus bulgaricus, and Streptococcus thermophilus for 21 days led to shorter average ICU stays (6.8 vs 10.7 days, P = .034) and a decrease in nosocomial infections (34.6% vs 57.7%, P = .095) vs placebo, although the latter difference was not statistically significant.28
A prospective RCT of 20 patients with brain injury29 found a similar impact of early enteral nutrition supplemented with Lactobacillus johnsonii and glutamine, 30 g, vs a standard enteral nutrition formula. The treatment group experienced fewer nosocomial infections (50% vs 100%, P = .03), shorter ICU stays (10 vs 22 days, P < .01), and fewer days on mechanical ventilation (7 vs 14, P = .04). Despite these studies, evidence for the use of glutamine in patients with TBI is scarce and inconclusive.
N-acetylcysteine (NAC) comes from the amino acid L-cysteine. NAC is an effective scavenger of free radicals and improves cerebral microcirculatory blood flow and tissue oxygenation.30 A randomized, double-blind, placebo-controlled study of oral NAC supplementation in 81 active duty service members with mild TBI found NAC had a significant effect on outcomes.31 Oral NAC supplementation led to improved neuropsychological test results, number of mild TBI symptoms, complete symptom resolution by day 7 of treatment compared with placebo, and NAC was well tolerated. Lack of replication studies and generalizability of findings to civilian, moderate, or chronic TBI populations are key limitations of this study.
Proposed mechanisms for the neuroprotective benefit of NAC include its antioxidant and inflammatory activation of cysteine/glutamate exchange, metabotropic glutamate receptor modulation, and glutathione synthesis.32 NAC has poor blood–brain permeability, but the vascular disruption seen in acute TBI might facilitate its delivery to affected neural sites.31 As such, the benefits of NAC in subacute or chronic TBI are questionable.
Neuropsychiatric outcomes of nutraceuticals
Enzogenol. This flavonoid-rich extract from the bark of the Monterey pine tree (Pinus radiata), known by the trade name Enzogenol, reportedly has antioxidant and anti-inflammatory properties that may counter oxidative damage and neuroinflammation following TBI. A phase II trial randomized participants to Enzogenol, 1,000 mg/d, or placebo for 6 weeks, then all participants received Enzogenol for 6 weeks followed by placebo for 4 weeks.33 Enzogenol was well tolerated with few side effects.
Compared with placebo, participants receiving Enzogenol showed no significant change in mood, as measured by the Hospital Anxiety and Depression Scale, and greater improvement in overall cognition as assessed by the Cognitive Failures Questionnaire. However, measures of working memory (digit span, arithmetic, and letter–number sequencing subtests of the Wechsler Adult Intelligence Scale) and episodic memory (California Verbal Learning Test) showed no benefit from Enzogenol.
Citicoline (CDP-choline) is an endogenous compound widely available as a nutraceutical that has been approved for TBI therapy in 59 countries.34 Animal studies indicate that it could possess neuroprotective properties. Proposed mechanisms for such effects have included stabilizing cell membranes, reducing inflammation, reducing the presence of free radicals, or stimulating production of acetylcholine.35,36 A study in rats found that CDP-choline was associated with increased levels of acetylcholine in the hippocampus and neocortex, which may help reduce neurobehavioral deficits.37
A study of 14 adults with mild to moderate closed head injury38 found that patients who received CDP-choline showed a greater reduction in post-concussion symptoms and improvement in recognition memory than controls who received placebo. However, the Citicoline Brain Injury Treatment Trial, a large randomized trial of 1,213 adults with complicated mild, moderate, or severe TBI, reported that CDP-choline did not improve functional and cognitive status.39
Physostigmine and lecithin. The cholinergic system is a key modulatory neurotransmitter system of the brain that mediates conscious awareness, attention, learning, and working memory.40 A double-blind, placebo-controlled study of 16 patients with moderate to severe closed head injury provided inconsistent evidence for the efficacy of physostigmine and lecithin in the treatment of memory and attention disturbances.41The results showed no differences between the physostigmine–lecithin combination vs lecithin alone, although sustained attention on the Continuous Performance Test was more efficient with physostigmine than placebo when the drug condition occurred first in the crossover design. The lack of encouraging data and concerns about its cardiovascular and proconvulsant properties in patients with TBI may explain the dearth of studies with physostigmine.
Cerebrolysin. A peptide preparation produced from purified pig brain proteins, known by the trade name Cerebrolysin, is popular in Asia and Europe for its nootropic properties. Cerebrolysin may activate cerebral mechanisms related to attention and memory processes,42 and some data have shown efficacy in improving cognitive symptoms and daily activities in patients with Alzheimer’s disease43 and TBI.44
A blinded 12-week study of 32 participants with acute mild TBI reported that those randomized to Cerebrolysin showed improvement in cognitive functioning vs the placebo group.45 The authors concluded that Cerebrolysin provides an advantage for patients with mild TBI and brain contusion if treatment starts within 24 hours of mild TBI onset. Cerebrolysin was well tolerated. Major limitations of this study were small sample size, lack of information regarding comorbid neuropsychiatric conditions and treatments, and short treatment duration.
A recent Cochrane review of 6 RCTs with 1,501 participants found no clinical benefit of Cerebrolysin for treating acute ischemic stroke, and found moderate-quality evidence of an increase with non-fatal serious adverse events but not in total serious adverse events.46 We do not recommend Cerebrolysin use in patients with TBI at this time until additional efficacy and safety data are available.
Nutraceuticals used in other populations
Other nutraceuticals with preclinical evidence of possible benefit in TBI but lacking evidence from human clinical trials include omega-3 fatty acids,47 curcumin,48 and resveratrol,49 providing further proof that results from experimental studies do not necessarily extend to clinical trials.50
Studies of nutraceuticals in other neurological and psychiatric populations have yielded some promising results. Significant interest has focused on the association between vitamin D deficiency, dementia, and neurodegenerative conditions such as Alzheimer’s disease, multiple sclerosis, and Parkinson’s disease.51 The role of vitamin D in regulation of calcium-mediated neuronal excitotoxicity and oxidative stress and in the induction of synaptic structural proteins, neurotrophic factors, and deficient neurotransmitters makes it an attractive candidate as a neuroprotective agent.52
RCTs of nutraceuticals also have reported positive findings for a variety of mood and anxiety disorders, such as St. John’s wort, S-adenosylmethionine, omega-3 fatty acids for major depression53 and bipolar depression,54 and kava for generalized anxiety disorder.55 More research, however, is needed in these areas.
The use of nonpharmacologic agents in TBI often relies on similar neuropsychiatric symptom profiles of idiopathic psychiatric disorders. Attention-deficit/hyperactivity disorder (ADHD) closely resembles TBI, but systemic reviews of studies of zinc, magnesium, and polyunsaturated fatty acids supplementation in ADHD provide no evidence of therapeutic benefit.56-58
Educate patients in role of nutraceuticals
Despite lack of FDA oversight and limited empirical support, nutraceuticals continue to be widely marketed and used for their putative health benefits59 and have gained increased attention among clinicians.60 Because nutritional deficiency may make the brain less able than other organs to recover from injury,61 supplementation is an option, especially in individuals who could be at greater risk of TBI (eg, athletes and military personnel).
Lacking robust scientific evidence to support the use of nutraceuticals either for enhancing TBI recovery or treating neuropsychiatric disturbances, clinicians must educate patients that these agents are not completely benign and can have significant side effects and drug interactions.62,63 Nutraceuticals may contain multiple ingredients, some of which can be toxic, particularly at higher doses. Many patients may not volunteer information about their nutraceutical use to their health care providers,64 so we must ask them about that and inform them of the potential for adverse events and drug interactions.
1. Centers for Disease Control and Prevention. Report to Congress on traumatic brain injury in the United States: epidemiology and rehabilitation. https://www.cdc.gov/traumaticbraininjury/pubs/congress_epi_rehab.html. Updated January 22, 2016. Accessed June 5, 2017.
2. Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99(1):4-9.
3. Vaishnavi S, Rao V, Fann JR. Neuropsychiatric problems after traumatic brain injury: unraveling the silent epidemic. Psychosomatics. 2009;50(3):198-205.
4. National Institutes of Health Office of Dietary Supplements. Dietary supplement fact sheets. https://ods.od.nih.gov/factsheets/list-all. Accessed June 5, 2017.
5. Institute of Medicine, Food and Nutrition Board. Dietary reference intakes for energy, carbohydrate, fiber, fat, fatty acids, cholesterol, protein, and amino acids. Washington, DC: National Academy of Sciences; 2002.
6. Rao V, Koliatsos V, Ahmed F, et al. Neuropsychiatric disturbances associated with traumatic brain injury: a practical approach to evaluation and management. Semin Neurol. 2015;35(1):64-82.
7. Chew E, Zafonte RD. Pharmacological management of neurobehavioral disorders following traumatic brain injury—a state-of-the-art review. J Rehabil Res Dev. 2009;46(6):851-879.
8. Petraglia AL, Maroon JC, Bailes JE. From the field of play to the field of combat: a review of the pharmacological management of concussion. Neurosurgery. 2012;70(6):1520-1533; discussion 1533.
9. Bengtsson M, Godbolt AK. Effects of acetylcholinesterase inhibitors on cognitive function in patients with chronic traumatic brain injury: a systematic review. J Rehabil Med. 2016;48(1):1-5.
10. Neurobehavioral Guidelines Working Group; Warden DL, Gordon B, McAllister TW, et al. Guidelines for the pharmacologic treatment of neurobehavioral sequelae of traumatic brain injury. J Neurotrauma. 2006;23(10):1468-1501.
11. DeFelice SL. The nutraceutical revolution: its impact on food industry R&D. Trends Food Sci Technol. 1995;6(2):59-61.
12. Cope EC, Morris DR, Levenson CW. Improving treatments and outcomes: an emerging role for zinc in traumatic brain injury. Nutr Rev. 2012;70(7):410-413.
13. Morris DR, Levenson CW. Zinc in traumatic brain injury: from neuroprotection to neurotoxicity. Curr Opin Clin Nutr Metab Care. 2013;16(6):708-711.
14. Young B, Ott L, Kasarskis E, et al. Zinc supplementation is associated with improved neurologic recovery rate and visceral protein levels of patients with severe closed head injury. J Neurotrauma. 1996;13(1):25-34.
15. Fernández-Gajardo R, Matamala JM, Carrasco R, et al. Novel therapeutic strategies for traumatic brain injury: acute antioxidant reinforcement. CNS Drugs. 2014;28(3):229-248.
16. Razmkon A, Sadidi A, Sherafat-Kazemzadeh E, et al. Administration of vitamin C and vitamin E in severe head injury: a randomized double-blind controlled trial. Clin Neurosurg. 2011;58:133-137.
17. Cekic M, Cutler SM, VanLandingham JW, et al. Vitamin D deficiency reduces the benefits of progesterone treatment after brain injury in aged rats. Neurobiol Aging. 2011;32(5):864-874.
18. Aminmansour B, Nikbakht H, Ghorbani A, et al. Comparison of the administration of progesterone versus progesterone and vitamin D in improvement of outcomes in patients with traumatic brain injury: a randomized clinical trial with placebo group. Adv Biomed Res. 2012;1:58.
19. Cernak I, Savic VJ, Kotur J, et al. Characterization of plasma magnesium concentration and oxidative stress following graded traumatic brain injury in humans. J Neurotrauma. 2000;17(1):53-68.
20. Dhandapani SS, Gupta A, Vivekanandhan S, et al. Randomized controlled trial of magnesium sulphate in severe closed traumatic brain injury. The Indian Journal of Neurotrauma. 2008;5(1):27-33.
21. Temkin NR, Anderson GD, Winn HR, et al. Magnesium sulfate for neuroprotection after traumatic brain injury: a randomised controlled trial. Lancet Neurol. 2007;6(1):29-38.
22. Jeter CB, Hergenroeder GW, Ward NH 3rd, et al. Human mild traumatic brain injury decreases circulating branched-chain amino acids and their metabolite levels. J Neurotrauma. 2013;30(8):671-679.
23. Cole JT, Mitala CM, Kundu S, et al. Dietary branched chain amino acids ameliorate injury-induced cognitive impairment. Proc Natl Acad Sci U S A. 2010;107(1):366-371.
24. Aquilani R, Iadarola P, Contardi A, et al. Branched-chain amino acids enhance the cognitive recovery of patients with severe traumatic brain injury. Arch Phys Med Rehabil. 2005;86(9):1729-1735.
25. Aquilani R, Boselli M, Boschi F, et al. Branched-chain amino acids may improve recovery from a vegetative or minimally conscious state in patients with traumatic brain injury: a pilot study. Arch Phys Med Rehabil. 2008;89(9):1642-1647.
26. Kang HJ, Im SH. Probiotics as an immune modulator. J Nutr Sci Vitaminol (Tokyo). 2015;61(suppl):S103-S105.
27. DiPiro JT, Howdieshell TR, Goddard JK, et al. Association of interleukin-4 plasma levels with traumatic injury and clinical course. Arch Surg. 1995;130(11):1159-1162; discussion 1162-1163.
28. Tan M, Zhu JC, Du J, et al. Effects of probiotics on serum levels of Th1/Th2 cytokine and clinical outcomes in severe traumatic brain-injured patients: a prospective randomized pilot study. Crit Care. 2011;15(6):R290.
29. Falcão de Arruda IS, de Aguilar-Nascimento JE. Benefits of early enteral nutrition with glutamine and probiotics in brain injury patients. Clin Sci (Lond). 2004;106(3):287-292.
30. Cuzzocrea S, Mazzon E, Costantino G, et al. Beneficial effects of n-acetylcysteine on ischaemic brain injury. Br J Pharmacol. 2000;130(6):1219-1226.
31. Hoffer ME, Balaban C, Slade MD, et al. Amelioration of acute sequelae of blast induced mild traumatic brain injury by N-acetyl cysteine: a double-blind, placebo controlled study. PLoS One. 2013;8(1):e54163.
32. Eakin K, Baratz-Goldstein R, Pick CG, et al. Efficacy of N-acetyl cysteine in traumatic brain injury. PLoS One. 2014;9(4):e90617.
33. Theadom A, Mahon S, Barker-Collo S, et al. Enzogenol for cognitive functioning in traumatic brain injury: a pilot placebo-controlled RCT. Eur J Neurol. 2013;20(8):1135-1144.
34. Arenth PM, Russell KC, Ricker JH, et al. CDP-choline as a biological supplement during neurorecovery: a focused review. PM R. 2011;3(6 suppl 1):S123-S131.
35. Clark WM. Efficacy of citicoline as an acute stroke treatment. Expert Opin Pharmacother. 2009;10(5):839-846.
36. Guseva MV, Hopkins DM, Scheff SW, et al. Dietary choline supplementation improves behavioral, histological, and neurochemical outcomes in a rat model of traumatic brain injury. J Neurotrauma. 2008;25(8):975-983.
37. Dixon CE, Ma X, Marion DW. Effects of CDP-choline treatment on neurobehavioral deficits after TBI and on hippocampal and neocortical acetylcholine release. J Neurotrauma. 1997;14(3):161-169.
38. Levin HS. Treatment of postconcussional symptoms with CDP-choline. J Neurol Sci. 1991;103(suppl):S39-S42.
39. Zafonte RD, Bagiella E, Ansel BM, et al. Effect of citicoline on functional and cognitive status among patients with traumatic brain injury: Citicoline Brain Injury Treatment Trial (COBRIT). JAMA. 2012;308(19):1993-2000.
40. Perry E, Walker M, Grace J, et al. Acetylcholine in mind: a neurotransmitter correlate of consciousness? Trends Neurosci. 1999;22(6):273-280.
41. Levin HS, Peters BH, Kalisky Z, et al. Effects of oral physostigmine and lecithin on memory and attention in closed head-injured patients. Cent Nerv Syst Trauma. 1986;3(4):333-342.
42. Alvarez XA, Lombardi VR, Corzo L, et al. Oral cerebrolysin enhances brain alpha activity and improves cognitive performance in elderly control subjects. J Neural Transm Suppl. 2000;59:315-328.
43. Ruether E, Husmann R, Kinzler E, et al. A 28-week, double-blind, placebo-controlled study with cerebrolysin in patients with mild to moderate Alzheimer’s disease. Int Clin Psychopharmacol. 2001;16(5):253-263.
44. Wong GK, Zhu XL, Poon WS. Beneficial effect of cerebrolysin on moderate and severe head injury patients: result of a cohort study. Acta Neurochir Suppl. 2005;95:59-60.
45. Chen CC, Wei ST, Tsaia SC, et al. Cerebrolysin enhances cognitive recovery of mild traumatic brain injury patients: double-blind, placebo-controlled, randomized study. Br J Neurosurg. 2013;27(6):803-807.
46. Ziganshina LE, Abakumova T, Vernay L. Cerebrolysin for acute ischaemic stroke. Cochrane Database Syst Rev. 2016;12:CD007026.
47. Barrett EC, McBurney MI, Ciappio ED. ω-3 fatty acid supplementation as a potential therapeutic aid for the recovery from mild traumatic brain injury/concussion. Adv Nutr. 2014;5(3):268-277.
48. Sharma S, Zhuang Y, Ying Z, et al. Dietary curcumin supplementation counteracts reduction in levels of molecules involved in energy homeostasis after brain trauma. Neuroscience. 2009;161(4):1037-1044.
49. Gatson JW, Liu MM, Abdelfattah K, et al. Resveratrol decreases inflammation in the brain of mice with mild traumatic brain injury. J Trauma Acute Care Surg. 2013;74(2):470-475; discussion 474-475.
50. Grey A, Bolland M. Clinical trial evidence and use of fish oil supplements. JAMA Intern Med. 2014;174(3):460-462.
51. Mpandzou G, Aït Ben Haddou E, Regragui W, et al. Vitamin D deficiency and its role in neurological conditions: a review. Rev Neurol (Paris). 2016;172(2):109-122.
52. Karakis I, Pase MP, Beiser A, et al. Association of serum vitamin D with the risk of incident dementia and subclinical indices of brain aging: The Framingham Heart Study. J Alzheimers Dis. 2016;51(2):451-461.
53. Sarris J, Papakostas GI, Vitolo O, et al. S-adenosyl methionine (SAMe) versus escitalopram and placebo in major depression RCT: efficacy and effects of histamine and carnitine as moderators of response. J Affect Disord. 2014;164:76-81.
54. Sarris J, Mischoulon D, Schweitzer I. Omega-3 for bipolar disorder: meta-analyses of use in mania and bipolar depression. J Clin Psychiatry. 2012;73(1):81-86.
55. Sarris J, Stough C, Bousman C, et al. Kava in the treatment of generalized anxiety disorder: a double-blind, randomized, placebo-controlled study. J Clin Psychopharmacol. 2013;33(5):643-648.
56. Hariri M, Azadbakht L. Magnesium, iron, and zinc supplementation for the treatment of attention deficit hyperactivity disorder: a systematic review on the recent literature. Int J Prev Med. 2015;6:83.
57. Gillies D, Sinn JKh, Lad SS, et al. Polyunsaturated fatty acids (PUFA) for attention deficit hyperactivity disorder (ADHD) in children and adolescents. Cochrane Database Syst Rev. 2012;7:CD007986.
58. Ghanizadeh A, Berk M. Zinc for treating of children and adolescents with attention-deficit hyperactivity disorder: a systematic review of randomized controlled clinical trials. Eur J Clin Nutr. 2013;67(1):122-124.
59. U.S. Food and Drug Administration. Can a dietary supplement treat a concussion? No! http://www.fda.gov/forconsumers/consumerupdates/ucm378845.htm. Updated February 13, 2015. Accessed June 5, 2017.
60. Sarris J, Logan AC, Akbaraly TN, et al; International Society for Nutritional Psychiatry Research. Nutritional medicine as mainstream in psychiatry. Lancet Psychiatry. 2015;2(3):271-274.
61. Desai A, Kevala K, Kim HY. Depletion of brain docosahexaenoic acid impairs recovery from traumatic brain injury. PLoS One. 2014;9(1):e86472.
62. Edie CF, Dewan N. Which psychotropics interact with four common supplements. Current Psychiatry. 2005;4(1):16-30.
63. Di Lorenzo C, Ceschi A, Kupferschmidt H, et al. Adverse effects of plant food supplements and botanical preparations: a systematic review with critical evaluation of causality. Br J Clin Pharmacol. 2015;79(4):578-592.
64. National Center for Complementary and Integrative Health. Complementary and alternative medicine: what people aged 50 and older discuss with their health care providers. https://nccih.nih.gov/research/statistics/2010. Published 2011. Accessed June 5, 2017.
Traumatic brain injury (TBI) affects more than 2 million people in the United States each year.1 TBI can trigger a cascade of secondary injury mechanisms, such as inflammation, hypoxic/ischemic injury, excitotoxicity, and oxidative stress,2 that could contribute to cognitive and behavioral changes. Although neuropsychiatric symptoms might not be obvious after a TBI, they have a high prevalence in these patients, can last long term, and may be difficult to treat.3 Despite research advances in understanding the biological basis of TBI and identifying potential therapeutic targets, treatment options for individuals with TBI remain limited.
As a result, clinicians have turned to alternative treatments for TBI, including nutraceuticals. In this article, we will:
- provide an overview of nutraceuticals used in treating TBI, first exploring outcomes soon after TBI, then concentrating on neuropsychiatric outcomes
- evaluate the existing evidence, including recommended dietary allowances (Table 1)4,5 and side effects (Table 2)
- review recommendations for their clinical use.
Pharmacologic approaches are limited
Nutraceuticals have gained attention for managing TBI-associated neuropsychiatric disorders because of the limited evidence supporting current approaches. Existing strategies encompass pharmacologic and non-pharmacologic interventions, psychoeducation, supportive and behavioral psychotherapies, and cognitive rehabilitation.6
Many pharmacologic options exist for specific neurobehavioral symptoms, but the evidence for their use is based on small studies, case reports, and knowledge extrapolated from their use in idiopathic psychiatric disorders.7,8 No FDA-approved drugs have been effective for treating neuropsychiatric disturbances after a TBI. Off-label use of antidepressants, anticonvulsants, dopaminergic agents, and cholinesterase inhibitors in TBI has been associated with inadequate clinical response and/or intolerable side effects.9,10
What are nutraceuticals?
DeFelice11 introduced the term “nutraceutical” to refer to “any substance that is a food or part of a food and provides medical or health benefits, including the prevention and treatment of disease.” The term has been expanded to include dietary supplements, such as vitamins, minerals, amino acids, herbal or other botanicals, and food products that provide health benefits beyond what they normally provide in food form. The FDA does not regulate the marketing or manufacturing of nutraceuticals; therefore, their bioavailability and metabolism can vary.
Despite their widespread use, the evidence supporting the efficacy of nutraceuticals for patients with TBI is limited. Their effects might vary by population and depend on dose, timing, TBI severity, and whether taken alone or in combination with other nutraceutical or pharmaceutical agents. Fourteen randomized controlled trials (RCTs) have addressed the use of nutraceuticals in TBI (Table 3), but further research is needed to clarify for which conditions they provide maximum benefit.
Nutraceuticals and their potential use in TBI
Zinc is considered essential for optimal CNS functioning. Patients with TBI might be at risk for zinc deficiency, which has been associated with increased cell death and behavioral deficits.12,13 A randomized, prospective, double-blinded controlled trial examined the effects of supplemental zinc administration (12 mg for 15 days) compared with standard zinc therapy (2.5 mg for 15 days) over 1 month in 68 adults with acute severe closed head injury.14 The supplemental zinc group showed improved visceral protein levels, lower mortality, and more favorable neurologic recovery based on higher adjusted mean Glasgow Coma Scale score on day 28 and mean motor score on days 15 and 21.
Rodent studies have shown that zinc supplementation could reduce deficits in spatial learning and memory and depression-like behaviors and help decrease stress and anxiety,12 although no human clinical trials have been conducted. Despite the potential neuroprotective effects of zinc supplementation, evidence exists that endogenous zinc release and accumulation following TBI can trigger cellular changes that result in neuronal death.13
Vitamins C and E. Oxidative damage is believed to play a significant role in secondary injury in TBI, so research has focused on the role of antioxidants, such as vitamins C and E, to promote post-TBI recovery.15 One RCT16 of 100 adults with acute severe head injury reported that vitamin E administration was associated with reduced mortality and lower Glasgow Outcome Scale (GOS) scores, and vitamin C was associated with stabilized or reduced perilesional edema/infarct on CT scan.
Vitamin D. An animal study reported that vitamin D supplementation can help reduce inflammation, oxidative stress, and cell death in TBI, and that vitamin D deficiency has been associated with increased inflammation and behavioral deficits.17 Further evidence suggests that vitamin D may have a synergistic effect when used in combination with the hormone progesterone. A RCT of 60 patients with severe TBI reported that 60% of those who received progesterone plus vitamin D had GOS scores of 4 (good recovery) or 5 (moderate disability) vs 45% receiving progesterone alone or 25% receiving placebo.18
Magnesium, one of the most widely used nutraceuticals, is considered essential for CNS functioning, including the regulation of N-methyl-
A RCT evaluated the safety and efficacy of magnesium supplementation in 60 patients with severe closed TBI, with one-half randomized to standard care and the other also receiving magnesium sulfate (MgSO4; initiation dose of 4 g IV and 10 g IM, continuation dose of 5 g IM every 4 hours for 24 hours).20 After 3 months, more patients in the MgSO4 group had higher GOS scores than controls (73.3% vs 40%), lower 1-month mortality rates (13.3% vs 43.3%), and lower rates of intraoperative brain swelling (29.4% vs 73.3%).
