User login
An unusual cause of bruising
A 61-year-old woman presented to our hematology clinic for evaluation of multiple episodes of bruising. The first episode occurred 8 months earlier, when she developed a large bruise after water skiing. Two months before coming to us, she went to her local emergency room because of new bruising and was found to have a prolonged activated partial thromboplastin time (aPTT) of 60 seconds (reference range 23.3–34.9), but she underwent no further testing at that time.
At presentation to our clinic, she reported having no fevers, night sweats, unintentional weight loss, swollen lymph nodes, joint pain, rashes, mouth sores, nosebleeds, or blood in the urine or stool. Her history was notable only for hypothyroidism, which was diagnosed in the previous year. Her medications included levothyroxine, vitamin D3, and vitamin C. She had been taking a baby aspirin daily for the past 10 years but had stopped 1 month earlier because of the bruising.

Ten years earlier she had been evaluated for a possible transient ischemic attack; laboratory results at that time included a normal aPTT of 25.1 seconds and a normal factor VIII level of 153% (reference range 50%–173%).


EVALUATION FOR AN ISOLATED PROLONGED aPTT
1. What is the appropriate next test to evaluate this patient’s prolonged aPTT?
- Lupus anticoagulant panel
- Coagulation factor levels
- Mixing studies
- Bethesda assay
Mixing studies
Once a prolonged aPTT is confirmed, the appropriate next step is a mixing study. This involves mixing the patient’s plasma with pooled normal plasma in a 1-to-1 ratio, then repeating the aPTT test immediately, and again after 1 hour of incubation at 37°C. If the patient does not have enough of one of the coagulation factors, the aPTT immediately returns to the normal range when plasma is mixed with the pooled plasma because the pooled plasma contains the factor that is lacking. If this happens, then factor assays should be performed to identify the deficient factor.1
Various antibodies that inhibit coagulation factors can also affect the aPTT. There are 2 general types: immediate-acting and delayed.
With an immediate-acting inhibitor, the aPTT does not correct into the normal range with initial mixing. Immediate-acting inhibitors are often seen together with lupus anticoagulants, which are nonspecific phospholipid antibodies. If an immediate-acting inhibitor is detected, further testing should focus on evaluation for lupus anticoagulant, including phospholipid-dependency studies.
With a delayed inhibitor, the aPTT initially comes down, but subsequently goes back up after incubation. Acquired factor VIII inhibitor is a classic delayed-type inhibitor and is also the most common factor inhibitor.1 If a delayed-acting inhibitor is found, specific intrinsic factor levels should be measured (factors VIII, IX, XI, and XII),2 and testing should also be done for lupus anticoagulant, as these inhibitors may occur together.
Bethesda assay

Case continued: Results of mixing and Bethesda studies

FACTOR VIII INHIBITOR EVALUATION
2. What is the most likely underlying condition associated with this patient’s factor VIII inhibitor?
- Autoimmune disease
- Malignancy
- A medication
- Unknown (idiopathic)
Acquired hemophilia A (AHA) is a rare disorder caused by autoantibodies against factor VIII. Its estimated incidence is about 1 person per million per year.4 It usually presents as unexplained bruising or bleeding and is only rarely diagnosed by an incidentally noted prolonged aPTT. The severity of bleeding is variable and can include subcutaneous, soft-tissue, retroperitoneal, gastrointestinal, and intracranial hemorrhage.5
AHA is considered idiopathic in more than half of cases. A study based on a European registry5 of 501 patients with AHA and a UK study6 of 172 patients found no underlying disease in 52% and 65% of patients, respectively. For patients with an identified cause, the most common causes were malignancy (12%5 and 15%6) and autoimmune disease (12%5 and 17%6).
Drugs have rarely been associated with factor VIII inhibitors. Such occurrences have been reported with interferon, blood thinners, antibiotics, and psychiatric medications, but no study yet has indicated causation. However, patients with congenital hemophilia A treated with factor VIII preparations have about a 15% chance of developing factor VIII inhibitors. In this setting, inhibitors develop in response to recombinant factor VIII exposure, unlike the autoimmune phenomena seen in AHA.
TREATMENT OF ACQUIRED HEMOPHILIA A
3. What is the most appropriate treatment for AHA?
- Desmopressin and prednisone
- Recombinant porcine factor VIII and prednisone plus cyclophosphamide
- Recombinant factor VIIa and rituximab
- Any of the above
Any of the above regimens can be used. In general, treatment of AHA has two purposes: to stop acute hemorrhage, and to reduce the level of factor VIII inhibitor. No standard treatment guidelines are available; evidence of the effectiveness of different drugs is based largely on data on congenital hemophilia A.3
Acute treatment to stop bleeding
Initial treatment of AHA often focuses on stopping an acute hemorrhage by either raising circulating levels of factor VIII or bypassing it in the coagulation cascade.
Desmopressin can temporarily raise factor VIII levels, but it is often ineffective in AHA unless the patient has very low inhibitor titers.3
Factor VIII concentrate (human or recombinant porcine factor VIII) may be effective in patients with low inhibitor titers (< 5 BU). Higher doses are often required than those used in congenital hemophilia A. Factor VIII concentrate is usually combined with immunosuppressive treatment to lower the factor VIII inhibitor level (described below).3
If these methods are ineffective or the patient has high inhibitor titers (> 5 BU), activated prothrombin complex concentrates, known as FEIBA (factor eight inhibitor bypassing activity), or recombinant factor VIIa is available. These agents bypass factor VIII in the clotting cascade.
Immunosuppression to reduce factor VIII inhibitor
Immunosuppressive agents are the mainstay of AHA treatment to lower the inhibitor level.
Regimens vary. A 2003 meta-analysis4 including 249 patients found that prednisone alone resulted in complete response in about 30% of patients, and the addition of cyclophosphamide increased the response rate to 60% to 100%. High-dose intravenous immunoglobulin led to conflicting results. Conclusions were limited by the variability of dosing and duration in treatment regimens among the 20 different studies included.
An analysis of 331 patients in the European Acquired Hemophilia Registry (EACH2)7 found that steroids alone produced remission in 48% of patients, while steroids combined with cyclophosphamide raised the rate to 70%. Rituximab-based regimens were successful in 59% but required twice as long to achieve remission as steroid or cyclophosphamide-based regimens. No benefit was noted from intravenous immunoglobulin.
Risks of disease and treatment
AHA is associated with significant risk of morbidity and death related to bleeding, complications of treatment, and underlying disease.
In EACH2, 16 of the 331 patients died of bleeding, 16 died of causes related to immunosuppression, and 45 died of causes related to the underlying condition.5 In the UK registry of 172 patients, 13 patients died of bleeding, and 12 died of sepsis related to immunosuppression.6
The factor VIII level and inhibitor titer are not necessarily useful in stratifying bleeding risk, as severe and fatal bleeding can occur at variable levels and patients remain at risk of bleeding as long as the inhibitor persists.6,7
CASE CONTINUED: TREATMENT, LYMPHOCYTOSIS
The patient was started on 60 mg daily of prednisone, resulting in a decrease in her aPTT, increase in factor VIII level, and lower Bethesda titer. On a return visit, her absolute lymphocyte count was 7.04 × 109/L (reference range 1.0–4.0). She reported no fevers, chills, or recent infections.
EVALUATING LYMPHOCYTOSIS
Lymphocytosis is defined in most laboratories as an absolute lymphocyte count greater than 4.0 × 109/L for adults. Normally, T cells (CD3+) make up 60% to 80% of lymphocytes, B cells (CD20+) 10% to 20%, and natural killer (NK) cells (CD3–, CD56+) 5% to 10%. Lymphocytosis is usually caused by infection, but it can have other causes, including malignancy.
Peripheral blood smear. If there is no clear cause of lymphocytosis, a peripheral blood smear can be used to assess lymphocyte morphology, providing clues to the underlying etiology. For example, atypical lymphocytes are often seen in infectious mononucleosis, while “smudge” lymphocytes are characteristic of chronic lymphocytic leukemia. If a peripheral smear shows abnormal morphology, further workup should include establishing whether the lymphocytes are polyclonal or clonal.8
CASE CONTINUED: LARGE GRANULAR LYMPHOCYTES

4. What is the next step to evaluate the patient’s lymphocytosis?
- Bone marrow biopsy
- Karyotype analysis
- Flow cytometry
- Fluorescence in situ hybridization
Flow cytometry with V-beta analysis is the best first test to determine the cause of lymphocytosis after review of the peripheral smear. For persistent lymphocytosis, flow cytometry should be done even if a peripheral smear shows normal lymphocyte morphology.
Most T cells possess receptors composed of alpha and beta chains, each encoded by variable (V), diversity (D), joining (J), and constant (C) gene segments. The V, D, and J segments undergo rearrangement during T-cell development in the thymus based on antigen exposure, producing a diverse T-cell receptor population.
In a polyclonal population of lymphocytes, the T-cell receptors have a variety of gene segment arrangements, indicating normal T-cell development. But in a clonal population of lymphocytes, the T-cell receptors have a single identical gene segment arrangement, indicating they all originated from a single clone.9 Lymphocytosis in response to an infection is typically polyclonal, while malignant lymphocytosis is clonal.
Monoclonal antibodies against many of the variable regions of the beta chain (V-beta) of T-cell receptors have been developed, enabling flow cytometry to establish clonality.
T-cell receptor gene rearrangement studies can also be performed using polymerase chain reaction and Southern blot techniques.9
Karyotype analysis is usually not performed for the finding of LGLs, because most leukemias (eg, T-cell and NK-cell leukemias) have cells with a normal karyotype.
Bone marrow biopsy is invasive and usually not required to evaluate LGLs. It can be especially risky for a patient with a bleeding disorder such as a factor VIII inhibitor.10
Case continued: Flow cytometry confirms clonality
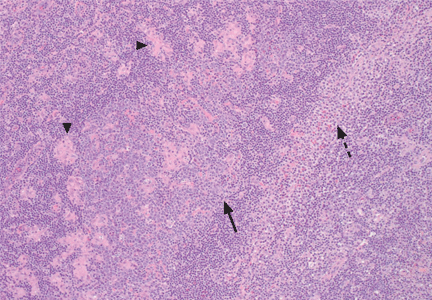
Subsequent flow cytometry found that more than 50% of the patient’s lymphocytes were LGLs that co-expressed CD3+, CD8+, CD56+, and CD57+, with aberrantly decreased CD7 expression. T-cell V-beta analysis demonstrated an expansion of the V-beta 17 family, and T-cell receptor gene analysis with polymerase chain reaction confirmed the presence of a clonal rearrangement.
LGL LEUKEMIA: CLASSIFICATION AND MANAGEMENT
LGLs normally account for 10% to 15% of peripheral mononuclear cells.11 LGL leukemia is caused by a clonal population of cytotoxic T cells or NK cells and involves an increased number of LGLs (usually > 2 × 109/L).10
LGL leukemia is divided into 3 categories according to the most recent World Health Organization classification10,12:
T-cell LGL leukemia (about 85% of cases) is considered indolent but can cause significant cytopenias and is often associated with autoimmune disease.13 Cells usually express a CD3+, CD8+, CD16+, and CD57+ phenotype. Survival is about 70% at 10 years.
Chronic NK-cell lymphocytosis (about 10%) also tends to have an indolent course with cytopenia and an autoimmune association, and with a similar prognosis to T-cell LGL leukemia. Cells express a CD3–, CD16+, and CD56+ phenotype.
Aggressive NK-cell LGL leukemia (about 5%) is associated with Epstein-Barr virus infection and occurs in younger patients. It is characterized by severe cytopenias, “B symptoms” (ie, fever, night sweats, weight loss), and has a very poor prognosis. Like chronic NK-cell lymphocytosis, cells express a CD3–, CD16+, and CD56+ phenotype. Fas (CD95) and Fas-ligand (CD178) are strongly expressed.10,13
Most cases of LGL leukemia can be diagnosed on the basis of classic morphology on peripheral blood smear and evidence of clonality on flow cytometry or gene rearrangement studies. T-cell receptor gene studies cannot be used to establish clonality in the NK subtypes, as NK cells do not express T-cell receptors.11
Case continued: Diagnosis, continued course
In our patient, T-cell LGL leukemia was diagnosed on the basis of the peripheral smear, flow cytometry results, and positive T-cell receptor gene studies for clonal rearrangement in the T-cell receptor beta region.
While her corticosteroid therapy was being tapered, her factor III inhibitor level increased, and she had a small episode of bleeding, prompting the start of cyclophosphamide 50 mg daily with lower doses of prednisone.

LGL LEUKEMIA AND AUTOIMMUNE DISEASE
Patients with LGL leukemia commonly have or develop autoimmune conditions. Immune-mediated cytopenias including pure red cell aplasia, aplastic anemia, and autoimmune hemolytic anemias can occur. Neutropenia, the most common cytopenia in LGL leukemia, is thought to be at least partly autoimmune, as the degree of neutropenia is often worse than would be expected solely from bone-marrow infiltration of LGL cells.10,14,15
Rheumatoid arthritis is the most common autoimmune condition associated with LGL leukemia, with a reported incidence between 11% and 36%.13–15
Felty syndrome (rheumatoid arthritis, splenomegaly, and neutropenia) is often associated with LGL leukemia and is thought by some to be part of the same disease process.15
Treat with immunosuppressives if needed
Indications for treating LGL leukemia include the development of cytopenias and associated autoimmune diseases. Immunosuppressive agents, such as methotrexate, cyclophosphamide, and cyclosporine, are commonly used.10,11,14 Most evidence of treatment efficacy is from retrospective studies and case reports, with widely variable response rates that overall are around 50%.10
ACQUIRED HEMOPHILIA A AND HEMATOLOGIC MALIGNANCY
A systematic review found 30 cases of AHA associated with hematologic malignancies.16 The largest case series17 in this analysis had 8 patients, and included diagnoses of chronic lymphocytic leukemia, erythroleukemia, myelofibrosis, multiple myeloma, and myelodysplastic syndrome. In 3 of these patients, the appearance of the inhibitor preceded the diagnosis of the underlying malignancy by an average of 3.5 months. In 1 patient with erythroleukemia and another with multiple myeloma, the activity of the inhibitor could be clearly correlated with the underlying malignancy. In the other 6 patients, no association between the two could be made.
In the same series, complete resolution of the inhibitor was related only to the level of Bethesda titer present at diagnosis, with those who achieved resolution having lower mean Bethesda titers.17 Similarly, in EACH2, lower inhibitor Bethesda titers and higher factor VIII levels at presentation were associated with faster inhibitor eradication and normalization of factor VIII levels.7
Murphy et al18 described a 62-year-old woman with Felty syndrome who developed a factor VIII inhibitor and was subsequently given a diagnosis of LGL leukemia. Treatment with immunosuppressive agents, including cyclophosphamide, azathioprine, and rituximab, successfully eradicated her factor VIII inhibitor, although the LGL leukemia persisted.
Case conclusion: Eradication of factor VIII inhibitor
Our patient, similar to the patient described by Murphy et al18 above, had eradication of the factor VIII inhibitor despite persistence of LGL leukemia. Between the time of diagnosis at our clinic, when she had 54% LGLs, and eradication of the inhibitor 3 months later, the LGL percentage ranged from 45% to 89%. No clear direct correlation between LGL and factor VIII inhibitor levels could be detected.
Given the strong association of LGL leukemia with autoimmune disease, it is tempting to believe that her factor VIII inhibitor was somehow related to her malignancy, although the exact mechanism remained unclear. The average age at diagnosis is 60 for LGL leukemia11 and over 70 for AHA,5,6 so advanced age may be the common denominator. Whether or not our patient will have recurrence of her factor VIII inhibitor or the development of other autoimmune diseases with the persistence of her LGL leukemia remains to be seen.
At last follow-up, our patient was off all therapy and continued to have normal aPTT and factor VIII levels. Repeat flow cytometry after treatment of her factor VIII inhibitor showed persistence of a clonal T-cell population, although reduced from 72% to 60%. It may be that the 2 entities were unrelated, and the clonal T-cell population was simply fluctuating over time. This can be determined only with further observation. As the patient had no symptoms from her LGL leukemia, she continued to be observed without treatment.
TAKE-HOME POINTS
- The coagulation assay is key to initially assessing a bleeding abnormality; whether the prothrombin time and aPTT are normal or prolonged narrows the differential diagnosis and determines next steps in evaluation.
- Mixing studies can help pinpoint the responsible deficient factor.
- Acquired factor VIII deficiency, also known as AHA, may be caused by autoimmune disease, malignancy, or medications, but it is usually idiopathic.
- AHA treatment is focused on achieving hemostasis and reducing factor VIII inhibitor.
- Lymphocytosis should be evaluated with a peripheral blood smear and flow cytometry to determine if the population is polyclonal (associated with infection) or clonal (associated with malignancy).
- LGL leukemia is usually a chronic, indolent disease, although an uncommon subtype has an aggressive course.
- The association between AHA and LGL leukemia is unclear, and both conditions must be monitored and managed.
- Kamal AH, Tefferi A, Pruthi RK. How to interpret and pursue an abnormal prothrombin time, activated partial thromboplastin time, and bleeding time in adults. Mayo Clin Proc 2007; 82(7):864–873. doi:10.4065/82.7.864
- Tcherniantchouk O, Laposata M, Marques MB. The isolated prolonged PTT. Am J Hematol 2013; 88(1):82–85. doi:10.1002/ajh.23285
- Ma AD, Carrizosa D. Acquired factor VIII inhibitors: pathophysiology and treatment. Hematology Am Soc Hematol Educ Program 2006:432–437. doi:10.1182/asheducation-2006.1.432
- Delgado J, Jimenez-Yuste V, Hernandez-Navarro F, Villar A. Acquired haemophilia: review and meta-analysis focused on therapy and prognostic factors. Br J Haematol 2003; 121(1):21–35. pmid:12670328
- Knoebl P, Marco P, Baudo F, et al; EACH2 Registry Contributors. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost 2012; 10(4):622–631. doi:10.1111/j.1538-7836.2012.04654.x
- Collins PW, Hirsch S, Baglin TP, et al; UK Haemophilia Centre Doctors’ Organisation. Acquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood 2007; 109(5):1870–1877. doi:10.1182/blood-2006-06-029850
- Collins P, Baudo F, Knoebl P, et al; EACH2 Registry Collaborators. Immunosuppression for acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). Blood 2012; 120(1):47–55. doi:10.1182/blood-2012-02-409185
- George TI. Malignant or benign leukocytosis. Hematology Am Soc Hematol Educ Program 2012; 2012:475–484. doi:10.1182/asheducation-2012.1.475
- Watters RJ, Liu X, Loughran TP Jr. T-cell and natural killer-cell large granular lymphocyte leukemia neoplasias. Leuk Lymphoma 2011; 52(12):2217–2225. doi:10.3109/10428194.2011.593276
- Lamy T, Moignet A, Loughran TP Jr. LGL leukemia: from pathogenesis to treatment. Blood 2017; 129(9):1082–1094. doi:10.1182/blood-2016-08-692590
- Zhang D, Loughran TP Jr. Large granular lymphocytic leukemia: molecular pathogenesis, clinical manifestations, and treatment. Hematology Am Soc Hematol Educ Program 2012; 2012:652–659. doi:10.1182/asheducation-2012.1.652
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016; 127(20):2375–2390. doi:10.1182/blood-2016-01-643569
- Rose MG, Berliner N. T-cell large granular lymphocyte leukemia and related disorders. Oncologist 2004; 9(3):247–258. pmid:15169980
- Bockorny B, Dasanu CA. Autoimmune manifestations in large granular lymphocyte leukemia. Clin Lymphoma Myeloma Leuk 2012; 12(6):400–405. doi:10.1016/j.clml.2012.06.006
- Liu X, Loughran TP Jr. The spectrum of large granular lymphocyte leukemia and Felty’s syndrome. Curr Opin Hematol 2011; 18(4):254–259. doi:10.1097/MOH.0b013e32834760fb
- Franchini M, Lippi G. Acquired factor V inhibitors: a systematic review. J Thromb Thrombolysis 2011; 31(4):449–457. doi:10.1007/s11239-010-0529-6
- Sallah S, Nguyen NP, Abdallah JM, Hanrahan LR. Acquired hemophilia in patients with hematologic malignancies. Arch Pathol Lab Med 2000; 124(5):730–734.
- Murphy PW, Brett LK, Verla-Tebit E, Macik BG, Loughran TP Jr. Acquired inhibitors to factor VIII and fibrinogen in the setting of T-cell large granular lymphocyte leukemia: a case report and review of the literature. Blood Coagul Fibrinolysis 2015; 26(2):211–213. doi:10.1097/MBC.0000000000000209
A 61-year-old woman presented to our hematology clinic for evaluation of multiple episodes of bruising. The first episode occurred 8 months earlier, when she developed a large bruise after water skiing. Two months before coming to us, she went to her local emergency room because of new bruising and was found to have a prolonged activated partial thromboplastin time (aPTT) of 60 seconds (reference range 23.3–34.9), but she underwent no further testing at that time.
At presentation to our clinic, she reported having no fevers, night sweats, unintentional weight loss, swollen lymph nodes, joint pain, rashes, mouth sores, nosebleeds, or blood in the urine or stool. Her history was notable only for hypothyroidism, which was diagnosed in the previous year. Her medications included levothyroxine, vitamin D3, and vitamin C. She had been taking a baby aspirin daily for the past 10 years but had stopped 1 month earlier because of the bruising.
