User login
Relapsing polychondritis (RP) is a recurrent inflammatory condition involving primarily cartilaginous structures. The disease, first described as a clinical entity in 1960 by Pearson et al,1 is rare with an estimated incidence of 3.5 cases per 1 million individuals.2 The pathogenesis of RP is widely accepted as being autoimmune in nature, largely due to the identification of circulating autoantibodies seen in the sera of patients with similar clinical pictures.3
Although in most patients the primary process involves inflammation of cartilage, a subset of patients experience involvement of noncartilaginous sites.4 The degree of systemic involvement varies from none to notable, affecting the cardiovascular and respiratory systems and potentially leading to life-threatening complications, including cardiac valve compromise and airway collapse. Relapsing polychondritis is considered to be a progressive disease with the ultimate potential to be life-threatening.5
Human immunodeficiency virus (HIV) infection leads to a profound state of immune dysregulation, affecting innate, adaptive, and natural killer components of the immune system.6 There is variability in the development of autoimmune disease in HIV patients depending on the stage of infection. The frequency of rheumatologic disease in HIV patients might be as high as 60%.6 Relapsing polychondritis is rare in patients with HIV.7-9 Of 4 reported cases, 2 patients had other coexisting autoimmune disease—sarcoidosis and Behçet disease.8,9
Case Report
A 36-year-old man presented to the clinic with a concern of recurrent ear pain and swelling of approximately 2 years’ duration. Onset was sudden without inciting event. Symptoms initially involved the right ear with eventual progression to both ears. Additional symptoms included an auditory perception of underwater submersion, intermittent vertigo, and 3 episodes of throat closure sensation.
The patient’s medical history was notable for asthma; gastritis; depression; and HIV infection, which was diagnosed 4 years earlier and adequately managed with highly active antiretroviral therapy. His family history was notable for systemic lupus erythematosus in his mother, maternal aunt, and maternal cousin.
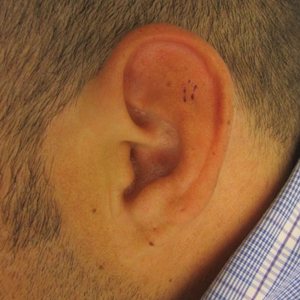
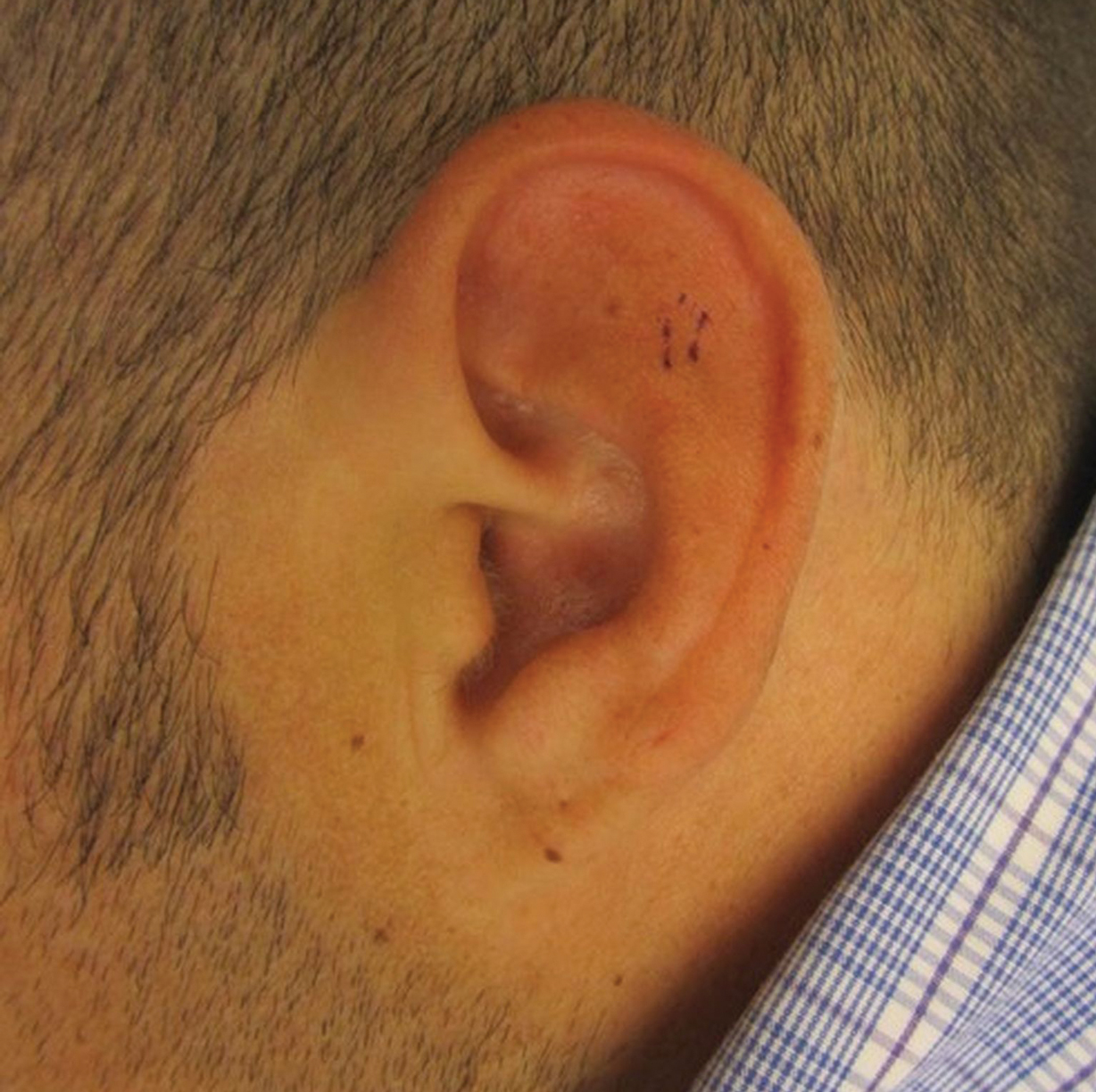
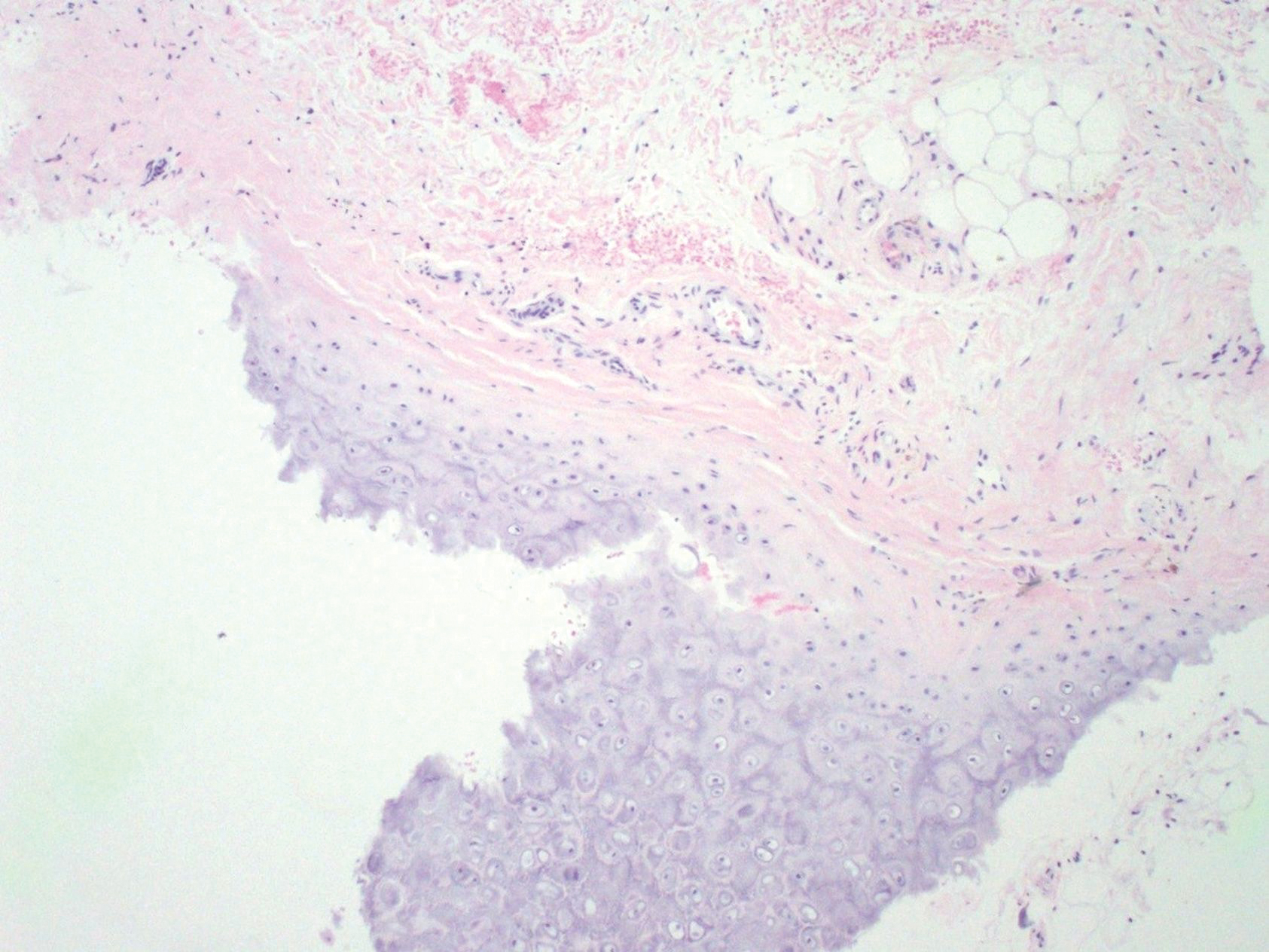
At presentation, the patient’s CD4 count was 799 cells/mm3 with an undetectable viral load. Medications included abacavir-dolutegravir-lamivudine, hydroxyzine, meclizine, mometasone, and quetiapine. Physical examination showed erythema, swelling, and tenderness of the left and right auricles with sparing of the earlobe that was more noticeable on the left ear (Figure 1). Bacterial culture from the external auditory meatus was positive for methicillin-resistant Staphylococcus aureus. Biopsy revealed chronic inflammatory perichondritis with mild to moderate fibrosis and chronic lymphocytic inflammation at the dermal cartilaginous junction (Figure 2). A direct immunofluorescent biopsy was unremarkable, but subsequent type II collagen antibodies were positive (35.5 endotoxin units/mL [reference range, <20 endotoxin units/mL]).
Comment
