User login
A Case-Based Review of Iron Overload With an Emphasis on Porphyria Cutanea Tarda, Hepatitis C, C282Y Heterozygosity, and Coronary Artery Disease
Sporadic porphyria cutanea tarda (PCT) is the most common cause of porphyria worldwide.1,2 Unlike other forms of porphyria, PCT usually is an acquired disease precipitated by extrinsic risk factors that commonly include excessive alcohol consumption, smoking, and chronic hepatitis C virus (HCV) infection. Additional risk factors include myeloproliferative disorders, exposure to polyhalogenated compounds, estrogen therapy, diseases of iron overload like hereditary hemochromatosis (HH), and potentially, HIV infection.1-3
In this case report, we present a patient with an iron overload (due in part to an HFE gene mutation) and concomitant PCT,
Case Presentation
Mr. M is a 59-year-old white male of Irish background with a medical history that includes coronary artery disease. He is status post ST-elevation myocardial infarction and percutaneous coronary intervention with placement of 2 drug-eluting stents. Additional medical issues include PCT and HCV infection with cirrhosis. He is an active smoker.
The patient has a long history of developing blisters with minor trauma, such as rubbing against his mattress/bed sheets or bumping into doors. These blisters primarily occur on his upper extremities, but also can occur on his face after shaving. Mr. M was diagnosed with HCV infection in 1979 while on active military duty. At that time, he had an acute HCV infection and jaundice that required a prolonged hospitalization. He reported no IV drug use and that many others on his military base had similar manifestations. He drinks 1 to 2 beers daily, but reports no binge drinking.
His laboratory studies were notable for ferritin, 2,069 ng/mL; serum iron, 317 mcg/dL; total iron binding capacity, 320 mcg/dL; transferrin, 239 mg/dl; liver function test alanine aminotransferase, 151 U/L; aspartate aminotransferase, 159 U/L; total bilirubin, 1.73 mg/dL; albumin, 3.6 g/dL; alkaline phosphatase, 119 U/L; INR, 1.1; and transferrin saturation, 99%. Mr. M’s HCV viral load was 28,700 IU/L with genotype 1b. Hemochromatosis genetic studies were notable for a heterozygous C282Y gene mutation and negative for H63D and S65C mutations. He repeatedly declined completing a 24-hour urine study of porphyrins. Ultrasonography was consistent with cirrhosis and splenomegaly. The patient was treatment naïve for HCV. He declined multiple offers for treatment of his HCV, citing financial considerations.
Porphyria Cutanea Tarda
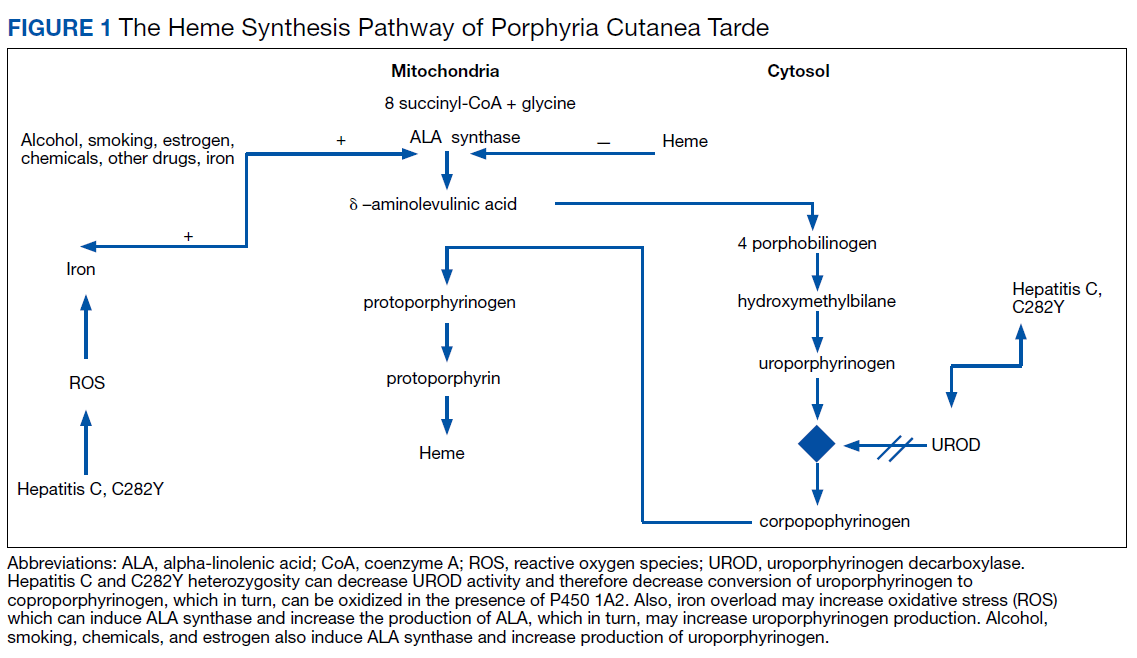
The pathogenesis of PCT is related to the intrahepatic deficiency of uroporphyrinogen decarboxylase (UROD), an enzyme in the heme biosynthetic pathway (Figure 1). Decreased activity of UROD leads to accumulation of uroporphyrinogen and its derivatives, which most likely are oxidized in presence of cytochrome P450 1A2. Up to 80% of PCT cases are sporadic, in which the deficiency of UROD is acquired by exogenous risk factors as mentioned above. However, the remaining 20% of PCT cases are due to an autosomal dominant mutation of UROD that causes the partial deficiency (up to 50%) of UROD. In these cases, additional risk factors are needed to decrease UROD activity to < 75% for symptoms to occur.
Clinical Manifestation
Patients with PCT typically develop blisters, skin fragility, and peeling with sun exposure or minor trauma. They also may experience delayed wound healing in sun-exposed skin.3 The photosensitivity of PCT is believed to be related to the saturation of highly carboxylated uroporphyrins in the liver, which are then released into the circulation. Sun exposure then activates these products facilitating an immune reaction and subsequent skin damage.2 In chronic cases, fibrotic reactions and scaring occur which can be mistaken for scleroderma. Other skin manifestations include hyperpigmentation, hypertrichosis, alopecia due to scaring and purplish heliotrope suffusion of periorbital areas.
Patients can develop cirrhosis due to accumulation of porphyria in the hepatocytes and subsequent parenchymal damage. Hepatocellular carcinoma surveillance is recommended for patients with PCT, although its incidence is rare in those patients.
Diagnosis and Treatment
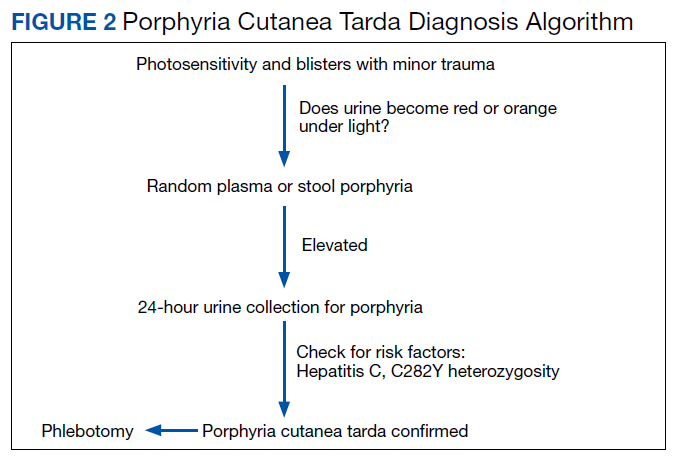
PCT is mainly a clinical diagnosis. Physicians should consider PCT in patients with photosensitivity and blisters after minor trauma (Figure 2). The urine of a patient with PCT is often pink or red when exposed to air or light due to its high concentration of porphyrin products. Mild elevation of liver enzymes and fatty liver on ultrasound are also noted. Evidence of iron overload is seen in most cases. Screening for risk factors like HCV, HIV, hepatitis B virus, and HH is recommended. Confirmation of PCT typically requires measurement of the porphyria level in a 24-hour urine collection.
Avoiding sun exposure is fundamental in decreasing the development of skin lesions and scaring. Additionally, patients should be advised about the adverse effects of alcohol, smoking, and estrogen therapy on PCT. Treatment of PCT is frequently focused on iron overload and subsequent increased porphyrin oxidation.1,2 Iron can increase reactive oxygen species (ROS), which, in turn, increases the rate of oxidation of uroporphyrinogens. Excess iron also decreases the activity of UROD and increases δ-aminolevulinic acid (ALA) production (the precursor of uroporphyrinogen). Phlebotomy to treat iron overload should be done to a target ferritin level of 20 ng/mL. Clinical manifestations, including skin lesions, typically will normalize before the laboratory findings. Therapeutic remission is expected after 6 to 7 phlebotomy attempts, while clinical improvement can occur after 2 to 3 phlebotomies.
In addition to phlebotomy, 4-aminoquinoline medications (chloroquine and hydroxychloroquine) can be used effectively to treat PCT. Hydroxychloroquine is generally preferred due to its better safety profile. Although the exact mechanism of action of 4-aminoquinolines is not clear, it has been suggested that they bind to porphyrins and form water-soluble products, which are then excreted in the urine. Again, clinical remission occurs much sooner than chemical remission, (3 months vs 12 months). A 4-aminoquinoline should not be used in patients with severe liver disease, renal insufficiency, pregnancy, or G6PD deficiency. When used, they should be used in lower than typical doses due to the rapid removal of accumulated porphyrin from the hepatocytes potentially causing necrosis and acute hepatitis.
Iron chelation also is effective, but it is slower in achieving remission and more expensive than phlebotomy. Treatment of PCT should be individualized. For example, 4-aminoquinolines are contraindicated for patients with end-stage renal disease (ESRD), while phlebotomy could present a problem for patients with preexisting anemia. In this instance, removing 50 cc of blood every 2 weeks may be safe and effective. Furthermore, 4-aminoquinolines in patients with severe iron overload and phlebotomy have been used together. Plasmapheresis is still another option in patients with ESRD.
The use of direct antiviral agents (DAA) in the treatment of HCV has shown promising results in maintaining undetectable viral loads and concurrent remission of PCT. Several studies have shown that treatment of HCV with a DAA obviates the need for treatment PCT.3-5 Treatment of HCV with interferon (IFN) and ribavirin have shown mixed results in controlling PCT, possibly due to their ineffectiveness in maintaining a suppressed viral load. Some studies even showed worsening of PCT with IFN/ribavirin.6
Hemochromatosis
Human cells need iron for aerobic respiration. The intestinal mucosa controls iron uptake and its transfer to the blood stream. Aside from variations in intestinal absorption with fecal excretion, humans do not have another pathway to excrete excess iron. HH is the most common genetic disorder in whites.7 It is an autosomal recessive disorder that increases the intestinal absorption of iron. The most common mutation in the hemochromatosis (HFE) gene results in a substitution of tyrosine for cysteine at amino acid number 282 and is referred to as the C282Y mutation. A second mutation changes histidine at position 63 to aspartic acid and is referred to as a H63D mutation. H63D is present in a minority of the patients with phenotypically expressed HH and its clinical impact is unknown.
Homozygosity of the C282Y mutation is the most common genotype associated with clinical hemochromatosis. While carriers of the C282Y gene heterozygote mutation typically do not develop enough iron overload to cause clinical hemochromatosis, they can if other risk factors, such as PCT, excess alcohol use, liver disease, or HCV, are present.8 Additionally, an associated genetic defect, like a compound heterozygotes C282Y/H63D mutation, a private HFE mutation in trans, or other iron-related genes, can cause manifestations of iron overload. Lastly, about 20% of patients that are heterozygous for both mutations can express the HH phenotype.8
Clinical Manifestation
Patients with HH absorb only a few extra milligrams of iron daily. The clinical manifestation begins to occur when the total body iron store reaches 15-40 g (normal, 4 g). While the genetic mutation is present from birth, iron stores start to rise slowly to around 10 g > age 15 years, at which point serum iron levels are elevated. After age 20 years, the speed with which the iron is stored increases, and by 30 years, liver damage and tissue injury will occur. Cirrhosis is possible by 40 years.7 Age, sex, dietary iron intake, blood loss (menstruation), pregnancy, and other unknown factors greatly influence the disease progression. Homozygote C282Y mutation is as common in women as it is in men, but women are less likely to express the HH phenotype, presumably due, in part, to menstruation. When diagnosed early, most of the clinical manifestations of HH are preventable. Additional manifestations of HH include hyperpigmentation, cardiomyopathy, diabetes mellitus, hypogonadism, hypothyroidism, and arthropathy due to pseudogout.
Iron overload due to HH should be distinguished from other causes of iron overload including exogenous iron overload, anemia (thalassemia, sideroblastic), and chronic liver diseases like PCT, viral hepatitis, nonalcoholic steatohepatitis, and alcoholic liver disease.
Diagnosis
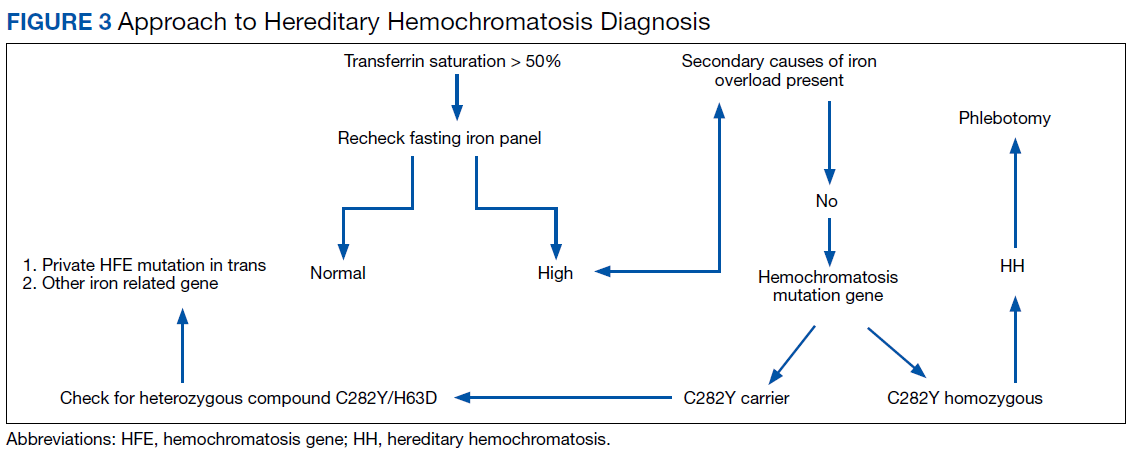
HH should be suspected in patients with a high serum transferrin saturation and elevated serum ferritin concentrations. Typically, transferrin saturation is > 50% and ferritin levels are > 300 ng/mL in men and > 200 ng/mL in women. In early stages of the disease, transferrin saturation can be normal. Additionally, in patients with chronic inflammation, ferritin may be high due to acute-phase reactants and the iron panel should be interpreted with caution. When the secondary causes of abnormalities in a patient’s iron studies are excluded, genetic testing for HFE gene is recommended.
The majority of patients (60-93%) with clinically evident hemochromatosis are homozygous for C282Y mutation. In a heterozygous C282Y mutation with a high transferrin saturation and HH phenotype, additional genetic testing for a heterozygous compound mutation C282Y/H63D is recommended.8 Additional studies could include evaluation for a private HFE mutation in trans or other iron-related genes. Liver biopsy is the gold standard for assessing the degree of hepatic fibrosis. Determining the degree of fibrosis by some means is needed due to the increased risk of hepatocellular carcinoma (HCC) in HH patients with advanced fibrosis and cirrhosis.9
Treatment
Iron depletion with phlebotomy is the cornerstone of treatment in HH. Phlebotomy initially is done weekly with goal of achieving a transferrin saturation < 50%, a serum ferritin level < 50 ng/mL, and a hemoglobin of 12 to 13 ng/mL. When these goals are achieved, patients typically need 4 to 8 phlebotomies per year to maintain a transferrin saturation < 50% (Figure 3).
Hemochromatosis and PCT
Many studies have investigated the relevance of C282Y and/or H63D mutations in patients with PCT.9,10 It appears that ≥ 1 mutation of the HFE gene in PCT may be an important susceptibility factor in the development of clinical PCT. Various studies have shown an incidence of C282Y mutations of 44 to 47% in patients with PCT, compared with 9 to 12% in control populations.9,10 The incidence of the H63D mutation in PCT has been more variable, with some studies showing no difference between patients with PCT and a control group, while other studies showed 31% incidence of H63D mutation in patients with PCT.9,10 A higher incidence of C282Y and H63D mutations in PCT may be a sign that the HFE mutation could be an important factor in developing PCT.
Hemochromatosis and Hepatitis C
Transferrin saturation is frequently elevated in patients with HCV. It is yet unclear whether the pathology of liver disease in patients with HCV is influenced by iron overload or limited to the direct cell damage from replication of the virus and subsequent inflammation. It is believed that the pathology of iron overload in the patients with HCV is different from HH. Like other secondary causes of iron overload, the excess iron is stored in the Kupffer cells of patients with HCV. In HH, excess iron is stored in hepatocytes.
The prevalence of the HFE mutation is the same in the patients with chronic HCV and healthy individuals.10,11 However, HFE mutations are more prevalent in 30 to 60% of the patients with chronic HCV who have elevated transferrin saturations. Alone, C282Y heterozygosity, H63D heterozygosity, or C282Y/H63D compound heterozygosity could not lead to clinically significant iron overload in otherwise healthy individuals; however, these could be a significant cause of iron overload in patients with chronic HCV. Theoretically, the combination of iron overload and HFE gene mutations could increase the rate of advanced fibrosis/cirrhosis in chronic HCV. An increase serum ferritin level of 200 ng/dL in women and 250 ng/dL in men has been observed in 32% of patients with chronic HCV. In this subset of patients, phlebotomy reduced the progression of their liver disease and reduction in their liver enzymes.
Iron Overload and Cardiovascular Risk
In 1987, a Framingham cohort of > 2,800 patients showed a higher incident of CAD in postmenopausal women when compared with premenopausal women.12 In the 1980s, Sullivan hypothesized that the reason for higher incidence of CAD in men when compared with premenopausal women was due to their higher body iron storage.13-16 A study of 1,930 Finnish men reported that the men with ferritin level ≥ 200 ng/dL had a risk 2.2 times higher of acute myocardial infarction when compared to men with lower serum ferritin level.17
A prospective study published in 1997 by Klechl showed the role of iron stores in early atherogenesis via promotion of lipid oxidation.18 Other epidemiological studies have shown a decreased risk of myocardial infarction in blood donors, and while arguments have been made that the blood donors tend to be healthier individuals, 2 studies were published in 1997 matching healthy blood donors to healthy nonblood donors, and both showed a lower risk of CVD in the donors when compared with nondonors.19,20 Furthermore, in an animal model of atherosclerosis, an iron depleted diet showed a reduction of atherosclerosis progression.21 Multiple studies have shown that the heterozygosity for HFE is significantly linked to the risk of cardiovascular events, including the fact that heterozygosity for C282Y has been shown to be a risk factor for myocardial infarction in men and cerebrovascular death in women.22-25
Conclusion
Multiple studies have shown an association between the elevated iron levels associated with the HFE genotype and the disease states of our patient. These include an increased risk of CAD, the increased risk of cirrhosis in HCV and the development of PCT. Indeed, in this case, our patient likely acquired PCT from the combined risks of HCV and his heterozygous HFE genetic mutation.
With regard to Mr. M’s treatment, the use of an antiviral agent in the treatment of his HCV is fundamental, along with avoidance of alcohol and smoking. If he were to accept HCV treatment, we would anticipate resolution of the PCT, but the ongoing progression of his liver and cardiovascular conditions, due perhaps in part, to relative iron overload from his heterozygous HFE mutation. In this situation, we expect that an ongoing course of therapeutic phlebotomy could help to delay the progression of his chronic liver and cardiovascular diseases.
1. Singal AK. Porphyria cutanea tarda: Recent update. Mol Genet Metab. 2019;128(3):271-281.
2. Ryan Caballes F, Sendi H, Bonkovsky HL. Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int. 2012;32(6):880-893.
3. Wiznia LE, Laird ME, Franks AG Jr. Hepatitis C virus and its cutaneous manifestations: treatment in the direct-acting antiviral era. J Eur Acad Dermatol Venereol. 2017;31(8):1260-1270.
4. Nihei T, Kiniwa Y, Mikoshiba Y, Joshita S, Okuyama R. Improvement of porphyria cutanea tarda following treatment of hepatitis C virus by direct-acting antivirals: a case report. J Dermatol. 2019;46(5):e149-e151.
5. Combalia A, To-Figueras J, Laguno M, Martínez-Rebollar M, Aguilera P. Direct-acting antivirals for hepatitis C virus induce a rapid clinical and biochemical remission of porphyria cutanea tarda. Br J Dermatol. 2017;177(5):e183-e184. 6. Singal AK, Venkata KVR, Jampana S, Islam FU, Anderson KE. Hepatitis C treatment in patients with porphyria cutanea tarda. Am J Med Sci. 2017;353 (6):523-528.
7. Brandhagen DJ, Fairbanks VF, Baldus W. Recognition and management of hereditary hemochromatosis. Am Fam Physician. 2002;65(5):853-860.
8. Aguilar-Martinez P, Grandchamp B, Cunat S, et al. Iron overload in HFE C282Y heterozygotes at first genetic testing: a strategy for identifying rare HFE variants. Haematologica. 2011;96(4):507-514.
9. Erhardt A, Maschner-Olberg A, Mellenthin C, et al. HFE mutations and chronic hepatitis C: H63D and C282Y heterozygosity are independent risk factors for liver fibrosis and cirrhosis. J Hepatol. 2003;38(3):335-342.
10. Mehrany K, Drage LA, Brandhagen DJ, Pittelkow MR. Association of porphyria cutanea tarda with hereditary hemochromatosis. J Am Acad Dermatol. 2004;51(2):205-211.
11. Pietrangelo A. Hemochromatosis gene modifies course of hepatitis C viral infection. Gastroenterology. 2003;124(5):1509-1523.
12. Gordon T, Kannel WB, Hjortland MC, McNamara PM. Menopause and coronary heart disease. The Framingham Study. Ann Intern Med. 1978;89(2):157-161.
13. Sullivan JL. Iron and the sex difference in heart disease risk. Lancet. 1981;1(8233):1293-1294.
14. Sullivan JL. The sex difference in ischemic heart disease. Perspect Biol Med. 1983;26(4):657-671.
15. Sullivan JL. The iron paradigm of ischemic heart disease. Am Heart J. 1989;117(5):1177-1188.
16. Sullivan JL. Stored iron and ischemic heart disease: empirical support for a new paradigm. Circulation. 1992;86(3):1036-1037.
17. Salonen JT, Nyyssönen K, Korpela H, Tuomilehto J, Seppänen R, Salonen R. High stored iron levels are associated with excess risk of myocardial infarction in eastern Finnish men. Circulation. 1992;86(3):803-811.
18. Kiechl S, Willeit J, Egger G, Poewe W, Oberhollenzer F. Body iron stores and the risk of carotid atherosclerosis: prospective results from the Bruneck study. Circulation. 1997;96(10):3300-3307.
19. Tuomainen TP, Salonen R, Nyyssönen K, Salonen JT. Cohort study of relation between donating blood and risk of myocardial infarction in 2682 men in eastern Finland. BMJ. 1997;314(7083):793-794.
20. Meyers DG, Strickland D, Maloley PA, Seburg JK, Wilson JE, McManus BF. Possible association of a reduction in cardiovascular events with blood donation. Heart. 1997;78(2):188-193.
21. Lee TS, Shiao MS, Pan CC, Chau LY. Iron-deficient diet reduces atherosclerotic lesions in apoE-deficient mice. Circulation. 1999;99(9):1222-1229.
22. Surber R, Sigusch HH, Kuehnert H, Figulla HR. Haemochromatosis (HFE) gene C282Y mutation and the risk of coronary artery disease and myocardial infarction: a study in 1279 patients undergoing coronary angiography. J Med Genet. 2003;40(5):e58.
23. Tuomainen TP, Kontula K, Nyyssönen K, Lakka TA, Heliö T, Salonen JT. Increased risk of acute myocardial infarction in carriers of the hemochromatosis gene Cys282Tyr mutation: a prospective cohort study in men in eastern Finland. Circulation. 1999;100(12):1274-1279.
24. Roest M, van der Schouw YT, de Valk B, et al. Heterozygosity for a hereditary hemochromatosis gene is associated with cardiovascular death in women. Circulation. 1999;100(12):1268-1273.
25. Pourmoghaddas A, Sanei H, Garakyaraghi M, Esteki-Ghashghaei F, Gharaati M. The relation between body iron store and ferritin, and coronary artery disease. ARYA Atheroscler. 2014;10(1):32-36.
Sporadic porphyria cutanea tarda (PCT) is the most common cause of porphyria worldwide.1,2 Unlike other forms of porphyria, PCT usually is an acquired disease precipitated by extrinsic risk factors that commonly include excessive alcohol consumption, smoking, and chronic hepatitis C virus (HCV) infection. Additional risk factors include myeloproliferative disorders, exposure to polyhalogenated compounds, estrogen therapy, diseases of iron overload like hereditary hemochromatosis (HH), and potentially, HIV infection.1-3
In this case report, we present a patient with an iron overload (due in part to an HFE gene mutation) and concomitant PCT,
Case Presentation
Mr. M is a 59-year-old white male of Irish background with a medical history that includes coronary artery disease. He is status post ST-elevation myocardial infarction and percutaneous coronary intervention with placement of 2 drug-eluting stents. Additional medical issues include PCT and HCV infection with cirrhosis. He is an active smoker.
The patient has a long history of developing blisters with minor trauma, such as rubbing against his mattress/bed sheets or bumping into doors. These blisters primarily occur on his upper extremities, but also can occur on his face after shaving. Mr. M was diagnosed with HCV infection in 1979 while on active military duty. At that time, he had an acute HCV infection and jaundice that required a prolonged hospitalization. He reported no IV drug use and that many others on his military base had similar manifestations. He drinks 1 to 2 beers daily, but reports no binge drinking.
His laboratory studies were notable for ferritin, 2,069 ng/mL; serum iron, 317 mcg/dL; total iron binding capacity, 320 mcg/dL; transferrin, 239 mg/dl; liver function test alanine aminotransferase, 151 U/L; aspartate aminotransferase, 159 U/L; total bilirubin, 1.73 mg/dL; albumin, 3.6 g/dL; alkaline phosphatase, 119 U/L; INR, 1.1; and transferrin saturation, 99%. Mr. M’s HCV viral load was 28,700 IU/L with genotype 1b. Hemochromatosis genetic studies were notable for a heterozygous C282Y gene mutation and negative for H63D and S65C mutations. He repeatedly declined completing a 24-hour urine study of porphyrins. Ultrasonography was consistent with cirrhosis and splenomegaly. The patient was treatment naïve for HCV. He declined multiple offers for treatment of his HCV, citing financial considerations.
Porphyria Cutanea Tarda
The pathogenesis of PCT is related to the intrahepatic deficiency of uroporphyrinogen decarboxylase (UROD), an enzyme in the heme biosynthetic pathway (Figure 1). Decreased activity of UROD leads to accumulation of uroporphyrinogen and its derivatives, which most likely are oxidized in presence of cytochrome P450 1A2. Up to 80% of PCT cases are sporadic, in which the deficiency of UROD is acquired by exogenous risk factors as mentioned above. However, the remaining 20% of PCT cases are due to an autosomal dominant mutation of UROD that causes the partial deficiency (up to 50%) of UROD. In these cases, additional risk factors are needed to decrease UROD activity to < 75% for symptoms to occur.
Clinical Manifestation
Patients with PCT typically develop blisters, skin fragility, and peeling with sun exposure or minor trauma. They also may experience delayed wound healing in sun-exposed skin.3 The photosensitivity of PCT is believed to be related to the saturation of highly carboxylated uroporphyrins in the liver, which are then released into the circulation. Sun exposure then activates these products facilitating an immune reaction and subsequent skin damage.2 In chronic cases, fibrotic reactions and scaring occur which can be mistaken for scleroderma. Other skin manifestations include hyperpigmentation, hypertrichosis, alopecia due to scaring and purplish heliotrope suffusion of periorbital areas.
Patients can develop cirrhosis due to accumulation of porphyria in the hepatocytes and subsequent parenchymal damage. Hepatocellular carcinoma surveillance is recommended for patients with PCT, although its incidence is rare in those patients.
Diagnosis and Treatment
PCT is mainly a clinical diagnosis. Physicians should consider PCT in patients with photosensitivity and blisters after minor trauma (Figure 2). The urine of a patient with PCT is often pink or red when exposed to air or light due to its high concentration of porphyrin products. Mild elevation of liver enzymes and fatty liver on ultrasound are also noted. Evidence of iron overload is seen in most cases. Screening for risk factors like HCV, HIV, hepatitis B virus, and HH is recommended. Confirmation of PCT typically requires measurement of the porphyria level in a 24-hour urine collection.
Avoiding sun exposure is fundamental in decreasing the development of skin lesions and scaring. Additionally, patients should be advised about the adverse effects of alcohol, smoking, and estrogen therapy on PCT. Treatment of PCT is frequently focused on iron overload and subsequent increased porphyrin oxidation.1,2 Iron can increase reactive oxygen species (ROS), which, in turn, increases the rate of oxidation of uroporphyrinogens. Excess iron also decreases the activity of UROD and increases δ-aminolevulinic acid (ALA) production (the precursor of uroporphyrinogen). Phlebotomy to treat iron overload should be done to a target ferritin level of 20 ng/mL. Clinical manifestations, including skin lesions, typically will normalize before the laboratory findings. Therapeutic remission is expected after 6 to 7 phlebotomy attempts, while clinical improvement can occur after 2 to 3 phlebotomies.
In addition to phlebotomy, 4-aminoquinoline medications (chloroquine and hydroxychloroquine) can be used effectively to treat PCT. Hydroxychloroquine is generally preferred due to its better safety profile. Although the exact mechanism of action of 4-aminoquinolines is not clear, it has been suggested that they bind to porphyrins and form water-soluble products, which are then excreted in the urine. Again, clinical remission occurs much sooner than chemical remission, (3 months vs 12 months). A 4-aminoquinoline should not be used in patients with severe liver disease, renal insufficiency, pregnancy, or G6PD deficiency. When used, they should be used in lower than typical doses due to the rapid removal of accumulated porphyrin from the hepatocytes potentially causing necrosis and acute hepatitis.
Iron chelation also is effective, but it is slower in achieving remission and more expensive than phlebotomy. Treatment of PCT should be individualized. For example, 4-aminoquinolines are contraindicated for patients with end-stage renal disease (ESRD), while phlebotomy could present a problem for patients with preexisting anemia. In this instance, removing 50 cc of blood every 2 weeks may be safe and effective. Furthermore, 4-aminoquinolines in patients with severe iron overload and phlebotomy have been used together. Plasmapheresis is still another option in patients with ESRD.
The use of direct antiviral agents (DAA) in the treatment of HCV has shown promising results in maintaining undetectable viral loads and concurrent remission of PCT. Several studies have shown that treatment of HCV with a DAA obviates the need for treatment PCT.3-5 Treatment of HCV with interferon (IFN) and ribavirin have shown mixed results in controlling PCT, possibly due to their ineffectiveness in maintaining a suppressed viral load. Some studies even showed worsening of PCT with IFN/ribavirin.6
Hemochromatosis
Human cells need iron for aerobic respiration. The intestinal mucosa controls iron uptake and its transfer to the blood stream. Aside from variations in intestinal absorption with fecal excretion, humans do not have another pathway to excrete excess iron. HH is the most common genetic disorder in whites.7 It is an autosomal recessive disorder that increases the intestinal absorption of iron. The most common mutation in the hemochromatosis (HFE) gene results in a substitution of tyrosine for cysteine at amino acid number 282 and is referred to as the C282Y mutation. A second mutation changes histidine at position 63 to aspartic acid and is referred to as a H63D mutation. H63D is present in a minority of the patients with phenotypically expressed HH and its clinical impact is unknown.