Ten years earlier she had been evaluated for a possible transient ischemic attack; laboratory results at that time included a normal aPTT of 25.1 seconds and a normal factor VIII level of 153% (reference range 50%–173%).
EVALUATION FOR AN ISOLATED PROLONGED aPTT
1. What is the appropriate next test to evaluate this patient’s prolonged aPTT?
- Lupus anticoagulant panel
- Coagulation factor levels
- Mixing studies
- Bethesda assay
Mixing studies
Once a prolonged aPTT is confirmed, the appropriate next step is a mixing study. This involves mixing the patient’s plasma with pooled normal plasma in a 1-to-1 ratio, then repeating the aPTT test immediately, and again after 1 hour of incubation at 37°C. If the patient does not have enough of one of the coagulation factors, the aPTT immediately returns to the normal range when plasma is mixed with the pooled plasma because the pooled plasma contains the factor that is lacking. If this happens, then factor assays should be performed to identify the deficient factor.1
Various antibodies that inhibit coagulation factors can also affect the aPTT. There are 2 general types: immediate-acting and delayed.
With an immediate-acting inhibitor, the aPTT does not correct into the normal range with initial mixing. Immediate-acting inhibitors are often seen together with lupus anticoagulants, which are nonspecific phospholipid antibodies. If an immediate-acting inhibitor is detected, further testing should focus on evaluation for lupus anticoagulant, including phospholipid-dependency studies.
With a delayed inhibitor, the aPTT initially comes down, but subsequently goes back up after incubation. Acquired factor VIII inhibitor is a classic delayed-type inhibitor and is also the most common factor inhibitor.1 If a delayed-acting inhibitor is found, specific intrinsic factor levels should be measured (factors VIII, IX, XI, and XII),2 and testing should also be done for lupus anticoagulant, as these inhibitors may occur together.
Bethesda assay
Case continued: Results of mixing and Bethesda studies
FACTOR VIII INHIBITOR EVALUATION
2. What is the most likely underlying condition associated with this patient’s factor VIII inhibitor?
- Autoimmune disease
- Malignancy
- A medication
- Unknown (idiopathic)
Acquired hemophilia A (AHA) is a rare disorder caused by autoantibodies against factor VIII. Its estimated incidence is about 1 person per million per year.4 It usually presents as unexplained bruising or bleeding and is only rarely diagnosed by an incidentally noted prolonged aPTT. The severity of bleeding is variable and can include subcutaneous, soft-tissue, retroperitoneal, gastrointestinal, and intracranial hemorrhage.5
AHA is considered idiopathic in more than half of cases. A study based on a European registry5 of 501 patients with AHA and a UK study6 of 172 patients found no underlying disease in 52% and 65% of patients, respectively. For patients with an identified cause, the most common causes were malignancy (12%5 and 15%6) and autoimmune disease (12%5 and 17%6).
Drugs have rarely been associated with factor VIII inhibitors. Such occurrences have been reported with interferon, blood thinners, antibiotics, and psychiatric medications, but no study yet has indicated causation. However, patients with congenital hemophilia A treated with factor VIII preparations have about a 15% chance of developing factor VIII inhibitors. In this setting, inhibitors develop in response to recombinant factor VIII exposure, unlike the autoimmune phenomena seen in AHA.
TREATMENT OF ACQUIRED HEMOPHILIA A
3. What is the most appropriate treatment for AHA?
- Desmopressin and prednisone
- Recombinant porcine factor VIII and prednisone plus cyclophosphamide
- Recombinant factor VIIa and rituximab
- Any of the above
Any of the above regimens can be used. In general, treatment of AHA has two purposes: to stop acute hemorrhage, and to reduce the level of factor VIII inhibitor. No standard treatment guidelines are available; evidence of the effectiveness of different drugs is based largely on data on congenital hemophilia A.3
Acute treatment to stop bleeding
Initial treatment of AHA often focuses on stopping an acute hemorrhage by either raising circulating levels of factor VIII or bypassing it in the coagulation cascade.
Desmopressin can temporarily raise factor VIII levels, but it is often ineffective in AHA unless the patient has very low inhibitor titers.3
Factor VIII concentrate (human or recombinant porcine factor VIII) may be effective in patients with low inhibitor titers (< 5 BU). Higher doses are often required than those used in congenital hemophilia A. Factor VIII concentrate is usually combined with immunosuppressive treatment to lower the factor VIII inhibitor level (described below).3
If these methods are ineffective or the patient has high inhibitor titers (> 5 BU), activated prothrombin complex concentrates, known as FEIBA (factor eight inhibitor bypassing activity), or recombinant factor VIIa is available. These agents bypass factor VIII in the clotting cascade.
Immunosuppression to reduce factor VIII inhibitor
Immunosuppressive agents are the mainstay of AHA treatment to lower the inhibitor level.
Regimens vary. A 2003 meta-analysis4 including 249 patients found that prednisone alone resulted in complete response in about 30% of patients, and the addition of cyclophosphamide increased the response rate to 60% to 100%. High-dose intravenous immunoglobulin led to conflicting results. Conclusions were limited by the variability of dosing and duration in treatment regimens among the 20 different studies included.
An analysis of 331 patients in the European Acquired Hemophilia Registry (EACH2)7 found that steroids alone produced remission in 48% of patients, while steroids combined with cyclophosphamide raised the rate to 70%. Rituximab-based regimens were successful in 59% but required twice as long to achieve remission as steroid or cyclophosphamide-based regimens. No benefit was noted from intravenous immunoglobulin.
Risks of disease and treatment
AHA is associated with significant risk of morbidity and death related to bleeding, complications of treatment, and underlying disease.
In EACH2, 16 of the 331 patients died of bleeding, 16 died of causes related to immunosuppression, and 45 died of causes related to the underlying condition.5 In the UK registry of 172 patients, 13 patients died of bleeding, and 12 died of sepsis related to immunosuppression.6
The factor VIII level and inhibitor titer are not necessarily useful in stratifying bleeding risk, as severe and fatal bleeding can occur at variable levels and patients remain at risk of bleeding as long as the inhibitor persists.6,7
CASE CONTINUED: TREATMENT, LYMPHOCYTOSIS
The patient was started on 60 mg daily of prednisone, resulting in a decrease in her aPTT, increase in factor VIII level, and lower Bethesda titer. On a return visit, her absolute lymphocyte count was 7.04 × 109/L (reference range 1.0–4.0). She reported no fevers, chills, or recent infections.
EVALUATING LYMPHOCYTOSIS
Lymphocytosis is defined in most laboratories as an absolute lymphocyte count greater than 4.0 × 109/L for adults. Normally, T cells (CD3+) make up 60% to 80% of lymphocytes, B cells (CD20+) 10% to 20%, and natural killer (NK) cells (CD3–, CD56+) 5% to 10%. Lymphocytosis is usually caused by infection, but it can have other causes, including malignancy.
Peripheral blood smear. If there is no clear cause of lymphocytosis, a peripheral blood smear can be used to assess lymphocyte morphology, providing clues to the underlying etiology. For example, atypical lymphocytes are often seen in infectious mononucleosis, while “smudge” lymphocytes are characteristic of chronic lymphocytic leukemia. If a peripheral smear shows abnormal morphology, further workup should include establishing whether the lymphocytes are polyclonal or clonal.8
CASE CONTINUED: LARGE GRANULAR LYMPHOCYTES
4. What is the next step to evaluate the patient’s lymphocytosis?
- Bone marrow biopsy
- Karyotype analysis
- Flow cytometry
- Fluorescence in situ hybridization
Flow cytometry with V-beta analysis is the best first test to determine the cause of lymphocytosis after review of the peripheral smear. For persistent lymphocytosis, flow cytometry should be done even if a peripheral smear shows normal lymphocyte morphology.
Most T cells possess receptors composed of alpha and beta chains, each encoded by variable (V), diversity (D), joining (J), and constant (C) gene segments. The V, D, and J segments undergo rearrangement during T-cell development in the thymus based on antigen exposure, producing a diverse T-cell receptor population.
In a polyclonal population of lymphocytes, the T-cell receptors have a variety of gene segment arrangements, indicating normal T-cell development. But in a clonal population of lymphocytes, the T-cell receptors have a single identical gene segment arrangement, indicating they all originated from a single clone.9 Lymphocytosis in response to an infection is typically polyclonal, while malignant lymphocytosis is clonal.
Monoclonal antibodies against many of the variable regions of the beta chain (V-beta) of T-cell receptors have been developed, enabling flow cytometry to establish clonality.
T-cell receptor gene rearrangement studies can also be performed using polymerase chain reaction and Southern blot techniques.9
Karyotype analysis is usually not performed for the finding of LGLs, because most leukemias (eg, T-cell and NK-cell leukemias) have cells with a normal karyotype.
Bone marrow biopsy is invasive and usually not required to evaluate LGLs. It can be especially risky for a patient with a bleeding disorder such as a factor VIII inhibitor.10
Case continued: Flow cytometry confirms clonality
Subsequent flow cytometry found that more than 50% of the patient’s lymphocytes were LGLs that co-expressed CD3+, CD8+, CD56+, and CD57+, with aberrantly decreased CD7 expression. T-cell V-beta analysis demonstrated an expansion of the V-beta 17 family, and T-cell receptor gene analysis with polymerase chain reaction confirmed the presence of a clonal rearrangement.
LGL LEUKEMIA: CLASSIFICATION AND MANAGEMENT
LGLs normally account for 10% to 15% of peripheral mononuclear cells.11 LGL leukemia is caused by a clonal population of cytotoxic T cells or NK cells and involves an increased number of LGLs (usually > 2 × 109/L).10
LGL leukemia is divided into 3 categories according to the most recent World Health Organization classification10,12:
T-cell LGL leukemia (about 85% of cases) is considered indolent but can cause significant cytopenias and is often associated with autoimmune disease.13 Cells usually express a CD3+, CD8+, CD16+, and CD57+ phenotype. Survival is about 70% at 10 years.
Chronic NK-cell lymphocytosis (about 10%) also tends to have an indolent course with cytopenia and an autoimmune association, and with a similar prognosis to T-cell LGL leukemia. Cells express a CD3–, CD16+, and CD56+ phenotype.
Aggressive NK-cell LGL leukemia (about 5%) is associated with Epstein-Barr virus infection and occurs in younger patients. It is characterized by severe cytopenias, “B symptoms” (ie, fever, night sweats, weight loss), and has a very poor prognosis. Like chronic NK-cell lymphocytosis, cells express a CD3–, CD16+, and CD56+ phenotype. Fas (CD95) and Fas-ligand (CD178) are strongly expressed.10,13
Most cases of LGL leukemia can be diagnosed on the basis of classic morphology on peripheral blood smear and evidence of clonality on flow cytometry or gene rearrangement studies. T-cell receptor gene studies cannot be used to establish clonality in the NK subtypes, as NK cells do not express T-cell receptors.11
Case continued: Diagnosis, continued course
In our patient, T-cell LGL leukemia was diagnosed on the basis of the peripheral smear, flow cytometry results, and positive T-cell receptor gene studies for clonal rearrangement in the T-cell receptor beta region.
While her corticosteroid therapy was being tapered, her factor III inhibitor level increased, and she had a small episode of bleeding, prompting the start of cyclophosphamide 50 mg daily with lower doses of prednisone.
LGL LEUKEMIA AND AUTOIMMUNE DISEASE
Patients with LGL leukemia commonly have or develop autoimmune conditions. Immune-mediated cytopenias including pure red cell aplasia, aplastic anemia, and autoimmune hemolytic anemias can occur. Neutropenia, the most common cytopenia in LGL leukemia, is thought to be at least partly autoimmune, as the degree of neutropenia is often worse than would be expected solely from bone-marrow infiltration of LGL cells.10,14,15
Rheumatoid arthritis is the most common autoimmune condition associated with LGL leukemia, with a reported incidence between 11% and 36%.13–15
Felty syndrome (rheumatoid arthritis, splenomegaly, and neutropenia) is often associated with LGL leukemia and is thought by some to be part of the same disease process.15
Treat with immunosuppressives if needed
Indications for treating LGL leukemia include the development of cytopenias and associated autoimmune diseases. Immunosuppressive agents, such as methotrexate, cyclophosphamide, and cyclosporine, are commonly used.10,11,14 Most evidence of treatment efficacy is from retrospective studies and case reports, with widely variable response rates that overall are around 50%.10
ACQUIRED HEMOPHILIA A AND HEMATOLOGIC MALIGNANCY
A systematic review found 30 cases of AHA associated with hematologic malignancies.16 The largest case series17 in this analysis had 8 patients, and included diagnoses of chronic lymphocytic leukemia, erythroleukemia, myelofibrosis, multiple myeloma, and myelodysplastic syndrome. In 3 of these patients, the appearance of the inhibitor preceded the diagnosis of the underlying malignancy by an average of 3.5 months. In 1 patient with erythroleukemia and another with multiple myeloma, the activity of the inhibitor could be clearly correlated with the underlying malignancy. In the other 6 patients, no association between the two could be made.
In the same series, complete resolution of the inhibitor was related only to the level of Bethesda titer present at diagnosis, with those who achieved resolution having lower mean Bethesda titers.17 Similarly, in EACH2, lower inhibitor Bethesda titers and higher factor VIII levels at presentation were associated with faster inhibitor eradication and normalization of factor VIII levels.7
Murphy et al18 described a 62-year-old woman with Felty syndrome who developed a factor VIII inhibitor and was subsequently given a diagnosis of LGL leukemia. Treatment with immunosuppressive agents, including cyclophosphamide, azathioprine, and rituximab, successfully eradicated her factor VIII inhibitor, although the LGL leukemia persisted.
Case conclusion: Eradication of factor VIII inhibitor
Our patient, similar to the patient described by Murphy et al18 above, had eradication of the factor VIII inhibitor despite persistence of LGL leukemia. Between the time of diagnosis at our clinic, when she had 54% LGLs, and eradication of the inhibitor 3 months later, the LGL percentage ranged from 45% to 89%. No clear direct correlation between LGL and factor VIII inhibitor levels could be detected.
Given the strong association of LGL leukemia with autoimmune disease, it is tempting to believe that her factor VIII inhibitor was somehow related to her malignancy, although the exact mechanism remained unclear. The average age at diagnosis is 60 for LGL leukemia11 and over 70 for AHA,5,6 so advanced age may be the common denominator. Whether or not our patient will have recurrence of her factor VIII inhibitor or the development of other autoimmune diseases with the persistence of her LGL leukemia remains to be seen.
At last follow-up, our patient was off all therapy and continued to have normal aPTT and factor VIII levels. Repeat flow cytometry after treatment of her factor VIII inhibitor showed persistence of a clonal T-cell population, although reduced from 72% to 60%. It may be that the 2 entities were unrelated, and the clonal T-cell population was simply fluctuating over time. This can be determined only with further observation. As the patient had no symptoms from her LGL leukemia, she continued to be observed without treatment.
TAKE-HOME POINTS
- The coagulation assay is key to initially assessing a bleeding abnormality; whether the prothrombin time and aPTT are normal or prolonged narrows the differential diagnosis and determines next steps in evaluation.
- Mixing studies can help pinpoint the responsible deficient factor.
- Acquired factor VIII deficiency, also known as AHA, may be caused by autoimmune disease, malignancy, or medications, but it is usually idiopathic.
- AHA treatment is focused on achieving hemostasis and reducing factor VIII inhibitor.
- Lymphocytosis should be evaluated with a peripheral blood smear and flow cytometry to determine if the population is polyclonal (associated with infection) or clonal (associated with malignancy).
- LGL leukemia is usually a chronic, indolent disease, although an uncommon subtype has an aggressive course.
- The association between AHA and LGL leukemia is unclear, and both conditions must be monitored and managed.
A 61-year-old woman presented to our hematology clinic for evaluation of multiple episodes of bruising. The first episode occurred 8 months earlier, when she developed a large bruise after water skiing. Two months before coming to us, she went to her local emergency room because of new bruising and was found to have a prolonged activated partial thromboplastin time (aPTT) of 60 seconds (reference range 23.3–34.9), but she underwent no further testing at that time.
At presentation to our clinic, she reported having no fevers, night sweats, unintentional weight loss, swollen lymph nodes, joint pain, rashes, mouth sores, nosebleeds, or blood in the urine or stool. Her history was notable only for hypothyroidism, which was diagnosed in the previous year. Her medications included levothyroxine, vitamin D3, and vitamin C. She had been taking a baby aspirin daily for the past 10 years but had stopped 1 month earlier because of the bruising.
Ten years earlier she had been evaluated for a possible transient ischemic attack; laboratory results at that time included a normal aPTT of 25.1 seconds and a normal factor VIII level of 153% (reference range 50%–173%).
EVALUATION FOR AN ISOLATED PROLONGED aPTT
1. What is the appropriate next test to evaluate this patient’s prolonged aPTT?
- Lupus anticoagulant panel
- Coagulation factor levels
- Mixing studies
- Bethesda assay
Mixing studies
Once a prolonged aPTT is confirmed, the appropriate next step is a mixing study. This involves mixing the patient’s plasma with pooled normal plasma in a 1-to-1 ratio, then repeating the aPTT test immediately, and again after 1 hour of incubation at 37°C. If the patient does not have enough of one of the coagulation factors, the aPTT immediately returns to the normal range when plasma is mixed with the pooled plasma because the pooled plasma contains the factor that is lacking. If this happens, then factor assays should be performed to identify the deficient factor.1
Various antibodies that inhibit coagulation factors can also affect the aPTT. There are 2 general types: immediate-acting and delayed.
With an immediate-acting inhibitor, the aPTT does not correct into the normal range with initial mixing. Immediate-acting inhibitors are often seen together with lupus anticoagulants, which are nonspecific phospholipid antibodies. If an immediate-acting inhibitor is detected, further testing should focus on evaluation for lupus anticoagulant, including phospholipid-dependency studies.
With a delayed inhibitor, the aPTT initially comes down, but subsequently goes back up after incubation. Acquired factor VIII inhibitor is a classic delayed-type inhibitor and is also the most common factor inhibitor.1 If a delayed-acting inhibitor is found, specific intrinsic factor levels should be measured (factors VIII, IX, XI, and XII),2 and testing should also be done for lupus anticoagulant, as these inhibitors may occur together.
Bethesda assay
Case continued: Results of mixing and Bethesda studies
FACTOR VIII INHIBITOR EVALUATION
2. What is the most likely underlying condition associated with this patient’s factor VIII inhibitor?
- Autoimmune disease
- Malignancy
- A medication
- Unknown (idiopathic)
Acquired hemophilia A (AHA) is a rare disorder caused by autoantibodies against factor VIII. Its estimated incidence is about 1 person per million per year.4 It usually presents as unexplained bruising or bleeding and is only rarely diagnosed by an incidentally noted prolonged aPTT. The severity of bleeding is variable and can include subcutaneous, soft-tissue, retroperitoneal, gastrointestinal, and intracranial hemorrhage.5
AHA is considered idiopathic in more than half of cases. A study based on a European registry5 of 501 patients with AHA and a UK study6 of 172 patients found no underlying disease in 52% and 65% of patients, respectively. For patients with an identified cause, the most common causes were malignancy (12%5 and 15%6) and autoimmune disease (12%5 and 17%6).
Drugs have rarely been associated with factor VIII inhibitors. Such occurrences have been reported with interferon, blood thinners, antibiotics, and psychiatric medications, but no study yet has indicated causation. However, patients with congenital hemophilia A treated with factor VIII preparations have about a 15% chance of developing factor VIII inhibitors. In this setting, inhibitors develop in response to recombinant factor VIII exposure, unlike the autoimmune phenomena seen in AHA.
TREATMENT OF ACQUIRED HEMOPHILIA A
3. What is the most appropriate treatment for AHA?
- Desmopressin and prednisone
- Recombinant porcine factor VIII and prednisone plus cyclophosphamide
- Recombinant factor VIIa and rituximab
- Any of the above
Any of the above regimens can be used. In general, treatment of AHA has two purposes: to stop acute hemorrhage, and to reduce the level of factor VIII inhibitor. No standard treatment guidelines are available; evidence of the effectiveness of different drugs is based largely on data on congenital hemophilia A.3
Acute treatment to stop bleeding
Initial treatment of AHA often focuses on stopping an acute hemorrhage by either raising circulating levels of factor VIII or bypassing it in the coagulation cascade.
Desmopressin can temporarily raise factor VIII levels, but it is often ineffective in AHA unless the patient has very low inhibitor titers.3
Factor VIII concentrate (human or recombinant porcine factor VIII) may be effective in patients with low inhibitor titers (< 5 BU). Higher doses are often required than those used in congenital hemophilia A. Factor VIII concentrate is usually combined with immunosuppressive treatment to lower the factor VIII inhibitor level (described below).3
If these methods are ineffective or the patient has high inhibitor titers (> 5 BU), activated prothrombin complex concentrates, known as FEIBA (factor eight inhibitor bypassing activity), or recombinant factor VIIa is available. These agents bypass factor VIII in the clotting cascade.
Immunosuppression to reduce factor VIII inhibitor
Immunosuppressive agents are the mainstay of AHA treatment to lower the inhibitor level.
