User login
Papular Mucinosis/Scleromyxedema
Papular mucinosis/scleromyxedema, also known as generalized lichen myxedematosus, is a rare dermal mucinosis characterized by a papular eruption that can have an associated IgG λ paraproteinemia. The clinical presentation is gradual with the development of firm, flesh-colored, 2- to 3-mm papules often involving the hands, face, and neck that can progress to plaques that cover the entire body. Skin stiffening also can be seen.1 Extracutaneous symptoms are common and include dysphagia, arthralgia, myopathy, and cardiac dysfunction.2 Occasionally, central nervous system involvement can lead to the often fatal dermato-neuro syndrome.3,4
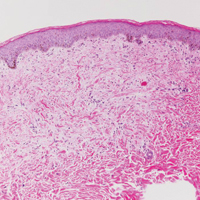
Histologically, papular mucinosis/scleromyxedema demonstrates increased, irregularly arranged fibroblasts in the reticular dermis with increased dermal mucin deposition (quiz image and Figure 1). The epidermis is normal or slightly thinned due to pressure from dermal changes. There may be a mild superficial perivascular lymphocytic infiltrate and atrophy of hair follicles.5 In this case, the clinical and histologic findings best supported a diagnosis of papular mucinosis/scleromyxedema.

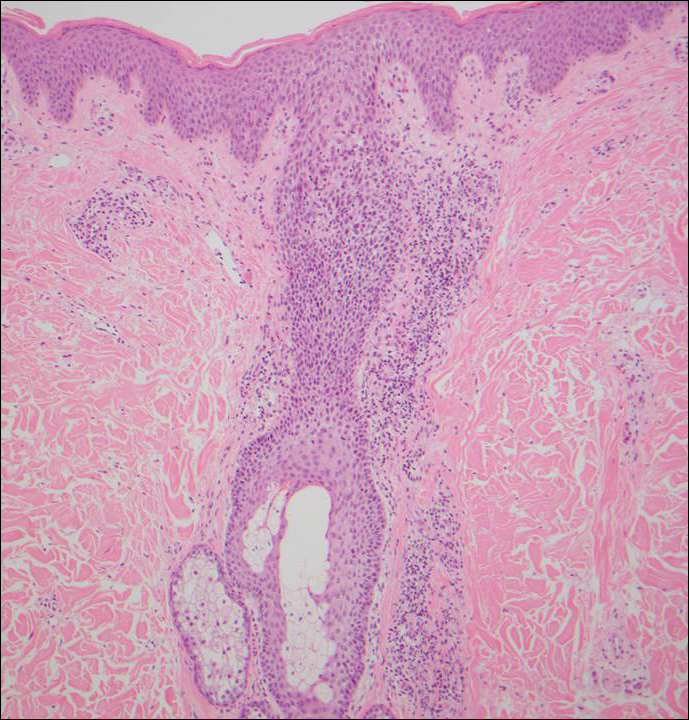
Infundibulofolliculitis is a pruritic follicular papular eruption typically involving the neck, trunk, and proximal upper arms and shoulders. It is most common in black men who reside in hot and humid climates. Although infundibulofolliculitis would be included in the clinical differential diagnosis for the current patient, the histopathologic findings were quite distinct for the correct diagnosis of papular mucinosis/scleromyxedema. Infundibulofolliculitis shows widening of the upper part of the hair follicle (infundibulum) and infundibular inflammatory infiltrate with follicular spongiosis (Figure 2). Neither mucin deposition nor fibroblast proliferation is appreciated in infundibulofolliculitis.6,7
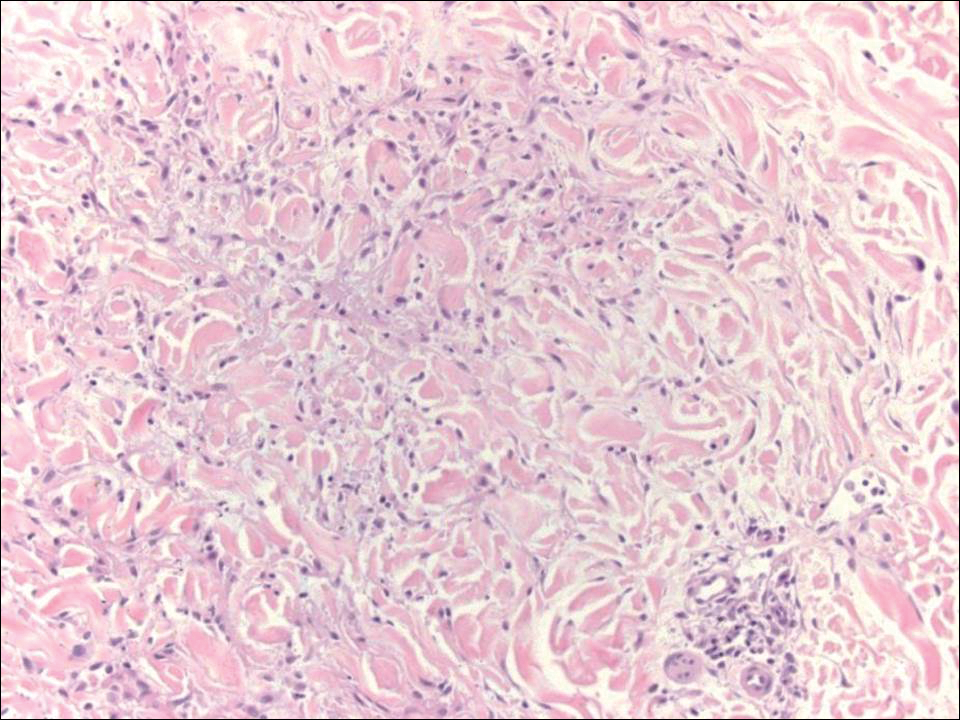
