User login
A 59‐year‐old man with a history of hypertension was admitted with 6 months of shortness of breath, night sweats, and debilitating fatigue. His symptoms were initially mild and would persist for weeks at a time, after which he would feel better for several days. Over the 2 weeks prior to admission his symptoms had progressed, and he had become dyspneic with minimal exertion.
Progressive dyspnea has a broad differential that includes diseases of the heart (eg, congestive heart failure, aortic stenosis, constrictive pericarditis), lung (eg, chronic obstructive pulmonary disease, interstitial lung disease, pulmonary hypertension, pleural effusion), and blood (eg, anemia).
Night sweats suggest an inflammatory condition, but do not help prioritize infection, malignancy, or autoimmunity. Any of those conditions can be relapsing and remitting, at least in their early phases, but the return to normalcy raises the possibility of hypersensitivity pneumonitis from a periodic exposure.
The 6‐month duration makes typical bacterial and viral infections less likely and suggests indolent infections such as mycobacteria, fungi, or human immunodeficiency virus. Lymphoma or chronic leukemia could cause dyspnea through pleural or pulmonary involvement or from anemia. Autoimmune conditions such as systemic lupus erythematosus or adult Still's disease could also present with this course.
On admission, he described progressive orthopnea, lower extremity edema, and a 15‐lb weight gain. He denied chest pain or palpitations. His symptoms did not correlate with environmental or occupational exposures. He had been diagnosed with essential hypertension a few years earlier but was not taking any medications. He worked as an editor for a newspaper and had traveled throughout California. He never used tobacco and drank alcohol in moderation. He previously smoked marijuana. His father died of Alzheimer's disease, and his mother and 2 siblings were healthy.
Orthopnea, lower extremity edema, and weight gain suggest volume overload, which can result from heart failure, cirrhosis, renal failure, or nephrotic syndrome. The untreated hypertension is a principal risk factor for heart failure. Subacute bacterial endocarditis is an important consideration in a patient with suspected heart failure and night sweats. Travel through the central valley of California may have exposed him to coccidiodomycosis, which can cause chronic pulmonary and extrapulmonary infection.
Physical examination revealed a chronically ill‐appearing man in mild respiratory distress. His temperature was 37.2C, heart rate was 83 bpm, and blood pressure was 168/81 mm Hg. His oxygen saturation was 97% with a respiratory rate of 17 while breathing ambient air. Bilateral chemosis was present. He had crackles at the lung bases. There was a 2/6 systolic murmur loudest at the left lower sternal border with apical radiation. His jugular venous pressure was 2 cm above the sternal angle at 45. He had mild pitting edema of both lower extremities. His abdomen was soft and nondistended. He demonstrated full range of motion of all extremities and had no rashes. He was alert and oriented to person, place, and time. There were no cranial nerve deficits. His strength, sensation and coordination were intact, and he had a normal gait.
Chemosis (conjunctival edema) usually represents conjunctival irritation from an allergic, infectious, or toxic process. It can also be seen in cases of increased ophthalmic venous pressure such as hyperthyroid ophthalmopathy, superior vena cava syndrome, or carotid‐cavernous sinus fistula. The crackles, weight gain, borderline jugular venous distention, and edema suggest some systemic volume overload, but not enough to produce chemosis.
The location and timing of the murmur suggests regurgitation through the mitral or tricuspid valve, a ventricular septal defect, or hypertrophic cardiomyopathy. Tricuspid regurgitation may indicate pulmonary hypertension with right ventricular failure. Despite the absence of fever, subacute bacterial endocarditis remains a concern.
Laboratory evaluation revealed a white blood cell count of 9600/L, hemoglobin of 8.7 g/dL, and platelet count of 522,000/L. Mean corpuscular volume was 88 fL. Serum chemistries were normal; serum creatinine was 1.2 mg/dL. Serum albumin was 2.6 g/dL. A urinalysis was normal. An electrocardiogram demonstrated normal sinus rhythm and left ventricular hypertrophy (LVH). A chest x‐ray revealed interstitial edema and small bilateral pleural effusions. A transthoracic echocardiogram demonstrated normal left ventricular systolic function, an ejection fraction of 65%, mild LVH, and mild diastolic dysfunction. Mild mitral regurgitation, a mildly dilated left atrium, and a minimal pericardial effusion were also noted. A renal ultrasound revealed an atrophic left kidney without arterial flow. He was treated with diuretics for presumed heart failure related to diastolic dysfunction. His dyspnea partially improved, and he was discharged.
Heart failure with preserved ejection fraction may be contributing to his dyspnea but is unlikely to be entirely explanatory given the laboratory abnormalities. The absence of valvular vegetations on transthoracic echocardiogram lowers the probability of bacterial endocarditis. The interstitial pulmonary markings may represent pulmonary edema but alternatively could reflect interstitial lung disease, lymphangitic spread of cancer, infection (eg, Pneumocystis jiroveci), or diffuse alveolar hemorrhage.
Anemia may also be contributing to his dyspnea. There is no evidence of bleeding on history, examination, or imaging. Hemolysis is unlikely given the absence of jaundice, splenomegaly, or a known predisposing condition. The normocytic anemia may also arise from chronic inflammation. Severe anemia can cause high output heart failure, but usually the hemoglobin level is much lower and the echocardiogram would have suggestive findings. Thrombocytosis suggests inflammation, a primary myeloproliferative disorder, or severe iron deficiency (not suspected here). His hypoalbuminemia is further evidence of chronic inflammation especially in the absence of nephropathy, hepatopathy, or a protein‐losing enteropathy.
An atrophic kidney may be congenital or result from long‐standing unilateral renal ischemia, infection, or obstruction. Diminished arterial flow in a middle‐aged man with hypertension may simply reflect atherosclerotic renal artery stenosis, but mass effect within the left renal artery from thrombus, infection, or cancer cannot be ruled out.
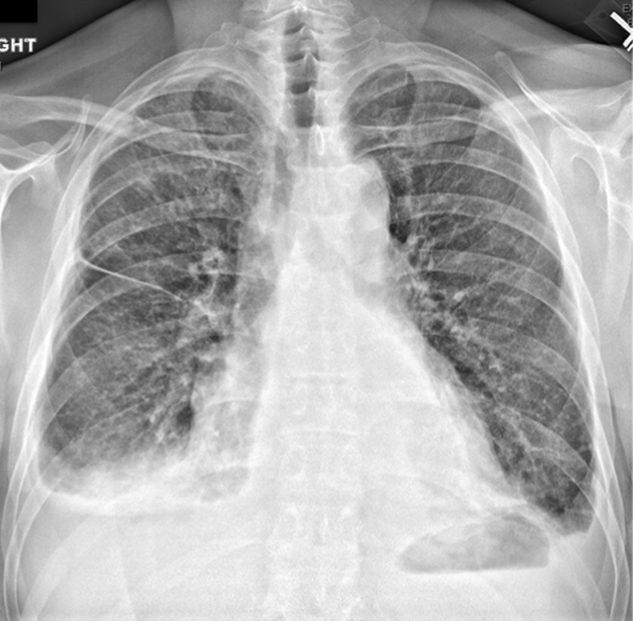
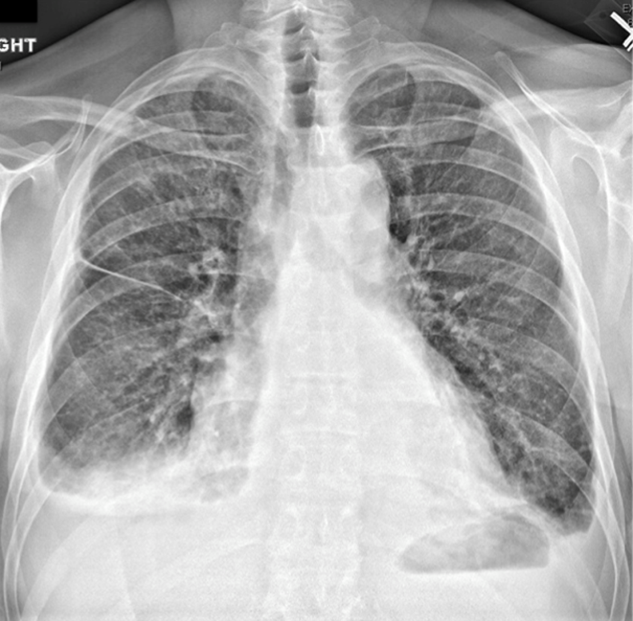
Four weeks later he was readmitted for progressive dyspnea and persistent night sweats. He was afebrile, fatigued, and in marked respiratory distress. The remainder of his physical examination was unchanged. Laboratory evaluation revealed a white blood cell count of 20,000/L with neutrophilic predominance, hemoglobin of 11 g/dL, and platelet count of 614,000/L. Creatinine was 1.4 mg/dL. Erythrocyte sedimentation rate (ESR) was greater than 100 mm/h, and C‐reactive protein (CRP) was 44 mg/L. Blood cultures were negative. Chest x‐ray (Figure 1) revealed persistent interstitial edema and increased bilateral pleural effusions.
