User login
Cellular Versus Acellular Grafts for Diabetic Foot Ulcers: Altering the Protocol to Improve Recruitment to a Comparative Efficacy Trial
Chronic diabetic foot ulcers (DFUs) remain a serious therapeutic challenge worldwide.1-2 Patients with DFUs are at higher risk for infections, which may lead to limb loss.1-5 In fact, 1 in 6 patients with DFUs will undergo an amputation.6 The long-term consequences of DFUs are numerous and can severely affect patients’ quality of life, including loss of productivity.7 The current standard of care for DFUs consists of debridement of the necrotic tissue, application of a moist dressing, and use of an off-loading device that protects the wound from pressure or trauma related to ambulation and other acts of daily living.4-6,8 Unfortunately, studies have shown that the best standard of care (SOC) only heals 30% of DFUs after 20 weeks of therapy.9 With the estimated cost per episode of care approaching $40,000, DFUs remain a costly and important problem.10
The altered extracellular matrix (ECM) in DFUs has been a target for the development of new therapeutic devices that provide a new matrix that is either devoid of cells or can be enriched with fibroblasts.8,11 These bioengineered skin substitutes stimulate the growth of new vessels and generate cytokines essential for tissue repair. In 2013, Lev-Tov et al12 published this study protocol (Dermagraft Oasis Longitudinal Comparative Efficacy [DOLCE] trial) to compare the effectiveness of 2 advanced wound care devices, specifically to evaluate the clinical efficacy of a cellular matrix versus an acellular matrix, which we have amended. The cellular matrix used in the study is a dermal substitute composed of viable newborn foreskin fibroblasts seeded onto a bioabsorbable polyglactin mesh on which fibroblasts generate an ECM.13,14 It is supplied frozen and requires specific thawing steps prior to application. The recommended regimen for treatment of DFUs for this cellular matrix is 8 weekly applications.13,14 In 2016, the cost of the product was reported as $1411 per 5.0×7.5-cm sheet.15 The acellular matrix product used in the study is a bioabsorbable ECM that is derived from porcine small intestinal submucosa.16,17 It is stored at room temperature and has a long shelf life, with a current price of $112.6 for a 3.0×3.5-cm single-layer fenestrated sheet ($1126.60 per box of 10 sheets). The industry-supported randomized controlled trials for each of these devices have reported a 20% added benefit in the rate of wound closure at week 12 compared to SOC.14,17
This article provides the interim report of the trial (registered at www.clinicaltrials.gov with the identifier NCT01450943) described in the published protocol and initiated in 2011,12 focusing on elements that required modification during the trial’s duration.
Methods
Study Protocol
The clinical trial was approved by the Veterans’ Affairs Institutional Research and Development Committee and their institutional review board. This study was funded by the Veteran’s Administration Merit Award (#10554640), which was awarded to 2 of the investigators (S.E.D. and R.R.I.). Eligible veterans were recruited from all 7 sites of the VA Northern California Healthcare System. This trial is a randomized, single-blinded, 3-armed, controlled clinical equivalence trial comparing the effectiveness of an SOC treatment, cellular ECM, and acellular ECM.
Study Products
The SOC dressing applied in the clinical trial included a sterile antimicrobial gel, a nonadherent dressing, and gauze.12 The SOC dressing also was used as a secondary dressing for the active treatment arms. Bacitracin antibiotic ointment was used as an alternative for patients with allergy to iodine.12
Randomization
The inclusion and exclusion criteria were previously outlined.12 After a 2-week screening phase to exclude rapid healers, patients were randomized into a treatment arm and entered the active phase for 12 weeks.
Primary and Secondary Outcomes
The primary outcome was complete wound closure by week 12.12 Complete healing was defined as full reepithelialization with no drainage or dressing requirement. The secondary outcomes included healing at 28 weeks, rate of healing, ulcer recurrence at week 20, association of wound healing with ulcer characteristics or patients’ characteristic, incidence of adverse events, and cost-effectiveness of each treatment compared to the SOC arm.12
Statistical Analysis
To detect a 25% difference in the incidence of ulcer closure between the 2 study groups and the SOC group, the estimation of the sample size was based on 80% power with a significance level of 0.05. Specifically, it was expected that 50% of the cellular and acellular matrix groups and 25% of the SOC group would reach complete wound closure. The protocol indicated that 57 participants would be enrolled in each arm (total of 171 participants). Lev-Tov et al12 discussed the statistical analysis in more detail.