Regimens vary. A 2003 meta-analysis4 including 249 patients found that prednisone alone resulted in complete response in about 30% of patients, and the addition of cyclophosphamide increased the response rate to 60% to 100%. High-dose intravenous immunoglobulin led to conflicting results. Conclusions were limited by the variability of dosing and duration in treatment regimens among the 20 different studies included.
An analysis of 331 patients in the European Acquired Hemophilia Registry (EACH2)7 found that steroids alone produced remission in 48% of patients, while steroids combined with cyclophosphamide raised the rate to 70%. Rituximab-based regimens were successful in 59% but required twice as long to achieve remission as steroid or cyclophosphamide-based regimens. No benefit was noted from intravenous immunoglobulin.
Risks of disease and treatment
AHA is associated with significant risk of morbidity and death related to bleeding, complications of treatment, and underlying disease.
In EACH2, 16 of the 331 patients died of bleeding, 16 died of causes related to immunosuppression, and 45 died of causes related to the underlying condition.5 In the UK registry of 172 patients, 13 patients died of bleeding, and 12 died of sepsis related to immunosuppression.6
The factor VIII level and inhibitor titer are not necessarily useful in stratifying bleeding risk, as severe and fatal bleeding can occur at variable levels and patients remain at risk of bleeding as long as the inhibitor persists.6,7
CASE CONTINUED: TREATMENT, LYMPHOCYTOSIS
The patient was started on 60 mg daily of prednisone, resulting in a decrease in her aPTT, increase in factor VIII level, and lower Bethesda titer. On a return visit, her absolute lymphocyte count was 7.04 × 109/L (reference range 1.0–4.0). She reported no fevers, chills, or recent infections.
EVALUATING LYMPHOCYTOSIS
Lymphocytosis is defined in most laboratories as an absolute lymphocyte count greater than 4.0 × 109/L for adults. Normally, T cells (CD3+) make up 60% to 80% of lymphocytes, B cells (CD20+) 10% to 20%, and natural killer (NK) cells (CD3–, CD56+) 5% to 10%. Lymphocytosis is usually caused by infection, but it can have other causes, including malignancy.
Peripheral blood smear. If there is no clear cause of lymphocytosis, a peripheral blood smear can be used to assess lymphocyte morphology, providing clues to the underlying etiology. For example, atypical lymphocytes are often seen in infectious mononucleosis, while “smudge” lymphocytes are characteristic of chronic lymphocytic leukemia. If a peripheral smear shows abnormal morphology, further workup should include establishing whether the lymphocytes are polyclonal or clonal.8
CASE CONTINUED: LARGE GRANULAR LYMPHOCYTES
4. What is the next step to evaluate the patient’s lymphocytosis?
- Bone marrow biopsy
- Karyotype analysis
- Flow cytometry
- Fluorescence in situ hybridization
Flow cytometry with V-beta analysis is the best first test to determine the cause of lymphocytosis after review of the peripheral smear. For persistent lymphocytosis, flow cytometry should be done even if a peripheral smear shows normal lymphocyte morphology.
Most T cells possess receptors composed of alpha and beta chains, each encoded by variable (V), diversity (D), joining (J), and constant (C) gene segments. The V, D, and J segments undergo rearrangement during T-cell development in the thymus based on antigen exposure, producing a diverse T-cell receptor population.
In a polyclonal population of lymphocytes, the T-cell receptors have a variety of gene segment arrangements, indicating normal T-cell development. But in a clonal population of lymphocytes, the T-cell receptors have a single identical gene segment arrangement, indicating they all originated from a single clone.9 Lymphocytosis in response to an infection is typically polyclonal, while malignant lymphocytosis is clonal.
Monoclonal antibodies against many of the variable regions of the beta chain (V-beta) of T-cell receptors have been developed, enabling flow cytometry to establish clonality.
T-cell receptor gene rearrangement studies can also be performed using polymerase chain reaction and Southern blot techniques.9
Karyotype analysis is usually not performed for the finding of LGLs, because most leukemias (eg, T-cell and NK-cell leukemias) have cells with a normal karyotype.
Bone marrow biopsy is invasive and usually not required to evaluate LGLs. It can be especially risky for a patient with a bleeding disorder such as a factor VIII inhibitor.10
Case continued: Flow cytometry confirms clonality
Subsequent flow cytometry found that more than 50% of the patient’s lymphocytes were LGLs that co-expressed CD3+, CD8+, CD56+, and CD57+, with aberrantly decreased CD7 expression. T-cell V-beta analysis demonstrated an expansion of the V-beta 17 family, and T-cell receptor gene analysis with polymerase chain reaction confirmed the presence of a clonal rearrangement.
LGL LEUKEMIA: CLASSIFICATION AND MANAGEMENT
LGLs normally account for 10% to 15% of peripheral mononuclear cells.11 LGL leukemia is caused by a clonal population of cytotoxic T cells or NK cells and involves an increased number of LGLs (usually > 2 × 109/L).10
LGL leukemia is divided into 3 categories according to the most recent World Health Organization classification10,12:
T-cell LGL leukemia (about 85% of cases) is considered indolent but can cause significant cytopenias and is often associated with autoimmune disease.13 Cells usually express a CD3+, CD8+, CD16+, and CD57+ phenotype. Survival is about 70% at 10 years.
Chronic NK-cell lymphocytosis (about 10%) also tends to have an indolent course with cytopenia and an autoimmune association, and with a similar prognosis to T-cell LGL leukemia. Cells express a CD3–, CD16+, and CD56+ phenotype.
Aggressive NK-cell LGL leukemia (about 5%) is associated with Epstein-Barr virus infection and occurs in younger patients. It is characterized by severe cytopenias, “B symptoms” (ie, fever, night sweats, weight loss), and has a very poor prognosis. Like chronic NK-cell lymphocytosis, cells express a CD3–, CD16+, and CD56+ phenotype. Fas (CD95) and Fas-ligand (CD178) are strongly expressed.10,13
Most cases of LGL leukemia can be diagnosed on the basis of classic morphology on peripheral blood smear and evidence of clonality on flow cytometry or gene rearrangement studies. T-cell receptor gene studies cannot be used to establish clonality in the NK subtypes, as NK cells do not express T-cell receptors.11
Case continued: Diagnosis, continued course
In our patient, T-cell LGL leukemia was diagnosed on the basis of the peripheral smear, flow cytometry results, and positive T-cell receptor gene studies for clonal rearrangement in the T-cell receptor beta region.
While her corticosteroid therapy was being tapered, her factor III inhibitor level increased, and she had a small episode of bleeding, prompting the start of cyclophosphamide 50 mg daily with lower doses of prednisone.
LGL LEUKEMIA AND AUTOIMMUNE DISEASE
Patients with LGL leukemia commonly have or develop autoimmune conditions. Immune-mediated cytopenias including pure red cell aplasia, aplastic anemia, and autoimmune hemolytic anemias can occur. Neutropenia, the most common cytopenia in LGL leukemia, is thought to be at least partly autoimmune, as the degree of neutropenia is often worse than would be expected solely from bone-marrow infiltration of LGL cells.10,14,15
Rheumatoid arthritis is the most common autoimmune condition associated with LGL leukemia, with a reported incidence between 11% and 36%.13–15
Felty syndrome (rheumatoid arthritis, splenomegaly, and neutropenia) is often associated with LGL leukemia and is thought by some to be part of the same disease process.15
Treat with immunosuppressives if needed
Indications for treating LGL leukemia include the development of cytopenias and associated autoimmune diseases. Immunosuppressive agents, such as methotrexate, cyclophosphamide, and cyclosporine, are commonly used.10,11,14 Most evidence of treatment efficacy is from retrospective studies and case reports, with widely variable response rates that overall are around 50%.10
ACQUIRED HEMOPHILIA A AND HEMATOLOGIC MALIGNANCY
A systematic review found 30 cases of AHA associated with hematologic malignancies.16 The largest case series17 in this analysis had 8 patients, and included diagnoses of chronic lymphocytic leukemia, erythroleukemia, myelofibrosis, multiple myeloma, and myelodysplastic syndrome. In 3 of these patients, the appearance of the inhibitor preceded the diagnosis of the underlying malignancy by an average of 3.5 months. In 1 patient with erythroleukemia and another with multiple myeloma, the activity of the inhibitor could be clearly correlated with the underlying malignancy. In the other 6 patients, no association between the two could be made.
In the same series, complete resolution of the inhibitor was related only to the level of Bethesda titer present at diagnosis, with those who achieved resolution having lower mean Bethesda titers.17 Similarly, in EACH2, lower inhibitor Bethesda titers and higher factor VIII levels at presentation were associated with faster inhibitor eradication and normalization of factor VIII levels.7
Murphy et al18 described a 62-year-old woman with Felty syndrome who developed a factor VIII inhibitor and was subsequently given a diagnosis of LGL leukemia. Treatment with immunosuppressive agents, including cyclophosphamide, azathioprine, and rituximab, successfully eradicated her factor VIII inhibitor, although the LGL leukemia persisted.
Case conclusion: Eradication of factor VIII inhibitor
Our patient, similar to the patient described by Murphy et al18 above, had eradication of the factor VIII inhibitor despite persistence of LGL leukemia. Between the time of diagnosis at our clinic, when she had 54% LGLs, and eradication of the inhibitor 3 months later, the LGL percentage ranged from 45% to 89%. No clear direct correlation between LGL and factor VIII inhibitor levels could be detected.
Given the strong association of LGL leukemia with autoimmune disease, it is tempting to believe that her factor VIII inhibitor was somehow related to her malignancy, although the exact mechanism remained unclear. The average age at diagnosis is 60 for LGL leukemia11 and over 70 for AHA,5,6 so advanced age may be the common denominator. Whether or not our patient will have recurrence of her factor VIII inhibitor or the development of other autoimmune diseases with the persistence of her LGL leukemia remains to be seen.
At last follow-up, our patient was off all therapy and continued to have normal aPTT and factor VIII levels. Repeat flow cytometry after treatment of her factor VIII inhibitor showed persistence of a clonal T-cell population, although reduced from 72% to 60%. It may be that the 2 entities were unrelated, and the clonal T-cell population was simply fluctuating over time. This can be determined only with further observation. As the patient had no symptoms from her LGL leukemia, she continued to be observed without treatment.
TAKE-HOME POINTS
- The coagulation assay is key to initially assessing a bleeding abnormality; whether the prothrombin time and aPTT are normal or prolonged narrows the differential diagnosis and determines next steps in evaluation.
- Mixing studies can help pinpoint the responsible deficient factor.
- Acquired factor VIII deficiency, also known as AHA, may be caused by autoimmune disease, malignancy, or medications, but it is usually idiopathic.
- AHA treatment is focused on achieving hemostasis and reducing factor VIII inhibitor.
- Lymphocytosis should be evaluated with a peripheral blood smear and flow cytometry to determine if the population is polyclonal (associated with infection) or clonal (associated with malignancy).
- LGL leukemia is usually a chronic, indolent disease, although an uncommon subtype has an aggressive course.
- The association between AHA and LGL leukemia is unclear, and both conditions must be monitored and managed.
- Kamal AH, Tefferi A, Pruthi RK. How to interpret and pursue an abnormal prothrombin time, activated partial thromboplastin time, and bleeding time in adults. Mayo Clin Proc 2007; 82(7):864–873. doi:10.4065/82.7.864
- Tcherniantchouk O, Laposata M, Marques MB. The isolated prolonged PTT. Am J Hematol 2013; 88(1):82–85. doi:10.1002/ajh.23285
- Ma AD, Carrizosa D. Acquired factor VIII inhibitors: pathophysiology and treatment. Hematology Am Soc Hematol Educ Program 2006:432–437. doi:10.1182/asheducation-2006.1.432
- Delgado J, Jimenez-Yuste V, Hernandez-Navarro F, Villar A. Acquired haemophilia: review and meta-analysis focused on therapy and prognostic factors. Br J Haematol 2003; 121(1):21–35. pmid:12670328
- Knoebl P, Marco P, Baudo F, et al; EACH2 Registry Contributors. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost 2012; 10(4):622–631. doi:10.1111/j.1538-7836.2012.04654.x
- Collins PW, Hirsch S, Baglin TP, et al; UK Haemophilia Centre Doctors’ Organisation. Acquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood 2007; 109(5):1870–1877. doi:10.1182/blood-2006-06-029850
- Collins P, Baudo F, Knoebl P, et al; EACH2 Registry Collaborators. Immunosuppression for acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). Blood 2012; 120(1):47–55. doi:10.1182/blood-2012-02-409185
- George TI. Malignant or benign leukocytosis. Hematology Am Soc Hematol Educ Program 2012; 2012:475–484. doi:10.1182/asheducation-2012.1.475
- Watters RJ, Liu X, Loughran TP Jr. T-cell and natural killer-cell large granular lymphocyte leukemia neoplasias. Leuk Lymphoma 2011; 52(12):2217–2225. doi:10.3109/10428194.2011.593276
- Lamy T, Moignet A, Loughran TP Jr. LGL leukemia: from pathogenesis to treatment. Blood 2017; 129(9):1082–1094. doi:10.1182/blood-2016-08-692590
- Zhang D, Loughran TP Jr. Large granular lymphocytic leukemia: molecular pathogenesis, clinical manifestations, and treatment. Hematology Am Soc Hematol Educ Program 2012; 2012:652–659. doi:10.1182/asheducation-2012.1.652
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016; 127(20):2375–2390. doi:10.1182/blood-2016-01-643569
- Rose MG, Berliner N. T-cell large granular lymphocyte leukemia and related disorders. Oncologist 2004; 9(3):247–258. pmid:15169980
- Bockorny B, Dasanu CA. Autoimmune manifestations in large granular lymphocyte leukemia. Clin Lymphoma Myeloma Leuk 2012; 12(6):400–405. doi:10.1016/j.clml.2012.06.006
- Liu X, Loughran TP Jr. The spectrum of large granular lymphocyte leukemia and Felty’s syndrome. Curr Opin Hematol 2011; 18(4):254–259. doi:10.1097/MOH.0b013e32834760fb
- Franchini M, Lippi G. Acquired factor V inhibitors: a systematic review. J Thromb Thrombolysis 2011; 31(4):449–457. doi:10.1007/s11239-010-0529-6
- Sallah S, Nguyen NP, Abdallah JM, Hanrahan LR. Acquired hemophilia in patients with hematologic malignancies. Arch Pathol Lab Med 2000; 124(5):730–734.
- Murphy PW, Brett LK, Verla-Tebit E, Macik BG, Loughran TP Jr. Acquired inhibitors to factor VIII and fibrinogen in the setting of T-cell large granular lymphocyte leukemia: a case report and review of the literature. Blood Coagul Fibrinolysis 2015; 26(2):211–213. doi:10.1097/MBC.0000000000000209
- Kamal AH, Tefferi A, Pruthi RK. How to interpret and pursue an abnormal prothrombin time, activated partial thromboplastin time, and bleeding time in adults. Mayo Clin Proc 2007; 82(7):864–873. doi:10.4065/82.7.864
- Tcherniantchouk O, Laposata M, Marques MB. The isolated prolonged PTT. Am J Hematol 2013; 88(1):82–85. doi:10.1002/ajh.23285
- Ma AD, Carrizosa D. Acquired factor VIII inhibitors: pathophysiology and treatment. Hematology Am Soc Hematol Educ Program 2006:432–437. doi:10.1182/asheducation-2006.1.432
- Delgado J, Jimenez-Yuste V, Hernandez-Navarro F, Villar A. Acquired haemophilia: review and meta-analysis focused on therapy and prognostic factors. Br J Haematol 2003; 121(1):21–35. pmid:12670328
- Knoebl P, Marco P, Baudo F, et al; EACH2 Registry Contributors. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost 2012; 10(4):622–631. doi:10.1111/j.1538-7836.2012.04654.x
- Collins PW, Hirsch S, Baglin TP, et al; UK Haemophilia Centre Doctors’ Organisation. Acquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood 2007; 109(5):1870–1877. doi:10.1182/blood-2006-06-029850
- Collins P, Baudo F, Knoebl P, et al; EACH2 Registry Collaborators. Immunosuppression for acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). Blood 2012; 120(1):47–55. doi:10.1182/blood-2012-02-409185
- George TI. Malignant or benign leukocytosis. Hematology Am Soc Hematol Educ Program 2012; 2012:475–484. doi:10.1182/asheducation-2012.1.475
- Watters RJ, Liu X, Loughran TP Jr. T-cell and natural killer-cell large granular lymphocyte leukemia neoplasias. Leuk Lymphoma 2011; 52(12):2217–2225. doi:10.3109/10428194.2011.593276
- Lamy T, Moignet A, Loughran TP Jr. LGL leukemia: from pathogenesis to treatment. Blood 2017; 129(9):1082–1094. doi:10.1182/blood-2016-08-692590
- Zhang D, Loughran TP Jr. Large granular lymphocytic leukemia: molecular pathogenesis, clinical manifestations, and treatment. Hematology Am Soc Hematol Educ Program 2012; 2012:652–659. doi:10.1182/asheducation-2012.1.652
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016; 127(20):2375–2390. doi:10.1182/blood-2016-01-643569
- Rose MG, Berliner N. T-cell large granular lymphocyte leukemia and related disorders. Oncologist 2004; 9(3):247–258. pmid:15169980
- Bockorny B, Dasanu CA. Autoimmune manifestations in large granular lymphocyte leukemia. Clin Lymphoma Myeloma Leuk 2012; 12(6):400–405. doi:10.1016/j.clml.2012.06.006
- Liu X, Loughran TP Jr. The spectrum of large granular lymphocyte leukemia and Felty’s syndrome. Curr Opin Hematol 2011; 18(4):254–259. doi:10.1097/MOH.0b013e32834760fb
- Franchini M, Lippi G. Acquired factor V inhibitors: a systematic review. J Thromb Thrombolysis 2011; 31(4):449–457. doi:10.1007/s11239-010-0529-6
- Sallah S, Nguyen NP, Abdallah JM, Hanrahan LR. Acquired hemophilia in patients with hematologic malignancies. Arch Pathol Lab Med 2000; 124(5):730–734.
- Murphy PW, Brett LK, Verla-Tebit E, Macik BG, Loughran TP Jr. Acquired inhibitors to factor VIII and fibrinogen in the setting of T-cell large granular lymphocyte leukemia: a case report and review of the literature. Blood Coagul Fibrinolysis 2015; 26(2):211–213. doi:10.1097/MBC.0000000000000209

Erythrocytosis due to presumed polycythemia vera
A 40-year-old woman with hypertrophic obstructive cardiomyopathy presents to the hematology clinic for a second opinion regarding a history of headaches and fatigue for the past 10 years. She has been diagnosed with idiopathic erythrocytosis, presumed to be due to polycythemia vera. She periodically undergoes phlebotomy to keep her hematocrit below 41%, and this markedly improves her headaches. She denies shortness of breath, cough, fever, weight loss, joint pain, and visual or other neurologic symptoms. She has never reported pruritus related to bathing or exposure to water.
She does not smoke, drink alcohol, or use illicit drugs. She works as a pharmacy technician. She says her father died of cancer (no further details available) and describes a family history of gastrointestinal malignancy in her grandfather and paternal aunt. She takes aspirin, metoprolol, and spironolactone for her cardiomyopathy.
Physical examination reveals generalized plethora, more marked on her cheeks and face, and mild bilateral pitting pedal edema. No lymphadenopathy or hepatosplenomegaly can be palpated. Other systems, including the cardiac, respiratory, and nervous systems, are normal.
ERYTHROCYTOSIS AND POLYCYTHEMIA VERA
1. In patients with erythrocytosis, which of the following is not characteristic of polycythemia vera?
- Erythromelalgia and postbathing pruritus
- Splenomegaly
- History of thrombosis
- Gout
- Hematuria

Erythrocytosis—an abnormally high concentration of red blood cells in the peripheral blood—is a laboratory finding. It often reflects an increase in the total quantity or mass of red blood cells in the body (polycythemia) but can sometimes be due to decreased plasma volume (spurious polycythemia).1 Erythrocytosis can be caused by a number of diseases, hereditary and acquired, and can be classified as primary or secondary (Table 1).
Symptoms arise from an increase in the total blood volume and red blood cell mass, often leading to dilated capillaries and other blood vessels. Symptoms can occur regardless of the cause and classically include headache (often described as diffuse heaviness), dizziness, and a tendency for bleeding or thrombosis.2 Symptoms are relieved when the hematocrit is lowered.
Several features in the history and physical examination of a patient being evaluated for erythrocytosis can suggest an underlying cause. Smoking, chronic respiratory insufficiency, and congenital cyanotic heart disease point to secondary erythrocytosis and can usually be identified at the outset. A history of occupational exposure to carbon monoxide (such as engine exhaust) should be elicited carefully. A family history of erythrocytosis should raise suspicion of a heritable condition such as a hemoglobinopathy associated with increased oxygen affinity or rare forms of primary erythrocytosis associated with endogenous overproduction of erythropoietin or activating mutations of the erythropoietin receptor.3 Iatrogenic causes such as androgen supplementation, erythropoietin abuse, and postrenal-transplant erythrocytosis should also be considered.
Secretion of erythropoietin or erythropoietinlike proteins by a malignant neoplasm is a rare but important cause of erythrocytosis. For example, renal cell carcinoma may present with erythrocytosis secondary to excessive erythropoietin production, and hematuria can be an early symptom.
