User login
EVI1 overexpression promotes leukemogenesis, study suggests

Preclinical research suggests the oncoprotein EVI1 can promote leukemogenesis by suppressing erythropoiesis and lymphopoiesis while shifting differentiation toward the expansion of myeloid cells.
Researchers developed a new mouse model that mimics chromosomal rearrangements at 3q26, which are associated with poor-prognosis acute myeloid leukemia (AML), myelodysplastic syndromes, and myeloproliferative neoplasms.
Using the mouse model, the team demonstrated that EVI1 overexpression distorts hematopoiesis and markedly expands premalignant myelopoiesis that eventually results in leukemic transformation.
Archibald Perkins, MD, PhD, of the University of Rochester Medical Center in New York, and his colleagues published these findings in Nature Communications.
The team demonstrated that the “myeloid-skewed phenotype” is dependent upon EVI1-binding DNA. This upregulates Spi1 and encodes the master myeloid regulator PU.1.
When the researchers knocked down Spi1, the myeloid skewing diminished.
“It’s not so pie-in-the-sky anymore,” Dr. Perkins said, “to think we can interrupt the process within the genome that leads to leukemia.”
The researchers first created a mouse model of 3q26 AML with a tetracycline-inducible allele of EVI1 by inserting tetracycline operons within the first exon. This allowed the induction of all three isoforms of EVI1.
These mice were viable and fertile but had no phenotype, which indicated that the allele functioned normally unless induced.
To assess the effect of EVI1 overexpression, the researchers transplanted oncogene-expressing bone marrow mixed 1:1 with wild-type bone marrow into recipient mice.
After confirming successful engraftment, the researchers fed the mice doxycycline-treated food to induce EVI1. The team analyzed cells in the peripheral blood and bone marrow at 10 weeks post-induction.
The researchers observed a more than two-fold expansion of the EVI1-overexpressing compartment in the mouse model.
Suppression of erythropoiesis
The researchers analyzed erythroid lineage in the transplanted mice at 2, 6, and 10 weeks post-induction and found the EVI1-overexpressing cells did not contribute effectively to erythropoiesis.
Using flow cytometry, the researchers quantitated apoptosis and proliferation in erythroid progenitors. They observed a six-fold increase in apoptosis within the erythroblasts compared to wild-type cells.
They also observed a drop in the proliferation of proerythroblasts and erythroblasts compared to wild-type.
Suppression of lymphopoiesis
The researchers observed significantly lower numbers of EVI1-overexpressing B-lineage cells within the bone marrow at 6 and 10 weeks.
And at 10 weeks post-induction, the team observed a decrease in peripheral T cells from approximately 1,800 cells/µL to approximately 750 cells/µL.
EVI1 nearly eliminated the peripheral B cells completely, they noted.
Expansion of myelopoiesis
The team reported that, at 2 weeks post-induction, the EVI1-overexpressing bone marrow and control bone marrow showed the same number of myeloid cells.
But at 6 and 10 weeks post-induction, the EVI1-overexpressing myeloid compartment expanded markedly.
The researchers aged a cohort of five mice transplanted with the 1:1 mix of wild-type and EVI1 bone marrow cells to determine if chronic overexpression of EVI1 results in leukemia.
All five mice died at 90 to 119 days of doxycycline treatment. Analysis revealed AML in all mice. Bone marrows were replete with blasts, and the peripheral blood revealed severe anemia.
The researchers then proceeded to establish the relationship between EVI1 and Spi1/PU.1 transcriptional regulation.
They documented binding of EVI1 to the regulatory element -14kbURE, which, together with EVI1., induced upregulation of PU.1.
When the team knocked down PU.1, myeloid skewing diminished. This, they say, indicates PU.1 is necessary for EVI1-induced myeloid expansion.
Funding for this research was provided by the National Institutes of Health, New York State Stem Cell Science, the Wilmot Cancer Institute, and the Clinical and Translational Science Institute at the University of Rochester.
The authors had no competing interests to disclose.
Preclinical research suggests the oncoprotein EVI1 can promote leukemogenesis by suppressing erythropoiesis and lymphopoiesis while shifting differentiation toward the expansion of myeloid cells.
Researchers developed a new mouse model that mimics chromosomal rearrangements at 3q26, which are associated with poor-prognosis acute myeloid leukemia (AML), myelodysplastic syndromes, and myeloproliferative neoplasms.
Using the mouse model, the team demonstrated that EVI1 overexpression distorts hematopoiesis and markedly expands premalignant myelopoiesis that eventually results in leukemic transformation.
Archibald Perkins, MD, PhD, of the University of Rochester Medical Center in New York, and his colleagues published these findings in Nature Communications.
The team demonstrated that the “myeloid-skewed phenotype” is dependent upon EVI1-binding DNA. This upregulates Spi1 and encodes the master myeloid regulator PU.1.
When the researchers knocked down Spi1, the myeloid skewing diminished.
“It’s not so pie-in-the-sky anymore,” Dr. Perkins said, “to think we can interrupt the process within the genome that leads to leukemia.”
The researchers first created a mouse model of 3q26 AML with a tetracycline-inducible allele of EVI1 by inserting tetracycline operons within the first exon. This allowed the induction of all three isoforms of EVI1.
These mice were viable and fertile but had no phenotype, which indicated that the allele functioned normally unless induced.
To assess the effect of EVI1 overexpression, the researchers transplanted oncogene-expressing bone marrow mixed 1:1 with wild-type bone marrow into recipient mice.
After confirming successful engraftment, the researchers fed the mice doxycycline-treated food to induce EVI1. The team analyzed cells in the peripheral blood and bone marrow at 10 weeks post-induction.
The researchers observed a more than two-fold expansion of the EVI1-overexpressing compartment in the mouse model.
Suppression of erythropoiesis
The researchers analyzed erythroid lineage in the transplanted mice at 2, 6, and 10 weeks post-induction and found the EVI1-overexpressing cells did not contribute effectively to erythropoiesis.
Using flow cytometry, the researchers quantitated apoptosis and proliferation in erythroid progenitors. They observed a six-fold increase in apoptosis within the erythroblasts compared to wild-type cells.
They also observed a drop in the proliferation of proerythroblasts and erythroblasts compared to wild-type.
Suppression of lymphopoiesis
The researchers observed significantly lower numbers of EVI1-overexpressing B-lineage cells within the bone marrow at 6 and 10 weeks.
And at 10 weeks post-induction, the team observed a decrease in peripheral T cells from approximately 1,800 cells/µL to approximately 750 cells/µL.
EVI1 nearly eliminated the peripheral B cells completely, they noted.
Expansion of myelopoiesis
The team reported that, at 2 weeks post-induction, the EVI1-overexpressing bone marrow and control bone marrow showed the same number of myeloid cells.
But at 6 and 10 weeks post-induction, the EVI1-overexpressing myeloid compartment expanded markedly.
The researchers aged a cohort of five mice transplanted with the 1:1 mix of wild-type and EVI1 bone marrow cells to determine if chronic overexpression of EVI1 results in leukemia.
All five mice died at 90 to 119 days of doxycycline treatment. Analysis revealed AML in all mice. Bone marrows were replete with blasts, and the peripheral blood revealed severe anemia.
The researchers then proceeded to establish the relationship between EVI1 and Spi1/PU.1 transcriptional regulation.
They documented binding of EVI1 to the regulatory element -14kbURE, which, together with EVI1., induced upregulation of PU.1.
When the team knocked down PU.1, myeloid skewing diminished. This, they say, indicates PU.1 is necessary for EVI1-induced myeloid expansion.
Funding for this research was provided by the National Institutes of Health, New York State Stem Cell Science, the Wilmot Cancer Institute, and the Clinical and Translational Science Institute at the University of Rochester.
The authors had no competing interests to disclose.
Preclinical research suggests the oncoprotein EVI1 can promote leukemogenesis by suppressing erythropoiesis and lymphopoiesis while shifting differentiation toward the expansion of myeloid cells.
Researchers developed a new mouse model that mimics chromosomal rearrangements at 3q26, which are associated with poor-prognosis acute myeloid leukemia (AML), myelodysplastic syndromes, and myeloproliferative neoplasms.
Using the mouse model, the team demonstrated that EVI1 overexpression distorts hematopoiesis and markedly expands premalignant myelopoiesis that eventually results in leukemic transformation.
Archibald Perkins, MD, PhD, of the University of Rochester Medical Center in New York, and his colleagues published these findings in Nature Communications.
The team demonstrated that the “myeloid-skewed phenotype” is dependent upon EVI1-binding DNA. This upregulates Spi1 and encodes the master myeloid regulator PU.1.
When the researchers knocked down Spi1, the myeloid skewing diminished.
“It’s not so pie-in-the-sky anymore,” Dr. Perkins said, “to think we can interrupt the process within the genome that leads to leukemia.”
The researchers first created a mouse model of 3q26 AML with a tetracycline-inducible allele of EVI1 by inserting tetracycline operons within the first exon. This allowed the induction of all three isoforms of EVI1.
These mice were viable and fertile but had no phenotype, which indicated that the allele functioned normally unless induced.
To assess the effect of EVI1 overexpression, the researchers transplanted oncogene-expressing bone marrow mixed 1:1 with wild-type bone marrow into recipient mice.
After confirming successful engraftment, the researchers fed the mice doxycycline-treated food to induce EVI1. The team analyzed cells in the peripheral blood and bone marrow at 10 weeks post-induction.
The researchers observed a more than two-fold expansion of the EVI1-overexpressing compartment in the mouse model.
Suppression of erythropoiesis
The researchers analyzed erythroid lineage in the transplanted mice at 2, 6, and 10 weeks post-induction and found the EVI1-overexpressing cells did not contribute effectively to erythropoiesis.
Using flow cytometry, the researchers quantitated apoptosis and proliferation in erythroid progenitors. They observed a six-fold increase in apoptosis within the erythroblasts compared to wild-type cells.
They also observed a drop in the proliferation of proerythroblasts and erythroblasts compared to wild-type.
Suppression of lymphopoiesis
The researchers observed significantly lower numbers of EVI1-overexpressing B-lineage cells within the bone marrow at 6 and 10 weeks.
And at 10 weeks post-induction, the team observed a decrease in peripheral T cells from approximately 1,800 cells/µL to approximately 750 cells/µL.
EVI1 nearly eliminated the peripheral B cells completely, they noted.
Expansion of myelopoiesis
The team reported that, at 2 weeks post-induction, the EVI1-overexpressing bone marrow and control bone marrow showed the same number of myeloid cells.
