User login
Acute myocardial infarction (MI) portends important and substantial consequences. Angioplasty or fibrinolytic therapy to open the blocked coronary artery is proven to improve the patient’s chances of surviving without consequent morbidity or death. But the diagnosis is not always straightforward. The presentation of acute MI can vary widely, and a number of other conditions—many of them equally serious emergencies—can mimic its symptoms, electrocardiographic signs, and biomarker patterns.
In an attempt to improve the accuracy of the diagnosis of MI, a multinational task force met in 1999 under the auspices of the European Society of Cardiology and the American College of Cardiology. The goal was to develop a simple, clinically oriented definition of MI that could be widely adopted. A document was created and published simultaneously in 2000 in the European Heart Journal and the Journal of the American College of Cardiology.1 These organizations updated their paper in 2007 with a new definition of acute MI to account for advances in diagnosis and management.2
In this article we will review the new definition and how to make the diagnosis of acute MI today. Specifically, the updated definition includes:
- Subtypes of acute MI
- Imaging tests supporting the diagnosis
- Biomarker thresholds after percutaneous coronary intervention or bypass grafting.
TROPONIN: BETTER THAN CK, BUT NOT PERFECT
The original 2000 paper1 and the 2007 update2 featured the use of the cardiac biomarker troponin, which is considerably more sensitive and specific for heart damage than total creatine kinase (CK) or its isoform, CK-MB.
The new, more-sensitive biomarker-based definition of MI resulted in more cases of MI being diagnosed, and this has attracted the attention and scrutiny of many, especially population scientists and interventional cardiologists.3 This change has caused some controversy, especially when dealing with small rises in troponin following percutaneous coronary intervention.
In addition, some confusion over terminology remains. For example, the phrase “troponin leak” is often used to describe cases in which serum troponin levels rise but there is no MI. However, most experts believe that a rise and fall in troponin is due to true myocardial cell death. Troponin I and T are such large molecules that they cannot “leak” from a cardiac cell unless there has been irreparable cellular damage—that is, cell death.

Creatine kinase still has a role
In some cases, CK and CK-MB may be helpful in determining the acuity of myocardial necrosis, but their use will vary by institution. These biomarkers typically rise 2 to 4 hours after the initial event and fall within 24 to 48 hours, whereas troponin levels stay elevated for days or weeks. Thus, the presence of troponin without CK and CK-MB in the right clinical context may indicate a past MI that is no longer acute.
INFARCTION: CELL DEATH DUE TO ISCHEMIA
MI is myocardial cell death due to prolonged ischemia. Under the microscope, it can be categorized as coagulation necrosis in which ghost-like cell structures remain after hypoxic insult (typical of most MIs) or contraction band necrosis with amorphous cells that cannot contract anymore, the latter often a hallmark of excessive catecholamine damage or reperfusion injury. Apoptosis occurs in the heart but is technically not considered necrosis and is thought not to be associated with elevated troponin levels.6,7
In experiments in animals, cell death can occur as little as 20 minutes after coronary artery occlusion, although completion of infarction is thought to take 2 to 4 hours. The time to infarct completion may be longer in patients with collateral circulation or when the culprit coronary artery has intermittent (“stuttering”) occlusion. Preconditioning of myocardial cells with intermittent ischemia can also influence the timing of myocardial necrosis by protecting against cell death to some extent. Alteration in myocardial demand can influence the time required for completion of infarction either favorably or unfavorably; hence, reducing myocardial demand is beneficial in acute MI.
Three pathologic phases of MI
MI can be categorized pathologically as acute, healing, or healed.
Acute MI. In the first 6 hours after coronary artery occlusion, coagulation necrosis can be seen with no cellular infiltration. After 6 hours, polymorphonuclear leukocytes infiltrate the infarcted area, and this may continue for up to 7 days if coronary perfusion does not increase or myocardial demand does not decrease.
Healing MI is characterized by mononuclear cells and fibroblasts and the absence of polymorphonuclear leukocytes. The entire healing process takes 5 to 6 weeks and can be altered by coronary reperfusion.
Healed MI refers to scar tissue without cellular infiltration.