Results
Study Protocol Amendments
Given the number of diabetic patients in the US veteran population, we anticipated that there would be enough participants meeting the inclusion and exclusion criteria; however, because of the difficulty with recruitment, the initial study criteria were modified. The study was initially designed to incorporate DFUs with a minimum size of 1.0 cm2.12
Another limiting criterion was the percentage of total hemoglobin level for hemoglobin A1C (HbA1C). The study was originally established to include participants with an HbA1C level of 10% of total hemoglobin or below.12 Unfortunately, the majority of the potential participants had values substantially higher, and thus could not be enrolled in the trial, requiring another amendment to the study protocol in 2014, which was approved to include patients with an HbA1C level less than 12% of total hemoglobin. This change contributed considerably to the noted increase in enrollment rates in 2015, which almost doubled relative to enrollment under the original exclusion criteria (Figure).
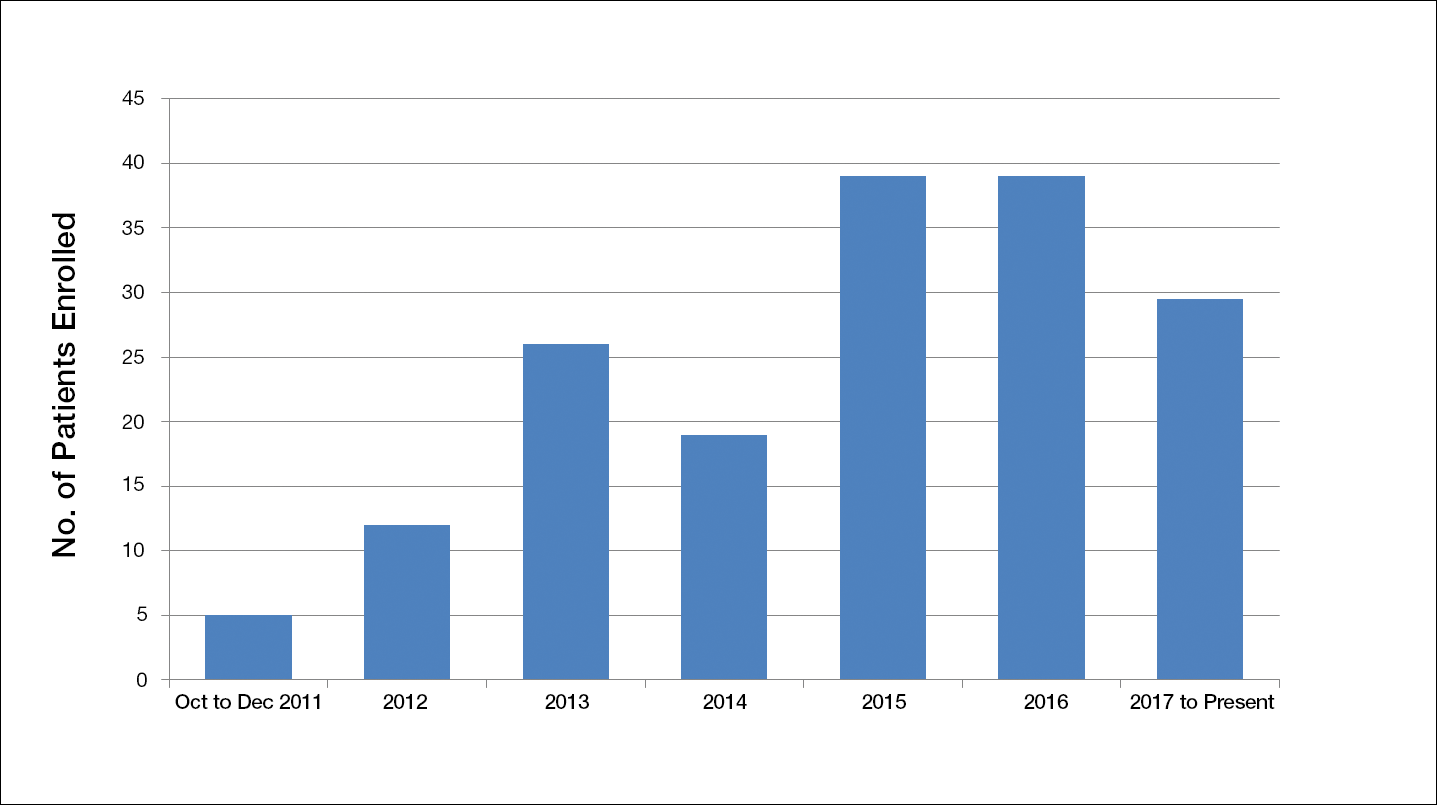
The study has screened more than 600 patients. Among them, 137 were assessed for eligibility; 71 were excluded for various reasons, including screen failure (eg, decrease in wound size by >40% during the 2-week screening phase), loss to follow-up, and adverse events. Sixty-six participants reached the primary outcome at week 12, while 55 participants completed the study (19 in the SOC group; 18 in the cellular matrix group; 18 in the acellular matrix group).