However, a larger RCT of 499 patients with moderate or severe TBI randomized to high-dose (1.25 to 2.5 mmol/L) or low-dose (1.0 to 1.85 mmol/L) IV MgSO4 or placebo provided conflicting results.21 Participants received MgSO4 8 hours after injury and continued for 5 days. After 6 months, patients in the high-dose MgSO4 and placebo groups had similar composite primary outcome measures (eg, seizures, neuropsychological measures, functional status measures), although the high-dose group had a higher mortality rate than the placebo group. Patients who received low-dose MgSO4 showed worse outcomes than those assigned to placebo.
Amino acids. Branched-chain amino acids (BCAAs), including valine, isoleucine, and leucine, are essential in protein and neurotransmitter synthesis. Reduced levels of endogenous BCAAs have been reported in patients with mild or severe TBI.22 Preclinical studies suggest that BCAAs can improve hippocampal-dependent cognitive functioning following TBI.23
Two RCTs of BCAAs have been conducted in humans. One study randomized 40 men with severe TBI to IV BCAAs or placebo.24 After 15 days, the BCAA group showed greater improvement in Disability Rating Scale scores. The study also found that supplementation increased total BCAA levels without negatively affecting plasma levels of neurotransmitter precursors tyrosine and tryptophan. A second study found that 41 patients in a vegetative or minimally conscious state who received BCAA supplementation for 15 days had higher Disability Rating Scale scores than those receiving placebo.25
Probiotics and glutamine. Probiotics are non-pathogenic microorganisms that have been shown to modulate the host’s immune system.26 TBI is associated with immunological changes, including a shift from T-helper type 1 (TH1) cells to T-helper type 2 (TH2) cells that increase susceptibility to infection.27
A RCT of 52 patients with severe TBI suggested a correlation between probiotic administration-modulated cytokine levels and TH1/TH2 balance.28 A 3-times daily probiotic mix of Bifidobacterium longum, Lactobacillus bulgaricus, and Streptococcus thermophilus for 21 days led to shorter average ICU stays (6.8 vs 10.7 days, P = .034) and a decrease in nosocomial infections (34.6% vs 57.7%, P = .095) vs placebo, although the latter difference was not statistically significant.28
A prospective RCT of 20 patients with brain injury29 found a similar impact of early enteral nutrition supplemented with Lactobacillus johnsonii and glutamine, 30 g, vs a standard enteral nutrition formula. The treatment group experienced fewer nosocomial infections (50% vs 100%, P = .03), shorter ICU stays (10 vs 22 days, P < .01), and fewer days on mechanical ventilation (7 vs 14, P = .04). Despite these studies, evidence for the use of glutamine in patients with TBI is scarce and inconclusive.
N-acetylcysteine (NAC) comes from the amino acid L-cysteine. NAC is an effective scavenger of free radicals and improves cerebral microcirculatory blood flow and tissue oxygenation.30 A randomized, double-blind, placebo-controlled study of oral NAC supplementation in 81 active duty service members with mild TBI found NAC had a significant effect on outcomes.31 Oral NAC supplementation led to improved neuropsychological test results, number of mild TBI symptoms, complete symptom resolution by day 7 of treatment compared with placebo, and NAC was well tolerated. Lack of replication studies and generalizability of findings to civilian, moderate, or chronic TBI populations are key limitations of this study.
Proposed mechanisms for the neuroprotective benefit of NAC include its antioxidant and inflammatory activation of cysteine/glutamate exchange, metabotropic glutamate receptor modulation, and glutathione synthesis.32 NAC has poor blood–brain permeability, but the vascular disruption seen in acute TBI might facilitate its delivery to affected neural sites.31 As such, the benefits of NAC in subacute or chronic TBI are questionable.
Neuropsychiatric outcomes of nutraceuticals
Enzogenol. This flavonoid-rich extract from the bark of the Monterey pine tree (Pinus radiata), known by the trade name Enzogenol, reportedly has antioxidant and anti-inflammatory properties that may counter oxidative damage and neuroinflammation following TBI. A phase II trial randomized participants to Enzogenol, 1,000 mg/d, or placebo for 6 weeks, then all participants received Enzogenol for 6 weeks followed by placebo for 4 weeks.33 Enzogenol was well tolerated with few side effects.
Compared with placebo, participants receiving Enzogenol showed no significant change in mood, as measured by the Hospital Anxiety and Depression Scale, and greater improvement in overall cognition as assessed by the Cognitive Failures Questionnaire. However, measures of working memory (digit span, arithmetic, and letter–number sequencing subtests of the Wechsler Adult Intelligence Scale) and episodic memory (California Verbal Learning Test) showed no benefit from Enzogenol.
Citicoline (CDP-choline) is an endogenous compound widely available as a nutraceutical that has been approved for TBI therapy in 59 countries.34 Animal studies indicate that it could possess neuroprotective properties. Proposed mechanisms for such effects have included stabilizing cell membranes, reducing inflammation, reducing the presence of free radicals, or stimulating production of acetylcholine.35,36 A study in rats found that CDP-choline was associated with increased levels of acetylcholine in the hippocampus and neocortex, which may help reduce neurobehavioral deficits.37
A study of 14 adults with mild to moderate closed head injury38 found that patients who received CDP-choline showed a greater reduction in post-concussion symptoms and improvement in recognition memory than controls who received placebo. However, the Citicoline Brain Injury Treatment Trial, a large randomized trial of 1,213 adults with complicated mild, moderate, or severe TBI, reported that CDP-choline did not improve functional and cognitive status.39
Physostigmine and lecithin. The cholinergic system is a key modulatory neurotransmitter system of the brain that mediates conscious awareness, attention, learning, and working memory.40 A double-blind, placebo-controlled study of 16 patients with moderate to severe closed head injury provided inconsistent evidence for the efficacy of physostigmine and lecithin in the treatment of memory and attention disturbances.41The results showed no differences between the physostigmine–lecithin combination vs lecithin alone, although sustained attention on the Continuous Performance Test was more efficient with physostigmine than placebo when the drug condition occurred first in the crossover design. The lack of encouraging data and concerns about its cardiovascular and proconvulsant properties in patients with TBI may explain the dearth of studies with physostigmine.
Cerebrolysin. A peptide preparation produced from purified pig brain proteins, known by the trade name Cerebrolysin, is popular in Asia and Europe for its nootropic properties. Cerebrolysin may activate cerebral mechanisms related to attention and memory processes,42 and some data have shown efficacy in improving cognitive symptoms and daily activities in patients with Alzheimer’s disease43 and TBI.44
A blinded 12-week study of 32 participants with acute mild TBI reported that those randomized to Cerebrolysin showed improvement in cognitive functioning vs the placebo group.45 The authors concluded that Cerebrolysin provides an advantage for patients with mild TBI and brain contusion if treatment starts within 24 hours of mild TBI onset. Cerebrolysin was well tolerated. Major limitations of this study were small sample size, lack of information regarding comorbid neuropsychiatric conditions and treatments, and short treatment duration.
A recent Cochrane review of 6 RCTs with 1,501 participants found no clinical benefit of Cerebrolysin for treating acute ischemic stroke, and found moderate-quality evidence of an increase with non-fatal serious adverse events but not in total serious adverse events.46 We do not recommend Cerebrolysin use in patients with TBI at this time until additional efficacy and safety data are available.
Nutraceuticals used in other populations
Other nutraceuticals with preclinical evidence of possible benefit in TBI but lacking evidence from human clinical trials include omega-3 fatty acids,47 curcumin,48 and resveratrol,49 providing further proof that results from experimental studies do not necessarily extend to clinical trials.50
Studies of nutraceuticals in other neurological and psychiatric populations have yielded some promising results. Significant interest has focused on the association between vitamin D deficiency, dementia, and neurodegenerative conditions such as Alzheimer’s disease, multiple sclerosis, and Parkinson’s disease.51 The role of vitamin D in regulation of calcium-mediated neuronal excitotoxicity and oxidative stress and in the induction of synaptic structural proteins, neurotrophic factors, and deficient neurotransmitters makes it an attractive candidate as a neuroprotective agent.52
RCTs of nutraceuticals also have reported positive findings for a variety of mood and anxiety disorders, such as St. John’s wort, S-adenosylmethionine, omega-3 fatty acids for major depression53 and bipolar depression,54 and kava for generalized anxiety disorder.55 More research, however, is needed in these areas.
The use of nonpharmacologic agents in TBI often relies on similar neuropsychiatric symptom profiles of idiopathic psychiatric disorders. Attention-deficit/hyperactivity disorder (ADHD) closely resembles TBI, but systemic reviews of studies of zinc, magnesium, and polyunsaturated fatty acids supplementation in ADHD provide no evidence of therapeutic benefit.56-58
Educate patients in role of nutraceuticals
Despite lack of FDA oversight and limited empirical support, nutraceuticals continue to be widely marketed and used for their putative health benefits59 and have gained increased attention among clinicians.60 Because nutritional deficiency may make the brain less able than other organs to recover from injury,61 supplementation is an option, especially in individuals who could be at greater risk of TBI (eg, athletes and military personnel).
Lacking robust scientific evidence to support the use of nutraceuticals either for enhancing TBI recovery or treating neuropsychiatric disturbances, clinicians must educate patients that these agents are not completely benign and can have significant side effects and drug interactions.62,63 Nutraceuticals may contain multiple ingredients, some of which can be toxic, particularly at higher doses. Many patients may not volunteer information about their nutraceutical use to their health care providers,64 so we must ask them about that and inform them of the potential for adverse events and drug interactions.
Traumatic brain injury (TBI) affects more than 2 million people in the United States each year.1 TBI can trigger a cascade of secondary injury mechanisms, such as inflammation, hypoxic/ischemic injury, excitotoxicity, and oxidative stress,2 that could contribute to cognitive and behavioral changes. Although neuropsychiatric symptoms might not be obvious after a TBI, they have a high prevalence in these patients, can last long term, and may be difficult to treat.3 Despite research advances in understanding the biological basis of TBI and identifying potential therapeutic targets, treatment options for individuals with TBI remain limited.
As a result, clinicians have turned to alternative treatments for TBI, including nutraceuticals. In this article, we will:
- provide an overview of nutraceuticals used in treating TBI, first exploring outcomes soon after TBI, then concentrating on neuropsychiatric outcomes
- evaluate the existing evidence, including recommended dietary allowances (Table 1)4,5 and side effects (Table 2)
- review recommendations for their clinical use.
Pharmacologic approaches are limited
Nutraceuticals have gained attention for managing TBI-associated neuropsychiatric disorders because of the limited evidence supporting current approaches. Existing strategies encompass pharmacologic and non-pharmacologic interventions, psychoeducation, supportive and behavioral psychotherapies, and cognitive rehabilitation.6
Many pharmacologic options exist for specific neurobehavioral symptoms, but the evidence for their use is based on small studies, case reports, and knowledge extrapolated from their use in idiopathic psychiatric disorders.7,8 No FDA-approved drugs have been effective for treating neuropsychiatric disturbances after a TBI. Off-label use of antidepressants, anticonvulsants, dopaminergic agents, and cholinesterase inhibitors in TBI has been associated with inadequate clinical response and/or intolerable side effects.9,10
What are nutraceuticals?
DeFelice11 introduced the term “nutraceutical” to refer to “any substance that is a food or part of a food and provides medical or health benefits, including the prevention and treatment of disease.” The term has been expanded to include dietary supplements, such as vitamins, minerals, amino acids, herbal or other botanicals, and food products that provide health benefits beyond what they normally provide in food form. The FDA does not regulate the marketing or manufacturing of nutraceuticals; therefore, their bioavailability and metabolism can vary.
Despite their widespread use, the evidence supporting the efficacy of nutraceuticals for patients with TBI is limited. Their effects might vary by population and depend on dose, timing, TBI severity, and whether taken alone or in combination with other nutraceutical or pharmaceutical agents. Fourteen randomized controlled trials (RCTs) have addressed the use of nutraceuticals in TBI (Table 3), but further research is needed to clarify for which conditions they provide maximum benefit.
Nutraceuticals and their potential use in TBI
Zinc is considered essential for optimal CNS functioning. Patients with TBI might be at risk for zinc deficiency, which has been associated with increased cell death and behavioral deficits.12,13 A randomized, prospective, double-blinded controlled trial examined the effects of supplemental zinc administration (12 mg for 15 days) compared with standard zinc therapy (2.5 mg for 15 days) over 1 month in 68 adults with acute severe closed head injury.14 The supplemental zinc group showed improved visceral protein levels, lower mortality, and more favorable neurologic recovery based on higher adjusted mean Glasgow Coma Scale score on day 28 and mean motor score on days 15 and 21.
Rodent studies have shown that zinc supplementation could reduce deficits in spatial learning and memory and depression-like behaviors and help decrease stress and anxiety,12 although no human clinical trials have been conducted. Despite the potential neuroprotective effects of zinc supplementation, evidence exists that endogenous zinc release and accumulation following TBI can trigger cellular changes that result in neuronal death.13
Vitamins C and E. Oxidative damage is believed to play a significant role in secondary injury in TBI, so research has focused on the role of antioxidants, such as vitamins C and E, to promote post-TBI recovery.15 One RCT16 of 100 adults with acute severe head injury reported that vitamin E administration was associated with reduced mortality and lower Glasgow Outcome Scale (GOS) scores, and vitamin C was associated with stabilized or reduced perilesional edema/infarct on CT scan.
Vitamin D. An animal study reported that vitamin D supplementation can help reduce inflammation, oxidative stress, and cell death in TBI, and that vitamin D deficiency has been associated with increased inflammation and behavioral deficits.17 Further evidence suggests that vitamin D may have a synergistic effect when used in combination with the hormone progesterone. A RCT of 60 patients with severe TBI reported that 60% of those who received progesterone plus vitamin D had GOS scores of 4 (good recovery) or 5 (moderate disability) vs 45% receiving progesterone alone or 25% receiving placebo.18
Magnesium, one of the most widely used nutraceuticals, is considered essential for CNS functioning, including the regulation of N-methyl-
A RCT evaluated the safety and efficacy of magnesium supplementation in 60 patients with severe closed TBI, with one-half randomized to standard care and the other also receiving magnesium sulfate (MgSO4; initiation dose of 4 g IV and 10 g IM, continuation dose of 5 g IM every 4 hours for 24 hours).20 After 3 months, more patients in the MgSO4 group had higher GOS scores than controls (73.3% vs 40%), lower 1-month mortality rates (13.3% vs 43.3%), and lower rates of intraoperative brain swelling (29.4% vs 73.3%).
However, a larger RCT of 499 patients with moderate or severe TBI randomized to high-dose (1.25 to 2.5 mmol/L) or low-dose (1.0 to 1.85 mmol/L) IV MgSO4 or placebo provided conflicting results.21 Participants received MgSO4 8 hours after injury and continued for 5 days. After 6 months, patients in the high-dose MgSO4 and placebo groups had similar composite primary outcome measures (eg, seizures, neuropsychological measures, functional status measures), although the high-dose group had a higher mortality rate than the placebo group. Patients who received low-dose MgSO4 showed worse outcomes than those assigned to placebo.
Amino acids. Branched-chain amino acids (BCAAs), including valine, isoleucine, and leucine, are essential in protein and neurotransmitter synthesis. Reduced levels of endogenous BCAAs have been reported in patients with mild or severe TBI.22 Preclinical studies suggest that BCAAs can improve hippocampal-dependent cognitive functioning following TBI.23
Two RCTs of BCAAs have been conducted in humans. One study randomized 40 men with severe TBI to IV BCAAs or placebo.24 After 15 days, the BCAA group showed greater improvement in Disability Rating Scale scores. The study also found that supplementation increased total BCAA levels without negatively affecting plasma levels of neurotransmitter precursors tyrosine and tryptophan. A second study found that 41 patients in a vegetative or minimally conscious state who received BCAA supplementation for 15 days had higher Disability Rating Scale scores than those receiving placebo.25
Probiotics and glutamine. Probiotics are non-pathogenic microorganisms that have been shown to modulate the host’s immune system.26 TBI is associated with immunological changes, including a shift from T-helper type 1 (TH1) cells to T-helper type 2 (TH2) cells that increase susceptibility to infection.27
A RCT of 52 patients with severe TBI suggested a correlation between probiotic administration-modulated cytokine levels and TH1/TH2 balance.28 A 3-times daily probiotic mix of Bifidobacterium longum, Lactobacillus bulgaricus, and Streptococcus thermophilus for 21 days led to shorter average ICU stays (6.8 vs 10.7 days, P = .034) and a decrease in nosocomial infections (34.6% vs 57.7%, P = .095) vs placebo, although the latter difference was not statistically significant.28
A prospective RCT of 20 patients with brain injury29 found a similar impact of early enteral nutrition supplemented with Lactobacillus johnsonii and glutamine, 30 g, vs a standard enteral nutrition formula. The treatment group experienced fewer nosocomial infections (50% vs 100%, P = .03), shorter ICU stays (10 vs 22 days, P < .01), and fewer days on mechanical ventilation (7 vs 14, P = .04). Despite these studies, evidence for the use of glutamine in patients with TBI is scarce and inconclusive.
N-acetylcysteine (NAC) comes from the amino acid L-cysteine. NAC is an effective scavenger of free radicals and improves cerebral microcirculatory blood flow and tissue oxygenation.30 A randomized, double-blind, placebo-controlled study of oral NAC supplementation in 81 active duty service members with mild TBI found NAC had a significant effect on outcomes.31 Oral NAC supplementation led to improved neuropsychological test results, number of mild TBI symptoms, complete symptom resolution by day 7 of treatment compared with placebo, and NAC was well tolerated. Lack of replication studies and generalizability of findings to civilian, moderate, or chronic TBI populations are key limitations of this study.
Proposed mechanisms for the neuroprotective benefit of NAC include its antioxidant and inflammatory activation of cysteine/glutamate exchange, metabotropic glutamate receptor modulation, and glutathione synthesis.32 NAC has poor blood–brain permeability, but the vascular disruption seen in acute TBI might facilitate its delivery to affected neural sites.31 As such, the benefits of NAC in subacute or chronic TBI are questionable.
Neuropsychiatric outcomes of nutraceuticals
Enzogenol. This flavonoid-rich extract from the bark of the Monterey pine tree (Pinus radiata), known by the trade name Enzogenol, reportedly has antioxidant and anti-inflammatory properties that may counter oxidative damage and neuroinflammation following TBI. A phase II trial randomized participants to Enzogenol, 1,000 mg/d, or placebo for 6 weeks, then all participants received Enzogenol for 6 weeks followed by placebo for 4 weeks.33 Enzogenol was well tolerated with few side effects.
Compared with placebo, participants receiving Enzogenol showed no significant change in mood, as measured by the Hospital Anxiety and Depression Scale, and greater improvement in overall cognition as assessed by the Cognitive Failures Questionnaire. However, measures of working memory (digit span, arithmetic, and letter–number sequencing subtests of the Wechsler Adult Intelligence Scale) and episodic memory (California Verbal Learning Test) showed no benefit from Enzogenol.
Citicoline (CDP-choline) is an endogenous compound widely available as a nutraceutical that has been approved for TBI therapy in 59 countries.34 Animal studies indicate that it could possess neuroprotective properties. Proposed mechanisms for such effects have included stabilizing cell membranes, reducing inflammation, reducing the presence of free radicals, or stimulating production of acetylcholine.35,36 A study in rats found that CDP-choline was associated with increased levels of acetylcholine in the hippocampus and neocortex, which may help reduce neurobehavioral deficits.37
A study of 14 adults with mild to moderate closed head injury38 found that patients who received CDP-choline showed a greater reduction in post-concussion symptoms and improvement in recognition memory than controls who received placebo. However, the Citicoline Brain Injury Treatment Trial, a large randomized trial of 1,213 adults with complicated mild, moderate, or severe TBI, reported that CDP-choline did not improve functional and cognitive status.39
Physostigmine and lecithin. The cholinergic system is a key modulatory neurotransmitter system of the brain that mediates conscious awareness, attention, learning, and working memory.40 A double-blind, placebo-controlled study of 16 patients with moderate to severe closed head injury provided inconsistent evidence for the efficacy of physostigmine and lecithin in the treatment of memory and attention disturbances.41The results showed no differences between the physostigmine–lecithin combination vs lecithin alone, although sustained attention on the Continuous Performance Test was more efficient with physostigmine than placebo when the drug condition occurred first in the crossover design. The lack of encouraging data and concerns about its cardiovascular and proconvulsant properties in patients with TBI may explain the dearth of studies with physostigmine.
Cerebrolysin. A peptide preparation produced from purified pig brain proteins, known by the trade name Cerebrolysin, is popular in Asia and Europe for its nootropic properties. Cerebrolysin may activate cerebral mechanisms related to attention and memory processes,42 and some data have shown efficacy in improving cognitive symptoms and daily activities in patients with Alzheimer’s disease43 and TBI.44
A blinded 12-week study of 32 participants with acute mild TBI reported that those randomized to Cerebrolysin showed improvement in cognitive functioning vs the placebo group.45 The authors concluded that Cerebrolysin provides an advantage for patients with mild TBI and brain contusion if treatment starts within 24 hours of mild TBI onset. Cerebrolysin was well tolerated. Major limitations of this study were small sample size, lack of information regarding comorbid neuropsychiatric conditions and treatments, and short treatment duration.
A recent Cochrane review of 6 RCTs with 1,501 participants found no clinical benefit of Cerebrolysin for treating acute ischemic stroke, and found moderate-quality evidence of an increase with non-fatal serious adverse events but not in total serious adverse events.46 We do not recommend Cerebrolysin use in patients with TBI at this time until additional efficacy and safety data are available.
Nutraceuticals used in other populations
Other nutraceuticals with preclinical evidence of possible benefit in TBI but lacking evidence from human clinical trials include omega-3 fatty acids,47 curcumin,48 and resveratrol,49 providing further proof that results from experimental studies do not necessarily extend to clinical trials.50
Studies of nutraceuticals in other neurological and psychiatric populations have yielded some promising results. Significant interest has focused on the association between vitamin D deficiency, dementia, and neurodegenerative conditions such as Alzheimer’s disease, multiple sclerosis, and Parkinson’s disease.51 The role of vitamin D in regulation of calcium-mediated neuronal excitotoxicity and oxidative stress and in the induction of synaptic structural proteins, neurotrophic factors, and deficient neurotransmitters makes it an attractive candidate as a neuroprotective agent.52
RCTs of nutraceuticals also have reported positive findings for a variety of mood and anxiety disorders, such as St. John’s wort, S-adenosylmethionine, omega-3 fatty acids for major depression53 and bipolar depression,54 and kava for generalized anxiety disorder.55 More research, however, is needed in these areas.
The use of nonpharmacologic agents in TBI often relies on similar neuropsychiatric symptom profiles of idiopathic psychiatric disorders. Attention-deficit/hyperactivity disorder (ADHD) closely resembles TBI, but systemic reviews of studies of zinc, magnesium, and polyunsaturated fatty acids supplementation in ADHD provide no evidence of therapeutic benefit.56-58
Educate patients in role of nutraceuticals
Despite lack of FDA oversight and limited empirical support, nutraceuticals continue to be widely marketed and used for their putative health benefits59 and have gained increased attention among clinicians.60 Because nutritional deficiency may make the brain less able than other organs to recover from injury,61 supplementation is an option, especially in individuals who could be at greater risk of TBI (eg, athletes and military personnel).
Lacking robust scientific evidence to support the use of nutraceuticals either for enhancing TBI recovery or treating neuropsychiatric disturbances, clinicians must educate patients that these agents are not completely benign and can have significant side effects and drug interactions.62,63 Nutraceuticals may contain multiple ingredients, some of which can be toxic, particularly at higher doses. Many patients may not volunteer information about their nutraceutical use to their health care providers,64 so we must ask them about that and inform them of the potential for adverse events and drug interactions.
1. Centers for Disease Control and Prevention. Report to Congress on traumatic brain injury in the United States: epidemiology and rehabilitation. https://www.cdc.gov/traumaticbraininjury/pubs/congress_epi_rehab.html. Updated January 22, 2016. Accessed June 5, 2017.
2. Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99(1):4-9.
3. Vaishnavi S, Rao V, Fann JR. Neuropsychiatric problems after traumatic brain injury: unraveling the silent epidemic. Psychosomatics. 2009;50(3):198-205.
4. National Institutes of Health Office of Dietary Supplements. Dietary supplement fact sheets. https://ods.od.nih.gov/factsheets/list-all. Accessed June 5, 2017.
5. Institute of Medicine, Food and Nutrition Board. Dietary reference intakes for energy, carbohydrate, fiber, fat, fatty acids, cholesterol, protein, and amino acids. Washington, DC: National Academy of Sciences; 2002.
6. Rao V, Koliatsos V, Ahmed F, et al. Neuropsychiatric disturbances associated with traumatic brain injury: a practical approach to evaluation and management. Semin Neurol. 2015;35(1):64-82.
7. Chew E, Zafonte RD. Pharmacological management of neurobehavioral disorders following traumatic brain injury—a state-of-the-art review. J Rehabil Res Dev. 2009;46(6):851-879.
8. Petraglia AL, Maroon JC, Bailes JE. From the field of play to the field of combat: a review of the pharmacological management of concussion. Neurosurgery. 2012;70(6):1520-1533; discussion 1533.
9. Bengtsson M, Godbolt AK. Effects of acetylcholinesterase inhibitors on cognitive function in patients with chronic traumatic brain injury: a systematic review. J Rehabil Med. 2016;48(1):1-5.
10. Neurobehavioral Guidelines Working Group; Warden DL, Gordon B, McAllister TW, et al. Guidelines for the pharmacologic treatment of neurobehavioral sequelae of traumatic brain injury. J Neurotrauma. 2006;23(10):1468-1501.