But at 6 and 10 weeks post-induction, the EVI1-overexpressing myeloid compartment expanded markedly.
The researchers aged a cohort of five mice transplanted with the 1:1 mix of wild-type and EVI1 bone marrow cells to determine if chronic overexpression of EVI1 results in leukemia.
All five mice died at 90 to 119 days of doxycycline treatment. Analysis revealed AML in all mice. Bone marrows were replete with blasts, and the peripheral blood revealed severe anemia.
The researchers then proceeded to establish the relationship between EVI1 and Spi1/PU.1 transcriptional regulation.
They documented binding of EVI1 to the regulatory element -14kbURE, which, together with EVI1., induced upregulation of PU.1.
When the team knocked down PU.1, myeloid skewing diminished. This, they say, indicates PU.1 is necessary for EVI1-induced myeloid expansion.
Funding for this research was provided by the National Institutes of Health, New York State Stem Cell Science, the Wilmot Cancer Institute, and the Clinical and Translational Science Institute at the University of Rochester.
The authors had no competing interests to disclose.
Team finds potential therapeutic target for AML
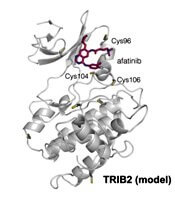
Researchers have found the cancer-associated pseudokinase Tribbles 2 (TRIB2) to be a potential therapeutic target in solid tumors and blood cancers, including acute myeloid leukemia (AML).
Previous research had described TRIB2 as a target of small-molecule protein kinase inhibitors originally designed to interfere with kinase domains of the epidermal growth factor receptor (EGFR) tyrosine kinase family.
Using a thermal shift assay, the team discovered TRIB2-binding compounds within the Published Kinase Inhibitor Set (PKIS). They then employed a biochemical drug repurposing approach to classify compounds that either stabilized or destabilized TRIB2 in vitro.
The researchers found that afatinib, which is already approved by the U.S. Food and Drug Administration to treat non-small cell lung cancer, led to rapid TRIB2 degradation in human AML cells.
Patrick A. Eyers, PhD, of the University of Liverpool in the U.K., and his colleagues published their findings in Science Signaling.
The team found afatinib to be relatively specific for EGFR and human epidermal growth factor receptor 2 (HER2) at nanomolar concentrations in cells.
The researchers confirmed that at least two TRIB2 Cys residues interact with afatinib in vitro.
The team also discovered TRIB2 could be destabilized by neratinib and osimertinib in vitro.
“Our data prove that the cellular mechanism by which TRIB2 stability is regulated by compounds is proteasome-based,” the researchers wrote, “and we speculate that an afatinib-induced conformational change might induce TRIB2 ubiquitination.”
The researchers plan to study further TRIB2 small-molecule interactions with dynamic changes in ubiquitination status.
Furthermore, they report their work demonstrates that covalent inhibitors such as afatinib have TRIB2-degrading activity in human cells at micromolar concentrations.
The researchers determined that afatinib has similar efficacy to the TRIB2-destabilizing quinazoline neratinib at similar ranges.
The team believes their data “raise the intriguing possibility that clinical inhibitors might be used as TRIB2-degrading agents in research, and possibly clinical, contexts.”
“A long-standing goal in cancer research is drug-induced degradation of oncogenic proteins,” Dr. Eyers commented. “Our study highlights how information obtained with ‘off-target’ effects of known drugs is potentially useful because it might be exploited in the future to help eliminate a protein that is involved in a completely different type of cancer.”
The TRIB proteins play many diverse roles in cell signaling, development, and cancer. According to a paper in Developmental Dynamics, they were named after the small, round, fictional organisms from the original Star Trek television series. Their major role was to eat and reproduce.
This work was funded by two U.K. Biotechnology and Biological Sciences Research Council Doctoral Training Partnership studentships, a Tools and Resources Development Fund award, Royal Society Research Grants, North West Cancer Research grants, and funding from the National Institutes of Health.
The authors disclosed no perceived conflicts of interest, although several authors are affiliated with the Structural Genomics Consortium at the University of North Carolina at Chapel Hill, which receives direct funds from various pharmaceutical companies but remains entirely independent.
Researchers have found the cancer-associated pseudokinase Tribbles 2 (TRIB2) to be a potential therapeutic target in solid tumors and blood cancers, including acute myeloid leukemia (AML).
Previous research had described TRIB2 as a target of small-molecule protein kinase inhibitors originally designed to interfere with kinase domains of the epidermal growth factor receptor (EGFR) tyrosine kinase family.
Using a thermal shift assay, the team discovered TRIB2-binding compounds within the Published Kinase Inhibitor Set (PKIS). They then employed a biochemical drug repurposing approach to classify compounds that either stabilized or destabilized TRIB2 in vitro.
The researchers found that afatinib, which is already approved by the U.S. Food and Drug Administration to treat non-small cell lung cancer, led to rapid TRIB2 degradation in human AML cells.
Patrick A. Eyers, PhD, of the University of Liverpool in the U.K., and his colleagues published their findings in Science Signaling.
The team found afatinib to be relatively specific for EGFR and human epidermal growth factor receptor 2 (HER2) at nanomolar concentrations in cells.
The researchers confirmed that at least two TRIB2 Cys residues interact with afatinib in vitro.
The team also discovered TRIB2 could be destabilized by neratinib and osimertinib in vitro.
“Our data prove that the cellular mechanism by which TRIB2 stability is regulated by compounds is proteasome-based,” the researchers wrote, “and we speculate that an afatinib-induced conformational change might induce TRIB2 ubiquitination.”
The researchers plan to study further TRIB2 small-molecule interactions with dynamic changes in ubiquitination status.
Furthermore, they report their work demonstrates that covalent inhibitors such as afatinib have TRIB2-degrading activity in human cells at micromolar concentrations.
The researchers determined that afatinib has similar efficacy to the TRIB2-destabilizing quinazoline neratinib at similar ranges.
The team believes their data “raise the intriguing possibility that clinical inhibitors might be used as TRIB2-degrading agents in research, and possibly clinical, contexts.”
“A long-standing goal in cancer research is drug-induced degradation of oncogenic proteins,” Dr. Eyers commented. “Our study highlights how information obtained with ‘off-target’ effects of known drugs is potentially useful because it might be exploited in the future to help eliminate a protein that is involved in a completely different type of cancer.”
The TRIB proteins play many diverse roles in cell signaling, development, and cancer. According to a paper in Developmental Dynamics, they were named after the small, round, fictional organisms from the original Star Trek television series. Their major role was to eat and reproduce.
This work was funded by two U.K. Biotechnology and Biological Sciences Research Council Doctoral Training Partnership studentships, a Tools and Resources Development Fund award, Royal Society Research Grants, North West Cancer Research grants, and funding from the National Institutes of Health.
The authors disclosed no perceived conflicts of interest, although several authors are affiliated with the Structural Genomics Consortium at the University of North Carolina at Chapel Hill, which receives direct funds from various pharmaceutical companies but remains entirely independent.
Researchers have found the cancer-associated pseudokinase Tribbles 2 (TRIB2) to be a potential therapeutic target in solid tumors and blood cancers, including acute myeloid leukemia (AML).
Previous research had described TRIB2 as a target of small-molecule protein kinase inhibitors originally designed to interfere with kinase domains of the epidermal growth factor receptor (EGFR) tyrosine kinase family.
Using a thermal shift assay, the team discovered TRIB2-binding compounds within the Published Kinase Inhibitor Set (PKIS). They then employed a biochemical drug repurposing approach to classify compounds that either stabilized or destabilized TRIB2 in vitro.
The researchers found that afatinib, which is already approved by the U.S. Food and Drug Administration to treat non-small cell lung cancer, led to rapid TRIB2 degradation in human AML cells.
Patrick A. Eyers, PhD, of the University of Liverpool in the U.K., and his colleagues published their findings in Science Signaling.
The team found afatinib to be relatively specific for EGFR and human epidermal growth factor receptor 2 (HER2) at nanomolar concentrations in cells.
The researchers confirmed that at least two TRIB2 Cys residues interact with afatinib in vitro.
The team also discovered TRIB2 could be destabilized by neratinib and osimertinib in vitro.
“Our data prove that the cellular mechanism by which TRIB2 stability is regulated by compounds is proteasome-based,” the researchers wrote, “and we speculate that an afatinib-induced conformational change might induce TRIB2 ubiquitination.”
The researchers plan to study further TRIB2 small-molecule interactions with dynamic changes in ubiquitination status.
Furthermore, they report their work demonstrates that covalent inhibitors such as afatinib have TRIB2-degrading activity in human cells at micromolar concentrations.
The researchers determined that afatinib has similar efficacy to the TRIB2-destabilizing quinazoline neratinib at similar ranges.
The team believes their data “raise the intriguing possibility that clinical inhibitors might be used as TRIB2-degrading agents in research, and possibly clinical, contexts.”
“A long-standing goal in cancer research is drug-induced degradation of oncogenic proteins,” Dr. Eyers commented. “Our study highlights how information obtained with ‘off-target’ effects of known drugs is potentially useful because it might be exploited in the future to help eliminate a protein that is involved in a completely different type of cancer.”
The TRIB proteins play many diverse roles in cell signaling, development, and cancer. According to a paper in Developmental Dynamics, they were named after the small, round, fictional organisms from the original Star Trek television series. Their major role was to eat and reproduce.
This work was funded by two U.K. Biotechnology and Biological Sciences Research Council Doctoral Training Partnership studentships, a Tools and Resources Development Fund award, Royal Society Research Grants, North West Cancer Research grants, and funding from the National Institutes of Health.
The authors disclosed no perceived conflicts of interest, although several authors are affiliated with the Structural Genomics Consortium at the University of North Carolina at Chapel Hill, which receives direct funds from various pharmaceutical companies but remains entirely independent.
Blood donated after mass shootings may go to waste
Public calls for blood donations in response to mass shootings may be unnecessary and result in waste, according to researchers.
Mass shootings often trigger a sharp increase in blood donations for affected communities.
In response to the recent mass shooting at the Tree Of Life Synagogue in Pittsburgh, Pennsylvania, multiple news outlets called for blood donations, and local blood centers extended their hours to compensate for the increase in donations.
However, a new study suggests that blood products donated in response to mass shootings may go unused and have to be discarded.
This study was published in The Journal of Trauma and Acute Care Surgery.