Homozygosity of the C282Y mutation is the most common genotype associated with clinical hemochromatosis. While carriers of the C282Y gene heterozygote mutation typically do not develop enough iron overload to cause clinical hemochromatosis, they can if other risk factors, such as PCT, excess alcohol use, liver disease, or HCV, are present.8 Additionally, an associated genetic defect, like a compound heterozygotes C282Y/H63D mutation, a private HFE mutation in trans, or other iron-related genes, can cause manifestations of iron overload. Lastly, about 20% of patients that are heterozygous for both mutations can express the HH phenotype.8
Clinical Manifestation
Patients with HH absorb only a few extra milligrams of iron daily. The clinical manifestation begins to occur when the total body iron store reaches 15-40 g (normal, 4 g). While the genetic mutation is present from birth, iron stores start to rise slowly to around 10 g > age 15 years, at which point serum iron levels are elevated. After age 20 years, the speed with which the iron is stored increases, and by 30 years, liver damage and tissue injury will occur. Cirrhosis is possible by 40 years.7 Age, sex, dietary iron intake, blood loss (menstruation), pregnancy, and other unknown factors greatly influence the disease progression. Homozygote C282Y mutation is as common in women as it is in men, but women are less likely to express the HH phenotype, presumably due, in part, to menstruation. When diagnosed early, most of the clinical manifestations of HH are preventable. Additional manifestations of HH include hyperpigmentation, cardiomyopathy, diabetes mellitus, hypogonadism, hypothyroidism, and arthropathy due to pseudogout.
Iron overload due to HH should be distinguished from other causes of iron overload including exogenous iron overload, anemia (thalassemia, sideroblastic), and chronic liver diseases like PCT, viral hepatitis, nonalcoholic steatohepatitis, and alcoholic liver disease.
Diagnosis
HH should be suspected in patients with a high serum transferrin saturation and elevated serum ferritin concentrations. Typically, transferrin saturation is > 50% and ferritin levels are > 300 ng/mL in men and > 200 ng/mL in women. In early stages of the disease, transferrin saturation can be normal. Additionally, in patients with chronic inflammation, ferritin may be high due to acute-phase reactants and the iron panel should be interpreted with caution. When the secondary causes of abnormalities in a patient’s iron studies are excluded, genetic testing for HFE gene is recommended.
The majority of patients (60-93%) with clinically evident hemochromatosis are homozygous for C282Y mutation. In a heterozygous C282Y mutation with a high transferrin saturation and HH phenotype, additional genetic testing for a heterozygous compound mutation C282Y/H63D is recommended.8 Additional studies could include evaluation for a private HFE mutation in trans or other iron-related genes. Liver biopsy is the gold standard for assessing the degree of hepatic fibrosis. Determining the degree of fibrosis by some means is needed due to the increased risk of hepatocellular carcinoma (HCC) in HH patients with advanced fibrosis and cirrhosis.9
Treatment
Iron depletion with phlebotomy is the cornerstone of treatment in HH. Phlebotomy initially is done weekly with goal of achieving a transferrin saturation < 50%, a serum ferritin level < 50 ng/mL, and a hemoglobin of 12 to 13 ng/mL. When these goals are achieved, patients typically need 4 to 8 phlebotomies per year to maintain a transferrin saturation < 50% (Figure 3).
Hemochromatosis and PCT
Many studies have investigated the relevance of C282Y and/or H63D mutations in patients with PCT.9,10 It appears that ≥ 1 mutation of the HFE gene in PCT may be an important susceptibility factor in the development of clinical PCT. Various studies have shown an incidence of C282Y mutations of 44 to 47% in patients with PCT, compared with 9 to 12% in control populations.9,10 The incidence of the H63D mutation in PCT has been more variable, with some studies showing no difference between patients with PCT and a control group, while other studies showed 31% incidence of H63D mutation in patients with PCT.9,10 A higher incidence of C282Y and H63D mutations in PCT may be a sign that the HFE mutation could be an important factor in developing PCT.
Hemochromatosis and Hepatitis C
Transferrin saturation is frequently elevated in patients with HCV. It is yet unclear whether the pathology of liver disease in patients with HCV is influenced by iron overload or limited to the direct cell damage from replication of the virus and subsequent inflammation. It is believed that the pathology of iron overload in the patients with HCV is different from HH. Like other secondary causes of iron overload, the excess iron is stored in the Kupffer cells of patients with HCV. In HH, excess iron is stored in hepatocytes.
The prevalence of the HFE mutation is the same in the patients with chronic HCV and healthy individuals.10,11 However, HFE mutations are more prevalent in 30 to 60% of the patients with chronic HCV who have elevated transferrin saturations. Alone, C282Y heterozygosity, H63D heterozygosity, or C282Y/H63D compound heterozygosity could not lead to clinically significant iron overload in otherwise healthy individuals; however, these could be a significant cause of iron overload in patients with chronic HCV. Theoretically, the combination of iron overload and HFE gene mutations could increase the rate of advanced fibrosis/cirrhosis in chronic HCV. An increase serum ferritin level of 200 ng/dL in women and 250 ng/dL in men has been observed in 32% of patients with chronic HCV. In this subset of patients, phlebotomy reduced the progression of their liver disease and reduction in their liver enzymes.
Iron Overload and Cardiovascular Risk
In 1987, a Framingham cohort of > 2,800 patients showed a higher incident of CAD in postmenopausal women when compared with premenopausal women.12 In the 1980s, Sullivan hypothesized that the reason for higher incidence of CAD in men when compared with premenopausal women was due to their higher body iron storage.13-16 A study of 1,930 Finnish men reported that the men with ferritin level ≥ 200 ng/dL had a risk 2.2 times higher of acute myocardial infarction when compared to men with lower serum ferritin level.17
A prospective study published in 1997 by Klechl showed the role of iron stores in early atherogenesis via promotion of lipid oxidation.18 Other epidemiological studies have shown a decreased risk of myocardial infarction in blood donors, and while arguments have been made that the blood donors tend to be healthier individuals, 2 studies were published in 1997 matching healthy blood donors to healthy nonblood donors, and both showed a lower risk of CVD in the donors when compared with nondonors.19,20 Furthermore, in an animal model of atherosclerosis, an iron depleted diet showed a reduction of atherosclerosis progression.21 Multiple studies have shown that the heterozygosity for HFE is significantly linked to the risk of cardiovascular events, including the fact that heterozygosity for C282Y has been shown to be a risk factor for myocardial infarction in men and cerebrovascular death in women.22-25
Conclusion
Multiple studies have shown an association between the elevated iron levels associated with the HFE genotype and the disease states of our patient. These include an increased risk of CAD, the increased risk of cirrhosis in HCV and the development of PCT. Indeed, in this case, our patient likely acquired PCT from the combined risks of HCV and his heterozygous HFE genetic mutation.
With regard to Mr. M’s treatment, the use of an antiviral agent in the treatment of his HCV is fundamental, along with avoidance of alcohol and smoking. If he were to accept HCV treatment, we would anticipate resolution of the PCT, but the ongoing progression of his liver and cardiovascular conditions, due perhaps in part, to relative iron overload from his heterozygous HFE mutation. In this situation, we expect that an ongoing course of therapeutic phlebotomy could help to delay the progression of his chronic liver and cardiovascular diseases.
Sporadic porphyria cutanea tarda (PCT) is the most common cause of porphyria worldwide.1,2 Unlike other forms of porphyria, PCT usually is an acquired disease precipitated by extrinsic risk factors that commonly include excessive alcohol consumption, smoking, and chronic hepatitis C virus (HCV) infection. Additional risk factors include myeloproliferative disorders, exposure to polyhalogenated compounds, estrogen therapy, diseases of iron overload like hereditary hemochromatosis (HH), and potentially, HIV infection.1-3
In this case report, we present a patient with an iron overload (due in part to an HFE gene mutation) and concomitant PCT,
Case Presentation
Mr. M is a 59-year-old white male of Irish background with a medical history that includes coronary artery disease. He is status post ST-elevation myocardial infarction and percutaneous coronary intervention with placement of 2 drug-eluting stents. Additional medical issues include PCT and HCV infection with cirrhosis. He is an active smoker.
The patient has a long history of developing blisters with minor trauma, such as rubbing against his mattress/bed sheets or bumping into doors. These blisters primarily occur on his upper extremities, but also can occur on his face after shaving. Mr. M was diagnosed with HCV infection in 1979 while on active military duty. At that time, he had an acute HCV infection and jaundice that required a prolonged hospitalization. He reported no IV drug use and that many others on his military base had similar manifestations. He drinks 1 to 2 beers daily, but reports no binge drinking.
His laboratory studies were notable for ferritin, 2,069 ng/mL; serum iron, 317 mcg/dL; total iron binding capacity, 320 mcg/dL; transferrin, 239 mg/dl; liver function test alanine aminotransferase, 151 U/L; aspartate aminotransferase, 159 U/L; total bilirubin, 1.73 mg/dL; albumin, 3.6 g/dL; alkaline phosphatase, 119 U/L; INR, 1.1; and transferrin saturation, 99%. Mr. M’s HCV viral load was 28,700 IU/L with genotype 1b. Hemochromatosis genetic studies were notable for a heterozygous C282Y gene mutation and negative for H63D and S65C mutations. He repeatedly declined completing a 24-hour urine study of porphyrins. Ultrasonography was consistent with cirrhosis and splenomegaly. The patient was treatment naïve for HCV. He declined multiple offers for treatment of his HCV, citing financial considerations.
Porphyria Cutanea Tarda
The pathogenesis of PCT is related to the intrahepatic deficiency of uroporphyrinogen decarboxylase (UROD), an enzyme in the heme biosynthetic pathway (Figure 1). Decreased activity of UROD leads to accumulation of uroporphyrinogen and its derivatives, which most likely are oxidized in presence of cytochrome P450 1A2. Up to 80% of PCT cases are sporadic, in which the deficiency of UROD is acquired by exogenous risk factors as mentioned above. However, the remaining 20% of PCT cases are due to an autosomal dominant mutation of UROD that causes the partial deficiency (up to 50%) of UROD. In these cases, additional risk factors are needed to decrease UROD activity to < 75% for symptoms to occur.
Clinical Manifestation
Patients with PCT typically develop blisters, skin fragility, and peeling with sun exposure or minor trauma. They also may experience delayed wound healing in sun-exposed skin.3 The photosensitivity of PCT is believed to be related to the saturation of highly carboxylated uroporphyrins in the liver, which are then released into the circulation. Sun exposure then activates these products facilitating an immune reaction and subsequent skin damage.2 In chronic cases, fibrotic reactions and scaring occur which can be mistaken for scleroderma. Other skin manifestations include hyperpigmentation, hypertrichosis, alopecia due to scaring and purplish heliotrope suffusion of periorbital areas.
Patients can develop cirrhosis due to accumulation of porphyria in the hepatocytes and subsequent parenchymal damage. Hepatocellular carcinoma surveillance is recommended for patients with PCT, although its incidence is rare in those patients.
Diagnosis and Treatment
PCT is mainly a clinical diagnosis. Physicians should consider PCT in patients with photosensitivity and blisters after minor trauma (Figure 2). The urine of a patient with PCT is often pink or red when exposed to air or light due to its high concentration of porphyrin products. Mild elevation of liver enzymes and fatty liver on ultrasound are also noted. Evidence of iron overload is seen in most cases. Screening for risk factors like HCV, HIV, hepatitis B virus, and HH is recommended. Confirmation of PCT typically requires measurement of the porphyria level in a 24-hour urine collection.
Avoiding sun exposure is fundamental in decreasing the development of skin lesions and scaring. Additionally, patients should be advised about the adverse effects of alcohol, smoking, and estrogen therapy on PCT. Treatment of PCT is frequently focused on iron overload and subsequent increased porphyrin oxidation.1,2 Iron can increase reactive oxygen species (ROS), which, in turn, increases the rate of oxidation of uroporphyrinogens. Excess iron also decreases the activity of UROD and increases δ-aminolevulinic acid (ALA) production (the precursor of uroporphyrinogen). Phlebotomy to treat iron overload should be done to a target ferritin level of 20 ng/mL. Clinical manifestations, including skin lesions, typically will normalize before the laboratory findings. Therapeutic remission is expected after 6 to 7 phlebotomy attempts, while clinical improvement can occur after 2 to 3 phlebotomies.
In addition to phlebotomy, 4-aminoquinoline medications (chloroquine and hydroxychloroquine) can be used effectively to treat PCT. Hydroxychloroquine is generally preferred due to its better safety profile. Although the exact mechanism of action of 4-aminoquinolines is not clear, it has been suggested that they bind to porphyrins and form water-soluble products, which are then excreted in the urine. Again, clinical remission occurs much sooner than chemical remission, (3 months vs 12 months). A 4-aminoquinoline should not be used in patients with severe liver disease, renal insufficiency, pregnancy, or G6PD deficiency. When used, they should be used in lower than typical doses due to the rapid removal of accumulated porphyrin from the hepatocytes potentially causing necrosis and acute hepatitis.
Iron chelation also is effective, but it is slower in achieving remission and more expensive than phlebotomy. Treatment of PCT should be individualized. For example, 4-aminoquinolines are contraindicated for patients with end-stage renal disease (ESRD), while phlebotomy could present a problem for patients with preexisting anemia. In this instance, removing 50 cc of blood every 2 weeks may be safe and effective. Furthermore, 4-aminoquinolines in patients with severe iron overload and phlebotomy have been used together. Plasmapheresis is still another option in patients with ESRD.
The use of direct antiviral agents (DAA) in the treatment of HCV has shown promising results in maintaining undetectable viral loads and concurrent remission of PCT. Several studies have shown that treatment of HCV with a DAA obviates the need for treatment PCT.3-5 Treatment of HCV with interferon (IFN) and ribavirin have shown mixed results in controlling PCT, possibly due to their ineffectiveness in maintaining a suppressed viral load. Some studies even showed worsening of PCT with IFN/ribavirin.6
Hemochromatosis
Human cells need iron for aerobic respiration. The intestinal mucosa controls iron uptake and its transfer to the blood stream. Aside from variations in intestinal absorption with fecal excretion, humans do not have another pathway to excrete excess iron. HH is the most common genetic disorder in whites.7 It is an autosomal recessive disorder that increases the intestinal absorption of iron. The most common mutation in the hemochromatosis (HFE) gene results in a substitution of tyrosine for cysteine at amino acid number 282 and is referred to as the C282Y mutation. A second mutation changes histidine at position 63 to aspartic acid and is referred to as a H63D mutation. H63D is present in a minority of the patients with phenotypically expressed HH and its clinical impact is unknown.
Homozygosity of the C282Y mutation is the most common genotype associated with clinical hemochromatosis. While carriers of the C282Y gene heterozygote mutation typically do not develop enough iron overload to cause clinical hemochromatosis, they can if other risk factors, such as PCT, excess alcohol use, liver disease, or HCV, are present.8 Additionally, an associated genetic defect, like a compound heterozygotes C282Y/H63D mutation, a private HFE mutation in trans, or other iron-related genes, can cause manifestations of iron overload. Lastly, about 20% of patients that are heterozygous for both mutations can express the HH phenotype.8
Clinical Manifestation
Patients with HH absorb only a few extra milligrams of iron daily. The clinical manifestation begins to occur when the total body iron store reaches 15-40 g (normal, 4 g). While the genetic mutation is present from birth, iron stores start to rise slowly to around 10 g > age 15 years, at which point serum iron levels are elevated. After age 20 years, the speed with which the iron is stored increases, and by 30 years, liver damage and tissue injury will occur. Cirrhosis is possible by 40 years.7 Age, sex, dietary iron intake, blood loss (menstruation), pregnancy, and other unknown factors greatly influence the disease progression. Homozygote C282Y mutation is as common in women as it is in men, but women are less likely to express the HH phenotype, presumably due, in part, to menstruation. When diagnosed early, most of the clinical manifestations of HH are preventable. Additional manifestations of HH include hyperpigmentation, cardiomyopathy, diabetes mellitus, hypogonadism, hypothyroidism, and arthropathy due to pseudogout.
Iron overload due to HH should be distinguished from other causes of iron overload including exogenous iron overload, anemia (thalassemia, sideroblastic), and chronic liver diseases like PCT, viral hepatitis, nonalcoholic steatohepatitis, and alcoholic liver disease.
Diagnosis
HH should be suspected in patients with a high serum transferrin saturation and elevated serum ferritin concentrations. Typically, transferrin saturation is > 50% and ferritin levels are > 300 ng/mL in men and > 200 ng/mL in women. In early stages of the disease, transferrin saturation can be normal. Additionally, in patients with chronic inflammation, ferritin may be high due to acute-phase reactants and the iron panel should be interpreted with caution. When the secondary causes of abnormalities in a patient’s iron studies are excluded, genetic testing for HFE gene is recommended.
The majority of patients (60-93%) with clinically evident hemochromatosis are homozygous for C282Y mutation. In a heterozygous C282Y mutation with a high transferrin saturation and HH phenotype, additional genetic testing for a heterozygous compound mutation C282Y/H63D is recommended.8 Additional studies could include evaluation for a private HFE mutation in trans or other iron-related genes. Liver biopsy is the gold standard for assessing the degree of hepatic fibrosis. Determining the degree of fibrosis by some means is needed due to the increased risk of hepatocellular carcinoma (HCC) in HH patients with advanced fibrosis and cirrhosis.9
Treatment
Iron depletion with phlebotomy is the cornerstone of treatment in HH. Phlebotomy initially is done weekly with goal of achieving a transferrin saturation < 50%, a serum ferritin level < 50 ng/mL, and a hemoglobin of 12 to 13 ng/mL. When these goals are achieved, patients typically need 4 to 8 phlebotomies per year to maintain a transferrin saturation < 50% (Figure 3).
Hemochromatosis and PCT
Many studies have investigated the relevance of C282Y and/or H63D mutations in patients with PCT.9,10 It appears that ≥ 1 mutation of the HFE gene in PCT may be an important susceptibility factor in the development of clinical PCT. Various studies have shown an incidence of C282Y mutations of 44 to 47% in patients with PCT, compared with 9 to 12% in control populations.9,10 The incidence of the H63D mutation in PCT has been more variable, with some studies showing no difference between patients with PCT and a control group, while other studies showed 31% incidence of H63D mutation in patients with PCT.9,10 A higher incidence of C282Y and H63D mutations in PCT may be a sign that the HFE mutation could be an important factor in developing PCT.
Hemochromatosis and Hepatitis C
Transferrin saturation is frequently elevated in patients with HCV. It is yet unclear whether the pathology of liver disease in patients with HCV is influenced by iron overload or limited to the direct cell damage from replication of the virus and subsequent inflammation. It is believed that the pathology of iron overload in the patients with HCV is different from HH. Like other secondary causes of iron overload, the excess iron is stored in the Kupffer cells of patients with HCV. In HH, excess iron is stored in hepatocytes.
The prevalence of the HFE mutation is the same in the patients with chronic HCV and healthy individuals.10,11 However, HFE mutations are more prevalent in 30 to 60% of the patients with chronic HCV who have elevated transferrin saturations. Alone, C282Y heterozygosity, H63D heterozygosity, or C282Y/H63D compound heterozygosity could not lead to clinically significant iron overload in otherwise healthy individuals; however, these could be a significant cause of iron overload in patients with chronic HCV. Theoretically, the combination of iron overload and HFE gene mutations could increase the rate of advanced fibrosis/cirrhosis in chronic HCV. An increase serum ferritin level of 200 ng/dL in women and 250 ng/dL in men has been observed in 32% of patients with chronic HCV. In this subset of patients, phlebotomy reduced the progression of their liver disease and reduction in their liver enzymes.
Iron Overload and Cardiovascular Risk
In 1987, a Framingham cohort of > 2,800 patients showed a higher incident of CAD in postmenopausal women when compared with premenopausal women.12 In the 1980s, Sullivan hypothesized that the reason for higher incidence of CAD in men when compared with premenopausal women was due to their higher body iron storage.13-16 A study of 1,930 Finnish men reported that the men with ferritin level ≥ 200 ng/dL had a risk 2.2 times higher of acute myocardial infarction when compared to men with lower serum ferritin level.17
A prospective study published in 1997 by Klechl showed the role of iron stores in early atherogenesis via promotion of lipid oxidation.18 Other epidemiological studies have shown a decreased risk of myocardial infarction in blood donors, and while arguments have been made that the blood donors tend to be healthier individuals, 2 studies were published in 1997 matching healthy blood donors to healthy nonblood donors, and both showed a lower risk of CVD in the donors when compared with nondonors.19,20 Furthermore, in an animal model of atherosclerosis, an iron depleted diet showed a reduction of atherosclerosis progression.21 Multiple studies have shown that the heterozygosity for HFE is significantly linked to the risk of cardiovascular events, including the fact that heterozygosity for C282Y has been shown to be a risk factor for myocardial infarction in men and cerebrovascular death in women.22-25
Conclusion
Multiple studies have shown an association between the elevated iron levels associated with the HFE genotype and the disease states of our patient. These include an increased risk of CAD, the increased risk of cirrhosis in HCV and the development of PCT. Indeed, in this case, our patient likely acquired PCT from the combined risks of HCV and his heterozygous HFE genetic mutation.
With regard to Mr. M’s treatment, the use of an antiviral agent in the treatment of his HCV is fundamental, along with avoidance of alcohol and smoking. If he were to accept HCV treatment, we would anticipate resolution of the PCT, but the ongoing progression of his liver and cardiovascular conditions, due perhaps in part, to relative iron overload from his heterozygous HFE mutation. In this situation, we expect that an ongoing course of therapeutic phlebotomy could help to delay the progression of his chronic liver and cardiovascular diseases.
1. Singal AK. Porphyria cutanea tarda: Recent update. Mol Genet Metab. 2019;128(3):271-281.
2. Ryan Caballes F, Sendi H, Bonkovsky HL. Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int. 2012;32(6):880-893.
3. Wiznia LE, Laird ME, Franks AG Jr. Hepatitis C virus and its cutaneous manifestations: treatment in the direct-acting antiviral era. J Eur Acad Dermatol Venereol. 2017;31(8):1260-1270.
4. Nihei T, Kiniwa Y, Mikoshiba Y, Joshita S, Okuyama R. Improvement of porphyria cutanea tarda following treatment of hepatitis C virus by direct-acting antivirals: a case report. J Dermatol. 2019;46(5):e149-e151.
5. Combalia A, To-Figueras J, Laguno M, Martínez-Rebollar M, Aguilera P. Direct-acting antivirals for hepatitis C virus induce a rapid clinical and biochemical remission of porphyria cutanea tarda. Br J Dermatol. 2017;177(5):e183-e184. 6. Singal AK, Venkata KVR, Jampana S, Islam FU, Anderson KE. Hepatitis C treatment in patients with porphyria cutanea tarda. Am J Med Sci. 2017;353 (6):523-528.
7. Brandhagen DJ, Fairbanks VF, Baldus W. Recognition and management of hereditary hemochromatosis. Am Fam Physician. 2002;65(5):853-860.
8. Aguilar-Martinez P, Grandchamp B, Cunat S, et al. Iron overload in HFE C282Y heterozygotes at first genetic testing: a strategy for identifying rare HFE variants. Haematologica. 2011;96(4):507-514.
9. Erhardt A, Maschner-Olberg A, Mellenthin C, et al. HFE mutations and chronic hepatitis C: H63D and C282Y heterozygosity are independent risk factors for liver fibrosis and cirrhosis. J Hepatol. 2003;38(3):335-342.
10. Mehrany K, Drage LA, Brandhagen DJ, Pittelkow MR. Association of porphyria cutanea tarda with hereditary hemochromatosis. J Am Acad Dermatol. 2004;51(2):205-211.
11. Pietrangelo A. Hemochromatosis gene modifies course of hepatitis C viral infection. Gastroenterology. 2003;124(5):1509-1523.
12. Gordon T, Kannel WB, Hjortland MC, McNamara PM. Menopause and coronary heart disease. The Framingham Study. Ann Intern Med. 1978;89(2):157-161.
13. Sullivan JL. Iron and the sex difference in heart disease risk. Lancet. 1981;1(8233):1293-1294.
14. Sullivan JL. The sex difference in ischemic heart disease. Perspect Biol Med. 1983;26(4):657-671.
15. Sullivan JL. The iron paradigm of ischemic heart disease. Am Heart J. 1989;117(5):1177-1188.
16. Sullivan JL. Stored iron and ischemic heart disease: empirical support for a new paradigm. Circulation. 1992;86(3):1036-1037.
17. Salonen JT, Nyyssönen K, Korpela H, Tuomilehto J, Seppänen R, Salonen R. High stored iron levels are associated with excess risk of myocardial infarction in eastern Finnish men. Circulation. 1992;86(3):803-811.
18. Kiechl S, Willeit J, Egger G, Poewe W, Oberhollenzer F. Body iron stores and the risk of carotid atherosclerosis: prospective results from the Bruneck study. Circulation. 1997;96(10):3300-3307.
19. Tuomainen TP, Salonen R, Nyyssönen K, Salonen JT. Cohort study of relation between donating blood and risk of myocardial infarction in 2682 men in eastern Finland. BMJ. 1997;314(7083):793-794.
20. Meyers DG, Strickland D, Maloley PA, Seburg JK, Wilson JE, McManus BF. Possible association of a reduction in cardiovascular events with blood donation. Heart. 1997;78(2):188-193.
21. Lee TS, Shiao MS, Pan CC, Chau LY. Iron-deficient diet reduces atherosclerotic lesions in apoE-deficient mice. Circulation. 1999;99(9):1222-1229.
22. Surber R, Sigusch HH, Kuehnert H, Figulla HR. Haemochromatosis (HFE) gene C282Y mutation and the risk of coronary artery disease and myocardial infarction: a study in 1279 patients undergoing coronary angiography. J Med Genet. 2003;40(5):e58.
23. Tuomainen TP, Kontula K, Nyyssönen K, Lakka TA, Heliö T, Salonen JT. Increased risk of acute myocardial infarction in carriers of the hemochromatosis gene Cys282Tyr mutation: a prospective cohort study in men in eastern Finland. Circulation. 1999;100(12):1274-1279.
24. Roest M, van der Schouw YT, de Valk B, et al. Heterozygosity for a hereditary hemochromatosis gene is associated with cardiovascular death in women. Circulation. 1999;100(12):1268-1273.
25. Pourmoghaddas A, Sanei H, Garakyaraghi M, Esteki-Ghashghaei F, Gharaati M. The relation between body iron store and ferritin, and coronary artery disease. ARYA Atheroscler. 2014;10(1):32-36.
1. Singal AK. Porphyria cutanea tarda: Recent update. Mol Genet Metab. 2019;128(3):271-281.
2. Ryan Caballes F, Sendi H, Bonkovsky HL. Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int. 2012;32(6):880-893.
3. Wiznia LE, Laird ME, Franks AG Jr. Hepatitis C virus and its cutaneous manifestations: treatment in the direct-acting antiviral era. J Eur Acad Dermatol Venereol. 2017;31(8):1260-1270.
4. Nihei T, Kiniwa Y, Mikoshiba Y, Joshita S, Okuyama R. Improvement of porphyria cutanea tarda following treatment of hepatitis C virus by direct-acting antivirals: a case report. J Dermatol. 2019;46(5):e149-e151.
5. Combalia A, To-Figueras J, Laguno M, Martínez-Rebollar M, Aguilera P. Direct-acting antivirals for hepatitis C virus induce a rapid clinical and biochemical remission of porphyria cutanea tarda. Br J Dermatol. 2017;177(5):e183-e184. 6. Singal AK, Venkata KVR, Jampana S, Islam FU, Anderson KE. Hepatitis C treatment in patients with porphyria cutanea tarda. Am J Med Sci. 2017;353 (6):523-528.
7. Brandhagen DJ, Fairbanks VF, Baldus W. Recognition and management of hereditary hemochromatosis. Am Fam Physician. 2002;65(5):853-860.
8. Aguilar-Martinez P, Grandchamp B, Cunat S, et al. Iron overload in HFE C282Y heterozygotes at first genetic testing: a strategy for identifying rare HFE variants. Haematologica. 2011;96(4):507-514.
9. Erhardt A, Maschner-Olberg A, Mellenthin C, et al. HFE mutations and chronic hepatitis C: H63D and C282Y heterozygosity are independent risk factors for liver fibrosis and cirrhosis. J Hepatol. 2003;38(3):335-342.
10. Mehrany K, Drage LA, Brandhagen DJ, Pittelkow MR. Association of porphyria cutanea tarda with hereditary hemochromatosis. J Am Acad Dermatol. 2004;51(2):205-211.
11. Pietrangelo A. Hemochromatosis gene modifies course of hepatitis C viral infection. Gastroenterology. 2003;124(5):1509-1523.
12. Gordon T, Kannel WB, Hjortland MC, McNamara PM. Menopause and coronary heart disease. The Framingham Study. Ann Intern Med. 1978;89(2):157-161.
13. Sullivan JL. Iron and the sex difference in heart disease risk. Lancet. 1981;1(8233):1293-1294.
14. Sullivan JL. The sex difference in ischemic heart disease. Perspect Biol Med. 1983;26(4):657-671.
15. Sullivan JL. The iron paradigm of ischemic heart disease. Am Heart J. 1989;117(5):1177-1188.
16. Sullivan JL. Stored iron and ischemic heart disease: empirical support for a new paradigm. Circulation. 1992;86(3):1036-1037.
17. Salonen JT, Nyyssönen K, Korpela H, Tuomilehto J, Seppänen R, Salonen R. High stored iron levels are associated with excess risk of myocardial infarction in eastern Finnish men. Circulation. 1992;86(3):803-811.
18. Kiechl S, Willeit J, Egger G, Poewe W, Oberhollenzer F. Body iron stores and the risk of carotid atherosclerosis: prospective results from the Bruneck study. Circulation. 1997;96(10):3300-3307.
19. Tuomainen TP, Salonen R, Nyyssönen K, Salonen JT. Cohort study of relation between donating blood and risk of myocardial infarction in 2682 men in eastern Finland. BMJ. 1997;314(7083):793-794.
20. Meyers DG, Strickland D, Maloley PA, Seburg JK, Wilson JE, McManus BF. Possible association of a reduction in cardiovascular events with blood donation. Heart. 1997;78(2):188-193.
21. Lee TS, Shiao MS, Pan CC, Chau LY. Iron-deficient diet reduces atherosclerotic lesions in apoE-deficient mice. Circulation. 1999;99(9):1222-1229.
22. Surber R, Sigusch HH, Kuehnert H, Figulla HR. Haemochromatosis (HFE) gene C282Y mutation and the risk of coronary artery disease and myocardial infarction: a study in 1279 patients undergoing coronary angiography. J Med Genet. 2003;40(5):e58.
23. Tuomainen TP, Kontula K, Nyyssönen K, Lakka TA, Heliö T, Salonen JT. Increased risk of acute myocardial infarction in carriers of the hemochromatosis gene Cys282Tyr mutation: a prospective cohort study in men in eastern Finland. Circulation. 1999;100(12):1274-1279.
24. Roest M, van der Schouw YT, de Valk B, et al. Heterozygosity for a hereditary hemochromatosis gene is associated with cardiovascular death in women. Circulation. 1999;100(12):1268-1273.
25. Pourmoghaddas A, Sanei H, Garakyaraghi M, Esteki-Ghashghaei F, Gharaati M. The relation between body iron store and ferritin, and coronary artery disease. ARYA Atheroscler. 2014;10(1):32-36.
Incidentally Discovered Ochronosis Explaining Decades of Chronic Pain
Alkaptonuria is a rare autosomal recessive disorder uniquely known for causing black, or darkened, urine when left standing due to the renal excretion of excess homogentisic acid (HGA). When this disorder goes undiagnosed, as demonstrated in this case, patients experience its many complications without a unifying explanation. The disorder has 3 clinical stages that occur in a predictable order: clinical silence, clinical ochronosis, and ochronotic arthropathy. These stages lead to multiple musculoskeletal, cardiovascular (CV), and renal complications that can be mitigated with management focused on decreasing homogentisic acid buildup, alleviating symptoms, and close monitoring for these complications.
Case Presentation
A 61-year-old African American male with a medical history of multiple traumatic fractures, right Achilles tendon injury, early-onset multijoint osteoarthritis, chronic low back pain, and recurrent nephrolithiasis presented to the emergency department with sudden onset of sharp left ankle pain while moving furniture. His physical exam revealed a positive Thompson test—lack of foot plantar flexion with calf squeeze—and a subsequent magnetic resonance image (MRI) showed evidence of an acute Achilles tendon rupture.
Given these findings the patient was treated with nonsteroidal anti-inflammatory drugs (NSAIDs) and rest to allow for resolution of swelling and inflammation, followed by elective surgery a month later to repair the ruptured tendon. An operative report following his surgery described “black ends to the area where the Achilles was ruptured…and tendinopathy of the flexor hallucis longus with blackening of the flexor.”
