User login
A 25-year-old man presented to his primary care physician with generalized malaise. His symptoms started around 2 months earlier with progressive fatigue, nausea, decreased appetite, and weight loss (15 lb in 2 months). He denied having fever, chills, night sweats, abdominal pain, diarrhea, melena, or hematochezia.
His medical history was remarkable only for depression, well controlled with sertraline (Zoloft), which he started taking 3 years ago. He was not taking any other prescribed, over-the-counter, or herbal medications.
He had no family history of cancer or liver disease. He did not smoke and rarely drank alcohol. He had never used recreational drugs. He was sexually active with one female partner, used condoms for protection, and had never been diagnosed with a sexually transmitted disease. He had not traveled recently and had not been exposed to any pet.
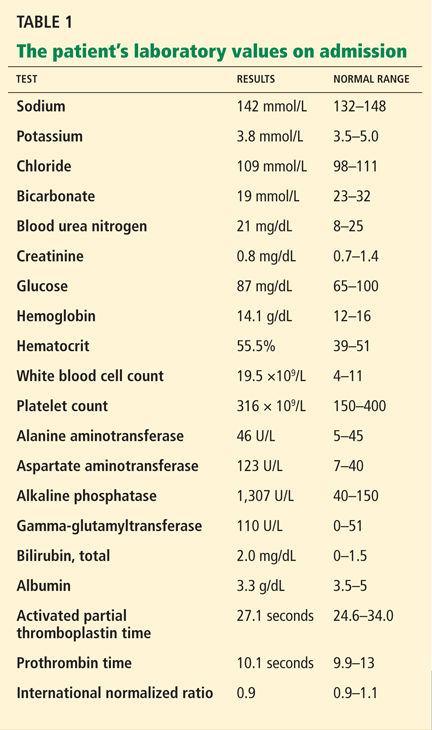
The patient’s laboratory values on admission are shown in Table 1. Of note, his serum alkaline phosphatase level was 1,307 U/L (reference range 40–150 U/L).
LIVER TESTS CAN NARROW THE DIAGNOSIS
The most commonly used laboratory tests of the liver can be classified into those that measure either:
- Liver synthetic function (eg, the serum albumin and bilirubin concentrations and the prothrombin time) or
- Liver damage, as reflected by the serum concentrations of the enzymes alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and gamma-glutamyltransferase (GGT).1,2
ALT and AST are normally concentrated in the hepatocytes and thus, when present in the serum in elevated concentrations, are markers of liver cell injury. The serum levels of these enzymes start to increase within a few hours of liver cell injury as they leak out of the cells via the damaged cell membrane. AST is less liver-specific than ALT, since AST levels can be elevated not only in liver injury but also in muscle, cardiac, and red blood cell injury.3,4
Alkaline phosphatase is actually a heterogeneous group of enzymes found mainly in liver and bone cells. Hepatic alkaline phosphatase is concentrated near the biliary canalicular membrane of the hepatocyte. Accordingly, increased levels of hepatic alkaline phosphatase are mainly seen in liver diseases that predominantly affect the biliary system.3
GGT is also concentrated in hepatic biliary epithelial cells, and thus GGT elevation is another marker of hepatobiliary disease. In fact, measuring the GGT level can help to determine whether an isolated elevation of alkaline phosphatase is due to liver injury.2,3
Accordingly, liver diseases can be classified into two broad categories:
- Hepatocellular injury, in which the primary injury occurs to the hepatocytes
- Cholestatic injury, in which the primary injury is to the bile ducts.
In the former, elevated levels of ALT and AST predominate, while in the latter, elevated alkaline phosphatase is the main finding.3
WHAT TEST NEXT FOR OUR PATIENT?
1. What is the next most appropriate diagnostic step for our patient?
- Liver biopsy
- Ultrasonography of the liver
- Computed tomography (CT) of the liver
- Observation
Our patient has an elevated GGT level, which suggests that his elevated alkaline phosphatase is of hepatic rather than bony origin. Moreover, a serum alkaline phosphatase level that is elevated out of proportion to the aminotransferase levels reflects cholestatic liver injury.
CASE CONTINUED: ULTRASONOGRAPHY IS MOSTLY NORMAL
Ultrasonography of the right upper quadrant revealed that the liver had normal echogenicity and was mildly enlarged. There was no focal hepatic lesion. The gallbladder appeared normal, with no stones or sludge. No dilated intrahepatic or extrahepatic biliary ducts were seen. The common bile duct measured 4 mm. A small amount of ascites not amenable to paracentesis was present.
Thus, in the absence of biliary dilation on ultrasonography, we are dealing with an intrahepatic cholestatic process.
CAUSES OF CHOLESTATIC LIVER DISEASE
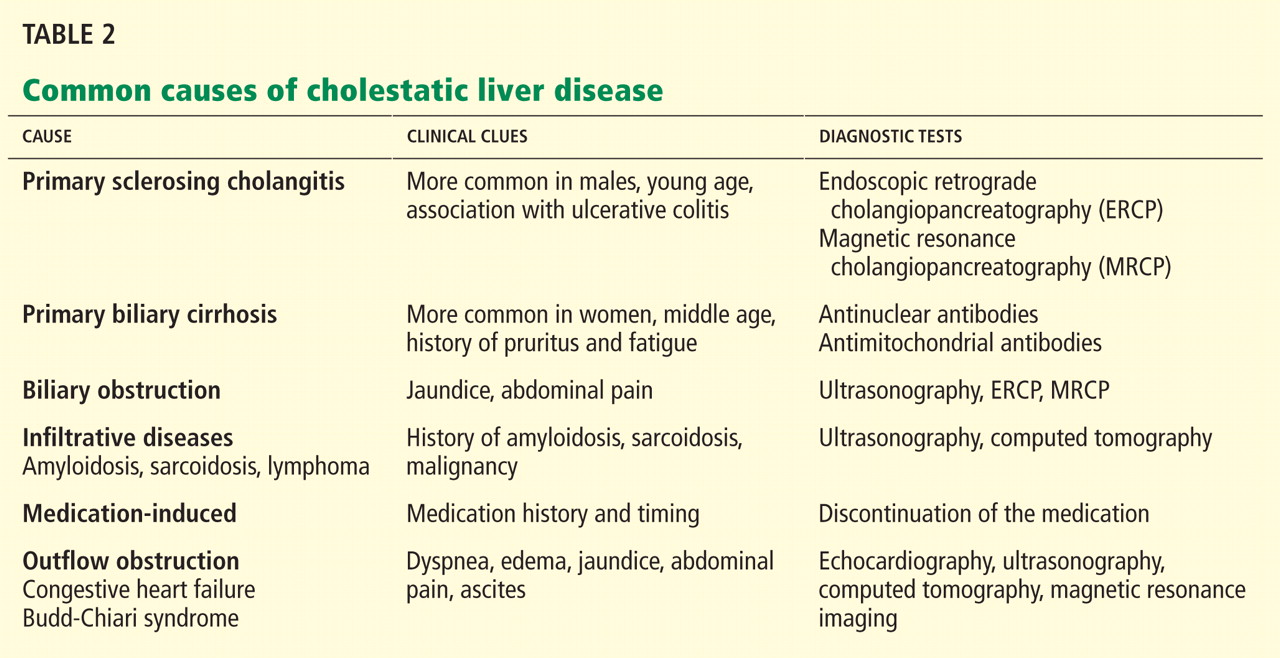
Viral hepatitis
Viral hepatitis most often produces a hepatocellular pattern of injury (ie, AST and ALT elevations predominate). However, in rare cases it can cause a cholestatic pattern of injury.