Although clinical and radiologic features continue to suggest heart failure, the marked respiratory distress and persistent chest x‐ray abnormalities imply that a superimposed process is affecting the lungs. The night sweats, neutrophilia, and elevated ESR and CRP strongly suggest an inflammatory state from infection, malignancy, or autoimmunity.
A computed tomography (CT) scan of the lungs would help assess for interstitial lung disease, lymphangitic carcinomatosis and septic emboli. Blood cultures should be repeated to definitively exclude subacute endocarditis. A peripheral blood smear is needed to evaluate for hematologic malignancy. Finally, human immunodeficiency virus antibody testing is indicated.
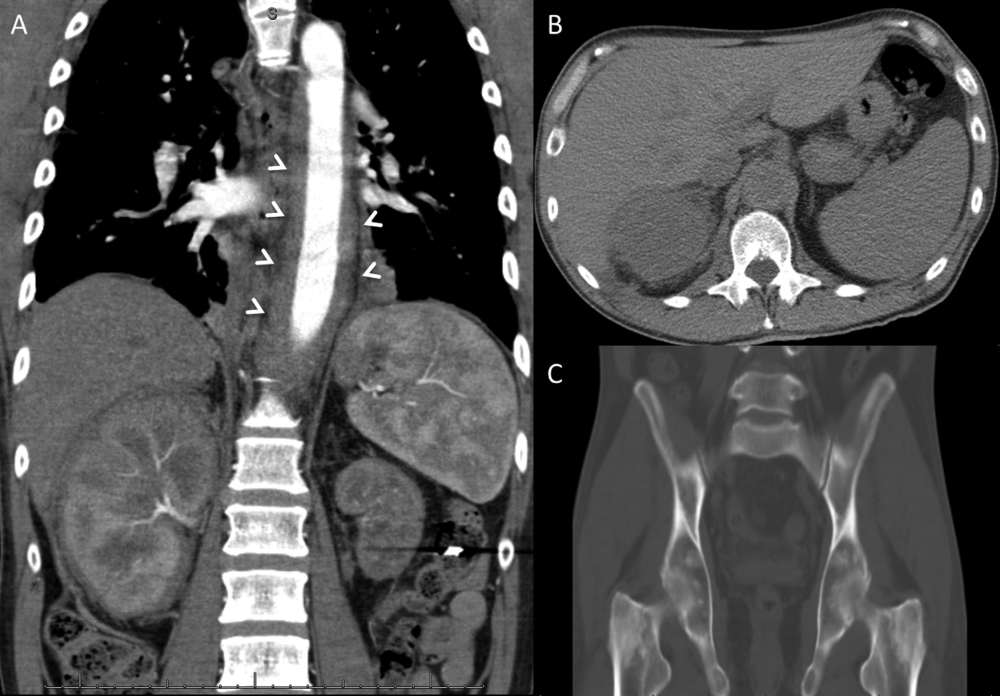
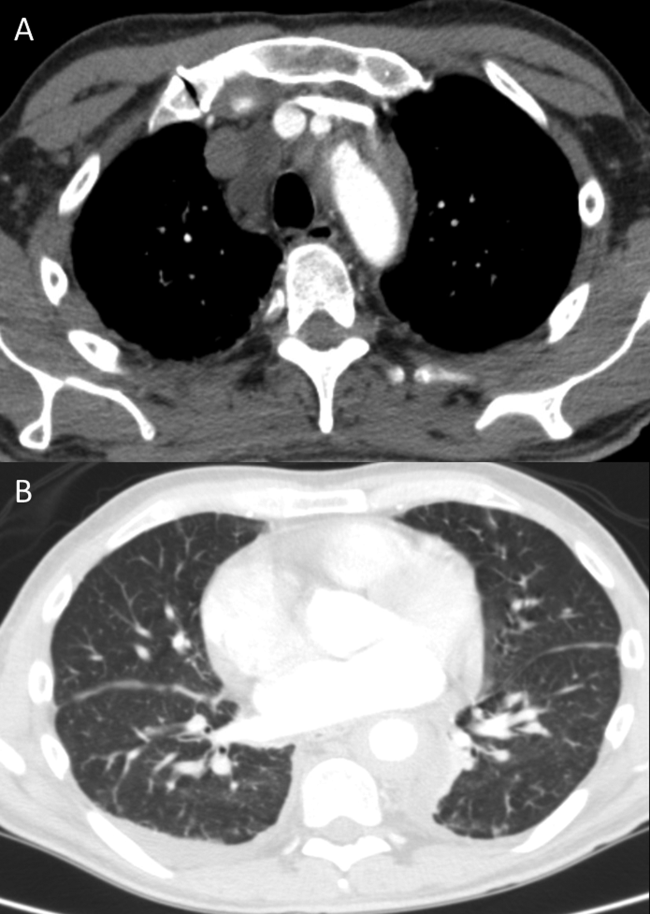
CT of the abdomen and pelvis demonstrated left renal artery stenosis, an atrophic left kidney, right kidney edema with mild perinephric stranding, and mild‐to‐moderate right hydroureter without an obstructing mass or calculus. There was mild splenomegaly and mesenteric lymphadenopathy up to 3 cm in diameter. The distal thoracic and suprarenal abdominal aorta had crescentic high‐density wall thickening. There were small sclerotic densities of the proximal femora, pelvic girdle, and thoracolumbar spine (Figure 2). Contrast chest CT demonstrated severe wall thickening of his entire thoracic aorta. There was also cardiomegaly, mild interlobular septal thickening, small bilateral pleural effusions, a 3.2‐cm right upper lobe paratracheal lymph node, and nodular pleural thickening (Figure 3).
Diffuse aortopathy is caused by inflammatory, infectious, or infiltrative processes. Large vessel vasculitides such as Behet's disease, giant cell arteritis, and Takayasu's arteritis are unlikely, as the patient lacks the associated clinical findings or epidemiology. Imaging does not reveal preexisting aortic pathology, such as an aneurysm or atherosclerotic plaque, which could predispose him to bacterial endovascular infection.
Urinary system dilation without an obvious obstruction could be explained by retroperitoneal fibrosis. Generalized lymphadenopathy, (suspected) retroperitoneal fibrosis, sclerotic bone lesions, and cardiopulmonary disease collectively suggest a widespread infiltrative process. Lymphoma may lead to lymphadenopathy and bone lesions but would not explain the aortopathy. He lacks risk factors for infections like tuberculosis or tertiary syphilis, a well‐known cause of aortopathy in the past.
Widespread multisystem involvement invites consideration of nonmalignant, noninfectious infiltrative disorders such as immunoglobulin G4‐related disease (IgG4‐RD), histiocytoses such as Erdheim‐Chester disease (ECD), systemic mastocytosis (SM), and sarcoidosis. ECD is a disorder of non‐Langerhans histiocytes that infiltrate the aorta, bones, retroperitoneum, lungs, myocardium, and periorbital structures. Perinephric stranding is sometimes seen in this condition. The lymphoplasmacytes in IgG4‐RD and noncaseating granulomas of sarcoidosis infiltrate many of the same organs. Common sites infiltrated by mast cells in SM include the bone and lymph nodes. Among these diseases, ECD and IgG4‐RD more commonly manifest with aortic and retroperitoneal infiltration and thus are prioritized on this differential diagnosis.