Granuloma annulare (GA) often can be distinguished clinically from papular mucinosis/scleromyxedema due to the annular appearance of papules and plaques in GA and the lack of stiffness of underlying skin. Interstitial granuloma annulare is a histologic variant of GA that can be included in the histologic differential diagnosis of papular mucinosis/scleromyxedema. Histologically, there is an interstitial infiltrate of cytologically bland histiocytes dissecting between collagen bundles in interstitial GA (Figure 3). Necrobiosis and collections of mucin often are inconspicuous. Occasionally, the presence of eosinophils can be a helpful clue.8 A fibroblast proliferation is not a feature of GA.
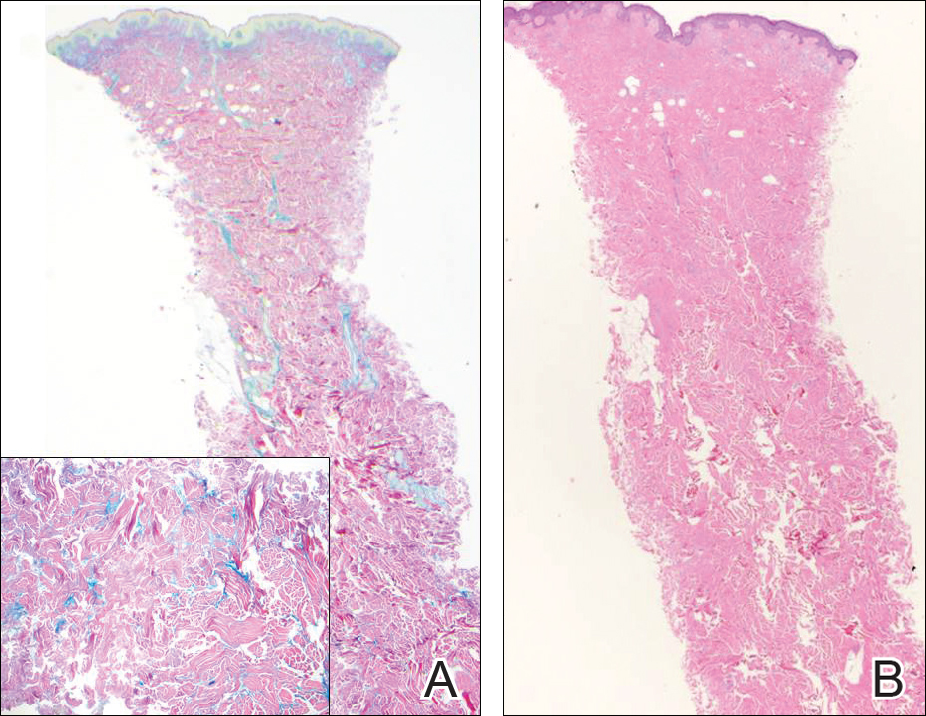
Reticular erythematous mucinosis also is a type of cutaneous mucinosis but with a classic clinical appearance of a reticulated erythematous plaque on the chest or back, making it clinically distinct from papular mucinosis/scleromyxedema and the presentation described in the current patient. Reticular erythematous mucinosis can be histologically distinguished from papular mucinosis/scleromyxedema by the presence of a superficial and deep perivascular lymphocytic infiltrate with increased dermal mucin deposition (Figure 4). It often shows a positive IgM deposition on the basement membrane on direct immunofluorescence.9

Similar to papular mucinosis/scleromyxedema, scleredema shows thickening of the skin with decreased movement of involved areas. Scleredema often involves the upper back, shoulders, and neck where affected areas often have a peau d'orange appearance. Scleredema is classified into 3 clinical forms based on clinical associations. Type 1 often is preceded by an infection, classically Streptococcus pyogenes. Type 2 is associated with a hematologic dyscrasia such as multiple myeloma, or it can have an associated paraproteinemia that is typically of the IgA κ type, which is distinct from papular mucinosis/scleromyxedema where IgG λ paraproteinemia typically is seen. Type 3 is associated with diabetes mellitus. Histologically, scleredema also is distinct from papular mucinosis/scleromyxedema. Although increased mucin is seen in the dermis, the mucin is classically more prominent in the deep reticular dermis as compared with papular mucinosis/scleromyxedema (Figure 5). Additionally, collagen bundles are thickened with clear separation between them. Hyperplasia of fibroblasts in the dermis that is a characteristic feature of papular mucinosis/scleromyxedema is not observed in scleredema.10
- Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Fleming KE, Virmani D, Sutton E, et al. Scleromyxedema and the dermato-neuro syndrome: case report and review of the literature. J Cutan Pathol. 2012;39:508-517.
- Hummers LK. Scleromyxedema. Curr Opin Rheumatol. 2014;26:658-662.
- Rongioleti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Owen WR, Wood C. Disseminate and recurrent infundibulofolliculitis. Arch Dermatol. 1979;5:174-175.
- Soyinka F. Recurrent disseminated infundibulofolliculitis. Int J Dermatol. 1973;12:314-317.
- Keimig EL. Granuloma annulare. Dermatol Clin. 2015;33:315-329.
- Thareja S, Paghdal K, Lein MH, et al. Reticular erythematous mucinosis--a review. Int J Dermatol. 2012;51:903-909.
- Beers WH, Ince AI, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
Papular Mucinosis/Scleromyxedema
Papular mucinosis/scleromyxedema, also known as generalized lichen myxedematosus, is a rare dermal mucinosis characterized by a papular eruption that can have an associated IgG λ paraproteinemia. The clinical presentation is gradual with the development of firm, flesh-colored, 2- to 3-mm papules often involving the hands, face, and neck that can progress to plaques that cover the entire body. Skin stiffening also can be seen.1 Extracutaneous symptoms are common and include dysphagia, arthralgia, myopathy, and cardiac dysfunction.2 Occasionally, central nervous system involvement can lead to the often fatal dermato-neuro syndrome.3,4
Histologically, papular mucinosis/scleromyxedema demonstrates increased, irregularly arranged fibroblasts in the reticular dermis with increased dermal mucin deposition (quiz image and Figure 1). The epidermis is normal or slightly thinned due to pressure from dermal changes. There may be a mild superficial perivascular lymphocytic infiltrate and atrophy of hair follicles.5 In this case, the clinical and histologic findings best supported a diagnosis of papular mucinosis/scleromyxedema.
