User login
Angioimmunoblastic T-Cell Lymphoma Mimicking Diffuse Large B-Cell Lymphoma
Angioimmunoblastic T-cell lymphoma (AITL) is a rare, often aggressive type of peripheral T-cell lymphoma. It comprises 18% of peripheral T-cell lymphomas and 1% to 2% of all non-Hodgkin lymphomas.1 The incidence of AITL in the United States is estimated to be 0.05 cases per 100,000 person-years,2 and there is a slight male predominance.1,3,4 It typically presents in the seventh decade of life; however, cases have been reported in adults ranging from 20 to 91 years of age.3
Angioimmunoblastic T-cell lymphoma presents with lymphadenopathy, hepatosplenomegaly, and systemic B symptoms (eg, fever, night sweats, weight loss, generalized pruritus).4-6 There are cutaneous manifestations in up to 50% of cases4,5,7 and frequently signs of autoimmune disorder.4,5 The diagnosis often is made by excisional lymph node biopsy. Lymph node specimens characteristically have a mixed inflammatory infiltrate that includes numerous B cells often infected with Epstein-Barr virus (EBV) and a relatively small population of atypical T lymphocytes.8 Identification of this neoplastic population of CD4+CD8− T lymphocytes expressing normal follicular helper T-cell markers CD10, chemokine CXCL13, programmed cell death protein 1 (PD-1), and B-cell lymphoma 6 (BCL-6) confirms the diagnosis of AITL.9,10 These malignant cells can be identified in skin specimens in cases of cutaneous metastatic disease.11,12 We present a case originally misdiagnosed as diffuse large B-cell lymphoma that was later identified as AITL on skin biopsy.
Case Report
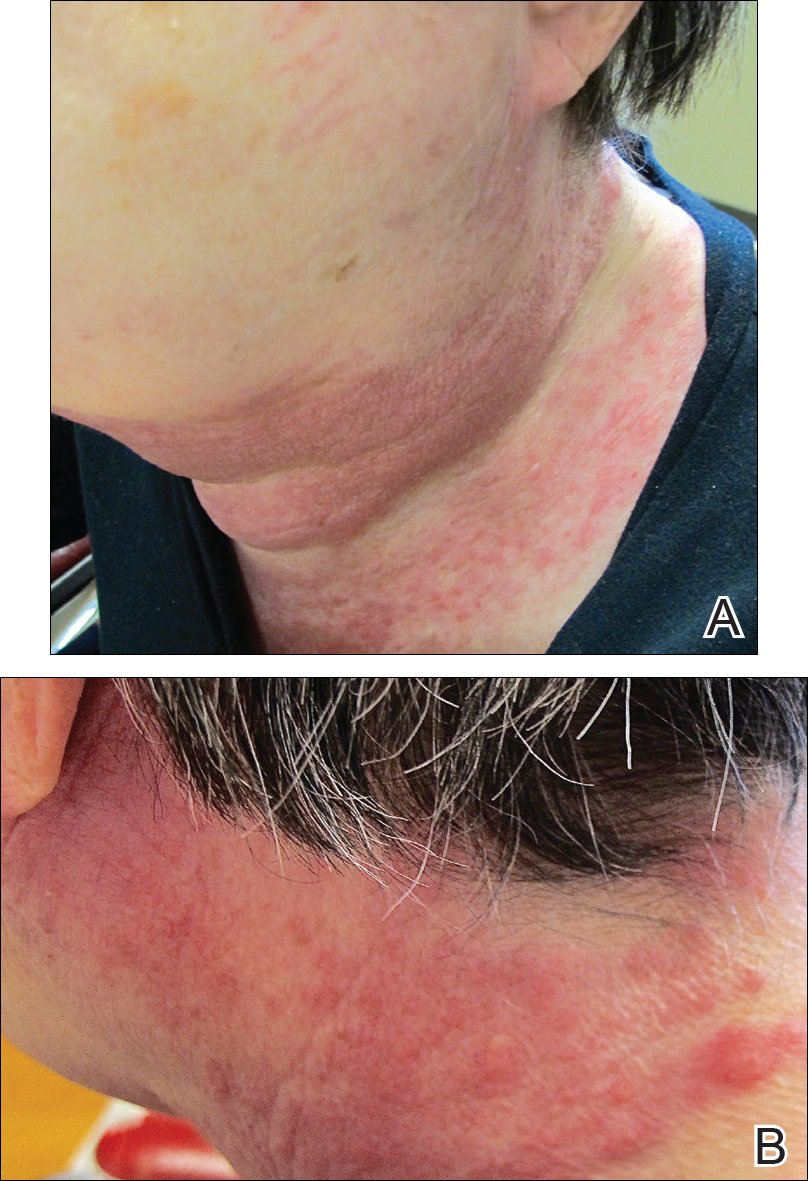
A 72-year-old woman presented with a pruritic erythematous eruption around the neck of 3 weeks’ duration (Figure 1). Her medical history was notable for diffuse large B-cell non-Hodgkin lymphoma diagnosed 3 months prior based on results from a right cervical lymph node biopsy. She was treated with bendamustine and rituximab. On physical examination there were erythematous edematous papules coalescing into indurated plaques around the neck. The differential diagnosis included drug hypersensitivity reaction, herpes zoster, urticaria, and cutaneous metastasis. Two punch biopsies were taken for hematoxylin and eosin and tissue culture.
