User login
Obesity Management: Clinical Review and Update of the Pharmacologic Treatment Options
Over the past decade the prevalence of obesity as defined by a body mass index (BMI) ≥ 30 kg/m2 has significantly increased. In the U.S. more than 78 million adults are estimated to be obese.1 The World Health Organization projects that by 2025 up to half the U.S. population will be obese. Cardiovascular disease (CVD) and diabetes mellitus (DM) are the main comorbid conditions that are complicated by obesity. Initial weight loss of 5% to 10% of total body weight reduces CVD risk factors, prevents or delays the development of type 2 DM (T2DM) and improves the health consequences of obesity.2
To date, public health initiatives that have focused on obesity prevention and lifestyle intervention have had marginal success. In recent years, anti-obesity drug therapies have had a limited role in clinical treatment algorithms. In 2013, the American Medical Association acknowledged obesity as a disease. In turn, this acknowledgement allowed the recognition of anti-obesity drugs as acceptable therapeutic adjuncts to intensive lifestyle intervention that could address the growing obesity endemic.
In the past, medications for weight reduction were limited. Several that were FDA approved had to be removed from the market due to safety concerns. With few approved options, clinicians often had to resort to off-label use of medications. However, the landscape has changed with 4 new medications gaining recent FDA approval. This review covers older available medications and the newer medications that are now available.
Sympathomimetics
Sympathomimetic drugs have been approved for use as a pharmacological method to lose weight since 1960. Of the many versions of this drug class that have been available since then, there are 4 major versions available today. These include diethylpropion3 and benzphetamine,4 both approved in 1960; phendimetrazine, approved in 1976;5 phentermine, approved in 1980;6 and phentermine hydrochloride, approved in 2012.7 Despite the existence of several other classes of drugs to treat obesity, phentermine remains the most often prescribed weight loss drug in the U.S.8
Although the mechanism of action (MOA) of sympathomimetic drugs is not particularly clear, weight loss from these medications is believed to be due to the increase in the release of biogenic amines (mainly norepinephrine, but also possibly dopamine), from storage sites in nerve terminals. It is possible that these drugs slow catecholamine metabolism by inhibiting the actions of monoamine oxidase. The resulting increase in amine availability, particularly in the lateral hypothalamic feeding center, is associated with reduced food intake. Interestingly, injection of these drugs into the ventromedial satiety center dooes not seem to suppress food intake, and the effects of biogenic amines on increasing metabolism does not seem to play a significant role in weight loss in patients on these medications.9
Each of these drugs is rapidly absorbed from the gastrointestinal (GI) tract except for phentermine hydrochloride, the newest of the medications in this class. Phentermine hydrochloride is a sublingual tablet that is readily absorbed through the buccal mucosa.5 All of the drugs in this class are excreted through the kidneys, with varying rates. Each drug’s excretion is highly dependent on the pH of the urine—more alkaline conditions result in less excretion and more acidic conditions result in more excretion. As a result, these drugs should be used with caution in patients with renal impairment; however, there are no specific contraindications listed for patients with poor renal function.
The adverse effects (AEs) for this drug class are to be expected from an increase in the release of biogenic amines in the central nervous system (CNS). The most common AEs include palpitations, tremors, restlessness, insomnia, dry mouth, constipation, diaphoresis, changes in libido, and irritability. The more dangerous AEs that have been observed include arrhythmias, hypertension, dependency/abuse, convulsions, acute transient ischemic colitis, and acute urinary retention secondary to increased bladder sphincter tone, transient hyperthyroxemia, and paranoia.10
Several contraindications exist for sympathomimetics, including the presence of advanced arteriosclerosis, symptomatic CVD, moderate to severe hypertension, hyperthyroidism, glaucoma, patients in an agitated state, or those with a history of amphetamine abuse. The warnings for prescribers include pulmonary hypertension and cardiomyopathy secondary to chronic use of sympathomimetics, and valvular heart disease secondary to use of sympathomimetics with additional anorectic agents.
Additional precautions should be considered in those with a history of anxiety/psychosis, those who operate machinery and motor vehicles, and even those with mild hypertension. The data surrounding the effects of sympathomimetics on blood pressure (BP) appears to be conflicting and the relationship does not seem to have been significantly studied in depth to warrant any definitive conclusions. The MOA of this drug class itself is enough to urge caution to prescribers.11 Special attention should be given to patients with diabetes when using sympathomimetics. A reduction of insulin dose or oral hypoglycemic dose may be necessary in some people with diabetes.
Only diethylpropion is pregnancy category B, whereas the others drugs in this class are pregnancy category X. It has been demonstrated that diethylpropion and benzphetamine are secreted into breastmilk; insufficient data exist to suggest whether or not phentermine and phendimetrazine are present in breastmilk. All drugs in this class should be used in caution with breastfeeding mothers.
Although all 4 drugs are registered as controlled substances, benzphetamine and phendimetrazine are schedule III and phentermine and diethylpropion are schedule IV, despite evidence suggesting the potential for abuse to be extremely low.12,13 Phentermine has been approved for adults aged > 18 years, phendimetrazine has been approved for those aged > 17 years, diethylpropion has been approved for those aged > 16 years, and benzphetamine has been approved for those aged > 12 years.
There is a wealth of literature surrounding the effectiveness of this drug class for weight loss. One of the longest trials of phentermine was recently conducted as part of the initial component of a FDA study for the newly approved topiramate-phentermine combination. Weight loss at 6 months in the phentermine-only group was significantly higher at -5.8% compared with -1.5% with the placebo group in the last observation carried forward-Intent to treat (LOCF-ITT) analysis.14 Similarly, a long-term study looking at diethylpropion examined the use of diethylpropion for up to a year vs placebo. Participants administered diethylpropion lost a mean 9.8% of original weight vs 3.7% in the placebo group in the first 6 months alone.15
Several meta-analyses and review papers have been authored that examine and analyze the published data on this drug class overall and comparatively within this class. Haddock and colleagues in 2002 reviewed the numerous clinical trials associated with each drug in this class, in addition to several other classes, and found that although each drug demonstrated a significant advantage vs placebo in weight loss, there was not a specific drug that was significantly superior to any of the others.16
These results seem to be in relative agreement with additional studies like that published by Suplicy and colleagues, which demonstrated that several sympathomimetics were better than placebo in weight loss, and that there was little difference between the specific drugs in the class.17 However, it should be noted that as highlighted in a review by Ioannides-Demos and colleagues in 2005, the vast majority of studies that had been performed on this drug class focused on short-term use (< 16 weeks) and none of the sympathomimetics listed here have been approved for long-term use.18
Orlistat
Orlistat 120 mg was approved in 1999 as a reversible inhibitor of GI lipases that specifically reduced the absorption of dietary fat due to the inhibition of triglyceride hydrolysis.19 Orlistat was later approved in 2007 for release in a reduced dosage form (60 mg) for over-the-counter sales.20
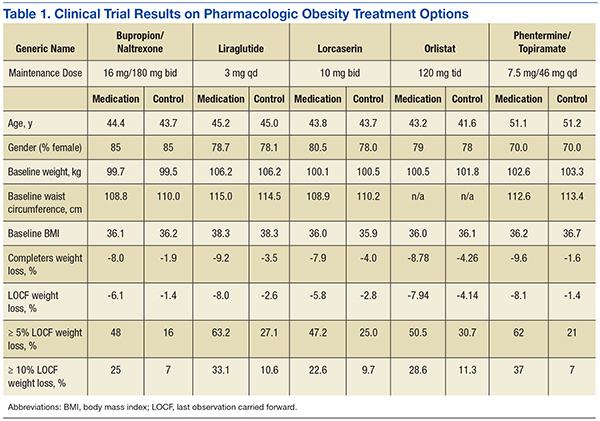
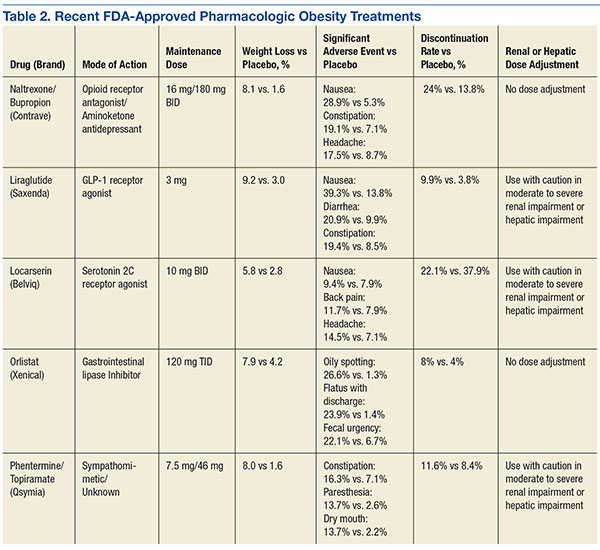
Orlistat forms a covalent bond with the active serine residue site of gastric and pancreatic lipases in the lumen of the stomach and small intestine. The inhibition of these enzymes causes dietary fat to remain undigested as triglycerides, which cannot be converted to absorbable free fatty acids and monoglycerides, leading to decreased calorie absorption. Orlistat is not systemically absorbed and is eliminated mainly through feces. Some metabolism occurs in the GI wall.21Orlistat is most known for its GI AEs. Because it is most active in the lumen of the GI system and reduces the absorption of triglycerides, many AEs are related to malabsorption. The most common issues 1 year after starting the drug were oily spotting (26.6% vs 1.3% placebo); flatus with discharge (23.9% vs 1.4% placebo); fecal urgency (22.1% vs 6.7% placebo); fatty/oily stool (20% vs 2.9% placebo); increased defecation (10.8% vs 4.1% placebo); and fecal incontinence (7.7% vs 0.9% placebo) (Table 1). Most of these AEs were greatly reduced after taking the drug for 2 years. Orlistat also has more serious AEs noted, including abdominal pain/discomfort; nausea; infectious diarrhea; rectal pain/discomfort; tooth disorder; gingival disorder; vomiting; upper respiratory infection; lower respiratory infection; ear, nose and throat symptoms; back pain; arthritis; myalgia; joint disorders; tendonitis; headache; dizziness; fatigue; sleep disorders; rash; dry skin; menstrual irregularity; vaginitis; urinary tract infection; and psychiatric disorders, although these did not differ markedly from placebo.
One of the most serious AEs reported was fulminate hepatic failure, though this AE is rare. Thirteen cases of liver injury were reported with the 120-mg prescription dose of orlistat and 1 case report in the U.S. involved the 60-mg over-the-counter dosage of orlistat.21,22 The FDA suggests that patients talk to their physicians about risks of liver failure, and that physicians should educate their patients about signs and symptoms of liver failure so that patients can stop taking orlistat and seek immediate medical help if symptoms occur.
One of the first published trials was the European Multicentre Orlistat Study Group, which included 743 participants with BMI between 28 kg/m2 and 47 kg/m2 from 15 different European centers. To test adherence, a 4-week single blind placebo lead-in was started with a hypocaloric diet. The first stage was completed by 688 patients who then proceeded to the double blind randomized control trial portion with a hypocaloric diet. From the start of lead-in to the end of year 1, the orlistat group weight decreased 10.2% (10.3 kg) vs 6.1% (6.1 kg) in the placebo group. The placebo subtracted difference between the groups was 3.9 kg (P < .001).23
A U.S.-based randomized double-blind placebo-controlled multicenter study included 796 obese patients with BMI between 30 kg/m2 and 44 kg/m2. Patients were assigned to 1 of 3 groups: placebo, orlistat 60 mg 3 times daily, or orlistat 120 mg 3 times daily. All groups were given a reduced energy diet. Patients in the orlistat 120 mg group lost significantly more weight than did the placebo group, -8.78% vs -4.26% respectively in year 1 in the completer analysis (P = .001). More participants who were treated with orlistat 120 mg lost 5% or more of their initial weight in year 1 compared with placebo, 50.5% vs 30.7% respectively (P < .001).24
In the XENDOS study the primary outcome measurement was time to onset of T2DM. Eligible participants were aged 30 to 60 years, with a BMI > 30 kg/m2. All patients had a 75-g oral glucose tolerance test and were required to have normal glucose tolerance or impaired glucose tolerance, but not T2DM. The double-blind randomized controlled trial included 3,305 subjects and compared a group taking 120 mg orlistat 3 times daily vs placebo. All patients were prescribed a reduced-calorie diet (800 kcal/d deficit) containing 30% of calories from fat. Patients were also encouraged to walk at least 1 kilometer daily in addition to their usual physical activity. Incidence of T2DM after 4 years was 6.2% in the orlistat group and 9.0% in the placebo group, reflecting a 37.3% risk reduction in the orlistat group (P = .0032).25,26
Lorcaserin
In 2012, lorcaserin HCl was FDA approved as a schedule IV drug for use as a weight loss medication as an adjunct to a reduced-calorie diet and increased physical activity. Lorcaserin is thought to act on 5-hydroxytryptamine-2c (5HT2c) receptors on the pro-opiomelanocortin (POMC) neurons in the arcuate nucleus, causing release of alpha-melanocortin-stimulating hormone (alpha-MSH), which in turn acts on melanocortin-4 receptors in the paraventricular nucleus to suppress appetite. At the maximum suggested dose of 10 mg twice daily, lorcaserin binds with 15 to 100 times greater affinity to 5HT2c receptors compared with 5HT2a and 5HT2b receptors respectively.
Indications for lorcaserin include patients with BMI ≥ 30 kg/m2 or ≥ 27 kg/m2 or greater with a weight-related comorbid condition such as hypertension, dyslipidemia, cardiovascular disease, impaired glucose tolerance, or sleep apnea.
The efficacy of lorcaserin for weight loss has been evaluated in 3 separate trials. The trials were randomized, double blinded and placebo controlled. The BLOOM trial, which included 3,182 patients with a mean BMI of 36.2 kg/m2, evaluated the efficacy of lorcaserin as a weight loss adjunct.27 Patients with pre-existing valvular disease, uncontrolled hypertension, or a major psychiatric condition were excluded. After initial randomization, patients were assigned to receive either lorcaserin 10 mg twice daily or a placebo. The primary endpoint was a 5% weight reduction from baseline by the end of 2 years. At 1 year, 47.5% of patients in the lorcaserin group and 20.3% in the placebo group had lost ≥ 5% of their body weight (P <.001). The average loss for the lorcaserin group was 5.8 ± 0.2 kg and 2.2 ± 0.1 kg for the placebo group at 1 year (P < .001).
The BLOSSOM trial was a 1-year study of 4,008 patients aged 18 to 65 years. The trial evaluated the effects of lorcaserin on body weight, CVD risk factors, and safety in obese and overweight patients.28 Patients were randomized in a 2:1:2 ratio to receive lorcaserin 10 mg twice daily, lorcaserin 10 mg once daily, or placebo. The primary endpoint was the proportion of patients achieving at least 5% reduction in body weight. Completer analysis showed weight reduction in the placebo group was 4.0% and 7.9% in the lorcaserin group (P < .001). In the modified intent-to-treat/last observation carried forward analysis (MITT/LOCF), a statistically significant 47.2% of patients receiving lorcaserin 10 mg twice daily and 40.2% of patients receiving lorcaserin 10 mg once daily lost at least 5% of baseline body weight; compared with 25% of patients receiving placebo (P < .001). Weight loss of at least 10% was achieved by 22.6% of patients receiving lorcaserin 10 mg twice daily, and 17.4% of patients receiving 10 mg daily compared with 9.7% of patients in the placebo group (P < .001).
The most common AEs noted were headache, nausea, and dizziness. Echocardiographic evidence of valvulopathy occurred in 2% of patients taking lorcaserin 10 mg twice daily and those taking the placebo. Lorcaserin administered in conjunction with a diet and exercise program was associated with an overall reduction in baseline BMI when compared with placebo over the year.
The BLOOM-DM study evaluated efficacy and safety of lorcaserin for weight loss in 604 patients with T2DM over the course of 1 year.29 Patients had a hemoglobin A1c (A1c) of 7% to 10% and were treated with metformin, a sulfonylurea, or both. The primary endpoint was a 5% weight reduction from baseline at the end of 1 year. Patients were randomized into 3 groups: 1 group received lorcaserin 10 mg twice daily, 1 group took lorcaserin 10 mg daily, and 1 group received the placebo. A statistically significant 37.5% of patients taking lorcaserin 10 mg twice daily achieved > 5% body weight reduction, compared with 44.7% in the lorcaserin 10 mg daily group, and 16.1% in the placebo group. Overall reductions in A1c and fasting glucose were observed in both lorcaserin groups taking as compared with placebo. Patient A1c decreased 0.9 ± 0.06 with lorcaserin 10 mg bid, 1.0 ± 0.09 with lorcaserin 10 mg qd, and 0.4 ± 0.06 with the placebo (P < .001). Fasting glucose in the lorcaserin bid, lorcaserin qd, and placebo groups decreased 27.4 ± 2.5 mg/dL, 28.4 ± 3.8 mg/dL, and 11.9 ± 2.5 mg/dL, respectively (P < .001). Symptomatic hypoglycemia occurred in 7.4% of patients on lorcaserin bid, 10.5% on lorcaserin qd, and 6.3% on placebo. Headache, back pain, nasopharyngitis, and nausea were among the most commonly reported AEs.
As lorcaserin is a serotonergic agonist, potential interactions exist when used with other medications affecting serotonin. Most notably, serotonin syndrome and neuroleptic malignant syndrome-like reactions may occur. Because of this, it is recommended to avoid selective serotonin re-uptake inhibitors (SSRIs), selective norepinephrine reuptake inhibitors, tricyclic antidepressants, bupropion, triptans, monoamine oxidase inhibitors, lithium, dextromethorphan, and dopamine agonists. Lorcaserin seems to be safe in those patient populations with mild hepatic as well as mild renal impairment; however, it is not recommended for those with severe renal impairment. Given the multiple enzymatic pathways used to metabolize lorcaserin, there is a low probability for cytochrome drug interactions. Safety has not been well evaluated in patients aged < 18 years and those that are pregnant (pregnancy category X).
Adverse events include headache, dizziness, fatigue, nausea, and dry mouth. Other notable AEs include nasopharyngitis and URI. Hypoglycemia appeared to be more common in patients with DM taking lorcaserin. Cognitive impairment and psychiatric disorders including euphoria and hallucinations were also reported. Notably, valvular heart disease has been reported in patients who take medications with 5HT2b activity. In a 1-year clinical trial, a small number of patients were found to develop valvular regurgitation. Furthermore, bradycardia, priapism, leucopenia, elevated prolactin, and pulmonary hypertension have also been observed. Caution is recommended if symptoms of any of the aforementioned conditions are noticed.
Qsymia
The schedule IV controlled substance Qsymia (Vivus, Mountain View, CA) is a combination of phentermine, an anorexigenic agent, and topiramate extended-release, an antiepileptic drug. In July of 2012 it was approved for chronic weight management as an addition to a reduced-calorie diet and exercise. The drug is approved for adults with a BMI ≥ 30 kg/m2 or adults with a BMI ≥ 27 kg/m2 who have at least 1 weight-related condition such as hypertension, T2DM, or dyslipidemia.30
In 1996 topiramate was approved by the FDA for the treatment of seizure disorders and was also approved for migraine prophylaxis in 2004. In patients who were treated with topiramate for seizure disorders and migraines, weight loss and a reduction in visceral body fat has been observed.31 The precise MOA of topiramate in regards to weight loss is not fully understood. It may be due to its effects on both appetite suppression and satiety enhancement. Topiramate exhibits a combination of properties including modulatory effects on sodium channels, enhancement of GABA-activated chloride channels, inhibition of excitatory neurotransmission through actions on kainite and AMPA receptors, and inhibition of carbonic anhydrase (CA) isoenzymes in particular CA II and IV.14
The combination of phentermine and topiramate is a once-daily formulation that is designed to provide an immediate release of phentermine and a delayed release of topiramate, allowing a peak exposure of the phentermine in the morning and a peak concentration of topiramate in the evening. It should be taken in the morning in order to avoid the possibility of insomnia that can occur if taken in the evening. It can be taken with or without food. The recommended dose is as follows: Start treatment with Qsymia 3.75 mg/23 mg extended-release daily for 14 days; after 14 days increase to the recommended dose of Qsymia 7.5 mg/46 mg once daily.
Weight loss should be evaluated after 12 weeks at the higher dose. If at least 3% of baseline body weight has not been lost at that time, discontinue or escalate the dose. To escalate the dose: Increase to Qsymia 11.25 mg/69 mg daily for 14 days; followed by Qsymia 15 mg/92 mg daily. Evaluate weight loss following dose escalation to Qsymia 15 mg/92 mg after an additional 12 weeks of treatment. If at least 5% of baseline body weight has not been lost on Qsymia 15 mg/92 mg, discontinue as directed. It is important not to suddenly discontinue, as this may cause seizures. Patients should be slowly titrated off the medication.
In vitro studies of phentermine and topiramate indicate that these drugs are not likely to cause clinically significant interactions with drugs using the cytochrome P450 enzyme pathways, or those involved in plasma protein binding displacement; however there is evidence suggesting that ethinyl estradiol levels may be decreased by 16%, thus raising a concern about the possibility of decreased contraceptive efficacy.31 In patients with moderate (creatine clearance ≥ 30 mL/min to < 50 mL/min) and severe renal dysfunction (< 30 mL/min), the maximum dose of should not exceed 7.5 mg/46 mg.
Qsymia was evaluated in 3 phase 3 trials for its long-term efficacy and safety. In all trials, diet and lifestyle counseling were provided for all patients. The first of these studies was OB-301, a 28-week confirmatory trial with a factorial design involving 7 treatment arms, tested 2 fixed-dose Qsymia combinations—regular dose (7.5 mg/46 mg) and maximum dose (15 mg/92 mg)—as well as regular and maximum doses of the individual constituent drugs vs placebo.32 The study randomized 756 obese patients with a BMI range of 30 kg/m2 to 45 kg/m2 to 1 of the 7 treatment arms for 28 weeks. Patients treated with maximum-dose Qsymia achieved an average weight change of -9.0%, vs -1.5% with placebo (P < .0001). Weight change with regular-dose Qsymia was -8.2%. Weight changes with monotherapies were: -6.1% with topiramate 92 mg, -4.9% with topiramate 46 mg, -5.8% with phentermine 15 mg, and -5.2% with phentermine 7.5 mg.
OB-302 was a 56-week trial that randomized 1,267 morbidly obese patients with a BMI ≥ 35 kg/m2 without significant comorbidities to low-dose Qsymia (3.7 mg/23 mg), maximum-dose Qsymia (15 mg/92 mg), or placebo.33 At baseline, the mean BMI for the entire study cohort was 42 kg/m2. Mean weight changes were -1.6% with placebo, -5.1% with low-dose Qsymia, and -10.9% with maximum-dose Qsymia. The proportions of patients achieving ≥ 5% weight loss were: 17% with placebo, 45% with low-dose Qsymia, and 67% with maximum-dose Qsymia.
CONQUER was the largest of the phase 3 trials. It randomized 2,487 overweight or obese patients with a BMI of 27 kg/m2 to 45 kg/m2 and ≥ 2 obesity-related comorbidities (hypertension, dyslipidemia, T2DM, prediabetes or abdominal obesity) to receive a placebo, regular-dose Qsymia, or maximum-dose Qsymia for 56 weeks.34 In the completer population, mean weight changes in the placebo, regular dose Qsymia, and maximum-dose Qsymia groups were -1.6%, -9.6% (P <.0001), and -12.4% (P < .0001); and weight loss of ≥ 5% was achieved by 21%, 62%, and 70%, respectively. Relative to placebo, there were greater reductions in systolic BP, triglycerides, and fasting insulin with both doses of Qsymia.
Patients should not take Qsymia if they are pregnant, planning to become pregnant, or become pregnant during Qsymia treatment as there is an increased risk of birth defects, namely cleft lip and cleft palate. Women who can become pregnant should have a negative pregnancy test before taking Qsymia and every month while on the medication. They should use effective birth control consistently while taking Qsymia.
Qsymia is contraindicated in patients with glaucoma and patients who have hyperthyroidism. Qsymia can cause an increase in resting heart rate and regular monitoring of resting heart rate is recommended, especially in patients with cardiac or cerebrovascular disease. It has not been studied in patients with recent or unstable cardiac or cerebrovascular disease and therefore use is not recommended.
Qsymia can cause mood disorders such as anxiety and depression and can increase the risk of suicidal thoughts. Patients should be monitored for worsening depression, suicidal thoughts or behavior, or any unusual changes in mood or behavior. It is not recommended in patients with a history of suicidal attempts or active suicidal ideation. Qsymia can cause cognitive dysfunction. It can cause confusion, problems with concentration, attention, memory, or speech. Patients should be cautioned about operating automobiles and hazardous machinery.
Normal anion gap hyperchloremic metabolic acidosis has been reported in patients treated with Qsymia. If this does develop and persists, consideration should be given to either reduce the dose or discontinue Qsymia.
Weight loss may increase the risk of hypoglycemia in patients with T2DM treated with insulin and/or insulin secretagogues (eg, sulfonylureas). Qsymia has not been studied in combination with insulin. A reduction in the dose of antidiabetic medications, which are nonglucose dependent, should be considered to reduce the risk of hypoglycemia.
The most common AEs in controlled clinical studies (≥ 5% and at least 1.5 times placebo) included paraesthesia in the hands, arms, feet or face, dizziness, dysgeusia, insomnia, constipation, and dry mouth.
Contrave
In 2014, the FDA approved Contrave (Takeda, Deerfield, IL) as treatment option for chronic weight management in addition to reduced-calorie diet and physical activity. The combination of naltrexone hydrochloride and bupropion hydrochloride was originally introduced for the treatment of opioid addiction and later expanded to include the treatment of alcoholism. The antidepressant bupropion was approved in the U.S. in 1989. It is structurally different from all other marketed antidepressants (ie, tricyclics, tetracyclics, and SSRIs), but closely resembles the structure of diethylpropion, an appetite depressant with minimal CNS effects.35
This drug is approved for adults with BMI ≥ 30 kg/m2 and for adults with BMI ≥ 27 kg/m2 with at least 1 weight-related risk factors such as hypertension, T2DM, or dyslipidemia. It should be used as an adjunct to diet and exercise and is not approved for use for depression even though it contains bupropion.
Naltrexone is a pure opioid antagonist with high affinity to μ-opioid receptor, which is implicated in eating behavior. Naltrexone is rapidly and nearly completely absorbed from the GI tract after oral administration. The time to peak plasma concentration is about 1 hour. Naltrexone is well absorbed but first pass extraction and metabolism by the liver decreases oral bioavailability to between 5% to 40%. Primary elimination of naltrexone and its metabolites is renal excretion.
Bupropion is a weak inhibitor of neuronal reuptake of dopamine and norepinephrine. This drug is used to treat depression and seasonal affective disorder, and aid in smoking cessation. Bupropion is absorbed rapidly after oral administration, but the absolute oral bioavailability of bupropion is not known because an IV preparation is not available. The time to peak plasma concentrations of bupropion is within 2 hours of oral administration. Bupropion is extensively metabolized by the liver to multiple metabolites. Primary elimination of bupropion is urinary excretion. However, hepatic and renal impairment may affect the elimination of bupropion and its metabolites. Patients with hepatic or renal impairment should use a reduced dosage.
Combination therapy has been found to have complementary actions on CNS to reduce food intake. They are believed to dampen CNS reward pathways, taking away the compulsive feeding behavior and pleasure of feeding, ultimately leading to weight loss. Bupropion stimulates hypothalamic pro-opiomelanocortin neurons (POMC), which results in reduced food intake and increased energy expenditure. Naltrexone blocks opioid-receptor mediated POMC auto-inhibition, blocks the increase in dopamine in nucleus accumbens that occurs when eating, and acting synergistically with bupropion in augmenting POMC firing.
The COR-I and COR-II trials compared Contrave to diet and exercise in patients who did not have DM. The COR-Diabetes trial included the same study design but focused on patients with DM. In all the studies the participants had a 4-week titration to Contrave (naltrexone 8 mg/bupropion 90 mg) to decrease nausea. The first week dosing was 1 tablet in the morning. Week 2 was 1 tablet in morning and 1 tablet in the evening. In week 3, patients took 2 tablets in the morning and 1 tablet in the evening. The final titration step was 2 tablets in the morning and 2 tablets in the evening.
The COR-1 study was a 56-week randomized, double-blind, placebo-controlled study. It compared Contrave 32 mg naltrexone/360 mg bupropion (NB32/360) with an active placebo of diet and exercise.36 To be included adults must be aged 18 to 65 years with a BMI 30 kg/m2 to 45 kg/m2 or a BMI 27 kg/m2 to 45 kg/m2 with dyslipidemia or hypertension. Patients were instructed on a hypocaloric diet that was a 500 kcal per day deficit based on World Health Organization algorithm for calculating metabolic rate and they were urged to increase physical activity.
The completer population results showed 8.0% weight loss in the NB32/360 group and 1.9% weight loss in the placebo group (P < .001). For the NB32/360 and placebo groups, weight loss of ≥ 5% was achieved by 48% and 16% (P <.001), respectively; and weight loss of ≥ 10% by 25% and 7% (P < .001), respectively. The most common AE was nausea—29.8% with NB32/360 vs 5.3% with placebo. Nausea generally occurred early and then diminished and the discontinuation rate from nausea was significantly lower (6.3%) then the overall reported nausea rates.
Contrave was also studied in patients with T2DM. The COR-Diabetes Trial was a 56-week randomized, double blind, placebo-controlled study. The trial compared Contrave 32 mg naltrexone/360 mg bupropion (NB32/360) with an active placebo of diet and exercise.37 Inclusion criteria for the trial were patients aged 18 to 70 years with T2DM and a BMI from 27 kg/m2 to 45 kg/m2, A1c between 7% and 10%, and fasting blood glucose < 270 mg/dL. Participants either were not taking a DM medication or were on stable doses of oral antidiabetes drugs ≥ 3 months prior to randomization. Patients were placed on a 500 kcal hypocaloric diet and advised to increase physical activity.
The results showed 5.0% weight loss in the NB32/360 group and 1.8% weight loss in the placebo group (P < .001). Weight loss of ≥ 5% and ≥ 10% was achieved by 44.5% and 18.5% of the NB32/360 group, respectively, and 18.9% and 5.7%, respectively (P < .001) of the placebo group. The NB32/360 and placebo showed a reduction of A1c of 0.6% and 0.1% respectively (P < .001). The most common AE was nausea (42.3% with NB32/360 vs 7.1% with placebo). Nausea generally occurred early and then diminished and the discontinuation rate from nausea was significantly lower (9.6%) then the overall reported nausea rates.37
Due to potential nausea caused by naltrexone, Contrave should be titrated over 4 weeks as described earlier. At maintenance dose, patients should be evaluated after 12 weeks to determine treatment benefits. If a patient has not lost at least 5% of baseline body weight, Contrave should be discontinued, because it would be unlikely that the patient will achieve and sustain clinically meaningful weight loss with continued treatment. Contrave should not be taken with high-fat meals that may result in significant increase in bupropion and naltrexone systemic exposure.
Since Contrave contains the antidepressant bupropion, it has a boxed warning similar to other antidepressants in its class of increased risk of suicidal thoughts and behaviors, especially in children, adolescents, and young adults.38Contrave can lower the seizure threshold; therefore it should not be used in people with a seizure disorder. It can also raise BP and heart rate; however the clinical significance of hypertension and elevated heart rate observed with Contrave treatment is unclear. Blood pressure rose on average by 1 point during the first 8 weeks of treatment and then returned to baseline.38 The heart rate also increased by about 1.7 beats per minute.38 Patients with uncontrolled hypertension should avoid Contrave.