We have stopped enrolling patients from all sites and the community, as we have reached our target enrollment.
Comment
One of the challenges of clinical trials is the recruitment of an adequate number of participants within an appropriate time frame, which is explained by Lasagna’s Law,18 a well-described phenomenon whereby the investigator overestimates the number of potential participants available to meet the inclusion criteria. This so-called funnel-effect was partly encountered in our selection process. A review of the veteran population with DFUs seemed to be more than adequate to fulfill the sample size; however, some important participant-related factors also played a substantial role.
In addition, the Veterans’ Affairs network centralizes health information, making it readily available to all providers participating in their care. As a result, patients with diabetes mellitus typically are seen by a primary care physician along with an endocrinologist, a diabetic nurse, and/or a dietician. Despite the collaboration with an interdisciplinary team, the glycemic control of the participants remains an issue along with other psychosocial factors that are deterrents in patient compliance. As a result, patients with poorly controlled diabetes and an HbA1C level above 10% (and less than 12%) of total hemoglobin who were initially excluded from the study were reincluded after modifying the inclusion criteria. Some patients were interested in joining the study, but physical limitations (eg, impaired mobility) prompted their decision not to join the trial, even though they met all the inclusion criteria.
As far as research-related factors that could affect participation, it is notable that most of the patients were retired; thus, the interventions did not cause additional burden of taking time off from work or loss of productivity. Although randomization could be a deterrent in many clinical trials, the majority of patients were willing to participate without demanding to be assigned to a particular treatment group.
There are many factors that are intertwined and can lead to enrollment and/or attrition rates. It was critical for our team to make some adjustment without compromising the controlled nature of a randomized trial.
Acknowledgment
The authors wish to acknowledge Huong Le, DPM, MPH, who was the coauthor of the study protocol.
- Sen CK, Gordillo GM, Roy S, et al. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen. 2009;17:763-771.
- Gurtner GC, Werner S, Barrandon Y, et al. Wound repair and regeneration. Nature. 2008;453:314-321.
- Falanga V. Wound healing and its impairment in the diabetic foot. Lancet. 2005;366:1736-1743.
- Boulton AJ. The diabetic foot: grand overview, epidemiology and pathogenesis. Diabetes Metab Res Rev. 2008;24(suppl 1):S3-S6.
- Singh N, Armstrong DG, Lipsky BA. Preventing foot ulcers in patients with diabetes. JAMA. 2005;293:217-228.
- Vuorisalo S, Venermo M, Lepäntalo M. Treatment of diabetic foot ulcers. J Cardiovasc Surg (Torino). 2009;50:275-291.
- Meijer JW, Trip J, Jaegers SM, et al. Quality of life in patients with diabetic foot ulcers. Disabil Rehabil. 2001;23:336-340.
- Santema TB, Poyck PP, Ubbink DT. Skin grafting and tissue replacement for treating foot ulcers in people with diabetes. Cochrane Database Syst Rev. 2016;2:CD011255.
- Margolis DJ, Kantor J, Berlin JA. Healing of diabetic neuropathic foot ulcers receiving standard treatment. a meta-analysis. Diabetes Care. 1999;22:692-695.
- Cavanagh P, Attinger C, Abbas Z, et al. Cost of treating diabetic foot ulcers in five different countries. Diabetes Metab Res Rev. 2012;2(suppl 1):107-111.
- Panuncialman J, Falanga V. The science of wound bed preparation. Surg Clin North Am. 2009;89:611-626.
- Lev-Tov H, Li CS, Dahle S, et al. Cellular versus acellular matrix devices in treatment of diabetic foot ulcers: study protocol for a comparative efficacy randomized controlled trial. Trials. 2013;14:8.
- Gentzkow GD, Iwasaki SD, Hershon KS, et al. Use of dermagraft, a cultured human dermis, to treat diabetic foot ulcers. Diabetes Care. 1996;19:350-354.
- Marston WA, Hanft J, Norwood P, et al; Dermagraft Diabetic Foot Ulcer Study Group. The efficacy and safety of Dermagraft in improving the healing of chronic diabetic foot ulcers: results of a prospective randomized trial. Diabetes Care. 2003;26:1701-1705.
