User login
A Case of Pruritis Rash

History of Present Illness
A55-year-old male presented with a one and one-half week history of a sore throat, shortness of breath on exertion, ankle edema, and arthralgias that began in the ankles and subsequently spread to involve the elbows and wrists.
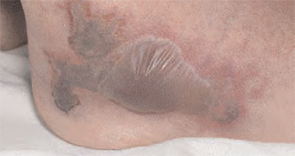
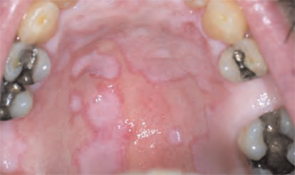
He had also developed a pruritic eruption involving the lower extremities, which consisted of erythematous palpable purpuric lesions and patches with superficial and central necrosis and ulceration, as well as a large 4-cm bulla of the right lateral ankle. (See Figures 1 and 2, below.) Other skin findings included petechiae of the palms and multiple ulcerations of the hard palate. Laboratory evaluation demonstrated c-ANCA antibody positivity (1:512), a proteinase 3 antibody level of greater than 100 U/ml, and a creatinine of 1.0mg/dl. TH
What is the most appropriate treatment for this condition?
- Prednisone;
- Azathioprine;
- Cyclophosphamide;
- Prednisone combined with cyclophosphamide; or
- Vancomycin combined with rifampin
Discussion
The answer is D: Wegener’s granulomatosis (WG) is a chronic granulomatous inflammatory response of unknown etiology that usually presents with the classic triad of systemic vasculitis, necrotizing granulomatous inflammation of the upper and lower respiratory tracts, and glomerulonephritis. The generalized or classic form of WG can progress rapidly to cause irreversible organ dysfunction and death. Although the pathogenesis remains unknown, it is felt that WG may result from an exaggerated cell-mediated response to an unknown antigen.1
The average age of onset for WG is 45.2 years, with 63.5% of patients male and 91% Caucasian.2 A WG diagnosis can be very difficult, and elements of the classic triad may not all be present initially. Pulmonary infiltrates or nodules are seen via chest X-ray or CT scan in just less than half of patients as an early manifestation of WG.
Occasionally WG presents with skin lesions (13%) or oral ulcers (6%), however, 40% of patients eventually develop skin involvement consisting of painful subcutaneous nodules, papules, vesicles or bullae, petechiae, palpable purpura, and pyoderma gangrenosum-like lesions.3 Histologic evaluation of these skin lesions reveals non-specific perivascular lymphocytic inflammation, leukocytoclastic vasculitis-like changes, palisading granulomas, and granulomatous vasculitis; however, it is rare to see granulomatous vasculitis or palisading necrotizing granulomas in skin specimens.4,5
WG can also affect the eyes, heart, respiratory system, nervous system, kidneys, and joints.6 The upper respiratory tract is involved in the majority of patients, and symptoms reflecting otitis, epistaxis, rhinorrhea, or sinusitis are common and may be the first manifestation of disease. When mucosal necrotizing granulomas occur, they can result in the typical saddle nose deformity seen in patients with WG. Lower respiratory tract involvement is also common and can present with cough, dyspnea, chest pain, and hemoptysis.1
Patients with WG usually have a positive c-ANCA, however this is not specific for WG and may also indicate Churg-Strauss Syndrome and microscopic polyarteritis. The median survival of patients with untreated WG is five months, and corticosteroids used alone do not change this median survival. When corticosteroids are combined with cytotoxic agents, such as cyclophosphamide, the prognosis significantly improves in greater than 90% of patients, with a 75% remission rate, and an 87% survival of patients followed from six months to 24 years.3 TH
References
- Hannon CW, Swerlick RA. Vasculitis. In: Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. Vol 1. New York: Elsevier Limited; 2003: 393-395.
- Cotch MF, Hoffman GS, Yerg DE, et al. The epidemiology of Wegener’s granulomatosis. Estimates of the five-year period prevalence, annual mortality, and geographic disease distribution from population-based data sources. Arthritis Rheum. 1996 Jan;39(1):87-92.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. [see comments]. Ann Int Med. 1992;116:488-498.
- Hu CH, O’Loughlin S, Winkelmann RK. Cutaneous manifestations of Wegener granulomatosis. Arch Dermatol. 1997;113(2):175-182.