Our patient subsequently had serologic tests for viral hepatitis, including hepatitis A, B, and C, and the results were negative.
Autoimmune liver disease
The three most common forms of autoimmune liver disease are autoimmune hepatitis, primary biliary cirrhosis, and primary sclerosing cholangitis.
Autoimmune hepatitis is characterized by high serum ALT and AST levels, whereas primary biliary cirrhosis and primary sclerosing cholangitis are associated with predominant elevations of alkaline phosphatase, since they are cholestatic disorders.
Our patient’s alkaline phosphatase level was much higher than his ALT and AST levels, making the latter two diseases more likely.
Primary biliary cirrhosis (and autoimmune hepatitis) are associated with autoantibodies in the serum, such as antinuclear antibody, smooth muscle antibody, and antimitochondrial antibody.
Our patient subsequently was tested for these antibodies, and the results were negative.
Primary sclerosing cholangitis usually affects the extrahepatic biliary system. Thus, if it is present, abnormalities should be seen on imaging.
As mentioned previously, no dilated intrahepatic or extrahepatic biliary ducts were seen on ultrasonography in our patient. Moreover, primary sclerosing cholangitis is associated with inflammatory bowel disease, particularly ulcerative colitis, which our patient did not have.
Drug-induced liver injury
Drug-induced liver injury is a common cause of cholestatic liver disease. However, our patient was not taking any prescribed, over-the-counter, or herbal medications. Additionally, he denied heavy alcohol use.
Infiltrative disorders
Infiltrative disorders such as amyloidosis, sarcoidosis, or lymphoma should be considered in the differential diagnosis of cholestatic liver disease. A clue to a possible infiltrative process is a markedly elevated level of alkaline phosphatase with a mildly increased serum bilirubin concentration, both of which our patient had.
AFTER ULTRASONOGRAPHY, WHAT IS THE NEXT STEP?
2. Which of the following is the next most appropriate diagnostic test for our patient?
- Endoscopic retrograde cholangiopancreatography (ERCP)
- Magnetic resonance cholangiopancreatography (MRCP)
- Liver biopsy
- CT of the abdomen
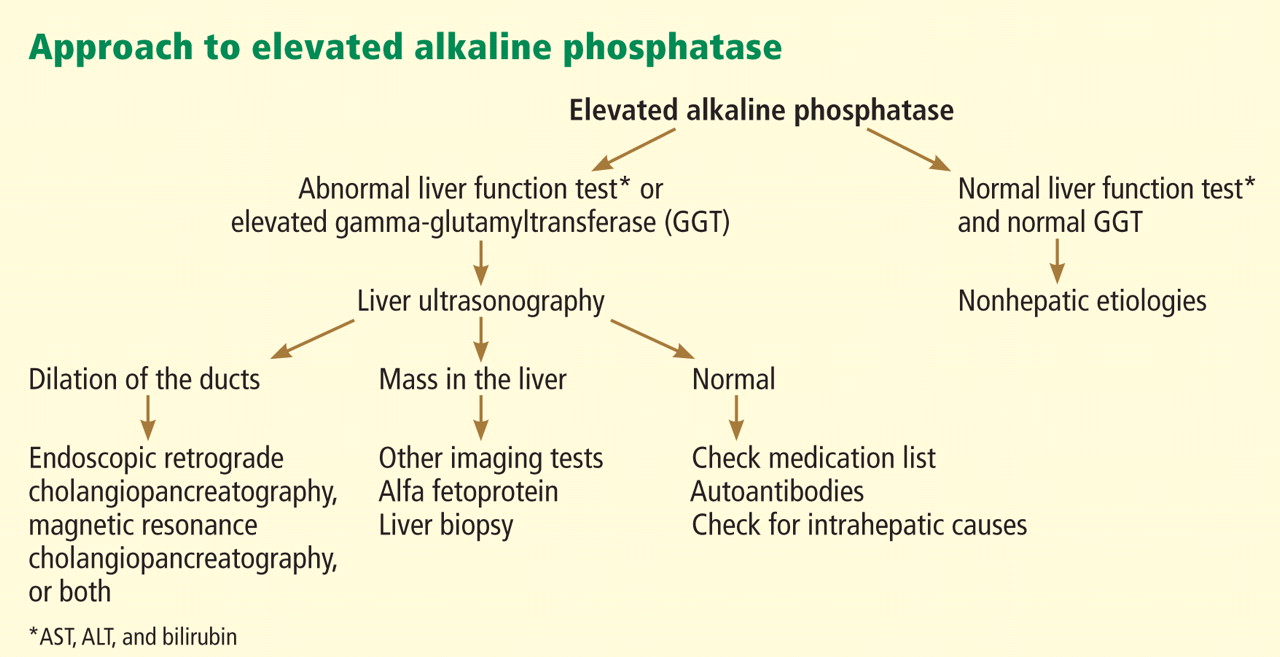
Figure 1 shows a proposed algorithm for evaluating increased alkaline phosphatase levels.
If there is no biliary duct dilation on ultrasonography, then abnormal levels of alkaline phosphatase most likely represent an intrahepatic pattern of cholestatic liver injury. Therefore, additional imaging with CT or magnetic resonance imaging is of limited diagnostic value. ERCP is used today for therapy rather than diagnosis, so its use is limited to patients known to have dilated biliary ducts on imaging. Liver biopsy, however, can provide useful findings.
Case continued: He undergoes biopsy
Our patient underwent transjugular liver biopsy. During the procedure, transjugular venography showed stenosis in the right, middle, and left hepatic veins and the hepatic portion of the inferior vena cava, consistent with Budd-Chiari syndrome.
The liver biopsy specimen was positive for extensive deposition of slight eosinophilic and amorphous material in a sinusoidal pattern in the liver parenchyma, as well as in the portal tracts, with markedly atrophic hepatocytes. Congo red birefringence confirmed the diagnosis of amyloidosis. The immunohistochemical phenotype was positive for kappa light chains, which is diagnostic for primary-type amyloidosis, also called amyloidosis of light chain composition, or AL.