A more in-depth patient history revealed that he underwent multiple invasive and noninvasive interventions for his chronic low back and joint pain with medical management of a prior right Achilles tendon injury. His medical history also included multiple nonspecific diagnoses, such as premature atherosclerosis (diagnosed in his third decade), severe lumbar degenerative disc disease, several tendonopathies and cartilage injuries (Figure 1), pseudogout (following calcium pyrophosphate dehydrate crystals found from a left knee aspirate), and chronic pain syndrome. Along this diagnostic journey, he had several health care providers (HCPs) in rheumatology, orthopedic surgery, pain management, and podiatry who offered a range of symptom management options, including physical therapy, NSAIDs, opioid agonists, tricyclic antidepressants, gabapentin, colchicine, and epidural steroid injections, all of which provided little or no relief of his pain. The patient reported that he told a HCP, “I’ll just live with [the pain].”

At the postsurgery follow-up, the patient reported that he had noticed dark urine and dark spots on his ears in the past. He also recounted that chronic joint pain was common in his family, with both his mother and brother receiving bilateral total knee replacements. Taking into consideration the surgical report and this new history, a urine assessment for HGA was ordered and yielded a diagnosis of alkaptonuria.
He later suffered an acute myocardial infarction leading to an incidental discovery of severe aortic stenosis on echocardiography, requiring coronary stent placements and transcatheter aortic valve replacement, respectively. He reported that with CV interventions and joint replacement surgeries, including bilateral knees and hips, his symptoms and quality of life began to significantly improve. However, he continued to have diffuse chronic joint pain unimproved with any single agent or intervention.
Discussion
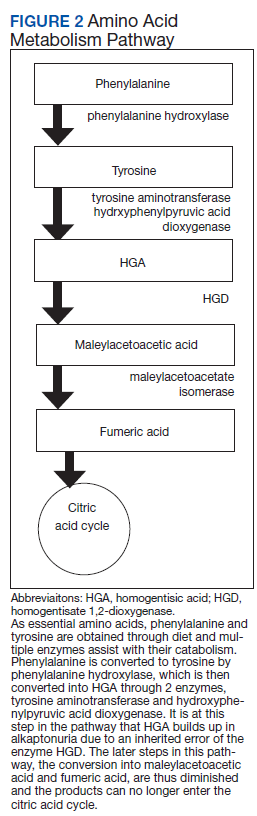
Alkaptonuria is a rare autosomal recessive disorder, with a prevalence of about 1 in 100,000 to 250,000, which results from an enzyme error in an essential amino acid metabolism pathway (Figure 2).1 This inheritable gene mutation leads to ineffective homogentisate 1,2-dioxygenase (HGD), an enzyme required to break down HGA—which is a product of phenylalanine and tyrosine metabolism.2 As these patients engage in normal dietary protein intake, which includes essential amino acid phenylalanine, they develop clinically evident manifestations of the buildup and deposition of HGA.
The rarity of alkaptonuria combined with the gradual buildup of HGA makes it difficult to diagnose. A common diagnostic technique is the visualization of discolored cartilage during surgical procedures, especially when discoloration in urine or skin is not immediately evident. A few case reports have noted surgical diagnosis of black or darkening tissue, known as ochronosis, following tendon rupture—a common complication of this disorder.3-5 Additional intervention-related case reports linked to the discovery of ochronosis include aortic valve replacement, lumbar discectomy, and bronchoscopy.6-9 Cases like these illustrate the complex, disabling, and unclear nature of this disorder when not diagnosed early in life.
The patient in this case communicated via e-mail about his tendon repair surgery. “Something very interesting was found during the surgery,” the patient explained. “I was diagnosed with the disease called ochronosis. I don’t know much about this disease but I am beginning to know why some of the things are happening to me and why I am always in constant pain.” This was the first recognized clue toward a diagnosis of alkaptonuria.
Pathophysiology
The pathophysiology of alkaptonuria is based on the extensive deposition of HGA throughout the body. Its progression is based on 3 clinical stages: clinical silence, clinical ochronosis, and ochronotic arthropathy.1 In the first stage the disorder is asymptomatic but includes its most notable feature—the gradual darkening of urine when exposed to air through oxidation of the renally excreted HGA. A similar process occurs in the blood through formed HGA-melanin compounds, which cause discoloration in cartilage.1 This internal metabolic disruption accounts for the disorder’s eventual second stage, clinical ochronosis, usually with an onset in the second or third decade. Prominent features noted on physical examination primarily include discoloration of ear pinnae and eye sclera but can involve the nose, gums, teeth, and hands. The third, final, and symptomatic stage, ochronotic arthropathy, occurs by the patient’s fourth to fifth decade and presents as joint pain, usually starting with the vertebrae and larger joints like hips, knees, and shoulders, that can appear as advanced early osteoarthritis on imaging.
Treatment
This clinical manifestation of alkaptonuria requires that HCPs manage patients with 3 strategies: decrease HGA buildup, alleviate symptoms, and monitor for disorder complications. Decreasing HGA buildup is a difficult aspect of management given the natural physiology of protein intake and metabolism. Three approaches to limit HGA buildup incorporate decreasing protein intake, inhibiting enzyme production of HGA, and increasing HGA excretion. Phenylalanine is an essential amino acid—meaning its levels are dependent on dietary protein intake. Patients should be advised to adhere to a low protein diet, especially phenylalanine and tyrosine, to lessen HGA concentrations.
Manipulating the metabolic pathway of phenylalanine with medication is a second option. An example of this is nitisinone, a US Food and Drug Administration-approved medication for treatment of tyrosinemia. It acts by inhibiting hydroxyphenylpyruvic acid dioxygenase, one of the enzymes that converts tyrosine into HGA, to prevent the buildup of damaging tyrosine byproducts. At low doses it has been effective in decreasing HGA concentrations in alkaptonuria and tyrosinemia.10,11 Due to this mechanism of action, nitisinone directly causes increased tyrosine levels. Therefore, tyrosine toxicity, usually not predicted by tyrosine levels, has been associated with eye-related adverse effects (AEs), including keratopathy, diminished visual acuity, and corneal tissue damage.1,2,10 Incidence of these AEs have not been clearly documented, but routine monitoring should include patient education on ocular symptoms and slit-lamp examinations.12
In addition, case reports have shown that high-dose ascorbic acid (vitamin C) promotes HGA, tyrosine, and phenylalanine excretion in urine, which may slow the progression of alkaptonuria, but clinical effect has not been proven.13 Additionally, high vitamin C intake is considered a risk factor for nephrolithiasis, which must be balanced with the increased risk of stone formation from HGA excretion.14 These dietary and medical options can be considered, especially in the setting of severe symptoms or complications, but the risks must be discussed with patients.
A second and commonly utilized strategy for caring for these patients is symptom management. As demonstrated through this case report, there is no clear medication that adequately addresses the pain caused by HGA deposition. Patients should be referred to a pain specialist to allow for single provider prescribing of pain medications. This patient found most relief and least AEs with tramadol but eventually self-discontinued due to diminishing pain relief. Given the eventual involvement of large joints, these patients will often require further symptom management with joint replacement surgery, usually much earlier than patients who undergo these surgeries for age-related osteoarthritis. The imperative aspect of symptom management is to engage patients in shared decision making with clear expectation setting.
Given the progressive nature of alkaptonuria, providers must monitor and address complications that are a result of this disorder. HGA becomes pathologic by binding to and weakening collagen fibers.5 This gradual buildup leads to degenerative changes in weight-bearing lower vertebrae and large joints that can become severe. Due to HGA’s interaction with collagen fibers, tendon involvement leading to inflammation, calcification, and rupture can result as patients enter the third stage, ochronotic arthropathy, of the disorder (Figure 3).15 Many of these arthropathies will require medical and surgical management and can be urgent in situations like tendon ruptures and meniscal tears. Understanding the pathophysiology of tendinopathies in patients with alkaptonuria also can aid orthopedic surgeons during the postoperative period where patients may be at risk for poor healing.5
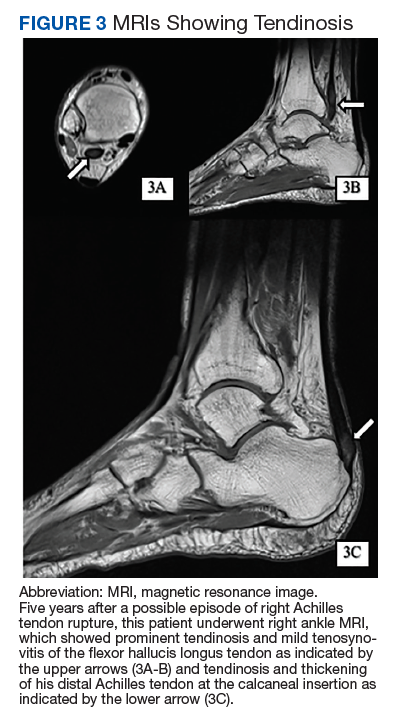
A second area of complications includes CV involvement. This patient was diagnosed with premature atherosclerosis and underwent cardiac interventions, including coronary stent placement and valve replacements at age 63 years. This early cardiac involvement was likely due in part to the deposition of HGA and collagen injury in CV tissue leading to damage of the endocardium, aortic intima, heart valves, and coronary arteries.1 HCPs should monitor for these manifestations with regular visits, chest computed tomography, and echocardiographic studies.2
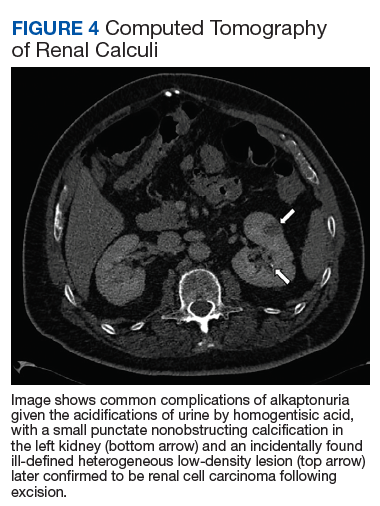
The most classic aspect of this rare disorder is urine darkening due to the renal excretion of HGA and comprises the third area of complications. This process leads to chronically acidic urine—every urinalysis in this patient’s chart displayed the lowest pH measurable—and an increased risk for calcification and precipitation of solutes within the kidney and urinary tract (Figure 4). Both X-ray and ultrasound imaging should be used to identify kidney and prostate stones in the setting of abdominal or genitourinary pain or infection. Patients with diminished renal function may manifest a more severe and rapidly progressing form of alkaptonuria that exacerbates symptoms and complications, but direct damage to the kidneys by HGA is not evident.
Conclusion
Alkaptonuria is a rare autosomal recessive metabolic disorder that has a progressively debilitating pathophysiologic course spanning decades of a patient’s life. Its low prevalence and gradually progressive nature make it a difficult diagnosis to make without clinical suspicion. In patients with early-onset degenerative joint disease, tendinopathy, and cartilage or skin discoloration, congenital metabolic disorders like alkaptonuria should be considered.
As this case shows, suspicion and diagnosis can occur during surgical intervention in which tendon discoloration is directly visualized, especially in patients without prominent skin or cartilage discoloration. Once the diagnosis is made through elevated levels of urine HGA, there are 3 management strategies, including decreasing homogentisic acid buildup, providing symptom management, and monitoring for arthropathic, CV, and genitourinary complications.
1. Aquaron R. Alkaptonuria: a very rare metabolic disorder. Indian J Biochem Biophys. 2013;50(5):339-344.
2. Phornphutkul C, Introne WJ, Perry MB, et al. Natural history of alkaptonuria. N Engl J Med. 2002;347(26):2111-2121.
3. Alajoulin OA, Alsbou MS, Ja’afreh SO, Kalbouneh HM. Spontaneous Achilles tendon rupture in alkaptonuria. Saudi Med J. 2015;36(12):1486-1489.
4. Manoj Kumar RV, Rajasekaran S. Spontaneous tendon ruptures in alkaptonuria. J Bone Joint Surg Br. 2003;85(6):883-886.
5. Tanoglu O, Arican G, Ozmeric A, Alemdaroglu KB, Caydere M. Calcaneal avulsion of an ochronotic Achilles tendon: a case report. J Foot Ankle Surg. 2018;57(1):179-183.
6. Schuuring MJ, Delemarre B, Keyhan-Falsafi AM, van der Bilt IA. Mending a darkened heart: alkaptonuria discovered during aortic valve replacement. Circulation. 2016;133(12):e444-445.
7. Hiroyoshi J, Saito A, Panthee N, et al. Aortic valve replacement for aortic stenosis caused by alkaptonuria. Ann Thorac Surg. 2013;95(3):1076-1079.
8. Parambil JG, Daniels CE, Zehr KJ, Utz JP. Alkaptonuria diagnosed by flexible bronchoscopy. Chest. 2005;128(5):3678-3680.
9. Farzannia A, Shokouhi G, Hadidchi S. Alkaptonuria and lumbar disc herniation. Report of three cases. J Neurosurg. 2003;98(suppl 1):87-89.
10. Introne WJ, Perry MB, Troendle J, et al. A 3-year randomized therapeutic trial of nitisinone in alkaptonuria. Mol Genet Metab. 2011;103(4):307-314.
11. Gissen P, Preece MA, Willshaw HA, McKiernan PJ. Ophthalmic follow-up of patients with tyrosinaemia type I on NTBC. J Inherit Metab Dis. 2003;26(1):13-16.
12. Khedr M, Judd S, Briggs MC, et al. Asymptomatic corneal keratopathy secondary to hypertyrosinaemia following low dose nitisinone and a literature review of tyrosine keratopathy in alkaptonuria. JIMD Rep. 2018;40:31-37.
13. Wolff JA, Barshop B, Nyhan WL, et al. Effects of ascorbic acid in alkaptonuria: alterations in benzoquinone acetic acid and an ontogenic effect in infancy. Pediatr Res. 1989;26(2):140-144.
14. Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. J Am Soc Nephrol. 2004;15(12):3225-3232.
15. Abate M, Salini V, Andia I. Tendons involvement in congenital metabolic disorders. Adv Exp Med Biol. 2016;920:117-122.
Alkaptonuria is a rare autosomal recessive disorder uniquely known for causing black, or darkened, urine when left standing due to the renal excretion of excess homogentisic acid (HGA). When this disorder goes undiagnosed, as demonstrated in this case, patients experience its many complications without a unifying explanation. The disorder has 3 clinical stages that occur in a predictable order: clinical silence, clinical ochronosis, and ochronotic arthropathy. These stages lead to multiple musculoskeletal, cardiovascular (CV), and renal complications that can be mitigated with management focused on decreasing homogentisic acid buildup, alleviating symptoms, and close monitoring for these complications.
Case Presentation
A 61-year-old African American male with a medical history of multiple traumatic fractures, right Achilles tendon injury, early-onset multijoint osteoarthritis, chronic low back pain, and recurrent nephrolithiasis presented to the emergency department with sudden onset of sharp left ankle pain while moving furniture. His physical exam revealed a positive Thompson test—lack of foot plantar flexion with calf squeeze—and a subsequent magnetic resonance image (MRI) showed evidence of an acute Achilles tendon rupture.
Given these findings the patient was treated with nonsteroidal anti-inflammatory drugs (NSAIDs) and rest to allow for resolution of swelling and inflammation, followed by elective surgery a month later to repair the ruptured tendon. An operative report following his surgery described “black ends to the area where the Achilles was ruptured…and tendinopathy of the flexor hallucis longus with blackening of the flexor.”
A more in-depth patient history revealed that he underwent multiple invasive and noninvasive interventions for his chronic low back and joint pain with medical management of a prior right Achilles tendon injury. His medical history also included multiple nonspecific diagnoses, such as premature atherosclerosis (diagnosed in his third decade), severe lumbar degenerative disc disease, several tendonopathies and cartilage injuries (Figure 1), pseudogout (following calcium pyrophosphate dehydrate crystals found from a left knee aspirate), and chronic pain syndrome. Along this diagnostic journey, he had several health care providers (HCPs) in rheumatology, orthopedic surgery, pain management, and podiatry who offered a range of symptom management options, including physical therapy, NSAIDs, opioid agonists, tricyclic antidepressants, gabapentin, colchicine, and epidural steroid injections, all of which provided little or no relief of his pain. The patient reported that he told a HCP, “I’ll just live with [the pain].”
At the postsurgery follow-up, the patient reported that he had noticed dark urine and dark spots on his ears in the past. He also recounted that chronic joint pain was common in his family, with both his mother and brother receiving bilateral total knee replacements. Taking into consideration the surgical report and this new history, a urine assessment for HGA was ordered and yielded a diagnosis of alkaptonuria.
He later suffered an acute myocardial infarction leading to an incidental discovery of severe aortic stenosis on echocardiography, requiring coronary stent placements and transcatheter aortic valve replacement, respectively. He reported that with CV interventions and joint replacement surgeries, including bilateral knees and hips, his symptoms and quality of life began to significantly improve. However, he continued to have diffuse chronic joint pain unimproved with any single agent or intervention.
Discussion
Alkaptonuria is a rare autosomal recessive disorder, with a prevalence of about 1 in 100,000 to 250,000, which results from an enzyme error in an essential amino acid metabolism pathway (Figure 2).1 This inheritable gene mutation leads to ineffective homogentisate 1,2-dioxygenase (HGD), an enzyme required to break down HGA—which is a product of phenylalanine and tyrosine metabolism.2 As these patients engage in normal dietary protein intake, which includes essential amino acid phenylalanine, they develop clinically evident manifestations of the buildup and deposition of HGA.
The rarity of alkaptonuria combined with the gradual buildup of HGA makes it difficult to diagnose. A common diagnostic technique is the visualization of discolored cartilage during surgical procedures, especially when discoloration in urine or skin is not immediately evident. A few case reports have noted surgical diagnosis of black or darkening tissue, known as ochronosis, following tendon rupture—a common complication of this disorder.3-5 Additional intervention-related case reports linked to the discovery of ochronosis include aortic valve replacement, lumbar discectomy, and bronchoscopy.6-9 Cases like these illustrate the complex, disabling, and unclear nature of this disorder when not diagnosed early in life.
The patient in this case communicated via e-mail about his tendon repair surgery. “Something very interesting was found during the surgery,” the patient explained. “I was diagnosed with the disease called ochronosis. I don’t know much about this disease but I am beginning to know why some of the things are happening to me and why I am always in constant pain.” This was the first recognized clue toward a diagnosis of alkaptonuria.
Pathophysiology
The pathophysiology of alkaptonuria is based on the extensive deposition of HGA throughout the body. Its progression is based on 3 clinical stages: clinical silence, clinical ochronosis, and ochronotic arthropathy.1 In the first stage the disorder is asymptomatic but includes its most notable feature—the gradual darkening of urine when exposed to air through oxidation of the renally excreted HGA. A similar process occurs in the blood through formed HGA-melanin compounds, which cause discoloration in cartilage.1 This internal metabolic disruption accounts for the disorder’s eventual second stage, clinical ochronosis, usually with an onset in the second or third decade. Prominent features noted on physical examination primarily include discoloration of ear pinnae and eye sclera but can involve the nose, gums, teeth, and hands. The third, final, and symptomatic stage, ochronotic arthropathy, occurs by the patient’s fourth to fifth decade and presents as joint pain, usually starting with the vertebrae and larger joints like hips, knees, and shoulders, that can appear as advanced early osteoarthritis on imaging.
Treatment
This clinical manifestation of alkaptonuria requires that HCPs manage patients with 3 strategies: decrease HGA buildup, alleviate symptoms, and monitor for disorder complications. Decreasing HGA buildup is a difficult aspect of management given the natural physiology of protein intake and metabolism. Three approaches to limit HGA buildup incorporate decreasing protein intake, inhibiting enzyme production of HGA, and increasing HGA excretion. Phenylalanine is an essential amino acid—meaning its levels are dependent on dietary protein intake. Patients should be advised to adhere to a low protein diet, especially phenylalanine and tyrosine, to lessen HGA concentrations.
Manipulating the metabolic pathway of phenylalanine with medication is a second option. An example of this is nitisinone, a US Food and Drug Administration-approved medication for treatment of tyrosinemia. It acts by inhibiting hydroxyphenylpyruvic acid dioxygenase, one of the enzymes that converts tyrosine into HGA, to prevent the buildup of damaging tyrosine byproducts. At low doses it has been effective in decreasing HGA concentrations in alkaptonuria and tyrosinemia.10,11 Due to this mechanism of action, nitisinone directly causes increased tyrosine levels. Therefore, tyrosine toxicity, usually not predicted by tyrosine levels, has been associated with eye-related adverse effects (AEs), including keratopathy, diminished visual acuity, and corneal tissue damage.1,2,10 Incidence of these AEs have not been clearly documented, but routine monitoring should include patient education on ocular symptoms and slit-lamp examinations.12
In addition, case reports have shown that high-dose ascorbic acid (vitamin C) promotes HGA, tyrosine, and phenylalanine excretion in urine, which may slow the progression of alkaptonuria, but clinical effect has not been proven.13 Additionally, high vitamin C intake is considered a risk factor for nephrolithiasis, which must be balanced with the increased risk of stone formation from HGA excretion.14 These dietary and medical options can be considered, especially in the setting of severe symptoms or complications, but the risks must be discussed with patients.
A second and commonly utilized strategy for caring for these patients is symptom management. As demonstrated through this case report, there is no clear medication that adequately addresses the pain caused by HGA deposition. Patients should be referred to a pain specialist to allow for single provider prescribing of pain medications. This patient found most relief and least AEs with tramadol but eventually self-discontinued due to diminishing pain relief. Given the eventual involvement of large joints, these patients will often require further symptom management with joint replacement surgery, usually much earlier than patients who undergo these surgeries for age-related osteoarthritis. The imperative aspect of symptom management is to engage patients in shared decision making with clear expectation setting.
Given the progressive nature of alkaptonuria, providers must monitor and address complications that are a result of this disorder. HGA becomes pathologic by binding to and weakening collagen fibers.5 This gradual buildup leads to degenerative changes in weight-bearing lower vertebrae and large joints that can become severe. Due to HGA’s interaction with collagen fibers, tendon involvement leading to inflammation, calcification, and rupture can result as patients enter the third stage, ochronotic arthropathy, of the disorder (Figure 3).15 Many of these arthropathies will require medical and surgical management and can be urgent in situations like tendon ruptures and meniscal tears. Understanding the pathophysiology of tendinopathies in patients with alkaptonuria also can aid orthopedic surgeons during the postoperative period where patients may be at risk for poor healing.5
A second area of complications includes CV involvement. This patient was diagnosed with premature atherosclerosis and underwent cardiac interventions, including coronary stent placement and valve replacements at age 63 years. This early cardiac involvement was likely due in part to the deposition of HGA and collagen injury in CV tissue leading to damage of the endocardium, aortic intima, heart valves, and coronary arteries.1 HCPs should monitor for these manifestations with regular visits, chest computed tomography, and echocardiographic studies.2
The most classic aspect of this rare disorder is urine darkening due to the renal excretion of HGA and comprises the third area of complications. This process leads to chronically acidic urine—every urinalysis in this patient’s chart displayed the lowest pH measurable—and an increased risk for calcification and precipitation of solutes within the kidney and urinary tract (Figure 4). Both X-ray and ultrasound imaging should be used to identify kidney and prostate stones in the setting of abdominal or genitourinary pain or infection. Patients with diminished renal function may manifest a more severe and rapidly progressing form of alkaptonuria that exacerbates symptoms and complications, but direct damage to the kidneys by HGA is not evident.
Conclusion
Alkaptonuria is a rare autosomal recessive metabolic disorder that has a progressively debilitating pathophysiologic course spanning decades of a patient’s life. Its low prevalence and gradually progressive nature make it a difficult diagnosis to make without clinical suspicion. In patients with early-onset degenerative joint disease, tendinopathy, and cartilage or skin discoloration, congenital metabolic disorders like alkaptonuria should be considered.
As this case shows, suspicion and diagnosis can occur during surgical intervention in which tendon discoloration is directly visualized, especially in patients without prominent skin or cartilage discoloration. Once the diagnosis is made through elevated levels of urine HGA, there are 3 management strategies, including decreasing homogentisic acid buildup, providing symptom management, and monitoring for arthropathic, CV, and genitourinary complications.
Alkaptonuria is a rare autosomal recessive disorder uniquely known for causing black, or darkened, urine when left standing due to the renal excretion of excess homogentisic acid (HGA). When this disorder goes undiagnosed, as demonstrated in this case, patients experience its many complications without a unifying explanation. The disorder has 3 clinical stages that occur in a predictable order: clinical silence, clinical ochronosis, and ochronotic arthropathy. These stages lead to multiple musculoskeletal, cardiovascular (CV), and renal complications that can be mitigated with management focused on decreasing homogentisic acid buildup, alleviating symptoms, and close monitoring for these complications.
Case Presentation
A 61-year-old African American male with a medical history of multiple traumatic fractures, right Achilles tendon injury, early-onset multijoint osteoarthritis, chronic low back pain, and recurrent nephrolithiasis presented to the emergency department with sudden onset of sharp left ankle pain while moving furniture. His physical exam revealed a positive Thompson test—lack of foot plantar flexion with calf squeeze—and a subsequent magnetic resonance image (MRI) showed evidence of an acute Achilles tendon rupture.
Given these findings the patient was treated with nonsteroidal anti-inflammatory drugs (NSAIDs) and rest to allow for resolution of swelling and inflammation, followed by elective surgery a month later to repair the ruptured tendon. An operative report following his surgery described “black ends to the area where the Achilles was ruptured…and tendinopathy of the flexor hallucis longus with blackening of the flexor.”
A more in-depth patient history revealed that he underwent multiple invasive and noninvasive interventions for his chronic low back and joint pain with medical management of a prior right Achilles tendon injury. His medical history also included multiple nonspecific diagnoses, such as premature atherosclerosis (diagnosed in his third decade), severe lumbar degenerative disc disease, several tendonopathies and cartilage injuries (Figure 1), pseudogout (following calcium pyrophosphate dehydrate crystals found from a left knee aspirate), and chronic pain syndrome. Along this diagnostic journey, he had several health care providers (HCPs) in rheumatology, orthopedic surgery, pain management, and podiatry who offered a range of symptom management options, including physical therapy, NSAIDs, opioid agonists, tricyclic antidepressants, gabapentin, colchicine, and epidural steroid injections, all of which provided little or no relief of his pain. The patient reported that he told a HCP, “I’ll just live with [the pain].”
At the postsurgery follow-up, the patient reported that he had noticed dark urine and dark spots on his ears in the past. He also recounted that chronic joint pain was common in his family, with both his mother and brother receiving bilateral total knee replacements. Taking into consideration the surgical report and this new history, a urine assessment for HGA was ordered and yielded a diagnosis of alkaptonuria.
He later suffered an acute myocardial infarction leading to an incidental discovery of severe aortic stenosis on echocardiography, requiring coronary stent placements and transcatheter aortic valve replacement, respectively. He reported that with CV interventions and joint replacement surgeries, including bilateral knees and hips, his symptoms and quality of life began to significantly improve. However, he continued to have diffuse chronic joint pain unimproved with any single agent or intervention.
Discussion
Alkaptonuria is a rare autosomal recessive disorder, with a prevalence of about 1 in 100,000 to 250,000, which results from an enzyme error in an essential amino acid metabolism pathway (Figure 2).1 This inheritable gene mutation leads to ineffective homogentisate 1,2-dioxygenase (HGD), an enzyme required to break down HGA—which is a product of phenylalanine and tyrosine metabolism.2 As these patients engage in normal dietary protein intake, which includes essential amino acid phenylalanine, they develop clinically evident manifestations of the buildup and deposition of HGA.
The rarity of alkaptonuria combined with the gradual buildup of HGA makes it difficult to diagnose. A common diagnostic technique is the visualization of discolored cartilage during surgical procedures, especially when discoloration in urine or skin is not immediately evident. A few case reports have noted surgical diagnosis of black or darkening tissue, known as ochronosis, following tendon rupture—a common complication of this disorder.3-5 Additional intervention-related case reports linked to the discovery of ochronosis include aortic valve replacement, lumbar discectomy, and bronchoscopy.6-9 Cases like these illustrate the complex, disabling, and unclear nature of this disorder when not diagnosed early in life.
The patient in this case communicated via e-mail about his tendon repair surgery. “Something very interesting was found during the surgery,” the patient explained. “I was diagnosed with the disease called ochronosis. I don’t know much about this disease but I am beginning to know why some of the things are happening to me and why I am always in constant pain.” This was the first recognized clue toward a diagnosis of alkaptonuria.
Pathophysiology
The pathophysiology of alkaptonuria is based on the extensive deposition of HGA throughout the body. Its progression is based on 3 clinical stages: clinical silence, clinical ochronosis, and ochronotic arthropathy.1 In the first stage the disorder is asymptomatic but includes its most notable feature—the gradual darkening of urine when exposed to air through oxidation of the renally excreted HGA. A similar process occurs in the blood through formed HGA-melanin compounds, which cause discoloration in cartilage.1 This internal metabolic disruption accounts for the disorder’s eventual second stage, clinical ochronosis, usually with an onset in the second or third decade. Prominent features noted on physical examination primarily include discoloration of ear pinnae and eye sclera but can involve the nose, gums, teeth, and hands. The third, final, and symptomatic stage, ochronotic arthropathy, occurs by the patient’s fourth to fifth decade and presents as joint pain, usually starting with the vertebrae and larger joints like hips, knees, and shoulders, that can appear as advanced early osteoarthritis on imaging.
Treatment
This clinical manifestation of alkaptonuria requires that HCPs manage patients with 3 strategies: decrease HGA buildup, alleviate symptoms, and monitor for disorder complications. Decreasing HGA buildup is a difficult aspect of management given the natural physiology of protein intake and metabolism. Three approaches to limit HGA buildup incorporate decreasing protein intake, inhibiting enzyme production of HGA, and increasing HGA excretion. Phenylalanine is an essential amino acid—meaning its levels are dependent on dietary protein intake. Patients should be advised to adhere to a low protein diet, especially phenylalanine and tyrosine, to lessen HGA concentrations.
Manipulating the metabolic pathway of phenylalanine with medication is a second option. An example of this is nitisinone, a US Food and Drug Administration-approved medication for treatment of tyrosinemia. It acts by inhibiting hydroxyphenylpyruvic acid dioxygenase, one of the enzymes that converts tyrosine into HGA, to prevent the buildup of damaging tyrosine byproducts. At low doses it has been effective in decreasing HGA concentrations in alkaptonuria and tyrosinemia.10,11 Due to this mechanism of action, nitisinone directly causes increased tyrosine levels. Therefore, tyrosine toxicity, usually not predicted by tyrosine levels, has been associated with eye-related adverse effects (AEs), including keratopathy, diminished visual acuity, and corneal tissue damage.1,2,10 Incidence of these AEs have not been clearly documented, but routine monitoring should include patient education on ocular symptoms and slit-lamp examinations.12
In addition, case reports have shown that high-dose ascorbic acid (vitamin C) promotes HGA, tyrosine, and phenylalanine excretion in urine, which may slow the progression of alkaptonuria, but clinical effect has not been proven.13 Additionally, high vitamin C intake is considered a risk factor for nephrolithiasis, which must be balanced with the increased risk of stone formation from HGA excretion.14 These dietary and medical options can be considered, especially in the setting of severe symptoms or complications, but the risks must be discussed with patients.
A second and commonly utilized strategy for caring for these patients is symptom management. As demonstrated through this case report, there is no clear medication that adequately addresses the pain caused by HGA deposition. Patients should be referred to a pain specialist to allow for single provider prescribing of pain medications. This patient found most relief and least AEs with tramadol but eventually self-discontinued due to diminishing pain relief. Given the eventual involvement of large joints, these patients will often require further symptom management with joint replacement surgery, usually much earlier than patients who undergo these surgeries for age-related osteoarthritis. The imperative aspect of symptom management is to engage patients in shared decision making with clear expectation setting.