- 2016 Dermagraft® Medicare Product and Related Procedure Payment. http://www.dermagraft.com/wp-content/uploads/sites/1/Dermagraft_Hotsheet%202016%20Q1%20HOSPITAL_FINAL.pdf. Accessed November 23, 2017.
- Oasis® Wound Matrix. http://www.oasiswoundmatrix.com/aboutowm. Accessed November 23, 2017.
- Niezgoda JA, Van Gils CC, Frykberg RG, et al. Randomized clinical trial comparing OASIS Wound Matrix to Regranex Gel for diabetic ulcers. Adv Skin Wound Care. 2005;18(5, pt 1):258-266.
- Torgerson JS, Arlinger K, Käppi M, et al. Principles for enhanced recruitment of subjects in a large clinical trial. the XENDOS (XENical in the prevention of Diabetes in Obese Subjects) study experience. Controlled Clin Trials. 2001;22:515-525.
Chronic diabetic foot ulcers (DFUs) remain a serious therapeutic challenge worldwide.1-2 Patients with DFUs are at higher risk for infections, which may lead to limb loss.1-5 In fact, 1 in 6 patients with DFUs will undergo an amputation.6 The long-term consequences of DFUs are numerous and can severely affect patients’ quality of life, including loss of productivity.7 The current standard of care for DFUs consists of debridement of the necrotic tissue, application of a moist dressing, and use of an off-loading device that protects the wound from pressure or trauma related to ambulation and other acts of daily living.4-6,8 Unfortunately, studies have shown that the best standard of care (SOC) only heals 30% of DFUs after 20 weeks of therapy.9 With the estimated cost per episode of care approaching $40,000, DFUs remain a costly and important problem.10
The altered extracellular matrix (ECM) in DFUs has been a target for the development of new therapeutic devices that provide a new matrix that is either devoid of cells or can be enriched with fibroblasts.8,11 These bioengineered skin substitutes stimulate the growth of new vessels and generate cytokines essential for tissue repair. In 2013, Lev-Tov et al12 published this study protocol (Dermagraft Oasis Longitudinal Comparative Efficacy [DOLCE] trial) to compare the effectiveness of 2 advanced wound care devices, specifically to evaluate the clinical efficacy of a cellular matrix versus an acellular matrix, which we have amended. The cellular matrix used in the study is a dermal substitute composed of viable newborn foreskin fibroblasts seeded onto a bioabsorbable polyglactin mesh on which fibroblasts generate an ECM.13,14 It is supplied frozen and requires specific thawing steps prior to application. The recommended regimen for treatment of DFUs for this cellular matrix is 8 weekly applications.13,14 In 2016, the cost of the product was reported as $1411 per 5.0×7.5-cm sheet.15 The acellular matrix product used in the study is a bioabsorbable ECM that is derived from porcine small intestinal submucosa.16,17 It is stored at room temperature and has a long shelf life, with a current price of $112.6 for a 3.0×3.5-cm single-layer fenestrated sheet ($1126.60 per box of 10 sheets). The industry-supported randomized controlled trials for each of these devices have reported a 20% added benefit in the rate of wound closure at week 12 compared to SOC.14,17
This article provides the interim report of the trial (registered at www.clinicaltrials.gov with the identifier NCT01450943) described in the published protocol and initiated in 2011,12 focusing on elements that required modification during the trial’s duration.
Methods
Study Protocol
The clinical trial was approved by the Veterans’ Affairs Institutional Research and Development Committee and their institutional review board. This study was funded by the Veteran’s Administration Merit Award (#10554640), which was awarded to 2 of the investigators (S.E.D. and R.R.I.). Eligible veterans were recruited from all 7 sites of the VA Northern California Healthcare System. This trial is a randomized, single-blinded, 3-armed, controlled clinical equivalence trial comparing the effectiveness of an SOC treatment, cellular ECM, and acellular ECM.
Study Products
The SOC dressing applied in the clinical trial included a sterile antimicrobial gel, a nonadherent dressing, and gauze.12 The SOC dressing also was used as a secondary dressing for the active treatment arms. Bacitracin antibiotic ointment was used as an alternative for patients with allergy to iodine.12
Randomization
The inclusion and exclusion criteria were previously outlined.12 After a 2-week screening phase to exclude rapid healers, patients were randomized into a treatment arm and entered the active phase for 12 weeks.
