User login
A 15-year-old African American girl presented to the emergency department with 3 days of fever, sore throat, nausea, vomiting, and poor appetite. She reported a 4-week history of fatigue, right hand pain and swelling, and a 6-kilogram weight loss for which she had seen her primary care provider several times. She reported no recent travel, sick contacts, or new medications.
It appears that there are potentially at least 2 separate problems: an acute one (past 3 days) and a more chronic one (past 4 weeks). These 2 problems may be directly related (ie, acute worsening of the more chronic problem), indirectly related (ie, the more chronic problem is leading to increased susceptibility to the acute problem, for instance, an evolving immunodeficiency predisposing to an opportunistic infection), or “true, true, but unrelated.” The clinical challenge is to keep one’s mind open to each of these potential scenarios and to avoid the tendency to focus on one of the problems and not pay enough attention to the other. Occam’s razor likely does not apply here.
Numerous common and typically transient diseases could cause the symptoms of the past 3 days, particularly infectious etiologies such as streptococcal pharyngitis or a viral infection. One cannot forget about these possibilities while contemplating the more worrisome symptoms of the past 4 weeks, especially weight loss in a growing adolescent. Patients may unintentionally lose weight for a variety of reasons, which can be broadly categorized by decreased caloric supply, gastrointestinal losses or malabsorption, and increased caloric demand; these categories are not mutually exclusive.
Lastly, 1 symptom may provide a more specific direction: the right hand pain and swelling of the past 4 weeks. More specifics, including the extent of the hand swelling, other areas of involvement, and the nature of her pain, will be helpful.
Her temperature was 99.5°F, heart rate 100 beats per minute, respiratory rate 18 breaths per minute, oxygen saturation 95% while breathing ambient air, blood pressure 99/56 mmHg, weight 44 kilograms, height 161 centimeters, and body mass index 17. She appeared generally ill and underweight. She had edematous and violaceous eyelids, dry cracked lips, and pharyngeal erythema with ulcerations of the hard palate. She had nontender cervical and inguinal lymphadenopathy. Her abdomen was tender to palpation in the lower quadrants without guarding or rebound; there was no organomegaly. A right knee effusion with overlying warmth was present without redness or decreased range of motion. She also had an enlarged third proximal interphalangeal joint and loss of palpable metacarpal phalangeal joint landmarks on her right hand. She was noted to be using her arms to move her legs when repositioning in bed.
These exam findings clearly point toward a systemic process but not 1 specific diagnosis. The presence of at least 2 inflamed joints points toward rheumatologic/inflammatory or infectious diseases. Localized edema (eyelids and right metacarpal phalangeal joints), oral ulcers, possible myositis, and arthritis point toward a systemic vasculitis (eg, granulomatosis with polyangiitis, Behçet disease). While Kawasaki disease is also a systemic vasculitis, the presence of oral ulcers and generalized lymphadenopathy argues against it. Inflammatory myopathies like polymyositis, and especially juvenile dermatomyositis, fit many aspects of this presentation with the violaceous eyelids and possible myositis, though no other cutaneous stigmata of this disease are evident (eg, no Gottron’s papules). Polyarthritis, violaceous eyelids, and possible myositis could be consistent with systemic lupus erythematosus (SLE).
The presence of oral ulcers and arthritis make other systemic inflammatory conditions, such as inflammatory bowel disease with arthritis and autoimmune- or infection-related hepatitis, possible. Infectious etiologies alone or in combination with a rheumatologic process are also possible given fevers and lymphadenopathy. In particular, herpesvirus infections (Epstein-Barr virus [EBV], cytomegalovirus [CMV], herpes simplex virus, or human herpes virus 6), human immunodeficiency virus (HIV), hepatitis C virus (HCV), and syphilis can cause oral ulcers and lymphadenopathy. Other potential infectious etiologies include subacute bacterial endocarditis and disseminated gonococcal infection given the presence of polyarthritis, but these infections are less likely as they do not explain all of the symptoms.
In summary, the differential diagnosis is broad and should be prioritized to consider systemic inflammatory conditions, including autoimmune and infectious (especially viral) syndromes, and initial work-up should focus on these etiologies.

The pancytopenia is obviously notable. It raises the possibility that the oral ulcerations are due to the neutropenia rather than a primary disease manifestation. Other possible causes of pancytopenia include SLE, antiphospholipid antibody syndrome, and related rheumatologic diagnoses, including hemophagocytic lymphohistiocytosis (HLH). Given her age and subacute presentation, secondary forms of HLH seem more likely than primary (genetic) forms, which typically present within the first few years of life. Secondary forms of HLH can occur in association with rheumatic diseases and are then referred to as Macrophage Activation Syndrome (MAS). The most common rheumatologic diseases associated with MAS are systemic juvenile idiopathic arthritis, SLE, and Kawasaki disease. Secondary HLH can also occur with infectious diseases, particularly viral infections such as EBV. It is also important to consider thrombotic thrombocytopenic purpura and other forms of thrombotic microangiopathy, especially if her violaceous eyelids actually represent purpura. The presence of pancytopenia also expands the differential diagnosis to include leukemia, lymphoma, and other oncologic diseases. After obtaining results from pending infectious disease studies, additional diagnostic work-up should include examination of the bone marrow and a peripheral blood smear to evaluate for hemophagocytosis and/or malignancy. Testing for double-stranded DNA antibodies and antinuclear antibodies (ANA) should be sent to evaluate for SLE, and antiphospholipid antibodies should also be checked. Renal function must also be evaluated.
Additional laboratory work-up revealed a reticulocyte count of 0.2%, a positive Coombs immunoglobulin G (IgG) test, haptoglobin less than 80 mg/L, and lactate dehydrogenase (LDH) 25.2 µkat/L (1509 units/L); coagulation studies were normal. Her chemistries showed electrolytes, blood urea nitrogen, and creatinine were within normal limits; her aspartate aminotransferase was 216 units/L, and alanine aminotransferase was 56 units/L. Her spot urine protein-to-creatinine ratio was 1.28. Complement and inflammatory studies showed C3 0.14 g/L (14 mg/dL, normal 83-151 mg/dL), C4 0.05 g/L (5 mg/dL, normal 13-37 mg/dL), erythrocyte sedimentation rate (ESR) 103 mm/hr (normal 0-20 mm/hr), and C-reactive protein (CRP) 3.2 mg/L (normal 0.7-1.7 mg/L). Additional studies showed elevated triglycerides (376 mg/dL), elevated creatine kinase (2437 units/L), and elevated ferritin (22,295.5 ng/mL). An ANA screen and specific autoantibody studies were sent, including antidouble stranded DNA antibody, antiribonucleoprotein antibody, anti-Smith antibody, anti-Ro antibody, and anti-La antibody. A bone marrow biopsy was performed.