CLINICAL FEATURES VARY WIDELY
Sir William Osler said, “Variability is the law of life, and as no two faces are the same, so no two bodies are alike, and no two individuals react alike and behave alike under the abnormal conditions which we know as disease.”8
Just so, patients with acute MI display a wide variety of presentations, from no symptoms (about 25%) to severe, crushing chest pain. Discomfort may occur in the upper back, neck, jaw, teeth, arms, wrist, and epigastrium. Shortness of breath, diaphoresis, nausea, vomiting, and even syncope may occur. Unlike in acute aortic dissection, the discomfort is not usually maximal at its onset: it builds up in a crescendo manner. It is not usually changed by position, but can lessen in intensity upon standing. The discomfort in the chest is deep and visceral, and typically not well localized. A pressure sensation, air hunger, or “gas buildup” can be described. The only symptom may be shortness of breath or severe diaphoresis. The symptoms can last from minutes to hours and can be relieved by sublingual nitroglycerin. Atypical or less-prominent symptoms may make the diagnosis more difficult in the elderly, patients with diabetes mellitus, and women.
The physical examination during acute MI usually finds no clear-cut distinguishing features. The patient may appear pale and diaphoretic, and the skin cool to the touch. Heart sounds are generally soft. A fourth heart sound may be audible. Blood pressure may be low, but it can vary widely. Tachycardia, particularly sinus tachycardia, and pulmonary edema are poor prognostic signs.
In view of the wide variation in presentations, the history and physical findings can raise the suspicion of acute MI, but sequential electrocardiograms and measurements of biomarkers (troponin) are always necessary.
ELECTROCARDIOGRAPHY: NECESSARY BUT NOT SUFFICIENT
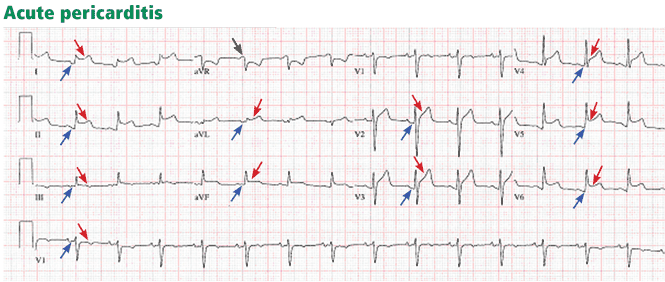

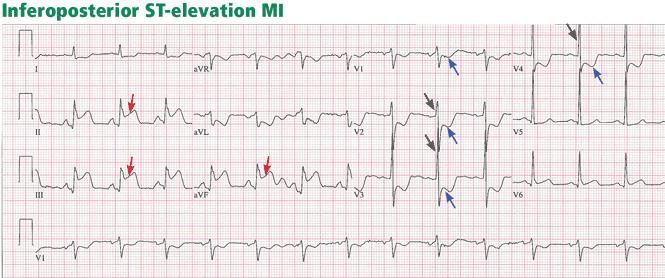
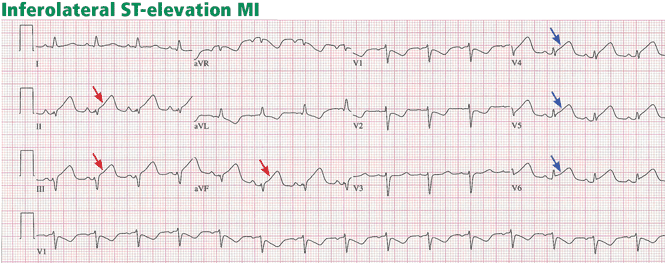
ST-elevation MI vs non-ST-elevation MI
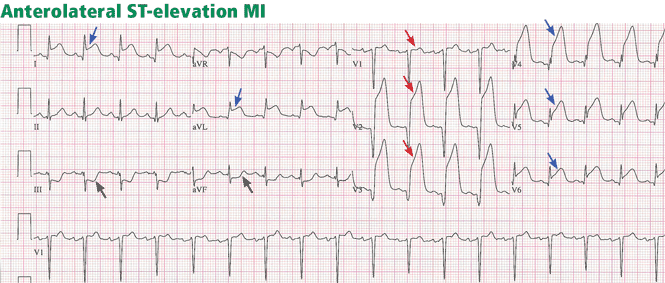
Changes in the ST segment can be very dynamic, making sequential tracings very useful. Rhythm disturbances and heart block are also more likely to be recorded when using sequential readings.