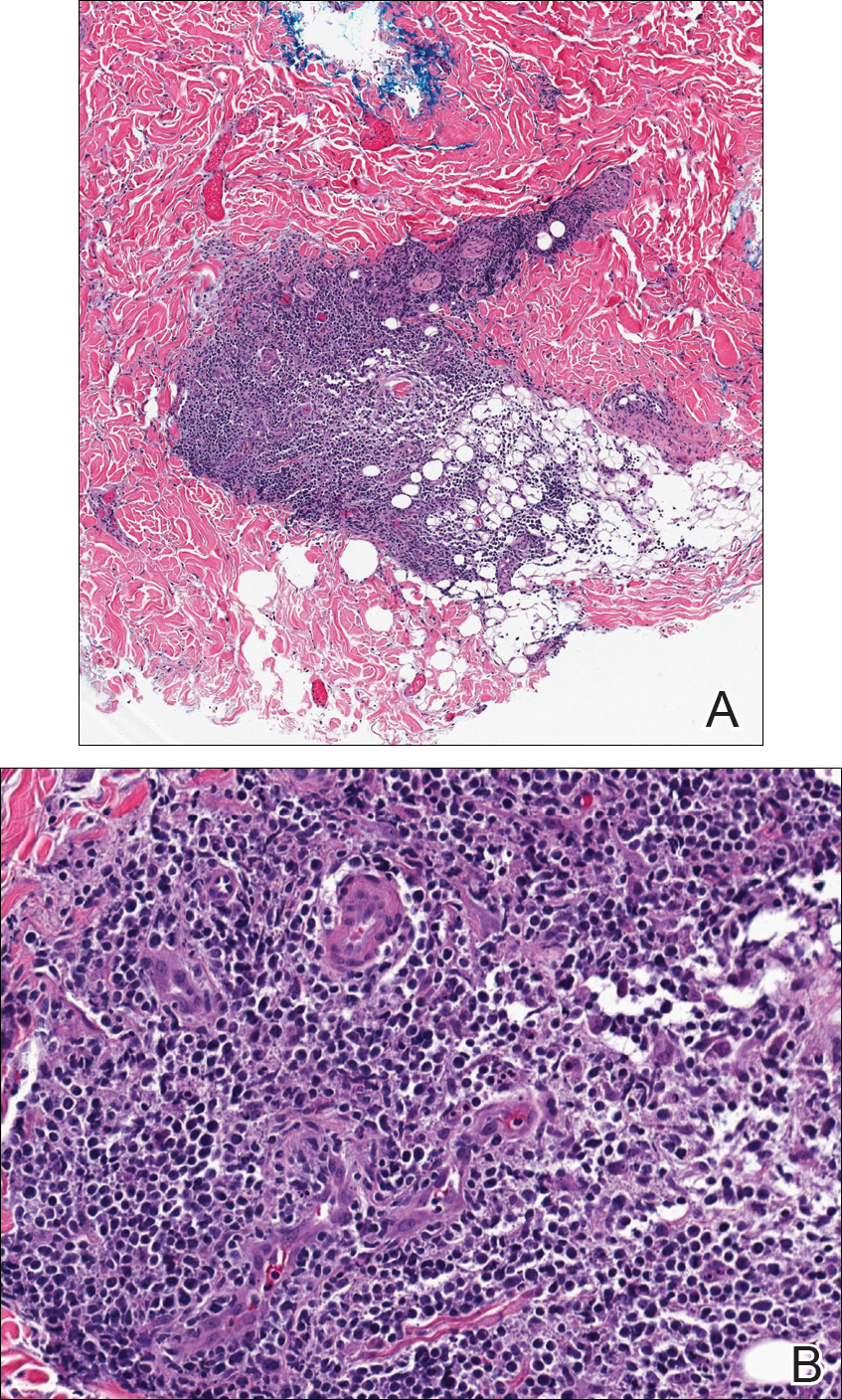
Tissue cultures and viral polymerase chain reaction were negative. Histopathologic examination revealed a scant atypical lymphoid infiltrate focally involving the deep dermis. The cells were medium to large in size and contained hyperchromatic pleomorphic nuclei (Figure 2). They were positive for CD3 and CD4, which was concerning for T-cell lymphoma. The histologic report of the excisional lymph node biopsy done 3 months prior described an atypical lymphoid neoplasm with extensive necrosis and extranodal spread that stained positively for CD20 (Figure 3).
Further staining of this cervical lymph node specimen revealed large atypical lymphoid cells positive for CD3, CD10, B-cell lymphoma 2 (BCL-2), BCL-6, and PD-1. There were intermixed mature B lymphocytes positive for CD20 and BCL-2. Chromogenic in situ hybridization with probes for EBV showed numerous positive cells throughout the infiltrate. Polymerase chain reaction demonstrated a T-cell population with clonally rearranged T-cell receptor genes. Primers for immunoglobulin heavy and light chains showed no evidence of a clonal B-cell population.
Additional staining of the atypical cutaneous lymphocytes revealed positivity for CD3, CD10, and PD-1. The morphologic and immunophenotypic findings of both specimens supported the diagnosis of AITL.
The patient declined further treatment and chose hospice care.
Comment
Etiology
Angioimmunoblastic T-cell lymphoma was originally named angioimmunoblastic lymphadenopathy with dysproteinemia. It was initially thought to be a benign hyperreactive immune process driven by B cells, and patients often died of infectious complications not long after the diagnosis was made.13 As more cases were reported with clonal rearrangements and signs of progressive lymphoma, AITL was recognized as a malignancy.
