User login
Acute atherothrombotic events associated with ischemic heart disease and stroke are the first and third most common causes of death in the United States, respectively.1 Despite an overall decrease in age‐adjusted mortality since 1970 in the United States, the worldwide prevalence of these diseases is anticipated to sharply increase by 2020.1, 2 Caring for patients with atherothrombosis is now within the purview of hospitalists to a larger extent than ever before. In recognition of the expanding role of these health care professionals and to reduce the risk of adverse cardiovascular events in the outpatient setting, the Society of Hospital Medicine held a symposium during its 10th Annual Meeting.
Rules of Engagement: The Hospitalist and Atherothrombosis took place on May 24, 2007, in Dallas, Texas. This supplement summarizes the highlights from this symposium and reviews the causes and polyvascular nature of atherothrombosis. The role of the hospitalist in managing atherothrombotic disease and evidence‐based practices for the evaluation and treatment of patients with various manifestations of atherothrombotic disease are also discussed.
ARTERIAL THROMBOSIS AND ITS POLYVASCULAR NATURE
Atherothrombosis refers to the formation of large and occlusive mural thrombi that arise from the rupture of an atherosclerotic plaque. Myocardial infarction (MI), ischemic stroke, and acute limb ischemia are the most severe manifestations of this disease.3, 4 This process begins when denuded or inflamed endothelial cells develop properties that permit platelet adhesion. At the site of endothelial dysfunction, activation of adherent platelet results in the release of inflammatory and mitogenic factors. After a series of dynamic and repetitive processes including amplified platelet activation, monocyte chemotaxis, adhesion, transmigration, and lipoprotein retention, plaque formation occurs.5 Consequently, the rupture or erosion of an atherosclerotic plaque produces a higher degree of platelet adhesion, activation, and aggregation, causing the fibrotic organization of a mural thrombus.3
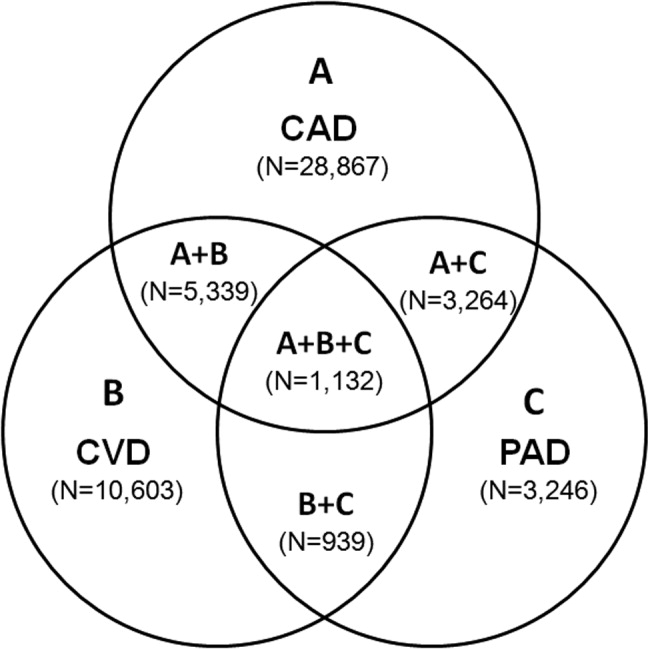
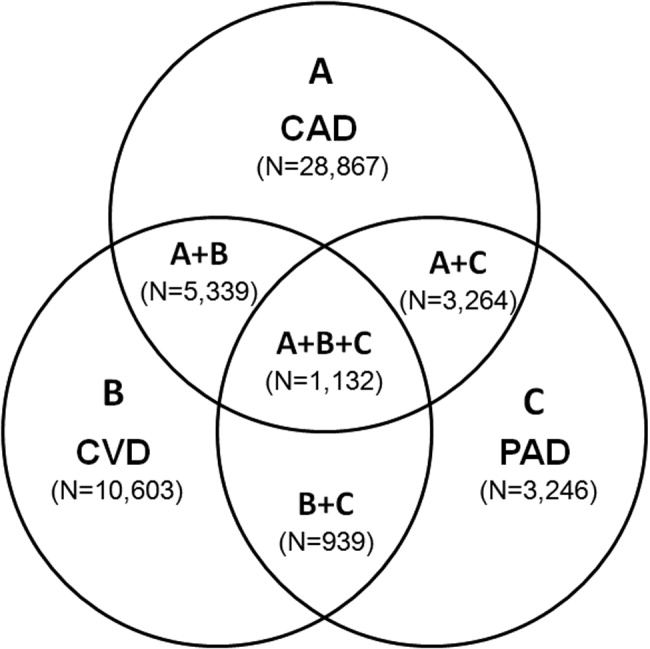
