User login
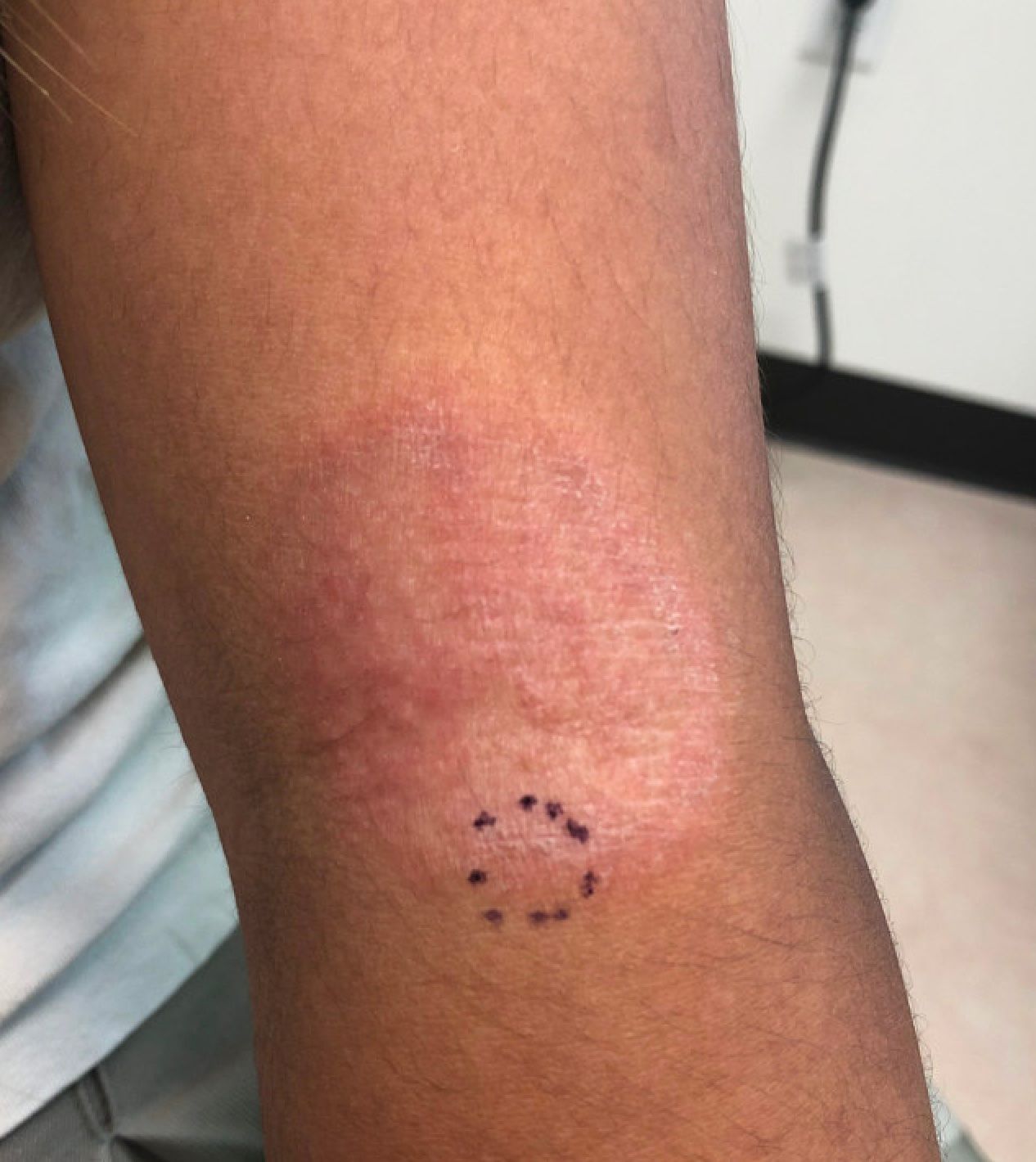
An infant with a tender bump on her ear
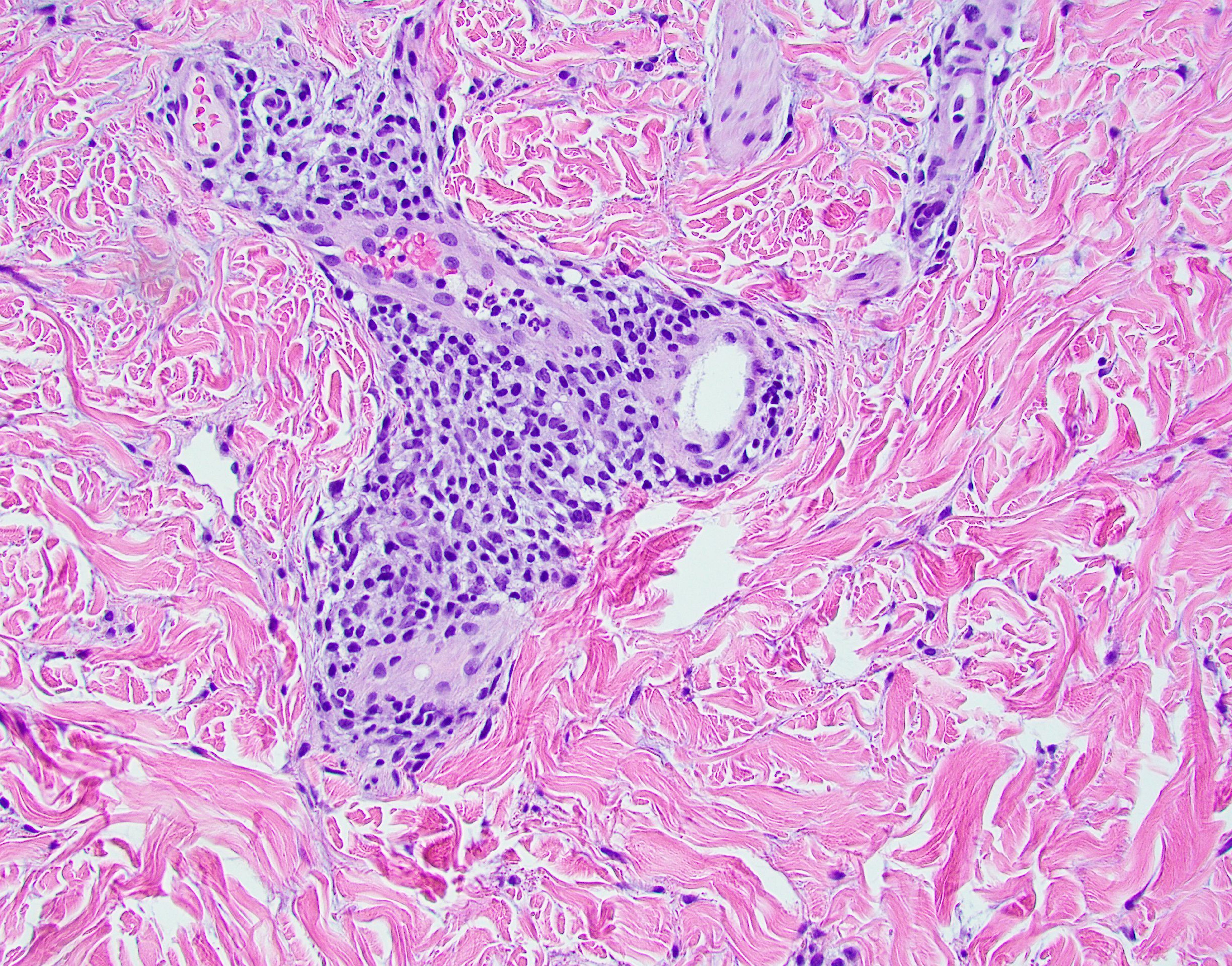
A biopsy of the lesion was performed that showed a well-defined nodulocystic tumor composed of nests of basaloid cells that are undergoing trichilemmal keratinization. Shadow cells are seen as well as small areas of calcification. There is also a histiocytic infiltrate with multinucleated giant cells. The histologic diagnosis is of a pilomatrixoma.
Pilomatrixoma, also known as calcifying epithelioma of Malherbe, was first described in 1880, as a tumor of sebaceous gland origin. Later, in 1961, Robert Forbis Jr, MD, and Elson B. Helwig, MD, coined the term pilomatrixoma to describe the hair follicle matrix as the source of the tumor. Pilomatrixomas are commonly seen in the pediatric population, usually in children between 8 and 13 years of age. Our patient is one of the youngest described. The lesions are commonly seen on the face and neck in about 70% of the cases followed by the upper extremities, back, and legs. Clinically, the lesions appear as a firm dermal papule or nodule, which moves freely and may have associated erythema on the skin surface or a blueish gray hue on the underlying skin.
Most pilomatrixomas that have been studied have shown a mutation in Exon 3 of the beta-catenin gene (CTNNB1). The beta-catenin molecule is a subunit of the cadherin protein, which is part of an important pathway in the terminal hair follicle differentiation. Beta-catenin also plays an important role in the Wnt pathway, which regulates cell fate as well as early embryonic patterning. Beta-catenin is responsible for forming adhesion junctions among cells. There have also been immunohistochemical studies that have shown a BCL2 proto-oncogene overexpression to pilomatrixoma.
There are several genetic syndromes that have been associated with the presence of pilomatrixomas: Turner syndrome (XO chromosome abnormality associated with short stature and cardiac defects), Gardner syndrome (polyposis coli and colon and rectal cancer), myotonic dystrophy, Rubinstein-Taybi syndrome (characterized by broad thumbs and toes, short stature, distinctive facial features, and varying degrees of intellectual disability), and trisomy 9. On physical examination our patient didn’t present with any of the typical features or history that could suggest any of these syndromes. A close follow-up and evaluation by a geneticist was recommended because after the initial visit she developed a second lesion on the forehead.
The differential diagnosis for this lesion includes other cysts that may occur on the ear such as epidermal inclusion cyst or dermoid cysts, though these lesions do not tend to be as firm as pilomatrixomas are, which can help with the diagnosis. Dermoid cysts are made of dermal and epidermal components. They are usually present at birth and are commonly seen on the scalp and the periorbital face.
Keloids are rubbery nodules of scar tissue that can form on sites of trauma, and although the lesion occurred after she had her ears pierced, the consistency and rapid growth of the lesion as well as the pathological description made this benign fibrous growth less likely.
When pilomatrixomas are inflamed they can be confused with vascular growths: in this particular case, a hemangioma or another vascular tumor such as a tufted angioma or kaposiform hemangioendothelioma. An ultrasound of the lesion could have helped in the differential diagnosis of the lesion.
Pilomatrixomas can grow significantly and in some cases get inflamed or infected. Surgical management of pilomatrixomas is often required because the lesions do not regress spontaneously.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Forbis R Jr and Helwig EB. Arch Dermatol 1961;83:606-18.
Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016 Jun;85:148-53.
A biopsy of the lesion was performed that showed a well-defined nodulocystic tumor composed of nests of basaloid cells that are undergoing trichilemmal keratinization. Shadow cells are seen as well as small areas of calcification. There is also a histiocytic infiltrate with multinucleated giant cells. The histologic diagnosis is of a pilomatrixoma.
Pilomatrixoma, also known as calcifying epithelioma of Malherbe, was first described in 1880, as a tumor of sebaceous gland origin. Later, in 1961, Robert Forbis Jr, MD, and Elson B. Helwig, MD, coined the term pilomatrixoma to describe the hair follicle matrix as the source of the tumor. Pilomatrixomas are commonly seen in the pediatric population, usually in children between 8 and 13 years of age. Our patient is one of the youngest described. The lesions are commonly seen on the face and neck in about 70% of the cases followed by the upper extremities, back, and legs. Clinically, the lesions appear as a firm dermal papule or nodule, which moves freely and may have associated erythema on the skin surface or a blueish gray hue on the underlying skin.
Most pilomatrixomas that have been studied have shown a mutation in Exon 3 of the beta-catenin gene (CTNNB1). The beta-catenin molecule is a subunit of the cadherin protein, which is part of an important pathway in the terminal hair follicle differentiation. Beta-catenin also plays an important role in the Wnt pathway, which regulates cell fate as well as early embryonic patterning. Beta-catenin is responsible for forming adhesion junctions among cells. There have also been immunohistochemical studies that have shown a BCL2 proto-oncogene overexpression to pilomatrixoma.
There are several genetic syndromes that have been associated with the presence of pilomatrixomas: Turner syndrome (XO chromosome abnormality associated with short stature and cardiac defects), Gardner syndrome (polyposis coli and colon and rectal cancer), myotonic dystrophy, Rubinstein-Taybi syndrome (characterized by broad thumbs and toes, short stature, distinctive facial features, and varying degrees of intellectual disability), and trisomy 9. On physical examination our patient didn’t present with any of the typical features or history that could suggest any of these syndromes. A close follow-up and evaluation by a geneticist was recommended because after the initial visit she developed a second lesion on the forehead.
The differential diagnosis for this lesion includes other cysts that may occur on the ear such as epidermal inclusion cyst or dermoid cysts, though these lesions do not tend to be as firm as pilomatrixomas are, which can help with the diagnosis. Dermoid cysts are made of dermal and epidermal components. They are usually present at birth and are commonly seen on the scalp and the periorbital face.
Keloids are rubbery nodules of scar tissue that can form on sites of trauma, and although the lesion occurred after she had her ears pierced, the consistency and rapid growth of the lesion as well as the pathological description made this benign fibrous growth less likely.
When pilomatrixomas are inflamed they can be confused with vascular growths: in this particular case, a hemangioma or another vascular tumor such as a tufted angioma or kaposiform hemangioendothelioma. An ultrasound of the lesion could have helped in the differential diagnosis of the lesion.
Pilomatrixomas can grow significantly and in some cases get inflamed or infected. Surgical management of pilomatrixomas is often required because the lesions do not regress spontaneously.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Forbis R Jr and Helwig EB. Arch Dermatol 1961;83:606-18.
Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016 Jun;85:148-53.
A biopsy of the lesion was performed that showed a well-defined nodulocystic tumor composed of nests of basaloid cells that are undergoing trichilemmal keratinization. Shadow cells are seen as well as small areas of calcification. There is also a histiocytic infiltrate with multinucleated giant cells. The histologic diagnosis is of a pilomatrixoma.
Pilomatrixoma, also known as calcifying epithelioma of Malherbe, was first described in 1880, as a tumor of sebaceous gland origin. Later, in 1961, Robert Forbis Jr, MD, and Elson B. Helwig, MD, coined the term pilomatrixoma to describe the hair follicle matrix as the source of the tumor. Pilomatrixomas are commonly seen in the pediatric population, usually in children between 8 and 13 years of age. Our patient is one of the youngest described. The lesions are commonly seen on the face and neck in about 70% of the cases followed by the upper extremities, back, and legs. Clinically, the lesions appear as a firm dermal papule or nodule, which moves freely and may have associated erythema on the skin surface or a blueish gray hue on the underlying skin.
Most pilomatrixomas that have been studied have shown a mutation in Exon 3 of the beta-catenin gene (CTNNB1). The beta-catenin molecule is a subunit of the cadherin protein, which is part of an important pathway in the terminal hair follicle differentiation. Beta-catenin also plays an important role in the Wnt pathway, which regulates cell fate as well as early embryonic patterning. Beta-catenin is responsible for forming adhesion junctions among cells. There have also been immunohistochemical studies that have shown a BCL2 proto-oncogene overexpression to pilomatrixoma.
There are several genetic syndromes that have been associated with the presence of pilomatrixomas: Turner syndrome (XO chromosome abnormality associated with short stature and cardiac defects), Gardner syndrome (polyposis coli and colon and rectal cancer), myotonic dystrophy, Rubinstein-Taybi syndrome (characterized by broad thumbs and toes, short stature, distinctive facial features, and varying degrees of intellectual disability), and trisomy 9. On physical examination our patient didn’t present with any of the typical features or history that could suggest any of these syndromes. A close follow-up and evaluation by a geneticist was recommended because after the initial visit she developed a second lesion on the forehead.
The differential diagnosis for this lesion includes other cysts that may occur on the ear such as epidermal inclusion cyst or dermoid cysts, though these lesions do not tend to be as firm as pilomatrixomas are, which can help with the diagnosis. Dermoid cysts are made of dermal and epidermal components. They are usually present at birth and are commonly seen on the scalp and the periorbital face.
Keloids are rubbery nodules of scar tissue that can form on sites of trauma, and although the lesion occurred after she had her ears pierced, the consistency and rapid growth of the lesion as well as the pathological description made this benign fibrous growth less likely.
When pilomatrixomas are inflamed they can be confused with vascular growths: in this particular case, a hemangioma or another vascular tumor such as a tufted angioma or kaposiform hemangioendothelioma. An ultrasound of the lesion could have helped in the differential diagnosis of the lesion.
Pilomatrixomas can grow significantly and in some cases get inflamed or infected. Surgical management of pilomatrixomas is often required because the lesions do not regress spontaneously.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Forbis R Jr and Helwig EB. Arch Dermatol 1961;83:606-18.
Schwarz Y et al. Int J Pediatr Otorhinolaryngol. 2016 Jun;85:148-53.
A 4-month-old female was referred to our clinic for evaluation of a bump on the right ear. The lesion was first noted at 2 months of age as a little pimple. She was evaluated by her pediatrician and was treated with topical and oral antibiotics without resolution of the lesion. The bump continued to grow and seemed tender to palpation, so she was referred to dermatology for evaluation.
She was born via normal vaginal delivery at 40 weeks. Her mother has no medical conditions and the pregnancy was uneventful. She has been growing and developing well. She takes vitamin D and is currently breast fed.
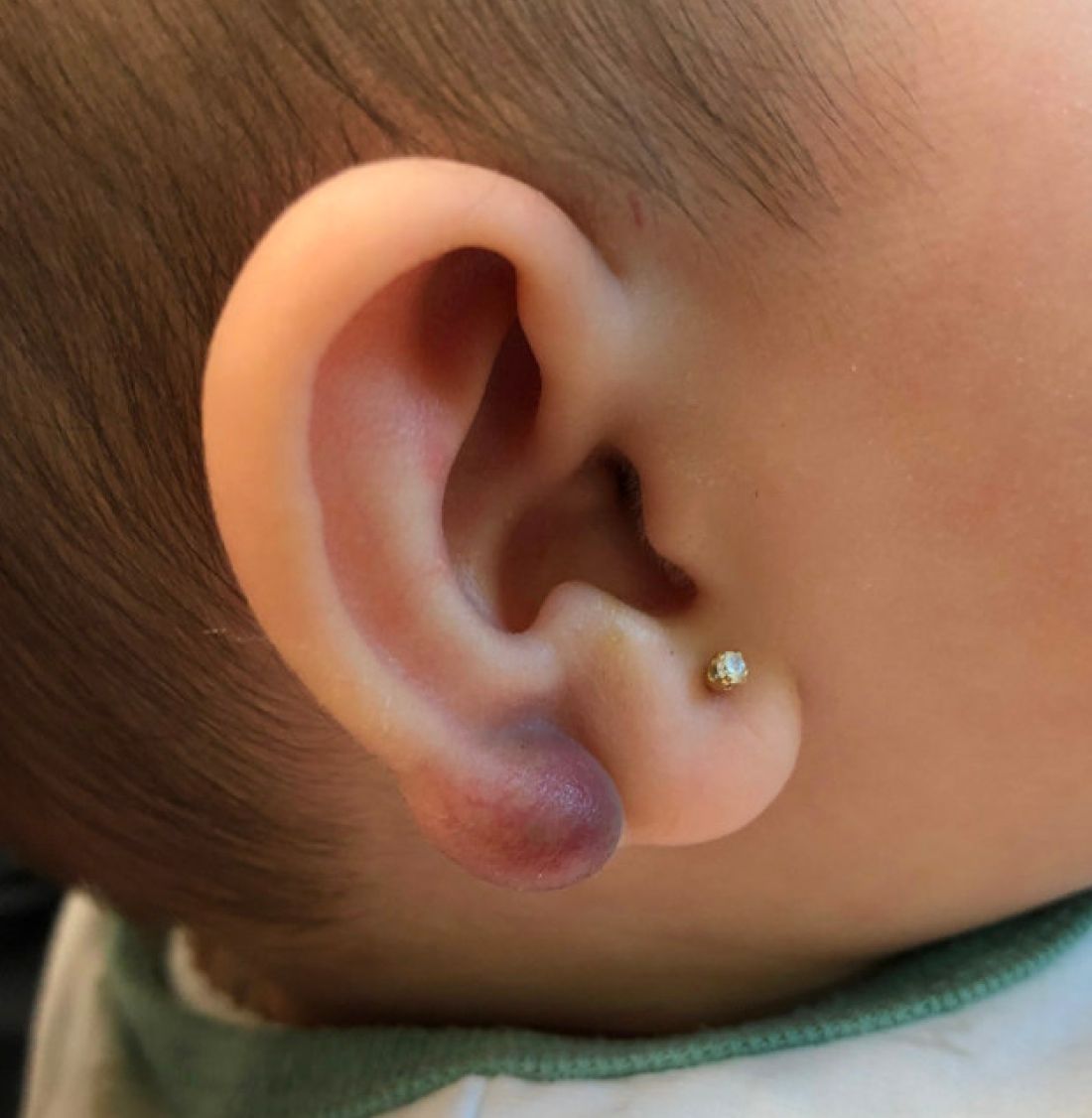
There have been no other family members with similar lesions. She had her ears pierced at a month of age without any complications.
On skin examination she has a firm red nodule on the right ear that appears slightly tender to touch. She has no other skin lesions of concern. She has normal muscle tone and there are no other abnormalities noted on the physical exam. She has no hepatomegaly, splenomegaly, or lymphadenopathy.
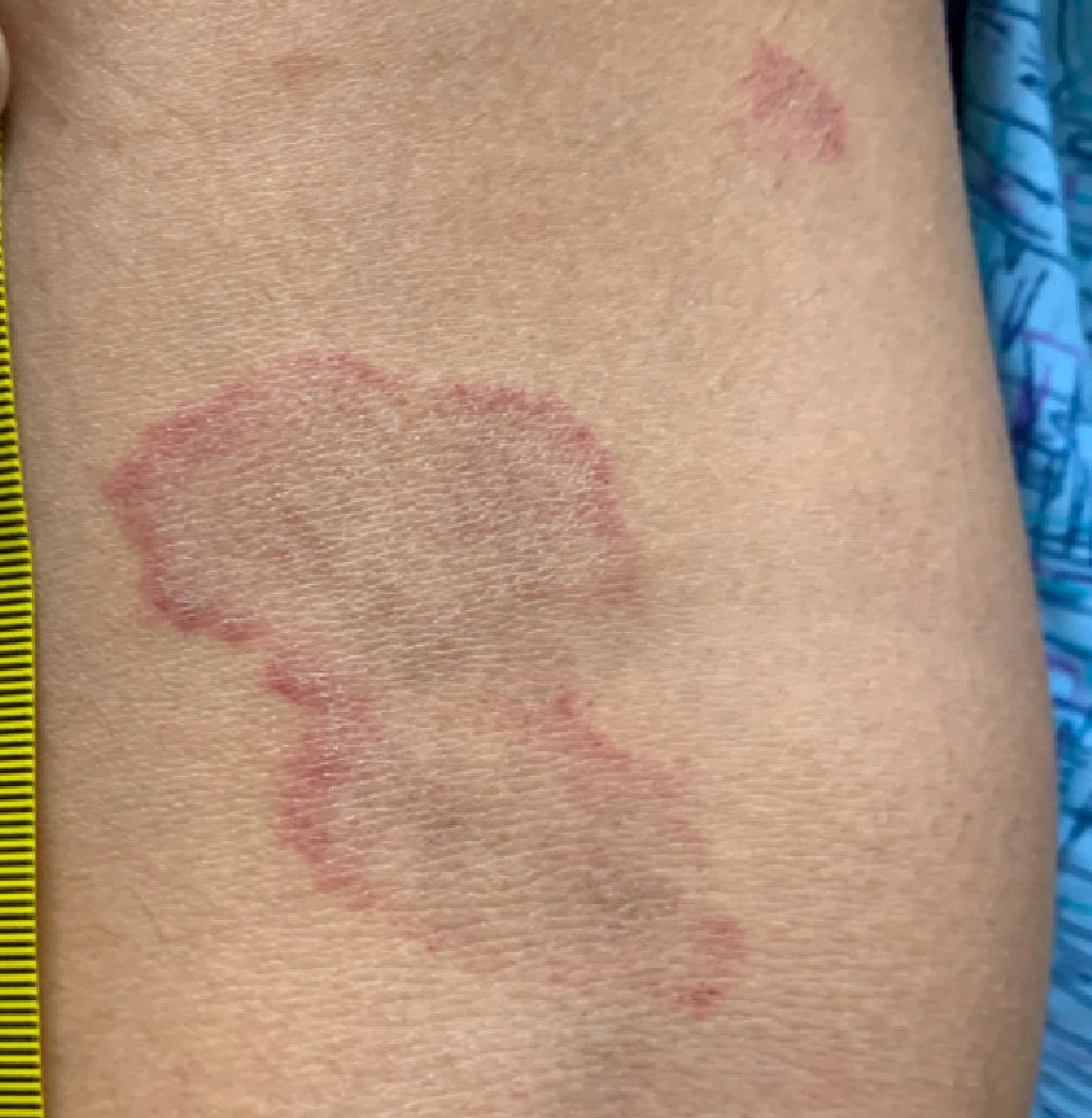
A 9-year-old girl was evaluated for a week-long history of rash on the feet
A complete body examination failed to reveal any other lesions suggestive of a fungal infection. A blood count and urinalysis were within normal limits. She had no lymphadenopathy or hepatosplenomegaly. She was diagnosed with cutaneous larva migrans (CLM) given the clinical appearance of the lesions and the recent travel history.