Polycythemia vera
Polycythemia vera, a myeloproliferative neoplasm, is characterized by increased red blood cell production independent of the mechanisms that normally regulate erythropoiesis. The bone marrow shows a panmyelosis that is often accompanied by leukocytosis or thrombocytosis, or both, in the peripheral blood.
Symptoms such as severe itching after exposure to hot water (aquagenic pruritus) and periodic attacks of redness, swelling, and pain in the hands or feet, or both (erythromelalgia), have been described in patients with polycythemia vera. Splenomegaly is relatively common, seen in approximately two-thirds of patients.4 Hyperuricemia (from increased cell turnover) and gout are also associated with polycythemia vera, as is a history of arterial and venous thrombosis.5
Hematuria is not commonly seen in polycythemia vera, although bleeding from the bladder, vagina, or uterus has been described.
CASE RESUMED: INITIAL LABORATORY TESTS
Results of our patient’s initial laboratory tests are:
- Hemoglobin 16.9 g/dL (reference range 11.5–15.5)
- Hematocrit 48.8% (36.0–46.0)
- Mean corpuscular volume 85.2 fL (80–100)
- Platelet count 328 × 109/L (150–400)
- White blood cell count 9.14 × 109/L (3.7–11.0)
- Absolute neutrophil count 5.95 × 109/L (1.45–7.5)
- Blood urea nitrogen 12 mg/dL (8–25)
- Creatinine 0.5 mg/dL (0.7–1.4)
- Lactate dehydrogenase 180 U/L (100–220)
- Uric acid 3.0 mg/dL (2.0–7.0)
- Thyroid-stimulating hormone 2.2 µU/mL (0.4–5.5).
The patient undergoes additional tests, including a serum erythropoietin level and hemoglobinopathy screen. Bone marrow aspiration and biopsy are performed, with cytogenetic analysis, chromosomal microarray analysis, and molecular testing for mutation of the Janus kinase 2 (JAK2) gene.
CONFIRMING SUSPECTED POLYCYTHEMIA VERA
2. In patients with suspected polycythemia vera, which of the following laboratory tests is most useful in making the diagnosis?
- Hemoglobin, hematocrit, and red blood cell mass
- Serum erythropoietin level
- Arterial blood gases with co-oximetry
- Testing for the JAK2 mutation
- Bone marrow aspiration and biopsy
The aim of the initial workup of erythrocytosis is to differentiate polycythemia vera from secondary causes of erythrocytosis.
Hemoglobin, hematocrit, red cell mass
Erythrocytosis is defined by an abnormal elevation in the hematocrit (> 48% in women or > 49% in men), hemoglobin concentration (> 16.0 g/dL in women or > 16.5 g/dL in men), or red blood cell mass. The red blood cell count should not be used as a surrogate for red blood cell mass, since some anemias (especially thalassemia minor) can be associated with an increase in the number of red blood cells but a low hemoglobin concentration.
Isotope dilution techniques to determine the red cell mass and plasma volume can differentiate true erythrocytosis from a spurious elevation due to a decrease in plasma volume.6,7 However, this is an expensive, time-consuming test that is not widely available and so is rarely performed.8
JAK2 mutation testing
The initial evaluation of a patient with erythrocytosis has changed significantly in the past 10 years with the discovery of the JAK2 gene and its role in the pathogenesis of polycythemia vera and other myeloproliferative neoplasms.
JAK2, located at 9p24, codes for a tyrosine kinase important for signal transduction in hematopoietic cells. Mutations in this gene have been shown to promote hypersensitivity to cytokines, including erythropoietin.9 The most common somatic mutation occurs within exon 14 at base pair 1849 and results in a phenylalanine-for-valine amino acid substitution in the JAK2 protein, designated V617F. Less commonly, mutations occur elsewhere in exons 12 to 15, with more than 50 different mutations described; nonpolymorphic mutations are assumed to have biologic effects similar to those of V617F.

Taken together, the JAK2 V617F and non-V617F mutations have a diagnostic sensitivity of 98% to 100% for polycythemia vera. For practical purposes, this means that the presence of a JAK2 mutation can be used as a clonal marker to distinguish polycythemia vera from reactive secondary causes of erythrocytosis. A JAK2 mutation is one of three major diagnostic criteria for polycythemia vera in the 2016 revision to the 2008 World Health Organization criteria (Table 2).10 Of note, this mutation is not specific for polycythemia vera and can also be found in other myeloproliferative neoplasms, including primary myelofibrosis and essential thrombocythemia.
Absence of a JAK2 mutation makes polycythemia vera unlikely, so this test is most useful in making the diagnosis.
Serum erythropoietin
Serum erythropoietin testing can be very useful to distinguish polycythemia vera from secondary erythrocytosis. Low levels suggest polycythemia vera, while high levels are seen in secondary processes.11
This test is best used along with JAK2 V617F mutation analysis as an initial step in evaluating patients with erythrocytosis. When JAK2 V617F mutation analysis is negative, a low serum erythropoietin level should prompt further testing for non-V617F JAK2 mutations, whereas a normal or elevated erythropoietin level should be evaluated further with tests to distinguish hereditary from acquired secondary causes of erythrocytosis.
Arterial blood gas analysis and co-oximetry
Arterial blood gas analysis can reveal hypoxemia, pointing to a cardiorespiratory process driving the erythrocytosis, whereas co-oximetry can be used to identify the presence and amount of carboxyhemoglobin in the blood.
Bone marrow biopsy
An increase in pleomorphic megakaryocytes in the bone marrow without stainable iron is often described as characteristic in polycythemia vera patients, but it is not diagnostic. Panmyelosis with increased cellularity is the norm but can be seen in other myeloproliferative neoplasms. The morphologic features of bone marrow are now included as one of the major diagnostic criteria for polycythemia vera (Table 2).
OUR PATIENT’S FURTHER WORKUP
Our patient’s erythropoietin level is 34.2 mIU/mL (reference range 4.7–28.6). Her oxygen saturation is 96%, and her carboxyhemoglobin level is 1.1% (0–5).
She undergoes bone marrow biopsy. Analysis finds that the marrow is normocellular (60%) with trilineage hematopoiesis and decreased stainable iron.
Cytogenetic analysis shows a 46,XX[20] karyotype. Chromosomal microarray analysis shows no pathogenic copy-number changes. There is no detectable JAK2 V617F or exon 12-to-15 mutation.
The patient’s erythrocytosis and abnormal hemoglobin electrophoresis study raise suspicion for a variant type of hemoglobin that has a higher affinity for oxygen than normal.
3. What is the next best step to evaluate this patient?
- Red-cell oxygen equilibrium curve to calculate the P50 (the partial pressure of oxygen that is required to saturate 50% of the hemoglobin.)
- High-performance liquid chromatography
- Globin gene DNA sequencing
- Testing 2,3-bisphosphoglycerate mutase (BPGM) activity
Nearly 200 mutational variants in alpha and beta globin chains that lead to an increased affinity of hemoglobin for oxygen have been reported.12 While not all mutations are clinically significant, increased oxygen affinity variants can lead to impaired oxygen delivery to tissues, especially the kidneys, resulting in a physiologic increase in erythropoietin and erythrocytosis.
In patients being evaluated for a high-oxygen-affinity hemoglobinopathy, a two-step approach has been outlined.13 The first involves measuring the oxygen-binding properties of a freshly collected sample of blood by directly measuring the oxygen saturation of the hemoglobin and pO2 using a co-oximeter. This information is used to create a red cell oxygen equilibrium curve and to calculate the P50. A low P50 correlates with an abnormally high affinity of hemoglobin for oxygen.
The second step is to identify the abnormal hemoglobin. High-performance liquid chromatography is now widely available as a screening test but does not detect all variants. For many years, sequencing of globin chain DNA has been a gold standard for identifying specific mutations. Subsequent to analyzing a catalog of known hemoglobin variants, mass spectrometry can serve as a screening and identification technique. Mass spectroscopy can also detect known rare variants with posttranslational modifications14 that are not recognized by DNA analysis. Mass spectroscopy and DNA sequencing are complementary techniques available only in specialized reference laboratories.
Erythrocytosis due to BPGM deficiency is very rare. Clinical and laboratory features mimic those of high-oxygen-affinity hemoglobin, but patients do not have a demonstrable mutation in alpha or beta globin genes. The level of BPGM is low, and the diagnosis is established by measuring BPGM levels and sequencing the BPGM gene.15
RESULTS OF THE ADDITIONAL WORKUP
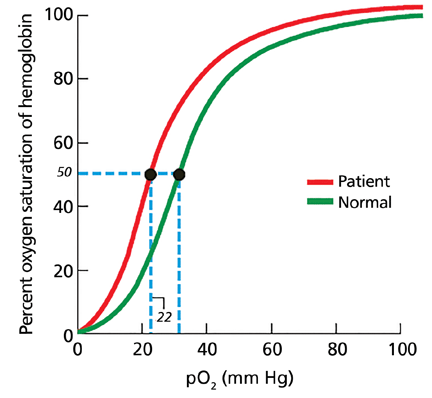
In our patient, hemoglobin electrophoresis reveals an abnormal hemoglobin variant. High-performance liquid chromatography reveals an abnormal peak that comprises approximately 23.7% of the total hemoglobin, consistent with an alpha globin variant. Further characterization (using a sample of venous blood) shows an oxygen dissociation P50 of 22 mm Hg (normal 24–30 mm Hg) (Figure 1).
Mass spectrometry identifies the variant as hemoglobin Tarrant. This variant is characterized by a substitution of asparagine for aspartic acid at position 126 of the alpha globin chain, a known site of contact between the alpha 1 and beta 1 chains.16 It has been seen in patients of Hispanic heritage and clinically correlates with mild erythrocytosis. Indeed, this woman’s mother was from Mexico.
EDUCATING PATIENTS
4. What should patients know about their high-oxygen-affinity hemoglobinopathy?
- High altitudes and air travel can be risky
- Pregnancy may have adverse outcomes
- Systemic anticoagulation may lower the risk of venous thromboembolism
- Periodic phlebotomy may help control symptoms
Most patients with high-oxygen-affinity hemoglobin do not require specific clinical management but only counseling and education about their condition. Establishing an accurate diagnosis is important in order to avoid further inappropriate, invasive, and expensive testing.
Although exposure to high altitudes may be associated with decreased ambient oxygen levels, hypoxia is usually not a problem because of hemoglobin’s high affinity for oxygen.
Impaired delivery of oxygen across the placenta may be anticipated in a mother with high-oxygen-affinity hemoglobin, but this has not been observed clinically.17
Compared with patients with polycythemia vera, patients with high-oxygen-affinity hemoglobin have fewer complications from hyperviscosity and thrombosis, even with comparable degrees of erythrocytosis.
Although patients usually do not require treatment, phlebotomy may be helpful for symptoms that can be attributed to the higher hemoglobin concentration.
Our patient continues to be seen in clinic for periodic blood counts and phlebotomy for her headaches, as required.
HEMOGLOBIN: RELAXED OR TENSE
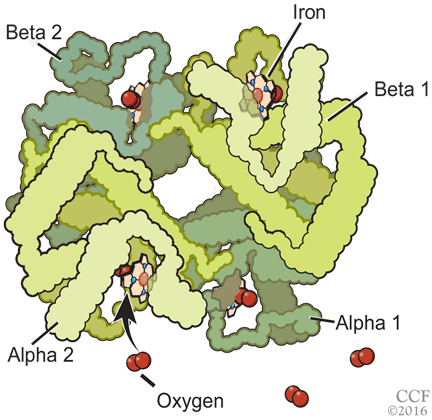
Normal adult hemoglobin is a tetramer composed of two pairs of globin polypeptide chains: alpha and beta (Figure 2). The intrinsic properties of the constituent globin chains and their allosteric conformation—as well as extrinsic factors including temperature, pH, and the binding of hydrogen ion and 2,3-BPG—play important roles in modifying the affinity of hemoglobin for oxygen. The major modulator of hemoglobin-oxygen affinity in human erythrocytes is 2,3-BPG.
The hemoglobin tetramer, consisting of two identical halves, alpha 1-beta 1 and alpha 2-beta 2, oscillates between two quaternary conformations, “relaxed” (fully oxygenated) and “tense” (fully deoxygenated).18 High-oxygen-affinity hemoglobins can result from factors that enhance the relaxed state, either by stabilizing the relaxed state or by destabilizing the tense state. Structural modifications in hemoglobin typically affect the main contacts involved in the transition from the deoxygenated to the oxygenated state, the 2,3-BPG binding sites, the heme pocket, or elongation of globin chains by various mutations. In hemoglobin Tarrant, the mutation prevents formation of noncovalent salt bridges in the alpha 1-beta 1 contact that normally stabilize the deoxygenated conformation of hemoglobin. As a result, the deoxygenated (tense) state is destabilized, shifting the allosteric equilibrium in favor of the oxygenated (relaxed) state with consequent high oxygen affinity.16
MORE ABOUT HIGH-OXYGEN-AFFINITY HEMOGLOBINS
The first case of erythrocytosis due to an abnormal hemoglobin was identified in 1966. This was an alpha chain variant with an arginine-to-leucine substitution at position 92, named hemoglobin Chesapeake.19
High-oxygen-affinity hemoglobin variants are usually transmitted as autosomal dominant traits. Patients are most often identified because of unexplained erythrocytosis detected on a routine blood cell count, as in our patient.
Not all high-oxygen-affinity hemoglobinopathies are associated with erythrocytosis. The degree of increased oxygen affinity may only be mild or the abnormal hemoglobin may be slightly unstable, thereby masking the usual clinical signs and symptoms.
Therapeutic phlebotomy should be used cautiously since it can decrease delivery of oxygen to tissues. A subset of patients whose symptoms are related to an elevated red cell mass may experience some relief, as did our patient.
- Kremyanskaya M, Mascarenhas J, Hoffman R. Why does my patient have erythrocytosis? Hematol Oncol Clin North Am 2012; 26:267–283.
- Keohane C, McMullin MF, Harrison C. The diagnosis and management of erythrocytosis. BMJ 2013; 347:f6667.
- Agarwal N, Gordeuk RV, Prchal JT. Genetic mechanisms underlying regulation of hemoglobin mass. Adv Exp Med Biol 2007; 618:195–210.
- Tefferi A. Polycythemia vera and essential thrombocythemia: 2012 update on diagnosis, risk stratification, and management. Am J Hematol 2012; 87:285–293.
- Landolfi R, Di Gennaro L, Falanga A. Thrombosis in myeloproliferative disorders: pathogenetic facts and speculation. Leukemia 2008; 22:2020–2028.
- Tefferi A, Spivak JL. Polycythemia vera: scientific advances and current practice. Semin Hematol 2005; 42:206–220.
- Ferrant A. What clinical and laboratory data are indicative of polycythemia and when are blood volume studies needed? Nouv Rev Fr Hematol 1994; 36:151–154.
- Fairbanks VF, Klee GG, Wiseman GA, et al. Measurement of blood volume and red cell mass: re-examination of 51Cr and 125I methods. Blood Cells Mol Dis 1996; 22:169–186; discussion 186a–186g.
- James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005; 434:1144–1148.
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127:2391–2405.
- Messinezy M, Westwood NB, El-Hemaidi I, Marsden JT, Sherwood RS, Pearson TC. Serum erythropoietin values in erythrocytosis and in primary thrombocythaemia. Br J Haematol 2002; 117:47–53.
- Hardison RC, Chui DHK, Giardine B, et al. HbVar: a relational database of human hemoglobin variants and thalassemia mutations at the globin gene server. Human Mutat 2002; 19:225–233.
- Percy MJ, Butt NN, Crotty GM, et al. Identification of high oxygen affinity hemoglobin variants in the investigation of patients with erythrocytosis. Haematologica 2009; 94:1321–1322.
- Kattamis AC, Kelly KM, Ohene-Frempong K, et al. Hb Osler [beta 145(HC2)Tyr-->Asp] results from posttranslational modification. Hemoglobin 1997; 21:109–120.
- Hoyer JD, Allen SL, Beutler E, Kubik K, West C, Fairbanks VF. Erythrocytosis due to bisphosphoglycerate mutase deficiency with concurrent glucose-6-phosphate dehydrogenase (G-6-PD) deficiency. Am J Hematol 2004; 75:205–208.
- Moo-Penn WF, Jue DL, Johnson MH, Wilson SM, Therrell B Jr, Schmidt RM. Hemoglobin Tarrant: alpha126(H9) asp leads to asn. A new hemoglobin variant in the alpha1beta1 contact region showing high oxygen affinity and reduced cooperativity. Biochim Biophys Acta 1977; 490:443–451.
- Bard H, Peri KG, Gagnon C. The biologic implications of a rare hemoglobin mutant that decreases oxygen affinity. Pediatr Res 2001; 49:69–73.
- Wajcman H, Galacteros F. Hemoglobins with high oxygen affinity leading to erythrocytosis: new variants and concepts. Hemoglobin 2005; 29:91–106.
- Clegg JB, Naughton MA, Weatherall DJ. Abnormal human haemoglobins. Separation and characterization of the alpha and beta chains by chromatography, and the determination of two new variants, hb Chesapeak and hb J (Bangkok). J Mol Biol 1966; 19:91–108.
A 40-year-old woman with hypertrophic obstructive cardiomyopathy presents to the hematology clinic for a second opinion regarding a history of headaches and fatigue for the past 10 years. She has been diagnosed with idiopathic erythrocytosis, presumed to be due to polycythemia vera. She periodically undergoes phlebotomy to keep her hematocrit below 41%, and this markedly improves her headaches. She denies shortness of breath, cough, fever, weight loss, joint pain, and visual or other neurologic symptoms. She has never reported pruritus related to bathing or exposure to water.
She does not smoke, drink alcohol, or use illicit drugs. She works as a pharmacy technician. She says her father died of cancer (no further details available) and describes a family history of gastrointestinal malignancy in her grandfather and paternal aunt. She takes aspirin, metoprolol, and spironolactone for her cardiomyopathy.
Physical examination reveals generalized plethora, more marked on her cheeks and face, and mild bilateral pitting pedal edema. No lymphadenopathy or hepatosplenomegaly can be palpated. Other systems, including the cardiac, respiratory, and nervous systems, are normal.
ERYTHROCYTOSIS AND POLYCYTHEMIA VERA
1. In patients with erythrocytosis, which of the following is not characteristic of polycythemia vera?
- Erythromelalgia and postbathing pruritus
- Splenomegaly
- History of thrombosis
- Gout
- Hematuria
Erythrocytosis—an abnormally high concentration of red blood cells in the peripheral blood—is a laboratory finding. It often reflects an increase in the total quantity or mass of red blood cells in the body (polycythemia) but can sometimes be due to decreased plasma volume (spurious polycythemia).1 Erythrocytosis can be caused by a number of diseases, hereditary and acquired, and can be classified as primary or secondary (Table 1).
Symptoms arise from an increase in the total blood volume and red blood cell mass, often leading to dilated capillaries and other blood vessels. Symptoms can occur regardless of the cause and classically include headache (often described as diffuse heaviness), dizziness, and a tendency for bleeding or thrombosis.2 Symptoms are relieved when the hematocrit is lowered.
Several features in the history and physical examination of a patient being evaluated for erythrocytosis can suggest an underlying cause. Smoking, chronic respiratory insufficiency, and congenital cyanotic heart disease point to secondary erythrocytosis and can usually be identified at the outset. A history of occupational exposure to carbon monoxide (such as engine exhaust) should be elicited carefully. A family history of erythrocytosis should raise suspicion of a heritable condition such as a hemoglobinopathy associated with increased oxygen affinity or rare forms of primary erythrocytosis associated with endogenous overproduction of erythropoietin or activating mutations of the erythropoietin receptor.3 Iatrogenic causes such as androgen supplementation, erythropoietin abuse, and postrenal-transplant erythrocytosis should also be considered.
Secretion of erythropoietin or erythropoietinlike proteins by a malignant neoplasm is a rare but important cause of erythrocytosis. For example, renal cell carcinoma may present with erythrocytosis secondary to excessive erythropoietin production, and hematuria can be an early symptom.
Polycythemia vera
Polycythemia vera, a myeloproliferative neoplasm, is characterized by increased red blood cell production independent of the mechanisms that normally regulate erythropoiesis. The bone marrow shows a panmyelosis that is often accompanied by leukocytosis or thrombocytosis, or both, in the peripheral blood.
Symptoms such as severe itching after exposure to hot water (aquagenic pruritus) and periodic attacks of redness, swelling, and pain in the hands or feet, or both (erythromelalgia), have been described in patients with polycythemia vera. Splenomegaly is relatively common, seen in approximately two-thirds of patients.4 Hyperuricemia (from increased cell turnover) and gout are also associated with polycythemia vera, as is a history of arterial and venous thrombosis.5
Hematuria is not commonly seen in polycythemia vera, although bleeding from the bladder, vagina, or uterus has been described.
CASE RESUMED: INITIAL LABORATORY TESTS
Results of our patient’s initial laboratory tests are:
- Hemoglobin 16.9 g/dL (reference range 11.5–15.5)
- Hematocrit 48.8% (36.0–46.0)
- Mean corpuscular volume 85.2 fL (80–100)
- Platelet count 328 × 109/L (150–400)
- White blood cell count 9.14 × 109/L (3.7–11.0)
- Absolute neutrophil count 5.95 × 109/L (1.45–7.5)
- Blood urea nitrogen 12 mg/dL (8–25)
- Creatinine 0.5 mg/dL (0.7–1.4)
- Lactate dehydrogenase 180 U/L (100–220)
- Uric acid 3.0 mg/dL (2.0–7.0)
- Thyroid-stimulating hormone 2.2 µU/mL (0.4–5.5).