Relapsing polychondritis is an uncommon progressive disease characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures.4 The most consistent clinical features of RP are ear inflammation that involves the auricle and spares the lobe, nasal chondritis, and arthralgia.10 Laryngotracheal compromise may occur from tracheal cartilage inflammation. The involvement of these specific structures is due to commonality between their component collagens.5 Although any organ system can be affected, as many as 50% of patients have respiratory tract involvement, which may affect any portion of the respiratory tree.11 If involving the larynx, this inflammation can lead to severe edema warranting intubation. Cardiovascular involvement is present in 24% to 52% of patients,10 which most commonly manifests as valvular impairment affecting the aortic valve more frequently than the mitral valve.5
Pathogenesis
Although the etiology of RP remains undetermined, multiple hypotheses have been proposed. One is that a certain subset of patients is predisposed to autoimmunity, and a secondary triggering event in the form of infection, malignancy, or medication catalyzes development of RP. A second hypothesis is that mechanical trauma to cartilage exposes the immune system to certain antigens that would have otherwise remained hidden, prompting autosensitization.12,13
Regardless of cause, an autoimmune pathogenesis is favored based on the following observations: RP is frequently associated with other autoimmune diseases in the same patient, glucocorticosteroids and other immunosuppressive therapies are effective for treatment, and histopathologic findings include an infiltrate of CD4+ T lymphocytes with detection of immunoglobulins and plasma cells in different lesions.5 The detection of autoantibodies against collagen in the serum of patients with RP further supports an autoimmune pathogenesis.3 The earliest identified autoantibodies in patients with RP were against type II collagen. Subsequent studies have identified autoantibodies against type IV and type XI collagens as well as other cartilage-related proteins such as matrilin 114 and cartilage oligomeric matrix proteins.15 Although circulating antibodies to type II collagen are present in a variable number of patients with the disease (30%–70%), levels likely correlate with disease activity and are highest at times of acute inflammation.3 Additionally, titers of type II collagen antibodies have been shown to decrease upon institution of immunosuppressive therapy.16
Although a humoral response dominates the picture of RP, there also is an associated T cell–mediated response.13 Histopathologically, biopsy of an active lesion of auricular cartilage shows a mixed inflammatory infiltrate composed primarily of lymphocytes, with variable numbers of polymorphonuclear cells, monocytes, and plasma cells. Loss of basophilia of the cartilage matrix can be observed, thought to be the result of proteoglycan depletion.13 Later, lesions classically display apoptosis of chondrocytes, focal calcification, or fibrosis.5
Diagnosis
Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge. Many sets of diagnostic criteria have been proposed. The most referenced remains the original criteria described by McAdam et al.17 In 2012, the Relapsing Polychondritis Disease Activity Index modified criteria set forth by Michet et al18 and might serve as the standard for diagnosis going forward.19
McAdam et al17 proposed that 3 of 6 clinical features are necessary for diagnosis: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation, respiratory tract chondritis, and audiovestibular damage. Michet et al18 proposed that 1 of 2 conditions are necessary for diagnosis of RP: (1) proven inflammation in 2 of 3 of the auricular, nasal, or laryngotracheal cartilages; or (2) proven inflammation in 1 of 3 of the auricular, nasal, or laryngotracheal cartilages, plus 2 other signs, including ocular inflammation, vestibular dysfunction, seronegative inflammatory arthritis, and hearing loss.
