User login
The Basement Flight
A 14-year-old girl with a history of asthma presented to the Emergency Department (ED) with three months of persistent, nonproductive cough, and progressive shortness of breath. She reported fatigue, chest tightness, orthopnea, and dyspnea with exertion. She denied fever, rhinorrhea, congestion, hemoptysis, or paroxysmal nocturnal dyspnea.
Her age and past medical history of asthma are incongruent with her new symptoms, as asthma is typified by intermittent exacerbations, not progressive symptoms. Thus, another process, in addition to asthma, is most likely present; it is also important to question the accuracy of previous diagnoses in light of new information. Her symptoms may signify an underlying cardiopulmonary process, such as infiltrative diseases (eg, lymphoma or sarcoidosis), atypical infections, genetic conditions (eg, variant cystic fibrosis), autoimmune conditions, or cardiomyopathy. A detailed symptom history, family history, and careful physical examination will help expand and then refine the differential diagnosis. At this stage, typical infections are less likely.
She had presented two months prior with nonproductive cough and dyspnea. At that presentation, her temperature was 36.3°C, heart rate 110 beats per minute, blood pressure 119/63 mm Hg, respiratory rate 43 breaths per minute, and oxygen saturation 86% while breathing ambient air. A chest CT with contrast demonstrated diffuse patchy multifocal ground-glass opacities in the bilateral lungs as well as a mixture of atelectasis and lobular emphysema in the dependent lobes bilaterally (Figure 1). Her main pulmonary artery was dilated at 3.6 cm (mean of 2.42 cm with SD 0.22). She was diagnosed with atypical pneumonia. She was administered azithromycin, weaned off oxygen, and discharged after a seven-day hospitalization.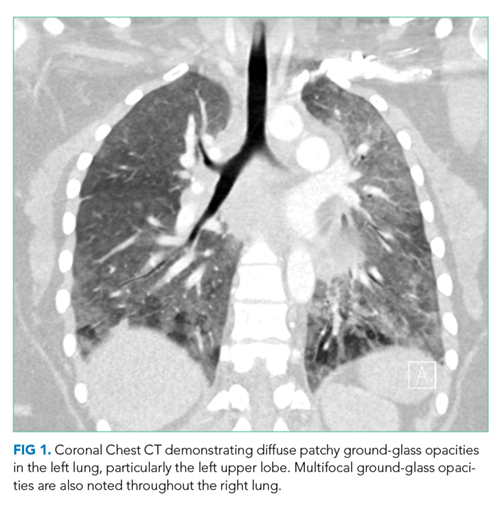
Two months prior, she had marked tachypnea, tachycardia, and hypoxemia, and imaging revealed diffuse ground-glass opacities. The differential diagnosis for this constellation of symptoms is extensive and includes many conditions that have an inflammatory component, such as atypical pneumonia caused by Mycoplasma or Chlamydia pneumoniae or a common respiratory virus such as rhinovirus or human metapneumovirus. However, two findings make an acute pneumonia unlikely to be the sole cause of her symptoms: underlying emphysema and an enlarged pulmonary artery. Emphysema is an uncommon finding in children and can be related to congenital or acquired causes; congenital lobar emphysema most often presents earlier in life and is focal, not diffuse. Alpha-1-anti-trypin deficiency and mutations in connective tissue genes such as those encoding for elastin and fibrillin can lead to pulmonary disease. While not diagnostic of pulmonary hypertension, her dilated pulmonary artery, coupled with her history, makes pulmonary hypertension a strong possibility. While her pulmonary hypertension is most likely secondary to chronic lung disease based on the emphysematous changes on CT, it could still be related to a cardiac etiology.
The patient had a history of seasonal allergies and well-controlled asthma. She was hospitalized at age six for an asthma exacerbation associated with a respiratory infection. She was discharged with an albuterol inhaler, but seldom used it. Her parents denied any regular coughing during the day or night. She was morbidly obese. Her tonsils and adenoids were removed to treat obstructive sleep apnea (OSA) at age seven, and a subsequent polysomnography was normal. Her medications included intranasal fluticasone propionate and oral iron supplementation. She had no known allergies or recent travels. She had never smoked. She had two pet cats and a dog. Her mother had a history of obesity, OSA, and eczema. Her father had diabetes and eczema.
The patient’s history prior to the recent few months sheds little light on the cause of her current symptoms. While it is possible that her current symptoms are related to the worsening of a process that had been present for many years which mimicked asthma, this seems implausible given the long period of time between her last asthma exacerbation and her present symptoms. Similarly, while tonsillar and adenoidal hypertrophy can be associated with infiltrative diseases (such as lymphoma), this is less common than the usual (and normal) disproportionate increase in size of the adenoids compared to other airway structures during growth in children.
She was admitted to the hospital. On initial examination, her temperature was 37.4°C, heart rate 125 beats per minute, blood pressure 143/69 mm Hg, respiratory rate 48 breaths per minute, and oxygen saturation 86% breathing ambient air. Her BMI was 58 kg/m2. Her exam demonstrated increased work of breathing with accessory muscle use, and decreased breath sounds at the bases. There were no wheezes or crackles. Cardiovascular, abdominal, and skin exams were normal except for tachycardia. At rest, later in the hospitalization, her oxygen saturation was 97% breathing ambient air and heart rate 110 bpm. After two minutes of walking, her oxygen saturation was 77% and heart rate 132 bpm. Two minutes after resting, her oxygen saturation increased to 91%.
Her white blood cell count was 11.9 x 10 9 /L (67% neutrophils, 24.2% lymphocytes, 6% monocytes, and 2% eosinophils), hemoglobin 11.2 g/dL, and platelet count 278,000/mm 3 . Her complete metabolic panel was normal. The C-reactive protein (CRP) was 24 mg/L (normal range, < 4.9) and erythrocyte sedimentation rate (ESR) 103 mm/hour (normal range, 0-32). A venous blood gas (VBG) showed a pH of 7.42 and pCO2 39. An EKG demonstrated sinus tachycardia.
The combination of the patient’s tachypnea, hypoxemia, respiratory distress, and obesity is striking. Her lack of adventitious lung sounds is surprising given her CT findings, but the sensitivity of chest auscultation may be limited in obese patients. Her laboratory findings help narrow the diagnostic frame: she has mild anemia and leukocytosis along with significant inflammation. The normal CO2 concentration on VBG is concerning given the degree of her tachypnea and reflects significant alveolar hypoventilation.
This marked inflammation with diffuse lung findings again raises the possibility of an inflammatory or, less likely, infectious disorder. Sjogren’s syndrome, systemic lupus erythematosus (SLE), and juvenile dermatomyositis can present in young women with interstitial lung disease. She does have exposure to pets and hypersensitivity pneumonitis can worsen rapidly with continued exposure. Another possibility is that she has an underlying immunodeficiency such as common variable immunodeficiency, although a history of recurrent infections such as pneumonia, bacteremia, or sinusitis is lacking.
An echocardiogram should be performed. In addition, laboratory evaluation for the aforementioned autoimmune causes of interstitial lung disease, immunoglobulin levels, pulmonary function testing (if available as an inpatient), and potentially a bronchoscopy with bronchoalveolar lavage (BAL), and biopsy should be pursued. The BAL and biopsy would be helpful in evaluating for infection and interstitial lung disease in an expeditious manner.
A chest CT without contrast was done and compared to the scan from two months prior. New diffuse, ill-defined centrilobular ground-glass opacities were evident throughout the lung fields; dilation of the main pulmonary artery was unchanged, and previously seen ground-glass opacities had resolved. There were patchy areas of air-trapping and mosaic attenuation in the lower lobes (Figure 2).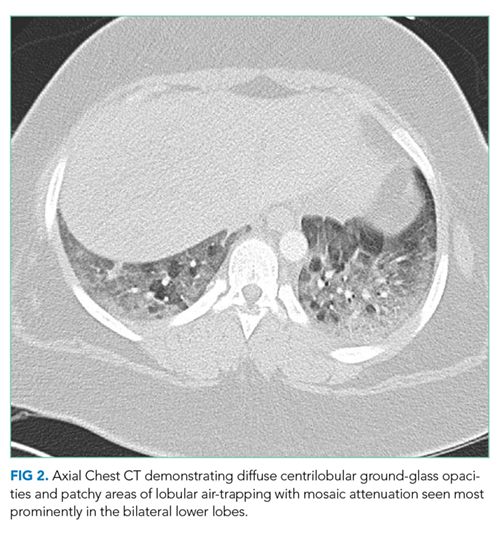
Transthoracic echocardiogram demonstrated a right ventricular systolic pressure of 58 mm Hg with flattened intraventricular septum during systole. Left and right ventricular systolic function were normal. The left ventricular diastolic function was normal. Pulmonary function testing demonstrated a FEV1/FVC ratio of 100 (112% predicted), FVC 1.07 L (35 % predicted) and FEV1 1.07 L (39% predicted), and total lung capacity was 2.7L (56% predicted) (Figure 3). Single-breath carbon monoxide uptake in the lung was not interpretable based on 2017 European Respiratory Society (ERS)/American Thoracic Society (ATS) technical standards.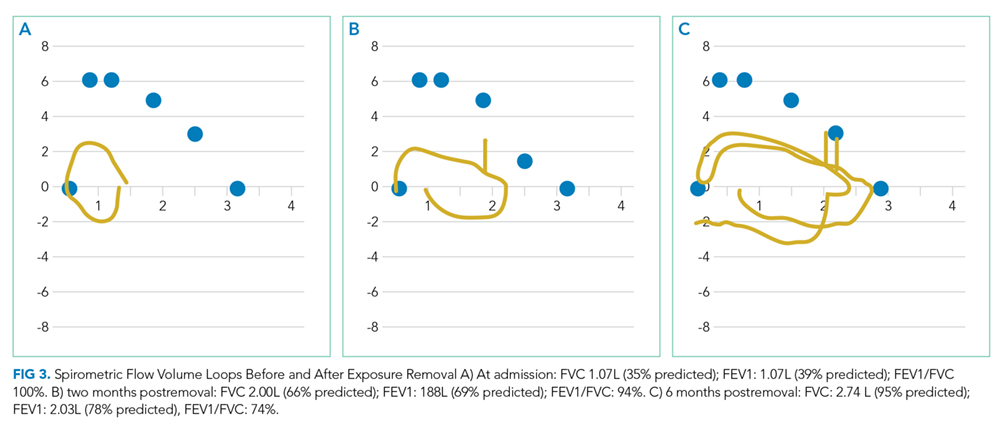
This information is helpful in classifying whether this patient’s primary condition is cardiac or pulmonary in nature. Her normal left ventricular systolic and diastolic function make a cardiac etiology for her pulmonary hypertension less likely. Further, the combination of pulmonary hypertension, a restrictive pattern on pulmonary function testing, and findings consistent with interstitial lung disease on cross-sectional imaging all suggest a primary pulmonary etiology rather than a cardiac, infectious, or thromboembolic condition. While chronic thromboembolic hypertension can result in nonspecific mosaic attenuation, it typically would not cause centrilobular ground-glass opacities nor restrictive lung disease. Thus, it seems most likely that this patient has a progressive pulmonary process resulting in hypoxia, pulmonary hypertension, centrilobular opacities, and lower-lobe mosaic attenuation. Considerations for this process can be broadly categorized as one of the childhood interstitial lung disease (chILD). While this differential diagnosis is broad, strong consideration should be given to hypersensitivity pneumonitis, chronic aspiration, sarcoidosis, and Sjogren’s syndrome. An intriguing possibility is that the patient’s “response to azithromycin” two months prior was due to the avoidance of an inhaled antigen while she was in the hospital; a detailed environmental history should be explored. The normal polysomnography after tonsilloadenoidectomy makes it unlikely that OSA is a major contributor to her current presentation. However, since the surgery was seven years ago, and her BMI is presently 58 kg/m2 she remains at risk for OSA and obesity-hypoventilation syndrome. Polysomnography should be done after her acute symptoms improve.
She was started on 5 mm Hg of continuous positive airway pressure (CPAP) at night after a sleep study on room air demonstrated severe OSA with a respiratory disturbance index of 13 events per hour. Antinuclear antibodies (ANA), anti-neutrophil cytoplasmic antibody (ANCA), anti-Jo-1 antibody, anti-RNP antibody, anti-Smith antibody, anti-Ro/SSA and anti-La/SSB antibody were negative as was the histoplasmin antibody. Serum angiotensin-converting enzyme (ACE) level was normal. Mycoplasma IgM and IgG were negative. IgE was 529 kU/L (normal range, <114).
This evaluation reduces the likelihood the patient has Sjogren’s syndrome, SLE, dermatomyositis, or ANCA-associated pulmonary disease. While many patients with dermatomyositis may have negative serologic evaluations, other findings usually present such as rash and myositis are lacking. The negative ANCA evaluation makes granulomatosis with polyangiitis and microscopic polyangiitis very unlikely given the high sensitivity of the ANCA assay for these conditions. ANCA assays are less sensitive for eosinophilic granulomatosis with polyangiitis (EGPA), but the lack of eosinophilia significantly decreases the likelihood of EGPA. ACE levels have relatively poor operating characteristics in the evaluation of sarcoidosis; however, sarcoidosis seems unlikely in this case, especially as patients with sarcoidosis tend to have low or normal IgE levels. Patients with asthma can have elevated IgE levels. However, very elevated IgE levels are more common in other conditions, including allergic bronchopulmonary aspergillosis (ABPA) and the Hyper-IgE syndrome. The latter manifests with recurrent infections and eczema, and is inherited in an autosomal dominant manner. However, both the Hyper-IgE syndrome and ABPA have much higher IgE levels than seen in this case. Allergen-specific IgE testing (including for antibodies to Aspergillus) should be sent. It seems that an interstitial lung disease is present; the waxing and waning pattern and clinical presentation, along with the lack of other systemic findings, make hypersensitivity pneumonitis most likely.
The family lived in an apartment building. Her symptoms started when the family’s neighbor recently moved his outdoor pigeon coop into his basement. The patient often smelled the pigeons and noted feathers coming through the holes in the wall.
One of the key diagnostic features of hypersensitivity pneumonitis (HP) is the history of exposure to a potential offending antigen—in this case likely bird feathers—along with worsening upon reexposure to that antigen. HP is primarily a clinical diagnosis, and testing for serum precipitants has limited value, given the high false negative rate and the frequent lack of clinical symptoms accompanying positive testing. Bronchoalveolar lavage fluid may reveal lymphocytosis and reduced CD4:CD8 ratio. Crackles are commonly heard on examination, but in this case were likely not auscultated due to her obese habitus. The most important treatment is withdrawal of the offending antigen. Limited data suggest that corticosteroid therapy may be helpful in certain HP cases, including subacute, chronic and severe cases as well as patients with hypoxemia, significant imaging findings, and those with significant abnormalities on pulmonary function testing (PFT).
A hypersensitivity pneumonitis precipitins panel was sent with positive antibodies to M. faeni, T. Vulgaris, A. Fumigatus 1 and 6, A. Flavus, and pigeon serum. Her symptoms gradually improved within five days of oral prednisone (60 mg). She was discharged home without dyspnea and normal oxygen saturation while breathing ambient air. A repeat echocardiogram after nighttime CPAP for 1 week demonstrated a right ventricular systolic pressure of 17 mm Hg consistent with improved pulmonary hypertension.
Three weeks later, she returned to clinic for follow up. She had re-experienced dyspnea, cough, and wheezing, which improved when she was outdoors. She was afebrile, tachypneic, tachycardic, and her oxygen saturation was 92% on ambient air.
Her steroid-responsive interstitial lung disease and rapid improvement upon avoidance of the offending antigen is consistent with HP. The positive serum precipitins assay lends further credence to the diagnosis of HP, although serologic analysis with such antibody assays is limited by false positives and false negatives; further, individuals exposed to pigeons often have antibodies present without evidence of HP. History taking at this visit should ask specifically about further pigeon exposure: were the pigeons removed from the home completely, were heating-cooling filters changed, carpets cleaned, and bedding laundered? An in-home evaluation may be helpful before conducting further diagnostic testing.
She was admitted for oxygen therapy and a bronchoscopy, which showed mucosal friability and cobblestoning, suggesting inflammation. BAL revealed a normal CD4:CD8 ratio of 3; BAL cultures were sterile. Her shortness of breath significantly improved following a prolonged course of systemic steroids and removal from the triggering environment. PFTs improved with a FEV1/FVC ratio of 94 (105% predicted), FVC of 2.00 L (66% predicted), FEV1 of 1.88L (69% predicted) (Figure 3B). Her presenting symptoms of persistent cough and progressive dyspnea on exertion, characteristic CT, sterile BAL cultures, positive serum precipitants against pigeon serum, and resolution of her symptoms with withdrawal of the offending antigen were diagnostic of hypersensitivity pneumonitis due to pigeon exposure, also known as bird fancier’s disease.
COMMENTARY
The patient’s original presentation of dyspnea, tachypnea, and hypoxia is commonly associated with pediatric pneumonia and asthma exacerbations.1 However, an alternative diagnosis was suggested by the lack of wheezing, absence of fever, and recurrent presentations with progressive symptoms.
Hypersensitivity pneumonitis (HP) represents an exaggerated T-cell meditated immune response to inhalation of an offending antigen that results in a restrictive ventilatory defect and interstitial infiltrates.2 Bird pneumonitis (also known as bird fancier’s disease) is a frequent cause of HP, accounting for approximately 65-70% of cases.3 HP, however, only manifests in a small number of subjects exposed to culprit antigens, suggesting an underlying genetic susceptibility.4 Prevalence estimates vary depending on bird species, county, climate, and other possible factors.
There are no standard criteria for the diagnosis of HP, though a combination of findings is suggestive. A recent prospective multicenter study created a scoring system for HP based on factors associated with the disease to aid in accurate diagnosis. The most relevant criteria included antigen exposure, recurrent symptoms noted within 4-8 hours after antigen exposure, weight loss, presence of specific IgG antibodies to avian antigens, and inspiratory crackles on exam. Using this rule, the probability that our patient has HP based on clinical characteristics was 93% with an area under the receiver operating curve of 0.93 (96% confidence interval: 0.90-0.95)5. Chest imaging (high resolution CT) often consists of a mosaic pattern of air trapping, as seen in this patient in combination with ground-glass opacities6. Bronchoalveolar lavage (BAL) is sensitive in detecting lung inflammation in a patient with suspected HP. On BAL, a lymphocytic alveolitis can be seen, but absence of this finding does not exclude HP.5,7,8 Pulmonary function tests (PFTs) may be normal in acute HP. When abnormal, PFTs may reveal a restrictive pattern and reduction in carbon monoxide diffusing capacity.7 However, BAL and PFT results are neither specific nor diagnostic of HP; it is important to consider results in the context of the clinical picture.
The respiratory response to inhalation of the avian antigen has traditionally been classified as acute, subacute, or chronic.9 The acute response occurs within hours of exposure to the offending agent and usually resolves within 24 hours after antigen withdrawal. The subacute presentation involves cough and dyspnea over several days to weeks, and can progress to chronic and permanent lung damage if unrecognized and untreated. In chronic presentations, lung abnormalities may persist despite antigen avoidance and pharmacologic interventions.4,10 The patient’s symptoms occurred over a six-month period which coincided with pigeon exposure and resolved during each hospitalization with steroid treatment and removal from the offending agent. Her presentation was consistent with a subacute time course of HP.
The dilated pulmonary artery, elevated right systolic ventricular pressure, and normal right ventricular function in our patient suggested pulmonary hypertension of chronic duration. Her risk factors for pulmonary hypertension included asthma, sleep apnea, possible obesity-hypoventilation syndrome, and HP-associated interstitial lung disease.11
The most important intervention in HP is avoidance of the causative antigen. Medical therapy without removal of antigen is inadequate. Systemic corticosteroids can help ameliorate acute symptoms though dosing and duration remains unclear. For chronic patients unresponsive to steroid therapy, lung transplantation can be considered.4
The key to diagnosis of HP in this patient—and to minimizing repeat testing upon the patient’s recrudescence of symptoms—was the clinician’s consideration that the major impetus for the patient’s improvement in the hospital was removal from the offending antigen in her home environment. As in this case, taking time to delve deeply into a patient’s environment—even by descending the basement stairs—may lead to the diagnosis.
LEARNING POINTS
- Consider hypersensitivity pneumonitis (HP) in patients with recurrent respiratory distress, offending exposure, and resolution of symptoms with removal of culprit antigen.
- The most important treatment of HP is removal of offending antigen; systemic and/or inhaled corticosteroids are indicated until the full resolution of respiratory symptoms.
- Prognosis is dependent on early diagnosis and removal of offending exposures.
- Failure to treat HP might result in end-stage lung disease from pulmonary fibrosis secondary to long-term inflammation.
Disclosures
Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The authors declare no conflicts of interests.
1. Ebell MH. Clinical diagnosis of pneumonia in children. Am Fam Physician. 2010;82(2):192-193. PubMed
2. Cormier Y, Lacasse Y. Hypersensitivity pneumonitis and organic dust toxic syndrome. In: Malo J-L, Chan-Yeung M, Bernstein DI, eds. Asthma in the Workplace. Vol 32. Boca Raton, FL: Fourth Informa Healthcare; 2013:392-405.
3. Chan AL, Juarez MM, Leslie KO, Ismail HA, Albertson TE. Bird fancier’s lung: a state-of-the-art review. Clin Rev Allergy Immunol. 2012;43(1-2):69-83. doi: 10.1007/s12016-011-8282-y. PubMed
4. Camarena A, Juárez A, Mejía M, et al. Major histocompatibility complex and tumor necrosis factor-α polymorphisms in pigeon breeder’s disease. Am J Respir Crit Care Med. 2001;163(7):1528-1533. https:/doi.org/10.1164/ajrccm.163.7.2004023. PubMed
5. Lacasse Y, Selman M, Costabel U, et al. Clinical diagnosis of hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2003;168(8):952-958. doi: 10.1164/rccm.200301-137OC. PubMed
6. Glazer CS, Rose CS, Lynch DA. Clinical and radiologic manifestations of hypersensitivity pneumonitis. J Thorac Imaging. 2002;17(4):261-272. PubMed
7. Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med. 2012;186(4):314-324. doi: 10.1164/rccm.201203-0513CI. PubMed
8. Calillad DM, Vergnon, JM, Madroszyk A, et al. Bronchoalveolar lavage in hypersensitivity pneumonitis: a series of 139 patients. Inflamm Allergy Drug Targets. 2012;11(1):15-19. doi: 10.2174/187152812798889330. PubMed
9. Richerson HB, Bernstein IL, Fink JN, et al. Guidelines for the clinical evaluation of hypersensitivity pneumonitis. Report of the Subcommittee on Hypersensitivity Pneumonitis. J Allergy Clin Immunol. 1989;84(5 Pt 2):839-844. doi: 10.1016/0091-6749(89)90349-7. PubMed
10. Zacharisen MC, Schlueter DP, Kurup VP, Fink JN. The long-term outcome in acute, subacute, and chronic forms of pigeon breeder’s disease hypersensitivity pneumonitis. Ann Allergy Asthma Immunol. 2002;88(2):175-182. doi: 10.1016/S1081-1206(10)61993-X. PubMed
11. Raymond TE, Khabbaza JE, Yadav R, Tonelli AR. Significance of main pulmonary artery dilation on imaging studies. Ann Am Thorac Soc. 2014;11(10):1623-1632. doi: 10.1513/AnnalsATS.201406-253PP. PubMed
A 14-year-old girl with a history of asthma presented to the Emergency Department (ED) with three months of persistent, nonproductive cough, and progressive shortness of breath. She reported fatigue, chest tightness, orthopnea, and dyspnea with exertion. She denied fever, rhinorrhea, congestion, hemoptysis, or paroxysmal nocturnal dyspnea.