The patient undergoes additional tests, including a serum erythropoietin level and hemoglobinopathy screen. Bone marrow aspiration and biopsy are performed, with cytogenetic analysis, chromosomal microarray analysis, and molecular testing for mutation of the Janus kinase 2 (JAK2) gene.
CONFIRMING SUSPECTED POLYCYTHEMIA VERA
2. In patients with suspected polycythemia vera, which of the following laboratory tests is most useful in making the diagnosis?
- Hemoglobin, hematocrit, and red blood cell mass
- Serum erythropoietin level
- Arterial blood gases with co-oximetry
- Testing for the JAK2 mutation
- Bone marrow aspiration and biopsy
The aim of the initial workup of erythrocytosis is to differentiate polycythemia vera from secondary causes of erythrocytosis.
Hemoglobin, hematocrit, red cell mass
Erythrocytosis is defined by an abnormal elevation in the hematocrit (> 48% in women or > 49% in men), hemoglobin concentration (> 16.0 g/dL in women or > 16.5 g/dL in men), or red blood cell mass. The red blood cell count should not be used as a surrogate for red blood cell mass, since some anemias (especially thalassemia minor) can be associated with an increase in the number of red blood cells but a low hemoglobin concentration.
Isotope dilution techniques to determine the red cell mass and plasma volume can differentiate true erythrocytosis from a spurious elevation due to a decrease in plasma volume.6,7 However, this is an expensive, time-consuming test that is not widely available and so is rarely performed.8
JAK2 mutation testing
The initial evaluation of a patient with erythrocytosis has changed significantly in the past 10 years with the discovery of the JAK2 gene and its role in the pathogenesis of polycythemia vera and other myeloproliferative neoplasms.
JAK2, located at 9p24, codes for a tyrosine kinase important for signal transduction in hematopoietic cells. Mutations in this gene have been shown to promote hypersensitivity to cytokines, including erythropoietin.9 The most common somatic mutation occurs within exon 14 at base pair 1849 and results in a phenylalanine-for-valine amino acid substitution in the JAK2 protein, designated V617F. Less commonly, mutations occur elsewhere in exons 12 to 15, with more than 50 different mutations described; nonpolymorphic mutations are assumed to have biologic effects similar to those of V617F.
Taken together, the JAK2 V617F and non-V617F mutations have a diagnostic sensitivity of 98% to 100% for polycythemia vera. For practical purposes, this means that the presence of a JAK2 mutation can be used as a clonal marker to distinguish polycythemia vera from reactive secondary causes of erythrocytosis. A JAK2 mutation is one of three major diagnostic criteria for polycythemia vera in the 2016 revision to the 2008 World Health Organization criteria (Table 2).10 Of note, this mutation is not specific for polycythemia vera and can also be found in other myeloproliferative neoplasms, including primary myelofibrosis and essential thrombocythemia.
Absence of a JAK2 mutation makes polycythemia vera unlikely, so this test is most useful in making the diagnosis.
Serum erythropoietin
Serum erythropoietin testing can be very useful to distinguish polycythemia vera from secondary erythrocytosis. Low levels suggest polycythemia vera, while high levels are seen in secondary processes.11
This test is best used along with JAK2 V617F mutation analysis as an initial step in evaluating patients with erythrocytosis. When JAK2 V617F mutation analysis is negative, a low serum erythropoietin level should prompt further testing for non-V617F JAK2 mutations, whereas a normal or elevated erythropoietin level should be evaluated further with tests to distinguish hereditary from acquired secondary causes of erythrocytosis.
Arterial blood gas analysis and co-oximetry
Arterial blood gas analysis can reveal hypoxemia, pointing to a cardiorespiratory process driving the erythrocytosis, whereas co-oximetry can be used to identify the presence and amount of carboxyhemoglobin in the blood.
Bone marrow biopsy
An increase in pleomorphic megakaryocytes in the bone marrow without stainable iron is often described as characteristic in polycythemia vera patients, but it is not diagnostic. Panmyelosis with increased cellularity is the norm but can be seen in other myeloproliferative neoplasms. The morphologic features of bone marrow are now included as one of the major diagnostic criteria for polycythemia vera (Table 2).
OUR PATIENT’S FURTHER WORKUP
Our patient’s erythropoietin level is 34.2 mIU/mL (reference range 4.7–28.6). Her oxygen saturation is 96%, and her carboxyhemoglobin level is 1.1% (0–5).
She undergoes bone marrow biopsy. Analysis finds that the marrow is normocellular (60%) with trilineage hematopoiesis and decreased stainable iron.
Cytogenetic analysis shows a 46,XX[20] karyotype. Chromosomal microarray analysis shows no pathogenic copy-number changes. There is no detectable JAK2 V617F or exon 12-to-15 mutation.
The patient’s erythrocytosis and abnormal hemoglobin electrophoresis study raise suspicion for a variant type of hemoglobin that has a higher affinity for oxygen than normal.
3. What is the next best step to evaluate this patient?
- Red-cell oxygen equilibrium curve to calculate the P50 (the partial pressure of oxygen that is required to saturate 50% of the hemoglobin.)
- High-performance liquid chromatography
- Globin gene DNA sequencing
- Testing 2,3-bisphosphoglycerate mutase (BPGM) activity
Nearly 200 mutational variants in alpha and beta globin chains that lead to an increased affinity of hemoglobin for oxygen have been reported.12 While not all mutations are clinically significant, increased oxygen affinity variants can lead to impaired oxygen delivery to tissues, especially the kidneys, resulting in a physiologic increase in erythropoietin and erythrocytosis.
In patients being evaluated for a high-oxygen-affinity hemoglobinopathy, a two-step approach has been outlined.13 The first involves measuring the oxygen-binding properties of a freshly collected sample of blood by directly measuring the oxygen saturation of the hemoglobin and pO2 using a co-oximeter. This information is used to create a red cell oxygen equilibrium curve and to calculate the P50. A low P50 correlates with an abnormally high affinity of hemoglobin for oxygen.
The second step is to identify the abnormal hemoglobin. High-performance liquid chromatography is now widely available as a screening test but does not detect all variants. For many years, sequencing of globin chain DNA has been a gold standard for identifying specific mutations. Subsequent to analyzing a catalog of known hemoglobin variants, mass spectrometry can serve as a screening and identification technique. Mass spectroscopy can also detect known rare variants with posttranslational modifications14 that are not recognized by DNA analysis. Mass spectroscopy and DNA sequencing are complementary techniques available only in specialized reference laboratories.
Erythrocytosis due to BPGM deficiency is very rare. Clinical and laboratory features mimic those of high-oxygen-affinity hemoglobin, but patients do not have a demonstrable mutation in alpha or beta globin genes. The level of BPGM is low, and the diagnosis is established by measuring BPGM levels and sequencing the BPGM gene.15
RESULTS OF THE ADDITIONAL WORKUP
In our patient, hemoglobin electrophoresis reveals an abnormal hemoglobin variant. High-performance liquid chromatography reveals an abnormal peak that comprises approximately 23.7% of the total hemoglobin, consistent with an alpha globin variant. Further characterization (using a sample of venous blood) shows an oxygen dissociation P50 of 22 mm Hg (normal 24–30 mm Hg) (Figure 1).
Mass spectrometry identifies the variant as hemoglobin Tarrant. This variant is characterized by a substitution of asparagine for aspartic acid at position 126 of the alpha globin chain, a known site of contact between the alpha 1 and beta 1 chains.16 It has been seen in patients of Hispanic heritage and clinically correlates with mild erythrocytosis. Indeed, this woman’s mother was from Mexico.
EDUCATING PATIENTS
4. What should patients know about their high-oxygen-affinity hemoglobinopathy?
- High altitudes and air travel can be risky
- Pregnancy may have adverse outcomes
- Systemic anticoagulation may lower the risk of venous thromboembolism
- Periodic phlebotomy may help control symptoms
Most patients with high-oxygen-affinity hemoglobin do not require specific clinical management but only counseling and education about their condition. Establishing an accurate diagnosis is important in order to avoid further inappropriate, invasive, and expensive testing.
Although exposure to high altitudes may be associated with decreased ambient oxygen levels, hypoxia is usually not a problem because of hemoglobin’s high affinity for oxygen.
Impaired delivery of oxygen across the placenta may be anticipated in a mother with high-oxygen-affinity hemoglobin, but this has not been observed clinically.17
Compared with patients with polycythemia vera, patients with high-oxygen-affinity hemoglobin have fewer complications from hyperviscosity and thrombosis, even with comparable degrees of erythrocytosis.
Although patients usually do not require treatment, phlebotomy may be helpful for symptoms that can be attributed to the higher hemoglobin concentration.
Our patient continues to be seen in clinic for periodic blood counts and phlebotomy for her headaches, as required.
HEMOGLOBIN: RELAXED OR TENSE
Normal adult hemoglobin is a tetramer composed of two pairs of globin polypeptide chains: alpha and beta (Figure 2). The intrinsic properties of the constituent globin chains and their allosteric conformation—as well as extrinsic factors including temperature, pH, and the binding of hydrogen ion and 2,3-BPG—play important roles in modifying the affinity of hemoglobin for oxygen. The major modulator of hemoglobin-oxygen affinity in human erythrocytes is 2,3-BPG.
The hemoglobin tetramer, consisting of two identical halves, alpha 1-beta 1 and alpha 2-beta 2, oscillates between two quaternary conformations, “relaxed” (fully oxygenated) and “tense” (fully deoxygenated).18 High-oxygen-affinity hemoglobins can result from factors that enhance the relaxed state, either by stabilizing the relaxed state or by destabilizing the tense state. Structural modifications in hemoglobin typically affect the main contacts involved in the transition from the deoxygenated to the oxygenated state, the 2,3-BPG binding sites, the heme pocket, or elongation of globin chains by various mutations. In hemoglobin Tarrant, the mutation prevents formation of noncovalent salt bridges in the alpha 1-beta 1 contact that normally stabilize the deoxygenated conformation of hemoglobin. As a result, the deoxygenated (tense) state is destabilized, shifting the allosteric equilibrium in favor of the oxygenated (relaxed) state with consequent high oxygen affinity.16
MORE ABOUT HIGH-OXYGEN-AFFINITY HEMOGLOBINS
The first case of erythrocytosis due to an abnormal hemoglobin was identified in 1966. This was an alpha chain variant with an arginine-to-leucine substitution at position 92, named hemoglobin Chesapeake.19
High-oxygen-affinity hemoglobin variants are usually transmitted as autosomal dominant traits. Patients are most often identified because of unexplained erythrocytosis detected on a routine blood cell count, as in our patient.
Not all high-oxygen-affinity hemoglobinopathies are associated with erythrocytosis. The degree of increased oxygen affinity may only be mild or the abnormal hemoglobin may be slightly unstable, thereby masking the usual clinical signs and symptoms.
Therapeutic phlebotomy should be used cautiously since it can decrease delivery of oxygen to tissues. A subset of patients whose symptoms are related to an elevated red cell mass may experience some relief, as did our patient.
A 40-year-old woman with hypertrophic obstructive cardiomyopathy presents to the hematology clinic for a second opinion regarding a history of headaches and fatigue for the past 10 years. She has been diagnosed with idiopathic erythrocytosis, presumed to be due to polycythemia vera. She periodically undergoes phlebotomy to keep her hematocrit below 41%, and this markedly improves her headaches. She denies shortness of breath, cough, fever, weight loss, joint pain, and visual or other neurologic symptoms. She has never reported pruritus related to bathing or exposure to water.
She does not smoke, drink alcohol, or use illicit drugs. She works as a pharmacy technician. She says her father died of cancer (no further details available) and describes a family history of gastrointestinal malignancy in her grandfather and paternal aunt. She takes aspirin, metoprolol, and spironolactone for her cardiomyopathy.
Physical examination reveals generalized plethora, more marked on her cheeks and face, and mild bilateral pitting pedal edema. No lymphadenopathy or hepatosplenomegaly can be palpated. Other systems, including the cardiac, respiratory, and nervous systems, are normal.
ERYTHROCYTOSIS AND POLYCYTHEMIA VERA
1. In patients with erythrocytosis, which of the following is not characteristic of polycythemia vera?
- Erythromelalgia and postbathing pruritus
- Splenomegaly
- History of thrombosis
- Gout
- Hematuria
Erythrocytosis—an abnormally high concentration of red blood cells in the peripheral blood—is a laboratory finding. It often reflects an increase in the total quantity or mass of red blood cells in the body (polycythemia) but can sometimes be due to decreased plasma volume (spurious polycythemia).1 Erythrocytosis can be caused by a number of diseases, hereditary and acquired, and can be classified as primary or secondary (Table 1).
Symptoms arise from an increase in the total blood volume and red blood cell mass, often leading to dilated capillaries and other blood vessels. Symptoms can occur regardless of the cause and classically include headache (often described as diffuse heaviness), dizziness, and a tendency for bleeding or thrombosis.2 Symptoms are relieved when the hematocrit is lowered.
Several features in the history and physical examination of a patient being evaluated for erythrocytosis can suggest an underlying cause. Smoking, chronic respiratory insufficiency, and congenital cyanotic heart disease point to secondary erythrocytosis and can usually be identified at the outset. A history of occupational exposure to carbon monoxide (such as engine exhaust) should be elicited carefully. A family history of erythrocytosis should raise suspicion of a heritable condition such as a hemoglobinopathy associated with increased oxygen affinity or rare forms of primary erythrocytosis associated with endogenous overproduction of erythropoietin or activating mutations of the erythropoietin receptor.3 Iatrogenic causes such as androgen supplementation, erythropoietin abuse, and postrenal-transplant erythrocytosis should also be considered.
Secretion of erythropoietin or erythropoietinlike proteins by a malignant neoplasm is a rare but important cause of erythrocytosis. For example, renal cell carcinoma may present with erythrocytosis secondary to excessive erythropoietin production, and hematuria can be an early symptom.
Polycythemia vera
Polycythemia vera, a myeloproliferative neoplasm, is characterized by increased red blood cell production independent of the mechanisms that normally regulate erythropoiesis. The bone marrow shows a panmyelosis that is often accompanied by leukocytosis or thrombocytosis, or both, in the peripheral blood.
Symptoms such as severe itching after exposure to hot water (aquagenic pruritus) and periodic attacks of redness, swelling, and pain in the hands or feet, or both (erythromelalgia), have been described in patients with polycythemia vera. Splenomegaly is relatively common, seen in approximately two-thirds of patients.4 Hyperuricemia (from increased cell turnover) and gout are also associated with polycythemia vera, as is a history of arterial and venous thrombosis.5
Hematuria is not commonly seen in polycythemia vera, although bleeding from the bladder, vagina, or uterus has been described.
CASE RESUMED: INITIAL LABORATORY TESTS
Results of our patient’s initial laboratory tests are:
- Hemoglobin 16.9 g/dL (reference range 11.5–15.5)
- Hematocrit 48.8% (36.0–46.0)
- Mean corpuscular volume 85.2 fL (80–100)
- Platelet count 328 × 109/L (150–400)
- White blood cell count 9.14 × 109/L (3.7–11.0)
- Absolute neutrophil count 5.95 × 109/L (1.45–7.5)
- Blood urea nitrogen 12 mg/dL (8–25)
- Creatinine 0.5 mg/dL (0.7–1.4)
- Lactate dehydrogenase 180 U/L (100–220)
- Uric acid 3.0 mg/dL (2.0–7.0)
- Thyroid-stimulating hormone 2.2 µU/mL (0.4–5.5).
The patient undergoes additional tests, including a serum erythropoietin level and hemoglobinopathy screen. Bone marrow aspiration and biopsy are performed, with cytogenetic analysis, chromosomal microarray analysis, and molecular testing for mutation of the Janus kinase 2 (JAK2) gene.
CONFIRMING SUSPECTED POLYCYTHEMIA VERA
2. In patients with suspected polycythemia vera, which of the following laboratory tests is most useful in making the diagnosis?
- Hemoglobin, hematocrit, and red blood cell mass
- Serum erythropoietin level
- Arterial blood gases with co-oximetry
- Testing for the JAK2 mutation
- Bone marrow aspiration and biopsy
The aim of the initial workup of erythrocytosis is to differentiate polycythemia vera from secondary causes of erythrocytosis.
Hemoglobin, hematocrit, red cell mass
Erythrocytosis is defined by an abnormal elevation in the hematocrit (> 48% in women or > 49% in men), hemoglobin concentration (> 16.0 g/dL in women or > 16.5 g/dL in men), or red blood cell mass. The red blood cell count should not be used as a surrogate for red blood cell mass, since some anemias (especially thalassemia minor) can be associated with an increase in the number of red blood cells but a low hemoglobin concentration.
Isotope dilution techniques to determine the red cell mass and plasma volume can differentiate true erythrocytosis from a spurious elevation due to a decrease in plasma volume.6,7 However, this is an expensive, time-consuming test that is not widely available and so is rarely performed.8
JAK2 mutation testing
The initial evaluation of a patient with erythrocytosis has changed significantly in the past 10 years with the discovery of the JAK2 gene and its role in the pathogenesis of polycythemia vera and other myeloproliferative neoplasms.
JAK2, located at 9p24, codes for a tyrosine kinase important for signal transduction in hematopoietic cells. Mutations in this gene have been shown to promote hypersensitivity to cytokines, including erythropoietin.9 The most common somatic mutation occurs within exon 14 at base pair 1849 and results in a phenylalanine-for-valine amino acid substitution in the JAK2 protein, designated V617F. Less commonly, mutations occur elsewhere in exons 12 to 15, with more than 50 different mutations described; nonpolymorphic mutations are assumed to have biologic effects similar to those of V617F.
Taken together, the JAK2 V617F and non-V617F mutations have a diagnostic sensitivity of 98% to 100% for polycythemia vera. For practical purposes, this means that the presence of a JAK2 mutation can be used as a clonal marker to distinguish polycythemia vera from reactive secondary causes of erythrocytosis. A JAK2 mutation is one of three major diagnostic criteria for polycythemia vera in the 2016 revision to the 2008 World Health Organization criteria (Table 2).10 Of note, this mutation is not specific for polycythemia vera and can also be found in other myeloproliferative neoplasms, including primary myelofibrosis and essential thrombocythemia.
Absence of a JAK2 mutation makes polycythemia vera unlikely, so this test is most useful in making the diagnosis.
Serum erythropoietin
Serum erythropoietin testing can be very useful to distinguish polycythemia vera from secondary erythrocytosis. Low levels suggest polycythemia vera, while high levels are seen in secondary processes.11
This test is best used along with JAK2 V617F mutation analysis as an initial step in evaluating patients with erythrocytosis. When JAK2 V617F mutation analysis is negative, a low serum erythropoietin level should prompt further testing for non-V617F JAK2 mutations, whereas a normal or elevated erythropoietin level should be evaluated further with tests to distinguish hereditary from acquired secondary causes of erythrocytosis.
Arterial blood gas analysis and co-oximetry
Arterial blood gas analysis can reveal hypoxemia, pointing to a cardiorespiratory process driving the erythrocytosis, whereas co-oximetry can be used to identify the presence and amount of carboxyhemoglobin in the blood.
Bone marrow biopsy
An increase in pleomorphic megakaryocytes in the bone marrow without stainable iron is often described as characteristic in polycythemia vera patients, but it is not diagnostic. Panmyelosis with increased cellularity is the norm but can be seen in other myeloproliferative neoplasms. The morphologic features of bone marrow are now included as one of the major diagnostic criteria for polycythemia vera (Table 2).
OUR PATIENT’S FURTHER WORKUP
Our patient’s erythropoietin level is 34.2 mIU/mL (reference range 4.7–28.6). Her oxygen saturation is 96%, and her carboxyhemoglobin level is 1.1% (0–5).
She undergoes bone marrow biopsy. Analysis finds that the marrow is normocellular (60%) with trilineage hematopoiesis and decreased stainable iron.
Cytogenetic analysis shows a 46,XX[20] karyotype. Chromosomal microarray analysis shows no pathogenic copy-number changes. There is no detectable JAK2 V617F or exon 12-to-15 mutation.
The patient’s erythrocytosis and abnormal hemoglobin electrophoresis study raise suspicion for a variant type of hemoglobin that has a higher affinity for oxygen than normal.
3. What is the next best step to evaluate this patient?
- Red-cell oxygen equilibrium curve to calculate the P50 (the partial pressure of oxygen that is required to saturate 50% of the hemoglobin.)
- High-performance liquid chromatography
- Globin gene DNA sequencing
- Testing 2,3-bisphosphoglycerate mutase (BPGM) activity
Nearly 200 mutational variants in alpha and beta globin chains that lead to an increased affinity of hemoglobin for oxygen have been reported.12 While not all mutations are clinically significant, increased oxygen affinity variants can lead to impaired oxygen delivery to tissues, especially the kidneys, resulting in a physiologic increase in erythropoietin and erythrocytosis.
