User login
A guide to diagnosing and managing ascites in cirrhosis
Liver cirrhosis is implicated in 75% to 85% of ascites cases in the Western world, with heart failure or malignancy accounting for fewer cases.1 Among patients who have decompensated cirrhosis with ascites, annual mortality is 20%.2 Another study showed a 3-year survival rate after onset of ascites of only 56%.3 It is vital for primary care physicians (PCPs) to be alert for ascites not only in patients who have risk factors for chronic liver disease and cirrhosis—eg, a history of alcohol use disorder, chronic viral infections (hepatitis B and C), or metabolic syndrome—but also in patients with abnormal liver function tests and thrombocytopenia. In this review, we discuss the initial assessment of ascites and its long-term management, concentrating on the role of the PCP.
Pathophysiology: Vasodilation leads to a cascade
Splanchnic vasodilation is the main underlying event triggering a pathologic cascade that leads to the development of ascites.4 Initially portal hypertension in the setting of liver inflammation and fibrosis causes the release of inflammatory cytokines such as nitric oxide and carbon monoxide. This, in turn, causes the pathologic dilation of splanchnic circulation that decreases effective circulating volume. Activation of the sympathetic nervous system, vasopressin, and renin-angiotensin-aldosterone system (RAAS) then causes the proximal and distal tubules to increase renal absorption of sodium and water.5 The resulting volume overload further decreases the heart’s ability to maintain circulating volume, leading to increased activation of compensating symptoms. This vicious cycle eventually manifests as ascites.6
A complex interplay of cirrhosis-associated immune dysfunction (CAID), gut dysbiosis, and increased translocation of microorganisms into ascitic fluid is also an important aspect of the pathogenesis.7 CAID (FIGURE 1)7,8 is an immunodeficient state due to cirrhosis with reduced phagocytic activity by neutrophils and macrophages, T- and B-cell hypoproliferation, and reduced cytotoxicity of natural killer cells. In parallel, there is increased production of inflammatory cytokines due to the effects of damage-associated molecular patterns (DAMPs) from hepatocytes and pathogen-associated molecular patterns (PAMPs) from the gut microbiota on the immune system, which leads to many of the manifestations of decompensated cirrhosis including ascites.8
")
Key in on these elementsof the history and exam
Each step of the basic work-up for ascites provides opportunities to refine or redirect the diagnostic inquiry (TABLE).

History
Generally, patients with ascites present with weight gain and symptoms of abdominal distension, such as early satiety, nausea, and vomiting. Besides cirrhosis, rule out other causes of ascites, as treatment differs based on the cause.9 Also ask about histories of cancer and cardiac, renal, or thyroid disease.10
Patients with ascites in the setting of liver disease usually are asymptomatic in its early stages. Common complaints are vague abdominal pain, generalized weakness, malaise, and fatigue.11 Ask patients about risk factors for liver disease such as obesity, diabetes, hypertension, alcohol use, unsafe sexual practices, recent travel, and needle sharing or drug use. Due to a strong association between obstructive sleep apnea and fatty liver disease, consider screening at-risk patients for sleep apnea.12
Physical exam
When there are risk factors for liver disease, examine the patient for stigmata of cirrhosis and ascites. Signs of liver disease, aside from ascites, may include spider angiomas on the upper trunk (33% of cirrhosis patients),13 gynecomastia (44% of cirrhosis patients),14 palmar erythema, jaundice, asterixis, and abdominal wall collaterals including caput medusa.15
Continue to: We suggest a systematic...
We suggest a systematic and targeted approach to using various physical exam maneuvers described in the literature. If the patient has a full/distended abdomen, percuss the flanks. If increased dullness at the flanks is detected, check for shifting dullness, which indicates at least 1500 mL of fluid in the abdomen.16 Keep in mind that a 10% chance of ascites exists even if shifting dullness is absent.17 Maneuvers such as the puddle sign and fluid thrill are less accurate than shifting dullness, which has 83% sensitivity and 56% specificity in detecting ascites.17 Patients with cirrhosis also have a high likelihood of complications from ascites such as inguinal, umbilical, and other hernias.
Diagnostic work-up includes blood tests and ultrasound
Blood tests. The initial work-up for ascites should include complete blood count, complete metabolic panel, and prothrombin time/international normalized ratio.18
Abdominal ultrasound is recommended as the first-line imaging test.19 Aside from detecting ascites, it can give an estimate of the volume of ascites and indicate whether it is amenable to paracentesis. A vascular exam added to the standard ultrasound can detect radiologic evidence of portal hypertension such as splenomegaly, portosystemic collaterals, splenorenal shunt, patency of the paraumbilical vein, and portal vein diameter. Patients with established cirrhosis also require abdominal ultrasound every 6 months to screen for hepatocellular cancer.20
Abdominal paracentesis is the cornerstone of ascites evaluation.21 It is indicated for every patient with new-onset ascites or for any patient with known ascites and clinical deterioration. Ascitic fluid analysis can be used to easily differentiate portal hypertension from other causes of ascites. It can also be used to rule out bacterial peritonitis. The recommended sites for evaluation are in the left lower quadrant, 3 cm cranially and 3 cm medially from the anterior superior iliac spine.22 A large cohort study showed that abdominal ultrasound-guided paracentesis reduced bleeding complications by 68% following the procedure and is strongly recommended (if available).23 Generally, paracentesis is a relatively safe procedure with a low risk of complications such as abdominal wall hematoma (1%), hemoperitoneum (< 0.1%), bowel perforation (< 0.1%), and infection (< 0.1%).24
Assess all ascitic fluid samples for color, consistency, cell count and differential, albumin, and total protein. These tests are usually sufficient to provide evidence regarding the cause of ascites. If there is suspicion of infection, order a gram stain and culture (80% sensitivity for detecting an infection if obtained prior to initiation of antibiotics)25 and glucose, lactate dehydrogenase (useful to differentiate primary from secondary bacterial peritonitis),26 and amylase tests. Other tests such as cytology, acid-fast bacilli smear and culture, and triglyceride level should only be obtained if specific conditions are suspected based on high pretest probabilities.
Continue to: Calculating serum ascites albumin gradient...
Calculating serum ascites albumin gradient (SAAG) is recommended as it has been shown to better characterize ascitic fluid than total protein-based tests.27 SAAG is calculated by subtracting the level of ascitic fluid albumin from serum albumin level (SAAG = serum albumin – ascitic fluid albumin). A SAAG ≥ 1.1 g/dL is consistent with portal hypertension,28 with approximately 97% accuracy.
After calculating SAAG, look at total protein levels in ascitic fluid. Total protein concentration ≥ 2.5 g/dL with SAAG ≥ 1.1 g/dL has a 78.3% diagnostic accuracy in determining heart failure as the cause of ascites, with a sensitivity of 53.3% and specificity of 86.7%.28 On the other hand, a value of total protein < 2.5 g/dL indicates cirrhosis, liver failure, or acute hepatitis as the cause of fluid build-up.29 Stepwise evaluation of SAAG and total protein and how they can point toward the most likely cause of ascites is presented in FIGURE 2.27-29

Management
Noninvasive measures
Sodium restriction. The aim of treatment for uncomplicated clinically apparent ascites is sodium restriction and removal of fluid from the body. Dietary salt restriction is complicated, and care should be taken to properly educate patients. Salt restriction advised in the literature has shifted from a strict measure of < 2 g/d30 to more moderate strategies (described below).18
The 2 main reasons for this easing of restriction are issues with patient compliance and concerns about adverse effects with aggressive salt-restricted diets. One study assessing patient compliance with a salt-restricted diet found that more than two-thirds of the patients were noncompliant,31 and 65% of the patients incorrectly assumed they were following the plan, which suggests poor dietary education.31 Of the group that was compliant, 20% actually decreased their caloric intake, which can be detrimental in liver disease.31 Concerns have been raised that aggressive salt restriction along with diuretic use can lead to diuretic-induced hyponatremia and renal failure.32 Current European Association for the Study of the Liver (EASL) guidelines recommend salt restriction to a more moderate degree (80-120 mmol/d of sodium). This is equivalent to 4.9-6.9 g of salt (1 tablespoon is roughly equivalent to 6 g or 104 mmol of sodium).18
Diuretics. Initiation and dosage of diuretic therapy is a matter of some controversy. Historically, simultaneous administration of a loop diuretic and mineralocorticoid receptor blocker were recommended: 40 mg furosemide and 100 mg spironolactone, keeping the ratio constant with any dosage increases. This was based on a randomized controlled trial (RCT) showing that the combined diuretic therapy effectively mobilized ascites in a shorter period of time and with less frequent adverse effects (eg, hyperkalemia) compared with initial monotherapy.33
Continue to: On the other hand...
On the other hand, another study with more stable patients and relatively normal renal function showed that starting with a mineralocorticoid receptor blocker alone with sequential dose increments had equivalent benefit with no increase in adverse effects.34 Since the patient population in this study was more in line with what a PCP might encounter, we recommend following this guideline initially and keeping a close watch on serum electrolytes.
Usual maximum doses are spironolactone 400 mg/d and furosemide 160 mg/d.21,35 Adequate weight loss for patients with diffuse edema is at least 1 kg/d, per EASL guidelines.36,37 However, this might not be practical in outpatient settings, and a more conservative target of 0.5 kg/d may be used for patients without significant edema.37
It is vital to get accurate daily weights and avoid excessive diuretic use, as it has been associated with intravascular volume depletion and acute kidney injury (25%), hyponatremia (28%),38,39 and hepatic encephalopathy (30%).40 Therefore, patients with acute kidney injury, hyponatremia, acute variceal hemorrhage, or infection should also have their diuretics held until their creatinine returns to baseline.
Invasive measures
Large-volume paracentesis. Patients with extensive and tense ascites should be treated initially with large-volume paracentesis, as this has been shown to predictably remove fluid more effectively than diuretics.38 This should be accompanied by albumin administration, 8 g for every liter of ascitic fluid removed if the total amount exceeds 5 L.41 Following large-volume paracentesis, manage patients with the standard salt restriction and diuretic regimen.38 Serial large-volume paracentesis is a temporary measure reserved for a select group of patients who are intolerant to diuretics and are not candidates for a shunt.
Transjugular intrahepatic portosystemic shunt (TIPS) is another option to control refractory ascites, but its benefit should be weighed against complications such as hepatic encephalopathy. An RCT found that TIPS with covered stents improved survival in patients with cirrhosis compared with regular large-volume paracentesis.42 Patients should be referred to hepatologists to make a determination about TIPS placement. Widely accepted contraindications for the placement of TIPS are decompensated cirrhosis (Child-Pugh > 11, model for end-stage liver disease [MELD] > 18), renal failure (serum creatinine > 3 mg/dL), heart failure, porto-pulmonary hypertension, and uncontrolled sepsis.43 Recurrent or persistent hepatic encephalopathy (West Haven grade ≥ 2) is also a contraindication. The West Haven scale is widely used to measure severity of hepatic encephalopathy, grading it from 1 to 4, with 1 being mild encephalopathy characterized by lack of awareness and shorter attention span, and 4 indicating unresponsiveness or coma.44
Continue to: How to manage refractory ascites
How to manage refractory ascites
Fragile patients are those with refractory ascites that is either unresponsive to standard salt restriction and maximum-dose diuretic therapy or that results in a re-accumulation of ascitic fluid soon after paracentesis.45 Specialist care is required to improve survival and quality of life for these patients. They should be referred to a hepatologist for consideration of TIPS placement or liver transplantation.18
Long-term use of albumin was tested in 2 trials for management of decompensated cirrhosis with ascites, yielding conflicting results. The ANSWER trial from Italy showed benefit with this treatment for prolonged survival.46 The other trial, from Spain, showed no benefit from albumin and midodrine administration for survival or for improving complications of cirrhosis.47 The contradictory results are likely due to heterogeneous populations in the 2 trials and differences in dose and duration of albumin administration. Hence, no clear recommendations can be made based on the available data; further research is needed.
Getting a handle on bacterial peritonitis
Bacterial peritonitis can be divided into spontaneous bacterial peritonitis (SBP) and secondary bacterial peritonitis. SBP is a common complication in patients with cirrhosis and occurs in around 16% of hospitalized patients, based on 1 study.48 SBP is defined as a polymorphonuclear leukocyte count ≥ 250 cells/μL in the absence of a surgically treatable source of infection.49 It is believed to be caused by bacterial translocation and is treated empirically with a third-generation cephalosporin. This treatment has been shown to be effective in 85% of patients.50
Patients with SBP are at a higher risk for renal impairment, likely resulting from increased cytokine production and decreased circulatory volume.51 Concomitant albumin administration has been shown to significantly improve outcomes and to reduce rates of hepatorenal syndrome in patients with serum creatinine > 1 mg/dL, blood urea nitrogen > 30 mg/dL, or total bilirubin > 4 mg/dL.52 The recommended amount of albumin is 1.5 g/kg given within 6 hours of SBP detection and repeat administration of 1 g/kg on Day 3.52
Guidelines from the American Association for the Study of Liver Diseases and from EASL recommend the long-term use of daily norfloxacin or trimethoprim-sulfamethoxazole as secondary prophylaxis in patients who have survived an episode of SBP.18
Continue to: Avoid these medications
Avoid these medications
Commonly used medications that should be avoided in patients with cirrhosis and ascites are angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. These agents block the action of angiotensin, which is a vital vasoconstrictor, and thereby cause a drop in blood pressure. This has independently been associated with poor outcomes in patients with cirrhosis.37
Nonsteroidal anti-inflammatory drugs (NSAIDs) are also relatively contraindicated in cirrhosis, as they can affect kidney function, induce azotemia, and reduce kidney sodium excretion. NSAIDs induce vasoconstriction of afferent arterioles in the kidneys, leading to a decreased glomerular filtration rate, further activating RAAS and sympathetic drive. This leads to increased sodium and water retention and worsening ascites.54
Improve outcomes by circling in a hepatologist
PCPs can play a vital role in the prevention, treatment, surveillance, and home care of patients with cirrhosis who are at risk for ascites.55 Referral of patients with hepatic impairment manifesting as unexplained abnormal liver function tests, new-onset ascites, and/or image findings consistent with cirrhosis to a hepatologist at least once is recommended. Such referrals have been shown to be associated with a better overall outcome.56 Patients with known cirrhosis leading to ascites can generally be managed at home with the assistance of specialists and specialized nurses.35
In a study from the University of Michigan, 69% of patients with cirrhosis had at least 1 nonelective readmission; 14% of patients were readmitted within 1 week, and 37% within 1 month.57 These are staggering statistics that highlight the gaps in care coordination and management of patients with cirrhosis in the outpatient setting. PCPs can play a vital role in bridging this gap.
A promising framework is suggested by a study from Italy by Morando et al in 2013.58 The researchers assessed a specialized health care model for cirrhotic patients and showed significant improvement in health care cost, readmission rate, and overall mortality when compared with the existing model of outpatient care.58
Continue to: This was not a blinded study...
This was not a blinded study and there were concerns raised by the scientific community about its design. Because it was conducted in Italy, the results might not be fully applicable to the United States health care setting. However, it did show that better coordination of care leads to significantly better patient outcomes and reduces health care expenditure. Therefore, a more complete understanding of the disease process and latest literature by PCPs, communication with specialists, and comprehensive coordination of care by all parties involved is vital for the management of this patient population.
CORRESPONDENCE
Muhammad Salman Faisal, MD, Cleveland Clinic Foundation, 9500 Euclid Avenue, Cleveland, OH 44195; faisalm@ccf.org
1. Runyon BA, Montano AA, Akriviadis EA, et al. The serum-ascites albumin gradient is superior to the exudate-transudate concept in the differential diagnosis of ascites. Ann Intern Med. 1992;117:215-220.
2. D’Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol. 2006;44:217-231.
3. Gordon FD. Ascites. Clin Liver Dis. 2012;16:285-299.
4. Schrier RW, Arroyo V, Bernardi M, et al. Peripheral arterial vasodilation hypothesis: a proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology. 1988;8:1151-1157.
5. Arroyo V, Terra C, Gines P. Advances in the pathogenesis and treatment of type-1 and type-2 hepatorenal syndrome. J Hepatol. 2007;46:935-946.
6. Bernardi M, Moreau R, Angeli P, et al. Mechanisms of decompensation and organ failure in cirrhosis: from peripheral arterial vasodilation to systemic inflammation hypothesis. J Hepatol. 2015;63:1272-1284.