Bone marrow aspiration and bone marrow biopsy were performed and showed 22% plasma cells, well above the normal range (0–2%), consistent with the diagnosis of multiple myeloma.
BUDD-CHIARI SYNDROME: A CHALLENGING DIAGNOSIS
Budd-Chiari syndrome is a rare condition characterized by obstruction of venous outflow from the liver at a site that may vary from the small hepatic veins up to the inferior vena cava or even the right atrium.5,6 Obstruction of hepatic venous outflow leads to sinusoidal congestion and hypoxic damage of the hepatocytes.7 Hypoxia and necrosis of the hepatocytes result in the release of free radicals. Cirrhosis can eventually occur secondary to ischemic necrosis of hepatocytes and hepatic fibrosis.8
The estimated incidence of this syndrome is 1 in 2.5 million persons per year.7 It is more prevalent in women and young adults.8
Heterogeneous in its causes and manifestations

Budd-Chiari syndrome is also heterogeneous in its manifestations, which depend on the extent of the occlusion, on the acuteness of the obstruction, and on whether venous collateral circulation has developed to decompress the liver sinusoids.9,12,13 Therefore, on the basis of its clinical manifestations, it can be classified as fulminant, acute, subacute, or chronic.12–16
The fulminant form presents with hepatic encephalopathy within 8 weeks after the development of jaundice. The subacute form, which is the most common, has a more insidious onset in which hepatic sinusoids are decompressed by portal and hepatic venous collateral circulation. The patient usually presents with abdominal pain, ascites, hepatomegaly, nausea, vomiting, and mild jaundice. Finally the chronic form presents as complications of cirrhosis.12–16
Imaging plays an important role in diagnosing Budd-Chiari syndrome
Imaging plays an important role in detecting and classifying Budd-Chiari syndrome.
Duplex ultrasonography is useful for detecting this syndrome and has a sensitivity and specificity of 85%.9
CT and magnetic resonance imaging can also help in the diagnosis by showing thrombosis, obstruction, or occlusion in the hepatic vein or the inferior vena cava.5
Venography is the gold standard for diagnosis. However, it should be performed only if noninvasive tests are negative or nondiagnostic and there is a high clinical suspicion of this disease.17 Budd-Chiari syndrome has a characteristic pattern on venography known as “spider web,” which is due to the formation of venous collaterals to bypass the occluded hepatic veins.9
Liver biopsy is not necessarily required to confirm the diagnosis of Budd-Chiari syndrome, but it can help in diagnosing the acute or subacute forms and also in ruling out other causes. Histologic findings can include centrizonal congestion, loss of hepatocytes, hemorrhage, and fibrosis.18,19 Regenerative nodules are found in about 25% of patients.19
TREATING BUDD-CHIARI SYNDROME
The primary goal of treatment is to prevent further extension of the venous thrombosis in the hepatic veins, in their collaterals, and in the intrahepatic and extrahepatic portal venous system. Resolution of hepatic congestion improves liver perfusion and preserves function of the hepatocytes.
Anticoagulation is recommended in the early stages. Heparin therapy should be initiated and subsequently switched to warfarin with the goal of achieving an international normalized ratio of the prothrombin time of 2.0 to 2.5.8,9,19
Thrombolysis is effective in the acute form.20,21 Recanalization, including percutaneous or transhepatic angioplasty of localized segments of the narrowed hepatic veins or inferior vena cava, has long-term patency rates of 80% to 90%.22
If thrombolytic therapy and angioplasty are unsuccessful, a transjugular intrahepatic portosystemic shunt or a surgical procedure (side-to-side portocaval shunt, central splenorenal shunt, or mesocaval shunt) should be considered.9
Liver transplantation is another treatment option in those with fulminant Budd-Chiari syndrome or advanced liver cirrhosis.8
PROGNOSIS HAS IMPROVED
The prognosis of Budd-Chiari syndrome has improved, thanks to both earlier diagnosis and new treatments. The 1-year survival rate, which was about 60% before 1985, has increased to more than 80% in recent cohort studies.19
Studies have shown that the Child-Pugh score, which is based on a combination of serum albumin, bilirubin, prothrombin time, encephalopathy, and ascites, can be considered as an independent prognostic factor. A lower Child-Pugh score and a younger age are associated with a good prognosis.19,23,24 (The Child-Pugh score cannot be applied to our patient because he does not have cirrhosis.)
What happened to our patient?
Our patient was started on anticoagulation for his Budd-Chiari syndrome and on bortezomib (Velcade) and dexamethasone for his multiple myeloma. He achieved remarkable improvement in his liver function tests. Follow-up duplex ultrasonography 1 month after discharge revealed that the stenosis in the hepatic veins had resolved. He is following up with the oncology clinic for management of his multiple myeloma.
- Folwaczny C. Efficient diagnostics for elevated transaminases. [Article in German] MMW Fortschr Med 2007; 149:44–48.
- Moussavian SN, Becker RC, Piepmeyer JL, Mezey E, Bozian RC. Serum gamma-glutamyl transpeptidase and chronic alcoholism. Influence of alcohol ingestion and liver disease. Dig Dis Sci 1985; 30:211–214.
- Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med 2010; 77:195–204.
- Lepper PM, Dufour JF. Elevated transaminases—what to do if everything was done?. [Article in German] Praxis (Bern 1994) 2009; 98:330–334.
- Buzas C, Sparchez Z, Cucuianu A, Manole S, Lupescu I, Acalovschi M. Budd-Chiari syndrome secondary to polycythemia vera. A case report. J Gastrointestin Liver Dis 2009; 18:363–366.
- Valla DC. Primary Budd-Chiari syndrome. J Hepatol 2009; 50:195–203.
- Rautou PE, Moucari R, Cazals-Hatem D, et al. Levels and initial course of serum alanine aminotransferase can predict outcome of patients with Budd-Chiari syndrome. Clin Gastroenterol Hepatol 2009; 7:1230–1235.
- Cura M, Haskal Z, Lopera J. Diagnostic and interventional radiology for Budd-Chiari syndrome. Radiographics 2009; 29:669–681.
- Menon KV, Shah V, Kamath PS. The Budd-Chiari syndrome. N Engl J Med 2004; 350:578–585.
- Darwish Murad S, Plessier A, Hernandez-Guerra M, et al; EN-Vie (European Network for Vascular Disorders of the Liver). Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med 2009; 151:167–175.
- Valla D, Le MG, Poynard T, Zucman N, Rueff B, Benhamou JP. Risk of hepatic vein thrombosis in relation to recent use of oral contraceptives. A case-control study. Gastroenterology 1986; 90:807–811.
- Bismuth H, Sherlock DJ. Portasystemic shunting versus liver transplantation for the Budd-Chiari syndrome. Ann Surg 1991; 214:581–589.