The number of persons with multiple, concomitant cardiovascular disease (CAD), cerebrovascular disease (CVD), and peripheral arterial disease (PAD) accentuates the polyvascular nature of atherothrombosis (Fig. 1). The international Reduction of Atherothrombosis for Continued Health (REACH) Registry demonstrated that 1‐year incidence rates of major cardiovascular events (eg, MI, stroke, death) were high in patients with an established atherothrombotic disease and increased with the number of concomitant vascular diseases.6 These data infer that the burden on the vascular system is considered extensive on diagnosis of a single atherothrombotic disease. Thus, aggressive therapies are needed to reduce the risk of recurrent or other cardiovascular events. The management of risk factors for atherothrombosis such as hypercholesterolemia, dyslipidemia, hypertension, and diabetes mellitus fall under specific disease‐specific guidelines for patients presenting with atherothrombotic diseases.712
ANTIPLATELET THERAPIES
Antiplatelet therapies are used for the acute and long‐term treatment of patients after a thrombic event. Antiplatelet agents target the molecular mechanisms responsible for platelet activation and aggregation, such as the synthesis of thromboxane A2. On platelet activation, free arachidonic acid is converted to prostaglandin H2 (PGH2) by cyclooxygenase‐1 (COX‐1; Fig. 2). Further metabolism of PGH2 by thromboxane synthase produces thromboxane A2, which induces vasoconstriction (Fig. 2). Fortunately, the ability of platelets to produce COX‐1 is limited, and irreversible inhibition of this enzyme can impair thromboxane A2 synthesis for approximately 10 days.
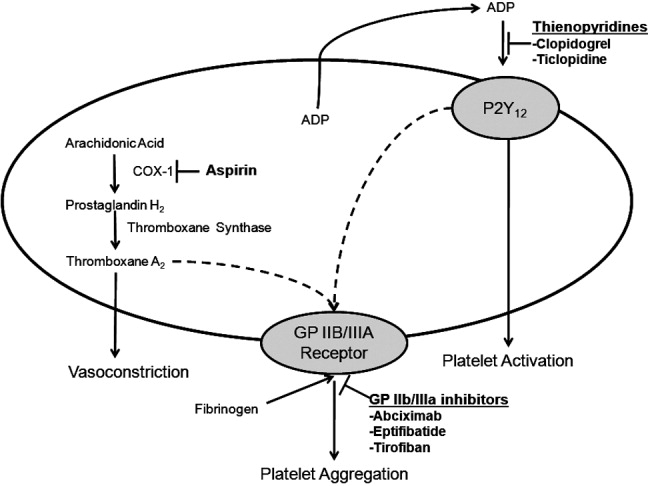
Aspirin is a potent COX‐1 inhibitor, whose effects are evident 1 hour after dosing (Fig. 2).4, 13 Aspirin effectively prevents fatal and nonfatal vascular events in healthy individuals and in patients who present with acute MI or ischemic stroke.13 Unfortunately, a proportion of patients are aspirin resistant. Recent studies have indicated that interactions with the nonsteroidal anti‐inflammatory drug (NSAID) ibuprofen may diminish the primary and secondary protective effects of aspirin and may contribute to aspirin resistance, although the origin of this remains unclear.