A positron emission tomography (PET) scan revealed abnormal fluordeoxyglucose uptake involving the thoracic aorta, right apical pleural surface, perinephric soft tissue, and various marrow spaces. Core needle biopsy of a sclerotic lesion in the right ischium demonstrated focal marrow replacement by a fibrohistiocytic process. No malignant cells or pathogenic organisms were identified. Biopsy of the right kidney revealed chronic interstitial nephritis with features of megalocytic interstitial nephritis (histiocytic inflammation) and arteriolar nephrosclerosis. A transbronchial biopsy demonstrated alveolar tissue with focal intra‐alveolar hemorrhage and organization, but no malignancy, atypia, or pathogenic organisms.
The biopsy results do not support infection, lymphoma, or carcinoma. The absence of noncaseating granulomas and mastocytes on multiple biopsies essentially rules out sarcoidosis and SM, respectively. None of the characteristic pathologic features of IgG4‐RDlymphoplasmacytic infiltrate, obliterative phlebitis, and fibrosiswere observed. The pulmonary pathology points to injury, but not the underlying cause. The bone and kidney tissue samples reveal histiocytic infiltration.
The abnormalities of the aorta, bone, lung, kidney, and retroperitoneum can be explained by the diffuse histiocytic involvement seen in ECD. The perinephric stranding detected on CT and perinephric inflammation on the PET scan may reflect the hairy kidney of ECD, which is a result of histiocytic infiltration. It is possible that the chemosis relates to exophthalmos from histiocytic orbital infiltrates. Sensitivity for detecting orbital pathology on a PET scan is limited because of the high signal from the adjacent brain.
ECD should be distinguished from Langerhans cell histiocytosis (LCH) by immunohistologic staining and microscopic characteristics of the histiocytes. LCH usually does not involve the aorta, and it more commonly involves the skin.
Serum IgG4 was within normal limits, and immunohistochemical staining of pathology specimens for IgG4 was negative. The BRAF V600E mutation, which is present in the majority of patients with ECD, was detected in a subsequent right perinephric biopsy specimen. The patient was diagnosed with ECD.
Prednisone and pegylated interferon‐ led to a rapid improvement in his symptoms. As the prednisone was tapered, he developed bilateral periorbital swelling. Magnetic resonance imaging (MRI) revealed well‐circumscribed, intraorbital soft tissue masses with partial encasement of his optic nerves and superior ophthalmic veins, as well as infiltration of his transverse sinuses, consistent with intracranial manifestations of ECD. There was no evidence of pituitary, hypothalamic, or other brain parenchymal infiltration. His dyspnea, night sweats, and hypertension improved; however, 3 months into therapy he developed an extensive rash. Interferon was discontinued. Vemurafinib, a serine kinase inhibitor that targets the BRAF mutation, was prescribed with subsequent resolution of the rash.
COMMENTARY
This patient suffered from a chronic, progressive, inflammatory illness. Although the disease initially appeared to be confined to the heart and lungs, laboratory testing signaled a more systemic condition, and subsequent imaging demonstrated involvement of a disparate group of organs. Subacute disease processes with elevated markers of inflammation and diffuse organ involvement often fall into 1 of 3 categories: infectious, autoimmune, or neoplastic. The histiocytoses inhabit a fourth and easily overlooked category that can be described as infiltrative. Infiltrative diseases are a heterogeneous group of conditions that cause illness when cells or substances not normally found in tissues lead to organ dysfunction.
Although traditional teaching has focused on sarcoidosis, amyloidosis, and hemochromatosis as the primary representatives of this category, the medical literature describes a number of other infiltrative disease processes. IgG4‐RD is a fibroinflammatory disorder characterized by space‐occupying lesions, a lymphoplasmacytic infiltrate of IgG4‐positive plasma cells, and storiform (matted and irregularly whorled microscopic pattern) fibrosis.[1] IgG4‐RD, like sarcoidosis, blurs the categorical line between infiltrative and autoimmune diseases. Other infiltrative cellular disorders, such as histiocytosis and mastocytosis, exist on a spectrum between monoclonal proliferation and neoplastic invasion.
The histiocytoses represent a diverse group of disorders with an evolving nomenclature, characterized by localized or diffuse infiltration of macrophages, monocytes, and dendritic cells (Table 1). ECD is a rare, non‐Langerhans histiocytosis characterized by excessive recruitment and activation of histiocytes through kinase signaling pathways.[2, 3] Immunohistochemical staining for CD68, CD163, and Factor XIIIa, with lack of staining for CD1a, S100, and CD207, supports the diagnosis.[3] Mutations in the BRAF V600E gene (a protein kinase involved in cell proliferation) represent the most likely etiology of this overactivation. An estimated 38% to 100% of patients with ECD harbor this mutation, with detection rates influenced by the sensitivity of testing techniques.[3] The serine kinase inhibitor vemurafinib targets this mutation, and early experience with this agent in ECD demonstrates encouraging results.[4]
| Dendritic cell disorders |
| Langerhans cell histiocytosis |
| Secondary dendritic cell processes |
| Juvenile xanthogranuloma and related disorders (including Erdheim‐Chester disease) |
| Solitary histiocytomas with a dendritic phenotype |
| Macrophage‐related disorders |
| Primary hemophagocytic lymphohistiocytosis (familial and sporadic) |
| Secondary hemophagocytic syndromes |
| Sinus histiocytosis with massive lymphadenopathy (Rosai‐Dorfman disease) |
| Solitary histiocytoma with a macrophage phenotype |
| Malignant histiocytic disorders |
| Monocyte‐related leukemias |
| Extramedullary monocytic tumor or sarcoma |
| Dendritic cell‐related histiocytic sarcoma |
| Macrophage‐related histiocytic sarcoma |
ECD presents heterogeneously, occurring most commonly between the ages of 40 and 70 years. Nonspecific symptoms include weakness, fatigue, fever, chills, weight loss, and night sweats. Typical sites of involvement include the bone, central nervous system, cardiovascular system, lungs, and retroperitoneum. Bone involvement is nearly universal, and bone pain is the most common presenting symptom. Symmetric diaphyseal and metaphyseal osteosclerotic lesions may be seen on x‐rays, bone scan, PET, CT, and MRI.[3] Approximately 50% of patients have extraskeletal involvement at diagnosis.[5] Neurologic manifestations may result from invasion of histiocytes into the facial bones, orbits, meninges, and intracranial vessels, as eventually developed in this patient. Diabetes insipidus is the most common neurologic manifestation of ECD, followed by exophthalmos, cerebellar ataxia, panhypopituitarism, and papilledema.[6, 7] Approximately 75% of patients eventually suffer from cardiovascular disease, including hypertension, congestive heart failure, acute myocardial infarction, valvular dysfunction, pericardial infiltration, and cardiac tamponade.[8] Vascular involvement includes perivascular infiltration and periaortic fibrosis, resulting in the coated aorta seen in 20% of patients with ECD.[3] Pulmonary manifestations of ECD include interstitial, pleural, and consolidative lung disease. A review of high‐resolution chest CTs of patients with ECD demonstrated that greater than half had evidence of parenchymal lung disease, with interlobular septal thickening being the most common finding.[9] Infiltration and fibrosis of retroperitoneal structures is common. Infiltration of perinephric fat creates irregular renal borders, appearing radiographically as hairy kidneys.
Arriving at the diagnosis in this case proved to be challenging because the early presentation was consistent with congestive heart failure. As the patient's conditioned deteriorated, imaging suggested multisystem involvement. It was the extensive aortopathy in particularnot the less specific bone, kidney, lymph node, or pulmonary findingsthat allowed the clinicians to hone the extensive differential diagnosis. The coated aorta is a finding that has been strongly associated with ECD; few other conditions coat the aorta in a similar fashion.[10] In most mysteries, the perpetrator's coat conceals his identity; however, in this story the coat gave it away.
KEY LEARNING POINTS
- Subacute, inflammatory, multiorgan disease is usually explained by 3 categoriesinfection, autoimmunity, and neoplasiabut a fourth category, infiltrative disorders, sometimes warrants consideration.