Given the progressive nature of alkaptonuria, providers must monitor and address complications that are a result of this disorder. HGA becomes pathologic by binding to and weakening collagen fibers.5 This gradual buildup leads to degenerative changes in weight-bearing lower vertebrae and large joints that can become severe. Due to HGA’s interaction with collagen fibers, tendon involvement leading to inflammation, calcification, and rupture can result as patients enter the third stage, ochronotic arthropathy, of the disorder (Figure 3).15 Many of these arthropathies will require medical and surgical management and can be urgent in situations like tendon ruptures and meniscal tears. Understanding the pathophysiology of tendinopathies in patients with alkaptonuria also can aid orthopedic surgeons during the postoperative period where patients may be at risk for poor healing.5
A second area of complications includes CV involvement. This patient was diagnosed with premature atherosclerosis and underwent cardiac interventions, including coronary stent placement and valve replacements at age 63 years. This early cardiac involvement was likely due in part to the deposition of HGA and collagen injury in CV tissue leading to damage of the endocardium, aortic intima, heart valves, and coronary arteries.1 HCPs should monitor for these manifestations with regular visits, chest computed tomography, and echocardiographic studies.2
The most classic aspect of this rare disorder is urine darkening due to the renal excretion of HGA and comprises the third area of complications. This process leads to chronically acidic urine—every urinalysis in this patient’s chart displayed the lowest pH measurable—and an increased risk for calcification and precipitation of solutes within the kidney and urinary tract (Figure 4). Both X-ray and ultrasound imaging should be used to identify kidney and prostate stones in the setting of abdominal or genitourinary pain or infection. Patients with diminished renal function may manifest a more severe and rapidly progressing form of alkaptonuria that exacerbates symptoms and complications, but direct damage to the kidneys by HGA is not evident.
Conclusion
Alkaptonuria is a rare autosomal recessive metabolic disorder that has a progressively debilitating pathophysiologic course spanning decades of a patient’s life. Its low prevalence and gradually progressive nature make it a difficult diagnosis to make without clinical suspicion. In patients with early-onset degenerative joint disease, tendinopathy, and cartilage or skin discoloration, congenital metabolic disorders like alkaptonuria should be considered.
As this case shows, suspicion and diagnosis can occur during surgical intervention in which tendon discoloration is directly visualized, especially in patients without prominent skin or cartilage discoloration. Once the diagnosis is made through elevated levels of urine HGA, there are 3 management strategies, including decreasing homogentisic acid buildup, providing symptom management, and monitoring for arthropathic, CV, and genitourinary complications.
1. Aquaron R. Alkaptonuria: a very rare metabolic disorder. Indian J Biochem Biophys. 2013;50(5):339-344.
2. Phornphutkul C, Introne WJ, Perry MB, et al. Natural history of alkaptonuria. N Engl J Med. 2002;347(26):2111-2121.
3. Alajoulin OA, Alsbou MS, Ja’afreh SO, Kalbouneh HM. Spontaneous Achilles tendon rupture in alkaptonuria. Saudi Med J. 2015;36(12):1486-1489.
4. Manoj Kumar RV, Rajasekaran S. Spontaneous tendon ruptures in alkaptonuria. J Bone Joint Surg Br. 2003;85(6):883-886.
5. Tanoglu O, Arican G, Ozmeric A, Alemdaroglu KB, Caydere M. Calcaneal avulsion of an ochronotic Achilles tendon: a case report. J Foot Ankle Surg. 2018;57(1):179-183.
6. Schuuring MJ, Delemarre B, Keyhan-Falsafi AM, van der Bilt IA. Mending a darkened heart: alkaptonuria discovered during aortic valve replacement. Circulation. 2016;133(12):e444-445.
7. Hiroyoshi J, Saito A, Panthee N, et al. Aortic valve replacement for aortic stenosis caused by alkaptonuria. Ann Thorac Surg. 2013;95(3):1076-1079.
8. Parambil JG, Daniels CE, Zehr KJ, Utz JP. Alkaptonuria diagnosed by flexible bronchoscopy. Chest. 2005;128(5):3678-3680.
9. Farzannia A, Shokouhi G, Hadidchi S. Alkaptonuria and lumbar disc herniation. Report of three cases. J Neurosurg. 2003;98(suppl 1):87-89.
10. Introne WJ, Perry MB, Troendle J, et al. A 3-year randomized therapeutic trial of nitisinone in alkaptonuria. Mol Genet Metab. 2011;103(4):307-314.
11. Gissen P, Preece MA, Willshaw HA, McKiernan PJ. Ophthalmic follow-up of patients with tyrosinaemia type I on NTBC. J Inherit Metab Dis. 2003;26(1):13-16.
12. Khedr M, Judd S, Briggs MC, et al. Asymptomatic corneal keratopathy secondary to hypertyrosinaemia following low dose nitisinone and a literature review of tyrosine keratopathy in alkaptonuria. JIMD Rep. 2018;40:31-37.
13. Wolff JA, Barshop B, Nyhan WL, et al. Effects of ascorbic acid in alkaptonuria: alterations in benzoquinone acetic acid and an ontogenic effect in infancy. Pediatr Res. 1989;26(2):140-144.
14. Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. J Am Soc Nephrol. 2004;15(12):3225-3232.
15. Abate M, Salini V, Andia I. Tendons involvement in congenital metabolic disorders. Adv Exp Med Biol. 2016;920:117-122.
1. Aquaron R. Alkaptonuria: a very rare metabolic disorder. Indian J Biochem Biophys. 2013;50(5):339-344.
2. Phornphutkul C, Introne WJ, Perry MB, et al. Natural history of alkaptonuria. N Engl J Med. 2002;347(26):2111-2121.
3. Alajoulin OA, Alsbou MS, Ja’afreh SO, Kalbouneh HM. Spontaneous Achilles tendon rupture in alkaptonuria. Saudi Med J. 2015;36(12):1486-1489.
4. Manoj Kumar RV, Rajasekaran S. Spontaneous tendon ruptures in alkaptonuria. J Bone Joint Surg Br. 2003;85(6):883-886.
5. Tanoglu O, Arican G, Ozmeric A, Alemdaroglu KB, Caydere M. Calcaneal avulsion of an ochronotic Achilles tendon: a case report. J Foot Ankle Surg. 2018;57(1):179-183.
6. Schuuring MJ, Delemarre B, Keyhan-Falsafi AM, van der Bilt IA. Mending a darkened heart: alkaptonuria discovered during aortic valve replacement. Circulation. 2016;133(12):e444-445.
7. Hiroyoshi J, Saito A, Panthee N, et al. Aortic valve replacement for aortic stenosis caused by alkaptonuria. Ann Thorac Surg. 2013;95(3):1076-1079.
8. Parambil JG, Daniels CE, Zehr KJ, Utz JP. Alkaptonuria diagnosed by flexible bronchoscopy. Chest. 2005;128(5):3678-3680.
9. Farzannia A, Shokouhi G, Hadidchi S. Alkaptonuria and lumbar disc herniation. Report of three cases. J Neurosurg. 2003;98(suppl 1):87-89.
10. Introne WJ, Perry MB, Troendle J, et al. A 3-year randomized therapeutic trial of nitisinone in alkaptonuria. Mol Genet Metab. 2011;103(4):307-314.
11. Gissen P, Preece MA, Willshaw HA, McKiernan PJ. Ophthalmic follow-up of patients with tyrosinaemia type I on NTBC. J Inherit Metab Dis. 2003;26(1):13-16.
12. Khedr M, Judd S, Briggs MC, et al. Asymptomatic corneal keratopathy secondary to hypertyrosinaemia following low dose nitisinone and a literature review of tyrosine keratopathy in alkaptonuria. JIMD Rep. 2018;40:31-37.
13. Wolff JA, Barshop B, Nyhan WL, et al. Effects of ascorbic acid in alkaptonuria: alterations in benzoquinone acetic acid and an ontogenic effect in infancy. Pediatr Res. 1989;26(2):140-144.
14. Taylor EN, Stampfer MJ, Curhan GC. Dietary factors and the risk of incident kidney stones in men: new insights after 14 years of follow-up. J Am Soc Nephrol. 2004;15(12):3225-3232.
15. Abate M, Salini V, Andia I. Tendons involvement in congenital metabolic disorders. Adv Exp Med Biol. 2016;920:117-122.
Transgender Care in the Primary Care Setting: A Review of Guidelines and Literature
Lesbian, gay, bisexual, and transgender (LGBT) individuals face significant difficulties in obtaining high-quality,compassionate medical care, much of which has been attributed to inadequate provider knowledge. In this article, the authors present a transgender patient seen in primary care and discuss the knowledge gleaned to inform future care of this patient as well as the care of other similar patients.
The following case discussion and review of the literature also seeks to improve the practice of other primary care providers (PCPs) who are inexperienced in this arena. This article aims to review the basics to permit PCPs to venture into transgender care, including a review of basic terminology; a few interactive tips; and basics in medical and hormonal treatment, follow-up, contraindications, and risk. More details can be obtained through electronic consultation (Transgender eConsult) in the VA.
Case Presentation
A 35-year-old patient who was assigned male sex at birth presented to the primary care clinic to discuss her desire to undergo male-to-female (MTF) transition. The patient stated that she had started taking female estrogen hormones 9 years previously purchased from craigslist without a prescription. She tried oral contraceptives as well as oral and injectable estradiol. While the patient was taking injectable estradiol she had breast growth, decreased anxiety, weight gain, and a feeling of peacefulness. The patient also reported that she had received several laser treatments for whole body hair removal, beginning 8 to 10 years before and more regularly in the past 2 to 3 years. She asked whether transition-related care could be provided, because she could no longer afford the hormones.
The patient wanted to transition because she felt that “Women are beautiful, the way they carry themselves, wear their hair, their nails, I want to be like that.” She also mentioned that when she watched TV, she envisioned herself as a woman. She reported that she enjoyed wearing her mother’s clothing since age 10, which made her feel more like herself. The patient noted that she had desired to remove her body hair since childhood but could not afford to do it until recently. She bought female clothing, shoes and makeup, and did her nails from a young age. The patient also reported that she did not “know what transgender was” until a decade ago.
The patient struggled with her identity growing up; however, she tried not to think about it or talk about it with anyone. She related that she was ashamed of her thoughts and that only recently had made peace with being transgender. Thus, she pursued talking to her medical provider about transitioning. The patient reported that she felt more energetic when taking female hormones and was better able to discuss the issue. Specifically, she noted that if she were not on estrogen now she would not be able to talk about transitioning.
The patient related that she has done extensive research about transitioning, including reading online about other transgender people. She noted that she was aware of “possible backlash with society,” but ultimately, she had decided that transitioning was the right decision for her.
She expressed a desire to have an orchiectomy and continue hormonal therapy to permit her “to have a more feminine face, soft skin, hairless body, big breasts, more fat around the hips, and a high-pitched voice.” She additionally related a desire to be in a stable relationship and be her true self. She also stated that she had not identified herself as a female to anyone yet but would like to soon. The patient reported a history of depression, especially during her military service when she wanted to be a woman but did not feel she understood what was going on or how to manage her feelings. She said that for the past 2 months she felt much happier since beginning to take estradiol 4 mg orally daily, which she had found online. She also tried to purchase anti-androgen medication but could not afford it. In addition, she said that she would like to eventually proceed with gender affirmation surgery.
She was currently having sex with men, primarily via anal receptive intercourse. She had no history of sexually transmitted infections but reported that she did not use condoms regularly. She had no history of physical or sexual abuse. The patient was offered referral to the HIV clinic to receive HIV preexposure prophylaxis therapy (emtricitabine + tenofovir), which she declined, but she was counseled on safe sex practice.
The patient was referred to psychiatry both for supportive mental health care and to clarify that her concomitant mental health issues would not preclude the prescription of gender-affirming hormone treatment. Based on the psychiatric evaluation, the patient was felt to be appropriate for treatment with feminizing hormone therapy. The psychiatric assessment also noted that although the patient had a history of psychosis, she was not exhibiting psychotic symptoms currently, and this would not be a contraindication to treatment.
After discussion of the risks and benefits of cross-sex hormone therapy, the patient was started on estradiol 4 mg orally daily, as well as spironolactone 50 mg daily. She was then switched to estradiol 10 mg intramuscular every 2 weeks with the aim of using a less thrombogenic route of administration.
Treatment Outcomes
The patient remains under care. She has had follow-up visits every 3 months to ensure appropriate signs of feminization and monitoring of adverse effects (AEs). The patient’s testosterone and estradiol levels are being checked every 3 months to ensure total testosterone is 1,2
After 12 months on therapy with estradiol and spironolactone, the patient notes that her mood has improved, she feels more energetic, she has gained some weight, and her skin is softer. Her voice pitch, with the help of speech therapy, is gradually changing to what she perceives as more feminine. Hormone levels and electrolytes are all in an acceptable range, and blood sugar and blood pressure (BP) are within normal range. The patient will be offered age-appropriate cancer screening at the appropriate time.
Discussion
The treatment of gender-nonconforming individuals has come a long way since Lili Elbe, the transgender artist depicted in The Danish Girl, underwent gender-affirmation surgery in the early 20th century. Lili and people like her paved the way for other transgender individuals by doggedly pursuing gender-affirming medical treatment although they faced rejection by society and forged a difficult path. In recent years, an increasing number of transgender individuals have begun to seek mainstream medical care; however, PCPs often lack the knowledge and training to properly interact with and care for transgender patients.3,4
Terminology
Although someone’s sex is typically assigned at birth based on the external appearance of their genitalia, gender identity refers to a person’s internal sense of self and how they fit in to the world. People often use these 2 terms interchangeably in everyday language, but these terms are different.1,2
Transgender refers to a person whose gender identity differs from the sex that was assigned at birth. A transgender man or transman, or female-to-male (FTM) transgender person, is an individual who is assigned female sex at birth but identifies as a male. A transgender woman, or transwoman or a male-to-female (MTF) transgender person, is an individual who is assigned male sex at birth but identifies as female. A nontransgender person may be referred to as cisgender.
Transsexual is a medical term and refers to a transgender individual who sought medical intervention to transition to their identified gender. 
Sexual orientation describes sexual attraction only and is not related to gender identity. The sexual orientation of a transgender person is determined by emotional and/or physical attraction and not gender identity.
Gender dysphoria refers to the distress experienced by an individual when one’s gender identity and sex are not completely congruent.
Improving Patient Interaction
Transgender patients might avoid seeking care due to previous negative experiences or a fear of being judged. It is very important to create a safe environment where the patients feel comfortable. Meeting patients “where they are” without judgment will enhance the patient-physician relationship. It is necessary to train all clinic staff about the importance of transgender health issues. All staff should address the patient with the name, pronouns, and gender identity that the patient prefers. For patients with a gender identity that is not strictly male or female (nonbinary patients), gender-neutral pronouns, such as they/them/their, may be chosen. It is helpful to be direct in asking: What is your preferred name? When I speak about you to other providers, what pronouns do you prefer I use, he, she, they? This information can then be documented in the electronic health record (EHR) so that all staff know from visit to visit. Thank the patient for the clarification.
The physical examination can be uncomfortable for both the patient and the physician. Experience and familiarity with the current recommendations can help. The physical examination should be relevant to the anatomy that is present, regardless of the gender presentation. An anatomic survey of the organs currently present in an individual can be useful.1 The physician should be sensitive in examining and obtaining information from the patient, focusing on only those issues relevant to the presenting concern. Chest and genital examinations may be particularly distressing for patients. If a chest or genital examination is indicated, the provider and patient should have a discussion explaining the importance of the examination and how the patient’s comfort can be optimized.
Medical Treatment
Gender-affirmation treatment should be multidisciplinary and include some or all of the following: diagnostic assessment, psychotherapy or counseling, real-life experience (RLE), hormone therapy, and surgical therapy..1,2,5 The World Professional Association for Transgender Health (WPATH) has established internationally accepted Standards of Care (SOC) for the treatment of gender dysphoria that provide detailed expert opinion reviewing the background and guidance for care of transgender individuals. Most commonly, the diagnosis of gender dysphoria is made by a mental health professional (MHP) based on the Diagnostic and Statistical Manual of Mental Disorders (DSM–5) criteria for gender dysphoria.1,2 The involvement of a MHP can be crucial in assessing potential psychological and social risk factors for unfavorable outcomes of medical interventions. In case of severe psychopathology, which can interfere with diagnosis and treatment, the psychopathology should be addressed first.1,2 The MHP also can confirm that the patient has the capacity to make an informed decision.
The 2017 Endocrine Society guidelines for the treatment of gender-dysphoric/gender-incongruent persons emphasize the utility of evaluation of these patients by an expert MHP before starting the treatment.2 However, the guidelines from WPATH and the Center for Transgender Excellence at University of California, San Francisco (UCSF) have stipulated that any provider who feels comfortable assessing the informed decision-making process with a patient can make this determination.
The WPATH SOC states that RLE is essential to transition to the gender role that is congruent with the patient’s gender identity. The RLE is defined as the act of fully adopting a new or evolving gender role or gender presentation in everyday life. In the RLE, the person should fully experience life in the desired gender role before irreversible physical treatment is undertaken. Newer guidelines note that it may be too challenging to adopt the desired gender role without the benefit of feminizing or masculinizing treatment, and therefore, the treatment can be offered at the same time as adopting the new gender role.1
Medical treatment involves administration of masculinizing or feminizing hormone therapy. There are 2 major goals of this hormonal therapy. 
For many transgender adults, genital reconstruction surgery and/or gonadectomy is a necessary step toward achieving their goal. 
Pretreatment screening and appropriate medical monitoring is recommended for both FTM and MTF transgender patients during the endocrine transition and periodically thereafter.2 The physician should monitor the patient’s weight, BP, directed physical examinations, routine health questions focused on risk factors and medications, complete blood count, renal and liver functions, lipid and blood sugar.2 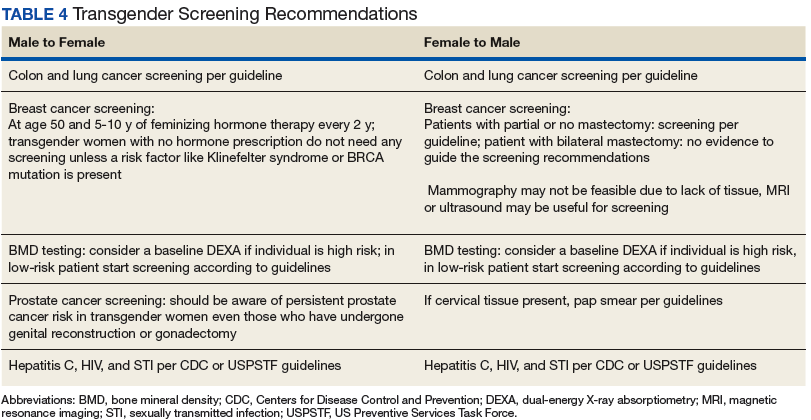
Physical Changes With Hormone Therapy
Transgender men. Physical changes that are expected to occur during the first 1 to 6 months of testosterone therapy include cessation of menses, increased sexual desire, increased facial and body hair, increased oiliness of skin, increased muscle, and redistribution of fat mass. Changes that occur within the first year of testosterone therapy include deepening of the voice, clitoromegaly, and male pattern hair loss (in some cases). Deepening of the voice, and clitoromegaly are not reversible with discontinuation of hormonal therapy.2
Transgender women. Physical changes that may occur in transgender females in the first 3 to 12 months of estrogen and anti-androgen therapy include decreased sexual desire, decreased spontaneous erections, decreased facial and body hair (usually mild), decreased oiliness of skin, increased breast tissue growth, and redistribution of fat mass. Breast development is generally maximal at 2 years after initiating estrogen, and it is irreversible.2 Effect on fertility may be permanent. Medical therapy has little effect on voice, and most transwomen will require speech therapy to achieve desired pitch.
Routine Health Maintenance
Breast Cancer Screening
Although there are limited data, it is thought that gender-affirming hormone therapy has similar risks as sex hormone replacement therapy in nontransgender males and females. Most AEs arise from use of supraphysiologic doses or inadequate doses.2 Therefore, regular clinical and laboratory monitoring is essential to cross-sex hormone therapy. Treatment with exogenous estrogen and anti-androgens result in transgender women developing breast tissue with ducts, lobules, and acini that is histologically identical to breast tissue in nontransgender females.6
Breast cancer is a concern in transgender women due to prolonged exposure to estrogen. However, the relationship between breast cancer and cross-sex hormone therapy is controversial.
Many factors contribute to breast cancer risk in patients of all genders. Studies of premenopausal and menopausal women taking exogenous estrogen alone have not shown an increase in breast cancer risk. However, the combination of estrogen and progesterone has shown an association with a significant increase in the incidence of breast cancer in postmenopausal women.2,7-10
A study of 5,136 veterans showed a statistically insignificant increased incidence of breast cancer in transgender women compared with data collected from the Surveillance, Epidemiology, and End Results database, although the sample size and duration of the observation were limiting factors.8 A European cohort study found decreased incidence of breast cancer in both MTF and FTM transgender patients, but these patients were an overall younger cohort with decreased risk in general. A cohort of 2,236 MTF individuals in the Netherlands in 1997 showed no increase in all-cause mortality related to hormone therapy at 30-year follow-up. Patients were exposed to exogenous estrogen from 2 months to 41 years.9 A follow-up of this study published in 2013, which included 2,307 MTF individuals taking estrogen for 5 years to > 30 years, revealed only 2 cases of breast cancer, which was the same incidence rate (4.1 per 100,000 person-years) as that of nontransgender women.10
In general, the incidence of breast cancer is rare in nontransgender men, and therefore there have not been a lot of clinical studies to assess risk factors and detection methods. The following risk factors can increase the risk of breast cancer in nontransgender patients: known presence of BRCA mutation, estrogen exposure/androgen insufficiency, Klinefelter syndrome, liver cirrhosis, and obesity.11
Guidelines from the Endocrine Society, WPATH, and UCSF suggest that MTF transgender individuals who have a known increased risk for breast cancer should follow screening guidelines recommended for nontransgender women if they are aged > 50 years and have had more than 5 years of hormone use.2 For FTM patients who have not had chest surgery, screening guidelines should follow those for nontransgender women. For those patients who have had chest reconstruction, small residual amounts of breast tissue may remain. Screening guidelines for these patients do not exist. For these patients, mammography can be technically difficult. Clinical chest wall examination, magnetic resonance imaging (MRI), and/or ultrasound may be helpful modalities. An individual risk vs benefit discussion with the patient is recommended.
Prostate Cancer Screening
Although the prostate gland will undergo atrophy with extended treatment with feminizing hormone therapy, there are case reports of prostate cancer in transgender women.12,13 Usually these patients have started hormone treatment after age 50 years. Therefore, prostate cancer screening is recommended in transgender women as per general guidelines. Because the prostate-specific antigen (PSA) level is expected to be reduced, a PSA > 1.0 should be considered abnormal.1
Cervical Cancer Screening
When a transgender man has a pap smear, it is essential to make it clear to the laboratory that the sample is a cervical pap smear (especially if the gender is marked as male) to avoid the sample being run incorrectly as an anal pap. Also, it is essential to indicate on the pap smear request form that the patient is on testosterone therapy and amenorrhea is present, because the lack of the female hormone can cause atrophy of cervix. This population has a high rate of inadequate specimens. Pretreatment with 1 to 2 weeks of vaginal estrogen can improve the success rate of inadequate specimens. Transgender women who have undergone vaginoplasty do not have a cervix, therefore, cervical cancer screening is not recommended. The anatomy of the neovagina has a more posterior orientation, and an anoscope is a more appropriate tool to examine the neovagina when necessary.
Hematology Health
Transgender women on cross-sex hormone therapy with estrogens may be at increased risk for a venous thromboembolism (VTE). In 2 European studies, patients treated with oral ethinyl estradiol as well as the anti-androgen cyproterone acetate were found to have up to 20 times increased risk of VTE. However, in later studies, oral ethinyl estradiol was changed to either oral conjugated estrogens or transdermal/intramuscular estradiol, and these studies did not show a significant increase in VTE risk.14-16 Tobacco use in combination with estrogen therapy is associated with an increased risk of deep vein thrombosis (DVT).1 All transgender women who smoke should be counseled on tobacco risks and cessation options at every visit.1 The transgender individuals who are not willing to quit smoking may be offered transdermal estrogen, which has lower risk of DVT.14-16
Sexual Health
Clinicians should assess the risks for sexually transmitted infection (STIs) or HIV for transgender patients based on current anatomy and sexual behaviors. Presentations of STIs can be atypical due to varied sexual practices and gender-affirming surgeries. Thus, providers must remain vigilant for symptoms consistent with common STIs and screen for asymptomatic STIs on the basis of behavior history and sexual practices.17 Preexposure prophylaxis for HIV should be considered when appropriate. Serologic screening recommendations for transgender people (eg, HIV, hepatitis B and C, syphilis) do not differ in recommendations from those for nontransgender people.
Cardiovascular Health
The effect of cross-hormone treatment on cardiovascular (CV) health is still unknown. There are no randomized controlled trials that have investigated the relationship between cross-hormone treatment and CV health. Evidence from several studies suggests that CV risk is unchanged among transgender men using testosterone compared with that of nontransgender women.18,19 There is conflicting evidence for transgender women with respect to CV risk and cross-sex hormone treatment.1,18,19 The current American College of Cardiology/American Heart Association guideline advises using the ASCVD risk calculator to determine the need for aspirin and statin treatment based on race, age, gender, and risk factors. There is no guideline on whether to use natal sex or affirmed gender while using the ASCVD calculator. It is reasonable to use the calculator based on natal sex if the cross-hormone treatment has started later in life, but if the cross-sex hormone treatment started at a young age, then one should consider using the affirmed gender to calculate the risk.
As with all patients, life style modifications, including smoking cessation, weight loss, physical activity, and management of BP and blood sugar, are important for CV health. For transgender women with CV risk factors or known CV disease, transdermal route of estrogen is preferred due to lower rates of VTE.18,19
Conclusion
In recent years, an increased number of transgender individuals are seeking mainstream medical care. However, PCPs often lack the knowledge and training to properly interact with and care for transgender patients. It is critical that clinicians understand the difference between sex, gender, and sexuality. For patients who desire transgender care, providers must be able to comfortably ask the patient about their preferred name and prior care, know the basics in cross-sex hormone therapy, including appropriate follow-up of hormonal levels as well as laboratory tests that delineate risk, and know possible complications and AEs. The VA offers significant resources, including electronic transgender care consultation for cases where the provider does not have adequate expertise in the care of these patients.
Both medical schools and residency training programs are starting to incorporate curricula regarding LGBT care. For those who have already completed training, this article serves as a brief guide to terminology, interactive tips, and management of this growing and underserved group of individuals.
1. Deutsch MB. Guidelines for the primary and gender-affirming care of transgender and gender nonbinary people. http://transhealth.ucsf.edu/protocols. Updated June 17, 2016. Accessed June 13, 2018.
2. Hembree WC, Cohen-Kettenis PT, Gooren L, et al. Endocrine treatment of gender-dysphoria/gender-incongruent persons: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2017;102(11):3869-3903.
3. Buchholz L. Transgender care moves into the mainstream. JAMA. 2015;314(17):1785-1787.
4. Sobralske M. Primary care needs of patients who have undergone gender reassignment. J Am Acad Nurse Pract. 2005;17(4):133-138.
5. Unger CA. Hormone therapy for transgender patients. Transl Androl Urol. 2016;5(6):877-884.
6. Kanhai RC, Hage JJ, van Diest PJ, Bloemena E, Mulder JW. Short-term and long-term histologic effects of castration and estrogen treatment on breast tissue of 14 male-to-female transsexuals in comparison with two chemically castrated men. Am J Surg Pathol. 2000;24(1):74-80.
7. Braun H, Nash R, Tangpricha V, Brockman J, Ward K, Goodman M. Cancer in transgender people: evidence and methodological consideration. Epidemiol Rev. 2017;39(1):93-107.
8. Brown GR, Jones KT. Incidence of breast cancer in a cohort of 5,135 transgender veterans. Breast Cancer Res Treat. 2015;149(1):191-198.
9. Van Kesteren PJ, Asscheman H, Megens JA, Gooren LJ. Mortality and morbidity in transsexual subjects treated with cross-sex hormones. Clin Endocrinol (Oxf). 1997;47(3):337-342.
10. Gooren LJ, van Trotsenburg MA, Giltay EJ, van Diest PJ. Breast cancer development in transsexual subjects receiving cross-sex hormone treatment. J Sex Med. 2013;10(12):3129-3134.
11. Johansen Taber KA, Morisy LR, Osbahr AJ III, Dickinson BD. Male breast cancer: risk factors, diagnosis and management (review). Oncol Rep. 2010;24(5):1115-1120.
12. Miksad RA, Bubley G, Church P, et al. Prostate cancer in a transgender woman, 41 years after initiation of feminization. JAMA. 2006;296(19):2316-2317.
13. Turo R, Jallad S, Prescott S, Cross WR. Metastatic prostate cancer in transsexual diagnosed after three decades of estrogen therapy. Can Urol Assoc J. 2013;7(7-8):E544-E546.
14. American College of Obstetricians and Gynecologists. ACOG committee opinion no. 556: postmenopausal estrogen therapy: route of administration and risk of venous thromboembolism. Obstet Gynecol. 2013;121(4):887-890.
15. Asscheman H, Gooren LJ, Eklund PL. Mortality and morbidity in transsexual patients with cross-gender treatment. Metabolism. 1989;38(9):869-873.
16. Asscheman H, Giltay EJ, Megens JA, de Ronde WP, van Trotsenburg MA, Gooren LJ. A long-term follow-up study of mortality in transsexuals receiving treatment with cross-sex hormones. Eur J Endocrinol. 2011;164(4):635-642.
17. Workowski KA, Bolan GA; Centers for Disease Control and Prevention. Sexually transmitted disease treatment guidelines, 2015. MMWR Recomm Rep. 2015;64(RR-03):1-137.
18. Gooren LJ, Wierckx K, Giltay EJ. Cardiovascular disease in transsexual persons treated with cross-sex hormones: reversal of the traditional sex difference in cardiovascular disease pattern. Eur J Endocrinol. 2014;170(6):809-819.
19. Streed CG Jr, Harfouch O, Marvel F, Blumenthal RS, Martin SS, Mukherjee M. Cardiovascular disease among transgender adults receiving hormone therapy: a narrative review. Ann Int Med. 2017;167(4):256-267.
Lesbian, gay, bisexual, and transgender (LGBT) individuals face significant difficulties in obtaining high-quality,compassionate medical care, much of which has been attributed to inadequate provider knowledge. In this article, the authors present a transgender patient seen in primary care and discuss the knowledge gleaned to inform future care of this patient as well as the care of other similar patients.
The following case discussion and review of the literature also seeks to improve the practice of other primary care providers (PCPs) who are inexperienced in this arena. This article aims to review the basics to permit PCPs to venture into transgender care, including a review of basic terminology; a few interactive tips; and basics in medical and hormonal treatment, follow-up, contraindications, and risk. More details can be obtained through electronic consultation (Transgender eConsult) in the VA.
Case Presentation
A 35-year-old patient who was assigned male sex at birth presented to the primary care clinic to discuss her desire to undergo male-to-female (MTF) transition. The patient stated that she had started taking female estrogen hormones 9 years previously purchased from craigslist without a prescription. She tried oral contraceptives as well as oral and injectable estradiol. While the patient was taking injectable estradiol she had breast growth, decreased anxiety, weight gain, and a feeling of peacefulness. The patient also reported that she had received several laser treatments for whole body hair removal, beginning 8 to 10 years before and more regularly in the past 2 to 3 years. She asked whether transition-related care could be provided, because she could no longer afford the hormones.
The patient wanted to transition because she felt that “Women are beautiful, the way they carry themselves, wear their hair, their nails, I want to be like that.” She also mentioned that when she watched TV, she envisioned herself as a woman. She reported that she enjoyed wearing her mother’s clothing since age 10, which made her feel more like herself. The patient noted that she had desired to remove her body hair since childhood but could not afford to do it until recently. She bought female clothing, shoes and makeup, and did her nails from a young age. The patient also reported that she did not “know what transgender was” until a decade ago.
The patient struggled with her identity growing up; however, she tried not to think about it or talk about it with anyone. She related that she was ashamed of her thoughts and that only recently had made peace with being transgender. Thus, she pursued talking to her medical provider about transitioning. The patient reported that she felt more energetic when taking female hormones and was better able to discuss the issue. Specifically, she noted that if she were not on estrogen now she would not be able to talk about transitioning.
The patient related that she has done extensive research about transitioning, including reading online about other transgender people. She noted that she was aware of “possible backlash with society,” but ultimately, she had decided that transitioning was the right decision for her.
She expressed a desire to have an orchiectomy and continue hormonal therapy to permit her “to have a more feminine face, soft skin, hairless body, big breasts, more fat around the hips, and a high-pitched voice.” She additionally related a desire to be in a stable relationship and be her true self. She also stated that she had not identified herself as a female to anyone yet but would like to soon. The patient reported a history of depression, especially during her military service when she wanted to be a woman but did not feel she understood what was going on or how to manage her feelings. She said that for the past 2 months she felt much happier since beginning to take estradiol 4 mg orally daily, which she had found online. She also tried to purchase anti-androgen medication but could not afford it. In addition, she said that she would like to eventually proceed with gender affirmation surgery.