- Orloff MJ, Daily PO, Orloff SL, Girard B, Orloff MS. A 27-year experience with surgical treatment of Budd-Chiari syndrome. Ann Surg 2000; 232:340–352.
- Dilawari JB, Bambery P, Chawla Y, et al. Hepatic outflow obstruction (Budd-Chiari syndrome). Experience with 177 patients and a review of the literature. Medicine (Baltimore) 1994; 73:21–36.
- Mahmoud AE, Mendoza A, Meshikhes AN, et al. Clinical spectrum, investigations and treatment of Budd-Chiari syndrome. QJM 1996; 89:37–43.
- Klein AS, Cameron JL. Diagnosis and management of the Budd-Chiari syndrome. Am J Surg 1990; 160:128–133.
- Plessier A, Valla DC. Budd-Chiari syndrome. Semin Liver Dis 2008; 28:259–269.
- Cazals-Hatem D, Vilgrain V, Genin P, et al. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: a study in 17 explanted livers. Hepatology 2003; 37:510–519.
- Hoekstra J, Janssen HL. Vascular liver disorders (I): diagnosis, treatment and prognosis of Budd-Chiari syndrome. Neth J Med 2008; 66:334–359.
- Frank JW, Kamath PS, Stanson AW. Budd-Chiari syndrome: early intervention with angioplasty and thrombolytic therapy. Mayo Clin Proc 1994; 69:877–881.
- Raju GS, Felver M, Olin JW, Satti SD. Thrombolysis for acute Budd-Chiari syndrome: case report and literature review. Am J Gastroenterol 1996; 91:1262–1263.
- Fisher NC, McCafferty I, Dolapci M, et al. Managing Budd-Chiari syndrome: a retrospective review of percutaneous hepatic vein angioplasty and surgical shunting. Gut 1999; 44:568–574.
- Zeitoun G, Escolano S, Hadengue A, et al. Outcome of Budd-Chiari syndrome: a multivariate analysis of factors related to survival including surgical portosystemic shunting. Hepatology 1999; 30:84–89.
- Darwish Murad S, Valla DC, de Groen PC, et al. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology 2004; 39:500–508.
A 25-year-old man presented to his primary care physician with generalized malaise. His symptoms started around 2 months earlier with progressive fatigue, nausea, decreased appetite, and weight loss (15 lb in 2 months). He denied having fever, chills, night sweats, abdominal pain, diarrhea, melena, or hematochezia.
His medical history was remarkable only for depression, well controlled with sertraline (Zoloft), which he started taking 3 years ago. He was not taking any other prescribed, over-the-counter, or herbal medications.
He had no family history of cancer or liver disease. He did not smoke and rarely drank alcohol. He had never used recreational drugs. He was sexually active with one female partner, used condoms for protection, and had never been diagnosed with a sexually transmitted disease. He had not traveled recently and had not been exposed to any pet.
The patient’s laboratory values on admission are shown in Table 1. Of note, his serum alkaline phosphatase level was 1,307 U/L (reference range 40–150 U/L).
LIVER TESTS CAN NARROW THE DIAGNOSIS
The most commonly used laboratory tests of the liver can be classified into those that measure either:
- Liver synthetic function (eg, the serum albumin and bilirubin concentrations and the prothrombin time) or
- Liver damage, as reflected by the serum concentrations of the enzymes alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and gamma-glutamyltransferase (GGT).1,2
ALT and AST are normally concentrated in the hepatocytes and thus, when present in the serum in elevated concentrations, are markers of liver cell injury. The serum levels of these enzymes start to increase within a few hours of liver cell injury as they leak out of the cells via the damaged cell membrane. AST is less liver-specific than ALT, since AST levels can be elevated not only in liver injury but also in muscle, cardiac, and red blood cell injury.3,4
Alkaline phosphatase is actually a heterogeneous group of enzymes found mainly in liver and bone cells. Hepatic alkaline phosphatase is concentrated near the biliary canalicular membrane of the hepatocyte. Accordingly, increased levels of hepatic alkaline phosphatase are mainly seen in liver diseases that predominantly affect the biliary system.3
GGT is also concentrated in hepatic biliary epithelial cells, and thus GGT elevation is another marker of hepatobiliary disease. In fact, measuring the GGT level can help to determine whether an isolated elevation of alkaline phosphatase is due to liver injury.2,3
Accordingly, liver diseases can be classified into two broad categories:
- Hepatocellular injury, in which the primary injury occurs to the hepatocytes
- Cholestatic injury, in which the primary injury is to the bile ducts.
In the former, elevated levels of ALT and AST predominate, while in the latter, elevated alkaline phosphatase is the main finding.3
WHAT TEST NEXT FOR OUR PATIENT?
1. What is the next most appropriate diagnostic step for our patient?
- Liver biopsy
- Ultrasonography of the liver
- Computed tomography (CT) of the liver
- Observation
Our patient has an elevated GGT level, which suggests that his elevated alkaline phosphatase is of hepatic rather than bony origin. Moreover, a serum alkaline phosphatase level that is elevated out of proportion to the aminotransferase levels reflects cholestatic liver injury.
CASE CONTINUED: ULTRASONOGRAPHY IS MOSTLY NORMAL
Ultrasonography of the right upper quadrant revealed that the liver had normal echogenicity and was mildly enlarged. There was no focal hepatic lesion. The gallbladder appeared normal, with no stones or sludge. No dilated intrahepatic or extrahepatic biliary ducts were seen. The common bile duct measured 4 mm. A small amount of ascites not amenable to paracentesis was present.
Thus, in the absence of biliary dilation on ultrasonography, we are dealing with an intrahepatic cholestatic process.
CAUSES OF CHOLESTATIC LIVER DISEASE
Viral hepatitis
Viral hepatitis most often produces a hepatocellular pattern of injury (ie, AST and ALT elevations predominate). However, in rare cases it can cause a cholestatic pattern of injury.
Our patient subsequently had serologic tests for viral hepatitis, including hepatitis A, B, and C, and the results were negative.
Autoimmune liver disease
The three most common forms of autoimmune liver disease are autoimmune hepatitis, primary biliary cirrhosis, and primary sclerosing cholangitis.
Autoimmune hepatitis is characterized by high serum ALT and AST levels, whereas primary biliary cirrhosis and primary sclerosing cholangitis are associated with predominant elevations of alkaline phosphatase, since they are cholestatic disorders.
Our patient’s alkaline phosphatase level was much higher than his ALT and AST levels, making the latter two diseases more likely.
Primary biliary cirrhosis (and autoimmune hepatitis) are associated with autoantibodies in the serum, such as antinuclear antibody, smooth muscle antibody, and antimitochondrial antibody.
Our patient subsequently was tested for these antibodies, and the results were negative.
Primary sclerosing cholangitis usually affects the extrahepatic biliary system. Thus, if it is present, abnormalities should be seen on imaging.