The study authors focused on blood donated in response to the mass shooting that took place in Las Vegas on October 1, 2017. This shooting resulted in 58 deaths, 869 injuries, 220 hospital admissions, and at least 68 critical care admissions.
Three healthcare systems provided data for the study. In all, 519 shooting victims were treated within these systems, and 185 were admitted to the hospitals. During the first 24 hours, these patients received 499 blood components, or 2.7 units per admission.
“From our data, it is likely that the total 1-day blood component transfusions needed in Las Vegas were more than in any mass shooting on record,” said study author M. James Lozada, DO, of Vanderbilt University Medical Center in Nashville, Tennessee.
However, the blood donations made in response to the shooting surpassed the need.
A public call for blood donations was issued during a press conference in the early hours of October 2, and that call was amplified in news stories. Stories about blood donation in Las Vegas increased from a daily average of 10 to more than 100 on October 2.
From October 2 to 4, the American Red Cross saw a 53% increase in blood donations nationwide.
The Las Vegas blood bank, United Blood Services, reported receiving 791 donations right after the shooting.
Unfortunately, 137 of these donations (17%) went unused and were subsequently discarded, compared to an average of 26 wasted donations per month at the blood bank.
Therefore, Dr. Lozada and his colleagues concluded that a call for immediate blood donation was unnecessary.
“There is an emotional desire after these events on the part of the public to immediately donate blood, but that’s not always necessary, and it’s not always the best immediate response,” Dr. Lozada said. “The best thing you can do is donate blood year-round.”
“One of the things that we propose in the paper is for cities to develop some protocols for these kind of scenarios, where instead of issuing a blanket call for blood donation, you would do it in a systematic way. As one suggestion, you might do it by ZIP code.”
“Our findings are important to help us prepare for the next mass shooting in the United States. It shows us the amount of blood components we likely will need. It will also help first responders adequately prepare to save lives.”
There was no funding for this research, and the study authors declared no conflicts of interest.
Public calls for blood donations in response to mass shootings may be unnecessary and result in waste, according to researchers.
Mass shootings often trigger a sharp increase in blood donations for affected communities.
In response to the recent mass shooting at the Tree Of Life Synagogue in Pittsburgh, Pennsylvania, multiple news outlets called for blood donations, and local blood centers extended their hours to compensate for the increase in donations.
However, a new study suggests that blood products donated in response to mass shootings may go unused and have to be discarded.
This study was published in The Journal of Trauma and Acute Care Surgery.
The study authors focused on blood donated in response to the mass shooting that took place in Las Vegas on October 1, 2017. This shooting resulted in 58 deaths, 869 injuries, 220 hospital admissions, and at least 68 critical care admissions.
Three healthcare systems provided data for the study. In all, 519 shooting victims were treated within these systems, and 185 were admitted to the hospitals. During the first 24 hours, these patients received 499 blood components, or 2.7 units per admission.
“From our data, it is likely that the total 1-day blood component transfusions needed in Las Vegas were more than in any mass shooting on record,” said study author M. James Lozada, DO, of Vanderbilt University Medical Center in Nashville, Tennessee.
However, the blood donations made in response to the shooting surpassed the need.
A public call for blood donations was issued during a press conference in the early hours of October 2, and that call was amplified in news stories. Stories about blood donation in Las Vegas increased from a daily average of 10 to more than 100 on October 2.
From October 2 to 4, the American Red Cross saw a 53% increase in blood donations nationwide.
The Las Vegas blood bank, United Blood Services, reported receiving 791 donations right after the shooting.
Unfortunately, 137 of these donations (17%) went unused and were subsequently discarded, compared to an average of 26 wasted donations per month at the blood bank.
Therefore, Dr. Lozada and his colleagues concluded that a call for immediate blood donation was unnecessary.
“There is an emotional desire after these events on the part of the public to immediately donate blood, but that’s not always necessary, and it’s not always the best immediate response,” Dr. Lozada said. “The best thing you can do is donate blood year-round.”
“One of the things that we propose in the paper is for cities to develop some protocols for these kind of scenarios, where instead of issuing a blanket call for blood donation, you would do it in a systematic way. As one suggestion, you might do it by ZIP code.”
“Our findings are important to help us prepare for the next mass shooting in the United States. It shows us the amount of blood components we likely will need. It will also help first responders adequately prepare to save lives.”
There was no funding for this research, and the study authors declared no conflicts of interest.
Public calls for blood donations in response to mass shootings may be unnecessary and result in waste, according to researchers.
Mass shootings often trigger a sharp increase in blood donations for affected communities.
In response to the recent mass shooting at the Tree Of Life Synagogue in Pittsburgh, Pennsylvania, multiple news outlets called for blood donations, and local blood centers extended their hours to compensate for the increase in donations.
However, a new study suggests that blood products donated in response to mass shootings may go unused and have to be discarded.
This study was published in The Journal of Trauma and Acute Care Surgery.
The study authors focused on blood donated in response to the mass shooting that took place in Las Vegas on October 1, 2017. This shooting resulted in 58 deaths, 869 injuries, 220 hospital admissions, and at least 68 critical care admissions.
Three healthcare systems provided data for the study. In all, 519 shooting victims were treated within these systems, and 185 were admitted to the hospitals. During the first 24 hours, these patients received 499 blood components, or 2.7 units per admission.
“From our data, it is likely that the total 1-day blood component transfusions needed in Las Vegas were more than in any mass shooting on record,” said study author M. James Lozada, DO, of Vanderbilt University Medical Center in Nashville, Tennessee.
However, the blood donations made in response to the shooting surpassed the need.
A public call for blood donations was issued during a press conference in the early hours of October 2, and that call was amplified in news stories. Stories about blood donation in Las Vegas increased from a daily average of 10 to more than 100 on October 2.
From October 2 to 4, the American Red Cross saw a 53% increase in blood donations nationwide.
The Las Vegas blood bank, United Blood Services, reported receiving 791 donations right after the shooting.
Unfortunately, 137 of these donations (17%) went unused and were subsequently discarded, compared to an average of 26 wasted donations per month at the blood bank.
Therefore, Dr. Lozada and his colleagues concluded that a call for immediate blood donation was unnecessary.
“There is an emotional desire after these events on the part of the public to immediately donate blood, but that’s not always necessary, and it’s not always the best immediate response,” Dr. Lozada said. “The best thing you can do is donate blood year-round.”
“One of the things that we propose in the paper is for cities to develop some protocols for these kind of scenarios, where instead of issuing a blanket call for blood donation, you would do it in a systematic way. As one suggestion, you might do it by ZIP code.”
“Our findings are important to help us prepare for the next mass shooting in the United States. It shows us the amount of blood components we likely will need. It will also help first responders adequately prepare to save lives.”
There was no funding for this research, and the study authors declared no conflicts of interest.
Dogs can sniff out malaria, team says

NEW ORLEANS—Dogs can be trained to sniff out malaria in humans, according to research presented at the American Society of Tropical Medicine and Hygiene Annual Meeting.
Researchers found that dogs could detect malaria by sniffing socks worn by children from a malaria-endemic area of West Africa.
“While our findings are at an early stage, in principle, we have shown that dogs could be trained to detect malaria-infected people by their odor with a credible degree of accuracy,” said study investigator Steve Lindsay, PhD, of Durham University in Durham City, U.K.
Dr. Lindsay presented these findings at the meeting as abstract 32.
The research began in The Gambia, where 600 school children were recruited to join the study. They were checked for overall general health, sampled for malaria parasites, and fitted with a pair of socks they were asked to wear overnight.
The next day, the socks were collected. The socks were sorted according to the malaria infection status of the children. The researchers only selected socks from uninfected children and children with malaria who did not have fever.
The socks were shipped to the United Kingdom, where they were stored in a freezer for several months while dogs were trained to sniff out malaria.
The dogs had to distinguish between socks from children with malaria parasites and socks from uninfected children. The animals were trained to sniff each sample, freeze if they thought they detected malaria, and move on if they did not.
In total, 175 sock samples were tested, including those from 30 malaria-positive children and those from 145 uninfected children.
The dogs correctly identified 70% of the infected children and 90% of the uninfected children.
Dr. Lindsay and his colleagues believe that, with more training and more samples, the dogs could provide a level of accuracy approaching that of a clinical test.
Now, the researchers are considering a follow-up study that would take samples from people in different parts of Africa to test whether parasites from one part of the continent present odors that are different from another part of the continent.
As for putting malaria-detecting dogs to work in the field, Dr. Lindsay said they could be helpful assistants in malaria elimination campaigns.
Currently, the only way to address the problem of asymptomatic malaria carriers is to test or treat an entire community. Dr. Lindsay said detection dogs could be useful for significantly narrowing the focus of clinical testing and treatment efforts.
Dr. Lindsay also believes detection dogs could be used at ports of entry into countries that have eliminated malaria or are close to elimination.
“This could provide a non-invasive way of screening for the disease at ports of entry in a similar way to how sniffer dogs are routinely used to detect fruit and vegetables or drugs at airports,” he said.
“This could help prevent the spread of malaria to countries that have been declared malaria-free and also ensure that people, many of whom might be unaware that they are infected with the malaria parasite, receive antimalarial drug treatment for the disease.”
Dr. Lindsay and his colleagues’ research was funded by the Bill & Melinda Gates Foundation. Dr. Lindsay reported no conflicts of interest.
NEW ORLEANS—Dogs can be trained to sniff out malaria in humans, according to research presented at the American Society of Tropical Medicine and Hygiene Annual Meeting.
Researchers found that dogs could detect malaria by sniffing socks worn by children from a malaria-endemic area of West Africa.
“While our findings are at an early stage, in principle, we have shown that dogs could be trained to detect malaria-infected people by their odor with a credible degree of accuracy,” said study investigator Steve Lindsay, PhD, of Durham University in Durham City, U.K.
Dr. Lindsay presented these findings at the meeting as abstract 32.
The research began in The Gambia, where 600 school children were recruited to join the study. They were checked for overall general health, sampled for malaria parasites, and fitted with a pair of socks they were asked to wear overnight.
The next day, the socks were collected. The socks were sorted according to the malaria infection status of the children. The researchers only selected socks from uninfected children and children with malaria who did not have fever.
The socks were shipped to the United Kingdom, where they were stored in a freezer for several months while dogs were trained to sniff out malaria.
The dogs had to distinguish between socks from children with malaria parasites and socks from uninfected children. The animals were trained to sniff each sample, freeze if they thought they detected malaria, and move on if they did not.
