User login
A 54-year-old woman with pancytopenia
A 54-year-old woman with a 1-month history of progressive weakness was transported to the emergency department of a local hospital when a family member found her unresponsive. Before this event, the patient had said she had been feeling tired and cold and looking pale for several weeks.
In the emergency department, her temperature was low. Cableomputed tomography (CT) of the head showed a 1.4-cm hyperdense extraaxial mass. Imaging of the chest showed focal consolidations within the anterior segment of the right upper lobe and the left and right lower lobes.
A urine toxicology screen was positive for acetaminophen (Tylenol), opiates, and benzodiazepines. She was given three doses of naloxone (Narcan), which raised her level of arousal; however, she later became obtunded again and was intubated and transferred to Cleveland Clinic.
A new CT scan of the head confirmed a small left temporal, extradural, calcified lesion with no mass effect or overt bleeding; it appeared most compatible with a solitary calcified meningioma—a likely benign finding.
Her medical history includes hypertension, type 2 diabetes (controlled with diet), and osteoarthritis of the spine. In 1999, she had undergone a hysterectomy that necessitated a blood transfusion. She has never smoked tobacco and does not consume alcohol or use illicit drugs. In the past she worked as a nurse’s aid in a nursing home. However, for the past several years she has stayed at home. Her only avocation of note is gardening.
Initial physical examination
The patient is intubated and sedated. Her temperature is 35.3°C (95.5°F), blood pressure 122/81 mm Hg, heart rate 83 beats per minute, and respiratory rate 14 on assist-controlled ventilator settings with an Fio2 of 100% and a positive end-expiratory pressure of 5 cm H2O.
Her pupils are round, equal, and reactive to light. Her face is symmetric and notable for hirsutism over the chin. Her neck is supple and without lymphadenopathy or thyromegaly.
Rhonchi can be heard at both lung bases. She has normal bowel sounds, and her abdomen is soft and nondistended, with no masses or palpable hepatosplenomegaly. She has no pedal edema on either side, and no clubbing or cyanosis. Her skin is intact, without rashes, lesions, or tattoos. She is able to withdraw from painful stimuli in all four extremities.
INITIAL TESTS PROVIDE A CLUE
1. Which of the following is the likely cause of this patient’s pancytopenia?
- Folate deficiency
- Gastrointestinal bleeding secondary to colon cancer
- Paroxysmal nocturnal hemoglobinuria
- Myelophthisis
- Other

Causes of pancytopenia are listed in Table 2.
Folate deficiency
Folate is necessary for thymidylate synthesis, a rate-limiting step in DNA synthesis. The minimum daily requirement for dietary folate intake is 50 μg.
Severe deficiency of folate has been reported to cause pancytopenia in alcoholics.1 Abuse of alcohol leads to an abrupt decrease in serum folate (within 2 to 4 days of ceasing intake of proper amounts of folate, as in an alcoholic binge) by inhibiting its absorption in the proximal jejunum as well as its metabolism in the liver.2 The resulting folate deficiency, if sustained, can develop into megaloblastosis in 5 to 10 weeks.
The duration of weakness and pallor reported by this patient would raise suspicion of folate deficiency if she had a history of malnutrition or of alcohol abuse, but she has neither. Further, her mean corpuscular volume is 82.5 fL, red blood cell folate 391 ng/mL (reference range 257–800 ng/mL), and serum vitamin B12 1,886 pg/mL (22–700 pg/mL), and she has no macro-ovalocytes or hypersegmented neutrophils on a peripheral blood smear. This makes folate or vitamin B12 deficiency less likely.
Gastrointestinal bleeding due to colon cancer
Iron-deficiency anemia, hematochezia, melena, a change in bowel habits, and abdominal pain may be manifestations of colon cancer. Cancers of the colon originate from adenomatous polyps arising from the colonic mucosa.
The quantity of occult blood loss depends on the site of the tumor. Patients with tumors in the cecum or ascending colon lose an average of 9 mL/day, whereas those with tumors in the transverse, descending, or sigmoid colon or rectum lose less than 2 mL/day.3
Pertinent laboratory findings in iron-deficiency anemia are a low iron concentration, a low transferrin saturation, a depleted serum ferritin, and a normal to high total iron-binding capacity. An initial microcytic normochromic anemia eventually progresses to a microcytic hypochromic anemia that has a tendency to increasingly demonstrate anisocytosis and poikilocytosis.
Our patient’s symptoms, signs, and laboratory values (with normocytic normochromic anemia) are inconsistent with symptomatic colon cancer leading to iron-deficiency anemia.
Acute myeloid leukemia
Acute myeloid leukemia generally manifests with symptoms related to pancytopenia, with weakness and fatigability being the most common.4
In this condition, genetic alterations in hematopoietic precursor cells result in reduced differentiation capacity and accumulation of leukemic blasts in the bone marrow, peripheral blood, and other tissues.
Peripheral blood analysis usually reveals normocytic normochromic anemia with blasts. To establish a diagnosis of acute myeloid leukemia, one must observe at least 20% myeloblasts in the blood, the bone marrow, or both.
No blasts are seen on our patient’s peripheral blood smear, making acute myeloid leukemia less likely.
Paroxysmal nocturnal hemoglobinuria
Paroxysmal nocturnal hemoglobinuria is a possibility in the setting of intravascular hemolytic anemia, bone marrow failure, and thrombosis.
These processes are due to a defect in the glycosyl phosphatidyl inositol (GPI) anchor caused by an abnormality in the PIG-A gene. Partial or complete absence of the GPI anchor allows for activation of complement-mediated hemolysis. A diminished rate of hematopoiesis is presumably responsible for reticulocytopenia, granulocytopenia, or thrombocytopenia, though reticulocytosis can also be seen.5,6 The highly thrombogenic state is believed to occur because of microparticles rich in phosphatidylserine.7
Our patient’s peripheral smear has rare fragmented red blood cells and lacks teardrop red cells. Although paroxysmal nocturnal hemoglobinuria does not have characteristic morphologic features in the peripheral blood, there are no signs of thrombosis in our patient. Her lactate dehydrogenase level is 395 U/L (reference range 100–220 U/L), and her haptoglobin level is less than 20 mg/dL (33–246). These findings could indicate a low level of intravascular hemolysis.
