User login
Strategies for maintaining resilience to the burnout threat
It sometimes seems that the pace of life, and its stresses, have spiraled out of control: There just never seems to be enough time to deal with all the directions in which we are pulled. This easily can lead to the exhaustion of physical or emotional strength or motivation, otherwise known as “burnout.” Burnout is physical or mental collapse caused by overwork or stress and we are all at risk of suffering it. Conflicting demands on our time, loss of control (real or imagined), and a diminishing sense of worth grind at us from every direction.
In general, having some control over schedule and hours worked is associated with reductions in burnout and improved job satisfaction.1 But this is not always the case. Well-intentioned efforts to reduce workload, such as the electronic medical records or physician order entry systems, have actually made the problem worse.2 The seeming level of control that comes with being the chair of an obstetrics and gynecology department does not necessarily reduce burnout rates,3 and neither does the perceived resilience of mental health professionals, who still report burnout rates that approach 25%.4
This article continues the focus on recalibrating work/life balance that began last month with “ObGyn burnout: ACOG takes aim,” by Lucia DiVenere, MA, and the peer-to-peer audiocast with Ms. DiVenere and myself titled “Is burnout on the rise and what are the signs ObGyns should be on the lookout for?” Here, I identify the causes and symptoms of burnout and provide specific tools to help you develop resilience.
Who is most at risk for burnout?
Estimates range from 40% to 75% of ObGyns currently suffer from professional burnout, making the lifetime risk a virtual certainty.1−3 The idea of professional burnout is not new, but wider recognition of the alarming rates of burnout is very current.4,5 A recent survey of gynecologic oncologists6 found that of those studied 30% scored high for emotional exhaustion, 10% high for depersonalization, and 11% low for personal accomplishment. Overall, 32% of physicians had scores indicating burnout. More worrisome was that 33% screened positive for depression, 13% had a history of suicidal ideation, 15% screened positive for alcohol abuse, and 34% reported impaired quality of life. Almost 40% would not encourage their children to enter medicine and more than 10% said that they would not enter medicine again if they had to do it over.
Residents and those at mid-career are particularly vulnerable,7 with resident burnout rates reported to be as high as 75%.8 Of surveyed residents in a 2012 study, 13% satisfied all 3 subscale scores for high burnout and greater than 50% had high levels of depersonalization and emotional exhaustion. Those with high levels of emotional exhaustion were less satisfied with their careers, regretted choosing obstetrics and gynecology, and had higher rates of depression—all findings consistent with older studies.
9,10
References
- Peckham C. Medscape Lifestyle Report 2016: Bias and Burnout. Medscape website. http://www.medscape.com/features/slideshow/lifestyle/2016/public/overview#page=1. Published January 13, 2016. Accessed July 7, 2016.
- Shanafelt TD, Boone, S, Tan L, et al. Burnout and satisfaction with work-life balance among US physicians relative to the general US population. Arch Intern Med. 2012;172(18):1377–1385.
- Martini S, Arfken CL, Churchill A, Balon R. Burnout comparison among residents in different medical specialties. Acad Psychiatry. 2004;28(3):240–242.
- Lee YY, Medford AR, Halim AS. Burnout in physicians. J R Coll Physicians Edinb. 2015;45(2):104–107.
- Shanafelt TD, Hasan O, Dyrbye LN, et al. Changes in burnout and satisfaction with work-life balance in physicians and the general US working population between 2011 and 2014. Mayo Clin Proc. 2015;90(12):1600–1613.
- Rath KS, Huffman LB, Phillips GS, Carpenter KM, Fowler JM. Burnout and associated factors among members of the Society of Gynecologic Oncology. Am J Obstet Gynecol. 2015;213(6):824.e1–e9.
- Dyrbye LN, Varkey P, Boone SL, Satele DV, Sloan JA, Shanafelt TD. Physician satisfaction and burnout at different career stages. Mayo Clin Proc. 2013;88(12):1358–1367.
- Govardhan LM, Pinelli V, Schnatz PF. Burnout, depression and job satisfaction in obstetrics and gynecology residents. Conn Med. 2012;76(7):389–395.
- Becker JL, Milad MP, Klock SC. Burnout, depression, and career satisfaction: cross-sectional study of obstetrics and gynecology residents. Am J Obstet Gynecol. 2006;195(5):1444–1449.
- Castelo-Branco C, Figueras F, Eixarch E, et al. Stress symptoms and burnout in obstetric and gynaecology residents. BJOG. 2007;114(1):94–98
Why burnout occurs
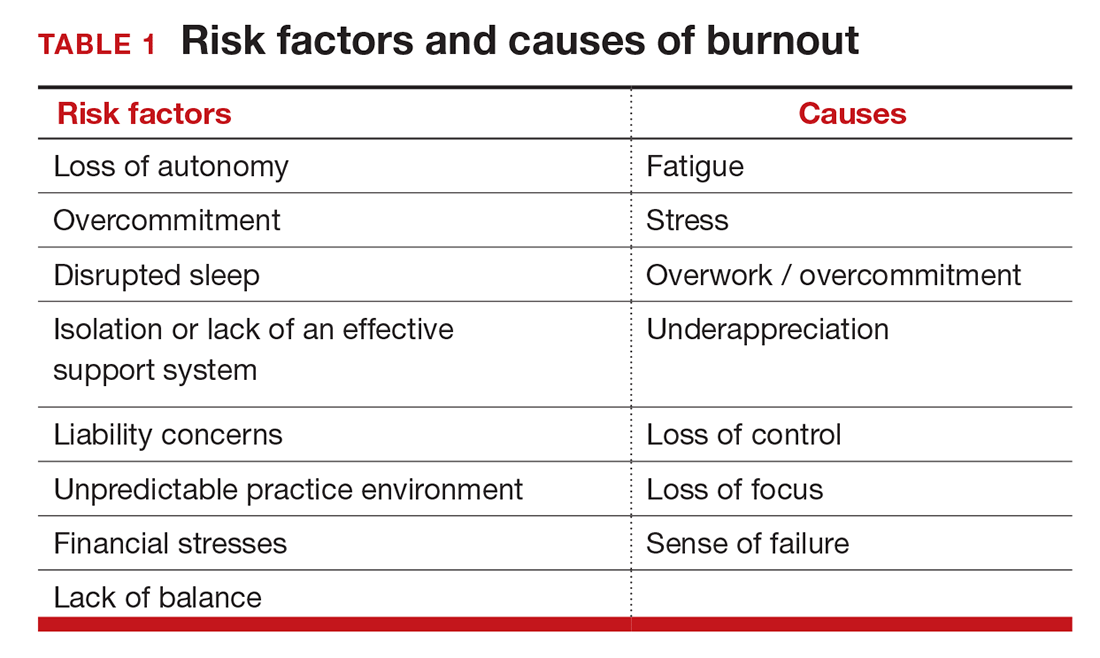
Simply identifying ourselves as professionals and the same attributes that make us successful as physicians (type-A behavior, obsessive-compulsive commitment to our profession) put us at risk for professional burnout (see “Who is most at risk for burnout?”). Those predilections combine with the forces from the world in which we live and practice to increase this threat (TABLE 1). Conditions in which there are weak retention rates, high turnover, heavy workloads, and low staffing levels or staffing shortages increase the risk of burnout and, when burnout is present, are associated with a degraded quality of care.5
Does stress cause burnout?
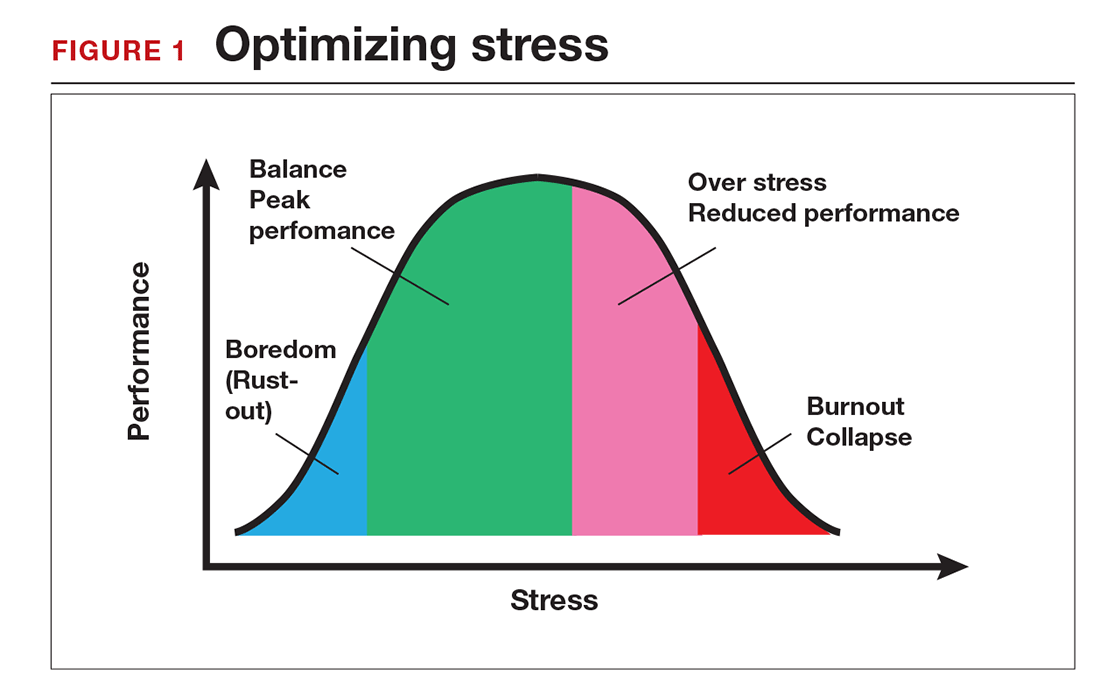
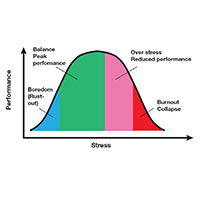
Stress is often seen as the reason for burnout. Research shows that there is no single source of burnout,6 however, and a number of factors combine to cause this physical or mental collapse. Stress can be a positive or negative factor in our performance. Too little stress and we feel underutilized; too much stress and we collapse from the strain.
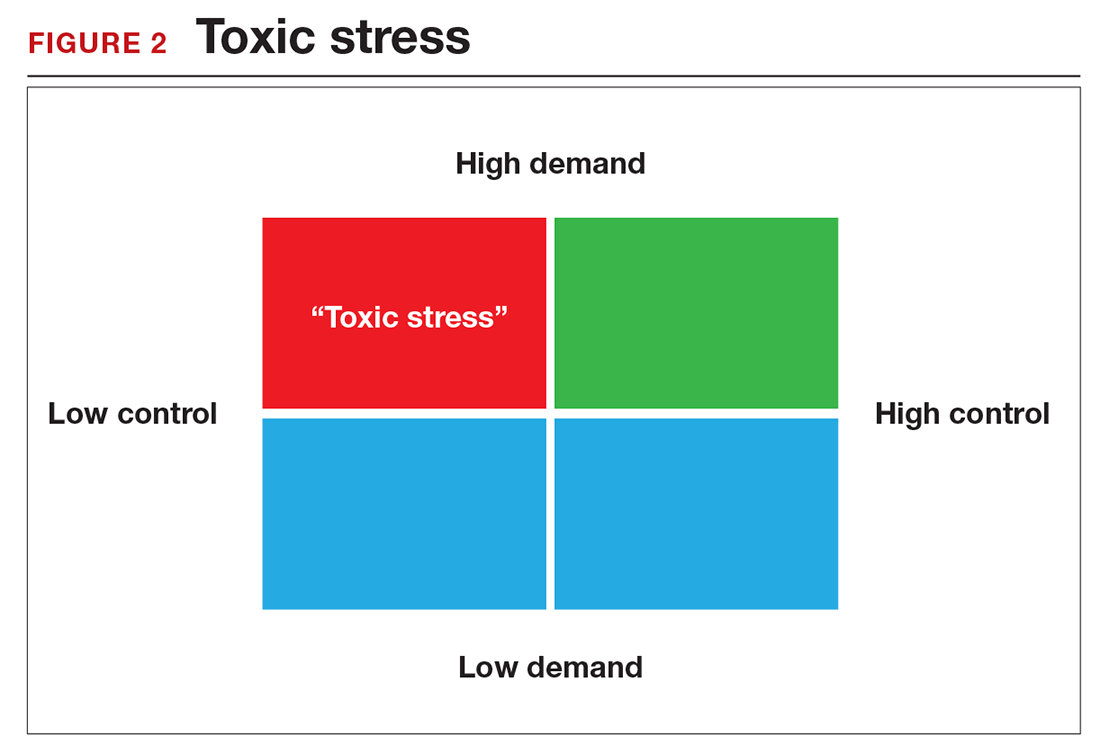
There is a middle ground where stress and expectations keep us focused and at peak productivity (FIGURE 1). The key is the balance between control and demand: When we have a greater level of control, we can handle high demands (FIGURE 2). It is when we lack that control that high demands result in what has been called “toxic stress,” and we collapse under the strain.
The impact of burnout
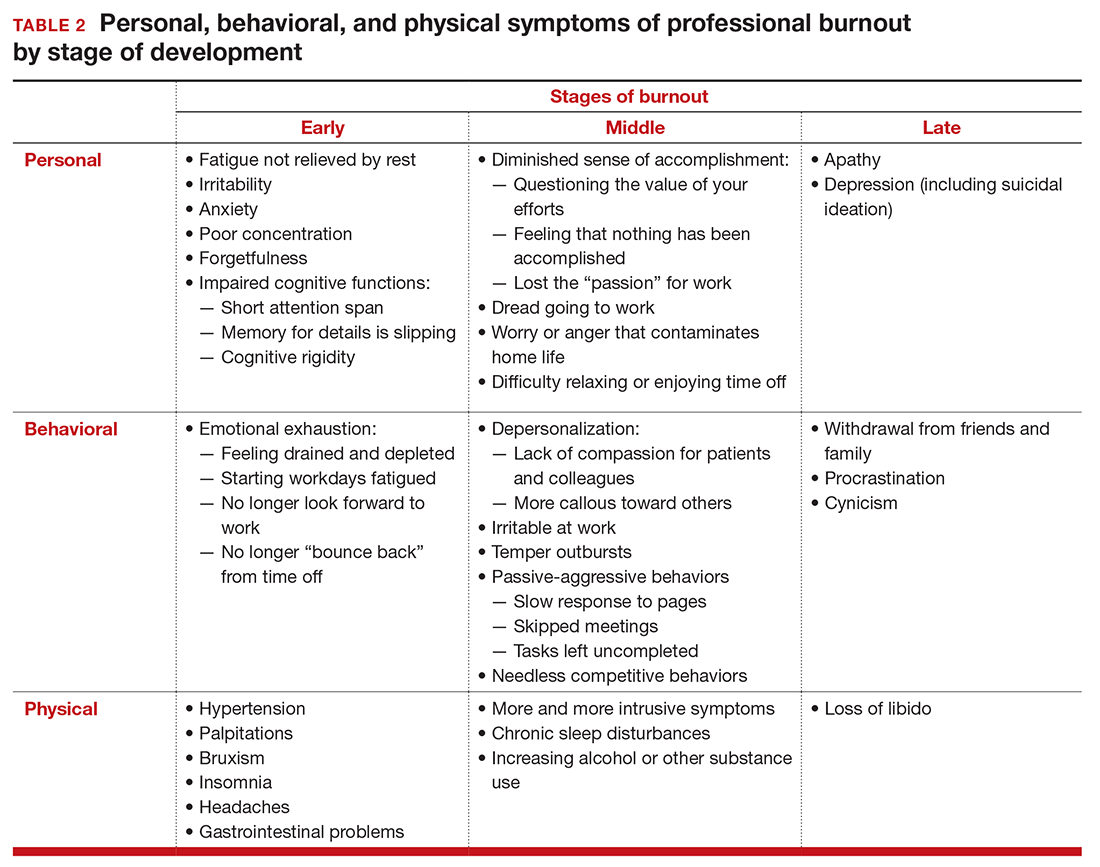
Burnout is associated with reduced performance and job satisfaction, increased rates of illness and absenteeism, accidents, premature retirement, and even premature death. Physically, stress induces the dry mouth, dilated pupils, and release of adrenalin and noradrenalin associated with the “fight-or-flight” reaction. The degree to which the physical, emotional, and professional symptoms are manifest depends on the depth or stage of burnout present (TABLE 2). Overall, burnout is associated with an increased risk for physical illness.7 Economically, the impact of physician burnout (for physicians practicing in Canada) has been estimated to be $213.1 million,8 which includes $185.2 million due to early retirement and $27.9 million due to reduced clinical hours.
“Do I have burnout?”
We all suffer from fatigue and have stress, but do we have burnout? With so many myths surrounding stress and burnout, it is sometimes hard to know where the truth lies. Some of those myths say that:
- you can leave your troubles at home
- mental stress does not affect physical performance
- stress is only for wimps
- stress and burnout are chemical imbalances that can be treated with medications
- stress is always bad
- burnout will get better if you just give it more time.
Maslach Burnout Inventory. The effective “gold standard” for diagnosing burnout is the Maslach Burnout Inventory,9 which operationalizes burnout as a 3-dimensional syndrome made up of exhaustion, cynicism, and inefficacy. Other diagnostic tools have been introduced10 but have not gained the wide acceptance of the Maslach Inventory. Some authors have argued that burnout and depression represent different, closely spaced points along a spectrum and that any effort to separate them may be artificial.11,12
The Maslach Burnout Inventory consists of a survey of 22 items; it requires a fee to take and is interpreted by a qualified individual. A simpler screening test consists of 10 questions (TABLE 3). If you answer “yes” to 5 or more of the questions, you probably have burnout. An even quicker test is to see, when you go on vacation, if your symptoms disappear. If so, you are not depressed; you have burnout. (If you cannot even go on vacation, then it is almost certain.)

12 stages of burnout. Psychologists Herbert Freudenberger and Gail North have theorized that the burnout process can be divided into 12 phases (TABLE 4).13 These stages are not necessarily sequential—some may be absent and others may present simultaneously. It is easy to see how these can represent stages in a potentially spiraling series of behaviors and changes that result in complete dysfunction. It is also easy to understand that the characteristics that are associated with success in medical school, clinical training, and practice, such as high expectations, placing the needs of others above our own, and a desire to prove oneself, virtually define the first 3 stages.

Approaches for burnout control and prevention
There are some simple steps we can take to reduce the risk of burnout or to reverse its effects. Because fatigue and stress are 2 of the greatest risk factors, reducing these is a good place to start.
Prioritize sleep. When it comes to fatigue, that one is easy: get some sleep. Physicians tend to sleep fewer hours than the general population and what we get is often not the type that is restful and restorative.14 Just reducing the number of hours worked is not enough, as a number of studies have found.15 The rest must result in relaxation.
e Stress reduction may seem a more difficult goal than getting more sleep. In reality, there are several simple approaches to use to reduce stress:
- Even though we all have busy clinical schedules, take short breaks to rest, sing, laugh, and exercise. Even breaks as short as 10 minutes can be effective.16
- Separate work from private life by taking a short break to resolve issues before heading home. Avoiding “baggage” or homework will go a long way to giving you the perspective you need from your time off. This may also mean that you have to delegate tasks, share chores, or get carry-out for dinner.
- Set meaningful and realistic goals for yourself professionally and personally. Do not expect or demand more than is possible. This will mean setting priorities and recognizing that some tasks may have to wait.
- Finally, do not forget to pay yourself with hobbies and activities that you enjoy.
Take action
If you feel the effects of burnout tugging at your coattails, you can reduce the effects, deal with the sources, and improve your attitude (TABLE 5). Rest and relaxation will go a long way to helping, but do not forget to take care of your physical well-being with a healthy diet, exercise, and health checkups. Deal with the sources of burnout by identifying the stressors, setting realistic priorities, and practicing good time management.