7. Jalan R, Fernandez J, Wiest R, et al. Bacterial infections in cirrhosis: a position statement based on the EASL Special Conference 2013. J Hepatol. 2014;60:1310-1324.
8. Albillos A, Lario M, Álvarez-Mon M. Cirrhosis-associated immune dysfunction: distinctive features and clinical relevance. J Hepatol. 2014;61:1385-1396.
9. Oey RC, van Buuren HR, de Man RA. The diagnostic work-up in patients with ascites: current guidelines and future prospects. Neth J Med. 2016;74:330-335.
10. de Kerguenec C, Hillaire S, Molinié V, et al. Hepatic manifestations of hemophagocytic syndrome: a study of 30 cases. Am J Gastroenterol. 2001;96:852-857.
11. Milić S, Lulić D, Štimac D. Non-alcoholic fatty liver disease and obesity: biochemical, metabolic and clinical presentations. World J Gastroenterol. 2014;20:9330-9337.
12. Aron-Wisnewsky J, Clement K, Pépin J-L. Nonalcoholic fatty liver disease and obstructive sleep apnea. Metabolism. 2016;65:1124-1135.
13. Li CP, Lee FY, Hwang SJ, et al. Spider angiomas in patients with liver cirrhosis: role of alcoholism and impaired liver function. Scand J Gastroenterol. 1999;34:520-523.
14. Cavanaugh J. Gynecomastia and cirrhosis of the liver. Arch Intern Med. 1990;150:563-565.
15. Karnath B. Stigmata of chronic liver disease. Hosp Phys. 2003;7:14-16,28.
16. Schipper HG, Godfried MH. [Physical diagnosis--ascites]. Ned Tijdschr Geneeskd. 2001;145:260-264.
17. Cattau EL, Jr., Benjamin SB, Knuff TE, et al. The accuracy of the physical examination in the diagnosis of suspected ascites. JAMA. 1982;247:1164-1166.
18. EASL clinical practice guidelines for the management of patients with decompensated cirrhosis. J Hepatol. 2018;69:406-460.
19. Runyon BA, AASLD Practice Guidelines Committee. Management of adult patients with ascites due to cirrhosis: an update. Hepatology 2009;49:2087-2107.
20. EASL Clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69:182-236.
21. Runyon BA. Care of patients with ascites. New Engl J Med. 1994;330:337-342.
22. Sakai H, Sheer TA, Mendler MH, et al. Choosing the location for non-image guided abdominal paracentesis. Liver Int. 2005;25:984-986.
23. Mercaldi CJ, Lanes SF. Ultrasound guidance decreases complications and improves the cost of care among patients undergoing thoracentesis and paracentesis. Chest. 2013;143:532-538.
24. Ennis J, Schultz G, Perera P, et al. Ultrasound for detection of ascites and for guidance of the paracentesis procedure: technique and review of the literature. Int J Clin Med. 2014;5:1277-1293.
25. Runyon BA, Canawati HN, Akriviadis EA. Optimization of ascitic fluid culture technique. Gastroenterology. 1988;95:1351-1355.
26. Akriviadis EA, Runyon BA. Utility of an algorithm in differentiating spontaneous from secondary bacterial peritonitis. Gastroenterology 1990;98:127-133.
27. Hoefs JC. Serum protein concentration and portal pressure determine the ascitic fluid protein concentration in patients with chronic liver disease. J Lab Clin Med. 1983;102:260-273.
28. Farias AQ, Silvestre OM, Garcia-Tsao G, et al. Serum B-type natriuretic peptide in the initial workup of patients with new onset ascites: a diagnostic accuracy study. Hepatology. 2014;59:1043-1051.
29. Gupta R, Misra SP, Dwivedi M, et al. Diagnosing ascites: value of ascitic fluid total protein, albumin, cholesterol, their ratios, serum-ascites albumin and cholesterol gradient. J Gastroenterol Hepatol. 1995;10:295-299.
30. Runyon BA. Management of adult patients with ascites due to cirrhosis: update 2012. AASLD Practice Guideline. Accessed April 28, 2021. www.aasld.org/sites/default/files/2019-06/AASLDPracticeGuidelineAsciteDuetoCirrhosisUpdate2012Edition4_.pdf
31. Morando F, Rosi S, Gola E, et al. Adherence to a moderate sodium restriction diet in outpatients with cirrhosis and ascites: a real-life cross-sectional study. Liver Int. 2015;35:1508-1515.
32. Bernardi M, Laffi G, Salvagnini M, et al. Efficacy and safety of the stepped care medical treatment of ascites in liver cirrhosis: a randomized controlled clinical trial comparing two diets with different sodium content. Liver. 1993;13:156-162.
33. Angeli P, Fasolato S, Mazza E, et al. Combined versus sequential diuretic treatment of ascites in non-azotaemic patients with cirrhosis: results of an open randomised clinical trial. Gut. 2010;59:98-104.
34. Santos J, Planas R, Pardo A, et al. Spironolactone alone or in combination with furosemide in the treatment of moderate ascites in nonazotemic cirrhosis. A randomized comparative study of efficacy and safety. J Hepatol. 2003;39:187–192.
35. Grattagliano I, Ubaldi E, Bonfrate L, et al. Management of liver cirrhosis between primary care and specialists. World J Gastroenterol. 2011;17:2273-2282.
36. Pockros PJ, Reynolds TB. Rapid diuresis in patients with ascites from chronic liver disease: the importance of peripheral edema. Gastroenterology. 1986;90:1827-1833.
37. EASL clinical practice guidelines on the management of ascites, spontaneous bacterial peritonitis, and hepatorenal syndrome in cirrhosis. J Hepatol. 2010;53:397-417.
38. Gines P, Arroyo V, Quintero E, et al. Comparison of paracentesis and diuretics in the treatment of cirrhotics with tense ascites. Results of a randomized study. Gastroenterology. 1987;93:234-241.
39. Salerno F, Badalamenti S, Incerti P, et al. Repeated paracentesis and i.v. albumin infusion to treat ‘tense’ ascites in cirrhotic patients. A safe alternative therapy. J Hepatol. 1987;5:102-108.
40. Sola R, Vila MC, Andreu M, et al. Total paracentesis with dextran 40 vs diuretics in the treatment of ascites in cirrhosis: a randomized controlled study. J Hepatol. 1994;20:282-288.
41. Bernardi M, Caraceni P, Navickis RJ, et al. Albumin infusion in patients undergoing large-volume paracentesis: a meta-analysis of randomized trials. Hepatology. 2012;55:1172-1181.
42. Bureau C, Thabut D, Oberti F, et al. Transjugular intrahepatic portosystemic shunts with covered stents increase transplant-free survival of patients with cirrhosis and recurrent ascites. Gastroenterology. 2017;152:157-163.
43. Fagiuoli S, Bruno R, Debernardi Venon W, et al. Consensus conference on TIPS management: techniques, indications, contraindications. Dig Liver Dis. 2017;49:121-137.
44. Ferenci P, Lockwood A, Mullen K, et al. Hepatic encephalopathy—definition, nomenclature, diagnosis, and quantification: final report of the working party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology. 2002;35:716-721.
45. Salerno F, Guevara M, Bernardi M, et al. Refractory ascites: pathogenesis, definition and therapy of a severe complication in patients with cirrhosis. Liver Int. 2010;30:937-947.
46. Caraceni P, Riggio O, Angeli P, et al. Long-term albumin administration in decompensated cirrhosis (ANSWER): an open-label randomised trial. Lancet. 2018;391:2417-2429.
47. Solà E, Solé C, Simón-Talero M, et al. Midodrine and albumin for prevention of complications in patients with cirrhosis awaiting liver transplantation. A randomized placebo-controlled trial. J Hepatol. 2018;69:1250-1259.
48. Fasolato S, Angeli P, Dallagnese L, et al. Renal failure and bacterial infections in patients with cirrhosis: epidemiology and clinical features. Hepatology. 2007;45:223-229.
49. Hoefs JC, Canawati HN, Sapico FL, et al. Spontaneous bacterial peritonitis. Hepatology. 2007;2:399-407.
50. Felisart J, Rimola A, Arroyo V, et al. Cefotaxime is more effective than is ampicillin-tobramycin in cirrhotics with severe infections. Hepatology. 1985;5:457-462.
51. Lenz K, Kapral C, Gegenhuber A, et al. Systemic, renal, and hepatic hemodynamic derangement in cirrhotic patients with spontaneous bacterial peritonitis. Hepatology. 2004;39:865-866.
52. Sigal SH, Stanca CM, Fernandez J, et al. Restricted use of albumin for spontaneous bacterial peritonitis. Gut. 2007;56:597-599.
53. Fernández J, Navasa M, Planas R, et al. Primary prophylaxis of spontaneous bacterial peritonitis delays hepatorenal syndrome and improves survival in cirrhosis. Gastroenterology. 2007;133:818-824.
54. Boyer TD, Zia P, Reynolds TB. Effect of indomethacin and prostaglandin A1 on renal function and plasma renin activity in alcoholic liver disease. Gastroenterology. 1979;77:215-222.
55. Grattagliano I, Ubaldi E, Portincasa P, et al. Liver disease: early signs you may be missing. J Fam Pract. 2009;58:514-521.
56. Bini EJ, Weinshel EH, Generoso R, et al. Impact of gastroenterology consultation on the outcomes of patients admitted to the hospital with decompensated cirrhosis. Hepatology. 2001;34:1089-1095.
57. Volk ML, Tocco RS, Bazick J, et al. Hospital readmissions among patients with decompensated cirrhosis. Am J Gastroenterol. 2012;107:247-252.
58. Morando F, Maresio G, Piano S, et al. How to improve care in outpatients with cirrhosis and ascites: a new model of care coordination by consultant hepatologists. J Hepatol. 2013;59:257-264.
Liver cirrhosis is implicated in 75% to 85% of ascites cases in the Western world, with heart failure or malignancy accounting for fewer cases.1 Among patients who have decompensated cirrhosis with ascites, annual mortality is 20%.2 Another study showed a 3-year survival rate after onset of ascites of only 56%.3 It is vital for primary care physicians (PCPs) to be alert for ascites not only in patients who have risk factors for chronic liver disease and cirrhosis—eg, a history of alcohol use disorder, chronic viral infections (hepatitis B and C), or metabolic syndrome—but also in patients with abnormal liver function tests and thrombocytopenia. In this review, we discuss the initial assessment of ascites and its long-term management, concentrating on the role of the PCP.
Pathophysiology: Vasodilation leads to a cascade
Splanchnic vasodilation is the main underlying event triggering a pathologic cascade that leads to the development of ascites.4 Initially portal hypertension in the setting of liver inflammation and fibrosis causes the release of inflammatory cytokines such as nitric oxide and carbon monoxide. This, in turn, causes the pathologic dilation of splanchnic circulation that decreases effective circulating volume. Activation of the sympathetic nervous system, vasopressin, and renin-angiotensin-aldosterone system (RAAS) then causes the proximal and distal tubules to increase renal absorption of sodium and water.5 The resulting volume overload further decreases the heart’s ability to maintain circulating volume, leading to increased activation of compensating symptoms. This vicious cycle eventually manifests as ascites.6
A complex interplay of cirrhosis-associated immune dysfunction (CAID), gut dysbiosis, and increased translocation of microorganisms into ascitic fluid is also an important aspect of the pathogenesis.7 CAID (FIGURE 1)7,8 is an immunodeficient state due to cirrhosis with reduced phagocytic activity by neutrophils and macrophages, T- and B-cell hypoproliferation, and reduced cytotoxicity of natural killer cells. In parallel, there is increased production of inflammatory cytokines due to the effects of damage-associated molecular patterns (DAMPs) from hepatocytes and pathogen-associated molecular patterns (PAMPs) from the gut microbiota on the immune system, which leads to many of the manifestations of decompensated cirrhosis including ascites.8
Key in on these elementsof the history and exam
Each step of the basic work-up for ascites provides opportunities to refine or redirect the diagnostic inquiry (TABLE).
History
Generally, patients with ascites present with weight gain and symptoms of abdominal distension, such as early satiety, nausea, and vomiting. Besides cirrhosis, rule out other causes of ascites, as treatment differs based on the cause.9 Also ask about histories of cancer and cardiac, renal, or thyroid disease.10
Patients with ascites in the setting of liver disease usually are asymptomatic in its early stages. Common complaints are vague abdominal pain, generalized weakness, malaise, and fatigue.11 Ask patients about risk factors for liver disease such as obesity, diabetes, hypertension, alcohol use, unsafe sexual practices, recent travel, and needle sharing or drug use. Due to a strong association between obstructive sleep apnea and fatty liver disease, consider screening at-risk patients for sleep apnea.12
Physical exam
When there are risk factors for liver disease, examine the patient for stigmata of cirrhosis and ascites. Signs of liver disease, aside from ascites, may include spider angiomas on the upper trunk (33% of cirrhosis patients),13 gynecomastia (44% of cirrhosis patients),14 palmar erythema, jaundice, asterixis, and abdominal wall collaterals including caput medusa.15
Continue to: We suggest a systematic...
We suggest a systematic and targeted approach to using various physical exam maneuvers described in the literature. If the patient has a full/distended abdomen, percuss the flanks. If increased dullness at the flanks is detected, check for shifting dullness, which indicates at least 1500 mL of fluid in the abdomen.16 Keep in mind that a 10% chance of ascites exists even if shifting dullness is absent.17 Maneuvers such as the puddle sign and fluid thrill are less accurate than shifting dullness, which has 83% sensitivity and 56% specificity in detecting ascites.17 Patients with cirrhosis also have a high likelihood of complications from ascites such as inguinal, umbilical, and other hernias.
Diagnostic work-up includes blood tests and ultrasound
Blood tests. The initial work-up for ascites should include complete blood count, complete metabolic panel, and prothrombin time/international normalized ratio.18
Abdominal ultrasound is recommended as the first-line imaging test.19 Aside from detecting ascites, it can give an estimate of the volume of ascites and indicate whether it is amenable to paracentesis. A vascular exam added to the standard ultrasound can detect radiologic evidence of portal hypertension such as splenomegaly, portosystemic collaterals, splenorenal shunt, patency of the paraumbilical vein, and portal vein diameter. Patients with established cirrhosis also require abdominal ultrasound every 6 months to screen for hepatocellular cancer.20
Abdominal paracentesis is the cornerstone of ascites evaluation.21 It is indicated for every patient with new-onset ascites or for any patient with known ascites and clinical deterioration. Ascitic fluid analysis can be used to easily differentiate portal hypertension from other causes of ascites. It can also be used to rule out bacterial peritonitis. The recommended sites for evaluation are in the left lower quadrant, 3 cm cranially and 3 cm medially from the anterior superior iliac spine.22 A large cohort study showed that abdominal ultrasound-guided paracentesis reduced bleeding complications by 68% following the procedure and is strongly recommended (if available).23 Generally, paracentesis is a relatively safe procedure with a low risk of complications such as abdominal wall hematoma (1%), hemoperitoneum (< 0.1%), bowel perforation (< 0.1%), and infection (< 0.1%).24
Assess all ascitic fluid samples for color, consistency, cell count and differential, albumin, and total protein. These tests are usually sufficient to provide evidence regarding the cause of ascites. If there is suspicion of infection, order a gram stain and culture (80% sensitivity for detecting an infection if obtained prior to initiation of antibiotics)25 and glucose, lactate dehydrogenase (useful to differentiate primary from secondary bacterial peritonitis),26 and amylase tests. Other tests such as cytology, acid-fast bacilli smear and culture, and triglyceride level should only be obtained if specific conditions are suspected based on high pretest probabilities.
Continue to: Calculating serum ascites albumin gradient...
Calculating serum ascites albumin gradient (SAAG) is recommended as it has been shown to better characterize ascitic fluid than total protein-based tests.27 SAAG is calculated by subtracting the level of ascitic fluid albumin from serum albumin level (SAAG = serum albumin – ascitic fluid albumin). A SAAG ≥ 1.1 g/dL is consistent with portal hypertension,28 with approximately 97% accuracy.