Primary and Secondary Outcomes
The primary outcome was complete wound closure by week 12.12 Complete healing was defined as full reepithelialization with no drainage or dressing requirement. The secondary outcomes included healing at 28 weeks, rate of healing, ulcer recurrence at week 20, association of wound healing with ulcer characteristics or patients’ characteristic, incidence of adverse events, and cost-effectiveness of each treatment compared to the SOC arm.12
Statistical Analysis
To detect a 25% difference in the incidence of ulcer closure between the 2 study groups and the SOC group, the estimation of the sample size was based on 80% power with a significance level of 0.05. Specifically, it was expected that 50% of the cellular and acellular matrix groups and 25% of the SOC group would reach complete wound closure. The protocol indicated that 57 participants would be enrolled in each arm (total of 171 participants). Lev-Tov et al12 discussed the statistical analysis in more detail.
Results
Study Protocol Amendments
Given the number of diabetic patients in the US veteran population, we anticipated that there would be enough participants meeting the inclusion and exclusion criteria; however, because of the difficulty with recruitment, the initial study criteria were modified. The study was initially designed to incorporate DFUs with a minimum size of 1.0 cm2.12
Another limiting criterion was the percentage of total hemoglobin level for hemoglobin A1C (HbA1C). The study was originally established to include participants with an HbA1C level of 10% of total hemoglobin or below.12 Unfortunately, the majority of the potential participants had values substantially higher, and thus could not be enrolled in the trial, requiring another amendment to the study protocol in 2014, which was approved to include patients with an HbA1C level less than 12% of total hemoglobin. This change contributed considerably to the noted increase in enrollment rates in 2015, which almost doubled relative to enrollment under the original exclusion criteria (Figure).
The study has screened more than 600 patients. Among them, 137 were assessed for eligibility; 71 were excluded for various reasons, including screen failure (eg, decrease in wound size by >40% during the 2-week screening phase), loss to follow-up, and adverse events. Sixty-six participants reached the primary outcome at week 12, while 55 participants completed the study (19 in the SOC group; 18 in the cellular matrix group; 18 in the acellular matrix group).
We have stopped enrolling patients from all sites and the community, as we have reached our target enrollment.
Comment
One of the challenges of clinical trials is the recruitment of an adequate number of participants within an appropriate time frame, which is explained by Lasagna’s Law,18 a well-described phenomenon whereby the investigator overestimates the number of potential participants available to meet the inclusion criteria. This so-called funnel-effect was partly encountered in our selection process. A review of the veteran population with DFUs seemed to be more than adequate to fulfill the sample size; however, some important participant-related factors also played a substantial role.
In addition, the Veterans’ Affairs network centralizes health information, making it readily available to all providers participating in their care. As a result, patients with diabetes mellitus typically are seen by a primary care physician along with an endocrinologist, a diabetic nurse, and/or a dietician. Despite the collaboration with an interdisciplinary team, the glycemic control of the participants remains an issue along with other psychosocial factors that are deterrents in patient compliance. As a result, patients with poorly controlled diabetes and an HbA1C level above 10% (and less than 12%) of total hemoglobin who were initially excluded from the study were reincluded after modifying the inclusion criteria. Some patients were interested in joining the study, but physical limitations (eg, impaired mobility) prompted their decision not to join the trial, even though they met all the inclusion criteria.
As far as research-related factors that could affect participation, it is notable that most of the patients were retired; thus, the interventions did not cause additional burden of taking time off from work or loss of productivity. Although randomization could be a deterrent in many clinical trials, the majority of patients were willing to participate without demanding to be assigned to a particular treatment group.
There are many factors that are intertwined and can lead to enrollment and/or attrition rates. It was critical for our team to make some adjustment without compromising the controlled nature of a randomized trial.
Acknowledgment
The authors wish to acknowledge Huong Le, DPM, MPH, who was the coauthor of the study protocol.