Presentation
Patients with AITL often present with advanced stage III or IV disease with extranodal and bone marrow involvement.3-6 Cutaneous disease occurs in up to half of patients and portends a poor prognosis.7 The rash often is a nonspecific erythematous macular and papular eruption mimicking a morbilliform viral exanthem or drug reaction. Urticarial, nodular, petechial, purpuric, eczematous, erythrodermic, and vesiculobullous presentations have been described.4,11,12 In up to one-third of cases, the eruption occurs in association with a new medication, often leading to an initial misdiagnosis of drug hypersensitivity reaction.4,11 In a review conducted by Balaraman et al,14 84% of patients with AITL reported having pruritus.
There is an association of autoimmune phenomena in patients with AITL, which is likely a result of immune dysregulation associated with poorly functioning follicular helper T cells. Patients may present with arthralgia, hemolytic anemia, or thrombocytopenic purpura. Hypergammaglobulinemia has been reported in 30% to 50% of AITL patients.4,6 Other pertinent immunologic findings include positive Coombs test, cold agglutinins, cryoglobulinemia, hypocomplementemia, and positive antinuclear antibodies.4-7
Gene Analysis
Affected lymph nodes have a characteristically effaced architecture with proliferative arborizing venules; a hyperplastic population of follicular dendritic cells; and a mixed inflammatory infiltrate that is comprised of atypical lymphocytes and variable numbers of reactive lymphocytes, histiocytes, eosinophils, and plasma cells. The malignant T lymphocytes often account for only a small portion of the infiltrate.8 T-cell gene rearrangement studies identify clonal cells with β and γ rearrangements in the majority of cases.4 These cells are predominantly CD4+CD8− and express normal follicular helper T-cell markers CD10, CXCL13, BCL-6,5,9 and PD-1.10 Numerous B cells are seen intermixed with follicular dendritic cells. They are frequently infected with EBV and can have an atypical Reed-Sternberg cell–like appearance.4,5,15 In the evaluation of AITL, polymerase chain reaction studies with primers for immunoglobulin heavy and light chain should be performed to look for clonal B-cell populations and rule out a possible secondary B-cell lymphoma.
Histology
Five histologic patterns have been described with cutaneous AITL: (1) superficial perivascular infiltrate of eosinophils and lymphocytes that lack atypia, (2) sparse perivascular infiltrate with atypical lymphocytes, (3) dense dermal infiltrate of pleomorphic lymphocytes, (4) leukocytoclastic vasculitis without atypical lymphocytes,11 and (5) necrotizing vasculitis.12 The finding of vascular hyperplasia, perivascular infiltrate, or vasculitis has been reported in 91% of cases in the literature. Although these findings are nonspecific, an analysis of cutaneous cases reported in the literature found that 87% demonstrated T-cell receptor gene rearrangements.14 Lymphoid cells are positive for CD10 and PD-1, as was demonstrated in our case, and are CXCL13 positive in the majority of cases.12 Atypical and EBV-infected B cells also can be found in the skin.11,12
Differential Diagnosis
Angioimmunoblastic T-cell lymphoma can mimic infectious, autoimmune, or allergic etiologies, and misdiagnosis of another type of lymphoma is not uncommon, as occurred in our case. Patients who have a delay in the correct diagnosis have similar outcomes to those correctly diagnosed at first presentation.4
Treatment
There are no effective therapies for AITL. Poor prognostic factors include age (>60 years), stages III to IV disease, male gender, elevated serum lactate dehydrogenase level,3,5,10 and cutaneous involvement.7 Corticosteroids, anthracycline-based chemotherapy, and autologous stem cell transplant are currently the mainstays of therapy. Initial response to chemotherapy is promising, but duration of response is poor overall and there is no increased survival.5,15 A large population-based study of 1207 cases by Xu and Liu3 showed the overall survival rate at 2 and 10 years was 46.8% and 21.9%, respectively. Ten-year disease-specific survival was 35.9%, and there was no demonstrable improvement in survival over the last 2 decades.3 Case reports have demonstrated that thalidomide,16 lenalidomide,17 and cyclosporine plus dexamethasone18 have been successfully used to achieve remission for up to 3 years.
Conclusion
Angioimmunoblastic T-cell lymphoma is difficult to diagnose due to nonspecific clinical and histologic findings. Cutaneous manifestations are seen in AITL in up to half of cases that may occur early or in advanced disease. Similar to all cutaneous metastases, the appearance of the lesions can greatly vary. Our case demonstrates that dermatologists and dermatopathologists can make this diagnosis in the appropriate clinicopathologic context utilizing appropriate immunohistochemical staining and gene rearrangement studies.
- Rudiger T, Weisenburger DD, Anderson JR, et al. Peripheral T-cell lymphoma (excluding anaplastic large-cell lymphoma): results from the Non-Hodgkins Lymphoma Classification Project. Ann Oncol. 2002;13:140-149.
- Morton LM, Wang SS, Devesa SS, et al. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood. 2006;107:265-276.
- Xu B, Liu P. No survival improvement for patients with angioimmunoblastic T-cell lymphoma over the past two decades: a population-based study of 1207 cases. PLoS One. 2014;9:e92585.
- Lachenal F, Berger F, Ghesquieres H, et al. Angioimmunoblastic T-cell lymphoma: clinical and laboratory features at diagnosis in 77 patients. Medicine (Baltimore). 2007;86:282-292.
- Mourad N, Mounier N, Briére J, et al. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l’Adulte (GELA) trials. Blood. 2008;111:4463-4470.
- Frederico M, Rudiger T, Bellei M, et al. Clinicopathologic characteristics of angioimmunoblastic T-cell lymphoma: analysis of the International Peripheral T-cell Lymphoma Project. J Clin Oncol. 2013;31:240-246.