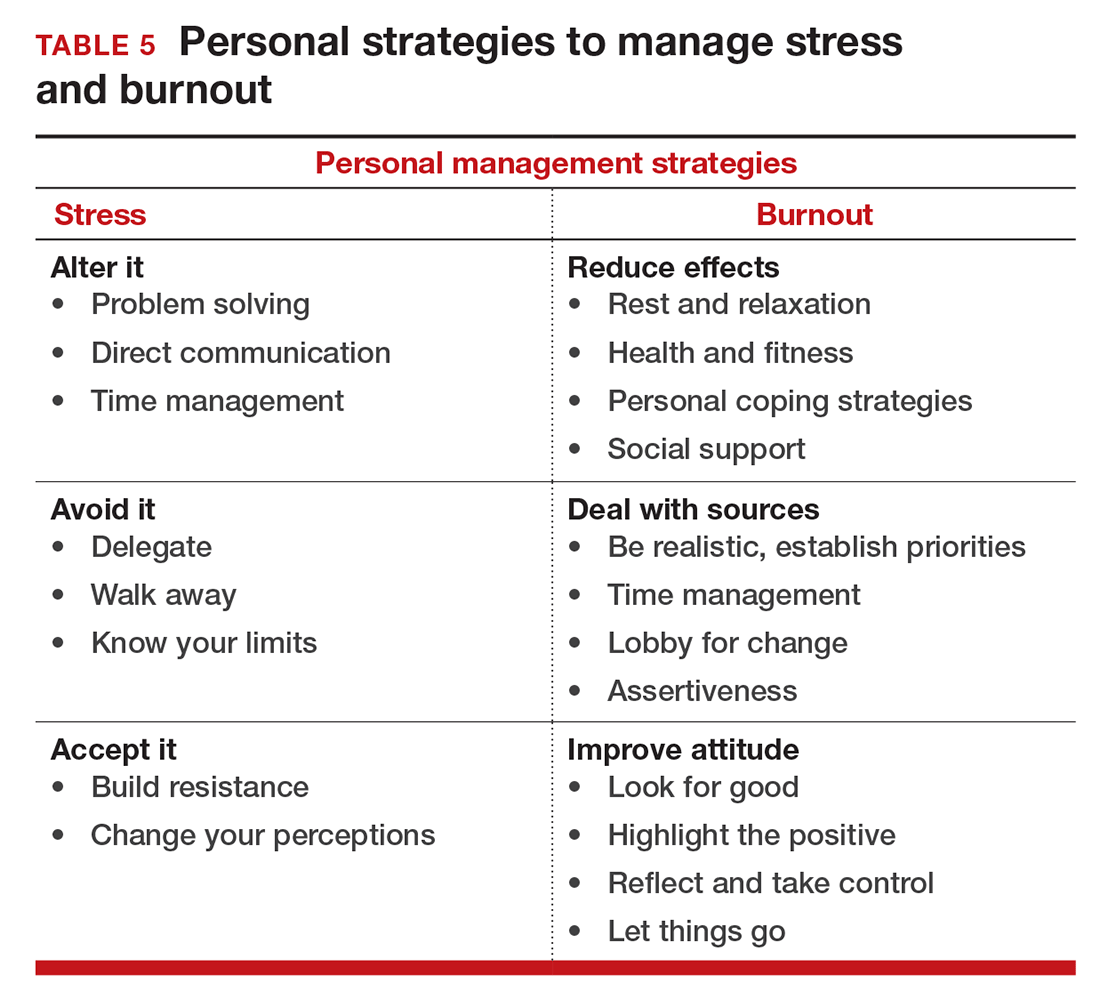
You also should lobby for changes that will increase your control and reduce unnecessary obstacles to completing your goals. Be your own best advocate. Look for the good and try to identify at least one instance during the day where your presence or acts made a difference. In the end, it is like Smokey the Bear says, “Only you can prevent burnout.”
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
- Keeton K, Fenner DE, Johnson TR, Hayward RA. Predictors of physician career satisfaction, work-life balance, and burnout. Obstet Gynecol. 2007;109(4):949-955.
- Shanafelt TD, Dyrbye LN, Sinsky C, et al. Relationship between clerical burden and characteristics of the electronic environment with physician burnout and professional satisfaction. Mayo Clin Proc. 2016;91(7):836-848.
- Gabbe SG, Melville J, Mandel L, Walker E. Burnout in chairs of obstetrics and gynecology: diagnosis, treatment, and prevention. Am J Obstet Gynecol. 2002;186(4):601-612.
- Kok BC, Herrell RK, Grossman SH, West JC, Wilk JE. Prevalence of professional burnout among military mental health service providers. Psychiatr Serv. 2016;67(1):137-140.
- Humphries N, Morgan K, Conry MC, McGowan Y, Montgomery A, McGee H. Quality of care and health professional burnout: narrative literature review. Int J Health Care Qual Assur. 2014;27(4):293-307.
- Streu R, Hansen J, Abrahamse P, Alderman AK. Professional burnout among US plastic surgeons: results of a national survey. Ann Plast Surg. 2014;72(3):346-350.
- Honkonen T, Ahola K, Pertovaara M, et al. The association between burnout and physical illness in the general population--results from the Finnish Health 2000 Study. J Psychosom Res. 2006;61(1):59-66.
- Dewa CS, Jacobs P, Thanh NX, Loong D. An estimate of the cost of burnout on early retirement and reduction in clinical hours of practicing physicians in Canada. BMC Health Serv Res. 2014;14:254.
- Maslach C, Jackson SE, Leiter MP. The Maslach Burnout Inventory Manual. Palo Alto, California: Consulting Psychologists Press, 1996.
- Kristensen TS, Borritz M, Villadsen E, Christensen KB. The Copenhagen Burnout Inventory: A new tool for the assessment of burnout. Work & Stress. 2005;19(3):192-207.
- Bianchi R, Boffy C, Hingray C, Truchot D, Laurent E. Comparative symptomatology of burnout and depression. J Health Psychol. 2013;18(6):782-787.
- Bianchi R, Schonfeld I S, Laurent E. Is burnout a depressive disorder? A re-examination with special focus on atypical depression. Intl J Stress Manag. 2014;21(4):307-324.
- Freudenberger HJ, North G. Women's burnout: How to spot it, how to reverse it, and how to prevent it. New York, New York: Doubleday, 1985.
- Abrams RM. Sleep deprivation. Obstet Gynecol Clin North Am. 2015;42(3):493-506.
- Williams D, Tricomi G, Gupta J, Janise A. Efficacy of burnout interventions in the medical education pipeline. Acad Psychiatry. 2015;39(1):47-54.
- Shanafelt TD, Oreskovich MR, Dyrbye LN, et al. Avoiding burnout: The personal health habits and wellness practices of US surgeons. Ann Surg. 2012;255(4):625-633.

Dr. Smith is Assistant Dean for Graduate Medical Education and Professor of Clinical Biologic Sciences, Charles E. Schmidt College of Medicine, Florida Atlantic University, Boca Raton, Florida.
The author reports no financial relationships relevant to this article.
Dr. Smith is Assistant Dean for Graduate Medical Education and Professor of Clinical Biologic Sciences, Charles E. Schmidt College of Medicine, Florida Atlantic University, Boca Raton, Florida.
The author reports no financial relationships relevant to this article.
Dr. Smith is Assistant Dean for Graduate Medical Education and Professor of Clinical Biologic Sciences, Charles E. Schmidt College of Medicine, Florida Atlantic University, Boca Raton, Florida.
The author reports no financial relationships relevant to this article.
It sometimes seems that the pace of life, and its stresses, have spiraled out of control: There just never seems to be enough time to deal with all the directions in which we are pulled. This easily can lead to the exhaustion of physical or emotional strength or motivation, otherwise known as “burnout.” Burnout is physical or mental collapse caused by overwork or stress and we are all at risk of suffering it. Conflicting demands on our time, loss of control (real or imagined), and a diminishing sense of worth grind at us from every direction.
In general, having some control over schedule and hours worked is associated with reductions in burnout and improved job satisfaction.1 But this is not always the case. Well-intentioned efforts to reduce workload, such as the electronic medical records or physician order entry systems, have actually made the problem worse.2 The seeming level of control that comes with being the chair of an obstetrics and gynecology department does not necessarily reduce burnout rates,3 and neither does the perceived resilience of mental health professionals, who still report burnout rates that approach 25%.4
This article continues the focus on recalibrating work/life balance that began last month with “ObGyn burnout: ACOG takes aim,” by Lucia DiVenere, MA, and the peer-to-peer audiocast with Ms. DiVenere and myself titled “Is burnout on the rise and what are the signs ObGyns should be on the lookout for?” Here, I identify the causes and symptoms of burnout and provide specific tools to help you develop resilience.
Who is most at risk for burnout?
Estimates range from 40% to 75% of ObGyns currently suffer from professional burnout, making the lifetime risk a virtual certainty.1−3 The idea of professional burnout is not new, but wider recognition of the alarming rates of burnout is very current.4,5 A recent survey of gynecologic oncologists6 found that of those studied 30% scored high for emotional exhaustion, 10% high for depersonalization, and 11% low for personal accomplishment. Overall, 32% of physicians had scores indicating burnout. More worrisome was that 33% screened positive for depression, 13% had a history of suicidal ideation, 15% screened positive for alcohol abuse, and 34% reported impaired quality of life. Almost 40% would not encourage their children to enter medicine and more than 10% said that they would not enter medicine again if they had to do it over.
Residents and those at mid-career are particularly vulnerable,7 with resident burnout rates reported to be as high as 75%.8 Of surveyed residents in a 2012 study, 13% satisfied all 3 subscale scores for high burnout and greater than 50% had high levels of depersonalization and emotional exhaustion. Those with high levels of emotional exhaustion were less satisfied with their careers, regretted choosing obstetrics and gynecology, and had higher rates of depression—all findings consistent with older studies.
9,10
References
- Peckham C. Medscape Lifestyle Report 2016: Bias and Burnout. Medscape website. http://www.medscape.com/features/slideshow/lifestyle/2016/public/overview#page=1. Published January 13, 2016. Accessed July 7, 2016.
- Shanafelt TD, Boone, S, Tan L, et al. Burnout and satisfaction with work-life balance among US physicians relative to the general US population. Arch Intern Med. 2012;172(18):1377–1385.
- Martini S, Arfken CL, Churchill A, Balon R. Burnout comparison among residents in different medical specialties. Acad Psychiatry. 2004;28(3):240–242.
- Lee YY, Medford AR, Halim AS. Burnout in physicians. J R Coll Physicians Edinb. 2015;45(2):104–107.
- Shanafelt TD, Hasan O, Dyrbye LN, et al. Changes in burnout and satisfaction with work-life balance in physicians and the general US working population between 2011 and 2014. Mayo Clin Proc. 2015;90(12):1600–1613.
- Rath KS, Huffman LB, Phillips GS, Carpenter KM, Fowler JM. Burnout and associated factors among members of the Society of Gynecologic Oncology. Am J Obstet Gynecol. 2015;213(6):824.e1–e9.
- Dyrbye LN, Varkey P, Boone SL, Satele DV, Sloan JA, Shanafelt TD. Physician satisfaction and burnout at different career stages. Mayo Clin Proc. 2013;88(12):1358–1367.
- Govardhan LM, Pinelli V, Schnatz PF. Burnout, depression and job satisfaction in obstetrics and gynecology residents. Conn Med. 2012;76(7):389–395.
- Becker JL, Milad MP, Klock SC. Burnout, depression, and career satisfaction: cross-sectional study of obstetrics and gynecology residents. Am J Obstet Gynecol. 2006;195(5):1444–1449.
- Castelo-Branco C, Figueras F, Eixarch E, et al. Stress symptoms and burnout in obstetric and gynaecology residents. BJOG. 2007;114(1):94–98
Why burnout occurs
Simply identifying ourselves as professionals and the same attributes that make us successful as physicians (type-A behavior, obsessive-compulsive commitment to our profession) put us at risk for professional burnout (see “Who is most at risk for burnout?”). Those predilections combine with the forces from the world in which we live and practice to increase this threat (TABLE 1). Conditions in which there are weak retention rates, high turnover, heavy workloads, and low staffing levels or staffing shortages increase the risk of burnout and, when burnout is present, are associated with a degraded quality of care.5
Does stress cause burnout?
Stress is often seen as the reason for burnout. Research shows that there is no single source of burnout,6 however, and a number of factors combine to cause this physical or mental collapse. Stress can be a positive or negative factor in our performance. Too little stress and we feel underutilized; too much stress and we collapse from the strain.
There is a middle ground where stress and expectations keep us focused and at peak productivity (FIGURE 1). The key is the balance between control and demand: When we have a greater level of control, we can handle high demands (FIGURE 2). It is when we lack that control that high demands result in what has been called “toxic stress,” and we collapse under the strain.
The impact of burnout
Burnout is associated with reduced performance and job satisfaction, increased rates of illness and absenteeism, accidents, premature retirement, and even premature death. Physically, stress induces the dry mouth, dilated pupils, and release of adrenalin and noradrenalin associated with the “fight-or-flight” reaction. The degree to which the physical, emotional, and professional symptoms are manifest depends on the depth or stage of burnout present (TABLE 2). Overall, burnout is associated with an increased risk for physical illness.7 Economically, the impact of physician burnout (for physicians practicing in Canada) has been estimated to be $213.1 million,8 which includes $185.2 million due to early retirement and $27.9 million due to reduced clinical hours.
“Do I have burnout?”
We all suffer from fatigue and have stress, but do we have burnout? With so many myths surrounding stress and burnout, it is sometimes hard to know where the truth lies. Some of those myths say that:
- you can leave your troubles at home
- mental stress does not affect physical performance
- stress is only for wimps
- stress and burnout are chemical imbalances that can be treated with medications
- stress is always bad
- burnout will get better if you just give it more time.
Maslach Burnout Inventory. The effective “gold standard” for diagnosing burnout is the Maslach Burnout Inventory,9 which operationalizes burnout as a 3-dimensional syndrome made up of exhaustion, cynicism, and inefficacy. Other diagnostic tools have been introduced10 but have not gained the wide acceptance of the Maslach Inventory. Some authors have argued that burnout and depression represent different, closely spaced points along a spectrum and that any effort to separate them may be artificial.11,12
The Maslach Burnout Inventory consists of a survey of 22 items; it requires a fee to take and is interpreted by a qualified individual. A simpler screening test consists of 10 questions (TABLE 3). If you answer “yes” to 5 or more of the questions, you probably have burnout. An even quicker test is to see, when you go on vacation, if your symptoms disappear. If so, you are not depressed; you have burnout. (If you cannot even go on vacation, then it is almost certain.)
12 stages of burnout. Psychologists Herbert Freudenberger and Gail North have theorized that the burnout process can be divided into 12 phases (TABLE 4).13 These stages are not necessarily sequential—some may be absent and others may present simultaneously. It is easy to see how these can represent stages in a potentially spiraling series of behaviors and changes that result in complete dysfunction. It is also easy to understand that the characteristics that are associated with success in medical school, clinical training, and practice, such as high expectations, placing the needs of others above our own, and a desire to prove oneself, virtually define the first 3 stages.
Approaches for burnout control and prevention
There are some simple steps we can take to reduce the risk of burnout or to reverse its effects. Because fatigue and stress are 2 of the greatest risk factors, reducing these is a good place to start.
Prioritize sleep. When it comes to fatigue, that one is easy: get some sleep. Physicians tend to sleep fewer hours than the general population and what we get is often not the type that is restful and restorative.14 Just reducing the number of hours worked is not enough, as a number of studies have found.15 The rest must result in relaxation.
e Stress reduction may seem a more difficult goal than getting more sleep. In reality, there are several simple approaches to use to reduce stress:
- Even though we all have busy clinical schedules, take short breaks to rest, sing, laugh, and exercise. Even breaks as short as 10 minutes can be effective.16
- Separate work from private life by taking a short break to resolve issues before heading home. Avoiding “baggage” or homework will go a long way to giving you the perspective you need from your time off. This may also mean that you have to delegate tasks, share chores, or get carry-out for dinner.
- Set meaningful and realistic goals for yourself professionally and personally. Do not expect or demand more than is possible. This will mean setting priorities and recognizing that some tasks may have to wait.
- Finally, do not forget to pay yourself with hobbies and activities that you enjoy.
Take action
If you feel the effects of burnout tugging at your coattails, you can reduce the effects, deal with the sources, and improve your attitude (TABLE 5). Rest and relaxation will go a long way to helping, but do not forget to take care of your physical well-being with a healthy diet, exercise, and health checkups. Deal with the sources of burnout by identifying the stressors, setting realistic priorities, and practicing good time management.
You also should lobby for changes that will increase your control and reduce unnecessary obstacles to completing your goals. Be your own best advocate. Look for the good and try to identify at least one instance during the day where your presence or acts made a difference. In the end, it is like Smokey the Bear says, “Only you can prevent burnout.”
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
It sometimes seems that the pace of life, and its stresses, have spiraled out of control: There just never seems to be enough time to deal with all the directions in which we are pulled. This easily can lead to the exhaustion of physical or emotional strength or motivation, otherwise known as “burnout.” Burnout is physical or mental collapse caused by overwork or stress and we are all at risk of suffering it. Conflicting demands on our time, loss of control (real or imagined), and a diminishing sense of worth grind at us from every direction.
In general, having some control over schedule and hours worked is associated with reductions in burnout and improved job satisfaction.1 But this is not always the case. Well-intentioned efforts to reduce workload, such as the electronic medical records or physician order entry systems, have actually made the problem worse.2 The seeming level of control that comes with being the chair of an obstetrics and gynecology department does not necessarily reduce burnout rates,3 and neither does the perceived resilience of mental health professionals, who still report burnout rates that approach 25%.4
This article continues the focus on recalibrating work/life balance that began last month with “ObGyn burnout: ACOG takes aim,” by Lucia DiVenere, MA, and the peer-to-peer audiocast with Ms. DiVenere and myself titled “Is burnout on the rise and what are the signs ObGyns should be on the lookout for?” Here, I identify the causes and symptoms of burnout and provide specific tools to help you develop resilience.
Who is most at risk for burnout?
Estimates range from 40% to 75% of ObGyns currently suffer from professional burnout, making the lifetime risk a virtual certainty.1−3 The idea of professional burnout is not new, but wider recognition of the alarming rates of burnout is very current.4,5 A recent survey of gynecologic oncologists6 found that of those studied 30% scored high for emotional exhaustion, 10% high for depersonalization, and 11% low for personal accomplishment. Overall, 32% of physicians had scores indicating burnout. More worrisome was that 33% screened positive for depression, 13% had a history of suicidal ideation, 15% screened positive for alcohol abuse, and 34% reported impaired quality of life. Almost 40% would not encourage their children to enter medicine and more than 10% said that they would not enter medicine again if they had to do it over.
Residents and those at mid-career are particularly vulnerable,7 with resident burnout rates reported to be as high as 75%.8 Of surveyed residents in a 2012 study, 13% satisfied all 3 subscale scores for high burnout and greater than 50% had high levels of depersonalization and emotional exhaustion. Those with high levels of emotional exhaustion were less satisfied with their careers, regretted choosing obstetrics and gynecology, and had higher rates of depression—all findings consistent with older studies.
9,10
References
- Peckham C. Medscape Lifestyle Report 2016: Bias and Burnout. Medscape website. http://www.medscape.com/features/slideshow/lifestyle/2016/public/overview#page=1. Published January 13, 2016. Accessed July 7, 2016.
- Shanafelt TD, Boone, S, Tan L, et al. Burnout and satisfaction with work-life balance among US physicians relative to the general US population. Arch Intern Med. 2012;172(18):1377–1385.
- Martini S, Arfken CL, Churchill A, Balon R. Burnout comparison among residents in different medical specialties. Acad Psychiatry. 2004;28(3):240–242.
- Lee YY, Medford AR, Halim AS. Burnout in physicians. J R Coll Physicians Edinb. 2015;45(2):104–107.
- Shanafelt TD, Hasan O, Dyrbye LN, et al. Changes in burnout and satisfaction with work-life balance in physicians and the general US working population between 2011 and 2014. Mayo Clin Proc. 2015;90(12):1600–1613.
- Rath KS, Huffman LB, Phillips GS, Carpenter KM, Fowler JM. Burnout and associated factors among members of the Society of Gynecologic Oncology. Am J Obstet Gynecol. 2015;213(6):824.e1–e9.