She was currently having sex with men, primarily via anal receptive intercourse. She had no history of sexually transmitted infections but reported that she did not use condoms regularly. She had no history of physical or sexual abuse. The patient was offered referral to the HIV clinic to receive HIV preexposure prophylaxis therapy (emtricitabine + tenofovir), which she declined, but she was counseled on safe sex practice.
The patient was referred to psychiatry both for supportive mental health care and to clarify that her concomitant mental health issues would not preclude the prescription of gender-affirming hormone treatment. Based on the psychiatric evaluation, the patient was felt to be appropriate for treatment with feminizing hormone therapy. The psychiatric assessment also noted that although the patient had a history of psychosis, she was not exhibiting psychotic symptoms currently, and this would not be a contraindication to treatment.
After discussion of the risks and benefits of cross-sex hormone therapy, the patient was started on estradiol 4 mg orally daily, as well as spironolactone 50 mg daily. She was then switched to estradiol 10 mg intramuscular every 2 weeks with the aim of using a less thrombogenic route of administration.
Treatment Outcomes
The patient remains under care. She has had follow-up visits every 3 months to ensure appropriate signs of feminization and monitoring of adverse effects (AEs). The patient’s testosterone and estradiol levels are being checked every 3 months to ensure total testosterone is 1,2
After 12 months on therapy with estradiol and spironolactone, the patient notes that her mood has improved, she feels more energetic, she has gained some weight, and her skin is softer. Her voice pitch, with the help of speech therapy, is gradually changing to what she perceives as more feminine. Hormone levels and electrolytes are all in an acceptable range, and blood sugar and blood pressure (BP) are within normal range. The patient will be offered age-appropriate cancer screening at the appropriate time.
Discussion
The treatment of gender-nonconforming individuals has come a long way since Lili Elbe, the transgender artist depicted in The Danish Girl, underwent gender-affirmation surgery in the early 20th century. Lili and people like her paved the way for other transgender individuals by doggedly pursuing gender-affirming medical treatment although they faced rejection by society and forged a difficult path. In recent years, an increasing number of transgender individuals have begun to seek mainstream medical care; however, PCPs often lack the knowledge and training to properly interact with and care for transgender patients.3,4
Terminology
Although someone’s sex is typically assigned at birth based on the external appearance of their genitalia, gender identity refers to a person’s internal sense of self and how they fit in to the world. People often use these 2 terms interchangeably in everyday language, but these terms are different.1,2
Transgender refers to a person whose gender identity differs from the sex that was assigned at birth. A transgender man or transman, or female-to-male (FTM) transgender person, is an individual who is assigned female sex at birth but identifies as a male. A transgender woman, or transwoman or a male-to-female (MTF) transgender person, is an individual who is assigned male sex at birth but identifies as female. A nontransgender person may be referred to as cisgender.
Transsexual is a medical term and refers to a transgender individual who sought medical intervention to transition to their identified gender.
Sexual orientation describes sexual attraction only and is not related to gender identity. The sexual orientation of a transgender person is determined by emotional and/or physical attraction and not gender identity.
Gender dysphoria refers to the distress experienced by an individual when one’s gender identity and sex are not completely congruent.
Improving Patient Interaction
Transgender patients might avoid seeking care due to previous negative experiences or a fear of being judged. It is very important to create a safe environment where the patients feel comfortable. Meeting patients “where they are” without judgment will enhance the patient-physician relationship. It is necessary to train all clinic staff about the importance of transgender health issues. All staff should address the patient with the name, pronouns, and gender identity that the patient prefers. For patients with a gender identity that is not strictly male or female (nonbinary patients), gender-neutral pronouns, such as they/them/their, may be chosen. It is helpful to be direct in asking: What is your preferred name? When I speak about you to other providers, what pronouns do you prefer I use, he, she, they? This information can then be documented in the electronic health record (EHR) so that all staff know from visit to visit. Thank the patient for the clarification.
The physical examination can be uncomfortable for both the patient and the physician. Experience and familiarity with the current recommendations can help. The physical examination should be relevant to the anatomy that is present, regardless of the gender presentation. An anatomic survey of the organs currently present in an individual can be useful.1 The physician should be sensitive in examining and obtaining information from the patient, focusing on only those issues relevant to the presenting concern. Chest and genital examinations may be particularly distressing for patients. If a chest or genital examination is indicated, the provider and patient should have a discussion explaining the importance of the examination and how the patient’s comfort can be optimized.
Medical Treatment
Gender-affirmation treatment should be multidisciplinary and include some or all of the following: diagnostic assessment, psychotherapy or counseling, real-life experience (RLE), hormone therapy, and surgical therapy..1,2,5 The World Professional Association for Transgender Health (WPATH) has established internationally accepted Standards of Care (SOC) for the treatment of gender dysphoria that provide detailed expert opinion reviewing the background and guidance for care of transgender individuals. Most commonly, the diagnosis of gender dysphoria is made by a mental health professional (MHP) based on the Diagnostic and Statistical Manual of Mental Disorders (DSM–5) criteria for gender dysphoria.1,2 The involvement of a MHP can be crucial in assessing potential psychological and social risk factors for unfavorable outcomes of medical interventions. In case of severe psychopathology, which can interfere with diagnosis and treatment, the psychopathology should be addressed first.1,2 The MHP also can confirm that the patient has the capacity to make an informed decision.
The 2017 Endocrine Society guidelines for the treatment of gender-dysphoric/gender-incongruent persons emphasize the utility of evaluation of these patients by an expert MHP before starting the treatment.2 However, the guidelines from WPATH and the Center for Transgender Excellence at University of California, San Francisco (UCSF) have stipulated that any provider who feels comfortable assessing the informed decision-making process with a patient can make this determination.
The WPATH SOC states that RLE is essential to transition to the gender role that is congruent with the patient’s gender identity. The RLE is defined as the act of fully adopting a new or evolving gender role or gender presentation in everyday life. In the RLE, the person should fully experience life in the desired gender role before irreversible physical treatment is undertaken. Newer guidelines note that it may be too challenging to adopt the desired gender role without the benefit of feminizing or masculinizing treatment, and therefore, the treatment can be offered at the same time as adopting the new gender role.1
Medical treatment involves administration of masculinizing or feminizing hormone therapy. There are 2 major goals of this hormonal therapy.
For many transgender adults, genital reconstruction surgery and/or gonadectomy is a necessary step toward achieving their goal.
Pretreatment screening and appropriate medical monitoring is recommended for both FTM and MTF transgender patients during the endocrine transition and periodically thereafter.2 The physician should monitor the patient’s weight, BP, directed physical examinations, routine health questions focused on risk factors and medications, complete blood count, renal and liver functions, lipid and blood sugar.2
Physical Changes With Hormone Therapy
Transgender men. Physical changes that are expected to occur during the first 1 to 6 months of testosterone therapy include cessation of menses, increased sexual desire, increased facial and body hair, increased oiliness of skin, increased muscle, and redistribution of fat mass. Changes that occur within the first year of testosterone therapy include deepening of the voice, clitoromegaly, and male pattern hair loss (in some cases). Deepening of the voice, and clitoromegaly are not reversible with discontinuation of hormonal therapy.2
Transgender women. Physical changes that may occur in transgender females in the first 3 to 12 months of estrogen and anti-androgen therapy include decreased sexual desire, decreased spontaneous erections, decreased facial and body hair (usually mild), decreased oiliness of skin, increased breast tissue growth, and redistribution of fat mass. Breast development is generally maximal at 2 years after initiating estrogen, and it is irreversible.2 Effect on fertility may be permanent. Medical therapy has little effect on voice, and most transwomen will require speech therapy to achieve desired pitch.
Routine Health Maintenance
Breast Cancer Screening
Although there are limited data, it is thought that gender-affirming hormone therapy has similar risks as sex hormone replacement therapy in nontransgender males and females. Most AEs arise from use of supraphysiologic doses or inadequate doses.2 Therefore, regular clinical and laboratory monitoring is essential to cross-sex hormone therapy. Treatment with exogenous estrogen and anti-androgens result in transgender women developing breast tissue with ducts, lobules, and acini that is histologically identical to breast tissue in nontransgender females.6
Breast cancer is a concern in transgender women due to prolonged exposure to estrogen. However, the relationship between breast cancer and cross-sex hormone therapy is controversial.
Many factors contribute to breast cancer risk in patients of all genders. Studies of premenopausal and menopausal women taking exogenous estrogen alone have not shown an increase in breast cancer risk. However, the combination of estrogen and progesterone has shown an association with a significant increase in the incidence of breast cancer in postmenopausal women.2,7-10
A study of 5,136 veterans showed a statistically insignificant increased incidence of breast cancer in transgender women compared with data collected from the Surveillance, Epidemiology, and End Results database, although the sample size and duration of the observation were limiting factors.8 A European cohort study found decreased incidence of breast cancer in both MTF and FTM transgender patients, but these patients were an overall younger cohort with decreased risk in general. A cohort of 2,236 MTF individuals in the Netherlands in 1997 showed no increase in all-cause mortality related to hormone therapy at 30-year follow-up. Patients were exposed to exogenous estrogen from 2 months to 41 years.9 A follow-up of this study published in 2013, which included 2,307 MTF individuals taking estrogen for 5 years to > 30 years, revealed only 2 cases of breast cancer, which was the same incidence rate (4.1 per 100,000 person-years) as that of nontransgender women.10
In general, the incidence of breast cancer is rare in nontransgender men, and therefore there have not been a lot of clinical studies to assess risk factors and detection methods. The following risk factors can increase the risk of breast cancer in nontransgender patients: known presence of BRCA mutation, estrogen exposure/androgen insufficiency, Klinefelter syndrome, liver cirrhosis, and obesity.11
Guidelines from the Endocrine Society, WPATH, and UCSF suggest that MTF transgender individuals who have a known increased risk for breast cancer should follow screening guidelines recommended for nontransgender women if they are aged > 50 years and have had more than 5 years of hormone use.2 For FTM patients who have not had chest surgery, screening guidelines should follow those for nontransgender women. For those patients who have had chest reconstruction, small residual amounts of breast tissue may remain. Screening guidelines for these patients do not exist. For these patients, mammography can be technically difficult. Clinical chest wall examination, magnetic resonance imaging (MRI), and/or ultrasound may be helpful modalities. An individual risk vs benefit discussion with the patient is recommended.
Prostate Cancer Screening
Although the prostate gland will undergo atrophy with extended treatment with feminizing hormone therapy, there are case reports of prostate cancer in transgender women.12,13 Usually these patients have started hormone treatment after age 50 years. Therefore, prostate cancer screening is recommended in transgender women as per general guidelines. Because the prostate-specific antigen (PSA) level is expected to be reduced, a PSA > 1.0 should be considered abnormal.1
Cervical Cancer Screening
When a transgender man has a pap smear, it is essential to make it clear to the laboratory that the sample is a cervical pap smear (especially if the gender is marked as male) to avoid the sample being run incorrectly as an anal pap. Also, it is essential to indicate on the pap smear request form that the patient is on testosterone therapy and amenorrhea is present, because the lack of the female hormone can cause atrophy of cervix. This population has a high rate of inadequate specimens. Pretreatment with 1 to 2 weeks of vaginal estrogen can improve the success rate of inadequate specimens. Transgender women who have undergone vaginoplasty do not have a cervix, therefore, cervical cancer screening is not recommended. The anatomy of the neovagina has a more posterior orientation, and an anoscope is a more appropriate tool to examine the neovagina when necessary.
Hematology Health
Transgender women on cross-sex hormone therapy with estrogens may be at increased risk for a venous thromboembolism (VTE). In 2 European studies, patients treated with oral ethinyl estradiol as well as the anti-androgen cyproterone acetate were found to have up to 20 times increased risk of VTE. However, in later studies, oral ethinyl estradiol was changed to either oral conjugated estrogens or transdermal/intramuscular estradiol, and these studies did not show a significant increase in VTE risk.14-16 Tobacco use in combination with estrogen therapy is associated with an increased risk of deep vein thrombosis (DVT).1 All transgender women who smoke should be counseled on tobacco risks and cessation options at every visit.1 The transgender individuals who are not willing to quit smoking may be offered transdermal estrogen, which has lower risk of DVT.14-16
Sexual Health
Clinicians should assess the risks for sexually transmitted infection (STIs) or HIV for transgender patients based on current anatomy and sexual behaviors. Presentations of STIs can be atypical due to varied sexual practices and gender-affirming surgeries. Thus, providers must remain vigilant for symptoms consistent with common STIs and screen for asymptomatic STIs on the basis of behavior history and sexual practices.17 Preexposure prophylaxis for HIV should be considered when appropriate. Serologic screening recommendations for transgender people (eg, HIV, hepatitis B and C, syphilis) do not differ in recommendations from those for nontransgender people.
Cardiovascular Health
The effect of cross-hormone treatment on cardiovascular (CV) health is still unknown. There are no randomized controlled trials that have investigated the relationship between cross-hormone treatment and CV health. Evidence from several studies suggests that CV risk is unchanged among transgender men using testosterone compared with that of nontransgender women.18,19 There is conflicting evidence for transgender women with respect to CV risk and cross-sex hormone treatment.1,18,19 The current American College of Cardiology/American Heart Association guideline advises using the ASCVD risk calculator to determine the need for aspirin and statin treatment based on race, age, gender, and risk factors. There is no guideline on whether to use natal sex or affirmed gender while using the ASCVD calculator. It is reasonable to use the calculator based on natal sex if the cross-hormone treatment has started later in life, but if the cross-sex hormone treatment started at a young age, then one should consider using the affirmed gender to calculate the risk.
As with all patients, life style modifications, including smoking cessation, weight loss, physical activity, and management of BP and blood sugar, are important for CV health. For transgender women with CV risk factors or known CV disease, transdermal route of estrogen is preferred due to lower rates of VTE.18,19
Conclusion
In recent years, an increased number of transgender individuals are seeking mainstream medical care. However, PCPs often lack the knowledge and training to properly interact with and care for transgender patients. It is critical that clinicians understand the difference between sex, gender, and sexuality. For patients who desire transgender care, providers must be able to comfortably ask the patient about their preferred name and prior care, know the basics in cross-sex hormone therapy, including appropriate follow-up of hormonal levels as well as laboratory tests that delineate risk, and know possible complications and AEs. The VA offers significant resources, including electronic transgender care consultation for cases where the provider does not have adequate expertise in the care of these patients.
Both medical schools and residency training programs are starting to incorporate curricula regarding LGBT care. For those who have already completed training, this article serves as a brief guide to terminology, interactive tips, and management of this growing and underserved group of individuals.
Lesbian, gay, bisexual, and transgender (LGBT) individuals face significant difficulties in obtaining high-quality,compassionate medical care, much of which has been attributed to inadequate provider knowledge. In this article, the authors present a transgender patient seen in primary care and discuss the knowledge gleaned to inform future care of this patient as well as the care of other similar patients.
The following case discussion and review of the literature also seeks to improve the practice of other primary care providers (PCPs) who are inexperienced in this arena. This article aims to review the basics to permit PCPs to venture into transgender care, including a review of basic terminology; a few interactive tips; and basics in medical and hormonal treatment, follow-up, contraindications, and risk. More details can be obtained through electronic consultation (Transgender eConsult) in the VA.
Case Presentation
A 35-year-old patient who was assigned male sex at birth presented to the primary care clinic to discuss her desire to undergo male-to-female (MTF) transition. The patient stated that she had started taking female estrogen hormones 9 years previously purchased from craigslist without a prescription. She tried oral contraceptives as well as oral and injectable estradiol. While the patient was taking injectable estradiol she had breast growth, decreased anxiety, weight gain, and a feeling of peacefulness. The patient also reported that she had received several laser treatments for whole body hair removal, beginning 8 to 10 years before and more regularly in the past 2 to 3 years. She asked whether transition-related care could be provided, because she could no longer afford the hormones.
The patient wanted to transition because she felt that “Women are beautiful, the way they carry themselves, wear their hair, their nails, I want to be like that.” She also mentioned that when she watched TV, she envisioned herself as a woman. She reported that she enjoyed wearing her mother’s clothing since age 10, which made her feel more like herself. The patient noted that she had desired to remove her body hair since childhood but could not afford to do it until recently. She bought female clothing, shoes and makeup, and did her nails from a young age. The patient also reported that she did not “know what transgender was” until a decade ago.
The patient struggled with her identity growing up; however, she tried not to think about it or talk about it with anyone. She related that she was ashamed of her thoughts and that only recently had made peace with being transgender. Thus, she pursued talking to her medical provider about transitioning. The patient reported that she felt more energetic when taking female hormones and was better able to discuss the issue. Specifically, she noted that if she were not on estrogen now she would not be able to talk about transitioning.
The patient related that she has done extensive research about transitioning, including reading online about other transgender people. She noted that she was aware of “possible backlash with society,” but ultimately, she had decided that transitioning was the right decision for her.
She expressed a desire to have an orchiectomy and continue hormonal therapy to permit her “to have a more feminine face, soft skin, hairless body, big breasts, more fat around the hips, and a high-pitched voice.” She additionally related a desire to be in a stable relationship and be her true self. She also stated that she had not identified herself as a female to anyone yet but would like to soon. The patient reported a history of depression, especially during her military service when she wanted to be a woman but did not feel she understood what was going on or how to manage her feelings. She said that for the past 2 months she felt much happier since beginning to take estradiol 4 mg orally daily, which she had found online. She also tried to purchase anti-androgen medication but could not afford it. In addition, she said that she would like to eventually proceed with gender affirmation surgery.
She was currently having sex with men, primarily via anal receptive intercourse. She had no history of sexually transmitted infections but reported that she did not use condoms regularly. She had no history of physical or sexual abuse. The patient was offered referral to the HIV clinic to receive HIV preexposure prophylaxis therapy (emtricitabine + tenofovir), which she declined, but she was counseled on safe sex practice.
The patient was referred to psychiatry both for supportive mental health care and to clarify that her concomitant mental health issues would not preclude the prescription of gender-affirming hormone treatment. Based on the psychiatric evaluation, the patient was felt to be appropriate for treatment with feminizing hormone therapy. The psychiatric assessment also noted that although the patient had a history of psychosis, she was not exhibiting psychotic symptoms currently, and this would not be a contraindication to treatment.
After discussion of the risks and benefits of cross-sex hormone therapy, the patient was started on estradiol 4 mg orally daily, as well as spironolactone 50 mg daily. She was then switched to estradiol 10 mg intramuscular every 2 weeks with the aim of using a less thrombogenic route of administration.
Treatment Outcomes
The patient remains under care. She has had follow-up visits every 3 months to ensure appropriate signs of feminization and monitoring of adverse effects (AEs). The patient’s testosterone and estradiol levels are being checked every 3 months to ensure total testosterone is 1,2
After 12 months on therapy with estradiol and spironolactone, the patient notes that her mood has improved, she feels more energetic, she has gained some weight, and her skin is softer. Her voice pitch, with the help of speech therapy, is gradually changing to what she perceives as more feminine. Hormone levels and electrolytes are all in an acceptable range, and blood sugar and blood pressure (BP) are within normal range. The patient will be offered age-appropriate cancer screening at the appropriate time.
Discussion
The treatment of gender-nonconforming individuals has come a long way since Lili Elbe, the transgender artist depicted in The Danish Girl, underwent gender-affirmation surgery in the early 20th century. Lili and people like her paved the way for other transgender individuals by doggedly pursuing gender-affirming medical treatment although they faced rejection by society and forged a difficult path. In recent years, an increasing number of transgender individuals have begun to seek mainstream medical care; however, PCPs often lack the knowledge and training to properly interact with and care for transgender patients.3,4
Terminology
Although someone’s sex is typically assigned at birth based on the external appearance of their genitalia, gender identity refers to a person’s internal sense of self and how they fit in to the world. People often use these 2 terms interchangeably in everyday language, but these terms are different.1,2
Transgender refers to a person whose gender identity differs from the sex that was assigned at birth. A transgender man or transman, or female-to-male (FTM) transgender person, is an individual who is assigned female sex at birth but identifies as a male. A transgender woman, or transwoman or a male-to-female (MTF) transgender person, is an individual who is assigned male sex at birth but identifies as female. A nontransgender person may be referred to as cisgender.
Transsexual is a medical term and refers to a transgender individual who sought medical intervention to transition to their identified gender.
Sexual orientation describes sexual attraction only and is not related to gender identity. The sexual orientation of a transgender person is determined by emotional and/or physical attraction and not gender identity.
Gender dysphoria refers to the distress experienced by an individual when one’s gender identity and sex are not completely congruent.
Improving Patient Interaction
Transgender patients might avoid seeking care due to previous negative experiences or a fear of being judged. It is very important to create a safe environment where the patients feel comfortable. Meeting patients “where they are” without judgment will enhance the patient-physician relationship. It is necessary to train all clinic staff about the importance of transgender health issues. All staff should address the patient with the name, pronouns, and gender identity that the patient prefers. For patients with a gender identity that is not strictly male or female (nonbinary patients), gender-neutral pronouns, such as they/them/their, may be chosen. It is helpful to be direct in asking: What is your preferred name? When I speak about you to other providers, what pronouns do you prefer I use, he, she, they? This information can then be documented in the electronic health record (EHR) so that all staff know from visit to visit. Thank the patient for the clarification.
The physical examination can be uncomfortable for both the patient and the physician. Experience and familiarity with the current recommendations can help. The physical examination should be relevant to the anatomy that is present, regardless of the gender presentation. An anatomic survey of the organs currently present in an individual can be useful.1 The physician should be sensitive in examining and obtaining information from the patient, focusing on only those issues relevant to the presenting concern. Chest and genital examinations may be particularly distressing for patients. If a chest or genital examination is indicated, the provider and patient should have a discussion explaining the importance of the examination and how the patient’s comfort can be optimized.
Medical Treatment
Gender-affirmation treatment should be multidisciplinary and include some or all of the following: diagnostic assessment, psychotherapy or counseling, real-life experience (RLE), hormone therapy, and surgical therapy..1,2,5 The World Professional Association for Transgender Health (WPATH) has established internationally accepted Standards of Care (SOC) for the treatment of gender dysphoria that provide detailed expert opinion reviewing the background and guidance for care of transgender individuals. Most commonly, the diagnosis of gender dysphoria is made by a mental health professional (MHP) based on the Diagnostic and Statistical Manual of Mental Disorders (DSM–5) criteria for gender dysphoria.1,2 The involvement of a MHP can be crucial in assessing potential psychological and social risk factors for unfavorable outcomes of medical interventions. In case of severe psychopathology, which can interfere with diagnosis and treatment, the psychopathology should be addressed first.1,2 The MHP also can confirm that the patient has the capacity to make an informed decision.
The 2017 Endocrine Society guidelines for the treatment of gender-dysphoric/gender-incongruent persons emphasize the utility of evaluation of these patients by an expert MHP before starting the treatment.2 However, the guidelines from WPATH and the Center for Transgender Excellence at University of California, San Francisco (UCSF) have stipulated that any provider who feels comfortable assessing the informed decision-making process with a patient can make this determination.
The WPATH SOC states that RLE is essential to transition to the gender role that is congruent with the patient’s gender identity. The RLE is defined as the act of fully adopting a new or evolving gender role or gender presentation in everyday life. In the RLE, the person should fully experience life in the desired gender role before irreversible physical treatment is undertaken. Newer guidelines note that it may be too challenging to adopt the desired gender role without the benefit of feminizing or masculinizing treatment, and therefore, the treatment can be offered at the same time as adopting the new gender role.1
Medical treatment involves administration of masculinizing or feminizing hormone therapy. There are 2 major goals of this hormonal therapy.
For many transgender adults, genital reconstruction surgery and/or gonadectomy is a necessary step toward achieving their goal.
Pretreatment screening and appropriate medical monitoring is recommended for both FTM and MTF transgender patients during the endocrine transition and periodically thereafter.2 The physician should monitor the patient’s weight, BP, directed physical examinations, routine health questions focused on risk factors and medications, complete blood count, renal and liver functions, lipid and blood sugar.2
Physical Changes With Hormone Therapy
Transgender men. Physical changes that are expected to occur during the first 1 to 6 months of testosterone therapy include cessation of menses, increased sexual desire, increased facial and body hair, increased oiliness of skin, increased muscle, and redistribution of fat mass. Changes that occur within the first year of testosterone therapy include deepening of the voice, clitoromegaly, and male pattern hair loss (in some cases). Deepening of the voice, and clitoromegaly are not reversible with discontinuation of hormonal therapy.2
Transgender women. Physical changes that may occur in transgender females in the first 3 to 12 months of estrogen and anti-androgen therapy include decreased sexual desire, decreased spontaneous erections, decreased facial and body hair (usually mild), decreased oiliness of skin, increased breast tissue growth, and redistribution of fat mass. Breast development is generally maximal at 2 years after initiating estrogen, and it is irreversible.2 Effect on fertility may be permanent. Medical therapy has little effect on voice, and most transwomen will require speech therapy to achieve desired pitch.
Routine Health Maintenance
Breast Cancer Screening
Although there are limited data, it is thought that gender-affirming hormone therapy has similar risks as sex hormone replacement therapy in nontransgender males and females. Most AEs arise from use of supraphysiologic doses or inadequate doses.2 Therefore, regular clinical and laboratory monitoring is essential to cross-sex hormone therapy. Treatment with exogenous estrogen and anti-androgens result in transgender women developing breast tissue with ducts, lobules, and acini that is histologically identical to breast tissue in nontransgender females.6
Breast cancer is a concern in transgender women due to prolonged exposure to estrogen. However, the relationship between breast cancer and cross-sex hormone therapy is controversial.
Many factors contribute to breast cancer risk in patients of all genders. Studies of premenopausal and menopausal women taking exogenous estrogen alone have not shown an increase in breast cancer risk. However, the combination of estrogen and progesterone has shown an association with a significant increase in the incidence of breast cancer in postmenopausal women.2,7-10
A study of 5,136 veterans showed a statistically insignificant increased incidence of breast cancer in transgender women compared with data collected from the Surveillance, Epidemiology, and End Results database, although the sample size and duration of the observation were limiting factors.8 A European cohort study found decreased incidence of breast cancer in both MTF and FTM transgender patients, but these patients were an overall younger cohort with decreased risk in general. A cohort of 2,236 MTF individuals in the Netherlands in 1997 showed no increase in all-cause mortality related to hormone therapy at 30-year follow-up. Patients were exposed to exogenous estrogen from 2 months to 41 years.9 A follow-up of this study published in 2013, which included 2,307 MTF individuals taking estrogen for 5 years to > 30 years, revealed only 2 cases of breast cancer, which was the same incidence rate (4.1 per 100,000 person-years) as that of nontransgender women.10
In general, the incidence of breast cancer is rare in nontransgender men, and therefore there have not been a lot of clinical studies to assess risk factors and detection methods. The following risk factors can increase the risk of breast cancer in nontransgender patients: known presence of BRCA mutation, estrogen exposure/androgen insufficiency, Klinefelter syndrome, liver cirrhosis, and obesity.11
Guidelines from the Endocrine Society, WPATH, and UCSF suggest that MTF transgender individuals who have a known increased risk for breast cancer should follow screening guidelines recommended for nontransgender women if they are aged > 50 years and have had more than 5 years of hormone use.2 For FTM patients who have not had chest surgery, screening guidelines should follow those for nontransgender women. For those patients who have had chest reconstruction, small residual amounts of breast tissue may remain. Screening guidelines for these patients do not exist. For these patients, mammography can be technically difficult. Clinical chest wall examination, magnetic resonance imaging (MRI), and/or ultrasound may be helpful modalities. An individual risk vs benefit discussion with the patient is recommended.
Prostate Cancer Screening
Although the prostate gland will undergo atrophy with extended treatment with feminizing hormone therapy, there are case reports of prostate cancer in transgender women.12,13 Usually these patients have started hormone treatment after age 50 years. Therefore, prostate cancer screening is recommended in transgender women as per general guidelines. Because the prostate-specific antigen (PSA) level is expected to be reduced, a PSA > 1.0 should be considered abnormal.1
Cervical Cancer Screening
When a transgender man has a pap smear, it is essential to make it clear to the laboratory that the sample is a cervical pap smear (especially if the gender is marked as male) to avoid the sample being run incorrectly as an anal pap. Also, it is essential to indicate on the pap smear request form that the patient is on testosterone therapy and amenorrhea is present, because the lack of the female hormone can cause atrophy of cervix. This population has a high rate of inadequate specimens. Pretreatment with 1 to 2 weeks of vaginal estrogen can improve the success rate of inadequate specimens. Transgender women who have undergone vaginoplasty do not have a cervix, therefore, cervical cancer screening is not recommended. The anatomy of the neovagina has a more posterior orientation, and an anoscope is a more appropriate tool to examine the neovagina when necessary.
Hematology Health
Transgender women on cross-sex hormone therapy with estrogens may be at increased risk for a venous thromboembolism (VTE). In 2 European studies, patients treated with oral ethinyl estradiol as well as the anti-androgen cyproterone acetate were found to have up to 20 times increased risk of VTE. However, in later studies, oral ethinyl estradiol was changed to either oral conjugated estrogens or transdermal/intramuscular estradiol, and these studies did not show a significant increase in VTE risk.14-16 Tobacco use in combination with estrogen therapy is associated with an increased risk of deep vein thrombosis (DVT).1 All transgender women who smoke should be counseled on tobacco risks and cessation options at every visit.1 The transgender individuals who are not willing to quit smoking may be offered transdermal estrogen, which has lower risk of DVT.14-16
Sexual Health
Clinicians should assess the risks for sexually transmitted infection (STIs) or HIV for transgender patients based on current anatomy and sexual behaviors. Presentations of STIs can be atypical due to varied sexual practices and gender-affirming surgeries. Thus, providers must remain vigilant for symptoms consistent with common STIs and screen for asymptomatic STIs on the basis of behavior history and sexual practices.17 Preexposure prophylaxis for HIV should be considered when appropriate. Serologic screening recommendations for transgender people (eg, HIV, hepatitis B and C, syphilis) do not differ in recommendations from those for nontransgender people.
Cardiovascular Health
The effect of cross-hormone treatment on cardiovascular (CV) health is still unknown. There are no randomized controlled trials that have investigated the relationship between cross-hormone treatment and CV health. Evidence from several studies suggests that CV risk is unchanged among transgender men using testosterone compared with that of nontransgender women.18,19 There is conflicting evidence for transgender women with respect to CV risk and cross-sex hormone treatment.1,18,19 The current American College of Cardiology/American Heart Association guideline advises using the ASCVD risk calculator to determine the need for aspirin and statin treatment based on race, age, gender, and risk factors. There is no guideline on whether to use natal sex or affirmed gender while using the ASCVD calculator. It is reasonable to use the calculator based on natal sex if the cross-hormone treatment has started later in life, but if the cross-sex hormone treatment started at a young age, then one should consider using the affirmed gender to calculate the risk.
As with all patients, life style modifications, including smoking cessation, weight loss, physical activity, and management of BP and blood sugar, are important for CV health. For transgender women with CV risk factors or known CV disease, transdermal route of estrogen is preferred due to lower rates of VTE.18,19
Conclusion
In recent years, an increased number of transgender individuals are seeking mainstream medical care. However, PCPs often lack the knowledge and training to properly interact with and care for transgender patients. It is critical that clinicians understand the difference between sex, gender, and sexuality. For patients who desire transgender care, providers must be able to comfortably ask the patient about their preferred name and prior care, know the basics in cross-sex hormone therapy, including appropriate follow-up of hormonal levels as well as laboratory tests that delineate risk, and know possible complications and AEs. The VA offers significant resources, including electronic transgender care consultation for cases where the provider does not have adequate expertise in the care of these patients.
Both medical schools and residency training programs are starting to incorporate curricula regarding LGBT care. For those who have already completed training, this article serves as a brief guide to terminology, interactive tips, and management of this growing and underserved group of individuals.
1. Deutsch MB. Guidelines for the primary and gender-affirming care of transgender and gender nonbinary people. http://transhealth.ucsf.edu/protocols. Updated June 17, 2016. Accessed June 13, 2018.
2. Hembree WC, Cohen-Kettenis PT, Gooren L, et al. Endocrine treatment of gender-dysphoria/gender-incongruent persons: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2017;102(11):3869-3903.