11. DeFelice SL. The nutraceutical revolution: its impact on food industry R&D. Trends Food Sci Technol. 1995;6(2):59-61.
12. Cope EC, Morris DR, Levenson CW. Improving treatments and outcomes: an emerging role for zinc in traumatic brain injury. Nutr Rev. 2012;70(7):410-413.
13. Morris DR, Levenson CW. Zinc in traumatic brain injury: from neuroprotection to neurotoxicity. Curr Opin Clin Nutr Metab Care. 2013;16(6):708-711.
14. Young B, Ott L, Kasarskis E, et al. Zinc supplementation is associated with improved neurologic recovery rate and visceral protein levels of patients with severe closed head injury. J Neurotrauma. 1996;13(1):25-34.
15. Fernández-Gajardo R, Matamala JM, Carrasco R, et al. Novel therapeutic strategies for traumatic brain injury: acute antioxidant reinforcement. CNS Drugs. 2014;28(3):229-248.
16. Razmkon A, Sadidi A, Sherafat-Kazemzadeh E, et al. Administration of vitamin C and vitamin E in severe head injury: a randomized double-blind controlled trial. Clin Neurosurg. 2011;58:133-137.
17. Cekic M, Cutler SM, VanLandingham JW, et al. Vitamin D deficiency reduces the benefits of progesterone treatment after brain injury in aged rats. Neurobiol Aging. 2011;32(5):864-874.
18. Aminmansour B, Nikbakht H, Ghorbani A, et al. Comparison of the administration of progesterone versus progesterone and vitamin D in improvement of outcomes in patients with traumatic brain injury: a randomized clinical trial with placebo group. Adv Biomed Res. 2012;1:58.
19. Cernak I, Savic VJ, Kotur J, et al. Characterization of plasma magnesium concentration and oxidative stress following graded traumatic brain injury in humans. J Neurotrauma. 2000;17(1):53-68.
20. Dhandapani SS, Gupta A, Vivekanandhan S, et al. Randomized controlled trial of magnesium sulphate in severe closed traumatic brain injury. The Indian Journal of Neurotrauma. 2008;5(1):27-33.
21. Temkin NR, Anderson GD, Winn HR, et al. Magnesium sulfate for neuroprotection after traumatic brain injury: a randomised controlled trial. Lancet Neurol. 2007;6(1):29-38.
22. Jeter CB, Hergenroeder GW, Ward NH 3rd, et al. Human mild traumatic brain injury decreases circulating branched-chain amino acids and their metabolite levels. J Neurotrauma. 2013;30(8):671-679.
23. Cole JT, Mitala CM, Kundu S, et al. Dietary branched chain amino acids ameliorate injury-induced cognitive impairment. Proc Natl Acad Sci U S A. 2010;107(1):366-371.
24. Aquilani R, Iadarola P, Contardi A, et al. Branched-chain amino acids enhance the cognitive recovery of patients with severe traumatic brain injury. Arch Phys Med Rehabil. 2005;86(9):1729-1735.
25. Aquilani R, Boselli M, Boschi F, et al. Branched-chain amino acids may improve recovery from a vegetative or minimally conscious state in patients with traumatic brain injury: a pilot study. Arch Phys Med Rehabil. 2008;89(9):1642-1647.
26. Kang HJ, Im SH. Probiotics as an immune modulator. J Nutr Sci Vitaminol (Tokyo). 2015;61(suppl):S103-S105.
27. DiPiro JT, Howdieshell TR, Goddard JK, et al. Association of interleukin-4 plasma levels with traumatic injury and clinical course. Arch Surg. 1995;130(11):1159-1162; discussion 1162-1163.
28. Tan M, Zhu JC, Du J, et al. Effects of probiotics on serum levels of Th1/Th2 cytokine and clinical outcomes in severe traumatic brain-injured patients: a prospective randomized pilot study. Crit Care. 2011;15(6):R290.
29. Falcão de Arruda IS, de Aguilar-Nascimento JE. Benefits of early enteral nutrition with glutamine and probiotics in brain injury patients. Clin Sci (Lond). 2004;106(3):287-292.
30. Cuzzocrea S, Mazzon E, Costantino G, et al. Beneficial effects of n-acetylcysteine on ischaemic brain injury. Br J Pharmacol. 2000;130(6):1219-1226.
31. Hoffer ME, Balaban C, Slade MD, et al. Amelioration of acute sequelae of blast induced mild traumatic brain injury by N-acetyl cysteine: a double-blind, placebo controlled study. PLoS One. 2013;8(1):e54163.
32. Eakin K, Baratz-Goldstein R, Pick CG, et al. Efficacy of N-acetyl cysteine in traumatic brain injury. PLoS One. 2014;9(4):e90617.
33. Theadom A, Mahon S, Barker-Collo S, et al. Enzogenol for cognitive functioning in traumatic brain injury: a pilot placebo-controlled RCT. Eur J Neurol. 2013;20(8):1135-1144.
34. Arenth PM, Russell KC, Ricker JH, et al. CDP-choline as a biological supplement during neurorecovery: a focused review. PM R. 2011;3(6 suppl 1):S123-S131.
35. Clark WM. Efficacy of citicoline as an acute stroke treatment. Expert Opin Pharmacother. 2009;10(5):839-846.
36. Guseva MV, Hopkins DM, Scheff SW, et al. Dietary choline supplementation improves behavioral, histological, and neurochemical outcomes in a rat model of traumatic brain injury. J Neurotrauma. 2008;25(8):975-983.
37. Dixon CE, Ma X, Marion DW. Effects of CDP-choline treatment on neurobehavioral deficits after TBI and on hippocampal and neocortical acetylcholine release. J Neurotrauma. 1997;14(3):161-169.
38. Levin HS. Treatment of postconcussional symptoms with CDP-choline. J Neurol Sci. 1991;103(suppl):S39-S42.
39. Zafonte RD, Bagiella E, Ansel BM, et al. Effect of citicoline on functional and cognitive status among patients with traumatic brain injury: Citicoline Brain Injury Treatment Trial (COBRIT). JAMA. 2012;308(19):1993-2000.
40. Perry E, Walker M, Grace J, et al. Acetylcholine in mind: a neurotransmitter correlate of consciousness? Trends Neurosci. 1999;22(6):273-280.
41. Levin HS, Peters BH, Kalisky Z, et al. Effects of oral physostigmine and lecithin on memory and attention in closed head-injured patients. Cent Nerv Syst Trauma. 1986;3(4):333-342.
42. Alvarez XA, Lombardi VR, Corzo L, et al. Oral cerebrolysin enhances brain alpha activity and improves cognitive performance in elderly control subjects. J Neural Transm Suppl. 2000;59:315-328.
43. Ruether E, Husmann R, Kinzler E, et al. A 28-week, double-blind, placebo-controlled study with cerebrolysin in patients with mild to moderate Alzheimer’s disease. Int Clin Psychopharmacol. 2001;16(5):253-263.
44. Wong GK, Zhu XL, Poon WS. Beneficial effect of cerebrolysin on moderate and severe head injury patients: result of a cohort study. Acta Neurochir Suppl. 2005;95:59-60.
45. Chen CC, Wei ST, Tsaia SC, et al. Cerebrolysin enhances cognitive recovery of mild traumatic brain injury patients: double-blind, placebo-controlled, randomized study. Br J Neurosurg. 2013;27(6):803-807.
46. Ziganshina LE, Abakumova T, Vernay L. Cerebrolysin for acute ischaemic stroke. Cochrane Database Syst Rev. 2016;12:CD007026.
47. Barrett EC, McBurney MI, Ciappio ED. ω-3 fatty acid supplementation as a potential therapeutic aid for the recovery from mild traumatic brain injury/concussion. Adv Nutr. 2014;5(3):268-277.
48. Sharma S, Zhuang Y, Ying Z, et al. Dietary curcumin supplementation counteracts reduction in levels of molecules involved in energy homeostasis after brain trauma. Neuroscience. 2009;161(4):1037-1044.
49. Gatson JW, Liu MM, Abdelfattah K, et al. Resveratrol decreases inflammation in the brain of mice with mild traumatic brain injury. J Trauma Acute Care Surg. 2013;74(2):470-475; discussion 474-475.
50. Grey A, Bolland M. Clinical trial evidence and use of fish oil supplements. JAMA Intern Med. 2014;174(3):460-462.
51. Mpandzou G, Aït Ben Haddou E, Regragui W, et al. Vitamin D deficiency and its role in neurological conditions: a review. Rev Neurol (Paris). 2016;172(2):109-122.
52. Karakis I, Pase MP, Beiser A, et al. Association of serum vitamin D with the risk of incident dementia and subclinical indices of brain aging: The Framingham Heart Study. J Alzheimers Dis. 2016;51(2):451-461.
53. Sarris J, Papakostas GI, Vitolo O, et al. S-adenosyl methionine (SAMe) versus escitalopram and placebo in major depression RCT: efficacy and effects of histamine and carnitine as moderators of response. J Affect Disord. 2014;164:76-81.
54. Sarris J, Mischoulon D, Schweitzer I. Omega-3 for bipolar disorder: meta-analyses of use in mania and bipolar depression. J Clin Psychiatry. 2012;73(1):81-86.
55. Sarris J, Stough C, Bousman C, et al. Kava in the treatment of generalized anxiety disorder: a double-blind, randomized, placebo-controlled study. J Clin Psychopharmacol. 2013;33(5):643-648.
56. Hariri M, Azadbakht L. Magnesium, iron, and zinc supplementation for the treatment of attention deficit hyperactivity disorder: a systematic review on the recent literature. Int J Prev Med. 2015;6:83.
57. Gillies D, Sinn JKh, Lad SS, et al. Polyunsaturated fatty acids (PUFA) for attention deficit hyperactivity disorder (ADHD) in children and adolescents. Cochrane Database Syst Rev. 2012;7:CD007986.
58. Ghanizadeh A, Berk M. Zinc for treating of children and adolescents with attention-deficit hyperactivity disorder: a systematic review of randomized controlled clinical trials. Eur J Clin Nutr. 2013;67(1):122-124.
59. U.S. Food and Drug Administration. Can a dietary supplement treat a concussion? No! http://www.fda.gov/forconsumers/consumerupdates/ucm378845.htm. Updated February 13, 2015. Accessed June 5, 2017.
60. Sarris J, Logan AC, Akbaraly TN, et al; International Society for Nutritional Psychiatry Research. Nutritional medicine as mainstream in psychiatry. Lancet Psychiatry. 2015;2(3):271-274.
61. Desai A, Kevala K, Kim HY. Depletion of brain docosahexaenoic acid impairs recovery from traumatic brain injury. PLoS One. 2014;9(1):e86472.
62. Edie CF, Dewan N. Which psychotropics interact with four common supplements. Current Psychiatry. 2005;4(1):16-30.
63. Di Lorenzo C, Ceschi A, Kupferschmidt H, et al. Adverse effects of plant food supplements and botanical preparations: a systematic review with critical evaluation of causality. Br J Clin Pharmacol. 2015;79(4):578-592.
64. National Center for Complementary and Integrative Health. Complementary and alternative medicine: what people aged 50 and older discuss with their health care providers. https://nccih.nih.gov/research/statistics/2010. Published 2011. Accessed June 5, 2017.
1. Centers for Disease Control and Prevention. Report to Congress on traumatic brain injury in the United States: epidemiology and rehabilitation. https://www.cdc.gov/traumaticbraininjury/pubs/congress_epi_rehab.html. Updated January 22, 2016. Accessed June 5, 2017.
2. Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99(1):4-9.
3. Vaishnavi S, Rao V, Fann JR. Neuropsychiatric problems after traumatic brain injury: unraveling the silent epidemic. Psychosomatics. 2009;50(3):198-205.
4. National Institutes of Health Office of Dietary Supplements. Dietary supplement fact sheets. https://ods.od.nih.gov/factsheets/list-all. Accessed June 5, 2017.
5. Institute of Medicine, Food and Nutrition Board. Dietary reference intakes for energy, carbohydrate, fiber, fat, fatty acids, cholesterol, protein, and amino acids. Washington, DC: National Academy of Sciences; 2002.
6. Rao V, Koliatsos V, Ahmed F, et al. Neuropsychiatric disturbances associated with traumatic brain injury: a practical approach to evaluation and management. Semin Neurol. 2015;35(1):64-82.
7. Chew E, Zafonte RD. Pharmacological management of neurobehavioral disorders following traumatic brain injury—a state-of-the-art review. J Rehabil Res Dev. 2009;46(6):851-879.
8. Petraglia AL, Maroon JC, Bailes JE. From the field of play to the field of combat: a review of the pharmacological management of concussion. Neurosurgery. 2012;70(6):1520-1533; discussion 1533.
9. Bengtsson M, Godbolt AK. Effects of acetylcholinesterase inhibitors on cognitive function in patients with chronic traumatic brain injury: a systematic review. J Rehabil Med. 2016;48(1):1-5.
10. Neurobehavioral Guidelines Working Group; Warden DL, Gordon B, McAllister TW, et al. Guidelines for the pharmacologic treatment of neurobehavioral sequelae of traumatic brain injury. J Neurotrauma. 2006;23(10):1468-1501.
11. DeFelice SL. The nutraceutical revolution: its impact on food industry R&D. Trends Food Sci Technol. 1995;6(2):59-61.
12. Cope EC, Morris DR, Levenson CW. Improving treatments and outcomes: an emerging role for zinc in traumatic brain injury. Nutr Rev. 2012;70(7):410-413.
13. Morris DR, Levenson CW. Zinc in traumatic brain injury: from neuroprotection to neurotoxicity. Curr Opin Clin Nutr Metab Care. 2013;16(6):708-711.
14. Young B, Ott L, Kasarskis E, et al. Zinc supplementation is associated with improved neurologic recovery rate and visceral protein levels of patients with severe closed head injury. J Neurotrauma. 1996;13(1):25-34.
15. Fernández-Gajardo R, Matamala JM, Carrasco R, et al. Novel therapeutic strategies for traumatic brain injury: acute antioxidant reinforcement. CNS Drugs. 2014;28(3):229-248.
16. Razmkon A, Sadidi A, Sherafat-Kazemzadeh E, et al. Administration of vitamin C and vitamin E in severe head injury: a randomized double-blind controlled trial. Clin Neurosurg. 2011;58:133-137.
17. Cekic M, Cutler SM, VanLandingham JW, et al. Vitamin D deficiency reduces the benefits of progesterone treatment after brain injury in aged rats. Neurobiol Aging. 2011;32(5):864-874.
18. Aminmansour B, Nikbakht H, Ghorbani A, et al. Comparison of the administration of progesterone versus progesterone and vitamin D in improvement of outcomes in patients with traumatic brain injury: a randomized clinical trial with placebo group. Adv Biomed Res. 2012;1:58.
19. Cernak I, Savic VJ, Kotur J, et al. Characterization of plasma magnesium concentration and oxidative stress following graded traumatic brain injury in humans. J Neurotrauma. 2000;17(1):53-68.
20. Dhandapani SS, Gupta A, Vivekanandhan S, et al. Randomized controlled trial of magnesium sulphate in severe closed traumatic brain injury. The Indian Journal of Neurotrauma. 2008;5(1):27-33.
21. Temkin NR, Anderson GD, Winn HR, et al. Magnesium sulfate for neuroprotection after traumatic brain injury: a randomised controlled trial. Lancet Neurol. 2007;6(1):29-38.
22. Jeter CB, Hergenroeder GW, Ward NH 3rd, et al. Human mild traumatic brain injury decreases circulating branched-chain amino acids and their metabolite levels. J Neurotrauma. 2013;30(8):671-679.
23. Cole JT, Mitala CM, Kundu S, et al. Dietary branched chain amino acids ameliorate injury-induced cognitive impairment. Proc Natl Acad Sci U S A. 2010;107(1):366-371.
24. Aquilani R, Iadarola P, Contardi A, et al. Branched-chain amino acids enhance the cognitive recovery of patients with severe traumatic brain injury. Arch Phys Med Rehabil. 2005;86(9):1729-1735.
25. Aquilani R, Boselli M, Boschi F, et al. Branched-chain amino acids may improve recovery from a vegetative or minimally conscious state in patients with traumatic brain injury: a pilot study. Arch Phys Med Rehabil. 2008;89(9):1642-1647.
26. Kang HJ, Im SH. Probiotics as an immune modulator. J Nutr Sci Vitaminol (Tokyo). 2015;61(suppl):S103-S105.
27. DiPiro JT, Howdieshell TR, Goddard JK, et al. Association of interleukin-4 plasma levels with traumatic injury and clinical course. Arch Surg. 1995;130(11):1159-1162; discussion 1162-1163.
28. Tan M, Zhu JC, Du J, et al. Effects of probiotics on serum levels of Th1/Th2 cytokine and clinical outcomes in severe traumatic brain-injured patients: a prospective randomized pilot study. Crit Care. 2011;15(6):R290.
29. Falcão de Arruda IS, de Aguilar-Nascimento JE. Benefits of early enteral nutrition with glutamine and probiotics in brain injury patients. Clin Sci (Lond). 2004;106(3):287-292.
30. Cuzzocrea S, Mazzon E, Costantino G, et al. Beneficial effects of n-acetylcysteine on ischaemic brain injury. Br J Pharmacol. 2000;130(6):1219-1226.
31. Hoffer ME, Balaban C, Slade MD, et al. Amelioration of acute sequelae of blast induced mild traumatic brain injury by N-acetyl cysteine: a double-blind, placebo controlled study. PLoS One. 2013;8(1):e54163.
32. Eakin K, Baratz-Goldstein R, Pick CG, et al. Efficacy of N-acetyl cysteine in traumatic brain injury. PLoS One. 2014;9(4):e90617.
33. Theadom A, Mahon S, Barker-Collo S, et al. Enzogenol for cognitive functioning in traumatic brain injury: a pilot placebo-controlled RCT. Eur J Neurol. 2013;20(8):1135-1144.
34. Arenth PM, Russell KC, Ricker JH, et al. CDP-choline as a biological supplement during neurorecovery: a focused review. PM R. 2011;3(6 suppl 1):S123-S131.
35. Clark WM. Efficacy of citicoline as an acute stroke treatment. Expert Opin Pharmacother. 2009;10(5):839-846.
36. Guseva MV, Hopkins DM, Scheff SW, et al. Dietary choline supplementation improves behavioral, histological, and neurochemical outcomes in a rat model of traumatic brain injury. J Neurotrauma. 2008;25(8):975-983.
37. Dixon CE, Ma X, Marion DW. Effects of CDP-choline treatment on neurobehavioral deficits after TBI and on hippocampal and neocortical acetylcholine release. J Neurotrauma. 1997;14(3):161-169.
38. Levin HS. Treatment of postconcussional symptoms with CDP-choline. J Neurol Sci. 1991;103(suppl):S39-S42.
39. Zafonte RD, Bagiella E, Ansel BM, et al. Effect of citicoline on functional and cognitive status among patients with traumatic brain injury: Citicoline Brain Injury Treatment Trial (COBRIT). JAMA. 2012;308(19):1993-2000.
40. Perry E, Walker M, Grace J, et al. Acetylcholine in mind: a neurotransmitter correlate of consciousness? Trends Neurosci. 1999;22(6):273-280.
41. Levin HS, Peters BH, Kalisky Z, et al. Effects of oral physostigmine and lecithin on memory and attention in closed head-injured patients. Cent Nerv Syst Trauma. 1986;3(4):333-342.
42. Alvarez XA, Lombardi VR, Corzo L, et al. Oral cerebrolysin enhances brain alpha activity and improves cognitive performance in elderly control subjects. J Neural Transm Suppl. 2000;59:315-328.
43. Ruether E, Husmann R, Kinzler E, et al. A 28-week, double-blind, placebo-controlled study with cerebrolysin in patients with mild to moderate Alzheimer’s disease. Int Clin Psychopharmacol. 2001;16(5):253-263.
44. Wong GK, Zhu XL, Poon WS. Beneficial effect of cerebrolysin on moderate and severe head injury patients: result of a cohort study. Acta Neurochir Suppl. 2005;95:59-60.
45. Chen CC, Wei ST, Tsaia SC, et al. Cerebrolysin enhances cognitive recovery of mild traumatic brain injury patients: double-blind, placebo-controlled, randomized study. Br J Neurosurg. 2013;27(6):803-807.
46. Ziganshina LE, Abakumova T, Vernay L. Cerebrolysin for acute ischaemic stroke. Cochrane Database Syst Rev. 2016;12:CD007026.
47. Barrett EC, McBurney MI, Ciappio ED. ω-3 fatty acid supplementation as a potential therapeutic aid for the recovery from mild traumatic brain injury/concussion. Adv Nutr. 2014;5(3):268-277.
48. Sharma S, Zhuang Y, Ying Z, et al. Dietary curcumin supplementation counteracts reduction in levels of molecules involved in energy homeostasis after brain trauma. Neuroscience. 2009;161(4):1037-1044.
49. Gatson JW, Liu MM, Abdelfattah K, et al. Resveratrol decreases inflammation in the brain of mice with mild traumatic brain injury. J Trauma Acute Care Surg. 2013;74(2):470-475; discussion 474-475.
50. Grey A, Bolland M. Clinical trial evidence and use of fish oil supplements. JAMA Intern Med. 2014;174(3):460-462.
51. Mpandzou G, Aït Ben Haddou E, Regragui W, et al. Vitamin D deficiency and its role in neurological conditions: a review. Rev Neurol (Paris). 2016;172(2):109-122.
52. Karakis I, Pase MP, Beiser A, et al. Association of serum vitamin D with the risk of incident dementia and subclinical indices of brain aging: The Framingham Heart Study. J Alzheimers Dis. 2016;51(2):451-461.
53. Sarris J, Papakostas GI, Vitolo O, et al. S-adenosyl methionine (SAMe) versus escitalopram and placebo in major depression RCT: efficacy and effects of histamine and carnitine as moderators of response. J Affect Disord. 2014;164:76-81.
54. Sarris J, Mischoulon D, Schweitzer I. Omega-3 for bipolar disorder: meta-analyses of use in mania and bipolar depression. J Clin Psychiatry. 2012;73(1):81-86.
55. Sarris J, Stough C, Bousman C, et al. Kava in the treatment of generalized anxiety disorder: a double-blind, randomized, placebo-controlled study. J Clin Psychopharmacol. 2013;33(5):643-648.
56. Hariri M, Azadbakht L. Magnesium, iron, and zinc supplementation for the treatment of attention deficit hyperactivity disorder: a systematic review on the recent literature. Int J Prev Med. 2015;6:83.
57. Gillies D, Sinn JKh, Lad SS, et al. Polyunsaturated fatty acids (PUFA) for attention deficit hyperactivity disorder (ADHD) in children and adolescents. Cochrane Database Syst Rev. 2012;7:CD007986.
58. Ghanizadeh A, Berk M. Zinc for treating of children and adolescents with attention-deficit hyperactivity disorder: a systematic review of randomized controlled clinical trials. Eur J Clin Nutr. 2013;67(1):122-124.
59. U.S. Food and Drug Administration. Can a dietary supplement treat a concussion? No! http://www.fda.gov/forconsumers/consumerupdates/ucm378845.htm. Updated February 13, 2015. Accessed June 5, 2017.
60. Sarris J, Logan AC, Akbaraly TN, et al; International Society for Nutritional Psychiatry Research. Nutritional medicine as mainstream in psychiatry. Lancet Psychiatry. 2015;2(3):271-274.
61. Desai A, Kevala K, Kim HY. Depletion of brain docosahexaenoic acid impairs recovery from traumatic brain injury. PLoS One. 2014;9(1):e86472.
62. Edie CF, Dewan N. Which psychotropics interact with four common supplements. Current Psychiatry. 2005;4(1):16-30.
63. Di Lorenzo C, Ceschi A, Kupferschmidt H, et al. Adverse effects of plant food supplements and botanical preparations: a systematic review with critical evaluation of causality. Br J Clin Pharmacol. 2015;79(4):578-592.
64. National Center for Complementary and Integrative Health. Complementary and alternative medicine: what people aged 50 and older discuss with their health care providers. https://nccih.nih.gov/research/statistics/2010. Published 2011. Accessed June 5, 2017.

Modafinil: Not just for sleep disorders?
Ms. B, a middle-aged mother of 3, is being monitored for bipolar disorder. She has a history of stimulant abuse but has been in remission for 5 years. She complains of excessive daytime sleepiness. Most days she wakes at 7 AM, but sleeps on several occasions during the day. She also complains of fatigue and lack of motivation.
She is being treated with lithium, venlafaxine, and zolpidem and reports good adherence. Basic laboratory work and serum lithium levels are within acceptable ranges. Her symptoms do not improve when venlafaxine is titrated from 225 mg/d to 300 mg/d. She also reports previously failed trials with bupropion and fluoxetine.
We decide to try a psychostimulant as an augmenting agent. Because of her past stimulant abuse, we add modafinil, 100 mg/d and increase to 200 mg/d. Ms. B reports improvement in her daytime sleepiness and fatigue and—except for a mild headache—tolerates the medication well.
Modafinil is being investigated for potential roles in managing inattention, excess sleepiness, fatigue, and cognitive dysfunction associated with:
- mood disorders (major depression and bipolar depression)
- attention-deficit/hyperactivity disorder (ADHD)
- schizophrenia
- cocaine dependence.
This article discusses how the drug promotes wakefulness, how it might improve cognitive function, and what the evidence reveals about off-label indications.