- Dyrbye LN, Varkey P, Boone SL, Satele DV, Sloan JA, Shanafelt TD. Physician satisfaction and burnout at different career stages. Mayo Clin Proc. 2013;88(12):1358–1367.
- Govardhan LM, Pinelli V, Schnatz PF. Burnout, depression and job satisfaction in obstetrics and gynecology residents. Conn Med. 2012;76(7):389–395.
- Becker JL, Milad MP, Klock SC. Burnout, depression, and career satisfaction: cross-sectional study of obstetrics and gynecology residents. Am J Obstet Gynecol. 2006;195(5):1444–1449.
- Castelo-Branco C, Figueras F, Eixarch E, et al. Stress symptoms and burnout in obstetric and gynaecology residents. BJOG. 2007;114(1):94–98
Why burnout occurs
Simply identifying ourselves as professionals and the same attributes that make us successful as physicians (type-A behavior, obsessive-compulsive commitment to our profession) put us at risk for professional burnout (see “Who is most at risk for burnout?”). Those predilections combine with the forces from the world in which we live and practice to increase this threat (TABLE 1). Conditions in which there are weak retention rates, high turnover, heavy workloads, and low staffing levels or staffing shortages increase the risk of burnout and, when burnout is present, are associated with a degraded quality of care.5
Does stress cause burnout?
Stress is often seen as the reason for burnout. Research shows that there is no single source of burnout,6 however, and a number of factors combine to cause this physical or mental collapse. Stress can be a positive or negative factor in our performance. Too little stress and we feel underutilized; too much stress and we collapse from the strain.
There is a middle ground where stress and expectations keep us focused and at peak productivity (FIGURE 1). The key is the balance between control and demand: When we have a greater level of control, we can handle high demands (FIGURE 2). It is when we lack that control that high demands result in what has been called “toxic stress,” and we collapse under the strain.
The impact of burnout
Burnout is associated with reduced performance and job satisfaction, increased rates of illness and absenteeism, accidents, premature retirement, and even premature death. Physically, stress induces the dry mouth, dilated pupils, and release of adrenalin and noradrenalin associated with the “fight-or-flight” reaction. The degree to which the physical, emotional, and professional symptoms are manifest depends on the depth or stage of burnout present (TABLE 2). Overall, burnout is associated with an increased risk for physical illness.7 Economically, the impact of physician burnout (for physicians practicing in Canada) has been estimated to be $213.1 million,8 which includes $185.2 million due to early retirement and $27.9 million due to reduced clinical hours.
“Do I have burnout?”
We all suffer from fatigue and have stress, but do we have burnout? With so many myths surrounding stress and burnout, it is sometimes hard to know where the truth lies. Some of those myths say that:
- you can leave your troubles at home
- mental stress does not affect physical performance
- stress is only for wimps
- stress and burnout are chemical imbalances that can be treated with medications
- stress is always bad
- burnout will get better if you just give it more time.
Maslach Burnout Inventory. The effective “gold standard” for diagnosing burnout is the Maslach Burnout Inventory,9 which operationalizes burnout as a 3-dimensional syndrome made up of exhaustion, cynicism, and inefficacy. Other diagnostic tools have been introduced10 but have not gained the wide acceptance of the Maslach Inventory. Some authors have argued that burnout and depression represent different, closely spaced points along a spectrum and that any effort to separate them may be artificial.11,12
The Maslach Burnout Inventory consists of a survey of 22 items; it requires a fee to take and is interpreted by a qualified individual. A simpler screening test consists of 10 questions (TABLE 3). If you answer “yes” to 5 or more of the questions, you probably have burnout. An even quicker test is to see, when you go on vacation, if your symptoms disappear. If so, you are not depressed; you have burnout. (If you cannot even go on vacation, then it is almost certain.)
12 stages of burnout. Psychologists Herbert Freudenberger and Gail North have theorized that the burnout process can be divided into 12 phases (TABLE 4).13 These stages are not necessarily sequential—some may be absent and others may present simultaneously. It is easy to see how these can represent stages in a potentially spiraling series of behaviors and changes that result in complete dysfunction. It is also easy to understand that the characteristics that are associated with success in medical school, clinical training, and practice, such as high expectations, placing the needs of others above our own, and a desire to prove oneself, virtually define the first 3 stages.
Approaches for burnout control and prevention
There are some simple steps we can take to reduce the risk of burnout or to reverse its effects. Because fatigue and stress are 2 of the greatest risk factors, reducing these is a good place to start.
Prioritize sleep. When it comes to fatigue, that one is easy: get some sleep. Physicians tend to sleep fewer hours than the general population and what we get is often not the type that is restful and restorative.14 Just reducing the number of hours worked is not enough, as a number of studies have found.15 The rest must result in relaxation.
e Stress reduction may seem a more difficult goal than getting more sleep. In reality, there are several simple approaches to use to reduce stress:
- Even though we all have busy clinical schedules, take short breaks to rest, sing, laugh, and exercise. Even breaks as short as 10 minutes can be effective.16
- Separate work from private life by taking a short break to resolve issues before heading home. Avoiding “baggage” or homework will go a long way to giving you the perspective you need from your time off. This may also mean that you have to delegate tasks, share chores, or get carry-out for dinner.
- Set meaningful and realistic goals for yourself professionally and personally. Do not expect or demand more than is possible. This will mean setting priorities and recognizing that some tasks may have to wait.
- Finally, do not forget to pay yourself with hobbies and activities that you enjoy.
Take action
If you feel the effects of burnout tugging at your coattails, you can reduce the effects, deal with the sources, and improve your attitude (TABLE 5). Rest and relaxation will go a long way to helping, but do not forget to take care of your physical well-being with a healthy diet, exercise, and health checkups. Deal with the sources of burnout by identifying the stressors, setting realistic priorities, and practicing good time management.
You also should lobby for changes that will increase your control and reduce unnecessary obstacles to completing your goals. Be your own best advocate. Look for the good and try to identify at least one instance during the day where your presence or acts made a difference. In the end, it is like Smokey the Bear says, “Only you can prevent burnout.”
Share your thoughts! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
- Keeton K, Fenner DE, Johnson TR, Hayward RA. Predictors of physician career satisfaction, work-life balance, and burnout. Obstet Gynecol. 2007;109(4):949-955.
- Shanafelt TD, Dyrbye LN, Sinsky C, et al. Relationship between clerical burden and characteristics of the electronic environment with physician burnout and professional satisfaction. Mayo Clin Proc. 2016;91(7):836-848.
- Gabbe SG, Melville J, Mandel L, Walker E. Burnout in chairs of obstetrics and gynecology: diagnosis, treatment, and prevention. Am J Obstet Gynecol. 2002;186(4):601-612.
- Kok BC, Herrell RK, Grossman SH, West JC, Wilk JE. Prevalence of professional burnout among military mental health service providers. Psychiatr Serv. 2016;67(1):137-140.
- Humphries N, Morgan K, Conry MC, McGowan Y, Montgomery A, McGee H. Quality of care and health professional burnout: narrative literature review. Int J Health Care Qual Assur. 2014;27(4):293-307.
- Streu R, Hansen J, Abrahamse P, Alderman AK. Professional burnout among US plastic surgeons: results of a national survey. Ann Plast Surg. 2014;72(3):346-350.
- Honkonen T, Ahola K, Pertovaara M, et al. The association between burnout and physical illness in the general population--results from the Finnish Health 2000 Study. J Psychosom Res. 2006;61(1):59-66.
- Dewa CS, Jacobs P, Thanh NX, Loong D. An estimate of the cost of burnout on early retirement and reduction in clinical hours of practicing physicians in Canada. BMC Health Serv Res. 2014;14:254.
- Maslach C, Jackson SE, Leiter MP. The Maslach Burnout Inventory Manual. Palo Alto, California: Consulting Psychologists Press, 1996.
- Kristensen TS, Borritz M, Villadsen E, Christensen KB. The Copenhagen Burnout Inventory: A new tool for the assessment of burnout. Work & Stress. 2005;19(3):192-207.
- Bianchi R, Boffy C, Hingray C, Truchot D, Laurent E. Comparative symptomatology of burnout and depression. J Health Psychol. 2013;18(6):782-787.
- Bianchi R, Schonfeld I S, Laurent E. Is burnout a depressive disorder? A re-examination with special focus on atypical depression. Intl J Stress Manag. 2014;21(4):307-324.
- Freudenberger HJ, North G. Women's burnout: How to spot it, how to reverse it, and how to prevent it. New York, New York: Doubleday, 1985.
- Abrams RM. Sleep deprivation. Obstet Gynecol Clin North Am. 2015;42(3):493-506.
- Williams D, Tricomi G, Gupta J, Janise A. Efficacy of burnout interventions in the medical education pipeline. Acad Psychiatry. 2015;39(1):47-54.
- Shanafelt TD, Oreskovich MR, Dyrbye LN, et al. Avoiding burnout: The personal health habits and wellness practices of US surgeons. Ann Surg. 2012;255(4):625-633.
- Keeton K, Fenner DE, Johnson TR, Hayward RA. Predictors of physician career satisfaction, work-life balance, and burnout. Obstet Gynecol. 2007;109(4):949-955.
- Shanafelt TD, Dyrbye LN, Sinsky C, et al. Relationship between clerical burden and characteristics of the electronic environment with physician burnout and professional satisfaction. Mayo Clin Proc. 2016;91(7):836-848.
- Gabbe SG, Melville J, Mandel L, Walker E. Burnout in chairs of obstetrics and gynecology: diagnosis, treatment, and prevention. Am J Obstet Gynecol. 2002;186(4):601-612.
- Kok BC, Herrell RK, Grossman SH, West JC, Wilk JE. Prevalence of professional burnout among military mental health service providers. Psychiatr Serv. 2016;67(1):137-140.
- Humphries N, Morgan K, Conry MC, McGowan Y, Montgomery A, McGee H. Quality of care and health professional burnout: narrative literature review. Int J Health Care Qual Assur. 2014;27(4):293-307.
- Streu R, Hansen J, Abrahamse P, Alderman AK. Professional burnout among US plastic surgeons: results of a national survey. Ann Plast Surg. 2014;72(3):346-350.
- Honkonen T, Ahola K, Pertovaara M, et al. The association between burnout and physical illness in the general population--results from the Finnish Health 2000 Study. J Psychosom Res. 2006;61(1):59-66.
- Dewa CS, Jacobs P, Thanh NX, Loong D. An estimate of the cost of burnout on early retirement and reduction in clinical hours of practicing physicians in Canada. BMC Health Serv Res. 2014;14:254.
- Maslach C, Jackson SE, Leiter MP. The Maslach Burnout Inventory Manual. Palo Alto, California: Consulting Psychologists Press, 1996.
- Kristensen TS, Borritz M, Villadsen E, Christensen KB. The Copenhagen Burnout Inventory: A new tool for the assessment of burnout. Work & Stress. 2005;19(3):192-207.
- Bianchi R, Boffy C, Hingray C, Truchot D, Laurent E. Comparative symptomatology of burnout and depression. J Health Psychol. 2013;18(6):782-787.
- Bianchi R, Schonfeld I S, Laurent E. Is burnout a depressive disorder? A re-examination with special focus on atypical depression. Intl J Stress Manag. 2014;21(4):307-324.
- Freudenberger HJ, North G. Women's burnout: How to spot it, how to reverse it, and how to prevent it. New York, New York: Doubleday, 1985.
- Abrams RM. Sleep deprivation. Obstet Gynecol Clin North Am. 2015;42(3):493-506.
- Williams D, Tricomi G, Gupta J, Janise A. Efficacy of burnout interventions in the medical education pipeline. Acad Psychiatry. 2015;39(1):47-54.
- Shanafelt TD, Oreskovich MR, Dyrbye LN, et al. Avoiding burnout: The personal health habits and wellness practices of US surgeons. Ann Surg. 2012;255(4):625-633.
In this Article
- Symptoms by stage of burnout
- Tips to reduce stress and burnout
- Who is most at risk for burnout?
Is burnout on the rise and what are the signs ObGyns should be on the lookout for?
In a peer-to-peer audiocast, Ms. DiVenere probed Dr. Smith for the problem areas that appear in the early to late stages of burnout. He spoke to the stressors that residents experience in the learning situation and that ObGyns must deal with in practice, as well as described strategies to deal with that stress.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Have you read "ObGyn burnout: ACOG takes aim," by Lucia DiVenere, MA (September 2016)?
In a peer-to-peer audiocast, Ms. DiVenere probed Dr. Smith for the problem areas that appear in the early to late stages of burnout. He spoke to the stressors that residents experience in the learning situation and that ObGyns must deal with in practice, as well as described strategies to deal with that stress.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Have you read "ObGyn burnout: ACOG takes aim," by Lucia DiVenere, MA (September 2016)?
In a peer-to-peer audiocast, Ms. DiVenere probed Dr. Smith for the problem areas that appear in the early to late stages of burnout. He spoke to the stressors that residents experience in the learning situation and that ObGyns must deal with in practice, as well as described strategies to deal with that stress.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Have you read "ObGyn burnout: ACOG takes aim," by Lucia DiVenere, MA (September 2016)?

NSAIDs: Is newer better for dysmenorrhea?
- Nonsteroidal anti-inflammatory drugs (NSAIDs) can prevent dysmenorrhea, unlike other agents that simply relieve symptoms.
- Although NSAIDs in one form or another have been used for centuries, agents introduced in the past 50 years have significantly improved efficacy and safety profiles.
- A greater understanding of the role of prostaglandins in physiologic and pathophysiologic processes can enhance the selection of appropriate therapeutic agents.
- Some drugs can selectively block the cyclooxygenase-2 (COX-2) isoform of the enzyme instrumental in the production of prostaglandins.
A major shift in the way menstrual pain is viewed and treated took place in the 1970s and ’80s, with a greater understanding of the role of prostaglandins and more effective nonsteroidal anti-inflammatory drugs (NSAIDs). Subjective studies of pain and objective studies of uterine activity established a firm connection between the two. These studies also amply demonstrated the ability of NSAIDs to alter the physiology of dysmenorrhea, making it possible to prevent—rather than simply relieve—pain.
Yet, these agents still are not universally used in the treatment of dysmenorrhea, despite more than 20 years of experience with them. Moreover, the introduction of new NSAIDs has clouded rather than clarified the issue of their relative efficacy. Drugs that are welldesigned for the suppression of chronic inflammation (e.g., arthritis therapies) are not very effective for dysmenorrhea, and vice versa. Even so, it is possible to apply the findings of published studies and an understanding of the pathophysiology of dysmenorrhea to demystify the range of options.
The therapeutic effects of NSAIDs come from their ability to inhibit the production of prostaglandin.
A brief history
The term “dysmenorrhea” is derived from a Greek root meaning “difficult monthly flow,” but it did not make its appearance in the English language until about 1810. Therapies for dysmenorrhea ranged from the plausible and somewhat effective to the outlandish and useless. Everything from cauterizing the middle turbinate of the nose,1 exercise programs,2 and presacral sympathectomy3,4 to uterine-relaxing factor,5 vasodilators,6,7 tranquilizers,8 and hormones9-11 have been tried. Today’s effective therapies against primary dysmenorrhea are an outgrowth of earlier observations of uterine activity and the presence of a menstrual “toxin.” That toxin was later identified as prostaglandin.
Endometrial prostaglandin production is tied to changes throughout the menstrual cycle. Prostaglandin is stored in the endometrium as it thickens in preparation for implantation or menstruation. With the onset of menstruation, preformed prostaglandins are liberated and large amounts of arachidonic acid are released from the cell walls of sloughed endometrial cells. This large increase in arachidonic acid substrate results in a tremendous rise in prostaglandin production, which augments the supplies of preformed prostaglandins liberated from the sloughed endometrial cells.
The causative role of prostaglandin F2α in dysmenorrhea was confirmed when researchers triggered dysmenorrhea-like pain and uterine activity after intravenous (IV) injection of prostaglandins.12 (Current evidence indicates that women with primary dysmenorrhea make 2 to 7 times the normal amount of prostaglandin F2α.) Excess prostaglandins also may be responsible for the smooth-muscle activity noted in the gastrointestinal (GI) tracts of these women. Hypermobility of the gut may be responsible for the frequent coexistence of nausea, vomiting, and diarrhea in these patients. In addition, prostaglandins appear to act as initiators and potentiators of nociceptive pain signals, further contributing to the symptoms of dysmenorrhea.
In 1967, Pickles demonstrated that prostaglandin levels were lower during anovulatory cycles, prompting the use of oral contraceptives (OCs) to suppress ovulation and relieve menstrual pain.13,14 Although this approach is usually successful, not all women want to—or can—use OCs. NSAIDs more directly alter the physiologic sequence leading to discomfort by inhibiting the production and/or action of prostaglandins. Moreover, NSAIDs generally are well-tolerated and need only be taken at the time of menstruation. While OCs act to reduce the substrate available to the reaction, NSAIDs act to block the pathway at 2 later enzymatic steps.
In 1979, Jacobson et al reported successful pain relief in 64% to 100% of patients from 16 studies of NSAIDs in dysmenorrhea.15 Unfortunately, few of those studies were double-blinded, and many failed to report the incidence of side effects. Dingfelder evaluated 23 trials published from 1970 to 1980 and found a 67% to 86% rate of pain relief.16 In a more thorough review, Owen presented data from 51 reports and attempted to analyze the diverse methods, designs, and outcomes.17 She found an 87% rate of “excellent” pain relief for the fenamates versus 56%, 68%, and 56% for ibuprofen, indomethacin, and naproxen, respectively. Unfortunately, she lumped together 2 different drugs (tolfenamic and mefenamic acids) and misinterpreted some primarily methodological reports. More recent attempts to analyze existing studies have failed to further clarify the issue.18,19
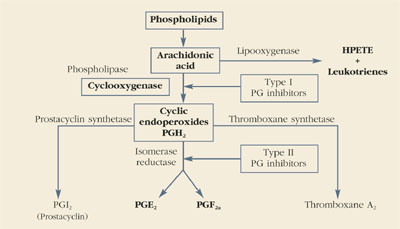
Prostaglandins are made throughout the body and are important autocrine and paracrine regulators of cellular and organ function. As depicted below, the main substrate for their production is arachidonic acid, a major constituent of cell walls. Under some circumstances, phospholipase A2 also can be used as a substrate for prostaglandin production.
Cyclooxygenase, also called prostaglandin H synthase, is the first enzymatic step in the conversion of arachidonic acid into prostaglandins. This enzyme folds the arachidonic acid molecule (cyclization) and oxygenates it to produce prostaglandin H2 (PGH2).
All other members of the prostaglandin family are then formed from PGH2. Arachidonic acid also is the substrate for the production of leukotrienes and 5-hydroxyeicosatetraenoic acid (5-HETE) through the 5-lipooxygenase pathway. Like prostaglandins F2α and E2, the products of the lipooxygenase pathway are potent vasoconstrictors and stimulators of uterine contractions.—Roger P. Smith, MD, and Jeffrey Ellis, MD
Increased uterine activity was first hypothesized as a cause of dysmenorrhea in 1932. By the late 1930s, objective findings began to support that hypothesis.1 The correlation between uterine activity and menstrual pain was strengthened when Jacobson et al studied simultaneous electrical and mechanical changes within the uterus.2,3 Using an intrauterine air-filled balloon system, Wilson and Kurzrok also noted the relationship between maximal uterine activity and pain.4,5 Despite the strength of these investigations, few changes occurred in the way dysmenorrhea was viewed or treated.
In the late 1940s, Liessé demonstrated that women with dysmenorrhea not only had a greater degree of uterine electrical and mechanical activity, but that this activity correlated with the pain of menstruation.6 Liessé found minimal activity between pains and as many as 30 irregular electrical discharges per second during pain, suggesting a cause (electrical) for dysmenorrhea but offering no clue to the underlying physiologic disturbance that might account for it.
The most detailed and influential studies of dysmenorrhea and uterine activity came in 1947 in a trial conducted by Woodbury.7 His findings of a direct correlation between pressure, pattern of contractions, resting tone, and pain became a standard reference.