CLM is a zoonotic infection caused by several hookworms such as Ancylostoma braziliense, Ancylostoma caninum, and Uncinaria stenocephala, as well as human hookworms such as Ancylostoma duodenale and Necator americanus. The hookworms can be present in contaminated soils and sandy beaches on the coastal regions of South America, the Caribbean, the Southeastern United States, Southeast Asia, and Africa.1-5
It is a common disease in the tourist population visiting tropical countries because of exposure to the hookworms in the soil without use of proper foot protection.
The clinical features are of an erythematous linear serpiginous plaque that is pruritic and can progress from millimeters to centimeters in size within a few days to weeks. Vesicles and multiple tracks can also be seen. The most common locations are the feet, buttocks, and thighs.
The larvae in the soil come from eggs excreted in the feces of infected cats and dogs. The infection is caused by direct contact of the larvae with the stratum corneum of the skin creating a burrow and an inflammatory response that will cause erythema, edema, track formation, and pruritus.
Diagnosis is made clinically. Rarely, a skin biopsy is warranted. The differential diagnosis includes tinea pedis, granuloma annulare, larva currens, contact dermatitis, and herpes zoster.

Tinea pedis is a fungal infection of the skin of the feet, commonly localized on the web spaces. The risk factors are a hot and humid environment, prolonged wear of occlusive footwear, excess sweating, and prolonged exposure to water.6 Diagnosis is confirmed by microscopic evaluation of skin scrapings with potassium hydroxide or a fungal culture. The infection is treated with topical antifungal creams and, in severe cases, systemic antifungals. Granuloma annulare is a benign chronic skin condition that presents with annular-shaped lesions. Its etiology is unknown. The lesions may be asymptomatic or mildly pruritic. Localized granuloma annulare typically presents as reddish-brown papules or plaques on the fingers, hands, elbows, dorsal feet, or ankles. The feature distinguishing granuloma annulare from other annular lesions is its absence of scale.
Allergic contact dermatitis is caused by skin exposure to an allergen and a secondary inflammatory response to this material on the skin causing inflammation, vesiculation, and pruritus. Lesions are treated with topical corticosteroids and avoidance of the allergen.
Herpes zoster is caused by a viral infection of the latent varicella-zoster virus. Its reactivation causes the presence of vesicles with an erythematous base that have a dermatomal distribution. The lesions are usually tender. Treatment is recommended to be started within 72 hours of the eruption with antivirals such as acyclovir or valacyclovir.
Cutaneous larva currens is caused by the cutaneous infection with Strongyloides stercoralis. In comparison with CLM, the lesions progress faster, at up to a centimeter within hours.
CLM is usually self-limited. If the patient has multiple lesions or more severe disease, oral albendazole or ivermectin can be prescribed. Other treatments, though not preferred, include freezing and topical thiabendazole solutions.
As our patient had several lesions, oral ivermectin was chosen as treatment and the lesions cleared within a week. Also, she was recommended to always wear shoes when walking on the beach.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Dr. Valderrama is a pediatric dermatologist at Fundación Cardioinfantil, Bogota, Colombia.
References
1. Feldmeier H and Schuster A. Eur J Clin Microbiol Infect Dis. 2012 Jun;31(6):915-8.
2. Jacobson CC and Abel EA. J Am Acad Dermatol. 2007 Jun;56(6):1026-43.
3. Kincaid L et al. Travel Med Infect Dis. 2015 Sep-Oct;13(5):382-7.
4. Gill N et al. Adv Skin Wound Care. 2020 Jul;33(7):356-9.
5. Rodenas-Herranz T et al. Dermatol Ther. 2020 May;33(3):e13316.
6. Pramod K et al. In: StatPearls [Internet]. Treasure Island (Fla): StatPearls Publishing; 2022 Jan.
A complete body examination failed to reveal any other lesions suggestive of a fungal infection. A blood count and urinalysis were within normal limits. She had no lymphadenopathy or hepatosplenomegaly. She was diagnosed with cutaneous larva migrans (CLM) given the clinical appearance of the lesions and the recent travel history.
CLM is a zoonotic infection caused by several hookworms such as Ancylostoma braziliense, Ancylostoma caninum, and Uncinaria stenocephala, as well as human hookworms such as Ancylostoma duodenale and Necator americanus. The hookworms can be present in contaminated soils and sandy beaches on the coastal regions of South America, the Caribbean, the Southeastern United States, Southeast Asia, and Africa.1-5
It is a common disease in the tourist population visiting tropical countries because of exposure to the hookworms in the soil without use of proper foot protection.
The clinical features are of an erythematous linear serpiginous plaque that is pruritic and can progress from millimeters to centimeters in size within a few days to weeks. Vesicles and multiple tracks can also be seen. The most common locations are the feet, buttocks, and thighs.
The larvae in the soil come from eggs excreted in the feces of infected cats and dogs. The infection is caused by direct contact of the larvae with the stratum corneum of the skin creating a burrow and an inflammatory response that will cause erythema, edema, track formation, and pruritus.
Diagnosis is made clinically. Rarely, a skin biopsy is warranted. The differential diagnosis includes tinea pedis, granuloma annulare, larva currens, contact dermatitis, and herpes zoster.
Tinea pedis is a fungal infection of the skin of the feet, commonly localized on the web spaces. The risk factors are a hot and humid environment, prolonged wear of occlusive footwear, excess sweating, and prolonged exposure to water.6 Diagnosis is confirmed by microscopic evaluation of skin scrapings with potassium hydroxide or a fungal culture. The infection is treated with topical antifungal creams and, in severe cases, systemic antifungals. Granuloma annulare is a benign chronic skin condition that presents with annular-shaped lesions. Its etiology is unknown. The lesions may be asymptomatic or mildly pruritic. Localized granuloma annulare typically presents as reddish-brown papules or plaques on the fingers, hands, elbows, dorsal feet, or ankles. The feature distinguishing granuloma annulare from other annular lesions is its absence of scale.
Allergic contact dermatitis is caused by skin exposure to an allergen and a secondary inflammatory response to this material on the skin causing inflammation, vesiculation, and pruritus. Lesions are treated with topical corticosteroids and avoidance of the allergen.
Herpes zoster is caused by a viral infection of the latent varicella-zoster virus. Its reactivation causes the presence of vesicles with an erythematous base that have a dermatomal distribution. The lesions are usually tender. Treatment is recommended to be started within 72 hours of the eruption with antivirals such as acyclovir or valacyclovir.
Cutaneous larva currens is caused by the cutaneous infection with Strongyloides stercoralis. In comparison with CLM, the lesions progress faster, at up to a centimeter within hours.
CLM is usually self-limited. If the patient has multiple lesions or more severe disease, oral albendazole or ivermectin can be prescribed. Other treatments, though not preferred, include freezing and topical thiabendazole solutions.
As our patient had several lesions, oral ivermectin was chosen as treatment and the lesions cleared within a week. Also, she was recommended to always wear shoes when walking on the beach.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Dr. Valderrama is a pediatric dermatologist at Fundación Cardioinfantil, Bogota, Colombia.
References
1. Feldmeier H and Schuster A. Eur J Clin Microbiol Infect Dis. 2012 Jun;31(6):915-8.
2. Jacobson CC and Abel EA. J Am Acad Dermatol. 2007 Jun;56(6):1026-43.
3. Kincaid L et al. Travel Med Infect Dis. 2015 Sep-Oct;13(5):382-7.
4. Gill N et al. Adv Skin Wound Care. 2020 Jul;33(7):356-9.
5. Rodenas-Herranz T et al. Dermatol Ther. 2020 May;33(3):e13316.
6. Pramod K et al. In: StatPearls [Internet]. Treasure Island (Fla): StatPearls Publishing; 2022 Jan.
A complete body examination failed to reveal any other lesions suggestive of a fungal infection. A blood count and urinalysis were within normal limits. She had no lymphadenopathy or hepatosplenomegaly. She was diagnosed with cutaneous larva migrans (CLM) given the clinical appearance of the lesions and the recent travel history.
CLM is a zoonotic infection caused by several hookworms such as Ancylostoma braziliense, Ancylostoma caninum, and Uncinaria stenocephala, as well as human hookworms such as Ancylostoma duodenale and Necator americanus. The hookworms can be present in contaminated soils and sandy beaches on the coastal regions of South America, the Caribbean, the Southeastern United States, Southeast Asia, and Africa.1-5
It is a common disease in the tourist population visiting tropical countries because of exposure to the hookworms in the soil without use of proper foot protection.
The clinical features are of an erythematous linear serpiginous plaque that is pruritic and can progress from millimeters to centimeters in size within a few days to weeks. Vesicles and multiple tracks can also be seen. The most common locations are the feet, buttocks, and thighs.
The larvae in the soil come from eggs excreted in the feces of infected cats and dogs. The infection is caused by direct contact of the larvae with the stratum corneum of the skin creating a burrow and an inflammatory response that will cause erythema, edema, track formation, and pruritus.
Diagnosis is made clinically. Rarely, a skin biopsy is warranted. The differential diagnosis includes tinea pedis, granuloma annulare, larva currens, contact dermatitis, and herpes zoster.
Tinea pedis is a fungal infection of the skin of the feet, commonly localized on the web spaces. The risk factors are a hot and humid environment, prolonged wear of occlusive footwear, excess sweating, and prolonged exposure to water.6 Diagnosis is confirmed by microscopic evaluation of skin scrapings with potassium hydroxide or a fungal culture. The infection is treated with topical antifungal creams and, in severe cases, systemic antifungals. Granuloma annulare is a benign chronic skin condition that presents with annular-shaped lesions. Its etiology is unknown. The lesions may be asymptomatic or mildly pruritic. Localized granuloma annulare typically presents as reddish-brown papules or plaques on the fingers, hands, elbows, dorsal feet, or ankles. The feature distinguishing granuloma annulare from other annular lesions is its absence of scale.
Allergic contact dermatitis is caused by skin exposure to an allergen and a secondary inflammatory response to this material on the skin causing inflammation, vesiculation, and pruritus. Lesions are treated with topical corticosteroids and avoidance of the allergen.
Herpes zoster is caused by a viral infection of the latent varicella-zoster virus. Its reactivation causes the presence of vesicles with an erythematous base that have a dermatomal distribution. The lesions are usually tender. Treatment is recommended to be started within 72 hours of the eruption with antivirals such as acyclovir or valacyclovir.
Cutaneous larva currens is caused by the cutaneous infection with Strongyloides stercoralis. In comparison with CLM, the lesions progress faster, at up to a centimeter within hours.
CLM is usually self-limited. If the patient has multiple lesions or more severe disease, oral albendazole or ivermectin can be prescribed. Other treatments, though not preferred, include freezing and topical thiabendazole solutions.
As our patient had several lesions, oral ivermectin was chosen as treatment and the lesions cleared within a week. Also, she was recommended to always wear shoes when walking on the beach.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Dr. Valderrama is a pediatric dermatologist at Fundación Cardioinfantil, Bogota, Colombia.
References
1. Feldmeier H and Schuster A. Eur J Clin Microbiol Infect Dis. 2012 Jun;31(6):915-8.
2. Jacobson CC and Abel EA. J Am Acad Dermatol. 2007 Jun;56(6):1026-43.
3. Kincaid L et al. Travel Med Infect Dis. 2015 Sep-Oct;13(5):382-7.
4. Gill N et al. Adv Skin Wound Care. 2020 Jul;33(7):356-9.
5. Rodenas-Herranz T et al. Dermatol Ther. 2020 May;33(3):e13316.
6. Pramod K et al. In: StatPearls [Internet]. Treasure Island (Fla): StatPearls Publishing; 2022 Jan.
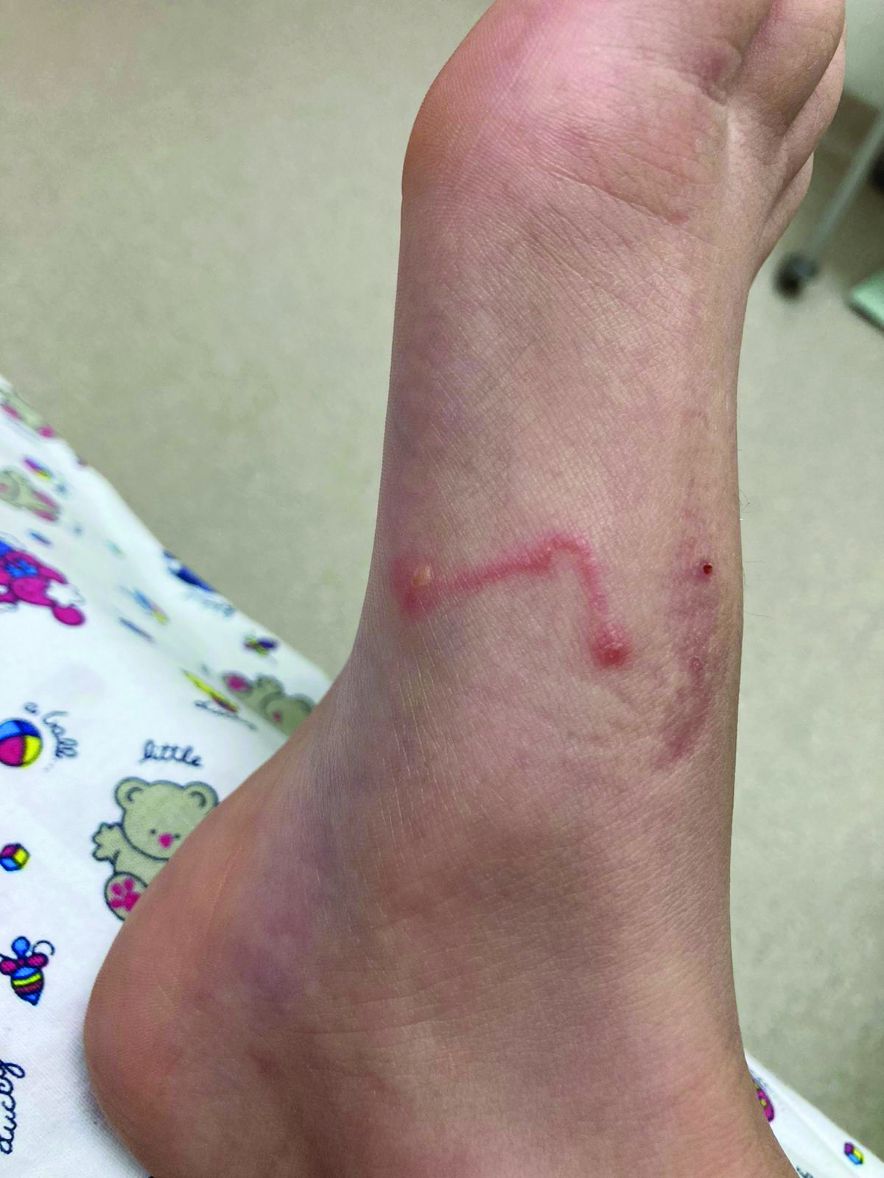
Her mother reported recent travel to a beachside city in Colombia. A review of systems was negative. She was not taking any other medications or vitamin supplements. There were no pets at home and no other affected family members. Physical exam was notable for an erythematous curvilinear plaque on the feet and a small vesicle.
Adolescent female with rash on the arms and posterior legs
Erythema annulare centrifugum
A thorough body examination failed to reveal any other rashes or lesions suggestive of a fungal infection. A blood count and urinalysis were within normal limits. She had no lymphadenopathy or hepatosplenomegaly. A potassium hydroxide analysis of skin scrapings was negative for fungal elements. Punch biopsy of the skin on the left arm revealed focal intermittent parakeratosis, mildly acanthotic and spongiotic epidermis, and a tight superficial perivascular chronic dermatitis consisting of lymphocytes and histiocytes (Figures). Given these findings, a diagnosis of erythema annulare centrifugum (EAC) was rendered.
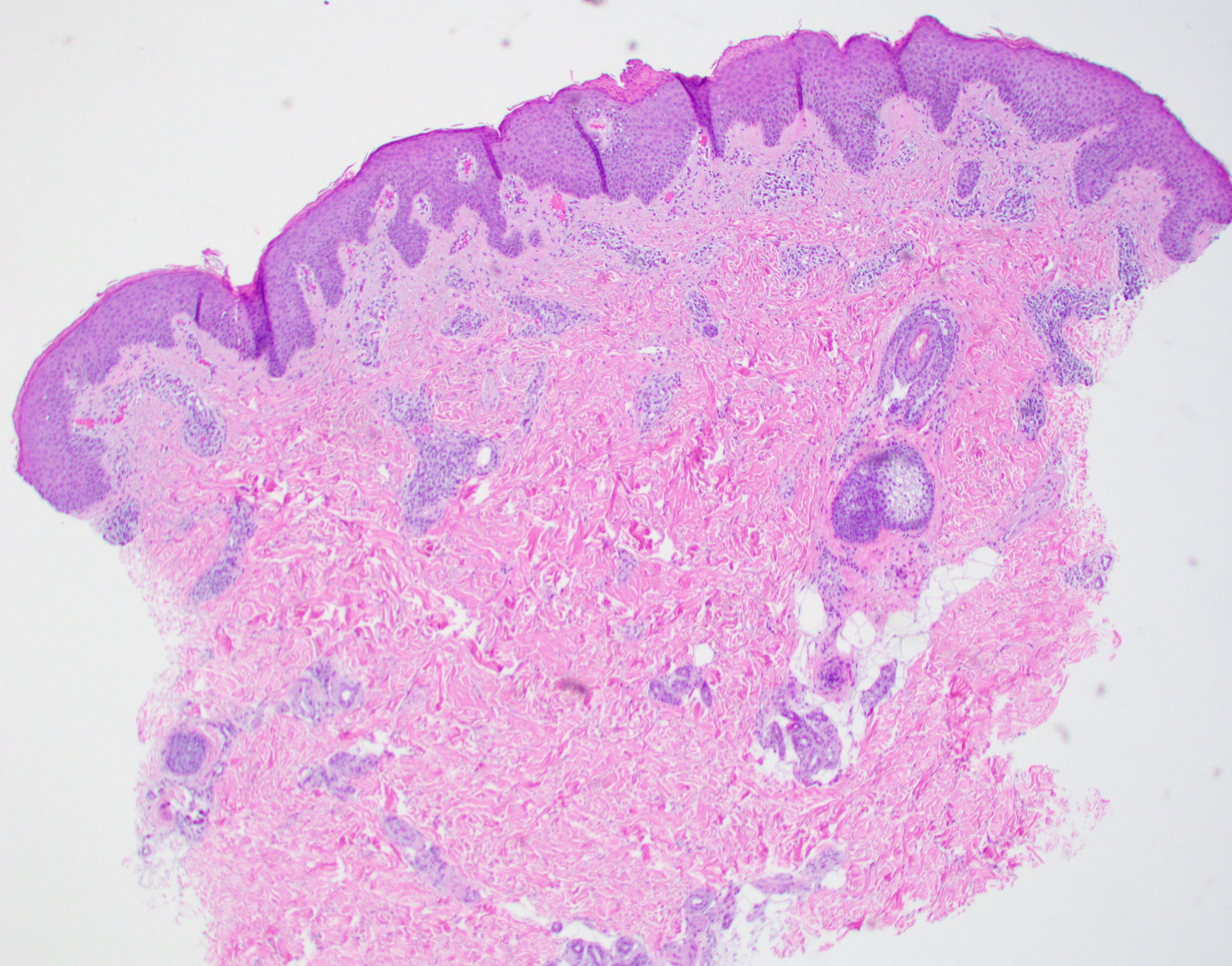
EAC is a rare, reactive skin rash characterized by redness (erythema) and ring-shaped lesions (annulare) that slowly spread from the center (centrifugum). The lesions present with a characteristic trailing scale on the inner border of the erythematous ring. Lesions may be asymptomatic or mildly pruritic and commonly involve the trunk, buttocks, hips, and upper legs. It is important to note that its duration is highly variable, ranging from weeks to decades, with most cases persisting for 9 months. EAC typically affects young or middle-aged adults but can occur at any age.
Although the etiology of EAC is unknown, it is believed to be a hypersensitivity reaction to a foreign antigen. Cutaneous fungal infections are commonly reported as triggers as well as other viral infections, medications, malignancy, underlying systemic disease, and certain foods. Treatment depends on the underlying condition and removing the implicated agent. However, most cases of EAC are idiopathic and self-limiting. It is possible that our patient’s prior history of tinea capitis could have triggered the skin lesions suggestive of EAC, but interestingly, these lesions did not go away after the fungal infection was cleared and have continued to recur. For patients with refractory lesions or treatment of patients without an identifiable cause, the use of oral antimicrobials has been proposed. Medications such as azithromycin, erythromycin, fluconazole, and metronidazole have been reported to be helpful in some patients with refractory EAC. Our patient wanted to continue topical treatment with betamethasone as needed and may consider antimicrobial therapy if the lesions continue to recur.
Tinea corporis refers to a superficial fungal infection of the skin. It may present as one or more asymmetrical, annular, pruritic plaques with a raised scaly leading edge rather than the trailing scale seen with EAC. Diagnosis is made by KOH examination of skin scrapings. Common risk factors include close contact with an infected person or animal, warm, moist environments, sharing personal items, and prolonged use of systemic corticosteroids. Our patient’s KOH analysis of skin scrapings was negative for fungal elements.
Erythema marginatum is a rare skin rash commonly seen with acute rheumatic fever secondary to streptococcal infection. It presents as annular erythematous lesions on the trunk and proximal extremities that are exacerbated by heat. It is often associated with active carditis related to rheumatic fever. This self-limited rash usually resolves in 2-3 days. Our patient was asymptomatic without involvement of other organs.
Like EAC, granuloma annulare is a benign chronic skin condition that presents with ring-shaped lesions. Its etiology is unknown, and lesions may be asymptomatic or mildly pruritic. Localized granuloma annulare typically presents as reddish-brown papules or plaques on the fingers, hands, elbows, dorsal feet, or ankles. The distinguishing feature of granuloma annulare from other annular lesions is its absence of scale.
Urticaria multiforme is an allergic hypersensitivity reaction commonly linked to viral infections, medications, and immunizations. Clinical features include blanchable annular/polycyclic lesions with a central purplish or dusky hue. Diagnostic pearls include the presence of pruritus, dermatographism, and individual lesions that resolve within 24 hours, all of which were not found in our patient’s case.
Ms. Laborada is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital. Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Ms. Laborada and Dr. Matiz have no relevant financial disclosures.
References
1. Paller A and Mancini AJ. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. 4th ed. Philadelphia: Elsevier Saunders, 2011.
2. McDaniel B and Cook C. “Erythema annulare centrifugum” 2021 Aug 27. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing, 2022 Jan. PMID: 29494101.
3. Leung AK e al. Drugs Context. 2020 Jul 20;9:5-6.
4. Majmundar VD and Nagalli S. “Erythema marginatum” 2022 May 8. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing, 2022 Jan.
5. Piette EW and Rosenbach M. J Am Acad Dermatol. 2016 Sep;75(3):467-79.
6. Barros M et al. BMJ Case Rep. 2021 Jan 28;14(1):e241011.
Erythema annulare centrifugum
A thorough body examination failed to reveal any other rashes or lesions suggestive of a fungal infection. A blood count and urinalysis were within normal limits. She had no lymphadenopathy or hepatosplenomegaly. A potassium hydroxide analysis of skin scrapings was negative for fungal elements. Punch biopsy of the skin on the left arm revealed focal intermittent parakeratosis, mildly acanthotic and spongiotic epidermis, and a tight superficial perivascular chronic dermatitis consisting of lymphocytes and histiocytes (Figures). Given these findings, a diagnosis of erythema annulare centrifugum (EAC) was rendered.