Myelophthisis
Myelophthisis refers to any disorder in which an abnormal cell process invades the bone marrow, damaging hematopoietic tissue. These processes include neoplastic diseases, storage disorders, and a variety of infections. A decrease in all three cell types may result, depending on the severity of invasion. Documented infectious causes include hepatitis viruses, Epstein-Barr virus, human immunodeficiency virus (HIV), mycobacteria, and fungi.
Our patient’s condition is likely due to a marrow-based process of uncertain etiology. In myelophthisic processes, one may see teardrop red cells, which are not seen in this patient’s smear. However, on her chest imaging, the finding of focal consolidations within the anterior segment of the right upper lobe and both lower lobes raises suspicion of an infectious cause.
CASE CONTINUED: SHE UNDERGOES DIAGNOSTIC TESTING
Let us recap some of the laboratory studies that document the extent of our patient’s pancytopenia and the pattern of her anemia:
- Hemoglobin 10.2 g/dL (reference range 11.5–15.5 g/dL)
- Platelet count 27 × 109/L (150–400)
- Leukopenia with profound T-cell lymphopenia
- Iron 59 μg/dL (30–140)
- Total iron-binding capacity 110 μg/dL (210–415)
- Ferritin 3,004 ng/mL (18–300)
- Transferrin saturation 54% (11%–46%).
2. Which of the following would be the best test to obtain next?
- Bone marrow examination
- Blood cultures
- Tuberculin skin test
- Liver biopsy
- Positron emission tomography and CT
Our patient has unexplained pancytopenia. While all the tests listed above might shed light on her condition, a bone marrow examination would be the best test to obtain next.
Urine histoplasma antigen studies are positive at greater than 39 ng/mL (normal 0, low positive < 0.6–3.9, moderate positive 4.0–19.9, high positive 20–39 ng/mL). A culture of the marrow subsequently grows this organism.
3. Which of the following tests would establish a definitive diagnosis in this patient?
- Methenamine silver stain of the marrow
- Serum antibody testing
- Fungal culture
- Peripheral blood smear
- Carbolfuchsin stain of marrow
- Urine histoplasma antigen
A prompt diagnosis is critical in patients with acute pulmonary histoplasmosis or progressive disseminated histoplasmosis because early treatment may shorten the clinical course and length of treatment and, in cases of disseminated histoplasmosis, prevent death.8–10
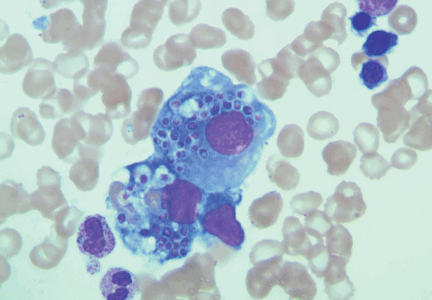
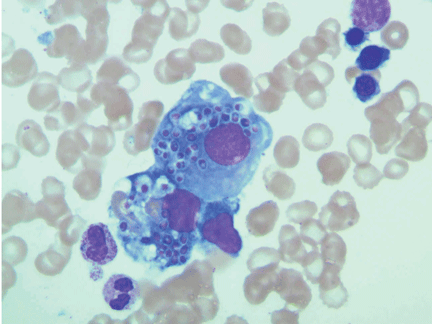
Histopathologic examination of the bone marrow gives the most rapid results, although biopsy to obtain the tissue is invasive. It can give a definitive diagnosis if it reveals the typical 2- to 4-μm yeast structures of H capsulatum. These are observed on an aspirate smear of the patient’s bone marrow biopsy (Figure 1) and can be confirmed by methenamine silver or periodic acid-Schiff staining of the tissue.
Antibody detection is less practical because the antibodies take 2 to 6 weeks after infection to form.11 Also, it is less useful in cases of disseminated infection because many of these patients are immunosuppressed.
Fungal culture remains the gold standard diagnostic test for histoplasmosis. However, results may take up to 1 month and may be falsely negative in less severe cases.
Histoplasma antigen testing is of greater utility in patients with severe disease, including cases of disseminated histoplasmosis. Rates of antigen detection approach 90% in urine specimens from non-AIDS patients with disseminated infection.12 The urine assay has a greater sensitivity and specificity than the serum assay. The rate of detection is lower (ie, around 82%) in patients with acute pulmonary histoplasmosis when both the serum and urine specimens are tested.13
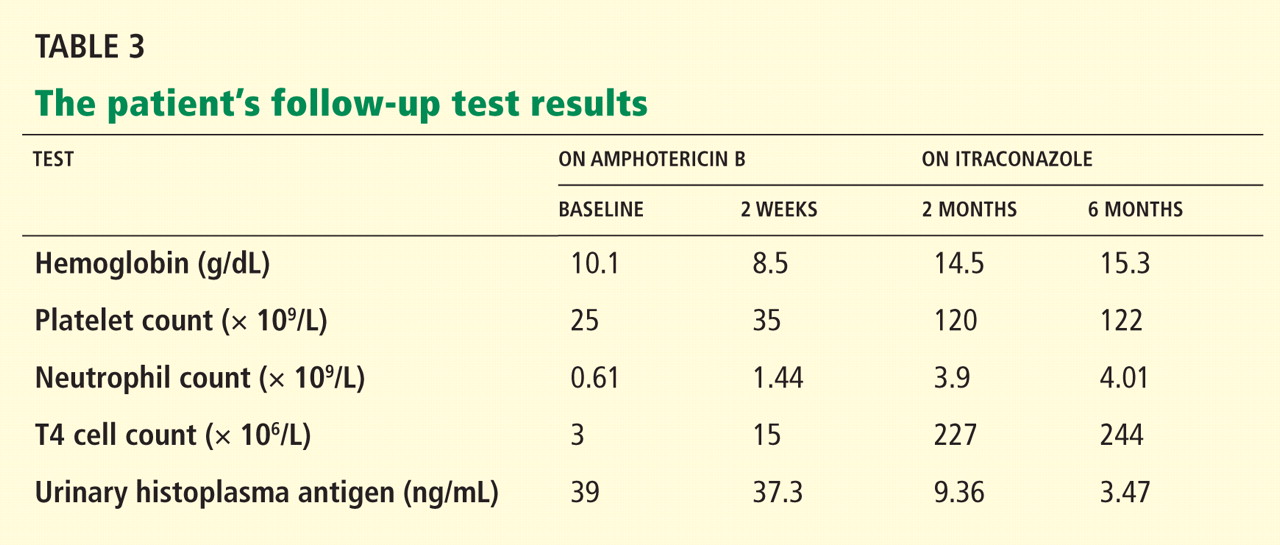
The immunoassay for histoplasma antigen is particularly useful for monitoring the response to therapy. Antigen levels should be measured before treatment is started and at 2 weeks, 1 month, and then approximately every 3 months during therapy.14 If the treatment is effective, antigens should decline by at least 20% in the first month of treatment and by another 20% in each of the following 3-month intervals. Antigen testing should be done every 3 months until a negative antigen level is achieved. The antigen level should also be followed for at least 6 months after treatment has stopped.14
HISTOPLASMA IS INHALED
H capsulatum is the cause of one of the most common pulmonary and systemic mycotic infections in the world, with hundreds of thousands of new cases annually. In areas where the soil is contaminated by bird or bat guano, the fungus is inhaled, resulting in an asymptomatic or a self-limiting influenza-like syndrome in an immunocompetent individual.15
An antigen-specific CD4+ T lymphocytemediated immunity occurs. The immune response of the host is thought to be fungistatic rather than fungicidal, resulting in a persistent inactive infection capable of reactivation in the presence of a host-pathogen imbalance.16
Most infections are asymptomatic or self-limited. For every 2,000 acute infections there is one that results in severe and progressive dissemination, usually in an immunocompromised host.17,18