Her age and past medical history of asthma are incongruent with her new symptoms, as asthma is typified by intermittent exacerbations, not progressive symptoms. Thus, another process, in addition to asthma, is most likely present; it is also important to question the accuracy of previous diagnoses in light of new information. Her symptoms may signify an underlying cardiopulmonary process, such as infiltrative diseases (eg, lymphoma or sarcoidosis), atypical infections, genetic conditions (eg, variant cystic fibrosis), autoimmune conditions, or cardiomyopathy. A detailed symptom history, family history, and careful physical examination will help expand and then refine the differential diagnosis. At this stage, typical infections are less likely.
She had presented two months prior with nonproductive cough and dyspnea. At that presentation, her temperature was 36.3°C, heart rate 110 beats per minute, blood pressure 119/63 mm Hg, respiratory rate 43 breaths per minute, and oxygen saturation 86% while breathing ambient air. A chest CT with contrast demonstrated diffuse patchy multifocal ground-glass opacities in the bilateral lungs as well as a mixture of atelectasis and lobular emphysema in the dependent lobes bilaterally (Figure 1). Her main pulmonary artery was dilated at 3.6 cm (mean of 2.42 cm with SD 0.22). She was diagnosed with atypical pneumonia. She was administered azithromycin, weaned off oxygen, and discharged after a seven-day hospitalization.
Two months prior, she had marked tachypnea, tachycardia, and hypoxemia, and imaging revealed diffuse ground-glass opacities. The differential diagnosis for this constellation of symptoms is extensive and includes many conditions that have an inflammatory component, such as atypical pneumonia caused by Mycoplasma or Chlamydia pneumoniae or a common respiratory virus such as rhinovirus or human metapneumovirus. However, two findings make an acute pneumonia unlikely to be the sole cause of her symptoms: underlying emphysema and an enlarged pulmonary artery. Emphysema is an uncommon finding in children and can be related to congenital or acquired causes; congenital lobar emphysema most often presents earlier in life and is focal, not diffuse. Alpha-1-anti-trypin deficiency and mutations in connective tissue genes such as those encoding for elastin and fibrillin can lead to pulmonary disease. While not diagnostic of pulmonary hypertension, her dilated pulmonary artery, coupled with her history, makes pulmonary hypertension a strong possibility. While her pulmonary hypertension is most likely secondary to chronic lung disease based on the emphysematous changes on CT, it could still be related to a cardiac etiology.
The patient had a history of seasonal allergies and well-controlled asthma. She was hospitalized at age six for an asthma exacerbation associated with a respiratory infection. She was discharged with an albuterol inhaler, but seldom used it. Her parents denied any regular coughing during the day or night. She was morbidly obese. Her tonsils and adenoids were removed to treat obstructive sleep apnea (OSA) at age seven, and a subsequent polysomnography was normal. Her medications included intranasal fluticasone propionate and oral iron supplementation. She had no known allergies or recent travels. She had never smoked. She had two pet cats and a dog. Her mother had a history of obesity, OSA, and eczema. Her father had diabetes and eczema.
The patient’s history prior to the recent few months sheds little light on the cause of her current symptoms. While it is possible that her current symptoms are related to the worsening of a process that had been present for many years which mimicked asthma, this seems implausible given the long period of time between her last asthma exacerbation and her present symptoms. Similarly, while tonsillar and adenoidal hypertrophy can be associated with infiltrative diseases (such as lymphoma), this is less common than the usual (and normal) disproportionate increase in size of the adenoids compared to other airway structures during growth in children.
She was admitted to the hospital. On initial examination, her temperature was 37.4°C, heart rate 125 beats per minute, blood pressure 143/69 mm Hg, respiratory rate 48 breaths per minute, and oxygen saturation 86% breathing ambient air. Her BMI was 58 kg/m2. Her exam demonstrated increased work of breathing with accessory muscle use, and decreased breath sounds at the bases. There were no wheezes or crackles. Cardiovascular, abdominal, and skin exams were normal except for tachycardia. At rest, later in the hospitalization, her oxygen saturation was 97% breathing ambient air and heart rate 110 bpm. After two minutes of walking, her oxygen saturation was 77% and heart rate 132 bpm. Two minutes after resting, her oxygen saturation increased to 91%.
Her white blood cell count was 11.9 x 10 9 /L (67% neutrophils, 24.2% lymphocytes, 6% monocytes, and 2% eosinophils), hemoglobin 11.2 g/dL, and platelet count 278,000/mm 3 . Her complete metabolic panel was normal. The C-reactive protein (CRP) was 24 mg/L (normal range, < 4.9) and erythrocyte sedimentation rate (ESR) 103 mm/hour (normal range, 0-32). A venous blood gas (VBG) showed a pH of 7.42 and pCO2 39. An EKG demonstrated sinus tachycardia.
The combination of the patient’s tachypnea, hypoxemia, respiratory distress, and obesity is striking. Her lack of adventitious lung sounds is surprising given her CT findings, but the sensitivity of chest auscultation may be limited in obese patients. Her laboratory findings help narrow the diagnostic frame: she has mild anemia and leukocytosis along with significant inflammation. The normal CO2 concentration on VBG is concerning given the degree of her tachypnea and reflects significant alveolar hypoventilation.
This marked inflammation with diffuse lung findings again raises the possibility of an inflammatory or, less likely, infectious disorder. Sjogren’s syndrome, systemic lupus erythematosus (SLE), and juvenile dermatomyositis can present in young women with interstitial lung disease. She does have exposure to pets and hypersensitivity pneumonitis can worsen rapidly with continued exposure. Another possibility is that she has an underlying immunodeficiency such as common variable immunodeficiency, although a history of recurrent infections such as pneumonia, bacteremia, or sinusitis is lacking.
An echocardiogram should be performed. In addition, laboratory evaluation for the aforementioned autoimmune causes of interstitial lung disease, immunoglobulin levels, pulmonary function testing (if available as an inpatient), and potentially a bronchoscopy with bronchoalveolar lavage (BAL), and biopsy should be pursued. The BAL and biopsy would be helpful in evaluating for infection and interstitial lung disease in an expeditious manner.
A chest CT without contrast was done and compared to the scan from two months prior. New diffuse, ill-defined centrilobular ground-glass opacities were evident throughout the lung fields; dilation of the main pulmonary artery was unchanged, and previously seen ground-glass opacities had resolved. There were patchy areas of air-trapping and mosaic attenuation in the lower lobes (Figure 2).
Transthoracic echocardiogram demonstrated a right ventricular systolic pressure of 58 mm Hg with flattened intraventricular septum during systole. Left and right ventricular systolic function were normal. The left ventricular diastolic function was normal. Pulmonary function testing demonstrated a FEV1/FVC ratio of 100 (112% predicted), FVC 1.07 L (35 % predicted) and FEV1 1.07 L (39% predicted), and total lung capacity was 2.7L (56% predicted) (Figure 3). Single-breath carbon monoxide uptake in the lung was not interpretable based on 2017 European Respiratory Society (ERS)/American Thoracic Society (ATS) technical standards.
This information is helpful in classifying whether this patient’s primary condition is cardiac or pulmonary in nature. Her normal left ventricular systolic and diastolic function make a cardiac etiology for her pulmonary hypertension less likely. Further, the combination of pulmonary hypertension, a restrictive pattern on pulmonary function testing, and findings consistent with interstitial lung disease on cross-sectional imaging all suggest a primary pulmonary etiology rather than a cardiac, infectious, or thromboembolic condition. While chronic thromboembolic hypertension can result in nonspecific mosaic attenuation, it typically would not cause centrilobular ground-glass opacities nor restrictive lung disease. Thus, it seems most likely that this patient has a progressive pulmonary process resulting in hypoxia, pulmonary hypertension, centrilobular opacities, and lower-lobe mosaic attenuation. Considerations for this process can be broadly categorized as one of the childhood interstitial lung disease (chILD). While this differential diagnosis is broad, strong consideration should be given to hypersensitivity pneumonitis, chronic aspiration, sarcoidosis, and Sjogren’s syndrome. An intriguing possibility is that the patient’s “response to azithromycin” two months prior was due to the avoidance of an inhaled antigen while she was in the hospital; a detailed environmental history should be explored. The normal polysomnography after tonsilloadenoidectomy makes it unlikely that OSA is a major contributor to her current presentation. However, since the surgery was seven years ago, and her BMI is presently 58 kg/m2 she remains at risk for OSA and obesity-hypoventilation syndrome. Polysomnography should be done after her acute symptoms improve.
She was started on 5 mm Hg of continuous positive airway pressure (CPAP) at night after a sleep study on room air demonstrated severe OSA with a respiratory disturbance index of 13 events per hour. Antinuclear antibodies (ANA), anti-neutrophil cytoplasmic antibody (ANCA), anti-Jo-1 antibody, anti-RNP antibody, anti-Smith antibody, anti-Ro/SSA and anti-La/SSB antibody were negative as was the histoplasmin antibody. Serum angiotensin-converting enzyme (ACE) level was normal. Mycoplasma IgM and IgG were negative. IgE was 529 kU/L (normal range, <114).
This evaluation reduces the likelihood the patient has Sjogren’s syndrome, SLE, dermatomyositis, or ANCA-associated pulmonary disease. While many patients with dermatomyositis may have negative serologic evaluations, other findings usually present such as rash and myositis are lacking. The negative ANCA evaluation makes granulomatosis with polyangiitis and microscopic polyangiitis very unlikely given the high sensitivity of the ANCA assay for these conditions. ANCA assays are less sensitive for eosinophilic granulomatosis with polyangiitis (EGPA), but the lack of eosinophilia significantly decreases the likelihood of EGPA. ACE levels have relatively poor operating characteristics in the evaluation of sarcoidosis; however, sarcoidosis seems unlikely in this case, especially as patients with sarcoidosis tend to have low or normal IgE levels. Patients with asthma can have elevated IgE levels. However, very elevated IgE levels are more common in other conditions, including allergic bronchopulmonary aspergillosis (ABPA) and the Hyper-IgE syndrome. The latter manifests with recurrent infections and eczema, and is inherited in an autosomal dominant manner. However, both the Hyper-IgE syndrome and ABPA have much higher IgE levels than seen in this case. Allergen-specific IgE testing (including for antibodies to Aspergillus) should be sent. It seems that an interstitial lung disease is present; the waxing and waning pattern and clinical presentation, along with the lack of other systemic findings, make hypersensitivity pneumonitis most likely.
The family lived in an apartment building. Her symptoms started when the family’s neighbor recently moved his outdoor pigeon coop into his basement. The patient often smelled the pigeons and noted feathers coming through the holes in the wall.
One of the key diagnostic features of hypersensitivity pneumonitis (HP) is the history of exposure to a potential offending antigen—in this case likely bird feathers—along with worsening upon reexposure to that antigen. HP is primarily a clinical diagnosis, and testing for serum precipitants has limited value, given the high false negative rate and the frequent lack of clinical symptoms accompanying positive testing. Bronchoalveolar lavage fluid may reveal lymphocytosis and reduced CD4:CD8 ratio. Crackles are commonly heard on examination, but in this case were likely not auscultated due to her obese habitus. The most important treatment is withdrawal of the offending antigen. Limited data suggest that corticosteroid therapy may be helpful in certain HP cases, including subacute, chronic and severe cases as well as patients with hypoxemia, significant imaging findings, and those with significant abnormalities on pulmonary function testing (PFT).
A hypersensitivity pneumonitis precipitins panel was sent with positive antibodies to M. faeni, T. Vulgaris, A. Fumigatus 1 and 6, A. Flavus, and pigeon serum. Her symptoms gradually improved within five days of oral prednisone (60 mg). She was discharged home without dyspnea and normal oxygen saturation while breathing ambient air. A repeat echocardiogram after nighttime CPAP for 1 week demonstrated a right ventricular systolic pressure of 17 mm Hg consistent with improved pulmonary hypertension.
Three weeks later, she returned to clinic for follow up. She had re-experienced dyspnea, cough, and wheezing, which improved when she was outdoors. She was afebrile, tachypneic, tachycardic, and her oxygen saturation was 92% on ambient air.
Her steroid-responsive interstitial lung disease and rapid improvement upon avoidance of the offending antigen is consistent with HP. The positive serum precipitins assay lends further credence to the diagnosis of HP, although serologic analysis with such antibody assays is limited by false positives and false negatives; further, individuals exposed to pigeons often have antibodies present without evidence of HP. History taking at this visit should ask specifically about further pigeon exposure: were the pigeons removed from the home completely, were heating-cooling filters changed, carpets cleaned, and bedding laundered? An in-home evaluation may be helpful before conducting further diagnostic testing.
She was admitted for oxygen therapy and a bronchoscopy, which showed mucosal friability and cobblestoning, suggesting inflammation. BAL revealed a normal CD4:CD8 ratio of 3; BAL cultures were sterile. Her shortness of breath significantly improved following a prolonged course of systemic steroids and removal from the triggering environment. PFTs improved with a FEV1/FVC ratio of 94 (105% predicted), FVC of 2.00 L (66% predicted), FEV1 of 1.88L (69% predicted) (Figure 3B). Her presenting symptoms of persistent cough and progressive dyspnea on exertion, characteristic CT, sterile BAL cultures, positive serum precipitants against pigeon serum, and resolution of her symptoms with withdrawal of the offending antigen were diagnostic of hypersensitivity pneumonitis due to pigeon exposure, also known as bird fancier’s disease.
COMMENTARY
The patient’s original presentation of dyspnea, tachypnea, and hypoxia is commonly associated with pediatric pneumonia and asthma exacerbations.1 However, an alternative diagnosis was suggested by the lack of wheezing, absence of fever, and recurrent presentations with progressive symptoms.
Hypersensitivity pneumonitis (HP) represents an exaggerated T-cell meditated immune response to inhalation of an offending antigen that results in a restrictive ventilatory defect and interstitial infiltrates.2 Bird pneumonitis (also known as bird fancier’s disease) is a frequent cause of HP, accounting for approximately 65-70% of cases.3 HP, however, only manifests in a small number of subjects exposed to culprit antigens, suggesting an underlying genetic susceptibility.4 Prevalence estimates vary depending on bird species, county, climate, and other possible factors.
There are no standard criteria for the diagnosis of HP, though a combination of findings is suggestive. A recent prospective multicenter study created a scoring system for HP based on factors associated with the disease to aid in accurate diagnosis. The most relevant criteria included antigen exposure, recurrent symptoms noted within 4-8 hours after antigen exposure, weight loss, presence of specific IgG antibodies to avian antigens, and inspiratory crackles on exam. Using this rule, the probability that our patient has HP based on clinical characteristics was 93% with an area under the receiver operating curve of 0.93 (96% confidence interval: 0.90-0.95)5. Chest imaging (high resolution CT) often consists of a mosaic pattern of air trapping, as seen in this patient in combination with ground-glass opacities6. Bronchoalveolar lavage (BAL) is sensitive in detecting lung inflammation in a patient with suspected HP. On BAL, a lymphocytic alveolitis can be seen, but absence of this finding does not exclude HP.5,7,8 Pulmonary function tests (PFTs) may be normal in acute HP. When abnormal, PFTs may reveal a restrictive pattern and reduction in carbon monoxide diffusing capacity.7 However, BAL and PFT results are neither specific nor diagnostic of HP; it is important to consider results in the context of the clinical picture.
The respiratory response to inhalation of the avian antigen has traditionally been classified as acute, subacute, or chronic.9 The acute response occurs within hours of exposure to the offending agent and usually resolves within 24 hours after antigen withdrawal. The subacute presentation involves cough and dyspnea over several days to weeks, and can progress to chronic and permanent lung damage if unrecognized and untreated. In chronic presentations, lung abnormalities may persist despite antigen avoidance and pharmacologic interventions.4,10 The patient’s symptoms occurred over a six-month period which coincided with pigeon exposure and resolved during each hospitalization with steroid treatment and removal from the offending agent. Her presentation was consistent with a subacute time course of HP.
The dilated pulmonary artery, elevated right systolic ventricular pressure, and normal right ventricular function in our patient suggested pulmonary hypertension of chronic duration. Her risk factors for pulmonary hypertension included asthma, sleep apnea, possible obesity-hypoventilation syndrome, and HP-associated interstitial lung disease.11
The most important intervention in HP is avoidance of the causative antigen. Medical therapy without removal of antigen is inadequate. Systemic corticosteroids can help ameliorate acute symptoms though dosing and duration remains unclear. For chronic patients unresponsive to steroid therapy, lung transplantation can be considered.4
The key to diagnosis of HP in this patient—and to minimizing repeat testing upon the patient’s recrudescence of symptoms—was the clinician’s consideration that the major impetus for the patient’s improvement in the hospital was removal from the offending antigen in her home environment. As in this case, taking time to delve deeply into a patient’s environment—even by descending the basement stairs—may lead to the diagnosis.
LEARNING POINTS
- Consider hypersensitivity pneumonitis (HP) in patients with recurrent respiratory distress, offending exposure, and resolution of symptoms with removal of culprit antigen.
- The most important treatment of HP is removal of offending antigen; systemic and/or inhaled corticosteroids are indicated until the full resolution of respiratory symptoms.
- Prognosis is dependent on early diagnosis and removal of offending exposures.
- Failure to treat HP might result in end-stage lung disease from pulmonary fibrosis secondary to long-term inflammation.
Disclosures
Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The authors declare no conflicts of interests.
A 14-year-old girl with a history of asthma presented to the Emergency Department (ED) with three months of persistent, nonproductive cough, and progressive shortness of breath. She reported fatigue, chest tightness, orthopnea, and dyspnea with exertion. She denied fever, rhinorrhea, congestion, hemoptysis, or paroxysmal nocturnal dyspnea.
Her age and past medical history of asthma are incongruent with her new symptoms, as asthma is typified by intermittent exacerbations, not progressive symptoms. Thus, another process, in addition to asthma, is most likely present; it is also important to question the accuracy of previous diagnoses in light of new information. Her symptoms may signify an underlying cardiopulmonary process, such as infiltrative diseases (eg, lymphoma or sarcoidosis), atypical infections, genetic conditions (eg, variant cystic fibrosis), autoimmune conditions, or cardiomyopathy. A detailed symptom history, family history, and careful physical examination will help expand and then refine the differential diagnosis. At this stage, typical infections are less likely.
She had presented two months prior with nonproductive cough and dyspnea. At that presentation, her temperature was 36.3°C, heart rate 110 beats per minute, blood pressure 119/63 mm Hg, respiratory rate 43 breaths per minute, and oxygen saturation 86% while breathing ambient air. A chest CT with contrast demonstrated diffuse patchy multifocal ground-glass opacities in the bilateral lungs as well as a mixture of atelectasis and lobular emphysema in the dependent lobes bilaterally (Figure 1). Her main pulmonary artery was dilated at 3.6 cm (mean of 2.42 cm with SD 0.22). She was diagnosed with atypical pneumonia. She was administered azithromycin, weaned off oxygen, and discharged after a seven-day hospitalization.
Two months prior, she had marked tachypnea, tachycardia, and hypoxemia, and imaging revealed diffuse ground-glass opacities. The differential diagnosis for this constellation of symptoms is extensive and includes many conditions that have an inflammatory component, such as atypical pneumonia caused by Mycoplasma or Chlamydia pneumoniae or a common respiratory virus such as rhinovirus or human metapneumovirus. However, two findings make an acute pneumonia unlikely to be the sole cause of her symptoms: underlying emphysema and an enlarged pulmonary artery. Emphysema is an uncommon finding in children and can be related to congenital or acquired causes; congenital lobar emphysema most often presents earlier in life and is focal, not diffuse. Alpha-1-anti-trypin deficiency and mutations in connective tissue genes such as those encoding for elastin and fibrillin can lead to pulmonary disease. While not diagnostic of pulmonary hypertension, her dilated pulmonary artery, coupled with her history, makes pulmonary hypertension a strong possibility. While her pulmonary hypertension is most likely secondary to chronic lung disease based on the emphysematous changes on CT, it could still be related to a cardiac etiology.
The patient had a history of seasonal allergies and well-controlled asthma. She was hospitalized at age six for an asthma exacerbation associated with a respiratory infection. She was discharged with an albuterol inhaler, but seldom used it. Her parents denied any regular coughing during the day or night. She was morbidly obese. Her tonsils and adenoids were removed to treat obstructive sleep apnea (OSA) at age seven, and a subsequent polysomnography was normal. Her medications included intranasal fluticasone propionate and oral iron supplementation. She had no known allergies or recent travels. She had never smoked. She had two pet cats and a dog. Her mother had a history of obesity, OSA, and eczema. Her father had diabetes and eczema.
The patient’s history prior to the recent few months sheds little light on the cause of her current symptoms. While it is possible that her current symptoms are related to the worsening of a process that had been present for many years which mimicked asthma, this seems implausible given the long period of time between her last asthma exacerbation and her present symptoms. Similarly, while tonsillar and adenoidal hypertrophy can be associated with infiltrative diseases (such as lymphoma), this is less common than the usual (and normal) disproportionate increase in size of the adenoids compared to other airway structures during growth in children.
She was admitted to the hospital. On initial examination, her temperature was 37.4°C, heart rate 125 beats per minute, blood pressure 143/69 mm Hg, respiratory rate 48 breaths per minute, and oxygen saturation 86% breathing ambient air. Her BMI was 58 kg/m2. Her exam demonstrated increased work of breathing with accessory muscle use, and decreased breath sounds at the bases. There were no wheezes or crackles. Cardiovascular, abdominal, and skin exams were normal except for tachycardia. At rest, later in the hospitalization, her oxygen saturation was 97% breathing ambient air and heart rate 110 bpm. After two minutes of walking, her oxygen saturation was 77% and heart rate 132 bpm. Two minutes after resting, her oxygen saturation increased to 91%.
Her white blood cell count was 11.9 x 10 9 /L (67% neutrophils, 24.2% lymphocytes, 6% monocytes, and 2% eosinophils), hemoglobin 11.2 g/dL, and platelet count 278,000/mm 3 . Her complete metabolic panel was normal. The C-reactive protein (CRP) was 24 mg/L (normal range, < 4.9) and erythrocyte sedimentation rate (ESR) 103 mm/hour (normal range, 0-32). A venous blood gas (VBG) showed a pH of 7.42 and pCO2 39. An EKG demonstrated sinus tachycardia.
The combination of the patient’s tachypnea, hypoxemia, respiratory distress, and obesity is striking. Her lack of adventitious lung sounds is surprising given her CT findings, but the sensitivity of chest auscultation may be limited in obese patients. Her laboratory findings help narrow the diagnostic frame: she has mild anemia and leukocytosis along with significant inflammation. The normal CO2 concentration on VBG is concerning given the degree of her tachypnea and reflects significant alveolar hypoventilation.
This marked inflammation with diffuse lung findings again raises the possibility of an inflammatory or, less likely, infectious disorder. Sjogren’s syndrome, systemic lupus erythematosus (SLE), and juvenile dermatomyositis can present in young women with interstitial lung disease. She does have exposure to pets and hypersensitivity pneumonitis can worsen rapidly with continued exposure. Another possibility is that she has an underlying immunodeficiency such as common variable immunodeficiency, although a history of recurrent infections such as pneumonia, bacteremia, or sinusitis is lacking.
An echocardiogram should be performed. In addition, laboratory evaluation for the aforementioned autoimmune causes of interstitial lung disease, immunoglobulin levels, pulmonary function testing (if available as an inpatient), and potentially a bronchoscopy with bronchoalveolar lavage (BAL), and biopsy should be pursued. The BAL and biopsy would be helpful in evaluating for infection and interstitial lung disease in an expeditious manner.