EAC is a rare, reactive skin rash characterized by redness (erythema) and ring-shaped lesions (annulare) that slowly spread from the center (centrifugum). The lesions present with a characteristic trailing scale on the inner border of the erythematous ring. Lesions may be asymptomatic or mildly pruritic and commonly involve the trunk, buttocks, hips, and upper legs. It is important to note that its duration is highly variable, ranging from weeks to decades, with most cases persisting for 9 months. EAC typically affects young or middle-aged adults but can occur at any age.
Although the etiology of EAC is unknown, it is believed to be a hypersensitivity reaction to a foreign antigen. Cutaneous fungal infections are commonly reported as triggers as well as other viral infections, medications, malignancy, underlying systemic disease, and certain foods. Treatment depends on the underlying condition and removing the implicated agent. However, most cases of EAC are idiopathic and self-limiting. It is possible that our patient’s prior history of tinea capitis could have triggered the skin lesions suggestive of EAC, but interestingly, these lesions did not go away after the fungal infection was cleared and have continued to recur. For patients with refractory lesions or treatment of patients without an identifiable cause, the use of oral antimicrobials has been proposed. Medications such as azithromycin, erythromycin, fluconazole, and metronidazole have been reported to be helpful in some patients with refractory EAC. Our patient wanted to continue topical treatment with betamethasone as needed and may consider antimicrobial therapy if the lesions continue to recur.
Tinea corporis refers to a superficial fungal infection of the skin. It may present as one or more asymmetrical, annular, pruritic plaques with a raised scaly leading edge rather than the trailing scale seen with EAC. Diagnosis is made by KOH examination of skin scrapings. Common risk factors include close contact with an infected person or animal, warm, moist environments, sharing personal items, and prolonged use of systemic corticosteroids. Our patient’s KOH analysis of skin scrapings was negative for fungal elements.
Erythema marginatum is a rare skin rash commonly seen with acute rheumatic fever secondary to streptococcal infection. It presents as annular erythematous lesions on the trunk and proximal extremities that are exacerbated by heat. It is often associated with active carditis related to rheumatic fever. This self-limited rash usually resolves in 2-3 days. Our patient was asymptomatic without involvement of other organs.
Like EAC, granuloma annulare is a benign chronic skin condition that presents with ring-shaped lesions. Its etiology is unknown, and lesions may be asymptomatic or mildly pruritic. Localized granuloma annulare typically presents as reddish-brown papules or plaques on the fingers, hands, elbows, dorsal feet, or ankles. The distinguishing feature of granuloma annulare from other annular lesions is its absence of scale.
Urticaria multiforme is an allergic hypersensitivity reaction commonly linked to viral infections, medications, and immunizations. Clinical features include blanchable annular/polycyclic lesions with a central purplish or dusky hue. Diagnostic pearls include the presence of pruritus, dermatographism, and individual lesions that resolve within 24 hours, all of which were not found in our patient’s case.
Ms. Laborada is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital. Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Ms. Laborada and Dr. Matiz have no relevant financial disclosures.
References
1. Paller A and Mancini AJ. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. 4th ed. Philadelphia: Elsevier Saunders, 2011.
2. McDaniel B and Cook C. “Erythema annulare centrifugum” 2021 Aug 27. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing, 2022 Jan. PMID: 29494101.
3. Leung AK e al. Drugs Context. 2020 Jul 20;9:5-6.
4. Majmundar VD and Nagalli S. “Erythema marginatum” 2022 May 8. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing, 2022 Jan.
5. Piette EW and Rosenbach M. J Am Acad Dermatol. 2016 Sep;75(3):467-79.
6. Barros M et al. BMJ Case Rep. 2021 Jan 28;14(1):e241011.
Erythema annulare centrifugum
A thorough body examination failed to reveal any other rashes or lesions suggestive of a fungal infection. A blood count and urinalysis were within normal limits. She had no lymphadenopathy or hepatosplenomegaly. A potassium hydroxide analysis of skin scrapings was negative for fungal elements. Punch biopsy of the skin on the left arm revealed focal intermittent parakeratosis, mildly acanthotic and spongiotic epidermis, and a tight superficial perivascular chronic dermatitis consisting of lymphocytes and histiocytes (Figures). Given these findings, a diagnosis of erythema annulare centrifugum (EAC) was rendered.
EAC is a rare, reactive skin rash characterized by redness (erythema) and ring-shaped lesions (annulare) that slowly spread from the center (centrifugum). The lesions present with a characteristic trailing scale on the inner border of the erythematous ring. Lesions may be asymptomatic or mildly pruritic and commonly involve the trunk, buttocks, hips, and upper legs. It is important to note that its duration is highly variable, ranging from weeks to decades, with most cases persisting for 9 months. EAC typically affects young or middle-aged adults but can occur at any age.
Although the etiology of EAC is unknown, it is believed to be a hypersensitivity reaction to a foreign antigen. Cutaneous fungal infections are commonly reported as triggers as well as other viral infections, medications, malignancy, underlying systemic disease, and certain foods. Treatment depends on the underlying condition and removing the implicated agent. However, most cases of EAC are idiopathic and self-limiting. It is possible that our patient’s prior history of tinea capitis could have triggered the skin lesions suggestive of EAC, but interestingly, these lesions did not go away after the fungal infection was cleared and have continued to recur. For patients with refractory lesions or treatment of patients without an identifiable cause, the use of oral antimicrobials has been proposed. Medications such as azithromycin, erythromycin, fluconazole, and metronidazole have been reported to be helpful in some patients with refractory EAC. Our patient wanted to continue topical treatment with betamethasone as needed and may consider antimicrobial therapy if the lesions continue to recur.
Tinea corporis refers to a superficial fungal infection of the skin. It may present as one or more asymmetrical, annular, pruritic plaques with a raised scaly leading edge rather than the trailing scale seen with EAC. Diagnosis is made by KOH examination of skin scrapings. Common risk factors include close contact with an infected person or animal, warm, moist environments, sharing personal items, and prolonged use of systemic corticosteroids. Our patient’s KOH analysis of skin scrapings was negative for fungal elements.
Erythema marginatum is a rare skin rash commonly seen with acute rheumatic fever secondary to streptococcal infection. It presents as annular erythematous lesions on the trunk and proximal extremities that are exacerbated by heat. It is often associated with active carditis related to rheumatic fever. This self-limited rash usually resolves in 2-3 days. Our patient was asymptomatic without involvement of other organs.
Like EAC, granuloma annulare is a benign chronic skin condition that presents with ring-shaped lesions. Its etiology is unknown, and lesions may be asymptomatic or mildly pruritic. Localized granuloma annulare typically presents as reddish-brown papules or plaques on the fingers, hands, elbows, dorsal feet, or ankles. The distinguishing feature of granuloma annulare from other annular lesions is its absence of scale.
Urticaria multiforme is an allergic hypersensitivity reaction commonly linked to viral infections, medications, and immunizations. Clinical features include blanchable annular/polycyclic lesions with a central purplish or dusky hue. Diagnostic pearls include the presence of pruritus, dermatographism, and individual lesions that resolve within 24 hours, all of which were not found in our patient’s case.
Ms. Laborada is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital. Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Ms. Laborada and Dr. Matiz have no relevant financial disclosures.
References
1. Paller A and Mancini AJ. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. 4th ed. Philadelphia: Elsevier Saunders, 2011.
2. McDaniel B and Cook C. “Erythema annulare centrifugum” 2021 Aug 27. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing, 2022 Jan. PMID: 29494101.
3. Leung AK e al. Drugs Context. 2020 Jul 20;9:5-6.
4. Majmundar VD and Nagalli S. “Erythema marginatum” 2022 May 8. In: StatPearls [Internet]. Treasure Island, Fla.: StatPearls Publishing, 2022 Jan.
5. Piette EW and Rosenbach M. J Am Acad Dermatol. 2016 Sep;75(3):467-79.
6. Barros M et al. BMJ Case Rep. 2021 Jan 28;14(1):e241011.
A review of systems was noncontributory. She was not taking any other medications or vitamin supplements. There were no pets at home and no other affected family members. Physical exam was notable for scattered, pink, annular plaques with central clearing, faint brownish pigmentation, and fine scale.
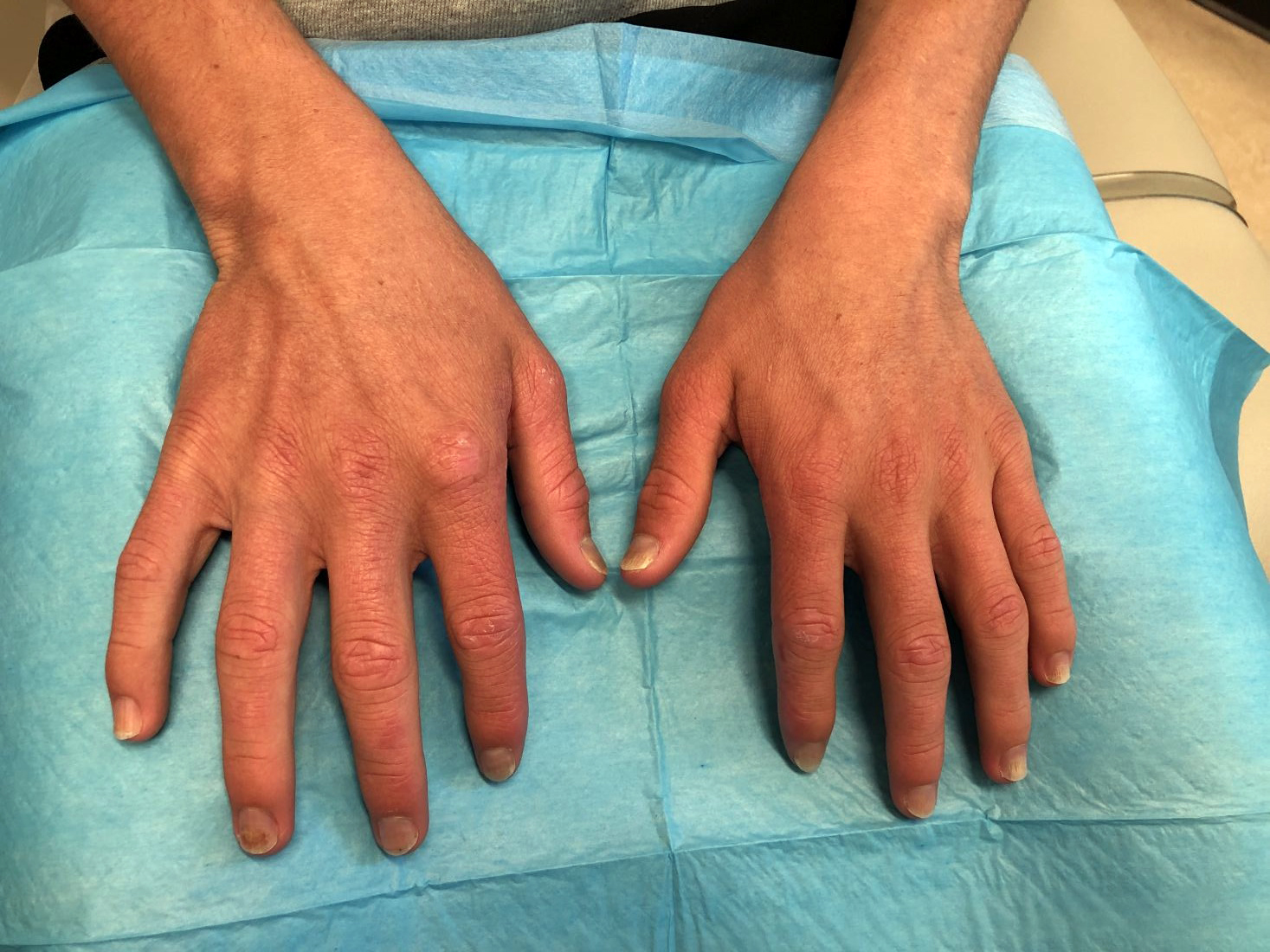
A 14-year-old male presents to clinic with a new-onset rash of the hands
Photosensitivity due to doxycycline
As the patient’s rash presented in sun-exposed areas with both skin and nail changes, our patient was diagnosed with a phototoxic reaction to doxycycline, the oral antibiotic used to treat his acne.
Photosensitive cutaneous drug eruptions are reactions that occur after exposure to a medication and subsequent exposure to UV radiation or visible light. Reactions can be classified into two ways based on their mechanism of action: phototoxic or photoallergic.1 Phototoxic reactions are more common and are a result of direct keratinocyte damage and cellular necrosis. Many classes of medications may cause this adverse effect, but the tetracycline class of antibiotics is a common culprit.2 Photoallergic reactions are less common and are a result of a type IV immune reaction to the offending agent.1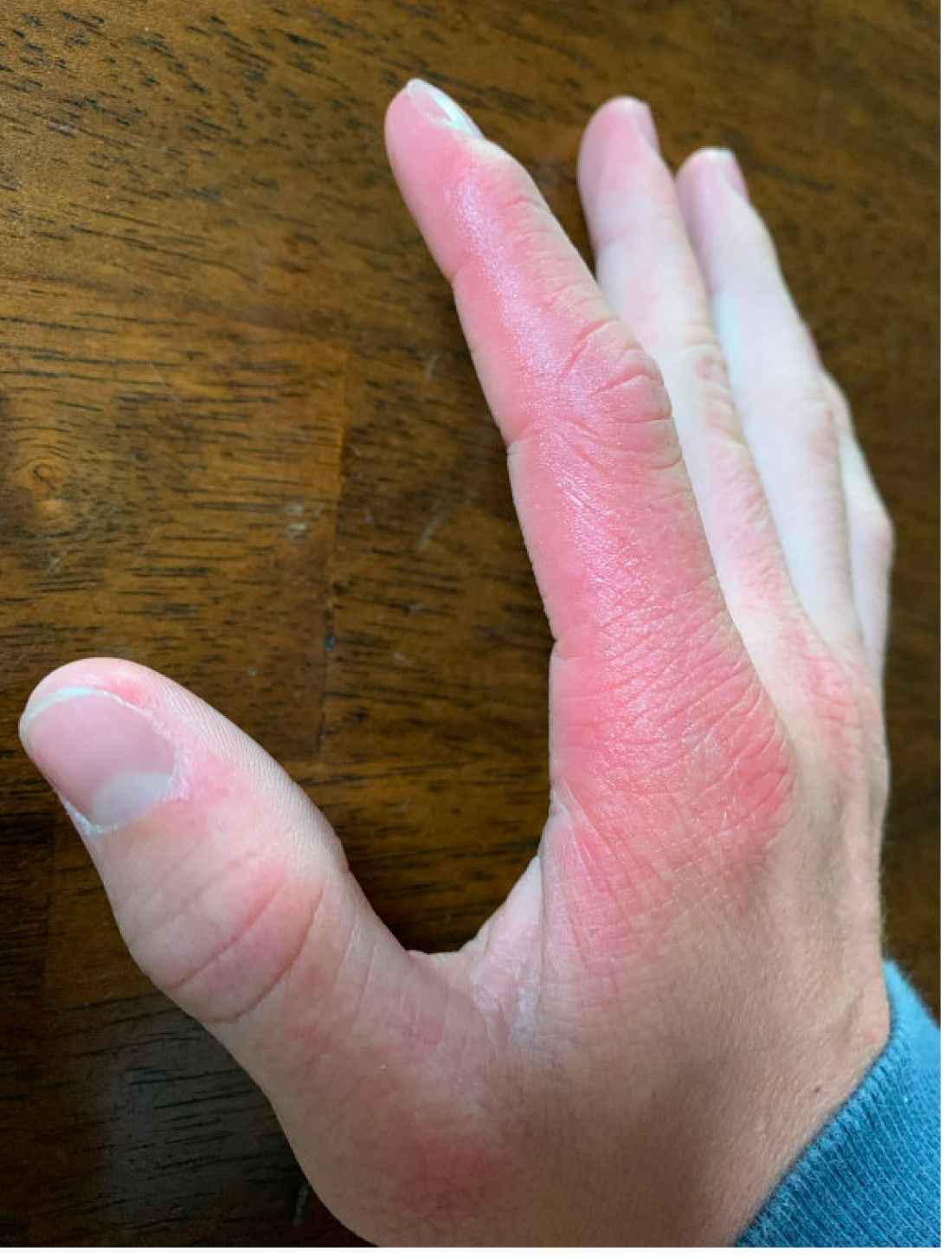
Phototoxic reactions generally present shortly after sun or UV exposure with a photo-distributed eruption pattern.3 Commonly involved areas include the face, the neck, and the extensor surfaces of extremities, with sparing of relatively protected skin such as the upper eyelids and the skin folds.2 Erythema may initially develop in the exposed skin areas, followed by appearance of edema, vesicles, or bullae.1-3 The eruption may be painful and itchy, with some patients reporting severe pain.3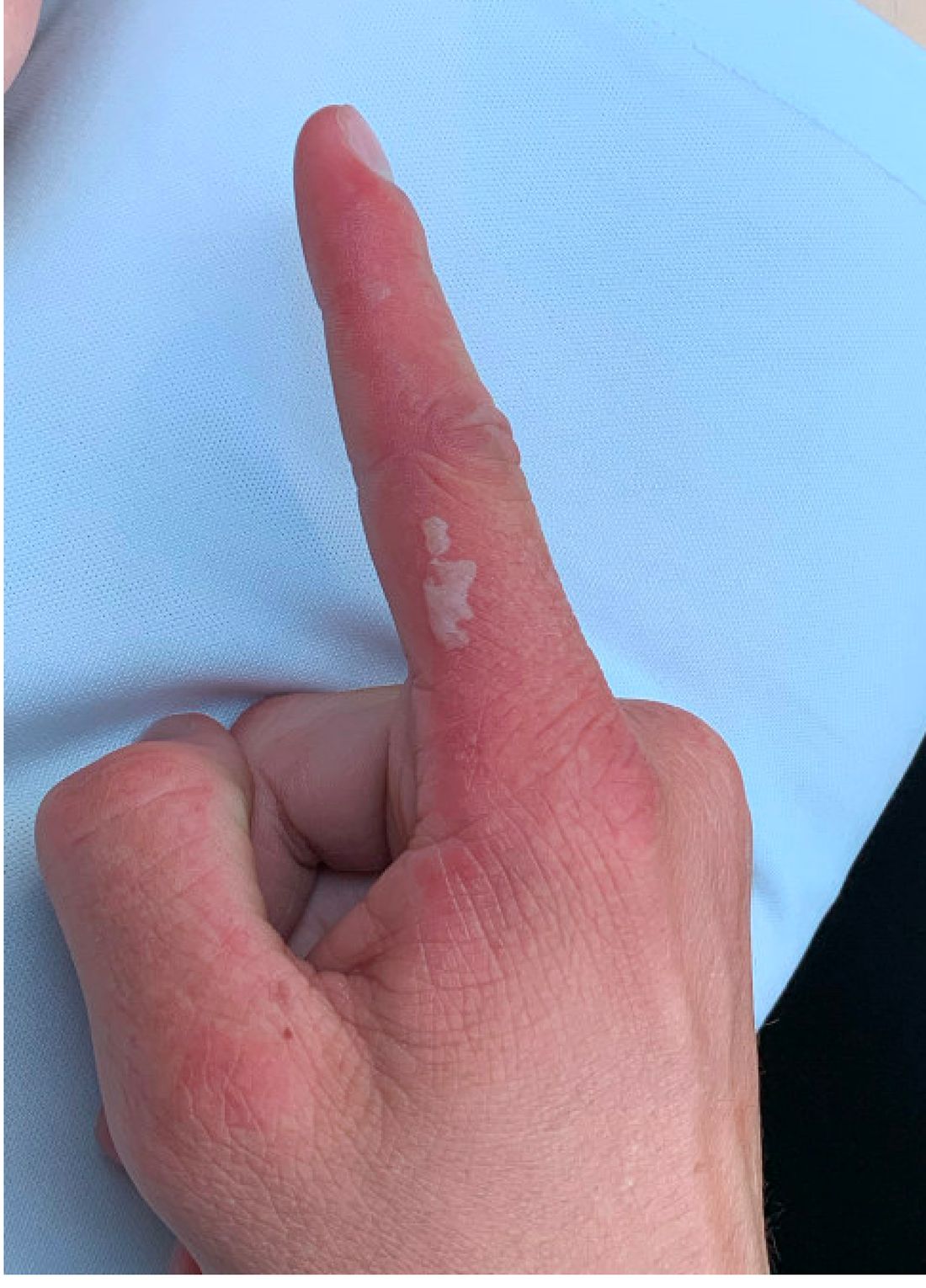
Doxycycline phototoxicity may also cause onycholysis of the nails.2 The reaction is dose dependent, with higher doses of medication leading to a higher likelihood of symptoms.1,2 It is also more prevalent in patients with Fitzpatrick skin type I and II. The usual UVA wavelength required to induce this reaction appears to be in the 320-400 nm range of the UV spectrum.4 By contrast, photoallergic reactions are dose independent, and require a sensitization period prior to the eruption.1 An eczematous eruption is most commonly seen with photoallergic reactions.3
Treatment of drug-induced photosensitivity reactions requires proper identification of the diagnosis and the offending agent, followed by cessation of the medication. If cessation is not possible, then lowering the dose can help to minimize worsening of the condition. However, for photoallergic reactions, the reaction is dose independent so switching to another tolerated agent is likely required. For persistent symptoms following medication withdrawal, topical or systemic steroids and oral antihistamine can help with symptom management.1 For patients with photo-onycholysis, treatment involves stopping the medication and waiting for the intact nail plate to grow.
Prevention is key in the management of photosensitivity reactions. Patients should be counseled about the increased risk of photosensitivity while on tetracycline medications and encouraged to engage in enhanced sun protection measures such as wearing sun protective hats and clothing, increasing use of sunscreen that provides mainly UVA but also UVB protection, and avoiding the sun during the midday when the UV index is highest.1-3
Dermatomyositis
Dermatomyositis is an autoimmune condition that presents with skin lesions as well as systemic findings such as myositis. The cutaneous findings are variable, but pathognomonic findings include Gottron papules of the hands, Gottron’s sign on the elbows, knees, and ankles, and the heliotrope rash of the face. Eighty percent of patients have myopathy presenting as muscle weakness, and commonly have elevated creatine kinase, aspartate transaminase, and alanine transaminase values.5 Diagnosis may be confirmed through skin or muscle biopsy, though antibody studies can also play a helpful role in diagnosis. Treatment is generally with oral corticosteroids or other immunosuppressants as well as sun protection.6 The rash seen in our patient could have been seen in patients with dermatomyositis, though it was not in the typical location on the knuckles (Gottron papules) as it also affected the lateral sides of the fingers.
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune condition characterized by systemic and cutaneous manifestations. Systemic symptoms may include weight loss, fever, fatigue, arthralgia, or arthritis; patients are at risk of renal, cardiovascular, pulmonary, and neurologic complications of SLE.7 The most common cutaneous finding is malar rash, though there are myriad dermatologic manifestations that can occur associated with photosensitivity. Diagnosis is made based on history, physical, and laboratory testing. Treatment options include NSAIDs, oral glucocorticoids, antimalarial drugs, and immunosuppressants.7 Though our patient exhibited photosensitivity, he had none of the systemic findings associated with SLE, making this diagnosis unlikely.
Allergic contact dermatitis
Allergic contact dermatitis (ACD) is a type IV hypersensitivity reaction, and may present as acute, subacute, or chronic dermatitis. The clinical findings vary based on chronicity. Acute ACD presents as pruritic erythematous papules and vesicles or bullae, similar to how it occurred in our patient, though our patient’s lesions were more tender than pruritic. Chronic ACD presents with erythematous lesions with pruritis, lichenification, scaling, and/or fissuring. Observing shapes or sharp demarcation of lesions may help with diagnosis. Patch testing is also useful in the diagnosis of ACD.

Treatment generally involves avoiding the offending agent with topical corticosteroids for symptom management.8
Polymorphous light eruption
Polymorphous light eruption (PLE) is a delayed, type IV hypersensitivity reaction to UV-induced antigens, though these antigens are unknown. PLE presents hours to days following solar or UV exposure and presents only in sun-exposed areas. Itching and burning are always present, but lesion morphology varies from erythema and papules to vesico-papules and blisters. Notably, PLE must be distinguished from drug photosensitivity through history. Treatment generally involves symptom management with topical steroids and sun protective measures for prevention.9 While PLE may present similarly to drug photosensitivity reactions, our patient’s use of a known phototoxic agent makes PLE a less likely diagnosis.
Ms. Appiah is a pediatric dermatology research associate and medical student at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Neither Dr. Matiz nor Ms. Appiah has any relevant financial disclosures.