These criteria were proposed originally in 197617 and modified in 1986.18 No further updates have been offered since then. As such, serologic findings, such as antibodies against type II collagen, are not included in the diagnostic criteria. Additionally, these antibodies are not specific for RP and can be seen in other conditions such as rheumatoid arthritis.20
More recently, imaging analysis has been employed in conjunction with clinical and serologic data to diagnose the disease and evaluate its severity. The use of imaging modalities for these purposes is most beneficial in patients with notable disease and respiratory involvement.21
Although the clinical picture is typified by the classic findings described above, the clinician must be aware of more subtle clues to diagnosis,11 which is of particular importance to the dermatologist because 35% of patients with RP alone will have skin manifestations that can precede onset of chondritis.10 Most commonly, dermatologic manifestations are nonspecific and can include nodules on the limbs, purpura, and urticarial lesions.22 Individual case reports have noted the coexistence of RP with erythema multiforme,18 erythema annulare centrifugum,23 pyoderma gangrenosum,24 and panniculitis,18 among other disorders.
Treatment
Standardized guidelines for treatment do not exist. Treatments should be chosen based on severity of disease. Mild disease, presenting with recurrent chondritis and arthritis without evidence of systemic involvement, can be treated with nonsteroidal anti-inflammatory drugs, dapsone, or colchicine. Refractory disease often requires high-dose systemic corticosteroids.5
Severe systemic involvement leads to increased mortality and warrants more aggressive treatment.22 Commonly used agents include the immunosuppressants cyclophosphamide, cyclosporine, and methotrexate. Tumor necrosis factor α inhibitors have been the most widely utilized immunomodulatory agent for treatment of RP.25,26 Abatacept and rituximab also have been used with variable efficacy in patients with severe disease. Recently, the IL-6 receptor blocker tocilizumab has been used with some success.27
Prognosis
The prognosis for patients with RP largely depends on the severity of disease and degree of internal involvement. With improved management, largely due to awareness and recognition of disease, the survival rate among RP patients has increased from 55% at 10 years to 94% at the end of 8 years.18 The main cause of death in RP patients is airway complications related to laryngotracheal involvement.10 The second most common cause of death is cardiovascular complications in which valvular disease predominates.5
Concomitant Illness
Thirty-five percent of RP patients have coexisting autoimmune disease, the most common being antineutrophil cytoplasmic antibody–associated vasculitis.5,28 Although this association with autoimmune disease is well described, reports of RP occurring in other states of immune dysfunction are sparse. One case of RP has been reported in a child with common variable immunodeficiency thought to be related to underlying abnormal immune regulation and immunodeficiency.29 Relapsing polychondritis has been described in 4 patients with HIV, 2 of whom had concomitant autoimmune disease.7-9
Human immunodeficiency virus infection is a well-established cause of immune dysregulation and has variable association with autoimmunity. This variability depends largely on the stage of infection. When divided into stages, autoimmune diseases develop predominantly in stage I, during acute infection with an intact immune system; in stage III, with immunosuppression, a low CD4 count, and development of AIDS; and in stage IV, when the immune system is restored with the institution of highly active antiretroviral therapy.6 The interplay between HIV infection and development of autoimmune disease is complex, and pathogenesis remains speculative.
Conclusion
Our patient represents a case of RP in an HIV-positive patient. Additionally, our patient had no other identifiable autoimmune conditions but did have a strong family history of them. It is important for providers to be aware of the potential for development of RP as well as other autoimmune disease in the setting of HIV infection. The implications of a missed diagnosis could be dire because the disease course of RP is progressive and has the potential to decrease survival.
- Pearson CM, Kline HM, Newcomer VD. Relapsing polychondritis. N Engl J Med. 1960;263:51-58.
- Kent PD, Michet CJ Jr, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol. 2004;16:56-61.
- Ebringer R, Rook G, Swana GT, et al. Autoantibodies to cartilage and type II collagen in relapsing polychondritis and other rheumatic diseases. Ann Rheum Dis. 1981;40:473-479.
- Sharma A, Law AD, Bambery P, et al. Relapsing polychondritis: clinical presentations, disease activity and outcomes. Orphanet J Rare Dis. 2014;9:198.
- Vitale A, Sota J, Rigante D, et al. Relapsing polychondritis: an update on pathogenesis, clinical features, diagnostic tools, and therapeutic perspectives. Curr Rheumatol Rep. 2016;18:3.
- Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329-337.
- Dolev JC, Maurer TA, Reddy SG, et al. Relapsing polychondritis in HIV-infected patients: a report of two cases. J Am Acad Dermatol. 2004;51:1023-1025.
- Zandman-Goddard G, Peeva E, Barland P. Combined autoimmune disease in a patient with AIDS. Clin Rheumatol. 2002;21:70-72.
- Belzunegui J, Cancio J, Pego JM, et al. Relapsing polychondritis and Behc¸et’s syndrome in a patient with HIV infection. Ann Rheum Dis. 1995;54:780.