Contrave should not be taken with products contain bupropion or naltrexone. It should not be taken by patient who are regularly taking opioids or who are opioid dependent, or who are experiencing opiate withdrawal. Pregnant women should also avoid Contrave. In patients with renal impairment the maximum dose is 1 tablet twice a day and in patients with hepatic impairment the maximum dose is 1 tablet a day.
Liraglutide
Liraglutide is the newest weight loss medication to be approved by the FDA for chronic weight management as an adjunct to a reduced calorie diet and increased physical activity in adult patients with BMI ≥ 30 kg/m2 or ≥ 27 kg/m2 with hypertension, diabetes, or dyslipidemia. The recommended dose of liraglutide is 3 mg daily. The initial dose is 0.6 mg daily for the first week, then titrated up by 0.6 each week for 4 weeks, until reaching 3 mg daily.
Liraglutide is an acylated human glucagon-like peptide-1 (GLP-1) receptor agonist, which are expressed in the brain and is involved in the control of appetite. It is also found in the beta cells of the pancreas, where GLP-1 receptors stimulate insulin release in response to elevated blood glucose concentrations and suppress glucagon secretion. Endogenous GLP-1 has a half-life of 1.5 to 2 minutes due to degradation by the DDP-4 enzyme, but liraglutide is stable against degradation by peptidases and has a half- life of 13 hours.
Liraglutide was studied in a 56-week randomized, double-blind, placebo-controlled trial, which compared liraglutide 3 mg with an active placebo of diet and physical activity.39 Inclusion criteria were adults aged ≥ 18 years old with a BMI 30 kg/m2 to 45 kg/m2 or BMI 27 kg/m2 to 45 kg/m2 with dyslipidemia and/or hypertension. Both groups received lifestyle modification counseling. Patients were excluded if they had DM.
In the trial, 3,731 participants enrolled, 2,487 in the liraglutide group and 1,244 in the placebo group; 78.7% of the participants were female and the average age was 45 years. Subjects in the liraglutide group had a weekly titration regimen. The starting dose at week 1 was 0.6 mg, week 2 was 1.2 mg, week 3 was 1.8 mg, week 4 was 2.4 mg, and week 5 was 3.0 kg.
The completer population showed 9.2% weight loss in the liraglutide group and 3.0% weight loss in the control group.39 Weight loss of ≥ 5% was seen in 63.2% and 27.1% of the liraglutide and placebo groups, respectively. Weight loss rates of ≥ 10% was seen by 33.1% and 10.6%, respectively. The most common AEs were nausea, diarrhea, and constipation. Nausea generally occurred early during the titration period and then diminished.
A second clinically relevant study was performed with liraglutide. Often patients are able to lose weight with diet and exercise and then plateau. This study examined participants who lost 5% percent of their initial body weight and then were randomized to liraglutide or placebo.40 Key inclusion criteria were people aged ≥ 18 years old with a BMI 30 kg/m2 to 45 kg/m2 or BMI 27 kg/m2 to 45 kg/m2 with dyslipidemia and/or hypertension. In order to be randomized, participants were required to lose at least 5% of their initial body weight on a 1,200 kcal to 1,400 kcal diet with increased physical activity during a 4 to 12 week run-in period.
Four hundred twenty-two participants were enrolled, 212 in the liraglutide group and 210 in the placebo group. Most of the participants were female (81%). The average BMI in the study was 35.6 kg/m2. Subjects in the liraglutide group had a weekly titration regimen.
After an average weight loss of 6% using a low calorie diet and increased physical activity the participants were randomized to continue diet and increased activity alone (placebo) or with liraglutide. At week 56 the results showed an additional 6.2% weight loss in the liraglutide group and 0.2% weight gain in the placebo group. The liraglutide group had a greater number of participants with ≥ 5% weight loss compared to placebo, 50.5% vs 21.8% (P < .0001).40 In the pooled data set from the registration trials the 3 most common GI AEs were nausea, diarrhea, and constipation occurring in 39.3%, 20.9%, and 19.4% of participants respectively. Discontinuation due to nausea for liraglutide was 2.9%.41
Clinicians should be aware that medications that can cause hypoglycemia such as sulfonylureas and insulin must be tapered as patients lose weight with liraglutide. Documented symptomatic hypoglycemia in patients with T2DM and with sulfonylurea background therapy was 43.6% with liraglutide vs 27.3% with placebo.
In the setting of renal impairment, patients treated with GLP-1 receptor agonists, including liraglutide, have had reports of acute renal failure and worsening of chronic renal failure usually associated with nausea, vomiting, diarrhea, or dehydration. Liraglutide causes thyroid C-cell tumors at clinically relevant exposures in rats and mice. It is unknown whether liraglutide causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans. As the human relevance of liraglutide-induced rodent thyroid C-cell tumors has not been determined liraglutide is contraindicated in patients with a personal or family history of MTC or in patients with multiple endocrine neoplasia syndrome type 2.
Acute pancreatitis, including fatal and nonfatal hemorrhagic or necrotizing pancreatitis, has been observed in patients treated with liraglutide in postmarketing reports. After initiation of liraglutide, observe patients carefully for signs and symptoms of pancreatitis (including persistent severe abdominal pain, sometimes radiating to the back, which may or may not be accompanied by vomiting). If pancreatitis is suspected, liraglutide should promptly be discontinued.
Conclusion
The treatment of obesity and overweight with comorbidities has always been a challenge. In the past there were few FDA-approved drugs and many drugs had to be used off-label. The toolbox of medications available for medical weight management is more robust than ever. The medications have different MOAs and can be used in a variety of patients. There are differences in the classes and some are controlled substances. Phentermine, lorcaserin, and Qsymia (phentermine/topiramate) are controlled substances whereas orlistat, naltrexone/bupropion and liraglutide are not. Other differences exist including duration of use. The sympathomimetic drugs have a limited window of use whereas orlistat, Qsymia (phentermine/topiramate), lorcaserin, naltrexone/bupropion, and liraglutide do not.
The medications that are available have a wide variety of MOAs. Therefore, if a patient fails one medication, then it is very reasonable to try a medication with a different MOA. In addition, there is the potential for weight regain when weight reduction medications are discontinued. As people lose weight their metabolic rate decreases about 15 kcal per pound of weight reduction.42
Another challenge of using these medications is managing patient expectations. The current metric used for FDA approval is a 5% weight loss that is greater in the study group compared with the diet and physical activity active control. However, many clinicians and patients do not find this weight reduction amount consistent with their expectations. In addition weight loss trajectory may also be too slow for patients and cause early discontinuation. Therefore, patient education and a discussion of reasonable expectations for weight reduction medications are necessary.
Clinicians must acknowledge that there are limitations to the use of these medications. Newer agents do have a higher cost and insurance reimbursement is somewhat limited. However, they offer the opportunity to prevent more expensive, protracted conditions such as diabetes and cardiovascular disease. In summary, clinicians now have a wider variety of medication options to be used with dietary and lifestyle changes in order to improve health and prevent chronic diseases.
1. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of obesity in the United States, 2009-2010. NCHS Data Brief. 2012(82):1-8.
2. Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. Circulation. 2014;129(25 suppl 2):S102-S138.
3. U.S. Food and Drug Administration. Drugs@FDA: diethylpropion hydrochloride. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=012546&DrugName=TENUATE%20DOSPAN&ActiveIngred=DIETHYLPROPION%20HYDROCHLORIDE&SponsorApplicant=ACTAVIS%20LABS%20UT%20INC&ProductMktStatus=1&goto=Search.DrugDetails. Accessed December 28, 2015.
4. U.S. Food and Drug Administration. Drugs@FDA: benzphetamine hydrochloride. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=012427&DrugName=DIDREX&ActiveIngred=BENZPHETAMINE%20HYDROCHLORIDE&SponsorApplicant=PHARMACIA%20AND%20UPJOHN&ProductMktStatus=3&goto=Search.DrugDetails. Accessed December 28, 2015.
5. U.S. Food and Drug Administration. Drugs@ FDA: phendimetrazine tartrate. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=088021&DrugName=BONTRIL&ActiveIngred=PHENDIMETRAZINE%20TARTRATE&SponsorApplicant=VALEANT&ProductMktStatus=3&goto=Search.DrugDetails. Accessed December 28, 2015.
6. U.S. Food and Drug Administration. Drugs@FDA: phentermine. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=085128&DrugName=ADIPEX%2DP&ActiveIngred=PHENTERMINE%20HYDROCHLORIDE&SponsorApplicant=TEVA&ProductMktStatus=1&goto=Search.DrugDetails. Accessed December 28, 2015.
7. U.S. Food and Drug Administration. Drugs @ FDA: phentermine hydrochloride. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=202088&DrugName=SUPRENZA&ActiveIngred=PHENTERMINE%20HYDROCHLORIDE&SponsorApplicant=CITIUS%20PHARMS&ProductMktStatus=1&goto=Search.DrugDetails. Accessed December 28, 2015.
8. Hendricks EJ, Rothman RB, Greenway FL. How Physician Obesity Specialists use drugs to treat obesity. Obesity (Silver Spring). 2009;17(9):1730-1735.
9. Gilman AG, Gilman A, Goodman LS, eds. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 8th ed. New York: Pergamon Press; 1990: 211.
10. Gilman AG, Gilman A, Goodman LS, eds. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 8th ed. New York: Pergamon Press; 1990:217.
11. Addy C, Jumes P, Rosko K, et al. Pharmacokinetics, safety, and tolerability of phentermine in health participants receiving taranabant, a novel cannabinoid-1 receptor (CB1R) inverse agonist. J Clin Pharmacol. 2009;49(10):1228-1232.
12. U.S. Department of Justice, Drug Enforcement Administration, Office of Diversion Control. Controlled substances. U.S. Department of Justice Website. http://www.deadiversion.usdoj.gov/schedules/orangebook/c_cs_alpha.pdf. Updated November 12, 2015. Accessed December 16, 2015.
13. Bray GA, Greenway FL. Pharmacological treatment of the overweight patient. Pharmacol Rev. 2007;59(2):151-184.
14. V1-0521 (QNEXA) Advisory committee briefing document. NDA 022580. Endocrinologic and Metabolic Drugs Advisory Committee meeting; June 17, 2010.
15. Cercato C, Roizenblatt VA, Leanca CC, et al. A randomized double-blind placebo-controlled study of the long-term efficacy and safety of diethylpropion in the treatment of obese subjects. Int J Obes (Lond). 2009;33(8):857-865.
16. Haddock CK, Poston WS, Dill PL, Foreyt JP, Ericsson M. Pharmacotherapy for obesity: a quantitative analysis of four decades of published randomized clinical trials. Int J Obes (Lond). 2002;26(2):262-273.
17. Suplicy H, Boquszewski CL, dos Santos CM, do Desterro de Fiqueiredo M, Cunha DR, Radominski R. A comparative study of five centrally acting drugs on pharmacological treatment of obesity. Int J Obes (Lond). 2014;38(8):1097-1103.
18. Ioannides-Demos LL, Proietto J, McNeill JJ. 2005. Pharmacotherapy for obesity. Drugs. 2005;65(10): 1391-1418.
19. Drent ML, van der Veen EA. Lipase inhibition: a novel concept in the treatment of obesity. Int J Obes Relat Metab Disord. 1993;17(4):241-244.
20. Xenical [package insert]. Nutley, NJ: Roche Laboratories Inc.; 1999.
21. U.S. Food and Drug Administration. FDA Drug Safety Communication: Completed safety review of Xenical/Alli and severe liver injury. U.S. Food and Drug Administration Website. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm213038.htm. Updated August 2, 2010. Accessed December 16, 2015.
22. Sall D, Wang J, Rashkin M, Welch M, Droege C, Schauer D. Orlistat-induced fulminant hepatic failure. Clin Obes. 2014;4(6):342-347.
23. Sjöström L, Rissanen A, Andersen T, et al. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. European Multicentre Orlistat Study Group. Lancet. 1998;352(9123):167-172.
24. Hauptman J, Lucas C, Boldrin MN, Collins H, Segal KR. Orlistat in the long-term treatment of obesity in primary care settings. Arch Fam Med. 2000;9(2):160-167.
25. Torgerson JS, Hauptman J, Boldrin MN, Sjöström L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care. 2004;27(1):155-161.
26. Torgerson JS, Arlinger K, Käppi M, Sjöström L. Principles for enhanced recruitment of subjects in a large clinical trial. the XENDOS (XENical in the prevention of Diabetes in Obese Subjects) study experience. Control Clin Trials. 2001;22(5):515-525.
27.Smith SR, Weissman NJ, Anderson CM, et al; Behavioral Modification and Lorcaserin for Overweight and Obesity Management (BLOOM) Study Group. Multicenter, placebo-controlled trial of lorcaserin for weight management. N Engl J Med. 2010;363(3):245-256.
28. Fidler MC, Sanchez M, Raether B, et al; BLOSSOM Clinical Trial Group. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab. 2011;96(10):3067-3077.
29. O’Neil PM, Smith SR, Weisserman NJ, et al. Randomized placebo-controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: the BLOOM-DM Study. Obesity (Silver Spring). 2012;20(7):1426-1436.
30. U.S. Food and Drug Administration. FDA approves weight-management drug Qsymia. U.S. Food and Drug Administration Website. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm312468.htm. Published July 17, 2012. Accessed December 16, 2015.
31. Shin J, Gadde KM. Clinical utility of phentermine/topiramate (Qsymia™) combination for the treatment of obesity. Diabetes Metab Syndr Obes. 2013;6:131-139.
32. Qsymia [package insert] Mountain View, CA: Vivus, Inc; 2012.
33. Allison DB, Gadde KM, Garvey WT, et al. Controlled-release phentermine/topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring). 2012;20(2):330-342.
34. Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377(9774):1341-1352.
35. Plodkowski RA, Nguyen Q, Sundaram U, Nguyen L, Chau DL, St Jeor S. Bupropion and naltrexone: a review of their use individually and in combination for the treatment of obesity. Expert Opin Pharmacother. 2009;10(6):1069-1081.
36. Greenway FL, Fujioka K, Plodkowski RA, et al; COR-I Study Group. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-1): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376(9741):595-605.
37. Hollander P, Gupta AK, Plodkowski R, et al; COR-Diabetes Study Group. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care. 2013;36(12):4022-4029.
38. Contrave [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc; 2014.
39. Pi-Sunyer X, Astrup A, Fujioka K, et al; SCALE Obesity and Prediabetes NN8022-1839 Study Group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Eng J Med. 2015;373(1):11-22.
40. Wadden TA, Hollander P, Klein S, et al; NN8022-1923 Investigators. Weight maintenance and additional weight loss with liraglutide after low-calorie-diet-induced weight loss: the SCALE Maintenance randomized study. Int J Obes (Lond). 2013;37(11):1443-1451
41. Saxenda [package insert]. Novo Nordisk: Plainsboro, NJ; 2015.
42.Schwartz A, Doucet E. Relative changes in resting energy expenditure during weight loss: a systemic review. Obes Rev. 2010;11(7): 531-547. ```````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````
Over the past decade the prevalence of obesity as defined by a body mass index (BMI) ≥ 30 kg/m2 has significantly increased. In the U.S. more than 78 million adults are estimated to be obese.1 The World Health Organization projects that by 2025 up to half the U.S. population will be obese. Cardiovascular disease (CVD) and diabetes mellitus (DM) are the main comorbid conditions that are complicated by obesity. Initial weight loss of 5% to 10% of total body weight reduces CVD risk factors, prevents or delays the development of type 2 DM (T2DM) and improves the health consequences of obesity.2
To date, public health initiatives that have focused on obesity prevention and lifestyle intervention have had marginal success. In recent years, anti-obesity drug therapies have had a limited role in clinical treatment algorithms. In 2013, the American Medical Association acknowledged obesity as a disease. In turn, this acknowledgement allowed the recognition of anti-obesity drugs as acceptable therapeutic adjuncts to intensive lifestyle intervention that could address the growing obesity endemic.
In the past, medications for weight reduction were limited. Several that were FDA approved had to be removed from the market due to safety concerns. With few approved options, clinicians often had to resort to off-label use of medications. However, the landscape has changed with 4 new medications gaining recent FDA approval. This review covers older available medications and the newer medications that are now available.
Sympathomimetics
Sympathomimetic drugs have been approved for use as a pharmacological method to lose weight since 1960. Of the many versions of this drug class that have been available since then, there are 4 major versions available today. These include diethylpropion3 and benzphetamine,4 both approved in 1960; phendimetrazine, approved in 1976;5 phentermine, approved in 1980;6 and phentermine hydrochloride, approved in 2012.7 Despite the existence of several other classes of drugs to treat obesity, phentermine remains the most often prescribed weight loss drug in the U.S.8
Although the mechanism of action (MOA) of sympathomimetic drugs is not particularly clear, weight loss from these medications is believed to be due to the increase in the release of biogenic amines (mainly norepinephrine, but also possibly dopamine), from storage sites in nerve terminals. It is possible that these drugs slow catecholamine metabolism by inhibiting the actions of monoamine oxidase. The resulting increase in amine availability, particularly in the lateral hypothalamic feeding center, is associated with reduced food intake. Interestingly, injection of these drugs into the ventromedial satiety center dooes not seem to suppress food intake, and the effects of biogenic amines on increasing metabolism does not seem to play a significant role in weight loss in patients on these medications.9
Each of these drugs is rapidly absorbed from the gastrointestinal (GI) tract except for phentermine hydrochloride, the newest of the medications in this class. Phentermine hydrochloride is a sublingual tablet that is readily absorbed through the buccal mucosa.5 All of the drugs in this class are excreted through the kidneys, with varying rates. Each drug’s excretion is highly dependent on the pH of the urine—more alkaline conditions result in less excretion and more acidic conditions result in more excretion. As a result, these drugs should be used with caution in patients with renal impairment; however, there are no specific contraindications listed for patients with poor renal function.
The adverse effects (AEs) for this drug class are to be expected from an increase in the release of biogenic amines in the central nervous system (CNS). The most common AEs include palpitations, tremors, restlessness, insomnia, dry mouth, constipation, diaphoresis, changes in libido, and irritability. The more dangerous AEs that have been observed include arrhythmias, hypertension, dependency/abuse, convulsions, acute transient ischemic colitis, and acute urinary retention secondary to increased bladder sphincter tone, transient hyperthyroxemia, and paranoia.10
Several contraindications exist for sympathomimetics, including the presence of advanced arteriosclerosis, symptomatic CVD, moderate to severe hypertension, hyperthyroidism, glaucoma, patients in an agitated state, or those with a history of amphetamine abuse. The warnings for prescribers include pulmonary hypertension and cardiomyopathy secondary to chronic use of sympathomimetics, and valvular heart disease secondary to use of sympathomimetics with additional anorectic agents.
Additional precautions should be considered in those with a history of anxiety/psychosis, those who operate machinery and motor vehicles, and even those with mild hypertension. The data surrounding the effects of sympathomimetics on blood pressure (BP) appears to be conflicting and the relationship does not seem to have been significantly studied in depth to warrant any definitive conclusions. The MOA of this drug class itself is enough to urge caution to prescribers.11 Special attention should be given to patients with diabetes when using sympathomimetics. A reduction of insulin dose or oral hypoglycemic dose may be necessary in some people with diabetes.
Only diethylpropion is pregnancy category B, whereas the others drugs in this class are pregnancy category X. It has been demonstrated that diethylpropion and benzphetamine are secreted into breastmilk; insufficient data exist to suggest whether or not phentermine and phendimetrazine are present in breastmilk. All drugs in this class should be used in caution with breastfeeding mothers.
Although all 4 drugs are registered as controlled substances, benzphetamine and phendimetrazine are schedule III and phentermine and diethylpropion are schedule IV, despite evidence suggesting the potential for abuse to be extremely low.12,13 Phentermine has been approved for adults aged > 18 years, phendimetrazine has been approved for those aged > 17 years, diethylpropion has been approved for those aged > 16 years, and benzphetamine has been approved for those aged > 12 years.
There is a wealth of literature surrounding the effectiveness of this drug class for weight loss. One of the longest trials of phentermine was recently conducted as part of the initial component of a FDA study for the newly approved topiramate-phentermine combination. Weight loss at 6 months in the phentermine-only group was significantly higher at -5.8% compared with -1.5% with the placebo group in the last observation carried forward-Intent to treat (LOCF-ITT) analysis.14 Similarly, a long-term study looking at diethylpropion examined the use of diethylpropion for up to a year vs placebo. Participants administered diethylpropion lost a mean 9.8% of original weight vs 3.7% in the placebo group in the first 6 months alone.15
Several meta-analyses and review papers have been authored that examine and analyze the published data on this drug class overall and comparatively within this class. Haddock and colleagues in 2002 reviewed the numerous clinical trials associated with each drug in this class, in addition to several other classes, and found that although each drug demonstrated a significant advantage vs placebo in weight loss, there was not a specific drug that was significantly superior to any of the others.16
These results seem to be in relative agreement with additional studies like that published by Suplicy and colleagues, which demonstrated that several sympathomimetics were better than placebo in weight loss, and that there was little difference between the specific drugs in the class.17 However, it should be noted that as highlighted in a review by Ioannides-Demos and colleagues in 2005, the vast majority of studies that had been performed on this drug class focused on short-term use (< 16 weeks) and none of the sympathomimetics listed here have been approved for long-term use.18
Orlistat
Orlistat 120 mg was approved in 1999 as a reversible inhibitor of GI lipases that specifically reduced the absorption of dietary fat due to the inhibition of triglyceride hydrolysis.19 Orlistat was later approved in 2007 for release in a reduced dosage form (60 mg) for over-the-counter sales.20
Orlistat forms a covalent bond with the active serine residue site of gastric and pancreatic lipases in the lumen of the stomach and small intestine. The inhibition of these enzymes causes dietary fat to remain undigested as triglycerides, which cannot be converted to absorbable free fatty acids and monoglycerides, leading to decreased calorie absorption. Orlistat is not systemically absorbed and is eliminated mainly through feces. Some metabolism occurs in the GI wall.21Orlistat is most known for its GI AEs. Because it is most active in the lumen of the GI system and reduces the absorption of triglycerides, many AEs are related to malabsorption. The most common issues 1 year after starting the drug were oily spotting (26.6% vs 1.3% placebo); flatus with discharge (23.9% vs 1.4% placebo); fecal urgency (22.1% vs 6.7% placebo); fatty/oily stool (20% vs 2.9% placebo); increased defecation (10.8% vs 4.1% placebo); and fecal incontinence (7.7% vs 0.9% placebo) (Table 1). Most of these AEs were greatly reduced after taking the drug for 2 years. Orlistat also has more serious AEs noted, including abdominal pain/discomfort; nausea; infectious diarrhea; rectal pain/discomfort; tooth disorder; gingival disorder; vomiting; upper respiratory infection; lower respiratory infection; ear, nose and throat symptoms; back pain; arthritis; myalgia; joint disorders; tendonitis; headache; dizziness; fatigue; sleep disorders; rash; dry skin; menstrual irregularity; vaginitis; urinary tract infection; and psychiatric disorders, although these did not differ markedly from placebo.
One of the most serious AEs reported was fulminate hepatic failure, though this AE is rare. Thirteen cases of liver injury were reported with the 120-mg prescription dose of orlistat and 1 case report in the U.S. involved the 60-mg over-the-counter dosage of orlistat.21,22 The FDA suggests that patients talk to their physicians about risks of liver failure, and that physicians should educate their patients about signs and symptoms of liver failure so that patients can stop taking orlistat and seek immediate medical help if symptoms occur.
One of the first published trials was the European Multicentre Orlistat Study Group, which included 743 participants with BMI between 28 kg/m2 and 47 kg/m2 from 15 different European centers. To test adherence, a 4-week single blind placebo lead-in was started with a hypocaloric diet. The first stage was completed by 688 patients who then proceeded to the double blind randomized control trial portion with a hypocaloric diet. From the start of lead-in to the end of year 1, the orlistat group weight decreased 10.2% (10.3 kg) vs 6.1% (6.1 kg) in the placebo group. The placebo subtracted difference between the groups was 3.9 kg (P < .001).23
A U.S.-based randomized double-blind placebo-controlled multicenter study included 796 obese patients with BMI between 30 kg/m2 and 44 kg/m2. Patients were assigned to 1 of 3 groups: placebo, orlistat 60 mg 3 times daily, or orlistat 120 mg 3 times daily. All groups were given a reduced energy diet. Patients in the orlistat 120 mg group lost significantly more weight than did the placebo group, -8.78% vs -4.26% respectively in year 1 in the completer analysis (P = .001). More participants who were treated with orlistat 120 mg lost 5% or more of their initial weight in year 1 compared with placebo, 50.5% vs 30.7% respectively (P < .001).24
In the XENDOS study the primary outcome measurement was time to onset of T2DM. Eligible participants were aged 30 to 60 years, with a BMI > 30 kg/m2. All patients had a 75-g oral glucose tolerance test and were required to have normal glucose tolerance or impaired glucose tolerance, but not T2DM. The double-blind randomized controlled trial included 3,305 subjects and compared a group taking 120 mg orlistat 3 times daily vs placebo. All patients were prescribed a reduced-calorie diet (800 kcal/d deficit) containing 30% of calories from fat. Patients were also encouraged to walk at least 1 kilometer daily in addition to their usual physical activity. Incidence of T2DM after 4 years was 6.2% in the orlistat group and 9.0% in the placebo group, reflecting a 37.3% risk reduction in the orlistat group (P = .0032).25,26
Lorcaserin
In 2012, lorcaserin HCl was FDA approved as a schedule IV drug for use as a weight loss medication as an adjunct to a reduced-calorie diet and increased physical activity. Lorcaserin is thought to act on 5-hydroxytryptamine-2c (5HT2c) receptors on the pro-opiomelanocortin (POMC) neurons in the arcuate nucleus, causing release of alpha-melanocortin-stimulating hormone (alpha-MSH), which in turn acts on melanocortin-4 receptors in the paraventricular nucleus to suppress appetite. At the maximum suggested dose of 10 mg twice daily, lorcaserin binds with 15 to 100 times greater affinity to 5HT2c receptors compared with 5HT2a and 5HT2b receptors respectively.
Indications for lorcaserin include patients with BMI ≥ 30 kg/m2 or ≥ 27 kg/m2 or greater with a weight-related comorbid condition such as hypertension, dyslipidemia, cardiovascular disease, impaired glucose tolerance, or sleep apnea.
The efficacy of lorcaserin for weight loss has been evaluated in 3 separate trials. The trials were randomized, double blinded and placebo controlled. The BLOOM trial, which included 3,182 patients with a mean BMI of 36.2 kg/m2, evaluated the efficacy of lorcaserin as a weight loss adjunct.27 Patients with pre-existing valvular disease, uncontrolled hypertension, or a major psychiatric condition were excluded. After initial randomization, patients were assigned to receive either lorcaserin 10 mg twice daily or a placebo. The primary endpoint was a 5% weight reduction from baseline by the end of 2 years. At 1 year, 47.5% of patients in the lorcaserin group and 20.3% in the placebo group had lost ≥ 5% of their body weight (P <.001). The average loss for the lorcaserin group was 5.8 ± 0.2 kg and 2.2 ± 0.1 kg for the placebo group at 1 year (P < .001).
The BLOSSOM trial was a 1-year study of 4,008 patients aged 18 to 65 years. The trial evaluated the effects of lorcaserin on body weight, CVD risk factors, and safety in obese and overweight patients.28 Patients were randomized in a 2:1:2 ratio to receive lorcaserin 10 mg twice daily, lorcaserin 10 mg once daily, or placebo. The primary endpoint was the proportion of patients achieving at least 5% reduction in body weight. Completer analysis showed weight reduction in the placebo group was 4.0% and 7.9% in the lorcaserin group (P < .001). In the modified intent-to-treat/last observation carried forward analysis (MITT/LOCF), a statistically significant 47.2% of patients receiving lorcaserin 10 mg twice daily and 40.2% of patients receiving lorcaserin 10 mg once daily lost at least 5% of baseline body weight; compared with 25% of patients receiving placebo (P < .001). Weight loss of at least 10% was achieved by 22.6% of patients receiving lorcaserin 10 mg twice daily, and 17.4% of patients receiving 10 mg daily compared with 9.7% of patients in the placebo group (P < .001).
The most common AEs noted were headache, nausea, and dizziness. Echocardiographic evidence of valvulopathy occurred in 2% of patients taking lorcaserin 10 mg twice daily and those taking the placebo. Lorcaserin administered in conjunction with a diet and exercise program was associated with an overall reduction in baseline BMI when compared with placebo over the year.
The BLOOM-DM study evaluated efficacy and safety of lorcaserin for weight loss in 604 patients with T2DM over the course of 1 year.29 Patients had a hemoglobin A1c (A1c) of 7% to 10% and were treated with metformin, a sulfonylurea, or both. The primary endpoint was a 5% weight reduction from baseline at the end of 1 year. Patients were randomized into 3 groups: 1 group received lorcaserin 10 mg twice daily, 1 group took lorcaserin 10 mg daily, and 1 group received the placebo. A statistically significant 37.5% of patients taking lorcaserin 10 mg twice daily achieved > 5% body weight reduction, compared with 44.7% in the lorcaserin 10 mg daily group, and 16.1% in the placebo group. Overall reductions in A1c and fasting glucose were observed in both lorcaserin groups taking as compared with placebo. Patient A1c decreased 0.9 ± 0.06 with lorcaserin 10 mg bid, 1.0 ± 0.09 with lorcaserin 10 mg qd, and 0.4 ± 0.06 with the placebo (P < .001). Fasting glucose in the lorcaserin bid, lorcaserin qd, and placebo groups decreased 27.4 ± 2.5 mg/dL, 28.4 ± 3.8 mg/dL, and 11.9 ± 2.5 mg/dL, respectively (P < .001). Symptomatic hypoglycemia occurred in 7.4% of patients on lorcaserin bid, 10.5% on lorcaserin qd, and 6.3% on placebo. Headache, back pain, nasopharyngitis, and nausea were among the most commonly reported AEs.
As lorcaserin is a serotonergic agonist, potential interactions exist when used with other medications affecting serotonin. Most notably, serotonin syndrome and neuroleptic malignant syndrome-like reactions may occur. Because of this, it is recommended to avoid selective serotonin re-uptake inhibitors (SSRIs), selective norepinephrine reuptake inhibitors, tricyclic antidepressants, bupropion, triptans, monoamine oxidase inhibitors, lithium, dextromethorphan, and dopamine agonists. Lorcaserin seems to be safe in those patient populations with mild hepatic as well as mild renal impairment; however, it is not recommended for those with severe renal impairment. Given the multiple enzymatic pathways used to metabolize lorcaserin, there is a low probability for cytochrome drug interactions. Safety has not been well evaluated in patients aged < 18 years and those that are pregnant (pregnancy category X).
Adverse events include headache, dizziness, fatigue, nausea, and dry mouth. Other notable AEs include nasopharyngitis and URI. Hypoglycemia appeared to be more common in patients with DM taking lorcaserin. Cognitive impairment and psychiatric disorders including euphoria and hallucinations were also reported. Notably, valvular heart disease has been reported in patients who take medications with 5HT2b activity. In a 1-year clinical trial, a small number of patients were found to develop valvular regurgitation. Furthermore, bradycardia, priapism, leucopenia, elevated prolactin, and pulmonary hypertension have also been observed. Caution is recommended if symptoms of any of the aforementioned conditions are noticed.