Pitfalls to electrocardiographic diagnosis
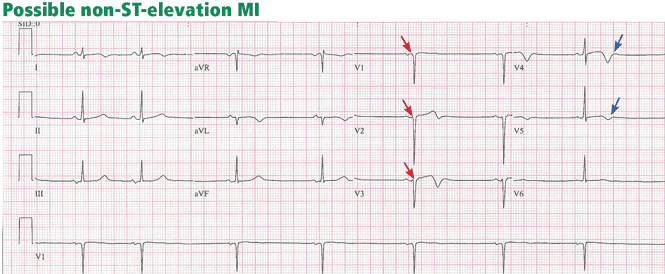
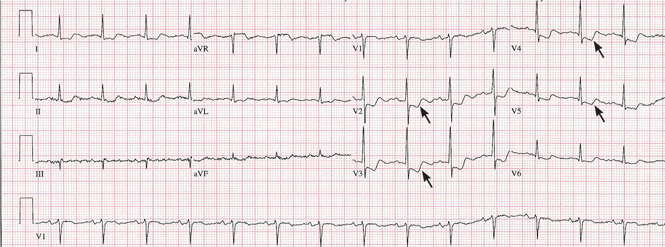
Prior MI. Q waves or QS complexes, when the Q wave is sufficiently wide (≥ 0.03 msec) or deep (≥ 1 mV), usually indicate a previous MI. However, many nuances that further raise or lower the suspicion for previous MI need to be considered. These are beyond the scope of this brief review but are available in the 2007 update.

Right ventricular infarction often requires the use of right-sided leads, which may reveal ST elevation in V4R.
ECHOCARDIOGRAPHY IF THE DIAGNOSIS IS IN DOUBT
Echocardiography is an excellent way to assess wall-motion abnormalities. In the absence of any wall-motion abnormality, a large ST-elevation MI is unlikely. A large wall-motion abnormality would verify the probability of ongoing acute MI and thus would help with rapid decision-making.
Furthermore, echocardiography can help determine the likelihood that the patient has aortic dissection or pulmonary embolism, either of which can mimic acute MI but requires very different treatment.
CLINICAL CLASSIFICATION OF ACUTE MI

The new classification scheme of the different types of MI is shown in Table 4.
The new classification scheme does not include myocardial necrosis from mechanical manipulation of the heart during open heart surgery, from cardioversion, or from toxic drugs.
As clinicians are aware, it is not unusual to see elevated biomarker levels in a host of conditions unrelated to acute myocardial ischemia or MI. The new classification of acute MI is most helpful in this regard. It will likely be even more helpful in guiding treatment and management when new ultrasensitive troponin assays are widely introduced into clinical practice.
The new classification also negotiates the controversy regarding elevated biomarker levels following percutaneous coronary intervention. In brief, elevation of biomarkers is not entirely avoidable even with a successful percutaneous coronary intervention, and furthermore, there is no scientific cutoff for biomarker elevations. So, by arbitrary convention, the troponin level must rise to more than three times the 99th percentile upper reference limit to make the diagnosis of type 4a MI. A separate type 4b MI is ascribed to angiographic or autopsy-proven stent thrombosis.
The new guidelines also suggest that troponin values be more than five times the 99th percentile of the normal reference range during the first 72 hours following coronary artery bypass graft surgery (CABG) when considering a CABG-related MI (type 5). Whenever new pathologic Q waves appear in territories other than those identified before the procedure, MI should be considered, especially if associated with elevated biomarkers, new wall-motion abnormalities, or hemodynamic instability.
Thus, the diagnosis of acute MI now has widely accepted global criteria that distinguish various types of acute MI that occur under multiple circumstances. It is expected that describing the type of acute MI according to the new criteria will further enhance our understanding of the event, its proper management, and its prognosis.
- The Joint European Society of Cardiology/American College of Cardiology Committee. Myocardial infarction redefined—a consensus document of the Joint European Society of Cardiology/American College of Cardiology Committee for the Redefinition of Myocardial Infarction. J Am Coll Cardiol 2000; 36:959–969.
- Thygesen K, Alpert JS, White HD, on behalf of the Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction. Universal definition of myocardial infarction. J Am Coll Cardiol 2007; 50:2173–2188.
- Roger VL, Killian JM, Weston SA, et al. Redefinition of myocardial infarction—prospective evaluation in the community. Circulation 2006; 114:790–797.
- Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease. J Am Coll Cardiol 2006; 48:1–11.