- Sharma A, Gnanapandithan K, Sharma K, et al. Relapsing polychondritis: a review. Clin Rheumatol. 2013;32:1575-1583.
- Cantarini L, Vitale A, Brizi MG, et al. Diagnosis and classification of relapsing polychondritis. J Autoimmun. 2014;48-49:53-59.
- Cañas CA, Bonilla Abadía F. Local cartilage trauma as a pathogenic factor in autoimmunity (one hypothesis based on patients with relapsing polychondritis triggered by cartilage trauma). Autoimmune Dis. 2012;2012:453698.
- Ouchi N, Uzuki M, Kamataki A, et al. Cartilage destruction is partly induced by the internal proteolytic enzymes and apoptotic phenomenon of chondrocytes in relapsing polychondritis. J Rheumatol. 2011;38:730-737.
- Buckner JH, Wu JJ, Reife RA, et al. Autoreactivity against matrilin-1 in a patient with relapsing polychondritis. Arthritis Rheum. 2000;43:939-943.
- Kempta Lekpa F, Piette JC, Bastuji-Garin S, et al. Serum cartilage oligomeric matrix protein (COMP) is a marker of disease activity in relapsing polychondritis. Clin Exp Rheumatol. 2010;28:553-555.
- Foidart JM, Abe S, Martin GR, et al. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med. 1978;299:1203-1207.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet CJ, McKenna CH, Luthra HS, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Arnaud L, Devilliers H, Peng SL, et al. The Relapsing Polychondritis Disease Activity Index: development of a disease activity score for relapsing polychondritis. Autoimmun Rev. 2012;12:204-209.
- Brand DD, Kang AH, Rosloniec EF. Immunopathogenesis of collagen arthritis. Springer Semin Immunopathol. 2003;25:3-18.
- Thaiss WM, Nikolaou K, Spengler W, et al. Imaging diagnosis in relapsing polychondritis and correlation with clinical and serological data. Skeletal Radiol. 2015;5:339-346.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Francès C, el Rassi R, Laporte JL, et al. Dermatologic manifestations of relapsing polychondritis. A study of 200 cases at a single center. Medicine (Baltimore). 2001;80:173-179.
- Chopra R, Chaudhary N, Kay J. Relapsing polychondritis. Rheum Dis Clin North Am. 2013;39:263-276.
- Moulis G, Sailler L, Pugnet G, et al. Biologics in relapsing polychondritis: a case series. Clin Exp Rheumatol. 2013;31:937-939.
- Henes CJ, Xenitidis T, Horger M. Tocilizumab for refractory relapsing polychondritis—long-term response monitoring by magnetic resonance imaging. Joint Bone Spine. 2016;83:365-366.
- Weinberger A, Myers AR. Relapsing polychondritis associated with cutaneous vasculitis. Arch Dermatol. 1979;115:980-981.
- Karaca NE, Aksu G, Yildiz B, et al. Relapsing polychondritis in a child with common variable immunodeficiency. Int J Dermatol. 2009;48:525-528.
Relapsing polychondritis (RP) is a recurrent inflammatory condition involving primarily cartilaginous structures. The disease, first described as a clinical entity in 1960 by Pearson et al,1 is rare with an estimated incidence of 3.5 cases per 1 million individuals.2 The pathogenesis of RP is widely accepted as being autoimmune in nature, largely due to the identification of circulating autoantibodies seen in the sera of patients with similar clinical pictures.3
Although in most patients the primary process involves inflammation of cartilage, a subset of patients experience involvement of noncartilaginous sites.4 The degree of systemic involvement varies from none to notable, affecting the cardiovascular and respiratory systems and potentially leading to life-threatening complications, including cardiac valve compromise and airway collapse. Relapsing polychondritis is considered to be a progressive disease with the ultimate potential to be life-threatening.5
Human immunodeficiency virus (HIV) infection leads to a profound state of immune dysregulation, affecting innate, adaptive, and natural killer components of the immune system.6 There is variability in the development of autoimmune disease in HIV patients depending on the stage of infection. The frequency of rheumatologic disease in HIV patients might be as high as 60%.6 Relapsing polychondritis is rare in patients with HIV.7-9 Of 4 reported cases, 2 patients had other coexisting autoimmune disease—sarcoidosis and Behçet disease.8,9
Case Report
A 36-year-old man presented to the clinic with a concern of recurrent ear pain and swelling of approximately 2 years’ duration. Onset was sudden without inciting event. Symptoms initially involved the right ear with eventual progression to both ears. Additional symptoms included an auditory perception of underwater submersion, intermittent vertigo, and 3 episodes of throat closure sensation.
The patient’s medical history was notable for asthma; gastritis; depression; and HIV infection, which was diagnosed 4 years earlier and adequately managed with highly active antiretroviral therapy. His family history was notable for systemic lupus erythematosus in his mother, maternal aunt, and maternal cousin.
At presentation, the patient’s CD4 count was 799 cells/mm3 with an undetectable viral load. Medications included abacavir-dolutegravir-lamivudine, hydroxyzine, meclizine, mometasone, and quetiapine. Physical examination showed erythema, swelling, and tenderness of the left and right auricles with sparing of the earlobe that was more noticeable on the left ear (Figure 1). Bacterial culture from the external auditory meatus was positive for methicillin-resistant Staphylococcus aureus. Biopsy revealed chronic inflammatory perichondritis with mild to moderate fibrosis and chronic lymphocytic inflammation at the dermal cartilaginous junction (Figure 2). A direct immunofluorescent biopsy was unremarkable, but subsequent type II collagen antibodies were positive (35.5 endotoxin units/mL [reference range, <20 endotoxin units/mL]).