After Woodbury, the stage was set for a connection to be made between uterine activity and prostaglandins. That happened in 1965, when Pickles reported elevated levels of prostaglandin F2αin the menstrual fluid of dysmenorrheic women.8
During the past 2 decades, more sophisticated analytic techniques have been applied to intrauterine pressure data,9,10 and strong correlations between uterine activity and pain have been reported.11,12 The basic assertion that menstrual pain is caused by increased intrauterine pressure, poor relaxation, and more frequent, irregular contractions appears to be valid.—Roger P. Smith, MD, and Jeffrey Ellis, MD
REFERENCES
1. Novac E, Reynolds SRM. The cause of primary dysmenorrhea. JAMA. 1932;99:1466.-
2. Jacobson E, Lackner JE, Sinykin MB. Electrical and mechanical activity of the human non-pregnant uterus. Am J Obstet Gynecol. 1939;38:1008.-
3. Jacobson E, Lackner JE, Sinykin MB. Activity of the human non-pregnant uterus. Am J Physiol. 1940;53:407.-
4. Wilson L, Kurzrok R. Studies on the motility of the human uterus in vivo. Endocrinology. 1938;23:79.-
5. Wilson L, Kurzrok R. Uterine contractility in functional dysmenorrhea. Endocrinology. 1940;27:23.-
6. Liessé A. L’Activité électrique de l’uterus dans la dysmenorrhee functionnelle. Gynec et Obstet. 1948;47:850.-
7. Woodbury RA, Torpin R, Child GP, Watson H, Jarboe M. Myometrial physiology and its relation to pelvic pain. JAMA. 1947;134:1081-1085.
8. Pickles VR, Hall WJ, Best FA, Smith GN. Prostaglandins in endometrium and menstrual fluid from normal and dysmenorrheic subjects. J Obstet Gynaecol Br Comm. 1965;72:185.-
9. Smith RP. Intrauterine pressure analysis in nonpregnant dysmenorrheic women. Med Instr. 1984;185:137-139.
10. Smith RP. Distribution analysis of intrauterine pressure in nonpregnant dysmenorrheic women. Am J Obstet Gynecol. 1984;150:271-273.
11. Smith RP. The dynamics of nonsteroidal anti-inflammatory therapy for primary dysmenorrhea. Obstet Gynecol. 1987;70:785-788.
12. Smith RP, Powell JR. Simultaneous objective and subjective evaluation of meclofenamate sodium in the treatment of primary dysmenorrhea. Am J Obstet Gynecol. 1987;157:611-616.
Classes of NSAIDs
Although aspirin was synthesized in 1853 and incorporated into medical practice in 1899, its history goes back even farther. That fact—along with the introduction of newer agents—ensured that NSAIDs became the mainstays of medical therapy for fever, pain, and inflammation. In the United States, NSAIDs are among the most widely prescribed drugs, with more than 70 million prescriptions and more than 30 billion over-the-counter tablets sold each year.20 Most of their therapeutic effects come from their ability to inhibit the production of prostaglandins.21
Interestingly, some drugs with the ability to inhibit prostaglandin synthesis have little clinical usefulness. Some have weak antiprostaglandin activity, require metabolic transformation to become active, or have side effects that limit their usefulness. While these drugs can be used to treat dysmenorrhea, they generally have been replaced by more effective agents.
There are 2 broad classes of NSAIDs: enolic acids and carboxylic acids. Each class can be further subcategorized (Table 1).
Enolic acids. With the exception of the newer cyclooxygenase-2 (COX-2) selective agents, drugs of the enolic-acid type are primarily type II inhibitors of prostaglandin synthesis. That means they impede the isomerase/reductase step in the formation of prostaglandins E2 (PGE2) and F2α (PGF2α). The most frequently used agents in the enolic-acid group are phenylbutazone and piroxicam.
Phenylbutazone was discovered in the 19th century during a search for a substitute for quinine. (Quinine had become popular for the treatment of fever; however, uncontrolled cutting of the Peruvian cinchona tree dramatically increased its cost.) While phenylbutazone is an effective shortterm analgesic for musculoskeletal pain (through antiprostaglandin activity), its relative toxicity has limited its use in dysmenorrhea and general therapy.
Piroxicam has a long half-life (50 hours), making once-daily dosing possible. Its action as an anti-inflammatory drug for the treatment of arthritis is well-established, but its use as an analgesic for acute pain therapy or for dysmenorrhea has not been fully evaluated. Based on the pharmacodynamics of drug absorption and action, one would anticipate its efficacy in that regard to be poor. In general, drugs in the pyrazolone group have a higher incidence of blood dyscrasias, limiting their broad utility.
Celecoxib is structurally similar to phenylbutazone and was the first selective COX-2 inhibitor approved by the Food and Drug Administration (FDA). It has been studied in dental pain models and the treatment of osteoarthritis. In these trials, celecoxib performed as well as naproxen and slow-release diclofenac, but with fewer GI side effects. No data on its use in dysmenorrhea is available.
Chemically related but less similar to the enolic acids is refecoxib. Like celecoxib, it has been studied in the treatment of osteoarthritis, where it was comparable in efficacy to ibuprofen and diclofenac, but with side-effect rates similar to placebo therapy. In studies of women with primary dysmenorrhea, rofecoxib has proved to be statistically superior to placebo but indistinguishable from naproxen (Figure 1).22 Peak blood levels are achieved in 2 to 3 hours (delayed by 1 to 2 hours by a fatty meal), but a steady state is not achieved until day 4 of continuous therapy. When the drug is used to treat women who have a rapid onset of symptoms, the significance of this delay is unclear but is a potential drawback. In addition, the dosage of rofecoxib for the treatment of pain or dysmenorrhea is generally much larger than that required for the treatment of arthritis. Thus, the risk of side effects may be increased.
A third selective COX-2 inhibitor, meloxicam, was introduced into the US market solely for the treatment of osteoarthritis. This drug is marketed on the basis of its more favorable side-effect profile rather than its selectivity in blocking the COX-2 isoform.
Carboxylic acids. The first of the carboxylic acid subgroups are the salicylic acids. The use of salicylates derived from the bark or leaves of the Salic alba or S. fragilis dates back to Biblical times. Salicylates were formally introduced into medical use in 1763 by the Reverend Edmund Stone.23
The relatively low potency of aspirin for reducing prostaglandin synthesis in the uterus limits its clinical utility in preventing moderate or severe dysmenorrhea. In contrast, diflunisal has a long half-life, making twice-daily dosing possible. However, its slow onset of action limits use for most patients with menstrual pain.
The indoleacetic acid subgroup offers increased potency against dysmenorrhea. Most of these drugs have a direct effect rather than requiring metabolism into an active form.
Indomethacin was one of the first NSAIDs widely used to treat dysmenorrhea, but a moderate incidence of side effects limits its use—and that of most of the other drugs in this family.
No NSAID completely eliminates gastric cyclooxygenase activity.
The most commonly used drugs for dysmenorrhea come from the 2 remaining carboxylate groups: arylalkanoic acids (propionic acid derivatives) and anthranilic acids (fenamates). Of the propionic acid derivatives currently available, ibuprofen and naproxen are commonly used for dysmenorrhea. Other drugs of this class have been used for pain relief or arthritis therapy, but are not currently FDA-approved for dysmenorrhea. The subset of drugs that are phenylpropionic acid derivatives are associated with a higher incidence of GI side effects and skin reactions.
Mefenamic acid was the first alternative synthesized in an effort to reduce the gastric irritation caused by acetylsalicylic acid.24 Mefenamic acid and meclofenamate sodium—both fenamates—are potent prostaglandin synthetase inhibitors. In addition, they have been shown to antagonize already formed prostaglandins, providing a dual mode of action.25-28 This ability to block the action of existing prostaglandins produces a more rapid onset of uterine relaxation. Some of the first studies of NSAIDs in dysmenorrhea used these agents.29,30 In fact, they are some of the most thoroughly investigated drugs for the treatment of dysmenorrhea, shown to be consistently effective in reducing its subjective discomfort.31-35
In the U.S., mefenamic acid was one of the first drugs approved for dysmenorrhea, and clinical studies have shown meclofenamate to be very effective in improving symptoms and altering the underlying pathophysiology.36,37 The dual mechanism of action likely gives these agents an edge in efficacy, although no clinical trials have been initiated to evaluate this presumed advantage. Intrauterine pressure changes have been documented as soon as 15 minutes after administration of the medications, suggesting a very rapid onset of action (obviating the need for preloading). In addition, in vitro studies have demonstrated meclofenamate’s ability to inhibit the activity of 5-lipooxygenase, unlike members of the propionic acid group, which have little or no inhibitory ability.38,39 The clinical significance of inhibiting the production of the extremely potent leukotrienes has yet to be fully explored.
TABLE 1
Families of NSAIDs
| Family | Example |
|---|---|
| ENOLIC ACIDS | |
| PYRAZOLONES | |
| Oxyphenbutazone | Azolid** |
| Phenylbutazone | Butazolidin** |
| Nabumetone | Relafen |
| Celecoxib | Celebrex |
| Rofecoxib* | Vioxx |
| OXICAMS | |
| Piroxicam | Feldene, Piroxicam |
| Meloxicam | Mobic |
| CARBOXYLIC ACIDS | |
| SALICYLIC ACIDS | |
| Acetylsalicylic acid | Aspirin (various) |
| Diflunisal | Dolobid |
| Salicylate | Disalcid, Trilisate |
| INDOLEACETIC ACIDS | |
| Diclofenac potassium | Cataflam |
| Diclofenac sodium | Voltaren, Arthrotec (combined with misoprostol) |
| Etodolac | Lodine |
| Indomethacin | Indocin |
| Ketorolac tromethamine | Acular, Toradol |
| Sulindac | Clinoril, Sulindac |
| Tolmetin | Tolectin, Tolmetin |
| PROPIONIC ACIDS | |
| Fenoprofen calcium | Fenoprofen |
| Flurbiprofen | Flurbiprofen |
| Ibuprofen* | Motrin |
| Ketoprofen | Orudis, Oruvail, Ketoprofen |
| Naproxen sodium* | Aleve, Anaprox, Naprelan |
| Naproxen* | Naprosyn |
| FENAMATES | |
| Meclofenamate sodium* | Meclofenamate |
| Mefenamic acid | Ponstel |
| *FDA-approved for primary dysmenorrhea | |
| **No longer available | |
| Adapted from: Smith RP.Gynecology in Primary Care. Baltimore, Md: Williams and Wilkins; 1996:399. | |
FIGURE 1 Comparison of rofecoxib, naproxen, and placebo in treatment of primary dysmenorrhea
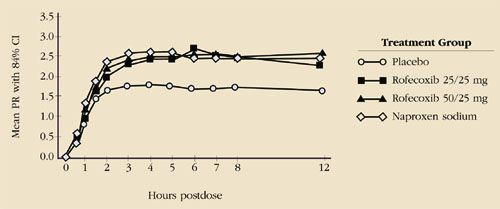
Reprinted with permission from The American College of Obstetricians and Gynecologists. Morrison BW, Daniels SE, Kotey P, Cantu N, Seidenberg B. Rofecoxib, a specific cyclooxygenase-2 inhibitor, in primary dysmenorrhea: a randomized-controlled trial. Obstet Gynecol. 1999;94:504-508.
COX-2 inhibitors
Although the short-term efficacy and safety of new selective COX-2 inhibitors appear to be good, several concerns remain. Despite a decrease in the incidence of GI side effects with these agents, their use by patients with active GI ulceration, infection with Helicobacter pylori, or inflammatory bowel disease has not been adequately studied. Indeed, recent studies suggest that, at therapeutic concentrations, no NSAID—not even the selective COX-2 inhibitors—completely eliminates gastric cyclooxygenase activity.40
In addition, questions have been raised about the speed and efficacy of the selective COX-2 inhibitors for the management of acute pain, compared with conventional NSAIDs.41 When used for postoperative analgesia, these agents theoretically could retard wound healing because of the role that COX-2 plays in healing and neovascularization. As more data emerge about the physiologic functions of COX-1 and -2 (Table 2), there are growing concerns that COX-2 is not restricted to inflammation and pathology, suggesting the possibility of unanticipated adverse effects associated with its use. For example, there is evidence of constitutive expression of COX-2 in the kidney and brain, and essential physiological functions in ovulation and implantation.42 In the treatment of dysmenorrhea, the increased cost of these drugs over the more commonly used agents, along with the lack of clinical efficacy studies, suggests a second-line role.
Like most NSAIDs, COX-2 inhibitors fall into pregnancy category C. Because of the risk of premature closure of the ductus arteriosus, they should not be used in the third trimester of pregnancy. In addition, drug levels in human milk mimic those in serum, so the decision to use these agents in nursing mothers must be made carefully.
Studies over the past 10 years have revealed that the cyclooxygenase enzyme (COX) is found in 2 isomeric forms, known as COX-1 and COX-2.1 Initial investigations suggested that COX-1 was present in most tissues and responsible for the homeostatic production of arachidonic acid metabolites. The COX-2 enzyme was thought to be induced in response to inflammatory stimuli such as cytokines and bacterial lipopolysaccharides, rather than expressed under normal cellular function. Thus, the COX-2 form was thought to be responsible for the large amounts of prostaglandins associated with inflammation. It was further hypothesized that COX-2 used intracellular arachidonic acid as a substrate almost exclusively, while COX-1 could use phospholipase A2 as an extracellular substrate under some conditions.2
Recent data suggest that COX-1 and -2 are more complex than originally assumed. There is increasing evidence that COX-2 is constitutively expressed in the brain, kidneys, and pancreatic islet cells and plays a role in intestinal tolerance to dietary antigens, ulcer healing, ovulation, and implantation. Transgenic mice with deletions of the COX-2 enzyme maintain the inflammatory response when only COX-1 is present.3
In the reproductive tract, COX-2 expression increases substantially in midcycle because of the surge in luteinizing hormone (LH). COX-2 and the resulting prostaglandins (released from granulosa cells) are thought to play an important role in rupture of the follicle.4,5 With fertilization, COX-2 expression increases in the endometrium surrounding the implantation site.6 The resultant prostaglandins are important for successful implantation and angiogenesis. Interestingly, transgenic mice that lack the COX-2 enzyme are infertile, while those missing only the COX-1 enzyme have normal reproduction.7,8
Studies also have demonstrated a rapid increase in COX-2 expression in the placenta and amnion immediately before and during labor.9 It is well established that prostaglandins have a pivotal role in myometrial contraction10 and inhibition of COX-2 has been shown to delay labor.11
The COX-1 and -2 enzymes share approximately 60% homology.12 Both have a long, narrow channel that is the active site of arachidonic acid folding and oxygenation. The critical difference between the 2 isoenzymes occurs at position 523, where the COX-1 enzyme has an isoleucine while COX-2 has a valine.13 This substitution causes the channel in the COX-2 form to be 17% wider; it also provides a side pocket that increases the volume of the active site by 8%. Drugs that are designed to block the COX-2 enzyme take advantage of this difference in channel size—they are too large to fit the normal channel. Nonselective NSAIDs are small enough to block the channel in both isoenzymes while, at least in theory, the selective agents can block only the larger channel.14 Most COX-2 inhibitors also exhibit some action against the COX-1 isoform.—Roger P. Smith, MD, and Jeffrey Ellis, MD
REFERENCES
1. Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthase (cyclooxygenase)-1 and -2. J Biol Chem. 1996;271:33157.-
2. Herschman HR. Prostaglandin synthase 2. Biochem Biophys Acta. 1996;1299:125-140.
3. Wallace JL, Bak A, McKnight W, Asfaha S, Sharkey KA, MacNaughton WK. Cyclooxygenase 1 contributes to inflammatory responses in rats and mice: implications for gastrointestinal toxicity. Gastroenterology. 1998;115:101-109.
4. Richards JS, Fitzpatrick SL, Clemens JW, Morris JK, Alliston T, Sirois J. Ovarian cell differentiation: a cascade of multiple hormones, cellular signals, and regulated genes. Recent Prog Horm Res. 1995;50:223-254.
5. Akil M, Amos RS, Stewart P. Infertility may sometimes be associated with NSAID consumption. Br J Rheumatol. 1996;35:76-78.
6. Chakraborty I, Das SK, Wang J, Dey SK. Developmental expression of the cyclo-oxygenase-1 and cyclooxygenase-2 genes in the peri-implantation mouse uterus and their differential regulation by the blastocyst and ovarian steroids. J Mol Endocrinol. 1996;16:107-122.
7. Lim H, Paria BC, Das SK, Dinchuk JE, Langenbach R, Trzaskos JM, et al. Multiple female reproductive failures in cyclooxygenase 2-deficient mice. Cell. 1997;91:197-208.
8. Langenbach R, Loftin C, Lee C, Tiano H. Cyclooxygenase knockout mice: models for elucidating isoform-specific functions. Biochem Pharmacol. 1999;58:1237-1246.
9. Gibb W, Sun M. Localization of prostaglandin H synthase type 2 protein and mRNA in term human fetal membranes and decidua. J Endocrinol. 1996;150:497-503.
10. O’Brien WF. The role of prostaglandins in labor and delivery. Clin Perinatol. 1995;22:973-984.
11. Mitchell MD, Romero RJ, Edwin SS, Trautman MS. Prostaglandins and parturition. Reprod Fertil Dev. 1995;7:623-632.
12. Kurumbail RG, Stevens AM, Gierse JK, et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384:644-4.(Published correction appears in Nature. 1997;385:555.)
13. Gierse JK, McDonald JJ, Hauser SD, Rangwala SH, Koboldt CM, Seibert K. A single amino acid difference between cyclooxygenase-1 (COX-1) and -2 (COX-2) reverses the selectivity of COX-2 specific inhibitors. J Biol Chem. 1996;271:15810.-
14. Lanzo CA, Beechem JM, Talley J, Marnett LJ. Investigation of the binding of isoform-selective inhibitors to prostaglandin endoperoxide synthases using fluorescence spectroscopy. Biochemistry. 1998;37:217-226.
TABLE 2
Comparison of COX-1 and COX-2 in human tissues
| Characteristic | COX-1 | COX-2 |
|---|---|---|
| Tonic expression | High | Low |
| Inducibility | Low | High |
| Predominant location | All (stomach, kidney, platelets, vasculature) | Sites of inflammation (neoplastic tissue, small intestine, ovary, uterus, brain) |
| Function | Homeostasis | Inflammation and repair, neoplasia, modulation of immune response |
Conclusion
It is apparent from the multitude of published studies and the breadth of clinical experience with NSAID therapy for primary dysmenorrhea that most of these drugs provide relief for the majority of dysmenorrheic women. The success rates reported from both subjective and objective studies of the use of mefenamic acid in dysmenorrhea support its potential superiority. The physiology of primary dysmenorrhea would suggest that the ability of the fenamates to block the action of preformed prostaglandins—as well as the 5-lipooxygenase pathway—gives them a therapeutic edge. Interestingly, one investigation of the level of selectivity of multiple NSAIDs found that mefenamic acid had a selectivity (preferential COX-2 inhibition) comparable to some of the newest selective COX-2 inhibitors.43 This may account for the low incidence of GI and other side effects reported with mefenamic acid.
The history of NSAIDs has involved the sequential introduction of agents designed to reduce side effects or increase efficacy, often through the modification of existing compounds. Many of these products have failed to meet expectations and have been withdrawn from the market. Notable recent examples include benoxaprofen, suprofen, oxyphenbutazone, and zomepirac.
Thus, it is ironic that mefenamic acid, the first agent introduced to overcome the drawbacks of aspirin, has proved to be so robust in efficacy and tolerability. In addition, it is one of the only agents objectively shown to reduce menstrual blood loss in women with menorrhagia,44-47 and to ease menstrual migraine48 and the physical and emotional symptoms of premenstrual syndrome.49-51 In fact, the combination of dysmenorrhea, premenstrual syndrome, and menorrhagia experienced by many women would seem to support the use of mefenamic acid over other agents.
Unfortunately, no direct comparisons of various drugs have definitively resolved the question of which agent is “best” for the treatment of dysmenorrhea. Until there is further refinement of our knowledge of uterine prostaglandins, menstrual function, and the physiology of dysmenorrhea, it would seem wise to rely on agents that have been studied most extensively.
Dr. Smith reports no financial relationship with any companies whose products are mentioned in this article. Dr. Ellis reports that he is on the speakers’ bureau for First Horizon Pharmaceutical Company.