A chest CT without contrast was done and compared to the scan from two months prior. New diffuse, ill-defined centrilobular ground-glass opacities were evident throughout the lung fields; dilation of the main pulmonary artery was unchanged, and previously seen ground-glass opacities had resolved. There were patchy areas of air-trapping and mosaic attenuation in the lower lobes (Figure 2).
Transthoracic echocardiogram demonstrated a right ventricular systolic pressure of 58 mm Hg with flattened intraventricular septum during systole. Left and right ventricular systolic function were normal. The left ventricular diastolic function was normal. Pulmonary function testing demonstrated a FEV1/FVC ratio of 100 (112% predicted), FVC 1.07 L (35 % predicted) and FEV1 1.07 L (39% predicted), and total lung capacity was 2.7L (56% predicted) (Figure 3). Single-breath carbon monoxide uptake in the lung was not interpretable based on 2017 European Respiratory Society (ERS)/American Thoracic Society (ATS) technical standards.
This information is helpful in classifying whether this patient’s primary condition is cardiac or pulmonary in nature. Her normal left ventricular systolic and diastolic function make a cardiac etiology for her pulmonary hypertension less likely. Further, the combination of pulmonary hypertension, a restrictive pattern on pulmonary function testing, and findings consistent with interstitial lung disease on cross-sectional imaging all suggest a primary pulmonary etiology rather than a cardiac, infectious, or thromboembolic condition. While chronic thromboembolic hypertension can result in nonspecific mosaic attenuation, it typically would not cause centrilobular ground-glass opacities nor restrictive lung disease. Thus, it seems most likely that this patient has a progressive pulmonary process resulting in hypoxia, pulmonary hypertension, centrilobular opacities, and lower-lobe mosaic attenuation. Considerations for this process can be broadly categorized as one of the childhood interstitial lung disease (chILD). While this differential diagnosis is broad, strong consideration should be given to hypersensitivity pneumonitis, chronic aspiration, sarcoidosis, and Sjogren’s syndrome. An intriguing possibility is that the patient’s “response to azithromycin” two months prior was due to the avoidance of an inhaled antigen while she was in the hospital; a detailed environmental history should be explored. The normal polysomnography after tonsilloadenoidectomy makes it unlikely that OSA is a major contributor to her current presentation. However, since the surgery was seven years ago, and her BMI is presently 58 kg/m2 she remains at risk for OSA and obesity-hypoventilation syndrome. Polysomnography should be done after her acute symptoms improve.
She was started on 5 mm Hg of continuous positive airway pressure (CPAP) at night after a sleep study on room air demonstrated severe OSA with a respiratory disturbance index of 13 events per hour. Antinuclear antibodies (ANA), anti-neutrophil cytoplasmic antibody (ANCA), anti-Jo-1 antibody, anti-RNP antibody, anti-Smith antibody, anti-Ro/SSA and anti-La/SSB antibody were negative as was the histoplasmin antibody. Serum angiotensin-converting enzyme (ACE) level was normal. Mycoplasma IgM and IgG were negative. IgE was 529 kU/L (normal range, <114).
This evaluation reduces the likelihood the patient has Sjogren’s syndrome, SLE, dermatomyositis, or ANCA-associated pulmonary disease. While many patients with dermatomyositis may have negative serologic evaluations, other findings usually present such as rash and myositis are lacking. The negative ANCA evaluation makes granulomatosis with polyangiitis and microscopic polyangiitis very unlikely given the high sensitivity of the ANCA assay for these conditions. ANCA assays are less sensitive for eosinophilic granulomatosis with polyangiitis (EGPA), but the lack of eosinophilia significantly decreases the likelihood of EGPA. ACE levels have relatively poor operating characteristics in the evaluation of sarcoidosis; however, sarcoidosis seems unlikely in this case, especially as patients with sarcoidosis tend to have low or normal IgE levels. Patients with asthma can have elevated IgE levels. However, very elevated IgE levels are more common in other conditions, including allergic bronchopulmonary aspergillosis (ABPA) and the Hyper-IgE syndrome. The latter manifests with recurrent infections and eczema, and is inherited in an autosomal dominant manner. However, both the Hyper-IgE syndrome and ABPA have much higher IgE levels than seen in this case. Allergen-specific IgE testing (including for antibodies to Aspergillus) should be sent. It seems that an interstitial lung disease is present; the waxing and waning pattern and clinical presentation, along with the lack of other systemic findings, make hypersensitivity pneumonitis most likely.
The family lived in an apartment building. Her symptoms started when the family’s neighbor recently moved his outdoor pigeon coop into his basement. The patient often smelled the pigeons and noted feathers coming through the holes in the wall.
One of the key diagnostic features of hypersensitivity pneumonitis (HP) is the history of exposure to a potential offending antigen—in this case likely bird feathers—along with worsening upon reexposure to that antigen. HP is primarily a clinical diagnosis, and testing for serum precipitants has limited value, given the high false negative rate and the frequent lack of clinical symptoms accompanying positive testing. Bronchoalveolar lavage fluid may reveal lymphocytosis and reduced CD4:CD8 ratio. Crackles are commonly heard on examination, but in this case were likely not auscultated due to her obese habitus. The most important treatment is withdrawal of the offending antigen. Limited data suggest that corticosteroid therapy may be helpful in certain HP cases, including subacute, chronic and severe cases as well as patients with hypoxemia, significant imaging findings, and those with significant abnormalities on pulmonary function testing (PFT).
A hypersensitivity pneumonitis precipitins panel was sent with positive antibodies to M. faeni, T. Vulgaris, A. Fumigatus 1 and 6, A. Flavus, and pigeon serum. Her symptoms gradually improved within five days of oral prednisone (60 mg). She was discharged home without dyspnea and normal oxygen saturation while breathing ambient air. A repeat echocardiogram after nighttime CPAP for 1 week demonstrated a right ventricular systolic pressure of 17 mm Hg consistent with improved pulmonary hypertension.
Three weeks later, she returned to clinic for follow up. She had re-experienced dyspnea, cough, and wheezing, which improved when she was outdoors. She was afebrile, tachypneic, tachycardic, and her oxygen saturation was 92% on ambient air.
Her steroid-responsive interstitial lung disease and rapid improvement upon avoidance of the offending antigen is consistent with HP. The positive serum precipitins assay lends further credence to the diagnosis of HP, although serologic analysis with such antibody assays is limited by false positives and false negatives; further, individuals exposed to pigeons often have antibodies present without evidence of HP. History taking at this visit should ask specifically about further pigeon exposure: were the pigeons removed from the home completely, were heating-cooling filters changed, carpets cleaned, and bedding laundered? An in-home evaluation may be helpful before conducting further diagnostic testing.
She was admitted for oxygen therapy and a bronchoscopy, which showed mucosal friability and cobblestoning, suggesting inflammation. BAL revealed a normal CD4:CD8 ratio of 3; BAL cultures were sterile. Her shortness of breath significantly improved following a prolonged course of systemic steroids and removal from the triggering environment. PFTs improved with a FEV1/FVC ratio of 94 (105% predicted), FVC of 2.00 L (66% predicted), FEV1 of 1.88L (69% predicted) (Figure 3B). Her presenting symptoms of persistent cough and progressive dyspnea on exertion, characteristic CT, sterile BAL cultures, positive serum precipitants against pigeon serum, and resolution of her symptoms with withdrawal of the offending antigen were diagnostic of hypersensitivity pneumonitis due to pigeon exposure, also known as bird fancier’s disease.
COMMENTARY
The patient’s original presentation of dyspnea, tachypnea, and hypoxia is commonly associated with pediatric pneumonia and asthma exacerbations.1 However, an alternative diagnosis was suggested by the lack of wheezing, absence of fever, and recurrent presentations with progressive symptoms.
Hypersensitivity pneumonitis (HP) represents an exaggerated T-cell meditated immune response to inhalation of an offending antigen that results in a restrictive ventilatory defect and interstitial infiltrates.2 Bird pneumonitis (also known as bird fancier’s disease) is a frequent cause of HP, accounting for approximately 65-70% of cases.3 HP, however, only manifests in a small number of subjects exposed to culprit antigens, suggesting an underlying genetic susceptibility.4 Prevalence estimates vary depending on bird species, county, climate, and other possible factors.
There are no standard criteria for the diagnosis of HP, though a combination of findings is suggestive. A recent prospective multicenter study created a scoring system for HP based on factors associated with the disease to aid in accurate diagnosis. The most relevant criteria included antigen exposure, recurrent symptoms noted within 4-8 hours after antigen exposure, weight loss, presence of specific IgG antibodies to avian antigens, and inspiratory crackles on exam. Using this rule, the probability that our patient has HP based on clinical characteristics was 93% with an area under the receiver operating curve of 0.93 (96% confidence interval: 0.90-0.95)5. Chest imaging (high resolution CT) often consists of a mosaic pattern of air trapping, as seen in this patient in combination with ground-glass opacities6. Bronchoalveolar lavage (BAL) is sensitive in detecting lung inflammation in a patient with suspected HP. On BAL, a lymphocytic alveolitis can be seen, but absence of this finding does not exclude HP.5,7,8 Pulmonary function tests (PFTs) may be normal in acute HP. When abnormal, PFTs may reveal a restrictive pattern and reduction in carbon monoxide diffusing capacity.7 However, BAL and PFT results are neither specific nor diagnostic of HP; it is important to consider results in the context of the clinical picture.
The respiratory response to inhalation of the avian antigen has traditionally been classified as acute, subacute, or chronic.9 The acute response occurs within hours of exposure to the offending agent and usually resolves within 24 hours after antigen withdrawal. The subacute presentation involves cough and dyspnea over several days to weeks, and can progress to chronic and permanent lung damage if unrecognized and untreated. In chronic presentations, lung abnormalities may persist despite antigen avoidance and pharmacologic interventions.4,10 The patient’s symptoms occurred over a six-month period which coincided with pigeon exposure and resolved during each hospitalization with steroid treatment and removal from the offending agent. Her presentation was consistent with a subacute time course of HP.
The dilated pulmonary artery, elevated right systolic ventricular pressure, and normal right ventricular function in our patient suggested pulmonary hypertension of chronic duration. Her risk factors for pulmonary hypertension included asthma, sleep apnea, possible obesity-hypoventilation syndrome, and HP-associated interstitial lung disease.11
The most important intervention in HP is avoidance of the causative antigen. Medical therapy without removal of antigen is inadequate. Systemic corticosteroids can help ameliorate acute symptoms though dosing and duration remains unclear. For chronic patients unresponsive to steroid therapy, lung transplantation can be considered.4
The key to diagnosis of HP in this patient—and to minimizing repeat testing upon the patient’s recrudescence of symptoms—was the clinician’s consideration that the major impetus for the patient’s improvement in the hospital was removal from the offending antigen in her home environment. As in this case, taking time to delve deeply into a patient’s environment—even by descending the basement stairs—may lead to the diagnosis.
LEARNING POINTS
- Consider hypersensitivity pneumonitis (HP) in patients with recurrent respiratory distress, offending exposure, and resolution of symptoms with removal of culprit antigen.
- The most important treatment of HP is removal of offending antigen; systemic and/or inhaled corticosteroids are indicated until the full resolution of respiratory symptoms.
- Prognosis is dependent on early diagnosis and removal of offending exposures.
- Failure to treat HP might result in end-stage lung disease from pulmonary fibrosis secondary to long-term inflammation.
Disclosures
Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME). The authors declare no conflicts of interests.
1. Ebell MH. Clinical diagnosis of pneumonia in children. Am Fam Physician. 2010;82(2):192-193. PubMed
2. Cormier Y, Lacasse Y. Hypersensitivity pneumonitis and organic dust toxic syndrome. In: Malo J-L, Chan-Yeung M, Bernstein DI, eds. Asthma in the Workplace. Vol 32. Boca Raton, FL: Fourth Informa Healthcare; 2013:392-405.
3. Chan AL, Juarez MM, Leslie KO, Ismail HA, Albertson TE. Bird fancier’s lung: a state-of-the-art review. Clin Rev Allergy Immunol. 2012;43(1-2):69-83. doi: 10.1007/s12016-011-8282-y. PubMed
4. Camarena A, Juárez A, Mejía M, et al. Major histocompatibility complex and tumor necrosis factor-α polymorphisms in pigeon breeder’s disease. Am J Respir Crit Care Med. 2001;163(7):1528-1533. https:/doi.org/10.1164/ajrccm.163.7.2004023. PubMed
5. Lacasse Y, Selman M, Costabel U, et al. Clinical diagnosis of hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2003;168(8):952-958. doi: 10.1164/rccm.200301-137OC. PubMed
6. Glazer CS, Rose CS, Lynch DA. Clinical and radiologic manifestations of hypersensitivity pneumonitis. J Thorac Imaging. 2002;17(4):261-272. PubMed
7. Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med. 2012;186(4):314-324. doi: 10.1164/rccm.201203-0513CI. PubMed
8. Calillad DM, Vergnon, JM, Madroszyk A, et al. Bronchoalveolar lavage in hypersensitivity pneumonitis: a series of 139 patients. Inflamm Allergy Drug Targets. 2012;11(1):15-19. doi: 10.2174/187152812798889330. PubMed
9. Richerson HB, Bernstein IL, Fink JN, et al. Guidelines for the clinical evaluation of hypersensitivity pneumonitis. Report of the Subcommittee on Hypersensitivity Pneumonitis. J Allergy Clin Immunol. 1989;84(5 Pt 2):839-844. doi: 10.1016/0091-6749(89)90349-7. PubMed
10. Zacharisen MC, Schlueter DP, Kurup VP, Fink JN. The long-term outcome in acute, subacute, and chronic forms of pigeon breeder’s disease hypersensitivity pneumonitis. Ann Allergy Asthma Immunol. 2002;88(2):175-182. doi: 10.1016/S1081-1206(10)61993-X. PubMed
11. Raymond TE, Khabbaza JE, Yadav R, Tonelli AR. Significance of main pulmonary artery dilation on imaging studies. Ann Am Thorac Soc. 2014;11(10):1623-1632. doi: 10.1513/AnnalsATS.201406-253PP. PubMed
1. Ebell MH. Clinical diagnosis of pneumonia in children. Am Fam Physician. 2010;82(2):192-193. PubMed
2. Cormier Y, Lacasse Y. Hypersensitivity pneumonitis and organic dust toxic syndrome. In: Malo J-L, Chan-Yeung M, Bernstein DI, eds. Asthma in the Workplace. Vol 32. Boca Raton, FL: Fourth Informa Healthcare; 2013:392-405.
3. Chan AL, Juarez MM, Leslie KO, Ismail HA, Albertson TE. Bird fancier’s lung: a state-of-the-art review. Clin Rev Allergy Immunol. 2012;43(1-2):69-83. doi: 10.1007/s12016-011-8282-y. PubMed
4. Camarena A, Juárez A, Mejía M, et al. Major histocompatibility complex and tumor necrosis factor-α polymorphisms in pigeon breeder’s disease. Am J Respir Crit Care Med. 2001;163(7):1528-1533. https:/doi.org/10.1164/ajrccm.163.7.2004023. PubMed
5. Lacasse Y, Selman M, Costabel U, et al. Clinical diagnosis of hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2003;168(8):952-958. doi: 10.1164/rccm.200301-137OC. PubMed
6. Glazer CS, Rose CS, Lynch DA. Clinical and radiologic manifestations of hypersensitivity pneumonitis. J Thorac Imaging. 2002;17(4):261-272. PubMed
7. Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med. 2012;186(4):314-324. doi: 10.1164/rccm.201203-0513CI. PubMed
8. Calillad DM, Vergnon, JM, Madroszyk A, et al. Bronchoalveolar lavage in hypersensitivity pneumonitis: a series of 139 patients. Inflamm Allergy Drug Targets. 2012;11(1):15-19. doi: 10.2174/187152812798889330. PubMed
9. Richerson HB, Bernstein IL, Fink JN, et al. Guidelines for the clinical evaluation of hypersensitivity pneumonitis. Report of the Subcommittee on Hypersensitivity Pneumonitis. J Allergy Clin Immunol. 1989;84(5 Pt 2):839-844. doi: 10.1016/0091-6749(89)90349-7. PubMed
10. Zacharisen MC, Schlueter DP, Kurup VP, Fink JN. The long-term outcome in acute, subacute, and chronic forms of pigeon breeder’s disease hypersensitivity pneumonitis. Ann Allergy Asthma Immunol. 2002;88(2):175-182. doi: 10.1016/S1081-1206(10)61993-X. PubMed
11. Raymond TE, Khabbaza JE, Yadav R, Tonelli AR. Significance of main pulmonary artery dilation on imaging studies. Ann Am Thorac Soc. 2014;11(10):1623-1632. doi: 10.1513/AnnalsATS.201406-253PP. PubMed
© 2019 Society of Hospital Medicine
A Howling Cause of Pancytopenia
A 15-year-old African American girl presented to the emergency department with 3 days of fever, sore throat, nausea, vomiting, and poor appetite. She reported a 4-week history of fatigue, right hand pain and swelling, and a 6-kilogram weight loss for which she had seen her primary care provider several times. She reported no recent travel, sick contacts, or new medications.
It appears that there are potentially at least 2 separate problems: an acute one (past 3 days) and a more chronic one (past 4 weeks). These 2 problems may be directly related (ie, acute worsening of the more chronic problem), indirectly related (ie, the more chronic problem is leading to increased susceptibility to the acute problem, for instance, an evolving immunodeficiency predisposing to an opportunistic infection), or “true, true, but unrelated.” The clinical challenge is to keep one’s mind open to each of these potential scenarios and to avoid the tendency to focus on one of the problems and not pay enough attention to the other. Occam’s razor likely does not apply here.
Numerous common and typically transient diseases could cause the symptoms of the past 3 days, particularly infectious etiologies such as streptococcal pharyngitis or a viral infection. One cannot forget about these possibilities while contemplating the more worrisome symptoms of the past 4 weeks, especially weight loss in a growing adolescent. Patients may unintentionally lose weight for a variety of reasons, which can be broadly categorized by decreased caloric supply, gastrointestinal losses or malabsorption, and increased caloric demand; these categories are not mutually exclusive.
Lastly, 1 symptom may provide a more specific direction: the right hand pain and swelling of the past 4 weeks. More specifics, including the extent of the hand swelling, other areas of involvement, and the nature of her pain, will be helpful.
Her temperature was 99.5°F, heart rate 100 beats per minute, respiratory rate 18 breaths per minute, oxygen saturation 95% while breathing ambient air, blood pressure 99/56 mmHg, weight 44 kilograms, height 161 centimeters, and body mass index 17. She appeared generally ill and underweight. She had edematous and violaceous eyelids, dry cracked lips, and pharyngeal erythema with ulcerations of the hard palate. She had nontender cervical and inguinal lymphadenopathy. Her abdomen was tender to palpation in the lower quadrants without guarding or rebound; there was no organomegaly. A right knee effusion with overlying warmth was present without redness or decreased range of motion. She also had an enlarged third proximal interphalangeal joint and loss of palpable metacarpal phalangeal joint landmarks on her right hand. She was noted to be using her arms to move her legs when repositioning in bed.
These exam findings clearly point toward a systemic process but not 1 specific diagnosis. The presence of at least 2 inflamed joints points toward rheumatologic/inflammatory or infectious diseases. Localized edema (eyelids and right metacarpal phalangeal joints), oral ulcers, possible myositis, and arthritis point toward a systemic vasculitis (eg, granulomatosis with polyangiitis, Behçet disease). While Kawasaki disease is also a systemic vasculitis, the presence of oral ulcers and generalized lymphadenopathy argues against it. Inflammatory myopathies like polymyositis, and especially juvenile dermatomyositis, fit many aspects of this presentation with the violaceous eyelids and possible myositis, though no other cutaneous stigmata of this disease are evident (eg, no Gottron’s papules). Polyarthritis, violaceous eyelids, and possible myositis could be consistent with systemic lupus erythematosus (SLE).
The presence of oral ulcers and arthritis make other systemic inflammatory conditions, such as inflammatory bowel disease with arthritis and autoimmune- or infection-related hepatitis, possible. Infectious etiologies alone or in combination with a rheumatologic process are also possible given fevers and lymphadenopathy. In particular, herpesvirus infections (Epstein-Barr virus [EBV], cytomegalovirus [CMV], herpes simplex virus, or human herpes virus 6), human immunodeficiency virus (HIV), hepatitis C virus (HCV), and syphilis can cause oral ulcers and lymphadenopathy. Other potential infectious etiologies include subacute bacterial endocarditis and disseminated gonococcal infection given the presence of polyarthritis, but these infections are less likely as they do not explain all of the symptoms.
In summary, the differential diagnosis is broad and should be prioritized to consider systemic inflammatory conditions, including autoimmune and infectious (especially viral) syndromes, and initial work-up should focus on these etiologies.

The pancytopenia is obviously notable. It raises the possibility that the oral ulcerations are due to the neutropenia rather than a primary disease manifestation. Other possible causes of pancytopenia include SLE, antiphospholipid antibody syndrome, and related rheumatologic diagnoses, including hemophagocytic lymphohistiocytosis (HLH). Given her age and subacute presentation, secondary forms of HLH seem more likely than primary (genetic) forms, which typically present within the first few years of life. Secondary forms of HLH can occur in association with rheumatic diseases and are then referred to as Macrophage Activation Syndrome (MAS). The most common rheumatologic diseases associated with MAS are systemic juvenile idiopathic arthritis, SLE, and Kawasaki disease. Secondary HLH can also occur with infectious diseases, particularly viral infections such as EBV. It is also important to consider thrombotic thrombocytopenic purpura and other forms of thrombotic microangiopathy, especially if her violaceous eyelids actually represent purpura. The presence of pancytopenia also expands the differential diagnosis to include leukemia, lymphoma, and other oncologic diseases. After obtaining results from pending infectious disease studies, additional diagnostic work-up should include examination of the bone marrow and a peripheral blood smear to evaluate for hemophagocytosis and/or malignancy. Testing for double-stranded DNA antibodies and antinuclear antibodies (ANA) should be sent to evaluate for SLE, and antiphospholipid antibodies should also be checked. Renal function must also be evaluated.
Additional laboratory work-up revealed a reticulocyte count of 0.2%, a positive Coombs immunoglobulin G (IgG) test, haptoglobin less than 80 mg/L, and lactate dehydrogenase (LDH) 25.2 µkat/L (1509 units/L); coagulation studies were normal. Her chemistries showed electrolytes, blood urea nitrogen, and creatinine were within normal limits; her aspartate aminotransferase was 216 units/L, and alanine aminotransferase was 56 units/L. Her spot urine protein-to-creatinine ratio was 1.28. Complement and inflammatory studies showed C3 0.14 g/L (14 mg/dL, normal 83-151 mg/dL), C4 0.05 g/L (5 mg/dL, normal 13-37 mg/dL), erythrocyte sedimentation rate (ESR) 103 mm/hr (normal 0-20 mm/hr), and C-reactive protein (CRP) 3.2 mg/L (normal 0.7-1.7 mg/L). Additional studies showed elevated triglycerides (376 mg/dL), elevated creatine kinase (2437 units/L), and elevated ferritin (22,295.5 ng/mL). An ANA screen and specific autoantibody studies were sent, including antidouble stranded DNA antibody, antiribonucleoprotein antibody, anti-Smith antibody, anti-Ro antibody, and anti-La antibody. A bone marrow biopsy was performed.