TREATMENT OF HISTOPLASMOSIS
4. What is the appropriate initial choice of treatment for a severe case of disseminated histoplasmosis?
- Amphotericin B in a lipid complex formulation (Abelcet)
- Itraconazole (Sporanox)
- Fluconazole (Diflucan)
- Ketoconazole (Nizoral)
Untreated, acute disseminated histoplasmosis can progress over a period of 2 to 12 weeks, ultimately killing the patient.17,19
The leading therapies include amphotericin B in a lipid formulation and azole drugs, in particular itraconazole. Fluconazole and ketoconazole are not first-line options in severe cases because they are less predictably effective, and ketoconazole has a higher rate of side effects.20–23 The current recommendation is to treat severely ill hospitalized patients with one of the liposomal formulations or the lipid complex formulation of amphotericin B. Itraconazole is used for patients who have mild to moderate symptoms and as a step-down therapy in patients who improve after initial use of amphotericin B.
CASE CONCLUDED: THE PATIENT RECOVERS
At the time of the initial patient encounter, there was no history of or obvious cause of immunosuppression in this patient. She was found to be HIV-negative and was subsequently diagnosed with “profound immunosuppression of unknown etiology” resulting in a low CD4 count.
The patient receives trimethoprim-sulfamethoxazole (Bactrim, Septra) and azithromycin (Zithromax) for prophylaxis against Pneumocystis carinii pneumonia and Mycobacterium avium intracellulare infection. Two months after the hospitalization, she recalls being at a corn maze 1 month before becoming ill.
- Clarke V, Weston-Smith S. Severe folate-deficiency pancytopenia. BMJ Case Reports 2010; published online.
- Anthony AC. Megaloblastic anemias. In:Hoffman R, Benz EJ, Shattil SJ, Furie B, Cohebn HJ, Silberstein LE, editors. Hematology: Basic Principles and Practice, 2nd ed. New York, NY: Churchill Livingston, 1995:552–586.
- Macrae FA, St John DJ. Relationship between patterns of bleeding and Hemoccult sensitivity in patients with colorectal cancers or adenomas. Gastroenterology 1982; 82:891–898.
- Meyers CA, Albitar M, Estey E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer 2005; 104:788–793.
- Parker CJ. Bone marrow failure syndromes: paroxysmal nocturnal hemoglobinuria. Hematol Oncol Clin North Am 2009; 23:333–346.
- Young NS, Maciejewski JP, Sloand E, et al. The relationship of aplastic anemia and PNH. Int J Hematol 2002; 76(suppl 2):168–172.
- Rosse W. A new way to prevent thrombosis? Blood 2007; 110:3821.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Meals LT, McKinney WP. Acute pulmonary histoplasmosis: progressive pneumonia resulting from high inoculum exposure. J Ky Med Assoc 1998; 96:258–260.
- Salomon J, Flament Saillour M, De Truchis P, et al. An outbreak of acute pulmonary histoplasmosis in members of a trekking trip in Martinique, French West Indies. J Travel Med 2003; 10:87–93.
- Joseph Wheat L. Current diagnosis of histoplasmosis. Trends Microbiol 2003; 11:488–494.
- Wheat LJ, Kauffman CA. Histoplasmosis. Infect Dis Clin North Am 2003; 17:1–19.
- Swartzentruber S, Rhodes L, Kurkjian K, et al. Diagnosis of acute pulmonary histoplasmosis by antigen detection. Clin Infect Dis 2009; 49:1878–1882.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Retallack DM, Woods JP. Molecular epidemiology, pathogenesis, and genetics of the dimorphic fungus Histoplasma capsulatum. Microbes Infect 1999; 1:817–825.
- Deepe GS. The immune response to Histoplasma capsulatum: unearthing its secrets. J Lab Clin Med 1994; 123:201–205.
- Goodwin RA, Shapiro JL, Thurman GH, Thurman SS, Des Prez RM. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore) 1980; 59:1–33.
- Wheat LJ, Connolly-Stringfield PA, Baker RL, et al. Disseminated histoplasmosis in the acquired immune deficiency syndrome: clinical findings, diagnosis and treatment, and review of the literature. Medicine (Baltimore) 1990; 69:361–374.
- Rubin H, Furcolow ML, Yates JL, Brasher CA. The course and prognosis of histoplasmosis. Am J Med 1959; 27:278–288.
- Wheat J, MaWhinney S, Hafner R, et al. Treatment of histoplasmosis with fluconazole in patients with acquired immunodeficiency syndrome. National Institute of Allergy and Infectious Diseases Acquired Immunodeficiency Syndrome Clinical Trials Group and Mycoses Study Group. Am J Med 1997; 103:223–232.
- McKinsey DS, Kauffman CA, Pappas PG, et al. Fluconazole therapy for histoplasmosis. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Clin Infect Dis 1996; 23:996–1001.
- Slama TG. Treatment of disseminated and progressive cavitary histoplasmosis with ketoconazole. Am J Med 1983; 74:70–73.
- Treatment of blastomycosis and histoplasmosis with ketoconazole. Results of a prospective randomized clinical trial. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Ann Intern Med 1985; 103:861–872.