- Siegert W, Nerl C, Agthe A, et al. Angioimmunoblastic lym-phadenopathy (AILD)-type T-cell lymphoma: prognostic impact of clinical observations and laboratory findings at presentation. The Kiel Lymphoma Study Group. Ann Oncol. 1995;6:659-664.
- Attygalle AD, Chuang SS, Diss TC, et al. Distinguishing angioimmunoblastic T-cell lymphoma from peripheral T-cell lymphoma, unspecified, using morphology, immunophenotype, and molecular genetics. Histopathology. 2007;50:498-508.
- Dupuis J, Boye K, Martin N, et al. Expression of CXCL13 by neoplastic cells in angioimmunoblastic T-cell lymphoma (AITL): a new diagnostic marker providing evidence that AITL derives from follicular helper cells. Am J Surg Pathol. 2006;30:490-494.
- Odejide O, Weigert O, Lane AA, et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123:1293-1296.
- Martel P, Laroche L, Courville P, et al. Cutaneous involvementin patients with angioimmunoblastic lymphadenopathy with dysproteinemia: a clinical, immunohistological, and molecular analysis. Arch Dermatol. 2000;136:881-886.
- Ortonne N, Dupuis J, Plonquet A, et al. Characterization of CXCL13+ neoplastic t cells in cutaneous lesions of angioimmunoblastic T-cell lymphoma (AITL). Am J Surg Pathol. 2007;31:1068-1076.
- Frizzera G, Moran E, Rappaport H. Angioimmunoblastic lymphadenopathy with dysproteinemia. Lancet. 1974;1:1070-1073.
- Balaraman B, Conley JA, Sheinbein DM. Evaluation of cutaneous angioimmunoblastic T-cell lymphoma [published online May 6, 2011]. J Am Acad Dermatol. 2011;65:855-862.
- Tokunaga T, Shimada K, Yamamoto K, et al. Retrospective analysis of prognostic factors for angioimmunoblastic T-cell lymphoma: a multicenter cooperative study in Japan. Blood. 2012;119:2837-2843.
- Dogan A, Ngu LSP, Ng SH, et al. Pathology and clinical features of angioimmunoblastic T-cell lymphoma after successful treatment with thalidomide. Leukemia. 2005;19:873-875.
- Fabbri A, Cencini E, Pietrini A, et al. Impressive activity of lenalidomide monotherapy in refractory angioimmunoblastic T-cell lymphoma: report of a case with long-term follow-up. Hematol Oncol. 2013;31:213-217.
- Kobayashi T, Kuroda J, Uchiyama H, et al. Successful treatment of chemotherapy-refractory angioimmunoblastic T cell lymphoma with cyclosporin A. Acta Haematol. 2012;127:10-15.
Angioimmunoblastic T-cell lymphoma (AITL) is a rare, often aggressive type of peripheral T-cell lymphoma. It comprises 18% of peripheral T-cell lymphomas and 1% to 2% of all non-Hodgkin lymphomas.1 The incidence of AITL in the United States is estimated to be 0.05 cases per 100,000 person-years,2 and there is a slight male predominance.1,3,4 It typically presents in the seventh decade of life; however, cases have been reported in adults ranging from 20 to 91 years of age.3
Angioimmunoblastic T-cell lymphoma presents with lymphadenopathy, hepatosplenomegaly, and systemic B symptoms (eg, fever, night sweats, weight loss, generalized pruritus).4-6 There are cutaneous manifestations in up to 50% of cases4,5,7 and frequently signs of autoimmune disorder.4,5 The diagnosis often is made by excisional lymph node biopsy. Lymph node specimens characteristically have a mixed inflammatory infiltrate that includes numerous B cells often infected with Epstein-Barr virus (EBV) and a relatively small population of atypical T lymphocytes.8 Identification of this neoplastic population of CD4+CD8− T lymphocytes expressing normal follicular helper T-cell markers CD10, chemokine CXCL13, programmed cell death protein 1 (PD-1), and B-cell lymphoma 6 (BCL-6) confirms the diagnosis of AITL.9,10 These malignant cells can be identified in skin specimens in cases of cutaneous metastatic disease.11,12 We present a case originally misdiagnosed as diffuse large B-cell lymphoma that was later identified as AITL on skin biopsy.
Case Report
A 72-year-old woman presented with a pruritic erythematous eruption around the neck of 3 weeks’ duration (Figure 1). Her medical history was notable for diffuse large B-cell non-Hodgkin lymphoma diagnosed 3 months prior based on results from a right cervical lymph node biopsy. She was treated with bendamustine and rituximab. On physical examination there were erythematous edematous papules coalescing into indurated plaques around the neck. The differential diagnosis included drug hypersensitivity reaction, herpes zoster, urticaria, and cutaneous metastasis. Two punch biopsies were taken for hematoxylin and eosin and tissue culture.
Tissue cultures and viral polymerase chain reaction were negative. Histopathologic examination revealed a scant atypical lymphoid infiltrate focally involving the deep dermis. The cells were medium to large in size and contained hyperchromatic pleomorphic nuclei (Figure 2). They were positive for CD3 and CD4, which was concerning for T-cell lymphoma. The histologic report of the excisional lymph node biopsy done 3 months prior described an atypical lymphoid neoplasm with extensive necrosis and extranodal spread that stained positively for CD20 (Figure 3).