The hematologic studies provide a mixed picture. There is evidence of an autoimmune hemolytic anemia (AIHA). Typically, AIHA is associated with reticulocytosis rather than reticulocytopenia. Reticulocytopenia can occur in AIHA, however, because of antibodies directed against erythroid precursors or if 2 processes are occurring simultaneously—ie, AIHA plus bone marrow destructive/failure process. The latter scenario is more likely here. Specifically, the pancytopenia, elevated triglycerides, and extreme hyperferritinemia strongly support the diagnosis of HLH. The very low C3 and C4 suggest a complement-consumptive process, and SLE is the most likely etiology. Proteinuria and Coombs-positive anemia are also features of SLE. The discordance between the ESR (markedly elevated) and CRP (mild elevation) is surprising in the setting of systemic inflammation. However, her other clinical features are consistent with marked systemic inflammation, and it is important not to dismiss a likely diagnosis simply on the basis of a few incongruous features. At this point, the diagnosis of SLE complicated by secondary HLH is favored, remembering that both these entities can be triggered by a viral infection. Therefore, diligent follow-up of the aforementioned specific autoantibody studies and the bone marrow biopsy is the next logical step, along with the still-pending infectious disease studies.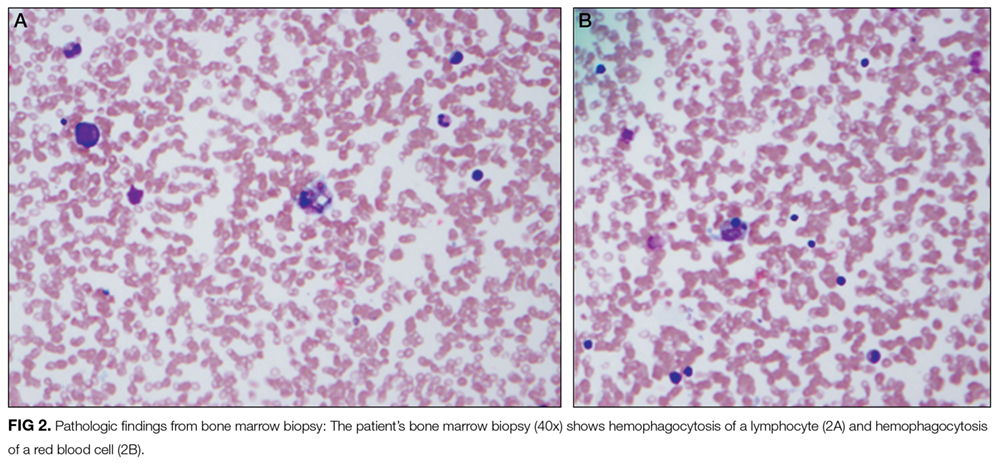
These findings are consistent with the diagnosis of SLE complicated by secondary HLH (ie, MAS). It remains possible, but unlikely, that the patient has genetic or familial HLH (fHLH), as this entity is exceedingly rare with distinct underlying genetic aberrations separate from SLE. Ideally, the NK cell function studies would be repeated after the current episode of HLH is controlled and the patient is off of immunosuppressive therapies, but this will likely not be possible given the underlying SLE. Patients with fHLH have reduced or absent NK cell function at baseline (ie, not only during an acute episode of HLH and not because of immunosuppressive medications). Alternatively, one could consider genetic testing for fHLH. The clinical importance of doing this is that patients with fHLH are candidates for bone marrow or stem cell transplantation. There currently is not a published standard of care for the work-up and management of MAS in children with rheumatic disease, so the decision to repeat NK cell function testing and/or genetic testing would be left to the discretion of the treating physician and would depend on the patient’s ongoing clinical course.
The patient required red blood cell and platelet transfusions. She received pulse dose intravenous methylprednisolone for treatment of SLE and MAS; she clinically improved within 48 hours of starting steroids. Cyclosporine was added for management of MAS. The patient was transitioned to oral corticosteroids and discharged home. All cell counts normalized within 1 month of discharge. She was weaned off corticosteroids and cyclosporine was discontinued. Her maintenance SLE therapy includes hydroxychloroquine and mycophenolate mofetil.
COMMENTARY
Because the differential diagnosis for new-onset pancytopenia encompasses many diseases across several medical subspecialties, a thorough history and physical exam are necessary to form a tailored clinical approach.1 The primary causes of pediatric pancytopenia vary depending on geographic location because of the local prevalence of infectious agents and nutritional deficiency patterns. A retrospective study investigating the primary cause of pancytopenia in children without existing malignancy presenting to a US tertiary care hospital found that 64% of cases were due to infection, 28% were due to hematologic disease (most frequently aplastic anemia), and 8% were due to miscellaneous etiologies, including adverse drug reactions and autoimmune diseases.2 In contrast, the most common cause of pancytopenia in pediatric patients presenting to a tertiary care hospital in India was megaloblastic anemia (28%), followed by infections (21%), acute leukemia (21%), and aplastic anemia (20%).3 While clinicians do (and should) consider malignancy as a cause of pancytopenia, there is sparse literature regarding the frequency of pancytopenia associated with the presentations of childhood malignancies.4 A study of pediatric patients with acute lymphoblastic anemia found that only 11% of newly diagnosed patients had pancytopenia at initial presentation.4
There are no official guidelines for the work-up of pediatric pancytopenia from any of the academic societies. Depending on the clinical history, initial laboratory investigation for pediatric pancytopenia may include complete blood cell count with differential, reticulocyte count, peripheral blood smear, complete metabolic panel, hemolysis labs (haptoglobin, LDH, Coombs test) and inflammatory markers (ESR, CRP, fibrinogen). Further investigation to clarify the specific etiology of pancytopenia can be guided by the results of these initial tests.
Hematologic abnormalities are present in greater than 70% of pediatric SLE cases.10,11 The pathogenesis of hematologic abnormalities in SLE is heterogeneous, involving actions of autoreactive lymphocytes, autoantibodies, and proinflammatory cytokines that can disrupt bone marrow production and cause peripheral blood cell destruction.12,13 While pancytopenia is common in children with SLE, other coexisting diagnoses should be considered in patients with SLE and pancytopenia. Concurrent diagnoses that can lead to pancytopenia in patients with SLE include infection, pharmacologic side effects, and secondary HLH,14,15 each of which has differing implications for prognosis and treatment.
Secondary HLH is a severe and often acute complication of systemic inflammatory disorders caused by the proliferation and activation of T cells and macrophages, leading to an enhanced inflammatory state. When HLH occurs in the setting of an underlying autoimmune or autoinflammatory process, it is typically termed MAS. MAS affects an estimated 0.9% to 4.6% of patients with SLE.16 Early diagnosis and treatment of MAS is important because MAS can be rapidly fatal, with a mortality rate of 8% to 20% in pediatric patients.17,18 Clinical features of MAS include physical exam findings of fever and splenomegaly as well as laboratory abnormalities, including pancytopenia, elevated ferritin, elevated triglycerides, and low fibrinogen.18 A bone marrow biopsy showing hemophagocytosis in the absence of malignancy is diagnostic of MAS. Although a bone marrow biopsy is not required to diagnose MAS, it is often obtained to exclude other etiologies of pancytopenia such as malignancy.19 Specialized diagnostic testing for MAS includes NK cell counts and functional studies, including expression of perforin and granzyme B (NK cell proteins triggering apoptosis in target cells), soluble IL-2R (marker of activated lymphocytes), and CD163 (transmembrane protein of hemophagocytic macrophages). There is no standardized protocol for treating MAS.20 It is most commonly treated with highdose corticosteroids; additional agents, including cyclosporine and biologic therapies, are also utilized.16,20
KEY POINTS
- Children with SLE tend to have greater involvement of major organ systems and more rapid accrual of organ damage than adults with SLE. Therefore, it is sometimes necessary to initiate immunosuppressive therapies before full diagnostic criteria are met, provided that malignancy and infection have been ruled out.
- While pancytopenia is common in pediatric patients with SLE, providers should make sure to consider coexisting diagnoses such as infection and MAS, both of which require different treatment strategies.
- It is important to consider HLH/MAS early in the work-up of pancytopenia, because early diagnosis and treatment improves clinical outcomes. Obtaining a ferritin level can aid in the work-up of pancytopenia because it is both a sensitive and specific marker of HLH/MAS when dramatically elevated.
Disclosure
The authors report no conflicts of interest.
1. Weinzierl EP, Arber DA. The Differential Diagnosis and Bone Marrow Evaluation of New-Onset Pancytopenia. Am J Clin Pathol. 2012;139(1):9-29. doi:10.1309/AJCP50AEEYGREWUZ. PubMed
2. Pine M, Walter AW. Pancytopenia in hospitalized children: a five-year review. J Pediatr Hematol Oncol. 2010;32(5):e192-e194. doi:10.1097/MPH.0b013e3181e03082. PubMed
3. Bhatnagar SK. Pancytopenia in Children: Etiological Profile. J Trop Pediatr. 2005;51(4):236-239. doi:10.1093/tropej/fmi010. PubMed
4. Kulkarni KP, Marwaha RK. Acute lymphoblastic leukemia with pancytopenia at presentation: clinical correlates, prognostic impact, and association with survival. J Pediatr Hematol Oncol. 2013;35(7):573-576. doi:10.1097/MPH.0b013e31829d46f3. PubMed
5. Holubar, K. Terminology and iconography of lupus erythematosus: A historical vignette. Am J Dermatopathol. 1980;2(3):239-242. PubMed
6. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. doi: 10.1002/art.1780400928. PubMed
7. Petri M, Orbai, A, Alarcon GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677-2686. doi:10.1002/art.34473. PubMed
8. Tucker L. Review: Making the diagnosis of systemic lupus erythematosus in children and adolescents. Lupus. 2007;16(8):546-549. doi:10.1177/0961203307078068. PubMed
9. Brunner HI, Gladman DD, Ibañez D, Urowitz MD, Silverman ED. Difference in disease features between childhood-onset and adult-onset systemic lupus erythematosus. Arthritis Rheum. 2008;58(2):556-562. doi:10.1002/art.23204. PubMed
10. Benseler SM, Silverman ED. Systemic Lupus Erythematosus. Rheum Dis Clin North Am. 2007;33(3):471-498. doi:10.1016/j.rdc.2007.07.008. PubMed
11. Gokce M, Bilginer Y, Besbas N, et al. Hematological features of pediatric systemic lupus erythematosus: suggesting management strategies in children. Lupus. 2012;21(8):878-884. doi:10.1177/0961203312443721. PubMed
12. Voulgarelis M, Giannouli S, Tasidou A, Anagnostou D, Ziakas PD, Tzioufas AG. Bone marrow histological findings in systemic lupus erythematosus with hematologic abnormalities: A clinicopathological study. Am J Hematol. 2006;81(8):590-597. doi:10.1002/ajh.20593. PubMed
13. Pereira RM, Velloso ER, Menezes Y, Gualandro S, Vassalo J, Yoshinari NH. Bone marrow findings in systemic lupus erythematosus patients with peripheral cytopenias. Clin Rheumatol. 1998;17(3):219-222. PubMed
14. Avčin T, Tse SML, Schneider R, Ngan B, Silverman ED. Macrophage activation syndrome as the presenting manifestation of rheumatic diseases in childhood. J Pediatr. 2006;148(5):683-686. doi:10.1016/j.jpeds.2005.12.070. PubMed
15. Lambotte O, Khellaf M, Harmouche H, et al. Characteristics and Long-Term Outcome of 15 Episodes of Systemic Lupus Erythematosus-Associated Hemophagocytic Syndrome. Medicine. 2006;85(3):169-182. doi:10.1097/01.md.0000224708.62510.d1. PubMed
16. Fukaya S, Yasuda S, Hashimoto T, et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30 cases. Rheumatology. 2008;47(11):1686-1691. doi:10.1093/rheumatology/ken342. PubMed
17. Stephan JL. Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology. 2001;40(11):1285-1292. doi:10.1093/rheumatology/40.11.1285. PubMed
18. Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child. 2001;85(5):421-426. PubMed
19. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. doi:10.1002/pbc.21039. PubMed
20. Lin CI, Yu HH, Lee JH, et al. Clinical analysis of macrophage activation syndrome in pediatric patients with autoimmune diseases. Clin Rheumatol. 2012;31(8):1223-1230. doi:10.1007/s10067-012-1998-0. PubMed
A 15-year-old African American girl presented to the emergency department with 3 days of fever, sore throat, nausea, vomiting, and poor appetite. She reported a 4-week history of fatigue, right hand pain and swelling, and a 6-kilogram weight loss for which she had seen her primary care provider several times. She reported no recent travel, sick contacts, or new medications.
It appears that there are potentially at least 2 separate problems: an acute one (past 3 days) and a more chronic one (past 4 weeks). These 2 problems may be directly related (ie, acute worsening of the more chronic problem), indirectly related (ie, the more chronic problem is leading to increased susceptibility to the acute problem, for instance, an evolving immunodeficiency predisposing to an opportunistic infection), or “true, true, but unrelated.” The clinical challenge is to keep one’s mind open to each of these potential scenarios and to avoid the tendency to focus on one of the problems and not pay enough attention to the other. Occam’s razor likely does not apply here.
Numerous common and typically transient diseases could cause the symptoms of the past 3 days, particularly infectious etiologies such as streptococcal pharyngitis or a viral infection. One cannot forget about these possibilities while contemplating the more worrisome symptoms of the past 4 weeks, especially weight loss in a growing adolescent. Patients may unintentionally lose weight for a variety of reasons, which can be broadly categorized by decreased caloric supply, gastrointestinal losses or malabsorption, and increased caloric demand; these categories are not mutually exclusive.
Lastly, 1 symptom may provide a more specific direction: the right hand pain and swelling of the past 4 weeks. More specifics, including the extent of the hand swelling, other areas of involvement, and the nature of her pain, will be helpful.
Her temperature was 99.5°F, heart rate 100 beats per minute, respiratory rate 18 breaths per minute, oxygen saturation 95% while breathing ambient air, blood pressure 99/56 mmHg, weight 44 kilograms, height 161 centimeters, and body mass index 17. She appeared generally ill and underweight. She had edematous and violaceous eyelids, dry cracked lips, and pharyngeal erythema with ulcerations of the hard palate. She had nontender cervical and inguinal lymphadenopathy. Her abdomen was tender to palpation in the lower quadrants without guarding or rebound; there was no organomegaly. A right knee effusion with overlying warmth was present without redness or decreased range of motion. She also had an enlarged third proximal interphalangeal joint and loss of palpable metacarpal phalangeal joint landmarks on her right hand. She was noted to be using her arms to move her legs when repositioning in bed.
These exam findings clearly point toward a systemic process but not 1 specific diagnosis. The presence of at least 2 inflamed joints points toward rheumatologic/inflammatory or infectious diseases. Localized edema (eyelids and right metacarpal phalangeal joints), oral ulcers, possible myositis, and arthritis point toward a systemic vasculitis (eg, granulomatosis with polyangiitis, Behçet disease). While Kawasaki disease is also a systemic vasculitis, the presence of oral ulcers and generalized lymphadenopathy argues against it. Inflammatory myopathies like polymyositis, and especially juvenile dermatomyositis, fit many aspects of this presentation with the violaceous eyelids and possible myositis, though no other cutaneous stigmata of this disease are evident (eg, no Gottron’s papules). Polyarthritis, violaceous eyelids, and possible myositis could be consistent with systemic lupus erythematosus (SLE).
The presence of oral ulcers and arthritis make other systemic inflammatory conditions, such as inflammatory bowel disease with arthritis and autoimmune- or infection-related hepatitis, possible. Infectious etiologies alone or in combination with a rheumatologic process are also possible given fevers and lymphadenopathy. In particular, herpesvirus infections (Epstein-Barr virus [EBV], cytomegalovirus [CMV], herpes simplex virus, or human herpes virus 6), human immunodeficiency virus (HIV), hepatitis C virus (HCV), and syphilis can cause oral ulcers and lymphadenopathy. Other potential infectious etiologies include subacute bacterial endocarditis and disseminated gonococcal infection given the presence of polyarthritis, but these infections are less likely as they do not explain all of the symptoms.
In summary, the differential diagnosis is broad and should be prioritized to consider systemic inflammatory conditions, including autoimmune and infectious (especially viral) syndromes, and initial work-up should focus on these etiologies.
The pancytopenia is obviously notable. It raises the possibility that the oral ulcerations are due to the neutropenia rather than a primary disease manifestation. Other possible causes of pancytopenia include SLE, antiphospholipid antibody syndrome, and related rheumatologic diagnoses, including hemophagocytic lymphohistiocytosis (HLH). Given her age and subacute presentation, secondary forms of HLH seem more likely than primary (genetic) forms, which typically present within the first few years of life. Secondary forms of HLH can occur in association with rheumatic diseases and are then referred to as Macrophage Activation Syndrome (MAS). The most common rheumatologic diseases associated with MAS are systemic juvenile idiopathic arthritis, SLE, and Kawasaki disease. Secondary HLH can also occur with infectious diseases, particularly viral infections such as EBV. It is also important to consider thrombotic thrombocytopenic purpura and other forms of thrombotic microangiopathy, especially if her violaceous eyelids actually represent purpura. The presence of pancytopenia also expands the differential diagnosis to include leukemia, lymphoma, and other oncologic diseases. After obtaining results from pending infectious disease studies, additional diagnostic work-up should include examination of the bone marrow and a peripheral blood smear to evaluate for hemophagocytosis and/or malignancy. Testing for double-stranded DNA antibodies and antinuclear antibodies (ANA) should be sent to evaluate for SLE, and antiphospholipid antibodies should also be checked. Renal function must also be evaluated.
Additional laboratory work-up revealed a reticulocyte count of 0.2%, a positive Coombs immunoglobulin G (IgG) test, haptoglobin less than 80 mg/L, and lactate dehydrogenase (LDH) 25.2 µkat/L (1509 units/L); coagulation studies were normal. Her chemistries showed electrolytes, blood urea nitrogen, and creatinine were within normal limits; her aspartate aminotransferase was 216 units/L, and alanine aminotransferase was 56 units/L. Her spot urine protein-to-creatinine ratio was 1.28. Complement and inflammatory studies showed C3 0.14 g/L (14 mg/dL, normal 83-151 mg/dL), C4 0.05 g/L (5 mg/dL, normal 13-37 mg/dL), erythrocyte sedimentation rate (ESR) 103 mm/hr (normal 0-20 mm/hr), and C-reactive protein (CRP) 3.2 mg/L (normal 0.7-1.7 mg/L). Additional studies showed elevated triglycerides (376 mg/dL), elevated creatine kinase (2437 units/L), and elevated ferritin (22,295.5 ng/mL). An ANA screen and specific autoantibody studies were sent, including antidouble stranded DNA antibody, antiribonucleoprotein antibody, anti-Smith antibody, anti-Ro antibody, and anti-La antibody. A bone marrow biopsy was performed.
The hematologic studies provide a mixed picture. There is evidence of an autoimmune hemolytic anemia (AIHA). Typically, AIHA is associated with reticulocytosis rather than reticulocytopenia. Reticulocytopenia can occur in AIHA, however, because of antibodies directed against erythroid precursors or if 2 processes are occurring simultaneously—ie, AIHA plus bone marrow destructive/failure process. The latter scenario is more likely here. Specifically, the pancytopenia, elevated triglycerides, and extreme hyperferritinemia strongly support the diagnosis of HLH. The very low C3 and C4 suggest a complement-consumptive process, and SLE is the most likely etiology. Proteinuria and Coombs-positive anemia are also features of SLE. The discordance between the ESR (markedly elevated) and CRP (mild elevation) is surprising in the setting of systemic inflammation. However, her other clinical features are consistent with marked systemic inflammation, and it is important not to dismiss a likely diagnosis simply on the basis of a few incongruous features. At this point, the diagnosis of SLE complicated by secondary HLH is favored, remembering that both these entities can be triggered by a viral infection. Therefore, diligent follow-up of the aforementioned specific autoantibody studies and the bone marrow biopsy is the next logical step, along with the still-pending infectious disease studies.
These findings are consistent with the diagnosis of SLE complicated by secondary HLH (ie, MAS). It remains possible, but unlikely, that the patient has genetic or familial HLH (fHLH), as this entity is exceedingly rare with distinct underlying genetic aberrations separate from SLE. Ideally, the NK cell function studies would be repeated after the current episode of HLH is controlled and the patient is off of immunosuppressive therapies, but this will likely not be possible given the underlying SLE. Patients with fHLH have reduced or absent NK cell function at baseline (ie, not only during an acute episode of HLH and not because of immunosuppressive medications). Alternatively, one could consider genetic testing for fHLH. The clinical importance of doing this is that patients with fHLH are candidates for bone marrow or stem cell transplantation. There currently is not a published standard of care for the work-up and management of MAS in children with rheumatic disease, so the decision to repeat NK cell function testing and/or genetic testing would be left to the discretion of the treating physician and would depend on the patient’s ongoing clinical course.
The patient required red blood cell and platelet transfusions. She received pulse dose intravenous methylprednisolone for treatment of SLE and MAS; she clinically improved within 48 hours of starting steroids. Cyclosporine was added for management of MAS. The patient was transitioned to oral corticosteroids and discharged home. All cell counts normalized within 1 month of discharge. She was weaned off corticosteroids and cyclosporine was discontinued. Her maintenance SLE therapy includes hydroxychloroquine and mycophenolate mofetil.
COMMENTARY
Because the differential diagnosis for new-onset pancytopenia encompasses many diseases across several medical subspecialties, a thorough history and physical exam are necessary to form a tailored clinical approach.1 The primary causes of pediatric pancytopenia vary depending on geographic location because of the local prevalence of infectious agents and nutritional deficiency patterns. A retrospective study investigating the primary cause of pancytopenia in children without existing malignancy presenting to a US tertiary care hospital found that 64% of cases were due to infection, 28% were due to hematologic disease (most frequently aplastic anemia), and 8% were due to miscellaneous etiologies, including adverse drug reactions and autoimmune diseases.2 In contrast, the most common cause of pancytopenia in pediatric patients presenting to a tertiary care hospital in India was megaloblastic anemia (28%), followed by infections (21%), acute leukemia (21%), and aplastic anemia (20%).3 While clinicians do (and should) consider malignancy as a cause of pancytopenia, there is sparse literature regarding the frequency of pancytopenia associated with the presentations of childhood malignancies.4 A study of pediatric patients with acute lymphoblastic anemia found that only 11% of newly diagnosed patients had pancytopenia at initial presentation.4
There are no official guidelines for the work-up of pediatric pancytopenia from any of the academic societies. Depending on the clinical history, initial laboratory investigation for pediatric pancytopenia may include complete blood cell count with differential, reticulocyte count, peripheral blood smear, complete metabolic panel, hemolysis labs (haptoglobin, LDH, Coombs test) and inflammatory markers (ESR, CRP, fibrinogen). Further investigation to clarify the specific etiology of pancytopenia can be guided by the results of these initial tests.
Hematologic abnormalities are present in greater than 70% of pediatric SLE cases.10,11 The pathogenesis of hematologic abnormalities in SLE is heterogeneous, involving actions of autoreactive lymphocytes, autoantibodies, and proinflammatory cytokines that can disrupt bone marrow production and cause peripheral blood cell destruction.12,13 While pancytopenia is common in children with SLE, other coexisting diagnoses should be considered in patients with SLE and pancytopenia. Concurrent diagnoses that can lead to pancytopenia in patients with SLE include infection, pharmacologic side effects, and secondary HLH,14,15 each of which has differing implications for prognosis and treatment.