A 54-year-old woman with a 1-month history of progressive weakness was transported to the emergency department of a local hospital when a family member found her unresponsive. Before this event, the patient had said she had been feeling tired and cold and looking pale for several weeks.
In the emergency department, her temperature was low. Cableomputed tomography (CT) of the head showed a 1.4-cm hyperdense extraaxial mass. Imaging of the chest showed focal consolidations within the anterior segment of the right upper lobe and the left and right lower lobes.
A urine toxicology screen was positive for acetaminophen (Tylenol), opiates, and benzodiazepines. She was given three doses of naloxone (Narcan), which raised her level of arousal; however, she later became obtunded again and was intubated and transferred to Cleveland Clinic.
A new CT scan of the head confirmed a small left temporal, extradural, calcified lesion with no mass effect or overt bleeding; it appeared most compatible with a solitary calcified meningioma—a likely benign finding.
Her medical history includes hypertension, type 2 diabetes (controlled with diet), and osteoarthritis of the spine. In 1999, she had undergone a hysterectomy that necessitated a blood transfusion. She has never smoked tobacco and does not consume alcohol or use illicit drugs. In the past she worked as a nurse’s aid in a nursing home. However, for the past several years she has stayed at home. Her only avocation of note is gardening.
Initial physical examination
The patient is intubated and sedated. Her temperature is 35.3°C (95.5°F), blood pressure 122/81 mm Hg, heart rate 83 beats per minute, and respiratory rate 14 on assist-controlled ventilator settings with an Fio2 of 100% and a positive end-expiratory pressure of 5 cm H2O.
Her pupils are round, equal, and reactive to light. Her face is symmetric and notable for hirsutism over the chin. Her neck is supple and without lymphadenopathy or thyromegaly.
Rhonchi can be heard at both lung bases. She has normal bowel sounds, and her abdomen is soft and nondistended, with no masses or palpable hepatosplenomegaly. She has no pedal edema on either side, and no clubbing or cyanosis. Her skin is intact, without rashes, lesions, or tattoos. She is able to withdraw from painful stimuli in all four extremities.
INITIAL TESTS PROVIDE A CLUE
1. Which of the following is the likely cause of this patient’s pancytopenia?
- Folate deficiency
- Gastrointestinal bleeding secondary to colon cancer
- Paroxysmal nocturnal hemoglobinuria
- Myelophthisis
- Other
Causes of pancytopenia are listed in Table 2.
Folate deficiency
Folate is necessary for thymidylate synthesis, a rate-limiting step in DNA synthesis. The minimum daily requirement for dietary folate intake is 50 μg.
Severe deficiency of folate has been reported to cause pancytopenia in alcoholics.1 Abuse of alcohol leads to an abrupt decrease in serum folate (within 2 to 4 days of ceasing intake of proper amounts of folate, as in an alcoholic binge) by inhibiting its absorption in the proximal jejunum as well as its metabolism in the liver.2 The resulting folate deficiency, if sustained, can develop into megaloblastosis in 5 to 10 weeks.
The duration of weakness and pallor reported by this patient would raise suspicion of folate deficiency if she had a history of malnutrition or of alcohol abuse, but she has neither. Further, her mean corpuscular volume is 82.5 fL, red blood cell folate 391 ng/mL (reference range 257–800 ng/mL), and serum vitamin B12 1,886 pg/mL (22–700 pg/mL), and she has no macro-ovalocytes or hypersegmented neutrophils on a peripheral blood smear. This makes folate or vitamin B12 deficiency less likely.
Gastrointestinal bleeding due to colon cancer
Iron-deficiency anemia, hematochezia, melena, a change in bowel habits, and abdominal pain may be manifestations of colon cancer. Cancers of the colon originate from adenomatous polyps arising from the colonic mucosa.
The quantity of occult blood loss depends on the site of the tumor. Patients with tumors in the cecum or ascending colon lose an average of 9 mL/day, whereas those with tumors in the transverse, descending, or sigmoid colon or rectum lose less than 2 mL/day.3
Pertinent laboratory findings in iron-deficiency anemia are a low iron concentration, a low transferrin saturation, a depleted serum ferritin, and a normal to high total iron-binding capacity. An initial microcytic normochromic anemia eventually progresses to a microcytic hypochromic anemia that has a tendency to increasingly demonstrate anisocytosis and poikilocytosis.
Our patient’s symptoms, signs, and laboratory values (with normocytic normochromic anemia) are inconsistent with symptomatic colon cancer leading to iron-deficiency anemia.
Acute myeloid leukemia
Acute myeloid leukemia generally manifests with symptoms related to pancytopenia, with weakness and fatigability being the most common.4
In this condition, genetic alterations in hematopoietic precursor cells result in reduced differentiation capacity and accumulation of leukemic blasts in the bone marrow, peripheral blood, and other tissues.
Peripheral blood analysis usually reveals normocytic normochromic anemia with blasts. To establish a diagnosis of acute myeloid leukemia, one must observe at least 20% myeloblasts in the blood, the bone marrow, or both.
No blasts are seen on our patient’s peripheral blood smear, making acute myeloid leukemia less likely.
Paroxysmal nocturnal hemoglobinuria
Paroxysmal nocturnal hemoglobinuria is a possibility in the setting of intravascular hemolytic anemia, bone marrow failure, and thrombosis.
These processes are due to a defect in the glycosyl phosphatidyl inositol (GPI) anchor caused by an abnormality in the PIG-A gene. Partial or complete absence of the GPI anchor allows for activation of complement-mediated hemolysis. A diminished rate of hematopoiesis is presumably responsible for reticulocytopenia, granulocytopenia, or thrombocytopenia, though reticulocytosis can also be seen.5,6 The highly thrombogenic state is believed to occur because of microparticles rich in phosphatidylserine.7
Our patient’s peripheral smear has rare fragmented red blood cells and lacks teardrop red cells. Although paroxysmal nocturnal hemoglobinuria does not have characteristic morphologic features in the peripheral blood, there are no signs of thrombosis in our patient. Her lactate dehydrogenase level is 395 U/L (reference range 100–220 U/L), and her haptoglobin level is less than 20 mg/dL (33–246). These findings could indicate a low level of intravascular hemolysis.