Qsymia
The schedule IV controlled substance Qsymia (Vivus, Mountain View, CA) is a combination of phentermine, an anorexigenic agent, and topiramate extended-release, an antiepileptic drug. In July of 2012 it was approved for chronic weight management as an addition to a reduced-calorie diet and exercise. The drug is approved for adults with a BMI ≥ 30 kg/m2 or adults with a BMI ≥ 27 kg/m2 who have at least 1 weight-related condition such as hypertension, T2DM, or dyslipidemia.30
In 1996 topiramate was approved by the FDA for the treatment of seizure disorders and was also approved for migraine prophylaxis in 2004. In patients who were treated with topiramate for seizure disorders and migraines, weight loss and a reduction in visceral body fat has been observed.31 The precise MOA of topiramate in regards to weight loss is not fully understood. It may be due to its effects on both appetite suppression and satiety enhancement. Topiramate exhibits a combination of properties including modulatory effects on sodium channels, enhancement of GABA-activated chloride channels, inhibition of excitatory neurotransmission through actions on kainite and AMPA receptors, and inhibition of carbonic anhydrase (CA) isoenzymes in particular CA II and IV.14
The combination of phentermine and topiramate is a once-daily formulation that is designed to provide an immediate release of phentermine and a delayed release of topiramate, allowing a peak exposure of the phentermine in the morning and a peak concentration of topiramate in the evening. It should be taken in the morning in order to avoid the possibility of insomnia that can occur if taken in the evening. It can be taken with or without food. The recommended dose is as follows: Start treatment with Qsymia 3.75 mg/23 mg extended-release daily for 14 days; after 14 days increase to the recommended dose of Qsymia 7.5 mg/46 mg once daily.
Weight loss should be evaluated after 12 weeks at the higher dose. If at least 3% of baseline body weight has not been lost at that time, discontinue or escalate the dose. To escalate the dose: Increase to Qsymia 11.25 mg/69 mg daily for 14 days; followed by Qsymia 15 mg/92 mg daily. Evaluate weight loss following dose escalation to Qsymia 15 mg/92 mg after an additional 12 weeks of treatment. If at least 5% of baseline body weight has not been lost on Qsymia 15 mg/92 mg, discontinue as directed. It is important not to suddenly discontinue, as this may cause seizures. Patients should be slowly titrated off the medication.
In vitro studies of phentermine and topiramate indicate that these drugs are not likely to cause clinically significant interactions with drugs using the cytochrome P450 enzyme pathways, or those involved in plasma protein binding displacement; however there is evidence suggesting that ethinyl estradiol levels may be decreased by 16%, thus raising a concern about the possibility of decreased contraceptive efficacy.31 In patients with moderate (creatine clearance ≥ 30 mL/min to < 50 mL/min) and severe renal dysfunction (< 30 mL/min), the maximum dose of should not exceed 7.5 mg/46 mg.
Qsymia was evaluated in 3 phase 3 trials for its long-term efficacy and safety. In all trials, diet and lifestyle counseling were provided for all patients. The first of these studies was OB-301, a 28-week confirmatory trial with a factorial design involving 7 treatment arms, tested 2 fixed-dose Qsymia combinations—regular dose (7.5 mg/46 mg) and maximum dose (15 mg/92 mg)—as well as regular and maximum doses of the individual constituent drugs vs placebo.32 The study randomized 756 obese patients with a BMI range of 30 kg/m2 to 45 kg/m2 to 1 of the 7 treatment arms for 28 weeks. Patients treated with maximum-dose Qsymia achieved an average weight change of -9.0%, vs -1.5% with placebo (P < .0001). Weight change with regular-dose Qsymia was -8.2%. Weight changes with monotherapies were: -6.1% with topiramate 92 mg, -4.9% with topiramate 46 mg, -5.8% with phentermine 15 mg, and -5.2% with phentermine 7.5 mg.
OB-302 was a 56-week trial that randomized 1,267 morbidly obese patients with a BMI ≥ 35 kg/m2 without significant comorbidities to low-dose Qsymia (3.7 mg/23 mg), maximum-dose Qsymia (15 mg/92 mg), or placebo.33 At baseline, the mean BMI for the entire study cohort was 42 kg/m2. Mean weight changes were -1.6% with placebo, -5.1% with low-dose Qsymia, and -10.9% with maximum-dose Qsymia. The proportions of patients achieving ≥ 5% weight loss were: 17% with placebo, 45% with low-dose Qsymia, and 67% with maximum-dose Qsymia.
CONQUER was the largest of the phase 3 trials. It randomized 2,487 overweight or obese patients with a BMI of 27 kg/m2 to 45 kg/m2 and ≥ 2 obesity-related comorbidities (hypertension, dyslipidemia, T2DM, prediabetes or abdominal obesity) to receive a placebo, regular-dose Qsymia, or maximum-dose Qsymia for 56 weeks.34 In the completer population, mean weight changes in the placebo, regular dose Qsymia, and maximum-dose Qsymia groups were -1.6%, -9.6% (P <.0001), and -12.4% (P < .0001); and weight loss of ≥ 5% was achieved by 21%, 62%, and 70%, respectively. Relative to placebo, there were greater reductions in systolic BP, triglycerides, and fasting insulin with both doses of Qsymia.
Patients should not take Qsymia if they are pregnant, planning to become pregnant, or become pregnant during Qsymia treatment as there is an increased risk of birth defects, namely cleft lip and cleft palate. Women who can become pregnant should have a negative pregnancy test before taking Qsymia and every month while on the medication. They should use effective birth control consistently while taking Qsymia.
Qsymia is contraindicated in patients with glaucoma and patients who have hyperthyroidism. Qsymia can cause an increase in resting heart rate and regular monitoring of resting heart rate is recommended, especially in patients with cardiac or cerebrovascular disease. It has not been studied in patients with recent or unstable cardiac or cerebrovascular disease and therefore use is not recommended.
Qsymia can cause mood disorders such as anxiety and depression and can increase the risk of suicidal thoughts. Patients should be monitored for worsening depression, suicidal thoughts or behavior, or any unusual changes in mood or behavior. It is not recommended in patients with a history of suicidal attempts or active suicidal ideation. Qsymia can cause cognitive dysfunction. It can cause confusion, problems with concentration, attention, memory, or speech. Patients should be cautioned about operating automobiles and hazardous machinery.
Normal anion gap hyperchloremic metabolic acidosis has been reported in patients treated with Qsymia. If this does develop and persists, consideration should be given to either reduce the dose or discontinue Qsymia.
Weight loss may increase the risk of hypoglycemia in patients with T2DM treated with insulin and/or insulin secretagogues (eg, sulfonylureas). Qsymia has not been studied in combination with insulin. A reduction in the dose of antidiabetic medications, which are nonglucose dependent, should be considered to reduce the risk of hypoglycemia.
The most common AEs in controlled clinical studies (≥ 5% and at least 1.5 times placebo) included paraesthesia in the hands, arms, feet or face, dizziness, dysgeusia, insomnia, constipation, and dry mouth.
Contrave
In 2014, the FDA approved Contrave (Takeda, Deerfield, IL) as treatment option for chronic weight management in addition to reduced-calorie diet and physical activity. The combination of naltrexone hydrochloride and bupropion hydrochloride was originally introduced for the treatment of opioid addiction and later expanded to include the treatment of alcoholism. The antidepressant bupropion was approved in the U.S. in 1989. It is structurally different from all other marketed antidepressants (ie, tricyclics, tetracyclics, and SSRIs), but closely resembles the structure of diethylpropion, an appetite depressant with minimal CNS effects.35
This drug is approved for adults with BMI ≥ 30 kg/m2 and for adults with BMI ≥ 27 kg/m2 with at least 1 weight-related risk factors such as hypertension, T2DM, or dyslipidemia. It should be used as an adjunct to diet and exercise and is not approved for use for depression even though it contains bupropion.
Naltrexone is a pure opioid antagonist with high affinity to μ-opioid receptor, which is implicated in eating behavior. Naltrexone is rapidly and nearly completely absorbed from the GI tract after oral administration. The time to peak plasma concentration is about 1 hour. Naltrexone is well absorbed but first pass extraction and metabolism by the liver decreases oral bioavailability to between 5% to 40%. Primary elimination of naltrexone and its metabolites is renal excretion.
Bupropion is a weak inhibitor of neuronal reuptake of dopamine and norepinephrine. This drug is used to treat depression and seasonal affective disorder, and aid in smoking cessation. Bupropion is absorbed rapidly after oral administration, but the absolute oral bioavailability of bupropion is not known because an IV preparation is not available. The time to peak plasma concentrations of bupropion is within 2 hours of oral administration. Bupropion is extensively metabolized by the liver to multiple metabolites. Primary elimination of bupropion is urinary excretion. However, hepatic and renal impairment may affect the elimination of bupropion and its metabolites. Patients with hepatic or renal impairment should use a reduced dosage.
Combination therapy has been found to have complementary actions on CNS to reduce food intake. They are believed to dampen CNS reward pathways, taking away the compulsive feeding behavior and pleasure of feeding, ultimately leading to weight loss. Bupropion stimulates hypothalamic pro-opiomelanocortin neurons (POMC), which results in reduced food intake and increased energy expenditure. Naltrexone blocks opioid-receptor mediated POMC auto-inhibition, blocks the increase in dopamine in nucleus accumbens that occurs when eating, and acting synergistically with bupropion in augmenting POMC firing.
The COR-I and COR-II trials compared Contrave to diet and exercise in patients who did not have DM. The COR-Diabetes trial included the same study design but focused on patients with DM. In all the studies the participants had a 4-week titration to Contrave (naltrexone 8 mg/bupropion 90 mg) to decrease nausea. The first week dosing was 1 tablet in the morning. Week 2 was 1 tablet in morning and 1 tablet in the evening. In week 3, patients took 2 tablets in the morning and 1 tablet in the evening. The final titration step was 2 tablets in the morning and 2 tablets in the evening.
The COR-1 study was a 56-week randomized, double-blind, placebo-controlled study. It compared Contrave 32 mg naltrexone/360 mg bupropion (NB32/360) with an active placebo of diet and exercise.36 To be included adults must be aged 18 to 65 years with a BMI 30 kg/m2 to 45 kg/m2 or a BMI 27 kg/m2 to 45 kg/m2 with dyslipidemia or hypertension. Patients were instructed on a hypocaloric diet that was a 500 kcal per day deficit based on World Health Organization algorithm for calculating metabolic rate and they were urged to increase physical activity.
The completer population results showed 8.0% weight loss in the NB32/360 group and 1.9% weight loss in the placebo group (P < .001). For the NB32/360 and placebo groups, weight loss of ≥ 5% was achieved by 48% and 16% (P <.001), respectively; and weight loss of ≥ 10% by 25% and 7% (P < .001), respectively. The most common AE was nausea—29.8% with NB32/360 vs 5.3% with placebo. Nausea generally occurred early and then diminished and the discontinuation rate from nausea was significantly lower (6.3%) then the overall reported nausea rates.
Contrave was also studied in patients with T2DM. The COR-Diabetes Trial was a 56-week randomized, double blind, placebo-controlled study. The trial compared Contrave 32 mg naltrexone/360 mg bupropion (NB32/360) with an active placebo of diet and exercise.37 Inclusion criteria for the trial were patients aged 18 to 70 years with T2DM and a BMI from 27 kg/m2 to 45 kg/m2, A1c between 7% and 10%, and fasting blood glucose < 270 mg/dL. Participants either were not taking a DM medication or were on stable doses of oral antidiabetes drugs ≥ 3 months prior to randomization. Patients were placed on a 500 kcal hypocaloric diet and advised to increase physical activity.
The results showed 5.0% weight loss in the NB32/360 group and 1.8% weight loss in the placebo group (P < .001). Weight loss of ≥ 5% and ≥ 10% was achieved by 44.5% and 18.5% of the NB32/360 group, respectively, and 18.9% and 5.7%, respectively (P < .001) of the placebo group. The NB32/360 and placebo showed a reduction of A1c of 0.6% and 0.1% respectively (P < .001). The most common AE was nausea (42.3% with NB32/360 vs 7.1% with placebo). Nausea generally occurred early and then diminished and the discontinuation rate from nausea was significantly lower (9.6%) then the overall reported nausea rates.37
Due to potential nausea caused by naltrexone, Contrave should be titrated over 4 weeks as described earlier. At maintenance dose, patients should be evaluated after 12 weeks to determine treatment benefits. If a patient has not lost at least 5% of baseline body weight, Contrave should be discontinued, because it would be unlikely that the patient will achieve and sustain clinically meaningful weight loss with continued treatment. Contrave should not be taken with high-fat meals that may result in significant increase in bupropion and naltrexone systemic exposure.
Since Contrave contains the antidepressant bupropion, it has a boxed warning similar to other antidepressants in its class of increased risk of suicidal thoughts and behaviors, especially in children, adolescents, and young adults.38Contrave can lower the seizure threshold; therefore it should not be used in people with a seizure disorder. It can also raise BP and heart rate; however the clinical significance of hypertension and elevated heart rate observed with Contrave treatment is unclear. Blood pressure rose on average by 1 point during the first 8 weeks of treatment and then returned to baseline.38 The heart rate also increased by about 1.7 beats per minute.38 Patients with uncontrolled hypertension should avoid Contrave.
Contrave should not be taken with products contain bupropion or naltrexone. It should not be taken by patient who are regularly taking opioids or who are opioid dependent, or who are experiencing opiate withdrawal. Pregnant women should also avoid Contrave. In patients with renal impairment the maximum dose is 1 tablet twice a day and in patients with hepatic impairment the maximum dose is 1 tablet a day.
Liraglutide
Liraglutide is the newest weight loss medication to be approved by the FDA for chronic weight management as an adjunct to a reduced calorie diet and increased physical activity in adult patients with BMI ≥ 30 kg/m2 or ≥ 27 kg/m2 with hypertension, diabetes, or dyslipidemia. The recommended dose of liraglutide is 3 mg daily. The initial dose is 0.6 mg daily for the first week, then titrated up by 0.6 each week for 4 weeks, until reaching 3 mg daily.
Liraglutide is an acylated human glucagon-like peptide-1 (GLP-1) receptor agonist, which are expressed in the brain and is involved in the control of appetite. It is also found in the beta cells of the pancreas, where GLP-1 receptors stimulate insulin release in response to elevated blood glucose concentrations and suppress glucagon secretion. Endogenous GLP-1 has a half-life of 1.5 to 2 minutes due to degradation by the DDP-4 enzyme, but liraglutide is stable against degradation by peptidases and has a half- life of 13 hours.
Liraglutide was studied in a 56-week randomized, double-blind, placebo-controlled trial, which compared liraglutide 3 mg with an active placebo of diet and physical activity.39 Inclusion criteria were adults aged ≥ 18 years old with a BMI 30 kg/m2 to 45 kg/m2 or BMI 27 kg/m2 to 45 kg/m2 with dyslipidemia and/or hypertension. Both groups received lifestyle modification counseling. Patients were excluded if they had DM.
In the trial, 3,731 participants enrolled, 2,487 in the liraglutide group and 1,244 in the placebo group; 78.7% of the participants were female and the average age was 45 years. Subjects in the liraglutide group had a weekly titration regimen. The starting dose at week 1 was 0.6 mg, week 2 was 1.2 mg, week 3 was 1.8 mg, week 4 was 2.4 mg, and week 5 was 3.0 kg.
The completer population showed 9.2% weight loss in the liraglutide group and 3.0% weight loss in the control group.39 Weight loss of ≥ 5% was seen in 63.2% and 27.1% of the liraglutide and placebo groups, respectively. Weight loss rates of ≥ 10% was seen by 33.1% and 10.6%, respectively. The most common AEs were nausea, diarrhea, and constipation. Nausea generally occurred early during the titration period and then diminished.
A second clinically relevant study was performed with liraglutide. Often patients are able to lose weight with diet and exercise and then plateau. This study examined participants who lost 5% percent of their initial body weight and then were randomized to liraglutide or placebo.40 Key inclusion criteria were people aged ≥ 18 years old with a BMI 30 kg/m2 to 45 kg/m2 or BMI 27 kg/m2 to 45 kg/m2 with dyslipidemia and/or hypertension. In order to be randomized, participants were required to lose at least 5% of their initial body weight on a 1,200 kcal to 1,400 kcal diet with increased physical activity during a 4 to 12 week run-in period.
Four hundred twenty-two participants were enrolled, 212 in the liraglutide group and 210 in the placebo group. Most of the participants were female (81%). The average BMI in the study was 35.6 kg/m2. Subjects in the liraglutide group had a weekly titration regimen.
After an average weight loss of 6% using a low calorie diet and increased physical activity the participants were randomized to continue diet and increased activity alone (placebo) or with liraglutide. At week 56 the results showed an additional 6.2% weight loss in the liraglutide group and 0.2% weight gain in the placebo group. The liraglutide group had a greater number of participants with ≥ 5% weight loss compared to placebo, 50.5% vs 21.8% (P < .0001).40 In the pooled data set from the registration trials the 3 most common GI AEs were nausea, diarrhea, and constipation occurring in 39.3%, 20.9%, and 19.4% of participants respectively. Discontinuation due to nausea for liraglutide was 2.9%.41
Clinicians should be aware that medications that can cause hypoglycemia such as sulfonylureas and insulin must be tapered as patients lose weight with liraglutide. Documented symptomatic hypoglycemia in patients with T2DM and with sulfonylurea background therapy was 43.6% with liraglutide vs 27.3% with placebo.
In the setting of renal impairment, patients treated with GLP-1 receptor agonists, including liraglutide, have had reports of acute renal failure and worsening of chronic renal failure usually associated with nausea, vomiting, diarrhea, or dehydration. Liraglutide causes thyroid C-cell tumors at clinically relevant exposures in rats and mice. It is unknown whether liraglutide causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans. As the human relevance of liraglutide-induced rodent thyroid C-cell tumors has not been determined liraglutide is contraindicated in patients with a personal or family history of MTC or in patients with multiple endocrine neoplasia syndrome type 2.
Acute pancreatitis, including fatal and nonfatal hemorrhagic or necrotizing pancreatitis, has been observed in patients treated with liraglutide in postmarketing reports. After initiation of liraglutide, observe patients carefully for signs and symptoms of pancreatitis (including persistent severe abdominal pain, sometimes radiating to the back, which may or may not be accompanied by vomiting). If pancreatitis is suspected, liraglutide should promptly be discontinued.
Conclusion
The treatment of obesity and overweight with comorbidities has always been a challenge. In the past there were few FDA-approved drugs and many drugs had to be used off-label. The toolbox of medications available for medical weight management is more robust than ever. The medications have different MOAs and can be used in a variety of patients. There are differences in the classes and some are controlled substances. Phentermine, lorcaserin, and Qsymia (phentermine/topiramate) are controlled substances whereas orlistat, naltrexone/bupropion and liraglutide are not. Other differences exist including duration of use. The sympathomimetic drugs have a limited window of use whereas orlistat, Qsymia (phentermine/topiramate), lorcaserin, naltrexone/bupropion, and liraglutide do not.
The medications that are available have a wide variety of MOAs. Therefore, if a patient fails one medication, then it is very reasonable to try a medication with a different MOA. In addition, there is the potential for weight regain when weight reduction medications are discontinued. As people lose weight their metabolic rate decreases about 15 kcal per pound of weight reduction.42
Another challenge of using these medications is managing patient expectations. The current metric used for FDA approval is a 5% weight loss that is greater in the study group compared with the diet and physical activity active control. However, many clinicians and patients do not find this weight reduction amount consistent with their expectations. In addition weight loss trajectory may also be too slow for patients and cause early discontinuation. Therefore, patient education and a discussion of reasonable expectations for weight reduction medications are necessary.
Clinicians must acknowledge that there are limitations to the use of these medications. Newer agents do have a higher cost and insurance reimbursement is somewhat limited. However, they offer the opportunity to prevent more expensive, protracted conditions such as diabetes and cardiovascular disease. In summary, clinicians now have a wider variety of medication options to be used with dietary and lifestyle changes in order to improve health and prevent chronic diseases.
Over the past decade the prevalence of obesity as defined by a body mass index (BMI) ≥ 30 kg/m2 has significantly increased. In the U.S. more than 78 million adults are estimated to be obese.1 The World Health Organization projects that by 2025 up to half the U.S. population will be obese. Cardiovascular disease (CVD) and diabetes mellitus (DM) are the main comorbid conditions that are complicated by obesity. Initial weight loss of 5% to 10% of total body weight reduces CVD risk factors, prevents or delays the development of type 2 DM (T2DM) and improves the health consequences of obesity.2
To date, public health initiatives that have focused on obesity prevention and lifestyle intervention have had marginal success. In recent years, anti-obesity drug therapies have had a limited role in clinical treatment algorithms. In 2013, the American Medical Association acknowledged obesity as a disease. In turn, this acknowledgement allowed the recognition of anti-obesity drugs as acceptable therapeutic adjuncts to intensive lifestyle intervention that could address the growing obesity endemic.
In the past, medications for weight reduction were limited. Several that were FDA approved had to be removed from the market due to safety concerns. With few approved options, clinicians often had to resort to off-label use of medications. However, the landscape has changed with 4 new medications gaining recent FDA approval. This review covers older available medications and the newer medications that are now available.
Sympathomimetics
Sympathomimetic drugs have been approved for use as a pharmacological method to lose weight since 1960. Of the many versions of this drug class that have been available since then, there are 4 major versions available today. These include diethylpropion3 and benzphetamine,4 both approved in 1960; phendimetrazine, approved in 1976;5 phentermine, approved in 1980;6 and phentermine hydrochloride, approved in 2012.7 Despite the existence of several other classes of drugs to treat obesity, phentermine remains the most often prescribed weight loss drug in the U.S.8
Although the mechanism of action (MOA) of sympathomimetic drugs is not particularly clear, weight loss from these medications is believed to be due to the increase in the release of biogenic amines (mainly norepinephrine, but also possibly dopamine), from storage sites in nerve terminals. It is possible that these drugs slow catecholamine metabolism by inhibiting the actions of monoamine oxidase. The resulting increase in amine availability, particularly in the lateral hypothalamic feeding center, is associated with reduced food intake. Interestingly, injection of these drugs into the ventromedial satiety center dooes not seem to suppress food intake, and the effects of biogenic amines on increasing metabolism does not seem to play a significant role in weight loss in patients on these medications.9
Each of these drugs is rapidly absorbed from the gastrointestinal (GI) tract except for phentermine hydrochloride, the newest of the medications in this class. Phentermine hydrochloride is a sublingual tablet that is readily absorbed through the buccal mucosa.5 All of the drugs in this class are excreted through the kidneys, with varying rates. Each drug’s excretion is highly dependent on the pH of the urine—more alkaline conditions result in less excretion and more acidic conditions result in more excretion. As a result, these drugs should be used with caution in patients with renal impairment; however, there are no specific contraindications listed for patients with poor renal function.
The adverse effects (AEs) for this drug class are to be expected from an increase in the release of biogenic amines in the central nervous system (CNS). The most common AEs include palpitations, tremors, restlessness, insomnia, dry mouth, constipation, diaphoresis, changes in libido, and irritability. The more dangerous AEs that have been observed include arrhythmias, hypertension, dependency/abuse, convulsions, acute transient ischemic colitis, and acute urinary retention secondary to increased bladder sphincter tone, transient hyperthyroxemia, and paranoia.10
Several contraindications exist for sympathomimetics, including the presence of advanced arteriosclerosis, symptomatic CVD, moderate to severe hypertension, hyperthyroidism, glaucoma, patients in an agitated state, or those with a history of amphetamine abuse. The warnings for prescribers include pulmonary hypertension and cardiomyopathy secondary to chronic use of sympathomimetics, and valvular heart disease secondary to use of sympathomimetics with additional anorectic agents.
Additional precautions should be considered in those with a history of anxiety/psychosis, those who operate machinery and motor vehicles, and even those with mild hypertension. The data surrounding the effects of sympathomimetics on blood pressure (BP) appears to be conflicting and the relationship does not seem to have been significantly studied in depth to warrant any definitive conclusions. The MOA of this drug class itself is enough to urge caution to prescribers.11 Special attention should be given to patients with diabetes when using sympathomimetics. A reduction of insulin dose or oral hypoglycemic dose may be necessary in some people with diabetes.
Only diethylpropion is pregnancy category B, whereas the others drugs in this class are pregnancy category X. It has been demonstrated that diethylpropion and benzphetamine are secreted into breastmilk; insufficient data exist to suggest whether or not phentermine and phendimetrazine are present in breastmilk. All drugs in this class should be used in caution with breastfeeding mothers.
Although all 4 drugs are registered as controlled substances, benzphetamine and phendimetrazine are schedule III and phentermine and diethylpropion are schedule IV, despite evidence suggesting the potential for abuse to be extremely low.12,13 Phentermine has been approved for adults aged > 18 years, phendimetrazine has been approved for those aged > 17 years, diethylpropion has been approved for those aged > 16 years, and benzphetamine has been approved for those aged > 12 years.
There is a wealth of literature surrounding the effectiveness of this drug class for weight loss. One of the longest trials of phentermine was recently conducted as part of the initial component of a FDA study for the newly approved topiramate-phentermine combination. Weight loss at 6 months in the phentermine-only group was significantly higher at -5.8% compared with -1.5% with the placebo group in the last observation carried forward-Intent to treat (LOCF-ITT) analysis.14 Similarly, a long-term study looking at diethylpropion examined the use of diethylpropion for up to a year vs placebo. Participants administered diethylpropion lost a mean 9.8% of original weight vs 3.7% in the placebo group in the first 6 months alone.15
Several meta-analyses and review papers have been authored that examine and analyze the published data on this drug class overall and comparatively within this class. Haddock and colleagues in 2002 reviewed the numerous clinical trials associated with each drug in this class, in addition to several other classes, and found that although each drug demonstrated a significant advantage vs placebo in weight loss, there was not a specific drug that was significantly superior to any of the others.16
These results seem to be in relative agreement with additional studies like that published by Suplicy and colleagues, which demonstrated that several sympathomimetics were better than placebo in weight loss, and that there was little difference between the specific drugs in the class.17 However, it should be noted that as highlighted in a review by Ioannides-Demos and colleagues in 2005, the vast majority of studies that had been performed on this drug class focused on short-term use (< 16 weeks) and none of the sympathomimetics listed here have been approved for long-term use.18
Orlistat
Orlistat 120 mg was approved in 1999 as a reversible inhibitor of GI lipases that specifically reduced the absorption of dietary fat due to the inhibition of triglyceride hydrolysis.19 Orlistat was later approved in 2007 for release in a reduced dosage form (60 mg) for over-the-counter sales.20
Orlistat forms a covalent bond with the active serine residue site of gastric and pancreatic lipases in the lumen of the stomach and small intestine. The inhibition of these enzymes causes dietary fat to remain undigested as triglycerides, which cannot be converted to absorbable free fatty acids and monoglycerides, leading to decreased calorie absorption. Orlistat is not systemically absorbed and is eliminated mainly through feces. Some metabolism occurs in the GI wall.21Orlistat is most known for its GI AEs. Because it is most active in the lumen of the GI system and reduces the absorption of triglycerides, many AEs are related to malabsorption. The most common issues 1 year after starting the drug were oily spotting (26.6% vs 1.3% placebo); flatus with discharge (23.9% vs 1.4% placebo); fecal urgency (22.1% vs 6.7% placebo); fatty/oily stool (20% vs 2.9% placebo); increased defecation (10.8% vs 4.1% placebo); and fecal incontinence (7.7% vs 0.9% placebo) (Table 1). Most of these AEs were greatly reduced after taking the drug for 2 years. Orlistat also has more serious AEs noted, including abdominal pain/discomfort; nausea; infectious diarrhea; rectal pain/discomfort; tooth disorder; gingival disorder; vomiting; upper respiratory infection; lower respiratory infection; ear, nose and throat symptoms; back pain; arthritis; myalgia; joint disorders; tendonitis; headache; dizziness; fatigue; sleep disorders; rash; dry skin; menstrual irregularity; vaginitis; urinary tract infection; and psychiatric disorders, although these did not differ markedly from placebo.
One of the most serious AEs reported was fulminate hepatic failure, though this AE is rare. Thirteen cases of liver injury were reported with the 120-mg prescription dose of orlistat and 1 case report in the U.S. involved the 60-mg over-the-counter dosage of orlistat.21,22 The FDA suggests that patients talk to their physicians about risks of liver failure, and that physicians should educate their patients about signs and symptoms of liver failure so that patients can stop taking orlistat and seek immediate medical help if symptoms occur.
One of the first published trials was the European Multicentre Orlistat Study Group, which included 743 participants with BMI between 28 kg/m2 and 47 kg/m2 from 15 different European centers. To test adherence, a 4-week single blind placebo lead-in was started with a hypocaloric diet. The first stage was completed by 688 patients who then proceeded to the double blind randomized control trial portion with a hypocaloric diet. From the start of lead-in to the end of year 1, the orlistat group weight decreased 10.2% (10.3 kg) vs 6.1% (6.1 kg) in the placebo group. The placebo subtracted difference between the groups was 3.9 kg (P < .001).23
A U.S.-based randomized double-blind placebo-controlled multicenter study included 796 obese patients with BMI between 30 kg/m2 and 44 kg/m2. Patients were assigned to 1 of 3 groups: placebo, orlistat 60 mg 3 times daily, or orlistat 120 mg 3 times daily. All groups were given a reduced energy diet. Patients in the orlistat 120 mg group lost significantly more weight than did the placebo group, -8.78% vs -4.26% respectively in year 1 in the completer analysis (P = .001). More participants who were treated with orlistat 120 mg lost 5% or more of their initial weight in year 1 compared with placebo, 50.5% vs 30.7% respectively (P < .001).24
In the XENDOS study the primary outcome measurement was time to onset of T2DM. Eligible participants were aged 30 to 60 years, with a BMI > 30 kg/m2. All patients had a 75-g oral glucose tolerance test and were required to have normal glucose tolerance or impaired glucose tolerance, but not T2DM. The double-blind randomized controlled trial included 3,305 subjects and compared a group taking 120 mg orlistat 3 times daily vs placebo. All patients were prescribed a reduced-calorie diet (800 kcal/d deficit) containing 30% of calories from fat. Patients were also encouraged to walk at least 1 kilometer daily in addition to their usual physical activity. Incidence of T2DM after 4 years was 6.2% in the orlistat group and 9.0% in the placebo group, reflecting a 37.3% risk reduction in the orlistat group (P = .0032).25,26
Lorcaserin
In 2012, lorcaserin HCl was FDA approved as a schedule IV drug for use as a weight loss medication as an adjunct to a reduced-calorie diet and increased physical activity. Lorcaserin is thought to act on 5-hydroxytryptamine-2c (5HT2c) receptors on the pro-opiomelanocortin (POMC) neurons in the arcuate nucleus, causing release of alpha-melanocortin-stimulating hormone (alpha-MSH), which in turn acts on melanocortin-4 receptors in the paraventricular nucleus to suppress appetite. At the maximum suggested dose of 10 mg twice daily, lorcaserin binds with 15 to 100 times greater affinity to 5HT2c receptors compared with 5HT2a and 5HT2b receptors respectively.