Further staining of this cervical lymph node specimen revealed large atypical lymphoid cells positive for CD3, CD10, B-cell lymphoma 2 (BCL-2), BCL-6, and PD-1. There were intermixed mature B lymphocytes positive for CD20 and BCL-2. Chromogenic in situ hybridization with probes for EBV showed numerous positive cells throughout the infiltrate. Polymerase chain reaction demonstrated a T-cell population with clonally rearranged T-cell receptor genes. Primers for immunoglobulin heavy and light chains showed no evidence of a clonal B-cell population.
Additional staining of the atypical cutaneous lymphocytes revealed positivity for CD3, CD10, and PD-1. The morphologic and immunophenotypic findings of both specimens supported the diagnosis of AITL.
The patient declined further treatment and chose hospice care.
Comment
Etiology
Angioimmunoblastic T-cell lymphoma was originally named angioimmunoblastic lymphadenopathy with dysproteinemia. It was initially thought to be a benign hyperreactive immune process driven by B cells, and patients often died of infectious complications not long after the diagnosis was made.13 As more cases were reported with clonal rearrangements and signs of progressive lymphoma, AITL was recognized as a malignancy.
Presentation
Patients with AITL often present with advanced stage III or IV disease with extranodal and bone marrow involvement.3-6 Cutaneous disease occurs in up to half of patients and portends a poor prognosis.7 The rash often is a nonspecific erythematous macular and papular eruption mimicking a morbilliform viral exanthem or drug reaction. Urticarial, nodular, petechial, purpuric, eczematous, erythrodermic, and vesiculobullous presentations have been described.4,11,12 In up to one-third of cases, the eruption occurs in association with a new medication, often leading to an initial misdiagnosis of drug hypersensitivity reaction.4,11 In a review conducted by Balaraman et al,14 84% of patients with AITL reported having pruritus.
There is an association of autoimmune phenomena in patients with AITL, which is likely a result of immune dysregulation associated with poorly functioning follicular helper T cells. Patients may present with arthralgia, hemolytic anemia, or thrombocytopenic purpura. Hypergammaglobulinemia has been reported in 30% to 50% of AITL patients.4,6 Other pertinent immunologic findings include positive Coombs test, cold agglutinins, cryoglobulinemia, hypocomplementemia, and positive antinuclear antibodies.4-7
Gene Analysis
Affected lymph nodes have a characteristically effaced architecture with proliferative arborizing venules; a hyperplastic population of follicular dendritic cells; and a mixed inflammatory infiltrate that is comprised of atypical lymphocytes and variable numbers of reactive lymphocytes, histiocytes, eosinophils, and plasma cells. The malignant T lymphocytes often account for only a small portion of the infiltrate.8 T-cell gene rearrangement studies identify clonal cells with β and γ rearrangements in the majority of cases.4 These cells are predominantly CD4+CD8− and express normal follicular helper T-cell markers CD10, CXCL13, BCL-6,5,9 and PD-1.10 Numerous B cells are seen intermixed with follicular dendritic cells. They are frequently infected with EBV and can have an atypical Reed-Sternberg cell–like appearance.4,5,15 In the evaluation of AITL, polymerase chain reaction studies with primers for immunoglobulin heavy and light chain should be performed to look for clonal B-cell populations and rule out a possible secondary B-cell lymphoma.
Histology
Five histologic patterns have been described with cutaneous AITL: (1) superficial perivascular infiltrate of eosinophils and lymphocytes that lack atypia, (2) sparse perivascular infiltrate with atypical lymphocytes, (3) dense dermal infiltrate of pleomorphic lymphocytes, (4) leukocytoclastic vasculitis without atypical lymphocytes,11 and (5) necrotizing vasculitis.12 The finding of vascular hyperplasia, perivascular infiltrate, or vasculitis has been reported in 91% of cases in the literature. Although these findings are nonspecific, an analysis of cutaneous cases reported in the literature found that 87% demonstrated T-cell receptor gene rearrangements.14 Lymphoid cells are positive for CD10 and PD-1, as was demonstrated in our case, and are CXCL13 positive in the majority of cases.12 Atypical and EBV-infected B cells also can be found in the skin.11,12
Differential Diagnosis
Angioimmunoblastic T-cell lymphoma can mimic infectious, autoimmune, or allergic etiologies, and misdiagnosis of another type of lymphoma is not uncommon, as occurred in our case. Patients who have a delay in the correct diagnosis have similar outcomes to those correctly diagnosed at first presentation.4
Treatment
There are no effective therapies for AITL. Poor prognostic factors include age (>60 years), stages III to IV disease, male gender, elevated serum lactate dehydrogenase level,3,5,10 and cutaneous involvement.7 Corticosteroids, anthracycline-based chemotherapy, and autologous stem cell transplant are currently the mainstays of therapy. Initial response to chemotherapy is promising, but duration of response is poor overall and there is no increased survival.5,15 A large population-based study of 1207 cases by Xu and Liu3 showed the overall survival rate at 2 and 10 years was 46.8% and 21.9%, respectively. Ten-year disease-specific survival was 35.9%, and there was no demonstrable improvement in survival over the last 2 decades.3 Case reports have demonstrated that thalidomide,16 lenalidomide,17 and cyclosporine plus dexamethasone18 have been successfully used to achieve remission for up to 3 years.
Conclusion
Angioimmunoblastic T-cell lymphoma is difficult to diagnose due to nonspecific clinical and histologic findings. Cutaneous manifestations are seen in AITL in up to half of cases that may occur early or in advanced disease. Similar to all cutaneous metastases, the appearance of the lesions can greatly vary. Our case demonstrates that dermatologists and dermatopathologists can make this diagnosis in the appropriate clinicopathologic context utilizing appropriate immunohistochemical staining and gene rearrangement studies.