Infundibulofolliculitis is a pruritic follicular papular eruption typically involving the neck, trunk, and proximal upper arms and shoulders. It is most common in black men who reside in hot and humid climates. Although infundibulofolliculitis would be included in the clinical differential diagnosis for the current patient, the histopathologic findings were quite distinct for the correct diagnosis of papular mucinosis/scleromyxedema. Infundibulofolliculitis shows widening of the upper part of the hair follicle (infundibulum) and infundibular inflammatory infiltrate with follicular spongiosis (Figure 2). Neither mucin deposition nor fibroblast proliferation is appreciated in infundibulofolliculitis.6,7
Granuloma annulare (GA) often can be distinguished clinically from papular mucinosis/scleromyxedema due to the annular appearance of papules and plaques in GA and the lack of stiffness of underlying skin. Interstitial granuloma annulare is a histologic variant of GA that can be included in the histologic differential diagnosis of papular mucinosis/scleromyxedema. Histologically, there is an interstitial infiltrate of cytologically bland histiocytes dissecting between collagen bundles in interstitial GA (Figure 3). Necrobiosis and collections of mucin often are inconspicuous. Occasionally, the presence of eosinophils can be a helpful clue.8 A fibroblast proliferation is not a feature of GA.
Reticular erythematous mucinosis also is a type of cutaneous mucinosis but with a classic clinical appearance of a reticulated erythematous plaque on the chest or back, making it clinically distinct from papular mucinosis/scleromyxedema and the presentation described in the current patient. Reticular erythematous mucinosis can be histologically distinguished from papular mucinosis/scleromyxedema by the presence of a superficial and deep perivascular lymphocytic infiltrate with increased dermal mucin deposition (Figure 4). It often shows a positive IgM deposition on the basement membrane on direct immunofluorescence.9
Similar to papular mucinosis/scleromyxedema, scleredema shows thickening of the skin with decreased movement of involved areas. Scleredema often involves the upper back, shoulders, and neck where affected areas often have a peau d'orange appearance. Scleredema is classified into 3 clinical forms based on clinical associations. Type 1 often is preceded by an infection, classically Streptococcus pyogenes. Type 2 is associated with a hematologic dyscrasia such as multiple myeloma, or it can have an associated paraproteinemia that is typically of the IgA κ type, which is distinct from papular mucinosis/scleromyxedema where IgG λ paraproteinemia typically is seen. Type 3 is associated with diabetes mellitus. Histologically, scleredema also is distinct from papular mucinosis/scleromyxedema. Although increased mucin is seen in the dermis, the mucin is classically more prominent in the deep reticular dermis as compared with papular mucinosis/scleromyxedema (Figure 5). Additionally, collagen bundles are thickened with clear separation between them. Hyperplasia of fibroblasts in the dermis that is a characteristic feature of papular mucinosis/scleromyxedema is not observed in scleredema.10
Papular Mucinosis/Scleromyxedema
Papular mucinosis/scleromyxedema, also known as generalized lichen myxedematosus, is a rare dermal mucinosis characterized by a papular eruption that can have an associated IgG λ paraproteinemia. The clinical presentation is gradual with the development of firm, flesh-colored, 2- to 3-mm papules often involving the hands, face, and neck that can progress to plaques that cover the entire body. Skin stiffening also can be seen.1 Extracutaneous symptoms are common and include dysphagia, arthralgia, myopathy, and cardiac dysfunction.2 Occasionally, central nervous system involvement can lead to the often fatal dermato-neuro syndrome.3,4
Histologically, papular mucinosis/scleromyxedema demonstrates increased, irregularly arranged fibroblasts in the reticular dermis with increased dermal mucin deposition (quiz image and Figure 1). The epidermis is normal or slightly thinned due to pressure from dermal changes. There may be a mild superficial perivascular lymphocytic infiltrate and atrophy of hair follicles.5 In this case, the clinical and histologic findings best supported a diagnosis of papular mucinosis/scleromyxedema.