References
1. Montgomery S et al. Clin Dermatol. 2022;40(1):57-63.
2. Blakely KM et al. Drug Saf. 2019;42(7):827-47.
3. Goetze S et al. Skin Pharmacol Physiol. 2017;30(2):76-80.
4. Odorici G et al. Dermatol Ther. 2021;34(4):e14978.
5. DeWane ME et al. J Am Acad Dermatol. 2020;82(2):267-81.
6. Waldman R et al. J Am Acad Dermatol. 2020;82(2):283-96.
7. Kiriakidou M et al. Ann Intern Med. 2020;172(11):ITC81-ITC96.
8. Nassau S et al. Med Clin North Am. 2020;104(1):61-76.
9. Guarrera M. Adv Exp Med Biol. 2017;996:61-70.
Photosensitivity due to doxycycline
As the patient’s rash presented in sun-exposed areas with both skin and nail changes, our patient was diagnosed with a phototoxic reaction to doxycycline, the oral antibiotic used to treat his acne.
Photosensitive cutaneous drug eruptions are reactions that occur after exposure to a medication and subsequent exposure to UV radiation or visible light. Reactions can be classified into two ways based on their mechanism of action: phototoxic or photoallergic.1 Phototoxic reactions are more common and are a result of direct keratinocyte damage and cellular necrosis. Many classes of medications may cause this adverse effect, but the tetracycline class of antibiotics is a common culprit.2 Photoallergic reactions are less common and are a result of a type IV immune reaction to the offending agent.1
Phototoxic reactions generally present shortly after sun or UV exposure with a photo-distributed eruption pattern.3 Commonly involved areas include the face, the neck, and the extensor surfaces of extremities, with sparing of relatively protected skin such as the upper eyelids and the skin folds.2 Erythema may initially develop in the exposed skin areas, followed by appearance of edema, vesicles, or bullae.1-3 The eruption may be painful and itchy, with some patients reporting severe pain.3
Doxycycline phototoxicity may also cause onycholysis of the nails.2 The reaction is dose dependent, with higher doses of medication leading to a higher likelihood of symptoms.1,2 It is also more prevalent in patients with Fitzpatrick skin type I and II. The usual UVA wavelength required to induce this reaction appears to be in the 320-400 nm range of the UV spectrum.4 By contrast, photoallergic reactions are dose independent, and require a sensitization period prior to the eruption.1 An eczematous eruption is most commonly seen with photoallergic reactions.3
Treatment of drug-induced photosensitivity reactions requires proper identification of the diagnosis and the offending agent, followed by cessation of the medication. If cessation is not possible, then lowering the dose can help to minimize worsening of the condition. However, for photoallergic reactions, the reaction is dose independent so switching to another tolerated agent is likely required. For persistent symptoms following medication withdrawal, topical or systemic steroids and oral antihistamine can help with symptom management.1 For patients with photo-onycholysis, treatment involves stopping the medication and waiting for the intact nail plate to grow.
Prevention is key in the management of photosensitivity reactions. Patients should be counseled about the increased risk of photosensitivity while on tetracycline medications and encouraged to engage in enhanced sun protection measures such as wearing sun protective hats and clothing, increasing use of sunscreen that provides mainly UVA but also UVB protection, and avoiding the sun during the midday when the UV index is highest.1-3
Dermatomyositis
Dermatomyositis is an autoimmune condition that presents with skin lesions as well as systemic findings such as myositis. The cutaneous findings are variable, but pathognomonic findings include Gottron papules of the hands, Gottron’s sign on the elbows, knees, and ankles, and the heliotrope rash of the face. Eighty percent of patients have myopathy presenting as muscle weakness, and commonly have elevated creatine kinase, aspartate transaminase, and alanine transaminase values.5 Diagnosis may be confirmed through skin or muscle biopsy, though antibody studies can also play a helpful role in diagnosis. Treatment is generally with oral corticosteroids or other immunosuppressants as well as sun protection.6 The rash seen in our patient could have been seen in patients with dermatomyositis, though it was not in the typical location on the knuckles (Gottron papules) as it also affected the lateral sides of the fingers.
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune condition characterized by systemic and cutaneous manifestations. Systemic symptoms may include weight loss, fever, fatigue, arthralgia, or arthritis; patients are at risk of renal, cardiovascular, pulmonary, and neurologic complications of SLE.7 The most common cutaneous finding is malar rash, though there are myriad dermatologic manifestations that can occur associated with photosensitivity. Diagnosis is made based on history, physical, and laboratory testing. Treatment options include NSAIDs, oral glucocorticoids, antimalarial drugs, and immunosuppressants.7 Though our patient exhibited photosensitivity, he had none of the systemic findings associated with SLE, making this diagnosis unlikely.
Allergic contact dermatitis
Allergic contact dermatitis (ACD) is a type IV hypersensitivity reaction, and may present as acute, subacute, or chronic dermatitis. The clinical findings vary based on chronicity. Acute ACD presents as pruritic erythematous papules and vesicles or bullae, similar to how it occurred in our patient, though our patient’s lesions were more tender than pruritic. Chronic ACD presents with erythematous lesions with pruritis, lichenification, scaling, and/or fissuring. Observing shapes or sharp demarcation of lesions may help with diagnosis. Patch testing is also useful in the diagnosis of ACD.
Treatment generally involves avoiding the offending agent with topical corticosteroids for symptom management.8
Polymorphous light eruption
Polymorphous light eruption (PLE) is a delayed, type IV hypersensitivity reaction to UV-induced antigens, though these antigens are unknown. PLE presents hours to days following solar or UV exposure and presents only in sun-exposed areas. Itching and burning are always present, but lesion morphology varies from erythema and papules to vesico-papules and blisters. Notably, PLE must be distinguished from drug photosensitivity through history. Treatment generally involves symptom management with topical steroids and sun protective measures for prevention.9 While PLE may present similarly to drug photosensitivity reactions, our patient’s use of a known phototoxic agent makes PLE a less likely diagnosis.
Ms. Appiah is a pediatric dermatology research associate and medical student at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Neither Dr. Matiz nor Ms. Appiah has any relevant financial disclosures.
References
1. Montgomery S et al. Clin Dermatol. 2022;40(1):57-63.
2. Blakely KM et al. Drug Saf. 2019;42(7):827-47.
3. Goetze S et al. Skin Pharmacol Physiol. 2017;30(2):76-80.
4. Odorici G et al. Dermatol Ther. 2021;34(4):e14978.
5. DeWane ME et al. J Am Acad Dermatol. 2020;82(2):267-81.
6. Waldman R et al. J Am Acad Dermatol. 2020;82(2):283-96.
7. Kiriakidou M et al. Ann Intern Med. 2020;172(11):ITC81-ITC96.
8. Nassau S et al. Med Clin North Am. 2020;104(1):61-76.
9. Guarrera M. Adv Exp Med Biol. 2017;996:61-70.
Photosensitivity due to doxycycline
As the patient’s rash presented in sun-exposed areas with both skin and nail changes, our patient was diagnosed with a phototoxic reaction to doxycycline, the oral antibiotic used to treat his acne.
Photosensitive cutaneous drug eruptions are reactions that occur after exposure to a medication and subsequent exposure to UV radiation or visible light. Reactions can be classified into two ways based on their mechanism of action: phototoxic or photoallergic.1 Phototoxic reactions are more common and are a result of direct keratinocyte damage and cellular necrosis. Many classes of medications may cause this adverse effect, but the tetracycline class of antibiotics is a common culprit.2 Photoallergic reactions are less common and are a result of a type IV immune reaction to the offending agent.1
Phototoxic reactions generally present shortly after sun or UV exposure with a photo-distributed eruption pattern.3 Commonly involved areas include the face, the neck, and the extensor surfaces of extremities, with sparing of relatively protected skin such as the upper eyelids and the skin folds.2 Erythema may initially develop in the exposed skin areas, followed by appearance of edema, vesicles, or bullae.1-3 The eruption may be painful and itchy, with some patients reporting severe pain.3
Doxycycline phototoxicity may also cause onycholysis of the nails.2 The reaction is dose dependent, with higher doses of medication leading to a higher likelihood of symptoms.1,2 It is also more prevalent in patients with Fitzpatrick skin type I and II. The usual UVA wavelength required to induce this reaction appears to be in the 320-400 nm range of the UV spectrum.4 By contrast, photoallergic reactions are dose independent, and require a sensitization period prior to the eruption.1 An eczematous eruption is most commonly seen with photoallergic reactions.3
Treatment of drug-induced photosensitivity reactions requires proper identification of the diagnosis and the offending agent, followed by cessation of the medication. If cessation is not possible, then lowering the dose can help to minimize worsening of the condition. However, for photoallergic reactions, the reaction is dose independent so switching to another tolerated agent is likely required. For persistent symptoms following medication withdrawal, topical or systemic steroids and oral antihistamine can help with symptom management.1 For patients with photo-onycholysis, treatment involves stopping the medication and waiting for the intact nail plate to grow.
Prevention is key in the management of photosensitivity reactions. Patients should be counseled about the increased risk of photosensitivity while on tetracycline medications and encouraged to engage in enhanced sun protection measures such as wearing sun protective hats and clothing, increasing use of sunscreen that provides mainly UVA but also UVB protection, and avoiding the sun during the midday when the UV index is highest.1-3
Dermatomyositis
Dermatomyositis is an autoimmune condition that presents with skin lesions as well as systemic findings such as myositis. The cutaneous findings are variable, but pathognomonic findings include Gottron papules of the hands, Gottron’s sign on the elbows, knees, and ankles, and the heliotrope rash of the face. Eighty percent of patients have myopathy presenting as muscle weakness, and commonly have elevated creatine kinase, aspartate transaminase, and alanine transaminase values.5 Diagnosis may be confirmed through skin or muscle biopsy, though antibody studies can also play a helpful role in diagnosis. Treatment is generally with oral corticosteroids or other immunosuppressants as well as sun protection.6 The rash seen in our patient could have been seen in patients with dermatomyositis, though it was not in the typical location on the knuckles (Gottron papules) as it also affected the lateral sides of the fingers.
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune condition characterized by systemic and cutaneous manifestations. Systemic symptoms may include weight loss, fever, fatigue, arthralgia, or arthritis; patients are at risk of renal, cardiovascular, pulmonary, and neurologic complications of SLE.7 The most common cutaneous finding is malar rash, though there are myriad dermatologic manifestations that can occur associated with photosensitivity. Diagnosis is made based on history, physical, and laboratory testing. Treatment options include NSAIDs, oral glucocorticoids, antimalarial drugs, and immunosuppressants.7 Though our patient exhibited photosensitivity, he had none of the systemic findings associated with SLE, making this diagnosis unlikely.
Allergic contact dermatitis
Allergic contact dermatitis (ACD) is a type IV hypersensitivity reaction, and may present as acute, subacute, or chronic dermatitis. The clinical findings vary based on chronicity. Acute ACD presents as pruritic erythematous papules and vesicles or bullae, similar to how it occurred in our patient, though our patient’s lesions were more tender than pruritic. Chronic ACD presents with erythematous lesions with pruritis, lichenification, scaling, and/or fissuring. Observing shapes or sharp demarcation of lesions may help with diagnosis. Patch testing is also useful in the diagnosis of ACD.
Treatment generally involves avoiding the offending agent with topical corticosteroids for symptom management.8
Polymorphous light eruption
Polymorphous light eruption (PLE) is a delayed, type IV hypersensitivity reaction to UV-induced antigens, though these antigens are unknown. PLE presents hours to days following solar or UV exposure and presents only in sun-exposed areas. Itching and burning are always present, but lesion morphology varies from erythema and papules to vesico-papules and blisters. Notably, PLE must be distinguished from drug photosensitivity through history. Treatment generally involves symptom management with topical steroids and sun protective measures for prevention.9 While PLE may present similarly to drug photosensitivity reactions, our patient’s use of a known phototoxic agent makes PLE a less likely diagnosis.
Ms. Appiah is a pediatric dermatology research associate and medical student at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Neither Dr. Matiz nor Ms. Appiah has any relevant financial disclosures.
References
1. Montgomery S et al. Clin Dermatol. 2022;40(1):57-63.
2. Blakely KM et al. Drug Saf. 2019;42(7):827-47.
3. Goetze S et al. Skin Pharmacol Physiol. 2017;30(2):76-80.
4. Odorici G et al. Dermatol Ther. 2021;34(4):e14978.
5. DeWane ME et al. J Am Acad Dermatol. 2020;82(2):267-81.
6. Waldman R et al. J Am Acad Dermatol. 2020;82(2):283-96.
7. Kiriakidou M et al. Ann Intern Med. 2020;172(11):ITC81-ITC96.
8. Nassau S et al. Med Clin North Am. 2020;104(1):61-76.
9. Guarrera M. Adv Exp Med Biol. 2017;996:61-70.
He reported no hiking or gardening, no new topical products such as new sunscreens or lotions, and no new medications. The patient had a history of acne, for which he used over-the-counter benzoyl peroxide wash, adapalene gel, and an oral antibiotic for 3 months. His review of systems was negative for fevers, chills, muscle weakness, mouth sores, or joint pain and no prior rashes following sun exposure.
On physical exam he presented with pink plaques with thin vesicles on the dorsum of the hands that were more noticeable on the lateral aspect of both the first and second fingers (Figures 1 and 2). His nails also had a yellow discoloration.
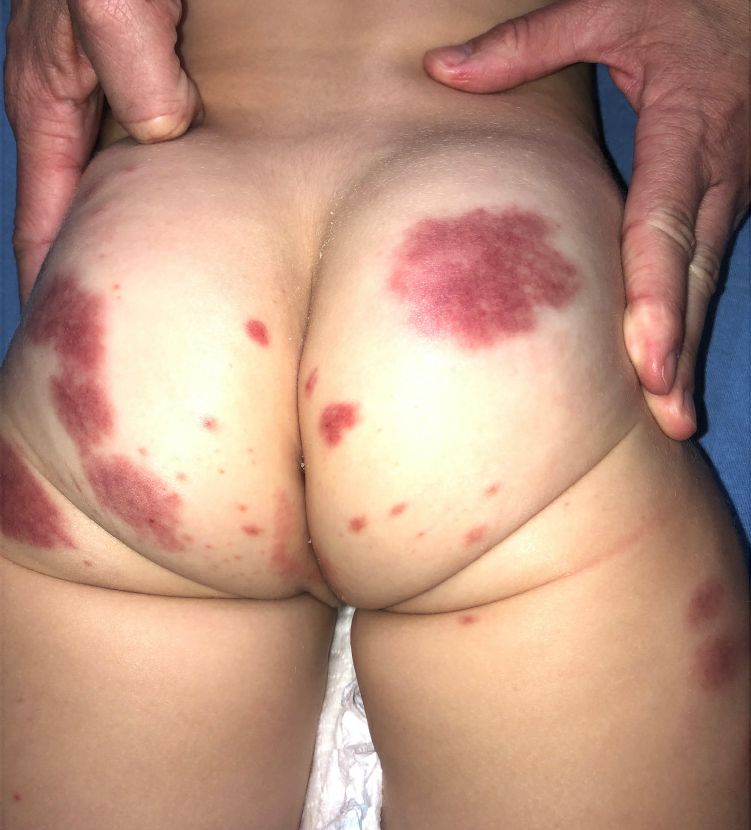
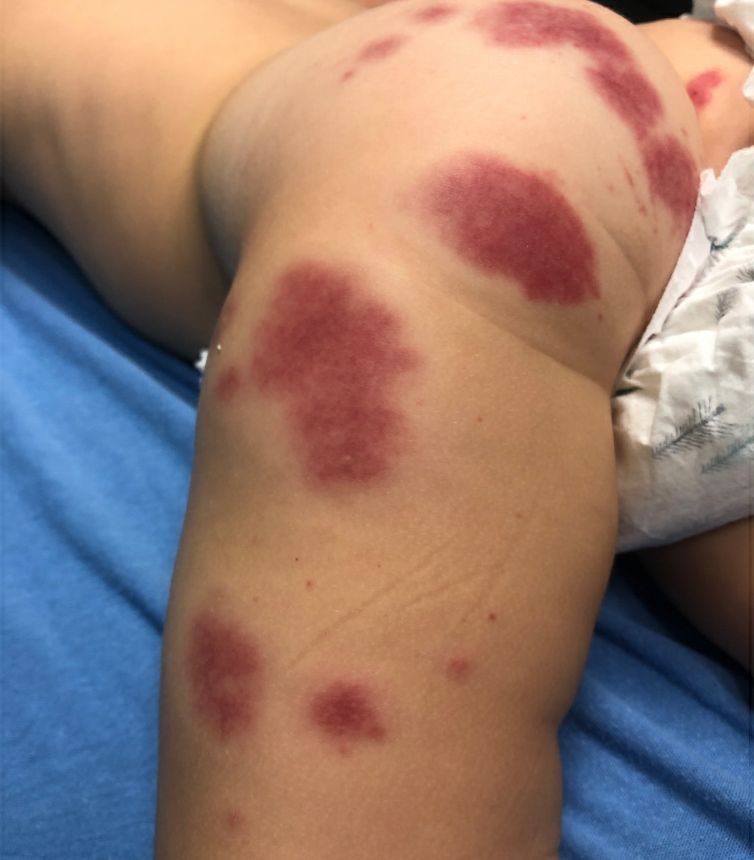
A 19-month-old vaccinated female with 2 days of rash
Acute hemorrhagic edema of infancy (AHEI) is a leukocytoclastic vasculitis that typically affects children between 4 months and 2 years of age.1 Etiology is unknown but the majority of cases are preceded by infections, vaccinations, or certain medications.2
AHEI is a self-limited disease that runs a benign course with spontaneous resolution within days to 3 weeks.3 Classic presentation involves acute onset of fever, purpura, ecchymosis, and inflammatory edema. Edema is often the first sign, and may involve the face, ears, scrotum, or extremities. Hemorrhagic lesions may vary in size but often coalesce and present in a distinctive “cockade” or rosette pattern with scalloped borders. Systemic manifestations are rare, but renal and joint involvement may occur.4 Despite the dramatic and sometimes extensive appearance of the dermatologic manifestations, patients with AHEI are usually not in significant distress.
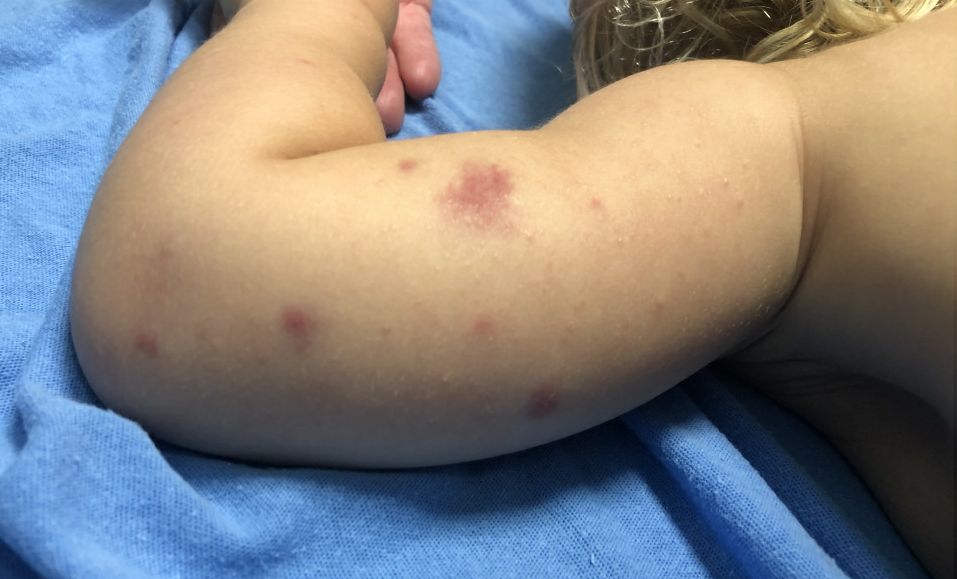
Diagnosis is clinical, but skin biopsy may show leukocytoclastic vasculitis of the superficial small vessels with infiltrations of neutrophils, extravasation of red blood cells, and fibrinoid necrosis.5 In most cases, immunofluorescence is negative for perivascular IgA deposition. Treatment is symptomatic as the disease resolves spontaneously. Recurrence is uncommon but may occur, and usually occurs early.
What is on the differential?
Kawasaki disease. Similar to AHEI, patients with Kawasaki disease also may present with facial and extremity edema. However, patients with Kawasaki disease appear sicker, have associated lymphadenopathy, conjunctivitis, and fever longer than 5 days. The lack of elevated inflammatory markers, acute-onset, classic dermatologic lesions, and nontoxic appearance in our patient rule out Kawasaki disease and make AHEI more likely.

IgA vasculitis/Henoch-Schönlein purpura. The distinction between AHEI and Henoch-Schönlein purpura is among the most challenging. AHEI commonly afflicts younger children ranging from 4 months to 2 years, whereas Henoch-Schönlein purpura occurs in older children from 3 to 6 years of age. Visceral involvement is rare in AHEI, but frequently presents in Henoch-Schönlein purpura with gastrointestinal and renal complications. Although our patient had both mild renal involvement and a distribution primarily on the buttocks and lower limbs, similar to the classic distribution of Henoch-Schönlein purpura, the younger age and lack of gastrointestinal and arthritic manifestations make AHEI more likely.
Gianotti-Crosti syndrome. Gianotti-Crosti syndrome, also known as papulovesicular acrodermatitis of childhood, mainly affects children between the ages of 6 months and 12 years. Like AHEI, Gianotti-Crosti is a self-limiting condition likely triggered by viral infection or immunization. However, Gianotti-Crosti is characterized by a papular rash that may last for several weeks. Neither AHEI nor Gianotti-Crosti are pruritic, but patients with Gianotti-Crosti tend to have either inguinal or axillary lymphadenopathy. Our patient’s large, coalescing dusky red patches and edematous plaques without lymphadenopathy are more consistent with AHEI.
Erythema multiforme. Erythema multiforme is an acute, immune-mediated condition characterized by distinctive target-like lesions on the skin often accompanied by erosions or bullae. Unlike AHEI, erythema multiforme can involve the oral, genital, and/or ocular mucosae. Erythema multiforme is rare before the age of 4 years. Although the targetoid or annular purpuric configuration of erythema multiforme may present similarly to AHEI in some cases, the young age of our patient and the lack of mucosal involvement make AHEI more likely.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Ms. Kleinman is a pediatric dermatology research associate at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Dr. Matiz nor Ms. Kleinman has any relevant financial disclosures.
References
1. Savino F et al. Pediatr Dermatol. 2013;30(6):e149-e152.
2. Carboni E et al. F1000Res. 2019;8:1771. 2019 Oct 17.
3. Fiore E et al. J Am Acad Dermatol. 2008;59(4):684-95.
4. Watanabe T and Sato Y. Pediatr Nephrol. 2007;22(11):1979-81.
5. Cunha DF et al. Autops Case Rep. 2015;5(3):37-41.
Acute hemorrhagic edema of infancy (AHEI) is a leukocytoclastic vasculitis that typically affects children between 4 months and 2 years of age.1 Etiology is unknown but the majority of cases are preceded by infections, vaccinations, or certain medications.2
AHEI is a self-limited disease that runs a benign course with spontaneous resolution within days to 3 weeks.3 Classic presentation involves acute onset of fever, purpura, ecchymosis, and inflammatory edema. Edema is often the first sign, and may involve the face, ears, scrotum, or extremities. Hemorrhagic lesions may vary in size but often coalesce and present in a distinctive “cockade” or rosette pattern with scalloped borders. Systemic manifestations are rare, but renal and joint involvement may occur.4 Despite the dramatic and sometimes extensive appearance of the dermatologic manifestations, patients with AHEI are usually not in significant distress.
Diagnosis is clinical, but skin biopsy may show leukocytoclastic vasculitis of the superficial small vessels with infiltrations of neutrophils, extravasation of red blood cells, and fibrinoid necrosis.5 In most cases, immunofluorescence is negative for perivascular IgA deposition. Treatment is symptomatic as the disease resolves spontaneously. Recurrence is uncommon but may occur, and usually occurs early.
What is on the differential?
Kawasaki disease. Similar to AHEI, patients with Kawasaki disease also may present with facial and extremity edema. However, patients with Kawasaki disease appear sicker, have associated lymphadenopathy, conjunctivitis, and fever longer than 5 days. The lack of elevated inflammatory markers, acute-onset, classic dermatologic lesions, and nontoxic appearance in our patient rule out Kawasaki disease and make AHEI more likely.
IgA vasculitis/Henoch-Schönlein purpura. The distinction between AHEI and Henoch-Schönlein purpura is among the most challenging. AHEI commonly afflicts younger children ranging from 4 months to 2 years, whereas Henoch-Schönlein purpura occurs in older children from 3 to 6 years of age. Visceral involvement is rare in AHEI, but frequently presents in Henoch-Schönlein purpura with gastrointestinal and renal complications. Although our patient had both mild renal involvement and a distribution primarily on the buttocks and lower limbs, similar to the classic distribution of Henoch-Schönlein purpura, the younger age and lack of gastrointestinal and arthritic manifestations make AHEI more likely.