As mentioned previously, no dilated intrahepatic or extrahepatic biliary ducts were seen on ultrasonography in our patient. Moreover, primary sclerosing cholangitis is associated with inflammatory bowel disease, particularly ulcerative colitis, which our patient did not have.
Drug-induced liver injury
Drug-induced liver injury is a common cause of cholestatic liver disease. However, our patient was not taking any prescribed, over-the-counter, or herbal medications. Additionally, he denied heavy alcohol use.
Infiltrative disorders
Infiltrative disorders such as amyloidosis, sarcoidosis, or lymphoma should be considered in the differential diagnosis of cholestatic liver disease. A clue to a possible infiltrative process is a markedly elevated level of alkaline phosphatase with a mildly increased serum bilirubin concentration, both of which our patient had.
AFTER ULTRASONOGRAPHY, WHAT IS THE NEXT STEP?
2. Which of the following is the next most appropriate diagnostic test for our patient?
- Endoscopic retrograde cholangiopancreatography (ERCP)
- Magnetic resonance cholangiopancreatography (MRCP)
- Liver biopsy
- CT of the abdomen
Figure 1 shows a proposed algorithm for evaluating increased alkaline phosphatase levels.
If there is no biliary duct dilation on ultrasonography, then abnormal levels of alkaline phosphatase most likely represent an intrahepatic pattern of cholestatic liver injury. Therefore, additional imaging with CT or magnetic resonance imaging is of limited diagnostic value. ERCP is used today for therapy rather than diagnosis, so its use is limited to patients known to have dilated biliary ducts on imaging. Liver biopsy, however, can provide useful findings.
Case continued: He undergoes biopsy
Our patient underwent transjugular liver biopsy. During the procedure, transjugular venography showed stenosis in the right, middle, and left hepatic veins and the hepatic portion of the inferior vena cava, consistent with Budd-Chiari syndrome.
The liver biopsy specimen was positive for extensive deposition of slight eosinophilic and amorphous material in a sinusoidal pattern in the liver parenchyma, as well as in the portal tracts, with markedly atrophic hepatocytes. Congo red birefringence confirmed the diagnosis of amyloidosis. The immunohistochemical phenotype was positive for kappa light chains, which is diagnostic for primary-type amyloidosis, also called amyloidosis of light chain composition, or AL.
Bone marrow aspiration and bone marrow biopsy were performed and showed 22% plasma cells, well above the normal range (0–2%), consistent with the diagnosis of multiple myeloma.
BUDD-CHIARI SYNDROME: A CHALLENGING DIAGNOSIS
Budd-Chiari syndrome is a rare condition characterized by obstruction of venous outflow from the liver at a site that may vary from the small hepatic veins up to the inferior vena cava or even the right atrium.5,6 Obstruction of hepatic venous outflow leads to sinusoidal congestion and hypoxic damage of the hepatocytes.7 Hypoxia and necrosis of the hepatocytes result in the release of free radicals. Cirrhosis can eventually occur secondary to ischemic necrosis of hepatocytes and hepatic fibrosis.8
The estimated incidence of this syndrome is 1 in 2.5 million persons per year.7 It is more prevalent in women and young adults.8
Heterogeneous in its causes and manifestations
Budd-Chiari syndrome is also heterogeneous in its manifestations, which depend on the extent of the occlusion, on the acuteness of the obstruction, and on whether venous collateral circulation has developed to decompress the liver sinusoids.9,12,13 Therefore, on the basis of its clinical manifestations, it can be classified as fulminant, acute, subacute, or chronic.12–16
The fulminant form presents with hepatic encephalopathy within 8 weeks after the development of jaundice. The subacute form, which is the most common, has a more insidious onset in which hepatic sinusoids are decompressed by portal and hepatic venous collateral circulation. The patient usually presents with abdominal pain, ascites, hepatomegaly, nausea, vomiting, and mild jaundice. Finally the chronic form presents as complications of cirrhosis.12–16
Imaging plays an important role in diagnosing Budd-Chiari syndrome
Imaging plays an important role in detecting and classifying Budd-Chiari syndrome.
Duplex ultrasonography is useful for detecting this syndrome and has a sensitivity and specificity of 85%.9
CT and magnetic resonance imaging can also help in the diagnosis by showing thrombosis, obstruction, or occlusion in the hepatic vein or the inferior vena cava.5
Venography is the gold standard for diagnosis. However, it should be performed only if noninvasive tests are negative or nondiagnostic and there is a high clinical suspicion of this disease.17 Budd-Chiari syndrome has a characteristic pattern on venography known as “spider web,” which is due to the formation of venous collaterals to bypass the occluded hepatic veins.9
Liver biopsy is not necessarily required to confirm the diagnosis of Budd-Chiari syndrome, but it can help in diagnosing the acute or subacute forms and also in ruling out other causes. Histologic findings can include centrizonal congestion, loss of hepatocytes, hemorrhage, and fibrosis.18,19 Regenerative nodules are found in about 25% of patients.19
TREATING BUDD-CHIARI SYNDROME
The primary goal of treatment is to prevent further extension of the venous thrombosis in the hepatic veins, in their collaterals, and in the intrahepatic and extrahepatic portal venous system. Resolution of hepatic congestion improves liver perfusion and preserves function of the hepatocytes.
Anticoagulation is recommended in the early stages. Heparin therapy should be initiated and subsequently switched to warfarin with the goal of achieving an international normalized ratio of the prothrombin time of 2.0 to 2.5.8,9,19
Thrombolysis is effective in the acute form.20,21 Recanalization, including percutaneous or transhepatic angioplasty of localized segments of the narrowed hepatic veins or inferior vena cava, has long-term patency rates of 80% to 90%.22
If thrombolytic therapy and angioplasty are unsuccessful, a transjugular intrahepatic portosystemic shunt or a surgical procedure (side-to-side portocaval shunt, central splenorenal shunt, or mesocaval shunt) should be considered.9
Liver transplantation is another treatment option in those with fulminant Budd-Chiari syndrome or advanced liver cirrhosis.8
PROGNOSIS HAS IMPROVED
The prognosis of Budd-Chiari syndrome has improved, thanks to both earlier diagnosis and new treatments. The 1-year survival rate, which was about 60% before 1985, has increased to more than 80% in recent cohort studies.19
Studies have shown that the Child-Pugh score, which is based on a combination of serum albumin, bilirubin, prothrombin time, encephalopathy, and ascites, can be considered as an independent prognostic factor. A lower Child-Pugh score and a younger age are associated with a good prognosis.19,23,24 (The Child-Pugh score cannot be applied to our patient because he does not have cirrhosis.)
What happened to our patient?