Chronic diabetic foot ulcers (DFUs) remain a serious therapeutic challenge worldwide.1-2 Patients with DFUs are at higher risk for infections, which may lead to limb loss.1-5 In fact, 1 in 6 patients with DFUs will undergo an amputation.6 The long-term consequences of DFUs are numerous and can severely affect patients’ quality of life, including loss of productivity.7 The current standard of care for DFUs consists of debridement of the necrotic tissue, application of a moist dressing, and use of an off-loading device that protects the wound from pressure or trauma related to ambulation and other acts of daily living.4-6,8 Unfortunately, studies have shown that the best standard of care (SOC) only heals 30% of DFUs after 20 weeks of therapy.9 With the estimated cost per episode of care approaching $40,000, DFUs remain a costly and important problem.10
The altered extracellular matrix (ECM) in DFUs has been a target for the development of new therapeutic devices that provide a new matrix that is either devoid of cells or can be enriched with fibroblasts.8,11 These bioengineered skin substitutes stimulate the growth of new vessels and generate cytokines essential for tissue repair. In 2013, Lev-Tov et al12 published this study protocol (Dermagraft Oasis Longitudinal Comparative Efficacy [DOLCE] trial) to compare the effectiveness of 2 advanced wound care devices, specifically to evaluate the clinical efficacy of a cellular matrix versus an acellular matrix, which we have amended. The cellular matrix used in the study is a dermal substitute composed of viable newborn foreskin fibroblasts seeded onto a bioabsorbable polyglactin mesh on which fibroblasts generate an ECM.13,14 It is supplied frozen and requires specific thawing steps prior to application. The recommended regimen for treatment of DFUs for this cellular matrix is 8 weekly applications.13,14 In 2016, the cost of the product was reported as $1411 per 5.0×7.5-cm sheet.15 The acellular matrix product used in the study is a bioabsorbable ECM that is derived from porcine small intestinal submucosa.16,17 It is stored at room temperature and has a long shelf life, with a current price of $112.6 for a 3.0×3.5-cm single-layer fenestrated sheet ($1126.60 per box of 10 sheets). The industry-supported randomized controlled trials for each of these devices have reported a 20% added benefit in the rate of wound closure at week 12 compared to SOC.14,17
This article provides the interim report of the trial (registered at www.clinicaltrials.gov with the identifier NCT01450943) described in the published protocol and initiated in 2011,12 focusing on elements that required modification during the trial’s duration.
Methods
Study Protocol
The clinical trial was approved by the Veterans’ Affairs Institutional Research and Development Committee and their institutional review board. This study was funded by the Veteran’s Administration Merit Award (#10554640), which was awarded to 2 of the investigators (S.E.D. and R.R.I.). Eligible veterans were recruited from all 7 sites of the VA Northern California Healthcare System. This trial is a randomized, single-blinded, 3-armed, controlled clinical equivalence trial comparing the effectiveness of an SOC treatment, cellular ECM, and acellular ECM.
Study Products
The SOC dressing applied in the clinical trial included a sterile antimicrobial gel, a nonadherent dressing, and gauze.12 The SOC dressing also was used as a secondary dressing for the active treatment arms. Bacitracin antibiotic ointment was used as an alternative for patients with allergy to iodine.12
Randomization
The inclusion and exclusion criteria were previously outlined.12 After a 2-week screening phase to exclude rapid healers, patients were randomized into a treatment arm and entered the active phase for 12 weeks.
Primary and Secondary Outcomes
The primary outcome was complete wound closure by week 12.12 Complete healing was defined as full reepithelialization with no drainage or dressing requirement. The secondary outcomes included healing at 28 weeks, rate of healing, ulcer recurrence at week 20, association of wound healing with ulcer characteristics or patients’ characteristic, incidence of adverse events, and cost-effectiveness of each treatment compared to the SOC arm.12
Statistical Analysis
To detect a 25% difference in the incidence of ulcer closure between the 2 study groups and the SOC group, the estimation of the sample size was based on 80% power with a significance level of 0.05. Specifically, it was expected that 50% of the cellular and acellular matrix groups and 25% of the SOC group would reach complete wound closure. The protocol indicated that 57 participants would be enrolled in each arm (total of 171 participants). Lev-Tov et al12 discussed the statistical analysis in more detail.
Results
Study Protocol Amendments
Given the number of diabetic patients in the US veteran population, we anticipated that there would be enough participants meeting the inclusion and exclusion criteria; however, because of the difficulty with recruitment, the initial study criteria were modified. The study was initially designed to incorporate DFUs with a minimum size of 1.0 cm2.12
Another limiting criterion was the percentage of total hemoglobin level for hemoglobin A1C (HbA1C). The study was originally established to include participants with an HbA1C level of 10% of total hemoglobin or below.12 Unfortunately, the majority of the potential participants had values substantially higher, and thus could not be enrolled in the trial, requiring another amendment to the study protocol in 2014, which was approved to include patients with an HbA1C level less than 12% of total hemoglobin. This change contributed considerably to the noted increase in enrollment rates in 2015, which almost doubled relative to enrollment under the original exclusion criteria (Figure).