Myelophthisis
Myelophthisis refers to any disorder in which an abnormal cell process invades the bone marrow, damaging hematopoietic tissue. These processes include neoplastic diseases, storage disorders, and a variety of infections. A decrease in all three cell types may result, depending on the severity of invasion. Documented infectious causes include hepatitis viruses, Epstein-Barr virus, human immunodeficiency virus (HIV), mycobacteria, and fungi.
Our patient’s condition is likely due to a marrow-based process of uncertain etiology. In myelophthisic processes, one may see teardrop red cells, which are not seen in this patient’s smear. However, on her chest imaging, the finding of focal consolidations within the anterior segment of the right upper lobe and both lower lobes raises suspicion of an infectious cause.
CASE CONTINUED: SHE UNDERGOES DIAGNOSTIC TESTING
Let us recap some of the laboratory studies that document the extent of our patient’s pancytopenia and the pattern of her anemia:
- Hemoglobin 10.2 g/dL (reference range 11.5–15.5 g/dL)
- Platelet count 27 × 109/L (150–400)
- Leukopenia with profound T-cell lymphopenia
- Iron 59 μg/dL (30–140)
- Total iron-binding capacity 110 μg/dL (210–415)
- Ferritin 3,004 ng/mL (18–300)
- Transferrin saturation 54% (11%–46%).
2. Which of the following would be the best test to obtain next?
- Bone marrow examination
- Blood cultures
- Tuberculin skin test
- Liver biopsy
- Positron emission tomography and CT
Our patient has unexplained pancytopenia. While all the tests listed above might shed light on her condition, a bone marrow examination would be the best test to obtain next.
Urine histoplasma antigen studies are positive at greater than 39 ng/mL (normal 0, low positive < 0.6–3.9, moderate positive 4.0–19.9, high positive 20–39 ng/mL). A culture of the marrow subsequently grows this organism.
3. Which of the following tests would establish a definitive diagnosis in this patient?
- Methenamine silver stain of the marrow
- Serum antibody testing
- Fungal culture
- Peripheral blood smear
- Carbolfuchsin stain of marrow
- Urine histoplasma antigen
A prompt diagnosis is critical in patients with acute pulmonary histoplasmosis or progressive disseminated histoplasmosis because early treatment may shorten the clinical course and length of treatment and, in cases of disseminated histoplasmosis, prevent death.8–10
Histopathologic examination of the bone marrow gives the most rapid results, although biopsy to obtain the tissue is invasive. It can give a definitive diagnosis if it reveals the typical 2- to 4-μm yeast structures of H capsulatum. These are observed on an aspirate smear of the patient’s bone marrow biopsy (Figure 1) and can be confirmed by methenamine silver or periodic acid-Schiff staining of the tissue.
Antibody detection is less practical because the antibodies take 2 to 6 weeks after infection to form.11 Also, it is less useful in cases of disseminated infection because many of these patients are immunosuppressed.
Fungal culture remains the gold standard diagnostic test for histoplasmosis. However, results may take up to 1 month and may be falsely negative in less severe cases.
Histoplasma antigen testing is of greater utility in patients with severe disease, including cases of disseminated histoplasmosis. Rates of antigen detection approach 90% in urine specimens from non-AIDS patients with disseminated infection.12 The urine assay has a greater sensitivity and specificity than the serum assay. The rate of detection is lower (ie, around 82%) in patients with acute pulmonary histoplasmosis when both the serum and urine specimens are tested.13
The immunoassay for histoplasma antigen is particularly useful for monitoring the response to therapy. Antigen levels should be measured before treatment is started and at 2 weeks, 1 month, and then approximately every 3 months during therapy.14 If the treatment is effective, antigens should decline by at least 20% in the first month of treatment and by another 20% in each of the following 3-month intervals. Antigen testing should be done every 3 months until a negative antigen level is achieved. The antigen level should also be followed for at least 6 months after treatment has stopped.14
HISTOPLASMA IS INHALED
H capsulatum is the cause of one of the most common pulmonary and systemic mycotic infections in the world, with hundreds of thousands of new cases annually. In areas where the soil is contaminated by bird or bat guano, the fungus is inhaled, resulting in an asymptomatic or a self-limiting influenza-like syndrome in an immunocompetent individual.15
An antigen-specific CD4+ T lymphocytemediated immunity occurs. The immune response of the host is thought to be fungistatic rather than fungicidal, resulting in a persistent inactive infection capable of reactivation in the presence of a host-pathogen imbalance.16
Most infections are asymptomatic or self-limited. For every 2,000 acute infections there is one that results in severe and progressive dissemination, usually in an immunocompromised host.17,18
TREATMENT OF HISTOPLASMOSIS
4. What is the appropriate initial choice of treatment for a severe case of disseminated histoplasmosis?
- Amphotericin B in a lipid complex formulation (Abelcet)
- Itraconazole (Sporanox)
- Fluconazole (Diflucan)
- Ketoconazole (Nizoral)
Untreated, acute disseminated histoplasmosis can progress over a period of 2 to 12 weeks, ultimately killing the patient.17,19
The leading therapies include amphotericin B in a lipid formulation and azole drugs, in particular itraconazole. Fluconazole and ketoconazole are not first-line options in severe cases because they are less predictably effective, and ketoconazole has a higher rate of side effects.20–23 The current recommendation is to treat severely ill hospitalized patients with one of the liposomal formulations or the lipid complex formulation of amphotericin B. Itraconazole is used for patients who have mild to moderate symptoms and as a step-down therapy in patients who improve after initial use of amphotericin B.
CASE CONCLUDED: THE PATIENT RECOVERS
At the time of the initial patient encounter, there was no history of or obvious cause of immunosuppression in this patient. She was found to be HIV-negative and was subsequently diagnosed with “profound immunosuppression of unknown etiology” resulting in a low CD4 count.
The patient receives trimethoprim-sulfamethoxazole (Bactrim, Septra) and azithromycin (Zithromax) for prophylaxis against Pneumocystis carinii pneumonia and Mycobacterium avium intracellulare infection. Two months after the hospitalization, she recalls being at a corn maze 1 month before becoming ill.
A 54-year-old woman with a 1-month history of progressive weakness was transported to the emergency department of a local hospital when a family member found her unresponsive. Before this event, the patient had said she had been feeling tired and cold and looking pale for several weeks.
In the emergency department, her temperature was low. Cableomputed tomography (CT) of the head showed a 1.4-cm hyperdense extraaxial mass. Imaging of the chest showed focal consolidations within the anterior segment of the right upper lobe and the left and right lower lobes.