Infundibulofolliculitis is a pruritic follicular papular eruption typically involving the neck, trunk, and proximal upper arms and shoulders. It is most common in black men who reside in hot and humid climates. Although infundibulofolliculitis would be included in the clinical differential diagnosis for the current patient, the histopathologic findings were quite distinct for the correct diagnosis of papular mucinosis/scleromyxedema. Infundibulofolliculitis shows widening of the upper part of the hair follicle (infundibulum) and infundibular inflammatory infiltrate with follicular spongiosis (Figure 2). Neither mucin deposition nor fibroblast proliferation is appreciated in infundibulofolliculitis.6,7
Granuloma annulare (GA) often can be distinguished clinically from papular mucinosis/scleromyxedema due to the annular appearance of papules and plaques in GA and the lack of stiffness of underlying skin. Interstitial granuloma annulare is a histologic variant of GA that can be included in the histologic differential diagnosis of papular mucinosis/scleromyxedema. Histologically, there is an interstitial infiltrate of cytologically bland histiocytes dissecting between collagen bundles in interstitial GA (Figure 3). Necrobiosis and collections of mucin often are inconspicuous. Occasionally, the presence of eosinophils can be a helpful clue.8 A fibroblast proliferation is not a feature of GA.
Reticular erythematous mucinosis also is a type of cutaneous mucinosis but with a classic clinical appearance of a reticulated erythematous plaque on the chest or back, making it clinically distinct from papular mucinosis/scleromyxedema and the presentation described in the current patient. Reticular erythematous mucinosis can be histologically distinguished from papular mucinosis/scleromyxedema by the presence of a superficial and deep perivascular lymphocytic infiltrate with increased dermal mucin deposition (Figure 4). It often shows a positive IgM deposition on the basement membrane on direct immunofluorescence.9
Similar to papular mucinosis/scleromyxedema, scleredema shows thickening of the skin with decreased movement of involved areas. Scleredema often involves the upper back, shoulders, and neck where affected areas often have a peau d'orange appearance. Scleredema is classified into 3 clinical forms based on clinical associations. Type 1 often is preceded by an infection, classically Streptococcus pyogenes. Type 2 is associated with a hematologic dyscrasia such as multiple myeloma, or it can have an associated paraproteinemia that is typically of the IgA κ type, which is distinct from papular mucinosis/scleromyxedema where IgG λ paraproteinemia typically is seen. Type 3 is associated with diabetes mellitus. Histologically, scleredema also is distinct from papular mucinosis/scleromyxedema. Although increased mucin is seen in the dermis, the mucin is classically more prominent in the deep reticular dermis as compared with papular mucinosis/scleromyxedema (Figure 5). Additionally, collagen bundles are thickened with clear separation between them. Hyperplasia of fibroblasts in the dermis that is a characteristic feature of papular mucinosis/scleromyxedema is not observed in scleredema.10
- Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Fleming KE, Virmani D, Sutton E, et al. Scleromyxedema and the dermato-neuro syndrome: case report and review of the literature. J Cutan Pathol. 2012;39:508-517.
- Hummers LK. Scleromyxedema. Curr Opin Rheumatol. 2014;26:658-662.
- Rongioleti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Owen WR, Wood C. Disseminate and recurrent infundibulofolliculitis. Arch Dermatol. 1979;5:174-175.