Indications for lorcaserin include patients with BMI ≥ 30 kg/m2 or ≥ 27 kg/m2 or greater with a weight-related comorbid condition such as hypertension, dyslipidemia, cardiovascular disease, impaired glucose tolerance, or sleep apnea.
The efficacy of lorcaserin for weight loss has been evaluated in 3 separate trials. The trials were randomized, double blinded and placebo controlled. The BLOOM trial, which included 3,182 patients with a mean BMI of 36.2 kg/m2, evaluated the efficacy of lorcaserin as a weight loss adjunct.27 Patients with pre-existing valvular disease, uncontrolled hypertension, or a major psychiatric condition were excluded. After initial randomization, patients were assigned to receive either lorcaserin 10 mg twice daily or a placebo. The primary endpoint was a 5% weight reduction from baseline by the end of 2 years. At 1 year, 47.5% of patients in the lorcaserin group and 20.3% in the placebo group had lost ≥ 5% of their body weight (P <.001). The average loss for the lorcaserin group was 5.8 ± 0.2 kg and 2.2 ± 0.1 kg for the placebo group at 1 year (P < .001).
The BLOSSOM trial was a 1-year study of 4,008 patients aged 18 to 65 years. The trial evaluated the effects of lorcaserin on body weight, CVD risk factors, and safety in obese and overweight patients.28 Patients were randomized in a 2:1:2 ratio to receive lorcaserin 10 mg twice daily, lorcaserin 10 mg once daily, or placebo. The primary endpoint was the proportion of patients achieving at least 5% reduction in body weight. Completer analysis showed weight reduction in the placebo group was 4.0% and 7.9% in the lorcaserin group (P < .001). In the modified intent-to-treat/last observation carried forward analysis (MITT/LOCF), a statistically significant 47.2% of patients receiving lorcaserin 10 mg twice daily and 40.2% of patients receiving lorcaserin 10 mg once daily lost at least 5% of baseline body weight; compared with 25% of patients receiving placebo (P < .001). Weight loss of at least 10% was achieved by 22.6% of patients receiving lorcaserin 10 mg twice daily, and 17.4% of patients receiving 10 mg daily compared with 9.7% of patients in the placebo group (P < .001).
The most common AEs noted were headache, nausea, and dizziness. Echocardiographic evidence of valvulopathy occurred in 2% of patients taking lorcaserin 10 mg twice daily and those taking the placebo. Lorcaserin administered in conjunction with a diet and exercise program was associated with an overall reduction in baseline BMI when compared with placebo over the year.
The BLOOM-DM study evaluated efficacy and safety of lorcaserin for weight loss in 604 patients with T2DM over the course of 1 year.29 Patients had a hemoglobin A1c (A1c) of 7% to 10% and were treated with metformin, a sulfonylurea, or both. The primary endpoint was a 5% weight reduction from baseline at the end of 1 year. Patients were randomized into 3 groups: 1 group received lorcaserin 10 mg twice daily, 1 group took lorcaserin 10 mg daily, and 1 group received the placebo. A statistically significant 37.5% of patients taking lorcaserin 10 mg twice daily achieved > 5% body weight reduction, compared with 44.7% in the lorcaserin 10 mg daily group, and 16.1% in the placebo group. Overall reductions in A1c and fasting glucose were observed in both lorcaserin groups taking as compared with placebo. Patient A1c decreased 0.9 ± 0.06 with lorcaserin 10 mg bid, 1.0 ± 0.09 with lorcaserin 10 mg qd, and 0.4 ± 0.06 with the placebo (P < .001). Fasting glucose in the lorcaserin bid, lorcaserin qd, and placebo groups decreased 27.4 ± 2.5 mg/dL, 28.4 ± 3.8 mg/dL, and 11.9 ± 2.5 mg/dL, respectively (P < .001). Symptomatic hypoglycemia occurred in 7.4% of patients on lorcaserin bid, 10.5% on lorcaserin qd, and 6.3% on placebo. Headache, back pain, nasopharyngitis, and nausea were among the most commonly reported AEs.
As lorcaserin is a serotonergic agonist, potential interactions exist when used with other medications affecting serotonin. Most notably, serotonin syndrome and neuroleptic malignant syndrome-like reactions may occur. Because of this, it is recommended to avoid selective serotonin re-uptake inhibitors (SSRIs), selective norepinephrine reuptake inhibitors, tricyclic antidepressants, bupropion, triptans, monoamine oxidase inhibitors, lithium, dextromethorphan, and dopamine agonists. Lorcaserin seems to be safe in those patient populations with mild hepatic as well as mild renal impairment; however, it is not recommended for those with severe renal impairment. Given the multiple enzymatic pathways used to metabolize lorcaserin, there is a low probability for cytochrome drug interactions. Safety has not been well evaluated in patients aged < 18 years and those that are pregnant (pregnancy category X).
Adverse events include headache, dizziness, fatigue, nausea, and dry mouth. Other notable AEs include nasopharyngitis and URI. Hypoglycemia appeared to be more common in patients with DM taking lorcaserin. Cognitive impairment and psychiatric disorders including euphoria and hallucinations were also reported. Notably, valvular heart disease has been reported in patients who take medications with 5HT2b activity. In a 1-year clinical trial, a small number of patients were found to develop valvular regurgitation. Furthermore, bradycardia, priapism, leucopenia, elevated prolactin, and pulmonary hypertension have also been observed. Caution is recommended if symptoms of any of the aforementioned conditions are noticed.
Qsymia
The schedule IV controlled substance Qsymia (Vivus, Mountain View, CA) is a combination of phentermine, an anorexigenic agent, and topiramate extended-release, an antiepileptic drug. In July of 2012 it was approved for chronic weight management as an addition to a reduced-calorie diet and exercise. The drug is approved for adults with a BMI ≥ 30 kg/m2 or adults with a BMI ≥ 27 kg/m2 who have at least 1 weight-related condition such as hypertension, T2DM, or dyslipidemia.30
In 1996 topiramate was approved by the FDA for the treatment of seizure disorders and was also approved for migraine prophylaxis in 2004. In patients who were treated with topiramate for seizure disorders and migraines, weight loss and a reduction in visceral body fat has been observed.31 The precise MOA of topiramate in regards to weight loss is not fully understood. It may be due to its effects on both appetite suppression and satiety enhancement. Topiramate exhibits a combination of properties including modulatory effects on sodium channels, enhancement of GABA-activated chloride channels, inhibition of excitatory neurotransmission through actions on kainite and AMPA receptors, and inhibition of carbonic anhydrase (CA) isoenzymes in particular CA II and IV.14
The combination of phentermine and topiramate is a once-daily formulation that is designed to provide an immediate release of phentermine and a delayed release of topiramate, allowing a peak exposure of the phentermine in the morning and a peak concentration of topiramate in the evening. It should be taken in the morning in order to avoid the possibility of insomnia that can occur if taken in the evening. It can be taken with or without food. The recommended dose is as follows: Start treatment with Qsymia 3.75 mg/23 mg extended-release daily for 14 days; after 14 days increase to the recommended dose of Qsymia 7.5 mg/46 mg once daily.
Weight loss should be evaluated after 12 weeks at the higher dose. If at least 3% of baseline body weight has not been lost at that time, discontinue or escalate the dose. To escalate the dose: Increase to Qsymia 11.25 mg/69 mg daily for 14 days; followed by Qsymia 15 mg/92 mg daily. Evaluate weight loss following dose escalation to Qsymia 15 mg/92 mg after an additional 12 weeks of treatment. If at least 5% of baseline body weight has not been lost on Qsymia 15 mg/92 mg, discontinue as directed. It is important not to suddenly discontinue, as this may cause seizures. Patients should be slowly titrated off the medication.
In vitro studies of phentermine and topiramate indicate that these drugs are not likely to cause clinically significant interactions with drugs using the cytochrome P450 enzyme pathways, or those involved in plasma protein binding displacement; however there is evidence suggesting that ethinyl estradiol levels may be decreased by 16%, thus raising a concern about the possibility of decreased contraceptive efficacy.31 In patients with moderate (creatine clearance ≥ 30 mL/min to < 50 mL/min) and severe renal dysfunction (< 30 mL/min), the maximum dose of should not exceed 7.5 mg/46 mg.
Qsymia was evaluated in 3 phase 3 trials for its long-term efficacy and safety. In all trials, diet and lifestyle counseling were provided for all patients. The first of these studies was OB-301, a 28-week confirmatory trial with a factorial design involving 7 treatment arms, tested 2 fixed-dose Qsymia combinations—regular dose (7.5 mg/46 mg) and maximum dose (15 mg/92 mg)—as well as regular and maximum doses of the individual constituent drugs vs placebo.32 The study randomized 756 obese patients with a BMI range of 30 kg/m2 to 45 kg/m2 to 1 of the 7 treatment arms for 28 weeks. Patients treated with maximum-dose Qsymia achieved an average weight change of -9.0%, vs -1.5% with placebo (P < .0001). Weight change with regular-dose Qsymia was -8.2%. Weight changes with monotherapies were: -6.1% with topiramate 92 mg, -4.9% with topiramate 46 mg, -5.8% with phentermine 15 mg, and -5.2% with phentermine 7.5 mg.
OB-302 was a 56-week trial that randomized 1,267 morbidly obese patients with a BMI ≥ 35 kg/m2 without significant comorbidities to low-dose Qsymia (3.7 mg/23 mg), maximum-dose Qsymia (15 mg/92 mg), or placebo.33 At baseline, the mean BMI for the entire study cohort was 42 kg/m2. Mean weight changes were -1.6% with placebo, -5.1% with low-dose Qsymia, and -10.9% with maximum-dose Qsymia. The proportions of patients achieving ≥ 5% weight loss were: 17% with placebo, 45% with low-dose Qsymia, and 67% with maximum-dose Qsymia.
CONQUER was the largest of the phase 3 trials. It randomized 2,487 overweight or obese patients with a BMI of 27 kg/m2 to 45 kg/m2 and ≥ 2 obesity-related comorbidities (hypertension, dyslipidemia, T2DM, prediabetes or abdominal obesity) to receive a placebo, regular-dose Qsymia, or maximum-dose Qsymia for 56 weeks.34 In the completer population, mean weight changes in the placebo, regular dose Qsymia, and maximum-dose Qsymia groups were -1.6%, -9.6% (P <.0001), and -12.4% (P < .0001); and weight loss of ≥ 5% was achieved by 21%, 62%, and 70%, respectively. Relative to placebo, there were greater reductions in systolic BP, triglycerides, and fasting insulin with both doses of Qsymia.
Patients should not take Qsymia if they are pregnant, planning to become pregnant, or become pregnant during Qsymia treatment as there is an increased risk of birth defects, namely cleft lip and cleft palate. Women who can become pregnant should have a negative pregnancy test before taking Qsymia and every month while on the medication. They should use effective birth control consistently while taking Qsymia.
Qsymia is contraindicated in patients with glaucoma and patients who have hyperthyroidism. Qsymia can cause an increase in resting heart rate and regular monitoring of resting heart rate is recommended, especially in patients with cardiac or cerebrovascular disease. It has not been studied in patients with recent or unstable cardiac or cerebrovascular disease and therefore use is not recommended.
Qsymia can cause mood disorders such as anxiety and depression and can increase the risk of suicidal thoughts. Patients should be monitored for worsening depression, suicidal thoughts or behavior, or any unusual changes in mood or behavior. It is not recommended in patients with a history of suicidal attempts or active suicidal ideation. Qsymia can cause cognitive dysfunction. It can cause confusion, problems with concentration, attention, memory, or speech. Patients should be cautioned about operating automobiles and hazardous machinery.
Normal anion gap hyperchloremic metabolic acidosis has been reported in patients treated with Qsymia. If this does develop and persists, consideration should be given to either reduce the dose or discontinue Qsymia.
Weight loss may increase the risk of hypoglycemia in patients with T2DM treated with insulin and/or insulin secretagogues (eg, sulfonylureas). Qsymia has not been studied in combination with insulin. A reduction in the dose of antidiabetic medications, which are nonglucose dependent, should be considered to reduce the risk of hypoglycemia.
The most common AEs in controlled clinical studies (≥ 5% and at least 1.5 times placebo) included paraesthesia in the hands, arms, feet or face, dizziness, dysgeusia, insomnia, constipation, and dry mouth.
Contrave
In 2014, the FDA approved Contrave (Takeda, Deerfield, IL) as treatment option for chronic weight management in addition to reduced-calorie diet and physical activity. The combination of naltrexone hydrochloride and bupropion hydrochloride was originally introduced for the treatment of opioid addiction and later expanded to include the treatment of alcoholism. The antidepressant bupropion was approved in the U.S. in 1989. It is structurally different from all other marketed antidepressants (ie, tricyclics, tetracyclics, and SSRIs), but closely resembles the structure of diethylpropion, an appetite depressant with minimal CNS effects.35
This drug is approved for adults with BMI ≥ 30 kg/m2 and for adults with BMI ≥ 27 kg/m2 with at least 1 weight-related risk factors such as hypertension, T2DM, or dyslipidemia. It should be used as an adjunct to diet and exercise and is not approved for use for depression even though it contains bupropion.
Naltrexone is a pure opioid antagonist with high affinity to μ-opioid receptor, which is implicated in eating behavior. Naltrexone is rapidly and nearly completely absorbed from the GI tract after oral administration. The time to peak plasma concentration is about 1 hour. Naltrexone is well absorbed but first pass extraction and metabolism by the liver decreases oral bioavailability to between 5% to 40%. Primary elimination of naltrexone and its metabolites is renal excretion.
Bupropion is a weak inhibitor of neuronal reuptake of dopamine and norepinephrine. This drug is used to treat depression and seasonal affective disorder, and aid in smoking cessation. Bupropion is absorbed rapidly after oral administration, but the absolute oral bioavailability of bupropion is not known because an IV preparation is not available. The time to peak plasma concentrations of bupropion is within 2 hours of oral administration. Bupropion is extensively metabolized by the liver to multiple metabolites. Primary elimination of bupropion is urinary excretion. However, hepatic and renal impairment may affect the elimination of bupropion and its metabolites. Patients with hepatic or renal impairment should use a reduced dosage.
Combination therapy has been found to have complementary actions on CNS to reduce food intake. They are believed to dampen CNS reward pathways, taking away the compulsive feeding behavior and pleasure of feeding, ultimately leading to weight loss. Bupropion stimulates hypothalamic pro-opiomelanocortin neurons (POMC), which results in reduced food intake and increased energy expenditure. Naltrexone blocks opioid-receptor mediated POMC auto-inhibition, blocks the increase in dopamine in nucleus accumbens that occurs when eating, and acting synergistically with bupropion in augmenting POMC firing.
The COR-I and COR-II trials compared Contrave to diet and exercise in patients who did not have DM. The COR-Diabetes trial included the same study design but focused on patients with DM. In all the studies the participants had a 4-week titration to Contrave (naltrexone 8 mg/bupropion 90 mg) to decrease nausea. The first week dosing was 1 tablet in the morning. Week 2 was 1 tablet in morning and 1 tablet in the evening. In week 3, patients took 2 tablets in the morning and 1 tablet in the evening. The final titration step was 2 tablets in the morning and 2 tablets in the evening.
The COR-1 study was a 56-week randomized, double-blind, placebo-controlled study. It compared Contrave 32 mg naltrexone/360 mg bupropion (NB32/360) with an active placebo of diet and exercise.36 To be included adults must be aged 18 to 65 years with a BMI 30 kg/m2 to 45 kg/m2 or a BMI 27 kg/m2 to 45 kg/m2 with dyslipidemia or hypertension. Patients were instructed on a hypocaloric diet that was a 500 kcal per day deficit based on World Health Organization algorithm for calculating metabolic rate and they were urged to increase physical activity.
The completer population results showed 8.0% weight loss in the NB32/360 group and 1.9% weight loss in the placebo group (P < .001). For the NB32/360 and placebo groups, weight loss of ≥ 5% was achieved by 48% and 16% (P <.001), respectively; and weight loss of ≥ 10% by 25% and 7% (P < .001), respectively. The most common AE was nausea—29.8% with NB32/360 vs 5.3% with placebo. Nausea generally occurred early and then diminished and the discontinuation rate from nausea was significantly lower (6.3%) then the overall reported nausea rates.
Contrave was also studied in patients with T2DM. The COR-Diabetes Trial was a 56-week randomized, double blind, placebo-controlled study. The trial compared Contrave 32 mg naltrexone/360 mg bupropion (NB32/360) with an active placebo of diet and exercise.37 Inclusion criteria for the trial were patients aged 18 to 70 years with T2DM and a BMI from 27 kg/m2 to 45 kg/m2, A1c between 7% and 10%, and fasting blood glucose < 270 mg/dL. Participants either were not taking a DM medication or were on stable doses of oral antidiabetes drugs ≥ 3 months prior to randomization. Patients were placed on a 500 kcal hypocaloric diet and advised to increase physical activity.
The results showed 5.0% weight loss in the NB32/360 group and 1.8% weight loss in the placebo group (P < .001). Weight loss of ≥ 5% and ≥ 10% was achieved by 44.5% and 18.5% of the NB32/360 group, respectively, and 18.9% and 5.7%, respectively (P < .001) of the placebo group. The NB32/360 and placebo showed a reduction of A1c of 0.6% and 0.1% respectively (P < .001). The most common AE was nausea (42.3% with NB32/360 vs 7.1% with placebo). Nausea generally occurred early and then diminished and the discontinuation rate from nausea was significantly lower (9.6%) then the overall reported nausea rates.37
Due to potential nausea caused by naltrexone, Contrave should be titrated over 4 weeks as described earlier. At maintenance dose, patients should be evaluated after 12 weeks to determine treatment benefits. If a patient has not lost at least 5% of baseline body weight, Contrave should be discontinued, because it would be unlikely that the patient will achieve and sustain clinically meaningful weight loss with continued treatment. Contrave should not be taken with high-fat meals that may result in significant increase in bupropion and naltrexone systemic exposure.
Since Contrave contains the antidepressant bupropion, it has a boxed warning similar to other antidepressants in its class of increased risk of suicidal thoughts and behaviors, especially in children, adolescents, and young adults.38Contrave can lower the seizure threshold; therefore it should not be used in people with a seizure disorder. It can also raise BP and heart rate; however the clinical significance of hypertension and elevated heart rate observed with Contrave treatment is unclear. Blood pressure rose on average by 1 point during the first 8 weeks of treatment and then returned to baseline.38 The heart rate also increased by about 1.7 beats per minute.38 Patients with uncontrolled hypertension should avoid Contrave.
Contrave should not be taken with products contain bupropion or naltrexone. It should not be taken by patient who are regularly taking opioids or who are opioid dependent, or who are experiencing opiate withdrawal. Pregnant women should also avoid Contrave. In patients with renal impairment the maximum dose is 1 tablet twice a day and in patients with hepatic impairment the maximum dose is 1 tablet a day.
Liraglutide
Liraglutide is the newest weight loss medication to be approved by the FDA for chronic weight management as an adjunct to a reduced calorie diet and increased physical activity in adult patients with BMI ≥ 30 kg/m2 or ≥ 27 kg/m2 with hypertension, diabetes, or dyslipidemia. The recommended dose of liraglutide is 3 mg daily. The initial dose is 0.6 mg daily for the first week, then titrated up by 0.6 each week for 4 weeks, until reaching 3 mg daily.
Liraglutide is an acylated human glucagon-like peptide-1 (GLP-1) receptor agonist, which are expressed in the brain and is involved in the control of appetite. It is also found in the beta cells of the pancreas, where GLP-1 receptors stimulate insulin release in response to elevated blood glucose concentrations and suppress glucagon secretion. Endogenous GLP-1 has a half-life of 1.5 to 2 minutes due to degradation by the DDP-4 enzyme, but liraglutide is stable against degradation by peptidases and has a half- life of 13 hours.
Liraglutide was studied in a 56-week randomized, double-blind, placebo-controlled trial, which compared liraglutide 3 mg with an active placebo of diet and physical activity.39 Inclusion criteria were adults aged ≥ 18 years old with a BMI 30 kg/m2 to 45 kg/m2 or BMI 27 kg/m2 to 45 kg/m2 with dyslipidemia and/or hypertension. Both groups received lifestyle modification counseling. Patients were excluded if they had DM.
In the trial, 3,731 participants enrolled, 2,487 in the liraglutide group and 1,244 in the placebo group; 78.7% of the participants were female and the average age was 45 years. Subjects in the liraglutide group had a weekly titration regimen. The starting dose at week 1 was 0.6 mg, week 2 was 1.2 mg, week 3 was 1.8 mg, week 4 was 2.4 mg, and week 5 was 3.0 kg.
The completer population showed 9.2% weight loss in the liraglutide group and 3.0% weight loss in the control group.39 Weight loss of ≥ 5% was seen in 63.2% and 27.1% of the liraglutide and placebo groups, respectively. Weight loss rates of ≥ 10% was seen by 33.1% and 10.6%, respectively. The most common AEs were nausea, diarrhea, and constipation. Nausea generally occurred early during the titration period and then diminished.
A second clinically relevant study was performed with liraglutide. Often patients are able to lose weight with diet and exercise and then plateau. This study examined participants who lost 5% percent of their initial body weight and then were randomized to liraglutide or placebo.40 Key inclusion criteria were people aged ≥ 18 years old with a BMI 30 kg/m2 to 45 kg/m2 or BMI 27 kg/m2 to 45 kg/m2 with dyslipidemia and/or hypertension. In order to be randomized, participants were required to lose at least 5% of their initial body weight on a 1,200 kcal to 1,400 kcal diet with increased physical activity during a 4 to 12 week run-in period.
Four hundred twenty-two participants were enrolled, 212 in the liraglutide group and 210 in the placebo group. Most of the participants were female (81%). The average BMI in the study was 35.6 kg/m2. Subjects in the liraglutide group had a weekly titration regimen.
After an average weight loss of 6% using a low calorie diet and increased physical activity the participants were randomized to continue diet and increased activity alone (placebo) or with liraglutide. At week 56 the results showed an additional 6.2% weight loss in the liraglutide group and 0.2% weight gain in the placebo group. The liraglutide group had a greater number of participants with ≥ 5% weight loss compared to placebo, 50.5% vs 21.8% (P < .0001).40 In the pooled data set from the registration trials the 3 most common GI AEs were nausea, diarrhea, and constipation occurring in 39.3%, 20.9%, and 19.4% of participants respectively. Discontinuation due to nausea for liraglutide was 2.9%.41
Clinicians should be aware that medications that can cause hypoglycemia such as sulfonylureas and insulin must be tapered as patients lose weight with liraglutide. Documented symptomatic hypoglycemia in patients with T2DM and with sulfonylurea background therapy was 43.6% with liraglutide vs 27.3% with placebo.
In the setting of renal impairment, patients treated with GLP-1 receptor agonists, including liraglutide, have had reports of acute renal failure and worsening of chronic renal failure usually associated with nausea, vomiting, diarrhea, or dehydration. Liraglutide causes thyroid C-cell tumors at clinically relevant exposures in rats and mice. It is unknown whether liraglutide causes thyroid C-cell tumors, including medullary thyroid carcinoma (MTC), in humans. As the human relevance of liraglutide-induced rodent thyroid C-cell tumors has not been determined liraglutide is contraindicated in patients with a personal or family history of MTC or in patients with multiple endocrine neoplasia syndrome type 2.
Acute pancreatitis, including fatal and nonfatal hemorrhagic or necrotizing pancreatitis, has been observed in patients treated with liraglutide in postmarketing reports. After initiation of liraglutide, observe patients carefully for signs and symptoms of pancreatitis (including persistent severe abdominal pain, sometimes radiating to the back, which may or may not be accompanied by vomiting). If pancreatitis is suspected, liraglutide should promptly be discontinued.
Conclusion
The treatment of obesity and overweight with comorbidities has always been a challenge. In the past there were few FDA-approved drugs and many drugs had to be used off-label. The toolbox of medications available for medical weight management is more robust than ever. The medications have different MOAs and can be used in a variety of patients. There are differences in the classes and some are controlled substances. Phentermine, lorcaserin, and Qsymia (phentermine/topiramate) are controlled substances whereas orlistat, naltrexone/bupropion and liraglutide are not. Other differences exist including duration of use. The sympathomimetic drugs have a limited window of use whereas orlistat, Qsymia (phentermine/topiramate), lorcaserin, naltrexone/bupropion, and liraglutide do not.
The medications that are available have a wide variety of MOAs. Therefore, if a patient fails one medication, then it is very reasonable to try a medication with a different MOA. In addition, there is the potential for weight regain when weight reduction medications are discontinued. As people lose weight their metabolic rate decreases about 15 kcal per pound of weight reduction.42
Another challenge of using these medications is managing patient expectations. The current metric used for FDA approval is a 5% weight loss that is greater in the study group compared with the diet and physical activity active control. However, many clinicians and patients do not find this weight reduction amount consistent with their expectations. In addition weight loss trajectory may also be too slow for patients and cause early discontinuation. Therefore, patient education and a discussion of reasonable expectations for weight reduction medications are necessary.
Clinicians must acknowledge that there are limitations to the use of these medications. Newer agents do have a higher cost and insurance reimbursement is somewhat limited. However, they offer the opportunity to prevent more expensive, protracted conditions such as diabetes and cardiovascular disease. In summary, clinicians now have a wider variety of medication options to be used with dietary and lifestyle changes in order to improve health and prevent chronic diseases.
1. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of obesity in the United States, 2009-2010. NCHS Data Brief. 2012(82):1-8.
2. Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. Circulation. 2014;129(25 suppl 2):S102-S138.
3. U.S. Food and Drug Administration. Drugs@FDA: diethylpropion hydrochloride. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=012546&DrugName=TENUATE%20DOSPAN&ActiveIngred=DIETHYLPROPION%20HYDROCHLORIDE&SponsorApplicant=ACTAVIS%20LABS%20UT%20INC&ProductMktStatus=1&goto=Search.DrugDetails. Accessed December 28, 2015.
4. U.S. Food and Drug Administration. Drugs@FDA: benzphetamine hydrochloride. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=012427&DrugName=DIDREX&ActiveIngred=BENZPHETAMINE%20HYDROCHLORIDE&SponsorApplicant=PHARMACIA%20AND%20UPJOHN&ProductMktStatus=3&goto=Search.DrugDetails. Accessed December 28, 2015.
5. U.S. Food and Drug Administration. Drugs@ FDA: phendimetrazine tartrate. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=088021&DrugName=BONTRIL&ActiveIngred=PHENDIMETRAZINE%20TARTRATE&SponsorApplicant=VALEANT&ProductMktStatus=3&goto=Search.DrugDetails. Accessed December 28, 2015.
6. U.S. Food and Drug Administration. Drugs@FDA: phentermine. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=085128&DrugName=ADIPEX%2DP&ActiveIngred=PHENTERMINE%20HYDROCHLORIDE&SponsorApplicant=TEVA&ProductMktStatus=1&goto=Search.DrugDetails. Accessed December 28, 2015.
7. U.S. Food and Drug Administration. Drugs @ FDA: phentermine hydrochloride. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=202088&DrugName=SUPRENZA&ActiveIngred=PHENTERMINE%20HYDROCHLORIDE&SponsorApplicant=CITIUS%20PHARMS&ProductMktStatus=1&goto=Search.DrugDetails. Accessed December 28, 2015.
8. Hendricks EJ, Rothman RB, Greenway FL. How Physician Obesity Specialists use drugs to treat obesity. Obesity (Silver Spring). 2009;17(9):1730-1735.
9. Gilman AG, Gilman A, Goodman LS, eds. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 8th ed. New York: Pergamon Press; 1990: 211.
10. Gilman AG, Gilman A, Goodman LS, eds. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 8th ed. New York: Pergamon Press; 1990:217.
11. Addy C, Jumes P, Rosko K, et al. Pharmacokinetics, safety, and tolerability of phentermine in health participants receiving taranabant, a novel cannabinoid-1 receptor (CB1R) inverse agonist. J Clin Pharmacol. 2009;49(10):1228-1232.
12. U.S. Department of Justice, Drug Enforcement Administration, Office of Diversion Control. Controlled substances. U.S. Department of Justice Website. http://www.deadiversion.usdoj.gov/schedules/orangebook/c_cs_alpha.pdf. Updated November 12, 2015. Accessed December 16, 2015.
13. Bray GA, Greenway FL. Pharmacological treatment of the overweight patient. Pharmacol Rev. 2007;59(2):151-184.
14. V1-0521 (QNEXA) Advisory committee briefing document. NDA 022580. Endocrinologic and Metabolic Drugs Advisory Committee meeting; June 17, 2010.
15. Cercato C, Roizenblatt VA, Leanca CC, et al. A randomized double-blind placebo-controlled study of the long-term efficacy and safety of diethylpropion in the treatment of obese subjects. Int J Obes (Lond). 2009;33(8):857-865.
16. Haddock CK, Poston WS, Dill PL, Foreyt JP, Ericsson M. Pharmacotherapy for obesity: a quantitative analysis of four decades of published randomized clinical trials. Int J Obes (Lond). 2002;26(2):262-273.
17. Suplicy H, Boquszewski CL, dos Santos CM, do Desterro de Fiqueiredo M, Cunha DR, Radominski R. A comparative study of five centrally acting drugs on pharmacological treatment of obesity. Int J Obes (Lond). 2014;38(8):1097-1103.
18. Ioannides-Demos LL, Proietto J, McNeill JJ. 2005. Pharmacotherapy for obesity. Drugs. 2005;65(10): 1391-1418.
19. Drent ML, van der Veen EA. Lipase inhibition: a novel concept in the treatment of obesity. Int J Obes Relat Metab Disord. 1993;17(4):241-244.
20. Xenical [package insert]. Nutley, NJ: Roche Laboratories Inc.; 1999.
21. U.S. Food and Drug Administration. FDA Drug Safety Communication: Completed safety review of Xenical/Alli and severe liver injury. U.S. Food and Drug Administration Website. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm213038.htm. Updated August 2, 2010. Accessed December 16, 2015.
22. Sall D, Wang J, Rashkin M, Welch M, Droege C, Schauer D. Orlistat-induced fulminant hepatic failure. Clin Obes. 2014;4(6):342-347.
23. Sjöström L, Rissanen A, Andersen T, et al. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. European Multicentre Orlistat Study Group. Lancet. 1998;352(9123):167-172.
24. Hauptman J, Lucas C, Boldrin MN, Collins H, Segal KR. Orlistat in the long-term treatment of obesity in primary care settings. Arch Fam Med. 2000;9(2):160-167.
25. Torgerson JS, Hauptman J, Boldrin MN, Sjöström L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care. 2004;27(1):155-161.
26. Torgerson JS, Arlinger K, Käppi M, Sjöström L. Principles for enhanced recruitment of subjects in a large clinical trial. the XENDOS (XENical in the prevention of Diabetes in Obese Subjects) study experience. Control Clin Trials. 2001;22(5):515-525.
27.Smith SR, Weissman NJ, Anderson CM, et al; Behavioral Modification and Lorcaserin for Overweight and Obesity Management (BLOOM) Study Group. Multicenter, placebo-controlled trial of lorcaserin for weight management. N Engl J Med. 2010;363(3):245-256.
28. Fidler MC, Sanchez M, Raether B, et al; BLOSSOM Clinical Trial Group. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab. 2011;96(10):3067-3077.