Comment
Relapsing polychondritis is an uncommon progressive disease characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures.4 The most consistent clinical features of RP are ear inflammation that involves the auricle and spares the lobe, nasal chondritis, and arthralgia.10 Laryngotracheal compromise may occur from tracheal cartilage inflammation. The involvement of these specific structures is due to commonality between their component collagens.5 Although any organ system can be affected, as many as 50% of patients have respiratory tract involvement, which may affect any portion of the respiratory tree.11 If involving the larynx, this inflammation can lead to severe edema warranting intubation. Cardiovascular involvement is present in 24% to 52% of patients,10 which most commonly manifests as valvular impairment affecting the aortic valve more frequently than the mitral valve.5
Pathogenesis
Although the etiology of RP remains undetermined, multiple hypotheses have been proposed. One is that a certain subset of patients is predisposed to autoimmunity, and a secondary triggering event in the form of infection, malignancy, or medication catalyzes development of RP. A second hypothesis is that mechanical trauma to cartilage exposes the immune system to certain antigens that would have otherwise remained hidden, prompting autosensitization.12,13
Regardless of cause, an autoimmune pathogenesis is favored based on the following observations: RP is frequently associated with other autoimmune diseases in the same patient, glucocorticosteroids and other immunosuppressive therapies are effective for treatment, and histopathologic findings include an infiltrate of CD4+ T lymphocytes with detection of immunoglobulins and plasma cells in different lesions.5 The detection of autoantibodies against collagen in the serum of patients with RP further supports an autoimmune pathogenesis.3 The earliest identified autoantibodies in patients with RP were against type II collagen. Subsequent studies have identified autoantibodies against type IV and type XI collagens as well as other cartilage-related proteins such as matrilin 114 and cartilage oligomeric matrix proteins.15 Although circulating antibodies to type II collagen are present in a variable number of patients with the disease (30%–70%), levels likely correlate with disease activity and are highest at times of acute inflammation.3 Additionally, titers of type II collagen antibodies have been shown to decrease upon institution of immunosuppressive therapy.16
Although a humoral response dominates the picture of RP, there also is an associated T cell–mediated response.13 Histopathologically, biopsy of an active lesion of auricular cartilage shows a mixed inflammatory infiltrate composed primarily of lymphocytes, with variable numbers of polymorphonuclear cells, monocytes, and plasma cells. Loss of basophilia of the cartilage matrix can be observed, thought to be the result of proteoglycan depletion.13 Later, lesions classically display apoptosis of chondrocytes, focal calcification, or fibrosis.5
Diagnosis
Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge. Many sets of diagnostic criteria have been proposed. The most referenced remains the original criteria described by McAdam et al.17 In 2012, the Relapsing Polychondritis Disease Activity Index modified criteria set forth by Michet et al18 and might serve as the standard for diagnosis going forward.19
McAdam et al17 proposed that 3 of 6 clinical features are necessary for diagnosis: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation, respiratory tract chondritis, and audiovestibular damage. Michet et al18 proposed that 1 of 2 conditions are necessary for diagnosis of RP: (1) proven inflammation in 2 of 3 of the auricular, nasal, or laryngotracheal cartilages; or (2) proven inflammation in 1 of 3 of the auricular, nasal, or laryngotracheal cartilages, plus 2 other signs, including ocular inflammation, vestibular dysfunction, seronegative inflammatory arthritis, and hearing loss.
These criteria were proposed originally in 197617 and modified in 1986.18 No further updates have been offered since then. As such, serologic findings, such as antibodies against type II collagen, are not included in the diagnostic criteria. Additionally, these antibodies are not specific for RP and can be seen in other conditions such as rheumatoid arthritis.20
More recently, imaging analysis has been employed in conjunction with clinical and serologic data to diagnose the disease and evaluate its severity. The use of imaging modalities for these purposes is most beneficial in patients with notable disease and respiratory involvement.21
Although the clinical picture is typified by the classic findings described above, the clinician must be aware of more subtle clues to diagnosis,11 which is of particular importance to the dermatologist because 35% of patients with RP alone will have skin manifestations that can precede onset of chondritis.10 Most commonly, dermatologic manifestations are nonspecific and can include nodules on the limbs, purpura, and urticarial lesions.22 Individual case reports have noted the coexistence of RP with erythema multiforme,18 erythema annulare centrifugum,23 pyoderma gangrenosum,24 and panniculitis,18 among other disorders.
Treatment
Standardized guidelines for treatment do not exist. Treatments should be chosen based on severity of disease. Mild disease, presenting with recurrent chondritis and arthritis without evidence of systemic involvement, can be treated with nonsteroidal anti-inflammatory drugs, dapsone, or colchicine. Refractory disease often requires high-dose systemic corticosteroids.5
Severe systemic involvement leads to increased mortality and warrants more aggressive treatment.22 Commonly used agents include the immunosuppressants cyclophosphamide, cyclosporine, and methotrexate. Tumor necrosis factor α inhibitors have been the most widely utilized immunomodulatory agent for treatment of RP.25,26 Abatacept and rituximab also have been used with variable efficacy in patients with severe disease. Recently, the IL-6 receptor blocker tocilizumab has been used with some success.27
Prognosis
The prognosis for patients with RP largely depends on the severity of disease and degree of internal involvement. With improved management, largely due to awareness and recognition of disease, the survival rate among RP patients has increased from 55% at 10 years to 94% at the end of 8 years.18 The main cause of death in RP patients is airway complications related to laryngotracheal involvement.10 The second most common cause of death is cardiovascular complications in which valvular disease predominates.5
Concomitant Illness
Thirty-five percent of RP patients have coexisting autoimmune disease, the most common being antineutrophil cytoplasmic antibody–associated vasculitis.5,28 Although this association with autoimmune disease is well described, reports of RP occurring in other states of immune dysfunction are sparse. One case of RP has been reported in a child with common variable immunodeficiency thought to be related to underlying abnormal immune regulation and immunodeficiency.29 Relapsing polychondritis has been described in 4 patients with HIV, 2 of whom had concomitant autoimmune disease.7-9
Human immunodeficiency virus infection is a well-established cause of immune dysregulation and has variable association with autoimmunity. This variability depends largely on the stage of infection. When divided into stages, autoimmune diseases develop predominantly in stage I, during acute infection with an intact immune system; in stage III, with immunosuppression, a low CD4 count, and development of AIDS; and in stage IV, when the immune system is restored with the institution of highly active antiretroviral therapy.6 The interplay between HIV infection and development of autoimmune disease is complex, and pathogenesis remains speculative.
Conclusion
Our patient represents a case of RP in an HIV-positive patient. Additionally, our patient had no other identifiable autoimmune conditions but did have a strong family history of them. It is important for providers to be aware of the potential for development of RP as well as other autoimmune disease in the setting of HIV infection. The implications of a missed diagnosis could be dire because the disease course of RP is progressive and has the potential to decrease survival.