After calculating SAAG, look at total protein levels in ascitic fluid. Total protein concentration ≥ 2.5 g/dL with SAAG ≥ 1.1 g/dL has a 78.3% diagnostic accuracy in determining heart failure as the cause of ascites, with a sensitivity of 53.3% and specificity of 86.7%.28 On the other hand, a value of total protein < 2.5 g/dL indicates cirrhosis, liver failure, or acute hepatitis as the cause of fluid build-up.29 Stepwise evaluation of SAAG and total protein and how they can point toward the most likely cause of ascites is presented in FIGURE 2.27-29
Management
Noninvasive measures
Sodium restriction. The aim of treatment for uncomplicated clinically apparent ascites is sodium restriction and removal of fluid from the body. Dietary salt restriction is complicated, and care should be taken to properly educate patients. Salt restriction advised in the literature has shifted from a strict measure of < 2 g/d30 to more moderate strategies (described below).18
The 2 main reasons for this easing of restriction are issues with patient compliance and concerns about adverse effects with aggressive salt-restricted diets. One study assessing patient compliance with a salt-restricted diet found that more than two-thirds of the patients were noncompliant,31 and 65% of the patients incorrectly assumed they were following the plan, which suggests poor dietary education.31 Of the group that was compliant, 20% actually decreased their caloric intake, which can be detrimental in liver disease.31 Concerns have been raised that aggressive salt restriction along with diuretic use can lead to diuretic-induced hyponatremia and renal failure.32 Current European Association for the Study of the Liver (EASL) guidelines recommend salt restriction to a more moderate degree (80-120 mmol/d of sodium). This is equivalent to 4.9-6.9 g of salt (1 tablespoon is roughly equivalent to 6 g or 104 mmol of sodium).18
Diuretics. Initiation and dosage of diuretic therapy is a matter of some controversy. Historically, simultaneous administration of a loop diuretic and mineralocorticoid receptor blocker were recommended: 40 mg furosemide and 100 mg spironolactone, keeping the ratio constant with any dosage increases. This was based on a randomized controlled trial (RCT) showing that the combined diuretic therapy effectively mobilized ascites in a shorter period of time and with less frequent adverse effects (eg, hyperkalemia) compared with initial monotherapy.33
Continue to: On the other hand...
On the other hand, another study with more stable patients and relatively normal renal function showed that starting with a mineralocorticoid receptor blocker alone with sequential dose increments had equivalent benefit with no increase in adverse effects.34 Since the patient population in this study was more in line with what a PCP might encounter, we recommend following this guideline initially and keeping a close watch on serum electrolytes.
Usual maximum doses are spironolactone 400 mg/d and furosemide 160 mg/d.21,35 Adequate weight loss for patients with diffuse edema is at least 1 kg/d, per EASL guidelines.36,37 However, this might not be practical in outpatient settings, and a more conservative target of 0.5 kg/d may be used for patients without significant edema.37
It is vital to get accurate daily weights and avoid excessive diuretic use, as it has been associated with intravascular volume depletion and acute kidney injury (25%), hyponatremia (28%),38,39 and hepatic encephalopathy (30%).40 Therefore, patients with acute kidney injury, hyponatremia, acute variceal hemorrhage, or infection should also have their diuretics held until their creatinine returns to baseline.
Invasive measures
Large-volume paracentesis. Patients with extensive and tense ascites should be treated initially with large-volume paracentesis, as this has been shown to predictably remove fluid more effectively than diuretics.38 This should be accompanied by albumin administration, 8 g for every liter of ascitic fluid removed if the total amount exceeds 5 L.41 Following large-volume paracentesis, manage patients with the standard salt restriction and diuretic regimen.38 Serial large-volume paracentesis is a temporary measure reserved for a select group of patients who are intolerant to diuretics and are not candidates for a shunt.
Transjugular intrahepatic portosystemic shunt (TIPS) is another option to control refractory ascites, but its benefit should be weighed against complications such as hepatic encephalopathy. An RCT found that TIPS with covered stents improved survival in patients with cirrhosis compared with regular large-volume paracentesis.42 Patients should be referred to hepatologists to make a determination about TIPS placement. Widely accepted contraindications for the placement of TIPS are decompensated cirrhosis (Child-Pugh > 11, model for end-stage liver disease [MELD] > 18), renal failure (serum creatinine > 3 mg/dL), heart failure, porto-pulmonary hypertension, and uncontrolled sepsis.43 Recurrent or persistent hepatic encephalopathy (West Haven grade ≥ 2) is also a contraindication. The West Haven scale is widely used to measure severity of hepatic encephalopathy, grading it from 1 to 4, with 1 being mild encephalopathy characterized by lack of awareness and shorter attention span, and 4 indicating unresponsiveness or coma.44
Continue to: How to manage refractory ascites
How to manage refractory ascites
Fragile patients are those with refractory ascites that is either unresponsive to standard salt restriction and maximum-dose diuretic therapy or that results in a re-accumulation of ascitic fluid soon after paracentesis.45 Specialist care is required to improve survival and quality of life for these patients. They should be referred to a hepatologist for consideration of TIPS placement or liver transplantation.18
Long-term use of albumin was tested in 2 trials for management of decompensated cirrhosis with ascites, yielding conflicting results. The ANSWER trial from Italy showed benefit with this treatment for prolonged survival.46 The other trial, from Spain, showed no benefit from albumin and midodrine administration for survival or for improving complications of cirrhosis.47 The contradictory results are likely due to heterogeneous populations in the 2 trials and differences in dose and duration of albumin administration. Hence, no clear recommendations can be made based on the available data; further research is needed.
Getting a handle on bacterial peritonitis
Bacterial peritonitis can be divided into spontaneous bacterial peritonitis (SBP) and secondary bacterial peritonitis. SBP is a common complication in patients with cirrhosis and occurs in around 16% of hospitalized patients, based on 1 study.48 SBP is defined as a polymorphonuclear leukocyte count ≥ 250 cells/μL in the absence of a surgically treatable source of infection.49 It is believed to be caused by bacterial translocation and is treated empirically with a third-generation cephalosporin. This treatment has been shown to be effective in 85% of patients.50
Patients with SBP are at a higher risk for renal impairment, likely resulting from increased cytokine production and decreased circulatory volume.51 Concomitant albumin administration has been shown to significantly improve outcomes and to reduce rates of hepatorenal syndrome in patients with serum creatinine > 1 mg/dL, blood urea nitrogen > 30 mg/dL, or total bilirubin > 4 mg/dL.52 The recommended amount of albumin is 1.5 g/kg given within 6 hours of SBP detection and repeat administration of 1 g/kg on Day 3.52
Guidelines from the American Association for the Study of Liver Diseases and from EASL recommend the long-term use of daily norfloxacin or trimethoprim-sulfamethoxazole as secondary prophylaxis in patients who have survived an episode of SBP.18
Continue to: Avoid these medications
Avoid these medications
Commonly used medications that should be avoided in patients with cirrhosis and ascites are angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. These agents block the action of angiotensin, which is a vital vasoconstrictor, and thereby cause a drop in blood pressure. This has independently been associated with poor outcomes in patients with cirrhosis.37
Nonsteroidal anti-inflammatory drugs (NSAIDs) are also relatively contraindicated in cirrhosis, as they can affect kidney function, induce azotemia, and reduce kidney sodium excretion. NSAIDs induce vasoconstriction of afferent arterioles in the kidneys, leading to a decreased glomerular filtration rate, further activating RAAS and sympathetic drive. This leads to increased sodium and water retention and worsening ascites.54
Improve outcomes by circling in a hepatologist
PCPs can play a vital role in the prevention, treatment, surveillance, and home care of patients with cirrhosis who are at risk for ascites.55 Referral of patients with hepatic impairment manifesting as unexplained abnormal liver function tests, new-onset ascites, and/or image findings consistent with cirrhosis to a hepatologist at least once is recommended. Such referrals have been shown to be associated with a better overall outcome.56 Patients with known cirrhosis leading to ascites can generally be managed at home with the assistance of specialists and specialized nurses.35
In a study from the University of Michigan, 69% of patients with cirrhosis had at least 1 nonelective readmission; 14% of patients were readmitted within 1 week, and 37% within 1 month.57 These are staggering statistics that highlight the gaps in care coordination and management of patients with cirrhosis in the outpatient setting. PCPs can play a vital role in bridging this gap.
A promising framework is suggested by a study from Italy by Morando et al in 2013.58 The researchers assessed a specialized health care model for cirrhotic patients and showed significant improvement in health care cost, readmission rate, and overall mortality when compared with the existing model of outpatient care.58
Continue to: This was not a blinded study...
This was not a blinded study and there were concerns raised by the scientific community about its design. Because it was conducted in Italy, the results might not be fully applicable to the United States health care setting. However, it did show that better coordination of care leads to significantly better patient outcomes and reduces health care expenditure. Therefore, a more complete understanding of the disease process and latest literature by PCPs, communication with specialists, and comprehensive coordination of care by all parties involved is vital for the management of this patient population.
CORRESPONDENCE
Muhammad Salman Faisal, MD, Cleveland Clinic Foundation, 9500 Euclid Avenue, Cleveland, OH 44195; faisalm@ccf.org
Liver cirrhosis is implicated in 75% to 85% of ascites cases in the Western world, with heart failure or malignancy accounting for fewer cases.1 Among patients who have decompensated cirrhosis with ascites, annual mortality is 20%.2 Another study showed a 3-year survival rate after onset of ascites of only 56%.3 It is vital for primary care physicians (PCPs) to be alert for ascites not only in patients who have risk factors for chronic liver disease and cirrhosis—eg, a history of alcohol use disorder, chronic viral infections (hepatitis B and C), or metabolic syndrome—but also in patients with abnormal liver function tests and thrombocytopenia. In this review, we discuss the initial assessment of ascites and its long-term management, concentrating on the role of the PCP.
Pathophysiology: Vasodilation leads to a cascade
Splanchnic vasodilation is the main underlying event triggering a pathologic cascade that leads to the development of ascites.4 Initially portal hypertension in the setting of liver inflammation and fibrosis causes the release of inflammatory cytokines such as nitric oxide and carbon monoxide. This, in turn, causes the pathologic dilation of splanchnic circulation that decreases effective circulating volume. Activation of the sympathetic nervous system, vasopressin, and renin-angiotensin-aldosterone system (RAAS) then causes the proximal and distal tubules to increase renal absorption of sodium and water.5 The resulting volume overload further decreases the heart’s ability to maintain circulating volume, leading to increased activation of compensating symptoms. This vicious cycle eventually manifests as ascites.6
A complex interplay of cirrhosis-associated immune dysfunction (CAID), gut dysbiosis, and increased translocation of microorganisms into ascitic fluid is also an important aspect of the pathogenesis.7 CAID (FIGURE 1)7,8 is an immunodeficient state due to cirrhosis with reduced phagocytic activity by neutrophils and macrophages, T- and B-cell hypoproliferation, and reduced cytotoxicity of natural killer cells. In parallel, there is increased production of inflammatory cytokines due to the effects of damage-associated molecular patterns (DAMPs) from hepatocytes and pathogen-associated molecular patterns (PAMPs) from the gut microbiota on the immune system, which leads to many of the manifestations of decompensated cirrhosis including ascites.8
Key in on these elementsof the history and exam
Each step of the basic work-up for ascites provides opportunities to refine or redirect the diagnostic inquiry (TABLE).
History
Generally, patients with ascites present with weight gain and symptoms of abdominal distension, such as early satiety, nausea, and vomiting. Besides cirrhosis, rule out other causes of ascites, as treatment differs based on the cause.9 Also ask about histories of cancer and cardiac, renal, or thyroid disease.10
Patients with ascites in the setting of liver disease usually are asymptomatic in its early stages. Common complaints are vague abdominal pain, generalized weakness, malaise, and fatigue.11 Ask patients about risk factors for liver disease such as obesity, diabetes, hypertension, alcohol use, unsafe sexual practices, recent travel, and needle sharing or drug use. Due to a strong association between obstructive sleep apnea and fatty liver disease, consider screening at-risk patients for sleep apnea.12
Physical exam
When there are risk factors for liver disease, examine the patient for stigmata of cirrhosis and ascites. Signs of liver disease, aside from ascites, may include spider angiomas on the upper trunk (33% of cirrhosis patients),13 gynecomastia (44% of cirrhosis patients),14 palmar erythema, jaundice, asterixis, and abdominal wall collaterals including caput medusa.15
Continue to: We suggest a systematic...
We suggest a systematic and targeted approach to using various physical exam maneuvers described in the literature. If the patient has a full/distended abdomen, percuss the flanks. If increased dullness at the flanks is detected, check for shifting dullness, which indicates at least 1500 mL of fluid in the abdomen.16 Keep in mind that a 10% chance of ascites exists even if shifting dullness is absent.17 Maneuvers such as the puddle sign and fluid thrill are less accurate than shifting dullness, which has 83% sensitivity and 56% specificity in detecting ascites.17 Patients with cirrhosis also have a high likelihood of complications from ascites such as inguinal, umbilical, and other hernias.
Diagnostic work-up includes blood tests and ultrasound
Blood tests. The initial work-up for ascites should include complete blood count, complete metabolic panel, and prothrombin time/international normalized ratio.18
Abdominal ultrasound is recommended as the first-line imaging test.19 Aside from detecting ascites, it can give an estimate of the volume of ascites and indicate whether it is amenable to paracentesis. A vascular exam added to the standard ultrasound can detect radiologic evidence of portal hypertension such as splenomegaly, portosystemic collaterals, splenorenal shunt, patency of the paraumbilical vein, and portal vein diameter. Patients with established cirrhosis also require abdominal ultrasound every 6 months to screen for hepatocellular cancer.20
Abdominal paracentesis is the cornerstone of ascites evaluation.21 It is indicated for every patient with new-onset ascites or for any patient with known ascites and clinical deterioration. Ascitic fluid analysis can be used to easily differentiate portal hypertension from other causes of ascites. It can also be used to rule out bacterial peritonitis. The recommended sites for evaluation are in the left lower quadrant, 3 cm cranially and 3 cm medially from the anterior superior iliac spine.22 A large cohort study showed that abdominal ultrasound-guided paracentesis reduced bleeding complications by 68% following the procedure and is strongly recommended (if available).23 Generally, paracentesis is a relatively safe procedure with a low risk of complications such as abdominal wall hematoma (1%), hemoperitoneum (< 0.1%), bowel perforation (< 0.1%), and infection (< 0.1%).24
Assess all ascitic fluid samples for color, consistency, cell count and differential, albumin, and total protein. These tests are usually sufficient to provide evidence regarding the cause of ascites. If there is suspicion of infection, order a gram stain and culture (80% sensitivity for detecting an infection if obtained prior to initiation of antibiotics)25 and glucose, lactate dehydrogenase (useful to differentiate primary from secondary bacterial peritonitis),26 and amylase tests. Other tests such as cytology, acid-fast bacilli smear and culture, and triglyceride level should only be obtained if specific conditions are suspected based on high pretest probabilities.
Continue to: Calculating serum ascites albumin gradient...
Calculating serum ascites albumin gradient (SAAG) is recommended as it has been shown to better characterize ascitic fluid than total protein-based tests.27 SAAG is calculated by subtracting the level of ascitic fluid albumin from serum albumin level (SAAG = serum albumin – ascitic fluid albumin). A SAAG ≥ 1.1 g/dL is consistent with portal hypertension,28 with approximately 97% accuracy.
After calculating SAAG, look at total protein levels in ascitic fluid. Total protein concentration ≥ 2.5 g/dL with SAAG ≥ 1.1 g/dL has a 78.3% diagnostic accuracy in determining heart failure as the cause of ascites, with a sensitivity of 53.3% and specificity of 86.7%.28 On the other hand, a value of total protein < 2.5 g/dL indicates cirrhosis, liver failure, or acute hepatitis as the cause of fluid build-up.29 Stepwise evaluation of SAAG and total protein and how they can point toward the most likely cause of ascites is presented in FIGURE 2.27-29
Management
Noninvasive measures
Sodium restriction. The aim of treatment for uncomplicated clinically apparent ascites is sodium restriction and removal of fluid from the body. Dietary salt restriction is complicated, and care should be taken to properly educate patients. Salt restriction advised in the literature has shifted from a strict measure of < 2 g/d30 to more moderate strategies (described below).18
The 2 main reasons for this easing of restriction are issues with patient compliance and concerns about adverse effects with aggressive salt-restricted diets. One study assessing patient compliance with a salt-restricted diet found that more than two-thirds of the patients were noncompliant,31 and 65% of the patients incorrectly assumed they were following the plan, which suggests poor dietary education.31 Of the group that was compliant, 20% actually decreased their caloric intake, which can be detrimental in liver disease.31 Concerns have been raised that aggressive salt restriction along with diuretic use can lead to diuretic-induced hyponatremia and renal failure.32 Current European Association for the Study of the Liver (EASL) guidelines recommend salt restriction to a more moderate degree (80-120 mmol/d of sodium). This is equivalent to 4.9-6.9 g of salt (1 tablespoon is roughly equivalent to 6 g or 104 mmol of sodium).18
Diuretics. Initiation and dosage of diuretic therapy is a matter of some controversy. Historically, simultaneous administration of a loop diuretic and mineralocorticoid receptor blocker were recommended: 40 mg furosemide and 100 mg spironolactone, keeping the ratio constant with any dosage increases. This was based on a randomized controlled trial (RCT) showing that the combined diuretic therapy effectively mobilized ascites in a shorter period of time and with less frequent adverse effects (eg, hyperkalemia) compared with initial monotherapy.33
Continue to: On the other hand...