29. O’Neil PM, Smith SR, Weisserman NJ, et al. Randomized placebo-controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: the BLOOM-DM Study. Obesity (Silver Spring). 2012;20(7):1426-1436.
30. U.S. Food and Drug Administration. FDA approves weight-management drug Qsymia. U.S. Food and Drug Administration Website. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm312468.htm. Published July 17, 2012. Accessed December 16, 2015.
31. Shin J, Gadde KM. Clinical utility of phentermine/topiramate (Qsymia™) combination for the treatment of obesity. Diabetes Metab Syndr Obes. 2013;6:131-139.
32. Qsymia [package insert] Mountain View, CA: Vivus, Inc; 2012.
33. Allison DB, Gadde KM, Garvey WT, et al. Controlled-release phentermine/topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring). 2012;20(2):330-342.
34. Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377(9774):1341-1352.
35. Plodkowski RA, Nguyen Q, Sundaram U, Nguyen L, Chau DL, St Jeor S. Bupropion and naltrexone: a review of their use individually and in combination for the treatment of obesity. Expert Opin Pharmacother. 2009;10(6):1069-1081.
36. Greenway FL, Fujioka K, Plodkowski RA, et al; COR-I Study Group. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-1): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376(9741):595-605.
37. Hollander P, Gupta AK, Plodkowski R, et al; COR-Diabetes Study Group. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care. 2013;36(12):4022-4029.
38. Contrave [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc; 2014.
39. Pi-Sunyer X, Astrup A, Fujioka K, et al; SCALE Obesity and Prediabetes NN8022-1839 Study Group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Eng J Med. 2015;373(1):11-22.
40. Wadden TA, Hollander P, Klein S, et al; NN8022-1923 Investigators. Weight maintenance and additional weight loss with liraglutide after low-calorie-diet-induced weight loss: the SCALE Maintenance randomized study. Int J Obes (Lond). 2013;37(11):1443-1451
41. Saxenda [package insert]. Novo Nordisk: Plainsboro, NJ; 2015.
42.Schwartz A, Doucet E. Relative changes in resting energy expenditure during weight loss: a systemic review. Obes Rev. 2010;11(7): 531-547. ```````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````
1. Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of obesity in the United States, 2009-2010. NCHS Data Brief. 2012(82):1-8.
2. Jensen MD, Ryan DH, Apovian CM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines; Obesity Society. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. Circulation. 2014;129(25 suppl 2):S102-S138.
3. U.S. Food and Drug Administration. Drugs@FDA: diethylpropion hydrochloride. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=012546&DrugName=TENUATE%20DOSPAN&ActiveIngred=DIETHYLPROPION%20HYDROCHLORIDE&SponsorApplicant=ACTAVIS%20LABS%20UT%20INC&ProductMktStatus=1&goto=Search.DrugDetails. Accessed December 28, 2015.
4. U.S. Food and Drug Administration. Drugs@FDA: benzphetamine hydrochloride. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=012427&DrugName=DIDREX&ActiveIngred=BENZPHETAMINE%20HYDROCHLORIDE&SponsorApplicant=PHARMACIA%20AND%20UPJOHN&ProductMktStatus=3&goto=Search.DrugDetails. Accessed December 28, 2015.
5. U.S. Food and Drug Administration. Drugs@ FDA: phendimetrazine tartrate. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=088021&DrugName=BONTRIL&ActiveIngred=PHENDIMETRAZINE%20TARTRATE&SponsorApplicant=VALEANT&ProductMktStatus=3&goto=Search.DrugDetails. Accessed December 28, 2015.
6. U.S. Food and Drug Administration. Drugs@FDA: phentermine. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=085128&DrugName=ADIPEX%2DP&ActiveIngred=PHENTERMINE%20HYDROCHLORIDE&SponsorApplicant=TEVA&ProductMktStatus=1&goto=Search.DrugDetails. Accessed December 28, 2015.
7. U.S. Food and Drug Administration. Drugs @ FDA: phentermine hydrochloride. U.S. Food and Drug Administration Website. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=202088&DrugName=SUPRENZA&ActiveIngred=PHENTERMINE%20HYDROCHLORIDE&SponsorApplicant=CITIUS%20PHARMS&ProductMktStatus=1&goto=Search.DrugDetails. Accessed December 28, 2015.
8. Hendricks EJ, Rothman RB, Greenway FL. How Physician Obesity Specialists use drugs to treat obesity. Obesity (Silver Spring). 2009;17(9):1730-1735.
9. Gilman AG, Gilman A, Goodman LS, eds. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 8th ed. New York: Pergamon Press; 1990: 211.
10. Gilman AG, Gilman A, Goodman LS, eds. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 8th ed. New York: Pergamon Press; 1990:217.
11. Addy C, Jumes P, Rosko K, et al. Pharmacokinetics, safety, and tolerability of phentermine in health participants receiving taranabant, a novel cannabinoid-1 receptor (CB1R) inverse agonist. J Clin Pharmacol. 2009;49(10):1228-1232.
12. U.S. Department of Justice, Drug Enforcement Administration, Office of Diversion Control. Controlled substances. U.S. Department of Justice Website. http://www.deadiversion.usdoj.gov/schedules/orangebook/c_cs_alpha.pdf. Updated November 12, 2015. Accessed December 16, 2015.
13. Bray GA, Greenway FL. Pharmacological treatment of the overweight patient. Pharmacol Rev. 2007;59(2):151-184.
14. V1-0521 (QNEXA) Advisory committee briefing document. NDA 022580. Endocrinologic and Metabolic Drugs Advisory Committee meeting; June 17, 2010.
15. Cercato C, Roizenblatt VA, Leanca CC, et al. A randomized double-blind placebo-controlled study of the long-term efficacy and safety of diethylpropion in the treatment of obese subjects. Int J Obes (Lond). 2009;33(8):857-865.
16. Haddock CK, Poston WS, Dill PL, Foreyt JP, Ericsson M. Pharmacotherapy for obesity: a quantitative analysis of four decades of published randomized clinical trials. Int J Obes (Lond). 2002;26(2):262-273.
17. Suplicy H, Boquszewski CL, dos Santos CM, do Desterro de Fiqueiredo M, Cunha DR, Radominski R. A comparative study of five centrally acting drugs on pharmacological treatment of obesity. Int J Obes (Lond). 2014;38(8):1097-1103.
18. Ioannides-Demos LL, Proietto J, McNeill JJ. 2005. Pharmacotherapy for obesity. Drugs. 2005;65(10): 1391-1418.
19. Drent ML, van der Veen EA. Lipase inhibition: a novel concept in the treatment of obesity. Int J Obes Relat Metab Disord. 1993;17(4):241-244.
20. Xenical [package insert]. Nutley, NJ: Roche Laboratories Inc.; 1999.
21. U.S. Food and Drug Administration. FDA Drug Safety Communication: Completed safety review of Xenical/Alli and severe liver injury. U.S. Food and Drug Administration Website. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm213038.htm. Updated August 2, 2010. Accessed December 16, 2015.
22. Sall D, Wang J, Rashkin M, Welch M, Droege C, Schauer D. Orlistat-induced fulminant hepatic failure. Clin Obes. 2014;4(6):342-347.
23. Sjöström L, Rissanen A, Andersen T, et al. Randomised placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. European Multicentre Orlistat Study Group. Lancet. 1998;352(9123):167-172.
24. Hauptman J, Lucas C, Boldrin MN, Collins H, Segal KR. Orlistat in the long-term treatment of obesity in primary care settings. Arch Fam Med. 2000;9(2):160-167.
25. Torgerson JS, Hauptman J, Boldrin MN, Sjöström L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care. 2004;27(1):155-161.
26. Torgerson JS, Arlinger K, Käppi M, Sjöström L. Principles for enhanced recruitment of subjects in a large clinical trial. the XENDOS (XENical in the prevention of Diabetes in Obese Subjects) study experience. Control Clin Trials. 2001;22(5):515-525.
27.Smith SR, Weissman NJ, Anderson CM, et al; Behavioral Modification and Lorcaserin for Overweight and Obesity Management (BLOOM) Study Group. Multicenter, placebo-controlled trial of lorcaserin for weight management. N Engl J Med. 2010;363(3):245-256.
28. Fidler MC, Sanchez M, Raether B, et al; BLOSSOM Clinical Trial Group. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab. 2011;96(10):3067-3077.
29. O’Neil PM, Smith SR, Weisserman NJ, et al. Randomized placebo-controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: the BLOOM-DM Study. Obesity (Silver Spring). 2012;20(7):1426-1436.
30. U.S. Food and Drug Administration. FDA approves weight-management drug Qsymia. U.S. Food and Drug Administration Website. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm312468.htm. Published July 17, 2012. Accessed December 16, 2015.
31. Shin J, Gadde KM. Clinical utility of phentermine/topiramate (Qsymia™) combination for the treatment of obesity. Diabetes Metab Syndr Obes. 2013;6:131-139.
32. Qsymia [package insert] Mountain View, CA: Vivus, Inc; 2012.
33. Allison DB, Gadde KM, Garvey WT, et al. Controlled-release phentermine/topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring). 2012;20(2):330-342.
34. Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377(9774):1341-1352.
35. Plodkowski RA, Nguyen Q, Sundaram U, Nguyen L, Chau DL, St Jeor S. Bupropion and naltrexone: a review of their use individually and in combination for the treatment of obesity. Expert Opin Pharmacother. 2009;10(6):1069-1081.
36. Greenway FL, Fujioka K, Plodkowski RA, et al; COR-I Study Group. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-1): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376(9741):595-605.
37. Hollander P, Gupta AK, Plodkowski R, et al; COR-Diabetes Study Group. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care. 2013;36(12):4022-4029.
38. Contrave [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc; 2014.
39. Pi-Sunyer X, Astrup A, Fujioka K, et al; SCALE Obesity and Prediabetes NN8022-1839 Study Group. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Eng J Med. 2015;373(1):11-22.
40. Wadden TA, Hollander P, Klein S, et al; NN8022-1923 Investigators. Weight maintenance and additional weight loss with liraglutide after low-calorie-diet-induced weight loss: the SCALE Maintenance randomized study. Int J Obes (Lond). 2013;37(11):1443-1451
41. Saxenda [package insert]. Novo Nordisk: Plainsboro, NJ; 2015.
42.Schwartz A, Doucet E. Relative changes in resting energy expenditure during weight loss: a systemic review. Obes Rev. 2010;11(7): 531-547. ```````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````````
SGLT2 Inhibitors for Type 2 Diabetes Mellitus Treatment
Over the past 2 decades, the treatment of type 2 diabetes mellitus (T2DM) has been an evolving science. With therapeutic advances, the prevalence of catastrophic complications such as amputations, renal failure requiring dialysis, and blindness due to retinopathy have significantly declined. Developed drugs have successfully met treatment goals; however, they are often associated with a higher risk of hypoglycemia and weight gain. Now that better glucose control is possible, the science of diabetes care continues to evolve. Newly developed drugs should control glucose without significant hypoglycemia and also promote weight reduction. The sodium-glucose transport protein 2 (SGLT2) inhibitor drug class has these characteristics, and the novel mechanism of action complements older medications used to treat T2DM.
Phlorizin is a plant-based compound originally discovered in 1935 when it was derived from the bark of apple trees.1 It is a naturally occurring botanical glucoside and is fairly nonselective between SGLT1 and SGLT2. Due to its poor bioavailability and its degradation in the gastrointestinal tract, it was not an ideal drug candidate in humans. 
Canagliflozin
Canagliflozin is a SGLT2 inhibitor and a low-potency SGLT1 inhibitor. It was the first SGLT2 inhibitor approved by the FDA (March 2013) to be used with diet and exercise to improve glycemic control in adults with T2DM. The recommended starting dose is 100 mg once daily for patients who have an estimated glomerular filtration rate (eGFR) > 60 mL/min/1.73m2 and can be increased to 300 mg once daily. It is also available in a fixed-dose combination with metformin. Canagliflozin SGLT-2 inhibition leads to increased glycosuria and osmotic diuresis that lowers plasma glucose concentrations. Lower blood pressure (BP) is likely an effect of the osmotic diuresis. Increased urinary excretion of glucose also leads to a loss of calories and weight loss. It was studied alone and in combination with metformin, sulfonylurea, pioglitazone, and insulin therapy.
The pharmacokinetics of canagliflozin is similar in healthy subjects and patients with T2DM. Peak plasma concentrations (Cmax) and area under the cover (AUC) of canagliflozin increased in a dose-proportional manner from 50 mg to 300 mg. Following single-dose oral administration of 100 mg and 300 mg of canagliflozin, time to Cmax (Tmax) of canagliflozin occurs within 1 to 2 hours postdose. The apparent terminal half-life (t1/2) was 10.6 hours and 13.1 hours for the 100 mg and 300 mg doses, respectively. Steady state was reached after 4 to 5 days of once-daily dosing with canagliflozin 100 mg to 300 mg. Glucuronidation is the major metabolic pathway. There is balanced renal and biliary excretion of metabolites, and there are no active metabolites.
Following oral doses of canagliflozin in patients with T2DM, dose-dependent decreases were seen in the renal threshold for glucose (RTG). From a starting value of about 240 mg/dL, the 300-mg dose suppressed the mean (RTG) to about 70 to 90 mg/dL in T2DM in phase 1 studies. The reduction in RTG led to increase in urinary excretion of glucose of about 100 g/d.
In addition to renal SGLT2 inhibition leading to increased urinary glucose excretion (UGE), canagliflozin has been shown to lower postprandial glucose excursion (PPGE) and insulin concentrations by delaying intestinal glucose absorption.2 A study was done in 20 healthy subjects who received either placebo or canagliflozin 300 mg 20 minutes before a 600 kcal mixed-meal tolerance test. Compared with placebo, canagliflozin reduced PPGE and insulin excursions (0-2 h) AUC by 35% and 43%, respectively (P < .001 for both). This may present a difference between canagliflozin and the other SGLT2 inhibitors.
Because of the potential differences due to canagliflozin’s inhibition of intestinal SGLT1, the pharmacodynamic differences between canagliflozin and dapagliflozin were studied.3 The randomized, double-blind, crossover study consisted of 54 subjects. The subjects received the maximum approved doses of canagliflozin 300 mg or dapagliflozin 10 mg a day. Each group was treated with the study drug for 2 days, and then a 600 kcal mixed-meal tolerance test was performed. The results of the PPGE 0- to 2-hour AUC analysis showed 3.66 mmol*h/L with canagliflozin 300 mg and 4.08 mmol*h/L with dapagliflozin 10 mg. There was a difference of 0.42 (P = .0122), which was a 10.3% reduction in AUC PPGE by canagliflozin compared with dapagliflozin.
Canagliflozin has been studied in patients with T2DM and stage 3 nephropathy. Data were pooled from 4 randomized, placebo-controlled, phase 3 studies in which subjects had baseline eGFR > 30 to < 60 mL/min/1.73 m2.4 In the setting of decreased eGFR associated with stage 3 chronic kidney disease, subjects treated with canagliflozin 100 mg and canagliflozin 300 mg had placebo-subtracted reductions in hemoglobin A1c (A1c) of -0.38% and -0.47%, respectively, and placebosubtracted reduction in weight of -1.6% and -1.9%, respectively. Decreases in eGFR were seen at week 6 but trended toward baseline over time with a mean change in eGFR of 0.7, -1.7, -2.2 mL/min/1.73 m2 for placebo, canagliflozin 100 mg, and canagliflozin 300 mg, respectively.
Clinical Efficacy Trials
Canagliflozin was studied as add-on therapy to metformin and compared with glimepiride (a sulfonylurea).5 The randomized, double-blind study included 1,450 subjects for a core study period of 52 weeks followed by a 52-week extension. Eligible subjects were aged > 18 and < 80 years, A1c > 7% and < 9.5%, and were receiving metformin > 2,000 mg/d or > 1,500 mg/d if unable to tolerate a higher dose. The study groups were canagliflozin 100 mg, canagliflozin 300 mg, and glimepiride, and baseline A1c measurements were 7.78%, 7.79%, and 7.83%, respectively. The glimepiride was titrated up if > 50% of fasting blood glucose measurements were > 108 mg/dL with no hypoglycemic events in the previous 2 weeks.
Over 104 weeks, canagliflozin 100 mg and 300 mg and glimepiride reduced A1c from mean baseline values by -0.65%, -0.74%, and -0.55%, respectively, and the proportions of patients achieving A1c < 7% at week 104 was 42.5%, 50.2%, and 43.9%, respectively. Weight fell over 104 weeks with canagliflozin 100 mg (-4.1%, -3.6 kg) and canagliflozin 300 mg (-4.2%, -3.6 kg). In contrast, glimepiride showed weight increase (0.9%, 0.8 kg). Documented hypoglycemia episodes were lower in canagliflozin 100 mg and 300 mg than with glimepiride (6.8%, 8.2%, and 40.9%, respectively).
A study was undertaken to compare canagliflozin with the dipeptidyl peptidase-4 (DPP-4) inhibitor sitagliptin in patients with T2DM on background therapy of metformin.6 This randomized, double-blind trial studied subjects aged > 18 and < 80 years with inadequate glucose control A1c > 7% and < 10.5%. Subjects received metformin > 2,000 mg/d or > 1,500 mg/d if unable to tolerate a higher dose. At week 52, canagliflozin showed noninferiority to sitagliptin, with both drugs lowering A1c by 0.73%. Canagliflozin 300 mg showed superiority to sitagliptin with -0.88% change in A1c. Both canagliflozin 100 mg and 300 mg were superior to sitagliptin 100 mg in weight reduction: -3.8%, -4.2%, and -1.3%, respectively. Genital mycotic infections were higher in the canagliflozin groups. Rates for mycotic infections for sitagliptin 100 mg, canagliflozin 100 mg, and canagliflozin 300 mg were 1.2%, 5.2%, and 2.4% in men, respectively, and 2.6%, 11.3%, and 9.9% in women, respectively.
Canagliflozin was also studied in combination with insulin to determine efficacy and safety in this setting.7 Subjects were randomized to receive placebo, canagliflozin 100 mg, or canagliflozin 300 mg. Subjects had a mean baseline A1c of 8.3%. The median daily insulin dose was 60 IU, and most individuals were using basal/bolus regimens. The primary endpoint was 18 weeks of therapy, and A1c was lowered 0.62% (P < .001) with canagliflozin 100 mg and 0.73% with canagliflozin 300 mg compared with placebo. Weight decreased 1.9% (P < .001) with canagliflozin 100 mg and 2.4% (P < .001) with canagliflozin 300 mg compared with placebo.
Adverse Effects and Precautions
Canagliflozin adverse effects (AEs) were generally low. In the aforementioned 104-week study comparing canagliflozin 100 mg, canagliflozin 300 mg, and glimepiride, AEs leading to discontinuation were low at 6.2%, 9.5%, and 7.3%, respectively. Serious AEs were lower in the canagliflozin 100 mg and 300 mg groups compared with glimepiride at 9.7%, 9.7%, 14.3%, respectively.5
Limitations for use of canagliflozin are T1DM and diabetic ketoacidosis (DKA). In mild renal impairment, there is no dose adjustment in patients with eGFR > 60 mL/min/1.73 m2. In moderate renal impairment (eGFR 45-60 mL/min/1.73 m2), dose is limited to 100 mg once daily. It is recommended not to initiate canagliflozin if eGFR is < 45 mL/min/1.73 m2. Canagliflozin is contraindicated if eGFR is < 30 mL/min/1.73 m2.8
Dapagliflozin
Dapagliflozin is a highly selective SGLT2 inhibitor. It is a 1,400-fold greater inhibitor of SGLT2 vs SGLT1 and was approved in January 2014. It is indicated as an adjunct to diet and exercise to improve glycemic control in adults with T2DM. The recommended starting dose is 5 mg once daily, taken in the morning, with or without food, and the dosage can be increased to 10 mg once daily in patients who require additional glycemic control. Dapagliflozin should not be initiated if eGFR is < 60 mL/min/1.73 m2, and it should be discontinued if eGFR is persistently < 60 mL/min/1.73 m2.
By inhibiting SGLT2, dapagliflozin reduces reabsorption of filtered glucose and lowers the renal threshold for glucose, thereby increasing UGE. Increased glucose secretion also leads to weight reduction. Dapagliflozin has been studied alone and in combination with glipizide, glimepiride, pioglitazone, and a DDP-4 inhibitor and as an add-on to insulin with and without other oral antidiabetic drugs. It is also available in a fixed-dose combination with metformin.
Following oral administration of dapagliflozin, the Tmax is usually attained within 2 hours under a fasting state. The Cmax and AUC values increase the dose proportionally with increase in dapagliflozin dose in the therapeutic dose range. The absolute oral bioavailability of dapagliflozin following the administration of a 10-mg dose is 78%. The mean plasma t1⁄2 for dapagliflozin is about 12.9 hours following a single oral dose of 10 mg. In patients with normal renal function, the renal glucose excretion was 85 g per day at maximal dose. In humans, 75% of the dose of dapagliflozin is primarily metabolized through the uridine diphosphate glucuronosyltransferase 1A9 pathway. Dapagliflozin and related metabolites are primarily eliminated via the renal pathway.9
Clinical Efficacy Trials
Dapagliflozin was studied as add-on therapy to metformin with glipizide (a sulfonylurea) as the comparator.10 This 52-week double-blind, multicenter, active-controlled, noninferiority trial randomized 801 patients. Eligible subjects were aged > 18 years with A1c > 6.5% and < 10% and were receiving metformin or metformin and 1 other oral antidiabetic drug up to half-maximal dose for at least 8 weeks. During an 18-week titration period, all patients started dapagliflozin 2.5 mg/d and glipizide 5 mg/d. At 21-day intervals, patients were titrated up to the next dosage level if fasting plasma glucose was > 110 mg/dL. Dapagliflozin could be titrated to 5 mg and then 10 mg per protocol. Glipizide could be titrated to 10 mg or 20 mg per protocol.
The A1c change with dapagliflozin was noninferior to glipizide at week 52. The A1c adjusted mean change from baseline was -0.52 for both dapagliflozin and glipizide. The secondary endpoint was weight change. The glipizide group had a +1.44 kg weight gain, whereas the dapagliflozin group had a -3.22 kg weight loss. The number of patients with ≥ 1 episode of hypoglycemia, either symptomatic or with no symptoms, with blood glucose ≤ 63 mg/dL was assessed. The dapagliflozin group had a 3.5% rate compared with the glipizide group, which had a 40.8% rate of patients with > 1 hypoglycemic episode.
A study examined the efficacy and safety of dapagliflozin in combination with and also vs a DPP-4 inhibitor with metformin background therapy. The study looked at add-ons of saxagliptin plus dapagliflozin vs saxagliptin or dapagliflozin added alone.11 This was a randomized, double-blind, 24-week study with patients aged > 18 years with inadequate glucose control A1c > 8.0% and < 12.0%. Patients had to be on a stable dose of metformin > 1,500 mg/d for at least 8 weeks. Patients were randomized 1:1:1 to receive either saxagliptin 5 mg/d and dapagliflozin 10 mg/d plus metformin, saxagliptin 5 mg/d and placebo plus metformin, or dapagliflozin 10 mg/d and placebo plus metformin.
The patients had a mean age of 54 years and a mean duration of T2DM of 7.6 years. The mean baseline A1c was 8.94%. The addition of saxagliptin plus dapagliflozin to metformin resulted in significantly greater A1c reduction compared with saxagliptin plus metformin or dapagliflozin plus metformin from baseline A1c levels: -1.47%, -0.88%, -1.2%, respectively.11
Dapagliflozin was also studied in combination with insulin to determine the safety and efficacy in this setting.12 This double-blind, placebo-controlled, parallel-group trial had an initial study period of 24 weeks followed by an extension period totaling 104 weeks. At 48 weeks, patients on dapagliflozin 5 mg were switched to 10 mg. Outcomes over 104 weeks were changed from baseline A1c, insulin dose, and body weight. Up-titration of insulin was permitted if at least 3 self-monitored blood glucose readings from the 7 days prior to the study visit were > 240 mg/dL up to week 12; > 220 mg/dL between weeks 12 and 24; > 178 mg/dL or if A1c was > 8% between weeks 24 and 48. Between weeks 52 and 65, insulin titrated up was allowed if A1c was > 7.5% and between weeks 78 and 104 if A1c was > 7%. Insulin could be titrated down if ≥ 2 self-monitored blood glucose readings were < 68 mg/dL.
The study group had a T2DM diagnosis for 13.6 years and a mean duration of insulin therapy for about 6 years. The mean daily insulin dose was 77.1 IU. The mean baseline A1c was 8.5%. At 104 weeks, the differences from placebo in A1c adjusted mean change from baseline were -4.0% (P = .0002) and -0.4% (P = .0007) in the dapagliflozin 5 mg (switched to 10 mg at week 48) and 10 mg groups, respectively. Insulin requirements increased progressively in the placebo group +18.3 IU/d at 104 weeks. Insulin requirements stayed stable over 104 weeks in the dapagliflozin groups. Body weight increased in the placebo group, whereas it decreased in the dapagliflozin groups. At 104 weeks, the weight changes from baseline in placebo, dapagliflozin 5 mg/10 mg, and dapagliflozin 10 mg were -0.99 kg, -1.03 kg (P < .001), and -1.5 kg (P < .0001), respectively. The frequency of ≥ 1 minor or major episodes of hypoglycemia was fairly balanced across placebo, dapagliflozin 5 mg/10 mg, and dapagliflozin 10 mg at 61.9%, 61.3%, and 60.7%, respectively.
Adverse Effects and Precautions
Dapagliflozin was generally well tolerated. In the trial of dapagliflozin compared with glipizide in the setting of metformin background therapy, AEs led to discontinuation rates of 9.1% vs 5.9%, respectively. Serious AEs were lower in the dapagliflozin group compared with glipizide at 8.6% vs 11.3%, respectively.10
Limitations for use of dapagliflozin are the treatment of T1DM or the treatment of DKA. Dapagliflozin should not be initiated in patients with an eGFR < 60 mL/min/1.73 m2. No dose adjustment is needed in patients with mild renal impairment (eGFR of ≥ 60 mL/ min/1.73 m2), and dapagliflozin should be discontinued when eGFR is persistently < 60 mL/min/1.73 m2. In the clinical trials, there was an imbalance in the number of bladder cancer reported with dapagliflozin compared with placebo. Across 22 clinical studies, newly diagnosed cases of bladder cancer were reported in 10 of 6,045 patients (0.17%) treated with dapagliflozin and 1 of 3,512 patients (0.03%) treated with placebo or a comparator.9 There were too few cases to determine whether the emergence of these events is related to dapagliflozin. Product labeling states dapagliflozin should not be used in patients with active bladder cancer and should be used with caution in those with a prior history of bladder cancer.
Empaglifilozin
Empagliflozin is the most recently FDA-approved SGLT2 inhibitor (August 2014) for improved glycemic control in T2DM. It has the highest SGLT2 selectivity: > 2,500-fold selectivity for SGLT2 over SGLT1.13 Empagliflozin regulates blood glucose levels by increased UGE, independent of endogenous insulin secretion. It is associated with modest reductions in body weight, visceral adiposity, and systolic BP.
Empagliflozin is available in 10-mg and 25-mg tablets, with a recommended initial dose of 10 mg daily.13 Dosing adjustments are not required for geriatric patients or for those patients with hepatic impairment.14 The use of empagliflozin is contraindicated in patients with eGFR < 45 mL/min/1.73 m2 or for those whose eGFR declines to < 45 mL/min/1.73 m2 during therapy.15,16 Empagliflozin has a pregnancy risk category C. Drug transference during lactation is unknown; therefore, empagliflozin during breast-feeding is not recommended. It is also available in a fixed-dose combination with metformin.
Empagliflozin has been studied alone and in combination therapy with other oral antidiabetic drugs as well as insulin therapy. Metformin, pioglitazone, sitagliptin, and linagliptin have been studied in combination with empagliflozin with sustained glycemic improvement without significantly increased risk of hypoglycemia.17-21 Empagliflozin/linagliptin combination was recently approved after phase 3 trials demonstrated 62% of patients achieved an A1c value < 7% on the 25/5-mg dose at 24 weeks.21 Empagliflozin, coadministered with multiple daily injections of insulin (MDI), has been shown to safely improve glycemic control and reduce total daily insulin requirements without an increased risk of hypoglycemia.22 Currently, it is not approved for use in patients with T1DM, but phase 3 trials are ongoing.
Pharmacokinetics of empagliflozin among healthy volunteers paralleled those of people with T2DM with rapid absorption. The plasma glucose lowering effect of empagliflozin was evident after the first dose and became more pronounced with treatment duration. The AUC and Cmax were dose-proportional over a range of empagliflozin doses in a single rising dose study, with maximum UGE of 90.8 g in healthy volunteers reached at the 400-mg dose.23 The Tmax was 1.5 to 2.1 hours after dosing, comparable to dapagliflozin.24 Steady state with once-daily dosing is reached by day 5 with t1/2 range of 10 to 19 hours.24-27 Plasma levels of empagliflozin declined in a biphasic pattern, with a rapid distribution phase and a slower elimination phase. Total urine volume did not differ significantly in the empagliflozin group compared with placebo.
Healthy subjects treated with placebo or empagliflozin had comparable plasma glucose concentrations. Patients with T2DM demonstrated a decrease in mean daily glucose of -37.0 mg/dL for the 10-mg dose compared with -13.5 mg/dL for placebo. Doses up to 10 mg were found to inhibit renal tubular reabsorption up to 40%, and higher doses inhibit up to 60% of filtered glucose.22-26 Empagliflozin has been shown to have similar efficacy independent of food.27 The pharmacodynamic response declines with increasing renal impairment in SGLT2 inhibitors. Empagliflozin has been associated with a decline from UGE of 97.6 g in normal renal function to 18.2 g in severe renal impairment.
Phase 1 studies of healthy male volunteers initially elucidated empagliflozin’s therapeutic potential of dosedependent increases in UGE and associated reduction in A1c with daily administration, respectively.24,28 Single rising dose studies further defined the linear pharmacokinetics and excellent tolerability of empagliflozin.23 Heise and colleagues demonstrated in 2 separate studies with multiple oral doses (2.5 mg, 10 mg, 25 mg, and 100 mg) of empagliflozin in people with T2DM similar pharmacokinetics and efficacy in UGE, tolerability, and reduction in plasma glucose.24
Ferrannini and colleagues, in a 12-week phase 2 study, demonstrated a statistically significant reduction in A1c of 0.5% and 0.6% with 10-mg and 25-mg doses, respectively, as well as a universal body weight decline by 2 kg.29
Clinical Efficacy Trials
A phase 3 trial of 1,549 randomized people with T2DM (aged > 18 years with baseline A1c of 7%-10%), comparing empagliflozin and glimepiride as an add-on to metformin, demonstrated noninferiority at 52 weeks and statistically significant superiority for the empagliflozin group at 104 weeks.30 The adjusted mean difference in change from baseline in A1c with empagliflozin vs glimepiride at week 104 was -0.11% (95% confidence interval, -0.19 to -0.02; P = .0153 for superiority). Despite 39% of both groups achieving a A1c < 7% at week 52, the empagliflozin group had significantly less hypoglycemia (2% vs 24%). In the body composition substudy, empagliflozin demonstrated a significant reduction of 3 kg compared with an increase of slightly over 1 kg in the glimepiride group at week 104. The majority of the empagliflozin-associated weight loss was found to be a reduction in fat mass.