A urine toxicology screen was positive for acetaminophen (Tylenol), opiates, and benzodiazepines. She was given three doses of naloxone (Narcan), which raised her level of arousal; however, she later became obtunded again and was intubated and transferred to Cleveland Clinic.
A new CT scan of the head confirmed a small left temporal, extradural, calcified lesion with no mass effect or overt bleeding; it appeared most compatible with a solitary calcified meningioma—a likely benign finding.
Her medical history includes hypertension, type 2 diabetes (controlled with diet), and osteoarthritis of the spine. In 1999, she had undergone a hysterectomy that necessitated a blood transfusion. She has never smoked tobacco and does not consume alcohol or use illicit drugs. In the past she worked as a nurse’s aid in a nursing home. However, for the past several years she has stayed at home. Her only avocation of note is gardening.
Initial physical examination
The patient is intubated and sedated. Her temperature is 35.3°C (95.5°F), blood pressure 122/81 mm Hg, heart rate 83 beats per minute, and respiratory rate 14 on assist-controlled ventilator settings with an Fio2 of 100% and a positive end-expiratory pressure of 5 cm H2O.
Her pupils are round, equal, and reactive to light. Her face is symmetric and notable for hirsutism over the chin. Her neck is supple and without lymphadenopathy or thyromegaly.
Rhonchi can be heard at both lung bases. She has normal bowel sounds, and her abdomen is soft and nondistended, with no masses or palpable hepatosplenomegaly. She has no pedal edema on either side, and no clubbing or cyanosis. Her skin is intact, without rashes, lesions, or tattoos. She is able to withdraw from painful stimuli in all four extremities.
INITIAL TESTS PROVIDE A CLUE
1. Which of the following is the likely cause of this patient’s pancytopenia?
- Folate deficiency
- Gastrointestinal bleeding secondary to colon cancer
- Paroxysmal nocturnal hemoglobinuria
- Myelophthisis
- Other
Causes of pancytopenia are listed in Table 2.
Folate deficiency
Folate is necessary for thymidylate synthesis, a rate-limiting step in DNA synthesis. The minimum daily requirement for dietary folate intake is 50 μg.
Severe deficiency of folate has been reported to cause pancytopenia in alcoholics.1 Abuse of alcohol leads to an abrupt decrease in serum folate (within 2 to 4 days of ceasing intake of proper amounts of folate, as in an alcoholic binge) by inhibiting its absorption in the proximal jejunum as well as its metabolism in the liver.2 The resulting folate deficiency, if sustained, can develop into megaloblastosis in 5 to 10 weeks.
The duration of weakness and pallor reported by this patient would raise suspicion of folate deficiency if she had a history of malnutrition or of alcohol abuse, but she has neither. Further, her mean corpuscular volume is 82.5 fL, red blood cell folate 391 ng/mL (reference range 257–800 ng/mL), and serum vitamin B12 1,886 pg/mL (22–700 pg/mL), and she has no macro-ovalocytes or hypersegmented neutrophils on a peripheral blood smear. This makes folate or vitamin B12 deficiency less likely.
Gastrointestinal bleeding due to colon cancer
Iron-deficiency anemia, hematochezia, melena, a change in bowel habits, and abdominal pain may be manifestations of colon cancer. Cancers of the colon originate from adenomatous polyps arising from the colonic mucosa.
The quantity of occult blood loss depends on the site of the tumor. Patients with tumors in the cecum or ascending colon lose an average of 9 mL/day, whereas those with tumors in the transverse, descending, or sigmoid colon or rectum lose less than 2 mL/day.3
Pertinent laboratory findings in iron-deficiency anemia are a low iron concentration, a low transferrin saturation, a depleted serum ferritin, and a normal to high total iron-binding capacity. An initial microcytic normochromic anemia eventually progresses to a microcytic hypochromic anemia that has a tendency to increasingly demonstrate anisocytosis and poikilocytosis.
Our patient’s symptoms, signs, and laboratory values (with normocytic normochromic anemia) are inconsistent with symptomatic colon cancer leading to iron-deficiency anemia.
Acute myeloid leukemia
Acute myeloid leukemia generally manifests with symptoms related to pancytopenia, with weakness and fatigability being the most common.4
In this condition, genetic alterations in hematopoietic precursor cells result in reduced differentiation capacity and accumulation of leukemic blasts in the bone marrow, peripheral blood, and other tissues.
Peripheral blood analysis usually reveals normocytic normochromic anemia with blasts. To establish a diagnosis of acute myeloid leukemia, one must observe at least 20% myeloblasts in the blood, the bone marrow, or both.
No blasts are seen on our patient’s peripheral blood smear, making acute myeloid leukemia less likely.
Paroxysmal nocturnal hemoglobinuria
Paroxysmal nocturnal hemoglobinuria is a possibility in the setting of intravascular hemolytic anemia, bone marrow failure, and thrombosis.
These processes are due to a defect in the glycosyl phosphatidyl inositol (GPI) anchor caused by an abnormality in the PIG-A gene. Partial or complete absence of the GPI anchor allows for activation of complement-mediated hemolysis. A diminished rate of hematopoiesis is presumably responsible for reticulocytopenia, granulocytopenia, or thrombocytopenia, though reticulocytosis can also be seen.5,6 The highly thrombogenic state is believed to occur because of microparticles rich in phosphatidylserine.7
Our patient’s peripheral smear has rare fragmented red blood cells and lacks teardrop red cells. Although paroxysmal nocturnal hemoglobinuria does not have characteristic morphologic features in the peripheral blood, there are no signs of thrombosis in our patient. Her lactate dehydrogenase level is 395 U/L (reference range 100–220 U/L), and her haptoglobin level is less than 20 mg/dL (33–246). These findings could indicate a low level of intravascular hemolysis.
Myelophthisis
Myelophthisis refers to any disorder in which an abnormal cell process invades the bone marrow, damaging hematopoietic tissue. These processes include neoplastic diseases, storage disorders, and a variety of infections. A decrease in all three cell types may result, depending on the severity of invasion. Documented infectious causes include hepatitis viruses, Epstein-Barr virus, human immunodeficiency virus (HIV), mycobacteria, and fungi.
Our patient’s condition is likely due to a marrow-based process of uncertain etiology. In myelophthisic processes, one may see teardrop red cells, which are not seen in this patient’s smear. However, on her chest imaging, the finding of focal consolidations within the anterior segment of the right upper lobe and both lower lobes raises suspicion of an infectious cause.