- ECD presents heterogeneously, ranging from localized disease to widespread organ infiltration. The classic presentation includes bone pain, diabetes insipidus, and exophthalmos.
- Characteristic radiological findings that suggest ECD include long bone osteosclerosis, a coated aorta from periaortic infiltration, and hairy kidneys from perinephric infiltration.
Disclosure
Nothing to report
- , , IgG4‐related disease. N Engl J Med. 2012;366(6):539–551.
- , , , Diagnosing Erdheim‐Chester disease. Ann Rheum Dis. 2013;72(7):e19.
- , , , et al. Consensus guidelines for the diagnosis and clinical management of Erdheim‐Chester disease. Blood. 2014;124(4):483–492.
- , , , et al. The efficacy of vemurafenib in Erdheim‐Chester Disease and Langerhans Cell Histiocytosis: preliminary results from VE‐Basket Study. Blood. 2014;124(21):635.
- , , , et al. Erdheim‐Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore). 1996;75(3):157–169.
- , , , et al. Neurological manifestations and neuroradiological presentation of Erdheim‐Chester disease: report of 6 cases and systematic review of the literature. J Neurol. 2006;253(10):1267–1277.
- , , , et al. Cerebral, facial, and orbital involvement in Erdheim‐Chester disease: CT and MR imaging findings. Radiology. 2010;255(2):586–594.
- , , , et al. Images in cardiovascular medicine. Cardiac involvement in Erdheim‐Chester disease: magnetic resonance and computed tomographic scan imaging in a monocentric series of 37 patients. Circulation. 2009;119(25):e597–e598.
- , , , et al. Pulmonary involvement in Erdheim‐Chester disease: a single‐center study of thirty‐four patients and a review of the literature. Arthritis Rheum. 2010;62(11):3504–3512.
- , , , et al. “Coated aorta”: a new sign of Erdheim‐Chester disease. J Rheumatol. 2000;27(6):1550–1553.
A 59‐year‐old man with a history of hypertension was admitted with 6 months of shortness of breath, night sweats, and debilitating fatigue. His symptoms were initially mild and would persist for weeks at a time, after which he would feel better for several days. Over the 2 weeks prior to admission his symptoms had progressed, and he had become dyspneic with minimal exertion.
Progressive dyspnea has a broad differential that includes diseases of the heart (eg, congestive heart failure, aortic stenosis, constrictive pericarditis), lung (eg, chronic obstructive pulmonary disease, interstitial lung disease, pulmonary hypertension, pleural effusion), and blood (eg, anemia).
Night sweats suggest an inflammatory condition, but do not help prioritize infection, malignancy, or autoimmunity. Any of those conditions can be relapsing and remitting, at least in their early phases, but the return to normalcy raises the possibility of hypersensitivity pneumonitis from a periodic exposure.
The 6‐month duration makes typical bacterial and viral infections less likely and suggests indolent infections such as mycobacteria, fungi, or human immunodeficiency virus. Lymphoma or chronic leukemia could cause dyspnea through pleural or pulmonary involvement or from anemia. Autoimmune conditions such as systemic lupus erythematosus or adult Still's disease could also present with this course.
On admission, he described progressive orthopnea, lower extremity edema, and a 15‐lb weight gain. He denied chest pain or palpitations. His symptoms did not correlate with environmental or occupational exposures. He had been diagnosed with essential hypertension a few years earlier but was not taking any medications. He worked as an editor for a newspaper and had traveled throughout California. He never used tobacco and drank alcohol in moderation. He previously smoked marijuana. His father died of Alzheimer's disease, and his mother and 2 siblings were healthy.
Orthopnea, lower extremity edema, and weight gain suggest volume overload, which can result from heart failure, cirrhosis, renal failure, or nephrotic syndrome. The untreated hypertension is a principal risk factor for heart failure. Subacute bacterial endocarditis is an important consideration in a patient with suspected heart failure and night sweats. Travel through the central valley of California may have exposed him to coccidiodomycosis, which can cause chronic pulmonary and extrapulmonary infection.
Physical examination revealed a chronically ill‐appearing man in mild respiratory distress. His temperature was 37.2C, heart rate was 83 bpm, and blood pressure was 168/81 mm Hg. His oxygen saturation was 97% with a respiratory rate of 17 while breathing ambient air. Bilateral chemosis was present. He had crackles at the lung bases. There was a 2/6 systolic murmur loudest at the left lower sternal border with apical radiation. His jugular venous pressure was 2 cm above the sternal angle at 45. He had mild pitting edema of both lower extremities. His abdomen was soft and nondistended. He demonstrated full range of motion of all extremities and had no rashes. He was alert and oriented to person, place, and time. There were no cranial nerve deficits. His strength, sensation and coordination were intact, and he had a normal gait.
Chemosis (conjunctival edema) usually represents conjunctival irritation from an allergic, infectious, or toxic process. It can also be seen in cases of increased ophthalmic venous pressure such as hyperthyroid ophthalmopathy, superior vena cava syndrome, or carotid‐cavernous sinus fistula. The crackles, weight gain, borderline jugular venous distention, and edema suggest some systemic volume overload, but not enough to produce chemosis.
The location and timing of the murmur suggests regurgitation through the mitral or tricuspid valve, a ventricular septal defect, or hypertrophic cardiomyopathy. Tricuspid regurgitation may indicate pulmonary hypertension with right ventricular failure. Despite the absence of fever, subacute bacterial endocarditis remains a concern.
Laboratory evaluation revealed a white blood cell count of 9600/L, hemoglobin of 8.7 g/dL, and platelet count of 522,000/L. Mean corpuscular volume was 88 fL. Serum chemistries were normal; serum creatinine was 1.2 mg/dL. Serum albumin was 2.6 g/dL. A urinalysis was normal. An electrocardiogram demonstrated normal sinus rhythm and left ventricular hypertrophy (LVH). A chest x‐ray revealed interstitial edema and small bilateral pleural effusions. A transthoracic echocardiogram demonstrated normal left ventricular systolic function, an ejection fraction of 65%, mild LVH, and mild diastolic dysfunction. Mild mitral regurgitation, a mildly dilated left atrium, and a minimal pericardial effusion were also noted. A renal ultrasound revealed an atrophic left kidney without arterial flow. He was treated with diuretics for presumed heart failure related to diastolic dysfunction. His dyspnea partially improved, and he was discharged.
Heart failure with preserved ejection fraction may be contributing to his dyspnea but is unlikely to be entirely explanatory given the laboratory abnormalities. The absence of valvular vegetations on transthoracic echocardiogram lowers the probability of bacterial endocarditis. The interstitial pulmonary markings may represent pulmonary edema but alternatively could reflect interstitial lung disease, lymphangitic spread of cancer, infection (eg, Pneumocystis jiroveci), or diffuse alveolar hemorrhage.
Anemia may also be contributing to his dyspnea. There is no evidence of bleeding on history, examination, or imaging. Hemolysis is unlikely given the absence of jaundice, splenomegaly, or a known predisposing condition. The normocytic anemia may also arise from chronic inflammation. Severe anemia can cause high output heart failure, but usually the hemoglobin level is much lower and the echocardiogram would have suggestive findings. Thrombocytosis suggests inflammation, a primary myeloproliferative disorder, or severe iron deficiency (not suspected here). His hypoalbuminemia is further evidence of chronic inflammation especially in the absence of nephropathy, hepatopathy, or a protein‐losing enteropathy.
An atrophic kidney may be congenital or result from long‐standing unilateral renal ischemia, infection, or obstruction. Diminished arterial flow in a middle‐aged man with hypertension may simply reflect atherosclerotic renal artery stenosis, but mass effect within the left renal artery from thrombus, infection, or cancer cannot be ruled out.
Four weeks later he was readmitted for progressive dyspnea and persistent night sweats. He was afebrile, fatigued, and in marked respiratory distress. The remainder of his physical examination was unchanged. Laboratory evaluation revealed a white blood cell count of 20,000/L with neutrophilic predominance, hemoglobin of 11 g/dL, and platelet count of 614,000/L. Creatinine was 1.4 mg/dL. Erythrocyte sedimentation rate (ESR) was greater than 100 mm/h, and C‐reactive protein (CRP) was 44 mg/L. Blood cultures were negative. Chest x‐ray (Figure 1) revealed persistent interstitial edema and increased bilateral pleural effusions.