Secondary HLH is a severe and often acute complication of systemic inflammatory disorders caused by the proliferation and activation of T cells and macrophages, leading to an enhanced inflammatory state. When HLH occurs in the setting of an underlying autoimmune or autoinflammatory process, it is typically termed MAS. MAS affects an estimated 0.9% to 4.6% of patients with SLE.16 Early diagnosis and treatment of MAS is important because MAS can be rapidly fatal, with a mortality rate of 8% to 20% in pediatric patients.17,18 Clinical features of MAS include physical exam findings of fever and splenomegaly as well as laboratory abnormalities, including pancytopenia, elevated ferritin, elevated triglycerides, and low fibrinogen.18 A bone marrow biopsy showing hemophagocytosis in the absence of malignancy is diagnostic of MAS. Although a bone marrow biopsy is not required to diagnose MAS, it is often obtained to exclude other etiologies of pancytopenia such as malignancy.19 Specialized diagnostic testing for MAS includes NK cell counts and functional studies, including expression of perforin and granzyme B (NK cell proteins triggering apoptosis in target cells), soluble IL-2R (marker of activated lymphocytes), and CD163 (transmembrane protein of hemophagocytic macrophages). There is no standardized protocol for treating MAS.20 It is most commonly treated with highdose corticosteroids; additional agents, including cyclosporine and biologic therapies, are also utilized.16,20
KEY POINTS
- Children with SLE tend to have greater involvement of major organ systems and more rapid accrual of organ damage than adults with SLE. Therefore, it is sometimes necessary to initiate immunosuppressive therapies before full diagnostic criteria are met, provided that malignancy and infection have been ruled out.
- While pancytopenia is common in pediatric patients with SLE, providers should make sure to consider coexisting diagnoses such as infection and MAS, both of which require different treatment strategies.
- It is important to consider HLH/MAS early in the work-up of pancytopenia, because early diagnosis and treatment improves clinical outcomes. Obtaining a ferritin level can aid in the work-up of pancytopenia because it is both a sensitive and specific marker of HLH/MAS when dramatically elevated.
Disclosure
The authors report no conflicts of interest.
A 15-year-old African American girl presented to the emergency department with 3 days of fever, sore throat, nausea, vomiting, and poor appetite. She reported a 4-week history of fatigue, right hand pain and swelling, and a 6-kilogram weight loss for which she had seen her primary care provider several times. She reported no recent travel, sick contacts, or new medications.
It appears that there are potentially at least 2 separate problems: an acute one (past 3 days) and a more chronic one (past 4 weeks). These 2 problems may be directly related (ie, acute worsening of the more chronic problem), indirectly related (ie, the more chronic problem is leading to increased susceptibility to the acute problem, for instance, an evolving immunodeficiency predisposing to an opportunistic infection), or “true, true, but unrelated.” The clinical challenge is to keep one’s mind open to each of these potential scenarios and to avoid the tendency to focus on one of the problems and not pay enough attention to the other. Occam’s razor likely does not apply here.
Numerous common and typically transient diseases could cause the symptoms of the past 3 days, particularly infectious etiologies such as streptococcal pharyngitis or a viral infection. One cannot forget about these possibilities while contemplating the more worrisome symptoms of the past 4 weeks, especially weight loss in a growing adolescent. Patients may unintentionally lose weight for a variety of reasons, which can be broadly categorized by decreased caloric supply, gastrointestinal losses or malabsorption, and increased caloric demand; these categories are not mutually exclusive.
Lastly, 1 symptom may provide a more specific direction: the right hand pain and swelling of the past 4 weeks. More specifics, including the extent of the hand swelling, other areas of involvement, and the nature of her pain, will be helpful.
Her temperature was 99.5°F, heart rate 100 beats per minute, respiratory rate 18 breaths per minute, oxygen saturation 95% while breathing ambient air, blood pressure 99/56 mmHg, weight 44 kilograms, height 161 centimeters, and body mass index 17. She appeared generally ill and underweight. She had edematous and violaceous eyelids, dry cracked lips, and pharyngeal erythema with ulcerations of the hard palate. She had nontender cervical and inguinal lymphadenopathy. Her abdomen was tender to palpation in the lower quadrants without guarding or rebound; there was no organomegaly. A right knee effusion with overlying warmth was present without redness or decreased range of motion. She also had an enlarged third proximal interphalangeal joint and loss of palpable metacarpal phalangeal joint landmarks on her right hand. She was noted to be using her arms to move her legs when repositioning in bed.
These exam findings clearly point toward a systemic process but not 1 specific diagnosis. The presence of at least 2 inflamed joints points toward rheumatologic/inflammatory or infectious diseases. Localized edema (eyelids and right metacarpal phalangeal joints), oral ulcers, possible myositis, and arthritis point toward a systemic vasculitis (eg, granulomatosis with polyangiitis, Behçet disease). While Kawasaki disease is also a systemic vasculitis, the presence of oral ulcers and generalized lymphadenopathy argues against it. Inflammatory myopathies like polymyositis, and especially juvenile dermatomyositis, fit many aspects of this presentation with the violaceous eyelids and possible myositis, though no other cutaneous stigmata of this disease are evident (eg, no Gottron’s papules). Polyarthritis, violaceous eyelids, and possible myositis could be consistent with systemic lupus erythematosus (SLE).
The presence of oral ulcers and arthritis make other systemic inflammatory conditions, such as inflammatory bowel disease with arthritis and autoimmune- or infection-related hepatitis, possible. Infectious etiologies alone or in combination with a rheumatologic process are also possible given fevers and lymphadenopathy. In particular, herpesvirus infections (Epstein-Barr virus [EBV], cytomegalovirus [CMV], herpes simplex virus, or human herpes virus 6), human immunodeficiency virus (HIV), hepatitis C virus (HCV), and syphilis can cause oral ulcers and lymphadenopathy. Other potential infectious etiologies include subacute bacterial endocarditis and disseminated gonococcal infection given the presence of polyarthritis, but these infections are less likely as they do not explain all of the symptoms.
In summary, the differential diagnosis is broad and should be prioritized to consider systemic inflammatory conditions, including autoimmune and infectious (especially viral) syndromes, and initial work-up should focus on these etiologies.
The pancytopenia is obviously notable. It raises the possibility that the oral ulcerations are due to the neutropenia rather than a primary disease manifestation. Other possible causes of pancytopenia include SLE, antiphospholipid antibody syndrome, and related rheumatologic diagnoses, including hemophagocytic lymphohistiocytosis (HLH). Given her age and subacute presentation, secondary forms of HLH seem more likely than primary (genetic) forms, which typically present within the first few years of life. Secondary forms of HLH can occur in association with rheumatic diseases and are then referred to as Macrophage Activation Syndrome (MAS). The most common rheumatologic diseases associated with MAS are systemic juvenile idiopathic arthritis, SLE, and Kawasaki disease. Secondary HLH can also occur with infectious diseases, particularly viral infections such as EBV. It is also important to consider thrombotic thrombocytopenic purpura and other forms of thrombotic microangiopathy, especially if her violaceous eyelids actually represent purpura. The presence of pancytopenia also expands the differential diagnosis to include leukemia, lymphoma, and other oncologic diseases. After obtaining results from pending infectious disease studies, additional diagnostic work-up should include examination of the bone marrow and a peripheral blood smear to evaluate for hemophagocytosis and/or malignancy. Testing for double-stranded DNA antibodies and antinuclear antibodies (ANA) should be sent to evaluate for SLE, and antiphospholipid antibodies should also be checked. Renal function must also be evaluated.
Additional laboratory work-up revealed a reticulocyte count of 0.2%, a positive Coombs immunoglobulin G (IgG) test, haptoglobin less than 80 mg/L, and lactate dehydrogenase (LDH) 25.2 µkat/L (1509 units/L); coagulation studies were normal. Her chemistries showed electrolytes, blood urea nitrogen, and creatinine were within normal limits; her aspartate aminotransferase was 216 units/L, and alanine aminotransferase was 56 units/L. Her spot urine protein-to-creatinine ratio was 1.28. Complement and inflammatory studies showed C3 0.14 g/L (14 mg/dL, normal 83-151 mg/dL), C4 0.05 g/L (5 mg/dL, normal 13-37 mg/dL), erythrocyte sedimentation rate (ESR) 103 mm/hr (normal 0-20 mm/hr), and C-reactive protein (CRP) 3.2 mg/L (normal 0.7-1.7 mg/L). Additional studies showed elevated triglycerides (376 mg/dL), elevated creatine kinase (2437 units/L), and elevated ferritin (22,295.5 ng/mL). An ANA screen and specific autoantibody studies were sent, including antidouble stranded DNA antibody, antiribonucleoprotein antibody, anti-Smith antibody, anti-Ro antibody, and anti-La antibody. A bone marrow biopsy was performed.
The hematologic studies provide a mixed picture. There is evidence of an autoimmune hemolytic anemia (AIHA). Typically, AIHA is associated with reticulocytosis rather than reticulocytopenia. Reticulocytopenia can occur in AIHA, however, because of antibodies directed against erythroid precursors or if 2 processes are occurring simultaneously—ie, AIHA plus bone marrow destructive/failure process. The latter scenario is more likely here. Specifically, the pancytopenia, elevated triglycerides, and extreme hyperferritinemia strongly support the diagnosis of HLH. The very low C3 and C4 suggest a complement-consumptive process, and SLE is the most likely etiology. Proteinuria and Coombs-positive anemia are also features of SLE. The discordance between the ESR (markedly elevated) and CRP (mild elevation) is surprising in the setting of systemic inflammation. However, her other clinical features are consistent with marked systemic inflammation, and it is important not to dismiss a likely diagnosis simply on the basis of a few incongruous features. At this point, the diagnosis of SLE complicated by secondary HLH is favored, remembering that both these entities can be triggered by a viral infection. Therefore, diligent follow-up of the aforementioned specific autoantibody studies and the bone marrow biopsy is the next logical step, along with the still-pending infectious disease studies.
These findings are consistent with the diagnosis of SLE complicated by secondary HLH (ie, MAS). It remains possible, but unlikely, that the patient has genetic or familial HLH (fHLH), as this entity is exceedingly rare with distinct underlying genetic aberrations separate from SLE. Ideally, the NK cell function studies would be repeated after the current episode of HLH is controlled and the patient is off of immunosuppressive therapies, but this will likely not be possible given the underlying SLE. Patients with fHLH have reduced or absent NK cell function at baseline (ie, not only during an acute episode of HLH and not because of immunosuppressive medications). Alternatively, one could consider genetic testing for fHLH. The clinical importance of doing this is that patients with fHLH are candidates for bone marrow or stem cell transplantation. There currently is not a published standard of care for the work-up and management of MAS in children with rheumatic disease, so the decision to repeat NK cell function testing and/or genetic testing would be left to the discretion of the treating physician and would depend on the patient’s ongoing clinical course.
The patient required red blood cell and platelet transfusions. She received pulse dose intravenous methylprednisolone for treatment of SLE and MAS; she clinically improved within 48 hours of starting steroids. Cyclosporine was added for management of MAS. The patient was transitioned to oral corticosteroids and discharged home. All cell counts normalized within 1 month of discharge. She was weaned off corticosteroids and cyclosporine was discontinued. Her maintenance SLE therapy includes hydroxychloroquine and mycophenolate mofetil.
COMMENTARY
Because the differential diagnosis for new-onset pancytopenia encompasses many diseases across several medical subspecialties, a thorough history and physical exam are necessary to form a tailored clinical approach.1 The primary causes of pediatric pancytopenia vary depending on geographic location because of the local prevalence of infectious agents and nutritional deficiency patterns. A retrospective study investigating the primary cause of pancytopenia in children without existing malignancy presenting to a US tertiary care hospital found that 64% of cases were due to infection, 28% were due to hematologic disease (most frequently aplastic anemia), and 8% were due to miscellaneous etiologies, including adverse drug reactions and autoimmune diseases.2 In contrast, the most common cause of pancytopenia in pediatric patients presenting to a tertiary care hospital in India was megaloblastic anemia (28%), followed by infections (21%), acute leukemia (21%), and aplastic anemia (20%).3 While clinicians do (and should) consider malignancy as a cause of pancytopenia, there is sparse literature regarding the frequency of pancytopenia associated with the presentations of childhood malignancies.4 A study of pediatric patients with acute lymphoblastic anemia found that only 11% of newly diagnosed patients had pancytopenia at initial presentation.4
There are no official guidelines for the work-up of pediatric pancytopenia from any of the academic societies. Depending on the clinical history, initial laboratory investigation for pediatric pancytopenia may include complete blood cell count with differential, reticulocyte count, peripheral blood smear, complete metabolic panel, hemolysis labs (haptoglobin, LDH, Coombs test) and inflammatory markers (ESR, CRP, fibrinogen). Further investigation to clarify the specific etiology of pancytopenia can be guided by the results of these initial tests.
Hematologic abnormalities are present in greater than 70% of pediatric SLE cases.10,11 The pathogenesis of hematologic abnormalities in SLE is heterogeneous, involving actions of autoreactive lymphocytes, autoantibodies, and proinflammatory cytokines that can disrupt bone marrow production and cause peripheral blood cell destruction.12,13 While pancytopenia is common in children with SLE, other coexisting diagnoses should be considered in patients with SLE and pancytopenia. Concurrent diagnoses that can lead to pancytopenia in patients with SLE include infection, pharmacologic side effects, and secondary HLH,14,15 each of which has differing implications for prognosis and treatment.
Secondary HLH is a severe and often acute complication of systemic inflammatory disorders caused by the proliferation and activation of T cells and macrophages, leading to an enhanced inflammatory state. When HLH occurs in the setting of an underlying autoimmune or autoinflammatory process, it is typically termed MAS. MAS affects an estimated 0.9% to 4.6% of patients with SLE.16 Early diagnosis and treatment of MAS is important because MAS can be rapidly fatal, with a mortality rate of 8% to 20% in pediatric patients.17,18 Clinical features of MAS include physical exam findings of fever and splenomegaly as well as laboratory abnormalities, including pancytopenia, elevated ferritin, elevated triglycerides, and low fibrinogen.18 A bone marrow biopsy showing hemophagocytosis in the absence of malignancy is diagnostic of MAS. Although a bone marrow biopsy is not required to diagnose MAS, it is often obtained to exclude other etiologies of pancytopenia such as malignancy.19 Specialized diagnostic testing for MAS includes NK cell counts and functional studies, including expression of perforin and granzyme B (NK cell proteins triggering apoptosis in target cells), soluble IL-2R (marker of activated lymphocytes), and CD163 (transmembrane protein of hemophagocytic macrophages). There is no standardized protocol for treating MAS.20 It is most commonly treated with highdose corticosteroids; additional agents, including cyclosporine and biologic therapies, are also utilized.16,20
KEY POINTS
- Children with SLE tend to have greater involvement of major organ systems and more rapid accrual of organ damage than adults with SLE. Therefore, it is sometimes necessary to initiate immunosuppressive therapies before full diagnostic criteria are met, provided that malignancy and infection have been ruled out.
- While pancytopenia is common in pediatric patients with SLE, providers should make sure to consider coexisting diagnoses such as infection and MAS, both of which require different treatment strategies.
- It is important to consider HLH/MAS early in the work-up of pancytopenia, because early diagnosis and treatment improves clinical outcomes. Obtaining a ferritin level can aid in the work-up of pancytopenia because it is both a sensitive and specific marker of HLH/MAS when dramatically elevated.
Disclosure
The authors report no conflicts of interest.
1. Weinzierl EP, Arber DA. The Differential Diagnosis and Bone Marrow Evaluation of New-Onset Pancytopenia. Am J Clin Pathol. 2012;139(1):9-29. doi:10.1309/AJCP50AEEYGREWUZ. PubMed
2. Pine M, Walter AW. Pancytopenia in hospitalized children: a five-year review. J Pediatr Hematol Oncol. 2010;32(5):e192-e194. doi:10.1097/MPH.0b013e3181e03082. PubMed
3. Bhatnagar SK. Pancytopenia in Children: Etiological Profile. J Trop Pediatr. 2005;51(4):236-239. doi:10.1093/tropej/fmi010. PubMed
4. Kulkarni KP, Marwaha RK. Acute lymphoblastic leukemia with pancytopenia at presentation: clinical correlates, prognostic impact, and association with survival. J Pediatr Hematol Oncol. 2013;35(7):573-576. doi:10.1097/MPH.0b013e31829d46f3. PubMed
5. Holubar, K. Terminology and iconography of lupus erythematosus: A historical vignette. Am J Dermatopathol. 1980;2(3):239-242. PubMed
6. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. doi: 10.1002/art.1780400928. PubMed
7. Petri M, Orbai, A, Alarcon GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677-2686. doi:10.1002/art.34473. PubMed
8. Tucker L. Review: Making the diagnosis of systemic lupus erythematosus in children and adolescents. Lupus. 2007;16(8):546-549. doi:10.1177/0961203307078068. PubMed
9. Brunner HI, Gladman DD, Ibañez D, Urowitz MD, Silverman ED. Difference in disease features between childhood-onset and adult-onset systemic lupus erythematosus. Arthritis Rheum. 2008;58(2):556-562. doi:10.1002/art.23204. PubMed
10. Benseler SM, Silverman ED. Systemic Lupus Erythematosus. Rheum Dis Clin North Am. 2007;33(3):471-498. doi:10.1016/j.rdc.2007.07.008. PubMed
11. Gokce M, Bilginer Y, Besbas N, et al. Hematological features of pediatric systemic lupus erythematosus: suggesting management strategies in children. Lupus. 2012;21(8):878-884. doi:10.1177/0961203312443721. PubMed
12. Voulgarelis M, Giannouli S, Tasidou A, Anagnostou D, Ziakas PD, Tzioufas AG. Bone marrow histological findings in systemic lupus erythematosus with hematologic abnormalities: A clinicopathological study. Am J Hematol. 2006;81(8):590-597. doi:10.1002/ajh.20593. PubMed
13. Pereira RM, Velloso ER, Menezes Y, Gualandro S, Vassalo J, Yoshinari NH. Bone marrow findings in systemic lupus erythematosus patients with peripheral cytopenias. Clin Rheumatol. 1998;17(3):219-222. PubMed
14. Avčin T, Tse SML, Schneider R, Ngan B, Silverman ED. Macrophage activation syndrome as the presenting manifestation of rheumatic diseases in childhood. J Pediatr. 2006;148(5):683-686. doi:10.1016/j.jpeds.2005.12.070. PubMed
15. Lambotte O, Khellaf M, Harmouche H, et al. Characteristics and Long-Term Outcome of 15 Episodes of Systemic Lupus Erythematosus-Associated Hemophagocytic Syndrome. Medicine. 2006;85(3):169-182. doi:10.1097/01.md.0000224708.62510.d1. PubMed
16. Fukaya S, Yasuda S, Hashimoto T, et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30 cases. Rheumatology. 2008;47(11):1686-1691. doi:10.1093/rheumatology/ken342. PubMed
17. Stephan JL. Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology. 2001;40(11):1285-1292. doi:10.1093/rheumatology/40.11.1285. PubMed
18. Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child. 2001;85(5):421-426. PubMed
19. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. doi:10.1002/pbc.21039. PubMed
20. Lin CI, Yu HH, Lee JH, et al. Clinical analysis of macrophage activation syndrome in pediatric patients with autoimmune diseases. Clin Rheumatol. 2012;31(8):1223-1230. doi:10.1007/s10067-012-1998-0. PubMed
1. Weinzierl EP, Arber DA. The Differential Diagnosis and Bone Marrow Evaluation of New-Onset Pancytopenia. Am J Clin Pathol. 2012;139(1):9-29. doi:10.1309/AJCP50AEEYGREWUZ. PubMed
2. Pine M, Walter AW. Pancytopenia in hospitalized children: a five-year review. J Pediatr Hematol Oncol. 2010;32(5):e192-e194. doi:10.1097/MPH.0b013e3181e03082. PubMed
3. Bhatnagar SK. Pancytopenia in Children: Etiological Profile. J Trop Pediatr. 2005;51(4):236-239. doi:10.1093/tropej/fmi010. PubMed
4. Kulkarni KP, Marwaha RK. Acute lymphoblastic leukemia with pancytopenia at presentation: clinical correlates, prognostic impact, and association with survival. J Pediatr Hematol Oncol. 2013;35(7):573-576. doi:10.1097/MPH.0b013e31829d46f3. PubMed
5. Holubar, K. Terminology and iconography of lupus erythematosus: A historical vignette. Am J Dermatopathol. 1980;2(3):239-242. PubMed
6. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. doi: 10.1002/art.1780400928. PubMed
7. Petri M, Orbai, A, Alarcon GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677-2686. doi:10.1002/art.34473. PubMed
8. Tucker L. Review: Making the diagnosis of systemic lupus erythematosus in children and adolescents. Lupus. 2007;16(8):546-549. doi:10.1177/0961203307078068. PubMed
9. Brunner HI, Gladman DD, Ibañez D, Urowitz MD, Silverman ED. Difference in disease features between childhood-onset and adult-onset systemic lupus erythematosus. Arthritis Rheum. 2008;58(2):556-562. doi:10.1002/art.23204. PubMed
10. Benseler SM, Silverman ED. Systemic Lupus Erythematosus. Rheum Dis Clin North Am. 2007;33(3):471-498. doi:10.1016/j.rdc.2007.07.008. PubMed
11. Gokce M, Bilginer Y, Besbas N, et al. Hematological features of pediatric systemic lupus erythematosus: suggesting management strategies in children. Lupus. 2012;21(8):878-884. doi:10.1177/0961203312443721. PubMed
12. Voulgarelis M, Giannouli S, Tasidou A, Anagnostou D, Ziakas PD, Tzioufas AG. Bone marrow histological findings in systemic lupus erythematosus with hematologic abnormalities: A clinicopathological study. Am J Hematol. 2006;81(8):590-597. doi:10.1002/ajh.20593. PubMed
13. Pereira RM, Velloso ER, Menezes Y, Gualandro S, Vassalo J, Yoshinari NH. Bone marrow findings in systemic lupus erythematosus patients with peripheral cytopenias. Clin Rheumatol. 1998;17(3):219-222. PubMed
14. Avčin T, Tse SML, Schneider R, Ngan B, Silverman ED. Macrophage activation syndrome as the presenting manifestation of rheumatic diseases in childhood. J Pediatr. 2006;148(5):683-686. doi:10.1016/j.jpeds.2005.12.070. PubMed
15. Lambotte O, Khellaf M, Harmouche H, et al. Characteristics and Long-Term Outcome of 15 Episodes of Systemic Lupus Erythematosus-Associated Hemophagocytic Syndrome. Medicine. 2006;85(3):169-182. doi:10.1097/01.md.0000224708.62510.d1. PubMed
16. Fukaya S, Yasuda S, Hashimoto T, et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30 cases. Rheumatology. 2008;47(11):1686-1691. doi:10.1093/rheumatology/ken342. PubMed
17. Stephan JL. Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology. 2001;40(11):1285-1292. doi:10.1093/rheumatology/40.11.1285. PubMed
18. Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child. 2001;85(5):421-426. PubMed
19. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. doi:10.1002/pbc.21039. PubMed
20. Lin CI, Yu HH, Lee JH, et al. Clinical analysis of macrophage activation syndrome in pediatric patients with autoimmune diseases. Clin Rheumatol. 2012;31(8):1223-1230. doi:10.1007/s10067-012-1998-0. PubMed
© 2018 Society of Hospital Medicine
Not a Textbook Case
A 25‐year‐old male presented to the emergency department with a 3‐day history of fever, chills, nausea, vomiting, diarrhea, and myalgias.