1. Mayer E. The intranasal treatment of dysmenorrhea. With a report of ninety-three cases. JAMA. 1914;62:6.-
2. Miller NF, et al. Dysmenorrhea. Am J Obstet Gynecol. 1953;65:505.-
3. Cotte G. La sympathectomie hypogastrique: A-t-elle sa place dans la therapeutique gynecologique? Presse Med. 1925;33:98.-
4. Black WT, Jr. Use of presacral sympathectomy in the treatment of dysmenorrhea: a second look after twenty-five years. Am J Obstet Gynecol. 1964;89:16.-
5. Hayden GE. Relief of primary dysmenorrhea. Obstet Gynecol. 1960;16:730.-
6. Schuck F. Pain and pain relief in essential dysmenorrhea. Am J Obstet Gynecol. 1951;62:559.-
7. Voulgaris DM. Dysmenorrhea. Treatment with isoxsuprine. Obstet Gynecol. 1960;15:220.-
8. Champlin WD, Corbit JP. Chlorpromazine and chlorpromazine combinations in the treatment of dysmenorrhea. Am J Obstet Gynecol. 1957;74:419.-
9. Filler W. The treatment of dysmenorrhea. With special reference to the primary type. Med Clin North Am. 1951;35:861.-
10. Menaker JS, Powers KD. Management of primary dysmenorrhea. Obstet Gynecol. 1962;20:66.-
11. Molla LA, Donald JF. A comparative study of ibuprofen and paracetamol in primary dysmenorrhea. J Int Med Res. 1974;2:395.-
12. Roth-Brandel U, Bygdman M, Wiqvist N. Effect of intravenous administration of prostaglandin E1 and F2 on the contractility of the nonpregnant human uterus in vivo. Acta Obstet Gynecol Scand Suppl. 1970;49(5):19.-
13. Filler W. The treatment of dysmenorrhea. With special reference to the primary type. Med Clin North Am. 1951;35:861.-
14. Menaker JS, Powers KD. Management of primary dysmenorrhea. Obstet Gynecol. 1962;20:66.-
15. Jacobson J, Cavalli-Bjorkman K, Lundstrom V, Nilsson B, Norbeck M. Prostaglandin synthetase inhibitors and dysmenorrhea. Acta Obstet Gynecol Scand. 1979;87(suppl):73.-
16. Dingfelder JR. Primary dysmenorrhea treatment with prostaglandin inhibitors: a review. Am J Obstet Gynecol. 1981;140:874.-
17. Owen PR. Prostaglandin synthetase inhibitors in the treatment of primary dysmenorrhea. Am J Obstet Gynecol. 1984;148:96.-
18. Dawood MY. Nonsteroidal anti-inflammatory drugs and reproduction. Am J Obstet Gynecol. 1993;169:1255-1265.
19. Zhang WY, Po ALW. Efficacy of minor analgesics in primary dysmenorrhoea: a systematic review. Br J Obstet Gynaecol. 1998;105:780-789.
20. Lichtenstein DR, Syngal S, Wolfe MM. Nonsteroidal anti-inflammatory drugs and the gastrointestinal tract: the double-edged sword. Arthritis Rheumk. 1995;38:5-18.
21. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for the aspirin-like drugs. Nature. 1971;231:232-235.
22. Morrison BW, Daniels SE, Kotey P, Cantu N, Seidenberg B. Rofecoxib, a specific cyclooxygenase-2 inhibitor, in primary dysmenorrhea: a randomized controlled trial. Obstet Gynecol. 1999;94:504-508.
23. Stone E. An account of the success of the bark of the willow in the cure of agues. Philosophical Transactions of the Royal Society. 1763;53:195-200.
24. Winder CV, Wax J, Scotti L, Scherrer RA, Jones EM, Short FW. Antiinflammatory, antipyretic, and antinociceptive properties of N-(2,3-xylyl) anthranilic acid (mefenamic acid). J Pharm Exp Ther. 1962;138:405-413.
25. Collier HOJ, Sweatmon WJF. Antagonism by fenamates of prostaglandin F 2α and of slow-reacting substance on human bronchial muscle. Nature. 1968;219:864.-
26. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature New Biol. 1971;231:232.-
27. Tolman EL, Partridge R. Multiple sites of interaction between prostaglandins and nonsteroidal anti-inflammatory drugs. Prostaglandins. 1975;9:349.-
28. Ferreira SH, Vane JR. New aspects of the mode of action of nonsteroidal anti-inflammatory drugs. Ann Rev Pharmacol. 1973;14:57.-
29. Schwartz A, Zor U, Lindner HR, Naor S. Primary dysmenorrhea. Alleviation by an inhibitor of prostaglandin synthesis and action. Obstet Gynecol. 1974;44:709.-
30. Kauppila A, Ylikorkala O. Indomethacin and tolfenamic acid in primary dysmenorrhea. Eur J Obstet Gynecol Reprod Biol. 1977;7:59.-
31. Anderson ABM, Haynes PJ, Fraser IS, Turnbull AC. Trial of prostaglandin-synthetase inhibitors in primary dysmenorrhea. Lancet. 1978;1:345.-
32. Kapadia L, Elder MG. Flufenamic acid in treatment of primary spasmodic dysmenorrhea. Lancet. 1978;1:348.-
33. Budoff PW. Use of mefenamic acid in the treatment of primary dysmenorrhea. JAMA. 1979;241:2713.-
34. Powell JR, Smith RP. Mefenamic acid (Ponstel) for treating primary spasmodic dysmenorrhea. Curr Ther Res. 1981;29:544.-
35. Roy S. A double-blind comparison of a propionic acid derivative (ibuprofen) and a fenamate (mefenamic acid) in the treatment of dysmenorrhea. Obstet Gynecol. 1983;61:628.-
36. Smith RP, Powell JR. Simultaneous objective and subjective evaluation of meclofenamate sodium in the treatment of primary dysmenorrhea. Am J Obstet Gynecol. 1987;157:611.-
37. Smith RP. The dynamics of nonsteroidal anti-inflammatory therapy for primary dysmenorrhea. Obstet Gynecol. 1987;70:785.-
38. Boctor AM, Eickholt M, Pugsley TA. Meclofenamate sodium is an inhibitor of both the 5-lipooxygenase and cyclooxygenase pathways of the arachidonic acid cascade in vitro. Prostaglandins, Leukotrienes & Med. 1986;23:229.-
39. Myers RF, Siegel MI. Differential effects of anti-inflammatory drugs on lipooxygenase and cyclooxygenase activities of neutrophils from a reverse passive Arthus reaction. Biochemical Biophysical Res Comm. 1983;112:586.-
40. Feldman M, McMahon AT. Do cyclooxygenase-2 inhibitors provide benefits similar to those of traditional nonsteroidal anti-inflammatory drugs, with less gastrointestinal toxicity? Ann Intern Med. 2000;132:134-143.
41. Bannwarth B. Are COX-2 inhibitors as effective as conventional NSAIDs in acute pain states? [letter] Arch Intern Med. 2001;161:127-128.
42. Lipsky PE, Brooks P, Crofford LJ, DuBois R, Graham D, Simon LS, van de Putte LBA, Abramson SB, et al. Unresolved issues in the role of cyclooxygenase-2 in normal physiologic processes and disease. Arch Intern Med. 2000;160(7):913-920.
43. Cryer B, Feldman M. Cyclooxygenase-1 and cyclooxygenase-2 selectivity of widely used nonsteroidal anti-inflammatory drugs. Am J Med. 1998;104(5):413-421.
44. Fraser IS, Pearse C, Shearman RP, Elliott PM, McIlveen J, Markham R. Efficacy of mefenamic acid in patients with a complaint of menorrhagia. Obstet Gynecol. 1981;58:543-551.
45. Fraser IS, McCarron G, Markham R, Robinson M, Smyth E. Long-term treatment of menorrhagia with mefenamic acid. Obstet Gynecol. 1983;61:109-112.
46. Vargyas JM, Campeau JD, Mishell DR Jr. Treatment of menorrhagia with meclofenamate sodium. Am J Obstet Gynecol. 1987;157:944-950.
47. Van Eijkeren MA, Christiaens GCML, Geuze HJ, Haspels AA, Sixma JJ. Effects of mefenamic acid on menstrual hemostasis in essential menorrhagia. Am J Obstet Gynecol. 1992;166:1419-1428.
48. Al-Waili NS. Treatment of menstrual migraine with prostaglandin synthesis inhibitor mefenamic acid: double-blind study with placebo. Eur J Med Res. 2000;5:176-182.
49. Wood C, Jakubowicz D. The treatment of premenstrual symptoms with mefenamic acid. Br J Obstet Gynaecol. 1980;87:627-630.
50. Jakubowicz DL, Godard E, Dewhurst J. The treatment of premenstrual tension with mefenamic acid: analysis of prostaglandin concentrations. Br J Obstet Gynaecol. 1984;91:78-84.
51. Mira M, McNeil D, Fraser IS, Vizzard J, Abraham S. Mefenamic acid in the treatment of premenstrual syndrome. Obstet Gynecol. 1986;68:395-398.

ROGER P. SMITH, MD
Jeffrey ELLIS, MD
Dr. Smith is professor of OBG at the University of Missouri–Kansas City Truman Medical Center in Kansas City, Mo. Dr. Ellis is in private practice in Plymouth, Ind.
ROGER P. SMITH, MD
Jeffrey ELLIS, MD
Dr. Smith is professor of OBG at the University of Missouri–Kansas City Truman Medical Center in Kansas City, Mo. Dr. Ellis is in private practice in Plymouth, Ind.
ROGER P. SMITH, MD
Jeffrey ELLIS, MD
Dr. Smith is professor of OBG at the University of Missouri–Kansas City Truman Medical Center in Kansas City, Mo. Dr. Ellis is in private practice in Plymouth, Ind.
- Nonsteroidal anti-inflammatory drugs (NSAIDs) can prevent dysmenorrhea, unlike other agents that simply relieve symptoms.
- Although NSAIDs in one form or another have been used for centuries, agents introduced in the past 50 years have significantly improved efficacy and safety profiles.
- A greater understanding of the role of prostaglandins in physiologic and pathophysiologic processes can enhance the selection of appropriate therapeutic agents.
- Some drugs can selectively block the cyclooxygenase-2 (COX-2) isoform of the enzyme instrumental in the production of prostaglandins.
A major shift in the way menstrual pain is viewed and treated took place in the 1970s and ’80s, with a greater understanding of the role of prostaglandins and more effective nonsteroidal anti-inflammatory drugs (NSAIDs). Subjective studies of pain and objective studies of uterine activity established a firm connection between the two. These studies also amply demonstrated the ability of NSAIDs to alter the physiology of dysmenorrhea, making it possible to prevent—rather than simply relieve—pain.
Yet, these agents still are not universally used in the treatment of dysmenorrhea, despite more than 20 years of experience with them. Moreover, the introduction of new NSAIDs has clouded rather than clarified the issue of their relative efficacy. Drugs that are welldesigned for the suppression of chronic inflammation (e.g., arthritis therapies) are not very effective for dysmenorrhea, and vice versa. Even so, it is possible to apply the findings of published studies and an understanding of the pathophysiology of dysmenorrhea to demystify the range of options.
The therapeutic effects of NSAIDs come from their ability to inhibit the production of prostaglandin.
A brief history
The term “dysmenorrhea” is derived from a Greek root meaning “difficult monthly flow,” but it did not make its appearance in the English language until about 1810. Therapies for dysmenorrhea ranged from the plausible and somewhat effective to the outlandish and useless. Everything from cauterizing the middle turbinate of the nose,1 exercise programs,2 and presacral sympathectomy3,4 to uterine-relaxing factor,5 vasodilators,6,7 tranquilizers,8 and hormones9-11 have been tried. Today’s effective therapies against primary dysmenorrhea are an outgrowth of earlier observations of uterine activity and the presence of a menstrual “toxin.” That toxin was later identified as prostaglandin.
Endometrial prostaglandin production is tied to changes throughout the menstrual cycle. Prostaglandin is stored in the endometrium as it thickens in preparation for implantation or menstruation. With the onset of menstruation, preformed prostaglandins are liberated and large amounts of arachidonic acid are released from the cell walls of sloughed endometrial cells. This large increase in arachidonic acid substrate results in a tremendous rise in prostaglandin production, which augments the supplies of preformed prostaglandins liberated from the sloughed endometrial cells.
The causative role of prostaglandin F2α in dysmenorrhea was confirmed when researchers triggered dysmenorrhea-like pain and uterine activity after intravenous (IV) injection of prostaglandins.12 (Current evidence indicates that women with primary dysmenorrhea make 2 to 7 times the normal amount of prostaglandin F2α.) Excess prostaglandins also may be responsible for the smooth-muscle activity noted in the gastrointestinal (GI) tracts of these women. Hypermobility of the gut may be responsible for the frequent coexistence of nausea, vomiting, and diarrhea in these patients. In addition, prostaglandins appear to act as initiators and potentiators of nociceptive pain signals, further contributing to the symptoms of dysmenorrhea.
In 1967, Pickles demonstrated that prostaglandin levels were lower during anovulatory cycles, prompting the use of oral contraceptives (OCs) to suppress ovulation and relieve menstrual pain.13,14 Although this approach is usually successful, not all women want to—or can—use OCs. NSAIDs more directly alter the physiologic sequence leading to discomfort by inhibiting the production and/or action of prostaglandins. Moreover, NSAIDs generally are well-tolerated and need only be taken at the time of menstruation. While OCs act to reduce the substrate available to the reaction, NSAIDs act to block the pathway at 2 later enzymatic steps.
In 1979, Jacobson et al reported successful pain relief in 64% to 100% of patients from 16 studies of NSAIDs in dysmenorrhea.15 Unfortunately, few of those studies were double-blinded, and many failed to report the incidence of side effects. Dingfelder evaluated 23 trials published from 1970 to 1980 and found a 67% to 86% rate of pain relief.16 In a more thorough review, Owen presented data from 51 reports and attempted to analyze the diverse methods, designs, and outcomes.17 She found an 87% rate of “excellent” pain relief for the fenamates versus 56%, 68%, and 56% for ibuprofen, indomethacin, and naproxen, respectively. Unfortunately, she lumped together 2 different drugs (tolfenamic and mefenamic acids) and misinterpreted some primarily methodological reports. More recent attempts to analyze existing studies have failed to further clarify the issue.18,19
Prostaglandins are made throughout the body and are important autocrine and paracrine regulators of cellular and organ function. As depicted below, the main substrate for their production is arachidonic acid, a major constituent of cell walls. Under some circumstances, phospholipase A2 also can be used as a substrate for prostaglandin production.
Cyclooxygenase, also called prostaglandin H synthase, is the first enzymatic step in the conversion of arachidonic acid into prostaglandins. This enzyme folds the arachidonic acid molecule (cyclization) and oxygenates it to produce prostaglandin H2 (PGH2).
All other members of the prostaglandin family are then formed from PGH2. Arachidonic acid also is the substrate for the production of leukotrienes and 5-hydroxyeicosatetraenoic acid (5-HETE) through the 5-lipooxygenase pathway. Like prostaglandins F2α and E2, the products of the lipooxygenase pathway are potent vasoconstrictors and stimulators of uterine contractions.—Roger P. Smith, MD, and Jeffrey Ellis, MD
Increased uterine activity was first hypothesized as a cause of dysmenorrhea in 1932. By the late 1930s, objective findings began to support that hypothesis.1 The correlation between uterine activity and menstrual pain was strengthened when Jacobson et al studied simultaneous electrical and mechanical changes within the uterus.2,3 Using an intrauterine air-filled balloon system, Wilson and Kurzrok also noted the relationship between maximal uterine activity and pain.4,5 Despite the strength of these investigations, few changes occurred in the way dysmenorrhea was viewed or treated.
In the late 1940s, Liessé demonstrated that women with dysmenorrhea not only had a greater degree of uterine electrical and mechanical activity, but that this activity correlated with the pain of menstruation.6 Liessé found minimal activity between pains and as many as 30 irregular electrical discharges per second during pain, suggesting a cause (electrical) for dysmenorrhea but offering no clue to the underlying physiologic disturbance that might account for it.
The most detailed and influential studies of dysmenorrhea and uterine activity came in 1947 in a trial conducted by Woodbury.7 His findings of a direct correlation between pressure, pattern of contractions, resting tone, and pain became a standard reference.
After Woodbury, the stage was set for a connection to be made between uterine activity and prostaglandins. That happened in 1965, when Pickles reported elevated levels of prostaglandin F2αin the menstrual fluid of dysmenorrheic women.8
During the past 2 decades, more sophisticated analytic techniques have been applied to intrauterine pressure data,9,10 and strong correlations between uterine activity and pain have been reported.11,12 The basic assertion that menstrual pain is caused by increased intrauterine pressure, poor relaxation, and more frequent, irregular contractions appears to be valid.—Roger P. Smith, MD, and Jeffrey Ellis, MD
REFERENCES
1. Novac E, Reynolds SRM. The cause of primary dysmenorrhea. JAMA. 1932;99:1466.-
2. Jacobson E, Lackner JE, Sinykin MB. Electrical and mechanical activity of the human non-pregnant uterus. Am J Obstet Gynecol. 1939;38:1008.-
3. Jacobson E, Lackner JE, Sinykin MB. Activity of the human non-pregnant uterus. Am J Physiol. 1940;53:407.-
4. Wilson L, Kurzrok R. Studies on the motility of the human uterus in vivo. Endocrinology. 1938;23:79.-
5. Wilson L, Kurzrok R. Uterine contractility in functional dysmenorrhea. Endocrinology. 1940;27:23.-
6. Liessé A. L’Activité électrique de l’uterus dans la dysmenorrhee functionnelle. Gynec et Obstet. 1948;47:850.-
7. Woodbury RA, Torpin R, Child GP, Watson H, Jarboe M. Myometrial physiology and its relation to pelvic pain. JAMA. 1947;134:1081-1085.
8. Pickles VR, Hall WJ, Best FA, Smith GN. Prostaglandins in endometrium and menstrual fluid from normal and dysmenorrheic subjects. J Obstet Gynaecol Br Comm. 1965;72:185.-
9. Smith RP. Intrauterine pressure analysis in nonpregnant dysmenorrheic women. Med Instr. 1984;185:137-139.
10. Smith RP. Distribution analysis of intrauterine pressure in nonpregnant dysmenorrheic women. Am J Obstet Gynecol. 1984;150:271-273.
11. Smith RP. The dynamics of nonsteroidal anti-inflammatory therapy for primary dysmenorrhea. Obstet Gynecol. 1987;70:785-788.
12. Smith RP, Powell JR. Simultaneous objective and subjective evaluation of meclofenamate sodium in the treatment of primary dysmenorrhea. Am J Obstet Gynecol. 1987;157:611-616.
Classes of NSAIDs
Although aspirin was synthesized in 1853 and incorporated into medical practice in 1899, its history goes back even farther. That fact—along with the introduction of newer agents—ensured that NSAIDs became the mainstays of medical therapy for fever, pain, and inflammation. In the United States, NSAIDs are among the most widely prescribed drugs, with more than 70 million prescriptions and more than 30 billion over-the-counter tablets sold each year.20 Most of their therapeutic effects come from their ability to inhibit the production of prostaglandins.21
Interestingly, some drugs with the ability to inhibit prostaglandin synthesis have little clinical usefulness. Some have weak antiprostaglandin activity, require metabolic transformation to become active, or have side effects that limit their usefulness. While these drugs can be used to treat dysmenorrhea, they generally have been replaced by more effective agents.
There are 2 broad classes of NSAIDs: enolic acids and carboxylic acids. Each class can be further subcategorized (Table 1).
Enolic acids. With the exception of the newer cyclooxygenase-2 (COX-2) selective agents, drugs of the enolic-acid type are primarily type II inhibitors of prostaglandin synthesis. That means they impede the isomerase/reductase step in the formation of prostaglandins E2 (PGE2) and F2α (PGF2α). The most frequently used agents in the enolic-acid group are phenylbutazone and piroxicam.
Phenylbutazone was discovered in the 19th century during a search for a substitute for quinine. (Quinine had become popular for the treatment of fever; however, uncontrolled cutting of the Peruvian cinchona tree dramatically increased its cost.) While phenylbutazone is an effective shortterm analgesic for musculoskeletal pain (through antiprostaglandin activity), its relative toxicity has limited its use in dysmenorrhea and general therapy.