The hematologic studies provide a mixed picture. There is evidence of an autoimmune hemolytic anemia (AIHA). Typically, AIHA is associated with reticulocytosis rather than reticulocytopenia. Reticulocytopenia can occur in AIHA, however, because of antibodies directed against erythroid precursors or if 2 processes are occurring simultaneously—ie, AIHA plus bone marrow destructive/failure process. The latter scenario is more likely here. Specifically, the pancytopenia, elevated triglycerides, and extreme hyperferritinemia strongly support the diagnosis of HLH. The very low C3 and C4 suggest a complement-consumptive process, and SLE is the most likely etiology. Proteinuria and Coombs-positive anemia are also features of SLE. The discordance between the ESR (markedly elevated) and CRP (mild elevation) is surprising in the setting of systemic inflammation. However, her other clinical features are consistent with marked systemic inflammation, and it is important not to dismiss a likely diagnosis simply on the basis of a few incongruous features. At this point, the diagnosis of SLE complicated by secondary HLH is favored, remembering that both these entities can be triggered by a viral infection. Therefore, diligent follow-up of the aforementioned specific autoantibody studies and the bone marrow biopsy is the next logical step, along with the still-pending infectious disease studies.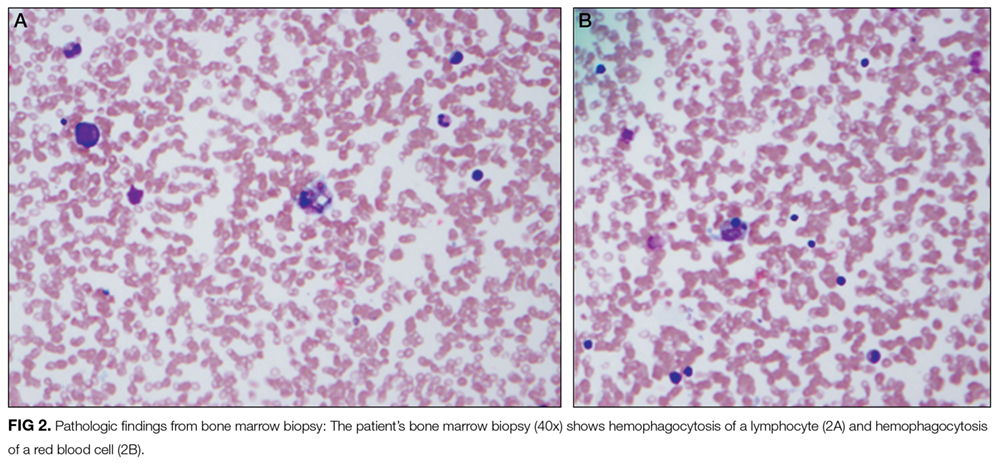
These findings are consistent with the diagnosis of SLE complicated by secondary HLH (ie, MAS). It remains possible, but unlikely, that the patient has genetic or familial HLH (fHLH), as this entity is exceedingly rare with distinct underlying genetic aberrations separate from SLE. Ideally, the NK cell function studies would be repeated after the current episode of HLH is controlled and the patient is off of immunosuppressive therapies, but this will likely not be possible given the underlying SLE. Patients with fHLH have reduced or absent NK cell function at baseline (ie, not only during an acute episode of HLH and not because of immunosuppressive medications). Alternatively, one could consider genetic testing for fHLH. The clinical importance of doing this is that patients with fHLH are candidates for bone marrow or stem cell transplantation. There currently is not a published standard of care for the work-up and management of MAS in children with rheumatic disease, so the decision to repeat NK cell function testing and/or genetic testing would be left to the discretion of the treating physician and would depend on the patient’s ongoing clinical course.
The patient required red blood cell and platelet transfusions. She received pulse dose intravenous methylprednisolone for treatment of SLE and MAS; she clinically improved within 48 hours of starting steroids. Cyclosporine was added for management of MAS. The patient was transitioned to oral corticosteroids and discharged home. All cell counts normalized within 1 month of discharge. She was weaned off corticosteroids and cyclosporine was discontinued. Her maintenance SLE therapy includes hydroxychloroquine and mycophenolate mofetil.
COMMENTARY
Because the differential diagnosis for new-onset pancytopenia encompasses many diseases across several medical subspecialties, a thorough history and physical exam are necessary to form a tailored clinical approach.1 The primary causes of pediatric pancytopenia vary depending on geographic location because of the local prevalence of infectious agents and nutritional deficiency patterns. A retrospective study investigating the primary cause of pancytopenia in children without existing malignancy presenting to a US tertiary care hospital found that 64% of cases were due to infection, 28% were due to hematologic disease (most frequently aplastic anemia), and 8% were due to miscellaneous etiologies, including adverse drug reactions and autoimmune diseases.2 In contrast, the most common cause of pancytopenia in pediatric patients presenting to a tertiary care hospital in India was megaloblastic anemia (28%), followed by infections (21%), acute leukemia (21%), and aplastic anemia (20%).3 While clinicians do (and should) consider malignancy as a cause of pancytopenia, there is sparse literature regarding the frequency of pancytopenia associated with the presentations of childhood malignancies.4 A study of pediatric patients with acute lymphoblastic anemia found that only 11% of newly diagnosed patients had pancytopenia at initial presentation.4
There are no official guidelines for the work-up of pediatric pancytopenia from any of the academic societies. Depending on the clinical history, initial laboratory investigation for pediatric pancytopenia may include complete blood cell count with differential, reticulocyte count, peripheral blood smear, complete metabolic panel, hemolysis labs (haptoglobin, LDH, Coombs test) and inflammatory markers (ESR, CRP, fibrinogen). Further investigation to clarify the specific etiology of pancytopenia can be guided by the results of these initial tests.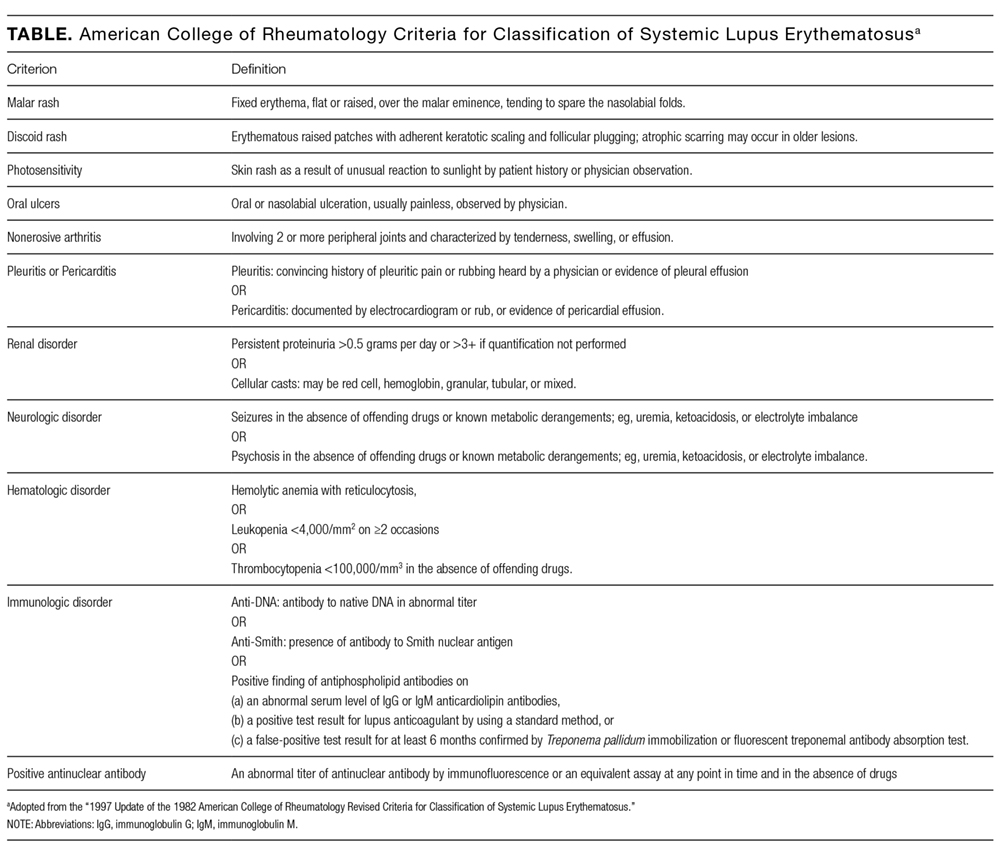
Hematologic abnormalities are present in greater than 70% of pediatric SLE cases.10,11 The pathogenesis of hematologic abnormalities in SLE is heterogeneous, involving actions of autoreactive lymphocytes, autoantibodies, and proinflammatory cytokines that can disrupt bone marrow production and cause peripheral blood cell destruction.12,13 While pancytopenia is common in children with SLE, other coexisting diagnoses should be considered in patients with SLE and pancytopenia. Concurrent diagnoses that can lead to pancytopenia in patients with SLE include infection, pharmacologic side effects, and secondary HLH,14,15 each of which has differing implications for prognosis and treatment.