On the other hand, another study with more stable patients and relatively normal renal function showed that starting with a mineralocorticoid receptor blocker alone with sequential dose increments had equivalent benefit with no increase in adverse effects.34 Since the patient population in this study was more in line with what a PCP might encounter, we recommend following this guideline initially and keeping a close watch on serum electrolytes.
Usual maximum doses are spironolactone 400 mg/d and furosemide 160 mg/d.21,35 Adequate weight loss for patients with diffuse edema is at least 1 kg/d, per EASL guidelines.36,37 However, this might not be practical in outpatient settings, and a more conservative target of 0.5 kg/d may be used for patients without significant edema.37
It is vital to get accurate daily weights and avoid excessive diuretic use, as it has been associated with intravascular volume depletion and acute kidney injury (25%), hyponatremia (28%),38,39 and hepatic encephalopathy (30%).40 Therefore, patients with acute kidney injury, hyponatremia, acute variceal hemorrhage, or infection should also have their diuretics held until their creatinine returns to baseline.
Invasive measures
Large-volume paracentesis. Patients with extensive and tense ascites should be treated initially with large-volume paracentesis, as this has been shown to predictably remove fluid more effectively than diuretics.38 This should be accompanied by albumin administration, 8 g for every liter of ascitic fluid removed if the total amount exceeds 5 L.41 Following large-volume paracentesis, manage patients with the standard salt restriction and diuretic regimen.38 Serial large-volume paracentesis is a temporary measure reserved for a select group of patients who are intolerant to diuretics and are not candidates for a shunt.
Transjugular intrahepatic portosystemic shunt (TIPS) is another option to control refractory ascites, but its benefit should be weighed against complications such as hepatic encephalopathy. An RCT found that TIPS with covered stents improved survival in patients with cirrhosis compared with regular large-volume paracentesis.42 Patients should be referred to hepatologists to make a determination about TIPS placement. Widely accepted contraindications for the placement of TIPS are decompensated cirrhosis (Child-Pugh > 11, model for end-stage liver disease [MELD] > 18), renal failure (serum creatinine > 3 mg/dL), heart failure, porto-pulmonary hypertension, and uncontrolled sepsis.43 Recurrent or persistent hepatic encephalopathy (West Haven grade ≥ 2) is also a contraindication. The West Haven scale is widely used to measure severity of hepatic encephalopathy, grading it from 1 to 4, with 1 being mild encephalopathy characterized by lack of awareness and shorter attention span, and 4 indicating unresponsiveness or coma.44
Continue to: How to manage refractory ascites
How to manage refractory ascites
Fragile patients are those with refractory ascites that is either unresponsive to standard salt restriction and maximum-dose diuretic therapy or that results in a re-accumulation of ascitic fluid soon after paracentesis.45 Specialist care is required to improve survival and quality of life for these patients. They should be referred to a hepatologist for consideration of TIPS placement or liver transplantation.18
Long-term use of albumin was tested in 2 trials for management of decompensated cirrhosis with ascites, yielding conflicting results. The ANSWER trial from Italy showed benefit with this treatment for prolonged survival.46 The other trial, from Spain, showed no benefit from albumin and midodrine administration for survival or for improving complications of cirrhosis.47 The contradictory results are likely due to heterogeneous populations in the 2 trials and differences in dose and duration of albumin administration. Hence, no clear recommendations can be made based on the available data; further research is needed.
Getting a handle on bacterial peritonitis
Bacterial peritonitis can be divided into spontaneous bacterial peritonitis (SBP) and secondary bacterial peritonitis. SBP is a common complication in patients with cirrhosis and occurs in around 16% of hospitalized patients, based on 1 study.48 SBP is defined as a polymorphonuclear leukocyte count ≥ 250 cells/μL in the absence of a surgically treatable source of infection.49 It is believed to be caused by bacterial translocation and is treated empirically with a third-generation cephalosporin. This treatment has been shown to be effective in 85% of patients.50
Patients with SBP are at a higher risk for renal impairment, likely resulting from increased cytokine production and decreased circulatory volume.51 Concomitant albumin administration has been shown to significantly improve outcomes and to reduce rates of hepatorenal syndrome in patients with serum creatinine > 1 mg/dL, blood urea nitrogen > 30 mg/dL, or total bilirubin > 4 mg/dL.52 The recommended amount of albumin is 1.5 g/kg given within 6 hours of SBP detection and repeat administration of 1 g/kg on Day 3.52
Guidelines from the American Association for the Study of Liver Diseases and from EASL recommend the long-term use of daily norfloxacin or trimethoprim-sulfamethoxazole as secondary prophylaxis in patients who have survived an episode of SBP.18
Continue to: Avoid these medications
Avoid these medications
Commonly used medications that should be avoided in patients with cirrhosis and ascites are angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. These agents block the action of angiotensin, which is a vital vasoconstrictor, and thereby cause a drop in blood pressure. This has independently been associated with poor outcomes in patients with cirrhosis.37
Nonsteroidal anti-inflammatory drugs (NSAIDs) are also relatively contraindicated in cirrhosis, as they can affect kidney function, induce azotemia, and reduce kidney sodium excretion. NSAIDs induce vasoconstriction of afferent arterioles in the kidneys, leading to a decreased glomerular filtration rate, further activating RAAS and sympathetic drive. This leads to increased sodium and water retention and worsening ascites.54
Improve outcomes by circling in a hepatologist
PCPs can play a vital role in the prevention, treatment, surveillance, and home care of patients with cirrhosis who are at risk for ascites.55 Referral of patients with hepatic impairment manifesting as unexplained abnormal liver function tests, new-onset ascites, and/or image findings consistent with cirrhosis to a hepatologist at least once is recommended. Such referrals have been shown to be associated with a better overall outcome.56 Patients with known cirrhosis leading to ascites can generally be managed at home with the assistance of specialists and specialized nurses.35
In a study from the University of Michigan, 69% of patients with cirrhosis had at least 1 nonelective readmission; 14% of patients were readmitted within 1 week, and 37% within 1 month.57 These are staggering statistics that highlight the gaps in care coordination and management of patients with cirrhosis in the outpatient setting. PCPs can play a vital role in bridging this gap.
A promising framework is suggested by a study from Italy by Morando et al in 2013.58 The researchers assessed a specialized health care model for cirrhotic patients and showed significant improvement in health care cost, readmission rate, and overall mortality when compared with the existing model of outpatient care.58
Continue to: This was not a blinded study...
This was not a blinded study and there were concerns raised by the scientific community about its design. Because it was conducted in Italy, the results might not be fully applicable to the United States health care setting. However, it did show that better coordination of care leads to significantly better patient outcomes and reduces health care expenditure. Therefore, a more complete understanding of the disease process and latest literature by PCPs, communication with specialists, and comprehensive coordination of care by all parties involved is vital for the management of this patient population.
CORRESPONDENCE
Muhammad Salman Faisal, MD, Cleveland Clinic Foundation, 9500 Euclid Avenue, Cleveland, OH 44195; faisalm@ccf.org
1. Runyon BA, Montano AA, Akriviadis EA, et al. The serum-ascites albumin gradient is superior to the exudate-transudate concept in the differential diagnosis of ascites. Ann Intern Med. 1992;117:215-220.
2. D’Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol. 2006;44:217-231.
3. Gordon FD. Ascites. Clin Liver Dis. 2012;16:285-299.
4. Schrier RW, Arroyo V, Bernardi M, et al. Peripheral arterial vasodilation hypothesis: a proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology. 1988;8:1151-1157.
5. Arroyo V, Terra C, Gines P. Advances in the pathogenesis and treatment of type-1 and type-2 hepatorenal syndrome. J Hepatol. 2007;46:935-946.
6. Bernardi M, Moreau R, Angeli P, et al. Mechanisms of decompensation and organ failure in cirrhosis: from peripheral arterial vasodilation to systemic inflammation hypothesis. J Hepatol. 2015;63:1272-1284.
7. Jalan R, Fernandez J, Wiest R, et al. Bacterial infections in cirrhosis: a position statement based on the EASL Special Conference 2013. J Hepatol. 2014;60:1310-1324.
8. Albillos A, Lario M, Álvarez-Mon M. Cirrhosis-associated immune dysfunction: distinctive features and clinical relevance. J Hepatol. 2014;61:1385-1396.
9. Oey RC, van Buuren HR, de Man RA. The diagnostic work-up in patients with ascites: current guidelines and future prospects. Neth J Med. 2016;74:330-335.
10. de Kerguenec C, Hillaire S, Molinié V, et al. Hepatic manifestations of hemophagocytic syndrome: a study of 30 cases. Am J Gastroenterol. 2001;96:852-857.
11. Milić S, Lulić D, Štimac D. Non-alcoholic fatty liver disease and obesity: biochemical, metabolic and clinical presentations. World J Gastroenterol. 2014;20:9330-9337.
12. Aron-Wisnewsky J, Clement K, Pépin J-L. Nonalcoholic fatty liver disease and obstructive sleep apnea. Metabolism. 2016;65:1124-1135.
13. Li CP, Lee FY, Hwang SJ, et al. Spider angiomas in patients with liver cirrhosis: role of alcoholism and impaired liver function. Scand J Gastroenterol. 1999;34:520-523.
14. Cavanaugh J. Gynecomastia and cirrhosis of the liver. Arch Intern Med. 1990;150:563-565.
15. Karnath B. Stigmata of chronic liver disease. Hosp Phys. 2003;7:14-16,28.
16. Schipper HG, Godfried MH. [Physical diagnosis--ascites]. Ned Tijdschr Geneeskd. 2001;145:260-264.
17. Cattau EL, Jr., Benjamin SB, Knuff TE, et al. The accuracy of the physical examination in the diagnosis of suspected ascites. JAMA. 1982;247:1164-1166.
18. EASL clinical practice guidelines for the management of patients with decompensated cirrhosis. J Hepatol. 2018;69:406-460.
19. Runyon BA, AASLD Practice Guidelines Committee. Management of adult patients with ascites due to cirrhosis: an update. Hepatology 2009;49:2087-2107.
20. EASL Clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69:182-236.
21. Runyon BA. Care of patients with ascites. New Engl J Med. 1994;330:337-342.
22. Sakai H, Sheer TA, Mendler MH, et al. Choosing the location for non-image guided abdominal paracentesis. Liver Int. 2005;25:984-986.
23. Mercaldi CJ, Lanes SF. Ultrasound guidance decreases complications and improves the cost of care among patients undergoing thoracentesis and paracentesis. Chest. 2013;143:532-538.
24. Ennis J, Schultz G, Perera P, et al. Ultrasound for detection of ascites and for guidance of the paracentesis procedure: technique and review of the literature. Int J Clin Med. 2014;5:1277-1293.
25. Runyon BA, Canawati HN, Akriviadis EA. Optimization of ascitic fluid culture technique. Gastroenterology. 1988;95:1351-1355.
26. Akriviadis EA, Runyon BA. Utility of an algorithm in differentiating spontaneous from secondary bacterial peritonitis. Gastroenterology 1990;98:127-133.
27. Hoefs JC. Serum protein concentration and portal pressure determine the ascitic fluid protein concentration in patients with chronic liver disease. J Lab Clin Med. 1983;102:260-273.
28. Farias AQ, Silvestre OM, Garcia-Tsao G, et al. Serum B-type natriuretic peptide in the initial workup of patients with new onset ascites: a diagnostic accuracy study. Hepatology. 2014;59:1043-1051.
29. Gupta R, Misra SP, Dwivedi M, et al. Diagnosing ascites: value of ascitic fluid total protein, albumin, cholesterol, their ratios, serum-ascites albumin and cholesterol gradient. J Gastroenterol Hepatol. 1995;10:295-299.
30. Runyon BA. Management of adult patients with ascites due to cirrhosis: update 2012. AASLD Practice Guideline. Accessed April 28, 2021. www.aasld.org/sites/default/files/2019-06/AASLDPracticeGuidelineAsciteDuetoCirrhosisUpdate2012Edition4_.pdf
31. Morando F, Rosi S, Gola E, et al. Adherence to a moderate sodium restriction diet in outpatients with cirrhosis and ascites: a real-life cross-sectional study. Liver Int. 2015;35:1508-1515.
32. Bernardi M, Laffi G, Salvagnini M, et al. Efficacy and safety of the stepped care medical treatment of ascites in liver cirrhosis: a randomized controlled clinical trial comparing two diets with different sodium content. Liver. 1993;13:156-162.
33. Angeli P, Fasolato S, Mazza E, et al. Combined versus sequential diuretic treatment of ascites in non-azotaemic patients with cirrhosis: results of an open randomised clinical trial. Gut. 2010;59:98-104.
34. Santos J, Planas R, Pardo A, et al. Spironolactone alone or in combination with furosemide in the treatment of moderate ascites in nonazotemic cirrhosis. A randomized comparative study of efficacy and safety. J Hepatol. 2003;39:187–192.
35. Grattagliano I, Ubaldi E, Bonfrate L, et al. Management of liver cirrhosis between primary care and specialists. World J Gastroenterol. 2011;17:2273-2282.
36. Pockros PJ, Reynolds TB. Rapid diuresis in patients with ascites from chronic liver disease: the importance of peripheral edema. Gastroenterology. 1986;90:1827-1833.
37. EASL clinical practice guidelines on the management of ascites, spontaneous bacterial peritonitis, and hepatorenal syndrome in cirrhosis. J Hepatol. 2010;53:397-417.
38. Gines P, Arroyo V, Quintero E, et al. Comparison of paracentesis and diuretics in the treatment of cirrhotics with tense ascites. Results of a randomized study. Gastroenterology. 1987;93:234-241.
39. Salerno F, Badalamenti S, Incerti P, et al. Repeated paracentesis and i.v. albumin infusion to treat ‘tense’ ascites in cirrhotic patients. A safe alternative therapy. J Hepatol. 1987;5:102-108.
40. Sola R, Vila MC, Andreu M, et al. Total paracentesis with dextran 40 vs diuretics in the treatment of ascites in cirrhosis: a randomized controlled study. J Hepatol. 1994;20:282-288.
41. Bernardi M, Caraceni P, Navickis RJ, et al. Albumin infusion in patients undergoing large-volume paracentesis: a meta-analysis of randomized trials. Hepatology. 2012;55:1172-1181.
42. Bureau C, Thabut D, Oberti F, et al. Transjugular intrahepatic portosystemic shunts with covered stents increase transplant-free survival of patients with cirrhosis and recurrent ascites. Gastroenterology. 2017;152:157-163.
43. Fagiuoli S, Bruno R, Debernardi Venon W, et al. Consensus conference on TIPS management: techniques, indications, contraindications. Dig Liver Dis. 2017;49:121-137.
44. Ferenci P, Lockwood A, Mullen K, et al. Hepatic encephalopathy—definition, nomenclature, diagnosis, and quantification: final report of the working party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology. 2002;35:716-721.
45. Salerno F, Guevara M, Bernardi M, et al. Refractory ascites: pathogenesis, definition and therapy of a severe complication in patients with cirrhosis. Liver Int. 2010;30:937-947.
46. Caraceni P, Riggio O, Angeli P, et al. Long-term albumin administration in decompensated cirrhosis (ANSWER): an open-label randomised trial. Lancet. 2018;391:2417-2429.
47. Solà E, Solé C, Simón-Talero M, et al. Midodrine and albumin for prevention of complications in patients with cirrhosis awaiting liver transplantation. A randomized placebo-controlled trial. J Hepatol. 2018;69:1250-1259.
48. Fasolato S, Angeli P, Dallagnese L, et al. Renal failure and bacterial infections in patients with cirrhosis: epidemiology and clinical features. Hepatology. 2007;45:223-229.
49. Hoefs JC, Canawati HN, Sapico FL, et al. Spontaneous bacterial peritonitis. Hepatology. 2007;2:399-407.
50. Felisart J, Rimola A, Arroyo V, et al. Cefotaxime is more effective than is ampicillin-tobramycin in cirrhotics with severe infections. Hepatology. 1985;5:457-462.