Our patient was started on anticoagulation for his Budd-Chiari syndrome and on bortezomib (Velcade) and dexamethasone for his multiple myeloma. He achieved remarkable improvement in his liver function tests. Follow-up duplex ultrasonography 1 month after discharge revealed that the stenosis in the hepatic veins had resolved. He is following up with the oncology clinic for management of his multiple myeloma.
A 25-year-old man presented to his primary care physician with generalized malaise. His symptoms started around 2 months earlier with progressive fatigue, nausea, decreased appetite, and weight loss (15 lb in 2 months). He denied having fever, chills, night sweats, abdominal pain, diarrhea, melena, or hematochezia.
His medical history was remarkable only for depression, well controlled with sertraline (Zoloft), which he started taking 3 years ago. He was not taking any other prescribed, over-the-counter, or herbal medications.
He had no family history of cancer or liver disease. He did not smoke and rarely drank alcohol. He had never used recreational drugs. He was sexually active with one female partner, used condoms for protection, and had never been diagnosed with a sexually transmitted disease. He had not traveled recently and had not been exposed to any pet.
The patient’s laboratory values on admission are shown in Table 1. Of note, his serum alkaline phosphatase level was 1,307 U/L (reference range 40–150 U/L).
LIVER TESTS CAN NARROW THE DIAGNOSIS
The most commonly used laboratory tests of the liver can be classified into those that measure either:
- Liver synthetic function (eg, the serum albumin and bilirubin concentrations and the prothrombin time) or
- Liver damage, as reflected by the serum concentrations of the enzymes alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and gamma-glutamyltransferase (GGT).1,2
ALT and AST are normally concentrated in the hepatocytes and thus, when present in the serum in elevated concentrations, are markers of liver cell injury. The serum levels of these enzymes start to increase within a few hours of liver cell injury as they leak out of the cells via the damaged cell membrane. AST is less liver-specific than ALT, since AST levels can be elevated not only in liver injury but also in muscle, cardiac, and red blood cell injury.3,4
Alkaline phosphatase is actually a heterogeneous group of enzymes found mainly in liver and bone cells. Hepatic alkaline phosphatase is concentrated near the biliary canalicular membrane of the hepatocyte. Accordingly, increased levels of hepatic alkaline phosphatase are mainly seen in liver diseases that predominantly affect the biliary system.3
GGT is also concentrated in hepatic biliary epithelial cells, and thus GGT elevation is another marker of hepatobiliary disease. In fact, measuring the GGT level can help to determine whether an isolated elevation of alkaline phosphatase is due to liver injury.2,3
Accordingly, liver diseases can be classified into two broad categories:
- Hepatocellular injury, in which the primary injury occurs to the hepatocytes
- Cholestatic injury, in which the primary injury is to the bile ducts.
In the former, elevated levels of ALT and AST predominate, while in the latter, elevated alkaline phosphatase is the main finding.3
WHAT TEST NEXT FOR OUR PATIENT?
1. What is the next most appropriate diagnostic step for our patient?
- Liver biopsy
- Ultrasonography of the liver
- Computed tomography (CT) of the liver
- Observation
Our patient has an elevated GGT level, which suggests that his elevated alkaline phosphatase is of hepatic rather than bony origin. Moreover, a serum alkaline phosphatase level that is elevated out of proportion to the aminotransferase levels reflects cholestatic liver injury.
CASE CONTINUED: ULTRASONOGRAPHY IS MOSTLY NORMAL
Ultrasonography of the right upper quadrant revealed that the liver had normal echogenicity and was mildly enlarged. There was no focal hepatic lesion. The gallbladder appeared normal, with no stones or sludge. No dilated intrahepatic or extrahepatic biliary ducts were seen. The common bile duct measured 4 mm. A small amount of ascites not amenable to paracentesis was present.
Thus, in the absence of biliary dilation on ultrasonography, we are dealing with an intrahepatic cholestatic process.
CAUSES OF CHOLESTATIC LIVER DISEASE
Viral hepatitis
Viral hepatitis most often produces a hepatocellular pattern of injury (ie, AST and ALT elevations predominate). However, in rare cases it can cause a cholestatic pattern of injury.
Our patient subsequently had serologic tests for viral hepatitis, including hepatitis A, B, and C, and the results were negative.
Autoimmune liver disease
The three most common forms of autoimmune liver disease are autoimmune hepatitis, primary biliary cirrhosis, and primary sclerosing cholangitis.
Autoimmune hepatitis is characterized by high serum ALT and AST levels, whereas primary biliary cirrhosis and primary sclerosing cholangitis are associated with predominant elevations of alkaline phosphatase, since they are cholestatic disorders.
Our patient’s alkaline phosphatase level was much higher than his ALT and AST levels, making the latter two diseases more likely.
Primary biliary cirrhosis (and autoimmune hepatitis) are associated with autoantibodies in the serum, such as antinuclear antibody, smooth muscle antibody, and antimitochondrial antibody.
Our patient subsequently was tested for these antibodies, and the results were negative.
Primary sclerosing cholangitis usually affects the extrahepatic biliary system. Thus, if it is present, abnormalities should be seen on imaging.
As mentioned previously, no dilated intrahepatic or extrahepatic biliary ducts were seen on ultrasonography in our patient. Moreover, primary sclerosing cholangitis is associated with inflammatory bowel disease, particularly ulcerative colitis, which our patient did not have.
Drug-induced liver injury
Drug-induced liver injury is a common cause of cholestatic liver disease. However, our patient was not taking any prescribed, over-the-counter, or herbal medications. Additionally, he denied heavy alcohol use.
Infiltrative disorders
Infiltrative disorders such as amyloidosis, sarcoidosis, or lymphoma should be considered in the differential diagnosis of cholestatic liver disease. A clue to a possible infiltrative process is a markedly elevated level of alkaline phosphatase with a mildly increased serum bilirubin concentration, both of which our patient had.
AFTER ULTRASONOGRAPHY, WHAT IS THE NEXT STEP?
2. Which of the following is the next most appropriate diagnostic test for our patient?
- Endoscopic retrograde cholangiopancreatography (ERCP)
- Magnetic resonance cholangiopancreatography (MRCP)
- Liver biopsy
- CT of the abdomen
Figure 1 shows a proposed algorithm for evaluating increased alkaline phosphatase levels.
If there is no biliary duct dilation on ultrasonography, then abnormal levels of alkaline phosphatase most likely represent an intrahepatic pattern of cholestatic liver injury. Therefore, additional imaging with CT or magnetic resonance imaging is of limited diagnostic value. ERCP is used today for therapy rather than diagnosis, so its use is limited to patients known to have dilated biliary ducts on imaging. Liver biopsy, however, can provide useful findings.
Case continued: He undergoes biopsy
Our patient underwent transjugular liver biopsy. During the procedure, transjugular venography showed stenosis in the right, middle, and left hepatic veins and the hepatic portion of the inferior vena cava, consistent with Budd-Chiari syndrome.