- French JK, White HD. Clinical implications of the new definition of myocardial infarction. Heart 2004; 90:99–106.
- James TN. The variable morphological coexistence of apoptosis and necrosis in human myocardial infarction: significance for understanding its pathogenesis, clinical course, diagnosis and prognosis. Coron Artery Dis 1998; 9:291–307.
- Sobel BE, LeWinter MM. Ingenuous interpretation of elevated blood levels of macromolecular markers of myocardial injury: a recipe for confusion. J Am Coll Cardiol 2000; 35:1355–1358.
- Osler W. Aequanimitas: With Other Addresses to Medical Students, Nurses and Practitioners of Medicine.Osler William Edition: 3, revised. Philadelphia: Blakiston’s, 1932.
- Wang F, Asinger RW, Marriott HJ. ST-segment elevation in conditions other than acute myocardial infarction. N Engl J Med 2003; 349:2128–2135.
Acute myocardial infarction (MI) portends important and substantial consequences. Angioplasty or fibrinolytic therapy to open the blocked coronary artery is proven to improve the patient’s chances of surviving without consequent morbidity or death. But the diagnosis is not always straightforward. The presentation of acute MI can vary widely, and a number of other conditions—many of them equally serious emergencies—can mimic its symptoms, electrocardiographic signs, and biomarker patterns.
In an attempt to improve the accuracy of the diagnosis of MI, a multinational task force met in 1999 under the auspices of the European Society of Cardiology and the American College of Cardiology. The goal was to develop a simple, clinically oriented definition of MI that could be widely adopted. A document was created and published simultaneously in 2000 in the European Heart Journal and the Journal of the American College of Cardiology.1 These organizations updated their paper in 2007 with a new definition of acute MI to account for advances in diagnosis and management.2
In this article we will review the new definition and how to make the diagnosis of acute MI today. Specifically, the updated definition includes:
- Subtypes of acute MI
- Imaging tests supporting the diagnosis
- Biomarker thresholds after percutaneous coronary intervention or bypass grafting.
TROPONIN: BETTER THAN CK, BUT NOT PERFECT
The original 2000 paper1 and the 2007 update2 featured the use of the cardiac biomarker troponin, which is considerably more sensitive and specific for heart damage than total creatine kinase (CK) or its isoform, CK-MB.
The new, more-sensitive biomarker-based definition of MI resulted in more cases of MI being diagnosed, and this has attracted the attention and scrutiny of many, especially population scientists and interventional cardiologists.3 This change has caused some controversy, especially when dealing with small rises in troponin following percutaneous coronary intervention.
In addition, some confusion over terminology remains. For example, the phrase “troponin leak” is often used to describe cases in which serum troponin levels rise but there is no MI. However, most experts believe that a rise and fall in troponin is due to true myocardial cell death. Troponin I and T are such large molecules that they cannot “leak” from a cardiac cell unless there has been irreparable cellular damage—that is, cell death.
Creatine kinase still has a role
In some cases, CK and CK-MB may be helpful in determining the acuity of myocardial necrosis, but their use will vary by institution. These biomarkers typically rise 2 to 4 hours after the initial event and fall within 24 to 48 hours, whereas troponin levels stay elevated for days or weeks. Thus, the presence of troponin without CK and CK-MB in the right clinical context may indicate a past MI that is no longer acute.
INFARCTION: CELL DEATH DUE TO ISCHEMIA
MI is myocardial cell death due to prolonged ischemia. Under the microscope, it can be categorized as coagulation necrosis in which ghost-like cell structures remain after hypoxic insult (typical of most MIs) or contraction band necrosis with amorphous cells that cannot contract anymore, the latter often a hallmark of excessive catecholamine damage or reperfusion injury. Apoptosis occurs in the heart but is technically not considered necrosis and is thought not to be associated with elevated troponin levels.6,7
In experiments in animals, cell death can occur as little as 20 minutes after coronary artery occlusion, although completion of infarction is thought to take 2 to 4 hours. The time to infarct completion may be longer in patients with collateral circulation or when the culprit coronary artery has intermittent (“stuttering”) occlusion. Preconditioning of myocardial cells with intermittent ischemia can also influence the timing of myocardial necrosis by protecting against cell death to some extent. Alteration in myocardial demand can influence the time required for completion of infarction either favorably or unfavorably; hence, reducing myocardial demand is beneficial in acute MI.
Three pathologic phases of MI
MI can be categorized pathologically as acute, healing, or healed.