Although clinical and radiologic features continue to suggest heart failure, the marked respiratory distress and persistent chest x‐ray abnormalities imply that a superimposed process is affecting the lungs. The night sweats, neutrophilia, and elevated ESR and CRP strongly suggest an inflammatory state from infection, malignancy, or autoimmunity.
A computed tomography (CT) scan of the lungs would help assess for interstitial lung disease, lymphangitic carcinomatosis and septic emboli. Blood cultures should be repeated to definitively exclude subacute endocarditis. A peripheral blood smear is needed to evaluate for hematologic malignancy. Finally, human immunodeficiency virus antibody testing is indicated.
CT of the abdomen and pelvis demonstrated left renal artery stenosis, an atrophic left kidney, right kidney edema with mild perinephric stranding, and mild‐to‐moderate right hydroureter without an obstructing mass or calculus. There was mild splenomegaly and mesenteric lymphadenopathy up to 3 cm in diameter. The distal thoracic and suprarenal abdominal aorta had crescentic high‐density wall thickening. There were small sclerotic densities of the proximal femora, pelvic girdle, and thoracolumbar spine (Figure 2). Contrast chest CT demonstrated severe wall thickening of his entire thoracic aorta. There was also cardiomegaly, mild interlobular septal thickening, small bilateral pleural effusions, a 3.2‐cm right upper lobe paratracheal lymph node, and nodular pleural thickening (Figure 3).
Diffuse aortopathy is caused by inflammatory, infectious, or infiltrative processes. Large vessel vasculitides such as Behet's disease, giant cell arteritis, and Takayasu's arteritis are unlikely, as the patient lacks the associated clinical findings or epidemiology. Imaging does not reveal preexisting aortic pathology, such as an aneurysm or atherosclerotic plaque, which could predispose him to bacterial endovascular infection.
Urinary system dilation without an obvious obstruction could be explained by retroperitoneal fibrosis. Generalized lymphadenopathy, (suspected) retroperitoneal fibrosis, sclerotic bone lesions, and cardiopulmonary disease collectively suggest a widespread infiltrative process. Lymphoma may lead to lymphadenopathy and bone lesions but would not explain the aortopathy. He lacks risk factors for infections like tuberculosis or tertiary syphilis, a well‐known cause of aortopathy in the past.
Widespread multisystem involvement invites consideration of nonmalignant, noninfectious infiltrative disorders such as immunoglobulin G4‐related disease (IgG4‐RD), histiocytoses such as Erdheim‐Chester disease (ECD), systemic mastocytosis (SM), and sarcoidosis. ECD is a disorder of non‐Langerhans histiocytes that infiltrate the aorta, bones, retroperitoneum, lungs, myocardium, and periorbital structures. Perinephric stranding is sometimes seen in this condition. The lymphoplasmacytes in IgG4‐RD and noncaseating granulomas of sarcoidosis infiltrate many of the same organs. Common sites infiltrated by mast cells in SM include the bone and lymph nodes. Among these diseases, ECD and IgG4‐RD more commonly manifest with aortic and retroperitoneal infiltration and thus are prioritized on this differential diagnosis.
A positron emission tomography (PET) scan revealed abnormal fluordeoxyglucose uptake involving the thoracic aorta, right apical pleural surface, perinephric soft tissue, and various marrow spaces. Core needle biopsy of a sclerotic lesion in the right ischium demonstrated focal marrow replacement by a fibrohistiocytic process. No malignant cells or pathogenic organisms were identified. Biopsy of the right kidney revealed chronic interstitial nephritis with features of megalocytic interstitial nephritis (histiocytic inflammation) and arteriolar nephrosclerosis. A transbronchial biopsy demonstrated alveolar tissue with focal intra‐alveolar hemorrhage and organization, but no malignancy, atypia, or pathogenic organisms.
The biopsy results do not support infection, lymphoma, or carcinoma. The absence of noncaseating granulomas and mastocytes on multiple biopsies essentially rules out sarcoidosis and SM, respectively. None of the characteristic pathologic features of IgG4‐RDlymphoplasmacytic infiltrate, obliterative phlebitis, and fibrosiswere observed. The pulmonary pathology points to injury, but not the underlying cause. The bone and kidney tissue samples reveal histiocytic infiltration.
The abnormalities of the aorta, bone, lung, kidney, and retroperitoneum can be explained by the diffuse histiocytic involvement seen in ECD. The perinephric stranding detected on CT and perinephric inflammation on the PET scan may reflect the hairy kidney of ECD, which is a result of histiocytic infiltration. It is possible that the chemosis relates to exophthalmos from histiocytic orbital infiltrates. Sensitivity for detecting orbital pathology on a PET scan is limited because of the high signal from the adjacent brain.
ECD should be distinguished from Langerhans cell histiocytosis (LCH) by immunohistologic staining and microscopic characteristics of the histiocytes. LCH usually does not involve the aorta, and it more commonly involves the skin.
Serum IgG4 was within normal limits, and immunohistochemical staining of pathology specimens for IgG4 was negative. The BRAF V600E mutation, which is present in the majority of patients with ECD, was detected in a subsequent right perinephric biopsy specimen. The patient was diagnosed with ECD.
Prednisone and pegylated interferon‐ led to a rapid improvement in his symptoms. As the prednisone was tapered, he developed bilateral periorbital swelling. Magnetic resonance imaging (MRI) revealed well‐circumscribed, intraorbital soft tissue masses with partial encasement of his optic nerves and superior ophthalmic veins, as well as infiltration of his transverse sinuses, consistent with intracranial manifestations of ECD. There was no evidence of pituitary, hypothalamic, or other brain parenchymal infiltration. His dyspnea, night sweats, and hypertension improved; however, 3 months into therapy he developed an extensive rash. Interferon was discontinued. Vemurafinib, a serine kinase inhibitor that targets the BRAF mutation, was prescribed with subsequent resolution of the rash.
COMMENTARY
This patient suffered from a chronic, progressive, inflammatory illness. Although the disease initially appeared to be confined to the heart and lungs, laboratory testing signaled a more systemic condition, and subsequent imaging demonstrated involvement of a disparate group of organs. Subacute disease processes with elevated markers of inflammation and diffuse organ involvement often fall into 1 of 3 categories: infectious, autoimmune, or neoplastic. The histiocytoses inhabit a fourth and easily overlooked category that can be described as infiltrative. Infiltrative diseases are a heterogeneous group of conditions that cause illness when cells or substances not normally found in tissues lead to organ dysfunction.
Although traditional teaching has focused on sarcoidosis, amyloidosis, and hemochromatosis as the primary representatives of this category, the medical literature describes a number of other infiltrative disease processes. IgG4‐RD is a fibroinflammatory disorder characterized by space‐occupying lesions, a lymphoplasmacytic infiltrate of IgG4‐positive plasma cells, and storiform (matted and irregularly whorled microscopic pattern) fibrosis.[1] IgG4‐RD, like sarcoidosis, blurs the categorical line between infiltrative and autoimmune diseases. Other infiltrative cellular disorders, such as histiocytosis and mastocytosis, exist on a spectrum between monoclonal proliferation and neoplastic invasion.