Gianotti-Crosti syndrome. Gianotti-Crosti syndrome, also known as papulovesicular acrodermatitis of childhood, mainly affects children between the ages of 6 months and 12 years. Like AHEI, Gianotti-Crosti is a self-limiting condition likely triggered by viral infection or immunization. However, Gianotti-Crosti is characterized by a papular rash that may last for several weeks. Neither AHEI nor Gianotti-Crosti are pruritic, but patients with Gianotti-Crosti tend to have either inguinal or axillary lymphadenopathy. Our patient’s large, coalescing dusky red patches and edematous plaques without lymphadenopathy are more consistent with AHEI.
Erythema multiforme. Erythema multiforme is an acute, immune-mediated condition characterized by distinctive target-like lesions on the skin often accompanied by erosions or bullae. Unlike AHEI, erythema multiforme can involve the oral, genital, and/or ocular mucosae. Erythema multiforme is rare before the age of 4 years. Although the targetoid or annular purpuric configuration of erythema multiforme may present similarly to AHEI in some cases, the young age of our patient and the lack of mucosal involvement make AHEI more likely.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Ms. Kleinman is a pediatric dermatology research associate at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Dr. Matiz nor Ms. Kleinman has any relevant financial disclosures.
References
1. Savino F et al. Pediatr Dermatol. 2013;30(6):e149-e152.
2. Carboni E et al. F1000Res. 2019;8:1771. 2019 Oct 17.
3. Fiore E et al. J Am Acad Dermatol. 2008;59(4):684-95.
4. Watanabe T and Sato Y. Pediatr Nephrol. 2007;22(11):1979-81.
5. Cunha DF et al. Autops Case Rep. 2015;5(3):37-41.
Acute hemorrhagic edema of infancy (AHEI) is a leukocytoclastic vasculitis that typically affects children between 4 months and 2 years of age.1 Etiology is unknown but the majority of cases are preceded by infections, vaccinations, or certain medications.2
AHEI is a self-limited disease that runs a benign course with spontaneous resolution within days to 3 weeks.3 Classic presentation involves acute onset of fever, purpura, ecchymosis, and inflammatory edema. Edema is often the first sign, and may involve the face, ears, scrotum, or extremities. Hemorrhagic lesions may vary in size but often coalesce and present in a distinctive “cockade” or rosette pattern with scalloped borders. Systemic manifestations are rare, but renal and joint involvement may occur.4 Despite the dramatic and sometimes extensive appearance of the dermatologic manifestations, patients with AHEI are usually not in significant distress.
Diagnosis is clinical, but skin biopsy may show leukocytoclastic vasculitis of the superficial small vessels with infiltrations of neutrophils, extravasation of red blood cells, and fibrinoid necrosis.5 In most cases, immunofluorescence is negative for perivascular IgA deposition. Treatment is symptomatic as the disease resolves spontaneously. Recurrence is uncommon but may occur, and usually occurs early.
What is on the differential?
Kawasaki disease. Similar to AHEI, patients with Kawasaki disease also may present with facial and extremity edema. However, patients with Kawasaki disease appear sicker, have associated lymphadenopathy, conjunctivitis, and fever longer than 5 days. The lack of elevated inflammatory markers, acute-onset, classic dermatologic lesions, and nontoxic appearance in our patient rule out Kawasaki disease and make AHEI more likely.
IgA vasculitis/Henoch-Schönlein purpura. The distinction between AHEI and Henoch-Schönlein purpura is among the most challenging. AHEI commonly afflicts younger children ranging from 4 months to 2 years, whereas Henoch-Schönlein purpura occurs in older children from 3 to 6 years of age. Visceral involvement is rare in AHEI, but frequently presents in Henoch-Schönlein purpura with gastrointestinal and renal complications. Although our patient had both mild renal involvement and a distribution primarily on the buttocks and lower limbs, similar to the classic distribution of Henoch-Schönlein purpura, the younger age and lack of gastrointestinal and arthritic manifestations make AHEI more likely.
Gianotti-Crosti syndrome. Gianotti-Crosti syndrome, also known as papulovesicular acrodermatitis of childhood, mainly affects children between the ages of 6 months and 12 years. Like AHEI, Gianotti-Crosti is a self-limiting condition likely triggered by viral infection or immunization. However, Gianotti-Crosti is characterized by a papular rash that may last for several weeks. Neither AHEI nor Gianotti-Crosti are pruritic, but patients with Gianotti-Crosti tend to have either inguinal or axillary lymphadenopathy. Our patient’s large, coalescing dusky red patches and edematous plaques without lymphadenopathy are more consistent with AHEI.
Erythema multiforme. Erythema multiforme is an acute, immune-mediated condition characterized by distinctive target-like lesions on the skin often accompanied by erosions or bullae. Unlike AHEI, erythema multiforme can involve the oral, genital, and/or ocular mucosae. Erythema multiforme is rare before the age of 4 years. Although the targetoid or annular purpuric configuration of erythema multiforme may present similarly to AHEI in some cases, the young age of our patient and the lack of mucosal involvement make AHEI more likely.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Ms. Kleinman is a pediatric dermatology research associate at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Dr. Matiz nor Ms. Kleinman has any relevant financial disclosures.
References
1. Savino F et al. Pediatr Dermatol. 2013;30(6):e149-e152.
2. Carboni E et al. F1000Res. 2019;8:1771. 2019 Oct 17.
3. Fiore E et al. J Am Acad Dermatol. 2008;59(4):684-95.
4. Watanabe T and Sato Y. Pediatr Nephrol. 2007;22(11):1979-81.
5. Cunha DF et al. Autops Case Rep. 2015;5(3):37-41.
What is the diagnosis?
As the lesion was growing, getting more violaceous and indurated, a biopsy was performed. The biopsy showed multiple discrete lobules of dermal capillaries with slight extension into the superficial subcutis. Capillary lobules demonstrate the “cannonball-like” architecture often associated with tufted angioma, and some lobules showed bulging into adjacent thin-walled vessels. Spindled endothelial cells lining slit-like vessels were present in the mid dermis, although this comprises a minority of the lesion. The majority of the subcutis was uninvolved. The findings are overall most consistent with a tufted angioma.
Kaposiform hemangioendothelioma (KHE) has been considered given the presence of occasional slit-like vascular spaces; however, the lesion is predominantly superficial and therefore the lesion is best classified as tufted angioma. GLUT–1 staining was negative.
At the time of biopsy, blood work was ordered, which showed a normal complete blood count with normal number of platelets, slightly elevated D-dimer, and slightly low fibrinogen. Several repeat blood counts and coagulation tests once a week for a few weeks revealed no changes.
The patient was started on aspirin at a dose of 5 mg/kg per day. After a week on the medication the lesion was starting to get smaller and less red.
Tufted angiomas are a rare type of vascular tumor within the spectrum of kaposiform hemangioendotheliomas. Most cases present within the first year of life; some occur at birth. They usually present as papules, plaques, or erythematous, violaceous indurated nodules on the face, neck, trunk, and extremities. The lesions can also be present with hyperhidrosis and hypertrichosis. Clinically, the lesions will have to be differentiated from other vascular tumors such as infantile hemangiomas, congenital hemangiomas, and Kaposi’s sarcoma, as well as subcutaneous fat necrosis of the newborn, cellulitis, and nonaccidental trauma.
Pathogenesis of tufted angiomas is poorly understood. A recent case report found a somatic mutation on GNA14.This protein regulates Ras activity and modulates endothelial cell permeability and migration in response to FGF2 and VEGFA. The p.205L mutation causes activation of GNA14, which upregulates pERK-MAPK pathway, suggesting MAPK inhibition as a potential target for therapy. Clinically, tufted angioma can present in three patterns: uncomplicated tufted angioma (most common type); tufted angioma without thrombocytopenia but with chronic coagulopathy, as it was seen in our patient; and tufted angioma associated with Kasabach-Merritt phenomenon (KMP). KMP is characterized by thrombocytopenia in association with microangiopathic hemolytic anemia, consumptive coagulopathy, and enlarging vascular tumor. Treatment of uncomplicated tufted angioma will depend on symptomatology, size, and location of the lesion. Smaller lesions in noncosmetically sensitive areas can be treated with surgical excision. Cases that are not amenable to excision can be treated with aspirin. There are also reports of response to topical modalities including tacrolimus and timolol. For complicated cases associated with KMP, sirolimus, systemic corticosteroids, ticlopidine, interferon, or vincristine are recommended. Some lesions may demonstrate spontaneous regression.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Cohen S et al. Dermatol Online J. 2019 Sep 15;25(9):13030/qt6pv254mc.
Lim YH et al. Pediatr Dermatol. 2019 Nov;36(6):963-4.
Prasuna A, Rao PN. Indian Dermatol Online J. 2015;6:266-8.
As the lesion was growing, getting more violaceous and indurated, a biopsy was performed. The biopsy showed multiple discrete lobules of dermal capillaries with slight extension into the superficial subcutis. Capillary lobules demonstrate the “cannonball-like” architecture often associated with tufted angioma, and some lobules showed bulging into adjacent thin-walled vessels. Spindled endothelial cells lining slit-like vessels were present in the mid dermis, although this comprises a minority of the lesion. The majority of the subcutis was uninvolved. The findings are overall most consistent with a tufted angioma.
Kaposiform hemangioendothelioma (KHE) has been considered given the presence of occasional slit-like vascular spaces; however, the lesion is predominantly superficial and therefore the lesion is best classified as tufted angioma. GLUT–1 staining was negative.
At the time of biopsy, blood work was ordered, which showed a normal complete blood count with normal number of platelets, slightly elevated D-dimer, and slightly low fibrinogen. Several repeat blood counts and coagulation tests once a week for a few weeks revealed no changes.
The patient was started on aspirin at a dose of 5 mg/kg per day. After a week on the medication the lesion was starting to get smaller and less red.
Tufted angiomas are a rare type of vascular tumor within the spectrum of kaposiform hemangioendotheliomas. Most cases present within the first year of life; some occur at birth. They usually present as papules, plaques, or erythematous, violaceous indurated nodules on the face, neck, trunk, and extremities. The lesions can also be present with hyperhidrosis and hypertrichosis. Clinically, the lesions will have to be differentiated from other vascular tumors such as infantile hemangiomas, congenital hemangiomas, and Kaposi’s sarcoma, as well as subcutaneous fat necrosis of the newborn, cellulitis, and nonaccidental trauma.
Pathogenesis of tufted angiomas is poorly understood. A recent case report found a somatic mutation on GNA14.This protein regulates Ras activity and modulates endothelial cell permeability and migration in response to FGF2 and VEGFA. The p.205L mutation causes activation of GNA14, which upregulates pERK-MAPK pathway, suggesting MAPK inhibition as a potential target for therapy. Clinically, tufted angioma can present in three patterns: uncomplicated tufted angioma (most common type); tufted angioma without thrombocytopenia but with chronic coagulopathy, as it was seen in our patient; and tufted angioma associated with Kasabach-Merritt phenomenon (KMP). KMP is characterized by thrombocytopenia in association with microangiopathic hemolytic anemia, consumptive coagulopathy, and enlarging vascular tumor. Treatment of uncomplicated tufted angioma will depend on symptomatology, size, and location of the lesion. Smaller lesions in noncosmetically sensitive areas can be treated with surgical excision. Cases that are not amenable to excision can be treated with aspirin. There are also reports of response to topical modalities including tacrolimus and timolol. For complicated cases associated with KMP, sirolimus, systemic corticosteroids, ticlopidine, interferon, or vincristine are recommended. Some lesions may demonstrate spontaneous regression.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Cohen S et al. Dermatol Online J. 2019 Sep 15;25(9):13030/qt6pv254mc.
Lim YH et al. Pediatr Dermatol. 2019 Nov;36(6):963-4.
Prasuna A, Rao PN. Indian Dermatol Online J. 2015;6:266-8.
As the lesion was growing, getting more violaceous and indurated, a biopsy was performed. The biopsy showed multiple discrete lobules of dermal capillaries with slight extension into the superficial subcutis. Capillary lobules demonstrate the “cannonball-like” architecture often associated with tufted angioma, and some lobules showed bulging into adjacent thin-walled vessels. Spindled endothelial cells lining slit-like vessels were present in the mid dermis, although this comprises a minority of the lesion. The majority of the subcutis was uninvolved. The findings are overall most consistent with a tufted angioma.
Kaposiform hemangioendothelioma (KHE) has been considered given the presence of occasional slit-like vascular spaces; however, the lesion is predominantly superficial and therefore the lesion is best classified as tufted angioma. GLUT–1 staining was negative.
At the time of biopsy, blood work was ordered, which showed a normal complete blood count with normal number of platelets, slightly elevated D-dimer, and slightly low fibrinogen. Several repeat blood counts and coagulation tests once a week for a few weeks revealed no changes.
The patient was started on aspirin at a dose of 5 mg/kg per day. After a week on the medication the lesion was starting to get smaller and less red.
Tufted angiomas are a rare type of vascular tumor within the spectrum of kaposiform hemangioendotheliomas. Most cases present within the first year of life; some occur at birth. They usually present as papules, plaques, or erythematous, violaceous indurated nodules on the face, neck, trunk, and extremities. The lesions can also be present with hyperhidrosis and hypertrichosis. Clinically, the lesions will have to be differentiated from other vascular tumors such as infantile hemangiomas, congenital hemangiomas, and Kaposi’s sarcoma, as well as subcutaneous fat necrosis of the newborn, cellulitis, and nonaccidental trauma.
Pathogenesis of tufted angiomas is poorly understood. A recent case report found a somatic mutation on GNA14.This protein regulates Ras activity and modulates endothelial cell permeability and migration in response to FGF2 and VEGFA. The p.205L mutation causes activation of GNA14, which upregulates pERK-MAPK pathway, suggesting MAPK inhibition as a potential target for therapy. Clinically, tufted angioma can present in three patterns: uncomplicated tufted angioma (most common type); tufted angioma without thrombocytopenia but with chronic coagulopathy, as it was seen in our patient; and tufted angioma associated with Kasabach-Merritt phenomenon (KMP). KMP is characterized by thrombocytopenia in association with microangiopathic hemolytic anemia, consumptive coagulopathy, and enlarging vascular tumor. Treatment of uncomplicated tufted angioma will depend on symptomatology, size, and location of the lesion. Smaller lesions in noncosmetically sensitive areas can be treated with surgical excision. Cases that are not amenable to excision can be treated with aspirin. There are also reports of response to topical modalities including tacrolimus and timolol. For complicated cases associated with KMP, sirolimus, systemic corticosteroids, ticlopidine, interferon, or vincristine are recommended. Some lesions may demonstrate spontaneous regression.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
Cohen S et al. Dermatol Online J. 2019 Sep 15;25(9):13030/qt6pv254mc.
Lim YH et al. Pediatr Dermatol. 2019 Nov;36(6):963-4.
Prasuna A, Rao PN. Indian Dermatol Online J. 2015;6:266-8.
A 35-day-old female was referred to our pediatric dermatology clinic for evaluation of a red lesion on the right arm. The lesion presented at about 4 days of life as a red plaque (image 1 at 8 days of life).
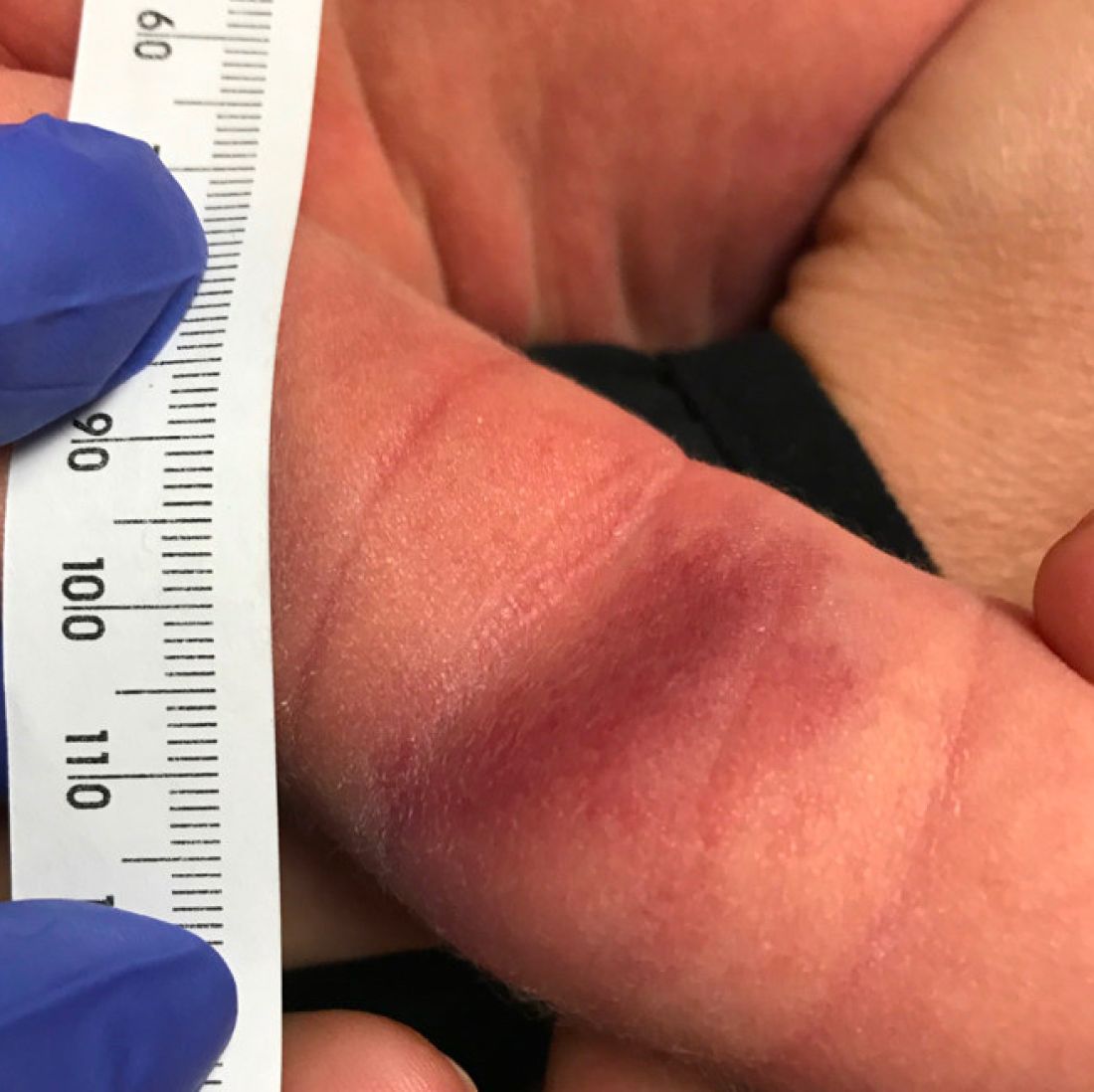
On the following days, the lesion started growing but it didn't seem to be tender or bothersome to the patient (image 2, at 35 days of life).
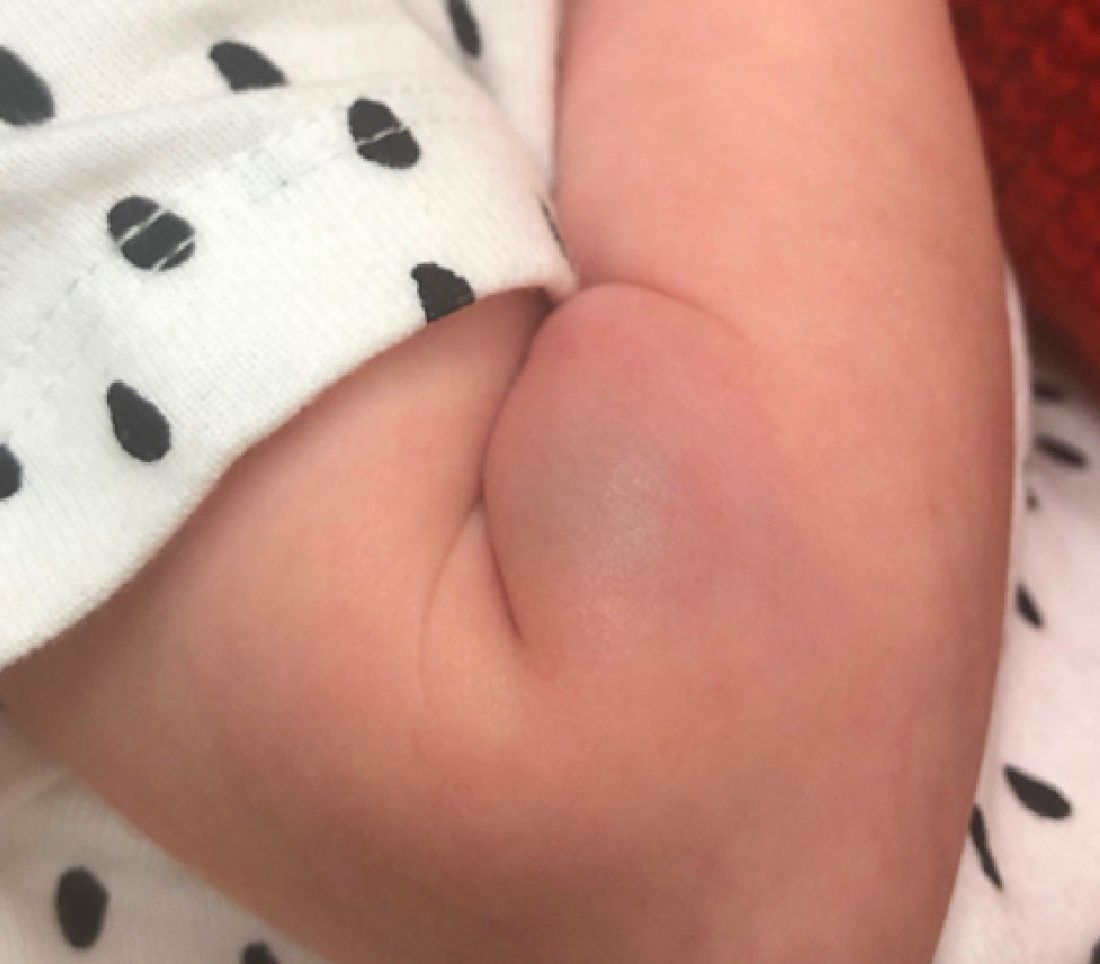
At a 2-week follow up the lesion was getting fuller and more violaceous. There was no history of fever and the lesion didn't appear tender to the touch.
She was born via normal spontaneous vaginal delivery. There were no complications and the mother received prenatal care.
On exam she had a red to violaceous nodule on the right arm (image 3 at 45 days of life).
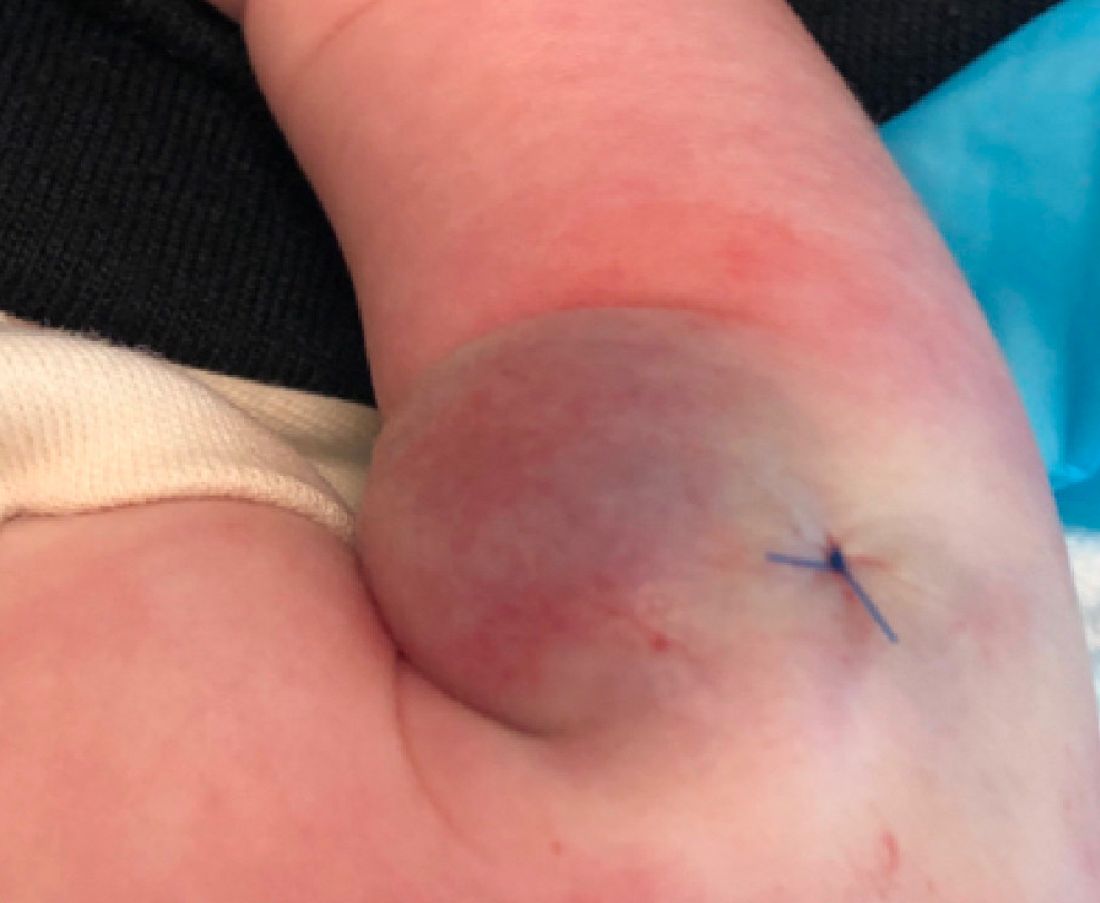
Teen boy’s knee lesion has changed
A biopsy of the lesion was performed which showed an increased number of eccrine glands and blood vessels within the dermis. Some areas showed an increase in adipocytes and smooth muscle bundles. The changes were consistent with eccrine angiomatous hamartoma (EAH).