The acute onset, combination of vomiting and diarrhea, and systemic symptoms are most characteristic of an acute gastrointestinal infection, such as viral gastroenteritis (eg, Norovirus or Rotavirus) or bacterial enteritis (eg, nontyphoidal Salmonella, Campylobacter jejuni, or Escherichia coli). A careful exposure history, taking into account travel, diet, sick contacts, and living situation, can help prioritize the likelihood of a given pathogen, although treatment is generally supportive in the absence of severe dehydration, abdominal pain, or vital sign abnormalities. Vomiting and diarrhea can also be nonspecific responses to severe, nongastrointestinal infections, such as influenza or staphylococcal bacteremia. A drug or toxin could prompt an allergic or inflammatory response similar to the syndrome observed here. Due to the acuity, other categories of disease, such as autoimmunity, metabolic derangement, or malignancy, seem unlikely at this point.
Aside from being treated for Trichomonas vaginalis urethritis 2 months prior, the patient had been in good health and took no medications until the onset of these symptoms. Upon review of systems, he complained of a sore throat and odynophagia but denied cough or rhinorrhea. On examination, he appeared comfortable. His temperature was 39.2C, blood pressure 137/64 mm Hg, heart rate 92 beats per minute, and respiratory rate 16 breaths per minute. His arterial oxygen saturation was 97% while breathing ambient air. The posterior oropharynx was erythematous without exudates. There was no cervical lymphadenopathy. He was tender in the epigastric region without rebound or guarding. The white blood cell count was 6800/mm3, hemoglobin 10.0 g/dL with a mean corpuscular volume of 81 fL, and platelet count 224,000/mm3. The aspartate aminotransferase (AST) was 60 U/L (reference range 045 U/L), and the total bilirubin was 3.6 mg/dL; electrolytes, alanine aminotransferase, alkaline phosphatase, albumin, and the international normalized ratio were normal. Rapid antigen testing for influenza A and B were negative, and a rapid test for group A Streptococcal (GAS) antigen was positive.
Vomiting and abdominal tenderness are less typical in adults than in children with routine GAS pharyngitis. His odynophagia could reflect a retropharyngeal or peritonsillar abscess. Influenza assays have limited sensitivity and cannot reliably exclude acute infection, especially when the prevalence is high during influenza season. Epstein‐Barr virus (EBV)‐associated mononucleosis and acute human immunodeficiency virus (HIV) can cause acute pharyngitis and hepatitis, but the lymphadenopathy that is characteristic of both infections was absent. His recent trichomonas infection indicates that he may be at risk for sexually transmitted diseases, including HIV, gonorrhea, and syphilis.
His elevated bilirubin and AST along with vomiting, epigastric tenderness, and fevers raise the possibility of cholecystitis or cholangitis, which should be explored further with abdominal imaging. Mild AST elevation alone could be explained by muscle damage (given his myalgias), viral or bacterial invasion of the liver, or alcohol or other toxins, including acetaminophen, which he may be taking to treat his pain and fever.
The combination of anemia and hyperbilirubinemia should prompt consideration of hemolysis, but the anemia could also be explained by an underlying chronic disease (eg, HIV or hematologic malignancy), preexisting iron deficiency, or thalassemia.
He was given intravenous ceftriaxone in the emergency department. Penicillin, ondansetron, and omeprazole were prescribed, and he was discharged home. He never took the penicillin because a family member told him that his throat swelled up in the past when he took it. He continued to have malaise, diarrhea, myalgias, fatigue, and fevers to 38.9C. He returned to the emergency department 2 days later. His temperature was 38.6C, and his remaining vital signs were normal. His posterior oropharynx was erythematous and his sclerae icteric; his abdomen was soft, nontender, and nondistended, without hepatosplenomegaly. His hemoglobin was 8.8 g/dL, bilirubin 3.6 mg/dL without conjugated bilirubin present, lactate dehydrogenase (LDH) 3077 U/L (reference range 325750 U/L), and AST 126 U/L; blood urea nitrogen and creatinine were normal. He was admitted to the hospital.
The progression of his systemic symptoms for an additional 2 days in the absence of directed treatment for acute pharyngitis is not unusual. However, his anemia is progressive, with features highly suggestive of hemolysis, including indirect hyperbilirubinemia, elevated LDH, and elevated AST. The single dose of ceftriaxone is unlikely to have triggered drug‐induced immune hemolysis, and his anemia predates the antibiotic regardless. Fever can accompany hemolysis when a malignancy (eg, lymphoma) or autoimmune condition (eg, systemic lupus erythematosus) triggers immune‐mediated hemolytic anemia. Microangiopathic processes (eg, thrombotic thrombocytopenic purpura and disseminated intravascular coagulation) can be associated with fever because of the underlying mechanism or an untreated infection, respectively. Some pathogens, such as Plasmodium, Babesia, and Clostridium species, directly invade erythrocytes, leading to their destruction. He may have an underlying predisposition for hemolysis (eg, glucose‐6‐phosphate dehydrogenase [G6PD] deficiency) that has been unmasked in the setting of acute infection.
At admission, intravenous azithromycin was administered for GAS infection; peripheral blood cultures were sterile. His hemoglobin decreased to 7.3 g/dL. The reticulocyte count was 1.2%, and the direct antiglobulin test (DAT) was negative. A normochromic, normocytic anemia with blister and bite cells, rare microspherocytes, and echinocytes was seen on the peripheral blood smear (Figure 1). A chest radiograph was normal, and polymerase chain reaction (PCR) tests for parvovirus and EBV DNA in peripheral blood were negative. Neither parvovirus IgM antibodies nor HIV antibodies were present. The ferritin level was >33,000 ng/mL (reference range 20300 ng/mL), serum iron 87 g/dL (reference range 35180 g/dL), iron binding capacity 200 g/dL (reference range 240430 g/dL), and iron saturation index 44% (reference range 15%46%).
His ongoing fevers suggest an untreated infection, autoimmune condition, or malignancy. The depressed reticulocyte count is unexpected in the setting of hemolysis in a young and previously healthy patient, raising the prospect of his bone marrow harboring a hematologic malignancy or infection (eg, mycobacterial, fungal, or viral). Alternatively, an immune or infectious process may be impeding erythropoiesis (eg, pure red cell aplasia or parvovirus infection). Hyperferritinemia is nonspecific and suggests systemic inflammation, but is also associated with Still's disease, histoplasmosis, hemochromatosis, and hemophagocytic syndromes. Still's disease causes high fevers and pharyngitis but typically features leukocytosis and arthralgias, both of which are absent. Hemophagocytosis in adults is typically due to a hyperinflammatory response to an underlying infection or malignancy caused by uncontrolled proliferation of activated lymphocytes and macrophages secreting large amounts of inflammatory cytokines.
The peripheral blood smear does not demonstrate a leukoerythroblastic profile seen with an infiltrated marrow and similarly does not reveal schistocytes that would suggest a microangiopathic hemolytic anemia. Echinocytes are generally seen in uremic states, although they can occasionally be seen in hemolysis as well. The presence of microspherocytes suggests autoimmune hemolytic anemia but a negative DAT suggests the hemolysis is not immune‐mediated. Vitamin B12 deficiency can cause marked intramedullary hemolysis with hypoproliferation, and thus the vitamin B12 level should be checked, even though macrocytosis and neurologic abnormalities are absent. The blister and bite cells present on the peripheral blood smear signal oxidative hemolysis. Testing for G6PD deficiency should be performed, and if negative, should be repeated in the convalescent phase once red cells of all ages are again present.
Cytomegalovirus and HIV‐1 viral loads were undetectable in the blood by PCR testing. The vitamin B12 level was 456 pg/mL (reference range >210 pg/mL). A Heinz body preparation (Figure 2) showed Heinz bodies in 6% of erythrocytes. A bone marrow biopsy (Figure 3) showed a cellularity of 80% to 90% with erythroid and megakaryocytic hyperplasia, left‐shifted erythropoiesis, and complete trilineage maturation without evidence of hemophagocytosis or excess blasts. Blood cultures remained sterile, and the patient defervesced 30 hours after receiving his first dose of azithromycin.
The vitamin B12 level is close to the lower limit of the normal range, and in light of the low reticulocyte count, warrants confirmation with methylmalonic acid and homocysteine measurement. The absence of macrocytic erythrocytes on his blood smear and megaloblastic changes in erythroid and myeloid precursors in the bone marrow make that nutritional deficiency less likely.
His marrow cellularity is high but near the upper range of normal given his age. Although his reticulocyte count is low, it appears that his bone marrow is starting to respond to his anemia, given the erythroid hyperplasia and left‐shifted erythropoiesis. The reticulocyte count should be repeated in 3 to 7 days, when it should be much higher.
Heinz bodies, which represent denatured hemoglobin, suggest that some erythrocytes have sustained oxidative stress that they could not defend against, typically because of a low G6PD level. Unstable hemoglobin variants are also vulnerable to oxidation. In addition, nonimmune causes of drug‐ and toxin‐induced hemolysis (eg, lead poisoning; Wilson's disease; or bites from insects, spiders, or snakes) should be considered.
It is possible that streptococcal pharyngitis triggered G6PD deficiency‐mediated hemolysis. Neither lymphoma nor hemophagocytosis was detected on the initial review of the bone marrow.
The hemoglobin decreased to 6.8 g/dL. One unit of packed red blood cells was transfused, and the next day the hemoglobin level was 7.8 g/dL. The family history was revisited, and the patient reported that a maternal uncle had G6PD deficiency. The G6PD activity was 3.2 U/g hemoglobin (reference range 7.020.5 U/g hemoglobin). One week later, the reticulocyte count was 16%, although the hemoglobin level remained relatively unchanged at 7.9 g/dL. The soluble interleukin‐2 receptor (sIL‐2R) level (sent to a reference laboratory during his hospitalization) was 1911 U/mL (reference range 451105 U/mL). At the 2‐week follow‐up appointment, his hemoglobin was 11.5 g/dL, LDH was 467 U/L, and ferritin was 277 ng/mL. Three months after his hospitalization, his hemolytic anemia had not recurred.
DISCUSSION
G6PD deficiency is the most common enzyme deficiency in humans, affecting more than 400 million people worldwide, with highest prevalence among Asian, African, and Mediterranean populations.[1] Oftentimes the characterization of an anemia as hemolytic and the identification of G6PD deficiency are straightforward. In this case, a more extensive evaluation was pursued on the basis of 2 conventional associations: reticulocytosis as an indicator of bone marrow response and the association of marked hyperferritinemia with a select group of diseases. More nuanced interpretation of these test results may have spared the patient a bone marrow biopsy and led to a less costly, more expeditious diagnosis.
One approach to anemia differentiates hypoproliferative anemias with an inappropriately low number of circulating reticulocytes for the degree of anemia (reflecting an inadequate bone marrow response) from regenerative anemias that have an appropriately elevated number of reticulocytes in circulation (reflecting adequate bone marrow response). This delineation can be a useful guide, but the variability of reticulocyte production, because of the presence of antibodies that inhibit erythroid colony forming units in the bone marrow,[2] viral infections,[3] or ineffective erythropoiesis,[4] can lead to misleading assumptions about the state of the bone marrow. In patients with G6PD deficiency, an increase in reticulocytes is often absent in the peripheral blood until 5 days after the acute onset of hemolysis and is not maximal until 7 to 10 days later.[5] Similarly, in a case series of patients with autoimmune hemolytic anemia, 37% of patients had an initial reticulocyte production index (RPI) <2, indicating hypoproliferation.[6] Of the 53% of these patients who underwent bone marrow examination, a majority (76%) showed erythroid hyperplasia despite the low RPI.[4] Malaria, the most prevalent worldwide cause of hemolytic anemia, can also present with relative reticulocytopenia. In 1 study, 75% of children with malaria‐related anemia had a reticulocyte production index <2.[7] These studies illustrate how classification of a patient's anemia solely on the basis of the reticulocyte count can lead to misdiagnosis.
In this case, the clinicians interpreted the low reticulocyte count as an indicator of a primary bone marrow disorder. The bone marrow biopsy instead demonstrated a brisk erythropoietic response that was not yet reflected in the peripheral blood. Given the absence of other cytopenias or myelophthisic findings on the peripheral smear and a strong suspicion of hemolysis, a reasonable approach would have been to instead repeat the reticulocyte count a few days into the evaluation to account for the transient lag in the bone marrow response to an acute episode of hemolysis. If the reticulocyte count remained suppressed 1 week later, it would have been appropriate to pursue a bone marrow biopsy at that time to investigate for a malignant, infectious, or nutritional etiology.
Iron studies revealed hyperferritinemia. This finding led the clinicians to consider HLH, a rare cause of multisystem organ failure and pancytopenia.[8] An elevated ferritin level (often in excess of 5000 ng/mL but at least >500 ng/mL) is a diagnostic criterion for HLH. However, the low probability of this rare condition is not meaningfully modified by hyperferritinemia, which has very limited specificity. In a case series of 23 patients with markedly elevated levels of serum ferritin (>10,000 ng/mL), malignancy, infection, liver disease, and chronic transfusions were common causes; there was 1 case of Still's disease and no cases of HLH.[9] In this case, the elevated ferritin and elevated sIL‐2R level, which was sent in response to the elevated ferritin to examine the remote possibility of HLH, reflected the inflammatory response to his GAS pharyngitis and acute hemolytic episode, not HLH.
G6PD deficiency leads to hemolysis due to an inability of the erythrocyte to resist oxidative stress. Drugs, including antimalarial, antibacterial, and other medications, are commonly considered major precipitants of G6PD deficiency‐mediated hemolysis.[1] However, a case series of patients with G6PD deficiency‐related hemolysis showed that most episodes were related to infection alone (53%, most commonly pneumonia) or to infection and drug therapy in combination (15%). Drug therapy alone accounted for only 32% of cases.[10] Another case series found infection caused nearly all cases of G6PD deficiency‐related hemolysis in children.[11] These findings suggest that clinicians should not implicate drugs as the cause of G6PD deficiency‐associated hemolysis unless infection has been excluded. One study demonstrated that infection with Streptococcus pneumoniae can lead to G6PD‐related hemolysis due to oxidative damage of red blood cells from binding of immune complexes to the red blood cell membrane.[12] An association between ‐hemolytic streptococcal pharyngitis and G6PD‐mediated hemolysis has been reported.[13] In this patient, G6PD‐related hemolysis was likely precipitated by his exaggerated inflammatory response to GAS pharyngitis.
Illness scripts are cognitive structures that allow clinicians to organize information about diseases into a useful framework for making clinical decisions.[14] Illness scripts are initially formed through our introduction to textbook cases, but they require constant revision throughout our careers to avoid reliance on outdated, incorrect, or biased information. Revision of illness scriptsthrough thoughtful reflection on patient casescreates more nuanced profiles of diseases and conditions that can be brought to bear on future cases. Through analysis of this case, clinicians will have the opportunity to update their illness scripts for anemia, reticulocytosis, hyperferritinemia, and G6PD‐associated hemolysis. When faced with similar cases, they will be better equipped to characterize anemia and avoid unnecessary testing (eg, sIL‐2R, bone marrow biopsy). This case reminds us that continual revision of our illness scripts is a cornerstone of delivering higher quality and less costly care for future patients.
TEACHING POINTS
- The reticulocyte count takes 7 to 10 days to peak in response to anemia. Classification of anemia solely based on an early reticulocyte count may lead to misdiagnosis.
- Hyperferritinemia in adults is most commonly seen in patients with malignancy, chronic transfusions, infection, and liver disease, and seldom signals a rare condition such as HLH or Still's disease.
- Infections are the most common triggers for G6PD‐related hemolysis and should be excluded diligently before ascribing the hemolysis to a drug.
Acknowledgements
The authors thank Wesley J. Miller, MD, for his review of an earlier version of the manuscript.
Disclosure: Nothing to report.
- . Glucose‐6‐phosphate dehydrogenase deficiency: a historical perspective. Blood. 2008;111(1):16–24.
- , , , , . Demonstration of two distinct antibodies in autoimmune hemolytic anemia with reticulocytopenia and red cell aplasia. Exp Hematol. 1984;12(10):788–793.
- , , . The acute and transient nature of idiopathic immune hemolytic anemia in childhood. J Pediatr. 1976;88(5):780–783.
- , , . Variability of the erythropoietic response in autoimmune hemolytic anemia: analysis of 109 cases. Blood. 1987;69(3):820–826.
- , , , , , . Mitigation of the haemolytic effect of primaquine and enhancement of its action against exoerythrocytic forms of the Chesson strain of Piasmodium vivax by intermittent regimens of drug administration: a preliminary report. Bull World Health Organ. 1960;22:621–631.
- . Characteristics of marrow production and reticulocyte maturation in normal man in response to anemia. J Clin Invest. 1969;48(3):443–453.
- , , , et al. Clinical predictors of severe malarial anaemia in a holoendemic Plasmodium falciparum transmission area. Br J Haematol. 2010;149(5):711–721.
- , , , et al. HLH‐2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48(2):124–131.
- , . Extreme hyperferritinaemia; clinical causes. J Clin Pathol. 2013;66(5):438–440.
- . Clinical spectrum of hemolytic anemia associated with glucose‐6‐phosphate dehydrogenase deficiency. Ann Intern Med. 1966;64(4):817.
- , . Severe hemolytic anemia in black children with glucose‐6‐phosphate dehydrogenase deficiency. Pediatrics. 1982;70(3):364–369.
- , , . G6PD‐deficiency infectious haemolysis: a complement dependent innocent bystander phenomenon. Br J Haematol. 1986;63(1):85–91.
- . Anemia during acute infections. Arch Intern Med. 1967;119(3):287.
- , , . Scripts and medical diagnostic knowledge: theory and applications for clinical reasoning instruction and research. Acad Med. 2000;75(2):182–190.
A 25‐year‐old male presented to the emergency department with a 3‐day history of fever, chills, nausea, vomiting, diarrhea, and myalgias.
The acute onset, combination of vomiting and diarrhea, and systemic symptoms are most characteristic of an acute gastrointestinal infection, such as viral gastroenteritis (eg, Norovirus or Rotavirus) or bacterial enteritis (eg, nontyphoidal Salmonella, Campylobacter jejuni, or Escherichia coli). A careful exposure history, taking into account travel, diet, sick contacts, and living situation, can help prioritize the likelihood of a given pathogen, although treatment is generally supportive in the absence of severe dehydration, abdominal pain, or vital sign abnormalities. Vomiting and diarrhea can also be nonspecific responses to severe, nongastrointestinal infections, such as influenza or staphylococcal bacteremia. A drug or toxin could prompt an allergic or inflammatory response similar to the syndrome observed here. Due to the acuity, other categories of disease, such as autoimmunity, metabolic derangement, or malignancy, seem unlikely at this point.
Aside from being treated for Trichomonas vaginalis urethritis 2 months prior, the patient had been in good health and took no medications until the onset of these symptoms. Upon review of systems, he complained of a sore throat and odynophagia but denied cough or rhinorrhea. On examination, he appeared comfortable. His temperature was 39.2C, blood pressure 137/64 mm Hg, heart rate 92 beats per minute, and respiratory rate 16 breaths per minute. His arterial oxygen saturation was 97% while breathing ambient air. The posterior oropharynx was erythematous without exudates. There was no cervical lymphadenopathy. He was tender in the epigastric region without rebound or guarding. The white blood cell count was 6800/mm3, hemoglobin 10.0 g/dL with a mean corpuscular volume of 81 fL, and platelet count 224,000/mm3. The aspartate aminotransferase (AST) was 60 U/L (reference range 045 U/L), and the total bilirubin was 3.6 mg/dL; electrolytes, alanine aminotransferase, alkaline phosphatase, albumin, and the international normalized ratio were normal. Rapid antigen testing for influenza A and B were negative, and a rapid test for group A Streptococcal (GAS) antigen was positive.
Vomiting and abdominal tenderness are less typical in adults than in children with routine GAS pharyngitis. His odynophagia could reflect a retropharyngeal or peritonsillar abscess. Influenza assays have limited sensitivity and cannot reliably exclude acute infection, especially when the prevalence is high during influenza season. Epstein‐Barr virus (EBV)‐associated mononucleosis and acute human immunodeficiency virus (HIV) can cause acute pharyngitis and hepatitis, but the lymphadenopathy that is characteristic of both infections was absent. His recent trichomonas infection indicates that he may be at risk for sexually transmitted diseases, including HIV, gonorrhea, and syphilis.
His elevated bilirubin and AST along with vomiting, epigastric tenderness, and fevers raise the possibility of cholecystitis or cholangitis, which should be explored further with abdominal imaging. Mild AST elevation alone could be explained by muscle damage (given his myalgias), viral or bacterial invasion of the liver, or alcohol or other toxins, including acetaminophen, which he may be taking to treat his pain and fever.
The combination of anemia and hyperbilirubinemia should prompt consideration of hemolysis, but the anemia could also be explained by an underlying chronic disease (eg, HIV or hematologic malignancy), preexisting iron deficiency, or thalassemia.
He was given intravenous ceftriaxone in the emergency department. Penicillin, ondansetron, and omeprazole were prescribed, and he was discharged home. He never took the penicillin because a family member told him that his throat swelled up in the past when he took it. He continued to have malaise, diarrhea, myalgias, fatigue, and fevers to 38.9C. He returned to the emergency department 2 days later. His temperature was 38.6C, and his remaining vital signs were normal. His posterior oropharynx was erythematous and his sclerae icteric; his abdomen was soft, nontender, and nondistended, without hepatosplenomegaly. His hemoglobin was 8.8 g/dL, bilirubin 3.6 mg/dL without conjugated bilirubin present, lactate dehydrogenase (LDH) 3077 U/L (reference range 325750 U/L), and AST 126 U/L; blood urea nitrogen and creatinine were normal. He was admitted to the hospital.
The progression of his systemic symptoms for an additional 2 days in the absence of directed treatment for acute pharyngitis is not unusual. However, his anemia is progressive, with features highly suggestive of hemolysis, including indirect hyperbilirubinemia, elevated LDH, and elevated AST. The single dose of ceftriaxone is unlikely to have triggered drug‐induced immune hemolysis, and his anemia predates the antibiotic regardless. Fever can accompany hemolysis when a malignancy (eg, lymphoma) or autoimmune condition (eg, systemic lupus erythematosus) triggers immune‐mediated hemolytic anemia. Microangiopathic processes (eg, thrombotic thrombocytopenic purpura and disseminated intravascular coagulation) can be associated with fever because of the underlying mechanism or an untreated infection, respectively. Some pathogens, such as Plasmodium, Babesia, and Clostridium species, directly invade erythrocytes, leading to their destruction. He may have an underlying predisposition for hemolysis (eg, glucose‐6‐phosphate dehydrogenase [G6PD] deficiency) that has been unmasked in the setting of acute infection.
At admission, intravenous azithromycin was administered for GAS infection; peripheral blood cultures were sterile. His hemoglobin decreased to 7.3 g/dL. The reticulocyte count was 1.2%, and the direct antiglobulin test (DAT) was negative. A normochromic, normocytic anemia with blister and bite cells, rare microspherocytes, and echinocytes was seen on the peripheral blood smear (Figure 1). A chest radiograph was normal, and polymerase chain reaction (PCR) tests for parvovirus and EBV DNA in peripheral blood were negative. Neither parvovirus IgM antibodies nor HIV antibodies were present. The ferritin level was >33,000 ng/mL (reference range 20300 ng/mL), serum iron 87 g/dL (reference range 35180 g/dL), iron binding capacity 200 g/dL (reference range 240430 g/dL), and iron saturation index 44% (reference range 15%46%).