- Lie JT. Wegener’s granulomatosis: histological documentation of common and uncommon manifestations in 216 patients. Vasa. 1997;26:261-270.
- Yi ES, Colby TV. Wegener’s granulomatosis. Semin Diagn Pathol. 2001 Feb;18(1):34-46.
History of Present Illness
A55-year-old male presented with a one and one-half week history of a sore throat, shortness of breath on exertion, ankle edema, and arthralgias that began in the ankles and subsequently spread to involve the elbows and wrists.
He had also developed a pruritic eruption involving the lower extremities, which consisted of erythematous palpable purpuric lesions and patches with superficial and central necrosis and ulceration, as well as a large 4-cm bulla of the right lateral ankle. (See Figures 1 and 2, below.) Other skin findings included petechiae of the palms and multiple ulcerations of the hard palate. Laboratory evaluation demonstrated c-ANCA antibody positivity (1:512), a proteinase 3 antibody level of greater than 100 U/ml, and a creatinine of 1.0mg/dl. TH
What is the most appropriate treatment for this condition?
- Prednisone;
- Azathioprine;
- Cyclophosphamide;
- Prednisone combined with cyclophosphamide; or
- Vancomycin combined with rifampin
Discussion
The answer is D: Wegener’s granulomatosis (WG) is a chronic granulomatous inflammatory response of unknown etiology that usually presents with the classic triad of systemic vasculitis, necrotizing granulomatous inflammation of the upper and lower respiratory tracts, and glomerulonephritis. The generalized or classic form of WG can progress rapidly to cause irreversible organ dysfunction and death. Although the pathogenesis remains unknown, it is felt that WG may result from an exaggerated cell-mediated response to an unknown antigen.1
The average age of onset for WG is 45.2 years, with 63.5% of patients male and 91% Caucasian.2 A WG diagnosis can be very difficult, and elements of the classic triad may not all be present initially. Pulmonary infiltrates or nodules are seen via chest X-ray or CT scan in just less than half of patients as an early manifestation of WG.
Occasionally WG presents with skin lesions (13%) or oral ulcers (6%), however, 40% of patients eventually develop skin involvement consisting of painful subcutaneous nodules, papules, vesicles or bullae, petechiae, palpable purpura, and pyoderma gangrenosum-like lesions.3 Histologic evaluation of these skin lesions reveals non-specific perivascular lymphocytic inflammation, leukocytoclastic vasculitis-like changes, palisading granulomas, and granulomatous vasculitis; however, it is rare to see granulomatous vasculitis or palisading necrotizing granulomas in skin specimens.4,5
WG can also affect the eyes, heart, respiratory system, nervous system, kidneys, and joints.6 The upper respiratory tract is involved in the majority of patients, and symptoms reflecting otitis, epistaxis, rhinorrhea, or sinusitis are common and may be the first manifestation of disease. When mucosal necrotizing granulomas occur, they can result in the typical saddle nose deformity seen in patients with WG. Lower respiratory tract involvement is also common and can present with cough, dyspnea, chest pain, and hemoptysis.1
Patients with WG usually have a positive c-ANCA, however this is not specific for WG and may also indicate Churg-Strauss Syndrome and microscopic polyarteritis. The median survival of patients with untreated WG is five months, and corticosteroids used alone do not change this median survival. When corticosteroids are combined with cytotoxic agents, such as cyclophosphamide, the prognosis significantly improves in greater than 90% of patients, with a 75% remission rate, and an 87% survival of patients followed from six months to 24 years.3 TH
References
- Hannon CW, Swerlick RA. Vasculitis. In: Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. Vol 1. New York: Elsevier Limited; 2003: 393-395.
- Cotch MF, Hoffman GS, Yerg DE, et al. The epidemiology of Wegener’s granulomatosis. Estimates of the five-year period prevalence, annual mortality, and geographic disease distribution from population-based data sources. Arthritis Rheum. 1996 Jan;39(1):87-92.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. [see comments]. Ann Int Med. 1992;116:488-498.
- Hu CH, O’Loughlin S, Winkelmann RK. Cutaneous manifestations of Wegener granulomatosis. Arch Dermatol. 1997;113(2):175-182.
- Lie JT. Wegener’s granulomatosis: histological documentation of common and uncommon manifestations in 216 patients. Vasa. 1997;26:261-270.