The histiocytoses represent a diverse group of disorders with an evolving nomenclature, characterized by localized or diffuse infiltration of macrophages, monocytes, and dendritic cells (Table 1). ECD is a rare, non‐Langerhans histiocytosis characterized by excessive recruitment and activation of histiocytes through kinase signaling pathways.[2, 3] Immunohistochemical staining for CD68, CD163, and Factor XIIIa, with lack of staining for CD1a, S100, and CD207, supports the diagnosis.[3] Mutations in the BRAF V600E gene (a protein kinase involved in cell proliferation) represent the most likely etiology of this overactivation. An estimated 38% to 100% of patients with ECD harbor this mutation, with detection rates influenced by the sensitivity of testing techniques.[3] The serine kinase inhibitor vemurafinib targets this mutation, and early experience with this agent in ECD demonstrates encouraging results.[4]
| Dendritic cell disorders |
| Langerhans cell histiocytosis |
| Secondary dendritic cell processes |
| Juvenile xanthogranuloma and related disorders (including Erdheim‐Chester disease) |
| Solitary histiocytomas with a dendritic phenotype |
| Macrophage‐related disorders |
| Primary hemophagocytic lymphohistiocytosis (familial and sporadic) |
| Secondary hemophagocytic syndromes |
| Sinus histiocytosis with massive lymphadenopathy (Rosai‐Dorfman disease) |
| Solitary histiocytoma with a macrophage phenotype |
| Malignant histiocytic disorders |
| Monocyte‐related leukemias |
| Extramedullary monocytic tumor or sarcoma |
| Dendritic cell‐related histiocytic sarcoma |
| Macrophage‐related histiocytic sarcoma |
ECD presents heterogeneously, occurring most commonly between the ages of 40 and 70 years. Nonspecific symptoms include weakness, fatigue, fever, chills, weight loss, and night sweats. Typical sites of involvement include the bone, central nervous system, cardiovascular system, lungs, and retroperitoneum. Bone involvement is nearly universal, and bone pain is the most common presenting symptom. Symmetric diaphyseal and metaphyseal osteosclerotic lesions may be seen on x‐rays, bone scan, PET, CT, and MRI.[3] Approximately 50% of patients have extraskeletal involvement at diagnosis.[5] Neurologic manifestations may result from invasion of histiocytes into the facial bones, orbits, meninges, and intracranial vessels, as eventually developed in this patient. Diabetes insipidus is the most common neurologic manifestation of ECD, followed by exophthalmos, cerebellar ataxia, panhypopituitarism, and papilledema.[6, 7] Approximately 75% of patients eventually suffer from cardiovascular disease, including hypertension, congestive heart failure, acute myocardial infarction, valvular dysfunction, pericardial infiltration, and cardiac tamponade.[8] Vascular involvement includes perivascular infiltration and periaortic fibrosis, resulting in the coated aorta seen in 20% of patients with ECD.[3] Pulmonary manifestations of ECD include interstitial, pleural, and consolidative lung disease. A review of high‐resolution chest CTs of patients with ECD demonstrated that greater than half had evidence of parenchymal lung disease, with interlobular septal thickening being the most common finding.[9] Infiltration and fibrosis of retroperitoneal structures is common. Infiltration of perinephric fat creates irregular renal borders, appearing radiographically as hairy kidneys.
Arriving at the diagnosis in this case proved to be challenging because the early presentation was consistent with congestive heart failure. As the patient's conditioned deteriorated, imaging suggested multisystem involvement. It was the extensive aortopathy in particularnot the less specific bone, kidney, lymph node, or pulmonary findingsthat allowed the clinicians to hone the extensive differential diagnosis. The coated aorta is a finding that has been strongly associated with ECD; few other conditions coat the aorta in a similar fashion.[10] In most mysteries, the perpetrator's coat conceals his identity; however, in this story the coat gave it away.
KEY LEARNING POINTS
- Subacute, inflammatory, multiorgan disease is usually explained by 3 categoriesinfection, autoimmunity, and neoplasiabut a fourth category, infiltrative disorders, sometimes warrants consideration.
- ECD presents heterogeneously, ranging from localized disease to widespread organ infiltration. The classic presentation includes bone pain, diabetes insipidus, and exophthalmos.
- Characteristic radiological findings that suggest ECD include long bone osteosclerosis, a coated aorta from periaortic infiltration, and hairy kidneys from perinephric infiltration.
Disclosure
Nothing to report
A 59‐year‐old man with a history of hypertension was admitted with 6 months of shortness of breath, night sweats, and debilitating fatigue. His symptoms were initially mild and would persist for weeks at a time, after which he would feel better for several days. Over the 2 weeks prior to admission his symptoms had progressed, and he had become dyspneic with minimal exertion.
Progressive dyspnea has a broad differential that includes diseases of the heart (eg, congestive heart failure, aortic stenosis, constrictive pericarditis), lung (eg, chronic obstructive pulmonary disease, interstitial lung disease, pulmonary hypertension, pleural effusion), and blood (eg, anemia).
Night sweats suggest an inflammatory condition, but do not help prioritize infection, malignancy, or autoimmunity. Any of those conditions can be relapsing and remitting, at least in their early phases, but the return to normalcy raises the possibility of hypersensitivity pneumonitis from a periodic exposure.
The 6‐month duration makes typical bacterial and viral infections less likely and suggests indolent infections such as mycobacteria, fungi, or human immunodeficiency virus. Lymphoma or chronic leukemia could cause dyspnea through pleural or pulmonary involvement or from anemia. Autoimmune conditions such as systemic lupus erythematosus or adult Still's disease could also present with this course.
On admission, he described progressive orthopnea, lower extremity edema, and a 15‐lb weight gain. He denied chest pain or palpitations. His symptoms did not correlate with environmental or occupational exposures. He had been diagnosed with essential hypertension a few years earlier but was not taking any medications. He worked as an editor for a newspaper and had traveled throughout California. He never used tobacco and drank alcohol in moderation. He previously smoked marijuana. His father died of Alzheimer's disease, and his mother and 2 siblings were healthy.
Orthopnea, lower extremity edema, and weight gain suggest volume overload, which can result from heart failure, cirrhosis, renal failure, or nephrotic syndrome. The untreated hypertension is a principal risk factor for heart failure. Subacute bacterial endocarditis is an important consideration in a patient with suspected heart failure and night sweats. Travel through the central valley of California may have exposed him to coccidiodomycosis, which can cause chronic pulmonary and extrapulmonary infection.
Physical examination revealed a chronically ill‐appearing man in mild respiratory distress. His temperature was 37.2C, heart rate was 83 bpm, and blood pressure was 168/81 mm Hg. His oxygen saturation was 97% with a respiratory rate of 17 while breathing ambient air. Bilateral chemosis was present. He had crackles at the lung bases. There was a 2/6 systolic murmur loudest at the left lower sternal border with apical radiation. His jugular venous pressure was 2 cm above the sternal angle at 45. He had mild pitting edema of both lower extremities. His abdomen was soft and nondistended. He demonstrated full range of motion of all extremities and had no rashes. He was alert and oriented to person, place, and time. There were no cranial nerve deficits. His strength, sensation and coordination were intact, and he had a normal gait.
Chemosis (conjunctival edema) usually represents conjunctival irritation from an allergic, infectious, or toxic process. It can also be seen in cases of increased ophthalmic venous pressure such as hyperthyroid ophthalmopathy, superior vena cava syndrome, or carotid‐cavernous sinus fistula. The crackles, weight gain, borderline jugular venous distention, and edema suggest some systemic volume overload, but not enough to produce chemosis.
The location and timing of the murmur suggests regurgitation through the mitral or tricuspid valve, a ventricular septal defect, or hypertrophic cardiomyopathy. Tricuspid regurgitation may indicate pulmonary hypertension with right ventricular failure. Despite the absence of fever, subacute bacterial endocarditis remains a concern.
Laboratory evaluation revealed a white blood cell count of 9600/L, hemoglobin of 8.7 g/dL, and platelet count of 522,000/L. Mean corpuscular volume was 88 fL. Serum chemistries were normal; serum creatinine was 1.2 mg/dL. Serum albumin was 2.6 g/dL. A urinalysis was normal. An electrocardiogram demonstrated normal sinus rhythm and left ventricular hypertrophy (LVH). A chest x‐ray revealed interstitial edema and small bilateral pleural effusions. A transthoracic echocardiogram demonstrated normal left ventricular systolic function, an ejection fraction of 65%, mild LVH, and mild diastolic dysfunction. Mild mitral regurgitation, a mildly dilated left atrium, and a minimal pericardial effusion were also noted. A renal ultrasound revealed an atrophic left kidney without arterial flow. He was treated with diuretics for presumed heart failure related to diastolic dysfunction. His dyspnea partially improved, and he was discharged.
Heart failure with preserved ejection fraction may be contributing to his dyspnea but is unlikely to be entirely explanatory given the laboratory abnormalities. The absence of valvular vegetations on transthoracic echocardiogram lowers the probability of bacterial endocarditis. The interstitial pulmonary markings may represent pulmonary edema but alternatively could reflect interstitial lung disease, lymphangitic spread of cancer, infection (eg, Pneumocystis jiroveci), or diffuse alveolar hemorrhage.