Piroxicam has a long half-life (50 hours), making once-daily dosing possible. Its action as an anti-inflammatory drug for the treatment of arthritis is well-established, but its use as an analgesic for acute pain therapy or for dysmenorrhea has not been fully evaluated. Based on the pharmacodynamics of drug absorption and action, one would anticipate its efficacy in that regard to be poor. In general, drugs in the pyrazolone group have a higher incidence of blood dyscrasias, limiting their broad utility.
Celecoxib is structurally similar to phenylbutazone and was the first selective COX-2 inhibitor approved by the Food and Drug Administration (FDA). It has been studied in dental pain models and the treatment of osteoarthritis. In these trials, celecoxib performed as well as naproxen and slow-release diclofenac, but with fewer GI side effects. No data on its use in dysmenorrhea is available.
Chemically related but less similar to the enolic acids is refecoxib. Like celecoxib, it has been studied in the treatment of osteoarthritis, where it was comparable in efficacy to ibuprofen and diclofenac, but with side-effect rates similar to placebo therapy. In studies of women with primary dysmenorrhea, rofecoxib has proved to be statistically superior to placebo but indistinguishable from naproxen (Figure 1).22 Peak blood levels are achieved in 2 to 3 hours (delayed by 1 to 2 hours by a fatty meal), but a steady state is not achieved until day 4 of continuous therapy. When the drug is used to treat women who have a rapid onset of symptoms, the significance of this delay is unclear but is a potential drawback. In addition, the dosage of rofecoxib for the treatment of pain or dysmenorrhea is generally much larger than that required for the treatment of arthritis. Thus, the risk of side effects may be increased.
A third selective COX-2 inhibitor, meloxicam, was introduced into the US market solely for the treatment of osteoarthritis. This drug is marketed on the basis of its more favorable side-effect profile rather than its selectivity in blocking the COX-2 isoform.
Carboxylic acids. The first of the carboxylic acid subgroups are the salicylic acids. The use of salicylates derived from the bark or leaves of the Salic alba or S. fragilis dates back to Biblical times. Salicylates were formally introduced into medical use in 1763 by the Reverend Edmund Stone.23
The relatively low potency of aspirin for reducing prostaglandin synthesis in the uterus limits its clinical utility in preventing moderate or severe dysmenorrhea. In contrast, diflunisal has a long half-life, making twice-daily dosing possible. However, its slow onset of action limits use for most patients with menstrual pain.
The indoleacetic acid subgroup offers increased potency against dysmenorrhea. Most of these drugs have a direct effect rather than requiring metabolism into an active form.
Indomethacin was one of the first NSAIDs widely used to treat dysmenorrhea, but a moderate incidence of side effects limits its use—and that of most of the other drugs in this family.
No NSAID completely eliminates gastric cyclooxygenase activity.
The most commonly used drugs for dysmenorrhea come from the 2 remaining carboxylate groups: arylalkanoic acids (propionic acid derivatives) and anthranilic acids (fenamates). Of the propionic acid derivatives currently available, ibuprofen and naproxen are commonly used for dysmenorrhea. Other drugs of this class have been used for pain relief or arthritis therapy, but are not currently FDA-approved for dysmenorrhea. The subset of drugs that are phenylpropionic acid derivatives are associated with a higher incidence of GI side effects and skin reactions.
Mefenamic acid was the first alternative synthesized in an effort to reduce the gastric irritation caused by acetylsalicylic acid.24 Mefenamic acid and meclofenamate sodium—both fenamates—are potent prostaglandin synthetase inhibitors. In addition, they have been shown to antagonize already formed prostaglandins, providing a dual mode of action.25-28 This ability to block the action of existing prostaglandins produces a more rapid onset of uterine relaxation. Some of the first studies of NSAIDs in dysmenorrhea used these agents.29,30 In fact, they are some of the most thoroughly investigated drugs for the treatment of dysmenorrhea, shown to be consistently effective in reducing its subjective discomfort.31-35
In the U.S., mefenamic acid was one of the first drugs approved for dysmenorrhea, and clinical studies have shown meclofenamate to be very effective in improving symptoms and altering the underlying pathophysiology.36,37 The dual mechanism of action likely gives these agents an edge in efficacy, although no clinical trials have been initiated to evaluate this presumed advantage. Intrauterine pressure changes have been documented as soon as 15 minutes after administration of the medications, suggesting a very rapid onset of action (obviating the need for preloading). In addition, in vitro studies have demonstrated meclofenamate’s ability to inhibit the activity of 5-lipooxygenase, unlike members of the propionic acid group, which have little or no inhibitory ability.38,39 The clinical significance of inhibiting the production of the extremely potent leukotrienes has yet to be fully explored.
TABLE 1
Families of NSAIDs
| Family | Example |
|---|---|
| ENOLIC ACIDS | |
| PYRAZOLONES | |
| Oxyphenbutazone | Azolid** |
| Phenylbutazone | Butazolidin** |
| Nabumetone | Relafen |
| Celecoxib | Celebrex |
| Rofecoxib* | Vioxx |
| OXICAMS | |
| Piroxicam | Feldene, Piroxicam |
| Meloxicam | Mobic |
| CARBOXYLIC ACIDS | |
| SALICYLIC ACIDS | |
| Acetylsalicylic acid | Aspirin (various) |
| Diflunisal | Dolobid |
| Salicylate | Disalcid, Trilisate |
| INDOLEACETIC ACIDS | |
| Diclofenac potassium | Cataflam |
| Diclofenac sodium | Voltaren, Arthrotec (combined with misoprostol) |
| Etodolac | Lodine |
| Indomethacin | Indocin |
| Ketorolac tromethamine | Acular, Toradol |
| Sulindac | Clinoril, Sulindac |
| Tolmetin | Tolectin, Tolmetin |
| PROPIONIC ACIDS | |
| Fenoprofen calcium | Fenoprofen |
| Flurbiprofen | Flurbiprofen |
| Ibuprofen* | Motrin |
| Ketoprofen | Orudis, Oruvail, Ketoprofen |
| Naproxen sodium* | Aleve, Anaprox, Naprelan |
| Naproxen* | Naprosyn |
| FENAMATES | |
| Meclofenamate sodium* | Meclofenamate |
| Mefenamic acid | Ponstel |
| *FDA-approved for primary dysmenorrhea | |
| **No longer available | |
| Adapted from: Smith RP.Gynecology in Primary Care. Baltimore, Md: Williams and Wilkins; 1996:399. | |
FIGURE 1 Comparison of rofecoxib, naproxen, and placebo in treatment of primary dysmenorrhea
Reprinted with permission from The American College of Obstetricians and Gynecologists. Morrison BW, Daniels SE, Kotey P, Cantu N, Seidenberg B. Rofecoxib, a specific cyclooxygenase-2 inhibitor, in primary dysmenorrhea: a randomized-controlled trial. Obstet Gynecol. 1999;94:504-508.
COX-2 inhibitors
Although the short-term efficacy and safety of new selective COX-2 inhibitors appear to be good, several concerns remain. Despite a decrease in the incidence of GI side effects with these agents, their use by patients with active GI ulceration, infection with Helicobacter pylori, or inflammatory bowel disease has not been adequately studied. Indeed, recent studies suggest that, at therapeutic concentrations, no NSAID—not even the selective COX-2 inhibitors—completely eliminates gastric cyclooxygenase activity.40
In addition, questions have been raised about the speed and efficacy of the selective COX-2 inhibitors for the management of acute pain, compared with conventional NSAIDs.41 When used for postoperative analgesia, these agents theoretically could retard wound healing because of the role that COX-2 plays in healing and neovascularization. As more data emerge about the physiologic functions of COX-1 and -2 (Table 2), there are growing concerns that COX-2 is not restricted to inflammation and pathology, suggesting the possibility of unanticipated adverse effects associated with its use. For example, there is evidence of constitutive expression of COX-2 in the kidney and brain, and essential physiological functions in ovulation and implantation.42 In the treatment of dysmenorrhea, the increased cost of these drugs over the more commonly used agents, along with the lack of clinical efficacy studies, suggests a second-line role.
Like most NSAIDs, COX-2 inhibitors fall into pregnancy category C. Because of the risk of premature closure of the ductus arteriosus, they should not be used in the third trimester of pregnancy. In addition, drug levels in human milk mimic those in serum, so the decision to use these agents in nursing mothers must be made carefully.
Studies over the past 10 years have revealed that the cyclooxygenase enzyme (COX) is found in 2 isomeric forms, known as COX-1 and COX-2.1 Initial investigations suggested that COX-1 was present in most tissues and responsible for the homeostatic production of arachidonic acid metabolites. The COX-2 enzyme was thought to be induced in response to inflammatory stimuli such as cytokines and bacterial lipopolysaccharides, rather than expressed under normal cellular function. Thus, the COX-2 form was thought to be responsible for the large amounts of prostaglandins associated with inflammation. It was further hypothesized that COX-2 used intracellular arachidonic acid as a substrate almost exclusively, while COX-1 could use phospholipase A2 as an extracellular substrate under some conditions.2
Recent data suggest that COX-1 and -2 are more complex than originally assumed. There is increasing evidence that COX-2 is constitutively expressed in the brain, kidneys, and pancreatic islet cells and plays a role in intestinal tolerance to dietary antigens, ulcer healing, ovulation, and implantation. Transgenic mice with deletions of the COX-2 enzyme maintain the inflammatory response when only COX-1 is present.3
In the reproductive tract, COX-2 expression increases substantially in midcycle because of the surge in luteinizing hormone (LH). COX-2 and the resulting prostaglandins (released from granulosa cells) are thought to play an important role in rupture of the follicle.4,5 With fertilization, COX-2 expression increases in the endometrium surrounding the implantation site.6 The resultant prostaglandins are important for successful implantation and angiogenesis. Interestingly, transgenic mice that lack the COX-2 enzyme are infertile, while those missing only the COX-1 enzyme have normal reproduction.7,8
Studies also have demonstrated a rapid increase in COX-2 expression in the placenta and amnion immediately before and during labor.9 It is well established that prostaglandins have a pivotal role in myometrial contraction10 and inhibition of COX-2 has been shown to delay labor.11
The COX-1 and -2 enzymes share approximately 60% homology.12 Both have a long, narrow channel that is the active site of arachidonic acid folding and oxygenation. The critical difference between the 2 isoenzymes occurs at position 523, where the COX-1 enzyme has an isoleucine while COX-2 has a valine.13 This substitution causes the channel in the COX-2 form to be 17% wider; it also provides a side pocket that increases the volume of the active site by 8%. Drugs that are designed to block the COX-2 enzyme take advantage of this difference in channel size—they are too large to fit the normal channel. Nonselective NSAIDs are small enough to block the channel in both isoenzymes while, at least in theory, the selective agents can block only the larger channel.14 Most COX-2 inhibitors also exhibit some action against the COX-1 isoform.—Roger P. Smith, MD, and Jeffrey Ellis, MD
REFERENCES
1. Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthase (cyclooxygenase)-1 and -2. J Biol Chem. 1996;271:33157.-
2. Herschman HR. Prostaglandin synthase 2. Biochem Biophys Acta. 1996;1299:125-140.
3. Wallace JL, Bak A, McKnight W, Asfaha S, Sharkey KA, MacNaughton WK. Cyclooxygenase 1 contributes to inflammatory responses in rats and mice: implications for gastrointestinal toxicity. Gastroenterology. 1998;115:101-109.
4. Richards JS, Fitzpatrick SL, Clemens JW, Morris JK, Alliston T, Sirois J. Ovarian cell differentiation: a cascade of multiple hormones, cellular signals, and regulated genes. Recent Prog Horm Res. 1995;50:223-254.
5. Akil M, Amos RS, Stewart P. Infertility may sometimes be associated with NSAID consumption. Br J Rheumatol. 1996;35:76-78.
6. Chakraborty I, Das SK, Wang J, Dey SK. Developmental expression of the cyclo-oxygenase-1 and cyclooxygenase-2 genes in the peri-implantation mouse uterus and their differential regulation by the blastocyst and ovarian steroids. J Mol Endocrinol. 1996;16:107-122.
7. Lim H, Paria BC, Das SK, Dinchuk JE, Langenbach R, Trzaskos JM, et al. Multiple female reproductive failures in cyclooxygenase 2-deficient mice. Cell. 1997;91:197-208.
8. Langenbach R, Loftin C, Lee C, Tiano H. Cyclooxygenase knockout mice: models for elucidating isoform-specific functions. Biochem Pharmacol. 1999;58:1237-1246.
9. Gibb W, Sun M. Localization of prostaglandin H synthase type 2 protein and mRNA in term human fetal membranes and decidua. J Endocrinol. 1996;150:497-503.
10. O’Brien WF. The role of prostaglandins in labor and delivery. Clin Perinatol. 1995;22:973-984.
11. Mitchell MD, Romero RJ, Edwin SS, Trautman MS. Prostaglandins and parturition. Reprod Fertil Dev. 1995;7:623-632.
12. Kurumbail RG, Stevens AM, Gierse JK, et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384:644-4.(Published correction appears in Nature. 1997;385:555.)
13. Gierse JK, McDonald JJ, Hauser SD, Rangwala SH, Koboldt CM, Seibert K. A single amino acid difference between cyclooxygenase-1 (COX-1) and -2 (COX-2) reverses the selectivity of COX-2 specific inhibitors. J Biol Chem. 1996;271:15810.-
14. Lanzo CA, Beechem JM, Talley J, Marnett LJ. Investigation of the binding of isoform-selective inhibitors to prostaglandin endoperoxide synthases using fluorescence spectroscopy. Biochemistry. 1998;37:217-226.
TABLE 2
Comparison of COX-1 and COX-2 in human tissues
| Characteristic | COX-1 | COX-2 |
|---|---|---|
| Tonic expression | High | Low |
| Inducibility | Low | High |
| Predominant location | All (stomach, kidney, platelets, vasculature) | Sites of inflammation (neoplastic tissue, small intestine, ovary, uterus, brain) |
| Function | Homeostasis | Inflammation and repair, neoplasia, modulation of immune response |
Conclusion
It is apparent from the multitude of published studies and the breadth of clinical experience with NSAID therapy for primary dysmenorrhea that most of these drugs provide relief for the majority of dysmenorrheic women. The success rates reported from both subjective and objective studies of the use of mefenamic acid in dysmenorrhea support its potential superiority. The physiology of primary dysmenorrhea would suggest that the ability of the fenamates to block the action of preformed prostaglandins—as well as the 5-lipooxygenase pathway—gives them a therapeutic edge. Interestingly, one investigation of the level of selectivity of multiple NSAIDs found that mefenamic acid had a selectivity (preferential COX-2 inhibition) comparable to some of the newest selective COX-2 inhibitors.43 This may account for the low incidence of GI and other side effects reported with mefenamic acid.
The history of NSAIDs has involved the sequential introduction of agents designed to reduce side effects or increase efficacy, often through the modification of existing compounds. Many of these products have failed to meet expectations and have been withdrawn from the market. Notable recent examples include benoxaprofen, suprofen, oxyphenbutazone, and zomepirac.
Thus, it is ironic that mefenamic acid, the first agent introduced to overcome the drawbacks of aspirin, has proved to be so robust in efficacy and tolerability. In addition, it is one of the only agents objectively shown to reduce menstrual blood loss in women with menorrhagia,44-47 and to ease menstrual migraine48 and the physical and emotional symptoms of premenstrual syndrome.49-51 In fact, the combination of dysmenorrhea, premenstrual syndrome, and menorrhagia experienced by many women would seem to support the use of mefenamic acid over other agents.
Unfortunately, no direct comparisons of various drugs have definitively resolved the question of which agent is “best” for the treatment of dysmenorrhea. Until there is further refinement of our knowledge of uterine prostaglandins, menstrual function, and the physiology of dysmenorrhea, it would seem wise to rely on agents that have been studied most extensively.
Dr. Smith reports no financial relationship with any companies whose products are mentioned in this article. Dr. Ellis reports that he is on the speakers’ bureau for First Horizon Pharmaceutical Company.
- Nonsteroidal anti-inflammatory drugs (NSAIDs) can prevent dysmenorrhea, unlike other agents that simply relieve symptoms.
- Although NSAIDs in one form or another have been used for centuries, agents introduced in the past 50 years have significantly improved efficacy and safety profiles.
- A greater understanding of the role of prostaglandins in physiologic and pathophysiologic processes can enhance the selection of appropriate therapeutic agents.
- Some drugs can selectively block the cyclooxygenase-2 (COX-2) isoform of the enzyme instrumental in the production of prostaglandins.
A major shift in the way menstrual pain is viewed and treated took place in the 1970s and ’80s, with a greater understanding of the role of prostaglandins and more effective nonsteroidal anti-inflammatory drugs (NSAIDs). Subjective studies of pain and objective studies of uterine activity established a firm connection between the two. These studies also amply demonstrated the ability of NSAIDs to alter the physiology of dysmenorrhea, making it possible to prevent—rather than simply relieve—pain.
Yet, these agents still are not universally used in the treatment of dysmenorrhea, despite more than 20 years of experience with them. Moreover, the introduction of new NSAIDs has clouded rather than clarified the issue of their relative efficacy. Drugs that are welldesigned for the suppression of chronic inflammation (e.g., arthritis therapies) are not very effective for dysmenorrhea, and vice versa. Even so, it is possible to apply the findings of published studies and an understanding of the pathophysiology of dysmenorrhea to demystify the range of options.
The therapeutic effects of NSAIDs come from their ability to inhibit the production of prostaglandin.
A brief history
The term “dysmenorrhea” is derived from a Greek root meaning “difficult monthly flow,” but it did not make its appearance in the English language until about 1810. Therapies for dysmenorrhea ranged from the plausible and somewhat effective to the outlandish and useless. Everything from cauterizing the middle turbinate of the nose,1 exercise programs,2 and presacral sympathectomy3,4 to uterine-relaxing factor,5 vasodilators,6,7 tranquilizers,8 and hormones9-11 have been tried. Today’s effective therapies against primary dysmenorrhea are an outgrowth of earlier observations of uterine activity and the presence of a menstrual “toxin.” That toxin was later identified as prostaglandin.
Endometrial prostaglandin production is tied to changes throughout the menstrual cycle. Prostaglandin is stored in the endometrium as it thickens in preparation for implantation or menstruation. With the onset of menstruation, preformed prostaglandins are liberated and large amounts of arachidonic acid are released from the cell walls of sloughed endometrial cells. This large increase in arachidonic acid substrate results in a tremendous rise in prostaglandin production, which augments the supplies of preformed prostaglandins liberated from the sloughed endometrial cells.
The causative role of prostaglandin F2α in dysmenorrhea was confirmed when researchers triggered dysmenorrhea-like pain and uterine activity after intravenous (IV) injection of prostaglandins.12 (Current evidence indicates that women with primary dysmenorrhea make 2 to 7 times the normal amount of prostaglandin F2α.) Excess prostaglandins also may be responsible for the smooth-muscle activity noted in the gastrointestinal (GI) tracts of these women. Hypermobility of the gut may be responsible for the frequent coexistence of nausea, vomiting, and diarrhea in these patients. In addition, prostaglandins appear to act as initiators and potentiators of nociceptive pain signals, further contributing to the symptoms of dysmenorrhea.
In 1967, Pickles demonstrated that prostaglandin levels were lower during anovulatory cycles, prompting the use of oral contraceptives (OCs) to suppress ovulation and relieve menstrual pain.13,14 Although this approach is usually successful, not all women want to—or can—use OCs. NSAIDs more directly alter the physiologic sequence leading to discomfort by inhibiting the production and/or action of prostaglandins. Moreover, NSAIDs generally are well-tolerated and need only be taken at the time of menstruation. While OCs act to reduce the substrate available to the reaction, NSAIDs act to block the pathway at 2 later enzymatic steps.