Angioimmunoblastic T-cell lymphoma (AITL) is a rare, often aggressive type of peripheral T-cell lymphoma. It comprises 18% of peripheral T-cell lymphomas and 1% to 2% of all non-Hodgkin lymphomas.1 The incidence of AITL in the United States is estimated to be 0.05 cases per 100,000 person-years,2 and there is a slight male predominance.1,3,4 It typically presents in the seventh decade of life; however, cases have been reported in adults ranging from 20 to 91 years of age.3
Angioimmunoblastic T-cell lymphoma presents with lymphadenopathy, hepatosplenomegaly, and systemic B symptoms (eg, fever, night sweats, weight loss, generalized pruritus).4-6 There are cutaneous manifestations in up to 50% of cases4,5,7 and frequently signs of autoimmune disorder.4,5 The diagnosis often is made by excisional lymph node biopsy. Lymph node specimens characteristically have a mixed inflammatory infiltrate that includes numerous B cells often infected with Epstein-Barr virus (EBV) and a relatively small population of atypical T lymphocytes.8 Identification of this neoplastic population of CD4+CD8− T lymphocytes expressing normal follicular helper T-cell markers CD10, chemokine CXCL13, programmed cell death protein 1 (PD-1), and B-cell lymphoma 6 (BCL-6) confirms the diagnosis of AITL.9,10 These malignant cells can be identified in skin specimens in cases of cutaneous metastatic disease.11,12 We present a case originally misdiagnosed as diffuse large B-cell lymphoma that was later identified as AITL on skin biopsy.
Case Report
A 72-year-old woman presented with a pruritic erythematous eruption around the neck of 3 weeks’ duration (Figure 1). Her medical history was notable for diffuse large B-cell non-Hodgkin lymphoma diagnosed 3 months prior based on results from a right cervical lymph node biopsy. She was treated with bendamustine and rituximab. On physical examination there were erythematous edematous papules coalescing into indurated plaques around the neck. The differential diagnosis included drug hypersensitivity reaction, herpes zoster, urticaria, and cutaneous metastasis. Two punch biopsies were taken for hematoxylin and eosin and tissue culture.
Tissue cultures and viral polymerase chain reaction were negative. Histopathologic examination revealed a scant atypical lymphoid infiltrate focally involving the deep dermis. The cells were medium to large in size and contained hyperchromatic pleomorphic nuclei (Figure 2). They were positive for CD3 and CD4, which was concerning for T-cell lymphoma. The histologic report of the excisional lymph node biopsy done 3 months prior described an atypical lymphoid neoplasm with extensive necrosis and extranodal spread that stained positively for CD20 (Figure 3).
Further staining of this cervical lymph node specimen revealed large atypical lymphoid cells positive for CD3, CD10, B-cell lymphoma 2 (BCL-2), BCL-6, and PD-1. There were intermixed mature B lymphocytes positive for CD20 and BCL-2. Chromogenic in situ hybridization with probes for EBV showed numerous positive cells throughout the infiltrate. Polymerase chain reaction demonstrated a T-cell population with clonally rearranged T-cell receptor genes. Primers for immunoglobulin heavy and light chains showed no evidence of a clonal B-cell population.
Additional staining of the atypical cutaneous lymphocytes revealed positivity for CD3, CD10, and PD-1. The morphologic and immunophenotypic findings of both specimens supported the diagnosis of AITL.
The patient declined further treatment and chose hospice care.
Comment
Etiology
Angioimmunoblastic T-cell lymphoma was originally named angioimmunoblastic lymphadenopathy with dysproteinemia. It was initially thought to be a benign hyperreactive immune process driven by B cells, and patients often died of infectious complications not long after the diagnosis was made.13 As more cases were reported with clonal rearrangements and signs of progressive lymphoma, AITL was recognized as a malignancy.
Presentation
Patients with AITL often present with advanced stage III or IV disease with extranodal and bone marrow involvement.3-6 Cutaneous disease occurs in up to half of patients and portends a poor prognosis.7 The rash often is a nonspecific erythematous macular and papular eruption mimicking a morbilliform viral exanthem or drug reaction. Urticarial, nodular, petechial, purpuric, eczematous, erythrodermic, and vesiculobullous presentations have been described.4,11,12 In up to one-third of cases, the eruption occurs in association with a new medication, often leading to an initial misdiagnosis of drug hypersensitivity reaction.4,11 In a review conducted by Balaraman et al,14 84% of patients with AITL reported having pruritus.
There is an association of autoimmune phenomena in patients with AITL, which is likely a result of immune dysregulation associated with poorly functioning follicular helper T cells. Patients may present with arthralgia, hemolytic anemia, or thrombocytopenic purpura. Hypergammaglobulinemia has been reported in 30% to 50% of AITL patients.4,6 Other pertinent immunologic findings include positive Coombs test, cold agglutinins, cryoglobulinemia, hypocomplementemia, and positive antinuclear antibodies.4-7
Gene Analysis
Affected lymph nodes have a characteristically effaced architecture with proliferative arborizing venules; a hyperplastic population of follicular dendritic cells; and a mixed inflammatory infiltrate that is comprised of atypical lymphocytes and variable numbers of reactive lymphocytes, histiocytes, eosinophils, and plasma cells. The malignant T lymphocytes often account for only a small portion of the infiltrate.8 T-cell gene rearrangement studies identify clonal cells with β and γ rearrangements in the majority of cases.4 These cells are predominantly CD4+CD8− and express normal follicular helper T-cell markers CD10, CXCL13, BCL-6,5,9 and PD-1.10 Numerous B cells are seen intermixed with follicular dendritic cells. They are frequently infected with EBV and can have an atypical Reed-Sternberg cell–like appearance.4,5,15 In the evaluation of AITL, polymerase chain reaction studies with primers for immunoglobulin heavy and light chain should be performed to look for clonal B-cell populations and rule out a possible secondary B-cell lymphoma.