51. Lenz K, Kapral C, Gegenhuber A, et al. Systemic, renal, and hepatic hemodynamic derangement in cirrhotic patients with spontaneous bacterial peritonitis. Hepatology. 2004;39:865-866.
52. Sigal SH, Stanca CM, Fernandez J, et al. Restricted use of albumin for spontaneous bacterial peritonitis. Gut. 2007;56:597-599.
53. Fernández J, Navasa M, Planas R, et al. Primary prophylaxis of spontaneous bacterial peritonitis delays hepatorenal syndrome and improves survival in cirrhosis. Gastroenterology. 2007;133:818-824.
54. Boyer TD, Zia P, Reynolds TB. Effect of indomethacin and prostaglandin A1 on renal function and plasma renin activity in alcoholic liver disease. Gastroenterology. 1979;77:215-222.
55. Grattagliano I, Ubaldi E, Portincasa P, et al. Liver disease: early signs you may be missing. J Fam Pract. 2009;58:514-521.
56. Bini EJ, Weinshel EH, Generoso R, et al. Impact of gastroenterology consultation on the outcomes of patients admitted to the hospital with decompensated cirrhosis. Hepatology. 2001;34:1089-1095.
57. Volk ML, Tocco RS, Bazick J, et al. Hospital readmissions among patients with decompensated cirrhosis. Am J Gastroenterol. 2012;107:247-252.
58. Morando F, Maresio G, Piano S, et al. How to improve care in outpatients with cirrhosis and ascites: a new model of care coordination by consultant hepatologists. J Hepatol. 2013;59:257-264.
1. Runyon BA, Montano AA, Akriviadis EA, et al. The serum-ascites albumin gradient is superior to the exudate-transudate concept in the differential diagnosis of ascites. Ann Intern Med. 1992;117:215-220.
2. D’Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol. 2006;44:217-231.
3. Gordon FD. Ascites. Clin Liver Dis. 2012;16:285-299.
4. Schrier RW, Arroyo V, Bernardi M, et al. Peripheral arterial vasodilation hypothesis: a proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology. 1988;8:1151-1157.
5. Arroyo V, Terra C, Gines P. Advances in the pathogenesis and treatment of type-1 and type-2 hepatorenal syndrome. J Hepatol. 2007;46:935-946.
6. Bernardi M, Moreau R, Angeli P, et al. Mechanisms of decompensation and organ failure in cirrhosis: from peripheral arterial vasodilation to systemic inflammation hypothesis. J Hepatol. 2015;63:1272-1284.
7. Jalan R, Fernandez J, Wiest R, et al. Bacterial infections in cirrhosis: a position statement based on the EASL Special Conference 2013. J Hepatol. 2014;60:1310-1324.
8. Albillos A, Lario M, Álvarez-Mon M. Cirrhosis-associated immune dysfunction: distinctive features and clinical relevance. J Hepatol. 2014;61:1385-1396.
9. Oey RC, van Buuren HR, de Man RA. The diagnostic work-up in patients with ascites: current guidelines and future prospects. Neth J Med. 2016;74:330-335.
10. de Kerguenec C, Hillaire S, Molinié V, et al. Hepatic manifestations of hemophagocytic syndrome: a study of 30 cases. Am J Gastroenterol. 2001;96:852-857.
11. Milić S, Lulić D, Štimac D. Non-alcoholic fatty liver disease and obesity: biochemical, metabolic and clinical presentations. World J Gastroenterol. 2014;20:9330-9337.
12. Aron-Wisnewsky J, Clement K, Pépin J-L. Nonalcoholic fatty liver disease and obstructive sleep apnea. Metabolism. 2016;65:1124-1135.
13. Li CP, Lee FY, Hwang SJ, et al. Spider angiomas in patients with liver cirrhosis: role of alcoholism and impaired liver function. Scand J Gastroenterol. 1999;34:520-523.
14. Cavanaugh J. Gynecomastia and cirrhosis of the liver. Arch Intern Med. 1990;150:563-565.
15. Karnath B. Stigmata of chronic liver disease. Hosp Phys. 2003;7:14-16,28.
16. Schipper HG, Godfried MH. [Physical diagnosis--ascites]. Ned Tijdschr Geneeskd. 2001;145:260-264.
17. Cattau EL, Jr., Benjamin SB, Knuff TE, et al. The accuracy of the physical examination in the diagnosis of suspected ascites. JAMA. 1982;247:1164-1166.
18. EASL clinical practice guidelines for the management of patients with decompensated cirrhosis. J Hepatol. 2018;69:406-460.
19. Runyon BA, AASLD Practice Guidelines Committee. Management of adult patients with ascites due to cirrhosis: an update. Hepatology 2009;49:2087-2107.
20. EASL Clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69:182-236.
21. Runyon BA. Care of patients with ascites. New Engl J Med. 1994;330:337-342.
22. Sakai H, Sheer TA, Mendler MH, et al. Choosing the location for non-image guided abdominal paracentesis. Liver Int. 2005;25:984-986.
23. Mercaldi CJ, Lanes SF. Ultrasound guidance decreases complications and improves the cost of care among patients undergoing thoracentesis and paracentesis. Chest. 2013;143:532-538.
24. Ennis J, Schultz G, Perera P, et al. Ultrasound for detection of ascites and for guidance of the paracentesis procedure: technique and review of the literature. Int J Clin Med. 2014;5:1277-1293.
25. Runyon BA, Canawati HN, Akriviadis EA. Optimization of ascitic fluid culture technique. Gastroenterology. 1988;95:1351-1355.
26. Akriviadis EA, Runyon BA. Utility of an algorithm in differentiating spontaneous from secondary bacterial peritonitis. Gastroenterology 1990;98:127-133.
27. Hoefs JC. Serum protein concentration and portal pressure determine the ascitic fluid protein concentration in patients with chronic liver disease. J Lab Clin Med. 1983;102:260-273.
28. Farias AQ, Silvestre OM, Garcia-Tsao G, et al. Serum B-type natriuretic peptide in the initial workup of patients with new onset ascites: a diagnostic accuracy study. Hepatology. 2014;59:1043-1051.
29. Gupta R, Misra SP, Dwivedi M, et al. Diagnosing ascites: value of ascitic fluid total protein, albumin, cholesterol, their ratios, serum-ascites albumin and cholesterol gradient. J Gastroenterol Hepatol. 1995;10:295-299.
30. Runyon BA. Management of adult patients with ascites due to cirrhosis: update 2012. AASLD Practice Guideline. Accessed April 28, 2021. www.aasld.org/sites/default/files/2019-06/AASLDPracticeGuidelineAsciteDuetoCirrhosisUpdate2012Edition4_.pdf
31. Morando F, Rosi S, Gola E, et al. Adherence to a moderate sodium restriction diet in outpatients with cirrhosis and ascites: a real-life cross-sectional study. Liver Int. 2015;35:1508-1515.
32. Bernardi M, Laffi G, Salvagnini M, et al. Efficacy and safety of the stepped care medical treatment of ascites in liver cirrhosis: a randomized controlled clinical trial comparing two diets with different sodium content. Liver. 1993;13:156-162.
33. Angeli P, Fasolato S, Mazza E, et al. Combined versus sequential diuretic treatment of ascites in non-azotaemic patients with cirrhosis: results of an open randomised clinical trial. Gut. 2010;59:98-104.
34. Santos J, Planas R, Pardo A, et al. Spironolactone alone or in combination with furosemide in the treatment of moderate ascites in nonazotemic cirrhosis. A randomized comparative study of efficacy and safety. J Hepatol. 2003;39:187–192.
35. Grattagliano I, Ubaldi E, Bonfrate L, et al. Management of liver cirrhosis between primary care and specialists. World J Gastroenterol. 2011;17:2273-2282.
36. Pockros PJ, Reynolds TB. Rapid diuresis in patients with ascites from chronic liver disease: the importance of peripheral edema. Gastroenterology. 1986;90:1827-1833.
37. EASL clinical practice guidelines on the management of ascites, spontaneous bacterial peritonitis, and hepatorenal syndrome in cirrhosis. J Hepatol. 2010;53:397-417.
38. Gines P, Arroyo V, Quintero E, et al. Comparison of paracentesis and diuretics in the treatment of cirrhotics with tense ascites. Results of a randomized study. Gastroenterology. 1987;93:234-241.
39. Salerno F, Badalamenti S, Incerti P, et al. Repeated paracentesis and i.v. albumin infusion to treat ‘tense’ ascites in cirrhotic patients. A safe alternative therapy. J Hepatol. 1987;5:102-108.
40. Sola R, Vila MC, Andreu M, et al. Total paracentesis with dextran 40 vs diuretics in the treatment of ascites in cirrhosis: a randomized controlled study. J Hepatol. 1994;20:282-288.
41. Bernardi M, Caraceni P, Navickis RJ, et al. Albumin infusion in patients undergoing large-volume paracentesis: a meta-analysis of randomized trials. Hepatology. 2012;55:1172-1181.
42. Bureau C, Thabut D, Oberti F, et al. Transjugular intrahepatic portosystemic shunts with covered stents increase transplant-free survival of patients with cirrhosis and recurrent ascites. Gastroenterology. 2017;152:157-163.
43. Fagiuoli S, Bruno R, Debernardi Venon W, et al. Consensus conference on TIPS management: techniques, indications, contraindications. Dig Liver Dis. 2017;49:121-137.
44. Ferenci P, Lockwood A, Mullen K, et al. Hepatic encephalopathy—definition, nomenclature, diagnosis, and quantification: final report of the working party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology. 2002;35:716-721.
45. Salerno F, Guevara M, Bernardi M, et al. Refractory ascites: pathogenesis, definition and therapy of a severe complication in patients with cirrhosis. Liver Int. 2010;30:937-947.
46. Caraceni P, Riggio O, Angeli P, et al. Long-term albumin administration in decompensated cirrhosis (ANSWER): an open-label randomised trial. Lancet. 2018;391:2417-2429.
47. Solà E, Solé C, Simón-Talero M, et al. Midodrine and albumin for prevention of complications in patients with cirrhosis awaiting liver transplantation. A randomized placebo-controlled trial. J Hepatol. 2018;69:1250-1259.
48. Fasolato S, Angeli P, Dallagnese L, et al. Renal failure and bacterial infections in patients with cirrhosis: epidemiology and clinical features. Hepatology. 2007;45:223-229.
49. Hoefs JC, Canawati HN, Sapico FL, et al. Spontaneous bacterial peritonitis. Hepatology. 2007;2:399-407.
50. Felisart J, Rimola A, Arroyo V, et al. Cefotaxime is more effective than is ampicillin-tobramycin in cirrhotics with severe infections. Hepatology. 1985;5:457-462.
51. Lenz K, Kapral C, Gegenhuber A, et al. Systemic, renal, and hepatic hemodynamic derangement in cirrhotic patients with spontaneous bacterial peritonitis. Hepatology. 2004;39:865-866.
52. Sigal SH, Stanca CM, Fernandez J, et al. Restricted use of albumin for spontaneous bacterial peritonitis. Gut. 2007;56:597-599.
53. Fernández J, Navasa M, Planas R, et al. Primary prophylaxis of spontaneous bacterial peritonitis delays hepatorenal syndrome and improves survival in cirrhosis. Gastroenterology. 2007;133:818-824.
54. Boyer TD, Zia P, Reynolds TB. Effect of indomethacin and prostaglandin A1 on renal function and plasma renin activity in alcoholic liver disease. Gastroenterology. 1979;77:215-222.
55. Grattagliano I, Ubaldi E, Portincasa P, et al. Liver disease: early signs you may be missing. J Fam Pract. 2009;58:514-521.
56. Bini EJ, Weinshel EH, Generoso R, et al. Impact of gastroenterology consultation on the outcomes of patients admitted to the hospital with decompensated cirrhosis. Hepatology. 2001;34:1089-1095.
57. Volk ML, Tocco RS, Bazick J, et al. Hospital readmissions among patients with decompensated cirrhosis. Am J Gastroenterol. 2012;107:247-252.
58. Morando F, Maresio G, Piano S, et al. How to improve care in outpatients with cirrhosis and ascites: a new model of care coordination by consultant hepatologists. J Hepatol. 2013;59:257-264.
PRACTICE RECOMMENDATIONS
› Calculate the serum ascites albumin gradient and measure the total ascites protein level to distinguish cirrhotic ascites from that caused by heart failure or other disorders. C
› Recommend sodium restriction of 4.9-6.9 g for patients with established ascites secondary to cirrhosis. C
› Avoid giving angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and nonsteroidal anti-inflammatory drugs in cirrhosis. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series

A 25-year-old man with very high alkaline phosphatase
A 25-year-old man presented to his primary care physician with generalized malaise. His symptoms started around 2 months earlier with progressive fatigue, nausea, decreased appetite, and weight loss (15 lb in 2 months). He denied having fever, chills, night sweats, abdominal pain, diarrhea, melena, or hematochezia.
His medical history was remarkable only for depression, well controlled with sertraline (Zoloft), which he started taking 3 years ago. He was not taking any other prescribed, over-the-counter, or herbal medications.
He had no family history of cancer or liver disease. He did not smoke and rarely drank alcohol. He had never used recreational drugs. He was sexually active with one female partner, used condoms for protection, and had never been diagnosed with a sexually transmitted disease. He had not traveled recently and had not been exposed to any pet.
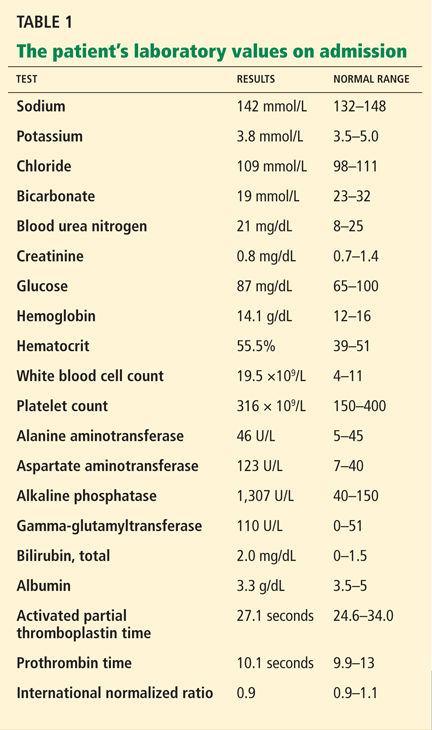
The patient’s laboratory values on admission are shown in Table 1. Of note, his serum alkaline phosphatase level was 1,307 U/L (reference range 40–150 U/L).
LIVER TESTS CAN NARROW THE DIAGNOSIS
The most commonly used laboratory tests of the liver can be classified into those that measure either:
- Liver synthetic function (eg, the serum albumin and bilirubin concentrations and the prothrombin time) or
- Liver damage, as reflected by the serum concentrations of the enzymes alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and gamma-glutamyltransferase (GGT).1,2
ALT and AST are normally concentrated in the hepatocytes and thus, when present in the serum in elevated concentrations, are markers of liver cell injury. The serum levels of these enzymes start to increase within a few hours of liver cell injury as they leak out of the cells via the damaged cell membrane. AST is less liver-specific than ALT, since AST levels can be elevated not only in liver injury but also in muscle, cardiac, and red blood cell injury.3,4
Alkaline phosphatase is actually a heterogeneous group of enzymes found mainly in liver and bone cells. Hepatic alkaline phosphatase is concentrated near the biliary canalicular membrane of the hepatocyte. Accordingly, increased levels of hepatic alkaline phosphatase are mainly seen in liver diseases that predominantly affect the biliary system.3
GGT is also concentrated in hepatic biliary epithelial cells, and thus GGT elevation is another marker of hepatobiliary disease. In fact, measuring the GGT level can help to determine whether an isolated elevation of alkaline phosphatase is due to liver injury.2,3
Accordingly, liver diseases can be classified into two broad categories:
- Hepatocellular injury, in which the primary injury occurs to the hepatocytes
- Cholestatic injury, in which the primary injury is to the bile ducts.
In the former, elevated levels of ALT and AST predominate, while in the latter, elevated alkaline phosphatase is the main finding.3
WHAT TEST NEXT FOR OUR PATIENT?
1. What is the next most appropriate diagnostic step for our patient?
- Liver biopsy
- Ultrasonography of the liver
- Computed tomography (CT) of the liver
- Observation
Our patient has an elevated GGT level, which suggests that his elevated alkaline phosphatase is of hepatic rather than bony origin. Moreover, a serum alkaline phosphatase level that is elevated out of proportion to the aminotransferase levels reflects cholestatic liver injury.