How it works
Although modafinil’s precise mechanism of action is unknown, it is believed to promote wakefulness more selectively than conventional stimulants such as amphetamine and methylphenidate. Modafinil does not bind to norepinephrine, serotonin, dopamine, or benzodiazepine receptors.1,2 It might target specific hypothalamic regions such as the tuberomammillary nucleus and orexin neurons, which are peptide neurotransmitters that promote wakefulness.3,4
Clinical trials found that modafinil has beneficial effects on:
- working memory, recognition memory, and sustained attention in healthy humans
- prefrontal-dependent cognitive functions in schizophrenia, major depression, and adult ADHD.5
Evidence for approved indications
Modafinil is indicated to improve wakefulness in patients who have excessive sleepiness associated with narcolepsy, obstructive sleep apnea, or shift work sleep disorder. It was approved for reducing excessive sleepiness in narcoleptic patients after two 9-week placebo-controlled clinical trials. The drug significantly reduced sleepiness and improved overall disease status as measured by the Clinical Global Impression of Change (CGI-C) scale.6,7
In patients with shift work sleep disorder, a 12-week placebo-controlled clinical trial found that modafinil significantly improved sleep latency and CGI-C scores.10
Dosage and side effects. For patients with narcolepsy or obstructive sleep apnea, the recommended dose is 200 mg given in the morning.11 For patients prescribed modafinil for work-time wakefulness, the dose is 200 mg 1 hour before their work shift. Lower doses are recommended for patients who are elderly or have hepatic impairment. Those with severe hepatic impairment typically are prescribed 100 mg/d.11 Modafinil is rapidly absorbed and is metabolized primarily by the liver (Table 1). A summary of potential drug-drug interactions appears in Table 2.11
In pivotal trials, adverse events that occurred more frequently with modafinil than with placebo and in >5% of the study population included headache, nausea, nervousness, rhinitis, diarrhea, back pain, insomnia, dizziness, and dyspepsia. Headache was most commonly reported; in most patients, it resolved soon after they started taking modafinil. Post-marketing reports have included cases of psychosis, mania, and suspected serious skin reactions, including Stevens-Johnson syndrome.11 Modafinil lacks euphorigenic properties and has minimal potential for abuse.12
Table 1
Modafinil’s pharmacokinetics
| Absorbed rapidly, with peak plasma concentrations at 2 to 4 hours |
| Apparent steady states reached after 2 to 4 days of dosing |
| Half-life: 15 hours |
| Major route of elimination (~90%) is metabolism, primarily by the liver |
Selected drug-drug interactions with modafinil
| Action of modafinil | Potential drug interactions |
|---|---|
| Increases elimination of CYP 3A4 substrates | Carbamazepine, phenytoin may decrease modafinil levels Azole antifungals, protease inhibitors, and erythromycin may increase modafinil levels |
| Inhibits CYP 2C19 enzyme | Modafinil may increase levels of citalopram, diazepam, and sertraline |
| Decreases absorption of ethinyl estradiol | Modafinil can decrease effectiveness of oral contraceptives |
| CYP: cytochrome P-450 | |
| Source: Reference 11 | |
Evidence for off-label uses
Major depressive disorder (MDD). The fatigue and excessive sleepiness often seen with MDD often persist after other depressive symptoms have remitted with antidepressant treatment.13 Patients with these symptoms might benefit from modafinil’s stimulating properties. Conventional stimulants such as methylphenidate have been used to improve neurovegetative symptoms of depression, but modafinil offers several advantages:
- decreased adverse CNS effects
- fewer drug-drug interactions
- minimal risk for dependence or abuse.
A 6-week open-label study of 25 depressed patients with residual fatigue and sleepiness showed that adjunctive modafinil, 100 to 200 mg/d, significantly improved these symptoms, as well as Hamilton Rating Scale for Depression (HAM-D) score, as early as week 2. Seventy-six percent of patients responded to treatment, defined as a >50% reduction in HAM-D scores.16
Several open-label studies and case re-ports have evaluated adjunctive modafinil use in patients with:
- depression characterized by ongoing lethargy or apathy17
- depression with atypical features18
- seasonal affective disorder19
- partial response to antidepressants.20,21
Bipolar depression. A 6-week, double-blind, placebo-controlled trial randomly assigned 85 patients with bipolar depression to adjunctive modafinil, 100 to 200 mg/d, or placebo for 6 weeks (Table 3).22 The number of patients receiving an antidepressant or mood stabilizer was not significantly different between the modafinil and placebo groups.
The primary outcome measure was change in the Inventory for Depressive Symptoms (IDS) score from baseline to endpoint. Forty-four percent of patients receiving modafinil achieved a ≥50% reduction in IDS score, compared with 23% of the placebo group; this difference was statistically significant (P=0.03).
In this study, modafinil was well tolerated and did not induce mania or hypomania. Cases of modafinil-induced mania have been reported elsewhere.23,24
The mechanisms of modafinil’s antidepressant effects are unclear. The drug does not cause release of norepinephrine or dopamine. One study proposed that modafinil acts by releasing histamine and activating noradrenaline receptors.25 Activation of these receptors increases dopamine and norepinephrine in these areas, and excites histaminergic tuberomammillary neurons, increasing histamine levels. Another trial suggested that modafinil may improve mood by mechanisms similar to the antidepressant effects induced by sleep deprivation.26
Summary. Modafinil may have a role in managing residual fatigue and excessive sleepiness associated with MDD and bipolar depression. Evidence for a mood-elevating effect is minimal; additional studies are needed. Adjunctive modafinil and conventional stimulants have not been compared head-to-head in patients with mood disorders. Modafinil’s tolerability profile and lack of euphorigenic and reinforcing properties make it a potentially attractive alternative, however.
ADHD. Approximately 30% of ADHD patients do not respond to or are unable to tolerate conventional stimulant medications such as methylphenidate and dextroamphetamine.27 Several studies have evaluated modafinil as a potential treatment for ADHD based on the drug’s action on arousal and attention systems. Although modafinil’s precise mechanism of action in ADHD is unknown, proposed mechanisms include:
- hypothalamic and cerebral cortex neuronal activation
- action on histamine that results in internal vigilance.28
Can modafinil help patients with mood disorders?
| Author | Study design | Modafinil dose | Conclusion |
|---|---|---|---|
| Major depressive disorder | |||
| Fava et al, 200514 | 8-week, double-blind, placebo-controlled; 331 subjects with partial or no response to SSRI monotherapy | 200 mg/d | No significant difference between modafinil and placebo at final visit |
| DeBattista et al, 200315 | 6-week, double-blind, placebo-controlled; 136 subjects with partial response to antidepressant therapy | 100 to 400 mg/d | Significant improvement in sleepiness by week 1 and fatigue by week 2, but differences between modafinil and placebo were not statistically significant by end of study |
| Konuk et al, 200616 | 6-week, open-label; 25 subjects with residual sleepiness or fatigue after SSRI therapy | 100 to 200 mg/d | All patients showed significant improvement in sleepiness, fatigue, and HAM-D scores |
| Bipolar depression | |||
| Frye et al, 200722 | 6-week, double-blind, placebo-controlled trial; 85 subjects who did not respond to a mood stabilizer with or without concomitant antidepressant therapy | 100 to 200 mg/d (mean 177 mg/d) | 44% of modafinil patients achieved ≥50% reduction in IDS score compared with 23% in placebo group (P=0.03) |
| HAM-D: Hamilton Rating Scale for Depression; IDS: Inventory for Depressive Symptoms; SSRI: selective serotonin reuptake inhibitor | |||
CASE 2: Another Tx for ADHD
Matt, age 8, is referred to our outpatient child psychiatric clinic after his parents noted declining school performance associated with increased aggression and irritability. Our assessment strongly supports a diagnosis of ADHD without comorbid conditions. We start Matt on methylphenidate, 5 mg twice daily, which quickly improves his ADHD symptoms. However, the medication causes GI side effects and profound sleep and weight changes.
Matt’s parents request that their son be treated with a different type of agent. A trial of atomoxetine is not as effective as the initial methylphenidate dosage and produces similar side effects. We then consider modafinil because of its side effect profile. We start Matt on 100 mg once daily and titrate up to 200 mg/d 4 weeks later. Matt and his parents notice an immediate improvement in his ADHD symptoms with no side effects.
In children and adolescents. Wigal et al29 reviewed pooled data from 3 randomized, double-blind, placebo-controlled studies of modafinil in pediatric ADHD (Table 4). Modafinil was well tolerated and improved ADHD symptoms and behaviors regardless of patients’ stimulant use history.
In a recent open-label study, 220 children and young adolescents with ADHD who had completed 4 weeks of a double-blind, placebo-controlled trial were evaluated for an additional 8 weeks. Modafinil improved ADHD symptoms and overall clinical condition as determined by the parent- or clinician-completed ADHD Rating Scale-IV Home Version, the parent-completed Conners’ ADHD/DSM-IV Scale Parent Version, and the clinician-rated CGI scale.30 Insomnia, headache, and decreased appetite were the most commonly reported adverse events.
In adults. The results of 2 double-blind, placebo-controlled trials of modafinil in adults with ADHD have been positive:
- In 1 study, modafinil (mean 206.8 mg/d) was more effective than placebo and comparable to dextroamphetamine in improving ADHD symptoms.31
- In another, modafinil (a single 200-mg dose) increased cognitive performance during treatment.32
Schizophrenia. Double-blind, randomized placebo-controlled studies have evaluated modafinil for improving cognitive function and reducing negative symptoms in patients with schizophrenia. Results have been inconsistent.
One double-blind, randomized, placebo-controlled crossover study of 20 patients with chronic schizophrenia found that modafinil, 200 mg/d, significantly improved short-term verbal memory span and attentional set shifting—the ability to discriminate and selectively attend to various stimulus dimensions (Table 5).33 Two other controlled studies showed no differences between the effects of modafinil and placebo on schizophrenia’s fatigue, cognition, or positive or negative symptoms.34,35
Summary. Although open-label studies have shown modafinil has beneficial effects on cognitive symptoms, controlled data are scarce. Reports of modafinil-induced psychosis or mania11 may limit the drug’s usefulness in schizophrenia patients.
Cocaine dependence. No medications are FDA-approved for treating cocaine dependence. A placebo-controlled, double-blind trial found that modafinil blunts cocaine euphoria under controlled conditions.36 This effect is hypothesized to be secondary to modafinil’s glutamate-enhancing and gamma-aminobutyric acid inhibitory effects.37
Summary. A single study supports using modafinil to improve outcomes in cocaine-dependent patients receiving standardized psychosocial treatment. More research is needed.
Table 4
Modafinil and ADHD: What the evidence says
| Author | Study design | Modafinil dosage | Conclusion |
|---|---|---|---|
| Wigal et al, 200629 | Analysis of data from 3 double-blind, placebo-controlled trials; total 638 children/adolescents, some of whom had received prior stimulant therapy | 170 to 425 mg/d | Whether or not patients received prior stimulant treatment, modafinil significantly improved ADHD symptoms and was well tolerated |
| Boellner et al, 200630 | 8-week, open-label extension of a 4-week double-blind, placebo-controlled trial; 220 subjects ages 6-14 | 100 to 400 mg/d | Modafinil improved ADHD symptoms and overall clinical condition |
| Taylor et al, 200031 | 2-week, double-blind, placebo-controlled crossover comparing modafinil with dextroamphetamine; 22 adults | Mean 206.8 mg/d | Both modafinil and dextroamphetamine significantly improved ADHD symptoms compared with placebo |
| Turner et al, 200432 | Double-blind, placebo-controlled crossover; 20 adults | Single 200-mg dose | Modafinil improved results on cognitive tests, including short-term memory span, visual memory, spatial planning, and sustained attention |
| ADHD: attention-deficit/hyperactivity disorder | |||
Table 5
Modafinil for schizophrenia or cocaine dependence:
More research is needed
| Author | Study design | Modafinil dosage | Conclusion |
|---|---|---|---|
| Schizophrenia | |||
| Turner et al, 200433 | Double-blind, placebo-controlled crossover; 20 adults | 200 mg/d | Modafinil significantly improved attentional set shifting and short-term verbal memory span |
| Sevy et al, 200534 | 8-week, double-blind, placebo-controlled; 24 subjects | Up to 200 mg/d | No significant difference between modafinil and placebo in reducing fatigue or positive or negative symptoms or in improving cognition |
| Pierre et al, 200735 | 8-week, double-blind, placebo-controlled; 20 subjects | 100 to 200 mg/d | Modafinil did not significantly improve neurocognitive or negative symptoms |
| Cocaine dependence | |||
| Dackis et al, 200538 | 8-week, double-blind, placebo-controlled; 62 cocaine-dependent subjects | 400 mg/d | Patients receiving modafinil provided significantly more cocaine-negative urine samples and were significantly more likely to achieve =3 weeks cocaine abstinence than those receiving placebo |
Related resource
- Ballon JS, Feifel D. A systematic review of modafinil: potential clinical uses and mechanisms of action. J Clin Psychiatry 2006;67(4):554-66.
- Atomoxetine • Strattera
- Bupropion • Wellbutrin
- Carbamazepine • Carbatrol, Tegretol, others
- Citalopram • Celexa
- Dextroamphetamine • Dexedrine, DextroStat
- Diazepam • Valium
- Erythromycin • Ery-Tab, Eryc, others
- Fluoxetine • Prozac
- Lithium • Eskalith, Lithobid
- Methylphenidate • Ritalin, others
- Modafinil • Provigil
- Phenytoin • Dilantin
- Sertraline • Zoloft
- Venlafaxine • Effexor
- Zolpidem • Ambien
Dr. Ramaswamy receives research support from Bristol-Myers Squibb, Shire, and Forest Pharmaceuticals and is a consultant to Dainippon Sumitomo Pharma.
Dr. Mattai reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Wilson receives research support from, is a consultant to, or is a speaker for the National Institute of Mental Health, the Substance Abuse and Mental Health Services Administration, the Veterans Administration, the State of Nebraska, the State of Ohio, Health Futures Foundation, Inc., Abbott Laboratories, Astra-Zeneca, Bristol-Myers Squibb, Elan, Eli Lilly and Company, GlaxoSmithKline, Janssen, Ortho-McNeil, Pfizer, and Wyeth.
1. Mignot E, Nishino S, Guilleminault C, et al. Modafinil binds to dopamine uptake carrier site with low affinity. Sleep 1994;17:436-7.
2. Lin J-S, Hou Y, Jouvet M. Potential brain neuronal targets for amphetamine, methylphenidate, and modafinil induced wakefulness, evidenced by c-fos immunocytochemistry in the cat. Proc Natl Acad Sci USA 1996;93:14128-33.
3. Scammell TE, Estabrooke IV, McCarthy MT, et al. Hypothalamic arousal regions are activated during modafinil-induced wakefulness. J Neurosci 2000;20(22):8620-8.
4. Stenberg D. Neuroanatomy and neurochemistry of sleep. Cell Mol Life Sci 2007;64(10):1187-204.
5. Minzenberg MJ, Carter CS. Modafinil: a review of neurochemical actions and effects on cognition. Neuropsychopharmacology In press.
6. U.S. Modafinil in Narcolepsy Multicenter Study Group. Randomized trial of modafinil for the treatment of pathological somnolence in narcolepsy. Ann Neurol 1998;43(1):88-97.
7. U.S. Modafinil in Narcolepsy Multicenter Study Group. Randomized trial of modafinil as a treatment for the excessive daytime somnolence of narcolepsy. Neurology 2000;54:1166-75.
8. Black JE, Hirshkowitz M. Modafinil for treatment of residual excessive sleepiness in nasal continuous positive airway pressure-treated obstructive sleep apnea/hypopnea syndrome. Sleep 2005;28(4):464-71.
9. Pack AI, Black JE, Schwartz JR, Matheson JK. Modafinil as adjunct therapy for daytime sleepiness in obstructive sleep apnea. Am J Respir Crit Care Med 2001;164(9):1675-81.
10. Czeisler CA, Walsh JK, Roth T, et al. and the U.S. Modafinil in Shift Work Sleep Disorder Study Group. Modafinil for excessive sleepiness associated with shift-work sleep disorder. N Engl J Med. 2005;353(5):476-86. Published correction appears in: N Engl J Med 2005;353(10):1078.-
11. Provigil [package insert] West Chester, PA: Cephalon Inc; 2004.
12. Myrick H, Malcolm R, Taylor B, et al. Modafinil: preclinical, clinical, and post-marketing surveillance—a review of abuse liability issues. Ann Clin Psychiatry 2004;16(2):101-9.
13. Baldwin DS, Papakostas GI. Symptoms of fatigue and sleepiness in major depressive disorder. J Clin Psychiatry 2006;67(suppl 6):9-15.
14. Fava M, Thase ME, DeBattista C. A multicenter, placebo-controlled study of modafinil augmentation in partial responders to selective serotonin reuptake inhibitors with persistent fatigue and sleepiness. J Clin Psychiatry 2005;66(1):85-93.
15. DeBattista C, Doghramji K, Menza MA, et al. and the Modafinil in Depression Study Group. Adjunct modafinil for the short-term treatment of fatigue and sleepiness in patients with major depressive disorder: a preliminary double-blind, placebo-controlled study. J Clin Psychiatry 2003;64(9):1057-64.
16. Konuk N, Atasoy N, Atik L, Akay O. Open-label study of adjunct modafinil for the treatment of patients with fatigue, sleepiness, and major depression treated with selective serotonin reuptake inhibitors. Adv Ther 2006;23(4):646-54.
17. Markovitz PJ, Wagner S. An open-label trial of modafinil augmentation in patients with partial response to antidepressant therapy. J Clin Psychopharmacol 2003;23(2):207-9.
18. Vaishnavi S, Gadde K, Alamy S, et al. Modafinil for atypical depression: effects of open-label and double-blind discontinuation treatment. J Clin Psychopharmacol 2006; 26(4): 373-8. Published correction appears in: J Clin Psychopharmacol. 2006;26(5):523.
19. Lundt L. Modafinil treatment in patients with seasonal affective disorder/winter depression: an open-label pilot study. J Affect Disord 2004;81(2):173-8.
20. DeBattista C, Lembke A, Solvason HB, et al. A prospective trial of modafinil as an adjunctive treatment of major depression. J Clin Psychopharmacol 2004;24(1):87-90.
21. Rasmussen NA, Schroder P, Olsen LR, et al. Modafinil augmentation in depressed patients with partial response to antidepressants: a pilot study on self-reported symptoms covered by the Major Depression Inventory (MDI) and the Symptom Checklist (SCL-92). Nord J Psychiatry 2005;59(3):173-8.
22. Frye MA, Grunze H, Suppes T, et al. A placebo-controlled evaluation of adjunctive modafinil in the treatment of bipolar depression. Am J Psychiatry 2007;164:1242-9.
23. Wolf J, Fiedler U, Anghelescu I, Schwertfeger N. Manic switch in a patient with treatment-resistant bipolar depression treated with modafinil. J Clin Psychiatry 2006;67(11):1817.-
24. Vorspan F, Warot D, Consoli A, et al. Mania in a boy treated with modafinil for narcolepsy. Am J Psychiatry 2005;162(4):813-4.
25. McClellan KJ, Spencer CM. Modafinil: a review of its pharmacology and clinical efficacy in the management of narcolepsy. CNS Drugs 1998;9(4):311-24.
26. Ballon JS, Feifel D. A systematic review of modafinil: potential clinical uses and mechanisms of action. J Clin Psychiatry 2006;67(4):554-66.
27. Dulcan M. Practice parameters for the assessment and treatment of children, adolescents and adults with attentiondeficit hyperactivity disorder. American Academy of Child and Adolescent Psychiatry. J Am Acad Child Adolesc Psychiatry 1997;36(suppl 10):85S-121S.
28. Lindsay SE, Gudelsky GA, Heaton PC. Use of modafinil for the treatment of attention deficit/hyperactivity disorder. Ann Pharmacother 2006;40(10):1829-33.
29. Wigal SB, Biederman J, Swanson JM, et al. Efficacy and safety of modafinil film-coated tablets in children and adolescents with or without prior stimulant treatment for attention-deficit/hyperactivity disorder: pooled analysis of 3 randomized, double-blind, placebo-controlled studies. Prim Care Companion J Clin Psychiatry 2006;8(6):352-60.
30. Boellner SW, Earl CQ, Arora S. Modafinil in children and adolescents with attention-deficit/hyperactivity disorder: a preliminary 8-week, open-label study. Curr Med Res Opin 2006;22(12):2457-65.
31. Taylor FB, Russo J. Efficacy of modafinil compared to dextroamphetamine for the treatment of attention deficit hyperactivity disorder in adults. J Child Adolesc Psychopharmacol 2000;10(4):311-20.
32. Turner DC, Clark L, Dowson J, et al. Modafinil improves cognition and response inhibition in adult attention-deficit/ hyperactivity disorder. Biol Psychiatry 2004;55(10):1031-40.
33. Turner DC, Clark L, Pomarol-Clotet E, et al. Modafinil improves cognition and attentional set shifting in patients with chronic schizophrenia. Neuropsychopharmacology 2004;29(7):1363-73.
34. Sevy S, Rosenthal MH, Alvir J, et al. Double-blind, placebo-controlled study of modafinil for fatigue and cognition in schizophrenia patients treated with psychotropic medications. J Clin Psychiatry 2005;66(7):839-43.
35. Pierre JM, Peloian JH, Wirshing DA, et al. A randomized, double-blind, placebo-controlled trial of modafinil for negative symptoms in schizophrenia. J Clin Psychiatry 2007;68(5):705-10.
36. Dackis CA, Lynch KG, Yu E, et al. Modafinil and cocaine: a double-blind, placebo-controlled drug interaction study. Drug Alcohol Depend 2003;70(1):29-37.
37. Perez de la Mora M, Aguilar-Garcia A, Ramon-Frias T, et al. Effects of the vigilance promoting drug modafinil on the synthesis of GABA and glutamate in slices of rat hypothalamus. Neurosci Lett 1999;259:181-5.
38. Dackis CA, Kampman KM, Lynch KG, et al. A double-blind, placebo-controlled trial of modafinil for cocaine dependence. Neuropsychopharmacology 2005;30(1):205-11.
Ms. B, a middle-aged mother of 3, is being monitored for bipolar disorder. She has a history of stimulant abuse but has been in remission for 5 years. She complains of excessive daytime sleepiness. Most days she wakes at 7 AM, but sleeps on several occasions during the day. She also complains of fatigue and lack of motivation.
She is being treated with lithium, venlafaxine, and zolpidem and reports good adherence. Basic laboratory work and serum lithium levels are within acceptable ranges. Her symptoms do not improve when venlafaxine is titrated from 225 mg/d to 300 mg/d. She also reports previously failed trials with bupropion and fluoxetine.
We decide to try a psychostimulant as an augmenting agent. Because of her past stimulant abuse, we add modafinil, 100 mg/d and increase to 200 mg/d. Ms. B reports improvement in her daytime sleepiness and fatigue and—except for a mild headache—tolerates the medication well.
Modafinil is being investigated for potential roles in managing inattention, excess sleepiness, fatigue, and cognitive dysfunction associated with:
- mood disorders (major depression and bipolar depression)
- attention-deficit/hyperactivity disorder (ADHD)
- schizophrenia
- cocaine dependence.
This article discusses how the drug promotes wakefulness, how it might improve cognitive function, and what the evidence reveals about off-label indications.
How it works
Although modafinil’s precise mechanism of action is unknown, it is believed to promote wakefulness more selectively than conventional stimulants such as amphetamine and methylphenidate. Modafinil does not bind to norepinephrine, serotonin, dopamine, or benzodiazepine receptors.1,2 It might target specific hypothalamic regions such as the tuberomammillary nucleus and orexin neurons, which are peptide neurotransmitters that promote wakefulness.3,4
Clinical trials found that modafinil has beneficial effects on:
- working memory, recognition memory, and sustained attention in healthy humans
- prefrontal-dependent cognitive functions in schizophrenia, major depression, and adult ADHD.5
Evidence for approved indications
Modafinil is indicated to improve wakefulness in patients who have excessive sleepiness associated with narcolepsy, obstructive sleep apnea, or shift work sleep disorder. It was approved for reducing excessive sleepiness in narcoleptic patients after two 9-week placebo-controlled clinical trials. The drug significantly reduced sleepiness and improved overall disease status as measured by the Clinical Global Impression of Change (CGI-C) scale.6,7
In patients with shift work sleep disorder, a 12-week placebo-controlled clinical trial found that modafinil significantly improved sleep latency and CGI-C scores.10
Dosage and side effects. For patients with narcolepsy or obstructive sleep apnea, the recommended dose is 200 mg given in the morning.11 For patients prescribed modafinil for work-time wakefulness, the dose is 200 mg 1 hour before their work shift. Lower doses are recommended for patients who are elderly or have hepatic impairment. Those with severe hepatic impairment typically are prescribed 100 mg/d.11 Modafinil is rapidly absorbed and is metabolized primarily by the liver (Table 1). A summary of potential drug-drug interactions appears in Table 2.11
In pivotal trials, adverse events that occurred more frequently with modafinil than with placebo and in >5% of the study population included headache, nausea, nervousness, rhinitis, diarrhea, back pain, insomnia, dizziness, and dyspepsia. Headache was most commonly reported; in most patients, it resolved soon after they started taking modafinil. Post-marketing reports have included cases of psychosis, mania, and suspected serious skin reactions, including Stevens-Johnson syndrome.11 Modafinil lacks euphorigenic properties and has minimal potential for abuse.12
Table 1
Modafinil’s pharmacokinetics
| Absorbed rapidly, with peak plasma concentrations at 2 to 4 hours |
| Apparent steady states reached after 2 to 4 days of dosing |
| Half-life: 15 hours |
| Major route of elimination (~90%) is metabolism, primarily by the liver |
Selected drug-drug interactions with modafinil
| Action of modafinil | Potential drug interactions |
|---|---|
| Increases elimination of CYP 3A4 substrates | Carbamazepine, phenytoin may decrease modafinil levels Azole antifungals, protease inhibitors, and erythromycin may increase modafinil levels |
| Inhibits CYP 2C19 enzyme | Modafinil may increase levels of citalopram, diazepam, and sertraline |
| Decreases absorption of ethinyl estradiol | Modafinil can decrease effectiveness of oral contraceptives |
| CYP: cytochrome P-450 | |
| Source: Reference 11 | |
Evidence for off-label uses
Major depressive disorder (MDD). The fatigue and excessive sleepiness often seen with MDD often persist after other depressive symptoms have remitted with antidepressant treatment.13 Patients with these symptoms might benefit from modafinil’s stimulating properties. Conventional stimulants such as methylphenidate have been used to improve neurovegetative symptoms of depression, but modafinil offers several advantages:
- decreased adverse CNS effects
- fewer drug-drug interactions
- minimal risk for dependence or abuse.