An international phase 2B randomized, controlled, open-label extension study of 659 people with T2DM (aged > 18 and < 79 years with a body mass index (BMI) < 40 kg/m2 and baseline A1c of 7%-10%) described the long-term safety and efficacy of empagliflozin monotherapy in combination with metformin as compared with sitagliptin monotherapy in combination with metformin. At week 90, the changes from baseline A1c were -0.34%/ -0.47% in the empagliflozin 10 mg/25 mg monotherapy group, -0.34%/0.63% in the empagliflozin 10 mg/25 mg/ metformin combination, -0.56% with metformin monotherapy, and -0.40% with sitagliptin/metformin combination.31 These data provided evidence that empagliflozin had sustained weight loss effects (-2.2 kg to -4.0 kg).
Empagliflozin monotherapy was studied with sitagliptin as an active comparator in a phase 3 trial of 899 people with T2DM (aged > 18 years with baseline A1c of 7%-10%), with the primary endpoint of change in baseline in A1c at week 24. Both the 10-mg and 25-mg dose sof empagliflozin were associated with greater reductions in A1c from baseline (-0.66%, -0.77%) at week 24 than that of sitagliptin (-1.04%). Additionally, both doses of empagliflozin were associated with greater reductions in bodyweight (-2.26 kg and -2.48 kg) as compared with sitagliptin 100 mg (+0.18 kg).20
Empagliflozin’s efficacy and safety in combination with MDI insulin as well as add-on to basal insulin was tested in 2 separate studies.23,32 People with T2DM (mean age 58.8 years, A1c 8.2%) on basal insulin were randomized to empagliflozin (10 mg or 25 mg) or placebo for 78 weeks with a constant basal insulin dose for the first 18 weeks and titration allowed thereafter. Empagliflozin was found to significantly reduce A1c at both 18 and 78 weeks (-0.48% and -0.64% for 10-mg/25-mg doses, respectively, vs -0.02% for placebo) and insulin dose at week 78 vs placebo (-8.8 IU on empagliflozin 10 mg and -11.2 IU on empagliflozin 25 mg).32 Similar rates of hypoglycemia were reported (36.1% of patients on empagliflozin and 35.3% on placebo)
Furthermore, empagliflozin was studied in obese people with uncontrolled, insulin-dependent T2DM (A1c 8.3%, BMI 34.8 kg/m2) on MDI insulin (average 92 IU/d) over a 52-week study.22 Patients were randomized to once-daily empagliflozin 10 mg, empagliflozin 25 mg, or placebo. The study demonstrated improved glycemic control (A1c reduction of -1.27% and -1.18% on empagliflozin 10 mg and 25 mg doses, respectively, compared with -0.81% on placebo) with lower insulin doses (-9 to -11 IU/d) and weight loss (-1.95 kg and -2.04 kg on empagliflozin 10-mg and 25-mg doses, respectively, compared with 0.44 kg on placebo.) There was no increased risk of hypoglycemia noted.
Adverse Effects and Precautions
Empagliflozin is generally well tolerated with low occurrence of AEs. Adverse effects reported in the pooled empagliflozin phase 3 studies were mild to moderate. Serious AEs reported were higher in the placebo group as compared with those of the empagliflozin subjects but did not result in study discontinuation.33
Drug-drug studies of empagliflozin co-administered with other commonly prescribed medication in T2DM showed very limited, if any, interaction. Empagliflozin had no effect on the pharmacokinetics of warfarin or on its anticoagulant activity; therefore, these 2 drugs were deemed safe to be co-administered. No AEs were reported when combining empagliflozin with the loop diuretic hydrochlorothiazide.33 Due to the mode of action of SGLT2 inhibitors, osmotic diuresis may result in a modest reduction in BP.34
The FDA recently released a warning that SGLT2 inhibitors may be associated with a higher risk of developing DKA.35 The FDA Adverse Event Reporting System identified 20 cases of DKA in patients treated with SGLT2 inhibitors from March 2013 to June 6, 2014. Diabetic ketoacidosis is typically accompanied by levels of ketone bodies > 3,000 μmol/L and develops almost exclusively in states of absolute insulin deficiency. The highest level of ketone bodies observed in patients receiving 25 mg of empagliflozin was 1,449 μmol/L compared with a mean of 1,300 μmol/L in a nondiabetic overnight fast.36 Therefore, it is unlikely that the modest empagliflozin-induced ketosis would increase the risk of developing DKA in the absence of absolute insulin deficiency or extreme ketogenic diet.37 Physiologic explanation at the present time is not clear.
Clinical Application
The SGLT2 inhibitors are the latest of 14 classes of drugs approved to treat T2DM. This class offers many beneficial characteristics besides blood sugar lowering. The drugs lower systolic BP, induce weight loss through diuresis of glucose (calories), and carry low risks for hypoglycemia. The American Association of Clinical Endocrinologists recently published their 2015 diabetes management algorithm.38 In this algorithm, they recognized metformin as first-line therapy along with diet and exercise intensification for the treatment of T2DM.
The glucagon-like peptide-1 (GLP-1) analogs and SGLT2 inhibitors are recognized as plausible second-line drugs. They go together well with metformin, providing powerful additional A1c-lowering effects while inducing weight loss without hypoglycemia. Because all GLP-1 analogs are currently available only in injectable forms, the SGLT2 inhibitors offer the additional advantage of being available in pill forms.
Conclusions
All 3 of the current SGLT2 inhibitors are effective tools for treating T2DM. The 3 drugs share many similar traits, and efficacy is generally similar. Empagliflozin has the highest SGLT2 selectivity, > 2,500-fold selectivity for SGLT2 over SGLT1. However, canagliflozin has mild SGLT1 activity, which may offer additional benefits with regards to attenuating PPGE excursions by delaying intestinal glucose absorption (Table).
The SGLT2 inhibitor class had been shown to be effective when used as monotherapy as well as in combination with other oral antidiabetic medications. All 3 SGLT2 inhibitors have also been shown to be effective in combination with insulin and have similar efficacy in these clinical settings (eTable).
All 3 SGLT2 inhibitors are generally well tolerated. There is less hypoglycemia compared with sulfonylureas. However, when the SGLT2 drugs are combined with drugs that can cause hypoglycemia, such as sulfonylureas and insulin, patients must be monitored for hypoglycemia, and titrating down the sulfonylurea or insulin may be necessary. Genital mycotic infections and urinary tract infections in men and woman are common AEs with SGLT2 drugs. Patients must be advised of these possible AEs, and treatment should be prompt if these AEs occur. Because of the mild osmotic diuresis, patients should be reminded to keep well hydrated. SGLT2s have a very mild effect on increasing low-density lipoprotein cholesterol (LDL-C), so care should be taken to ensure that patients’ LDL-C stays at goal.
The FDA recently released a warning that SGLT2 inhibitors may be associated with a higher risk of developing DKA. Although these reports are rare, clinicians should be vigilant. The FDA has suggested that patients should pay close attention for any signs of DKA and seek medical attention immediately if they experience difficulty breathing, nausea, vomiting, abdominal pain, confusion, and unusual fatigue or sleepiness.35
There are subtle differences in the eGFR thresholds for the use of the 3 drugs. It should be kept in mind that with all 3 drugs, the efficacy decreases as the eGFR decreases. It is recommended not to initiate canagliflozin if the patient’s eGFR is < 45 mL/min/1.73 m2. Dapagliflozin should not be initiated in patients with an eGFR < 60 mL/min/1.73 m2. Empagliflozin should not be initiated in patients with an eGFR < 45 mL/min/1.73 m2.
The SGLT2 inhibitors are useful tools to lower blood glucose levels in people with T2DM. They can be used as monotherapy or in combination. They also cause weight reduction. Thus, their unique mechanism of action is complementary to the other oral antidiabetic medications and insulin, so a wide variety of patients can benefit from this class.
Author disclosures
Dr. Nguyen is affiliated with the Astra Zeneca Speakers Bureau and Janssen Pharmaceutical Speakers Bureau. Dr. Plodkowski is a Janssen Pharmaceutical consultant. The remaining authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
1. Ehrenkranz JR, Lewis NG, Kahn CR, Roth J. Phlorizin: a review. Diabetes Metab Res Rev. 2005;21(1):31-38.
2. Polidori D, Sha S, Mudaliar S, et al. Canagliflozin lowers postprandial glucose and insulin by delaying intestinal glucose absorption in addition to increasing urinary glucose excretion: results of a randomized, placebo-controlled study. Diabetes Care. 2013;36(8):2154-2161.
3. Sha S, Polidori D, Farrell, et al. Pharmacodynamic differences between canagliflozin and dapagliflozin: results of a randomized, double-blind, crossover study. Diabetes Obes Metab. 2015;17(2):188-197.
4. Yamout H, Perkovic V, Davies M, et al. Efficacy and safety of canagliflozin in patients with type 2 diabetes and stage 3 nephropathy. Am J Nephrol. 2014;40(1):64-74.
5. Leiter LA, Yoon KH, Arias P, et al. Canagliflozin provides durable glycemic improvements and body weight reduction over 104 weeks versus glimepiride in patients with type 2 diabetes on metformin: a randomized, double-blind, phase 3 study. Diabetes Care. 2015;38(3):355-364.
6. Lavalle-González FJ, Januszewicz A, Davidson J, et al. Efficacy and safety of canagliflozin compared with placebo and sitagliptin in patients with type 2 diabetes on background metformin monotherapy: a randomised trial. Diabetologia. 2013;56(12):2582-2592.
7. Neal B, Perkovic V, de Zeeuw D, et al; CANVAS Trial Collaborative Group. Efficacy and safety of canagliflozin, an inhibitor of sodium-glucose cotransporter 2, when used in conjunction with insulin therapy in patients with type 2 diabetes. Diabetes Care. 2015;38(3):403-411.
8. INVOKANA [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2013.
9. FARXIGA [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2015.
10. Nauck MA, Del Prato S, Meier JJ, et al. Dapagliflozin versus glipizide as add-on therapy in patients with type 2 diabetes who have inadequate glycemic control with metformin: a randomized, 52-week, double-blind, active-controlled noninferiority trial. Diabetes Care. 2011;34(9):2015-2022.
11. Rosenstock J, Hansen L, Zee P, et al. Dual Add-on therapy in type 2 diabetes poorly controlled with metformin monotherapy: a randomized double-blind trial of saxagliptin plus dapagliflozin addition versus single addition of saxagliptin or dapagliflozin to metformin. Diabetes Care. 2015;38(3):376-383.
12. Wilding JP, Woo V, Rohwedder K, Sugg J, Parikh S; Dapagliflozin 006 Study Group. Dapagliflozin in patients with type 2 diabetes receiving high doses of insulin: efficacy and safety over 2 years. Diabetes Obes Metab. 2014;16(2):124-136.
13. Heise T, Seewaldt-Becker E, Macha S, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics following 4 weeks’ treatment with empagliflozin once daily in patients with type 2 diabetes. Diabetes Obes Metab. 2013;15(7):613-621.
14. Macha S, Rose P, Mattheus M, et al. Pharmacokinetics, safety and tolerability of empagliflozin, a sodium glucose cotransporter 2 inhibitor, in patients with hepatic impairment. Diabetes Obes Metab. 2014;16(2):118-123.
15. Barnett A, Mithal A, Manassie J, et al; EMPA-REG RENAL trial investigators. Efficacy and safety of empagliflozin added to existing antidiabetes treatment in patients with type 2 diabetes and chronic kidney disease: a randomised, doubleblind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2(5):369-384.
16. Macha S, Mattheus M, Halabi A, Pinnetti S, Woerle HJ, Broedl UC. Pharmacokinetics, pharmacodynamics and safety of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, in subjects with renal impairment. Diabetes Obes Metab. 2014;16(3):215-222.
17. Häring HU, Merker L, Seewaldt-Becker E, et al; EMPA-REG MET Trial Investigators.
Empagliflozin as add-on to metformin in patients with type 2 diabetes: a 24-week, randomized, double-blind, placebo-controlled trial. Diabetes Care. 2014;37(6):1650-1699.
18. Ridderstråle M, Svaerd R, Zeller C, Kim G, Woerle HJ, Broedi UC; EMPA-REG H2H-SU trial investigators. Rational, design and baseline characteristics of a 4-year (208-week) phase III trial of empagliflozin, an SGLT2 inhibitor, versus glimepiride as add-on to metformin in patients with type 2 diabetes mellitus with insufficient glycemic control. Cardiovasc Diabetol. 2013;12:129.
19. Kovacs CS, Seshiah V, Swallow R, et al; EMPA-REG PIO trial investigators. Empagliflozin improves glycaemic and weight control as add-on therapy to pioglitazone or pioglitazone plus metformin in patients with type 2 diabetes: a 24-week, randomized, placebo-controlled trial. Diabetes Obes Metab. 2014;16(2):147-158.
20. Roden M, Weng J, Eilbracht J, et al; EMPA-REG MONO trial investigators. Empagliflozin monotherapy with sitagliptin as an active comparator in patients with type 2 diabetes: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2013;1(3):208-219.
21. Friedrich C, Metzmann K, Rose P, Mattheus M, Pinnetti S, Woerle HJ. A randomized, open-label, crossover study to evaluate the pharmacokinetics of empagliflozin and linagliptin after coadministration in healthy male volunteers. Clin Ther. 2013;35(1):A33-A42.
22. Rosenstock J, Jelaska A, Frappin G, et al; EMPA-REG MDI Trial Investigators. Improved glucose control with weight loss, lower insulin doses, and no increased hypoglycemia with empagliflozin added to titrated multiple daily injections of insulin in obese inadequately controlled type 2 diabetes. Diabetes Care. 2014;37(3):1815-1823.
23. Seman L, Macha S, Nehmiz G, et al. Empagliflozin (BI 10773), a potent and selective SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin Pharmacol Drug Dev. 2013;2(2):152-161.
24. Heise T, Seman L, Macha S, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple rising doses of empagliflozin in patients with type 2 diabetes mellitus. Diabetes Ther. 2013;4(2):331-345.
25. Inzucchi SE, Zinman B, Wanner C, et al. SGLT-2 inhibitors and cardiovascular risk: proposed pathways and review of ongoing outcome trials. Diab Vasc Dis Res. 2015;12(2):90-100.
26. Scheen AJ. Pharmacokinetic and pharmacodynamic profile of empagliflozin, a sodium glucose co-transporter 2 inhibitor. Clin Pharmacokinet. 2014;53(3):213-225.
27. Macha S, Jungnik A, Hohl K, Hobson D, Salsali A, Woerle HJ. Effect of food on the pharmacokinetics of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, and assessment of dose proportionality in healthy volunteers. Int J Clinical Pharmacol Therapy. 2013;51(11):873-879.
28. Sarashina A, Koiwai K, Seman LJ, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of single doses of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, in healthy Japanese subjects. Drug Metab Pharmacokinet. 2013;28(3):213-219.
29. Ferrannini E, Seman L, Seewaldt-Becker E, Hantel S, Pinnetti S, Woerle HJ. A phase IIb, randomized, placebo-controlled study of the SGLT2 inhibitor empagliflozin in patients with type 2 diabetes. Diabetes Obes Metab. 2013;15(8):721-728.
30. Ridderstråle M, Andersen KR, Zeller C, Kim G, Woerle HJ, Broedl UC; EMPAREG H2H-SU trial investigators. Comparison of empagliflozin and glimepiride as add-on to metformin in patients with type 2 diabetes: a 104-week randomised, active-controlled, double-blind, phase 3 trial. Lancet Diabetes Endocrinol. 2014;2(9):691-700.
31. Ferrannini E, Berk A, Hantel S, et al. Long-term safety and efficacy of empagliflozin, sitagliptin, and metformin: an active-controlled, parallel-group, randomized, 78-week open-label extension study in patients with type 2 diabetes. Diabetes Care. 2013;36(12):4015-4021.
32. Rosenstock J, Jelaska A, Kim G, et al. Empagliflozin as add-on to basal insulin for 78 weeks improves glycemic control with weight loss in insulin-treated (T2DM) [Abstract 1102-P]. Diabetes. 2013;62(suppl 1):A285.
33. JARDIANCE [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals,
Inc.; 2015.
34. Häring HU, Merker L, Seewaldt-Becker E, et al; EMPA-REG METSU Trial Investigators. Empagliflozin as add-on to metformin plus sulfonylurea in patients with type 2 diabetes: a 24-week, randomized, double-blind, placebo-controlled trial. Diabetes Care. 2013;36(11):3396-3404.
35. FDA Drug Safety Communication: FDA Warns that SGLT2 inhibitors for diabetes may result in a serious condition of too much acid in the blood. U.S. Food and Drug Administration Website. http://www.fda.gov/Drugs/DrugSafety/ucm446845.htm. Updated May 19, 2015. Accessed September 23, 2015.
36. Nishimura R, Tanaka Y, Koiwai K, et al. Effect of empagliflozin monotherapy on postprandial glucose and 24-hour glucose variability in Japanese patients with type 2 diabetes mellitus: a randomized, double-blind, placebo-controlled, 4-week study. Cardiovasc Diabetol. 2015;14:11.
37. Tikkanen I, Narko K, Zeller C, et al. Empagliflozin reduces blood pressure in patients
with type 2 diabetes and hypertension. Diabetes Care. 2015;38(3):420-428.
38. Garber AJ, Abrahamson MJ, Barzilay JI, et al. AACE/ACE comprehensive diabetes management algorithm 2015. Endocr Pract. 2015;21(4):438-447.
Over the past 2 decades, the treatment of type 2 diabetes mellitus (T2DM) has been an evolving science. With therapeutic advances, the prevalence of catastrophic complications such as amputations, renal failure requiring dialysis, and blindness due to retinopathy have significantly declined. Developed drugs have successfully met treatment goals; however, they are often associated with a higher risk of hypoglycemia and weight gain. Now that better glucose control is possible, the science of diabetes care continues to evolve. Newly developed drugs should control glucose without significant hypoglycemia and also promote weight reduction. The sodium-glucose transport protein 2 (SGLT2) inhibitor drug class has these characteristics, and the novel mechanism of action complements older medications used to treat T2DM.
Phlorizin is a plant-based compound originally discovered in 1935 when it was derived from the bark of apple trees.1 It is a naturally occurring botanical glucoside and is fairly nonselective between SGLT1 and SGLT2. Due to its poor bioavailability and its degradation in the gastrointestinal tract, it was not an ideal drug candidate in humans.
Canagliflozin
Canagliflozin is a SGLT2 inhibitor and a low-potency SGLT1 inhibitor. It was the first SGLT2 inhibitor approved by the FDA (March 2013) to be used with diet and exercise to improve glycemic control in adults with T2DM. The recommended starting dose is 100 mg once daily for patients who have an estimated glomerular filtration rate (eGFR) > 60 mL/min/1.73m2 and can be increased to 300 mg once daily. It is also available in a fixed-dose combination with metformin. Canagliflozin SGLT-2 inhibition leads to increased glycosuria and osmotic diuresis that lowers plasma glucose concentrations. Lower blood pressure (BP) is likely an effect of the osmotic diuresis. Increased urinary excretion of glucose also leads to a loss of calories and weight loss. It was studied alone and in combination with metformin, sulfonylurea, pioglitazone, and insulin therapy.
The pharmacokinetics of canagliflozin is similar in healthy subjects and patients with T2DM. Peak plasma concentrations (Cmax) and area under the cover (AUC) of canagliflozin increased in a dose-proportional manner from 50 mg to 300 mg. Following single-dose oral administration of 100 mg and 300 mg of canagliflozin, time to Cmax (Tmax) of canagliflozin occurs within 1 to 2 hours postdose. The apparent terminal half-life (t1/2) was 10.6 hours and 13.1 hours for the 100 mg and 300 mg doses, respectively. Steady state was reached after 4 to 5 days of once-daily dosing with canagliflozin 100 mg to 300 mg. Glucuronidation is the major metabolic pathway. There is balanced renal and biliary excretion of metabolites, and there are no active metabolites.
Following oral doses of canagliflozin in patients with T2DM, dose-dependent decreases were seen in the renal threshold for glucose (RTG). From a starting value of about 240 mg/dL, the 300-mg dose suppressed the mean (RTG) to about 70 to 90 mg/dL in T2DM in phase 1 studies. The reduction in RTG led to increase in urinary excretion of glucose of about 100 g/d.
In addition to renal SGLT2 inhibition leading to increased urinary glucose excretion (UGE), canagliflozin has been shown to lower postprandial glucose excursion (PPGE) and insulin concentrations by delaying intestinal glucose absorption.2 A study was done in 20 healthy subjects who received either placebo or canagliflozin 300 mg 20 minutes before a 600 kcal mixed-meal tolerance test. Compared with placebo, canagliflozin reduced PPGE and insulin excursions (0-2 h) AUC by 35% and 43%, respectively (P < .001 for both). This may present a difference between canagliflozin and the other SGLT2 inhibitors.
Because of the potential differences due to canagliflozin’s inhibition of intestinal SGLT1, the pharmacodynamic differences between canagliflozin and dapagliflozin were studied.3 The randomized, double-blind, crossover study consisted of 54 subjects. The subjects received the maximum approved doses of canagliflozin 300 mg or dapagliflozin 10 mg a day. Each group was treated with the study drug for 2 days, and then a 600 kcal mixed-meal tolerance test was performed. The results of the PPGE 0- to 2-hour AUC analysis showed 3.66 mmol*h/L with canagliflozin 300 mg and 4.08 mmol*h/L with dapagliflozin 10 mg. There was a difference of 0.42 (P = .0122), which was a 10.3% reduction in AUC PPGE by canagliflozin compared with dapagliflozin.
Canagliflozin has been studied in patients with T2DM and stage 3 nephropathy. Data were pooled from 4 randomized, placebo-controlled, phase 3 studies in which subjects had baseline eGFR > 30 to < 60 mL/min/1.73 m2.4 In the setting of decreased eGFR associated with stage 3 chronic kidney disease, subjects treated with canagliflozin 100 mg and canagliflozin 300 mg had placebo-subtracted reductions in hemoglobin A1c (A1c) of -0.38% and -0.47%, respectively, and placebosubtracted reduction in weight of -1.6% and -1.9%, respectively. Decreases in eGFR were seen at week 6 but trended toward baseline over time with a mean change in eGFR of 0.7, -1.7, -2.2 mL/min/1.73 m2 for placebo, canagliflozin 100 mg, and canagliflozin 300 mg, respectively.
Clinical Efficacy Trials
Canagliflozin was studied as add-on therapy to metformin and compared with glimepiride (a sulfonylurea).5 The randomized, double-blind study included 1,450 subjects for a core study period of 52 weeks followed by a 52-week extension. Eligible subjects were aged > 18 and < 80 years, A1c > 7% and < 9.5%, and were receiving metformin > 2,000 mg/d or > 1,500 mg/d if unable to tolerate a higher dose. The study groups were canagliflozin 100 mg, canagliflozin 300 mg, and glimepiride, and baseline A1c measurements were 7.78%, 7.79%, and 7.83%, respectively. The glimepiride was titrated up if > 50% of fasting blood glucose measurements were > 108 mg/dL with no hypoglycemic events in the previous 2 weeks.
Over 104 weeks, canagliflozin 100 mg and 300 mg and glimepiride reduced A1c from mean baseline values by -0.65%, -0.74%, and -0.55%, respectively, and the proportions of patients achieving A1c < 7% at week 104 was 42.5%, 50.2%, and 43.9%, respectively. Weight fell over 104 weeks with canagliflozin 100 mg (-4.1%, -3.6 kg) and canagliflozin 300 mg (-4.2%, -3.6 kg). In contrast, glimepiride showed weight increase (0.9%, 0.8 kg). Documented hypoglycemia episodes were lower in canagliflozin 100 mg and 300 mg than with glimepiride (6.8%, 8.2%, and 40.9%, respectively).
A study was undertaken to compare canagliflozin with the dipeptidyl peptidase-4 (DPP-4) inhibitor sitagliptin in patients with T2DM on background therapy of metformin.6 This randomized, double-blind trial studied subjects aged > 18 and < 80 years with inadequate glucose control A1c > 7% and < 10.5%. Subjects received metformin > 2,000 mg/d or > 1,500 mg/d if unable to tolerate a higher dose. At week 52, canagliflozin showed noninferiority to sitagliptin, with both drugs lowering A1c by 0.73%. Canagliflozin 300 mg showed superiority to sitagliptin with -0.88% change in A1c. Both canagliflozin 100 mg and 300 mg were superior to sitagliptin 100 mg in weight reduction: -3.8%, -4.2%, and -1.3%, respectively. Genital mycotic infections were higher in the canagliflozin groups. Rates for mycotic infections for sitagliptin 100 mg, canagliflozin 100 mg, and canagliflozin 300 mg were 1.2%, 5.2%, and 2.4% in men, respectively, and 2.6%, 11.3%, and 9.9% in women, respectively.
Canagliflozin was also studied in combination with insulin to determine efficacy and safety in this setting.7 Subjects were randomized to receive placebo, canagliflozin 100 mg, or canagliflozin 300 mg. Subjects had a mean baseline A1c of 8.3%. The median daily insulin dose was 60 IU, and most individuals were using basal/bolus regimens. The primary endpoint was 18 weeks of therapy, and A1c was lowered 0.62% (P < .001) with canagliflozin 100 mg and 0.73% with canagliflozin 300 mg compared with placebo. Weight decreased 1.9% (P < .001) with canagliflozin 100 mg and 2.4% (P < .001) with canagliflozin 300 mg compared with placebo.
Adverse Effects and Precautions
Canagliflozin adverse effects (AEs) were generally low. In the aforementioned 104-week study comparing canagliflozin 100 mg, canagliflozin 300 mg, and glimepiride, AEs leading to discontinuation were low at 6.2%, 9.5%, and 7.3%, respectively. Serious AEs were lower in the canagliflozin 100 mg and 300 mg groups compared with glimepiride at 9.7%, 9.7%, 14.3%, respectively.5
Limitations for use of canagliflozin are T1DM and diabetic ketoacidosis (DKA). In mild renal impairment, there is no dose adjustment in patients with eGFR > 60 mL/min/1.73 m2. In moderate renal impairment (eGFR 45-60 mL/min/1.73 m2), dose is limited to 100 mg once daily. It is recommended not to initiate canagliflozin if eGFR is < 45 mL/min/1.73 m2. Canagliflozin is contraindicated if eGFR is < 30 mL/min/1.73 m2.8
Dapagliflozin
Dapagliflozin is a highly selective SGLT2 inhibitor. It is a 1,400-fold greater inhibitor of SGLT2 vs SGLT1 and was approved in January 2014. It is indicated as an adjunct to diet and exercise to improve glycemic control in adults with T2DM. The recommended starting dose is 5 mg once daily, taken in the morning, with or without food, and the dosage can be increased to 10 mg once daily in patients who require additional glycemic control. Dapagliflozin should not be initiated if eGFR is < 60 mL/min/1.73 m2, and it should be discontinued if eGFR is persistently < 60 mL/min/1.73 m2.
By inhibiting SGLT2, dapagliflozin reduces reabsorption of filtered glucose and lowers the renal threshold for glucose, thereby increasing UGE. Increased glucose secretion also leads to weight reduction. Dapagliflozin has been studied alone and in combination with glipizide, glimepiride, pioglitazone, and a DDP-4 inhibitor and as an add-on to insulin with and without other oral antidiabetic drugs. It is also available in a fixed-dose combination with metformin.
Following oral administration of dapagliflozin, the Tmax is usually attained within 2 hours under a fasting state. The Cmax and AUC values increase the dose proportionally with increase in dapagliflozin dose in the therapeutic dose range. The absolute oral bioavailability of dapagliflozin following the administration of a 10-mg dose is 78%. The mean plasma t1⁄2 for dapagliflozin is about 12.9 hours following a single oral dose of 10 mg. In patients with normal renal function, the renal glucose excretion was 85 g per day at maximal dose. In humans, 75% of the dose of dapagliflozin is primarily metabolized through the uridine diphosphate glucuronosyltransferase 1A9 pathway. Dapagliflozin and related metabolites are primarily eliminated via the renal pathway.9
Clinical Efficacy Trials
Dapagliflozin was studied as add-on therapy to metformin with glipizide (a sulfonylurea) as the comparator.10 This 52-week double-blind, multicenter, active-controlled, noninferiority trial randomized 801 patients. Eligible subjects were aged > 18 years with A1c > 6.5% and < 10% and were receiving metformin or metformin and 1 other oral antidiabetic drug up to half-maximal dose for at least 8 weeks. During an 18-week titration period, all patients started dapagliflozin 2.5 mg/d and glipizide 5 mg/d. At 21-day intervals, patients were titrated up to the next dosage level if fasting plasma glucose was > 110 mg/dL. Dapagliflozin could be titrated to 5 mg and then 10 mg per protocol. Glipizide could be titrated to 10 mg or 20 mg per protocol.
The A1c change with dapagliflozin was noninferior to glipizide at week 52. The A1c adjusted mean change from baseline was -0.52 for both dapagliflozin and glipizide. The secondary endpoint was weight change. The glipizide group had a +1.44 kg weight gain, whereas the dapagliflozin group had a -3.22 kg weight loss. The number of patients with ≥ 1 episode of hypoglycemia, either symptomatic or with no symptoms, with blood glucose ≤ 63 mg/dL was assessed. The dapagliflozin group had a 3.5% rate compared with the glipizide group, which had a 40.8% rate of patients with > 1 hypoglycemic episode.
A study examined the efficacy and safety of dapagliflozin in combination with and also vs a DPP-4 inhibitor with metformin background therapy. The study looked at add-ons of saxagliptin plus dapagliflozin vs saxagliptin or dapagliflozin added alone.11 This was a randomized, double-blind, 24-week study with patients aged > 18 years with inadequate glucose control A1c > 8.0% and < 12.0%. Patients had to be on a stable dose of metformin > 1,500 mg/d for at least 8 weeks. Patients were randomized 1:1:1 to receive either saxagliptin 5 mg/d and dapagliflozin 10 mg/d plus metformin, saxagliptin 5 mg/d and placebo plus metformin, or dapagliflozin 10 mg/d and placebo plus metformin.
The patients had a mean age of 54 years and a mean duration of T2DM of 7.6 years. The mean baseline A1c was 8.94%. The addition of saxagliptin plus dapagliflozin to metformin resulted in significantly greater A1c reduction compared with saxagliptin plus metformin or dapagliflozin plus metformin from baseline A1c levels: -1.47%, -0.88%, -1.2%, respectively.11
Dapagliflozin was also studied in combination with insulin to determine the safety and efficacy in this setting.12 This double-blind, placebo-controlled, parallel-group trial had an initial study period of 24 weeks followed by an extension period totaling 104 weeks. At 48 weeks, patients on dapagliflozin 5 mg were switched to 10 mg. Outcomes over 104 weeks were changed from baseline A1c, insulin dose, and body weight. Up-titration of insulin was permitted if at least 3 self-monitored blood glucose readings from the 7 days prior to the study visit were > 240 mg/dL up to week 12; > 220 mg/dL between weeks 12 and 24; > 178 mg/dL or if A1c was > 8% between weeks 24 and 48. Between weeks 52 and 65, insulin titrated up was allowed if A1c was > 7.5% and between weeks 78 and 104 if A1c was > 7%. Insulin could be titrated down if ≥ 2 self-monitored blood glucose readings were < 68 mg/dL.