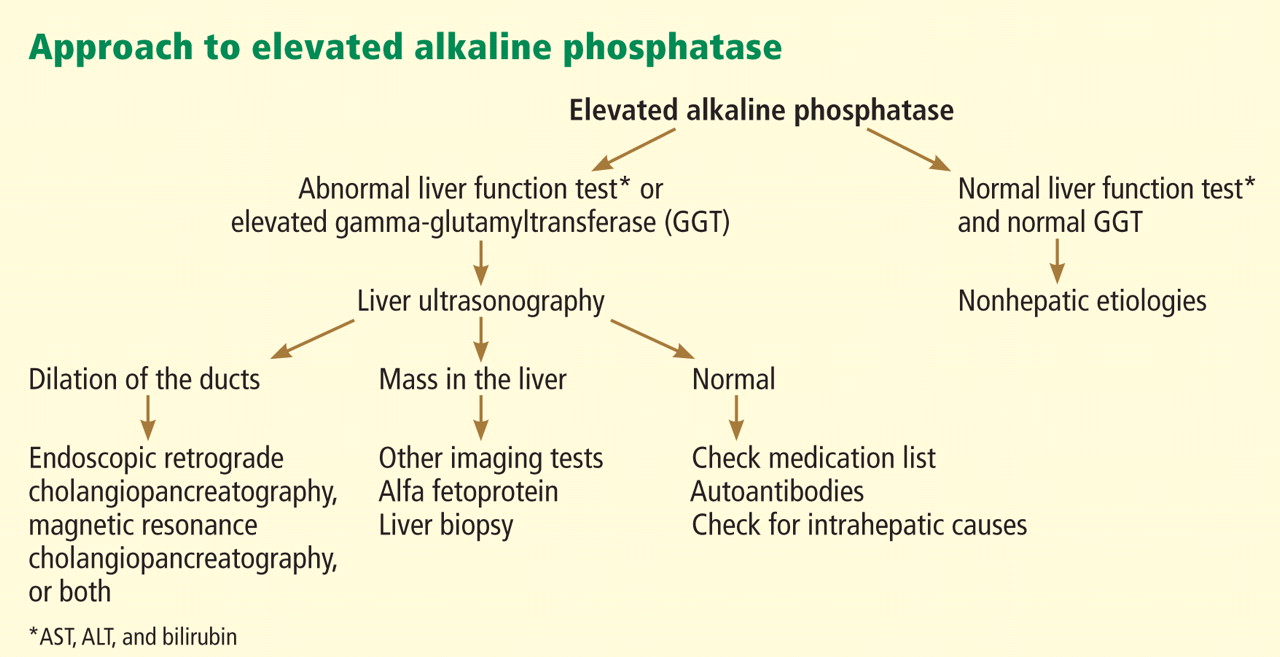
CASE CONTINUED: ULTRASONOGRAPHY IS MOSTLY NORMAL
Ultrasonography of the right upper quadrant revealed that the liver had normal echogenicity and was mildly enlarged. There was no focal hepatic lesion. The gallbladder appeared normal, with no stones or sludge. No dilated intrahepatic or extrahepatic biliary ducts were seen. The common bile duct measured 4 mm. A small amount of ascites not amenable to paracentesis was present.
Thus, in the absence of biliary dilation on ultrasonography, we are dealing with an intrahepatic cholestatic process.
CAUSES OF CHOLESTATIC LIVER DISEASE
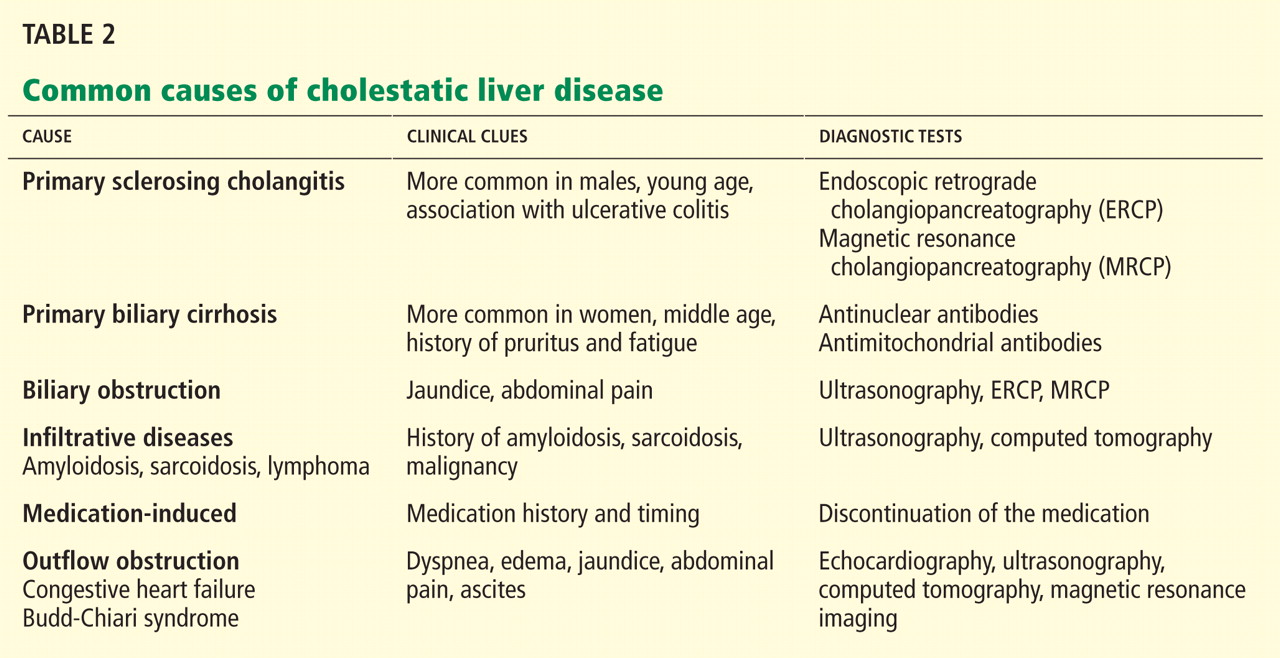
Viral hepatitis
Viral hepatitis most often produces a hepatocellular pattern of injury (ie, AST and ALT elevations predominate). However, in rare cases it can cause a cholestatic pattern of injury.
Our patient subsequently had serologic tests for viral hepatitis, including hepatitis A, B, and C, and the results were negative.
Autoimmune liver disease
The three most common forms of autoimmune liver disease are autoimmune hepatitis, primary biliary cirrhosis, and primary sclerosing cholangitis.
Autoimmune hepatitis is characterized by high serum ALT and AST levels, whereas primary biliary cirrhosis and primary sclerosing cholangitis are associated with predominant elevations of alkaline phosphatase, since they are cholestatic disorders.
Our patient’s alkaline phosphatase level was much higher than his ALT and AST levels, making the latter two diseases more likely.
Primary biliary cirrhosis (and autoimmune hepatitis) are associated with autoantibodies in the serum, such as antinuclear antibody, smooth muscle antibody, and antimitochondrial antibody.
Our patient subsequently was tested for these antibodies, and the results were negative.
Primary sclerosing cholangitis usually affects the extrahepatic biliary system. Thus, if it is present, abnormalities should be seen on imaging.
As mentioned previously, no dilated intrahepatic or extrahepatic biliary ducts were seen on ultrasonography in our patient. Moreover, primary sclerosing cholangitis is associated with inflammatory bowel disease, particularly ulcerative colitis, which our patient did not have.
Drug-induced liver injury
Drug-induced liver injury is a common cause of cholestatic liver disease. However, our patient was not taking any prescribed, over-the-counter, or herbal medications. Additionally, he denied heavy alcohol use.
Infiltrative disorders
Infiltrative disorders such as amyloidosis, sarcoidosis, or lymphoma should be considered in the differential diagnosis of cholestatic liver disease. A clue to a possible infiltrative process is a markedly elevated level of alkaline phosphatase with a mildly increased serum bilirubin concentration, both of which our patient had.
AFTER ULTRASONOGRAPHY, WHAT IS THE NEXT STEP?
2. Which of the following is the next most appropriate diagnostic test for our patient?
- Endoscopic retrograde cholangiopancreatography (ERCP)
- Magnetic resonance cholangiopancreatography (MRCP)
- Liver biopsy
- CT of the abdomen
Figure 1 shows a proposed algorithm for evaluating increased alkaline phosphatase levels.
If there is no biliary duct dilation on ultrasonography, then abnormal levels of alkaline phosphatase most likely represent an intrahepatic pattern of cholestatic liver injury. Therefore, additional imaging with CT or magnetic resonance imaging is of limited diagnostic value. ERCP is used today for therapy rather than diagnosis, so its use is limited to patients known to have dilated biliary ducts on imaging. Liver biopsy, however, can provide useful findings.
Case continued: He undergoes biopsy
Our patient underwent transjugular liver biopsy. During the procedure, transjugular venography showed stenosis in the right, middle, and left hepatic veins and the hepatic portion of the inferior vena cava, consistent with Budd-Chiari syndrome.
The liver biopsy specimen was positive for extensive deposition of slight eosinophilic and amorphous material in a sinusoidal pattern in the liver parenchyma, as well as in the portal tracts, with markedly atrophic hepatocytes. Congo red birefringence confirmed the diagnosis of amyloidosis. The immunohistochemical phenotype was positive for kappa light chains, which is diagnostic for primary-type amyloidosis, also called amyloidosis of light chain composition, or AL.
Bone marrow aspiration and bone marrow biopsy were performed and showed 22% plasma cells, well above the normal range (0–2%), consistent with the diagnosis of multiple myeloma.
BUDD-CHIARI SYNDROME: A CHALLENGING DIAGNOSIS
Budd-Chiari syndrome is a rare condition characterized by obstruction of venous outflow from the liver at a site that may vary from the small hepatic veins up to the inferior vena cava or even the right atrium.5,6 Obstruction of hepatic venous outflow leads to sinusoidal congestion and hypoxic damage of the hepatocytes.7 Hypoxia and necrosis of the hepatocytes result in the release of free radicals. Cirrhosis can eventually occur secondary to ischemic necrosis of hepatocytes and hepatic fibrosis.8
The estimated incidence of this syndrome is 1 in 2.5 million persons per year.7 It is more prevalent in women and young adults.8
Heterogeneous in its causes and manifestations

Budd-Chiari syndrome is also heterogeneous in its manifestations, which depend on the extent of the occlusion, on the acuteness of the obstruction, and on whether venous collateral circulation has developed to decompress the liver sinusoids.9,12,13 Therefore, on the basis of its clinical manifestations, it can be classified as fulminant, acute, subacute, or chronic.12–16
The fulminant form presents with hepatic encephalopathy within 8 weeks after the development of jaundice. The subacute form, which is the most common, has a more insidious onset in which hepatic sinusoids are decompressed by portal and hepatic venous collateral circulation. The patient usually presents with abdominal pain, ascites, hepatomegaly, nausea, vomiting, and mild jaundice. Finally the chronic form presents as complications of cirrhosis.12–16
Imaging plays an important role in diagnosing Budd-Chiari syndrome
Imaging plays an important role in detecting and classifying Budd-Chiari syndrome.
Duplex ultrasonography is useful for detecting this syndrome and has a sensitivity and specificity of 85%.9
CT and magnetic resonance imaging can also help in the diagnosis by showing thrombosis, obstruction, or occlusion in the hepatic vein or the inferior vena cava.5
Venography is the gold standard for diagnosis. However, it should be performed only if noninvasive tests are negative or nondiagnostic and there is a high clinical suspicion of this disease.17 Budd-Chiari syndrome has a characteristic pattern on venography known as “spider web,” which is due to the formation of venous collaterals to bypass the occluded hepatic veins.9
Liver biopsy is not necessarily required to confirm the diagnosis of Budd-Chiari syndrome, but it can help in diagnosing the acute or subacute forms and also in ruling out other causes. Histologic findings can include centrizonal congestion, loss of hepatocytes, hemorrhage, and fibrosis.18,19 Regenerative nodules are found in about 25% of patients.19
TREATING BUDD-CHIARI SYNDROME
The primary goal of treatment is to prevent further extension of the venous thrombosis in the hepatic veins, in their collaterals, and in the intrahepatic and extrahepatic portal venous system. Resolution of hepatic congestion improves liver perfusion and preserves function of the hepatocytes.
Anticoagulation is recommended in the early stages. Heparin therapy should be initiated and subsequently switched to warfarin with the goal of achieving an international normalized ratio of the prothrombin time of 2.0 to 2.5.8,9,19
Thrombolysis is effective in the acute form.20,21 Recanalization, including percutaneous or transhepatic angioplasty of localized segments of the narrowed hepatic veins or inferior vena cava, has long-term patency rates of 80% to 90%.22
If thrombolytic therapy and angioplasty are unsuccessful, a transjugular intrahepatic portosystemic shunt or a surgical procedure (side-to-side portocaval shunt, central splenorenal shunt, or mesocaval shunt) should be considered.9
Liver transplantation is another treatment option in those with fulminant Budd-Chiari syndrome or advanced liver cirrhosis.8
PROGNOSIS HAS IMPROVED
The prognosis of Budd-Chiari syndrome has improved, thanks to both earlier diagnosis and new treatments. The 1-year survival rate, which was about 60% before 1985, has increased to more than 80% in recent cohort studies.19
Studies have shown that the Child-Pugh score, which is based on a combination of serum albumin, bilirubin, prothrombin time, encephalopathy, and ascites, can be considered as an independent prognostic factor. A lower Child-Pugh score and a younger age are associated with a good prognosis.19,23,24 (The Child-Pugh score cannot be applied to our patient because he does not have cirrhosis.)
What happened to our patient?
Our patient was started on anticoagulation for his Budd-Chiari syndrome and on bortezomib (Velcade) and dexamethasone for his multiple myeloma. He achieved remarkable improvement in his liver function tests. Follow-up duplex ultrasonography 1 month after discharge revealed that the stenosis in the hepatic veins had resolved. He is following up with the oncology clinic for management of his multiple myeloma.
- Folwaczny C. Efficient diagnostics for elevated transaminases. [Article in German] MMW Fortschr Med 2007; 149:44–48.
- Moussavian SN, Becker RC, Piepmeyer JL, Mezey E, Bozian RC. Serum gamma-glutamyl transpeptidase and chronic alcoholism. Influence of alcohol ingestion and liver disease. Dig Dis Sci 1985; 30:211–214.
- Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med 2010; 77:195–204.
- Lepper PM, Dufour JF. Elevated transaminases—what to do if everything was done?. [Article in German] Praxis (Bern 1994) 2009; 98:330–334.
- Buzas C, Sparchez Z, Cucuianu A, Manole S, Lupescu I, Acalovschi M. Budd-Chiari syndrome secondary to polycythemia vera. A case report. J Gastrointestin Liver Dis 2009; 18:363–366.
- Valla DC. Primary Budd-Chiari syndrome. J Hepatol 2009; 50:195–203.
- Rautou PE, Moucari R, Cazals-Hatem D, et al. Levels and initial course of serum alanine aminotransferase can predict outcome of patients with Budd-Chiari syndrome. Clin Gastroenterol Hepatol 2009; 7:1230–1235.
- Cura M, Haskal Z, Lopera J. Diagnostic and interventional radiology for Budd-Chiari syndrome. Radiographics 2009; 29:669–681.
- Menon KV, Shah V, Kamath PS. The Budd-Chiari syndrome. N Engl J Med 2004; 350:578–585.
- Darwish Murad S, Plessier A, Hernandez-Guerra M, et al; EN-Vie (European Network for Vascular Disorders of the Liver). Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med 2009; 151:167–175.
- Valla D, Le MG, Poynard T, Zucman N, Rueff B, Benhamou JP. Risk of hepatic vein thrombosis in relation to recent use of oral contraceptives. A case-control study. Gastroenterology 1986; 90:807–811.
- Bismuth H, Sherlock DJ. Portasystemic shunting versus liver transplantation for the Budd-Chiari syndrome. Ann Surg 1991; 214:581–589.
- Orloff MJ, Daily PO, Orloff SL, Girard B, Orloff MS. A 27-year experience with surgical treatment of Budd-Chiari syndrome. Ann Surg 2000; 232:340–352.
- Dilawari JB, Bambery P, Chawla Y, et al. Hepatic outflow obstruction (Budd-Chiari syndrome). Experience with 177 patients and a review of the literature. Medicine (Baltimore) 1994; 73:21–36.