Acute MI. In the first 6 hours after coronary artery occlusion, coagulation necrosis can be seen with no cellular infiltration. After 6 hours, polymorphonuclear leukocytes infiltrate the infarcted area, and this may continue for up to 7 days if coronary perfusion does not increase or myocardial demand does not decrease.
Healing MI is characterized by mononuclear cells and fibroblasts and the absence of polymorphonuclear leukocytes. The entire healing process takes 5 to 6 weeks and can be altered by coronary reperfusion.
Healed MI refers to scar tissue without cellular infiltration.
CLINICAL FEATURES VARY WIDELY
Sir William Osler said, “Variability is the law of life, and as no two faces are the same, so no two bodies are alike, and no two individuals react alike and behave alike under the abnormal conditions which we know as disease.”8
Just so, patients with acute MI display a wide variety of presentations, from no symptoms (about 25%) to severe, crushing chest pain. Discomfort may occur in the upper back, neck, jaw, teeth, arms, wrist, and epigastrium. Shortness of breath, diaphoresis, nausea, vomiting, and even syncope may occur. Unlike in acute aortic dissection, the discomfort is not usually maximal at its onset: it builds up in a crescendo manner. It is not usually changed by position, but can lessen in intensity upon standing. The discomfort in the chest is deep and visceral, and typically not well localized. A pressure sensation, air hunger, or “gas buildup” can be described. The only symptom may be shortness of breath or severe diaphoresis. The symptoms can last from minutes to hours and can be relieved by sublingual nitroglycerin. Atypical or less-prominent symptoms may make the diagnosis more difficult in the elderly, patients with diabetes mellitus, and women.
The physical examination during acute MI usually finds no clear-cut distinguishing features. The patient may appear pale and diaphoretic, and the skin cool to the touch. Heart sounds are generally soft. A fourth heart sound may be audible. Blood pressure may be low, but it can vary widely. Tachycardia, particularly sinus tachycardia, and pulmonary edema are poor prognostic signs.
In view of the wide variation in presentations, the history and physical findings can raise the suspicion of acute MI, but sequential electrocardiograms and measurements of biomarkers (troponin) are always necessary.
ELECTROCARDIOGRAPHY: NECESSARY BUT NOT SUFFICIENT
ST-elevation MI vs non-ST-elevation MI
Changes in the ST segment can be very dynamic, making sequential tracings very useful. Rhythm disturbances and heart block are also more likely to be recorded when using sequential readings.
Pitfalls to electrocardiographic diagnosis
Prior MI. Q waves or QS complexes, when the Q wave is sufficiently wide (≥ 0.03 msec) or deep (≥ 1 mV), usually indicate a previous MI. However, many nuances that further raise or lower the suspicion for previous MI need to be considered. These are beyond the scope of this brief review but are available in the 2007 update.
Right ventricular infarction often requires the use of right-sided leads, which may reveal ST elevation in V4R.
ECHOCARDIOGRAPHY IF THE DIAGNOSIS IS IN DOUBT
Echocardiography is an excellent way to assess wall-motion abnormalities. In the absence of any wall-motion abnormality, a large ST-elevation MI is unlikely. A large wall-motion abnormality would verify the probability of ongoing acute MI and thus would help with rapid decision-making.
Furthermore, echocardiography can help determine the likelihood that the patient has aortic dissection or pulmonary embolism, either of which can mimic acute MI but requires very different treatment.
CLINICAL CLASSIFICATION OF ACUTE MI
The new classification scheme of the different types of MI is shown in Table 4.
The new classification scheme does not include myocardial necrosis from mechanical manipulation of the heart during open heart surgery, from cardioversion, or from toxic drugs.
As clinicians are aware, it is not unusual to see elevated biomarker levels in a host of conditions unrelated to acute myocardial ischemia or MI. The new classification of acute MI is most helpful in this regard. It will likely be even more helpful in guiding treatment and management when new ultrasensitive troponin assays are widely introduced into clinical practice.
The new classification also negotiates the controversy regarding elevated biomarker levels following percutaneous coronary intervention. In brief, elevation of biomarkers is not entirely avoidable even with a successful percutaneous coronary intervention, and furthermore, there is no scientific cutoff for biomarker elevations. So, by arbitrary convention, the troponin level must rise to more than three times the 99th percentile upper reference limit to make the diagnosis of type 4a MI. A separate type 4b MI is ascribed to angiographic or autopsy-proven stent thrombosis.