Anemia may also be contributing to his dyspnea. There is no evidence of bleeding on history, examination, or imaging. Hemolysis is unlikely given the absence of jaundice, splenomegaly, or a known predisposing condition. The normocytic anemia may also arise from chronic inflammation. Severe anemia can cause high output heart failure, but usually the hemoglobin level is much lower and the echocardiogram would have suggestive findings. Thrombocytosis suggests inflammation, a primary myeloproliferative disorder, or severe iron deficiency (not suspected here). His hypoalbuminemia is further evidence of chronic inflammation especially in the absence of nephropathy, hepatopathy, or a protein‐losing enteropathy.
An atrophic kidney may be congenital or result from long‐standing unilateral renal ischemia, infection, or obstruction. Diminished arterial flow in a middle‐aged man with hypertension may simply reflect atherosclerotic renal artery stenosis, but mass effect within the left renal artery from thrombus, infection, or cancer cannot be ruled out.
Four weeks later he was readmitted for progressive dyspnea and persistent night sweats. He was afebrile, fatigued, and in marked respiratory distress. The remainder of his physical examination was unchanged. Laboratory evaluation revealed a white blood cell count of 20,000/L with neutrophilic predominance, hemoglobin of 11 g/dL, and platelet count of 614,000/L. Creatinine was 1.4 mg/dL. Erythrocyte sedimentation rate (ESR) was greater than 100 mm/h, and C‐reactive protein (CRP) was 44 mg/L. Blood cultures were negative. Chest x‐ray (Figure 1) revealed persistent interstitial edema and increased bilateral pleural effusions.
Although clinical and radiologic features continue to suggest heart failure, the marked respiratory distress and persistent chest x‐ray abnormalities imply that a superimposed process is affecting the lungs. The night sweats, neutrophilia, and elevated ESR and CRP strongly suggest an inflammatory state from infection, malignancy, or autoimmunity.
A computed tomography (CT) scan of the lungs would help assess for interstitial lung disease, lymphangitic carcinomatosis and septic emboli. Blood cultures should be repeated to definitively exclude subacute endocarditis. A peripheral blood smear is needed to evaluate for hematologic malignancy. Finally, human immunodeficiency virus antibody testing is indicated.
CT of the abdomen and pelvis demonstrated left renal artery stenosis, an atrophic left kidney, right kidney edema with mild perinephric stranding, and mild‐to‐moderate right hydroureter without an obstructing mass or calculus. There was mild splenomegaly and mesenteric lymphadenopathy up to 3 cm in diameter. The distal thoracic and suprarenal abdominal aorta had crescentic high‐density wall thickening. There were small sclerotic densities of the proximal femora, pelvic girdle, and thoracolumbar spine (Figure 2). Contrast chest CT demonstrated severe wall thickening of his entire thoracic aorta. There was also cardiomegaly, mild interlobular septal thickening, small bilateral pleural effusions, a 3.2‐cm right upper lobe paratracheal lymph node, and nodular pleural thickening (Figure 3).
Diffuse aortopathy is caused by inflammatory, infectious, or infiltrative processes. Large vessel vasculitides such as Behet's disease, giant cell arteritis, and Takayasu's arteritis are unlikely, as the patient lacks the associated clinical findings or epidemiology. Imaging does not reveal preexisting aortic pathology, such as an aneurysm or atherosclerotic plaque, which could predispose him to bacterial endovascular infection.
Urinary system dilation without an obvious obstruction could be explained by retroperitoneal fibrosis. Generalized lymphadenopathy, (suspected) retroperitoneal fibrosis, sclerotic bone lesions, and cardiopulmonary disease collectively suggest a widespread infiltrative process. Lymphoma may lead to lymphadenopathy and bone lesions but would not explain the aortopathy. He lacks risk factors for infections like tuberculosis or tertiary syphilis, a well‐known cause of aortopathy in the past.
Widespread multisystem involvement invites consideration of nonmalignant, noninfectious infiltrative disorders such as immunoglobulin G4‐related disease (IgG4‐RD), histiocytoses such as Erdheim‐Chester disease (ECD), systemic mastocytosis (SM), and sarcoidosis. ECD is a disorder of non‐Langerhans histiocytes that infiltrate the aorta, bones, retroperitoneum, lungs, myocardium, and periorbital structures. Perinephric stranding is sometimes seen in this condition. The lymphoplasmacytes in IgG4‐RD and noncaseating granulomas of sarcoidosis infiltrate many of the same organs. Common sites infiltrated by mast cells in SM include the bone and lymph nodes. Among these diseases, ECD and IgG4‐RD more commonly manifest with aortic and retroperitoneal infiltration and thus are prioritized on this differential diagnosis.
A positron emission tomography (PET) scan revealed abnormal fluordeoxyglucose uptake involving the thoracic aorta, right apical pleural surface, perinephric soft tissue, and various marrow spaces. Core needle biopsy of a sclerotic lesion in the right ischium demonstrated focal marrow replacement by a fibrohistiocytic process. No malignant cells or pathogenic organisms were identified. Biopsy of the right kidney revealed chronic interstitial nephritis with features of megalocytic interstitial nephritis (histiocytic inflammation) and arteriolar nephrosclerosis. A transbronchial biopsy demonstrated alveolar tissue with focal intra‐alveolar hemorrhage and organization, but no malignancy, atypia, or pathogenic organisms.
The biopsy results do not support infection, lymphoma, or carcinoma. The absence of noncaseating granulomas and mastocytes on multiple biopsies essentially rules out sarcoidosis and SM, respectively. None of the characteristic pathologic features of IgG4‐RDlymphoplasmacytic infiltrate, obliterative phlebitis, and fibrosiswere observed. The pulmonary pathology points to injury, but not the underlying cause. The bone and kidney tissue samples reveal histiocytic infiltration.
The abnormalities of the aorta, bone, lung, kidney, and retroperitoneum can be explained by the diffuse histiocytic involvement seen in ECD. The perinephric stranding detected on CT and perinephric inflammation on the PET scan may reflect the hairy kidney of ECD, which is a result of histiocytic infiltration. It is possible that the chemosis relates to exophthalmos from histiocytic orbital infiltrates. Sensitivity for detecting orbital pathology on a PET scan is limited because of the high signal from the adjacent brain.
ECD should be distinguished from Langerhans cell histiocytosis (LCH) by immunohistologic staining and microscopic characteristics of the histiocytes. LCH usually does not involve the aorta, and it more commonly involves the skin.
Serum IgG4 was within normal limits, and immunohistochemical staining of pathology specimens for IgG4 was negative. The BRAF V600E mutation, which is present in the majority of patients with ECD, was detected in a subsequent right perinephric biopsy specimen. The patient was diagnosed with ECD.
Prednisone and pegylated interferon‐ led to a rapid improvement in his symptoms. As the prednisone was tapered, he developed bilateral periorbital swelling. Magnetic resonance imaging (MRI) revealed well‐circumscribed, intraorbital soft tissue masses with partial encasement of his optic nerves and superior ophthalmic veins, as well as infiltration of his transverse sinuses, consistent with intracranial manifestations of ECD. There was no evidence of pituitary, hypothalamic, or other brain parenchymal infiltration. His dyspnea, night sweats, and hypertension improved; however, 3 months into therapy he developed an extensive rash. Interferon was discontinued. Vemurafinib, a serine kinase inhibitor that targets the BRAF mutation, was prescribed with subsequent resolution of the rash.
COMMENTARY
This patient suffered from a chronic, progressive, inflammatory illness. Although the disease initially appeared to be confined to the heart and lungs, laboratory testing signaled a more systemic condition, and subsequent imaging demonstrated involvement of a disparate group of organs. Subacute disease processes with elevated markers of inflammation and diffuse organ involvement often fall into 1 of 3 categories: infectious, autoimmune, or neoplastic. The histiocytoses inhabit a fourth and easily overlooked category that can be described as infiltrative. Infiltrative diseases are a heterogeneous group of conditions that cause illness when cells or substances not normally found in tissues lead to organ dysfunction.