Secondary HLH is a severe and often acute complication of systemic inflammatory disorders caused by the proliferation and activation of T cells and macrophages, leading to an enhanced inflammatory state. When HLH occurs in the setting of an underlying autoimmune or autoinflammatory process, it is typically termed MAS. MAS affects an estimated 0.9% to 4.6% of patients with SLE.16 Early diagnosis and treatment of MAS is important because MAS can be rapidly fatal, with a mortality rate of 8% to 20% in pediatric patients.17,18 Clinical features of MAS include physical exam findings of fever and splenomegaly as well as laboratory abnormalities, including pancytopenia, elevated ferritin, elevated triglycerides, and low fibrinogen.18 A bone marrow biopsy showing hemophagocytosis in the absence of malignancy is diagnostic of MAS. Although a bone marrow biopsy is not required to diagnose MAS, it is often obtained to exclude other etiologies of pancytopenia such as malignancy.19 Specialized diagnostic testing for MAS includes NK cell counts and functional studies, including expression of perforin and granzyme B (NK cell proteins triggering apoptosis in target cells), soluble IL-2R (marker of activated lymphocytes), and CD163 (transmembrane protein of hemophagocytic macrophages). There is no standardized protocol for treating MAS.20 It is most commonly treated with highdose corticosteroids; additional agents, including cyclosporine and biologic therapies, are also utilized.16,20
KEY POINTS
- Children with SLE tend to have greater involvement of major organ systems and more rapid accrual of organ damage than adults with SLE. Therefore, it is sometimes necessary to initiate immunosuppressive therapies before full diagnostic criteria are met, provided that malignancy and infection have been ruled out.
- While pancytopenia is common in pediatric patients with SLE, providers should make sure to consider coexisting diagnoses such as infection and MAS, both of which require different treatment strategies.
- It is important to consider HLH/MAS early in the work-up of pancytopenia, because early diagnosis and treatment improves clinical outcomes. Obtaining a ferritin level can aid in the work-up of pancytopenia because it is both a sensitive and specific marker of HLH/MAS when dramatically elevated.
Disclosure
The authors report no conflicts of interest.
1. Weinzierl EP, Arber DA. The Differential Diagnosis and Bone Marrow Evaluation of New-Onset Pancytopenia. Am J Clin Pathol. 2012;139(1):9-29. doi:10.1309/AJCP50AEEYGREWUZ. PubMed
2. Pine M, Walter AW. Pancytopenia in hospitalized children: a five-year review. J Pediatr Hematol Oncol. 2010;32(5):e192-e194. doi:10.1097/MPH.0b013e3181e03082. PubMed
3. Bhatnagar SK. Pancytopenia in Children: Etiological Profile. J Trop Pediatr. 2005;51(4):236-239. doi:10.1093/tropej/fmi010. PubMed
4. Kulkarni KP, Marwaha RK. Acute lymphoblastic leukemia with pancytopenia at presentation: clinical correlates, prognostic impact, and association with survival. J Pediatr Hematol Oncol. 2013;35(7):573-576. doi:10.1097/MPH.0b013e31829d46f3. PubMed
5. Holubar, K. Terminology and iconography of lupus erythematosus: A historical vignette. Am J Dermatopathol. 1980;2(3):239-242. PubMed
6. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. doi: 10.1002/art.1780400928. PubMed
7. Petri M, Orbai, A, Alarcon GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677-2686. doi:10.1002/art.34473. PubMed
8. Tucker L. Review: Making the diagnosis of systemic lupus erythematosus in children and adolescents. Lupus. 2007;16(8):546-549. doi:10.1177/0961203307078068. PubMed
9. Brunner HI, Gladman DD, Ibañez D, Urowitz MD, Silverman ED. Difference in disease features between childhood-onset and adult-onset systemic lupus erythematosus. Arthritis Rheum. 2008;58(2):556-562. doi:10.1002/art.23204. PubMed
10. Benseler SM, Silverman ED. Systemic Lupus Erythematosus. Rheum Dis Clin North Am. 2007;33(3):471-498. doi:10.1016/j.rdc.2007.07.008. PubMed
11. Gokce M, Bilginer Y, Besbas N, et al. Hematological features of pediatric systemic lupus erythematosus: suggesting management strategies in children. Lupus. 2012;21(8):878-884. doi:10.1177/0961203312443721. PubMed
12. Voulgarelis M, Giannouli S, Tasidou A, Anagnostou D, Ziakas PD, Tzioufas AG. Bone marrow histological findings in systemic lupus erythematosus with hematologic abnormalities: A clinicopathological study. Am J Hematol. 2006;81(8):590-597. doi:10.1002/ajh.20593. PubMed
13. Pereira RM, Velloso ER, Menezes Y, Gualandro S, Vassalo J, Yoshinari NH. Bone marrow findings in systemic lupus erythematosus patients with peripheral cytopenias. Clin Rheumatol. 1998;17(3):219-222. PubMed
14. Avčin T, Tse SML, Schneider R, Ngan B, Silverman ED. Macrophage activation syndrome as the presenting manifestation of rheumatic diseases in childhood. J Pediatr. 2006;148(5):683-686. doi:10.1016/j.jpeds.2005.12.070. PubMed
15. Lambotte O, Khellaf M, Harmouche H, et al. Characteristics and Long-Term Outcome of 15 Episodes of Systemic Lupus Erythematosus-Associated Hemophagocytic Syndrome. Medicine. 2006;85(3):169-182. doi:10.1097/01.md.0000224708.62510.d1. PubMed
16. Fukaya S, Yasuda S, Hashimoto T, et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30 cases. Rheumatology. 2008;47(11):1686-1691. doi:10.1093/rheumatology/ken342. PubMed
17. Stephan JL. Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology. 2001;40(11):1285-1292. doi:10.1093/rheumatology/40.11.1285. PubMed
18. Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child. 2001;85(5):421-426. PubMed
19. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. doi:10.1002/pbc.21039. PubMed
20. Lin CI, Yu HH, Lee JH, et al. Clinical analysis of macrophage activation syndrome in pediatric patients with autoimmune diseases. Clin Rheumatol. 2012;31(8):1223-1230. doi:10.1007/s10067-012-1998-0. PubMed
A 15-year-old African American girl presented to the emergency department with 3 days of fever, sore throat, nausea, vomiting, and poor appetite. She reported a 4-week history of fatigue, right hand pain and swelling, and a 6-kilogram weight loss for which she had seen her primary care provider several times. She reported no recent travel, sick contacts, or new medications.