The study group had a T2DM diagnosis for 13.6 years and a mean duration of insulin therapy for about 6 years. The mean daily insulin dose was 77.1 IU. The mean baseline A1c was 8.5%. At 104 weeks, the differences from placebo in A1c adjusted mean change from baseline were -4.0% (P = .0002) and -0.4% (P = .0007) in the dapagliflozin 5 mg (switched to 10 mg at week 48) and 10 mg groups, respectively. Insulin requirements increased progressively in the placebo group +18.3 IU/d at 104 weeks. Insulin requirements stayed stable over 104 weeks in the dapagliflozin groups. Body weight increased in the placebo group, whereas it decreased in the dapagliflozin groups. At 104 weeks, the weight changes from baseline in placebo, dapagliflozin 5 mg/10 mg, and dapagliflozin 10 mg were -0.99 kg, -1.03 kg (P < .001), and -1.5 kg (P < .0001), respectively. The frequency of ≥ 1 minor or major episodes of hypoglycemia was fairly balanced across placebo, dapagliflozin 5 mg/10 mg, and dapagliflozin 10 mg at 61.9%, 61.3%, and 60.7%, respectively.
Adverse Effects and Precautions
Dapagliflozin was generally well tolerated. In the trial of dapagliflozin compared with glipizide in the setting of metformin background therapy, AEs led to discontinuation rates of 9.1% vs 5.9%, respectively. Serious AEs were lower in the dapagliflozin group compared with glipizide at 8.6% vs 11.3%, respectively.10
Limitations for use of dapagliflozin are the treatment of T1DM or the treatment of DKA. Dapagliflozin should not be initiated in patients with an eGFR < 60 mL/min/1.73 m2. No dose adjustment is needed in patients with mild renal impairment (eGFR of ≥ 60 mL/ min/1.73 m2), and dapagliflozin should be discontinued when eGFR is persistently < 60 mL/min/1.73 m2. In the clinical trials, there was an imbalance in the number of bladder cancer reported with dapagliflozin compared with placebo. Across 22 clinical studies, newly diagnosed cases of bladder cancer were reported in 10 of 6,045 patients (0.17%) treated with dapagliflozin and 1 of 3,512 patients (0.03%) treated with placebo or a comparator.9 There were too few cases to determine whether the emergence of these events is related to dapagliflozin. Product labeling states dapagliflozin should not be used in patients with active bladder cancer and should be used with caution in those with a prior history of bladder cancer.
Empaglifilozin
Empagliflozin is the most recently FDA-approved SGLT2 inhibitor (August 2014) for improved glycemic control in T2DM. It has the highest SGLT2 selectivity: > 2,500-fold selectivity for SGLT2 over SGLT1.13 Empagliflozin regulates blood glucose levels by increased UGE, independent of endogenous insulin secretion. It is associated with modest reductions in body weight, visceral adiposity, and systolic BP.
Empagliflozin is available in 10-mg and 25-mg tablets, with a recommended initial dose of 10 mg daily.13 Dosing adjustments are not required for geriatric patients or for those patients with hepatic impairment.14 The use of empagliflozin is contraindicated in patients with eGFR < 45 mL/min/1.73 m2 or for those whose eGFR declines to < 45 mL/min/1.73 m2 during therapy.15,16 Empagliflozin has a pregnancy risk category C. Drug transference during lactation is unknown; therefore, empagliflozin during breast-feeding is not recommended. It is also available in a fixed-dose combination with metformin.
Empagliflozin has been studied alone and in combination therapy with other oral antidiabetic drugs as well as insulin therapy. Metformin, pioglitazone, sitagliptin, and linagliptin have been studied in combination with empagliflozin with sustained glycemic improvement without significantly increased risk of hypoglycemia.17-21 Empagliflozin/linagliptin combination was recently approved after phase 3 trials demonstrated 62% of patients achieved an A1c value < 7% on the 25/5-mg dose at 24 weeks.21 Empagliflozin, coadministered with multiple daily injections of insulin (MDI), has been shown to safely improve glycemic control and reduce total daily insulin requirements without an increased risk of hypoglycemia.22 Currently, it is not approved for use in patients with T1DM, but phase 3 trials are ongoing.
Pharmacokinetics of empagliflozin among healthy volunteers paralleled those of people with T2DM with rapid absorption. The plasma glucose lowering effect of empagliflozin was evident after the first dose and became more pronounced with treatment duration. The AUC and Cmax were dose-proportional over a range of empagliflozin doses in a single rising dose study, with maximum UGE of 90.8 g in healthy volunteers reached at the 400-mg dose.23 The Tmax was 1.5 to 2.1 hours after dosing, comparable to dapagliflozin.24 Steady state with once-daily dosing is reached by day 5 with t1/2 range of 10 to 19 hours.24-27 Plasma levels of empagliflozin declined in a biphasic pattern, with a rapid distribution phase and a slower elimination phase. Total urine volume did not differ significantly in the empagliflozin group compared with placebo.
Healthy subjects treated with placebo or empagliflozin had comparable plasma glucose concentrations. Patients with T2DM demonstrated a decrease in mean daily glucose of -37.0 mg/dL for the 10-mg dose compared with -13.5 mg/dL for placebo. Doses up to 10 mg were found to inhibit renal tubular reabsorption up to 40%, and higher doses inhibit up to 60% of filtered glucose.22-26 Empagliflozin has been shown to have similar efficacy independent of food.27 The pharmacodynamic response declines with increasing renal impairment in SGLT2 inhibitors. Empagliflozin has been associated with a decline from UGE of 97.6 g in normal renal function to 18.2 g in severe renal impairment.
Phase 1 studies of healthy male volunteers initially elucidated empagliflozin’s therapeutic potential of dosedependent increases in UGE and associated reduction in A1c with daily administration, respectively.24,28 Single rising dose studies further defined the linear pharmacokinetics and excellent tolerability of empagliflozin.23 Heise and colleagues demonstrated in 2 separate studies with multiple oral doses (2.5 mg, 10 mg, 25 mg, and 100 mg) of empagliflozin in people with T2DM similar pharmacokinetics and efficacy in UGE, tolerability, and reduction in plasma glucose.24
Ferrannini and colleagues, in a 12-week phase 2 study, demonstrated a statistically significant reduction in A1c of 0.5% and 0.6% with 10-mg and 25-mg doses, respectively, as well as a universal body weight decline by 2 kg.29
Clinical Efficacy Trials
A phase 3 trial of 1,549 randomized people with T2DM (aged > 18 years with baseline A1c of 7%-10%), comparing empagliflozin and glimepiride as an add-on to metformin, demonstrated noninferiority at 52 weeks and statistically significant superiority for the empagliflozin group at 104 weeks.30 The adjusted mean difference in change from baseline in A1c with empagliflozin vs glimepiride at week 104 was -0.11% (95% confidence interval, -0.19 to -0.02; P = .0153 for superiority). Despite 39% of both groups achieving a A1c < 7% at week 52, the empagliflozin group had significantly less hypoglycemia (2% vs 24%). In the body composition substudy, empagliflozin demonstrated a significant reduction of 3 kg compared with an increase of slightly over 1 kg in the glimepiride group at week 104. The majority of the empagliflozin-associated weight loss was found to be a reduction in fat mass.
An international phase 2B randomized, controlled, open-label extension study of 659 people with T2DM (aged > 18 and < 79 years with a body mass index (BMI) < 40 kg/m2 and baseline A1c of 7%-10%) described the long-term safety and efficacy of empagliflozin monotherapy in combination with metformin as compared with sitagliptin monotherapy in combination with metformin. At week 90, the changes from baseline A1c were -0.34%/ -0.47% in the empagliflozin 10 mg/25 mg monotherapy group, -0.34%/0.63% in the empagliflozin 10 mg/25 mg/ metformin combination, -0.56% with metformin monotherapy, and -0.40% with sitagliptin/metformin combination.31 These data provided evidence that empagliflozin had sustained weight loss effects (-2.2 kg to -4.0 kg).
Empagliflozin monotherapy was studied with sitagliptin as an active comparator in a phase 3 trial of 899 people with T2DM (aged > 18 years with baseline A1c of 7%-10%), with the primary endpoint of change in baseline in A1c at week 24. Both the 10-mg and 25-mg dose sof empagliflozin were associated with greater reductions in A1c from baseline (-0.66%, -0.77%) at week 24 than that of sitagliptin (-1.04%). Additionally, both doses of empagliflozin were associated with greater reductions in bodyweight (-2.26 kg and -2.48 kg) as compared with sitagliptin 100 mg (+0.18 kg).20
Empagliflozin’s efficacy and safety in combination with MDI insulin as well as add-on to basal insulin was tested in 2 separate studies.23,32 People with T2DM (mean age 58.8 years, A1c 8.2%) on basal insulin were randomized to empagliflozin (10 mg or 25 mg) or placebo for 78 weeks with a constant basal insulin dose for the first 18 weeks and titration allowed thereafter. Empagliflozin was found to significantly reduce A1c at both 18 and 78 weeks (-0.48% and -0.64% for 10-mg/25-mg doses, respectively, vs -0.02% for placebo) and insulin dose at week 78 vs placebo (-8.8 IU on empagliflozin 10 mg and -11.2 IU on empagliflozin 25 mg).32 Similar rates of hypoglycemia were reported (36.1% of patients on empagliflozin and 35.3% on placebo)
Furthermore, empagliflozin was studied in obese people with uncontrolled, insulin-dependent T2DM (A1c 8.3%, BMI 34.8 kg/m2) on MDI insulin (average 92 IU/d) over a 52-week study.22 Patients were randomized to once-daily empagliflozin 10 mg, empagliflozin 25 mg, or placebo. The study demonstrated improved glycemic control (A1c reduction of -1.27% and -1.18% on empagliflozin 10 mg and 25 mg doses, respectively, compared with -0.81% on placebo) with lower insulin doses (-9 to -11 IU/d) and weight loss (-1.95 kg and -2.04 kg on empagliflozin 10-mg and 25-mg doses, respectively, compared with 0.44 kg on placebo.) There was no increased risk of hypoglycemia noted.
Adverse Effects and Precautions
Empagliflozin is generally well tolerated with low occurrence of AEs. Adverse effects reported in the pooled empagliflozin phase 3 studies were mild to moderate. Serious AEs reported were higher in the placebo group as compared with those of the empagliflozin subjects but did not result in study discontinuation.33
Drug-drug studies of empagliflozin co-administered with other commonly prescribed medication in T2DM showed very limited, if any, interaction. Empagliflozin had no effect on the pharmacokinetics of warfarin or on its anticoagulant activity; therefore, these 2 drugs were deemed safe to be co-administered. No AEs were reported when combining empagliflozin with the loop diuretic hydrochlorothiazide.33 Due to the mode of action of SGLT2 inhibitors, osmotic diuresis may result in a modest reduction in BP.34
The FDA recently released a warning that SGLT2 inhibitors may be associated with a higher risk of developing DKA.35 The FDA Adverse Event Reporting System identified 20 cases of DKA in patients treated with SGLT2 inhibitors from March 2013 to June 6, 2014. Diabetic ketoacidosis is typically accompanied by levels of ketone bodies > 3,000 μmol/L and develops almost exclusively in states of absolute insulin deficiency. The highest level of ketone bodies observed in patients receiving 25 mg of empagliflozin was 1,449 μmol/L compared with a mean of 1,300 μmol/L in a nondiabetic overnight fast.36 Therefore, it is unlikely that the modest empagliflozin-induced ketosis would increase the risk of developing DKA in the absence of absolute insulin deficiency or extreme ketogenic diet.37 Physiologic explanation at the present time is not clear.
Clinical Application
The SGLT2 inhibitors are the latest of 14 classes of drugs approved to treat T2DM. This class offers many beneficial characteristics besides blood sugar lowering. The drugs lower systolic BP, induce weight loss through diuresis of glucose (calories), and carry low risks for hypoglycemia. The American Association of Clinical Endocrinologists recently published their 2015 diabetes management algorithm.38 In this algorithm, they recognized metformin as first-line therapy along with diet and exercise intensification for the treatment of T2DM.
The glucagon-like peptide-1 (GLP-1) analogs and SGLT2 inhibitors are recognized as plausible second-line drugs. They go together well with metformin, providing powerful additional A1c-lowering effects while inducing weight loss without hypoglycemia. Because all GLP-1 analogs are currently available only in injectable forms, the SGLT2 inhibitors offer the additional advantage of being available in pill forms.
Conclusions
All 3 of the current SGLT2 inhibitors are effective tools for treating T2DM. The 3 drugs share many similar traits, and efficacy is generally similar. Empagliflozin has the highest SGLT2 selectivity, > 2,500-fold selectivity for SGLT2 over SGLT1. However, canagliflozin has mild SGLT1 activity, which may offer additional benefits with regards to attenuating PPGE excursions by delaying intestinal glucose absorption (Table).
The SGLT2 inhibitor class had been shown to be effective when used as monotherapy as well as in combination with other oral antidiabetic medications. All 3 SGLT2 inhibitors have also been shown to be effective in combination with insulin and have similar efficacy in these clinical settings (eTable).
All 3 SGLT2 inhibitors are generally well tolerated. There is less hypoglycemia compared with sulfonylureas. However, when the SGLT2 drugs are combined with drugs that can cause hypoglycemia, such as sulfonylureas and insulin, patients must be monitored for hypoglycemia, and titrating down the sulfonylurea or insulin may be necessary. Genital mycotic infections and urinary tract infections in men and woman are common AEs with SGLT2 drugs. Patients must be advised of these possible AEs, and treatment should be prompt if these AEs occur. Because of the mild osmotic diuresis, patients should be reminded to keep well hydrated. SGLT2s have a very mild effect on increasing low-density lipoprotein cholesterol (LDL-C), so care should be taken to ensure that patients’ LDL-C stays at goal.
The FDA recently released a warning that SGLT2 inhibitors may be associated with a higher risk of developing DKA. Although these reports are rare, clinicians should be vigilant. The FDA has suggested that patients should pay close attention for any signs of DKA and seek medical attention immediately if they experience difficulty breathing, nausea, vomiting, abdominal pain, confusion, and unusual fatigue or sleepiness.35
There are subtle differences in the eGFR thresholds for the use of the 3 drugs. It should be kept in mind that with all 3 drugs, the efficacy decreases as the eGFR decreases. It is recommended not to initiate canagliflozin if the patient’s eGFR is < 45 mL/min/1.73 m2. Dapagliflozin should not be initiated in patients with an eGFR < 60 mL/min/1.73 m2. Empagliflozin should not be initiated in patients with an eGFR < 45 mL/min/1.73 m2.
The SGLT2 inhibitors are useful tools to lower blood glucose levels in people with T2DM. They can be used as monotherapy or in combination. They also cause weight reduction. Thus, their unique mechanism of action is complementary to the other oral antidiabetic medications and insulin, so a wide variety of patients can benefit from this class.
Author disclosures
Dr. Nguyen is affiliated with the Astra Zeneca Speakers Bureau and Janssen Pharmaceutical Speakers Bureau. Dr. Plodkowski is a Janssen Pharmaceutical consultant. The remaining authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
Over the past 2 decades, the treatment of type 2 diabetes mellitus (T2DM) has been an evolving science. With therapeutic advances, the prevalence of catastrophic complications such as amputations, renal failure requiring dialysis, and blindness due to retinopathy have significantly declined. Developed drugs have successfully met treatment goals; however, they are often associated with a higher risk of hypoglycemia and weight gain. Now that better glucose control is possible, the science of diabetes care continues to evolve. Newly developed drugs should control glucose without significant hypoglycemia and also promote weight reduction. The sodium-glucose transport protein 2 (SGLT2) inhibitor drug class has these characteristics, and the novel mechanism of action complements older medications used to treat T2DM.
Phlorizin is a plant-based compound originally discovered in 1935 when it was derived from the bark of apple trees.1 It is a naturally occurring botanical glucoside and is fairly nonselective between SGLT1 and SGLT2. Due to its poor bioavailability and its degradation in the gastrointestinal tract, it was not an ideal drug candidate in humans.
Canagliflozin
Canagliflozin is a SGLT2 inhibitor and a low-potency SGLT1 inhibitor. It was the first SGLT2 inhibitor approved by the FDA (March 2013) to be used with diet and exercise to improve glycemic control in adults with T2DM. The recommended starting dose is 100 mg once daily for patients who have an estimated glomerular filtration rate (eGFR) > 60 mL/min/1.73m2 and can be increased to 300 mg once daily. It is also available in a fixed-dose combination with metformin. Canagliflozin SGLT-2 inhibition leads to increased glycosuria and osmotic diuresis that lowers plasma glucose concentrations. Lower blood pressure (BP) is likely an effect of the osmotic diuresis. Increased urinary excretion of glucose also leads to a loss of calories and weight loss. It was studied alone and in combination with metformin, sulfonylurea, pioglitazone, and insulin therapy.
The pharmacokinetics of canagliflozin is similar in healthy subjects and patients with T2DM. Peak plasma concentrations (Cmax) and area under the cover (AUC) of canagliflozin increased in a dose-proportional manner from 50 mg to 300 mg. Following single-dose oral administration of 100 mg and 300 mg of canagliflozin, time to Cmax (Tmax) of canagliflozin occurs within 1 to 2 hours postdose. The apparent terminal half-life (t1/2) was 10.6 hours and 13.1 hours for the 100 mg and 300 mg doses, respectively. Steady state was reached after 4 to 5 days of once-daily dosing with canagliflozin 100 mg to 300 mg. Glucuronidation is the major metabolic pathway. There is balanced renal and biliary excretion of metabolites, and there are no active metabolites.
Following oral doses of canagliflozin in patients with T2DM, dose-dependent decreases were seen in the renal threshold for glucose (RTG). From a starting value of about 240 mg/dL, the 300-mg dose suppressed the mean (RTG) to about 70 to 90 mg/dL in T2DM in phase 1 studies. The reduction in RTG led to increase in urinary excretion of glucose of about 100 g/d.
In addition to renal SGLT2 inhibition leading to increased urinary glucose excretion (UGE), canagliflozin has been shown to lower postprandial glucose excursion (PPGE) and insulin concentrations by delaying intestinal glucose absorption.2 A study was done in 20 healthy subjects who received either placebo or canagliflozin 300 mg 20 minutes before a 600 kcal mixed-meal tolerance test. Compared with placebo, canagliflozin reduced PPGE and insulin excursions (0-2 h) AUC by 35% and 43%, respectively (P < .001 for both). This may present a difference between canagliflozin and the other SGLT2 inhibitors.
Because of the potential differences due to canagliflozin’s inhibition of intestinal SGLT1, the pharmacodynamic differences between canagliflozin and dapagliflozin were studied.3 The randomized, double-blind, crossover study consisted of 54 subjects. The subjects received the maximum approved doses of canagliflozin 300 mg or dapagliflozin 10 mg a day. Each group was treated with the study drug for 2 days, and then a 600 kcal mixed-meal tolerance test was performed. The results of the PPGE 0- to 2-hour AUC analysis showed 3.66 mmol*h/L with canagliflozin 300 mg and 4.08 mmol*h/L with dapagliflozin 10 mg. There was a difference of 0.42 (P = .0122), which was a 10.3% reduction in AUC PPGE by canagliflozin compared with dapagliflozin.
Canagliflozin has been studied in patients with T2DM and stage 3 nephropathy. Data were pooled from 4 randomized, placebo-controlled, phase 3 studies in which subjects had baseline eGFR > 30 to < 60 mL/min/1.73 m2.4 In the setting of decreased eGFR associated with stage 3 chronic kidney disease, subjects treated with canagliflozin 100 mg and canagliflozin 300 mg had placebo-subtracted reductions in hemoglobin A1c (A1c) of -0.38% and -0.47%, respectively, and placebosubtracted reduction in weight of -1.6% and -1.9%, respectively. Decreases in eGFR were seen at week 6 but trended toward baseline over time with a mean change in eGFR of 0.7, -1.7, -2.2 mL/min/1.73 m2 for placebo, canagliflozin 100 mg, and canagliflozin 300 mg, respectively.
Clinical Efficacy Trials
Canagliflozin was studied as add-on therapy to metformin and compared with glimepiride (a sulfonylurea).5 The randomized, double-blind study included 1,450 subjects for a core study period of 52 weeks followed by a 52-week extension. Eligible subjects were aged > 18 and < 80 years, A1c > 7% and < 9.5%, and were receiving metformin > 2,000 mg/d or > 1,500 mg/d if unable to tolerate a higher dose. The study groups were canagliflozin 100 mg, canagliflozin 300 mg, and glimepiride, and baseline A1c measurements were 7.78%, 7.79%, and 7.83%, respectively. The glimepiride was titrated up if > 50% of fasting blood glucose measurements were > 108 mg/dL with no hypoglycemic events in the previous 2 weeks.
Over 104 weeks, canagliflozin 100 mg and 300 mg and glimepiride reduced A1c from mean baseline values by -0.65%, -0.74%, and -0.55%, respectively, and the proportions of patients achieving A1c < 7% at week 104 was 42.5%, 50.2%, and 43.9%, respectively. Weight fell over 104 weeks with canagliflozin 100 mg (-4.1%, -3.6 kg) and canagliflozin 300 mg (-4.2%, -3.6 kg). In contrast, glimepiride showed weight increase (0.9%, 0.8 kg). Documented hypoglycemia episodes were lower in canagliflozin 100 mg and 300 mg than with glimepiride (6.8%, 8.2%, and 40.9%, respectively).
A study was undertaken to compare canagliflozin with the dipeptidyl peptidase-4 (DPP-4) inhibitor sitagliptin in patients with T2DM on background therapy of metformin.6 This randomized, double-blind trial studied subjects aged > 18 and < 80 years with inadequate glucose control A1c > 7% and < 10.5%. Subjects received metformin > 2,000 mg/d or > 1,500 mg/d if unable to tolerate a higher dose. At week 52, canagliflozin showed noninferiority to sitagliptin, with both drugs lowering A1c by 0.73%. Canagliflozin 300 mg showed superiority to sitagliptin with -0.88% change in A1c. Both canagliflozin 100 mg and 300 mg were superior to sitagliptin 100 mg in weight reduction: -3.8%, -4.2%, and -1.3%, respectively. Genital mycotic infections were higher in the canagliflozin groups. Rates for mycotic infections for sitagliptin 100 mg, canagliflozin 100 mg, and canagliflozin 300 mg were 1.2%, 5.2%, and 2.4% in men, respectively, and 2.6%, 11.3%, and 9.9% in women, respectively.
Canagliflozin was also studied in combination with insulin to determine efficacy and safety in this setting.7 Subjects were randomized to receive placebo, canagliflozin 100 mg, or canagliflozin 300 mg. Subjects had a mean baseline A1c of 8.3%. The median daily insulin dose was 60 IU, and most individuals were using basal/bolus regimens. The primary endpoint was 18 weeks of therapy, and A1c was lowered 0.62% (P < .001) with canagliflozin 100 mg and 0.73% with canagliflozin 300 mg compared with placebo. Weight decreased 1.9% (P < .001) with canagliflozin 100 mg and 2.4% (P < .001) with canagliflozin 300 mg compared with placebo.
Adverse Effects and Precautions
Canagliflozin adverse effects (AEs) were generally low. In the aforementioned 104-week study comparing canagliflozin 100 mg, canagliflozin 300 mg, and glimepiride, AEs leading to discontinuation were low at 6.2%, 9.5%, and 7.3%, respectively. Serious AEs were lower in the canagliflozin 100 mg and 300 mg groups compared with glimepiride at 9.7%, 9.7%, 14.3%, respectively.5
Limitations for use of canagliflozin are T1DM and diabetic ketoacidosis (DKA). In mild renal impairment, there is no dose adjustment in patients with eGFR > 60 mL/min/1.73 m2. In moderate renal impairment (eGFR 45-60 mL/min/1.73 m2), dose is limited to 100 mg once daily. It is recommended not to initiate canagliflozin if eGFR is < 45 mL/min/1.73 m2. Canagliflozin is contraindicated if eGFR is < 30 mL/min/1.73 m2.8
Dapagliflozin
Dapagliflozin is a highly selective SGLT2 inhibitor. It is a 1,400-fold greater inhibitor of SGLT2 vs SGLT1 and was approved in January 2014. It is indicated as an adjunct to diet and exercise to improve glycemic control in adults with T2DM. The recommended starting dose is 5 mg once daily, taken in the morning, with or without food, and the dosage can be increased to 10 mg once daily in patients who require additional glycemic control. Dapagliflozin should not be initiated if eGFR is < 60 mL/min/1.73 m2, and it should be discontinued if eGFR is persistently < 60 mL/min/1.73 m2.
By inhibiting SGLT2, dapagliflozin reduces reabsorption of filtered glucose and lowers the renal threshold for glucose, thereby increasing UGE. Increased glucose secretion also leads to weight reduction. Dapagliflozin has been studied alone and in combination with glipizide, glimepiride, pioglitazone, and a DDP-4 inhibitor and as an add-on to insulin with and without other oral antidiabetic drugs. It is also available in a fixed-dose combination with metformin.
Following oral administration of dapagliflozin, the Tmax is usually attained within 2 hours under a fasting state. The Cmax and AUC values increase the dose proportionally with increase in dapagliflozin dose in the therapeutic dose range. The absolute oral bioavailability of dapagliflozin following the administration of a 10-mg dose is 78%. The mean plasma t1⁄2 for dapagliflozin is about 12.9 hours following a single oral dose of 10 mg. In patients with normal renal function, the renal glucose excretion was 85 g per day at maximal dose. In humans, 75% of the dose of dapagliflozin is primarily metabolized through the uridine diphosphate glucuronosyltransferase 1A9 pathway. Dapagliflozin and related metabolites are primarily eliminated via the renal pathway.9
Clinical Efficacy Trials
Dapagliflozin was studied as add-on therapy to metformin with glipizide (a sulfonylurea) as the comparator.10 This 52-week double-blind, multicenter, active-controlled, noninferiority trial randomized 801 patients. Eligible subjects were aged > 18 years with A1c > 6.5% and < 10% and were receiving metformin or metformin and 1 other oral antidiabetic drug up to half-maximal dose for at least 8 weeks. During an 18-week titration period, all patients started dapagliflozin 2.5 mg/d and glipizide 5 mg/d. At 21-day intervals, patients were titrated up to the next dosage level if fasting plasma glucose was > 110 mg/dL. Dapagliflozin could be titrated to 5 mg and then 10 mg per protocol. Glipizide could be titrated to 10 mg or 20 mg per protocol.
The A1c change with dapagliflozin was noninferior to glipizide at week 52. The A1c adjusted mean change from baseline was -0.52 for both dapagliflozin and glipizide. The secondary endpoint was weight change. The glipizide group had a +1.44 kg weight gain, whereas the dapagliflozin group had a -3.22 kg weight loss. The number of patients with ≥ 1 episode of hypoglycemia, either symptomatic or with no symptoms, with blood glucose ≤ 63 mg/dL was assessed. The dapagliflozin group had a 3.5% rate compared with the glipizide group, which had a 40.8% rate of patients with > 1 hypoglycemic episode.
A study examined the efficacy and safety of dapagliflozin in combination with and also vs a DPP-4 inhibitor with metformin background therapy. The study looked at add-ons of saxagliptin plus dapagliflozin vs saxagliptin or dapagliflozin added alone.11 This was a randomized, double-blind, 24-week study with patients aged > 18 years with inadequate glucose control A1c > 8.0% and < 12.0%. Patients had to be on a stable dose of metformin > 1,500 mg/d for at least 8 weeks. Patients were randomized 1:1:1 to receive either saxagliptin 5 mg/d and dapagliflozin 10 mg/d plus metformin, saxagliptin 5 mg/d and placebo plus metformin, or dapagliflozin 10 mg/d and placebo plus metformin.
The patients had a mean age of 54 years and a mean duration of T2DM of 7.6 years. The mean baseline A1c was 8.94%. The addition of saxagliptin plus dapagliflozin to metformin resulted in significantly greater A1c reduction compared with saxagliptin plus metformin or dapagliflozin plus metformin from baseline A1c levels: -1.47%, -0.88%, -1.2%, respectively.11
Dapagliflozin was also studied in combination with insulin to determine the safety and efficacy in this setting.12 This double-blind, placebo-controlled, parallel-group trial had an initial study period of 24 weeks followed by an extension period totaling 104 weeks. At 48 weeks, patients on dapagliflozin 5 mg were switched to 10 mg. Outcomes over 104 weeks were changed from baseline A1c, insulin dose, and body weight. Up-titration of insulin was permitted if at least 3 self-monitored blood glucose readings from the 7 days prior to the study visit were > 240 mg/dL up to week 12; > 220 mg/dL between weeks 12 and 24; > 178 mg/dL or if A1c was > 8% between weeks 24 and 48. Between weeks 52 and 65, insulin titrated up was allowed if A1c was > 7.5% and between weeks 78 and 104 if A1c was > 7%. Insulin could be titrated down if ≥ 2 self-monitored blood glucose readings were < 68 mg/dL.
The study group had a T2DM diagnosis for 13.6 years and a mean duration of insulin therapy for about 6 years. The mean daily insulin dose was 77.1 IU. The mean baseline A1c was 8.5%. At 104 weeks, the differences from placebo in A1c adjusted mean change from baseline were -4.0% (P = .0002) and -0.4% (P = .0007) in the dapagliflozin 5 mg (switched to 10 mg at week 48) and 10 mg groups, respectively. Insulin requirements increased progressively in the placebo group +18.3 IU/d at 104 weeks. Insulin requirements stayed stable over 104 weeks in the dapagliflozin groups. Body weight increased in the placebo group, whereas it decreased in the dapagliflozin groups. At 104 weeks, the weight changes from baseline in placebo, dapagliflozin 5 mg/10 mg, and dapagliflozin 10 mg were -0.99 kg, -1.03 kg (P < .001), and -1.5 kg (P < .0001), respectively. The frequency of ≥ 1 minor or major episodes of hypoglycemia was fairly balanced across placebo, dapagliflozin 5 mg/10 mg, and dapagliflozin 10 mg at 61.9%, 61.3%, and 60.7%, respectively.
Adverse Effects and Precautions
Dapagliflozin was generally well tolerated. In the trial of dapagliflozin compared with glipizide in the setting of metformin background therapy, AEs led to discontinuation rates of 9.1% vs 5.9%, respectively. Serious AEs were lower in the dapagliflozin group compared with glipizide at 8.6% vs 11.3%, respectively.10
Limitations for use of dapagliflozin are the treatment of T1DM or the treatment of DKA. Dapagliflozin should not be initiated in patients with an eGFR < 60 mL/min/1.73 m2. No dose adjustment is needed in patients with mild renal impairment (eGFR of ≥ 60 mL/ min/1.73 m2), and dapagliflozin should be discontinued when eGFR is persistently < 60 mL/min/1.73 m2. In the clinical trials, there was an imbalance in the number of bladder cancer reported with dapagliflozin compared with placebo. Across 22 clinical studies, newly diagnosed cases of bladder cancer were reported in 10 of 6,045 patients (0.17%) treated with dapagliflozin and 1 of 3,512 patients (0.03%) treated with placebo or a comparator.9 There were too few cases to determine whether the emergence of these events is related to dapagliflozin. Product labeling states dapagliflozin should not be used in patients with active bladder cancer and should be used with caution in those with a prior history of bladder cancer.