His ongoing fevers suggest an untreated infection, autoimmune condition, or malignancy. The depressed reticulocyte count is unexpected in the setting of hemolysis in a young and previously healthy patient, raising the prospect of his bone marrow harboring a hematologic malignancy or infection (eg, mycobacterial, fungal, or viral). Alternatively, an immune or infectious process may be impeding erythropoiesis (eg, pure red cell aplasia or parvovirus infection). Hyperferritinemia is nonspecific and suggests systemic inflammation, but is also associated with Still's disease, histoplasmosis, hemochromatosis, and hemophagocytic syndromes. Still's disease causes high fevers and pharyngitis but typically features leukocytosis and arthralgias, both of which are absent. Hemophagocytosis in adults is typically due to a hyperinflammatory response to an underlying infection or malignancy caused by uncontrolled proliferation of activated lymphocytes and macrophages secreting large amounts of inflammatory cytokines.
The peripheral blood smear does not demonstrate a leukoerythroblastic profile seen with an infiltrated marrow and similarly does not reveal schistocytes that would suggest a microangiopathic hemolytic anemia. Echinocytes are generally seen in uremic states, although they can occasionally be seen in hemolysis as well. The presence of microspherocytes suggests autoimmune hemolytic anemia but a negative DAT suggests the hemolysis is not immune‐mediated. Vitamin B12 deficiency can cause marked intramedullary hemolysis with hypoproliferation, and thus the vitamin B12 level should be checked, even though macrocytosis and neurologic abnormalities are absent. The blister and bite cells present on the peripheral blood smear signal oxidative hemolysis. Testing for G6PD deficiency should be performed, and if negative, should be repeated in the convalescent phase once red cells of all ages are again present.
Cytomegalovirus and HIV‐1 viral loads were undetectable in the blood by PCR testing. The vitamin B12 level was 456 pg/mL (reference range >210 pg/mL). A Heinz body preparation (Figure 2) showed Heinz bodies in 6% of erythrocytes. A bone marrow biopsy (Figure 3) showed a cellularity of 80% to 90% with erythroid and megakaryocytic hyperplasia, left‐shifted erythropoiesis, and complete trilineage maturation without evidence of hemophagocytosis or excess blasts. Blood cultures remained sterile, and the patient defervesced 30 hours after receiving his first dose of azithromycin.
The vitamin B12 level is close to the lower limit of the normal range, and in light of the low reticulocyte count, warrants confirmation with methylmalonic acid and homocysteine measurement. The absence of macrocytic erythrocytes on his blood smear and megaloblastic changes in erythroid and myeloid precursors in the bone marrow make that nutritional deficiency less likely.
His marrow cellularity is high but near the upper range of normal given his age. Although his reticulocyte count is low, it appears that his bone marrow is starting to respond to his anemia, given the erythroid hyperplasia and left‐shifted erythropoiesis. The reticulocyte count should be repeated in 3 to 7 days, when it should be much higher.
Heinz bodies, which represent denatured hemoglobin, suggest that some erythrocytes have sustained oxidative stress that they could not defend against, typically because of a low G6PD level. Unstable hemoglobin variants are also vulnerable to oxidation. In addition, nonimmune causes of drug‐ and toxin‐induced hemolysis (eg, lead poisoning; Wilson's disease; or bites from insects, spiders, or snakes) should be considered.
It is possible that streptococcal pharyngitis triggered G6PD deficiency‐mediated hemolysis. Neither lymphoma nor hemophagocytosis was detected on the initial review of the bone marrow.
The hemoglobin decreased to 6.8 g/dL. One unit of packed red blood cells was transfused, and the next day the hemoglobin level was 7.8 g/dL. The family history was revisited, and the patient reported that a maternal uncle had G6PD deficiency. The G6PD activity was 3.2 U/g hemoglobin (reference range 7.020.5 U/g hemoglobin). One week later, the reticulocyte count was 16%, although the hemoglobin level remained relatively unchanged at 7.9 g/dL. The soluble interleukin‐2 receptor (sIL‐2R) level (sent to a reference laboratory during his hospitalization) was 1911 U/mL (reference range 451105 U/mL). At the 2‐week follow‐up appointment, his hemoglobin was 11.5 g/dL, LDH was 467 U/L, and ferritin was 277 ng/mL. Three months after his hospitalization, his hemolytic anemia had not recurred.
DISCUSSION
G6PD deficiency is the most common enzyme deficiency in humans, affecting more than 400 million people worldwide, with highest prevalence among Asian, African, and Mediterranean populations.[1] Oftentimes the characterization of an anemia as hemolytic and the identification of G6PD deficiency are straightforward. In this case, a more extensive evaluation was pursued on the basis of 2 conventional associations: reticulocytosis as an indicator of bone marrow response and the association of marked hyperferritinemia with a select group of diseases. More nuanced interpretation of these test results may have spared the patient a bone marrow biopsy and led to a less costly, more expeditious diagnosis.
One approach to anemia differentiates hypoproliferative anemias with an inappropriately low number of circulating reticulocytes for the degree of anemia (reflecting an inadequate bone marrow response) from regenerative anemias that have an appropriately elevated number of reticulocytes in circulation (reflecting adequate bone marrow response). This delineation can be a useful guide, but the variability of reticulocyte production, because of the presence of antibodies that inhibit erythroid colony forming units in the bone marrow,[2] viral infections,[3] or ineffective erythropoiesis,[4] can lead to misleading assumptions about the state of the bone marrow. In patients with G6PD deficiency, an increase in reticulocytes is often absent in the peripheral blood until 5 days after the acute onset of hemolysis and is not maximal until 7 to 10 days later.[5] Similarly, in a case series of patients with autoimmune hemolytic anemia, 37% of patients had an initial reticulocyte production index (RPI) <2, indicating hypoproliferation.[6] Of the 53% of these patients who underwent bone marrow examination, a majority (76%) showed erythroid hyperplasia despite the low RPI.[4] Malaria, the most prevalent worldwide cause of hemolytic anemia, can also present with relative reticulocytopenia. In 1 study, 75% of children with malaria‐related anemia had a reticulocyte production index <2.[7] These studies illustrate how classification of a patient's anemia solely on the basis of the reticulocyte count can lead to misdiagnosis.
In this case, the clinicians interpreted the low reticulocyte count as an indicator of a primary bone marrow disorder. The bone marrow biopsy instead demonstrated a brisk erythropoietic response that was not yet reflected in the peripheral blood. Given the absence of other cytopenias or myelophthisic findings on the peripheral smear and a strong suspicion of hemolysis, a reasonable approach would have been to instead repeat the reticulocyte count a few days into the evaluation to account for the transient lag in the bone marrow response to an acute episode of hemolysis. If the reticulocyte count remained suppressed 1 week later, it would have been appropriate to pursue a bone marrow biopsy at that time to investigate for a malignant, infectious, or nutritional etiology.
Iron studies revealed hyperferritinemia. This finding led the clinicians to consider HLH, a rare cause of multisystem organ failure and pancytopenia.[8] An elevated ferritin level (often in excess of 5000 ng/mL but at least >500 ng/mL) is a diagnostic criterion for HLH. However, the low probability of this rare condition is not meaningfully modified by hyperferritinemia, which has very limited specificity. In a case series of 23 patients with markedly elevated levels of serum ferritin (>10,000 ng/mL), malignancy, infection, liver disease, and chronic transfusions were common causes; there was 1 case of Still's disease and no cases of HLH.[9] In this case, the elevated ferritin and elevated sIL‐2R level, which was sent in response to the elevated ferritin to examine the remote possibility of HLH, reflected the inflammatory response to his GAS pharyngitis and acute hemolytic episode, not HLH.
G6PD deficiency leads to hemolysis due to an inability of the erythrocyte to resist oxidative stress. Drugs, including antimalarial, antibacterial, and other medications, are commonly considered major precipitants of G6PD deficiency‐mediated hemolysis.[1] However, a case series of patients with G6PD deficiency‐related hemolysis showed that most episodes were related to infection alone (53%, most commonly pneumonia) or to infection and drug therapy in combination (15%). Drug therapy alone accounted for only 32% of cases.[10] Another case series found infection caused nearly all cases of G6PD deficiency‐related hemolysis in children.[11] These findings suggest that clinicians should not implicate drugs as the cause of G6PD deficiency‐associated hemolysis unless infection has been excluded. One study demonstrated that infection with Streptococcus pneumoniae can lead to G6PD‐related hemolysis due to oxidative damage of red blood cells from binding of immune complexes to the red blood cell membrane.[12] An association between ‐hemolytic streptococcal pharyngitis and G6PD‐mediated hemolysis has been reported.[13] In this patient, G6PD‐related hemolysis was likely precipitated by his exaggerated inflammatory response to GAS pharyngitis.
Illness scripts are cognitive structures that allow clinicians to organize information about diseases into a useful framework for making clinical decisions.[14] Illness scripts are initially formed through our introduction to textbook cases, but they require constant revision throughout our careers to avoid reliance on outdated, incorrect, or biased information. Revision of illness scriptsthrough thoughtful reflection on patient casescreates more nuanced profiles of diseases and conditions that can be brought to bear on future cases. Through analysis of this case, clinicians will have the opportunity to update their illness scripts for anemia, reticulocytosis, hyperferritinemia, and G6PD‐associated hemolysis. When faced with similar cases, they will be better equipped to characterize anemia and avoid unnecessary testing (eg, sIL‐2R, bone marrow biopsy). This case reminds us that continual revision of our illness scripts is a cornerstone of delivering higher quality and less costly care for future patients.
TEACHING POINTS
- The reticulocyte count takes 7 to 10 days to peak in response to anemia. Classification of anemia solely based on an early reticulocyte count may lead to misdiagnosis.
- Hyperferritinemia in adults is most commonly seen in patients with malignancy, chronic transfusions, infection, and liver disease, and seldom signals a rare condition such as HLH or Still's disease.
- Infections are the most common triggers for G6PD‐related hemolysis and should be excluded diligently before ascribing the hemolysis to a drug.
Acknowledgements
The authors thank Wesley J. Miller, MD, for his review of an earlier version of the manuscript.
Disclosure: Nothing to report.
A 25‐year‐old male presented to the emergency department with a 3‐day history of fever, chills, nausea, vomiting, diarrhea, and myalgias.
The acute onset, combination of vomiting and diarrhea, and systemic symptoms are most characteristic of an acute gastrointestinal infection, such as viral gastroenteritis (eg, Norovirus or Rotavirus) or bacterial enteritis (eg, nontyphoidal Salmonella, Campylobacter jejuni, or Escherichia coli). A careful exposure history, taking into account travel, diet, sick contacts, and living situation, can help prioritize the likelihood of a given pathogen, although treatment is generally supportive in the absence of severe dehydration, abdominal pain, or vital sign abnormalities. Vomiting and diarrhea can also be nonspecific responses to severe, nongastrointestinal infections, such as influenza or staphylococcal bacteremia. A drug or toxin could prompt an allergic or inflammatory response similar to the syndrome observed here. Due to the acuity, other categories of disease, such as autoimmunity, metabolic derangement, or malignancy, seem unlikely at this point.
Aside from being treated for Trichomonas vaginalis urethritis 2 months prior, the patient had been in good health and took no medications until the onset of these symptoms. Upon review of systems, he complained of a sore throat and odynophagia but denied cough or rhinorrhea. On examination, he appeared comfortable. His temperature was 39.2C, blood pressure 137/64 mm Hg, heart rate 92 beats per minute, and respiratory rate 16 breaths per minute. His arterial oxygen saturation was 97% while breathing ambient air. The posterior oropharynx was erythematous without exudates. There was no cervical lymphadenopathy. He was tender in the epigastric region without rebound or guarding. The white blood cell count was 6800/mm3, hemoglobin 10.0 g/dL with a mean corpuscular volume of 81 fL, and platelet count 224,000/mm3. The aspartate aminotransferase (AST) was 60 U/L (reference range 045 U/L), and the total bilirubin was 3.6 mg/dL; electrolytes, alanine aminotransferase, alkaline phosphatase, albumin, and the international normalized ratio were normal. Rapid antigen testing for influenza A and B were negative, and a rapid test for group A Streptococcal (GAS) antigen was positive.
Vomiting and abdominal tenderness are less typical in adults than in children with routine GAS pharyngitis. His odynophagia could reflect a retropharyngeal or peritonsillar abscess. Influenza assays have limited sensitivity and cannot reliably exclude acute infection, especially when the prevalence is high during influenza season. Epstein‐Barr virus (EBV)‐associated mononucleosis and acute human immunodeficiency virus (HIV) can cause acute pharyngitis and hepatitis, but the lymphadenopathy that is characteristic of both infections was absent. His recent trichomonas infection indicates that he may be at risk for sexually transmitted diseases, including HIV, gonorrhea, and syphilis.
His elevated bilirubin and AST along with vomiting, epigastric tenderness, and fevers raise the possibility of cholecystitis or cholangitis, which should be explored further with abdominal imaging. Mild AST elevation alone could be explained by muscle damage (given his myalgias), viral or bacterial invasion of the liver, or alcohol or other toxins, including acetaminophen, which he may be taking to treat his pain and fever.
The combination of anemia and hyperbilirubinemia should prompt consideration of hemolysis, but the anemia could also be explained by an underlying chronic disease (eg, HIV or hematologic malignancy), preexisting iron deficiency, or thalassemia.
He was given intravenous ceftriaxone in the emergency department. Penicillin, ondansetron, and omeprazole were prescribed, and he was discharged home. He never took the penicillin because a family member told him that his throat swelled up in the past when he took it. He continued to have malaise, diarrhea, myalgias, fatigue, and fevers to 38.9C. He returned to the emergency department 2 days later. His temperature was 38.6C, and his remaining vital signs were normal. His posterior oropharynx was erythematous and his sclerae icteric; his abdomen was soft, nontender, and nondistended, without hepatosplenomegaly. His hemoglobin was 8.8 g/dL, bilirubin 3.6 mg/dL without conjugated bilirubin present, lactate dehydrogenase (LDH) 3077 U/L (reference range 325750 U/L), and AST 126 U/L; blood urea nitrogen and creatinine were normal. He was admitted to the hospital.
The progression of his systemic symptoms for an additional 2 days in the absence of directed treatment for acute pharyngitis is not unusual. However, his anemia is progressive, with features highly suggestive of hemolysis, including indirect hyperbilirubinemia, elevated LDH, and elevated AST. The single dose of ceftriaxone is unlikely to have triggered drug‐induced immune hemolysis, and his anemia predates the antibiotic regardless. Fever can accompany hemolysis when a malignancy (eg, lymphoma) or autoimmune condition (eg, systemic lupus erythematosus) triggers immune‐mediated hemolytic anemia. Microangiopathic processes (eg, thrombotic thrombocytopenic purpura and disseminated intravascular coagulation) can be associated with fever because of the underlying mechanism or an untreated infection, respectively. Some pathogens, such as Plasmodium, Babesia, and Clostridium species, directly invade erythrocytes, leading to their destruction. He may have an underlying predisposition for hemolysis (eg, glucose‐6‐phosphate dehydrogenase [G6PD] deficiency) that has been unmasked in the setting of acute infection.
At admission, intravenous azithromycin was administered for GAS infection; peripheral blood cultures were sterile. His hemoglobin decreased to 7.3 g/dL. The reticulocyte count was 1.2%, and the direct antiglobulin test (DAT) was negative. A normochromic, normocytic anemia with blister and bite cells, rare microspherocytes, and echinocytes was seen on the peripheral blood smear (Figure 1). A chest radiograph was normal, and polymerase chain reaction (PCR) tests for parvovirus and EBV DNA in peripheral blood were negative. Neither parvovirus IgM antibodies nor HIV antibodies were present. The ferritin level was >33,000 ng/mL (reference range 20300 ng/mL), serum iron 87 g/dL (reference range 35180 g/dL), iron binding capacity 200 g/dL (reference range 240430 g/dL), and iron saturation index 44% (reference range 15%46%).
His ongoing fevers suggest an untreated infection, autoimmune condition, or malignancy. The depressed reticulocyte count is unexpected in the setting of hemolysis in a young and previously healthy patient, raising the prospect of his bone marrow harboring a hematologic malignancy or infection (eg, mycobacterial, fungal, or viral). Alternatively, an immune or infectious process may be impeding erythropoiesis (eg, pure red cell aplasia or parvovirus infection). Hyperferritinemia is nonspecific and suggests systemic inflammation, but is also associated with Still's disease, histoplasmosis, hemochromatosis, and hemophagocytic syndromes. Still's disease causes high fevers and pharyngitis but typically features leukocytosis and arthralgias, both of which are absent. Hemophagocytosis in adults is typically due to a hyperinflammatory response to an underlying infection or malignancy caused by uncontrolled proliferation of activated lymphocytes and macrophages secreting large amounts of inflammatory cytokines.
The peripheral blood smear does not demonstrate a leukoerythroblastic profile seen with an infiltrated marrow and similarly does not reveal schistocytes that would suggest a microangiopathic hemolytic anemia. Echinocytes are generally seen in uremic states, although they can occasionally be seen in hemolysis as well. The presence of microspherocytes suggests autoimmune hemolytic anemia but a negative DAT suggests the hemolysis is not immune‐mediated. Vitamin B12 deficiency can cause marked intramedullary hemolysis with hypoproliferation, and thus the vitamin B12 level should be checked, even though macrocytosis and neurologic abnormalities are absent. The blister and bite cells present on the peripheral blood smear signal oxidative hemolysis. Testing for G6PD deficiency should be performed, and if negative, should be repeated in the convalescent phase once red cells of all ages are again present.
Cytomegalovirus and HIV‐1 viral loads were undetectable in the blood by PCR testing. The vitamin B12 level was 456 pg/mL (reference range >210 pg/mL). A Heinz body preparation (Figure 2) showed Heinz bodies in 6% of erythrocytes. A bone marrow biopsy (Figure 3) showed a cellularity of 80% to 90% with erythroid and megakaryocytic hyperplasia, left‐shifted erythropoiesis, and complete trilineage maturation without evidence of hemophagocytosis or excess blasts. Blood cultures remained sterile, and the patient defervesced 30 hours after receiving his first dose of azithromycin.
The vitamin B12 level is close to the lower limit of the normal range, and in light of the low reticulocyte count, warrants confirmation with methylmalonic acid and homocysteine measurement. The absence of macrocytic erythrocytes on his blood smear and megaloblastic changes in erythroid and myeloid precursors in the bone marrow make that nutritional deficiency less likely.
His marrow cellularity is high but near the upper range of normal given his age. Although his reticulocyte count is low, it appears that his bone marrow is starting to respond to his anemia, given the erythroid hyperplasia and left‐shifted erythropoiesis. The reticulocyte count should be repeated in 3 to 7 days, when it should be much higher.
Heinz bodies, which represent denatured hemoglobin, suggest that some erythrocytes have sustained oxidative stress that they could not defend against, typically because of a low G6PD level. Unstable hemoglobin variants are also vulnerable to oxidation. In addition, nonimmune causes of drug‐ and toxin‐induced hemolysis (eg, lead poisoning; Wilson's disease; or bites from insects, spiders, or snakes) should be considered.
It is possible that streptococcal pharyngitis triggered G6PD deficiency‐mediated hemolysis. Neither lymphoma nor hemophagocytosis was detected on the initial review of the bone marrow.
The hemoglobin decreased to 6.8 g/dL. One unit of packed red blood cells was transfused, and the next day the hemoglobin level was 7.8 g/dL. The family history was revisited, and the patient reported that a maternal uncle had G6PD deficiency. The G6PD activity was 3.2 U/g hemoglobin (reference range 7.020.5 U/g hemoglobin). One week later, the reticulocyte count was 16%, although the hemoglobin level remained relatively unchanged at 7.9 g/dL. The soluble interleukin‐2 receptor (sIL‐2R) level (sent to a reference laboratory during his hospitalization) was 1911 U/mL (reference range 451105 U/mL). At the 2‐week follow‐up appointment, his hemoglobin was 11.5 g/dL, LDH was 467 U/L, and ferritin was 277 ng/mL. Three months after his hospitalization, his hemolytic anemia had not recurred.
DISCUSSION
G6PD deficiency is the most common enzyme deficiency in humans, affecting more than 400 million people worldwide, with highest prevalence among Asian, African, and Mediterranean populations.[1] Oftentimes the characterization of an anemia as hemolytic and the identification of G6PD deficiency are straightforward. In this case, a more extensive evaluation was pursued on the basis of 2 conventional associations: reticulocytosis as an indicator of bone marrow response and the association of marked hyperferritinemia with a select group of diseases. More nuanced interpretation of these test results may have spared the patient a bone marrow biopsy and led to a less costly, more expeditious diagnosis.
One approach to anemia differentiates hypoproliferative anemias with an inappropriately low number of circulating reticulocytes for the degree of anemia (reflecting an inadequate bone marrow response) from regenerative anemias that have an appropriately elevated number of reticulocytes in circulation (reflecting adequate bone marrow response). This delineation can be a useful guide, but the variability of reticulocyte production, because of the presence of antibodies that inhibit erythroid colony forming units in the bone marrow,[2] viral infections,[3] or ineffective erythropoiesis,[4] can lead to misleading assumptions about the state of the bone marrow. In patients with G6PD deficiency, an increase in reticulocytes is often absent in the peripheral blood until 5 days after the acute onset of hemolysis and is not maximal until 7 to 10 days later.[5] Similarly, in a case series of patients with autoimmune hemolytic anemia, 37% of patients had an initial reticulocyte production index (RPI) <2, indicating hypoproliferation.[6] Of the 53% of these patients who underwent bone marrow examination, a majority (76%) showed erythroid hyperplasia despite the low RPI.[4] Malaria, the most prevalent worldwide cause of hemolytic anemia, can also present with relative reticulocytopenia. In 1 study, 75% of children with malaria‐related anemia had a reticulocyte production index <2.[7] These studies illustrate how classification of a patient's anemia solely on the basis of the reticulocyte count can lead to misdiagnosis.
In this case, the clinicians interpreted the low reticulocyte count as an indicator of a primary bone marrow disorder. The bone marrow biopsy instead demonstrated a brisk erythropoietic response that was not yet reflected in the peripheral blood. Given the absence of other cytopenias or myelophthisic findings on the peripheral smear and a strong suspicion of hemolysis, a reasonable approach would have been to instead repeat the reticulocyte count a few days into the evaluation to account for the transient lag in the bone marrow response to an acute episode of hemolysis. If the reticulocyte count remained suppressed 1 week later, it would have been appropriate to pursue a bone marrow biopsy at that time to investigate for a malignant, infectious, or nutritional etiology.
Iron studies revealed hyperferritinemia. This finding led the clinicians to consider HLH, a rare cause of multisystem organ failure and pancytopenia.[8] An elevated ferritin level (often in excess of 5000 ng/mL but at least >500 ng/mL) is a diagnostic criterion for HLH. However, the low probability of this rare condition is not meaningfully modified by hyperferritinemia, which has very limited specificity. In a case series of 23 patients with markedly elevated levels of serum ferritin (>10,000 ng/mL), malignancy, infection, liver disease, and chronic transfusions were common causes; there was 1 case of Still's disease and no cases of HLH.[9] In this case, the elevated ferritin and elevated sIL‐2R level, which was sent in response to the elevated ferritin to examine the remote possibility of HLH, reflected the inflammatory response to his GAS pharyngitis and acute hemolytic episode, not HLH.
G6PD deficiency leads to hemolysis due to an inability of the erythrocyte to resist oxidative stress. Drugs, including antimalarial, antibacterial, and other medications, are commonly considered major precipitants of G6PD deficiency‐mediated hemolysis.[1] However, a case series of patients with G6PD deficiency‐related hemolysis showed that most episodes were related to infection alone (53%, most commonly pneumonia) or to infection and drug therapy in combination (15%). Drug therapy alone accounted for only 32% of cases.[10] Another case series found infection caused nearly all cases of G6PD deficiency‐related hemolysis in children.[11] These findings suggest that clinicians should not implicate drugs as the cause of G6PD deficiency‐associated hemolysis unless infection has been excluded. One study demonstrated that infection with Streptococcus pneumoniae can lead to G6PD‐related hemolysis due to oxidative damage of red blood cells from binding of immune complexes to the red blood cell membrane.[12] An association between ‐hemolytic streptococcal pharyngitis and G6PD‐mediated hemolysis has been reported.[13] In this patient, G6PD‐related hemolysis was likely precipitated by his exaggerated inflammatory response to GAS pharyngitis.