It appears that there are potentially at least 2 separate problems: an acute one (past 3 days) and a more chronic one (past 4 weeks). These 2 problems may be directly related (ie, acute worsening of the more chronic problem), indirectly related (ie, the more chronic problem is leading to increased susceptibility to the acute problem, for instance, an evolving immunodeficiency predisposing to an opportunistic infection), or “true, true, but unrelated.” The clinical challenge is to keep one’s mind open to each of these potential scenarios and to avoid the tendency to focus on one of the problems and not pay enough attention to the other. Occam’s razor likely does not apply here.
Numerous common and typically transient diseases could cause the symptoms of the past 3 days, particularly infectious etiologies such as streptococcal pharyngitis or a viral infection. One cannot forget about these possibilities while contemplating the more worrisome symptoms of the past 4 weeks, especially weight loss in a growing adolescent. Patients may unintentionally lose weight for a variety of reasons, which can be broadly categorized by decreased caloric supply, gastrointestinal losses or malabsorption, and increased caloric demand; these categories are not mutually exclusive.
Lastly, 1 symptom may provide a more specific direction: the right hand pain and swelling of the past 4 weeks. More specifics, including the extent of the hand swelling, other areas of involvement, and the nature of her pain, will be helpful.
Her temperature was 99.5°F, heart rate 100 beats per minute, respiratory rate 18 breaths per minute, oxygen saturation 95% while breathing ambient air, blood pressure 99/56 mmHg, weight 44 kilograms, height 161 centimeters, and body mass index 17. She appeared generally ill and underweight. She had edematous and violaceous eyelids, dry cracked lips, and pharyngeal erythema with ulcerations of the hard palate. She had nontender cervical and inguinal lymphadenopathy. Her abdomen was tender to palpation in the lower quadrants without guarding or rebound; there was no organomegaly. A right knee effusion with overlying warmth was present without redness or decreased range of motion. She also had an enlarged third proximal interphalangeal joint and loss of palpable metacarpal phalangeal joint landmarks on her right hand. She was noted to be using her arms to move her legs when repositioning in bed.
These exam findings clearly point toward a systemic process but not 1 specific diagnosis. The presence of at least 2 inflamed joints points toward rheumatologic/inflammatory or infectious diseases. Localized edema (eyelids and right metacarpal phalangeal joints), oral ulcers, possible myositis, and arthritis point toward a systemic vasculitis (eg, granulomatosis with polyangiitis, Behçet disease). While Kawasaki disease is also a systemic vasculitis, the presence of oral ulcers and generalized lymphadenopathy argues against it. Inflammatory myopathies like polymyositis, and especially juvenile dermatomyositis, fit many aspects of this presentation with the violaceous eyelids and possible myositis, though no other cutaneous stigmata of this disease are evident (eg, no Gottron’s papules). Polyarthritis, violaceous eyelids, and possible myositis could be consistent with systemic lupus erythematosus (SLE).
The presence of oral ulcers and arthritis make other systemic inflammatory conditions, such as inflammatory bowel disease with arthritis and autoimmune- or infection-related hepatitis, possible. Infectious etiologies alone or in combination with a rheumatologic process are also possible given fevers and lymphadenopathy. In particular, herpesvirus infections (Epstein-Barr virus [EBV], cytomegalovirus [CMV], herpes simplex virus, or human herpes virus 6), human immunodeficiency virus (HIV), hepatitis C virus (HCV), and syphilis can cause oral ulcers and lymphadenopathy. Other potential infectious etiologies include subacute bacterial endocarditis and disseminated gonococcal infection given the presence of polyarthritis, but these infections are less likely as they do not explain all of the symptoms.
In summary, the differential diagnosis is broad and should be prioritized to consider systemic inflammatory conditions, including autoimmune and infectious (especially viral) syndromes, and initial work-up should focus on these etiologies.
The pancytopenia is obviously notable. It raises the possibility that the oral ulcerations are due to the neutropenia rather than a primary disease manifestation. Other possible causes of pancytopenia include SLE, antiphospholipid antibody syndrome, and related rheumatologic diagnoses, including hemophagocytic lymphohistiocytosis (HLH). Given her age and subacute presentation, secondary forms of HLH seem more likely than primary (genetic) forms, which typically present within the first few years of life. Secondary forms of HLH can occur in association with rheumatic diseases and are then referred to as Macrophage Activation Syndrome (MAS). The most common rheumatologic diseases associated with MAS are systemic juvenile idiopathic arthritis, SLE, and Kawasaki disease. Secondary HLH can also occur with infectious diseases, particularly viral infections such as EBV. It is also important to consider thrombotic thrombocytopenic purpura and other forms of thrombotic microangiopathy, especially if her violaceous eyelids actually represent purpura. The presence of pancytopenia also expands the differential diagnosis to include leukemia, lymphoma, and other oncologic diseases. After obtaining results from pending infectious disease studies, additional diagnostic work-up should include examination of the bone marrow and a peripheral blood smear to evaluate for hemophagocytosis and/or malignancy. Testing for double-stranded DNA antibodies and antinuclear antibodies (ANA) should be sent to evaluate for SLE, and antiphospholipid antibodies should also be checked. Renal function must also be evaluated.
Additional laboratory work-up revealed a reticulocyte count of 0.2%, a positive Coombs immunoglobulin G (IgG) test, haptoglobin less than 80 mg/L, and lactate dehydrogenase (LDH) 25.2 µkat/L (1509 units/L); coagulation studies were normal. Her chemistries showed electrolytes, blood urea nitrogen, and creatinine were within normal limits; her aspartate aminotransferase was 216 units/L, and alanine aminotransferase was 56 units/L. Her spot urine protein-to-creatinine ratio was 1.28. Complement and inflammatory studies showed C3 0.14 g/L (14 mg/dL, normal 83-151 mg/dL), C4 0.05 g/L (5 mg/dL, normal 13-37 mg/dL), erythrocyte sedimentation rate (ESR) 103 mm/hr (normal 0-20 mm/hr), and C-reactive protein (CRP) 3.2 mg/L (normal 0.7-1.7 mg/L). Additional studies showed elevated triglycerides (376 mg/dL), elevated creatine kinase (2437 units/L), and elevated ferritin (22,295.5 ng/mL). An ANA screen and specific autoantibody studies were sent, including antidouble stranded DNA antibody, antiribonucleoprotein antibody, anti-Smith antibody, anti-Ro antibody, and anti-La antibody. A bone marrow biopsy was performed.