In patients being evaluated for a high-oxygen-affinity hemoglobinopathy, a two-step approach has been outlined.13 The first involves measuring the oxygen-binding properties of a freshly collected sample of blood by directly measuring the oxygen saturation of the hemoglobin and pO2 using a co-oximeter. This information is used to create a red cell oxygen equilibrium curve and to calculate the P50. A low P50 correlates with an abnormally high affinity of hemoglobin for oxygen.
The second step is to identify the abnormal hemoglobin. High-performance liquid chromatography is now widely available as a screening test but does not detect all variants. For many years, sequencing of globin chain DNA has been a gold standard for identifying specific mutations. Subsequent to analyzing a catalog of known hemoglobin variants, mass spectrometry can serve as a screening and identification technique. Mass spectroscopy can also detect known rare variants with posttranslational modifications14 that are not recognized by DNA analysis. Mass spectroscopy and DNA sequencing are complementary techniques available only in specialized reference laboratories.
Erythrocytosis due to BPGM deficiency is very rare. Clinical and laboratory features mimic those of high-oxygen-affinity hemoglobin, but patients do not have a demonstrable mutation in alpha or beta globin genes. The level of BPGM is low, and the diagnosis is established by measuring BPGM levels and sequencing the BPGM gene.15
RESULTS OF THE ADDITIONAL WORKUP
In our patient, hemoglobin electrophoresis reveals an abnormal hemoglobin variant. High-performance liquid chromatography reveals an abnormal peak that comprises approximately 23.7% of the total hemoglobin, consistent with an alpha globin variant. Further characterization (using a sample of venous blood) shows an oxygen dissociation P50 of 22 mm Hg (normal 24–30 mm Hg) (Figure 1).
Mass spectrometry identifies the variant as hemoglobin Tarrant. This variant is characterized by a substitution of asparagine for aspartic acid at position 126 of the alpha globin chain, a known site of contact between the alpha 1 and beta 1 chains.16 It has been seen in patients of Hispanic heritage and clinically correlates with mild erythrocytosis. Indeed, this woman’s mother was from Mexico.
EDUCATING PATIENTS
4. What should patients know about their high-oxygen-affinity hemoglobinopathy?
- High altitudes and air travel can be risky
- Pregnancy may have adverse outcomes
- Systemic anticoagulation may lower the risk of venous thromboembolism
- Periodic phlebotomy may help control symptoms
Most patients with high-oxygen-affinity hemoglobin do not require specific clinical management but only counseling and education about their condition. Establishing an accurate diagnosis is important in order to avoid further inappropriate, invasive, and expensive testing.
Although exposure to high altitudes may be associated with decreased ambient oxygen levels, hypoxia is usually not a problem because of hemoglobin’s high affinity for oxygen.
Impaired delivery of oxygen across the placenta may be anticipated in a mother with high-oxygen-affinity hemoglobin, but this has not been observed clinically.17
Compared with patients with polycythemia vera, patients with high-oxygen-affinity hemoglobin have fewer complications from hyperviscosity and thrombosis, even with comparable degrees of erythrocytosis.
Although patients usually do not require treatment, phlebotomy may be helpful for symptoms that can be attributed to the higher hemoglobin concentration.
Our patient continues to be seen in clinic for periodic blood counts and phlebotomy for her headaches, as required.
HEMOGLOBIN: RELAXED OR TENSE
Normal adult hemoglobin is a tetramer composed of two pairs of globin polypeptide chains: alpha and beta (Figure 2). The intrinsic properties of the constituent globin chains and their allosteric conformation—as well as extrinsic factors including temperature, pH, and the binding of hydrogen ion and 2,3-BPG—play important roles in modifying the affinity of hemoglobin for oxygen. The major modulator of hemoglobin-oxygen affinity in human erythrocytes is 2,3-BPG.
The hemoglobin tetramer, consisting of two identical halves, alpha 1-beta 1 and alpha 2-beta 2, oscillates between two quaternary conformations, “relaxed” (fully oxygenated) and “tense” (fully deoxygenated).18 High-oxygen-affinity hemoglobins can result from factors that enhance the relaxed state, either by stabilizing the relaxed state or by destabilizing the tense state. Structural modifications in hemoglobin typically affect the main contacts involved in the transition from the deoxygenated to the oxygenated state, the 2,3-BPG binding sites, the heme pocket, or elongation of globin chains by various mutations. In hemoglobin Tarrant, the mutation prevents formation of noncovalent salt bridges in the alpha 1-beta 1 contact that normally stabilize the deoxygenated conformation of hemoglobin. As a result, the deoxygenated (tense) state is destabilized, shifting the allosteric equilibrium in favor of the oxygenated (relaxed) state with consequent high oxygen affinity.16
MORE ABOUT HIGH-OXYGEN-AFFINITY HEMOGLOBINS
The first case of erythrocytosis due to an abnormal hemoglobin was identified in 1966. This was an alpha chain variant with an arginine-to-leucine substitution at position 92, named hemoglobin Chesapeake.19
High-oxygen-affinity hemoglobin variants are usually transmitted as autosomal dominant traits. Patients are most often identified because of unexplained erythrocytosis detected on a routine blood cell count, as in our patient.
Not all high-oxygen-affinity hemoglobinopathies are associated with erythrocytosis. The degree of increased oxygen affinity may only be mild or the abnormal hemoglobin may be slightly unstable, thereby masking the usual clinical signs and symptoms.
Therapeutic phlebotomy should be used cautiously since it can decrease delivery of oxygen to tissues. A subset of patients whose symptoms are related to an elevated red cell mass may experience some relief, as did our patient.
- Kremyanskaya M, Mascarenhas J, Hoffman R. Why does my patient have erythrocytosis? Hematol Oncol Clin North Am 2012; 26:267–283.
- Keohane C, McMullin MF, Harrison C. The diagnosis and management of erythrocytosis. BMJ 2013; 347:f6667.
- Agarwal N, Gordeuk RV, Prchal JT. Genetic mechanisms underlying regulation of hemoglobin mass. Adv Exp Med Biol 2007; 618:195–210.
- Tefferi A. Polycythemia vera and essential thrombocythemia: 2012 update on diagnosis, risk stratification, and management. Am J Hematol 2012; 87:285–293.
- Landolfi R, Di Gennaro L, Falanga A. Thrombosis in myeloproliferative disorders: pathogenetic facts and speculation. Leukemia 2008; 22:2020–2028.
- Tefferi A, Spivak JL. Polycythemia vera: scientific advances and current practice. Semin Hematol 2005; 42:206–220.
- Ferrant A. What clinical and laboratory data are indicative of polycythemia and when are blood volume studies needed? Nouv Rev Fr Hematol 1994; 36:151–154.
- Fairbanks VF, Klee GG, Wiseman GA, et al. Measurement of blood volume and red cell mass: re-examination of 51Cr and 125I methods. Blood Cells Mol Dis 1996; 22:169–186; discussion 186a–186g.
- James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005; 434:1144–1148.
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127:2391–2405.
- Messinezy M, Westwood NB, El-Hemaidi I, Marsden JT, Sherwood RS, Pearson TC. Serum erythropoietin values in erythrocytosis and in primary thrombocythaemia. Br J Haematol 2002; 117:47–53.
- Hardison RC, Chui DHK, Giardine B, et al. HbVar: a relational database of human hemoglobin variants and thalassemia mutations at the globin gene server. Human Mutat 2002; 19:225–233.
- Percy MJ, Butt NN, Crotty GM, et al. Identification of high oxygen affinity hemoglobin variants in the investigation of patients with erythrocytosis. Haematologica 2009; 94:1321–1322.
- Kattamis AC, Kelly KM, Ohene-Frempong K, et al. Hb Osler [beta 145(HC2)Tyr-->Asp] results from posttranslational modification. Hemoglobin 1997; 21:109–120.
- Hoyer JD, Allen SL, Beutler E, Kubik K, West C, Fairbanks VF. Erythrocytosis due to bisphosphoglycerate mutase deficiency with concurrent glucose-6-phosphate dehydrogenase (G-6-PD) deficiency. Am J Hematol 2004; 75:205–208.
- Moo-Penn WF, Jue DL, Johnson MH, Wilson SM, Therrell B Jr, Schmidt RM. Hemoglobin Tarrant: alpha126(H9) asp leads to asn. A new hemoglobin variant in the alpha1beta1 contact region showing high oxygen affinity and reduced cooperativity. Biochim Biophys Acta 1977; 490:443–451.
- Bard H, Peri KG, Gagnon C. The biologic implications of a rare hemoglobin mutant that decreases oxygen affinity. Pediatr Res 2001; 49:69–73.
- Wajcman H, Galacteros F. Hemoglobins with high oxygen affinity leading to erythrocytosis: new variants and concepts. Hemoglobin 2005; 29:91–106.
- Clegg JB, Naughton MA, Weatherall DJ. Abnormal human haemoglobins. Separation and characterization of the alpha and beta chains by chromatography, and the determination of two new variants, hb Chesapeak and hb J (Bangkok). J Mol Biol 1966; 19:91–108.
- Kremyanskaya M, Mascarenhas J, Hoffman R. Why does my patient have erythrocytosis? Hematol Oncol Clin North Am 2012; 26:267–283.
- Keohane C, McMullin MF, Harrison C. The diagnosis and management of erythrocytosis. BMJ 2013; 347:f6667.
- Agarwal N, Gordeuk RV, Prchal JT. Genetic mechanisms underlying regulation of hemoglobin mass. Adv Exp Med Biol 2007; 618:195–210.
- Tefferi A. Polycythemia vera and essential thrombocythemia: 2012 update on diagnosis, risk stratification, and management. Am J Hematol 2012; 87:285–293.
- Landolfi R, Di Gennaro L, Falanga A. Thrombosis in myeloproliferative disorders: pathogenetic facts and speculation. Leukemia 2008; 22:2020–2028.
- Tefferi A, Spivak JL. Polycythemia vera: scientific advances and current practice. Semin Hematol 2005; 42:206–220.
- Ferrant A. What clinical and laboratory data are indicative of polycythemia and when are blood volume studies needed? Nouv Rev Fr Hematol 1994; 36:151–154.
- Fairbanks VF, Klee GG, Wiseman GA, et al. Measurement of blood volume and red cell mass: re-examination of 51Cr and 125I methods. Blood Cells Mol Dis 1996; 22:169–186; discussion 186a–186g.
- James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005; 434:1144–1148.
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127:2391–2405.
- Messinezy M, Westwood NB, El-Hemaidi I, Marsden JT, Sherwood RS, Pearson TC. Serum erythropoietin values in erythrocytosis and in primary thrombocythaemia. Br J Haematol 2002; 117:47–53.
- Hardison RC, Chui DHK, Giardine B, et al. HbVar: a relational database of human hemoglobin variants and thalassemia mutations at the globin gene server. Human Mutat 2002; 19:225–233.
- Percy MJ, Butt NN, Crotty GM, et al. Identification of high oxygen affinity hemoglobin variants in the investigation of patients with erythrocytosis. Haematologica 2009; 94:1321–1322.
- Kattamis AC, Kelly KM, Ohene-Frempong K, et al. Hb Osler [beta 145(HC2)Tyr-->Asp] results from posttranslational modification. Hemoglobin 1997; 21:109–120.
- Hoyer JD, Allen SL, Beutler E, Kubik K, West C, Fairbanks VF. Erythrocytosis due to bisphosphoglycerate mutase deficiency with concurrent glucose-6-phosphate dehydrogenase (G-6-PD) deficiency. Am J Hematol 2004; 75:205–208.
- Moo-Penn WF, Jue DL, Johnson MH, Wilson SM, Therrell B Jr, Schmidt RM. Hemoglobin Tarrant: alpha126(H9) asp leads to asn. A new hemoglobin variant in the alpha1beta1 contact region showing high oxygen affinity and reduced cooperativity. Biochim Biophys Acta 1977; 490:443–451.
- Bard H, Peri KG, Gagnon C. The biologic implications of a rare hemoglobin mutant that decreases oxygen affinity. Pediatr Res 2001; 49:69–73.
- Wajcman H, Galacteros F. Hemoglobins with high oxygen affinity leading to erythrocytosis: new variants and concepts. Hemoglobin 2005; 29:91–106.
- Clegg JB, Naughton MA, Weatherall DJ. Abnormal human haemoglobins. Separation and characterization of the alpha and beta chains by chromatography, and the determination of two new variants, hb Chesapeak and hb J (Bangkok). J Mol Biol 1966; 19:91–108.
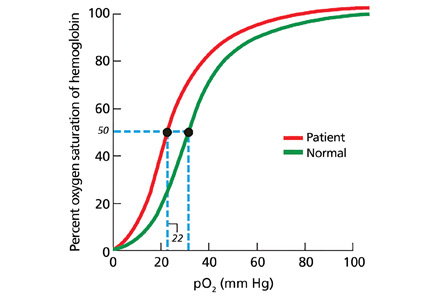
A young woman with enlarged lymph nodes
A previously healthy 25-year-old woman presents to her primary care physician with a “lump in the neck”—a painless, swollen area under the lower part of her left jaw that she noticed several weeks ago and that continues to enlarge. She has also noted a recent increase in fatigue, as well as the onset of generalized headaches and mild sinus congestion. Presumed by another physician to have sinusitis, she had already received a 2-week course of an antibiotic (she could not recall which antibiotic), with no improvement in her symptoms. She has been trying to lose weight and has lost 5 pounds in the last 4 months. She reports no fevers, chills, or night sweats.
She works as a special-education teacher and lives in a rural area. She has not travelled during the past year, inside or outside the United States. When she was an adolescent, she underwent tonsillectomy and had her wisdom teeth extracted. Her family has no history of hematologic dyscrasia or malignancy. She has two dogs, which are indoor pets, and she utilizes a city water supply.
1. Which of the following causes of a lump in the neck is most important to exclude?
- Viral or bacterial infection
- Lymphoma
- Oral cavity abscess
- Infectious mononucleosis
- Congenital anomaly
A lump in the neck can be broadly categorized as congenital, inflammatory, or malignant. Congenital causes include branchial cleft cyst (anterior to the sternocleidomastoid muscle) and thyroglossal duct cyst (usually in the midline between the hyoid bone and the isthmus of the thyroid gland). Other possibilities include lipoma and, less frequently, a salivary gland disorder such as sialadenitis.
A complaint of a neck lump very often correlates with the physical finding of lymphadenopathy, and a standard approach in evaluation should be undertaken, based on the mnemonic “PAAA”—ie, palpation, age, area, and associated symptoms.
Palpation. In palpation of the lymph node group, one should note the size and tactile quality of the lymph nodes and assess for abnormal temperature, tenderness, fluctuance, and mobility. In general, lymph nodes larger than 1.5 cm by 1.5 cm are more likely to be of granulomatous or neoplastic origin.1 Nodes that are tender, warm, or fluctuant are likely reactive to a local infectious process; nodes that are firm, matted, and fixed are most characteristic of malignancy; and rubbery, mobile nodes may represent either granulomatous disease or lymphoma.2
Age helps stratify the risk of malignancy as an underlying cause, which is increased in people over age 50 presenting with lymphadenopathy.1
Area. An assessment of the extent of the lymphadenopathy can guide the search either for a cause of generalized lymphadenopathy or for pathology in the anatomic area drained by the particular lymph node group, including the scalp (occipital or preauricular); external ear (posterior auricular); oral cavity (submandibular, submental); soft tissues of the face and neck (superficial cervical); upper respiratory tract and thyroid (deep cervical); and thoracic cavity and abdominal cavity (supraclavicular).

Asking the patient about occupational, environmental, and behavioral risk factors and associated signs and symptoms such as fever, rash, diaphoresis, unintentional weight loss, and splenomegaly helps to narrow the differential diagnosis. Common diagnoses to consider in the evaluation of peripheral lymphadenopathy are listed in Table 1.
A viral or bacterial upper respiratory infection is one of the most common causes of cervical lymphadenopathy, although this usually does not persist for many weeks. Mononucleosis more commonly involves the posterior cervical chain and is often accompanied by splenomegaly. Because of the prolonged presence of the lump, malignancy, including lymphoma, is the most important of the answer choices to consider and rule out in a timely fashion.
INITIAL PHYSICAL EXAMINATION
The woman appears to be well and is in no acute distress. Her oral temperature is 98.1°F (36.7°C), blood pressure 119/72 mm Hg, heart rate 86 beats per minute, and respiratory rate 18 breaths per minute.
The head and neck appear normal. The nares are patent with normal mucosa and no visible drainage. There is no tenderness during palpation of the facial sinuses. The ear canals, tympanic membranes, oropharynx, and tongue appear normal. Several firm, mobile, nontender lymph nodes about 1 cm in diameter are palpable in the left submandibular and right supraclavicular area. No other occipital, submental, axillary, or inguinal lymphadenopathy is noted. There is no overlying erythema or warmth. The cardiac examination is normal, and the lungs are clear on auscultation. The abdomen is soft, nontender, and nondistended, with no organomegaly. The skin appears normal, and the neurologic examination is normal.
INITIAL LABORATORY TESTS
Results of initial laboratory tests are as follows:
- White blood cell count 5.74 × 109/L (reference range 3.70–11.00)
- Red blood cell count 4.49 × 109/L (3.90–5.20)
- Hemoglobin 13.0 g/dL (11.5–15.5)
- Hematocrit 38.4% (36.0–46.0)
- Platelet count 210 × 109/L (150–400)
- Mean corpuscular volume 85 fL (80–100)
- Absolute neutrophil count 3.26 × 109/L (1.45–7.50)
- Blood urea nitrogen 8 mg/dL (8–25)
- Creatinine 0.65 mg/dL (0.70–1.40)
- Lactate dehydrogenase 146 U/L (100–220)
- Uric acid 4.0 mg/dL (2.0–7.0)
- Thyrotropin (thyroid-stimulating hormone) 1.86 μIU/mL (0.4–5.5).
A recent tuberculin skin test obtained as part of her employment screening was negative, and so was a test for antibody to human immunodeficiency virus (HIV), obtained recently before donating plasma. A urine pregnancy test done in the office was also negative. A peripheral blood smear showed slight toxic granulation with rare reactive lymphocytes.
2. Which test would provide the greatest diagnostic yield at this point?
- Needle aspiration biopsy of lymph node
- Excisional lymph node biopsy
- Polymerase chain reaction (PCR) testing for HIV
- Antistreptolysin-O (ASO) titer
Because of the persistent enlargement of the patient’s lymph nodes despite several weeks of antibiotic treatment, and because submandibular and supraclavicular nodes were involved, excisional lymph node biopsy would be the best of these choices to evaluate for malignancy. Compared with needle aspiration biopsy, it is the gold standard, preserving the nodal architecture and providing ample tissue for immunostaining and additional studies.
Needle aspiration biopsy is safe, inexpensive, and easy to do and can be useful in situations of limited resources, but it does not reliably distinguish between a reactive and a neoplastic process.1 Its collection and interpretation are highly variable and personnel-dependent, and its sensitivity for detecting lymphoma is reported to be as low as 7.1% (95% confidence interval 0.9% to 23.5%).2
An acute retroviral syndrome can cause adenopathy, especially before seroconversion is evident, but it is usually associated with an influenza-like illness and monocytosis. Although this patient had no apparent risk factors for HIV, ordering PCR testing for HIV is also an important step when the clinical situation is suggestive. In the absence of an abnormal-appearing oropharynx, tonsillar exudate, or high fever, the pretest probability of streptococcal pharyngitis is low, and an ASO titer is unlikely to be diagnostic in this case.
CASE CONTINUED: BIOPSY PERFORMED
An incisional lymph node biopsy was obtained (Figure 1).
3. Which can confirm the suspected diagnosis?
- Tissue culture
- Test for immunoglobulin (Ig) G and IgM antibodies to Toxoplasma gondii
- Serum PCR testing for T gondii
- T gondii IgG avidity testing
DIAGNOSIS OF TOXOPLASMOSIS
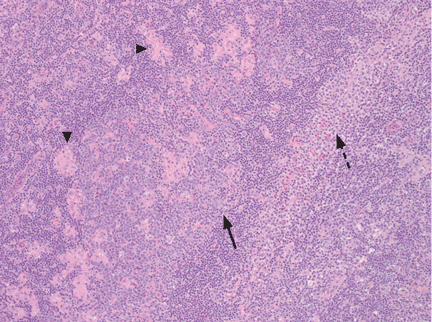
In this patient, acute toxoplasmosis was suspected based on recognition of the morphologic triad seen in Toxoplasma lymphadenitis— ie, follicular hyperplasia, abundance of monocytoid cells, and clusters of epithelioid lymphocytes.3,4
Detection and measurement of IgM antibodies against T gondii is the most widely used serologic test for acute toxoplasmosis and is often considered the reference standard among the most common commercially available agglutination screening assays. It has a sensitivity between 93.7% and 100% and a specificity of 97.1% to 99.2%.5 Confirmation is generally done with enzyme-linked immunoassay or chemiluminescent-based tests, which can detect lower levels of IgG and IgM.5
A positive serum IgG confirms seroconversion but by itself cannot distinguish between acute and chronic infection, although it is commonly obtained in conjunction with IgM levels.6 Since both IgG and IgM can be elevated months after initial infection, serum IgA and IgE levels can more accurately suggest the timing of infection if clarification is needed.6,7 In addition, IgG avidity testing can distinguish acute infection from chronic infection: a high avidity index suggests the acute infection occurred at least 3 to 5 months ago, whereas the avidity index may be low or zero if acute infection occurred within the past 4 weeks.7 The sensitivity of avidity testing is 91.3% to 94.4%, and the specificity 87.8% to 98.5%.7
Serum PCR testing for T gondii is useful when toxoplasmosis is suspected in patients whose immune system may not be able to mount an adequate antibody response or in patients in the hyperacute phase of infection, even before a detectable antibody response can be formed.8,9 However, because of limitations of equipment, expertise, and overall cost, this method is not universally available. Additionally, blood cultures and PCR testing or tissue culture of pathologic specimens cannot routinely be relied on for diagnosis, as often the burden of microorganisms present in these specimens is low. When positive, culture specimens may yield bradyzoites or tachyzoites, but only after considerable latency of many days to weeks.10
How people acquire acute toxoplasmosis
T gondii is an obligate intracellular protozoan parasite. Sexual replication of the organism takes place within the intestines of cats (the definitive host), with subsequent excretion of infective oocysts in feces.11 These hardy oocysts can contaminate soil or water supplies and can survive for months, depending on ambient temperature and humidity. Ingestion of oocysts can lead to infection of a variety of mammals, including sheep, pigs, chicken, and cattle.