The results of a post hoc subgroup analysis of 22,071 apparently healthy male physicians randomized to take aspirin or placebo for 5 years indicated that individuals who used NSAIDs for at least 60 days/year increased their risk of MI by more than 2‐fold compared with those who did not use NSAIDs.14 A second study conducted in patients following a major adverse cardiovascular event showed that the combination of aspirin plus ibuprofen increased the adjusted relative risk of cardiovascular mortality over an 8‐year period compared with aspirin alone.15 However, the effects of NSAIDS on aspirin's ability to inhibit COX‐1 are reversible and only last for the dosing interval and body clearance time of the drug.16
Adeonsine diphosphate (ADP)dependent stimulation of the P2Y12 receptor is another target for antiplatelet therapy. On its release, ADP binds to the P2Y12 receptor on platelets, resulting in activation and aggregation (Fig. 2). Ticlopidine and clopidogrel are thienopyridines that may irreversibly modify the P2Y12 receptor (Fig. 2).13 Safety concerns associated with ticlopidine use, including severe neutropenia, have limited its administration. Conversely, clopidogrel is relatively well‐tolerated and can prevent cardiovascular events in patients with CAD, ischemic stroke, and PAD. This agent is an orally administered prodrug requiring activation by hepatic cytochrome P450 enzymes.13
Aspirin and thienopyridines do not inhibit platelet aggregation induced by the binding of fibrinogen to the platelet glycoprotein (GP) IIb/IIIa receptor (Fig. 2).4, 13 However, there are 3 commonly administered GP IIb/IIIa inhibitors: abciximab, eptifibatide, and tirofiban (Fig. 2).4 Abciximab is the fab fragment of the chimeric monoclonal antibody 7E3 and irreversibly inhibits the GP IIb/IIIa receptor. By contrast, eptifibatide is a cyclic heptapeptide, tirofiban is a nonpeptide, and both agents are reversible inhibitors. These agents are administered intravenously, and boluses are reserved for the short‐term treatment of atherothrombosis in patients undergoing percutaneous coronary intervention.13
CONCLUSIONS
Atherothrombosis is a systemic disease that often affects coronary, intracranial, and peripheral arterial beds concomitantly, which increases the probability of a thrombotic event. Aggressive treatments, including acute and long‐term antiplatelet therapies, are required to reduce the risks associated with atherothrombosis. This supplement reviews the evidence‐based approaches for managing atherothrombosis. It will provide hospitalists with the knowledge needed to treat patients with PAD, stroke, and acute coronary syndrome. First, the administration of antiplatelet therapies to patients with acute coronary syndrome will be described. Then, guidelines for the management of patients with acute ischemic stroke and the use of antiplatelet therapies to reduce mortality due to primary and secondary ischemic events will be reviewed. Finally, the role of the hospitalist in the diagnosis of PAD in asymptomatic patients and in those with confirmed atherothrombosis will be discussed.
- ,,,.Trends in the leading causes of death in the United States, 1970‐2002.JAMA.2005;294:1255–1259.
- ,.The global burden of disease, 1990‐2020.Nat Med.1998:4:1241–1243.
- ,,,.The pathogenesis of coronary artery disease and the acute coronary syndromes.N Engl J Med.1992;326:242–250.
- .Antiplatelet therapy.Am J Med.1996;101:199–209.
- ,,.Platelets in inflammation and atherogenesis.J Clin Invest.2005;115:3378–3384.
- ,,, et al.One‐year cardiovascular event rates in outpatients with atherothrombosis.JAMA.2007;297:1197–1206.
- ,,, et al.ACC/AHA 2000 guidelines for management of patients with unstable angina and non‐ST‐segment elevation myocardial infarction.J Am Coll Cardiol.2000;36:970–1062.