In 1979, Jacobson et al reported successful pain relief in 64% to 100% of patients from 16 studies of NSAIDs in dysmenorrhea.15 Unfortunately, few of those studies were double-blinded, and many failed to report the incidence of side effects. Dingfelder evaluated 23 trials published from 1970 to 1980 and found a 67% to 86% rate of pain relief.16 In a more thorough review, Owen presented data from 51 reports and attempted to analyze the diverse methods, designs, and outcomes.17 She found an 87% rate of “excellent” pain relief for the fenamates versus 56%, 68%, and 56% for ibuprofen, indomethacin, and naproxen, respectively. Unfortunately, she lumped together 2 different drugs (tolfenamic and mefenamic acids) and misinterpreted some primarily methodological reports. More recent attempts to analyze existing studies have failed to further clarify the issue.18,19
Prostaglandins are made throughout the body and are important autocrine and paracrine regulators of cellular and organ function. As depicted below, the main substrate for their production is arachidonic acid, a major constituent of cell walls. Under some circumstances, phospholipase A2 also can be used as a substrate for prostaglandin production.
Cyclooxygenase, also called prostaglandin H synthase, is the first enzymatic step in the conversion of arachidonic acid into prostaglandins. This enzyme folds the arachidonic acid molecule (cyclization) and oxygenates it to produce prostaglandin H2 (PGH2).
All other members of the prostaglandin family are then formed from PGH2. Arachidonic acid also is the substrate for the production of leukotrienes and 5-hydroxyeicosatetraenoic acid (5-HETE) through the 5-lipooxygenase pathway. Like prostaglandins F2α and E2, the products of the lipooxygenase pathway are potent vasoconstrictors and stimulators of uterine contractions.—Roger P. Smith, MD, and Jeffrey Ellis, MD
Increased uterine activity was first hypothesized as a cause of dysmenorrhea in 1932. By the late 1930s, objective findings began to support that hypothesis.1 The correlation between uterine activity and menstrual pain was strengthened when Jacobson et al studied simultaneous electrical and mechanical changes within the uterus.2,3 Using an intrauterine air-filled balloon system, Wilson and Kurzrok also noted the relationship between maximal uterine activity and pain.4,5 Despite the strength of these investigations, few changes occurred in the way dysmenorrhea was viewed or treated.
In the late 1940s, Liessé demonstrated that women with dysmenorrhea not only had a greater degree of uterine electrical and mechanical activity, but that this activity correlated with the pain of menstruation.6 Liessé found minimal activity between pains and as many as 30 irregular electrical discharges per second during pain, suggesting a cause (electrical) for dysmenorrhea but offering no clue to the underlying physiologic disturbance that might account for it.
The most detailed and influential studies of dysmenorrhea and uterine activity came in 1947 in a trial conducted by Woodbury.7 His findings of a direct correlation between pressure, pattern of contractions, resting tone, and pain became a standard reference.
After Woodbury, the stage was set for a connection to be made between uterine activity and prostaglandins. That happened in 1965, when Pickles reported elevated levels of prostaglandin F2αin the menstrual fluid of dysmenorrheic women.8
During the past 2 decades, more sophisticated analytic techniques have been applied to intrauterine pressure data,9,10 and strong correlations between uterine activity and pain have been reported.11,12 The basic assertion that menstrual pain is caused by increased intrauterine pressure, poor relaxation, and more frequent, irregular contractions appears to be valid.—Roger P. Smith, MD, and Jeffrey Ellis, MD
REFERENCES
1. Novac E, Reynolds SRM. The cause of primary dysmenorrhea. JAMA. 1932;99:1466.-
2. Jacobson E, Lackner JE, Sinykin MB. Electrical and mechanical activity of the human non-pregnant uterus. Am J Obstet Gynecol. 1939;38:1008.-
3. Jacobson E, Lackner JE, Sinykin MB. Activity of the human non-pregnant uterus. Am J Physiol. 1940;53:407.-
4. Wilson L, Kurzrok R. Studies on the motility of the human uterus in vivo. Endocrinology. 1938;23:79.-
5. Wilson L, Kurzrok R. Uterine contractility in functional dysmenorrhea. Endocrinology. 1940;27:23.-
6. Liessé A. L’Activité électrique de l’uterus dans la dysmenorrhee functionnelle. Gynec et Obstet. 1948;47:850.-
7. Woodbury RA, Torpin R, Child GP, Watson H, Jarboe M. Myometrial physiology and its relation to pelvic pain. JAMA. 1947;134:1081-1085.
8. Pickles VR, Hall WJ, Best FA, Smith GN. Prostaglandins in endometrium and menstrual fluid from normal and dysmenorrheic subjects. J Obstet Gynaecol Br Comm. 1965;72:185.-
9. Smith RP. Intrauterine pressure analysis in nonpregnant dysmenorrheic women. Med Instr. 1984;185:137-139.
10. Smith RP. Distribution analysis of intrauterine pressure in nonpregnant dysmenorrheic women. Am J Obstet Gynecol. 1984;150:271-273.
11. Smith RP. The dynamics of nonsteroidal anti-inflammatory therapy for primary dysmenorrhea. Obstet Gynecol. 1987;70:785-788.
12. Smith RP, Powell JR. Simultaneous objective and subjective evaluation of meclofenamate sodium in the treatment of primary dysmenorrhea. Am J Obstet Gynecol. 1987;157:611-616.
Classes of NSAIDs
Although aspirin was synthesized in 1853 and incorporated into medical practice in 1899, its history goes back even farther. That fact—along with the introduction of newer agents—ensured that NSAIDs became the mainstays of medical therapy for fever, pain, and inflammation. In the United States, NSAIDs are among the most widely prescribed drugs, with more than 70 million prescriptions and more than 30 billion over-the-counter tablets sold each year.20 Most of their therapeutic effects come from their ability to inhibit the production of prostaglandins.21
Interestingly, some drugs with the ability to inhibit prostaglandin synthesis have little clinical usefulness. Some have weak antiprostaglandin activity, require metabolic transformation to become active, or have side effects that limit their usefulness. While these drugs can be used to treat dysmenorrhea, they generally have been replaced by more effective agents.
There are 2 broad classes of NSAIDs: enolic acids and carboxylic acids. Each class can be further subcategorized (Table 1).
Enolic acids. With the exception of the newer cyclooxygenase-2 (COX-2) selective agents, drugs of the enolic-acid type are primarily type II inhibitors of prostaglandin synthesis. That means they impede the isomerase/reductase step in the formation of prostaglandins E2 (PGE2) and F2α (PGF2α). The most frequently used agents in the enolic-acid group are phenylbutazone and piroxicam.
Phenylbutazone was discovered in the 19th century during a search for a substitute for quinine. (Quinine had become popular for the treatment of fever; however, uncontrolled cutting of the Peruvian cinchona tree dramatically increased its cost.) While phenylbutazone is an effective shortterm analgesic for musculoskeletal pain (through antiprostaglandin activity), its relative toxicity has limited its use in dysmenorrhea and general therapy.
Piroxicam has a long half-life (50 hours), making once-daily dosing possible. Its action as an anti-inflammatory drug for the treatment of arthritis is well-established, but its use as an analgesic for acute pain therapy or for dysmenorrhea has not been fully evaluated. Based on the pharmacodynamics of drug absorption and action, one would anticipate its efficacy in that regard to be poor. In general, drugs in the pyrazolone group have a higher incidence of blood dyscrasias, limiting their broad utility.
Celecoxib is structurally similar to phenylbutazone and was the first selective COX-2 inhibitor approved by the Food and Drug Administration (FDA). It has been studied in dental pain models and the treatment of osteoarthritis. In these trials, celecoxib performed as well as naproxen and slow-release diclofenac, but with fewer GI side effects. No data on its use in dysmenorrhea is available.
Chemically related but less similar to the enolic acids is refecoxib. Like celecoxib, it has been studied in the treatment of osteoarthritis, where it was comparable in efficacy to ibuprofen and diclofenac, but with side-effect rates similar to placebo therapy. In studies of women with primary dysmenorrhea, rofecoxib has proved to be statistically superior to placebo but indistinguishable from naproxen (Figure 1).22 Peak blood levels are achieved in 2 to 3 hours (delayed by 1 to 2 hours by a fatty meal), but a steady state is not achieved until day 4 of continuous therapy. When the drug is used to treat women who have a rapid onset of symptoms, the significance of this delay is unclear but is a potential drawback. In addition, the dosage of rofecoxib for the treatment of pain or dysmenorrhea is generally much larger than that required for the treatment of arthritis. Thus, the risk of side effects may be increased.
A third selective COX-2 inhibitor, meloxicam, was introduced into the US market solely for the treatment of osteoarthritis. This drug is marketed on the basis of its more favorable side-effect profile rather than its selectivity in blocking the COX-2 isoform.
Carboxylic acids. The first of the carboxylic acid subgroups are the salicylic acids. The use of salicylates derived from the bark or leaves of the Salic alba or S. fragilis dates back to Biblical times. Salicylates were formally introduced into medical use in 1763 by the Reverend Edmund Stone.23
The relatively low potency of aspirin for reducing prostaglandin synthesis in the uterus limits its clinical utility in preventing moderate or severe dysmenorrhea. In contrast, diflunisal has a long half-life, making twice-daily dosing possible. However, its slow onset of action limits use for most patients with menstrual pain.
The indoleacetic acid subgroup offers increased potency against dysmenorrhea. Most of these drugs have a direct effect rather than requiring metabolism into an active form.
Indomethacin was one of the first NSAIDs widely used to treat dysmenorrhea, but a moderate incidence of side effects limits its use—and that of most of the other drugs in this family.
No NSAID completely eliminates gastric cyclooxygenase activity.
The most commonly used drugs for dysmenorrhea come from the 2 remaining carboxylate groups: arylalkanoic acids (propionic acid derivatives) and anthranilic acids (fenamates). Of the propionic acid derivatives currently available, ibuprofen and naproxen are commonly used for dysmenorrhea. Other drugs of this class have been used for pain relief or arthritis therapy, but are not currently FDA-approved for dysmenorrhea. The subset of drugs that are phenylpropionic acid derivatives are associated with a higher incidence of GI side effects and skin reactions.
Mefenamic acid was the first alternative synthesized in an effort to reduce the gastric irritation caused by acetylsalicylic acid.24 Mefenamic acid and meclofenamate sodium—both fenamates—are potent prostaglandin synthetase inhibitors. In addition, they have been shown to antagonize already formed prostaglandins, providing a dual mode of action.25-28 This ability to block the action of existing prostaglandins produces a more rapid onset of uterine relaxation. Some of the first studies of NSAIDs in dysmenorrhea used these agents.29,30 In fact, they are some of the most thoroughly investigated drugs for the treatment of dysmenorrhea, shown to be consistently effective in reducing its subjective discomfort.31-35
In the U.S., mefenamic acid was one of the first drugs approved for dysmenorrhea, and clinical studies have shown meclofenamate to be very effective in improving symptoms and altering the underlying pathophysiology.36,37 The dual mechanism of action likely gives these agents an edge in efficacy, although no clinical trials have been initiated to evaluate this presumed advantage. Intrauterine pressure changes have been documented as soon as 15 minutes after administration of the medications, suggesting a very rapid onset of action (obviating the need for preloading). In addition, in vitro studies have demonstrated meclofenamate’s ability to inhibit the activity of 5-lipooxygenase, unlike members of the propionic acid group, which have little or no inhibitory ability.38,39 The clinical significance of inhibiting the production of the extremely potent leukotrienes has yet to be fully explored.
TABLE 1
Families of NSAIDs
| Family | Example |
|---|---|
| ENOLIC ACIDS | |
| PYRAZOLONES | |
| Oxyphenbutazone | Azolid** |
| Phenylbutazone | Butazolidin** |
| Nabumetone | Relafen |
| Celecoxib | Celebrex |
| Rofecoxib* | Vioxx |
| OXICAMS | |
| Piroxicam | Feldene, Piroxicam |
| Meloxicam | Mobic |
| CARBOXYLIC ACIDS | |
| SALICYLIC ACIDS | |
| Acetylsalicylic acid | Aspirin (various) |
| Diflunisal | Dolobid |
| Salicylate | Disalcid, Trilisate |
| INDOLEACETIC ACIDS | |
| Diclofenac potassium | Cataflam |
| Diclofenac sodium | Voltaren, Arthrotec (combined with misoprostol) |
| Etodolac | Lodine |
| Indomethacin | Indocin |
| Ketorolac tromethamine | Acular, Toradol |
| Sulindac | Clinoril, Sulindac |
| Tolmetin | Tolectin, Tolmetin |
| PROPIONIC ACIDS | |
| Fenoprofen calcium | Fenoprofen |
| Flurbiprofen | Flurbiprofen |
| Ibuprofen* | Motrin |
| Ketoprofen | Orudis, Oruvail, Ketoprofen |
| Naproxen sodium* | Aleve, Anaprox, Naprelan |
| Naproxen* | Naprosyn |
| FENAMATES | |
| Meclofenamate sodium* | Meclofenamate |
| Mefenamic acid | Ponstel |
| *FDA-approved for primary dysmenorrhea | |
| **No longer available | |
| Adapted from: Smith RP.Gynecology in Primary Care. Baltimore, Md: Williams and Wilkins; 1996:399. | |
FIGURE 1 Comparison of rofecoxib, naproxen, and placebo in treatment of primary dysmenorrhea
Reprinted with permission from The American College of Obstetricians and Gynecologists. Morrison BW, Daniels SE, Kotey P, Cantu N, Seidenberg B. Rofecoxib, a specific cyclooxygenase-2 inhibitor, in primary dysmenorrhea: a randomized-controlled trial. Obstet Gynecol. 1999;94:504-508.
COX-2 inhibitors
Although the short-term efficacy and safety of new selective COX-2 inhibitors appear to be good, several concerns remain. Despite a decrease in the incidence of GI side effects with these agents, their use by patients with active GI ulceration, infection with Helicobacter pylori, or inflammatory bowel disease has not been adequately studied. Indeed, recent studies suggest that, at therapeutic concentrations, no NSAID—not even the selective COX-2 inhibitors—completely eliminates gastric cyclooxygenase activity.40
In addition, questions have been raised about the speed and efficacy of the selective COX-2 inhibitors for the management of acute pain, compared with conventional NSAIDs.41 When used for postoperative analgesia, these agents theoretically could retard wound healing because of the role that COX-2 plays in healing and neovascularization. As more data emerge about the physiologic functions of COX-1 and -2 (Table 2), there are growing concerns that COX-2 is not restricted to inflammation and pathology, suggesting the possibility of unanticipated adverse effects associated with its use. For example, there is evidence of constitutive expression of COX-2 in the kidney and brain, and essential physiological functions in ovulation and implantation.42 In the treatment of dysmenorrhea, the increased cost of these drugs over the more commonly used agents, along with the lack of clinical efficacy studies, suggests a second-line role.
Like most NSAIDs, COX-2 inhibitors fall into pregnancy category C. Because of the risk of premature closure of the ductus arteriosus, they should not be used in the third trimester of pregnancy. In addition, drug levels in human milk mimic those in serum, so the decision to use these agents in nursing mothers must be made carefully.
Studies over the past 10 years have revealed that the cyclooxygenase enzyme (COX) is found in 2 isomeric forms, known as COX-1 and COX-2.1 Initial investigations suggested that COX-1 was present in most tissues and responsible for the homeostatic production of arachidonic acid metabolites. The COX-2 enzyme was thought to be induced in response to inflammatory stimuli such as cytokines and bacterial lipopolysaccharides, rather than expressed under normal cellular function. Thus, the COX-2 form was thought to be responsible for the large amounts of prostaglandins associated with inflammation. It was further hypothesized that COX-2 used intracellular arachidonic acid as a substrate almost exclusively, while COX-1 could use phospholipase A2 as an extracellular substrate under some conditions.2
Recent data suggest that COX-1 and -2 are more complex than originally assumed. There is increasing evidence that COX-2 is constitutively expressed in the brain, kidneys, and pancreatic islet cells and plays a role in intestinal tolerance to dietary antigens, ulcer healing, ovulation, and implantation. Transgenic mice with deletions of the COX-2 enzyme maintain the inflammatory response when only COX-1 is present.3
In the reproductive tract, COX-2 expression increases substantially in midcycle because of the surge in luteinizing hormone (LH). COX-2 and the resulting prostaglandins (released from granulosa cells) are thought to play an important role in rupture of the follicle.4,5 With fertilization, COX-2 expression increases in the endometrium surrounding the implantation site.6 The resultant prostaglandins are important for successful implantation and angiogenesis. Interestingly, transgenic mice that lack the COX-2 enzyme are infertile, while those missing only the COX-1 enzyme have normal reproduction.7,8
Studies also have demonstrated a rapid increase in COX-2 expression in the placenta and amnion immediately before and during labor.9 It is well established that prostaglandins have a pivotal role in myometrial contraction10 and inhibition of COX-2 has been shown to delay labor.11
The COX-1 and -2 enzymes share approximately 60% homology.12 Both have a long, narrow channel that is the active site of arachidonic acid folding and oxygenation. The critical difference between the 2 isoenzymes occurs at position 523, where the COX-1 enzyme has an isoleucine while COX-2 has a valine.13 This substitution causes the channel in the COX-2 form to be 17% wider; it also provides a side pocket that increases the volume of the active site by 8%. Drugs that are designed to block the COX-2 enzyme take advantage of this difference in channel size—they are too large to fit the normal channel. Nonselective NSAIDs are small enough to block the channel in both isoenzymes while, at least in theory, the selective agents can block only the larger channel.14 Most COX-2 inhibitors also exhibit some action against the COX-1 isoform.—Roger P. Smith, MD, and Jeffrey Ellis, MD
REFERENCES
1. Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthase (cyclooxygenase)-1 and -2. J Biol Chem. 1996;271:33157.-
2. Herschman HR. Prostaglandin synthase 2. Biochem Biophys Acta. 1996;1299:125-140.
3. Wallace JL, Bak A, McKnight W, Asfaha S, Sharkey KA, MacNaughton WK. Cyclooxygenase 1 contributes to inflammatory responses in rats and mice: implications for gastrointestinal toxicity. Gastroenterology. 1998;115:101-109.
4. Richards JS, Fitzpatrick SL, Clemens JW, Morris JK, Alliston T, Sirois J. Ovarian cell differentiation: a cascade of multiple hormones, cellular signals, and regulated genes. Recent Prog Horm Res. 1995;50:223-254.
5. Akil M, Amos RS, Stewart P. Infertility may sometimes be associated with NSAID consumption. Br J Rheumatol. 1996;35:76-78.
6. Chakraborty I, Das SK, Wang J, Dey SK. Developmental expression of the cyclo-oxygenase-1 and cyclooxygenase-2 genes in the peri-implantation mouse uterus and their differential regulation by the blastocyst and ovarian steroids. J Mol Endocrinol. 1996;16:107-122.
7. Lim H, Paria BC, Das SK, Dinchuk JE, Langenbach R, Trzaskos JM, et al. Multiple female reproductive failures in cyclooxygenase 2-deficient mice. Cell. 1997;91:197-208.
8. Langenbach R, Loftin C, Lee C, Tiano H. Cyclooxygenase knockout mice: models for elucidating isoform-specific functions. Biochem Pharmacol. 1999;58:1237-1246.
9. Gibb W, Sun M. Localization of prostaglandin H synthase type 2 protein and mRNA in term human fetal membranes and decidua. J Endocrinol. 1996;150:497-503.
10. O’Brien WF. The role of prostaglandins in labor and delivery. Clin Perinatol. 1995;22:973-984.
11. Mitchell MD, Romero RJ, Edwin SS, Trautman MS. Prostaglandins and parturition. Reprod Fertil Dev. 1995;7:623-632.
12. Kurumbail RG, Stevens AM, Gierse JK, et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384:644-4.(Published correction appears in Nature. 1997;385:555.)
13. Gierse JK, McDonald JJ, Hauser SD, Rangwala SH, Koboldt CM, Seibert K. A single amino acid difference between cyclooxygenase-1 (COX-1) and -2 (COX-2) reverses the selectivity of COX-2 specific inhibitors. J Biol Chem. 1996;271:15810.-
14. Lanzo CA, Beechem JM, Talley J, Marnett LJ. Investigation of the binding of isoform-selective inhibitors to prostaglandin endoperoxide synthases using fluorescence spectroscopy. Biochemistry. 1998;37:217-226.