The liver biopsy specimen was positive for extensive deposition of slight eosinophilic and amorphous material in a sinusoidal pattern in the liver parenchyma, as well as in the portal tracts, with markedly atrophic hepatocytes. Congo red birefringence confirmed the diagnosis of amyloidosis. The immunohistochemical phenotype was positive for kappa light chains, which is diagnostic for primary-type amyloidosis, also called amyloidosis of light chain composition, or AL.
Bone marrow aspiration and bone marrow biopsy were performed and showed 22% plasma cells, well above the normal range (0–2%), consistent with the diagnosis of multiple myeloma.
BUDD-CHIARI SYNDROME: A CHALLENGING DIAGNOSIS
Budd-Chiari syndrome is a rare condition characterized by obstruction of venous outflow from the liver at a site that may vary from the small hepatic veins up to the inferior vena cava or even the right atrium.5,6 Obstruction of hepatic venous outflow leads to sinusoidal congestion and hypoxic damage of the hepatocytes.7 Hypoxia and necrosis of the hepatocytes result in the release of free radicals. Cirrhosis can eventually occur secondary to ischemic necrosis of hepatocytes and hepatic fibrosis.8
The estimated incidence of this syndrome is 1 in 2.5 million persons per year.7 It is more prevalent in women and young adults.8
Heterogeneous in its causes and manifestations
Budd-Chiari syndrome is also heterogeneous in its manifestations, which depend on the extent of the occlusion, on the acuteness of the obstruction, and on whether venous collateral circulation has developed to decompress the liver sinusoids.9,12,13 Therefore, on the basis of its clinical manifestations, it can be classified as fulminant, acute, subacute, or chronic.12–16
The fulminant form presents with hepatic encephalopathy within 8 weeks after the development of jaundice. The subacute form, which is the most common, has a more insidious onset in which hepatic sinusoids are decompressed by portal and hepatic venous collateral circulation. The patient usually presents with abdominal pain, ascites, hepatomegaly, nausea, vomiting, and mild jaundice. Finally the chronic form presents as complications of cirrhosis.12–16
Imaging plays an important role in diagnosing Budd-Chiari syndrome
Imaging plays an important role in detecting and classifying Budd-Chiari syndrome.
Duplex ultrasonography is useful for detecting this syndrome and has a sensitivity and specificity of 85%.9
CT and magnetic resonance imaging can also help in the diagnosis by showing thrombosis, obstruction, or occlusion in the hepatic vein or the inferior vena cava.5
Venography is the gold standard for diagnosis. However, it should be performed only if noninvasive tests are negative or nondiagnostic and there is a high clinical suspicion of this disease.17 Budd-Chiari syndrome has a characteristic pattern on venography known as “spider web,” which is due to the formation of venous collaterals to bypass the occluded hepatic veins.9
Liver biopsy is not necessarily required to confirm the diagnosis of Budd-Chiari syndrome, but it can help in diagnosing the acute or subacute forms and also in ruling out other causes. Histologic findings can include centrizonal congestion, loss of hepatocytes, hemorrhage, and fibrosis.18,19 Regenerative nodules are found in about 25% of patients.19
TREATING BUDD-CHIARI SYNDROME
The primary goal of treatment is to prevent further extension of the venous thrombosis in the hepatic veins, in their collaterals, and in the intrahepatic and extrahepatic portal venous system. Resolution of hepatic congestion improves liver perfusion and preserves function of the hepatocytes.
Anticoagulation is recommended in the early stages. Heparin therapy should be initiated and subsequently switched to warfarin with the goal of achieving an international normalized ratio of the prothrombin time of 2.0 to 2.5.8,9,19
Thrombolysis is effective in the acute form.20,21 Recanalization, including percutaneous or transhepatic angioplasty of localized segments of the narrowed hepatic veins or inferior vena cava, has long-term patency rates of 80% to 90%.22
If thrombolytic therapy and angioplasty are unsuccessful, a transjugular intrahepatic portosystemic shunt or a surgical procedure (side-to-side portocaval shunt, central splenorenal shunt, or mesocaval shunt) should be considered.9
Liver transplantation is another treatment option in those with fulminant Budd-Chiari syndrome or advanced liver cirrhosis.8
PROGNOSIS HAS IMPROVED
The prognosis of Budd-Chiari syndrome has improved, thanks to both earlier diagnosis and new treatments. The 1-year survival rate, which was about 60% before 1985, has increased to more than 80% in recent cohort studies.19
Studies have shown that the Child-Pugh score, which is based on a combination of serum albumin, bilirubin, prothrombin time, encephalopathy, and ascites, can be considered as an independent prognostic factor. A lower Child-Pugh score and a younger age are associated with a good prognosis.19,23,24 (The Child-Pugh score cannot be applied to our patient because he does not have cirrhosis.)
What happened to our patient?
Our patient was started on anticoagulation for his Budd-Chiari syndrome and on bortezomib (Velcade) and dexamethasone for his multiple myeloma. He achieved remarkable improvement in his liver function tests. Follow-up duplex ultrasonography 1 month after discharge revealed that the stenosis in the hepatic veins had resolved. He is following up with the oncology clinic for management of his multiple myeloma.
- Folwaczny C. Efficient diagnostics for elevated transaminases. [Article in German] MMW Fortschr Med 2007; 149:44–48.
- Moussavian SN, Becker RC, Piepmeyer JL, Mezey E, Bozian RC. Serum gamma-glutamyl transpeptidase and chronic alcoholism. Influence of alcohol ingestion and liver disease. Dig Dis Sci 1985; 30:211–214.
- Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med 2010; 77:195–204.
- Lepper PM, Dufour JF. Elevated transaminases—what to do if everything was done?. [Article in German] Praxis (Bern 1994) 2009; 98:330–334.
- Buzas C, Sparchez Z, Cucuianu A, Manole S, Lupescu I, Acalovschi M. Budd-Chiari syndrome secondary to polycythemia vera. A case report. J Gastrointestin Liver Dis 2009; 18:363–366.
- Valla DC. Primary Budd-Chiari syndrome. J Hepatol 2009; 50:195–203.
- Rautou PE, Moucari R, Cazals-Hatem D, et al. Levels and initial course of serum alanine aminotransferase can predict outcome of patients with Budd-Chiari syndrome. Clin Gastroenterol Hepatol 2009; 7:1230–1235.
- Cura M, Haskal Z, Lopera J. Diagnostic and interventional radiology for Budd-Chiari syndrome. Radiographics 2009; 29:669–681.
- Menon KV, Shah V, Kamath PS. The Budd-Chiari syndrome. N Engl J Med 2004; 350:578–585.
- Darwish Murad S, Plessier A, Hernandez-Guerra M, et al; EN-Vie (European Network for Vascular Disorders of the Liver). Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med 2009; 151:167–175.