In total, 175 sock samples were tested, including those from 30 malaria-positive children and those from 145 uninfected children.
The dogs correctly identified 70% of the infected children and 90% of the uninfected children.
Dr. Lindsay and his colleagues believe that, with more training and more samples, the dogs could provide a level of accuracy approaching that of a clinical test.
Now, the researchers are considering a follow-up study that would take samples from people in different parts of Africa to test whether parasites from one part of the continent present odors that are different from another part of the continent.
As for putting malaria-detecting dogs to work in the field, Dr. Lindsay said they could be helpful assistants in malaria elimination campaigns.
Currently, the only way to address the problem of asymptomatic malaria carriers is to test or treat an entire community. Dr. Lindsay said detection dogs could be useful for significantly narrowing the focus of clinical testing and treatment efforts.
Dr. Lindsay also believes detection dogs could be used at ports of entry into countries that have eliminated malaria or are close to elimination.
“This could provide a non-invasive way of screening for the disease at ports of entry in a similar way to how sniffer dogs are routinely used to detect fruit and vegetables or drugs at airports,” he said.
“This could help prevent the spread of malaria to countries that have been declared malaria-free and also ensure that people, many of whom might be unaware that they are infected with the malaria parasite, receive antimalarial drug treatment for the disease.”
Dr. Lindsay and his colleagues’ research was funded by the Bill & Melinda Gates Foundation. Dr. Lindsay reported no conflicts of interest.
NEW ORLEANS—Dogs can be trained to sniff out malaria in humans, according to research presented at the American Society of Tropical Medicine and Hygiene Annual Meeting.
Researchers found that dogs could detect malaria by sniffing socks worn by children from a malaria-endemic area of West Africa.
“While our findings are at an early stage, in principle, we have shown that dogs could be trained to detect malaria-infected people by their odor with a credible degree of accuracy,” said study investigator Steve Lindsay, PhD, of Durham University in Durham City, U.K.
Dr. Lindsay presented these findings at the meeting as abstract 32.
The research began in The Gambia, where 600 school children were recruited to join the study. They were checked for overall general health, sampled for malaria parasites, and fitted with a pair of socks they were asked to wear overnight.
The next day, the socks were collected. The socks were sorted according to the malaria infection status of the children. The researchers only selected socks from uninfected children and children with malaria who did not have fever.
The socks were shipped to the United Kingdom, where they were stored in a freezer for several months while dogs were trained to sniff out malaria.
The dogs had to distinguish between socks from children with malaria parasites and socks from uninfected children. The animals were trained to sniff each sample, freeze if they thought they detected malaria, and move on if they did not.
In total, 175 sock samples were tested, including those from 30 malaria-positive children and those from 145 uninfected children.
The dogs correctly identified 70% of the infected children and 90% of the uninfected children.
Dr. Lindsay and his colleagues believe that, with more training and more samples, the dogs could provide a level of accuracy approaching that of a clinical test.
Now, the researchers are considering a follow-up study that would take samples from people in different parts of Africa to test whether parasites from one part of the continent present odors that are different from another part of the continent.
As for putting malaria-detecting dogs to work in the field, Dr. Lindsay said they could be helpful assistants in malaria elimination campaigns.
Currently, the only way to address the problem of asymptomatic malaria carriers is to test or treat an entire community. Dr. Lindsay said detection dogs could be useful for significantly narrowing the focus of clinical testing and treatment efforts.
Dr. Lindsay also believes detection dogs could be used at ports of entry into countries that have eliminated malaria or are close to elimination.
“This could provide a non-invasive way of screening for the disease at ports of entry in a similar way to how sniffer dogs are routinely used to detect fruit and vegetables or drugs at airports,” he said.
“This could help prevent the spread of malaria to countries that have been declared malaria-free and also ensure that people, many of whom might be unaware that they are infected with the malaria parasite, receive antimalarial drug treatment for the disease.”
Dr. Lindsay and his colleagues’ research was funded by the Bill & Melinda Gates Foundation. Dr. Lindsay reported no conflicts of interest.
Team tracks changes in height, weight in pediatric ALL

New research suggests several factors may be associated with the risk of short stature and excess weight gain in children with acute lymphoblastic leukemia (ALL).
Researchers found that patients who were younger at ALL diagnosis had an increased risk of becoming overweight or obese both during and after therapy.
Patients had an increased risk of short stature after therapy if they were older at diagnosis or had standard or high-risk disease, higher white blood cell counts at diagnosis, and central nervous system disease.
The researchers reported these findings in Cancer.
The team looked at 372 children with ALL, reviewing changes in their body mass index (BMI), weight, and height from diagnosis to 5 years after treatment ended.
The patients were treated with the Total XV protocol between 2000 and 2007 (NCT00137111). They received 6 weeks of induction therapy, 8 weeks of consolidation, and continuation for 120 weeks in females and 146 weeks in males.
BMI changes
Roughly a quarter of patients were overweight or obese at diagnosis, but that increased to roughly half of patients by the time they had been off therapy for 5 years.
“Over the whole population that was studied, we found statistically significant weight gain even during remission-induction therapy,” said study author Hiroto Inaba, MD, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
Patients’ median BMI z scores increased significantly during induction (P<0.001) and reinduction (P=0.001) with glucocorticoid therapy as well as in the first year after therapy ended (P=0.006).
At various points during treatment, there were significant differences in BMI z scores according to sex, race, and disease risk group. However, these differences were not present after therapy.
On the other hand, there were significant differences in BMI z scores according to age both during and after therapy.
Between week 21 of treatment and 3 years after therapy ended, patients who were ages 2 to 9 at diagnosis had median BMI z scores that were significantly higher than scores of patients who were age 10 or older at diagnosis (P≤0.033 for all time points).
The researchers also found that patients who were of a healthy weight or underweight at the time of diagnosis had a significantly higher risk of becoming overweight or obese during or after therapy if they were ages 2 to 9 at diagnosis, compared to the older patients (P=0.001).
Height changes
The researchers found that height z scores declined during treatment and improved after it ended, although z scores “never improved to the levels noted at the time of diagnosis.”
Median height z scores at the end of induction and in continuation weeks 1 to 21 were significantly higher in patients age 10 or older at diagnosis than in patients ages to 2 to 9 at diagnosis (P≤0.038 for all time points).
However, the median height z scores at 5 years off therapy were significantly higher for the younger patients than for the older patients (P=0.011).
The median height z scores were higher for patients with low-risk disease than for standard- or high-risk patients in weeks 17, 21, 48, and 146 of treatment and at 1 to 3 years after therapy ended (P≤0.024 for all time points).
At 3 years to 5 years after treatment ended, the median height z scores were significantly higher among patients with white blood cell counts below 50 × 109/L at diagnosis (P≤0.018 for all time points).
Patients without central nervous system disease had significantly higher median height z scores at 3 years after treatment ended (P=0.029).
Males had significantly higher median height z scores than females in weeks 96 and 120 of therapy (P≤0.009 for both time points).
And white patients had higher median height z scores than black patients at 2 to 4 years after treatment ended (P≤0.027 for all time points).
Implications
To address the issue of excess weight gain in ALL patients, the researchers suggested early interventions, such as education about proper diet and exercise.
“When you look at the literature of childhood obesity prevention for the general population, there are interventions that could also help ALL patients,” said study author Emily Browne, of St. Jude.
“But we need to adapt those recommendations to take the cancer therapy into account.”
For the issue of height, the researchers recommended evaluating certain patients for growth hormone deficiency.
The team also noted that further study is needed to determine whether emerging therapeutic approaches can reduce toxicities without compromising antileukemic effects.
“We are hoping new therapeutic options can decrease intensity of chemotherapy and keep normal tissues intact,” Dr. Inaba said. “But until then, we’re collaborating with multiple clinical departments to help ensure a good, quality cure and a good quality of life in survivorship.”
This research was supported by grants from the National Institutes of Health and ALSAC, the fundraising and awareness organization of St. Jude.
New research suggests several factors may be associated with the risk of short stature and excess weight gain in children with acute lymphoblastic leukemia (ALL).
Researchers found that patients who were younger at ALL diagnosis had an increased risk of becoming overweight or obese both during and after therapy.
Patients had an increased risk of short stature after therapy if they were older at diagnosis or had standard or high-risk disease, higher white blood cell counts at diagnosis, and central nervous system disease.
The researchers reported these findings in Cancer.
The team looked at 372 children with ALL, reviewing changes in their body mass index (BMI), weight, and height from diagnosis to 5 years after treatment ended.
The patients were treated with the Total XV protocol between 2000 and 2007 (NCT00137111). They received 6 weeks of induction therapy, 8 weeks of consolidation, and continuation for 120 weeks in females and 146 weeks in males.
BMI changes
Roughly a quarter of patients were overweight or obese at diagnosis, but that increased to roughly half of patients by the time they had been off therapy for 5 years.
“Over the whole population that was studied, we found statistically significant weight gain even during remission-induction therapy,” said study author Hiroto Inaba, MD, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
Patients’ median BMI z scores increased significantly during induction (P<0.001) and reinduction (P=0.001) with glucocorticoid therapy as well as in the first year after therapy ended (P=0.006).
At various points during treatment, there were significant differences in BMI z scores according to sex, race, and disease risk group. However, these differences were not present after therapy.
On the other hand, there were significant differences in BMI z scores according to age both during and after therapy.
Between week 21 of treatment and 3 years after therapy ended, patients who were ages 2 to 9 at diagnosis had median BMI z scores that were significantly higher than scores of patients who were age 10 or older at diagnosis (P≤0.033 for all time points).
The researchers also found that patients who were of a healthy weight or underweight at the time of diagnosis had a significantly higher risk of becoming overweight or obese during or after therapy if they were ages 2 to 9 at diagnosis, compared to the older patients (P=0.001).
Height changes
The researchers found that height z scores declined during treatment and improved after it ended, although z scores “never improved to the levels noted at the time of diagnosis.”
Median height z scores at the end of induction and in continuation weeks 1 to 21 were significantly higher in patients age 10 or older at diagnosis than in patients ages to 2 to 9 at diagnosis (P≤0.038 for all time points).
However, the median height z scores at 5 years off therapy were significantly higher for the younger patients than for the older patients (P=0.011).
The median height z scores were higher for patients with low-risk disease than for standard- or high-risk patients in weeks 17, 21, 48, and 146 of treatment and at 1 to 3 years after therapy ended (P≤0.024 for all time points).