A 6-week open-label study of 25 depressed patients with residual fatigue and sleepiness showed that adjunctive modafinil, 100 to 200 mg/d, significantly improved these symptoms, as well as Hamilton Rating Scale for Depression (HAM-D) score, as early as week 2. Seventy-six percent of patients responded to treatment, defined as a >50% reduction in HAM-D scores.16
Several open-label studies and case re-ports have evaluated adjunctive modafinil use in patients with:
- depression characterized by ongoing lethargy or apathy17
- depression with atypical features18
- seasonal affective disorder19
- partial response to antidepressants.20,21
Bipolar depression. A 6-week, double-blind, placebo-controlled trial randomly assigned 85 patients with bipolar depression to adjunctive modafinil, 100 to 200 mg/d, or placebo for 6 weeks (Table 3).22 The number of patients receiving an antidepressant or mood stabilizer was not significantly different between the modafinil and placebo groups.
The primary outcome measure was change in the Inventory for Depressive Symptoms (IDS) score from baseline to endpoint. Forty-four percent of patients receiving modafinil achieved a ≥50% reduction in IDS score, compared with 23% of the placebo group; this difference was statistically significant (P=0.03).
In this study, modafinil was well tolerated and did not induce mania or hypomania. Cases of modafinil-induced mania have been reported elsewhere.23,24
The mechanisms of modafinil’s antidepressant effects are unclear. The drug does not cause release of norepinephrine or dopamine. One study proposed that modafinil acts by releasing histamine and activating noradrenaline receptors.25 Activation of these receptors increases dopamine and norepinephrine in these areas, and excites histaminergic tuberomammillary neurons, increasing histamine levels. Another trial suggested that modafinil may improve mood by mechanisms similar to the antidepressant effects induced by sleep deprivation.26
Summary. Modafinil may have a role in managing residual fatigue and excessive sleepiness associated with MDD and bipolar depression. Evidence for a mood-elevating effect is minimal; additional studies are needed. Adjunctive modafinil and conventional stimulants have not been compared head-to-head in patients with mood disorders. Modafinil’s tolerability profile and lack of euphorigenic and reinforcing properties make it a potentially attractive alternative, however.
ADHD. Approximately 30% of ADHD patients do not respond to or are unable to tolerate conventional stimulant medications such as methylphenidate and dextroamphetamine.27 Several studies have evaluated modafinil as a potential treatment for ADHD based on the drug’s action on arousal and attention systems. Although modafinil’s precise mechanism of action in ADHD is unknown, proposed mechanisms include:
- hypothalamic and cerebral cortex neuronal activation
- action on histamine that results in internal vigilance.28
Can modafinil help patients with mood disorders?
| Author | Study design | Modafinil dose | Conclusion |
|---|---|---|---|
| Major depressive disorder | |||
| Fava et al, 200514 | 8-week, double-blind, placebo-controlled; 331 subjects with partial or no response to SSRI monotherapy | 200 mg/d | No significant difference between modafinil and placebo at final visit |
| DeBattista et al, 200315 | 6-week, double-blind, placebo-controlled; 136 subjects with partial response to antidepressant therapy | 100 to 400 mg/d | Significant improvement in sleepiness by week 1 and fatigue by week 2, but differences between modafinil and placebo were not statistically significant by end of study |
| Konuk et al, 200616 | 6-week, open-label; 25 subjects with residual sleepiness or fatigue after SSRI therapy | 100 to 200 mg/d | All patients showed significant improvement in sleepiness, fatigue, and HAM-D scores |
| Bipolar depression | |||
| Frye et al, 200722 | 6-week, double-blind, placebo-controlled trial; 85 subjects who did not respond to a mood stabilizer with or without concomitant antidepressant therapy | 100 to 200 mg/d (mean 177 mg/d) | 44% of modafinil patients achieved ≥50% reduction in IDS score compared with 23% in placebo group (P=0.03) |
| HAM-D: Hamilton Rating Scale for Depression; IDS: Inventory for Depressive Symptoms; SSRI: selective serotonin reuptake inhibitor | |||
CASE 2: Another Tx for ADHD
Matt, age 8, is referred to our outpatient child psychiatric clinic after his parents noted declining school performance associated with increased aggression and irritability. Our assessment strongly supports a diagnosis of ADHD without comorbid conditions. We start Matt on methylphenidate, 5 mg twice daily, which quickly improves his ADHD symptoms. However, the medication causes GI side effects and profound sleep and weight changes.
Matt’s parents request that their son be treated with a different type of agent. A trial of atomoxetine is not as effective as the initial methylphenidate dosage and produces similar side effects. We then consider modafinil because of its side effect profile. We start Matt on 100 mg once daily and titrate up to 200 mg/d 4 weeks later. Matt and his parents notice an immediate improvement in his ADHD symptoms with no side effects.
In children and adolescents. Wigal et al29 reviewed pooled data from 3 randomized, double-blind, placebo-controlled studies of modafinil in pediatric ADHD (Table 4). Modafinil was well tolerated and improved ADHD symptoms and behaviors regardless of patients’ stimulant use history.
In a recent open-label study, 220 children and young adolescents with ADHD who had completed 4 weeks of a double-blind, placebo-controlled trial were evaluated for an additional 8 weeks. Modafinil improved ADHD symptoms and overall clinical condition as determined by the parent- or clinician-completed ADHD Rating Scale-IV Home Version, the parent-completed Conners’ ADHD/DSM-IV Scale Parent Version, and the clinician-rated CGI scale.30 Insomnia, headache, and decreased appetite were the most commonly reported adverse events.
In adults. The results of 2 double-blind, placebo-controlled trials of modafinil in adults with ADHD have been positive:
- In 1 study, modafinil (mean 206.8 mg/d) was more effective than placebo and comparable to dextroamphetamine in improving ADHD symptoms.31
- In another, modafinil (a single 200-mg dose) increased cognitive performance during treatment.32
Schizophrenia. Double-blind, randomized placebo-controlled studies have evaluated modafinil for improving cognitive function and reducing negative symptoms in patients with schizophrenia. Results have been inconsistent.
One double-blind, randomized, placebo-controlled crossover study of 20 patients with chronic schizophrenia found that modafinil, 200 mg/d, significantly improved short-term verbal memory span and attentional set shifting—the ability to discriminate and selectively attend to various stimulus dimensions (Table 5).33 Two other controlled studies showed no differences between the effects of modafinil and placebo on schizophrenia’s fatigue, cognition, or positive or negative symptoms.34,35
Summary. Although open-label studies have shown modafinil has beneficial effects on cognitive symptoms, controlled data are scarce. Reports of modafinil-induced psychosis or mania11 may limit the drug’s usefulness in schizophrenia patients.
Cocaine dependence. No medications are FDA-approved for treating cocaine dependence. A placebo-controlled, double-blind trial found that modafinil blunts cocaine euphoria under controlled conditions.36 This effect is hypothesized to be secondary to modafinil’s glutamate-enhancing and gamma-aminobutyric acid inhibitory effects.37
Summary. A single study supports using modafinil to improve outcomes in cocaine-dependent patients receiving standardized psychosocial treatment. More research is needed.
Table 4
Modafinil and ADHD: What the evidence says
| Author | Study design | Modafinil dosage | Conclusion |
|---|---|---|---|
| Wigal et al, 200629 | Analysis of data from 3 double-blind, placebo-controlled trials; total 638 children/adolescents, some of whom had received prior stimulant therapy | 170 to 425 mg/d | Whether or not patients received prior stimulant treatment, modafinil significantly improved ADHD symptoms and was well tolerated |
| Boellner et al, 200630 | 8-week, open-label extension of a 4-week double-blind, placebo-controlled trial; 220 subjects ages 6-14 | 100 to 400 mg/d | Modafinil improved ADHD symptoms and overall clinical condition |
| Taylor et al, 200031 | 2-week, double-blind, placebo-controlled crossover comparing modafinil with dextroamphetamine; 22 adults | Mean 206.8 mg/d | Both modafinil and dextroamphetamine significantly improved ADHD symptoms compared with placebo |
| Turner et al, 200432 | Double-blind, placebo-controlled crossover; 20 adults | Single 200-mg dose | Modafinil improved results on cognitive tests, including short-term memory span, visual memory, spatial planning, and sustained attention |
| ADHD: attention-deficit/hyperactivity disorder | |||
Table 5
Modafinil for schizophrenia or cocaine dependence:
More research is needed
| Author | Study design | Modafinil dosage | Conclusion |
|---|---|---|---|
| Schizophrenia | |||
| Turner et al, 200433 | Double-blind, placebo-controlled crossover; 20 adults | 200 mg/d | Modafinil significantly improved attentional set shifting and short-term verbal memory span |
| Sevy et al, 200534 | 8-week, double-blind, placebo-controlled; 24 subjects | Up to 200 mg/d | No significant difference between modafinil and placebo in reducing fatigue or positive or negative symptoms or in improving cognition |
| Pierre et al, 200735 | 8-week, double-blind, placebo-controlled; 20 subjects | 100 to 200 mg/d | Modafinil did not significantly improve neurocognitive or negative symptoms |
| Cocaine dependence | |||
| Dackis et al, 200538 | 8-week, double-blind, placebo-controlled; 62 cocaine-dependent subjects | 400 mg/d | Patients receiving modafinil provided significantly more cocaine-negative urine samples and were significantly more likely to achieve =3 weeks cocaine abstinence than those receiving placebo |
Related resource
- Ballon JS, Feifel D. A systematic review of modafinil: potential clinical uses and mechanisms of action. J Clin Psychiatry 2006;67(4):554-66.
- Atomoxetine • Strattera
- Bupropion • Wellbutrin
- Carbamazepine • Carbatrol, Tegretol, others
- Citalopram • Celexa
- Dextroamphetamine • Dexedrine, DextroStat
- Diazepam • Valium
- Erythromycin • Ery-Tab, Eryc, others
- Fluoxetine • Prozac
- Lithium • Eskalith, Lithobid
- Methylphenidate • Ritalin, others
- Modafinil • Provigil
- Phenytoin • Dilantin
- Sertraline • Zoloft
- Venlafaxine • Effexor
- Zolpidem • Ambien
Dr. Ramaswamy receives research support from Bristol-Myers Squibb, Shire, and Forest Pharmaceuticals and is a consultant to Dainippon Sumitomo Pharma.
Dr. Mattai reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Wilson receives research support from, is a consultant to, or is a speaker for the National Institute of Mental Health, the Substance Abuse and Mental Health Services Administration, the Veterans Administration, the State of Nebraska, the State of Ohio, Health Futures Foundation, Inc., Abbott Laboratories, Astra-Zeneca, Bristol-Myers Squibb, Elan, Eli Lilly and Company, GlaxoSmithKline, Janssen, Ortho-McNeil, Pfizer, and Wyeth.
Ms. B, a middle-aged mother of 3, is being monitored for bipolar disorder. She has a history of stimulant abuse but has been in remission for 5 years. She complains of excessive daytime sleepiness. Most days she wakes at 7 AM, but sleeps on several occasions during the day. She also complains of fatigue and lack of motivation.
She is being treated with lithium, venlafaxine, and zolpidem and reports good adherence. Basic laboratory work and serum lithium levels are within acceptable ranges. Her symptoms do not improve when venlafaxine is titrated from 225 mg/d to 300 mg/d. She also reports previously failed trials with bupropion and fluoxetine.
We decide to try a psychostimulant as an augmenting agent. Because of her past stimulant abuse, we add modafinil, 100 mg/d and increase to 200 mg/d. Ms. B reports improvement in her daytime sleepiness and fatigue and—except for a mild headache—tolerates the medication well.
Modafinil is being investigated for potential roles in managing inattention, excess sleepiness, fatigue, and cognitive dysfunction associated with:
- mood disorders (major depression and bipolar depression)
- attention-deficit/hyperactivity disorder (ADHD)
- schizophrenia
- cocaine dependence.
This article discusses how the drug promotes wakefulness, how it might improve cognitive function, and what the evidence reveals about off-label indications.
How it works
Although modafinil’s precise mechanism of action is unknown, it is believed to promote wakefulness more selectively than conventional stimulants such as amphetamine and methylphenidate. Modafinil does not bind to norepinephrine, serotonin, dopamine, or benzodiazepine receptors.1,2 It might target specific hypothalamic regions such as the tuberomammillary nucleus and orexin neurons, which are peptide neurotransmitters that promote wakefulness.3,4
Clinical trials found that modafinil has beneficial effects on:
- working memory, recognition memory, and sustained attention in healthy humans
- prefrontal-dependent cognitive functions in schizophrenia, major depression, and adult ADHD.5
Evidence for approved indications
Modafinil is indicated to improve wakefulness in patients who have excessive sleepiness associated with narcolepsy, obstructive sleep apnea, or shift work sleep disorder. It was approved for reducing excessive sleepiness in narcoleptic patients after two 9-week placebo-controlled clinical trials. The drug significantly reduced sleepiness and improved overall disease status as measured by the Clinical Global Impression of Change (CGI-C) scale.6,7
In patients with shift work sleep disorder, a 12-week placebo-controlled clinical trial found that modafinil significantly improved sleep latency and CGI-C scores.10
Dosage and side effects. For patients with narcolepsy or obstructive sleep apnea, the recommended dose is 200 mg given in the morning.11 For patients prescribed modafinil for work-time wakefulness, the dose is 200 mg 1 hour before their work shift. Lower doses are recommended for patients who are elderly or have hepatic impairment. Those with severe hepatic impairment typically are prescribed 100 mg/d.11 Modafinil is rapidly absorbed and is metabolized primarily by the liver (Table 1). A summary of potential drug-drug interactions appears in Table 2.11
In pivotal trials, adverse events that occurred more frequently with modafinil than with placebo and in >5% of the study population included headache, nausea, nervousness, rhinitis, diarrhea, back pain, insomnia, dizziness, and dyspepsia. Headache was most commonly reported; in most patients, it resolved soon after they started taking modafinil. Post-marketing reports have included cases of psychosis, mania, and suspected serious skin reactions, including Stevens-Johnson syndrome.11 Modafinil lacks euphorigenic properties and has minimal potential for abuse.12
Table 1
Modafinil’s pharmacokinetics
| Absorbed rapidly, with peak plasma concentrations at 2 to 4 hours |
| Apparent steady states reached after 2 to 4 days of dosing |
| Half-life: 15 hours |
| Major route of elimination (~90%) is metabolism, primarily by the liver |
Selected drug-drug interactions with modafinil
| Action of modafinil | Potential drug interactions |
|---|---|
| Increases elimination of CYP 3A4 substrates | Carbamazepine, phenytoin may decrease modafinil levels Azole antifungals, protease inhibitors, and erythromycin may increase modafinil levels |
| Inhibits CYP 2C19 enzyme | Modafinil may increase levels of citalopram, diazepam, and sertraline |
| Decreases absorption of ethinyl estradiol | Modafinil can decrease effectiveness of oral contraceptives |
| CYP: cytochrome P-450 | |
| Source: Reference 11 | |
Evidence for off-label uses
Major depressive disorder (MDD). The fatigue and excessive sleepiness often seen with MDD often persist after other depressive symptoms have remitted with antidepressant treatment.13 Patients with these symptoms might benefit from modafinil’s stimulating properties. Conventional stimulants such as methylphenidate have been used to improve neurovegetative symptoms of depression, but modafinil offers several advantages:
- decreased adverse CNS effects
- fewer drug-drug interactions
- minimal risk for dependence or abuse.
A 6-week open-label study of 25 depressed patients with residual fatigue and sleepiness showed that adjunctive modafinil, 100 to 200 mg/d, significantly improved these symptoms, as well as Hamilton Rating Scale for Depression (HAM-D) score, as early as week 2. Seventy-six percent of patients responded to treatment, defined as a >50% reduction in HAM-D scores.16
Several open-label studies and case re-ports have evaluated adjunctive modafinil use in patients with:
- depression characterized by ongoing lethargy or apathy17
- depression with atypical features18
- seasonal affective disorder19
- partial response to antidepressants.20,21
Bipolar depression. A 6-week, double-blind, placebo-controlled trial randomly assigned 85 patients with bipolar depression to adjunctive modafinil, 100 to 200 mg/d, or placebo for 6 weeks (Table 3).22 The number of patients receiving an antidepressant or mood stabilizer was not significantly different between the modafinil and placebo groups.
The primary outcome measure was change in the Inventory for Depressive Symptoms (IDS) score from baseline to endpoint. Forty-four percent of patients receiving modafinil achieved a ≥50% reduction in IDS score, compared with 23% of the placebo group; this difference was statistically significant (P=0.03).
In this study, modafinil was well tolerated and did not induce mania or hypomania. Cases of modafinil-induced mania have been reported elsewhere.23,24
The mechanisms of modafinil’s antidepressant effects are unclear. The drug does not cause release of norepinephrine or dopamine. One study proposed that modafinil acts by releasing histamine and activating noradrenaline receptors.25 Activation of these receptors increases dopamine and norepinephrine in these areas, and excites histaminergic tuberomammillary neurons, increasing histamine levels. Another trial suggested that modafinil may improve mood by mechanisms similar to the antidepressant effects induced by sleep deprivation.26
Summary. Modafinil may have a role in managing residual fatigue and excessive sleepiness associated with MDD and bipolar depression. Evidence for a mood-elevating effect is minimal; additional studies are needed. Adjunctive modafinil and conventional stimulants have not been compared head-to-head in patients with mood disorders. Modafinil’s tolerability profile and lack of euphorigenic and reinforcing properties make it a potentially attractive alternative, however.
ADHD. Approximately 30% of ADHD patients do not respond to or are unable to tolerate conventional stimulant medications such as methylphenidate and dextroamphetamine.27 Several studies have evaluated modafinil as a potential treatment for ADHD based on the drug’s action on arousal and attention systems. Although modafinil’s precise mechanism of action in ADHD is unknown, proposed mechanisms include:
- hypothalamic and cerebral cortex neuronal activation
- action on histamine that results in internal vigilance.28
Can modafinil help patients with mood disorders?
| Author | Study design | Modafinil dose | Conclusion |
|---|---|---|---|
| Major depressive disorder | |||
| Fava et al, 200514 | 8-week, double-blind, placebo-controlled; 331 subjects with partial or no response to SSRI monotherapy | 200 mg/d | No significant difference between modafinil and placebo at final visit |
| DeBattista et al, 200315 | 6-week, double-blind, placebo-controlled; 136 subjects with partial response to antidepressant therapy | 100 to 400 mg/d | Significant improvement in sleepiness by week 1 and fatigue by week 2, but differences between modafinil and placebo were not statistically significant by end of study |
| Konuk et al, 200616 | 6-week, open-label; 25 subjects with residual sleepiness or fatigue after SSRI therapy | 100 to 200 mg/d | All patients showed significant improvement in sleepiness, fatigue, and HAM-D scores |
| Bipolar depression | |||
| Frye et al, 200722 | 6-week, double-blind, placebo-controlled trial; 85 subjects who did not respond to a mood stabilizer with or without concomitant antidepressant therapy | 100 to 200 mg/d (mean 177 mg/d) | 44% of modafinil patients achieved ≥50% reduction in IDS score compared with 23% in placebo group (P=0.03) |
| HAM-D: Hamilton Rating Scale for Depression; IDS: Inventory for Depressive Symptoms; SSRI: selective serotonin reuptake inhibitor | |||
CASE 2: Another Tx for ADHD
Matt, age 8, is referred to our outpatient child psychiatric clinic after his parents noted declining school performance associated with increased aggression and irritability. Our assessment strongly supports a diagnosis of ADHD without comorbid conditions. We start Matt on methylphenidate, 5 mg twice daily, which quickly improves his ADHD symptoms. However, the medication causes GI side effects and profound sleep and weight changes.
Matt’s parents request that their son be treated with a different type of agent. A trial of atomoxetine is not as effective as the initial methylphenidate dosage and produces similar side effects. We then consider modafinil because of its side effect profile. We start Matt on 100 mg once daily and titrate up to 200 mg/d 4 weeks later. Matt and his parents notice an immediate improvement in his ADHD symptoms with no side effects.
In children and adolescents. Wigal et al29 reviewed pooled data from 3 randomized, double-blind, placebo-controlled studies of modafinil in pediatric ADHD (Table 4). Modafinil was well tolerated and improved ADHD symptoms and behaviors regardless of patients’ stimulant use history.
In a recent open-label study, 220 children and young adolescents with ADHD who had completed 4 weeks of a double-blind, placebo-controlled trial were evaluated for an additional 8 weeks. Modafinil improved ADHD symptoms and overall clinical condition as determined by the parent- or clinician-completed ADHD Rating Scale-IV Home Version, the parent-completed Conners’ ADHD/DSM-IV Scale Parent Version, and the clinician-rated CGI scale.30 Insomnia, headache, and decreased appetite were the most commonly reported adverse events.
In adults. The results of 2 double-blind, placebo-controlled trials of modafinil in adults with ADHD have been positive:
- In 1 study, modafinil (mean 206.8 mg/d) was more effective than placebo and comparable to dextroamphetamine in improving ADHD symptoms.31
- In another, modafinil (a single 200-mg dose) increased cognitive performance during treatment.32
Schizophrenia. Double-blind, randomized placebo-controlled studies have evaluated modafinil for improving cognitive function and reducing negative symptoms in patients with schizophrenia. Results have been inconsistent.
One double-blind, randomized, placebo-controlled crossover study of 20 patients with chronic schizophrenia found that modafinil, 200 mg/d, significantly improved short-term verbal memory span and attentional set shifting—the ability to discriminate and selectively attend to various stimulus dimensions (Table 5).33 Two other controlled studies showed no differences between the effects of modafinil and placebo on schizophrenia’s fatigue, cognition, or positive or negative symptoms.34,35
Summary. Although open-label studies have shown modafinil has beneficial effects on cognitive symptoms, controlled data are scarce. Reports of modafinil-induced psychosis or mania11 may limit the drug’s usefulness in schizophrenia patients.
Cocaine dependence. No medications are FDA-approved for treating cocaine dependence. A placebo-controlled, double-blind trial found that modafinil blunts cocaine euphoria under controlled conditions.36 This effect is hypothesized to be secondary to modafinil’s glutamate-enhancing and gamma-aminobutyric acid inhibitory effects.37
Summary. A single study supports using modafinil to improve outcomes in cocaine-dependent patients receiving standardized psychosocial treatment. More research is needed.
Table 4
Modafinil and ADHD: What the evidence says
| Author | Study design | Modafinil dosage | Conclusion |
|---|---|---|---|
| Wigal et al, 200629 | Analysis of data from 3 double-blind, placebo-controlled trials; total 638 children/adolescents, some of whom had received prior stimulant therapy | 170 to 425 mg/d | Whether or not patients received prior stimulant treatment, modafinil significantly improved ADHD symptoms and was well tolerated |
| Boellner et al, 200630 | 8-week, open-label extension of a 4-week double-blind, placebo-controlled trial; 220 subjects ages 6-14 | 100 to 400 mg/d | Modafinil improved ADHD symptoms and overall clinical condition |
| Taylor et al, 200031 | 2-week, double-blind, placebo-controlled crossover comparing modafinil with dextroamphetamine; 22 adults | Mean 206.8 mg/d | Both modafinil and dextroamphetamine significantly improved ADHD symptoms compared with placebo |
| Turner et al, 200432 | Double-blind, placebo-controlled crossover; 20 adults | Single 200-mg dose | Modafinil improved results on cognitive tests, including short-term memory span, visual memory, spatial planning, and sustained attention |
| ADHD: attention-deficit/hyperactivity disorder | |||
Table 5
Modafinil for schizophrenia or cocaine dependence:
More research is needed
| Author | Study design | Modafinil dosage | Conclusion |
|---|---|---|---|
| Schizophrenia | |||
| Turner et al, 200433 | Double-blind, placebo-controlled crossover; 20 adults | 200 mg/d | Modafinil significantly improved attentional set shifting and short-term verbal memory span |
| Sevy et al, 200534 | 8-week, double-blind, placebo-controlled; 24 subjects | Up to 200 mg/d | No significant difference between modafinil and placebo in reducing fatigue or positive or negative symptoms or in improving cognition |
| Pierre et al, 200735 | 8-week, double-blind, placebo-controlled; 20 subjects | 100 to 200 mg/d | Modafinil did not significantly improve neurocognitive or negative symptoms |
| Cocaine dependence | |||
| Dackis et al, 200538 | 8-week, double-blind, placebo-controlled; 62 cocaine-dependent subjects | 400 mg/d | Patients receiving modafinil provided significantly more cocaine-negative urine samples and were significantly more likely to achieve =3 weeks cocaine abstinence than those receiving placebo |
Related resource
- Ballon JS, Feifel D. A systematic review of modafinil: potential clinical uses and mechanisms of action. J Clin Psychiatry 2006;67(4):554-66.