Infection in humans can occur with consumption of raw or undercooked foods contaminated with oocysts, and inadequate hand-washing and poor kitchen hygiene substantially increase the risk of infection.12 Activities such as gardening can expose humans to oocysts in contaminated water and soil. In addition, direct contact with cat feces, such as when cleaning the litterbox, is a known exposure risk. Vertical transmission can manifest as congenital toxoplasmosis in a fetus when transmitted from an infected pregnant mother.
Eating raw or undercooked food is considered to be the greatest risk factor for acquired toxoplasmosis and is believed to be responsible for about 50% of all cases.12 However, in pooled data from 14 case-control studies, no clear risk factor for Toxoplasma infection could be identified in up to 60% of affected people, leading many experts to believe contaminated water may play a larger role in acquisition than previously surmised.12
Toxoplasma cysts have a predilection for muscle and neural tissue, resulting in myositis, myocarditis, encephalitis, and chorioretinitis. Severe systemic manifestations are seen in people with impaired T-cell immunity, such as those with HIV infection and acquired immunodeficiency syndrome; or hematologic malignancy; in recipients of solid-organ transplants; or in people taking corticosteroids or cytotoxic drugs. Congenital infection can result in stillbirth, microcephaly, developmental delay, or deafness in the developing fetus and is an important cause of infant morbidity and death worldwide.13
Infection in immunocompetent people is usually asymptomatic.14 However, up to 20% of immunocompetent patients develop symptoms that tend to be nonspecific and include muscle aches and lymphadenopathy, and these are often mistaken for an influenza-like illness.14,15 Other symptoms include malaise, fevers, night sweats, pharyngitis, abdominal pain, hepatosplenomegaly, maculopapular rash, and atypical lymphocytosis (less than 10% of peripheral blood).11 The most common physical manifestation of acute toxoplasmosis is isolated cervical lymphadenopathy, although any lymph node group can be affected.14 Lymph nodes are not fixed or matted and generally are neither tender nor suppurative.16
4. What is the correct treatment strategy for acute toxoplasmosis in this case?
- Symptomatic treatment only
- Trimethoprim 160 mg and sulfamethoxazole 800 mg daily
- Combination of atovaquone and clindamycin
- Combination pyrimethamine, sulfadiazine, and folinic acid
TREATMENT AND PROGNOSIS OF ACUTE TOXOPLASMOSIS
No antimicrobial treatment is required for most immunocompetent patients. Symptoms are self-limited and resolve within 1 to 2 months in 60% of patients.14 A substantial proportion of patients—25%—will have lingering symptoms at 2 to 4 months, and some (10%) can have mild symptoms for 6 months or longer.16
Symptomatic treatment with analgesics such as nonsteroidal anti-inflammatory drugs (NSAIDs) is appropriate.
Immunocompromised and critically ill patients and those with ocular manifestations require combination therapy with pyrimethamine, sulfadiazine, and folinic acid.17 Trimethoprim-sulfamethoxazole is effective as prophylaxis against T gondii infection in immunocompromised patients at a dosage of 160 mg trimethoprim/800 mg sulfamethoxazole daily, but it is also an alternative for treatment at higher dosages (5 mg/kg trimethoprim and 25 mg/kg sulfamethoxazole twice daily).
Atovaquone and clindamycin can be used in sulfa-sensitive patients17 and also in those with latent toxoplasmosis for better penetration of tissue cysts. Corticosteroids are used as adjuncts in those with ocular involvement.
Spiramycin is the treatment of choice in pregnant women and can be given throughout the pregnancy.17,18 A recent comparative study by Hotop et al18 reported a reduction in the rate of fetal transmission (1.6% vs 4.8%) when spiramycin was given from the time of diagnosis through the 16th week of pregnancy, followed by a minimum of 4 weeks of combination therapy with pyrimethamine, sulfadiazine, and folinic acid.18
CASE CONCLUDED
Serologic testing was positive for IgM and IgG antibodies to T gondii, which suggested subacute infection. The patient received no antimicrobial therapy and her lymphadenopathy eventually resolved. Her generalized fatigue gradually resolved over the next year without antimicrobial treatment.
A thorough re-review of potential exposures was done at subsequent office visits to help elucidate how she may have acquired the infection. She recalled no recent exposure to cats or rodents, nor consumption of raw meat. We could only suppose that there may have been inadvertent exposure to oocyst-containing soil or water or to undercooked meat products. Thus, the diagnosis of acute toxoplasmosis should be kept in mind in the evaluation of lymphadenopathy, even in the absence of a clear history of exposure.
- Habermann TM, Steensma DP. Lymphadenopathy. Mayo Clin Proc 2000; 75:723–732.
- Khillan R, Sidhu G, Axiotis C, Braverman AS. Fine needle aspiration (FNA) cytology for diagnosis of cervical lymphadenopathy. Int J Hematol 2012; 95:282–284.
- Dorfman RF, Remington JS. Value of lymph-node biopsy in the diagnosis of acute acquired toxoplasmosis. N Engl J Med 1973; 289:878–881.
- Eapen M, Mathew CF, Aravindan KP. Evidence based criteria for the histopathological diagnosis of toxoplasmic lymphadenopathy. J Clin Pathol 2005; 58:1143–1146.
- Villard O, Cimon B, Franck J, et al; Network from the French National Reference Center for Toxoplasmosis. Evaluation of the usefulness of six commercial agglutination assays for serologic diagnosis of toxoplasmosis. Diagn Microbiol Infect Dis 2012; 73:231–235.
- Suzuki LA, Rocha RJ, Rossi CL. Evaluation of serological markers for the immunodiagnosis of acute acquired toxoplasmosis. J Med Microbiol 2001; 50:62–70.
- Lachaud L, Calas O, Picot MC, Albaba S, Bourgeois N, Pratlong F. Value of 2 IgG avidity commercial tests used alone or in association to date toxoplasmosis contamination. Diagn Microbiol Infect Dis 2009; 64:267–274.
- Rahumatullah A, Khoo BY, Noordin R. Triplex PCR using new primers for the detection of Toxoplasma gondii. Exp Parasitol 2012; 131:231–238.
- Contini C, Giuliodori M, Cultrera R, Seraceni S. Detection of clinical-stage specific molecular Toxoplasma gondii gene patterns in patients with toxoplasmic lymphadenitis. J Med Microbiol 2006; 55:771–774.
- Silveira C, Vallochi AL, Rodrigues da Silva U, et al. Toxoplasma gondii in the peripheral blood of patients with acute and chronic toxoplasmosis. Br J Ophthalmol 2011; 95:396–400.
- Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet 2004; 363:1965–1976.
- Petersen E, Vesco G, Villari S, Buffolano W. What do we know about risk factors for infection in humans with Toxoplasma gondii and how can we prevent infections? Zoonoses Public Health 2010; 57:8–17.
- Feldman DM, Timms D, Borgida AF. Toxoplasmosis, parvovirus, and cytomegalovirus in pregnancy. Clin Lab Med 2010; 30:709–720.
- Weiss LM, Dubey JP. Toxoplasmosis: a history of clinical observations. Int J Parasitol 2009; 39:895–901.
- Remington JS. Toxoplasmosis in the adult. Bull NY Acad Med 1974; 50:211–227.
- McCabe RE, Brooks RG, Dorfman RF, Remington JS. Clinical spectrum in 107 cases of toxoplasmic lymphadenopathy. Rev Infect Dis 1987; 9:754–774.
- Toxoplasmosis. The Medical Letter, Drugs for Parasitic Infections. New Rochelle, NY: The Medical Letter Inc, June 1, 2010:57–58.
- Hotop A, Hlobil H, Gross U. Efficacy of rapid treatment initiation following primary Toxoplasma gondii infection during pregnancy. Clin Infect Dis 2012; 54:1545–1552.
A previously healthy 25-year-old woman presents to her primary care physician with a “lump in the neck”—a painless, swollen area under the lower part of her left jaw that she noticed several weeks ago and that continues to enlarge. She has also noted a recent increase in fatigue, as well as the onset of generalized headaches and mild sinus congestion. Presumed by another physician to have sinusitis, she had already received a 2-week course of an antibiotic (she could not recall which antibiotic), with no improvement in her symptoms. She has been trying to lose weight and has lost 5 pounds in the last 4 months. She reports no fevers, chills, or night sweats.
She works as a special-education teacher and lives in a rural area. She has not travelled during the past year, inside or outside the United States. When she was an adolescent, she underwent tonsillectomy and had her wisdom teeth extracted. Her family has no history of hematologic dyscrasia or malignancy. She has two dogs, which are indoor pets, and she utilizes a city water supply.
1. Which of the following causes of a lump in the neck is most important to exclude?
- Viral or bacterial infection
- Lymphoma
- Oral cavity abscess
- Infectious mononucleosis
- Congenital anomaly
A lump in the neck can be broadly categorized as congenital, inflammatory, or malignant. Congenital causes include branchial cleft cyst (anterior to the sternocleidomastoid muscle) and thyroglossal duct cyst (usually in the midline between the hyoid bone and the isthmus of the thyroid gland). Other possibilities include lipoma and, less frequently, a salivary gland disorder such as sialadenitis.
A complaint of a neck lump very often correlates with the physical finding of lymphadenopathy, and a standard approach in evaluation should be undertaken, based on the mnemonic “PAAA”—ie, palpation, age, area, and associated symptoms.
Palpation. In palpation of the lymph node group, one should note the size and tactile quality of the lymph nodes and assess for abnormal temperature, tenderness, fluctuance, and mobility. In general, lymph nodes larger than 1.5 cm by 1.5 cm are more likely to be of granulomatous or neoplastic origin.1 Nodes that are tender, warm, or fluctuant are likely reactive to a local infectious process; nodes that are firm, matted, and fixed are most characteristic of malignancy; and rubbery, mobile nodes may represent either granulomatous disease or lymphoma.2
Age helps stratify the risk of malignancy as an underlying cause, which is increased in people over age 50 presenting with lymphadenopathy.1
Area. An assessment of the extent of the lymphadenopathy can guide the search either for a cause of generalized lymphadenopathy or for pathology in the anatomic area drained by the particular lymph node group, including the scalp (occipital or preauricular); external ear (posterior auricular); oral cavity (submandibular, submental); soft tissues of the face and neck (superficial cervical); upper respiratory tract and thyroid (deep cervical); and thoracic cavity and abdominal cavity (supraclavicular).
Asking the patient about occupational, environmental, and behavioral risk factors and associated signs and symptoms such as fever, rash, diaphoresis, unintentional weight loss, and splenomegaly helps to narrow the differential diagnosis. Common diagnoses to consider in the evaluation of peripheral lymphadenopathy are listed in Table 1.
A viral or bacterial upper respiratory infection is one of the most common causes of cervical lymphadenopathy, although this usually does not persist for many weeks. Mononucleosis more commonly involves the posterior cervical chain and is often accompanied by splenomegaly. Because of the prolonged presence of the lump, malignancy, including lymphoma, is the most important of the answer choices to consider and rule out in a timely fashion.
INITIAL PHYSICAL EXAMINATION
The woman appears to be well and is in no acute distress. Her oral temperature is 98.1°F (36.7°C), blood pressure 119/72 mm Hg, heart rate 86 beats per minute, and respiratory rate 18 breaths per minute.
The head and neck appear normal. The nares are patent with normal mucosa and no visible drainage. There is no tenderness during palpation of the facial sinuses. The ear canals, tympanic membranes, oropharynx, and tongue appear normal. Several firm, mobile, nontender lymph nodes about 1 cm in diameter are palpable in the left submandibular and right supraclavicular area. No other occipital, submental, axillary, or inguinal lymphadenopathy is noted. There is no overlying erythema or warmth. The cardiac examination is normal, and the lungs are clear on auscultation. The abdomen is soft, nontender, and nondistended, with no organomegaly. The skin appears normal, and the neurologic examination is normal.
INITIAL LABORATORY TESTS
Results of initial laboratory tests are as follows:
- White blood cell count 5.74 × 109/L (reference range 3.70–11.00)
- Red blood cell count 4.49 × 109/L (3.90–5.20)
- Hemoglobin 13.0 g/dL (11.5–15.5)
- Hematocrit 38.4% (36.0–46.0)
- Platelet count 210 × 109/L (150–400)
- Mean corpuscular volume 85 fL (80–100)
- Absolute neutrophil count 3.26 × 109/L (1.45–7.50)
- Blood urea nitrogen 8 mg/dL (8–25)
- Creatinine 0.65 mg/dL (0.70–1.40)
- Lactate dehydrogenase 146 U/L (100–220)
- Uric acid 4.0 mg/dL (2.0–7.0)
- Thyrotropin (thyroid-stimulating hormone) 1.86 μIU/mL (0.4–5.5).
A recent tuberculin skin test obtained as part of her employment screening was negative, and so was a test for antibody to human immunodeficiency virus (HIV), obtained recently before donating plasma. A urine pregnancy test done in the office was also negative. A peripheral blood smear showed slight toxic granulation with rare reactive lymphocytes.
2. Which test would provide the greatest diagnostic yield at this point?
- Needle aspiration biopsy of lymph node
- Excisional lymph node biopsy
- Polymerase chain reaction (PCR) testing for HIV
- Antistreptolysin-O (ASO) titer
Because of the persistent enlargement of the patient’s lymph nodes despite several weeks of antibiotic treatment, and because submandibular and supraclavicular nodes were involved, excisional lymph node biopsy would be the best of these choices to evaluate for malignancy. Compared with needle aspiration biopsy, it is the gold standard, preserving the nodal architecture and providing ample tissue for immunostaining and additional studies.
Needle aspiration biopsy is safe, inexpensive, and easy to do and can be useful in situations of limited resources, but it does not reliably distinguish between a reactive and a neoplastic process.1 Its collection and interpretation are highly variable and personnel-dependent, and its sensitivity for detecting lymphoma is reported to be as low as 7.1% (95% confidence interval 0.9% to 23.5%).2
An acute retroviral syndrome can cause adenopathy, especially before seroconversion is evident, but it is usually associated with an influenza-like illness and monocytosis. Although this patient had no apparent risk factors for HIV, ordering PCR testing for HIV is also an important step when the clinical situation is suggestive. In the absence of an abnormal-appearing oropharynx, tonsillar exudate, or high fever, the pretest probability of streptococcal pharyngitis is low, and an ASO titer is unlikely to be diagnostic in this case.
CASE CONTINUED: BIOPSY PERFORMED
An incisional lymph node biopsy was obtained (Figure 1).
3. Which can confirm the suspected diagnosis?
- Tissue culture
- Test for immunoglobulin (Ig) G and IgM antibodies to Toxoplasma gondii
- Serum PCR testing for T gondii
- T gondii IgG avidity testing
DIAGNOSIS OF TOXOPLASMOSIS
In this patient, acute toxoplasmosis was suspected based on recognition of the morphologic triad seen in Toxoplasma lymphadenitis— ie, follicular hyperplasia, abundance of monocytoid cells, and clusters of epithelioid lymphocytes.3,4
Detection and measurement of IgM antibodies against T gondii is the most widely used serologic test for acute toxoplasmosis and is often considered the reference standard among the most common commercially available agglutination screening assays. It has a sensitivity between 93.7% and 100% and a specificity of 97.1% to 99.2%.5 Confirmation is generally done with enzyme-linked immunoassay or chemiluminescent-based tests, which can detect lower levels of IgG and IgM.5
A positive serum IgG confirms seroconversion but by itself cannot distinguish between acute and chronic infection, although it is commonly obtained in conjunction with IgM levels.6 Since both IgG and IgM can be elevated months after initial infection, serum IgA and IgE levels can more accurately suggest the timing of infection if clarification is needed.6,7 In addition, IgG avidity testing can distinguish acute infection from chronic infection: a high avidity index suggests the acute infection occurred at least 3 to 5 months ago, whereas the avidity index may be low or zero if acute infection occurred within the past 4 weeks.7 The sensitivity of avidity testing is 91.3% to 94.4%, and the specificity 87.8% to 98.5%.7
Serum PCR testing for T gondii is useful when toxoplasmosis is suspected in patients whose immune system may not be able to mount an adequate antibody response or in patients in the hyperacute phase of infection, even before a detectable antibody response can be formed.8,9 However, because of limitations of equipment, expertise, and overall cost, this method is not universally available. Additionally, blood cultures and PCR testing or tissue culture of pathologic specimens cannot routinely be relied on for diagnosis, as often the burden of microorganisms present in these specimens is low. When positive, culture specimens may yield bradyzoites or tachyzoites, but only after considerable latency of many days to weeks.10
How people acquire acute toxoplasmosis
T gondii is an obligate intracellular protozoan parasite. Sexual replication of the organism takes place within the intestines of cats (the definitive host), with subsequent excretion of infective oocysts in feces.11 These hardy oocysts can contaminate soil or water supplies and can survive for months, depending on ambient temperature and humidity. Ingestion of oocysts can lead to infection of a variety of mammals, including sheep, pigs, chicken, and cattle.
Infection in humans can occur with consumption of raw or undercooked foods contaminated with oocysts, and inadequate hand-washing and poor kitchen hygiene substantially increase the risk of infection.12 Activities such as gardening can expose humans to oocysts in contaminated water and soil. In addition, direct contact with cat feces, such as when cleaning the litterbox, is a known exposure risk. Vertical transmission can manifest as congenital toxoplasmosis in a fetus when transmitted from an infected pregnant mother.
Eating raw or undercooked food is considered to be the greatest risk factor for acquired toxoplasmosis and is believed to be responsible for about 50% of all cases.12 However, in pooled data from 14 case-control studies, no clear risk factor for Toxoplasma infection could be identified in up to 60% of affected people, leading many experts to believe contaminated water may play a larger role in acquisition than previously surmised.12
Toxoplasma cysts have a predilection for muscle and neural tissue, resulting in myositis, myocarditis, encephalitis, and chorioretinitis. Severe systemic manifestations are seen in people with impaired T-cell immunity, such as those with HIV infection and acquired immunodeficiency syndrome; or hematologic malignancy; in recipients of solid-organ transplants; or in people taking corticosteroids or cytotoxic drugs. Congenital infection can result in stillbirth, microcephaly, developmental delay, or deafness in the developing fetus and is an important cause of infant morbidity and death worldwide.13
Infection in immunocompetent people is usually asymptomatic.14 However, up to 20% of immunocompetent patients develop symptoms that tend to be nonspecific and include muscle aches and lymphadenopathy, and these are often mistaken for an influenza-like illness.14,15 Other symptoms include malaise, fevers, night sweats, pharyngitis, abdominal pain, hepatosplenomegaly, maculopapular rash, and atypical lymphocytosis (less than 10% of peripheral blood).11 The most common physical manifestation of acute toxoplasmosis is isolated cervical lymphadenopathy, although any lymph node group can be affected.14 Lymph nodes are not fixed or matted and generally are neither tender nor suppurative.16
4. What is the correct treatment strategy for acute toxoplasmosis in this case?
- Symptomatic treatment only
- Trimethoprim 160 mg and sulfamethoxazole 800 mg daily
- Combination of atovaquone and clindamycin
- Combination pyrimethamine, sulfadiazine, and folinic acid
TREATMENT AND PROGNOSIS OF ACUTE TOXOPLASMOSIS
No antimicrobial treatment is required for most immunocompetent patients. Symptoms are self-limited and resolve within 1 to 2 months in 60% of patients.14 A substantial proportion of patients—25%—will have lingering symptoms at 2 to 4 months, and some (10%) can have mild symptoms for 6 months or longer.16
Symptomatic treatment with analgesics such as nonsteroidal anti-inflammatory drugs (NSAIDs) is appropriate.
Immunocompromised and critically ill patients and those with ocular manifestations require combination therapy with pyrimethamine, sulfadiazine, and folinic acid.17 Trimethoprim-sulfamethoxazole is effective as prophylaxis against T gondii infection in immunocompromised patients at a dosage of 160 mg trimethoprim/800 mg sulfamethoxazole daily, but it is also an alternative for treatment at higher dosages (5 mg/kg trimethoprim and 25 mg/kg sulfamethoxazole twice daily).