The hematologic studies provide a mixed picture. There is evidence of an autoimmune hemolytic anemia (AIHA). Typically, AIHA is associated with reticulocytosis rather than reticulocytopenia. Reticulocytopenia can occur in AIHA, however, because of antibodies directed against erythroid precursors or if 2 processes are occurring simultaneously—ie, AIHA plus bone marrow destructive/failure process. The latter scenario is more likely here. Specifically, the pancytopenia, elevated triglycerides, and extreme hyperferritinemia strongly support the diagnosis of HLH. The very low C3 and C4 suggest a complement-consumptive process, and SLE is the most likely etiology. Proteinuria and Coombs-positive anemia are also features of SLE. The discordance between the ESR (markedly elevated) and CRP (mild elevation) is surprising in the setting of systemic inflammation. However, her other clinical features are consistent with marked systemic inflammation, and it is important not to dismiss a likely diagnosis simply on the basis of a few incongruous features. At this point, the diagnosis of SLE complicated by secondary HLH is favored, remembering that both these entities can be triggered by a viral infection. Therefore, diligent follow-up of the aforementioned specific autoantibody studies and the bone marrow biopsy is the next logical step, along with the still-pending infectious disease studies.
These findings are consistent with the diagnosis of SLE complicated by secondary HLH (ie, MAS). It remains possible, but unlikely, that the patient has genetic or familial HLH (fHLH), as this entity is exceedingly rare with distinct underlying genetic aberrations separate from SLE. Ideally, the NK cell function studies would be repeated after the current episode of HLH is controlled and the patient is off of immunosuppressive therapies, but this will likely not be possible given the underlying SLE. Patients with fHLH have reduced or absent NK cell function at baseline (ie, not only during an acute episode of HLH and not because of immunosuppressive medications). Alternatively, one could consider genetic testing for fHLH. The clinical importance of doing this is that patients with fHLH are candidates for bone marrow or stem cell transplantation. There currently is not a published standard of care for the work-up and management of MAS in children with rheumatic disease, so the decision to repeat NK cell function testing and/or genetic testing would be left to the discretion of the treating physician and would depend on the patient’s ongoing clinical course.
The patient required red blood cell and platelet transfusions. She received pulse dose intravenous methylprednisolone for treatment of SLE and MAS; she clinically improved within 48 hours of starting steroids. Cyclosporine was added for management of MAS. The patient was transitioned to oral corticosteroids and discharged home. All cell counts normalized within 1 month of discharge. She was weaned off corticosteroids and cyclosporine was discontinued. Her maintenance SLE therapy includes hydroxychloroquine and mycophenolate mofetil.
COMMENTARY
Because the differential diagnosis for new-onset pancytopenia encompasses many diseases across several medical subspecialties, a thorough history and physical exam are necessary to form a tailored clinical approach.1 The primary causes of pediatric pancytopenia vary depending on geographic location because of the local prevalence of infectious agents and nutritional deficiency patterns. A retrospective study investigating the primary cause of pancytopenia in children without existing malignancy presenting to a US tertiary care hospital found that 64% of cases were due to infection, 28% were due to hematologic disease (most frequently aplastic anemia), and 8% were due to miscellaneous etiologies, including adverse drug reactions and autoimmune diseases.2 In contrast, the most common cause of pancytopenia in pediatric patients presenting to a tertiary care hospital in India was megaloblastic anemia (28%), followed by infections (21%), acute leukemia (21%), and aplastic anemia (20%).3 While clinicians do (and should) consider malignancy as a cause of pancytopenia, there is sparse literature regarding the frequency of pancytopenia associated with the presentations of childhood malignancies.4 A study of pediatric patients with acute lymphoblastic anemia found that only 11% of newly diagnosed patients had pancytopenia at initial presentation.4
There are no official guidelines for the work-up of pediatric pancytopenia from any of the academic societies. Depending on the clinical history, initial laboratory investigation for pediatric pancytopenia may include complete blood cell count with differential, reticulocyte count, peripheral blood smear, complete metabolic panel, hemolysis labs (haptoglobin, LDH, Coombs test) and inflammatory markers (ESR, CRP, fibrinogen). Further investigation to clarify the specific etiology of pancytopenia can be guided by the results of these initial tests.
Hematologic abnormalities are present in greater than 70% of pediatric SLE cases.10,11 The pathogenesis of hematologic abnormalities in SLE is heterogeneous, involving actions of autoreactive lymphocytes, autoantibodies, and proinflammatory cytokines that can disrupt bone marrow production and cause peripheral blood cell destruction.12,13 While pancytopenia is common in children with SLE, other coexisting diagnoses should be considered in patients with SLE and pancytopenia. Concurrent diagnoses that can lead to pancytopenia in patients with SLE include infection, pharmacologic side effects, and secondary HLH,14,15 each of which has differing implications for prognosis and treatment.
Secondary HLH is a severe and often acute complication of systemic inflammatory disorders caused by the proliferation and activation of T cells and macrophages, leading to an enhanced inflammatory state. When HLH occurs in the setting of an underlying autoimmune or autoinflammatory process, it is typically termed MAS. MAS affects an estimated 0.9% to 4.6% of patients with SLE.16 Early diagnosis and treatment of MAS is important because MAS can be rapidly fatal, with a mortality rate of 8% to 20% in pediatric patients.17,18 Clinical features of MAS include physical exam findings of fever and splenomegaly as well as laboratory abnormalities, including pancytopenia, elevated ferritin, elevated triglycerides, and low fibrinogen.18 A bone marrow biopsy showing hemophagocytosis in the absence of malignancy is diagnostic of MAS. Although a bone marrow biopsy is not required to diagnose MAS, it is often obtained to exclude other etiologies of pancytopenia such as malignancy.19 Specialized diagnostic testing for MAS includes NK cell counts and functional studies, including expression of perforin and granzyme B (NK cell proteins triggering apoptosis in target cells), soluble IL-2R (marker of activated lymphocytes), and CD163 (transmembrane protein of hemophagocytic macrophages). There is no standardized protocol for treating MAS.20 It is most commonly treated with highdose corticosteroids; additional agents, including cyclosporine and biologic therapies, are also utilized.16,20
KEY POINTS
- Children with SLE tend to have greater involvement of major organ systems and more rapid accrual of organ damage than adults with SLE. Therefore, it is sometimes necessary to initiate immunosuppressive therapies before full diagnostic criteria are met, provided that malignancy and infection have been ruled out.
- While pancytopenia is common in pediatric patients with SLE, providers should make sure to consider coexisting diagnoses such as infection and MAS, both of which require different treatment strategies.
- It is important to consider HLH/MAS early in the work-up of pancytopenia, because early diagnosis and treatment improves clinical outcomes. Obtaining a ferritin level can aid in the work-up of pancytopenia because it is both a sensitive and specific marker of HLH/MAS when dramatically elevated.
Disclosure
The authors report no conflicts of interest.
A 15-year-old African American girl presented to the emergency department with 3 days of fever, sore throat, nausea, vomiting, and poor appetite. She reported a 4-week history of fatigue, right hand pain and swelling, and a 6-kilogram weight loss for which she had seen her primary care provider several times. She reported no recent travel, sick contacts, or new medications.