- Mahmoud AE, Mendoza A, Meshikhes AN, et al. Clinical spectrum, investigations and treatment of Budd-Chiari syndrome. QJM 1996; 89:37–43.
- Klein AS, Cameron JL. Diagnosis and management of the Budd-Chiari syndrome. Am J Surg 1990; 160:128–133.
- Plessier A, Valla DC. Budd-Chiari syndrome. Semin Liver Dis 2008; 28:259–269.
- Cazals-Hatem D, Vilgrain V, Genin P, et al. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: a study in 17 explanted livers. Hepatology 2003; 37:510–519.
- Hoekstra J, Janssen HL. Vascular liver disorders (I): diagnosis, treatment and prognosis of Budd-Chiari syndrome. Neth J Med 2008; 66:334–359.
- Frank JW, Kamath PS, Stanson AW. Budd-Chiari syndrome: early intervention with angioplasty and thrombolytic therapy. Mayo Clin Proc 1994; 69:877–881.
- Raju GS, Felver M, Olin JW, Satti SD. Thrombolysis for acute Budd-Chiari syndrome: case report and literature review. Am J Gastroenterol 1996; 91:1262–1263.
- Fisher NC, McCafferty I, Dolapci M, et al. Managing Budd-Chiari syndrome: a retrospective review of percutaneous hepatic vein angioplasty and surgical shunting. Gut 1999; 44:568–574.
- Zeitoun G, Escolano S, Hadengue A, et al. Outcome of Budd-Chiari syndrome: a multivariate analysis of factors related to survival including surgical portosystemic shunting. Hepatology 1999; 30:84–89.
- Darwish Murad S, Valla DC, de Groen PC, et al. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology 2004; 39:500–508.
A 25-year-old man presented to his primary care physician with generalized malaise. His symptoms started around 2 months earlier with progressive fatigue, nausea, decreased appetite, and weight loss (15 lb in 2 months). He denied having fever, chills, night sweats, abdominal pain, diarrhea, melena, or hematochezia.
His medical history was remarkable only for depression, well controlled with sertraline (Zoloft), which he started taking 3 years ago. He was not taking any other prescribed, over-the-counter, or herbal medications.
He had no family history of cancer or liver disease. He did not smoke and rarely drank alcohol. He had never used recreational drugs. He was sexually active with one female partner, used condoms for protection, and had never been diagnosed with a sexually transmitted disease. He had not traveled recently and had not been exposed to any pet.
The patient’s laboratory values on admission are shown in Table 1. Of note, his serum alkaline phosphatase level was 1,307 U/L (reference range 40–150 U/L).
LIVER TESTS CAN NARROW THE DIAGNOSIS
The most commonly used laboratory tests of the liver can be classified into those that measure either:
- Liver synthetic function (eg, the serum albumin and bilirubin concentrations and the prothrombin time) or
- Liver damage, as reflected by the serum concentrations of the enzymes alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and gamma-glutamyltransferase (GGT).1,2
ALT and AST are normally concentrated in the hepatocytes and thus, when present in the serum in elevated concentrations, are markers of liver cell injury. The serum levels of these enzymes start to increase within a few hours of liver cell injury as they leak out of the cells via the damaged cell membrane. AST is less liver-specific than ALT, since AST levels can be elevated not only in liver injury but also in muscle, cardiac, and red blood cell injury.3,4
Alkaline phosphatase is actually a heterogeneous group of enzymes found mainly in liver and bone cells. Hepatic alkaline phosphatase is concentrated near the biliary canalicular membrane of the hepatocyte. Accordingly, increased levels of hepatic alkaline phosphatase are mainly seen in liver diseases that predominantly affect the biliary system.3
GGT is also concentrated in hepatic biliary epithelial cells, and thus GGT elevation is another marker of hepatobiliary disease. In fact, measuring the GGT level can help to determine whether an isolated elevation of alkaline phosphatase is due to liver injury.2,3
Accordingly, liver diseases can be classified into two broad categories:
- Hepatocellular injury, in which the primary injury occurs to the hepatocytes
- Cholestatic injury, in which the primary injury is to the bile ducts.
In the former, elevated levels of ALT and AST predominate, while in the latter, elevated alkaline phosphatase is the main finding.3
WHAT TEST NEXT FOR OUR PATIENT?
1. What is the next most appropriate diagnostic step for our patient?
- Liver biopsy
- Ultrasonography of the liver
- Computed tomography (CT) of the liver
- Observation
Our patient has an elevated GGT level, which suggests that his elevated alkaline phosphatase is of hepatic rather than bony origin. Moreover, a serum alkaline phosphatase level that is elevated out of proportion to the aminotransferase levels reflects cholestatic liver injury.
CASE CONTINUED: ULTRASONOGRAPHY IS MOSTLY NORMAL
Ultrasonography of the right upper quadrant revealed that the liver had normal echogenicity and was mildly enlarged. There was no focal hepatic lesion. The gallbladder appeared normal, with no stones or sludge. No dilated intrahepatic or extrahepatic biliary ducts were seen. The common bile duct measured 4 mm. A small amount of ascites not amenable to paracentesis was present.
Thus, in the absence of biliary dilation on ultrasonography, we are dealing with an intrahepatic cholestatic process.
CAUSES OF CHOLESTATIC LIVER DISEASE
Viral hepatitis
Viral hepatitis most often produces a hepatocellular pattern of injury (ie, AST and ALT elevations predominate). However, in rare cases it can cause a cholestatic pattern of injury.
Our patient subsequently had serologic tests for viral hepatitis, including hepatitis A, B, and C, and the results were negative.
Autoimmune liver disease
The three most common forms of autoimmune liver disease are autoimmune hepatitis, primary biliary cirrhosis, and primary sclerosing cholangitis.
Autoimmune hepatitis is characterized by high serum ALT and AST levels, whereas primary biliary cirrhosis and primary sclerosing cholangitis are associated with predominant elevations of alkaline phosphatase, since they are cholestatic disorders.
Our patient’s alkaline phosphatase level was much higher than his ALT and AST levels, making the latter two diseases more likely.
Primary biliary cirrhosis (and autoimmune hepatitis) are associated with autoantibodies in the serum, such as antinuclear antibody, smooth muscle antibody, and antimitochondrial antibody.
Our patient subsequently was tested for these antibodies, and the results were negative.
Primary sclerosing cholangitis usually affects the extrahepatic biliary system. Thus, if it is present, abnormalities should be seen on imaging.
As mentioned previously, no dilated intrahepatic or extrahepatic biliary ducts were seen on ultrasonography in our patient. Moreover, primary sclerosing cholangitis is associated with inflammatory bowel disease, particularly ulcerative colitis, which our patient did not have.
Drug-induced liver injury
Drug-induced liver injury is a common cause of cholestatic liver disease. However, our patient was not taking any prescribed, over-the-counter, or herbal medications. Additionally, he denied heavy alcohol use.
Infiltrative disorders
Infiltrative disorders such as amyloidosis, sarcoidosis, or lymphoma should be considered in the differential diagnosis of cholestatic liver disease. A clue to a possible infiltrative process is a markedly elevated level of alkaline phosphatase with a mildly increased serum bilirubin concentration, both of which our patient had.
AFTER ULTRASONOGRAPHY, WHAT IS THE NEXT STEP?
2. Which of the following is the next most appropriate diagnostic test for our patient?
- Endoscopic retrograde cholangiopancreatography (ERCP)
- Magnetic resonance cholangiopancreatography (MRCP)
- Liver biopsy
- CT of the abdomen
Figure 1 shows a proposed algorithm for evaluating increased alkaline phosphatase levels.
If there is no biliary duct dilation on ultrasonography, then abnormal levels of alkaline phosphatase most likely represent an intrahepatic pattern of cholestatic liver injury. Therefore, additional imaging with CT or magnetic resonance imaging is of limited diagnostic value. ERCP is used today for therapy rather than diagnosis, so its use is limited to patients known to have dilated biliary ducts on imaging. Liver biopsy, however, can provide useful findings.
Case continued: He undergoes biopsy
Our patient underwent transjugular liver biopsy. During the procedure, transjugular venography showed stenosis in the right, middle, and left hepatic veins and the hepatic portion of the inferior vena cava, consistent with Budd-Chiari syndrome.
The liver biopsy specimen was positive for extensive deposition of slight eosinophilic and amorphous material in a sinusoidal pattern in the liver parenchyma, as well as in the portal tracts, with markedly atrophic hepatocytes. Congo red birefringence confirmed the diagnosis of amyloidosis. The immunohistochemical phenotype was positive for kappa light chains, which is diagnostic for primary-type amyloidosis, also called amyloidosis of light chain composition, or AL.
Bone marrow aspiration and bone marrow biopsy were performed and showed 22% plasma cells, well above the normal range (0–2%), consistent with the diagnosis of multiple myeloma.
BUDD-CHIARI SYNDROME: A CHALLENGING DIAGNOSIS
Budd-Chiari syndrome is a rare condition characterized by obstruction of venous outflow from the liver at a site that may vary from the small hepatic veins up to the inferior vena cava or even the right atrium.5,6 Obstruction of hepatic venous outflow leads to sinusoidal congestion and hypoxic damage of the hepatocytes.7 Hypoxia and necrosis of the hepatocytes result in the release of free radicals. Cirrhosis can eventually occur secondary to ischemic necrosis of hepatocytes and hepatic fibrosis.8
The estimated incidence of this syndrome is 1 in 2.5 million persons per year.7 It is more prevalent in women and young adults.8
Heterogeneous in its causes and manifestations
Budd-Chiari syndrome is also heterogeneous in its manifestations, which depend on the extent of the occlusion, on the acuteness of the obstruction, and on whether venous collateral circulation has developed to decompress the liver sinusoids.9,12,13 Therefore, on the basis of its clinical manifestations, it can be classified as fulminant, acute, subacute, or chronic.12–16
The fulminant form presents with hepatic encephalopathy within 8 weeks after the development of jaundice. The subacute form, which is the most common, has a more insidious onset in which hepatic sinusoids are decompressed by portal and hepatic venous collateral circulation. The patient usually presents with abdominal pain, ascites, hepatomegaly, nausea, vomiting, and mild jaundice. Finally the chronic form presents as complications of cirrhosis.12–16
Imaging plays an important role in diagnosing Budd-Chiari syndrome
Imaging plays an important role in detecting and classifying Budd-Chiari syndrome.
Duplex ultrasonography is useful for detecting this syndrome and has a sensitivity and specificity of 85%.9
CT and magnetic resonance imaging can also help in the diagnosis by showing thrombosis, obstruction, or occlusion in the hepatic vein or the inferior vena cava.5
Venography is the gold standard for diagnosis. However, it should be performed only if noninvasive tests are negative or nondiagnostic and there is a high clinical suspicion of this disease.17 Budd-Chiari syndrome has a characteristic pattern on venography known as “spider web,” which is due to the formation of venous collaterals to bypass the occluded hepatic veins.9
Liver biopsy is not necessarily required to confirm the diagnosis of Budd-Chiari syndrome, but it can help in diagnosing the acute or subacute forms and also in ruling out other causes. Histologic findings can include centrizonal congestion, loss of hepatocytes, hemorrhage, and fibrosis.18,19 Regenerative nodules are found in about 25% of patients.19
TREATING BUDD-CHIARI SYNDROME
The primary goal of treatment is to prevent further extension of the venous thrombosis in the hepatic veins, in their collaterals, and in the intrahepatic and extrahepatic portal venous system. Resolution of hepatic congestion improves liver perfusion and preserves function of the hepatocytes.
Anticoagulation is recommended in the early stages. Heparin therapy should be initiated and subsequently switched to warfarin with the goal of achieving an international normalized ratio of the prothrombin time of 2.0 to 2.5.8,9,19
Thrombolysis is effective in the acute form.20,21 Recanalization, including percutaneous or transhepatic angioplasty of localized segments of the narrowed hepatic veins or inferior vena cava, has long-term patency rates of 80% to 90%.22
If thrombolytic therapy and angioplasty are unsuccessful, a transjugular intrahepatic portosystemic shunt or a surgical procedure (side-to-side portocaval shunt, central splenorenal shunt, or mesocaval shunt) should be considered.9
Liver transplantation is another treatment option in those with fulminant Budd-Chiari syndrome or advanced liver cirrhosis.8
PROGNOSIS HAS IMPROVED
The prognosis of Budd-Chiari syndrome has improved, thanks to both earlier diagnosis and new treatments. The 1-year survival rate, which was about 60% before 1985, has increased to more than 80% in recent cohort studies.19
Studies have shown that the Child-Pugh score, which is based on a combination of serum albumin, bilirubin, prothrombin time, encephalopathy, and ascites, can be considered as an independent prognostic factor. A lower Child-Pugh score and a younger age are associated with a good prognosis.19,23,24 (The Child-Pugh score cannot be applied to our patient because he does not have cirrhosis.)
What happened to our patient?
Our patient was started on anticoagulation for his Budd-Chiari syndrome and on bortezomib (Velcade) and dexamethasone for his multiple myeloma. He achieved remarkable improvement in his liver function tests. Follow-up duplex ultrasonography 1 month after discharge revealed that the stenosis in the hepatic veins had resolved. He is following up with the oncology clinic for management of his multiple myeloma.
A 25-year-old man presented to his primary care physician with generalized malaise. His symptoms started around 2 months earlier with progressive fatigue, nausea, decreased appetite, and weight loss (15 lb in 2 months). He denied having fever, chills, night sweats, abdominal pain, diarrhea, melena, or hematochezia.
His medical history was remarkable only for depression, well controlled with sertraline (Zoloft), which he started taking 3 years ago. He was not taking any other prescribed, over-the-counter, or herbal medications.
He had no family history of cancer or liver disease. He did not smoke and rarely drank alcohol. He had never used recreational drugs. He was sexually active with one female partner, used condoms for protection, and had never been diagnosed with a sexually transmitted disease. He had not traveled recently and had not been exposed to any pet.
The patient’s laboratory values on admission are shown in Table 1. Of note, his serum alkaline phosphatase level was 1,307 U/L (reference range 40–150 U/L).
LIVER TESTS CAN NARROW THE DIAGNOSIS
The most commonly used laboratory tests of the liver can be classified into those that measure either:
- Liver synthetic function (eg, the serum albumin and bilirubin concentrations and the prothrombin time) or
- Liver damage, as reflected by the serum concentrations of the enzymes alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and gamma-glutamyltransferase (GGT).1,2
ALT and AST are normally concentrated in the hepatocytes and thus, when present in the serum in elevated concentrations, are markers of liver cell injury. The serum levels of these enzymes start to increase within a few hours of liver cell injury as they leak out of the cells via the damaged cell membrane. AST is less liver-specific than ALT, since AST levels can be elevated not only in liver injury but also in muscle, cardiac, and red blood cell injury.3,4
Alkaline phosphatase is actually a heterogeneous group of enzymes found mainly in liver and bone cells. Hepatic alkaline phosphatase is concentrated near the biliary canalicular membrane of the hepatocyte. Accordingly, increased levels of hepatic alkaline phosphatase are mainly seen in liver diseases that predominantly affect the biliary system.3
GGT is also concentrated in hepatic biliary epithelial cells, and thus GGT elevation is another marker of hepatobiliary disease. In fact, measuring the GGT level can help to determine whether an isolated elevation of alkaline phosphatase is due to liver injury.2,3
Accordingly, liver diseases can be classified into two broad categories:
- Hepatocellular injury, in which the primary injury occurs to the hepatocytes
- Cholestatic injury, in which the primary injury is to the bile ducts.
In the former, elevated levels of ALT and AST predominate, while in the latter, elevated alkaline phosphatase is the main finding.3
WHAT TEST NEXT FOR OUR PATIENT?
1. What is the next most appropriate diagnostic step for our patient?
- Liver biopsy
- Ultrasonography of the liver
- Computed tomography (CT) of the liver
- Observation
Our patient has an elevated GGT level, which suggests that his elevated alkaline phosphatase is of hepatic rather than bony origin. Moreover, a serum alkaline phosphatase level that is elevated out of proportion to the aminotransferase levels reflects cholestatic liver injury.
CASE CONTINUED: ULTRASONOGRAPHY IS MOSTLY NORMAL
Ultrasonography of the right upper quadrant revealed that the liver had normal echogenicity and was mildly enlarged. There was no focal hepatic lesion. The gallbladder appeared normal, with no stones or sludge. No dilated intrahepatic or extrahepatic biliary ducts were seen. The common bile duct measured 4 mm. A small amount of ascites not amenable to paracentesis was present.