- Valla D, Le MG, Poynard T, Zucman N, Rueff B, Benhamou JP. Risk of hepatic vein thrombosis in relation to recent use of oral contraceptives. A case-control study. Gastroenterology 1986; 90:807–811.
- Bismuth H, Sherlock DJ. Portasystemic shunting versus liver transplantation for the Budd-Chiari syndrome. Ann Surg 1991; 214:581–589.
- Orloff MJ, Daily PO, Orloff SL, Girard B, Orloff MS. A 27-year experience with surgical treatment of Budd-Chiari syndrome. Ann Surg 2000; 232:340–352.
- Dilawari JB, Bambery P, Chawla Y, et al. Hepatic outflow obstruction (Budd-Chiari syndrome). Experience with 177 patients and a review of the literature. Medicine (Baltimore) 1994; 73:21–36.
- Mahmoud AE, Mendoza A, Meshikhes AN, et al. Clinical spectrum, investigations and treatment of Budd-Chiari syndrome. QJM 1996; 89:37–43.
- Klein AS, Cameron JL. Diagnosis and management of the Budd-Chiari syndrome. Am J Surg 1990; 160:128–133.
- Plessier A, Valla DC. Budd-Chiari syndrome. Semin Liver Dis 2008; 28:259–269.
- Cazals-Hatem D, Vilgrain V, Genin P, et al. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: a study in 17 explanted livers. Hepatology 2003; 37:510–519.
- Hoekstra J, Janssen HL. Vascular liver disorders (I): diagnosis, treatment and prognosis of Budd-Chiari syndrome. Neth J Med 2008; 66:334–359.
- Frank JW, Kamath PS, Stanson AW. Budd-Chiari syndrome: early intervention with angioplasty and thrombolytic therapy. Mayo Clin Proc 1994; 69:877–881.
- Raju GS, Felver M, Olin JW, Satti SD. Thrombolysis for acute Budd-Chiari syndrome: case report and literature review. Am J Gastroenterol 1996; 91:1262–1263.
- Fisher NC, McCafferty I, Dolapci M, et al. Managing Budd-Chiari syndrome: a retrospective review of percutaneous hepatic vein angioplasty and surgical shunting. Gut 1999; 44:568–574.
- Zeitoun G, Escolano S, Hadengue A, et al. Outcome of Budd-Chiari syndrome: a multivariate analysis of factors related to survival including surgical portosystemic shunting. Hepatology 1999; 30:84–89.
- Darwish Murad S, Valla DC, de Groen PC, et al. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology 2004; 39:500–508.
- Folwaczny C. Efficient diagnostics for elevated transaminases. [Article in German] MMW Fortschr Med 2007; 149:44–48.
- Moussavian SN, Becker RC, Piepmeyer JL, Mezey E, Bozian RC. Serum gamma-glutamyl transpeptidase and chronic alcoholism. Influence of alcohol ingestion and liver disease. Dig Dis Sci 1985; 30:211–214.
- Aragon G, Younossi ZM. When and how to evaluate mildly elevated liver enzymes in apparently healthy patients. Cleve Clin J Med 2010; 77:195–204.
- Lepper PM, Dufour JF. Elevated transaminases—what to do if everything was done?. [Article in German] Praxis (Bern 1994) 2009; 98:330–334.
- Buzas C, Sparchez Z, Cucuianu A, Manole S, Lupescu I, Acalovschi M. Budd-Chiari syndrome secondary to polycythemia vera. A case report. J Gastrointestin Liver Dis 2009; 18:363–366.
- Valla DC. Primary Budd-Chiari syndrome. J Hepatol 2009; 50:195–203.
- Rautou PE, Moucari R, Cazals-Hatem D, et al. Levels and initial course of serum alanine aminotransferase can predict outcome of patients with Budd-Chiari syndrome. Clin Gastroenterol Hepatol 2009; 7:1230–1235.
- Cura M, Haskal Z, Lopera J. Diagnostic and interventional radiology for Budd-Chiari syndrome. Radiographics 2009; 29:669–681.
- Menon KV, Shah V, Kamath PS. The Budd-Chiari syndrome. N Engl J Med 2004; 350:578–585.
- Darwish Murad S, Plessier A, Hernandez-Guerra M, et al; EN-Vie (European Network for Vascular Disorders of the Liver). Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med 2009; 151:167–175.
- Valla D, Le MG, Poynard T, Zucman N, Rueff B, Benhamou JP. Risk of hepatic vein thrombosis in relation to recent use of oral contraceptives. A case-control study. Gastroenterology 1986; 90:807–811.
- Bismuth H, Sherlock DJ. Portasystemic shunting versus liver transplantation for the Budd-Chiari syndrome. Ann Surg 1991; 214:581–589.
- Orloff MJ, Daily PO, Orloff SL, Girard B, Orloff MS. A 27-year experience with surgical treatment of Budd-Chiari syndrome. Ann Surg 2000; 232:340–352.
- Dilawari JB, Bambery P, Chawla Y, et al. Hepatic outflow obstruction (Budd-Chiari syndrome). Experience with 177 patients and a review of the literature. Medicine (Baltimore) 1994; 73:21–36.
- Mahmoud AE, Mendoza A, Meshikhes AN, et al. Clinical spectrum, investigations and treatment of Budd-Chiari syndrome. QJM 1996; 89:37–43.
- Klein AS, Cameron JL. Diagnosis and management of the Budd-Chiari syndrome. Am J Surg 1990; 160:128–133.
- Plessier A, Valla DC. Budd-Chiari syndrome. Semin Liver Dis 2008; 28:259–269.
- Cazals-Hatem D, Vilgrain V, Genin P, et al. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: a study in 17 explanted livers. Hepatology 2003; 37:510–519.
- Hoekstra J, Janssen HL. Vascular liver disorders (I): diagnosis, treatment and prognosis of Budd-Chiari syndrome. Neth J Med 2008; 66:334–359.
- Frank JW, Kamath PS, Stanson AW. Budd-Chiari syndrome: early intervention with angioplasty and thrombolytic therapy. Mayo Clin Proc 1994; 69:877–881.
- Raju GS, Felver M, Olin JW, Satti SD. Thrombolysis for acute Budd-Chiari syndrome: case report and literature review. Am J Gastroenterol 1996; 91:1262–1263.
- Fisher NC, McCafferty I, Dolapci M, et al. Managing Budd-Chiari syndrome: a retrospective review of percutaneous hepatic vein angioplasty and surgical shunting. Gut 1999; 44:568–574.
- Zeitoun G, Escolano S, Hadengue A, et al. Outcome of Budd-Chiari syndrome: a multivariate analysis of factors related to survival including surgical portosystemic shunting. Hepatology 1999; 30:84–89.
- Darwish Murad S, Valla DC, de Groen PC, et al. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology 2004; 39:500–508.