CASE CONTINUED: SHE UNDERGOES DIAGNOSTIC TESTING
Let us recap some of the laboratory studies that document the extent of our patient’s pancytopenia and the pattern of her anemia:
- Hemoglobin 10.2 g/dL (reference range 11.5–15.5 g/dL)
- Platelet count 27 × 109/L (150–400)
- Leukopenia with profound T-cell lymphopenia
- Iron 59 μg/dL (30–140)
- Total iron-binding capacity 110 μg/dL (210–415)
- Ferritin 3,004 ng/mL (18–300)
- Transferrin saturation 54% (11%–46%).
2. Which of the following would be the best test to obtain next?
- Bone marrow examination
- Blood cultures
- Tuberculin skin test
- Liver biopsy
- Positron emission tomography and CT
Our patient has unexplained pancytopenia. While all the tests listed above might shed light on her condition, a bone marrow examination would be the best test to obtain next.
Urine histoplasma antigen studies are positive at greater than 39 ng/mL (normal 0, low positive < 0.6–3.9, moderate positive 4.0–19.9, high positive 20–39 ng/mL). A culture of the marrow subsequently grows this organism.
3. Which of the following tests would establish a definitive diagnosis in this patient?
- Methenamine silver stain of the marrow
- Serum antibody testing
- Fungal culture
- Peripheral blood smear
- Carbolfuchsin stain of marrow
- Urine histoplasma antigen
A prompt diagnosis is critical in patients with acute pulmonary histoplasmosis or progressive disseminated histoplasmosis because early treatment may shorten the clinical course and length of treatment and, in cases of disseminated histoplasmosis, prevent death.8–10
Histopathologic examination of the bone marrow gives the most rapid results, although biopsy to obtain the tissue is invasive. It can give a definitive diagnosis if it reveals the typical 2- to 4-μm yeast structures of H capsulatum. These are observed on an aspirate smear of the patient’s bone marrow biopsy (Figure 1) and can be confirmed by methenamine silver or periodic acid-Schiff staining of the tissue.
Antibody detection is less practical because the antibodies take 2 to 6 weeks after infection to form.11 Also, it is less useful in cases of disseminated infection because many of these patients are immunosuppressed.
Fungal culture remains the gold standard diagnostic test for histoplasmosis. However, results may take up to 1 month and may be falsely negative in less severe cases.
Histoplasma antigen testing is of greater utility in patients with severe disease, including cases of disseminated histoplasmosis. Rates of antigen detection approach 90% in urine specimens from non-AIDS patients with disseminated infection.12 The urine assay has a greater sensitivity and specificity than the serum assay. The rate of detection is lower (ie, around 82%) in patients with acute pulmonary histoplasmosis when both the serum and urine specimens are tested.13
The immunoassay for histoplasma antigen is particularly useful for monitoring the response to therapy. Antigen levels should be measured before treatment is started and at 2 weeks, 1 month, and then approximately every 3 months during therapy.14 If the treatment is effective, antigens should decline by at least 20% in the first month of treatment and by another 20% in each of the following 3-month intervals. Antigen testing should be done every 3 months until a negative antigen level is achieved. The antigen level should also be followed for at least 6 months after treatment has stopped.14
HISTOPLASMA IS INHALED
H capsulatum is the cause of one of the most common pulmonary and systemic mycotic infections in the world, with hundreds of thousands of new cases annually. In areas where the soil is contaminated by bird or bat guano, the fungus is inhaled, resulting in an asymptomatic or a self-limiting influenza-like syndrome in an immunocompetent individual.15
An antigen-specific CD4+ T lymphocytemediated immunity occurs. The immune response of the host is thought to be fungistatic rather than fungicidal, resulting in a persistent inactive infection capable of reactivation in the presence of a host-pathogen imbalance.16
Most infections are asymptomatic or self-limited. For every 2,000 acute infections there is one that results in severe and progressive dissemination, usually in an immunocompromised host.17,18
TREATMENT OF HISTOPLASMOSIS
4. What is the appropriate initial choice of treatment for a severe case of disseminated histoplasmosis?
- Amphotericin B in a lipid complex formulation (Abelcet)
- Itraconazole (Sporanox)
- Fluconazole (Diflucan)
- Ketoconazole (Nizoral)
Untreated, acute disseminated histoplasmosis can progress over a period of 2 to 12 weeks, ultimately killing the patient.17,19
The leading therapies include amphotericin B in a lipid formulation and azole drugs, in particular itraconazole. Fluconazole and ketoconazole are not first-line options in severe cases because they are less predictably effective, and ketoconazole has a higher rate of side effects.20–23 The current recommendation is to treat severely ill hospitalized patients with one of the liposomal formulations or the lipid complex formulation of amphotericin B. Itraconazole is used for patients who have mild to moderate symptoms and as a step-down therapy in patients who improve after initial use of amphotericin B.
CASE CONCLUDED: THE PATIENT RECOVERS
At the time of the initial patient encounter, there was no history of or obvious cause of immunosuppression in this patient. She was found to be HIV-negative and was subsequently diagnosed with “profound immunosuppression of unknown etiology” resulting in a low CD4 count.
The patient receives trimethoprim-sulfamethoxazole (Bactrim, Septra) and azithromycin (Zithromax) for prophylaxis against Pneumocystis carinii pneumonia and Mycobacterium avium intracellulare infection. Two months after the hospitalization, she recalls being at a corn maze 1 month before becoming ill.
- Clarke V, Weston-Smith S. Severe folate-deficiency pancytopenia. BMJ Case Reports 2010; published online.
- Anthony AC. Megaloblastic anemias. In:Hoffman R, Benz EJ, Shattil SJ, Furie B, Cohebn HJ, Silberstein LE, editors. Hematology: Basic Principles and Practice, 2nd ed. New York, NY: Churchill Livingston, 1995:552–586.
- Macrae FA, St John DJ. Relationship between patterns of bleeding and Hemoccult sensitivity in patients with colorectal cancers or adenomas. Gastroenterology 1982; 82:891–898.
- Meyers CA, Albitar M, Estey E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer 2005; 104:788–793.
- Parker CJ. Bone marrow failure syndromes: paroxysmal nocturnal hemoglobinuria. Hematol Oncol Clin North Am 2009; 23:333–346.