Histology
Five histologic patterns have been described with cutaneous AITL: (1) superficial perivascular infiltrate of eosinophils and lymphocytes that lack atypia, (2) sparse perivascular infiltrate with atypical lymphocytes, (3) dense dermal infiltrate of pleomorphic lymphocytes, (4) leukocytoclastic vasculitis without atypical lymphocytes,11 and (5) necrotizing vasculitis.12 The finding of vascular hyperplasia, perivascular infiltrate, or vasculitis has been reported in 91% of cases in the literature. Although these findings are nonspecific, an analysis of cutaneous cases reported in the literature found that 87% demonstrated T-cell receptor gene rearrangements.14 Lymphoid cells are positive for CD10 and PD-1, as was demonstrated in our case, and are CXCL13 positive in the majority of cases.12 Atypical and EBV-infected B cells also can be found in the skin.11,12
Differential Diagnosis
Angioimmunoblastic T-cell lymphoma can mimic infectious, autoimmune, or allergic etiologies, and misdiagnosis of another type of lymphoma is not uncommon, as occurred in our case. Patients who have a delay in the correct diagnosis have similar outcomes to those correctly diagnosed at first presentation.4
Treatment
There are no effective therapies for AITL. Poor prognostic factors include age (>60 years), stages III to IV disease, male gender, elevated serum lactate dehydrogenase level,3,5,10 and cutaneous involvement.7 Corticosteroids, anthracycline-based chemotherapy, and autologous stem cell transplant are currently the mainstays of therapy. Initial response to chemotherapy is promising, but duration of response is poor overall and there is no increased survival.5,15 A large population-based study of 1207 cases by Xu and Liu3 showed the overall survival rate at 2 and 10 years was 46.8% and 21.9%, respectively. Ten-year disease-specific survival was 35.9%, and there was no demonstrable improvement in survival over the last 2 decades.3 Case reports have demonstrated that thalidomide,16 lenalidomide,17 and cyclosporine plus dexamethasone18 have been successfully used to achieve remission for up to 3 years.
Conclusion
Angioimmunoblastic T-cell lymphoma is difficult to diagnose due to nonspecific clinical and histologic findings. Cutaneous manifestations are seen in AITL in up to half of cases that may occur early or in advanced disease. Similar to all cutaneous metastases, the appearance of the lesions can greatly vary. Our case demonstrates that dermatologists and dermatopathologists can make this diagnosis in the appropriate clinicopathologic context utilizing appropriate immunohistochemical staining and gene rearrangement studies.
- Rudiger T, Weisenburger DD, Anderson JR, et al. Peripheral T-cell lymphoma (excluding anaplastic large-cell lymphoma): results from the Non-Hodgkins Lymphoma Classification Project. Ann Oncol. 2002;13:140-149.
- Morton LM, Wang SS, Devesa SS, et al. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood. 2006;107:265-276.
- Xu B, Liu P. No survival improvement for patients with angioimmunoblastic T-cell lymphoma over the past two decades: a population-based study of 1207 cases. PLoS One. 2014;9:e92585.
- Lachenal F, Berger F, Ghesquieres H, et al. Angioimmunoblastic T-cell lymphoma: clinical and laboratory features at diagnosis in 77 patients. Medicine (Baltimore). 2007;86:282-292.
- Mourad N, Mounier N, Briére J, et al. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l’Adulte (GELA) trials. Blood. 2008;111:4463-4470.
- Frederico M, Rudiger T, Bellei M, et al. Clinicopathologic characteristics of angioimmunoblastic T-cell lymphoma: analysis of the International Peripheral T-cell Lymphoma Project. J Clin Oncol. 2013;31:240-246.
- Siegert W, Nerl C, Agthe A, et al. Angioimmunoblastic lym-phadenopathy (AILD)-type T-cell lymphoma: prognostic impact of clinical observations and laboratory findings at presentation. The Kiel Lymphoma Study Group. Ann Oncol. 1995;6:659-664.
- Attygalle AD, Chuang SS, Diss TC, et al. Distinguishing angioimmunoblastic T-cell lymphoma from peripheral T-cell lymphoma, unspecified, using morphology, immunophenotype, and molecular genetics. Histopathology. 2007;50:498-508.
- Dupuis J, Boye K, Martin N, et al. Expression of CXCL13 by neoplastic cells in angioimmunoblastic T-cell lymphoma (AITL): a new diagnostic marker providing evidence that AITL derives from follicular helper cells. Am J Surg Pathol. 2006;30:490-494.
- Odejide O, Weigert O, Lane AA, et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123:1293-1296.
- Martel P, Laroche L, Courville P, et al. Cutaneous involvementin patients with angioimmunoblastic lymphadenopathy with dysproteinemia: a clinical, immunohistological, and molecular analysis. Arch Dermatol. 2000;136:881-886.
- Ortonne N, Dupuis J, Plonquet A, et al. Characterization of CXCL13+ neoplastic t cells in cutaneous lesions of angioimmunoblastic T-cell lymphoma (AITL). Am J Surg Pathol. 2007;31:1068-1076.