- ,,, et al.ACC/AHA guidelines for the management of patients with ST‐elevation myocardial infarction.Circulation.2004;110:82–292.
- ,,, et al.Guidelines for the prevention of stroke in patients with ischemic stroke or transient ischemic attack. A statement for healthcare professionals from the American Heart Association/American Stroke Association Council on Stroke.Stroke.2006;37:557–617.
- ,,, et al.ACC/AHA 2005 practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines.Circulation.2006;113:463–654.
- ,,.Inflammation and atherosclerosis.Circulation.2002;105:1135–1143.
- ,,, et al.AHA/ACC guidelines for secondary prevention for patients with coronary and other atherosclerotic vascular disease: 2006 update.J Am Coll Cardiol.2006;47:2130–2139.
- ,,, et al.Platelet‐active drugs: the relationships among dose, effectiveness, and side effects.Chest.2001;119:39–63.
- ,,, et al.Inhibition of clinical benefits of aspirin on first myocardial infarction by nonsteroidal anti‐inflammatory drugs.Circulation.2003;108:1191–1195.
- ,.The effect of ibuprofen on cardioprotective effects of aspirin.Lancet.2003;361:573–574.
- ,,, et al.Cyclooxygenase inhibitors and the antiplatelet effects of aspirin.N Engl J Med.2001;345:1809–1817.
Acute atherothrombotic events associated with ischemic heart disease and stroke are the first and third most common causes of death in the United States, respectively.1 Despite an overall decrease in age‐adjusted mortality since 1970 in the United States, the worldwide prevalence of these diseases is anticipated to sharply increase by 2020.1, 2 Caring for patients with atherothrombosis is now within the purview of hospitalists to a larger extent than ever before. In recognition of the expanding role of these health care professionals and to reduce the risk of adverse cardiovascular events in the outpatient setting, the Society of Hospital Medicine held a symposium during its 10th Annual Meeting.
Rules of Engagement: The Hospitalist and Atherothrombosis took place on May 24, 2007, in Dallas, Texas. This supplement summarizes the highlights from this symposium and reviews the causes and polyvascular nature of atherothrombosis. The role of the hospitalist in managing atherothrombotic disease and evidence‐based practices for the evaluation and treatment of patients with various manifestations of atherothrombotic disease are also discussed.
ARTERIAL THROMBOSIS AND ITS POLYVASCULAR NATURE
Atherothrombosis refers to the formation of large and occlusive mural thrombi that arise from the rupture of an atherosclerotic plaque. Myocardial infarction (MI), ischemic stroke, and acute limb ischemia are the most severe manifestations of this disease.3, 4 This process begins when denuded or inflamed endothelial cells develop properties that permit platelet adhesion. At the site of endothelial dysfunction, activation of adherent platelet results in the release of inflammatory and mitogenic factors. After a series of dynamic and repetitive processes including amplified platelet activation, monocyte chemotaxis, adhesion, transmigration, and lipoprotein retention, plaque formation occurs.5 Consequently, the rupture or erosion of an atherosclerotic plaque produces a higher degree of platelet adhesion, activation, and aggregation, causing the fibrotic organization of a mural thrombus.3
The number of persons with multiple, concomitant cardiovascular disease (CAD), cerebrovascular disease (CVD), and peripheral arterial disease (PAD) accentuates the polyvascular nature of atherothrombosis (Fig. 1). The international Reduction of Atherothrombosis for Continued Health (REACH) Registry demonstrated that 1‐year incidence rates of major cardiovascular events (eg, MI, stroke, death) were high in patients with an established atherothrombotic disease and increased with the number of concomitant vascular diseases.6 These data infer that the burden on the vascular system is considered extensive on diagnosis of a single atherothrombotic disease. Thus, aggressive therapies are needed to reduce the risk of recurrent or other cardiovascular events. The management of risk factors for atherothrombosis such as hypercholesterolemia, dyslipidemia, hypertension, and diabetes mellitus fall under specific disease‐specific guidelines for patients presenting with atherothrombotic diseases.712
ANTIPLATELET THERAPIES
Antiplatelet therapies are used for the acute and long‐term treatment of patients after a thrombic event. Antiplatelet agents target the molecular mechanisms responsible for platelet activation and aggregation, such as the synthesis of thromboxane A2. On platelet activation, free arachidonic acid is converted to prostaglandin H2 (PGH2) by cyclooxygenase‐1 (COX‐1; Fig. 2). Further metabolism of PGH2 by thromboxane synthase produces thromboxane A2, which induces vasoconstriction (Fig. 2). Fortunately, the ability of platelets to produce COX‐1 is limited, and irreversible inhibition of this enzyme can impair thromboxane A2 synthesis for approximately 10 days.