The study has screened more than 600 patients. Among them, 137 were assessed for eligibility; 71 were excluded for various reasons, including screen failure (eg, decrease in wound size by >40% during the 2-week screening phase), loss to follow-up, and adverse events. Sixty-six participants reached the primary outcome at week 12, while 55 participants completed the study (19 in the SOC group; 18 in the cellular matrix group; 18 in the acellular matrix group).
We have stopped enrolling patients from all sites and the community, as we have reached our target enrollment.
Comment
One of the challenges of clinical trials is the recruitment of an adequate number of participants within an appropriate time frame, which is explained by Lasagna’s Law,18 a well-described phenomenon whereby the investigator overestimates the number of potential participants available to meet the inclusion criteria. This so-called funnel-effect was partly encountered in our selection process. A review of the veteran population with DFUs seemed to be more than adequate to fulfill the sample size; however, some important participant-related factors also played a substantial role.
In addition, the Veterans’ Affairs network centralizes health information, making it readily available to all providers participating in their care. As a result, patients with diabetes mellitus typically are seen by a primary care physician along with an endocrinologist, a diabetic nurse, and/or a dietician. Despite the collaboration with an interdisciplinary team, the glycemic control of the participants remains an issue along with other psychosocial factors that are deterrents in patient compliance. As a result, patients with poorly controlled diabetes and an HbA1C level above 10% (and less than 12%) of total hemoglobin who were initially excluded from the study were reincluded after modifying the inclusion criteria. Some patients were interested in joining the study, but physical limitations (eg, impaired mobility) prompted their decision not to join the trial, even though they met all the inclusion criteria.
As far as research-related factors that could affect participation, it is notable that most of the patients were retired; thus, the interventions did not cause additional burden of taking time off from work or loss of productivity. Although randomization could be a deterrent in many clinical trials, the majority of patients were willing to participate without demanding to be assigned to a particular treatment group.
There are many factors that are intertwined and can lead to enrollment and/or attrition rates. It was critical for our team to make some adjustment without compromising the controlled nature of a randomized trial.
Acknowledgment
The authors wish to acknowledge Huong Le, DPM, MPH, who was the coauthor of the study protocol.
- Sen CK, Gordillo GM, Roy S, et al. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen. 2009;17:763-771.
- Gurtner GC, Werner S, Barrandon Y, et al. Wound repair and regeneration. Nature. 2008;453:314-321.
- Falanga V. Wound healing and its impairment in the diabetic foot. Lancet. 2005;366:1736-1743.
- Boulton AJ. The diabetic foot: grand overview, epidemiology and pathogenesis. Diabetes Metab Res Rev. 2008;24(suppl 1):S3-S6.
- Singh N, Armstrong DG, Lipsky BA. Preventing foot ulcers in patients with diabetes. JAMA. 2005;293:217-228.
- Vuorisalo S, Venermo M, Lepäntalo M. Treatment of diabetic foot ulcers. J Cardiovasc Surg (Torino). 2009;50:275-291.
- Meijer JW, Trip J, Jaegers SM, et al. Quality of life in patients with diabetic foot ulcers. Disabil Rehabil. 2001;23:336-340.
- Santema TB, Poyck PP, Ubbink DT. Skin grafting and tissue replacement for treating foot ulcers in people with diabetes. Cochrane Database Syst Rev. 2016;2:CD011255.
- Margolis DJ, Kantor J, Berlin JA. Healing of diabetic neuropathic foot ulcers receiving standard treatment. a meta-analysis. Diabetes Care. 1999;22:692-695.
- Cavanagh P, Attinger C, Abbas Z, et al. Cost of treating diabetic foot ulcers in five different countries. Diabetes Metab Res Rev. 2012;2(suppl 1):107-111.
- Panuncialman J, Falanga V. The science of wound bed preparation. Surg Clin North Am. 2009;89:611-626.
- Lev-Tov H, Li CS, Dahle S, et al. Cellular versus acellular matrix devices in treatment of diabetic foot ulcers: study protocol for a comparative efficacy randomized controlled trial. Trials. 2013;14:8.
- Gentzkow GD, Iwasaki SD, Hershon KS, et al. Use of dermagraft, a cultured human dermis, to treat diabetic foot ulcers. Diabetes Care. 1996;19:350-354.
- Marston WA, Hanft J, Norwood P, et al; Dermagraft Diabetic Foot Ulcer Study Group. The efficacy and safety of Dermagraft in improving the healing of chronic diabetic foot ulcers: results of a prospective randomized trial. Diabetes Care. 2003;26:1701-1705.