It appears that there are potentially at least 2 separate problems: an acute one (past 3 days) and a more chronic one (past 4 weeks). These 2 problems may be directly related (ie, acute worsening of the more chronic problem), indirectly related (ie, the more chronic problem is leading to increased susceptibility to the acute problem, for instance, an evolving immunodeficiency predisposing to an opportunistic infection), or “true, true, but unrelated.” The clinical challenge is to keep one’s mind open to each of these potential scenarios and to avoid the tendency to focus on one of the problems and not pay enough attention to the other. Occam’s razor likely does not apply here.
Numerous common and typically transient diseases could cause the symptoms of the past 3 days, particularly infectious etiologies such as streptococcal pharyngitis or a viral infection. One cannot forget about these possibilities while contemplating the more worrisome symptoms of the past 4 weeks, especially weight loss in a growing adolescent. Patients may unintentionally lose weight for a variety of reasons, which can be broadly categorized by decreased caloric supply, gastrointestinal losses or malabsorption, and increased caloric demand; these categories are not mutually exclusive.
Lastly, 1 symptom may provide a more specific direction: the right hand pain and swelling of the past 4 weeks. More specifics, including the extent of the hand swelling, other areas of involvement, and the nature of her pain, will be helpful.
Her temperature was 99.5°F, heart rate 100 beats per minute, respiratory rate 18 breaths per minute, oxygen saturation 95% while breathing ambient air, blood pressure 99/56 mmHg, weight 44 kilograms, height 161 centimeters, and body mass index 17. She appeared generally ill and underweight. She had edematous and violaceous eyelids, dry cracked lips, and pharyngeal erythema with ulcerations of the hard palate. She had nontender cervical and inguinal lymphadenopathy. Her abdomen was tender to palpation in the lower quadrants without guarding or rebound; there was no organomegaly. A right knee effusion with overlying warmth was present without redness or decreased range of motion. She also had an enlarged third proximal interphalangeal joint and loss of palpable metacarpal phalangeal joint landmarks on her right hand. She was noted to be using her arms to move her legs when repositioning in bed.
These exam findings clearly point toward a systemic process but not 1 specific diagnosis. The presence of at least 2 inflamed joints points toward rheumatologic/inflammatory or infectious diseases. Localized edema (eyelids and right metacarpal phalangeal joints), oral ulcers, possible myositis, and arthritis point toward a systemic vasculitis (eg, granulomatosis with polyangiitis, Behçet disease). While Kawasaki disease is also a systemic vasculitis, the presence of oral ulcers and generalized lymphadenopathy argues against it. Inflammatory myopathies like polymyositis, and especially juvenile dermatomyositis, fit many aspects of this presentation with the violaceous eyelids and possible myositis, though no other cutaneous stigmata of this disease are evident (eg, no Gottron’s papules). Polyarthritis, violaceous eyelids, and possible myositis could be consistent with systemic lupus erythematosus (SLE).
The presence of oral ulcers and arthritis make other systemic inflammatory conditions, such as inflammatory bowel disease with arthritis and autoimmune- or infection-related hepatitis, possible. Infectious etiologies alone or in combination with a rheumatologic process are also possible given fevers and lymphadenopathy. In particular, herpesvirus infections (Epstein-Barr virus [EBV], cytomegalovirus [CMV], herpes simplex virus, or human herpes virus 6), human immunodeficiency virus (HIV), hepatitis C virus (HCV), and syphilis can cause oral ulcers and lymphadenopathy. Other potential infectious etiologies include subacute bacterial endocarditis and disseminated gonococcal infection given the presence of polyarthritis, but these infections are less likely as they do not explain all of the symptoms.
In summary, the differential diagnosis is broad and should be prioritized to consider systemic inflammatory conditions, including autoimmune and infectious (especially viral) syndromes, and initial work-up should focus on these etiologies.
The pancytopenia is obviously notable. It raises the possibility that the oral ulcerations are due to the neutropenia rather than a primary disease manifestation. Other possible causes of pancytopenia include SLE, antiphospholipid antibody syndrome, and related rheumatologic diagnoses, including hemophagocytic lymphohistiocytosis (HLH). Given her age and subacute presentation, secondary forms of HLH seem more likely than primary (genetic) forms, which typically present within the first few years of life. Secondary forms of HLH can occur in association with rheumatic diseases and are then referred to as Macrophage Activation Syndrome (MAS). The most common rheumatologic diseases associated with MAS are systemic juvenile idiopathic arthritis, SLE, and Kawasaki disease. Secondary HLH can also occur with infectious diseases, particularly viral infections such as EBV. It is also important to consider thrombotic thrombocytopenic purpura and other forms of thrombotic microangiopathy, especially if her violaceous eyelids actually represent purpura. The presence of pancytopenia also expands the differential diagnosis to include leukemia, lymphoma, and other oncologic diseases. After obtaining results from pending infectious disease studies, additional diagnostic work-up should include examination of the bone marrow and a peripheral blood smear to evaluate for hemophagocytosis and/or malignancy. Testing for double-stranded DNA antibodies and antinuclear antibodies (ANA) should be sent to evaluate for SLE, and antiphospholipid antibodies should also be checked. Renal function must also be evaluated.
Additional laboratory work-up revealed a reticulocyte count of 0.2%, a positive Coombs immunoglobulin G (IgG) test, haptoglobin less than 80 mg/L, and lactate dehydrogenase (LDH) 25.2 µkat/L (1509 units/L); coagulation studies were normal. Her chemistries showed electrolytes, blood urea nitrogen, and creatinine were within normal limits; her aspartate aminotransferase was 216 units/L, and alanine aminotransferase was 56 units/L. Her spot urine protein-to-creatinine ratio was 1.28. Complement and inflammatory studies showed C3 0.14 g/L (14 mg/dL, normal 83-151 mg/dL), C4 0.05 g/L (5 mg/dL, normal 13-37 mg/dL), erythrocyte sedimentation rate (ESR) 103 mm/hr (normal 0-20 mm/hr), and C-reactive protein (CRP) 3.2 mg/L (normal 0.7-1.7 mg/L). Additional studies showed elevated triglycerides (376 mg/dL), elevated creatine kinase (2437 units/L), and elevated ferritin (22,295.5 ng/mL). An ANA screen and specific autoantibody studies were sent, including antidouble stranded DNA antibody, antiribonucleoprotein antibody, anti-Smith antibody, anti-Ro antibody, and anti-La antibody. A bone marrow biopsy was performed.
The hematologic studies provide a mixed picture. There is evidence of an autoimmune hemolytic anemia (AIHA). Typically, AIHA is associated with reticulocytosis rather than reticulocytopenia. Reticulocytopenia can occur in AIHA, however, because of antibodies directed against erythroid precursors or if 2 processes are occurring simultaneously—ie, AIHA plus bone marrow destructive/failure process. The latter scenario is more likely here. Specifically, the pancytopenia, elevated triglycerides, and extreme hyperferritinemia strongly support the diagnosis of HLH. The very low C3 and C4 suggest a complement-consumptive process, and SLE is the most likely etiology. Proteinuria and Coombs-positive anemia are also features of SLE. The discordance between the ESR (markedly elevated) and CRP (mild elevation) is surprising in the setting of systemic inflammation. However, her other clinical features are consistent with marked systemic inflammation, and it is important not to dismiss a likely diagnosis simply on the basis of a few incongruous features. At this point, the diagnosis of SLE complicated by secondary HLH is favored, remembering that both these entities can be triggered by a viral infection. Therefore, diligent follow-up of the aforementioned specific autoantibody studies and the bone marrow biopsy is the next logical step, along with the still-pending infectious disease studies.