At 3 years to 5 years after treatment ended, the median height z scores were significantly higher among patients with white blood cell counts below 50 × 109/L at diagnosis (P≤0.018 for all time points).
Patients without central nervous system disease had significantly higher median height z scores at 3 years after treatment ended (P=0.029).
Males had significantly higher median height z scores than females in weeks 96 and 120 of therapy (P≤0.009 for both time points).
And white patients had higher median height z scores than black patients at 2 to 4 years after treatment ended (P≤0.027 for all time points).
Implications
To address the issue of excess weight gain in ALL patients, the researchers suggested early interventions, such as education about proper diet and exercise.
“When you look at the literature of childhood obesity prevention for the general population, there are interventions that could also help ALL patients,” said study author Emily Browne, of St. Jude.
“But we need to adapt those recommendations to take the cancer therapy into account.”
For the issue of height, the researchers recommended evaluating certain patients for growth hormone deficiency.
The team also noted that further study is needed to determine whether emerging therapeutic approaches can reduce toxicities without compromising antileukemic effects.
“We are hoping new therapeutic options can decrease intensity of chemotherapy and keep normal tissues intact,” Dr. Inaba said. “But until then, we’re collaborating with multiple clinical departments to help ensure a good, quality cure and a good quality of life in survivorship.”
This research was supported by grants from the National Institutes of Health and ALSAC, the fundraising and awareness organization of St. Jude.
New research suggests several factors may be associated with the risk of short stature and excess weight gain in children with acute lymphoblastic leukemia (ALL).
Researchers found that patients who were younger at ALL diagnosis had an increased risk of becoming overweight or obese both during and after therapy.
Patients had an increased risk of short stature after therapy if they were older at diagnosis or had standard or high-risk disease, higher white blood cell counts at diagnosis, and central nervous system disease.
The researchers reported these findings in Cancer.
The team looked at 372 children with ALL, reviewing changes in their body mass index (BMI), weight, and height from diagnosis to 5 years after treatment ended.
The patients were treated with the Total XV protocol between 2000 and 2007 (NCT00137111). They received 6 weeks of induction therapy, 8 weeks of consolidation, and continuation for 120 weeks in females and 146 weeks in males.
BMI changes
Roughly a quarter of patients were overweight or obese at diagnosis, but that increased to roughly half of patients by the time they had been off therapy for 5 years.
“Over the whole population that was studied, we found statistically significant weight gain even during remission-induction therapy,” said study author Hiroto Inaba, MD, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
Patients’ median BMI z scores increased significantly during induction (P<0.001) and reinduction (P=0.001) with glucocorticoid therapy as well as in the first year after therapy ended (P=0.006).
At various points during treatment, there were significant differences in BMI z scores according to sex, race, and disease risk group. However, these differences were not present after therapy.
On the other hand, there were significant differences in BMI z scores according to age both during and after therapy.
Between week 21 of treatment and 3 years after therapy ended, patients who were ages 2 to 9 at diagnosis had median BMI z scores that were significantly higher than scores of patients who were age 10 or older at diagnosis (P≤0.033 for all time points).
The researchers also found that patients who were of a healthy weight or underweight at the time of diagnosis had a significantly higher risk of becoming overweight or obese during or after therapy if they were ages 2 to 9 at diagnosis, compared to the older patients (P=0.001).
Height changes
The researchers found that height z scores declined during treatment and improved after it ended, although z scores “never improved to the levels noted at the time of diagnosis.”
Median height z scores at the end of induction and in continuation weeks 1 to 21 were significantly higher in patients age 10 or older at diagnosis than in patients ages to 2 to 9 at diagnosis (P≤0.038 for all time points).
However, the median height z scores at 5 years off therapy were significantly higher for the younger patients than for the older patients (P=0.011).
The median height z scores were higher for patients with low-risk disease than for standard- or high-risk patients in weeks 17, 21, 48, and 146 of treatment and at 1 to 3 years after therapy ended (P≤0.024 for all time points).
At 3 years to 5 years after treatment ended, the median height z scores were significantly higher among patients with white blood cell counts below 50 × 109/L at diagnosis (P≤0.018 for all time points).
Patients without central nervous system disease had significantly higher median height z scores at 3 years after treatment ended (P=0.029).
Males had significantly higher median height z scores than females in weeks 96 and 120 of therapy (P≤0.009 for both time points).
And white patients had higher median height z scores than black patients at 2 to 4 years after treatment ended (P≤0.027 for all time points).
Implications
To address the issue of excess weight gain in ALL patients, the researchers suggested early interventions, such as education about proper diet and exercise.
“When you look at the literature of childhood obesity prevention for the general population, there are interventions that could also help ALL patients,” said study author Emily Browne, of St. Jude.
“But we need to adapt those recommendations to take the cancer therapy into account.”
For the issue of height, the researchers recommended evaluating certain patients for growth hormone deficiency.
The team also noted that further study is needed to determine whether emerging therapeutic approaches can reduce toxicities without compromising antileukemic effects.
“We are hoping new therapeutic options can decrease intensity of chemotherapy and keep normal tissues intact,” Dr. Inaba said. “But until then, we’re collaborating with multiple clinical departments to help ensure a good, quality cure and a good quality of life in survivorship.”
This research was supported by grants from the National Institutes of Health and ALSAC, the fundraising and awareness organization of St. Jude.
Fusion protein identified as new target in AML

Researchers have identified a promising therapeutic target for t(8;21) acute myeloid leukemia (AML), according to preclinical data published in Cancer Cell.
The fusion protein RUNX1/ETO drives t(8;21) AML by promoting cell-cycle progression.
Using an RNAi screen, the team recognized the cell-cycle regulator cyclin D2 (CCND2) as having critical involvement in RUNX1/ETO-driven leukemia propagation.
And when they knocked down CCND2 with palbociclib, a drug already approved for breast cancer, leukemic expansion of human AML cells and engraftment in murine models were significantly impaired.
"Our discovery that this treatment can be effective in AML is an important step towards a more effective and less toxic treatment for patients with this form of leukemia,” said study author Olaf Heidenreich, PhD, from the Wolfson Childhood Cancer Research Centre at Newcastle University in the U.K.
After identifying the fusion protein with the RNAi screen, the investigators determined that RUNX1/ETO regulates CCND2 transcription. They knocked down the fusion protein and found the expression of CCND2 was diminished in primary AML blasts. They therefore concluded that RUNX1/ETO maintains CCND2 expression.
The team then examined the significance of CCND2 in engraftment, proliferation, and clonal expansion of AML cells and its impact on the accumulation of cells in the G1 phase of the cell cycle. They found that depletion of CCND2 inhibited cell proliferation and clonogenic capacity and arrested the cell cycle in G0/G1 without increasing apoptosis.
They also confirmed that knockdown of RUNX1/ETO or CCND2 did not affect the expression of other D cyclins and G1 cyclin-dependent kinase (CDK)-CCND complexes, such as CDK4/6.
Next, they explored whether RUNX1/ETO-expressing cells were sensitive to the CDK4/6 inhibitor palbociclib. AML cells were highly sensitive to palbociclib and did not proliferate during drug exposure.
The researchers cultured cells from t(8;21)-positive and -negative AML patients and found palbociclib to cause a dose-dependent inhibition of proliferation of AML blasts.
They also tested palbociclib on a sample from a relapsed t(8;21) AML patient. The sample was highly sensitive to palbociclib, with a five-fold reduction in cell numbers using 300 nM of drug.
The investigators conducted in vivo experiments with palbociclib in mice transplanted with AML cells. Mice treated with palbociclib at doses of 100–150 mg/kg had a significantly longer survival than control mice.
Finally, the team examined whether interference with G1 CDK activity would create other vulnerabilities, such as activating KIT mutations, which are frequent secondary mutations found in t(8;21) AML.
They found that G1 CDK inhibition sensitized AML cells toward KIT inhibition, suggesting that “concurrent targeting of the two mutations may offer substantial therapeutic benefit.”
The team plans to conduct experiments that will refine the precise palbociclib dose in AML either as a single agent or in combination.
This study was supported by grants from Bloodwise, Children with Cancer, North of England Children’s Cancer Research Fund, Children's Cancer and Leukaemia Group, and a CRUK program grant in addition to an Aga Khan PhD studentship, a University Sains Malaysia PhD studentship, and an NC3R fellowship.
The authors had no competing interests to disclose.
Researchers have identified a promising therapeutic target for t(8;21) acute myeloid leukemia (AML), according to preclinical data published in Cancer Cell.
The fusion protein RUNX1/ETO drives t(8;21) AML by promoting cell-cycle progression.
Using an RNAi screen, the team recognized the cell-cycle regulator cyclin D2 (CCND2) as having critical involvement in RUNX1/ETO-driven leukemia propagation.
And when they knocked down CCND2 with palbociclib, a drug already approved for breast cancer, leukemic expansion of human AML cells and engraftment in murine models were significantly impaired.
"Our discovery that this treatment can be effective in AML is an important step towards a more effective and less toxic treatment for patients with this form of leukemia,” said study author Olaf Heidenreich, PhD, from the Wolfson Childhood Cancer Research Centre at Newcastle University in the U.K.
After identifying the fusion protein with the RNAi screen, the investigators determined that RUNX1/ETO regulates CCND2 transcription. They knocked down the fusion protein and found the expression of CCND2 was diminished in primary AML blasts. They therefore concluded that RUNX1/ETO maintains CCND2 expression.
The team then examined the significance of CCND2 in engraftment, proliferation, and clonal expansion of AML cells and its impact on the accumulation of cells in the G1 phase of the cell cycle. They found that depletion of CCND2 inhibited cell proliferation and clonogenic capacity and arrested the cell cycle in G0/G1 without increasing apoptosis.
They also confirmed that knockdown of RUNX1/ETO or CCND2 did not affect the expression of other D cyclins and G1 cyclin-dependent kinase (CDK)-CCND complexes, such as CDK4/6.
Next, they explored whether RUNX1/ETO-expressing cells were sensitive to the CDK4/6 inhibitor palbociclib. AML cells were highly sensitive to palbociclib and did not proliferate during drug exposure.
The researchers cultured cells from t(8;21)-positive and -negative AML patients and found palbociclib to cause a dose-dependent inhibition of proliferation of AML blasts.
They also tested palbociclib on a sample from a relapsed t(8;21) AML patient. The sample was highly sensitive to palbociclib, with a five-fold reduction in cell numbers using 300 nM of drug.