Aspirin is a potent COX‐1 inhibitor, whose effects are evident 1 hour after dosing (Fig. 2).4, 13 Aspirin effectively prevents fatal and nonfatal vascular events in healthy individuals and in patients who present with acute MI or ischemic stroke.13 Unfortunately, a proportion of patients are aspirin resistant. Recent studies have indicated that interactions with the nonsteroidal anti‐inflammatory drug (NSAID) ibuprofen may diminish the primary and secondary protective effects of aspirin and may contribute to aspirin resistance, although the origin of this remains unclear.
The results of a post hoc subgroup analysis of 22,071 apparently healthy male physicians randomized to take aspirin or placebo for 5 years indicated that individuals who used NSAIDs for at least 60 days/year increased their risk of MI by more than 2‐fold compared with those who did not use NSAIDs.14 A second study conducted in patients following a major adverse cardiovascular event showed that the combination of aspirin plus ibuprofen increased the adjusted relative risk of cardiovascular mortality over an 8‐year period compared with aspirin alone.15 However, the effects of NSAIDS on aspirin's ability to inhibit COX‐1 are reversible and only last for the dosing interval and body clearance time of the drug.16
Adeonsine diphosphate (ADP)dependent stimulation of the P2Y12 receptor is another target for antiplatelet therapy. On its release, ADP binds to the P2Y12 receptor on platelets, resulting in activation and aggregation (Fig. 2). Ticlopidine and clopidogrel are thienopyridines that may irreversibly modify the P2Y12 receptor (Fig. 2).13 Safety concerns associated with ticlopidine use, including severe neutropenia, have limited its administration. Conversely, clopidogrel is relatively well‐tolerated and can prevent cardiovascular events in patients with CAD, ischemic stroke, and PAD. This agent is an orally administered prodrug requiring activation by hepatic cytochrome P450 enzymes.13
Aspirin and thienopyridines do not inhibit platelet aggregation induced by the binding of fibrinogen to the platelet glycoprotein (GP) IIb/IIIa receptor (Fig. 2).4, 13 However, there are 3 commonly administered GP IIb/IIIa inhibitors: abciximab, eptifibatide, and tirofiban (Fig. 2).4 Abciximab is the fab fragment of the chimeric monoclonal antibody 7E3 and irreversibly inhibits the GP IIb/IIIa receptor. By contrast, eptifibatide is a cyclic heptapeptide, tirofiban is a nonpeptide, and both agents are reversible inhibitors. These agents are administered intravenously, and boluses are reserved for the short‐term treatment of atherothrombosis in patients undergoing percutaneous coronary intervention.13
CONCLUSIONS
Atherothrombosis is a systemic disease that often affects coronary, intracranial, and peripheral arterial beds concomitantly, which increases the probability of a thrombotic event. Aggressive treatments, including acute and long‐term antiplatelet therapies, are required to reduce the risks associated with atherothrombosis. This supplement reviews the evidence‐based approaches for managing atherothrombosis. It will provide hospitalists with the knowledge needed to treat patients with PAD, stroke, and acute coronary syndrome. First, the administration of antiplatelet therapies to patients with acute coronary syndrome will be described. Then, guidelines for the management of patients with acute ischemic stroke and the use of antiplatelet therapies to reduce mortality due to primary and secondary ischemic events will be reviewed. Finally, the role of the hospitalist in the diagnosis of PAD in asymptomatic patients and in those with confirmed atherothrombosis will be discussed.