The boy was referred to vascular laser therapy for treatment of the lesion.
EAH is a rare benign vascular growth characterized by an increased number of mature eccrine glands and blood vessels in the dermis and subcutis. The lesions are mostly present on the extremities, but cases of diffuse congenital lesions and lesions on the face and trunk have also been described. The lesions can be seen at birth or during the first years of life in about half of the cases, and the others tend to occur later in puberty and rarely in adulthood.1
Clinically, EAH lesions present as red, yellow to brown papules and plaques. Different dermoscopic patterns have been described which include the popcorn pattern that presents as yellow, confluent nodules with popcornlike shapes over a background of erythema, and linear arborizing vessels. The spitzoid pattern are brown globules on a background of erythema and pseudoreticular pigmentation around the globules. The verrucous hemangiomalike pattern has a bluish-white hue, reddish-blue or bluish lacunae, as seen in our patient.2-4
Most of the lesions are asymptomatic, but in some patients, they can be associated with pain, hyperhidrosis, and sometimes bleeding. Hyperhidrosis has been reported early in the presentation or during puberty or pregnancy. Our patient had started on amphetamines when hyperhidrosis occurred. Hyperhidrosis is a knowns side effect of this type of medication and may have had a role in the increased sweating noted on the hamartoma.
EAH can clinically look like verrucous hemangiomas, angiokeratomas, and vascular malformations, and histopathology may be needed to differentiate between them. Eccrine nevi and EAH can be similar. Hyperhidrosis is an early and predominant component of eccrine nevi, compared with one-third of EAH.
The exact etiology of this lesion is not known. It is thought to be caused by an abnormal differentiation of the epithelium, adnexal structure, and the mesenchyme during organogenesis.3 No other associated conditions have been described with EAH.
EAH are benign lesions that rarely require treatment. If the lesions are symptomatic or because of cosmetic reasons, they can be removed surgically. There are some reports of successful treatment with pulse dual-wavelength sequential 595- and 1064-nm lasers.5 Botulinum toxin has also been used in cases of symptomatic hyperhidrosis.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She has no conflicts. Email her at pdnews@mdedge.com.
References
1. Smith SD et al. Pediatr Dermatol. 2019 Nov;36(6):909-12.
2. Patterson AT et al. Am J Dermatopathol. 2016;38:413-7.
3. Garcıa-Garcıa SC et al. JAAD Case Rep. 2018;4(2):165-7.
4. Awatef Kelati et al. JAAD Case Rep. 2018;4(8)835-6.
5. Felgueiras J et al. Dermatol Surg. 2015 Mar;41(3):428-30.
A biopsy of the lesion was performed which showed an increased number of eccrine glands and blood vessels within the dermis. Some areas showed an increase in adipocytes and smooth muscle bundles. The changes were consistent with eccrine angiomatous hamartoma (EAH).
The boy was referred to vascular laser therapy for treatment of the lesion.
EAH is a rare benign vascular growth characterized by an increased number of mature eccrine glands and blood vessels in the dermis and subcutis. The lesions are mostly present on the extremities, but cases of diffuse congenital lesions and lesions on the face and trunk have also been described. The lesions can be seen at birth or during the first years of life in about half of the cases, and the others tend to occur later in puberty and rarely in adulthood.1
Clinically, EAH lesions present as red, yellow to brown papules and plaques. Different dermoscopic patterns have been described which include the popcorn pattern that presents as yellow, confluent nodules with popcornlike shapes over a background of erythema, and linear arborizing vessels. The spitzoid pattern are brown globules on a background of erythema and pseudoreticular pigmentation around the globules. The verrucous hemangiomalike pattern has a bluish-white hue, reddish-blue or bluish lacunae, as seen in our patient.2-4
Most of the lesions are asymptomatic, but in some patients, they can be associated with pain, hyperhidrosis, and sometimes bleeding. Hyperhidrosis has been reported early in the presentation or during puberty or pregnancy. Our patient had started on amphetamines when hyperhidrosis occurred. Hyperhidrosis is a knowns side effect of this type of medication and may have had a role in the increased sweating noted on the hamartoma.
EAH can clinically look like verrucous hemangiomas, angiokeratomas, and vascular malformations, and histopathology may be needed to differentiate between them. Eccrine nevi and EAH can be similar. Hyperhidrosis is an early and predominant component of eccrine nevi, compared with one-third of EAH.
The exact etiology of this lesion is not known. It is thought to be caused by an abnormal differentiation of the epithelium, adnexal structure, and the mesenchyme during organogenesis.3 No other associated conditions have been described with EAH.
EAH are benign lesions that rarely require treatment. If the lesions are symptomatic or because of cosmetic reasons, they can be removed surgically. There are some reports of successful treatment with pulse dual-wavelength sequential 595- and 1064-nm lasers.5 Botulinum toxin has also been used in cases of symptomatic hyperhidrosis.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She has no conflicts. Email her at pdnews@mdedge.com.
References
1. Smith SD et al. Pediatr Dermatol. 2019 Nov;36(6):909-12.
2. Patterson AT et al. Am J Dermatopathol. 2016;38:413-7.
3. Garcıa-Garcıa SC et al. JAAD Case Rep. 2018;4(2):165-7.
4. Awatef Kelati et al. JAAD Case Rep. 2018;4(8)835-6.
5. Felgueiras J et al. Dermatol Surg. 2015 Mar;41(3):428-30.
A biopsy of the lesion was performed which showed an increased number of eccrine glands and blood vessels within the dermis. Some areas showed an increase in adipocytes and smooth muscle bundles. The changes were consistent with eccrine angiomatous hamartoma (EAH).
The boy was referred to vascular laser therapy for treatment of the lesion.
EAH is a rare benign vascular growth characterized by an increased number of mature eccrine glands and blood vessels in the dermis and subcutis. The lesions are mostly present on the extremities, but cases of diffuse congenital lesions and lesions on the face and trunk have also been described. The lesions can be seen at birth or during the first years of life in about half of the cases, and the others tend to occur later in puberty and rarely in adulthood.1
Clinically, EAH lesions present as red, yellow to brown papules and plaques. Different dermoscopic patterns have been described which include the popcorn pattern that presents as yellow, confluent nodules with popcornlike shapes over a background of erythema, and linear arborizing vessels. The spitzoid pattern are brown globules on a background of erythema and pseudoreticular pigmentation around the globules. The verrucous hemangiomalike pattern has a bluish-white hue, reddish-blue or bluish lacunae, as seen in our patient.2-4
Most of the lesions are asymptomatic, but in some patients, they can be associated with pain, hyperhidrosis, and sometimes bleeding. Hyperhidrosis has been reported early in the presentation or during puberty or pregnancy. Our patient had started on amphetamines when hyperhidrosis occurred. Hyperhidrosis is a knowns side effect of this type of medication and may have had a role in the increased sweating noted on the hamartoma.
EAH can clinically look like verrucous hemangiomas, angiokeratomas, and vascular malformations, and histopathology may be needed to differentiate between them. Eccrine nevi and EAH can be similar. Hyperhidrosis is an early and predominant component of eccrine nevi, compared with one-third of EAH.
The exact etiology of this lesion is not known. It is thought to be caused by an abnormal differentiation of the epithelium, adnexal structure, and the mesenchyme during organogenesis.3 No other associated conditions have been described with EAH.
EAH are benign lesions that rarely require treatment. If the lesions are symptomatic or because of cosmetic reasons, they can be removed surgically. There are some reports of successful treatment with pulse dual-wavelength sequential 595- and 1064-nm lasers.5 Botulinum toxin has also been used in cases of symptomatic hyperhidrosis.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She has no conflicts. Email her at pdnews@mdedge.com.
References
1. Smith SD et al. Pediatr Dermatol. 2019 Nov;36(6):909-12.
2. Patterson AT et al. Am J Dermatopathol. 2016;38:413-7.
3. Garcıa-Garcıa SC et al. JAAD Case Rep. 2018;4(2):165-7.
4. Awatef Kelati et al. JAAD Case Rep. 2018;4(8)835-6.
5. Felgueiras J et al. Dermatol Surg. 2015 Mar;41(3):428-30.
A 14-year-old male was referred to our pediatric dermatology clinic for evaluation of a lesion on the left knee that appeared at 1 year of age. The lesion has been growing with him and was not symptomatic until 6 months prior to the consultation, when it started bleeding and feeling wet.
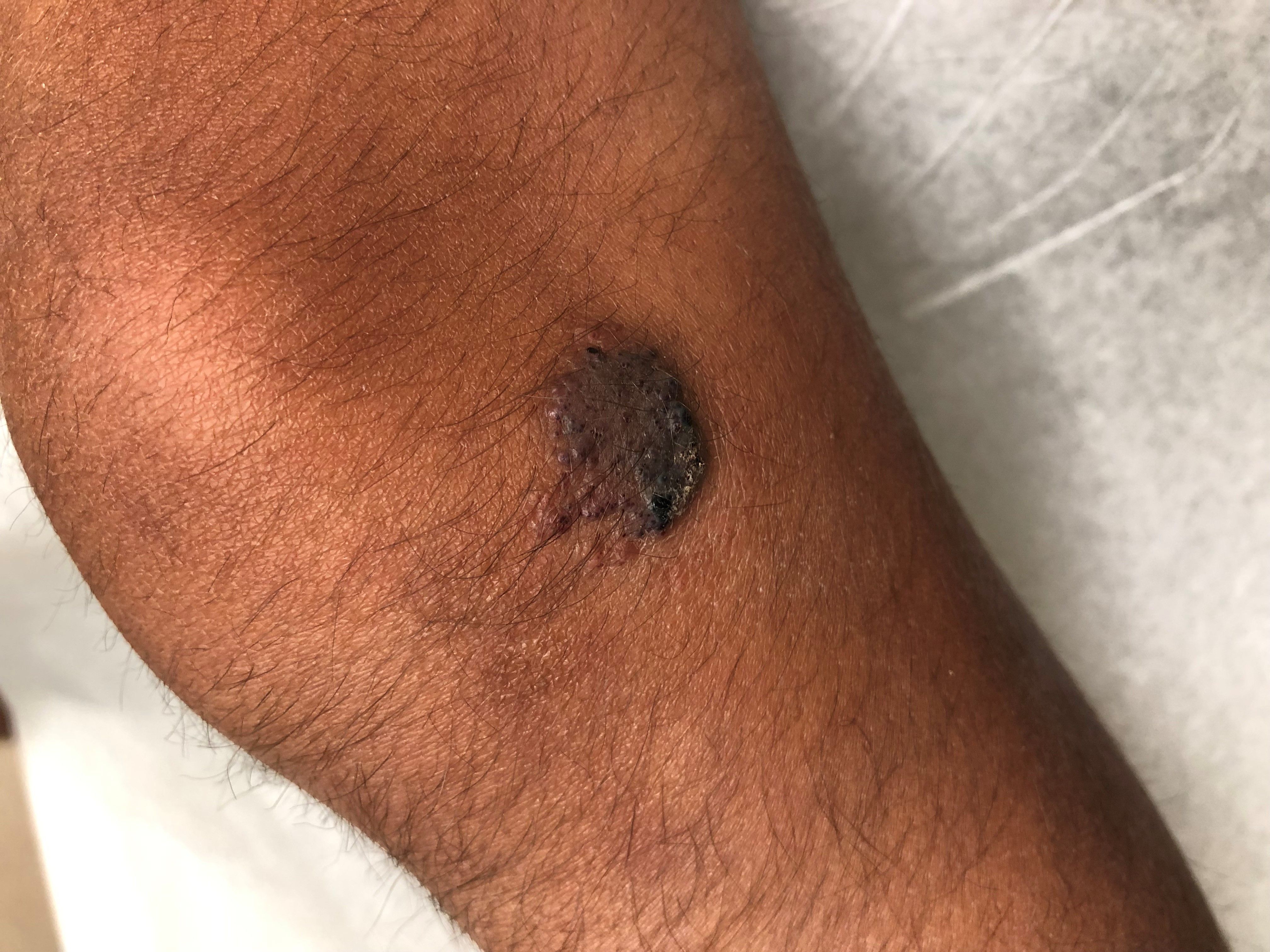
He has a history of attention-deficit/hyperactivity disorder managed with dextroamphetamine-amphetamine. The changes noted on the knee lesion seem to occur at the same time that his ADHD medication was started.
On physical exam he had a violaceous circular plaque on the left knee.
On dermoscopy the lesion showed multiple dilated red and violaceous lacunae and whitish blue hue.
What’s under my toenail?
After the teledermatology consultation, an x-ray was recommended. The x-ray showed an elongated irregular radiopaque mass projecting from the anterior medial aspect of the midshaft of the distal phalanx of the great toe (Picture 3). With these findings, subungual exostosis was suspected, and she was referred to orthopedic surgery for excision of the lesion. Histopathology showed a stack of trabecular bone with a fibrocartilaginous cap, confirming the diagnosis of subungual exostosis.

Subungual exostosis is a benign osteocartilaginous tumor, first described by Dupuytren in 1874. These lesions are rare and are seen mainly in children and young adults. Females appear to be affected more often than males.1 In a systematic review by DaCambra and colleagues, 55% of the cases occur in patients aged younger than 18 years, and the hallux was the most commonly affected digit, though any finger or toe can be affected.2 There are reported case of congenital multiple exostosis delineated to translocation t(X;6)(q22;q13-14).3
The exact cause of these lesions is unknown, but there are multiple theories, which include a reactive process secondary to trauma, infection, or genetic causes. Pathologic examination of the lesions shows an osseous center covered by a fibrocartilaginous cap. There is proliferation of spindle cells that generate cartilage, which later forms trabecular bone.4
On physical examination, subungual exostosis appear like a firm, fixed nodule with a hyperkeratotic smooth surface at the distal end of the nail bed, that slowly grows and can distort and lift up the nail. Dermoscopy features of these lesions include vascular ectasia, hyperkeratosis, onycholysis, and ulceration.
The differential diagnosis of subungual growths includes osteochondromas, which can present in a similar way but are rarer. Pathologic examination is usually required to differentiate between both lesions.5 In exostoses, bone is formed directly from fibrous tissue, whereas in osteochondromas they derive from enchondral ossification.6 The cartilaginous cap of this lesion is what helps to differentiate it in histopathology. In subungual exostosis, the cap is composed of fibrocartilage, while in osteochondromas it is made of hyaline cartilage similar to what is seen in normal growing epiphysis.5 Subungual exostosis can be confused with pyogenic granulomas and verruca, and often are treated as such, which delays appropriate surgical management.
Firm, slow-growing tumors in the fingers or toes of children should raise suspicion for underlying bony lesions like subungual exostosis and osteochondromas. X-rays of the lesion should be performed in order to clarify the diagnosis. Referral to orthopedic surgery is needed for definitive surgical management.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Zhang W et al. JAAD Case Rep. 2020 Jun 1;6(8):725-6.
2. DaCambra MP et al. Clin Orthop Relat Res. 2014 Apr;472(4):1251-9.
3. Torlazzi C et al. Int J Cancer. 2006;118:1972-6.
4. Calonje E et al. McKee’s pathology of the skin: With clinical correlations. (4th ed.) Philadelphia: Elsevier/Saunders, 2012.
5. Lee SK et al. Foot Ankle Int. 2007 May;28(5):595-601.
6. Mavrogenis A et al. Orthopedics. 2008 Oct;31(10).
After the teledermatology consultation, an x-ray was recommended. The x-ray showed an elongated irregular radiopaque mass projecting from the anterior medial aspect of the midshaft of the distal phalanx of the great toe (Picture 3). With these findings, subungual exostosis was suspected, and she was referred to orthopedic surgery for excision of the lesion. Histopathology showed a stack of trabecular bone with a fibrocartilaginous cap, confirming the diagnosis of subungual exostosis.
Subungual exostosis is a benign osteocartilaginous tumor, first described by Dupuytren in 1874. These lesions are rare and are seen mainly in children and young adults. Females appear to be affected more often than males.1 In a systematic review by DaCambra and colleagues, 55% of the cases occur in patients aged younger than 18 years, and the hallux was the most commonly affected digit, though any finger or toe can be affected.2 There are reported case of congenital multiple exostosis delineated to translocation t(X;6)(q22;q13-14).3
The exact cause of these lesions is unknown, but there are multiple theories, which include a reactive process secondary to trauma, infection, or genetic causes. Pathologic examination of the lesions shows an osseous center covered by a fibrocartilaginous cap. There is proliferation of spindle cells that generate cartilage, which later forms trabecular bone.4
On physical examination, subungual exostosis appear like a firm, fixed nodule with a hyperkeratotic smooth surface at the distal end of the nail bed, that slowly grows and can distort and lift up the nail. Dermoscopy features of these lesions include vascular ectasia, hyperkeratosis, onycholysis, and ulceration.
The differential diagnosis of subungual growths includes osteochondromas, which can present in a similar way but are rarer. Pathologic examination is usually required to differentiate between both lesions.5 In exostoses, bone is formed directly from fibrous tissue, whereas in osteochondromas they derive from enchondral ossification.6 The cartilaginous cap of this lesion is what helps to differentiate it in histopathology. In subungual exostosis, the cap is composed of fibrocartilage, while in osteochondromas it is made of hyaline cartilage similar to what is seen in normal growing epiphysis.5 Subungual exostosis can be confused with pyogenic granulomas and verruca, and often are treated as such, which delays appropriate surgical management.
Firm, slow-growing tumors in the fingers or toes of children should raise suspicion for underlying bony lesions like subungual exostosis and osteochondromas. X-rays of the lesion should be performed in order to clarify the diagnosis. Referral to orthopedic surgery is needed for definitive surgical management.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Zhang W et al. JAAD Case Rep. 2020 Jun 1;6(8):725-6.
2. DaCambra MP et al. Clin Orthop Relat Res. 2014 Apr;472(4):1251-9.
3. Torlazzi C et al. Int J Cancer. 2006;118:1972-6.
4. Calonje E et al. McKee’s pathology of the skin: With clinical correlations. (4th ed.) Philadelphia: Elsevier/Saunders, 2012.
5. Lee SK et al. Foot Ankle Int. 2007 May;28(5):595-601.
6. Mavrogenis A et al. Orthopedics. 2008 Oct;31(10).
After the teledermatology consultation, an x-ray was recommended. The x-ray showed an elongated irregular radiopaque mass projecting from the anterior medial aspect of the midshaft of the distal phalanx of the great toe (Picture 3). With these findings, subungual exostosis was suspected, and she was referred to orthopedic surgery for excision of the lesion. Histopathology showed a stack of trabecular bone with a fibrocartilaginous cap, confirming the diagnosis of subungual exostosis.
Subungual exostosis is a benign osteocartilaginous tumor, first described by Dupuytren in 1874. These lesions are rare and are seen mainly in children and young adults. Females appear to be affected more often than males.1 In a systematic review by DaCambra and colleagues, 55% of the cases occur in patients aged younger than 18 years, and the hallux was the most commonly affected digit, though any finger or toe can be affected.2 There are reported case of congenital multiple exostosis delineated to translocation t(X;6)(q22;q13-14).3
The exact cause of these lesions is unknown, but there are multiple theories, which include a reactive process secondary to trauma, infection, or genetic causes. Pathologic examination of the lesions shows an osseous center covered by a fibrocartilaginous cap. There is proliferation of spindle cells that generate cartilage, which later forms trabecular bone.4
On physical examination, subungual exostosis appear like a firm, fixed nodule with a hyperkeratotic smooth surface at the distal end of the nail bed, that slowly grows and can distort and lift up the nail. Dermoscopy features of these lesions include vascular ectasia, hyperkeratosis, onycholysis, and ulceration.
The differential diagnosis of subungual growths includes osteochondromas, which can present in a similar way but are rarer. Pathologic examination is usually required to differentiate between both lesions.5 In exostoses, bone is formed directly from fibrous tissue, whereas in osteochondromas they derive from enchondral ossification.6 The cartilaginous cap of this lesion is what helps to differentiate it in histopathology. In subungual exostosis, the cap is composed of fibrocartilage, while in osteochondromas it is made of hyaline cartilage similar to what is seen in normal growing epiphysis.5 Subungual exostosis can be confused with pyogenic granulomas and verruca, and often are treated as such, which delays appropriate surgical management.
Firm, slow-growing tumors in the fingers or toes of children should raise suspicion for underlying bony lesions like subungual exostosis and osteochondromas. X-rays of the lesion should be performed in order to clarify the diagnosis. Referral to orthopedic surgery is needed for definitive surgical management.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Zhang W et al. JAAD Case Rep. 2020 Jun 1;6(8):725-6.
2. DaCambra MP et al. Clin Orthop Relat Res. 2014 Apr;472(4):1251-9.
3. Torlazzi C et al. Int J Cancer. 2006;118:1972-6.
4. Calonje E et al. McKee’s pathology of the skin: With clinical correlations. (4th ed.) Philadelphia: Elsevier/Saunders, 2012.
5. Lee SK et al. Foot Ankle Int. 2007 May;28(5):595-601.
6. Mavrogenis A et al. Orthopedics. 2008 Oct;31(10).
A 13-year-old female was seen by her pediatrician for a lesion that had been on her right toe for about 6 months. She is unaware of any trauma to the area. The lesion has been growing slowly and recently it started lifting up the nail, became tender, and was bleeding, which is the reason why she sought care.
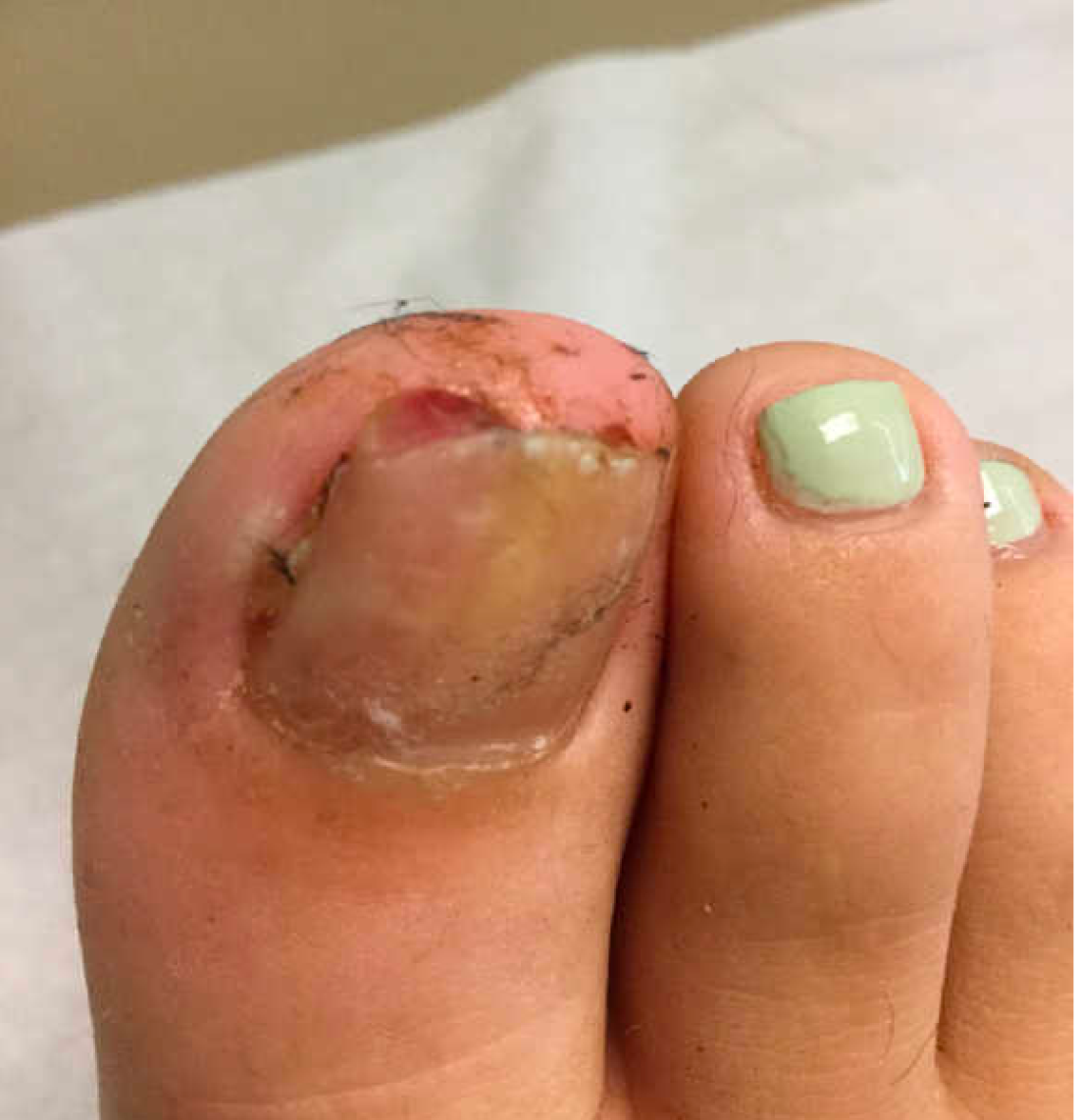
At the pediatrician's office, he noted a pink crusted papule under the nail. The nail was lifting up and was tender to the touch. She is a healthy girl who is not taking any medications and has no allergies. There is no family history of similar lesions.