The investigators conducted in vivo experiments with palbociclib in mice transplanted with AML cells. Mice treated with palbociclib at doses of 100–150 mg/kg had a significantly longer survival than control mice.
Finally, the team examined whether interference with G1 CDK activity would create other vulnerabilities, such as activating KIT mutations, which are frequent secondary mutations found in t(8;21) AML.
They found that G1 CDK inhibition sensitized AML cells toward KIT inhibition, suggesting that “concurrent targeting of the two mutations may offer substantial therapeutic benefit.”
The team plans to conduct experiments that will refine the precise palbociclib dose in AML either as a single agent or in combination.
This study was supported by grants from Bloodwise, Children with Cancer, North of England Children’s Cancer Research Fund, Children's Cancer and Leukaemia Group, and a CRUK program grant in addition to an Aga Khan PhD studentship, a University Sains Malaysia PhD studentship, and an NC3R fellowship.
The authors had no competing interests to disclose.
Researchers have identified a promising therapeutic target for t(8;21) acute myeloid leukemia (AML), according to preclinical data published in Cancer Cell.
The fusion protein RUNX1/ETO drives t(8;21) AML by promoting cell-cycle progression.
Using an RNAi screen, the team recognized the cell-cycle regulator cyclin D2 (CCND2) as having critical involvement in RUNX1/ETO-driven leukemia propagation.
And when they knocked down CCND2 with palbociclib, a drug already approved for breast cancer, leukemic expansion of human AML cells and engraftment in murine models were significantly impaired.
"Our discovery that this treatment can be effective in AML is an important step towards a more effective and less toxic treatment for patients with this form of leukemia,” said study author Olaf Heidenreich, PhD, from the Wolfson Childhood Cancer Research Centre at Newcastle University in the U.K.
After identifying the fusion protein with the RNAi screen, the investigators determined that RUNX1/ETO regulates CCND2 transcription. They knocked down the fusion protein and found the expression of CCND2 was diminished in primary AML blasts. They therefore concluded that RUNX1/ETO maintains CCND2 expression.
The team then examined the significance of CCND2 in engraftment, proliferation, and clonal expansion of AML cells and its impact on the accumulation of cells in the G1 phase of the cell cycle. They found that depletion of CCND2 inhibited cell proliferation and clonogenic capacity and arrested the cell cycle in G0/G1 without increasing apoptosis.
They also confirmed that knockdown of RUNX1/ETO or CCND2 did not affect the expression of other D cyclins and G1 cyclin-dependent kinase (CDK)-CCND complexes, such as CDK4/6.
Next, they explored whether RUNX1/ETO-expressing cells were sensitive to the CDK4/6 inhibitor palbociclib. AML cells were highly sensitive to palbociclib and did not proliferate during drug exposure.
The researchers cultured cells from t(8;21)-positive and -negative AML patients and found palbociclib to cause a dose-dependent inhibition of proliferation of AML blasts.
They also tested palbociclib on a sample from a relapsed t(8;21) AML patient. The sample was highly sensitive to palbociclib, with a five-fold reduction in cell numbers using 300 nM of drug.
The investigators conducted in vivo experiments with palbociclib in mice transplanted with AML cells. Mice treated with palbociclib at doses of 100–150 mg/kg had a significantly longer survival than control mice.
Finally, the team examined whether interference with G1 CDK activity would create other vulnerabilities, such as activating KIT mutations, which are frequent secondary mutations found in t(8;21) AML.
They found that G1 CDK inhibition sensitized AML cells toward KIT inhibition, suggesting that “concurrent targeting of the two mutations may offer substantial therapeutic benefit.”
The team plans to conduct experiments that will refine the precise palbociclib dose in AML either as a single agent or in combination.
This study was supported by grants from Bloodwise, Children with Cancer, North of England Children’s Cancer Research Fund, Children's Cancer and Leukaemia Group, and a CRUK program grant in addition to an Aga Khan PhD studentship, a University Sains Malaysia PhD studentship, and an NC3R fellowship.
The authors had no competing interests to disclose.
Ruxolitinib under priority review for acute GVHD

The U.S. Food and Drug Administration (FDA) has accepted for priority review a supplemental new drug application (sNDA) for the JAK1/JAK2 inhibitor ruxolitinib (Jakafi®).
With this sNDA, Incyte Corporation is seeking approval for ruxolitinib as a treatment for patients with acute graft-versus-host-disease (GVHD) who have had an inadequate response to corticosteroids.
“If approved, ruxolitinib will be the first and only treatment available in the U.S. for patients with acute GVHD who have not responded adequately to corticosteroid therapy,” said Steven Stein, MD, chief medical officer at Incyte.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions. The agency intends to take action on a priority review application within 6 months of receiving it rather than the standard 10 months.
In addition to priority review, ruxolitinib has received breakthrough therapy and orphan drug designations from the FDA as a treatment for acute GVHD.
The sNDA submission for ruxolitinib in acute GVHD is based on data from the phase 2 REACH1 trial (NCT02953678).
In this ongoing trial, researchers are evaluating ruxolitinib in combination with corticosteroids in patients who have steroid-refractory acute GVHD.
Incyte announced topline results from REACH1 in June, reporting on outcomes in 71 patients.
The study’s primary endpoint—overall response rate at day 28—was met. Ruxolitinib produced an overall response rate of 55% (39/71) at that time.
However, 73% of patients (52/71) responded to ruxolitinib at some point during the trial.
Incyte said the most common treatment-emergent adverse events were anemia (61%), thrombocytopenia (61%), and neutropenia (56%).
The U.S. Food and Drug Administration (FDA) has accepted for priority review a supplemental new drug application (sNDA) for the JAK1/JAK2 inhibitor ruxolitinib (Jakafi®).
With this sNDA, Incyte Corporation is seeking approval for ruxolitinib as a treatment for patients with acute graft-versus-host-disease (GVHD) who have had an inadequate response to corticosteroids.
“If approved, ruxolitinib will be the first and only treatment available in the U.S. for patients with acute GVHD who have not responded adequately to corticosteroid therapy,” said Steven Stein, MD, chief medical officer at Incyte.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions. The agency intends to take action on a priority review application within 6 months of receiving it rather than the standard 10 months.
In addition to priority review, ruxolitinib has received breakthrough therapy and orphan drug designations from the FDA as a treatment for acute GVHD.
The sNDA submission for ruxolitinib in acute GVHD is based on data from the phase 2 REACH1 trial (NCT02953678).
In this ongoing trial, researchers are evaluating ruxolitinib in combination with corticosteroids in patients who have steroid-refractory acute GVHD.
Incyte announced topline results from REACH1 in June, reporting on outcomes in 71 patients.
The study’s primary endpoint—overall response rate at day 28—was met. Ruxolitinib produced an overall response rate of 55% (39/71) at that time.
However, 73% of patients (52/71) responded to ruxolitinib at some point during the trial.
Incyte said the most common treatment-emergent adverse events were anemia (61%), thrombocytopenia (61%), and neutropenia (56%).
The U.S. Food and Drug Administration (FDA) has accepted for priority review a supplemental new drug application (sNDA) for the JAK1/JAK2 inhibitor ruxolitinib (Jakafi®).
With this sNDA, Incyte Corporation is seeking approval for ruxolitinib as a treatment for patients with acute graft-versus-host-disease (GVHD) who have had an inadequate response to corticosteroids.
“If approved, ruxolitinib will be the first and only treatment available in the U.S. for patients with acute GVHD who have not responded adequately to corticosteroid therapy,” said Steven Stein, MD, chief medical officer at Incyte.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions. The agency intends to take action on a priority review application within 6 months of receiving it rather than the standard 10 months.
In addition to priority review, ruxolitinib has received breakthrough therapy and orphan drug designations from the FDA as a treatment for acute GVHD.
The sNDA submission for ruxolitinib in acute GVHD is based on data from the phase 2 REACH1 trial (NCT02953678).
In this ongoing trial, researchers are evaluating ruxolitinib in combination with corticosteroids in patients who have steroid-refractory acute GVHD.
Incyte announced topline results from REACH1 in June, reporting on outcomes in 71 patients.
The study’s primary endpoint—overall response rate at day 28—was met. Ruxolitinib produced an overall response rate of 55% (39/71) at that time.
However, 73% of patients (52/71) responded to ruxolitinib at some point during the trial.
Incyte said the most common treatment-emergent adverse events were anemia (61%), thrombocytopenia (61%), and neutropenia (56%).
Lnk deficiency boosts HSC replication in Fanconi anemia

A novel approach can restore hematopoietic stem cell (HSC) function in Fanconi anemia (FA), according to preclinical research published in Nature Communications.
The investigators showed that Lnk (Sh2b3) deficiency restores HSC proliferation and survival in Fancd2-deficient mice.
And it does so, in part, by alleviating blocks to cytokine-mediated JAK2 signaling.
These findings, the researchers wrote, “highlight a new role for cytokine/JAK signaling” and have therapeutic implications for FA.
The investigators noted that FA is caused by mutations in genes that are essential for DNA interstrand crosslink repair and replication stress tolerance.
Allogeneic transplant can replace defective HSCs in patients with FA, the researchers said, but there are no interventions that mitigate defects in HSCs. So the investigators decided to test whether Lnk deficiency ameliorates HSC defects in FA.
Using a model of FA in which mice lacked Fancd2, the researchers inhibited the regulatory protein Sh2b3/Lnk.
The investigators said Lnk deficiency restored the proliferation and survival of Fancd2−/− HSCs while also reducing replication stress and genomic instability. Lnk deficiency did not, however, affect DNA interstrand crosslink repair.
“We expected that inhibiting Lnk would restore the ability of FA cells to repair DNA damage, but this was not the case,” said study author Wei Tong, PhD, of Children’s Hospital of Philadelphia in Pennsylvania.
“Instead, inhibiting Lnk stabilized the stalled replication machinery, allowing affected cells to continue to copy DNA, and to prevent small errors from cascading into a catastrophic failure. The most exciting aspect of this discovery is that Lnk is part of a well-known growth pathway that can be manipulated by existing drugs in other diseases.”
That pathway is the TPO/MPL/JAK2 pathway, which is already targeted by eltrombopag and romiplostin for immune thrombocytopenia and eltrombopag for aplastic anemia.
The researchers plan to continue their work with animal models of FA and Lnk.
“Our ultimate goal is to translate our knowledge into treatments for both Fanconi anemia and for the broader problem of bone marrow failure,” Dr. Tong said.