Acute atherothrombotic events associated with ischemic heart disease and stroke are the first and third most common causes of death in the United States, respectively.1 Despite an overall decrease in age‐adjusted mortality since 1970 in the United States, the worldwide prevalence of these diseases is anticipated to sharply increase by 2020.1, 2 Caring for patients with atherothrombosis is now within the purview of hospitalists to a larger extent than ever before. In recognition of the expanding role of these health care professionals and to reduce the risk of adverse cardiovascular events in the outpatient setting, the Society of Hospital Medicine held a symposium during its 10th Annual Meeting.
Rules of Engagement: The Hospitalist and Atherothrombosis took place on May 24, 2007, in Dallas, Texas. This supplement summarizes the highlights from this symposium and reviews the causes and polyvascular nature of atherothrombosis. The role of the hospitalist in managing atherothrombotic disease and evidence‐based practices for the evaluation and treatment of patients with various manifestations of atherothrombotic disease are also discussed.
ARTERIAL THROMBOSIS AND ITS POLYVASCULAR NATURE
Atherothrombosis refers to the formation of large and occlusive mural thrombi that arise from the rupture of an atherosclerotic plaque. Myocardial infarction (MI), ischemic stroke, and acute limb ischemia are the most severe manifestations of this disease.3, 4 This process begins when denuded or inflamed endothelial cells develop properties that permit platelet adhesion. At the site of endothelial dysfunction, activation of adherent platelet results in the release of inflammatory and mitogenic factors. After a series of dynamic and repetitive processes including amplified platelet activation, monocyte chemotaxis, adhesion, transmigration, and lipoprotein retention, plaque formation occurs.5 Consequently, the rupture or erosion of an atherosclerotic plaque produces a higher degree of platelet adhesion, activation, and aggregation, causing the fibrotic organization of a mural thrombus.3
The number of persons with multiple, concomitant cardiovascular disease (CAD), cerebrovascular disease (CVD), and peripheral arterial disease (PAD) accentuates the polyvascular nature of atherothrombosis (Fig. 1). The international Reduction of Atherothrombosis for Continued Health (REACH) Registry demonstrated that 1‐year incidence rates of major cardiovascular events (eg, MI, stroke, death) were high in patients with an established atherothrombotic disease and increased with the number of concomitant vascular diseases.6 These data infer that the burden on the vascular system is considered extensive on diagnosis of a single atherothrombotic disease. Thus, aggressive therapies are needed to reduce the risk of recurrent or other cardiovascular events. The management of risk factors for atherothrombosis such as hypercholesterolemia, dyslipidemia, hypertension, and diabetes mellitus fall under specific disease‐specific guidelines for patients presenting with atherothrombotic diseases.712
ANTIPLATELET THERAPIES
Antiplatelet therapies are used for the acute and long‐term treatment of patients after a thrombic event. Antiplatelet agents target the molecular mechanisms responsible for platelet activation and aggregation, such as the synthesis of thromboxane A2. On platelet activation, free arachidonic acid is converted to prostaglandin H2 (PGH2) by cyclooxygenase‐1 (COX‐1; Fig. 2). Further metabolism of PGH2 by thromboxane synthase produces thromboxane A2, which induces vasoconstriction (Fig. 2). Fortunately, the ability of platelets to produce COX‐1 is limited, and irreversible inhibition of this enzyme can impair thromboxane A2 synthesis for approximately 10 days.