- Frizzera G, Moran E, Rappaport H. Angioimmunoblastic lymphadenopathy with dysproteinemia. Lancet. 1974;1:1070-1073.
- Balaraman B, Conley JA, Sheinbein DM. Evaluation of cutaneous angioimmunoblastic T-cell lymphoma [published online May 6, 2011]. J Am Acad Dermatol. 2011;65:855-862.
- Tokunaga T, Shimada K, Yamamoto K, et al. Retrospective analysis of prognostic factors for angioimmunoblastic T-cell lymphoma: a multicenter cooperative study in Japan. Blood. 2012;119:2837-2843.
- Dogan A, Ngu LSP, Ng SH, et al. Pathology and clinical features of angioimmunoblastic T-cell lymphoma after successful treatment with thalidomide. Leukemia. 2005;19:873-875.
- Fabbri A, Cencini E, Pietrini A, et al. Impressive activity of lenalidomide monotherapy in refractory angioimmunoblastic T-cell lymphoma: report of a case with long-term follow-up. Hematol Oncol. 2013;31:213-217.
- Kobayashi T, Kuroda J, Uchiyama H, et al. Successful treatment of chemotherapy-refractory angioimmunoblastic T cell lymphoma with cyclosporin A. Acta Haematol. 2012;127:10-15.
- Rudiger T, Weisenburger DD, Anderson JR, et al. Peripheral T-cell lymphoma (excluding anaplastic large-cell lymphoma): results from the Non-Hodgkins Lymphoma Classification Project. Ann Oncol. 2002;13:140-149.
- Morton LM, Wang SS, Devesa SS, et al. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood. 2006;107:265-276.
- Xu B, Liu P. No survival improvement for patients with angioimmunoblastic T-cell lymphoma over the past two decades: a population-based study of 1207 cases. PLoS One. 2014;9:e92585.
- Lachenal F, Berger F, Ghesquieres H, et al. Angioimmunoblastic T-cell lymphoma: clinical and laboratory features at diagnosis in 77 patients. Medicine (Baltimore). 2007;86:282-292.
- Mourad N, Mounier N, Briére J, et al. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l’Adulte (GELA) trials. Blood. 2008;111:4463-4470.
- Frederico M, Rudiger T, Bellei M, et al. Clinicopathologic characteristics of angioimmunoblastic T-cell lymphoma: analysis of the International Peripheral T-cell Lymphoma Project. J Clin Oncol. 2013;31:240-246.
- Siegert W, Nerl C, Agthe A, et al. Angioimmunoblastic lym-phadenopathy (AILD)-type T-cell lymphoma: prognostic impact of clinical observations and laboratory findings at presentation. The Kiel Lymphoma Study Group. Ann Oncol. 1995;6:659-664.
- Attygalle AD, Chuang SS, Diss TC, et al. Distinguishing angioimmunoblastic T-cell lymphoma from peripheral T-cell lymphoma, unspecified, using morphology, immunophenotype, and molecular genetics. Histopathology. 2007;50:498-508.
- Dupuis J, Boye K, Martin N, et al. Expression of CXCL13 by neoplastic cells in angioimmunoblastic T-cell lymphoma (AITL): a new diagnostic marker providing evidence that AITL derives from follicular helper cells. Am J Surg Pathol. 2006;30:490-494.
- Odejide O, Weigert O, Lane AA, et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123:1293-1296.
- Martel P, Laroche L, Courville P, et al. Cutaneous involvementin patients with angioimmunoblastic lymphadenopathy with dysproteinemia: a clinical, immunohistological, and molecular analysis. Arch Dermatol. 2000;136:881-886.
- Ortonne N, Dupuis J, Plonquet A, et al. Characterization of CXCL13+ neoplastic t cells in cutaneous lesions of angioimmunoblastic T-cell lymphoma (AITL). Am J Surg Pathol. 2007;31:1068-1076.
- Frizzera G, Moran E, Rappaport H. Angioimmunoblastic lymphadenopathy with dysproteinemia. Lancet. 1974;1:1070-1073.
- Balaraman B, Conley JA, Sheinbein DM. Evaluation of cutaneous angioimmunoblastic T-cell lymphoma [published online May 6, 2011]. J Am Acad Dermatol. 2011;65:855-862.
- Tokunaga T, Shimada K, Yamamoto K, et al. Retrospective analysis of prognostic factors for angioimmunoblastic T-cell lymphoma: a multicenter cooperative study in Japan. Blood. 2012;119:2837-2843.
- Dogan A, Ngu LSP, Ng SH, et al. Pathology and clinical features of angioimmunoblastic T-cell lymphoma after successful treatment with thalidomide. Leukemia. 2005;19:873-875.
- Fabbri A, Cencini E, Pietrini A, et al. Impressive activity of lenalidomide monotherapy in refractory angioimmunoblastic T-cell lymphoma: report of a case with long-term follow-up. Hematol Oncol. 2013;31:213-217.
- Kobayashi T, Kuroda J, Uchiyama H, et al. Successful treatment of chemotherapy-refractory angioimmunoblastic T cell lymphoma with cyclosporin A. Acta Haematol. 2012;127:10-15.
Practice Points
- Angioimmunoblastic T-cell lymphoma (AITL) is a rare, often aggressive type of peripheral T-cell lymphoma.
- Cutaneous manifestations have been seen in up to 50% of cases.
- Immunohistochemical markers for normal follicular helper T cells—CD-10, chemokine CXCL-13, and programmed cell death protein 1 (PD-1)—can be used to differentiate AITL from other types of lymphoma.
- The prognosis of AITL is poor.