- Young NS, Maciejewski JP, Sloand E, et al. The relationship of aplastic anemia and PNH. Int J Hematol 2002; 76(suppl 2):168–172.
- Rosse W. A new way to prevent thrombosis? Blood 2007; 110:3821.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Meals LT, McKinney WP. Acute pulmonary histoplasmosis: progressive pneumonia resulting from high inoculum exposure. J Ky Med Assoc 1998; 96:258–260.
- Salomon J, Flament Saillour M, De Truchis P, et al. An outbreak of acute pulmonary histoplasmosis in members of a trekking trip in Martinique, French West Indies. J Travel Med 2003; 10:87–93.
- Joseph Wheat L. Current diagnosis of histoplasmosis. Trends Microbiol 2003; 11:488–494.
- Wheat LJ, Kauffman CA. Histoplasmosis. Infect Dis Clin North Am 2003; 17:1–19.
- Swartzentruber S, Rhodes L, Kurkjian K, et al. Diagnosis of acute pulmonary histoplasmosis by antigen detection. Clin Infect Dis 2009; 49:1878–1882.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Retallack DM, Woods JP. Molecular epidemiology, pathogenesis, and genetics of the dimorphic fungus Histoplasma capsulatum. Microbes Infect 1999; 1:817–825.
- Deepe GS. The immune response to Histoplasma capsulatum: unearthing its secrets. J Lab Clin Med 1994; 123:201–205.
- Goodwin RA, Shapiro JL, Thurman GH, Thurman SS, Des Prez RM. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore) 1980; 59:1–33.
- Wheat LJ, Connolly-Stringfield PA, Baker RL, et al. Disseminated histoplasmosis in the acquired immune deficiency syndrome: clinical findings, diagnosis and treatment, and review of the literature. Medicine (Baltimore) 1990; 69:361–374.
- Rubin H, Furcolow ML, Yates JL, Brasher CA. The course and prognosis of histoplasmosis. Am J Med 1959; 27:278–288.
- Wheat J, MaWhinney S, Hafner R, et al. Treatment of histoplasmosis with fluconazole in patients with acquired immunodeficiency syndrome. National Institute of Allergy and Infectious Diseases Acquired Immunodeficiency Syndrome Clinical Trials Group and Mycoses Study Group. Am J Med 1997; 103:223–232.
- McKinsey DS, Kauffman CA, Pappas PG, et al. Fluconazole therapy for histoplasmosis. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Clin Infect Dis 1996; 23:996–1001.
- Slama TG. Treatment of disseminated and progressive cavitary histoplasmosis with ketoconazole. Am J Med 1983; 74:70–73.
- Treatment of blastomycosis and histoplasmosis with ketoconazole. Results of a prospective randomized clinical trial. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Ann Intern Med 1985; 103:861–872.
- Clarke V, Weston-Smith S. Severe folate-deficiency pancytopenia. BMJ Case Reports 2010; published online.
- Anthony AC. Megaloblastic anemias. In:Hoffman R, Benz EJ, Shattil SJ, Furie B, Cohebn HJ, Silberstein LE, editors. Hematology: Basic Principles and Practice, 2nd ed. New York, NY: Churchill Livingston, 1995:552–586.
- Macrae FA, St John DJ. Relationship between patterns of bleeding and Hemoccult sensitivity in patients with colorectal cancers or adenomas. Gastroenterology 1982; 82:891–898.
- Meyers CA, Albitar M, Estey E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer 2005; 104:788–793.
- Parker CJ. Bone marrow failure syndromes: paroxysmal nocturnal hemoglobinuria. Hematol Oncol Clin North Am 2009; 23:333–346.
- Young NS, Maciejewski JP, Sloand E, et al. The relationship of aplastic anemia and PNH. Int J Hematol 2002; 76(suppl 2):168–172.
- Rosse W. A new way to prevent thrombosis? Blood 2007; 110:3821.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Meals LT, McKinney WP. Acute pulmonary histoplasmosis: progressive pneumonia resulting from high inoculum exposure. J Ky Med Assoc 1998; 96:258–260.
- Salomon J, Flament Saillour M, De Truchis P, et al. An outbreak of acute pulmonary histoplasmosis in members of a trekking trip in Martinique, French West Indies. J Travel Med 2003; 10:87–93.
- Joseph Wheat L. Current diagnosis of histoplasmosis. Trends Microbiol 2003; 11:488–494.
- Wheat LJ, Kauffman CA. Histoplasmosis. Infect Dis Clin North Am 2003; 17:1–19.
- Swartzentruber S, Rhodes L, Kurkjian K, et al. Diagnosis of acute pulmonary histoplasmosis by antigen detection. Clin Infect Dis 2009; 49:1878–1882.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Retallack DM, Woods JP. Molecular epidemiology, pathogenesis, and genetics of the dimorphic fungus Histoplasma capsulatum. Microbes Infect 1999; 1:817–825.
- Deepe GS. The immune response to Histoplasma capsulatum: unearthing its secrets. J Lab Clin Med 1994; 123:201–205.
- Goodwin RA, Shapiro JL, Thurman GH, Thurman SS, Des Prez RM. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore) 1980; 59:1–33.
- Wheat LJ, Connolly-Stringfield PA, Baker RL, et al. Disseminated histoplasmosis in the acquired immune deficiency syndrome: clinical findings, diagnosis and treatment, and review of the literature. Medicine (Baltimore) 1990; 69:361–374.
- Rubin H, Furcolow ML, Yates JL, Brasher CA. The course and prognosis of histoplasmosis. Am J Med 1959; 27:278–288.
- Wheat J, MaWhinney S, Hafner R, et al. Treatment of histoplasmosis with fluconazole in patients with acquired immunodeficiency syndrome. National Institute of Allergy and Infectious Diseases Acquired Immunodeficiency Syndrome Clinical Trials Group and Mycoses Study Group. Am J Med 1997; 103:223–232.
- McKinsey DS, Kauffman CA, Pappas PG, et al. Fluconazole therapy for histoplasmosis. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Clin Infect Dis 1996; 23:996–1001.
- Slama TG. Treatment of disseminated and progressive cavitary histoplasmosis with ketoconazole. Am J Med 1983; 74:70–73.
- Treatment of blastomycosis and histoplasmosis with ketoconazole. Results of a prospective randomized clinical trial. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Ann Intern Med 1985; 103:861–872.