Although traditional teaching has focused on sarcoidosis, amyloidosis, and hemochromatosis as the primary representatives of this category, the medical literature describes a number of other infiltrative disease processes. IgG4‐RD is a fibroinflammatory disorder characterized by space‐occupying lesions, a lymphoplasmacytic infiltrate of IgG4‐positive plasma cells, and storiform (matted and irregularly whorled microscopic pattern) fibrosis.[1] IgG4‐RD, like sarcoidosis, blurs the categorical line between infiltrative and autoimmune diseases. Other infiltrative cellular disorders, such as histiocytosis and mastocytosis, exist on a spectrum between monoclonal proliferation and neoplastic invasion.
The histiocytoses represent a diverse group of disorders with an evolving nomenclature, characterized by localized or diffuse infiltration of macrophages, monocytes, and dendritic cells (Table 1). ECD is a rare, non‐Langerhans histiocytosis characterized by excessive recruitment and activation of histiocytes through kinase signaling pathways.[2, 3] Immunohistochemical staining for CD68, CD163, and Factor XIIIa, with lack of staining for CD1a, S100, and CD207, supports the diagnosis.[3] Mutations in the BRAF V600E gene (a protein kinase involved in cell proliferation) represent the most likely etiology of this overactivation. An estimated 38% to 100% of patients with ECD harbor this mutation, with detection rates influenced by the sensitivity of testing techniques.[3] The serine kinase inhibitor vemurafinib targets this mutation, and early experience with this agent in ECD demonstrates encouraging results.[4]
| Dendritic cell disorders |
| Langerhans cell histiocytosis |
| Secondary dendritic cell processes |
| Juvenile xanthogranuloma and related disorders (including Erdheim‐Chester disease) |
| Solitary histiocytomas with a dendritic phenotype |
| Macrophage‐related disorders |
| Primary hemophagocytic lymphohistiocytosis (familial and sporadic) |
| Secondary hemophagocytic syndromes |
| Sinus histiocytosis with massive lymphadenopathy (Rosai‐Dorfman disease) |
| Solitary histiocytoma with a macrophage phenotype |
| Malignant histiocytic disorders |
| Monocyte‐related leukemias |
| Extramedullary monocytic tumor or sarcoma |
| Dendritic cell‐related histiocytic sarcoma |
| Macrophage‐related histiocytic sarcoma |
ECD presents heterogeneously, occurring most commonly between the ages of 40 and 70 years. Nonspecific symptoms include weakness, fatigue, fever, chills, weight loss, and night sweats. Typical sites of involvement include the bone, central nervous system, cardiovascular system, lungs, and retroperitoneum. Bone involvement is nearly universal, and bone pain is the most common presenting symptom. Symmetric diaphyseal and metaphyseal osteosclerotic lesions may be seen on x‐rays, bone scan, PET, CT, and MRI.[3] Approximately 50% of patients have extraskeletal involvement at diagnosis.[5] Neurologic manifestations may result from invasion of histiocytes into the facial bones, orbits, meninges, and intracranial vessels, as eventually developed in this patient. Diabetes insipidus is the most common neurologic manifestation of ECD, followed by exophthalmos, cerebellar ataxia, panhypopituitarism, and papilledema.[6, 7] Approximately 75% of patients eventually suffer from cardiovascular disease, including hypertension, congestive heart failure, acute myocardial infarction, valvular dysfunction, pericardial infiltration, and cardiac tamponade.[8] Vascular involvement includes perivascular infiltration and periaortic fibrosis, resulting in the coated aorta seen in 20% of patients with ECD.[3] Pulmonary manifestations of ECD include interstitial, pleural, and consolidative lung disease. A review of high‐resolution chest CTs of patients with ECD demonstrated that greater than half had evidence of parenchymal lung disease, with interlobular septal thickening being the most common finding.[9] Infiltration and fibrosis of retroperitoneal structures is common. Infiltration of perinephric fat creates irregular renal borders, appearing radiographically as hairy kidneys.
Arriving at the diagnosis in this case proved to be challenging because the early presentation was consistent with congestive heart failure. As the patient's conditioned deteriorated, imaging suggested multisystem involvement. It was the extensive aortopathy in particularnot the less specific bone, kidney, lymph node, or pulmonary findingsthat allowed the clinicians to hone the extensive differential diagnosis. The coated aorta is a finding that has been strongly associated with ECD; few other conditions coat the aorta in a similar fashion.[10] In most mysteries, the perpetrator's coat conceals his identity; however, in this story the coat gave it away.
KEY LEARNING POINTS
- Subacute, inflammatory, multiorgan disease is usually explained by 3 categoriesinfection, autoimmunity, and neoplasiabut a fourth category, infiltrative disorders, sometimes warrants consideration.
- ECD presents heterogeneously, ranging from localized disease to widespread organ infiltration. The classic presentation includes bone pain, diabetes insipidus, and exophthalmos.
- Characteristic radiological findings that suggest ECD include long bone osteosclerosis, a coated aorta from periaortic infiltration, and hairy kidneys from perinephric infiltration.
Disclosure
Nothing to report
- , , IgG4‐related disease. N Engl J Med. 2012;366(6):539–551.
- , , , Diagnosing Erdheim‐Chester disease. Ann Rheum Dis. 2013;72(7):e19.
- , , , et al. Consensus guidelines for the diagnosis and clinical management of Erdheim‐Chester disease. Blood. 2014;124(4):483–492.
- , , , et al. The efficacy of vemurafenib in Erdheim‐Chester Disease and Langerhans Cell Histiocytosis: preliminary results from VE‐Basket Study. Blood. 2014;124(21):635.
- , , , et al. Erdheim‐Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore). 1996;75(3):157–169.
- , , , et al. Neurological manifestations and neuroradiological presentation of Erdheim‐Chester disease: report of 6 cases and systematic review of the literature. J Neurol. 2006;253(10):1267–1277.
- , , , et al. Cerebral, facial, and orbital involvement in Erdheim‐Chester disease: CT and MR imaging findings. Radiology. 2010;255(2):586–594.
- , , , et al. Images in cardiovascular medicine. Cardiac involvement in Erdheim‐Chester disease: magnetic resonance and computed tomographic scan imaging in a monocentric series of 37 patients. Circulation. 2009;119(25):e597–e598.
- , , , et al. Pulmonary involvement in Erdheim‐Chester disease: a single‐center study of thirty‐four patients and a review of the literature. Arthritis Rheum. 2010;62(11):3504–3512.
- , , , et al. “Coated aorta”: a new sign of Erdheim‐Chester disease. J Rheumatol. 2000;27(6):1550–1553.
- , , IgG4‐related disease. N Engl J Med. 2012;366(6):539–551.
- , , , Diagnosing Erdheim‐Chester disease. Ann Rheum Dis. 2013;72(7):e19.
- , , , et al. Consensus guidelines for the diagnosis and clinical management of Erdheim‐Chester disease. Blood. 2014;124(4):483–492.
- , , , et al. The efficacy of vemurafenib in Erdheim‐Chester Disease and Langerhans Cell Histiocytosis: preliminary results from VE‐Basket Study. Blood. 2014;124(21):635.
- , , , et al. Erdheim‐Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore). 1996;75(3):157–169.
- , , , et al. Neurological manifestations and neuroradiological presentation of Erdheim‐Chester disease: report of 6 cases and systematic review of the literature. J Neurol. 2006;253(10):1267–1277.
- , , , et al. Cerebral, facial, and orbital involvement in Erdheim‐Chester disease: CT and MR imaging findings. Radiology. 2010;255(2):586–594.
- , , , et al. Images in cardiovascular medicine. Cardiac involvement in Erdheim‐Chester disease: magnetic resonance and computed tomographic scan imaging in a monocentric series of 37 patients. Circulation. 2009;119(25):e597–e598.
- , , , et al. Pulmonary involvement in Erdheim‐Chester disease: a single‐center study of thirty‐four patients and a review of the literature. Arthritis Rheum. 2010;62(11):3504–3512.
- , , , et al. “Coated aorta”: a new sign of Erdheim‐Chester disease. J Rheumatol. 2000;27(6):1550–1553.