Aspirin is a potent COX‐1 inhibitor, whose effects are evident 1 hour after dosing (Fig. 2).4, 13 Aspirin effectively prevents fatal and nonfatal vascular events in healthy individuals and in patients who present with acute MI or ischemic stroke.13 Unfortunately, a proportion of patients are aspirin resistant. Recent studies have indicated that interactions with the nonsteroidal anti‐inflammatory drug (NSAID) ibuprofen may diminish the primary and secondary protective effects of aspirin and may contribute to aspirin resistance, although the origin of this remains unclear.
The results of a post hoc subgroup analysis of 22,071 apparently healthy male physicians randomized to take aspirin or placebo for 5 years indicated that individuals who used NSAIDs for at least 60 days/year increased their risk of MI by more than 2‐fold compared with those who did not use NSAIDs.14 A second study conducted in patients following a major adverse cardiovascular event showed that the combination of aspirin plus ibuprofen increased the adjusted relative risk of cardiovascular mortality over an 8‐year period compared with aspirin alone.15 However, the effects of NSAIDS on aspirin's ability to inhibit COX‐1 are reversible and only last for the dosing interval and body clearance time of the drug.16
Adeonsine diphosphate (ADP)dependent stimulation of the P2Y12 receptor is another target for antiplatelet therapy. On its release, ADP binds to the P2Y12 receptor on platelets, resulting in activation and aggregation (Fig. 2). Ticlopidine and clopidogrel are thienopyridines that may irreversibly modify the P2Y12 receptor (Fig. 2).13 Safety concerns associated with ticlopidine use, including severe neutropenia, have limited its administration. Conversely, clopidogrel is relatively well‐tolerated and can prevent cardiovascular events in patients with CAD, ischemic stroke, and PAD. This agent is an orally administered prodrug requiring activation by hepatic cytochrome P450 enzymes.13
Aspirin and thienopyridines do not inhibit platelet aggregation induced by the binding of fibrinogen to the platelet glycoprotein (GP) IIb/IIIa receptor (Fig. 2).4, 13 However, there are 3 commonly administered GP IIb/IIIa inhibitors: abciximab, eptifibatide, and tirofiban (Fig. 2).4 Abciximab is the fab fragment of the chimeric monoclonal antibody 7E3 and irreversibly inhibits the GP IIb/IIIa receptor. By contrast, eptifibatide is a cyclic heptapeptide, tirofiban is a nonpeptide, and both agents are reversible inhibitors. These agents are administered intravenously, and boluses are reserved for the short‐term treatment of atherothrombosis in patients undergoing percutaneous coronary intervention.13
CONCLUSIONS
Atherothrombosis is a systemic disease that often affects coronary, intracranial, and peripheral arterial beds concomitantly, which increases the probability of a thrombotic event. Aggressive treatments, including acute and long‐term antiplatelet therapies, are required to reduce the risks associated with atherothrombosis. This supplement reviews the evidence‐based approaches for managing atherothrombosis. It will provide hospitalists with the knowledge needed to treat patients with PAD, stroke, and acute coronary syndrome. First, the administration of antiplatelet therapies to patients with acute coronary syndrome will be described. Then, guidelines for the management of patients with acute ischemic stroke and the use of antiplatelet therapies to reduce mortality due to primary and secondary ischemic events will be reviewed. Finally, the role of the hospitalist in the diagnosis of PAD in asymptomatic patients and in those with confirmed atherothrombosis will be discussed.
- ,,,.Trends in the leading causes of death in the United States, 1970‐2002.JAMA.2005;294:1255–1259.
- ,.The global burden of disease, 1990‐2020.Nat Med.1998:4:1241–1243.
- ,,,.The pathogenesis of coronary artery disease and the acute coronary syndromes.N Engl J Med.1992;326:242–250.
- .Antiplatelet therapy.Am J Med.1996;101:199–209.
- ,,.Platelets in inflammation and atherogenesis.J Clin Invest.2005;115:3378–3384.