This research was supported by the National Institutes of Health, the Fanconi Anemia Research Fund, the Department of Defense, the Basser Center for BRCA Team Science Grant, the Leukemia Lymphoma Society Scholar Award, and the Patel Family Award.
The researchers declared no competing interests.
A novel approach can restore hematopoietic stem cell (HSC) function in Fanconi anemia (FA), according to preclinical research published in Nature Communications.
The investigators showed that Lnk (Sh2b3) deficiency restores HSC proliferation and survival in Fancd2-deficient mice.
And it does so, in part, by alleviating blocks to cytokine-mediated JAK2 signaling.
These findings, the researchers wrote, “highlight a new role for cytokine/JAK signaling” and have therapeutic implications for FA.
The investigators noted that FA is caused by mutations in genes that are essential for DNA interstrand crosslink repair and replication stress tolerance.
Allogeneic transplant can replace defective HSCs in patients with FA, the researchers said, but there are no interventions that mitigate defects in HSCs. So the investigators decided to test whether Lnk deficiency ameliorates HSC defects in FA.
Using a model of FA in which mice lacked Fancd2, the researchers inhibited the regulatory protein Sh2b3/Lnk.
The investigators said Lnk deficiency restored the proliferation and survival of Fancd2−/− HSCs while also reducing replication stress and genomic instability. Lnk deficiency did not, however, affect DNA interstrand crosslink repair.
“We expected that inhibiting Lnk would restore the ability of FA cells to repair DNA damage, but this was not the case,” said study author Wei Tong, PhD, of Children’s Hospital of Philadelphia in Pennsylvania.
“Instead, inhibiting Lnk stabilized the stalled replication machinery, allowing affected cells to continue to copy DNA, and to prevent small errors from cascading into a catastrophic failure. The most exciting aspect of this discovery is that Lnk is part of a well-known growth pathway that can be manipulated by existing drugs in other diseases.”
That pathway is the TPO/MPL/JAK2 pathway, which is already targeted by eltrombopag and romiplostin for immune thrombocytopenia and eltrombopag for aplastic anemia.
The researchers plan to continue their work with animal models of FA and Lnk.
“Our ultimate goal is to translate our knowledge into treatments for both Fanconi anemia and for the broader problem of bone marrow failure,” Dr. Tong said.
This research was supported by the National Institutes of Health, the Fanconi Anemia Research Fund, the Department of Defense, the Basser Center for BRCA Team Science Grant, the Leukemia Lymphoma Society Scholar Award, and the Patel Family Award.
The researchers declared no competing interests.
A novel approach can restore hematopoietic stem cell (HSC) function in Fanconi anemia (FA), according to preclinical research published in Nature Communications.
The investigators showed that Lnk (Sh2b3) deficiency restores HSC proliferation and survival in Fancd2-deficient mice.
And it does so, in part, by alleviating blocks to cytokine-mediated JAK2 signaling.
These findings, the researchers wrote, “highlight a new role for cytokine/JAK signaling” and have therapeutic implications for FA.
The investigators noted that FA is caused by mutations in genes that are essential for DNA interstrand crosslink repair and replication stress tolerance.
Allogeneic transplant can replace defective HSCs in patients with FA, the researchers said, but there are no interventions that mitigate defects in HSCs. So the investigators decided to test whether Lnk deficiency ameliorates HSC defects in FA.
Using a model of FA in which mice lacked Fancd2, the researchers inhibited the regulatory protein Sh2b3/Lnk.
The investigators said Lnk deficiency restored the proliferation and survival of Fancd2−/− HSCs while also reducing replication stress and genomic instability. Lnk deficiency did not, however, affect DNA interstrand crosslink repair.
“We expected that inhibiting Lnk would restore the ability of FA cells to repair DNA damage, but this was not the case,” said study author Wei Tong, PhD, of Children’s Hospital of Philadelphia in Pennsylvania.
“Instead, inhibiting Lnk stabilized the stalled replication machinery, allowing affected cells to continue to copy DNA, and to prevent small errors from cascading into a catastrophic failure. The most exciting aspect of this discovery is that Lnk is part of a well-known growth pathway that can be manipulated by existing drugs in other diseases.”
That pathway is the TPO/MPL/JAK2 pathway, which is already targeted by eltrombopag and romiplostin for immune thrombocytopenia and eltrombopag for aplastic anemia.
The researchers plan to continue their work with animal models of FA and Lnk.
“Our ultimate goal is to translate our knowledge into treatments for both Fanconi anemia and for the broader problem of bone marrow failure,” Dr. Tong said.
This research was supported by the National Institutes of Health, the Fanconi Anemia Research Fund, the Department of Defense, the Basser Center for BRCA Team Science Grant, the Leukemia Lymphoma Society Scholar Award, and the Patel Family Award.
The researchers declared no competing interests.
Aspirin prevents VTE as well as anticoagulants, study suggests
Putting patients on aspirin following a knee replacement is a safe, cost-effective alternative to treatment with anticoagulants, according to researchers.
A study of more than 40,000 patients revealed a similar incidence of venous thromboembolism (VTE) or death after total knee arthroplasty (TKA) in patients receiving aspirin and patients receiving anticoagulants.
“Aspirin alone may provide similar protection compared to anticoagulation treatments,” said Brian R. Hallstrom, MD, of the University of Michigan in Ann Arbor.
He and his colleagues compared aspirin to anticoagulants after TKA and detailed their findings in JAMA Surgery.
The researchers evaluated 41,537 patients who underwent TKA and received:
- No pharmacological VTE prophylaxis (n=668; 1.6%)
- Aspirin only (n=12,831; 30.9%)
- Only an anticoagulant (n=22,620; 54.5%)
- Both aspirin and an anticoagulant (n=5418; 13.0%).
Anticoagulants included low-molecular-weight heparin, warfarin, and factor Xa inhibitors.
About 87% of all patients (n=36,113) were also using intermittent pneumatic compression stockings.
Safety and efficacy
The study’s primary outcome was a composite of VTE and death. It occurred in:
- 4.79% (32/668) of patients not on pharmacological prophylaxis
- 1.16% (149/12,831) of patients who received aspirin alone
- 1.42% (321/22,620) of patients on anticoagulation alone
- 1.31% (71/5418) of patients who received both aspirin and anticoagulation.
In a multivariable analysis adjusted for confounding, aspirin was noninferior to the other prophylaxis regimens for the outcome of VTE or death. The adjusted odds ratio was 0.85 (P=0.007).
Bleeding was a secondary outcome of this study, and it occurred in:
- 1.50% (10/668) of patients not on pharmacological prophylaxis
- 0.90% (116/12,831) of patients who received aspirin alone
- 1.14% (258/22,620) of patients who received anticoagulation
- 1.35% (73/5,418) of patients who received both aspirin and anticoagulation.
In an adjusted analysis, aspirin was noninferior to the other prophylaxis regimens for the outcome of bleeding. The adjusted odds ratio was 0.80 (P<0.001).
Other benefits
Aspirin provides other benefits aside from similar safety and efficacy as anticoagulants, according to the researchers.
“Aspirin is easy to take and much less expensive,” Dr. Hallstrom noted. “Patients can get it over the counter for pennies, while the other anticoagulants require monitoring, injections, frequent dose adjustments and are extremely expensive.”
Dr. Hallstrom and his colleagues said the reported cost for a 30-day supply of rivaroxaban is approximately $379 to $450, and the estimated cost of heparin is $450 to $890.
Although warfarin costs a few dollars for a 30-day supply, its cost approaches that of the other anticoagulants when doctor visits for monitoring are factored in, the researchers noted.
In contrast, aspirin costs approximately $2 a month.
Dr. Hallstrom and his colleagues did note that, although this study suggests aspirin can prevent VTE as well as anticoagulants, doctors need to consider factors such as a patient’s history of clots, obesity, and ability to mobilize after surgery.
Putting patients on aspirin following a knee replacement is a safe, cost-effective alternative to treatment with anticoagulants, according to researchers.
A study of more than 40,000 patients revealed a similar incidence of venous thromboembolism (VTE) or death after total knee arthroplasty (TKA) in patients receiving aspirin and patients receiving anticoagulants.
“Aspirin alone may provide similar protection compared to anticoagulation treatments,” said Brian R. Hallstrom, MD, of the University of Michigan in Ann Arbor.
He and his colleagues compared aspirin to anticoagulants after TKA and detailed their findings in JAMA Surgery.
The researchers evaluated 41,537 patients who underwent TKA and received:
- No pharmacological VTE prophylaxis (n=668; 1.6%)
- Aspirin only (n=12,831; 30.9%)
- Only an anticoagulant (n=22,620; 54.5%)
- Both aspirin and an anticoagulant (n=5418; 13.0%).
Anticoagulants included low-molecular-weight heparin, warfarin, and factor Xa inhibitors.
About 87% of all patients (n=36,113) were also using intermittent pneumatic compression stockings.
Safety and efficacy
The study’s primary outcome was a composite of VTE and death. It occurred in:
- 4.79% (32/668) of patients not on pharmacological prophylaxis
- 1.16% (149/12,831) of patients who received aspirin alone
- 1.42% (321/22,620) of patients on anticoagulation alone
- 1.31% (71/5418) of patients who received both aspirin and anticoagulation.
In a multivariable analysis adjusted for confounding, aspirin was noninferior to the other prophylaxis regimens for the outcome of VTE or death. The adjusted odds ratio was 0.85 (P=0.007).
Bleeding was a secondary outcome of this study, and it occurred in:
- 1.50% (10/668) of patients not on pharmacological prophylaxis
- 0.90% (116/12,831) of patients who received aspirin alone
- 1.14% (258/22,620) of patients who received anticoagulation
- 1.35% (73/5,418) of patients who received both aspirin and anticoagulation.
In an adjusted analysis, aspirin was noninferior to the other prophylaxis regimens for the outcome of bleeding. The adjusted odds ratio was 0.80 (P<0.001).
Other benefits
Aspirin provides other benefits aside from similar safety and efficacy as anticoagulants, according to the researchers.
“Aspirin is easy to take and much less expensive,” Dr. Hallstrom noted. “Patients can get it over the counter for pennies, while the other anticoagulants require monitoring, injections, frequent dose adjustments and are extremely expensive.”
Dr. Hallstrom and his colleagues said the reported cost for a 30-day supply of rivaroxaban is approximately $379 to $450, and the estimated cost of heparin is $450 to $890.
Although warfarin costs a few dollars for a 30-day supply, its cost approaches that of the other anticoagulants when doctor visits for monitoring are factored in, the researchers noted.