TABLE 2
Comparison of COX-1 and COX-2 in human tissues
| Characteristic | COX-1 | COX-2 |
|---|---|---|
| Tonic expression | High | Low |
| Inducibility | Low | High |
| Predominant location | All (stomach, kidney, platelets, vasculature) | Sites of inflammation (neoplastic tissue, small intestine, ovary, uterus, brain) |
| Function | Homeostasis | Inflammation and repair, neoplasia, modulation of immune response |
Conclusion
It is apparent from the multitude of published studies and the breadth of clinical experience with NSAID therapy for primary dysmenorrhea that most of these drugs provide relief for the majority of dysmenorrheic women. The success rates reported from both subjective and objective studies of the use of mefenamic acid in dysmenorrhea support its potential superiority. The physiology of primary dysmenorrhea would suggest that the ability of the fenamates to block the action of preformed prostaglandins—as well as the 5-lipooxygenase pathway—gives them a therapeutic edge. Interestingly, one investigation of the level of selectivity of multiple NSAIDs found that mefenamic acid had a selectivity (preferential COX-2 inhibition) comparable to some of the newest selective COX-2 inhibitors.43 This may account for the low incidence of GI and other side effects reported with mefenamic acid.
The history of NSAIDs has involved the sequential introduction of agents designed to reduce side effects or increase efficacy, often through the modification of existing compounds. Many of these products have failed to meet expectations and have been withdrawn from the market. Notable recent examples include benoxaprofen, suprofen, oxyphenbutazone, and zomepirac.
Thus, it is ironic that mefenamic acid, the first agent introduced to overcome the drawbacks of aspirin, has proved to be so robust in efficacy and tolerability. In addition, it is one of the only agents objectively shown to reduce menstrual blood loss in women with menorrhagia,44-47 and to ease menstrual migraine48 and the physical and emotional symptoms of premenstrual syndrome.49-51 In fact, the combination of dysmenorrhea, premenstrual syndrome, and menorrhagia experienced by many women would seem to support the use of mefenamic acid over other agents.
Unfortunately, no direct comparisons of various drugs have definitively resolved the question of which agent is “best” for the treatment of dysmenorrhea. Until there is further refinement of our knowledge of uterine prostaglandins, menstrual function, and the physiology of dysmenorrhea, it would seem wise to rely on agents that have been studied most extensively.
Dr. Smith reports no financial relationship with any companies whose products are mentioned in this article. Dr. Ellis reports that he is on the speakers’ bureau for First Horizon Pharmaceutical Company.
1. Mayer E. The intranasal treatment of dysmenorrhea. With a report of ninety-three cases. JAMA. 1914;62:6.-
2. Miller NF, et al. Dysmenorrhea. Am J Obstet Gynecol. 1953;65:505.-
3. Cotte G. La sympathectomie hypogastrique: A-t-elle sa place dans la therapeutique gynecologique? Presse Med. 1925;33:98.-
4. Black WT, Jr. Use of presacral sympathectomy in the treatment of dysmenorrhea: a second look after twenty-five years. Am J Obstet Gynecol. 1964;89:16.-
5. Hayden GE. Relief of primary dysmenorrhea. Obstet Gynecol. 1960;16:730.-
6. Schuck F. Pain and pain relief in essential dysmenorrhea. Am J Obstet Gynecol. 1951;62:559.-
7. Voulgaris DM. Dysmenorrhea. Treatment with isoxsuprine. Obstet Gynecol. 1960;15:220.-
8. Champlin WD, Corbit JP. Chlorpromazine and chlorpromazine combinations in the treatment of dysmenorrhea. Am J Obstet Gynecol. 1957;74:419.-
9. Filler W. The treatment of dysmenorrhea. With special reference to the primary type. Med Clin North Am. 1951;35:861.-
10. Menaker JS, Powers KD. Management of primary dysmenorrhea. Obstet Gynecol. 1962;20:66.-
11. Molla LA, Donald JF. A comparative study of ibuprofen and paracetamol in primary dysmenorrhea. J Int Med Res. 1974;2:395.-
12. Roth-Brandel U, Bygdman M, Wiqvist N. Effect of intravenous administration of prostaglandin E1 and F2 on the contractility of the nonpregnant human uterus in vivo. Acta Obstet Gynecol Scand Suppl. 1970;49(5):19.-
13. Filler W. The treatment of dysmenorrhea. With special reference to the primary type. Med Clin North Am. 1951;35:861.-
14. Menaker JS, Powers KD. Management of primary dysmenorrhea. Obstet Gynecol. 1962;20:66.-
15. Jacobson J, Cavalli-Bjorkman K, Lundstrom V, Nilsson B, Norbeck M. Prostaglandin synthetase inhibitors and dysmenorrhea. Acta Obstet Gynecol Scand. 1979;87(suppl):73.-
16. Dingfelder JR. Primary dysmenorrhea treatment with prostaglandin inhibitors: a review. Am J Obstet Gynecol. 1981;140:874.-
17. Owen PR. Prostaglandin synthetase inhibitors in the treatment of primary dysmenorrhea. Am J Obstet Gynecol. 1984;148:96.-
18. Dawood MY. Nonsteroidal anti-inflammatory drugs and reproduction. Am J Obstet Gynecol. 1993;169:1255-1265.
19. Zhang WY, Po ALW. Efficacy of minor analgesics in primary dysmenorrhoea: a systematic review. Br J Obstet Gynaecol. 1998;105:780-789.
20. Lichtenstein DR, Syngal S, Wolfe MM. Nonsteroidal anti-inflammatory drugs and the gastrointestinal tract: the double-edged sword. Arthritis Rheumk. 1995;38:5-18.
21. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for the aspirin-like drugs. Nature. 1971;231:232-235.
22. Morrison BW, Daniels SE, Kotey P, Cantu N, Seidenberg B. Rofecoxib, a specific cyclooxygenase-2 inhibitor, in primary dysmenorrhea: a randomized controlled trial. Obstet Gynecol. 1999;94:504-508.
23. Stone E. An account of the success of the bark of the willow in the cure of agues. Philosophical Transactions of the Royal Society. 1763;53:195-200.
24. Winder CV, Wax J, Scotti L, Scherrer RA, Jones EM, Short FW. Antiinflammatory, antipyretic, and antinociceptive properties of N-(2,3-xylyl) anthranilic acid (mefenamic acid). J Pharm Exp Ther. 1962;138:405-413.
25. Collier HOJ, Sweatmon WJF. Antagonism by fenamates of prostaglandin F 2α and of slow-reacting substance on human bronchial muscle. Nature. 1968;219:864.-
26. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature New Biol. 1971;231:232.-
27. Tolman EL, Partridge R. Multiple sites of interaction between prostaglandins and nonsteroidal anti-inflammatory drugs. Prostaglandins. 1975;9:349.-
28. Ferreira SH, Vane JR. New aspects of the mode of action of nonsteroidal anti-inflammatory drugs. Ann Rev Pharmacol. 1973;14:57.-
29. Schwartz A, Zor U, Lindner HR, Naor S. Primary dysmenorrhea. Alleviation by an inhibitor of prostaglandin synthesis and action. Obstet Gynecol. 1974;44:709.-
30. Kauppila A, Ylikorkala O. Indomethacin and tolfenamic acid in primary dysmenorrhea. Eur J Obstet Gynecol Reprod Biol. 1977;7:59.-
31. Anderson ABM, Haynes PJ, Fraser IS, Turnbull AC. Trial of prostaglandin-synthetase inhibitors in primary dysmenorrhea. Lancet. 1978;1:345.-
32. Kapadia L, Elder MG. Flufenamic acid in treatment of primary spasmodic dysmenorrhea. Lancet. 1978;1:348.-
33. Budoff PW. Use of mefenamic acid in the treatment of primary dysmenorrhea. JAMA. 1979;241:2713.-
34. Powell JR, Smith RP. Mefenamic acid (Ponstel) for treating primary spasmodic dysmenorrhea. Curr Ther Res. 1981;29:544.-
35. Roy S. A double-blind comparison of a propionic acid derivative (ibuprofen) and a fenamate (mefenamic acid) in the treatment of dysmenorrhea. Obstet Gynecol. 1983;61:628.-
36. Smith RP, Powell JR. Simultaneous objective and subjective evaluation of meclofenamate sodium in the treatment of primary dysmenorrhea. Am J Obstet Gynecol. 1987;157:611.-
37. Smith RP. The dynamics of nonsteroidal anti-inflammatory therapy for primary dysmenorrhea. Obstet Gynecol. 1987;70:785.-
38. Boctor AM, Eickholt M, Pugsley TA. Meclofenamate sodium is an inhibitor of both the 5-lipooxygenase and cyclooxygenase pathways of the arachidonic acid cascade in vitro. Prostaglandins, Leukotrienes & Med. 1986;23:229.-
39. Myers RF, Siegel MI. Differential effects of anti-inflammatory drugs on lipooxygenase and cyclooxygenase activities of neutrophils from a reverse passive Arthus reaction. Biochemical Biophysical Res Comm. 1983;112:586.-
40. Feldman M, McMahon AT. Do cyclooxygenase-2 inhibitors provide benefits similar to those of traditional nonsteroidal anti-inflammatory drugs, with less gastrointestinal toxicity? Ann Intern Med. 2000;132:134-143.
41. Bannwarth B. Are COX-2 inhibitors as effective as conventional NSAIDs in acute pain states? [letter] Arch Intern Med. 2001;161:127-128.
42. Lipsky PE, Brooks P, Crofford LJ, DuBois R, Graham D, Simon LS, van de Putte LBA, Abramson SB, et al. Unresolved issues in the role of cyclooxygenase-2 in normal physiologic processes and disease. Arch Intern Med. 2000;160(7):913-920.
43. Cryer B, Feldman M. Cyclooxygenase-1 and cyclooxygenase-2 selectivity of widely used nonsteroidal anti-inflammatory drugs. Am J Med. 1998;104(5):413-421.
44. Fraser IS, Pearse C, Shearman RP, Elliott PM, McIlveen J, Markham R. Efficacy of mefenamic acid in patients with a complaint of menorrhagia. Obstet Gynecol. 1981;58:543-551.
45. Fraser IS, McCarron G, Markham R, Robinson M, Smyth E. Long-term treatment of menorrhagia with mefenamic acid. Obstet Gynecol. 1983;61:109-112.
46. Vargyas JM, Campeau JD, Mishell DR Jr. Treatment of menorrhagia with meclofenamate sodium. Am J Obstet Gynecol. 1987;157:944-950.
47. Van Eijkeren MA, Christiaens GCML, Geuze HJ, Haspels AA, Sixma JJ. Effects of mefenamic acid on menstrual hemostasis in essential menorrhagia. Am J Obstet Gynecol. 1992;166:1419-1428.
48. Al-Waili NS. Treatment of menstrual migraine with prostaglandin synthesis inhibitor mefenamic acid: double-blind study with placebo. Eur J Med Res. 2000;5:176-182.
49. Wood C, Jakubowicz D. The treatment of premenstrual symptoms with mefenamic acid. Br J Obstet Gynaecol. 1980;87:627-630.
50. Jakubowicz DL, Godard E, Dewhurst J. The treatment of premenstrual tension with mefenamic acid: analysis of prostaglandin concentrations. Br J Obstet Gynaecol. 1984;91:78-84.
51. Mira M, McNeil D, Fraser IS, Vizzard J, Abraham S. Mefenamic acid in the treatment of premenstrual syndrome. Obstet Gynecol. 1986;68:395-398.
1. Mayer E. The intranasal treatment of dysmenorrhea. With a report of ninety-three cases. JAMA. 1914;62:6.-
2. Miller NF, et al. Dysmenorrhea. Am J Obstet Gynecol. 1953;65:505.-
3. Cotte G. La sympathectomie hypogastrique: A-t-elle sa place dans la therapeutique gynecologique? Presse Med. 1925;33:98.-
4. Black WT, Jr. Use of presacral sympathectomy in the treatment of dysmenorrhea: a second look after twenty-five years. Am J Obstet Gynecol. 1964;89:16.-
5. Hayden GE. Relief of primary dysmenorrhea. Obstet Gynecol. 1960;16:730.-
6. Schuck F. Pain and pain relief in essential dysmenorrhea. Am J Obstet Gynecol. 1951;62:559.-
7. Voulgaris DM. Dysmenorrhea. Treatment with isoxsuprine. Obstet Gynecol. 1960;15:220.-
8. Champlin WD, Corbit JP. Chlorpromazine and chlorpromazine combinations in the treatment of dysmenorrhea. Am J Obstet Gynecol. 1957;74:419.-
9. Filler W. The treatment of dysmenorrhea. With special reference to the primary type. Med Clin North Am. 1951;35:861.-
10. Menaker JS, Powers KD. Management of primary dysmenorrhea. Obstet Gynecol. 1962;20:66.-
11. Molla LA, Donald JF. A comparative study of ibuprofen and paracetamol in primary dysmenorrhea. J Int Med Res. 1974;2:395.-
12. Roth-Brandel U, Bygdman M, Wiqvist N. Effect of intravenous administration of prostaglandin E1 and F2 on the contractility of the nonpregnant human uterus in vivo. Acta Obstet Gynecol Scand Suppl. 1970;49(5):19.-
13. Filler W. The treatment of dysmenorrhea. With special reference to the primary type. Med Clin North Am. 1951;35:861.-
14. Menaker JS, Powers KD. Management of primary dysmenorrhea. Obstet Gynecol. 1962;20:66.-
15. Jacobson J, Cavalli-Bjorkman K, Lundstrom V, Nilsson B, Norbeck M. Prostaglandin synthetase inhibitors and dysmenorrhea. Acta Obstet Gynecol Scand. 1979;87(suppl):73.-
16. Dingfelder JR. Primary dysmenorrhea treatment with prostaglandin inhibitors: a review. Am J Obstet Gynecol. 1981;140:874.-
17. Owen PR. Prostaglandin synthetase inhibitors in the treatment of primary dysmenorrhea. Am J Obstet Gynecol. 1984;148:96.-
18. Dawood MY. Nonsteroidal anti-inflammatory drugs and reproduction. Am J Obstet Gynecol. 1993;169:1255-1265.
19. Zhang WY, Po ALW. Efficacy of minor analgesics in primary dysmenorrhoea: a systematic review. Br J Obstet Gynaecol. 1998;105:780-789.
20. Lichtenstein DR, Syngal S, Wolfe MM. Nonsteroidal anti-inflammatory drugs and the gastrointestinal tract: the double-edged sword. Arthritis Rheumk. 1995;38:5-18.
21. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for the aspirin-like drugs. Nature. 1971;231:232-235.
22. Morrison BW, Daniels SE, Kotey P, Cantu N, Seidenberg B. Rofecoxib, a specific cyclooxygenase-2 inhibitor, in primary dysmenorrhea: a randomized controlled trial. Obstet Gynecol. 1999;94:504-508.
23. Stone E. An account of the success of the bark of the willow in the cure of agues. Philosophical Transactions of the Royal Society. 1763;53:195-200.
24. Winder CV, Wax J, Scotti L, Scherrer RA, Jones EM, Short FW. Antiinflammatory, antipyretic, and antinociceptive properties of N-(2,3-xylyl) anthranilic acid (mefenamic acid). J Pharm Exp Ther. 1962;138:405-413.
25. Collier HOJ, Sweatmon WJF. Antagonism by fenamates of prostaglandin F 2α and of slow-reacting substance on human bronchial muscle. Nature. 1968;219:864.-
26. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature New Biol. 1971;231:232.-
27. Tolman EL, Partridge R. Multiple sites of interaction between prostaglandins and nonsteroidal anti-inflammatory drugs. Prostaglandins. 1975;9:349.-
28. Ferreira SH, Vane JR. New aspects of the mode of action of nonsteroidal anti-inflammatory drugs. Ann Rev Pharmacol. 1973;14:57.-
29. Schwartz A, Zor U, Lindner HR, Naor S. Primary dysmenorrhea. Alleviation by an inhibitor of prostaglandin synthesis and action. Obstet Gynecol. 1974;44:709.-
30. Kauppila A, Ylikorkala O. Indomethacin and tolfenamic acid in primary dysmenorrhea. Eur J Obstet Gynecol Reprod Biol. 1977;7:59.-
31. Anderson ABM, Haynes PJ, Fraser IS, Turnbull AC. Trial of prostaglandin-synthetase inhibitors in primary dysmenorrhea. Lancet. 1978;1:345.-
32. Kapadia L, Elder MG. Flufenamic acid in treatment of primary spasmodic dysmenorrhea. Lancet. 1978;1:348.-
33. Budoff PW. Use of mefenamic acid in the treatment of primary dysmenorrhea. JAMA. 1979;241:2713.-
34. Powell JR, Smith RP. Mefenamic acid (Ponstel) for treating primary spasmodic dysmenorrhea. Curr Ther Res. 1981;29:544.-
35. Roy S. A double-blind comparison of a propionic acid derivative (ibuprofen) and a fenamate (mefenamic acid) in the treatment of dysmenorrhea. Obstet Gynecol. 1983;61:628.-
36. Smith RP, Powell JR. Simultaneous objective and subjective evaluation of meclofenamate sodium in the treatment of primary dysmenorrhea. Am J Obstet Gynecol. 1987;157:611.-
37. Smith RP. The dynamics of nonsteroidal anti-inflammatory therapy for primary dysmenorrhea. Obstet Gynecol. 1987;70:785.-
38. Boctor AM, Eickholt M, Pugsley TA. Meclofenamate sodium is an inhibitor of both the 5-lipooxygenase and cyclooxygenase pathways of the arachidonic acid cascade in vitro. Prostaglandins, Leukotrienes & Med. 1986;23:229.-
39. Myers RF, Siegel MI. Differential effects of anti-inflammatory drugs on lipooxygenase and cyclooxygenase activities of neutrophils from a reverse passive Arthus reaction. Biochemical Biophysical Res Comm. 1983;112:586.-
40. Feldman M, McMahon AT. Do cyclooxygenase-2 inhibitors provide benefits similar to those of traditional nonsteroidal anti-inflammatory drugs, with less gastrointestinal toxicity? Ann Intern Med. 2000;132:134-143.
41. Bannwarth B. Are COX-2 inhibitors as effective as conventional NSAIDs in acute pain states? [letter] Arch Intern Med. 2001;161:127-128.
42. Lipsky PE, Brooks P, Crofford LJ, DuBois R, Graham D, Simon LS, van de Putte LBA, Abramson SB, et al. Unresolved issues in the role of cyclooxygenase-2 in normal physiologic processes and disease. Arch Intern Med. 2000;160(7):913-920.
43. Cryer B, Feldman M. Cyclooxygenase-1 and cyclooxygenase-2 selectivity of widely used nonsteroidal anti-inflammatory drugs. Am J Med. 1998;104(5):413-421.
44. Fraser IS, Pearse C, Shearman RP, Elliott PM, McIlveen J, Markham R. Efficacy of mefenamic acid in patients with a complaint of menorrhagia. Obstet Gynecol. 1981;58:543-551.
45. Fraser IS, McCarron G, Markham R, Robinson M, Smyth E. Long-term treatment of menorrhagia with mefenamic acid. Obstet Gynecol. 1983;61:109-112.
46. Vargyas JM, Campeau JD, Mishell DR Jr. Treatment of menorrhagia with meclofenamate sodium. Am J Obstet Gynecol. 1987;157:944-950.
47. Van Eijkeren MA, Christiaens GCML, Geuze HJ, Haspels AA, Sixma JJ. Effects of mefenamic acid on menstrual hemostasis in essential menorrhagia. Am J Obstet Gynecol. 1992;166:1419-1428.
48. Al-Waili NS. Treatment of menstrual migraine with prostaglandin synthesis inhibitor mefenamic acid: double-blind study with placebo. Eur J Med Res. 2000;5:176-182.
49. Wood C, Jakubowicz D. The treatment of premenstrual symptoms with mefenamic acid. Br J Obstet Gynaecol. 1980;87:627-630.
50. Jakubowicz DL, Godard E, Dewhurst J. The treatment of premenstrual tension with mefenamic acid: analysis of prostaglandin concentrations. Br J Obstet Gynaecol. 1984;91:78-84.
51. Mira M, McNeil D, Fraser IS, Vizzard J, Abraham S. Mefenamic acid in the treatment of premenstrual syndrome. Obstet Gynecol. 1986;68:395-398.