Illness scripts are cognitive structures that allow clinicians to organize information about diseases into a useful framework for making clinical decisions.[14] Illness scripts are initially formed through our introduction to textbook cases, but they require constant revision throughout our careers to avoid reliance on outdated, incorrect, or biased information. Revision of illness scriptsthrough thoughtful reflection on patient casescreates more nuanced profiles of diseases and conditions that can be brought to bear on future cases. Through analysis of this case, clinicians will have the opportunity to update their illness scripts for anemia, reticulocytosis, hyperferritinemia, and G6PD‐associated hemolysis. When faced with similar cases, they will be better equipped to characterize anemia and avoid unnecessary testing (eg, sIL‐2R, bone marrow biopsy). This case reminds us that continual revision of our illness scripts is a cornerstone of delivering higher quality and less costly care for future patients.
TEACHING POINTS
- The reticulocyte count takes 7 to 10 days to peak in response to anemia. Classification of anemia solely based on an early reticulocyte count may lead to misdiagnosis.
- Hyperferritinemia in adults is most commonly seen in patients with malignancy, chronic transfusions, infection, and liver disease, and seldom signals a rare condition such as HLH or Still's disease.
- Infections are the most common triggers for G6PD‐related hemolysis and should be excluded diligently before ascribing the hemolysis to a drug.
Acknowledgements
The authors thank Wesley J. Miller, MD, for his review of an earlier version of the manuscript.
Disclosure: Nothing to report.
- . Glucose‐6‐phosphate dehydrogenase deficiency: a historical perspective. Blood. 2008;111(1):16–24.
- , , , , . Demonstration of two distinct antibodies in autoimmune hemolytic anemia with reticulocytopenia and red cell aplasia. Exp Hematol. 1984;12(10):788–793.
- , , . The acute and transient nature of idiopathic immune hemolytic anemia in childhood. J Pediatr. 1976;88(5):780–783.
- , , . Variability of the erythropoietic response in autoimmune hemolytic anemia: analysis of 109 cases. Blood. 1987;69(3):820–826.
- , , , , , . Mitigation of the haemolytic effect of primaquine and enhancement of its action against exoerythrocytic forms of the Chesson strain of Piasmodium vivax by intermittent regimens of drug administration: a preliminary report. Bull World Health Organ. 1960;22:621–631.
- . Characteristics of marrow production and reticulocyte maturation in normal man in response to anemia. J Clin Invest. 1969;48(3):443–453.
- , , , et al. Clinical predictors of severe malarial anaemia in a holoendemic Plasmodium falciparum transmission area. Br J Haematol. 2010;149(5):711–721.
- , , , et al. HLH‐2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48(2):124–131.
- , . Extreme hyperferritinaemia; clinical causes. J Clin Pathol. 2013;66(5):438–440.
- . Clinical spectrum of hemolytic anemia associated with glucose‐6‐phosphate dehydrogenase deficiency. Ann Intern Med. 1966;64(4):817.
- , . Severe hemolytic anemia in black children with glucose‐6‐phosphate dehydrogenase deficiency. Pediatrics. 1982;70(3):364–369.
- , , . G6PD‐deficiency infectious haemolysis: a complement dependent innocent bystander phenomenon. Br J Haematol. 1986;63(1):85–91.
- . Anemia during acute infections. Arch Intern Med. 1967;119(3):287.
- , , . Scripts and medical diagnostic knowledge: theory and applications for clinical reasoning instruction and research. Acad Med. 2000;75(2):182–190.
- . Glucose‐6‐phosphate dehydrogenase deficiency: a historical perspective. Blood. 2008;111(1):16–24.
- , , , , . Demonstration of two distinct antibodies in autoimmune hemolytic anemia with reticulocytopenia and red cell aplasia. Exp Hematol. 1984;12(10):788–793.
- , , . The acute and transient nature of idiopathic immune hemolytic anemia in childhood. J Pediatr. 1976;88(5):780–783.
- , , . Variability of the erythropoietic response in autoimmune hemolytic anemia: analysis of 109 cases. Blood. 1987;69(3):820–826.
- , , , , , . Mitigation of the haemolytic effect of primaquine and enhancement of its action against exoerythrocytic forms of the Chesson strain of Piasmodium vivax by intermittent regimens of drug administration: a preliminary report. Bull World Health Organ. 1960;22:621–631.
- . Characteristics of marrow production and reticulocyte maturation in normal man in response to anemia. J Clin Invest. 1969;48(3):443–453.
- , , , et al. Clinical predictors of severe malarial anaemia in a holoendemic Plasmodium falciparum transmission area. Br J Haematol. 2010;149(5):711–721.
- , , , et al. HLH‐2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48(2):124–131.
- , . Extreme hyperferritinaemia; clinical causes. J Clin Pathol. 2013;66(5):438–440.
- . Clinical spectrum of hemolytic anemia associated with glucose‐6‐phosphate dehydrogenase deficiency. Ann Intern Med. 1966;64(4):817.
- , . Severe hemolytic anemia in black children with glucose‐6‐phosphate dehydrogenase deficiency. Pediatrics. 1982;70(3):364–369.
- , , . G6PD‐deficiency infectious haemolysis: a complement dependent innocent bystander phenomenon. Br J Haematol. 1986;63(1):85–91.
- . Anemia during acute infections. Arch Intern Med. 1967;119(3):287.
- , , . Scripts and medical diagnostic knowledge: theory and applications for clinical reasoning instruction and research. Acad Med. 2000;75(2):182–190.
Evaluation of Hemostasis
A previously healthy 25‐year‐old Guatemalan man presented to the emergency department with 1 day of fever, nausea, vomiting, and right lower quadrant abdominal pain. A computed tomography (CT) scan revealed acute appendicitis. The patient underwent an uncomplicated laparoscopic appendectomy and was discharged in stable condition after 48 hours.
Five days after the operation he returned to the emergency department with abdominal pain, nausea, vomiting, and lightheadedness. He was tachycardic, and his hemoglobin was 9.5 g/dL (normal, 13.3‐17.7 g/dL), decreased from 14.4 g/dL prior to his appendectomy. A CT scan showed intraperitoneal blood with active extravasation of contrast at the site of the appendectomy.
Additional laboratory testing revealed an activated partial thromboplastin time (aPTT) of 52 seconds (normal, <37 seconds) and protime (also prothrombin time [PT]) of 14 seconds (normal, <14.1 seconds). The platelet count was 449,000/L (normal, 150‐400,000/L) and the fibrinogen level was 337 mg/dL (normal, 170‐440 mg/dL). Crystalloid and packed red blood cells were administered. Since further laboratory evaluation of the prolonged aPTT was not immediately available, the patient was empirically treated with fresh frozen plasma (FFP), cryoprecipitate, and Factor VIII/von Willebrand factor concentrate. At laparotomy, bleeding was observed at the previous operative site, and 2 L of intraperitoneal blood was evacuated.
The next morning, Factor VIII and Factor IX (FIX) activities and the ristocetin cofactor study performed on specimens obtained immediately prior to the second operation were normal, but the FIX activity was 5% of normal. The diagnosis of FXI deficiency was made and 2 to 3 units of FFP (the amount necessary to maintain the patient's measured FXI activity near 20% of normal) were transfused daily. Nine days of FFP infusions were required to achieve complete wound hemostasis. The patient had no further bleeding episodes after discharge.
Upon further interviewing, the patient revealed that 2 months prior he sustained a small laceration on his arm that bled for a long time and that his brother had experienced prolonged bleeding after a dental extraction.
Commentary
Routine performance of preprocedural laboratory testing, and complete reliance on the results as a means of excluding a propensity to bleeding, may not only lead to excessive testing and delayed procedures, but also provides false reassurance because normal routine laboratory studies cannot be used to exclude some bleeding disorders (Table 1).
| Von Willebrand disease |
| Mild hemophilia A (Factor VIII deficiency) |
| Mild hemophilia B (Factor IX deficiency) |
| Mild hemophilia C (Factor XI deficiency) |
| Qualitative platelet disorders (congenital or acquired) |
| Factor XIII deficiency |
| Disorders of fibrinolysis (eg, antiplasmin deficiency, plasminogen activator inhibitor type 1 deficiency) |
| Disorders of the vasculature or integument (hereditary hemorrhagic telangiectasia, Ehlers‐Danlos syndrome) |
Most studies evaluating routine laboratory testing of hemostatic variables prior to invasive procedures come from patients undergoing elective general surgery. A 1988 study concluded that there is no benefit in the routine preoperative use of the PT, aPTT, platelet count, and bleeding time in the absence of clinical evidence of a hemostatic defect, as assessed by a patient questionnaire and a thorough physical examination.1 A subsequent European, prospective, multicenter study confirmed that abnormalities of preoperative laboratory screening in the absence of a history of bleeding or clinical abnormality were not associated with worse surgical morbidity or mortality, compared to patients with normal screening laboratory studies.2 A recent systematic review has also confirmed the poor positive predictive value of screening tests when used in isolation, and recommended a history‐based and physical exam‐based approach.3 Questionnaires have been shown to be particularly important tools for eliciting clinically significant bleeding disorders that may require revision of the surgical plan.1,4
FXI is a serine protease whose activity is crucial for robust fibrin clot formation and inhibition of fibrinolysis at sites of vascular injury.5 FXI deficiency is an autosomal recessive disorder with an incidence of 1 per 1,000,000 in the general population, with a significantly higher incidence in the Ashkenazi Jewish population. While the risk of spontaneous hemorrhage is typically low, life‐threatening bleeding may occur after surgery or trauma. The severity of the measured FXI level deficiency does not always correlate with risk of bleeding. Periprocedural prophylaxis and treatment of bleeding aim to replace FXI to the low‐normal range by administering FXI concentrate, (not available in the United States) or FFP. Antifibrinolytic agents such as tranexamic acid or ϵ‐aminocaproic acid may be used adjunctively in cases of mucosal bleeding.5
In this case, preoperative screening, either using a questionnaire or careful history‐taking, would have identified the patient's personal and family history of bleeding and prompted appropriate preoperative coagulation testing, which could have exposed the hemostatic defect, allowing for modification of the perioperative medical plan.
In summary, preoperative bleeding evaluations should be performed routinely and should begin with a careful history (use of a questionnaire may be considered) and physical examination. Excessive bleeding after prior surgery, trauma, dental extractions, parturition, or circumcision; bleeding tendency in family members; current use of medications that may increase bleeding risk (such as anticoagulants or aspirin); and physical signs associated with bleeding should be assessed. If clinical details fail to expose a potential bleeding disorder, it is safe and cost‐effective1 to proceed with surgery without performing additional laboratory testing. In contrast, any abnormality on the clinical assessment should trigger preoperative laboratory analysis of basic hemostatic parameters, which may prompt further testing or hematology consultation.
- ,,.A prospective evaluation of the efficacy of preoperative coagulation testing.Ann Surg.1988;208(5):554–557.
- ,,,,.A prospective multicenter evaluation of preoperative hemostatic screening tests. The French Associations for Surgical Research.Am J Surg.1995;170(1):19–23.
- ,,,.Guidelines on the assessment of bleeding risk prior to surgery or invasive procedures.Br J Haematol.2008;140:496–504.
- ,,, et al.A practical concept for preoperative identification of patients with impaired primary hemostasis.Clin Appl Thrombosis Haemost.2004;10(3):195–204.
- ,.Factor XI deficiency.Haemophilia.2008;14(6):1183–1189.
A previously healthy 25‐year‐old Guatemalan man presented to the emergency department with 1 day of fever, nausea, vomiting, and right lower quadrant abdominal pain. A computed tomography (CT) scan revealed acute appendicitis. The patient underwent an uncomplicated laparoscopic appendectomy and was discharged in stable condition after 48 hours.
Five days after the operation he returned to the emergency department with abdominal pain, nausea, vomiting, and lightheadedness. He was tachycardic, and his hemoglobin was 9.5 g/dL (normal, 13.3‐17.7 g/dL), decreased from 14.4 g/dL prior to his appendectomy. A CT scan showed intraperitoneal blood with active extravasation of contrast at the site of the appendectomy.
Additional laboratory testing revealed an activated partial thromboplastin time (aPTT) of 52 seconds (normal, <37 seconds) and protime (also prothrombin time [PT]) of 14 seconds (normal, <14.1 seconds). The platelet count was 449,000/L (normal, 150‐400,000/L) and the fibrinogen level was 337 mg/dL (normal, 170‐440 mg/dL). Crystalloid and packed red blood cells were administered. Since further laboratory evaluation of the prolonged aPTT was not immediately available, the patient was empirically treated with fresh frozen plasma (FFP), cryoprecipitate, and Factor VIII/von Willebrand factor concentrate. At laparotomy, bleeding was observed at the previous operative site, and 2 L of intraperitoneal blood was evacuated.
The next morning, Factor VIII and Factor IX (FIX) activities and the ristocetin cofactor study performed on specimens obtained immediately prior to the second operation were normal, but the FIX activity was 5% of normal. The diagnosis of FXI deficiency was made and 2 to 3 units of FFP (the amount necessary to maintain the patient's measured FXI activity near 20% of normal) were transfused daily. Nine days of FFP infusions were required to achieve complete wound hemostasis. The patient had no further bleeding episodes after discharge.
Upon further interviewing, the patient revealed that 2 months prior he sustained a small laceration on his arm that bled for a long time and that his brother had experienced prolonged bleeding after a dental extraction.
Commentary
Routine performance of preprocedural laboratory testing, and complete reliance on the results as a means of excluding a propensity to bleeding, may not only lead to excessive testing and delayed procedures, but also provides false reassurance because normal routine laboratory studies cannot be used to exclude some bleeding disorders (Table 1).
| Von Willebrand disease |
| Mild hemophilia A (Factor VIII deficiency) |
| Mild hemophilia B (Factor IX deficiency) |
| Mild hemophilia C (Factor XI deficiency) |
| Qualitative platelet disorders (congenital or acquired) |
| Factor XIII deficiency |
| Disorders of fibrinolysis (eg, antiplasmin deficiency, plasminogen activator inhibitor type 1 deficiency) |
| Disorders of the vasculature or integument (hereditary hemorrhagic telangiectasia, Ehlers‐Danlos syndrome) |
Most studies evaluating routine laboratory testing of hemostatic variables prior to invasive procedures come from patients undergoing elective general surgery. A 1988 study concluded that there is no benefit in the routine preoperative use of the PT, aPTT, platelet count, and bleeding time in the absence of clinical evidence of a hemostatic defect, as assessed by a patient questionnaire and a thorough physical examination.1 A subsequent European, prospective, multicenter study confirmed that abnormalities of preoperative laboratory screening in the absence of a history of bleeding or clinical abnormality were not associated with worse surgical morbidity or mortality, compared to patients with normal screening laboratory studies.2 A recent systematic review has also confirmed the poor positive predictive value of screening tests when used in isolation, and recommended a history‐based and physical exam‐based approach.3 Questionnaires have been shown to be particularly important tools for eliciting clinically significant bleeding disorders that may require revision of the surgical plan.1,4
FXI is a serine protease whose activity is crucial for robust fibrin clot formation and inhibition of fibrinolysis at sites of vascular injury.5 FXI deficiency is an autosomal recessive disorder with an incidence of 1 per 1,000,000 in the general population, with a significantly higher incidence in the Ashkenazi Jewish population. While the risk of spontaneous hemorrhage is typically low, life‐threatening bleeding may occur after surgery or trauma. The severity of the measured FXI level deficiency does not always correlate with risk of bleeding. Periprocedural prophylaxis and treatment of bleeding aim to replace FXI to the low‐normal range by administering FXI concentrate, (not available in the United States) or FFP. Antifibrinolytic agents such as tranexamic acid or ϵ‐aminocaproic acid may be used adjunctively in cases of mucosal bleeding.5
In this case, preoperative screening, either using a questionnaire or careful history‐taking, would have identified the patient's personal and family history of bleeding and prompted appropriate preoperative coagulation testing, which could have exposed the hemostatic defect, allowing for modification of the perioperative medical plan.
In summary, preoperative bleeding evaluations should be performed routinely and should begin with a careful history (use of a questionnaire may be considered) and physical examination. Excessive bleeding after prior surgery, trauma, dental extractions, parturition, or circumcision; bleeding tendency in family members; current use of medications that may increase bleeding risk (such as anticoagulants or aspirin); and physical signs associated with bleeding should be assessed. If clinical details fail to expose a potential bleeding disorder, it is safe and cost‐effective1 to proceed with surgery without performing additional laboratory testing. In contrast, any abnormality on the clinical assessment should trigger preoperative laboratory analysis of basic hemostatic parameters, which may prompt further testing or hematology consultation.
A previously healthy 25‐year‐old Guatemalan man presented to the emergency department with 1 day of fever, nausea, vomiting, and right lower quadrant abdominal pain. A computed tomography (CT) scan revealed acute appendicitis. The patient underwent an uncomplicated laparoscopic appendectomy and was discharged in stable condition after 48 hours.
Five days after the operation he returned to the emergency department with abdominal pain, nausea, vomiting, and lightheadedness. He was tachycardic, and his hemoglobin was 9.5 g/dL (normal, 13.3‐17.7 g/dL), decreased from 14.4 g/dL prior to his appendectomy. A CT scan showed intraperitoneal blood with active extravasation of contrast at the site of the appendectomy.
Additional laboratory testing revealed an activated partial thromboplastin time (aPTT) of 52 seconds (normal, <37 seconds) and protime (also prothrombin time [PT]) of 14 seconds (normal, <14.1 seconds). The platelet count was 449,000/L (normal, 150‐400,000/L) and the fibrinogen level was 337 mg/dL (normal, 170‐440 mg/dL). Crystalloid and packed red blood cells were administered. Since further laboratory evaluation of the prolonged aPTT was not immediately available, the patient was empirically treated with fresh frozen plasma (FFP), cryoprecipitate, and Factor VIII/von Willebrand factor concentrate. At laparotomy, bleeding was observed at the previous operative site, and 2 L of intraperitoneal blood was evacuated.
The next morning, Factor VIII and Factor IX (FIX) activities and the ristocetin cofactor study performed on specimens obtained immediately prior to the second operation were normal, but the FIX activity was 5% of normal. The diagnosis of FXI deficiency was made and 2 to 3 units of FFP (the amount necessary to maintain the patient's measured FXI activity near 20% of normal) were transfused daily. Nine days of FFP infusions were required to achieve complete wound hemostasis. The patient had no further bleeding episodes after discharge.
Upon further interviewing, the patient revealed that 2 months prior he sustained a small laceration on his arm that bled for a long time and that his brother had experienced prolonged bleeding after a dental extraction.
Commentary
Routine performance of preprocedural laboratory testing, and complete reliance on the results as a means of excluding a propensity to bleeding, may not only lead to excessive testing and delayed procedures, but also provides false reassurance because normal routine laboratory studies cannot be used to exclude some bleeding disorders (Table 1).
| Von Willebrand disease |
| Mild hemophilia A (Factor VIII deficiency) |
| Mild hemophilia B (Factor IX deficiency) |
| Mild hemophilia C (Factor XI deficiency) |
| Qualitative platelet disorders (congenital or acquired) |
| Factor XIII deficiency |
| Disorders of fibrinolysis (eg, antiplasmin deficiency, plasminogen activator inhibitor type 1 deficiency) |
| Disorders of the vasculature or integument (hereditary hemorrhagic telangiectasia, Ehlers‐Danlos syndrome) |
Most studies evaluating routine laboratory testing of hemostatic variables prior to invasive procedures come from patients undergoing elective general surgery. A 1988 study concluded that there is no benefit in the routine preoperative use of the PT, aPTT, platelet count, and bleeding time in the absence of clinical evidence of a hemostatic defect, as assessed by a patient questionnaire and a thorough physical examination.1 A subsequent European, prospective, multicenter study confirmed that abnormalities of preoperative laboratory screening in the absence of a history of bleeding or clinical abnormality were not associated with worse surgical morbidity or mortality, compared to patients with normal screening laboratory studies.2 A recent systematic review has also confirmed the poor positive predictive value of screening tests when used in isolation, and recommended a history‐based and physical exam‐based approach.3 Questionnaires have been shown to be particularly important tools for eliciting clinically significant bleeding disorders that may require revision of the surgical plan.1,4
FXI is a serine protease whose activity is crucial for robust fibrin clot formation and inhibition of fibrinolysis at sites of vascular injury.5 FXI deficiency is an autosomal recessive disorder with an incidence of 1 per 1,000,000 in the general population, with a significantly higher incidence in the Ashkenazi Jewish population. While the risk of spontaneous hemorrhage is typically low, life‐threatening bleeding may occur after surgery or trauma. The severity of the measured FXI level deficiency does not always correlate with risk of bleeding. Periprocedural prophylaxis and treatment of bleeding aim to replace FXI to the low‐normal range by administering FXI concentrate, (not available in the United States) or FFP. Antifibrinolytic agents such as tranexamic acid or ϵ‐aminocaproic acid may be used adjunctively in cases of mucosal bleeding.5
In this case, preoperative screening, either using a questionnaire or careful history‐taking, would have identified the patient's personal and family history of bleeding and prompted appropriate preoperative coagulation testing, which could have exposed the hemostatic defect, allowing for modification of the perioperative medical plan.
In summary, preoperative bleeding evaluations should be performed routinely and should begin with a careful history (use of a questionnaire may be considered) and physical examination. Excessive bleeding after prior surgery, trauma, dental extractions, parturition, or circumcision; bleeding tendency in family members; current use of medications that may increase bleeding risk (such as anticoagulants or aspirin); and physical signs associated with bleeding should be assessed. If clinical details fail to expose a potential bleeding disorder, it is safe and cost‐effective1 to proceed with surgery without performing additional laboratory testing. In contrast, any abnormality on the clinical assessment should trigger preoperative laboratory analysis of basic hemostatic parameters, which may prompt further testing or hematology consultation.
- ,,.A prospective evaluation of the efficacy of preoperative coagulation testing.Ann Surg.1988;208(5):554–557.
- ,,,,.A prospective multicenter evaluation of preoperative hemostatic screening tests. The French Associations for Surgical Research.Am J Surg.1995;170(1):19–23.
- ,,,.Guidelines on the assessment of bleeding risk prior to surgery or invasive procedures.Br J Haematol.2008;140:496–504.
- ,,, et al.A practical concept for preoperative identification of patients with impaired primary hemostasis.Clin Appl Thrombosis Haemost.2004;10(3):195–204.
- ,.Factor XI deficiency.Haemophilia.2008;14(6):1183–1189.
- ,,.A prospective evaluation of the efficacy of preoperative coagulation testing.Ann Surg.1988;208(5):554–557.
- ,,,,.A prospective multicenter evaluation of preoperative hemostatic screening tests. The French Associations for Surgical Research.Am J Surg.1995;170(1):19–23.
- ,,,.Guidelines on the assessment of bleeding risk prior to surgery or invasive procedures.Br J Haematol.2008;140:496–504.
- ,,, et al.A practical concept for preoperative identification of patients with impaired primary hemostasis.Clin Appl Thrombosis Haemost.2004;10(3):195–204.
- ,.Factor XI deficiency.Haemophilia.2008;14(6):1183–1189.