- ,,, et al.One‐year cardiovascular event rates in outpatients with atherothrombosis.JAMA.2007;297:1197–1206.
- ,,, et al.ACC/AHA 2000 guidelines for management of patients with unstable angina and non‐ST‐segment elevation myocardial infarction.J Am Coll Cardiol.2000;36:970–1062.
- ,,, et al.ACC/AHA guidelines for the management of patients with ST‐elevation myocardial infarction.Circulation.2004;110:82–292.
- ,,, et al.Guidelines for the prevention of stroke in patients with ischemic stroke or transient ischemic attack. A statement for healthcare professionals from the American Heart Association/American Stroke Association Council on Stroke.Stroke.2006;37:557–617.
- ,,, et al.ACC/AHA 2005 practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines.Circulation.2006;113:463–654.
- ,,.Inflammation and atherosclerosis.Circulation.2002;105:1135–1143.
- ,,, et al.AHA/ACC guidelines for secondary prevention for patients with coronary and other atherosclerotic vascular disease: 2006 update.J Am Coll Cardiol.2006;47:2130–2139.
- ,,, et al.Platelet‐active drugs: the relationships among dose, effectiveness, and side effects.Chest.2001;119:39–63.
- ,,, et al.Inhibition of clinical benefits of aspirin on first myocardial infarction by nonsteroidal anti‐inflammatory drugs.Circulation.2003;108:1191–1195.
- ,.The effect of ibuprofen on cardioprotective effects of aspirin.Lancet.2003;361:573–574.
- ,,, et al.Cyclooxygenase inhibitors and the antiplatelet effects of aspirin.N Engl J Med.2001;345:1809–1817.
- ,,,.Trends in the leading causes of death in the United States, 1970‐2002.JAMA.2005;294:1255–1259.
- ,.The global burden of disease, 1990‐2020.Nat Med.1998:4:1241–1243.
- ,,,.The pathogenesis of coronary artery disease and the acute coronary syndromes.N Engl J Med.1992;326:242–250.
- .Antiplatelet therapy.Am J Med.1996;101:199–209.
- ,,.Platelets in inflammation and atherogenesis.J Clin Invest.2005;115:3378–3384.
- ,,, et al.One‐year cardiovascular event rates in outpatients with atherothrombosis.JAMA.2007;297:1197–1206.
- ,,, et al.ACC/AHA 2000 guidelines for management of patients with unstable angina and non‐ST‐segment elevation myocardial infarction.J Am Coll Cardiol.2000;36:970–1062.
- ,,, et al.ACC/AHA guidelines for the management of patients with ST‐elevation myocardial infarction.Circulation.2004;110:82–292.
- ,,, et al.Guidelines for the prevention of stroke in patients with ischemic stroke or transient ischemic attack. A statement for healthcare professionals from the American Heart Association/American Stroke Association Council on Stroke.Stroke.2006;37:557–617.
- ,,, et al.ACC/AHA 2005 practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines.Circulation.2006;113:463–654.
- ,,.Inflammation and atherosclerosis.Circulation.2002;105:1135–1143.
- ,,, et al.AHA/ACC guidelines for secondary prevention for patients with coronary and other atherosclerotic vascular disease: 2006 update.J Am Coll Cardiol.2006;47:2130–2139.
- ,,, et al.Platelet‐active drugs: the relationships among dose, effectiveness, and side effects.Chest.2001;119:39–63.
- ,,, et al.Inhibition of clinical benefits of aspirin on first myocardial infarction by nonsteroidal anti‐inflammatory drugs.Circulation.2003;108:1191–1195.
- ,.The effect of ibuprofen on cardioprotective effects of aspirin.Lancet.2003;361:573–574.
- ,,, et al.Cyclooxygenase inhibitors and the antiplatelet effects of aspirin.N Engl J Med.2001;345:1809–1817.