The new guidelines also suggest that troponin values be more than five times the 99th percentile of the normal reference range during the first 72 hours following coronary artery bypass graft surgery (CABG) when considering a CABG-related MI (type 5). Whenever new pathologic Q waves appear in territories other than those identified before the procedure, MI should be considered, especially if associated with elevated biomarkers, new wall-motion abnormalities, or hemodynamic instability.
Thus, the diagnosis of acute MI now has widely accepted global criteria that distinguish various types of acute MI that occur under multiple circumstances. It is expected that describing the type of acute MI according to the new criteria will further enhance our understanding of the event, its proper management, and its prognosis.
Acute myocardial infarction (MI) portends important and substantial consequences. Angioplasty or fibrinolytic therapy to open the blocked coronary artery is proven to improve the patient’s chances of surviving without consequent morbidity or death. But the diagnosis is not always straightforward. The presentation of acute MI can vary widely, and a number of other conditions—many of them equally serious emergencies—can mimic its symptoms, electrocardiographic signs, and biomarker patterns.
In an attempt to improve the accuracy of the diagnosis of MI, a multinational task force met in 1999 under the auspices of the European Society of Cardiology and the American College of Cardiology. The goal was to develop a simple, clinically oriented definition of MI that could be widely adopted. A document was created and published simultaneously in 2000 in the European Heart Journal and the Journal of the American College of Cardiology.1 These organizations updated their paper in 2007 with a new definition of acute MI to account for advances in diagnosis and management.2
In this article we will review the new definition and how to make the diagnosis of acute MI today. Specifically, the updated definition includes:
- Subtypes of acute MI
- Imaging tests supporting the diagnosis
- Biomarker thresholds after percutaneous coronary intervention or bypass grafting.
TROPONIN: BETTER THAN CK, BUT NOT PERFECT
The original 2000 paper1 and the 2007 update2 featured the use of the cardiac biomarker troponin, which is considerably more sensitive and specific for heart damage than total creatine kinase (CK) or its isoform, CK-MB.
The new, more-sensitive biomarker-based definition of MI resulted in more cases of MI being diagnosed, and this has attracted the attention and scrutiny of many, especially population scientists and interventional cardiologists.3 This change has caused some controversy, especially when dealing with small rises in troponin following percutaneous coronary intervention.
In addition, some confusion over terminology remains. For example, the phrase “troponin leak” is often used to describe cases in which serum troponin levels rise but there is no MI. However, most experts believe that a rise and fall in troponin is due to true myocardial cell death. Troponin I and T are such large molecules that they cannot “leak” from a cardiac cell unless there has been irreparable cellular damage—that is, cell death.
Creatine kinase still has a role
In some cases, CK and CK-MB may be helpful in determining the acuity of myocardial necrosis, but their use will vary by institution. These biomarkers typically rise 2 to 4 hours after the initial event and fall within 24 to 48 hours, whereas troponin levels stay elevated for days or weeks. Thus, the presence of troponin without CK and CK-MB in the right clinical context may indicate a past MI that is no longer acute.
INFARCTION: CELL DEATH DUE TO ISCHEMIA
MI is myocardial cell death due to prolonged ischemia. Under the microscope, it can be categorized as coagulation necrosis in which ghost-like cell structures remain after hypoxic insult (typical of most MIs) or contraction band necrosis with amorphous cells that cannot contract anymore, the latter often a hallmark of excessive catecholamine damage or reperfusion injury. Apoptosis occurs in the heart but is technically not considered necrosis and is thought not to be associated with elevated troponin levels.6,7
In experiments in animals, cell death can occur as little as 20 minutes after coronary artery occlusion, although completion of infarction is thought to take 2 to 4 hours. The time to infarct completion may be longer in patients with collateral circulation or when the culprit coronary artery has intermittent (“stuttering”) occlusion. Preconditioning of myocardial cells with intermittent ischemia can also influence the timing of myocardial necrosis by protecting against cell death to some extent. Alteration in myocardial demand can influence the time required for completion of infarction either favorably or unfavorably; hence, reducing myocardial demand is beneficial in acute MI.
Three pathologic phases of MI
MI can be categorized pathologically as acute, healing, or healed.