The pediatrician took a picture of the lesion and he send it to our pediatric teledermatology service for consultation.
An otherwise healthy 1-month-old female presents with lesions on the face, scalp, and chest
A potassium hydroxide preparation (KOH) from skin scrapings from the scalp lesions demonstrated no fungal elements. Further laboratory work up revealed a normal blood cell count, normal liver enzymes, an antinuclear antibody (ANA) titer of less than 1:80, a positive anti–Sjögren’s syndrome type B (SSB) antibody but negative anti–Sjögren’s syndrome type A (SSA) antibody and anti-U1RNP antibody. An electrocardiogram revealed no abnormalities. Liver function tests were normal. The complete blood count showed mild thrombocytopenia. Given the typical skin lesions and the positive SSB test and associated thrombocytopenia, the baby was diagnosed with neonatal lupus erythematosus.
Because of the diagnosis of neonatal lupus the mother was also tested and was found to have an elevated ANA of 1:640, positive SSB and antiphospholipid antibodies. The mother was healthy and her review of systems was negative for any collagen vascular disease–related symptoms.
Discussion
Neonatal lupus erythematosus (NLE) is a rare form of systemic lupus erythematosus (SLE) believed to be caused by transplacental transfer of anti-Ro (Sjögren’s syndrome antigen A, SSA), or, less commonly, anti-La (Sjögren’s syndrome antigen B, SSB) from mothers who are positive for these antibodies. Approximately 95% of NLE is associated with maternal anti-SSA; of these cases, 40% are also associated with maternal anti-SSB.1 Only about 2% of children of mothers who have anti-SSA or anti-SSB develop NLE, a finding that has led some researchers to postulate that maternal factors, fetal genetic factors, and environmental factors determine which children of anti-SSA or SSB positive mothers develop NLE.
A recent review found no association between the development of NLE and fetal birth weight, prematurity, or age.3 Over half of mothers of children who develop NLE are asymptomatic at the time of diagnosis of the neonate,3 though many become symptomatic in following years. Of mothers who are symptomatic, SLE and undifferentiated autoimmune syndrome are the most common diagnoses, though NLE has been rarely reported in the offspring of mothers with Sjögren’s syndrome, rheumatoid arthritis, and psoriasis.4,5
Fetal genetics are not an absolute determinant of development of NLE, as discordance in the development of NLE in twins has been reported. However, certain genetic relationships have been established. Fetal mutations in tumor necrosis factor–alpha appear to increase the likelihood of cutaneous manifestations. Mutations in transforming growth factor beta appear to increase the likelihood of cardiac manifestations, and experiments in cultured mouse cardiocytes have shown anti-SSB antibodies to impair macrophage phagocytosis of apoptotic cells in the developing fetal heart. These observations taken together suggest a fibroblast-mediated response to unphagocytosed cardiocyte debris may account for conduction abnormalities in neonates with NLE-induced heart block.6
Cutaneous disease in NLE is possible at birth, but more skin findings develop upon exposure to the sun. Nearly 80% of neonates affected by NLE develop cutaneous manifestations in the first few months of life. The head, neck, and extensor surfaces of the arms are most commonly affected, presumably because they are most likely to be exposed to the sun. Erythematous, annular, or discoid lesions are most common, and periorbital erythema with or without scale (“raccoon eyes”) should prompt consideration of NLE. However, annular, or discoid lesions are sometimes not present in NLE; telangiectasias, bullae, atrophic divots (“ice-pick scars”) or ulcerations may be seen instead. Lesions in the genital area have been described in fewer than 5% of patients with NLE.
The differential diagnosis of annular, scaly lesions in neonates includes annular erythema of infancy, tinea corporis, and seborrheic dermatitis. Annular erythema of infancy is a rare skin condition characterized by a cyclical eruption of erythematous annular lesions with minimal scaling which resolve spontaneously within a few weeks to months without leaving scaring or pigment changes. There is no treatment needed as the lesions self-resolve.7 Acute urticaria can sometimes appear similar to NLE but these are not scaly and also the lesions will disappear within 24-36 hours, compared with NLE lesions, which may take weeks to months to go away. Seborrheic dermatitis is a common skin condition seen in newborns with in the first few weeks of life and can present as scaly annular erythematous plaques on the face, scalp, torso, and the diaper area. Seborrheic dermatitis usually responds well to a combination of an antiyeast cream and a low-potency topical corticosteroid medication.
When NLE is suspected, diagnostic testing for lupus antibodies (anti-SSA, anti-SSB, and anti-U1RNP) in both maternal and neonatal serum should be undertaken. The presence of a characteristic rash plus maternal or neonatal antibodies is sufficient to make the diagnosis. If the rash is less characteristic, a biopsy showing an interface dermatitis can help solidify the diagnosis. Neonates with cutaneous manifestations of lupus may also have systemic disease. The most common and serious complication is heart block, whose pathophysiology is described above. Neonates with evidence of first-, second-, or third-degree heart block should be referred to a pediatric cardiologist for careful monitoring and management. Hepatic involvement has been reported, but is usually mild. Hematologic abnormalities have also been described that include anemia, neutropenia, and thrombocytopenia, which resolve by 9 months of age. Central nervous system involvement may rarely occur. The mainstay of treatment for the rash in NLE is diligent sun avoidance and sun protection. Topical corticosteroids may be used, but are not needed as the rash typically resolves by 9 months to 1 year without treatment. Mothers who have one child with NLE should be advised that they are more likely to have another with NLE – the risk is as high as 30%-40% in the second child. Hydroxychloroquine taken during subsequent pregnancies can reduce the incidence of cardiac complications,8 as can the so-called “triple therapy” of plasmapheresis, steroids, and IVIg.9
The cutaneous manifestations of NLE are usually self-limiting. However, they can serve as important clues that can prompt diagnosis of SLE in the mother, investigation of cardiac complications in the infant, and appropriate preventative care in future pregnancies.
Dr. Matiz is with the department of dermatology, Southern California Permanente Medical Group, San Diego. Mr. Kusari is with the department of dermatology, University of California, San Francisco.
References
1. Moretti D et al. Int J Dermatol. 2014;53(12):1508-12.
2. Buyon JP et al. Nature Clin Prac Rheum. 2009;5(3):139-48.
3. Li Y-Q et al. Int J Rheum Dis. 2015;18(7):761-7.
4. Rivera TL et al. Annals Rheum Dis. 2009;68(6):828-35.
5. Li L et al. Zhonghua er ke za zhi 2011;49(2):146-50.
6. Izmirly PM et al. Clin Rheumatol. 2011;30(12):1641-5.
7. Toledo-Alberola F and Betlloch-Mas I. Actas Dermosifiliogr. 2010 Jul;101(6):473-84.
8. Izmirly PM et al. Circulation. 2012;126(1):76-82.
9. Martinez-Sanchez N et al. Autoimmun Rev. 2015;14(5):423-8.
A potassium hydroxide preparation (KOH) from skin scrapings from the scalp lesions demonstrated no fungal elements. Further laboratory work up revealed a normal blood cell count, normal liver enzymes, an antinuclear antibody (ANA) titer of less than 1:80, a positive anti–Sjögren’s syndrome type B (SSB) antibody but negative anti–Sjögren’s syndrome type A (SSA) antibody and anti-U1RNP antibody. An electrocardiogram revealed no abnormalities. Liver function tests were normal. The complete blood count showed mild thrombocytopenia. Given the typical skin lesions and the positive SSB test and associated thrombocytopenia, the baby was diagnosed with neonatal lupus erythematosus.
Because of the diagnosis of neonatal lupus the mother was also tested and was found to have an elevated ANA of 1:640, positive SSB and antiphospholipid antibodies. The mother was healthy and her review of systems was negative for any collagen vascular disease–related symptoms.
Discussion
Neonatal lupus erythematosus (NLE) is a rare form of systemic lupus erythematosus (SLE) believed to be caused by transplacental transfer of anti-Ro (Sjögren’s syndrome antigen A, SSA), or, less commonly, anti-La (Sjögren’s syndrome antigen B, SSB) from mothers who are positive for these antibodies. Approximately 95% of NLE is associated with maternal anti-SSA; of these cases, 40% are also associated with maternal anti-SSB.1 Only about 2% of children of mothers who have anti-SSA or anti-SSB develop NLE, a finding that has led some researchers to postulate that maternal factors, fetal genetic factors, and environmental factors determine which children of anti-SSA or SSB positive mothers develop NLE.
A recent review found no association between the development of NLE and fetal birth weight, prematurity, or age.3 Over half of mothers of children who develop NLE are asymptomatic at the time of diagnosis of the neonate,3 though many become symptomatic in following years. Of mothers who are symptomatic, SLE and undifferentiated autoimmune syndrome are the most common diagnoses, though NLE has been rarely reported in the offspring of mothers with Sjögren’s syndrome, rheumatoid arthritis, and psoriasis.4,5
Fetal genetics are not an absolute determinant of development of NLE, as discordance in the development of NLE in twins has been reported. However, certain genetic relationships have been established. Fetal mutations in tumor necrosis factor–alpha appear to increase the likelihood of cutaneous manifestations. Mutations in transforming growth factor beta appear to increase the likelihood of cardiac manifestations, and experiments in cultured mouse cardiocytes have shown anti-SSB antibodies to impair macrophage phagocytosis of apoptotic cells in the developing fetal heart. These observations taken together suggest a fibroblast-mediated response to unphagocytosed cardiocyte debris may account for conduction abnormalities in neonates with NLE-induced heart block.6
Cutaneous disease in NLE is possible at birth, but more skin findings develop upon exposure to the sun. Nearly 80% of neonates affected by NLE develop cutaneous manifestations in the first few months of life. The head, neck, and extensor surfaces of the arms are most commonly affected, presumably because they are most likely to be exposed to the sun. Erythematous, annular, or discoid lesions are most common, and periorbital erythema with or without scale (“raccoon eyes”) should prompt consideration of NLE. However, annular, or discoid lesions are sometimes not present in NLE; telangiectasias, bullae, atrophic divots (“ice-pick scars”) or ulcerations may be seen instead. Lesions in the genital area have been described in fewer than 5% of patients with NLE.
The differential diagnosis of annular, scaly lesions in neonates includes annular erythema of infancy, tinea corporis, and seborrheic dermatitis. Annular erythema of infancy is a rare skin condition characterized by a cyclical eruption of erythematous annular lesions with minimal scaling which resolve spontaneously within a few weeks to months without leaving scaring or pigment changes. There is no treatment needed as the lesions self-resolve.7 Acute urticaria can sometimes appear similar to NLE but these are not scaly and also the lesions will disappear within 24-36 hours, compared with NLE lesions, which may take weeks to months to go away. Seborrheic dermatitis is a common skin condition seen in newborns with in the first few weeks of life and can present as scaly annular erythematous plaques on the face, scalp, torso, and the diaper area. Seborrheic dermatitis usually responds well to a combination of an antiyeast cream and a low-potency topical corticosteroid medication.
When NLE is suspected, diagnostic testing for lupus antibodies (anti-SSA, anti-SSB, and anti-U1RNP) in both maternal and neonatal serum should be undertaken. The presence of a characteristic rash plus maternal or neonatal antibodies is sufficient to make the diagnosis. If the rash is less characteristic, a biopsy showing an interface dermatitis can help solidify the diagnosis. Neonates with cutaneous manifestations of lupus may also have systemic disease. The most common and serious complication is heart block, whose pathophysiology is described above. Neonates with evidence of first-, second-, or third-degree heart block should be referred to a pediatric cardiologist for careful monitoring and management. Hepatic involvement has been reported, but is usually mild. Hematologic abnormalities have also been described that include anemia, neutropenia, and thrombocytopenia, which resolve by 9 months of age. Central nervous system involvement may rarely occur. The mainstay of treatment for the rash in NLE is diligent sun avoidance and sun protection. Topical corticosteroids may be used, but are not needed as the rash typically resolves by 9 months to 1 year without treatment. Mothers who have one child with NLE should be advised that they are more likely to have another with NLE – the risk is as high as 30%-40% in the second child. Hydroxychloroquine taken during subsequent pregnancies can reduce the incidence of cardiac complications,8 as can the so-called “triple therapy” of plasmapheresis, steroids, and IVIg.9
The cutaneous manifestations of NLE are usually self-limiting. However, they can serve as important clues that can prompt diagnosis of SLE in the mother, investigation of cardiac complications in the infant, and appropriate preventative care in future pregnancies.
Dr. Matiz is with the department of dermatology, Southern California Permanente Medical Group, San Diego. Mr. Kusari is with the department of dermatology, University of California, San Francisco.
References
1. Moretti D et al. Int J Dermatol. 2014;53(12):1508-12.
2. Buyon JP et al. Nature Clin Prac Rheum. 2009;5(3):139-48.
3. Li Y-Q et al. Int J Rheum Dis. 2015;18(7):761-7.
4. Rivera TL et al. Annals Rheum Dis. 2009;68(6):828-35.
5. Li L et al. Zhonghua er ke za zhi 2011;49(2):146-50.
6. Izmirly PM et al. Clin Rheumatol. 2011;30(12):1641-5.
7. Toledo-Alberola F and Betlloch-Mas I. Actas Dermosifiliogr. 2010 Jul;101(6):473-84.
8. Izmirly PM et al. Circulation. 2012;126(1):76-82.
9. Martinez-Sanchez N et al. Autoimmun Rev. 2015;14(5):423-8.
A potassium hydroxide preparation (KOH) from skin scrapings from the scalp lesions demonstrated no fungal elements. Further laboratory work up revealed a normal blood cell count, normal liver enzymes, an antinuclear antibody (ANA) titer of less than 1:80, a positive anti–Sjögren’s syndrome type B (SSB) antibody but negative anti–Sjögren’s syndrome type A (SSA) antibody and anti-U1RNP antibody. An electrocardiogram revealed no abnormalities. Liver function tests were normal. The complete blood count showed mild thrombocytopenia. Given the typical skin lesions and the positive SSB test and associated thrombocytopenia, the baby was diagnosed with neonatal lupus erythematosus.
Because of the diagnosis of neonatal lupus the mother was also tested and was found to have an elevated ANA of 1:640, positive SSB and antiphospholipid antibodies. The mother was healthy and her review of systems was negative for any collagen vascular disease–related symptoms.
Discussion
Neonatal lupus erythematosus (NLE) is a rare form of systemic lupus erythematosus (SLE) believed to be caused by transplacental transfer of anti-Ro (Sjögren’s syndrome antigen A, SSA), or, less commonly, anti-La (Sjögren’s syndrome antigen B, SSB) from mothers who are positive for these antibodies. Approximately 95% of NLE is associated with maternal anti-SSA; of these cases, 40% are also associated with maternal anti-SSB.1 Only about 2% of children of mothers who have anti-SSA or anti-SSB develop NLE, a finding that has led some researchers to postulate that maternal factors, fetal genetic factors, and environmental factors determine which children of anti-SSA or SSB positive mothers develop NLE.
A recent review found no association between the development of NLE and fetal birth weight, prematurity, or age.3 Over half of mothers of children who develop NLE are asymptomatic at the time of diagnosis of the neonate,3 though many become symptomatic in following years. Of mothers who are symptomatic, SLE and undifferentiated autoimmune syndrome are the most common diagnoses, though NLE has been rarely reported in the offspring of mothers with Sjögren’s syndrome, rheumatoid arthritis, and psoriasis.4,5
Fetal genetics are not an absolute determinant of development of NLE, as discordance in the development of NLE in twins has been reported. However, certain genetic relationships have been established. Fetal mutations in tumor necrosis factor–alpha appear to increase the likelihood of cutaneous manifestations. Mutations in transforming growth factor beta appear to increase the likelihood of cardiac manifestations, and experiments in cultured mouse cardiocytes have shown anti-SSB antibodies to impair macrophage phagocytosis of apoptotic cells in the developing fetal heart. These observations taken together suggest a fibroblast-mediated response to unphagocytosed cardiocyte debris may account for conduction abnormalities in neonates with NLE-induced heart block.6
Cutaneous disease in NLE is possible at birth, but more skin findings develop upon exposure to the sun. Nearly 80% of neonates affected by NLE develop cutaneous manifestations in the first few months of life. The head, neck, and extensor surfaces of the arms are most commonly affected, presumably because they are most likely to be exposed to the sun. Erythematous, annular, or discoid lesions are most common, and periorbital erythema with or without scale (“raccoon eyes”) should prompt consideration of NLE. However, annular, or discoid lesions are sometimes not present in NLE; telangiectasias, bullae, atrophic divots (“ice-pick scars”) or ulcerations may be seen instead. Lesions in the genital area have been described in fewer than 5% of patients with NLE.
The differential diagnosis of annular, scaly lesions in neonates includes annular erythema of infancy, tinea corporis, and seborrheic dermatitis. Annular erythema of infancy is a rare skin condition characterized by a cyclical eruption of erythematous annular lesions with minimal scaling which resolve spontaneously within a few weeks to months without leaving scaring or pigment changes. There is no treatment needed as the lesions self-resolve.7 Acute urticaria can sometimes appear similar to NLE but these are not scaly and also the lesions will disappear within 24-36 hours, compared with NLE lesions, which may take weeks to months to go away. Seborrheic dermatitis is a common skin condition seen in newborns with in the first few weeks of life and can present as scaly annular erythematous plaques on the face, scalp, torso, and the diaper area. Seborrheic dermatitis usually responds well to a combination of an antiyeast cream and a low-potency topical corticosteroid medication.
When NLE is suspected, diagnostic testing for lupus antibodies (anti-SSA, anti-SSB, and anti-U1RNP) in both maternal and neonatal serum should be undertaken. The presence of a characteristic rash plus maternal or neonatal antibodies is sufficient to make the diagnosis. If the rash is less characteristic, a biopsy showing an interface dermatitis can help solidify the diagnosis. Neonates with cutaneous manifestations of lupus may also have systemic disease. The most common and serious complication is heart block, whose pathophysiology is described above. Neonates with evidence of first-, second-, or third-degree heart block should be referred to a pediatric cardiologist for careful monitoring and management. Hepatic involvement has been reported, but is usually mild. Hematologic abnormalities have also been described that include anemia, neutropenia, and thrombocytopenia, which resolve by 9 months of age. Central nervous system involvement may rarely occur. The mainstay of treatment for the rash in NLE is diligent sun avoidance and sun protection. Topical corticosteroids may be used, but are not needed as the rash typically resolves by 9 months to 1 year without treatment. Mothers who have one child with NLE should be advised that they are more likely to have another with NLE – the risk is as high as 30%-40% in the second child. Hydroxychloroquine taken during subsequent pregnancies can reduce the incidence of cardiac complications,8 as can the so-called “triple therapy” of plasmapheresis, steroids, and IVIg.9
The cutaneous manifestations of NLE are usually self-limiting. However, they can serve as important clues that can prompt diagnosis of SLE in the mother, investigation of cardiac complications in the infant, and appropriate preventative care in future pregnancies.
Dr. Matiz is with the department of dermatology, Southern California Permanente Medical Group, San Diego. Mr. Kusari is with the department of dermatology, University of California, San Francisco.
References
1. Moretti D et al. Int J Dermatol. 2014;53(12):1508-12.
2. Buyon JP et al. Nature Clin Prac Rheum. 2009;5(3):139-48.
3. Li Y-Q et al. Int J Rheum Dis. 2015;18(7):761-7.
4. Rivera TL et al. Annals Rheum Dis. 2009;68(6):828-35.
5. Li L et al. Zhonghua er ke za zhi 2011;49(2):146-50.
6. Izmirly PM et al. Clin Rheumatol. 2011;30(12):1641-5.
7. Toledo-Alberola F and Betlloch-Mas I. Actas Dermosifiliogr. 2010 Jul;101(6):473-84.
8. Izmirly PM et al. Circulation. 2012;126(1):76-82.
9. Martinez-Sanchez N et al. Autoimmun Rev. 2015;14(5):423-8.
A 1-month-old, full-term female, born via normal vaginal delivery, presented to the dermatology clinic with a 3-week history of recurrent skin lesions on the scalp, face, and chest. The mother has been treating the lesions with breast milk and most recently with clotrimazole cream without resolution. 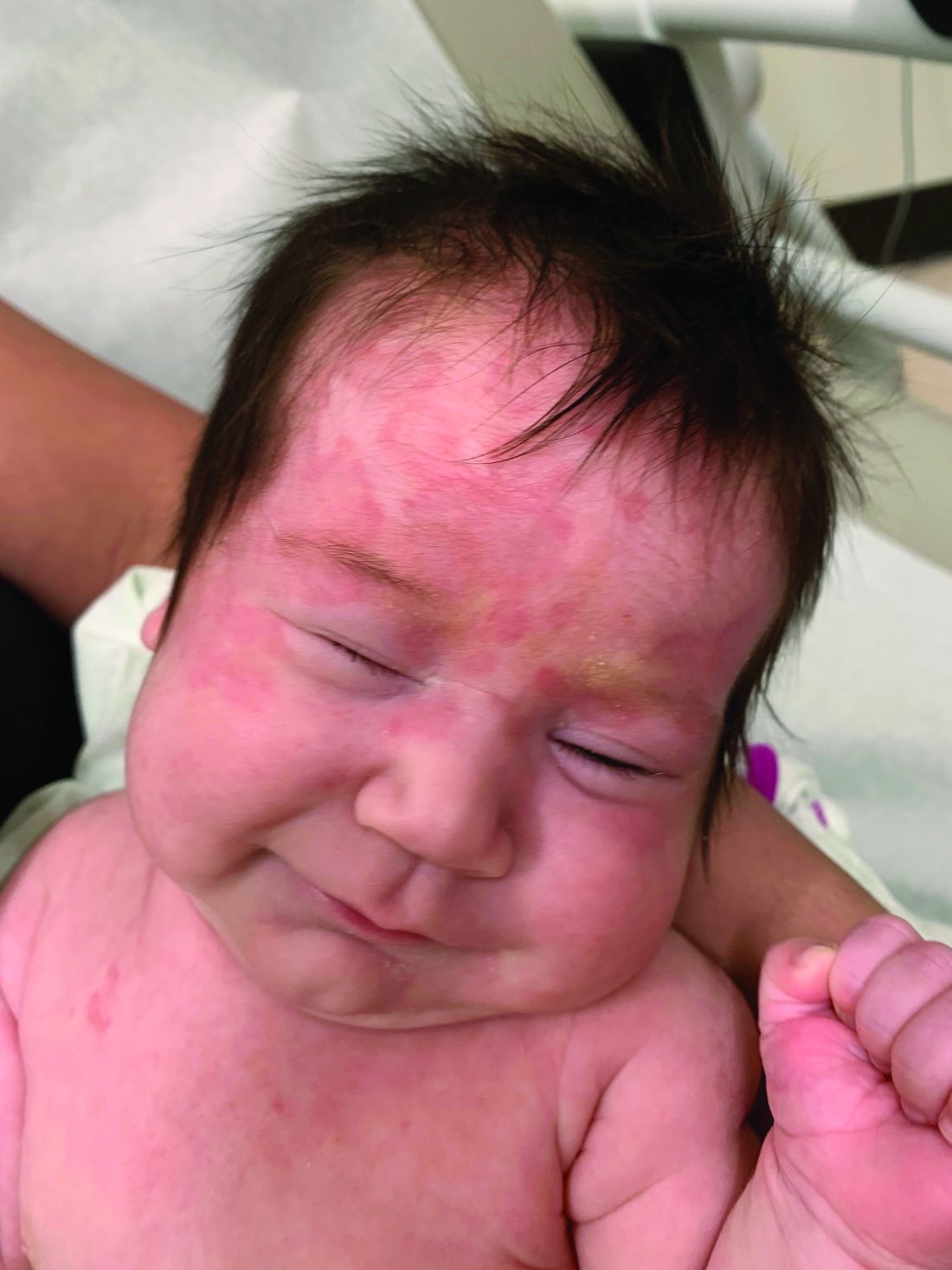
The mother of the baby is a healthy 32-year-old female with no past medical history. She had adequate prenatal care, and all the prenatal infectious and genetic tests were normal. The baby has been healthy and growing well. There is no history of associated fevers, chills, or any other symptoms. The family took no recent trips, and the parents are not affected. There are no other children at home and they have a cat and a dog. The family history is noncontributory.