3. Buchholz L. Transgender care moves into the mainstream. JAMA. 2015;314(17):1785-1787.
4. Sobralske M. Primary care needs of patients who have undergone gender reassignment. J Am Acad Nurse Pract. 2005;17(4):133-138.
5. Unger CA. Hormone therapy for transgender patients. Transl Androl Urol. 2016;5(6):877-884.
6. Kanhai RC, Hage JJ, van Diest PJ, Bloemena E, Mulder JW. Short-term and long-term histologic effects of castration and estrogen treatment on breast tissue of 14 male-to-female transsexuals in comparison with two chemically castrated men. Am J Surg Pathol. 2000;24(1):74-80.
7. Braun H, Nash R, Tangpricha V, Brockman J, Ward K, Goodman M. Cancer in transgender people: evidence and methodological consideration. Epidemiol Rev. 2017;39(1):93-107.
8. Brown GR, Jones KT. Incidence of breast cancer in a cohort of 5,135 transgender veterans. Breast Cancer Res Treat. 2015;149(1):191-198.
9. Van Kesteren PJ, Asscheman H, Megens JA, Gooren LJ. Mortality and morbidity in transsexual subjects treated with cross-sex hormones. Clin Endocrinol (Oxf). 1997;47(3):337-342.
10. Gooren LJ, van Trotsenburg MA, Giltay EJ, van Diest PJ. Breast cancer development in transsexual subjects receiving cross-sex hormone treatment. J Sex Med. 2013;10(12):3129-3134.
11. Johansen Taber KA, Morisy LR, Osbahr AJ III, Dickinson BD. Male breast cancer: risk factors, diagnosis and management (review). Oncol Rep. 2010;24(5):1115-1120.
12. Miksad RA, Bubley G, Church P, et al. Prostate cancer in a transgender woman, 41 years after initiation of feminization. JAMA. 2006;296(19):2316-2317.
13. Turo R, Jallad S, Prescott S, Cross WR. Metastatic prostate cancer in transsexual diagnosed after three decades of estrogen therapy. Can Urol Assoc J. 2013;7(7-8):E544-E546.
14. American College of Obstetricians and Gynecologists. ACOG committee opinion no. 556: postmenopausal estrogen therapy: route of administration and risk of venous thromboembolism. Obstet Gynecol. 2013;121(4):887-890.
15. Asscheman H, Gooren LJ, Eklund PL. Mortality and morbidity in transsexual patients with cross-gender treatment. Metabolism. 1989;38(9):869-873.
16. Asscheman H, Giltay EJ, Megens JA, de Ronde WP, van Trotsenburg MA, Gooren LJ. A long-term follow-up study of mortality in transsexuals receiving treatment with cross-sex hormones. Eur J Endocrinol. 2011;164(4):635-642.
17. Workowski KA, Bolan GA; Centers for Disease Control and Prevention. Sexually transmitted disease treatment guidelines, 2015. MMWR Recomm Rep. 2015;64(RR-03):1-137.
18. Gooren LJ, Wierckx K, Giltay EJ. Cardiovascular disease in transsexual persons treated with cross-sex hormones: reversal of the traditional sex difference in cardiovascular disease pattern. Eur J Endocrinol. 2014;170(6):809-819.
19. Streed CG Jr, Harfouch O, Marvel F, Blumenthal RS, Martin SS, Mukherjee M. Cardiovascular disease among transgender adults receiving hormone therapy: a narrative review. Ann Int Med. 2017;167(4):256-267.
1. Deutsch MB. Guidelines for the primary and gender-affirming care of transgender and gender nonbinary people. http://transhealth.ucsf.edu/protocols. Updated June 17, 2016. Accessed June 13, 2018.
2. Hembree WC, Cohen-Kettenis PT, Gooren L, et al. Endocrine treatment of gender-dysphoria/gender-incongruent persons: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2017;102(11):3869-3903.
3. Buchholz L. Transgender care moves into the mainstream. JAMA. 2015;314(17):1785-1787.
4. Sobralske M. Primary care needs of patients who have undergone gender reassignment. J Am Acad Nurse Pract. 2005;17(4):133-138.
5. Unger CA. Hormone therapy for transgender patients. Transl Androl Urol. 2016;5(6):877-884.
6. Kanhai RC, Hage JJ, van Diest PJ, Bloemena E, Mulder JW. Short-term and long-term histologic effects of castration and estrogen treatment on breast tissue of 14 male-to-female transsexuals in comparison with two chemically castrated men. Am J Surg Pathol. 2000;24(1):74-80.
7. Braun H, Nash R, Tangpricha V, Brockman J, Ward K, Goodman M. Cancer in transgender people: evidence and methodological consideration. Epidemiol Rev. 2017;39(1):93-107.
8. Brown GR, Jones KT. Incidence of breast cancer in a cohort of 5,135 transgender veterans. Breast Cancer Res Treat. 2015;149(1):191-198.
9. Van Kesteren PJ, Asscheman H, Megens JA, Gooren LJ. Mortality and morbidity in transsexual subjects treated with cross-sex hormones. Clin Endocrinol (Oxf). 1997;47(3):337-342.
10. Gooren LJ, van Trotsenburg MA, Giltay EJ, van Diest PJ. Breast cancer development in transsexual subjects receiving cross-sex hormone treatment. J Sex Med. 2013;10(12):3129-3134.
11. Johansen Taber KA, Morisy LR, Osbahr AJ III, Dickinson BD. Male breast cancer: risk factors, diagnosis and management (review). Oncol Rep. 2010;24(5):1115-1120.
12. Miksad RA, Bubley G, Church P, et al. Prostate cancer in a transgender woman, 41 years after initiation of feminization. JAMA. 2006;296(19):2316-2317.
13. Turo R, Jallad S, Prescott S, Cross WR. Metastatic prostate cancer in transsexual diagnosed after three decades of estrogen therapy. Can Urol Assoc J. 2013;7(7-8):E544-E546.
14. American College of Obstetricians and Gynecologists. ACOG committee opinion no. 556: postmenopausal estrogen therapy: route of administration and risk of venous thromboembolism. Obstet Gynecol. 2013;121(4):887-890.
15. Asscheman H, Gooren LJ, Eklund PL. Mortality and morbidity in transsexual patients with cross-gender treatment. Metabolism. 1989;38(9):869-873.
16. Asscheman H, Giltay EJ, Megens JA, de Ronde WP, van Trotsenburg MA, Gooren LJ. A long-term follow-up study of mortality in transsexuals receiving treatment with cross-sex hormones. Eur J Endocrinol. 2011;164(4):635-642.
17. Workowski KA, Bolan GA; Centers for Disease Control and Prevention. Sexually transmitted disease treatment guidelines, 2015. MMWR Recomm Rep. 2015;64(RR-03):1-137.
18. Gooren LJ, Wierckx K, Giltay EJ. Cardiovascular disease in transsexual persons treated with cross-sex hormones: reversal of the traditional sex difference in cardiovascular disease pattern. Eur J Endocrinol. 2014;170(6):809-819.
19. Streed CG Jr, Harfouch O, Marvel F, Blumenthal RS, Martin SS, Mukherjee M. Cardiovascular disease among transgender adults receiving hormone therapy: a narrative review. Ann Int Med. 2017;167(4):256-267.
The Clinical Pathophysiology of Chronic Systemic Sclerosis
Systemic sclerosis (SSc), also called scleroderma, is a rare but serious autoimmune connective tissue disease that has multiple fluctuating pathologic manifestations throughout its temporal course. Estimates have shown that the incidence is 10 to 20 cases per 1 million, and the prevalence is 4 to 253 cases per 1 million.1,2 Given the rarity of this incurable condition, it is vital that primary care providers (PCPs) are able to recognize its unique features early to limit and prevent acute and chronic complications. This case report discusses a patient’s journey with late-diagnosed scleroderma in order to convey these broad manifestations and what providers can do to manage it with their patients.
Case Presentation
Mr. P is a 60-year-old African American male with a history of hypertension, recurrent digital ulcers, pulmonary hypertension (PH), interstitial lung disease (ILD), kidney involvement, congestive heart failure (CHF), and gastroesophageal reflux disease (GERD). Mr. P’s workup began in his late 40s with resistant hypertension, resistant GERD, and multiple hospitalizations for hypertensive urgency. It was not until he was 54 years old that he was diagnosed with mixed connective tissue disorder with sclerodermatous predominance.
Review of systems throughout his medical examinations in his 50s were notable for skin tightening over his hands and shoulders, skin hypopigmentation over his scalp and face, and hair loss. Mr. P was found to have Raynaud phenomenon beginning with his original presentation and digital ulceration without complications of gangrene or autoamputation. Aggregate physical examinations were notable for digital ulceration, skin tightening/sclerodactyly, and telangiectasia. Serologic markers were notable for the following:
- Positive ANA (antinuclear antibody) with titer of 1:1,280/homogenous pattern;
- Positive anti-RNP (antiribonucleoprotein) with titer of 171.2;
- Positive anti-Scl-70 (antitopoisomerase I) with titer of 108.1;
- Positive anti-SM (anti-Smith antibody) with titer of 30.2;
- Positive anti-Ro (SSA) with titer of 107.6;
- Negative anti-La (SSB) with titer of 1.3;
- Negative anti-dsDNA (anti-double stranded DNA) with titer of 9; and
- Negative ACA (anticentromere antibody).
Early transthoracic echocardiograms revealed an ejection fraction (EF) initially at 55% with evidence of left ventricular hypertrophy. Following treatment with phosphodiesterase-5 inhibitors (PDE-5 inhibitors) and endothelin-1 antagonists for pulmonary hypertension, serial transthoracic echocardiograms showed improvement in his EF. 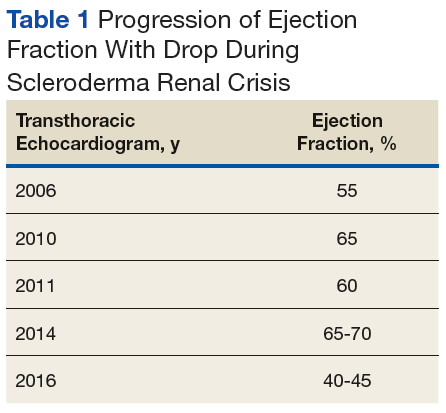
A chest X-ray did not show signs of ILD, but a subsequent high-resolution computed tomography (HRCT) scan was consistent with chronic ILD with a main pulmonary artery diameter of 3 cm (Figure 1). 
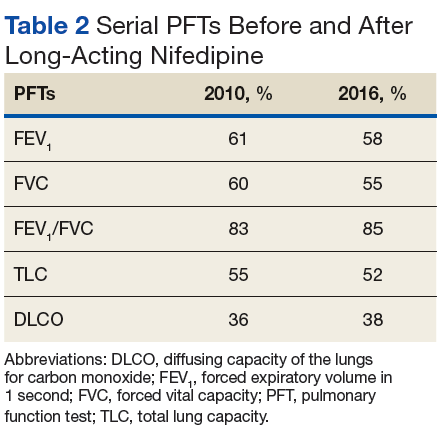
In subsequent years, Mr. P was hospitalized several times, secondary to digit pain, ulcerations, and osteomyelitis. His first episode was 1 year after his scleroderma diagnosis, when he was hospitalized for 6 days for complications of SSc and finger pain. The following year, he had a 3-day hospitalization for hypertensive urgency and right third-digit osteomyelitis, treated initially with IV fluids, levofloxacin, and vancomycin, and then ceftaroline for 1 month. Throughout the next 6 years, Mr. P presented multiple times with fingertip ulcerations and was followed in the Infectious Disease clinic for recurrent osteomyelitis. He found some relief with systemic antibiotics, including augmentin, minocycline, moxifloxacin, and doxycycline.
At age 59, he was hospitalized for scleroderma renal crisis (SRC). Early in his disease, his kidney function was normal, but the SRC was discovered after an abrupt rise in his blood pressure (BP) and an increase in serum creatinine (SCr) from 1.2 mg/dL at baseline to 3.06 mg/dL. The presence of brown granular cast in his urine prompted a renal biopsy that showed thrombotic microangiopathy with schistocytes. Mr. P was started on captopril and remained stable with outpatient follow-up for this renal complication.
Discussion
Initial presentation of SSc can occur along a spectrum of its pathophysiology. A more severe presentation, like the one seen in Mr. P, seems to occur more frequently in African American patients relative to white patients.3 Differentiating between the 2 types—diffuse vs limited SSc—is vital to managing patients and disease progression. Limited-type SSc is more common (60%) and less severe with slower progression than is diffuse SSc. 
Diffuse-type SSc (35%) includes features such as skin thickening and tightening, ILD, SRC, tendon friction rubs (palpable crepitus over tendons), and skin pigment changes.4 The specific involvement of the renal and cardiopulmonary systems accounts for the higher mortality rates in the diffuse-type.5
Many patients with SSc require periodic hospitalizations throughout their life for the acute complications of the disease. Hospitalized patients often range from age 45 to 64 years and are more often female. However, of hospitalized patients with SSc, in-hospital death rates are higher among men.3,6 Although these rates have decreased as the pathogenesis of SSc has become better understood, it is important to note that in-hospital mortality in 1995 for all patients with SSc was 7.1% and mean length of stay was 7.5 days, and in 2002 to 2003, 6.3% and 6.6 days, respectively.3 Though the burden of this disease has decreased, mortality and hospitalizations continue to persist at high rates. Understanding the pathogenesis, progression, and treatments of SSc are essential to aiding patients with this diagnosis.
Skin Involvement
A common finding and presentation for patients with SSc is related to skin involvement. Common patient complaints and exam findings include calcinosis along extensor tendons and digits, Raynaud phenomenon (seen in more than 95% of patients), sclerodactyly, telangiectasias, hyper/hypopigmentation, and pruritus.4 These findings are useful in diagnosing and monitoring patients for disease progression.
Many of the listed skin manifestations affect patients’ quality of life (QOL) but are not directly associated with mortality. However, a common and feared complication includes skin ulcers and osteomyelitis, seen in 48% and 7.7% of patients, respectively.7 Digit ulcers, areas with loss of dermis and epidermis distal to the proximal interphalangeal joints (Figures 2A and 2B), are significant because they parallel a more rapid progression of internal organ involvement.8 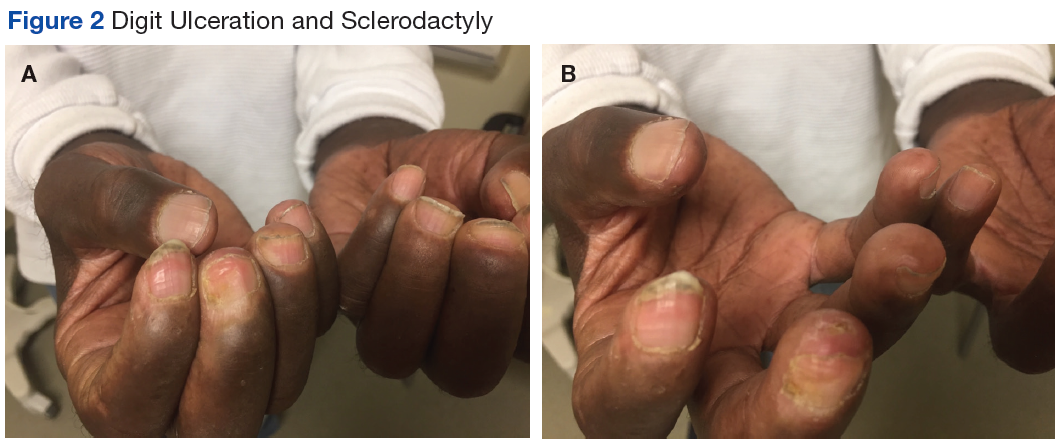
Mr. P required multiple hospitalizations and antibiotic regimens for painful digit ulcers complicated by osteomyelitis (Figure 3). 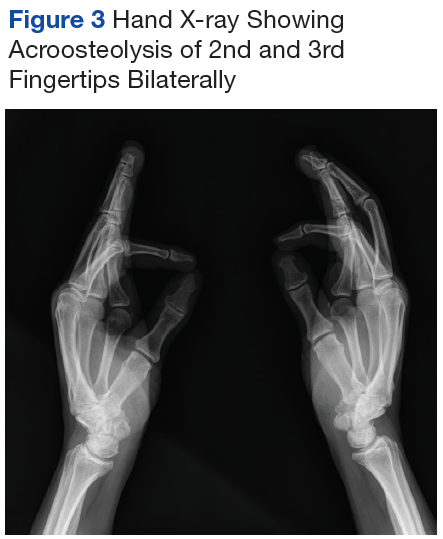
Treatment usually is aimed at infections that complicate these skin ulcers and are based on site-specific cultures. Preventive measures are aimed at the risk factors associated with digit ulcers, including decreased whole-body warmth, direct trauma to digits, smoking, and vasoconstrictors (eg, cocaine, sympathomimetics).8 Some patients may prevent ulcers by using D-penicillamine, mycophenolate mofetil, and cyclophosphamide, although definitive treatment has not been found.4 Calcium channel blockers, PDE-5 inhibitors, endothelin receptor antagonists, and prostacyclin analogues also have been used to reduce the severity of Raynaud phenomenon attacks and to decrease the number of digital ulcers (in addition to their beneficial effects on pulmonary involvement).8 Additional pharmacologic agents that have been linked to an improvement in Raynaud phenomenon and digital ulcers include statins, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, intravenous N-acetylcysteine, vitamin E gel, and surgical options (eg, revascularization and sympathectomy).8
Pulmonary Involvement
Systemic sclerosis can lead to 2 complications in the lungs, both present in Mr. P: PAH (mean pulmonary arterial pressure > 25 mm Hg) and ILD, which together make lung involvement the leading cause of death for these patients. Limited-type SSc usually is restricted to PAH, whereas diffuse-type SSc usually leads to ILD.4
On diagnosis of SSc, an HRCT is indicated to assess the degree of lung involvement. Mr. P’s HRCT showed a 3-cm main pulmonary artery, suggestive of PAH, in addition to evidence of ILD seen on the chest X-ray (Figure 4). 
Pulmonary arterial hypertension is a common finding in patients with SSc and carries a severe prognosis. Risk factors for the development of PAH, when not present on initial diagnosis, include limited-type SSc; late age of onset; Raynaud phenomenon; decreased DLCO, FVC/DLCO < 1.6; increased N-terminal pro-B-type natriuretic peptide (NT-proBNP) serum levels; and the presence of antibodies.9 Patients with both SSc and PAH have a 50% to 87% 1-year survival, whereas patients with idiopathic PAH without connective tissue disease have an 88% 1-year survival.9
Many patients with lung involvement are asymptomatic, but some have findings of crackles and interstitial thickening on chest X-ray that can progress to cyanosis and right heart failure (cor pulmonale).10 On discovery of lung involvement (Figure 5), it is important to follow up with a PFT, primarily because the outcomes and prognosis for patients with SSc are correlated with the presenting severity of ILD and the subsequent progression of their DLCO.4,10 
Treatment is directed at PAH and ILD separately. For PAH, a PDE-5 inhibitor, such as tadalafil or sildenafil, and an endothelin-1 receptor antagonist, such as ambrisentan or bosentan, are indicated. Other PAH treatments include diuretics and prostacyclin analogues (eg, epoprostenol, treprostinil, or iloprost) in addition to warfarin if patients have a history of thrombotic events.4 Mr. P’s pulmonologists deferred treatment with prostacyclin analogues given the potential for adverse effects with endothelin-1 receptor inhibitors, PDE-5 inhibitors, and calcium-channel blockers, although combination studies with bosentan and inhaled iloprost have shown promise.11
Like the skin manifestations of SSc, ILD lacks definitive treatment to prevent disease progression. However, some patients benefit from cyclophosphamide, followed by mycophenolate mofetil—which is usually better tolerated—azathioprine, haematopoietic stem cell transplant as rescue therapy, and lung transplant for life-saving treatment.10
Renal Involvement
Renal involvement in patients with SSc can have profound effects on QOL. Even without clinical renal involvement, glomerular filtration rate (GFR) usually decreases with the progression of vascular damage correlated with age and disease duration.12 Patients with a history of digital ulcers, like Mr. P, usually have a lower GFR than that of patients without digital ulcers.12 Monitoring renal function is vital in caring for these patients, because SRC occurs in 2% to 15% of all patients with SSc.13,14
Scleroderma renal crisis is generally defined as an abruptly elevated BP (> 140/90 mm Hgor > 30 mm Hg rise from baseline) with acute renal failure (elevated SCr) and decreased urine output.14 Given the rarity of SSc in the population, diagnosis of SRC requires high clinical suspicion. In the case of Mr. P, a workup involving serum analysis, urinalysis, and renal biopsy allowed for a definitive diagnosis. A renal biopsy can show microangiopathic hemolytic anemia and confirm SRC, although it may not be necessary in patients with known SSc presenting with new hypertension, rising creatinine, and unremarkable urine sediment on microscopy.14,15
Although an acute SRC can be difficult to predict, monitoring renal function and attention to key factors can assist in discovering this SSc complication. Scleroderma renal crisis usually occurs within the first 4 years of SSc diagnosis, often paralleling rapid progression of skin thickening and tightening with higher rates in both African Americans and males.13,14 Additional predictive factors include diffuse skin involvement, rapid progression of skin involvement, positive anti-RNA polymerase III antibodies, new anemia, new cardiac events (eg, pericardial effusion, pericarditis, left ventricular insufficiency), CHF, tendon friction rubs, arthritis, and recent (within 3 months) high-dose glucocorticoid use.4,13-15
The presentation of SRC can be nonspecific, often resembling findings related to acute kidney injury. Patients may report malaise, fatigue, fever, headache, seizure, blurred vision, or dyspnea.13 Clinical parametersto examine include systolic BP > 140 mm Hgand/or diastolic BP > 90 mm Hg (or an abrupt rise of > 30 or > 20, respectively), SCr increase by 50%+ or > 120% of the upper normal limit, proteinuria at 2+, high protein:creatinine ratio, hematuria > 2+ or > 10 red blood cells (RBCs), platelets < 100,000/mm3, and hypertensive encephalopathy.13 Mr. P presented with fatigue, dyspnea, an abrupt rise in BP (156/96 mm Hg), foamy urine, bilateral lower extremity edema (3.9 g/dL albumin), 2+ RBCs, and a SCr of 2.46 mg/dL on admission (a 205% increase from his last baseline of 1.2 mg/dL).
Treatments have had a large impact on the mortality rates of SRC. Following the introduction of ACE inhibitors, mortality from SRC has decreased from 76% to < 10% over recent decades.13 In addition to improving survival, these medications also have improved hospitalization outcomes for these patients.3 Captopril is often the medication of choice given its rapid onset and short duration of action relative to other medications in the same class, such as benazepril, enalapril, fosinopril, lisinopril, quinapril, and ramipril.4,13 Current clinical trials are assessing the specific renal benefits of endothelin-1 receptor antagonists, which often are used for pulmonary and skin involvement.14 For patients presenting with acute SRC and uremia, dialysis may be necessary (typically predicted by elevated NT-proBNP serum levels), while the decision for transplantation may be indicated at least 2 years after SRC onset and resolution.4,13-15
Once the acute crisis resolves, it is important to discuss prognosis with patients. The 1-, 2-, 3-, 5-, and 10-year survival rates are 82%, 74%, 71%, 59%, and 47%, respectively; however, when looking at only male patients at 10 years, the survival rate is 17%.13 Prevention of SRC can be addressed with daily BP checks and advising patients to seek medical care if they notice a consecutive 2-day abrupt rise.13
Cardiac Involvement
Systemic sclerosis has significant effects on patients’ heart function. The introduction of ACE inhibitors has shifted mortality in SSc patients from predominantly SRC to cardiac causes.6 Cardiac involvement can occur through a range of processes, including abnormal cardiac conduction, CHF, diastolic dysfunction, mitral valve nodular thickening, and pericardial effusion.3 Even though patients often present with skin findings, an initial cardiac workup is crucial to understanding disease progression and patient prognosis. The severity of the cutaneous manifestations often predicts the degree of diastolic dysfunction.16
Clinical evidence of cardiac involvement is seen in 20% to 25% of patients with SSc and is associated with a 70% mortality at 5 years when symptoms are evident.16 Additionally, right ventricular dysfunction at presentation is the strongest marker for all-mortality prognosis, representing the degree of pulmonary involvement, and includes findings such as progressive shortness of breath and systemic edema.9
Given the increasing survival of patients with SSc, cardiac involvement is becoming more evident and prominent. Direct treatments for cardiac manifestations are based on the causative feature, namely, focusing on pulmonary and renal involvement, which can be assessed with periodic echocardiograms evaluating left ventricular EF.
Gastrointestinal Involvement
Along with skin manifestations, the gastrointestinal (GI) involvement of SSc can have a significant impact on patients’ QOL without direct contribution to mortality. One of Mr. P’s earliest symptoms that led to a diagnosis of SSc was GERD, which caused a chronic cough, dental erosions, esophageal erosions, duodenal ulcers, dysphagia, abdominal pain, halitosis, pharyngitis, and weight loss. Esophageal involvement occurs in up to 96% of patients with SSc and can include motility abnormalities (eg, strictures and/or muscle dysfunction), lower esophageal sphincter abnormalities, and Barrett esophagus.17
Additional symptoms of the GI system linked with SSc can occur anywhere along the GI tract and include gastric antral vascular ectasia, causing GI bleeds and pernicious anemia, gastroparesis, bacterial overgrowth, intestinal malabsorption, pseudo-obstruction due to hypomotility, fecal incontinence due to anorectal involvement, and rarely, primary biliary cirrhosis.4,17 Decreased mobility of the oral aperture secondary to skin thickening and tightening also can contribute to malnutrition by decreasing oral intake.
Treatments are supportive and target symptom relief. Chronic treatment of GERD is often necessary and includes antacids, histamine-2 receptor blockers, and proton pump inhibitors.4 Other medications that can help with symptom relief include motility agents (such as metoclopramide, domperidone, prucalopride, tegaserod, and macrolides), osmotic laxatives, and ursodeoxycholic acid for primary biliary cirrhosis.17 Surgical intervention should be considered depending on the severity and progression of involvement within the GI tract. Behavioral changes that can improve patient symptoms include facial grimacing and other mouth-stretching exercises, frequent smaller meals followed by maintaining a vertical posture, and high fiber diets.17
Conclusion
Systemic sclerosis is an autoimmune and connective tissue disease with a pathophysiology that can manifest throughout the body. The organ systems that impact patient outcomes include skin, pulmonary, renal, cardiac, and GI. Primary care providers caring for patients diagnosed with SSc should monitor acute management and disease progression in all these systems. Important acute events that can impact morbidity, mortality, and/or QOL include Raynaud phenomenon, SRC, and pericardial effusion. Chronic manifestations that may be present on diagnosis of SSc or may develop while a patient is under a provider’s care include sclerodactyly, tendon calcinosis, PAH, ILD, chronic kidney injury, chronic cardiac damage, GERD, and esophageal dysmotility. While this discussion serves as a pertinent overview of patients with SSc, it is summative, and providers are encouraged to seek a stronger understanding of both the common and rarer manifestations within each of their patients.
1. Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41(5):778-799.
2. Silman AG. Scleroderma. In: Epidemiology of the Rheumatic Diseases. 2nd ed. Silman AJ, Hochberg MC, eds. Oxford, UK: Oxford University Press. 1993:chap 8.
3. Chung L, Krishnan E, Chakravarty EF. Hospitalizations and mortality in systemic sclerosis: results from the Nationwide Inpatient Sample. Rheumatology (Oxford). 2007;46(12):1808-1813.
4. Hinchcliff M, Varga J. Systemic sclerosis/scleroderma: a treatable multisystem disease. Am Fam Physician. 2008;78(8):961-968.
5. Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809-1815.
6. Piga M, Casula L, Sanna S, et al. Population-based analysis of hospitalizations for patients with systemic sclerosis in a West-European region over the period 2001-2012. Rheumatol Int. 2016;36(1):73-81.
7. Giuggioli D, Manfredi A, Colaci M, Lumetti F, Ferri C. Osteomyelitis complicating scleroderma digital ulcers. Clin Rheumatol. 2013;32(5):623-627.
8. Schiopu E, Impens AJ, Phillips K. Digital ischemia in scleroderma spectrum of diseases. Int J Rheumatol. 2010;2010:923743.
9. Hassoun PM. Therapies for scleroderma-related pulmonary arterial hypertension. Expert Rev Respir Med. 2009;3(2):187-196.
10. Giacomelli R, Liakouli V, Berardicurti O, et al. Interstitial lung disease in systemic sclerosis: current and future treatment. Rheumatol Int. 2017;37(6):853-863.
11. McLaughlin V, Humbert M, Coghlan G, Nash P, Steen V. Pulmonary arterial hypertension: the most devastating vascular complication of systemic sclerosis. Rheumatology (Oxford). 2009; (suppl 3):iii25-iii31.
12. Gigante A, Barbano B, Granata G, et al. Evaluation of estimated glomerular filtration rate and clinical variables in systemic sclerosis patients. Clin Nephrol. 2016;85(6):326-331.
13. Bose N, Chiesa-Vottero A, Chatterjee S. Scleroderma renal crisis. Semin Arthritis Rheum. 2015;44(6):687-694.
14. Woodworth TG, Suliman YA, Furst DE, Clements P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat Rev Nephrol. 2016;12(11):678-691.
15. Mouthon L, Bérezné A, Bussone G, Noël LH, Villiger PM, Guillevin L. Scleroderma renal crisis: a rare but severe complication of systemic sclerosis. Clin Rev Allergy Immunol. 2011;40(2):84-91.
16. Champion HC. The heart in scleroderma. Rheum Dis Clin North Am. 2008;34(1):181-90.
17. Tian XP, Zhang X. Gastrointestinal complications of systemic sclerosis. World J Gastroenterol. 2013;19(41):7062-7068.
Systemic sclerosis (SSc), also called scleroderma, is a rare but serious autoimmune connective tissue disease that has multiple fluctuating pathologic manifestations throughout its temporal course. Estimates have shown that the incidence is 10 to 20 cases per 1 million, and the prevalence is 4 to 253 cases per 1 million.1,2 Given the rarity of this incurable condition, it is vital that primary care providers (PCPs) are able to recognize its unique features early to limit and prevent acute and chronic complications. This case report discusses a patient’s journey with late-diagnosed scleroderma in order to convey these broad manifestations and what providers can do to manage it with their patients.
Case Presentation
Mr. P is a 60-year-old African American male with a history of hypertension, recurrent digital ulcers, pulmonary hypertension (PH), interstitial lung disease (ILD), kidney involvement, congestive heart failure (CHF), and gastroesophageal reflux disease (GERD). Mr. P’s workup began in his late 40s with resistant hypertension, resistant GERD, and multiple hospitalizations for hypertensive urgency. It was not until he was 54 years old that he was diagnosed with mixed connective tissue disorder with sclerodermatous predominance.
Review of systems throughout his medical examinations in his 50s were notable for skin tightening over his hands and shoulders, skin hypopigmentation over his scalp and face, and hair loss. Mr. P was found to have Raynaud phenomenon beginning with his original presentation and digital ulceration without complications of gangrene or autoamputation. Aggregate physical examinations were notable for digital ulceration, skin tightening/sclerodactyly, and telangiectasia. Serologic markers were notable for the following:
- Positive ANA (antinuclear antibody) with titer of 1:1,280/homogenous pattern;
- Positive anti-RNP (antiribonucleoprotein) with titer of 171.2;
- Positive anti-Scl-70 (antitopoisomerase I) with titer of 108.1;
- Positive anti-SM (anti-Smith antibody) with titer of 30.2;
- Positive anti-Ro (SSA) with titer of 107.6;
- Negative anti-La (SSB) with titer of 1.3;
- Negative anti-dsDNA (anti-double stranded DNA) with titer of 9; and
- Negative ACA (anticentromere antibody).
Early transthoracic echocardiograms revealed an ejection fraction (EF) initially at 55% with evidence of left ventricular hypertrophy. Following treatment with phosphodiesterase-5 inhibitors (PDE-5 inhibitors) and endothelin-1 antagonists for pulmonary hypertension, serial transthoracic echocardiograms showed improvement in his EF.
A chest X-ray did not show signs of ILD, but a subsequent high-resolution computed tomography (HRCT) scan was consistent with chronic ILD with a main pulmonary artery diameter of 3 cm (Figure 1).