Empaglifilozin
Empagliflozin is the most recently FDA-approved SGLT2 inhibitor (August 2014) for improved glycemic control in T2DM. It has the highest SGLT2 selectivity: > 2,500-fold selectivity for SGLT2 over SGLT1.13 Empagliflozin regulates blood glucose levels by increased UGE, independent of endogenous insulin secretion. It is associated with modest reductions in body weight, visceral adiposity, and systolic BP.
Empagliflozin is available in 10-mg and 25-mg tablets, with a recommended initial dose of 10 mg daily.13 Dosing adjustments are not required for geriatric patients or for those patients with hepatic impairment.14 The use of empagliflozin is contraindicated in patients with eGFR < 45 mL/min/1.73 m2 or for those whose eGFR declines to < 45 mL/min/1.73 m2 during therapy.15,16 Empagliflozin has a pregnancy risk category C. Drug transference during lactation is unknown; therefore, empagliflozin during breast-feeding is not recommended. It is also available in a fixed-dose combination with metformin.
Empagliflozin has been studied alone and in combination therapy with other oral antidiabetic drugs as well as insulin therapy. Metformin, pioglitazone, sitagliptin, and linagliptin have been studied in combination with empagliflozin with sustained glycemic improvement without significantly increased risk of hypoglycemia.17-21 Empagliflozin/linagliptin combination was recently approved after phase 3 trials demonstrated 62% of patients achieved an A1c value < 7% on the 25/5-mg dose at 24 weeks.21 Empagliflozin, coadministered with multiple daily injections of insulin (MDI), has been shown to safely improve glycemic control and reduce total daily insulin requirements without an increased risk of hypoglycemia.22 Currently, it is not approved for use in patients with T1DM, but phase 3 trials are ongoing.
Pharmacokinetics of empagliflozin among healthy volunteers paralleled those of people with T2DM with rapid absorption. The plasma glucose lowering effect of empagliflozin was evident after the first dose and became more pronounced with treatment duration. The AUC and Cmax were dose-proportional over a range of empagliflozin doses in a single rising dose study, with maximum UGE of 90.8 g in healthy volunteers reached at the 400-mg dose.23 The Tmax was 1.5 to 2.1 hours after dosing, comparable to dapagliflozin.24 Steady state with once-daily dosing is reached by day 5 with t1/2 range of 10 to 19 hours.24-27 Plasma levels of empagliflozin declined in a biphasic pattern, with a rapid distribution phase and a slower elimination phase. Total urine volume did not differ significantly in the empagliflozin group compared with placebo.
Healthy subjects treated with placebo or empagliflozin had comparable plasma glucose concentrations. Patients with T2DM demonstrated a decrease in mean daily glucose of -37.0 mg/dL for the 10-mg dose compared with -13.5 mg/dL for placebo. Doses up to 10 mg were found to inhibit renal tubular reabsorption up to 40%, and higher doses inhibit up to 60% of filtered glucose.22-26 Empagliflozin has been shown to have similar efficacy independent of food.27 The pharmacodynamic response declines with increasing renal impairment in SGLT2 inhibitors. Empagliflozin has been associated with a decline from UGE of 97.6 g in normal renal function to 18.2 g in severe renal impairment.
Phase 1 studies of healthy male volunteers initially elucidated empagliflozin’s therapeutic potential of dosedependent increases in UGE and associated reduction in A1c with daily administration, respectively.24,28 Single rising dose studies further defined the linear pharmacokinetics and excellent tolerability of empagliflozin.23 Heise and colleagues demonstrated in 2 separate studies with multiple oral doses (2.5 mg, 10 mg, 25 mg, and 100 mg) of empagliflozin in people with T2DM similar pharmacokinetics and efficacy in UGE, tolerability, and reduction in plasma glucose.24
Ferrannini and colleagues, in a 12-week phase 2 study, demonstrated a statistically significant reduction in A1c of 0.5% and 0.6% with 10-mg and 25-mg doses, respectively, as well as a universal body weight decline by 2 kg.29
Clinical Efficacy Trials
A phase 3 trial of 1,549 randomized people with T2DM (aged > 18 years with baseline A1c of 7%-10%), comparing empagliflozin and glimepiride as an add-on to metformin, demonstrated noninferiority at 52 weeks and statistically significant superiority for the empagliflozin group at 104 weeks.30 The adjusted mean difference in change from baseline in A1c with empagliflozin vs glimepiride at week 104 was -0.11% (95% confidence interval, -0.19 to -0.02; P = .0153 for superiority). Despite 39% of both groups achieving a A1c < 7% at week 52, the empagliflozin group had significantly less hypoglycemia (2% vs 24%). In the body composition substudy, empagliflozin demonstrated a significant reduction of 3 kg compared with an increase of slightly over 1 kg in the glimepiride group at week 104. The majority of the empagliflozin-associated weight loss was found to be a reduction in fat mass.
An international phase 2B randomized, controlled, open-label extension study of 659 people with T2DM (aged > 18 and < 79 years with a body mass index (BMI) < 40 kg/m2 and baseline A1c of 7%-10%) described the long-term safety and efficacy of empagliflozin monotherapy in combination with metformin as compared with sitagliptin monotherapy in combination with metformin. At week 90, the changes from baseline A1c were -0.34%/ -0.47% in the empagliflozin 10 mg/25 mg monotherapy group, -0.34%/0.63% in the empagliflozin 10 mg/25 mg/ metformin combination, -0.56% with metformin monotherapy, and -0.40% with sitagliptin/metformin combination.31 These data provided evidence that empagliflozin had sustained weight loss effects (-2.2 kg to -4.0 kg).
Empagliflozin monotherapy was studied with sitagliptin as an active comparator in a phase 3 trial of 899 people with T2DM (aged > 18 years with baseline A1c of 7%-10%), with the primary endpoint of change in baseline in A1c at week 24. Both the 10-mg and 25-mg dose sof empagliflozin were associated with greater reductions in A1c from baseline (-0.66%, -0.77%) at week 24 than that of sitagliptin (-1.04%). Additionally, both doses of empagliflozin were associated with greater reductions in bodyweight (-2.26 kg and -2.48 kg) as compared with sitagliptin 100 mg (+0.18 kg).20
Empagliflozin’s efficacy and safety in combination with MDI insulin as well as add-on to basal insulin was tested in 2 separate studies.23,32 People with T2DM (mean age 58.8 years, A1c 8.2%) on basal insulin were randomized to empagliflozin (10 mg or 25 mg) or placebo for 78 weeks with a constant basal insulin dose for the first 18 weeks and titration allowed thereafter. Empagliflozin was found to significantly reduce A1c at both 18 and 78 weeks (-0.48% and -0.64% for 10-mg/25-mg doses, respectively, vs -0.02% for placebo) and insulin dose at week 78 vs placebo (-8.8 IU on empagliflozin 10 mg and -11.2 IU on empagliflozin 25 mg).32 Similar rates of hypoglycemia were reported (36.1% of patients on empagliflozin and 35.3% on placebo)
Furthermore, empagliflozin was studied in obese people with uncontrolled, insulin-dependent T2DM (A1c 8.3%, BMI 34.8 kg/m2) on MDI insulin (average 92 IU/d) over a 52-week study.22 Patients were randomized to once-daily empagliflozin 10 mg, empagliflozin 25 mg, or placebo. The study demonstrated improved glycemic control (A1c reduction of -1.27% and -1.18% on empagliflozin 10 mg and 25 mg doses, respectively, compared with -0.81% on placebo) with lower insulin doses (-9 to -11 IU/d) and weight loss (-1.95 kg and -2.04 kg on empagliflozin 10-mg and 25-mg doses, respectively, compared with 0.44 kg on placebo.) There was no increased risk of hypoglycemia noted.
Adverse Effects and Precautions
Empagliflozin is generally well tolerated with low occurrence of AEs. Adverse effects reported in the pooled empagliflozin phase 3 studies were mild to moderate. Serious AEs reported were higher in the placebo group as compared with those of the empagliflozin subjects but did not result in study discontinuation.33
Drug-drug studies of empagliflozin co-administered with other commonly prescribed medication in T2DM showed very limited, if any, interaction. Empagliflozin had no effect on the pharmacokinetics of warfarin or on its anticoagulant activity; therefore, these 2 drugs were deemed safe to be co-administered. No AEs were reported when combining empagliflozin with the loop diuretic hydrochlorothiazide.33 Due to the mode of action of SGLT2 inhibitors, osmotic diuresis may result in a modest reduction in BP.34
The FDA recently released a warning that SGLT2 inhibitors may be associated with a higher risk of developing DKA.35 The FDA Adverse Event Reporting System identified 20 cases of DKA in patients treated with SGLT2 inhibitors from March 2013 to June 6, 2014. Diabetic ketoacidosis is typically accompanied by levels of ketone bodies > 3,000 μmol/L and develops almost exclusively in states of absolute insulin deficiency. The highest level of ketone bodies observed in patients receiving 25 mg of empagliflozin was 1,449 μmol/L compared with a mean of 1,300 μmol/L in a nondiabetic overnight fast.36 Therefore, it is unlikely that the modest empagliflozin-induced ketosis would increase the risk of developing DKA in the absence of absolute insulin deficiency or extreme ketogenic diet.37 Physiologic explanation at the present time is not clear.
Clinical Application
The SGLT2 inhibitors are the latest of 14 classes of drugs approved to treat T2DM. This class offers many beneficial characteristics besides blood sugar lowering. The drugs lower systolic BP, induce weight loss through diuresis of glucose (calories), and carry low risks for hypoglycemia. The American Association of Clinical Endocrinologists recently published their 2015 diabetes management algorithm.38 In this algorithm, they recognized metformin as first-line therapy along with diet and exercise intensification for the treatment of T2DM.
The glucagon-like peptide-1 (GLP-1) analogs and SGLT2 inhibitors are recognized as plausible second-line drugs. They go together well with metformin, providing powerful additional A1c-lowering effects while inducing weight loss without hypoglycemia. Because all GLP-1 analogs are currently available only in injectable forms, the SGLT2 inhibitors offer the additional advantage of being available in pill forms.
Conclusions
All 3 of the current SGLT2 inhibitors are effective tools for treating T2DM. The 3 drugs share many similar traits, and efficacy is generally similar. Empagliflozin has the highest SGLT2 selectivity, > 2,500-fold selectivity for SGLT2 over SGLT1. However, canagliflozin has mild SGLT1 activity, which may offer additional benefits with regards to attenuating PPGE excursions by delaying intestinal glucose absorption (Table).
The SGLT2 inhibitor class had been shown to be effective when used as monotherapy as well as in combination with other oral antidiabetic medications. All 3 SGLT2 inhibitors have also been shown to be effective in combination with insulin and have similar efficacy in these clinical settings (eTable).
All 3 SGLT2 inhibitors are generally well tolerated. There is less hypoglycemia compared with sulfonylureas. However, when the SGLT2 drugs are combined with drugs that can cause hypoglycemia, such as sulfonylureas and insulin, patients must be monitored for hypoglycemia, and titrating down the sulfonylurea or insulin may be necessary. Genital mycotic infections and urinary tract infections in men and woman are common AEs with SGLT2 drugs. Patients must be advised of these possible AEs, and treatment should be prompt if these AEs occur. Because of the mild osmotic diuresis, patients should be reminded to keep well hydrated. SGLT2s have a very mild effect on increasing low-density lipoprotein cholesterol (LDL-C), so care should be taken to ensure that patients’ LDL-C stays at goal.
The FDA recently released a warning that SGLT2 inhibitors may be associated with a higher risk of developing DKA. Although these reports are rare, clinicians should be vigilant. The FDA has suggested that patients should pay close attention for any signs of DKA and seek medical attention immediately if they experience difficulty breathing, nausea, vomiting, abdominal pain, confusion, and unusual fatigue or sleepiness.35
There are subtle differences in the eGFR thresholds for the use of the 3 drugs. It should be kept in mind that with all 3 drugs, the efficacy decreases as the eGFR decreases. It is recommended not to initiate canagliflozin if the patient’s eGFR is < 45 mL/min/1.73 m2. Dapagliflozin should not be initiated in patients with an eGFR < 60 mL/min/1.73 m2. Empagliflozin should not be initiated in patients with an eGFR < 45 mL/min/1.73 m2.
The SGLT2 inhibitors are useful tools to lower blood glucose levels in people with T2DM. They can be used as monotherapy or in combination. They also cause weight reduction. Thus, their unique mechanism of action is complementary to the other oral antidiabetic medications and insulin, so a wide variety of patients can benefit from this class.
Author disclosures
Dr. Nguyen is affiliated with the Astra Zeneca Speakers Bureau and Janssen Pharmaceutical Speakers Bureau. Dr. Plodkowski is a Janssen Pharmaceutical consultant. The remaining authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
1. Ehrenkranz JR, Lewis NG, Kahn CR, Roth J. Phlorizin: a review. Diabetes Metab Res Rev. 2005;21(1):31-38.
2. Polidori D, Sha S, Mudaliar S, et al. Canagliflozin lowers postprandial glucose and insulin by delaying intestinal glucose absorption in addition to increasing urinary glucose excretion: results of a randomized, placebo-controlled study. Diabetes Care. 2013;36(8):2154-2161.
3. Sha S, Polidori D, Farrell, et al. Pharmacodynamic differences between canagliflozin and dapagliflozin: results of a randomized, double-blind, crossover study. Diabetes Obes Metab. 2015;17(2):188-197.
4. Yamout H, Perkovic V, Davies M, et al. Efficacy and safety of canagliflozin in patients with type 2 diabetes and stage 3 nephropathy. Am J Nephrol. 2014;40(1):64-74.
5. Leiter LA, Yoon KH, Arias P, et al. Canagliflozin provides durable glycemic improvements and body weight reduction over 104 weeks versus glimepiride in patients with type 2 diabetes on metformin: a randomized, double-blind, phase 3 study. Diabetes Care. 2015;38(3):355-364.
6. Lavalle-González FJ, Januszewicz A, Davidson J, et al. Efficacy and safety of canagliflozin compared with placebo and sitagliptin in patients with type 2 diabetes on background metformin monotherapy: a randomised trial. Diabetologia. 2013;56(12):2582-2592.
7. Neal B, Perkovic V, de Zeeuw D, et al; CANVAS Trial Collaborative Group. Efficacy and safety of canagliflozin, an inhibitor of sodium-glucose cotransporter 2, when used in conjunction with insulin therapy in patients with type 2 diabetes. Diabetes Care. 2015;38(3):403-411.
8. INVOKANA [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2013.
9. FARXIGA [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2015.
10. Nauck MA, Del Prato S, Meier JJ, et al. Dapagliflozin versus glipizide as add-on therapy in patients with type 2 diabetes who have inadequate glycemic control with metformin: a randomized, 52-week, double-blind, active-controlled noninferiority trial. Diabetes Care. 2011;34(9):2015-2022.
11. Rosenstock J, Hansen L, Zee P, et al. Dual Add-on therapy in type 2 diabetes poorly controlled with metformin monotherapy: a randomized double-blind trial of saxagliptin plus dapagliflozin addition versus single addition of saxagliptin or dapagliflozin to metformin. Diabetes Care. 2015;38(3):376-383.
12. Wilding JP, Woo V, Rohwedder K, Sugg J, Parikh S; Dapagliflozin 006 Study Group. Dapagliflozin in patients with type 2 diabetes receiving high doses of insulin: efficacy and safety over 2 years. Diabetes Obes Metab. 2014;16(2):124-136.
13. Heise T, Seewaldt-Becker E, Macha S, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics following 4 weeks’ treatment with empagliflozin once daily in patients with type 2 diabetes. Diabetes Obes Metab. 2013;15(7):613-621.
14. Macha S, Rose P, Mattheus M, et al. Pharmacokinetics, safety and tolerability of empagliflozin, a sodium glucose cotransporter 2 inhibitor, in patients with hepatic impairment. Diabetes Obes Metab. 2014;16(2):118-123.
15. Barnett A, Mithal A, Manassie J, et al; EMPA-REG RENAL trial investigators. Efficacy and safety of empagliflozin added to existing antidiabetes treatment in patients with type 2 diabetes and chronic kidney disease: a randomised, doubleblind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2(5):369-384.
16. Macha S, Mattheus M, Halabi A, Pinnetti S, Woerle HJ, Broedl UC. Pharmacokinetics, pharmacodynamics and safety of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, in subjects with renal impairment. Diabetes Obes Metab. 2014;16(3):215-222.
17. Häring HU, Merker L, Seewaldt-Becker E, et al; EMPA-REG MET Trial Investigators.
Empagliflozin as add-on to metformin in patients with type 2 diabetes: a 24-week, randomized, double-blind, placebo-controlled trial. Diabetes Care. 2014;37(6):1650-1699.
18. Ridderstråle M, Svaerd R, Zeller C, Kim G, Woerle HJ, Broedi UC; EMPA-REG H2H-SU trial investigators. Rational, design and baseline characteristics of a 4-year (208-week) phase III trial of empagliflozin, an SGLT2 inhibitor, versus glimepiride as add-on to metformin in patients with type 2 diabetes mellitus with insufficient glycemic control. Cardiovasc Diabetol. 2013;12:129.
19. Kovacs CS, Seshiah V, Swallow R, et al; EMPA-REG PIO trial investigators. Empagliflozin improves glycaemic and weight control as add-on therapy to pioglitazone or pioglitazone plus metformin in patients with type 2 diabetes: a 24-week, randomized, placebo-controlled trial. Diabetes Obes Metab. 2014;16(2):147-158.
20. Roden M, Weng J, Eilbracht J, et al; EMPA-REG MONO trial investigators. Empagliflozin monotherapy with sitagliptin as an active comparator in patients with type 2 diabetes: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2013;1(3):208-219.
21. Friedrich C, Metzmann K, Rose P, Mattheus M, Pinnetti S, Woerle HJ. A randomized, open-label, crossover study to evaluate the pharmacokinetics of empagliflozin and linagliptin after coadministration in healthy male volunteers. Clin Ther. 2013;35(1):A33-A42.
22. Rosenstock J, Jelaska A, Frappin G, et al; EMPA-REG MDI Trial Investigators. Improved glucose control with weight loss, lower insulin doses, and no increased hypoglycemia with empagliflozin added to titrated multiple daily injections of insulin in obese inadequately controlled type 2 diabetes. Diabetes Care. 2014;37(3):1815-1823.
23. Seman L, Macha S, Nehmiz G, et al. Empagliflozin (BI 10773), a potent and selective SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin Pharmacol Drug Dev. 2013;2(2):152-161.
24. Heise T, Seman L, Macha S, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple rising doses of empagliflozin in patients with type 2 diabetes mellitus. Diabetes Ther. 2013;4(2):331-345.
25. Inzucchi SE, Zinman B, Wanner C, et al. SGLT-2 inhibitors and cardiovascular risk: proposed pathways and review of ongoing outcome trials. Diab Vasc Dis Res. 2015;12(2):90-100.
26. Scheen AJ. Pharmacokinetic and pharmacodynamic profile of empagliflozin, a sodium glucose co-transporter 2 inhibitor. Clin Pharmacokinet. 2014;53(3):213-225.
27. Macha S, Jungnik A, Hohl K, Hobson D, Salsali A, Woerle HJ. Effect of food on the pharmacokinetics of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, and assessment of dose proportionality in healthy volunteers. Int J Clinical Pharmacol Therapy. 2013;51(11):873-879.
28. Sarashina A, Koiwai K, Seman LJ, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of single doses of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, in healthy Japanese subjects. Drug Metab Pharmacokinet. 2013;28(3):213-219.
29. Ferrannini E, Seman L, Seewaldt-Becker E, Hantel S, Pinnetti S, Woerle HJ. A phase IIb, randomized, placebo-controlled study of the SGLT2 inhibitor empagliflozin in patients with type 2 diabetes. Diabetes Obes Metab. 2013;15(8):721-728.
30. Ridderstråle M, Andersen KR, Zeller C, Kim G, Woerle HJ, Broedl UC; EMPAREG H2H-SU trial investigators. Comparison of empagliflozin and glimepiride as add-on to metformin in patients with type 2 diabetes: a 104-week randomised, active-controlled, double-blind, phase 3 trial. Lancet Diabetes Endocrinol. 2014;2(9):691-700.
31. Ferrannini E, Berk A, Hantel S, et al. Long-term safety and efficacy of empagliflozin, sitagliptin, and metformin: an active-controlled, parallel-group, randomized, 78-week open-label extension study in patients with type 2 diabetes. Diabetes Care. 2013;36(12):4015-4021.
32. Rosenstock J, Jelaska A, Kim G, et al. Empagliflozin as add-on to basal insulin for 78 weeks improves glycemic control with weight loss in insulin-treated (T2DM) [Abstract 1102-P]. Diabetes. 2013;62(suppl 1):A285.
33. JARDIANCE [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals,
Inc.; 2015.
34. Häring HU, Merker L, Seewaldt-Becker E, et al; EMPA-REG METSU Trial Investigators. Empagliflozin as add-on to metformin plus sulfonylurea in patients with type 2 diabetes: a 24-week, randomized, double-blind, placebo-controlled trial. Diabetes Care. 2013;36(11):3396-3404.
35. FDA Drug Safety Communication: FDA Warns that SGLT2 inhibitors for diabetes may result in a serious condition of too much acid in the blood. U.S. Food and Drug Administration Website. http://www.fda.gov/Drugs/DrugSafety/ucm446845.htm. Updated May 19, 2015. Accessed September 23, 2015.
36. Nishimura R, Tanaka Y, Koiwai K, et al. Effect of empagliflozin monotherapy on postprandial glucose and 24-hour glucose variability in Japanese patients with type 2 diabetes mellitus: a randomized, double-blind, placebo-controlled, 4-week study. Cardiovasc Diabetol. 2015;14:11.
37. Tikkanen I, Narko K, Zeller C, et al. Empagliflozin reduces blood pressure in patients
with type 2 diabetes and hypertension. Diabetes Care. 2015;38(3):420-428.
38. Garber AJ, Abrahamson MJ, Barzilay JI, et al. AACE/ACE comprehensive diabetes management algorithm 2015. Endocr Pract. 2015;21(4):438-447.
1. Ehrenkranz JR, Lewis NG, Kahn CR, Roth J. Phlorizin: a review. Diabetes Metab Res Rev. 2005;21(1):31-38.
2. Polidori D, Sha S, Mudaliar S, et al. Canagliflozin lowers postprandial glucose and insulin by delaying intestinal glucose absorption in addition to increasing urinary glucose excretion: results of a randomized, placebo-controlled study. Diabetes Care. 2013;36(8):2154-2161.
3. Sha S, Polidori D, Farrell, et al. Pharmacodynamic differences between canagliflozin and dapagliflozin: results of a randomized, double-blind, crossover study. Diabetes Obes Metab. 2015;17(2):188-197.
4. Yamout H, Perkovic V, Davies M, et al. Efficacy and safety of canagliflozin in patients with type 2 diabetes and stage 3 nephropathy. Am J Nephrol. 2014;40(1):64-74.
5. Leiter LA, Yoon KH, Arias P, et al. Canagliflozin provides durable glycemic improvements and body weight reduction over 104 weeks versus glimepiride in patients with type 2 diabetes on metformin: a randomized, double-blind, phase 3 study. Diabetes Care. 2015;38(3):355-364.
6. Lavalle-González FJ, Januszewicz A, Davidson J, et al. Efficacy and safety of canagliflozin compared with placebo and sitagliptin in patients with type 2 diabetes on background metformin monotherapy: a randomised trial. Diabetologia. 2013;56(12):2582-2592.
7. Neal B, Perkovic V, de Zeeuw D, et al; CANVAS Trial Collaborative Group. Efficacy and safety of canagliflozin, an inhibitor of sodium-glucose cotransporter 2, when used in conjunction with insulin therapy in patients with type 2 diabetes. Diabetes Care. 2015;38(3):403-411.
8. INVOKANA [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2013.
9. FARXIGA [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2015.
10. Nauck MA, Del Prato S, Meier JJ, et al. Dapagliflozin versus glipizide as add-on therapy in patients with type 2 diabetes who have inadequate glycemic control with metformin: a randomized, 52-week, double-blind, active-controlled noninferiority trial. Diabetes Care. 2011;34(9):2015-2022.
11. Rosenstock J, Hansen L, Zee P, et al. Dual Add-on therapy in type 2 diabetes poorly controlled with metformin monotherapy: a randomized double-blind trial of saxagliptin plus dapagliflozin addition versus single addition of saxagliptin or dapagliflozin to metformin. Diabetes Care. 2015;38(3):376-383.
12. Wilding JP, Woo V, Rohwedder K, Sugg J, Parikh S; Dapagliflozin 006 Study Group. Dapagliflozin in patients with type 2 diabetes receiving high doses of insulin: efficacy and safety over 2 years. Diabetes Obes Metab. 2014;16(2):124-136.
13. Heise T, Seewaldt-Becker E, Macha S, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics following 4 weeks’ treatment with empagliflozin once daily in patients with type 2 diabetes. Diabetes Obes Metab. 2013;15(7):613-621.
14. Macha S, Rose P, Mattheus M, et al. Pharmacokinetics, safety and tolerability of empagliflozin, a sodium glucose cotransporter 2 inhibitor, in patients with hepatic impairment. Diabetes Obes Metab. 2014;16(2):118-123.
15. Barnett A, Mithal A, Manassie J, et al; EMPA-REG RENAL trial investigators. Efficacy and safety of empagliflozin added to existing antidiabetes treatment in patients with type 2 diabetes and chronic kidney disease: a randomised, doubleblind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2(5):369-384.
16. Macha S, Mattheus M, Halabi A, Pinnetti S, Woerle HJ, Broedl UC. Pharmacokinetics, pharmacodynamics and safety of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, in subjects with renal impairment. Diabetes Obes Metab. 2014;16(3):215-222.
17. Häring HU, Merker L, Seewaldt-Becker E, et al; EMPA-REG MET Trial Investigators.
Empagliflozin as add-on to metformin in patients with type 2 diabetes: a 24-week, randomized, double-blind, placebo-controlled trial. Diabetes Care. 2014;37(6):1650-1699.
18. Ridderstråle M, Svaerd R, Zeller C, Kim G, Woerle HJ, Broedi UC; EMPA-REG H2H-SU trial investigators. Rational, design and baseline characteristics of a 4-year (208-week) phase III trial of empagliflozin, an SGLT2 inhibitor, versus glimepiride as add-on to metformin in patients with type 2 diabetes mellitus with insufficient glycemic control. Cardiovasc Diabetol. 2013;12:129.
19. Kovacs CS, Seshiah V, Swallow R, et al; EMPA-REG PIO trial investigators. Empagliflozin improves glycaemic and weight control as add-on therapy to pioglitazone or pioglitazone plus metformin in patients with type 2 diabetes: a 24-week, randomized, placebo-controlled trial. Diabetes Obes Metab. 2014;16(2):147-158.
20. Roden M, Weng J, Eilbracht J, et al; EMPA-REG MONO trial investigators. Empagliflozin monotherapy with sitagliptin as an active comparator in patients with type 2 diabetes: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2013;1(3):208-219.
21. Friedrich C, Metzmann K, Rose P, Mattheus M, Pinnetti S, Woerle HJ. A randomized, open-label, crossover study to evaluate the pharmacokinetics of empagliflozin and linagliptin after coadministration in healthy male volunteers. Clin Ther. 2013;35(1):A33-A42.
22. Rosenstock J, Jelaska A, Frappin G, et al; EMPA-REG MDI Trial Investigators. Improved glucose control with weight loss, lower insulin doses, and no increased hypoglycemia with empagliflozin added to titrated multiple daily injections of insulin in obese inadequately controlled type 2 diabetes. Diabetes Care. 2014;37(3):1815-1823.
23. Seman L, Macha S, Nehmiz G, et al. Empagliflozin (BI 10773), a potent and selective SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin Pharmacol Drug Dev. 2013;2(2):152-161.
24. Heise T, Seman L, Macha S, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple rising doses of empagliflozin in patients with type 2 diabetes mellitus. Diabetes Ther. 2013;4(2):331-345.
25. Inzucchi SE, Zinman B, Wanner C, et al. SGLT-2 inhibitors and cardiovascular risk: proposed pathways and review of ongoing outcome trials. Diab Vasc Dis Res. 2015;12(2):90-100.
26. Scheen AJ. Pharmacokinetic and pharmacodynamic profile of empagliflozin, a sodium glucose co-transporter 2 inhibitor. Clin Pharmacokinet. 2014;53(3):213-225.
27. Macha S, Jungnik A, Hohl K, Hobson D, Salsali A, Woerle HJ. Effect of food on the pharmacokinetics of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, and assessment of dose proportionality in healthy volunteers. Int J Clinical Pharmacol Therapy. 2013;51(11):873-879.
28. Sarashina A, Koiwai K, Seman LJ, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of single doses of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, in healthy Japanese subjects. Drug Metab Pharmacokinet. 2013;28(3):213-219.
29. Ferrannini E, Seman L, Seewaldt-Becker E, Hantel S, Pinnetti S, Woerle HJ. A phase IIb, randomized, placebo-controlled study of the SGLT2 inhibitor empagliflozin in patients with type 2 diabetes. Diabetes Obes Metab. 2013;15(8):721-728.
30. Ridderstråle M, Andersen KR, Zeller C, Kim G, Woerle HJ, Broedl UC; EMPAREG H2H-SU trial investigators. Comparison of empagliflozin and glimepiride as add-on to metformin in patients with type 2 diabetes: a 104-week randomised, active-controlled, double-blind, phase 3 trial. Lancet Diabetes Endocrinol. 2014;2(9):691-700.
31. Ferrannini E, Berk A, Hantel S, et al. Long-term safety and efficacy of empagliflozin, sitagliptin, and metformin: an active-controlled, parallel-group, randomized, 78-week open-label extension study in patients with type 2 diabetes. Diabetes Care. 2013;36(12):4015-4021.
32. Rosenstock J, Jelaska A, Kim G, et al. Empagliflozin as add-on to basal insulin for 78 weeks improves glycemic control with weight loss in insulin-treated (T2DM) [Abstract 1102-P]. Diabetes. 2013;62(suppl 1):A285.
33. JARDIANCE [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals,
Inc.; 2015.
34. Häring HU, Merker L, Seewaldt-Becker E, et al; EMPA-REG METSU Trial Investigators. Empagliflozin as add-on to metformin plus sulfonylurea in patients with type 2 diabetes: a 24-week, randomized, double-blind, placebo-controlled trial. Diabetes Care. 2013;36(11):3396-3404.
35. FDA Drug Safety Communication: FDA Warns that SGLT2 inhibitors for diabetes may result in a serious condition of too much acid in the blood. U.S. Food and Drug Administration Website. http://www.fda.gov/Drugs/DrugSafety/ucm446845.htm. Updated May 19, 2015. Accessed September 23, 2015.
36. Nishimura R, Tanaka Y, Koiwai K, et al. Effect of empagliflozin monotherapy on postprandial glucose and 24-hour glucose variability in Japanese patients with type 2 diabetes mellitus: a randomized, double-blind, placebo-controlled, 4-week study. Cardiovasc Diabetol. 2015;14:11.
37. Tikkanen I, Narko K, Zeller C, et al. Empagliflozin reduces blood pressure in patients
with type 2 diabetes and hypertension. Diabetes Care. 2015;38(3):420-428.
38. Garber AJ, Abrahamson MJ, Barzilay JI, et al. AACE/ACE comprehensive diabetes management algorithm 2015. Endocr Pract. 2015;21(4):438-447.