- Atomoxetine • Strattera
- Bupropion • Wellbutrin
- Carbamazepine • Carbatrol, Tegretol, others
- Citalopram • Celexa
- Dextroamphetamine • Dexedrine, DextroStat
- Diazepam • Valium
- Erythromycin • Ery-Tab, Eryc, others
- Fluoxetine • Prozac
- Lithium • Eskalith, Lithobid
- Methylphenidate • Ritalin, others
- Modafinil • Provigil
- Phenytoin • Dilantin
- Sertraline • Zoloft
- Venlafaxine • Effexor
- Zolpidem • Ambien
Dr. Ramaswamy receives research support from Bristol-Myers Squibb, Shire, and Forest Pharmaceuticals and is a consultant to Dainippon Sumitomo Pharma.
Dr. Mattai reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Wilson receives research support from, is a consultant to, or is a speaker for the National Institute of Mental Health, the Substance Abuse and Mental Health Services Administration, the Veterans Administration, the State of Nebraska, the State of Ohio, Health Futures Foundation, Inc., Abbott Laboratories, Astra-Zeneca, Bristol-Myers Squibb, Elan, Eli Lilly and Company, GlaxoSmithKline, Janssen, Ortho-McNeil, Pfizer, and Wyeth.
1. Mignot E, Nishino S, Guilleminault C, et al. Modafinil binds to dopamine uptake carrier site with low affinity. Sleep 1994;17:436-7.
2. Lin J-S, Hou Y, Jouvet M. Potential brain neuronal targets for amphetamine, methylphenidate, and modafinil induced wakefulness, evidenced by c-fos immunocytochemistry in the cat. Proc Natl Acad Sci USA 1996;93:14128-33.
3. Scammell TE, Estabrooke IV, McCarthy MT, et al. Hypothalamic arousal regions are activated during modafinil-induced wakefulness. J Neurosci 2000;20(22):8620-8.
4. Stenberg D. Neuroanatomy and neurochemistry of sleep. Cell Mol Life Sci 2007;64(10):1187-204.
5. Minzenberg MJ, Carter CS. Modafinil: a review of neurochemical actions and effects on cognition. Neuropsychopharmacology In press.
6. U.S. Modafinil in Narcolepsy Multicenter Study Group. Randomized trial of modafinil for the treatment of pathological somnolence in narcolepsy. Ann Neurol 1998;43(1):88-97.
7. U.S. Modafinil in Narcolepsy Multicenter Study Group. Randomized trial of modafinil as a treatment for the excessive daytime somnolence of narcolepsy. Neurology 2000;54:1166-75.
8. Black JE, Hirshkowitz M. Modafinil for treatment of residual excessive sleepiness in nasal continuous positive airway pressure-treated obstructive sleep apnea/hypopnea syndrome. Sleep 2005;28(4):464-71.
9. Pack AI, Black JE, Schwartz JR, Matheson JK. Modafinil as adjunct therapy for daytime sleepiness in obstructive sleep apnea. Am J Respir Crit Care Med 2001;164(9):1675-81.
10. Czeisler CA, Walsh JK, Roth T, et al. and the U.S. Modafinil in Shift Work Sleep Disorder Study Group. Modafinil for excessive sleepiness associated with shift-work sleep disorder. N Engl J Med. 2005;353(5):476-86. Published correction appears in: N Engl J Med 2005;353(10):1078.-
11. Provigil [package insert] West Chester, PA: Cephalon Inc; 2004.
12. Myrick H, Malcolm R, Taylor B, et al. Modafinil: preclinical, clinical, and post-marketing surveillance—a review of abuse liability issues. Ann Clin Psychiatry 2004;16(2):101-9.
13. Baldwin DS, Papakostas GI. Symptoms of fatigue and sleepiness in major depressive disorder. J Clin Psychiatry 2006;67(suppl 6):9-15.
14. Fava M, Thase ME, DeBattista C. A multicenter, placebo-controlled study of modafinil augmentation in partial responders to selective serotonin reuptake inhibitors with persistent fatigue and sleepiness. J Clin Psychiatry 2005;66(1):85-93.
15. DeBattista C, Doghramji K, Menza MA, et al. and the Modafinil in Depression Study Group. Adjunct modafinil for the short-term treatment of fatigue and sleepiness in patients with major depressive disorder: a preliminary double-blind, placebo-controlled study. J Clin Psychiatry 2003;64(9):1057-64.
16. Konuk N, Atasoy N, Atik L, Akay O. Open-label study of adjunct modafinil for the treatment of patients with fatigue, sleepiness, and major depression treated with selective serotonin reuptake inhibitors. Adv Ther 2006;23(4):646-54.
17. Markovitz PJ, Wagner S. An open-label trial of modafinil augmentation in patients with partial response to antidepressant therapy. J Clin Psychopharmacol 2003;23(2):207-9.
18. Vaishnavi S, Gadde K, Alamy S, et al. Modafinil for atypical depression: effects of open-label and double-blind discontinuation treatment. J Clin Psychopharmacol 2006; 26(4): 373-8. Published correction appears in: J Clin Psychopharmacol. 2006;26(5):523.
19. Lundt L. Modafinil treatment in patients with seasonal affective disorder/winter depression: an open-label pilot study. J Affect Disord 2004;81(2):173-8.
20. DeBattista C, Lembke A, Solvason HB, et al. A prospective trial of modafinil as an adjunctive treatment of major depression. J Clin Psychopharmacol 2004;24(1):87-90.
21. Rasmussen NA, Schroder P, Olsen LR, et al. Modafinil augmentation in depressed patients with partial response to antidepressants: a pilot study on self-reported symptoms covered by the Major Depression Inventory (MDI) and the Symptom Checklist (SCL-92). Nord J Psychiatry 2005;59(3):173-8.
22. Frye MA, Grunze H, Suppes T, et al. A placebo-controlled evaluation of adjunctive modafinil in the treatment of bipolar depression. Am J Psychiatry 2007;164:1242-9.
23. Wolf J, Fiedler U, Anghelescu I, Schwertfeger N. Manic switch in a patient with treatment-resistant bipolar depression treated with modafinil. J Clin Psychiatry 2006;67(11):1817.-
24. Vorspan F, Warot D, Consoli A, et al. Mania in a boy treated with modafinil for narcolepsy. Am J Psychiatry 2005;162(4):813-4.
25. McClellan KJ, Spencer CM. Modafinil: a review of its pharmacology and clinical efficacy in the management of narcolepsy. CNS Drugs 1998;9(4):311-24.
26. Ballon JS, Feifel D. A systematic review of modafinil: potential clinical uses and mechanisms of action. J Clin Psychiatry 2006;67(4):554-66.
27. Dulcan M. Practice parameters for the assessment and treatment of children, adolescents and adults with attentiondeficit hyperactivity disorder. American Academy of Child and Adolescent Psychiatry. J Am Acad Child Adolesc Psychiatry 1997;36(suppl 10):85S-121S.
28. Lindsay SE, Gudelsky GA, Heaton PC. Use of modafinil for the treatment of attention deficit/hyperactivity disorder. Ann Pharmacother 2006;40(10):1829-33.
29. Wigal SB, Biederman J, Swanson JM, et al. Efficacy and safety of modafinil film-coated tablets in children and adolescents with or without prior stimulant treatment for attention-deficit/hyperactivity disorder: pooled analysis of 3 randomized, double-blind, placebo-controlled studies. Prim Care Companion J Clin Psychiatry 2006;8(6):352-60.
30. Boellner SW, Earl CQ, Arora S. Modafinil in children and adolescents with attention-deficit/hyperactivity disorder: a preliminary 8-week, open-label study. Curr Med Res Opin 2006;22(12):2457-65.
31. Taylor FB, Russo J. Efficacy of modafinil compared to dextroamphetamine for the treatment of attention deficit hyperactivity disorder in adults. J Child Adolesc Psychopharmacol 2000;10(4):311-20.
32. Turner DC, Clark L, Dowson J, et al. Modafinil improves cognition and response inhibition in adult attention-deficit/ hyperactivity disorder. Biol Psychiatry 2004;55(10):1031-40.
33. Turner DC, Clark L, Pomarol-Clotet E, et al. Modafinil improves cognition and attentional set shifting in patients with chronic schizophrenia. Neuropsychopharmacology 2004;29(7):1363-73.
34. Sevy S, Rosenthal MH, Alvir J, et al. Double-blind, placebo-controlled study of modafinil for fatigue and cognition in schizophrenia patients treated with psychotropic medications. J Clin Psychiatry 2005;66(7):839-43.
35. Pierre JM, Peloian JH, Wirshing DA, et al. A randomized, double-blind, placebo-controlled trial of modafinil for negative symptoms in schizophrenia. J Clin Psychiatry 2007;68(5):705-10.
36. Dackis CA, Lynch KG, Yu E, et al. Modafinil and cocaine: a double-blind, placebo-controlled drug interaction study. Drug Alcohol Depend 2003;70(1):29-37.
37. Perez de la Mora M, Aguilar-Garcia A, Ramon-Frias T, et al. Effects of the vigilance promoting drug modafinil on the synthesis of GABA and glutamate in slices of rat hypothalamus. Neurosci Lett 1999;259:181-5.
38. Dackis CA, Kampman KM, Lynch KG, et al. A double-blind, placebo-controlled trial of modafinil for cocaine dependence. Neuropsychopharmacology 2005;30(1):205-11.
1. Mignot E, Nishino S, Guilleminault C, et al. Modafinil binds to dopamine uptake carrier site with low affinity. Sleep 1994;17:436-7.
2. Lin J-S, Hou Y, Jouvet M. Potential brain neuronal targets for amphetamine, methylphenidate, and modafinil induced wakefulness, evidenced by c-fos immunocytochemistry in the cat. Proc Natl Acad Sci USA 1996;93:14128-33.
3. Scammell TE, Estabrooke IV, McCarthy MT, et al. Hypothalamic arousal regions are activated during modafinil-induced wakefulness. J Neurosci 2000;20(22):8620-8.
4. Stenberg D. Neuroanatomy and neurochemistry of sleep. Cell Mol Life Sci 2007;64(10):1187-204.
5. Minzenberg MJ, Carter CS. Modafinil: a review of neurochemical actions and effects on cognition. Neuropsychopharmacology In press.
6. U.S. Modafinil in Narcolepsy Multicenter Study Group. Randomized trial of modafinil for the treatment of pathological somnolence in narcolepsy. Ann Neurol 1998;43(1):88-97.
7. U.S. Modafinil in Narcolepsy Multicenter Study Group. Randomized trial of modafinil as a treatment for the excessive daytime somnolence of narcolepsy. Neurology 2000;54:1166-75.
8. Black JE, Hirshkowitz M. Modafinil for treatment of residual excessive sleepiness in nasal continuous positive airway pressure-treated obstructive sleep apnea/hypopnea syndrome. Sleep 2005;28(4):464-71.
9. Pack AI, Black JE, Schwartz JR, Matheson JK. Modafinil as adjunct therapy for daytime sleepiness in obstructive sleep apnea. Am J Respir Crit Care Med 2001;164(9):1675-81.
10. Czeisler CA, Walsh JK, Roth T, et al. and the U.S. Modafinil in Shift Work Sleep Disorder Study Group. Modafinil for excessive sleepiness associated with shift-work sleep disorder. N Engl J Med. 2005;353(5):476-86. Published correction appears in: N Engl J Med 2005;353(10):1078.-
11. Provigil [package insert] West Chester, PA: Cephalon Inc; 2004.
12. Myrick H, Malcolm R, Taylor B, et al. Modafinil: preclinical, clinical, and post-marketing surveillance—a review of abuse liability issues. Ann Clin Psychiatry 2004;16(2):101-9.
13. Baldwin DS, Papakostas GI. Symptoms of fatigue and sleepiness in major depressive disorder. J Clin Psychiatry 2006;67(suppl 6):9-15.
14. Fava M, Thase ME, DeBattista C. A multicenter, placebo-controlled study of modafinil augmentation in partial responders to selective serotonin reuptake inhibitors with persistent fatigue and sleepiness. J Clin Psychiatry 2005;66(1):85-93.
15. DeBattista C, Doghramji K, Menza MA, et al. and the Modafinil in Depression Study Group. Adjunct modafinil for the short-term treatment of fatigue and sleepiness in patients with major depressive disorder: a preliminary double-blind, placebo-controlled study. J Clin Psychiatry 2003;64(9):1057-64.
16. Konuk N, Atasoy N, Atik L, Akay O. Open-label study of adjunct modafinil for the treatment of patients with fatigue, sleepiness, and major depression treated with selective serotonin reuptake inhibitors. Adv Ther 2006;23(4):646-54.
17. Markovitz PJ, Wagner S. An open-label trial of modafinil augmentation in patients with partial response to antidepressant therapy. J Clin Psychopharmacol 2003;23(2):207-9.
18. Vaishnavi S, Gadde K, Alamy S, et al. Modafinil for atypical depression: effects of open-label and double-blind discontinuation treatment. J Clin Psychopharmacol 2006; 26(4): 373-8. Published correction appears in: J Clin Psychopharmacol. 2006;26(5):523.
19. Lundt L. Modafinil treatment in patients with seasonal affective disorder/winter depression: an open-label pilot study. J Affect Disord 2004;81(2):173-8.
20. DeBattista C, Lembke A, Solvason HB, et al. A prospective trial of modafinil as an adjunctive treatment of major depression. J Clin Psychopharmacol 2004;24(1):87-90.
21. Rasmussen NA, Schroder P, Olsen LR, et al. Modafinil augmentation in depressed patients with partial response to antidepressants: a pilot study on self-reported symptoms covered by the Major Depression Inventory (MDI) and the Symptom Checklist (SCL-92). Nord J Psychiatry 2005;59(3):173-8.
22. Frye MA, Grunze H, Suppes T, et al. A placebo-controlled evaluation of adjunctive modafinil in the treatment of bipolar depression. Am J Psychiatry 2007;164:1242-9.
23. Wolf J, Fiedler U, Anghelescu I, Schwertfeger N. Manic switch in a patient with treatment-resistant bipolar depression treated with modafinil. J Clin Psychiatry 2006;67(11):1817.-
24. Vorspan F, Warot D, Consoli A, et al. Mania in a boy treated with modafinil for narcolepsy. Am J Psychiatry 2005;162(4):813-4.
25. McClellan KJ, Spencer CM. Modafinil: a review of its pharmacology and clinical efficacy in the management of narcolepsy. CNS Drugs 1998;9(4):311-24.
26. Ballon JS, Feifel D. A systematic review of modafinil: potential clinical uses and mechanisms of action. J Clin Psychiatry 2006;67(4):554-66.
27. Dulcan M. Practice parameters for the assessment and treatment of children, adolescents and adults with attentiondeficit hyperactivity disorder. American Academy of Child and Adolescent Psychiatry. J Am Acad Child Adolesc Psychiatry 1997;36(suppl 10):85S-121S.
28. Lindsay SE, Gudelsky GA, Heaton PC. Use of modafinil for the treatment of attention deficit/hyperactivity disorder. Ann Pharmacother 2006;40(10):1829-33.
29. Wigal SB, Biederman J, Swanson JM, et al. Efficacy and safety of modafinil film-coated tablets in children and adolescents with or without prior stimulant treatment for attention-deficit/hyperactivity disorder: pooled analysis of 3 randomized, double-blind, placebo-controlled studies. Prim Care Companion J Clin Psychiatry 2006;8(6):352-60.
30. Boellner SW, Earl CQ, Arora S. Modafinil in children and adolescents with attention-deficit/hyperactivity disorder: a preliminary 8-week, open-label study. Curr Med Res Opin 2006;22(12):2457-65.
31. Taylor FB, Russo J. Efficacy of modafinil compared to dextroamphetamine for the treatment of attention deficit hyperactivity disorder in adults. J Child Adolesc Psychopharmacol 2000;10(4):311-20.
32. Turner DC, Clark L, Dowson J, et al. Modafinil improves cognition and response inhibition in adult attention-deficit/ hyperactivity disorder. Biol Psychiatry 2004;55(10):1031-40.
33. Turner DC, Clark L, Pomarol-Clotet E, et al. Modafinil improves cognition and attentional set shifting in patients with chronic schizophrenia. Neuropsychopharmacology 2004;29(7):1363-73.
34. Sevy S, Rosenthal MH, Alvir J, et al. Double-blind, placebo-controlled study of modafinil for fatigue and cognition in schizophrenia patients treated with psychotropic medications. J Clin Psychiatry 2005;66(7):839-43.
35. Pierre JM, Peloian JH, Wirshing DA, et al. A randomized, double-blind, placebo-controlled trial of modafinil for negative symptoms in schizophrenia. J Clin Psychiatry 2007;68(5):705-10.
36. Dackis CA, Lynch KG, Yu E, et al. Modafinil and cocaine: a double-blind, placebo-controlled drug interaction study. Drug Alcohol Depend 2003;70(1):29-37.
37. Perez de la Mora M, Aguilar-Garcia A, Ramon-Frias T, et al. Effects of the vigilance promoting drug modafinil on the synthesis of GABA and glutamate in slices of rat hypothalamus. Neurosci Lett 1999;259:181-5.
38. Dackis CA, Kampman KM, Lynch KG, et al. A double-blind, placebo-controlled trial of modafinil for cocaine dependence. Neuropsychopharmacology 2005;30(1):205-11.
Tips to manage and prevent discontinuation syndromes
Abruptly stopping common psychotropics—particularly antidepressants, benzodiazepines, or atypical antipsychotics—can trigger a discontinuation syndrome, with:
- rebound or relapse of original symptoms
- uncomfortable new physical and psychological symptoms
- physiologic withdrawal at times.
To increase health professionals’ awareness of the risk of these adverse effects,1 this article describes discontinuation syndromes associated with various psychotropics and offers strategies to anticipate, recognize, and manage them.
Antidepressant Discontinuation Syndromes
Discontinuation syndromes can occur with tricyclic and tetracyclic antidepressants (TCAs), monoamine oxidase inhibitors (MAOIs), selective serotonin reuptake inhibitors (SSRIs), and other newer antidepressants. Symptoms usually start within a few days of stopping a drug—or less commonly, reducing its dosage—and are usually mild and self-limited. Serious outcomes have been reported.
Distinguishing antidepressant discontinuation symptoms from depression recurrence is important. Discontinuation symptoms emerge within 1 to 3 days, whereas depressive symptoms usually occur 2 to 3 weeks after an antidepressant is stopped. Discontinuation reactions remit within a few days, especially if the antidepressant is re-instituted.
TCAs block serotonin and norepinephrine reuptake, increasing the availability of these biogenic amines at receptor sites in the brain and other tissues. Abrupt discontinuation can cause physical symptoms—such as lethargy, headache, and tremor—and psychological symptoms including irritability, anxiety, agitation, and low mood (Table 1).2
Long-term use of TCAs with potent anticholinergic properties leads to upregulation of postsynaptic muscarinic receptors, creating a “supersensitive” state. Abrupt discontinuation can cause cholinergic rebound, with symptoms emerging as soon as 12 hours—but typically 24 to 48 hours—after the last dose.
Table 1
Discontinuation symptoms seen with TCAs
| Physical symptoms | Lethargy, headache, tremor, sweating, anorexia, insomnia, nausea, vomiting, diarrhea, akathisia (rare), parkinsonism (rare) |
| Psychological symptoms | Irritability, anxiety/agitation, low mood, excessive dreaming, nightmares, paradoxical activation resulting in manic/hypomanic symptoms (rare) |
| TCA: Tricyclic antidepressants | |
| Source: Reference 2 | |
MAOIs such as phenelzine and tranylcypromine inhibit the enzyme monoamine oxidase, which is responsible for monoamine degradation and increases synaptic monoamine concentrations. Discontinuation syndromes may include acute confusional states, paranoid delusions, hallucinations, or worsening of depressive symptoms.3 These problems rarely occur in clinical practice, however, because MAOIs’ serious side effects discourage doctors from prescribing them.
SSRIs and other agents. SSRIs block synaptic reuptake of serotonin. Serotonin-norepinephrine reuptake inhibitors (SNRIs) such as venlafaxine and duloxetine inhibit both serotonin and norepinephrine reuptake. Mirtazapine—an alpha2-adrenergic and heteroreceptor antagonist—increases serotonin and norepinephrine at the synapse. Bupropion increases dopamine and norepinephrine turnover in the CNS and also blocks serotonin.
Up to 30% of patients who stop taking SSRIs develop discontinuation symptoms.4 Six symptom clusters—disequilibrium, sensory symptoms, general somatic symptoms, sleep disturbance, GI symptoms, and affective symptoms—characterize the SSRI discontinuation syndrome (Table 2).5 The four most common symptoms—in decreasing order of frequency—are dizziness, nausea, lethargy, and headache.6 Ataxia, sensory abnormalities, and possibly aggressive and impulsive behavior differentiate this discontinuation syndrome from that of the TCAs.
Table 2
Discontinuation symptoms seen with SSRIs
| Type | Symptoms |
|---|---|
| Disequilibrium | Lightheadedness/dizziness, vertigo, ataxia |
| Sensory symptoms | Paraesthesia, numbness, electric shock-like sensations |
| General somatic symptoms | Lethargy, headache, tremor, sweating, anorexia |
| Sleep disturbance | Insomnia, nightmares, excessive dreaming |
| GI symptoms | Nausea, vomiting, diarrhea |
| Affective symptoms | Irritability, anxiety/agitation, low mood |
| SSRIs: Selective serotonin reuptake inhibitors | |
| Source: Reference 5 | |
Risk factors. Risk factors for SSRI discontinuation syndrome have been identified (Table 3).7 Symptoms usually begin 1 to 3 days after an SSRI is abruptly stopped and are usually mild. However, some patients report falls, inability to work, and difficulty walking and driving. Untreated symptoms are short-lived and remit within 1 to 2 weeks. They also remit if the original antidepressant is reintroduced or a pharmacologically similar agent is substituted.
Discontinuation syndrome risk among SSRIs is highest for paroxetine, intermediate for sertraline and fluvoxamine, and lowest for fluoxetine.4 Citalopram may cause a mild and transient discontinuation syndrome.8 Citalopram’s long elimination half-life (30 to 35 hours) and fewer and much less-potent active metabolites9 may explain its relatively low risk of discontinuation symptoms.
Discontinuation reactions have been reported to occur 100 times more frequently with paroxetine than with fluoxetine.10 Fluoxetine’s lower rate could be explained by its 2- to 3-day half-life, compared with half-lives of 33 hours or less for paroxetine, sertraline, citalopram, and fluvoxamine. A longer half-life might protect against a discontinuation syndrome.
Among other newer antidepressants:
- venlafaxine’s discontinuation syndrome is similar to the SSRI syndrome11
- no discontinuation symptoms have been reported with mirtazapine, bupropion, or duloxetine.
Table 3
SSRI discontinuation syndrome: The patient at risk…
| Is taking an SSRI with a relatively short half-life |
| Has received antidepressant treatment > 4 weeks |
| Has history of treatment-emergent anxiety, discontinuation symptoms, nonadherence |
| SSRI: Selective serotonin reuptake inhibitor |
| Source: Reference 7 |
Causes. Theories to explain SSRI discontinuation syndrome include cholinergic rebound,12 as described with TCAs, or a decrease in available synaptic serotonin coinciding with down-regulated serotonin receptors.13 Paroxetine’s pharmacologic properties—cholinergic effects, short halflife, and high potency of serotonin reuptake blockade—may explain its relatively high frequency of discontinuation symptoms.
Atypical Antipsychotic Discontinuation Syndromes
Except for aripiprazole—which is a partial dopamine receptor agonist—most atypical antipsychotics are serotonin-dopamine antagonists. Discontinuation syndrome occurs most commonly with clozapine.
Clozapine. Abruptly stopping clozapine can exacerbate psychosis or cause delirium, agitation, confusion, and diaphoresis. Less-common symptoms may include extrapyramidal effects, nausea, diarrhea, headache, or restlessness.14 Clozapine is a weak dopamine D2 antagonist and a potent antagonist at the serotonin 5HT2, alpha adrenergic, histaminergic, and anticholinergic receptors. Thus, rebound from cholinergic, serotonin, dopamine and/or adrenergic receptor supersensitivity is thought to cause its discontinuation syndrome.15
Other atypicals. Case reports describe tics and withdrawal-emergent dyskinesia with risperidone16 and supersensitivity psychosis and a cholinergic/serotonergic syndrome with olanzapine.17,18 Anecdotal reports suggest that abruptly discontinuing quetiapine can cause nausea, emesis, lightheadedness, diaphoresis, orthostasis, tachycardia, and nervousness.19,20 Although discontinuation syndromes have not been reported with ziprasidone or aripiprazole, tapering any atypical antipsychotic during discontinuation is prudent.
Benzodiazepine Discontinuation Syndromes
Benzodiazepines modulate the neurotransmitter activity of gamma-aminobutyric acid (GABA). They differ in their pharmacokinetic properties and have varying half-lives:
- chlordiazepoxide and diazepam have long half-lives (48 hours)
- clonazepam has an intermediate half-life (10 to 24 hours)
- alprazolam, lorazepam, and oxazepam have short half-lives (10 hours).