- 2016 Dermagraft® Medicare Product and Related Procedure Payment. http://www.dermagraft.com/wp-content/uploads/sites/1/Dermagraft_Hotsheet%202016%20Q1%20HOSPITAL_FINAL.pdf. Accessed November 23, 2017.
- Oasis® Wound Matrix. http://www.oasiswoundmatrix.com/aboutowm. Accessed November 23, 2017.
- Niezgoda JA, Van Gils CC, Frykberg RG, et al. Randomized clinical trial comparing OASIS Wound Matrix to Regranex Gel for diabetic ulcers. Adv Skin Wound Care. 2005;18(5, pt 1):258-266.
- Torgerson JS, Arlinger K, Käppi M, et al. Principles for enhanced recruitment of subjects in a large clinical trial. the XENDOS (XENical in the prevention of Diabetes in Obese Subjects) study experience. Controlled Clin Trials. 2001;22:515-525.
- Sen CK, Gordillo GM, Roy S, et al. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen. 2009;17:763-771.
- Gurtner GC, Werner S, Barrandon Y, et al. Wound repair and regeneration. Nature. 2008;453:314-321.
- Falanga V. Wound healing and its impairment in the diabetic foot. Lancet. 2005;366:1736-1743.
- Boulton AJ. The diabetic foot: grand overview, epidemiology and pathogenesis. Diabetes Metab Res Rev. 2008;24(suppl 1):S3-S6.
- Singh N, Armstrong DG, Lipsky BA. Preventing foot ulcers in patients with diabetes. JAMA. 2005;293:217-228.
- Vuorisalo S, Venermo M, Lepäntalo M. Treatment of diabetic foot ulcers. J Cardiovasc Surg (Torino). 2009;50:275-291.
- Meijer JW, Trip J, Jaegers SM, et al. Quality of life in patients with diabetic foot ulcers. Disabil Rehabil. 2001;23:336-340.
- Santema TB, Poyck PP, Ubbink DT. Skin grafting and tissue replacement for treating foot ulcers in people with diabetes. Cochrane Database Syst Rev. 2016;2:CD011255.
- Margolis DJ, Kantor J, Berlin JA. Healing of diabetic neuropathic foot ulcers receiving standard treatment. a meta-analysis. Diabetes Care. 1999;22:692-695.
- Cavanagh P, Attinger C, Abbas Z, et al. Cost of treating diabetic foot ulcers in five different countries. Diabetes Metab Res Rev. 2012;2(suppl 1):107-111.
- Panuncialman J, Falanga V. The science of wound bed preparation. Surg Clin North Am. 2009;89:611-626.
- Lev-Tov H, Li CS, Dahle S, et al. Cellular versus acellular matrix devices in treatment of diabetic foot ulcers: study protocol for a comparative efficacy randomized controlled trial. Trials. 2013;14:8.
- Gentzkow GD, Iwasaki SD, Hershon KS, et al. Use of dermagraft, a cultured human dermis, to treat diabetic foot ulcers. Diabetes Care. 1996;19:350-354.
- Marston WA, Hanft J, Norwood P, et al; Dermagraft Diabetic Foot Ulcer Study Group. The efficacy and safety of Dermagraft in improving the healing of chronic diabetic foot ulcers: results of a prospective randomized trial. Diabetes Care. 2003;26:1701-1705.
- 2016 Dermagraft® Medicare Product and Related Procedure Payment. http://www.dermagraft.com/wp-content/uploads/sites/1/Dermagraft_Hotsheet%202016%20Q1%20HOSPITAL_FINAL.pdf. Accessed November 23, 2017.
- Oasis® Wound Matrix. http://www.oasiswoundmatrix.com/aboutowm. Accessed November 23, 2017.
- Niezgoda JA, Van Gils CC, Frykberg RG, et al. Randomized clinical trial comparing OASIS Wound Matrix to Regranex Gel for diabetic ulcers. Adv Skin Wound Care. 2005;18(5, pt 1):258-266.
- Torgerson JS, Arlinger K, Käppi M, et al. Principles for enhanced recruitment of subjects in a large clinical trial. the XENDOS (XENical in the prevention of Diabetes in Obese Subjects) study experience. Controlled Clin Trials. 2001;22:515-525.
Resident Pearl
- Deciding on the appropriate wound care regimen for diabetic foot ulcers is difficult given the vast amount of wound products on the market. This head-to-head clinical trial compared the use of an expensive cellular matrix and an inexpensive acellular matrix relative to the standard of care. We hope that this study will help to guide therapy based on cost-effectiveness of wound adjuncts without compromising patient care.