Relapsing polychondritis (RP) is a recurrent inflammatory condition involving primarily cartilaginous structures. The disease, first described as a clinical entity in 1960 by Pearson et al,1 is rare with an estimated incidence of 3.5 cases per 1 million individuals.2 The pathogenesis of RP is widely accepted as being autoimmune in nature, largely due to the identification of circulating autoantibodies seen in the sera of patients with similar clinical pictures.3
Although in most patients the primary process involves inflammation of cartilage, a subset of patients experience involvement of noncartilaginous sites.4 The degree of systemic involvement varies from none to notable, affecting the cardiovascular and respiratory systems and potentially leading to life-threatening complications, including cardiac valve compromise and airway collapse. Relapsing polychondritis is considered to be a progressive disease with the ultimate potential to be life-threatening.5
Human immunodeficiency virus (HIV) infection leads to a profound state of immune dysregulation, affecting innate, adaptive, and natural killer components of the immune system.6 There is variability in the development of autoimmune disease in HIV patients depending on the stage of infection. The frequency of rheumatologic disease in HIV patients might be as high as 60%.6 Relapsing polychondritis is rare in patients with HIV.7-9 Of 4 reported cases, 2 patients had other coexisting autoimmune disease—sarcoidosis and Behçet disease.8,9
Case Report
A 36-year-old man presented to the clinic with a concern of recurrent ear pain and swelling of approximately 2 years’ duration. Onset was sudden without inciting event. Symptoms initially involved the right ear with eventual progression to both ears. Additional symptoms included an auditory perception of underwater submersion, intermittent vertigo, and 3 episodes of throat closure sensation.
The patient’s medical history was notable for asthma; gastritis; depression; and HIV infection, which was diagnosed 4 years earlier and adequately managed with highly active antiretroviral therapy. His family history was notable for systemic lupus erythematosus in his mother, maternal aunt, and maternal cousin.
At presentation, the patient’s CD4 count was 799 cells/mm3 with an undetectable viral load. Medications included abacavir-dolutegravir-lamivudine, hydroxyzine, meclizine, mometasone, and quetiapine. Physical examination showed erythema, swelling, and tenderness of the left and right auricles with sparing of the earlobe that was more noticeable on the left ear (Figure 1). Bacterial culture from the external auditory meatus was positive for methicillin-resistant Staphylococcus aureus. Biopsy revealed chronic inflammatory perichondritis with mild to moderate fibrosis and chronic lymphocytic inflammation at the dermal cartilaginous junction (Figure 2). A direct immunofluorescent biopsy was unremarkable, but subsequent type II collagen antibodies were positive (35.5 endotoxin units/mL [reference range, <20 endotoxin units/mL]).
Comment
Relapsing polychondritis is an uncommon progressive disease characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures.4 The most consistent clinical features of RP are ear inflammation that involves the auricle and spares the lobe, nasal chondritis, and arthralgia.10 Laryngotracheal compromise may occur from tracheal cartilage inflammation. The involvement of these specific structures is due to commonality between their component collagens.5 Although any organ system can be affected, as many as 50% of patients have respiratory tract involvement, which may affect any portion of the respiratory tree.11 If involving the larynx, this inflammation can lead to severe edema warranting intubation. Cardiovascular involvement is present in 24% to 52% of patients,10 which most commonly manifests as valvular impairment affecting the aortic valve more frequently than the mitral valve.5
Pathogenesis
Although the etiology of RP remains undetermined, multiple hypotheses have been proposed. One is that a certain subset of patients is predisposed to autoimmunity, and a secondary triggering event in the form of infection, malignancy, or medication catalyzes development of RP. A second hypothesis is that mechanical trauma to cartilage exposes the immune system to certain antigens that would have otherwise remained hidden, prompting autosensitization.12,13
Regardless of cause, an autoimmune pathogenesis is favored based on the following observations: RP is frequently associated with other autoimmune diseases in the same patient, glucocorticosteroids and other immunosuppressive therapies are effective for treatment, and histopathologic findings include an infiltrate of CD4+ T lymphocytes with detection of immunoglobulins and plasma cells in different lesions.5 The detection of autoantibodies against collagen in the serum of patients with RP further supports an autoimmune pathogenesis.3 The earliest identified autoantibodies in patients with RP were against type II collagen. Subsequent studies have identified autoantibodies against type IV and type XI collagens as well as other cartilage-related proteins such as matrilin 114 and cartilage oligomeric matrix proteins.15 Although circulating antibodies to type II collagen are present in a variable number of patients with the disease (30%–70%), levels likely correlate with disease activity and are highest at times of acute inflammation.3 Additionally, titers of type II collagen antibodies have been shown to decrease upon institution of immunosuppressive therapy.16
Although a humoral response dominates the picture of RP, there also is an associated T cell–mediated response.13 Histopathologically, biopsy of an active lesion of auricular cartilage shows a mixed inflammatory infiltrate composed primarily of lymphocytes, with variable numbers of polymorphonuclear cells, monocytes, and plasma cells. Loss of basophilia of the cartilage matrix can be observed, thought to be the result of proteoglycan depletion.13 Later, lesions classically display apoptosis of chondrocytes, focal calcification, or fibrosis.5
Diagnosis
Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge. Many sets of diagnostic criteria have been proposed. The most referenced remains the original criteria described by McAdam et al.17 In 2012, the Relapsing Polychondritis Disease Activity Index modified criteria set forth by Michet et al18 and might serve as the standard for diagnosis going forward.19
McAdam et al17 proposed that 3 of 6 clinical features are necessary for diagnosis: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation, respiratory tract chondritis, and audiovestibular damage. Michet et al18 proposed that 1 of 2 conditions are necessary for diagnosis of RP: (1) proven inflammation in 2 of 3 of the auricular, nasal, or laryngotracheal cartilages; or (2) proven inflammation in 1 of 3 of the auricular, nasal, or laryngotracheal cartilages, plus 2 other signs, including ocular inflammation, vestibular dysfunction, seronegative inflammatory arthritis, and hearing loss.
These criteria were proposed originally in 197617 and modified in 1986.18 No further updates have been offered since then. As such, serologic findings, such as antibodies against type II collagen, are not included in the diagnostic criteria. Additionally, these antibodies are not specific for RP and can be seen in other conditions such as rheumatoid arthritis.20
More recently, imaging analysis has been employed in conjunction with clinical and serologic data to diagnose the disease and evaluate its severity. The use of imaging modalities for these purposes is most beneficial in patients with notable disease and respiratory involvement.21
Although the clinical picture is typified by the classic findings described above, the clinician must be aware of more subtle clues to diagnosis,11 which is of particular importance to the dermatologist because 35% of patients with RP alone will have skin manifestations that can precede onset of chondritis.10 Most commonly, dermatologic manifestations are nonspecific and can include nodules on the limbs, purpura, and urticarial lesions.22 Individual case reports have noted the coexistence of RP with erythema multiforme,18 erythema annulare centrifugum,23 pyoderma gangrenosum,24 and panniculitis,18 among other disorders.