Abruptly discontinuing benzodiazepines can cause relapse or rebound of pretreatment symptoms. Rebound—with symptoms exceeding pretreatment levels—sometimes occurs after 4 weeks of therapy. The syndrome may last 1 to 3 weeks and is more common with agents having relatively short half-lives.21
Withdrawal. During benzodiazepine withdrawal, new symptoms emerge and pre-existing symptoms worsen. An autonomic component differentiates withdrawal from relapse or rebound. Prominent symptoms include excess sensitivity to light and sound, insomnia, tachycardia, mild systolic hypertension, anxiety, nausea, irritability, tremors, sweating, and abdominal distress. Less-common but serious symptoms include confusion, paranoid delusions, hallucinations, and seizures.22
Withdrawal symptoms are more likely to occur after 6 months of benzodiazepine therapy, when physical dependence also can develop. More-severe benzodiazepine discontinuation syndrome is associated with higher dosages, longer duration of therapy, shorter half-lives, and rapid tapers. Patient factors associated with withdrawal symptoms include:
- personality traits such as dependency and neuroticism
- high pretreatment anxious and depressive symptoms
- history of substance abuse or dependence.23
Preventing discontinuation syndromes
Antidepressants. For TCAs, no discontinuation protocols exist, although some experts suggest tapering regimens over 4 weeks to 3 months. For MAOIs, reducing dosages 10% per week has been suggested.24 The SSRI taper rate depends on the drug’s pharmacologic profile, how long the patient has been taking the SSRI, and the dosage.25
With paroxetine, for example, a gradual reduction of 10 mg/d per week is recommended, based on clinical trial experience. When you reach 20 mg/d, continue this dosage for 1 week before stopping treatment. If reducing a dosage or discontinuing paroxetine causes intolerable symptoms, consider resuming the previously prescribed dosage and then taper more gradually.26
Also gradually taper other SSRIs with short half-lives. Suggested taper regimens for sertraline and fluvoxamine call for weekly reductions of 50 mg/d until you reach 25 to 50 mg. It is not unusual for this final dosage to be lower than the starting dosage.25 Substituting fluoxetine—with its longer half-life—for other SSRIs at the end of treatment has been suggested to suppress withdrawal symptoms,27 although the safety and efficacy of this approach is unknown.5 With venlafaxine, taper over a minimum of 2 to 4 weeks.28
Antipsychotics. To prevent psychotic relapse when discontinuing clozapine, some experts advocate starting a new antipsychotic in a therapeutic dosage before stopping clozapine. When highdose clozapine must be withdrawn immediately, hospitalize the patient and consider using cholinergics to prevent cholinergic rebound.15
Data on managing discontinuation syndromes associated with risperidone, olanzapine, or quetiapine are limited. In some cases, reinstituting the original drug, gradually tapering the antipsychotic,18,19 or using prochlorperazine20 have been useful.
Benzodiazepines. Taper oral benzodiazepines if a patient has taken them >4 to 6 weeks. Also taper IV midazolam used >7 days to sedate a critically ill patient. For the elderly, an 8- to 10-week taper may be required to discontinue benzodiazepines used >3 months for insomnia.
The American Psychiatric Association practice guideline for patients with panic disorder29 recommends tapering benzodiazepines across 2 to 4 months, reducing dosages not more than 10% weekly. Another option is to reduce the daily dosage by 25% per week, but close monitoring and flexibility are required during this taper.
Outcomes when tapering benzodiazepines, according to Rickels et al,23 depend less on pharmacologic adjuvant treatment than on benzodiazepine dosage before the taper, initial psychopathology severity, and patient personality traits (such as passivity/dependency). Before tapering, those authors recommend that you:
- establish a stable patient-physician relationship
- aggressively treat clinically significant anxiety and depression symptoms with medication or other means while the patient continues the established benzodiazepine dosage.
When the taper is nearly complete, maintain the reduced benzodiazepine dosage several months before the final taper.23 Carbamazepine, imipramine, valproate, or trazodone may help alleviate benzodiazepine discontinuation symptoms in select patients.21
When discontinuation occurs
Medical comorbidity. Common medical conditions, including pregnancy or acute surgical procedures, may necessitate abrupt psychotropic discontinuation (Table 4).
Because up to 30% of medical patients have a psychiatric disorder,30 primary care physicians often start psychotropics to manage anxiety and depressive symptoms and may seek psychiatric advice when switching or stopping medications. Moreover, 10% to 15% of hospitalized medically ill patients require dosage reduction or discontinuation of psychotropics that are contributing to the clinical presentation.31
Table 4
Common conditions requiring abrupt psychotropic discontinuation
|
Switching. When switching psychotropics, effects from the first psychotropic may appear to be adverse effects of the new psychotropic. Thus, unrecognized discontinuation syndromes may lead to unnecessary treatment changes.
In our experience, a general rule is to cross-taper and decrease the psychotropic being discontinued by 10% every 1 to 2 weeks. Prescribe adequate dosages of the new psychotropic, closely monitor vital signs, and watch for emerging discontinuation symptoms.
Pregnancy. For women who become pregnant while taking psychotropics, consider the patient’s psychiatric stability, week of pregnancy, psychotropic agent, and treatment preferences when adjusting the treatment plan. In one study of 34 women who stopped psychotropics abruptly for fear of harming the fetus:
- 26 (70%) reported physical and psychological adverse effects
- 11 (30%) reported suicidal ideation, and 4 were hospitalized.32
Patient education. In the study described above, some of the pregnant women’s physicians were unaware of the risks associated with abrupt psychotropic discontinuation and others were aware but failed to inform their patients.32 Thus, patient and family/caregiver education is important. When stopping psychotropics, discuss their risks/benefits, address unrealistic expectations, and individualize therapy by tapering and providing adequate dosing. Watch for suicidality; a weekly telephone call might be useful.
Related resource
- Hardman JG, Limbird LE, Gilman AG. Goodman & Gilman’s the pharmacological basis of therapeutics (10th ed). New York: McGraw-Hill, 2001.
Drug Brand Names
- Alprazolam • Xanax
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Carbamazepine • Equetro, Tegretol
- Chlordiazepoxide • Librium
- Citalopram • Celexa
- Clonazepam • Klonopin
- Clozapine • Clozaril
- Diazepam • Valium
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Imipramine • Tofranil
- Lorazepam • Ativan
- Mirtazapine • Remeron
- Oxazepam • Serax
- Paroxetine • Paxil
- Phenelzine • Nardil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Tranylcypromine • Parnate
- Trazodone • Desyrel
- Sertraline • Zoloft
- Valproate • Depakene
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Young AH, Currie A. Physicians’ knowledge of antidepressant withdrawal effects: a survey. J Clin Psychiatry 1997;58(7):28-30.
2. Dilsaver SC, Greden JF, Snider RM. Antidepressant withdrawal syndromes: phenomenology and pathophysiology. Int Clin Psychopharmacol 1987;2(1):1-19.
3. Liskin B, Roose S, Walsh T. Acute psychosis following phenelzine discontinuation. J Clin Psychopharmacol 1985;5:46-7.
4. Coupland NJ, Bell CJ, Potokar JP. Serotonin reuptake inhibitor withdrawal. J Clin Psychopharmacol 1996;16(5):356-62.
5. Haddad PM. Antidepressant discontinuation syndromes. Drug Safety 2001;24(3):183-97.
6. Haddad P. The SSRI discontinuation syndrome. J Psychopharmacol 1998;2(3):305-13.
7. Schatzberg AF, Haddad P, Kaplan EM, et al. for the Discontinuation Consensus Panel Serotonin reuptake inhibitor discontinuation syndrome: a hypothetical definition. J Clin Psychiatry 1997;58(S7):5-10.
8. Markowitz JS, DeVane CL, Liston HL, et al. An assessment of selective serotonin reuptake inhibitor discontinuation symptoms with citalopram. Int Clin Psychopharmacol 2000;15(6):329-33.
9. Bezchlibnyk-Butler K, Aleksic I, Kennedy SH. Citalopram—a review of pharmacological and clinical effects. J Psychiatry Neurosci 2000;25(3):241-54.
10. Price JS, Waller PC, Wood SM, et al. A comparison of the post-marketing safety of four selective serotonin reuptake inhibitors, including the investigation of symptoms occurring on withdrawal. Br J Clin Pharmacol 1996;42:757-63.
11. Fava M, Mulroy R, Alpert J, et al. Emergence of adverse events following discontinuation of treatment with extended-release venlafaxine. Am J Psychiatry 1997;154(12):1760-2.
12. Barr LC, Goodman WK, Price LH. Physical symptoms associated with paroxetine discontinuation. Am J Psychiatry 1994;151(2):289.-
13. Schatzberg AF, Haddad P, Kaplan EM, et al. for the Discontinuation Consensus Panel Possible biological mechanisms of the serotonin reuptake inhibitor discontinuation syndrome. J Clin Psychiatry 1997;58(S7):23-7.
14. Shore D. Clinical implications of clozapine discontinuation: report of an NIMH workshop. Schizophr Bull 1995;21(2):333-8.
15. de Leon J, Stanilla JK, White AO, Simpson GM. Anticholinergics to treat clozapine withdrawal. J Clin Psychiatry 1994;55(3):119-20.
16. Rosebush PI, Kennedy K, Dalton B, Mazurek MF. Protracted akathisia after risperidone withdrawal. Am J Psychiatry 1997;154(3):437-8.
17. Llorca PM, Vaiva G, Lancon C. Supersensitivity psychosis in patients with schizophrenia after sudden olanzapine withdrawal. Can J Psychiatry 2001;46(1):87-8.
18. Nayudu SK, Scheftner WA. Case report of withdrawal syndrome after olanzapine discontinuation. J Clin Psychopharmacol 2000;20:489-90.
19. Thurstone CC, Alahi P. A possible case of quetiapine withdrawal syndrome. J Clin Psychiatry 2000;61:602-3.
20. Kim DR, Staab JP. Quetiapine discontinuation syndrome. Am J Psychiatry 2005 May;162(5):1020.-
21. McLean W, Ariano R. Benzodiazepine withdrawal schedule and symptoms In: Klasco RK (ed). DRUGDEX® System (vol. 124). Greenwood Village, CO: Thomson Micromedex, 2005.
22. Greenblatt DJ, Miller LG, Shader RI. Benzodiazepine discontinuation syndromes. J Psychiatr Res 1990;24(S2):73-9.
23. Rickels K, Schweizer E, Case WG, Greenblatt DJ. Long-term therapeutic use of benzodiazepines. I. Effects of abrupt discontinuation. Arch Gen Psychiatry 1990;47(10):899-907.
24. Lejoyeux M, Ades J, Mourad I, et al. Antidepressant withdrawal syndrome: recognition, prevalence and management. CNS Drugs 1996;5:278-92.
25. Rosenbaum JF, Zajecka J. Clinical management of antidepressant discontinuation. J Clin Psychiatry 1997;58(S7):37-40.
26. Paxil (paroxetine) package labeling GlaxoSmithKline, 2002.
27. Keuthen NJ, Cyr P, Ricciardi JA, et al. Medication withdrawal symptoms in obsessive-compulsive disorder patients treated with paroxetine. J Clin Psychopharmacol 1994;14(3):206-7.
28. Dallal A, Chouinard G. Withdrawal and rebound symptoms associated with abrupt discontinuation of venlafaxine. J Clin Psychopharmacol 1998;18(4):343-4.
29. American Psychiatric Association Work Group on Panic Disorder Practice guideline for the treatment of patients with panic disorder. Am J Psychiatry 1998;155(S5):1-34.
30. Spitzer RL, Williams JB, Kroenke K, et al. Utility of a new procedure for diagnosing mental disorders in primary care. The PRIME-MD 1000 study. JAMA 1994;272(22):1749-56.
31. Bronheim HE, Fulop G, Kunkel EJ, et al. The Academy of Psychosomatic Medicine practice guidelines for psychiatric consultation in the general medical setting. Psychosomatics 1998;39(4):S8-30.
32. Einarson A, Selby P, Koren G. Abrupt discontinuation of psychotropic drugs during pregnancy: fear of teratogenic risk and impact of counseling. J Psychiatry Neurosci 2001;26(1):44-8.
Abruptly stopping common psychotropics—particularly antidepressants, benzodiazepines, or atypical antipsychotics—can trigger a discontinuation syndrome, with:
- rebound or relapse of original symptoms
- uncomfortable new physical and psychological symptoms
- physiologic withdrawal at times.
To increase health professionals’ awareness of the risk of these adverse effects,1 this article describes discontinuation syndromes associated with various psychotropics and offers strategies to anticipate, recognize, and manage them.
Antidepressant Discontinuation Syndromes
Discontinuation syndromes can occur with tricyclic and tetracyclic antidepressants (TCAs), monoamine oxidase inhibitors (MAOIs), selective serotonin reuptake inhibitors (SSRIs), and other newer antidepressants. Symptoms usually start within a few days of stopping a drug—or less commonly, reducing its dosage—and are usually mild and self-limited. Serious outcomes have been reported.
Distinguishing antidepressant discontinuation symptoms from depression recurrence is important. Discontinuation symptoms emerge within 1 to 3 days, whereas depressive symptoms usually occur 2 to 3 weeks after an antidepressant is stopped. Discontinuation reactions remit within a few days, especially if the antidepressant is re-instituted.
TCAs block serotonin and norepinephrine reuptake, increasing the availability of these biogenic amines at receptor sites in the brain and other tissues. Abrupt discontinuation can cause physical symptoms—such as lethargy, headache, and tremor—and psychological symptoms including irritability, anxiety, agitation, and low mood (Table 1).2
Long-term use of TCAs with potent anticholinergic properties leads to upregulation of postsynaptic muscarinic receptors, creating a “supersensitive” state. Abrupt discontinuation can cause cholinergic rebound, with symptoms emerging as soon as 12 hours—but typically 24 to 48 hours—after the last dose.
Table 1
Discontinuation symptoms seen with TCAs
| Physical symptoms | Lethargy, headache, tremor, sweating, anorexia, insomnia, nausea, vomiting, diarrhea, akathisia (rare), parkinsonism (rare) |
| Psychological symptoms | Irritability, anxiety/agitation, low mood, excessive dreaming, nightmares, paradoxical activation resulting in manic/hypomanic symptoms (rare) |
| TCA: Tricyclic antidepressants | |
| Source: Reference 2 | |
MAOIs such as phenelzine and tranylcypromine inhibit the enzyme monoamine oxidase, which is responsible for monoamine degradation and increases synaptic monoamine concentrations. Discontinuation syndromes may include acute confusional states, paranoid delusions, hallucinations, or worsening of depressive symptoms.3 These problems rarely occur in clinical practice, however, because MAOIs’ serious side effects discourage doctors from prescribing them.
SSRIs and other agents. SSRIs block synaptic reuptake of serotonin. Serotonin-norepinephrine reuptake inhibitors (SNRIs) such as venlafaxine and duloxetine inhibit both serotonin and norepinephrine reuptake. Mirtazapine—an alpha2-adrenergic and heteroreceptor antagonist—increases serotonin and norepinephrine at the synapse. Bupropion increases dopamine and norepinephrine turnover in the CNS and also blocks serotonin.
Up to 30% of patients who stop taking SSRIs develop discontinuation symptoms.4 Six symptom clusters—disequilibrium, sensory symptoms, general somatic symptoms, sleep disturbance, GI symptoms, and affective symptoms—characterize the SSRI discontinuation syndrome (Table 2).5 The four most common symptoms—in decreasing order of frequency—are dizziness, nausea, lethargy, and headache.6 Ataxia, sensory abnormalities, and possibly aggressive and impulsive behavior differentiate this discontinuation syndrome from that of the TCAs.
Table 2
Discontinuation symptoms seen with SSRIs
| Type | Symptoms |
|---|---|
| Disequilibrium | Lightheadedness/dizziness, vertigo, ataxia |
| Sensory symptoms | Paraesthesia, numbness, electric shock-like sensations |
| General somatic symptoms | Lethargy, headache, tremor, sweating, anorexia |
| Sleep disturbance | Insomnia, nightmares, excessive dreaming |
| GI symptoms | Nausea, vomiting, diarrhea |
| Affective symptoms | Irritability, anxiety/agitation, low mood |
| SSRIs: Selective serotonin reuptake inhibitors | |
| Source: Reference 5 | |
Risk factors. Risk factors for SSRI discontinuation syndrome have been identified (Table 3).7 Symptoms usually begin 1 to 3 days after an SSRI is abruptly stopped and are usually mild. However, some patients report falls, inability to work, and difficulty walking and driving. Untreated symptoms are short-lived and remit within 1 to 2 weeks. They also remit if the original antidepressant is reintroduced or a pharmacologically similar agent is substituted.
Discontinuation syndrome risk among SSRIs is highest for paroxetine, intermediate for sertraline and fluvoxamine, and lowest for fluoxetine.4 Citalopram may cause a mild and transient discontinuation syndrome.8 Citalopram’s long elimination half-life (30 to 35 hours) and fewer and much less-potent active metabolites9 may explain its relatively low risk of discontinuation symptoms.
Discontinuation reactions have been reported to occur 100 times more frequently with paroxetine than with fluoxetine.10 Fluoxetine’s lower rate could be explained by its 2- to 3-day half-life, compared with half-lives of 33 hours or less for paroxetine, sertraline, citalopram, and fluvoxamine. A longer half-life might protect against a discontinuation syndrome.
Among other newer antidepressants:
- venlafaxine’s discontinuation syndrome is similar to the SSRI syndrome11
- no discontinuation symptoms have been reported with mirtazapine, bupropion, or duloxetine.
Table 3
SSRI discontinuation syndrome: The patient at risk…
| Is taking an SSRI with a relatively short half-life |
| Has received antidepressant treatment > 4 weeks |
| Has history of treatment-emergent anxiety, discontinuation symptoms, nonadherence |
| SSRI: Selective serotonin reuptake inhibitor |
| Source: Reference 7 |
Causes. Theories to explain SSRI discontinuation syndrome include cholinergic rebound,12 as described with TCAs, or a decrease in available synaptic serotonin coinciding with down-regulated serotonin receptors.13 Paroxetine’s pharmacologic properties—cholinergic effects, short halflife, and high potency of serotonin reuptake blockade—may explain its relatively high frequency of discontinuation symptoms.
Atypical Antipsychotic Discontinuation Syndromes
Except for aripiprazole—which is a partial dopamine receptor agonist—most atypical antipsychotics are serotonin-dopamine antagonists. Discontinuation syndrome occurs most commonly with clozapine.
Clozapine. Abruptly stopping clozapine can exacerbate psychosis or cause delirium, agitation, confusion, and diaphoresis. Less-common symptoms may include extrapyramidal effects, nausea, diarrhea, headache, or restlessness.14 Clozapine is a weak dopamine D2 antagonist and a potent antagonist at the serotonin 5HT2, alpha adrenergic, histaminergic, and anticholinergic receptors. Thus, rebound from cholinergic, serotonin, dopamine and/or adrenergic receptor supersensitivity is thought to cause its discontinuation syndrome.15
Other atypicals. Case reports describe tics and withdrawal-emergent dyskinesia with risperidone16 and supersensitivity psychosis and a cholinergic/serotonergic syndrome with olanzapine.17,18 Anecdotal reports suggest that abruptly discontinuing quetiapine can cause nausea, emesis, lightheadedness, diaphoresis, orthostasis, tachycardia, and nervousness.19,20 Although discontinuation syndromes have not been reported with ziprasidone or aripiprazole, tapering any atypical antipsychotic during discontinuation is prudent.
Benzodiazepine Discontinuation Syndromes
Benzodiazepines modulate the neurotransmitter activity of gamma-aminobutyric acid (GABA). They differ in their pharmacokinetic properties and have varying half-lives:
- chlordiazepoxide and diazepam have long half-lives (48 hours)
- clonazepam has an intermediate half-life (10 to 24 hours)
- alprazolam, lorazepam, and oxazepam have short half-lives (10 hours).
Abruptly discontinuing benzodiazepines can cause relapse or rebound of pretreatment symptoms. Rebound—with symptoms exceeding pretreatment levels—sometimes occurs after 4 weeks of therapy. The syndrome may last 1 to 3 weeks and is more common with agents having relatively short half-lives.21
Withdrawal. During benzodiazepine withdrawal, new symptoms emerge and pre-existing symptoms worsen. An autonomic component differentiates withdrawal from relapse or rebound. Prominent symptoms include excess sensitivity to light and sound, insomnia, tachycardia, mild systolic hypertension, anxiety, nausea, irritability, tremors, sweating, and abdominal distress. Less-common but serious symptoms include confusion, paranoid delusions, hallucinations, and seizures.22
Withdrawal symptoms are more likely to occur after 6 months of benzodiazepine therapy, when physical dependence also can develop. More-severe benzodiazepine discontinuation syndrome is associated with higher dosages, longer duration of therapy, shorter half-lives, and rapid tapers. Patient factors associated with withdrawal symptoms include:
- personality traits such as dependency and neuroticism
- high pretreatment anxious and depressive symptoms
- history of substance abuse or dependence.23
Preventing discontinuation syndromes
Antidepressants. For TCAs, no discontinuation protocols exist, although some experts suggest tapering regimens over 4 weeks to 3 months. For MAOIs, reducing dosages 10% per week has been suggested.24 The SSRI taper rate depends on the drug’s pharmacologic profile, how long the patient has been taking the SSRI, and the dosage.25
With paroxetine, for example, a gradual reduction of 10 mg/d per week is recommended, based on clinical trial experience. When you reach 20 mg/d, continue this dosage for 1 week before stopping treatment. If reducing a dosage or discontinuing paroxetine causes intolerable symptoms, consider resuming the previously prescribed dosage and then taper more gradually.26
Also gradually taper other SSRIs with short half-lives. Suggested taper regimens for sertraline and fluvoxamine call for weekly reductions of 50 mg/d until you reach 25 to 50 mg. It is not unusual for this final dosage to be lower than the starting dosage.25 Substituting fluoxetine—with its longer half-life—for other SSRIs at the end of treatment has been suggested to suppress withdrawal symptoms,27 although the safety and efficacy of this approach is unknown.5 With venlafaxine, taper over a minimum of 2 to 4 weeks.28
Antipsychotics. To prevent psychotic relapse when discontinuing clozapine, some experts advocate starting a new antipsychotic in a therapeutic dosage before stopping clozapine. When highdose clozapine must be withdrawn immediately, hospitalize the patient and consider using cholinergics to prevent cholinergic rebound.15
Data on managing discontinuation syndromes associated with risperidone, olanzapine, or quetiapine are limited. In some cases, reinstituting the original drug, gradually tapering the antipsychotic,18,19 or using prochlorperazine20 have been useful.
Benzodiazepines. Taper oral benzodiazepines if a patient has taken them >4 to 6 weeks. Also taper IV midazolam used >7 days to sedate a critically ill patient. For the elderly, an 8- to 10-week taper may be required to discontinue benzodiazepines used >3 months for insomnia.
The American Psychiatric Association practice guideline for patients with panic disorder29 recommends tapering benzodiazepines across 2 to 4 months, reducing dosages not more than 10% weekly. Another option is to reduce the daily dosage by 25% per week, but close monitoring and flexibility are required during this taper.
Outcomes when tapering benzodiazepines, according to Rickels et al,23 depend less on pharmacologic adjuvant treatment than on benzodiazepine dosage before the taper, initial psychopathology severity, and patient personality traits (such as passivity/dependency). Before tapering, those authors recommend that you:
- establish a stable patient-physician relationship
- aggressively treat clinically significant anxiety and depression symptoms with medication or other means while the patient continues the established benzodiazepine dosage.
When the taper is nearly complete, maintain the reduced benzodiazepine dosage several months before the final taper.23 Carbamazepine, imipramine, valproate, or trazodone may help alleviate benzodiazepine discontinuation symptoms in select patients.21
When discontinuation occurs
Medical comorbidity. Common medical conditions, including pregnancy or acute surgical procedures, may necessitate abrupt psychotropic discontinuation (Table 4).
Because up to 30% of medical patients have a psychiatric disorder,30 primary care physicians often start psychotropics to manage anxiety and depressive symptoms and may seek psychiatric advice when switching or stopping medications. Moreover, 10% to 15% of hospitalized medically ill patients require dosage reduction or discontinuation of psychotropics that are contributing to the clinical presentation.31
Table 4
Common conditions requiring abrupt psychotropic discontinuation
|
Switching. When switching psychotropics, effects from the first psychotropic may appear to be adverse effects of the new psychotropic. Thus, unrecognized discontinuation syndromes may lead to unnecessary treatment changes.
In our experience, a general rule is to cross-taper and decrease the psychotropic being discontinued by 10% every 1 to 2 weeks. Prescribe adequate dosages of the new psychotropic, closely monitor vital signs, and watch for emerging discontinuation symptoms.
Pregnancy. For women who become pregnant while taking psychotropics, consider the patient’s psychiatric stability, week of pregnancy, psychotropic agent, and treatment preferences when adjusting the treatment plan. In one study of 34 women who stopped psychotropics abruptly for fear of harming the fetus:
- 26 (70%) reported physical and psychological adverse effects
- 11 (30%) reported suicidal ideation, and 4 were hospitalized.32
Patient education. In the study described above, some of the pregnant women’s physicians were unaware of the risks associated with abrupt psychotropic discontinuation and others were aware but failed to inform their patients.32 Thus, patient and family/caregiver education is important. When stopping psychotropics, discuss their risks/benefits, address unrealistic expectations, and individualize therapy by tapering and providing adequate dosing. Watch for suicidality; a weekly telephone call might be useful.
Related resource
- Hardman JG, Limbird LE, Gilman AG. Goodman & Gilman’s the pharmacological basis of therapeutics (10th ed). New York: McGraw-Hill, 2001.