These findings are consistent with the diagnosis of SLE complicated by secondary HLH (ie, MAS). It remains possible, but unlikely, that the patient has genetic or familial HLH (fHLH), as this entity is exceedingly rare with distinct underlying genetic aberrations separate from SLE. Ideally, the NK cell function studies would be repeated after the current episode of HLH is controlled and the patient is off of immunosuppressive therapies, but this will likely not be possible given the underlying SLE. Patients with fHLH have reduced or absent NK cell function at baseline (ie, not only during an acute episode of HLH and not because of immunosuppressive medications). Alternatively, one could consider genetic testing for fHLH. The clinical importance of doing this is that patients with fHLH are candidates for bone marrow or stem cell transplantation. There currently is not a published standard of care for the work-up and management of MAS in children with rheumatic disease, so the decision to repeat NK cell function testing and/or genetic testing would be left to the discretion of the treating physician and would depend on the patient’s ongoing clinical course.
The patient required red blood cell and platelet transfusions. She received pulse dose intravenous methylprednisolone for treatment of SLE and MAS; she clinically improved within 48 hours of starting steroids. Cyclosporine was added for management of MAS. The patient was transitioned to oral corticosteroids and discharged home. All cell counts normalized within 1 month of discharge. She was weaned off corticosteroids and cyclosporine was discontinued. Her maintenance SLE therapy includes hydroxychloroquine and mycophenolate mofetil.
COMMENTARY
Because the differential diagnosis for new-onset pancytopenia encompasses many diseases across several medical subspecialties, a thorough history and physical exam are necessary to form a tailored clinical approach.1 The primary causes of pediatric pancytopenia vary depending on geographic location because of the local prevalence of infectious agents and nutritional deficiency patterns. A retrospective study investigating the primary cause of pancytopenia in children without existing malignancy presenting to a US tertiary care hospital found that 64% of cases were due to infection, 28% were due to hematologic disease (most frequently aplastic anemia), and 8% were due to miscellaneous etiologies, including adverse drug reactions and autoimmune diseases.2 In contrast, the most common cause of pancytopenia in pediatric patients presenting to a tertiary care hospital in India was megaloblastic anemia (28%), followed by infections (21%), acute leukemia (21%), and aplastic anemia (20%).3 While clinicians do (and should) consider malignancy as a cause of pancytopenia, there is sparse literature regarding the frequency of pancytopenia associated with the presentations of childhood malignancies.4 A study of pediatric patients with acute lymphoblastic anemia found that only 11% of newly diagnosed patients had pancytopenia at initial presentation.4
There are no official guidelines for the work-up of pediatric pancytopenia from any of the academic societies. Depending on the clinical history, initial laboratory investigation for pediatric pancytopenia may include complete blood cell count with differential, reticulocyte count, peripheral blood smear, complete metabolic panel, hemolysis labs (haptoglobin, LDH, Coombs test) and inflammatory markers (ESR, CRP, fibrinogen). Further investigation to clarify the specific etiology of pancytopenia can be guided by the results of these initial tests.
Hematologic abnormalities are present in greater than 70% of pediatric SLE cases.10,11 The pathogenesis of hematologic abnormalities in SLE is heterogeneous, involving actions of autoreactive lymphocytes, autoantibodies, and proinflammatory cytokines that can disrupt bone marrow production and cause peripheral blood cell destruction.12,13 While pancytopenia is common in children with SLE, other coexisting diagnoses should be considered in patients with SLE and pancytopenia. Concurrent diagnoses that can lead to pancytopenia in patients with SLE include infection, pharmacologic side effects, and secondary HLH,14,15 each of which has differing implications for prognosis and treatment.
Secondary HLH is a severe and often acute complication of systemic inflammatory disorders caused by the proliferation and activation of T cells and macrophages, leading to an enhanced inflammatory state. When HLH occurs in the setting of an underlying autoimmune or autoinflammatory process, it is typically termed MAS. MAS affects an estimated 0.9% to 4.6% of patients with SLE.16 Early diagnosis and treatment of MAS is important because MAS can be rapidly fatal, with a mortality rate of 8% to 20% in pediatric patients.17,18 Clinical features of MAS include physical exam findings of fever and splenomegaly as well as laboratory abnormalities, including pancytopenia, elevated ferritin, elevated triglycerides, and low fibrinogen.18 A bone marrow biopsy showing hemophagocytosis in the absence of malignancy is diagnostic of MAS. Although a bone marrow biopsy is not required to diagnose MAS, it is often obtained to exclude other etiologies of pancytopenia such as malignancy.19 Specialized diagnostic testing for MAS includes NK cell counts and functional studies, including expression of perforin and granzyme B (NK cell proteins triggering apoptosis in target cells), soluble IL-2R (marker of activated lymphocytes), and CD163 (transmembrane protein of hemophagocytic macrophages). There is no standardized protocol for treating MAS.20 It is most commonly treated with highdose corticosteroids; additional agents, including cyclosporine and biologic therapies, are also utilized.16,20
KEY POINTS
- Children with SLE tend to have greater involvement of major organ systems and more rapid accrual of organ damage than adults with SLE. Therefore, it is sometimes necessary to initiate immunosuppressive therapies before full diagnostic criteria are met, provided that malignancy and infection have been ruled out.
- While pancytopenia is common in pediatric patients with SLE, providers should make sure to consider coexisting diagnoses such as infection and MAS, both of which require different treatment strategies.
- It is important to consider HLH/MAS early in the work-up of pancytopenia, because early diagnosis and treatment improves clinical outcomes. Obtaining a ferritin level can aid in the work-up of pancytopenia because it is both a sensitive and specific marker of HLH/MAS when dramatically elevated.
Disclosure
The authors report no conflicts of interest.