Treatment
Standardized guidelines for treatment do not exist. Treatments should be chosen based on severity of disease. Mild disease, presenting with recurrent chondritis and arthritis without evidence of systemic involvement, can be treated with nonsteroidal anti-inflammatory drugs, dapsone, or colchicine. Refractory disease often requires high-dose systemic corticosteroids.5
Severe systemic involvement leads to increased mortality and warrants more aggressive treatment.22 Commonly used agents include the immunosuppressants cyclophosphamide, cyclosporine, and methotrexate. Tumor necrosis factor α inhibitors have been the most widely utilized immunomodulatory agent for treatment of RP.25,26 Abatacept and rituximab also have been used with variable efficacy in patients with severe disease. Recently, the IL-6 receptor blocker tocilizumab has been used with some success.27
Prognosis
The prognosis for patients with RP largely depends on the severity of disease and degree of internal involvement. With improved management, largely due to awareness and recognition of disease, the survival rate among RP patients has increased from 55% at 10 years to 94% at the end of 8 years.18 The main cause of death in RP patients is airway complications related to laryngotracheal involvement.10 The second most common cause of death is cardiovascular complications in which valvular disease predominates.5
Concomitant Illness
Thirty-five percent of RP patients have coexisting autoimmune disease, the most common being antineutrophil cytoplasmic antibody–associated vasculitis.5,28 Although this association with autoimmune disease is well described, reports of RP occurring in other states of immune dysfunction are sparse. One case of RP has been reported in a child with common variable immunodeficiency thought to be related to underlying abnormal immune regulation and immunodeficiency.29 Relapsing polychondritis has been described in 4 patients with HIV, 2 of whom had concomitant autoimmune disease.7-9
Human immunodeficiency virus infection is a well-established cause of immune dysregulation and has variable association with autoimmunity. This variability depends largely on the stage of infection. When divided into stages, autoimmune diseases develop predominantly in stage I, during acute infection with an intact immune system; in stage III, with immunosuppression, a low CD4 count, and development of AIDS; and in stage IV, when the immune system is restored with the institution of highly active antiretroviral therapy.6 The interplay between HIV infection and development of autoimmune disease is complex, and pathogenesis remains speculative.
Conclusion
Our patient represents a case of RP in an HIV-positive patient. Additionally, our patient had no other identifiable autoimmune conditions but did have a strong family history of them. It is important for providers to be aware of the potential for development of RP as well as other autoimmune disease in the setting of HIV infection. The implications of a missed diagnosis could be dire because the disease course of RP is progressive and has the potential to decrease survival.
- Pearson CM, Kline HM, Newcomer VD. Relapsing polychondritis. N Engl J Med. 1960;263:51-58.
- Kent PD, Michet CJ Jr, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol. 2004;16:56-61.
- Ebringer R, Rook G, Swana GT, et al. Autoantibodies to cartilage and type II collagen in relapsing polychondritis and other rheumatic diseases. Ann Rheum Dis. 1981;40:473-479.
- Sharma A, Law AD, Bambery P, et al. Relapsing polychondritis: clinical presentations, disease activity and outcomes. Orphanet J Rare Dis. 2014;9:198.
- Vitale A, Sota J, Rigante D, et al. Relapsing polychondritis: an update on pathogenesis, clinical features, diagnostic tools, and therapeutic perspectives. Curr Rheumatol Rep. 2016;18:3.
- Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329-337.
- Dolev JC, Maurer TA, Reddy SG, et al. Relapsing polychondritis in HIV-infected patients: a report of two cases. J Am Acad Dermatol. 2004;51:1023-1025.
- Zandman-Goddard G, Peeva E, Barland P. Combined autoimmune disease in a patient with AIDS. Clin Rheumatol. 2002;21:70-72.
- Belzunegui J, Cancio J, Pego JM, et al. Relapsing polychondritis and Behc¸et’s syndrome in a patient with HIV infection. Ann Rheum Dis. 1995;54:780.
- Sharma A, Gnanapandithan K, Sharma K, et al. Relapsing polychondritis: a review. Clin Rheumatol. 2013;32:1575-1583.
- Cantarini L, Vitale A, Brizi MG, et al. Diagnosis and classification of relapsing polychondritis. J Autoimmun. 2014;48-49:53-59.
- Cañas CA, Bonilla Abadía F. Local cartilage trauma as a pathogenic factor in autoimmunity (one hypothesis based on patients with relapsing polychondritis triggered by cartilage trauma). Autoimmune Dis. 2012;2012:453698.
- Ouchi N, Uzuki M, Kamataki A, et al. Cartilage destruction is partly induced by the internal proteolytic enzymes and apoptotic phenomenon of chondrocytes in relapsing polychondritis. J Rheumatol. 2011;38:730-737.
- Buckner JH, Wu JJ, Reife RA, et al. Autoreactivity against matrilin-1 in a patient with relapsing polychondritis. Arthritis Rheum. 2000;43:939-943.
- Kempta Lekpa F, Piette JC, Bastuji-Garin S, et al. Serum cartilage oligomeric matrix protein (COMP) is a marker of disease activity in relapsing polychondritis. Clin Exp Rheumatol. 2010;28:553-555.
- Foidart JM, Abe S, Martin GR, et al. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med. 1978;299:1203-1207.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet CJ, McKenna CH, Luthra HS, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Arnaud L, Devilliers H, Peng SL, et al. The Relapsing Polychondritis Disease Activity Index: development of a disease activity score for relapsing polychondritis. Autoimmun Rev. 2012;12:204-209.
- Brand DD, Kang AH, Rosloniec EF. Immunopathogenesis of collagen arthritis. Springer Semin Immunopathol. 2003;25:3-18.