Acute MI. In the first 6 hours after coronary artery occlusion, coagulation necrosis can be seen with no cellular infiltration. After 6 hours, polymorphonuclear leukocytes infiltrate the infarcted area, and this may continue for up to 7 days if coronary perfusion does not increase or myocardial demand does not decrease.
Healing MI is characterized by mononuclear cells and fibroblasts and the absence of polymorphonuclear leukocytes. The entire healing process takes 5 to 6 weeks and can be altered by coronary reperfusion.
Healed MI refers to scar tissue without cellular infiltration.
CLINICAL FEATURES VARY WIDELY
Sir William Osler said, “Variability is the law of life, and as no two faces are the same, so no two bodies are alike, and no two individuals react alike and behave alike under the abnormal conditions which we know as disease.”8
Just so, patients with acute MI display a wide variety of presentations, from no symptoms (about 25%) to severe, crushing chest pain. Discomfort may occur in the upper back, neck, jaw, teeth, arms, wrist, and epigastrium. Shortness of breath, diaphoresis, nausea, vomiting, and even syncope may occur. Unlike in acute aortic dissection, the discomfort is not usually maximal at its onset: it builds up in a crescendo manner. It is not usually changed by position, but can lessen in intensity upon standing. The discomfort in the chest is deep and visceral, and typically not well localized. A pressure sensation, air hunger, or “gas buildup” can be described. The only symptom may be shortness of breath or severe diaphoresis. The symptoms can last from minutes to hours and can be relieved by sublingual nitroglycerin. Atypical or less-prominent symptoms may make the diagnosis more difficult in the elderly, patients with diabetes mellitus, and women.
The physical examination during acute MI usually finds no clear-cut distinguishing features. The patient may appear pale and diaphoretic, and the skin cool to the touch. Heart sounds are generally soft. A fourth heart sound may be audible. Blood pressure may be low, but it can vary widely. Tachycardia, particularly sinus tachycardia, and pulmonary edema are poor prognostic signs.
In view of the wide variation in presentations, the history and physical findings can raise the suspicion of acute MI, but sequential electrocardiograms and measurements of biomarkers (troponin) are always necessary.
ELECTROCARDIOGRAPHY: NECESSARY BUT NOT SUFFICIENT
ST-elevation MI vs non-ST-elevation MI
Changes in the ST segment can be very dynamic, making sequential tracings very useful. Rhythm disturbances and heart block are also more likely to be recorded when using sequential readings.
Pitfalls to electrocardiographic diagnosis
Prior MI. Q waves or QS complexes, when the Q wave is sufficiently wide (≥ 0.03 msec) or deep (≥ 1 mV), usually indicate a previous MI. However, many nuances that further raise or lower the suspicion for previous MI need to be considered. These are beyond the scope of this brief review but are available in the 2007 update.
Right ventricular infarction often requires the use of right-sided leads, which may reveal ST elevation in V4R.
ECHOCARDIOGRAPHY IF THE DIAGNOSIS IS IN DOUBT
Echocardiography is an excellent way to assess wall-motion abnormalities. In the absence of any wall-motion abnormality, a large ST-elevation MI is unlikely. A large wall-motion abnormality would verify the probability of ongoing acute MI and thus would help with rapid decision-making.
Furthermore, echocardiography can help determine the likelihood that the patient has aortic dissection or pulmonary embolism, either of which can mimic acute MI but requires very different treatment.
CLINICAL CLASSIFICATION OF ACUTE MI
The new classification scheme of the different types of MI is shown in Table 4.
The new classification scheme does not include myocardial necrosis from mechanical manipulation of the heart during open heart surgery, from cardioversion, or from toxic drugs.
As clinicians are aware, it is not unusual to see elevated biomarker levels in a host of conditions unrelated to acute myocardial ischemia or MI. The new classification of acute MI is most helpful in this regard. It will likely be even more helpful in guiding treatment and management when new ultrasensitive troponin assays are widely introduced into clinical practice.
The new classification also negotiates the controversy regarding elevated biomarker levels following percutaneous coronary intervention. In brief, elevation of biomarkers is not entirely avoidable even with a successful percutaneous coronary intervention, and furthermore, there is no scientific cutoff for biomarker elevations. So, by arbitrary convention, the troponin level must rise to more than three times the 99th percentile upper reference limit to make the diagnosis of type 4a MI. A separate type 4b MI is ascribed to angiographic or autopsy-proven stent thrombosis.