- Soyinka F. Recurrent disseminated infundibulofolliculitis. Int J Dermatol. 1973;12:314-317.
- Keimig EL. Granuloma annulare. Dermatol Clin. 2015;33:315-329.
- Thareja S, Paghdal K, Lein MH, et al. Reticular erythematous mucinosis--a review. Int J Dermatol. 2012;51:903-909.
- Beers WH, Ince AI, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
- Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Fleming KE, Virmani D, Sutton E, et al. Scleromyxedema and the dermato-neuro syndrome: case report and review of the literature. J Cutan Pathol. 2012;39:508-517.
- Hummers LK. Scleromyxedema. Curr Opin Rheumatol. 2014;26:658-662.
- Rongioleti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-281.
- Owen WR, Wood C. Disseminate and recurrent infundibulofolliculitis. Arch Dermatol. 1979;5:174-175.
- Soyinka F. Recurrent disseminated infundibulofolliculitis. Int J Dermatol. 1973;12:314-317.
- Keimig EL. Granuloma annulare. Dermatol Clin. 2015;33:315-329.
- Thareja S, Paghdal K, Lein MH, et al. Reticular erythematous mucinosis--a review. Int J Dermatol. 2012;51:903-909.
- Beers WH, Ince AI, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
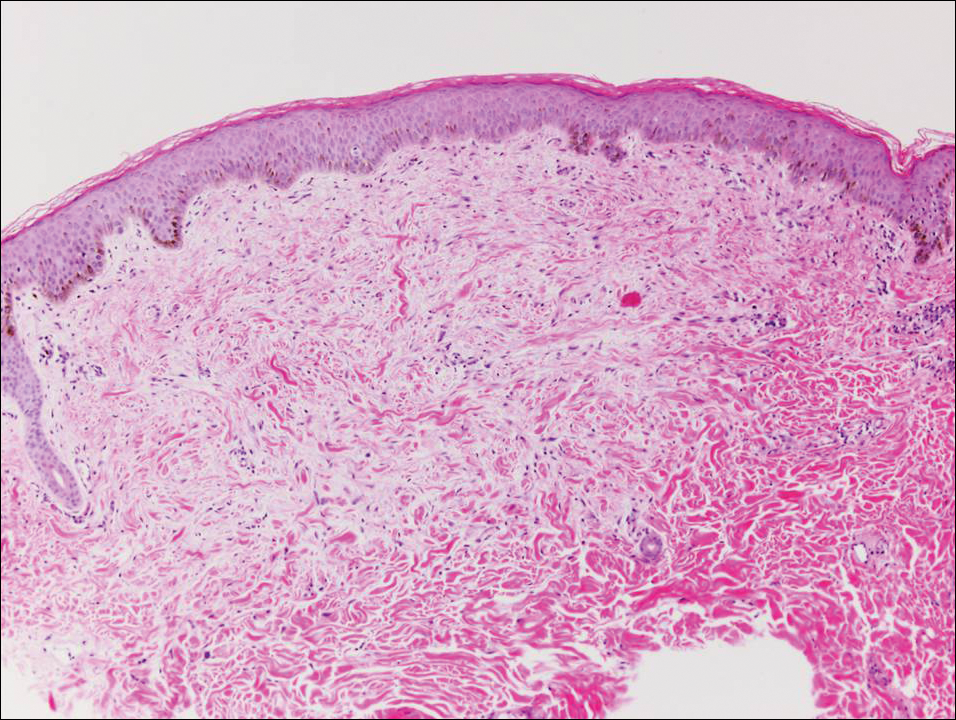
A 48-year-old black man presented with a rash of 7 months' duration that started on the face and spread to the body. He had extreme pruritus, increased stiffness in the hands and joints, and paresthesia. Physical examination revealed an eruption of 2- to 4-mm, flesh-colored papules with follicular accentuation on the face, neck, bilateral upper extremities, back, and thighs.