- Thaiss WM, Nikolaou K, Spengler W, et al. Imaging diagnosis in relapsing polychondritis and correlation with clinical and serological data. Skeletal Radiol. 2015;5:339-346.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Francès C, el Rassi R, Laporte JL, et al. Dermatologic manifestations of relapsing polychondritis. A study of 200 cases at a single center. Medicine (Baltimore). 2001;80:173-179.
- Chopra R, Chaudhary N, Kay J. Relapsing polychondritis. Rheum Dis Clin North Am. 2013;39:263-276.
- Moulis G, Sailler L, Pugnet G, et al. Biologics in relapsing polychondritis: a case series. Clin Exp Rheumatol. 2013;31:937-939.
- Henes CJ, Xenitidis T, Horger M. Tocilizumab for refractory relapsing polychondritis—long-term response monitoring by magnetic resonance imaging. Joint Bone Spine. 2016;83:365-366.
- Weinberger A, Myers AR. Relapsing polychondritis associated with cutaneous vasculitis. Arch Dermatol. 1979;115:980-981.
- Karaca NE, Aksu G, Yildiz B, et al. Relapsing polychondritis in a child with common variable immunodeficiency. Int J Dermatol. 2009;48:525-528.
- Pearson CM, Kline HM, Newcomer VD. Relapsing polychondritis. N Engl J Med. 1960;263:51-58.
- Kent PD, Michet CJ Jr, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol. 2004;16:56-61.
- Ebringer R, Rook G, Swana GT, et al. Autoantibodies to cartilage and type II collagen in relapsing polychondritis and other rheumatic diseases. Ann Rheum Dis. 1981;40:473-479.
- Sharma A, Law AD, Bambery P, et al. Relapsing polychondritis: clinical presentations, disease activity and outcomes. Orphanet J Rare Dis. 2014;9:198.
- Vitale A, Sota J, Rigante D, et al. Relapsing polychondritis: an update on pathogenesis, clinical features, diagnostic tools, and therapeutic perspectives. Curr Rheumatol Rep. 2016;18:3.
- Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002;1:329-337.
- Dolev JC, Maurer TA, Reddy SG, et al. Relapsing polychondritis in HIV-infected patients: a report of two cases. J Am Acad Dermatol. 2004;51:1023-1025.
- Zandman-Goddard G, Peeva E, Barland P. Combined autoimmune disease in a patient with AIDS. Clin Rheumatol. 2002;21:70-72.
- Belzunegui J, Cancio J, Pego JM, et al. Relapsing polychondritis and Behc¸et’s syndrome in a patient with HIV infection. Ann Rheum Dis. 1995;54:780.
- Sharma A, Gnanapandithan K, Sharma K, et al. Relapsing polychondritis: a review. Clin Rheumatol. 2013;32:1575-1583.
- Cantarini L, Vitale A, Brizi MG, et al. Diagnosis and classification of relapsing polychondritis. J Autoimmun. 2014;48-49:53-59.
- Cañas CA, Bonilla Abadía F. Local cartilage trauma as a pathogenic factor in autoimmunity (one hypothesis based on patients with relapsing polychondritis triggered by cartilage trauma). Autoimmune Dis. 2012;2012:453698.
- Ouchi N, Uzuki M, Kamataki A, et al. Cartilage destruction is partly induced by the internal proteolytic enzymes and apoptotic phenomenon of chondrocytes in relapsing polychondritis. J Rheumatol. 2011;38:730-737.
- Buckner JH, Wu JJ, Reife RA, et al. Autoreactivity against matrilin-1 in a patient with relapsing polychondritis. Arthritis Rheum. 2000;43:939-943.
- Kempta Lekpa F, Piette JC, Bastuji-Garin S, et al. Serum cartilage oligomeric matrix protein (COMP) is a marker of disease activity in relapsing polychondritis. Clin Exp Rheumatol. 2010;28:553-555.
- Foidart JM, Abe S, Martin GR, et al. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med. 1978;299:1203-1207.
- McAdam LP, O’Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and review of the literature. Medicine (Baltimore). 1976;55:193-215.
- Michet CJ, McKenna CH, Luthra HS, et al. Relapsing polychondritis: survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104:74-78.
- Arnaud L, Devilliers H, Peng SL, et al. The Relapsing Polychondritis Disease Activity Index: development of a disease activity score for relapsing polychondritis. Autoimmun Rev. 2012;12:204-209.
- Brand DD, Kang AH, Rosloniec EF. Immunopathogenesis of collagen arthritis. Springer Semin Immunopathol. 2003;25:3-18.
- Thaiss WM, Nikolaou K, Spengler W, et al. Imaging diagnosis in relapsing polychondritis and correlation with clinical and serological data. Skeletal Radiol. 2015;5:339-346.
- Lahmer T, Treiber M, von Werder A, et al. Relapsing polychondritis: an autoimmune disease with many faces. Autoimmun Rev. 2010;9:540-546.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Francès C, el Rassi R, Laporte JL, et al. Dermatologic manifestations of relapsing polychondritis. A study of 200 cases at a single center. Medicine (Baltimore). 2001;80:173-179.
- Chopra R, Chaudhary N, Kay J. Relapsing polychondritis. Rheum Dis Clin North Am. 2013;39:263-276.
- Moulis G, Sailler L, Pugnet G, et al. Biologics in relapsing polychondritis: a case series. Clin Exp Rheumatol. 2013;31:937-939.
- Henes CJ, Xenitidis T, Horger M. Tocilizumab for refractory relapsing polychondritis—long-term response monitoring by magnetic resonance imaging. Joint Bone Spine. 2016;83:365-366.
- Weinberger A, Myers AR. Relapsing polychondritis associated with cutaneous vasculitis. Arch Dermatol. 1979;115:980-981.
- Karaca NE, Aksu G, Yildiz B, et al. Relapsing polychondritis in a child with common variable immunodeficiency. Int J Dermatol. 2009;48:525-528.
Practice Points
- Relapsing polychondritis (RP) is characterized by recurrent inflammatory insults to cartilaginous and proteoglycan-rich structures, most often manifesting as ear inflammation that involves the auricle but spares the lobe, nasal chondritis, and arthralgia.
- Relapsing polychondritis acts classically as an autoimmune disease with a variable presentation, making diagnosis a challenge.
- One-third of RP patients have coexisting autoimmune disease.
- Treatment of RP depends on severity of disease.
- Dermatologists must be aware of the potential for development of RP in the setting of human immunodeficiency virus infection; a missed diagnosis of this progressive disease has the potential to be life-threatening.