The new guidelines also suggest that troponin values be more than five times the 99th percentile of the normal reference range during the first 72 hours following coronary artery bypass graft surgery (CABG) when considering a CABG-related MI (type 5). Whenever new pathologic Q waves appear in territories other than those identified before the procedure, MI should be considered, especially if associated with elevated biomarkers, new wall-motion abnormalities, or hemodynamic instability.
Thus, the diagnosis of acute MI now has widely accepted global criteria that distinguish various types of acute MI that occur under multiple circumstances. It is expected that describing the type of acute MI according to the new criteria will further enhance our understanding of the event, its proper management, and its prognosis.
- The Joint European Society of Cardiology/American College of Cardiology Committee. Myocardial infarction redefined—a consensus document of the Joint European Society of Cardiology/American College of Cardiology Committee for the Redefinition of Myocardial Infarction. J Am Coll Cardiol 2000; 36:959–969.
- Thygesen K, Alpert JS, White HD, on behalf of the Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction. Universal definition of myocardial infarction. J Am Coll Cardiol 2007; 50:2173–2188.
- Roger VL, Killian JM, Weston SA, et al. Redefinition of myocardial infarction—prospective evaluation in the community. Circulation 2006; 114:790–797.
- Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease. J Am Coll Cardiol 2006; 48:1–11.
- French JK, White HD. Clinical implications of the new definition of myocardial infarction. Heart 2004; 90:99–106.
- James TN. The variable morphological coexistence of apoptosis and necrosis in human myocardial infarction: significance for understanding its pathogenesis, clinical course, diagnosis and prognosis. Coron Artery Dis 1998; 9:291–307.
- Sobel BE, LeWinter MM. Ingenuous interpretation of elevated blood levels of macromolecular markers of myocardial injury: a recipe for confusion. J Am Coll Cardiol 2000; 35:1355–1358.
- Osler W. Aequanimitas: With Other Addresses to Medical Students, Nurses and Practitioners of Medicine.Osler William Edition: 3, revised. Philadelphia: Blakiston’s, 1932.
- Wang F, Asinger RW, Marriott HJ. ST-segment elevation in conditions other than acute myocardial infarction. N Engl J Med 2003; 349:2128–2135.
- The Joint European Society of Cardiology/American College of Cardiology Committee. Myocardial infarction redefined—a consensus document of the Joint European Society of Cardiology/American College of Cardiology Committee for the Redefinition of Myocardial Infarction. J Am Coll Cardiol 2000; 36:959–969.
- Thygesen K, Alpert JS, White HD, on behalf of the Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction. Universal definition of myocardial infarction. J Am Coll Cardiol 2007; 50:2173–2188.
- Roger VL, Killian JM, Weston SA, et al. Redefinition of myocardial infarction—prospective evaluation in the community. Circulation 2006; 114:790–797.
- Jaffe AS, Babuin L, Apple FS. Biomarkers in acute cardiac disease. J Am Coll Cardiol 2006; 48:1–11.
- French JK, White HD. Clinical implications of the new definition of myocardial infarction. Heart 2004; 90:99–106.
- James TN. The variable morphological coexistence of apoptosis and necrosis in human myocardial infarction: significance for understanding its pathogenesis, clinical course, diagnosis and prognosis. Coron Artery Dis 1998; 9:291–307.
- Sobel BE, LeWinter MM. Ingenuous interpretation of elevated blood levels of macromolecular markers of myocardial injury: a recipe for confusion. J Am Coll Cardiol 2000; 35:1355–1358.
- Osler W. Aequanimitas: With Other Addresses to Medical Students, Nurses and Practitioners of Medicine.Osler William Edition: 3, revised. Philadelphia: Blakiston’s, 1932.
- Wang F, Asinger RW, Marriott HJ. ST-segment elevation in conditions other than acute myocardial infarction. N Engl J Med 2003; 349:2128–2135.
KEY POINTS
- The clinical presentation of acute MI varies considerably from patient to patient. Therefore, one must consider the symptoms, serial electrocardiographic findings, and serial biomarker results in concert.
- Troponin I or T is now the preferred biomarker of myocardial necrosis. Still, troponin can be elevated in many conditions other than ischemic heart disease.
- Electrocardiographic signs of acute ischemia have been precisely defined, but electrocardiography can give false-positive or false-negative results in a number of conditions.
- MI is now categorized into five types depending on cause.