Thus, in the absence of biliary dilation on ultrasonography, we are dealing with an intrahepatic cholestatic process.
CAUSES OF CHOLESTATIC LIVER DISEASE
Viral hepatitis
Viral hepatitis most often produces a hepatocellular pattern of injury (ie, AST and ALT elevations predominate). However, in rare cases it can cause a cholestatic pattern of injury.
Our patient subsequently had serologic tests for viral hepatitis, including hepatitis A, B, and C, and the results were negative.
Autoimmune liver disease
The three most common forms of autoimmune liver disease are autoimmune hepatitis, primary biliary cirrhosis, and primary sclerosing cholangitis.
Autoimmune hepatitis is characterized by high serum ALT and AST levels, whereas primary biliary cirrhosis and primary sclerosing cholangitis are associated with predominant elevations of alkaline phosphatase, since they are cholestatic disorders.
Our patient’s alkaline phosphatase level was much higher than his ALT and AST levels, making the latter two diseases more likely.
Primary biliary cirrhosis (and autoimmune hepatitis) are associated with autoantibodies in the serum, such as antinuclear antibody, smooth muscle antibody, and antimitochondrial antibody.
Our patient subsequently was tested for these antibodies, and the results were negative.
Primary sclerosing cholangitis usually affects the extrahepatic biliary system. Thus, if it is present, abnormalities should be seen on imaging.
As mentioned previously, no dilated intrahepatic or extrahepatic biliary ducts were seen on ultrasonography in our patient. Moreover, primary sclerosing cholangitis is associated with inflammatory bowel disease, particularly ulcerative colitis, which our patient did not have.
Drug-induced liver injury
Drug-induced liver injury is a common cause of cholestatic liver disease. However, our patient was not taking any prescribed, over-the-counter, or herbal medications. Additionally, he denied heavy alcohol use.
Infiltrative disorders
Infiltrative disorders such as amyloidosis, sarcoidosis, or lymphoma should be considered in the differential diagnosis of cholestatic liver disease. A clue to a possible infiltrative process is a markedly elevated level of alkaline phosphatase with a mildly increased serum bilirubin concentration, both of which our patient had.
AFTER ULTRASONOGRAPHY, WHAT IS THE NEXT STEP?
2. Which of the following is the next most appropriate diagnostic test for our patient?
- Endoscopic retrograde cholangiopancreatography (ERCP)
- Magnetic resonance cholangiopancreatography (MRCP)
- Liver biopsy
- CT of the abdomen
Figure 1 shows a proposed algorithm for evaluating increased alkaline phosphatase levels.
If there is no biliary duct dilation on ultrasonography, then abnormal levels of alkaline phosphatase most likely represent an intrahepatic pattern of cholestatic liver injury. Therefore, additional imaging with CT or magnetic resonance imaging is of limited diagnostic value. ERCP is used today for therapy rather than diagnosis, so its use is limited to patients known to have dilated biliary ducts on imaging. Liver biopsy, however, can provide useful findings.
Case continued: He undergoes biopsy
Our patient underwent transjugular liver biopsy. During the procedure, transjugular venography showed stenosis in the right, middle, and left hepatic veins and the hepatic portion of the inferior vena cava, consistent with Budd-Chiari syndrome.
The liver biopsy specimen was positive for extensive deposition of slight eosinophilic and amorphous material in a sinusoidal pattern in the liver parenchyma, as well as in the portal tracts, with markedly atrophic hepatocytes. Congo red birefringence confirmed the diagnosis of amyloidosis. The immunohistochemical phenotype was positive for kappa light chains, which is diagnostic for primary-type amyloidosis, also called amyloidosis of light chain composition, or AL.
Bone marrow aspiration and bone marrow biopsy were performed and showed 22% plasma cells, well above the normal range (0–2%), consistent with the diagnosis of multiple myeloma.
BUDD-CHIARI SYNDROME: A CHALLENGING DIAGNOSIS
Budd-Chiari syndrome is a rare condition characterized by obstruction of venous outflow from the liver at a site that may vary from the small hepatic veins up to the inferior vena cava or even the right atrium.5,6 Obstruction of hepatic venous outflow leads to sinusoidal congestion and hypoxic damage of the hepatocytes.7 Hypoxia and necrosis of the hepatocytes result in the release of free radicals. Cirrhosis can eventually occur secondary to ischemic necrosis of hepatocytes and hepatic fibrosis.8
The estimated incidence of this syndrome is 1 in 2.5 million persons per year.7 It is more prevalent in women and young adults.8
Heterogeneous in its causes and manifestations
Budd-Chiari syndrome is also heterogeneous in its manifestations, which depend on the extent of the occlusion, on the acuteness of the obstruction, and on whether venous collateral circulation has developed to decompress the liver sinusoids.9,12,13 Therefore, on the basis of its clinical manifestations, it can be classified as fulminant, acute, subacute, or chronic.12–16
The fulminant form presents with hepatic encephalopathy within 8 weeks after the development of jaundice. The subacute form, which is the most common, has a more insidious onset in which hepatic sinusoids are decompressed by portal and hepatic venous collateral circulation. The patient usually presents with abdominal pain, ascites, hepatomegaly, nausea, vomiting, and mild jaundice. Finally the chronic form presents as complications of cirrhosis.12–16
Imaging plays an important role in diagnosing Budd-Chiari syndrome
Imaging plays an important role in detecting and classifying Budd-Chiari syndrome.
Duplex ultrasonography is useful for detecting this syndrome and has a sensitivity and specificity of 85%.9
CT and magnetic resonance imaging can also help in the diagnosis by showing thrombosis, obstruction, or occlusion in the hepatic vein or the inferior vena cava.5
Venography is the gold standard for diagnosis. However, it should be performed only if noninvasive tests are negative or nondiagnostic and there is a high clinical suspicion of this disease.17 Budd-Chiari syndrome has a characteristic pattern on venography known as “spider web,” which is due to the formation of venous collaterals to bypass the occluded hepatic veins.9
Liver biopsy is not necessarily required to confirm the diagnosis of Budd-Chiari syndrome, but it can help in diagnosing the acute or subacute forms and also in ruling out other causes. Histologic findings can include centrizonal congestion, loss of hepatocytes, hemorrhage, and fibrosis.18,19 Regenerative nodules are found in about 25% of patients.19
TREATING BUDD-CHIARI SYNDROME
The primary goal of treatment is to prevent further extension of the venous thrombosis in the hepatic veins, in their collaterals, and in the intrahepatic and extrahepatic portal venous system. Resolution of hepatic congestion improves liver perfusion and preserves function of the hepatocytes.
Anticoagulation is recommended in the early stages. Heparin therapy should be initiated and subsequently switched to warfarin with the goal of achieving an international normalized ratio of the prothrombin time of 2.0 to 2.5.8,9,19
Thrombolysis is effective in the acute form.20,21 Recanalization, including percutaneous or transhepatic angioplasty of localized segments of the narrowed hepatic veins or inferior vena cava, has long-term patency rates of 80% to 90%.22
If thrombolytic therapy and angioplasty are unsuccessful, a transjugular intrahepatic portosystemic shunt or a surgical procedure (side-to-side portocaval shunt, central splenorenal shunt, or mesocaval shunt) should be considered.9
Liver transplantation is another treatment option in those with fulminant Budd-Chiari syndrome or advanced liver cirrhosis.8
PROGNOSIS HAS IMPROVED
The prognosis of Budd-Chiari syndrome has improved, thanks to both earlier diagnosis and new treatments. The 1-year survival rate, which was about 60% before 1985, has increased to more than 80% in recent cohort studies.19
Studies have shown that the Child-Pugh score, which is based on a combination of serum albumin, bilirubin, prothrombin time, encephalopathy, and ascites, can be considered as an independent prognostic factor. A lower Child-Pugh score and a younger age are associated with a good prognosis.19,23,24 (The Child-Pugh score cannot be applied to our patient because he does not have cirrhosis.)
What happened to our patient?
Our patient was started on anticoagulation for his Budd-Chiari syndrome and on bortezomib (Velcade) and dexamethasone for his multiple myeloma. He achieved remarkable improvement in his liver function tests. Follow-up duplex ultrasonography 1 month after discharge revealed that the stenosis in the hepatic veins had resolved. He is following up with the oncology clinic for management of his multiple myeloma.
- Folwaczny C. Efficient diagnostics for elevated transaminases. [Article in German] MMW Fortschr Med 2007; 149:44–48.
- Moussavian SN, Becker RC, Piepmeyer JL, Mezey E, Bozian RC. Serum gamma-glutamyl transpeptidase and chronic alcoholism. Influence of alcohol ingestion and liver disease. Dig Dis Sci 1985; 30:211–214.
- Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med 2010; 77:195–204.
- Lepper PM, Dufour JF. Elevated transaminases—what to do if everything was done?. [Article in German] Praxis (Bern 1994) 2009; 98:330–334.
- Buzas C, Sparchez Z, Cucuianu A, Manole S, Lupescu I, Acalovschi M. Budd-Chiari syndrome secondary to polycythemia vera. A case report. J Gastrointestin Liver Dis 2009; 18:363–366.
- Valla DC. Primary Budd-Chiari syndrome. J Hepatol 2009; 50:195–203.
- Rautou PE, Moucari R, Cazals-Hatem D, et al. Levels and initial course of serum alanine aminotransferase can predict outcome of patients with Budd-Chiari syndrome. Clin Gastroenterol Hepatol 2009; 7:1230–1235.
- Cura M, Haskal Z, Lopera J. Diagnostic and interventional radiology for Budd-Chiari syndrome. Radiographics 2009; 29:669–681.
- Menon KV, Shah V, Kamath PS. The Budd-Chiari syndrome. N Engl J Med 2004; 350:578–585.
- Darwish Murad S, Plessier A, Hernandez-Guerra M, et al; EN-Vie (European Network for Vascular Disorders of the Liver). Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med 2009; 151:167–175.
- Valla D, Le MG, Poynard T, Zucman N, Rueff B, Benhamou JP. Risk of hepatic vein thrombosis in relation to recent use of oral contraceptives. A case-control study. Gastroenterology 1986; 90:807–811.
- Bismuth H, Sherlock DJ. Portasystemic shunting versus liver transplantation for the Budd-Chiari syndrome. Ann Surg 1991; 214:581–589.
- Orloff MJ, Daily PO, Orloff SL, Girard B, Orloff MS. A 27-year experience with surgical treatment of Budd-Chiari syndrome. Ann Surg 2000; 232:340–352.
- Dilawari JB, Bambery P, Chawla Y, et al. Hepatic outflow obstruction (Budd-Chiari syndrome). Experience with 177 patients and a review of the literature. Medicine (Baltimore) 1994; 73:21–36.
- Mahmoud AE, Mendoza A, Meshikhes AN, et al. Clinical spectrum, investigations and treatment of Budd-Chiari syndrome. QJM 1996; 89:37–43.
- Klein AS, Cameron JL. Diagnosis and management of the Budd-Chiari syndrome. Am J Surg 1990; 160:128–133.
- Plessier A, Valla DC. Budd-Chiari syndrome. Semin Liver Dis 2008; 28:259–269.
- Cazals-Hatem D, Vilgrain V, Genin P, et al. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: a study in 17 explanted livers. Hepatology 2003; 37:510–519.
- Hoekstra J, Janssen HL. Vascular liver disorders (I): diagnosis, treatment and prognosis of Budd-Chiari syndrome. Neth J Med 2008; 66:334–359.
- Frank JW, Kamath PS, Stanson AW. Budd-Chiari syndrome: early intervention with angioplasty and thrombolytic therapy. Mayo Clin Proc 1994; 69:877–881.
- Raju GS, Felver M, Olin JW, Satti SD. Thrombolysis for acute Budd-Chiari syndrome: case report and literature review. Am J Gastroenterol 1996; 91:1262–1263.
- Fisher NC, McCafferty I, Dolapci M, et al. Managing Budd-Chiari syndrome: a retrospective review of percutaneous hepatic vein angioplasty and surgical shunting. Gut 1999; 44:568–574.
- Zeitoun G, Escolano S, Hadengue A, et al. Outcome of Budd-Chiari syndrome: a multivariate analysis of factors related to survival including surgical portosystemic shunting. Hepatology 1999; 30:84–89.
- Darwish Murad S, Valla DC, de Groen PC, et al. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology 2004; 39:500–508.
- Folwaczny C. Efficient diagnostics for elevated transaminases. [Article in German] MMW Fortschr Med 2007; 149:44–48.
- Moussavian SN, Becker RC, Piepmeyer JL, Mezey E, Bozian RC. Serum gamma-glutamyl transpeptidase and chronic alcoholism. Influence of alcohol ingestion and liver disease. Dig Dis Sci 1985; 30:211–214.
- Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med 2010; 77:195–204.
- Lepper PM, Dufour JF. Elevated transaminases—what to do if everything was done?. [Article in German] Praxis (Bern 1994) 2009; 98:330–334.
- Buzas C, Sparchez Z, Cucuianu A, Manole S, Lupescu I, Acalovschi M. Budd-Chiari syndrome secondary to polycythemia vera. A case report. J Gastrointestin Liver Dis 2009; 18:363–366.
- Valla DC. Primary Budd-Chiari syndrome. J Hepatol 2009; 50:195–203.
- Rautou PE, Moucari R, Cazals-Hatem D, et al. Levels and initial course of serum alanine aminotransferase can predict outcome of patients with Budd-Chiari syndrome. Clin Gastroenterol Hepatol 2009; 7:1230–1235.
- Cura M, Haskal Z, Lopera J. Diagnostic and interventional radiology for Budd-Chiari syndrome. Radiographics 2009; 29:669–681.
- Menon KV, Shah V, Kamath PS. The Budd-Chiari syndrome. N Engl J Med 2004; 350:578–585.
- Darwish Murad S, Plessier A, Hernandez-Guerra M, et al; EN-Vie (European Network for Vascular Disorders of the Liver). Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med 2009; 151:167–175.
- Valla D, Le MG, Poynard T, Zucman N, Rueff B, Benhamou JP. Risk of hepatic vein thrombosis in relation to recent use of oral contraceptives. A case-control study. Gastroenterology 1986; 90:807–811.
- Bismuth H, Sherlock DJ. Portasystemic shunting versus liver transplantation for the Budd-Chiari syndrome. Ann Surg 1991; 214:581–589.
- Orloff MJ, Daily PO, Orloff SL, Girard B, Orloff MS. A 27-year experience with surgical treatment of Budd-Chiari syndrome. Ann Surg 2000; 232:340–352.
- Dilawari JB, Bambery P, Chawla Y, et al. Hepatic outflow obstruction (Budd-Chiari syndrome). Experience with 177 patients and a review of the literature. Medicine (Baltimore) 1994; 73:21–36.
- Mahmoud AE, Mendoza A, Meshikhes AN, et al. Clinical spectrum, investigations and treatment of Budd-Chiari syndrome. QJM 1996; 89:37–43.
- Klein AS, Cameron JL. Diagnosis and management of the Budd-Chiari syndrome. Am J Surg 1990; 160:128–133.
- Plessier A, Valla DC. Budd-Chiari syndrome. Semin Liver Dis 2008; 28:259–269.
- Cazals-Hatem D, Vilgrain V, Genin P, et al. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: a study in 17 explanted livers. Hepatology 2003; 37:510–519.
- Hoekstra J, Janssen HL. Vascular liver disorders (I): diagnosis, treatment and prognosis of Budd-Chiari syndrome. Neth J Med 2008; 66:334–359.
- Frank JW, Kamath PS, Stanson AW. Budd-Chiari syndrome: early intervention with angioplasty and thrombolytic therapy. Mayo Clin Proc 1994; 69:877–881.
- Raju GS, Felver M, Olin JW, Satti SD. Thrombolysis for acute Budd-Chiari syndrome: case report and literature review. Am J Gastroenterol 1996; 91:1262–1263.
- Fisher NC, McCafferty I, Dolapci M, et al. Managing Budd-Chiari syndrome: a retrospective review of percutaneous hepatic vein angioplasty and surgical shunting. Gut 1999; 44:568–574.
- Zeitoun G, Escolano S, Hadengue A, et al. Outcome of Budd-Chiari syndrome: a multivariate analysis of factors related to survival including surgical portosystemic shunting. Hepatology 1999; 30:84–89.
- Darwish Murad S, Valla DC, de Groen PC, et al. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology 2004; 39:500–508.