In subsequent years, Mr. P was hospitalized several times, secondary to digit pain, ulcerations, and osteomyelitis. His first episode was 1 year after his scleroderma diagnosis, when he was hospitalized for 6 days for complications of SSc and finger pain. The following year, he had a 3-day hospitalization for hypertensive urgency and right third-digit osteomyelitis, treated initially with IV fluids, levofloxacin, and vancomycin, and then ceftaroline for 1 month. Throughout the next 6 years, Mr. P presented multiple times with fingertip ulcerations and was followed in the Infectious Disease clinic for recurrent osteomyelitis. He found some relief with systemic antibiotics, including augmentin, minocycline, moxifloxacin, and doxycycline.
At age 59, he was hospitalized for scleroderma renal crisis (SRC). Early in his disease, his kidney function was normal, but the SRC was discovered after an abrupt rise in his blood pressure (BP) and an increase in serum creatinine (SCr) from 1.2 mg/dL at baseline to 3.06 mg/dL. The presence of brown granular cast in his urine prompted a renal biopsy that showed thrombotic microangiopathy with schistocytes. Mr. P was started on captopril and remained stable with outpatient follow-up for this renal complication.
Discussion
Initial presentation of SSc can occur along a spectrum of its pathophysiology. A more severe presentation, like the one seen in Mr. P, seems to occur more frequently in African American patients relative to white patients.3 Differentiating between the 2 types—diffuse vs limited SSc—is vital to managing patients and disease progression. Limited-type SSc is more common (60%) and less severe with slower progression than is diffuse SSc.
Diffuse-type SSc (35%) includes features such as skin thickening and tightening, ILD, SRC, tendon friction rubs (palpable crepitus over tendons), and skin pigment changes.4 The specific involvement of the renal and cardiopulmonary systems accounts for the higher mortality rates in the diffuse-type.5
Many patients with SSc require periodic hospitalizations throughout their life for the acute complications of the disease. Hospitalized patients often range from age 45 to 64 years and are more often female. However, of hospitalized patients with SSc, in-hospital death rates are higher among men.3,6 Although these rates have decreased as the pathogenesis of SSc has become better understood, it is important to note that in-hospital mortality in 1995 for all patients with SSc was 7.1% and mean length of stay was 7.5 days, and in 2002 to 2003, 6.3% and 6.6 days, respectively.3 Though the burden of this disease has decreased, mortality and hospitalizations continue to persist at high rates. Understanding the pathogenesis, progression, and treatments of SSc are essential to aiding patients with this diagnosis.
Skin Involvement
A common finding and presentation for patients with SSc is related to skin involvement. Common patient complaints and exam findings include calcinosis along extensor tendons and digits, Raynaud phenomenon (seen in more than 95% of patients), sclerodactyly, telangiectasias, hyper/hypopigmentation, and pruritus.4 These findings are useful in diagnosing and monitoring patients for disease progression.
Many of the listed skin manifestations affect patients’ quality of life (QOL) but are not directly associated with mortality. However, a common and feared complication includes skin ulcers and osteomyelitis, seen in 48% and 7.7% of patients, respectively.7 Digit ulcers, areas with loss of dermis and epidermis distal to the proximal interphalangeal joints (Figures 2A and 2B), are significant because they parallel a more rapid progression of internal organ involvement.8
Mr. P required multiple hospitalizations and antibiotic regimens for painful digit ulcers complicated by osteomyelitis (Figure 3).
Treatment usually is aimed at infections that complicate these skin ulcers and are based on site-specific cultures. Preventive measures are aimed at the risk factors associated with digit ulcers, including decreased whole-body warmth, direct trauma to digits, smoking, and vasoconstrictors (eg, cocaine, sympathomimetics).8 Some patients may prevent ulcers by using D-penicillamine, mycophenolate mofetil, and cyclophosphamide, although definitive treatment has not been found.4 Calcium channel blockers, PDE-5 inhibitors, endothelin receptor antagonists, and prostacyclin analogues also have been used to reduce the severity of Raynaud phenomenon attacks and to decrease the number of digital ulcers (in addition to their beneficial effects on pulmonary involvement).8 Additional pharmacologic agents that have been linked to an improvement in Raynaud phenomenon and digital ulcers include statins, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, intravenous N-acetylcysteine, vitamin E gel, and surgical options (eg, revascularization and sympathectomy).8
Pulmonary Involvement
Systemic sclerosis can lead to 2 complications in the lungs, both present in Mr. P: PAH (mean pulmonary arterial pressure > 25 mm Hg) and ILD, which together make lung involvement the leading cause of death for these patients. Limited-type SSc usually is restricted to PAH, whereas diffuse-type SSc usually leads to ILD.4
On diagnosis of SSc, an HRCT is indicated to assess the degree of lung involvement. Mr. P’s HRCT showed a 3-cm main pulmonary artery, suggestive of PAH, in addition to evidence of ILD seen on the chest X-ray (Figure 4).
Pulmonary arterial hypertension is a common finding in patients with SSc and carries a severe prognosis. Risk factors for the development of PAH, when not present on initial diagnosis, include limited-type SSc; late age of onset; Raynaud phenomenon; decreased DLCO, FVC/DLCO < 1.6; increased N-terminal pro-B-type natriuretic peptide (NT-proBNP) serum levels; and the presence of antibodies.9 Patients with both SSc and PAH have a 50% to 87% 1-year survival, whereas patients with idiopathic PAH without connective tissue disease have an 88% 1-year survival.9
Many patients with lung involvement are asymptomatic, but some have findings of crackles and interstitial thickening on chest X-ray that can progress to cyanosis and right heart failure (cor pulmonale).10 On discovery of lung involvement (Figure 5), it is important to follow up with a PFT, primarily because the outcomes and prognosis for patients with SSc are correlated with the presenting severity of ILD and the subsequent progression of their DLCO.4,10
Treatment is directed at PAH and ILD separately. For PAH, a PDE-5 inhibitor, such as tadalafil or sildenafil, and an endothelin-1 receptor antagonist, such as ambrisentan or bosentan, are indicated. Other PAH treatments include diuretics and prostacyclin analogues (eg, epoprostenol, treprostinil, or iloprost) in addition to warfarin if patients have a history of thrombotic events.4 Mr. P’s pulmonologists deferred treatment with prostacyclin analogues given the potential for adverse effects with endothelin-1 receptor inhibitors, PDE-5 inhibitors, and calcium-channel blockers, although combination studies with bosentan and inhaled iloprost have shown promise.11
Like the skin manifestations of SSc, ILD lacks definitive treatment to prevent disease progression. However, some patients benefit from cyclophosphamide, followed by mycophenolate mofetil—which is usually better tolerated—azathioprine, haematopoietic stem cell transplant as rescue therapy, and lung transplant for life-saving treatment.10
Renal Involvement
Renal involvement in patients with SSc can have profound effects on QOL. Even without clinical renal involvement, glomerular filtration rate (GFR) usually decreases with the progression of vascular damage correlated with age and disease duration.12 Patients with a history of digital ulcers, like Mr. P, usually have a lower GFR than that of patients without digital ulcers.12 Monitoring renal function is vital in caring for these patients, because SRC occurs in 2% to 15% of all patients with SSc.13,14
Scleroderma renal crisis is generally defined as an abruptly elevated BP (> 140/90 mm Hgor > 30 mm Hg rise from baseline) with acute renal failure (elevated SCr) and decreased urine output.14 Given the rarity of SSc in the population, diagnosis of SRC requires high clinical suspicion. In the case of Mr. P, a workup involving serum analysis, urinalysis, and renal biopsy allowed for a definitive diagnosis. A renal biopsy can show microangiopathic hemolytic anemia and confirm SRC, although it may not be necessary in patients with known SSc presenting with new hypertension, rising creatinine, and unremarkable urine sediment on microscopy.14,15
Although an acute SRC can be difficult to predict, monitoring renal function and attention to key factors can assist in discovering this SSc complication. Scleroderma renal crisis usually occurs within the first 4 years of SSc diagnosis, often paralleling rapid progression of skin thickening and tightening with higher rates in both African Americans and males.13,14 Additional predictive factors include diffuse skin involvement, rapid progression of skin involvement, positive anti-RNA polymerase III antibodies, new anemia, new cardiac events (eg, pericardial effusion, pericarditis, left ventricular insufficiency), CHF, tendon friction rubs, arthritis, and recent (within 3 months) high-dose glucocorticoid use.4,13-15
The presentation of SRC can be nonspecific, often resembling findings related to acute kidney injury. Patients may report malaise, fatigue, fever, headache, seizure, blurred vision, or dyspnea.13 Clinical parametersto examine include systolic BP > 140 mm Hgand/or diastolic BP > 90 mm Hg (or an abrupt rise of > 30 or > 20, respectively), SCr increase by 50%+ or > 120% of the upper normal limit, proteinuria at 2+, high protein:creatinine ratio, hematuria > 2+ or > 10 red blood cells (RBCs), platelets < 100,000/mm3, and hypertensive encephalopathy.13 Mr. P presented with fatigue, dyspnea, an abrupt rise in BP (156/96 mm Hg), foamy urine, bilateral lower extremity edema (3.9 g/dL albumin), 2+ RBCs, and a SCr of 2.46 mg/dL on admission (a 205% increase from his last baseline of 1.2 mg/dL).
Treatments have had a large impact on the mortality rates of SRC. Following the introduction of ACE inhibitors, mortality from SRC has decreased from 76% to < 10% over recent decades.13 In addition to improving survival, these medications also have improved hospitalization outcomes for these patients.3 Captopril is often the medication of choice given its rapid onset and short duration of action relative to other medications in the same class, such as benazepril, enalapril, fosinopril, lisinopril, quinapril, and ramipril.4,13 Current clinical trials are assessing the specific renal benefits of endothelin-1 receptor antagonists, which often are used for pulmonary and skin involvement.14 For patients presenting with acute SRC and uremia, dialysis may be necessary (typically predicted by elevated NT-proBNP serum levels), while the decision for transplantation may be indicated at least 2 years after SRC onset and resolution.4,13-15
Once the acute crisis resolves, it is important to discuss prognosis with patients. The 1-, 2-, 3-, 5-, and 10-year survival rates are 82%, 74%, 71%, 59%, and 47%, respectively; however, when looking at only male patients at 10 years, the survival rate is 17%.13 Prevention of SRC can be addressed with daily BP checks and advising patients to seek medical care if they notice a consecutive 2-day abrupt rise.13
Cardiac Involvement
Systemic sclerosis has significant effects on patients’ heart function. The introduction of ACE inhibitors has shifted mortality in SSc patients from predominantly SRC to cardiac causes.6 Cardiac involvement can occur through a range of processes, including abnormal cardiac conduction, CHF, diastolic dysfunction, mitral valve nodular thickening, and pericardial effusion.3 Even though patients often present with skin findings, an initial cardiac workup is crucial to understanding disease progression and patient prognosis. The severity of the cutaneous manifestations often predicts the degree of diastolic dysfunction.16
Clinical evidence of cardiac involvement is seen in 20% to 25% of patients with SSc and is associated with a 70% mortality at 5 years when symptoms are evident.16 Additionally, right ventricular dysfunction at presentation is the strongest marker for all-mortality prognosis, representing the degree of pulmonary involvement, and includes findings such as progressive shortness of breath and systemic edema.9
Given the increasing survival of patients with SSc, cardiac involvement is becoming more evident and prominent. Direct treatments for cardiac manifestations are based on the causative feature, namely, focusing on pulmonary and renal involvement, which can be assessed with periodic echocardiograms evaluating left ventricular EF.
Gastrointestinal Involvement
Along with skin manifestations, the gastrointestinal (GI) involvement of SSc can have a significant impact on patients’ QOL without direct contribution to mortality. One of Mr. P’s earliest symptoms that led to a diagnosis of SSc was GERD, which caused a chronic cough, dental erosions, esophageal erosions, duodenal ulcers, dysphagia, abdominal pain, halitosis, pharyngitis, and weight loss. Esophageal involvement occurs in up to 96% of patients with SSc and can include motility abnormalities (eg, strictures and/or muscle dysfunction), lower esophageal sphincter abnormalities, and Barrett esophagus.17
Additional symptoms of the GI system linked with SSc can occur anywhere along the GI tract and include gastric antral vascular ectasia, causing GI bleeds and pernicious anemia, gastroparesis, bacterial overgrowth, intestinal malabsorption, pseudo-obstruction due to hypomotility, fecal incontinence due to anorectal involvement, and rarely, primary biliary cirrhosis.4,17 Decreased mobility of the oral aperture secondary to skin thickening and tightening also can contribute to malnutrition by decreasing oral intake.
Treatments are supportive and target symptom relief. Chronic treatment of GERD is often necessary and includes antacids, histamine-2 receptor blockers, and proton pump inhibitors.4 Other medications that can help with symptom relief include motility agents (such as metoclopramide, domperidone, prucalopride, tegaserod, and macrolides), osmotic laxatives, and ursodeoxycholic acid for primary biliary cirrhosis.17 Surgical intervention should be considered depending on the severity and progression of involvement within the GI tract. Behavioral changes that can improve patient symptoms include facial grimacing and other mouth-stretching exercises, frequent smaller meals followed by maintaining a vertical posture, and high fiber diets.17
Conclusion
Systemic sclerosis is an autoimmune and connective tissue disease with a pathophysiology that can manifest throughout the body. The organ systems that impact patient outcomes include skin, pulmonary, renal, cardiac, and GI. Primary care providers caring for patients diagnosed with SSc should monitor acute management and disease progression in all these systems. Important acute events that can impact morbidity, mortality, and/or QOL include Raynaud phenomenon, SRC, and pericardial effusion. Chronic manifestations that may be present on diagnosis of SSc or may develop while a patient is under a provider’s care include sclerodactyly, tendon calcinosis, PAH, ILD, chronic kidney injury, chronic cardiac damage, GERD, and esophageal dysmotility. While this discussion serves as a pertinent overview of patients with SSc, it is summative, and providers are encouraged to seek a stronger understanding of both the common and rarer manifestations within each of their patients.
Systemic sclerosis (SSc), also called scleroderma, is a rare but serious autoimmune connective tissue disease that has multiple fluctuating pathologic manifestations throughout its temporal course. Estimates have shown that the incidence is 10 to 20 cases per 1 million, and the prevalence is 4 to 253 cases per 1 million.1,2 Given the rarity of this incurable condition, it is vital that primary care providers (PCPs) are able to recognize its unique features early to limit and prevent acute and chronic complications. This case report discusses a patient’s journey with late-diagnosed scleroderma in order to convey these broad manifestations and what providers can do to manage it with their patients.
Case Presentation
Mr. P is a 60-year-old African American male with a history of hypertension, recurrent digital ulcers, pulmonary hypertension (PH), interstitial lung disease (ILD), kidney involvement, congestive heart failure (CHF), and gastroesophageal reflux disease (GERD). Mr. P’s workup began in his late 40s with resistant hypertension, resistant GERD, and multiple hospitalizations for hypertensive urgency. It was not until he was 54 years old that he was diagnosed with mixed connective tissue disorder with sclerodermatous predominance.
Review of systems throughout his medical examinations in his 50s were notable for skin tightening over his hands and shoulders, skin hypopigmentation over his scalp and face, and hair loss. Mr. P was found to have Raynaud phenomenon beginning with his original presentation and digital ulceration without complications of gangrene or autoamputation. Aggregate physical examinations were notable for digital ulceration, skin tightening/sclerodactyly, and telangiectasia. Serologic markers were notable for the following:
- Positive ANA (antinuclear antibody) with titer of 1:1,280/homogenous pattern;
- Positive anti-RNP (antiribonucleoprotein) with titer of 171.2;
- Positive anti-Scl-70 (antitopoisomerase I) with titer of 108.1;
- Positive anti-SM (anti-Smith antibody) with titer of 30.2;
- Positive anti-Ro (SSA) with titer of 107.6;
- Negative anti-La (SSB) with titer of 1.3;
- Negative anti-dsDNA (anti-double stranded DNA) with titer of 9; and
- Negative ACA (anticentromere antibody).
Early transthoracic echocardiograms revealed an ejection fraction (EF) initially at 55% with evidence of left ventricular hypertrophy. Following treatment with phosphodiesterase-5 inhibitors (PDE-5 inhibitors) and endothelin-1 antagonists for pulmonary hypertension, serial transthoracic echocardiograms showed improvement in his EF.
A chest X-ray did not show signs of ILD, but a subsequent high-resolution computed tomography (HRCT) scan was consistent with chronic ILD with a main pulmonary artery diameter of 3 cm (Figure 1).
In subsequent years, Mr. P was hospitalized several times, secondary to digit pain, ulcerations, and osteomyelitis. His first episode was 1 year after his scleroderma diagnosis, when he was hospitalized for 6 days for complications of SSc and finger pain. The following year, he had a 3-day hospitalization for hypertensive urgency and right third-digit osteomyelitis, treated initially with IV fluids, levofloxacin, and vancomycin, and then ceftaroline for 1 month. Throughout the next 6 years, Mr. P presented multiple times with fingertip ulcerations and was followed in the Infectious Disease clinic for recurrent osteomyelitis. He found some relief with systemic antibiotics, including augmentin, minocycline, moxifloxacin, and doxycycline.
At age 59, he was hospitalized for scleroderma renal crisis (SRC). Early in his disease, his kidney function was normal, but the SRC was discovered after an abrupt rise in his blood pressure (BP) and an increase in serum creatinine (SCr) from 1.2 mg/dL at baseline to 3.06 mg/dL. The presence of brown granular cast in his urine prompted a renal biopsy that showed thrombotic microangiopathy with schistocytes. Mr. P was started on captopril and remained stable with outpatient follow-up for this renal complication.
Discussion
Initial presentation of SSc can occur along a spectrum of its pathophysiology. A more severe presentation, like the one seen in Mr. P, seems to occur more frequently in African American patients relative to white patients.3 Differentiating between the 2 types—diffuse vs limited SSc—is vital to managing patients and disease progression. Limited-type SSc is more common (60%) and less severe with slower progression than is diffuse SSc.
Diffuse-type SSc (35%) includes features such as skin thickening and tightening, ILD, SRC, tendon friction rubs (palpable crepitus over tendons), and skin pigment changes.4 The specific involvement of the renal and cardiopulmonary systems accounts for the higher mortality rates in the diffuse-type.5
Many patients with SSc require periodic hospitalizations throughout their life for the acute complications of the disease. Hospitalized patients often range from age 45 to 64 years and are more often female. However, of hospitalized patients with SSc, in-hospital death rates are higher among men.3,6 Although these rates have decreased as the pathogenesis of SSc has become better understood, it is important to note that in-hospital mortality in 1995 for all patients with SSc was 7.1% and mean length of stay was 7.5 days, and in 2002 to 2003, 6.3% and 6.6 days, respectively.3 Though the burden of this disease has decreased, mortality and hospitalizations continue to persist at high rates. Understanding the pathogenesis, progression, and treatments of SSc are essential to aiding patients with this diagnosis.
Skin Involvement
A common finding and presentation for patients with SSc is related to skin involvement. Common patient complaints and exam findings include calcinosis along extensor tendons and digits, Raynaud phenomenon (seen in more than 95% of patients), sclerodactyly, telangiectasias, hyper/hypopigmentation, and pruritus.4 These findings are useful in diagnosing and monitoring patients for disease progression.
Many of the listed skin manifestations affect patients’ quality of life (QOL) but are not directly associated with mortality. However, a common and feared complication includes skin ulcers and osteomyelitis, seen in 48% and 7.7% of patients, respectively.7 Digit ulcers, areas with loss of dermis and epidermis distal to the proximal interphalangeal joints (Figures 2A and 2B), are significant because they parallel a more rapid progression of internal organ involvement.8
Mr. P required multiple hospitalizations and antibiotic regimens for painful digit ulcers complicated by osteomyelitis (Figure 3).
Treatment usually is aimed at infections that complicate these skin ulcers and are based on site-specific cultures. Preventive measures are aimed at the risk factors associated with digit ulcers, including decreased whole-body warmth, direct trauma to digits, smoking, and vasoconstrictors (eg, cocaine, sympathomimetics).8 Some patients may prevent ulcers by using D-penicillamine, mycophenolate mofetil, and cyclophosphamide, although definitive treatment has not been found.4 Calcium channel blockers, PDE-5 inhibitors, endothelin receptor antagonists, and prostacyclin analogues also have been used to reduce the severity of Raynaud phenomenon attacks and to decrease the number of digital ulcers (in addition to their beneficial effects on pulmonary involvement).8 Additional pharmacologic agents that have been linked to an improvement in Raynaud phenomenon and digital ulcers include statins, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, intravenous N-acetylcysteine, vitamin E gel, and surgical options (eg, revascularization and sympathectomy).8
Pulmonary Involvement
Systemic sclerosis can lead to 2 complications in the lungs, both present in Mr. P: PAH (mean pulmonary arterial pressure > 25 mm Hg) and ILD, which together make lung involvement the leading cause of death for these patients. Limited-type SSc usually is restricted to PAH, whereas diffuse-type SSc usually leads to ILD.4
On diagnosis of SSc, an HRCT is indicated to assess the degree of lung involvement. Mr. P’s HRCT showed a 3-cm main pulmonary artery, suggestive of PAH, in addition to evidence of ILD seen on the chest X-ray (Figure 4).
Pulmonary arterial hypertension is a common finding in patients with SSc and carries a severe prognosis. Risk factors for the development of PAH, when not present on initial diagnosis, include limited-type SSc; late age of onset; Raynaud phenomenon; decreased DLCO, FVC/DLCO < 1.6; increased N-terminal pro-B-type natriuretic peptide (NT-proBNP) serum levels; and the presence of antibodies.9 Patients with both SSc and PAH have a 50% to 87% 1-year survival, whereas patients with idiopathic PAH without connective tissue disease have an 88% 1-year survival.9
Many patients with lung involvement are asymptomatic, but some have findings of crackles and interstitial thickening on chest X-ray that can progress to cyanosis and right heart failure (cor pulmonale).10 On discovery of lung involvement (Figure 5), it is important to follow up with a PFT, primarily because the outcomes and prognosis for patients with SSc are correlated with the presenting severity of ILD and the subsequent progression of their DLCO.4,10
Treatment is directed at PAH and ILD separately. For PAH, a PDE-5 inhibitor, such as tadalafil or sildenafil, and an endothelin-1 receptor antagonist, such as ambrisentan or bosentan, are indicated. Other PAH treatments include diuretics and prostacyclin analogues (eg, epoprostenol, treprostinil, or iloprost) in addition to warfarin if patients have a history of thrombotic events.4 Mr. P’s pulmonologists deferred treatment with prostacyclin analogues given the potential for adverse effects with endothelin-1 receptor inhibitors, PDE-5 inhibitors, and calcium-channel blockers, although combination studies with bosentan and inhaled iloprost have shown promise.11
Like the skin manifestations of SSc, ILD lacks definitive treatment to prevent disease progression. However, some patients benefit from cyclophosphamide, followed by mycophenolate mofetil—which is usually better tolerated—azathioprine, haematopoietic stem cell transplant as rescue therapy, and lung transplant for life-saving treatment.10
Renal Involvement
Renal involvement in patients with SSc can have profound effects on QOL. Even without clinical renal involvement, glomerular filtration rate (GFR) usually decreases with the progression of vascular damage correlated with age and disease duration.12 Patients with a history of digital ulcers, like Mr. P, usually have a lower GFR than that of patients without digital ulcers.12 Monitoring renal function is vital in caring for these patients, because SRC occurs in 2% to 15% of all patients with SSc.13,14
Scleroderma renal crisis is generally defined as an abruptly elevated BP (> 140/90 mm Hgor > 30 mm Hg rise from baseline) with acute renal failure (elevated SCr) and decreased urine output.14 Given the rarity of SSc in the population, diagnosis of SRC requires high clinical suspicion. In the case of Mr. P, a workup involving serum analysis, urinalysis, and renal biopsy allowed for a definitive diagnosis. A renal biopsy can show microangiopathic hemolytic anemia and confirm SRC, although it may not be necessary in patients with known SSc presenting with new hypertension, rising creatinine, and unremarkable urine sediment on microscopy.14,15
Although an acute SRC can be difficult to predict, monitoring renal function and attention to key factors can assist in discovering this SSc complication. Scleroderma renal crisis usually occurs within the first 4 years of SSc diagnosis, often paralleling rapid progression of skin thickening and tightening with higher rates in both African Americans and males.13,14 Additional predictive factors include diffuse skin involvement, rapid progression of skin involvement, positive anti-RNA polymerase III antibodies, new anemia, new cardiac events (eg, pericardial effusion, pericarditis, left ventricular insufficiency), CHF, tendon friction rubs, arthritis, and recent (within 3 months) high-dose glucocorticoid use.4,13-15
The presentation of SRC can be nonspecific, often resembling findings related to acute kidney injury. Patients may report malaise, fatigue, fever, headache, seizure, blurred vision, or dyspnea.13 Clinical parametersto examine include systolic BP > 140 mm Hgand/or diastolic BP > 90 mm Hg (or an abrupt rise of > 30 or > 20, respectively), SCr increase by 50%+ or > 120% of the upper normal limit, proteinuria at 2+, high protein:creatinine ratio, hematuria > 2+ or > 10 red blood cells (RBCs), platelets < 100,000/mm3, and hypertensive encephalopathy.13 Mr. P presented with fatigue, dyspnea, an abrupt rise in BP (156/96 mm Hg), foamy urine, bilateral lower extremity edema (3.9 g/dL albumin), 2+ RBCs, and a SCr of 2.46 mg/dL on admission (a 205% increase from his last baseline of 1.2 mg/dL).
Treatments have had a large impact on the mortality rates of SRC. Following the introduction of ACE inhibitors, mortality from SRC has decreased from 76% to < 10% over recent decades.13 In addition to improving survival, these medications also have improved hospitalization outcomes for these patients.3 Captopril is often the medication of choice given its rapid onset and short duration of action relative to other medications in the same class, such as benazepril, enalapril, fosinopril, lisinopril, quinapril, and ramipril.4,13 Current clinical trials are assessing the specific renal benefits of endothelin-1 receptor antagonists, which often are used for pulmonary and skin involvement.14 For patients presenting with acute SRC and uremia, dialysis may be necessary (typically predicted by elevated NT-proBNP serum levels), while the decision for transplantation may be indicated at least 2 years after SRC onset and resolution.4,13-15
Once the acute crisis resolves, it is important to discuss prognosis with patients. The 1-, 2-, 3-, 5-, and 10-year survival rates are 82%, 74%, 71%, 59%, and 47%, respectively; however, when looking at only male patients at 10 years, the survival rate is 17%.13 Prevention of SRC can be addressed with daily BP checks and advising patients to seek medical care if they notice a consecutive 2-day abrupt rise.13
Cardiac Involvement
Systemic sclerosis has significant effects on patients’ heart function. The introduction of ACE inhibitors has shifted mortality in SSc patients from predominantly SRC to cardiac causes.6 Cardiac involvement can occur through a range of processes, including abnormal cardiac conduction, CHF, diastolic dysfunction, mitral valve nodular thickening, and pericardial effusion.3 Even though patients often present with skin findings, an initial cardiac workup is crucial to understanding disease progression and patient prognosis. The severity of the cutaneous manifestations often predicts the degree of diastolic dysfunction.16
Clinical evidence of cardiac involvement is seen in 20% to 25% of patients with SSc and is associated with a 70% mortality at 5 years when symptoms are evident.16 Additionally, right ventricular dysfunction at presentation is the strongest marker for all-mortality prognosis, representing the degree of pulmonary involvement, and includes findings such as progressive shortness of breath and systemic edema.9
Given the increasing survival of patients with SSc, cardiac involvement is becoming more evident and prominent. Direct treatments for cardiac manifestations are based on the causative feature, namely, focusing on pulmonary and renal involvement, which can be assessed with periodic echocardiograms evaluating left ventricular EF.
Gastrointestinal Involvement
Along with skin manifestations, the gastrointestinal (GI) involvement of SSc can have a significant impact on patients’ QOL without direct contribution to mortality. One of Mr. P’s earliest symptoms that led to a diagnosis of SSc was GERD, which caused a chronic cough, dental erosions, esophageal erosions, duodenal ulcers, dysphagia, abdominal pain, halitosis, pharyngitis, and weight loss. Esophageal involvement occurs in up to 96% of patients with SSc and can include motility abnormalities (eg, strictures and/or muscle dysfunction), lower esophageal sphincter abnormalities, and Barrett esophagus.17
Additional symptoms of the GI system linked with SSc can occur anywhere along the GI tract and include gastric antral vascular ectasia, causing GI bleeds and pernicious anemia, gastroparesis, bacterial overgrowth, intestinal malabsorption, pseudo-obstruction due to hypomotility, fecal incontinence due to anorectal involvement, and rarely, primary biliary cirrhosis.4,17 Decreased mobility of the oral aperture secondary to skin thickening and tightening also can contribute to malnutrition by decreasing oral intake.
Treatments are supportive and target symptom relief. Chronic treatment of GERD is often necessary and includes antacids, histamine-2 receptor blockers, and proton pump inhibitors.4 Other medications that can help with symptom relief include motility agents (such as metoclopramide, domperidone, prucalopride, tegaserod, and macrolides), osmotic laxatives, and ursodeoxycholic acid for primary biliary cirrhosis.17 Surgical intervention should be considered depending on the severity and progression of involvement within the GI tract. Behavioral changes that can improve patient symptoms include facial grimacing and other mouth-stretching exercises, frequent smaller meals followed by maintaining a vertical posture, and high fiber diets.17
Conclusion
Systemic sclerosis is an autoimmune and connective tissue disease with a pathophysiology that can manifest throughout the body. The organ systems that impact patient outcomes include skin, pulmonary, renal, cardiac, and GI. Primary care providers caring for patients diagnosed with SSc should monitor acute management and disease progression in all these systems. Important acute events that can impact morbidity, mortality, and/or QOL include Raynaud phenomenon, SRC, and pericardial effusion. Chronic manifestations that may be present on diagnosis of SSc or may develop while a patient is under a provider’s care include sclerodactyly, tendon calcinosis, PAH, ILD, chronic kidney injury, chronic cardiac damage, GERD, and esophageal dysmotility. While this discussion serves as a pertinent overview of patients with SSc, it is summative, and providers are encouraged to seek a stronger understanding of both the common and rarer manifestations within each of their patients.
1. Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41(5):778-799.
2. Silman AG. Scleroderma. In: Epidemiology of the Rheumatic Diseases. 2nd ed. Silman AJ, Hochberg MC, eds. Oxford, UK: Oxford University Press. 1993:chap 8.
3. Chung L, Krishnan E, Chakravarty EF. Hospitalizations and mortality in systemic sclerosis: results from the Nationwide Inpatient Sample. Rheumatology (Oxford). 2007;46(12):1808-1813.
4. Hinchcliff M, Varga J. Systemic sclerosis/scleroderma: a treatable multisystem disease. Am Fam Physician. 2008;78(8):961-968.
5. Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809-1815.
6. Piga M, Casula L, Sanna S, et al. Population-based analysis of hospitalizations for patients with systemic sclerosis in a West-European region over the period 2001-2012. Rheumatol Int. 2016;36(1):73-81.
7. Giuggioli D, Manfredi A, Colaci M, Lumetti F, Ferri C. Osteomyelitis complicating scleroderma digital ulcers. Clin Rheumatol. 2013;32(5):623-627.
8. Schiopu E, Impens AJ, Phillips K. Digital ischemia in scleroderma spectrum of diseases. Int J Rheumatol. 2010;2010:923743.
9. Hassoun PM. Therapies for scleroderma-related pulmonary arterial hypertension. Expert Rev Respir Med. 2009;3(2):187-196.
10. Giacomelli R, Liakouli V, Berardicurti O, et al. Interstitial lung disease in systemic sclerosis: current and future treatment. Rheumatol Int. 2017;37(6):853-863.
11. McLaughlin V, Humbert M, Coghlan G, Nash P, Steen V. Pulmonary arterial hypertension: the most devastating vascular complication of systemic sclerosis. Rheumatology (Oxford). 2009; (suppl 3):iii25-iii31.
12. Gigante A, Barbano B, Granata G, et al. Evaluation of estimated glomerular filtration rate and clinical variables in systemic sclerosis patients. Clin Nephrol. 2016;85(6):326-331.
13. Bose N, Chiesa-Vottero A, Chatterjee S. Scleroderma renal crisis. Semin Arthritis Rheum. 2015;44(6):687-694.
14. Woodworth TG, Suliman YA, Furst DE, Clements P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat Rev Nephrol. 2016;12(11):678-691.