Atovaquone and clindamycin can be used in sulfa-sensitive patients17 and also in those with latent toxoplasmosis for better penetration of tissue cysts. Corticosteroids are used as adjuncts in those with ocular involvement.
Spiramycin is the treatment of choice in pregnant women and can be given throughout the pregnancy.17,18 A recent comparative study by Hotop et al18 reported a reduction in the rate of fetal transmission (1.6% vs 4.8%) when spiramycin was given from the time of diagnosis through the 16th week of pregnancy, followed by a minimum of 4 weeks of combination therapy with pyrimethamine, sulfadiazine, and folinic acid.18
CASE CONCLUDED
Serologic testing was positive for IgM and IgG antibodies to T gondii, which suggested subacute infection. The patient received no antimicrobial therapy and her lymphadenopathy eventually resolved. Her generalized fatigue gradually resolved over the next year without antimicrobial treatment.
A thorough re-review of potential exposures was done at subsequent office visits to help elucidate how she may have acquired the infection. She recalled no recent exposure to cats or rodents, nor consumption of raw meat. We could only suppose that there may have been inadvertent exposure to oocyst-containing soil or water or to undercooked meat products. Thus, the diagnosis of acute toxoplasmosis should be kept in mind in the evaluation of lymphadenopathy, even in the absence of a clear history of exposure.
A previously healthy 25-year-old woman presents to her primary care physician with a “lump in the neck”—a painless, swollen area under the lower part of her left jaw that she noticed several weeks ago and that continues to enlarge. She has also noted a recent increase in fatigue, as well as the onset of generalized headaches and mild sinus congestion. Presumed by another physician to have sinusitis, she had already received a 2-week course of an antibiotic (she could not recall which antibiotic), with no improvement in her symptoms. She has been trying to lose weight and has lost 5 pounds in the last 4 months. She reports no fevers, chills, or night sweats.
She works as a special-education teacher and lives in a rural area. She has not travelled during the past year, inside or outside the United States. When she was an adolescent, she underwent tonsillectomy and had her wisdom teeth extracted. Her family has no history of hematologic dyscrasia or malignancy. She has two dogs, which are indoor pets, and she utilizes a city water supply.
1. Which of the following causes of a lump in the neck is most important to exclude?
- Viral or bacterial infection
- Lymphoma
- Oral cavity abscess
- Infectious mononucleosis
- Congenital anomaly
A lump in the neck can be broadly categorized as congenital, inflammatory, or malignant. Congenital causes include branchial cleft cyst (anterior to the sternocleidomastoid muscle) and thyroglossal duct cyst (usually in the midline between the hyoid bone and the isthmus of the thyroid gland). Other possibilities include lipoma and, less frequently, a salivary gland disorder such as sialadenitis.
A complaint of a neck lump very often correlates with the physical finding of lymphadenopathy, and a standard approach in evaluation should be undertaken, based on the mnemonic “PAAA”—ie, palpation, age, area, and associated symptoms.
Palpation. In palpation of the lymph node group, one should note the size and tactile quality of the lymph nodes and assess for abnormal temperature, tenderness, fluctuance, and mobility. In general, lymph nodes larger than 1.5 cm by 1.5 cm are more likely to be of granulomatous or neoplastic origin.1 Nodes that are tender, warm, or fluctuant are likely reactive to a local infectious process; nodes that are firm, matted, and fixed are most characteristic of malignancy; and rubbery, mobile nodes may represent either granulomatous disease or lymphoma.2
Age helps stratify the risk of malignancy as an underlying cause, which is increased in people over age 50 presenting with lymphadenopathy.1
Area. An assessment of the extent of the lymphadenopathy can guide the search either for a cause of generalized lymphadenopathy or for pathology in the anatomic area drained by the particular lymph node group, including the scalp (occipital or preauricular); external ear (posterior auricular); oral cavity (submandibular, submental); soft tissues of the face and neck (superficial cervical); upper respiratory tract and thyroid (deep cervical); and thoracic cavity and abdominal cavity (supraclavicular).
Asking the patient about occupational, environmental, and behavioral risk factors and associated signs and symptoms such as fever, rash, diaphoresis, unintentional weight loss, and splenomegaly helps to narrow the differential diagnosis. Common diagnoses to consider in the evaluation of peripheral lymphadenopathy are listed in Table 1.
A viral or bacterial upper respiratory infection is one of the most common causes of cervical lymphadenopathy, although this usually does not persist for many weeks. Mononucleosis more commonly involves the posterior cervical chain and is often accompanied by splenomegaly. Because of the prolonged presence of the lump, malignancy, including lymphoma, is the most important of the answer choices to consider and rule out in a timely fashion.
INITIAL PHYSICAL EXAMINATION
The woman appears to be well and is in no acute distress. Her oral temperature is 98.1°F (36.7°C), blood pressure 119/72 mm Hg, heart rate 86 beats per minute, and respiratory rate 18 breaths per minute.
The head and neck appear normal. The nares are patent with normal mucosa and no visible drainage. There is no tenderness during palpation of the facial sinuses. The ear canals, tympanic membranes, oropharynx, and tongue appear normal. Several firm, mobile, nontender lymph nodes about 1 cm in diameter are palpable in the left submandibular and right supraclavicular area. No other occipital, submental, axillary, or inguinal lymphadenopathy is noted. There is no overlying erythema or warmth. The cardiac examination is normal, and the lungs are clear on auscultation. The abdomen is soft, nontender, and nondistended, with no organomegaly. The skin appears normal, and the neurologic examination is normal.
INITIAL LABORATORY TESTS
Results of initial laboratory tests are as follows:
- White blood cell count 5.74 × 109/L (reference range 3.70–11.00)
- Red blood cell count 4.49 × 109/L (3.90–5.20)
- Hemoglobin 13.0 g/dL (11.5–15.5)
- Hematocrit 38.4% (36.0–46.0)
- Platelet count 210 × 109/L (150–400)
- Mean corpuscular volume 85 fL (80–100)
- Absolute neutrophil count 3.26 × 109/L (1.45–7.50)
- Blood urea nitrogen 8 mg/dL (8–25)
- Creatinine 0.65 mg/dL (0.70–1.40)
- Lactate dehydrogenase 146 U/L (100–220)
- Uric acid 4.0 mg/dL (2.0–7.0)
- Thyrotropin (thyroid-stimulating hormone) 1.86 μIU/mL (0.4–5.5).
A recent tuberculin skin test obtained as part of her employment screening was negative, and so was a test for antibody to human immunodeficiency virus (HIV), obtained recently before donating plasma. A urine pregnancy test done in the office was also negative. A peripheral blood smear showed slight toxic granulation with rare reactive lymphocytes.
2. Which test would provide the greatest diagnostic yield at this point?
- Needle aspiration biopsy of lymph node
- Excisional lymph node biopsy
- Polymerase chain reaction (PCR) testing for HIV
- Antistreptolysin-O (ASO) titer
Because of the persistent enlargement of the patient’s lymph nodes despite several weeks of antibiotic treatment, and because submandibular and supraclavicular nodes were involved, excisional lymph node biopsy would be the best of these choices to evaluate for malignancy. Compared with needle aspiration biopsy, it is the gold standard, preserving the nodal architecture and providing ample tissue for immunostaining and additional studies.
Needle aspiration biopsy is safe, inexpensive, and easy to do and can be useful in situations of limited resources, but it does not reliably distinguish between a reactive and a neoplastic process.1 Its collection and interpretation are highly variable and personnel-dependent, and its sensitivity for detecting lymphoma is reported to be as low as 7.1% (95% confidence interval 0.9% to 23.5%).2
An acute retroviral syndrome can cause adenopathy, especially before seroconversion is evident, but it is usually associated with an influenza-like illness and monocytosis. Although this patient had no apparent risk factors for HIV, ordering PCR testing for HIV is also an important step when the clinical situation is suggestive. In the absence of an abnormal-appearing oropharynx, tonsillar exudate, or high fever, the pretest probability of streptococcal pharyngitis is low, and an ASO titer is unlikely to be diagnostic in this case.
CASE CONTINUED: BIOPSY PERFORMED
An incisional lymph node biopsy was obtained (Figure 1).
3. Which can confirm the suspected diagnosis?
- Tissue culture
- Test for immunoglobulin (Ig) G and IgM antibodies to Toxoplasma gondii
- Serum PCR testing for T gondii
- T gondii IgG avidity testing
DIAGNOSIS OF TOXOPLASMOSIS
In this patient, acute toxoplasmosis was suspected based on recognition of the morphologic triad seen in Toxoplasma lymphadenitis— ie, follicular hyperplasia, abundance of monocytoid cells, and clusters of epithelioid lymphocytes.3,4
Detection and measurement of IgM antibodies against T gondii is the most widely used serologic test for acute toxoplasmosis and is often considered the reference standard among the most common commercially available agglutination screening assays. It has a sensitivity between 93.7% and 100% and a specificity of 97.1% to 99.2%.5 Confirmation is generally done with enzyme-linked immunoassay or chemiluminescent-based tests, which can detect lower levels of IgG and IgM.5
A positive serum IgG confirms seroconversion but by itself cannot distinguish between acute and chronic infection, although it is commonly obtained in conjunction with IgM levels.6 Since both IgG and IgM can be elevated months after initial infection, serum IgA and IgE levels can more accurately suggest the timing of infection if clarification is needed.6,7 In addition, IgG avidity testing can distinguish acute infection from chronic infection: a high avidity index suggests the acute infection occurred at least 3 to 5 months ago, whereas the avidity index may be low or zero if acute infection occurred within the past 4 weeks.7 The sensitivity of avidity testing is 91.3% to 94.4%, and the specificity 87.8% to 98.5%.7
Serum PCR testing for T gondii is useful when toxoplasmosis is suspected in patients whose immune system may not be able to mount an adequate antibody response or in patients in the hyperacute phase of infection, even before a detectable antibody response can be formed.8,9 However, because of limitations of equipment, expertise, and overall cost, this method is not universally available. Additionally, blood cultures and PCR testing or tissue culture of pathologic specimens cannot routinely be relied on for diagnosis, as often the burden of microorganisms present in these specimens is low. When positive, culture specimens may yield bradyzoites or tachyzoites, but only after considerable latency of many days to weeks.10
How people acquire acute toxoplasmosis
T gondii is an obligate intracellular protozoan parasite. Sexual replication of the organism takes place within the intestines of cats (the definitive host), with subsequent excretion of infective oocysts in feces.11 These hardy oocysts can contaminate soil or water supplies and can survive for months, depending on ambient temperature and humidity. Ingestion of oocysts can lead to infection of a variety of mammals, including sheep, pigs, chicken, and cattle.
Infection in humans can occur with consumption of raw or undercooked foods contaminated with oocysts, and inadequate hand-washing and poor kitchen hygiene substantially increase the risk of infection.12 Activities such as gardening can expose humans to oocysts in contaminated water and soil. In addition, direct contact with cat feces, such as when cleaning the litterbox, is a known exposure risk. Vertical transmission can manifest as congenital toxoplasmosis in a fetus when transmitted from an infected pregnant mother.
Eating raw or undercooked food is considered to be the greatest risk factor for acquired toxoplasmosis and is believed to be responsible for about 50% of all cases.12 However, in pooled data from 14 case-control studies, no clear risk factor for Toxoplasma infection could be identified in up to 60% of affected people, leading many experts to believe contaminated water may play a larger role in acquisition than previously surmised.12
Toxoplasma cysts have a predilection for muscle and neural tissue, resulting in myositis, myocarditis, encephalitis, and chorioretinitis. Severe systemic manifestations are seen in people with impaired T-cell immunity, such as those with HIV infection and acquired immunodeficiency syndrome; or hematologic malignancy; in recipients of solid-organ transplants; or in people taking corticosteroids or cytotoxic drugs. Congenital infection can result in stillbirth, microcephaly, developmental delay, or deafness in the developing fetus and is an important cause of infant morbidity and death worldwide.13
Infection in immunocompetent people is usually asymptomatic.14 However, up to 20% of immunocompetent patients develop symptoms that tend to be nonspecific and include muscle aches and lymphadenopathy, and these are often mistaken for an influenza-like illness.14,15 Other symptoms include malaise, fevers, night sweats, pharyngitis, abdominal pain, hepatosplenomegaly, maculopapular rash, and atypical lymphocytosis (less than 10% of peripheral blood).11 The most common physical manifestation of acute toxoplasmosis is isolated cervical lymphadenopathy, although any lymph node group can be affected.14 Lymph nodes are not fixed or matted and generally are neither tender nor suppurative.16
4. What is the correct treatment strategy for acute toxoplasmosis in this case?
- Symptomatic treatment only
- Trimethoprim 160 mg and sulfamethoxazole 800 mg daily
- Combination of atovaquone and clindamycin
- Combination pyrimethamine, sulfadiazine, and folinic acid
TREATMENT AND PROGNOSIS OF ACUTE TOXOPLASMOSIS
No antimicrobial treatment is required for most immunocompetent patients. Symptoms are self-limited and resolve within 1 to 2 months in 60% of patients.14 A substantial proportion of patients—25%—will have lingering symptoms at 2 to 4 months, and some (10%) can have mild symptoms for 6 months or longer.16
Symptomatic treatment with analgesics such as nonsteroidal anti-inflammatory drugs (NSAIDs) is appropriate.
Immunocompromised and critically ill patients and those with ocular manifestations require combination therapy with pyrimethamine, sulfadiazine, and folinic acid.17 Trimethoprim-sulfamethoxazole is effective as prophylaxis against T gondii infection in immunocompromised patients at a dosage of 160 mg trimethoprim/800 mg sulfamethoxazole daily, but it is also an alternative for treatment at higher dosages (5 mg/kg trimethoprim and 25 mg/kg sulfamethoxazole twice daily).
Atovaquone and clindamycin can be used in sulfa-sensitive patients17 and also in those with latent toxoplasmosis for better penetration of tissue cysts. Corticosteroids are used as adjuncts in those with ocular involvement.
Spiramycin is the treatment of choice in pregnant women and can be given throughout the pregnancy.17,18 A recent comparative study by Hotop et al18 reported a reduction in the rate of fetal transmission (1.6% vs 4.8%) when spiramycin was given from the time of diagnosis through the 16th week of pregnancy, followed by a minimum of 4 weeks of combination therapy with pyrimethamine, sulfadiazine, and folinic acid.18
CASE CONCLUDED
Serologic testing was positive for IgM and IgG antibodies to T gondii, which suggested subacute infection. The patient received no antimicrobial therapy and her lymphadenopathy eventually resolved. Her generalized fatigue gradually resolved over the next year without antimicrobial treatment.
A thorough re-review of potential exposures was done at subsequent office visits to help elucidate how she may have acquired the infection. She recalled no recent exposure to cats or rodents, nor consumption of raw meat. We could only suppose that there may have been inadvertent exposure to oocyst-containing soil or water or to undercooked meat products. Thus, the diagnosis of acute toxoplasmosis should be kept in mind in the evaluation of lymphadenopathy, even in the absence of a clear history of exposure.
- Habermann TM, Steensma DP. Lymphadenopathy. Mayo Clin Proc 2000; 75:723–732.
- Khillan R, Sidhu G, Axiotis C, Braverman AS. Fine needle aspiration (FNA) cytology for diagnosis of cervical lymphadenopathy. Int J Hematol 2012; 95:282–284.
- Dorfman RF, Remington JS. Value of lymph-node biopsy in the diagnosis of acute acquired toxoplasmosis. N Engl J Med 1973; 289:878–881.
- Eapen M, Mathew CF, Aravindan KP. Evidence based criteria for the histopathological diagnosis of toxoplasmic lymphadenopathy. J Clin Pathol 2005; 58:1143–1146.
- Villard O, Cimon B, Franck J, et al; Network from the French National Reference Center for Toxoplasmosis. Evaluation of the usefulness of six commercial agglutination assays for serologic diagnosis of toxoplasmosis. Diagn Microbiol Infect Dis 2012; 73:231–235.
- Suzuki LA, Rocha RJ, Rossi CL. Evaluation of serological markers for the immunodiagnosis of acute acquired toxoplasmosis. J Med Microbiol 2001; 50:62–70.
- Lachaud L, Calas O, Picot MC, Albaba S, Bourgeois N, Pratlong F. Value of 2 IgG avidity commercial tests used alone or in association to date toxoplasmosis contamination. Diagn Microbiol Infect Dis 2009; 64:267–274.
- Rahumatullah A, Khoo BY, Noordin R. Triplex PCR using new primers for the detection of Toxoplasma gondii. Exp Parasitol 2012; 131:231–238.
- Contini C, Giuliodori M, Cultrera R, Seraceni S. Detection of clinical-stage specific molecular Toxoplasma gondii gene patterns in patients with toxoplasmic lymphadenitis. J Med Microbiol 2006; 55:771–774.
- Silveira C, Vallochi AL, Rodrigues da Silva U, et al. Toxoplasma gondii in the peripheral blood of patients with acute and chronic toxoplasmosis. Br J Ophthalmol 2011; 95:396–400.
- Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet 2004; 363:1965–1976.
- Petersen E, Vesco G, Villari S, Buffolano W. What do we know about risk factors for infection in humans with Toxoplasma gondii and how can we prevent infections? Zoonoses Public Health 2010; 57:8–17.
- Feldman DM, Timms D, Borgida AF. Toxoplasmosis, parvovirus, and cytomegalovirus in pregnancy. Clin Lab Med 2010; 30:709–720.
- Weiss LM, Dubey JP. Toxoplasmosis: a history of clinical observations. Int J Parasitol 2009; 39:895–901.
- Remington JS. Toxoplasmosis in the adult. Bull NY Acad Med 1974; 50:211–227.
- McCabe RE, Brooks RG, Dorfman RF, Remington JS. Clinical spectrum in 107 cases of toxoplasmic lymphadenopathy. Rev Infect Dis 1987; 9:754–774.
- Toxoplasmosis. The Medical Letter, Drugs for Parasitic Infections. New Rochelle, NY: The Medical Letter Inc, June 1, 2010:57–58.
- Hotop A, Hlobil H, Gross U. Efficacy of rapid treatment initiation following primary Toxoplasma gondii infection during pregnancy. Clin Infect Dis 2012; 54:1545–1552.
- Habermann TM, Steensma DP. Lymphadenopathy. Mayo Clin Proc 2000; 75:723–732.
- Khillan R, Sidhu G, Axiotis C, Braverman AS. Fine needle aspiration (FNA) cytology for diagnosis of cervical lymphadenopathy. Int J Hematol 2012; 95:282–284.
- Dorfman RF, Remington JS. Value of lymph-node biopsy in the diagnosis of acute acquired toxoplasmosis. N Engl J Med 1973; 289:878–881.
- Eapen M, Mathew CF, Aravindan KP. Evidence based criteria for the histopathological diagnosis of toxoplasmic lymphadenopathy. J Clin Pathol 2005; 58:1143–1146.
- Villard O, Cimon B, Franck J, et al; Network from the French National Reference Center for Toxoplasmosis. Evaluation of the usefulness of six commercial agglutination assays for serologic diagnosis of toxoplasmosis. Diagn Microbiol Infect Dis 2012; 73:231–235.
- Suzuki LA, Rocha RJ, Rossi CL. Evaluation of serological markers for the immunodiagnosis of acute acquired toxoplasmosis. J Med Microbiol 2001; 50:62–70.
- Lachaud L, Calas O, Picot MC, Albaba S, Bourgeois N, Pratlong F. Value of 2 IgG avidity commercial tests used alone or in association to date toxoplasmosis contamination. Diagn Microbiol Infect Dis 2009; 64:267–274.
- Rahumatullah A, Khoo BY, Noordin R. Triplex PCR using new primers for the detection of Toxoplasma gondii. Exp Parasitol 2012; 131:231–238.
- Contini C, Giuliodori M, Cultrera R, Seraceni S. Detection of clinical-stage specific molecular Toxoplasma gondii gene patterns in patients with toxoplasmic lymphadenitis. J Med Microbiol 2006; 55:771–774.
- Silveira C, Vallochi AL, Rodrigues da Silva U, et al. Toxoplasma gondii in the peripheral blood of patients with acute and chronic toxoplasmosis. Br J Ophthalmol 2011; 95:396–400.
- Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet 2004; 363:1965–1976.
- Petersen E, Vesco G, Villari S, Buffolano W. What do we know about risk factors for infection in humans with Toxoplasma gondii and how can we prevent infections? Zoonoses Public Health 2010; 57:8–17.
- Feldman DM, Timms D, Borgida AF. Toxoplasmosis, parvovirus, and cytomegalovirus in pregnancy. Clin Lab Med 2010; 30:709–720.
- Weiss LM, Dubey JP. Toxoplasmosis: a history of clinical observations. Int J Parasitol 2009; 39:895–901.
- Remington JS. Toxoplasmosis in the adult. Bull NY Acad Med 1974; 50:211–227.
- McCabe RE, Brooks RG, Dorfman RF, Remington JS. Clinical spectrum in 107 cases of toxoplasmic lymphadenopathy. Rev Infect Dis 1987; 9:754–774.
- Toxoplasmosis. The Medical Letter, Drugs for Parasitic Infections. New Rochelle, NY: The Medical Letter Inc, June 1, 2010:57–58.
- Hotop A, Hlobil H, Gross U. Efficacy of rapid treatment initiation following primary Toxoplasma gondii infection during pregnancy. Clin Infect Dis 2012; 54:1545–1552.