On physical examination the baby was not in acute distress and her vital signs were normal. On skin examination she had several erythematous annular plaques and patches on the face, scalp, and upper chest (Fig. 1). There was no liver or spleen enlargement and no lymphadenopathy was palpated on exam.
A 12-year-old male has persistent purple toes and new red lesions on his hands
A punch biopsy from one of the lesions on the feet showed subtle basal vacuolar interface inflammation on the epidermis and rare apoptotic keratinocytes. There was an underlying dermal lymphocytic inflammatory infiltrate around the vascular plexus. Dermal mucin appeared slightly increased. The histologic findings are consistent with pernio. He had a negative direct immunofluorescence study.
Laboratory work-up showed an elevated antinuclear antibody (ANA) of 1:620; positive anticardiolipin IgM was at 15.2. A complete blood count showed no anemia or lymphopenia, he had normal complement C3 and C4 levels, normal urinalysis, negative cryoglobulins and cold agglutinins, and a normal protein electrophoresis.
Given the chronicity of his lesions, the lack of improvement with weather changes, the histopathologic findings of a vacuolar interface dermatitis and the positive ANA titer he was diagnosed with chilblain lupus.
Chilblain lupus erythematosus (CLE) is an uncommon form of chronic cutaneous lupus erythematosus that presents with tender pink to violaceous macules, papules, and/or nodules that sometimes can ulcerate and are present on the fingers, toes, and sometimes the nose and ears. The lesions are usually triggered by cold exposure.1 These patients also have clinical and laboratory findings consistent with lupus erythematosus.
Even though more studies are needed to clarify the clinical and histopathologic features of chilblain lupus, compared with idiopathic pernio, some authors suggest several characteristics: CLE lesions tend to persist in summer months, as occurred in our patient, and histopathologic evaluation usually shows vacuolar and interface inflammation on the basal cell layer and may also have a positive lupus band on direct immunofluorescence.2 About 20% of patient with CLE may later develop systemic lupus erythematosus.3
There is also a familial form of CLE which is usually inherited as an autosomal-dominant trait. Mutations in TREX1, SAMHD1, and STING have been described in these patients.4 Affected children present with skin lesions at a young age and those with TREX1 mutations are at a higher risk to develop systemic lupus erythematosus.
The differential diagnosis of chilblain lupus includes idiopathic pernio or pernio secondary to other conditions. Other conditions that are thought to be associated with pernio, besides lupus erythematosus, include infectious causes (hepatitis B, COVID-19 infection),5 autoimmune conditions, malignancy and hematologic disorders (paraproteinemia).6 In histopathology, pernio lesions present with dermal edema and superficial and deep lymphocytic infiltrate.
The pathogenesis of pernio is not fully understood but is thought be related to vasospasm with secondary poor perfusion and ischemia and type I interferon (INF1) immune response. A recent review of the published studies trying to explain the causality between COVID 19 and pernio-like lesions, from January 2020 to December 2020, speculate several possible mechanisms: an increase in the vasoconstrictive, prothrombotic, and proinflammatory effects of the angiotensin II pathway through activation of the ACE2 by the virus; COVID-19 triggers a robust INF1 immune response in predisposed patients; pernio as a sign of mild disease, may be explained by genetic and hormonal differences in the patients affected.7
Another condition that can be confused with CLE is Raynaud phenomenon, were patients present with white to purple to red patches on the fingers and toes after exposure to cold, but in comparison with pernio, the lesions improve within minutes to hours after rewarming. Secondary Raynaud phenomenon can be seen in patients with systemic lupus erythematosus and in patients with other connective tissue disorders. The skin lesions in our patient were persistent and were not triggered by cold exposure, making Raynaud phenomenon less likely. Children with vasculitis can present with painful red, violaceous, or necrotic lesions on the extremities, which can mimic pernio. Vasculitis lesions tend to be more purpuric and angulated, compared with pernio lesions, though in severe cases of pernio with ulceration it may be difficult to distinguish between the two entities and a skin biopsy may be needed.
Sweet syndrome, also known as acute febrile neutrophilic dermatosis, is a rare skin condition in which children present with edematous tender nodules on the hands and with less frequency in other parts of the body with associated fever, malaise, conjunctivitis, or joint pain and it is usually associated with infection or malignancy. Our patient denied any systemic symptoms and had no conjunctivitis nor arthritis.
Most patients with idiopathic pernio do not require a biopsy or further laboratory evaluation unless the lesions are atypical, chronic, or there is a suspected associated condition. The workup for patients with prolonged or atypical pernio-like lesions include a skin biopsy with direct immunofluorescence, ANA, complete blood count, complement levels, antiphospholipid antibodies, cold agglutinins, and cryoglobulins.
Treatment of mild CLE is with moderate- to high-potency topical corticosteroids. In those patients not responding to topical measures and keeping the extremities warm, the use of hydroxychloroquine has been reported to be beneficial in some patients as well as the use of calcium-channel blockers.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Su WP et al. Cutis. 1994 Dec;54(6):395-9.
2. Boada A et al. Am J Dermatopathol. 2010 Feb;32(1):19-23.
3. Patel et al. SBMJ Case Rep. 2013;2013:bcr2013201165.
4. Genes Yi et al. BMC. 2020 Apr 15;18(1):32.
5. Battesti G et al. J Am Acad Dermatol. 2020;83(4):1219-22.
6. Cappel JA et al. Mayo Clin Proc. 2014 Feb;89(2):207-15.
7. Cappel MA et al. Mayo Clin Proc. 2021;96(4):989-1005.
A punch biopsy from one of the lesions on the feet showed subtle basal vacuolar interface inflammation on the epidermis and rare apoptotic keratinocytes. There was an underlying dermal lymphocytic inflammatory infiltrate around the vascular plexus. Dermal mucin appeared slightly increased. The histologic findings are consistent with pernio. He had a negative direct immunofluorescence study.
Laboratory work-up showed an elevated antinuclear antibody (ANA) of 1:620; positive anticardiolipin IgM was at 15.2. A complete blood count showed no anemia or lymphopenia, he had normal complement C3 and C4 levels, normal urinalysis, negative cryoglobulins and cold agglutinins, and a normal protein electrophoresis.
Given the chronicity of his lesions, the lack of improvement with weather changes, the histopathologic findings of a vacuolar interface dermatitis and the positive ANA titer he was diagnosed with chilblain lupus.
Chilblain lupus erythematosus (CLE) is an uncommon form of chronic cutaneous lupus erythematosus that presents with tender pink to violaceous macules, papules, and/or nodules that sometimes can ulcerate and are present on the fingers, toes, and sometimes the nose and ears. The lesions are usually triggered by cold exposure.1 These patients also have clinical and laboratory findings consistent with lupus erythematosus.
Even though more studies are needed to clarify the clinical and histopathologic features of chilblain lupus, compared with idiopathic pernio, some authors suggest several characteristics: CLE lesions tend to persist in summer months, as occurred in our patient, and histopathologic evaluation usually shows vacuolar and interface inflammation on the basal cell layer and may also have a positive lupus band on direct immunofluorescence.2 About 20% of patient with CLE may later develop systemic lupus erythematosus.3
There is also a familial form of CLE which is usually inherited as an autosomal-dominant trait. Mutations in TREX1, SAMHD1, and STING have been described in these patients.4 Affected children present with skin lesions at a young age and those with TREX1 mutations are at a higher risk to develop systemic lupus erythematosus.
The differential diagnosis of chilblain lupus includes idiopathic pernio or pernio secondary to other conditions. Other conditions that are thought to be associated with pernio, besides lupus erythematosus, include infectious causes (hepatitis B, COVID-19 infection),5 autoimmune conditions, malignancy and hematologic disorders (paraproteinemia).6 In histopathology, pernio lesions present with dermal edema and superficial and deep lymphocytic infiltrate.
The pathogenesis of pernio is not fully understood but is thought be related to vasospasm with secondary poor perfusion and ischemia and type I interferon (INF1) immune response. A recent review of the published studies trying to explain the causality between COVID 19 and pernio-like lesions, from January 2020 to December 2020, speculate several possible mechanisms: an increase in the vasoconstrictive, prothrombotic, and proinflammatory effects of the angiotensin II pathway through activation of the ACE2 by the virus; COVID-19 triggers a robust INF1 immune response in predisposed patients; pernio as a sign of mild disease, may be explained by genetic and hormonal differences in the patients affected.7
Another condition that can be confused with CLE is Raynaud phenomenon, were patients present with white to purple to red patches on the fingers and toes after exposure to cold, but in comparison with pernio, the lesions improve within minutes to hours after rewarming. Secondary Raynaud phenomenon can be seen in patients with systemic lupus erythematosus and in patients with other connective tissue disorders. The skin lesions in our patient were persistent and were not triggered by cold exposure, making Raynaud phenomenon less likely. Children with vasculitis can present with painful red, violaceous, or necrotic lesions on the extremities, which can mimic pernio. Vasculitis lesions tend to be more purpuric and angulated, compared with pernio lesions, though in severe cases of pernio with ulceration it may be difficult to distinguish between the two entities and a skin biopsy may be needed.
Sweet syndrome, also known as acute febrile neutrophilic dermatosis, is a rare skin condition in which children present with edematous tender nodules on the hands and with less frequency in other parts of the body with associated fever, malaise, conjunctivitis, or joint pain and it is usually associated with infection or malignancy. Our patient denied any systemic symptoms and had no conjunctivitis nor arthritis.
Most patients with idiopathic pernio do not require a biopsy or further laboratory evaluation unless the lesions are atypical, chronic, or there is a suspected associated condition. The workup for patients with prolonged or atypical pernio-like lesions include a skin biopsy with direct immunofluorescence, ANA, complete blood count, complement levels, antiphospholipid antibodies, cold agglutinins, and cryoglobulins.
Treatment of mild CLE is with moderate- to high-potency topical corticosteroids. In those patients not responding to topical measures and keeping the extremities warm, the use of hydroxychloroquine has been reported to be beneficial in some patients as well as the use of calcium-channel blockers.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Su WP et al. Cutis. 1994 Dec;54(6):395-9.
2. Boada A et al. Am J Dermatopathol. 2010 Feb;32(1):19-23.
3. Patel et al. SBMJ Case Rep. 2013;2013:bcr2013201165.
4. Genes Yi et al. BMC. 2020 Apr 15;18(1):32.
5. Battesti G et al. J Am Acad Dermatol. 2020;83(4):1219-22.
6. Cappel JA et al. Mayo Clin Proc. 2014 Feb;89(2):207-15.
7. Cappel MA et al. Mayo Clin Proc. 2021;96(4):989-1005.
A punch biopsy from one of the lesions on the feet showed subtle basal vacuolar interface inflammation on the epidermis and rare apoptotic keratinocytes. There was an underlying dermal lymphocytic inflammatory infiltrate around the vascular plexus. Dermal mucin appeared slightly increased. The histologic findings are consistent with pernio. He had a negative direct immunofluorescence study.
Laboratory work-up showed an elevated antinuclear antibody (ANA) of 1:620; positive anticardiolipin IgM was at 15.2. A complete blood count showed no anemia or lymphopenia, he had normal complement C3 and C4 levels, normal urinalysis, negative cryoglobulins and cold agglutinins, and a normal protein electrophoresis.
Given the chronicity of his lesions, the lack of improvement with weather changes, the histopathologic findings of a vacuolar interface dermatitis and the positive ANA titer he was diagnosed with chilblain lupus.
Chilblain lupus erythematosus (CLE) is an uncommon form of chronic cutaneous lupus erythematosus that presents with tender pink to violaceous macules, papules, and/or nodules that sometimes can ulcerate and are present on the fingers, toes, and sometimes the nose and ears. The lesions are usually triggered by cold exposure.1 These patients also have clinical and laboratory findings consistent with lupus erythematosus.
Even though more studies are needed to clarify the clinical and histopathologic features of chilblain lupus, compared with idiopathic pernio, some authors suggest several characteristics: CLE lesions tend to persist in summer months, as occurred in our patient, and histopathologic evaluation usually shows vacuolar and interface inflammation on the basal cell layer and may also have a positive lupus band on direct immunofluorescence.2 About 20% of patient with CLE may later develop systemic lupus erythematosus.3
There is also a familial form of CLE which is usually inherited as an autosomal-dominant trait. Mutations in TREX1, SAMHD1, and STING have been described in these patients.4 Affected children present with skin lesions at a young age and those with TREX1 mutations are at a higher risk to develop systemic lupus erythematosus.
The differential diagnosis of chilblain lupus includes idiopathic pernio or pernio secondary to other conditions. Other conditions that are thought to be associated with pernio, besides lupus erythematosus, include infectious causes (hepatitis B, COVID-19 infection),5 autoimmune conditions, malignancy and hematologic disorders (paraproteinemia).6 In histopathology, pernio lesions present with dermal edema and superficial and deep lymphocytic infiltrate.
The pathogenesis of pernio is not fully understood but is thought be related to vasospasm with secondary poor perfusion and ischemia and type I interferon (INF1) immune response. A recent review of the published studies trying to explain the causality between COVID 19 and pernio-like lesions, from January 2020 to December 2020, speculate several possible mechanisms: an increase in the vasoconstrictive, prothrombotic, and proinflammatory effects of the angiotensin II pathway through activation of the ACE2 by the virus; COVID-19 triggers a robust INF1 immune response in predisposed patients; pernio as a sign of mild disease, may be explained by genetic and hormonal differences in the patients affected.7
Another condition that can be confused with CLE is Raynaud phenomenon, were patients present with white to purple to red patches on the fingers and toes after exposure to cold, but in comparison with pernio, the lesions improve within minutes to hours after rewarming. Secondary Raynaud phenomenon can be seen in patients with systemic lupus erythematosus and in patients with other connective tissue disorders. The skin lesions in our patient were persistent and were not triggered by cold exposure, making Raynaud phenomenon less likely. Children with vasculitis can present with painful red, violaceous, or necrotic lesions on the extremities, which can mimic pernio. Vasculitis lesions tend to be more purpuric and angulated, compared with pernio lesions, though in severe cases of pernio with ulceration it may be difficult to distinguish between the two entities and a skin biopsy may be needed.
Sweet syndrome, also known as acute febrile neutrophilic dermatosis, is a rare skin condition in which children present with edematous tender nodules on the hands and with less frequency in other parts of the body with associated fever, malaise, conjunctivitis, or joint pain and it is usually associated with infection or malignancy. Our patient denied any systemic symptoms and had no conjunctivitis nor arthritis.
Most patients with idiopathic pernio do not require a biopsy or further laboratory evaluation unless the lesions are atypical, chronic, or there is a suspected associated condition. The workup for patients with prolonged or atypical pernio-like lesions include a skin biopsy with direct immunofluorescence, ANA, complete blood count, complement levels, antiphospholipid antibodies, cold agglutinins, and cryoglobulins.
Treatment of mild CLE is with moderate- to high-potency topical corticosteroids. In those patients not responding to topical measures and keeping the extremities warm, the use of hydroxychloroquine has been reported to be beneficial in some patients as well as the use of calcium-channel blockers.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Su WP et al. Cutis. 1994 Dec;54(6):395-9.
2. Boada A et al. Am J Dermatopathol. 2010 Feb;32(1):19-23.
3. Patel et al. SBMJ Case Rep. 2013;2013:bcr2013201165.
4. Genes Yi et al. BMC. 2020 Apr 15;18(1):32.
5. Battesti G et al. J Am Acad Dermatol. 2020;83(4):1219-22.
6. Cappel JA et al. Mayo Clin Proc. 2014 Feb;89(2):207-15.
7. Cappel MA et al. Mayo Clin Proc. 2021;96(4):989-1005.
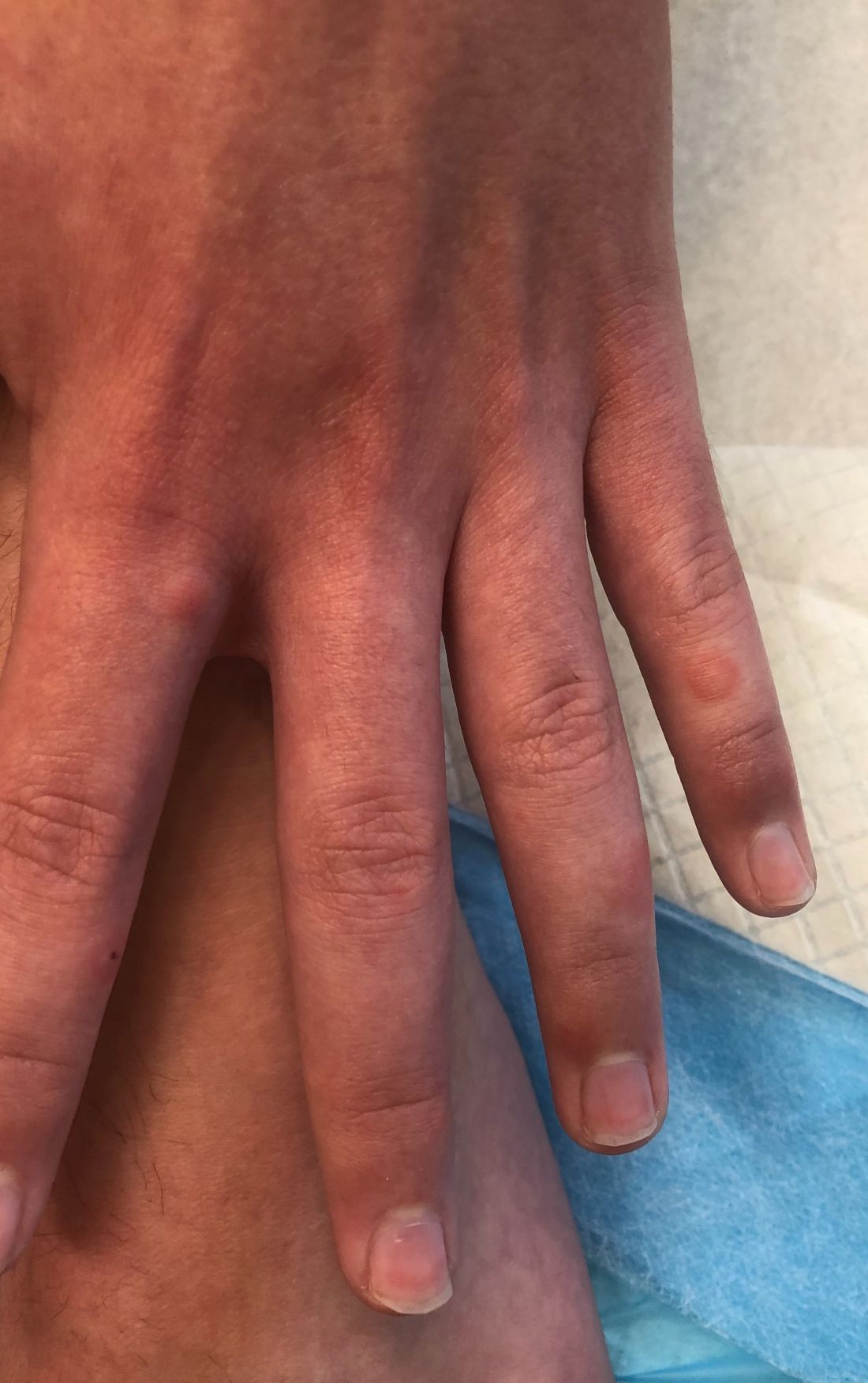
He denied any hair loss, mouth sores, sun sensitivity, headaches, gastrointestinal complaints, joint pain, or muscle weakness.
He is not taking any medications.
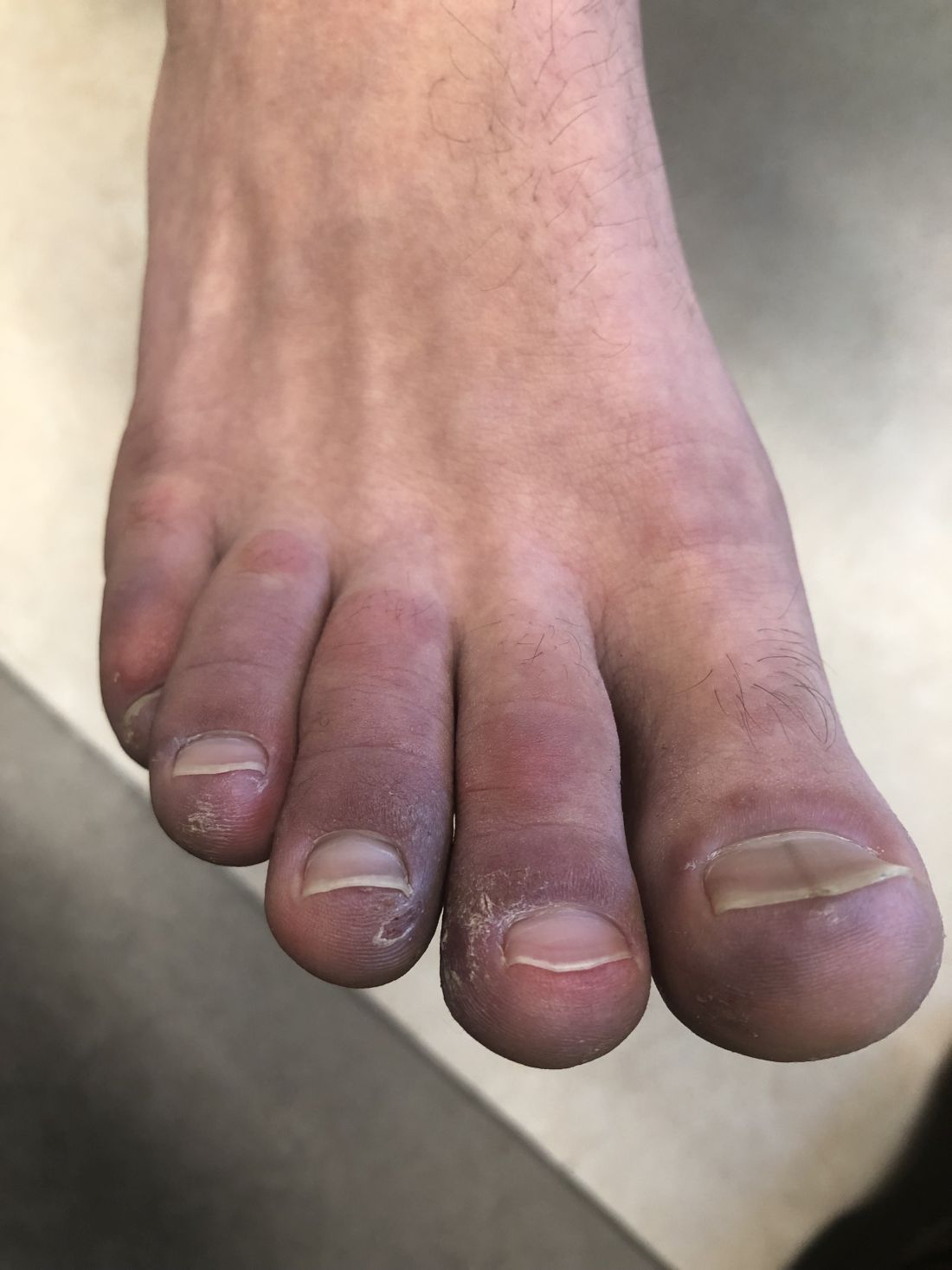
He has been at home doing virtual school and has not traveled. He likes to play the piano. There is no family history of similar lesions, connective tissue disorder, or autoimmunity.
On physical exam he has purple discoloration on the toes with some violaceous and pink papules. On the fingers he has pink to violaceous papules and macules.
There is no joint edema or pain.