15. Mouthon L, Bérezné A, Bussone G, Noël LH, Villiger PM, Guillevin L. Scleroderma renal crisis: a rare but severe complication of systemic sclerosis. Clin Rev Allergy Immunol. 2011;40(2):84-91.
16. Champion HC. The heart in scleroderma. Rheum Dis Clin North Am. 2008;34(1):181-90.
17. Tian XP, Zhang X. Gastrointestinal complications of systemic sclerosis. World J Gastroenterol. 2013;19(41):7062-7068.
1. Lawrence RC, Helmick CG, Arnett FC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41(5):778-799.
2. Silman AG. Scleroderma. In: Epidemiology of the Rheumatic Diseases. 2nd ed. Silman AJ, Hochberg MC, eds. Oxford, UK: Oxford University Press. 1993:chap 8.
3. Chung L, Krishnan E, Chakravarty EF. Hospitalizations and mortality in systemic sclerosis: results from the Nationwide Inpatient Sample. Rheumatology (Oxford). 2007;46(12):1808-1813.
4. Hinchcliff M, Varga J. Systemic sclerosis/scleroderma: a treatable multisystem disease. Am Fam Physician. 2008;78(8):961-968.
5. Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809-1815.
6. Piga M, Casula L, Sanna S, et al. Population-based analysis of hospitalizations for patients with systemic sclerosis in a West-European region over the period 2001-2012. Rheumatol Int. 2016;36(1):73-81.
7. Giuggioli D, Manfredi A, Colaci M, Lumetti F, Ferri C. Osteomyelitis complicating scleroderma digital ulcers. Clin Rheumatol. 2013;32(5):623-627.
8. Schiopu E, Impens AJ, Phillips K. Digital ischemia in scleroderma spectrum of diseases. Int J Rheumatol. 2010;2010:923743.
9. Hassoun PM. Therapies for scleroderma-related pulmonary arterial hypertension. Expert Rev Respir Med. 2009;3(2):187-196.
10. Giacomelli R, Liakouli V, Berardicurti O, et al. Interstitial lung disease in systemic sclerosis: current and future treatment. Rheumatol Int. 2017;37(6):853-863.
11. McLaughlin V, Humbert M, Coghlan G, Nash P, Steen V. Pulmonary arterial hypertension: the most devastating vascular complication of systemic sclerosis. Rheumatology (Oxford). 2009; (suppl 3):iii25-iii31.
12. Gigante A, Barbano B, Granata G, et al. Evaluation of estimated glomerular filtration rate and clinical variables in systemic sclerosis patients. Clin Nephrol. 2016;85(6):326-331.
13. Bose N, Chiesa-Vottero A, Chatterjee S. Scleroderma renal crisis. Semin Arthritis Rheum. 2015;44(6):687-694.
14. Woodworth TG, Suliman YA, Furst DE, Clements P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat Rev Nephrol. 2016;12(11):678-691.
15. Mouthon L, Bérezné A, Bussone G, Noël LH, Villiger PM, Guillevin L. Scleroderma renal crisis: a rare but severe complication of systemic sclerosis. Clin Rev Allergy Immunol. 2011;40(2):84-91.
16. Champion HC. The heart in scleroderma. Rheum Dis Clin North Am. 2008;34(1):181-90.
17. Tian XP, Zhang X. Gastrointestinal complications of systemic sclerosis. World J Gastroenterol. 2013;19(41):7062-7068.
Long-Term Survival of a Patient With Late-Stage Non-Small Cell Lung Cancer
Lung cancer is the leading cause of cancer death in the world, with non-small cell lung cancer (NSCLC) a significant component of those deaths.1,2 Treatments for advanced-stage NSCLC, however, are limited. Erlotinib, a small-molecule tyrosine kinase inhibitor of epidermal growth factor receptor (EGFR), has aided in advancing NSCLC therapy. Erlotinib has been shown to increase survival by 2 months compared with placebo in a phase 3, randomized controlled trial when used as second- or third-line therapy.3 The authors present a case of a man surviving almost 8 years with late-stage NSCLC on treatment with erlotinib at the VA West Los Angeles Medical Center (WLAMC).
Case Report
Mr. J is a 59-year-old man with a medical history of hepatitis C. He smoked 2 packs of cigarettes a day for 25 years and quit in 2003. He also had a known history of IV drug use. He was unaware of his family history because he was adopted, but his twin sister, who has no known medical problems, is also a smoker. In 2005, when Mr. J was evaluated for treatment options of long-standing hepatitis C with liver ultrasound, a large, irregular right adrenal mass was found, measuring 6.4 x 3.5 cm. A subsequent positron emission tomography (PET) scan identified a lung nodule measuring 3.8 x 3.6 x 3.3 cm. A biopsy guided by computed tomography (CT) showed NSCLC. Subsequently, metastases to the liver and adrenal glands were noted, and Mr. J was started on chemotherapy. He received 4 cycles of carboplatin 725 mg and gemcitabine 2,000 mg as well as a thoracotomy and left upper lung wedge resection in December 2005. His pain was controlled with slow-release morphine 15 mg 2 times per day and oxycodone 5 mg and acetaminophen 325 mg 4 times per day as needed for breakthrough pain.
In 2006, after 4 cycles of chemotherapy, the size of the adrenal mass and the lung mass had decreased; however, he developed new abdominal pain. A CT scan showed new intrahepatic and extrahepatic biliary dilatation and worsening pancreatic function. He could not tolerate the recommended endoscopic ultrasound and left WLAMC, later presenting to an outside hospital for his abdominal pain.
At the outside hospital, 2 masses that were surgically removed from the head of the pancreas were confirmed to be EGFR-positive NSCLC, and he was given 4 cycles of cisplatin and irinotecan at unknown doses. The only adverse effects (AEs) Mr. J reported during this period were nausea and vomiting immediately after chemotherapy. He failed to respond to this treatment and was started on bevacizumab, also at an unknown dose. The patient again did not respond and was transitioned to erlotinib 150 mg daily. The patient showed remarkable response, with lesions decreasing in size.
The patient returned to the WLAMC with multiple ulcerated lesions on his face, chest, back, and extremities and hair loss, which he reported all began within weeks of starting erlotinib. Later, he also developed trichomegaly, also presumed to be a consequence of erlotinib. Despite these AEs, erlotinib was continued at the same dose, given his impressive response to this treatment, the absence of response to other therapy, and the patient’s insistence on continuing the medication.
Of note, after his transition to the outside hospital, Mr. J and his family paid all his medical expenses because he had no insurance. His family was very supportive, and the patient described their motivation and support as paramount in his receiving treatment.
In 2008, Mr. J presented to a dermatologist and was treated with cleocin solution. Although this helped to control his symptoms, the rash persisted. As a complication of these lesions, he also experienced several superinfections for which he was treated with cephalexin. At this same time, a PET scan showed no evidence of disease. He presented to the pain service for persistent chest wall pain around the surgical site, and his pain regimen was changed to slow-release morphine 200 mg 3 times per day and morphine sulfate solution 20 mg/mL 80 mg every 4 hours for breakthrough pain.
The PET scans, which were repeated every 3 months after Mr. J resumed treatment at WLAMC, showed continued absence of disease. In 2009, when he presented to the hospital with pneumonia, a PET scan showed 2 new areas of tracer uptake measuring 1 cm. His chest wall was irradiated, but radiation therapy was stopped after the biopsy returned benign. In 2015, an annual PET scan showed only evidence of postsurgical changes.
Discussion
The benefits of EGFR therapy have been established for treatment of late-stage NSCLC, but such therapy has limitations. For advanced-stage NSCLC, erlotinib has been shown to improve disease-free progression by 2.7 to 3.25 months and overall survival by 6.7 to 7.9 months.4-6 However, 1-year survival estimates remain as low as 35.0 to 37.7%,5,6 and its utility as first-line therapy has been questioned; randomized control trials have shown EGFR therapy to be of benefit only as secondor third-line therapy, when used with platinum-based chemotherapeutics.3,4 The few reports of complete response, however, have not included a definition of duration of survival.5,6
Occasionally, there have been reports of patients surviving for significantly longer periods, including 1 report of a patient who survived with complete remission for 2 years.7 In another case report, a patient experienced partial remission for more than 1 year with erlotinib as a third-line therapy.8 Although several reports indicated prolonged survival with erlotinib, or induction of complete remission of metastasis, survival has not been longer than 2 years.9-12
Important considerations for use of erlotinib are factors that predict a positive treatment response, including female sex, no previous exposure to tobacco, Asian origin, and adenocarcinoma on histologic examination.3,13 Mr. J did not meet any of these criteria. Interestingly, one study examining characteristics predictive of a positive response to erlotinib did not show that EGFR gene mutations were associated with response, although other studies have shown this to be a significant predictor of response.3,14,15
In this patient, his impressive response to erlotinib was most likely augmented by the presence of the EGFR mutation. Additionally, some reports indicate that pretreatment with platinum-based therapy can induce genetic changes resulting in EGFR mutations, thus enabling the benefit of erlotinib.10 Given that his biopsy results were not tested for the EGFR mutation prior to initiating carboplatin, this is a possibility.
Other factors specific to Mr. J that may have influenced his response to therapy include his personal wealth, which may have given him direct access to physicians outside the VA. His family support also may have motivated him to pursue and continue treatment, thus augmenting his survival. This support likely contributed in large part to his continuing erlotinib therapy despite the severe rash, hair loss, and trichomegaly. Other AEs associated with long-term erlotinib therapy include folliculitis, diarrhea, fatigue, and paronychia,although Mr. J did not experience these.16
Conclusion
Mr. J continues to follow up regularly at WLAMC. To the authors’ knowledge, this patient’s 8 year survival is the longest length of survival for any patient with NSCLC on erlotinib therapy. While the therapeutic benefits of erlotinib as a second-line therapy have been shown, EGFR therapy may be more effective than previously thought. Further research is needed to fully understand the benefits of erlotinib.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
1. Ridge CA, McErlean AM, Ginsberg MS. Epidemiology of lung cancer. Semin Intervent Radiol. 2013;30(2):93-98.
2. Gridelli C, Bareschino MA, Schettino C, Rossi A, Maione P, Ciardiello F. Erlotinib in non-small cell lung cancer treatment: current status and future development. Oncologist. 2007;12(7):840-849.
3. Shepherd FA, Pereira JR, Ciuleanu T, et al; National Cancer Institute of Canada Clinical Trials Group. Erlotinib in previously treated non–small-cell lung cancer. N Engl J Med. 2005;353(2):123-132.
4. Smith J. Erlotinib: small-molecule targeted therapy in the treatment of non-smallcell lung cancer. Clin Ther. 2005;27(10):1513-1534.
5. Boyer M, Horwood K, Pavlakis N, et al. Efficacy of erlotinib in patients with advanced non-small-cell lung cancer (NSCLC): analysis of the Australian subpopulation of the TRUST study. Asia Pac J Clin Oncol. 2012;8(3):248-254.
6. Reck M, van Zandwijk N, Gridelli C, et al. Erlotinib in advanced non-small cell lung cancer: efficacy and safety findings of the global phase IV Tarceva Lung Cancer Survival Treatment study. J Thorac Oncol. 2010;5(10):1616-1622.
7. Vitale MG, Riccardi F, Mocerino C, et al. Erlotinib-induced complete response in a patient with epidermal growth factor receptor wild-type lung adenocarcinoma after chemotherapy failure: a case report. J Med Case Rep. 2014;8:102.
8. Duchnowska R, Siemiatkowska A, Grala B, Smoter M. Long-term remission after erlotinib therapy in an elderly patient with advanced non-small-cell lung cancer. Case report and conclusions for clinical practice [in Polish]. Pneumonol Alergol Pol. 2008;76(6):451-455.
9. Lai CSL, Boshoff C, Falzon M, Lee SM. Complete response to erlotinib treatment in brain metastases from recurrent NSCLC. Thorax. 2006;61(1):91.
10. Karam I, Melosky B. Response to second-line erlotinib in an EGFR mutationnegative patient with non-small-cell lung cancer: make no assumptions. Curr Oncol. 2012;19(1):42-46.
11. Kobayashi T, Koizumi T, Agatsuma T, et al. A phase II trial of erlotinib in patients with EGFR wild-type advanced non-small-cell lung cancer. Cancer Chemother Pharmacol. 2012;69(5):1241-1246.
12. Gridelli C, Maione P, Galetta D, et al. Three cases of long-lasting tumor control with erlotinib after progression with gefitinib in advanced non-small cell lung cancer. J Thorac Oncol. 2007;2(8):758-761.
13. Fukuoka M, Yano S, Giaccone G, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. J Clin Oncol. 2003;21(12):2237-2246.
14. Tsao MS, Sakurada A, Cutz JC, et al. Erlotinib in lung cancer—molecular and clinical predictors of outcome. N Engl J Med. 2005;353(2):133-144.
15. Sequist LV, Bell DW, Lynch TJ, Haber DA. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J Clin Oncol. 2007;25(5):587-595.
16. Becker A, van Wijk A, Smit EF, Postmus PE. Side-effects of long-term administration of erlotinib in patients with non-small cell lung cancer. J Thorac Oncol. 2010;5(9):1477-1480.
Note: Page numbers differ between the print issue and digital edition.
Lung cancer is the leading cause of cancer death in the world, with non-small cell lung cancer (NSCLC) a significant component of those deaths.1,2 Treatments for advanced-stage NSCLC, however, are limited. Erlotinib, a small-molecule tyrosine kinase inhibitor of epidermal growth factor receptor (EGFR), has aided in advancing NSCLC therapy. Erlotinib has been shown to increase survival by 2 months compared with placebo in a phase 3, randomized controlled trial when used as second- or third-line therapy.3 The authors present a case of a man surviving almost 8 years with late-stage NSCLC on treatment with erlotinib at the VA West Los Angeles Medical Center (WLAMC).
Case Report
Mr. J is a 59-year-old man with a medical history of hepatitis C. He smoked 2 packs of cigarettes a day for 25 years and quit in 2003. He also had a known history of IV drug use. He was unaware of his family history because he was adopted, but his twin sister, who has no known medical problems, is also a smoker. In 2005, when Mr. J was evaluated for treatment options of long-standing hepatitis C with liver ultrasound, a large, irregular right adrenal mass was found, measuring 6.4 x 3.5 cm. A subsequent positron emission tomography (PET) scan identified a lung nodule measuring 3.8 x 3.6 x 3.3 cm. A biopsy guided by computed tomography (CT) showed NSCLC. Subsequently, metastases to the liver and adrenal glands were noted, and Mr. J was started on chemotherapy. He received 4 cycles of carboplatin 725 mg and gemcitabine 2,000 mg as well as a thoracotomy and left upper lung wedge resection in December 2005. His pain was controlled with slow-release morphine 15 mg 2 times per day and oxycodone 5 mg and acetaminophen 325 mg 4 times per day as needed for breakthrough pain.
In 2006, after 4 cycles of chemotherapy, the size of the adrenal mass and the lung mass had decreased; however, he developed new abdominal pain. A CT scan showed new intrahepatic and extrahepatic biliary dilatation and worsening pancreatic function. He could not tolerate the recommended endoscopic ultrasound and left WLAMC, later presenting to an outside hospital for his abdominal pain.
At the outside hospital, 2 masses that were surgically removed from the head of the pancreas were confirmed to be EGFR-positive NSCLC, and he was given 4 cycles of cisplatin and irinotecan at unknown doses. The only adverse effects (AEs) Mr. J reported during this period were nausea and vomiting immediately after chemotherapy. He failed to respond to this treatment and was started on bevacizumab, also at an unknown dose. The patient again did not respond and was transitioned to erlotinib 150 mg daily. The patient showed remarkable response, with lesions decreasing in size.
The patient returned to the WLAMC with multiple ulcerated lesions on his face, chest, back, and extremities and hair loss, which he reported all began within weeks of starting erlotinib. Later, he also developed trichomegaly, also presumed to be a consequence of erlotinib. Despite these AEs, erlotinib was continued at the same dose, given his impressive response to this treatment, the absence of response to other therapy, and the patient’s insistence on continuing the medication.
Of note, after his transition to the outside hospital, Mr. J and his family paid all his medical expenses because he had no insurance. His family was very supportive, and the patient described their motivation and support as paramount in his receiving treatment.
In 2008, Mr. J presented to a dermatologist and was treated with cleocin solution. Although this helped to control his symptoms, the rash persisted. As a complication of these lesions, he also experienced several superinfections for which he was treated with cephalexin. At this same time, a PET scan showed no evidence of disease. He presented to the pain service for persistent chest wall pain around the surgical site, and his pain regimen was changed to slow-release morphine 200 mg 3 times per day and morphine sulfate solution 20 mg/mL 80 mg every 4 hours for breakthrough pain.
The PET scans, which were repeated every 3 months after Mr. J resumed treatment at WLAMC, showed continued absence of disease. In 2009, when he presented to the hospital with pneumonia, a PET scan showed 2 new areas of tracer uptake measuring 1 cm. His chest wall was irradiated, but radiation therapy was stopped after the biopsy returned benign. In 2015, an annual PET scan showed only evidence of postsurgical changes.
Discussion
The benefits of EGFR therapy have been established for treatment of late-stage NSCLC, but such therapy has limitations. For advanced-stage NSCLC, erlotinib has been shown to improve disease-free progression by 2.7 to 3.25 months and overall survival by 6.7 to 7.9 months.4-6 However, 1-year survival estimates remain as low as 35.0 to 37.7%,5,6 and its utility as first-line therapy has been questioned; randomized control trials have shown EGFR therapy to be of benefit only as secondor third-line therapy, when used with platinum-based chemotherapeutics.3,4 The few reports of complete response, however, have not included a definition of duration of survival.5,6
Occasionally, there have been reports of patients surviving for significantly longer periods, including 1 report of a patient who survived with complete remission for 2 years.7 In another case report, a patient experienced partial remission for more than 1 year with erlotinib as a third-line therapy.8 Although several reports indicated prolonged survival with erlotinib, or induction of complete remission of metastasis, survival has not been longer than 2 years.9-12
Important considerations for use of erlotinib are factors that predict a positive treatment response, including female sex, no previous exposure to tobacco, Asian origin, and adenocarcinoma on histologic examination.3,13 Mr. J did not meet any of these criteria. Interestingly, one study examining characteristics predictive of a positive response to erlotinib did not show that EGFR gene mutations were associated with response, although other studies have shown this to be a significant predictor of response.3,14,15
In this patient, his impressive response to erlotinib was most likely augmented by the presence of the EGFR mutation. Additionally, some reports indicate that pretreatment with platinum-based therapy can induce genetic changes resulting in EGFR mutations, thus enabling the benefit of erlotinib.10 Given that his biopsy results were not tested for the EGFR mutation prior to initiating carboplatin, this is a possibility.
Other factors specific to Mr. J that may have influenced his response to therapy include his personal wealth, which may have given him direct access to physicians outside the VA. His family support also may have motivated him to pursue and continue treatment, thus augmenting his survival. This support likely contributed in large part to his continuing erlotinib therapy despite the severe rash, hair loss, and trichomegaly. Other AEs associated with long-term erlotinib therapy include folliculitis, diarrhea, fatigue, and paronychia,although Mr. J did not experience these.16
Conclusion
Mr. J continues to follow up regularly at WLAMC. To the authors’ knowledge, this patient’s 8 year survival is the longest length of survival for any patient with NSCLC on erlotinib therapy. While the therapeutic benefits of erlotinib as a second-line therapy have been shown, EGFR therapy may be more effective than previously thought. Further research is needed to fully understand the benefits of erlotinib.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
Lung cancer is the leading cause of cancer death in the world, with non-small cell lung cancer (NSCLC) a significant component of those deaths.1,2 Treatments for advanced-stage NSCLC, however, are limited. Erlotinib, a small-molecule tyrosine kinase inhibitor of epidermal growth factor receptor (EGFR), has aided in advancing NSCLC therapy. Erlotinib has been shown to increase survival by 2 months compared with placebo in a phase 3, randomized controlled trial when used as second- or third-line therapy.3 The authors present a case of a man surviving almost 8 years with late-stage NSCLC on treatment with erlotinib at the VA West Los Angeles Medical Center (WLAMC).
Case Report
Mr. J is a 59-year-old man with a medical history of hepatitis C. He smoked 2 packs of cigarettes a day for 25 years and quit in 2003. He also had a known history of IV drug use. He was unaware of his family history because he was adopted, but his twin sister, who has no known medical problems, is also a smoker. In 2005, when Mr. J was evaluated for treatment options of long-standing hepatitis C with liver ultrasound, a large, irregular right adrenal mass was found, measuring 6.4 x 3.5 cm. A subsequent positron emission tomography (PET) scan identified a lung nodule measuring 3.8 x 3.6 x 3.3 cm. A biopsy guided by computed tomography (CT) showed NSCLC. Subsequently, metastases to the liver and adrenal glands were noted, and Mr. J was started on chemotherapy. He received 4 cycles of carboplatin 725 mg and gemcitabine 2,000 mg as well as a thoracotomy and left upper lung wedge resection in December 2005. His pain was controlled with slow-release morphine 15 mg 2 times per day and oxycodone 5 mg and acetaminophen 325 mg 4 times per day as needed for breakthrough pain.
In 2006, after 4 cycles of chemotherapy, the size of the adrenal mass and the lung mass had decreased; however, he developed new abdominal pain. A CT scan showed new intrahepatic and extrahepatic biliary dilatation and worsening pancreatic function. He could not tolerate the recommended endoscopic ultrasound and left WLAMC, later presenting to an outside hospital for his abdominal pain.
At the outside hospital, 2 masses that were surgically removed from the head of the pancreas were confirmed to be EGFR-positive NSCLC, and he was given 4 cycles of cisplatin and irinotecan at unknown doses. The only adverse effects (AEs) Mr. J reported during this period were nausea and vomiting immediately after chemotherapy. He failed to respond to this treatment and was started on bevacizumab, also at an unknown dose. The patient again did not respond and was transitioned to erlotinib 150 mg daily. The patient showed remarkable response, with lesions decreasing in size.
The patient returned to the WLAMC with multiple ulcerated lesions on his face, chest, back, and extremities and hair loss, which he reported all began within weeks of starting erlotinib. Later, he also developed trichomegaly, also presumed to be a consequence of erlotinib. Despite these AEs, erlotinib was continued at the same dose, given his impressive response to this treatment, the absence of response to other therapy, and the patient’s insistence on continuing the medication.
Of note, after his transition to the outside hospital, Mr. J and his family paid all his medical expenses because he had no insurance. His family was very supportive, and the patient described their motivation and support as paramount in his receiving treatment.
In 2008, Mr. J presented to a dermatologist and was treated with cleocin solution. Although this helped to control his symptoms, the rash persisted. As a complication of these lesions, he also experienced several superinfections for which he was treated with cephalexin. At this same time, a PET scan showed no evidence of disease. He presented to the pain service for persistent chest wall pain around the surgical site, and his pain regimen was changed to slow-release morphine 200 mg 3 times per day and morphine sulfate solution 20 mg/mL 80 mg every 4 hours for breakthrough pain.
The PET scans, which were repeated every 3 months after Mr. J resumed treatment at WLAMC, showed continued absence of disease. In 2009, when he presented to the hospital with pneumonia, a PET scan showed 2 new areas of tracer uptake measuring 1 cm. His chest wall was irradiated, but radiation therapy was stopped after the biopsy returned benign. In 2015, an annual PET scan showed only evidence of postsurgical changes.
Discussion
The benefits of EGFR therapy have been established for treatment of late-stage NSCLC, but such therapy has limitations. For advanced-stage NSCLC, erlotinib has been shown to improve disease-free progression by 2.7 to 3.25 months and overall survival by 6.7 to 7.9 months.4-6 However, 1-year survival estimates remain as low as 35.0 to 37.7%,5,6 and its utility as first-line therapy has been questioned; randomized control trials have shown EGFR therapy to be of benefit only as secondor third-line therapy, when used with platinum-based chemotherapeutics.3,4 The few reports of complete response, however, have not included a definition of duration of survival.5,6
Occasionally, there have been reports of patients surviving for significantly longer periods, including 1 report of a patient who survived with complete remission for 2 years.7 In another case report, a patient experienced partial remission for more than 1 year with erlotinib as a third-line therapy.8 Although several reports indicated prolonged survival with erlotinib, or induction of complete remission of metastasis, survival has not been longer than 2 years.9-12
Important considerations for use of erlotinib are factors that predict a positive treatment response, including female sex, no previous exposure to tobacco, Asian origin, and adenocarcinoma on histologic examination.3,13 Mr. J did not meet any of these criteria. Interestingly, one study examining characteristics predictive of a positive response to erlotinib did not show that EGFR gene mutations were associated with response, although other studies have shown this to be a significant predictor of response.3,14,15
In this patient, his impressive response to erlotinib was most likely augmented by the presence of the EGFR mutation. Additionally, some reports indicate that pretreatment with platinum-based therapy can induce genetic changes resulting in EGFR mutations, thus enabling the benefit of erlotinib.10 Given that his biopsy results were not tested for the EGFR mutation prior to initiating carboplatin, this is a possibility.
Other factors specific to Mr. J that may have influenced his response to therapy include his personal wealth, which may have given him direct access to physicians outside the VA. His family support also may have motivated him to pursue and continue treatment, thus augmenting his survival. This support likely contributed in large part to his continuing erlotinib therapy despite the severe rash, hair loss, and trichomegaly. Other AEs associated with long-term erlotinib therapy include folliculitis, diarrhea, fatigue, and paronychia,although Mr. J did not experience these.16
Conclusion
Mr. J continues to follow up regularly at WLAMC. To the authors’ knowledge, this patient’s 8 year survival is the longest length of survival for any patient with NSCLC on erlotinib therapy. While the therapeutic benefits of erlotinib as a second-line therapy have been shown, EGFR therapy may be more effective than previously thought. Further research is needed to fully understand the benefits of erlotinib.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
1. Ridge CA, McErlean AM, Ginsberg MS. Epidemiology of lung cancer. Semin Intervent Radiol. 2013;30(2):93-98.
2. Gridelli C, Bareschino MA, Schettino C, Rossi A, Maione P, Ciardiello F. Erlotinib in non-small cell lung cancer treatment: current status and future development. Oncologist. 2007;12(7):840-849.
3. Shepherd FA, Pereira JR, Ciuleanu T, et al; National Cancer Institute of Canada Clinical Trials Group. Erlotinib in previously treated non–small-cell lung cancer. N Engl J Med. 2005;353(2):123-132.
4. Smith J. Erlotinib: small-molecule targeted therapy in the treatment of non-smallcell lung cancer. Clin Ther. 2005;27(10):1513-1534.
5. Boyer M, Horwood K, Pavlakis N, et al. Efficacy of erlotinib in patients with advanced non-small-cell lung cancer (NSCLC): analysis of the Australian subpopulation of the TRUST study. Asia Pac J Clin Oncol. 2012;8(3):248-254.
6. Reck M, van Zandwijk N, Gridelli C, et al. Erlotinib in advanced non-small cell lung cancer: efficacy and safety findings of the global phase IV Tarceva Lung Cancer Survival Treatment study. J Thorac Oncol. 2010;5(10):1616-1622.
7. Vitale MG, Riccardi F, Mocerino C, et al. Erlotinib-induced complete response in a patient with epidermal growth factor receptor wild-type lung adenocarcinoma after chemotherapy failure: a case report. J Med Case Rep. 2014;8:102.
8. Duchnowska R, Siemiatkowska A, Grala B, Smoter M. Long-term remission after erlotinib therapy in an elderly patient with advanced non-small-cell lung cancer. Case report and conclusions for clinical practice [in Polish]. Pneumonol Alergol Pol. 2008;76(6):451-455.
9. Lai CSL, Boshoff C, Falzon M, Lee SM. Complete response to erlotinib treatment in brain metastases from recurrent NSCLC. Thorax. 2006;61(1):91.
10. Karam I, Melosky B. Response to second-line erlotinib in an EGFR mutationnegative patient with non-small-cell lung cancer: make no assumptions. Curr Oncol. 2012;19(1):42-46.
11. Kobayashi T, Koizumi T, Agatsuma T, et al. A phase II trial of erlotinib in patients with EGFR wild-type advanced non-small-cell lung cancer. Cancer Chemother Pharmacol. 2012;69(5):1241-1246.
12. Gridelli C, Maione P, Galetta D, et al. Three cases of long-lasting tumor control with erlotinib after progression with gefitinib in advanced non-small cell lung cancer. J Thorac Oncol. 2007;2(8):758-761.
13. Fukuoka M, Yano S, Giaccone G, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. J Clin Oncol. 2003;21(12):2237-2246.
14. Tsao MS, Sakurada A, Cutz JC, et al. Erlotinib in lung cancer—molecular and clinical predictors of outcome. N Engl J Med. 2005;353(2):133-144.
15. Sequist LV, Bell DW, Lynch TJ, Haber DA. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J Clin Oncol. 2007;25(5):587-595.
16. Becker A, van Wijk A, Smit EF, Postmus PE. Side-effects of long-term administration of erlotinib in patients with non-small cell lung cancer. J Thorac Oncol. 2010;5(9):1477-1480.
Note: Page numbers differ between the print issue and digital edition.
1. Ridge CA, McErlean AM, Ginsberg MS. Epidemiology of lung cancer. Semin Intervent Radiol. 2013;30(2):93-98.
2. Gridelli C, Bareschino MA, Schettino C, Rossi A, Maione P, Ciardiello F. Erlotinib in non-small cell lung cancer treatment: current status and future development. Oncologist. 2007;12(7):840-849.
3. Shepherd FA, Pereira JR, Ciuleanu T, et al; National Cancer Institute of Canada Clinical Trials Group. Erlotinib in previously treated non–small-cell lung cancer. N Engl J Med. 2005;353(2):123-132.
4. Smith J. Erlotinib: small-molecule targeted therapy in the treatment of non-smallcell lung cancer. Clin Ther. 2005;27(10):1513-1534.
5. Boyer M, Horwood K, Pavlakis N, et al. Efficacy of erlotinib in patients with advanced non-small-cell lung cancer (NSCLC): analysis of the Australian subpopulation of the TRUST study. Asia Pac J Clin Oncol. 2012;8(3):248-254.
6. Reck M, van Zandwijk N, Gridelli C, et al. Erlotinib in advanced non-small cell lung cancer: efficacy and safety findings of the global phase IV Tarceva Lung Cancer Survival Treatment study. J Thorac Oncol. 2010;5(10):1616-1622.
7. Vitale MG, Riccardi F, Mocerino C, et al. Erlotinib-induced complete response in a patient with epidermal growth factor receptor wild-type lung adenocarcinoma after chemotherapy failure: a case report. J Med Case Rep. 2014;8:102.
8. Duchnowska R, Siemiatkowska A, Grala B, Smoter M. Long-term remission after erlotinib therapy in an elderly patient with advanced non-small-cell lung cancer. Case report and conclusions for clinical practice [in Polish]. Pneumonol Alergol Pol. 2008;76(6):451-455.
9. Lai CSL, Boshoff C, Falzon M, Lee SM. Complete response to erlotinib treatment in brain metastases from recurrent NSCLC. Thorax. 2006;61(1):91.
10. Karam I, Melosky B. Response to second-line erlotinib in an EGFR mutationnegative patient with non-small-cell lung cancer: make no assumptions. Curr Oncol. 2012;19(1):42-46.
11. Kobayashi T, Koizumi T, Agatsuma T, et al. A phase II trial of erlotinib in patients with EGFR wild-type advanced non-small-cell lung cancer. Cancer Chemother Pharmacol. 2012;69(5):1241-1246.
12. Gridelli C, Maione P, Galetta D, et al. Three cases of long-lasting tumor control with erlotinib after progression with gefitinib in advanced non-small cell lung cancer. J Thorac Oncol. 2007;2(8):758-761.
13. Fukuoka M, Yano S, Giaccone G, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. J Clin Oncol. 2003;21(12):2237-2246.
14. Tsao MS, Sakurada A, Cutz JC, et al. Erlotinib in lung cancer—molecular and clinical predictors of outcome. N Engl J Med. 2005;353(2):133-144.
15. Sequist LV, Bell DW, Lynch TJ, Haber DA. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J Clin Oncol. 2007;25(5):587-595.
16. Becker A, van Wijk A, Smit EF, Postmus PE. Side-effects of long-term administration of erlotinib in patients with non-small cell lung cancer. J Thorac Oncol. 2010;5(9):1477-1480.
Note: Page numbers differ between the print issue and digital edition.