Drug Brand Names
- Alprazolam • Xanax
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Carbamazepine • Equetro, Tegretol
- Chlordiazepoxide • Librium
- Citalopram • Celexa
- Clonazepam • Klonopin
- Clozapine • Clozaril
- Diazepam • Valium
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Imipramine • Tofranil
- Lorazepam • Ativan
- Mirtazapine • Remeron
- Oxazepam • Serax
- Paroxetine • Paxil
- Phenelzine • Nardil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Tranylcypromine • Parnate
- Trazodone • Desyrel
- Sertraline • Zoloft
- Valproate • Depakene
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Abruptly stopping common psychotropics—particularly antidepressants, benzodiazepines, or atypical antipsychotics—can trigger a discontinuation syndrome, with:
- rebound or relapse of original symptoms
- uncomfortable new physical and psychological symptoms
- physiologic withdrawal at times.
To increase health professionals’ awareness of the risk of these adverse effects,1 this article describes discontinuation syndromes associated with various psychotropics and offers strategies to anticipate, recognize, and manage them.
Antidepressant Discontinuation Syndromes
Discontinuation syndromes can occur with tricyclic and tetracyclic antidepressants (TCAs), monoamine oxidase inhibitors (MAOIs), selective serotonin reuptake inhibitors (SSRIs), and other newer antidepressants. Symptoms usually start within a few days of stopping a drug—or less commonly, reducing its dosage—and are usually mild and self-limited. Serious outcomes have been reported.
Distinguishing antidepressant discontinuation symptoms from depression recurrence is important. Discontinuation symptoms emerge within 1 to 3 days, whereas depressive symptoms usually occur 2 to 3 weeks after an antidepressant is stopped. Discontinuation reactions remit within a few days, especially if the antidepressant is re-instituted.
TCAs block serotonin and norepinephrine reuptake, increasing the availability of these biogenic amines at receptor sites in the brain and other tissues. Abrupt discontinuation can cause physical symptoms—such as lethargy, headache, and tremor—and psychological symptoms including irritability, anxiety, agitation, and low mood (Table 1).2
Long-term use of TCAs with potent anticholinergic properties leads to upregulation of postsynaptic muscarinic receptors, creating a “supersensitive” state. Abrupt discontinuation can cause cholinergic rebound, with symptoms emerging as soon as 12 hours—but typically 24 to 48 hours—after the last dose.
Table 1
Discontinuation symptoms seen with TCAs
| Physical symptoms | Lethargy, headache, tremor, sweating, anorexia, insomnia, nausea, vomiting, diarrhea, akathisia (rare), parkinsonism (rare) |
| Psychological symptoms | Irritability, anxiety/agitation, low mood, excessive dreaming, nightmares, paradoxical activation resulting in manic/hypomanic symptoms (rare) |
| TCA: Tricyclic antidepressants | |
| Source: Reference 2 | |
MAOIs such as phenelzine and tranylcypromine inhibit the enzyme monoamine oxidase, which is responsible for monoamine degradation and increases synaptic monoamine concentrations. Discontinuation syndromes may include acute confusional states, paranoid delusions, hallucinations, or worsening of depressive symptoms.3 These problems rarely occur in clinical practice, however, because MAOIs’ serious side effects discourage doctors from prescribing them.
SSRIs and other agents. SSRIs block synaptic reuptake of serotonin. Serotonin-norepinephrine reuptake inhibitors (SNRIs) such as venlafaxine and duloxetine inhibit both serotonin and norepinephrine reuptake. Mirtazapine—an alpha2-adrenergic and heteroreceptor antagonist—increases serotonin and norepinephrine at the synapse. Bupropion increases dopamine and norepinephrine turnover in the CNS and also blocks serotonin.
Up to 30% of patients who stop taking SSRIs develop discontinuation symptoms.4 Six symptom clusters—disequilibrium, sensory symptoms, general somatic symptoms, sleep disturbance, GI symptoms, and affective symptoms—characterize the SSRI discontinuation syndrome (Table 2).5 The four most common symptoms—in decreasing order of frequency—are dizziness, nausea, lethargy, and headache.6 Ataxia, sensory abnormalities, and possibly aggressive and impulsive behavior differentiate this discontinuation syndrome from that of the TCAs.
Table 2
Discontinuation symptoms seen with SSRIs
| Type | Symptoms |
|---|---|
| Disequilibrium | Lightheadedness/dizziness, vertigo, ataxia |
| Sensory symptoms | Paraesthesia, numbness, electric shock-like sensations |
| General somatic symptoms | Lethargy, headache, tremor, sweating, anorexia |
| Sleep disturbance | Insomnia, nightmares, excessive dreaming |
| GI symptoms | Nausea, vomiting, diarrhea |
| Affective symptoms | Irritability, anxiety/agitation, low mood |
| SSRIs: Selective serotonin reuptake inhibitors | |
| Source: Reference 5 | |
Risk factors. Risk factors for SSRI discontinuation syndrome have been identified (Table 3).7 Symptoms usually begin 1 to 3 days after an SSRI is abruptly stopped and are usually mild. However, some patients report falls, inability to work, and difficulty walking and driving. Untreated symptoms are short-lived and remit within 1 to 2 weeks. They also remit if the original antidepressant is reintroduced or a pharmacologically similar agent is substituted.
Discontinuation syndrome risk among SSRIs is highest for paroxetine, intermediate for sertraline and fluvoxamine, and lowest for fluoxetine.4 Citalopram may cause a mild and transient discontinuation syndrome.8 Citalopram’s long elimination half-life (30 to 35 hours) and fewer and much less-potent active metabolites9 may explain its relatively low risk of discontinuation symptoms.
Discontinuation reactions have been reported to occur 100 times more frequently with paroxetine than with fluoxetine.10 Fluoxetine’s lower rate could be explained by its 2- to 3-day half-life, compared with half-lives of 33 hours or less for paroxetine, sertraline, citalopram, and fluvoxamine. A longer half-life might protect against a discontinuation syndrome.
Among other newer antidepressants:
- venlafaxine’s discontinuation syndrome is similar to the SSRI syndrome11
- no discontinuation symptoms have been reported with mirtazapine, bupropion, or duloxetine.
Table 3
SSRI discontinuation syndrome: The patient at risk…
| Is taking an SSRI with a relatively short half-life |
| Has received antidepressant treatment > 4 weeks |
| Has history of treatment-emergent anxiety, discontinuation symptoms, nonadherence |
| SSRI: Selective serotonin reuptake inhibitor |
| Source: Reference 7 |
Causes. Theories to explain SSRI discontinuation syndrome include cholinergic rebound,12 as described with TCAs, or a decrease in available synaptic serotonin coinciding with down-regulated serotonin receptors.13 Paroxetine’s pharmacologic properties—cholinergic effects, short halflife, and high potency of serotonin reuptake blockade—may explain its relatively high frequency of discontinuation symptoms.
Atypical Antipsychotic Discontinuation Syndromes
Except for aripiprazole—which is a partial dopamine receptor agonist—most atypical antipsychotics are serotonin-dopamine antagonists. Discontinuation syndrome occurs most commonly with clozapine.
Clozapine. Abruptly stopping clozapine can exacerbate psychosis or cause delirium, agitation, confusion, and diaphoresis. Less-common symptoms may include extrapyramidal effects, nausea, diarrhea, headache, or restlessness.14 Clozapine is a weak dopamine D2 antagonist and a potent antagonist at the serotonin 5HT2, alpha adrenergic, histaminergic, and anticholinergic receptors. Thus, rebound from cholinergic, serotonin, dopamine and/or adrenergic receptor supersensitivity is thought to cause its discontinuation syndrome.15
Other atypicals. Case reports describe tics and withdrawal-emergent dyskinesia with risperidone16 and supersensitivity psychosis and a cholinergic/serotonergic syndrome with olanzapine.17,18 Anecdotal reports suggest that abruptly discontinuing quetiapine can cause nausea, emesis, lightheadedness, diaphoresis, orthostasis, tachycardia, and nervousness.19,20 Although discontinuation syndromes have not been reported with ziprasidone or aripiprazole, tapering any atypical antipsychotic during discontinuation is prudent.
Benzodiazepine Discontinuation Syndromes
Benzodiazepines modulate the neurotransmitter activity of gamma-aminobutyric acid (GABA). They differ in their pharmacokinetic properties and have varying half-lives:
- chlordiazepoxide and diazepam have long half-lives (48 hours)
- clonazepam has an intermediate half-life (10 to 24 hours)
- alprazolam, lorazepam, and oxazepam have short half-lives (10 hours).
Abruptly discontinuing benzodiazepines can cause relapse or rebound of pretreatment symptoms. Rebound—with symptoms exceeding pretreatment levels—sometimes occurs after 4 weeks of therapy. The syndrome may last 1 to 3 weeks and is more common with agents having relatively short half-lives.21
Withdrawal. During benzodiazepine withdrawal, new symptoms emerge and pre-existing symptoms worsen. An autonomic component differentiates withdrawal from relapse or rebound. Prominent symptoms include excess sensitivity to light and sound, insomnia, tachycardia, mild systolic hypertension, anxiety, nausea, irritability, tremors, sweating, and abdominal distress. Less-common but serious symptoms include confusion, paranoid delusions, hallucinations, and seizures.22
Withdrawal symptoms are more likely to occur after 6 months of benzodiazepine therapy, when physical dependence also can develop. More-severe benzodiazepine discontinuation syndrome is associated with higher dosages, longer duration of therapy, shorter half-lives, and rapid tapers. Patient factors associated with withdrawal symptoms include:
- personality traits such as dependency and neuroticism
- high pretreatment anxious and depressive symptoms
- history of substance abuse or dependence.23
Preventing discontinuation syndromes
Antidepressants. For TCAs, no discontinuation protocols exist, although some experts suggest tapering regimens over 4 weeks to 3 months. For MAOIs, reducing dosages 10% per week has been suggested.24 The SSRI taper rate depends on the drug’s pharmacologic profile, how long the patient has been taking the SSRI, and the dosage.25
With paroxetine, for example, a gradual reduction of 10 mg/d per week is recommended, based on clinical trial experience. When you reach 20 mg/d, continue this dosage for 1 week before stopping treatment. If reducing a dosage or discontinuing paroxetine causes intolerable symptoms, consider resuming the previously prescribed dosage and then taper more gradually.26
Also gradually taper other SSRIs with short half-lives. Suggested taper regimens for sertraline and fluvoxamine call for weekly reductions of 50 mg/d until you reach 25 to 50 mg. It is not unusual for this final dosage to be lower than the starting dosage.25 Substituting fluoxetine—with its longer half-life—for other SSRIs at the end of treatment has been suggested to suppress withdrawal symptoms,27 although the safety and efficacy of this approach is unknown.5 With venlafaxine, taper over a minimum of 2 to 4 weeks.28
Antipsychotics. To prevent psychotic relapse when discontinuing clozapine, some experts advocate starting a new antipsychotic in a therapeutic dosage before stopping clozapine. When highdose clozapine must be withdrawn immediately, hospitalize the patient and consider using cholinergics to prevent cholinergic rebound.15
Data on managing discontinuation syndromes associated with risperidone, olanzapine, or quetiapine are limited. In some cases, reinstituting the original drug, gradually tapering the antipsychotic,18,19 or using prochlorperazine20 have been useful.
Benzodiazepines. Taper oral benzodiazepines if a patient has taken them >4 to 6 weeks. Also taper IV midazolam used >7 days to sedate a critically ill patient. For the elderly, an 8- to 10-week taper may be required to discontinue benzodiazepines used >3 months for insomnia.
The American Psychiatric Association practice guideline for patients with panic disorder29 recommends tapering benzodiazepines across 2 to 4 months, reducing dosages not more than 10% weekly. Another option is to reduce the daily dosage by 25% per week, but close monitoring and flexibility are required during this taper.
Outcomes when tapering benzodiazepines, according to Rickels et al,23 depend less on pharmacologic adjuvant treatment than on benzodiazepine dosage before the taper, initial psychopathology severity, and patient personality traits (such as passivity/dependency). Before tapering, those authors recommend that you:
- establish a stable patient-physician relationship
- aggressively treat clinically significant anxiety and depression symptoms with medication or other means while the patient continues the established benzodiazepine dosage.
When the taper is nearly complete, maintain the reduced benzodiazepine dosage several months before the final taper.23 Carbamazepine, imipramine, valproate, or trazodone may help alleviate benzodiazepine discontinuation symptoms in select patients.21
When discontinuation occurs
Medical comorbidity. Common medical conditions, including pregnancy or acute surgical procedures, may necessitate abrupt psychotropic discontinuation (Table 4).
Because up to 30% of medical patients have a psychiatric disorder,30 primary care physicians often start psychotropics to manage anxiety and depressive symptoms and may seek psychiatric advice when switching or stopping medications. Moreover, 10% to 15% of hospitalized medically ill patients require dosage reduction or discontinuation of psychotropics that are contributing to the clinical presentation.31
Table 4
Common conditions requiring abrupt psychotropic discontinuation
|
Switching. When switching psychotropics, effects from the first psychotropic may appear to be adverse effects of the new psychotropic. Thus, unrecognized discontinuation syndromes may lead to unnecessary treatment changes.
In our experience, a general rule is to cross-taper and decrease the psychotropic being discontinued by 10% every 1 to 2 weeks. Prescribe adequate dosages of the new psychotropic, closely monitor vital signs, and watch for emerging discontinuation symptoms.
Pregnancy. For women who become pregnant while taking psychotropics, consider the patient’s psychiatric stability, week of pregnancy, psychotropic agent, and treatment preferences when adjusting the treatment plan. In one study of 34 women who stopped psychotropics abruptly for fear of harming the fetus:
- 26 (70%) reported physical and psychological adverse effects
- 11 (30%) reported suicidal ideation, and 4 were hospitalized.32
Patient education. In the study described above, some of the pregnant women’s physicians were unaware of the risks associated with abrupt psychotropic discontinuation and others were aware but failed to inform their patients.32 Thus, patient and family/caregiver education is important. When stopping psychotropics, discuss their risks/benefits, address unrealistic expectations, and individualize therapy by tapering and providing adequate dosing. Watch for suicidality; a weekly telephone call might be useful.
Related resource
- Hardman JG, Limbird LE, Gilman AG. Goodman & Gilman’s the pharmacological basis of therapeutics (10th ed). New York: McGraw-Hill, 2001.
Drug Brand Names
- Alprazolam • Xanax
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Carbamazepine • Equetro, Tegretol
- Chlordiazepoxide • Librium
- Citalopram • Celexa
- Clonazepam • Klonopin
- Clozapine • Clozaril
- Diazepam • Valium
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Imipramine • Tofranil
- Lorazepam • Ativan
- Mirtazapine • Remeron
- Oxazepam • Serax
- Paroxetine • Paxil
- Phenelzine • Nardil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Tranylcypromine • Parnate
- Trazodone • Desyrel
- Sertraline • Zoloft
- Valproate • Depakene
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Young AH, Currie A. Physicians’ knowledge of antidepressant withdrawal effects: a survey. J Clin Psychiatry 1997;58(7):28-30.
2. Dilsaver SC, Greden JF, Snider RM. Antidepressant withdrawal syndromes: phenomenology and pathophysiology. Int Clin Psychopharmacol 1987;2(1):1-19.
3. Liskin B, Roose S, Walsh T. Acute psychosis following phenelzine discontinuation. J Clin Psychopharmacol 1985;5:46-7.
4. Coupland NJ, Bell CJ, Potokar JP. Serotonin reuptake inhibitor withdrawal. J Clin Psychopharmacol 1996;16(5):356-62.
5. Haddad PM. Antidepressant discontinuation syndromes. Drug Safety 2001;24(3):183-97.
6. Haddad P. The SSRI discontinuation syndrome. J Psychopharmacol 1998;2(3):305-13.
7. Schatzberg AF, Haddad P, Kaplan EM, et al. for the Discontinuation Consensus Panel Serotonin reuptake inhibitor discontinuation syndrome: a hypothetical definition. J Clin Psychiatry 1997;58(S7):5-10.
8. Markowitz JS, DeVane CL, Liston HL, et al. An assessment of selective serotonin reuptake inhibitor discontinuation symptoms with citalopram. Int Clin Psychopharmacol 2000;15(6):329-33.
9. Bezchlibnyk-Butler K, Aleksic I, Kennedy SH. Citalopram—a review of pharmacological and clinical effects. J Psychiatry Neurosci 2000;25(3):241-54.
10. Price JS, Waller PC, Wood SM, et al. A comparison of the post-marketing safety of four selective serotonin reuptake inhibitors, including the investigation of symptoms occurring on withdrawal. Br J Clin Pharmacol 1996;42:757-63.
11. Fava M, Mulroy R, Alpert J, et al. Emergence of adverse events following discontinuation of treatment with extended-release venlafaxine. Am J Psychiatry 1997;154(12):1760-2.
12. Barr LC, Goodman WK, Price LH. Physical symptoms associated with paroxetine discontinuation. Am J Psychiatry 1994;151(2):289.-
13. Schatzberg AF, Haddad P, Kaplan EM, et al. for the Discontinuation Consensus Panel Possible biological mechanisms of the serotonin reuptake inhibitor discontinuation syndrome. J Clin Psychiatry 1997;58(S7):23-7.
14. Shore D. Clinical implications of clozapine discontinuation: report of an NIMH workshop. Schizophr Bull 1995;21(2):333-8.
15. de Leon J, Stanilla JK, White AO, Simpson GM. Anticholinergics to treat clozapine withdrawal. J Clin Psychiatry 1994;55(3):119-20.
16. Rosebush PI, Kennedy K, Dalton B, Mazurek MF. Protracted akathisia after risperidone withdrawal. Am J Psychiatry 1997;154(3):437-8.
17. Llorca PM, Vaiva G, Lancon C. Supersensitivity psychosis in patients with schizophrenia after sudden olanzapine withdrawal. Can J Psychiatry 2001;46(1):87-8.
18. Nayudu SK, Scheftner WA. Case report of withdrawal syndrome after olanzapine discontinuation. J Clin Psychopharmacol 2000;20:489-90.
19. Thurstone CC, Alahi P. A possible case of quetiapine withdrawal syndrome. J Clin Psychiatry 2000;61:602-3.
20. Kim DR, Staab JP. Quetiapine discontinuation syndrome. Am J Psychiatry 2005 May;162(5):1020.-
21. McLean W, Ariano R. Benzodiazepine withdrawal schedule and symptoms In: Klasco RK (ed). DRUGDEX® System (vol. 124). Greenwood Village, CO: Thomson Micromedex, 2005.
22. Greenblatt DJ, Miller LG, Shader RI. Benzodiazepine discontinuation syndromes. J Psychiatr Res 1990;24(S2):73-9.
23. Rickels K, Schweizer E, Case WG, Greenblatt DJ. Long-term therapeutic use of benzodiazepines. I. Effects of abrupt discontinuation. Arch Gen Psychiatry 1990;47(10):899-907.
24. Lejoyeux M, Ades J, Mourad I, et al. Antidepressant withdrawal syndrome: recognition, prevalence and management. CNS Drugs 1996;5:278-92.
25. Rosenbaum JF, Zajecka J. Clinical management of antidepressant discontinuation. J Clin Psychiatry 1997;58(S7):37-40.
26. Paxil (paroxetine) package labeling GlaxoSmithKline, 2002.
27. Keuthen NJ, Cyr P, Ricciardi JA, et al. Medication withdrawal symptoms in obsessive-compulsive disorder patients treated with paroxetine. J Clin Psychopharmacol 1994;14(3):206-7.
28. Dallal A, Chouinard G. Withdrawal and rebound symptoms associated with abrupt discontinuation of venlafaxine. J Clin Psychopharmacol 1998;18(4):343-4.
29. American Psychiatric Association Work Group on Panic Disorder Practice guideline for the treatment of patients with panic disorder. Am J Psychiatry 1998;155(S5):1-34.
30. Spitzer RL, Williams JB, Kroenke K, et al. Utility of a new procedure for diagnosing mental disorders in primary care. The PRIME-MD 1000 study. JAMA 1994;272(22):1749-56.
31. Bronheim HE, Fulop G, Kunkel EJ, et al. The Academy of Psychosomatic Medicine practice guidelines for psychiatric consultation in the general medical setting. Psychosomatics 1998;39(4):S8-30.
32. Einarson A, Selby P, Koren G. Abrupt discontinuation of psychotropic drugs during pregnancy: fear of teratogenic risk and impact of counseling. J Psychiatry Neurosci 2001;26(1):44-8.
1. Young AH, Currie A. Physicians’ knowledge of antidepressant withdrawal effects: a survey. J Clin Psychiatry 1997;58(7):28-30.
2. Dilsaver SC, Greden JF, Snider RM. Antidepressant withdrawal syndromes: phenomenology and pathophysiology. Int Clin Psychopharmacol 1987;2(1):1-19.
3. Liskin B, Roose S, Walsh T. Acute psychosis following phenelzine discontinuation. J Clin Psychopharmacol 1985;5:46-7.
4. Coupland NJ, Bell CJ, Potokar JP. Serotonin reuptake inhibitor withdrawal. J Clin Psychopharmacol 1996;16(5):356-62.
5. Haddad PM. Antidepressant discontinuation syndromes. Drug Safety 2001;24(3):183-97.
6. Haddad P. The SSRI discontinuation syndrome. J Psychopharmacol 1998;2(3):305-13.
7. Schatzberg AF, Haddad P, Kaplan EM, et al. for the Discontinuation Consensus Panel Serotonin reuptake inhibitor discontinuation syndrome: a hypothetical definition. J Clin Psychiatry 1997;58(S7):5-10.
8. Markowitz JS, DeVane CL, Liston HL, et al. An assessment of selective serotonin reuptake inhibitor discontinuation symptoms with citalopram. Int Clin Psychopharmacol 2000;15(6):329-33.
9. Bezchlibnyk-Butler K, Aleksic I, Kennedy SH. Citalopram—a review of pharmacological and clinical effects. J Psychiatry Neurosci 2000;25(3):241-54.
10. Price JS, Waller PC, Wood SM, et al. A comparison of the post-marketing safety of four selective serotonin reuptake inhibitors, including the investigation of symptoms occurring on withdrawal. Br J Clin Pharmacol 1996;42:757-63.
11. Fava M, Mulroy R, Alpert J, et al. Emergence of adverse events following discontinuation of treatment with extended-release venlafaxine. Am J Psychiatry 1997;154(12):1760-2.
12. Barr LC, Goodman WK, Price LH. Physical symptoms associated with paroxetine discontinuation. Am J Psychiatry 1994;151(2):289.-
13. Schatzberg AF, Haddad P, Kaplan EM, et al. for the Discontinuation Consensus Panel Possible biological mechanisms of the serotonin reuptake inhibitor discontinuation syndrome. J Clin Psychiatry 1997;58(S7):23-7.
14. Shore D. Clinical implications of clozapine discontinuation: report of an NIMH workshop. Schizophr Bull 1995;21(2):333-8.
15. de Leon J, Stanilla JK, White AO, Simpson GM. Anticholinergics to treat clozapine withdrawal. J Clin Psychiatry 1994;55(3):119-20.
16. Rosebush PI, Kennedy K, Dalton B, Mazurek MF. Protracted akathisia after risperidone withdrawal. Am J Psychiatry 1997;154(3):437-8.
17. Llorca PM, Vaiva G, Lancon C. Supersensitivity psychosis in patients with schizophrenia after sudden olanzapine withdrawal. Can J Psychiatry 2001;46(1):87-8.
18. Nayudu SK, Scheftner WA. Case report of withdrawal syndrome after olanzapine discontinuation. J Clin Psychopharmacol 2000;20:489-90.
19. Thurstone CC, Alahi P. A possible case of quetiapine withdrawal syndrome. J Clin Psychiatry 2000;61:602-3.
20. Kim DR, Staab JP. Quetiapine discontinuation syndrome. Am J Psychiatry 2005 May;162(5):1020.-
21. McLean W, Ariano R. Benzodiazepine withdrawal schedule and symptoms In: Klasco RK (ed). DRUGDEX® System (vol. 124). Greenwood Village, CO: Thomson Micromedex, 2005.
22. Greenblatt DJ, Miller LG, Shader RI. Benzodiazepine discontinuation syndromes. J Psychiatr Res 1990;24(S2):73-9.
23. Rickels K, Schweizer E, Case WG, Greenblatt DJ. Long-term therapeutic use of benzodiazepines. I. Effects of abrupt discontinuation. Arch Gen Psychiatry 1990;47(10):899-907.
24. Lejoyeux M, Ades J, Mourad I, et al. Antidepressant withdrawal syndrome: recognition, prevalence and management. CNS Drugs 1996;5:278-92.
25. Rosenbaum JF, Zajecka J. Clinical management of antidepressant discontinuation. J Clin Psychiatry 1997;58(S7):37-40.
26. Paxil (paroxetine) package labeling GlaxoSmithKline, 2002.
27. Keuthen NJ, Cyr P, Ricciardi JA, et al. Medication withdrawal symptoms in obsessive-compulsive disorder patients treated with paroxetine. J Clin Psychopharmacol 1994;14(3):206-7.
28. Dallal A, Chouinard G. Withdrawal and rebound symptoms associated with abrupt discontinuation of venlafaxine. J Clin Psychopharmacol 1998;18(4):343-4.
29. American Psychiatric Association Work Group on Panic Disorder Practice guideline for the treatment of patients with panic disorder. Am J Psychiatry 1998;155(S5):1-34.
30. Spitzer RL, Williams JB, Kroenke K, et al. Utility of a new procedure for diagnosing mental disorders in primary care. The PRIME-MD 1000 study. JAMA 1994;272(22):1749-56.
31. Bronheim HE, Fulop G, Kunkel EJ, et al. The Academy of Psychosomatic Medicine practice guidelines for psychiatric consultation in the general medical setting. Psychosomatics 1998;39(4):S8-30.
32. Einarson A, Selby P, Koren G. Abrupt discontinuation of psychotropic drugs during pregnancy: fear of teratogenic risk and impact of counseling. J Psychiatry Neurosci 2001;26(1):44-8.