In contrast, aspirin costs approximately $2 a month.
Dr. Hallstrom and his colleagues did note that, although this study suggests aspirin can prevent VTE as well as anticoagulants, doctors need to consider factors such as a patient’s history of clots, obesity, and ability to mobilize after surgery.
Putting patients on aspirin following a knee replacement is a safe, cost-effective alternative to treatment with anticoagulants, according to researchers.
A study of more than 40,000 patients revealed a similar incidence of venous thromboembolism (VTE) or death after total knee arthroplasty (TKA) in patients receiving aspirin and patients receiving anticoagulants.
“Aspirin alone may provide similar protection compared to anticoagulation treatments,” said Brian R. Hallstrom, MD, of the University of Michigan in Ann Arbor.
He and his colleagues compared aspirin to anticoagulants after TKA and detailed their findings in JAMA Surgery.
The researchers evaluated 41,537 patients who underwent TKA and received:
- No pharmacological VTE prophylaxis (n=668; 1.6%)
- Aspirin only (n=12,831; 30.9%)
- Only an anticoagulant (n=22,620; 54.5%)
- Both aspirin and an anticoagulant (n=5418; 13.0%).
Anticoagulants included low-molecular-weight heparin, warfarin, and factor Xa inhibitors.
About 87% of all patients (n=36,113) were also using intermittent pneumatic compression stockings.
Safety and efficacy
The study’s primary outcome was a composite of VTE and death. It occurred in:
- 4.79% (32/668) of patients not on pharmacological prophylaxis
- 1.16% (149/12,831) of patients who received aspirin alone
- 1.42% (321/22,620) of patients on anticoagulation alone
- 1.31% (71/5418) of patients who received both aspirin and anticoagulation.
In a multivariable analysis adjusted for confounding, aspirin was noninferior to the other prophylaxis regimens for the outcome of VTE or death. The adjusted odds ratio was 0.85 (P=0.007).
Bleeding was a secondary outcome of this study, and it occurred in:
- 1.50% (10/668) of patients not on pharmacological prophylaxis
- 0.90% (116/12,831) of patients who received aspirin alone
- 1.14% (258/22,620) of patients who received anticoagulation
- 1.35% (73/5,418) of patients who received both aspirin and anticoagulation.
In an adjusted analysis, aspirin was noninferior to the other prophylaxis regimens for the outcome of bleeding. The adjusted odds ratio was 0.80 (P<0.001).
Other benefits
Aspirin provides other benefits aside from similar safety and efficacy as anticoagulants, according to the researchers.
“Aspirin is easy to take and much less expensive,” Dr. Hallstrom noted. “Patients can get it over the counter for pennies, while the other anticoagulants require monitoring, injections, frequent dose adjustments and are extremely expensive.”
Dr. Hallstrom and his colleagues said the reported cost for a 30-day supply of rivaroxaban is approximately $379 to $450, and the estimated cost of heparin is $450 to $890.
Although warfarin costs a few dollars for a 30-day supply, its cost approaches that of the other anticoagulants when doctor visits for monitoring are factored in, the researchers noted.
In contrast, aspirin costs approximately $2 a month.
Dr. Hallstrom and his colleagues did note that, although this study suggests aspirin can prevent VTE as well as anticoagulants, doctors need to consider factors such as a patient’s history of clots, obesity, and ability to mobilize after surgery.
OMS721 receives orphan designation for HSCT-TMA

The U.S. Food and Drug Administration (FDA) has granted OMS721 orphan designation for the treatment of hematopoietic stem cell transplant-associated thrombotic microangiopathy (HSCT-TMA).
OMS721 is a monoclonal antibody targeting MASP-2, the effector enzyme of the lectin pathway of the complement system.
The FDA previously granted OMS721 breakthrough therapy designation for HSCT-TMA and orphan designation for the prevention of complement-mediated TMA, including HSCT-TMA.
Omeros Corporation, the company developing OMS721, has established a compassionate-use program for OMS721, which is active in the United States and Europe.
Clinical trials
Phase 3 clinical programs are in progress for OMS721 in atypical hemolytic uremic syndrome, immunoglobulin A nephropathy, and HSCT-TMA. Two phase 2 trials of OMS721—one in TMA and one in immunoglobulin A nephropathy—are ongoing.
Omeros released results from the phase 2 TMA trial (NCT02222545) in February.
The study includes adults with HSCT-TMA persisting for at least 2 weeks following immunosuppressive regimen modification or more than 30 days post-transplant. Patients receive weekly OMS721 treatments for 4 to 8 weeks at the discretion of the investigator.
At the time of Omeros’s announcement, 18 HSCT-TMA patients had been treated on this study.
These patients had a significantly longer median overall survival than historical controls—347 days and 21 days, respectively (P<0.0001).
Omeros also reported that markers of TMA activity significantly improved following OMS721 treatment.
The mean platelet count increased from 18,100 x 106/mL at baseline to 52,300 x 106/mL (P=0.017). The mean lactate dehydrogenase decreased from 591 U/L to 250 U/L (P<0.001). And the mean haptoglobin increased from 8 mg/dL to 141 mg/dL (P=0.003).
Mean creatinine remained stable—at approximately 120 μmol/L—but a majority of patients had co-existing conditions for which they were receiving nephrotoxic medications. These conditions included graft-versus-host disease, cytomegalovirus and human herpes virus 6 infections, prior sepsis, diffuse alveolar hemorrhage, and residual underlying malignancies.
The most commonly reported adverse events were diarrhea and neutropenia.
Four deaths occurred. One of these—due to acute renal and respiratory failure—was considered possibly related to OMS721. The other deaths were due to progression of acute myeloid leukemia (n=1) and neutropenic sepsis (n=2).
About orphan and breakthrough designations
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the United States.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
The U.S. Food and Drug Administration (FDA) has granted OMS721 orphan designation for the treatment of hematopoietic stem cell transplant-associated thrombotic microangiopathy (HSCT-TMA).
OMS721 is a monoclonal antibody targeting MASP-2, the effector enzyme of the lectin pathway of the complement system.
The FDA previously granted OMS721 breakthrough therapy designation for HSCT-TMA and orphan designation for the prevention of complement-mediated TMA, including HSCT-TMA.
Omeros Corporation, the company developing OMS721, has established a compassionate-use program for OMS721, which is active in the United States and Europe.
Clinical trials
Phase 3 clinical programs are in progress for OMS721 in atypical hemolytic uremic syndrome, immunoglobulin A nephropathy, and HSCT-TMA. Two phase 2 trials of OMS721—one in TMA and one in immunoglobulin A nephropathy—are ongoing.
Omeros released results from the phase 2 TMA trial (NCT02222545) in February.
The study includes adults with HSCT-TMA persisting for at least 2 weeks following immunosuppressive regimen modification or more than 30 days post-transplant. Patients receive weekly OMS721 treatments for 4 to 8 weeks at the discretion of the investigator.
At the time of Omeros’s announcement, 18 HSCT-TMA patients had been treated on this study.
These patients had a significantly longer median overall survival than historical controls—347 days and 21 days, respectively (P<0.0001).
Omeros also reported that markers of TMA activity significantly improved following OMS721 treatment.
The mean platelet count increased from 18,100 x 106/mL at baseline to 52,300 x 106/mL (P=0.017). The mean lactate dehydrogenase decreased from 591 U/L to 250 U/L (P<0.001). And the mean haptoglobin increased from 8 mg/dL to 141 mg/dL (P=0.003).
Mean creatinine remained stable—at approximately 120 μmol/L—but a majority of patients had co-existing conditions for which they were receiving nephrotoxic medications. These conditions included graft-versus-host disease, cytomegalovirus and human herpes virus 6 infections, prior sepsis, diffuse alveolar hemorrhage, and residual underlying malignancies.
The most commonly reported adverse events were diarrhea and neutropenia.
Four deaths occurred. One of these—due to acute renal and respiratory failure—was considered possibly related to OMS721. The other deaths were due to progression of acute myeloid leukemia (n=1) and neutropenic sepsis (n=2).
About orphan and breakthrough designations
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the United States.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
The U.S. Food and Drug Administration (FDA) has granted OMS721 orphan designation for the treatment of hematopoietic stem cell transplant-associated thrombotic microangiopathy (HSCT-TMA).
OMS721 is a monoclonal antibody targeting MASP-2, the effector enzyme of the lectin pathway of the complement system.
The FDA previously granted OMS721 breakthrough therapy designation for HSCT-TMA and orphan designation for the prevention of complement-mediated TMA, including HSCT-TMA.
Omeros Corporation, the company developing OMS721, has established a compassionate-use program for OMS721, which is active in the United States and Europe.
Clinical trials
Phase 3 clinical programs are in progress for OMS721 in atypical hemolytic uremic syndrome, immunoglobulin A nephropathy, and HSCT-TMA. Two phase 2 trials of OMS721—one in TMA and one in immunoglobulin A nephropathy—are ongoing.
Omeros released results from the phase 2 TMA trial (NCT02222545) in February.
The study includes adults with HSCT-TMA persisting for at least 2 weeks following immunosuppressive regimen modification or more than 30 days post-transplant. Patients receive weekly OMS721 treatments for 4 to 8 weeks at the discretion of the investigator.
At the time of Omeros’s announcement, 18 HSCT-TMA patients had been treated on this study.
These patients had a significantly longer median overall survival than historical controls—347 days and 21 days, respectively (P<0.0001).
Omeros also reported that markers of TMA activity significantly improved following OMS721 treatment.
The mean platelet count increased from 18,100 x 106/mL at baseline to 52,300 x 106/mL (P=0.017). The mean lactate dehydrogenase decreased from 591 U/L to 250 U/L (P<0.001). And the mean haptoglobin increased from 8 mg/dL to 141 mg/dL (P=0.003).
Mean creatinine remained stable—at approximately 120 μmol/L—but a majority of patients had co-existing conditions for which they were receiving nephrotoxic medications. These conditions included graft-versus-host disease, cytomegalovirus and human herpes virus 6 infections, prior sepsis, diffuse alveolar hemorrhage, and residual underlying malignancies.
The most commonly reported adverse events were diarrhea and neutropenia.
Four deaths occurred. One of these—due to acute renal and respiratory failure—was considered possibly related to OMS721. The other deaths were due to progression of acute myeloid leukemia (n=1) and neutropenic sepsis (n=2).
About orphan and breakthrough designations
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the United States.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.