1. Weinzierl EP, Arber DA. The Differential Diagnosis and Bone Marrow Evaluation of New-Onset Pancytopenia. Am J Clin Pathol. 2012;139(1):9-29. doi:10.1309/AJCP50AEEYGREWUZ. PubMed
2. Pine M, Walter AW. Pancytopenia in hospitalized children: a five-year review. J Pediatr Hematol Oncol. 2010;32(5):e192-e194. doi:10.1097/MPH.0b013e3181e03082. PubMed
3. Bhatnagar SK. Pancytopenia in Children: Etiological Profile. J Trop Pediatr. 2005;51(4):236-239. doi:10.1093/tropej/fmi010. PubMed
4. Kulkarni KP, Marwaha RK. Acute lymphoblastic leukemia with pancytopenia at presentation: clinical correlates, prognostic impact, and association with survival. J Pediatr Hematol Oncol. 2013;35(7):573-576. doi:10.1097/MPH.0b013e31829d46f3. PubMed
5. Holubar, K. Terminology and iconography of lupus erythematosus: A historical vignette. Am J Dermatopathol. 1980;2(3):239-242. PubMed
6. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. doi: 10.1002/art.1780400928. PubMed
7. Petri M, Orbai, A, Alarcon GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677-2686. doi:10.1002/art.34473. PubMed
8. Tucker L. Review: Making the diagnosis of systemic lupus erythematosus in children and adolescents. Lupus. 2007;16(8):546-549. doi:10.1177/0961203307078068. PubMed
9. Brunner HI, Gladman DD, Ibañez D, Urowitz MD, Silverman ED. Difference in disease features between childhood-onset and adult-onset systemic lupus erythematosus. Arthritis Rheum. 2008;58(2):556-562. doi:10.1002/art.23204. PubMed
10. Benseler SM, Silverman ED. Systemic Lupus Erythematosus. Rheum Dis Clin North Am. 2007;33(3):471-498. doi:10.1016/j.rdc.2007.07.008. PubMed
11. Gokce M, Bilginer Y, Besbas N, et al. Hematological features of pediatric systemic lupus erythematosus: suggesting management strategies in children. Lupus. 2012;21(8):878-884. doi:10.1177/0961203312443721. PubMed
12. Voulgarelis M, Giannouli S, Tasidou A, Anagnostou D, Ziakas PD, Tzioufas AG. Bone marrow histological findings in systemic lupus erythematosus with hematologic abnormalities: A clinicopathological study. Am J Hematol. 2006;81(8):590-597. doi:10.1002/ajh.20593. PubMed
13. Pereira RM, Velloso ER, Menezes Y, Gualandro S, Vassalo J, Yoshinari NH. Bone marrow findings in systemic lupus erythematosus patients with peripheral cytopenias. Clin Rheumatol. 1998;17(3):219-222. PubMed
14. Avčin T, Tse SML, Schneider R, Ngan B, Silverman ED. Macrophage activation syndrome as the presenting manifestation of rheumatic diseases in childhood. J Pediatr. 2006;148(5):683-686. doi:10.1016/j.jpeds.2005.12.070. PubMed
15. Lambotte O, Khellaf M, Harmouche H, et al. Characteristics and Long-Term Outcome of 15 Episodes of Systemic Lupus Erythematosus-Associated Hemophagocytic Syndrome. Medicine. 2006;85(3):169-182. doi:10.1097/01.md.0000224708.62510.d1. PubMed
16. Fukaya S, Yasuda S, Hashimoto T, et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30 cases. Rheumatology. 2008;47(11):1686-1691. doi:10.1093/rheumatology/ken342. PubMed
17. Stephan JL. Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology. 2001;40(11):1285-1292. doi:10.1093/rheumatology/40.11.1285. PubMed
18. Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child. 2001;85(5):421-426. PubMed
19. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. doi:10.1002/pbc.21039. PubMed
20. Lin CI, Yu HH, Lee JH, et al. Clinical analysis of macrophage activation syndrome in pediatric patients with autoimmune diseases. Clin Rheumatol. 2012;31(8):1223-1230. doi:10.1007/s10067-012-1998-0. PubMed
1. Weinzierl EP, Arber DA. The Differential Diagnosis and Bone Marrow Evaluation of New-Onset Pancytopenia. Am J Clin Pathol. 2012;139(1):9-29. doi:10.1309/AJCP50AEEYGREWUZ. PubMed
2. Pine M, Walter AW. Pancytopenia in hospitalized children: a five-year review. J Pediatr Hematol Oncol. 2010;32(5):e192-e194. doi:10.1097/MPH.0b013e3181e03082. PubMed
3. Bhatnagar SK. Pancytopenia in Children: Etiological Profile. J Trop Pediatr. 2005;51(4):236-239. doi:10.1093/tropej/fmi010. PubMed
4. Kulkarni KP, Marwaha RK. Acute lymphoblastic leukemia with pancytopenia at presentation: clinical correlates, prognostic impact, and association with survival. J Pediatr Hematol Oncol. 2013;35(7):573-576. doi:10.1097/MPH.0b013e31829d46f3. PubMed
5. Holubar, K. Terminology and iconography of lupus erythematosus: A historical vignette. Am J Dermatopathol. 1980;2(3):239-242. PubMed
6. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. doi: 10.1002/art.1780400928. PubMed
7. Petri M, Orbai, A, Alarcon GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677-2686. doi:10.1002/art.34473. PubMed
8. Tucker L. Review: Making the diagnosis of systemic lupus erythematosus in children and adolescents. Lupus. 2007;16(8):546-549. doi:10.1177/0961203307078068. PubMed
9. Brunner HI, Gladman DD, Ibañez D, Urowitz MD, Silverman ED. Difference in disease features between childhood-onset and adult-onset systemic lupus erythematosus. Arthritis Rheum. 2008;58(2):556-562. doi:10.1002/art.23204. PubMed
10. Benseler SM, Silverman ED. Systemic Lupus Erythematosus. Rheum Dis Clin North Am. 2007;33(3):471-498. doi:10.1016/j.rdc.2007.07.008. PubMed
11. Gokce M, Bilginer Y, Besbas N, et al. Hematological features of pediatric systemic lupus erythematosus: suggesting management strategies in children. Lupus. 2012;21(8):878-884. doi:10.1177/0961203312443721. PubMed
12. Voulgarelis M, Giannouli S, Tasidou A, Anagnostou D, Ziakas PD, Tzioufas AG. Bone marrow histological findings in systemic lupus erythematosus with hematologic abnormalities: A clinicopathological study. Am J Hematol. 2006;81(8):590-597. doi:10.1002/ajh.20593. PubMed
13. Pereira RM, Velloso ER, Menezes Y, Gualandro S, Vassalo J, Yoshinari NH. Bone marrow findings in systemic lupus erythematosus patients with peripheral cytopenias. Clin Rheumatol. 1998;17(3):219-222. PubMed
14. Avčin T, Tse SML, Schneider R, Ngan B, Silverman ED. Macrophage activation syndrome as the presenting manifestation of rheumatic diseases in childhood. J Pediatr. 2006;148(5):683-686. doi:10.1016/j.jpeds.2005.12.070. PubMed
15. Lambotte O, Khellaf M, Harmouche H, et al. Characteristics and Long-Term Outcome of 15 Episodes of Systemic Lupus Erythematosus-Associated Hemophagocytic Syndrome. Medicine. 2006;85(3):169-182. doi:10.1097/01.md.0000224708.62510.d1. PubMed
16. Fukaya S, Yasuda S, Hashimoto T, et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: analysis of 30 cases. Rheumatology. 2008;47(11):1686-1691. doi:10.1093/rheumatology/ken342. PubMed
17. Stephan JL. Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology. 2001;40(11):1285-1292. doi:10.1093/rheumatology/40.11.1285. PubMed
18. Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child. 2001;85(5):421-426. PubMed
19. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. doi:10.1002/pbc.21039. PubMed
20. Lin CI, Yu HH, Lee JH, et al. Clinical analysis of macrophage activation syndrome in pediatric patients with autoimmune diseases. Clin Rheumatol. 2012;31(8):1223-1230. doi:10.1007/s10067-012-1998-0. PubMed
© 2018 Society of Hospital Medicine