- Yi ES, Colby TV. Wegener’s granulomatosis. Semin Diagn Pathol. 2001 Feb;18(1):34-46.
History of Present Illness
A55-year-old male presented with a one and one-half week history of a sore throat, shortness of breath on exertion, ankle edema, and arthralgias that began in the ankles and subsequently spread to involve the elbows and wrists.
He had also developed a pruritic eruption involving the lower extremities, which consisted of erythematous palpable purpuric lesions and patches with superficial and central necrosis and ulceration, as well as a large 4-cm bulla of the right lateral ankle. (See Figures 1 and 2, below.) Other skin findings included petechiae of the palms and multiple ulcerations of the hard palate. Laboratory evaluation demonstrated c-ANCA antibody positivity (1:512), a proteinase 3 antibody level of greater than 100 U/ml, and a creatinine of 1.0mg/dl. TH
What is the most appropriate treatment for this condition?
- Prednisone;
- Azathioprine;
- Cyclophosphamide;
- Prednisone combined with cyclophosphamide; or
- Vancomycin combined with rifampin
Discussion
The answer is D: Wegener’s granulomatosis (WG) is a chronic granulomatous inflammatory response of unknown etiology that usually presents with the classic triad of systemic vasculitis, necrotizing granulomatous inflammation of the upper and lower respiratory tracts, and glomerulonephritis. The generalized or classic form of WG can progress rapidly to cause irreversible organ dysfunction and death. Although the pathogenesis remains unknown, it is felt that WG may result from an exaggerated cell-mediated response to an unknown antigen.1
The average age of onset for WG is 45.2 years, with 63.5% of patients male and 91% Caucasian.2 A WG diagnosis can be very difficult, and elements of the classic triad may not all be present initially. Pulmonary infiltrates or nodules are seen via chest X-ray or CT scan in just less than half of patients as an early manifestation of WG.
Occasionally WG presents with skin lesions (13%) or oral ulcers (6%), however, 40% of patients eventually develop skin involvement consisting of painful subcutaneous nodules, papules, vesicles or bullae, petechiae, palpable purpura, and pyoderma gangrenosum-like lesions.3 Histologic evaluation of these skin lesions reveals non-specific perivascular lymphocytic inflammation, leukocytoclastic vasculitis-like changes, palisading granulomas, and granulomatous vasculitis; however, it is rare to see granulomatous vasculitis or palisading necrotizing granulomas in skin specimens.4,5
WG can also affect the eyes, heart, respiratory system, nervous system, kidneys, and joints.6 The upper respiratory tract is involved in the majority of patients, and symptoms reflecting otitis, epistaxis, rhinorrhea, or sinusitis are common and may be the first manifestation of disease. When mucosal necrotizing granulomas occur, they can result in the typical saddle nose deformity seen in patients with WG. Lower respiratory tract involvement is also common and can present with cough, dyspnea, chest pain, and hemoptysis.1
Patients with WG usually have a positive c-ANCA, however this is not specific for WG and may also indicate Churg-Strauss Syndrome and microscopic polyarteritis. The median survival of patients with untreated WG is five months, and corticosteroids used alone do not change this median survival. When corticosteroids are combined with cytotoxic agents, such as cyclophosphamide, the prognosis significantly improves in greater than 90% of patients, with a 75% remission rate, and an 87% survival of patients followed from six months to 24 years.3 TH
References
- Hannon CW, Swerlick RA. Vasculitis. In: Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. Vol 1. New York: Elsevier Limited; 2003: 393-395.
- Cotch MF, Hoffman GS, Yerg DE, et al. The epidemiology of Wegener’s granulomatosis. Estimates of the five-year period prevalence, annual mortality, and geographic disease distribution from population-based data sources. Arthritis Rheum. 1996 Jan;39(1):87-92.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. [see comments]. Ann Int Med. 1992;116:488-498.
- Hu CH, O’Loughlin S, Winkelmann RK. Cutaneous manifestations of Wegener granulomatosis. Arch Dermatol. 1997;113(2):175-182.
- Lie JT. Wegener’s granulomatosis: histological documentation of common and uncommon manifestations in 216 patients. Vasa. 1997;26:261-270.
- Yi ES, Colby TV. Wegener’s granulomatosis. Semin Diagn Pathol. 2001 Feb;18(1):34-46.